Настоящее изобретение относится к новым антимикробным пептидам и их применению в медицине.
Антимикробные пептиды, которые сокращенно обозначают также как «АМП», представляют собой часть врожденной иммунной системы, и они имеют жизненно важное значение для эпителиальной защиты от заражения микроорганизмами.
У здорового человека кожа и слизистая оболочка образуют физический барьер для заражения микроорганизмами. Физический барьер в здоровой коже состоит из рогового слоя и слизистого слоя в слизистой оболочке, в которых посредством десквамации и секреции слизи происходит непрерывное обновление поверхностей, при этом одновременно непрерывно удаляются микроорганизмы, прикрепившиеся к поверхностям. Благодаря взаимодействию с липидами, которые также присутствуют в коже, указанный физический барьер препятствует проникновению микроорганизмов в живой эпидермис.
Однако помимо указанного физического барьера необходимы также и другие факторы для успешной защиты здоровой кожи и слизистой оболочки от заражения; к указанным факторам относятся, среди прочего, эндогенные антимикробные пептиды. Так, например, лизоцим, представляющий собой антимикробный пептид, который присутствует в назальных секретах, обладает способностью уничтожать, прежде всего, грамположительные бактерии. Кроме того, в качестве антимикробных пептидов известны также дефензины, присутствующие в слизистой оболочке кишечника, присутствие которых, по-видимому, является необходимым, прежде всего, по той причине, что эпителий кишечника подвергается воздействию огромного количества бактерий. Помимо того, что она формирует слизистый слой, трудно проницаемый для микроорганизмов, слизистая оболочка кишечника содержит клетки Панета, которые секретируют человеческий дефензин-5 и, среди прочего, защищают стволовые клетки, важные для непрерывного обновления слизистой оболочки кишечника.
Другими известными АМП являются пептид, известный под названием псориазин, а также РНКаза-7, которая представляет собой эффективный эндогенный антибиотик широкого спектра действия, присутствующий в организме человека.
Помимо известных эндогенных антимикробных пептидов, в данной области известны также многочисленные антибиотики; к ним относятся как субстанции биологического происхождения, так и субстанции, созданные путем синтеза, таким образом, они представляют собой (с точки зрения происхождения) либо образовавшиеся в естественных условиях низкомолекулярные продукты метаболизма грибов или бактерий, либо синтезированные химическим путем терапевтические средства.
Принимая во внимание, прежде всего, тот факт, что вследствие развития устойчивости к природным и синтетическим антибиотикам становится все более трудно лечить вызываемые микроорганизмами инфекционные заболевания, постоянно возникает необходимость в новых обладающих антимикробной активностью средствах, которые отличаются небольшими побочными действиями и простотой их производства и манипуляций с ними.
Таким образом, в основу настоящего изобретения была положена задача разработать новую антимикробную субстанцию, которую можно применять для лечения вызываемых микроорганизмами инфекционных заболеваний.
Указанная задача решается с помощью предлагаемого в настоящем изобретении пептида, который обладает антимикробной активностью и имеет С-конец и N-конец, и который состоит как минимум из восьми и как максимум из 12 последовательно расположенных аминокислот, где пептид имеет последовательность, представленную следующей формулой I:
Ter1-X1-B1-X2-B2-X3-Z1-Z2-X4-Ter2 (формула I),
в которой
Теr1 обозначает свободную N-концевую аминогруппу N-концевой аминокислоты X1 или модифицированную N-концевую аминогруппу;
X1, Х2 и Х3 каждый являются одинаковыми или различными, и их выбирают в каждом случае независимо друг от друга из аминокислот с основными боковыми цепями, предпочтительно их выбирают из следующих аминокислот: аргинин, лизин, 6-гидроксилизин, гомоаргинин, 2,4-диаминомасляная кислота, β-гомоаргинин, D-аргинин, аргинал, 2-амино-3-гуанидинопропионовая кислота, нитроаргинин, н-метиларгинин, ε-н-метиллизин, алло-гидроксилизин, 2,3-диаминопропионовая кислота, 2,2'-диаминопимелиновая кислота, орнитин, сим-диметиларгинин, асим-диметиларгинин;
B1 и В2 являются одинаковыми или различными, и их выбирают в каждом случае независимо друг от друга из аминокислот, имеющих алифатическую или основную боковую цепь, и предпочтительно выбирают из аланина или глицина;
Z1 и Z2 либо каждый обозначает цистеин, либо они обозначают собой цистеин и аланин; и
Теr2 представляет собой свободную С-концевую карбоксильную группу С-концевой аминокислоты Х4 или представляет собой модифицированную С-концевую карбоксильную группу.
При этом предпочтительно пептид состоит из 8 аминокислот и имеет последовательность, представленную формулой I.
При этом, как уже указано, Z1 и Z2 либо каждый обозначает цистеин, либо они обозначают цистеин и аланин; т.е., если Z1 обозначает цистеин, то Z2 обозначает аланин, а если Z1 обозначает аланин, то Z2 обозначает цистеин.
Пептид предпочтительно получают путем синтеза, получают путем рекомбинации, получают путем ферментативного расщепления и/или выделяют. Поскольку пептид, предлагаемый в настоящем изобретении, представляет собой относительно короткий пептид, то предпочтительно пептид, предлагаемый в настоящем изобретении, получают путем синтеза; методы получения путем синтеза достаточно хорошо известны из существующего уровня техники и включают, прежде всего, методы жидкофазного и твердофазного химического синтеза. В качестве ссылки можно указать, например, обзорную статью/монографию S. Kent, «Chemical Synthesis of Peptides and Proteins», Annual Review of Biochemistry 57, 1988, cc. 957-989. В настоящее время в рассматриваемой области работают также многочисленные фирмы, которые занимаются коммерческим производством синтетических пептидов.
Помимо восьми аминокислот, указанных в формуле I, пептид, предлагаемый в настоящем изобретении, может иметь как на N-конце, так и на С-конце, и другие аминокислоты, которые не оказывают негативного воздействия или оказывают лишь незначительное воздействие на эффективность и стабильность пептида, предлагаемого в настоящем изобретении; из структуры указанного пептида, предлагаемого в настоящем изобретении, специалисту в данной области должно быть очевидно, какие аминокислоты или аминокислотные остатки можно дополнительно присоединять к С- или N-концу для достижения антимикробного действия, идентичного или очень близкого к тому, которым обладает пептид, состоящий из восьми аминокислот.
В проведенных при создании изобретения экспериментах было установлено, что пептид, предлагаемый в настоящем изобретении, обладает очень высокой эффективностью в отношении целого ряда бактериальных и грибных штаммов.
В контексте настоящего описания под понятием «пептид» следует понимать последовательность аминокислот, сцепленных друг с другом посредством пептидных связей; аминокислоты предпочтительно выбирают из двадцати встречающихся в естественных условиях аминокислот, и при этом аминокислоты могут присутствовать в L-конфигурации или в D-конфигурации. В качестве альтернативы пептиду, и исходя из его механизма действия и структуры, можно получать также пептидомиметики, которые в контексте настоящего изобретения также подпадают под объем настоящего изобретения. По определению пептидомиметики представляют собой в данном случае низкомолекулярные химические соединения, основные структурные элементы которых воспроизводят соответствующие элементы пептида, предлагаемого в настоящем изобретении. При этом пептид, предлагаемый в настоящем изобретении, может находиться, например, в выделенной, синтетической или рекомбинантной форме или может быть создан в соответствующей форме.
Следует понимать, что в контексте настоящего описания понятие «антимикробный» обозначает свойство, позволяющее снижать способность к размножению или инфекционность микроорганизмов, или уничтожать или инактивировать их. В контексте настоящего описания понятие «микроорганизмы» относится к организмам или единицам микроскопически малых размеров, которые, как правило, неразличимы невооруженным глазом, и в контексте настоящего описания оно относится, прежде всего, к бактериям, вирусам и грибам, которые вызывают вредные для здоровья процессы (заболевания) в других организмах, прежде всего у людей или других млекопитающих.
В предпочтительном варианте осуществления изобретения пептид, предлагаемый в настоящем изобретении, выбирают из пептидов, имеющих последовательности SEQ ID NO: 1-6, или их производных, где производные получают путем замены по меньшей мере одной аминокислоты на производное аминокислоты. Наиболее предпочтительными являются следующие пептиды: пептид, имеющий последовательность RGKAKCCK (SEQ ID NO: 1), пептид, имеющий последовательность RGKAKCAK (SEQ ID NO: 2), и пептид, имеющий последовательность RGKAKACK (SEQ ID NO: 3), прежде всего в немодифицированной форме, т.е. с немодифицированными концами, или в модифицированной форме, т.е. по меньшей мере с одним модифицированным (С- или N-) концом, или в модифицированной форме с модифицированным N-концом и модифицированным С-концом (см. SEQ ID NO: 4, 5 и 6).
В контексте настоящего описания понятие «производное конкретной/любой аминокислоты» относится ко всем аминокислотным остаткам, выведенным из соответствующей аминокислоты, которые получают из соответствующей аминокислоты, например, путем структурной модификации функциональной группы.
В контексте настоящего описания понятия «модифицированная N-концевая аминогруппа» и «модифицированная С-концевая карбоксильная группа» обозначают измененную аминогруппу или карбоксигруппу. Примерами N-концевых модификаций являются ацетилированные, формилированные или гуанилированные N-концы. Примерами С-концевых модификаций являются амидированные С-концы.
Наиболее предпочтительно, когда пептид в каждом случае состоит полностью из D-аминокислот или L-аминокислот, или их смесей. В данном случае понятие «D-аминокислоты» или «L-аминокислоты» означает, что применяемые встречающиеся в естественных условиях аминокислоты, не встречающиеся в естественных условиях аминокислоты или аминокислотные производные (такие как иминокислоты), могут присутствовать в L- или D-конфигурации.
В другом варианте осуществления изобретения пептид предпочтительно модифицируют на С-конце и/или на N-конце, и модификацию осуществляют прежде всего путем ацетилирования, амидирования, формилирования или гуанилирования.
Преимущество модификации С- и/или N-концов пептидов, предлагаемых в настоящем изобретении, заключается в том, что в результате пептиды становятся более стабильными в отношении расщепления пептидазами и протеазами; таким образом, пептиды, предлагаемые в настоящем изобретении, обладают более продолжительным временем полужизни, например, в сыворотке. Модификации N- и С-концов дают также возможность пептидам связываться с другими группами, например, с другими аминокислотными последовательностями или другими биомолекулами.
В другом варианте осуществления изобретения пептид, предлагаемый в настоящем изобретении, является восстановленным или находится в окисленном состоянии.
Согласно настоящему изобретению пептид применяют для лечения и/или профилактики воспалительных или инфекционных заболеваний, вызываемых микроорганизмами.
Таким образом, согласно настоящему изобретению применение осуществляют в случае воспалительных и/или инфекционных заболеваний, вызываемых бактериями, вирусами или грибами.
Применение согласно настоящему изобретению осуществляют прежде всего в случае воспалительных или инфекционных заболеваний, которые вызываются микроорганизмом, выбранным из Bifidobacterium sp., Lactobacillus sp., Escherichia coli, Streptococcus sp., Staphylococcus sp., Bacteroides sp., Candida sp., Pseudomonas sp., Propionibacterium sp., Treponema sp., Enterobacter sp., Salmonella sp., Legionella sp., при этом следует понимать, что этот перечень не является исчерпывающим и что пептид обладает эффективностью также и в отношении бактериальных и/или грибных штаммов, не указанных в настоящем описании, и прежде всего в отношении бактерий, относящихся в целом к семейству Neisseriaceae, Enterobacteriaceae.
Наиболее предпочтительно, когда пептид, предлагаемый в настоящем изобретении, применяют в случае хронических воспалительных заболеваний кишечника, воспалительных заболеваний ротоглоточной полости, например, кариеса и воспалений десен, легочных заболеваний, заболеваний мочеполового тракта, заболеваний поджелудочной железы, заболеваний женской репродуктивной системы, заболеваний и/или повреждений кожи (дерматологических заболеваний).
Соответственно, настоящее изобретение относится также к фармацевтической композиции, которая содержит по меньшей мере один пептид, предлагаемый в настоящем изобретении, а также необязательно фармацевтически приемлемый носитель и другие, используемые для приготовления композиции субстанции и адъюванты, известные из существующего уровня техники, и к способу лечения млекопитающих, которые страдают воспалительными инфекционными заболеваниями, вызываемыми микроорганизмами, в котором вводят в терапевтически эффективном количестве пептид, предлагаемый в настоящем изобретении, или фармацевтическую композицию, предлагаемую в настоящем изобретении. В контексте настоящего описания понятие «терапевтически эффективный» или «терапевтически эффективное количество» обозначают количество по меньшей мере одного пептида, предлагаемого в настоящем изобретении, или фармацевтической композиции, которая содержит по меньшей мере один пептид, предлагаемый в настоящем изобретении, которое позволяет снижать или полностью прекращать размножение и формирование колоний бактерий и/или грибов, или достигать поддающегося оценке терапевтического или профилактического успеха. Точное эффективное количество для индивидуума зависит от его размера и состояния здоровья, от природы и степени заболевания, и от по меньшей мере одного пептида или фармацевтической композиции, или от комбинации нескольких из указанных выше факторов.
Препараты/лекарственные средства, предлагаемые в настоящем изобретении, можно применять как in vitro, так и in vivo.
Фармацевтические композиции, предлагаемые в настоящем изобретении, можно вводить пациенту в различных формах, пригодных для выбранного пути введения, а именно, парентерального, орального, внутрибрюшинного, трансдермального и т.д. В контексте настоящего описания парентеральное введение включает введение, осуществляемое следующими путями: внутривенное, внутримышечное, интерстициальное, внутриартериальное, подкожное, интрасиновиальное, трансэпителиальное, включая трансдермальное, внутрилегочное введение посредством ингаляции, офтальмическое, подъязычное и трансбуккальное, местное применение, включая офтальмическое, дермальное, глазное, ректальное и назальную ингаляцию посредством инсуффляции.
Введение можно осуществлять в форме растворов, настоек, мазей, порошков, суспензий, кремов и других твердых или жидких препаратов, таких как таблетки, капсулы, спрей.
К заболеваниям кожи, которые можно лечить с помощью предлагаемого в настоящем изобретении пептида или содержащего его лекарственного средства, относятся, например, угри, дерматит, ожоги и другие заболевания кожи, вызываемые микроорганизмами, или повреждения кожи, при которых возникает риск заражения микроорганизмами.
Согласно предпочтительному варианту осуществления изобретения фармацевтическую композицию вводят через кожу или наносят на кожу, что представляет собой неинвазивное и удобное для пациента введение, которое по сравнению с оральным введением обладает тем преимуществом, что при этом не нужно учитывать среду в пищеварительной системе. Поглощение через кожу можно осуществлять, например, в носу, на щеке, под языком, на деснах или во влагалище. С помощью известных методов можно приготавливать соответствующие препаративные формы; они могут быть приготовлены в виде назальных капель, назального спрея, вставок, пленок, пластырей, гелей, суппозиториев, мазей или таблеток. Предпочтительно носитель, обеспечивающий поглощение через кожу, должен содержать один или несколько компонентов, который(е) прилипает(ют) к коже и тем самым приводит(ят) к увеличению времени контакта препаративной формы с адсорбирующей поверхностью, для того, чтобы повысить поглощение посредством абсорбции. Так, можно, например, включать по меньшей мере один пептид, предлагаемый в настоящем изобретении, в состав липосом, что способствует проникновению пептида в кожу.
Кроме того, пептид, предлагаемый в настоящем изобретении, можно применять для лечения заболеваний ротоглоточной полости, и для таких применений его можно включать в такие формы, как зубные пасты, зубные эликсиры, гели, и/или наносить на зубную нить.
Как уже указывалось выше, фармацевтическая композиция может содержать также наряду с по меньшей мере одним пептидом, предлагаемым в настоящем изобретении, два или большее количество пептидов, предлагаемых в настоящем изобретении. Кроме того, фармацевтическая композиция может содержать также наряду с по меньшей мере одним пептидом, предлагаемым в настоящем изобретении, одну или несколько других активных субстанций, например, антибиотики, известные из существующего уровня техники (например, стрептомицин, пенициллин, тетрациклин), или другие обладающие антимикробной активностью соединения, такие как фунгициды, например, миконазол, или другие субстанции, с помощью которых обычно осуществляют лечение симптомов, ассоциированных с инфекцией, таких, например, как лихорадка или кожная сыпь.
Лекарственное средство может дополнительно содержать также фармацевтически приемлемые носители, связующие агенты, эксципиенты или адъюванты. Фармацевтический носитель, эксципиент или разбавитель можно выбирать с учетом предполагаемого пути введения и на основе стандартизированной фармацевтической практики. В качестве фармацевтически приемлемых носителей можно применять растворители, разбавители или другие жидкие связующие средства, такие как адъюванты для дисперсий или суспензий, поверхностно-активные вещества, средства для придания изотоничности, загустители или эмульгаторы, консерванты, капсулирующие агенты, твердые связующие вещества или вещества, повышающие скольжение, в зависимости от того, какие из них являются наиболее пригодными для конкретной лекарственной формы и в то же время совместимыми с пептидом. Фармацевтическая композиция может содержать также буферы, разбавители и/или добавки. Пригодными буферами являются, например, Трис-HCl, глицин и фосфат, а пригодными разбавителями могут служить, например, водные растворы NaCl, лактоза или маннит. Пригодными добавками являются, например, детергенты, растворители, антиоксиданты и консерванты, и защитные коллоиды, например, гомологичный альбумин или биосовместимые гидрогели. Обзор таких дополнительных ингредиентов можно найти, например, в справочное A. Kibbe, «Handbook of Pharmaceutical Excipients», 3-е изд., изд-во American Pharmaceutical Association and Pharmaceutical Press, 2000.
Кроме того, фармацевтическая композиция, предлагаемая в настоящем изобретении, может содержать также фармацевтически приемлемые соли, например, соли минеральных кислот, такие как гидрохлориды, гидробромиды, фосфаты, сульфаты и аналогичные им соли; а также соли органических кислот, такие как ацетаты, пропионаты, малонаты, бензоаты и аналогичные им соли.
В целом, терапевтически эффективная суточная доза предположительно должна составлять от 0,01 до 50 мг/кг веса тела индивидуума, подлежащего лечению, предпочтительно от 0,1 до 20 мг/кг. Как уже отмечалось выше, лекарственное средство можно приготавливать в форме таблеток или капсул, которые можно вводить по одной или их можно одновременно вводить по две или в большем количестве. Лекарственное средство можно приготавливать также в виде препаративной формы с замедленным высвобождением.
Как правило, врач должен определять суточную дозу, пригодную для конкретного пациента, которая должна зависеть от его или ее возраста, веса и общего состояния здоровья пациента.
В зависимости от применения лекарственное средство можно вводить путем ингаляции, в форме суппозитория или пессария, применять местно в виде раствора, лосьона, мази, крема или присыпки, с использованием кожного пластыря, орально в форме таблеток или капсул, эликсиров, растворов или суспензий, которые необязательно могут содержать ароматизаторы или красители.
Помимо терапевтического применения для лечения инфекций по меньшей мере один пептид, предлагаемый в настоящем изобретении, можно применять также в дезинфицирующих или очищающих средствах, которые можно использовать для дезинфекции или очистки поверхностей или предметов. Еще одной областью применения являются упаковки, в которых пептиды могут быть связаны с упаковочным материалом или включены в него, или служить в качестве консервантов для других материалов, которые могут легко разрушаться микроорганизмами.
Помимо применения пептида, предлагаемого в настоящем изобретении, в медицине, его можно применять также в ветеринарии.
Изобретение относится также к выделенным молекулам нуклеиновой кислоты, последовательности которых кодируют пептид, предлагаемый в настоящем изобретении, и прежде всего пептид, имеющий SEQ ID NO: 1-6, представляющий собой антимикробный, т.е. антибактериальный или противогрибковый пептид, или кодирующей нуклеиновой кислоте, предлагаемой в настоящем изобретении, функционально связанной с регуляторными последовательностями, которые контролируют ее экспрессию в клетке-хозяине. Следующим объектом изобретения является клетка-хозяин, трансфектированная или трансформированная вышеуказанной молекулой нуклеиновой кислоты.
Дополнительные преимущества должны стать очевидными после ознакомления с представленным ниже описанием и прилагаемыми чертежами.
Следует понимать, что те отличительные признаки, которые указаны выше и те, которые еще будут пояснены ниже, можно рассматривать не только в соответствующей указанной комбинации, но также и в других комбинациях или по отдельности, не выходя при этом за пределы объема настоящего изобретения.
Примеры вариантов осуществления изобретения представлены на чертежах и более подробно пояснены в представленном ниже описании.
На чертежах показано:
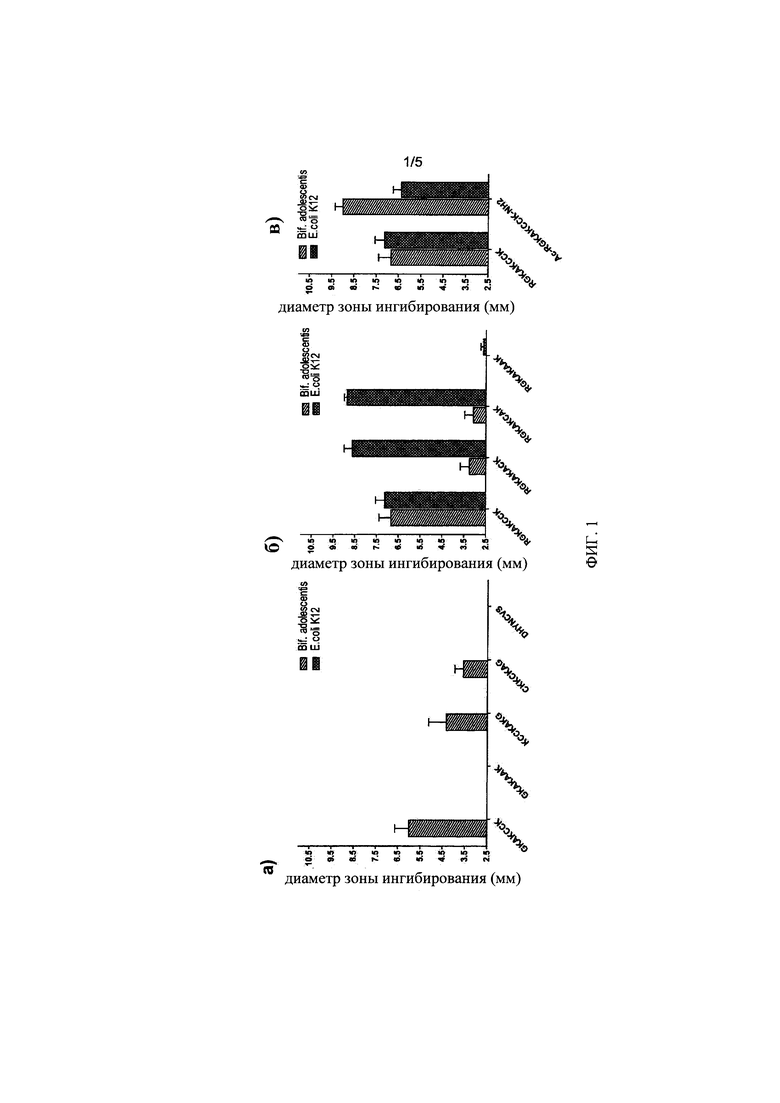
на фиг. 1 - результаты исследований антимикробного действия различных пептидов (гептапептиды (а); октапептиды (б)(в)) в отношении бактерий Bifidobacterium adolescentis или Escherichia coli. Буквами обозначены аминокислоты, использован однобуквенный код. Пептид ac-RGKAKCCK-NH2 (в) (SEQ ID NO: 4) имеет ацетилированный аминоконец и амидированный карбоксиконец. Диаметр зон ингибирования служит характеристикой антимикробной активности; диаметр, равный 2,5 мм, представляет собой диаметр пустой сделанной с помощью штампа лунки в агаровой пластине, которая заполнена только жидкостью-носителем (отрицательный контроль). Эксперименты повторяли по меньшей мере по три раза, результаты представлены в виде средних значений плюс стандартные отклонения;
на фиг. 2 - результаты исследования эффективности различных вариантов пептида, предлагаемого в настоящем изобретении, в качестве антибиотика в отношении различных патогенов. Исследовали следующие пептиды: тестировали модифицированный октапептид (SEQ ID NO: 4), октапептид дикого типа (SEQ ID NO: 1), подвергнутый мутации аланином октапептид (SEQ ID NO: 7) и гептапептид (SEQ ID NO: 8) (каждый в концентрации 50 мкг/мл), проводя анализ антимикробной эффективности методом проточной цитометрии в отношении Escherichia coli, Staphylococcus aureus, Candida albicans и Bacteroides fragilis. И в этом случае также буквами обозначены соответствующие аминокислоты с использованием однобуквенного кода. Эксперименты повторяли дважды для двух партий, результаты представлены в виде средних значений плюс стандартные отклонения;
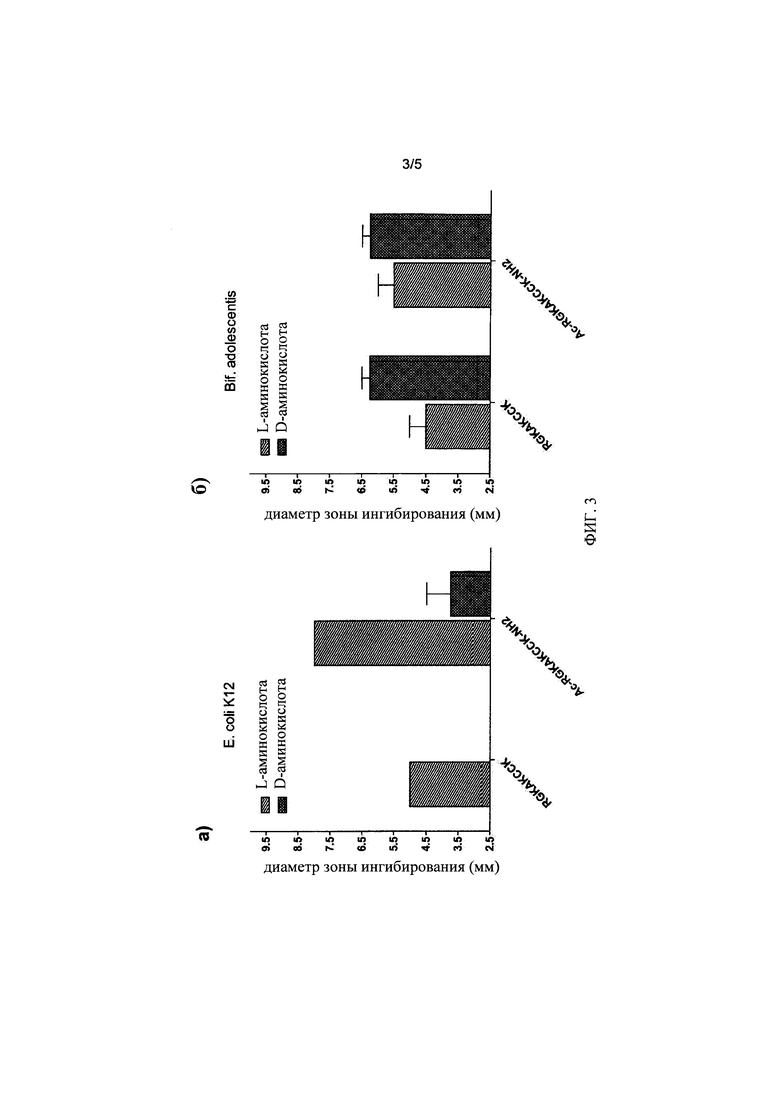
на фиг. 3 - результаты исследований октапептида, предлагаемого в настоящем изобретении, который состоял из D-аминокислот, в сравнении с октапептидом, состоящим из L-аминокислот, проведенных на Е. coli К12 (а) и Bifidobacterium adolescentis (б);
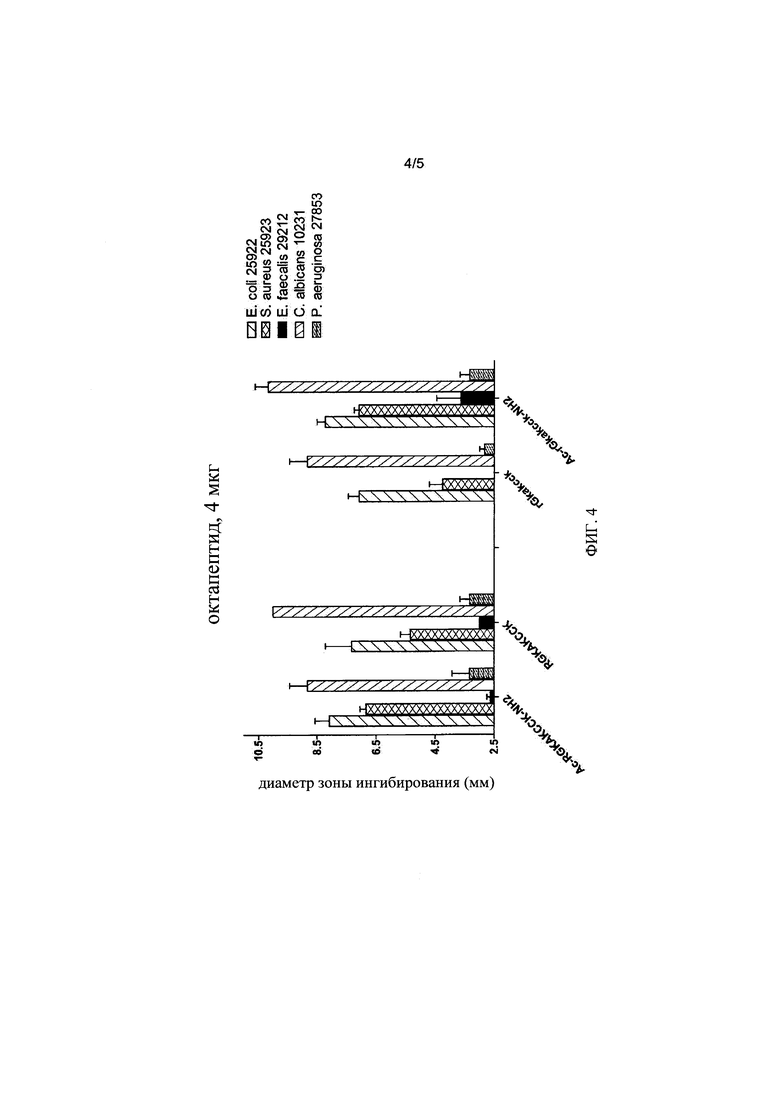
на фиг. 4 - результаты дополнительных исследований активности различных октапептидов, предлагаемых в настоящем изобретении, в отношении патогенных бактерий и грибов, полученных в анализе методом радиальной диффузии; и
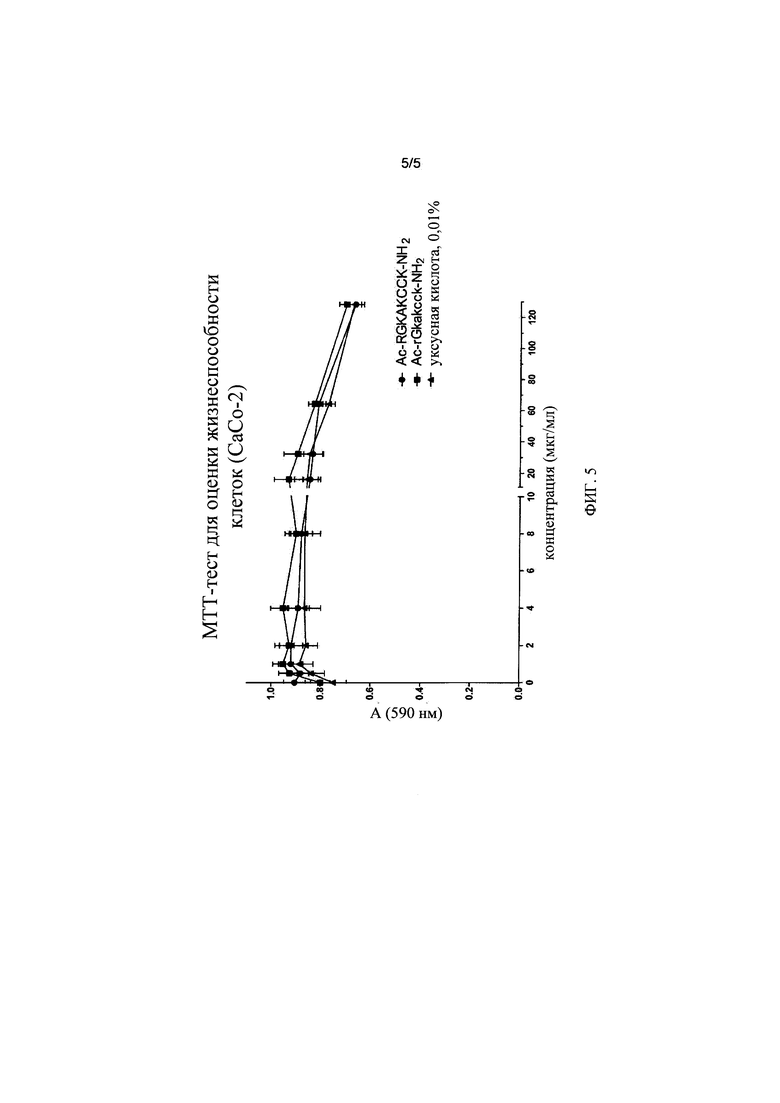
на фиг. 5 - результаты исследований клеточной токсичности октапептидов, полученные на клетках кишечника линии СаСо-2.
Как установлено ранее, антимикробные пептиды (АМП) продуцируются практически всеми организмами, и они представляют собой первый барьер для микробного заражения. Многие АМП обладают антимикробной активностью в отношении как грамположительных, так и грамотрицательных бактерий, а также в отношении грибов и некоторых вирусов, имеющих оболочку. В организме человека продуцируются различные классы АМП, одним из которых, как уже указано выше, являются дефензины. Они отличаются небольшим размером (от 3 до 5 кДа), чистым катионным зарядом и наличием шести консервативных остатков цистеина, которые соединены друг с другом тремя дисульфидными мостиками. Дефензины подразделяют на альфа- и бета-дефензины в зависимости от структуры мостикового связывания. До настоящего времени только четыре бета-дефензина (hBD-1 - hBD-4) были исследованы с точки зрения их функциональных свойств, в том числе в качестве кандидатов, обладающих антибиотической эффективностью.
Однако до настоящего времени химический синтез бета-дефензинов, которые, как уже указано выше, имеют три нативных дисульфидных мостика, был сопряжен со значительными проблемами, как с точки зрения стоимости, так и сложности метода производства.
Пептид, созданный впервые согласно настоящему изобретению, представляет собой октапептид С-концевого участка дефензина hBD-1, который содержит два свободных цистеина и который, как было установлено с помощью описанных ниже экспериментов, обладает более высокой антимикробной активностью по сравнению с hBD-1 и с более короткими пептидными последовательностями, выведенными из С-конца hBD-1.
Бактериальные и грибные штаммы
Бактериальные штаммы Bifidobacterium adolescentis Ni3, 29с (клинический изолят), Bifidobacterium breve PZ1343, Bifidobacterium longum DSM 20219T (клинический изолят), Lactobacillus acidophilus PZ1138 (клинический изолят), Lactobacillus fermentum PZ1162 (клинический изолят) и Streptococcus salivarius spp.thermophilus DSM20617 получали от фирмы Ardeypharm (Германия), a Bacteroides vulgatus DSM 1447 был предоставлен DSMZ (Немецкая коллекция микроорганизмов и клеточных культур). Штамм Candida albicans 526 был выделен из фекалий и предоставлен клиникой на Эйхерте Института лабораторной медицины (Геппинген, Германия). Референс-штаммы из Американской коллекции типовых культур (АТСС) Escherichia coli АТСС25922, Staphylococcus aureus ATCC25923 и Bacteroides fragilis ATCC25285 были предоставлены клиникой на Эйхерте Института лабораторной медицины (Геппинген, Германия). Были протестированы также штаммы Enterococcus faecalis АТСС29212, Candida albicans ATCFC 10231 и Pseudomonas aeruginosa ATCC27853, которые можно получать из Американской коллекции типовых культур под указанными номерами.
Пептиды
Человеческие бета-дефензины получали от фирмы Peptide Institute Inc., Осака, Япония; карбоксиконцевые гептапептиды и октапептиды, а также восстановленный hBD-1, синтезировали химическим путем (фирма EMC Micro Collections, Тюбинген, Германия).
Анализы антимикробной активности
Анализы антимикробной активности в отношении анаэробных бактерий методом радиальной диффузии осуществляли согласно описанной ранее процедуре (см. Schröder и др., «Reduction of disulfide bonds unmasks potent antimicrobial activity of human beta-defensin 1», Nature, 469, 2011, cc. 419-423). В целом метод заключался в следующем. Бактерии культивировали в анаэробных условиях (Oxoid AnaeroGen™, Великобритания) в течение 24 ч при 37°С на агаровых планшетах типа Columbia, затем инокулировали жидкую среду TSB («триптиказо-соевый бульон») и снова культивировали в течение 24 ч. Затем бактериальные культуры промывали и разводили до достижения оптической плотности (ОП620 нм), равной 0,1, образец объемом 150 мкл использовали для анализа эффективности. Инкубацию осуществляли в течение 3 ч в анаэробных условиях в 10 мл 10 мМ фосфата натрия, рН 7,4, с добавлением 0,3 мг/мл порошкообразного TSB и 1% (мас./об.) low-EEO агарозы (агароза с очень низкой величиной ЕЕО (электроэндоосмос))(фирма Appli-Chem) с добавлением 2 мМ дитиотреитола (ДТТ, фирма Sigma Aldrich) или без него, 1 мкг синтетического окисленного hBD-1 (фирма Peptide Institute, Япония) или синтетических пептидов. На планшеты наносили гелевое покрытие, состоящее из 6% (мас./об.) порошкообразного TSB, 1% агарозы и 10мМ натрий-фосфатного буфера (рН 7,4 или 5,7), с добавлением ДТТ или без него. После инкубации в течение 48 ч при 37°С измеряли диаметры зон ингибирования. Эксперименты повторяли по меньшей мере три раза.
Анализ антимикробной активности методом проточной цитометрии, в котором измеряли деполяризацию мембран бактерий и грибов, осуществляли согласно описанной ранее процедуре (см. Nuding и др., «А flow cytometric assay to monitor antimicrobial activity of defensins and cationic tissue extracts», Journal of Microbiological Methods, 65, 2006, cc. 335-380).
В целом метод состоял в следующем. 1,5×106 клеток/мл инкубировали в разведенной в соотношении 1:6 среде Шедлера при 37°С с пептидами в конечном объеме 50 мкл. Дефензины растворяли в 0,01%-ной уксусной кислоте и добавляли к бактериальным/грибным суспензиям в указанных конечных концентрациях. Бактериальные или грибные суспензии, которые были проинкубированы с растворителем (0,01%-ная уксусная кислота), служили в качестве контролей при оценке жизнеспособности. Спустя 90 мин суспензии инкубировали в течение 10 мин с 1 мг/мл чувствительным к мембранному потенциалу красителем DiBAC4(3) ([бис-(1,3-дибутилбарбитурат)триметиноксанол]) (фирма Invitrogen, США). Суспензии центрифугировали и осадки ресуспендировали в 300 мл забуференного фосфатом физиологического раствора. Процент деполяризованных флуоресцирующих бактерий или грибов в суспензии определяли с помощью проточного цитометра FACSCalibur (фирма Becton-Dickinson, США) с использованием программного обеспечения Cell Quest (фирма Becton-Dickinson). Эксперименты повторяли дважды, каждый раз с дублированием.
ЖХВР-анализ
Для анализа методом жидкостной хроматографии высокого разрешения (ЖХВР) октапептиды смешивали с 0,1 об.% трифторуксусной кислоты (ТФК) и анализировали с помощью системы Agilent 1200 (фирма Agilent) и колонки с обращенной фазой (ОФ) типа Synergi (250×4,6 мм, 4 мкм, фирма Phenomenex, Германия). Градиент возрастал от 0% В до 12% В в течение 24 мин (растворитель А: вода+0,18 об.% ТФК; растворитель В: ацетонитрил +0,15 об.% ТФК), хроматографию осуществляли при 25°С и 0,8 мл/мин.
Анализ ионного ингибирования
0,25 мкг/мл пептидов или дефензинов инкубировали при комнатной температуре в течение 45 мин в 4,5 мМ NaCl, с хлоридом магния MgCl2, хлоридом железа FeCl2, хлоридом цинка ZnCl2 или сульфатом цинка ZnSO4 Затем смесь анализировали методом радиальной диффузии для оценки антимикробной активности в отношении Bifidobacterium adolescentis и Escherichia coli. Эксперименты повторяли по меньшей мере три раза.
Результаты
В современной публикации Schröder и др., «Reduction of disulfide bonds unmasks potent antimicrobial activity of human beta-defensin 1», Nature, 469, 2011, cc. 419-423 было продемонстрировано, что человеческий бета-дефензин 1 обладает повышенной антимикробной активностью в восстанавливающих условиях.
При создании настоящего изобретения были проведены дополнительные исследования hBD-1 и его антимикробной активности. Для этой цели тестировали антимикробную активность трех человеческих бета-дефензинов hBD-1, -2 и -3 в отношении комменсальных бактерий, присутствующих во флоре человеческого кишечника, в стандартных условиях (рН 7,4) и слабо кислых условиях (рН 5,7); при этом в обоих условиях тестирование проводили также и в восстанавливающих условиях, добавляя в питательную среду 2 мМ химический восстановитель дитиотреитол (ДТТ). При этом было установлено (данные не представлены), что активность бета-дефензинов в отношении большинства бактерий была наивысшей в стандартных условиях, за исключением hBD-1, который в этих условиях был выражено неактивным и становился активным при восстановлении. Однако такая активация не наблюдалась при рН 5,7. В отличие от этого, восстановление не оказывало влияния на hBD-2, при этом изменение значения рН оказывало сильное негативное воздействие на его антимикробную активность.
В большинстве случаев hBD-3 обладал наиболее сильной активностью в отношении протестированных бактерий-комменсалов по сравнению с двумя другими дефензинами.
В целом, можно констатировать, что факторы окружающей среды, например, окислительно-восстановительный потенциал и значение рН, могут модулировать антимикробную активность бета-дефензинов в отношении кишечных бактерий-комменсалов. Однако такая модуляция, по-видимому, является специфической для конкретных взаимодействий дефензин-бактерии и не коррелирует ни с грам-статусом, ни с родом бактерий.
Эксперименты, проведенные с гептапептидом, имеющим семь концевых аминокислот hBD-1, который уже обладал антимикробной активностью в отношении Bifidobacterium adolescentis, продемонстрировали, что карбоксиконцевой гептапептид дикого типа обладал наибольшей активностью в отношении Bifidobacterium adolescentis, в то время как пептиды с обратной аминокислотной последовательностью оказались менее активными. Замена остатков цистеина на аланин приводила к полному подавлению активности. Выделенный аминоконец hBD-1 не обладал активностью.
Затем при создании настоящего изобретения был протестирован пептид, предлагаемый в настоящем изобретении, а именно октапептид, который содержит восемь концевых аминокислот карбоксиконца hBD-1: было установлено, что он обладал более высокой антимикробной активностью, чем протестированный ранее гептапептид (см. фиг. 1). После замены Cys6 или Cys7 происходило одинаково сильное снижение активности в отношении Bifidobacterium adolescentis, в то время как замена обоих цистеинов приводила к полному элиминированию активности. В отличие от протестированного ранее гептапептида, октапептид обладал антимикробной активностью также и в отношении Escherichia coli (см. фиг. 16). При создании изобретения неожиданно было установлено, что в этом случае замена Cys6 или Cys7 на аланин приводила к повышению антимикробной активности, в то время как замена обоих цистеинов и в этом случае приводила к почти полному «выключению» антимикробной активности.
На следующей стадии с целью оптимизации октапептида и улучшения его стабильности к действию протеаз осуществляли стабилизацию посредством ацетилирования аминоконца и амидирования карбоксиконца октапептида. В то время как активность в отношении Escherichia coli не претерпевала существенного изменения по сравнению с пептидом дикого типа, активность в отношении Bifidobacterium adolescentis резко возрастала (см. фиг. 1в).
С использованием нового идентифицированного и полученного октапептида можно просто и экономически эффективно создавать пептид, обладающий антибиотическими действиями, который можно применять в качестве терапевтического средства. Поэтому было проведено исследование как модифицированных, так и немодифицированных пептидов в отношении их способности уничтожать (условно патогенные) патогенные микроорганизмы. Для этой цели проводили анализы методом проточной цитометрии (см. фиг. 2), которые продемонстрировали, что эффективность октапептида дикого типа и подвергнутого мутации аланином пептида, и гептапептида дикого типа во многих случаях была лишь незначительной. В отличие от этого, модифицированный октапептид обладал выраженной активностью в отношении патогенных микроорганизмов Staphylococcus aureus и Candida albicans, но не в отношении Escherichia coli и Bacteroides fragilis.
При этом было установлено, что стабилизация концов приводила к повышению антимикробной активности не только в отношении кишечной комменсальной бактерии Bifidobacterium adolescentis, но также и в отношении по меньшей мере двух патогенных микроорганизмов, имеющих клиническое значение.
В других экспериментах проводили анализ методом ЖХВР с обращенной фазой с целью изучения гидрофобности тестируемых пептидов (данные не представлены). Модифицированный пептид элюировался из колонки последним, это свидетельствует о том, что он обладал наиболее высокой гидрофобностью; перед ним элюировались пептид дикого типа, варианты с одиночными заменами аминокислот на аланин и варианты с двойной заменой аминокислот на аланин (данные не представлены).
Для дальнейшего исследования роли заряда и ионных взаимодействий осуществляли инкубацию окисленного и восстановленного hBD-1 и октапептида дикого типа и модифицированного октапептида с одновалентными и двухвалентными катионами (данные не представлены). Активность полного дефензина в отношении Escherichia coli полностью «выключалась» после предварительной инкубации с хлоридом магния или хлоридом железа, в то время как для карбоксиконцевых пептидов имело место сильное ингибирование активности, но она еще оставалась на обнаруживаемом уровне после предварительной инкубации с указанными ионами металлов. В отличие от этого, NaCl не оказывал влияния на антимикробную активность в отношении Е. coli.
При исследовании на Bifidobacterium adolescentis также было установлено, что предварительная инкубация с хлоридом натрия не оказывала сильного воздействия на активность, в то время как присутствие хлорида железа, хлорида цинка и сульфата цинка полностью элиминировало или значительно снижало активность. В отличие от Е. coli инкубация с хлоридом магния не оказывала влияния на антибиотическую активность в отношении Bifidobacterium.
В других экспериментах исследовали октапептид, предлагаемый в настоящем изобретении, который имел последовательность RGKAKCCK (SEQ ID NO: 1), состоящую из D-аминокислот (за исключением глицина), в сравнении с октапептидом, который имел последовательность RGKAKCCK (SEQ ID NO: 1), состоящую из L-аминокислот, для вариантов с модифицированными и немодифицированными концами (фиг. 3). В отношении штамма Е. coli К12 (см. фиг. 3а) пептид, состоящий из D-аминокислот и имеющий модифицированные концы (N-конец: ацетилированный; С-конец: амидированный), обладал более слабой активностью, в то время как в отношении Bifidobacterium adolescentis указанный пептид как в немодифицированной форме, так и в модифицированной форме, обладал более высокой активностью по сравнению с пептидом, состоящим из L-аминокислот (см. фиг. 36).
Таким образом, в целом представленные данные свидетельствуют о том, что взаимодействие между пептидом и катионами базируется не только на положительном заряде, а, напротив, конкретные ионы могут оказывать влияние на активность в отношении определенных бактерий.
Активность октапептидов, предлагаемых в настоящем изобретении, в отношении патогенных бактерий и грибов была подтверждена также с помощью дополнительных анализов методом радиальной диффузии: активность тестировали в отношении штаммов Escherichia coli 25922, Staphylococcus aureus 25923, Enterococcus faecalis 29212, Candida albicans 10231 и Pseudomonas aeruginosa 27853 (см. фиг. 4) с использованием октапептида, предлагаемого в настоящем изобретении, который имел последовательность RGKAKCCK (SEQ ID NO: 1), состоящую либо из L-аминокислот (см. фиг. 4; пять столбиков в левой части диаграммы), либо из D-аминокислот (за исключением глицина) (см. фиг. 4; пять столбиков в правой части диаграммы), для вариантов с модифицированными и немодифицированными концами (N-конец: ацетилированный; С-конец: амидированный). Помимо высокой активности в отношении Escherichia coli и Staphylococcus aureus, в этом эксперименте была выявлена также очень высокая активность в отношении Candida albicans всех вариантов протестированного октапептида.
Клеточную токсичность стабилизированных на концах октапептидов исследовали также в дополнительных экспериментах с использованием МТТ-((3-[4,5-диметилтиазол-2-ил]-2,5-дифенилтетразолия бромид; тиазолиловый синий), прежде всего в отношении человеческой клеточной линии СаСо2 (АТСС НТВ-37), применяя возрастающие концентрации соответствующих октапептидов (SEQ ID NO: 1; состоящая из L- или D-аминокислот (за исключением глицина), имеющая модифицированные концы (N-конец: ацетилированный; и С-конец: амидированный)), в сравнении с взятой в возрастающих количествах 0,01% уксусной кислотой. Результаты, представленные на фиг. 5, свидетельствуют о том, что стабилизированные октапептиды не обладали клеточной токсичностью, которая превышала бы токсичность растворителя, представляющего собой 0,01% уксусную кислоту. Таким образом, октапептиды, предлагаемые в настоящем изобретении, пригодны для применения в терапии.
Представленные выше результаты и данные четко демонстрируют, что октапептид, впервые предложенный в настоящем изобретении, в форме дикого типа, имеющий аминокислотную замену, и/или находящийся в стабилизированной форме, представляет собой перспективное средство, обладающее антибиотической эффективностью. Полученные результаты являются неожиданными, и их нельзя было предсказать на основе известного в настоящее время уровня техники.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБЫ МОДУЛЯЦИИ КИШЕЧНОЙ МИКРОБИОТЫ | 2017 |
|
RU2738265C2 |
| АНТИМИКРОБНОЕ ВЕЩЕСТВО | 2010 |
|
RU2447896C1 |
| ПОЛИПЕПТИДЫ ЛИЗИНА, АКТИВНЫЕ ПРОТИВ ГРАМОТРИЦАТЕЛЬНЫХ БАКТЕРИЙ | 2016 |
|
RU2724545C2 |
| ПОЛИПЕПТИДЫ С ПРОТИВОМИКРОБНОЙ АКТИВНОСТЬЮ И КОДИРУЮЩИЕ ИХ ПОЛИНУКЛЕОТИДЫ | 2005 |
|
RU2393224C2 |
| ЛЕЧЕНИЕ НАРУШЕНИЙ СО СТОРОНЫ ПЕЧЕНИ, ЖЕЛЧНЫХ ПУТЕЙ И ПОДЖЕЛУДОЧНОЙ ЖЕЛЕЗЫ | 2017 |
|
RU2740913C2 |
| Варианты антимикробного пептида и кодирующие их полинуклеотиды | 2011 |
|
RU2611173C2 |
| ПРОДУЦИРУЕМЫЙ БАКТЕРИЯМИ МИКРОЦИН С, НОВЫЙ АНТИМИКРОБНЫЙ ПЕПТИД, ЭФФЕКТИВНЫЙ ПРОТИВ ПАТОГЕННЫХ МИКРООРГАНИЗМОВ, НАПРИМЕР ЭНТЕРОГЕМОРРАГИЧЕСКОЙ Escherichia coli (EHEC) | 2012 |
|
RU2651478C2 |
| ПОЛИПЕПТИДЫ, ОБЛАДАЮЩИЕ ПРОТИВОМИКРОБНЫМ ДЕЙСТВИЕМ, И КОДИРУЮЩИЕ ИХ ПОЛИНУКЛЕОТИДЫ | 2006 |
|
RU2403260C2 |
| ПОЛИПЕПТИДЫ, ОБЛАДАЮЩИЕ АНТИМИКРОБНОЙ АКТИВНОСТЬЮ, И ПОЛИНУКЛЕОТИДЫ, КОДИРУЮЩИЕ ИХ | 2006 |
|
RU2415150C2 |
| ПРОТИВОКАРИЕСНЫЕ КОМПОЗИЦИИ И ПРОБИОТИКИ/ПРЕБИОТИКИ | 2011 |
|
RU2650866C2 |

Группа изобретений относится к медицине и касается пептида, который обладает антимикробной активностью, выбранного из пептидов, имеющих SEQ ID NO: 1-4. Группа изобретений также касается применения указанного пептида в качестве антибиотика и/или в составе дезинфицирующего средства или очистительного средства; фармацевтической композиции, обладающей антимикробной активностью, содержащей по меньшей мере один указанный пептид. Группа изобретений обеспечивает высокую антимикробную активность. 4 н. и 5 з.п. ф-лы, 1 пр., 5 ил.
1. Пептид, который обладает антимикробной активностью, выбранный из пептидов, имеющих SEQ ID NO: 1-4.
2. Пептид по п. 1,
отличающийся тем, что он состоит из D-аминокислот или L-аминокислот, или их смесей.
3. Применение пептида по одному из пп. 1 и 2 в качестве антибиотика и/или в составе дезинфицирующего средства или очистительного средства.
4. Применение по п. 3 для лечения и/или профилактики воспалительных или инфекционных заболеваний, вызываемых микроорганизмами.
5. Применение по п. 4,
отличающееся тем, что воспалительное или инфекционное заболевание вызывается микроорганизмом, представляющим собой бактерию, вирус или дрожжи.
6. Применение по одному из пп. 3-5,
отличающееся тем, что воспалительное или инфекционное заболевание вызывается микроорганизмом, выбранным из Bifidobacterium sp., Lactobacillus sp., Escherichia coli, Streptococcus sp., Staphylococcus sp., Bacteroides sp., Candida sp., Pseudomonas sp., Propionibacterium sp., Treponema sp.
7. Применение по одному из пп. 3-5,
отличающееся тем, что воспалительное или инфекционное заболевание выбрано из хронических воспалительных заболеваний кишечника, воспалительных заболеваний ротоглоточной полости, легочных заболеваний, заболеваний мочеполового тракта, заболеваний поджелудочной железы, заболеваний женской репродуктивной системы, заболеваний или повреждений, или ожогов кожи.
8. Фармацевтическая композиция, обладающая антимикробной активностью, содержащая по меньшей мере один пептид по одному из пп. 1 и 2, а также фармацевтически приемлемый носитель.
9. Полинуклеотид, кодирующий пептид по одному из пп. 1 и 2.
| Топка с несколькими решетками для твердого топлива | 1918 |
|
SU8A1 |
| GIANNECCHINI S | |||
| et al | |||
| Antiviral activity and conformational features of an octapeptide derived from the membrane-proximal ectodomain of the feline immunodeficiency virus transmembrane glycoprotein.J Virol | |||
| Способ и приспособление для нагревания хлебопекарных камер | 1923 |
|
SU2003A1 |
| TAM JP | |||
| et al | |||
| Antimicrobial dendrimeric peptides.Eur J Biochem | |||
| Топчак-трактор для канатной вспашки | 1923 |
|
SU2002A1 |
| TINCU JA | |||
| et al | |||
| Plicatamide, an antimicrobial octapeptide from Styela plicata hemocytes.J Biol Chem | |||
| Способ и приспособление для нагревания хлебопекарных камер | 1923 |
|
SU2003A1 |
| Способ и приспособление для нагревания хлебопекарных камер | 1923 |
|
SU2003A1 |
| БЕЛИКОВ В.Г | |||
| Фармацевтическая химия, М., Высшая школа, 1993. | |||

