Данное изобретение касается терапии аллергических, аутоиммунных, онкологических и прочих заболеваний с помощью новых семейств химических соединений, обладающих повышенной эффективностью в ингибировании SYK-киназы и ее мутантных форм, а также других терапевтически значимых киназ, повышенной селективностью и биодоступностью.
Уровень техники
Протеинкиназы являются важным семейством белков, участвующим в регуляции ключевых клеточных процессов, нарушение активности которых может приводить к онкологическим заболеваниям, аутоимунным заболеваниям, хроническим воспалительным заболеваниям, заболеваниям, связанным с разрушением тканей и т.д. Список киназ, терапевтическая значимость которых к настоящему времени имеет доклиническую или клиническую валидацию, включает: ABL1, ALK, АКТ, АКТ2, AURKA, BRAF, BCR-ABL, BLK, BRK, C-KIT, С-МЕТ, С-SRC, САМК2 В, CDK1, CDK2, CDK3, CDK4, CDK5, CDK6, CDK7, CDK8, CDK9, CRAF1, СНЕК1, СНЕК2, CLK1, CLK3, CSF1R, CSK, CSNK1G2, CSNK1G3, CSNK2A1, DAPK1, DAPK2, DAPK3, EGFR, ЕРНА2, ЕРНА3, ЕРНА5, ERBB2, ERBB3, ERBB4, ERK, ERK2, ERK3, FES, FGFR1, FGFR2, FGFR3, FGFR4, FGFR5, FGR, FLT-1, FYN, GSK3B, НСК, IGF1R, INSR, ITK, JAK1, JAK2, JAK3, JNK1, JNK2, JNK3, KIT, LCK, LOK, МАР3К5, МАРКАРК2, MARK1, МЕК1, МЕК2, MET, MKNK2, MST1, NEK2, Р38α, Р38δ, Р38γ, РАК1, РАК4, РАК6, РАК7, PDPK1, PDGFR, PIK3CG, PIM1, PIM2, РКС, PLK1, PLK4, PRKCQ, PRKR, РТК2, РТК2В, RET, ROCK1, ROS, RPS6KA1, SLK, SRC, SRPK1, STK16, SYK, TAK1, TGFBR1, TIE, TIE2, TNK2, TRK, VEGFR2, WEE1, ZAP70 (Vieth, M. et. al., Drug Discov Today, 2005, 10, 839-46; Fedorov, O. et. al., Nat Chem Biol, 2010, 6, 166-169; Gaestel, M. et. al., Nat Rev Drug Discov, 2009, 8, 480-99; Karaman, M. W. et. al., Nat Biotechnol, 2008, 26, 127-32; Fabian, M.A. et. al., Nat Biotechnol, 2005, 23, 329-36; Bhagwat, S. S., Purinergic Signal, 2009, 5, 107-15; Grimminger, F. et. al., Nat Rev Drug Discov, 2010, 9, 956-70). С появлением новых экспериментальных данных этот список постоянно растет.
Перспективным подходом для терапии заболеваний, ассоциированных с нарушенной активностью протеинкиназ, является применение низкомолекулярных химических соединений для ингибирования их активности. Примерами таких ингибиторов, одобренных для применения в клинической практике, являются: Иматиниб (Imatinib), Нилотиниб (Nilotinib), Дазатитниб (Dasatinib), Сунитиниб (Sunitinib), Сорафениб (Sorafenib), Лапатиниб (Lapatinib), Гефитиниб (Gefitinib), Эрлотиниб (Erlotinib), Кризотиниб (Crizotinib). Большое количество лекарственных кандидатов, ингибиторов киназ, находятся в настоящее время в клинических испытаниях и в предклинической разработке.
SYK-киназа (Spleen tyrosine kinase) - нерецепторная цитоплазматическая тирозинкиназа, участвующая в передаче сигнала антигенными и Fc рецепторами, BCR и другими рецепторами. Наболее интенсивно SYK киназа экспрессируется в гематопоэтических клетках (таких как макрофаги, тучные клетки, лейкоциты, тромбоциты и эритроциты), в меньшей степени - в эпителиальных клетках, фибробластах, нейрональных клетках, гепатоцитах и т.д. (Yanagi, S., et al., Biochem Biophys Res Commun, 2001, 288, 495-8).
Активация SYK-киназы запускает клеточный каскад, приводящий к синтезу и высвобождению большого количества модуляторов воспаления, которые могут вызвать развитие острых аллергических реакций (Valent et al., Intl J Hematol, 2002, 75, 257-362). Кроме того, SYK-киназа играет одну из ключевых ролей в иммунном ответе, развитие аутоиммунных (Lee, DM. et al., Lancet, 2001, 358, 903-11) и онкологических заболеваний, например лимфом (Young, RM., et al., Blood, 2009, 113, 2508-16).
Ингибирование SYK-киназы является перспективной стратегией борьбы с различными аллергическими, аутоиммунными, онкологическими заболеваниями, причиной которых является абберантная активность клеток, экспрессирующих SYK-киназу. Установление функции SYK-киназы в развитии аутоимунных заболеваний и лимфом привело к активному поиску ее селективных ингибиторов. На сегодняшний день известно более ста низкомолекулярных веществ, способных ингибировать SYK (Xie, H.Z., et al., Bioorg Med Chem Lett, 2009, 19, 1944-9). Наибольший интерес из существующих ингибиторов SYK-киназы представляют вещества R112, R343 (разработан на основе R112 в качестве удобной для ингаляции формы), R406 и R788, находящиеся на различных этапах клинических исследований. Эти структурно схожие производные 2,4-диаминопиримидина были разработаны компанией Rigel Pharmaceuticals, Inc. Первый представитель этого семейства, R112, связывается с SYK Ki=96 нМ (WO0306379). R788 разработан на основе R406 для улучшения оральной биодоступности (Bajpai, М., IDrugs, 2009, 12, 174-85). Стоит отметить, что R788 в организме распадается, и действующим веществом, по сути, является R406. В настоящее время R788 проходит клинические испытания в ревматоидном артрите (II фаза) (Weinblatt, М.Е., et al., N Engl J Med, 2010, 363, 1303-12) и неходжкинской лимфоме (II фаза) (Friedberg, J.W., et al., Blood, 2010, 115, 2578-85).
Применение низкомолекулярных ингибиторов SYK в терапевтической практике выявило несколько серьезных проблем, связанных с их биодоступностью и эффективностью. Например, клинические испытания R112 против аллергических ринитов были прекращены, поскольку вторая фаза не показала эффективности препарата по сравнению с плацебо, а не удовлетворительные фармакокинетическими параметры R406 вынуждает применять его в виде пролекарства. Таким образом, создание новых соединений, способных ингибировать киназы, и обладающих хорошими фармакокинетическими параметрами, является практически важной задачей.
В качестве ближайшего аналога может быть указана международная заявка WO 2009143389, описывающая новые ингибиторы киназ, представляющие собой фосфорные производные, в ряде альтернатив содержащие диамино-триазиновые группы. Соединения обладают активностью в отношении целой группы киназ.
Данное изобретение касается новых химических соединений - ингибиторов киназ, обладающих повышенной эффективностью в ингибировании SYK-киназы и ее мутантных форм, и перспективных для применения в терапии онкологических, аутоиммунных, воспалительных и прочих заболеваний.
Предметом настоящего изобретения являются ингибиторы протеинкиназ, характеризующийся тем, что представляет собой соединения общей формулы I, их фармацевтически приемлемые соли, сольваты или гидраты, а также пролекарства в виде сложных эфиров, амидов, карбаматов, N-гидроксиметил производных, О-гидроксиметилпроизводных, эфиров серной или фосфорной кислот, метилфосфонатов или их солей:

Где:
Y представляет собой СН2, CHR′, CRD, C(R′)2, NR′, O, S, S(O) или S(O)2.
X1, X2, X3 выбираются независимо из групп CH, CR′ или N.
R1 представляет собой алифатическую группу, арильную группу, этилендиоксифенил, метилендиоксифенил, опционально замещенную одной или более одинаковых или разных групп R″
R′ представляет собой водород, OH, галоген, такой как F, Cl, Br, I, или карбоксил или карбоксамид или циано или гало(C1-8)алкил, (C1-8) алкил, (C1-8) алкокси, (C1-8) галоалкокси
R″ представляет собой R′ или RD
R21, R22, R23, R24 выбираются независимо из групп F, Cl, Br, I, CN, (C1-16) алкил, кроме того R21 и R22 или/и R23 и R24 могут быть объединены и представлять собой одну оксо (=O) группу или совместно с атомом углерода образовывать спироцикл содержащий от 3 до 7 атомов углерода, кроме того R21 и R24 могут совместно с двумя атомом углерода образовывать алифатический или ароматический цикл содержащий от 4 до 8 атомов опционально замещенный одной или несколькими группами R′.
RD представляет собой оксо группу =O, =S
2. Предпочтительные варианты воплощения изобретения
2.1. Отдельный класс соединений, представляющих интерес, включают соединения по формуле I, в которых R1 представляют алифатическую, опционально замещенную одной или более одинаковых или разных групп R″. Иллюстративными примерами этого класса являются следующие соединения:
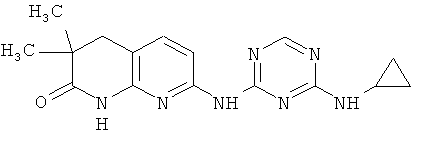
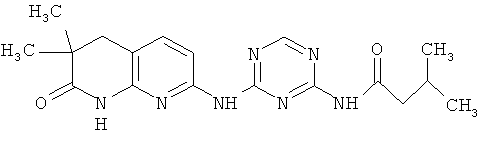

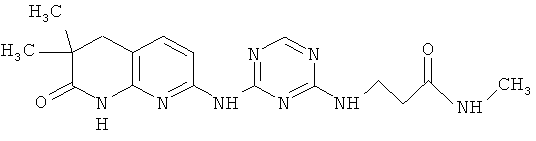
2.2. Отдельный класс соединений, представляющих интерес, включают соединения по формуле I, в которых R1 представляют арильную, этилендионсифенильную группу, опционально замещенную одной или более одинаковых или разных групп R″. Иллюстративными примерами этого класса являются следующие соединения:
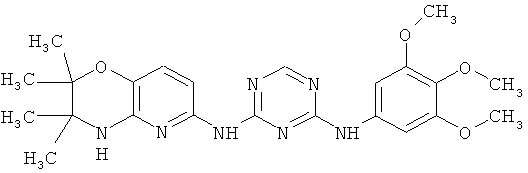
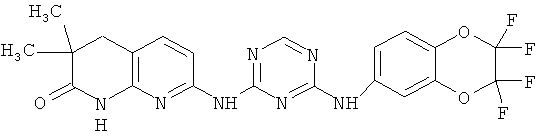
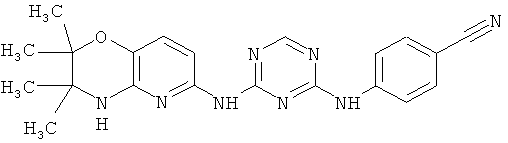
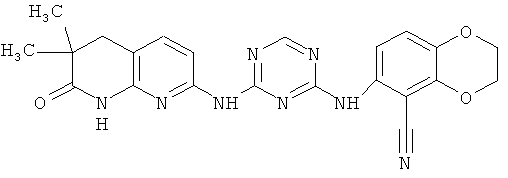
2.3. Одним из предпочтительных вариантов воплощения изобретения являются соединения по формуле
IA-1, в которых R1 представляет собой фенильную группу, опционально замещенную одной или более одинаковых или разных групп R″.
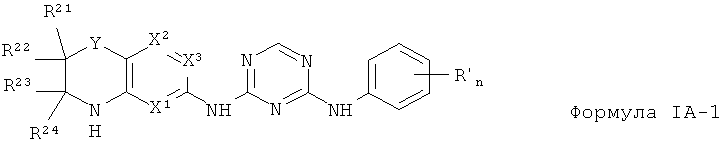
Где n - 0, 1, 2, 3, 4 или 5
2.4. Особо предпочтительный вариант воплощения изобретения составляют соединения по формуле IA-1, в которых R представляет собой алкокси группу. Иллюстративными примерами этого класса являются следующие соединения:
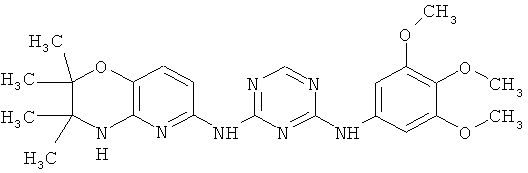
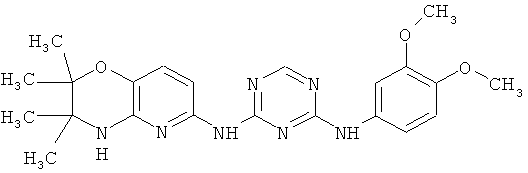

2.5. Отдельный предпочтительный вариант воплощения изобретения составляют соединения по формуле IA-1, в которых R′ представляет собой галоген или -CF3. Иллюстративными примерами этого класса являются следующие соединения:
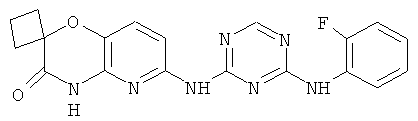
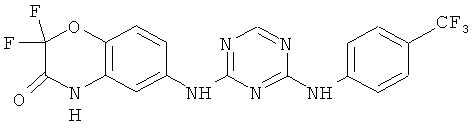
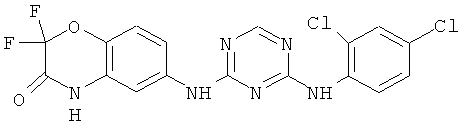
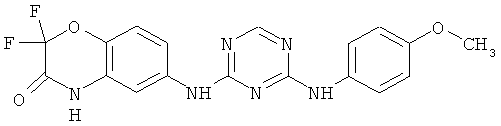
2.6. Одним из предпочтительных вариантов воплощения изобретения являются соединения по формуле
IA-2, в которых R1 представляет собой 3,4-этилендиоксифенильную группу, опционально замещенную одной или более одинаковых или разных групп R″.

Где n - 0, 1, 2, 3 или 4; m - 1, 2 или 3
2.7. Особо предпочтительный вариант воплощения изобретения составляют соединения по формуле IA-2, в которых R′ представляет собой алкокси группу или галоген или галоалкил. Иллюстративными примерами этого класса являются следующие соединения:

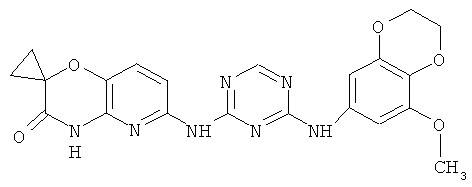


2.8. Одним из предпочтительных вариантов воплощения изобретения являются соединения по формуле
IA-3, в которых R1 представляет собой 3,4-метилендиоксифенильную группу, опционально замещенную одной или более одинаковых или разных групп R″.

Где n - 0,1 или 2; m -1,2 или 3
2.9. Особо предпочтительный вариант воплощения изобретения составляют соединения по формуле IA-3, в которых R″ представляет собой алкокси группу или галоген или галоалкил.
Иллюстративными примерами этого класса являются следующие соединения:
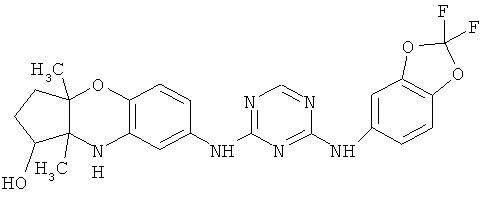
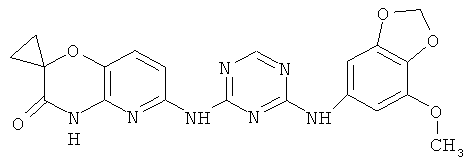
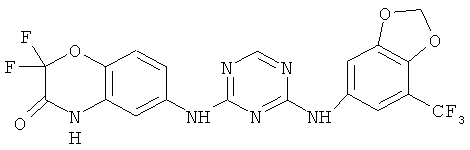
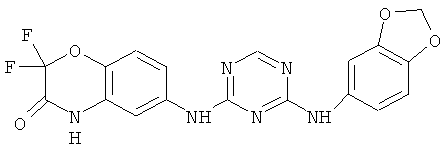
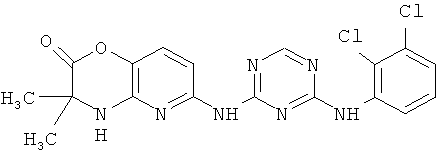
2.10. Отдельный класс соединений, представляющих интерес, включают соединения по формуле I, в которых X2 или X3 представляет собой атом N. Иллюстративными примерами этого класса являются следующие соединения:
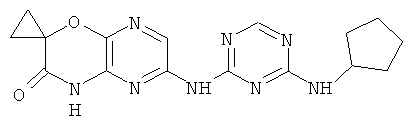
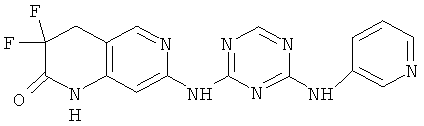
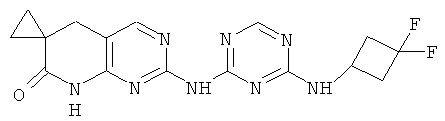
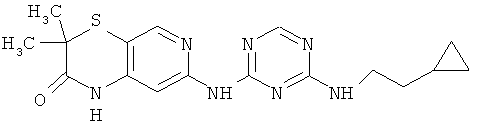
2.11. Отдельный класс соединений, представляющих интерес, включают соединения по формуле I, в которых X2 и X3 представляет собой CH или CR′ группу. Иллюстративными примерами этого класса являются следующие соединения:

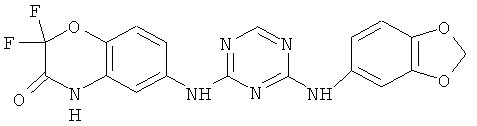
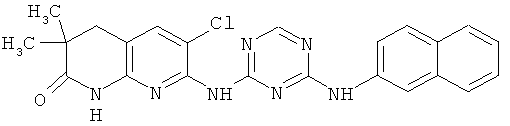
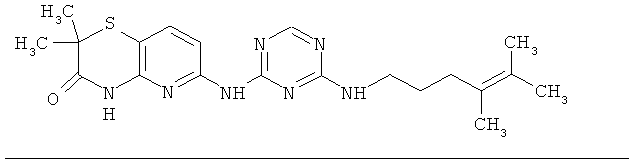
2.12. Отдельный класс соединений, представляющих интерес, включают соединения по формуле I, в которых Y представляет собой СНг, CHR′, CRD, C(R′)2 группу. Иллюстративными примерами этого класса являются следующие соединения:
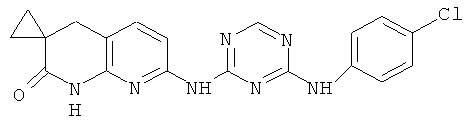
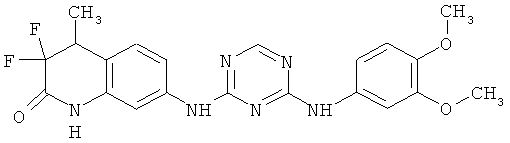

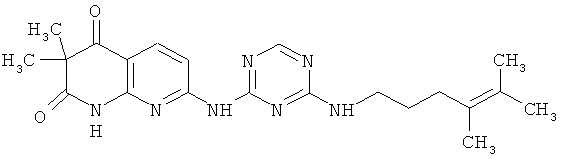
2.13. Отдельный класс соединений, представляющих интерес, включают соединения по формуле I, в которых Y представляет собой NR′ S, S(O) или S(O)2 группу. Иллюстративными примерами этого класса являются следующие соединения:

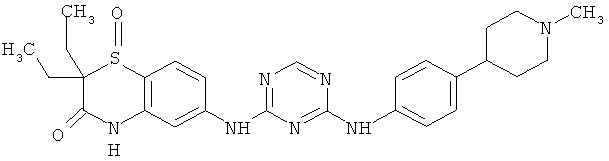
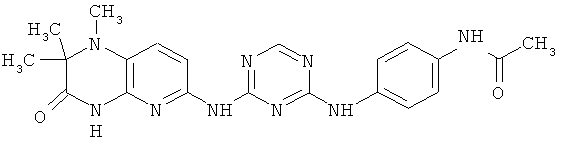
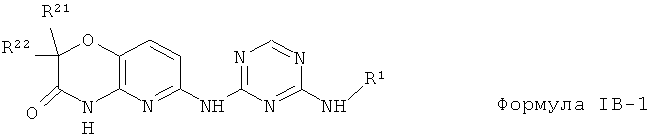
2.14. Одним из предпочтительных вариантов воплощения изобретения являются соединения по формуле
IB-1, в которых в которых X2 и X3 представляет собой СН группу, X1 представляет собой N группу, Y представляет собой атом O R23 и R24 объединены и представляют собой одну оксо (=O) группу.
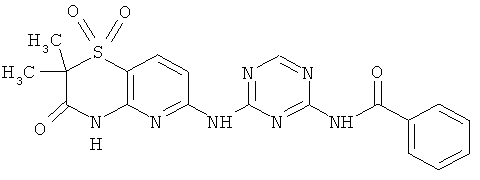
2.15. Особо предпочтительный вариант воплощения изобретения составляют соединения по формуле IB-1, в которых R21 и R22 представляет собой алкильную (обычно метальную) группу. Иллюстративными примерами этого класса являются следующие соединения:
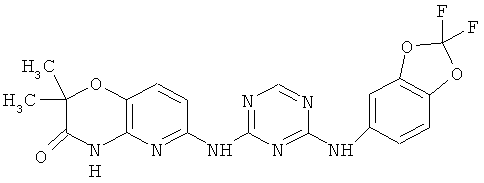
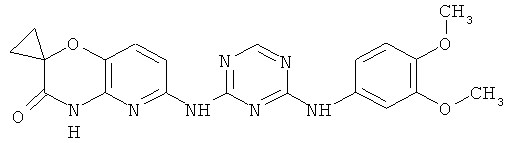
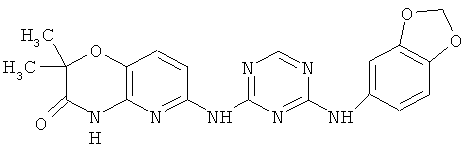
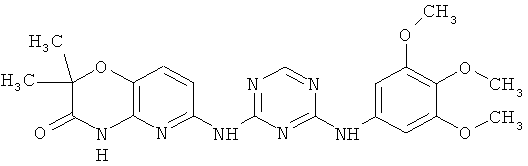
2.16. Одним из предпочтительных вариантов воплощения изобретения являются соединения по формуле
IB-2, в которых в которых X1, X2 и X3 представляет собой СН группу, Y представляет собой кислород О группу R23 и R24 объединены и представляют собой одну оксо (=O) группу.
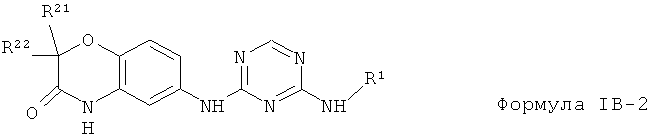
2.17. Особо предпочтительный вариант воплощения изобретения составляют соединения по формуле IB-2, в которых R21 и R22 представляет собой галоген (обычно фтор). Иллюстративными примерами этого класса являются следующие соединения:

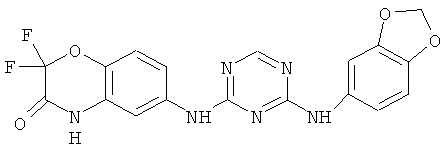


Соединения настоящего изобретения, представляющие особый интерес, обладают одной или несколькими из следующих характеристик:
- молекулярная масса менее 1000, предпочтительно менее 750, и наиболее предпочтительно менее 500 г/моль (не включая массу каких-либо сольватирующих или совместно кристаллизующихся веществ, а также противоионов в случае соли); или
- ингибиторная активность по отношению к нативным или мутантным (особенно клинически значимым мутантным) киназам, особенно к киназам SYK, ZAP-70, JAK1 JAK2, JAK3, ВТК, TYK1 и LYN, или другим киназам, представляющим интерес, со значением IC50 5 мкМ или менее (полученного с помощью любого научно обоснованного эксперимента по определению ингибирования киназ), предпочтительно с IC50 1 мкМ или ниже, и оптимально с IC50 500 нМ или ниже; или
- ингибиторная активность по отношению к SYK-киназе с IC50 как минимум в 100 раз меньшим, чем соответствующие значения IC50 для других киназ, представляющих интерес; или
- ингибитораня активность в отношении сигнального пути Fc или BCR рецепторов (особо предпочтительно Fes и Fey рецептора), ответственных за развитие восталительного ответа, определенная в экспирементах in vitro, или в исследованиях на животных, с использованием научно приемлемой модели (предпочтительны соединения, ингибирую-щие абберантную активность клеток имунной системы (например, тучных и гематопо-этических клеток) и соединения, ингибирующие абберантную пролиферацию и рост культуры клеток Daudi, NAMALVA, P3H3, P3H3-1, Raji).
Кроме того, изобретением предусматриваются композиции, содержащие как минимум одно соединение, являющееся предметом изобретения или соль, гидрат или другой сольват такового, и как минимум один фармакологически приемлемый наполнитель или добавку. Такие композиции могут быть введены объекту, нуждающемуся в подавлении абберантной активности и/или аберрантной пролиферации клеток иммунной системы.
Фармацевтические композиции могут быть полезны для лечения аллергических, аутоиммунных и онкологических заболеваний согласно настоящему изобретению, которое включает в себя введение (в качестве монотерапии или в комбинации с одним или несколькими имунодепресантами или противораковыми агентами, одним или несколькими агентами для смягчения побочных эффектов, облучением и т.п.) терапевтически эффективного количества соединения, являющегося предметом изобретения, в организм человека или животного, нуждающегося в подавлении абберантной активности или абберантной пролиферации клеток иммунной системы. Такое введение представляет собой метод лечения или профилактики заболеваний, вызываемых одной или несколькими киназами, ингибируемыми одним из раскрываемых соединений или их фармакологически приемлемых производных. «Введение» в организм соединения настоящего изобретения включает доставку к реципиенту соединения, описанного в настоящем изобретении, пролекарства, или другого фармакологически приемлемого производного такого соединения, используя любые допустимые препараты или пути введения в организм, как описано в настоящем документе. Обычно, соединение вводится в организм пациента один или несколько раз в неделю, например, ежедневно, через день, 5 дней в неделю и т.п. Пероральное и внутривенное введение представляет особый интерес.
Термин «фармакологически приемлемое производное» в настоящем документе означает любую фармакологически приемлемую соль, эфир или соль эфира соединения, а также любой другой аддукт или производное, которое, при введении пациенту, способно обеспечить (прямо или косвенно) доставку соединения, его метаболита или продукта его распада (имеющего молекулярную массу больше 300). Фармакологически приемлемые производные, таким образом, включают, в частности, пролекарства. Пролекарство - это производное соединения, обычно со значительно уменьшенной фармакологической активностью, которое содержит дополнительную химическую группу, способную к удалению в условиях in vivo, с образованием исходной фармакологически активной молекулы соединения. Например, пролекарством может быть сложный эфир или амид, который разлагается in vivo с образованием нужного соединения. Пролекарства различных соединений, материалы и методы получения производного исходного соединения хорошо известны и могут быть адаптированы для настоящего изобретения.
Особенно благоприятными производными и пролекарствами исходных соединений являются такие производные и пролекарства, которые увеличивают биодоступность соединения при введении в организм млекопитающего (например, увеличением абсорбции в крови при пероральном приеме), или те, которые увеличивают доставку в интересующую часть организма относительно исходного соединения. Предпочтительные пролекарства включают производные соединения, являющегося предметом изобретения, с повышенной растворимостью в воде или более активно проникающие через оболочку кишечника по сравнению с исходным соединением.
Важным аспектом изобретения является метод профилактики и лечения заболеваний человека и животных, основанный на ингибировании одной или нескольких протеинкиназ включающих, но не ограниченных SYK, ZAP-70, JAK1 JAK2, JAK3, ВТК, TYK1 и LYN киназами терапевтически эффективным количеством композиции, включающей в себя соединение изобретения. Лечение может проводиться в сочетании с одним или несколькими другими лекарственными средствами и нелекарственной терапией, включая хирургию, облучение (например, гамма-облучение, нейтронную, электронную, протонную лучевую терапию, брахите-рапию, и т.п.), гормональную терапию, модификаторы биологического отклика, гипертермию, криотерапию, агенты для смягчения каких-либо неблагоприятных последствий (например, противорвотные средства), а также другими противораковыми химиотерапевтическими препаратами. Прочие агент(ы) могут быть введены в организм пациента с использованием как сходных, так и отличные от тех, что использовались для соединений настоящего изобретения, технологий приготовления препарата, путей введения и схем дозирования.
Изобретение также включает получение соединений по любой из формул I, IA-1, IA-2, IA-3, IB-1, IB-2 или любых других соединений настоящего изобретения.
Изобретение также включает использование соединения изобретения или его фармакологически приемлемого производного в производстве лекарственного средства для лечения аллергических, аутоимунных, онкологических заболеваний, а также заболеваний, связанных с разрушением или воспалениием тканей. Соединения, составляющие суть настоящего изобретения, могут быть использованы в производстве лекарств для профилактики и лечения заболеваний, связанных с абберантной активностью клеток иммунной системы. Соединения, составляющие суть настоящего изобретения, могут быть использованы в производстве противораковых препаратов. Соединения настоящего изобретения также могут быть использованы в производстве лекарственных препаратов для ослабления или предотвращения различных расстройств путем ингибирования одной или нескольких киназ, таких как SYK, ZAP-70, JAK1 JAK2, JAK3, ВТК, TYK1 и LYN и т.д.
Изобретение также охватывает композиции, содержащие соединения настоящего изобретения, включая соединения любого из описанных классов или подклассов, в том числе любой из формул, описанных выше, помимо прочего, предпочтительно в терапевтически эффективном количестве, в соединении с по крайней мере одним терапевтически допустимым носителем, адъювантом или растворителем.
Соединения настоящего изобретения также могут быть использованы в качестве стандартов и реагентов для характеристики различных киназ, в особенности, но не ограничиваясь киназами SYK, ZAP-70, JAK1 JAK2, JAK3, ВТК, TYK1 и LYN, также как и для изучения роли таких киназ в биологических и патологических явлениях; для изучения сигнальных путей, осуществляемого с помощью таких киназ, для сравнительной оценки новых киназных ингибиторов; а также для изучения различных видов рака в моделях линий клеток и животных.
Следующие определения применяются в данном документе, если иное не указано явно. Кроме того, если не указано иное, все вхождения функциональных групп выбираются независимо.
Термин «алифатический» в настоящем документе означает как насыщенную, так и ненасыщенную (но не ароматическую) прямую (т.е. неразветвленную), разветвленную, циклическую или полициклическую неароматическую углеводородную цепь - остаток, который может быть опционально замещен одной или более функциональной группой. Если иное не указано явно, алкил, другие алифатические, алкокси и ацильные группы обычно содержат 1-8 (т.е. C1-8), а в большинстве случаев 1-6 (C1-6) смежных алифатических атомов углерода. В качестве примера, такие алифатические группы включают метил, этил, н-пропил, изопропил, циклопропил, метилен, циклопропил аллил,н-бутил, втор-бутл, циклобутил, циклобутилметил, н-пентил, 2-циклопентенил, циклопентил, трет-пентил, изопентил, циклопентилметил, н-гексил, втор-тексил, циклогексил, циклогексилметил, пропаргил, аллил, гомоаллил, гомо-пропаргил, адамантил производные и т.п., которые могут содержать один или несколько заместителей. Термин «алифатический», таким образом, подразумевает включение алкил, алкенил, алкинил, циклоалкил и циклоалкенил фрагментов.
Термин «алкил» в настоящем документе означает как неразветвленные, так и разветвленные, циклические или полициклические алкильные группы. Аналогичные условности применяются и к другим общим терминам, таким как «алкенил», «алкинил» и т.п. Кроме того, «алкил», «алкенил», «алкинил» и подобные группы могут быть как замещенными, так и незамещенными.
Термин «алкил» в настоящем документе относится к группам, обычно имеющим от одного до восьми, предпочтительно от одного до шести атомов углерода. Например, «алкил» может означать метил, этил, н-пропил, изопропил, циклопропил, бутил, изобутил, втор-бутил, циклобутил, трет-бутил, циклобутил, пентил, циклопентил, трет-пентил, изопентил, гексил, изогексил, циклогексил и т.д. В качестве иллюстрации, замещенные алкильные группы включают, но не ограничиваются, следующими группами: фторметил, дифторметил, трифторметил, 2-фторэтил, 3-фторпропил, гидроксиметил, 2-гидроксиэтил, 3-гидроксипропил, бензил, замещенный бензил, 2-фенилэтил, замещенный 2-фенилэтил и т.д. Термин C1-6 алкил означает алкил, содержащий от 1 до 6 атомов углерода, и включает C1, С2, С3, С4, C5 и C6-алкильные группы.
Термин «алкокси» относится к алкильным группам, соответствующим определению, приведенному выше, и которые присоединяются к молекуле посредством мостикового атома кислорода. Например, термин «алкокси» означает -O-алкил, где алкильная группа содержит от 1 до 8 атомов углерода в виде линейной (неразветвленной) или разветвленной цепи или в виде цикла. В качестве иллюстрации алкокси группы включают, но не ограничиваются, следующими группами: метокси, этокси, н-пропокси, н-бутокси, трет-бутокси, аллилокси, циклобутокси и т.д.
Термин «галоалкил» включает разветвленные и линейные насыщенные углеводородные цепи, в которых один или несколько атомов водорода замещены на галоген. Примеры га-лоалкильных групп включают, но не ограничиваются, следующие группы: дифторметил, трифторметил, трихлорметил, пентафторэтил, 1,1,1,3,3,3-гексафтор-2-метилпропан-2-ил и т.п.
Термин «алкенил» относится к группам, обычно имеющим от двух до восьми, чаще от двух до шести атомов углерода. Например, «алкенил» может означать проп-2-енил, бут-2-енил, бут-3-енил, 2-метилпроп-2-енил, гекс-5-енил, 2,3-диметилбут-2-енил и т.п. Термин «алкинил» также относится к группам, обычно имеющим от двух до восьми, чаще от двух до шести атомов углерода, включая, но не ограничиваясь, следующими группами: проп-2-инил, бут-2-инил, бут-3-инил, пент-2-инил, 3-метилпент-4-инил, гекс-2-инил, гекс-5-инил и т.д.
Термин «циклоалкил» относится к группам, имеющим от трех до 12, обычно от трех до десяти атомов углерода в моно-, ди- или полициклической (т.е. кольцевой) структуре. В качестве иллюстрации, циклоалкилы включают, но не ограничиваются, следующими радикалами: циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, норборнил и т.п., которые, как и в случае других алифатических или гетероалифатических или гетероциклических заместителей, могут быть замещенными. Термины «циклоалкил» и «карбоцикл» являются эквивалентными.
Термин «арил», используемый самостоятельно, или как часть большего фрагмента, такого как «арилалкил», «арокси» или «арилоксиалкил», означает группы, содержащие ароматический цикл, и имеющие от шести до четырнадцати атомов углерода в таком цикле, такие как фенил, 1-нафтил, 2-нафтил, 1-антрацил и 2-антрацил. «Арильные» циклы могут содержать один или несколько заместителей. Термин «арил» может использоваться взаимозаменяемо с термином «арильное кольцо». «Арил» также включает в себя полициклические ароматические системы, в которых ароматический цикл объединяет в себе один или несколько циклов. Примеры используемых арильных циклических групп включают в себя, но не ограничиваются, фенил, гидроксифенил, галгенфенил, алкоксифенил, диалкоксифенил, триалкоксифенил, алкилендиоксифенил, нафтил, фенантрил, антраценилил, фенантроценил и т.п., так же как и 1-нафтил, 2-нафтил, 1-антрацил и 2-антрацил. Кроме того, в значение термина «арил», так, как оно используется здесь, входят группы, в которых ароматический цикл соединен с одним или более неароматическими циклами или гетероциклами, такие как инданил, фенантридинил или тетрагидронафтил, 3,4-этилендиоксифенил, в которых радикальный атом или место соединения принадлежит ароматическому циклу.
Данное изобретение содержит только такие комбинации заместителей и производных, которые образуют стабильное или химически возможное соединение. Стабильным или химически возможным соединением называется такое соединение, время жизки которого достаточно для его синтеза и аналитического детектирования. Предпочтительные соединения данного изобретения являются достаточно стабильными и не разлагаются при температуре до 40°C в отсутствие влаги или других химически активных условий, в течение, по крайней мере, одной недели.
Некоторые соединения данного изобретения могут существовать в таутомерных формах, и это изобретение включает в себя все такие таутомерные формы таких соединений, если не указано иное.
Если не указано иначе, изображенные здесь структуры также подразумевают и все стереохимические формы, то есть R- и S-изомеры для каждого ассиметричного центра. Кроме того, отдельные стереохимические изомеры, равно как и энантиомеры и диастереомернНые смеси настоящих соединений, также являются предметом данного изобретения. Таким образом, данное изобретение охватывает каждый диастереомер или энантиомер, свободный в значительной степени от других изомеров (>90%, а предпочтительно >95% мольной чистоты), так же как и смесь таких изомеров.
Конкретный оптический изомер может быть получен разделением рацемической смеси в соответствии со стандартной процедурой, например путем получения диастереоизомерных солей путем обработки оптически активной кислотой или основанием с последующим разделением смеси диастереомеров кристаллизацией с последующим выделением оптически активных оснований или кислот из этих солей. Примерами соответствующих кислот являются винная, диацетилвинная, дибензоилвинная, дитолуолвинная и камфорсульфоновая кислота. Другая методика разделения оптических изомеров заключается в использовании хиральной хроматографической колонки. Кроме того, другой метод разделения включает синтез ковалентных диастереомерных молекул путем реакции соединений изобретения с оптически чистой кислотой в активированной форме или оптически чистым основанием. Полученные диастереомеры можно разделить обычными способами, например, хроматографией, ректификацией, кристаллизации или сублимацией, а затем гидролизовать для получения энантиомерно чистого соединения.
Оптически активные соединения данного изобретения могут быть получены с использованием оптически активных исходных материалов. Такие изомеры могут находиться в форме свободной кислоты, свободного основания, эфира или соли.
Соединения, составляющие суть данного изобретения, могут существовать в меченой радиоизотопом форме, т.е. указанные соединения могут содержать один или несколько атомов, чья атомная масса или массовое число отличается от атомной массы или массового числа, наиболее распространенного в природе. Например, радиоизотопы водорода, углерода, фосфора, хлора включают 3Н, 14С, 32Р, 35S, и 36Cl, соответственно. Соединения данного изобретения, которые содержат такие радиоизотопы и/или другие радиоизотопы других атомов, находятся в сфере настоящего изобретения. Дейтериевае, т.е. 2H, тритиевые, т.е. зн и углеродные, т.е. 14С радиоизотопы являются особенно предпочтительными благодаря простоте приготовления и обнаружения.
Радиоизотопно меченые соединения настоящего изобретения могут быть получены с помощью методов, хорошо известных специалистам в данной области. Меченые соединения могут быть получены с помощью процедур, описанными здесь, простой заменой немеченых реагентов соответствующими мечеными реагентами.
Способы получения соединений изобретения
Соединения, являющиеся предметом настоящего изобретения, могут быть получены с использованием различных общеизвестных синтетических методик, в том числе с использованием описанных ниже синтетических методик. Синтез химических соединений, являющиеся предметом настоящего изобретения, может быть проведен из коммерчески доступных исходных реагентов или исходных реагентов, которые могут быть получены по методикам, описанным в работах (Вейганд-Хильгетаг. Методы эксперимента в органической химии. Под ред. проф. Н.Н.Суворова. М., Химия, 1968, «Синтезы гетероциклических соединений» вып.1-16 Ереван 1956-1987; "Синтезы органических препаратов" 4.1-12 М. 1949-1964; «Синтезы органических соединений с изотопами водорода» Мэррей А., Уильяме Д.Л. Москва, Издательство ИЛ, 1961; «Препаративная органическая химия» под ред. Вульфсона Н.С., М. 1959; «Избранные методы синтеза органических соединений» Репинская И.Б., Шварцберг М.С., Новосибирск Издательство Новосибирского Университета 2000; «Реакции и методы исследования органических соединений» тт.1-26, 1951-1986; «Organic Syntheses Based on Name Reactions and Unnamed reactions)) Hassner A., Stumer C. Pergamon, Oxford, 1994) полное раскрытие которых включено в настоящее описание в качестве ссылки. Перечисленные методы не являются исчерпывающими и допускают введение разумных модификаций. Указанные реакции должны проводиться с использованием подходящих растворителей и материалов. При реализации данных общих методик для синтеза конкретных веществ необходимо учитывать присутствующие в веществах функциональные группы и их влияние на протекание реакции. Для получения некоторых веществ необходимо изменить порядок стадий либо отдать предпочтение одной из нескольких альтернативных схем синтеза.
Синтез соединений, являющихся предметом данного изобретения, может быть осуществлен в соответствии со схемами I-IV по стандартным методикам.

Где XX представляет собой галоген.
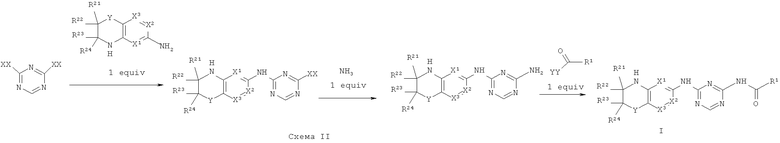
Где XX и YY представляет собой галоген.
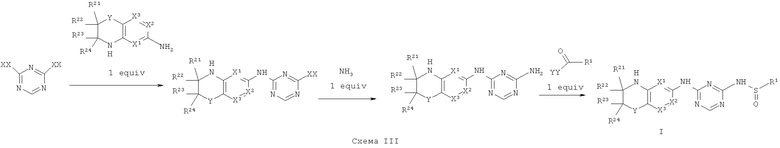
Где XX и YY представляет собой галоген
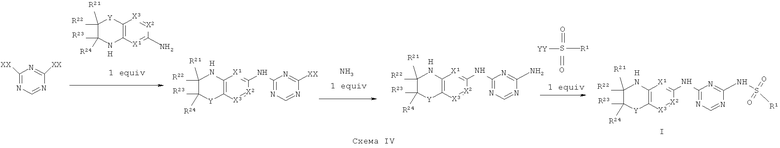
Где XX и YY представляет собой галоген
Все соединения, являющиеся предметом данного изобретения, могут быть получены на основании вышеизложенных синтетических подходов, примеров экспериментальных методик и общеизвестных методик и материалов.
Применение химических соединений изобретения
Применение соединений по медицинским показаниям
Соединения, описанные в данном изобретении могут применяться для терапии заболеваний, в патогенезе которых участвуют протеинкиназы. В частности, соединения, описанные в данном изобретении способны ингибировать тирозинкиназы SYK, ZAP-70, JAK1 JAK2, JAK3, ВТК, TYK1 и LYN. Кроме того показано, что ряд соединений, составляющих настоящее изобретение, подавляют абберантную активность клеток имунной системы в экспериментах in vitro и обладают антипролиферативной активностью in vitro по отношению к раковым клеточным линиям, таким, как например Daudi, NAMALVA, P3H3, P3HR-1, Raji. Такие соединения представляют интерес для лечения аутоиммунных, аллергических и онкологических заболеваний, и в особенности для лечения заболеваний, резистентных к другим способам терапии. К заболеваниям, для лечения которых могут применяться соединения данного изобретения, относятся аллергические заболевания (например, конъюнктивиты, риниты, астма, атопический дерматит, пищевая аллергия и т.д), аутоиммунные заболевания (например, тиреоидит Хашимото, аутоиммунная гемолитическая анемия, аутоиммунная атрофический гастрит, злокачественная анемия, аутоиммунный энцефаломиелит, аутоиммунный орхит, синдром Гудпасчера, аутоиммунная тромбоцитопении, симпатическая офтальмия, миастения, болезнь Грейвса, первичный билиарный цирроз печени, хронический агрессивный гепатит, неспецифический язвенный колит, мембранная гломерулопатия, системная красная волчанка, ревматоидный артрит, синдром Шегрена, синдром Рейтера, полимиозит, дерматомиозит, системнаая склеродермия, узелковый полиартрит, рассеянный склероз и т.д.) и онкологические заболевания (например, неходжкинская лимфома и другие формы рака, патогенез которых связан с активностью SYK, ZAP-70, JAK1 JAK2, JAK3, ВТК, TYK1 и LYN киназ).
SYK-киназа (Spleen tyrosine kinase) - нерецепторНая цитоплазматическая тирозинкиназа, участвующая в передаче сигнала антигенными и Fc рецепторами, BCR и другими рецепторами. Наболее интенсивно SYK киназа экспрессируется в гематопоэтических клетках (таких как макрофаги, тучные клетки, лейкоциты, тромбоциты и эритроциты), в меньшей степени в эпителиальных клетках, фибробластах, нейрональных клетках, гепатоцитах и т.д. (Yanagi, S., et al., Biochem Biophys Res Commun, 2001, 288, 495-8). Активация SYK-киназы запускает клеточный каскад, приводящий к синтезу и высвобождению большого количества модуляторов воспаления, которые могут вызвать развитие острых аллергических реакций (Valent et al., Intl J Hematol, 2002, 75, 257-362). Кроме того, SYK-киназа играет одну из ключевых ролей в иммунном ответе, развитие аутоиммунных (Lee, DM. et al., Lancet, 2001, 358, 903-11) и онкологических заболеваний, например лимфом (Young, RM., et al., Blood, 2009, 113, 2508-16). Ингибирование SYK-киназы является перспективной стратегией борьбы с различными аллергическими, аутоиммунными, онкологическими заболеваниями, причиной которых является абберантная активность клеток, экспрессирующих SYK-киназу.
Таким образом, мы предполагаем, что использование ингибитора SYK как препарата для монотерапии или в сочетании с текущими средствами химиотерапии против различных онкологических, аутоиммунных и алергических заболеваний, позволит достигнуть существенной и длительной ремиссии, или ингибитор SYK может быть использован как средство поддерживающей терапии, предназначенное для предотвращения возможных рецидивов у пациентов, нуждающихся в таком лечении.
Способ терапевтического применения соединений
Предмет данного изобретения включает введение субъекту, нуждающемуся в соответствующем лечении, терапевтически эффективного количества соединения из данного изобретения.
«Терапевтически эффективным количеством» называется такое количество соединения, которое необходимо для детектируемого уничтожения раковых клеток или ингибирования их роста или скорости распространения по организму, размера или количества опухолей, или других характеристик онкологического заболевания. Точное требуемое количество может меняться от субъекта к субъекту в зависимости от вида, возраста и общего состояния пациента, тяжести заболевания, особенностей противоракового агента, методики введения препарата, комбинированного лечения с другими препаратами и т.п.
Вещество, или фармацевтическая композиция, содержащая вещество, может быть введена в организм пациента в любом количестве и любым путем введения, эффективным для подавления абберантной активности или/и подавления абберантной пролиферации клеток иммунной системы.
Разовые дозы лекарственных соединений, состалвяющих предмет настоящего изобретения предпочтительно формулируются в виде, удобном для введения в организм пациента. Выражение «разовая доза»в терминах настоящего изобретения означает порцию препарата, подходящую для лечения пациента. Согласно существующей практике, совокупная дневная доза соединений и композиций, описанных в настоящем изобретении, назначается лечащим врачом с опорой на тщательное медицинское заключение. Конкретный терапевтически эффективный уровень дозировки для каждого конкретного пациента или организма зависит от ряда факторов, включая тип расстройства, тяжесть заболевания, активность конкретного используемого препарата, особенности фармацевтической композиции, возраст, массу тела, общее состояние здоровья, пол и диету пациента, способ и график введения, скорость метаболизма и/или выведения соединения, продолжительность лечения, лекарственные препараты, используемые в комбинации или совместно с введением соединения из изобретения, и тому подобные факторы, хорошо известные в медицине.
После смешения лекарственного препарата с конкретным подходящим фармацевтически допустимым носителем в желаемой дозировке, композиции, составляющие суть изобретения, могут быть введены в организм человека или других животных перорально, ректально, парентерально, интрацистернально, интравагинально, интраперитонально, местно (с помощью кожных пластырей, порошков, мазей или капель), сублингвально, буккально, в виде спрея для рта или носа и т.п.
Эффективная системная дозировка соединения, вводимая разово или в виде нескольких отдельных доз, как правило, лежит в диапазоне от 0.05 до 500 мг соединения на кг массы тела пациента, предпочтительно от 0.1 до 150 мг/кг. Обычно соединение вводится пациенту, нуждающемуся в таком лечении, в дневной дозировке ориентировочно от 50 до 2000 мг на пациента. Введение может осуществляться как разово, так и несколько раз в день, неделю (или любой другой временной интервал), или время от времени. Например, соединение может быть введено в организм пациента один или несколько раз в день на недельной основе (например, каждый понедельник) в течение неопределенного времени или в течение нескольких недель (например, 4-10 недель). Кроме того, соединение может вводиться в организм пациента ежедневно в течение определенного периода дней (например, 2-10 дней), а затем следует период без приема вещества (например, 1-30 дней). Такой цикл может повторяться неопределенное время или в течение заданного числа циклов, например 4-10 циклов. В качестве примера, соединение настоящего изобретения может вводиться в организм пациента ежедневно в течение 5 дней, затем следует перерыв на 2 дня, и так далее, повторяя цикл неопределенное число раз, или в течение 4-10 циклов.
Количество соединения, которое будет эффективным в лечении или профилактике конкретного расстройства или состояния, зависит, в частности, от хорошо известных факторов, влияющих на эффективную дозировку препаратов. Кроме того, опционально могут применяться измерения in vitro или in vivo для определения оптимального дозового диапазона. Грубым путем определения эффективной дозы может стать экстраполяция кривых доза - отклик, которые будут зависеть от модели тестирования in vitro или на животных. Точный уровень дозировки, определяемый лечащим врачом, зависит от хорошо известных факторов, включающих способ введения препарата, а также возраста, массы тела, пола и общего состояния здоровья пациента; характера, тяжести и клинического состояния заболевания; использования (или неиспользования) сопутствующей терапии; а также характера и степени генетических изменений в клетках пациента.
При приеме внутрь для лечения или подавления конкретного состояния заболевания или расстройства, эффективная дозировка соединения данного изобретения может изменяться в зависимости от конкретного применяемого соединения, пути введения препарата в организм, условий и тяжести такого введения; состояния болезни, а также различного числа физических факторов, связанных с пациентом, проходящем лечение. В большинстве случаев, удовлетворительный результат, может быть, достигнуть при введении пациенту соединения в дневной дозировке от около 0.05 мг/кг до 500 мг/кг, обычно между 0.1 и 150 мг/кг. Предполагаемая дневная дозировка, как ожидается, может изменяться в зависимости от способа введения в организм пациента. Так, уровень дозировки при парентеральном введении часто составляет от 10 до 20% уровня пероральной дозировки.
В том случаев когда соединение данного изобретения используется как часть режима комбинированной терапии, доза каждого из компонентов комбинированной терапии вводится в течение требуемого периода лечения. Соединения, составляющие комбинированную терапию, могут вводиться в организм пациента единовременно в виде дозировки, содержащей все компоненты, так и в виде индивидуальных дозировок компонентов; кроме того, соединения комбинации могут быть введены в организм пациента в разное время в течение периода лечения, или одно из них может быть введено в качестве предварительной терапии для другого.
Фармацевтически приемлемые производные соединений
Соединения данного изобретения могут существовать в свободной форме в процессе обработки, или, если требуется, в виде фармацевтически приемлемой соли или другого производного. Используемый здесь термин «фармацевтически приемлемая соль» относится к таким солям, которые, в рамках проведенного медицинского заключения, пригодны для использования в контакте с тканями человека и животных без излишней токсичности, раздражения, аллергической реакции и т.д., и отвечают разумному соотношению пользы и риска. Фармацевтически приемлемые соли аминов, карбоновых кислот, фосфонатов и другие типы соединений хорошо известны в медицине. Подробное описание свойств таких солей дано Berge S.M., et al., в "Pharmaceutical Salts" J. Pharmaceutical Science, 66: 1-19 (1977), приведенном здесь в качестве ссылки. Соли могут быть получены in situ в процессе выделения или очистки соединений изобретения, а также могут быть получены отдельно, путем взаимодействия свободной кислоты или свободного основания соединения изобретения с подходящим основанием или кислотой, соответственно. Примером фармацевтически приемлемых, нетоксичных солей кислот могут служить соли аминогруппы, образованные неорганическими кислотами, такими как соляная, бромоводородная, фосфорная, серная кислоты, или органическими кислотами, такими как уксусная, щавелевая, малеиновая, винная, янтарная, метансульфокислота или малоновая кислоты, или полученные другими методами, используемыми в данной области, например, с помощью ионного обмена. К другим фармацевтически приемлемым солям относятся адипинат, альгинат, аскорбат, аспартат, бензолсульфонат, бензоат, бисульфат, борат, бутират, камфорат, камфорсульфонат, цитрат, циклопентанпропионат, диглюконат, додецилсульфат, этансульфонат, формиат, фумарат, глюкогептонат, глицерофосфат, глюконат, гемисульфат, гептанат, гексанат, гидройодид, 2-гидроксиэтансульфонат, лактобионат, лактат, лаурат, лаурил сульфат, малат, малеат, малонат, метансульфонат, 2-нафталинсульфонат, никотинат, нитрат, олеат, пальмитат, памоат, пектинат, персульфат, 3-фенилпропионат, фосфат, пикрат, пивалат, про-пионат, стеарат, сукцинат, сульфат, тартрат, тиоцианат, п-толуолсульфонат, ундеканат, вале-риат и подобные. Типичные соли щелочных и щелочноземельных металлов содержат натрий, литий, калий, кальций, магний и другие. Кроме того, фармацевтически приемлемые соли могут содержать, если требуется, нетоксичные катионы аммония, четвертичного аммония и амина, полученные с использованием таких противоионов, как галогениды, гидроксиды, карбоксилаты, сульфаты, фосфаты, нитраты, низшие алкил сульфонаты и арил сульфонаты.
Кроме того, термин «фармацевтически приемлемый сложный эфир», как он используется здесь, обозначает гидролизующийся in vivo сложный эфир, который легко разлагается в теле человека до исходных соединений или их солей. Подходящая эфирная группа включает, например, производные фармацевтически приемлемых алифатических карбоновых кислот, в частности алкановых, алкеновых, циклоалкановых и алкандиеновых кислот, в которых каждый алкильный или алкенильный компонент обычно имеет не более 8 углеродных атомов.
Примерами конкретных эфиров могут служить производные формиатов, ацетатов, пропионатов, бутиратов, акрилатов и этилсукцинатов. Очевидно, что эфиры также могут быть образованы гидроксильной группой или группой карбоновой кислоты соединения изобретения.
Термин «фармацевтически приемлемая пролекарственная форма», в контексте данного изобретения, означает такие пролекарства из числа соединений, составляющих суть данного изобретения, которые пригодны для использования человеком и животными без излишней токсичности, раздражения, аллергической реакции и т.д., отвечают разумному соотношению пользы и риска. Термин «пролекарства» означает соединения, которые трансформируются in vivo с образованием исходного соединения указанной выше формулы, например, при гидролизе в крови (См. работы Т.Higuchi and V.Stella, Pro-drugs as Novel Delivery Systems, Vol.14 of the A.C.S. Symposium Series, и Edward B. Roche, ed., Bioreversible Carriers in Drug Design, American Pharmaceutical Association and Pergamon Press, 1987).
Фармацевтические композиции
Заявляются также фармацевтические композиции, которые содержат одно из описанных здесь соединений (или пролекарственную форму, фармацевтически приемлемую соль или другое фармацевтически приемлемое производное) и одно или несколько фармацевтически приемлемых носителей или наполнителей. Данные композиции также могут содержать один или несколько дополнительных терапевтических агентов. С другой стороны, соединение данного изобретения может быть введено пациенту, нуждающемуся в соответствующей терапии, в комбинации с одним или более других терапевтических режимов (например, совместно с другими ингибиторами киназ, антителами, блокирующими связывание TNF с TNFR, метотрексатом и т.д.).
Фармацевтические композиции, заявляемые в данном изобретении, содержат соединения данного изобретения совместно с фармацевтически приемлемыми носителями, которые, могут включать в себя любые растворители, разбавители, дисперсии или суспензии, поверхностно-активные вещества, изотонические агенты, загустители и эмульгаторы, консерванты, вяжущие вещества, смазочные материалы и т.д., подходящие для конкретной формы дозирования. Remington's Pharmaceutical sciences (15th edition, E.W. Martin, Mack Publishing Co., Easton, Pa., 1975) раскрывает различные носители, использованные при разработке фармацевтических композиций и известные методы их приготовления. За исключением таких случаев, когда среда обычных носителей несовместима с соединением изобретения, например, при появлении любых нежелательных биологических эффектов и иных нежелательных взаимодействий с любым другим компонентом (компонентами) фармацевтической композиции, использование таких композиций находится в рамках данного изобретения. Материалы, которые могут служить фармацевтически приемлемыми носителями, включают, но не ограничиваются, моно-и олигосахаридами, а также их производными; солодом, желатином; тальком; эксципиентами, такими как: какао-масло и воск для суппозиториев; масла, такие как арахисовое, хлопковое, сафроловое, кунжутное, оливковое, кукурузное и соевое масло; гликоли, такие как пропиленгликоль; сложные эфиры, такие как этилолеат и этиллаурат; агар; буферные вещества, такие как гидроксид магния и гидроксид алюминия; альгиновая кислота; апирогенная вода; изотонический раствор, раствор Рингера; этиловый спирт и фосфатные буферные растворы. Также в составе композиции могут быть другие нетоксичные совместимые смазочные вещества, такие как лаурилсульфат натрия и стеарат магния, а также красители, разделительные жидкости, пленкообразователи, подсластители, вкусовые добавки и ароматизаторы, консерванты и антиоксиданты.
Лекарственные формы
Предметом данного изобретения являются также лекарственные формы - класс фармацевтических композиций, состав которых оптимизирован для определенного пути введения в организм в терапевтически эффективной дозе. Лекарственные композиции данного изобретения могут быть введены в организм орально, местно, ректально, внутриглазным способом, пульмональным, например, в виде ингаляционного спрея, или внутрисосудистым способом, интраназально, интраперитонально, подкожно, внутримышечно, интрастернально, а также инфузионным способом, в рекомендованных дозировках.
Фармакологически активные соединения, составляющие суть данного изобретения, могут быть обработаны в соответствии с общепринятыми методами фармацевтического производства для получения соответствующих лекарственных форм для введения пациенту, в том числе человеку и другим млекопитающим.
При пероральном введении, лекарственная форма может быть, например, в виде таблетки, капсулы, суспензии или жидкости. Лекарственная форма предпочтительно изготавливается в виде единичной дозы, содержащей определенное количество активного ингредиента.
Примером такой единичной дозы являются таблетки или капсулы, которые могут содержать от 1 до 2000 мг активного ингредиента, предпочтительно от 1 до 500 мг, обыкновенно от 5 до 200 мг. Подходящая дневная доза для человека или другого млекопитающего зависит от состояния пациента и других факторов.
Для терапевтических целей активные соединения, составляющие суть изобретения, обычно сочетают с одним или более адъювантом, наполнителем или носителем, подходящем для выбранного пути введения в организм. Для перорального введения, соединение может быть смешано с лактозой, сахарозой, порошком крахмала, эфирами целлюлозы и алкановых кислот, алкильными эфирами целлюлозы, талька, стеариновой кислоты, стеаратом магния, оксидом магния, натриевыми и кальциевыми солями фосфорной и серной кислот, желатином, гуммиарабиком, альгинатом натрия, поливинилпирролидоном и/или поливиниловым спиртом, а затем таблетироваться или инкапсулироваться для удобного введения в организм пациента. Такие капсулы или таблетки могут обладать свойством контролируемого выделения активного соединения, при условии дисперсии активного соединения в гидроксипропилметил-целлюлозе.
В случае кожных заболеваний предпознительнее местное применение соединения данного изобретения на пораженный участок от одного до четырех раз в день.
Лекарственные формы, подходящие для местного применения, включают жидкие или полужидкие препараты, пригодные для проникновения через кожу (например, мази, лосьоны, притирания, крема или пасты), а также капли, подходящие для введения через глаз, нос или ухо. Типичная дозировка активного соединения данного изобретения лежит в диапазоне от 0,1 до 150 мг, вводимых ежедневно от одного до четырех раз, предпочтительно один или два раза в день. Для местного введения в организм пациента, содержание активного ингредиента в лекарственной форме может составлять от 0,001 до 10 масс.%, например, от 1% до 2% от массы препарата. Массовая доля активного ингредиента может также составлять до 10 масс.%, желательно не более 5 масс.%, и еще более желательно от 0,1 до 1% от массы препарата.
При составлении мазей, активный ингредиент может использоваться с любой парафиновой или водорастворимой основой. Кроме того, при сочетании активного ингредиента с эмульсией типа «масло в воде» может быть приготовлен крем. По желанию, водная фаза кремовой основы может включать, например, от 30 масс.% многоатомных спиртов, таких как пропиленгликоль, бутил-1,3-диол, маннит, сорбит, глицерин, полиэтиленгликоль или их смеси. Местный препарат может также включать соединения, облегчающие всасывание или проникновение активного ингредиента через кожу или другие участки. Примерами таких соединений для усиления проникновения через кожу являются диметилсульфоксид и аналогичные соединения.
Соединения, составляющие суть данного изобретения, также могут быть введены в организм пациента с помощью устройств для трансдермального ввода. Предпочтительно, трансдермальное введение осуществляется с помощью наклейки или пластыря с резервуаром и пористой мембраной, либо с твердофазным носителем. В любом случае, активный ингредиент доставляется непрерывно из емкости или микрокапсул через мембрану в проницаемый для активного агента слой, который контактирует с кожей или слизистой оболочкой пациента. Если активный агент всасывается кожей, то пациенту вводится контролируемое и предопределенное количество активного агента. В случае микрокапсул, инкапсулирующий материал может служить в качестве мембраны.
Масляная фаза эмульсии, входящей в данное изобретение, может быть создана из известных ингредиентов известным способом. Масляная фаза может состоять только из эмульгатора, или может содержать смесь хотя бы одного эмульгатора с жиром или маслом, или и с тем и с другим одновременно. Предпочтительно, гидрофильный эмульгатор используется совместно с липофильным эмульгатором, который действует как стабилизатор. Также желательно использование одновременно и жира, и масла. Совместно, эмульгатор(ы) вместе с или без стабилизатора(ов) образуют так называемый эмульсионный воск, а воск вместе с жиром и маслом образует так называемую эмульгирующую мазевую основу, которая составляет масляную фазу крема. Эмульгаторы и эмульсионные стабилизаторы, пригодные для использования в фармакологическом составе на основе соединений настоящего изобретения, включают Твин 60, Спан 80, цетостеариловый спирт, миристиловый спирт, глицерилмоностеарат, лаурилсульфат натрия, глицерина дистеарат, сами по себе или вместе с воском или другими материалами, известными в фармакологии.
Выбор подходящих масел или жиров для оптимального состава основывается на желаемых косметических свойствах. Крем предпочтительно должен быть нежирным, не оставляющим пятен и легко смываемым, в консистенции, позволяющей избежать вытекания из тюбика или другого контейнера. Можно использовать моно- или диосновные алкильные эфиры с прямой или разветвленной цепью, такие как диизоадипат, изоацетил стеарат, диэфиры пропиленгликоля и кокосовых жирных кислот, изопропилмиристат, децилолеат, изопропилпальмитат, бутилстеарат, 2-этилгексилпальмитат или смесь эфиров с разветвленной цепью. Эти вещества могут использоваться отдельно или в комбинации, в зависимости от требуемых свойств. Кроме того, можно использовать липиды с высокой температурой плавления, а также белый мягкий парафин и/или жидкий парафин или другие минеральные масла.
Составы, подходящие для местного глазного применения, также включают глазные капли, в которых активные ингредиенты находятся в растворенном или взвешенном состоянии в подходящем носителе, особенно водном растворителе. Обычно в таких составах; выбираемая ~ концентрация активного ингредиента составляет от 0,5 до 20%, преимущественно от 0,5 до 10%, в частности около 1,5 масс.%.
Препараты для парентерального введения могут быть в форме водных или неводных изотонических стерильных инъекционных растворов или суспензий. Такие растворы или суспензии могут быть приготовлены из стерильных порошков или гранул, используя один или более носителей или растворителей, перечисленных для использования в составах для перорального введения, или с помощью других подходящих диспергирующих или смачивающих или суспендирующих веществ. Соединения могут быть растворены в воде, полиэтиленгликоле, пропиленгликоле, этаноле, кукурузном, хлопковом, арахисовом, кунжутном масле, бензиловом спирте, хлориде натрия, и прочих буферных растворах.
Активный ингредиент также может быть введен в организм пациента путем инъекций в композиции с подходящими носителями, включающими физиологический раствор, раствор декстрозы, солюбилизирующий растворитель (например, пропиленгликоль), или мицеллярный солюбилизатор (например, Твин 80). Кроме того, в качестве растворителей или суспендирующих средств часто применяют стерильные масла. Для этих целей подойдет любое нелетучее масло, в том числе синтетические моно- и диглицериды. Кроме того, при подготовке инъекций можно использовать жирные кислоты, такие как олеиновая кислота.
Для внутрилегочного введения лекарственная форма может представлять собой аэрозоль (в том числе порошковый) и вводиться при помощи ингалятора.
Суппозитории для ректального введения лекарства могут быть выполнены смешением лекарства с подходящим нераздражающим наполнителем, таким как масло какао и полиэтиленгликоль, твердым при обычной температуре, но жидким при ректальной температуре, так что наполнитель расплавляется в прямой кишке и высвобождает лекарство.
Лекарственная форма может быть подвергнута обычным фармацевтическим операциям, таким как стерилизация, и/или может содержать общепринятые добавки, такие как консерванты, стабилизаторы, увлажнители, эмульгаторы, буферы и т.д. Таблетки и пилюли могут дополнительно иметь энтеросолюбильное покрытие. Такие композиции могут также содержать вспомогательные вещества, такие как увлажнители, подсластители, ароматизаторы, дезодорирующие агенты.
Лекарственная форма данного изобретения может содержать соединение описанной здесь формулы или его фармацевтически приемлемую соль, и дополнительный препарат, например, выбранный из числа следующих: ингибитор киназы, антидепрессант, противоопухолевый препарат, противовирусный препарат, противовоспалительный препарат, противогрибковый препарат или соединение против сосудистой гиперпролиферации, и любой фармацевтически приемлемый носитель, адъювант или растворитель.
Термин «фармацевтически допустимый носитель или адъювант» означает носитель или адъювант, который может быть введен в организм пациента совместно с соединением, составляющем суть данного изобретения, и который не разрушает фармакологической активности этого соединения, и является нетоксичным при введении в дозах, достаточных для доставки терапевтического количества соединения.
Фармацевтически допустимые носители, адъюванты и растворители, которые могут быть использованы в фармацевтической композиции данного изобретения, включают, но не ограничиваются, ионитами, оксидом алюминия, стеарат алюминия, лецитин, самоэмульгирующиеся системы доставки лекарств (SEDDS), такие как полиэтиленгликольсукцинат d-альфа-токоферола, поверхностно-активные вещества в фармацевтических формах, такие как Твины, или другие аналогичные полимерные матрицы доставки, сывороточные белки, такие как человеческий сывороточный альбумин, буферы, такие как фосфонаты, глицин, сорбиновая кислота, сорбат калия, парциальные смеси глицеридов растительных насыщенных жирных кислот, воду, соли или электролиты, такие как протаминсульфат, гидрофосфат натрия, гидрофосфат калия, хлорид натрия, цинковые соли, коллоидный диоксид кремния, силикат магния, поливинилпирролидон, вещества на основе целлюлозы, полиэтиленгликоль, натрий карбокси-метилцеллюлоза, полиакрилаты, воск, полиэтиленовые-полиоксипропиленовые блок-сополимеры, полиэтиленгликоль и ланолин. Цикл о декстрины, такие как u-, Р- и y-циклодекстрин или химически модифицированные производные, такие как гидроксиалкил-циклодекстрины, включая 2- и 3-гидроксипропил-циклодекстрины или другие растворимые производные, также могут быть с успехом использованы для повышения доставки соединения описанной здесь формулы.
Фармацевтические композиции настоящего изобретения могут быть введены в организм пациента пероральным путем в любой доступной форме дозирования, включая, но не ограничиваясь, капсулами, таблетками, эмульсиями, водными суспензиями, дисперсиями и растворами. В случае таблеток для перорального употребления, часто используемые носители включают лактозу и кукурузный крахмал. Кроме того, обычно добавляют смазывающие агенты, такие как стеарат магния. Для приема внутрь в форме капсул, используемые разбавители включают лактозу и сухой кукурузный крахмал. В случае водных суспензий и/или эмульсий для перорального введения, активный ингредиент может суспендироваться или растворяться в масляной фазе в сочетании с эмульгирующим или суспердирующим агентом.
Лекарственные формы данного изобретения могут содержать подсластители и/или ароматизаторы и/или красители.
Лекарственные формы данного изобретения могут содержать составы, полученные методами использования липосом или микрокапсуляционные методы, методами приготовления наноформ препарата, и прочие примеры, известные в фармацевтике.
Применение соединений в комбинированной терапии
Несмотря на то, что соединения данного изобретения могут вводиться в качестве индивидуального активного фармацевтического средства, их также можно использовать в сочетании с одним или несколькими соединениями изобретения, или одним или несколькими другими агентами. При совместном приеме внутрь терапевтические агенты могут представлять собой разные лекарственные формы, которые вводятся одновременно или последовательно в разное время, либо терапевтические агенты могут быть объединены в одну лекарственную форму.
Фраза «комбинированная терапия» в отношении соединений данного изобретения в сочетании с другими фармацевтическими агентами, означает одновременный или последовательный прием всех агентов, который так или иначе обеспечит благоприятное воздействие сочетания лекарств. Совместное введение подразумевает, в частности, совместную доставку, например, в одной таблетке, капсуле, инъекции или в другой форме, имеющий фиксированное соотношение активных веществ, также как и одновременную доставку в нескольких, отдельных лекарственных формах для каждого соединения соответственно.
Таким образом, введение соединений данного изобретения может быть осуществлено в сочетании с дополнительными методами лечения, известными специалистам в области профилактики и лечения аллергических, аутоиммунных, онкологических и других заболеваний, включающими лучевую терапию, применение имунодепресантов, цитостатических и цитотоксических препаратов и препаратов для подавления симптомов или побочных эффектов одного из лекарств.
Если лекарственная форма представляет собой фиксированную дозу, такая комбинация использует соединения данного изобретения в приемлемом дозовом диапазоне. Вещества данного изобретения также могут быть введены в организм пациента последовательно с другими противоопухолевыми, цитотоксическими или подавляющими имунитет агентами, в том случае, когда комбинация этих препаратов невозможна. Изобретение не ограничено последовательностью введения; соединения данного изобретения могут быть введены в организм пациента совместно, до или после введения другого противоопухолевого или цитотоксического препарата.
Терапевтические наборы
Предметом настоящего изобретения являются также фармацевтические наборы для осуществления удобной и эффективной терапии. В общем случае, фармацевтический набор или комплект включает один или несколько контейнеров, заполненных одной или более фармацевтической композицией, описанной в изобретении. Такие наборы особенно удобны для доставки твердых форм для перорального введения, таких как таблетки или капсулы. Такой набор обычно содержит определенное число единичных доз, а также может включать карточки с дозами, расположенными в порядке их предполагаемого использования. По желанию, также может быть дополнительно предоставлена памятка, например в виде чисел, букв или другой маркировки, или календаря, обозначающего график лечения с отмеченными днями для приема дозы. Кроме того, для каждодневного применения, в предоставляемый фармацевтический набор могут быть включены дозы плацебо, кальциевые добавки, в той же или другой форме. Опционально, вместе с таким контейнером(ами), может прилагаться уведомление в форме, установленной правительственным учреждением, регулирующим производство, использование или сбыт фармацевтической продукции, в котором отражены разрешения этого учреждения для производства, использования или сбыта препарата для использования на людях.
Возможность осуществления изобретения может быть проиллюстрирована следующими примерами.
Примеры
Синтез дихлор-1,3,5-триазина.
В соответствии со схемой I из коммерчески доступных исходных веществ осуществляется синтез
2,4-дихлор-1,3,5-триазина
Синтез N-цианохлорформамидина

6.40 г (71 ммоль) Дицианамида натрия растворяют в воде и быстро добавляют к 50 мл концентрированной соляной кислоты, охлажденной до -30°C. Реакционную смесь перемешивают при данной температуре 15 минут, затем нагревают до 35°C и через 5 минут снова охлаждают до 4°C после чего перемешивают при заданной температуре еще 45 минут. Образовавшийся осадок отфильтровывают, промывают небольшим количеством воды и сушат в вакууме 20 часов. Получают 2.90 г (39%) N-цианохлорформамидина.
Синтез 2,4-дихлор-1,3,5-триазина

К раствору 2.5 мл (34.0 ммоль) диметилформамида в 50 мл безводного дихлорметана при комнатной температуре прибавляют 2.8 мл (30.0 ммоль) POCl3, реакционную смесь перемешивают 10 минут, прибавляют 2.9 г (29.0 ммоль) N-цианохлороформамидина и оставляют при перемешивании на ночь, затем, к реакционной смеси прибавляют еще 100 мл дихлорметана и промывают органическую фазу водой (4×15 мл), сушат над Na2SO4, растворитель удаляют. Получают 1.7 г (40%) 2,4-дихлор-1,3,5-триазина.
Синтез 6-амино-2,2-дифтор-2H-бензо[8][1,4]-оксазин-3(4H)-она
В соответствии со схемой V из коммерчески доступных исходных веществ осуществляется синтез
6-амино-2,2-дифтор-2H-бензо[8][1,4]-оксазин-3(4H)-она.
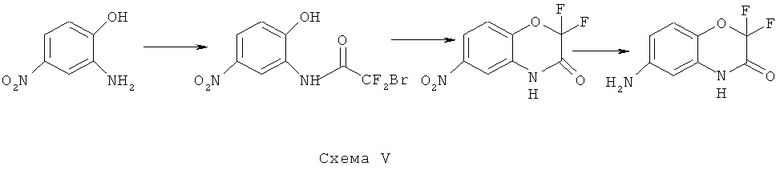
Синтез 2-бром-2,2-дифтор-N-(2-гидрокси-5-нитрофенол)ацетамида

К раствору 25.0 г (162 ммоль) 2-амино-4-нитрофенола и 21.9 г (170 ммоль) основания Хьюнинга в 250 мл безводного ацетонитрила прибавляют по каплям 29.7 г (170 ммоль) хлорангидрида бромдифторуксусной кислоты, поддерживая температуру реакционной смеси около 0°C. После окончания прибавления реакционную смесь перемешивают 4 ч поддерживая заданную температуру и оставляют на ночь. Растворитель удаляют в вакууме, к остатку прибавляют 500 мл этилацетата и промывают полученный раствор насыщенным раствором NaCl (3×70 мл), органическую фазу сушат над MgSO4, растворитель удаляют в вакууме. Получают 41.8 г (83%) продукта в виде темно коричневого масла, которое используется в следующей стадии без дополнительной очистки.
Синтез 2,2-дифтор-6-нитро-2Н-бензо[8][1,4]-оксазин-3(4Н)-она
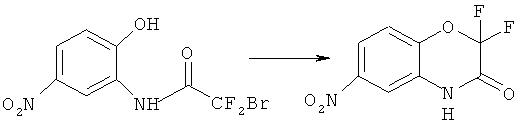
41.8 г (134 ммоль) дифторбромацетанилида, полученного на предыдущей стадии, растворяют в 200 мл безводного дегазированного ДМФА и прибавляют 20.5 г (148 ммоль) прокаленного в вакууме К2СО3, реакционную смесь нагревают до 85°C и выдерживают при этой температуре и энергичном перемешивании в течении 40 ч. Растворитель удаляют в вакууме, осадок экстрагируют этилацетатом (3×800 мл), экстракты объединяют, растворитель удаляют в вакууме роторного испарителя. Остаток разделяют хроматографически используя систему пентан-эфир нарастающей полярности. Получают 19.1 г (62%) продукта в виде бурых кристаллов.
Синтез 6-амино-2,2-дифтор-2H-бензо[8][1,4]-оксазин-3(4Н)-она
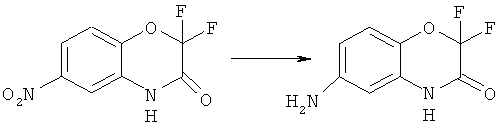
К раствору 19.1 г (83 ммоль) нитропроизводного 2,2-дифтор-6-нитро-2H-бензо[8][1,4]-оксазин-3(4H)-она в 200 мл метанола добавляют 840 мг 10% палладия на угле и гидрируют при 1 атм. водорода в течение 20 часов. После поглощения рассчитанного количества водорода катализатор отфильтровывают, растворитель удаляют в вакууме. Остаток разделяют хроматографически используя систему хлороформ-метанол нарастающей полярности Получают: 14.5 г (87%) продукта 6-амино-2,2-дифтор-2H-бензо[8][1,4]-оксазин-3(4H)-она.
Синтез 6-амино-2,2-диметил-2Н-пирид[3,2-b][1,4]оксазин-3(4Н)-она
В соответствии со схемой VI из коммерчески доступных исходных веществ осуществляется синтез
6-амино-2,2-диметил-2Н-пирид[3,2-b][1,4]оксазин-3(4Н)-она.
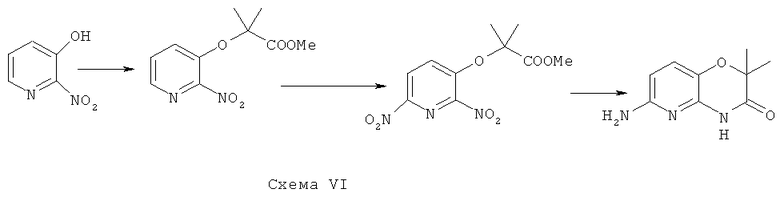
Синтез метилового эфира 2-метил-2-[(2-нитропиридин-3-ил)окси]пропановой кислоты
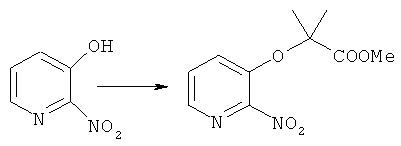
К раствору 0.31 г (2.2 ммоль) 2-нитро-3-гидроксипиридина в 5 мл безводного ДМФА прибавляют 1.10 г (8 ммоль) K2CO3 и 0.52 г (2.5 ммоль) метил диметилбромацетата, реакционную смесь перемешивают при 50°C 80 часов, затем растворитель удаляют в вакууме, остаток экстрагируют этилацетатом (3×30 мл) промывают на фильтре водой (3×10 мл), высушивают и перекристаллизовывают из бензола. Получают: 0.46 г (87%) продукта.
Синтез метилового эфира 2-метил-2-[(2,6-динитропиридин-3-ил)окси]пропановой кислоты
0.46 г (1.8 ммоль) нитропроизводного растворяют в 5 мл концентрированной серной кислоты и прибавляют 0.36 г (3.5 ммоль) сухого нитрата калия. Реакционную смесь перемешивают 4 часа при 20°C, выливают в воду, образующийся осадок отфильтровывают, многократно промывают на фильтре холодной водой до нейтральной реакции промывных вод и сушат. Получают: 0.39 г (71%) продукта метилового эфира 2-метил-2-[(2,6-динитропиридин-3-ил)окси]пропановой кислоты.
Синтез 6-амино-2,2-диметил-2Н-пирид[3,2-b][1,4]оксазин-3(4Н)-она
Раствор 0.39 г (1.37 ммоль) динитропроизводного метилового эфира 2-метил-2-[(2,6-динитропиридин-3-ил)окси]пропановой кислоты в 20 мл ДМФА гидрируют на 80 мг 5% палладия на угле при 1 атм. водорода в течение 6 часов. После поглощения рассчитанного количества водорода катализатор отфильтровывают, растворитель удаляют в вакууме при 70°С.Полученный остаток переносят на фильтр, промывают водой (2x10 мл), сушат и перекристал-лизовывают из в этанола. Получают: 0.22 г (84%) продукта 6-амино-2,2-диметил-2H-пирид [3,2-b][1,4] оксазин-3 (4H)-она.
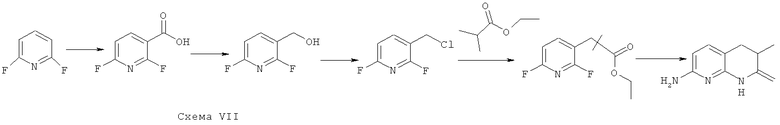
Синтез 7-амино-3,3-диметил-3,4-дигидро-1,8-нафтаридин-2(1H)-она
В соответствии со схемой VII из коммерчески доступных исходных веществ осуществляется синтез
7-амино-3,3-диметил-3,4-дигидро-1,8-нафтаридин-2(1H)-она.
Синтез 2,6-дифторникотновой кислоты
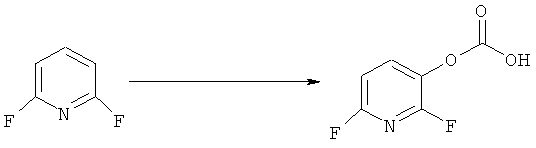
К раствору 337 мл (0.54 моль) 1.6М раствора н-бутиллития в гексане в 600 мл ТГФ в атмосфере аргона прибавляют раствор 67.5 г (0.54 моль) диизопропиламина в 150 мл ТГФ. Реакционную смесь перемешивают 40 мин при 10°C, после при энергичном перемешивании охлаждают до -78°C и прибавляют раствор 57.5 г (0.5 моль) 2,6-дифторпиридина в 150 мл ТГФ поддерживая температуру около -78°C. Реакционную смесь перемешивают 2.5 часа и прибавляют ~80 г колотого «сухого» льда, после чего перемешивают еще 45 минут, затем медленно дают нагреться до 5°С и выливают в смесь 5 л воды со льдом. Полученный раствор трижды экстрагируют этилацетатом (3×200 мл), водный слой подкисляют 6М соляной кислотой до pH ~4 и экстрагируют хлороформом (4×500 мл). Объедененные органические экстракты промывают рассолом (3×200 мл) и сушат над MgSO4. Растворитель удаляют в вакууме, остаток перекристаллизовывают из смеси эфиртескан 1:1. Получают: 66.0 г (83%) продукта в виде бесцветных кристаллов.
Синтез 2,6-дифтор-3-гидроксиметилпиридина

К охлажденному до 0°C раствору 55.65 г (0.35 моль) 2,6-дифторникотновой кислоты в 400 мл безводного ТГФ, в атмосфере аргона, прибавляют 1500 мл (1.2 моль) 0.8М растор борана в ТГФ. Полученную смесь перемешивают 4 часа при комнатной температуре, после чего кипятят 140 часов, охлаждают до -10°C и аккуратно, по каплям, не допуская роста температуры выше 5°C добавляют 920 мл 4М раствора NaOH в воде. Водный слой дополнительно насыщают NaCl, органическую фазу отделяют, водную экстрагируют эфиром (3×500 мл). Органические фракции объединяют, промывают рассолом (2x200 мл) и сушат над MgSO4, фильтруют через слой силикагеля, растворитель удаляют. Получают: 34.0 г (67%) 2,6-дифтор-3-гидроксиметилпиридина, который используют в следующей стадии без дополнительной очистки.
Синтез 2,6-дифтор-3-хлорметилпиридина

14.5 г (0.1 моль) Полученного на предыдущей стадии 2,6-дифтор-3-гидроксиметилпиридина растворяют в 200 мл сухого хлороформа и прибавляют, поддерживая температуру около 0°C, 8.8 мл (~0.12 моль) тионилхлорида. Реакционую смесь перемешивают 4.5 часа, после чего удаляют растворители в вакууме, а остоток промывают эфиром (3×100 мл). Получают: 16.0 г.(98%) 2,6-дифтор-3-хлорметилпиридина.
Синтез этил 3-(2,6-дифторпиридин-3-ил)-2,2-диметилпропаноата

К раствору 50 мл (0.08 моль) 1.6М раствора н-бутиллития в гексане в 100 мл безводного ТГФ в атмосфере аргона прибавляют раствор 13.9 мл (0.08 моль) диизопропиламина в 150 мл безводного ТГФ. Реакционную смесь перемешивают 1 час при 10°C, после при энергичном перемешивании охлаждают до -78°C и прибавляют раствор 11.6 г (0.1 моль) этил изобутирата в 100 мл ТГФ. Реакционную смесь перемешивают в течение часа при -78°C, затем еще в течение часа при -50°C и поддерживая заданную температуру прибавляют 9.8 г (0.06 моль) 2,6-дифтор-3-хлорметилпиридина, перемешивают реакционную смесь еще 2 часа при -50--40°C, после чего оставляю на ночь, выливают в 1 л холодной воды и подкисляют до рн~4, экстрагируют этилацетатом (3×300 мл). Объединенные органические фазы промывают водой до нейтральной реакции, затем рассолом и сушат над MgSO4, растворитель удаляют, остаток сушат и разделяют хроматографически (эфирггексан). Получают 3.5 г (24%) этил 3-(2,6-дифторпиридин-3-ил)-2,2-диметилпропаноата.
Синтез 7-амино-3,3-диметил-3,4-дигидро-1,8-нафтаридин-2(1Н)-она
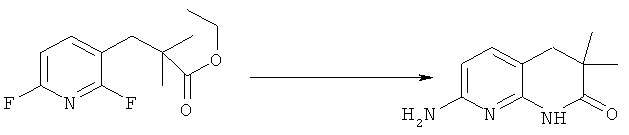
К раствору 4.87 г (0.02 моль) этил 3-(2,6-дифторпиридин-3-ил)-2,2-диметилпропаноата в 20 мл безводного этанола прибавляют 15 мл 6М раствора аммиака в безводного этаноле. Реакционную смесь нагревают в автоклаве 6 часов при 60°C, затем добавляют еще 10 мл 6М раствора аммиака в безводного этаноле и нагревают в автоклаве еще 6 часов при 120°C, охлаждают, растворитель удаляют, остаток разделяют хроматографически (дихлорметан:метанол 9:1->4:1). Получают: 0.61 г (16%) продукта.
Синтез 6-амино-2,2-диметил-2Н-пиридо[3,2-b][1,4]тиазин-3(4H)-она
В соответствии со схемой VIII из коммерчески доступных исходных веществ осуществляется синтез
6-амино-2,2-диметил-2H-пиридо[3,2-b][1,4]тиазин-3(4H)-она.

Синтез N-(6-фторпиридин-2-ил)пивалоиламида
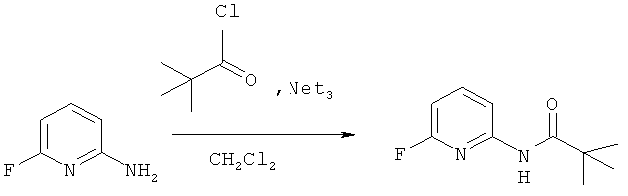
Раствор 13.8 г (0.11 моль) триметилацетилхлорида в 150 мл дихлорметана прибавляют охлажденному до 0°C раствору 11.2 г (0.1 моль) 2-амино-6-фторпиридина в и 15.3 мл (0.11 моль) триэтиламина в 200 мл дихлорметана. Полученную смесь перемешивают 2 часа при 0°C и оставляют на ночь, после чего выливают в воду. Органический слой промывают насыщенным раствором NaHCO3 (4×70 мл) и сушат над MgSO4, растворитель удаляют, остаток перекриста-лизовывают из гексана. Получают 17.2 г (88%) продукта.
Синтез изопропил 2-((2-фтор-6-пивалоиламидопиридин-3-ил)тио)-2-метилпропаноата
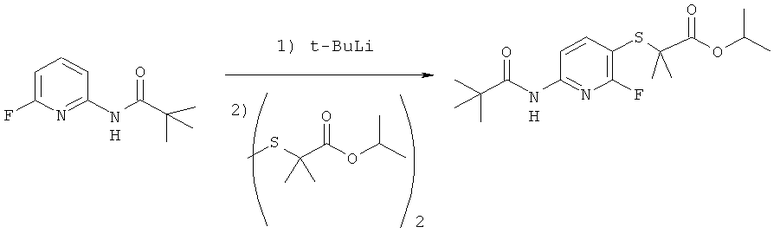
Раствор 14.7 г (0.075 моль) N-(6-фторпиридин-2-ил)пивалоиламида в 400 мл эфира охлаждают до -78°C и прибавляют ПО мл (0.19 моль) 1.7М раствора трет-бутиллития в пентане, после чего перемешивают реакционную смесь 4 часа поддерживая заданную температуру, затем добавляют 51.6 г (0.16 моль) дисульфида, дают реакционной смеси натрется до -35°C и перемешивают при этой температуре 4 часа, после чего дают медленно нагреется до комнатной температуры при которой выдерживают реакционную смесь в течение еще 4 часов. После чего выливают в 300 мл 10% раствора хлорида аммония в воде, органическую фазу отделяют, водную экстрагируют эфиром (2×200 мл) объединенные органические экстракты промывают насыщенным раствором NaHCO3 (3×100 мл), затем водой и сушат над MgSO4. Растворитель удаляют, остаток разделяют хроматографически (гексан:эфир 9.1->эфир). Получают: 15.3 г (57%) продукта в виде золотисто-желтого масла.
Синтез изопропил 2-((2-амино-6-пивалоиламидопиридин-3-ил)тио)-2-метилпропаноата

10.7 г (0.03 моль) изопропил 2-((2-фтор-6-пивалоиламидопиридин-3-ил)тио)-2-метилпропаноата растворяют в 100 мл 4М раствора аммиака в метаноле и нагревают реакционную смесь в автоклаве при 90°C в течение 70 часов, охлаждают, растворитель удаляют, остаток разделяют хроматографически (дихлорметан->дихлорметан:метанол 4:1). Получают: 3.08 г (29%) продукта.
Синтез изопропил 2-((2-амино-6-пивалоиламидопиридин-3-ил)тио)-2-метилпропаноата
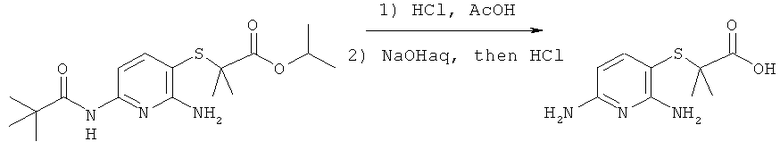
К 1.77 г (0.005 моль) изопропил 2-((2-амино-6-пивалоиламидопиридин-3-ил)тио)-2-метилпропаноата прибавляют 15 мл уксусной кислоты и 2 мл соляной кислоты, полученную смесь кипятят 4 часа, растворители удаляют в вакууме, к остатку прибавляют 8 мл 6М раствора NaOH в воде и выдерживают полученную смесь 8 часов при 80°C, охлаждают, подкисляют до нейтральной реакции, осадок отфильтровывают, промывают на фильтре холодной водой (3×2 мл) и сушат. Получают 0.42 г (37%) продукта.
Синтез 6-амино-2,2-диметил-2Н-пиридо[3,2-b][1,4]тиазин-3(4Н)-она
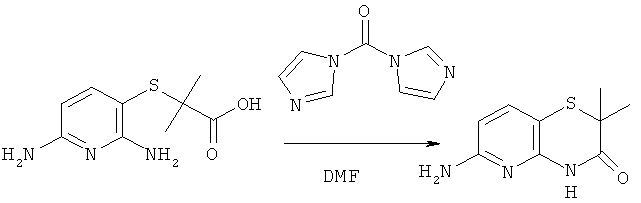
К раствору 1.14 г (0.005 моль) 2-((2,6-диаминопиридин-3-ил)тио)-2-метилприпионовой кислоты в 30 мл ДМФА прибавляют 1.30 г (0.008 моль) карбонилдиимидазола, реакционную смесь перемешивают при комнатной температуре 4 часа, растворитель удаляют в вакууме, остаток разделяют хроматографически (дихлорметан:метанол 19:1->4:1. Получают 0.71 г (68%) 6-амино-2,2-диметил-2H-пиридо[3,2-b][1,4]тиазин-3(4H)-она.
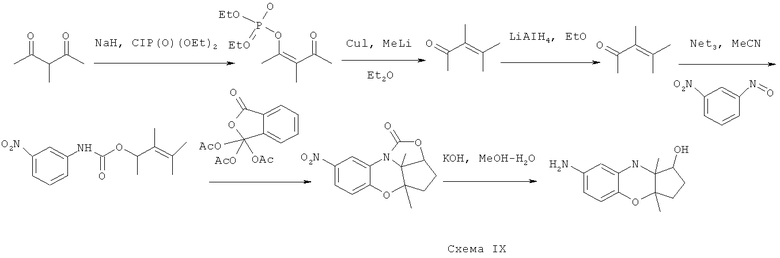
Синтез 7-амино-3a,9а-диметил-1,2,3,3a,9,9а-гексагидробензо[b]циклопента[e][1,4]оксазин-1-ола
В соответствии со схемой IX из коммерчески доступных исходных веществ осуществляется синтез
7-амино-3a,9а-диметил-1,2,3,3a,9,9а-гексагидробензо[b]циклопента[e][1,4]оксазин-1-ола.
Синтез диэтил(3-[(1)-3-метилпентен-2-он-4-ил] фосфат

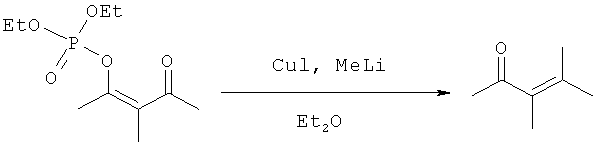
К суспензии 18.0 г (0.45 моль) гидрида натрия (60% суспензия в минеральном масле) в 180 мл диэтилового эфира, в атмосфере аргона, по каплям, поддерживая температуру около 0°C прибавляют 46.8 мл (0.40 моль) 3-метил-2,4-пентандиона в 200 мл эфира. Реакционную смесь перемешивают при 18°C 30 минут до прекращения выделения газа и добавляют раствор 77.6 г (0.45 моль) диэтилхлорфосфата в 200 мл эфира, поддерживая температуру около 0°C, после чего реакционную смесь перемешивали еще 4 часа при 18°C, после чего по каплям, поддерживая температуру около 0°C, прибавляют 15 мл насыщенного водного раствора хлорида аммония и перемешивают поддерживая заданную температуру до прекращения выделения газа (около 20 минут), органический слой отделяют, промывают насыщенным водным раствором NaHCO3 (1×100 мл), затем ледяной водой (2×100 мл) и сушат над MgSO4. Растворитель удаляют в вакууме при температуре не выше 10°C. Получают: 92.1 г (92%) продукта, который немедленно использовали в следующей стадии.
Синтез (Z)-3,4-диметилпентен-3--2
К суспензии 19.0 г (0.1 моль) сухого йодида меди (I) в 450 мл диэтилового эфира, в атмосфере аргона, прибавляют 420 мл (0.21 моль) 0.5М раствора метиллития в эфире, поддерживая температуру около -10°C. Реакционную смесь перемешивают до полного растворения осадка и образования слабо-желтого прозрачного раствора который охлаждают до -78°C и прибавляют раствор 9.4 г (0.04 моль) полученного ранее фосфоната в 120 мл диэтилового эфира. Реакционную смесь перемешивают при заданной температуре 8 часов, дают нагреться до комнатной температуры и прибавляют 100 мл насыщенного водного раствора хлорида аммония и перемешивают поддерживая температуру до прекращения выделения газа. Осадок отфильтровывают, промывают эфиром (3×100 мл), фильтрат промывают 20% водным раствором аммиака (3×100 мл), а затем водой (3×200 мл), сушат над Na2SO4, эфир удаляют, а остаток перегоняют в вакууме (76-79°С/50 торр). Получают: 4.0 г (89%) продукта.
Синтез (Z)-3,4-диметилпентен-3-ола-2
К раствору 2.3 г (~0.06 моль) алюмогидрида лития в 220 мл эфира, в атмосфере аргона, при 0°C, прибавляют 6.7 г (0.06 моль) (Z)-3,4-диметилпентен-3-она-2 в 80 мл диэтилового эфира, реакционную смесь перемешивают 4 часа, охлаждают до -5°C и прибавляют, по каплям воду, до тех пор пока прибавление новой порции не будет вызывать выделение газа, после чего перемешивают реакционную смесь еще 20 минут, осадок отфильтровывают, промывают эфиром (4×100 мл), органическую фазу отделяют, промывают водой (3×50 мл), сушат над Na2SO4, эфир удаляют, а остаток перегоняют (65-69°C/25 торр). Получают: 5.34 г (78%) продукта.
Синтез 3,4-Диметилпент-3-ен-2-ил(3-нитрофенил)карбамата. NEt3, MeCN

К раствору 2.28 г (0.02 моль) (г)-3,4-диметилпентен-3-ола-2 в ацетонитриле прибавляют 3.04 г (0.02 моль) 3-нитрофенилизоцианата и 1 каплю триэтиламина. Реакционную смесь перемешивают 5 часов при 50°C, растворитель удаляют, остаток разделяют хроматографически (гексан:этилацетат 9:1->1:1). Получают: 6.91 г (88%) продукта.
Синтез 2а1,4а-диметил-8-нитро-2а,3,4,4а-тетрагидро-2,5-диокса-9b-азапенталено[1,6-ab]нафталин-1(2а1Н)-она
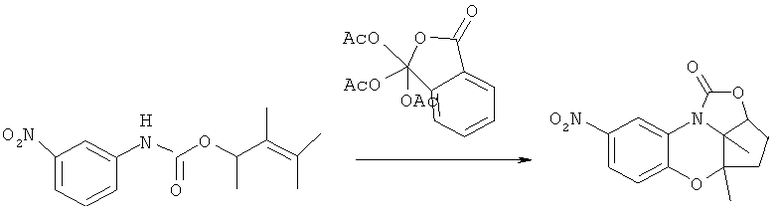
К раствору 5.57 г (0.02 моль) полученного на предыдущей стадии карбамата в 200 мл бензола прибавляют 17.00 г (~0.04 моль) перйодината Десс-Мартина и кипятят полученный раствор в течении 1.5 часов, охлаждают, прибавляют 300 мл этилацетата, промывают насыщенным водным раствором NaHCO3 (4×150 мл), затем рассолом (2×100 мл) и сушат над Na2S04. Растворители удаляют в вакууме остаток остаток разделяют хроматографически (гексан:этилацетат 9:1->1:1). Получают: 2.49 г (43%) продукта.
Синтез 8-амино-2а14а-диметил-2а,3,4,4а-тетрагидро-2,5-диокса-9b-азапенталено[1,6-ab]нафталин-1 (2а1Н)-она
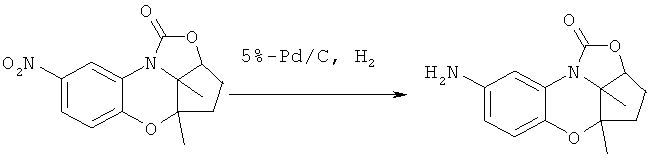
К раствору 2.90 г (0.01 моль) 2а',4а-диметил-8-нитро-2а,3,4,4а-тетрагидро-2,5-диокса-9b-азапенталено[1,6-ab]нафталин-1(2a1H)-она в 200 мл этанола прибавляют 300 мг 5% палладия на угле и гидрируют при 1 атм. водорода и температуре 20°C в течение 12 часов. После стандартной процедуры обработки реакционной смеси получают: 2.44 г (94%) продукта.
Синтез 7-амино-3a,9а-диметил-1,2)3,3a,9,9а-гексагидробензо[b]циклопента[e][1,4]окса-зин-1-ола
 К раствору 5.0 г гидроксида калия в 30 мл смеси метанол-вода (4:1 v/v) прибавляют 1.30 г (0.005 моль) 2а1,4а-диметил-8-амино-2а,3,4,4а-тетрагидро-2,5-диокса-9b-азапенталено[1,6-ab] нафталин-1(2a1H)-она и кипятят 60 часов в атмосфере аргона. Растворитель удаляют, к остатку прибавляют 600 мл эфира и 200 мл воды, органическую фазу отделяют, промывают рассолом (4×100 мл), сушат над MgSO4, растворитель удаляют, остаток разделяют хроматографи-чески (дихлорметан->дихлорметан:метанол 4:1). Получают: 0.90 г (77%) 7-амино-3a,9а-диметил-1,2,3,3a,9,9а-гексагидробензо[b]циклопента[e][1,4]оксазин-1-ола.
К раствору 5.0 г гидроксида калия в 30 мл смеси метанол-вода (4:1 v/v) прибавляют 1.30 г (0.005 моль) 2а1,4а-диметил-8-амино-2а,3,4,4а-тетрагидро-2,5-диокса-9b-азапенталено[1,6-ab] нафталин-1(2a1H)-она и кипятят 60 часов в атмосфере аргона. Растворитель удаляют, к остатку прибавляют 600 мл эфира и 200 мл воды, органическую фазу отделяют, промывают рассолом (4×100 мл), сушат над MgSO4, растворитель удаляют, остаток разделяют хроматографи-чески (дихлорметан->дихлорметан:метанол 4:1). Получают: 0.90 г (77%) 7-амино-3a,9а-диметил-1,2,3,3a,9,9а-гексагидробензо[b]циклопента[e][1,4]оксазин-1-ола.
Синтез 7′-аминоспиро[циклопропан-1,3′-пиридо[3,4-b][1,4]оксазина]-2′(1′H)-она
В соответствии со схемой X из коммерчески доступных исходных веществ осуществляется синтез
7′-аминоспиро [циклопропан-1,3′-пири до [3,4-b] [1,4]оксазина]-2′(1′′H)-она.

Синтез 5-((4-бензилкарбонилокси-1-метокси-1-оксобутан-2-ил)окси)-2-хлорпиридина
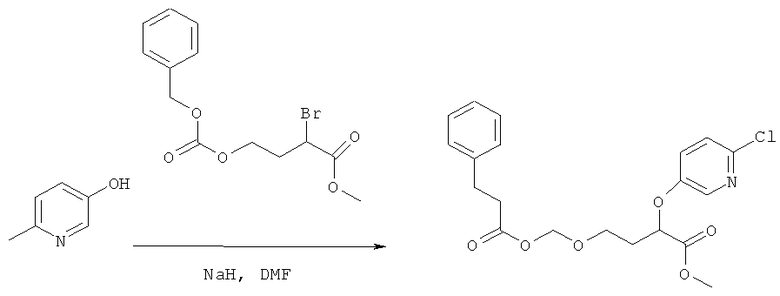
К раствору 23.3 г (0.18 моль) 3-гидрокси-5-хлорпиридина в 300 мл ДМФА прибавляют 4.8 г (0.20 моль) гидрида натрия (в виде 60% суспензии в минеральном масле). Реакционную смесь перемешивают 2 часа и прибавляют, поддерживая температуру около 30°C, 66.2 мл (0.2 моль) метил-4-(бензилоксикарбонилокси)-2-бромбутаноат.Реакционную смесь перемешивают при комнатной температуре 6 часов и оставляют на ночь. Выливают в 1.5 л ледяной воды и экстрагируют продукт дихлорметаном (3×300 мл). Объедененные экстракты промывают водой (3×100 мл) и сушат над Na2SO4. Получают: 58.0 г (79%) 5-((4-бензилкарбонилокси-1-метокси-1-оксобутан-2-ил)окси)-2-хлорпиридина.
Синтез 5-((4-бензилкарбонилокси-1-метокси-1-оксобутан-2-ил)окси)-2-хлорпиридин-1-оксида

К раствору 56.4 г (0.14 моль) метил 2-(6-хлорпиридин-3-илокси)пропионата в 500 мл пиридина прибавляют 35 мл (-0.3 моль) 30% водного раствора перекиси водорода и 35 мг метил три-оксорения. Смесь перемешивают 4 часа при комнатной темепратуре, охлаждают до 0°C и по каплям прибавляют 100 мл насыщенного водного раствора NaHCO3, органический слой отделяют, промывают насыщенным водным раствором NaHCCh (2×160 мл), а затем водой (2×100 мл). Органическую фазу сушат над MgSO4, растворитель удаляют. Получают: 57.55 г (97%) 5-((4-бензилкарбонилокси-1-метокси-1-оксобутан-2-ил)окси)-2-хлорпиридин-1-оксида.
Синтез 5-((4-бензилкарбонилокси-1-метокси-1-оксобутан-2-ил)окси)-2-хлор-4-нитропиридин 1-оксида
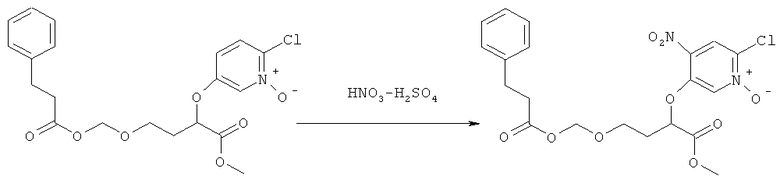
8.48 г (0.02 моль) N-оксида растворяют в 40 мл серной кислоты, охлаждают до 0°C и прибавляют 2 мл азотной кислоты. Реакционную смесь наргевают до 60°C, выдерживают при этой температуре 8 часов и выпивают в смесь воды и льда, нейтрализуют добавляя небольшими порциями гидрокарбонат натрия до рн~5 и экстрагируют этилацетатом (3×200 мл), органические фазы объединяют и промывают водой (3×100 мл), сушат над Na2SO4 и удаляют растворитель. Получают: 7.31 г (78%) 5-((4-бензилкарбонилокси-1-метокси-1-оксобутан-2-ил)окси)-2-хлор-4-нитропиридин-1-оксида.
Синтез 5-((4-бензилкарбонилокси-1-метокси-1-оксобутан-2-ил)окси)-2-хлор-4-нитропиридин 1-оксида
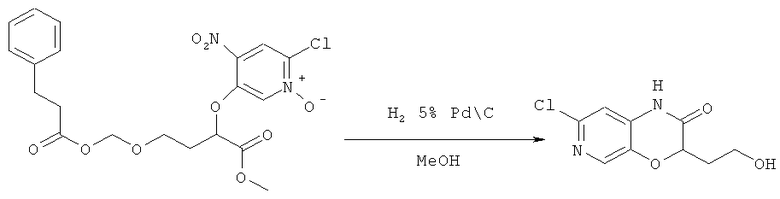
7.03 г (0.015 моль) нитросоединения растворяют в 100 мл метанола и гидрируют на скелетном никеле при 1атм. водорода и 10°C в течение 1.5 часов. Катализатор отфильтровывают, к фильтрату прибавляют 50 мл хлороформа и фильтруют через тонкий (~1 см) слой силикагеля, растворители удаляют, к остатку прибавляют 200 мл толуола и кипятят 4 часа, толуол удаляют, остаток разделяют хроматографически. Получают: 2.50 г (73%) 6-хлор-2-(2-гидроксиэтил)-2H-бензо[b][1,4]оксазина-3(4H)-она.
Синтез гидрохлорида 6-хлор-2-(2-хлорэтил)-2Н-бензо[b][1,4]оксазина-3(4Н)-она
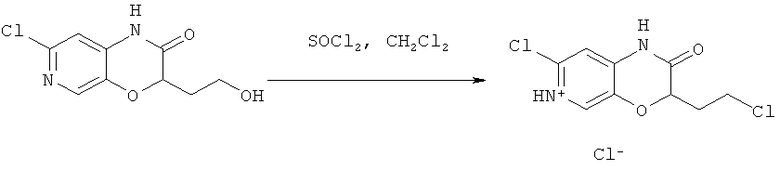
К раствору 11.43 г (0.05 моль) спирта в 200 мл дихлорметана прибавляют 4.4 мл (0.06 моль) тионил хлорида. Полученную смесь кипятят 4 часа, растворитель удаляют, остаток сушат. Получают: 13.75 г (97%) гидрохлорида 6-хлор-2-(2-хлорэтил)-2H-бензо[b][1,4]оксазина-3(4H)-она.
Синтез 7′-хлорспиро[циклопропан-1,3′-пиридо[3,4-b][1,4]оксазаина]-2′(1′Н)-она
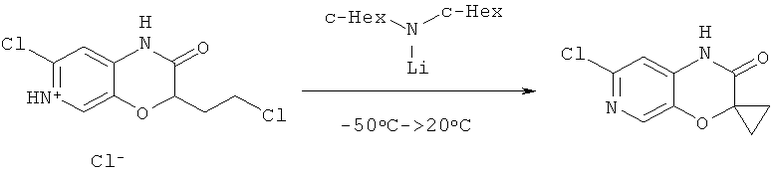
К раствору 43.8 мл (0.07 моль) 1.6М раствора м-бутиллития в гексане в 120 мл безводного ТГФ в атмосфере аргона прибавляют раствор 16 мл (0.07 моль) дициклогексиламина в 150 мл безводного ТГФ. Реакционную смесь перемешивают 2 часа при 20°C, после охлаждают до -50°С и прибавляют небольшими порциями 8.51 г (0.03 моль) гидрохлорида 6-хлор-2-(2-хлорэтил)-2H-бензо[b][1,4]оксазина-3(4H)-она. Реакционную смесь перемешивают 1 час при -50°C, после чего оставляю при перемешивании на ночь, после чего выливают в 800 мл холодной воды и подкисляют до рн~6, экстрагируют этилацетатом (3×200 мл). Объединенные органические фазы промывают водой до нейтральной реакции, затем рассолом и сушат над MgS04, растворитель удаляют, остаток сушат в вакууме и разделяют хромтографически (эфир:гексан). Получают 1.28 г (20%) 7′-хлорспиро[циклопропан-1,3′-пиридо[3,4-b][1,4]оксазина]-2′(1′H)-она.
Синтез 7′-аминоспиро[циклопропан-1,3′-пиридо[3,4-b][1,4]оксазина]-2′(1′Н)-она

Смесь 2.17 г (0.012 моль) бензофенонимина, трет-бутилат натрия 1.15 г (0.012 моль), 200 мг Pd2(dba)3 и 2.11 (0.01 моль) 7′-хлорспиро[циклопропан-1,3′-пиридо[3,4-b][1,4]оксазина]-2′(1′H)-она и 337 мг (0.6 ммоль) 2,2′-бис(дифенилфосфино)-1,Гбинафтила (в 40 мл толуола перемешивают при 80°C в течение 14 часов, охлаждают до комнатной температуры, прибавляют 100 мл 1М раствора соляной кислоты в смеси ТГФ:вода 1:1, перемешивают 2 часа, экстрагируют этилацетатом (3×200 мл), экстракты объединяют, промывают насыщенным водным раствором NaHCO3 (2×150 мл), сушат над MgSO4, растворитель удаляют, остаток разделяют хроматографически. Получают: 1.22 г (53%) 7′-аминоспиро[циклопропан-1,3′-пиридо[3,4-b][1,4]оксазина]-2′(1′H)-она.
Синтез 3′-амино-5′Н-спиро[иклопропан-1,7′-пиридо[2,3-b]пиразин]-6′ (8′H)-она
В соответствии со схемой XI из коммерчески доступных исходных веществ осуществляется синтез
3′-амино-5′Н-[иклопропан-1,7′-пиридо[2,3-b]пиразин]-6′(8′H)-она.
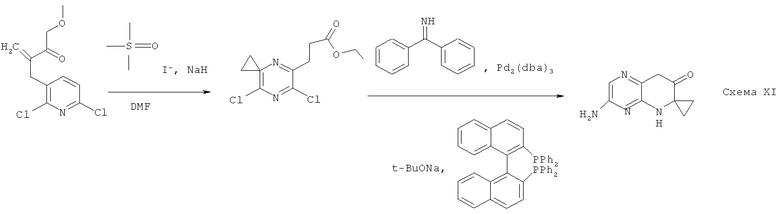
Синтез этил 1-((3,5-дихлорпиразин-2-ил)метил)циклопропиилкарбоксилата
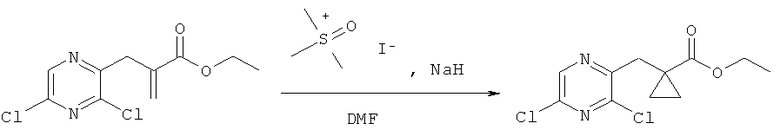
К суспензии 1.25 г (0.052 моль) гидрида натрия (60% суспензия в минеральном масле) в безводном ДМФА прибавляют 11.44 г (0.052 моль) йодида триметилсульфония и перемешивают полученную смесь 40 минут при комнатной температуре, затем прибавляют раствор 5.74 г (0.022 моль) этил 2-((3,5-дихлорпиразинил-2)метил)акрилата в 40 мл ДМФА. Реакционную смесь перемешивают 4 часа, выливают в 200 мл ледяной воды и экстагируют этилацетатом (3×250 мл). Объединенные экстракты промывают рассолом (3×100 мл) и сушат над MgSO4. Остаток растирают в смеси гексанэфир в результате чего происходит кристаллизация продукта, который отфильтровывают, промывают на фильтре семесью гексан:эфир (2×10 мл) и сушат. Получают: 4.90 г (81%) этил 1-((3,5-дихлорпиразин-2-ил)метил)циклопропиилкарбоксилата.
Синтез 7′-аминоспиро[циклопропан-1,3′-пиридо[3,4-b][1,4]оксазина]-2′(1′Н)-она

Смесь 5.80 г (0.032 моль) бензофенонимина, трет-бутилат натрия 3.07 г (0.032 моль), 356 мг Pd2(dba)3, 995 мг 2,2′-бис(дифенилфосфино)-1,1′бинафтила и 3.30 г (0.012 моль) полученного на предыдущей стадии этил 1-((3,5-дихлорпиразин-2-ил)метил)циклопропиилкарбоксилата в 40 мл толуола перемешивают при 90°C в течение 42 часов, охлаждают до комнатной температуры, прибавляют 100 мл 0.5М раствора соляной кислоты в смеси ТГФ:вода 1:1, перемешивают 2 часа, прибавляют 20 г твердого NaHCO3, перемешивают в течении 20 минут и экстрагируют этилацетатом (3×200 мл), экстракты объединяют, промывают насыщенным водным раствором NaHCO3 (2×150 мл), сушат над MgSO4, растворитель удаляют, остаток разделяют хро-матографически. Получают: 1.11 г (49%) 3′-амино-5′H-спиро[иклопропан-1,7′-пиридо[2,3-b]пиразин]-6′(8′H)-она.
1. Получение 2,2-дифтор-6-({4-[(3,4,5-триметоксифенил)амино]-1,3,5-триазин-2-ил} амино)-2H-бензо [8][1,4]-оксази н-3(4H)-она.
Ситнез 2,2-дифтор-6-({4-[(3,4,5-триметоксифенил)амино]-1,3,5-триазин-2-ил}амино)-2H-бензо[8][1,4]-оксазин-3(4H)-она осуществляется в соответствии со схемой I. При этом используется коммерчески доступный 3,4,5-триметоксианилин, и 6-амино-[(4-хлор-1,3,5-триазин-2-ил)амино]-2,2-дифтор-2H-бензо[8][1,4]-оксазин-3(4H)-он, синтезированный в соответствии со схемой V
Синтез 6-амино-[(4-хлор-1,3,5-триазин-2-ил)амино]-2,2-дифтор-2Н-бензо[8][1,4]-оксазин-3(4Н)-она
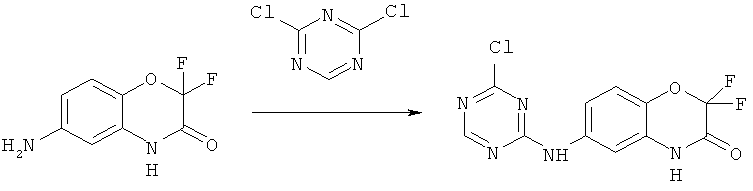
0.17 г (1.15 ммоль) 2,4-дихлор-1,3,5-триазина растворяют в 5 мл безводного дегазированного ДМФА, прибавляют 0.23 г (1.15 ммоль) 6-амино-2,2-дифтор-2H-бензо[8][1,4]-оксазин-3(4H)-она и перемешивают в атмосфере аргона при 60°C 8 часов. Растворитель удаляют в вакууме (при 5 мм. рт.ст.), остаток промывают холодной водой (2×5 мл), сушат и разделяют хроматографически используя в качестве элюента систему хлороформ:метанол нарастающей полярности. Получают: 0.29 г (81%) продукта 6-амино-[(4-хлор-1,3,5-триазин-2-ил)амино]-2,2-дифтор-2H-бензо[8][1,4]-оксазин-3(4H)-она.
Синтез 2,2-дифтор-6-({4-[(3,4,5-триметоксифенил)амино]-1,3,5-триазин-2-ил}амино)-2Н-бензо[8][1,4]-оксазин-3(4Н)-она
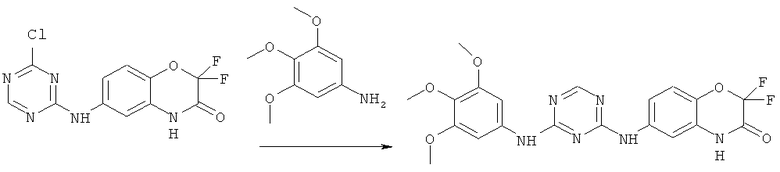
0.29 г (0.92 ммоль) соединения 6-амино-[(4-хлор-1,3,5-триазин-2-ил)амино]-2,2-дифтор-2H-бензо[8][1,4]-оксазин-3(4H)-она растворяют в 5 мл безводного дегазированного ДМФА, прибавляют 0.34 г (1.84 ммоль) 3,4,5-триметоксианилина и перемешивают в атмосфере аргона 6 часов при 110°C. Растворитель удаляют в вакууме (при 5 мм. рт.ст.), остаток промывают холодной водой (2×5 мл), сушат и разделяют хроматографически используя в качестве элюента систему хлороформ: метанол нарастающей полярности. Получают: 0.29 г (67%) 2,2-ди-фтор-6-({4-[(3,4,5-триметоксифенил)амино]-1,3,5-триазин-2-ил}амино)-2H-бензо [8] [1,4]-оксазин-3 (4H)-она.
1Н (500 MHz, DMSO-d6): 3.52 (c, зн, СН3), 3.70 (с, 6Н, 2СН3), 6.28 (с, 2Н, 2HAr), 7.10 (д. J=9.0Hz, 1H, HAr), 7.47 (д. J=9.1Hz, 1Н, HAr), 7.88 (уш.с, 1H, NH), 8.21 (уш.с, 1Н, NH), 8.00 (с, 1Н, HAr), 8.42 (c, 1Н, НАг), 9.26 (c, 1Н, C(O)NH).
2. Получение 2,2-дифтор-6-({4-[(1,3-бензодиоксол-5-ил)амино]-1,3,5-триазин-2-ил}амино)-2Н-бензо[8][1,4]-оксазин-3(4Н)-она.
Синтез 2,2-дифтор-6-({4-[(1,3-бензодиоксол-5-ил)амино]-1,3,5-триазин-2-ил}амино)-2Н-бензо[8][1,4]-оксазин-3(4Н)-она осуществляется в соответствии со схемой I. При этом используется коммерчески доступный 3,4-метилендиоксианилин и 2,2-дифтор-6-нитро-2H-бензо[8][1,4]-оксазин-3(4H)-он, синтезированный в соответствии со схемой V
Синтез 6-[(4-хлор-1,3,5-триазин-2-ил)амино]-2,2-дифтор-2Н-бензо[8][1,4]-оксазин-3(4Н)-она
0.17 г (1.15 ммоль) 2,4-дихлор-1,3,5-триазина растворяют в 5 мл безводного дегазированного ДМФА, прибавляют 0.20 г (1 ммоль) 6-амино-2,2-дифтор-2H-бензо[8][1,4]-оксазин-3(4H)-она и 0.2 мл (1.15 ммоль) основания Хюнинга и перемешивают в атмосфере аргона при 60°C 8 часов. Растворитель удаляют в вакууме (при 5 мм. рт.ст.), остаток промывают холодной водой (2×5 мл), сушат и разделяют хроматографически используя в качестве элюента систему хлороформ-.метанол нарастающей полярности. Получают: 0.29 г (81%) продукта 6-[(4-хлор-1,3,5-триазин-2-ил)амино]-2,2-дифтор-2H-бензо[8][1,4]-оксазин-3(4H)-она.
Синтез 2,2- дифтор-6-({4-[(1,3-бензодиоксол-5-ил)амино]-1,3,5-триазин-2-ил}амино)-2Н-бензо[8][1,4]-оксазин-3(4Н)-она
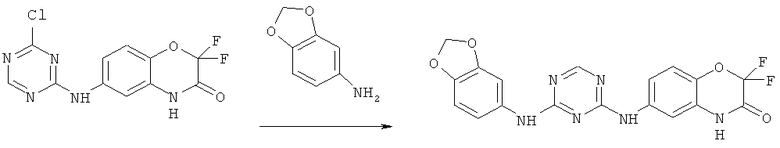
0.29 г (0.92 ммоль) 6-[(4-хлор-1,3,5-триазин-2-ил)амино]-2,2-дифтор-2H-бензо[8][1,4]-оксазин-3(4H)-она растворяют в 5 мл безводного дегазированного ДМФА, прибавляют 0.25 г (1.85 ммоль) 3,4-метилендиоксианилина и перемешивают в атмосфере аргона при 110°C 6 часов. Растворитель удаляют в вакууме (при 5 мм. рт.ст.), остаток промывают холодной водой (2×5 мл), сушат и разделяют хроматографически используя в качестве элюента систему хлороформ: метанол нарастающей полярности. Получают: 0.22 г (59%) 2,2- дифтор-6-({4-[(1,3-бензодиоксол-5-ил)амино]-1,3,5-триазин-2-ил}амино)-2H-бензо[8][1,4]-оксазин-3(4H)-она.
1Н (500 MHz, DMSO-d6): 6.01 (с, 2Н, СН2), 6.55 (с, 1Н, HAr), 6.58 (д. J=8.2 Hz, 1Н, HAr), 6.84 (д. J=8.2Hz, 1Н, HAr), 7.10 (д. J=9.0Hz, 1Н, HAr), 7.47 (д. J=9.0Hz, 1Н, HAr), 7.85 (уш.с, 1Н, NH), 8.21 (уш.с, 1Н, NH), 8.00 (с, 1H, НАг), 8.42 (с, 1H, HAr), 9.26 (с, 1H, C(O)NH).
3. Получение 2,2-диметил-6-({4-[(3,4,5-триметоксифенил)амино]-пиримидин-4-ил}амино)-2H-пирид[3,2-Ь][1,4]оксазин-3(4H)-она.
Ситнез 2,2-диметил-6-({4-[(3,4,5-триметоксифенил)амино]-пиримидин-4-ил}амино)-2H-пирид[3,2-b][1,4]оксазин-3(4H)-она осуществляется в соответствии со схемой I. При этом используется коммерчески доступный 2,2-дифтор-3,4-метилендиоксианилин, и 6-амино-2,2-диметил-2H-пирид[3,2-b][1,4]оксазин-3(4H)-она, синтезированный в соответствии со схемой VI
Синтез 6-[(4-хлор-1,3,5-триазин-2-ил)амино]-2,2-диметил-2Н-пирид[3,2-b][1,41оксазин-3(4Н)-она
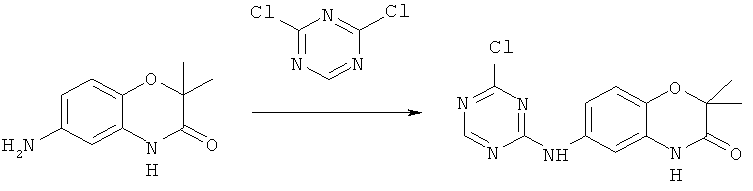
0.17 г (1.15 ммоль) 2,4-дихлор-1,3,5-триазина растворяют в 5 мл безводного дегазированного ДМФА, прибавляют 0.22 г (1.15 ммоль) 6-амино-2,2-диметил-2H-пирид[3,2-b][1,4]оксазин-3(4H)-она и 0.2 мл (1.15 ммоль) основания Хюнинга и перемешивают в атмосфере аргона при 60°C 8 часов. Растворитель удаляют в вакууме (при 5 мм. рт.ст.), остаток промывают холодной водой (2×5 мл), промывают этанолом (3×2 мл) и сушат. Получают: 0.28 г (80%) продукта 6-[(4-хлор-1,3,5-триазин-2-ил)амино]-2,2-диметил-2H-пирид[3,2-b][1,4]оксазин-3(4H)-она.
Синтез 2,2-диметил-6-({4-[(3,4,5-триметоксифенил)амино]-1,3,5-триазин-2-ил}амино)-2Н-пирид[3,2-b][1,4]оксазин-3(4Н)-она.
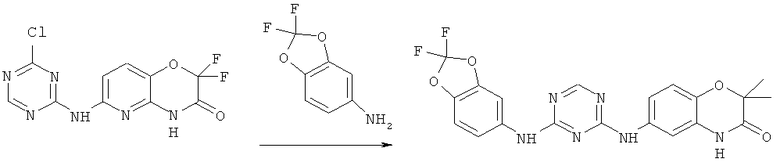
0.28 г (0.92 ммоль) полученного в предыдущей методике 6-[(4-хлор-1,3,5-триазин-2-ил)амино]-2,2-диметил-2H-пирид[3,2-b][1,4]оксазин-3(4H)-она растворяют в 5 мл безводного дегазированного ДМФА, прибавляют 0.32 г (1.84 ммоль) 2,2-дифтор-5-амино-1,3-бенздоксолана и перемешивают в атмосфере аргона при 110°C 6 часов. Растворитель удаляют в вакууме (при 5 мм. рт.ст.), остаток промывают холодной водой (2×5 мл), сушат и разделяют хроматографически используя в качестве элюента систему хлороформ: метанол нарастающей полярности. После стандартной процедуры обработки получают: 0.28 г (65%) целевого соединения 2,2-диметил-6-({4-[(3,4,5-триметоксифенил)амино]-1,3,5-триазин-2-ил }амино)-2H-пирид[3,2-b] [1,4]оксазин-3(4H)-она.
1Н (500 MHz, DMSO-d6): 1.47 (с, 6Н, 2СНЗ), 6.81 (с, 1H, HAr), 7.03 (д. J=8.2Hz, 1Н, HAr), 7.09 (д. J=8.2Hz, 1H, HAr), 7.31 (д. J=8.6Hz, 1Н, HAr), 7.43 (д. J=8.6Hz, 1Н, НАг), 7.84 (уш.с, 1Н, NH), 8.42 (с, 1Н, HAr), 8.39 (уш.с, 1H, NH), 9.28 (с, 1H, C(O)NH).
4. Получение 7-((4-(циклопропиламино)-1,3,5-триазин-2-ил)амино)-3,3-диметил-3,4-дигидро-1,8-нафтраридин-2(1H)-она.
Ситнез 7-((4-(циклопропиламино)-1,3,5-триазин-2-ил)амино)-3,3-диметил-3,4-дигидро-1,8-нафтраридин-2(1H)-она осуществляется в соответствии со схемой I. При этом используется коммерчески доступный 7-амино-3,3-диметил-3,4-дигидро-1,8-нафтаридин-2(1H)-она, синтезированный в соответствии со схемой VI
Синтез 7-((4-хлор-1,3,5-триазин-2-ил)амино)-3,3-диметил-3,4-дигидро-1,8-нафтраридин-2(1H)-она
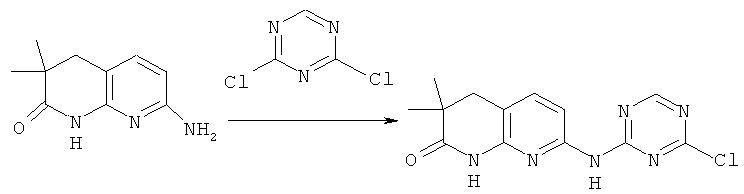
12.0 г (8 ммоль) 2,4-дихлор-1,3,5-триазина растворяют в 12 мл безводного дегазированного ДМФА, охлаждают до -5°С и прибавляют 11.5 г (6 ммоль) 7-амино-3,3-диметил-3,4-дигидро-1,8-нафтаридин-2(1H)-она и перемешивают 2 часа при 20°С, затем снова охлаждают до -5°C и прибавляют 11.8 мл (6.8 ммоль) основания Хюнинга, переменивают 2 часа при комнатной температуре, а затем еще 5 часов при 40°C. Растворитель удаляют в вакууме (при 5 мм. рт.ст.), остаток промывают холодной водой (2×5 мл), изопропанолом (2×5 мл), эфиром (2×10 мл) и сушат. Получают: 16.6 г (91%) 7-((4-хлор-1,3,5-триазин-2-ил)амино)-3,3-диметил-3,4-дигидро-1,8-нафтраридин-2(1H)-она.
Синтез 7-((4-(циклопропиламино)-1,3,5-триазин-2-ил)амино)-3,3-диметил-3,4-дигидро-1,8-нафтраридин-2(1Н)-она
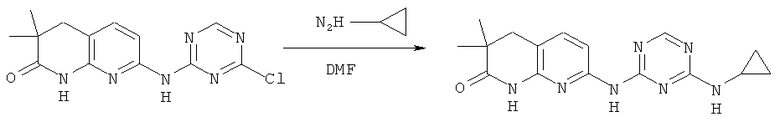
3.66 г (1.2 ммоль) полученного в предыдущей методике монохлорпроизводного растворяют в 5 мл безводного дегазированного ДМФА, прибавляют 1.71 г (3.0 ммоль) циклопропиламина и нагревают в автоклаве при 90°C в течение 6 часов. Растворитель удаляют в вакууме (при 5 мм. рт.ст.), остаток промывают холодной водой (2×5 мл), сушат и разделяют хроматографически используя в качестве элюента систему хлороформ: метанол нарастающей полярности. После стандартной процедуры обработки получают: 3.04 г (78%) 7-((4-(циклопропиламино)-1,3,5-триазин-2-ил)амино)-3,3 -диметил-3,4-дигидро-1,8-нафтраридин-2(1H)-она.
1Н (500 MHz, DMSO-d6): 0.91 (с, 6Н, 2СНЗ), 1.11-1.19 (м, 4Н, 2СН2), 2.28 (с, 2Н, СН2), 3.77-3.82 (м, 1H, СН), 7.14 (д. J=5.6Hz, 1H, HAr), 7.45 (д. J=5.7Hz, 1H, HAr), 7.58 (уш.с, 1H, NH), 8.26 (с, 1Н, HAr), 8.50 (уш.с, 1H, NH), 9.47 (с, 1Н, C(O)NH).
Элементный состав: C(58.93%), H(5.81%), N(30.22%).
5. Получение 3,3-диметил-7-((4-((2,2,3,3-тетрафтор-2,3-Дигидробензо[b] [1,4]диоксин-6-ил)амино)-1,3,5-триазин-2-ил)амино)-3,4-дигидро1,8-нафтраридин-2(1H)-она.
Ситнез 3,3-диметил-7-((4-((2,2,3,3-тетрафтор-2,3-дигидробензо[b] [1,4]диоксин-6-ил)амино)-1,3,5-триазин-2-ил)амино)-3,4-дигидро1,8-нафтраридин-2(1Н)-она осуществляется в соответствии со схемой I. При этом используется коммерчески доступный 7-амино-3,3-диметил-3,4-дигидро-1,8-нафтаридин-2(1Н)-она, синтезированный в соответствии со схемой VI
Синтез 7-((4-хлор-1,3,5-триазин-2-ил)амино)-3,3-диметил-3,4-дигидро-1,8-нафтраридин-2(1Н)-она
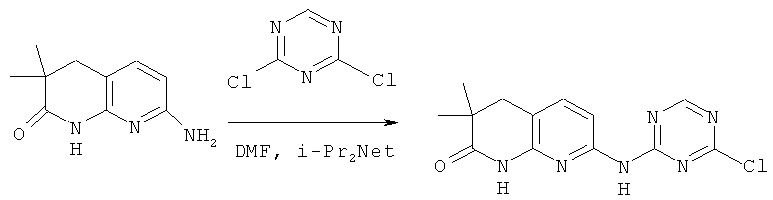
12.0 г (8 ммоль) 2,4-дихлор-1,3,5-триазина растворяют в 12 мл безводного дегазированного ДМФА, охлаждают до -5°C и прибавляют 11.5 г (6 ммоль) 7-амино-3,3-диметил-3,4-дигидро-1,8-нафтаридин-2(1H)-она и перемешивают 2 часа при 20°C, затем снова охлаждают до -5°C и прибавляют 11.8 мл (6.8 ммоль) основания Хюнинга, переменивают 2 часа при комнатной температуре, а затем еще 5 часов при 40°C. Растворитель удаляют в вакууме (при 5 мм. рт.ст.), остаток промывают холодной водой (2×5 мл), изопропанолом (2×5 мл), эфиром (2×10 мл) и сушат. Получают: 16.6 г (91%) 7-((4-хлор-1,3,5-триазин-2-ил)амино)-3,3-диметил-3,4-дигидро-1,8-нафтраридин-2(1H)-она.
Синтез 3,3-диметил-7-((4-((2,2,3,3-тетрафтор-2,3-дигидробензо[b][1,4]диоксин-6-ил)амино)-1,3,5-триазин-2-ил)амино)-3,4-дигидро1,8-нафтраридин-2(1Н)-она
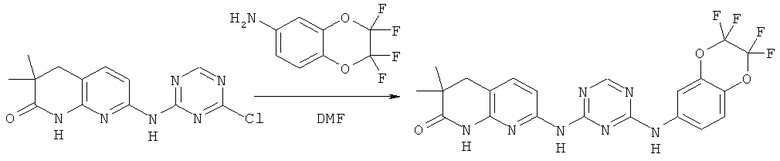
2.59 г (0.85 ммоль) полученного в предыдущей методике монохлорпроизводного растворяют в 5 мл безводного дегазированного ДМФА, прибавляют 4.13 г (1.85 ммоль) 2,2,3,3-тетрафтор-6-амино-1,4-бенздиоксана и нагревают в течение 6 часов при 100°С.Растворитель удаляют в вакууме (при 5 мм. рт.ст.), остаток промывают холодной водой (2x5 мл), сушат и разделяют хроматографически используя в качестве элюента систему хлороформ: метанол нарастающей полярности. После стандартной процедуры обработки получают: 2.21 г (53%) 3,3-диметил-7-((4-((2,2,3,3-тетрафтор-2,3-дигидробензо[Ь][1,4]диоксин-6-ил)амино)-1,3,5-триазин-2-ил)амино)-3,4-дигидро 1,8-нафтраридин-2(1 Н)-она.
Ш (500 MHz, DMSO-d6): 0.91 (с, 6Н, 2СН3), 2.27 (с, 2Н, СН2), 7.02 (д. J=8.1Hz, 1H, HAr, 7.30 (д. J=5.7Hz, 1Н, HAr, 7.38 (д. J=8.2Hz, 1Н, НАг), 7.45 (д. J=5.7Hz, 1H, Н^), 7.55 (с, 1H, НАг), 7.87 (уш.с, 1H, NH), 8.43 (с, 1Н, HAr, 8.50 (уш.с, 1Н, NH), 9.46 (с, 1H, C(O)NH).
Элементный состав: С(51.46%), Н(3.63%), N(19.79%)
6. Получение 6-((4-((4-метоксифенил)амино)-1,3,5-триазин-2-ил)амино)-2,2-диметил-2Н-пиридо[3,2-b][1,4]тиазин-3(4Н)-она.
Ситнез 6-((4-((4-метоксифенил)амино)-1,3,5-триазин-2-ил)амино)-2,2-диметил-2Н-пиридо[3,2-b][1,4]тиазин-3(4Н)-она осуществляется в соответствии со схемой I. При этом используется 76-амино-2,2-диметил-2Н-пиридо[3,2-b][1,4]тиазин-3(4Н)-он, синтезированный в соответствии со схемой VII
Синтез 6-((4-хлор-1,3,5-триазин-2-ил)амино)-2,2-диметил-2Н-пиридо[3,2-b][1,4]тиазин-3(4Н)-она
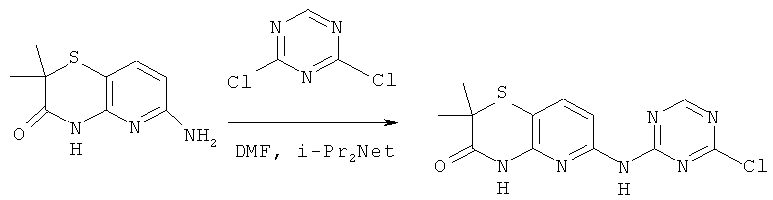
675 мг (0.45 ммоль) 2,4-дихлор-1,3,5-триазина растворяют в 12 мл безводного дегазированного ДМФА, охлаждают до -5°C и прибавляют 627 г (0.3 ммоль) 6-амино-2,2-диметил-2Н-пиридо[3,2-b][1,4]тиазин-3(4Н)-она и перемешивают 2 часа при 20°C, затем снова охлаждают до -5°C и прибавляют 0.7 мл (~0.4 ммоль) основания Хюнинга, переменивают 2 часа при комнатной температуре, а затем еще 5 часов при 45°C. Растворитель удаляют в вакууме (при 5 мм. рт.ст.), остаток промывают холодной водой (2x1 мл), изопропанолом (2×1 мл), эфиром (2×2 мл) и сушат.Получают: 859 мг (89%) 6-((4-хлор-1,3,5-триазин-2-ил)амино)-2,2-диметил-2Н-пиридо[3,2-b] [1,4]тиазин-3(4Н)-она.
Синтез 6-((4-((4-метоксифенил)амино)-1,3,5-триазин-2-ил)амино)-2,2-диметил-2Н-пиридо[3,2-b][1,4]тиазин-3(4Н)-она
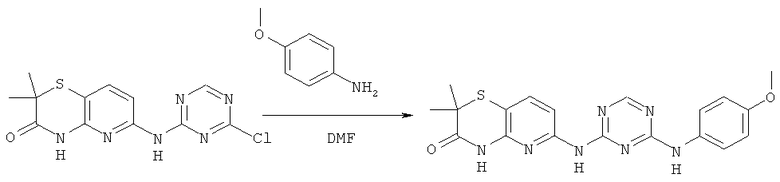
260 г (0.08 ммоль) полученного в предыдущей методике монохлорпроизводного растворяют в 3 мл безводного дегазированного ДМФА, прибавляют 220 г (0.18 ммоль) n-анизидина и нагревают в течение 6 часов при 100°С. Растворитель удаляют в вакууме (при 5 мм. рт.ст.), остаток промывают холодной водой (2×5 мл), сушат и перекристализовавыют из водного спирта Получают: 210 г (64%) 6-((4-((4-метоксифенил)амино)-1,3,5-триазин-2-ил)амино)- 2,2-диметил-2Н-пиридо[3,2-b][1,4]тиазин-3(4Н)-она.
1Н (500 MHz, DMSO-d6): 1.43 (с, 6Н, 2СНЗ), 3.73 (с, зн, ОСНЗ), 6.93 (д. J=8.6Hz, 2Н, HAr, 7.14 (д. J=9.2Hz, 1H, HAr, 7.41 (д. J=8.7Hz, 2Н, HAr, 7.43 (д. J=9.2Hz, 1H, HAr, 7.84 (уш.с, 1Н, NH), 8.16 (уш.с, 1Н, NH), 8.42 (с, 1H, HAr, 9.19 (с, 1Н, C(O)NH).
Элементный состав: С(55.47%), Н(4.61%), N(23.74%)
7. Получение 7-((4-((2,2-дифторбензо[d]1,3]диоксол-5-ил)амино)-1,3,5-триазин-2-ил)амино)-3a,9а-диметил-1,2,3)3a,9,9а-гексагидробензо[b]циклопента[e][1,4]оксаззин-1-ола
Ситнез 7′-((4-((3,3-дифторциклобутил)амино)-1,3,5-триазин-2-ил)амино)спиро[циклопропан-1,3′-пиридо[3,4-b][1,4]оксазина]-2′(1′H)-она осуществляется в соответствии со схемой I. При этом используется 7′-хлорспиро[циклопропан-1,3′-пиридо [3,4-b] [1,4]оксазина]-2′(1 'Н)-она, синтезированный в соответствии со схемой VIII
Синтез 7-((4-хлор-1,3,5-триазин-2-ил)амино)-3a,9а-диметил-1,2,3,3a,9,9а-гексагидробензо[b]циклопента[e][1,4]оксаззин-1-ола
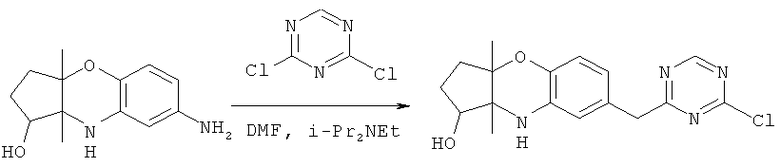
1.20 г (8 ммоль) 2,4-дихлор-1,3,5-триазина растворяют в 12 мл безводного дегазированного ДМФА, охлаждают до -5°C и прибавляют 1.40 г (6 ммоль) 7-амино-3,3-диметил-3,4-дигидро-1,8-нафтаридин-2(1H)-она и перемешивают 2 часа при 20°C, затем снова охлаждают до -5°C и прибавляют 1.2 мл (~6.8 ммоль) основания Хюнинга, переменивают 2 часа при комнатной температуре, а затем еще 5 часов при 40°C. Растворитель удаляют в вакууме (при 5 мм. рт.ст.), остаток промывают холодной водой (2×5 мл), эфиром (2×5 мл) и сушат сушат и разделяют хроматографически используя в качестве элюента систему хлороформ: метанол нарастающей полярности. После стандартной процедуры обработки получают 1.27 г (61%) 7-((4-хлор-1,3,5-триазин-2-ил)амино)-3a,9а-диметил-1,2,3,3a,9,9а-гексагидробензо[b] циклопента [e][1,4]оксаззин-1-ола.
Синтез 7-((4-((2,2-дифторбензо[й][1,3]диоксол-5-ил)амино)- 1,3,5-триазин-2-ил)амино)-3a,9а-диметил-1,2,3,3a,9,9а-гексагидробензо[b]цитопента[e][1,4]оксаззин-1-ола

Смесь 1.55 г (9 ммоль) 2,2-дифтор-5-амино-1,3-бенздиоксолана, трет-бутилат натрия 864 мг (9 ммоль), 280 мг 2,2′-бис(дифенилфосфино)-1,1′бинафтила (X моль, 1.2% молн.), 89 мг Pd2(dba)3 и Хг 2.43 г (0.007 моль) монохлорпроизводного полученного на предыдущей стадии в 15 мл толуола перемешивают при 60°C в течение 38 часов, охлаждают до комнатной температуры, прибавляют 100 мл 1М раствора соляной кислоты в смеси ТГФ:вода 1:1, перемешивают 2 часа, экстрагируют этилацетатом (3×200 мл), экстракты объединяют, промывают насыщенным водным раствором NaHCO3 (2×150 мл), сушат над MgSO4, растворитель удаляют, остаток разделяют хроматографически. Получают: 768 мг (31%) 7-((4-((2,2-дифторбензо[(1][1,3]диоксол-5-ил)амино)- 1,3,5-триазин-2-ил)амино)-3a,9а-диметил-1,2,3,3a,9,9а-гексагидробензо[b]циклопента[e][1,4]оксаззин-1-ола.
1Н (500 MHz, DMSO-d6): 1.30 (с, зн, CH3), 1.37 (с, зн, CH3), 1.85-2.43 (м, 4H, 2CH2), 4.24-4.29 (м, 1H, CH), 4.61 (уш. с, 1H, OH), 6.44 (д, J=8.7Hz, 1H, HAr), 6.68 (с, 1H, HAr), 6.81 (с, 1H, HAr), 7.03 (д, J=8.2Hz, 1H, HAr), 7.07 (д, J=8.7Hz, 1Н, HAr), 7.09 (д, J=8.2Hz, 1H, HAr), 7.31 (уш.с, 1Н, NH), 7.48 (уш.с, 1H, NH), 7.84 (уш.с, 1H, NH), 8.42 (c, 1H, HAr).
Элементный состав: C(57.13%), H(4.51%), N(17.26%)
8. Получение 7′-((4-((3,3-дифторциклобутил)амино)-1,3,5-триазин-2-ил)амино)спиро[циклопропан-1,3′-пиридо[3,4-b][1,4]оксазина]-2′(1′H)-она
Ситнез 7′-((4-((3,3-дифторциклобутил)амино)-1,3,5-триазин-2-ил)амино)спиро[циклопропан-1,3′-пиридо [3,4-b][1,4] оксазина]-2′(1′H)-она осуществляется в соответствии со схемой I. При этом используется 7′-хлорспиро [циклопропан-1,3′-пиридо [3,4-b] [1,4] оксазина]-2′(1′H)-она, синтезированный в соответствии со схемой IX
Синтез 7′-((4-хлор-1,3,5-триазин-2-ил)амино)спиро[циклопропан-1,3′-пиридо[3,4-b][1,4]оксазина]-2′(1′H)-она

750 мг (5 ммоль) 2,4-дихлор-1,3,5-триазина растворяют в 5 мл безводного дегазированного ДМФА, охлаждают до -5°C и прибавляют 764 мг (4 ммоль) 7′-хлорспиро[циклопропан-1,3′-пиридо[3,4-b][1,4]оксазина]-2′(1′H)-она и перемешивают 2 часа при 20°C, затем снова охлаждают до -5°C и прибавляют 0.9 мл (~5 ммоль) основания Хюнинга, переменивают 2 часа при комнатной температуре, а затем еще 5 часов при 55°C. Растворитель удаляют в вакууме (при 5 мм. рт.ст.), остаток промывают холодной водой (3×5 мл), изопропанолом (2×2 мл), эфиром (2×5 мл) и сушат. Получают: 938 мг (77%) 7′-((4-хлор-1,3,5-триазин-2-ил)амино)спиро[циклопропан-1,3′′-пиридо[3,4-b][1,4]оксазина]-2′(1′H)-она.
Синтез 7′-((4-((3,3-дифторциклобутил)амино)-1,3,5-триазин-2-ил)амино)спиро[циклопропан-1,3′-пиридо[3,4-b][1,4]оксазина]-2′(1′Н)-она
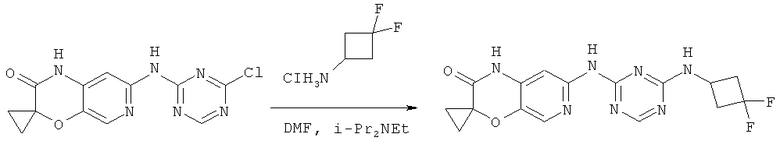
360 мг (0.12 ммоль) полученного в предыдущей методике монохлорпроизводного растворяют в 5 мл безводного дегазированного ДМФА, прибавляют 230 мг (0.16 ммоль) гидрохлорида 3,3-дифторциклобутиламина и 0.6 мл (~0.33 ммоль) основания Хюнинга и нагревают в запаяной ампуле в течение 9 часов при 80°C. Растворитель удаляют в вакууме (при 5 мм. рт.ст.), остаток промывают холодной водой (2×5 мл), сушат и разделяют хроматографически используя в качестве элюента систему хлороформ: метанол нарастающей полярности. После стандартной процедуры обработки получают: 441 мг (59%) 7′-((4-((3,3-дифторциклобутил)амино)-1,3,5-триазин-2-ил)амино)спиро[циклопропан-1,3′-пиридо[3,4-b][1,4]оксазина]-2′(1′H)-она.
1Н (500 MHz, DMSO-d6): 1.76-2.16 (м, 8H, 4CH2), 3.92-4.06 (м, 1H, CH), 7.17 (д, J=2.1Hz, 1H, Hat), 7.62 (уш.с, 1H, NH), 7.95 (д., J=2.1Hz, 1H, HAr), 8.34 (уш.с, 1H, NH), 8.53 (с, 1H, HAr).
Элементный состав: C(51.39%), H(4.22%), N(30.12%).
9. Получение 6′-((4-циклопентиламино-1,3,5-триазин-2-ил)амино)спиро[циклопропан-1,2,-пиразино[2,3-b][1,4]оксазин]-3′(4′Н)-она.
Ситнез 6′-((4-циклопентиламино-1,3,5-триазин-2-ил)амино)спиро[циклопропан-1,2′-пиразино[2,3-b][1,4]оксазин]-3′(4′H)-она осуществляется в соответствии со схемой I. При этом используется 6′-хлорспиро[циклопропан-1,2,-пиразино[2,3-b][1,4]оксазин]-3′(4′H)-она, синтезированный в соответствии со схемой XI
Синтез 6′-((4-хлор-1,3,5-триазин-2-ил)амино)спиро[циклопропан-1,2′-пиразино[2,3-b][1,4]оксазин]-3′(4′Н)-она
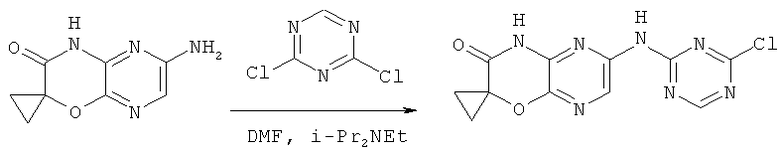
660 мг (4.4 ммоль) 2,4-дихлор-1,3,5-триазина растворяют в 3 мл безводного дегазированного ДМФА, охлаждают до -5°C и прибавляют 769 мг (4 ммоль) 6′-хлорспиро [циклопропан-1,2′-пиразино[2,3-b][1,4]оксазин]-3′(4′H)-она и перемешивают 2 часа при 20°C, затем снова охлаждают до -5°C и прибавляют 0.73 мл (4.2 ммоль) основания Хюнинга, переменивают 2 часа при комнатной температуре, а затем еще 5 часов при 40°C. Растворитель удаляют в вакууме (при 5 мм. рт.ст.), остаток промывают холодной водой (2×5 мл), изопропанолом (2×5 мл), эфиром (2×10 мл) и сушат. Получают: 1.02 г (83%) 6′-((4-хлор-1,3,5-триазин-2-ил)амино)спиро[циклопропан-1,2′-пиразино[2,3-b][1,4]оксазин]-3′(4′H)-она.
Синтез 6′-((4-цитопентиламино-1,3,5-триазин-2-ил)амино)спиро[циклопропан-1,2′-пиразино[2,3-b][1,4]оксазин]-3′(4′Н)-она
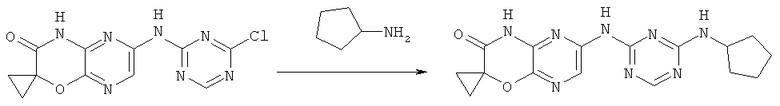
367 мг (1.2 ммоль) полученного в предыдущей методике монохлорпроизводного растворяют в 3 мл безводного дегазированного ДМФА, прибавляют 255 мг (3.0 ммоль) циклопентиламина и выдерживают в течение 8 часов при 90°C. Растворитель.удаляют в вакууме (при 5 мм. рт.ст.), остаток промывают холодной водой (4×5 мл), сушат и разделяют хроматографически используя в качестве элюента систему хлороформ: метанол нарастающей полярности. После стандартной процедуры обработки получают: 208 мг (49%) 6′-((4-циклопентиламино-1,3,5-триазин-2-ил)амино)спиро[циклопропан-1,2′-пиразино[2,3-b][1,4]оксазин]-3′(4′H)-она.
1Н (500 MHz, DMSO-d6): 1.32-1.68 (м, 8Н, 4СН2), 1.75-2.07 (м, 4Н, 2СН2), 3.75-3.83 (м,1Н, СН), 7.78 (уш.с, 1Н, NH), 8.13 (с, 1H, HAr), 8.33 (с, 1Н, HAr), 8.37 (уш.с, 1Н, NH), 9.46 (с, 1Н, C(O)NH).
Элементный состав: С (54.46%), Н (4.21%), N (31.48%)
10. Получение 6-((4-((4-метоксифенил)амино)-1,3,5-триазин-2-ил)амино)-2,2-диметил-2Н-пиридо[3,2-b][1,4]тиазин-3(4Н)-она сульфоксида

К охлажденному до 0°C раствору 409 мг (1 ммоль) исходного соединения в 50 мл ДМФА по каплям при интенсивном перемешивании прибавляют раствор 223 мг (1 ммоль) 77% м-хлорпербензойной кислоты и перемешивают при 0°C в течение 4 часов. Затем реакционную смесь выливают в 300 мл 3% водного раствора NaHCO3. Выпавший осадок отфильтровывают, промывают на фильтре водой (5×10 мл) и сушат в вакууме. Получают: 386 мг (67%) продукта.
1Н (500 MHz, DMSO-d6): 1.43 (с, 6Н, 2СН3), 3.73 (с, 3Н, ОСН3), 6.93 (д, J=8.6 Hz, 2Н, HAr), 7.14 (д. J=9.2 Hz, 1H, HAr), 7.41 (д. J=8.7 Hz, 2Н, HAr), 7.43 (д. J=9.2Hz, 1Н, HAr), 7.84 (уш.с, 1H, NH), 8.16 (уш.с, 1H, NH), 8.42 (с, 1Н, HAr), 9.19 (с, 1Н, C(O)NH).
Элементный анализ: С, 53.75%; Н, 4.48%; N, 22.83%; S, 6.99%
11. Получение 6-((4-((4-метоксифенил)амино)-1,3,5-триазин-2-ил)амино)-2,2-диметил-2Н-пиридо[3,2-b][1,4]тиазин-3(4Н)-она сульфона моногидрата

К раствору 409 мг (1 ммоль) исходного соединения в 20 мл ДМФА прибавляют 470 мг (2.1 ммоль) 77% м-хлорпербензойной кислоты. Реакционную смесь перемешивают при 40°C в течение 4 часов, затем выливают в 100 мл 3% водного раствора NaHCO3. Выпавший осадок отфильтровывают, промывают на фильтре водой (5×10 мл) и сушат в вакууме. Получают: 238 мг (53%) продукта в виде моногидрата.
1H (500 MHz, DMSO-d6): 1.7 (с, 6Н, СН3), 3.81 (с, 3Н, СН3), 6.78 (д, J=7,5 Гц, 1Н, СН), 6.93 (д, J=7.5 Гц, 2Н, СН), 7.44 (с, 1H, СН), 7.64 (д, J=7.5 Гц, 2Н, CH), 8.3 (д, J=7.5 Гц, 1Н, СН), 9.43 (уш.с., 1Н, NH), 10.18 (уш.с., 1Н, NH), 11.26 (уш.с., 1Н, NH)
Элементный состав: С, 49.92%; Н, 4.52%; N, 21.03%; S, 6.72%
12. Получение пролекарств активных соединений в виде сложных эфиров, амидов, N-гидроксиметил производных, О-гидроксиметилпроизводных, эфиров серной или фосфорной кислот, метилфосфонатов
Синтез этил 3-((4-((2,2-диметил-3-оксо-3,4-дигидро-2Н-пиридо[3,2-b][1,4]оксазин-6-ил)амино)-1,3,5-триазинил-2-)амино)пропионата
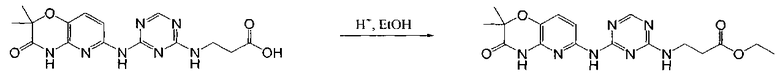
Раствор 359 мг (1 ммоль) исходной кислоты растворили в 5 мл безводного N-метилпирролидона прибавляют 2 мл безводного этанола и 100 мг ионообменной смолы КУ-2-8 и перемешивают при 50°C 8 часов. Ионообменную смолу отфильтровывают, фильтрат отделяют, удаляют в вакууме растворитель, остаток промывают на фильтре водой (2×5 мл), насыщенным раствором NaHCO3 (4×5 мл), водой до нейтральной реакции промывных вод, сушат. Получают 221 мг (57%) этил 3-((4-((2,2-диметил-3-оксо-3,4-дигидро-2H-пиридо[3,2-b][1,4]оксазин-6-ил)амино)-1,3,5-триазинил-2-)амино)пропионата.
1Н (500 MHz, DMSO-d6): 1.07 (м, J=8.0 Гц, 1H, СН3), 2.62 (с, 6Н, СН2), 3.56 (д, J=7.5 Гц, 1Н, СН2), 4.01 (д, J=7.5 Гц, 1H, СН2), 7.01 (с, 2Н, NH), 7.14 (с, 1H, СН), 7.44 (уш.с, 1Н, СН), 10.18 (уш.с, 1Н, NH), 11.26 (уш.с, 1H, NH).
Элементный анализ: С: 52.59%, Н: 5.39%; N:25.06%.
Синтез 4-((4-((2,2-дифтор-3-оксо-3,4-дигидро-2Н-pyrido[3,2-b][1,4]оксазин-6-ил)амино)-1,3,5-триазин-2-ил)амино)-2,6-диметоксифенил изобутирата

К раствору 447 мг (1 ммоль) исходного соединения в 4 мл безводного N-метилпирролидона прибавляют 146 мг (1.2 ммоль) 4-диметиламинопиридина и 125 мкл (1.2 ммоль) хлорангидрида изомасляной кислоты. Раекционную смесь перемешивают при 25-30°C 5 часов. Растворитель удаляют в вакууме, остаток промывают на фильтре водой (2×5 мл), насыщенным раствором NaHCO3 (4×5 мл), водой до нейтральной реакции промывных вод, сушат и разделяют хроматографически. Получают: 403 мг (78%) 4-((4-((2,2-дифтор-3-оксо-3,4-дигидро-2H-pyrido[3,2-b][1,4]оксазин-6-ил)амино)-1,3,5-триазин-2-ил)амино)-2,6-диметоксифенил изобутирата.
1Н (500 MHz, DMSO-d6): 1.19 (с, 6Н, СН3), 2.53 (м, 1H, СН), 3.83 (с, 6Н, СН3), 6.5 (с, 2Н, СН), 6.58 (д, J=7.5 Гц, 1H, CH), 7.14 (д, J=7.5 Гц, 1Н, СН), 7.44 (с, 1H, CH), 9.43 (уш.с, 1Н, NH), 10.18 (уш.с, 1Н, NH), 11.26 (уш.с, 1H, NH).
Элементный анализ: С: 49.98%, H: 4.02%; N: 19.03%.
Синтез 4-((4-((2,2-диметил-3-оксо-3,4-дигидро-2Н-пиридо[3,2-b][1,4]оксазин-6-ил)амино)-1,3,5-триазинил-2-ил)амино)-2,6-диметоксифенил этилкарбоната.

К раствору 439 мг (1 ммоль) исходного соединения в 4 мл безводного N-метилпирролидона прибавляют 146 мг (1.2 ммоль) 4-диметиламинопиридина и 115 мкл (1.2 ммоль) этил хлорформиата. Реакционную смесь перемешивают при 25-30°C 7 часов. Растворитель удаляют в вакууме, остаток промывают на фильтре водой (2×5 мл), насыщенным раствором NaHCO3 (4×5 мл), водой до нейтральной реакции промывных вод, сушат растворяют в 25 мл горячего этанола, раствор декантируют, растворитель удаляют, остаток сушат. Получают: 353 мг (69%) 4-((4-((2,2-диметил-3-оксо-3,4-дигидро-2H-пиридо[3,2-b][1,4]оксазин-6-ил)амино)-1,3,5-триазинил-2-ил)амино)-2,6-диметоксифенил этилкарбоната.
1H (500 MHz, DMSO-d6): J=8 Гц.0 1.55 (с, 6Н, СН3), 3.83 (с, J=7.5 Гц, 6Н, СН3), 4.22 (кв, J=7.5 Гц, 2Н, СН2), 6.47 (с, 2Н, CH), 7.14 (с, J=7.5 Гц, 1H, СН), 7.44 (с, 1Н, СН), 9.43 (уш.с, 1Н, NH), 10.18 (уш.с, 1H, NH), 11.26 (уш.с, 1H, NH).
Элементный анализ: С: 54.11%, Н: 4.89%; N: 21.77%.
Синтез 4-((4-((2,2-дифтор-3-оксо-3,4-дигидро-2Н-бензо[b][1,4]оксазин-6-ил)амино)-1,3,5-триазин-2-ил)амино)-2,6-диметоксифенил диметилкарбамата.

К раствору 446 мг (1 ммоль) исходного соединения в 4 мл безводного N-метилпирролидона прибавляют 146 мг (1.2 ммоль) 4-диметиламинопиридина и 110 мкл (1.2 ммоль) этил хлорформиата. Реакционную смесь перемешивают при 25-30°C 7 часов. Растворитель удаляют в вакууме, остаток промывают на фильтре водой (2×5 мл), насыщенным раствором NaHCO3 (4×5 мл), водой до нейтральной реакции промывных вод, сушат и разделяют хроматографически. Получают: 419 мг (81%) 4-((4-((2,2-дифтор-3-оксо-3,4-дигидро-2H-бензо[b][1,4]оксазин-6-ил)амино)-1,3,5-триазин-2-ил)амино)-2,6-диметоксифенил диметилкарбамата.
1Н (500 MHz, DMSO-d6): 3.27 (с, 6Н, СН3), 3.83 (с, 6Н, СН3), 6.47 (с, 2Н, СН), 7.02 (д, J=7.5 Гц, 1Н, СН), 7.3 (с, 1Н, СН), 7.39 (д, J=7.5 Гц, 1Н, СН), 7.44 (с, 1Н, СН), 9.43 (уш. с, 1H, NH), 9.43 (уш.с, 1Н, NH), 10.66 (уш.с, 1H, NH)
Элементный анализ: С: 49.90%, Н: 4.01%; N: 18.83%.
Синтез 2,2-дифтор-4-(гидроксиметил)-6-((4-((3-метокси-4,5-метелендиоксифенил-5)амино)-1,3,5-триазин-2-ил)амино)-2Н-benzo[b][1,4]оксазин-3(4Н)-она

К раствору 444 мг (1 ммоль) исходного соединения в 12 мл безводного диоксана, прибавляют 100 мг поташа и 92 мкл (1.2 ммоль) 36% водного раствора формальдегида. Реакционную смесь перемешивают при комнатной температуре 6 часов прибавляют 100 мл этилацетата и промывают полученный раствор водой (4×30 мл), растворитель удаляют, остаток сушат. Получают 346 мг (73%) 2,2-дифтор-4-(гидроксиметил)-6-((4-((3-метокси-4,5-метелендиоксифенил-5)амино)-1,3,5-триазин-2-ил)амино)-2H-benzo[b][1,4]оксазин-3(4H)-она содержащего согласно данным ВЭЖХ ~6% изомерных продуктов.
1Н (500 MHz, DMSO-d6): 3.83 (с, 3Н, СН3), 4.12 (уш. с, 1Н, ОН), 5.62 (с, 2Н, СН2), 6.07 (с, 2Н, СН2), 6.5 (с, 2Н, СН), 6.89 (с, 1H, СН), 6.97 (д, J=7.5 Гц, 1Н, СН), 7.44 (с, 1Н, СН), 7.53 (д, J=7.5 Гц, 1Н, СН), 9.43 (уш. с, 1H, NH), 9.84 (уш. с, 1H, NH)
Элементный анализ: С: 50.69%, Н: 3.42%; F: 7.97%, N: 17.83%.
Синтез 7-((4-((2,2-дифтор-3-оксо-3,4-дигидро-2Н-пиридо[3,2-b][1,4]оксазин-6-ил)амино)-1,3,5-триазин-2-ил)амино)-2,3-дигидробензо[b][1,4]диоксин-5-ил гидросульфата

К раствору 890 мг (2 ммоль) исходного соединения в смеси 8 мл смеси безводных пиридина и ДМФА (1:2 об./об.) прибавляют 333 мг (2.2 ммоль) комплекса триоксида серы с ДМФА. Реакционную смесь перемешивают 12 часов при комнатной температуре, растворители удаляют в вакууме при температуре не выше 30°C. Остаток переносят на фильтр и промывают диэтиловым эфиром (5×8 мл) и сушат. Получают:956 мг (91%) 7-((4-((2,2-дифтор-3-оксо-3,4-дигидро-2H-пиридо[3,2-b][1,4]оксазин-6-ил)амино)-1,3,5-триазин-2-ил)амино)-2,3-дигидробензо[b][1,4]диоксин-5-ил гидросульфата.
1Н (500 MHz, DMSO-d6): 4.28 (т, J=7.1 Гц, 2Н, СН2), 4.32 (т, J=7.1 Гц, 2Н, СН2), 6.35 (с, 1Н, CH), 6.38 (с, 1Н, СН), 7.44 (с, 1H, СН), 7.77 (д, J=7.5 Гц, 1Н, СН), 8.47 (с, 1Н, NH), 8.52 (д, J=7.5 Гц, 1Н, СН), 9.43 (с, 1H, NH), 10.56 (с, 1Н, NH)
Элементный анализ: С: 40.36%, H: 2.41%; F: 7.17%, N:17.06%.
Синтез 4-((4-((2,2-дифтор-3-оксо-3,4-дигидро-2Н-пирида[3,2-b][1,4]оксазин-6-ил)амино)-1,3,5-триазин-2-ил)amino)-2,6-диметоксифенил фосфата в форме сольвата
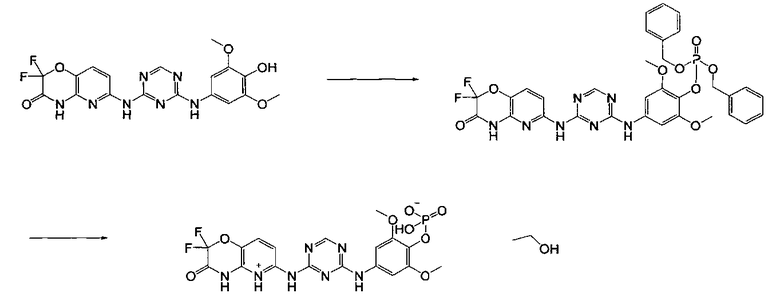
К раствору 446 мг (1 ммоль) исходного соединения в 4 мл безводного N-метилпирролидона прибавляют 146 мг (1.2 ммоль) 4-диметиламинопиридина и 346 мг (1.2 ммоль) дибензилхлорфосфата. Реакционную смесь перемешивают при 25-30°C 7 часов. Растворитель удаляют в вакууме, остаток растворяют в 100 мл этилацетата, промывают водой (2×30 мл) и сушат. Растворитель удаляют в вакууме, остаток растворяют в этаноле. К полученному раствору прибавляют 300 мг 10% Pd/C и перемешивают в атмосфере водорода 8 часов. Катализатор отфильтровывают, фильтрат упаривают. Получают: 353 мг (67%) 4-((4-((2,2-дифтор-3-оксо-3,4-дигидро-2Н-пирида[3,2-b][1,4]оксазин-6-ил)амино)-1,3,5-триазин-2-ил)amino)-2,6-диметоксифенил фосфата в форме сольвата.
1H (500 MHz, DMSO-d6): 1.06 (т, J=7.2 Гц, 3Н, СН3-EtOH), 3.44 (кв, J=7.2 Гц, 2Н, СН2-EtOH), 3.83 (с, 6Н, СН3), 4.63 (с, 1Н, ОН-EtOH), 4.8 (уш.с., 1Н, ОН), 6.51 (с, 2Н, СН), 7.44 (с, 1H, СН), 7.77 (д, J=7.5 Гц, 1H, СН), 8.47 (уш.с., 1H, NH), 8.52 (д, J=7.5 Гц, 1Н, СН), 9.43 (уш.с., 1H, NH), 10.56 (уш.с., 1H, NH).
Элементный анализ: С: 41.81, Н: 3.84; F: 6.67, N:17.01.
Синтез (4-((4-((2,2-дифтор-3-оксо-3,4-дигидро-2Н-бензо[b][1,4]оксазин-6-ил)амино)-1,3,5-триазин-2-ил)амино)-2,6-диметоксифенокси)метилацетата
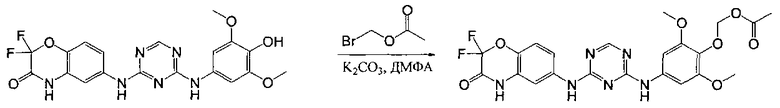
К раствору 446 мг (1 ммоль) исходного соединения в 5 мл безводного ДМФА прибавляют 276 мг (2 ммоль) безводного свежепрокаленного поташа и 184 мг (1.2 ммоль) бромметилацетата. Реакционную смесь перемешивают при 0°C 10 часов, затем выливают в 20 мл воды. Экстрагируют этилацетатом (2×50 мл), органические фазы объединяют, промывают водой (3×50 мл), сушат над Na2SO4, растворитель удаляют, остаток высушивают в вакууме. Получают: 454 мг (90%) (4-((4-((2,2-дифтор-3-оксо-3,4-дигидро-2Н-бензо[b][1,4]оксазин-6-ил)амино)-1,3,5-триазин-2-ил)амино)-2,6-диметоксифенокси)метилацетата.
1Н (500 MHz, DMSO-d6): 2.19 (с, 3H, CH3), 3.83 (с, 6H, CH3), 6.5 (с, 2H, CH), 6.73 (с, 2H, CH2), 7.02 (д, J=7.5 Гц, 1H, CH), 7.3 (c, 1H, CH), 7.39 (д, J=7.5 Гц, 1H, CH), 7.44 (c, 1H, CH), 9.43 (c, 1H, NH), 9.43 (c, 1H, NH), 10.66 (c, 1H, NH)
Элементный анализ: С: 51.06%, H: 3.91%; F: 7.28%, N: 16.07%.
Синтез N-(4-((2,2-диэтил-3-оксо-3,4-дигидро-2Н-пиридо[3,2-b][1,4]оксазин-6-ил)амино)-1,3,5-триазин-2-ил)-N-(3,4,5-триметоксифенил)ацетамида

К раствору 481 мг (1 ммоль) исходного соединения в 5 мл безводного дегазированного ДМФА в атмосфере аргона прибавляют 122 мг (1 ммоль) N,N-диметиламинопиридина и 0.7 мл (1,1 ммоль) ацетилхлорида. Реакционную смесь перемешивают при 50°C в инертной атмосфере 8 часов. Растворитель удаляют в вакууме, остаток растворяют в 150 мл хлороформа, промывают насыщенным раствором NaHCO3 (2×25 мл), водой (3×25 мл), сушат, растворитель удаляют, остаток разделяют хроматографически. Получают: 351 мг (67%) N-(4-((2,2-диэтил-3-оксо-3,4-дигидро-2Н-пиридо[3,2-b][1,4]оксазин-6-ил)амино)-1,3,5-триазин-2-ил)-N-(3,4,5-триметоксифенил)ацетамида.
1Н (500 MHz, DMSO-d6): 0.69 (т, J=8.0 Гц, 6Н, СН3), 1.93 (кв, J=8.0 Гц, 4Н, СН2), 2.07 (с, J=7.8 Гц, 3Н, СН3), 3.71 (с, 3H, СН3), 3.83 (с, 6Н, СН3), 6.5 (с, 2Н, СН), 7.61 (д, J=7.5 Гц, 1H, СН), 7.91 (с, 1H, СН), 8.2 (д, J=7.5 Гц, 1H, СН), 9.43 (уш.с., 1Н, NH), 11.26 (уш.с., 1Н, NH)
Элементный анализ: С: 57.30%, Н: 5.54%; N: 18.62%.
Синтез 7-((4-((4-(1-ацетилпиперидин-4-ил)фенил)амино)-1,3,5-триазин-2-ил)амино)-3,3-диметил-3,4-дигидро-1,8-нафтиридин-2(1Н)-она

К раствору 444 мг (1 ммоль) исходного соединения в 10 мл пиридина, охлажденному до 0°C, прибавляют 0,14 мл уксусного ангидрида, и перемешивают при комнатной температуре 8 часов. Реакционную смесь выливают в 200 мл воды, экстрагируют этилацетатом (3×100 мл). Органические фракции объединяют, промывают насыщенным раствором NaHCO3 (2×25 мл), водой (2×25 мл), растворитель удаляют, остаток высушивают в вакууме. Получают: 450 мг (93%) 7-((4-((4-(1-ацетилпиперидин-4-ил)фенил)амино)-1,3,5-триазин-2-ил)амино)-3,3-диметил-3,4-дигидро-1,8-нафтиридин-2(1Н)-она.
1Н (500 MHz, DMSO-d6): 1.34 (с, 6Н, СН3), 1.63-1.89 (м, 4Н, СН2), 2.1 (с, 3Н, СН3), 2.65-2.72 (м, 1Н, СН), 2.71 (с, 2Н, СН2), 3.5-3.6 (м, 4Н, СН2), 6.34 (д, J=7.5 Гц, 1Н, СН), 7.06 (с, 2Н, СН), 7.38 (с, 2Н, СН), 7.42 (д, J=7.5 Гц, 1Н, СН), 7.44 (с, 1H, СН), 9.43 (уш.с., 1Н, NH), 10.18 (уш.с., 1Н, NH), 11.26 (уш.с., 1Н, NH)
Элементный анализ: С: 64.15%, Н: 6.24%; N:22.89%.
13. Характеристика биологической активности соединений
Биологическая активность соединений, являющихся предметом настоящего изобретения, была изучена различными методами. Например, было исследовано ингибирование киназной активности данными соединениями. Некоторые соединения показали существенную ингибиторную активность в наномолярной концентрации по отношению к киназе SYK. Также некоторые соедиения показали существенную антипролиферативную активность на клетках неходжкинской лимфомы Daudi, NAMALVA, Р3Н3. P3HR-1. Raji (чуствительных к ингибированию SYK киназы) в концентрациях 10-10000 нМ. Кроме того, ряд соенинений, являющихся предметом настоящего изобретения, показали высокую активность в клеточных моделях ревматоидного артрита. Эффективность ингибиторов Syk-киназы исследовалась путем определения ингибирования продукции цитокинов дифференцированными моноцитами и было показано, что химических соединения, являющихся предметом настоящего изобретения, проявили ингибирующую способность по отношению к продукции цитокинов дифференцированными моноцитами (клеточная линия ТНР-1) с значениями ЕС50 0.1-100 мкМ.
Иллюстративные примеры соединений, обладающих высокой ингибиторной и антипролиферативной активностью, представлены ниже










13.1. Ингибирование киназ
Для соединений, являющихся предметом данного изобретения, была изучена способность ингибировать киназы, представляющие интерес для терапии аллергических, аутоиммунных, онкологических, хронических воспалительных и прочих заболеваний. Список киназ, ингибирование которых изучалось в соответствии с описанной методикой, включал, (но принципиально не ограничен) киназами ALK, JAK2, BRAF, MET, TIE-2, FLT3, ABL, LCK, LYN, SRC, FYN, SYK, ZAP70, ITK, ТЕС, ВТК, EGFR, ERB2, PDGFRa, PDGFRb, KFR, IGF-1R, FLT1, ТЕК, АКТ, ROS, EPHA1, а также их мутантными формами, в том числе придающими резистентность к существующим методам терапии аутоиммунных, воспалительных и онкологических заболеваний.
Киназы, в виде киназного домена или полноразмерного белка, соединенного с глутатион-S-трансферазой (GST) или поли-гистидиновым фрагментов, экспрессировались в зараженных бакуловирусом клетках насекомых (например, Sf21) или в клетках E.Coli. После выделения из клеток белки очищались до практически полной гомогенности с помощью афинной хроматографии по известным методикам (Lehr, R.V. et. al., Gene, 1996, 169, 275-9; Gish, G. et. al.., Protein Eng, 1995, 8, 609-14). В некоторых случаях киназы ко-экспрессировались или смешивались с очищенными или частично очищенными регуляторными полипептидами до измерения активности.
Активность и ингибирование киназ определялось в соответствии с известными протоколами (Braunwaler, A.F. et. al., Anal Biochem, 1996, 234, 23-6). Мерой ферментативной активности служила скорость переноса меченого 33PO4 с АТФ на синтетический субстрат поли(Glu, Tyr) 4:1, присоединенный к биоактивной поверхности микротитрационной подложки. По окончании периода инкубации (120 мин) подложку промывали 0,5% фосфорной кислотой, добавляли жидкий сцинтиллянт, и на основании подсчета количества сцинтилляций на жидкостном сцинтилляционном детекторе определяли количество перенесенного фосфата. IC50 соответствовало концентрации вещества, уменьшающей на 50% количество 33Р, перенесенного на субстрат, связанный с подложкой.
Для определения ингибирования киназ могут использоваться и другие методики, основанные на измерении степени переноса фосфата на пептид или полипептид, содержащий тирозин, серии или треонин и присутствующий в растворенном или иммобилизованном виде.
Соединения, описанные в данном изобретении, имеют наномолярные или микромолярные значения IC50 в отношении различных киназ, в том числе SYK, ZAP-70, JAK1 JAK2, JAK3, ВТК, TYK1 и LYN. Также соединения, описанные в данном изобретении, являются селективными, и в концентрации до 1000 нМ значимо не ингибируют такие киназы, как ABL, АКТ2, AURA, AURC, AXL, CDK2, СТК, FAK, IGF1R, IR, IRR, ITK, mTOR, MUSK, РКА, РКСθ, RON, SRC, TYRO3.
Ниже приведен список соединений, ингибирующих SYK киназу со значениями IC50 менее 100 нМ





Список соединений, ингибирующих SYK киназу со значениями IC50 менее 10 мкМ





Список соединений, ингибирующих ZAP-70 с значениями IC50 менее 10 мкМ







Список соединений, ингибирующих JAK3 и ВТК киназы со значениями IC50 менее 10 мкМ





Список соединений, ингибирующих JAK1 и JAK2 киназы со значениями IC50 менее 10 мкМ




Список соединений, ингибирующих TYK1 киназу со значениями IC50 менее 10 мкМ



Список соединений, ингибирующих LYN киназу со значениями IC50 менее 10 мкМ



13.2. Эксперименты с клеточными культурами
Соединения, являющиеся предметом данного изобретения, подавляют аберрантную активность и пролифирацию клеток иммунной системы и, таким образом, могут быть применены для лечения воспалительных, аутоиммунных, онкологических и других заболеваний.
Клеточные методы определения эффективности подавления абберантной активности клеток иммунной системы хорошо известны и могут быть использованы для сравнительной характеристики соединений, описанных в данном изобретении. Согласно современным представлениям развитие многих аутоиммунных заболеваний связано с активацией врожденной иммунной системы внешними агентами различной природы с последующим сохранением самоподдерживающейся аутоиммунной реакции. Среди антигенов, вызывающих аутоиммунный ответ, выделяют Fc фрагменты иммуноглобулина G (IgG), которые наряду с прочими антигенами приводят к активации клеток иммунной системы и выбросу цитокинов, ведущему в свою очередь к развитию аутоиммунного заболевания. Изучение механизма активации клеток иммунной системы показало, что распространение сигнала идет через Fc рецепторы, располагающиеся на поверхности макрофагов и нейтрофилов. Воздействие на эти рецепторы приводит к продукции цитокинов, важной для начала и распространения заболевания. Syk-киназа является нерецепторной тирозинкиназой и участвует в передаче сигнала антигенными и Fc рецепторами. Для оценки эффективности ингибиторов Syk-киназы могут быть использованы различные in vitro модели и, в частности модели, основанные на стимуляции рецепторов FcγR, расположенных на поверхности макрофагов.
Ниже приведен пример определения активности соединения в клеточных моделях аутоиммунных заболеваний. В данном эксперименте использовалась методика, основанная на стимуляции макрофагов иммуноглобулином IgG, и последующим определением интенсивности выброса цитокинов. Методика определения ингибирования сигнального пути FcγR рецепторов макрофагов, полученных дифференцировкой моноцитов, описана в работе (Braselmann, S., et al., J Pharmacol Exp Ther, 2006. 319, 998-1008) и патенте (WO 2004/014382 A1). Клеточная линия THP-1 инкубируются в культуральной среде RPMI 1640 (5% CO2 увлажненная атмосфера, 37°C). Для индукции дифференциации ТНР-1 в макрофаги клетки подвергают воздействию IFN-γ в течение 6 дней. 96-луночные планшеты покрываются объединенным человеческим IgG, и выдерживается в течение ночи при 4°C, или в течение 1 часа при 37°C. Чтобы оценить фоновую стимуляцию клеток часть лунок в качестве отрицательного контроля покрывают фрагментами антител F(ab')2. Несвязавшиеся антитела удаляются с помощью натрий-фосфатного буфера. В каждую лунку вносится раствор исследуемого соединения до конечной концентрации 0,01-50 мкМ и клетки дифференцированных макрофагов в культуральной среде. Каждую концентрацию препаратов исследуют в 3-х параллелях. Клетки инкубируют при 37°C, после чего в супернатанте определяют концентрацию TNFα.
Иллюстративные примеры соединений, обладающих активностью (со значениями ЕС50 0.1-100 мкМ.) в in vitro для подавления абберантной активности клеток иммунной системмы, представлены ниже












В целом, эксперименты по определению пролиферации клеток и количества жизнеспособных клеток дают детектируемый сигнал, пропорциональный количеству метаболически активных клеток. Противоопухолевая активность соединений может быть определена с использованием любой характеристики, отражающей уменьшение метаболической активности клеток после воздействия соединения. Традиционно используются методы, в которых в качестве меры жизнеспособности клетки выступают целостность мембраны (например, анализ элиминирования трипанового голубого) и синтез ДНК (например, определение включения BrdU или 3Н-тимидина).
В некоторых методах по определению пролиферации клеток используются реагенты, которые превращаются в детектируемые соединения в ходе пролиферации клеток. Предпочтительными реагентами для такого определения являются соли тетразолия, включающие, например, МТТ (3-(4,5-диметилтиазол-2-ил)-2,5-дифенилтетразолий бромид), MTS (3-(4,5-диметилтиазол-2-ил)-5-(3-карбоксиметоксифенил)-2-(4-сульфофенил)-2Н-тетразолий), ХТТ (2,3-бис(2-метокси-4-нитро-5-сульфофенил)-2Н-тетразолий-5-карбоксанилид), INT (2-(4-йодофенил)-3-(4-нитрофенил)-5-фенил тетразолий), NBT (2Н-Тетразолй, 2,2'-(3,3'-диметокси[1,1'-бифенил]-4,4'-диил1)бис[3-(4-нитрофенил)-5-фенил, дихлорид) (Bernas, Т. et. al., Biochim Biophys Acta, 1999, 1451, 73-81). Для определения пролиферации клеток проводят измерение количества продукта ферментативного превращения солей тетразолия в синие производные формазана, которые легко детектируются спектроскопическими методами (Mosmann, Т., J Immunol Methods, 1983, 65, 55-63).
В целом, предпочтительные методы определения пролиферации клеток включают инкубацию клеток в выбранной для роста среде в присутствии тестируемого вещества или без него. Условия роста для различных прокариотических и эукариотических клеток подробно описаны (Ausubel et al. Current Protocols in Molecular Biology. Wiley and Sons. 1999; Bonifacio et al. Current Protocols in Cell Biology. Wiley and Sons. 1999). Для определения пролиферации клеток, после инкубации к ним добавляется соль тетразолия и затем определяется количество образовавшегося формазана. Количество образовавшихся производных формазана определяется по оптической плотности обработанных клеток.
Для определения антипролиферативной активности соединений могут использоваться раковые клеточные линии, например Daudi, NAMALVA, Р3Н3, P3HR-1, Raji (неходжкинская лимфома), COLO 205, DLD-1, НСТ-15, НТ29 (рак толстой кишки); HEP G2 (гепатома); K-562 (лейкемия); А549, NCI-Н249, NCI-H2228, NCI-H3122 (рак легкого); Karpas-299, SU-DHL-1 (лимфома); MCF7, MDA-MB-231 (рак молочной железы); SAOS-2 (остеосаркома); OVCAR-3 (рак яичника); PANC-1 (рак поджелудочной железы); DU-145, РС-3 (рак простаты); ACHN, CAKI-1 (рак почек); MG-63 (саркома).
Хотя для определения антипролиферативной активности соединений предпочтительно используются клетки млекопитающих, для этой цели также могут быть использованы клетки низших эукариотов, например дрожжей. Предпочтительно использование клеточных линии человека, крыс, мышей, кроликов, низших обезьян, хомяков и морских свинок, поскольку клеточные линии из этих организмов наиболее хорошо изучены и полно охарактеризованы.
Ниже приведен пример определения активности соединения в клеточных моделях онкологических заболеваний. В данном эксперименте использовалась методика измерений цитотоксичности соединений на основе измерения активности клеточных дегидрогеназ по оптической плотности многократно описана в научной литературе и практически является стандартом определения цитотоксичности. Биохимический процесс, лежащий в основе метода, состоит в восстановлении желтого бромида 3-(4,5-диметилтиазол-2-ил)-2,5-тетразолия (МТТ) в нерастворимые пурпурно-синие внутриклеточные кристаллы МТТ-формазана (МТТ-ф) под действием дегидрогеназ, локализованных в митохондриях. Нежизнеспособные мертвые клетки такой способностью не обладают. Интенсивность превращения МТТ в МТТ-ф отражает общий уровень дегидрогеназной активности исследуемых клеток и модулируется активностью сопряженных ферментных систем, например, дыхательной цепи переноса электронов и т.д. Активность дегидрогеназ снижается на наиболее поздних стадиях гибели клеток, поэтому метод пригоден для скрининга цитотоксичности и токсичных, и малотоксичных соединений.
Навески образцов растворяют в диметилсульфоксиде до конечной концентрации 10 мМ. Из этих растворов приготовляются разведения каждого препарата в среде для культивирования.
Клетки неходжкинской лимфомы (клеточные линии Raji, NAMALVA, Daudi, Р3Н3, Р3Н3-1) высеиваются в 96-луночные планшеты. В лунки добавляют исследуемые препараты до конечных концентраций 0,01-50 мкМ. В контрольные лунки добавляют культуральную среду. Каждую концентрацию препаратов исследуют в трех параллелях. Клетки инкубируют при 37°C, 5% CO2 в увлажненной атмосфере, затем в лунки вносят раствор МТТ, инкубируют в CO2-инкубаторе, образовавшийся формазан растворяют в диметилсульфоксиде и измеряют оптическую плотность при длине волны 540 нм. Оптическая плотность в лунках без испытуемого соединения принимается за 100% выживаемости. Оптическая плотность в лунках с испытуемым соединением характеризует процент торможения пролиферации.
Иллюстративные примеры соединений, обладающих активностью (со значениями IC50 10-10000 нМ) в in vitro для подавления абберантной пролиферации клеток неходжкинсокой лимфомы.








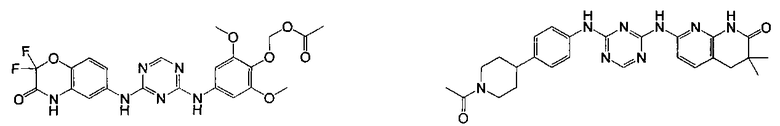
14. Эксперименты на животных моделях
Соединения, являющиеся предметом данного изобретения, подавляют аберрантную активность и пролифирацию клеток иммунной системы и, таким образом, могут быть применены для лечения воспалительных, аутоиммунных, онкологических и других заболеваний.
In vivo модели определения эффективности химических соединений в лечении различных аутоимунных заболеваний хорошо известны и могут быть использованы для сравнительной характеристики соединений, описанных в данном изобретении. Ниже приведен пример определения активности соединения в in vivo модели адъювантного артрита у крыс (хроническое иммунное воспаление).
Хроническое иммунное воспаление у крыс (предпочтительно использовать крыс Льюиса возрастом 6-7 недель) вызывают субплантарным введением в правую заднюю лапу 0,1 мл полного адъюванта Фрейнда (ПАФ). Оценивают вторичную иммунологическую реакцию (отек на левой лапе). Измерение объема лапы и введение исследуемых веществ начинют с 14 суток (после вовлечения в воспалительный процесс контрлатеральной лапы). Исследуемые соединения вводят внутрижелудочно дважды в день в течение 11 дней в концентрациях 30, 60 и 100 миллиграмов на килограмм массы животных. Первую оценку действия веществ проводят через 24 часа после первого внутрижелудочного введения, последние измерения объема лапы проводят на 25 день эксперимента (через 24 часа после последнего введения веществ) и на 27 день (через 2 дня после прекращения введения). Эффект терапевтического воздействия исследуемых соединений оценивают по степени угнетения воспалительной реакции и по предотвращению разрушения кости в сравнении с контрольной группой, не получавшей лечения. Для оценки повреждений кости у каждо животного использовали следующую шкалу: 1. Патологическая установка ногтевых фаланг; 2. Наличие отека мягких тканей; 3. Наличие содружественного поражения конечности; 4. Наличие субхондрального склероза; 5 Деформация плюсневых костей; 6. Сужение суставной щели проксимальных суставов; 7. Сужение суставной щели дистальных суставов; 8. Наличие специфических признаков ревматоидного полиартрита (ревматоидные узелки). Для анализа полученные данные ранжировали в зависимости от значимости признака в патогенезе.
Иллюстративные примеры соединений, обладающих активностью in vivo в терапевтической дозе 60 мг/кг и более для подавления воспалительной реакции и разрушения костной ткани в модели адъювантного артрита у крыс, представлены ниже. Данные соединения позволяют полностью предотвратить содружественное поражения конечности и другие более серьезные повреждения суставов. Средняя оценка по повреждений кости по группе из 20 крыс, получавших лечения была меньше 2.5, а для группы из 20 крыс не получавших препаратов была более 7.





Соединения, являющиеся предметом данного изобретения, показавшие антипролиферативную активность в клеточных экспериментах, затем исследовались in vivo на организмах млекопитающих. Как правило, эксперименты in vivo проводились на грызунах, таких как мыши и крысы.
Линии клеток нехождкенской лимфомы (Raji, NAMALVA, Daudi, Р3Н3, Р3Н3-1) в колличестве 5*106 клеток в бессывороточной среде вводились подкожно в правый бок бестимусных голых мышей (NCr) возрастом 3-4 недели. По достижении опухолью объема ~200 мм3 мышей разбивали на 2 группы: контрольную и терапевтическую. Мышам контрольной группы с помощью орального зонда вводили 0,3 мл 0,5% раствора метилцеллюлозы в воде, мышам терапевтической группы - 0,3 мл 0,5% раствора метилцеллюлозы, в котором было суспендировано терапевтическое вещество. Объем опухоли (мм3) рассчитывали по следующей формуле: V=L×W2×0,5, где L - длина опухоли в мм, W - ширнина в мм. Для определения эффективности ингибирования роста опухоли по окончании лечения (20 дней) рассчитывалось отношение среднего объема для терапавтической/контрольной групп (% Т/С). Для полученных данных был проведен анализ статистической достоверности с помощью теста Даннета. Примеры соединений, обладающих высокой активностью (% Т/С<40) в терапевтической дозе 50 мг/кг, представлены ниже






15. Примеры фармацевтической композиции
Вещества, описанные в данном изобретении, могут быть использованы для профилактики и лечения болезней человека в виде следующих составов (под «Веществом» понимается активный ингредиент):
1М раствор соляной кислоты до pH 7.6
Вода для инъекций до 100%
Вода для инъекций до 100%
Примечание: данные составы могут быть приготовлены в соответствии со стандартными фармацевтическими методиками. Таблетки (1)-(3) могут быть покрыты кишечнорастворимой оболочкой с использованием, например, фталата ацетата целлюлозы. Аэрозольные составы (8)-(11) могут быть использованы в сочетании со стандартными диспенсерами; в качестве суспендирующего агента вместо триолеата сорбитана и соевого лецитина может быть использован моноолеат сорбитана, полуолеат сорбитана, полисорбат 80, олеат полиглицерина или олеиновая кислота.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВАЯ КРИСТАЛЛИЧЕСКАЯ СОЛЕВАЯ ФОРМА 2,2-ДИМЕТИЛ-6-((4-((3,4,5-ТРИМЕТОКСИФЕНИЛ)АМИНО)-1,3,5-ТРИАЗИН-2-ИЛ)АМИНО)-2Н-ПИРИДО[3,2-В][1,4]ОКСАЗИН-3(4Н)-ОНА ДЛЯ МЕДИЦИНСКОГО ПРИМЕНЕНИЯ | 2016 |
|
RU2621187C1 |
| БИЦИКЛИЧЕСКИЕ ПИРИДИНЫ И АНАЛОГИ В КАЧЕСТВЕ МОДУЛЯТОРОВ СИРТУИНА | 2010 |
|
RU2550821C2 |
| СПОСОБЫ ЛЕЧЕНИЯ ИЛИ ПРОФИЛАКТИКИ АУТОИММУННЫХ ЗАБОЛЕВАНИЙ С ПОМОЩЬЮ СОЕДИНЕНИЙ 2,4-ПИРИМИДИНДИАМИНА | 2003 |
|
RU2376992C2 |
| АНАЛОГ ПИРИДИНО[1,2-А]ПИРИМИДОНА, ИСПОЛЬЗУЕМЫЙ В КАЧЕСТВЕ ИНГИБИТОРА mTOR/PI3K | 2015 |
|
RU2658912C1 |
| ИНГИБИТОРЫ ПРОТЕИНКИНАЗЫ MKK4 ДЛЯ СТИМУЛЯЦИИ РЕГЕНЕРАЦИИ ПЕЧЕНИ ИЛИ УМЕНЬШЕНИЯ ИЛИ ПРЕДОТВРАЩЕНИЯ ГИБЕЛИ ГЕПАТОЦИТОВ | 2019 |
|
RU2788000C2 |
| Замещенный 3,4,12,12а-тетрагидро-1Н-[1,4]оксазино[3,4-c]пиридо[2,1-f] [1,2,4]триазин-6,8-дион, фармацевтическая композиция, способы их получения и применения | 2019 |
|
RU2720305C1 |
| 7',9'-ДИМЕТИЛ-3'-АРИЛ-1'-СПИРО[ИНДЕН-2,2'-ПИРИДО[3',2':4,5]ТИЕНО]3,2-D]ПИРИМИДИН]-1,3,4'(3'H)ТРИОНЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ В КАЧЕСТВЕ АНТИДОТОВ 2,4-Д НА ПОДСОЛНЕЧНИКЕ | 2020 |
|
RU2754220C1 |
| ПРОЛЕКАРСТВА СОЕДИНЕНИЙ 2,4-ПИРИМИДИНДИАМИНА И ИХ ПРИМЕНЕНИЯ | 2006 |
|
RU2416616C2 |
| ИНГИБИТОР ГЕКСОН-ГЛЮКОКИНАЗЫ И ЕГО ПРИМЕНЕНИЕ | 2020 |
|
RU2797185C2 |
| АЗАИНДОЛЬНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ФАКТОРА Xa | 2004 |
|
RU2330853C2 |
Изобретение относится к производному 2,4-диамино-1,3,5-триазина общей формулы I, обладающему свойствами ингибитора протеинкиназ, его применению, а также к фармацевтической композиции на его основе. Изобретение может быть использовано для лечения аутоиммунных или онкологических заболеваний, ревматоидного артрита, неходжкинской лимфомы. В общей формуле I

Y представляет собой СН2, CHR', О, S, S(O) или S(O)2; Х1, X2, X3 выбираются независимо из групп СН или N; R1 представляет собой C1-8 алифатическую группу, С3-8 циклоалкил, С6-10 арил, этилен-диоксифенил, метилендиоксифенил, пиридил, каждое из которых оптимально замещено одной или более одинаковыми или разными группами R”; R' представляет собой водород, ОН, галоген, такой как F, Cl, Вr, I, или карбоксил или карбоксамид, оптимально N-замещенный (С1-6)алкилом, или циано или гало(С1-8)алкил, (С1-8)алкокси, пиперидинил, отимально замещенный метилом; R” представляет собой R' или RD; R21, R22, R23, R24 выбираются независимо из групп F, Cl, Вr, I, CN, (C1-16)алкил, кроме того R21 и R22 или/и R23 и R24 могут быть объединены и представлять собой одну оксо (=O) группу или совместно с атомом углерода образовывать спироцикл, содержащий от 3 до 7 атомов углерода, кроме того R21 и R24 могут совместно с двумя атомами углерода образовывать алифатический или ароматический цикл, содержащий от 4 до 8 атомов, опционально замещенный одной или несколькими группами R'; RD представляет собой оксо группу =O или =S. 5 н. и 8 з.п. ф-лы, 12 пр.
1. Производное 2,4-диамино-1,3,5-триазина - общей формулы I, или его фармацевтически приемлемая соль, гидрат или фармацевтически приемлемый сложный эфир, гидролизующийся in vivo ÷
где:
Y представляет собой СН2, CHR', О, S, S(O) или S(O)2,
Х1, X2, X3 выбираются независимо из групп СН или N,
R1 представляет собой C1-8 алифатическую группу, С3-8 циклоалкил, С6-10 арил, этилен-диоксифенил, метилендиоксифенил, пиридил, каждое из которых оптимально замещено одной или более одинаковыми или разными группами R”,
R' представляет собой водород, ОН, галоген, такой как F, Cl, Вr, I, или карбоксил или карбоксамид, оптимально N-замещенный (С1-6)алкилом, или циано или гало(С1-8)алкил, (С1-8)алкокси, пиперидинил, отимально замещенный метилом,
R” представляет собой
R' или RD
R21, R22, R23, R24 выбираются независимо из групп F, Cl, Вr, I, CN, (C1-16)алкил, кроме того R21 и R22 или/и R23 и R24 могут быть объединены и представлять собой одну оксо (=O) группу или совместно с атомом углерода образовывать спироцикл содержащий от 3 до 7 атомов углерода, кроме того R21 и R24 могут совместно с двумя атомами углерода образовывать алифатический или ароматический цикл, содержащий от 4 до 8 атомов, опционально замещенный одной или несколькими группами R',
RD представляет собой оксо группу =O или =S.
2. Производное по п.1, где X2 и X3 представляют собой СН.
3. Производное по п.1, где Y представляет собой О.
4. Производное по п.1, где R1 представляет собой фенил, оптимально замещенный одной или более одинаковыми или разными группами R'.
5. Производное по п.4, где R1 представляет собой 3,4,5-триметокси фенил.
6. Производное по п.1, где X1 представляет собой N, R21, R22 представляет собой -СН3, R23, R24 объединены и представляют собой одну оксо (=O) группу.
7. Производное по п.1, где X1 представляет собой СН, R21, R22 представляет собой -F, R23, R24 объединены и представляют собой одну оксо (=O) группу.
8. Производное по любому из пп.1-7, обладающее эффективностью в ингибировании SYK, ZAP-70, JAK1 JAK2, JAK3, BTK, TYK1 и LYN киназ.
9. Применение производных по любому из пп.1-7 для лечения заболеваний, вызванных аберрантной активностью протеинкиназ SYK, ZAP-70, JAK1, JAK2, JAK3, ВТК, TYK1 и LYN.
10. Применение производных по любому из пп.1-7 в лечении аутоиммунных или онкологических заболеваний.
11. Применение химических соединений по любому из пп.1-7 для лечения ревматоидного артрита и неходжкинской лимфомы.
12. Фармацевтическая композиция для лечения аутоимунных и онкологических заболеваний, характеризующаяся тем, что она содержит эффективное количество производного по любому из пп.1-7 и фармацевтически приемлемый носитель.
13. Фармацевтическая композиция по п.12, пригодная для лечения ревматоидного артрита и неходжкинской лимфомы.
| WO 2003070710 A1, 28.08.2003 | |||
| US 0007858782 B2, 28.12.2010 | |||
| WO 2009143389 A1, 26.11.2009 | |||
| ИММУНОДЕПРЕССАНТ И ПРОТИВООПУХОЛЕВОЕ СРЕДСТВО, СОДЕРЖАЩЕЕ ГЕТЕРОЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ В КАЧЕСТВЕ АКТИВНОГО ИНГРЕДИЕНТА | 2006 |
|
RU2443441C2 |

