Настоящее изобретение относится к 5'-деокси-5-фтор-N4-(пентилоксикарбонил)цитидину, которое является производным N4-(замещенный оксикарбонил)-5'-дезокси-5-фторцитидина общей формулы (I):
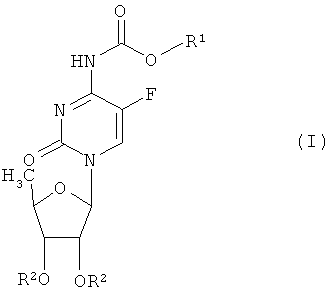
в которой R1 представляет насыщенный или ненасыщенный, нормальный или разветвленный углеводородный радикал (в котором число атомов углерода в самой длинной нормальной цепи находится в интервале три-семь) или радикал формулы -(CH2)n-Y (где n=0-4, если Y представляет циклогексил, или n=2-4, если Y представляет низшую алкоксигруппу с 1-4 атомами углерода или фенил) и R2 представляет атом водорода или легко гидролизуемый в физиологических условиях радикал, а также гидратам или сольватам соединений общей формулы (I) и к содержащему их фармацевтическому препарату, характеризующемуся прекрасными фармакокинетическими свойствами при лечении опухолей с высоким уровнем безопасности.
Известно, что многие предшественники 5-фторурацила (5-ФУ) применимы в качестве противоопухолевых средств, но в целом эффективность их биоконверсии все еще остается недостаточной при лечении страдающих опухолями больных, и являются причиной желудочной токсичности и иммуносупрессорной токсичности, и такая токсичность в основном и ограничивает их дозировки.
В патенте США 4966891 раскрыты предшественники 5-ФУ, улучшенные с точки зрения вышеупомянутых эффективности биоконверсии и токсичности. Под действием ациламидаз эти предшественники превращаются в 5'-дезокси-5-фторцитидин (5'-ДФЦТ), а под действием цитидиндезаминазы - в 5'дезокси-5-фторуридин (5'-ДФУР) и затем под действием пиримидиннуклеотидной фосфорилазы - в 5-ФУ (in vivo), которая преимущественно локализуется в печени, тонкой кишке и тканях опухоли. В ходе интенсивных исследований фармакокинетических показателей предшественников 5-ФУ, в частности производных N4-(замещенный оксикарбонил)-5'-дезокси-5-фторцитидина, создатели настоящего изобретения обнаружили, что определенные специфичные предшественники под действием изофермента ациламидазы, преимущественно обнаруживаемого в печени, но не в других органах человека, селективно превращаются в 5'-ДФЦТ и характеризуются лучшими фармакокинетическими показателями, чем другие испытанные соединения. Дальнейшие исследования, основанные на указанном открытии, позволили создателям настоящего изобретения выяснить, что особые производные N4-(замещенные оксикарбонил)-5'-дезокси-5-фторцитидина (далее N4-(замещенный оксикарбонил)-5'-ДФЦТ) вышеприведенной общей формулы (1) отличаются улучшенными фармакокинетическими показателями селективности на обезьянах, а именно в 4-7 раз более высокой максимальной концентрацией (Cмакс) 5'-ДФУР и в 4 раза большей площадью под кривой (ППК) в крови по сравнению с другими соединениями и меньшей кишечной токсичностью, в результате чего и возникло настоящее изобретение.
5'-деокси-5-фтор-N4-(пентилоксикарбонил)цитидин, относящийся к N4-(замещенным оксикарбонил)-5'-ДФЦТ общей формулы (I) может быть получен реакцией соединения общей формулы (II):
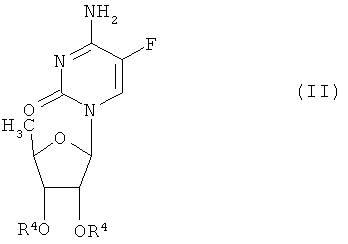
в которой R4 представляет радикал для защиты гидроксигруппы, такой как: ацетил, бензоил, триметилсилил, трет-бутилдиметилсилил и т.п.,
с соединением общей формулы (III):
R1OCOCl
в которой R1 принимает вышеуказанные значения, с последующим, если необходимо, удалением защитного радикала.
Соединения вышеприведенной общей формулы (II) могут быть получены 2', 3'-ди-O-ацилированием или -силилированием 5'-дезокси-5-фторцитидина (J. Med. Chem, 22. 1330 (1979)) по методике, приведенной в патенте США 4966891, или прямым соединением 5-фторцитозина с 1,2,3-три-O-ацетил-5-дезоксифуранозой по методике, аналогичной методике, приведенной в литературе (Synthesis, 748 (1981)).
Реакция соединения вышеприведенной общей формулы (II) с соединением вышеприведенной общей формулы (III) может быть проведена в растворителе, таком как: пиридин, диоксан, тетрагидрофуран, ацетонитрил, хлороформ, дихлорметан и т.п., в присутствии акцептора кислоты, такого как: триэтиламин, пиридин, пиколин, 4-(N,N-диметиламино)пиридин, лутидин и т.п. Реакция может быть проведена в температурном интервале 0-30°С.
Защитный радикал может быть, если необходимо, удален по окончании реакции по известным специалисту методикам (Защитные группы в органическом синтезе, Джон Вили и сыновья, Нью-Йорк, Can. J. Chem., 49, 493 (1971) и патент США 4966891), например, щелочным или кислотным гидролизом.
Соединения вышеприведенной формулы (I) могут существовать как в несольватированном, так и в сольватированном виде, в том числе в гидратированной форме. Гидратация может быть осуществлена в ходе процесса приготовления или же может произойти постепенно вследствие гигроскопичных свойств первоначально безводного продукта. Сольваты с фармацевтически приемлемыми растворителями, такими как этанол, могут быть получены, например, в ходе кристаллизации.
Производные N4-(замещенный оксикарбонил)-5'-ДФЦТ общей формулы (I), также как сольваты или гидраты соединений общей формулы (I), полученные способом изобретения, проявляют активность по отношению к ксенотрансплантантам рака ободочной кишки человека CXF280 и рака желудка GXF97, карциномы 26 ободочной кишки мышей, легочной карциномы Льюиса мышей и т.п. на мышах в очень широком интервале дозировок, как перорально, так и парентерально, вследствие чего применимы в качестве противоопухолевых средств. Под действием изофермента ациламидазы эти соединения успешно превращаются в 5'-ДФЦТ, под действием цитидиндезаминазы - в 5'-ДФУР и затем под действием пиридиннуклеозидной фосфорилазы превращаются в активный метаболит 5-ФУ.
Настоящее изобретение, кроме того, относится к фармацевтическим препаратам, в частности препаратам для лечения опухолей, содержащим соединения вышеприведенной общей формулы (I).
N4-(Замещенный оксикарбонил)-5'-ДФЦТ настоящего изобретения могут быть введены пероральным путем или непероральным путем человеку различными обычными методами введения. Кроме того, N4-(замещенный оксикарбонил)-5'-ДФЦТ настоящего изобретения используют в чистом виде или вводят в состав с совместимыми фармацевтическими носителями. Такой носитель может представлять собой органический или неорганический инертный материал, пригодный для энтерального, чрескожного или парентерального введения, такой как: вода, желатин, гумиарабик, лактоза, крахмал, стеарат магния, тальк, растительные масла, полиалкиленгликоли или петролатум. Фармацевтический препарат может быть приготовлен в твердом виде (например: в таблетках, драже, таблетках с желудочным покрытием, гранул, свеч, капсул или желудочных капсул), в полужидком виде (например, в виде мазей) или в жидкой форме (например, в виде растворов, суспензий или эмульсий). Фармацевтический препарат может быть стерилизован и/или может содержать дополнительные вспомогательные добавки, такие как: стабилизаторы, консерванты, осадители, эмульгаторы, улучшающие вкус добавки, соли для изменения осмотического давления или действующие в качестве буфера вещества. Фармацевтический препарат может быть получен обычным путем.
N4-(замещенный оксикарбонил)-5'-ДФЦТ настоящего изобретения могут быть использованы по отдельности или в виде смеси двух или более различных N4-(замещенный оксикарбонил)-5'-ДФЦТ, при этом количество N4-(замещенный оксикарбонил)-5'-ДФЦТ составляет 0,1-99,5%, предпочтительно 0,5-95% в пересчете на массу фармацевтического препарата.
Фармацевтический препарат настоящего изобретения может быть приготовлен в сочетании с другими обычными противоопухолевыми средствами.
Подверженность действию ациламидазы N4-(замещенный оксикарбонил)-5'-ДФЦТ настоящего изобретения и их фармакокинетические показатели приведены ниже.
1. Подверженность действию ациламидаз обезьяны и человека
N4-(замещенный оксикарбонил)-5'-ДФЦТ настоящего изобретения инкубируют 60 мин при 37°C с сырыми экстрактами печени обезьяны и человека в присутствии в качестве ингибитора цитидиндезаминазы тетрагидроуридина (0,4 Мм). Затем в качестве продукта с помощью ВЭЖХ выделяют 5'-ДФЦТ и подверженность к действию фермента подсчитывают по количеству продукта. Как видно из Таблицы 1, соединения настоящего изобретения в высшей степени подвержены действию ациламидазы печени человека, что предполагает их эффективное биопревращение в 5'-ДФЦТ в человеке (см. в конце описания).
2. Фармакокинетические показатели на обезьянах
Соединения настоящего изобретения вводят перорально группе из 2-5 циномолгичных обезьян (3-4 кг). Через различное время после введения отбирают плазму на определение в крови концентрации непревращенных молекул и их активного метаболита (5'-ДФУР).
С помощью ВЭЖХ из плазмы выделяют метаболиты и подсчитывают их концентрацию. Как видно из Таблицы 2, соединения настоящего изобретения характеризуются высоким уровнем Cмакс и ППК активного метаболита 5'-ДФУР в плазме. Полученные результаты показывают, что соединения настоящего изобретения могут быть эффективно использованы для лечения различных опухолей человека (см. в конце описания).
Противоопухолевая активность соединений изобретения показана ниже.
3. Противоопухолевые испытания на ксенотрансплантанте CXF280 рака ободочной кишки человека
Опухоль CXF280 (размером 2×2 мм) имплантируют подкожно мышам линии BALB/c nu/nu (21-22 г) в день 0. Когда размер опухоли достигает 100 мм3 (примерно на 14-й день), мышам ежедневно в течение 3 недель перорально вводят соединения настоящего изобретения. В один из дней после последнего введения подсчитывают объем опухоли.
Приведенный в Таблице 3 (см. в конце описания) процент ингибирования роста опухоли подсчитывают по формуле:
% ингибирования = {1-(T-V0)/(C-V0)}·100, где V0 - объем опухоли перед началом лечения, T - объем опухолей в подвергаемой лечению группе, C - объем опухоли из контрольной группы.
Как видно из Таблицы 3, соединения настоящего изобретения характеризуются безопасным введением, не вызывают при этом кишечной токсичности и гораздо более эффективны, чем 5-ФУ.
4. Противоопухолевая активность и активность против общего истощения по отношению к карциноме 26 ободочной кишки мышей
Противоопухолевую активность представительного соединения (пример 13) настоящего изобретения определяют следующим образом. Мышам (CDF1) подкожно инокулируют карциному 26 ободочной кишки (106 клеток) в день 0. Через 21 день, когда зверьки находятся в состоянии общего истощения, им ежедневно 7 раз дают испытуемое соединение. В день после последнего введения определяют прирост массы опухоли, прирост массы остова, массу жировой ткани, концентрацию глюкозы и острого фазового реагента ИКБ (иммуносупрессивного кислотного белка) в сыворотке. Как видно из Таблицы 4 (см. в конце описания), мыши, получавшие носитель, были ненормальны с точки зрения общего истощения, характеризующегося такими параметрами, как: масса жировой ткани, содержание в сыворотке глюкозы и ИКБ, в то время как лечение соединением примера 13 подавляет рост опухоли и ведет к улучшению параметров общего истощения.
Токсичность (ЛД50) представительных соединений (примеры 13, 14 и 17) настоящего изобретения выявлялась при пероральном введении ежедневно в течение 21 дня на мышах. Характерные значения ЛД50, полученные в таких опытах, составляют более 500 мг/кг/день.
Суточная дозировка больному N4-(замещенный оксикарбонил)-5'-ДФЦТ настоящего изобретения может меняться в зависимости от массы и состояния больного, но, как правило, находится в интервале 0,5-500 мг на 1 кг массы, предпочтительно 2-200 мг. Следует отметить, что для соединений настоящего изобретения можно ожидать в 3-5 раз более высокой активности при лечении человека по сравнению с соединениями, раскрытыми в патенте США 4966891, если эту активность рассматривать с точки зрения Смакс и ППК для 5'-ДФУР после перорального введения соединений настоящего изобретения обезьяне. По той же причине можно ожидать, что соединения настоящего изобретения покажут достаточную активность при дозировках, в 3-5 раз более низких по сравнению с дозировками соединений указанного патента США. Настоящим изобретением может быть получен фармацевтический препарат с высоким уровнем безопасности.
Нижеследующие примеры предназначены для более подробной иллюстрации настоящего изобретения, но ни в коей мере для ограничения его объема.
Ссылочный пример. Получение исходных соединений
Получение 2',3'-ди-O-ацетил-5'-дезокси-5-фторцитидина
(a) Из 5'-дезокси-5-фторцитидина
В сухом пиридине (1,3 мл) растворяют 5'-дезокси-5-фторцитидин (50 мг). К раствору при перемешивании при 0°C добавляют уксусный ангидрид (39 мл). Реакционную смесь перемешивают 3 часа при 0°C. После удаления при пониженном давлении растворителя остаток распределяют между этилацетатом и охлажденной льдом водой. Этилацетатный слой сушат над сульфатом магния и концентрируют при пониженном давлении. Остаток очищают колоночной хроматографией на силикагеле с элюированием смесью дихлорметан-метанол (9:1) и после перекристаллизации из изопропанола получают 37 мг 2',3'-ди-O-ацетил-5'-дезокси-5-фторцитидина, т.пл. 191,5-193°C, ББА-МС m/z 330 (МН+).
(б) Из 5-фторцитозина и 1,2,3-три-O-ацетил-5-дезокси-β-D-рибофуранозы
Раствор иодида натрия (3,6 г) и хлорметилсилана (794 мл) в сухом ацетонитриле (15 мл) перемешивают молекулярными ситами 4A (200 мг) при 0°C в течение 5 минут (при перемешивании осаждается бесцветный хлорид натрия). Добавляют 1,2,3-три-O-ацетил-5-дезокси-β-D-рибофуранозу (2,0 г) и смесь перемешивают при 0°C в течение 30 минут. Затем добавляют при 0°C свежеприготовленный из 5-фторцитозина (1,12 г) раствор триметилсилилованного 5-фторцитозина в сухом ацетонитриле (5 мл) и продолжают перемешивание в течение 3 часов при комнатной температуре. Смесь фильтруют, фильтрат концентрируют в вакууме, и остаток распределяют между дихлорметаном и насыщенным водным раствором бикарбоната натрия. Водный слой экстрагируют смесью CH2Cl2/MeOH (10:1). Объединенные органические слои сушат над безводным сульфатом натрия и испаряют при пониженном давлении. Остаток очищают хроматографией на силикагеле с применением в качестве элюента смеси CH2Cl2/MeOH (10:1) с последующей рекристаллизацией из изопропанола с получением 1,24 г 2',3'-ди-O-ацетил-5'-дезокси-5-фторцитидина.
Пример 1
Получение 2',3'-ди-O-ацетил-5'-дезокси-5-фтор-N4(пропоксикарбонил)-цитидина
К раствору 2',3'-ди-O-ацетил-5'-дезокси-5-фторцитидина (2 г) CH2Cl2 (15 мл) и сухом пиридине при перемешивании и охлаждении в бане со льдом по каплям добавляют n-пропилхлорформат (957 мл). После перемешивания в течение 30 минут при комнатной температуре смесь испаряют досуха при пониженном давлении. Остаток распределяют между эфиром и насыщенным водным раствором бикарбоната натрия. Органический слой промывают рассолом, сушат над безводным сульфатом натрия и фильтруют.
Испарением фильтрата получают 2',3'-ди-O-ацетил-5'-дезокси-5-фтор-N4-(пропоксикарбонил)цитидин (2,5 г)>E1-MC m/z 415 (М+). 1H ЯМР (ДМСО-d6) δ: 0,92 (3Н, т, J=7,3 Гц), 1,37 (3Н, д, J=6,3 Гц), 1,63 (2Н, секс, J=7,3 Гц), 4,06-4,14 (3Н, м), 5,11 (1H, т, J=6,3 Гц), 5,47 (1Н, дв. д. J=4,6 и 6,3 Гц), 5,81 (1Н, д, J=4,6 Гц), 8,31 (1Н, ш.с), 10,63 (1Н, ш.с).
Нижеприведенные соединения (см. табл.5) получены по методике, аналогичной методике примера 1 (R1 и R2 принимают значения, указанные для общей формулы (I)). Соединение примера 9 получено из известного 2',3'-ди-O-бензоил-5'-дезокси-5-фторцитидина (патент США 4966891) по методике, аналогичной методике примера 1.
Пример 10
Получение 5'-дезокси-5-фтор-N4-(пропоксикарбонил)цитидина
К раствору 2',3'-ди-O-ацетил-5'-дезокси-5-фтор-N4-(пропоксикарбонил) цитидина (2,5 г) в CH2Cl2 (17 мл) при перемешивании и охлаждении в бане со льдом по каплям прибавляют 1 н. NaOH (17 мл). После перемешивания 1 ч при 0°C к смеси добавляют MeOH (0,9 мл). Добавлением концентрированной HCl в реакционной смеси устанавливают pH 6 и слои разделяют. Водный слой экстрагируют смесью растворителей CH2Cl2-MeOH (95:5) (40 мл × 10). Соединенные органические слои сушат над безводным сульфатом натрия и фильтруют. Раствор испаряют и кристаллизацией из этилацетата получают 5'-дезокси-5-фтор-N4-(пропоксикарбонил)цитидин (1,6 г, выход 79,8%), т.пл. 125-126,5°C. E1-MC m/z 331 (M+).
Нижеприведенные соединения (см. табл.6) получены по методике, аналогичной методике примера 10 (R1 и R2 принимают значения, указанные для общей формулы (I)).
Пример 19
Получение N4-(циклогексилоксикарбонил)-5'-дезокси-5-фторцитидина
В 20 мл сухого пиридина растворяют 5'-дезокси-5-фторцитидин (2,5 г), к раствору по каплям при 0°C добавляют триметилсилилхлорид (3,4 мл) и смесь перемешивают 30 мин при комнатной температуре. Одной порцией при 0°C к реакционной смеси добавляют циклогексилхлорформат. После перемешивания смеси 1 час при комнатной температуре пиридин испаряют при пониженном давлении. Остаток затем распределяют между насыщенным водным раствором NaHCO3 и эфиром. Органический слой промывают рассолом, сушат над безводным MgSO4 и концентрируют при пониженном давлении. К остатку добавляют лимонную кислоту (2 г) и метанол (50 мл). Смесь перемешивают примерно сутки при комнатной температуре. После удаления растворителя при пониженном давлении остаток растворяют в смеси CH2Cl2-MeOH (95:5) и нейтрализуют водным раствором NaOH. Органический слой сушат над безводным Na2SO4 и концентрируют при пониженном давлении. Остаток очищают хроматографией на силикагеле с применением в качестве элюента смеси CH2Cl2-MeOH (20:1) и после перекристаллизации из этилацетата получают N4-(циклогексилоксикарбонил)-5'-дезокси-5-фторцитидин (3,47 г, выход 92%), т.пл. 134-136°C. ББА-МС m/z 372 (MH+).
Нижеприведенные соединения (см. табл.7) получены по методике, аналогичной методике примера 19 (R1 и R2 принимают значения, указанные для общей формулы (I)).
Пример 28
Получение 5'-дезокси-5-фтор-N4-(неопентилоксикарбонил)-цитидина
В 15 мл сухого дихлорметана растворяют 5'-дезокси-2',3'-ди-O- ацетил-5-фторцитидин (1,5 г) и сухой пиридин (0,74 мл). К полученной смеси при 0°C по каплям прибавляют толуольный раствор неопентилхлорформата (3 экв.) и перемешивают 1 ч при комнатной температуре. После удаления растворителя при пониженном давлении остаток распределяют между эфиром и насыщенным водным раствором карбоната натрия. Органический слой последовательно промывают водой и рассолом, сушат над безводным сульфатом натрия и концентрированием при пониженном давлении получают сырой 2',3'-ди-O-ацетил-5'-дезокси-5-фтор-N4-(неопентилоксикарбонил) цитидин в виде бледно-желтого масла. Сырой продукт растворяют в этаноле (15 мл) и охлаждают в бане со льдом. При температуре ниже 15°C по каплям прибавляют 1 н. водный раствор гидроокиси натрия. По окончании прибавления реакционную смесь нейтрализуют при 0°C конц. соляной кислотой. Раствор концентрируют при пониженном давлении, и концентрат распределяют между водой и смесью растворителей CH2>Cl2-MeOH (95:5). Водный слой вновь экстрагируют десять раз смесью CH2Cl2-MeOH (95:5) по 20 мл каждый раз. Все органические слои объединяют, сушат над безводным сульфатом натрия и концентрируют при пониженном давлении. Очисткой остатка хроматографией на колонке с силикагелем с применением в качестве элюента смеси CH2Cl2-MeOH (20:1) в виде аморфного порошка 1,37 г (выход 84%) 5'-дезокси-5-фтор-N4-(неопентилоксикарбонил)-цитидина. ББА-МС m/z 360 (MH+). 1H-ЯМР (ДМСО-d6) δ: 0,93 (9Н, с), 1,31 (3Н, д, J=6,3 Гц), 3,68 (1Н, к, J=5,9 Гц), 3,81 (2Н, ш. с), 3,87-3,92 (1Н, м), 4,04-4,09 (1Н, м), 5,05 (1Н, д, J=5,9 Гц), 5,41 (1Н, ш. д, J=5,3 Гц), 5,67 (1Н, дв. д. J=1,3 и 3,6 Гц), 8,04 (1Н, ш. с), 10,53 (~1Н, ш. с).
Пример 29
5'-Дезокси-N4[(3,3-диметилбутоксикарбонил]-5-фторцитидин
Заглавное соединение получено по методике, аналогичной методике примера 28, за исключением того, что в качестве ацилирующего средства используют 3,3-диметилбутилхлорформат, аморфный порошок (выход 71%), ББА-МС m/z 374 (MH+). 1H-ЯМР (ДМСО-d6) δ: 0,93 (9Н, с), 1,31 (3Н, д, J=6,3 Гц), 1,55 (2Н, т, J=7,3 Гц), 3,68 (1Н, к, J=5,9 Гц), 3,84-3,93 (1Н, м), 4,03-4,09 (1Н, м), 4,15 (2Н, т, J=7,3 Гц), 5,05 (1Н, д, J=5,9 Гц), 5,4 (1Н, ш. д, J=5,3 Гц), 5.67 (1Н, дв. д, J=1,3 и 4 Гц), 8 (1Н, ш. с), 10,53 (~1Н, ш. с).
Нижеследующие примеры иллюстрируют фармацевтические препараты, содержащие соединение настоящего изобретения.
Пример A
Замкнутые желатиновые капсулы, каждая из которых содержит нижеперечисленные компоненты, получают известными методами
N4-(Бутоксикарбонил)-5'-дезокси-5-фторцитидин -100 мг
Зерновой крахмал - 20 мг
Двуокись титана - 385 мг
Стеарат магния - 5 мг
Пленка - 20 мг
ПЭГ 6000 - 3 мг
Тальк - 10 мг - 543 мг
Пример B
Таблетки, каждая из которых содержит нижеперечисленные компоненты, получают известными методами.
N4-(Бутоксикарбонил)-5'-дезокси-5-фторцитидин - 100 мг
Лактоза - 25 мг
Зерновой крахмал - 20,2 мг
Гидроксипропилметилцеллюлоза - 4 мг
Стеарат магния - 0,8 мг
Пленка - 10 мг
ПЭГ 6000 - 1,5 мг
Тальк - 4,5 мг - 166 мг
Пример C
Сухие парентеральные дозировочные формы приготовляют известными методами.
(1) В общей сложности 5 г N4-(Бутоксикарбонил)-5'-дезокси-5-фторцитидина растворяют в 75 мл дистиллированной воды, раствор подвергают бактериологическому фильтрованию и затем в асептических условиях разливают по сосудикам. Затем раствор сушат вымораживанием с получением в каждом сосудике по 500 мг стерильного сухого твердого вещества.
(2) Чистый N4-(бутоксикарбонил)-5'-дезокси-5-фторцитидин в количестве 500 мг на сосудик или ампулу герметизируют в сосуде и стерилизуют нагреванием.
Указанные сухие дозировочные формы перед употреблением восстанавливают добавлением приемлемого стерильного водного растворителя, такого как вода, для инъекций, или изотонического раствора хлорида натрия, или 5%-ной декстразы для парентерального введения.
Примеры D-I
Процедура:
1. Смешивают 5'-деокси-5-фтор-N4-(пентилоксикарбонил)цитидин с безводной лактозой и частью кросповидона.
2. Растворяют гипромеллозу в очищенной воде.
3. Гранулируют смесь, полученную на стадии 1, с раствором для грануляции, полученным на стадии 2.
4. Подвергают мокрому размалыванию гранулированный продукт, полученный на стадии 3.
5. Сушат и размалывают гранулы, полученные на стадии 4.
6. Смешивают гранулы, полученные на стадии 5, с Pharmaburst С, оставшейся частью кросповидона, маннитом, микрокристаллической целлюлозой, аспартамом, сахарином натрия, ванилином, смесью для маскировки горького вкуса и клубничным ароматизатором.
7. Просеивают стеарат магния, добавляют его к смеси, полученной на стадии 6, и перемешивают.
8. Прессуют смесь для таблеток, полученную на стадии 7, с получением ядер.
9. Приготавливают суспензию пленочного покрытия путем диспергирования смеси для пленочного покрытия в очищенной воде.
10. Наносят пленочное покрытие на ядра, полученные на стадии 8 с использованием суспензии пленочного покрытия, полученной на стадии 9.
Примеры J-O
Ниже представлены составы (мг/таблетку) предпочтительных лекарственных форм, в которых лактоза заменена на маннит.
Процедура: аналогичная процедуре, описанной для примеров D-I, за исключением того, что на стадии 1 безводную лактозу заменяют на маннит.
Примеры P-U
Ниже представлены составы (мг/таблетку) предпочтительных лекарственных форм, в которых отсутствуют Pharmaburst C и лактоза.
Процедура: аналогична процедуре, описанной для примеров D-I, за исключением того, что на стадии 1 безводную лактозу заменяют на маннит и на стадии 6 не применяют Pharmaburst С.
| название | год | авторы | номер документа |
|---|---|---|---|
| N-ОКСИКАРБОНИЛЗАМЕЩЕННЫЕ 5'-ДЕОКСИ-5-ФТОРЦИТИДИНЫ И ФАРМАЦЕВТИЧЕСКИЙ ПРЕПАРАТ | 1993 |
|
RU2458932C2 |
| ПРОИЗВОДНЫЕ 5'-ДЕЗОКСИЦИТИДИНА И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 1998 |
|
RU2238278C2 |
| α- ИЛИ β-КРИСТАЛЛИЧЕСКИЕ МОДИФИКАЦИИ 5'-ДЕЗОКСИ-N-КАРБОПЕНТИЛОКСИ-5-ФТОРЦИТИДИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ НА ИХ ОСНОВЕ | 2008 |
|
RU2393164C1 |
| СПОСОБЫ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ N-АЦИЛ-5'-ДЕЗОКСИ-5-ФТОРЦИТИДИНА | 1993 |
|
RU2131879C1 |
| НОВЫЕ НУКЛЕОЗИДЫ, ИМЕЮЩИЕ БИЦИКЛИЧЕСКУЮ САХАРНУЮ ГРУППИРОВКУ, И СОДЕРЖАЩИЕ ИХ ОЛИГОНУКЛЕОТИДЫ | 1999 |
|
RU2211223C2 |
| СПОСОБ ИДЕНТИФИКАЦИИ ПРОТИВООПУХОЛЕВЫХ ЦЕЛЕВЫХ ФЕРМЕНТОВ | 2002 |
|
RU2319482C2 |
| АНТРАЦИКЛИН ГЛИКОЗИДЫ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, ОБЛАДАЮЩИЕ ПРОТИВООПУХОЛЕВЫМИ СВОЙСТВАМИ, И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1990 |
|
RU2043360C1 |
| СПОСОБ СОЕДИНЕНИЯ НУКЛЕОЗИДОВ 3'-5'-МЕЖНУКЛЕОТИДНЫМ СИЛИЛЬНЫМ ЗВЕНОМ | 1991 |
|
RU2079508C1 |
| Способ получения производных 5 @ -дезокси-5-фторцитидина | 1988 |
|
SU1736342A3 |
| СПОСОБ ПОЛУЧЕНИЯ КАПЕЦИТАБИНА И ИСПОЛЬЗУЕМОГО ПРИ ЭТОМ ОБОГАЩЕННОГО β-АНОМЕРОМ ТРИАЛКИЛКАРБОНАТНОГО СОЕДИНЕНИЯ | 2008 |
|
RU2439064C1 |
Изобретение относится к 5'-деокси-5-фтор-N4-(пентилоксикарбонил)цитидину, обладающему противоопухолевой активностью с высоким уровнем безопасности, а также к фармацевтической композиции на его основе. 2 н.п. ф-лы, 7 табл.
1. Соединение 5'-деокси-5-фтор-N4-пентилоксикарбонил)цитидин, обладающее противоопухолевой активностью.
2. Фармацевтический препарат, обладающий противоопухолевым действием, содержащий активный компонент и фармацевтически приемлемый носитель, отличающийся тем, что в качестве активного компонента он содержит соединение по п.1.
| ЗЕРКАЛЬНЫЙ ЭЛЕКТРОННЫЙ МИКРОСКОП | 0 |
|
SU184365A1 |
| US 4340729 А, 20.07.1982 | |||
| J | |||
| Med | |||
| Chem., 22, p.1330, 1979. | |||

