Изобретение относится к области химико-фармацевтической промышленности и медицины.
Известно, что наиболее используемыми в медицинской практике средствами для лечения воспалительных патологий являются нестероидные противовоспалительные средства, такие как кислота ацетилсалициловая (аспирин), индометацин, вольтарен (диклофенак натрия), ибупрофен (бруфен) и др. [1]. С влиянием на биосинтез простагландинов связан основной побочный эффект нестероидных противовоспалительных препаратов - ульцерогенное действие (способность повреждать слизистую оболочку желудка и двенадцатиперстной кишки, вплоть до развития язвенных состояний). Это осложнение связано с ингибированием биосинтеза простагландинов, являющихся физиологическими (эндогенными) гастроцитопротекторными веществами.
Вследствие этого актуальной задачей является поиск новых соединений, обладающих системным противовоспалительным эффектом, но лишенных указанного ульцерогенного действия.
С этой точки зрения представлялось перспективным изыскать вещество, проявляющее значительный противовоспалительный эффект, который не связан с ингибированием синтеза простагландинов, а воздействует на организм по иному механизму.
Хорошо известно, что целый ряд производных гуанидина проявляют выраженный антагонизм к NO-синтазам. И в этом отношении выделяются N-аминогуанидин (1) [2], а также некоторые его производные.
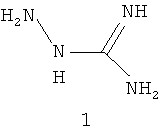
В большинстве in vitro-систем аминогуанидин и известный ингибитор синтаз оксида азота L-NMMA (NG-монометил-L-аргинин) равноэффективны при ингибировании индуцибельного изофермента, но первый на порядок менее активен в отношении конститутивных форм, т.е. первый существенно более селективен [3].
На моделях животных аминогуанидин уменьшает тяжесть протекания болезни при воспалениях, септическом шоке, повышает выживаемость при введении эндотоксинов.
Профиль активности аминогуанидина благоприятен для больного, в плане лечения воспалительных заболеваний.
В качестве ближайшего аналога могут быть указаны соли производных амидина и ингибитора циклооксигеназы общей формулы AB, в которой A - ингибитор циклооксигеназы с карбоксильной функцией; B - соединение общей формулы
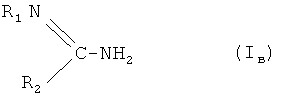
Соединения по изобретению обладают двойным биологическим эффектом, при котором они ингибируют образование окиси азота (NO) и активность циклооксигеназ и могут применяться в качестве противовоспалительных препаратов [8]. Однако им присущи, хотя менее выраженные, побочные эффекты, характерные для вышеупомянутых нестероидных противовоспалительных средств.
Задачей заявляемого изобретения является изыскание нового противовоспалительного средства, не проявляющего ульцерогенного действия.
Исходя из этого, представляется оптимальным синтез такого соединения, которое не только содержало бы аминогуанидиновый фрагмент в своей структуре, но обладало свойством метаболизироваться в живом организме с выделением аминогуанидина.
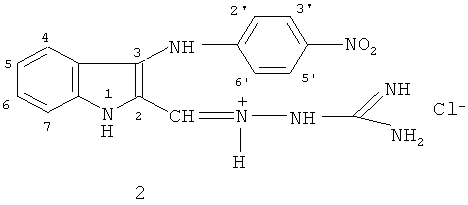
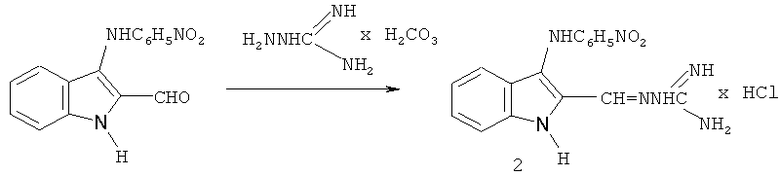
Именно это качество является вполне вероятным для заявляемого в данной работе соединения - N-[3-(4-нитрофениламино)-индол-2-илметилен]аминогуанидина гидрохлорид формулы (2):
Иминоструктура этого соединения предусматривает, что в водной среде оно может гидролизоваться (как это характерно для иминов) по схеме 1 с образованием 2-формил-3-п-нитрофенилиндола (3) и гидрохлорида аминогуанидина:
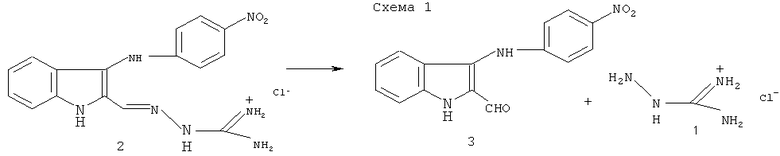
Способ получения N-[3-(4-нитрофениламино)-индол-2-илметилен] аминогуанидина гидрохлорида формулы (2) основан на взаимодействии 2-формил-3-(4-нитрофенил)аминоиндола [9, 10] с аминогуанидином в присутствии соляной кислоты при нагревании в спирте ректификате, обеспечивающий технологически приемлемые условия получения целевого продукта из доступного сырья и без применения особых условий проведения процесса.
Новое соединение проявляет ценные фармакологические свойства: оказывает системное противовоспалительное действие, а также проявляет свойства хондропротективного агента.
Возможность осуществления изобретения может быть продемонстрирована следующими конкретными примерами выполнения:
Пример 1. Синтез N-[3-(4-нитрофениламино)-индол-2-илметилен] аминогуанидина гидрохлорида (2).
Смесь 1 г (0.356 ммолей) 2-формил-3-(4-нитрофенил)аминоиндола, 0.58 г (0.43 ммолей) аминогуанидина карбоната, 0.87 мл (0.86 ммолей) конц. соляной кислоты, 0.87 мл воды и 26 мл этанола ректификата кипятят 1.5 ч. Охлаждают, осадок отфильтровывают, промывают спиртом и ацетоном. Получают 1.23 г гидрохлорида, который перекристаллизовывают в смеси этанол-вода (3:1). Выход 0.8 г (65%). Т.пл. 310-312°C.
При больших загрузках вещество кристаллизовали из водного N,N-диметилформамида (ДМФА).
Полученное соединение характеризуется следующими данными ЯМР-спектра:
Спектр 1Н (ДМСО-d6, δ, м.д.): 6,75 и 8,03
7,02, 7,26, 7,44, (все м. по 1Н, 2Н, 1Н, 4Н, 7Н)
7,83 (уш.с., 4Н, HN-CH=NH2)
8,18 (с. 1Н-α-Н)
9,21, 11,69, 11,98 (уш.с. по 1H, NH(Ph), NH (индол), NH+)
Пример 2. Изучение токсических свойств соединения.
Исследование соединения формулы 2 проводилось in vivo в сравнении с самыми эффективными нестероидными противовоспалительными препаратами - вольтареном и индометацином, главным недостатком которых является указанный выше ульцерогенный эффект. Для определения доз, в которых данное соединение необходимо изучать по фармакологическим показателям, в первую очередь необходимо определение его острой токсичности. Опыты проведены на мышах-самцах массой 18-20 г. Соединение 2 вводили внутрь в виде суспензии с водой. Каждую дозу вводили пяти животным. Наблюдали за поведением и состоянием животных в течение 5 дней. Величину показателя ЛД50, характеризующего дозу, вызывающую гибель 50% испытуемых животных, рассчитывали по методу Кербера.
Учитывая гибель одного животного из 5-и только при внутривенном введении препарата в дозе 2000,0 мг/кг, можно утверждать, что исследуемое соединение относится по ГОСТ 12.1.007-76 (Классификация производственных вредных веществ по степени опасности) как минимум к 3 классу - умеренно опасные вещества, а учитывая, что при введении внутрь ЛД50 может превышать 5000 мг/кг, данное соединение формулы 2 можно отнести к 4 классу - малоопасные вещества, т.е. соединение практически не токсично.
Пример 3. Изучение противовоспалительной активности.
Активность изучена по методикам, представленным в «Методических рекомендациях по экспериментальному (доклиническому) изучению новых нестероидных противовоспалительных средств» для скринингового изучения новых соединений, на моделях перитонита у мышей, вызванного липополисахаридом (ЛПС) и каррагенином [4].
1. Перитонит у мышей, вызванный каррагенином.
Исследование выполнено согласно методу, описанному в работе [5]. Опыты проведены на мышах-самцах массой 23-24 г, в каждой группе по 10 животных. Исследуемые соединения вводили зондом в желудок за 1,5 часа до внутрибрюшинного введения 0,2 мл 1% λ-каррагенина, через 4 часа животных забивали и измеряли в мл количество экссудата в перитониальной полости. Результаты экспериментов обрабатывали методами вариационной статистики для биологических исследований (определение средней арифметической, стандартной ошибки, при сопоставлении средних использовали критерий t Стьюдента).
2. Перитонит у мышей, вызванный ЛПС.
Антиэкссудативное действие изучали на мышах-самцах массой 22,0-23,0 г при перитоните, вызванном внутрибрюшинным введением липополисахарида (ЛПС), выделенного из Escherichia coli (Sigma) 1,0 мг/кг, как описано в [6]: через 4 часа животных забивали (ингаляцией СО2), вскрывали брюшную полость и измеряли объем экссудата в мл. Исследуемые соединения и препараты сравнения вводили внутрь (per os) за час до ЛПС, контролем в исследовании были мыши, получавшие по 0,3 мл физиологического раствора внутрь до введения ЛПС. Каждая исследуемая группа состояла из 10 животных.
Результаты экспериментов обрабатывали методами вариационной статистики для биологических исследований (определение средней арифметической, стандартной ошибки, при сопоставлении средних использовали критерий t Стьюдента).
Результаты исследования.
Результаты, полученные в ходе эксперимента, отражены и оценка противовоспалительной активности соединения 2 представлена в таблице 2.
вызванного каррагенином, в % от контроля
Перитонит у крыс, вызванный ЛПС.
Антиэкссудативное действие изучали на крысах-самцах массой 170,0-180,0 г при перитоните, вызванном внутрибрюшинным введением липополисахарида (ЛПС), выделенного из Escherichia coli (Sigma) - 1,0 мг/кг. Метод выполнен согласно [6]: через 4 часа животных умерщвляли (ингаляцией СО2), вскрывали брюшную полость и измеряли объем экссудата в мл. Исследуемые соединения и препараты сравнения вводили внутрь (per os) за час до ЛПС, контролем в исследовании были крысы, получавшие по 0,3 мл физиологического раствора внутрь до введения ЛПС. Каждая исследуемая группа состояла из 7 животных.
Результаты экспериментов обрабатывали методами вариационной статистики для биологических исследований (определение средней арифметической, стандартной ошибки, при сопоставлении средних использовали критерий t Стьюдента).
Результаты исследования.
Оценка противовоспалительной активности соединения 2 в сравнении с вольтареном и аминогуанидином представлена в таблице 3.
ЛПС, в % от контроля
Изучена противовоспалительная активность соединения формулы 2. Как видно из результатов, приведенных в таблицах 2 и 3, обсуждаемый здесь потенциальный лекарственный препарат формулы 2 на использованных фармакологических моделях несколько уступает вольтарену, но превосходит аминогуанидин.
Соединение 2 обладает противовоспалительным действием. Для большинства нестероидных противовоспалительных препаратов характерно часто встречающееся побочное действие - повреждение слизистой оболочки желудка и проявление ульцерогенного действия.
В этой связи изучение язвообразующего действия у потенциальных нестероидных противовоспалительных препаратов дает возможность, с одной стороны, выявить наличие и выраженность ульцерогенного эффекта, а с другой - косвенно судить о влиянии изучаемого вещества на биосинтез простогландинов.
Методы исследования.
Исследование язвообразующего действия соединения 2 выполнено в соответствии с Методическими рекомендациями по экспериментальному (доклиническому) изучению новых нестероидных противовоспалительных препаратов [4] и выполнены по следующей схеме:
1. Исследование раздражающего действия соединений на желудок у мышей в дозе в 5 раз выше фармакологической при однократном введении.
2. Исследование язвообразующего действия соединений в дозе, равной фармакологической, при хроническом введении в течение 7 дней.
3. Усиление язвообразующего действия исследуемых соединений в сочетании с 0,6 н. соляной кислотой.
4. Усиление язвообразующего действия исследуемых соединений в сочетании с известными нестероидными противовоспалительными средствами (индометацином).
1. Исследование раздражающего действия соединения 2.
Мышей-самцов массой 23-24 г лишали пищи в течение 24 часов, доступ к воде не ограничивали. Количество мышей в группе - 10.
Исследуемое соединение 2 в дозе 500 мг/кг вводили зондом в желудок, через 6 часов животных забивали, извлекали желудки и подсчитывали число язв, а также рассчитывали ульцерогенный индекс в баллах. Оценку ульцерогенного эффекта проводят по 4-бальной шкале: 0 - отсутствие повреждений, 0,5 - гиперемия, 1 - единичные незначительные повреждения (1 или 2 точечных кровоизлияний), 2 - множественные повреждения (эрозии, точечные кровоизлияния), 3 - значительные и множественные повреждения слизистой (эрозии, кровоизлияния), 4 - грубые повреждения, охватывающие всю поверхность слизистой (массивные кровоизлияния, эрозии, перфорации).
2. Исследование раздражающего действия соединения 2.
Мышам-самцам массой 23-24 г в течение 7 дней вводили соединение 2 в дозе 200,0 мг/кг внутрь. На 7 дней животных умерщвляли ингаляцией CO2, извлекали желудки и подсчитывали число язв. В качестве препаратов сравнения применяли внутрь: индометацин в дозе 20 мг/кг, вольтарен - 50 мг/кг и аминогуанидин - 200 мг/кг. Каждая исследуемая группа состояла из 10 животных.
3. Усиление язвообразующего действия исследуемых соединений в сочетании с 0,6 н. соляной кислотой.
Мышей-самцов массой 23-24 г лишали пищи в течение 24 часов, доступ к воде не ограничивали. Соединение 2 в дозе 100 мг/кг вводили зондом в желудок за 1 час до введения 0,6 н. соляной кислоты (5 мл/кг), через 4 часа животных забивали, извлекали желудки и подсчитывали число язв.
В качестве препаратов сравнения применяли внутрь: индометацин в дозе 20 мг/кг, вольтарен - 50 мг/кг и аминогуанидин - 200 мг/кг. Каждая исследуемая группа состояла из 10 животных.
4. Усиление язвообразующего действия соединения 2 известными нестероидными противовоспалительными средствами [1, 4].
Мышей-самцов массой 23-24 г лишали пищи в течение 24 часов, доступ к воде не ограничивали. Соединение 2 в дозе 100 мг/кг вводили зондом в желудок за 1 час до введения индометацина (20 мг/кг), через 5 часов животных забивали, извлекали желудки и подсчитывали число язв. В качестве препаратов сравнения применяли вольтарен в дозе 50 мг/кг и аминогуанидин в дозе 200 мг/кг; все препараты вводили животным внутрь. Каждая исследуемая группа состояла из 10 животных. Полученные результаты.
Результаты, полученные в ходе исследований, и оценка ульцерогенного действия соединения формулы 2 в сравнении с вольтареном, индометацином и аминогуанидином представлена в таблице 4.
Как видно из полученных результатов, соединение формулы 2 не обладает раздражающим действием на слизистую оболочку желудка как при однократном введении внутрь в дозе 500,0 мг/кг, так и при хроническом введении в дозе 200,0 мг/кг внутрь; также соединение формулы 2 не усиливает язвообразования при сочетанном его применении с 0,6 н. соляной кислотой или индометацином в дозе 20 мг/кг. Препараты сравнения вольтарен и индометацин проявили ульцерогенные эффекты, характерные для нестероидных противовоспалительных средств.
Таким образом, соединение формулы 2 исследовано на наличие у него ульцерогенного действия и показано, что соединение 2 не оказывает раздражающего и ульцерогенного действия на ряде моделей язвообразования, также как аминогуанидин и в отличие от вольтарена и индометацина.
Пример 4. Изучение хондропротекторной активности.
Соединение формулы 2 было изучено также на модели хронического (адъювантный артрит) артрита у крыс линии Вистар. Установлено, что исследуемое соединение препятствовало генерализации воспалительного процесса, уменьшая лейкоцитоз, уровень фактора некроза опухолей - TNFα в плазме крови и выраженность проявления реакции гиперчувствительности замедленного типа (по данным рентгенологического исследования мелких пястнофаланговых и дистальных межфаланговых суставов задней пораженной конечности). Соединение в дозе 50 мг/кг снижало уровень TNFα и по своей эффективности было сопоставимо с препаратом сравнения - индометацином. Соединение формулы 2 в дозировке 50 мг/кг наиболее эффективно препятствовало образованию склероза суставных поверхностей и сужения межфаланговых суставов задней стопы, превосходя по эффективности препарат сравнения.
Снижение толщины хряща в группах с применением разных концентраций нового соединения по сравнению с группой интактных крыс было достоверным, хондропротекторное воздействие соединение формулы 2 проявило в дозе 50 мг/кг.
На основании вышеприведенных экспериментальных исследований был сделан вывод, что новое соединение обладает при внутрижелудочном введении (аналог перорального) системным противовоспалительным действием (уменьшает лейкоцитоз и выработку противовоспалительного цитокина TNFα). Исследуемый препарат в дозах 50 и 75 мг/кг уменьшал выраженность отека задней конечности, а в дозе 50 мг/кг частично препятствовал развитию дистрофии межсуставного хряща скакательного сустава. В этой же дозе препарат проявлял наиболее выраженное (превышающее индометацин) хондропротективное действие в отношении межфаланговых суставов пораженной конечности (рентгенологические данные). При внутрижелудочном введении (аналог перорального) препарат проявлял системное противовоспалительное действие (снижение отека, лейкоцитоза, уровня TNFα, хондропротективное действие), что можно связать с купированием развития реакции гиперчувствительности замедленного типа.
Оценка других видов фармакологической активности соединения 2 показала, что оно обладает противоаллергической, антигипоксической и анальгетической активностью. Все выявленные виды фармакологической активности соединения 2 сохранялись при использовании его не только в виде гидрохлорида, но и в виде других фармацевтически приемлемых солей (например Na, K, Ca, Mg), а также в виде комплексных соединений с приемлемыми в фармацевтике комплексонами (например, с глицирризиновой кислотой).
Литература
1. Машковский М.Д. // Лекарственные средства, изд. 13, Харьков, Торгсин, 1997.
2. Saavedra J.E., Billiar T.R., Williams D.L. et al. // J. Med. Chem. 1997, 40, 1947.
3. Граник В.Г., Григорьев Н.Б. // Оксид азота, М.: Вузовская книга, 2004.
4. Шварц Г.Я., Сюбаев Р.Д. / Методические рекомендации по экспериментальному (доклиническому) изучению новых нестероидных противовоспалительных препаратов // Ведомости научного центра экспертизы и государственного контроля ЛС МЗ России, 2000, №1, с.44-50.
5. Cuzzocrea S., Mazzon Е., Dugo L. et al. / Protective effects of n-acetylcysteine on lung injury and red blood cell modification induced by carrageenan in the rat // FASEB J., 2001, 15(7), p.1187-1200.
6. Freyria A.M., Paul J., Belleville J. et al. / Rat peritoneal macrophage procoagulant and fibrinolytic activities. An expression of the local inflammatory response // Соmр. Biochem. Physiol. A. 1991, V.99, N4, p.517-524.
7. Vogel H.G. // Drug Discovery and evaluation: Pharmacological assays. Springer, 2008.
8. Патент РФ RU 2167856.
9. Рябова С.Ю., Тугушева H.З., Алексеева Л.М., Граник В.Г. // Хим. фарм. журн. 1996, т.30, №7, С.42-46.
10. Рябова С.Ю., Расторгуева Н.А., Лисица Е.А. и др. // Известия АН, Сер. хим., 2003, №6, с.1312-1323.
| название | год | авторы | номер документа |
|---|---|---|---|
| БИОЛОГИЧЕСКИ АКТИВНАЯ ДОБАВКА К ПИЩЕ "БИОСИНОЛ", ОБЛАДАЮЩАЯ ГАСТРОПРОТЕКТОРНОЙ АКТИВНОСТЬЮ | 2009 |
|
RU2397775C1 |
| Средство, обладающее гастропротекторной активностью | 2016 |
|
RU2629090C1 |
| АМИД β -ГЛИЦИРРИЗИНОВОЙ КИСЛОТЫ С ФЕНИЛБОРОНОВОЙ КИСЛОТОЙ, ПРОЯВЛЯЮЩИЙ ПРОТИВОВОСПАЛИТЕЛЬНУЮ И ПРОТИВОЯЗВЕННУЮ АКТИВНОСТЬ | 1991 |
|
RU2032694C1 |
| ГЛИКОПЕПТИД β-ГЛИЦИРРИЗИНОВОЙ КИСЛОТЫ С ДИБУТИЛОВЫМ ЭФИРОМ L-ГЛУТАМИНОВОЙ КИСЛОТЫ, ПРОЯВЛЯЮЩИЙ ПРОТИВОВОСПАЛИТЕЛЬНУЮ И ПРОТИВОЯЗВЕННУЮ АКТИВНОСТЬ | 1991 |
|
RU2024548C1 |
| 3-(2-ДИЭТИЛАМИНОЭТИЛТИО)-1,2,4-ТРИАЗИНО [6,5-B]ИНДОЛА ГИДРОХЛОРИД, ОБЛАДАЮЩИЙ ПРОТИВОВОСПАЛИТЕЛЬНОЙ АКТИВНОСТЬЮ И ПОВЫШАЮЩИЙ ФИЗИЧЕСКУЮ ВЫНОСЛИВОСТЬ И УСТОЙЧИВОСТЬ ОРГАНИЗМА К ГИПОКСИИ | 1981 |
|
SU1014249A1 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ НА ОСНОВЕ НЕСТЕРОИДНЫХ ПРОТИВОВОСПАЛИТЕЛЬНЫХ СРЕДСТВ | 2000 |
|
RU2191582C2 |
| Производные 5-этил-2-амино-1, 3, 4-тиадиазола, обладающие обезболивающей, противовоспалительной, противоаллергической и анальгетической активностями | 2016 |
|
RU2651572C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ С ПРОТИВОВОСПАЛИТЕЛЬНОЙ, КАРДИО- И ХОНДРОПРОТЕКТОРНОЙ АКТИВНОСТЬЮ, ДЕЙСТВИЕМ ПРОТИВ ГАСТРОПАТИЙ, ВЫЗЫВАЕМЫХ НПВП, И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2011 |
|
RU2502507C2 |
| СПОСОБ ПОЛУЧЕНИЯ СРЕДСТВА, ОБЛАДАЮЩЕГО ПРОТИВОЯЗВЕННЫМ ДЕЙСТВИЕМ | 2013 |
|
RU2533228C2 |
| КОРИЧНЫЙ ЭФИР β -ГЛИЦИРРИЗИНОВОЙ КИСЛОТЫ, ПРОЯВЛЯЮЩИЙ ПРОТИВОВОСПАЛИТЕЛЬНУЮ И ПРОТИВОЯЗВЕННУЮ АКТИВНОСТИ | 1989 |
|
SU1626663A1 |
Изобретение относится к новому соединению -N-[3-(4-нитрофениламино)-индол-2-илметилен] аминогуанидин гидрохлориду формулы (2)
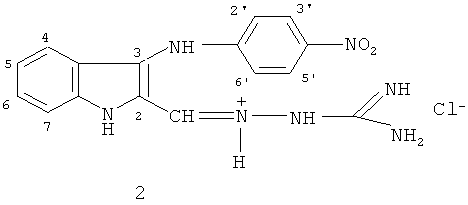
или его фармацевтически приемлемой соли, которое проявляет системное противовоспалительное и хондропротективное действие и не проявляет побочных эффектов. 2 з.п. ф-лы, 4 табл., 4 пр.
1. N-[3-(4-нитрофениламино)-индол-2-илметилен] аминогуанидина гидрохлорид формулы (2)
или его фармацевтически приемлемая соль.
2. N-[3-(4-нитрофениламино)-индол-2-илметилен] аминогуанидина гидрохлорид формулы (2) по п.1, проявляющий системное противовоспалительное действие.
3. N-[3-(4-нитрофениламино)-индол-2-илметилен] аминогуанидина гидрохлорид формулы (2) по п.1, проявляющий хондропротективное действие.
| СОЛИ ПРОИЗВОДНЫХ АМИДИНА И ИНГИБИТОРА ЦИКЛООКСИГЕНАЗЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ НА ИХ ОСНОВЕ | 1996 |
|
RU2167856C2 |
| АМИНОГУАНИДИНЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1992 |
|
RU2095347C1 |
| Известия АН, серия Химическая, 2003, №6, с.1312-1323. | |||

