Область техники, к которой относится изобретение
Настоящее изобретение относится к способам получения 4-алкокси-3-гидроксипиколиновых кислот. Точнее, настоящее изобретение относится к способу получения 4-алкокси-3-гидроксипиколиновых кислот из фурфураля.
Уровень техники
В патенте U.S. 6521622 B1 и заявке U.S. № 61/747723 описаны, в частности, некоторые гетероциклические ароматические амиды общей формулы
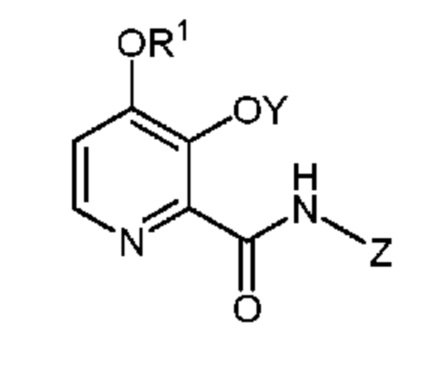
и их применение в качестве фунгицидов.
В настоящем изобретении также описано получение 4-алкокси-3-гидроксипиколиновых кислот и их производных в качестве ключевых промежуточных продуктов для получения этих гетероциклических ароматических амидов. Был бы полезен эффективный и масштабируемый способ получения 4-алкокси-3-гидроксипиколиновых кислот из недорогого сырья.
Сущность изобретения
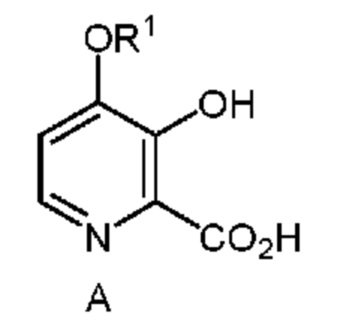
Настоящее изобретение относится к способам получения 4-алкокси-3-гидроксипиколиновых кислот формулы A
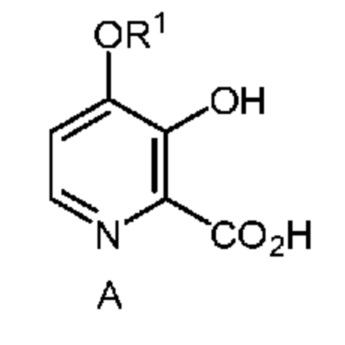
в которой R1 означает C1-C3 алкил;
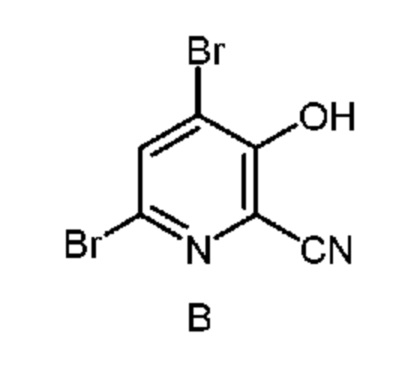
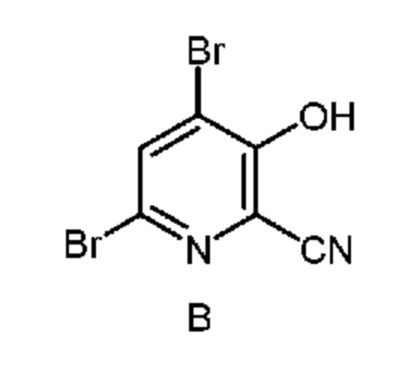
из соединения формулы B
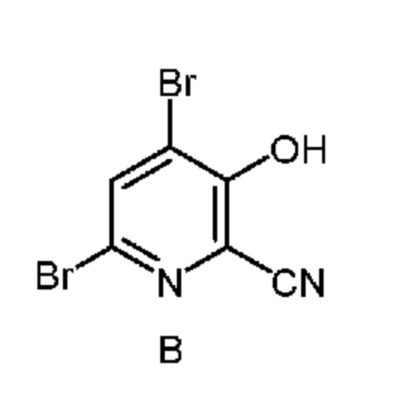
Соединение формулы A можно получить однореакторным способом, который включает следующие стадии:
a) получение первой смеси, содержащей алкоксид щелочного металла формулы C
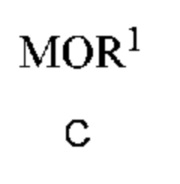
в которой M означает Na или K и R1 означает C1-C3 алкил;
и соединение формулы B, и нагревание первой смеси;
b) получение второй смеси путем добавления воды, сильного основания и металлического цинка к первой смеси;
c) нагревание второй смеси; и
d) выделение соединения формулы A.
Настоящее изобретение также относится к способу получения соединения формулы B
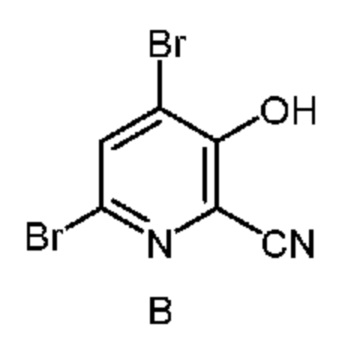
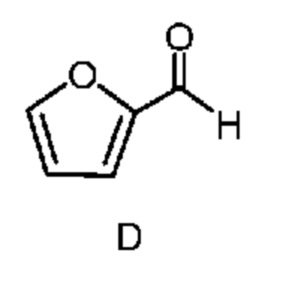
из соединения формулы D
Соединение формулы B можно получить способом, который включает следующие стадии:
a) получение первой смеси путем объединения 2-фазной системы вода-органический растворитель, источника аммония, источника цианида и соединения формулы D;
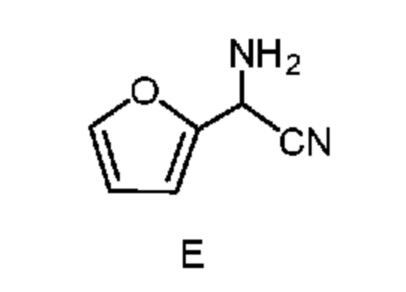
b) отделение второй смеси от первой смеси, содержащей соединение формулы E в виде раствора в органическом растворителе;
c) добавление водного раствора неорганической кислоты ко второй смеси с получением третьей смеси;
в которой неорганической кислотой является HCl, HBr, H2SO4, HNO3 или H3PO4;
d) отделение четвертой смеси от третьей смеси, которой является водная смесь, содержащая соединение формулы F;
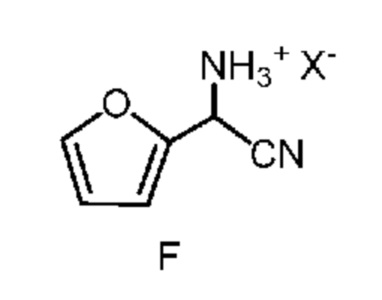
в которой X означает Cl, Br, HSO4, NO3 или H2PO4;
e) добавление бромирующего реагента, выбранного из группы, включающей:
i) бром и бромсодержащее соединение с окислителем; и
ii) бромсодержащее соединение с окислителем,
к четвертой смеси с получением пятой смеси; и
f) выделение соединения формулы B из пятой смеси.
Настоящее изобретение также относится к способу получения соединения формулы G
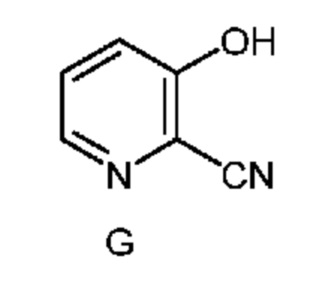
из соединения формулы D
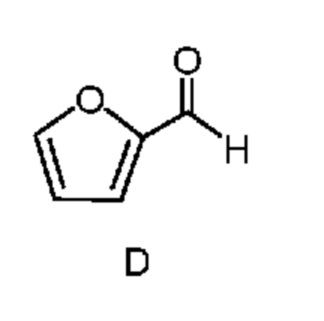
Соединение формулы G можно получить способом, который включает следующие стадии:
a) получение первой смеси путем объединения воды, органического растворителя, источника аммония, источника цианида и соединения формулы D;
b) отделение второй смеси от первой смеси, содержащей соединение формулы E в виде раствора в органическом растворителе;
c) добавление водного раствора неорганической кислоты ко второй смеси с получением третьей смеси;
в которой неорганической кислотой является HCl, HBr, H2SO4, HNO3 или H3PO4;
d) отделение четвертой смеси от третьей смеси, которой является водная смесь, содержащая соединение формулы F;
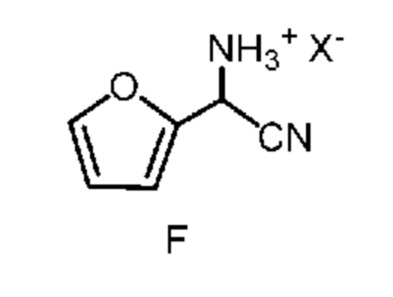
в которой X означает Cl, Br, HSO4, NO3 или H2PO4;
e) добавление бромирующего реагента, т. е. бромсодержащего соединения, с окислителем к четвертой смеси с получением пятой смеси; и
f) выделение соединения формулы G из пятой смеси.
Подробное описание изобретения
Термины "выделять", "выделение" при использовании в настоящем изобретении означают частичное или полное удаление или отделение искомого продукта от других компонентов готовой полученной химическим способом смеси по стандартным методикам, таким как, но не ограничиваясь только ими, фильтрование, экстракция, перегонка, кристаллизация, центрифугирование, истирание, разделение жидких фаз или другие методики, известные специалистам с общей подготовкой в данной области техники. Выделенный продукт может обладать чистотой в диапазоне от <50% до > 50% и его можно очистить до более высокой чистоты по стандартным методикам очистки. Выделенный продукт также можно использовать на последующей стадии способа с очисткой или без нее.
В способе, описанном в настоящем изобретении, 4-алкокси-3-гидроксипиколиновые кислоты получают из 4,6-дибром-3-гидроксипиколинонитрил с помощью последовательности химических стадий, включающих замещение брома, гидролиз нитрила и восстановление галогена. В настоящем изобретении описан улучшенный способ получения 4-алкокси-3-гидроксипиколиновых кислот из 4,6-дибром-3-гидроксипиколинонитрила с использованием более эффективного однореакторного способа.
В настоящем изобретении также описаны улучшенные способы получения 4,6-дибром-3-гидроксипиколинонитрила из фурфураля. В способах используют частичное или полное замещение бромом с помощью пары реагентов бромид/окислитель, которая образует бром in situ. При таком улучшении способа уменьшается потребность использования элементарного брома и увеличивается эффективность использования брома.
Образование брома in situ при получении 4,6-дибром-3-гидроксипиколинонитрила из фурфураля, описанное в настоящем изобретении, эквивалентно использованию элементарного брома и неожиданно установлено, что наличие окислителя не оказывает неблагоприятного влияния на реакции Штрекера или перегруппировки. Кроме того, также неожиданно установлено, что окислитель не приводит к разложению или окислению пиридинового кольца или нитрильной группы 4,6-дибром-3-гидроксипиколинонитрила.
A. Получение соединения формулы A
Описан улучшенный способ получения 4-алкокси-3-гидроксипиколиновых кислот формулы A из 4,6-дибром-3-гидроксипиколинонитрил (соединение B) с использованием более эффективного однореакторного способа. Способ включает обработку соединения формулы B сначала алкоксидом натрия и затем металлическим цинком, водным раствором сильного основания и необязательно добавление дополнительного водного раствора сильного основания и в заключение подкисление конечной реакционной смеси водным раствором сильной кислоты с получением соединения формулы A (в которой R1 означает C1-C3 алкил).
В одном варианте осуществления этого способа реакцию соединения формулы B с метоксидом натрия можно провести в диполярном, апротонном растворителе, таком как DMSO (диметилсульфоксид) или сульфолан, необязательно с добавлением метанола или в метаноле в качестве растворителя. Используют не менее 2 молярных эквивалентов метоксида натрия, предпочтительно 2,5-3 молярных эквивалентов, и нагревание при температуре от примерно 50 до примерно 80°C проводят в течение примерно 1 ч до примерно 24 ч до завершения замещения 4-бромидной группы метоксидной. Затем полученную реакционную смесь можно разбавить
Схема I
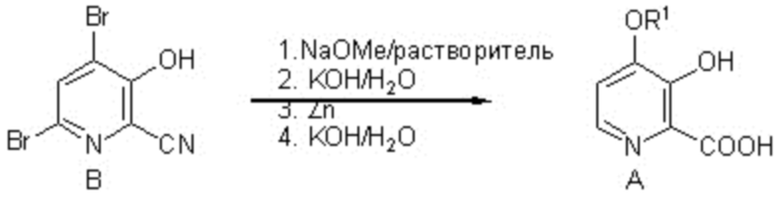
водой и водным раствором сильного основания, такого как гидроксид калия или гидроксид натрия (2-3 молярных эквивалента), обработать с помощью от примерно 1 до примерно 3 молярных эквивалентов металлического цинка (т. е. цинковой пыли с частицами размером < 10 мкм, порошкообразного цинка с частицами размером < 150 мкм или другого твердого препарата Zn с большой площадью поверхности) и перемешивать при температуре от примерно 20°C до примерно 70°C до завершения восстановления 6-бромидной группы. Затем можно дополнительно добавить водный раствор сильного основания (2-3 молярных эквивалента) и полученную смесь нагревать при температуре от примерно 80°C до примерно 95°C в течение от примерно 4 до примерно 24 ч. Искомое соединение формулы A (в которой R1 означает метил) можно выделить путем подкисления реакционной смеси и с использованием стандартных методик выделения и очистки.
В другом варианте осуществления этого способа после завершения реакции соединения формулы B с метоксидом натрия полученную реакционную смесь можно разбавить водой, добавить водный раствор сильного основания (4-6 молярных эквивалентов) и металлический цинк и затем поддерживать температуру в диапазоне от примерно 20°C до примерно 95°C в течение от примерно 2 до примерно 48 ч. После завершения реакций восстановления цинком и щелочного гидролиза искомый продукт можно выделить путем подкисления реакционной смеси и с использованием стандартных методик выделения и очистки.
В другом варианте осуществления этого способа после завершения реакции соединения формулы B с метоксидом натрия полученную реакционную смесь можно разбавить водой и добавить водный раствор сильного основания (4-6 молярных эквивалентов) и полученную смесь нагревать при температуре от примерно 80°C до примерно 95°C в течение от примерно 4 до примерно 24 ч до завершения гидролиза нитрильной группы. Затем полученную смесь можно обработать металлическим цинком и затем поддерживать температуру в диапазоне от примерно 20°C до примерно 70°C до завершения восстановления 6-бромидной группы. После завершения реакций восстановления цинком и щелочного гидролиза искомый продукт можно выделить путем подкисления реакционной смеси и с использованием стандартных методик выделения и очистки.
В другом варианте осуществления этого способа после завершения реакции соединения формулы B с метоксидом натрия гидролиз нитрильной группы и восстановление 6-бромидной группы можно провести одновременно путем добавления воды, водного раствора сильного основания и металлического цинка (одной порцией или путем добавления в течение некоторого времени) в сосуд с реакционной смесью и ее нагревания при температуре от примерно 80°C до примерно 95°C в течение времени, необходимого для завершения гидролиза нитрильной группы и восстановления 6-бромидной группы.
B. Получение соединения формулы B
Как показано на схеме II, фурфураль (формула D) в способе с использованием химических стадий a, b и c можно превратить в 4,6-дибром-3-гидроксипиколинонитрил (формула B).
Схема II
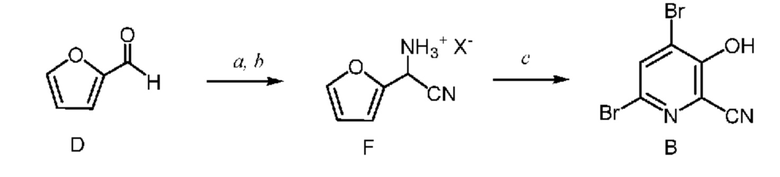
Циано(фуран-2-ил)метанаминийгалогенид формулы F получают двухфазным способом (2-фазная система вода-органический растворитель) путем проводимой сначала реакции фурфураля (формула D) не менее, чем с одним эквивалентом источника аммония и источника цианида (стадия a), по реакции,
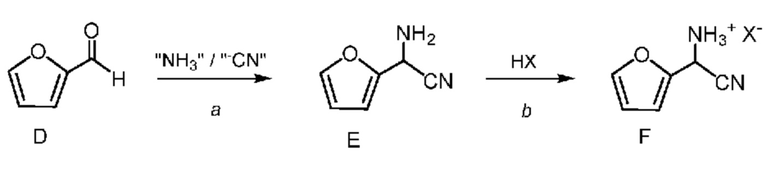
известной в данной области техники, как синтез α-аминонитрилов по Штрекеру, который описан в Organic Syntheses, Coll. Vol. I, page 21 and Coll. Vol. III, pages 84 and 88 с получением амино(фуран-2-ил)ацетонитрила формулы E. Подходящие источники аммония включают: соли аммония такие как, но не ограничиваясь только ими, ацетат аммония, бромид аммония, хлорид аммония, формиат аммония, сульфат аммония и цианид аммония; аммиак, растворенный в органическом растворителе, такой как, например, аммиак в метаноле, аммиак в этаноле и аммиак в диоксане; аммиак в воде (т. е. гидроксид аммония); и жидкий, безводный или газообразный аммиак. Подходящие источники цианида включают: цианиды, такие как, но не ограничиваясь только ими, цианид натрия, цианид калия и цианид аммония; и цианид водорода, который путем непрерывного добавления можно добавлять вместе с аммиаком к фурфуралю. Реакцию (стадию a) проводят в 2-фазной системе растворителей, включающей воду и смешивающийся с водой растворитель, выбранный из группы, включающей: простые эфиры, такие как диэтиловый эфир, метил-трет-бутиловый эфир (MTBE), тетрагидрофуран (THF) и 2-метилтетрагидрофуран (2-MeTHF); сложные эфиры, такие как этилацетат и изопропилацетат; алканы, такие как гексан, циклогексан, гептан и октан; ароматические соединения, такие как анизол, толуол и ксилол или смесь ксилолов, и их смеси. такая реакция была описана в заявке WO 2000049008, page 55. Реакцию, предлагаемую в настоящем изобретении, обычно проводят при перемешивании, достаточном для поддержания в основном однородной смеси реагентов. Такую реакцию можно провести в течение примерно 1 до примерно 50 ч при температуре от примерно 15°C до примерно 30°C.
После завершения реакции получения амино(фуран-2-ил)ацетонитрила формулы E органическую фазу 2-фазной системы растворителей, содержащую соединение формулы E, легко отделить от водной фазы по стандартным методикам разделения фаз и экстракции. Соединение формулы E в виде раствора в органической фазе после этого превращают в соль формулы F путем обработки водным раствором неорганической кислоты. Подходящие неорганические кислоты включают, но не ограничиваются только ими, бромистоводородную кислоту (HBr), хлористоводородную кислоту (HCl), азотную кислоту (HNO3), серную кислоту (H2SO4) и фосфорную кислоту (H3PO4). Реакцию, предлагаемую в настоящем изобретении, можно провести при температуре от примерно 0°C до примерно 25°C. После соответствующего смешивания органической фазы, содержащей соединение формулы E, и водного раствора неорганической кислоты, водный раствор кислоты, содержащий циано(фуран-2-ил)метанаминийгалогенид формулы F, отделяют от органической фазы по стандартным методикам разделения фаз и экстракции и он готов для проведения конечной реакции бромирование/перегруппировка (схема II, стадия c) получения соединения формулы B.
На стадии бромирование/перегруппировка реакции способа соль циано(фуран-2-ил)метанаминия формулы F обрабатывают бромирующим реагентом, таким как бром, и получают продукт формулы B. Исходное вещество формулы F, в которой X означает Br, Cl,
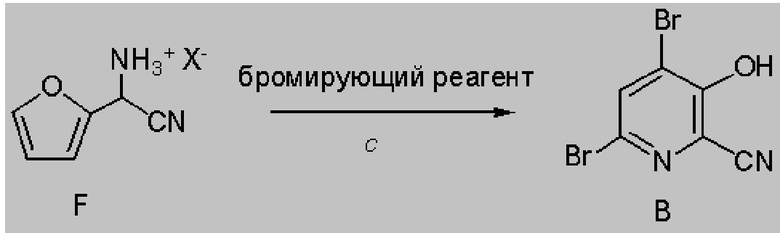
NO3, HSO4 или H2PO4, можно обработать подходящим бромирующим реагентом. Можно использовать от примерно 3 до примерно 6 молярных эквивалентов брома можно использовать. Реакцию предпочтительно проводят с использованием примерно 3-5 молярных эквивалентов брома и бромида соединения формулы F (X=Br). Часто удобно использовать избыток бромирующего реагента, такой как составляющий 5%, 10% или 15% молярный избыток, чтобы реакция протекала до конца. Реакцию предпочтительно проводят в протонном растворителе или реакционной среде, такой как вода, или смеси воды и растворимого в воде органического растворителя, такого как, например, метанол, этанол, тетрагидрофуран, диоксан или ацетонитрил. Температура, при которой проводят реакцию, обычно равна от примерно 10°C до примерно 25°C. После завершения добавления брома реакционной смеси можно дать медленно нагреться до комнатной температуры и перемешивать в течение 10-48 ч или реакционную смесь можно нагревать примерно при 30-40°C для завершения реакции. Продолжительность реакции необязательно можно сократить путем добавления к реакционной смеси основания, такого как, например, 2-4 молярных эквивалента ацетата натрия. После завершения реакции искомый продукт выделяют с использованием стандартных методик выделения и очистки.
В некоторых вариантах осуществления настоящего изобретения, бромирование/перегруппировка соединения формулы F может включать использование одного или большего количества бромирующих реагентов, выбранных из группы, включающей: (1) бром и (2) бромсодержащее соединение, в паре с окислителем. Из литературы известно, что бромиды, такие как, например, HBr, KBr и NaBr, при объединении с окислителем, таким как, например, пероксид водорода, пероксимоносульфат калия (т. е. оксон®), DMSO или трет-бутилгидропероксид, при проведении реакции в подходящих условиях могут образовывать бром (в настоящем изобретении это называется образованием брома in situ). Для использования бромсодержащего соединения, которое является солью, такой как, например, NaBr или KBr, для образования брома in situ также необходимо использование кислоты для образования брома). Кислоту можно выбрать из группы, включающей HBr, HCl, H2SO4, HNO3, H3PO4, уксусную кислоту и их смеси. Такой подход, который включает образование брома in situ, брома обеспечивает следующие преимущества: ограничение или исключение использования элементарного брома, повышение эффективности использования брома в способе и уменьшение образования и удаления потоков содержащих бромиды отходов.
В некоторых вариантах осуществления настоящего изобретения использование бромсодержащего соединения, такого как, например, HBr, KBr, или NaBr, в паре с окислителем, таким как пероксид водорода, в способе получения соединения формулы B из соединения формулы F (X=Br) можно осуществить путем медленного добавления пероксида водорода (окислитель) к соединению формулы F и бромсодержащему соединению (т. е. к KBr или NaBr в качестве бромсодержащего соединения, для чего необходимо использование кислоты для образования брома in situ) при температуре окружающей среды и поддержании во время добавления температуры равной ниже примерно 50°C. От примерно 3-5 молярных эквивалентов пероксида водорода в пересчете на соединение формулы B можно использовать в присутствии в способе достаточного количества бромсодержащего соединения (2-5 молярных эквивалентов) и кислоты.
Химическая литература, в которой описано использование бромидов с окислителями для проведения реакций бромирования включает: a) "Simple and Practical Halogenation of Arenes, Alkenes, and Alkynes with Hydrohalic Acid/H2O2 (or TBHP)", Tetrahedron, 55, (1999) 1127-1142, b) "Oxidative Halogenation with "Green" Oxidants: Oxygen and Hydrogen Peroxide", Angew. Chem. Int. Ed., 2009, 48, 8424 и цитированную в ней литературу. Патенты, в которых описано получение брома по реакции бромидов или HBr с пероксидом водорода, включают U.S. 5266295, U.S. 4029732 и U.S. 2772302.
C. Получение соединения формулы G
Другой вариант осуществления настоящего изобретения включает способ получения соединения формулы G фурфураля. На первой стадии этого способа фурфураль превращают в циано(фуран-2-ил)метанаминийбромид формулы F (X означает Br) двухфазным способом, описанным в настоящем изобретении. На следующей стадии способа бромид формулы F объединяют с дополнительным количеством водного раствора HBr (1,5 экв.) и затем вводят в реакцию с равным от примерно 3 до примерно 4 количеством молярных эквивалентов пероксида водорода (в пересчете на бромид формулы F) и получают 3-гидроксипиколинонитрил (формула G).
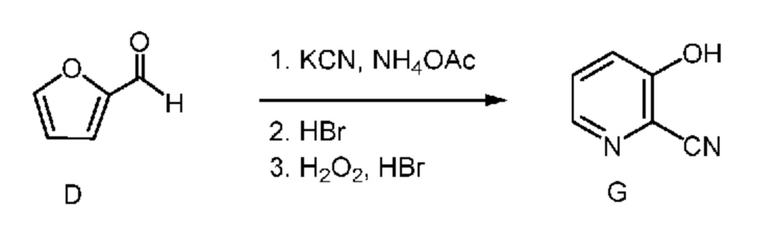
Температура, при которой можно провести добавление пероксида водорода, равна от примерно 0°C до примерно 50°C. После завершения добавления пероксида водорода реакционную смесь перемешивают при комнатной температуре в течение от примерно 1 до примерно 24 ч. После завершения реакции искомый продукт выделяют с использованием стандартных методик выделения и очистки.
D. Получение соединения формулы H
Превращение 4-алкокси-3-гидроксипиколиновой кислоты формулы A в 3-ацэтоксисоединение формулы H можно провести путем ацетилирования соединения формулы A одним или большим количеством ацетилирующих реагентов, выбранных из группы, включающей уксусный ангидрид и ацетилхлорид, оснований, выбранных из группы, включающей пиридин, алкилзамещеные пиридины и триалкиламины, или при использовании условий проведения реакции Шоттена-Баумана.

Продукт, полученный любым из этих способов, можно выделить обычными средствами, такими как выпаривание, фильтрование или экстракция, и можно очистить по стандартным методиками, таким как перекристаллизация или хроматография.
Приведенные ниже примеры представлены для иллюстрации настоящего изобретения.
Примеры
Пример 1a. 3-Гидрокси-4-метоксипиколиновая кислота
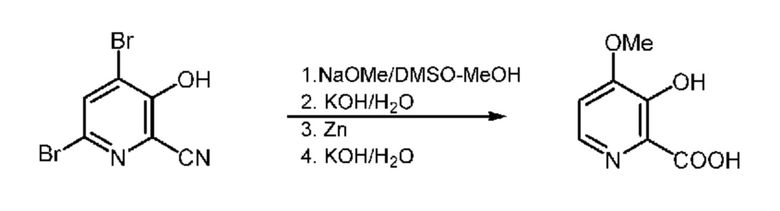
Суспензию метоксида натрия (25 г, 0,45 моля) получали с использованием 50 мл безводного DMSO и 1 мл MeOH. К этой суспензии добавляли раствор 4,6-дибром-3-гидрокси-2-пиколинонитрила (50 г, 0,181 моля) и примерно 50 мл безводного DMSO, который добавляли в течение 30 мин. Во время добавления реакционной смеси температуру поддерживали равной 50-65°C. После завершения добавления, реакционную смесь перемешивали в течение еще 1 ч при температуре >50°C. Завершение реакции устанавливали путем анализа с помощью 1H ЯМР. Реакционной смеси давали охладиться до 35°C и затем к раствору реакционной смеси добавляли 100 мл воды, затем 45% KOH (40 мл, 468 ммоля). Затем порциями по 5 г с интервалами по 15 мин добавляли цинковую пыль (15,4 г 234 ммоля; размер частиц < 10 мкм), что приводило к повышению температуры примерно до 45°C. Реакционную смесь перемешивали в течение ночи при температуре окружающей среды. Реакция не завершалась, поэтому реакционную смесь нагревали при 50°C и затем дополнительно добавляли цинковую пыль (4,8 г, 74 ммоля). Реакция завершалась через 3 ч. К реакционной смеси добавляли водный раствор KOH (45% водный раствор, 40 мл, 468 ммоля). Затем реакционную смесь нагревали при температуре 94°C в течение 12 ч до завершения гидролиза. Реакционную смесь охлаждали до температуры окружающей среды и затем фильтровали для удаления твердых веществ. Твердые вещества промывали путем добавления примерно 100 мл воды к раствору реакционной смеси. Затем pH объединенного фильтрата и промывочного раствора устанавливали равным 0,4 с помощью 12 н. HCl. Полученную смесь перемешивали в течение примерно 1 ч для обеспечения стабильного значения pH и затем твердые вещества собирали фильтрованием. Полученные почти белые твердые вещества промывали ацетоном. Вещество сушили в вакуумном сушильном шкафу при температуре 50°C и получали 4-метокси-3-гидроксипиколиновую кислоту в виде очень бледно-желтого порошка (19,22 г, выход 63,2% при чистоте 96%, что соответствует выходу 60,7%). По данным определения с помощью ВЭЖХ чистота органического соединения составляла 99,75%. 1H ЯМР (400 МГц, DMSO-d6) δ 8,03 (d, J=6,4 Гц, 1H), 7,39 (d, J=6,4 Гц, 1H), 4,04 (s, 3H).
Пример 1b. 3-Гидрокси-4-метоксипиколиновая кислота
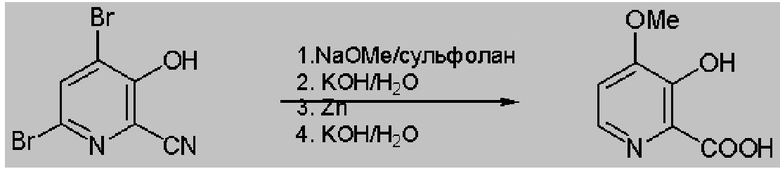
Неразведенный метоксид натрия (14,7 г, 271 ммоля) добавляли к раствору 4,6-дибром-3-гидроксипиколинонитрила (30,2 г, 109 ммоля) и сульфолана (120 г) в течение 30 мин, что приводило к повышению температуры до 50°C. Затем реакционную смесь нагревали при 60°C в течение 18 ч. Раствору реакционной смеси давали охладиться до температуры окружающей среды и затем к реакционной смеси добавляли 150 мл ДИ (деионизированной) воды, затем 50 мл 45 мас.% KOH (5,4 экв., 586 ммоль). Добавляли цинковую пыль (113 ммоля, 7,5 г) и затем реакционную смесь нагревали при 40°C. Через 2 ч дополнительно добавляли цинковую пыль (2,5 г, 38 ммоля) и затем реакционную смесь нагревали при 60°C в течение еще 2 ч. Реакционную смесь перемешивали в течение ночи при температуре окружающей среды. Анализ реакционной смеси с помощью 1H ЯМР показал, что дебромирование завершилось. Реакционную смесь фильтровали для удаления твердых веществ, к фильтрату добавляли 45% KOH (50 мл, 596 ммоля) и затем полученный раствор нагревали примерно до 90°C. Реакционную смесь перемешивали примерно при 90°C в течение 5,5 ч, что приводило к почти полному превращению. Реакционную смесь перемешивали примерно при 90°C в течение ночи. Реакционную смесь охлаждали до < 30°C и затем значение pH устанавливали равным 0,8 с помощью 40% серной кислоты, что приводило к образованию твердых веществ. Твердые вещества выделяли фильтрованием и затем сушили и получали твердое вещество, количество которого соответствовало выходу. превышающему 100%. Вещество суспендировали в течение ночи в 0,5 pH хлористоводородной кислоте. Затем вещество выделяли фильтрованием и сушили и получали 10,3 г 4-метокси-3-гидроксипиколиновой кислоты в виде почти белого порошкообразного вещества, которое по данным ВЭЖХ обладало чистотой 94% (выход 53%).
Пример 1c. 3-Гидрокси-4-метоксипиколиновая кислота
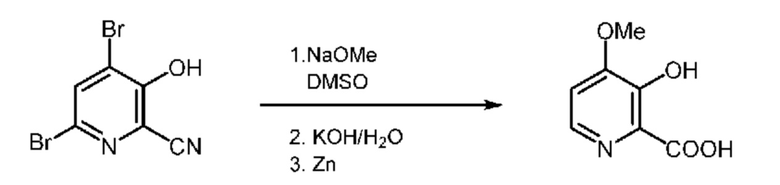
В 3-горлую колбу объемом 500 мл помещали метоксид натрия (25 г, 0,462 моля) и 25 мл диметилсульфоксида. Смесь метоксид натрия/DMSO помещали в атмосферу инертного газа и механически перемешивании и получали текучую суспензию. В отдельном сосуде получали раствор 4,6-дибром-3-гидроксипиколинонитрила (50,3 г, 0,181 моля, DBHP (2,5-диметил-2,5-ди-трет-бутилпероксигексан), чистота 96,2 мас.%) примерно в 25 мл безводного DMSO. Раствор DBHP шприцевым насосом добавляли к смеси метоксид натрия/DMSO в течение 50 мин. Во время добавления температуру поддерживали равной ниже 60°C. После завершения добавления реакционную смесь перемешивали в течение еще 1 ч. За это время реакционная смесь затвердевала. К затвердевшей реакционной смеси добавляли 100 мл воды, затем 50% KOH (50 мл, 941 ммоля). Полученную смесь перемешивали в течение примерно 1,5 ч для разрушения твердых веществ с образованием густой дисперсии. Затем порциями примерно по 5 г с интервалами примерно по 20 мин добавляли цинковую пыль (14,8 г, 226 ммоля), что приводило к повышению температуры примерно до 40°C. Во время растворения Zn реакционная смесь разжижалась с образованием легкое перемешиваемой дисперсии. Реакционную смесь перемешивали в течение ночи при температуре окружающей среды. Затем реакционную смесь нагревали до 95°C в течение 24 ч. Реакционную смесь охлаждали до <20°C и затем значение pH раствора устанавливали равным 0,6 водным раствором HCl (12 н.), что приводило к осаждению продукта. Твердые вещества выделяли фильтрованием, промывали с помощью примерно 50 мл воды и затем промывали с помощью примерно 25 мл ацетона. Полученному желтоватому порошку давали высыхать в вытяжном шкафу и получали 23,3 г продукта. Продукт обладал чистотой 96% по данным 1H ЯМР (относительно внутреннего стандарта), что соответствовало равному 76% выходу искомого продукта в пересчете на чистоту исходного вещества и конечного продукта. 1H ЯМР (400 МГц, DMSO-d6) δ 8,03 (d, J=6,4 Гц, 1H), 7,39 (d, J=6,4 Гц, 1H), 4,04 (s, 3H).
Пример 1d. Циано(фуран-2-ил)метанаминийбромид
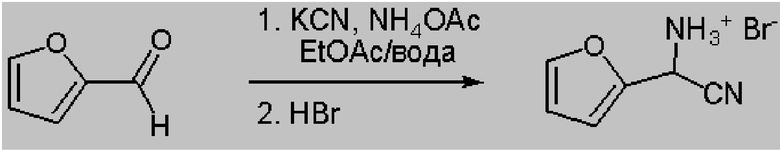
В колбу объемом 500 мл, снабженную стержнем для перемешивания, добавляли 33,65 г ацетата аммония (436 ммоля), 150 мл этилацетата, 30 мл ДИ воды и 10 г KCN (154 ммоля). Затем в реактор шприцем добавляли фурфураль (14 г, 145 ммоля). Температура в реакторе повышалась примерно от 15°C до 24°C. Реакционную смесь перемешивали в течение ночи при температуре окружающей среды. Анализ этилацетатной фазы с помощью 1H ЯМР показывал, что степень превращения составила > 95%. В реактор добавляли 75 мл 20% водного раствора карбоната натрия и перемешивали в течение 10 мин. Раствор карбоната натрия удаляли и затем реакционную смесь промывали с помощью 40 мл насыщенного рассола. 1H ЯМР (400 МГц, DMSO-d6) δ 7,53 (dd, J=2,0, 1,0 Гц, 1H), 6,47 (dd, J=3,4, 1,1 Гц, 1H), 6,42 (dd, J=3,3, 1,7 Гц, 1H), 5,08 (s, 1H).
После удаления фазы рассола к реакционной смеси добавляли 24,5 мл 48% водного раствора HBr (1 экв., 145 ммоля), разбавленного примерно в 130 мл ДИ воды. Реакционную смесь перемешивали в течение 15 мин. Водный слой удаляли и помещали в отдельный сосуд. Затем органический слой промывали с помощью 2×25 мл ДИ воды. Каждый промывочный раствор добавляли в сосуд для хранения, содержащий начальную содержащую HBr экстрагированную фазу. Получали всего 210,5 г водной фазы, содержащей примерно 14,06 мас.% циано(фуран-2-ил)метанаминийбромида.
Пример 1e. 4,6-Дибром-3-гидроксипиколинонитрил
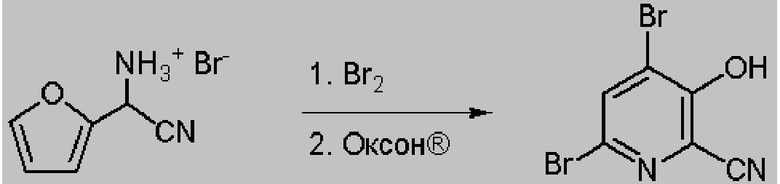
52,5 г Водной фазы, содержащей 7,38 г (36 ммоля) циано(фуран-2-ил)метанаминийбромида (14,06 мас.% в воде) помещали в колбу объемом 250 мл, снабженную стержнем для перемешивания. Затем колбу помещали в баню со льдом. После охлаждения до <10°C к реакционной смеси по каплям в течение 15 мин добавляли 5,8 г брома (36 ммоля), что приводило к образованию твердых веществ. После перемешивания в течение 1 ч реакционной смеси давали нагреться до температуры окружающей среды. К реакционной смеси порциями добавляли оксон® (27 г, 87,8 ммоля), что приводило к растворению твердых веществ и образованию красновато-коричневой жидкой фазы, которая после перемешивания в течение 1 ч медленно превращалась в круглое пеллетоподобное вещество. Реакцию останавливали насыщенным водным раствором бисульфита натрия. Затем твердые вещества выделяли фильтрованием, промывали ДИ водой и затем сушили в течение ночи и получали 6,25 г желтовато-коричневого порошка. Анализ с помощью 1H ЯМР показывал, что продукт содержал 4,6-дибром-3-гидроксипиколинонитрил (96,6 мол.%, выход 60,3%) и 6-бром-3-гидроксипиколинонитрил (3,4 мол.%, выход 2,2%). 1H ЯМР (400 МГц, DMSO-d6) δ 8,27 (s, 1,00H), 7,75 (d, J=8,9 Гц, 0,034H), 7,44 (d, J=8,9 Гц, 0,034H). HRMS (масс-спектроскопия высокого разрешения) (m/z) в режиме положительных ионов [M+1] рассчитано для C6H3Br2N2O 276,8612; найдено 276,8611.
Пример 1f. 4,6-Дибром-3-гидроксипиколинонитрил
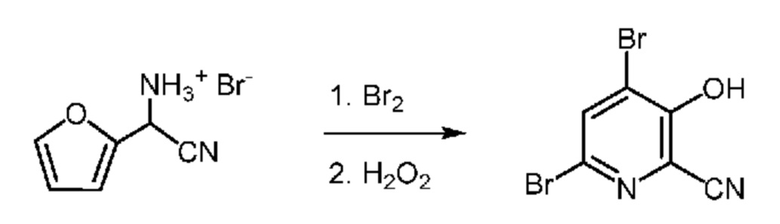
52,5 г Водной фазы, содержащей 7,38 г (36 ммоля) циано(фуран-2-ил)метанаминийбромида (14,06 мас.% в воде) помещали в колбу объемом 250 мл, снабженную стержнем для перемешивания. Затем колбу помещали в баню со льдом. После охлаждения до <10°C к реакционной смеси по каплям в течение примерно 15 мин добавляли 5,8 г брома (36 ммоля), что приводило к образованию твердых веществ. После перемешивания в течение 1 ч к реакционной смеси шприцем в течение 20-30 мин добавляли 30% пероксида водорода (9,4 мл). Это приводило к растворению твердых веществ и затем осаждению мелкодисперсного порока в течение 1-2 ч. Реакцию останавливали насыщенным раствором бисульфита натрия. Затем твердые вещества выделяли фильтрованием, промывали ДИ водой и затем сушили в течение ночи и получали 6,03 г желтовато-коричневого порошка. Анализ с помощью 1H ЯМР показывал, что продукт содержал 4,6-дибром-3-гидроксипиколинонитрил (94,5 мол.%, выход 57,3%) и 6-бром-3-гидроксипиколинонитрил (5,5 мол.%, выход 3,2%). 1H ЯМР (400 МГц, DMSO-d6) δ 8,27 (s, 1,00H), 7,75 (d, J=8,9 Гц, 0,075H), 7,44 (d, J=8,9 Гц, 0,075H). 13C ЯМР (101 МГц, DMSO-d6) δ 157,65, 141,95, 135,55, 128,76, 124,37, 120,34, 115,97. HRMS (m/z) в режиме положительных ионов [M+1] рассчитано для C6H3Br2N2O+ 276,8612; найдено 276,8609.
Пример 1g. 3-Гидроксипиколинонитрил
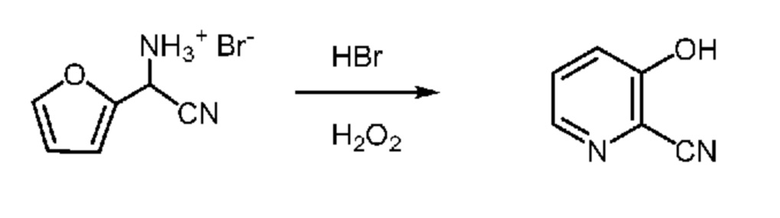
52,5 г Водной фазы, содержащей 7,38 г (36 ммоля) циано(фуран-2-ил)метанаминийбромид (14,06 мас.% в воде), помещали в колбу объемом 250 мл, снабженную стержнем для перемешивания. В колбу при перемешивании добавляли 48% HBr (6,2 мл, 55 ммоля). Затем колбу помещали в баню со льдом. После охлаждения до <5°C к реакционной смеси шприцем в течение 20-30 мин добавляли примерно 7 мл 30% пероксида водорода. Это приводило к выделению очень небольшого количества тепла. Реакционной смеси давали нагреться до температуры окружающей среды, затем реакционная смесь начинала самопроизвольно нагреваться примерно до 50°C. Реакционную смесь охлаждали до 20°C и затем добавляли 7 мл 30% пероксида, что приводило к образованию осадка. Реакционную смесь перемешивали в течение примерно 20 мин и затем реакцию останавливали насыщенным раствором бисульфита натрия, что приводило к повышению температуры примерно до 40°C. Во время повышения температуры твердые вещества растворялись. Затем реакционную смесь помещали в баню со льдом. После перемешивания в течение примерно 45 мин образовывались твердые вещества. Твердые вещества собирали фильтрованием и промывали ДИ водой. 3-Гидроксипиколинонитрил (1,63 г) выделяли в виде желтовато-коричневого кристаллического твердого вещества (выход 37,3%). 1H ЯМР (400 МГц, DMSO-d6) δ 11,67 (s, 1H), 8,19 (dd, J=4,4, 1,3 Гц, 1H), 7,56 (dd, J=8,6, 4,4 Гц, 1H), 7,47 (dd, J=8,7, 1,4 Гц, 1H). 13C ЯМР (101 МГц, DMSO-d6) δ 157,67, 141,93, 135,56, 128,75, 125,99, 124,37, 120,34, 115,97. HRMS (m/z) в режиме отрицательных ионов [M-1] рассчитано для C6H4N2O 119,0246; найдено 119,0240.
Пример 1h. 4,6-Дибром-3-гидроксипиколинонитрил
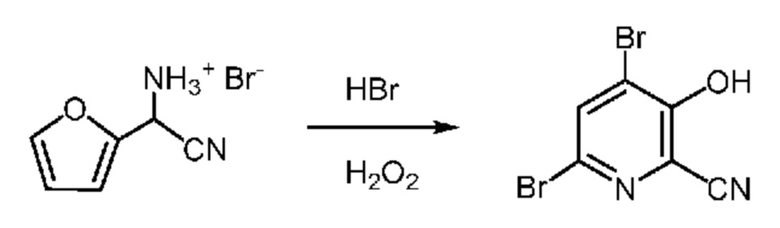
53 г Водной фазы, содержащей 7,45 г (37 ммоля) циано(фуран-2-ил)метанаминийбромид (14,06 мас.% в воде) помещали в колбу объемом 250 мл, снабженную стержнем для перемешивания. В колбу при перемешивании добавляли 48% HBr (8,2 мл, 73 ммоля). Колбу помещали в баню со льдом. После охлаждения до <5°C к реакционной смеси шприцем в течение 20-30 мин добавляли от 6 до 7 мл 30% пероксида водорода. Это приводило к выделению очень небольшого количества тепла. Реакционной смеси давали нагреться до температуры окружающей среды, затем реакционная смесь начинала самопроизвольно нагреваться примерно до 46-48°C и приобретала желто-оранжевый цвет (равномерный). Реакционную смесь охлаждали до 20°C и затем шприцем в течение 15-20 мин добавляли еще 7 мл 30% пероксида водорода, что приводило к образованию осадка. Реакционную смесь перемешивали в течение ночи. Реакцию останавливали бисульфитом натрия и получали желтоватый раствор с твердыми веществами. Полоски для пероксидного теста указывали на отсутствие остаточных пероксидов. Твердые вещества собирали фильтрованием, промывали водой и сушили и получали 6,22 г желтовато-коричневого порошка. Анализ с помощью 1H ЯМР показывал, что продукт содержал 4,6-дибром-3-гидроксипиколинонитрил (выход 58,1%) и 6-бром-3-гидроксипиколинонитрил (выход 3,8%). 1H ЯМР (400 МГц, DMSO-d6) δ 8,27 (s, 1,00H), 7,75 (d, J=8,9 Гц, 0,064H), 7,44 (d, J=8,9 Гц, 0,064H).
Пример 1i. 4,6-Дибром-3-гидроксипиколинонитрил
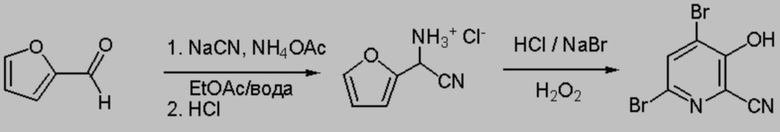
Исходный раствор циано(фуран-2-ил)метанаминийхлорида получали с использованием 39,21 г фурфураля, 20 г цианида натрия, 96 г ацетата аммония в 300 мл этилацетата и 260 мл воды. После образования α-аминонитрила к смеси добавляли 75 мл насыщенного раствора карбоната натрия и перемешивали в течение 20-30 мин. Водную фазу удаляли и затем органическую фазу промывали с помощью 2×50 мл насыщенного рассола. К органической фазе добавляли 34 мл 12 н. водного раствора HCl (1 экв., 408 ммоля), разбавленной примерно в 260 мл ДИ воды. Полученную смесь перемешивали (>500 об/мин) в течение 15 мин. После осаждения водный слой, содержащий циано(фуран-2-ил)метанаминийхлорид удаляли и помещали в пластмассовый сосуд для хранения в качестве исходного раствора. Затем органический слой экстрагировали с помощью 44 мл ДИ воды, затем с помощью 46 мл ДИ воды. Каждый водный экстракт помещали в сосуд для хранения и получали примерно 460 г водной фазы. Водную фазу разбавляли всего до 480 г и она содержала примерно 64,70 г (13,5 мас.%) циано(фуран-2-ил)метанаминийхлорида.
60 г Исходного раствора, содержащего примерно 8,1 г (51 ммоля циано(фуран-2-ил)метанаминийхлорида) помещали круглодонную колбу объемом 250 мл, снабженную стержнем для перемешивания. В колбу добавляли примерно 4,2 мл (50,4 ммоля) 12 н. HCl и 10,4 г (101 ммоля) NaBr. В течение 50 мин в колбу по каплям добавляли 30% пероксид водорода (20 г, 176 ммоля). В течение 25 мин во время добавления (когда было добавлено примерно 7,5 г пероксида) реакционная смесь самопроизвольно нагревалась до 56°C, затем реакционную смесь охлаждали примерно до 36°C. Через 40 мин начинались образовываться твердые вещества. Реакционную смесь перемешивали в течение еще 6 ч. Твердые вещества собирали фильтрованием, промывали ДИ водой и затем сушили. Получали 6,28 г сыпучего светло-желтовато-коричневого порошка, по данным 1H ЯМР представляющего собой смесь 4,6-дибром-3-гидроксипиколинонитрила (выход 39,1 мас.%), 6-бром-3-гидроксипиколинонитрила (3,0 мас.% выход), 6-хлор-3-гидроксипиколинонитрила (выход 6,23 мас.%) и одного из изомеров 4/6-хлор/бром-3-гидроксипиколинонитрила (выход 0,4 мас.%). Полный выход составлял 51%. 1H ЯМР (400 МГц, DMSO-d6) искомого продукта: δ 8,27 (s, 1,00H), δ 8,18 (s, 0,11H), δ 7,75 (d, J=8,9 Гц, 0,64H), 7,65 (d, J=8,9 Гц, 0,01H), 7,53 (d, J=8,9 Гц, 0,01H), 7,44 (d, J=8,9 Гц, 0,064H).
Пример 1j. 4,6-Дибром-3-гидроксипиколинонитрил
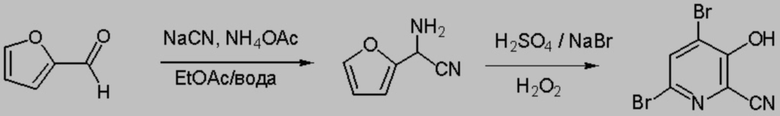
В колбу объемом 500 мл, снабженную стержнем для перемешивания, добавляли 36 г ацетата аммония (467 ммоля), 200 мл этилацетата и 7,5 г NaCN (153 ммоля). 75 мл Воды использовали для смывания остаточного цианида натрия в колбу и из воронки. Затем в реактор шприцем быстро добавляли фурфураль (12,7 мл, 14,7 г, 153 ммоля). Температура в реакторе повышалась примерно от 15°C до 24°C. Реакционную смесь перемешивали в течение ночи при температуре окружающей среды (18°C). Перемешивание прекращали, чтобы могли разделиться две жидкие фазы. Затем отбирали образец органической фазы для анализа с помощью 1H ЯМР и устанавливали, что степень превращения составила лишь примерно 80%. Затем реакционную смесь перемешивали при 25°C (на водной бане) в течение еще 6 ч и с помощью 1H ЯМР устанавливали, что степень превращения составила примерно 90%. В реактор добавляли 75 мл 20% водного раствора карбоната натрия и перемешивали в течение 30 мин, и затем смесь выдерживали без перемешивания в течение 20-30 мин. Водную фазу удаляли и затем органическую фазу, содержащую α-аминонитрил фурфураля в этилацетате, промывали с помощью 2×50 мл насыщенного рассола.
10 н. Серную кислоту (15 мл, 1 экв., 153 ммоля) разбавляли примерно в 225 мл ДИ воды. Раствор в этилацетате, содержащий α-аминонитрил фурфураля, экстрагировали разбавленным раствором серной кислоты порциями примерно по 1/3. Каждый экстракт помещали в круглодонную колбу объемом 500 мл, снабженную стержнем для перемешивания. Органический раствор экстрагировали с помощью еще 5 мл ДИ воды. К объединенным водным кислотным экстрактам добавляли 47 г бромида натрия (459 ммоля) и затем в течение 2 ч добавляли пероксид водорода (30%, 360 ммоля), что приводило к повышению температуры от 19°C примерно до 50°C. Реакционную смесь перемешивали в течение ночи. 1 Анализ с помощью 1H ЯМР показывал, что реакционная смесь представляла собой 1:1 смесь 6-бром-3-гидроксипиколинонитрила и 4,6-дибром-3-гидроксипиколинонитрила. К раствору реакционной смеси добавляли еще 15 мл 10 н. серной кислоты и 13,5 г 30% пероксида (107 ммоля) и реакционную смесь нагревали при 45°C. По данным анализа с помощью 1H ЯМР через 2 ч реакция завершалась. Твердые вещества собирали фильтрованием, промывали водой и сушили и получали 21,9 г светло-желтовато-коричневого порошка. Анализ с помощью 1H ЯМР показывал, что порошок содержал 4,6-дибром-3-гидроксипиколинонитрил (выход 49,8%) и 6-бром-3-гидроксипиколинонитрил (выход 2,4%).
Пример 1k. 3-(Ацетилокси)-4-метоксипиколиновая кислота
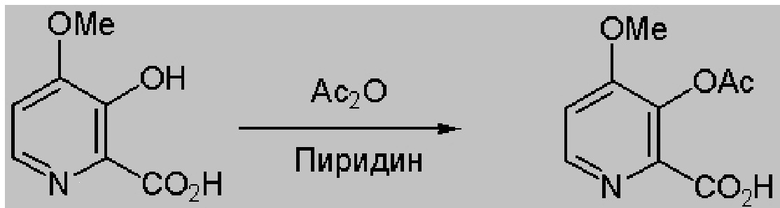
3-Гидрокси-4-метоксипиколиновую кислоту (5,0 г, 29,6 ммоля) диспергировали в 50 мл пиридина и 50 мл уксусного ангидрида при температуре окружающей среды. Через 1 ч образовывался желтый раствор, который затем перемешивали в течение ночи. Раствор выпаривали при 45°C (2 мм рт. ст.) и получали 6,28 г желтовато-коричневого твердого вещества (выход 99%, температура плавления=132-134°C). 1H ЯМР (400 МГц, DMSO-d6) δ 13,32 (s, 1H), 8,43 (d, J=5,5 Гц, 1H), 7,40 (d, J=5,5 Гц, 1H), 3,91 (s, 3H), 2,27 (s, 3H). 13C ЯМР (101 МГц, DMSO-d6) δ 167,95, 164,81, 158,34, 147,87, 142,77, 136,18, 110,87, 56,59, 20,27. HRMS (m/z) рассчитано для C9H9NO5 211,0478, найдено 211,0481 ([M]+).
| название | год | авторы | номер документа |
|---|---|---|---|
| ЗАМЕЩЕННЫЕ БЕНЗОЛЬНЫЕ СОЕДИНЕНИЯ | 2012 |
|
RU2629118C2 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛО[1,5-А]ПИРАЗИН-4-ИЛА В КАЧЕСТВЕ JAK-ИНГИБИТОРОВ | 2017 |
|
RU2718902C2 |
| СПОСОБ ЛЕЧЕНИЯ | 2012 |
|
RU2621148C2 |
| ПРОИЗВОДНОЕ 5-(2-ИМИДАЗОЛИНИЛАМИНО)БЕНЗИМИДАЗОЛА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1998 |
|
RU2193562C2 |
| БИАРИЛЬНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ БЕЛОК-БЕЛКОВОГО ВЗАИМОДЕЙСТВИЯ YAP/TAZ-TEAD | 2021 |
|
RU2830596C1 |
| АРИЛ-ИЛИ ГЕТЕРОАРИЛЗАМЕЩЕННЫЕ БЕНЗОЛЬНЫЕ СОЕДИНЕНИЯ | 2012 |
|
RU2632193C2 |
| ПРОИЗВОДНОЕ ТИОФЕНА И ЕГО ПРИМЕНЕНИЕ | 2019 |
|
RU2781643C2 |
| СОЕДИНЕНИЕ БЕНЗОИНДАЗОЛОНА И ЕГО ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ | 2020 |
|
RU2782882C1 |
| Соединения бензотиа(ди)азепина и их применение в качестве модуляторов желчных кислот | 2020 |
|
RU2838220C1 |
| СПОСОБ ПОЛУЧЕНИЯ АНТАГОНИСТОВ РЕЦЕПТОРОВ CGRP | 2013 |
|
RU2672056C2 |
Изобретение относится к способам получения соединения формулы А, в частности к способу получения соединения формулы А, в которой R1 означает C1-C3 алкил, из соединения формулы B, который включает следующие стадии a) получение первой смеси, содержащей алкоксид щелочного металла формулы C, в которой M означает Na или K и R1 означает C1-C3 алкил, растворитель, выбранный из группы, включающей DMSO, метанол, сульфолан и их смеси; и соединение формулы B, и нагревание первой смеси; b) получение второй смеси путем добавления воды, сильного основания и металлического цинка к первой смеси; c) нагревание второй смеси; и d) выделение соединения формулы A. Таким образом 4-алкокси-3-гидроксипиколиновые кислоты можно легко получить из 4,6-дибром-3-гидроксипиколинонитрила с помощью последовательности химических стадий, выбранных из группы, включающей замещение брома, гидролиз нитрила и восстановление галогена, которые проводят однореакторным способом. 4,6-Дибром-3-гидроксипиколинонитрил можно получить из фурфураля с помощью последовательности химических стадий, выбранных из группы, включающей цианоаминирование, получение соли амина и бромирование-перегруппировку. 2 н. и 8 з.п. ф-лы, 11 пр.
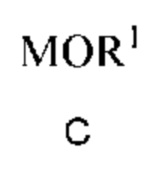
1. Способ получения соединения формулы A
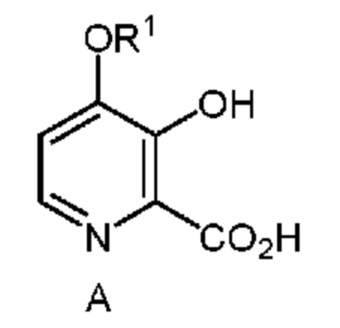
в которой R1 означает C1-C3 алкил;
из соединения формулы B
который включает следующие стадии:
a) получение первой смеси, содержащей алкоксид щелочного металла формулы C
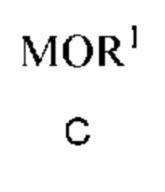
в которой M означает Na или K и R1 означает C1-C3 алкил,
растворитель, выбранный из группы, включающей DMSO, метанол, сульфолан и их смеси;
и соединение формулы B, и нагревание первой смеси;
b) получение второй смеси путем добавления воды, сильного основания и металлического цинка к первой смеси;
c) нагревание второй смеси; и
d) выделение соединения формулы A.
2. Способ по п. 1, в котором M означает Na и R1 означает метил.
3. Способ по п. 1, в котором сильное основание выбрано из группы, включающей гидроксид натрия или гидроксид калия.
4. Способ по п. 1, дополнительно включающий стадию удаления солей цинка или металлического цинка из второй смеси до нагревания второй смеси.
5. Способ получения соединения формулы A
в которой R1 означает C1-C3 алкил;
из соединения формулы D
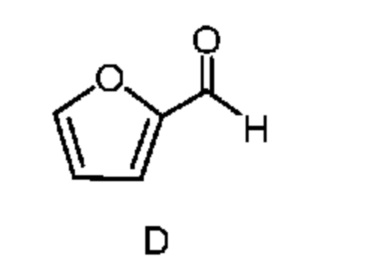
который включает следующие стадии:
a) получение первой смеси путем объединения 2-фазной системы вода-органический растворитель, источника аммония, источника цианида и соединения формулы D;
b) отделение второй смеси от первой смеси, содержащей соединение формулы E в виде раствора в органическом растворителе;
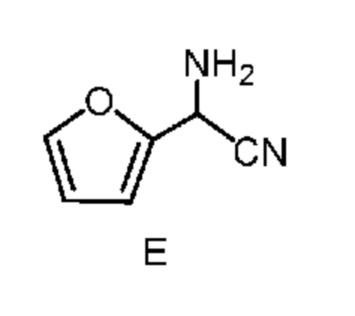
c) добавление водного раствора неорганической кислоты ко второй смеси с получением третьей смеси, в которой неорганической кислотой является HCl, HBr, H2SO4, HNO3 или H3PO4;
d) отделение четвертой смеси от третьей смеси, которой является водная смесь, содержащая соединение формулы F;
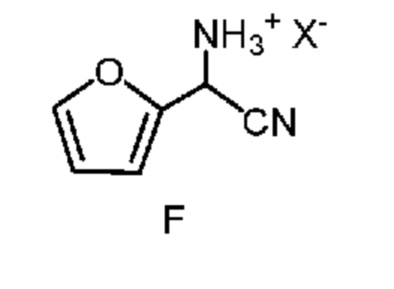
в которой X означает Cl, Br, HSO4, NO3 или H2PO4;
e) добавление бромирующего реагента, включающего бром, бромсодержащее соединение с окислителем и их смеси, к четвертой смеси с получением пятой смеси;
f) выделение соединения формулы B из пятой смеси;
g) получение первой смеси, содержащей алкоксид щелочного металла формулы C
в которой M означает Na или K и R1 означает C1-C3 алкил;
растворитель, выбранный из группы, включающей DMSO, метанол, сульфолан и их смеси;
и соединение формулы B, и нагревание первой смеси;
h) получение второй смеси путем добавления воды, сильного основания и металлического цинка к первой смеси;
i) нагревание второй смеси; и
j) выделение соединения формулы A.
6. Способ по п. 5, в котором органический растворитель выбран из группы, включающей MTBE, этилацетат, изопропилацетат, THF, 2-MeTHF, толуол, ксилол или смесь ксилолов и их смеси.
7. Способ по п. 5, в котором неорганической кислотой является бромистоводородная кислота.
8. Способ по п. 5, в котором X означает Br.
9. Способ по п. 5, в котором бромсодержащим соединением является бромистоводородная кислота или бромид, выбранный из группы, включающей NaBr и KBr, в комбинации с кислотой.
10. Способ по п. 5, в котором окислитель выбран из группы, включающей пероксид водорода и пероксимоносульфат калия.
| Токарный резец | 1924 |
|
SU2016A1 |
| WO 00/49008 A1, 24.08.2000 | |||
| Способ отсолки и обезвоживания гидросульфита натрия | 1939 |
|
SU61836A1 |
| RU 2002120924 A, 10.01.2004. | |||

