Настоящее изобретение относится к новым ингибиторам дипептидилпептидазы IV (DPP-IV), эффективным при лечении состояний, опосредованных действием DPP-IV. Как было обнаружено, DPP-IV ответственна за инактивацию глюкагоноподобного пептида-1 (GLP-1). Поскольку GLP-1 является главным стимулятором секреции инсулина в поджелудочной железе и обладает непосредственным благотворным воздействием в отношении утилизации глюкозы, ингибирование DPP-IV представляет собой, как выясняется, привлекательный подход к лечению таких состояний, как инсулиннезависимый сахарный диабет (ИНСД).
Объектом настоящего изобретения является соединение формулы (IA), (IB), (XA), (XB), (YA) или (YB) (соединения по настоящему изобретению)
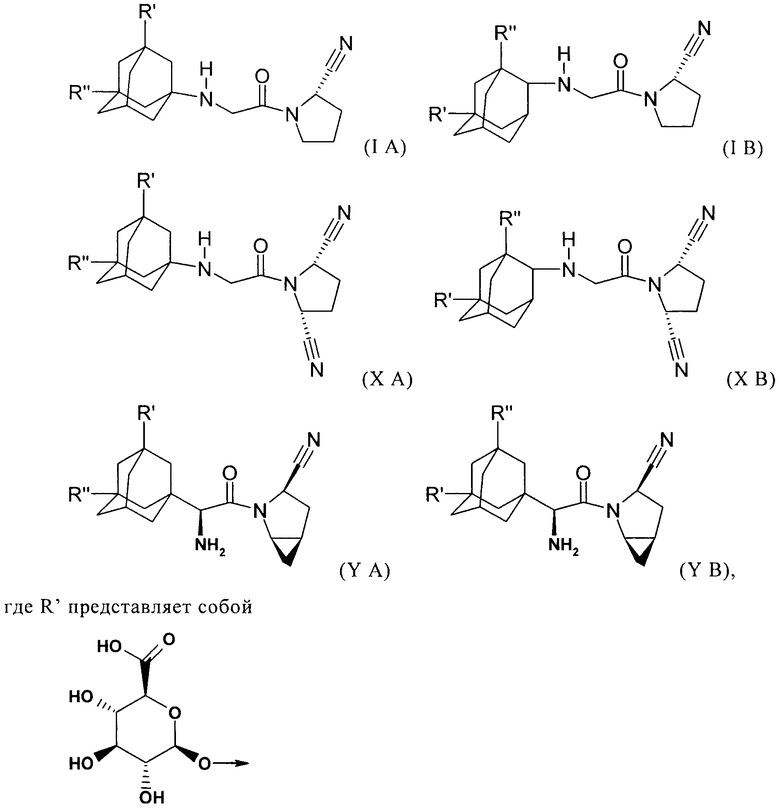
и R'' представляет собой водород, гидроксигруппу, (C1-C7)алкоксигруппу, (C1-C8)алканоилоксигруппу или группу R5R4N-CO-O-, где R4 и R5 независимо друг от друга представляют собой (C1-C7)алкил или фенил, незамещенный или замещенный заместителями, выбранными из (C1-C7)алкила, (C1-C7)алкоксигруппы, галоида и трифторметила, и где R4 дополнительно может представлять собой водород, или R4 и R5 совместно представляют собой (C3-C6)алкилен,
в свободной форме или в форме фармацевтически приемлемой кислотно-аддитивной соли.
Объектом настоящего изобретения также является соединение формулы (IA), (IB), (XA), (XB), (YA) или (YB), как оно описано выше, в котором R" представляет собой водород.
В предпочтительном варианте осуществления настоящего изобретения соединения формулы (IA), (IB), (XA), (XB), (YA) или (YB), как они описаны выше, находятся в химически чистом виде.
Соединения по настоящему изобретению могут существовать в свободной форме или в форме кислотно-аддитивной соли. Предпочтительными являются фармацевтически приемлемые (т.е. нетоксичные, физиологически приемлемые) соли, хотя и другие соли могут также быть полезны, например, для выделения или очистки соединений по настоящему изобретению. Хотя предпочтительные кислотно-аддитивные соли являются гидрохлоридами, также могут использоваться и соли метансульфокислоты, серной, фосфорной, лимонной молочной и уксусной кислоты.
Соединения по настоящему изобретению могут существовать в виде оптически активных изомеров или диастереоизомеров и могут быть разделены и выделены с помощью общепринятых способов, таких как хроматография.
Ниже перечислены определения различных терминов, употребляемых для описания настоящего изобретения. Эти определения действительны для соответствующих терминов, если в конкретных случаях не оговорено иное, при их употреблении на всем протяжении данного описания, как по отдельности, так и в качестве составляющей большей группы.
Термин «алкил» относится к неразветвленным или разветвленным углеводородным группам, содержащим от 1 до 10 атомов углерода, предпочтительно, от 1 до 7 атомов углерода, наиболее предпочтительно, от 1 до 5 атомов углерода. Примеры алкильных групп включают метил, этил, пропил, изопропил, н-бутил, трет-бутил, изобутил, пентил, гексил и им подобные.
Термин «алканоил» относится к группе алкил-C(O)-.
Термин «замещенный адамантил» относится к адамантилу, т.е. 1- или 2-адамантилу, замещенному одним или более, например двумя заместителями, выбранными из алкила, -OR1 или -NR2R3, где R1, R2 и R3 независимо друг от друга являются водородом, алкилом, (C1-C8)алканоилом, карбамилом или -CO-NR4R5, где R4 и R5 независимо друг от друга являются алкилом, незамещенным или замещенным арилом и где один из заместителей R4 и R5 дополнительно может являться водородом, или же R4 и R5 совместно представляют собой (C2-C7)алкилен.
Термин «арил» предпочтительно обозначает фенил. Замещенный фенил предпочтительно является фенилом, замещенным одним или более, например, двумя, заместителями, выбранными из, например, алкила, алкоксигруппы, галоида и трифторметила.
Термин «алкоксигруппа» относится к группе алкил-O-.
Термин «галоид» или «галоген» относится ко фтору, хлору, брому и иоду.
Термин «алкилен» относится к неразветвленному мостику, содержащему от 2 до 7 атомов углерода, предпочтительно, от 3 до 6 атомов углерода, наиболее предпочтительно, 5 атомов углерода.
Термин «химически чистый» следует понимать в контексте настоящего изобретения как означающий существенную степень чистоты от биологического материала, такого как присутствующий в крови, в особенности, менее 10%, предпочтительно, менее 1%, а наиболее предпочтительно - чистоту от подобного биологического материала.
Соединения по настоящему изобретению могут быть получены, например, с помощью способа, включающего сочетание реакционноспособного производного (2-цианопирролидино)карбонилметилена с соответствующим замещенным амином. Более конкретно, при получении соединений формулы I он включает взаимодействие соединения формулы II
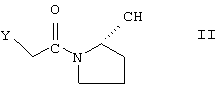
в котором Y является реакционноспособной группой (предпочтительно, галоидом, таким как бром, хлор или иод) с соединением формулы III
 ,
,
в котором R отвечает вышеприведенным определениям, и выделение получаемого таким образом соединения формулы (I A), (I B), (X A), (X B), (Y A) или (Y B) в свободной форме или в форме кислотно-аддитивной соли.
Альтернативные способы получения прекурсоров соединений формулы Y A или Y B описаны в международной заявке на изобретение WO 01/068603, а для соединений формулы X A или X B в WO 05/095339. Описанный ниже способ может быть применен для получения O-глюкуронидной формы соединений, описанных в международной заявке на изобретение WO 01/068603 или WO 05/095339 и в которых R' является -OH.
Остаток R' может быть присоединен к химической структуре любым способом, известным специалисту в соответствующей области, или посредством описанного ниже для структур химической формулы IA или IB способа. С помощью препаративной биоконверсии с использованием в качестве катализатора гомогентата печени крысы получают О-глюкуронид вилдаглиптина, как это описано ниже.
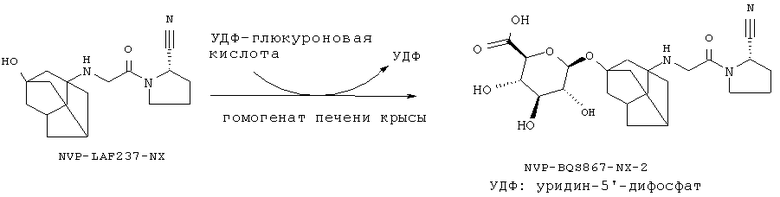
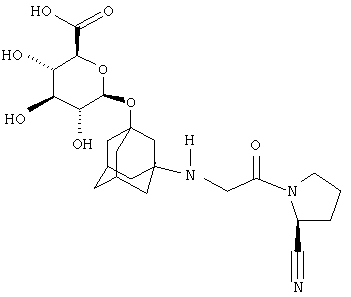
Вилдаглиптин (NVP-LAF237-NX или Галвус®) является новым гипогликемическим (противодиабетическим) лекарственным средством для перорального применения из класса ингибиторов дипептидилпептидазы-4 (DPP-4), проходящим в настоящее время регуляционное тестирование Управлением по контролю за продуктами и лекарствами США. Как было неожиданно обнаружено заявителем, O-глюкуронид вилдаглиптина (Фигура АА) остается столь же активным, сколь и сам вилдаглиптин. Как также было неожиданно обнаружено заявителем, O-глюкуронид вилдаглиптина, помимо одинаковой с вилдаглиптином действенности в отношении ингибирования фермента DPP-4, менее активен в отношении ингибирования ферментов DPP-8 или DPP-9. Особенной неожиданностью оказалось то, что замещение адамантильной группь: в ингибиторах DPP-4 приведет к улучшенному фармакологическому профилю. O-глюкуронид вилдаглиптина может обеспечить дополнительные фармакокинетические или фармакологические преимущества, например, снижение побочных эффектов, лучшую биодоступность.
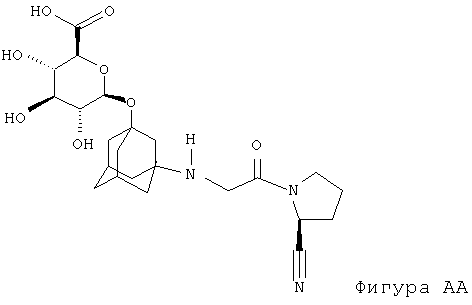
В наиболее предпочтительном варианте осуществления настоящего изобретения О-глюкуронид вилдаглиптина (Фигура АА) находится в химически чистом виде.
Способ по настоящему изобретению может быть осуществлен общепринятым образом. Например, соединение формулы II вводят во взаимодействие с 1-3 эквивалентами, предпочтительно, 3 эквивалентами, первичного амина формулы III. Взаимодействие легко осуществимо в присутствии инертного органического растворителя, такого как хлористый метилен или циклический простой эфир, такой как тетрагидрофуран. Температура предпочтительно находится в диапазоне от, приблизительно, 0° до, приблизительно, 35°C, предпочтительно, между, приблизительно, 0° и, приблизительно, 25°C.
Соединения по настоящему изобретению могут быть выделены из реакционной смеси и очищены общепринятым образом, например, хроматографически.
Исходные вещества могут также быть получены общепринятым образом. Соединения формулы II могут быть получены согласно следующей двухстадийной схеме реакции:
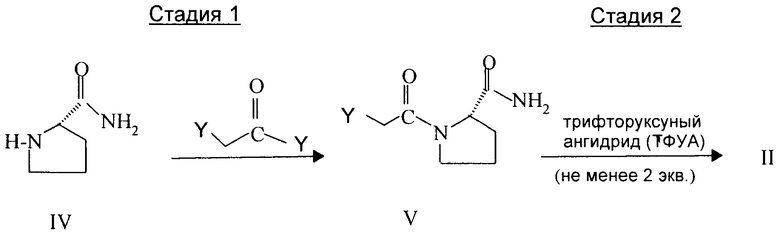
Стадия 1 включает взаимодействие производного пирролидина формулы IV с небольшим мольным избытком галоидангидрида галоидуксусной кислоты, такого как бромангидрид бромуксусной кислоты или хлорангидрид хлоруксусной кислоты, и основанием, таким как карбонат калия или триэтиламин. Реакция легко осуществима в присутствии инертного органического растворителя, такого как тетрагидрофуран или хлорированный алифатический углеводород, такой как хлористый метилен, при температуре от приблизительно 0° до приблизительно 25°C, предпочтительно при температуре между приблизительно 0° и приблизительно 15°C.
Стадия 2 состоит в дегидратации соединения формулы V, получаемого на стадии 1, с помощью 1-2 эквивалентов трифторуксусного ангидрида (ТФУА). Дегидратацию предпочтительно осуществляют в присутствии инертного органического растворителя, такого как тетрагидрофуран или хлорированный алифатический углеводород, такой как хлористый метилен, при температуре от приблизительно 0° до приблизительно 25°C, предпочтительно при температуре между приблизительно 0° и приблизительно 15°C.
Соединение, используемое в качестве исходного вещества, в той мере, в какой его получение не описано специально в контексте, является известным или может быть получено известным способом, или аналогично известным способам, или аналогично способам, описанным в Примере, из известных соединений.
Например, первичные аминосоединения формулы III являются известными и могут быть получены согласно методикам, описанным в литературе, например, в Khim.-Farm. Zh. (1986), 20(7), 810-15.
Наконец, соединения по настоящему изобретению получают либо в свободной форме, либо в виде их солей, если наличествуют солеобразующие группы.
Соединения по настоящему изобретению, включающие основные группы, могут быть преобразованы в кислотно-аддитивные соли, в особенности, в фармацевтически приемлемые кислотно-аддитивные соли. Таковые образуются, например, с неорганическими кислотами, такими как минеральные кислоты, наприме, серная кислота, фосфорная или галоидводородная кислота, или с органическими карбоновыми кислотами. Предпочтительными являются соли, образуемые с соляной кислотой.
Ввиду тесного родства между соединениями в свободной форме и соединениями в форме своих солей, при всяком упоминании соединения в соответствующем контексте подразумевается также и соль, при условии, что это возможно или уместно в соответствующих обстоятельствах.
Соединения, включая их соли, могут также быть получены в форме своих гидратов или могут включать другие растворители, задействованные для их кристаллизации.
Настоящее изобретение также включает фармацевтические композиции, например, полезные для ингибирования DPP-IV, включающие фармацевтически приемлемый носитель или разбавитель и терапевтически эффективное количество соединения формулы I или его фармацевтически приемлемой кислотно-аддитивной соли.
Еще в одном варианте осуществления настоящего изобретения его объектом является способ ингибирования DPP-IV, включающий введение нуждающемуся в таком лечении млекопитающему терапевтически эффективного количества соединения по настоящему изобретению или его фармацевтически приемлемой кислотно-аддитивной соли.
Еще в одном варианте осуществления настоящего изобретения его объектом является способ лечения состояний, опосредуемых ингибированием DPP-IV, включающий введение нуждающемуся в таковом лечении млекопитающему терапевтически эффективного количества соединении по настоящему изобретению или его фармацевтически приемлемой кислотно-аддитивной соли.
Объектом настоящего изобретения также является применение соединения согласно настоящему изобретению или его фармацевтически приемлемой соли, например для изготовления лекарственного средства, предназначенного для профилактики или лечения заболеваний или состояний, связанных с повышенными уровнями DPP-IV.
Как было указано выше, все соединения формулы (I A), (I B), (X A), (X B), (Y A) или (Y B) и их соответствующие фармацевтически приемлемые кислотно-аддитивные соли являются полезными для ингибирования DPP-IV. Способность соединений по настоящему изобретению и их соответствующих фармацевтически приемлемых кислотно-аддитивных солей ингибировать DPP-IV, может быть продемонстрирована посредством анализа DPP-IV, использующего клетки линии Сасо-2, в котором измеряется способность исследуемых соединений к ингибированию активности DPP-IV из экстрактов клеток карциномы ободочной кишки человека. Линию клеток карциномы ободочной кишки человека Сасо-2 получают от Американской коллекции типовых культур (АТСС НТВ 37). Дифференциацию клеток с целью стимуляции экспрессии DPP-IV осуществляют согласно описанию Reisher и др. в статье «Повышенная экспрессия кишечной линии клеток Сасо-2» в журнале Proc. Natl. Acad. Sci., т.90, стр.5757-5761 (1993). Клеточный экстракт получают из клеток, солюбилизированных в 10 мМ Трис-HCl, 0,15 М NaCl, 0,04 трипсин-ингибиторной единицы (t.i.u.) апротинина, 0,5% неионного детергента Нонидет-Р40 при pH 8,0, в результате центрифугирования при 35,000 g в течении 30 минут при температуре 4°C для удаления клеточных остатков. Анализ осуществляют посредством добавления 20 мкг солюбилизированного белка Сасо-2, растворенного до конечного объема 125 мкл в буфере для анализа (25 мМ Трис-HCl, pH 7,4, 140 MM NaCl, 10 мМ KCl, 1% бычьего сывороточного альбумина) в ячейки планшетов для микротитрования. После инкубирования при комнатной температуре в течение 60 минут инициируют взаимодействие посредством добавления 25 мкл 1 мМ субстрата (Н-аланин-пролин-n-НА; n-НА обозначает n-нитроанилин). Реакцию осуществляют при комнатной температуре в течение 10 минут, по прошествии какового времени для остановки реакции добавляют 19 мкл 25% ледяной уксусной кислоты. Исследуемые соединения добавляют, как правило, в виде 30 мкл порций, а объем буфера для анализа понижают до 95 мкл. Стандартную кривую для свободного я-нитроанилина строят с использованием 0-500 мкМ растворов свободного и-НА в буфере для анализа. Получаемая кривая является линейной и используется для интерполяции степени расхода субстрата (каталитическая активность в количестве наномолей расщепленного субстрата в минуту). Конечную точку определяют посредством измерения оптической плотности при длине волны 405 нм в спектрофотометре для планшетов для микротитрования Molecular Devices UV Max.
Эффективность исследуемых соединений в качестве ингибиторов DPP-IV, выраженную в виде IC50 (концентрация, обеспечивающая пятидесятипроцентное ингибирование), рассчитывают из кривых доза-отклик, построенных по восьми точкам с помощью четырехпараметрической логистической функции.
Способность соединений по настоящему изобретению и их соответствующих фармацевтически приемлемых кислотно-аддитивных солей ингибировать DPP-IV также может быть продемонстрирована посредством измерения влияния исследуемых соединений на активность DPP-IV в плазме крови человека или крысы с помощью модифицированного варианта анализа, описанного Kubota и др. в статье «Участие дипептидилпептидазы IV в иммунной реакции in vivo» в журнале Clin. Exp.Immunol., т.89, стр.192-197 (1992). Вкратце, 5 мкл плазмы добавляют в 96-ячеечные планшеты для микротитрования с плоским дном (Falcon), после чего добавляют 5 мкл 80 мМ MgCl2 в буфере для инкубирования (25 мМ HEPES (4-(2-гидроксиэтил)-1-пиперазинэтансульфокислота), 140 мМ NaCl, 1% бычьего сывороточного альбумина степени чистоты - для радиоиммунологического анализа, pH 7,8). После инкубирования при комнатной температуре в течение 60 минут инициируют взаимодействие посредством добавления 10 мкл буфера для инкубирования, содержащего 0,1 мМ субстрата (Н-глицин-пролин-АМК; АМК обозначает 7-амино-4-метилкумарин). Планшеты накрывают алюминиевой фольгой (или содержат в темноте) и инкубируют при комнатной температуре в течение 20 минут. По прошествии 20 минут взаимодействия измеряют флуоресценцию с помощью флуориметра CytoFluor 2350 (длина волны возбуждения 380 нм, испускания - 460 нм; режим чувствительности 4). Как правило, исследуемые соединения добавляют 2 мкл порциями и понижают объем буфера для анализа до 13 мкл. Кривую интенсивности флуоресценции в зависимости от концентрации для чистого АМК строят по данным для 0-50 мкМ растворов АМК в буфере для анализа. Получаемая кривая является линейной, ее используют для интерполяции степени расхода субстрата (каталитическая активность в количестве наномолей расщепленного субстрата в минуту). Как и для предшествующего анализа, эффективность исследуемых соединений в качестве ингибиторов DPP-IV, выраженную в виде IC50, рассчитывают из кривых доза-отклик, построенных по восьми точкам с помощью четырехпараметрической логистической функции.
Ввиду их способности ингибировать DPP-IV, соединения по настоящему изобретению и их соответствующие фармацевтически приемлемые кислотно-аддитивные соли, являются полезными для лечения состояний, опосредуемых ингибированием DPP-IV. На основании вышеизложенного и литературных сведений ожидается, что описанные в настоящей заявке соединения применимы в лечении состояний, таких как инсулиннезависимый сахарный диабет, артрит, ожирение, трансплантация аллотрансплантата и остеопороз. Кроме того, учитывая роль глюкагоноподобных пептидов (таких как GLP-1 и GLP-2) и их связь с ингибированием DPP-IV, ожидается, что описываемые в настоящей заявке соединения применимы, например для обеспечения седативного или анксиолитического эффекта, или для смягчения постоперационных катаболических изменений и гормональных реакции на стресс, или для снижения смертности и заболеваемости после инфаркта миокарда, или для лечения состояний, связанных с вышеуказанными эффектами, которые могут быть опосредованы уровнями GLP-1 и/или GLP-2.
Более конкретно, например соединения по настоящему изобретению и их соответствующие фармацевтически приемлемые кислотно-аддитивные соли улучшают ранний инсулиновый ответ на пероральную провокацию глюкозой и, таким образом, применимы для лечения инсулиннезависимого сахарного диабета. Способность соединений по настоящему изобретению и их соответствующих фармацевтически приемлемых кислотно-аддитивных солей улучшать ранний инсулиновый ответ на пероральную провокацию глюкозой, может быть измерена на инсулинрезистентных крысах нижеприведенным способом.
Самцов крыс породы Спрэг-Доули, выдерживавшихся на высокожировой диете (насыщенные жиры составляют 57% калорий) в течение 2-3 недель, оставляют голодать примерно на 2 часа в день опыта, разделяют на группы по 8-10 и вводят им перорально 10 мкмоль/кг исследуемого соединения в карбоксиметилцеллюлозе. Через 30 минут после исследуемого соединения прямо в желудок подопытных животных вводят пероральный болюс с 1 г/кг глюкозы. Образцы крови, получаемые в различные моменты времени через хронические катетеры из яремной вены, анализируют на содержание глюкозы и иммунореактивного инсулина (IRI) в плазме, а также на активность DPP-IV в плазме. Уровни инсулина в плазме анализируют методом радиоиммунологического анализа (RIA) с использованием двойных антител, в котором применяют специфичное крысиное антиинсулиновое антитело от компании Linco Research (St. Louis, MO). RIA обеспечивает нижний предел обнаружения 0,5 мкЕд/мл с внутрианалитическими и межаналитическими отклонениями менее 5%. Данные выражают в виде процентного увеличения от среднего для контрольных животных. После перорального введения все исследованные соединения обеспечили усиление раннего инсулинового ответа, что привело к повышению толерантности к глюкозе у подопытных инсулинрезистентных животных. Были получены следующие результаты.
Точная дозировка соединений по настоящему изобретению и их соответствующих фармацевтически приемлемых кислотно-аддитивных солей, для лечения состояний, опосредуемых ингибированием DPP-IV, зависит от нескольких факторов, включая конкретного пациента, природу и тяжесть подвергаемого лечению состояния, режим введения и конкретное используемое соединение. Как правило, однако, эффективное лечение состояний, опосредуемых ингибированием DPP-IV, имеет место при энтеральном, например, пероральном, или парэнтеральном, например, внутривенном, введении соединения по настоящему изобретению или соответствующей фармацевтически приемлемой кислотно-аддитивной соли, предпочтительно, при пероральном введении с суточной дозировкой 0,002-5, предпочтительно 0,02-2,5 миллиграмм на килограмм массы тела, или для большинства крупных приматов с суточной дозировкой 0,1-250, предпочтительно 1-100 мг.Типичная стандартная доза для перорального введения составляет 0,01-0,75 мг/кг, от одного до трех раз в сутки. Как правило, вначале вводят меньшую дозу, после чего дозировку постепенно повышают, пока не будет определена оптимальная дозировка для подвергаемого лечению пациента. Верхняя граница дозировки определяется побочными эффектами и может быть определена для подвергаемого лечению пациента посредством испытаний.
Соединения по настоящему изобретению и их соответствующие фармацевтически приемлемые кислотно-аддитивные соли могут быть объединены с одним или несколькими фармацевтически приемлемыми носителями и, необязательно, одним или несколькими иными общепринятыми фармацевтическими адъювантами и могут вводиться энтерально, например, перорально, в форме таблеток, капсул, каплет и т.п., или парэнтерально, например, внутривенно, в форме стерильных растворов или суспензий для инъекций. Энтеральные и парэнтеральные композиции могут быть изготовлены общепринятыми способами. Примеры подходящих препаратов, включающих соединения по настоящему изобретению, описаны в международных заявках на изобретение WO 2005/067976 или WO 2006/078593.
Таким образом, объектом настоящего изобретения также является фармацевтическая композиция, включающая соединение по любому из п.п.1-4 формулы изобретения или описанные в настоящей заявке соединения в свободной форме или в форме фармацевтически приемлемой кислотно-аддитивной соли совместно с по меньшей мере одним фармацевтически приемлемым носителем или разбавителем.
Фармацевтическими композициями согласно настоящему изобретению являются композиции, пригодные для энтерального, такого как пероральное или перректальное, трансдермального и парэнтерального введения млекопитающим, включая человека, для лечения состояний, опосредованных активностью DPP-4. Такие состояния включают пониженную толерантность к глюкозе, диабет типа 2 и ожирение.
Так, фармакологически активные соединения по настоящему изобретению могут быть использованы для изготовления фармацевтических композиций, включающих эффективное количество таковых соединений в сочетании или смеси с наполнителями или носителями, подходящими для энтерального или парэнтерального введения. Предпочтительными являются таблетки или желатиновые капсулы, содержащие активный ингредиент совместно с:
а) разбавителями, например, лактозой, декстрозой, сахарозой, маннитом, сорбитом, целлюлозой и/или глицином,
б) скользящими веществами, например, оксидом кремния, тальком, стеариновой кислотой, ее магниевой или кальциевой солью и/или полиэтиленгликолем, а для таблеток также
в) связующими веществами, например, алюмосиликатом магния, крахмальной пастой, желатином, трагакантом, метилцеллюлозой, натрий-карбоксиметилцеллюлозой и/или поливинилпирролидоном, и, если то требуется
г) веществами, способствующими распадению, например, крахмалами, агаром, альгиновой кислотой или ее натриевой солью, или же шипучими смесями, и/или
д) поглотителями, красителями, ароматизаторами и подсластителями.
Композиции для инъекций предпочтительно являются изотоническим водными растворами или суспензиями, а суппозитории предпочтительнее всего изготавливать из жировых эмульсий или суспензий.
Упомянутые композиции могут быть стерилизованы и/или содержать адъюванты, такие как консерванты, стабилизаторы, смачивающие вещества или эмульгаторы, вещества, способствующие растворению, соли для регулирования осмотического давления и/или буферы. Кроме того, они могут также содержать другие терапевтически ценные вещества. Таковые композиции получают согласно общепринятым способам смешения, грануляции и покрытия, соответственно, и содержат приблизительно 0,1-75%, предпочтительно приблизительно 1-50%, активного ингредиента.
Подходящие препараты для трансдермального применения включают терапевтически эффективное количество соединения по настоящему изобретению совместно с носителем. Предпочтительные носители включают всасываемые фармакологически приемлемые растворители, способствующие проникновению через кожу пациента. Как правило, трансдермальные устройства имеют форму пластыря, включающего основу, резервуар, содержащий соединение, необязательно в смеси с носителями, необязательно барьер, контролирующий скорость, для доставки соединения к коже пациента с контролируемой и заданной заранее скоростью в течение продолжительного периода времени, а также средства закрепления устройства на коже.
В соответствии с изложенным, объектом настоящего изобретения являются фармацевтические композиции, как они описаны выше, для лечения состояний, опосредованных ингибированием дипептидилпептидазы IV, предпочтительно пониженной толерантности к глюкозе, диабета типа 2 и ожирения.
Объектом настоящего изобретения также является применение фармацевтических композиций, как они описаны выше, для лечения состояний, опосредованных ингибированием дипептидилпептидазы IV, предпочтительно пониженной толерантности к глюкозе, диабета типа 2 и ожирения, где соединение по настоящему изобретению (как оно определено в любом из п.п.1-4 формулы изобретения) вводят в сочетании с другим терапевтическим средством, например, одним или двумя дополнительными средствами, как это описано ниже.
Фармацевтические композиции могут содержать терапевтически эффективное количество соединения по настоящему изобретению, как оно определено в его контексте (например, в п.п.1-4 формулы изобретения), как отдельно, так и в сочетании с другим терапевтическим средством, причем каждое может присутствовать, например, в терапевтически эффективной дозе, известной в соответствующей области технике. Подобные терапевтические средства включают:
а) противодиабетические средства, такие как инсулин, производные и миметики инсулина, стимуляторы секреции инсулина, такие как производные сульфонилмочевины, например, Глипизид, глибурид и амарил, инсулинотропные лиганды рецепторов сульфонилмочевины, такие как меглитиниды, например, натеглинид и репаглинид, ингибиторы протеинтирозинфосфатазы-1В (РТР-1В), такие как РТР-112, ингибиторы GSK3 (киназы-3 гликогенсинтазы), такие как SB-517955, SB-4195052, SB-216763, NN-57-05441 и NN-57-05445, лиганды RXR (ретиноидного Х-рецептора), такие как GW-0791 и AGN-194204, ингибиторы натрий-зависимого котранспортера глюкозы, такие как Т-1095; ингибиторы гликогенфосфорилазы А, такие как BAY R3401, бигуаниды, такие как метформин, ингибиторы альфа-глюкозидазы, такие как акарбоза, GLP-1 (глюкагоноподобный пептид-1), аналоги GLP-1, такие как эксендин-4 и миметики GLP-1, а также ингибиторы DPP-IV (дипептидилпептидазы-IV), такие как вилдаглиптин;
б) гиполипидемические средства, такие как ингибиторы редуктазы 3-гидрокси-3-метилглутарил-коэнзим-А-редуктазы (HMG-CoA), например ловастатин, питавастатин, симвастатин, правастатин, церивастатин, мевастатин, велостатин, флувастатин, далвастатин, аторвастатин, розувастатин и ривастатин, ингибиторы скваленсинтазы, лиганды FXR (фарнезоидного Х-рецептора) и LXR (Х-рецептора печени), холестирамин, фибраты, смолы, связывающие желчные кислоты, такие как холестирамин, никотиновая кислота и другие агонисты связанного с G-белком рецептора GPR109, ингибиторы всасывания холестерина, такие как эзетимиб, ингибиторы СЕТР (белка-переносчика сложных эфиров холестерина), а также аспирин;
в) средства против ожирения, такие как орлистат, сибутрамин и антагонисты каннабиноидного рецептора 1 (СВ1), например римонабант; и
г) средства против повышенного давления, например петлевые диуретики, такие как этакриновая кислота, фуросемид и торсемид, ингибиторы ангиотензин-превращающего фермента (АСЕ), такие как беназеприл, каптоприл, эналаприл, фозиноприл, лизиноприл, моэксиприл, перинодоприл, хинаприл, рамиприл и трандолаприл, ингибиторы Na-K-АТФазного (Na-K-аденозинтрифосфатазного) мембранного насоса, такие как дигоксин, ингибиторы нейтральной эндопептидазы (NEP), ингибиторы ACE/NEP, такие как омапатрилат, сампатрилат и фазидотрил, антагонисты ангиотензина II, такие как кандесартан, эпросартан, ирбесартан, лосартан, телмисартан и валсартан, в частности, валсартан, ингибиторы ренина, такие как дитекирен, занкирен, терлакирен, алискирен, RO 66-1132 и RO-66-1168, блокаторы β-адренергических рецепторов, такие как ацебутолол, атенолол, бетаксолол, бисопролол, метопролол, надолол, пропанолол, соталол и тимолол, инотропные средства, такие как дигоксин, добутамин и милринон, блокаторы кальциевых каналов, такие как амлодипин, бепридил, дилтиазем, фелодипин, никардипин, нимодипин, нифедипин, нисолдипин и верапамил, антагонисты альдостероновых рецепторов и ингибиторы альдостеронсинтазы;
д) агонисты пролифератор-активирующих рецепторов пероксисом, такие как фенофибрат, пиоглитазон, розиглитазоы, тезаглитазар, BMS-298585, L-796449, соединения, непосредственно описанные в международной заявке на изобретение WO 2004/103995, т.е. соединения согласно примерам 1-35 или соединения, непосредственно перечисленные в п.21 формулы изобретения, или соединения, непосредственно описанные в международной заявке на изобретение WO 03/043985, т.е. соединения согласно примерам 1-7, или соединения, непосредственно перечисленные в п.19 формулы изобретения, и в особенности (R)-1-{4-[5-метил-2-(4-трифторметилфенил)оксазол-4-илметокси]бензолсульфонил}-2,3-дигидро-1Н-индол-2-карбоновая кислота или ее соль.
В каждом отдельном случае указанные в пунктах формулы изобретения соединения и конечные продукты рабочих примеров, содержание конечных продуктов, фармацевтических препаратов и пунктов формулы изобретения, включены в настоящую заявку посредством ссылки на соответствующие публикации и заявки на изобретения.
Таким образом, настоящее изобретение включает в свой объем фармацевтические композиции, содержащие:
i) соединение по любому из пп.1-4 формулы изобретения, и
ii) по крайней мере одно соединение, выбранное из
a) противодиабетических средств,
b) гиполипидемических средств,
c) средств против ожирения,
d) средств против повышенного давления,
e) агонистов пролифератор-активирующих рецепторов пероксисом,
iii) один или несколько фармацевтически приемлемых носителей.
Другие примеры противодиабетических соединений описаны Patel Mona в Expert Opin Investig Drugs, 2003, 12(4), 623-633 на фигурах 1-7, которые включены в настоящую заявку посредством ссылки. Соединение по настоящему изобретению может вводиться как одновременно с другим активным ингредиентом, до или после него, так и по отдельности, одинаковым или разным путем введения, или же совместно, в том же фармацевтическом препарате.
Структура терапевтических средств, обозначаемых кодовыми названиями, родовыми или торговыми наименованиями, может быть взята из текущего издания стандартного справочника «The Merck Index» или из баз данных, например, «Patents International)) (например, издательства IMS World Publications). Соответствующее их содержание включено в настоящую заявку посредством ссылки.
В соответствии с изложенным, объектом настоящего изобретения являются фармацевтические композиции, включающие терапевтически эффективное количество соединения по настоящему изобретению в сочетании с терапевтически эффективным количеством другого терапевтического средства, предпочтительно, выбранного среди противодиабетических средств, гиполипидемических средств, средств против ожирения или средств против повышенного давления, наиболее предпочтительно, из противодиабетических средств или гиполипидемических средств, как те описаны выше.
Объектом настоящего изобретения также являются фармацевтические композиции, как они описаны выше, для применения в качестве лекарственного средства.
Объектом настоящего изобретения также является применение фармацевтических композиций или комбинаций, как они описаны выше, для изготовления лекарственного средства для лечения состояний, опосредуемых ингибированием дипептидилпептидазы-IV, предпочтительно пониженной толерантности к глюкозе, диабета типа 2 и ожирения.
Соединения по настоящему изобретению и их соответствующие фармацевтически приемлемые кислотно-аддитивные соли могут быть введены в фармацевтические композиции для энтерального и парентерального применения, содержащие такое количество активного вещества, которое эффективно для лечения состояний, опосредуемых ингибированием DPP-IV, подобные композиции в форме стандартной дозировки и подобные композиции, включающие фармацевтически приемлемый носитель.
Соединения по настоящему изобретению (включая относящиеся к любому из его отдельных аспектов и любому из примеров) могут вводиться в энантиомерно чистой форме (например, энантиомерная чистота >98%, предпочтительно, >99%) или совместно с R-энантиомером, например, в рацемической форме. Приведенные выше диапазоны дозировок основаны на соединениях по настоящему изобретению (исключая количество R-энантиомера).
Нижеследующие примеры описывают характерные соединения, входящие в объем настоящего изобретения, и их синтез. Следует, однако, четко понимать, что они приведены только в иллюстративных целях.
I. Пример 1
1-[(3-Гидрокси-1 -адамантил)амино]ацетил-2-цианопирролидин (S) (международное непатентованное название (МНН): вилдаглиптин) или NVP-LAF237
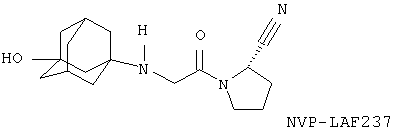
А. 1-Аминоадамантан-3-ол:
Может быть задействован синтез, описанный в KJiim.-Farm. Zh. (1986), 20(7), 810-15 с небольшими модификациями.
К быстро перемешиваемой охлаждаемой смесью льда с водой прозрачной и бесцветной смеси концентрированной серной кислоты 96% (210 мл, 3,943 ммоля) и 65% азотной кислоты (21,0 мл, 217,0 ммоля) добавляют небольшими порциями в продолжение 30 минут 21,0 г (112,0 ммоля) гидрохлорида 1-адамантиламина (99%). Во время добавления гидрохлорида адамантиламина имеет место легкое пузырение и реакция протекает слегка экзотермично. Данный пузырящийся раствор желтого цвета перемешивают при температуре смеси льда с водой в течение около 2 часов, а затем при комнатной температуре в течение 30 часов. После этого прозрачную светло-желтую реакционную смесь выливают в около 100 г льда, и получаемый таким образом раствор приобретает прозрачным зелено-синий вид.
Раствор помещают на баню из воды со льдом и оставляют перемешиваться в течение 30 минут. После этого добавляют небольшими порциями в продолжение 45 минут примерно 550 г 89% чистого КОН (8,74 моля). В процессе этого добавления реакция протекает экзотермично, причем температура достигает 80°C, и выделяются большие количества коричневого газообразного NO2. В конце добавления реакционная смесь густеет и образуются твердые вещества белого цвета (как продукты, так и соли). После этого получаемую белую пасту выливают на воронку Бюхнера с целитовым фильтром и промывают 1,2 л CH2Cl2. Затем слой CH2Cl2 г экстрагируют от водного слоя и сушат над Na2SO4. После этого раствор фильтруют и концентрируют (роторный испаритель/насос), что приводит к 1-аминоадамантан-3-олу в виде твердого вещества белого цвета.
B. 1-Хлорацетил-2-цианопирролидин
К перемешиваемому механической мешалкой раствору 20,0 г (180,0 ммоля) хлорангидрида хлоруксусной кислоты и 97 г (0,70 ммоля) карбоната калия в 150 мл тетрагидрофурана добавляют по каплям в продолжение 45 минут раствор 20,0 г (180,0 ммоля) L-пролинамида в 500 мл тетрагидрофурана. После этого данную реакционную смесь перемешивают механической мешалкой в течение еще двух часов при комнатной температуре. Затем реакционную смесь фильтруют для удаления солей калия и сушат фильтрат над Na2SO4. После этого удаляют Na2SO4 посредством фильтрования и добавляют к получаемому бесцветному фильтрату трифторуксусный ангидрид (25,0 мл, 0,180 ммоля) одной порцией. Затем перемешивают реакционную смесь магнитной мешалкой в течение 1 часа при комнатной температуре и концентрируют получаемый таким образом прозрачный раствор желтого/оранжевого цвета на роторном испарителе. Избыток трифторуксусного ангидрида удаляют посредством добавления к сконцентрированному маслянистому веществу этилацетата и повторного концентрирования на роторном испарителе. Эти действия по удалению повторяют трижды.
Получаемое таким образом маслянистое вещество распределяют между фазами этилацетата и воды. После этого экстрагируют продукт этилацетатом и дважды промывают этилацетатом водный слой. Затем последовательно промывают объединенные органические фракции водой и соляным раствором, сушат их над сульфатом магния, фильтруют и концентрируют, что приводит к 1-хлорацетил-2-цианопирролидину в виде твердого вещества желтого цвета.
C. 1-[(3-Гидрокси-1-адамантил)амино]ацетил-2-цианопирролидин, (S)
К гетерогенному раствору соединения А (1-аминоадамантан-3-ол (5,80 г, 34,7 ммоля) в CH2Cl2 (68,0 мл) добавляют 9,6 г (69 ммолей) K2CO3. Затем охлаждают данную гетерогенную смесь на бане из воды со льдом и добавляют по каплям в продолжение 30 минут раствор 3,0 г (17 ммолей) соединения Б (1-хлорацетил-2-цианопирролидин) в 25,0 мл CH2Cl2. Получаемую таким образом
смесь перемешивают в течение 2 часов при температуре 0°C и в течение 6 суток при комнатной температуре. После этого реакционную смесь концентрируют, что приводит к пастообразному веществу желтого цвета, которое очищают на силикагеле с помощью флэш-хроматогарфической системы SIMS/Biotage и 7% раствора метанола в хлористом метилене в качестве элюента, что приводит к указанному в заглавии соединению в форме свободного основания в виде твердого кристаллического вещества белого цвета (температура плавления 138°C-140°C, 13С ЯМР (м.д.)=119,59).
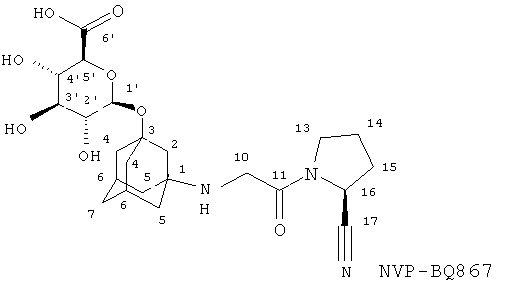
A.D. Биокаталитический синтез NVP-BQS867 (О-глюкуронида вилдаглиптина) и NVP-BRU563 (меченого О-глюкуронида вилдаглиптина)
Схема реакции ферментативного глюкуронидирования NVP-LAF237 при катализе гомогенатом печени крысы представлена на фигуре 1-1.
Фигура I-1. Биотрансформация NVP-LAF237
1. Получение гомогената печени крысы
Две порции по 15 г и одну по 11 г замороженной печени крысы размораживали и нарезали на мелкие кусочки. После добавления к каждой порции печени 0,5 объемных эквивалента ледяного 0,9% раствора NaCl и смешения в блендере Dispomix, ткань гомогенизировали в гомогенизаторе для тканей «Potter S» (Braun Biotech Inc., Melsungen, Germany) при охлаждении в смеси льда с водой посредством троекратного перемещения тефлонового песта вверх и вниз при 100% скорости перемешивания. Гомогенат загружали до достижения конечной массы 81 г и центрифугировали его при температуре 4-6°C в течение 30 минут при 10,000 об./мин. (=17,000×g) в центрифуге фирмы Beckmann Coulter (Fullerton, СА, USA) типа Avanti J-HC, оборудованной ротором JA-10. Надосадочная жидкость служила в качестве источника ферментов.
2. Биопреобразование в препаративном масштабе
NVP-LAF237 (вилдаглиптин) глюкуронидируют в масштабе 340 мг при следующих условиях: NVP-LAF237 5 мМ, УДФ-глюкуронат (Yamasa Co., Tokyo, JP) 20 мМ, MgCl2 20 MM, HEPES, 160 мМ, pH 8,5, гомогенат печени крысы 20% об./об., общий объем 224 мл, инкубирование в течение 5 часов при температуре 37°C и еще в течение 14 часов при температуре 30°C. Взаимодействия осуществляют в 50 мл пробирках марки Nunc, в каждую из которых помещают 20 мл реакционной смеси, встряхиваемых при 170 об./мин. в микробиологическом лабораторном встряхивателе с радиусом вращения 5 см.
Препаративную реакцию останавливают посредством добавления 224 мл ацетонитрила и смешивания в течение 10 минут при температуре 20°C. После центрифугирования в течение 15 минут при 8000 об./мин. (ротор JA-10) гранулу суспендируют в 50 мл ацетонитрила в деионизированной воде 50% об./об. и центрифугируют повторно. Объединенные надосадочные жидкости концентрируют при пониженном давлении при температуре 25°C до достижения объема 50 мл. Мутный концентрат центрифугируют при 5000 об./мин., и, после фильтрования надосадочной жидкости через стекловолокно, подвергают ее препаративной высокоэффективной жидкостной хроматографии (ВЭЖХ).
Для глюкуронидирования [l3C5 15N]-меченого NVP-LAF237 (50 мг масштаб) условия биотрансформации те же, что и для NVP-LAF237 в масштабе 340 мг, за тем исключением, что концентрация УДФ-глюкуроната составляет 80 мМ, гомогенат печени крысы - 30% об./об., объем заполнения пробирок Nunc составляет 16,5 мл, общий объем равен 33 мл, время взаимодействия 4 часа, а скорость встряхивания 200 об./мин.
Реакцию останавливают посредством добавления в каждую пробирку 16,5 мл ацетонитрила, инкубирования в течение 15 минут на льду и последующего смешения. После центрифугирования в течение 15 минут при 17,000 об./мин. (ротор JA-10) осадок заново суспендируют в 20 мл ацетонитрила в деионизированной воде 50% об./об. и повторно центрифугируют. Объединенные надосадочные жидкости концентрируют при пониженном давлении при температуре 30°C до достижения объема 10 мл, снова разбавляют до конечного объема 25 мл и центрифугируют до достижения окончательной прозрачности. Гранулу заново суспендируют в 5 мл деионизированной воды, после центрифугирования объединяют два последних надосадочных раствора и подвергают их препаративной ВЭЖХ.
Измерение степени преобразования / аналитическая ВЭЖХ с детектором на основе диодной матрицы (DAD)
По 200 мкл 50% ацетонитрила в воде смешивали с 50 мкл девятнадцатичасового образца 340 мг загрузки NVP-LAF237 и с 25 мкл четырехчасового образца загрузки с [l3C5 15N]-меченым NVP-LAF237 и центрифугировали в охлаждаемой центрифуге Sigma 4К15С. Надосадочные жидкости анализировали с помощью аналитической обращено-фазовой ВЭЖХ-DAD (хроматограф ВЭЖХ-DAD серии 1100 компании Agilent Technologies, Basel, Switzerland, две соединенных колонки типа Chromolith Performance RP-18e 100×4,6 мм с картриджем Chromolith Guard Cartridge RP-18e 5×4,6 мм (Merck), объемная скорость 2 мл/мин, подвижная фаза А: 3 мМ водный раствор H3PO4, подвижная фаза Б: ацетонитрил, градиент от 3% к 15% Б в течение 5 минут, детекция с помощью диодной матрицы в диапазоне 200-400 нм). Степень преобразования оценивают на основе площадей пиков поглощения в УФ-области при 205 нм глюкуронида и NVP-LAF237.
5. Е. Очистка метаболитов
a) a) NVP-BQS867-NX-2
Часть биопреобразованного сырого вещества (5 мл или примерно 10%) используют для получения образца NVP-BQS867-NX-2. К этому объему добавляют 35 мл 10 мМ водного раствора NH4AC и 8 мл метанола. Доводят pH до 7 с помощью разбавленной уксусной кислоты. Получаемую смесь затем используют в качестве исходного раствора для препаративной жидкостной хроматографии.
Препаративная жидкостная хроматографическая система с масс-спектральным детектором состояла из системы самоочистки компании Waters (Waters Corp.Milford, MA, USA) с насосом 2525, системы ввода и сбора образцов 2767, фотодиодным детектором 2996, подпиточным насосом 515, масс-спектрометром ZQ2000 и программным обеспечением MassLynx 4.0 и FractionLynx 4.0.
Использовали колонку Atlantis dC18, 5 мкм, 19 мм×100 мм (Waters), объемная скорость составляет 20 мл/мин., подвижная фаза А: 10 мМ водный раствор NH4Ac, pH 7,0, подвижная фаза Б: CH3CN/CH3OH 4:1, 10 мМ NH4Ac, программируемый градиент: 0 мин. - 5% Б, 2 мин. - 5% Б, 7 мин. - 20% Б, 7,1 -12 мин. - 95% Б, 12,1 - 14 мин. - 5% Б, объем пробы 1 - 5 мл. Вытекающий из колонки элюат разделяли на две части, очень малую часть смешивали непосредственно в системе с добавочной жидкостью 2-пропанол/H2O/HCOOH 400:100:1 и вводили в ионный источник масс-спектрометра, а остальное направляли в детектор на основе диодной матрицы с регистрацией в диапазоне 200-600 нм, а затем в систему ввода и сбора образцов для сбора фракций.
Масс-спектрометр был оборудован комбинированным интерфейсом для ионизации электрораспылением и химической ионизации при атмосферном давлении и применяется в варианте ионизации электрораспылением (ИЭР) в режиме положительных ионов. Устанавливают напряжение на капилляре 3 кВ и напряжение на входном сопле 30 В, диапазон масс составляет 250-550 Да.
Целевую фракцию собирают исходя из времени удерживания в диапазоне 5,3-6,7 мин. Объединяют целевые фракции из 12 циклов ВЭЖХ, отгоняют органический растворитель при пониженном давлении при температуре 30°C и сушат оставшийся раствор посредством сублимационной сушки при температуре -80°C и давлении 0,2 мбар. Было получено 15 мг практически бесцветного лиофилизата.
Чистоту определяли с помощью ВЭЖХ-DAD и ВЭЖХ-МС (ВЭЖХ с масс-спектральным детектором) (см. раздел 2.2.2): за исключением солей (ацетат аммония) и воды образец NVP-BQS867-NX-2 имел чистоту >97%.
b) b) NVP-BRU563-NX-1 (стабильный меченый глюкуронид)
К водному экстракту добавляют равный объем 20 мМ раствора NH4Ac, чтобы концентрация NH4Ac составила 10 мМ. Данную смесь доводят до pH 7,0 разбавленной уксусной кислотой, а затем используют в качестве исходного раствора для препаративной жидкостной хроматографии.
Препаративная жидкостная хроматографическая система с масс-спектральным детектором описана в разделе 2.1.3.1.
Использовали колонку Atlantis Prep dC18 OBD, 5 мкм, 19 мм×100 мм (Waters), объемная скорость составляет 40 мл/мин., подвижная фаза А: 10 мМ водный раствор NH4Ac, подвижная фаза Б: CH3CN/CH3OH 4:1+10 мМ NH4AC, программируемый градиент: 0 мин. - 5% Б, 2 мин. - 5% Б, 7 мин. - 20% Б, 7,1-12 мин. - 95% Б, 12,1-14 мин. - 5% Б, объем пробы 2-6 мл. Вытекающий из колонки элюат разделяли на две части таким же образом, как это описано в разделе 2.1.3.1.
Масс-спектрометр был оборудован комбинированным интерфейсом для ионизации электрораспылением и химической ионизации при атмосферном давлении и применяется в варианте ионизации электрораспылением (ИЭР) в режиме положительных ионов. Устанавливают напряжение на капилляре 3 кВ и напряжение на входном сопле 30 В, диапазон масс составляет 100-1000 Да.
Целевую фракцию собирают исходя из времени удерживания в диапазоне 7,0-9,0 мин. Объединяют целевые фракции из 12 циклов ВЭЖХ, отгоняют органический растворитель при пониженном давлении при температуре 30°C и сушат оставшийся раствор посредством сублимационной сушки при температуре -80°C и давлении 0,2 мбар. Было получено 63 мг практически бесцветного лиофилизата.
Чистоту определяли с помощью ВЭЖХ-DAD и ВЭЖХ-МС (см. раздел 2.2.2): за исключением солей (ацетат аммония) и воды образец NVP-BRU563-NX-1 имел чистоту >98%.
В. F. Анализы для определения структуры
1. Спектроскопия ядерного магнитного резонанса (ЯМР) Соединения растворяют в 5 мкл пердейтерированного диметилсульфоксида (ДМСО-d6) и заливают в ампулы для ЯМР диаметром 1 мм. Для NVP-BQS867 регистрируют одномерный спектр на ядрах 1H и двумерные гомо- и гетероядерные спектры (COSY (корреляционная спектроскопия), HSQC (гетероядерная спектроскопия с одноквантовым переносом когерренции), НМВС (спектроскопия гетероядерной корреляции через несколько связей), ROESY (спектроскопия ядерного эффекта Оверхаузера во вращающейся системе координат)). Для NVP-BRU563 получают спектр на ядрах 1H. Все спектры регистрировали при температуре 300°K с помощью спектрометра Bruker DRX600, с использованием 1H {l3C5 15N} датчика Microliterprobe. Для NVP-BQS867 также регистрировали спектр на ядрах 13C с помощью спектрометра BRUKER DRX600, с использованием 5 мм 13С {1H} датчика Cryoprobe.
2. 2. ВЭЖХ-масс-спектрометрия
Жидкостный хроматограф состоял из сверхпроизводительно жидкостного хроматографа Waters UPLC Acquity (Waters), оборудованного детектором на основе фотодиодной матрицы Waters Acquity 2996. Колонка: Acquity UPLC ВЕН С18, 1,7 мкм, 1,0×150 мм (Waters), объемная скорость 0,1 мл/мин., элюент А: H2O/ТФУК (трифторуксусная кислота) 100:0,1, элюент Б: ацетонитрил/ТФУК 100:0,1, градиент: 0 мин. 5% Б, 1 мин. 5% Б, 11 мин. 40% Б, 13-16 мин. 95% Б, 30°C, УФ-детекция в диапазоне 200-350 нм, разрешение 2,4 нм, объем пробы 5 мкл. Вещество растворяют в смеси вода/ацетонитрил 9:1 с концентрацией примерно 0,3 мг/мл. Оценку чистоты производят исходя из УФ-сигнала при 210 нм как процент от площади искомого пика. Вытекающий из колонки элюат непосредственно направляют в ионный источник масс-спектрометра.
Использовали тройной квадрупольный масс-спектрометр TSQ Quantum AM (Thermo, San Jose, CA, USA), оборудованный интерфейсом для ионизации электрораспылением, используемым в режиме положительных ионов. Прибор управляется программным обеспечением Xcalibur версии 2.0. Устанавливают поток газа-распылителя 25 единиц, поток встречного газа 5 единиц и напряжение распыления 3 кВ. Температура нагреваемого металлического капилляра поддерживается при 300°C, диапазон масс составляет от 200 до 800 Да. Параметры тандемной масс-спектрометрии (МС/МС): столкновительный газ - аргон, 1,5 мторр, энергия столкновений 25 В.
II. G. Результаты
А. 1. Биокаталитический синтез
Для двух препаративных реакций методом аналитической ВЭЖХ-DAD были получены следующие величины степени преобразования: 66% для образца с 340 мг NVP-LAF237 и 94% для образца с [l3C5 15N]-меченым NVP-LAF237 (50 мг).
2. Очистка глюкуронидов
Как было описано в предшествующем разделе, 15 мг О-глюкуронида NVP-BQS867-NX-2 были получены с помощью препаративной ВЭЖХ-МС из 5 мл водного культурального экстракта, полученного после биотрансформации. После всей препаративной реакции с 340 мг NVP-LAF237 были выделены четыре порции NVP-BQS867-NX (от NX-1 до NX-4), в целом составившее 295 мг О-глюкуронида.
3. Определение структуры
Масс-спектр NVP-BQS867-NX-2 содержит ион [M+H]+ массой 480,1, свидетельствующий о молекулярной массе 479. Это согласуется с глюкуронидом NVP-LAF237. Спектр ионов после столкновительной диссоциации (МС/МС) демонстрирует потерю глюкуронида (m/z 304), равно как и глюкуроновой кислоты (m/z 286).
Структура NVP-BQS867-NX-2 (фигура II-1) была однозначно выяснена на основе данных ЯМР-спектроскопии. Основные отличия ЯМР-спектра NVP-BQS867 по сравнению с NVP-LAF237 преимущественно связаны с резонансами, относящимися к глюкуронидному остатку. Химические сдвиги 1H (4,42 м.д.) и 13C (96,4 м.д.) аномерного резонанса положения 1' глюкуроновой кислоты свидетельствует о том, что последняя присоединена к кислороду, а не к азоту. Корреляция в спектре НМВС между H-1' и С-3 и корреляции в спектре ROESY между H-1' и Н-2 и Н-4 однозначно подтверждают структуру, представленную на фигуре II-1.
Интерпретация всех данных и корреляций ЯМР приводит к единственной структуре, согласующейся с масс-спектральным результатом (молекулярная масса 479, m/z MH+=480,1). Сводка всех химических сдвигов 1H и 13C и соответствующих гомо- и гетероядерных корреляций приведена в таблице II-1.
Соединение NVP-BRU563-NX-3, биосинтезированное из [U-пирролидин, циано- 13С5, пирролидин-15N]-меченого NVP-LAF237, демонстрирует спектральные свойства, идентичные NVP-BQS867-NX за ожидаемыми исключениями для стабильных меток: масса иона MN+ составляет 486,2, что на 6 Да выше, чем у NVP-BQS867-NX, причем в спектре МС/МС некоторые фрагменты также сдвинуты.
(i) Фигура II-1. Структура NVP-BOS867 со схемой нумерации, используемой в таблице II-1 и спектре ЯМР на ядрах 1H
13C
Другим предпочтительным соединением является:
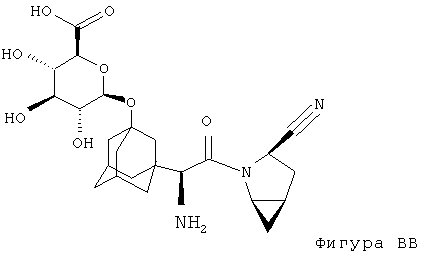
В наиболее предпочтительном варианте осуществления настоящего изобретения соединение согласно фигуре ВВ находится в химически чистом виде.
Так, O-глюкуронидное соединение согласно фигуре ВВ может быть получено посредством модификации описанного в настоящей заявке способа. Способ получения исходного соединения из, в котором R' является -ОН описан в международной заявке на изобретение WO 01/068603 или WO 05/095339.
Соль может являться солью с HCl, т.е. гидрохлоридом. Все соли конечных продуктов с HCl получают посредством пропускания газообразного HCl через 0,1 М раствор свободного основания в тетрагидрофуране до достижении я выраженной кислой реакции раствора с последующей отгонкой растворителя (роторный испаритель/насос).
Исходные аминоадамантановые производные известны в литературе или могут быть получены нижеописанными способами.
Получение 3,5-диметил-1-адамантиламина описано в J. Med. Chem, 25, 1, 1982, 51-56.
Получение 3-этил-адамантиламина описано в J. Med. Chem, 25, 1, 1982, 51-56.
3-Метокси-1-адамантиламин может быть получен следующим способом:
К охлаждаемой смесью льда с водой перемешиваемой суспензии гидрида натрия (0,680 г, 5,95 ммоля) в 15.0 мл тетрагидрофурана добавляют по каплям в продолжение 30 минут смесь 1-аминоадамантан-3-ола (1,00 г, 5,95 ммоля) и 15,0 мл тетрагидрофурана. Затем получаемую смесь перемешивают в течение еще 30 минут, после чего добавляют по каплям в продолжение одной минуты иодметан (0,370 мл, 5,95 ммоля). После этого получаемую таким образом белую мутную реакционную смесь перемешивают при комнатной температуре в течение 18 часов. Затем смесь разбавляют 50 мл хлористого метилен и фильтруют с целью удаления неорганических примесей. После этого фильтрат концентрируют и очищают на силикагеле с помощью аппарата SIMS/Biotage и 19% метанола и 1% гидроксида аммония в хлористом метилене в качестве элюента, что приводит к 3-метокси-1-адамантиламину в качестве мутного маслянистого вещества.
Синтез 3-[[(трет-бутиламино)карбонил]окси]1-1-аминоадамантана:
К смеси 1-аминоадамантан-3-ола (5,00 г, 30,0 ммоля) и карбоната калия (6,20 г, 45 ммолей) в 150 мл тетрагидрофурана добавляют по каплям в продолжение 10 минут бензиловый эфир хлормуравьиной кислоты (4,70 г, 33,0 ммоля). Затем смесь перемешивают при комнатной температуре в течение 2 часов, а затем распределяют между фазами этилацетата и воды. После этого продукт экстрагируют в этилацетат и дважды промывают водный слой этилацетатом (100 мл). Затем объединенные органические фракции последовательно промывают 100 мл 2н. водного раствора гидроксида натрия, водой и соляным раствором, сушат над сульфатом натрия, фильтруют и концентрируют (роторный испаритель/насос), что приводит к 1-бензилкарбамоиладамантан-3-олу в виде твердого вещества белого цвета с выходом 85%.
К прозрачному раствору 1-бензилкарбамоиладамантан-3-ола (1,00 г, 3,32 ммоля) и трет-бутилизоцианата (380 мкл, 3,32 ммоля) в 30 мл хлористого метилена добавляют через шприц триметилсилилхлорид (20,0 мкл, 0,17 ммоля). После этого данную реакционную смесь перемешивают при комнатной температуре в течение 18 часов, концентрируют (роторный испаритель) и очищают на силикагеле с помощью аппарата SIMS/Biotage и 20% раствора этилацетата в гексане в качестве элюента, что приводит к 3-[[(трет-бутиламино)карбонил]окси]-1-бензилкарбамоиладамантану в виде твердого вещества белого цвета с количественным выходом.
К смеси 3-[[(трет-бутиламино)карбонил]окси]-1-бензилкарбамоиладамантана (1,50 г, 3,75 ммоля) и 10% палладия на угле (400 мг) в этаноле (150 мл) в однолитровом аппарате Парра для гидрирования добавляют водород (давление 50 фунтов на кв. дюйм). Затем встряхивают получаемую мутную черную смесь в течение 24 часов. После этого фильтруют данную реакционную смесь через целит с целью удаления палладиевого катализатора и концентрируют (роторный испаритель/насос), что приводит к 3-[[(трет-бутиламино)карбонил]окси]-1-аминоадамантану в виде прозрачного маслянистого вещества с выходом 99%.
Способ синтеза 4-[[[(метоксифенил)амино]карбонил]окси]-1-аминоадамантана в основном совпадает с методикой для 3-[[(трет-бутиламино)карбонил]окси]-1-аминоадамантана, за исключением второй стадии, на которой трет-бутилизоцианат заменяют эквивалентом 4-метоксифенилизоцианата, в качестве растворителя вместо хлористого метилена используют 1,2-дихлорэтан и перемешивают реакционную смесь при температуре 50°C в течение 18 часов. В конце получают промежуточное аминосоединение в виде маслянистого вещества.
Способ синтеза 3-[[(фениламино)карбонил]окси]-1-аминоадамантана в основном совпадает с методикой для 3-[[(трет-бутиламино)карбонил]окси]-1-аминоадамантана, за исключением второй стадии, на которой трет-бутилизоцианат заменяют эквивалентом фенилизоцианата, в качестве растворителя вместо хлористого метилена используют 1,2-дихлорэтан и перемешивают реакционную смесь при температуре 50°C в течение 18 часов. В конце получают промежуточное аминосоединение в виде прозрачного маслянистого вещества.
Способ получения 2-аминоадамантан-5-ола совпадает с приведенным в примере 1, за исключением того, что исходным веществом является 2-аминоадамантан вместо 1-аминоадамантана.
Способ получения нуклеофила 3-ацетокси-1-аминоадамантана в основном совпадает с методикой для 3-[[(трет-бутиламино)карбонил]окси]-1-аминоадамантана, за исключением стандартного процесса ацилирования 1-бензилкарбамоиладамантан-3-ола с помощью 1,2 экв. ацетилхлорида, 3,0 экв. пиридина, 0,1 экв. 4-диметиламинопиридина и 1,2-дихлорэтана, каковые совместно перемешивают при комнатной температуре в течение 24 часов. Конечное аминосоединение получают в виде густого маслянистого вещества.
Способ получения 3-[[[(диизопропил)амино]карбонил]окси]-1-аминоадамантана в основном совпадает с методикой для 3-[[(трет-бутиламино)карбонил]окси]-1-аминоадамантана, за исключением второй стадии, на которой трет-бутилизоцианат заменяют эквивалентом диизопропилкарбамоилхлорида, в качестве растворителя вместо хлористого метилена используют 1,2-дихлорэтан и перемешивают реакционную смесь при температуре 85°C в течение 18 часов. В конце получают промежуточное аминосоединение в виде твердого вещества серого цвета.
Способ получения 3-[[[(циклогексил)амино]карбонил]окси]-1-аминоадамантана в основном совпадает с методикой для 3-[[(трет-бутиламино)карбонил]окси]-1-аминоадамантана, за исключением второй стадии, на которой трет-бутилизоцианат заменяют эквивалентом 4-циклогексилизоцианата, в качестве растворителя вместо хлористого метилена используют 1,2-дихлорэтан и перемешивают реакционную смесь при температуре 50°C в течение 18 часов. В конце получают промежуточное аминосоединение в виде густого прозрачного маслянистого вещества.
Способ получения 3-этокси-1-адамантиламина (прозрачное маслянистое вещество) совпадает с приведенным для 3-метокси-1-адамантиламина, за исключением того, что вместо подметана используют иодэтан (1,3 эквивалента).
Пример препарата
Таблетки, каждая из которых содержит 50 мг активного ингредиента, NVP-BQS867, могут быть изготовлены нижеизложенным способом.
Состав (на 10000 таблеток)
Активный ингредиент смешивают с лактозой и 292 г картофельного крахмала, смачивают смесь спиртовым раствором желатина и гранулируют ее с помощью сита. После сушки примешивают остаток картофельного крахмала, тальк, стеарат магния и высокодисперсный оксид кремния и запрессовывают смесь в таблетки весом по 145,0 мг с содержанием активного ингредиента 50,0 мг, которые если то требуется, могут быть снабжены насечками для разламывания для более тонкого подбора дозы.
III. Пример 2: Биологические эксперименты
IV. Вещества и методы
А. А. Оборудование
Все растворы, содержащие белки и пептиды содержали в силиконизированных пробирках (Life Systems Design, Merenschwand, Switzerland). Растворы соединений, равно как и растворы ферментов и субстратов переносили на 384-ячеечные планшеты (черные планшеты Cliniplate, номер по каталогу. 95040020 Labsystems Оу, Finland) с помощью 96-канальной дозирующей системы CyBi-Well (CyBio AG, Jena, Germany).
1. 1. Оборудование для измерения интенсивности флуоресценции Для измерений интенсивности флуоресценции (ИФ) с АМК в качестве красителя применяли детектор Ultra Evolution (TECAN, Maennedorf, Switzerland). Прибор оборудован комбинацией полосовых фильтров для 350 нм (ширина полосы 20 нм) и 500 нм (ширина полосы 25 нм) для возбуждения флуоресценции и измерения испускания, соответственно. Для повышения соотношения сигнал:фон применяли соответствующее дихроичное зеркало. Все фильтры и дихроичное зеркало были приобретены у TECAN.
Измерения ИФ с использованием красителя Rhl 10 осуществляли с детектором Safire2 (TECAN, Maennedorf, Switzerland). Safire2 является прибором на основе монохроматора, для возбуждения флуоресценции и измерения испускания были выбраны длины волн 485 нм 535 нм, соответственно. И для возбуждения, и для испускания была установлена ширина полосы 20 нм.
В каждой ячейке флуорофоры возбуждали тремя импульсами на каждое измерение.
2. Расчет величин IC50 из усредненных данных
Данные из независимых анализов были усреднены и представлены графически с помощью программы Origin 7.5SR6 (OriginLab Corporation, Northampton, MA, USA). Применяли встроенную процедуру нелинейной регрессии программы Origin для аппроксимации усредненных данных логистической функцией

где y является степенью ингибирования в процентах для концентрации ингибитора x. A1 является наинизшей степенью ингибирования, т.е. 0%, а A2 максимальной степенью ингибирования, т.е. 100%. Показатель экспоненты, p, является коэффициентом Хилла.
B. Определение величин IC50
Анализы для определения величин IC50 проводят при комнатной температуре в 384-ячеечных планшетах. Для всех анализов конечный объем составляет 30 мкл. Исследуемые соединения растворяют в 90% (об./об.) ДМСО в воде и разбавляют водой (содержащей 0,05% (масс./об.) ХАПС) до концентрации, троекратно превышающей требуемую для анализа. Для каждого анализа добавляют по 10 мкл воды/ХАПС (± исследуемое соединение) на ячейку, а затем 10 мкл раствора протеазы (разбавленного буфером для анализа, в отношении конечной концентрации в анализе см. раздел «Условия анализов»). После инкубирования при комнатной температуре в течение 1 часа инициируют взаимодействие посредством добавления 10 мкл раствора субстрата (в отношении конечных концентраций см. раздел «Условия анализов»). Одиннадцать конечных концентраций соединений составляют либо 0.9 нМ, 3 нМ, 9 нМ, 30 нМ, 90 нМ, 300 нМ, 900 нМ, 3 мкМ, 9 мкМ, 30 мкМ и 90 мкМ, либо 3 нМ, 10 нМ, 30 нМ, 100 нМ, 300 нМ, 1 мкМ, 3 мкМ, 10 мкМ, 30 мкМ, 100 мкМ и 300 мкМ. Влияние соединение на активность фермента получают исходя из линейных кинетических кривых и определяют из двух считываний, первое из которых осуществляют непосредственно после добавления субстрата (t=0 мин.), а второе по прошествии 1 часа (t=60 мин.). Величину IC50 рассчитывают исходя из графика процента ингибирования от концентрации ингибитора с помощью программ для нелинейного регрессионного анализа (XLfit, версия 4.0, ID Business Solution Ltd., Guildford, Surrey, UK).
C. Условия анализов
Все условия, относящиеся к ферменту, субстрату и буферу, перечислены ниже для индивидуальных анализов.
1. DPP-2 (дипептидилпептидаза-2)
Фермент: человеческая DPP-2, включающая аминокислоты 30-492, экспрессируемая в клетках насекомых и очищенная от них (бакуловирусная система экспрессии)
Субстрат: норлейцин-пролин-АМК, приобретенный у компании Biosyntan (www.biosyntan.de), номер продукта 4572
Концентрация фермента: 0,03 нМ Концентрация субстрата: 2 мкМ
Буфер для анализа: 100 мМ раствор цитрата натрия, pH 5,5, 0,05% (масс/об.) ХАПС
2. DPP-IV (дипептидилпептидаза-IV)
Фермент: человеческая DPP-IV, включающая аминокислоты 39-766, экспрессируемая в клетках насекомых и очищенная от них (бакуловирусная система экспрессии)
Субстрат: глицин-пролин-АМК, приобретенный у компании Bachem (www.bachem.com), номер по каталогу I-1225
Концентрация фермента: 0,01 нМ
Концентрация субстрата: 10 мкМ
Буфер для анализа: 25 мМ Трис, pH 7,4, 140 мМ NaCl, 10 мМ KCl, 0,05% (масс/об.) ХАПС
3. DPP-IV из плазмы крови человека (эндогенная дипептидилпептидаза-IV в плазме крови человека)
Фермент: эндогенная человеческая DPP-IV из образцов плазмы доноров-мужчин
Субстрат: (Н-аланин-пролин)2-Rh110, самостоятельный синтез Концентрация фермента: примерно 5 нМ
Концентрация субстрата: 10 мкМ
Раствор для анализа: плазма крови человека, разбавленная до 50% содержания плазмы буфером (25 мМ Трис, pH 7,4, 140 мМ NaCl, 10 мМ KCl, 0,05% (масс/об.) ХАПС)
4. DPP-8 (дипептидилпептидаза-8)
Фермент: человеческая DPP-8, включающая аминокислоты 1-882, экспрессируемая в клетках насекомых и очищенная от них (бакуловирусная система экспрессии)
Субстрат: глицин-пролин-АМК, приобретенный у компании Bachem (www.bachem.com), номер по каталогу 1-1225
Концентрация фермента: 0,05 нМ
Концентрация субстрата: 10 мкМ
Буфер для анализа: 25 мМ Трис, pH 7,4, 140 мМ NaCl, 10 мМ KCl, 0,05% (масс./об.) ХАПС
5. DPP-9 (дипептидилпептидаза-9)
Фермент: человеческая DPP-9, включающая аминокислоты 1-863, экспрессируемая в дрожжевых клетках Pichia pastoris и очищенная от них
Субстрат: глицин-пролин-АМК, приобретенный у компании Bachem (www.bachem.com), номер по каталогу I-1225
Концентрация фермента: 1 нМ
Концентрация субстрата: 10 мкМ
Буфер для анализа: 25 мМ Трис, pH 7,4, 140 мМ NaCl, 10 мМ KCl, 0,05% (масс./об.) ХАПС
6. FAP (белок-активатор фибробластов, альфа-разновидность) Фермент: человеческий FAPα, включающий аминокислоты 27-760, исключая цитоплазматический и трансмембранный домены, экспрессируемый в клетках насекомых и очищенный от них (бакуловирусная система экспрессии)
Субстрат: бензилоксикарбонил-глицин-пролин-АМК, приобретенный у компании Bachem (www.bachem.com), номер по каталогу 1-1145
Концентрация фермента: 0,1 нМ
Концентрация субстрата: 8 мкМ
Буфер для анализа: 25 мМ Трис, pH 7,4, 1 мМ этилендиаминтетрауксусной кислоты (ЭДТК), 0,05% (масс./об.) ХАПС
7. PEP (пролил-эндопептидаза)
Фермент: человеческая PEP, включающая аминокислоты 1-710, экспрессируемая в дрожжевых клетках Pichia pastoris и очищенная от них
Субстрат: бензилоксикарбонил-глицин-пролин-АМК, приобретенный у компании Bachem (www.bachem.com), номер по каталогу I-1145
Концентрация фермента: 0,03 нМ
Концентрация субстрата: 10 мкМ
Буфер для анализа: 25 мМ Трис, pH 7,4, 1 мМ ЭДТК, 0,05% (масс./об.) ХАПС, 0,1% (масс./об.) БСА.
V. D. Результаты
Результаты всех измерений IC50 для соединения NVP-BQS867 сведены в нижеприведенные таблицы 3-1 - 3-7. Они представлены для человеческой DPP-IV, эндогенной DPP-IV в плазме крови человека, человеческой DPP-8, человеческой DPP-9, человеческом FAP и человеческой PEP.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЙ СОСТАВ | 2006 |
|
RU2483716C2 |
| N-ЗАМЕЩЕННЫЕ 2-ЦИАНПИРРОЛИДИНЫ | 1999 |
|
RU2251544C2 |
| НОВЫЙ СОСТАВ | 2006 |
|
RU2821230C2 |
| ПРИМЕНЕНИЕ ИНГИБИТОРА DPP-IV ДЛЯ СНИЖЕНИЯ ПРИСТУПОВ ГЛИКЕМИИ | 2006 |
|
RU2440143C2 |
| ТВЕРДАЯ ДОЗИРОВАННАЯ ЛЕКАРСТВЕННАЯ ФОРМА ДЛЯ ОРАЛЬНОГО ВВЕДЕНИЯ, СОДЕРЖАЩАЯ КОМБИНАЦИЮ ВИЛДАГЛИПТИНА И ГЛИКВИДОНА | 2014 |
|
RU2585378C1 |
| ИНГИБИРУЮЩЕЕ ДИПЕПТИДИЛПЕПТИДАЗУ IV СРЕДСТВО И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ЕГО ОСНОВЕ | 2011 |
|
RU2485952C2 |
| ПРОИЗВОДНОЕ КСАНТИНА | 2016 |
|
RU2709348C2 |
| НОВЫЕ ИНГИБИТОРЫ ДИПЕПТИДИЛПЕПТИДАЗЫ IV И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ПРОТИВОРАКОВЫХ АГЕНТОВ | 2002 |
|
RU2299066C2 |
| ПРИМЕНЕНИЕ ИНГИБИТОРОВ ДИПЕПТИДИЛПЕПТИДАЗЫ IV | 2004 |
|
RU2385723C2 |
| СРЕДСТВО ПЕПТИДНОЙ СТРУКТУРЫ, ИНГИБИРУЮЩЕЕ ДИПЕПТИДИЛПЕПТИДАЗУ-4, И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ЕГО ОСНОВЕ | 2015 |
|
RU2589258C1 |
Настоящее изобретение относится к соединению формулы (IA), где R' представляет собой указанную ниже формулу и R'' представляет собой водород, в свободной форме или в форме фармацевтически приемлемой кислотно-аддитивной соли. Заявленные соединения формулы (IA) ингибируют активность DPP-IV (дипептидилпептидазы-IV). Вследствие этого, они показаны для применения в качестве лекарственных средств, предназначенных для ингибирования DPP-IV и для лечения состояний, опосредованных DPP-IV, таких как инсулиннезависимый сахарный диабет. 6 н. и 5 з.п. ф-лы, 2 пр., 2 ил., 8 табл.
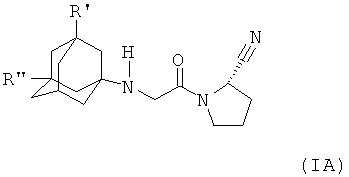
1. Соединение формулы (IА)
где R' представляет собой
и R'' представляет собой водород;
в свободной форме или в форме фармацевтически приемлемой кислотно-аддитивной соли.
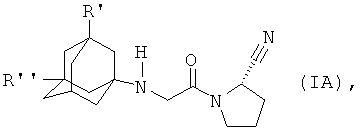
2. Соединение по п.1, которое представляет собой
его фармацевтически приемлемая кислотно-аддитивная соль.
3. Фармацевтическая композиция, обладающая ингибирующей активностью в отношении дипептидилпептидазы-IV, содержащая эффективное количество соединения по любому из пп.1, 2 в свободной форме или в форме фармацевтически приемлемой кислотно-аддитивной соли, совместно с по меньшей мере одним фармацевтически приемлемым носителем или разбавителем.
4. Применение соединения по любому из пп.1, 2 или его фармацевтически приемлемой кислотно-аддитивной соли для изготовления лекарственного средства, предназначенного для ингибирования дипептидилпептидазы-IV.
5. Применение фармацевтической композиции по п.3 для изготовления лекарственного средства, предназначенного для ингибирования дипептидилпептидазы-IV.
6. Применение по п.4 или 5, где лекарственное средство используется для лечения состояний, опосредованных ингибированием дипептидилпептидазы-IV.
7. Применение по п.6, где лекарственное средство используется для лечения инсулин-независимого сахарного диабета.
8. Способ ингибирования дипептидилпептидазы-IV, включающий введение нуждающемуся в таком лечении млекопитающему соединения по любому из пп.1, 2 или его фармацевтически приемлемой кислотно-аддитивной соли в терапевтически эффективном количестве.
9. Способ ингибирования дипептидилпептидазы-IV, включающий введение нуждающемуся в таком лечении млекопитающему фармацевтической композиции по п.3 в терапевтически эффективном количестве.
10. Способ по п.8 или 9, где лекарственное средство используется для лечения состояний, опосредованных ингибированием дипептидилпептидазы-IV.
11. Способ по п.10, в котором подвергаемое лечению состояние является инсулин-независимым сахарным диабетом.
| WO 2005012249 А2, 10.02.2005 | |||
| Приспособление для смазки холостых шкивов | 1930 |
|
SU34241A1 |
| WO 2007066805 A1, 14.06.2007 | |||
| WO 2001074835 A1, 11.10.2001 | |||
| О-АРИЛГЛЮКОЗИДНЫЕ ИНГИБИТОРЫ SGLT2 И СПОСОБ ИХ ПРИМЕНЕНИЯ | 2001 |
|
RU2269540C2 |

