Настоящее изобретение относится к новому способу получения 1,6:2,3-диангидро-β-D-маннопиранозы, называемой далее "эпоксидом Черни" или "соединением (I)" и соответствующей следующей формуле, в которой жирная черта означает связь, находящуюся выше плоскости пиранозного цикла:
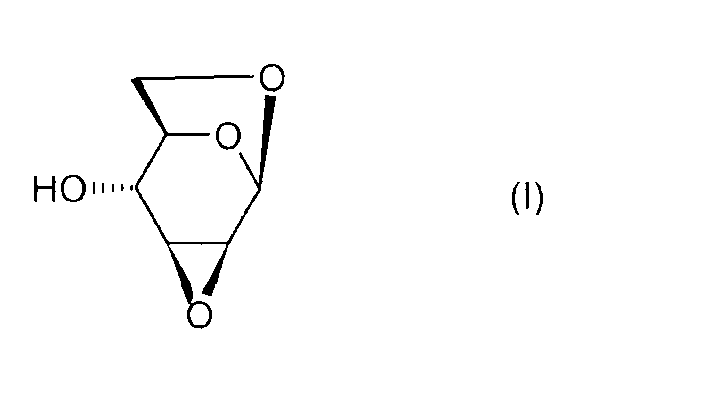
или же, согласно другому представлению:
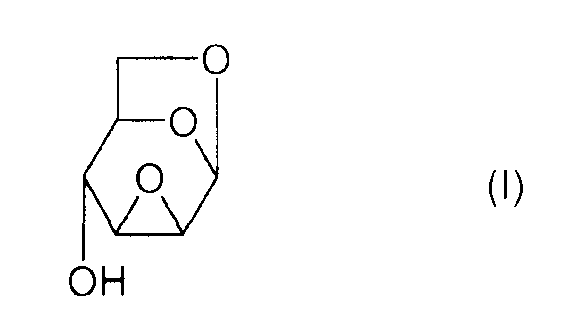
Соединение (I) и, в общем, соединения семейства 1,6:(2,3 и 3,4)-диангидро-β-D-гексопираноз были описаны в основном чешским химиком Милославом Черни. В литературе имеется три способа получения эпоксида Черни (I) из соединения 1 (1,6:3,4-диангидро-4-O-тозил-β-D-галактопираноза):
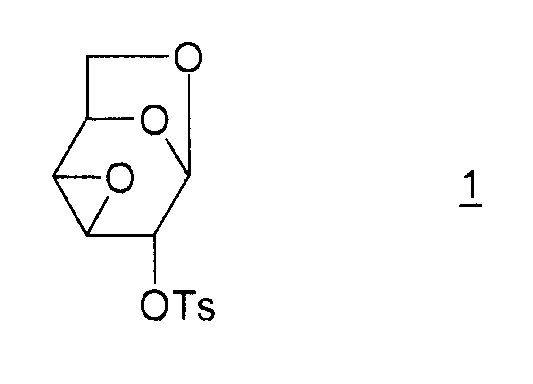
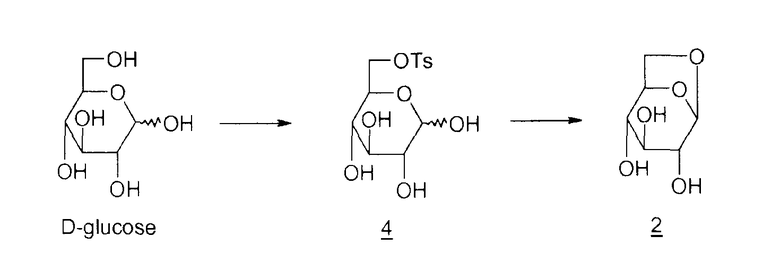
Соединение 1 получают из левоглюкозана 2 (или 1,6-ангидро-β-D-глюкопиранозы), как показано ниже (M. Cerny et al., Collect. Czech. Chem. Commun., 1961, vol. 26, p. 2542-2550):
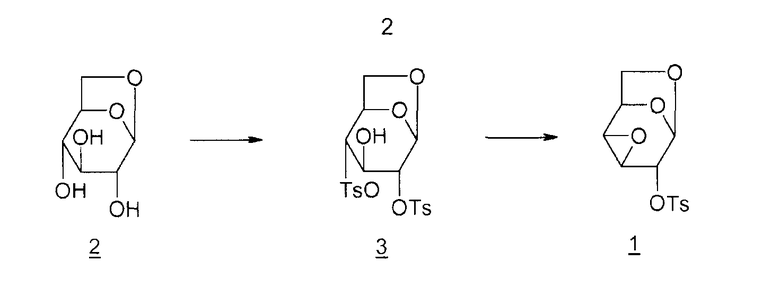
Дитозильное производное 3 (1,6-ангидро-2,4-ди-O-тозил-β-D-глюкопираноза) получают селективно (80%). Остальные 20% состоят в основном из тритозильного производного. Общий выход для превращения соединения 2 в соединение 1 составляет 55%.
Способов получения левоглюкозана 2 много; наиболее распространенными в промышленности, помимо пиролиза крахмала и целлюлозы, описанного еще в 1960-ые, являются циклизация D-глюкозы в основной или кислой среде, представленная ниже
Циклизация в основной среде
4: 6-0-тозил-D-глюкопираноза
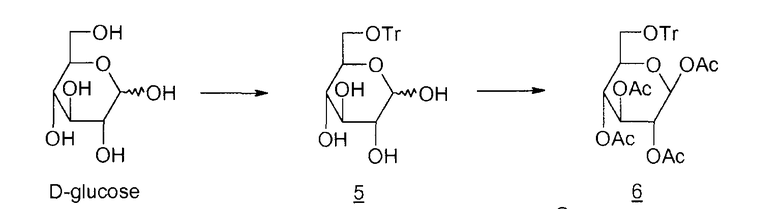
Циклизация в кислой среде
5: 6-О-тритил-D-глюкопираноза
6: 1,2,3,4-тетра-O-ацетил-6-O-тритил-β-D-глюкопираноза
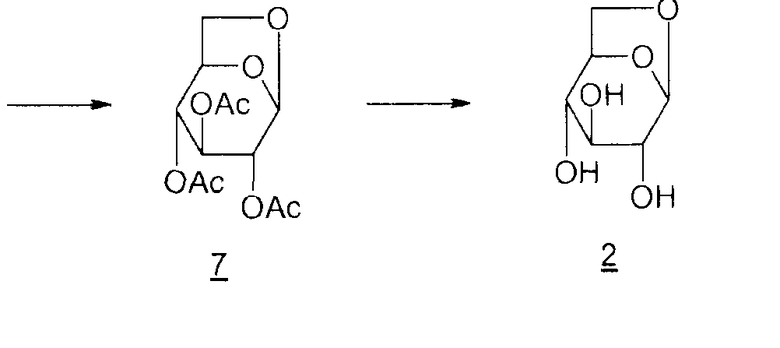
7: 1,6-ангидро-2,3,4-три-O-ацетил-β-D-глюкопираноза
Циклизация в основной среде (M.A. Zottola et al., J. Org. Chem., 1989, vol. 54, p.6123-6125 ; M. Akagi et al., Chem. Pharm. Bull., 1962, vol. 10, p.905-909) выражается в низком выходе (15%). Кроме того, необходимо ацетилировать неочищенный левоглюкозан 2 для его выделения. Что касается способа циклизации в кислой среде (M.V. Rao et al., Carbohydrate Research, 1987, vol. 162, 141-144; R.L. Wistler et al., Methods Carbohydr. Chem., 1972, vol. 6, p.411-412; E. Zara-Kaczian et al., 1982, vol. 111, no. 3, p.271-283; E. Zara-Kaczian et al., Acta Chemica Acad. Scient. Hung., 1978, vol. 96, no. 3, p.311-313), описано, что он имеет лучший выход (70%), но содержит на две стадии больше.
Три способа получения эпоксида Черни (I) из соединения 1 следующие.
Способ 1:

8: 1,6-ангидро-2-O-тозил-β-D-глюкопираноза
9: 1,6-ангидро-2-O-тозил-4-O-тритил-β-D-глюкопираноза
10: 1,6:2,3-диангидро-4-O-тритил-β-D-глюкопираноза
Наряду с последовательностью этапов, необходимых, чтобы получить эпоксид Черни (I), в этой цепочке трудно, кроме прочего, провести селективный гидролиз группы ангидро-3,4 на первом этапе. Гидроксил в положении 4 монотозильного производного 8 защищают затем тритильной группой (Tr), чтобы предотвратить миграцию эпоксида во время циклизации в присутствии этилата натрия (EtONa).
Способ 2:
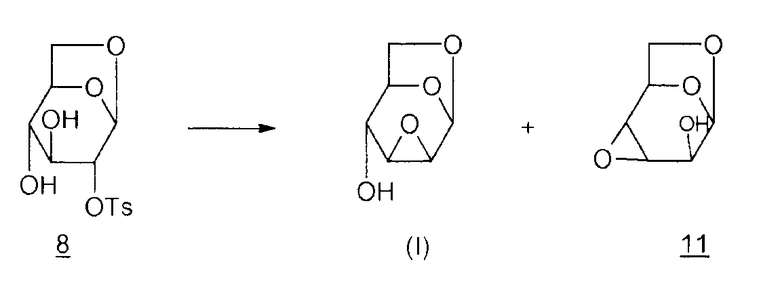
Согласно M. Cerny et al. (Synthesis, 1972, 698-699), эпоксид Черни (I) может быть получен из производного 8 в присутствии смолы амберлит IRA 400/OH-. Однако продолжительный контакт со смолой приводит к миграции эпоксида в положение 3,4 и образование производного 11 (1,6:3,4-диангидро-β-D-альтропираноза). Таким образом, остается сложным получить избирательно соединение (I). Также трудно избирательно получить исходное соединение 8, как упоминалось ранее.
Способ 3
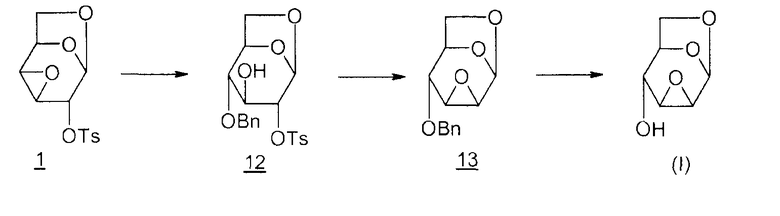
12: 1,6-ангидро-4-O-бензил-2-O-тозил-β-D-глюкопираноза
13: 1,6:2,3-диангидро-4-O-бензил-β-D-маннопираноза
Этот вариант позволяет провести циклизацию в диангидро производное 13 без миграции эпоксида (T. Trnka et al., Collect. Czech. Chem. Commun., 1971, vol. 36, p.2216-2225; M. Cerny et al., Collect. Czech. Chem. Commun., 1968, vol. 33, p.1143-1156). Тем не менее, он содержит большое число этапов получения эпоксида Черни (I) из D-глюкозы.
В заключение отметим, что три описанных выше маршрута получения эпоксида Черни (I) насчитывают соответственно 10, 8 и 9 этапов, если исходить из D-глюкозы (используя для получения левоглюкозана 2 циклизацию в кислой среде, что, как описано, является способом с наилучшим выходом), и полный выход для способов 1, 2 и 3 равен 0,5%, 10% и 13%, соответственно.
Кроме того, V. Bailliez et al. описали в Synthesis, 2003, No. 7, 1015-1017 способ получения 1,6:3,4-диангидро-β-D-альтропиранозы, который сопровождается поточным образованием 1,6:2,3-диангидро-β-D-маннопиранозы в качестве примесного продукта. Согласно этим авторам, эпоксид Черни может быть получен из предшественника, предварительно циклизованного между положениями 1 и 6, или же (N-1)-ый предшественник эпоксида Черни, ацетилированный в положении 4, может быть получен, с уровнем выхода 5%, за несколько этапов, исходя из 1,3,4-три-O-ацетил-2,6-ди-O-тозилглюкозы, подвергнутой действию оксида алюминия, микроволновому облучению и пер-O-ацетилированию.
Учитывая трудозатраты и расходы на исходные материалы, и для получения соединения (I) в промышленном масштабе необходимо разработать более короткий и, следовательно, более рентабельный синтез. Так, авторы настоящего изобретения нашли способ получения соединения (I) в два этапа, исходя из D-глюкозы, который удовлетворяет указанным выше потребностям.
Способ согласно изобретению включает в себя этапы, представленные ниже на схеме 1.
Схема 1:
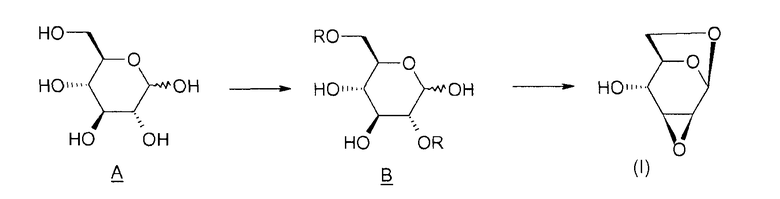
Таким образом, объектом изобретения является способ получения соединения (I), отличающийся тем, что он содержит этап циклизации соединения B, в котором R означает активатор, в присутствии основания.
Под "активатором" понимается агент, позволяющий убрать уходящую группу -OR и облегчающий реакцию циклизации между положениями 1 и 6 соединения B, например, тозил-, бензил-, бензолсульфонилгалогенид или галогенид бензолсульфонильного производного, такой как п-галогенобензилсульфонилгалогенид. Таким образом, соединение B таково, что R означает тозильную, бензильную, бензолсульфонильную или п-галогенобензилсульфонильную группу.
В случае тозилхлорида (TsCl) группа -OTs является отличной уходящей группой. Таким образом, выгодно использовать тозилхлорид в качестве активатора в растворителе, таком как пиридин.
Основание, используемое на определенном выше этапе циклизации, выбрано из гидроксидов аммония и неорганических оснований. Подходящими неорганическими основаниями могут быть сильные неорганические основания (например, гидроксид натрия или калия), слабые неорганические основания, в частности, твердого типа (например, карбонат калия, натрия или цезия).
Под "гидроксидом аммония" понимается соединение формулы
N+(R1)(R2)(R3)(R4)OH-, в которой R1, R2, R3 и R4, одинаковые или разные, означают алкильные группы, причем указанные алкильные группы являются линейными или разветвленными насыщенными алифатическими группами, содержащими от 1 до 4 атомов углерода. Гидроксид аммония, используемый в реакции циклизации соединения B, может состоять, например, из гидроксида тетрабутиламмония.
Этап циклизации соединения B проводится в подходящем растворителе, выбираемом в зависимости от природы используемого основания, в зависимости от познаний специалиста в этой области. Реакция может быть проведена, например, в изопропаноле, дихлорметане или ацетонитриле, или же в бинарной смеси этих растворителей.
В качестве растворителя используют, например, смесь изопропанол/дихлорметан, содержащую, например, около 5 объемных % дихлорметана, когда применяющимся основанием является гидроксид тетрабутиламмония. В этом случае реакция циклизации благоприятно реализуется при низкой температуре, в частности, при температуре меньше или равной 0°C, причем присутствие дихлорметана позволяет растворить соединение B при низкой температуре. Можно, например, осуществить реакцию циклизации при температуре в интервале от -10°C до 0°C, например, при примерно -5°C.
Когда основанием, используемым в реакции циклизации, является карбонат цезия, предпочтительно в качестве растворителя использовать ацетонитрил при температуре около 40°C.
Как известно специалисту, температура реакционной среды на этапе циклизации подбирается в зависимости от используемой пары растворитель/основание, чтобы оптимизировать кинетику реакции.
Согласно изобретению, соединение (I) получается с селективностью 65-85%, если исходить из интермедиата B. Химический выход на этом этапе, рассчитанный по выделенному продукту, составляет по меньшей мере примерно 60%.
Объектом изобретения является также способ получения соединения (I), отличающийся тем, что он содержит этап активации соединения A (D-глюкоза), позволяющий получить соединение B, затем этап циклизации соединения B в присутствии основания, какое определено ранее.
Этап активации соединения A может быть осуществлен с помощью активатора, какой определен ранее. Этот этап благоприятно проводить с помощью тозил-, бензил-, бензолсульфонилгалогенида или галогенида бензолсульфонильного производного, такого как п-галогенобензилсульфонилгалогенид, в растворителе, таком как пиридин.
Изобретение иллюстрируется в следующих примерах, которые детализируют способ получения соединения (I) согласно изобретению, следуя приведенной ниже схеме 2.
Схема 2:
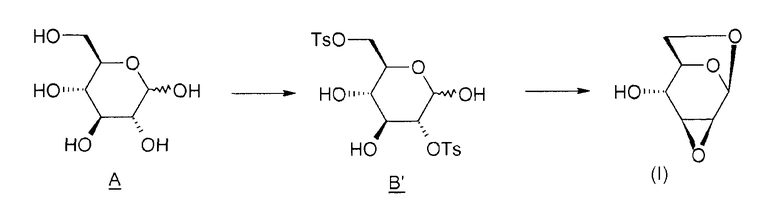
1) Получение соединения B' (2,6-ди-O-тозил-глюкопираноза)
В реактор на 2 литра, оборудованный системой перемешивания, помещают 100 г (0,55 моль) D-глюкозы и 500 г пиридина. Реакционную среду охлаждают до -10°C. В другом реакторе на 1 литр готовят раствор тозилхлорида: вводят 212 г (1,12 моль) тозилхлорида и 667 г пиридина и перемешивают при 20°C до полного растворения. Постепенно (за 4-5 ч) переносят содержимое однолитрового реактора в двухлитровый реактор, поддерживая температуру -10°C. Промывают 39 граммами пиридина и продолжают перемешивать реакционную среду в течение 17 ч при -11°C.
Проводят замену растворителя путем перегонки с помощью деминерализованной воды. Пиридина в реакционной среде должно быть ≤20%; если это не так, снова проводят перегонку с 400 мл воды.
Реакционную среду охлаждают до 20°C и вводят 400 мл деминерализованной воды. После удаления монотозильного соединения вводят 400 мл дихлорметана, 48 г соляной кислоты и 33 мл воды. Выдерживают 30 мин и измеряют pH, который должен быть меньше или равен 1; если это не так, по каплям добавляют соляную кислоту до получения pH≤1. Затем промывают раствором хлорида натрия (400 мл воды + 40 г хлорида натрия) до получения pH примерно 5-5,5.
Наконец, на роторном испарителе концентрируют фазу дихлорметана. Получают концентрат со 150 мл дихлорметана, который снова концентрируют. После проведения трех таких операций получают соединение B' в форме бежевой (кремовой) пены.
Ожидаемая масса продукта=271 г
Полученная масса продукта=184 г
Органическая чистота=81,6%, измеренная по ВЭЖХ (высокоэффективная жидкостная хроматография)
Химический выход=55%
2) Получение соединения (I)
2.1: Циклизация, осуществленная с помощью гидроксида тетрабутиламмония
В реактор объемом 1 литр вводят 10 г соединения B', полученного на предыдущем этапе, 100 мл изопропанола и 5 мл дихлорметана. Реакционную среду охлаждают до температуры -5°C при перемешивании со скоростью 400 об/мин. Медленно вливают (в течение примерно 30 мин) 26,6 г гидроксида тетрабутиламмония (40% в воде). Оставляют перемешиваться на 30 минут и реакцию останавливают, нейтрализуя реакционную среду 12%-ной соляной кислоты, до получения pH примерно 6-7. Меняют растворитель (замена изопропанола на этилацетат) на роторном испарителе с 6 мл этилацетата. Расчетный химический выход на этом этапе составляет около 60%, органическая чистота соединения (I), измеренная, исходя из пиков газовой хроматографии, равна 68%.
Спектры ЯМР H' и С-13 соединения (I) сняты на приборе Bruker 300 МГц. Химические сдвиги выражены относительно тетраметилсилана, с точностью 0,01 ч/млн для протонного спектра и точностью 0,1 ч/млн для спектра С-13. Константы взаимодействия указаны в абсолютных величинах в Гц, точность 0,5 Гц.
1H-ЯМР (CDCl3): 2,67 (д, 1H, OH, J4,OH 5,5 Гц), 3,12 (д, 1H, H3, J2,3 3,4 Гц), 3,42 (дд, 1H, H2, J2,3=J2,1=3,0 Гц), 3,69-3,77 (м, 2H, H6, H6), 3,89 (д, 1H, H4, J4,OH 5,5 Гц), 4,40 (дм, 1H, H1, J1,2 3,0 Гц).
13C-ЯМР: 49,3: C3; 54,3: C2; 65,6: C6,6'; 67,1: C4; 97,7: C1; 74,2: C5.
2.2: Циклизация, осуществленная с помощью карбоната цезия
Как вариант, действуют, как указано в примере 2.1, но используя карбонат цезия в качестве основания для реакции циклизации соединения B'.
Используют 2 эквивалента карбоната цезия в расчете на количество соединения B', а именно 1,133 г карбоната цезия на 1 г соединения B', в 10,5 мл ацетонитрила и при температуре около 40°C. Химический выход, оцененный для этого этапа, составляет около 80%, причем органическая чистота полученного так соединения (I), измеренная из площади пиков в газовой хроматографии, равна 87%.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ОРГАНИЧЕСКИХ АЗИДОВ | 2000 |
|
RU2241714C2 |
| НОВЫЕ ПЕНТАСАХАРИДЫ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 1999 |
|
RU2193040C2 |
| СИНТЕЗ β-L-2'-ДЕЗОКСИНУКЛЕОЗИДОВ | 2004 |
|
RU2361875C2 |
| ПРОИЗВОДНЫЕ 3-ДЕЗОКСИОЛИГОСАХАРИДОВ, СПОСОБ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ), ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1994 |
|
RU2133752C1 |
| N-СУЛЬФАТИРОВАННЫЕ ОЛИГОСАХАРИДЫ, АКТИВИРУЮЩИЕ РЕЦЕПТОРЫ FGF, ИХ ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ В ТЕРАПИИ | 2010 |
|
RU2559629C2 |
| СИНТЕТИЧЕСКИЕ ПОЛИСАХАРИДЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1997 |
|
RU2167163C2 |
| Производные 1,6-ангидро- @ - @ -глюкопиранозы для трехмерной полимеризации и способ их получения | 1977 |
|
SU1038344A1 |
| СПОСОБ ПОЛУЧЕНИЯ АЛЬДОНОВЫХ КИСЛОТ И ИХ ПРОИЗВОДНЫХ | 2007 |
|
RU2455296C2 |
| ПРОИЗВОДНЫЕ СПИРОКЕТАЛЕЙ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ЛЕКАРСТВЕННОГО СРЕДСТВА ПРОТИВ ДИАБЕТА | 2006 |
|
RU2416617C2 |
| СПОСОБ ПОЛУЧЕНИЯ ЗАМЕЩЕННЫХ 1-О-АЦИЛ-2-ДЕЗОКСИ-2-ФТОР-4-ТИО-β-D-АРАБИНОФУРАНОЗ | 2010 |
|
RU2559364C2 |
Способ получения 1,6:2,3-диангидро-β-D-маннопиранозы (соединения I), отличающийся тем, что он содержит этап циклизации соединения В (указанного ниже), в котором R означает активатор в присутствии основания, выбранного из гидроксидов аммония и неорганических оснований. Данный способ предварительно включает стадию активации соединения А (см. ниже), позволяющий получить соединение В, за которым идет этап циклизации соединения В. 11 з.п. ф-лы.



1. Способ получения соединения (I):
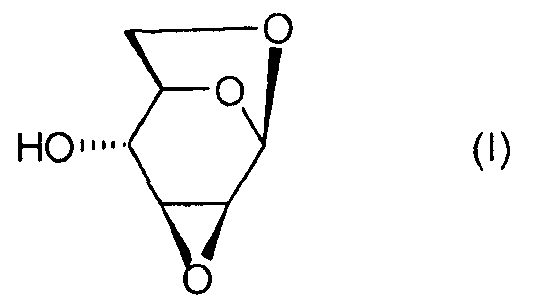
отличающийся тем, что он содержит этап циклизации соединения В:
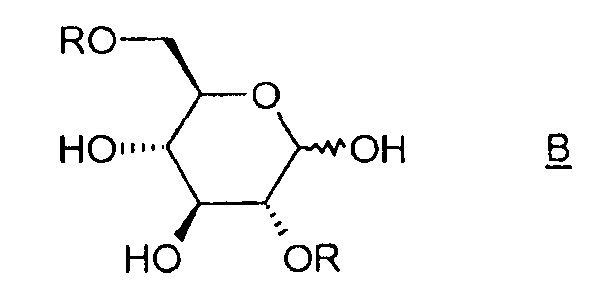
в котором R означает активатор; в присутствии основания, выбранного из гидроксидов аммония и неорганических оснований.
2. Способ по п.1, отличающийся тем, что основание выбрано из гидроксида аммония, гидроксида натрия или калия и карбоната калия, натрия или цезия.
3. Способ по п.1 или 2, отличающийся тем, что основание является гидроксидом тетрабутиламмония.
4. Способ по п.1 или 2, отличающийся тем, что основание является карбонатом цезия.
5. Способ по любому из пп.1 и 2, отличающийся тем, что этап циклизации проводят в изопропаноле, дихлорметане или ацетонитриле или же в бинарной смеси этих растворителей.
6. Способ по п.3, отличающийся тем, что этап циклизации проводят в смеси изопропанол/дихлорметан.
7. Способ по п.6, отличающийся тем, что этап циклизации проводят при температуре от -10°С до 0°С.
8. Способ по п.4, отличающийся тем, что этап циклизации проводят в ацетонитриле.
9. Способ по п.8, отличающийся тем, что этап циклизации проводят при температуре около 40°С.
10. Способ по любому из пп.1 и 2, отличающийся тем, что соединение В таково, что R означает тозильную, бензильную, бензолсульфонильную или п-галогенобензилсульфонильную группу.
11. Способ по любому из пп.1 и 2, отличающийся тем, что он включает:
- этап активации соединения А:
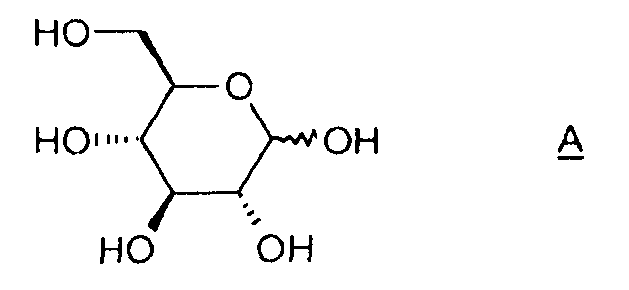
позволяющий получить соединение В, какое описано в п.1,
- за которым идет этап циклизации соединения В, какое определено в любом из пп.1-9.
12. Способ по п.11, отличающийся тем, что этап активации соединения А проводят с помощью тозил-, бензил-, бензолсульфонилгалогенида или п-галогенобензилсульфонилгалогенида.
| Bailliez V | |||
| et al., "A partical large-scale access to l,6-anhydro-[beta]-D-hexopyranoses by a solid-supported solvent-free microwave-assisted procedure", Synthesis, 2003, no.7, p.1015-1017 | |||
| Miljkovic D | |||
| et al., "A convenient route to 6 functionalized derivatives of D glucal", Carbohydrate Research, 1989, vol.193, p.275-278 | |||
| US 20050010023 A1, 13.01.2005 | |||
| СПОСОБ ПОЛУЧЕНИЯ ОРГАНИЧЕСКИХ АЗИДОВ | 2000 |
|
RU2241714C2 |

