Предпосылки изобретения
Модуляция метаболизма простагландинов является основным моментом в современной противовоспалительной терапии. Ингибиторы нестероидных противовоспалительных лекарственных средств (НСПВЛС) и СОХ-2 блокируют активность циклооксигеназ и их способность преобразовывать арахидоновую кислоту (АА) в простагландин (PG) H2. PGH2 может затем метаболизироваться под действием терминальных простагландинсинтаз в соответствующие биологически активные PG, а именно, PGI2, тромбоксан (Тх) А2, PGD2, PGF2α и PGE2. Сочетание фармакологических, генетических подходов и подходов на основе нейтрализующих антител демонстрирует важную роль PGE2 в воспалении. Во многих отношениях прерывание РСЕ2-зависимой передачи сигнала в животных моделях воспаления может быть столь же эффективным, как и лечение ингибиторами НСПВЛС или СОХ-2. Поэтому преобразование PGH2 в PGE2 посредством простагландин Е синтаз (PGES) может представлять собой основную стадию в распространении воспалительных стимулов.
Микросомальная простагландин Е синтаза-1 (mPGES-1) представляет собой индуцируемую PGES после воздействия на нее воспалительных стимулов. mPGES-1 индуцируется на периферии и в ЦНС в результате воспаления и поэтому представляет новую мишень для острых и хронических воспалительных расстройств. Логическое обоснование разработки специфических ингибиторов mPGES-1 сводится к гипотезе, что терапевтическая польза ингибиторов НСПВЛС и COX-2, в основном, является результатом ингибирования провоспалительного PGE2, тогда как профиль побочных эффектов, в основном, зависит от ингибирования других простагландинов.
Настоящее изобретение направлено на новые соединения, которые являются селективными ингибиторами фермента микросомальной простагландин E синтазы-1 и поэтому будут полезными для лечения боли и воспаления при различных заболеваниях или состояниях, таких как остеоартрит, ревматоидный артрит и острая или хроническая боль. Кроме того, считают, что соединения по настоящему изобретению как селективные ингибиторы провоспалительного PGE2 должны иметь меньший потенциал побочных эффектов, связанных с ингибированием других простагландинов, по сравнению с традиционными нестероидными противовоспалительными лекарственными средствами, таких как желудочно-кишечная и почечная токсичность.
Краткое описание изобретения
Настоящее изобретение охватывает новые соединения формулы I
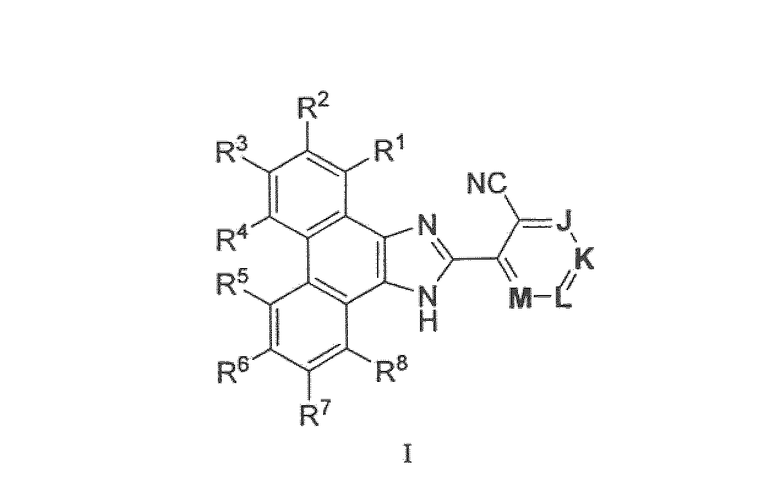
или их фармацевтически приемлемые соли. Эти соединения являются ингибиторами фермента микросомальной простагландин E синтазы-1 (mPGES-1) и поэтому являются полезными для лечения боли и/или воспаления при различных заболеваниях или состояниях, таких как остеоартрит, ревматоидный артрит и острая или хроническая боль. Охватываются также способы лечения заболеваний или состояний, опосредованных ферментом mPGES-1, и фармацевтические композиции.
Подробное описание изобретения
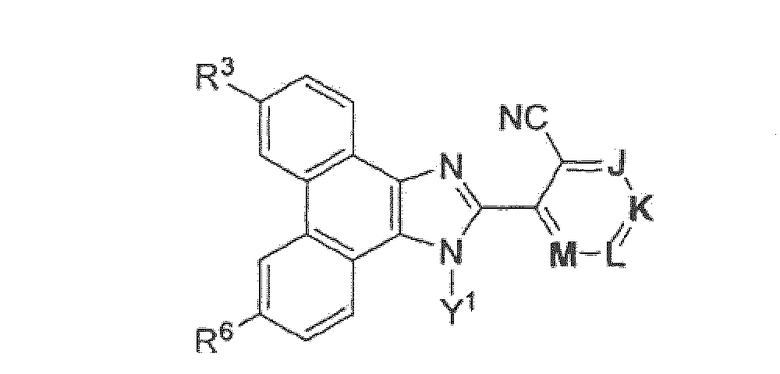
Настоящее изобретение охватывает группу соединений, представленных формулой B
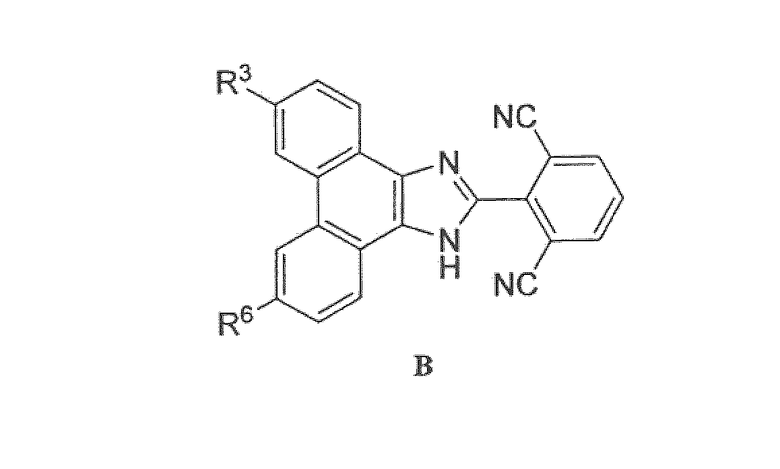
или их пролекарства, или фармацевтически приемлемые соли указанных соединений или пролекарств, где
R3 представляет собой 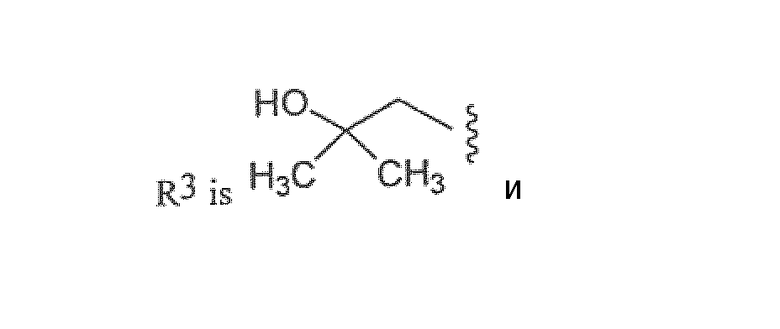
R6 выбран из группы, состоящей из (1) H; (2) F; (3) Cl; (4) Br; (5) I; (6) -CN; (7) С1-10алкила или C2-10алкенила, где один или несколько атомов водорода, связанных с указанным С1-10алкилом или C2-10алкенилом, могут быть заменены атомом фтора, или два атома водорода на смежных атомах углерода могут быть объединены вместе и замещены группой -CH2- с образованием циклопропильной группы, или два атома водорода по одному и тому же атому углерода могут быть замещены и объединены вместе с образованием спиро C3-6циклоалкильной группы, и где указанный С1-10алкил или C2-10алкенил, необязательно, может быть замещен одним-тремя заместителями, независимо выбранными из группы, состоящей из -OH, ацетила, метокси, этенила, R11-O-C(O)-, R35-N(R36)-; R37-N(R38)-C(O)-, циклопропила, пирролила, имидазолила, пиридила и фенила, при этом указанный пирролил, имидазолил, пиридил и фенил необязательно замещены C1-4алкилом или моногидроксизамещенным C1-4алкилом; (8) C3-6циклоалкила; (9) R12-O-; (10) Rl3-S(O)k-, (11) R14-S(O)k-N(Rl5)-; (12) R16-C(O)-; (13) R17-N(R18)-; (14) R19-N(R20)-C(O)-; (15) R21-N(R22)-S(O)k-; (16) R23-C(O)-N(R24)-; (17) Z-C≡С; (18) -(CH3)C=N-ОH или -(CH3)C=N-OCH3; (19) R34-O-C(O)-; (20) R39-C(O)-O- и (21) фенила, нафтила, пиридила, пиридазинила, пиримидинила, пиразинила, пирролила, пиразолила, имидазолила, триазолила, тетразолила, оксазолила, изоксазолила, оксадиазолила, тиенила или фурила, где каждый необязательно замещен заместителем, независимо выбранным из группы, состоящей из F, Cl, Br, I, C1-4алкила, фенила, метилсульфонила, метилсульфониламино, R25-O-C(O)- и R26-N(R27)-, при этом указанный C1-4алкил необязательно замещен 1-3 группами, независимо выбранными из галогена и гидрокси;
каждый Z независимо выбран из группы, состоящей из (1) H; (2) C1-6алкила, где один или несколько атомов водорода, связанных с указанным С1-6алкилом, могут быть замещены атомом фтора, и где указанный C1-6алкил необязательно замещен одним-тремя заместителями, независимо выбранными из гидрокси, метокси, циклопропила, фенила, пиридила, пирролила, R28-N(R29)- и R30-O-C(O)-; (3) -(CH3)C=N-OH или -(CH3)C=N-OCH3; (4) R31-C(O)-; (5) фенила; (6) пиридила или его N-оксида; (7) C3-6циклоалкила, необязательно замещенного гидрокси; (8) тетрагидропиранила, необязательно замещенного гидрокси; и (9) пятичленного ароматического гетероцикла, содержащего от 1 до 3 атомов, независимо выбранных из O, N или S, и необязательно замещенного метилом;
каждый R15, R24 и R32 независимо выбран из группы, состоящей из (1) H и (2) C1-4алкила;
каждый R11, R12, R13; R14, R16, R23, R25, R30; R31, R34 и R39 независимо выбран из группы, состоящей из (1) H; (2) C1-4алкила, (3) C3-6циклоалкила; (4) C3-6циклоалкил-C1-4алкил-; (5) фенила, (6) бензила и (7) пиридила; при этом указанный C1-4алкил, C3-6циклоалкил, C3-6циклоалкил-C1-4алкил-, фенил, бензил и пиридил, каждый, может быть необязательно замещен 1-3 заместителями, независимо выбранными из группы, состоящей из OH, F, Cl, Br и I, и где указанный C1-4алкил может быть дополнительно замещен оксо или метокси или обоими такими заместителями;
каждый R17, R18, R19, R20, R21; R22, R26, R27, R28, R29, R35, R36, R37 и R38 независимо выбран из группы, состоящей из (1) H; (2) C1-6алкила; (3) C1-6алкокси; (4) OH и (5) бензила или 1-фенилэтила; и R17 и R18, R19 и R20, R21 и R22, R26 и R27, и R28 и R29, R35 и R36, и R37 и R38 могут быть объединены вместе с атомом азота, с которым они связаны, с образованием моноциклического кольца из 5 или 6 атомов углерода, необязательно содержащего один или два атома, независимо выбранных из -O-, -S(O)k- и -N(R32)-; и
каждый k, независимо, имеет значение 0, 1 или 2.
В рамках указанной группы настоящее изобретение охватывает первую подгруппу соединений формулы B, где R6 представляет собой R12-O.
В рамках первой подгруппы настоящее изобретение охватывает класс соединений формулы B, где R12 выбран из группы, состоящей из (1) C1-4алкила и (2) C3-6циклоалкил-C1-4алкил-, где указанный C1-4алкил и C3-6циклоалкил, каждый, может быть необязательно замещен 1-3 заместителями, независимо выбранными из группы, состоящей из OH, F, Cl, Br и I.
Также в рамках указанной группы настоящее изобретение охватывает вторую подгруппу соединений формулы B, где R6 выбран из F, Cl, Br и I.
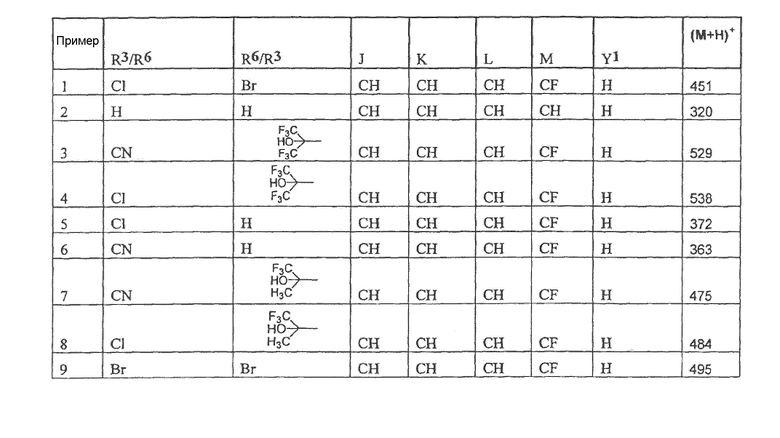
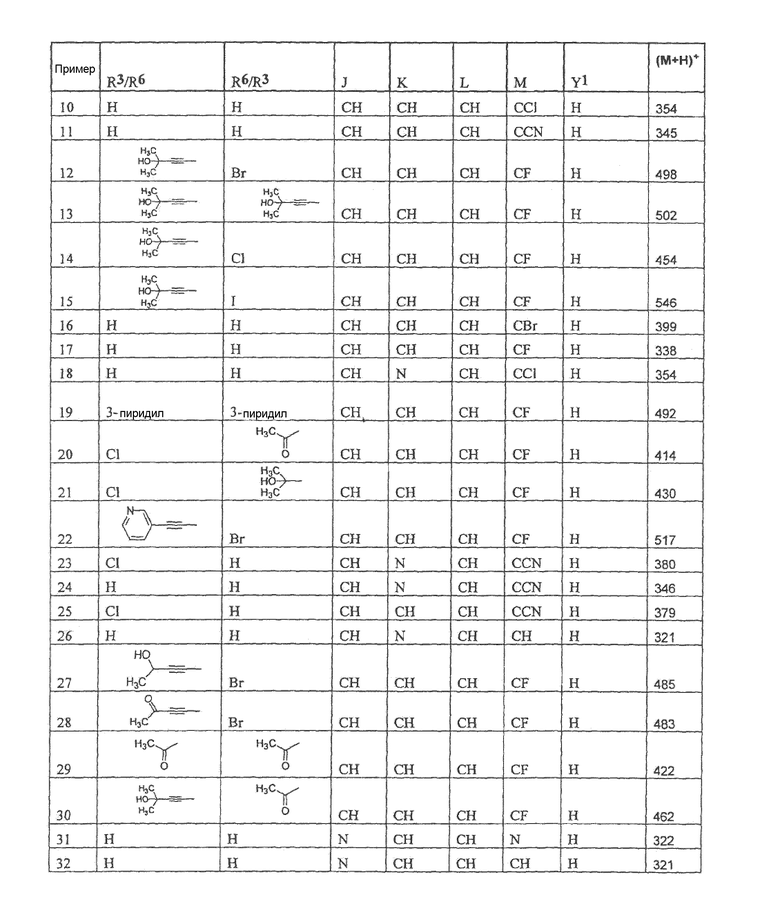
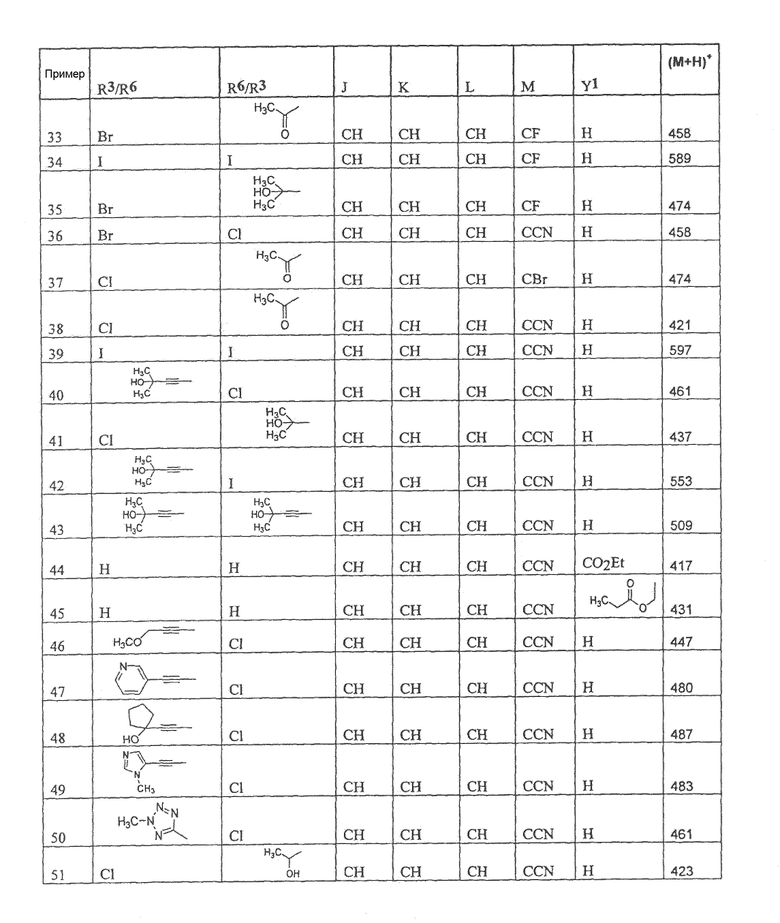
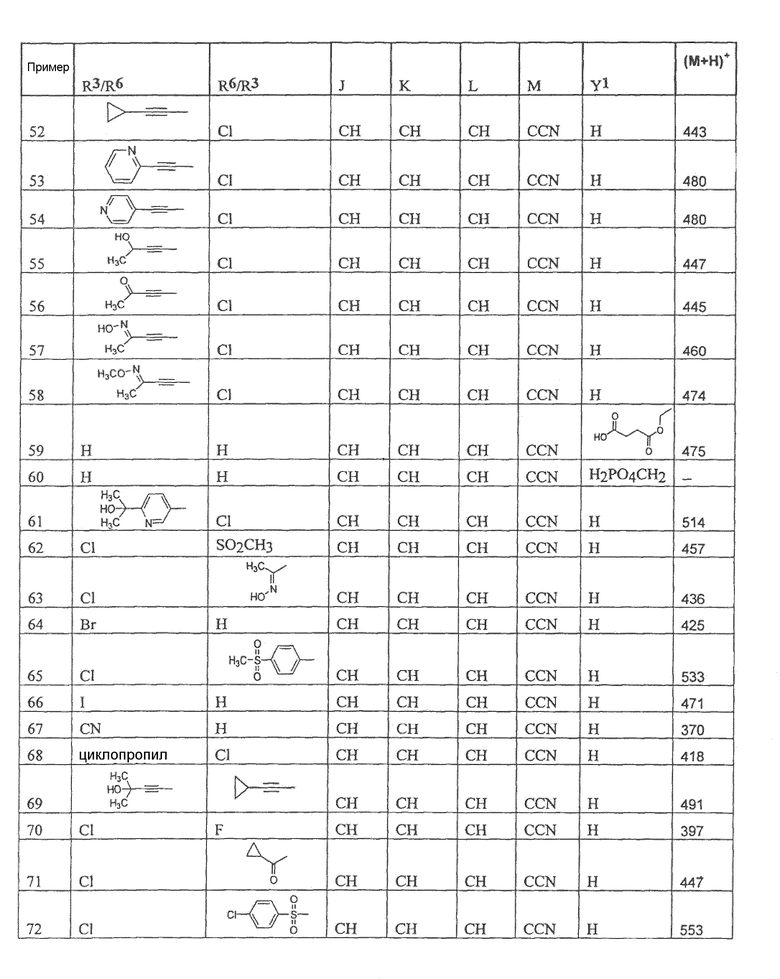
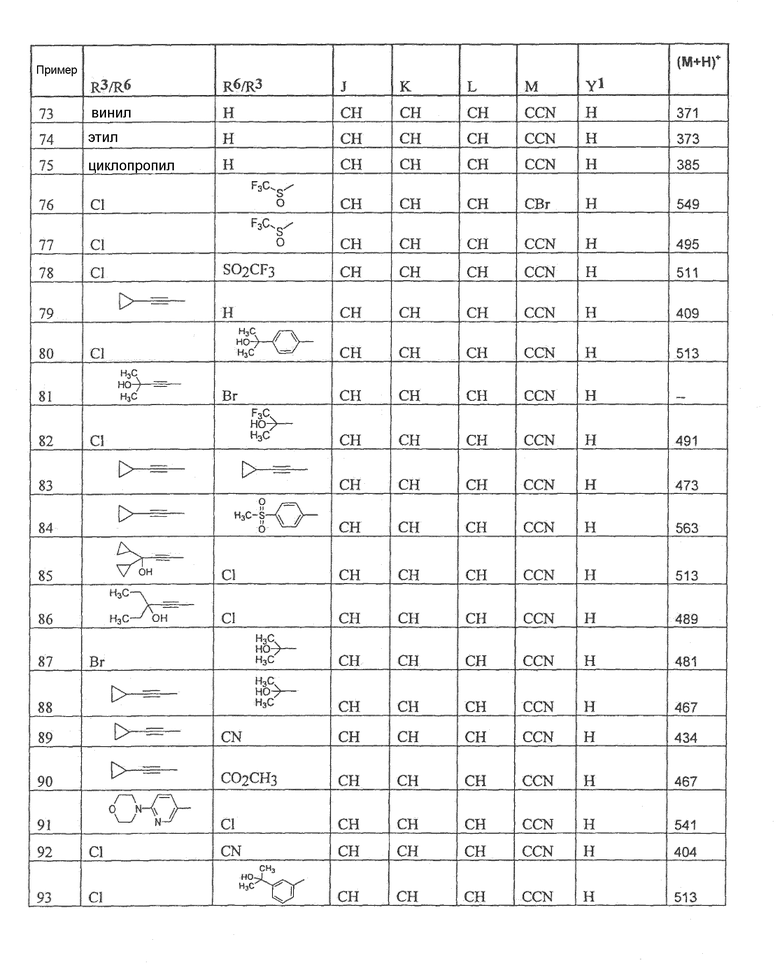
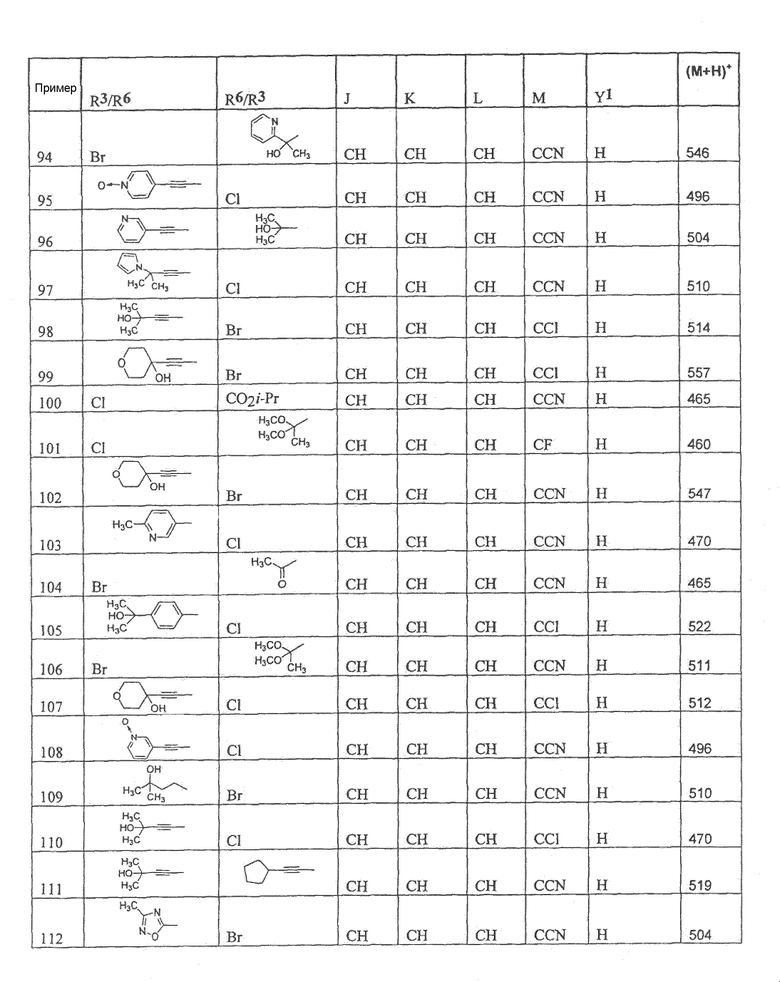
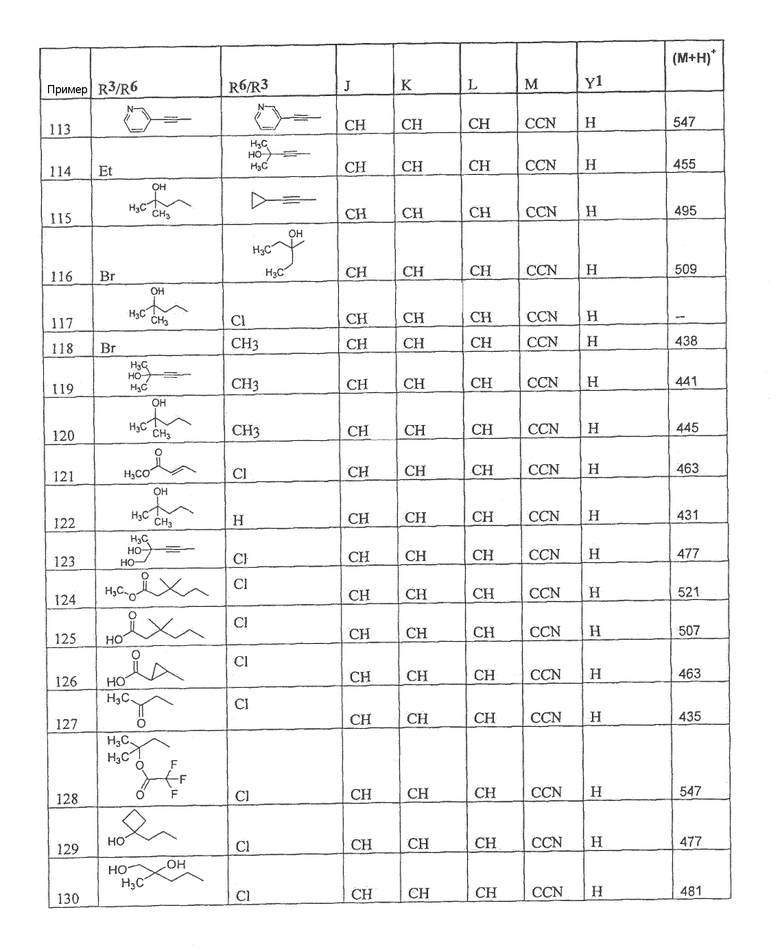
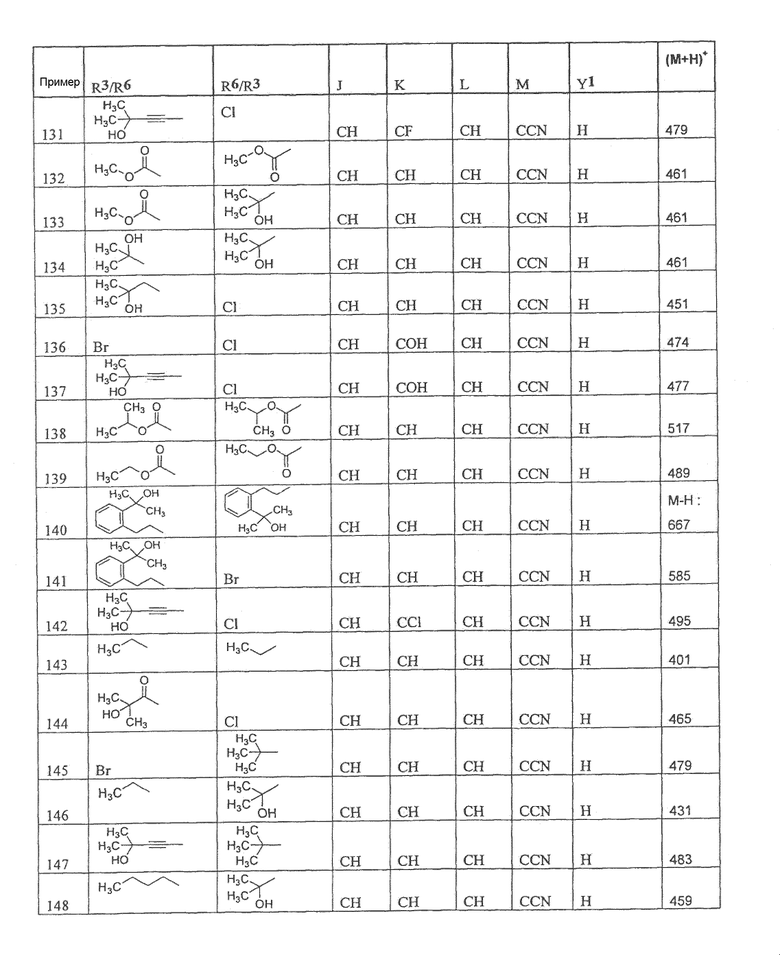
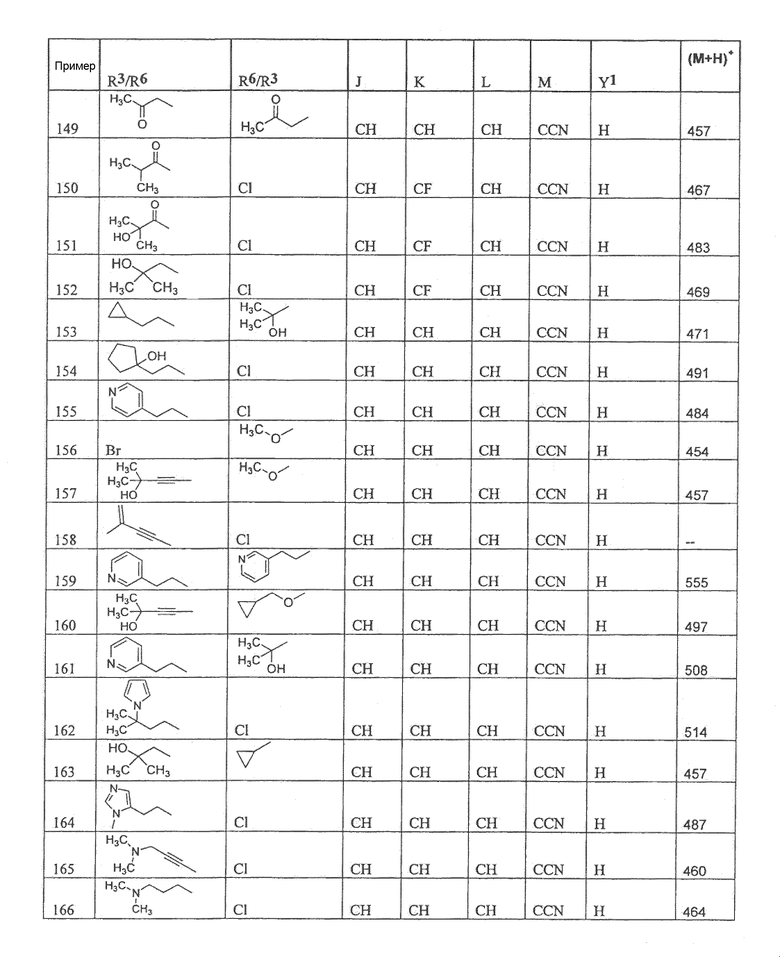
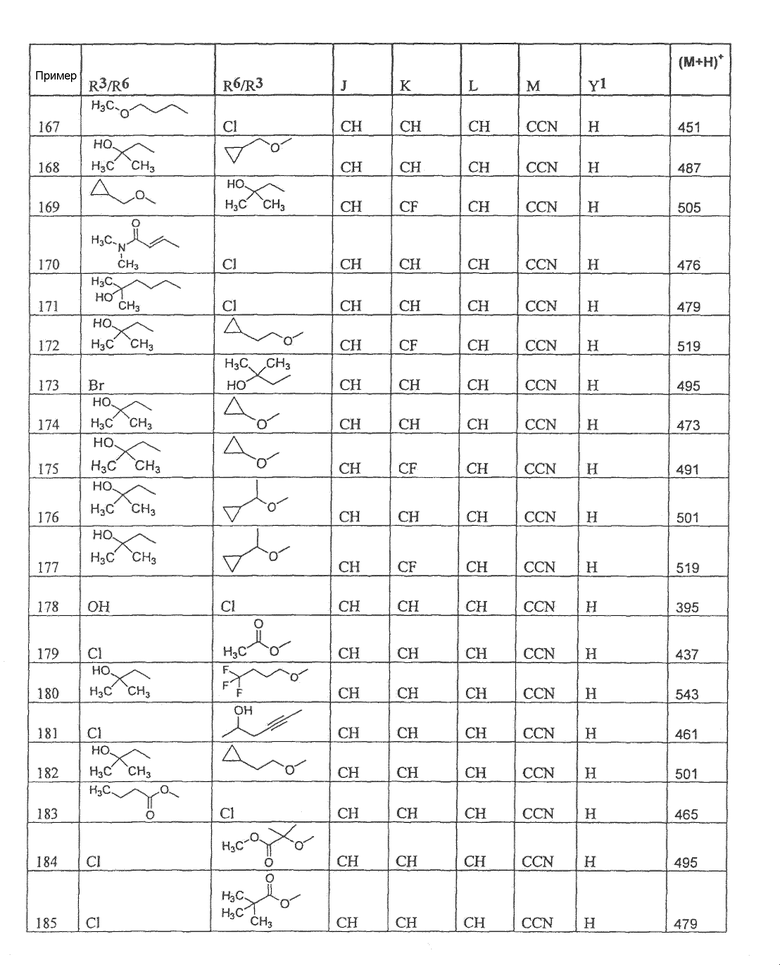
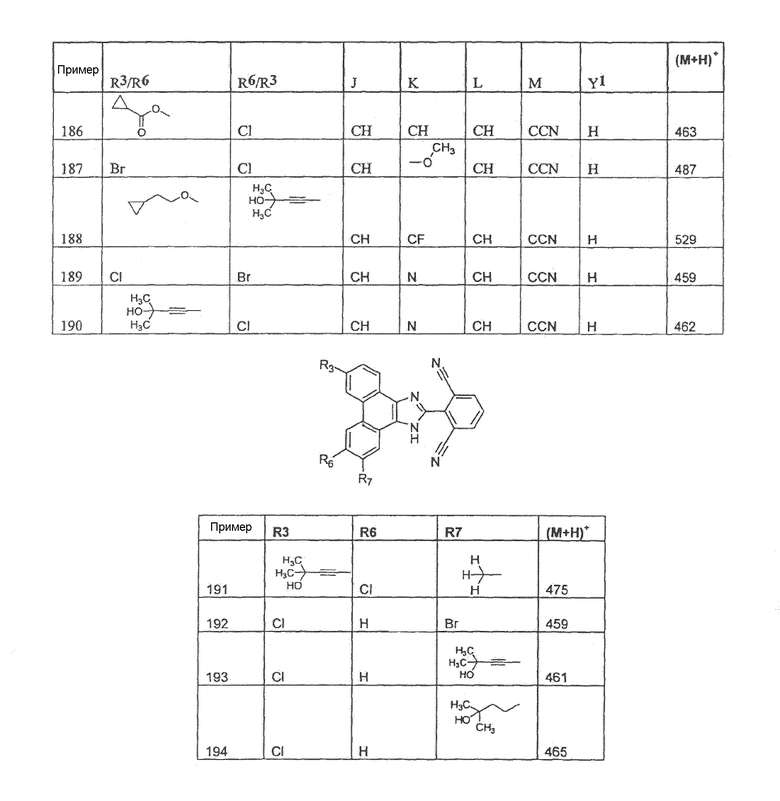
Настоящее изобретение охватывает соединение, выбранное из следующей таблицы:
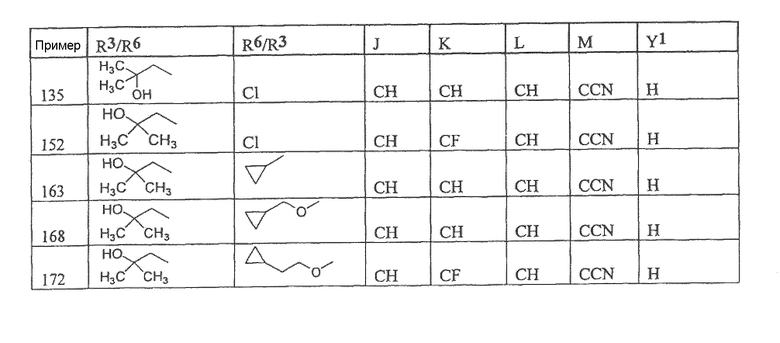
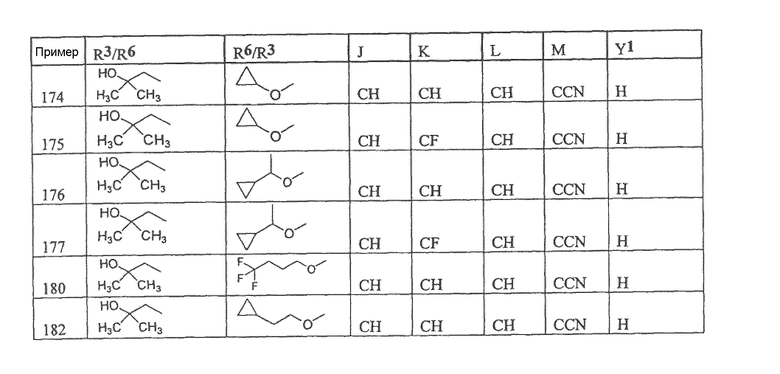
или фармацевтически приемлемую соль любого из представленных выше соединений.
Настоящее изобретение также охватывает фармацевтическую композицию, включающую соединение формулы B в сочетании с фармацевтически приемлемым носителем.
В другом варианте воплощения настоящее изобретение охватывает способ лечения опосредованных микросомальной простагландин E синтазой-1 заболеваний или состояний у пациента-человека, нуждающегося в таком лечении, включающий введение указанному пациенту соединения формулы B в количестве, эффективном для лечения опосредованных микросомальной простагландин E синтазой-1 заболеваний или состояний. Этот вариант воплощения настоящего изобретения включает заболевание или состояние, выбранное из группы, состоящей из острой или хронической боли, остеоартрита, ревматоидного артрита, бурсита, алкилозирующего спондилита и первичной дисменореи.
Настоящее изобретение также охватывает соединение, которое представляет собой
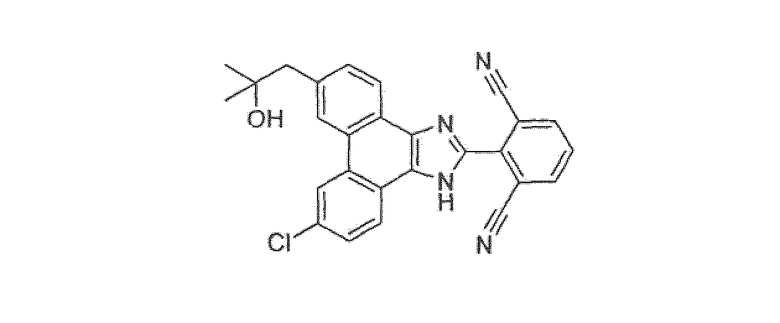
или его фармацевтически приемлемую соль.
Настоящее изобретение также охватывает соединение, которое представляет собой
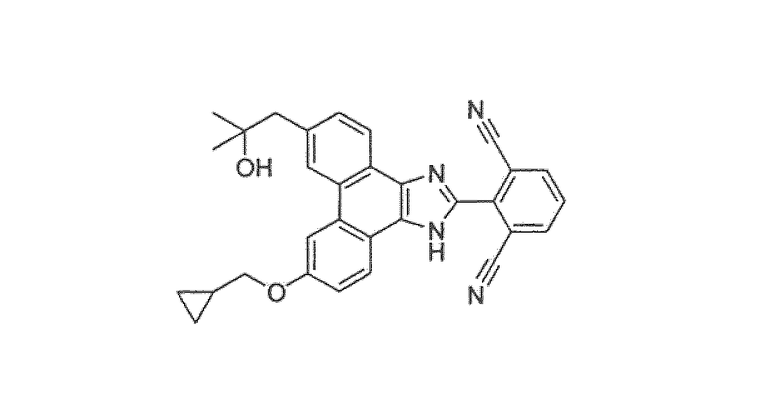
или его фармацевтически приемлемую соль.
Настоящее изобретение также охватывает соединение, которое представляет собой
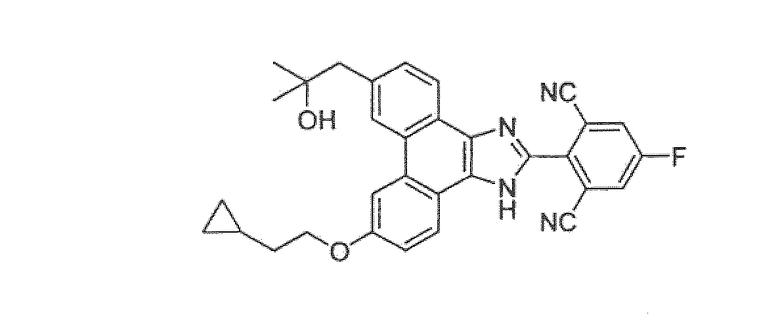
или его фармацевтически приемлемую соль.
Настоящее изобретение также охватывает соединение, которое представляет собой
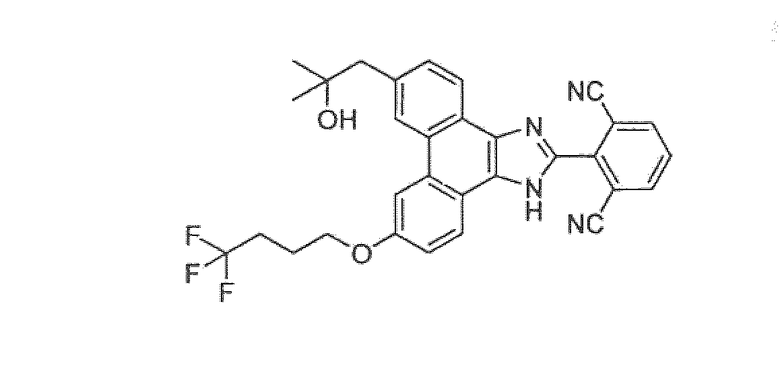
или его фармацевтически приемлемую соль.
Настоящее изобретение включает, если это является подходящим, фармацевтически приемлемые соли любого из указанных выше соединений. В целях настоящего описания заголовок "R3/R6" означает, что заместитель, указанный в этой графе, замещен в положении, представленном либо R3, либо R6. В соседней графе заголовок "R6/R3" означает, что указанный заместитель замещен в положении R3 или R6, не замещенном в предыдущей графе. В качестве примера, пример 6 представляет R3=CN и R6=H, или R3=H и R6=CN, представляя оба таутомера.
Термин "галоген" включает F, Cl, Br и I.
Термин "алкил" означает линейные или разветвленные структуры и их сочетания, содержащие указанное количество атомов углерода. Так, например, С1-10алкил включает метил, этил, пропил, 2-пропил, втор- и трет-бутил, бутил, пентил, гексил и 1,1-диметилэтил.
Термин "алкенил" означает линейные или разветвленные структуры и их сочетания, включающие указанное количество атомов углерода, содержащие, по меньшей мере, одну углерод-углеродную двойную связь, где водород может быть замещен дополнительной углерод-углеродной двойной связью. C2-6алкенил, например, включает этенил, пропенил, 1-метилэтенил, бутенил и т.п.
Термин "алкинил" означает линейные или разветвленные структуры и их сочетания, включающие указанное количество атомов углерода, содержащие, по меньшей мере, одну углерод-углеродную тройную связь. C3-6алкинил, например, включает пропенил, 1-метилэтенил, бутенил и т.п.
Термин "алкокси" означает алкоксигруппы линейной, разветвленной или циклической конфигурации, содержащие указанное количество атомов углерода. C1-6алкокси, например, включает метокси, этокси, пропокси, изопропокси и т.п.
Термин "циклоалкил" означает моно-, би- или трициклические структуры, необязательно объединенные с линейными или разветвленными структурами, которые содержат указанное количество атомов углерода. Примеры циклоалкильных групп включают циклопропил, циклопентил, циклогептил, адамантил, циклододецилметил, 2-этил-1-бицикло[4.4.0]децил, циклобутилметил, циклопропилметил и т.п.
Соединения, описанные в настоящей заявке, могут содержать асимметричный центр и, таким образом, могут существовать в виде энантиомеров. Когда соединения по настоящему изобретению содержат два или более асимметричных центра, они также могут существовать в виде диастереомеров. Настоящее изобретение включает все такие возможные стереоизомеры в виде по существу чистых разделенных энантиомеров, их рацемических смесей, а также смесей диастереомеров. Представленная выше формула I показана без определенной стереохимии в определенных положениях. Настоящее изобретение включает все стереоизомеры формулы I и их фармацевтически приемлемые соли. Диастереоизомерные пары энантиомеров можно разделить, например, при помощи фракционированной кристаллизации из подходящего растворителя, и пару энантиомеров, полученную таким образом, можно разделить на индивидуальные стереоизомеры традиционными способами, например, с использованием оптически активной кислоты или основания в качестве агента разделения или на хиральной ВЭЖХ-колонке. Кроме того, любой энантиомер или диастереомер соединения общей Формулы I можно получить путем стереоспецифического синтеза с использованием оптически чистых исходных веществ или реагентов известной конфигурации.
Некоторые из соединений, описанных в настоящей заявке, содержат олефиновые двойные связи и, если не указано иное, они включают как E, так и Z геометрические изомеры.
Некоторые из соединений, описанных в настоящей заявке, могут существовать с различными точками присоединения водорода, и их называют таутомерами. Соединение формулы I существует в следующих таутомерных формах:
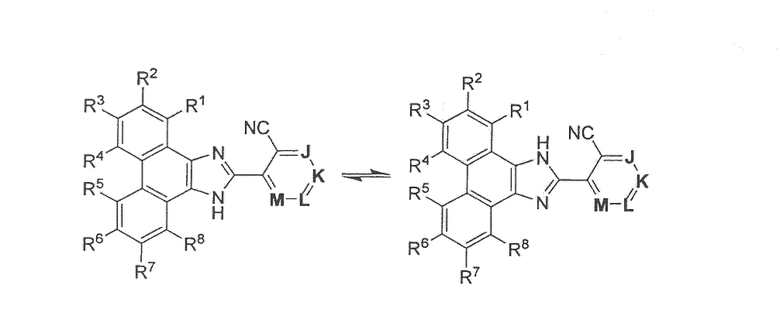
Индивидуальные таутомеры, а также их смеси охватываются формулой I.
В объем настоящего изобретения включены пролекарства соединений по настоящему изобретению. Как правило, такие пролекарства представляют собой функциональные производные соединений по настоящему изобретению, которые легко преобразуются in vivo в нужное соединение. Так, в способах лечения по настоящему изобретению термин "введение" охватывает лечение различных описанных состояний при помощи соединения, конкретно раскрытого, или соединения, которое может быть не раскрыто конкретным образом, но которое преобразуется в раскрываемое в настоящей заявке соединение in vivo после введения пациенту. Традиционные процедуры для выбора и получения подходящих пролекарственных производных описаны, например, в "Design of Prodrugs," ed. Н. Bundgaard, Elsevier, 1985. Метаболиты этих соединений включают активные группы, получаемые при введении соединений по настоящему изобретению в биологическую среду. Примерами пролекарств по настоящему изобретению являются соединения формулы C.
Термин "лечение опосредованных микросомальной простагландин E синтазой-1 заболеваний или состояний" означает лечение или предотвращение любого заболевания или состояния, которое можно с выгодным преимуществом лечить или предотвратить путем ингибирования фермента микросомальной простагландин E синтазы-1 (mPGES-1). Этот термин включает облегчение боли, лихорадки и воспаления при различных состояниях, включая ревматическую лихорадку, симптомы, связанные с гриппом или другими вирусными инфекциями, обычную простуду, боль в спине и шейную боль, дисменорею, головную боль, мигрень (лечение острых приступов и профилактическое лечение), зубную боль, растяжения и напряжения при нагрузках, миозит, невралгию, синовит, артрит, включая ревматоидный артрит, дегенеративные заболевания суставов (остеоартрит), подагру и алкилозирующий спондилит, острые, подострые и хронические мышечно-скелетные болевые синдромы, такие как бурсит, ожоги, травмы и боли после хирургических и стоматологических процедур, а также предупреждение хирургической боли. Кроме того, этот термин включает ингибирование клеточных опухолевых преобразований и роста метастазов опухоли и, следовательно, лечение рака. Термин также включает лечение эндометриоза и болезни Паркинсона, а также лечение опосредованных mPGES-1 пролиферативных расстройств, которые могут возникать при диабетической ретинопатии и ангиогенезе опухоли. Термин "лечение" охватывает не только лечение пациента для облегчения у пациента признаков и симптомов заболевания или состояния, но также профилактическое лечение асимптоматического пациента для предотвращения начала развития или прогрессирования заболевания или состояния.
Термин "количества, которые являются эффективными для лечения" означает такое количество лекарственного средства или фармацевтического средства, которое будет вызывать такой биологический или медицинский ответ ткани, системы, животного или человека, которого добивается исследователь, ветеринар, врач или другой клиницист. Этот термин также охватывает количество фармацевтического средства, которое будет препятствовать или снижать риск возникновения биологического или медицинского события, которое пытаются предотвратить в ткани, системе, у животного или человека исследователь, ветеринар, врач или другой клиницист. Подходящие дозы соединения формулы I по настоящему изобретению описаны ниже. Соединение можно вводить по схеме раз или два раза в день.
Фармацевтические композиции по настоящему изобретению включают соединение формулы I в качестве активного ингредиента или его фармацевтически приемлемую соль и также могут содержать фармацевтически приемлемый носитель и, необязательно, другие терапевтические ингредиенты. Термин "фармацевтически приемлемые соли" включает соли, полученные из оснований, которые образуют нетоксичные фармацевтически приемлемые соли, включая неорганические основания и органические основания. Соли, полученные из неорганических оснований, включают соли алюминия, аммония, кальция, меди, трехвалентного железа, двухвалентного железа, лития, магния, трехвалентного марганца, двухвалентного марганца, калия, натрия, цинка и т.п. Особенно предпочтительными являются соли аммония, кальция, магния, калия и натрия. Соли, полученные из фармацевтически приемлемых органических нетоксичных оснований, включают соли первичного, вторичного и третичного аминов, замещенных аминов, включая природные замещенные амины, циклические амины и основные ионообменные смолы, такие как аргинин, бетаин, кофеин, холин, N,N-дибензилэтилендиамин, диэтиламин, 2-диэтиламиноэтанол, 2-диметиламиноэтанол, этаноламин, этилендиамин, N-этилморфолин, N-этилпиперидин, глюкамин, глюкозамин, гистидин, гидрабамин, изопропиламин, лизин, метилглюкамин, морфолин, пиперазин, пиперидин, полиаминные смолы, прокаин, пурины, теобромин, триэтиламин, триметиламин, трипропиламин, трометамин и т.п.
Когда соединение по настоящему изобретению является основным, соли можно получить из кислот, которые образуют фармацевтически приемлемые соли, включая неорганические и органические кислоты. Такие кислоты включают уксусную, адипиновую, аспарагиновую, 1,5-нафталиндисульфоновую, бензолсульфоновую, бензойную, камфорсульфоновую, лимонную, 1,2-этандисульфоновую, этансульфоновую, этилендиаминтетрауксусную, фумаровую, глюкoгептоновую, глюкoновую, глутаминовую, йодистоводородную, бромистоводородную, хлористоводородную, изетионовую, молочную, малеиновую, яблочную, миндальную, метансульфоновую, слизевую, 2-нафталинсульфоновую, азотную, щавелевую, памовую, пантотеновую, фосфорную, пивалиновую, пропионовую, салициловую, стеариновую, янтарную, серную, винную, п-толуолсульфоновую кислоту, ундекановую, 10-ундеценовую и т.п.
Благодаря mPGES-1 ингибиторной активности соединений по настоящему изобретению, соединения формулы I являются полезными для облегчения боли, лихорадки и воспаления при различных состояниях, включая ревматическую лихорадку, симптомы, связанные с гриппом или другими вирусными инфекциями, обычную простуду, боль в спине и шейную боль, дисменорею, головную боль, мигрень (лечение острых приступов и профилактическое лечение), зубную боль, растяжения и напряжения при нагрузках, миозит, невралгию, синовит, артрит, включая ревматоидный артрит, юношеский ревматоидный артрит, дегенеративные заболевания суставов (остеоартрит), острую подагру и алкилозирующий спондилит, острые, подострые и хронические мышечно-скелетные болевые синдромы, такие как бурсит, ожоги, травмы и боли после хирургических и стоматологических процедур, а также предупреждение хирургической боли. Кроме того, такое соединение может ингибировать клеточные опухолевые преобразования и рост метастазов опухоли и, следовательно, его можно использовать для лечения рака. Соединения формулы I также могут быть полезными для лечения или предотвращения эндометриоза, гемофилической артропатии и болезни Паркинсона.
Соединения формулы I также ингибируют простаноид-индуцированное сокращение гладких мышц, препятствуя синтезу контрактильных простаноидов и, следовательно, могут быть полезными для лечения дисменореи, преждевременных родов и астмы.
Благодаря селективному ингибированию фермента mPGES-1, соединения формулы I могут быть полезны в качестве альтернативы традиционным нестероидным противовоспалительным лекарственным средствам (НСПВЛС), особенно когда такие нестероидные противовоспалительные лекарственные средства могут быть противопоказаны, как у пациентов с пептической язвой, гастритом, региональным энтеритом, язвенным колитом, дивертикулитом или рецидивирующими желудочно-кишечными поражениями; желудочно-кишечным кровотечением, нарушениями свертывания крови, включая анемию, такую как гипопротромбинемия, гемофилию или другие проблемы кровотечения (включая те, которые связаны с пониженной или нарушенной функцией тромбоцитов); заболеванием почек (например, нарушение функции почек); у пациентов перед хирургической операцией или принимающих антикоагулянты; и у пациентов, подверженных НСПВЛС-индуцируемой астме.
Подобным образом, соединения формулы I будут полезны в качестве частичной или полной замены традиционных НСПВЛС в существующих в настоящее время препаратах для их совместного введения с другими средствами или ингредиентами. Таким образом, в следующих аспектах изобретение охватывает фармацевтические композиции для лечения опосредованных mPGES-1 заболеваний, указанных выше, включающие нетоксичное терапевтически эффективное количество соединения формулы I, указанное выше, и один или несколько ингредиентов, таких как другие средства для облегчения боли, включая ацетаминофен или фенацетин; опиоидные аналгетики, такие как кодеин, фентанил, гидроморфон, леворфанол, меперидин, метадон, морфин, оксикодон, оксиморфин, пропоксифен, бупренорфин, буторфанол, дезоцин, налбуфин и пентазоцин; потенцирующее средство, включая кофеин; H2-антагонист; гидроксид алюминия или магния; симетикон; противоотечное средство, включая фенилэфрин, фенилпропаноламин, псевдофедрин, оксиметазолин, эпинефрин, нафазолин, ксилометазолин, пропилгекседрин или леводезоксиэфедрин; средство против кашля, включая кодеин, гидрокодон, карамифен, карбетапентан или декстраметорфан; диуретик; седативное или неседативное антигистаминное средство; ингибитор протонного насоса, такой как омепразол; антагонист брадикинина-1; антагонист VR1 рецептора и блокатор натриевых каналов (NAV1). Для лечения или предотвращения мигрени настоящее изобретение также охватывает совместное введение с агонистом 5-HT, таким как ризатриптан, суматриптан, золмитриптан и наратриптан, или антагонистом CGRP. Кроме того, настоящее изобретение охватывает способ лечения опосредованных mPGES-1 заболеваний, включающий введение пациенту, нуждающемуся в таком лечении, нетоксичного терапевтически эффективного количества соединения формулы I, необязательно совместно введенного с одним или несколькими такими ингредиентами, которые перечислены непосредственно выше.
Как указано выше, фармацевтические композиции для лечения указанных опосредованных mPGES-1 заболеваний, необязательно, могут включать один или несколько ингредиентов, перечисленных выше.
В другом аспекте изобретение охватывает совместное введение ингибитора протонного насоса с соединением формулы I. Ингибиторы протонного насоса, которые можно использовать в этом аспекте настоящего изобретения, включают омепразол, лансопразол, рабепразол, пантопразол и эзомепразол, или фармацевтически приемлемую соль любого из указанных выше. Такие ингибиторы протонного насоса являются коммерчески доступными, например, омепразол (PRILOSEC, AstraZeneca), лансопразол (PREVACID, TAP Pharmaceuticals), рабепразол (ACIPHEX, Janssen Pharmaceutica), пантопразол (PROTONIX, Wyeth-Ayerst) и эзомепразол (NEXIUM, AstraZeneca). Указанные ингибиторы протонного насоса можно вводить в обычных дозах. Например, омепразол или омепразол магний можно вводить при дозе 10 мг, 20 мг или 40 мг. Лансопразол можно вводить при дозе 15 мг или 30 мг. Рабепразол натрий можно вводить при дозе 20 мг. Пантопразол можно вводить при дозе 20 мг или 40 мг. Эзомепразол можно вводить при дозе 20 мг или 40 мг. Соединение формулы I и ингибитор протонного насоса можно вводить одновременно в одной фармацевтической лекарственной форме или в виде двух отдельных лекарственных форм, принимаемых пациентом по существу одновременно. Альтернативно, соединение формулы I и ингибитор протонного насоса можно принимать последовательно, раздельно по времени, при условии, что фармацевтические эффекты этих двух средств реализуются у пациента одновременно.
Фармацевтические композиции, содержащие активный ингредиент, могут быть в форме, подходящей для перорального применения, например, в виде таблеток, драже, лепешек, водных или масляных суспензий, диспергируемых порошков или гранул, эмульсий, твердых или мягких капсул, или сиропов или эликсиров. Композиции, предназначенные для перорального применения, можно получить в соответствии с любым известным из уровня техники способом получения фармацевтических композиций, и такие композиции могут содержать одно или несколько средств, выбранных из группы, включающей подсластители, отдушки, красители и консерванты, для получения фармацевтически привлекательных и приятных на вкус препаратов. Таблетки содержат активный ингредиент в смеси с нетоксичными фармацевтически приемлемыми эксципиентами, которые являются подходящими для получения таблеток. Эти эксципиенты могут представлять собой, например, инертные разбавители, такие как карбонат кальция, карбонат натрия, лактоза, фосфат кальция или фосфат натрия; агенты гранулирования и разрыхлители, например, кукурузный крахмал или альгиновая кислота; связующие, например, крахмал, желатин или аравийская камедь, и смазывающие вещества, например, стеарат магния, стеариновая кислота или тальк. Таблетки могут быть без покрытия, или на них может быть нанесено покрытие с использованием известных способов для замедления их разложения и абсорбции в желудочно-кишечном тракте с обеспечением, таким образом, замедленного действия в течение более длительного периода времени. Например, можно использовать вещества, замедляющие действие, такие как глицерилмоностеарат или глицерилдистеарат. На таблетки также можно нанести покрытие способом, описанным в патентах США № 4256108; 4166452 и 4265874, с получением осмотических терапевтических таблеток для контролируемого высвобождения.
Композиции для перорального применения также могут быть представлены в виде твердых желатиновых капсул, где активный ингредиент смешан с инертным твердым разбавителем, например, карбонатом кальция, фосфатом кальция или каолином, или в виде мягких желатиновых капсул, где активные ингредиенты смешаны с водой или масляной средой, например, такой как арахисовое масло, жидкий парафин или оливковое масло. Примером композиции по настоящему изобретению являются сухие наполненные капсулы, содержащие 50/50 смесь микрокристаллической целлюлозы и лактозы и 1 мг, 10 мг или 100 мг соединения формулы I.
Водные суспензии содержат активное вещество в смеси с эксципиентами, подходящими для получения водных суспензий. Такие эксципиенты представляют собой суспендирующие вещества, например, натриевую соль карбоксиметилцеллюлозы, метилцеллюлозу, гидроксипропилметилцеллюлозу, альгинат натрия, поливинилпирролидон, смолу трагаканта и аравийскую камедь; диспергирующие или смачивающие вещества могут представлять собой природные фосфатиды, например, лецитин, или продукты конденсации алкиленоксида с жирными кислотами, например, полиоксиэтиленстеарат, или продукты конденсации этиленоксида с длинноцепочечными алифатическими спиртами, например, гептадекаэтиленоксиэтанол, или продукты конденсации этиленоксида с неполными эфирами, полученными из жирных кислот и гексита, такие как полиоксиэтиленсорбитолмоноолеат, или продукты конденсации этиленоксида с неполными эфирами, полученными из жирных кислот и ангидридов гексита, например полиэтиленсорбитанмоноолеат. Водные суспензии также могут содержать один или несколько консервантов, например, этил- или н-пропил-пара-гидроксибензоат, один или несколько красителей, одну или несколько отдушек и один или несколько подсластителей, таких как сахароза, сахарин или аспартам.
Для жидких композиций используют самоэмульгирующиеся системы доставки лекарственного средства и технологию NanoCrystal®. Также можно использовать комплексы включения с циклодекстрином.
Масляные суспензии можно сформулировать путем суспендирования активного ингредиента в растительном масле, например, арахисовом масле, оливковом масле, кунжутном масле или масле кокосового ореха, или в минеральном масле, таком как жидкий парафин. Масляные суспензии могут содержать загуститель, например, пчелиный воск, твердый парафин или цетиловый спирт. Можно добавить подсластители, такие как указанные выше, и отдушки для получения приятного на вкус препарата. Такие композиции можно сохранять путем добавления антиоксиданта, такого как аскорбиновая кислота.
Диспергируемые порошки и гранулы, подходящие для получения водных суспензий путем добавления воды, обеспечивают активный ингредиент в смеси с диспергирующими или смачивающими веществами, суспендирующими веществами и одним или несколькими консервантами. Подходящие диспергирующие или смачивающие вещества и суспендирующие вещества представлены выше. Дополнительные эксципиенты, например, подсластители, отдушки и красители, также могут присутствовать.
Фармацевтические композиции по настоящему изобретению также могут быть в форме эмульсий масло-в-воде. Масляная фаза может представлять собой растительное масло, например, оливковое масло или арахисовое масло, или минеральное масло, например, жидкий парафин, или их смеси. Подходящими эмульгаторами могут быть природные фосфатиды, например, соевые бобы, лецитин и эфиры или неполные эфиры, полученные из жирных кислот и ангидридов гексита, например, сорбитанмоноолеат, и продукты конденсации указанных неполных эфиров с этиленоксидом, например, полиоксиэтиленсорбитанмоноолеат. Эмульсии также могут содержать подсластители и отдушки.
Композиции сиропов и эликсиров можно сформулировать с подсластителями, например, с глицерином, пропиленгликолем, сорбитом или сахарозой. Такие композиции также могут содержать смягчающее вещество, консервант и отдушки и красители. Фармацевтические композиции могут быть в форме стерильных водных или масляных суспензий для инъекций. Такие суспензии можно сформулировать в соответствии со способами, известными из уровня техники, с использованием указанных выше подходящих диспергирующих или смачивающих веществ и суспендирующих веществ. Стерильные препараты для инъекций также могут представлять собой стерильный раствор или суспензию для инъекций в нетоксичном парентерально приемлемом разбавителе или растворителе, например, в виде раствора в 1,3-бутандиоле. Из приемлемых носителей и растворителей, которые можно использовать, можно указать воду, раствор Рингера и изотонический раствор хлорида натрия. Кроме того, стерильные нелетучие масла традиционно используют в качестве растворителей или среды для суспендирования. Для этих целей можно использовать любое не раздражающее нелетучее масло, включая синтетические моно- или диглицериды. Кроме того, жирные кислоты, такие как олеиновая кислота, находят применение в препаратах для инъекций.
Соединения формулы I также можно вводить в форме суппозиториев для ректального введения лекарственного средства. Такие композиции можно получить путем смешивания лекарственного средства с подходящим не раздражающим эксципиентом, который является твердым при обычных температурах, но жидким при ректальной температуре, и поэтому расплавляется в прямой кишке с высвобождением лекарственного средства. Такие вещества представляют собой масло какао и полиэтиленгликоли.
Для местного применения используют кремы, мази, желе, растворы или суспензии и т.д., содержащие соединение формулы I. (Для целей, указанных в настоящей заявке, местное применение включает растворы или жидкости для полоскания рта или горла.)
В фармацевтических композициях по настоящему изобретению также можно использовать усилители абсорбции, такие как Tween 80, Tween 20, витамин E TPGS (d-альфа-токоферил полиэтиленгликоль 1000 сукцинат) и Gelucire®.
Дозы на уровне от около 0,01 мг до около 140 мг/кг массы тела в день являются полезными для лечения указанных выше состояний или, альтернативно, от около 0,5 мг до около 7 г на пациента в день. Например, воспаление можно эффективно лечить путем введения от около 0,01 до 50 мг соединения на килограмм массы тела в день или, альтернативно, от около 0,5 мг до около 3,5 г на пациента в день, предпочтительно от 2,5 мг до 1 г на пациента в день.
Количество активного ингредиента, которое можно сочетать с веществами, используемыми в качестве носителя, для получения стандартной лекарственной формы, варьирует в зависимости от хозяина, которого лечат, и конкретного способа введения. Например, композиция, предназначенная для перорального введения человеку, может содержать от 0,5 мг до 5 г активного соединения с подходящим и удобным количеством вещества-носителя, которое может варьировать от около 5 до около 95 процентов от общего количества композиции. Стандартные лекарственные формы обычно содержат от около 1 мг до около 500 мг активного ингредиента, типично 25 мг, 50 мг, 100 мг, 200 мг, 300 мг, 400 мг, 500 мг, 600 мг, 800 мг или 1000 мг. Дозируемые количества 4 мг, 8 мг, 18 мг, 20 мг, 36 мг, 40 мг, 80 мг, 160 мг, 320 мг и 640 мг также можно использовать. Стандартные лекарственные формы, содержащие 1, 10 или 100 мг, также охватываются настоящим изобретением.
Однако должно быть понятно, что конкретный уровень доз для любого конкретного пациента зависит от различных факторов, включая возраст, массу тела, общее состояние здоровья, пол, режим питания, время введения, путь введения, скорость выведения из организма, сочетание лекарственных средств и тяжесть конкретного заболевания, которое лечат.
Следующие соединения приводятся в качестве примеров настоящего изобретения. Эти соединения были синтезированы в соответствии со схемами и примерами, описанными ниже.
Способы синтеза
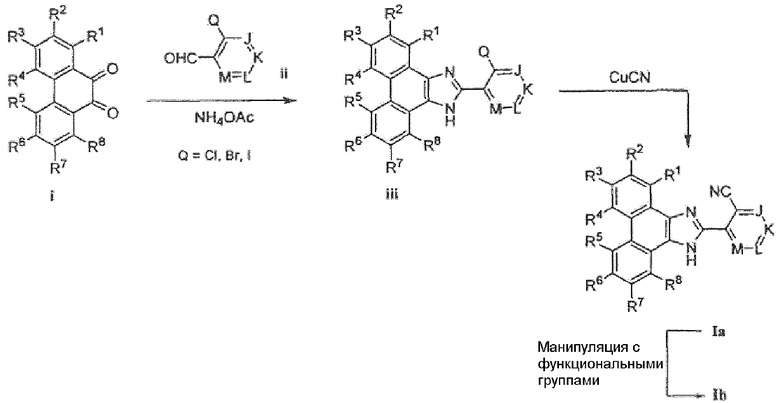
Соединения формулы I по настоящему изобретению можно получить в соответствии со способами синтеза, в общем виде представленными на схемах 1 и 4 ниже, и в соответствии со способами, описанными в настоящей заявке. Имидазол формулы I можно получить в несколько последовательных стадий, исходя из имеющегося фенантренхинона i. Фенантренимидазол iii получают путем обработки фенантренхинона i и соответствующе замещенного альдегида ii реагентом, таким как NH4OAC или NH4HCO3, в растворителе, таком как уксусная кислота. Обработка имидазола iii при помощи CuCN в растворителе, таком как ДМФА или ДМСО, дает моно- или бис-нитрил (M=CCN) Ia. Последующее взаимопреобразование функциональных групп можно осуществить в любых положениях R1-R8. Например, когда один или несколько из R1-R8 заместителей представляют собой Cl, Br или I и когда M является отличным от CBr или CI, Ia можно преобразовать в Ib, помещая Ia в присутствии монозамещенного алкинила, станнана, бороновой кислоты, борана или бороната в условия, которые промотируют реакцию перекрестного связывания, такие как нагревание в присутствии катализатора, такого как Pd(PPh3)4 и CuI, в присутствии основания, такого как карбонат натрия или диизопропиламин, и в подходящем растворителе, таком как ТГФ, ДМФА или DME. Эту последнюю указанную стадию, или любое другое преобразование подходящей функциональной группы можно итерирующим образом повторять на R1-R8.
Схема 1
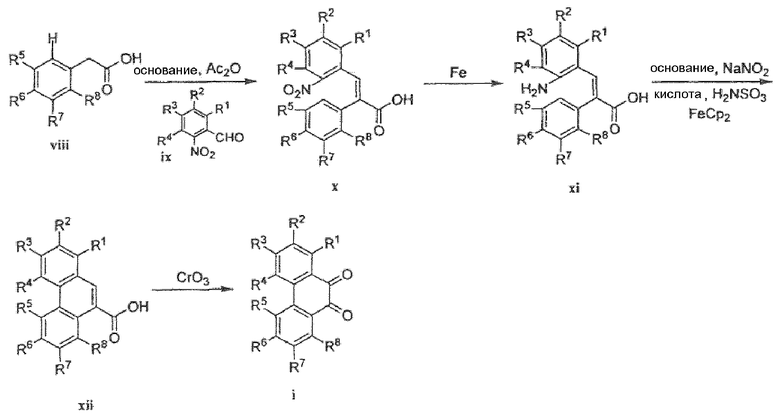
Фенантренхинон i можно получить в несколько последовательных стадий, как показано на схемах 2 и 3. Депротонирование фосфониевой соли iv (схема 2) в присутствии основания, такого как гидрид натрия или метоксид натрия, в растворителе, таком как ДМФА, а затем добавление альдегида v дает стильбен vi в виде смеси E- и Z-изомеров. Внутримолекулярная циклизация этой смеси при воздействии УФ света, в присутствии окислителя, такого как йод, и акцептора кислоты, такого как пропиленоксид, в подходящем растворителе, таком как циклогексан, дает фенантрен vii. Этот фенантрен viia может быть непосредственно окислен при помощи окислителя, такого как CrО3, в подходящем растворителе, таком как уксусная кислота, с получением фенантренхинона i, или, необязательно, фенантрен viia может быть далее преобразован в фенантрен viib путем соответствующего взаимопреобразования любой из функциональных групп R1-R8, такого как трансметаллирование с использованием металлоорганического реагента, такого как бутиллитий, в подходящем растворителе, таком как ТГФ, с последующим добавлением электрофила, такого как йод или диоксид углерода. Альтернативно (схема 3), фенилуксусную кислоту viii можно конденсировать с альдегидом ix в присутствии основания, такого как карбонат калия, и в присутствии уксусного ангидрида с получением нитростильбена x. Этот нитроарил x затем восстанавливают с использованием подходящего восстановителя, такого как железо или сульфат железа, в присутствии гидроксида аммония, в подходящем растворителе, таком как уксусная кислота, с получением амина xi. Диазотизация этого амина xi при помощи нитрита натрия, в присутствии водного гидроксида, такого как гидроксид натрия с последующим подкислением кислотой, такой как серная кислота и сульфаминовая кислота, и циклизация в присутствии катализатора, такого как медь или ферроцен, дает фенантренкарбоновую кислоту xii. Этот фенантрен может быть окислен и одновременно декарбоксилирован с использованием подходящего окислителя, такого как триоксид хрома, в подходящем растворителе, таком как уксусная кислота, с получением фенантренхинона i.
Схема 2
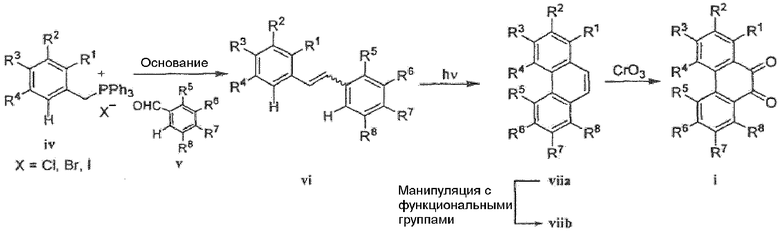
Схема 3
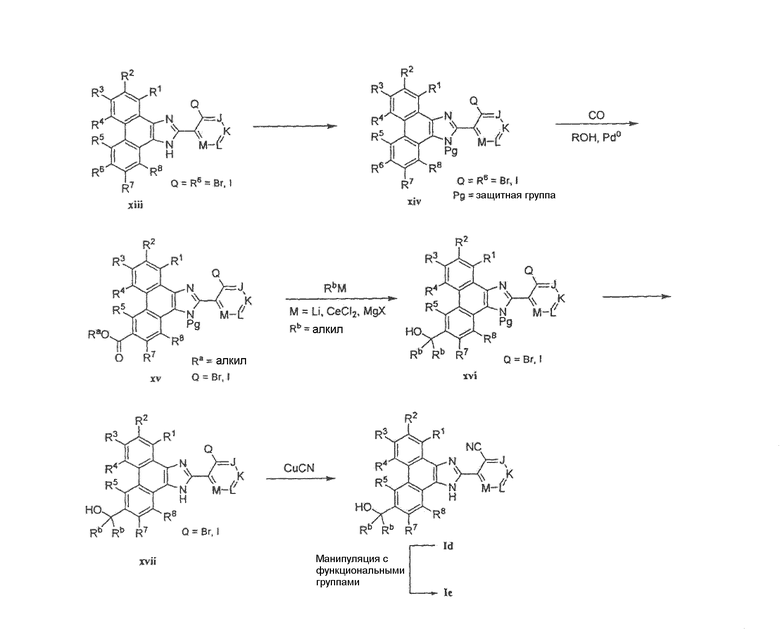
Как показано на схеме 4, защита галогенфенантрена xiii подходящей защитной группой, такой как 2-(триметилсилил)этоксиметил, в присутствии основания, такого как гидрид натрия или диизопропилэтиламин, в подходящем растворителе, таком как ДМФА или метиленхлорид, дает защищенный фенантренимидазол xiv. Этот фенантренимидазол xiv затем подвергают карбонилированию при помощи моноксида углерода в присутствии катализатора, такого как Pd(OAc)2, и в присутствии основания, такого как триэтиламин, в смеси спиртового растворителя, такого как метанол, и ДМФА или любого другого подходящего органического растворителя. Обработка сложного эфира xv нуклеофильным реагентом, таким как литийорганический, церийорганический реагент или реагент Гриньяра, в органическом растворителе, таком как простой эфир, ТГФ или метиленхлорид (реагент Гриньяра), дает третичный спирт xvi. Удаление защитной группы у имидазола, например путем обработки xvi минеральной кислотой, такой как хлористоводородная кислота, или в присутствии источника фтора, такого как TBAF, в органическом растворителе, таком как ТГФ, дает незащищенный имидазол xvii. Обработка этого фенантренимидазола xvii при помощи CuCN в растворителе, таком как ДМФА или ДМСО, дает моно- или бис-нитрил (M=CCN) Id. Последующее взаимопреобразование функциональных групп можно осуществить по любому положению R1-R8. Например, когда один или несколько из заместителей R1-R8 представляет собой Cl, Br или I и когда M является отличным от CBr или CI, соединение Id может быть преобразовано в Ie путем помещения соединения Id в присутствии монозамещенного алкинила, станнана, бороновой кислоты, борана или бороната в условия, которые промотируют реакцию перекрестного связывания, такие как нагревание в присутствии катализатора, такого как Pd(PPh3)4 и CuI, в присутствии основания, такого как карбонат натрия или диизопропиламин, и в подходящем растворителе, таком как ТГФ, ДМФА или DME. Эту последнюю указанную стадию или любое другое преобразование подходящей функциональной группы можно итерирующим образом повторять на R1-R8.
Схема 4
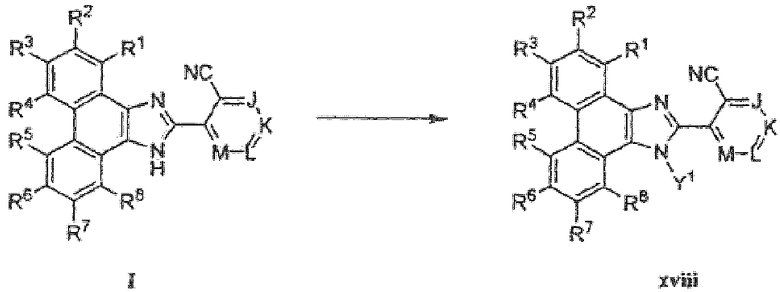
Вторичный амин имидазола может быть замещен, как описано на схеме 5, путем обработки соответствующе функционализированного фенантренимидазола I реагентом, таким как ацилирующий агент или алкилирующий агент, такой как метилиодид, в присутствии основания, такого как гидрид натрия, в подходящем растворителе, таком как ДМФА.
Схема 5
ПРИМЕРЫ
Настоящее изобретение представлено следующими не ограничивающими примерами:
ПРИМЕР 14
2-[9-хлор-6-(3-гидрокси-3-метилбутил-1-ин-1-ил)-1H-фенантро[9,10-d]имидазол-2-ил]-3-фторбензонитрил
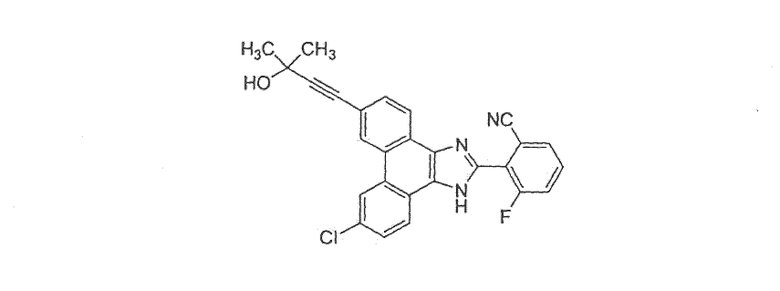
Стадия 1: 6,9-дибром-2-(2-хлор-6-фторфенил)-1H-фенантро[9,10-d]имидазол
К раствору 30 г (82 ммоль) 3,6-дибромфенантрен-9,10-диона (Bhatt, Tetrahedron, 1963, 20, 803) в 1,0 л уксусной кислоты добавляли 25,9 г (328 ммоль) NН4НCO3 с последующим добавлением 26 г (164 ммоль) 2-фтор-6-хлорбензальдегида. Раствор перемешивали в течение ночи при 130єC, охлаждали до комнатной температуры и выливали в 2,5 л воды. Смесь фильтровали, промывали водой с последующим добавлением гексана и диэтилового эфира. Полученное твердое вещество кипятили с обратным холодильником в 1,0 л толуола с использованием аппарата Дина-Старка и примерно 100 мл воды удаляли за 3 часа. После охлаждения до комнатной температуры происходила кристаллизация бежевого твердого вещества из раствора. Это твердое вещество фильтровали, промывали толуолом и откачивали при пониженном давлении с получением 32 г (80%) 6,9-дибром-2-(2-хлор-6-фторфенил)-1H-фенантро[9,10-d]имидазола.
Стадия 2: 2-(6-бром-9-хлор-1H-фенантро[9,10-d]имидазол-2-ил)-3-фторбензонитрил
К раствору в ДМФА (10 мл) 3,0 г 6,9-дибром-2-(2-хлор-6-фторфенил)-1H-фенантро[9,10-d]имидазола со стадии 1 добавляли 587 мг CuCN и раствор перемешивали в течение ночи при 130°C. Раствор охлаждали до комнатной температуры с последующим добавлением водного раствора гидроксида аммония и этилацетата. Слои разделяли и органический слой промывали насыщенным солевым раствором, сушили над сульфатом натрия и летучие вещества удаляли при пониженном давлении. Остаток очищали флэш-хроматографией на силикагеле с использованием градиента от 30% до 50% этилацетата/гексан с получением 500 мг 2-(6-бром-9-хлор-1H-фенантро[9,10-d]имидазол-2-ил)-3-фторбензонитрила.
Стадия 3: 2-[9-хлор-6-(3-гидрокси-3-метилбутил-1-ин-1-ил)-1H-фенантро[9,10-d]имидазол-2-ил]-3-фторбензонитрил
К раствору в ДМФА (2 мл) 2-(6-бром-9-хлор-1H-фенантро[9,10-d]имидазол-2-ил)-3-фторбензонитрила (320 мг) со стадии 2 добавляли 5 мл триэтиламина, 0,1 мл 2-метил-3-бутин-2-ола, 20 мг CuI и 82 мг Pd(PPh3)4. Полученную смесь перемешивали в течение ночи при 80°C, охлаждали до комнатной температуры и разбавляли этилацетатом/водой. Органический слой промывали насыщенным солевым раствором, сушили над сульфатом натрия и летучие вещества удаляли при пониженном давлении. Остаток очищали флэш-хроматографией на силикагеле с использованием градиента от 30% до 50% этилацетата/гексан с получением 85 мг 2-[9-хлор-6-(3-гидрокси-3-метилбутил-1-ин-1-ил)-1H-фенантро[9,10-d]имидазол-2-ил]-3-фторбензонитрила.
1Н ЯМР (ацетон-d6): δ 8,89 (с, 2Н), 8,71 (ушир.с, 1Н), 8,51 (ушир.с, 1Н), 7,93 (д, 1Н), 8,88-8,72 (м, 4Н), 4,55 (с, 1Н), 1,65 (с, 6Н).
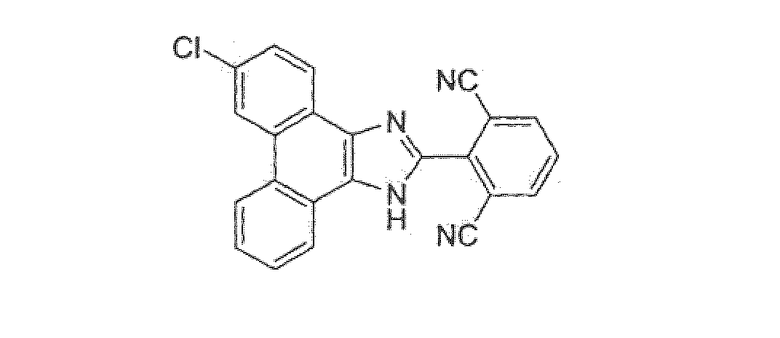
ПРИМЕР 25
2-(6-хлор-1H-фенантро[9,10-d]имидазол-2-ил)изофталонитрил
Стадия 1: 1-(3-фенантрил)этаноноксим
В 200 мл абсолютного этанола объединяли смесь 50 г (0,23 моль) 1-(3-фенантрил)этанона и 40 г гидрохлорида гидроксиламина. Раствор нагревали до температуры кипения с обратным холодильником с последующим добавлением 70 мл пиридина. Через 3 часа реакционную смесь охлаждали до комнатной температуры и раствор концентрировали на роторном испарителе. К остатку добавляли смесь лед/вода и смесь перемешивали в течение 1 часа. Полученное не совсем белое твердое вещество фильтровали, промывали водой и сушили на воздухе с получением после перекристаллизации в диэтиловом эфире 32 г 1-(3-фенантрил)этаноноксима.
Стадия 2: 3-фенантриламин
К 385 г полифосфорной кислоты при 100°C добавляли 32 г (0,14 моль) 1-(3-фенантрил)этаноноксима со стадии 1 в течение 30 минут. Смесь перемешивали при 100°C в течение 2 часов, охлаждали до комнатной температуры с последующим добавлением смеси вода/лед. Смесь перемешивали 30 минут, фильтровали и промывали водой. Полученное белое твердое вещество затем помещали в 500 мл метанола и 40 мл концентрированной HCl. Реакционную смесь кипятили с обратным холодильником в течение ночи, охлаждали до комнатной температуры и концентрировали. К остатку добавляли смесь этилацетат/вода и полученный раствор подщелачивали при помощи 10 н. раствора KOH. Водный слой экстрагировали этилацетатом и объединенные органические слои промывали водой, насыщенным солевым раствором, сушили над сульфатом натрия и летучие вещества удаляли при пониженном давлении с получением 25 г 3-фенантриламина в виде бежевого твердого вещества.
Стадия 3: 3-хлорфенантрен
CuCl2 (21 г) сушили в высоком вакууме при 115°C в течение 90 минут, затем охлаждали до 65°C с последующим добавлением 250 мл безводного ацетoнитрила и 26 г трет-бутилнитрита. Добавляли 3-фенантриламин (25 г) со стадии 2 в течение 30 минут в виде раствора в 100 мл ацетoнитрила. Реакционную смесь перемешивали в течение 45 минут при 65єC, охлаждали до комнатной температуры с последующим добавлением 1 л 1 н. раствора HCl. Водный слой экстрагировали метиленхлоридом и объединенные органические слои промывали водой, насыщенным солевым раствором, сушили над сульфатом натрия и летучие вещества удаляли при пониженном давлении. Остаток очищали флэш-хроматографией на силикагеле с использованием гексана в качестве элюента с получением белого твердого вещества, которое перекристаллизовывали из гексана, с получением 14,4 г 3-хлорфенантрена в виде белого твердого вещества.
Стадия 4: 3-хлорфенантрен-9,10-дион
К раствору 12,5 г (58,7 ммоль) 3-хлорфенантрена со стадии 3 в 350 мл уксусной кислоты добавляли 23,5 г (0,23 моль) CrO3. Реакционную смесь перемешивали в течение 2 часов при 100єC, охлаждали до комнатной температуры и выливали в 2 л воды. Суспензию перемешивали в течение 1 часа, фильтровали и промывали водой. Остаток сушили в высоком вакууме с получением 12,5 г (88%) 3-хлорфенантрен-9,10-диона.
Стадия 5: 6-хлор-2-(2,6-дибромфенил)-1H-фенантро[9,10-d]имидазол
Этот имидазол получали, следуя процедуре, описанной в примере 14, стадия 1, но с замещением 3,6-дибромфенантрен-9,10-диона 3-хлорфенантрен-9,10-дионом и с замещением 2-фтор-6-хлорбензальдегида 2,6-дибромбензальдегидом, с получением 27 г 6-хлор-2-(2,6-дибромфенил)-1H-фенантро[9,10-d]имидазола в виде не совсем белого твердого вещества.
Стадия 6: 2-(6-хлор-1Н-фенантро[9,10-d]имидазол-2-ил)изофталонитрил
К раствору в ДМФА (300 мл) 32 г (65,7 ммоль) 6-хлор-2-(2,6-дибромфенил)-1Н-фенантро[9,10-d]имидазола со стадии 5 добавляли 14,7 г CuCN. Реакционную смесь перемешивали в течение ночи при 80°C, охлаждали до комнатной температуры, выливали в смесь 1,5 л воды, 1,5 л этилацетата и 200 мл концентрированного гидроксида аммония и перемешивали в течение 1 часа при комнатной температуре. Водный слой экстрагировали этилацетатом и объединенные органические слои промывали 10% раствором гидроксида аммония, водой, насыщенным солевым раствором, сушили над сульфатом натрия и летучие вещества удаляли при пониженном давлении. Остаток быстро переносили в толуол (2×200 мл) и этилацетат (1 л). Полученное твердое вещество очищали флэш-хроматографией на силикагеле 5 порциями с использованием градиента от 60% до 80% до 100% этилацетата/гексана с получением 19,9 г 2-(6-хлор-1H-фенантро[9,10-d]имидазол-2-ил)изофталонитрила в виде бледно-желтого твердого вещества.
1Н ЯМР (400 МГц, ДМСО): δ 14,32 (с, 1Н), 9,0-8,9 (м, 2Н), 8,55-8,45 (м, 4Н), 7,99 (т, 1Н), 7,85-7,78 (м, 2Н), 7,72 (т, 1Н).
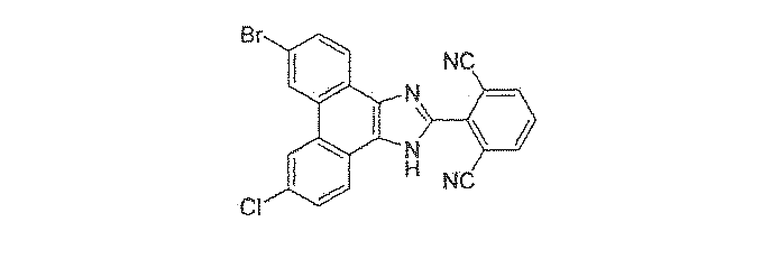
ПРИМЕР 36
2-(6-бром-9-хлор-1H-фенантро[9,10-d]имидазол-2-ил)изофталонитрил
Стадия 1: 1-бром-4-[2-(4-хлорфенил)винил]бензол
К раствору (4-бромбензил)трифенилфосфонийбромида (396 г; 0,77 моль) в 2,5 л ДМФА при 0°C добавляли 37 г (0,92 моль) NaH (60% в масле) четырьмя порциями. Раствор перемешивали в течение 1 часа при 0°C с последующим добавлением 109 г (0,77 моль) 4-хлорбензальдегида двумя порциями. Полученную смесь нагревали до комнатной температуры, перемешивали в течение 1 часа и гасили, выливая реакционную смесь в 5єC смесь 10 л воды и 2,5 л Et2О. Водный слой экстрагировали при помощи Et2О, объединенные органические слои промывали насыщенным солевым раствором и сушили над Na2SO4. Летучие вещества удаляли при пониженном давлении и остаток растворяли в 1,5 л циклогексана и фильтровали через слой силикагеля (промывка циклогексаном). 16 г одного изомера кристаллизовали из раствора в виде белого твердого вещества и после выпаривания летучих веществ выделяли 166 г другого изомера 1-бром-4-[2-(4-хлорфенил)винил]бензола.
Стадия 2: 3-бром-6-хлорфенантрен
В 2-л сосуд, снабженный внутренним водоохлаждаемым кожухом из пирекса, загружали 5,16 г (17 ммоль) 1-бром-4-[2-(4-хлорфенил)винил]бензола со стадии 1, 2 л циклогексана, 25 мл ТГФ, 25 мл пропиленоксида и 6,7 г (26 ммоль) йода. Перемешиваемый раствор дегазировали путем барботирования азота и подвергали УФ облучению в течение 24 часов путем введения внутрь ртутной лампы среднего давления 450 ватт. Реакционную смесь гасили при помощи 10% раствора Na2S2О3 и водный слой экстрагировали этилацетатом. Объединенные органические слои промывали насыщенным солевым раствором, сушили над Na2SO4 и летучие вещества удаляли при пониженном давлении. Остаток переносили в минимальное количество этилацетата с получением примерно 5 г 3-бром-6-хлорфенантрена в виде твердого вещества.
Стадия 3: 3-Бром-6-хлорфенантрен-9,10-дион
К раствору 3-бром-6-хлорфенантрена со стадии 2 (1,71 г; 5,86 ммоль) в 35 мл уксусной кислоты добавляли 2,3 г (23,5 ммоль) CrО3. Смесь перемешивали в течение 2 часов при 100єC, охлаждали до комнатной температуры, выливали в 300 мл воды и перемешивали в течение 1 часа. Суспензию фильтровали, промывали водой и Et2О и откачивали при пониженном давлении с получением 1,67 г 3-бром-6-хлорфенантрен-9,10-диона в виде твердого вещества.
Стадия 4: 9-бром-6-хлор-2-(2,6-дибромфенил)-1H-фенантро[9,10-d]имидазол
К раствору 15,5 г 3-бром-6-хлорфенантрен-9,10-диона со стадии 3 в 400 мл уксусной кислоты добавляли 74,2 г ацетата аммония и 19,1 г 2,6-дибромбензальдегида. Смесь перемешивали в течение ночи при 120єC, охлаждали до комнатной температуры, разбавляли в 4 л воды и фильтровали. Полученное твердое вещество кипятили с обратным холодильником в течение 2 часов в толуоле с использованием аппарата Дина-Старка. После охлаждения до комнатной температуры суспензию фильтровали, твердое вещество промывали толуолом и полученное бежевое твердое вещество сушили в высоком вакууме с получением 26 г 9-бром-6-хлор-2-(2,6-дибромфенил)-1H-фенантро[9,10-d]имидазола.
Стадия 5: 2-(9-бром-6-хлор-1H-фенантро[9,10-d]имидазол-2-ил)изофталонитрил
К раствору 26 г 9-бром-6-хлор-2-(2,6-дибромфенил)-1H-фенантро[9,10-d]имидазола со стадии 4 в 200 мл безводного ДМФА добавляли 14,2 г CuCN. Реакционную смесь перемешивали в течение ночи при 85єC, охлаждали до комнатной температуры, добавляли насыщенный солевой раствор и смесь перемешивали в течение 30 минут. Раствор разбавляли в этилацетате, промывали 10% раствором гидроксида аммония, насыщенным солевым раствором, сушили над сульфатом натрия и летучие вещества удаляли при пониженном давлении с получением 26 г 2-(9-бром-6-хлор-1H-фенантро[9,10-d]имидазол-2-ил)изофталонитрила в виде твердого вещества.
1Н ЯМР (ацетон-d6) δ 9,19 (с, 1Н), 9,02 (с, 1Н), 9,71 (ушир.с, 1Н), 8,49 (ушир.с, 1Н), 8,39 (д, 2Н), 8,07 (т, 1Н), 7,97 (д, 1Н), 8,81 (д, 1Н).
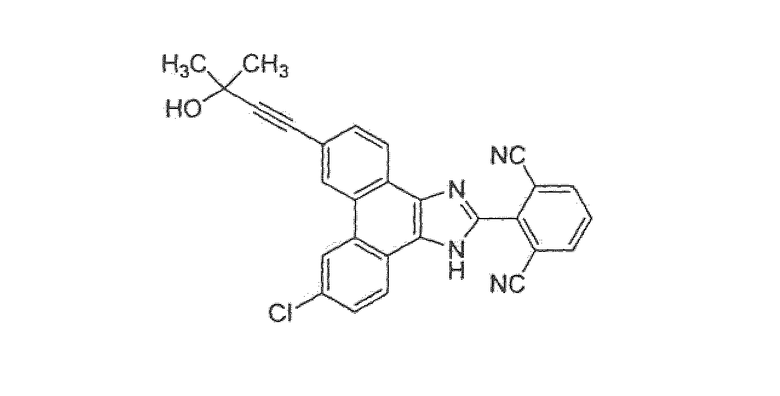
ПРИМЕР 40
2-[9-хлор-6-(3-гидрокси-3-метилбут-1-ин-1-ил)-1Н-фенантро[9,10-d]имидазол-2-ил]изофталонитрил
Стадия 1: (2Е)-2-(4-бромфенил)-3-(4-хлор-2-нитрофенил)акриловая кислота
В 2-л колбу, снабженную механической мешалкой, загружали 183 г 2-нитро-4-хлорбензальдегида, 212 г 4-бромфенилуксусной кислоты и 233 мл уксусного ангидрида. К этому раствору добавляли 82 г карбоната калия и реакционную смесь перемешивали в течение ночи при 100єC. Полученную темную смесь охлаждали до комнатной температуры и добавляли 1,6 л воды с последующим добавлением 800 мл 10% раствора HCl. Раствор декантировали и переносили в воду/этилацетат. Слои разделяли, органическую фазу промывали насыщенным солевым раствором, сушили над сульфатом магния и летучие вещества удаляли при пониженном давлении. Остаток растирали в EtOH и маточный раствор еще 4 раза растирали с EtOH с получением 219 г желаемой (2Е)-2-(4-бромфенил)-3-(4-хлор-2-нитрофенил)акриловой кислоты.
Стадия 2: (2Е)-3-(2-амино-4-хлорфенил)-2-(4-бромфенил)акриловая кислота
К 50°C раствору 135 г (2Е)-2-(4-бромфенил)-3-(4-хлор-2-нитрофенил)акриловой кислоты со стадии 1 в 1,2 л уксусной кислоты и 80 мл воды добавляли 98 г железа (порошок) по порциям, поддерживая температуру ниже 50єC. Смесь перемешивали в течение 2 часов при 50єC, охлаждали до комнатной температуры, разбавляли этилацетатом (1 л) и фильтровали через слой целита. Добавляли воду (1 л), слои разделяли и органический слой промывали 2 раза водой, насыщенным солевым раствором, сушили над сульфатом магния и летучие вещества удаляли при пониженном давлении. Остаточную уксусную кислоту удаляли добавлением к неочищенной смеси 1 л H2O, раствор фильтровали и промывали при помощи еще 1 л H2O и в завершение твердое вещество сушили в высоком вакууме с получением 130 г (2Е)-3-(2-амино-4-хлорфенил)-2-(4-бромфенил)акриловой кислоты.
Стадия 3: 3-Бром-6-хлорфенантрен-9,10-дион
Этот хинон можно получить, следуя процедуре, описанной в примере 36, стадии 1-3, или с использованием следующей процедуры: к 0єC раствору 118 мл концентрированной серной кислоты в 1,0 л воды добавляли по каплям раствор, полученный следующим способом: 65 г (2Е)-3-(2-амино-4-хлорфенил)-2-(4-бромфенил)акриловой кислоты со стадии 2 в 1 л воды, с последующим добавлением 11 г NaOH при перемешивании в течение 10 минут при 0єC, добавлением NaNО2 (15 г) и перемешиванием полученного раствора при 0°C в течение 20 минут. Через 30 минут к полученной смеси добавляли сульфаминовую кислоту (12,5 г) и после прекращения выделения газа добавляли 1,3 л ацетона и раствор перемешивали при 0°C в течение 10 минут. Полученную смесь затем добавляли к раствору ферроцена (6,9 г) в 480 мл ацетона, в результате чего происходило образование зеленого осадка. После перемешивания в течение 20 минут добавляли воду (2,0 л), твердое вещество фильтровали и получали 6-бром-3-хлорфенантрен-9-карбоновую кислоту, подвергая ее сушке на воздухе. Полученный неочищенный фенантрен помещали в 2,0 л уксусной кислоты с последующим добавлением 54 г CrO3. Реакционную смесь помещали в температурные условия 110°C и после перемешивания в течение 1 часа добавляли 18 г CrO3. Реакцию отслеживали при помощи ТСХ и каждый час добавляли 18 г CrO3 в течение 3 часов, после чего наблюдали 100% конверсию по данным анализа 1H ЯМР. Смесь охлаждали до комнатной температуры, разбавляли в воде (2,0 л), фильтровали и промывали водой (1,0 л) с получением после сушки 37 г 3-бром-6-хлорфенантрен-9,10-диона в виде желтого твердого вещества.
Стадия 4: 9-бром-6-хлор-2-(2,6-дибромфенил)-1H-фенантро[9,10-d]имидазол
Этот имидазол получали, следуя процедуре, описанной для примера 36, стадия 4.
Стадия 5: 2-(9-бром-6-хлор-1H-фенантро[9,10-d]имидазол-2-ил)изофталонитрил
Этот имидазол получали, следуя процедуре, описанной для примера 36, стадия 5.
Стадия 6: 2-[9-хлор-6-(3-гидрокси-3-метилбут-1-ин-1-ил)-1Н-фенантро[9,10-d]имидазол-2-ил]изофталонитрил
К раствору 13 г 2-(9-бром-6-хлор-1H-фенантро[9,10-d]имидазол-2-ил)изофталонитрила в 240 мл ДМФА добавляли 5,5 мл 2-метил-3-бутин-2-ола, 2,0 г тетракис(трифенилфосфин)палладия, 1,1 г иодида меди и 5,6 мл диизопропиламина. Смесь перемешивали при 55°C в течение 1 часа, затем охлаждали до комнатной температуры и разбавляли этилацетатом (250 мл). Добавляли воду (250 мл) и слои разделяли, органическую фазу промывали насыщенным солевым раствором, сушили над сульфатом магния и летучие вещества удаляли при пониженном давлении. Неочищенную смесь затем очищали на силикагеле с использованием смеси 50% гексан/этилацетат. Продукт затем перекристаллизовывали в ТГФ и растирали в горячей смеси этилацетат/простой эфир с получением 5,4 г [9-хлор-6-(3-гидрокси-3-метилбут-1-ин-1-ил)-1Н-фенантро[9,10-d]имидазол-2-ил]изофталонитрила в виде светло-желтого твердого вещества.
1Н ЯМР (ацетон-d6): δ 8,93 (с, 2Н), 8,53 (м, 2Н), 8,36 (д, 2Н), 8,01 (т, 1Н), 7,78 (д, 2Н), 4,53 (с, 1Н), 1,61 (с, 6Н).
ПРИМЕР 60
2-(1-{[дигидрокси(диоксидо)фосфино]метил}-1H-фенантро[9,10-d]имидазол-2-ил)изофталонитрил
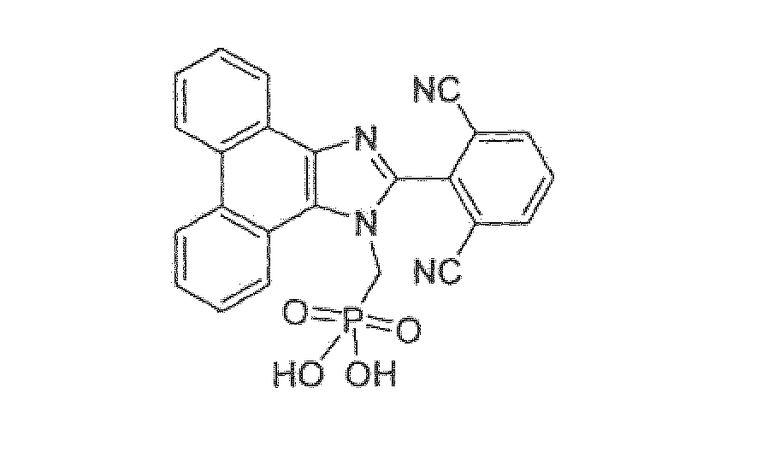
Стадия 1: 2-(2,6-дибромфенил)-1H-фенантро[9,10-d]имидазол
Этот имидазол получали, следуя процедуре, описанной в примере 36, стадия 4, но с замещением 3-бром-6-хлорфенантрен-9,10-диона фенантрен-9,10-дионом, с получением 2-(2,6-дибромфенил)-1H-фенантро[9,10-d]имидазола
Стадия 2: 2-(1H-фенантро[9,10-d]имидазол-2-ил)изофталонитрил
Это соединение получали с использованием процедуры, описанной в примере 36, стадия 5, но с замещением 9-бром-6-хлор-2-(2,6-дибромфенил)-1H-фенантро[9,10-d]имидазола 2-(2,6-дибромфенил)-1H-фенантро[9,10-d]имидазолом, с получением желаемого 2-(1Н-фенантро[9,10-d]имидазол-2-ил)изофталонитрила.
Стадия 3: 2-[1-(хлорметил)-1Н-фенантро[9,10-d]имидазол-2-ил]изофталонитрил
2-(1Н-фенантро[9,10-d]имидазол-2-ил)изофталонитрил со стадии 2 (1 г, 2,91 ммоль) смешивали карбонатом цезия (1,14 г, 3,49 ммоль) в хлорйодметане (10 мл). Смесь нагревали до 80°C в течение ночи. Реакционную смесь охлаждали до комнатной температуры и выливали в 200 мл воды и 500 мл этилацетата. Слои разделяли и органический слой промывали при помощи 200 мл воды, 200 мл насыщенного водного раствора бикарбоната натрия, 100 мл насыщенного солевого раствора и сушили над безводным сульфатом магния. Растворитель удаляли при пониженном давлении. Неочищенное твердое вещество очищали колоночной флэш-хроматографией с использованием 40% этилацетата в гексане с получением 357 мг 2-[1-(хлорметил)-1Н-фенантро[9,10-d]имидазол-2-ил]изофталонитрила (31%) плюс 650 мг смеси продукта и исходного вещества.
Стадия 4: 2-(1-{[дигидрокси(диоксидо)фосфино]метил}-1Н-фенантро[9,10-d]имидазол-2-ил)изофталонитрил
2-[1-(хлорметил)-1Н-фенантро[9,10-d]имидазол-2-ил] изофталонитрил со стадии 3 (200 мг, 0,509 ммоль) смешивали с ди(трет-бутил)фосфатом тетраметиламмония (288 мг, 1,02 ммоль) в ДМФА (5 мл) и нагревали при 50°C в течение 8 часов. Смесь охлаждали до комнатной температуры и выливали в 15 мл воды и 35 мл этилацетата. Слои разделяли и органический слой промывали при помощи 10 мл воды (два раза), 10 мл насыщенного водного раствора бикарбоната натрия, насыщенного солевого раствора и сушили над безводным сульфатом магния. Растворитель удаляли при пониженном давлении. Неочищенное твердое вещество очищали колоночной флэш-хроматографией с использованием 50-70% этилацетата в гексане с получением 221 мг защищенного фосфата (77%). 155 мг этого твердого вещества растворяли в 10% ТГФ/толуол (3 мл) и перемешивали при комнатной температуре в течение ночи. Растворитель удаляли при пониженном давлении. Полученный неочищенный продукт очищали полупрепаративной ОФ-ВЭЖХ с использованием колонки C18 и с элюированием градиентом 44-49% ацетoнитрила + 0,2% TFA в течение 8 минут. Фракции, содержащие продукт, объединяли и лиофилизировали с получением 80 мг желаемого 2-(1-{[дигидрокси(диоксидо)фосфино]метил}-1H-фенантро[9,10-d]имидазол-2-ил)изофталонитрила.
1Н ЯМР (ДМСО): δ 9,05 (д, 1Н), 8,95 (д, 1Н), 8,54-8,61 (м, 2Н), 8,47 (д, 2Н), 8,06 (т, 1Н), 8,70-8,85 (м, 4Н), 6,21 (д, 2Н).
ПРИМЕР 87
2-[6-бром-9-(1-гидрокси-1-метилэтил)-1H-фенантро[9,10-d]имидазол-2-ил]изофталонитрил
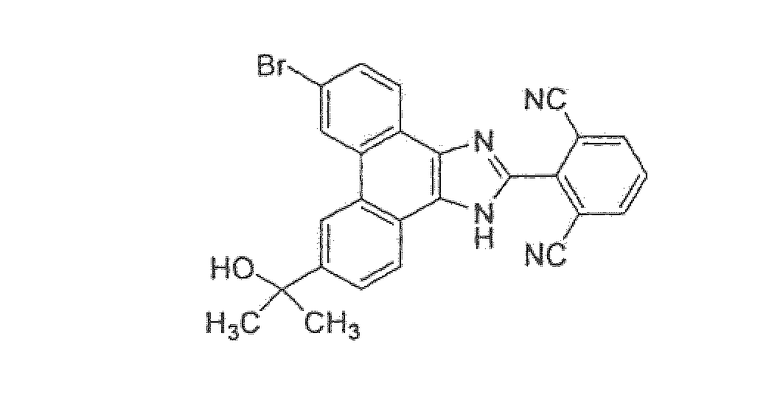
Стадия 1: 6,9-дибром-2-(2,6-дибромфенил)-1H-фенантро[9,10-d]имидазол
Cуспензию дибромхинона (38,6 г, 0,1 моль), ацетата аммония (165 г, 2,1 моль) и дибромбензальдегида (45 г, 0,1 моль) в уксусной кислоте (1,5 л) нагревали при температуре кипения с обратным холодильником в течение 16 часов. Реакционную смесь гасили, выливая ее в воду (2,2 л), с последующим перемешиванием в течение 2 часов. Полученное твердое вещество фильтровали и промывали последовательно водой и гексанами. Твердое вещество затем нагревали при температуре кипения с обратным холодильником в толуоле (600 мл) с использованием аппарата Дина-Старка в течение 4 часов и затем фильтровали с получением желаемого 6,9-дибром-2-(2,6-дибромфенил)-1H-фенантро[9,10-d]имидазола в виде бежевого порошка (62,3 г, 97%).
Стадия 2: 6,9-дибром-2-(2,6-дибромфенил)-1-{[2-(триметилсилил)этокси]метил}-1Н-фенантро[9,10-d]имидазол
К суспензии 6,9-дибром-2-(2,6-дибромфенил)-1Н-фенантро[9,10-d]имидазола со стадии 1 (61,8 г, 0,1 моль) в ТГФ (980 мл) при 0°C добавляли гидрид натрия (60% дисперсия в минеральном масле, 10 г, 0,25 моль). Суспензию перемешивали при 0°C в течение 15 минут с последующим добавлением SEMCl (45 мл, 0,25 моль). Смесь нагревали до комнатной температуры и перемешивали в течение 3 часов, после чего ее выливали в воду. Водную фазу экстрагировали этилацетатом, органический слой промывали один раз насыщенным солевым раствором, сушили над Na2SO4, фильтровали и концентрировали. Неочищенное вещество переносили в смесь гексан/диэтиловый эфир на 4 часа, затем фильтровали с получением 6,9-дибром-2-(2,6-дибромфенил)-1-{[2-(триметилсилил)этокси]метил}-1H-фенантро[9,10-d]имидазола в виде бежевого порошка (71,5 г, 95%).
Стадия 3: метил 6-бром-2-(2,6-дибромфенил)-1-{[2-(триметилсилил)этокси]метил}-1H-фенантро[9,10-d]имидазол-9-карбоксилат
К раствору 6,9-дибром-2-(2,6-дибромфенил)-1-{[2-
(триметилсилил)этокси]метил}-1H-фенантро[9,10-d]имидазола со стадии 2 (22,8 г, 30,8 ммоль) в ДМФА (150 мл) и MeOH (150 мл) в 3-горлую 1-л круглодонную колбу добавляли Pd(OAc)2 (350 мг, 1,5 ммоль) и dppf (1,7 г, 3,0 ммоль). Смесь дегазировали три раза и снова наполняли моноксидом углерода. Затем добавляли триэтиламин (9,5 мл, 43 ммоль) и реакционную смесь нагревали при 60°C в атмосфере моноксида углерода в течение 1 часа. Реакционную смесь гасили, выливая ее в воду и этилацетат. Затем смесь фильтровали через целит, водную фазу экстрагировали этилацетатом, органический слой промывали один раз насыщенным солевым раствором, сушили над Na2SO4, фильтровали и концентрировали. Неочищенное вещество очищали флэш-хроматографией на диоксиде кремния (0-5% этилацетата в толуоле) с получением изомеров желаемого метил 6-бром-2-(2,6-дибромфенил)-1-{[2-(триметилсилил)этокси]метил}-1Н-фенантро[9,10-d]имидазол-9-карбоксилата в виде твердых веществ бежевого цвета (9,8 г, 44%).
Стадия 4: 2-[6-бром-2-(2,6-дибромфенил)-1Н-фенантро[9,10-d]имидазол-9-ил]пропан-2-ол
К -78°C раствору изомерного метил 6-бром-2-(2,6-дибромфенил)-1-{[2-(триметилсилил)этокси]метил}-1H-фенантро[9,10-d]имидазол-9-карбоксилата со стадии 3 (9,9 г, 13,8 ммоль) в CН2Cl2 (200 мл) добавляли метилмагнийбромид (3,0 M в Et2О, 33 мл) через капельную воронку. Смесь затем нагревали до -40°C, перемешивали при этой температуре в течение 0,5 часа, затем нагревали до температуры в пределах от -30 до -35°C и перемешивали при этой температуре в течение 2 часов. Реакционную смесь затем нагревали до -25°C, перемешивали в течение 3 часов и затем перемешивали при 0°C в течение 1,5 часов. Реакцию гасили, выливая реакционную смесь в воду и этилацетат. Водную фазу экстрагировали этилацетатом, органический слой промывали один раз насыщенным солевым раствором, сушили над Na2SO4, фильтровали и концентрировали. Неочищенный продукт растворяли в ТГФ (150 мл) и охлаждали до 0°C. Затем добавляли TBAF (1,0 M в ТГФ, 35 мл) и смесь нагревали при температуре кипения с обратным холодильником в течение 17 часов, затем гасили при помощи 25% NH4OAC, водную фазу экстрагировали этилацетатом, органический слой промывали один раз насыщенным солевым раствором, сушили над Na2SO4, фильтровали и концентрировали. Вещество, полученное после очистки флэш-хроматографией на диоксиде кремния (5-30% ТГФ в толуоле), переносили в толуол на 5 часов и затем фильтровали с получением 2-[6-бром-2-(2,6-дибромфенил)-1H-фенантро[9,10-d]имидазол-9-ил]пропан-2-ола в виде белого порошка (4,53 г, 56%, 2 стадии).
Стадия 5: 2-[6-бром-9-(1-гидрокси-1-метилэтил)-1Н-фенантро[9,10-d]имидазол-2-ил]изофталонитрил
Цианид меди (420 мг, 4,7 ммоль) добавляли при комнатной температуре к раствору 2-[6-бром-2-(2,6-дибромфенил)-1H-фенантро[9,10-d]имидазол-9-ил]пропан-2-ола со стадии 4 (1,25 г, 2,1 ммоль) в ДМФА (100 мл) и смесь нагревали при 80°C в течение 18 часов, после чего ее выливали в смесь NН4OН и этилацетата и перемешивали в течение 1 часа. Водную фазу экстрагировали этилацетатом, органический слой промывали один раз водой, один раз насыщенным солевым раствором, сушили над Na2SO4, фильтровали и концентрировали. Вещество, полученное после очистки флэш-хроматографией на диоксиде кремния (20-80% этилацетата в толуоле), переносили в этилацетат и ТГФ на 2 часа и затем фильтровали с получением 2-[6-бром-9-(1-гидрокси-1-метилэтил)-1Н-фенантро[9,10-d]имидазол-2-ил]изофталонитрила в виде желтого твердого вещества (250 мг, 25%).
1Н ЯМР δ (м.д.) (ДМСО с добавленным TFA) 9,08 (1Н, с), 8,90 (1Н, с), 8,45-8,39 (4Н, м), 7,99-7,91 (3Н, м), 1,61 (6Н, с).
ПРИМЕР 88
2-[6-(циклопропилэтинил)-9-(1-гидрокси-1-метилэтил)-1H-фенантро[9,10-d]имидазол-2-ил]изофталонитрил
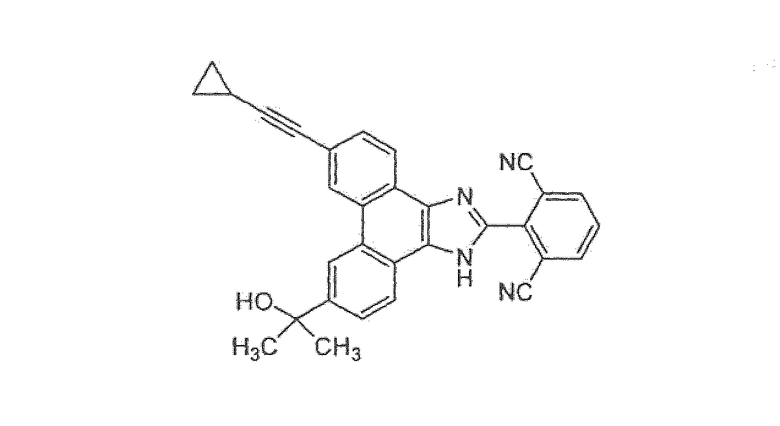
Стадия 1: 2-[6-(циклопропилэтинил)-9-(1-гидрокси-1-метилэтил)-1Н-фенантро[9,10-d]имидазол-2-ил]изофталонитрил
Круглодонную колбу, содержащую 2-[6-бром-9-(l-гидрокси-1-метилэтил)-1H-фенантро[9,10-d]имидазол-2-ил]изофталонитрил из примера 87 (1,26 г, 2,62 ммоль), Pd(PPh3)4 (190 мг, 0,27 ммоль) и иодид меди (100 мг, 0,52 ммоль), продували азотом в течение 15 минут с последующим добавлением ДМФА (50 мл), циклопропилацетилена (1,4 мл, 21 ммоль) и диизопропиламина (560 мкл, 4 ммоль). Полученную смесь нагревали при 60-65°C в течение 3,5 часов, охлаждали до комнатной температуры и затем выливали в смесь NН4OН и этилацетата и перемешивали в течение 1 часа. Водную фазу экстрагировали этилацетатом, органический слой промывали один раз водой, один раз насыщенным солевым раствором, сушили над Na2SO4, фильтровали и концентрировали. Вещество, полученное после очистки флэш-хроматографией на диоксиде кремния (30-100% этилацетата в толуоле), переносили в толуол на 2 часа и затем фильтровали с получением 2-[6-(циклопропилэтинил)-9-(1-гидрокси-1-метилэтил)-1Н-фенантро[9,10-d]имидазол-2-ил]изофталонитрила в виде желтого твердого вещества (350 мг). Маточный раствор объединяли со смешанными фракциями и снова очищали флэш-хроматографией на диоксиде кремния (3-40% ацетoнитрила в толуоле) с получением 286 мг бис-нитрила (общий выход 52%).
1Н ЯМР δ (м.д.) (ДМСО с добавленным TFA): 8,92 (1Н, с), 8,87 (1Н, с), 8,43-8,39 (4Н, м), 7,96 (1Н, т), 7,90 (1Н, д), 7,71 (1Н, д), 1,60 (7Н, с), 0,90 (2Н, т), 0,84 (2Н, д).
ПРИМЕР 117
2-[9-хлор-6-(3-гидрокси-3-метилбутил)-1H-фенантро[9,10-d]имидазол-2-ил)изофталонитрил
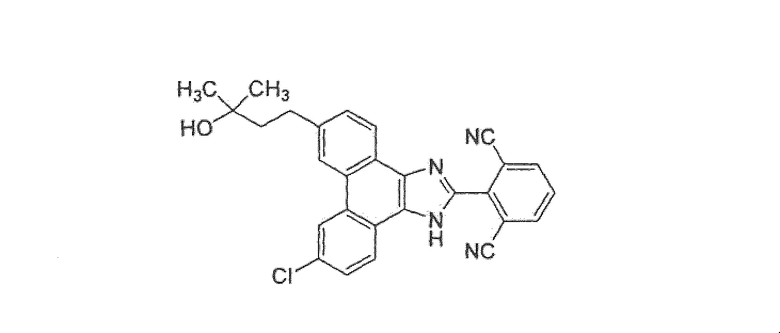
Стадия 1: 2-[9-хлор-6-(3-гидрокси-3-метилбутил)-1H-фенантро[9,10-d]имидазол-2-ил)изофталонитрил
К раствору 9-BBN в ТГФ (24 мл, 12 ммоль, 0,5 M) добавляли 2-метил-3-бутен-2-ол (345 мг, 4,0 ммоль) и полученный раствор перемешивали в атмосфере N2 при комнатной температуре в течение ночи. Во вторую колбу, содержащую PdCl2(dppf) (324 мг, 0,40 ммоль), Cs2CO3 (2,4 г, 8,0 ммоль) и Ph3As (124 мг, 0,4 ммоль), добавляли 2-(6-бром-9-хлор-1H-фенантро[9,10-d]имидазол-2-ил)изофталонитрил из примера 36, ДМФА (24 мл) и Н2O (0,88 мл) и смесь перемешивали в атмосфере N2 в течение 5 минут. Смесь для гидроборирования затем переносили во вторую колбу и полученную реакционную суспензию перемешивали при комнатной температуре в атмосфере N2 в течение 5 дней. После обработки насыщенным солевым раствором водную фазу экстрагировали при помощи EtOAc и объединенный органический раствор промывали водой и насыщенным солевым раствором, сушили над MgSO4. После удаления осушителя при помощи фильтрации раствор концентрировали при пониженном давлении и остаток очищали хроматографией на силикагеле (50% EtOAc/гексан) с получением 600 мг 2-[9-хлор-6-(3-гидрокси-3-метилбутил)-1H-фенантро[9,10-d]имидазол-2-ил)изофталонитрила в виде желтого твердого вещества.
1Н ЯМР (400 МГц, ацетон):δ 13,10 (с, ушир., 1Н); 8,94 (с, 1Н); 8,77 (с, 1Н); 8,70-8,60 (м, ушир., 2Н); 8,39 (д, 2Н); 8,03 (т, 1Н); 7,75 (дд, 1Н); 7,69 (дд, 1Н); 4,92 (с, 1Н); 3,05 (м, 2Н); 1,95 (м, 2Н).
ПРИМЕР 123
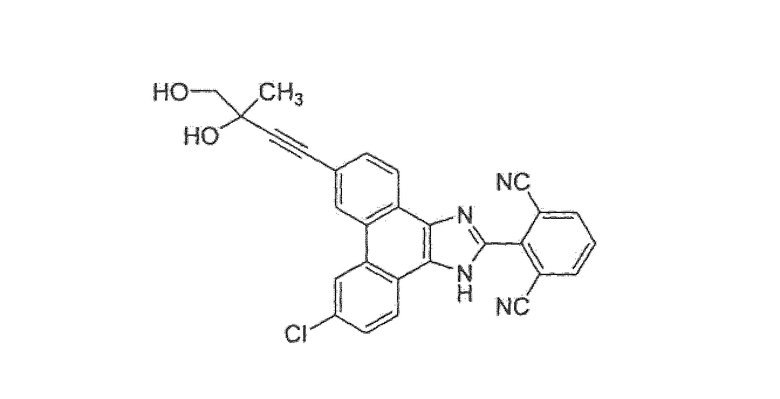
(±)-2-[9-хлор-6-(3,4-дигидрокси-3-метилбут-1-ин-1-ил)-1H-фенантро[9,10-d]имидазол-2-ил]изофталонитрил
Стадия 1: 2-[6-хлор-9-(3-метилбут-3-ен-1-ин-1-ил)-1H-фенантро[9,10-d]имидазол-2-ил]изофталонитрил
К перемешиваемой суспензии 2-[9-хлор-6-(3-гидрокси-3-метилбут-1-ин-1-ил)-1H-фенантро[9,10-d]имидазол-2-ил]изофталонитрила из примера 40 (120 мг, 0,26 ммоль) в бензоле (4 мл) добавляли реагент Burgess (70 мг, 0,29 ммоль) и кипятили с обратным холодильником в течение 2 часов в атмосфере N2. Полученную реакционную смесь разбавляли EtOAc (20 мл). Полученный EtOAc раствор промывали водой, насыщенным солевым раствором и сушили над MgSO4. После удаления осушителя при помощи фильтрации органический раствор концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюировали 50/50 EtOAc/гексан) с получением 90 мг 2-[6-хлор-9-(3-метилбут-3-ен-1-ин-1-ил)-1H-фенантро[9,10-d]имидазол-2-ил]изофталонитрила в виде желтого твердого вещества.
Стадия 2: (±)-2-[9-хлор-6-(3,4-дигидрокси-3-метилбут-1-ин-1-ил)-1H-фенантро[9,10-d]имидазол-2-ил]изофталонитрил
К перемешиваемой суспензии 2-[6-хлор-9-(3-метилбут-3-ен-1-ин-1-ил)-1H-фенантро[9,10-d]имидазол-2-ил]изофталонитрила со стадии 1 (22 мг, 0,05 ммоль) в 50/50 t-BuOН/Н2О (0,5 мл) добавляли AD-mix-α (70 мг) при 0°C. Смесь оставляли для перемешивания при 0°C в течение 24 часов. Полученную реакционную смесь обрабатывали насыщенным водным раствором Na2S2О3 и перемешивали в течение 10 минут, разбавляли водой и экстрагировали при помощи EtOAc. Полученный EtOAc раствор промывали водой, насыщенным солевым раствором и сушили над MgSО4. После удаления осушителя при помощи фильтрации органический раствор концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (элюировали 50/50 EtOAc/гексан до 95/5 EtOAc/MeOH) с получением 19 мг желтого твердого вещества. Эту же процедуру повторяли с AD-mix-β с получением еще 19 мг желтого твердого вещества. Эти два желтых твердых вещества объединяли с получением рацемического 2-[9-хлор-6-(3,4-дигидрокси-3-метилбут-1-ин-1-ил)-1H-фенантро[9,10-d]имидазол-2-ил]изофталонитрила
1Н ЯМР (400 МГц, ацетон):δ 8,84 (д, 1Н); 8,80 (с, 1Н); 8,57 (д, 1Н); 8,47 (д, 1Н); 8,39 (д, 2Н); 8,03 (т, 1Н); 7,77 (дд, 8,6 Гц, 1Н); 7,71 (дд, 1Н), 4,56 (с, 1Н); 4,30 (с, 1Н); 3,67 (кв., 2Н); 1,56 (с, 3Н).
ПРИМЕР 135
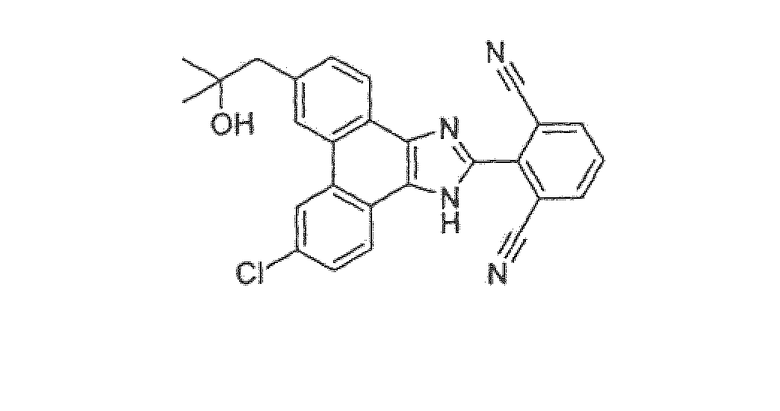
2-[9-хлор-6-(2-гидрокси-2-метилпропил)-1H-фенантро[9,10-d]имидазол-2-ил]изофталонитрил
Стадия 1: 2-(6-бром-9-хлор-1-{[2-(триметилсилил)этокси]метил}-1H-фенантро[9,10-d]имидазол-2-ил)изофталонитрил
К раствору 2-(6-бром-9-хлор-1H-фенантро[9,10-d]имидазол-2-ил)изофталонитрила (5 г, 10,9 ммоль) из примера 36 в ТГФ (30 мл) добавляли NaH (60% дисперсия в масле, 1,31 г, 32,7 ммоль). Смесь перемешивали при комнатной температуре в течение 10 минут, после чего добавляли 2-(триметилсилил)этоксиметилхлорид (5,8 мл, 32,7 ммоль). Через 1 час реакционную смесь гасили медленным добавлением воды. Водный слой экстрагировали этилацетатом, органический слой промывали один раз водой, один раз насыщенным солевым раствором, сушили над безводным MgSO4 и концентрировали с получением неочищенного 2-(6-бром-9-хлор-1-{[2-(триметилсилил)этокси]метил}-1Н-фенантро[9,10-d]имидазол-2-ил)изофталонитрила (6,06 г).
Стадия 2: 2-(9-хлор-6-(2-оксопропил)-1-{[2-(триметилсилил)этокси]метил}-1H-фенантро[9,10-d]имидазол-2-ил)изофталонитрил
Раствор трибутил(метокси)станнана (4,5 мл, 15,5 ммоль), изопропенилацетата (1,7 мл, 15,5 ммоль), 2-(6-бром-9-хлор-1-{[2-(триметилсилил)этокси]метил}-1H-фенантро[9,10-d]имидазол-2-ил)изофталонитрила со стадии 1 выше (6,06 г, 10,3 ммоль), ацетата палладия(II) (0,232 г, 1,03 ммоль) и три-o-толилфосфина (0,628 г, 2,07 ммоль) в толуоле (50 мл) нагревали при 100°C в течение ночи. Реакционную смесь гасили водой и этилацетатом. После обычной обработки и хроматографии на диоксиде кремния (50% этилацетата в гексане) выделяли 2-(9-хлор-6-(2-оксопропил)-1-{[2-(триметилсилил)этокси]метил}-1H-фенантро[9,10-d]имидазол-2-ил)изофталонитрил (2,8 г) в виде желто-оранжевого твердого вещества.
Стадия 3: 2-(9-хлор-6-(2-гидрокси-2-метилпропил)-1-{[2-(триметилсилил)этокси]метил}-1H-фенантро[9,10-d]имидазол-2-ил)изофталонитрил
В круглодонную колбу при -78°C, содержащую TiCl4 (1 M в CH2Cl2, 20 мл), добавляли метиллитий (1,6 M в диэтиловом эфире, 12,5 мл). Полученный темно-красный раствор перемешивали при -78°C в течение 15 минут и затем добавляли через канюлю к 0°C раствору 2-(9-хлор-6-(2-оксопропил)-1-{[2-(триметилсилил)этокси]метил}-1H-фенантро[9,10-d]имидазол-2-ил)изофталонитрила (2,8 г, 5,0 ммоль) со стадии 2 выше в диэтиловом эфире (10 мл). Полученную смесь перемешивали при 0°C в течение 3 часов, затем гасили насыщенным раствором хлорида аммония. Водный слой экстрагировали этилацетатом. Органический слой промывали насыщенным солевым раствором, сушили над MgSO4, фильтровали и концентрировали. Неочищенное вещество очищали флэш-хроматографией на диоксиде кремния (50% этилацетата в гексане) с получением 2-(9-хлор-6-(2-гидрокси-2-метилпропил)-1-{[2-(триметилсилил)этокси]метил}-1H-фенантро[9,10-d]имидазол-2-ил)изофталонитрила (1,94 г),
Стадия 4: 2-[9-хлор-6-(2-гидрокси-2-метилпропил)-1H-фенантро[9,10-d]имидазол-2-ил]изофталонитрил
2-(9-хлор-6-(2-гидрокси-2-метилпропил)-1-{[2-(триметилсилил)этокси]метил}-1Н-фенантро[9,10-d]имидазол-2-ил)изофталонитрил (1,94 г) со стадии 3 выше растворяли в TBAF (1 M в ТГФ, 20 мл). Смесь нагревали при температуре кипения с обратным холодильником в течение 5 часов и затем гасили водой. Водный слой экстрагировали этилацетатом. Органический слой промывали насыщенным солевым раствором, сушили над MgSO4, фильтровали и концентрировали. Неочищенное вещество очищали флэш-хроматографией на диоксиде кремния (50% этилацетата в гексане) с получением 2-[9-хлор-6-(2-гидрокси-2-метилпропил)-1H-фенантро[9,10-d]имидазол-2-ил]изофталонитрила (500 мг) в виде желтого твердого вещества.
1Н ЯМР δ (м.д.) (400 МГц, ацетон-d6): 13,13 (1Н, ушир.с), 8,87 (1Н, с), 8,77 (1Н, с), 8,58 (1Н, м), 8,43 (1Н, м), 8,35 (2Н, д, J=7,9 Гц), 7,99 (1Н, т, J=7,9 Гц), 7,73 (2Н, дд, J=1,9, 8,6 Гц), 3,51 (1Н, ушир.с); 3,08 (2Н, с), 1,26 (6Н, с).
ПРИМЕР 160
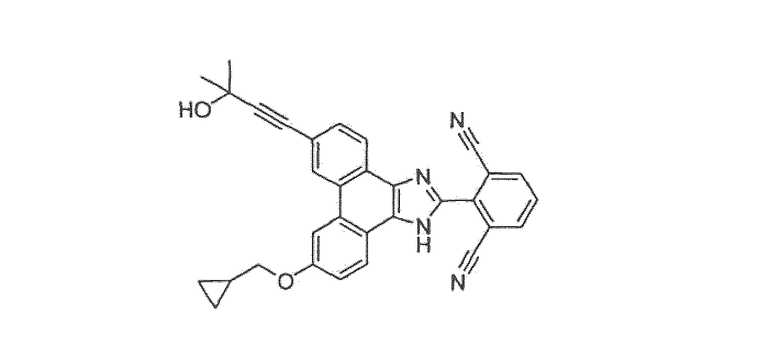
2-[9-(циклопропилметокси)-6-(3-гидрокси-3-метилбут-1-ин-1-ил)-1Н-фенантро[9,10-d]имидазол-2-ил]изофталонитрил
Стадия 1: 1-бром-4-[2-(4-метоксифенил)винил]бензол
Этот стильбен получали, как описано для стадии 1 примера 36, с замещением 4-хлорбензальдегида п-анисальдегидом.
Стадия 2: 3-бром-6-метоксифенантрен
Этот фенантрен получали, как описано для стадии 2 примера 36, с замещением 1-бром-4-[2-(4-хлорфенил)винил]бензола 1-бром-4-[2-(4-метоксифенил)винил]бензолом со стадии 1 выше и с воздействием на реакционную смесь облучения в течение 4 дней.
Стадия 3: 3-бром-6-метоксифенантрен-9,10-дион
Этот хинон получали, как описано для стадии 3, пример 36, с замещением 3-бром-6-хлорфенантрена 3-бром-6-метоксифенантреном со стадии 2 выше.
Стадия 4: 3-бром-6-гидроксифенантрен-9,10-дион
Смесь 3-бром-6-метоксифенантрен-9,10-диона со стадии 3 выше и избыточного количества BBr3 в CH2Cl2 перемешивали при комнатной температуре с получением 3-бром-6-гидроксифенантрен-9,10-диона, который использовали непосредственно на следующей стадии (стадия 5 ниже).
Стадия 5: 3-бром-6-(циклопропилметокси)фенантрен-9,10-дион
Раствор 3-бром-6-гидроксифенантрен-9,10-диона со стадии 4 в ацетоне обрабатывали избыточным количеством карбоната калия, иодида калия и (бромметил)циклопропана. Смесь нагревали при температуре кипения с обратным холодильником в течение ночи с последующей стандартной обработкой с получением 3-бром-6-(циклопропилметокси)фенантрен-9,10-диона.
Стадия 6: 6-бром-9-(циклопропилметокси)-2-(2,6-дибромфенил)-1H-фенантро[9,10-d]имидазол
Этот имидазол получали, как описано для стадии 4 примера 36, с замещением 3-бром-6-хлорфенантрен-9,10-диона 3-бром-6-(циклопропилметокси)фенантрен-9,10-дионом со стадии 5 выше.
Стадия 7: 2-[6-бром-9-(циклопропилметокси)-1H-фенантро[9,10-d]имидазол-2-ил]изофталонитрил
Этот имидазол получали, как описано для стадии 5 примера 36, с замещением 9-бром-6-хлор-2-(2,6-дибромфенил)-1H-фенантро[9,10-d]имидазола 6-бром-9-(циклопропилметокси)-2-(2,6-дибромфенил)-1H-фенантро[9,10-d]имидазолом со стадии 6 выше. Примеси, присутствующие в продукте, удаляли методом дигидроксилирования Sharpless.
Стадия 8: 2-[9-(циклопропилметокси)-6-(3-гидрокси-3-метилбут-1-ин-1-ил)-1H-фенантро[9,10-d]имидазол-2-ил]изофталонитрил
Этот имидазол получали, как описано для стадии 6 примера 40, с замещением 2-(9-бром-6-хлор-1H-фенантро[9,10-d]имидазол-2-ил)изофталонитрила 2-[6-бром-9-(циклопропилметокси)-1H-фенантро[9,10-d]имидазол-2-ил]изофталонитрилом со стадии 7 выше.
1Н ЯМР δ (м.д.) (400 МГц, ацетон-d6): 13,04 (1Н, ушир.с), 8,88 (1Н, д, J=5,7 Гц), 8,49 (2Н, м), 8,33 (3Н, м), 7,99 (1Н, т, J=8,0 Гц), 7,73 (1Н, д, J=8,2 Гц), 7,43 (1Н, д, J=8,8 Гц), 4,54 (1Н, ушир.с), 4,17 (2Н, д, J=6,8 Гц), 1,63 (6Н, с), 1,48-1,36 (1Н, м), 0,68 (1Н, м), 0,49-0,45 (1Н, м).
ПРИМЕР 168
2-[9-(циклопропилметокси)-6-(2-гидрокси-2-метилпропил)-1H-фенантро[9,10-d]имидазол-2-ил]изофталонитрил
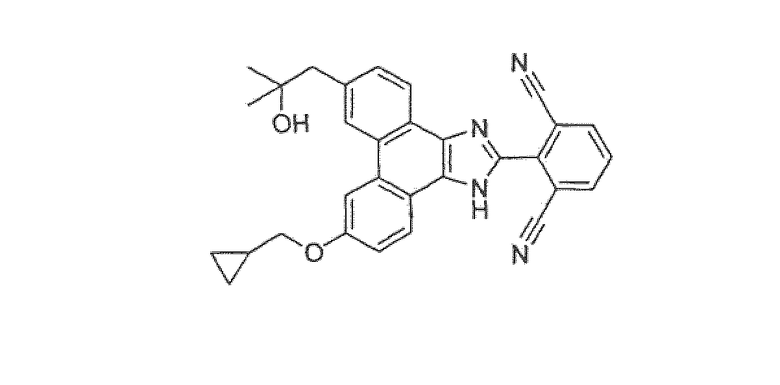
Это соединение получали двумя путями, как описано ниже:
Путь A:
Стадия 1: 6-бромфенантрен-3-ол
В колбу, содержащую BBr3 (1 M в CH2Cl2, 17 мл), при 0°C добавляли раствор 3-бром-6-метоксифенантрена (1 г, 3,5 ммоль) со стадии 2 примера 160 в CH2Cl2 (10 мл). Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 30 минут, после чего реакцию гасили водой. Водный слой экстрагировали при помощи CH2Cl2. Органический слой сушили над MgSO4, фильтровали и концентрировали с получением неочищенного 6-бромфенантрен-3-ола.
Стадия 2: 3-бром-6-(циклопропилметокси)фенантрен
Смесь 6-бромфенантрен-3-ола (0,823 г, 3,02 ммоль) со стадии 1 выше, (бромметил)циклопропана (0,5 мл, 5,4 ммоль), карбоната калия (2,5 г, 18 ммоль) и иодида калия (5 мг) в ацетоне (50 мл) нагревали при температуре кипения с обратным холодильником в течение 3 дней. Затем добавляли воду и реакционную смесь экстрагировали этилацетатом. Органический слой промывали насыщенным солевым раствором, сушили над MgSO4, фильтровали и концентрировали. Неочищенное вещество очищали флэш-хроматографией на диоксиде кремния (100% гексаны) с получением 3-бром-6-(циклопропилметокси)фенантрена (0,859 г, 87%).
Стадия 3: 1-[6-(циклопропилметокси)-3-фенантрил]ацетон
Этот фенантрен получали, как описано для стадии 2 примера 135, с замещением 2-(6-бром-9-хлор-1-([2-{триметилсилил)этокси]метил}-1H-фенантро[9,10-d]имидазол-2-ил)изофталонитрила 3-бром-6-(циклопропилметокси)фенантреном со стадии 2 выше.
Стадия 4: 1-[6-(циклопропилметокси)-3-фенантрил]-2-метилпропан-2-ол
Этот фенантрен получали, как описано для стадии 3 примера 135, с замещением 2-(9-хлор-6-(2-оксопропил)-1-{[2-(триметилсилил)этокси]метил)-1H-фенантро[9,10-d]имидазол-2-ил)изофталонитрила 1-[6-(циклопропилметокси)-3-фенантрил]ацетоном со стадии 3 выше. Неочищенный продукт использовали непосредственно в следующей реакции.
Стадия 5: трет-бутил(2-[6-(циклопропилметокси)-3-фенантрил]-1,1-диметилэтокси)диметилсилан
К раствору неочищенного 1-[6-(циклопропилметокси)-3-фенантрил]-2-метилпропан-2-ола со стадии 4 выше в ТГФ (10 мл) добавляли гидрид натрия (60% дисперсия в масле, 0,27 г, 6,79 ммоль). Смесь нагревали при температуре кипения с обратным холодильником в течение 2 минут, затем охлаждали до комнатной температуры. Добавляли трет-бутилдиметилсилилхлорид (0,512 г, 3,39 ммоль) и реакционную смесь нагревали при температуре кипения с обратным холодильником в течение 2 часов. После обычной обработки реакционной смеси получали трет-бутил(2-[6-(циклопропилметокси)-3-фенантрил]-1,1-диметилэтокси)диметилсилан (0,5 г), который использовали неочищенным на следующей стадии.
Стадия 6: 3-(2-{[трет-бутил(диметил)силил]окси}-2-метилпропил)-6-(циклопропилметокси)фенантрен-9,10-дион
К раствору трет-бутил(2-[6-(циклопропилметокси)-3-фенантрил]-1,1-диметилэтокси)диметилсилана (0,5 г, 1,15 ммоль) со стадии 5 выше в уксусной кислоте (10 мл) добавляли CrO3 (0,346 г, 3,46 ммоль). Смесь перемешивали при 50°C в течение 30 минут, охлаждали до комнатной температуры, выливали в воду и перемешивали в течение 15 минут. Суспензию фильтровали, промывали водой и откачивали при пониженном давлении с получением 3-(2-{[трет-бутил(диметил)силил]окси}-2-метилпропил)-6-(циклопропилметокси)фенантрен-9,10-диона.
Стадия 7: 6-(2-{[трет-бутил(диметил)силил]окси}-2-метилпропил)-9-(циклопропилметокси)-2-(2,6-дибромфенил)-1Н-фенантро[9,10-d]имидазол
К раствору 3-(2-{[трет-бутил(диметил)силил]окси}-2-метилпропил)-6-(циклопропилметокси)фенантрен-9,10-диона (1,15 ммоль) со стадии 6 выше в уксусной кислоте (10 мл) добавляли ацетат аммония (1,78 г, 23 ммоль) и дибромбензальдегид (0,42 г, 1,5 ммоль). Смесь перемешивали при 70°C в течение 1 часа, охлаждали до комнатной температуры, выливали в воду и перемешивали в течение 5 минут. Полученное твердое вещество промывали водой и диэтиловым эфиром. Неочищенное вещество очищали флэш-хроматографией на диоксиде кремния (30% этилацетата в гексанах) с получением 6-(2-{[трет-бутил(диметил)силил]окси}-2-метилпропил)-9-(циклопропилметокси)-2-(2,6-дибромфенил)-1H-фенантро[9,10-d]имидазола (0,223 г) в виде желтого твердого вещества.
Стадия 8: 1-[9-(циклопропилметокси)-2-(2,6-дибромфенил)-1H-фенантро[9,10-d]имидазол-6-ил]-2-метилпропан-2-ол
TBAF (1 M в ТГФ, 10 мл) добавляли в колбу, содержащую 6-(2-{[трет-бутил(диметил)силил]окси}-2-метилпропил)-9-(циклопропилметокси)-2-(2,6-дибромфенил)-1H-фенантро[9,10-d]имидазол (0,223 г, 0,31 ммоль) со стадии 7 выше, при комнатной температуре. Полученный раствор нагревали при температуре кипения с обратным холодильником в течение 36 часов, после чего к реакционной смеси добавляли воду. Водный слой экстрагировали этилацетатом, органический слой сушили над MgSO4, фильтровали и концентрировали. Неочищенный продукт использовали непосредственно в следующей реакции (стадия 9 ниже).
Стадия 9: 2-[9-(циклопропилметокси)-6-(2-гидрокси-2-метилпропил)-1H-фенантро[9,10-d]имидазол-2-ил] изофталонитрил
Этот имидазол получали, как описано для стадии 5 примера 36, с замещением 9-бром-6-хлор-2-(2,6-дибромфенил)-1H-фенантро[9,10-d]имидазола неочищенным 1-[9-(циклопропилметокси)-2-(2,6-дибромфенил)-1H-фенантро[9,10-d]имидазол-6-ил]-2-метилпропан-2-олом со стадии 8 выше.
1Н ЯМР δ (м.д.) (400 МГц, ацетон-d6): 12,96 (1Н, ушир.с), 8,70 (1Н, м), 8,59 (1Н, м), 8,32 (3Н, д, J=8,0 Гц), 8,28 (1Н, м), 7,95 (1Н, т, J=7,9 Гц), 7,67 (1Н, д, J=8,1 Гц), 7,38 (1Н, д, J=8,7 Гц), 4,09 (2Н, д, J=6,9 Гц), 3,46 (1Н, ушир.с), 3,05 (2Н, с), 1,38-1,34 (1Н, м), 1,25 (6Н, с), 0,67-0,63 (2Н, м), 0,45-0,41 (2Н, м).
Путь B:
Стадия 1: 3-бром-6-(циклопропилметокси)фенантрен-9,10-дион
Этот хинон получали либо как описано для стадии 5 примера 160, либо следуя процедуре, описанной для стадии 3 примера 36, с замещением 3-бром-6-хлорфенантрена 3-бром-6-(циклопропилметокси)фенантреном со стадии 2 пути A выше.
Стадия 2: 6-бром-9-(циклопропилметокси)-2-(2,6-дибромфенил)-1H-фенантро[9,10-d]имидазол
Этот имидазол получали, как описано для стадии 6 примера 160.
Стадия 3: 2-[6-бром-9-(циклопропилметокси)-1H-фенантро[9,10-d]имидазол-2-ил]изофталонитрил
Этот имидазол получали, как описано для стадии 7 примера 160.
Стадия 4: 2-(6-бром-9-(циклопропилметокси)-1-{[2-(триметилсилил)этокси]метил}-1H-фенантро[9,10-d]имидазол-2-ил)изофталонитрил
Этот SEM-защищенный имидазол получали, как описано для стадии 2 примера 87, с замещением 6,9-дибром-2-(2,6-дибромфенил)-1H-фенантро[9,10-d]имидазола 2-[6-бром-9-(циклопропилметокси)-1H-фенантро[9,10-d]имидазол-2-ил]изофталонитрилом со стадии 3 выше.
Стадия 5: 2-(9-(циклопропилметокси)-6-(2-оксопропил)-1-{[2-(триметилсилил)этокси]метил}-1H-фенантро[9,10-d]имидазол-2-ил)изофталонитрил
Этот имидазол получали, как описано для стадии 2 примера 135, с замещением 2-(6-бром-9-хлор-1-([2-(триметилсилил)этокси]метил)-1H-фенантро[9,10-d]имидазол-2-ил)изофталонитрила 2-(6-бром-9-(циклопропилметокси)-1-{[2-(триметилсилил)этокси]метил}-1Н-фенантро[9,10-d]имидазол-2-ил)изофталонитрилом со стадии 4 выше.
Стадия 6: 2-(9-(циклопропилметокси)-6-(2-гидрокси-2-метилпропил)-1-{[2-(триметилсилил)этокси]метил}-1Н-фенантро[9,10-d]имидазол-2-ил)изофталонитрил
Этот имидазол получали, как описано для стадии 3 примера 135, с замещением 2-(9-хлор-6-(2-оксопропил)-1-{[2-(триметилсилил)этокси]метил)-1H-фенантро[9,10-d]имидазол-2-ил)изофталонитрила 2-(9-(циклопропилметокси)-6-(2-оксопропил)-1-{[2-(триметилсилил)этокси]метил}-1Н-фенантро[9,10-d]имидазол-2-ил)изофталонитрилом со стадии 5 выше.
Стадия 7: 2-[9-(циклопропилметокси)-6-(2-гидрокси-2-метилпропил)-1H-фенантро[9,10-d]имидазол-2-ил] изофталонитрил
Неочищенный 2-(9-(циклопропилметокси)-6-(2-гидрокси-2-метилпропил)-1-{[2-(триметилсилил)этокси]метил}-1H-фенантро[9,10-d]имидазол-2-ил)изофталонитрил (1,37 ммоль) со стадии 6 выше растворяли в TBAF (1 M в ТГФ, 10 мл) и смесь нагревали при температуре кипения с обратным холодильником в течение 1,5 часов. Добавляли воду и водный слой экстрагировали этилацетатом. Органический слой сушили над MgSO4, фильтровали и концентрировали. Вещество очищали флэш-хроматографией на диоксиде кремния (70% этилацетата в гексанах) с получением 2-[9-(циклопропилметокси)-6-(2-гидрокси-2-метилпропил)-1H-фенантро[9,10-d]имидазол-2-ил]изофталонитрила (240 мг).
ПРИМЕР 172
2-[9-(2-циклопропилэтокси)-6-(2-гидрокси-2-метилпропил)-1Н-фенантро[9,10-d]имидазол-2-ил]-5-фторизофталонитрил
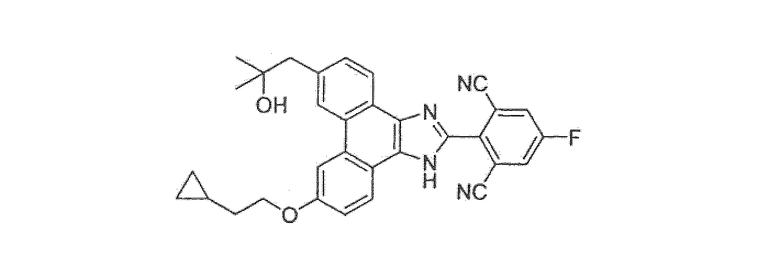
Стадия 1: 3-бром-6-(2-циклопропилэтокси)фенантрен
К смеси 6-бромфенантрен-3-ола (3 г, 11 ммоль) со стадии 1 пути A примера 168, 2-циклопропилэтанола (2,85 г, 33 ммоль) и трифенилфосфина (5,78 г, 22 ммоль) в ТГФ (50 мл) добавляли ди-трет-бутилазодикарбоксилат (5,08 г, 22 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи, затем гасили водой. Водный слой экстрагировали этилацетатом. Объединенные органические слои промывали насыщенным солевым раствором, сушили над MgSO4, фильтровали и концентрировали. Вещество очищали флэш-хроматографией на диоксиде кремния (100% гексан) с получением 3-бром-6-(2-циклопропилэтокси)фенантрена.
Стадия 2: 1-[6-(2-циклопропилэтокси)-3-фенантрил]-2-метилпропан-2-ол
Этот фенантрен можно получить либо двухстадийным способом, как описано для стадий 3 и 4 пути A примера 168, с замещением 3-бром-6-(циклопропилметокси)фенантрена 3-бром-6-(2-циклопропилэтокси)фенантреном со стадии 1 выше, либо следуя процедуре, описанной ниже:
К раствору 3-бром-6-(2-циклопропилэтокси)фенантрена (11 ммоль) со стадии 1 выше в ТГФ (75 мл) при -78°C последовательно добавляли метиллитий (1,6 M в диэтиловом эфире, 1 мл) и бутиллитий (2,5 M в гексане, 5,3 мл). Смесь перемешивали при -78°C в течение 30 минут, после чего добавляли изобутиленоксид (2,9 мл, 33 ммоль) с последующим добавлением BF3.OEt2 (4,2 мл, 33 ммоль). Реакционную смесь перемешивали при -78°C в течение 1 часа, затем гасили при помощи 1 M HCl. Водный слой экстрагировали этилацетатом. Объединенные органические слои промывали насыщенным солевым раствором, сушили над MgSO4, фильтровали и концентрировали. Вещество очищали флэш-хроматографией на диоксиде кремния (10% этилацетата в гексанах) с получением 1-[6-(2-циклопропилэтокси)-3-фенантрил]-2-метилпропан-2-ола (1,33 г) в виде желтого масла.
Стадия 3: трет-бутил(2-[6-(2-циклопропилэтокси)-3-фенантрил]-1,1-диметилэтокси)диметилсилан
Этот фенантрен получали, как описано для стадии 5 пути A примера 168, с замещением 1-[6-(циклопропилметокси)-3-фенантрил]-2-метилпропан-2-ола 1-[6-(2-циклопропилэтокси)-3-фенантрил]-2-метилпропан-2-олом со стадии 2 выше.
Стадия 4: 3-(2-{[трет-бутил(диметил)силил]окси}-2-метилпропил)-6-(2-циклопропилэтокси)фенантрен-9,10-дион
Этот хинон получали, как описано для стадии 6 пути A примера 168, с замещением трет-бутил(2-[6-(циклопропилметокси)-3-фенантрил]-1,1-диметилэтокси)диметилсилана трет-бутил(2-[6-(2-циклопропилэтокси)-3-фенантрил]-1,1-диметилэтокси)диметилсиланом со стадии 3 выше.
Стадия 5: 6-(2-{[трет-бутил(диметил)силил]окси}-2-метилпропил)-9-(2-циклопропилэтокси)-2-(2,6-дибром-4-фторфенил)-1H-фенантро[9,10-d]имидазол
Этот имидазол получали, как описано для стадии 7 пути A примера 168, с замещением 3-(2-{[трет-бутил(диметил)силил]окси}-2-метилпропил)-6-(циклопропилметокси)фенантрен-9,10-диона 3-(2-{[трет-бутил(диметил)силил]окси}-2-метилпропил)-6-(2-циклопропилэтокси)фенантрен-9,10-дионом со стадии 4 выше и с замещением дибромбензальдегида 2,6-дибром-4-фторбензальдегидом.
Стадия 6: 1-[9-(2-циклопропилэтокси)-2-(2,6-дибром-4-фторфенил)-1H-фенантро[9,10-d]имидазол-6-ил]-2-метилпропан-2-ол
Этот имидазол получали, как описано для стадии 8 пути A примера 168, с замещением 6-(2-{[трет-бутил(диметил)силил]окси}-2-метилпропил)-9-(циклопропилметокси)-2-(2,6-дибромфенил)-1H-фенантро[9,10-d]имидазола 6-(2-{[трет-бутил(диметил)силил]окси}-2-метилпропил)-9-(2-циклопропилэтокси)-2-(2,6-дибром-4-фторфенил)-1H-фенантро[9,10-d]имидазолом со стадии 5 выше.
Стадия 7: 2-[9-(2-циклопропилэтокси)-6-(2-гидрокси-2-метилпропил)-1Н-фенантро[9,10-d]имидазол-2-ил]-5-фторизофталонитрил
Этот имидазол получали, как описано для стадии 5 примера 36, с замещением 9-бром-6-хлор-2-(2,6-дибромфенил)-1H-фенантро[9,10-d]имидазола 1-[9-(2-циклопропилэтокси)-2-(2,6-дибром-4-фторфенил)-1H-фенантро[9,10-d]имидазол-6-ил]-2-метилпропан-2-олом со стадии 6 выше.
1Н ЯМР δ (м.д.) (400 МГц, ацетон-d6): 12,95 (1Н, ушир.с), 8,70 (1Н, м), 8,58 (1Н, м), 8,28 (4Н, м), 7,67 (1Н, д, J=8,1 Гц), 7,40 (1Н, д, J=9,1 Гц), 4,31 (2Н, т, J=6,5 Гц), 3,43 (1Н, ушир.с), 3,05 (2Н, с), 1,78 (1Н, кв., J=6,7 Гц), 1,26 (6Н, с), 0,98 (1Н, м), 0,54-0,48 (2Н, м), 0,20-0,18 (2Н, м).
ПРИМЕР 180
2-[6-(2-гидрокси-2-метилпропил)-9-(4,4,4-трифторбутокси)-1Н-фенантро[9,10-d] имидазол-2-ил]изофталонитрил
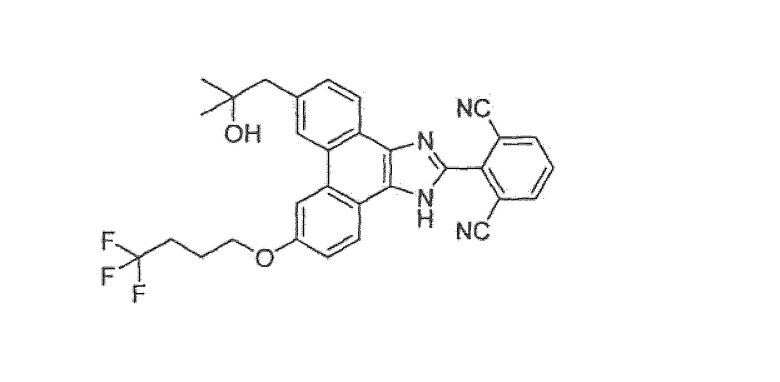
Стадия 1: 3-бром-6-(4,4,4-трифторбутокси)фенантрен
Этот фенантрен получали, как описано для стадии 2 пути A примера 168, с замещением (бромметил)циклопропана 4,4,4-трифтор-1-йодбутаном.
Стадия 2: 2-метил-1-[6-(4,4,4-трифторбутокси)-3-фенантрил]пропан-2-ол
Этот фенантрен получали, как описано для стадии 2 примера 172, с замещением 3-бром-6-(2-циклопропилэтокси)фенантрена 3-бром-6-(4,4,4-трифторбутокси)фенантреном со стадии 1 выше.
Стадия 3: трет-бутил(1,1-диметил-2-[6-(4,4,4-трифторбутокси)-3-фенантрил]этокси)диметилсилан
Этот фенантрен получали, как описано для стадии 5 пути A примера 168, с замещением 1-[6-(циклопропилметокси)-3-фенантрил]-2-метилпропан-2-ола 2-метил-1-[6-(4,4,4-трифторбутокси)-3-фенантрил]пропан-2-олом со стадии 2 выше.
Стадия 4: 3-(2-{[трет-бутил(диметил)силил]окси}-2-метилпропил)-6-(4,4,4-трифторбутокси)фенантрен-9,10-дион
Этот хинон получали, как описано для стадии 6 пути A примера 168, с замещением трет-бутил(2-[6-(циклопропилметокси)-3-фенантрил]-1,1-диметилэтокси)диметилсилана трет-бутил(1,1-диметил-2-[6-(4,4,4-трифторбутокси)-3-фенантрил]этокси)диметилсиланом со стадии 3 выше.
Стадия 5: 6-(2-([трет-бутил(диметил)силил]окси)-2-метилпропил)-2-(2,6-дибромфенил)-9-(4,4,4-трифторбутокси)-1H-фенантро[9,10-d]имидазол
Этот имидазол получали, как описано для стадии 7 пути A примера 168, с замещением 3-(2-{[трет-бутил(диметил)силил]окси}-2-метилпропил)-6-(циклопропилметокси)фенантрен-9,10-диона 3-(2-{[трет-бутил(диметил)силил]окси}-2-метилпропил)-6-(4,4,4-трифторбутокси)фенантрен-9,10-дионом со стадии 4 выше.
Стадия 6: 1-[2-(2,6-дибромфенил)-9-(4,4,4-трифторбутокси)-1H-фенантро[9,10-d]имидазол-6-ил]-2-метилпропан-2-ол
Этот имидазол получали, как описано для стадии 8 пути A примера 168, с замещением 6-(2-{[трет-бутил(диметил)силил]окси}-2-метилпропил)-9-(циклопропилметокси)-2-(2,6-дибромфенил)-1H-фенантро[9,10-d]имидазола 6-(2-([трет-бутил(диметил)силил]окси)-2-метилпропил)-2-(2,6-дибромфенил)-9-(4,4,4-трифторбутокси)-1H-фенантро[9,10-d]имидазолом со стадии 5 выше.
Стадия 7: 2-[6-(2-гидрокси-2-метилпропил)-9-(4,4,4-трифторбутокси)-1H-фенантро[9,10-d]имидазол-2-ил]изофталонитрил
Этот имидазол получали, как описано для стадии 5 примера 36, с замещением 9-бром-6-хлор-2-(2,6-дибромфенил)-1H-фенантро[9,10-d]имидазола 1-[2-(2,6-дибромфенил)-9-(4,4,4-трифторбутокси)-1H-фенантро[9,10-d]имидазол-6-ил]-2-метилпропан-2-олом со стадии 6 выше.
1Н ЯМР δ (м.д.) (400 МГц, ацетон-d6): 12,95 (1Н, ушир.с), 8,72 (2Н, м), 8,33 (4Н, м), 7,96 (1Н, т, J=7,9 Гц), 7,68 (1Н, д, J=8,1 Гц), 7,42 (1Н, д, J=9,5 Гц), 4,36 (2Н, т, J=6,0 Гц), 3,45 (1Н, ушир.с), 3,05 (2Н, с), 2,57-2,51 (2Н, м), 2,20-2,12 (2Н, м), 1,25 (6Н, с).
Анализы для определения биологической активности
Ингибирование активности простагландин E синтазы
Соединения были испытаны на их действие в качестве ингибиторов активности простагландин E синтазы в цельноклеточных in vivo анализах с использованием микросомальных простагландин E синтаз. В этих анализах определяли уровень синтеза простагландина E2 (PGE2) с использованием либо ферментного иммуноанализа (EIA), либо масс-спектрометрии. Клетки, используемые для получения препарата микросом, представляли собой клетки CHO-K1, транзиторно трансфицированные плазмидами, кодирующими кДНК mPGES-1 человека. Клетки, используемые для клеточных анализов, представляли собой клетки A549 человека (которые экспрессируют mPGES-1 человека). Для испытания активности избранных соединений in vivo использовали морских свинок. Во всех этих анализах 100% активность определяли как продукцию PGE2 в образцах, обрабатываемых носителем. IC50 и ED50 представляют концентрацию или дозу ингибитора, необходимую для ингибирования синтеза PGE2 на 50% по сравнению с не ингибированным контролем.
Анализ микросомальной простагландин E синтазы
Микросомальные фракции простагландин E синтазы получали из CHO-Kl клеток, транзиторно трансфицированных плазмидой, кодирующей кДНК mPGES-1 человека. Затем подготавливали микросомы и начинали анализ PGES с инкубации 5 мкг/мл микросомальной PGES-1 с соединением или ДМСО (конечная концентрация 1%) в течение 20-30 минут при комнатной температуре. Ферментные реакции осуществляли в 200 мМ KPi pH 7,0, 2 мМ EDTA и 2,5 мМ GSH-восстановленной формы. Ферментную реакцию затем инициировали добавлением 1 мкМ конечного PGH2 субстрата, подготовленного в изопропаноле (3,5% конечная концентрация в лунке для анализа) и инкубировали при комнатной температуре в течение 30 секунд. Реакцию останавливали добавлением SnCl2 в 1 н. растворе HCl (конечное разведение 1 мг/мл). Определение продукции PGE2 в аликвотах ферментной реакции осуществляли при помощи EIA с использованием стандартного коммерчески доступного набора (Cat #: 901-001 от Assay Designs).
Данные этого анализа для репрезентативных соединений представлены в таблице ниже. Активность выражена в виде значений IC50, и указанное значение представляет собой среднее значение при, по меньшей мере, n=3.
Цельноклеточный анализ простагландин E синтазы с использованием A549 человека
План анализа
Цельные клетки обеспечивают интактную клеточную среду для исследования клеточной проницаемости и биохимической специфичности противовоспалительных соединений, таких как ингибиторы простагландин E синтазы. Для исследования ингибиторной активности этих соединений клетки A549 человека стимулировали 10 нг/мл рекомбинантным IL-1β человека в течение 24 часов. Продукцию PGE2 и PGF2α определяли при помощи EIA в конце инкубации как показатели селективности и эффективности против mPGES-1-зависимой продукции PGE2.
Методы
Клетки A549 человека специфически экспрессировали микросомальную простагландин E синтазу-1 человека и индуцировали ее экспрессию после обработки при помощи IL-1β в течение 24 часов. 2,5×104 клеток высевали в лунки при плотности 100 мкл/лунка (96-луночный планшет) и инкубировали в течение ночи в стандартных условиях. К клеткам затем добавляли 100 мкл клеточной культуральной среды, содержащей 10 нг/мл IL-1β, с последующим добавлением либо RPMI, содержащей 2% FBS, либо RPMI, содержащей 50% FBS. Затем добавляли 2 мкл лекарственных средств или носитель (ДМСО) и образцы сразу же смешивали. Клетки инкубировали в течение 24 часов и после инкубации 175 мкл среды собирали и анализировали на содержание PGE2 и PGF2α при помощи EIA.
Анализ простагландин E синтазы с использованием цельной крови человека
План анализа
Цельная кровь обеспечивает белок и клеточно-обогащенную среду для исследования биохимической специфичности противовоспалительных соединений, таких как ингибиторы простагландин E синтазы. Для исследования ингибиторной активности этих соединений кровь человека стимулируют липополисахаридом (LPS) в течение 24 часов для индукции mPGES-1 экспрессии. Продукцию простагландина E2 (PGE2) и тромбоксана B2 (TxB2) измеряют при помощи EIA в конце инкубации как показатели селективности и эффективности против mPGES-1-зависимой продукции PGE2.
Методы
Анализы цельной крови человека на mPGES-1 активность, которые были описаны (Brideau, et al., Inflamm. Res., vol. 45, p. 68, 1996), осуществляли, как описано ниже.
Свежесобранную у добровольцев венозную кровь человека собирали в гепаринизированные пробирки. У этих субъектов не было явного состояния воспаления, и они не принимали никакие НСПВЛС в течение, по меньшей мере, 7 дней до забора крови. 250 мкл крови предварительно инкубировали с 1 мкл носителя (ДМСО) или 1 мкл испытываемого соединения. Затем добавляли бактериальные LPS при 100 мкг/мл (E.сoli серотип 0111:B4, разбавленный в 0,1% масс./об. бычьего сывороточного альбумина в забуференном фосфатом физиологическом растворе) и образцы инкубировали в течение 24 часов при 37°C. Не стимулированную контрольную кровь во время ноль (без LPS) использовали в качестве контроля. По окончании 24-часовой инкубации кровь центрифугировали при 3000 об/мин в течение 10 минут при 4°C. Плазму анализировали на PGE2 и TxB2 с использованием набора EIA, как указано выше.
Определение противовоспалительной активности in vivo
План анализа
Интактное животное обеспечивает цельную физиологическую систему для подтверждения противовоспалительной активности испытываемых соединений, определенной in vitro. Для определения активности ингибиторов простагландин E синтазы in vivo животным вводят соединения либо до, либо после воспалительных стимулов, LPS. LPS вводят при помощи инъекции в заднюю лапу морских свинок и показатели гипералгезии определяют через 4,5 и/или 6 часов после инъекции.
Композиция испытываемых соединений для перорального введения
Испытываемое соединение измельчали и делали его аморфным с использованием системы с шаровой мельницей. Соединение помещали в агатовый сосуд, содержащий агатовые шарики, и осуществляли измельчение при высокоскоростном вращении в течение 10 минут в аппарате, таком как Planetary Micro Mill Pulverisette 7 system. Сосуд затем открывали и к измельченному твердому веществу добавляли 0,5% раствор Метоцел. Полученную смесь снова подвергали измельчению при высокоскоростном вращении в течение 10 минут. Полученную суспензию переносили в сцинтилляционный флакон, разбавляли подходящим количеством 0,5% раствора Метоцел, обрабатывали ультразвуком в течение 2 минут и перемешивали до получения гомогенной суспензии. Альтернативно, испытываемое соединение можно сформулировать в композицию с использованием аморфного вещества, полученного любым подходящим химическим или механическим способом. Полученное аморфное твердое вещество затем смешивали и перемешивали в течение определенного периода времени, такого как 12 часов, с подходящим носителем, таким как 0,5% Метоцел с 0,02-0,2% додецилсульфата натрия перед введением.
Методы
Использовали самцов морских свинок Hartley с массой тела 200-250 г. LPS (30 мг/кг) вводили путем субплантарной инъекции в левую заднюю лапу морской свинки, чтобы вызвать гипералгезию в лапе, куда вводили инъекцию. Ректальную температуру и латентность отдергивания лапы, как меру гиперчувствительности к боли (гипералгезия), определяли перед LPS инъекцией и использовали в качестве базовой линии. Латентность отдергивания лапы определяли с использованием инструмента для определения термальной гипералгезии (Ugo Basile Corp.). В процессе исследования животных помещали 8"×8" боксы из плексигласа над стеклянным основанием. Слабый (223 мВт/см2) инфракрасный свет направляли на нижнюю сторону задней лапы. Регистрировали время, которое требовалось животному, чтобы убрать лапу (показание, что животное ощущает боль, вызванную воздействием тепла). Инфракрасный свет сразу же отключали, когда животное отдергивало лапу от этого участка. Свет также отключался автоматически по прошествии 20 секунд.
Парадигма предварительно определенной дозы:
Испытываемые соединения перорально вводили при 5 мл/кг с использованием иглы размера 18. LPS (серотип 0111:B4, 10 мкг) или 0,9% физиологический раствор вводят в плантарную область левой задней лапы путем инъекции объемом 100 мкл с использованием иглы размера 26 через 1 час после введения соединения. Ректальную температуру и латентность отдергивания лапы определяли через 4,5 часа после введения LPS. После проведения измерений животных умерщвляли при помощи CO2 и брали у них люмбальный спинной мозг, заднюю лапу и образцы крови.
Обратная парадигма:
Отдергивание лапы под воздействием тепла у каждого животного определяли до и через 3 часа после субплантарной инъекции LPS. Животных, которые получали LPS и не показали снижение латентности отдергивания лапы через 3 часа, удаляли из испытания и умерщвляли. Испытываемые соединения вводили перорально при дозе 5 мл/кг сразу после проведения измерений отдергивания лапы под воздействием тепла. Латентность отдергивания лапы под воздействием тепла определяли через 1,5 и 3 часа после введения соединения (4,5 и 6 часов после введения LPS). После снятия последних показаний животных умерщвляли с использованием CO2 и брали у них люмбальный спинной мозг и образцы крови для определения простагландина методом масс-спектрометрии и уровня лекарственного средства, соответственно.
Альтернативный способ получения примера 40 представляет собой следующий:
АЛЬТЕРНАТИВНЫЙ ПРИМЕР 40
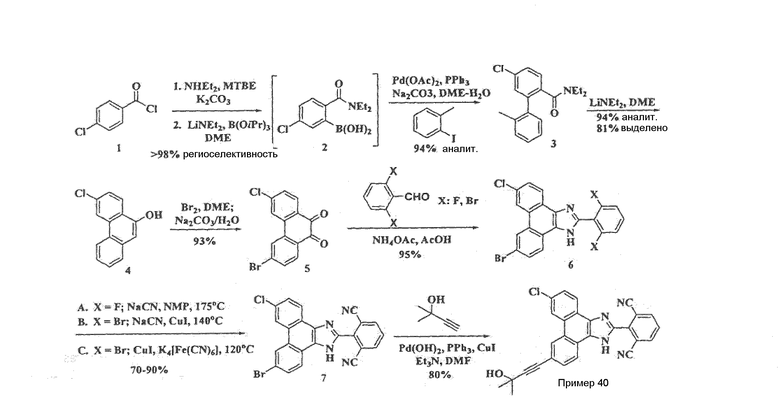
Процедура эксперимента
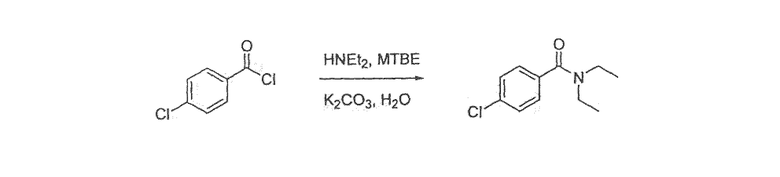
В круглодонную колбу загружали карбонат калия (65 г, 469,7 ммоль), H2O (400 мл), MTBE (800) и диэтиламин (81 мл, 861,1 ммоль). Затем добавляли п-хлорбензоилхлорид (100 мл, 782,8 ммоль) в течение 30 минут, поддерживая температуру ниже 25°C. После добавления фазы разделяли и органическую фазу промывали насыщенным солевым раствором (200 мл). Затем растворитель в этом растворе заменяли на DME с получением неочищенного раствора амида, который использовали непосредственно на следующей стадии.
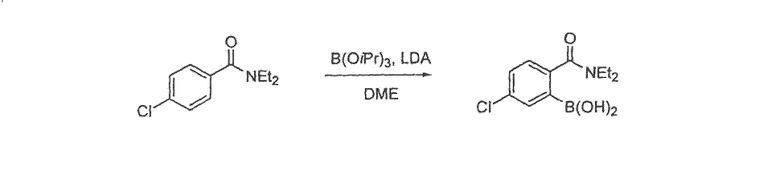
К неочищенному раствору амида (10 г, 47,3 ммоль) в 7,5 мл/г DME (75 мл) добавляли триизопропилборат (19,5 мл, 85,1 ммоль) и полученный раствор охлаждали до -25°C. Затем добавляли по каплям в течение 30 минут свежеприготовленный раствор 1,45 M диэтиламида лития (45,6 мл, 66,2 ммоль). [Примечание: Диэтиламид лития получали путем обработки диэтиламина в ТГФ раствором 2,5 M н-бутиллития в гексанах, поддерживая в процессе добавления температуру ниже 0°C]. По завершении добавления смесь подвергали старению еще в течение 15 минут, при этом все исходное вещество было израсходовано, с получением соответствующей бороновой кислоты с >98% региоселективностью. Неочищенный раствор затем использовали непосредственно на следующей стадии.
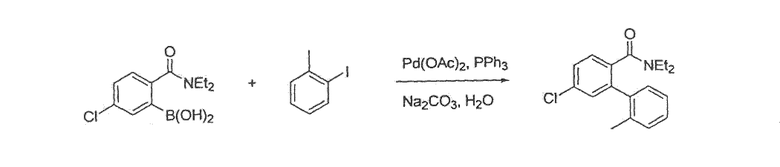
К неочищенному раствору бороновой кислоты, полученному выше, добавляли дегазированную воду (95 мл) при 0°C и твердый Na2CO3 (13,5 г, 127,7 ммоль). К полученной суспензии последовательно добавляли PPh3 (223 мг, 0,85 ммоль), 2-йодтолуол (5,4 мл, 42,6 ммоль) и Pd(OAc)2 (95,5 мг, 0,43 ммоль) и смесь дегазировали, нагревали до 70°C и подвергали старению в течение 6 часов, при этом обычно наблюдали полное расходование 2-йодтолуола. В конце реакции добавляли MTBE (75 мл) и полученную суспензию фильтровали. К бифазному фильтрату добавляли хлорид натрия для облегчения разделения и слои разделяли. Органическую фазу промывали один раз водой (20 мл) и насыщенным солевым раствором (2×30 мл). Неочищенный раствор затем концентрировали, заменяли в нем растворитель на DME и использовали непосредственно на следующей стадии. Типичный аналитический выход: 90-94%.
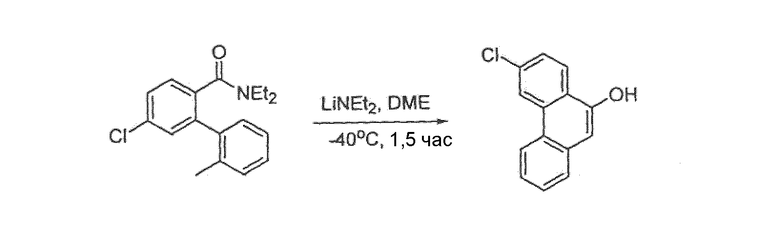
К неочищенному раствору амида (13,9 г, 46,2 ммоль) в 7,5 мл/г DME (104 мл), поддерживаемому при -45°C, добавляли свежеприготовленный раствор 1,44 M LiNEt2 в ТГФ (41,7 мл, 60 ммоль) в течение 15 минут. Полученный коричневый раствор подвергали старению в течение 75 минут, при этом наблюдали полное расходование исходного вещества, согласно данным ВЭЖХ. Добавляли MTBE (120 мл) с последующим медленным добавлением 6н. раствора HCl (30,8 мл, 184,7 ммоль). Полученной смеси давали нагреться до комнатной температуры и слои разделяли (pH водного раствора слоя должен быть 2-3). Органический слой промывали один раз H2O (55 мл), насыщенным солевым раствором (60 мл), концентрировали и растворитель заменяли на толуол для кристаллизации. Когда было получено примерно 4 мл/г продукта в 3:1 смеси толуол:DME, суспензию подвергали кипячению с обратным холодильником для растворения всех твердых веществ, медленно охлаждали до 60°C и обрабатывали при помощи 5 мл/г метилциклогексана (кристаллы типично образуются при 75-80°C) в течение 1 часа, давая при этом смеси охладиться до комнатной температуры. Суспензию затем концентрировали с получением объема 3,5 мл/г продукта и затем снова обрабатывали при помощи 2 мл/г метилциклогексана в течение 0,5 часа. Суспензию подвергали старению при 0°C в течение 0,5 часа, фильтровали и мокрую лепешку промывали холодной смесью 3:1 толуол:метилциклогексан с последующей сушкой под постоянным потоком N2. Желаемый продукт получали в виде светло-бурого твердого вещества с выходом 81%.
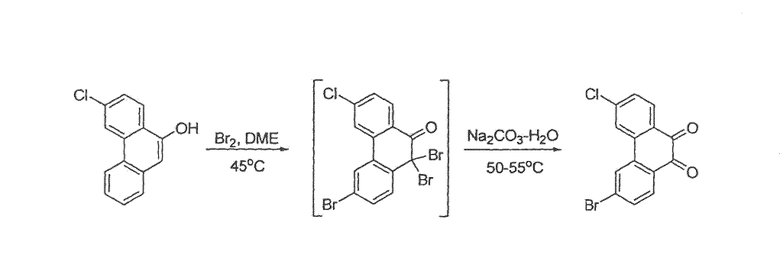
К раствору хлорфенантрола (41 г, 179,8 ммоль) в безводном DME (600 мл, KF = 25 ч/млн, KF раствора = 1000 ч/млн) при 15°C добавляли Br2 (32,3 мл, 629,4 ммоль) в течение 20 минут, при этом в процессе добавления наблюдали 15°C экзотерму. Полученную суспензию затем нагревали до 40-45°C и подвергали старению в течение 4 часов с получением прозрачного красного раствора. Добавляли раствор Na2SO3 (4,4 г, 36 ммоль) в 30 мл H2O с последующим добавлением раствора Na2CO3 (57 г, 539,4 ммоль) в 250 мл H2O. Полученную суспензию нагревали до 55°C и подвергали старению в течение 5 часов, при этом наблюдали полный гидролиз (может потребоваться дополнительное количество H2O для повторного растворения осадка Na2CO3). Реакционную смесь затем концентрировали при 35-40°C (35-40 Торр) до около трети ее объема и суспензию фильтровали, промывали при помощи H2O (80-100 мл), а затем 1:1 DME:Н2O (100 мл) и сушили под постоянным потоком N2. Полученное твердое вещество, в основном, было достаточно чистым для использования на следующей стадии; типичный выход: 93%.
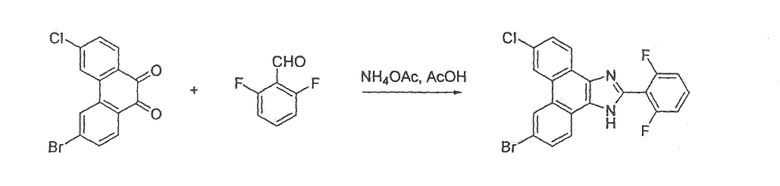
Хлорбромдикетон (4,54 г, 14,12 ммоль), дифторбензальдегид (1,5 мл, 14,12 ммоль) и ацетат аммония (21,77 г, 282,38 ммоль) загружали в 250-мл круглодонную трехгорлую колбу в атмосфере азота. Добавляли уксусную кислоту (90 мл) при перемешивании и суспензию нагревали до 120°C в течение 1 часа. Суспензию затем охлаждали до комнатной температуры и добавляли воду (90 мл) в течение 30 минут. По завершении добавления воды реакционную смесь фильтровали, промывали водой (45 мл) и сушили в течение ночи в атмосфере азота и в вакууме с получением соли уксусной кислоты в виде желтого твердого вещества.
Для получения свободного основания неочищенный продукт растворяли в 1:1 смеси ТГФ/MTBE (90 мл) и загружали в 250-мл колбу вместе с 1н. раствором NaOH (45 мл). Смесь затем нагревали до 40°C в течение одного часа. Фазы разделяли при 40°C и органический слой промывали при помощи 1н. раствора NaOH (45 мл). Органический слой затем концентрировали, растворитель заменяли на MTBE и доводили до конечного объема 45 мл. Реакционную смесь суспендировали при 35°C в течение одного часа, охлаждали до комнатной температуры, фильтровали, промывали при помощи MTBE (23 мл) и сушили в атмосфере азота. Свободное основание дифторимидазола (5,97 г) получали в виде светло-желтого твердого вещества с выходом 95%.
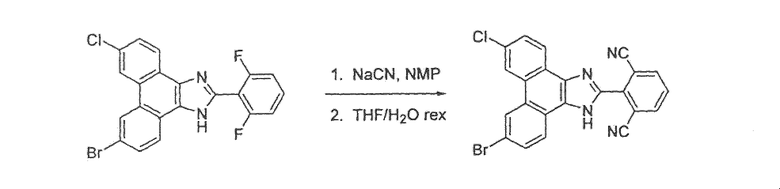
Способ A: Дифторимидазол (6,79 г, 13,39 ммоль) и цианид натрия (3,28 г, 66,95 ммоль) загружали в 500-мл круглодонную колбу в атмосфере азота. Добавляли при перемешивании N-метилпирролидон (NMP, 60 мл) и суспензию нагревали до 175°C в течение 28 часов. Реакционную смесь затем охлаждали до комнатной температуры. Добавляли воду (240 мл) в течение 2 часов и суспензию оставляли для перемешивания в течение 48 часов. К суспензии добавляли хлорид натрия (36 г) и перемешивали еще в течение 2 часов. Суспензию затем охлаждали до 0°C, перемешивали в течение 1 часа, фильтровали и промывали водой (30 мл). Мокрую лепешку затем сушили в атмосфере азота с получением желаемого продукта в виде NMP сольвата.
Твердое вещество суспендировали в ТГФ (42 мл, 7,5 мл/г) при 65°C в течение 1 часа. Смесь затем охлаждали до комнатной температуры с последующим добавлением воды (14 мл, 2,5 мл/г) в течение 1 часа. Суспензию затем концентрировали в вакууме, удаляли 14 мл растворителя и полученную суспензию фильтровали. Мокрую лепешку промывали смесью 1:1 ТГФ/H2O (14 мл) и сушили в атмосфере азота. Желаемый продукт (3,83 г) получали в виде сольвата в ТГФ с выходом 54%.
Способ B:
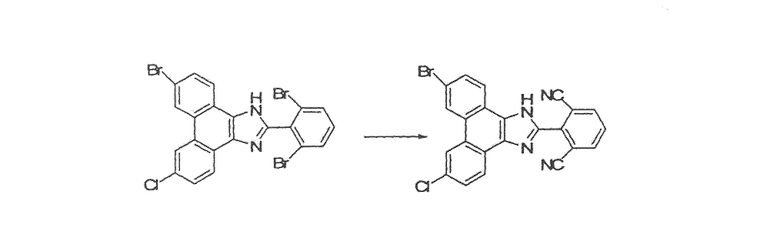
1,0 г свободного основания трибромимидазола (1,8 ммоль), 260 мг NaCN (5,3 ммоль), 135 мг CuI (0,71 ммоль) и 7 мл ДМФА объединяли и дегазировали, затем нагревали до 120°C в течение 45 часов. Добавляли 7 мл 6:1 смеси вода:NH4OH и неочищенный продукт выделяли при помощи фильтрации. После сушки полученное вещество перекристаллизовывали из смеси 1:1 ТГФ:MTBE (16 мл) с получением 870 мг дицианопродукта в виде сольвата в ТГФ (97%).
Способ C: AcOH соль трибромимидазола (1,30 г, 87% масс в расчете на свободное основание, 2 ммоль) обрабатывали K4[Fe(CN)6]·3H2О (845 мг, 2 ммоль, тонкодисперсный порошок), CuI (76,2 мг, 0,4 ммоль) и 1,2-фенилендиамином (43,3 мг, 0,4 ммоль) в ДМФА (5,7 мл). Реакционную смесь нагревали до 135°C в течение 36 часов, разбавляли при помощи ДМФА (5,7 мл) и фильтровали в горячем состоянии. Твердое вещество тщательно промывали ацетоном и промывки объединяли с фильтратом. Органический раствор концентрировали для удаления ацетона и добавляли H2O (2,8 мл) в течение 15 минут при комнатной температуре. Образовавшееся твердое вещество собирали путем фильтрования, промывали при помощи H2O и получали коричневое твердое вещество (1,06 г). Неочищенное твердое вещество затем перемешивали в ТГФ (4 мл) при 60°C в течение 1 часа и давали охладиться до комнатной температуры. Полученное твердое вещество собирали при помощи фильтрации, промывали гексаном и сушили с получением дицианида, сольвата в ТГФ, в виде не совсем белого порошка (864 мг, 89,5% масс.).
Для описанных выше способов B и C соединение трибромимидазола получают, следуя процедуре, описанной выше для получения соединения дифторимидазола, но с замещением дифторбензальдегида дибромбензальдегидом.
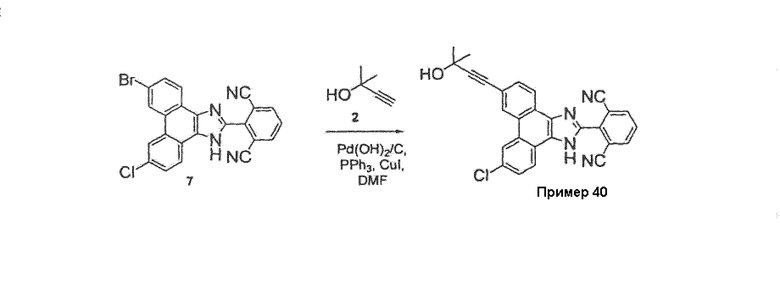
В 7-мл флакон, снабженный стержнем для перемешивания и навинчивающимся колпачком с изолирующей прокладкой, загружали 6,2 мг 20% масс Pd(OH)2 на углероде, содержащего около 16% масс воды (около 1,0 мг Pd(OH)2 с поправкой на твердую подложку и воду), 69 мг соединения 7, 8 мг трифенилфосфина и 6 мг иодида меди(I). Флакон переносили в заполненный азотом небольшой бокс, куда добавляли остальную продутую азотом реакционную смесь. Добавляли N,N-диметилформамид (0,68 мл) с последующим добавлением 2-метил-3-бутин-2-ола (0,022 мл) и триэтиламина (0,031 мл). Флакон герметично закрывали, удаляли из бокса, помещали в нагревательный блок, снабженный продутым азотом кожухом, и нагревали до внешней температуры 52°C. Реакционную смесь перемешивали при нагревании в течение около 17 часов. ВЭЖХ анализ реакционной смеси в это время показал около 95% LCAP конверсии в соединение примера 40 с использованием внешнего стандарта с >99 LCAP конверсией бромида 7 при 210 нм.
Следующие примеры описывают способы получения примера 40 в виде аморфного вещества.
ПРИМЕР A
2 г твердого соединения примера 40 и 10 мл растворителя диметилсульфоксида (ДМСО) загружали в стеклянную колбу при комнатной температуре. Все твердые вещества растворяли. Раствор быстро смешивали с 20-30 мл воды (в качестве антирастворителя) с использованием устройства с бьющей струей, аналогичного раскрытому в патенте США № 5314506, выданному 24 мая 1994 года, для осаждения соединения примера 40 в виде аморфного вещества. Соотношение ДМСО и воды при впрыскивании находилось в пределах от 1/2 до 1/3. Полученную суспензию переносили в снабженный кожухом кристаллизатор, который содержал 30-20 мл воды, при перемешивании. Конечное соотношение ДМСО/вода поддерживали при 1/5. Температуру этой партии поддерживали на уровне от -5°C до 5°C для сохранения стабильности аморфного твердого вещества примера 40 в суспензии. Суспензию фильтровали и промывали водой при температуре от 0°C до -5°C. Мокрую лепешку сушили в вакууме. Кристалличность лепешки определяли при помощи рентгено-структурного анализа и оптического микроскопа. Остаточный растворитель в лепешке анализировали методом ГХ.
Изображение аморфного твердого вещества, полученное при помощи оптического микроскопа, в основном не было двоякопреломляющим с некоторыми двоякопреломляющими кристаллами. ГХ анализ аморфного твердого вещества показал <0,5% масс. остаточного ДМСО в твердом веществе.
ПРИМЕР B
В 125-мл имеющий кожух кристаллизатор, снабженный гомогенизатором с ротором/статором EKA-Works (модель T25 с мелкорассеивающим элементом) в качестве смесителя, загружали 50 мл деионизированной воды. Включали скорость гомогенизатора при 9,1 м/сек и температуру кожуха регулировали, пока температура воды в сосуде не достигала от 0°C до -2°C. В отдельной 50-мл стеклянной колбе растворяли 1 г соединения примера 40 в 5 мл ТГФ, затем этот раствор добавляли в указанный выше 125-мл кристаллизатор в течение 5 минут. После загрузки температуру кожуха указанного выше кристаллизатора регулировали так, чтобы температура партии достигала 0-2°C. Партию фильтровали и промывали холодной водой. Высушенный образец анализировали при помощи рентгено-структурного анализа, который подтвердил, что вещество является аморфным.
| название | год | авторы | номер документа |
|---|---|---|---|
| ФОТОХРОМНЫЕ ОКСАЗИНОВЫЕ СОЕДИНЕНИЯ И СПОСОБЫ ИХ ПРОИЗВОДСТВА | 2002 |
|
RU2315042C2 |
| ПРОИЗВОДНЫЕ 1-(4-БЕНЗИЛПИПЕРАЗИН-1-ИЛ)-3-ФЕНИЛПРОПЕНОНА И ИХ ПРИМЕНЕНИЕ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ИНГИБИРОВАНИЯ РЕЦЕПТОРОВ ХЕМОКИНА (CCR-1) | 2003 |
|
RU2347782C2 |
| АМИДНЫЕ ПРОИЗВОДНЫЕ И ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, СПОСОБ ИХ ПОЛУЧЕНИЯ И МЕДИЦИНСКОЕ ПРИМЕНЕНИЕ | 2015 |
|
RU2681537C2 |
| ИНГИБИТОРЫ КИНАЗЫ, ИХ ПРОЛЕКАРСТВЕННЫЕ ФОРМЫ И ИХ ПРИМЕНЕНИЕ В ТЕРАПИИ | 2010 |
|
RU2568639C2 |
| РЕГУЛЯТОРЫ-ПРОИЗВОДНЫЕ СТЕРОИДОВ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2019 |
|
RU2797408C2 |
| ФОТОХРОМНЫЕ ОКСАЗИНОВЫЕ СОЕДИНЕНИЯ И СПОСОБЫ ИХ ПРОИЗВОДСТВА | 2002 |
|
RU2295521C2 |
| РЕГУЛЯТОРЫ ПРОИЗВОДНЫХ СТЕРОИДОВ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2019 |
|
RU2803499C1 |
| ПРОИЗВОДНЫЕ ИМИДАЗОЛА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ИНГИБИРУЮЩАЯ АКТИВНОСТЬ АНГИОТЕНЗИНА II, И СПОСОБ ЛЕЧЕНИЯ ГИПЕРТОНИИ У МЛЕКОПИТАЮЩИХ | 1991 |
|
RU2111208C1 |
| ИНГИБИТОРЫ ФАКТОРА XIa | 2016 |
|
RU2728783C2 |
| СОЕДИНЕНИЕ ГИДРОКСИТРИАЗИНА И ЕГО ПРИМЕНЕНИЕ В МЕДИЦИНСКИХ ЦЕЛЯХ | 2016 |
|
RU2772907C2 |
Изобретение относится к новым производным 2-(фенил)-1Н-фенантро[9,10-d]имидазола общей формулы В, или к его фармацевтически приемлемым солям, где R3 представляет собой 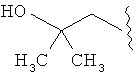 и R6 выбран из группы, состоящей из (2) F; (3) Cl; (4) Br; (5) I; (8) С3-6циклоалкила; (9) R12-O-; R12 выбран из группы, состоящей из (2) С1-4алкила, (3) С3-6циклоалкила; (4) С3-6циклоалкил-С1-4алкил-; при этом указанный С1-4алкил необязательно замещен 1-3 заместителями, независимо выбранными из группы, состоящей из F, Cl, Br и I. Также изобретение относится к конкретным соединениям, описываемым общей формулой В, к фармацевтической композиции на основе соединения формулы В, к способу лечения с использованием соединения формулы В. Технический результат: получены новые производные 2-(фенил)-1Н-фенантро[9,10-d]имидазола, проявляющие ингибирующую активность микросомальной простагландин Е синтазы-1 (mPGES-1). 8 н. и 4 з.п. ф-лы.
и R6 выбран из группы, состоящей из (2) F; (3) Cl; (4) Br; (5) I; (8) С3-6циклоалкила; (9) R12-O-; R12 выбран из группы, состоящей из (2) С1-4алкила, (3) С3-6циклоалкила; (4) С3-6циклоалкил-С1-4алкил-; при этом указанный С1-4алкил необязательно замещен 1-3 заместителями, независимо выбранными из группы, состоящей из F, Cl, Br и I. Также изобретение относится к конкретным соединениям, описываемым общей формулой В, к фармацевтической композиции на основе соединения формулы В, к способу лечения с использованием соединения формулы В. Технический результат: получены новые производные 2-(фенил)-1Н-фенантро[9,10-d]имидазола, проявляющие ингибирующую активность микросомальной простагландин Е синтазы-1 (mPGES-1). 8 н. и 4 з.п. ф-лы.
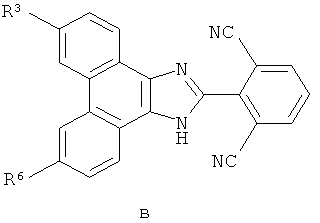
1. Соединение формулы В
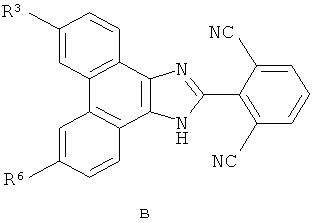
или фармацевтически приемлемая соль, где
R3 представляет собой и
R6 выбран из группы, состоящей из (2) F; (3) Cl; (4) Br; (5) I; (8) С3-6циклоалкила; (9) R12-O-;
R12 выбран из группы, состоящей из (2) С1-4алкила, (3) С3-6циклоалкила; (4) С3-6циклоалкил-С1-4алкил-; при этом указанный С1-4алкил необязательно замещен 1-3 заместителями, независимо выбранными из группы, состоящей из F, Cl, Br и I.
2. Соединение по п.1, где R6 представляет собой R12-O.
3. Соединение по п.2, где R12 выбран из группы, состоящей из (1) C1-4алкила и (2) С3-6циклоалкил-С1-4алкил-, где указанный С1-4алкил необязательно замещен 1-3 заместителями, независимо выбранными из группы, состоящей из F, Cl, Br и I.
4. Соединение по п.1, где R6 выбран из F, Cl, Br и I.
5. Соединение, выбранное из следующей таблицы:
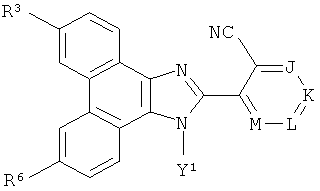
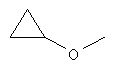
или фармацевтически приемлемая соль любого из указанных соединений.
6. Фармацевтическая композиция, ингибирующая активность фермента микросомальной простагландин Е синтазы (mPGES-1), включающая эффективное количество соединения по п.1 в сочетании с фармацевтически приемлемым носителем.
7. Способ лечения опосредованных микросомальной простагландин Е синтазой-1 заболеваний или состояний у пациента-человека, нуждающегося таком лечении, включающий введение указанному пациенту соединения по п.1 в количестве, эффективном для лечения опосредованных микросомальной простагландин Е синтазой-1 заболеваний или состояний.
8. Способ по п.7, где заболевание или состояние выбрано из группы, включающей острую или хроническую боль, остеоартрит, ревматоидный артрит, бурсит, алкилозирующий спондилит и первичную дисменорею.
9. Соединение, которое представляет собой
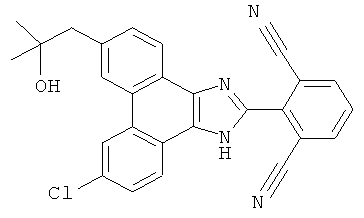
или его фармацевтически приемлемая соль.
10. Соединение, которое представляет собой
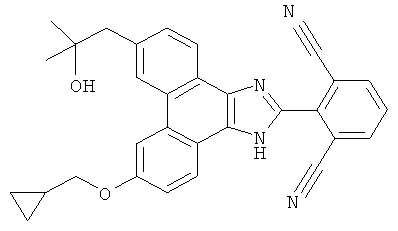
или его фармацевтически приемлемая соль.
11. Соединение, которое представляет собой
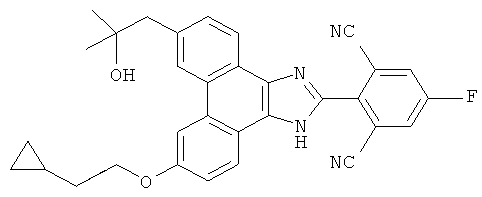
или его фармацевтически приемлемая соль.
12. Соединение, которое представляет собой
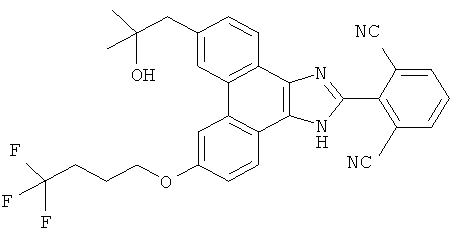
или его фармацевтически приемлемая соль.
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| JP 63287963 А, 25.11.1988 | |||
| Производные имидазо [1,2= @ ]=фенатрено=[9,10= @ ]-1,2,4-триазино и способ их получения | 1975 |
|
SU552792A1 |

