ОБЛАСТЬ ТЕХНИКИ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к способу получения (3S,4S)-4-((R)-2-(бензилокси)тридецил)-3-гексил-2-оксетанона и используемого для этого нового промежуточного соединения.
ПРЕДЫДУЩИЙ УРОВЕНЬ ТЕХНИКИ
(3S,4S)-4-((R)-2-(бензилокси)тридецил)-3-гексил-2-оксетанон формулы I известен своей полезностью в качестве промежуточного соединения для получения тетрагидролипстатина (орлистата) (патенты США №5245056 и 5399720, и Mark A. Schwindt et al., Org. Process Research, Dec. 2007, 524).
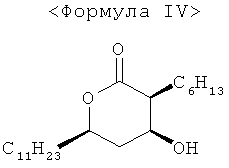
(2S,3S,5R)-3-гексил-4-гидрокси-6-ундецилтетрагидропиран-2-он формулы IV использовали в качестве основного исходного вещества при получении оптически чистого (3S,4S)-4-((R)-2-(бензилокси)тридецил)-3-гексил-2-оксетанона формулы I или его производных, имеющих легкоудаляемую алкильную защитную группу.
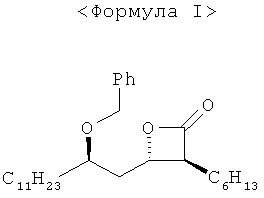
Такая методика получения, как та, что описана в патенте США №5245056, включает несколько стадий, как показано на схеме I:
<Схема I>
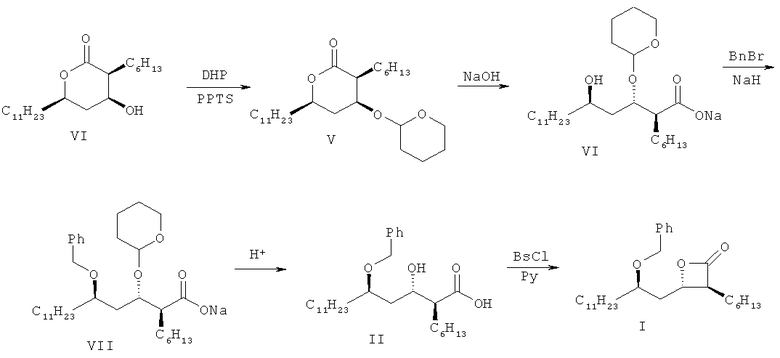
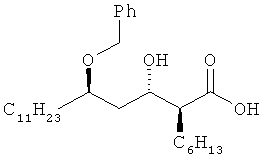
То есть соединение формулы V получают, вводя тетрагидропиранильную группу для защиты гидроксильной группы соединения формулы IV. Затем соединение формулы V гидролизуют до получения соединения формулы VI, которое обрабатывают бензилирующим реагентом до получения соединения формулы VII. Реакцию снятия защитной группы проводят путем обработки соединения формулы VII кислотой, за которой следует разделение оптических изомеров получившегося в результате соединения до получения оптически чистой (2S,3S,5R)-5-бензилокси-2-гексил-3-гидроксигексадекановой кислоты формулы II. Наконец, проводят реакцию циклизации соединения формулы II до получения (3S,4S)-4-(R)-2-(бензилокси)тридецил)-3-гексил-2-оксетанона формулы I.
Однако способ имеет трудности, связанные с использованием усложненных реакционных стадий, которые приводят к низкому общему выходу.
Что касается альтернативного способа, то корейский патент №191365 раскрывает методику получения соединения формулы II, исходя из соединения формулы IV, как показано на схеме II:
<Формула II>
<Схема II>
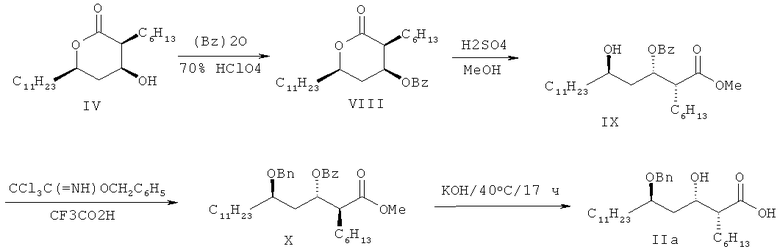
Вышеупомянутая методика включает защиту гидроксильной группы соединения формулы IV путем введения в нее бензоильной группы до получения соединения формулы VIII, гидролиз соединения формулы VIII до получения соединения формулы IX, которое подвергают реакции бензилирования до получения соединения формулы X, а соединение формулы X гидролизуют до получения соединения формулы II. Вышеописанный способ также дает низкий выход.
Однако этот способ имеет трудности, связанные с тем, что нужно проводить введение защитной группы и последующее снятие защиты с гидроксильной группы соединения формулы IV; из-за структуры соединения формулы VIII, которая содержит стерически большую бензоильную группу по соседству с гидроксильной группой в 5'-положении, возникают стерические затруднения и для реакции бензилирования требуются жесткие реакционные условия; и его общий выход составляет всего 41,2%.
Патент США №5399720 раскрывает способ получения бензиламиновой соли (2S,3S,5R)-5-бензилокси-2-гексил-гидроксигексадекановой кислоты формулы IIа путем использования соединения формулы IV в качестве исходного вещества, как показано на схеме III:
<Формула IIa>
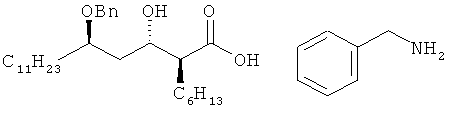
<Схема III>
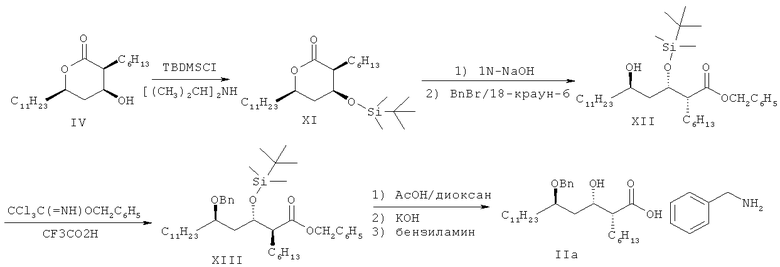
Однако этот способ также страдает от проблем низкого общего выхода (43%) из-за необходимости строгих условий бензилирования и необходимости проводить после бензилирования стадии снятия защитной группы и гидролиза.
Изобретатели настоящего изобретения стремились решить вышеупомянутые проблемы способами предыдущего уровня техники и обнаружили, что рассматриваемое соединение можно получить с высоким выходом и высокой степенью чистоты путем гидролиза соединения формулы IV без защиты его гидроксильной группы и подвергания получающегося в результате соединения селективному бензилированию и циклизации.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Соответственно, целью настоящего изобретения является обеспечение улучшенного способа получения (3S,4S)-4-((R)-2-(бензилокси)тридецил)-3-гексил-2-оксетанона формулы I, используя новое промежуточное соединение.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение обеспечивает способ получения с высоким выходом высокочистого (3S,4S)-4-((R)-2-(бензилокси)тридецил)-3-гексил-2-оксетанона формулы I, включающий
1) обработку соединения формулы IV гидроксидом металла в растворителе до получения соединения формулы III;
2) обработку соединения формулы III бензилирующим реагентом в растворителе в присутствии основания до получения соединения формулы II; и
3) подвергание соединения формулы II реакции циклизации до получения соединения формулы I:
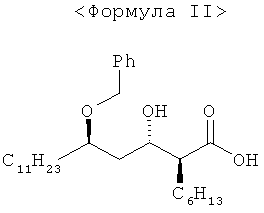
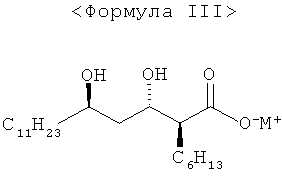
где
М представляет собой Na, K или Li.
Согласно другому аспекту настоящего изобретения обеспечивается соль металла (2S,3S,5R)-2-гексил-3,5-дигидроксигексадекановой кислоты, которая полезна в качестве промежуточного соединения для вышеупомянутого способа.
(3S,4S)-4-((R)-2-(бензилокси)тридецил)-3-гексил-2-оксетанон формулы I согласно настоящему изобретению можно получить синтетическим способом, как показано на схеме IV:
<Схема IV>
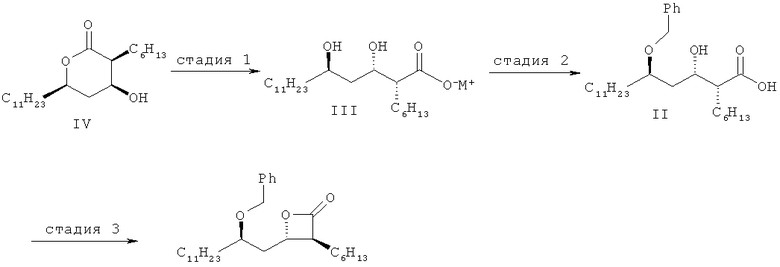
где
М представляет собой Na, K или Li,
Подробным описанием, касающимся способа по настоящему изобретению, показанного на схеме IV, является следующее.
Стадия 1
(3S,4S,6R)-3-гексил-4-гидрокси-6-ундецилтетрагидропиран-2-он формулы IV можно получить по способу, раскрытому в публикации выложенной корейской патентной заявки №2009-0044817. На стадии 1 схемы IV, соль металла (2S,3S,5R)-2-гексил-3,5-дигидроксигексадекановой кислоты (Формула III) можно получить путем гидролиза соединения формулы IV гидроксидом металла, таким как гидроксид натрия, гидроксид калия и гидроксид лития, в растворителе.
Растворитель, как использовано здесь, может представлять собой апротонный растворитель, выбираемый из группы, состоящей из простого диэтилового эфира, простого метил-трет-бутилового эфира, тетрагидрофурана, 1,4-диоксана, 1,2-диметоксиэтана, ацетонитрила, метилацетата, этилацетата, толуола и их смеси, или смеси апротонного растворителя и воды.
Гидроксид металла, такой как гидроксид натрия, гидроксид калия и гидроксид лития, можно превратить в солевую форму кальция или магния. Количество гидроксида металла, которое следует использовать, лежит в пределах от 1 до 50 моль-эквивалентов, предпочтительно от 1 до 3 моль-эквивалентов, более предпочтительно от 1,05 до 1,2 моль-эквивалентов, из расчета на соединение формулы IV.
Реакцию на стадии 1 можно проводить при температуре в пределах от 0°С до температуры кипения растворителя, предпочтительно от 30°С до 80°С.
Получаемое таким образом соединение формулы III можно использовать на стадии 2 без дополнительной очистки, и оно полезно в качестве промежуточного соединения для получения (3S,4S)-4-((R)-2-(бензилокси)тридецил)-3-гексил-2-оксетанона формулы I.
Стадия 2
На стадии 2 соединение формулы III, полученное на вышеупомянутой стадии 1, подвергают бензилированию без введения в него защитной группы, в растворителе в присутствии основания, до получения (2S,3S,5R)-5-бензилокси-2-гексил-3-гидроксигексадекановой кислоты формулы II, в которой в 5'-положение селективно вводят бензильную группу.
Растворитель, как использовано здесь, может представлять собой апротонный растворитель, выбираемый из группы, состоящей из тетрагидрофурана, простого диэтилового эфира, простого метил-трет-бутилового эфира, 1,2-диметоксиэтана, 1,4-диоксана, дихлорметана, дихлорэтана, ацетонитрила, метилацетата, этилацетата, толуола, N,N-диметилформамида, диметилсульфоксида и их смеси, или смеси апротонного растворителя и воды. Предпочтительным растворителем, как использовано здесь, является тетрагидрофуран, простой метил-трет-бутиловый эфир или 1,2-диметоксиэтан.
Между тем, основание, как использовано здесь, может представлять собой гидроксид лития, гидроксид натрия, гидроксид калия, трет-бутоксид лития, трет-бутоксид натрия или трет-бутоксид калия, и количество основания, которое следует использовать, лежит в пределах от 1 до 5 моль-эквивалентов, предпочтительно от 1 до 3 моль-эквивалентов, более предпочтительно от 1,05 до 2,0 моль-эквивалентов из расчета на соединение формулы III.
Бензилирующий реагент, как использовано здесь, для бензилирования может представлять собой необязательно замещенный бензилгалогенид, такой как бензилхлорид, бензилбромид и бензилйодид, и, принимая во внимание реакционную способность, бензилбромид является предпочтительным. Количество бензилирующего реагента, которое следует использовать, лежит в пределах от 1 до 5 моль-эквивалентов, предпочтительно от 1 до 3 моль-эквивалентов, более предпочтительно от 1,05 до 2,0 моль-эквивалентов из расчета на соединение формулы III.
Реакцию на стадии 2 можно проводить при температуре в пределах от 0°С до температуры кипения растворителя, предпочтительно от 10°С до 100°С. Для повышения скорости реакции можно использовать добавки, такие как йодид тетрабутиламмония.
Кроме того, для того чтобы легко получить (2S,3S,5R)-5-бензилокси-2-гексил-3-гидроксигексадекановую кислоту формулы II в виде кристаллов в ходе процесса очистки, к реакции можно добавить аминное основание для превращения (2S,3S,5R)-5-бензилокси-2-гексил-3-гидроксигексадекановой кислоты в ее аминную соль.
Стадия 3
На стадии 3 (3S,4S)-4-((R)-2-(бензилокси)тридецил)-3-гексил-2-оксетанон формулы I можно получить путем циклизации (2S,3S,5R)-5-бензилокси-2-гексил-3-гидроксигексадекановой кислоты формулы II с реагентом для циклизации. Вышеупомянутую 3-гидроксигексадекановую кислоту, содержащую защитную группу в 5'-положении, можно превратить в оксетанон, используя бензолсульфонилхлорид в пиридине, как описано в патентах США №4983746 и 5245056.
(3S,4S)-4-((R)-2-(бензилокси)тридецил)-3-гексил-2-оксетанон формулы I, полученный в упомянутой выше 3 стадии, можно подвергнуть дополнительному дебензилированию до получения (3S,4S)-3-гексил-4-((R)-2-гидрокситридецил)-2-оксетанона, который можно использовать для получения орлистата.
Настоящее изобретение будет описано с дополнительными подробностями со ссылкой на следующие ниже примеры. Однако следует понимать, что настоящее изобретение не ограничивается конкретными примерами.
Пример 1. Получение натрий (2S,3S,5R)-3,5-дигидрокси-2-гексилгексадеканоата (соединение формулы III) - (1)
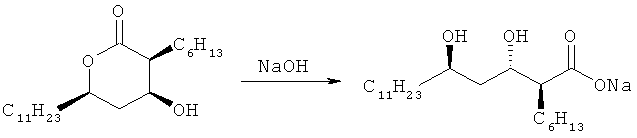
25,5 г (2S,3S,5R)-3-гексил-4-гидрокси-6-ундецилтетрагидропиран-2-она растворяли в 178,5 мл простого метил-трет-бутилового эфира, добавляли 90 мл 2 н. раствора NaOH и получающийся в результате раствор перемешивали в течение 3 часов, в то время как температуру смешанного раствора поддерживали при 50°С. Затем реакционную смесь выдерживали до тех пор, пока не происходило фазовое разделение, водный слой удаляли и оставшуюся органическую фазу дважды промывали порциями по 100 мл соленой воды. Полученный таким образом органический раствор сушили над безводным сульфатом натрия, и фильтровали, и перегоняли для удаления растворителя при пониженном давлении до получения 28,4 г указанного в заголовке соединения (выход: 100%) в виде масла.
1H-ЯМР, 300 МГц (CD3OD, м.д.): δ 0,89 (дд, 6Н, J=5,8, 1,2 Гц), 1,24-1,80 (м, 32Н), 2,10-2,49 (м, 2Н), 3,72-3,87 (м, 1Н).
Пример 2. Получение метилбензиламинной соли (2S,3S,5R)-5-бензилокси-2-гексил-3-гидроксигексадекановой кислоты (соединение формулы IIа) - (1)
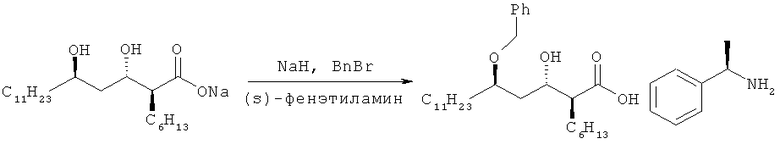
98 мл натрий (2S,3S,5R)-3,5-дигидрокси-2-гексилгексадеканоата, полученного по вышеупомянутому примеру 1, растворяли в смешанном растворителе из 98 мл тетрагидрофурана и 328 мл простого метил-трет-бутилового эфира. Реакционную смесь охлаждали до 0°С и к смешанному раствору последовательно добавляли 7,19 г гидрида натрия и 30,7 г бензилбромида. Затем температуру реакционной смеси повышали до 60°С, и реакционную смесь кипятили с обратным холодильником в течение 5 дней (стадия (а)). После охлаждения реакционной смеси до комнатной температуры ее рН доводили до 1, и раствор перемешивали при комнатной температуре в течение 3 часов. Затем рН реакционной смеси доводили до 4 и смешанный раствор выдерживали до тех пор, пока не происходило фазовое разделение. Водный слой удаляли и оставшуюся органическую фазу дважды промывали порциями по 120 мл соленой воды. Полученный вышеупомянутый органический раствор сушили над безводным сульфатом магния, фильтровали и перегоняли для удаления растворителя при пониженном давлении. Медленно при перемешивании реакционной смеси добавляли 438 мл метилацетата и 8,7 г (S)-α-метилбензиламина. После перемешивания в течение 3 часов смесь охлаждали до 5°С и дополнительно перемешивали в течение 1 часа (стадия (b)). Смесь сушили и фильтровали до получения 31,1 г указанного в заголовке соединения (выход: 74%) в виде твердого вещества белого цвета.
Температура плавления 104-106°С.
1Н-ЯМР (300 МГц, CDCl3): 0,85-0,91 (м, 6Н), 1,24 (м, 26Н), 1,54 (д, 3Н), 1,71-1,39 (м, 6Н), 2,09-2,16 (м, 1Н), 3,70-3,72 (м, 1Н), 3,85-3,90 (м, 1Н), 4,18 (кв, 1Н), 4,52 (д, 2Н), 6,10 (ушир.с, 5,0), 7,36-7,22 (м, 10Н).
Пример 3: Получение натрий (2S,3S,5R)-3,5-дигидрокси-2-гексилгексадеканоата (соединения формулы III) - (2)
300 г (2S,3S,5R)-3-гексил-4-гидрокси-6-ундецилтетрагидропиран-2-она растворяли в 1,8 л простого метил-трет-бутилового эфира. К реакционной смеси добавляли 9 л 2 н. раствора NaOH, температуру реакционной смеси медленно повышали, и водный слой отделяли и удаляли из реакционной смеси. Органический слой отделяли от остатков реакционной смеси и промывали порциями по 450 мл насыщенной соленой воды, и растворитель удаляли при пониженном давлении. К остатку добавляли 900 мл толуола и растворитель и влагу удаляли азеотропной перегонкой при пониженном давлении. К остатку добавляли 1,8 л гептана и 1,8 л простого метил-трет-бутилового эфира, и температуру реакционной смеси повышали до 40°С. Затем реакционную смесь охлаждали до комнатной температуры и добавляли 50 мл метанола. Когда начиналось образование кристаллов, реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь затем охлаждали до 10°С и перемешивали в течение 30 минут. Затем реакционную смесь фильтровали и сушили в теплом воздушном потоке до получения 534 г указанного в заголовке соединения (выход: 96%).
Дифференциальная сканирующая калориметрия (ДСК): 109,29-117,30°С.
1Н-ЯМР, 300 МГц (CD3OD, м.д.): δ 0,89 (дд, 6Н, J=5,8, 1,2 Гц), 1,24-1,80 (м, 32Н), 2,10-2,49 (м, 2Н), 3,72-3,87 (м, 1Н).
Пример 4. Получение метилбензаминной соли (2S,3S,5R)-5-бензилокси-2-гексил-3-гидроксигексадекановой кислоты (соединение формулы IIа) - (2)
300 г натрий (2S,3S,5R)-3,5-дигидрокси-2-гексилгексадеканоата, полученного по вышеупомянутому примеру 3, растворяли в смешанном растворителе из 405 мл толуола и 45 мл диметилсульфоксида. Реакционную смесь охлаждали до 5°С и добавляли 49,8 г (1,5 эквивалента) 55% гидрида натрия и перемешивали при той же температуре в течение 30 минут. Затем добавляли 325 г бензилбромида. Температуру реакционной смеси поддерживали при 15°С и реакционную смесь перемешивали в течение 18 часов. Затем проводили ту же реакцию, что описана на стадии (b) примера 2, и твердое вещество, полученное в результате вышеупомянутой реакции, фильтровали и сушили до получения 332,9 г указанного в заголовке соединения (выход: 75%) в виде твердого вещества белого цвета.
Были получены те же самые результаты анализа, что и в примере 2.
Пример 5. Получение метилбензаминной соли (2S,3S,5R)-5-бензилокси-2-гексил-3-гидроксигексадекановой кислоты (соединение формулы IIа) - (3)
100 г натрий (2S,3S,5R)-3,5-дигидрокси-2-гексилгексадеканоата, полученного по вышеупомянутому примеру 3, растворяли в 1,5 л толуола. 108,4 г бензилбромида и 48,3 г трет-бутоксида калия последовательно добавляли к реакционной смеси. Затем ее температуру медленно повышали до 60-75°С и реакционную смесь нагревали в течение 12 часов. Затем проводили ту же реакцию, которая описана на стадии (b) примера 2, и твердое вещество, полученное в результате вышеупомянутой реакции, фильтровали и сушили до получения 106 г указанного в заголовке соединения (выход: 72%) в виде твердого вещества белого цвета.
Были получены те же результаты анализа, что и в примере 2.
Пример 6. Получение (3S,4S)-4-((R)-2(бензилокси)тридецил)-3-гексил-2-оксетанона (соединение формулы I)
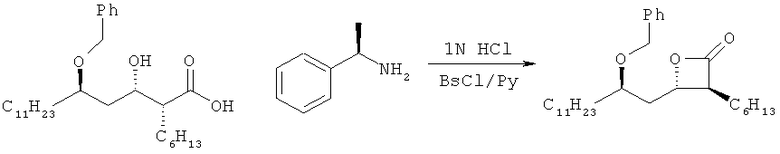
330 г метилбензиламинной соли (2S,3S,5R)-5-бензилокси-2-гексил-3-гидроксигексадекановой кислоты, полученной по вышеупомянутому примеру 4, растворяли в смешанном растворителе из 1,65 л гексана и 1,65 л 2 н. водного раствора гидрохлорида. После перемешивания в течение 2 часов водный слой удаляли и оставшуюся органическую фазу дважды промывали порциями по 1,65 л дистиллированной воды. Полученный выше органический раствор сушили над безводным сульфатом магния, фильтровали и перегоняли при пониженном давлении. Затем добавляли 2,61 л пиридина и реакционную смесь охлаждали до 0°С и перемешивали. Затем медленно, в течение 2 часов, добавляли 1,44 л бензолсульфонилхлорида и реакционную смесь перемешивали в течение дополнительных 20 часов при той же температуре. После добавления 2,61 л дистиллированной воды и 2,61 л гексана реакционную смесь сильно перемешивали. Затем реакционную смесь выдерживали до тех пор, пока не происходило фазовое разделение, и водный слой удаляли из реакционной смеси. Полученный выше органический раствор дважды промывали 2 н. гидрохлоридом, порциями по 2,61 л, и дополнительно дважды промывали дистиллированной водой, порциями по 2,61 л. Полученный выше органический раствор сушили над безводным сульфатом магния и фильтровали, и затем растворитель удаляли при пониженном давлении. Соответственно получали 270,1 г указанного в заголовке соединения (выход: 101,2%) в виде твердого вещества белого цвета.
1Н-ЯМР (300 МГц, CDCl3): 0,80-0,95 (м, 6Н), 1,15-1,85 (м, 30Н), 1,94 (т, 2Н, J=Гц), 3,15-3,26 (м, 1Н), 3,55-3,65 (м, 1Н), 4,35-4,63 (м, 3Н), 7,25-7,40 (м, 5Н).
Референсный пример 1. Получение (3S,4S)-3-гексил-4-((R)-2-гидрокситридецил)-2-оксетанона
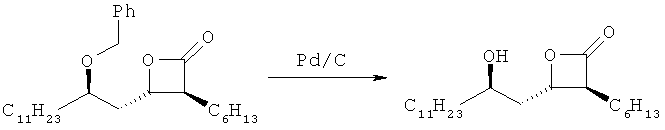
270 г (3S,4S)-4-((R)-2(бензилокси)тридецил)-3-гексил-2-оксетанона, полученного по вышеупомянутому примеру 6, растворяли в 1,89 л этил ацетата. Добавляли 19 г 5% Pd/c (палладия/на углероде), и реакционную смесь перемешивали в течение 3 часов, в то время как давление водорода составляло 6 бар. Удаляли Pd путем фильтрования через Celite® и удаляли растворитель при пониженном давлении. Добавляли 2,15 л гексана и температуру реакционной смеси повышали до 40°С для расплавления полученного выше твердого вещества. Затем реакционную смесь охлаждали до комнатной температуры, и при 20°С в реакционной смеси формировались кристаллы. После перемешивания в течение 8 часов реакционную смесь охлаждали до 5°С, перемешивали в течение 2 часов и фильтровали до получения 166,3 г указанного в заголовке соединения (выход: 83%) в виде твердого вещества белого цвета. Температура плавления 60-61°С.
1H-ЯМР (300 МГц, CDCl3): 4,54-4, 44 (м, 1Н), 3,87-3,74 (м, 1Н), 3,3-3,16 (м, 1Н), 1,95-1,12 (м, 32Н), 0,88 (т-образный, 6Н).
Референсный пример 2: Синтез орлистата
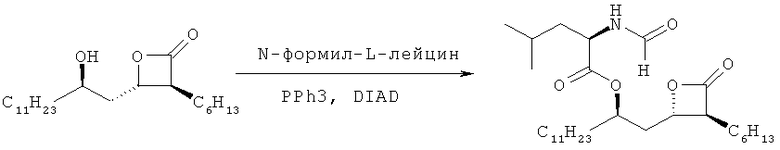
166 г (3S,4S)-3-гексил-4-((R)-2-гидрокситридецил)-2-оксетанона, полученного по референсному примеру 1, растворяли в 830 мл тетрагидрофурана. После добавления 160 г трифенилфосфина (PPh3) и 86 г N-формил-L-лейцина, реакционную смесь охлаждали до 0°С. В течение 1,5 часов непрерывно добавляли смешанный раствор, в котором 120 мл диизопропилазодикарбоксилата (DIAD) разбавляли в 332 мл тетрагидрофурана. После перемешивания в течение дополнительных 30 минут реакционную смесь перемешивали в течение 4 часов, в то время как температуру медленно повышали. После удаления растворителя добавляли 832 мл гексана и реакционную смесь перемешивали в течение 5 часов. Образовавшееся твердое вещество удаляли путем фильтрования, и гексановый слой промывали три раза 499 мл 55% смеси метанол/дистиллированная вода. Оставшийся органический слой сушили над безводным сульфатом магния и фильтровали, и затем удаляли растворитель при пониженном давлении. Затем добавляли 2,5 л гексана, и реакционную смесь охлаждали до 10°С и перемешивали в течение 1 часа с последующим добавлением затравочного кристалла орлистата. Реакционную смесь снова медленно охлаждали до 0°С и перемешивали в течение ночи. Полученное твердое вещество фильтровали, промывали 500 мл холодного гексана и сушили до получения 192,5 г указанного в заголовке соединения (выход: 83%) в виде твердого вещества белого цвета.
Температура плавления 42,4-44,5°С.
1Н-ЯМР (300 МГц, CDCl3): 8,22 (с, 1Н), 5,99 (д, 1Н), 5,06-4,98 (м, 1Н), 4,77-4,60 (м, 1Н), 4,32-4,22 (м, 1Н), 3,27-3,16 (м, 1Н), 2,28-1,98 (м, 2Н), 1,90-1,15 (м, 30Н), 1,03-0,82 (м, 12Н).
Несмотря на то что изобретение было описано в отношении вышеупомянутых специфических вариантов осуществления, следует отдавать отчет в том, что специалисты в данной области могут внести различные модификации и изменения, которые также подпадают под объем изобретения, как определено прилагаемой формулой изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ВОРИКОНАЗОЛА | 2008 |
|
RU2434009C1 |
| СПОСОБ ПОЛУЧЕНИЯ (6R)-3-ГЕКСИЛ-4-ГИДРОКСИ-6-УНДЕЦИЛ-5,6-ДИГИДРОПИРАН-2-ОНА И ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ, ПРИМЕНЯЕМОГО В ДАННОМ СПОСОБЕ | 2008 |
|
RU2434860C1 |
| ПРОИЗВОДНЫЕ БЕНЗОПИРАНА, ЗАМЕЩЕННЫЕ ВТОРИЧНЫМИ АМИНАМИ, ВКЛЮЧАЮЩИМИ В СЕБЯ ТЕТРАЗОЛ, СПОСОБ ИХ ПОЛУЧЕНИЯ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2003 |
|
RU2283312C2 |
| СПОСОБ ПОЛУЧЕНИЯ ГИДРОХЛОРИДА 1-(3,4-ДИХЛОР-2-ФТОРФЕНИЛАМИНО)-7-МЕТОКСИХИНАЗОЛИН-6-ИЛОКСИ)ПИПЕРИДИН-1-ИЛ)ПРОП-2-ЕН-1-ОНА И ИСПОЛЬЗУЕМЫХ В НЕМ ПРОМЕЖУТОЧНЫХ СОЕДИНЕНИЙ | 2012 |
|
RU2563630C1 |
| КОМБИНАЦИЯ, СОДЕРЖАЩАЯ НОВОЕ ПРОИЗВОДНОЕ 3-(4-(БЕНЗИЛОКСИ)ФЕНИЛ)ГЕКС-4-ИНОВОЙ КИСЛОТЫ И ДРУГОЙ АКТИВНЫЙ ИНГРЕДИЕНТ, ДЛЯ АКТИВИРОВАНИЯ ФЕРМЕНТА РЕЦЕПТОРА G-БЕЛКА 40 | 2015 |
|
RU2680248C1 |
| НОВОЕ АМИДНОЕ ПРОИЗВОДНОЕ ДЛЯ ИНГИБИРОВАНИЯ РОСТА РАКОВЫХ КЛЕТОК | 2008 |
|
RU2434010C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ ДЛЯ ПОЛУЧЕНИЯ АВИБАКТАМА | 2018 |
|
RU2722932C1 |
| НОВОЕ ПРОИЗВОДНОЕ 3-(4-(БЕНЗИЛОКСИ)ФЕНИЛ)ГЕКС-4-ИНОВОЙ КИСЛОТЫ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ПРОФИЛАКТИКИ И ЛЕЧЕНИЯ МЕТАБОЛИЧЕСКОГО ЗАБОЛЕВАНИЯ, ВКЛЮЧАЮЩАЯ ЕГО В КАЧЕСТВЕ ЭФФЕКТИВНОГО ИНГРЕДИЕНТА | 2014 |
|
RU2628077C2 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ ПОЗАКОНАЗОЛА И СПОСОБ ПОЛУЧЕНИЯ АМОРФНОГО ПОЗАКОНАЗОЛА | 2016 |
|
RU2750898C2 |
| СПОСОБ ПОЛУЧЕНИЯ (R)-1-АРИЛ-2-ТЕТРАЗОЛИЛЭТИЛОВОГО ЭФИРА КАРБАМИНОВОЙ КИСЛОТЫ | 2009 |
|
RU2508290C2 |
Данное изобретение относится к способу получения с высоким выходом высокочистого (3S,4S)-4-((R)-2-(бензилокси)тридецил)-3-гексил-2-оксетанона формулы I:
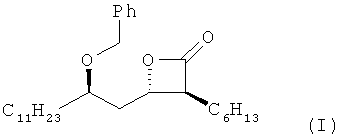
используя соль металла (2S,3S,5R)-2-гексил-3,5-дигидроксигексадекановой кислоты формулы III:
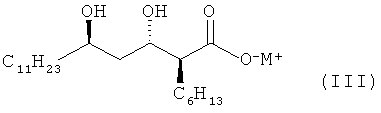
в качестве промежуточного соединения. Соединение формулы 1 является промежуточным продуктом для получения тетрагидролипстатина. Данный способ позволяет получить соединение формулы 1 с высоким выходом и высокой степенью чистоты. 4 н. и 7 з.п. ф-лы, 6 пр.
1. Способ получения (3S,4S)-4-((R)-2-(бензилокси)тридецил)-3-гексил-2-оксетанона формулы I, включающий
1) обработку соединения формулы IV гидроксидом металла в растворителе для получения соединения формулы III;
2) обработку соединения формулы III бензилирующим реагентом в растворителе в присутствии основания для получения соединения формулы II; и
3) подвергание соединения формулы II реакции циклизации для получения соединения формулы I:
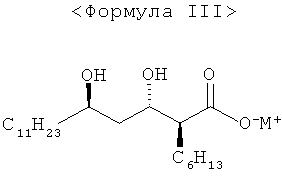
где М представляет собой Na, К или Li.
2. Способ по п.1, где растворитель, используемый на стадии 1), является апротонным растворителем, выбираемым из группы, состоящей из простого диэтилового эфира, простого метил-трет-бутилового эфира, тетрагидрофурана, 1,4-диоксана, 1,2-диметоксиэтана, ацетонитрила, метилацетата, этилацетата, толуола и их смеси, или смеси апротонного растворителя с водой.
3. Способ по п.1, где гидроксид металла на стадии 1) используют в количестве от 1 до 50 моль-эквивалентов из расчета на соединение формулы IV.
4. Способ по п.1, где растворитель, используемый на стадии 2), является апротонным растворителем, выбираемым из группы, состоящей из тетрагидрофурана, простого диэтилового эфира, простого метил-трет-бутилового эфира, 1,2-диметоксиэтана, 1,4-диоксана, дихлорметана, дихлорэтана, ацетонитрила, метилацетата, этилацетата, толуола, N,N-диметилформамида, диметилсульфоксида и их смеси, или смеси апротонного растворителя с водой.
5. Способ по п.1, где основание, используемое на стадии 2), выбирают из группы, состоящей из гидроксида лития, гидроксида натрия, гидроксида калия, трет-бутоксида лития, трет-бутоксида натрия и трет-бутоксида калия.
6. Способ по п.5, где основание используют в количестве от 1 до 5 моль-эквивалентов из расчета на соединение формулы III.
7. Способ по п.1, где бензилирующий реагент, используемый на стадии 2), является бензилгалогенидом, выбираемым из группы, состоящей из бензилхлорида, бензилбромида и бензилйодида.
8. Способ по п.1, где бензилирующий реагент на стадии 2) используют в количестве от 1 до 5 моль-эквивалентов из расчета на соединение формулы III.
9. Соединение формулы III;
где М представляет собой Na, К или Li.
10. Способ получения соединения формулы III, включающий стадию обработки (3S,4S,6R)-3-гексил-4-гидрокси-6-ундецилтетрагидропиран-2-она формулы IV гидроксидом металла в растворителе:
где М представляет собой Na, К или Li.
11. Способ получения соединения формулы II, включающий стадию обработки соли металла (2S,3S,5R)-3,5-дигидрокси-2-гексилгексадекановой кислоты формулы III бензилирующим реагентом в растворителе в присутствии основания:
где М представляет собой Na, К или Li.
| US 5245056 A, 14.09.1993 | |||
| US 5399720 A, 21.03.1995 | |||
| RU 2001121139 A, 20.07.2003 | |||
| Gil Ma et al, Amer | |||
| Chem | |||
| Soc, 2006, v.8, №20, p.4497-4500 | |||
| Arun K | |||
| et al, Organ.Letters, 2000, v.2, №16, p.2405-2407 | |||
| Christine Wedler et al, J.Org.Chem., 1999, v.64, p.5301-5303. |

