Область техники, к которой относится изобретение
Перекрестная ссылка на родственные заявки
Настоящая заявка испрашивает приоритет заявки на патент Японии JP № 2009-246400, поданной 27 октября 2009 г., полное содержание которой включено в настоящую заявку путем ссылки.
Настоящее изобретение относится к новому производному 5-фторурацила или его соли и к его применению.
Предшествующий уровень техники
5-Фторурацил (далее в настоящем описании именуемый 5-FU) широко применяется при лечении различных злокачественных заболеваний, главным образом злокачественных заболеваний желудочно-кишечного тракта, отдельно или в комбинации с другими противораковыми средствами. Однако сам 5-FU обладает лишь слабым противоопухолевым эффектом и вызывает различные побочные эффекты, такие как диарея, стоматит и другие, ввиду токсического действия на желудочно-кишечный тракт и миелосуппрессии. Поэтому трудно сказать, что 5-FU всегда легко применять у пациентов со злокачественными заболеваниями. Для разрешения указанных проблем в настоящее время разрабатываются различные перорально вводимые производные 5-FU; однако удовлетворительные клинические эффекты еще не были получены. Возможными причинами этого являются следующие. 5-FU быстро разрушается in vivo дигидропиримидин-дегидрогеназой (далее в настоящем описании именуемой DPD), которая содержится, в частности, в печени и опухолевых тканях; поэтому трудно достичь достаточного противоопухолевого эффекта, соответствующего его дозировке. 5-FU захватывается не только в раковые клетки, но также в нормальные клетки, такие как клетки костного мозга и клетки желудочно-кишечного тракта, и превращается в активные метаболиты действием оротат-фосфорибозил-трансферазой (далее в настоящем описании именуемой OPRT). Такие активные метаболиты вызывают повреждение клеток, т.е. они обладают цитотоксичностью; поэтому их противоопухолевый эффект и побочные эффекты недостаточно сбалансированы.
Сообщалось о соединениях, обладающих активностью ингибирования DPD и противоопухолевой активностью, в качестве примеров производных 5-FU (см. патентные ссылки 1-3). Среди них, в патентной ссылке 2, в частности, описывается Соединение (1), показанное ниже, которое представляет собой соединение, в целом известное как Эмитефур (также именуемое BOF-A2). Было проведено клиническое испытание для оценки Эмитефура; однако его разработка была прекращена, поскольку он имеет сильные побочные эффекты.

Как описано выше, еще не были разработаны производные 5-FU, которые могут усилить противоопухолевый эффект путем подавления разрушения 5-FU in vivo и в то же время уменьшить побочные эффекты. Поэтому необходима разработка нового производного 5-FU, имеющего усиленный противоопухолевый эффект и низкую токсичность для улучшения терапевтического эффекта при лечении пациентов со злокачественными заболеваниями.
Как описано выше, не было сообщений о производном, обладающем противоопухолевой активностью в дополнение к активности, ингибирующей DPD, и активности, ингибирующей OPRT, в одном соединении. Поэтому требуется разработка лекарственного средства, которое достигает баланса между эффектами и токсичностью, т.е. обладает сильным противоопухолевым эффектом на злокачественные опухоли у людей и сниженным повреждением желудочно-кишечного тракта, и которое улучшает качество жизни пациентов с онкологическими заболеваниями.
Список ссылок
Патентная литература
1: Не прошедшая экспертизу заявка на патент Японии № S63-201127.
2: Не прошедшая экспертизу заявка на патент Японии № S63-301880.
3: WO87/06582.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Техническая проблема
Целью настоящего изобретения является получение нового антиметаболического противоракового средства, проявляющего сильный противоопухолевый эффект на опухолевые клетки, в то же время уменьшая повреждение желудочно-кишечного тракта, т.е. имеющего сбалансированные эффекты и токсичность, за счет наличия активности, ингибирующей DPD, а также активности, ингибирующей OPRT in vivo.
Решение проблемы
Заявители провели обширные исследования для решения указанной выше проблемы. В результате они обнаружили, что производное 5-фторурацила, представленное ниже формулой (I) (далее в настоящем описании именуемое Соединением (I) по настоящему изобретению) или его солью, имеет (1) активность, ингибирующую DPD, и (2) активность, ингибирующую OPRT, и, в результате, (3) достигает баланса между сильным противоопухолевым эффектом и уменьшенным повреждением желудочно-кишечного тракта, т.е. превосходит известные производные 5-FU с точки зрения баланса между эффектами и токсичностью.
Настоящее изобретение было осуществлено на основании указанных полученных данных.
Конкретнее, настоящее изобретение относится к следующим предметам изобретения.
Пункт 1. Производное 5-фторурацила, представленное ниже формулой (I), или его соль:
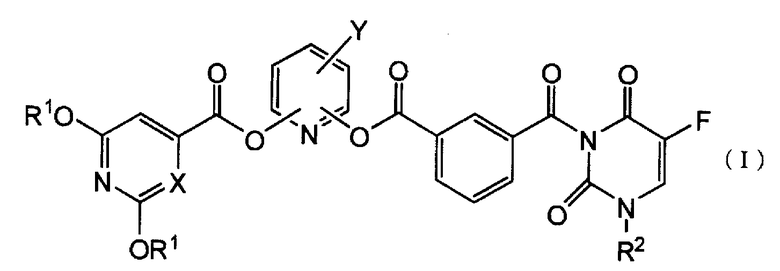
где R1 представляет атом водорода или защитную группу гидроксигруппы, R2 представляет группу низшего алкоксинизшего аликила или тетрагидрофуранильную группу, X представляет атом углерода или атом азота и Y представляет атом галогена или цианогруппу.
Пункт 2. Производное 5-фторурацила или его соль по п.1, где группа, представленная следующей формулой в формуле (I), представляет собой:

группу, представленную формулой:
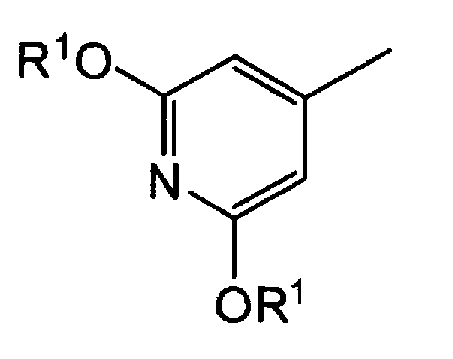
где R1 представляет атом водорода, аллильную группу, или замещенную, или незамещенную бензильную группу,
группу, представленную формулой:
группу, представленную формулой:
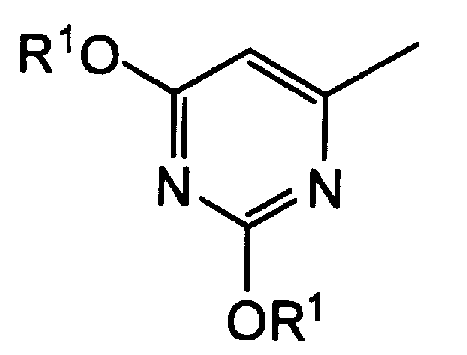
где R1 представляет атом водорода, аллильную группу, или замещенную, или незамещенную бензильную группу, или
группу, представленную формулой:
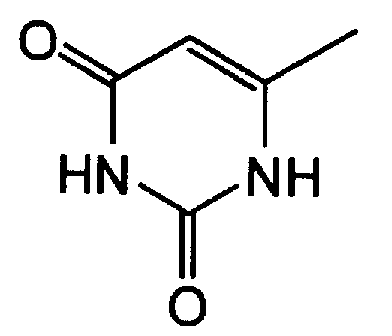
Пункт 3. Производное 5-фторурацила или его соль по п.1 или 2, где R1 представляет атом водорода, аллильную группу, бензильную группу, алифатическую ацильную группу, ароматическую ацильную группу или алициклическую ацильную группу, R2 представляет низшую алкоксиметильную группу, в которой часть низшего алкокси имеет от 1 до 6 атомов углерода или тетрагидрофуранильную группу, X представляет атом углерода или атом азота и Y представляет атом фтора или атом хлора.
Пункт 4. Производное 5-фторурацила или его соль по любому из пп.1-3, где R1 представляет атом водорода, бензильную группу, ацетильную группу, пропионильную группу, изобутирильную группу, пивалоильную группу, бензоильную группу, п-хлорбензоильную или циклопентанкарбонильную группу, R2 представляет низшую алкоксиметильную группу, в которой часть низшего алкокси имеет от 1 до 6 атомов углерода, X представляет атом углерода или атом азота и Y представляет атом фтора или атом хлора.
Пункт 5. Производное 5-фторурацила или его соль по любому из пп.1-4, где R1 представляет атом водорода или ацетильную группу, R2 представляет низшую алкоксиметильную группу, в которой часть низшего алкокси имеет от 1 до 6 атомов углерода, X представляет атом углерода или атом азота и Y представляет атом хлора.
Пункт 6. Производное 5-фторурацила или его соль по любому из пп.1-5, где R1 представляет атом водорода или ацетильную группу, R2 представляет низшую алкоксиметильную группу, в которой часть низшего алкокси имеет от 1 до 6 атомов углерода, X представляет атом углерода и Y представляет атом хлора.
Пункт 7. Производное 5-фторурацила или его соль по любому из пп.1-5, где R1 представляет атом водорода или ацетильную группу, R2 представляет этоксиметильную группу, X представляет атом углерода и Y представляет атом хлора.
Пункт 8. Лекарственное средство, содержащее производное 5-фторурацила или его соль по любому из пп.1-7 в качестве активного ингредиента.
Пункт 9. Противоопухолевое средство, содержащее производное 5-фторурацила или его соль по любому из пп.1-7 в качестве активного ингредиента.
Пункт 10. Противоопухолевое средство по п.9, где противоопухолевое средство применяется для лечения, по меньшей мере, одного злокачественного заболевания, выбранного из группы, состоящей из злокачественных опухолей головы и шеи, злокачественных опухолей пищевода, злокачественных опухолей желудка, злокачественных опухолей ободочной кишки, злокачественных опухолей прямой кишки, злокачественных опухолей печени, злокачественных опухолей желчного пузыря и желчных протоков, злокачественных опухолей желчевыводящих путей, злокачественных опухолей поджелудочной железы, злокачественных опухолей легких, злокачественных опухолей молочных желез, злокачественных опухолей яичников, злокачественных опухолей шейки матки, злокачественных опухолей эндометрия, злокачественных опухолей почек, злокачественных опухолей мочевого пузыря, злокачественных опухолей предстательной железы, злокачественных опухолей семенников, саркомы костей и мягких тканей, лейкоза, злокачественных лимфом, множественных миелом, злокачественных заболеваний кожи, опухолей головного мозга и мезотелиом.
Пункт 11. Способ лечения злокачественного заболевания, включающий введение эффективного количества производного 5-фторурацила или его соли по любому из пп.1-7 пациенту, страдающему злокачественным заболеванием.
Пункт 12. Применение производного 5-фторурацила или его соли по любому из пп.1-7 при получении противоопухолевого средства.
Преимущественные эффекты изобретения
Соединение (I) по настоящему изобретению или его соль проявляет превосходные противоопухолевые эффекты при уменьшенных побочных эффектах, таких как повреждение желудочно-кишечного тракта; поэтому оно может применяться в качестве противоопухолевого средства.
Примеры заболеваний, которые можно лечить введением лекарственного средства, содержащего соединение по настоящему изобретению, включают злокачественные опухоли головы и шеи, пищевода, желудка, ободочной кишки, прямой кишки, печени, желчного пузыря и желчных протоков, желчевыводящих путей, поджелудочной железы, легких, молочных желез, яичников, шейки матки, эндометрия, почек, мочевого пузыря, предстательной железы, семенников, саркомы костей и мягких тканей, лейкоза, злокачественных лимфом, множественных миелом, злокачественных поражений кожи, опухолей мозга и мезотелиом.
Описание вариантов осуществления изобретения
Настоящее изобретение относится к производному 5-фторурацила, представленному ниже формулой (I), или его соли:
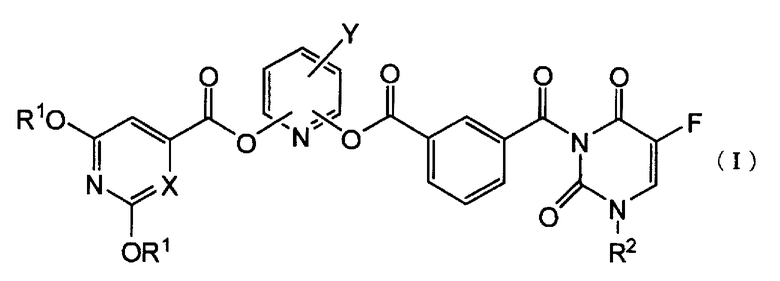
где R1 представляет атом водорода или защитную группу гидроксигруппы, R2 представляет низшую алкоксинизшую алкильную группу или тетрагидрофуранильную группу, X представляет атом углерода или атом азота и Y представляет атом галогена или цианогруппу.
В настоящем изобретении производное 5-фторурацила, представленное выше формулой (I), или его соль, включает их таутомеры.
Конкретнее, настоящее изобретение включает производное 5-фторурацила, представленное выше формулой (I), или его соль, где группа, представленная следующей формулой:
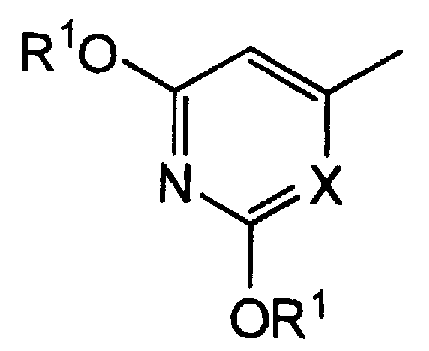
представляет собой группу, представленную формулой:

где R1 представляет атом водорода или защитную группу гидроксигруппы;
группу, представленную формулой:
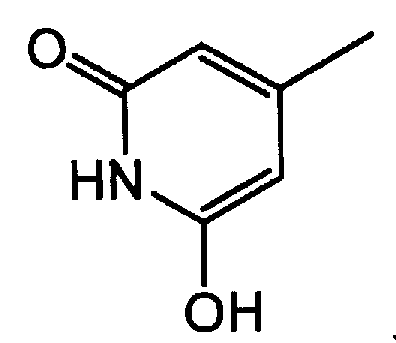
группу, представленную формулой:
где R1 представляет атом водорода или защитную группу гидрокси группы; или
группу, представленную формулой:
Определенными примерами групп, представленных формулой (I), являются следующие.
В формуле (I) R1 представляет атом водорода или защитную группу гидрокси группы.
Защитная группа гидроксигруппы, представленная R1, может представлять собой любую защитную группу, пока она может расщепляться химической процедурой, такой как гидрогенолиз, гидролиз, электролиз и фотолиз, такой как гидролиз, осуществляемый в организме человека. Ее определенные примеры включают ацильные группы, такие как замещенные или незамещенные алифатические ацильные группы, и замещенные или незамещенные ароматические ацильные группы или алициклические ацильные группы; низшие алкоксикарбонильные группы; низшие алкоксикарбамоильные группы; замещенные или незамещенные низшие алкильные группы; низшие алкенильные группы; замещенные или незамещенные алкенильные группы; силильные защитные группы; и аминокислотные остатки.
Примеры алифатических ацильных групп включают формильную группу, ацетильную группу, пропионильную группу, бутирильную группу, изобутирильную группу, пентаноильную группу, изовалерильную группу, пивалоильную группу, гексаноильную группу и тому подобные C1-6 линейные или разветвленные ацильные группы. Примеры ароматических ацильных групп включают бензоильную группу, α-нафтоильную группу и β-нафтоильную группу. Эти группы могут иметь от 1 до 3 заместителей, выбранных из низшей алкильной группы, низшей алкоксигруппы, атома галогена, нитрогруппы, карбоксигруппы и тому подобных.
Примеры алициклических ацильных групп включают циклобутанкарбонильную группу, циклопентанкарбонильную группу, циклогексанкарбонильную группу и тому подобные C3-6 циклоалкилкарбонильные группы.
Примеры низших алкоксикарбонильных групп включают метоксикарбонильную группу, этоксикарбонильную группу, н-пропоксикарбонильную группу, изопропоксикарбонильную группу, н-бутоксикарбонильную группу, изобутоксикарбонильную группу, втор-бутоксикарбонильную группу, трет-бутоксикарбонильную группу, пентилоксикарбонильную группу, гексилоксикарбонильную группу и тому подобные C2-7 линейные или разветвленные алкоксикарбонильные группы.
Примеры низших алкилкарбамоильных групп включают метилкарбамоильную группу, этилкарбамоильную группу, пропилкарбамоильную группу, бутилкарбамоильную группу, пентилкарбамоильную группу, гексилкарбамоильную группу, диметилкарбамоильную группу, диэтилкарбамоильную группу и подобные карбамоильные группы, моно- или дизамещенные C1-6 низшей алкильной группой.
Примеры низших алкильных групп включают метильную группу, этильную группу, н-пропильную группу, изопропильную группу, н-бутильную группу, изобутильную группу, втор-бутильную группу, трет-бутильную группу, н-пентильную группу, изопентильную группу, н-гексильную группу, изогексильную группу и тому подобные C1-6 линейные или разветвленные алкильные группы. Эти группы могут иметь от 1 до 3 заместителей, таких как атом галогена и низшая алкоксигруппа. Их определенные примеры также включают хлорметильную группу, метоксиметильную группу, этоксиметильную группу, метоксиэтильную группу, этоксиэтильную группу и подобные замещенные алкильные группы.
Примеры низших алкенильных групп включают этенильную группу, аллильную группу, бутенильную группу, бутадиенильную группу, гексатриенильную группу и подобные C2-6 линейные или разветвленные алкенильные группы.
Примеры арилалкильных групп включают бензильную группу, бензгидрильную группу и тритильную группу. Эти группы могут иметь от 1 до 5 или предпочтительно от 1 до 3 заместителей, таких как низшая алкильная группа, низшая алкоксигруппа, атом галогена и нитрогруппа.
Примеры силильных защитных групп включают триметилсилильную группу, трет-бутилдиметилсилильную группу, метилдиизопропилсилильную группу, триизопропилсилильную группу, тетраизопропилдисилоксильную группу (TIPDS) и дифенилметилсилильную группу.
Примеры аминокислотных остатков включают те, которые образованы удалением гидроксигруппы из карбоксигруппы аминокислоты. Определенные примеры подходящих для использования аминокислот включают глицин, аланин, β-аланин, валин и изолейцин; и могут использоваться любые аминокислотные остатки, описанные в не прошедшей экспертизу заявке на патент Японии № H1-104093.
Примеры низших алкильных групп, которые могут использоваться в качестве заместителя, включают те, которые перечислены выше.
Примеры низших алкоксигрупп включают метоксигруппу, этоксигруппу, н-пропоксигруппу, изопропилоксигруппу, н-бутилоксигруппу, изобутоксигруппу, втор-бутоксигруппу, трет-бутоксигруппу, н-пентилоксигруппу, н-гексилоксигруппу и тому подобные C1-6 линейные или разветвленные алкоксигруппы.
Примеры атомов галогена включают атом фтора, атом хлора, атом брома и атом йода.
R2 представляет низшую алкоксинизшую алкильную группу или тетрагидрофуранильную группу.
В формуле (I) примеры «низшей алкокси» части в «низшей алкоксинизшей алкильной группе», представленной R2, включают метоксигруппу, этоксигруппу, н-пропоксигруппу, изопропилоксигруппу, н-бутилоксигруппу, втор-бутилоксигруппу, трет-бутилоксигруппу, н-пентилоксигруппу, н-гексилоксигруппу и тому подобные C1-6 линейные или разветвленные алкоксигруппы. Примеры низших алкокси частей включают предпочтительно C1-3 алкоксигруппу, предпочтительнее, метоксигруппу и этоксигруппу и еще предпочтительнее этоксигруппу. Примеры «низшей алкильной группы» в «группе низшего алкоксинизшего алкила» включают метильную группу, этильную группу, н-пропильную группу, изопропильную группу, н-бутильную группу, изобутильную группу, втор-бутильную группу, трет-бутильную группу, н-пентильную группу, изопентильную группу, н-гексильную группу, изогексильную группу и подобные C1-6 линейные или разветвленные алкильные группы. Примеры низших алкильных частей включают предпочтительно C1-3 алкильную группу, предпочтительнее метильную группу и этильную группу, а еще предпочтительнее метильную группу.
Примеры «низших алкоксинизших алкильных групп» включают указанные выше низшие алкильные группы, имеющие «низкие алкоксичасти», описанные выше. Их определенные примеры включают алкоксиалкильные группы, такие как метоксиметильная группа, этоксиметильная группа, пропоксиметильная группа, метоксиэтильная группа, этоксиэтильная группа, пропоксиэтильная группа, 3-метоксипропильная группа, 4-этоксибутильная группа, 6-пропоксигексильная группа, 5-изопропоксипентильная группа, 1,1-диметил-2-бутоксиэтильная группа, 2-метил-3-трет-бутоксипропильная группа, 2-пентилоксиэтильная группа и 2-гексилоксиэтильная группа. Низшая алкоксинизшая алкильная группа представляет собой предпочтительно метоксиметильную группу, этоксиметильную группу или пропоксиметильную группу, а предпочтительнее этоксиметильную группу.
Примеры тетрагидрофуранильных групп включают 2-тетрагидрофуранильную группу и 3-тетрагидрофуранильную группу. Среди них предпочтительной является 2-тетрагидрофуранильная группа.
X представляет атом углерода или атом азота.
Y представляет атом галогена или цианогруппу. В формуле (I) примеры атомов галогенов, представленных Y, включают атом фтора, атом хлора, атом брома и атом йода.
Примеры групп в особенно предпочтительном варианте осуществления описаны ниже.
Предпочтительно, R1 представляет собой атом водорода, аллильную группу, бензильную группу, алифатическую ацильную группу, ароматическую ацильную группу или алициклическую ацильную группу, предпочтительнее атом водорода, бензильную группу, ацетильную группу, пропионильную группу, изобутирильную группу, пивалоильную группу, бензоильную группу, п-хлорбензоильную группу или циклопентанкарбонильную группу, а еще предпочтительнее атом водорода или ацетильную группу.
Предпочтительно, R2 представляет собой низшую алкоксиметильную группу, в которой низшая алкоксичасть имеет от 1 до 6 атомов углерода или 2-тетрагидрофуранильную группу, предпочтительнее низшую алкоксиметильную группу, в которой низшая алкоксичасть имеет от 1 до 6 атомов углерода, а еще предпочтительнее этоксиметильную группу.
Предпочтительно, X представляет собой атом углерода.
Предпочтительно, Y представляет собой атом фтора или атом хлора, а предпочтительнее атом хлора.
Предпочтительные варианты осуществления R1, R2, X и Y могут использоваться в любой комбинации.
Производное 5-фторурацила, представленное формулой (I) по настоящему изобретению, включает стереоизомеры, оптические изомеры, сольваты, такие как гидраты, и кристаллические полиморфизмы.
Производное 5-фторурацила, представленное формулой (I) по настоящему изобретению, может представлять собой соль. По существу, предпочтительна фармацевтически приемлемая соль. Ее примеры включают соли с неорганическими кислотами и соли с органическими кислотами.
Их определенные примеры включают соли с неорганическими кислотами, такими как хлористоводородная кислота, серная кислота, бромистоводородная кислота, йодистоводородная кислота, азотная кислота и фосфорная кислота.
Их определенные примеры включают соли с органическими кислотами, такими как муравьиная кислота, уксусная кислота, пропионовая кислота, малоновая кислота, янтарная кислота, глутаровая кислота, фумаровая кислота, малеиновая кислота, молочная кислота, яблочная кислота, лимонная кислота, винная кислота, бензолсульфоновая кислота, п-толуолсульфоновая кислота и метансульфоновая кислота.
Соединение по настоящему изобретению может быть получено различными способами, и примером такого способа является тот, который иллюстрируется схемой, показанной ниже. Материалы, необходимые для синтеза соединения по настоящему изобретению, могут быть легко получены из коммерчески доступных продуктов или в соответствии со способом получения, описанным в статье, или тому подобным. Заместители в формуле (I) такие же, как заместители, определенные выше.
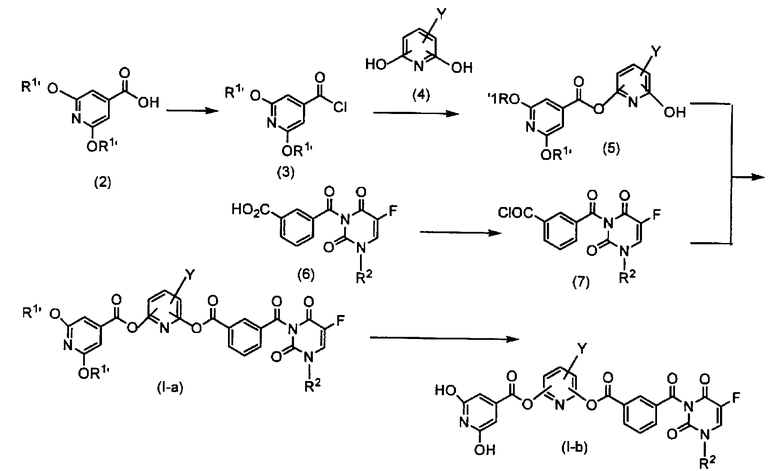
Схема 1
где R1' представляет аллильную группу, или замещенную, или незамещенную бензильную группу, R2 представляет низшую алкоксинизшую алкильную группу или тетрагидрофуранильную группу и Y представляет атом галогена или циано группу.
Синтез производного изоникотиновой кислоты (2)
Натриевую или калиевую соль аллилалкоголя, бензилового спирта или замещенного бензилового спирта растворяют в растворителе, который не воздействует на реакцию, таком как тетрагидрофуран, толуол и диметилформамид, и предпочтительно диметилформамид. Натриевую соль 2,6-дихлоризоникотиновой кислоты добавляют к полученному раствору при комнатной температуре. Полученную смесь перемешивают при 60-100ºC в течение 2-24 часов. Смеси дают возможность вступить в реакцию, предпочтительно при 80ºC в течение 4 часов. В данном случае от 2 до 10 эквивалентов, а предпочтительно 4 эквивалента количества алкоголята используют к натриевой соли 2,6-дихлоризоникотиновой кислоты. После завершения реакции к продукту реакции добавляют воду и водный слой отделяют с использованием этилацетата или подобного растворителя; pH водного слоя доводят до 5-6, используя 1N хлористоводородную кислоту или уксусную кислоту. Полученный в результате продукт подвергают экстракции с использованием этилацетата, смешанного растворителя из этилацетата и н-гексана, толуола или тому подобных. Экстракт сушат над сульфатом натрия, сульфатом магния и т.д. и затем концентрируют для получения производного изоникотиновой кислоты аллилокси, бензилокси или замещенного бензилокси (2) (в настоящем описании производное изоникотиновой кислоты аллилокси, бензилокси или замещенного бензилокси (2) может именоваться производным изоникотиновой кислоты (2)).
Стадия 1. Синтез производного изоникотиновой кислоты-хлорангидрида (3)
Производное изоникотиновой кислоты (2), полученное выше, растворяют в растворителе, который не воздействует на реакцию, такой как хлороформ, 1,2-дихлорэтан и толуол и предпочтительно толуол. Затем к полученному таким образом раствору по каплям добавляют тионилхлорид при комнатной температуре. Используемое количество тионилхлорида составляет от 1 до 10 эквивалентов и предпочтительно 5 эквивалентов относительно раствора. После завершения добавления по каплям полученную смесь перемешивают при кипячении в сосуде с обратным холодильником в течение 2-8 часов, а предпочтительно в течение 4 часов. После завершения реакции смесь концентрируют и остаток используют на следующей стадии без дополнительной обработки.
Синтез производного пиридина (4)
Производное пиридина (4) может быть получено способом, описанным в не прошедшей экспертизу заявке на патент Японии № H05-80451, в публикации Heterocycles, Vol. 36, No. 1, 145-148, 1993, и т.д.
Эти производные пиридина (4) присутствуют в виде таутомеров гидроксипиридиновой структуры и структуры 2(1H)-пиридона.
Стадия 2. Синтез вещества, связанного со сложным эфиром (5)
Производное пиридина (4), полученное выше, растворяют в соли органического амина, такой как триэтиламин, диизопропилэтиламин и диметиланилин, предпочтительно смеси триэтиламина с растворителем, который не воздействует на реакцию, таким как дихлорметан, ацетонитрил, диметилформамид, диметилацетамид и предпочтительно смесь триэтиламина с диметилацетамидом. Раствор диметилацетамида (3), полученный на стадии 1, по каплям добавляют к полученному веществу при охлаждении льдом. В данном случае используют от 1,0 до 1,2 эквивалентов производного изоникотиновой кислоты-хлорангидрида (3) и от 1,0 до 1,2 эквивалентов соли органического амина на производное пиридина (4). После предоставления возможности смеси прореагировать при комнатной температуре в течение 1-4 часов к смеси добавляют воду с последующей экстракцией с использованием этилацетата, смешанного растворителя этилацетата и н-гексана, толуола или тому подобных. Экстракт сушат над сульфатом натрия, сульфатом магния или тому подобными веществами, концентрируют до перекристаллизации, очищают колоночной хроматографией и затем доставляют на следующую стадию.
Синтез моноамида изофталевой кислоты 5-фторурацила (6)
Моноамид изофталевой кислоты 5-фторурацила (6) может быть получен способом, описанным в не прошедшей экспертизу заявке на патент Японии № H02-164871.
Стадия 3. Синтез моноамида изофтелевой кислоты 5-фторурацила-хлорангидрида (7)
Моноамид изофталевой кислоты 5-фторурацила (6) растворяют в растворителе, который не воздействует на реакцию, таком как дихлорметан, хлороформ, 1,2-дихлорэтан и толуол, и предпочтительно дихлорметан. К полученному раствору по каплям добавляют тионилхлорид при комнатной температуре. Количество тионилхлорида составляет от 1 до 4 эквивалентов и предпочтительно 1,2 эквивалента относительно раствора. После завершения добавления по каплям полученную смесь перемешивают в условиях кипячения в сосуде с обратным холодильником в течение 2-8 часов и предпочтительно 4 часов. После завершения реакции смесь концентрируют и остаток подают на следующую стадию без дополнительной обработки.
Стадия 4. Синтез соединения по настоящему изобретению (I-a)
Связанное со сложным эфиром вещество (5), полученное на стадии 2, растворяют в соли органического амина, такой как триэтиламин, диизопропилэтиламин и диметиланилин и предпочтительно триэтиламин, и растворителе, который не воздействует на реакцию, таком как дихлорметан, ацетонитрил и диметилформамид. К полученному таким образом раствору добавляют по каплям раствор дихлорметана моноамида изофталевой кислоты 5-фторурацила-хлорангидрида (7), полученный на стадии 3 в условиях охлаждения льдом. В данном случае используют от 1,0 до 1,2 эквивалента моноамида изофталевой кислоты 5-фторурацила-хлорангидрида (7) и от 1,0 до 1,2 эквивалента соли органического амина относительно связанного со сложным эфиром вещества (5). После предоставления смеси возможности прореагировать при комнатной температуре в течение 1-4 часов к продукту реакции добавляют воду с последующей экстракцией с использованием дихлорметана или подобного соединения. Экстракт сушат над сульфатом натрия, сульфатом магния или тому подобным веществом, концентрируют до перекристаллизации, очищают колоночной хороматографией и затем доставляют на следующую стадию.
Стадия 5. Синтез соединения по настоящему изобретению (I-b)
Снятие защиты выполняют в соответствии со способом, описанным в руководстве Green «Protective Group in Organic Synthesis» («Защитная группа в органическом синтезе»).
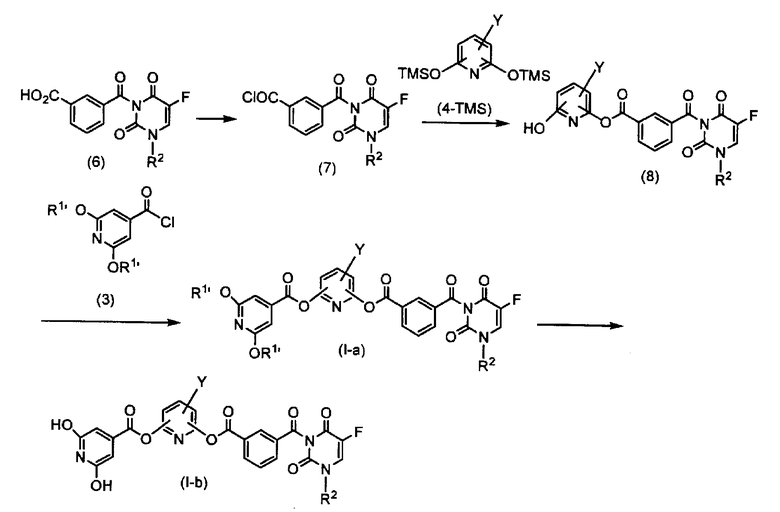
Схема 2
где группы R1', R2 и Y такие же, как определено выше.
Стадия 6. Синтез моноамида-моноэфира изофталевой кислоты (8)
Производное пиридина (4-TMS), которое было триметилсилилировано (TMS) в соответствии со способом, описанным в публикации Chem. Pharm. Bull. Vol. 41, No. 9, 1498-1506, 1993, растворяют в растворителе, который не воздействует на реакцию, таком как дихлорметан, ацетонитрил и диметилформамид и предпочтительно ацетонитрил. К полученному таким образом раствору добавляют по каплям раствор моноамида изофталевой кислоты 5-фторурацила-хлорангидрида (7) в условиях охлаждения льдом. В последующем к нему по каплям добавляют кислоту Льюиса, такую как хлорид четырехвалентного олова и тетрахлорид титана. В данном случае используют от 0,8 до 1,0 эквивалента моноамида изофталевой кислоты 5-фторурацила-хлорангидрида (7) и количество кислоты Льюиса относительно производного пиридина (4-TMS). После предоставления возможности смеси прореагировать при комнатной температуре в течение 1-4 часов к реакционной жидкости добавляют воду с последующей экстракцией с использованием дихлорметана или тому подобного соединения. Экстракт сушат над сульфатом натрия, сульфатом магния или тому подобным веществом, концентрируют до перекристаллизации, очищают колоночной хроматографией и затем доставляют на следующую стадию.
Стадия 7. Синтез соединения по настоящему изобретению (I-a)
Моноамид-моноэфир изофталевой кислоты (8) растворяют в смеси соли органического амина, такого как триэтиламин, диизопропилэтиламин и диметиланилин, предпочтительно триэтиламин, и растворителе, который не воздействует на реакцию, таком как дихлорметан, ацетонитрил и диметилформамид и предпочтительно дихлорметан. К полученной смеси по каплям добавляют раствор в дихлорметане производного изоникотиновой кислоты-хлорангидрида (3). В данном случае используют от 1,0 до 1,2 эквивалента производного изоникотиновой кислоты-хлорангидрида (3) и от 1,0 до 1,2 эквивалента соли органического амина относительно моноамида-моноэфира изофталевой кислоты (8). После представления возможности смеси прореагировать при комнатной температуре в течение 1-4 часов к реакционной жидкости добавляют воду с последующей экстракцией с использованием дихлорметана или тому подобного соединения. Экстракт сушат над сульфатом натрия, сульфатом магния или тому подобным веществом, концентрируют до перекристаллизации, очищают колоночной хроматографией и затем доставляют на стадию 5, показанную на схеме 1.
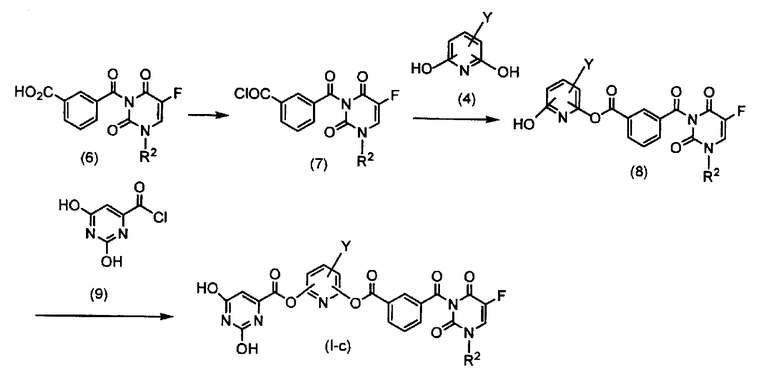
Схема 3
где R2 и Y представляют собой, как определено выше.
Синтез оротовой кислоты-хлорангидрида (9)
Тионилхлорид по каплям добавляют к оротовой кислоте при комнатной температуре вместе с растворителем, который не воздействует на реакцию, таким как хлороформ, 1,2-дихлорэтан и толуол, или без растворителя. В данном случае используют от 1 до 5 эквивалентов, предпочтительно 4 эквивалента относительно оротовой кислоты. После завершения добавления по каплям полученную смесь перемешивают в условиях кипячения в сосуде с обратным холодильником в течение 2-8 часов и предпочтительно в течение 4 часов. После завершения реакции, смесь концентрируют и остаток доставляют на следующую стадию без дополнительной обработки.
Стадия 8. Синтез соединения по настоящему изобретению (I-c)
Моноамид-моноэфир изофталевой кислоты (8) растворяют в смешанной жидкости из соли органического амина, такой как триэтиламин, диизопропилэтиламин и диметиланилин, предпочтительно триэтиламин, с растворителем, который не воздействует на реакцию, таким как дихлорметан, ацетонитрил и диметилформамид, и предпочтительно в смешанной жидкости триэтиламина с дихлорметаном. Раствор в дихлорметане оротовой кислоты-хлорангидрида (9) по каплям добавляют к полученному раствору при охлаждением льдом. В данном случае используют от 1,0 до 1,2 эквивалента оротовой кислоты-хлорангидрида (9) и от 1,0 до 1,2 эквивалента соли органического амина относительно моноамида-моноэфира изофталевой кислоты (8). После предоставления смеси возможности прореагировать при комнатной температуре в течение 1-4 часов к продукту реакции добавляют воду с последующей экстракцией с использованием дихлорметана или подобного соединения. Экстракт сушат над сульфатом натрия, сульфатом магния или подобным веществом, концентрируют до перекристаллизации и затем очищают колоночной хроматографией.
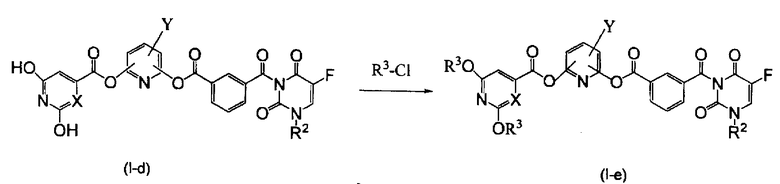
Схема 4
где X представляет атом углерода или атом азота, R3 представляет защитную группу гидроксигруппы, и R2 и Y являются такими же, как определено выше.
Стадия 9. Синтез соединения по настоящему изобретению (I-e), где R 1 представляет собой защитную группу гидроксигруппы
Производное пиридина (I-d), полученное по схемам 2 и 3, и хлорангидрид, представленный формулой R3-Cl, растворяют в растворителе, который не воздействует на реакцию, таком как диметоксиэтан, дихлорметан, ацетонитрил, диметилформамид и диметилацетамид и предпочтительно диметоксиэтан. К полученному раствору добавляют соль органического амина, такую как пиридин, триэтиламин, диизопропилэтиламин и диметиланилин, при охлаждении льдом. В данном случае используют от 2,2 до 4,0 эквивалентов хлорангидрида и от 2,2 до 4,0 эквивалентов соли органического амина относительно производного пиридина (I-d). После предоставления смеси возможности прореагировать в условиях охлаждения льдом в течение 0,5-4 часов, к реакционной жидкости добавляют воду с последующей экстракцией с использованием этилацетата, смешанного растворителя этилацетата и н-гексана, толуола или тому подобного соединения. Экстракт сушат над сульфатом натрия, сульфатом магния и т.д., концентрируют до перекристаллизации и затем очищают колоночной хроматографией и т.д.
Как описано выше, соединение (I) по настоящему изобретению и его соли проявляют превосходный противоопухолевый эффект при уменьшенных побочных эффектах, таких как повреждение желудочно-кишечного тракта; поэтому они могут применяться в качестве противоопухолевого средства. Соответственно, соединение (I) по настоящему изобретению и его соли эффективны при лечении злокачественных заболеваний. В настоящем изобретении способы лечения злокачественных заболеваний включают введение соединения (I) по настоящему изобретению или его соли для предотвращения рецидива злокачественного заболевания после его лечения лучевой терапией, хирургической операции и т.д.
Когда соединение (I) по настоящему изобретению или его соль применяется для лечения указанных выше заболеваний млекопитающих, включая людей, дозировка, число введений и тому подобные параметры должны варьироваться в зависимости от условий и тяжести подлежащего лечению заболевания и пути введения соединения (I) по настоящему изобретению. Дозировка, число введений и тому подобные параметры также варьируются в зависимости от возраста, массы тела, общего состояния здоровья, пола, рациона, времени введения, скорости выведения, сопутствующей медикаментозной терапии, реакции на лечение и тому подобных показателей. Соединение (I) по настоящему изобретению и его соли вводятся обычно перорально или парентерально. Дозировка в целом представляет собой терапевтически эффективное количество для лечения указанных выше заболеваний, т.е. от примерно 0,001 до 100 мг и предпочтительно от 0,01 до 50 мг/кг в день на кг массы тела млекопитающего, такого как человек. Однако дозировка вне указанного диапазона может применяться в зависимости от конкретного случая.
Путем смешивания эффективного количества соединения по настоящему изобретению с физиологически приемлемыми носителями соединение по настоящему изобретению может вводиться перорально или парентерально (например, наружным применением, ингаляцией, подкожной инъекцией, внутриартериальной и внутривенной инъекцией, внутримышечной инъекцией, инстилляцией в мочевой пузырь, интрацеребральной инстилляцией, промыванием носовой полости и в виде глазных капель) в виде твердых препаратов, таких как таблетка, капсула, гранула и порошок; жидких препаратов, таких как сироп и инъекционная лекарственная форма; и препаратов для наружного применения, таких как мазь, лосьон, гель и крем.
Различные обычные органические или неорганические носители, используемые в качестве препаратов для получения, используются в качестве фармакологически приемлемых носителей в настоящем изобретении. Их определенные примеры включают эксципиенты, смазывающие вещества, связывающие агенты и разрыхлители, используемые в твердых препаратах; и растворители, солюбилизирующие агенты, суспендирующие агенты, агенты, регулирующие осмоляльность, буферы и смягчающие агенты, используемые в жидких препаратах. При необходимости могут использоваться добавки к препаратам, такие как антисептики, антиоксиданты, красящие агенты и подсластители. Предпочтительные примеры эксципиентов включают лактозу, D-маннит, крахмал, кристаллическую целлюлозу и легкую безводную кремниевую кислоту. Предпочтительные примеры смазывающих веществ включают стеарат магния, стеарат кальция, тальк и коллоидный диоксид кремния. Предпочтительные примеры связывающих агентов включают кристаллическую целлюлозу, белый сахар, D-маннит, декстрин, гидроксипропилцеллюлозу, гидроксиметилцеллюлозу и поливинилпирролидон. Предпочтительные примеры разрыхлителей включают крахмал, карбоксиметилцеллюлозу, карбоксиметилцеллюлозу кальция и карбоксиметилкрахмал натрия. Предпочтительные примеры растворителей включают воду для инъекций, спирт, пропиленгликоль, макрогол, кунжутное масло и кукурузное масло. Предпочтительные примеры солюбилизаторов включают полиэтиленгликоль, пропиленгликоль, D-маннит, бензилбензоат, этанол, трисаминометан, холестерин, триэтаноламин, карбонат натрия и цитрат натрия. Предпочтительные примеры суспендирующих агентов включают поверхностно-активные вещества, такие как стеарилтриэтаноламин, лаурилсульфатнатрия, лауриламинопропионовая кислота, лецитин, бензалконий хлорид и глицеринмоностеарат; и гидрофильные полимеры, такие как поливинилалкоголь, поливинилпирролидон, карбоксиметилцеллюлоза натрия, метилцеллюлоза, гидроксиметилцеллюлоза, гидроксиэтилцеллюлоза и гидроксипропилцеллюлоза. Предпочтительные примеры буферов включают буферные растворы фосфата, ацетата, карбоната и цитрата. Предпочтительные примеры смягчающих агентов включают бензиловый спирт. Предпочтительные примеры антисептиков включают сложные эфиры парагидроксибензойной кислоты, хлорбутанол, бензиловый спирт, фенэтиловый спирт, дегидроуксусные кислоты и сорбиновые кислоты. Предпочтительные примеры антиоксидантов включают сульфиты и соли аскорбиновой кислоты.
ПРИМЕРЫ
Ниже настоящее изобретение конкретно описано со ссылкой на сравнительные примеры, примеры и примеры тестирования. Однако настоящее изобретение не ограничивается описанными конкретными вариантами осуществления.
Контрольный пример 1
Синтез 2,6-дибензилоксиизоникотиновой кислоты (Соединение 2a)
В атмосфере аргона 2,6-дихлоризоникотиновую кислоту (57,6 г) постепенно добавляли к 55% гидриду натрия (52,5 г) и диметилформамиду (1 л) при охлаждении и перемешивании. В последующем, к продукту реакции постепенно по каплям добавляли бензиловый спирт (93 мл) при той же температуре. После того как водород больше не образовывался, продукт реакции перемешивали в течение 4 часов при 80ºC, и затем к нему добавляли воду (1 л). Смесь разделяли смешанным растворителем (1 л) этилацетата и н-гексана (1:1). Кислотность водного слоя снижали уксусной кислотой (75,5 мл) и к ней далее добавляли воду (1,7 л). Осадок фильтровали и сушили для получения соединения 2a.
Выход: 77,17 г (77%).
1H-ЯМР (DMSOd6, δPPM): 13,60 (1H, ушир.с), 7,43-7,29 (10H, м) 6,81 (2H, с), 5,36 (4H, с).
Точка плавления: 145-147ºC.
Контрольный пример 2
Синтез 2,6-ди-п-метоксибензилоксиизоникотиновой кислоты (Соединение 2b)
Соединение 2b синтезировали в соответствии со способом контрольного примера 1 за исключением того, что вместо бензилового спирта использовали п-метоксибензиловый спирт.
1H-ЯМР (DMSOd6, δPPM): 13,56 (1H, ушир.с), 7,39 (4H, д, J=8,4 Гц), 6,95 (4H, д, J=8,6 Гц), 5,33 (4H, с), 3,77 (6H, с).
Контрольный пример 3
Синтез 2,6-диаллилоксиизоникотиновой кислоты (Соединение 2c)
Соединение 2c синтезировали в соответствии со способом контрольного примера 1 за исключением того, что вместо бензилового спирта использовали аллиловый спирт.
1H-ЯМР (DMSOd6, δPPM): 13,60 (1H, ушир.с), 6,77 (2H, с), 6,15-5,99 (2H, м), 5,38 (2H, дд, J=17,2 Гц, J=1,5 Гц), 5,23 (2H, д, J=10,4 Гц), 4,82 (4H, д, J=5,4 Гц).
Контрольный пример 4
Синтез 4-{2,6-дибензилоксиизоникотиноилокси}-5-хлор-2-гидроксипиридина (Соединение 5a), 2-{2,6- дибензилоксиизоникотиноилокси}-5-хлор-4-гидроксипиридина (Соединение 5b) и 2,4-ди{2,6-дибензилоксиизоникотиноилокси}-5-хлорпиридина (Соединение 5c)
Тионилхлорид (98,5 мл) и диметилформамид (1,7 мл) добавляли по каплям к раствору 2,6-дибензилоксиизоникотиновой кислоты (Соединение 2a) (75,45 г), полученной в контрольном примере 1 и толуоле (800 мл), и смесь перемешивали при 80ºC в течение 4,5 часов. После охлаждения растворители выпаривали. Без очистки остаточный хлорангидрид растворяли в диметилацетамиде (100 мл) для получения раствора хлорангидрида в диметилацетамиде, подлежащего использованию в следующей реакции. Раствор хлорангидрида в диметилацетамиде добавляли по каплям при охлаждении льдом к раствору 2,4-дигидрокси-5-хлорпиридина (31,9 г), триэтиламина (31,19 мл) и диметилацетамида (1,6 л). Полученную смесь перемешивали при комнатной температуре в течение 1 часа, и затем к ней добавляли воду (1,7 л). Смесь разделяли смешанным растворителем (1 л) из этилацетата и н-гексана (3:1). После сушки раствора сульфатом натрия растворитель выпаривали. Осадок фильтровали и сушили для получения Соединения 5a.
Выход: 44,8 г (45%).
Тем временем фильтрат концентрировали, и полученный остаток очищали хроматографией на колонке силикагеля (элюировали этилацетатом/н-гексаном (1:1)) для получения Соединения 5b (18,94 г) и Соединения 5c (4,74 г).
Соединение 5a
1H-ЯМР (DMSOd6, δPPM): 12,09 (1H, ушир.с), 7,91 (1H, с), 7,50-7,30 (10H, м), 6,99 (2H, с), 6,58 (1H, с), 5,42 (4H, с).
Точка плавления: 149-150ºC.
Соединение 5b
1H-ЯМР (DMSOd6, δPPM): 12,26 (1H, ушир.с), 8,27 (1H, с), 7,50-7,30 (10H, м), 6,98 (2H, с), 6,86 (1H, с), 5,42 (4H, с).
Точка плавления: 136-138°C (температура разрушения).
Соединение 5c
1H-ЯМР (DMSOd6, δPPM): 8,75 (1H, с), 7,79 (1H, с), 7,50-7,30 (20H, м), 7,05 (2H, с), 7,02 (2H, с), 5,42 (8H, с).
Контрольный пример 5
Синтез 2,6-дигидрокси-3-фторпиридина (Соединение 4a)
2,6-Дибензилокси-5-фторникотиновую кислоту (13,30 г) получали с использованием 2,6-дихлор-5-фторникотиновой кислоты (15,15 г) в качестве исходного соединения таким же способом, как в контрольном примере 1.
1H-ЯМР (DMSOd6, δPPM): 8,02 (1H, д, J=10,2 Гц), 7,49-7,28 (10H, м), 5,48 (2H, с), 5,45 (2H, с).
Затем 2,6-дибензилокси-5-фторникотиновую кислоту (5,00 г) растворяли в диоксане (50 мл) и к ней добавляли 20% гидроксид палладия (50% во влажном состоянии, 500 мг), и реакцию осуществляли в атмосфере водорода в течение 1 часа. Гидроксид палладия отфильтровывали через целит от полученного продукта реакции, и растворитель выпаривали для получения соединения, т.е. 2,6-дигидрокси-5-фторникотиновой кислоты (2,58 г). Выход: 88%.
1H-ЯМР (DMSOd6, δPPM): 7,44 (1H, д, J=11,4 Гц).
2,6-Дигидрокси-5-фторникотиновую кислоту (2,25 г) растворяли в диоксане (25 мл) и кипятили в сосуде с обратным холодильником в течение 15 минут. После охлаждения растворитель выпаривали для получения Соединения 4a (1,65 г). Выход: 99%.
1H-ЯМР (DMSOd6, δPPM): 7,44 (1H, д, J=11,4 Гц).
Контрольный пример 6
6-{2,6-дибензилоксиизоникотиноилокси}-3-фтор-2-гидроксипиридин (Соединение 5d)
Соединение 5d синтезировали в соответствии со способом контрольного примера 4 за исключением того, что использовали соединение 4a, полученное в контрольном примере 5, вместо 2,4-дигидрокси-5-хлорпиридина.
1H-ЯМР (DMSOd6, δPPM): 7,79 (1H, т, J=9,0 Гц), 7,47-7,30 (11H, м), 6,98 (2H, с), 5,42 (4H, с).
Контрольный пример 7
Синтез 3-{3-[4-гидрокси-5-хлор-2-пиридилоксикарбонил]бензоил}-1-этоксиметил-5-фторурацила (Соединение 8a) и 3-{3-[2-гидрокси-5-хлор-4-пиридилоксикарбонил]бензоил}-1-этоксиметил-5-фторурацила (Соединение 8b)
1-Этоксиметил-3-м-гидроксикарбонилбензоил-5-фторурацил (3,46 г) растворяли в метиленхлориде (50 мл). Затем к раствору добавляли тионилхлорид (0,9 мл) и диметилформамид (0,04 мл). Полученный продукт реакции кипятили в сосуде с обратным холодильником в течение 2 часов, и затем растворители выпаривали. Остаток растворяли в метиленхлориде (12 мл) для получения раствора хлорангидрида в метиленхлориде. После разжижения 2,4-дигидрокси-5-хлорпиридина (1,5 г) в гексаметилдисилазане (15 мл) в течение 6 часов растворитель выпаривали, и полученный остаток растворяли в метиленхлориде (30 мл). К нему по каплям добавляли указанный выше раствор хлорангидрида в метиленхлориде в условиях охлаждения льдом и в последующем к нему добавляли безводный хлорид четырехвалентного олова (0,15 мл). Смесь перемешивали при комнатной температуре в течение 15 часов. После нейтрализации триэтиламином остаток, полученный выпариванием растворителей, очищали хроматографией на колонке силикагеля (элюировали этилацетатом/н-гексаном (1:2)) для получения Соединения 8a (2,15 г; выход: 45%) и Соединения 8b (496 мг; выход: 10%).
Соединение 8a
1H-ЯМР (DMSOd6, δPPM): 8,63 (1H, т, J=1,6 Гц), 8,49 (2H, д, J=8,2 Гц), 8,45 (1H, д, J=6,7 Гц), 8,28 (1H, с), 7,86 (1H, т, J=7,8 Гц), 6,91 (1H, с), 5,11 (2H, с), 3,58 (2H, кв, J=6,9 Гц), 1,11 (3H, т, J=7,0 Гц).
Соединение 8b
1H-ЯМР (DMSOd6, δPPM): 8,65 (1H, т, J=3,3 Гц), 8,51 (2H, дт, J=1,7, 7,7 Гц), 8,46 (1H, д, J=6,6 Гц), 7,93 (1H, с), 7,89 (1H, т, J=7,8 Гц), 6,63 (1H, с), 5,11 (2H, с), 3,58 (2H, кв, J=7,0 Гц), 1,12 (3H, т, J=7,1 Гц).
Контрольный пример 8
Альтернативный синтез 3-{3-[4-гидрокси-5-хлор-2-пиридилоксикарбонил]бензоил}-1-этоксиметил-5-фторурацила (Соединение 8a)
1-Этоксиметил-3-м-гидроксикарбонилбензоил-5-фторурацил (3,46 г) растворяли в метиленхлориде (50 мл). Затем к нему добавляли тионилхлорид (0,9 мл) и диметилформамид (0,04 мл). Полученный продукт реакции кипятили в сосуде с обратным холодильником в течение 2,5 часов, и растворители затем выпаривали. Остаток растворяли в диметилацетамиде (12 мл) для получения раствора хлорангидрида в диацетамиде. Раствор хлорангидрида в диацетамиде по каплям добавляли в условиях охлаждения льдом к раствору 2,4-дигидрокси-5-хлорпиридина (1,5 г), триэтиламина (1,57 мл) и диметилацетамида (15 мл). Полученный продукт реакции перемешивали при комнатной температуре в течение 8 часов. К нему добавляли воду, и смесь разделяли смешанным растворителем этилацетата и н-гексана (1:1). Органический слой сушили сульфатом натрия, и растворитель затем выпаривали. Остаток очищали хроматографией на колонке силикагеля (элюировали этилацетатом/н-гексаном (1:1)) для получения Соединения 8a (668 мг). Выход: 14%.
Контрольный пример 9
Альтернативный синтез 2-{2,6-дибензилоксиизоникотиноилокси}-5-хлор-4-гидроксипиридина (Соединение 5b), 4-{2,6-дибензилоксиизоникотиноилокси}-5-хлор-2-гидроксипиридина (Соединение 5a) и 2,4-ди{2,6-дибензилоксиизоникотиноилокси}-5-хлорпиридина (Соединение 5c)
Хлорангидрид получали используя Соединение 2a (1 г), полученное в контрольном примере 1, в качестве исходного соединения, таким же способом, как способ, использованный в контрольном примере 8. Далее, как и в случае с контрольным примером 7, 2,4-дигидрокси-5-хлорпиридин (1,0 г) триметилсилилировали и затем проводили реакцию с хлорангидридом. После нейтрализации триэтиламином остаток, полученный выпариванием растворителем, очищали хроматографией на колонке силикагеля (элюировали этилацетатом/н-гексаном (1:1)) для получения Соединения 5b (1,38 г; выход: 44%), Соединения 5a (675 мг; выход: 21%) и Соединения 5c (647 мг; выход: 12%).
Пример 1
Синтез 3-{3-[4-(2,6-дибензилоксиизоникотиноилокси)-5-хлор-2-пиридилоксикарбонил]бензоил}-1-этоксиметил-5-фторурацила (Соединение I-1)
1-Этоксиметил-3-м-гидроксикарбонилбензоил-5-фторурацил (17 г) растворяли в метиленхлориде (200 мл). Затем к нему добавляли тионилхлорид (5,5 мл) и диметилформамид (0,4 мл). Полученный продукт реакции кипятили в сосуде с обратным холодильником в течение 2,5 часов, и затем растворители выпаривали. Остаток растворяли в метиленхлориде (60 мл) для получения раствора хлорангидрида в метиленхлориде. Этот раствор добавляли по каплям в условиях охлаждения льдом к раствору Соединения 5a (21,27 г), полученного в контрольном примере 4, триэтиламина (7,3 мл) и метиленхлорида (180 мл). Полученный продукт реакции перемешивали при комнатной температуре в течение 1 часа, и растворители затем выпаривали. Полученный остаток очищали хроматографией на колонке силикагеля (элюировали этилацетатом/н-гексаном (1:1)) для получения Соединения I-1 (25,5 г). Выход: 71%.
1H-ЯМР (DMSOd6, δPPM): 8,77 (1H, с), 8,67 (1H, т, J=1,6 Гц), 8,54-8,50 (2H, м), 8,45 (1H, д, J=6,6 Гц), 7,88 (1H, т, J=7,9 Гц), 7,83 (1H, с), 7,50-7,30 (10H, м), 7,06 (2H, с), 5,43 (4H, с), 5,11 (2H, с), 3,58 (2H, кв, J=7,0 Гц), 1,11 (3H, т, J=7,0 Гц).
Точка плавления: 66-69ºC.
Пример 2
Синтез 3-{3-[4-(2,6-дигидроксиизоникотиноилокси)-5-хлор-2-пиридилоксикарбонил]бензоил}-1-этоксиметил-5-фторурацила (Соединение I-2)
Соединение I-1 (25,3 г), полученное в примере 1, растворяли в диоксане (800 мл), и к нему добавляли 20% гидроксид палладия (50% влажности, 2,53 г), и реакцию проводили в атмосфере водорода в течение 1 часа. Гидроксид палладия отфильтровывали через целит от полученного продукта реакции и промывали ацетоном (200 мл). Фильтрат (т.е. раствор диоксана и ацетона) концентрировали, и полученный остаток кристаллизовали из ацетона-н-гексана (1:1) для получения Соединения I-2 (13,14 г). Выход: 68%.
1H-ЯМР (DMSOd6, δPPM): 11,57 (2H, ушир.с), 8,77 (1H, с), 8,67 (1H, т, J=1,6 Гц), 8,55-8,49 (2H, м), 8,45 (1H, д, J=6,6 Гц), 7,89 (1H, т, J=7,9 Гц), 7,83 (1H, с), 6,47 (2H, ушир.с), 5,12 (2H, с), 3,59 (2H, кв, J=7,0 Гц), 1,12 (3H, т, J=7,0 Гц).
Точка плавления: 125-129ºC.
Пример 3
Синтез 3-{3-[4-(2,6-дибензилоксиизоникотиноилокси)-5-хлор-4-пиридилоксикарбонил]бензоил}-1-этоксиметил-5-фторурацила (Соединение I-3)
Соединение I-3 синтезировали в соответствии со способом примера 1 за исключением того, что Соединение 5b, полученное в контрольном примере 4, использовали вместо соединения 5a.
1H-ЯМР (DMSOd6, δPPM): 8,77 (1H, с), 8,71 (1H, т, J=1,6 Гц), 8,58-8,53 (2H, м), 8,46 (1H, д, J=6,6 Гц), 7,91 (1H, т, J=7,9 Гц), 7,83 (1H, с), 7,50-7,30 (10H, м), 7,03 (2H, с), 5,43 (4H, с), 5,11 (2H, с), 3,58 (2H, кв, J=7,0 Гц), 1,12 (3H, т, J=7,0 Гц).
Пример 4
Синтез 3-{3-[2-(2,6-дигидроксиизоникотиноилокси)-5-хлор-4-пиридилоксикарбонил]бензоил}-1-этоксиметил-5-фторурацила (Соединение I-4)
Соединение I-4 синтезировали с использованием Соединения I-3, полученного в примере 3, в соответствии со способом примера 2.
1H-ЯМР (DMSOd6, δPPM): 8,77 (1H, с), 8,71 (1H, ушир.с), 8,57-8,53 (2H, м), 8,47 (1H, д, J=6,4 Гц), 7,92 (1H, т, J=7,9 Гц), 7,83 (1H, с), 6,41 (2H, ушир.с), 5,12 (2H, с), 3,59 (2H, кв, J=6,3 Гц), 1,12 (3H, т, J=7,1 Гц).
Пример 5
Синтез 3-{3-[6-(2,6-дибензилоксиизоникотиноилокси)-5-фтор-2-пиридилоксикарбонил]бензоил}-1-этоксиметил-5-фторурацила (Соединение I-5)
Соединение I-5 синтезировали в соответствии со способом примера 1 за исключением того, что соединение 5d, полученное в контрольном примере 6, использовали вместо соединения 5a.
1H-ЯМР (DMSOd6, δPPM): 8,71 (1H, т, J=1,5 Гц), 8,57-8,53 (м, 2H), 8,44 (1H, д, J=6,6 Гц), 8,33 (1H, т, J=8,6 Гц), 7,90 (1H, т, J=7,9 Гц), 7,62 (1H, дд, J=2,7 Гц, J=8,7 Гц), 7,46-7,32 (10H, м), 5,42 (4H, с), 5,12 (2H, с), 3,58 (2H, кв, J=7,0 Гц), 1,12 (3H, т, J=7,0 Гц).
Пример 6
Синтез 3-{3-[6-(2,6-гидроксиизоникотиноилокси)-3-фтор-2-пиридилоксикарбонил]бензоил}-1-этоксиметил-5-фторурацила (Соединение I-6)
Соединение I-6 синтезировали с использованием соединения I-5, полученного в примере 5, в соответствии со способом примера 2.
1H-ЯМР (DMSOd6, δPPM): 8,71 (1H, т, J=1,6 Гц), 8,58-8,53 (2H, м), 8,44 (1H, д, J=6,8 Гц), 8,32 (1H, т, J=8,6 Гц), 7,91 (1H, т, J=7,8 Гц), 7,60 (1H, дд, J=2,8 Гц, J=8,6 Гц), 6,41 (2H, ушир.с), 5,11 (2H, с), 3,58 (2H, кв, J=7,0 Гц), 1,11 (3H, т, J=7,0 Гц).
Пример 7
Синтез 3-{3-[4-(2,6-дибензилоксиизоникотиноилокси)-5-хлор-2-пиридилоксикарбонил]бензоил}-1-этоксиметил-5-фторурацила (Соединение I-1)
Тионилхлорид (0,23 мл) и диметилформамид (0,01 мл) по каплям добавляли к раствору соединения 2a (217 мг), полученному в контрольном примере 1, и толуолу (4 мл), и смесь перемешивали при 80ºC в течение 4,5 часов. После охлаждения полученной смеси растворители выпаривали. Без очистки остаточный хлорангидрид растворяли в диоксане (2 мл) для получения раствора хлорангидрида в диоксане, подлежащего использованию в следующей реакции. Раствор хлорангидрида в диоксане добавляли по каплям в условиях охлаждения льдом к раствору соединения 8a (100 мг), полученного в контрольном примере 7 или контрольном примере 8, триэтиламина (0,01 мл) и диоксана (10 мл). Полученную смесь перемешивали при комнатной температуре в течение 8 часов, и затем растворители выпаривали. Полученный остаток очищали хроматографией на колонке силикагеля (элюировали этилацетатом/н-гексаном (1:1)) для получения соединения I-1 (119 мг). Выход: 92%.
Пример 8
Синтез 3-{3-[2-(оротиноилокси)-5-хлор-4-пиридилоксикарбонил]бензоил}-1-этоксиметил-5-фторурацила (Соединение I-7)
Тионилхлорид (35 мл) и пиридин (0,17 мл) добавляли к оротовой кислоте (6 г) и смесь кипятили в сосуде с обратным холодильником в течение 20 часов. В последующем тионилхлорид выпаривали из полученного продукта реакции, и остаток получали в виде хлорангидрида оротовой кислоты без очистки. Полученный в результате хлорангидрид (0,59 г) и соединение 8b (0,78 г), синтезированное в контрольном примере 7, растворяли в диоксане (10 мл). Затем к указанному выше раствору добавляли раствор триэтиламина (0,46 мл) и диоксана (5 мл) и перемешивали при комнатной температуре в течение 30 минут. Нежелательные материалы удаляли фильтрацией из полученного в результате продукта реакции, и растворители затем выпаривали. Раствор разделяли этилацетатом и очищенной водой. Полученный в результате остаток сушили сульфатом магния, и затем растворитель выпаривали. Полученные в результате неочищенные кристаллы промывали смешанным растворителем (10 мл) н-гексана и этилацетата (3:1) для получения соединения I-7 (859 мг; выход: 85%).
1H-ЯМР (DMSOd6, δPPM): 11,53 (1H, с), 11,49 (1H, с), 8,78 (1H, с), 8,71 (1H, с), 8,55 (2H, д, J=7,9 Гц), 8,47 (1H, д, J=6,6 Гц), 7,92 (1H, т, J=7,9 Гц), 7,82 (1H, с), 6,35 (1H, с), 5,11 (2H, с), 3,58 (2H, кв, J=7,0 Гц), 1,12 (3H, т, J=7,0 Гц).
Пример 9
Синтез 3-{3-[4-(2,6-диизобутирилоксиизоникотиноилокси)-5-хлор-2-пиридилоксикарбонил]бензоил}-1-этоксиметил-5-фторурацила (Соединение I-8)
Соединение I-2 (5,0 г), полученное в примере 2, растворяли в 1,2-диметоксиэтане (75 мл). К нему добавляли изобутирилхлорид (3,07 мл) и пиридин (1,99 мл) в условиях охлаждения льдом, и проводили реакцию при той же температуре в течение 30 минут. Растворители выпаривали из полученного в результате продукта реакции. В последующем к продукту реакции добавляли воду, и смесь разделяли этилацетатом. Органический слой сушили сульфатом натрия, и затем растворитель выпаривали. Остаток очищали хроматографией на колонке силикагеля (элюировали хлороформом) для получения Соединения I-8 (5,37 г; выход: 87%).
1H-ЯМР (CDCl3, δPPM): 8,64 (1H, т, J=1,5 Гц), 8,56 (1H, с), 8,52 (1H, тд, J=7,9 Гц, J=1,5 Гц), 8,29 (1H, тд, J=8,0 Гц, J=1,5 Гц), 7,74 (2H, с), 7,73 (1H, т, J=7,7 Гц), 7,52 (1H, д, J=5,3 Гц), 7,34 (1H, с), 5,18 (2H, с), 3,64 (2H, кв, J=7,1 Гц), 2,89 (2H, гепт, J=6,9 Гц), 1,36 (12H, д, J=7,1 Гц), 1,21 (3H, т, J=6,3 Гц).
Пример 10
Синтез 3-{3-[4-(2,6-дициклопентанкарбонилоксиизоникотиноилокси)-5-хлор-2-пиридилоксикарбонил]бензоил}-1-этоксиметил-5-фторурацила (Соединение I-9)
Соединение I-9 получали таким же способом, как способ, использованный в примере 9, за исключением того, что циклопентанкарбонилхлорид использовали вместо изобутирилхлорида.
1H-ЯМР (CDCl3, δPPM): 8,64 (1H, т, J=1,5 Гц), 8,55 (1H, с), 8,52 (1H, тд, J=1,5 Гц, J=7,8 Гц), 8,29 (1H, тд, J=1,6 Гц, J=8,4 Гц), 7,74 (2H, с), 7,73 (1H, т, J=7,8 Гц), 7,51 (1H, д, J=5,1 Гц), 7,33 (1H, с), 5,18 (2H, с), 3,65 (2H, кв, J=7,1 Гц), 3,06 (2H, кв, J=8,1 Гц), 2,20-1,40 (16H, м), 1,24 (3H, т, J=7,1 Гц).
Пример 11
Синтез 3-{3-[4-(2,6-диацетилоксиизоникотиноилокси)-5-хлор-2-пиридилоксикарбонил]бензоил}-1-этоксиметил-5-фторурацила (Соединение I-10)
Соединение I-10 получали таким же способом, как способ, использованный в примере 9, за исключением того, что ацетилхлорид использовали вместо изобутирилхлорида.
1H-ЯМР (CDCl3, δPPM): 8,64 (1H, т, J=1,5 Гц), 8,56 (1H, с), 8,52 (1H, тд, J=8,0 Гц, J=1,4 Гц), 8,29 (1H, тд, J=8,0 Гц, J=1,6 Гц), 7,78 (2H, с), 7,73 (1H, т, J=7,7 Гц), 7,51 (1H, д, J=5,3 Гц), 7,35 (1H, с), 5,18 (2H, с), 3,64 (2H, кв, J=7,0 Гц), 2,38 (6H, с), 1,24 (3H, т, J=7,0 Гц).
Пример 12
Синтез 3-{3-[4-(2,6-дипропионилоксиизоникотиноилокси)-5-хлор-2-пиридилоксикарбонил]бензоил}-1-этоксиметил-5-фторурацила (Соединение I-11)
Соединение I-11 получали таким же способом, как способ, использованный в примере 9, за исключением того, что пропионилхлорид использовали вместо изобутирилхлорида.
1H-ЯМР (CDCl3, δPPM): 8,63 (1H, т, J=1,8 Гц), 8,55 (1H, с), 8,52 (1H, д, J=7,9 Гц), 8,29 (1H, д, J=7,9 Гц), 7,77 (2H, с), 7,73 (1H, т, J=7,8 Гц), 7,51 (1H, д, J=5,3 Гц), 7,35 (1H, с), 5,18 (2H, с), 3,64 (2H, кв, J=7,0 Гц), 2,69 (4H, кв, J=7,5 Гц), 1,32-1,21 (9H, м).
Пример 13
Синтез 3-{3-[4-(2,6-дипивалоилоксиизоникотиноилокси)-5-хлор-2-пиридилоксикарбонил]бензоил}-1-этоксиметил-5-фторурацила (Соединение I-12)
Соединение I-12 получали таким же способом, как способ, использованный в примере 9, за исключением того, что пивалоилхлорид использовали вместо изобутирилхлорида.
1H-ЯМР (CDCl3, δPPM): 8,64 (1H, т, J=1,5 Гц), 8,56 (1H, с), 8,52 (1H, дт, J=1,5 Гц, 7,7 Гц), 8,29 (1H, дт, J=1,6 Гц, J=7,9 Гц), 7,73 (1H, т, J=7,9 Гц), 7,69 (2H, с), 7,51 (1H, д, J=5,3 Гц), 7,33 (1H, с), 5,19 (2H, с), 3,65 (2H, кв, J=7,0 Гц), 1,41 (18H, с), 1,24 (3H, т, J=7,0 Гц).
Пример 14
Синтез 3-{3-[4-(2,6-дибензоилоксиизоникотиноилокси)-5-хлор-2-пиридилоксикарбонил]бензоил}-1-этоксиметил-5-фторурацила (Соединение I-13)
Соединение I-13 получали таким же способом, как способ, использованный в примере 9, за исключением того, что бензоилхлорид использовали вместо изобутирилхлорида.
1H-ЯМР (CDCl3, δPPM): 8,64 (1H, с), 8,56 (1H, с), 8,53 (1H, д, J=7,9 Гц), 8,30 (1H, д, J=8,2 Гц), 8,24 (5H, д, J=6,6 Гц), 7,91 (2H, с), 7,77―7,65 (3H, м), 7,56―7,39 (6H, м), 5,18 (2H, с), 3,64 (2H, кв, J=7,0 Гц), 1,24 (3H, т, J=7,0 Гц).
Пример 15
Синтез 3-{3-[4-(2,6-ди-п-хлорбензоилоксиизоникотиноилокси)-5-хлор-2-пиридилоксикарбонил]бензоил}-1-этоксиметил-5-фторурацила (Соединение I-14)
Соединение I-14 получали таким же способом, как способ, использованный в примере 9, за исключением того, что п-хлорбензоилхлорид использовали вместо изобутирилхлорида.
1H-ЯМР (CDCl3, δPPM): 8,64 (1H, с), 8,57 (1H, с), 8,52 (1H, д, J=7,9 Гц), 8,29 (1H, д, J=7,9 Гц), 8,18 (4H, д, J=8,6 Гц), 8,03 (1H, д, J=8,4 Гц), 7,96 (2H, с), 7,74 (1H, т, J=7,8 Гц), 7,65―7,50 (4H, м), 7,38 (1H, с), 5,18 (2H, с), 3,64 (2H, кв, J=7,0 Гц), 1,24 (3H, т, J=7,0 Гц).
Пример тестирования 1
Действие, ингибирующее DPD, и действие, ингибирующие OPRT, in vitro
(a) Тестируемый жидкий препарат (1): Соединение I-2 по настоящему изобретению растворяли до концентрации 10 мМ в ацетонитриле. Полученный в результате продукт разбавляли до величин концентрации 1 мМ, 100 мкМ, 10 мкМ и 1 мкМ буферным раствором с 10 мкМ фосфата калия (pH 6,0), посредством этого получая тестируемую жидкость 1. Полученные тестируемые жидкости 1 отдельно добавляли к описанному выше раствору для ферментной реакции с получением величин конечной концентрации 200 мкМ, 20 мкМ, 2 мкМ и 0,2 мкМ.
(b) Тестируемый жидкий препарат (2): В качестве ингибитора DPD 5-хлор-2,4-дигидроксипиридин (CDHP; гимерацил) растворяли до концентрации 10 мМ в буферном растворе с 10 мМ фосфата калия (pH 6,0). Полученный в результате продукт разбавляли до величин концентрации 1 мМ, 100 мкМ, 10 мкМ и 1 мкМ, посредством этого получая тестируемые жидкости 2. Полученные тестируемые жидкости 2 отдельно добавляли к раствору для ферментной реакции с получением величин конечной концентрации 200 мкМ, 20 мкМ, 2 мкМ и 0,2 мкМ.
(c) Тестируемый жидкий препарат (3): В качестве ингибитора OPRT цитразиновую кислоту (Citra. A.) растворяли до концентрации 10 мМ в 20 мМ трисгидроксиаминометан-хлористоводородном буферном растворе (pH 8,0). Затем полученный в результате продукт разбавляли до величин концентрации 1 мМ, 100 мкМ и 10 мкМ буферным раствором с 10 мМ фосфата калия, посредством этого получая тестируемые жидкости (3). Полученные тестируемые жидкости 3 отдельно добавляли к раствору для ферментной реакции с получением величин конечной концентрации 200 мкМ, 20 мкМ и 2 мкМ.
(d) Получение ферментного раствора: Печень 8-недельной крысы SD использовали в качестве источника фермента DPD, в то время как человеческие опухолевые клетки, которые были трансплантированы «голым» мышам и пролиферировали в них, использовали в качестве источника фермента OPRT. В частности, сразу после экстракции клеток печени крыс или последовательно трансплантированных человеческих опухолевых клеток к ним добавляли буферный раствор с 50 мМ трис-хлористоводородной кислоты (pH 8,0), содержащий 0,25 M сахарозы, 5 мМ хлорида магния и 1 мМ дитреитола до концентрации 25% (масс./об.), и гомогенизировали. Затем выполняли центрифугирование при 105000 g в течение 60 минут, и полученный супернатант использовали в качестве раствора фермента DPD или раствора OPRT.
(e) Ферментная реакция: Реакцию фермента DPD проводили в соответствии со способом Tatsumi et al. (Gann, 78: 748-755 (1987)), используя 5-FU, меченный тритием, в качестве субстрата. Реакцию фермента OPRT проводили в соответствии со способом Shirasaka et al. (Cancer Res., 53: 4004-4009 (1993)), используя 5-FU, меченный тритием, в качестве субстрата. Затем измеряли активность в контрольной группе (без тестируемого соединения) и в группе тестируемого соединения. Ингибирование в процентах тестируемым соединением относительно DPD или OPRT рассчитывали используя следующую формулу: [1-(активность фермента с тестируемым соединением/активность фермента контрольной группы)]×100 (%). Результаты показаны в таблицах 1 и 2.
средство
(f) Результаты тестирования: Соединения I-2, I-9 и I-10 по настоящему изобретению ингибировали активность DPD и активность OPRT, вызванную на 5-FU в качестве субстрата, в системе ферментной реакции in vitro. Ингибирующая активность соединений по настоящему изобретению была почти равной активности гимерацила или цитразиновой кислоты, которые служили в качестве контроля. Хотя гимерацил или цитразиновая кислота не обладают противоопухолевой активностью, они обладают активностью ингибирования OPRT. Это подтвердило, что противоопухолевые средства по настоящему изобретению обладают высокой активностью ингибирования DPD и высокой активностью ингибирования OPRT.
Пример тестирования 2
Ингибирующий эффект соединения I-2 на активацию (фосфорилирование) 5-FU в раковых клетках
(a) Тестируемый жидкий препарат 1: Соединение I-2 или I-10 по настоящему изобретению растворяли до концентрации 1 мМ в холодном физиологическом солевом растворе. Полученный продукт дополнительно разбавляли в 10 раз физиологическим солевым раствором, получая раствор с концентрацией 100 мкМ.
(b) Тестируемый жидкий препарат 2: В качестве ингибитора OPRT, который ингибирует фосфорилирование 5-FU, цитразиновую кислоту (Citra.A.) разбавляли до концентрации 1 мМ в холодном физиологическом солевом растворе и дополнительно разбавляли в 10 раз физиологическим солевым раствором, получая раствор с концентрацией 100 мкМ.
(c) Получение суспензии раковых клеток: Клетки саркомы асцитического типа 180 предварительно трансплантировали внутрибрюшинно мышам ICR, у которых они пролиферировали, для использования в тесте в качестве необработанных (интактных) клеток. Пролиферировавшие клетки выделяли и затем промывали физиологическим солевым раствором. Осажденные центрифугированием клеточные массы собирали, суспендировали в концентрации 1,25×107 клеток/мл в среде Хэнкса и сразу использовали в тесте.
(d) Эксперимент по ингибированию внутриклеточного фосфорилирования 5-FU: 0,1 мл каждой тестируемой жидкости и 0,1 мл раствора, содержащего 10 мкМ 5-FU, добавляли к 0,8 мл клеток саркомы 180 во льду и инкубировали при 37ºC в течение 30 минут. Сразу после завершения реакции к реакционному раствору добавляли 4 мл холодного раствора Хэнкса, и клетки промывали и отделяли центрифугированием. После двукратного повторения этой процедуры к клеточному осадку после центрифугирования добавляли 2 мл холодной 5% трихлоруксусной кислоты (TCA) для разрушения клеток и экстрагировали 5-FU и его нуклеотидные метаболиты. В последующем часть экстракта нуклеиновых кислот наносили на платину силикагеля 60F254 и проявляли смешанной жидкостью, содержащей хлороформ-метанол-уксусную кислоту (17:3:1) для выделения нуклеотидной части. Измеряли ее радиоактивность и измеряли концентрацию фосфорилированного 5-FU. В описанных выше условиях рассчитывали процентное ингибирование тестируемых жидкостей 1 и 2 на фосфорилирование 5-FU относительно контрольной группы (без тестируемой жидкости). В таблице 3 показаны результаты.
(e) Результаты тестирования: Соединения I-2 и I-10 по настоящему изобретению зависимым от концентрации образом ингибировали внутриклеточное фосфорилирование 5-FU во время реакции в течение 30 минут; в концентрации 10 мкМ процентное ингибирование составляло примерно от 20 до 30%, и в концентрации 100 мкМ процентное ингибирование составляло от 50 до 60%. Напротив, цитразиновая кислота, которая служила в качестве контроля, почти не проявляла ингибирования внутриклеточного фосфорилирования 5-FU посредством реакции в течение 30 минут; даже в концентрации 100 мкМ процентное ингибирование составило только от 5 до 15%. Описанные выше результаты подтверждают, что соединения по настоящему изобретению после захвата клетками ингибируют фосфорилирование 5-FU, вызванное OPRT, дозозависимым образом.
Пример тестирования 3
Противоопухолевые эффекты и побочные эффекты у мышей с трансплантированными человеческими опухолевыми клетками
(a) Тестируемый жидкий препарат 1: Соединение I-2 по настоящему изобретению суспендировали в концентрации 1,5, 2,25, или 3,0 мг/мл в 0,5% (масс./об.) растворе гидроксипропилметилцеллюлозы (далее именуемой «HPMC») и перемешивали мешалкой при комнатной температуре в течение примерно 10 минут. Затем ультразвуковую обработку выполняли в условиях охлаждения льдом в течение примерно 5 минут, посредством этого получая жидкое лекарственное средство, Соединение I-2, для применения в дозе 15 мг/кг/день, 22,5 мг/кг/день или 30 мг/кг/день.
(b) Тестируемый жидкий препарат 2: Соединение I-10 по настоящему изобретению суспендировали в концентрации 2,57, 3,42 или 42,8 мг/мл в 0,5% (масс./об.) растворе HPMC и перемешивали мешалкой при комнатной температуре в течение примерно 10 минут. Затем выполняли ультразвуковую обработку в условиях охлаждения льдом в течение 5 минут, посредством этого получая жидкое лекарственное средство, Соединение I-10, подлежащее применению в дозе 25,7 мг/кг/день, 34,2 мг/кг/день или 42,8 мг/кг/день.
(c) Тестируемый жидкий препарат 3: Среди соединений по настоящему изобретению тестируемое вещество (Соединение 8a), которое не содержит часть цитразиновой кислоты, суспендировали в концентрации 1,15, 1,73 или 2,3 мг/мл в 0,5% (масс./об.) растворе HPMC и перемешивали мешалкой при комнатной температуре в течение примерно 10 минут. Затем выполняли ультразвуковую обработку в условиях охлаждения льдом в течение примерно 5 минут, посредством этого получая жидкое лекарственное средство, Соединение 8а, подлежащее применению в дозе 11,5 мг/кг/день, 17,3 мг/кг/день или 23 мг/кг/день.
(d) Тестируемый жидкий препарат 4: S-1 (патент № 2614164), который представляет собой комбинированное средство, содержащее тегафур-гимерацил-отерацил калия, в молярном соотношении 1:0,4:1, суспендировали в 0,5% растворе (масс./об.) HPMC так, что количество тегафура составляло 0,5, 0,75 или 1,0 мг/мл. Полученный продукт перемешивали мешалкой при комнатной температуре в течение примерно 10 минут. Затем выполняли обработку ультразвуком в условиях охлаждения льдом в течение примерно 5 минут, посредством этого получая жидкость лекарственного средства S-1, подлежащую применению в дозе 5,0 мг/кг/день, 7,5 мг/кг/день или 10 мг/кг/день.
(e) Тестируемый жидкий препарат 5: Тестируемое вещество BOF-A2 суспендировали в концентрации 1,4, 2,1 или 2,8 мг/мл в 0,5% (масс./об.) растворе HPMC и перемешивали мешалкой при комнатной температуре в течение примерно 10 минут. Затем выполняли обработку ультразвуком в условиях охлаждения льдом в течение примерно 5 минут, посредством этого получая жидкость лекарственного средства BOF-A2, подлежащую применению в дозе 14 мг/кг/день, 21 мг/кг/день или 28 мг/кг/день.
(f) Эксперимент: Клетки линии клеток рака желудка человека (SC-2) подкожно трансплантировали в спину мышей BALB/cA-nu, где они предварительно пролиферировали. Опухолевое образование из пролиферированных клеток экстрагировали, разрезали на квадратные кусочки размером примерно 2 мм ножницами в физиологическом солевом растворе и подкожно инокулировали в правую подмышечную область мышей той же линии в возрасте 5-6 недель, используя трансплантационную иглу. Подготовленных таким образом мышей держали для адаптации, по меньшей мере, в течение 1-2 недель и делили на контрольную группу и группы тестируемых лекарственных средств в трех различных дозах (3 группы для Соединения I-2; 3 группы для Соединения I-10; 3 группы для Соединения 8a; 3 группы для Соединения S-1 и 3 группы для Соединения BOF-A3) с тем, чтобы средний объем опухолей и среднее стандартное отклонение (S. D.) были насколько возможно равны между группами (5-6 мышей на группу) (день 0). Затем начинали введение лекарственного средства со следующего дня. Каждую из тестируемых жидкостей, показанных выше в пунктах с (a) по (e), вводили перорально мышам в каждой из групп тестируемых лекарственных средств с использованием зонда для перорального введения в дозе 0,1 мл на 10 г массы тела один раз в день в течение 14 последовательных дней. Таким же образом, как указано выше, имеющим раковую опухоль мышам в контрольной группе перорально вводили только жидкость с 0,5% HPMC в течение 14 последовательных дней.
Объем опухоли у каждой мыши в каждой группе перед началом лечения, на 3, 5, 8 (через 1 неделю), 11 и 15-й день (через 2 недели), т.е. после окончания введения, рассчитывали используя указанное ниже уравнение 1 и рассчитывали соответствующий относительный объем опухоли (RTV) относительно объема опухоли во время начала лечения. В отношении противоопухолевого эффекта среднюю величину степени ингибирования пролиферации опухоли (IR; %) рассчитывали по среднему объему опухоли каждой группы лечения на 15-й день (после окончания лечения) относительно среднего объема опухоли в контрольной группе на 15-й день с использованием уравнения 2. Кроме того, частоту случаев диареи и смерти наблюдали в течение всего периода лечения длительностью 15 дней. Число случаев показано в таблицах. Наряду с ними, изменение массы тела рассчитывали по массе тела мышей во время окончания введения лекарственного средства относительно массы тела мышей во время начала введения с использованием уравнения 3. Результаты показаны в таблице 4.
Уравнение 1:
Объем опухоли (мм3) = (большая ось) × (малая ось)2 × 1/2
Уравнение 2:
Коэффициент ингибирования пролиферации опухоли (IR, %) = [1 - (средний объем опухоли в группе лечения)/(средний объем опухоли в контрольной группе)] × 100
Уравнение 3:
Средняя степень изменения массы тела (%) = [(средняя масса тела на 15-й день)-(средняя масса тела на 1-й день)]/(средняя масса тела на 1-й день) × 100
Результаты тестирования: Соединения I-2 и I-10 по настоящему изобретению проявили высокую противоопухолевую эффективность дозозависимым образом. Кроме того, значимая потеря массы тела у мышей не наблюдалась, и диарея или случаи смерти в результате токсического действия также не наблюдались. Это подтверждает, что соединения I-2 и I-10 по настоящему изобретению проявляют высокую противоопухолевую эффективность при сниженной токсичности. В отличие от этого противоопухолевый эффект Соединения 8a, которое аналогично соединениям по настоящему изобретению, но не содержит цитразиновую кислоту, увеличивался дозозависимым образом в диапазоне дозировок, использованных в этом тесте, но у мышей наблюдалась значительная потеря массы тела, диарея и случаи смерти. BOF-A2 проявил противоопухолевый эффект и токсичность, аналогичные таковым Соединения 8a, которое подобно соединениям по настоящему изобретению, но не содержит цитразиновую кислоту. В частности, BOF-A2 в дозе, которая проявляет высокую противоопухолевую эффективность, вызывал у мышей значительную потерю массы тела, диарею и смерть в результате токсического действия. S-1, в котором гимерацил (аингибитор DPD) и отерацил калия (средство, которое снижает токсическое действие на желудочно-кишечный тракт) добавляются к тегафуру (производному 5-FU), проявлял противоопухолевый эффект дозозависимым образом и не вызывал у мышей значительную потерю массы тела или диарею при введении в дозах меньше, чем 12,5 мг/кг/день, которые считаются передозировкой.
Как понятно из сказанного выше, соединения по настоящему изобретению обладают превосходным противоопухолевым эффектом и эффектом уменьшения побочных воздействий. Такие эффекты почти равны эффектам S-1, полезность которого широко признана в области противоопухолевых средств.
В отличие от S-1, который представляет собой комбинированное лекарственное средство, содержащее три агента, соединение по настоящему изобретению представлено в форме одного соединения. Поэтому ожидается, что вариабельность фармакокинетики метаболита среди пациентов будет небольшой. В частности, в случае, когда применяется комбинация трех агентов, которая нацелена на увеличение противоопухолевого эффекта 5-FU при одновременном облегчении побочных эффектов, в частности, токсического действия на желудочно-кишечный тракт, то каждый компонент в целом независимо абсорбируется и распределяется, что вызывает варьирование фармакокинетики среди пациентов. Это варьирование вызывает изменения концентрации 5-FU, возможно, приводя к ситуации, когда не может установиться благоприятный баланс между противоопухолевыми эффектами и токсичностью. Напротив, лекарственное средство в форме одного соединения, которое предназначено для достижения указанных выше целей, абсорбируется через ткань желудочно-кишечного тракта, после чего оно быстро активируется в организме и выполнит свои функции (высвобождение 5-FU из замаскированной формы, ингибирование DPD, подавление желудочно-кишечных расстройств). Поэтому предполагается, что различие варьирования фармакокинетики метаболизма активных метаболитов in vivo среди пациентов меньше, чем у комбинированного лекарственного средства.
| название | год | авторы | номер документа |
|---|---|---|---|
| УСИЛИТЕЛЬ ДЕЙСТВИЯ ПРОТИВООПУХОЛЕВОГО СРЕДСТВА | 2010 |
|
RU2548913C2 |
| НОВЫЕ АМИНОПИРИДИНОВЫЕ ИЛИ АМИНОПИРАЗИНОВЫЕ ПРОИЗВОДНЫЕ С СЕЛЕКТИВНОЙ ИНГИБИРУЮЩЕЙ АКТИВНОСТЬЮ В ОТНОШЕНИИ АВРОРЫ А | 2007 |
|
RU2437880C2 |
| ПРОИЗВОДНЫЕ ДИГИДРОПИРАЗОЛОПИРИМИДИНОНА | 2007 |
|
RU2437885C2 |
| КОМБИНАЦИЯ, СОДЕРЖАЩАЯ ПИРИМИДИНОВОЕ СОЕДИНЕНИЕ, ДЛЯ ПРИМЕНЕНИЯ В ЛЕЧЕНИИ ОПУХОЛЕЙ | 2021 |
|
RU2827100C1 |
| КОНДЕНСИРОВАННЫЕ ПРОИЗВОДНЫЕ ИНДАНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ ПРОТИВООПУХОЛЕВЫМ ДЕЙСТВИЕМ | 1995 |
|
RU2124017C1 |
| АНАЛОГ 3-ФЕНИЛЦИННОЛИНА И ПРОТИВООПУХОЛЕВОЕ СРЕДСТВО НА ЕГО ОСНОВЕ | 2003 |
|
RU2324683C2 |
| ПРОИЗВОДНЫЕ 2,4-ОКСАЗОЛИДИНДИОНА ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМАЯ СОЛЬ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ЛЕКАРСТВЕННАЯ КОМПОЗИЦИЯ, ОКАЗЫВАЮЩАЯ ГИПОГЛИКЕМИЧЕСКОЕ И ГИПОЛИПИДЕМИЧЕСКОЕ ДЕЙСТВИЕ, СПОСОБ СНИЖЕНИЯ СОДЕРЖАНИЯ САХАРА И ЛИПИДОВ В КРОВИ ПРИ ЛЕЧЕНИИ МЛЕКОПИТАЮЩИХ, СТРАДАЮЩИХ ДИАБЕТОМ ИЛИ ГИПЕРЛИПИДИМИЕЙ | 1994 |
|
RU2126797C1 |
| НОВЫЕ ПРОИЗВОДНЫЕ АМИНОПИРИМИДИНА В КАЧЕСТВЕ ИНГИБИТОРОВ PLK1 | 2007 |
|
RU2458062C2 |
| ПРОИЗВОДНЫЕ КАМПТОТЕЦИНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 1996 |
|
RU2138503C1 |
| ТИЕНОДИАЗЕПИНОВЫЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ | 2018 |
|
RU2795005C2 |
Изобретение относится к новым производным 5-фторурацила общей формулы (I) или их фармацевтически приемлемым солям. Соединения обладают противоопухолевым действием, сбалансированным с уровнем токсичности, и могут быть использованы в качестве активного ингредиента для получения лекарственного средства для лечения злокачественных заболеваний. В общей формуле (I):
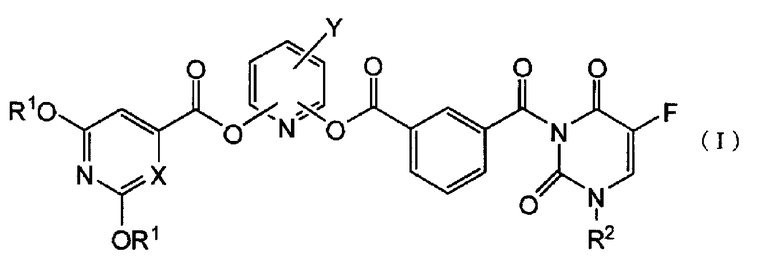
R1 представляет атом водорода или защитную группу гидроксигруппы, которую выбирают из C1-C6алифатической ацильной группы; C5-C6алициклической ацильной группы; ароматической ацильной группы, которую выбирают из бензоильной группы или галогенбензоильной группы, R2 представляет группу низшего алкоксинизшего алкила; Х представляет СН или атом азота и Y представляет атом галогена. 5 н. и 6 з.п. ф-лы, 4 cх., 4 табл., 24 пр.
1. Производное 5-фторурацила, представленное формулой (I), или его фармацевтически приемлемая соль:
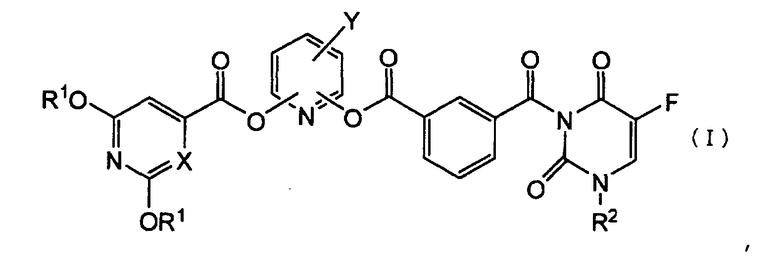
где R1 представляет атом водорода или защитную группу гидроксигруппы, которую выбирают из C1-C6алифатической ацильной группы; C5-C6 алициклической ацильной группы; ароматической ацильной группы, которую выбирают из бензоильной группы или галогенбензоильной группы,
R2 представляет группу низшего алкоксинизшего алкила;
Х представляет СН или атом азота, и
Y представляет атом галогена.
2. Производное 5-фторурацила или его фармацевтически приемлемая соль по п.1, где группа, представленная следующей формулой в формуле (I), представляет собой:
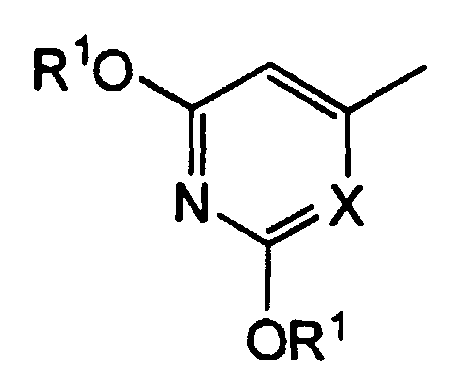
группу, представленную формулой:
где R1 представляет атом водорода,
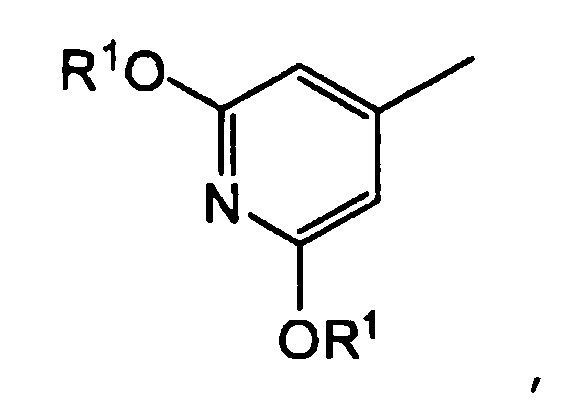
группу, представленную формулой:
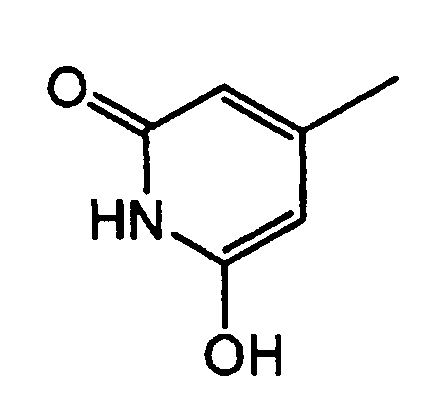
группу, представленную формулой:
где R1 представляет атом водорода или
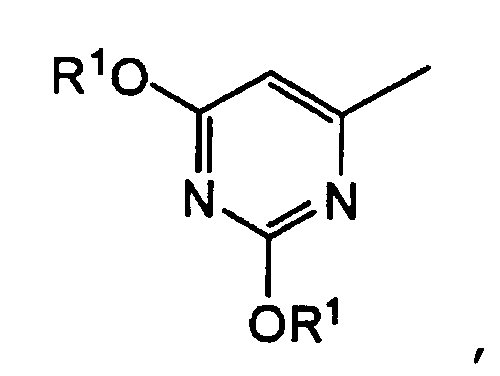
группу, представленную формулой:
3. Производное 5-фторурацила или его фармацевтически приемлемая соль по п.1, где R1 представляет атом водорода, C1-С6алифатическую ацильную группу, ароматическую ацильную группу, которую выбирают из бензоильной группы или галогенбензоильной группы, или С5-С6алициклическую ацильную группу, R2 представляет низшую алкоксиметильную группу, в которой часть низшего алкокси имеет от 1 до 6 атомов углерода, Х представляет СН или атом азота, и Y представляет атом фтора или атом хлора.
4. Производное 5-фторурацила или его фармацевтически приемлемая соль по п.1, где R1 представляет атом водорода, ацетильную группу, пропионильную группу, изобутирильную группу, пивалоильную группу, бензоильную группу, п-хлорбензоильную или циклопентанкарбонильную группу, R2 представляет низшую алкоксиметильную группу, в которой часть низшего алкокси имеет от 1 до 6 атомов углерода, Х представляет СH или атом азота, и Y представляет атом фтора или атом хлора.
5. Производное 5-фторурацила или его фармацевтически приемлемая соль по п.1, где R1 представляет атом водорода или ацетильную группу, R2 представляет низшую алкоксиметильную группу, в которой часть низшего алкокси имеет от 1 до 6 атомов углерода, Х представляет СН или атом азота, и Y представляет атом хлора.
6. Производное 5-фторурацила или его фармацевтически приемлемая соль по п.1, где R1 представляет атом водорода или ацетильную группу, R2 представляет низшую алкоксиметильную группу, в которой часть низшего алкокси имеет от 1 до 6 атомов углерода, Х представляет СН, и Y представляет атом хлора.
7. Производное 5-фторурацила или его фармацевтически приемлемая соль по п.1, где R1 представляет атом водорода или ацетильную группу, R2 представляет этоксиметильную группу, Х представляет СН, и Y представляет атом хлора.
8. Лекарственное средство, обладающее противоопухолевым действием, содержащее эффективное количество производного 5-фторурацила или его фармацевтически приемлемой соли по п.1 в качестве активного ингредиента.
9. Противоопухолевое средство, представляющее собой производное 5-фторурацила или его фармацевтически приемлемую соль по п.1 в эффективном количестве в качестве активного ингредиента.
10. Способ лечения злокачественного заболевания, включающий введение эффективного количества производного 5-фторурацила или его фармацевтически приемлемой соли по п.1 пациенту, страдающему злокачественным заболеванием.
11. Применение производного 5-фторурацила или его фармацевтически приемлемой соли по п.1 для получения противоопухолевого средства.
| CH 671578 A5, 15.09.1989 | |||
| СПОСОБ ЛЕЧЕНИЯ ОТГРАНИЧЕННЫХ ГНОЙНО-ВОСПАЛИТЕЛЬНЫХ ЗАБОЛЕВАНИЙ ЛЕГКИХ И ПЛЕВРЫ СТАФИЛОКОККОВОЙ ЭТИОЛОГИИ | 2000 |
|
RU2192880C2 |
| US 5047581 A1, 10.09.1991 | |||
| Способ получения 1-ацил-5-фторурацила | 1976 |
|
SU574155A3 |
| СПОСОБ ЛЕЧЕНИЯ ЭКСПЕРИМЕНТАЛЬНОГО ТУБЕРКУЛЕЗА | 2000 |
|
RU2165761C1 |

