Данная заявка заявляет приоритет предварительной заявки на патент № 60/843595, зарегистрированной 8 сентября 2006, которая включена здесь в качестве ссылки во всей ее полноте.
1. Область техники, к которой относится изобретение
Изобретение относится к способам получения соединений, применимых для лечения, профилактики или устранения заболеваний, связанных с нонсенс-мутацией. Более конкретно, здесь предложены способы синтеза 1,2,4-оксадиазолов. В частности, здесь предложены способы, применимые для получения 3-[5-(2-фторфенил-[1,2,4]оксадиазол-3-ил]бензойной кислоты.
2. Уровень техники
Производные 1,2,4-оксадиазола, применимые для лечения, профилактики или устранения заболеваний, интенсивность симптомов которых уменьшают модуляцией преждевременной терминации трансляции или разрушением нонсенс-опосредуемой мРНК, описаны в патенте США № 6992096 В2, зарегистрированном 31 января 2006, который включен здесь в качестве ссылки во всей его полноте. Одним таким соединением является 3-[5-(2-фторфенил[1,2,4]оксадиазол-3-ил]бензойная кислота.
Существующие способы синтеза 1,2,4-оксадиазолбензойных кислот описаны в патенте США № 6992096 В2, зарегистрированном 31 января 2006 (см. колонку 57, строчку 40, схему В и пример 2). В частности, указанные способы включают в себя многочисленные стадии реакций, за каждой из которых следует выделение нужного промежуточного продукта.
Хотя такие способы являются адаптируемыми и применимыми для получения 1,2,4-оксадиазолбензойных кислот, имеются возможности для изменений, которые могут привести к более эффективному синтезу. В частности, синтетические способы с меньшим числом стадий выделения, в которых можно применять меньше растворителей, могут быть более эффективными и менее дорогими.
Цитирование любой ссылки в разделе 2 данной заявки не следует истолковывать как допущение, что такая ссылка является прототипом настоящей заявки.
3. Сущность изобретения
Здесь предложены способы, применимые для получения 1,2,4-оксадиазолбензойных кислот, которые являются эффективными, экономичными по стоимости и легко адаптируемыми по масштабам реакции к коммерческим реагентам.
В одном варианте осуществления здесь предложены способы, применимые для получения 1,2,4-оксадиазолбензойной кислоты и включающие в себя следующие стадии: (1) реакцию цианобензоатного эфира с гидроксиламином; (2) ацилирование галогенбензоилхлоридом; (3) конденсацию и (4) гидролиз бензоатного эфира.
В конкретном варианте осуществления стадии (1)-(3) проводят в том же самом органическом растворителе.
В другом конкретном варианте осуществления стадии (1)-(3) проводят в одном органическом растворителе.
В другом варианте осуществления стадии (1)-(4) проводят в том же самом органическом растворителе.
В другом варианте осуществления стадии (1)-(4) проводят в одном органическом растворителе.
В другом варианте осуществления стадии (1)-(3) проводят в том же самом водном растворителе.
В другом варианте осуществления стадии (1)-(3) проводят в одном водном растворителе.
В другом варианте осуществления стадии (1)-(4) проводят в том же самом водном растворителе.
В другом варианте осуществления стадии (1)-(4) проводят в одном водном растворителе.
В другом варианте осуществления стадии (1)-(3) проводят без выделения промежуточного продукта.
В другом варианте осуществления стадии (1)-(4) проводят без выделения промежуточного продукта.
В другом варианте осуществления за стадиями (1)-(4) следует стадия микронизации.
Еще в одном другом варианте осуществления способы, предложенные здесь, являются применимыми для получения 1,2,4-оксадиазолбензойных кислот и их фармацевтически приемлемых солей, гидратов, сольватов или полиморфов. Еще в одном другом варианте осуществления способы, предложенные здесь, являются применимыми для получения 1,2,4-оксадиазолбензойных кислот и их фармацевтически приемлемых солей, гидратов, сольватов или полиморфов, применимых для лечения, профилактики или устранения заболеваний или состояний, связанных с нонсенс-мутацией. Еще в одном варианте осуществления предложенные здесь способы являются применимыми для получения 1,2,4-оксадиазолбензойных кислот и их фармацевтически приемлемых солей, гидратов, сольватов или полиморфов, применимых для лечения, профилактики или устранения генетических заболеваний и нарушений.
4. Подробное описание изобретения
4.1 Терминология
Применяемый здесь, если не указано иначе, термин “галоген” или тому подобное означает -F, -Cl, -Br или -I.
Если не указано иначе, описанные здесь соединения, в том числе промежуточные продукты, применимые для получения таких соединений, которые содержат реакционно-способные функциональные группы (такие, как, но без ограничения указанным, карбокси-, гидрокси- и аминогруппы), включают в себя также их защищенные производные. «Защищенными производными» являются такие соединения, у которых реакционно-способный центр или центры блокированы одной или несколькими защитными группами (известными также как блокирующие группы). Подходящие защитные группы для карбоксигрупп включают в себя бензил, трет-бутил и тому подобное. Подходящие защитные группы для амино- и амидогрупп включают в себя ацетил, трет-бутилоксикарбонил, бензилоксикарбонил и тому подобное. Подходящие защитные группы для гидрокси включают в себя бензил и тому подобное. Другие подходящие защитные группы хорошо известны среднему специалисту в данной области. Выбор и применение защитных групп и условий реакции для введения и удаления защитных групп описаны в публикации T.W. Green, “Protective Groups in Organic Synthesis”, Third Ed., Wiley, New York, 1999, которая включена здесь в качестве ссылки во всей ее полноте.
Применяемая здесь фраза, если не оговорено особо, композиция, которая “по существу не содержит” соединение, означает, что композиция содержит меньше чем приблизительно 20 масс.%, более предпочтительно меньше чем приблизительно 10 масс.%, еще более предпочтительно меньше чем приблизительно 5 масс.% и наиболее предпочтительно меньше чем приблизительно 3 масс.% соединения.
Применяемый здесь термин, если не оговорено особо, «способ(ы)» относится к способам, описанным здесь, которые являются применимыми для получения производного 1,2,4-оксадиазолбензойной кислоты.
Применяемые здесь, если не оговорено особо, термины «добавляемый» или «добавление», или тому подобное означает контактирование одного реагирующего вещества, реагента, растворителя, катализатора или тому подобное с другим реагирующим веществом, реагентом, растворителем, катализатором или тому подобное. Реагирующие вещества, реагенты, растворители, катализаторы или тому подобное можно добавлять по отдельности, одновременно или раздельно и можно добавлять в любом порядке. Их можно добавлять в присутствии или в отсутствие нагрева и можно необязательно добавлять в инертной атмосфере.
Применяемая здесь фраза, если не оговорено особо, реакция, которая «по существу завершена» или доведена «по существу до завершения», означает, что реакция имеет выход более чем приблизительно 80%, более чем приблизительно 90% или более чем приблизительно 97% требуемого продукта.
Применяемый здесь, если не оговорено особо, термин «без выделения» означает, что реакционную смесь, являющуюся результатом одной стадии, подвергают превращению в следующей стадии без выделения требуемого продукта. В некоторых вариантах осуществления проведение многих стадий реакций «без выделения» включает в себя способы, которые включают в себя перенос реакционной смеси, являющейся результатом одной стадии, в другой реакционный сосуд перед началом следующей реакции.
Применяемый здесь, если не оговорено особо, термин «конденсация» означает химическую реакцию, в которой два химических соединения взаимодействуют и становятся ковалентно связанными друг с другом с одновременной потерей маленькой молекулы, например, воды.
Применяемый здесь, если не оговорено особо, термин «фармацевтически приемлемая соль» относится к соли соединения настоящего изобретения, которая является безопасной и эффективной для применения пациентом. Примерные фармацевтически приемлемые соли получают с применением металлов, неорганических оснований или органических оснований. Подходящие соли оснований включают в себя, но не ограничиваются перечисленным, соли алюминия, кальция, лития, магния, калия, натрия, аммония и цинка. Подходящей солью органического основания является соль триэтиламина.
Применяемый здесь, если не оговорено особо, термин «гидрат» означает соединение настоящего изобретения или его соль, которая дополнительно включает в себя стехиометрическое или нестехиометрическое количество воды, связанной нековалентными межмолекулярными связями.
Применяемый здесь, если не оговорено особо, термин «сольват» означает сольват, образованный ассоциацией одной или нескольких молекул растворителя с соединением настоящего изобретения. Термин «сольват» включает в себя гидраты (например, полугидрат, моногидрат, дигидрат, тригидрат, тетрагидрат и тому подобное).
Применяемый здесь, если не оговорено особо, термин «полиморф» означает твердые кристаллические формы соединения настоящего изобретения или его комплекса. Различные полиморфы одного и того же соединения могут проявлять различные физические, химические и/или спектроскопические свойства.
Применяемая здесь, если не оговорено особо, фраза «заболевания или нарушения, связанные с нонсенс-мутацией» означает заболевания или нарушения, которые не могут возникать, протекать или вызывать симптомы, если не присутствовала нонсенс-мутация.
Применяемый здесь, если не оговорено особо, термин «лечат», «лечение», «подвергаемый лечению» или тому подобное относится к уменьшению или снижению развития, тяжести и/или продолжительности заболевания, или нарушения, или уменьшению одного или нескольких симптомов (предпочтительно, одного или нескольких явных симптомов) заболевания или состояния, являющегося результатом проведения одной или нескольких терапий (например, введения одного или нескольких лекарственных средств, таких как 1,2,4-оксадиазолбензойная кислота).
Применяемый здесь, если не оговорено особо, термин «проводить профилактику», «профилактика», «подвергаемый профилактике» или тому подобное относится к уменьшению риска приобретения или развития данного заболевания или нарушения или уменьшению или ингибированию рецидива, начала появления или развития одного или нескольких симптомов данного заболевания или нарушения.
Если имеется расхождение между изображенной структурой и названием, данным такой структуре, изображенная структура должна представляться более весомой. Кроме того, если стереохимия структуры или ее части не указана, например, сплошными или пунктирными линиями, структура или часть ее должна быть интерпретирована как включающая в себя все ее стереоизомеры.
Предложенные здесь варианты осуществления можно понять более полно обращением к нижеследующему подробному описанию и иллюстративным примерам, которые предназначаются для иллюстрации неограничивающих вариантов осуществления.
4.2 Способы
Здесь предложены эффективные по стоимости и экономичные способы, применимые для получения 1,2,4-оксадиазолбензойных кислот.
В одном варианте осуществления способы включают в себя применение м-цианобензойной кислоты.
В другом варианте осуществления способы включают в себя применение фторбензоилхлорида.
В другом варианте осуществления способы включают в себя применение о-фторбензоилхлорида.
В другом варианте осуществления стадии (1)-(3) проводят в одном органическом растворителе или том же самом органическом растворителе и без выделения промежуточного продукта между стадиями.
В другом варианте осуществления стадии (1)-(4) проводят в одном органическом растворителе или том же самом органическом растворителе и без выделения промежуточного продукта между стадиями (1)-(3).
В другом варианте осуществления стадии (1)-(4) проводят в одном органическом растворителе или том же самом органическом растворителе и без выделения промежуточного продукта между стадиями (1)-(4).
В другом варианте осуществления стадии (1)-(3) или стадии (1)-(4) проводят в одном водном растворителе или том же самом водном растворителе и без выделения промежуточного продукта между стадиями (1)-(3) или в другом варианте осуществления стадиями (1)-(4).
В одном варианте осуществления растворителем, применяемым в описанных здесь способах, является полярный растворитель, такой как тетрагидрофуран, диоксан, изобутилацетат, изопропилацетат и этилацетат.
В другом варианте осуществления растворителем, применяемым в описанных здесь способах, является спиртовый растворитель, такой как метанол, этанол, изопропанол, изобутанол, пропанол, бутанол и трет-амиловый спирт.
В другом варианте осуществления растворителем, применяемым в описанных здесь способах, является трет-бутанол.
В одном варианте осуществления способы, описанные здесь, являются применимыми для получения партии 1,2,4-оксадиазолбензойной кислоты в количестве приблизительно 500 мг или больше, приблизительно 1 кг или больше, приблизительно 5 кг или больше, приблизительно 10 кг или больше, приблизительно 25 кг или больше, приблизительно 50 кг или больше, приблизительно 75 кг или больше, приблизительно 100 кг или больше, приблизительно 125 кг или больше, приблизительно 150 кг или больше, приблизительно 175 кг или больше, приблизительно 200 кг или больше, приблизительно 225 кг или больше, приблизительно 250 кг или больше, приблизительно 275 кг или больше, приблизительно 300 кг или больше, приблизительно 325 кг или больше, приблизительно 350 кг или больше, приблизительно 375 кг или больше, приблизительно 400 кг или больше, приблизительно 425 кг или больше, приблизительно 450 кг или больше, приблизительно 475 кг или больше, или приблизительно 500 кг, или приблизительно 600 кг, или приблизительно 700 кг, или приблизительно 800 кг, или приблизительно 900 кг, или приблизительно 1000 кг, или больше.
В одном варианте осуществления 1,2,4-оксадиазолбензойную кислоту получают в виде одной из партий описанных выше количеств с общим выходом приблизительно 50% или больше, приблизительно 55% или больше, приблизительно 60% или больше, приблизительно 65% или больше, приблизительно 70% или больше, приблизительно 75% или больше, приблизительно 80% или больше, приблизительно 85% или больше, приблизительно 90% или больше или приблизительно 95% или больше.
В одном варианте осуществления за стадиями (1)-(4) следует стадия микронизации. В конкретном варианте осуществления микронизированная 1,2,4-оксадиазолбензойная кислота имеет распределение размера частиц D (v, 0,1): от приблизительно 0,5 мкм до приблизительно 1,0 мкм; D (v, 0,5): от приблизительно 1,5 мкм до приблизительно 5,0 мкм и D (v, 0,9): от приблизительно 5,5 мкм до приблизительно 10,0 мкм.
В одном варианте осуществления предложенные здесь способы являются применимыми для получения соединения формулы I:
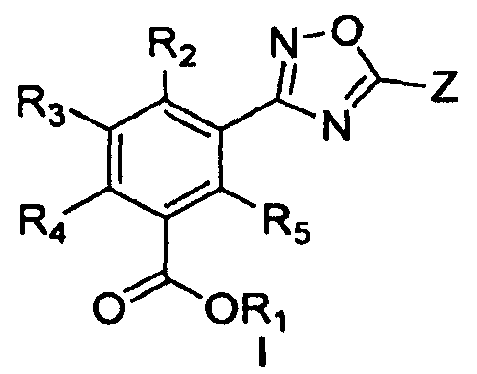
или его фармацевтически приемлемых солей, гидратов, клатратов, пролекарств, полиморфов, стереоизомеров, в том числе энантиомеров, диастереомеров, рацематов или смесей стереоизомеров, где
Z представляет собой замещенный или незамещенный арил, замещенный или незамещенный гетероарил, замещенный или незамещенный циклоалкил, замещенный или незамещенный алкил, замещенный или незамещенный алкенил, замещенный или незамещенный гетероцикл, замещенный или незамещенный арилалкил;
R1 представляет собой водород, замещенный или незамещенный алкил, замещенный или незамещенный циклоалкил, замещенный или незамещенный гетероциклоалкил, замещенный или незамещенный арил, замещенный или незамещенный гетероарил, -(CH2CH2O)nR6 или любую биогидролизуемую группу;
R2, R3, R4, R5 и R6 независимо представляют собой водород, замещенный или незамещенный алкил, замещенный или незамещенный алкенил, замещенный или незамещенный алкинил, замещенный или незамещенный циклоалкил, замещенный или незамещенный гетероциклоалкил, замещенный или незамещенный арил, замещенный или незамещенный гетероарил, алкокси, арилокси, гетероарилокси, галоген, CF3, OCF3, OCHF2, CN, COOH, COOR7, SO2R7, NO2, NH2 или N(R7)2;
в каждом случае R7 независимо представляет собой водород, замещенный или незамещенный алкил, замещенный или незамещенный алкенил, замещенный или незамещенный алкинил, замещенный или незамещенный циклоалкил, замещенный или незамещенный гетероциклоалкил, замещенный или незамещенный арил, замещенный или незамещенный гетероарил, алкокси, арилокси, гетероарилокси, галоген или CF3 и
n равно целому числу от 1 до 7,
и включают в себя следующие стадии:
(1) реакцию необязательно замещенного цианобензоатного эфира с гидроксиламином;
(2) ацилирование хлорангидридом кислоты;
(3) конденсацию и
(4) необязательный гидролиз бензоатного эфира.
В одном варианте осуществления стадии (1)-(3) проводят в одном органическом растворителе.
В другом варианте осуществления стадии (1)-(3) проводят в том же самом органическом растворителе.
В другом варианте осуществления стадии (1)-(4) проводят в одном органическом растворителе.
В другом варианте осуществления стадии (1)-(4) проводят в том же самом органическом растворителе.
В другом варианте осуществления стадии (1)-(3) проводят без выделения промежуточного продукта.
В другом варианте осуществления стадии (1)-(4) проводят без выделения промежуточного продукта.
В другом варианте осуществления стадии (1)-(3) проводят в одном органическом растворителе или в том же самом органическом растворителе и без выделения промежуточного продукта между стадиями.
В другом варианте осуществления стадии (1)-(4) проводят в одном органическом растворителе или в том же самом органическом растворителе и без выделения промежуточного продукта между стадиями (1)-(3).
В другом варианте осуществления стадии (1)-(4) проводят в одном органическом растворителе или в том же самом органическом растворителе и без выделения промежуточного продукта между стадиями (1)-(4).
В одном варианте осуществления растворителем, применяемым в описанных здесь способах, является трет-бутанол.
В одном варианте осуществления предложенные здесь способы являются применимыми для получения соединения формулы I, имеющего структуру формулы II:
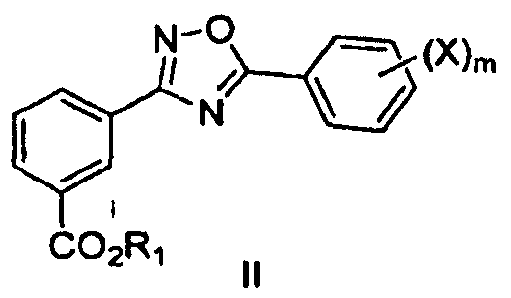
или его фармацевтически приемлемых солей, гидратов, клатратов, пролекарств, полиморфов, стереоизомеров, в том числе энантиомеров, диастереомеров, рацематов или смесей стереоизомеров, где
R1 представляет собой водород, замещенный или незамещенный алкил, замещенный или незамещенный циклоалкил, замещенный или незамещенный гетероциклоалкил, замещенный или незамещенный арил, замещенный или незамещенный гетероарил, -(CH2CH2O)nR6 или любую биогидролизуемую группу;
Х в каждом случае независимо представляет собой F, Cl, Br или I и
m равно целому числу от 1 до 5,
и включают в себя следующие стадии:
(1) реакцию метилового эфира цианобензойной кислоты с гидроксиламином;
(2) ацилирование галогенбензоилхлоридом;
(3) конденсацию и
(4) гидролиз метилового сложного эфира.
В одном варианте осуществления Х представляет собой F.
В другом варианте осуществления m равно 1.
В другом варианте осуществления Х представляет собой F и m равно 1.
В другом варианте осуществления m равно 1 и Х представляет собой F в орто-положении.
В другом варианте осуществления m равно 1 и Х представляет собой F в мета-положении.
В другом варианте осуществления m равно 1 и Х представляет собой F в пара-положении.
В другом варианте осуществления R1 представляет собой Н.
В одном варианте осуществления здесь предложены способы получения соединений формулы I, в том числе соединений, имеющих структуру формул II и IIa, включающие в себя стадии, указанные на схеме I:
Схема 1
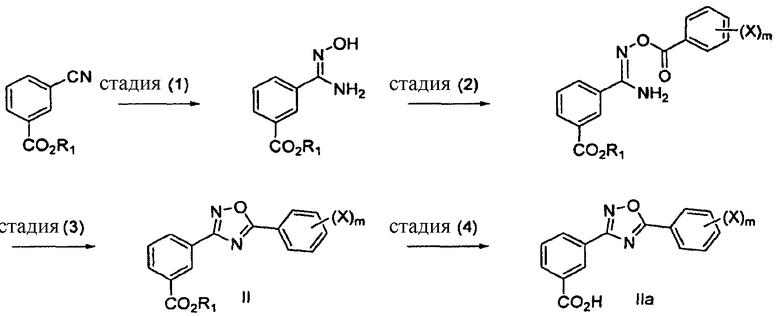
где R1 представляет собой водород, замещенный или незамещенный алкил, замещенный или незамещенный циклоалкил, замещенный или незамещенный гетероциклоалкил, замещенный или незамещенный арил, замещенный или незамещенный гетероарил, -(CH2CH2O)nR6 или любую биогидролизуемую группу, или любую подходящую блокирующую группу, известную специалисту в данной области, где стадия (4) является необязательной стадией гидролиза, когда R1 является другим, чем Н;
Х в каждом случае независимо представляет собой F, Cl, Br или I и
m равно целому числу от 1 до 5.
В одном варианте осуществления схемы 1 Х представляет собой F.
В другом варианте осуществления схемы 1 m равно 1.
В другом варианте осуществления схемы 1 Х представляет собой F и m равно 1.
В другом варианте осуществления m равно 1 и Х представляет собой F в орто-положении.
В другом варианте осуществления m равно 1 и Х представляет собой F в мета-положении.
В другом варианте осуществления m равно 1 и Х представляет собой F в пара-положении.
В другом варианте осуществления схемы 1 R1 представляет собой метил.
В другом варианте осуществления схемы 1 стадии (1)-(3) проводят в одном органическом растворителе.
В другом варианте осуществления схемы 1 стадии (1)-(3) проводят в том же самом органическом растворителе.
В одном варианте осуществления схемы 1 стадии (1)-(4) проводят в одном органическом растворителе.
В одном варианте осуществления схемы 1 стадии (1)-(4) проводят в том же самом органическом растворителе.
В другом варианте осуществления схемы 1 стадии (1)-(3) проводят без выделения промежуточного продукта.
В другом варианте осуществления схемы 1 стадии (1)-(4) проводят без выделения промежуточного продукта.
В другом варианте осуществления схемы 1 стадии (1)-(3) проводят в одном органическом растворителе или в том же самом органическом растворителе и без выделения промежуточного продукта между стадиями.
В другом варианте осуществления схемы 1 стадии (1)-(4) проводят в одном органическом растворителе или в том же самом органическом растворителе и без выделения промежуточного продукта между стадиями (1)-(3).
В другом варианте осуществления стадии (1)-(4) проводят в одном органическом растворителе или в том же самом органическом растворителе и без выделения промежуточного продукта между стадиями (1)-(4).
В одном варианте осуществления схемы 1 применяемым растворителем является тетрагидрофуран, диоксан, изобутилацетат, изопропилацетат, этилацетат, метанол, этанол, изопропанол, изобутанол, пропанол, бутанол или трет-амиловый спирт.
В конкретном варианте осуществления схемы 1 применяемым растворителем является трет-бутанол.
В одном варианте осуществления схемы 1 стадия (1) содержит реакцию метилового эфира 3-цианобензойной кислоты с водным гидроксиламином в трет-бутаноле. В конкретном варианте осуществления в стадии (1) применяют 50% водный гидроксиламин. В другом варианте осуществления в стадии (1) применяют расплавленный трет-бутанол. В другом варианте осуществления к метиловому эфиру 3-цианобензойной кислоты и трет-бутанола приблизительно при 40-45°С добавляют водный гидроксиламин. В другом варианте осуществления реакционную смесь стадии (1) перемешивают в течение приблизительно 2 часов.
В другом варианте осуществления схемы 1 стадия (2) включает в себя реакцию продукта из стадии (1) с галогенбензоилхлоридом в триэтиламине и трет-бутаноле. В конкретном варианте осуществления галогенбензоилхлоридом является фторбензоилхлорид, в частности, 2-фторбензоилхлорид. В другом варианте осуществления реакционную смесь стадии (2) далее разбавляют расплавленным трет-бутанолом. В другом варианте осуществления реакцию стадии (2) проводят при температуре ниже 40°С и в конкретном варианте осуществления приблизительно при 30-35°С. В другом варианте осуществления реакционную смесь стадии (2) перемешивают в течение по меньшей мере 2 часов. В некоторых вариантах осуществления для доведения реакции до завершения к реакционной смеси (2) добавляют дополнительный триэтиламин или галогенбензоилхлорид.
В другом варианте осуществления схемы 1 стадия (3) включает в себя кипячение с обратным холодильником продукта из стадии (2) в трет-бутаноле. В конкретном варианте осуществления стадия (3) включает в себя кипячение с обратным холодильником продукта из стадии (2) в трет-бутаноле приблизительно при 82°С. В другом варианте осуществления стадия (3) включает в себя кристаллизацию продукта с замкнутым циклом добавлением воды приблизительно при 60-65°С. В другом варианте осуществления образовавшуюся суспензию охлаждают до комнатной температуры, фильтруют, промывают смесью трет-бутанол/вода (50/50, об./об.) и сушат в вакууме.
В другом варианте осуществления схемы 1 стадия (4) включает в себя гидролиз метилового сложного эфира продукта из стадии (3) в соответствующую натриевую соль добавлением водного гидроксида натрия в трет-бутаноле. В одном варианте осуществления гидролиз метилового сложного эфира продукта стадии (3) проводят в водном гидроксиде натрия и трет-бутаноле приблизительно при 68-72°С. В следующем варианте осуществления стадия (4) включает в себя превращение натриевой соли в свободную кислоту фильтрованием горячего раствора натриевой соли через находящийся в ряду системы фильтр (например, через находящийся в ряду системы фильтр с отверстиями 5 микронов) и подкислением серной кислотой до рН приблизительно 1-3. Еще в одном следующем варианте осуществления стадия (4) включает в себя превращение натриевой соли в свободную кислоту фильтрованием горячего раствора натриевой соли через находящийся в ряду системы фильтр (например, через находящийся в ряду системы фильтр с отверстиями 1 микрон) и подкислением приблизительно 10-15% хлористоводородной кислотой до рН приблизительно 1-3 с последующим перемешиванием приблизительно при 70°С в течение приблизительно 1 часа. В следующем варианте осуществления свободную кислоту выделяют применением фильтра Розенмунда и промывают водным трет-бутанолом и водой с последующей сушкой (например, лопастной сушилкой или сушилкой с двумя конусами) или центрифугированием.
Мониторинг развития описанной здесь реакции можно проводить любым способом, известным специалисту в данной области, включающим в себя, но не ограничивающимся перечисленным, тонкослойную хроматографию (ТСХ), высокоэффективную жидкостную хроматографию (ВЭЖХ) или спектроскопические способы (например, способы 1Н ЯМР, 13С ЯМР, ИК-спектроскопии, спектроскопии Рамана, МС).
В одном варианте осуществления здесь предложены способы получения 3-[5-(2-фторфенил-[1,2,4]оксадиазол-3-ил]бензойной кислоты, включающие в себя стадии, указанные на схеме 2:
Схема 2
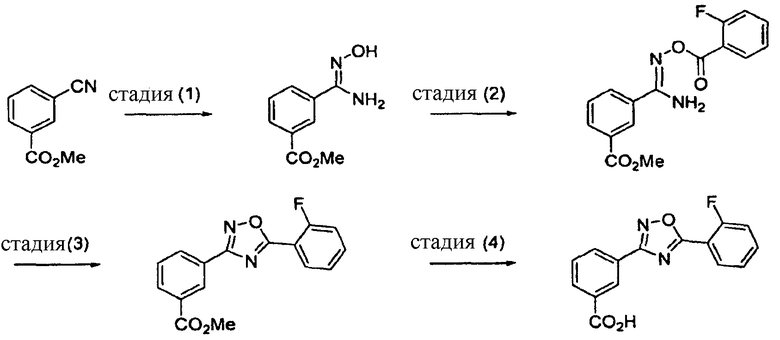
В одном варианте осуществления здесь предложены способы, применимые для получения соединения формулы II и включающие в себя проведение следующих стадий:
(1) реакцию метилового эфира цианобензойной кислоты
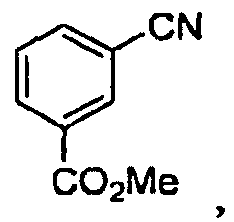
с гидроксиламином с получением соединения

последующее (2) ацилирование галогенбензоилхлоридом с получением соединения
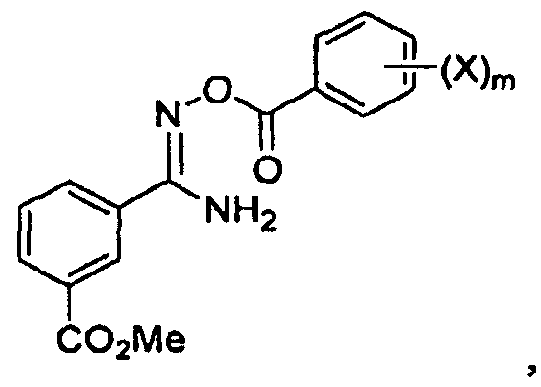
и последующую (3) конденсацию с получением соединения
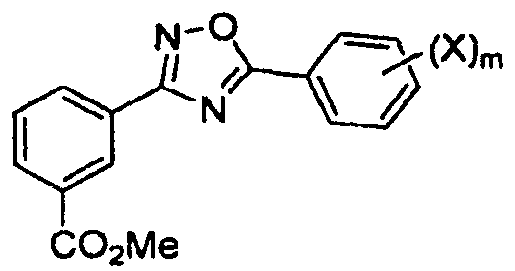
В другом варианте осуществления предложенные здесь способы дополнительно включают в себя (4) гидролиз метилового сложного эфира с получением соединения
В одном варианте осуществления Х представляет собой F.
В другом варианте осуществления m равно 1.
В другом варианте осуществления Х представляет собой F и m равно 1.
В другом варианте осуществления m равно 1 и Х представляет собой F в орто-положении.
В другом варианте осуществления m равно 1 и Х представляет собой F в мета-положении.
В другом варианте осуществления m равно 1 и Х представляет собой F в пара-положении.
В другом варианте осуществления стадии (1)-(3) проводят в одном органическом растворителе.
В другом варианте осуществления стадии (1)-(3) проводят в том же самом органическом растворителе.
В другом варианте осуществления стадии (1)-(4) проводят в одном органическом растворителе.
В другом варианте осуществления стадии (1)-(4) проводят в том же самом органическом растворителе.
В другом варианте осуществления стадии (1)-(3) или стадии (1)-(4) проводят в трет-бутаноле.
В другом варианте осуществления стадии (1)-(3) проводят без выделения промежуточного продукта.
В другом варианте осуществления стадии (1)-(4) проводят без выделения промежуточного продукта.
В другом варианте осуществления стадии (1)-(3) или стадии (1)-(4) проводят в одном органическом растворителе или в том же самом органическом растворителе и без выделения промежуточного продукта.
В одном варианте осуществления применяемым растворителем является тетрагидрофуран, диоксан, изобутилацетат, изопропилацетат, этилацетат, метанол, этанол, изопропанол, изобутанол, пропанол, бутанол или трет-амиловый спирт.
В конкретном варианте осуществления применяемым растворителем является трет-бутанол.
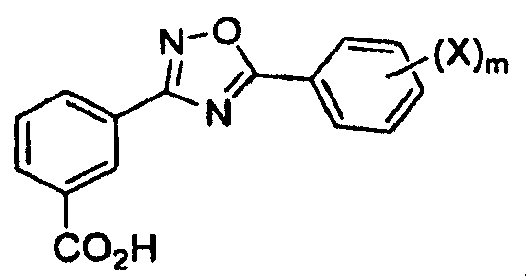
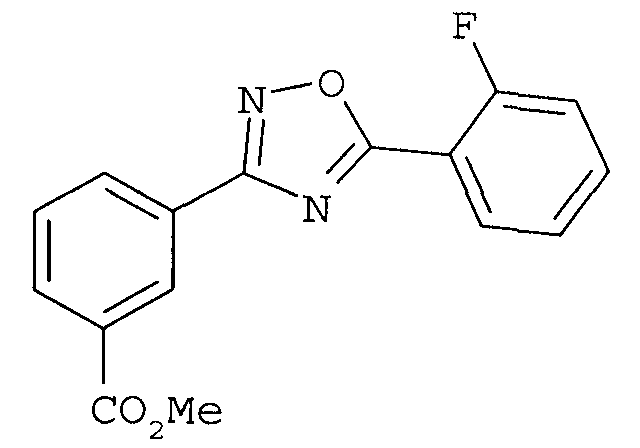
В одном варианте осуществления соединением формулы II является 3-[5-(2-фторфенил-[1,2,4]оксадиазол-3-ил]бензойная кислота.
В другом варианте осуществления синтез проводят в одном реакционном сосуде (т.е. синтез «в одном сосуде»).
В одном варианте осуществления здесь предложены способы, применимые для получения соединения формулы II, включающие в себя проведение нижеследующих стадий в одном реакционном сосуде:
реакцию цианобензойной кислоты
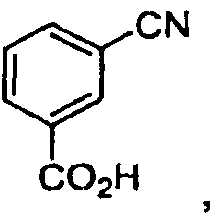
с гидроксиламином с получением соединения
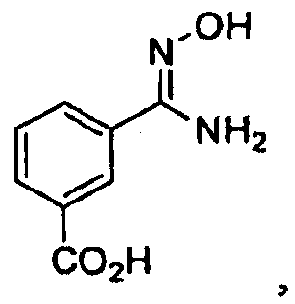
последующее ацилирование его галогенбензоилхлоридом с получением соединения
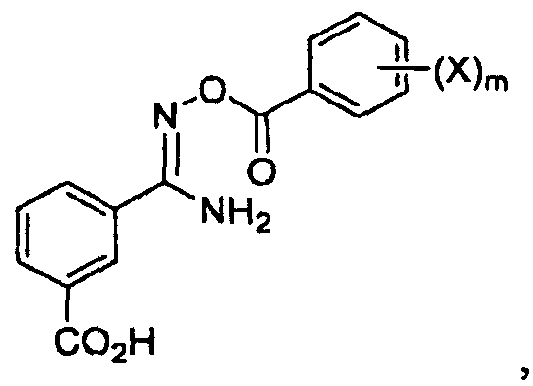
и последующее кипячение с обратным холодильником с получением
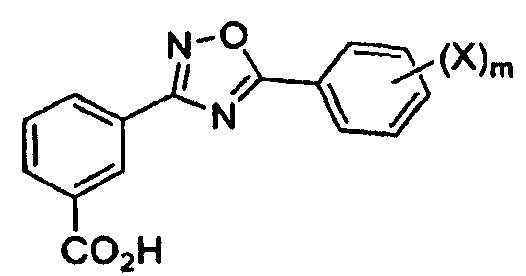
В одном варианте осуществления соединением является 3-[5-(2-фторфенил-[1,2,4]оксадиазол-3-ил]бензойная кислота.
Описанные здесь варианты осуществления далее иллюстрируются примерами, указанными ниже, которые не должны истолковываться как ограничение объема описанных здесь вариантов осуществления.
Исходные вещества и реагенты, применяемые в описанных здесь способах, можно получить из коммерческих источников или получить с применением способов, известных специалисту в данной области.
5. Примеры
Метил-3-[5-(2-фторфенил-[1,2,4]оксадиазол-3-ил]бензоат
Партия 1
Метил-3-цианобензоат (105 кг) и расплавленный трет-бутанол загружали в сухой реактор. К прозрачному раствору метил-3-цианобензоата в расплавленном трет-бутаноле в инертной атмосфере на протяжении приблизительно 2 часов и 48 минут добавляли 50% водный гидроксиламин (43 л, 47,4 кг). Максимальная температура раствора партии во время добавления 50% водного гидроксиламина была приблизительно 43°С. Скорость добавления 50% водного гидроксиламина варьировали от приблизительно 9 л в час в начале добавления до приблизительно 30 л в час. Температуру смеси партии поддерживали изменением температуры установленного места рубашки на реакторе. В частности, температуру установленного места изменяли от приблизительно 40,5°С в начале добавления до приблизительно 29,6°С, когда скорость добавления повышали. Реакцию считали завершенной (т.е. осталось меньше приблизительно 0,5% сложного эфира) после перемешивания в течение приблизительно 4 часов приблизительно при 40-45°С.
Смесь партии переносили в сухой реактор и в реактор загружали приблизительно 10 л расплавленного трет-бутанола. Температуру установленного места рубашки снижали от приблизительно 33°С, когда смесь партии переносили в сухой реактор, до приблизительно 27°С после того, как перенос был завершен. Наблюдали частичную кристаллизацию продукта партии, на которую перемешивание не оказывало неблагоприятное влияние. Продукт партии охлаждали до приблизительно 34,4°С и в реактор загружали триэтиламин (72,6 кг, 100 л). Температуру установленного места рубашки повышали от приблизительно 20,4°С до приблизительно 31,0°С для поддержания температуры смеси партии в диапазоне приблизительно 30-35°С. После промывания линии расплавленным трет-бутанолом (10 л) в смесь партии загружали 2-фторбензоилхлорид (113,7 кг, 86,0 л). Скорость добавления на протяжении первой трети загрузки была приблизительно 25 л в час. Температуру ввода в рубашку снижали приблизительно до 15°С во время этого периода и температура смеси партии оставалась приблизительно на уровне 34,6°С. Добавление завершали после приблизительно 5,5 часов. Максимальная температура смеси партии во время добавления была приблизительно 38,8°С. Скорость добавления замедляли к концу добавления, причем последние 27 литров 2-фторбензоилхлорида добавляли при скорости 11 л в час. Реакцию считали завершенной (т.е. оставалось меньше приблизительно 0,5% метил-3-амидинобензоата) после перемешивания в течение приблизительно 2 часов при 30-35°С.
Смесь партии затем нагревали до температуры флегмы (приблизительно 82°С) на протяжении приблизительно 1 часа и 42 минут и перемешивали в течение приблизительно дополнительных 18 часов. Во время перемешивания некоторое количество продукта частично кристаллизовывалось с образованием суспензии. Суспензию охлаждали до приблизительно 40°С, чтобы дать возможность взять пробу, причем во время такого охлаждения происходила полная кристаллизация. Смесь партии снова нагревали до температуры флегмы и перемешивали в течение приблизительно 1 часа и 50 минут. Смесь затем охлаждали на протяжении приблизительно 2 часов до приблизительно 69°С и на протяжении приблизительно 4 часов и 15 минут медленно добавляли 630 л очищенной воды, в то время как температуру смеси партии поддерживали между приблизительно 66-69°С. Суспензию охлаждали до приблизительно 22,4°С на протяжении приблизительно 3 часов и 14 минут и переносили в керамические фильтры 2×200 л, снабженные полипропиленовыми фильтровальными тканями с отверстиями 25-30 мк. Перенос вещества из сосуда на фильтры завершали спустя приблизительно 55 минут. Фильтровальные осадки промывали 50% водным трет-бутанолом (210 л), предоставляя приблизительно 10 минут для пропитывания каждого фильтровального осадка промывочной жидкостью. Фильтровальные осадки затем сушили в вакууме на протяжении приблизительно 5-10 минут. Очищенную воду добавляли к фильтровальным осадкам в качестве второй промывочной жидкости (158 л на осадок) для удаления остаточного трет-бутанола и соли хлорида триэтиламмония. Жидкости удаляли после сушки в вакууме в течение приблизительно 5 минут. Фильтровальные осадки сушили в вакууме в течение приблизительно 2 часов и затем отбирали пробу и анализировали с применением жидкостной хроматографии. Жидкостной хроматографией определили, что чистота осадков была приблизительно 99,6%.
После сушки осадков в вакууме в течение приблизительно 8 часов и 25 минут сырой осадок (207,4 кг) переносили в воздушный сушильный шкаф. Сушку в воздушном сушильном шкафу проводили приблизительно при 50-55°С в течение приблизительно 52 часов. Общий выход выделенного продукта был приблизительно 89,9% (174,65 кг), который можно корректировать до приблизительно 90,7% после учета вещества, израсходованного для взятия пробы.
Партия 2
Метил-3-цианобензоат (105 кг) и расплавленный трет-бутанол загружали в сухой реактор. В реактор в инертной атмосфере на протяжении приблизительно 3 часов и 29 минут загружали 50% водный гидроксиламин (47,85 кг). Во время добавления температуру поддерживали приблизительно при 40-45°С. Реакцию считали завершенной (т.е. оставалось меньше, чем приблизительно 0,5% сложного эфира) после перемешивания в течение приблизительно 3 часов и 16 минут при 40-45°С.
Смесь партии переносили в сухой реактор, как описано для партии 1. Смесь охлаждали приблизительно до 34,4°С и загружали триэтиламин (72,6 кг, 100 л). Добавление проводили на протяжении периода приблизительно 45 минут при поддержании температуры смеси партии приблизительно при 30-35°С. Во время добавления температуру места ввода в рубашку повышали от приблизительно 31,4°С до приблизительно 32,6°С. После промывания линии расплавленным трет-бутанолом в смесь партии загружали 2-фторбензоилхлорид (113,7 кг, 86,0 л). Хлорангидрид кислоты добавляли на протяжении приблизительно 3 часов и 27 минут. После перемешивания в течение приблизительно 8 часов при 35°С считали, что реакция не была завершена (т.е. оставалось более чем приблизительно 0,5% метил-3-амидинобензоата). К смеси партии затем добавляли 1,5 масс.% первоначальных загрузок триэтиламина и 2-фторбензоилхлорида. Каждую из дополнительных загрузок выполняли промыванием линии трет-бутанолом (10 л). Во время добавления хлорангидрида кислоты дополнительное охлаждение не проводили. Температуру партии поддерживали приблизительно при 30-35°С с температурой у ввода в рубашку, составляющей от приблизительно 30,3°С до приблизительно 33,0°С. Реакцию считали завершенной (т.е. оставалось меньше, чем приблизительно 0,5% метил-3-амидинобензоата) после перемешивания в течение приблизительно 2 часов при 30-35°С.
Смесь партии нагревали до температуры флегмы (приблизительно 83°С) на протяжении приблизительно 1 часа и 44 минут и перемешивали в течение приблизительно 18 часов. Как в случае партии 1, твердые вещества полностью кристаллизовались во время охлаждения для взятия пробы. Смесь снова нагревали до температуры флегмы и перемешивали в течение приблизительно 1 часа и 2 минут. Смесь затем охлаждали на протяжении приблизительно 2 часов и 20 минут до приблизительно 69,2°С и на протяжении приблизительно 4 часов и 30 минут медленно добавляли 630 л очищенной воды, в то время как температуру смеси поддерживали между приблизительно 65,6-69,2°С. Суспензию охлаждали до приблизительно 23,4°С на протяжении приблизительно 3 часов и 30 минут и содержимое переносили в двойные керамические фильтры, как описано для партии 1. Перенос вещества завершали после приблизительно 5 часов и 6 минут. Фильтровальные осадки промывали приблизительно 50% водным трет-бутанолом (2 объема на осадок), предоставляя приблизительно 10 минут для пропитывания каждого осадка промывочной жидкостью перед сушкой в вакууме. Фильтрование завершали спустя приблизительно 1 час и 40 минут. Очищенную воду добавляли к осадкам в качестве последней промывочной жидкости. Жидкости удаляли сушкой в вакууме в течение приблизительно 10 минут. Осадки сушили в вакууме в течение приблизительно дополнительных 2 часов и 5 минут и затем отбирали пробу и анализировали с применением жидкостной хроматографии. Жидкостной хроматографией определили, что чистота осадков была приблизительно 99,5% и 99,6% соответственно.
После сушки осадков в вакууме в течение приблизительно 2 часов и 5 минут сырой осадок (191,5 кг) переносили в воздушный сушильный шкаф. Сушку в воздушном сушильном шкафу проводили приблизительно при 50-55°С в течение приблизительно 48 часов. Общий выход выделенного продукта был приблизительно 92,5% (179,7 кг).
Партия 3
В реакционный сосуд загружали метиловый эфир 3-цианобензойной кислоты (52,5 кг) и расплавленный трет-бутанол (228 кг). Сосуд герметизировали и устанавливали температуру смеси партии до приблизительно 40-45°С и запускали мешалку. В реактор в инертной атмосфере на протяжении 2 часов и 40 минут загружали 50% водный гидроксиламин (24 кг). Во время добавления поддерживали температуру приблизительно при 40-45°С. Реакцию завершали после перемешивания в течение приблизительно дополнительных 5 часов приблизительно при 42°С.
Смесь партии охлаждали до 30-35°С и в нее загружали триэтиламин (36 кг) на протяжении 15 минут. На протяжении приблизительно 2 часов и 44 минут добавляли 2-фторбензоилхлорид (57 кг). Во время добавления поддерживали температуру смеси приблизительно 30-35°С. Смесь перемешивали в течение дополнительных 2 часов и 10 минут при 32°С и реакцию завершали.
Смесь партии нагревали до температуры флегмы (приблизительно 83-86°С) на протяжении приблизительно 50 минут и перемешивали в течение приблизительно 18 часов приблизительно при 81°С. Смесь затем охлаждали на протяжении приблизительно 2 часов до приблизительно 65-70°С и на протяжении приблизительно 6 часов и 25 минут медленно добавляли очищенную воду (315 л), в то время как температуру смеси поддерживали между приблизительно 65-70°С. Суспензию охлаждали до приблизительно 22°С на протяжении приблизительно 2 часов и 15 минут и содержимое переносили в центробежный фильтр (2 порции). Фильтрование завершали спустя приблизительно 1 час и 40 минут. Фильтровальные осадки промывали приблизительно 50% водным трет-бутанолом (90 кг на осадок) на протяжении приблизительно 20 минут. Очищенную воду (79 кг на осадок) добавляли к осадкам в качестве последней промывочной жидкости. Осадки сушили при скорости вращения 900 об./мин в течение приблизительно 1 часа и 5 минут и затем выгружали в барабан. Жидкостной хроматографией определили, что чистота сырого осадка (91,5 кг, LOD=5 масс./масс.%) была приблизительно 99,75% площади (хроматограммы).
3-[5-(2-Фторфенил)-[1,2,4]оксадиазол-3-ил]бензойная кислота
Партия 1
В реакционный сосуд загружали метил-3-[5-(2-фторфенил)-[1,2,4]оксадиазол-3-ил]бензоат (74,0 кг), сосуд герметизировали, создавали вакуум и продували. Температуру установленного места рубашки устанавливали до приблизительно 35°С и в сосуде запускали мешалку. В сосуд загружали расплавленный трет-бутанол (222 л, 3 объема) и очищенную воду (355 л, 4,8 объема). За этими загрузками следовало добавление 25,1 масс./масс.% водного раствора гидроксида натрия (43,5 кг, 1,1 мол. экв.) и линию промывали дополнительной очищенной водой (100 л, 1,35 моль). Во время добавления температуру смеси партии снижали от приблизительно 39,0°С до приблизительно 38,8°С. Температуру смеси партии повышали до приблизительно 63-67°С на протяжении приблизительно 1 часа и 54 минут и затем ее регулировали до приблизительно 68-72°С на протяжении 30 минут. Смесь перемешивали в течение приблизительно 3 часов при приблизительно 68-72°С. Раствор затем охлаждали до приблизительно 40-45°С на протяжении приблизительно 5 часов и 11 минут. Раствор затем снова нагревали до приблизительно 68-72°С по указанной выше методике на протяжении приблизительно 3 часов и 33 минуты.
Температуру рубашки на реакционном сосуде устанавливали до приблизительно 60°С, запускали мешалку и горячую жидкость пропускали через фильтр с отверстиями 1 микрон приблизительно при 70°С при слабом избыточном давлении азота (10,341-38,606 кПа). Температуру продукта снижали до приблизительно 64,3°С во время пропускания, которое завершали через приблизительно 45 минут. В сосуд загружали очищенную воду (61 л, 0,82 объема) и содержимое нагревали до приблизительно 68-72°С.
Температуру смеси регулировали до приблизительно 69,4°С и обрабатывали 13,9 масс./масс.% серной кислоты (100,7 кг, 1,15 мол. экв.) на протяжении приблизительно 4 часов и 18 минут. Во время добавления температуру смеси поддерживали приблизительно 68,0-70,8°С. За добавлением кислоты следовало промывание линии очищенной водой (50 л, 0,68 объема) и перемешивание приблизительно при 68-72°С продолжали в течение приблизительно дополнительных 31 минут.
Смесь охлаждали линейным образом от приблизительно 69,2°С до приблизительно 41,2°С на протяжении приблизительно 4 часов и 10 минут. Мешалку на системе Розенмунда фильтр/сушилка поднимали до самого высокого положения и температуру заданного места рубашки устанавливали приблизительно у 40°С. Суспензию в виде двух частей переносили в систему фильтр/сушилка. Для первой части применяли постоянное давление азота (меньше чем приблизительно 10,341 кПа). Во время переноса давление составляло от приблизительно 16,477 до приблизительно 19,855 кПа, перенос завершали через приблизительно 1 час и 5 минут. Вторую часть суспензии переносили на верх фильтровального осадка и композит быстро перемешивали для гомогенизации части. Вторую часть фильтровали с применением давления азота от приблизительно 17,993 до приблизительно 20,061 кПа и осадок освобождали от жидкости спустя приблизительно 3 часа. Осадок промывали горячим водным раствором трет-бутанола (352 кг, 5 объемов) приблизительно при 38-42°С и 3 раза горячей очищенной водой (370 л, 5 объемов) приблизительно при 65-70°С.
Температуру рубашки системы фильтр/сушилка устанавливали приблизительно при 43°С и продукт сушили в вакууме с периодическим перемешиванием на протяжении приблизительно 26 часов. Определили, что чистота была приблизительно 99,7%. Общий выход выделенного продукта был приблизительно 74,4% (52,45 кг).
Партия 2
В сосуд реактора загружали метил-3-[5-(2-фторфенил)-[1,2,4]оксадиазол-3-ил]бензоат (47 кг, влажный осадок)) и расплавленный трет-бутанол (111,4 кг). Сосуд герметизировали и устанавливали температуру смеси партии до 30-40°С и запускали мешалку. В сосуд загружали очищенную воду (51,6 кг). За этой загрузкой следовало добавление 3,47 масс./масс.% водного раствора гидроксида натрия (202,4 кг). Температуру смеси партии повышали до приблизительно 67-73°С на протяжении приблизительно 1 часа и затем перемешивали в течение приблизительно 3 часов приблизительно при 70°С.
Смесь партии фильтровали через полипропиленовый фильтровальный мешок с отверстиями 1 микрон под слабым избыточным давлением азота и затем переносили в другой реактор. В сосуд загружали очищенную воду (146 кг) и смесь нагревали до приблизительно 68-72°С.
В партию загружали 10,7% водную хлористоводородную кислоту на протяжении приблизительно 4 часов. Температуру смеси партии во время добавления поддерживали при приблизительно 68-72°С. рН-метром определили, что рН партии было приблизительно 2,2, и перемешивание продолжали приблизительно при 70°С в течение приблизительно дополнительного 1 часа.
Смесь партии охлаждали линейным образом от 70°С до приблизительно 60°С на протяжении приблизительно 2 часов. Партию при приблизительно 60°С охлаждали линейным образом от 60°С до приблизительно 40°С на протяжении приблизительно 2 часов. Смесь перемешивали в течение дополнительных 2 часов при 40°С и суспензию переносили в центрифужный фильтр. Фильтрование завершали спустя приблизительно 30 минут. Фильтровальные осадки промывали приблизительно 42 масс./масс.% водным трет-бутанолом (165 кг) на протяжении приблизительно 30 минут. К осадкам в качестве последней промывочной жидкости добавляли очищенную воду (118 кг, 40°С). Осадки сушили приблизительно при вращении 900 об./мин в течение приблизительно 1 часа и затем выгружают в барабан.
Сырые осадки переносили в лопастную сушилку (для этой стадии подходящей является также сушилка с двумя конусами) и температуру рубашки устанавливали приблизительно 70°С. Продукт сушили в вакууме при приблизительно 70°С с периодическим перемешиванием на протяжении приблизительно 48 часов. Определили, что чистота была приблизительно 99,8°С. Общий выход выделенного продукта был приблизительно 74% (68,5 кг).
Партия 3
В реакционный сосуд загружали метил-3-[5-(2-фторфенил)-[1,2,4]оксадиазол-3-ил]бензоат (10 г) и расплавленный трет-бутанол (128 мл). Температуру смеси партии устанавливали до 30-40°С и запускали мешалку. В сосуд на протяжении приблизительно 30 минут загружали 4,48 масс./масс.% водного раствора гидроксида натрия (32,5 г). Температуру смеси партии поддерживали приблизительно 40-50°С. Температуру смеси повышали до приблизительно 78-82°С на протяжении приблизительно 1 часа и затем перемешивали в течение приблизительно дополнительного 1 часа приблизительно при 78-82°С. Партию фильтровали через полиэтиленовый фильтр с отверстиями 5 мкм при слабом избыточном давлении азота и затем переносили в другой реактор. Температуру партии поддерживали приблизительно при 78-82°С.
Другой сосуд загружали 37% водной хлористоводородной кислотой (4 мл) и расплавленным трет-бутанолом (8 мл). Температуру поддерживали приблизительно при 30-40°С и смесь перемешивали в течение приблизительно 30 минут.
В смесь партии загружали хлористоводородную кислоту в трет-бутаноле на протяжении приблизительно 4 часов с применением дозирующего насоса. Первую половину загрузки добавляли на протяжении приблизительно 20-30 минут. Скорость мешалки устанавливали до приблизительно 200 об./мин. Оставшуюся часть загрузки добавляли на протяжении приблизительно 3,5 часа. Скорость мешалки устанавливали до приблизительно 100 об./мин. Температуру партии во время добавления поддерживали приблизительно при 78-82°С. рН конечной партии регулировали до приблизительно 1,2 с применением рН-метра и перемешивание продолжали приблизительно при 78-82°С в течение приблизительно дополнительного 1 часа.
Партию охлаждали линейным образом от 78-82°С до приблизительно 70°С на протяжении приблизительно 1 часа. Партию при приблизительно 70°С охлаждали линейным образом от 70°С до приблизительно 50°С на протяжении приблизительно 4 часов и скорость мешалки устанавливали при приблизительно 80 об./мин. Партию при приблизительно 50°С охлаждали линейным образом от 50°С до приблизительно 40°С на протяжении приблизительно 4 часов и скорость мешалки устанавливали при приблизительно 60 об./мин. Партию перемешивали в течение дополнительных 4 часов при 40°С.
Температуру фильтра устанавливали до приблизительно 40-45°С. Суспензию переносили на фильтр. Фильтрование завершили спустя приблизительно 1 минуту. Фильтровальные осадки промывали трет-бутанолом (50 мл, 50°С) на протяжении 2 минут. В качестве последней промывочной жидкости к осадкам добавляли очищенную воду (100 мл × 2, 60°С). Осадки сушили приблизительно при 60-70°С в вакууме в течение приблизительно 12 часов и затем разгружали в контейнер.
Определили, что чистота по ВЭЖХ была приблизительно 99,9% площади (хроматограммы). Выход выделенного продукта был приблизительно 94% (9,0 г).
3-[5-(2-Фторфенил)-[1,2,4]оксадиазол-3-ил]бензойная кислота: способ получения в одном реакторе
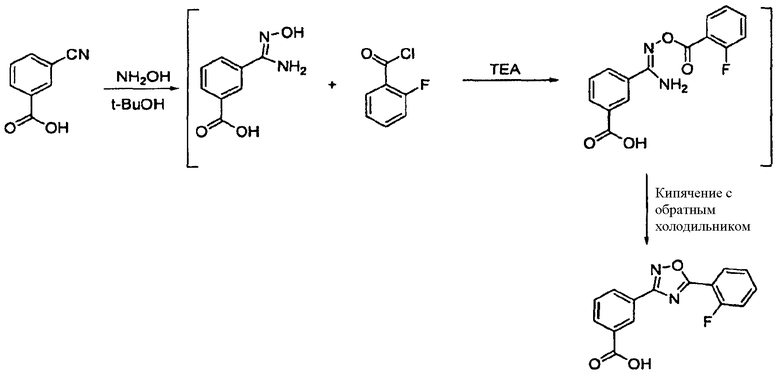
В сосуд реактора загружали 3-цианобензойную кислоту (7,35 г) и расплавленный трет-бутанол (100 мл). Сосуд герметизировали и температуру смеси загрузки устанавливали до 60°С и запускали смеситель. Суспензию перемешивали в течение 1 час и затем температуру смеси устанавливали до 40°С. В реактор в инертной атмосфере на протяжении 3 часов загружали 50% водный гидроксиламин (3,63 г). Во время добавления температуру смеси поддерживали при 38-41°С. Реакцию завершали после перемешивания в течение 18 часов при 40°С.
Партию охлаждали до 27°С и загружали триэтиламин (5,56 г) на протяжении 2 минут. На протяжении 3 часов добавляли 2-фторбензоилхлорид (7,82 г). Во время добавления температуру смеси поддерживали при 24-27°С. Смесь перемешивали в течение дополнительных 4 часов при 40°С.
Партию нагревали до 79°С на протяжении 30 минут и перемешивали в течение 16 часов приблизительно при 79°С. К белой суспензии на протяжении 3 часов добавляли воду (100 мл) при поддержании температуры партии при 70°С. В смесь загружали 37% водную хлористоводородную кислоту на протяжении 20 минут. рН-метром определили, что значение рН смеси было приблизительно 2,2, и перемешивание продолжали приблизительно при 70°С в течение приблизительно дополнительного 1 часа.
Партию охлаждали линейным образом от 70°С до 30°С на протяжении 3 часов и суспензию переносили в фильтр. Фильтрацию завершили через 5 минут. Фильтровальные осадки промывали трет-бутанолом (50 мл, 40°С) на протяжении 5 минут. В качестве последней промывочной жидкости к осадкам добавляли очищенную воду (100 мл, 60°С). Осадки сушили в вакуумном сушильном шкафу при 70°С в течение 18 часов и затем выгружали. Определили, что чистота была приблизительно 98,68%. Общий выход выделенного продукта был приблизительно 76% (10,8 г).
Специалисту в данной области должно быть понятно, что с применением не более чем обычного экспериментирования можно предложить много эквивалентов к определенным вариантам осуществления описанного здесь изобретения или он сам способен предложить такие варианты. Предполагается, что такие эквиваленты включаются нижеследующей формулой изобретения. Все публикации, патенты и заявки на патенты, указанные в данном описании, включены здесь в качестве ссылки в описание до такой степени, что как если бы каждая отдельная публикация, патент или заявка на патент была бы специально и отдельно указана как включенная здесь в качестве ссылки.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ СИНТЕЗА АНАЛОГОВ ГОРМОНОВ ЩИТОВИДНОЙ ЖЕЛЕЗЫ И ИХ ПОЛИМОРФОВ | 2013 |
|
RU2668960C2 |
| ПОЛУЧЕНИЕ 2-(5-БРОМ-4-(4-ЦИКЛОПРОПИЛНАФТАЛИН-1-ИЛ)-4H-1,2,4-ТРИАЗОЛ-3-ИЛТИО)УКСУСНОЙ КИСЛОТЫ | 2013 |
|
RU2666549C2 |
| СПОСОБ ПОЛУЧЕНИЯ АРИЛТРИАЗОЛИНОНА | 1993 |
|
RU2119918C1 |
| СПОСОБ СИНТЕЗА АНАЛОГОВ ГОРМОНОВ ЩИТОВИДНОЙ ЖЕЛЕЗЫ И ИХ ПОЛИМОРФОВ | 2021 |
|
RU2802282C2 |
| Новый способ получения эверолимуса | 2019 |
|
RU2716714C1 |
| СПОСОБЫ ПОЛУЧЕНИЯ ФУНКЦИОНАЛЬНОГО БЕЛКА ИЗ ДНК, ИМЕЮЩЕЙ НОНСЕНС-МУТАЦИЮ, И ЛЕЧЕНИЯ НАРУШЕНИЙ, АССОЦИИРОВАННЫХ С НЕЙ | 2007 |
|
RU2462246C2 |
| ФАРМАЦЕВТИЧЕСКИЕ ПРОДУКТЫ ДЛЯ ЛЕЧЕНИЯ И ПРОФИЛАКТИКИ ЗАБОЛЕВАНИЙ, ПОЯВЛЯЮЩИХСЯ В РЕЗУЛЬТАТЕ ПОВРЕЖДЕНИЯ ЭНДОТЕЛИАЛЬНЫХ КЛЕТОК СОСУДОВ | 1997 |
|
RU2195273C2 |
| СПОСОБЫ СИНТЕЗА 2-АМИНО-4,6-ДИМЕТОКСИБЕНЗАМИДА И ДРУГИХ БЕНЗАМИДНЫХ СОЕДИНЕНИЙ | 2013 |
|
RU2650110C2 |
| СПОСОБ ПОЛУЧЕНИЯ [(3-ГИДРОКСИПИРИДИН-2-КАРБОНИЛ)АМИНО]АЛКАНОВЫХ КИСЛОТ, СЛОЖНЫХ ЭФИРОВ И АМИДОВ | 2012 |
|
RU2764667C2 |
| СПОСОБ ПОЛУЧЕНИЯ [(3-ГИДРОКСИПИРИДИН-2-КАРБОНИЛ)АМИНО]АЛКАНОВЫХ КИСЛОТ, СЛОЖНЫХ ЭФИРОВ И АМИДОВ | 2012 |
|
RU2602083C2 |
Изобретение относится к способам синтеза 1,2,4-оксадиазолов формулы:
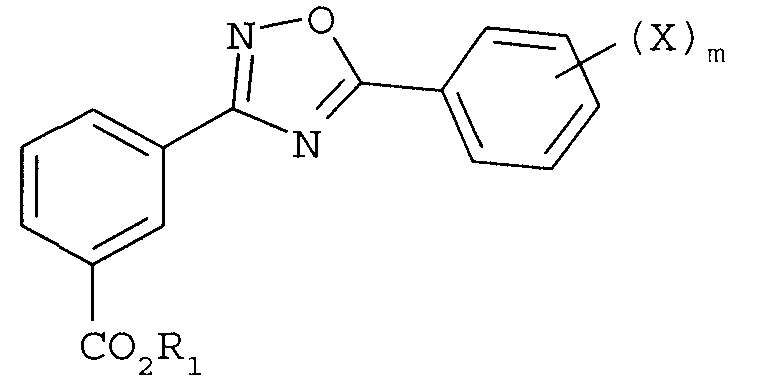
где R1 представляет собой замещенный или незамещенный алкил или -(CH2CH2O)nR6; R6 представляет собой водород, замещенный или незамещенный алкил, X в каждом случае независимо представляет собой F, Cl, Br или I, n равно целому числу от 1 до 7 и m равно целому числу от 1 до 5, которые применяют для получения 3-[5-(2-фторфенил-[1,2,4]оксадиазол-3-ил]бензойной кислоты. 3 н. и 17 з.п. ф-лы.
1. Способ получения соединения формулы
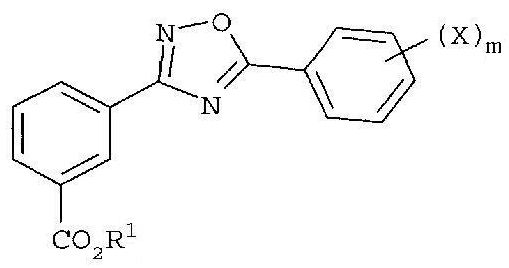
или его фармацевтически приемлемой соли, где
R1 представляет собой замещенный или незамещенный алкил или -(CH2CH2O)nR6;
R6 представляют собой водород, замещенный или незамещенный алкил,
X в каждом случае независимо представляет собой F, Cl, Br или I,
n равно целому числу от 1 до 7, и
m равно целому числу от 1 до 5,
включающий в себя проведение следующих стадий в том же самом растворителе:
(1) реакцию эфира цианобензойной кислоты
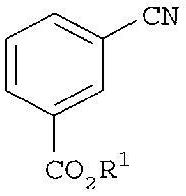
с гидроксиламином с получением
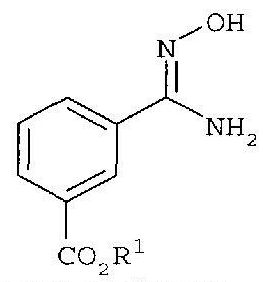
последующее (2) ацилирование галогенбензоилхлоридом с получением
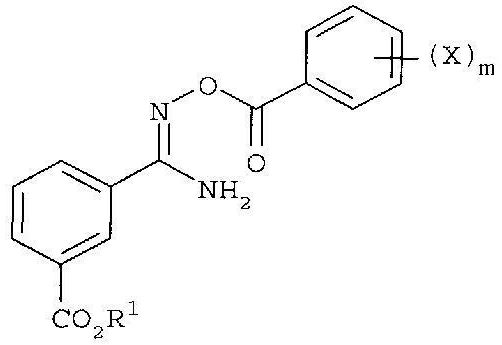
и с последующей (3) конденсацией.
2. Способ по п.1, где X представляет собой F.
3. Способ по п.1, где m равно 1.
4. Способ по п.1, где X представляет собой F и m равно 1.
5. Способ по п.1, где R1 представляет собой метил.
6. Способ по п.1, где стадии (1)-(3) проводят без выделения промежуточного продукта.
7. Способ по п.6, где растворителем является трет-бутанол.
8. Способ по п.1, где галогенбензоилхлоридом является 2-фторбензоилхлорид.
9. Способ по п.1, дополнительно включающий (4) гидролиз сложного эфира соединения формулы
с получением соединения
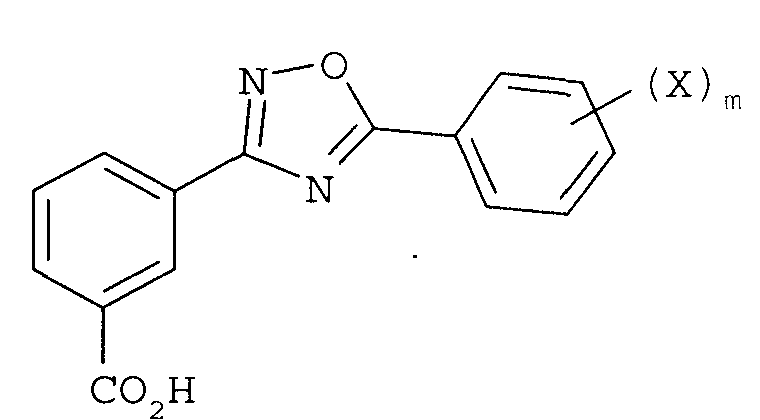
или его фармацевтически приемлемой соли, где
R1 представляет собой замещенный или незамещенный алкил или -(CH2CH2O)nR6;
R6 представляют собой водород, замещенный или незамещенный алкил,
X в каждом случае независимо представляет собой F, Cl, Br или I,
n равно целому числу от 1 до 7, и
m равно целому числу от 1 до 5.
10. Способ по п.9, где гидролиз проводят в трет-бутаноле.
11. Способ по п.10, где R1 представляет собой метил.
12. Способ получения соединения формулы
или его фармацевтически приемлемой соли,
включающий в себя проведение нижеследующих стадий без выделения промежуточного продукта:
(1) реакцию метилового эфира цианобензойной кислоты
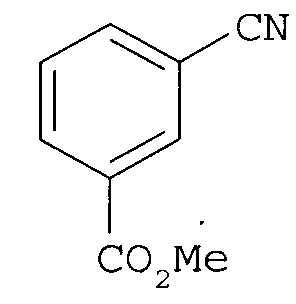
с гидроксиламином с получением
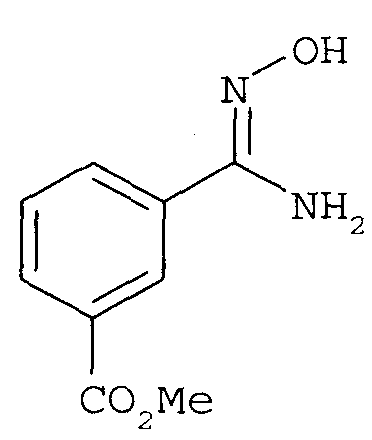
последующее (2) ацилирование 2-фторбензоилхлоридом с получением
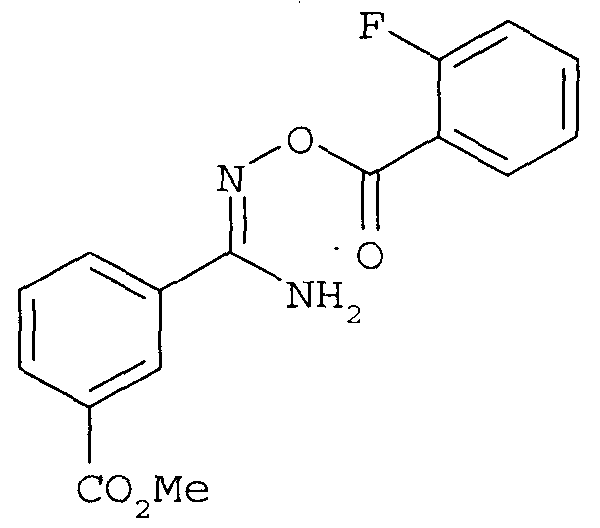
с последующей (3) конденсацией.
13. Способ по п.12, где стадии (1)-(3) проводят в том же самом органическом растворителе.
14. Способ по п.13, где растворителем является трет-бутанол.
15. Способ по п.12, дополнительно включающий
(4) гидролиз сложного метилового эфира соединения формулы
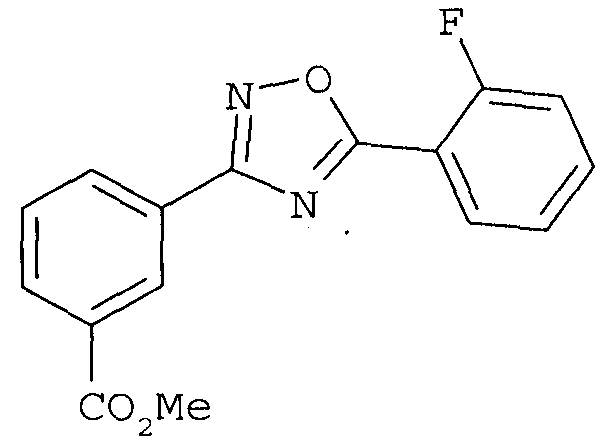
с получением соединения
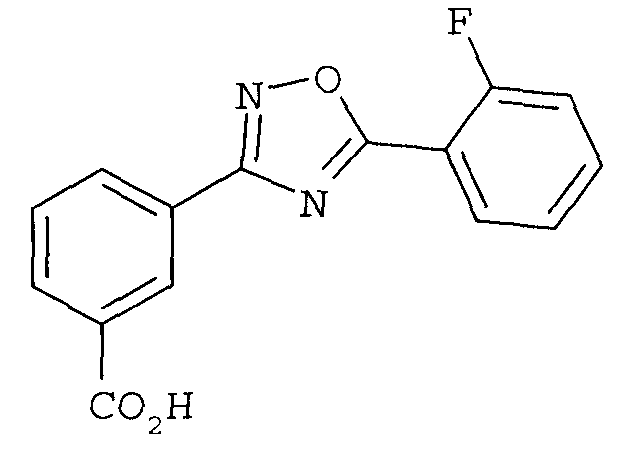
или его фармацевтически приемлемой соли.
16. Способ получения соединения формулы
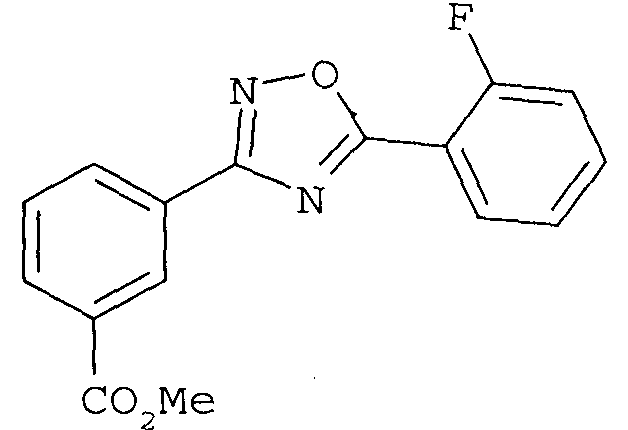
или его фармацевтически приемлемой соли,
включающий проведение нижеследующих стадий в одном и том же органическом растворителе:
(1) реакцию метилового эфира цианобензойной кислоты
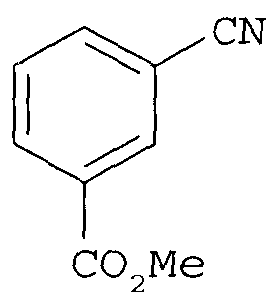
с гидроксиламином с получением
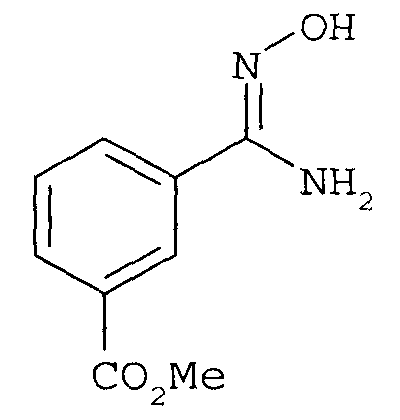
последующее (2) ацилирование 2-фторбензоилхлоридом с получением
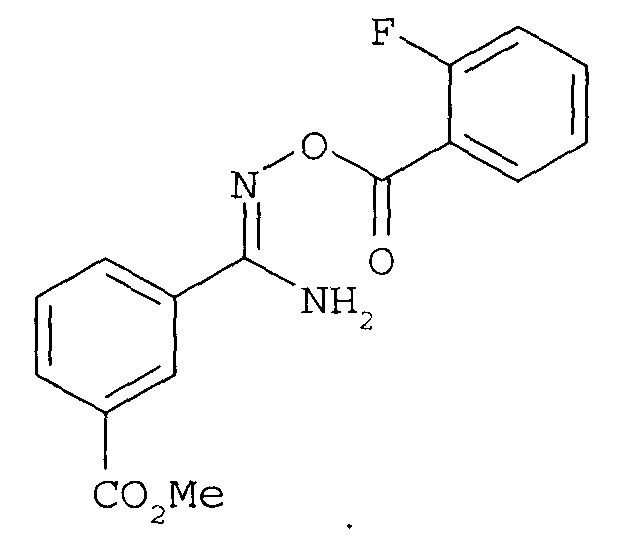
с последующей (3) конденсацией.
17. Способ по п.16, где растворителем является трет-бутанол.
18. Способ по п.16, где стадии (1)-(3) проводят без выделения промежуточного продукта.
19. Способ по п.16, дополнительно включающий (4) гидролиз метилового сложного эфира формулы
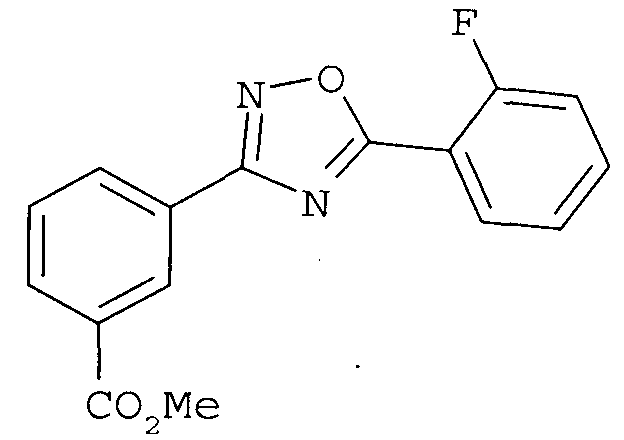
с получением соединения
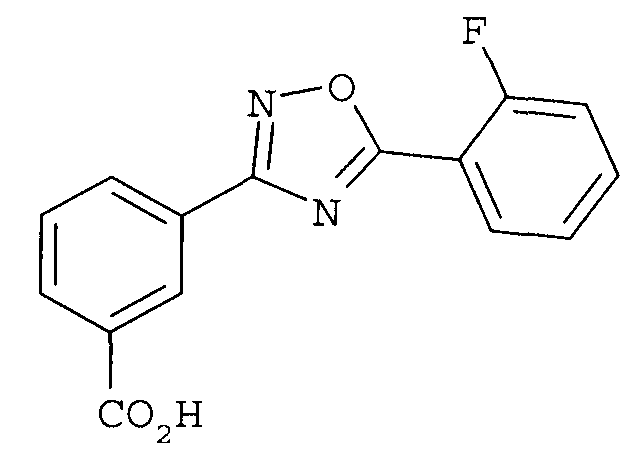
или его фармацевтически приемлемой соли.
20. Способ по п.19, где гидролиз проводят в трет-бутаноле.
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| Способ получения производных 1,2,4-оксадиазола (его вариант) | 1979 |
|
SU969162A3 |

