ОБЛАСТЬ ТЕХНИКИ
[0001] Настоящее изобретение относится к способам синтеза 2-амино-4,6-диметоксибензамида и других бензамидных соединений.
УРОВЕНЬ ТЕХНИКИ
[0002] Бензамидные соединения используют в качестве промежуточных соединений для синтеза многих фармацевтических терапевтических лекарственных средств. В частности, 2-амино-4,6-диметоксибензамид отмечен в качестве промежуточного соединения в патенте США № 3966965 при синтезе производных оксаминовой кислоты для предотвращения при аллергических реакциях гиперчувствительности. В последних патентах это соединение описано как чрезвычайно важное промежуточное соединение для новых сердечно-сосудистых средств (U.S. 2008/0188467 и WO 2008/92231, принадлежащие Resverlogix Corp.).
[0003] 2-амино-4,6-диметоксибензамид получен из ангидрида 4,6-диметокси-N-карбоксиантраниловой кислоты. Ангидрид 4,6-диметокси-N-карбоксиантраниловой кислоты, в свою очередь, получали посредством реакции 4,6-диметоксиантраниловой кислоты с фосгеном (патент США № 4191840 и Org. Synth. 1947, 27, 45). По другому способу 3,5-диметоксианилин преобразуют в его гидрохлоридную соль, после этого соль вступает в реакцию с оксалилхлоридом для получения 4,6-диметокси-изатина. Изатин преобразуют в целевое соединение с помощью нестабильного карбоксильного промежуточного соединения посредством реакции с гидроксидом натрия и пероксидом водорода с последующим EDCI/HOBt-опосредованным связыванием для получения 2-амино-4,6-диметоксибензамида (WO 2008/92231).
[0004] Известные способы синтеза бензамидных соединений и их производных для коммерческого применения часто включают нестабильные промежуточные соединения, неэффективные способы и в некоторых случаях большое количество стадий, которые приводят к неприемлемо низкому выходу и неприемлемо высоким издержкам производства. Существует необходимость в данной области в коммерчески целесообразных способах получения бензамидных соединений и их производных.
ИЗОБРЕТЕНИЕ
[0005] Настоящее изобретение соответствует вышеуказанным требованиям посредством обеспечения способов получения, по меньшей мере, одного
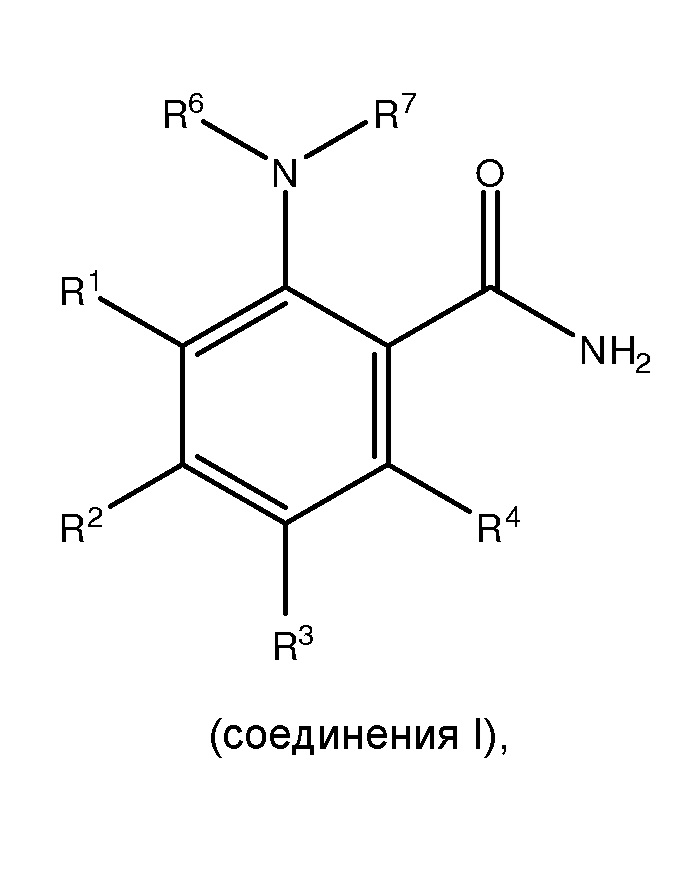 ,
,
где способ включает:
(i) сочетание, по меньшей мере, одного
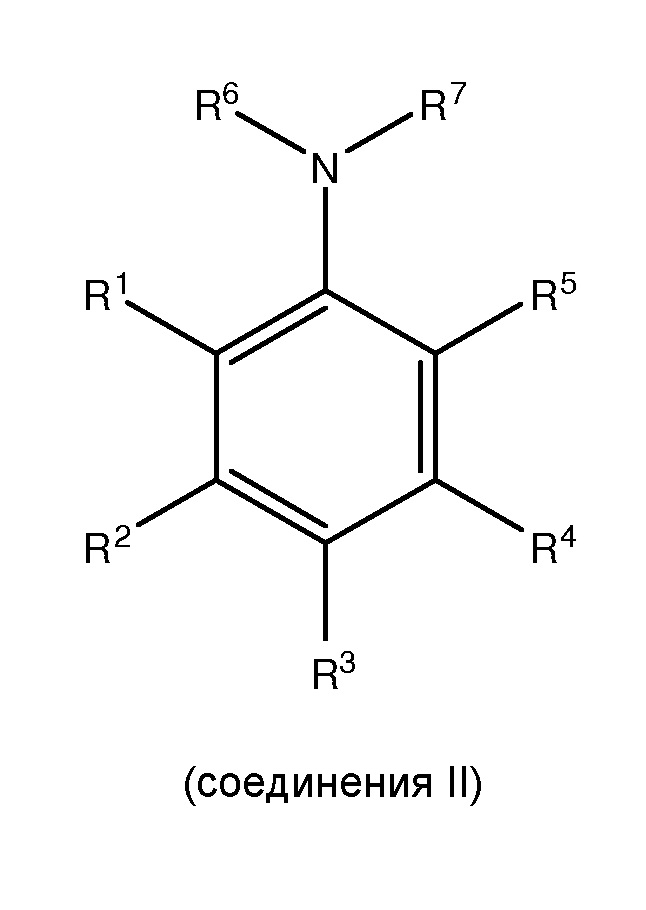
с, по меньшей мере, одним галогенирующим агентом для получения, по меньшей мере, одного
 ;
;
(ii) сочетание, по меньшей мере, одного соединения III с, по меньшей мере, одним цианирующим агентом для получения, по меньшей мере, одного
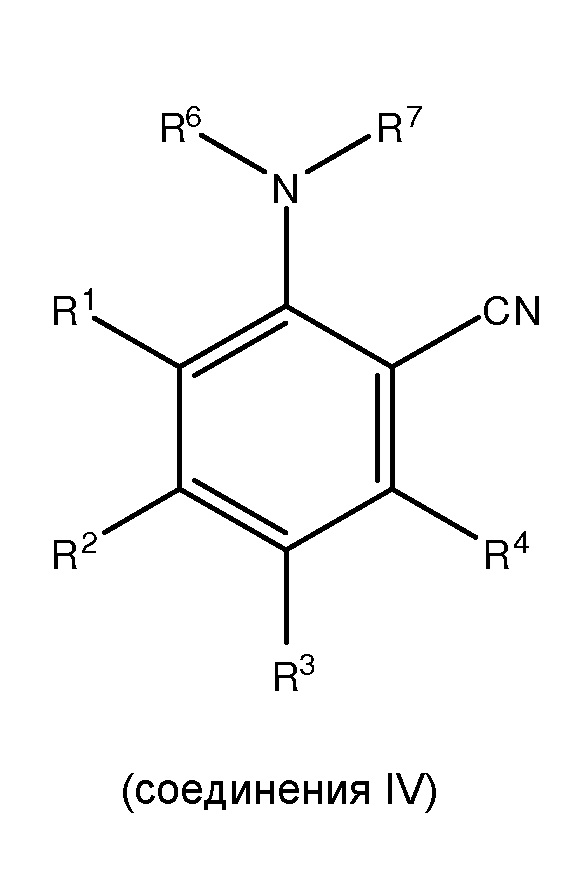 ;
;
и
(iii) сочетание, по меньшей мере, одного соединения IV с, по меньшей мере, одним гидратирующим агентом и/или, по меньшей мере, одним гидратирующим катализатором для получения, по меньшей мере, одного соединения I,
где каждый из R1 и R5 независимо представляет собой водород, C1-C6 алкил, гидрокси или C1-C6 алкокси;
где каждый из R2, R3 и R4 независимо представляет собой водород, C1-C6 алкил или C1-C6 алкокси;
где каждый из R6 и R7 независимо представляет собой водород, C1-C6 алкил, защитную группу или направляющую группу;
где X представляет собой молекулу галогена или галогеноподобную молекулу;
где, по меньшей мере, один из R1 и R5 представляет собой водород; и
при условии, что в соединении II R7 представляет собой защитную группу или направляющую группу, и или
(1) перед началом стадии (ii), по меньшей мере, одно соединение III сочетают с, по меньшей мере, одним агентом для снятия защитной группы так, чтобы защитная группа или направляющая группа, представленная R7, была заменена водородом или C1-C6 алкилом, или
(2) перед началом стадии (iii), по меньшей мере, одно соединение IV сочетают с, по меньшей мере, одним агентом для снятия защитной группы, так, чтобы защитная группа или направляющая группа, представленная R7, была заменена водородом или C1-C6 алкилом.
[0006] Настоящее изобретение также соответствует вышеуказанным требованиям посредством обеспечения способов для получения, по меньшей мере, одного
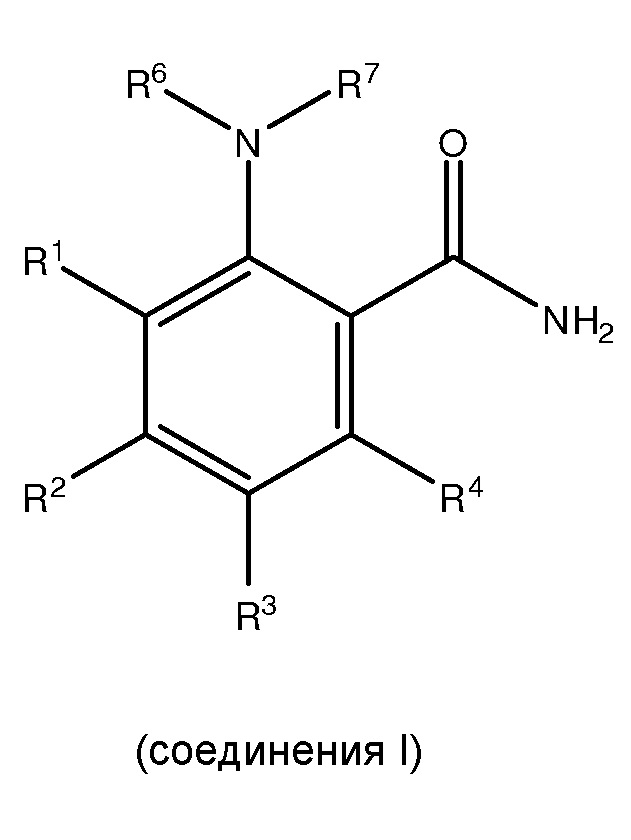 ,
,
где способ включает: (i) сочетание, по меньшей мере, одного
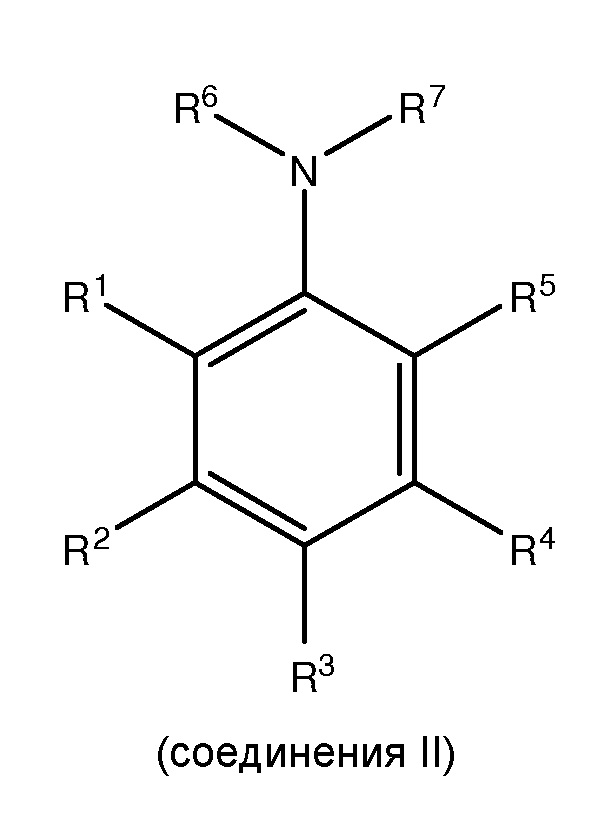
с, по меньшей мере, одним галогенирующим агентом для получения, по меньшей мере, одного
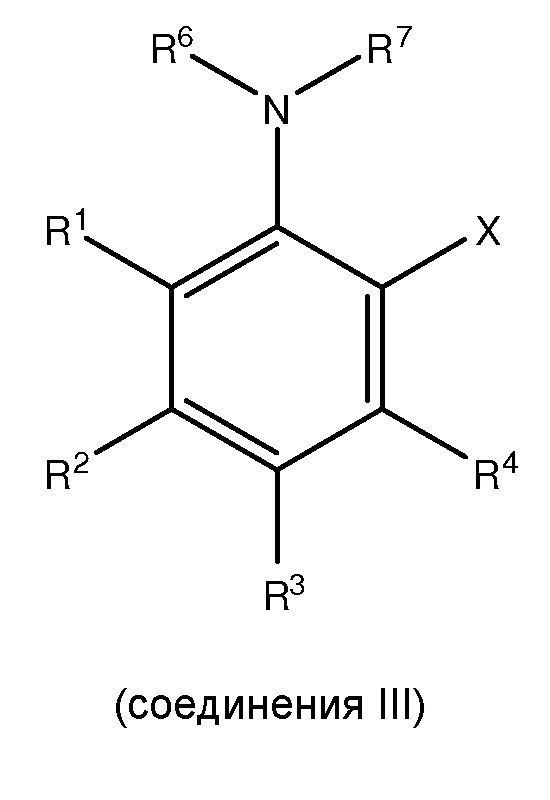 ;
;
(ii) сочетание, по меньшей мере, одного соединения III с, по меньшей мере, одним цианирующим агентом для получения, по меньшей мере, одного
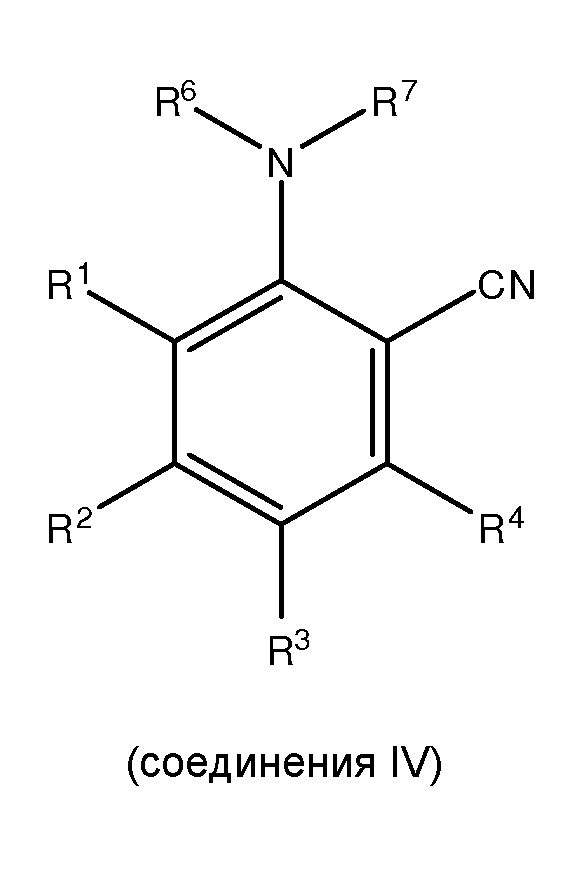 ;
;
(iii) сочетание, по меньшей мере, одного соединения IV с, по меньшей мере, одним первым осаждающим агентом; и
(iv) сочетание, по меньшей мере, одного соединения IV с, по меньшей мере, одним гидратирующим агентом и/или, по меньшей мере, одним гидратирующим катализатором для получения, по меньшей мере, одного соединения I,
где каждый из R1 и R5 независимо представляет собой водород, C1-C6 алкил, гидрокси или C1-C6 алкокси;
где каждый из R2, R3 и R4 независимо представляет собой водород, C1-C6 алкил или C1-C6 алкокси;
где каждый из R6 и R7 независимо представляет собой водород, C1-C6 алкил, защитную группу или направляющую группу;
где X представляет собой молекулу галогена или галогеноподобную молекулу; где, по меньшей мере, один из R1 и R5 представляет собой водород; и при условии, что в соединении II R представляет собой защитную группу или направляющую группу, и или
(1) перед началом стадии (ii), по меньшей мере, одно соединение III сочетают с, по меньшей мере, одним агентом для снятия защитной группы так, чтобы защитная группа или направляющая группа, представленная R7, была заменена водородом или C1-C6 алкилом, или
(2) перед началом стадии (iii), по меньшей мере, одно соединение IV сочетают с, по меньшей мере, одним агентом для снятия защитной группы так, чтобы защитная группа или направляющая группа, представленная R7, была заменена водородом или C1-C6 алкилом.
[0007] Способы, где перед началом стадии (iv), по меньшей мере, одно соединение IV сочетают с, по меньшей мере, одним вторым осаждающим агентом для осаждения, по меньшей мере, одного соединения IV.
[0008] Кроме того, представлены такие способы, где перед началом стадии (ii), по меньшей мере, одно соединение III сочетают с, по меньшей мере, одним агентом для снятия защитной группы так, чтобы защитная группа или направляющая группа, представленная R7, была заменена водородом или C1-C6 алкилом, и, по меньшей мере, одно соединение III сочетают с, по меньшей мере, одним третьим осаждающим агентом для осаждения, по меньшей мере, одного соединения III.
[0009] Также представлены такие способы, где до начала стадии (iii), по меньшей мере, одно соединение III сочетают с, по меньшей мере, одним агентом для снятия защитной группы так, чтобы защитная группа или направляющая группа, представленная R7, была заменена водородом или C1-C6 алкилом, и по меньшей мере, одно соединение IV сочетают с, по меньшей мере, одним третьим осаждающим агентом для осаждения, по меньшей мере, одного соединения IV.
[0010] Также представлены такие способы, где, по меньшей мере, одно соединение II получают посредством способа, включающего сочетание, по меньшей мере, одного
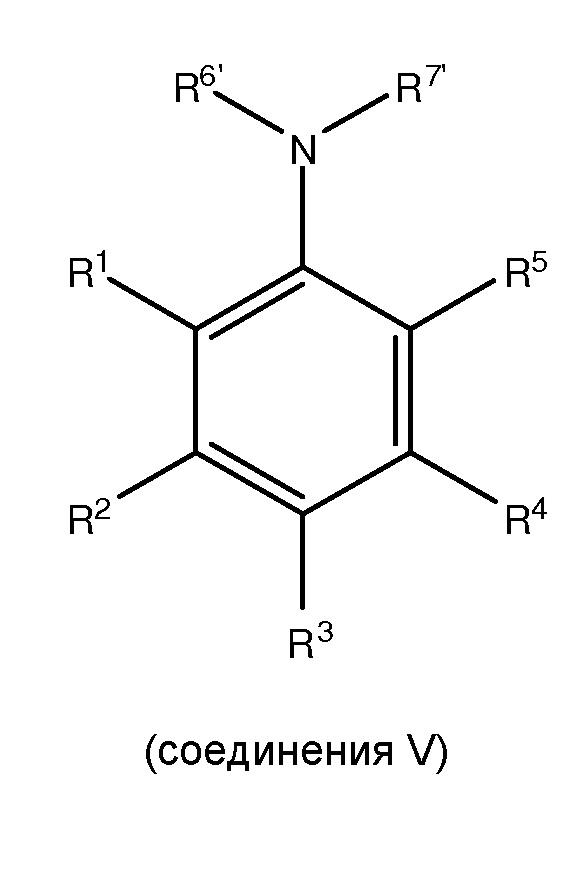
с, по меньшей мере, одним защитным агентом для получения, по меньшей мере, одного соединения II,
где в соединении V каждый из R1 и R5 независимо представляет собой водород, C1-C6 алкил, гидрокси или C1-C6 алкокси; каждый из R2, R3 и R4 независимо представляет собой водород, C1-C6 алкил или C1-C6 алкокси; и каждый из R6’ и R7’ независимо представляет собой водород или C1-C6 алкил.
[0011] В одном из вариантов осуществления настоящего изобретения X представляет собой молекулу галогена или галогеноподобную молекулу.
[0012] В другом варианте осуществления, по меньшей мере, один из R1, R2, R3, R4 или R5 представляет собой C1-C6 алкокси.
[0013] В еще одном варианте осуществления, по меньшей мере, два из R1, R2, R3, R4 и R5 каждый независимо представляет собой C1-C6 алкокси.
[0014] В еще одном варианте осуществления, по меньшей мере, три из R1, R2, R3, R4 и R5 каждый независимо представляет собой C1-C6 алкокси.
[0015] В другом варианте осуществления каждый из R2 и R4 независимо представляет собой C1-C6 алкокси, и каждый из R1 и R3 представляет собой водород.
[0016] В одном из вариантов осуществления C1-C6 алкокси является метокси.
[0017] В одном из вариантов осуществления в соединении II каждый из R2 и R4 независимо представляет собой C1-C6 алкокси, каждый из R1 и R3 представляет собой водород, и R7 представляет собой трифторацетил.
[0018] В одном из вариантов осуществления способ по настоящему изобретению включает:
(i) присоединение защитной группы к 3,5-диметоксианилину с помощью защитного агента для получения 3,5-диметоксианилина с присоединенной защитной группой;
(ii) галогенирование 3,5-диметоксианилина с присоединенной защитной группой с помощью галогенирующего агента для получения галогенированного 3,5-диметоксианилина с присоединенной защитной группой;
(iii) цианирование галогенированного 3,5-диметоксианилина с присоединенной защитной группой с помощью цианирующего агента для получения цианированного 3,5-диметоксианилина с присоединенной защитной группой;
(iv) снятие защитной группы с цианированного 3,5-диметоксианилина с присоединенной защитной группой; и
(v) гидратирование цианированного 3,5-диметоксианилина со снятой защитной группой для получения 2-амино-4,6-диметоксибензамида.
[0019] В одном из вариантов осуществления способ по настоящему изобретению включает:
(i) присоединение защитной группы к 3,5-диметоксианилину с помощью защитного агента для получения 3,5-диметоксианилина с присоединенной защитной группой;
(ii) галогенирование 3,5-диметоксианилина с присоединенной защитной группой с помощью галогенирующего агента для получения галогенированного 3,5-диметоксианилина с присоединенной защитной группой;
(iii) цианирование галогенированного 3,5-диметоксианилина с присоединенной защитной группой с помощью цианирующего агента для получения цианированного 3,5-диметоксианилина с присоединенной защитной группой;
(iv) снятие защитной группы с цианированного 3,5-диметоксианилина с присоединенной защитной группой и индуцирование осаждения для получения осадка, содержащего цианированный 3,5-диметоксианилин; и
(v) гидратирование осадка, содержащего цианированный 3,5-диметоксианилин, для получения 2-амино-4,6-диметоксибензамида.
[0020] В одном из вариантов осуществления способ по настоящему изобретению включает:
(i) присоединение защитной группы к 3,5-диметоксианилину с помощью защитного агента для получения 3,5-диметоксианилина с присоединенной защитной группой;
(ii) галогенирование 3,5-диметоксианилина с присоединенной защитной группой с помощью галогенирующего агента для получения галогенированного 3,5-диметоксианилина с присоединенной защитной группой;
(iii) цианирование галогенированного 3,5-диметоксианилина с присоединенной защитной группой с помощью цианирующего агента для получения цианированного 3,5-диметоксианилина с присоединенной защитной группой;
(iv) снятие защитной группы с и осаждение цианированного 3,5-диметоксианилина с присоединенной защитной группой для получения первичного осадка, содержащего цианированный 3,5-диметоксианилин;
(v) повторное осаждение первичного осадка, содержащего цианированный 3,5-диметоксианилин, для получения вторичного осадка, содержащего цианированный 3,5-диметоксианилин; и
(vi) гидратирование вторичного осадка, содержащего цианированный 3,5-диметоксианилин, для получения 2-амино-4,6-диметоксибензамида.
[0021] В другом варианте осуществления способ по настоящему изобретению включает:
(i) присоединение защитной группы к 3,5-диметоксианилину с помощью защитного агента для получения 3,5-диметоксианилина с присоединенной защитной группой;
(ii) галогенирование 3,5-диметоксианилина с присоединенной защитной группой с помощью галогенирующего средства для получения галогенированного 3,5-диметоксианилина с присоединенной защитной группой;
(iii) снятие защитной группы с галогенированного 3,5-диметоксианилина с присоединенной защитной группой для получения галогенированного 3,5-диметоксианилина;
(iv) осаждение галогенированного 3,5-диметоксианилина;
(v) цианирование галогенированного 3,5-диметоксианилина с помощью цианирующего агента для получения цианированного 3,5-диметоксианилина;
(vi) осаждение цианированного 3,5-диметоксианилина и
(vii) гидратирование осажденного цианированного 3,5-диметоксианилина для получения 2-амино-4,6-диметоксибензамида.
[0022] В рамках изобретения «циано» или «нитрил» относится к -CN.
[0023] В рамках изобретения молекула галогена или галогеноподобная молекула относится к хлору (Cl), брому (Br), йоду (I), трифлату (-OTf), тозилату (-OTs) или мезилату (-OMs).
[0024] В рамках изобретения «алкил» относится к насыщенному неразветвленному, разветвленному или циклическому, первичному, вторичному или третичному углеводороду.
[0025] В рамках изобретения «гидрокси» относится к гидроксильной группе (-OH).
[0026] В рамках изобретения, «алкокси» или «алкилокси» относится к молекуле алкила с присоединенной к ней молекулой кислорода, например, метокси, этокси, н-пропокси, сек-бутокси, т-бутокси, пентокси, н-гексилокси и т.п.
[0027] В рамках изобретения «карбоксамид» или «амид» относится к -CONH2.
[0028] В рамках изобретения «промежуточное соединение» относится к любому из соединений II, III или IV.
[0029] В другом аспекте изобретение относится к способам осаждения и/или повторного осаждения, по меньшей мере, одного промежуточного соединения, полученного при синтезе соединения I.
[0030] Изобретение также относится к способу синтеза соединения I, включающему проведение двух или более стадий однореакторного способа.
[0031] В одном из вариантов осуществления стадии присоединение защитной группы, галогенирование, цианирование, и снятие защитной группы, как описано в настоящем документе, проводят в однореакторном способе.
[0032] Следует понимать, что специалист в данной области органического синтеза может руководствоваться способами, описанными или приведенными в качестве примера в настоящем документе, для получения аналогов или производных 2-амино-4,6-диметоксибензамида. Цель настоящего изобретения заключается в обеспечении нового и усовершенствованного способа синтеза 2-амино-4,6-диметоксибензамида, пригодного в качестве промежуточного соединения для различных терапевтических фармацевтических соединений, таких как сердечно-сосудистые средства.
[0033] Эти и другие отличительные признаки настоящего изобретения будут очевидны из следующего описания, чертежей и приложенной формулы изобретения.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
[0034] Фиг. 1 представляет собой иллюстративный пример способа по настоящему изобретению для получения 2-амино-4,6-диметоксибензамида.
[0035] Фиг. 2 представляет собой другой иллюстративный пример способа по настоящему изобретению для получения 2-амино-4,6-диметоксибензамида.
[0036] На каждой из вышеуказанных фигур, схожие числа используют для обозначения схожих или функционально схожих частей нескольких фигур.
Стадия присоединения защитной группы
[0037] Стадия способа, где по меньшей мере одно соединение II получают посредством способа, включающего сочетание, по меньшей мере, одного соединения V с, по меньшей мере, одним защитным агентом для получения, по меньшей мере, одного соединения II, может быть обозначена как «стадия присоединения защитной группы».
[0038] Термин «защитный агент» относится к любому соединению или группам соединений, которые защищают функциональные группы от нежелательных реакций. Например, защитный агент может защитить R5 соединения V во время получения соединения II, или может защитить R5 соединения II во время стадии галогенирования.
[0039] Термин «защищающая группа» относится к защитному агенту или части защитного агента, прикрепленному к соединениям II, III, IV или I. Защитная группа выборочно блокирует реакционноспособный участок в многофункциональном соединении так, чтобы химическую реакцию можно было проводить выборочно в другом незащищенном реакционноспособном участке в соответствующем значении, общепринятом в синтетической химии. Определенные способы по настоящему изобретению основаны на защитных группах для блокирования реактивного азота, присутствующего в веществах, участвующих в реакции. Желательно, чтобы защитная группа проявляла следующие характеристики: (i) выборочно вступает в реакцию с необходимой функциональной группой с хорошим выходом для получения защищенного субстрата, являющегося стабильным к прогнозируемым реакциям, для которых необходима защита; (ii) может быть выборочно удалена из защищенного субстрата для получения необходимой функциональной группы; и (iii) может быть удалена с хорошим выходом с помощью реагентов, совместимых с другой(ими) функциональной(ыми) группой(ами), присутствующими или сгенерированными в таких прогнозируемых реакциях. Термин «направляющая группа» относится к защитному агенту или части защитного агента, влияющим на реактивность соединения так, что химическая реакция, в которой участвует соединение, может возникнуть с измененной региоселективностью соединения. Специалистам в данной области органического синтеза хорошо известен широкий спектр защитных групп/направляющих групп. Неограничивающие примеры подходящих защитных групп/направляющих групп можно найти в Wuts et al. (2007) Greene's Protective Groups in Organic Synthesis, 4е изд. (John Wiley & Sons, Inc., Нью-Йорк). Например, термины «амино-защитная группа» и «азот-защитная группа» используют взаимозаменяемо в настоящем документе, и они относятся к тем органическим группам, которые, как предполагается, защищают атом азота от нежелательных реакций во время синтетических способов. При указании защитного агента специалисту в данной области будет понятно, какая часть защитного агента, т.е. защитная группа, будет прикреплена к соединению, подлежащему защите. Альтернативно, при указании защитной группы специалист в данной области может сделать вывод о необходимости защитного агента для достижения получаемой в результате защитной группы. Например, трифторуксусный ангидрид может быть обозначен как защитный агент, и трифторацетил является защитной группой. В другом примере хлорангидрид может быть обозначен как защитный агент; например, бензоилхлорид может быть обозначен как защитный агент, в этом случае бензоил является защитной группой. Таким образом, в целях настоящего изобретения термины «защитная группа», «направляющая группа» или «защитный агент» можно использовать взаимозаменяемо, и специалисту в данной области будет понятна взаимосвязь защитного агента с защитной группой или направляющей группой. Указание защитного агента включает все из получаемых в результате защитных групп или направляющих групп, и указание защитной группы или направляющей группы также включает все из защитных агентов для создания получаемой в результате защитной группы. Неограничивающий иллюстративный защитный агент или его соответствующие защитные группы или направляющие группы включают ацетил, моногалоацетил, дигалоацетил и тригалоацетил (где молекула галогена может быть той же самой или различной для дигалоацетильной и тригалоацетильной группы), ацетамидо, бензил (Bn), бензоил (Bz), бензилоксикарбонил (карбобензилокси, CBZ), п-метоксибензилоксикарбонил, п-нитробензилоксикарбонил, трет-бутоксикарбонил (BOC), трет-бутилдиметилсилил (TBDMS), 9-флуоренилметилоксикарбонил (Fmoc), другую защитную группу подобного и их сочетания. Термин «с присоединенной защитной группой» относится к соединению, где, по меньшей мере, одна из его функциональных групп является защитной группой или направляющей группой.
[0040] В одном из вариантов осуществления защитной группой или направляющей группой является ацетил.
[0041] В другом варианте осуществления защитной группой или направляющей группой является моногалоацетил.
[0042] В другом варианте осуществления защитной группой или направляющей группой является дигалоацетил.
[0043] В другом варианте осуществления защитной группой или направляющей группой является тригалоацетил.
[0044] В другом варианте осуществления защитной группой или направляющей группой является трифторацетил. Было обнаружено, что защитная/направляющая группа трифторацетила приводит к повышению выборочности по сравнению с защитной/направляющей группой ацетила во время стадии галогенирования.
[0045] В одном из вариантов осуществления получение синтеза соединения I включает сочетание соединения V с, по меньшей мере, одним защитным агентом для получения соединения II с присоединенной защитной группой. Следует понимать, что специалист в данной области органического синтеза может руководствоваться способами, описанными или приведенными в качестве примера в настоящем документе, для определения необходимости обеспечения соединения II с присоединенной защитной группой или присоединения защитной группы к соединению V для получения соединения II с присоединенной защитной группой для синтезирования соединения I. Реакцию стадии присоединения защитной группы можно проводить при любом времени реакции, давлении, температуре, растворителе, условиях pH, концентрации, соотношении/количестве реагентов и любых других условиях химической реакции, подходящих для обеспечения необходимого защитного/направляющего эффекта. Следует понимать, что специалист в данной области, принимая во внимание теорию настоящего описания, может определить и/или оптимизировать время реакции, давление, температуру, растворитель, условия pH, концентрацию, соотношение/количество реагентов и любые другие условия химической реакции, подходящие для стадии присоединения защитной группы.
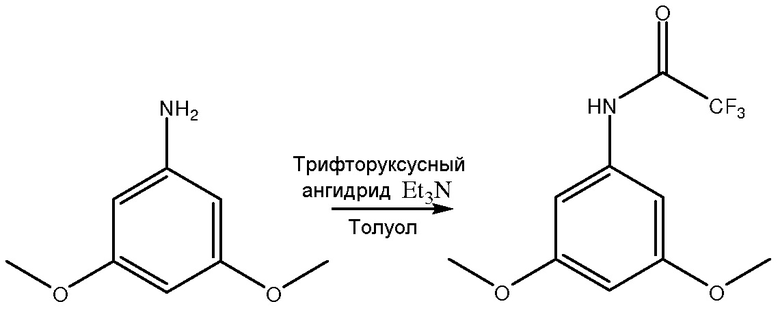
[0046] В одном примере соединением I является 2-амино-4,6-диметоксибензамид, и соединением V является 3,5-диметоксианилин. Как показано на фиг. 1, синтез 2-амино-4,6-диметоксибензамида включает присоединение защитной группы к 3,5-диметоксианилину с помощью, по меньшей мере, одного защитного агента для получения соединения II с присоединенной защитной группой.
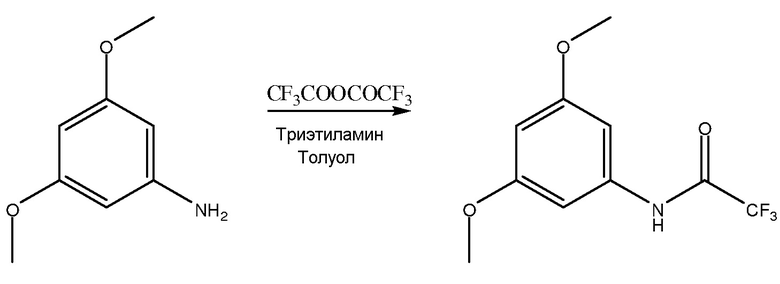
Стадия присоединения защитной группы в этом примере включает сочетание 3,5-диметоксианилина с трифторуксусным ангидридом и использованием триэтиламина в толуоле для получения соединения II с присоединенной защитной группой, или в этом примере из 3,5-диметоксианилина получают 3,5-диметокситрифторацетанилид. Метил-трет-бутиловый эфир (МТБЭ) можно использовать в качестве растворителя вместо или в дополнение к толуолу. Также можно использовать другие подходящие растворители. Принимая во внимания теорию настоящего описания, специалист в данной области может выбрать один или несколько других подходящих растворителей.
[0047] В одном из вариантов осуществления раствор, содержащий толуол или другой растворитель, переносят непосредственно на следующую стадию, т.е. стадию галогенирования после водных промывок.
[0048] В другом варианте осуществления раствор, содержащий толуол или другой растворитель, переносят непосредственно на следующую стадию, т.е. стадию галогенирования без каких-либо водных промывок.
[0049] Альтернативно, специалист в данной области может удалить, снизить или повысить содержание толуола или другого растворителя и/или других промежуточных соединений и/или удалить воду перед галогенированием соединения II, например, удалить воду посредством азеотропной дистилляции толуола или другого растворителя и воды.
[0050] В зависимости от исходного соединения II и необходимого бензамидного соединения I, подлежащего синтезированию, может потребоваться присоединение защитной группы к определенным функциональным группам. Специалист в данной области может использовать любые известные способы для защиты функциональных(ой) групп(ы) соединения II от нежелательных реакций во время стадий галогенирования, цианирования и/или гидратирования.
[0051] Может потребоваться получение соединения II с присоединенной защитной группой.
Стадия галогенирования
[0052] Стадия способа, где, по меньшей мере, одно соединение III получают посредством способа, включающего сочетание, по меньшей мере, одного соединения II с, по меньшей мер, одним галогенирующим агентом для получения, по меньшей мере, одного соединения III, может обозначаться как стадия галогенирования.
[0053] Термин «галогенирующий агент» относится к любому соединению, способному вступать в реакцию с соединением II для добавления, по меньшей мере, одной молекулы галогена, галогеноподобной молекулы, или замещения, по меньшей мере, одного R1, R2, R3, R4 или R5 соединения II с, по меньшей мере, одной молекулой галогена или галогеноподобной молекулой.
[0054] Термин «галогеноподобная» молекула относится к любой группе, поведение которой аналогично молекуле галогена в отношении реактивности. Примеры галогеноподобных молекул включают трифлат (-OTf), мезилат (-OMs) и тозилат (-OTs).
[0055] Примеры подходящих галогенирующих агентов включают I2, ICl, ICl3, IBr, Br2, BrCl, Cl2, N-хлорсукцинимид, N-бромосукцинимид (NBS), 1,3-дибромо-5,5-диметилгадинтоин (1,3-дибром-5,5-диметилгидантоин), TsCl, тозильный ангидрид, MsCl, трифторметансульфохлорид и трифторметансульфоновый ангидрид. Специалист в данной области органического синтеза может использовать альтернативный галогенирующий агент. Желательно, чтобы используемое количество галогенирующего агента было стехиометрично необходимому продукту, как хорошо известно специалистам в данной области.
[0056] В одном из вариантов осуществления галогенирующим агентом является NBS.
[0057] В одном из вариантов осуществления галогенирующим агентом является N-хлорсукцинимид.
[0058] В другом варианте осуществления галогенирующим агентом является 1,3-дибром-5,5-диметилгидантоин.
[0059] В другом варианте осуществления галогенирующий агент выбран из I2, ICl, ICl3, IBr, Br2, BrCl и Cl2.
[0060] В другом варианте осуществления галогенирующий агент выбран из TsCl, тозильного ангидрида, MsCl, трифторметансульфохлорид и трифторметансульфонового ангидрида.
[0061] Следует понимать, что, принимая во внимание теорию настоящего описания, специалист в данной области может определить и/или оптимизировать время реакции, давление, температуру, растворитель, условия pH, концентрацию, соотношение/количество реагентов и любые другие условия химической реакции, подходящие для стадии галогенирования, для добавления, по меньшей мере, одной молекулы галогена или галогеноподобной молекулы к соединению II.
[0062] В одном из вариантов осуществления стадию галогенирования можно проводить при любом температурном диапазоне выше температуры замерзания и ниже температуры кипения выбранного растворителя. Необходимо оптимизировать условия температуры и растворителя с соответствием с учетом растворимости соединения II при воздействии галогенирующего агента.
[0063] В одном из вариантов осуществления температурный диапазон составляет от приблизительно -65°C до приблизительно 100°C.
[0064] В другом варианте осуществления температурный диапазон составляет от приблизительно -65°C до приблизительно 50°C.
[0065] В другом варианте осуществления температурный диапазон составляет от приблизительно -65°C до приблизительно 10°C.
[0066] В другом варианте осуществления температурный диапазон составляет от приблизительно -10°C до приблизительно 10°C.
[0067] В еще одном варианте осуществления температурный диапазон составляет от приблизительно -10°C до приблизительно 5°C.
[0068] В еще одном варианте осуществления температурный диапазон составляет от приблизительно -5°C до приблизительно 5°C.
[0069] В еще одном варианте осуществления температурный диапазон составляет от приблизительно 0°C до приблизительно 5°C.
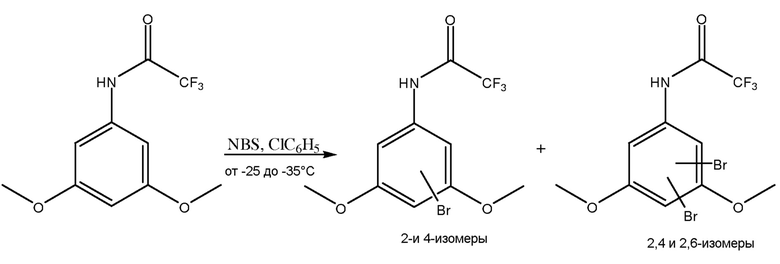
[0070] В одном из вариантов осуществления, как показано на фиг. 1, соединение анилина (3,5-диметокситрифторацетанилида) с присоединенной защитной группой вступает в реакцию с, по меньшей мере, одним галогенирующим агентом, как показано, N-бромосукцинимидом (NBS), при температурном диапазоне от приблизительно -5°C до 0°C для получения изомеров галогена (например, изомеров бромо-3,5-диметокситрифторацетанилида).
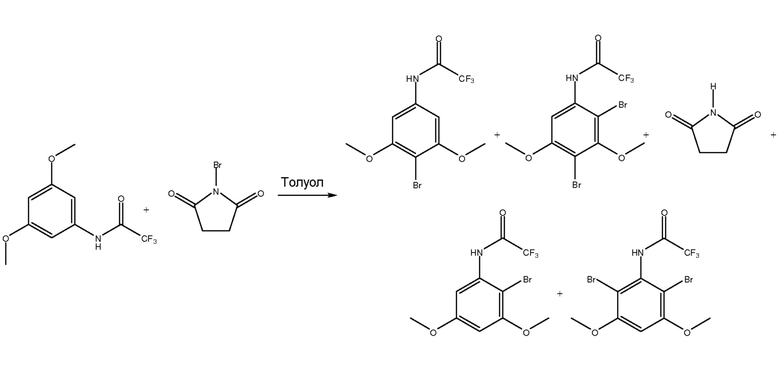
[0071] Как показано, растворитель на стадии галогенирования может содержать толуол. МТБЭ и/или диметилацетамид (DMAc) можно использовать в качестве растворителя на стадии галогенирования вместо или в дополнение к толуолу. Также можно использовать другие подходящие растворители стадии галогенирования. Принимая во внимание теорию настоящего описания, специалист в данной области может выбрать один или несколько других подходящих растворителей.
[0072] На стадии галогенирования получают различные изомеры галогенов. Например, как показано на фиг. 1, на стадии галогенирования можно получить изомеры, где галоген или, как показано, бром, находится в положении 4, положении 2, как в положении 2, так и 4, и, как в положении 2, так и в положении 6. Следует понимать, что могут быть получены другие изомеры в зависимости от используемого соединения II, галогенирующих агентов средства и условий.
[0073] Преимущество способов по настоящему изобретению заключается в том, что, несмотря на присутствие любого или всех изомеров, полученных на стадии галогенирования, необходимый 2-циано изомер (соединение IV) получают во время стадии цианирования.
[0074] В одном из вариантов осуществления по завершении реакции галогенирования сукцинимидные побочные продукты удаляют посредством любого известного способа в данной области, такого как промывка партии водой.
[0075] В другом варианте осуществления растворитель, например, толуол, можно удалить с использованием любых известных способов, таких как дистилляция или вакуум.
[0076] В другом варианте осуществления обмен растворителя завершают N',N'-диметилформамидом (ДМФА).
[0077] В другом варианте осуществления раствор бромированного, анилина (бромо-3,5-диметокситрифторацетанилида) с присоединенной защитной группой в ДМФА используют на стадии цианирования.
[0078] В другом варианте осуществления стадия галогенирования включает вступление в реакцию 3,5-диметокситрифторацетанилида с, по меньшей мере, одним галогенирующим агентом, таким как N-бромосукцинимид (NBS), в хлорбензоле для получения 2-бромо-3,5-диметокситрифторацетанилида и его изомеров.
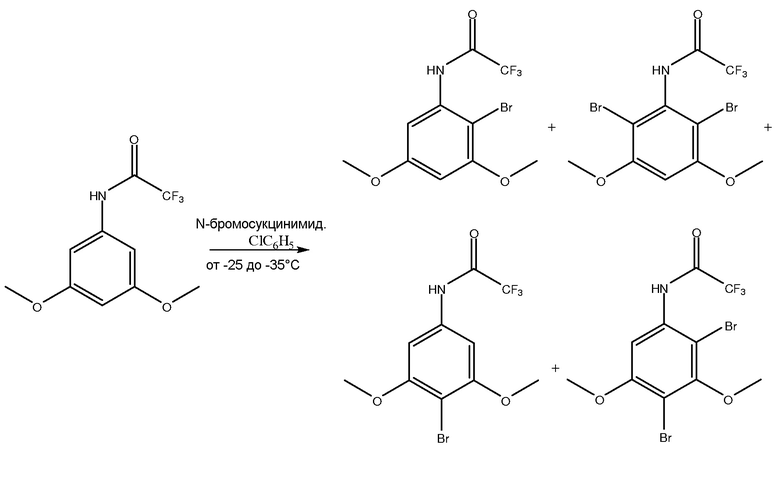
[0079] В одном из вариантов осуществления изомеры можно удалять или отделять из раствора перед стадией цианирования.
[0080] В другом варианте осуществления изомеры не удаляют или не отделяют от раствора перед проведением последующего реакционного способа.
[0081] Выявлено, что стадия присоединение защитной группы и выбор защитного агента может повысить региоселективность соединения III. В одном из вариантов осуществления защитный агент повышает или приводит к повышенной региоселективности изомера 2-галогена соединения III по сравнению с другими изомерами, такими как изомер 4-галогена.
[0082] В одном из вариантов осуществления молекулой галогена является бром.
Стадия цианирования
[0083] Стадия способа, где соединение IV получают посредством способа, включающего сочетание, по меньшей мере, одного соединения III с, по меньшей мере, одним цианирующим агентом для получения, по меньшей мере, одного соединения IV, обозначена в настоящем документе как стадия цианирования.
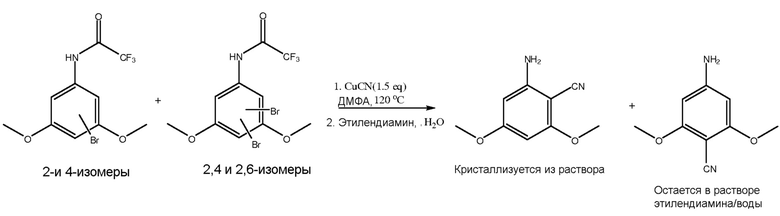
[0084] В одном из вариантов осуществления стадия цианирования содержит сочетание изомеров галогена стадии галогенирования с, по меньшей мере, одним цианирующим агентом для получения циано-изомеров.
[0085] В одном из вариантов осуществления, пример которого представлен на фиг. 1, изомеры брома соединения III, включая 2-бромо-3,5-диметокситрифторацетанилид, в ДМФА сочетают с цианидом меди (I) при температурном диапазоне от приблизительно 98°C до приблизительно 120°C для получения 2-амино-4,6-диметоксибензонитрила. Можно также получать другие изомеры. В другом варианте осуществления K4Fe(CN)6, вместе с подходящим катализатором, таким как CuI, при более чем 100 моль%, используют в качестве цианирующего агента.
[0086] В одном из вариантов осуществления ДМФА удаляют с использованием любого известного способа, такого как дистилляция, для концентрирования партии.
[0087] В другом варианте осуществления концентрированный раствор ДМФА переносят в раствор этилендиамина и воды для проведения стадии снятия защитной группы и удаления солей меди.
[0088] Было обнаружено, что выделение соединения IV при температуре около высшей границы диапазона, описываемого в настоящем документе, приводит к получению выхода на нижней границе диапазона, описываемого в настоящем документе.
[0089] В другом варианте осуществления соединение IV, такое как 2-амино-4,6-диметоксибензонитрил, выделяют посредством фильтрации и сушат под паром азота.
[0090] С использованием методов в настоящем документе в отношении способов по настоящему изобретению происходит выделение 2-изомера в растворе или его выпадение в осадок, при этом не происходит выделение в растворе неподходящих изомеров. Преимущество настоящего изобретения заключается в выделении необходимого изомера и невыделении нежелательных изомеров (4-, 2-/4- и 2-/6- изомеров), которые считаются примесями.
[0091] В одном из вариантов осуществления стадия цианирования включает осаждение 2-амино-4,6-диметоксибензонитрила. В результате способа получили осаждение необходимого промежуточного продукта 2-амино-4,6-диметоксибензонитрила, вследствие чего было получено более низкое содержание примесей изомеров. В сущности, однореакторный способ улучшает факторы стоимости и времени для повышения эффективности без необходимости очистки и выделения при каждой промежуточной реакции.
[0092] В одном из вариантов осуществления соединение IV сочетают с, по меньшей мере, одним агентом для снятия защитной группы и первым осаждающим агентом, таким как этилендиамин и вода. В этом варианте осуществления после стадии снятия защитной группы или удаления трифторацетиловой группы подходящий изомер, т.е. 2-амино-4,6-диметоксибензонитрил, выпадает в осадок в растворе, в то время как другие изомеры остаются в растворе.
[0093] Стадию цианирования можно проводить при любом времени реакции, давлении, температуре, растворителе, условиях pH, концентрации, соотношении/количестве реагентов и любых других условиях химической реакции, подходящих для добавления, по меньшей мере, одной группы циано (-CN) к соединению III или замене, по меньшей мере, одной молекулы галогена, по меньшей мере, одной цианогруппой в соединении III. Следует понимать, что специалист в данной области может определить и/или оптимизировать время реакции, давление, температуру, растворитель, условия pH, концентрацию, соотношение/количество реагентов и любые другие условия химической реакции, подходящие для стадии цианирования.
[0094] В одном из вариантов осуществления температурный диапазон для стадии цианирования может составлять от приблизительно 50°C до приблизительно 155°C.
[0095] В другом варианте осуществления температурный диапазон для стадии цианирования может составлять от приблизительно 50°C до приблизительно 120°C.
[0096] В еще одном варианте осуществления температурный диапазон для стадии цианирования может составлять от приблизительно 50°C до приблизительно 105°C.
[0097] В еще одном варианте осуществления температурный диапазон для стадии цианирования может составлять от приблизительно 98°C до приблизительно 105°C.
[0098] Термин «цианирующий агент» относится к любому соединению, которое при вступлении в реакцию с соединением III может заменить, по меньшей мере, одну молекулу галогена цианогруппой или добавить, по меньшей мере, одну группу циано к соединению III. Неограничивающие примеры цианирующего агента включают Zn(CN)2, CuCN, NaCN, KCN, Cu(CN)2, Ni(CN)2, цианид железа и другие подобные агенты. Цианирование можно проводить в присутствии или в отсутствие катализатора, например, цианирующий агент может содержать катализатор.
[0099] В одном из вариантов осуществления цианирующим агентом является CuCN.
[00100] В другом варианте осуществления цианирующий агент содержит K4Fe(CN)6 и CuI.
[00101] В другом варианте осуществления цианирующий агент содержит Na4Fe(CN)6 и CuBr.
[00102] В другом варианте осуществления цианирующий агент содержит K4Fe(CN)6 и CuBr.
[00103] В другом варианте осуществления цианирующий агент содержит Na4Fe(CN)6 и CuI.
Стадия снятия защитной группы
[00104] Стадия способа, где защитная группа или направляющая группа, представленная R7, заменена так, что R представляет собой водород или C1-C6 алкил, обозначена в настоящем документе как стадия снятия защитной группы.
[00105] Термин «снятие защитной группы» относится к удалению, по меньшей мере, одной защитной группы или направляющей группы. Термин «агент для снятия защитной группы» относится к любому соединению, которое может удалить, по меньшей мере, одну защитную группу или направляющую группу. Как правило, специалистам в данной области известны различные способы и условия снятия защитной группы с соединения. Неограничивающие агенты для снятия защитной группы включают этилендиамин, аммиак, этаноламин и метиламин.
[00106] В одном из вариантов осуществления стадию снятия защитной группы проводят перед стадией цианирования.
[00107] В одном из вариантов осуществления стадию снятия защитной группы проводят после стадии цианирования.
[00108] В одном из вариантов осуществления стадию снятия защитной группы проводят одномоментно со стадией цианирования.
[00109] Стадию снятия защитной группы также моно проводить частично перед стадией цианирования и/или частично во время стадии цианирования и/или частично после стадии цианирования.
[00110] Преимущество настоящего изобретения заключается в том, что любую из стадий цианирования, снятия защитной группы и осаждения можно проводить в однореакторном способе.
Стадия осаждения
[00111] Стадия способа, где, по меньшей мере, одно соединение III сочетают с, по меньшей мере, одним третьим осаждающим агентом, или где, по меньшей мере, одно соединение IV сочетают с, по меньшей мере, одним первым или третьим осаждающим агентом, обозначена в настоящем документе как стадия осаждения.
[0012] В рамках изобретения «первый осаждающий агент» относится к любому веществу, способствующему или вызывающему выпадение в осадок соединения IV в растворе. Полученный в результате осадок может содержать кристаллические структуры и/или аморфные структуры. Неограничивающим примером подходящего первого осаждающего агента является вода. В свете теории настоящего описания другие подходящие первые осаждающие агенты хорошо известны специалистам в данной области.
[00113] В рамках изобретения «третий осаждающий агент» относится к любому подходящему неполярному растворителю или другому подходящему веществу, способствующему или вызывающему выпадение в осадок соединения III или соединения IV в растворе. Полученный в результате осадок может содержать кристаллические структуры и/или аморфные структуры. Неограничивающим примером подходящего третьего осаждающего агента является гептан. В свете теории настоящего описания другие подходящие третьи осаждающие агенты хорошо известны специалистам в данной области.
[00114] В одном из вариантов осуществления снятие защитной группы и осаждение осуществляют на одной стадии.
[00115] В одном из вариантов осуществления для способствования снятию защитной группы и осаждения соединения III используют этилендиамин и гептан.
[00116] В одном из вариантов осуществления для способствования снятию защитной группы и осаждения соединения III используют аммиак и гептан.
[00117] В одном из вариантов осуществления для способствования снятию защитной группы и осаждения соединения III используют этаноламин и гептан.
[00118] В одном из вариантов осуществления для способствования снятию защитной группы и осаждения соединения III используют метиламин и гептан.
[00119] В одном из вариантов осуществления для способствования снятию защитной группы и осаждения соединения IV используют этилендиамин и воду.
[00120] В одном из вариантов осуществления для способствования снятию защитной группы и осаждения соединения IV используют аммиак и воду.
[00121] В одном из вариантов осуществления для способствования снятию защитной группы и осаждения соединения IV используют этаноламин и воду.
[00122] В одном из вариантов осуществления для способствования снятию защитной группы и осаждения соединения IV используют метиламин и воду.
[00123] В одном из вариантов осуществления стадию осаждения проводят после стадий цианирования и снятия защитной группы.
[00124] Альтернативно, специалист в данной области может осуществлять осаждение и/или выделение полученных промежуточных соединений III или IV при любом необходимом времени в ходе способов по настоящему изобретению.
Стадия повторного осаждения
[00125] Дополнительная стадия способа, где, по меньшей мере, одно соединение IV сочетают с, по меньшей мере, одним вторым осаждающим агентом, обозначена в настоящем документе как «стадия повторного осаждения».
[00126] В рамках изобретения термин «второй осаждающий агент» относится к любому веществу, способствующему или вызывающему выпадение в осадок соединения IV в растворе после, по меньшей мере, одного предыдущего осаждения соединения IV с помощью или первого осаждающего агента, или третьего осаждающего агента в соответствии с настоящим изобретением. Полученный в результате осадок может содержать кристаллические структуры и/или аморфные структуры. Неограничивающим примером подходящего второго осаждающего агента является изопропиловый спирт. В свете теории настоящего описания другие подходящие вторые осаждающие агенты хорошо известны специалистам в данной области.
[00127] В способах по настоящему изобретению стадия повторного осаждения может также привести к очистке осадка соединения IV.
Стадия гидратирования
[00128] Стадия способа, где соединение I получают посредством способа, включающего сочетание, по меньшей мере, одного соединения IV с, по меньшей мере, одним гидратирующим агентом и/или, по меньшей мере, одним гидратирующим катализатором для получения, по меньшей мере, одного соединения I, обозначена в настоящем документе как стадия гидратирования.
[00129] В одном из вариантов осуществления стадия гидратирования включает сочетание соединения IV с, по меньшей мере, одним гидратирующим агентом и/или, по меньшей мере, одним гидратирующим катализатором для получения, по меньшей мере, одного соединения I, обозначена в настоящем документе как стадия гидратирования.
[00130] Реакцию на стадии гидратирования можно проводить при любом времени реакции, давлении, температуре, растворителе, условиях pH, концентрации, соотношении/количестве реагентов и любых других условиях химической реакции, подходящих для удаления или замещения, по меньшей мере, одной циано молекулы из соединения IV. Следует понимать, что специалист в данной области может определить и/или оптимизировать время реакции, давление, температуру, растворитель, условия pH, концентрацию, соотношение/количество реагента и любые другие условия химической реакции, подходящие для стадии гидратирования.
[00131] В одном из вариантов осуществления стадия гидратирования содержит преобразование, по меньшей мере, одной группы циано в, по меньшей мере, один карбоксамид.
[00132] В одном из вариантов осуществления стадию гидратирования можно проводить при температурном диапазоне, составляющем от приблизительно 70°C до приблизительно 150°C.
[00133] В другом варианте осуществления стадию гидратирования можно проводить при температурном диапазоне, составляющем от приблизительно 100°C до приблизительно 115°C.
[00134] Следует понимать, что используемая температура оказывает влияние на скорость реакции. Более длительный период времени реакции гидратирования при низкой температуре может привести к повышению содержания примесей, но, тем не менее, в ходе представленного способа все же можно получить необходимый продукт. Кроме того, как правило, более высокая температура обуславливает более высокую скорость реакции, а также профиль с более высоким содержанием примеси. Таким образом, оптимизация времени и температуры приводит к улучшенному профилю содержания примесей.
[00135] В одном из вариантов осуществления способ синтеза 2-амино-4,6-диметоксибензамида включает гидратирование 2-амино-4,6-диметоксибензонитрила с помощью, по меньшей мере, одного гидратирующего агента, такого как метансульфоновая кислота

[00136] В одном из вариантов осуществления стадия гидратирования дополнительно включает повторное осаждение 2-амино-4,6-диметоксибензонитрила перед его взаимодействием с гидратирующим агентом. Реакцию проводят в течение приблизительно двух часов нагревания (приблизительно 1-2 часов при температуре 100-115°C).
[00137] В другом варианте осуществления гидратирование нитрильного соединения дополнительно включает добавление воды и дихлорметана (ДХМ) к кислотной смеси. Выбор растворителя не ограничен предыдущими примерами, но зависит от структуры исследуемых соединений, а также от других факторов. Специалист в данной области может использовать альтернативные растворители, не вступающие в реакцию с исследуемым соединением, включая в качестве неограничивающих примеров этилацетат, изопропил ацетат, простой эфир, такой как 2-метилтетрагидрофуран, спирт, такой как изопропиловый спирт, и другие растворители или комбинации подобных растворителей.
[00138] В другом варианте осуществления стадия гидратирования дополнительно включает нейтрализацию партии посредством взаимодействия с приблизительно 50% каустиком и корректировки диапазона pH от приблизительно 3 до приблизительно 12. Специалист в данной области может использовать альтернативный нейтрализующий агент для корректировки необходимого уровня pH.
[00139] В одном из вариантов осуществления диапазон pH может составлять от приблизительно 6,0 до приблизительно 8,0. В другом варианте осуществления диапазон pH может составлять от приблизительно 6,5 до приблизительно 7,3.
[00140] В дополнительном варианте осуществления стадия гидратирования дополнительно включает стадию экстрагирования. Стадия экстрагирования включает экстрагирование водного слоя с помощью ДХМ три раза, и органический слой промывают водой для удаления метансульфонатных солей. Происходит дистилляция дихлорметана для снижения общего объема партии, и партию медленно охлаждают до температурного диапазона от приблизительно 23°C до приблизительно 28°C. МТБЭ загружают в партию и также охлаждают до температурного диапазона от приблизительно -5°C до приблизительно 0°C. Полученный 2-амино-4,6-диметоксибензамид выделяют любым способом, таким как фильтрация и сушка под паром азота. Для экстрагирования специалист в данной области может использовать другой тип растворителя, который не оказывает негативного воздействия на реакции, такой как изопропилацетат, этилацетат, изопропиловый спирт, 2-метилтетрагидрофуран или их сочетания. Если необходима партия, в которой отсутствует галогенированный растворитель, проходит обмен растворителя до достижения менее 1% галогенированного растворителя, оставшегося в реакционном сосуде. Альтернативно, можно использовать негалогенированный растворитель после нейтрализации, таким образом потенциально сводя к минимуму необходимость в обмене растворителя. Объем снижают посредством дистилляции до получения необходимого диапазона объема изопропилацетата, изофталевой кислоты, этилацетата, метил-ТГФ, или альтернативного растворителя. Необязательным является использование соответствующего антирастворителя, такого как МТБЭ или гептан. Однако повышение выхода наблюдают с соответствующим антирастворителем.
[00141] В другом варианте осуществления получение 2-амино-4,6-диметоксибензамид включает гидратирование 2-циано-3,5-диметокситрифторацетанилида с помощью гидратирующего агента, такого как трифторуксусная кислота, как показано ниже.
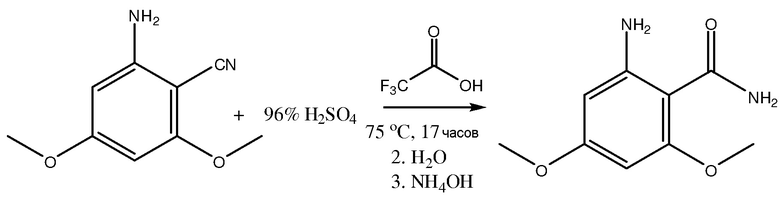
В одном из вариантов осуществления получение 2-амино-4,6-диметоксибензамида содержит снятие защитной группы с 2-циано-3,5-диметокситрифторацетанилида для получения 2-амино-4,6-диметоксибензонитрила.
[00142] Термин «гидратирующий агент» относится к любому соединению, которое при сочетании с соединением IV может преобразовать, по меньшей мере, одну цианогруппу в карбоксамидную группу или гидратировать тройную связь цианогруппы. Гидратирующий агент также включает гидратирующие катализаторы. Термин «гидратирующий катализатор» относится к любому соединению, способствующему и/или ускоряющему гидратирование соединения IV в соединение I. Неограничивающие примеры гидратирующих агентов включают воду, нитрат железа, спирты, кислоты, такие как серная кислота, трифторуксусная кислота, метансульфоновая кислота, фосфорная кислота, такая как полифосфорная кислота, основы, такие как NaOH, KOH, гидроксид цезия, гидроксид бария, металлический катализатор и т.п., и их сочетания.
[00143] В одном из вариантов осуществления на стадии гидратирования осуществляется преобразование цианогруппы в карбоксамидную группу при сведении к минимуму количества получаемой карбоновой кислоты.
[00144] В другом варианте осуществления стадию гидратирование проводят при кислотных условиях реакции.
[00145] В другом варианте осуществления стадию гидратирования можно также проводить при основных условиях реакции.
[00146] Реакции синтетических способов, заявленные в настоящем документе, проводят в подходящих растворителях, которые легко может выбрать специалист в данной области органического синтеза; указанным подходящим растворителем, как правило, является любой растворитель, который по существу не вступает в реакцию с исходными веществами (веществами, участвующими в реакции), промежуточными соединениями или продуктами при температуре проведения реакций, т.е., температуре, которая может варьироваться от температуры замерзания растворителя до температуры кипения растворителя. Указанную реакцию можно проводить в одном растворителе или в комбинации двух или более растворителей. В зависимости от конкретной стадии реакции, специалист в данной области может выбрать подходящие растворители для конкретной стадии реакции.
[00147] Подразумевается, что способ, представленный на фиг. 1, служит в качестве иллюстрации настоящего изобретения, и его не следует рассматривать как ограничение объема изобретения. Специалисту в данной области понятно, что в ходе способа, представленного в настоящем документе, можно получить различные побочные продукты, которые также можно создать с помощью реакций по настоящему изобретению. Таким образом, способ по изобретению обеспечивает направленную обработку, так как промежуточные соединения после каждой стадии можно использовать непосредственно в последующих реакциях без необходимости их выделения или очистки. Альтернативно, специалист в данной области может произвести очистку и выделение промежуточных соединений перед последующими реакциями.
[00148] Функциональные группы или молекулы по настоящему изобретению включают замещенные или незамещенные, защищенные или незащищенные молекулы. При «замещении» конкретной группы она может иметь один или несколько заместителей, от одного до пяти заместителей, от одного до трех заместителей, от одного до двух заместителей, независимо выбранных из перечня заместителей.
[00149] Следует понимать, что определение любого заместителя или переменной при конкретном положении в молекуле не зависит от определений в каком-либо другом месте в этой молекуле. Следует понимать, что специалист в данной области может выбирать заместители и схемы замещения соединений по настоящему изобретению для получения соединений, которые являются химически стабильными и которые легко можно синтезировать посредством методов, известных в данной области, а также тех способов, которые указаны в настоящем документе.
[00150] В соответствии со стандартной номенклатурой, первой описана концевая часть указанной боковой цепи, а затем соседняя функциональная группа по направлению к точке присоединения. Номенклатура, используемая в настоящем документе для функциональных группы или молекул, не ограничена стандартным расположением групп, но может быть представлена в любом порядке при условии, что она не изменяет какие-либо химические свойства.
[00151] Кроме того, некоторые из кристаллических форм соединений по настоящему изобретению могут существовать в качестве полиморфов и по существу предназначены для включения в настоящее изобретение. Кроме того, некоторые соединения по настоящему изобретению могут образовывать сольваты с водой (т.е., гидраты) или общепринятые органические растворители, и, предполагается, что такие сольваты также включены в объем настоящего изобретения.
[00152] Любая из функциональных групп, представленных в настоящем документе, может быть также замещена любой другой известной функциональной группой, известной в данной области, которая не оказывает негативного воздействия на стабильность соединений или реакции, представленные в настоящем документе.
[00153] Также следует понимать, что перечисленные растворители и реагенты можно использовать по отдельности или в комбинации с другими растворителями и реагентами при условии, что комбинации не оказывают негативного воздействия на стабильность соединений или реакции, представленные в настоящем документе.
[00154] Следующие примеры представлены с иллюстративной целью и предполагается, что они не накладывают ограничений на объем настоящего изобретения.
ПРИМЕР 1
3,5-Диметокситрифторацетанилид
[00155] В 2-литровую колбу с рубашкой загружали 3,5-диметоксианилин (120 г), толуол (1335 г) и триэтиламин (87 г). Смесь перемешивали при 18-20°C до растворения всех твердых веществ. Трифторуксусный ангидрид (185 г) добавляли на протяжении, по меньшей мере, 1 часа, при этом поддерживая температуру реакции 18-25°C. Реакцию перемешивали в течение, по меньшей мере, 1 часа, а затем завершение реакции проверяли с помощью ВЭЖХ. Воду (250 г) загружали в партию и реакцию нагревали до 40-45°C и перемешивали в течение, по меньшей мере, 10 минут. Встряхивание прекращали, и разделяли фазы. Нижнюю водную фазу удаляли, и воду (250 г) загружали в слой полученного толуола. Партию перемешивали при 40-45°C в течение, по меньшей мере, 10 минут, и фазы разделяли посредством удаления нижней водной фазы. Затем раствор полученного 3,5-диметокситрифторацетанилида в толуоле охлаждали до менее чем 0°C для подготовки к стадии способа получения бромо-3,5-диметокситрифторацетанилида.
ПРИМЕР 2
Бромо-3,5-диметокситрифторацетанилид
[00156] Раствор 3,5-диметокситрифторацетанилида в толуоле, помещенный в 2-литровую колбу с рубашкой, охлаждали от -5 до 0°C. N-бромосукцинимид в твердом состоянии (145 г) загружали в холодную суспензию 3,5-диметокситрифторацетанилида частями от 5 до 10 грамм на протяжении, по меньшей мере, 1 часа.
Во время добавления поддерживали температуру менее 0°C. По завершению добавления партию оставляли для нагревания до 15-23°C и перемешивали в течение, по меньшей мере, 1 часа. Завершение реакции проверяли с помощью ВЭЖХ. После завершения реакции воду (235 г) загружали в партию и реакцию нагревали до 35-45°C и выдерживали в течение, по меньшей мере, 10 минут. Встряхивание прекращали, и оставляли для разделения фаз. Нижнюю водную фазу удаляли и воду (235 г) загружали в раствор бромо-3,5-диметокситрифторацетанилида в толуоле. Партию встряхивали при 35-45°C в течение, по меньшей мере, 10 минут, и фазы разделяли посредством удаления нижней водной фазы. Раствор бромо-3,5-диметокситрифторацетанилида в толуоле переносили в 2-литровую четырехгорлую круглодонную колбу, оснащенную дистилляционным аппаратом и колбонагревателем. Раствор нагревали до кипения с обратным холодильником, и толуол дистиллировали до достижения температуры реакционного сосуда 125-140°C. Партию охлаждали до менее 80°C под азотом, и в реакционный сосуд загружали N',N'-диметилформамид (ДМФА) (1215 г). Партию встряхивали и охлаждали до менее 80°C. Этот раствор использовали на стадии способа получения 2-амино-4,6-диметоксибензонитрила.
ПРИМЕР 3
Цианирование бромо-3,5-диметокситрифторацетанилида
[00157] В раствор бромо-3,5-диметокситрифторацетанилида/ДМФА в 2-литровой круглодонной колбе загружали 89 граммов цианида меди (CuCN). Партию нагревали до 98-120°C и выдерживали в течение, по меньшей мере, 6 часов. Завершение реакции проверяли посредством анализа ВЭЖХ. После завершения реакцию охлаждали до менее 60°C и к сосуду применяли вакуум и дистиллировали ДМФА. Дистилляцию продолжали до получения объема реакционного сосуда приблизительно 570 мл. Остаток в реакционном сосуде охлаждали до менее 40°C.
Снятие защитной группы с 2-циано-3,5-диметокситрифторацетанилида
[00158] Для разделения в 2-литровую колбу с рубашкой загружали воду (1065 г) и этилендиамин (390 г). Водный раствор нагревали до 50-55°C и оставляли. Остаток 2-циано-3,5-диметокситрифторацетанилид/ДМФА в реакционном сосуде от предыдущей стадии загружали в водную смесь в течение, по меньшей мере, 15 минут. Реакционный раствор перемешивали при 50-55°C в течение, по меньшей мере, 2 часов. Анализ завершения реакции проводили с помощью ВЭЖХ. По завершении реакции партию корректировали до 35-37°C и оставляли для образования суспензии) Полученную в результате суспензию охлаждали медленно до 5-15°C в течение, по меньшей мере, 2 часов. Партию выдерживали при 5-15°C в течение 2 часов, а затем полученный 2-амино-4,6-диметоксибензонитрил выделяли посредством фильтрации. Осадок 2-амино-4,6-диметоксибензонитрила отмывали водой для удаления маточного раствора. Конечный отфильтрованный осадок сушили и анализировали с помощью ВЭЖХ. В результате способа получили 123 граммов 2-амино-4,6-диметоксибензонитрила с выходом 88% от исходного 3,5-диметоксианилина.
ПРИМЕР 4
Повторное осаждение 2-амино-4,6-диметоксибензонитрила
[00159] В 1-литровую четырехгорлую круглодонную колбу загружали 2-амино-4,6-диметоксибензонитрил (90 г) и изопропиловый спирт (720 мл). Колбу оснастили конденсатором и колбонагревателем. К встряхиваемой смеси добавляли углерод (1,8 г), и партию нагревали до кипения с обратным холодильником (82-83°C). Партию выдерживали в течение 1 часа при кипячении с обратным холодильником, а затем охлаждали до 75-77°C и оставляли на, по меньшей мере, 6 часов. Затем отфильтровывали углерод, и фильтрат собирали в чистую 1-литровую четырехгорлую круглодонную колбу. Фильтрат медленно охлаждали до 60-62°C и оставляли до возникновения повторного осаждения. Полученную в результате суспензию медленно охлаждали до 0-5°C в течение, по меньшей мере, 2 часов. Партию выдерживали при 0-5°C в течение, по меньшей мере, 0,5 часов и фильтровали для сбора продукта. Осадок 2-амино-4,6-диметоксибензонитрила отмывали с помощью изопропилового спирта и сушили в вакуумной печи при 50°C при 22 дюймах вакуума. В результате способа получили 83,8 граммов очищенного 2-амино-4,6-диметоксибензонитрила с выходом 84%.
ПРИМЕР 5
2-Амино-4,6-диметоксибензамид
[00160] 2-Амино-4,6-диметоксибензонитрил (10,0 г, 0,056 моль, 1 экв.) загружали в 1-литровый стеклянный сосуд в среде N2 и начинали встряхивание. В сосуд загружали метансульфоновую кислоту (120 мл, 1,848 моль, 33 экв.) и реакцию нагревали при 100-115°C в течение 1-2 часов. После проверки завершения реакции посредством ВЭЖХ партию охлаждали до 20-30°C. При охлаждении добавляли дихлорметан (67 мл) и прохладную воду (164 мл) (5-10°C) для поддержания температуры в диапазоне 10-30°C. В сосуд загружали 50% каустик (94 мл) с использованием капельной воронки при одновременном поддержании 0-30°C. Точную установку pH от 6,5 до 7,3 осуществляли с использованием или 32% HCl, или 50% каустика. При уравновешивании осуществляли разделение первой органической фазы. Водный слой экстрагировали дополнительно два раза с помощью ДХМ (74 мл и 57 мл соответственно). К объединенным органическим слоям добавляли дихлорметан (54 мл), и органический слой отмывали водой (143 мл) для удаления метансульфонатных солей. К объединенным органическим слоям добавляли дихлорметан (16 мл), и органические слой отмывали еще раз водой (143 мл) для удаления метансульфонатных солей. Партию дистиллировали с использованием (5-10 дюймов Hg) вакуума применительно к объему реакционного сосуда, составлявшему 4,5 объема ДХМ (45 мл). Содержимое сосуда перемешивали в течение 1 часа при 38°C. Сосуд охлаждали до 23-28°C в течение 1 часа при медленном перемешивании. При наблюдении помутнения в сосуд медленно вводили 14,4 объема МТБЭ (144 мл) и партию встряхивали в течение 30 минут при 25°C (при соотношении ДХМ/МТБЭ 1:3,2; при общем объеме растворителя, составляющем 18,9). Партию медленно охлаждали от -5°C до 5°C в течение, по меньшей мере, 3 часов. Партию выдерживали в течение, по меньшей мере, 1 часа при температуре от -5°C до 0°C. Осаждение проверяли посредством сбора, по меньшей мере, двух образцов жидкости для определения количества 2-амино-4,6-диметоксибензамида, оставшегося в растворе. Партию выделяли посредством фильтрации и отфильтрованный осадок промывали с помощью холодной (0°C) смеси 1:4 дхм/мтбэ (51 мл). Отфильтрованный осадок 2-амино-4,6-диметоксибензамида сушили в вакуумной печи (40-45°C, 25 дюймов Hg) для получения 2-амино-4,6-диметоксибензамида (8,33 г, 0,0425 моль, с выходом 75,7%; с выходом 53% после 5 стадий, начиная с 3,5-диметоксианилина). Было исследовано использование альтернативного дихлорметану растворителя, действие которого аналогично действию дихлорметана. Примеры альтернативных растворителей в качестве неограничивающих примеров включают сложные эфиры, такие как изопропилацетат и этилацетат, и простые эфиры, такие как 2-метилтетрагидрофуран. Кроме того, также были изучены альтернативные системы осаждения с или без использования антирастворителя. В качестве неограничивающих примеров системы осаждения могут включать сложные эфиры, такие как изопропилацетат, простые эфиры, такие как 2-метилтетрагидрофуран, и спирты, такие ка изопропиловый спирт. Выход варьируется в зависимости от этих модификаций в экспериментальном способе и составляет от 72 до 79%. Кроме того, чистота необходимого соединения неизменно составляет более 99%.
ПРИМЕР 6
2-амино-4,6-диметоксибензамид
[00161] 2-Амино-4,6-диметоксибензонитрил (50,0 г, 0,2805 моль, 1 eq.) загружали в 3-литровый стеклянный сосуд в среде N2 и начинали встряхивание. В сосуд загружали метансульфоновую кислоту (537 мл, 8,29 моль, 29,5 eq.) и реакцию нагревали при 100-115°C в течение 1-2 часов. После проверки завершения реакции посредством ВЭЖХ, партию охлаждали до 20-30°C. При охлаждении добавляли дихлорметан (334 мл) и прохладную воду (807 мл) (5-10°C) для поддержания температуры в диапазоне 10-30°C. В сосуд загружали 50% каустик (423 мл) с использованием капельной воронки при поддержании температуры на уровне 0-30°C. Точную корректировку pH от 6,5 до 7,5 осуществляли с использованием или 32% HCl, или 50% каустика, или их разбавленных растворов. При уравновешивании осуществляли разделение первой органической фазы. Водный слой дополнительно экстрагировали два раза с помощью ДХМ (371 мл и 286 мл соответственно). К объединенным органическим слоям добавляли дихлорметан (271 мл) и органический слой отмывали водой (714 мл) для удаления метансульфонатных солей. К объединенным органическим слоям добавляли дихлорметан (79 мл) и органический слой отмывали еще раз водой (714 мл) для удаления метансульфонатных солей. Партию дистиллировали с использованием вакуума (5-10 дюймов Hg) применительно к объему реакционного сосуда, составлявшему 6 объемов ДХМ (300 мл). Обмен растворителя с изопропилацетатом (2 обмена 402 мл и 484 мл соответственно) завершали при остатке ДХМ менее 1-2% и объеме реакционного сосуда, составляющем 400-450 мл. Содержимое сосуда перемешивали в течение 1 часа при 85-90°C. Сосуд охлаждали до 50°C в течение, по меньшей мере, 2 часов при медленном перемешивании. В сосуд медленно загружали МТБЭ (250 мл) и партию встряхивали в течение 30 минут при 50°C. Партию медленно охлаждали до 30°C в течение, по меньшей мере, 1 часа. Партию охлаждали до -5°C и 5°C в течение, по меньшей мере, 2 часов. Партию выдерживали при -5°C и 5°C в течение, по меньшей мере, 1 часа. Осаждение проверяли посредством сбора, по меньшей мере, двух образцов жидкости для определения количества 2-амино-4,6-диметоксибензамида, оставшегося в растворе. Партию выделяли посредством фильтрации и отфильтрованный осадок промывали с помощью холодного (0°C) МТБЭ (100 мл). Отфильтрованный осадок 2-амино-4,6-диметоксибензамида сушили в вакуумной печи (40-45°C, 25 дюймов Hg) для получения 2-амино-4,6-диметоксибензамида (42,79 г, 0,218 моль, с выходом 77,7%; с выходом 54,5% после 5 стадий, начиная с 3,5-диметоксианилина). Изменения объема растворителя, использованного во время экстрагирования, и части осаждения в этом примере могут привести к повышению или снижению выхода и/или содержания примесей. Специалист в данной области может изменять эти условия для получения необходимого результата.
ПРИМЕР 7
3,5-Диметокситрифтор ацетанилид
[00162] В 1-листровую колбу с рубашкой, оснащенную механической мешалкой и термопарой загружали тонкоизмельченный 3,5-диметоксианилин (51,20 г, 0,33 моль), триэтиламин (38,21 г, 0,38 моль) и толуол (246,70 г). Охлаждающую установку настроили на 15°C, и, когда температура реакции достигла этого значения, добавляли трифторуксусный ангидрид (73,51 г, 0,35 моль) в течение 1,5 часов с поддержанием температуры реакции около 20°C. Затем к охлажденному раствору (15°C) добавляли воду (124,69 г), и когда реакционную смесь охлаждали до 12°C, в реакции твердые вещества выпали в осадок. Двухфазный раствор фильтровали и твердые вещества перемешивали с водой (326,38 г) в течение 15 минут. Продукт фильтровали и сушили для получения белого с металлическим оттенком твердого вещества (71,15 г, с выходом 85,4%).
ПРИМЕР 8
Бромо-3,5-диметокситрифторацетанилид
[00163] В 1-листровую колбу, оснащенную механической мешалкой и термопарой, добавляли раствор хлорбензола (434,04 г) и 3,5-диметокситрифторацетанилида (50,03 г, 0,20 моль, 1,0 эквивалентная молярная масса). Колбу охлаждали в ванне с изопропиловым спиртом и добавляли небольшое количество сухого льда для поддержания температуры раствора в колбе приблизительно -25°C. NBS (37,07 г, 0,20 моль, 1,04 эквивалентная молярная масса) небольшими частями добавляли к реакционной смеси на протяжении 1,2 часов при поддержании температуры реакции от -25 до -35°C. Затем раствор нагревали до 0-1°C на протяжении 3,5 часов. Реакционный образец добавляли к раствору тиосульфата натрия и органический слой анализировали посредством ГХ. К реакционной смеси добавляли воду (100,01 г) и смесь встряхивали. Водный слой удаляли и затем органический слой дважды отмывали дополнительным количеством воды (101,08 и 100,94 г). Растворитель удаляли посредством испарения с применением роторного испарителя в вакууме для получения белого с металлическим оттенком твердого вещества (63,04 г).
ПРИМЕР 9
2-циано-3,5-диметокситрифторацетанилид
[00164] Неочищенную бромированную смесь изомеров (40,06 г) растворяли в ДМФА (234,79 г) в 500 мл колбе, оснащенной термопарой и механической мешалкой. К раствору добавляли цианид меди (16,95 г) при комнатной температуре. Затем темный раствор нагревали до 120°C и оставляли при такой температуре реакции в течение 4,25 часов. После охлаждения неочищенной реакционной смеси до комнатной температуры смесь выливали в колбу Эрленмейера, содержащую воду (222,27 г), которой быстро придавали вращательное движение при добавлении раствора. Твердые вещества, выпавшие в осадок, фильтровали и промывали водой (121,94 г). При объединении воды после промывки твердых веществ с раствором воды/ДМФА получали дополнительные твердые вещества. Эти твердые вещества отмывали водой (122,16 г). В 1-литровую колбу, оснащенную механической мешалкой и термопарой, загружали воду (292,32 г). К перемешанному раствору медленно добавляли этилендиамин (106,92 г). Объединенные реакционные твердые вещества вводили в перемешанный раствор. По завершению добавления температуру реакции поднимали до 50°C и такую температуру поддерживали в течение 0,5 часа. По мере охлаждения раствора до комнатной температуры твердые вещества выпали в осадок в растворе. Эти твердые вещества фильтровали для получения продукта (15,29 г).
ПРИМЕР 10
2-амино-4,6-диметоксибензамид

[00165] Трифторуксусную кислоту (6,52 г, 4,4 мл) загружали в колбу, оснащенную магнитной мешалкой и термопарой. Серную кислоту (96%, 9,2 г, 5 мл) загружали в колбу. 2-амино-4,6-диметоксибензонитрил (1,01 г, с чистотой -76%) загружали маленькими частями в смешанный кислотный раствор в колбе. Добавляли конденсатор, и раствор нагревали при 75°C в течение 17 часов. После охлаждения темной смеси до комнатной температуры ее загружали в воду (9,79 г), охлажденную в большой ванне со льдом с поддержанием температуры реакции ниже 10°C. К охлажденному раствору (с поддерживаемой температурой реакции ниже 15°C) добавляли капельно концентрированный гидроксид аммония (19,37 г). Затем раствор экстрагировали два раза с помощью дихлорметана. Органические экстракты сушили над MgSО4 и выпаривали для получения необходимого продукта (0,45 г) в виде светло-коричневого твердого вещества.
ПРИМЕР 11
3,5-диметокситрифторацетанилид
[00166] В 1-литровую трехгорлую круглодонную колбу с верхнеприводной мешалкой загружали 3,5-диметоксианилин (50 г), триэтиламин (50 мл) и МТБЭ (450 мл). Смесь перемешивали и охлаждали с помощью метанол/ванны со льдом при -20°C. Добавляли трифторуксусный ангидрид (50 мл) при поддержании температуры реакции менее 30°C. Завершение реакции проверяли посредством ВЭЖХ. Затем раствор полученного 3,5-диметокситрифторацетанилида в МТБЭ охлаждали до менее 0°C при подготовке к стадии способа получения бромо-3,5-диметокситрифторацетанилида.
ПРИМЕР 12
Бромо-3,5-диметокситрифторацетанилид
[00167] Раствор 3,5-диметокситрифторацетанилида в МТБЭ, помещенный в 1-литровую трехгорлую круглодонную колбу охлаждали до -5 до 0°C. В 125-мл колбу Эрленмейера загружали 1,3-дибром-5,5-диметилгидантоин (47,5 г) и N,N-диметилацетамид (55 мл). Смесь 1,3-дибром-5,5-диметилгидантоина перемешивали до растворения и загружали в холодный раствор 3,5-диметокситрифторацетанилида. Во время добавления поддерживали температуру менее 0°C. Партию проверяли посредством ВЭЖХ на завершение реакции. По завершении реакции смесь нагревали до 40°C, и в партию загружали воду (100 г). Встряхивание прекращали и оставляли для разделения фаз. Нижнюю водную фаза удаляли, и раствору бромо-3,5-диметокситрифторацетанилида в МТБЭ загружали воду (25 г). Партию встряхивали при 40°C и фазы разделяли посредством удаления нижней водной фазы. Колбу оснащали насадкой Дина-Старка. Раствор бромо-3,5-диметокситрифторацетанилида в МТБЭ нагревали до кипения с обратным холодильником и остаток воды собирали в насадку. После прекращения сбора воды в насадку МТБЭ дистиллировали до достижения температуры в реакционном сосуде 75°C. В смесь загружали гептан (500 мл), партию встряхивали и нагревали до 80°C. Удаляли внешний нагрев и партию охлаждали до 20-25°C. Осажденный бромо-3,5-диметокситрифторацетанилид выделяли посредством фильтрации, промывали с помощью гептана (100 мл) и сушили в вакуумной печи при 50°C. В результате способа получали 99,6 граммов бромо-3,5-диметокситрифторацетанилида с выходом 93%.
ПРИМЕР 13
[00168] В этом примере представлены различные условия в соответствии с настоящим изобретением для преобразования 2-амино-4,6-диметоксибензонитрила в 2-амино-4,6-диметоксибензамид. Основными способами гидратирования являются: кислотный, основной и каталитический (включающий катализатор в дополнение к другому реагенту), или использование различных групп металлов для ускорения реакции. Время реакции варьируется. При следующих условиях оптимальная температура реакции находится в диапазоне от приблизительно 100°C до приблизительно 115°C, с завершением реакции через 1-2 часа.
[00169] 2-Амино-4,6-диметоксибензонитрил (2-амино-4,6-диметоксибензонитрил), CH3SO3H, с или без Al2O3, обработка с помощью KOH и/или 50% каустического и/или фосфатного буфера и/или 30% карбоната калия при 120°C в течение 2-4 часов. Без выделения продукта. Преобразование в 2-амино-4,6-диметоксибензамид: 78-88% (согласно ВЭЖХ).
[00170] 2-Амино-4,6-диметоксибензонитрил, CH3SO3H. Обработка с помощью 50% каустика; осаждение в 1-4 ДХМ/1-6 МТБЭ при 120°C в течение 0,5-4 часов. С практическим выходом 28-64%; с чистотой 2-амино-4,6-диметоксибензамида, составляющей 97,5-98,2%;
[00171] 2-Амино-4,6-диметоксибензонитрил, CH3SO3H. Обработка с помощью 50% каустика; осаждение в 1:3,2 ДХМ/МТБЭ при 120°C в течение 1 часа. С практическим выходом 78,5%; с чистотой 2-амино-4,6-диметоксибензамида, составляющей 99,64%.
[00172] 2-Амино-4,6-диметоксибензонитрил, CH3SO3H. Обработка с помощью 50% каустика; осаждение в 1:3,2 ДХМ/МТБЭ при 100-115°C в течение 1,5 часов. С практическим выходом 75,7%; с чистотой 2-амино-4,6-диметоксибензамида, составляющей 99,82%.
[00173] 2-Амино-4,6-диметоксибензонитрил, CH3SO3H. Обработка с помощью 50% каустика; осаждение в 1,6:1 изопропилацетата/МТБЭ при 105-110°C в течение 2 часов. С практическим выходом 77,7%; с чистотой 2-амино-4,6-диметоксибензамида, составляющей 99,58%.
[00174] 2-Амино-4,6-диметоксибензонитрил, CH3SO3H. Обработка с помощью 50% каустика (без ДХМ - альтернативного растворителя); Осаждение в 1,38:1 изопропилацетате/МТБЭ при 105-110°C в течение 2 часов. С практическим выходом 77,1%; с чистотой 2-амино-4,6-диметоксибензамида составляющей 99,61%.
[00175] 2-Амино-4,6-диметоксибензонитрил, порошковый 85% гидроксид калия, т-бутанол при 85°C в течение 20,4 часов. Без выделения продукта. Преобразование в 2-амино-4,6-диметоксибензамид 76% (посредством ГХ).
[00176] 2-Амино-4,6-диметоксибензонитрил, полифосфорная кислота при 115°C в течение 6,9 часов. Без выделения продукта. Преобразование в 2-амино-4,6-диметоксибензамид 100% (посредством ГХ).
[00177] 2-Амино-4,6-диметоксибензонитрил, 85% гидроксид натрия, 1,2-пропандиол, вода, разогревали в микроволновой печи при 150°C в течение 0,16-58 часов. Без выделения продукта. Преобразование в 2-амино-4,6-диметоксибензамид 60-84% (посредством ГХ).
[00178] 2-Амино-4,6-диметоксибензонитрил, 85% гидроксид калия, т-бутанол, разогревали в микроволновой печи. 150°C. 0,08-1,08 часов. Без выделения продукта. Преобразование в 2-амино-4,6-диметоксибензамид 62-84% (посредством ГХ).
[00179] 2-Амино-4,6-диметоксибензонитрил, NaBO3⋅H2О, MeOH, вода при 50°C в течение 48 часов. Без выделения продукта. Преобразование в 2-амино-4,6-диметоксибензамид -5% (посредством ГХ).
[00180] 2-Амино-4,6-диметоксибензонитрил, ацетальдоксим, Pd-катализатор, PPh3, водный раствор этанола при 75°C в течение 22-144 часов. Без выделения продукта. Преобразование в 2-амино-4,6-диметоксибензамид 20-36% (посредством ГХ).
[00181] 2-Амино-4,6-диметоксибензонитрил, ацетальдоксим, Pd-катализатор, PPh3, толуол при 75°C в течение 144 часов. Без выделения продукта. Преобразование в 2-амино-4,6-диметоксибензамид 12-18% (посредством ГХ).
[00182] 2-Амино-4,6-диметоксибензонитрил, KOH (хлопья), т-амиловый спирт, H2О, кислота Льюиса (кат.) при 98°C в течение 22-130 часов. Без выделения продукта. Преобразование в 2-амино-4,6-диметоксибензамид 9-49% (посредством ГХ).
[00183] 2-Амино-4,6-диметоксибензонитрил, гидроксид цезия или бария, т-амиловый спирт при 106°C в течение l9-20 часов. Без выделения продукта. Преобразование в 2-амино-4,6-диметоксибензамид ~2-3% (посредством ГХ).
[00184] 2-Амино-4,6-диметоксибензонитрил, метансульфоновая кислота, ледяная уксусная кислота при 120°C в течение 8,7 часов. Без выделения продукта. Преобразование в 2-амино-4,6-диметоксибензамид 1,8% (посредством ГХ).
[00185] 2-Амино-4,6-диметоксибензонитрил, нитрат железа*9 H2О, вода при 100°C в течение 21 часов. Без выделения продукта. Преобразование в 2-амино-4,6-диметоксибензамид 0,4% (посредством ГХ).
[00186] Следует понимать, что вещества, участвующие в реакции, и компоненты, обозначаемые химическим названием или формулой в любом месте настоящего документа, в единственном или множественном числе, определены такими, какими они являются перед взаимодействием с другим веществом, обозначенным химическим названием или химическим типом (например, другим веществом, участвующим в реакции, растворителем и т.д.). Имеют значение не то, что предварительные химические изменения, трансформации и/или реакции, в случае их присутствия, возникают в получаемой в результате смеси или растворе или реакционной среде, по существу такие изменения, трансформации и/или реакции являются естественным результатом объединения указанных веществ участвующих в реакции, и/или компонентов в необходимых условиях в соответствии с настоящим описанием. Таким образом, вещества участвующие в реакции, и компоненты указаны как ингредиенты, которые требуется объединить в связи с проведением необходимого химического способа или реакции или с образованием смеси для использования при проведении необходимого способа или реакции. Кроме того, несмотря на то, что вариант осуществления может относиться к веществам, компонентам и/или ингредиентам в настоящем времени («состоит из», «содержит», «является» и т.д.), вещество, компонент или ингредиент указывается таким, каким оно является в момент непосредственно перед его первым взаимодействием, сочетанием или смешиванием с одним или несколькими веществами, компонентами и/или ингредиентами в соответствии с настоящим описанием.
[00187] Кроме того, несмотря на то что пункты формулы изобретения могут относиться к веществам в настоящем времени (например, «содержит», «является» и т.д.), вещество указывается таким, каким оно является в момент непосредственно перед его первым взаимодействием, сочетанием или смешиванием с одним или несколькими веществами в соответствии с настоящим описанием.
[00188] За исключением иных прямо указанных случаев, предполагается, что при использовании в рамках изобретения форма единственного числа не носит ограничительный характер, и ее не следует считать ограничивающей описание или пункт формулы изобретения единичным элементом, к которому относится форма единственного числа. Напротив, при использовании в рамках изобретения форма единственного числа предназначена для включения одного или нескольких таких элементов, за исключением иных случаев, прямо указанных в тексте.
[00189] Настоящее изобретение подвержено значительным изменениям в пределах существа и объема приложенной формулы изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| СОЕДИНЕНИЯ, ПРИГОДНЫЕ ДЛЯ СИНТЕЗА БЕНЗАМИДНЫХ СОЕДИНЕНИЙ | 2013 |
|
RU2593752C1 |
| СПОСОБ ПОЛУЧЕНИЯ АМИНОФЕНИЛСУЛЬФОНИЛМОЧЕВИН (ВАРИАНТЫ), ПРОМЕЖУТОЧНЫЕ ПРОДУКТЫ | 1996 |
|
RU2177003C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ЦИКЛОПРОПАНА | 2011 |
|
RU2593202C2 |
| ГАЛОГЕНИДЫ ГАЛОГЕНСУЛЬФОНИЛБЕНЗОЙНОЙ КИСЛОТЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ ДЛЯ ПОЛУЧЕНИЯ ЗАМЕЩЕННЫХ ФЕНИЛСУЛЬФОНИЛМОЧЕВИН | 2003 |
|
RU2330027C2 |
| АЗОТСОДЕРЖАЩЕЕ ГЕТЕРОЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2011 |
|
RU2533708C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПЕПТИДА | 2008 |
|
RU2478105C2 |
| СПОСОБЫ ПОЛУЧЕНИЯ ЭПЛЕРЕНОНА | 2003 |
|
RU2339642C9 |
| ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ 3-БЕНЗОФУРАНИЛ-ИНДОЛ-2-ОН-3-АЦЕТАМИДОПИПЕРАЗИНОВ, ИХ ПОЛУЧЕНИЕ И ИХ ПРИМЕНЕНИЕ В ТЕРАПИИ | 2010 |
|
RU2542980C2 |
| СПОСОБ ПОЛУЧЕНИЯ 5-ЗАМЕЩЕННЫХ ПИРРОЛО (2,3-α)ПИРИМИДИНОВ | 1993 |
|
RU2127274C1 |
| СПОСОБ ПОЛУЧЕНИЯ ТРОВАФЛОКСАЦИНА | 1999 |
|
RU2167867C2 |
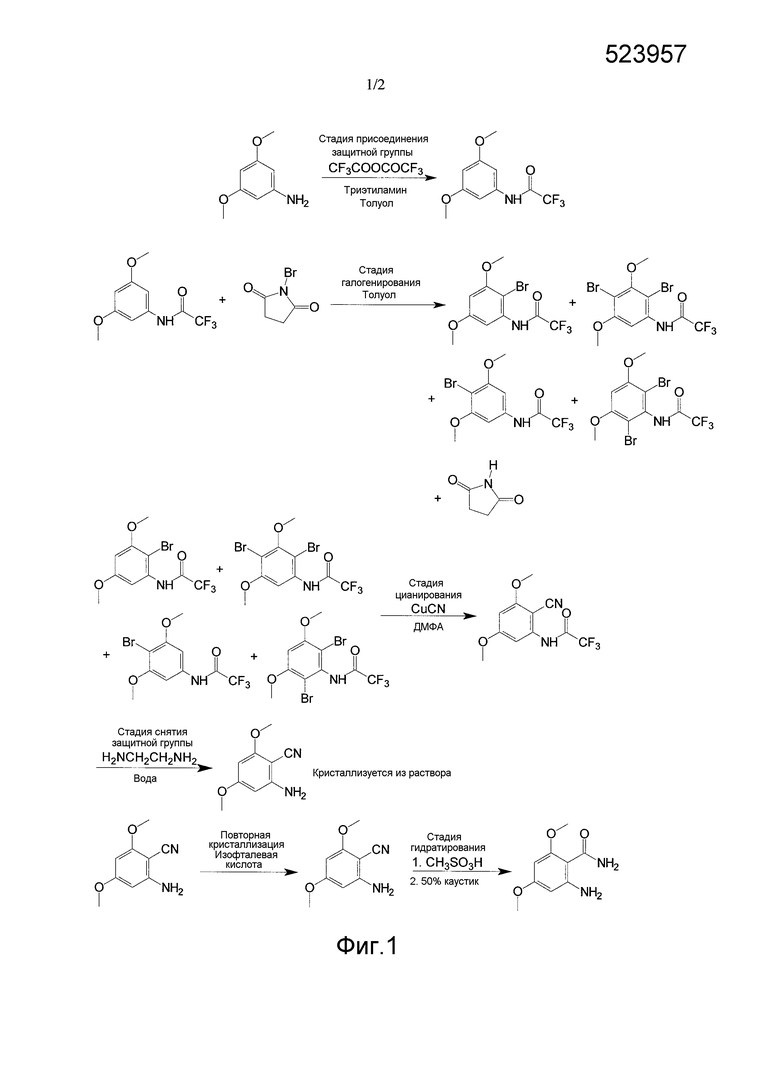
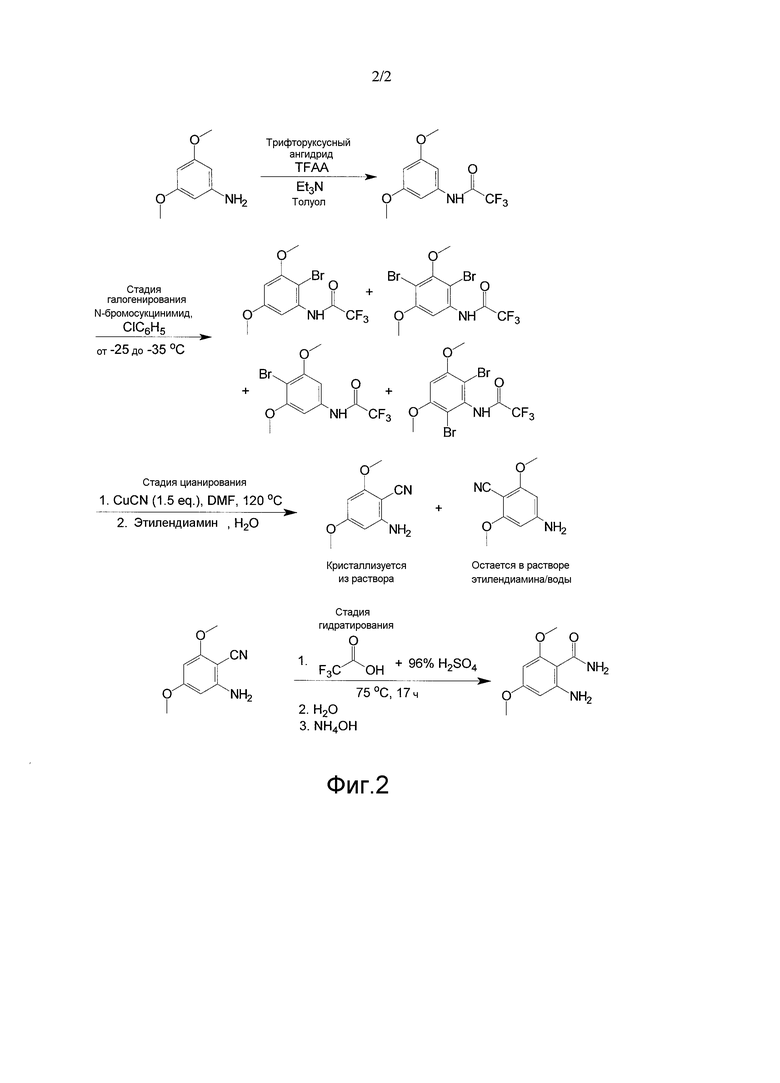
Изобретение относится к способу получения соединения I. Предлагаемый способ включает стадии (i)-(iii). На стадии (i) осуществляют сочетание по меньшей мере одного соединения II с по меньшей мере одним галогенирующим агентом для получения по меньшей мере одного соединения III. На стадии (ii) осуществляют сочетание по меньшей мере одного соединения III с по меньшей мере одним цианирующим агентом для получения по меньшей мере одного соединения IV. На стадии (iii) осуществляют сочетание по меньшей мере одного соединения IV с по меньшей мере одним гидратирующим агентом и/или гидратирующим катализатором для получения по меньшей мере одного соединения I. В формулах I-IV R1 и R5 представляют собой водород, C1-C6 алкил, гидрокси или C1-C6 алкокси; R2, R3 и R4 представляют собой водород, C1-C6 алкил или C1-C6 алкокси; R6 и R7 представляют собой водород, C1-C6 алкил, защитную или направляющую группу; X представляет собой Cl, Br, I, трифлат, тозилат или мезилат; причем по меньшей мере один из R1 и R5 представляет собой водород. При этом в соединении II R7 представляет собой защитную или направляющую группу и или (1) перед началом стадии (ii) по меньшей мере одно соединение III сочетают с по меньшей мере одним агентом для снятия защитной группы так, чтобы защитная или направляющая группа R7 была заменена водородом или C1-C6 алкилом, или (2) перед началом стадии (iii) по меньшей мере одно соединение IV сочетают с по меньшей мере одним агентом для снятия защитной группы так, чтобы защитная или направляющая группа R7 была заменена водородом или C1-C6 алкилом. Предлагаемый способ позволяет получать соединения I, пригодные в качестве промежуточных соединений для синтеза различных терапевтических средств. Изобретение относится также к различным вариантам способа получения соединения I, в частности 2-амино-4,6-диметоксибензамида. 6 н. и 32 з.п. ф-лы, 2 ил., 13 пр.
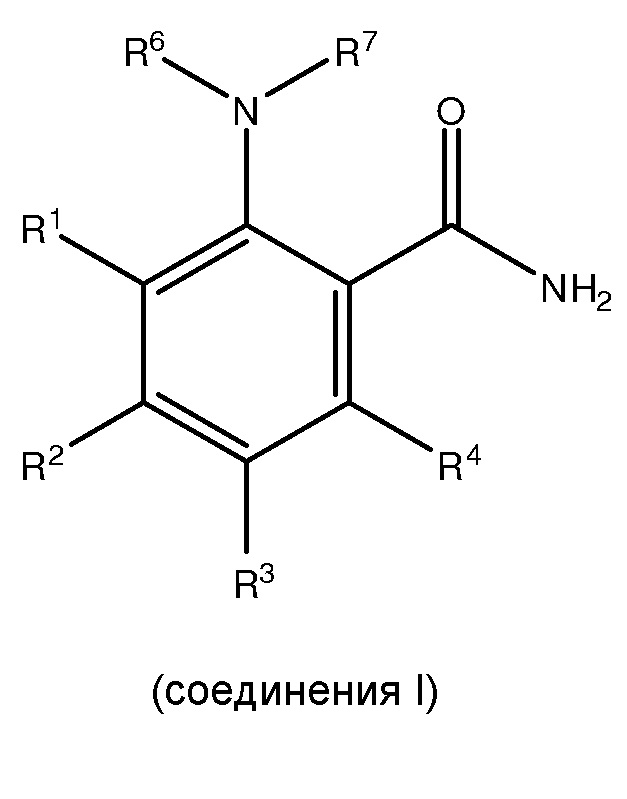 (I)
(I) 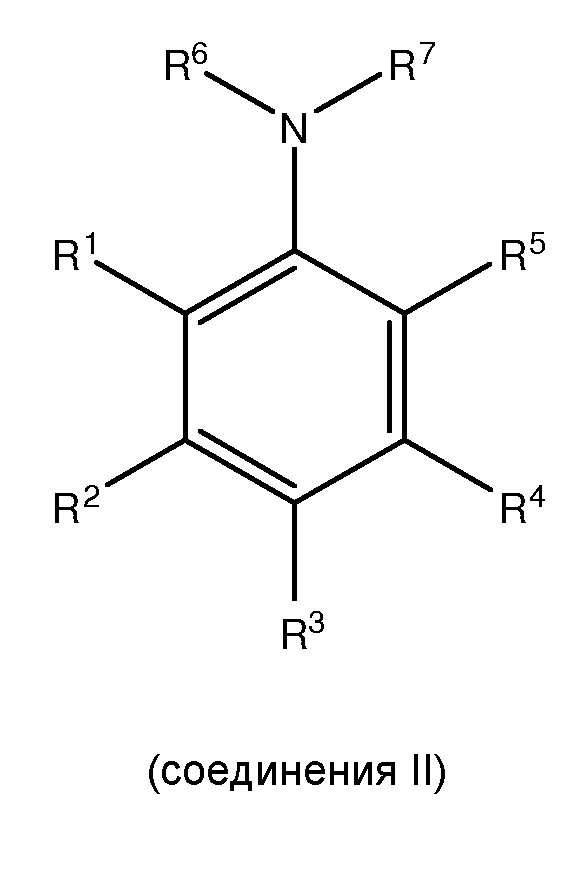 (II)
(II)
 (III)
(III) 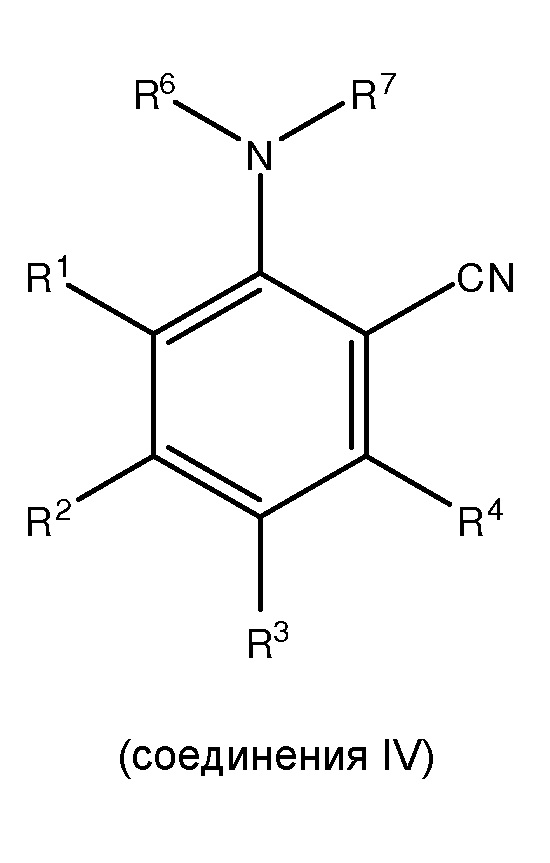 (IV)
(IV)
1. Способ получения по меньшей мере одного
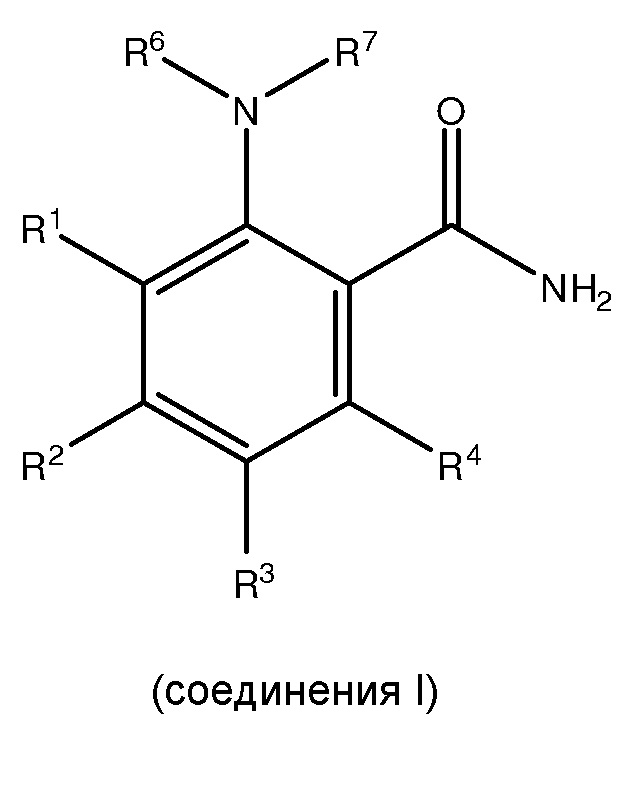 ,
,
где способ включает:
(i) сочетание по меньшей мере одного
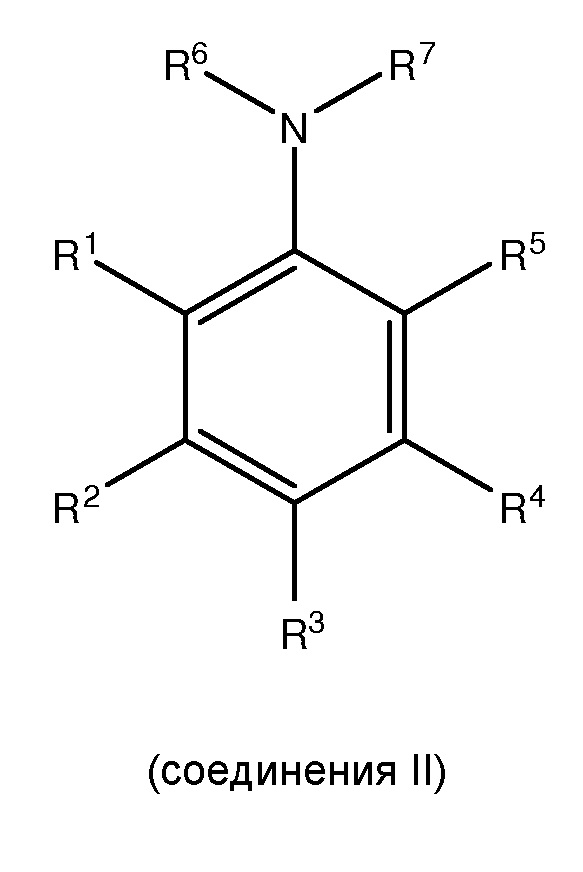
с по меньшей мере одним галогенирующим агентом для получения по меньшей мере одного
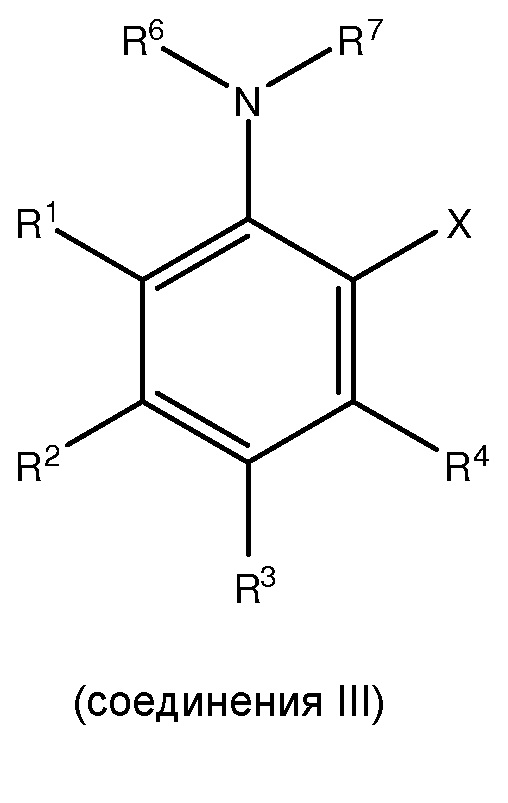 ;
;
(ii) сочетание по меньшей мере одного соединения III с по меньшей мере одним цианирующим агентом для получения по меньшей мере одного
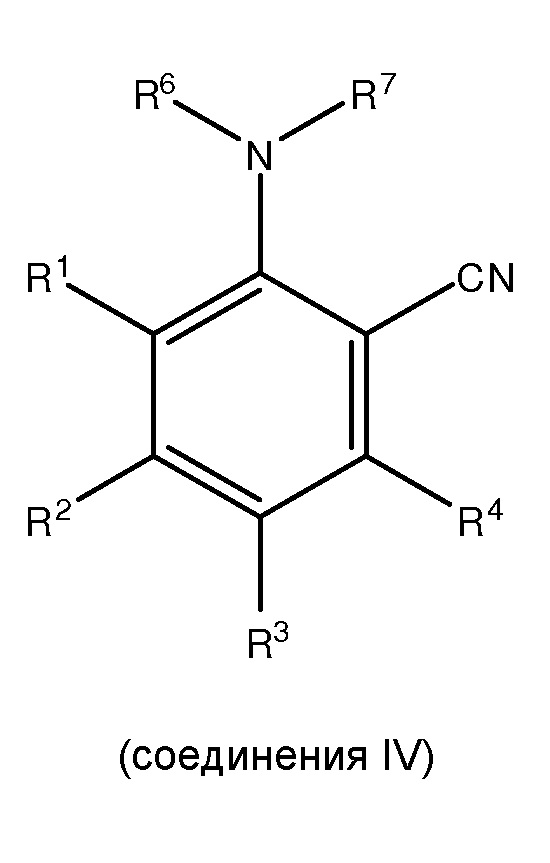 ;
;
и
(iii) сочетание по меньшей мере одного соединения IV с по меньшей мере одним гидратирующим агентом и/или по меньшей мере одним гидратирующим катализатором для получения по меньшей мере одного соединения I,
где каждый из R1 и R5 независимо представляет собой водород, C1-C6 алкил, гидрокси или C1-C6 алкокси;
где каждый из R2, R3 и R4 независимо представляет собой водород, C1-C6 алкил или C1-C6 алкокси;
где каждый из R6 и R7 независимо представляет собой водород, C1-C6 алкил, защитную группу или направляющую группу;
где X представляет собой хлор (Cl), бром (Br), йод (I), трифлат (-OTf), тозилат (-OTs) или мезилат (-OMs);
где по меньшей мере один из R1 и R5 представляет собой водород; и
при условии, что в соединении II R7 представляет собой защитную группу или направляющую группу, и или
(1) перед началом стадии (ii) по меньшей мере одно соединение III сочетают с по меньшей мере одним агентом для снятия защитной группы так, чтобы защитная группа или направляющая группа, представленная R7, была заменена водородом или C1-C6 алкилом, или
(2) перед началом стадии (iii) по меньшей мере одно соединение IV сочетают с по меньшей мере одним агентом для снятия защитной группы так, чтобы защитная группа или направляющая группа, представленная R7, была заменена водородом или C1-C6 алкилом.
2. Способ получения по меньшей мере одного
 ,
,
где способ включает:
(i) сочетание по меньшей мере одного
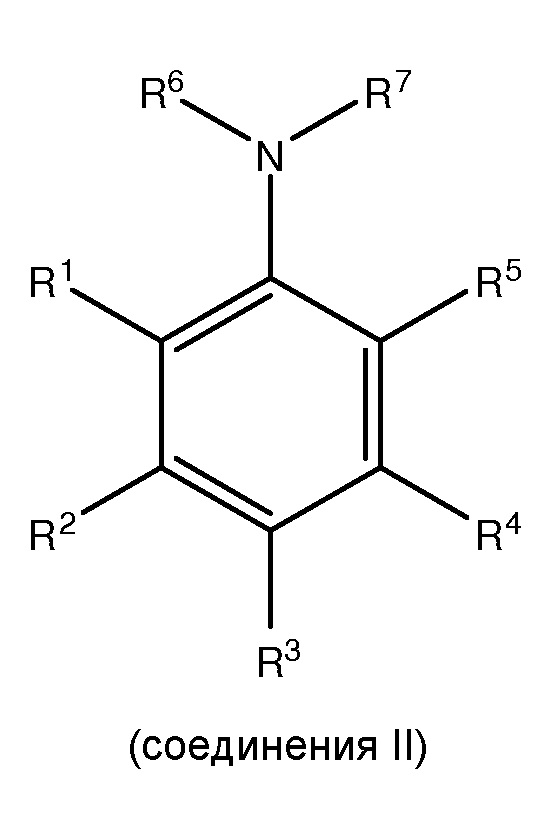
с по меньшей мере одним галогенирующим агентом для получения по меньшей мере одного
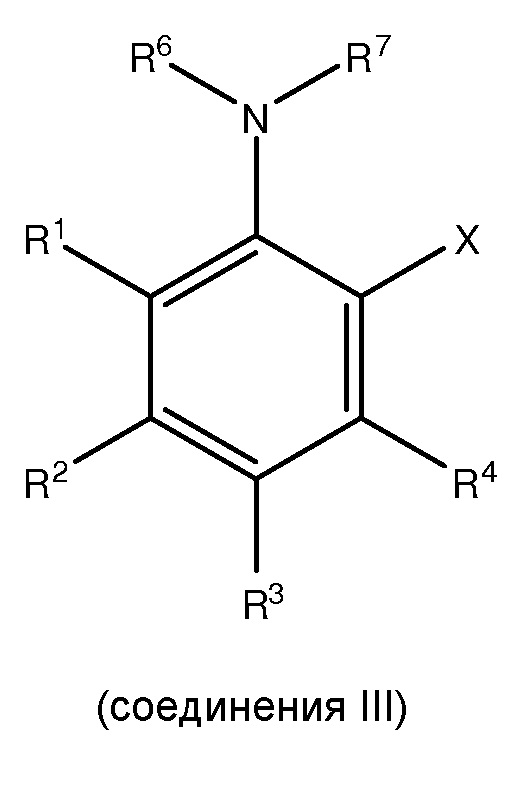 ;
;
(ii) сочетание по меньшей мере одного соединения III с по меньшей мере одним цианирующим агентом для получения по меньшей мере одного
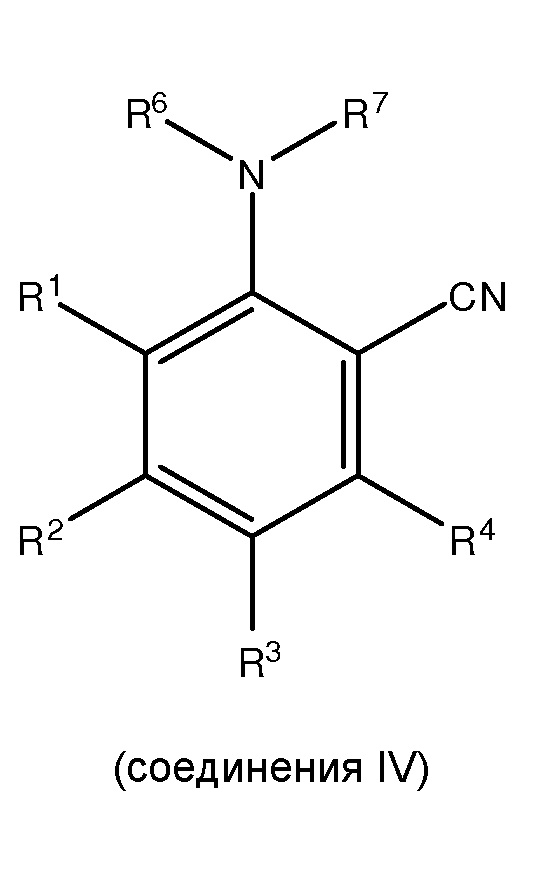 ;
;
(iii) осаждение по меньшей мере одного соединения IV по меньшей мере одним первым осаждающим агентом; и
(iv) сочетание по меньшей мере одного соединения IV с по меньшей мере одним гидратирующим агентом и/или по меньшей мере одним гидратирующим катализатором для получения по меньшей мере одного соединения I,
где каждый из R1 и R5 независимо представляет собой водород, C1-C6 алкил, гидрокси или C1-C6 алкокси;
где каждый из R2, R3 и R4 независимо представляет собой водород, C1-C6 алкил или C1-C6 алкокси;
где каждый из R6 и R7 независимо представляет собой водород, C1-C6 алкил, защитную группу или направляющую группу;
где X представляет собой хлор (Cl), бром (Br), йод (I), трифлат (-OTf), тозилат (-OTs) или мезилат (-OMs);
где по меньшей мере один из R1 и R5 представляет собой водород; и
при условии, что в соединении II R7 представляет собой защитную группу или направляющую группу, и или
(1) перед началом стадии (ii) по меньшей мере одно соединение III сочетают с по меньшей мере одним агентом для снятия защитной группы так, чтобы защитная группа или направляющая группа, представленная R7, была заменена водородом или C1-C6 алкилом, или
(2) перед началом стадии (iii) по меньшей мере одно соединение IV сочетают с по меньшей мере одним агентом для снятия защитной группы так, чтобы защитная группа или направляющая группа, представленная R7, была заменена водородом или C1-C6 алкилом.
3. Способ по п.2, где перед началом стадии (iv) по меньшей мере одно соединение IV осаждают по меньшей мере одним вторым осаждающим агентом.
4. Способ по п.2, где перед началом стадии (ii) по меньшей мере одно соединение III сочетают с по меньшей мере одним агентом для снятия защитной группы так, чтобы защитная группа или направляющая группа, представленная R7, была заменена водородом или C1-C6 алкилом.
5. Способ по п.2, где перед началом стадии (iii) по меньшей мере одно соединение IV сочетают с по меньшей мере одним агентом для снятия защитной группы так, чтобы защитная группа или направляющая группа, представленная R7, была заменена водородом или C1-C6 алкилом.
6. Способ по п.2, где по меньшей мере одно соединение II получают посредством способа, включающего сочетание по меньшей мере одного
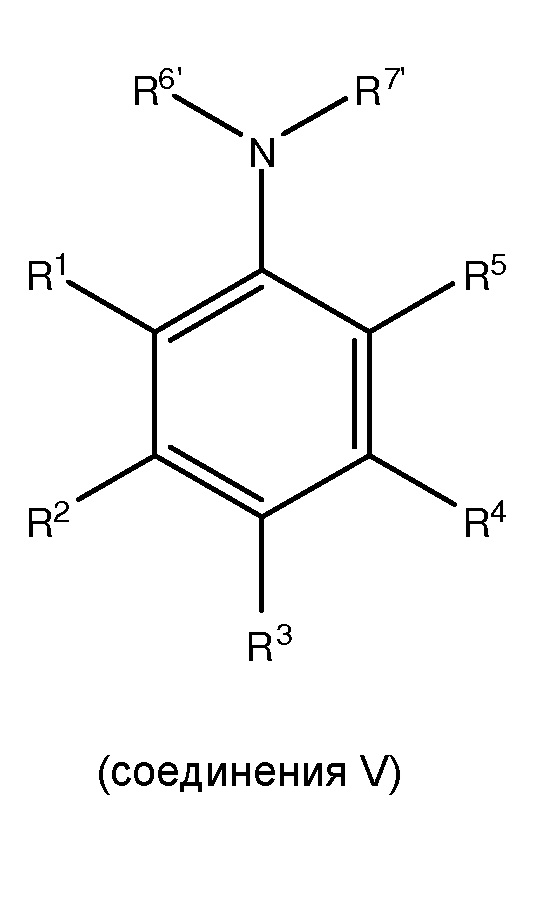
с по меньшей мере одним защитным агентом для получения по меньшей мере одного соединения II, где каждый из R6’ и R7’ независимо представляет собой водород или C1-C6 алкил.
7. Способ по п.2, где по меньшей мере один из R1, R2, R3, R4 или R5 представляет собой C1-C6 алкокси.
8. Способ по п.2, где по меньшей мере два из R1, R2, R3, R4 или R5, каждый из которых независимо представляет собой C1-C6 алкокси.
9. Способ по п.2, где по меньшей мере три из R1, R2, R3, R4 или R5, каждый из которых независимо представляет собой C1-C6 алкокси.
10. Способ по п.2, где каждый из R2 и R4 независимо представляет собой C1-C6 алкокси и каждый из R1 и R3 представляет собой водород.
11. Способ по п.10, где C1-C6 алкокси является метокси.
12. Способ по п.6, где защитный агент содержит ангидрид.
13. Способ по п.12, где ангидридом является трифторуксусный ангидрид.
14. Способ по п.2, где по меньшей мере один галогенирующий агент содержит 1,3-дибром-5,5-диметилгидантоин (DBDMN).
15. Способ по п.2, где гидратирующий агент содержит воду.
16. Способ по п.2, где гидратирующий катализатор содержит по меньшей мере одну кислоту.
17. Способ по п.16, где по меньшей мере одна кислота выбрана из трифторуксусной, серной и метансульфоновой кислоты.
18. Способ по п.2, также включающий осуществление стадий (i)-(iii) способа в реакции, проводимой в одном реакционном сосуде.
19. Способ по п.2, где в соединении II каждый из R2 и R4 независимо представляет собой C1-C6 алкокси, каждый из R1 и R3 представляет собой водород и R7 представляет собой трифторацетил.
20. Способ по п.2, где по меньшей мере один галогенирующий агент содержит N-бромосукцинимид.
21. Способ по п.2, где по меньшей мере один цианирующий агент содержит CuCN.
22. Способ по п.2, где по меньшей мере один цианирующий агент содержит K4Fe(CN)6 и CuI.
23. Способ по п.2, где соединением I является 2-амино-4,6-диметоксибензамид.
24. Способ получения 2-амино-4,6-диметоксибензамида, включающий:
(i) присоединение защитной группы к 3,5-диметоксианилину с помощью защитного агента для получения 3,5-диметоксианилина с присоединенной защитной группой;
(ii) галогенирование 3,5-диметоксианилина с присоединенной защитной группой с помощью галогенирующего агента для получения галогенированного 3,5-диметоксианилина с присоединенной защитной группой;
(iii) цианирование галогенированного 3,5-диметоксианилина с присоединенной защитной группой с помощью цианирующего агента для получения цианированного 3,5-диметоксианилина с присоединенной защитной группой;
(iv) снятие защитной группы с цианированного 3,5-диметоксианилина с присоединенной защитной группой; и
(v) гидратирование осадка, содержащего цианированный 3,5-диметоксианилин, для получения 2-амино-4,6-диметоксибензамида.
25. Способ получения 2-амино-4,6-диметоксибензамида, включающий:
(i) присоединение защитной группы к 3,5-диметоксианилину с помощью защитного агента для получения 3,5-диметоксианилина с присоединенной защитной группой;
(ii) галогенирование 3,5-диметоксианилина с присоединенной защитной группой с помощью галогенирующего агента для получения галогенированного 3,5-диметоксианилина с присоединенной защитной группой;
(iii) цианирование галогенированного 3,5-диметоксианилина с присоединенной защитной группой с помощью цианирующего агента для получения цианированного 3,5-диметоксианилина с присоединенной защитной группой;
(iv) снятие защитной группы с цианированного 3,5-диметоксианилина с присоединенной защитной группой и индуцирование осаждения для получения осадка, содержащего цианированный 3,5-диметоксианилин; и
(v) гидратирование осадка, содержащего цианированный 3,5-диметоксианилин, для получения 2-амино-4,6-диметоксибензамида.
26. Способ получения 2-амино-4,6-диметоксибензамида, включающий:
(i) присоединение защитной группы к 3,5-диметоксианилину с помощью защитного агента для получения 3,5-диметоксианилина с присоединенной защитной группой;
(ii) галогенирование 3,5-диметоксианилина с присоединенной защитной группой с помощью галогенирующего агента для получения галогенированного 3,5-диметоксианилина с присоединенной защитной группой;
(iii) цианирование галогенированного 3,5-диметоксианилина с присоединенной защитной группой с помощью цианирующего агента для получения цианированного 3,5-диметоксианилина с присоединенной защитной группой;
(iv) снятие защитной группы с цианированного 3,5-диметоксианилина с присоединенной защитной группой и индуцирование осаждения для получения первичного осадка, содержащего цианированный 3,5-диметоксианилин;
(v) повторное осаждение первичного осадка, содержащего цианированный 3,5-диметоксианилин, для получения вторичного осадка, содержащего цианированный 3,5-диметоксианилин; и
(vi) гидратирование вторичного осадка, содержащего цианированный 3,5-диметоксианилин, для получения 2-амино-4,6-диметоксибензамида.
27. Способ получения 2-амино-4,6-диметоксибензамида, включающий:
(i) присоединение защитной группы к 3,5-диметоксианилину с помощью защитного агента для получения 3,5-диметоксианилина с присоединенной защитной группой;
(ii) галогенирование 3,5-диметоксианилина с присоединенной защитной группой с помощью галогенирующего агента для получения галогенированного 3,5-диметоксианилина с присоединенной защитной группой;
(iii) снятие защитной группы с галогенированного 3,5-диметоксианилина с присоединенной защитной группой для получения галогенированного 3,5-диметоксианилина;
(iv) осаждение галогенированного 3,5-диметоксианилина;
(v) цианирование галогенированного 3,5-диметоксианилина с помощью цианирующего агента для получения цианированного 3,5-диметоксианилина;
(vi) осаждение цианированного 3,5-диметоксианилина; и
(vii) гидратирование осажденного цианированного 3,5-диметоксианилина для получения 2-амино-4,6-диметоксибензамида.
28. Способ по п.26 или 27, где 3,5-диметоксианилином с присоединенной защитной группой является 3,5-диметокситрифторацетанилид.
29. Способ по п.26 или 27, где галогенированный 3,5-диметоксианилин с присоединенной защитной группой содержит изомеры бромо-3,5-диметокситрифторацетанилида.
30. Способ по п.26 или 27, где цианированный 3,5-диметоксианилин с присоединенной защитной группой содержит изомеры циано-3,5-диметокситрифторацетанилида.
31. Способ по п.26 или 27, где защитный агент содержит ангидрид.
32. Способ по п.31, где ангидридом является трифторуксусный ангидрид.
33. Способ по п.26 или 27, где защитным агентом является хлорангидрид.
34. Способ по п.26 или 27, где галогенирующий агент содержит N-бромосукцинимид.
35. Способ по п.26 или 27, где цианирующий агент содержит CuCN.
36. Способ по п.26 или 27, где цианирующий агент содержит K4Fe(CN)6 и CuI.
37. Способ по п.26 или 27, где гидратирующий агент содержит воду.
38. Способ по п.26 или 27, где гидратирующий катализатор содержит по меньшей мере одну кислоту, выбранную из трифторуксусной кислоты, серной кислоты и метансульфоновой кислоты.
| Станок для изготовления деревянных ниточных катушек из цилиндрических, снабженных осевым отверстием, заготовок | 1923 |
|
SU2008A1 |
| US 3966965 A, 29.06.1976 | |||
| US 4137325 A, 30.01.1979 | |||
| Способ получения замещенных бензамидов или их гидрохлоридов | 1973 |
|
SU618039A3 |

