Область техники
Настоящее изобретение относится к способу получения капецитабина, а также к способу получения используемого при этом обогащенного β-аномером триалкилкарбонатного соединения.
Предшествующий уровень техники
Капецитабин является вводимым перорально противораковым агентом, широко используемым при лечении метастатических раков груди и прямой кишки. Капецитабин является нуклеозидом на основе рибофуранозы и имеет стереохимическую структуру рибофуранозы, содержащей β-ориентированный 5-фторцитозиновый фрагмент в положении С-1.
Патенты США № 5472949 и 5453497 описывают способ получения капецитабина путем гликозилирования три-О-ацетил-5-дезокси-β-D-рибофуранозы формулы I с использованием 5-фторцитозина для получения цитидина формулы II; и карбамоилирования и гидролиза полученного соединения, как показано на схеме реакции 1
Схема реакции 1
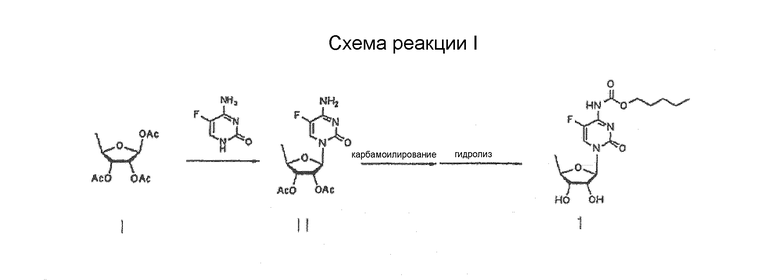
Соединение формулы I, применяемое в качестве промежуточного в схеме реакции I, представляет собой изомер, имеющий β-ориентированную ацетильную группу в положении 1 по той причине, что 5-фторцитозин является более реакционноспособным по отношению к β-изомеру, чем α-изомер в реакции гликозилирования вследствие возникновения существенного участия соседней группы, которое имеет место, когда защитной группой для 2-гидроксигруппы является ацил.
Соответственно, β-ориентированную три-О-ацетил-5-дезокси-β-D-рибофуранозу (формула 1) рассматривали в традиционной практике как незаменимое промежуточное соединение для получения капецитабина. Однако такая реакция дает смесь β- и α-изомеров, из которой цитидин (формула II) должен быть выделен путем неэкономичной стадии.
Между тем патент США № 4340729 раскрывает способ получения капецитабина по методике, показанной на схеме реакции 2, которая включает гидролиз 1-метилацетонида формулы III для получения тиола формулы IV; ацетилирование соединения формулы IV с использованием безводного уксусного ангидрида в пиридине для получения β-/α-аномерной смеси три-О-ацетил-5-дезокси-D-рибофуранозы формулы V; проведение вакуумной дистилляции для очистки β-/α-аномерной смеси и выделение из нее β-аномера формулы I
Схема реакций 2
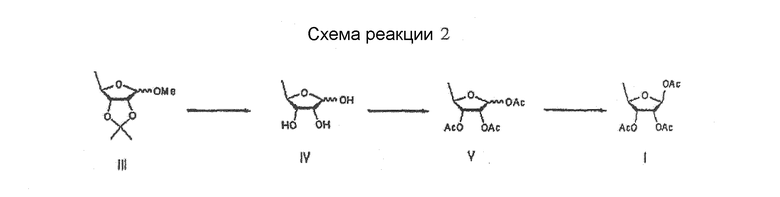
Однако вышеуказанный способ также затруднен требованием проводить стадии неэкономичной и усложненной перекристаллизации для выделения β-аномера из смеси β-/α-изомеров формулы V, что приводит к низкому выходу в только примерно 35-40% (Guangyi Wang et al., J. Med. Chem., 2000, vol. 43, 2566-2574; Pothukuchii Sairam et al., Carbohydrate Research, 2003, vol. 338, 303-306; Xiangshu Fei et al., Nuclear Medicine and Biology, 2004, vol. 31, 1033-1041 и Henry M. Kissman et al., J. Am. Chem. Soc., 1957, vol. 79, 5534-5540).
Далее, патент США № 5476932 описывает способ получения капецитабина реакцией 5'-дезокси-5-фторцитидина формулы VI с пентилхлорформиатом для получения соединения формулы VII, имеющего аминогруппы и 2-,3-гидроксигруппы, защищенные группами С5Н11СО2, и удаления гидроксизащищенных групп из полученного соединения, как показано на схеме реакций 3
Схема реакций 3
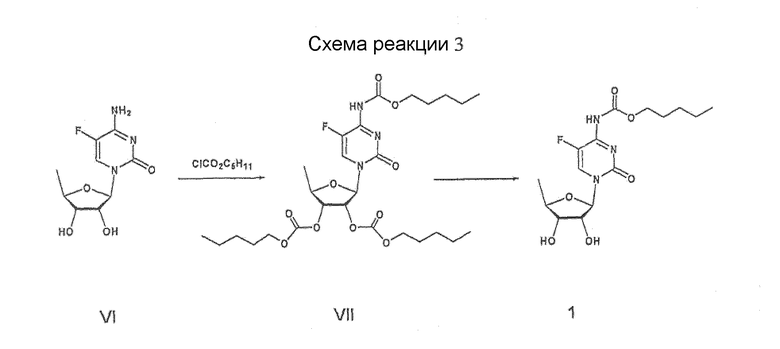
Однако этот способ сопряжен с большими производственными затратами и также требует проведения нескольких усложненных стадий для получения 5'-дезокси-5-фторцитидина формулы VI, защиты 2-,3-гидроксигрупп, проведения его взаимодействия с 5-фторцитозином и снятия защиты 2-,3-гидроксигрупп.
Соответственно, авторы настоящего изобретения пытались разработать эффективный способ получения капецитабина и неожиданно нашли новый эффективный способ получения высокочистого капецитабина с использованием триалкилкарбонатного промежуточного соединения, который не требует проведения неэкономичных стадий выделения β-аномера.
Сущность изобретения
Соответственно, задача настоящего изобретения состоит в разработке усовершенствованного способа получения капецитабина, а также способа получения обогащенного β-аномером триалкилкарбоната, который может быть использован как промежуточное соединение в указанном способе.
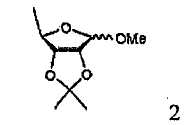
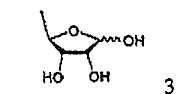
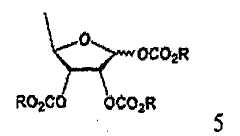
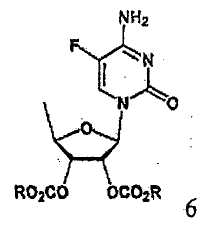
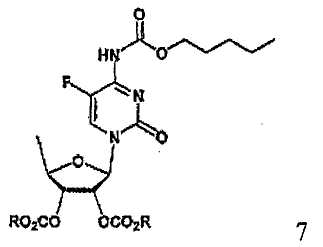
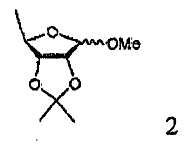
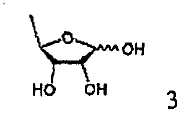
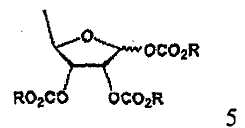
В соответствии с одним аспектом настоящего изобретения предложен способ получения капецитабина формулы 1, включающий стадии (1) гидролиза метилацетонидного соединения формулы 2 для получения триольного соединения формулы 3; (2) взаимодействия соединения формулы 3 с галогеналкилформиатом формулы 4 в присутствии смеси пиридина и триэтиламина для получения обогащенного β-аномером триалкилкарбонатного соединения формулы 5; (3) проведения гликозилирования соединения формулы 5 с использованием 5-фторцитозина в присутствии кислоты для получения диалкоксикарбонилцитидинового соединения формулы 6; (4) проведения карбамоилирования соединения формулы 6 с использованием н-пентилхлорформиата для получения карбамоилцитидинового соединения формулы 7; и (5) удаления карбонатных групп, защищающих гидроксигруппы соединения формулы (7)
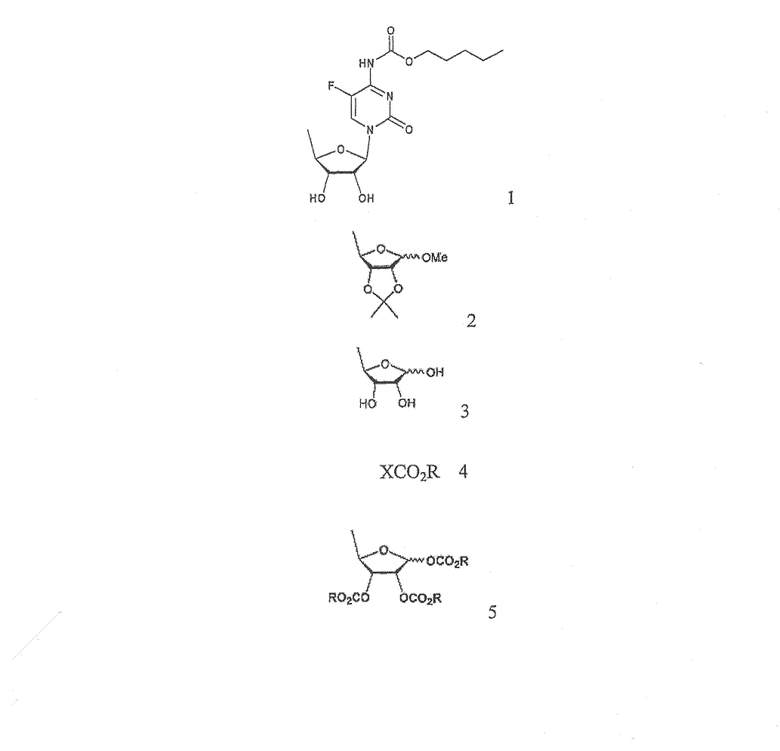
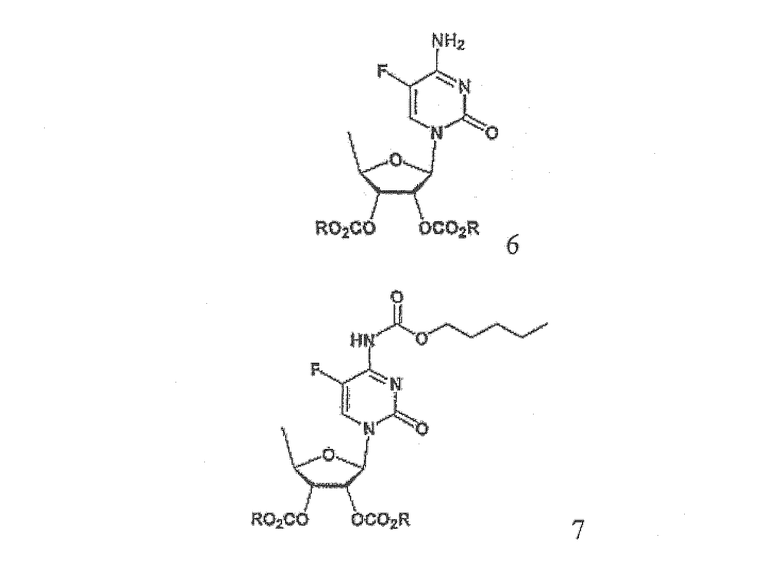
где Х представляет хлор, бром или йод и R представляет метил или этил.
Согласно другому аспекту настоящего изобретения предложен способ получения триалкилкарбонатного соединения формулы 5, используемого в качестве промежуточного соединения в указанном способе
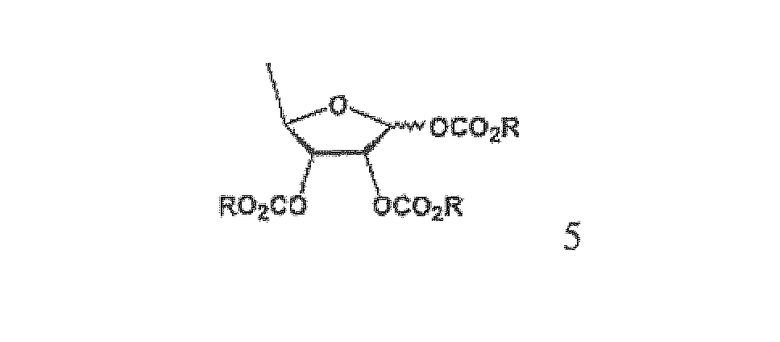
где R имеет такое же значение, как определено выше.
Подробное описание изобретения
В настоящем изобретении триалкилкарбонатное соединение формулы 5 представляет собой смесь в соотношении от 2:1 до 4:1 β- и α-аномеров, которая может быть использована для получения высокочистого капецитабина формулы 1 с высоким выходом по усовершенствованному методу гликозилирования триалкилкарбонатного промежуточного соединения с использованием 5-фторцитозина.
Способ получения капецитабина по изобретению суммирован в схеме реакций 4.
Схема реакций 4
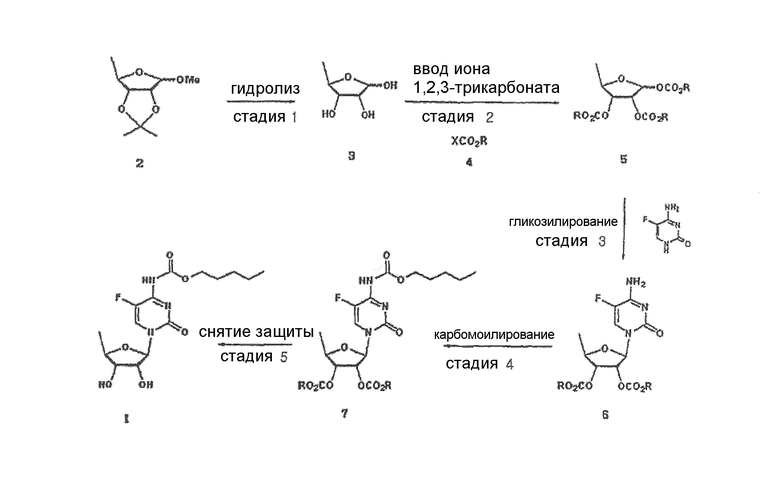
где Х и R имеют такие же значения, как определено выше.
Далее стадии способа по изобретению, показанного на схеме реакций 4, будут описаны подробно, как следует ниже.
Стадия 1
На стадии 1 триольное соединение формулы 3 может быть получено гидролизом метилацетонидного соединения формулы 2 в растворителе, таком как водная серная кислота, согласно обычному способу, описанному в патенте США № 4340729. Способ по настоящему изобретению может дополнительно, необязательно, включать процесс выделения каждого из аномеров полученного триольного соединения.
Стадия 2
На стадии 2 обогащенное β-аномером триалкилкарбонатное соединение может быть получено путем предоставления полученному на стадии 1 триольному соединению возможности реагировать с галогеналкилформиатным соединением формулы 4 в растворителе в присутствии основания, предпочтительно органического основания, такого как пиридин, триэтиламин и их смесь. Полученное в результате соединение представляет собой обогащенный β-аномером триалкилкарбонат формулы 5, который подвергается быстрому гликозилированию на стадии 3, потому что β-аномер является более реакционноспособным, чем α-аномер.
Когда карбонизацию триольного соединения проводят в присутствии только пиридина, получаемое в результате соединение может быть в форме смеси 1:1 α- и β-аномеров или обогащенной α-аномером смеси. Далее, если карбонизацию проводят в присутствии только триэтиламина, получаемое в результате соединение может быть сильно обогащенной β-аномером смесью, имеющей соотношение β-аномер:α-аномер такое высокое, как 6:1, в зависимости от температуры реакции и ее эквивалента. Однако такая карбонизация с использованием только триэтиламина осложнена тем, что побочный продукт - соединение формулы 1а - может образовываться в избыточном количестве
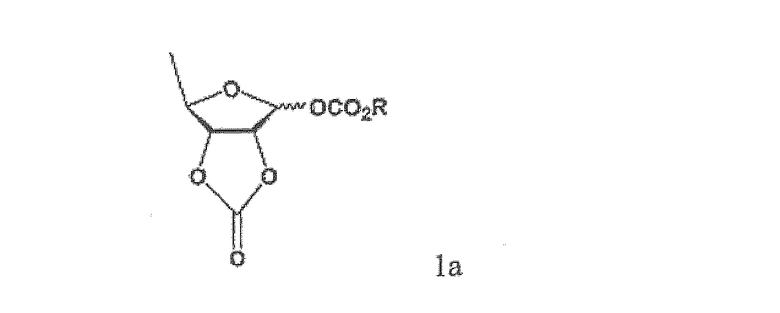
где R имеет такое же значение, как определено выше.
Согласно настоящему изобретению в качестве основания в этой реакции карбонизации триольного соединения может быть применена смесь пиридина и триэтиламина, имеющая специфическое соотношение компонентов, которое делает возможным получение обогащенного β-аномером соединения формулы 5, содержание которого более чем в два раза превышает содержание α-аномера, в то же время минимизируя образование загрязнений, например, циклического карбонатного соединения формулы 1а. В особенности, когда реакцию проводят в присутствии пиридина и триэтиламина при низкой температуре, содержание циклического карбонатного соединения в продукте реакции может быть уменьшено до менее чем 0,2%.
Согласно настоящему изобретению пиридин, используемый в смеси, может быть применен в количестве, составляющем от 1 до 2 эквивалентов, предпочтительно от 1,3 до 1,6 эквивалентов, в расчете на триэтиламин. Далее, смесь пиридина и триэтиламина может быть применена в количестве, составляющем от 4 до 10 эквивалентов, предпочтительно от 4 до 6 эквивалентов, в расчете на триольное соединение.
Растворителем может быть дихлорметан, дихлорэтан, хлороформ, тетрагидрофуран, ацетонитрил, диметилформамид или их смесь, предпочтительно дихлорметан.
Галогеноалкилформиатное соединение формулы 4 может быть применено в количестве, составляющем от 3 до 10 эквивалентов, предпочтительно от 5 до 7 эквивалентов в расчете на триольное соединение.
Предпочтительно вышеуказанную реакцию проводят при температуре от -50 до -30°С, предпочтительно от -35 до 30°С, так как в случае проведения реакции при температуре выше -30°С может образоваться циклическое карбонатное соединение в избыточном количестве.
Стадия 3
На стадии 3 диалкоксикарбонилцитидиновое соединение (формула 6) может быть получено путем гликозилирования соединения, полученного на стадии 2, с использованием 5-фторцитозина в растворителе в присутствии кислоты.
В вышеуказанной реакции для того, чтобы подавить конкурирующую реакцию аминогрупп в 1-аномерном положении, предпочтительно использовать вместо 5-фторцитозина силилированное производное 5-фторцитозина, полученное реакцией 5-фторцитозина с силилирующим агентом, таким как гексаметилдисилазан, согласно обычному методу. 5-Фторцитозин или его силилированное производное могут быть применены в количестве, составляющем от 1 до 2 эквивалентов, предпочтительно одного эквивалента в расчете на триалкилкарбонатное соединение формулы 5.
Кислоту используют для ускорения гликозилирования, и репрезентативные примеры кислот могут включать этилалюминийдихлорид, метилалюминийдихлорид, SnCl4, триметилсилилтрифторметансульфоновую кислоту и трифторметансульфоновую кислоту, предпочтительно триметилтрифторметансульфоновую кислоту. Далее, кислота может применяться в количестве, составляющем от 0,5 до 3 эквивалентов, предпочтительно одного эквивалента в расчете на триалкилкарбонатное соединение формулы 5.
Согласно настоящему изобретению растворителем, используемым в вышеуказанной реакции, может быть этилацетат, дихлорметан, дихлорэтан, хлороформ, тетрагидрофуран, ацетонитрил или диметилформамид, предпочтительно ацетонитрил, и реакция может проводиться при температуре от 0 до 50°С, предпочтительно от 20 до 35°С.
Согласно настоящему изобретению соединение диалкилоксикарбонилцитидина формулы 6 может быть получено из обогащенного β-аномером триалкилкарбонатного соединения формулы 5 с повышенным более чем на 10% выходом относительно обычного способа с использованием три-О-ацетил-5-дезокси-β-D-рибофуранозы (формула I), например, с высоким выходом более 90% путем гликозилирования. В особенности, соединение формулы 6, полученное способом по изобретению, имеет высокую чистоту более 98,5%. Далее, благодаря использованию такого высокочистого соединения с высоким выходом на последующих стадиях способа по изобретению можно получить конечный продукт, капецитабин, имеющий высокую чистоту 99,5%.
Стадия 4
На стадии 4 карбамоилцитидиновое соединение формулы 7 может быть получено осуществлением карбамоилирования диалкоксикарбонилцитидинового соединения, полученного на стадии 3, с использрванием н-пентилхлорформиата в растворителе в соответствии с обычным способом.
В этой реакции н-пентилхлорформиат может быть применен в количестве, составляющем от 1 до 3 эквивалентов, предпочтительно от 1,1 до 1,5 эквивалента в расчете на диалкоксикарбонилцитидиновое соединение формулы 6.
Растворителем может быть органический растворитель, такой как хлороформ, дихлорметан, дихлорэтан, тетрагидрофуран и ацетонитрил, предпочтительно дихлорметан.
Между тем, во время карбамоилирования органическое основание, такое как триэтиламин и пиридин, может быть добавлено в реакционную смесь, чтобы нейтрализовать хлористоводородную кислоту, образующуюся при реакции, и органическое основание может быть применено в количестве, составляющем от 1 до 5 эквивалентов, предпочтительно, от 1,3 до 2,5 эквивалента в расчете на диалкоксикарбонилцитидиновое соединение формулы 6.
Вышеуказанную реакцию можно проводить при температуре от -10 до 10°С, предпочтительно от -5 до 5°С.
Карбамоилирование может быть проведено количественно, и предпочтительно, чтобы его продукт использовался на последующей стадии, не подвергаясь процессу выделения.
Стадия 5
На стадии 5 капецитабин формулы 1 может быть получен путем удаления карбонатных групп, защищающих гидроксильные группы, из карбамоилцитидинового соединения, полученного на стадии 4, согласно обычному методу.
В соответствии с обычным методом, описанным в книге Theodora W. Green, Green's Protective Groups in Organic Synthesis, 4th Ed., 2007, pp. 280, 998 and 1022, Wiley-Interscience, в случае сосуществования в соединении карбонатных групп, защищающих гидрокси, с карбаматными защитными группами карбонатные защитные группы могут быть селективно удалены путем регулирования температуры реакции и концентрации используемого здесь основания. Это селективное снятие защиты основано на том различии между реакционной способностью карбонатных и карбаматных защитных групп, что карбонатные группы могут быть удалены даже при рН 10 и комнатной температуре, тогда как удаление карбаматных групп требует высоких значений рН, выше 12, и высокой температуры более 100°С.
В настоящем изобретении селективное снятие защиты может быть проведено в органическом растворителе, таком как смесь метанола и воды (2:1 об./об.) в присутствии основания, включающего гидроксид натрия и карбонат натрия, при температуре от -10 до 0°С, предпочтительно от -5 до 0°С.
Соответственно, согласно способу по настоящему изобретению, используя в качестве промежуточного соединения, обогащенного β-аномером триалкилкарбонатного соединения, содержащего β-аномера более чем в два раза больше, чем α-аномера, можно получить капецитабин, имеющий высокую чистоту, более 99%, избегая неэкономичный процесс выделения β-аномера. Далее, способ по изобретению обеспечивает высокий суммарный выход в 90% на стадии 4 и стадии 5.
Следующие примеры предназначены для того, чтобы дополнительно пояснить изобретение без ограничения его объема.
Пример 1. Получение 1,2,3-три-О-метиксикарбонил-5-дезокси-D-рибофуранозы (соединение формулы 5)
20 г метил-2,3-О-изопропилиден-5-дезокси-D-рибофуранозы растворяли в 100 мл 2% мол. водной серной кислоты, и смесь перемешивали при 80-85°С в течение 2 часов. Реакционную смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении, чтобы удалить примерно от одной трети до половины растворителя. К полученному в результате концентрату добавляли 100 мл 2% мол. водной серной кислоты, полученную смесь перемешивали при 80-85°С в течение 1 часа, охлаждали до комнатной температуры и добавляли к ней кислый карбонат натрия до тех пор, пока рН смеси не становился равным 3,0-3,5. Полученный в результате раствор концентрировали при пониженном давлении, смешивали с 100 мл ацетонитрила и 20 г безводного сульфата натрия с последующим перемешиванием в течение 30 мин, фильтровали и фильтрат концентрировали при пониженном давлении для получения 5-дезокси-D-рибофуранозы.
14,3 г (0,107 моля) 5-дезокси-D-рибофуранозы добавляли к 200 мл дихлорметана, добавляли к ним 30,1 мл (0,372 моль) пиридина и 37 мл (0,266 моль) триэтиламина и смесь охлаждали до -30°С. 49,1 мл (0,638 моль) метилхлорформиата добавляли к ней по каплям при -30°С за 30 минут, реакционную смесь нагревали до 10°С, добавляли к ней 100 мл воды и полученную смесь перемешивали в течение 30 минут. Органический слой отделяли и промывали последовательно 200 мл 1N HCl, водным бикарбонатом натрия и водным NaCl. Полученный в результате органический слой сушили над безводным сульфатом натрия, фильтровали и удаляли из него растворитель, получая 27,7 г указанного в заголовке соединения.
β-аномер:α-аномер = 2,7:1
ЯМР-характеристика β-аномера: 1Н ЯМР (300 МГц, CDCl3): δ 1,42 (д, 3H), 3,82 (с, 9H), 4,34-4,41 (м, 1H), 5,00 (дд, 1H), 5,28 (дд, 1H), 6,07 (д, 1H)
ЯМР-характеристика α-аномера: 1Н ЯМР (300 МГц, CDCl3): δ 1,37 (д, 3H), 3,81 (с, 9H), 4,40-4,48 (м, 1H), 4,90 (дд, 1H), 5,17 (дд, 1H), 6,29 (д, 1H)
Пример 2. Получение 2',3'-ди-О-метоксикарбонил-5'-дезокси-5-фторцитидина (соединение формулы 6)
Смешивали 11,6 г (0,090 моль) 5-фторцитидина, 19 мл гексаметилдисилазана и 24 мл ацетонитрила и 0,2 г сульфата аммония добавляли к смеси, которую кипятили с обратным холодильником в течение 1 ч. После охлаждения реакционной смеси до комнатной температуры к ней добавляли 72 мл ацетонитрила, после чего подвергали полученную в результате смесь дистилляции для удаления примерно 60 мл растворителя. Полученный раствор охлаждали до комнатной температуры, смешивали с 27,7 г (0,090 моль) соединения, полученного в примере 1, и 72 мл ацетонитрила, и полученную в результате смесь охлаждали до 20°С. После добавления к ней по каплям при 25°С 16,3 мл (0,090 моль) триметилсилилтрифторметансульфоната реакционную смесь перемешивали при комнатной температуре в течение ночи, охлаждали до 10°С, смешивали с 45,4 г кислого карбоната натрия и перемешивали в течение 30 мин. К ней добавляли по каплям 9,8 г воды и 72 мл дихлорметана, и полученный в результате раствор перемешивали в течение 2 ч, фильтровали, и выделенное твердое вещество промывали 72 мл дихлорметана. Фильтрат промывали 120 мл 4% бикарбоната натрия, сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении, получая 35,8 г указанного в заголовке соединения.
1Н ЯМР (CDCl3): δ 1,47 (д, 3H), 3,79 (с, 3H), 3,81 (с, 3H), 4,22~4,30 (м, 1H), 4,94 (дд, 1H), 5,39 (дд, 1H), 5,76 (д, 1H), 6,00 (ущир.с, 1H), 7,37 (д, 1H), 8,78 (ушир.с, 1H)
Пример 3. Получение 2',3'-ди-О-метоксикарбонил-5'-дезокси-5-фтор-N 4 -(пентилоксикарбонил)цитидина (соединение формулы 7)
35,8 г (0,099 моль) соединения, полученного в примере 2, смешивали с 163 мл дихлорметана и 11 мл (0,136 моль) пиридина и перемешивали. После охлаждения полученной в результате смеси до температуры от -5 до 0°С к ней добавляли по каплям 15,7 мл (0,109 моль) н-пентилхлорформиата, поддерживая в то же время температуру реакционной смеси ниже 0°С, с последующим добавлением к ней, после того как смесь нагревали до комнатной температуры и перемешивали в течение 2 ч, 1N HCl. Органический слой отделяли, последовательно промывали 163 мл насыщенного бикарбоната натрия и 163 мл воды, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении, получая 42,9 г указанного в заголовке соединения.
1Н ЯМР (CDCl3): δ 0,91 (т, 3H), 1,33~1,40 (м, 4H), 1,48 (д, 3H), 1,69~1,74 (м, 2H), 3,82 (с, 6H), 4,16 (т, 2H), 4,27~4,32 (м, 1H), 4,93 (дд, 1H), 5,32 (дд, 1H), 5,83 (д, 1H), 7,40 (с, 1H), 12,2 (ушир.с, 1H)
Пример 4. Получение 5'-дезокси-5-фтор-N 4 -(пентилоксикарбонил)цитидина (соединение формулы 1)
42,9 г соединения, полученного в примере 3, добавляли к 215 мл метанола, и смесь перемешивали и охлаждали до температуры от -5 до 0°С. 10,8 г NaOH растворяли в 107 мл воды, и раствор NaOH добавляли к ним, поддерживая температуру реакционной смеси ниже 0°С. Полученную в результате смесь перемешивали в течение 30 мин и добавляли к ней по каплям 48 мл 6N HCl, пока рН реакционной смеси не становился равным 5,3. Полученную в результате смесь последовательно промывали дважды 215 мл дихлорметана и один раз 108 мл дихлорметана, и объединенный органический слой промывали 215 мл воды, сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. После добавления к ней 129 мл этилацетата остаток смешивали с 97 мл этилацетата перемешиванием для кристаллизации. К нему добавляли по каплям 97 мл гексана, чтобы дать кристаллам вызреть, и полученную смесь перемешивали в течение 1 ч, охлаждали до 0°С и опять перемешивали в течение 1 ч. Полученное в результате твердое вещество отфильтровывали, промывали 86 мл смеси этилацетата и гексана (1:1 об./об.), охлаждали до 0°С и сушили при 35°С в вакуумном сушильном шкафу в течение ночи, получая 28,6 г соединения, указанного в заголовке.
1Н ЯМР (CD3OD): δ 0,91 (т, 3H), 1,36~1,40 (м, 4H), 1,41 (д, 3H), 1,68~1,73 (м, 2H), 3,72 (дд, 1H), 4,08 (дд, 3H), 4,13~4,21 (м, 3H), 5,70 (с, 1H), 7,96 (д, 1H)
Несмотря на то, что изобретение было описано со ссылкой на вышеприведенные конкретные примеры осуществления, различные модификации изобретения, которые могут быть сделаны специалистами, также попадают в сферу изобретения, как оно определено в прилагаемой формуле изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ 1-α-ГАЛОГЕН-2,2-ДИФТОР-2-ДЕЗОКСИ-D-РИБОФУРАНОЗЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2005 |
|
RU2346948C2 |
| СПОСОБ ПОЛУЧЕНИЯ 2'-ДЕЗОКСИ-2', 2'-ДИФТОРЦИТИДИНА | 2005 |
|
RU2360919C2 |
| СПОСОБ ПОЛУЧЕНИЯ ОБОГАЩЕННЫХ БЕТА-АНОМЕРОМ НУКЛЕОЗИДОВ | 1993 |
|
RU2131880C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНОГО D-ЭРИТРО-2,2-ДИФТОРО-2-ДЕЗОКСИ-1-ОКСОРИБОЗЫ | 2005 |
|
RU2337917C1 |
| СПОСОБ ПОЛУЧЕНИЯ МОНТЕЛУКАСТА И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ, ИСПОЛЬЗУЕМЫЕ В НЕМ | 2007 |
|
RU2408583C1 |
| СПОСОБ ПОЛУЧЕНИЯ 2'-ФТОР-2'-АЛКИЛЗАМЕЩЕННЫХ ИЛИ ДРУГИХ ЗАМЕЩЕННЫХ РИБОФУРАНОЗИЛПИРИМИДИНОВ И ПУРИНОВ И ИХ ПРОИЗВОДНЫХ | 2005 |
|
RU2433124C2 |
| СПОСОБ СИНТЕЗА КЛОФАРАБИНА | 2015 |
|
RU2663584C2 |
| МОНОЦИКЛИЧЕСКИЕ L-НУКЛЕОЗИДЫ, ИХ АНАЛОГИ И ПРИМЕНЕНИЯ | 1997 |
|
RU2188828C2 |
| МИМЕТИКИ ПОЛИ (ADP-РИБОЗЫ) И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2011 |
|
RU2559873C2 |
| ПРОИЗВОДНЫЕ 5,6-ДИХЛОРБЕНЗИМИДАЗОЛА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКИЙ СОСТАВ И СПОСОБ ЛЕЧЕНИЯ И ПРОФИЛАКТИКИ ВИРУСНЫХ ИНФЕКЦИЙ | 1995 |
|
RU2145963C1 |
Настоящее изобретение относится к способу получения капецитабина, включающему стадии: 1) гидролиза метилацетонидного соединения для получения триольного соединения формулы 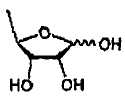 , 2) взаимодействия триольного соединения с галогеналкилформиатом формулы в присутствии смеси пиридина и триэтиламина для получения обогащенного β-аномером триалкилкарбонатного соединения формулы
, 2) взаимодействия триольного соединения с галогеналкилформиатом формулы в присутствии смеси пиридина и триэтиламина для получения обогащенного β-аномером триалкилкарбонатного соединения формулы 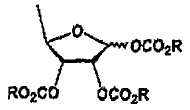 , 3) гликозилирования триалкилкарбонатного соединения с использованием 5-фторцитозина в присутствии кислоты, 4) карбамоилирования полученного соединения формулы
, 3) гликозилирования триалкилкарбонатного соединения с использованием 5-фторцитозина в присутствии кислоты, 4) карбамоилирования полученного соединения формулы 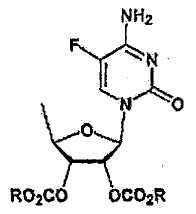 с использованием н-пентилхлорформиата с последующим удалением карбонатных гидроксизащитных групп фуранового цикла. 2 н. и 5 з.п. ф-лы.
с использованием н-пентилхлорформиата с последующим удалением карбонатных гидроксизащитных групп фуранового цикла. 2 н. и 5 з.п. ф-лы.
1. Способ получения капецитабина формулы 1, включающий стадии:
(1) гидролиза метилацетонидного соединения формулы 2 для получения триольного соединения формулы 3;
(2) взаимодействия соединения формулы 3 с галогеналкилформиатом формулы 4 в присутствии смеси пиридина и триэтиламина для получения обогащенного β-аномером триалкилкарбонатного соединения формулы 5;
(3) гликозилирования соединения формулы 5 с использованием 5-фторцитозина в присутствии кислоты для получения диалкоксикарбонилцитидинового соединения формулы 6;
(4) карбамоилирования соединения формулы 6 с использованием н- пентилхлорформиата для получения карбамоилцитидинового соединения формулы 7; и
(5) удаления карбонатных гидроксизащитных групп в соединениях формулы (7):
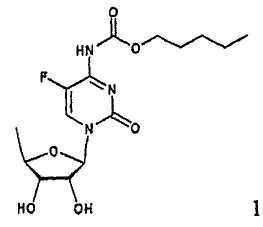
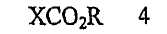
где X представляет хлор, бром или йод, и R представляет метил или этил.
2. Способ по п.1, в котором пиридин применяют в количестве, составляющем от 1 до 2 эквивалентов в расчете на триэтиламин.
3. Способ по п.1, в котором смесь пиридина и триэтиламина применяют в количестве, составляющем от 4 до 10 эквивалентов в расчете на соединение формулы 3.
4. Способ по п.1, в котором реакцию на стадии (2) проводят при температуре от -50°С до -30°С.
5. Способ по п.1, в котором кислота, используемая на стадии (3) представляет собой этилалюминийдихлорид, метилалюминийдихлорид, SnCl4, триметилсилилтрифторметансульфоновую кислоту или трифторметансульфоновую кислоту.
6. Способ по п.5, в котором кислоту применяют в количестве, составляющем от 0,5 до 3 эквивалентов в расчете на соединение формулы 5.
7. Способ получения триалкилкарбонатного соединения формулы 5, включающий стадии:
(1) гидролиза метилацетонидного соединения формулы 2 для получения триольного соединения формулы 3; и
(2) взаимодействия соединения формулы 3 с галогеналкилформиатом формулы 4 в присутствии смеси пиридина и триэтиламина для получения обогащенного β-аномером триалкилкарбоната формулы 5:
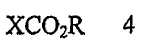
где X и R имеют значения, определенные в п.1.
| US 5453497 А, 26.09.1995 | |||
| US 4340729 A 20.06.1982 | |||
| Nobuo Shimma et al | |||
| The design and synthesis of a new tumor-selective fluoropyrimidine carbamate, Capecitabine, Bioirganic & Medicinal Chemistry, 2000, vol.8, no.7, pp.1697-1706 | |||
| ПРОИЗВОДНЫЕ 5'-ДЕЗОКСИЦИТИДИНА И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 1998 |
|
RU2238278C2 |

