Область изобретения
Областью изобретения являются нуклеозидные и олигонуклеотидные аналоги и способы их получения.
Предпосылки
Нуклеозидные и олигонуклеотидные аналоги долгое время применяли в качестве фармацевтических ингредиентов против ряда вирусов и некоторых видов рака. В настоящее время целый ряд нуклеозидных и нуклеотидных аналогов находится в клинических испытаниях для лечения некоторых болезней.
В клетке нуклеозиды и нуклеотиды фосфорилируются или дополнительно фосфорилируются до соответствующих нуклеозидтрифосфатов. Нуклеозидтрифосфаты являются ингибиторами ДНК- или РНК-полимераз. Нуклеозидтрифосфаты также могут встраиваться в ДНК или РНК, что препятствует элонгации ДНК или РНК.
Активные нуклеозидные аналоги обычно быстро фосфорилируются в клетке-мишени. Соответствующие нуклеозидтрифосфаты имеют высокое сродство к каталитическим сайтам полимераз и конкурируют с природными нуклеозидтрифосфатами в качестве субстрата полимераз. Определенные нуклеозидные аналоги работают на нуклеозидном или монофосфатном уровне. Одна группа перспективных нуклеозидных аналогов представляет собой нуклеозиды с конформационно замкнутыми сахарными группировками. Сообщалось, что определенные конформационно замкнутые карбоциклические нуклеозидные аналоги продемонстрировали потенциальную активность против HCMV (человеческого цитомегаловируса), HSV (вируса простого герпеса) и EBV (вируса Эпштейна-Барра) (Siddiqui et al. Nucleosides Nucleotides 1996, 15, 235-250; Marquez et al. J. Med. Chem. 1996, 39, 3739-3747). Было описано, что конформационно замкнутый карбоциклический АZТ-5'-трифосфат (азидотимидин-5'-трифосфат) является эквипотентным ингибитором обратной транскриптазы HIV (вируса иммунодефицита человека) (Marquez et al. J. Am. Chem. Soc. 1998, 120, 2780-2789). Были также получены другие нуклеозиды с бициклическими сахарными группировками, даже если у них не была обнаружена активность (Chao et al. Tetrahedron 1997, 53, 1957-1970; Okabe et al. Tetrahedron Lett. 1989, 30, 2203-2206, Hong, et al. Tetrahedron Lett. 1998, 39, 225-228).
Ожидают, что подходящие конформационно замкнутые нуклеозиды имеют положительное воздействие на антисмысловые олигонуклеотиды. В течение двух последних десятилетий олигонуклеотиды рассматривают и применяют в качестве потенциальных лекарств на основе антисмысловых олигонуклеотидов. Олигонуклеотиды способны образовывать двойную или тройную спираль с комплементарными ДНК или РНК и обладают способностью взаимодействовать со специфическими последовательностями в вирусном и раковом геноме. Специфическое связывание олигонуклеотидов с интересующими ДНК- или РНК-мишенями, по-видимому, инактивирует функцию, ассоциированную с ДНК или РНК, такую как репликация, транскрипция и трансляция. Вследствие этого можно прервать вирусные циклы или злокачественные процессы, не затрагивая нормальных клеточных циклов.
Поскольку природные олигонуклеотиды чувствительны к клеточным и внеклеточным нуклеазам, было предпринято много усилий в изучении олигонуклеотидных модификаций, особенно тех модификаций, которые направлены на повышение устойчивости к нуклеазам и аффинности связывания. Было показано, что олигонуклеотиды, содержащие определенные бициклические нуклеозиды, демонстрируют повышенную нуклеазную стабильность (Leumann et al. Bioorg. Med. Chem. Letts. 1995, 5, 1231-4; Altmann et al. Tetrahedron Lett. 1994, 35, 2331-2334, 7625-7628). Недавно были синтезированы и встроены в олигонуклеотиды 2'-O, 4'-С-метиленрибонуклеозиды, которые имеют замкнутую 3'-эндо-сахарную "складку". Гибридизационный анализ показывает, что конформационно замкнутые нуклеозиды могут существенно увеличить гибридизацию модифицированных олигонуклеотидов с комплементарными РНК и ДНК (Obika et al. Tetrahedron Lett. 1997, 38, 8735-8738; Koshkin et al. Tetrahedron 1998, 4, 3607-3630).
Существует необходимость в новых конформационно замкнутых нуклеозидах с бициклическими сахарными группировками. Такие новые нуклеозиды будут полезны для противовирусного, противоопухолевого и другого лечения. Кроме того, олигонуклеотиды, состоящие из этих новых модифицированных нуклеозидов, могут обладать желаемой стабильностью по отношению к клеточным нуклеазам и высокой аффинностью связывания с нуклеиновыми кислотами-мишенями. Поэтому эти олигонуклеотиды могут быть потенциально полезными для лечения и диагностики.
Краткое изложение сущности изобретения
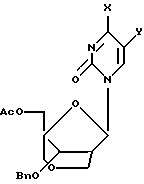
Описаны конформационно замкнутые бициклические сахарные нуклеозиды, которые имеют общую геометрическую форму, а также способы получения конформационно замкнутых бициклических сахарных нуклеозидов. Предложены нуклеозиды, имеющие бициклические сахарные группировки, и олигонуклеозиды, имеющие следующую формулу: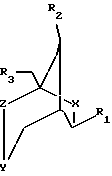
где X, Y и Z независимо выбраны из группы О, S, CH2, NR, С=O, С=СН2 или ничего, где R выбран из группы, содержащей водород, алкил, алкенил, алкинил, ацил;
R1 выбран из группы, содержащей аденин, цитозин, гуанин, гипоксантин, урацил, тимин, гетероциклы, Н, ОСН3, ОАс (Ac - ацетил), галоген, сульфонат;
R2, R3 независимо выбраны из группы Н, ОН, DMTO (DMT - диметокситритил), TBDMSO (TBDMS - трет-бутилдиметилсилил), BnO (Bn - бензил), ТНРО (ТНР - тетрагидропиранил), АсО, BzO (Bz - бензоил), OP(NiPr2)O(CH2)2CN (iPr - изопропил), ОРО3Н, РО3Н, дифосфата, трифосфата, R2 и R3 вместе могут образовывать PhCHO2 (Ph - фенил), TIPDSO2 (TIPDS -тетраизопропилдисилил) или DTBSO2 (DTBS - ди(трет-бутил)силил).
Предполагается, что описанные здесь новые нуклеозиды будут полезными для противовирусного, противоопухолевого и другого лечения. Олигонуклеотиды, состоящие из этих модифицированных нуклеозидов, имеют желаемую физиологическую стабильность и аффинность связывания, что делает их полезными для лечения и диагностики.
Детальное описание
Предложены конформационно замкнутые нуклеозиды, которые имеют 3'-эндо-сахарную "складку", а также способы их получения. Способы получения ранее описанных бициклических нуклеозидных аналогов нельзя применять к новым нуклеозидным аналогам, описанным здесь. Описанные здесь аналоги получены в результате удачного связывания между С2' и С4' положениями рибозы в нуклеозидных аналогах.
Используемая здесь аббревиатура "Ас" относится к ацетилу; аббревиатура "Вn" относится бензилу; аббревиатура "Bz" относится к бензоилу; аббревиатура "DMT" относится к диметокситритилу; аббревиатура "ТНР" относится к тетрагидропиранилу; аббревиатура "TBDMS" относится к тpeт-бутилдиметилсилилу; аббревиатура "TIPDS" относится к тетраизопропилдисилилу и аббревиатура "DTBS" относится к ди(трет-бутил)силилу.
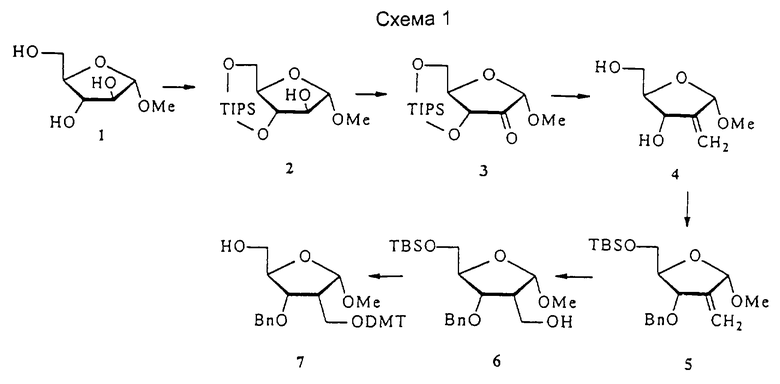
Синтез производных рибофуранозы с 2,4-мостиковой связью
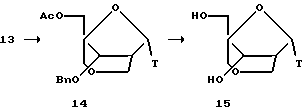
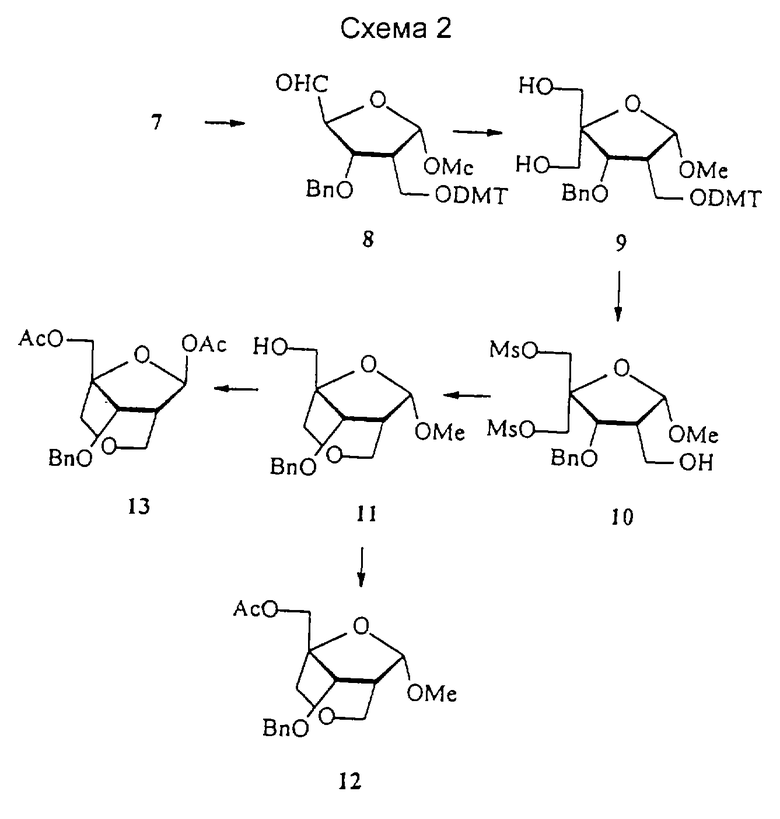
1-α-Метиларабиноза 1, полученная в соответствии с опубликованной методикой (Tejima et al. J. Org. Chem. 1963, 28, 2999-3003), была защищена 1,1,3,3, -тетраизопропилдисилоксанилом (TIPS) при O3 и O5, с получением соединения 2, которое преобразовывали в кетон 3 посредством обработки смесью DMSO/DCC/TFA (диметилсульфоксид/дициклогексилкарбодиимид/трифторуксусная кислота). Последующая реакция Виттига и отщепление TIPS давали алкен 4 с очень хорошим выходом. Соединение 4 было защищено трет-бутилдиметилсилилом (TBS) при O5 и бензилом (Вn) при O3 с получением 5. Гидроборирование 5 осуществляли с помощью 9-BBN (9-борабицикло[3.3.1]нонана), получая исключительно 2-дезокси-2-гидроксиметилпроизводное 6 с очень высоким выходом. 2-Дезокси-2-гидроксиметилпроизводное 6 подвергали тритилированию с помощью 4,4'-O-диметокситритила (DMT) хлорида и отщеплению TBS с помощью тетрабутиламмония фторида (TBAF) с получением 7.
Соединение 7 окисляли, получая альдегид 8, который подвергали обработке формальдегидом и гидроксидом натрия с получением 4-гидроксиметилпроизводного 9 с высоким выходом. Мезилирование 9 и последующее отщепление DMT давали 10. Циклизация, проведенная с NaH в THF (тетрагидрофуране), и последующее удаление мезила давали бициклический сахар 11. Обработка соединения 11 уксусным ангидридом в присутствии DMAP (диметиламинопиридина) дает 12, тогда как обработка смесью уксусный ангидрид/уксусная кислота в присутствии серной кислоты дает 13, где ацетокси при С1 имеет обращенную ориентацию (1-β) по сравнению с метокси в 11.
Синтез бициклонуклеозидов с 2',4'-мостиковой связью
Бициклонуклеозиды, имеющие сахарную группировку с 2',4'-мостиковой связью, синтезировали из продуктов конденсации силилированных нуклеозидных оснований и бициклических сахаров, как показано ниже. Конденсация 13 с бис(триметилсилил)тимином давала продукт 14, представляющий собой α-аномер с высоким выходом. Путем обработки 14 с помощью ВСl3 одновременно отщепляли ацетил и бензил, с получением бициклического α-тимидина 15.
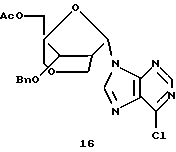
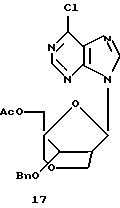
Конденсация 13 с 6-хлор-9-триметилсилилпурином давала смесь α- и β-пуриновых нуклеозидов 16 и 17 (отношение α:β от 1:1 до 2:3), которые можно разделить путем хроматографии.
Обработка 17 и 16 аммиаком в метаноле с последующим гидрогенолизом давала аналоги аденозина 18 и 19 соответственно. Гидрогенолиз требовал большого количества каталитического материала, а также длительного времени реакции вследствие повышенного стерического препятствия из-за наличия сахарной группировки. Обработка 17 и 16 меркаптоэтанолом в присутствии метилата натрия с последующим гидрогенолизом дает аналоги инозина 20 и 21 соответственно.
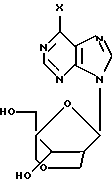
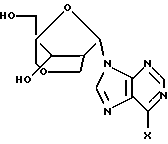
18, X = NH2;
19, X = NH2;
20, X = OH;
21, X = OH.
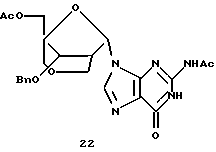
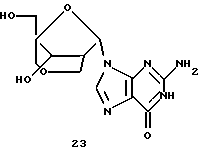
Конденсация 13 с силилированным N2-ацетилгуанином дает производное α-гуанозина 22 в виде основного продукта (30%), небольшое количество β-изомера и N7-связанные продукты. Обработка производного α-гуанозина аммиаком в метаноле с последующим гидрогенолизом давала бициклический α-гуанозин 23.
Как описано выше, реакции конденсации давали либо исключительно α-нуклеозид, либо смесь α- и β-нуклеозидов без предпочтения для β-аномеров. Для того чтобы увеличить отношение β-нуклеозидов, были изучены различные условия конденсации. Температура оказывала незначительный эффект на отношение α- и β-аномеров. Однако реагент сочетания и функциональная группа при С1 сахара действительно оказывали существенные эффекты на отношение α- и β-нуклеозидов.
Конденсация 12 с бис- либо три(триметилсилил)пиримидинами в присутствии хлорида олова (IV) давала β-нуклеозиды в виде основных продуктов с хорошим выходом. Так, реакция 12 с силилированным тимином давала производное тимидина 24 с отношением β:α, приблизительно равным 4:1. Конденсация 12 с силилированным урацилом и N4-бензоилцитозином давала соответствующие нуклеозиды 25 и 26 соответственно, с отношением β:α, приблизительно равным 9:1 в обеих реакциях. Обработка 24-26 трихлоридом бора давала пиримидиновые бициклонуклеозиды 27-29 соответственно. В случае производного цитидина бензоильную группу 29 удаляли путем обработки аммиаком с получением 30. Альтернативный путь (не показан) получения 30 начинали от 28, которое ацетилировали при O3' и O5' с последующим взаимодействием с триазолом и последующей обработкой аммиаком. В этом случае 30 получали с невысоким выходом.
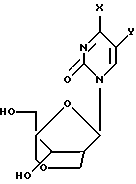
24, X = OH, Y = Me;
25, X = OH, Y = H;
26, X = NHBz, Y = H;
27, X = OH, Y = Me;
28, X = OH, Y = H;
29, X = NHBz, Y = H;
30, X = NH2, Y = H.
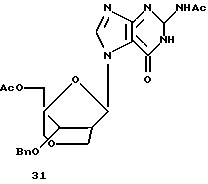
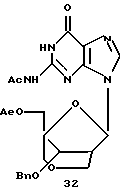
Также была исследована конденсация 12 с силилированными пуринами вместе с хлоридом олова (IV) в качестве реагента сочетания. В отличие от реакций с пиримидинами конденсация силилированного 6-хлорпурина с 12 давала не только α- и β-нуклеозиды 16 и 17, но также и N7-связанный продукт (не показано). Аналогично конденсация силилированного N2-ацетилгуанина с 12 давала смесь трех продуктов, представляющую собой N7-связанный β-нуклеозид 31 (42%), желаемый β-нуклеозид 32 (10%) и α-нуклеозид 22 (6%). Однако при нагревании с силилированным N2-ацетилгуанином в присутствии триметилсилилтрифлата N7-связанный продукт 31 частично превращался в N9-связанные α- и β-бициклонуклеозиды 22 (~ 22%) и 32 (~25%). Выделенный 32 подвергали таким же обработкам, как и 22, с получением бициклического β-гуанозина 33.
Стереохимические распределения производного 2,6-диоксабицикло[3,2,1] октана 11 и бициклонуклеозидов, образованных при конденсации бициклических сахаров с силилированными нуклеозидными основаниями, можно определить с помощью NОЕ (ядерного эффекта Оверхаузера), протонного ЯМР. Как показано с помощью указанной модели, жесткая кольцевая система диоксабицикло[3,2,1] октана заставляет протоны (Н1' и Н2') при С1' и С2' α-бициклонуклеозидов становиться почти параллельно, тогда как Н1' и Н2' в β-бициклонуклеозидах направлены в противоположные стороны. Например, угол закручивания Н1'-С1'-С2'-Н2' бициклического α-тимидина 15 после геометрической оптимизации равен 37o, и в соответствии с этим определили константу взаимодействия 3,9 Гц в протонном ЯМР. Угол закручивания Н1'-С1'-С2'-Н2' в бициклическом β-тимидине 27 равен 96o после геометрической оптимизации, и как ожидалось, не было обнаружено связывания между Н1' и Н2'. Действительно протон при С1' во всех измеренных β-бициклонуклеозидах представлен в виде единичного пика. В противоположность этому у всех измеренных α-бициклонуклеозидов протон при С1' представлен в виде дублета с константой взаимодействия, приблизительно равной 4,0 Гц.
Стереохимические распределения бициклонуклеозидов далее были подтверждены с помощью рентгеноструктурной кристаллографии бициклических тимидинов 15 и 27. Кольцо рибозы сахарной группировки диоксабицикло[3,2,1]октана в обоих соединениях принимает вид типичной С3'-эндо-сахарной "складки", тогда как шестичленное кольцо в сахарной группировке принимает форму "кресла". Тиминовое основание в обоих соединениях имеет противоположную ориентацию.
Синтез фосфорамидитов бициклонуклеозидов с 2,4-мостиковой связью
Бициклический β-тимидин 27, бициклический β-N4-бензоилцитидин 29 и бициклический β-N4-ацетилцитидин 29 были защищены с помощью DMT и затем преобразованы в соответствующие фосфорамидиты соответственно. Из-за стерического препятствия требовалось более длительное время реакции.
34, X = OH, Y = Me;
35, X = NMAc, Y = H;
36, X = NHBz, Y = H;
37, X = OH, Y = Me;
38, X = NHAc, Y = H;
39, X = NHBz, Y = H.
ПРИМЕРЫ
Способы синтеза, которые были использованы для получения описанных соединений, можно также использовать для синтеза других заявленных соединений. Настоящее изобретение включает в себя, но не ограничивается этим, соединения, полученные в следующих примерах. Номера в скобках, указанные после названий соединений в примерах, соответствуют номерам структур в разделе детального описания.
Пример 1
Получение 1-α--метил-3,5-O-(1,1,3,3-тетраизопропил-1,3-дисилоксандиил)-D-рибофуранозы (2)
αМетиларабинозу получали в соответствии с опубликованной процедурой (Tejima S., Fletcher Jr. H. G. J. Org. Chem. 1963, 28, 2999-3003) и отделяли от ее β-аномера (второстепенный продукт) с использованием хроматографии на диоксиде кремния. К перемешиваемому раствору α-метиларабинозы (19,27 г, 119,9 ммоль) в безводном пиридине (200 мл) при 0oС добавили 1,3-дихлор-1,1,3,3-тетраизопропилдисилоксан (38,4 мл, 119,9 ммоль). Полученный раствор перемешивали при 0oС в течение 1 часа и затем при комнатной температуре в течение 1,5 часов. Раствор охладили до 0oС и добавили воду (20 мл). Смесь перемешивали в течение 10 минут и разбавили с помощью ЕtOАс. Водный слой экстрагировали с помощью ЕtOАс. Объединенный органический слой высушивали (Na2SO4) и концентрировали до сухого состояния. Хроматография на диоксиде кремния с использованием 15%-ного ЕtOАс в гексанах дала 42,7 г (88%) указанного в заголовке соединения в виде бесцветного сиропа.
Пример 2
Получение 2-С,2-O-дидегидро-α-метил-3,5-O-(1,1,3,3-тетраизопропил-1,3-дисилоксандиил)-O-рибофуранозы (3)
К перемешиваемому раствору 1-α-метил-3,5-O-(1,1,3,3-тетраизопропил-1,3-дисилоксан-диил)-D-рибофуранозы (42,6 г, 104,9 ммоль) и DCC (43,4 г, 209,8 ммоль) в безводном DMSO (250 мл) и эфире (100 мл) при 0oС в атмосфере аргона добавили раствор трифторуксусной кислоты (4,04 мл, 52,5 ммоль) и пиридина (8,44 мл, 105 ммоль) в DMSO (30 мл). Полученную реакционную смесь нагревали до комнатной температуры, перемешивали в течение 5 часов и затем охлаждали до 0oС. Добавили щавелевую кислоту (21,3 г, 236 ммоль) в метаноле (60 мл) с последующим добавлением воды (30 мл). Полученную смесь перемешивали при комнатной температуре в течение 1 часа и осадок отфильтровали и тщательно промыли гексанами. Далее фильтрат разбавили гексанами, пятикратно промывали водой, высушивали (Na2SO4) и концентрировали до сухого состояния. Хроматография на диоксиде кремния с использованием 2%-ного МеОН в метиленхлорид-гексановой смеси (1:2) дала 37,6 г (89%) указанного в заголовке соединения в виде бесцветного сиропа.
1H-ЯМР (CDCl3): δ 1,00-1,12 (m, 28H, TIPDS), 3,47 (s, 3Н, ОСН3), 4,05-4,19 (m, 3Н, Н4, Н5а, Н5b), 4,51 (dd, J=9,3 Гц, 1,5 Гц, 1Н, Н3), 4,89 (t, J= 1,5 Гц, 1Н, Н1).
Пример 3
Получение 2-дезокси-2-метилен-1-α-метил-3,5-O-(1,1,3,3-тетраизопропил-1,3-дисилоксандиил)-D-рибофуранозы
К перемешиваемой суспензии метилтрифенилфосфония бромида (21,5 г, 60,1 ммоль) в безводном эфире (1380 мл) при комнатной температуре в атмосфере аргона добавили раствор трет-пентаоксида натрия (5,97 г, 54,0 ммоль) в безводном бензоле (50 мл). Полученную светло-желтую смесь перемешивали при комнатной температуре в течение 6 часов и охлаждали до -10oС, затем добавили раствор 2-С, 2-О-дидегидро-α-метил-3,5-О-(1,1,3,3-тетраизопропил-1,3-дисилоксандиил)-D-рибофуранозы (12,1 г, 30,1 ммоль) в эфире (35 мл). Реакционную смесь перемешивали при -10oС в течение 1 часа, дважды промывали солевым раствором, высушивали (Na2SO4) и концентрировали. Хроматография на диоксиде кремния с использованием 5%-ного ЕtOАс в гексанах дала 11,0 г (91%) указанного в заголовке соединения в виде бесцветного сиропа.
1H-ЯМР (CDCl3): δ 1,00-1,12 (m, 28H, TIPDS), 3,45 (s, 3Н, ОСН3), 3,73 (dt, J=9,0 Гц, 3,0 Гц, 1Н, Н4), 4,02, 4,03 (2s, 2H, Н5), 4,62 (dt, J=9,0 Гц, 2,7 Гц, 1Н, Н3), 5,27 (m, 1Н, Н1), 5,32-5,36 (m, 2H, Н2').
Пример 4
Получение 2-дезокси-2-метилен-1-α-метил-D-рибофуранозы (4)
К перемешиваемому раствору 2-дезокси-2-метилен-1-α-метил-3,5-O-(1,1,3,3-тетраизо-пропил-1,3-дисилоксандиил)-D-рибофуранозы (35,0 г, 87,1 ммоль) в THF (200 мл) добавили 1,0 M TBAF в THF (180 мл). Полученный раствор выдерживали при комнатной температуре в течение 1 часа. THF выпаривали, а остаток подвергали хроматографии на диоксиде кремния с использованием 10%-ного ЕtOН в метиленхлориде с получением 14,6 г (88%) указанного в заголовке соединения в виде сиропа.
Пример 5
Получение 3-O-бензил-5-O-(трет-бутилдиметилсилил)-2-дезокси-2-метилен-1-α-метил-D-рибофуранозы (5)
Раствор 2-дезокси-2-метилен-1-α-метил-D-рибофуранозы (13,7 г, 85,5 ммоль) и TBDMS-CI (13,5 г, 89,6 ммоль) в безводном пиридине (130 мл) выдерживали при комнатной температуре в течение 15 часов. После охлаждения до 0oС и добавления воды (2 мл) полученную смесь перемешивали при комнатной температуре в течение 1 часа, концентрировали до половины объема, разбавляли ЕtOАс, промывали солевым раствором, высушивали (Na2SO4) и концентрировали до сухого состояния. Тщательно высушенный сырой продукт растворили в THF (70 мл) и добавили к перемешиваемой смеси NaH (60% в минеральном масле, 5,6 г, 140 ммоль) в THF (350 мл) при 0oС. После перемешивания при комнатной температуре в течение 40 минут добавили бензилбромид (10,75 мл, 90,5 ммоль). Реакционную смесь перемешивали в течение 4 часов и охладили до 0oС с последующим медленным добавлением воды (2 мл) и затем 10%-ного АсОН в воде до достижения рН, равного 7. Смесь разбавляли ЕtOАс, промывали солевым раствором, затем разбавленным бикарбонатом натрия, высушивали (Na2SO4) и концентрировали до сухого состояния. Хроматография на диоксиде кремния с использованием 0-10%-ного ЕtOАс в гексанах дала 23,8 г (76%) указанного в заголовке соединения в виде бесцветной жидкости.
1H-ЯМР (CDCl3): δ 0,01 (s, 3Н, SiCН3), 0,02 (s, 3Н, SiCH3), 0,85 (s, 9H, t-Bu), 3,41 (s, 3Н, ОСН3), 3,60-3,72 (m, 2Н, Н5а, H5b), 4,20 (dd, J=8,7 Гц, 4,5 Гц, 1Н, Н3), 4,57, 4,66 (АВ, J=12,0 Гц, 2Н, Bn), 5,22 (t, J=1,2 Гц, 1Н, Н1), 5,38 (t, J=1,5 Гц, 1Н, Н2а'), 5,43 (m, J=1,2 Гц, 1Н, H2b'), 7,23-7,37 (m, 5H, Bn).
Аналитически рассчитано для C20H32O4Si,%: С 65,89; Н 8,85.
Обнаружено,%: С 65,92; Н 9,22.
Пример 6
Получение 3-O-бензил-5-O-(трет-бутилдиметилсилил)-2-дезокси-2-гидроксиметил-1-α-метил-О-рибофуранозы (6)
К перемешиваемому раствору 3-О-бензил-5-О-(трет-бутилдиметилсилил)-2-дезокси-2-метилен-1-α-метил-D-рибофуранозы (5,28 г, 14,50 ммоль) в атмосфере аргона добавили 9-BBN (0,5 М в THF, 87 мл). Полученный раствор перемешивали при температуре окружающей среды в течение 1 часа, затем при 40oС в течение ночи, охлаждали до комнатной температуры, а затем перенесли в колбу, содержащую тетрагидрат пербората натрия (13,39 г, 87 ммоль) в воде (85 мл) и этанол (85 мл). Полученную смесь интенсивно перемешивали при 50oС в течение 4 часов, охладили до 0oС, нейтрализовали с помощью АсОН до рН 8 и сконцентрировали до небольшого объема. Оставшийся объем разбавили водой (20 мл) и трижды экстрагировали метиленхлоридом. Объединенный органический слой дважды промывали солевым раствором, высушивали (Na2SO4) и концентрировали до сухого состояния. Хроматография на диоксиде кремния с использованием смеси ЕtOАс-гексаны (1: 2) дала 5,17 г (93%) указанного в заголовке соединения в виде бесцветного сиропа.
1H-ЯМР (CDCl3): δ 0,03 (s, 6H, SiCН3), 0,87 (s, 9H, трет-бутил), 2,34-2,43 (m, 1H, Н2), 3,39 (s, 3Н, ОСН3), 3,48 (dd, J=10,5 Гц, 6,0 Гц, 1Н, Н5а), 3,60 (dd, J= 10,5 Гц, 3,6 Гц, 1H, H5b), 3,88 (d, J=7,2 Гц, 2Н, Н2'), 3,98 (dd, J=7,2 Гц, 2,7 Гц, 1H, Н3), 4,17 (m, 1H, Н4), 4,44, 4,66 (AB, J=12,3 Гц, 2Н, Bn), 4,95 (d, J=5,4 Гц, 1H, Н1), 7,23-7,36 (m, 5H, Bn).
Аналитически рассчитано для С20Н34O5Si,%: С 62,79; Н 8,96.
Обнаружено,%: С 62,92; Н 9,21.
Пример 7
Получение 3-O-бензил-2-дезокси-2-(4,4'-диметокситритилоксиметил)-1-α-метил-D-рибофуранозы (7)
Раствор 3-O-бензил-5-O-(трет-бутилдиметилсилил)-2-дезокси-2-гидроксиметил-1-α-метил-D-рибофуранозы (6,60 г, 17,28 ммоль) и DMT-CI (7,03 г, 20,74 ммоль) в безводном пиридине (50 мл) выдерживали при комнатной температуре в течение ночи и реакцию резко остановили посредством добавления воды (8 мл). Полученный раствор выдерживали в течение 10 минут и затем разбавили с помощью ЕtOАс, трижды промывали солевым раствором, высушивали (Na2SO4) и концентрировали с получением сырого продукта 9, который растворили в THF (52 мл). Добавили TBAF (1,0 М в THF, 26 мл) и полученный раствор выдерживали при комнатной температуре в течение 30 минут. THF выпаривали, а остаток подвергали хроматографии на диоксиде кремния с использованием смеси ЕtOАс-гексаны (1: 1), получили указанное в заголовке соединение в виде белой пены.
1H-ЯМР (CDCl3): δ 2,33-2,42 (m, 1H, Н2), 3,26-3,63 (m, 7H, Н5а, H5b, H2a', H2b', ОСН3), 3,79 (d, J=1,2 Гц, 6Н, ДМТ), 3,91 (dd, J=7,5 Гц, 2,4 Гц, 1H, Н3), 4,13 (m, 1H, H4), 4,41, 4,50 (AB, J=12,9 Гц, 2Н, Bn), 5,05 (d, J= 5,1 Гц, 1H, Н1), 6,78-6,85 (m, 4H, DMT), 7,14-7,47 (m, 14H, Bn, DMT).
Аналитически рассчитано для С35Н38О7,%: С 73,66; Н 6,71.
Обнаружено,%: С 73,57; Н 6,76.
Пример 8
Получение 3-O-бензил-2-дезокси-2-(4,4'-диметокситритилоксиметил)-5-С, 5-О-дидегидро-1-α-метил-D-рибофуранозы (8)
К перемешиваемому раствору 3-O-бензил-2-дезокси-2-(4,4'-диметокситритилоксиметил)-1-α-метил-D-рибофуранозы (9,18 г, 16,16 ммоль) и DCC (10,0 г, 48,49 ммоль) в безводном DMSO (60 мл) при 10oС добавили раствор трифторуксусной кислоты (0,622 мл, 8,08 ммоль) и пиридин (1,95 мл, 24,24 ммоль) в DMSO (15 мл). Полученную реакционную смесь перемешивали при 10oС в течение 1 часа, при комнатной температуре в течение 6 часов и затем охлаждали до 0oС. После добавления воды (8 мл) смесь перемешивали в течение ночи и разбавили с помощью ЕtOАс. Осадок отфильтровали и тщательно промыли с помощью ЕtOАс. Объединенный фильтрат промывали солевым раствором пять раз, высушивали (Na2SO4) и концентрировали до сухого состояния. Хроматография на диоксиде кремния с использованием смеси ЕtOАс-гексаны (1:1) дала 8,26 г (90%) указанного в заголовке соединения в виде белой пены.
Пример 9
Получение 3-O-бензил-2-дезокси-2-(4,4'-диметокситритилоксиметил)-4-С-гидроксиметил-1-α-метил-D-рибофуранозы (9)
К перемешиваемому раствору 3-O-бензил-2-дезокси-2-(4,4'-диметокситритилоксиметил)-5-С, 5-O-дидегидро-1-α-метил-D-рибофуранозы (8,0 г, 14,08 ммоль) и формальдегида (37% в воде, 85 мл) в диоксане (420 мл) при 0oС добавляли по каплям водный раствор NaOH (2,0 М, 210 мл) в течение 15 минут. Полученный мутный раствор перемешивали при комнатной температуре в течение 2 дней до получения прозрачного раствора. После охлаждения до 0oС раствор нейтрализовали 10%-ной уксусной кислотой до рН 8, сконцентрировали до малого объема, разбавили водой (100 мл) и трижды экстрагировали метиленхлоридом. Объединенный органический слой промывали солевым раствором, высушивали (Na2SO4) и концентрировали до сухого состояния. Хроматография на диоксиде кремния с использованием 4-5%-ного этанола в метиленхлориде дала 8,11 г (94%) указанного в заголовке соединения в виде белой пены.
1H-ЯМР (CDCl3): δ 2,46-2,57 (m, 1H, Н2), 3,23-3,73 (m, 9H, Н5, Н4', Н2', ОСН3), 3,79 (d, J=1,8 Гц, 6Н, DMT), 4,14 (d, J=6,9 Гц, 1H, Н3), 4,43, 4,47 (АВ, J= 12 Гц, 2Н, Bn), 4,97 (d, J=4,8 Гц, 1H, Н1), 6,77-6,85 (m, 4H, DMT), 7,11-7,46 (m, 14H, Bn, DMT).
Пример 10
Получение 3-O-бензил-2-дезокси-2-гидроксиметил-5-O-мезил-4-мезилоксиметил-1-α-метил-D-рибофуранозы (10)
К перемешиваемому раствору 3-O-бензил-2-дезокси-2-(4,4'-диметокситритилоксиметил)-4-С-гидроксиметил-1-α-метил-D-рибофуранозы (7,80 г, 13,0 ммоль) в безводном пиридине (60 мл) при 0oС в атмосфере аргона добавляли по каплям метансульфонилхлорид (3,03 мл, 39 ммоль). Полученную реакционную смесь перемешивали при комнатной температуре в течение 45 минут, затем охладили и разбавили путем добавления воды (5 мл). Полученную смесь перемешивали при комнатной температуре в течение 15 минут, разбавили с помощью ЕtOАс, трижды промывали солевым раствором, высушивали (Na2SO4) и концентрировали, с получением сырого продукта в виде белой пены, который растворили в смеси АсОН-вода (80:20, 400 мл). Полученный раствор выдерживали при комнатной температуре в течение 2 часов и разбавили водой (200 мл), затем сконцентрировали до приблизительно одной четверти от исходного объема. Добавили воду (100 мл) и сконцентрировали смесь до сухого состояния. Хроматография на диоксиде кремния смесью ЕtOАс-гексаны (от 3:1 до 1:0) дала 5,32 г (90%) указанного в заголовке соединения в виде полутвердого вещества.
1H-ЯМР (CDCl3): δ 2,43-2,54 (m, 1Н, Н2), 3,01 (s, 3Н, OMs), 3,03 (s, 3H, OMs), 3,41 (s, 3H, ОСН3), 3,81 (d, J=4,8 Гц, 2Н, Н2'), 4,01, 4,04 (АВ, J= 10,5 Гц, 2Н, Н4'), 4,21 (d, J=7,5 Гц, 1Н, Н3), 4,30, 4,50 (АВ, J=1,8 Гц, 2Н, Н5), 4,56, 4,63 (АВ, J=12,0 Гц, 2Н, Вn), 4,99 (d, J=5,1 Гц, 1Н, Н1), 7,30-7,42 (m, 5H, Вn).
Аналитически рассчитано для C17H27O10S2,%: С 44,82; Н 5,97. Обнаружено, %: С 44,68; Н 6,00.
Пример 11
Получение (1S, 3S,4R,8S)-8-бензилокси-1-гидроксиметил-3-метокси-2,6-диоксабицикло[3,2,1]октана (11)
К перемешиваемой смеси NaH (60% в минеральном масле, 1,83 г, 22,90 ммоль) в безводном THF (200 мл) добавили раствор 3-O-бензил-2-дезокси-2-гидроксиметил-5-O-мезил-4-мезилоксиметил-1-α-метил-D-рибофуранозы (5,20 г, 11,45 ммоль) в THF (30 мл). Полученную реакционную смесь перемешивали при 55oС в течение 42 часов и реакцию резко остановили путем добавления воды при 0oС. THF выпаривали и затем добавили водный NaOH (0,5 M, 250 мл). Полученную смесь нагревали с обратным холодильником в течение 24 часов, затем охладили до 0oС, нейтрализовали разбавленной хлорной кислотой до рН 8, экстрагировали метиленхлоридом четыре раза. Объединенный органический слой высушивали (Na2SO4) и концентрировали до сухого состояния. Хроматография на диоксиде кремния с использованием смеси ЕtOАс-гексаны (от 2:1 до 1:0) дала 3,16 г (98%) указанного в заголовке соединения в виде бесцветного сиропа.
1H-ЯМР (CDCl3): δ 2,32 (m, 1H, Н2), 3,41 (d, J=11,4 Гц, 1Н, Н4а'), 3,46-3,60 (m, 2Н, 5H, Н5, ОСН3), 3,91 (d, J=11,1 Гц, 1H, Н4b'), 3,92 (dd, J= 10,8 Гц, 2,4 Гц, 1H, Н2а'), 4,01 (d, J=5,4 Гц, 1H, Н3), 4,04 (d, J=10,5 Гц, 1H, H2b'), 4,58, 4,64 (АВ, J= 12,0 Гц, Вn), 5,07 (d, J=3,9 Гц, 1H, Н1), 7,28-7,40 (m, 5H, Вn).
Пример 12
Получение (1R, 3S,4R,8S)-1-ацетоксиметил-8-бензилокси-3-метокси-2,6-диоксабицикло[3,2,1]октана (12)
Раствор (1S, 3S,4R,8S)-8-бензилокси-1-гидроксиметил-3-метокси-2,6-диоксабицикло[3,2,1] октана (1,60 г, 5,71 ммоль), уксусный ангидрид (1,08 мл, 11,42 ммоль) и DMAP (2,09 г, 17,13 ммоль) в безводном метиленхлориде (10 мл) перемешивали при комнатной температуре в течение 2 часов, затем охладили до 0oС и разбавили метанолом (4 мл). Смесь перемешивали при комнатной температуре в течение 15 минут, разбавили метиленхлоридом, промывали солевым раствором и затем 10%-ным NaHCO3, высушивали (Na2SO4) и концентрировали до сухого состояния. Хроматография на диоксиде кремния с использованием смеси этилацетат-гексаны (1:1) дала 1,82 г (99%) указанного в заголовке соединения в виде бесцветного сиропа.
1H-ЯМР (CDCl3): δ 2,02 (s, 3H, ОАс), 2,33 (m, 1H, Н2), 3,50 (d, J=10,8 Гц, 1Н, Н4а'), 3,57 (s, 3H, ОСН3), 3,86-4,04 (m, 5Н, Н2а', H2b', Н3, H4b', Н5а), 4,14 (d, J=12,0 Гц, 1H, H5b), 4,50, 4,64 (АВ, J=12,0 Гц, 1H, Bn), 5,09 (d, J=3,9 Гц, 1H, Н1), 7,29-7,42 (m, 5H, Bn),
Аналитически рассчитано для C17H22O6,%: С 63,34; Н 6,88. Обнаружено,%: С 63,41; Н 6,94.
Пример 13
Получение (1R, 3S, 4R,8S)-3-ацетокси-1-ацетоксиметил-8-бензилокси-2,6-диоксабицикло[3,2,1]октана (13)
К перемешиваемому раствору (1S,3S,4R,8S)-8-бензилокси-1-гидроксиметил-3-метокси-2,6-диоксабицикло[3,2,1] октана (600 мг, 2,14 ммоль) в смеси уксусной кислоты (6 мл) и уксусного ангидрида (0,6 мл) при 0oС добавляли по каплям концентрированную серную кислоту (57 мкл, 1,07 ммоль). Полученную реакционную смесь перемешивали при 0oС в течение 10 минут и затем при комнатной температуре в течение 2 часов. После охлаждения до 0oС раствор разбавили с помощью ЕtOАс, трижды промывали солевым раствором и затем 10%-ным бикарбонатом натрия, высушивали (Na2SO4) и концентрировали до сухого состояния. Хроматография на диоксиде кремния с использованием смеси ЕtOАс-гексаны (2:3) дала 696 мг (93%) указанного в заголовке соединения (β-аномера) и 31 мг (3%) α-аномера, оба в виде бесцветного сиропа. β-Аномер затвердевал после выдерживания при комнатной температуре в течение нескольких дней, т.пл. 55-58oС.
1H-ЯМР (CDCl3): δ 2,03 (s, 3Н, ОАс), 2,08 (s, 3Н, ОАс), 2,36-2,39 (m, 1H, Н2), 3,49 (d, J=10,8 Гц, Н4а'), 3,73 (d, J=11,1 Гц, 2,7 Гц, 1H, Н2а'), 3,89 (d, J=11,1 Гц, 1H, H4b'), 4,01 (d, J=11,1 Гц, 1H, H2b'), 4,03 (d, J=9,3 Гц, 1H, Н5а), 4,14 (d, J=5,1 Гц, 1H, Н3), 4,55 (d, J=9,6 Гц, 1H, H5b), 4,55, 4,64 (AB, J=11,7 Гц, 2Н, Вn), 6,39 (s, 1H, Н1), 7,29-7,42 (m, 5H, Вn).
Аналитически рассчитано для C18H22O7,%: С 61,70; Н 6,33.
Обнаружено,%: С 61,74; Н 6,46.
Пример 14
Получение (1R, 3S, 4R,8S)-1-ацетоксиметил-8-бензилокси-3-(тимин-1-ил)-2,6-диоксабицикло[3,2,1]октана (14)
Смесь тимина (189 мг, 1,5 ммоль) и безводного сульфата аммония (15 мг) в HMDS (6 мл) нагревали с обратным холодильником в течение ночи. После удаления HMDS осадок выпаривали совместно с безводным м-ксилолом, высушивали под вакуумом в течение 30 минут и растворяли в растворе (1R,3S,4R,8S)-3-ацетокси-1-ацетоксиметил-8-бензилокси-2,6-диоксабицикло[3,2,1]октана (306 мг, 0,87 ммоль) в 1,2-дихлорэтане (5 мл). К этому перемешиваемому раствору в атмосфере аргона добавляли по каплям триметилсилилтрифлат (0,38 мл) в 1,2-дихлорэтане (2 мл). Полученный раствор нагревали с обратным холодильником в течение 2 часов, затем охладили до 0oС, разбавили хлороформом и нейтрализовали 10%-ным NаНСО3 (10 мл). Органический слой отделяли, а водный слой дважды экстрагировали хлороформом. Объединенный органический слой высушивали (Na2SO4) и концентрировали до сухого состояния. Кристаллизация из EtOAc-CH2Cl2 дала указанное в заголовке соединение (303 мг, 83%) в виде бесцветного твердого вещества, т.пл. 198-200oС.
1H-ЯМР (CDCl3): δ 1,94 (d, J=1,2 Гц, 1H, АrСН3), 2,04 (s, 3Н, ОАс), 2,93 (m, 1H, H2'), 3,50 (dd, J=11,8 Гц, 2,1 Гц, 1H, Н2а''), 3,59 (d, J=11,4 Гц, 1H, Н4а''), 4,016 (d, J=11,7 Гц, 1H, H4b''), 4,022 (d, J=12,6 Гц, 1H, Н5а'), 4,09 (d, J=12,0 Гц, 1H, H2b''), 4,11 (d, J=4,5 Гц, 1H, Н3'), 4,27 (d, J=12,6 Гц, 1H, H5b'), 4,53, 4,70 (AB, J=11,7 Гц, 2Н, Вn), 5,88 (d, J=3,6 Гц, 1H, Н1'), 7,30-7,42 (m, 5H, Вn), 7,74 (d, J=1,5 Гц, 1H, Н6), 8,79 (s, 1H, NH).
Аналитически рассчитано для C21H24N2O7,%: С 60,57; Н 5,81; N 6,73.
Обнаружено,%: С 60,55; Н 5,84; N 6,69.
Пример 15
Получение (1S,3S,4R,8S)-8-гидрокси-1-гидроксиметил-3-(тимин-1-ил)-2,6-диоксабицикло[3,2,1]октана (15)
К раствору (1R,3S,4R,8S)-1-ацетоксиметил-8-бензилокси-3-(тимин-1-ил)-2,6-диоксабицикло[3,2,1] октана в безводном метиленхлориде (3 мл) при 10oС добавили трихлорид бора (1,0 М в CH2Cl2, 6 мл). Полученную реакционную смесь перемешивали при температуре от 15oС до комнатной температуры в течение ночи и затем охладили до 0oС. Добавляли по каплям метанол (1,5 мл) и полученную смесь перемешивали при 0oС в течение 15 минут с последующим добавлением триэтиламина (2 мл). Растворитель выпаривали, а осадок тщательно экстрагировали теплым ацетоном. Ацетоновый раствор высушивали (Na2SO4) и концентрировали до сухого состояния. Хроматография на диоксиде кремния с использованием 10%-ного метанола в хлороформе дала 99 мг соединения 20 в виде белой пены. Кристаллизация из ацетона дала 95 мг (93%) указанного в заголовке соединения в виде бесцветного твердого вещества, т.пл. 225-226oС.
1H-ЯМР (DMSO-d6): δ 1,76 (d, J=0,9 Гц, 1Н, АrСН3), 2,45 (m, 1Н, Н2'), 3,25 (dd, J=11,4 Гц, 2,1 Гц, 1Н, Н2а''), 3,32-3,52 (m, 2H, Н5'), 3,53 (d, J= 11,4 Гц, 1Н, Н4а''), 3,72 (d, J=11,1 Гц, 1Н, H4b''), 3,93 (d, J=11,1 Гц, 1Н, H2b''), 4,16 (m, 1H, H3'), 4,84 (t, J=6,0 Гц, 1H, ОН), 5,74 (d, J=4,2 Гц, 1H, Н1'), 5,84 (d, J=3,9 Гц, 1Н, ОН), 7,76 (d, J=1,2 Гц, 1H, Н6), 11,32 (s, 1H, NH).
MS: m/z 285 (МН+).
Аналитически рассчитано для C12H16N2O6,%: С 50,70; Н 5,67; N 9,85.
Обнаружено,%: С 50,85; Н 5,68; N 9,75.
Пример 16
Получение (1R,3R,4R,8S)-1-ацетоксиметил-8-бензилокси-3-(6-хлорпурин-9-ил)-2,6-диоксабицикло[3,2,1] октана (17) и (1R,3S,4R,8S)-1-ацетоксиметил-8-бензилокси-3-(6-хлорпурин-9-ил)-2,6-диоксабицикло[3,2,1]октана (16)
Смесь 6-хлорпурина (246 мг, 1,6 ммоль) и HMDS (8,0 мл) кипятили с обратным холодильником в атмосфере аргона в течение 2 часов. HMDS выпаривали, осадок высушивали под вакуумом в течение 30 минут и затем растворяли в растворе (1R, 3S,4R,8S)-3-ацетокси-1-ацетоксиметил-8-бензилокси-2,6-диоксабицикло[3,2,1] октана (302 мг, 0,83 ммоль) в безводном 1,2-дихлорэтане (5,0 мл) с последующим добавлением триметилсилилтрифлата (0,38 мл, 2,25 ммоль) в 1,2-дихлорэтане (2,0 мл). Полученный раствор нагревали с обратным холодильником в атмосфере аргона в течение 45 минут. Способ получения аналогичен способу, описанному ранее. Хроматография на диоксиде кремния с использованием смеси ЕtOАс-гексаны (1: 1) дала (1R,3S,4R,8S)-1-ацетоксиметил-8-бензилокси-3-(6-хлорпурин-9-ил)-2,6-диоксабицикло[3,2,1] октан (122 мг, α-аномер) и (1R, 3R, 4R,8S)-1-ацетоксиметил-8-бензилокси-3-(6-хлорпурин-9-ил)-2,6-диоксабицикло[3,2,1] октан (157 мг, β-аномер), оба в виде бесцветного твердого вещества. Общий выход составил 75%.
α-Изомер: 1H-ЯМР (CDCl3): δ 2,05 (s, 3H, ОАс), 2,89 (m, 1H, Н2'), 3,23 (dd, J=12,0 Гц, 2,4 Гц, 1Н, Н2а''), 3,72 (d, J=11,7 Гц, Н4а''), 4,09 (d, J= 12,3 Гц, 2Н, Н4'', Н5а'), 4,13 (d, J=13,2 Гц, 1H, H2b''), 4,24 (d, J=4,8 Гц, Н3'), 4,29 (d, J= 12,3 Гц, 1H, H5b'), 4,60, 4,74 (АВ, J=11,7 Гц, 2Н, Bn), 6,50 (d, J= 4,2 Гц, 1H, Н1'), 7,32-7,44 (m, 5H, Bn), 8,69 (s, 1H, Н8) 8,78 (s, 1H, Н2).
β-Изомер: т. пл. 124-125oС (ЕtOАс-гексаны). 1H-ЯМР (CDCl3): δ 2,05 (s, 3H, ОАс), 2,90 (m, 1H, H2'), 3,55 (d, J=11,1 Гц, Н4а''), 3,95-4,03 (m, 2H, Н2а'', Н4b''), 4,18-4,24 (m, 3H, Н5', H2b''), 4,32 (d, J=4,8 Гц, Н3'), 4,47, 4,63 (АВ, J=11,7 Гц, 2H, Bn), 6,52 (s, 1H, Н1'), 7,24-7,35 (m, 5H, Bn), 8,40 (s, 1H, H8), 8,72 (s, 1H, H2).
Аналитически рассчитано для C21H21N4O5Cl,%: С 56,70; Н 4,76; N 12,59.
Обнаружено,%: С 56,36; Н 4,56; N 12,37.
Пример 17
Получение (1R, 3S, 4R, 8S)-1-ацетоксиметил-3-(N2-ацетилгуанин-9-ил)-8-бензилокси-2,6-диоксабицикло[3,2,1]октана (22)
Смесь N2-ацетилгуанина (193 мг, 1,0 ммоль) и сульфата аммония (20 мг) в пиридине (1,0 мл) и HMDS (5,0 мл) кипятили с обратным холодильником в атмосфере аргона в течение 3 часов. Полученный прозрачный раствор концентрировали и выпаривали вместе с ксилолом (10 мл, сухой натрий). Осадок высушивали под вакуумом при 50oС в течение 1 часа и растворили в растворе (1R,3S, 4R, 8S)-3-ацетокси-1 -ацетоксиметил-8-бензилокси-2,6-диоксабицикло[3,2,1]октана (175 мг, 0,5 ммоль) в безводном 1,2-дихлорэтане (5 мл) с последующим добавлением триметилсилилтрифлата (0,27 мл, 1,5 ммоль) в 1,2-дихлорэтане (1,0 мл). Полученный раствор перемешивали при комнатной температуре в атмосфере аргона в течение 30 минут, затем нагревали при 70-75oС в течение 2 часов, охладили до 0oС и нейтрализовали 10%-ным бикарбонатом натрия (10 мл). Полученную смесь перемешивали в течение 15 минут и органический слой отделили. Водный слой дважды экстрагировали хлороформом. Объединенные органические слои высушивали (Na2SO4) и концентрировали до сухого состояния. Хроматография на диоксиде кремния с использованием 10%-ного этанола в СНСl3-ЕtOАс (1: 1) дала указанное в заголовке соединение (72 мг, 30%) в виде бесцветного твердого вещества, т.пл. 249oС (с разложением, ЕtOАс).
1H-ЯМР (CDCl3): δ 2,01 (s, 3Н, ОАс), 2,29 (s, 3Н, NAc), 2,75 (m, 1Н, Н2'), 3,29 (dd, J= 11,7 Гц, 1,8 Гц, 1Н, Н2а''), 3,66 (d, J=11,4 Гц, 1Н, Н4а''), 4,03 (d, J=11,4 Гц, 1Н, H4b''), 4,05 (d, J=11,7 Гц, 1Н, H2b''), 4,70 (d, J= 12,3 Гц, 1Н, Н5а'), 4,13 (d, J=4,8 Гц, Н3'), 4,23 (d, J=12,3 Гц, 1Н, H5b'), 4,53, 4,67 (AB, J= 11,7 Гц, 2Н, Bn), 6,17 (d, J=4,2 Гц, 1Н, Н1'), 7,28-7,40 (m, 5H, Вn), 8,32 (s, 1Н, Н8), 9,80 (s, 1H, NH), 12,12 (s, 1H, NH).
Пример 18
Получение (1S, 3R, 4R, 8S)-3-(аденин-9-ил)-8-гидрокси-1-гидроксиметил-2,6-диоксабицикло[3,2,1]октана (18)
Раствор (1R, 3R, 4R,8S)-1-ацетоксиметил-8-бензилокси-3-(6-хлорпурин-9-ил)-2,6-диоксабицикло[3,2,1] октана (100 мг, 0,225 ммоль) в смеси диоксана (20 мл) и 30%-ного водного гидроксида аммония (20 мл) нагревали в стальном автоклаве при 100oС в течение 16 часов. Растворители выпаривали и осадок растворили в метаноле с последующим добавлением 20%-ного гидроксида палладия на угле (~50% воды, 3 х 250 мг, добавляемой ежедневно). Гидрогенолиз проводили при комнатной температуре под давлением 379,22 кПа водорода в течение 4 дней. Катализатор отфильтровывали и промывали метанолом. Объединенный метанольный раствор сконцентрировали и осадок подвергли хроматографии на диоксиде кремния с использованием 20%-ного метанола в метиленхлориде с получением указанного в заголовке соединения (39 мг, 59%) в виде бесцветного твердого вещества, которое кристаллизовали из метанола, т.пл. 250oС (с разложением).
1H-ЯМР (DMSO-d6 + D2O): δ 2,53 (m, 1H, Н2'), 3,33 (d, J=11,1 Гц, 1H, Н2а''), 3,40 (d, J= 12,3 Гц, 1H, Н5а'), 3,50 (d, J=12,6 Гц, 1H, H5b'), 3,69-3,76 (m, 2H, H2b'', Н4а''), 4,05 (d, J=10,2 Гц, H4b''), 4,45 (d, J=5,1 Гц, 1H, Н3'), 6,26 (s, 1H, Н1'), 7,28 (m, 2H, NH2), 8,12 (s, 1H, H8), 8,33 (s, 1H, Н2).
MS: 294 (МН+).
Аналитически рассчитано для C12H15N5O4,%: С 49,14; Н 5,16; N 23,88.
Обнаружено,%: С 49,01; Н 4,97; N 23,92.
Пример 19
Получение (1S, 3S, 4R, 8S)-3-(аденин-9-ил)-8-гидрокси-1-гидроксиметил-2,6-диоксабицикло[3,2,1]октана (19)
Способ, аналогичный описанному в примере 18, дал указанное в заголовке соединение (43 мг, 65%) в виде бесцветного твердого вещества из (1R,3S,4R, 8S)-1-ацетоксиметил-8-бензилокси-3-(6-хлорпурин-9-ил)-2,6-диоксабицикло[3,2,1]октана (100 мг).
1H-ЯМР (CD3OD): δ 2,71 (m, 1H, Н2'), 3,13 (dd, J=11,7 Гц, 2,4 Гц, 1H, Н2а''), 3,57 (d, J=12,6 Гц, 1H, Н5а'), 3,64 (d, J=11,1 Гц, Н4а''), 3,68 (d, J= 12,3 Гц, 1H, H5b'), 3,96 (d, J=11,1 Гц, 1H, H4b''), 4,14 (d, J=11,7 Гц, 1 H, H2b''), 6,39 (d, J=4,2 Гц, 1Н, Н1'), 8,04 (s, 1Н, Н8), 8,44 (s, 1 Н, Н2).
MS: m/z 294 (МН+).
Пример 20
Получение (1S, 3R, 4R,8S)-8-гидрокси-1-гидроксиметил-3-(гипоксантин-9-ил)-2,6-диоксабицикло[3,2,1]октана (20)
К раствору (1R,3R,4R,8S)-1-ацетоксиметил-8-бензилокси-3-(6-хлорпурин-9-ил)-2,6-диоксабицикло[3,2,1] октана (150 мг, 0,34 ммоль) и меркаптоэтанола (0,19 мл, 2,7 ммоль) в метаноле (20 мл) добавили метилат натрия (0,37 мл 5,4 М раствора в метаноле, 2,0 ммоль). Полученный раствор нагревали с обратным холодильником в течение 6 часов, охладили до комнатной температуры, нейтрализовали 10%-ным АсОН до рН 7. Метанол выпаривали и осадок разбавили 1,0 М NаНСО3 (15 мл) с последующей экстракцией 10%-ным метанолом в хлороформе до полного выделения продукта из водной фазы. Объединенный органический слой высушивали (Na2SO4) и концентрировали до сухого состояния. Хроматография на диоксиде кремния с использованием 10-15%-ного метанола в хлороформе дала 109 мг (84%) производного инозина (не показано) в виде бесцветного твердого вещества, 100 мг (0,26 ммоль) которого растворили в метаноле с последующим добавлением 20%-ного гидроксида палладия на угле (50% воды, 600 мг). Гидрогенолиз проводили при комнатной температуре под давлением 344,75 кПа водорода в течение 3 дней. Катализатор отфильтровывали и промывали метанолом. Объединенный метанольный раствор сконцентрировали и осадок подвергли хроматографии на диоксиде кремния с использованием 20-25%-ного метанола в метиленхлориде с получением 61 мг (61%) указанного в заголовке соединения в виде бесцветного твердого вещества, которое кристаллизовали из смеси метанол-этилацетат, т.пл. 228oС (с разложением).
1Н-ЯМР (DMSO-d6): δ 2,52 (m, 1H, Н2'), 3,30-3,55 (m, 3Н, Н5', Н4а''), 3,69 (dd, J= 11,1 Гц, 2,7 Гц, 1H, Н2а''), 3,73 (d, J=10,8 Гц, H4b''), 4,05 (d, 10,8 Гц, 1H, H2b''), 4,40 (m, 1H, H2b''), 5,03 (t, J=6,0 Гц, 1H, OH), 5,74 (d, J= 4,2 Гц, 1H, ОН), 6,24 (s, 1H, H1'), 8,06 (s, 1H, H8), 8,30 (s, 1H, H2), 12,40 (s, 1H, NH).
MS: m/z 295 (MH+).
Пример 21
Получение (1S, 3S, 4R,8S)-8-гидрокси-1-гидроксиметил-3-(гипоксантин-9-ил)-2,6-диоксабицикло[3,2,1]октана (21)
К раствору (1R,3S,4R,8S)-1-ацетоксиметил-8-бензилокси-3-(6-хлорпурин-9-ил)-2,6-диоксабицикло[3,2,1] октана (120 мг, 0,27 ммоль), меркаптоэтанолу (0,15 мл, 2,1 ммоль) в метаноле (16 мл) добавили метилат натрия (1,62 ммоль, 0,30 мл 5,4 М раствора в метаноле). Способ, аналогичный описанному для примера 20, дал 37 мг (47%) указанного в заголовке соединения в виде гигроскопичного твердого вещества.
1H-ЯМР (DMSO-d6): δ 2,52 (m, 1H, H2'), 3,06 (dd, J=11,7 Гц, 2,4 Гц, 1H, Н2а''), 3,34-3,53 (m, 2H, Н5'), 3,56 (d, J=11,1 Гц, 1H, Н4а''), 3,79 (d, J= 11,4 Гц, 1H, H4b''), 3,98 (d, J=11,4 Гц, 1H, H2b''), 4,31 (d, J=4,5 Гц, 1H, Н3'), 4,89 (br, 1H, ОН), 5,99 (br, 1H, ОН), 6,28 (d, J=4,2 Гц, 1H, Н1'), 8,03 (s, 1H, H8), 8,27 (s, 1H, H2), 12,30 (br, 1H, NH).
Пример 22
Получение (1S, 3S, 4R, 8S)-3-(гуанин-9-ил)-8-гидрокси-1-гидроксиметил-2,6-диоксабицикло[3,2,1]октана (23)
Способ, аналогичный описанному для примера 18, дал указанное в заголовке соединение (41 мг, 66%) в виде беловатого твердого вещества из (1R,3S,4R, 8S)-1-ацетоксиметил-3-(N2-ацетилгуанин-9-ил)-8-бензилокси-2,6-диоксабицикло[3,2,1]октана (100 мг).
1H-ЯМР (DMSO-d6 + D2O): δ 2,42 (m, 1H, H2'), 3,15 (dd, J=11,4 Гц, 2,1 Гц, 1H, Н2а''), 3,34 (d, J= 11,4 Гц, 1H, Н5а'), 3,47 (d, J=12,6 Гц, 1H, H5b'), 3,51 (d, J=12,0 Гц, 1H, Н4а''), 3,77 (d, J=10,8 Гц, 1H, H4b''), 3,98 (d, J= 11,7 Гц, 1H, H2b''), 4,23 (d, J=4,8 Гц, 1H, Н3'), 4,80 (br, 1H, ОН), 5,90 (br, 1H, ОН), 6,05 (d, J=4,2 Гц, 1H, Н1'), 6,52 (br, 2H, NH2), 7,93 (s, 1H, H8), 12,30 (br, 1H, NH).
MS: m/z 310 (MH+).
Пример 23
Получение (1R, 3R, 4R,8S)-1-ацетоксиметил-8-бензилокси-3-(тимин-1-ил)-2,6-диоксабицикло[3,2,1]октана (24)
Реакцию проводили тем же способом, который описан для примера 14, за исключением того, что связывающим реагентом являлся хлорид олова (IV) (0,45 мл) и сахарный субстрат представлял собой (1R,3S,4R,8S)-1-ацетоксиметил-8-бензилокси-3-метокси-2,6-диоксабицикло[3,2,1] октан (202 мг, 0,63 ммоль). Хроматография на диоксиде кремния с использованием 5%-ного ЕtOН в СН2Сl2 дала смесь (233 мг, 89%) из указанного в заголовке соединения (β-аномера) и его α-аномера (отношение β:α приблизительно 4:1) в виде бесцветного твердого вещества.
1H-ЯМР (CDCl3) β-аномера (из спектра смеси α- и β-аномеров): δ 1,93 (d, J= 0,9 Гц, 1Н, АrСН3), 2,05 (s, 3Н, ОАс), 2,66 (m, 1H, Н2'), 3,48 (d, J=11,1 Гц, Н4а''), 3,86-4,12 (m, 5Н, Н2а'', H2b'', H3', Н4b'', Н5а'), 4,26 (d, J= 12,6 Гц, H5b'), 4,44, 4,64 (AB, J=11,4 Гц, 2Н, Вn), 6,06 (s, 1H, Н1'), 7,26-7,42 (m, 5Н, Вn), 7,59 (d, J=1,2 Гц, 1H, Н6), 8,94 (s, 1H, NH).
Пример 24
Получение (1R, 3R,4R,8S)-1-ацетоксиметил-8-бензилокси-3-(урацил-1-ил)-2,6-диоксабицикло[3,2,1]октана (25)
Способ, аналогичный описанному для примера 23, после хроматографии на диоксиде кремния с использованием 5%-ного ЕtOН в метиленхлориде дал смесь (267 мг, 87%) указанного в заголовке соединения и его α-аномера (отношение β:α приблизительно 9:1) в виде бесцветного твердого вещества из (1R,3S,4R, 8S)-1-ацетоксиметил-8-бензилокси-3-метокси-2,6-диоксабицикло[3,2,1] октана (230 мг, 0,71 ммоль) и силилированного урацила (2,0 ммоль). Указанное в заголовке соединение (β-аномер) было частично выделено путем хроматографии на диоксиде кремния, т.пл. 145-147oС (ЕtOАс-гексаны).
1H-ЯМР (CDCl3): δ 2,02 (s, 3Н, ОАс), 2,67 (m, 1H, Н2'), 3,49 (d, J=11,4 Гц, 1H, Н4а''), 3,86-3,97 (m, 3Н, Н2а'', H3', H4b''), 4,08 (d, J=12,3 Гц, 1H, Н5а'), 4,09 (d, J=10,5 Гц, 1H, H2b''), 4,25 (d, J=12,3 Гц, 1H, H5b'), 4,44, 4,64 (AB, J= 11,7 Гц, 2Н, Вn), 6,05 (s, 1H, Н1'), 7,26-7,40 (m, 5H, Вn), 5,69 (d, J=8,1 Гц, 1Н, Н5), 7,79 (d, J=8,4 Гц, 1H, Н6), 8,92 (s, 1H, NH),
Аналитически рассчитано для C20H22N2O7,%: С 59,69; Н 5,51; N, 6,96.
Обнаружено,%: С 59,45; Н 5,56; N 6,91.
Пример 25
Получение (1R,3R,4R,8S)-1-ацетоксиметил-8-бензилокси-3-(N4-бензоилцитозин-1-ил)-2,6-диоксабицикло[3,2,1]октана (26)
Способ, аналогичный описанному для примера 23, после хроматографии на диоксиде кремния с использованием 5%-ного ЕtOН в метиленхлориде дал 910 мг (90%) указанного в заголовке соединения (β-аномера) в виде бесцветного твердого вещества, в результате взаимодействия (1R,3S,4R,8S)-1-ацетоксиметил-8-бензилокси-3-метокси-2,6-диоксабицикло[3,2,1] октана (645 мг, 2,0 ммоль) с силилированным N4-бензоилцитозином (4,0 ммоль), т.пл. 173-174oС (ЕtOАс).
1H-ЯМР (CDCl3): δ 2,07 (s, 3Н, ОАс), 2,83 (m, 1H, Н2'), 3,51 (d, J=11,1 Гц, Н4а''), 3,86 (d, J=5,4 Гц, 1H, Н3'), 3,97 (d, J=11,1 Гц, 1H, Н4b''), 3,99-4,13 (m, 3Н, Н2а'', H2b'', H5a'), 4,27 (d, J=12,3 Гц, 1H, H5b'), 4,38, 4,61 (AB, J= 11,4 Гц, 2Н, Вn), 6,15 (s, 1H, Н1'), 7,24-7,38 (m, 5Н, Вn), 7,50-7,66 (m, 4Н, Н5, Bz), 7,90 (m, 2Н, Bz), 8,28 (d, J=7,5 Гц, 1H, Н6), 8,84 (br, 1H, NH).
Аналитически рассчитано для C27H27N3O7,%: С 64,15; Н 5,38; N 8,31.
Обнаружено,%: С 64,10; Н 5,20; N 8,43.
Пример 26
Получение (1S,3R,4R,8S)-8-гидрокси-1-гидроксиметил-3-(тимин-1-ил)-2,6-диоксабицикло[3,2,1]октана (27)
К раствору смеси (1R,3R,4R,8S)-1-ацетоксиметил-8-бензилокси-3-(тимин-1-ил)-2,6-диоксабицикло[3,2,1] октана и его α-аномера (приблизительно 4:1, 200 мг, 0,48 ммоль) в безводном метиленхлориде (4 мл) при 0oС добавили трихлорид бора (1,0 М в CH2CH2, 8 мл). Полученную реакционную смесь перемешивали при комнатной температуре в течение 8 часов, при 15oС в течение ночи и затем охладили до 0oС. Метанол (5,0 мл) добавляли по каплям с последующим добавлением 1,0 M NaOMe в МеОН до достижения рН 8. Раствор разделили и осадок тщательно экстрагировали 20%-ным метанолом в метиленхлориде. Объединенный фильтрат высушивали (Na2SO4) и концентрировали до сухого состояния. Хроматография на диоксиде кремния с использованием 10-15%-ного метанола в этилацетате дала указанное в заголовке соединение (78 мг), смесь указанного в заголовке соединения и его α-аномера (24 мг) и α-аномер (23 мг), все в виде бесцветного твердого вещества. Общий выход составил 91%. Кристаллизация указанного в заголовке соединения из смеси метанол-этилацетат дала кристаллическое вещество, т.пл. 217-218oС.
1H-ЯМР (DMSO-d6): δ 1,75 (d, J=1,2 Гц, 1Н, АrСН3), 2,24 (m, 1H, Н2'), 3,20 (d, J=10,8 Гц, 1H, Н4а''), 3,33-3,58 (m, 3Н, Н2а'', Н5'), 3,66 (d, J= 10,8 Гц, H4b''), 3,97 (d, J=10,5 Гц, 1H, H2b''), 4,14 (m, 1H, Н3'), 5,24 (t, J= 5,1 Гц, 1H, ОН), 5,67 (d, J=2,4 Гц, 1H, ОН), 5,82 (s, 1H, Н1'), 7,95 (d, J=0,9 Гц, 1H, Н6), 11,32 (s, 1H, NH).
MS: m/z 285 (MH+).
Аналитически рассчитано для C12H16N2O6,%: С 50,70; Н 5,67; N 9,85.
Обнаружено,%: С 50,65; Н 5,57; N 9,73.
Пример 27
Получение (1S, 3R, 4R, 8S)-8-гидрокси-1-гидроксиметил-3-(урацил-1-ил)-2,6-диоксабицикло[3,2,1]октана (28)
Способ, аналогичный описанному для примера 26, после хроматографии на диоксиде кремния с использованием 10%-ного метанола в метиленхлориде дал 110 мг (76%) указанного в заголовке соединения в виде белого твердого вещества из (1R, 3R, 4R,8S)-1-ацетоксиметил-8-бензилокси-3-(урацил-1-ил)-2,6-диоксабицикло[3,2,1] октана (215 мг, 0,53 ммоль). Указанное в заголовке соединение содержало незначительное количество его α-аномера. Чистое указанное в заголовке соединение было получено путем перекристаллизации из смеси ацетон-этилацетат, т.пл. 218-219oС.
1H-ЯМР (ацетон-d6): δ 2,42 (m, 1H, Н2'), 3,27 (d, J=10,8 Гц, 1H, Н4а''), 3,58-3,72 (m, 3Н, Н2а'', Н5'), 3,83 (d, J=10,8 Гц, 1H, H4b''), 4,13 (d, J= 10,5 Гц, 1H, H2b''), 4,37 (t, J=5,1 Гц, 1H, ОН), 4,42 (m, 1H, Н3'), 4,88 (d, J= 3,9 Гц, 1H, ОН), 5,52 (d, J=7,8 Гц, 1H, Н5), 5,95 (s, 1H, Н1'), 8,17 (d, J=7,8 Гц, 1H, Н6), 10,02 (s, 1H, NH).
MS: m/z 271 (MH+).
Аналитически рассчитано для C11H14N2O6,%: С 48,89; Н 5,22; N 10,37. Обнаружено,%: С 48,60; Н 5,64; N 10,21.
Пример 28
Получение (1S, 3R, 4R,8S)-3-(цитозин-1-ил)-8-гидрокси-1-гидроксиметил-2,6-диоксабицикло[3,2,1]октана (30)
Способ, аналогичный описанному для примера 26, после хроматографии на диоксиде кремния с использованием 10%-ного МеОН в метиленхлориде дал из (1R, 3R, 4R, 8S)-1-ацетоксиметил-8-бензилокси-3-(N4-бензоилцитозин-1-ил)-2,6-диоксабицикло[3,2,1]октана 364 мг (65%) (1S,3R,4R,8S)-3-(N4-бензоилцитозин-1-ил)-8-гидрокси-1-гидроксиметил-2,6-диоксабицикло[3,2,1] октана (760 мг), 120 мг (0,32 ммоль) которого растворили в насыщенном растворе аммиака в метаноле, и раствор перемешивали при комнатной температуре в течение 24 часов. Аммиак и метанол выпаривали, а осадок растворили в воде с последующей тщательной экстракцией хлороформом (5 раз) и затем толуолом (2 раза). Воду выпарили, и кристаллизация из метанола дала 62 мг указанного в заголовке соединения (45 мг кристаллического твердого вещества и 17 мг некристаллического твердого вещества), т.пл. 250oС (с разложением).
1H-ЯМР (CD3OD): δ 2,33 (m, 1Н, Н2'), 3,31 (d, J=11,1 Гц, 1Н, Н4а''), 3,57 (d, J=12,3 Гц, 1Н, Н5а'), 3,65 (d, J=12,3 Гц, 1Н, Н5b'), 3,78 (dd, J= 10,5 Гц, 2,7 Гц, Н2а''), 3,84 (d, J=11,1 Гц, 1Н, H4b''), 4,14 (d, J=10,5 Гц, 1Н, H2b''), 4,20 (d, J=5,1 Гц, 1Н, Н3'), 5,86 (d, J=7,5 Гц, 1Н, Н5), 5,96 (s, 1H, Н1'), 8,22 (d, J=7,8 Гц, 1H, Н6).
MS: m/z 270 (МH+).
Аналитически рассчитано для C11H15N3O5,%: С 49,07; Н 5,62; N 15,61.
Обнаружено,%: С 48,93; Н 5,55; N 15,64.
Аналогичным образом получили (1S,3R,4R,8S)-3-(N4-ацетилцитозин-1-ил)-8-гидрокси-1-гидроксиметил-2,6-диоксабицикло[3,2,1]октан.
Альтернативный способ. Смесь (1S,3R,4R,8S)-8-гидрокси-1-гидроксиметил-3-(урацил-1-ил)-2,6-диоксабицикло[3,2,1]октана (170 мг, 0,63 ммоль), уксусного ангидрида (2,16 мл, 20,1 ммоль) и пиридина (0,29 мл, 3,5 ммоль) в безводном DMF (N,N-диметилформамиде) (2,5 мл) перемешивали при комнатной температуре в течение ночи, разбавили метиленхлоридом, промыли солевым раствором и 10%-ным NaHCO3, высушили (Na2SO4), сконцентрировали до сухого состояния. Хроматография на диоксиде кремния с использованием смеси этилацетат-гексаны (2: 1) дала 117 мг (77%) 3',5'-диацетилпроизводного (1S,3R,4R,8S)-8-ацетокси-1-ацетоксиметил-3-(урацил-1-ил)-2,6-диоксабицикло[3,2,1]октана.
(1S, 3R, 4R,8S)-8-ацетокси-1-ацетоксиметил-3-(урацил-1-ил)-2,6-диоксабицикло[3,2,1] октан (175 мг, 0,58 ммоль) растворили в безводном пиридине (1,5 мл) и полученный раствор охладили до 0oС в атмосфере аргона с последующим добавлением 4-хлорфенилдихлорфосфата (0,29 мл, 1,75 ммоль). Полученный раствор нагрели до комнатной температуры и перенесли во флакон с разделительной мембраной, содержащий 1,2,4-триазол (120 мг, 1,75 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 3 дней, разбавили CH2Cl2, промыли солевым раствором и 5%-ным NaHCO3, высушили (Na2SO4) и сконцентрировали до сухого состояния. Осадок растворили в диоксане (7 мл) и 30%-ном гидроксиде аммония (10 мл). Раствор находился при комнатной температуре в течение 16 часов, и затем растворители выпаривали. Осадок подвергали хроматографии на диоксиде кремния с использованием Еt3N-МеОН-СНСl3 (5:30:65) с получением 74 мг (55%) указанного в заголовке соединения в виде светло-желтого твердого вещества.
Пример 29
Получение (1R, 3R, 4R, 8S)-1-ацетоксиметил-3-(N2-ацетилгуанин-7-ил)-8-бензилокси-2,6-диоксабицикло[3,2,1]октана (31)
Силилированное основание получили по способу, описанному для примера 17, из N2-ацетилгуанина (386 мг, 2,0 ммоль) и растворили в растворе (1R,3S,4R, 8S)-1-ацетоксиметил-8-бензилокси-3-метокси-2,6-диоксабицикло[3,2,1] октана (477 мг, 1,48 ммоль) в безводном 1,2-дихлорэтане (10 мл) с последующим добавлением хлорида олова (IV) (0,75 мл) в 1,2-дихлорэтане (2,0 мл). Полученную смесь нагревали с обратным холодильником в течение 3 часов, затем при 70oС в течение ночи и охладили до 0oС. Смесь нейтрализовали 2,0 М карбонатом натрия, профильтровали через целит и тщательно экстрагировали хлороформом. Объединенный фильтрат высушивали (Na2SO4) и концентрировали до сухого состояния. Хроматография на диоксиде кремния с использованием 5%-ного ЕtOН в хлороформе дала 297 мг (42%) указанного в заголовке соединения, 73 мг (10%) N9-связанного β-аномера указанного в заголовке соединения и 46 мг (6%) N9-связанного α-аномера, все в виде белого твердого вещества. Указанное в заголовке соединение: т.пл. 176-178oС (CH3Cl-EtOAc).
1H-ЯМР (CDCl3): δ 2,09 (s, 3H, ОАс), 2,40 (s, 3H, NAc), 2,78 (m, 1H, Н2'), 3,53 (d, J=11,4 Гц, 1Н, Н4а''), 3,99 (d, J=11,1 Гц, H4b''), 4,03-4,18 (m, 4H, H2a'', H2b'', H3', Н5а'), 4,26 (d, J=12,6 Гц, 1H, H5b'), 4,39, 4,58 (АВ, J=11,7 Гц, 2Н, Bn), 6,62 (s, 1H, Н1'), 7,22-7,40 (m, 5H, Bn), 8,21 (s, 1H, Н8), 10,60 (s, 1H, NH), 12,34 (s, 1H, NH).
Аналитически рассчитано для C23H25N5O8,%: С 55,31; Н 5,05; N 14,02.
Обнаружено,%: С 55,35; Н 4,83; N 13,80.
Пример 30
Получение (1R, 3R, 4R, 8S)-1-ацетоксиметил-3-(N2-ацетилгуанин-9-ил)-8-бензилокси-2,6-диоксабицикло[3,2,1]октана (32)
Такое же количество силилированного N2-ацетилгуанина, как было описано для примера 29, растворили в растворе (1R,3R,4R,8S)-1-ацетоксиметил-3-(N2-ацетилгуанин-7-ил)-8-бензилокси-2,6-диоксабицикло[3,2,1] октана (370 мг, 0,76 ммоль) в безводном 1,2-дихлорэтане (10 мл) и добавили триметилсилилтрифлат (TMSOTf) (0,54 мг, 3,0 ммоль) в 1,2-дихлорэтане (3 мл). Полученный раствор нагревали с обратным холодильником в течение ночи. Добавили дополнительное количество TMSOTf (0,54 мл) и смесь кипятили с обратным холодильником еще в течение двух дней. Применение того же способа, который был описан для примера 29, после хроматографии на диоксиде кремния с использованием 5%-ного этанола в хлороформе дало 104 мг (28%) интактного исходного материала, 91 мг (25%) указанного в заголовке соединения и 80 мг (22%) α-аномера указанного в заголовке соединения, все в виде белого твердого вещества. Указанное в заголовке соединение: т.пл. 128-131oС (СН3Сl-ЕtOАс).
1H-ЯМР (CDCl3): δ 2,02 (s, 3Н, ОАс), 2,30 (s, 3Н, NAc), 2,67 (m, 1Н, Н2'), 3,50 (d, J= 10,8 Гц, 1Н, Н4а''), 3,78 (dd, J=10,8 Гц, 2,7 Гц, 1Н, Н2а''), 3,99 (d, J=10,8 Гц, H4b''), 4,12 (d, J=12,3 Гц, 1Н, Н5а'), 4,14 (d, J=10,8 Гц, 1Н, H2b''), 4,27 (d, J=12,3 Гц, 1Н, H5b'), 4,33 (d, J=5,1 Гц, 1Н, Н3'), 4,49, 4,62 (АВ, J=11,7 Гц, 2Н, Вn), 6,25 (s, 1H, Н1'), 7,26-7,38 (m, 5H, Bn), 7,83 (s, 1H, Н8), 9,0 (s, 1H, NH), 11,95 (s, 1H, NH).
MS: m/z 310 (MH+).
Аналитически рассчитано для C23H25N5O8,%: С 55,31; Н 5,05; N 14,02.
Обнаружено,%: С 55,70; Н 5,00; N 13,95.
Пример 31
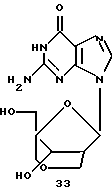
Получение (1S, 3R, 4R, 8S)-3-(гуанин-9-ил)-8-гидрокси-1-гидроксиметил-2,6-диоксабицикло[3,2,1]октана (33)
Способ, аналогичный описанному для примера 22, после хроматографии дал 52 мг (45%) указанного в заголовке соединения в виде бесцветного твердого вещества, полученного из (1R, 3R, 4R, 8S)-1-ацетоксиметил-3-(N2-ацетилгуанин-9-ил)-8-бензилокси-2,6-диоксабицикло[3,2,1] октана (180 мг). Кристаллизация из смеси вода-этанол (9:1) дала кристаллическое вещество, т.пл. 258oС (с разложением).
1H-ЯМР (DMSO): δ 2,45 (m, 1H, Н2'), 3,31 (d, J=10,8 Гц, 1Н, Н4а''), 3,36-3,50 (m, 2H, H5a', H5b'), 3,60 (dd, J=10,2 Гц, 2,7 Гц, 1Н, Н2а''), 3,1 (d, J= 11,1 Гц, H4b''), 4,03 (d, J=10,5 Гц, 1Н, H2b''), 4,36 (m, 1H, Н3'), 4,95 (t, J=5,7 Гц, 1H, ОН), 5,70 (d, J=3,9 Гц, 1H, ОН), 6,06 (s, 1H, Н1'), 6,55 (br, 2H, NH2), 7,90 (s, 1H, Н8), 10,68 (s, 1H, NH).
MS: m/z 310 (MH+).
Пример 32
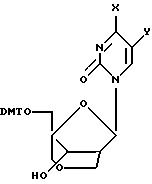
Получение (1S, 3R,4R,8S)-8-гидрокси-1-(4,4'-диметокситритилоксиметил)-3-(N4-ацетилцитозин-ил)-2,6-диоксабицикло[3,2,1]октана (35)
Раствор (1S, 3R, 4R, 8S)-8-гидрокси-1-гидроксиметил-3-(N4-ацетилцитозинил)-2,6-диоксабицикло[3,2,1] октана (200 мг, 0,64 ммоль) и 4,4'-диметокситритилхлорида (548 мг, 0,61 ммоль) в безводном пиридине (7 мл) выдерживали при комнатной температуре в течение ночи, затем разбавили этилацетатом, промыли солевым раствором и 10%-ным NаНСО3, высушили над сульфатом натрия и сконцентрировали. Хроматография на диоксиде кремния с использованием 10%-ного этанола в хлороформе дала 342 мг (87%) указанного в заголовке соединение в виде бесцветной пены.
Аналогичным образом получили (1S,3R,4R,8S)-8-гидрокси-1-(4,4'-диметокситритилоксиметил)-3-(N4-бензоилцитозин-1-ил)-2,6-диоксабицикло[3,2,1] октан (36) и (1S, 3R, 4R, 8S)-8-гидрокси-1-(4,4'-диметокситритилоксиметил)-3-(тимин-ил)-2,6-диоксабицикло[3,2,1]октан (34).
Пример 33
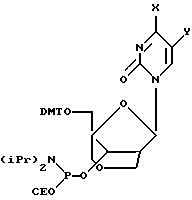
Получение (1S, 3R,4R,8S)-8-гидрокси-1-(4,4'-диметокситритилоксиметил)-3-(N4-ацетилцитозин-ил)-2,6-диоксабицикпо[3,2,1] октана 8-O-(2-цианоэтил-N, N-диизопропилфосфорамидита) (38)
К перемешиваемому раствору (1S, 3R,4R,8S)-8-гидрокси-1-(4,4'-диметокситритилоксиметил)-3-(N4-ацетилцитозин-ил)-2,6-диоксабицикло[3,2,1] октана (320 мг, 0,52 ммоль) и диизопропилэтиламина (0,36 мл, 2,08 ммоль) в безводном дихлорметане (6 мл) при 0oС в атмосфере аргона добавляли по каплям 2-цианоэтил-N, N-диизопропилхлорфосфорамидит (0,23 мл, 1,04 ммоль). Полученный раствор перемешивали при температуре окружающей среды в течение 4 часов, охладили во льду, разбавили этилацетатом, промыли охлажденным 10%-ным NаНСО3, высушили над сульфатом натрия и сконцентрировали при комнатной температуре. Хроматография на диоксиде кремния с использованием 5%-ного триэтиламина и 5%-ного ацетона в метиленхлориде дала 376 мг (89%) указанного в заголовке соединения в виде бесцветной пены.
Аналогичным образом получили (1S,3R,4R,8S)-8-гидрокси-1-(4,4'-диметокситритилоксиметил)-3-(N4-бензоилцитозин-1-ил)-2,6-диоксабицикло[3,2,1]октана 8-O-(2-цианоэтил-N,N-диизопропилфосфорамидит) (39) и (1S,3R,4R,8S)-8-гидрокси-1-(4,4'-диметокситритилоксиметил)-3-(тимин-1-ил)-2,6-диоксабицикло[3,2,1] октана 8-O-(2-цианоэтил-N,N-диизопропилфосфорамидит) (37).
Пример 34
Получение олигонуклеотидов, содержащих 2,4-бициклонуклеотиды
Этот пример иллюстрирует применение фосфорамидитов бициклонуклеозидов 37-39 для синтеза олигонуклетида, содержащего бициклонуклеозиды с 2'-С,4'-С-мостиковой связью. Олигонуклеотиды в этом примере синтезировали с использованием фосфорамидитного способа. Модифицированные олигонуклеотиды синтезировали согласно стандартной процедуре (протокол для ABI 394 Synthesizer от Perkin-Elmer, 1994) за исключением того, что использовали более концентрированный раствор и более продолжительное время реакции сочетания. Растворы для модифицированных фосфорамидитов имели молярность 0,13 М, что является на 30% более концентрированным по сравнению с немодифицированными фосфорамидитами (0,1 М). Для модифицированных фосфорамидитов использовали десять минут времени реакции сочетания, а для немодифицированных фосфорамидитов использовали время сочетания на пять минут дольше по сравнению с модифицированными фосфорамидитами. Выходы продуктов сочетания для модифицированных фосфорамидитов сопоставимы с выходами для немодифицированных фосфорамидитов (98-99%). Модифицированные олигодезоксирибонуклеотиды подвергали очистке с помощью обращенно-фазовой ВЭЖХ и характеризовали с помощью масс-спектрометрии.
Следующие синтезированные последовательности приведены в качестве примеров.
5'-d(ATCTCTCCGCTTCCTTTC)-3'
5'-d(ATCTCTCCGCTTCCTTTC)-3'
5'-d(ATCTCTCCGCTTCCTTTC)-3'
5'-d(ATCTCTCCGCTTCCTTTC)-3'
5'-d(ATCTCTCCGCTTCCTTTC)-3'
5'-d(CTTCCTGTCTGATGGCTTC)-3'
5'-d(CTTCCTGTCTGATGGCTTC)-3'
5'-d(CTTCCTGTCTGATGGCTTC)-3'
5'-d(CTTCCTGTCTGATGGCTTC)-3'
5'-d(CTTCCTGTCTGATGGCTTC)-3'-
5'-d(CTTCCTGTCTGATGGCTTC)-3'
где А, С, G и Т - немодифицированные дезоксирибонуклеозиды,
Т - тимидин с 2',4'-С-мостиковой связью,
С - дезоксицитидин с 2',4'-С-мостиковой связью.
Пример 35
Гибридизационные свойства олигонуклеотидов, содержащих бициклонуклеотиды с 2'-С,4'-С-мостиковой связью
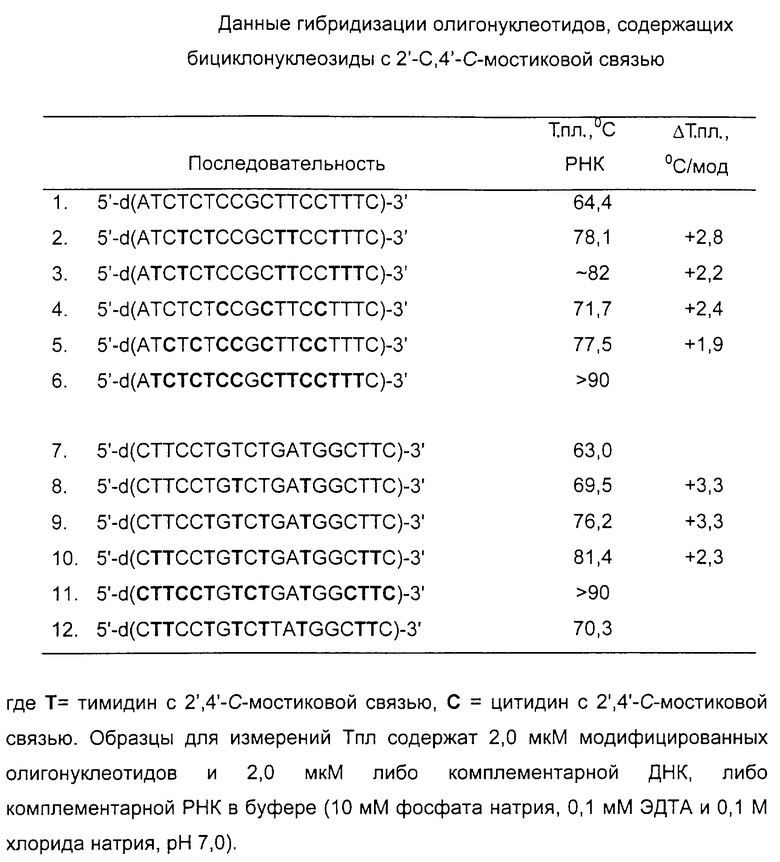
Гибридизацию модифицированных олигонуклеотидов с комплементарными ДНК и РНК исследовали с помощью измерений термодинамического плавления (Wang et al. Nucleosides Nucleotides 1997, 16, 445). Как видно из таблицы , модификации значительно усиливают гибридизацию с РНК. Для последовательностей, содержащих бициклический тимидин Т, увеличение показателей т.пл. находится в диапазоне 2,2-3,3 градуса на модификацию. Последовательность, содержащая бициклический цитидин С, также имеет более высокие показатели т.пл. по сравнению с немодифицированными олигонуклеотидами, на 2,4o выше на модификацию для последовательности 4 и на 1,9o выше на модификацию для последовательности 5. Последовательность 12, которая содержит ошибочно спаренный нуклеозид (G в середине последовательности заменен на Т), имеет показатель т. пл. на одиннадцать градусов ниже, чем последовательность 10, что характеризует специфичность последовательности. Для последовательностей, в которых все Т и С заменены на Т и С, показатели т.пл. (>90o) были выше настолько, что было невозможно получить точные значения в данной системе измерений.
Таким образом, здесь описаны специфические воплощения и использования примеров, а также способы получения новых нуклеозидов и олигонуклеотидов с бициклическими сахарными группировками. Однако для специалистов в данной области будет очевидно, что кроме уже описанных модификаций возможно получение значительно большего количества модификаций без отклонения от сущности изобретения. Поэтому предмет изобретения не должен быть ограничен, кроме как содержанием прилагаемой формулы. Более того, при интерпретации и описания изобретения, и формулы изобретения все термины следует интерпретировать в самом широком смысле сообразно с контекстом. В частности, термины "содержит" и "содержащий" по отношению к элементам, компонентам или стадиям следует трактовать нестрогим образом, показывая, что упоминаемые элементы, компоненты или стадии могут присутствовать, или использоваться, или объединяться с другими элементами, компонентами или стадиями, на которые специально не ссылались.
Изобретение относится к нуклеозидам формулы (I), имеющим бициклическую сахарную группировку, содержащую 2',4'-мостиковую связь, и олигонуклеотидам на их основе. Олигонуклеотиды по изобретению обладают высокой аффинностью по отношению к нуклеиновым кислотам-мишеням и могут найти применение в медицине. 3 с. и 1 з.п.ф-лы., 1 табл. 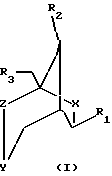
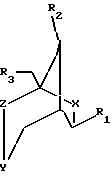
где Х и Y представляют собой О, и где Z представляет собой СН2;
R1 выбран из группы, содержащей аденин, цитозин, гуанин, гипоксантин, урацил и тимин;
R2, R3 независимо выбраны из группы, содержащей ОН, DMTO, TBDMSO, ВnО, ТНРО, АсО, BzO, OP(NiPr2)O(CH2)2CN, или R2 и R3 вместе образуют PhCHO2, (тетраизопропилдисилил)O2 или (ди(трет-бутил)силил)O2.

