Область техники, к которой относится изобретение
Изобретение относится к области биотехнологии, в частности позволяет методом конструирования и скрининга банка делеционных производных получать укороченные варианты практически важных генов с улучшенными биотехнологическими характеристиками. Метод позволяет снижать токсичность рекомбинантного продукта для клеток продуцента и добиваться улучшения фолдинга целевого продукта в гетерологичной системе. Актуальность разработки обусловлена высокой склонностью многих практически важных белков, прежде всего, вирусного происхождения, образовывать агрегаты при попытках их продукции в клетках бактерий, а также токсичностью многих таких белков по отношению к клеткам рекомбинантного продуцента. Предполагается, что общей причиной такого поведения большинства вирусных белков, а также многих бактериальных антигенов, рецепторов и поверхностных белков клеток эукариот является наличие в их структуре двух и более доменов с выраженной связывающей способностью в отношении клеточных структур: липидных мембран, нуклеиновых кислот, белков, полисахаридов и других. Такая организация с большой вероятностью приводит к образованию в клетке рекомбинантного продуцента нежелательной сети физических контактов, нарушающих нормальную передачу сигналов в цитоплазме. С точки зрения этой гипотезы расчленение генов природных белков на фрагменты, кодирующие изолированные глобулярные домены, способно снизить общую токсичность мультидоменных белков, обеспечить повышение уровня их экспрессии в гетерологичных системах. Однако до настоящего времени не предложен универсальный эвристический подход к расчленению белка на изолированные домены, содержащие на флангах линкерные последовательности аминокислот небольшой протяженности, оптимальные для трансляции, ретрансляционного фолдинга и релизинга продуктов из рибосом.
Предлагаемый нами подход нацелен на решение задачи по расчленению целевого гена на фрагменты, оптимальные для экспрессии в бактериальных клетках, методом конструирования и скрининга банка делеционных производных. Этот подход позволяет работать с неохарактеризованными генами с неизвестной пространственной организацией продуктов. Эта задача особенно актуальна для большинства структурных и неструктурных вирусных антигенов, предварительные кристаллографические исследования которых затруднены невозможностью получения этих белков в нативной водорастворимой форме. Таким образом, рациональный подход к вычленению в их составе глобулярных доменов дополнительно осложнен. В то же время делеционные производные вирусных антигенов, сохраняющие иммуногенность и антигенность, при условии хорошей совместимости с рекомбинантными системами экспрессии являются оптимальным инструментом для создания диагностических систем и вакцин, существенно облегчают исследования структурно-функциональной организации таких белков in vivo и in vitro.
Уровень техники
Получение серийных делеций является актуальной задачей на протяжении последних двадцати лет. Делеционные производные используются при секвенировании протяженных участков ДНК, выявлении полиморфизма генов с целью исследования структурно-функциональной организации белков и повышения уровня их продукции в гетерологичных системах. Предложен целый ряд методов создания серийных делеций: с использованием экзонуклеазы III, нуклеазы Bal31, транспозонов и ДНКазы.
Исторически первым и в то же время популярным методом является применение экзонуклеазы III [1]. Существенными недостатками данного метода являются односторонний характер делений и большой расход исходной ДНК.
Использование нуклеазы Bal31 позволяет получить двухсторонние симметричные делении, но также требует значительного количество исходной ДНК.
Метод получения серийных делений, основанный на транспозонном принципе [2], позволяет получить банк с равномерной представленностью делеций. В качестве недостатков метода следует отметить недостаточную воспроизводимость и трудность регуляции интенсивности транспозиции in vivo.
В литературе [3] также описан способ получения серийных делеций с помощью ПЦР в сочетании с удалением образующихся димеров случайных праймеров гель-фильтрацией на колонке с носителем Сефадекс. Этот подход лишен недостатков ранее описанных методик, но его практическое применение затрудняется высокой степенью контаминации рабочей зоны ПЦР-продуктами в момент проведения хроматографии.
Раскрытие изобретения
Большинство перечисленных методов используются для получения наборов клонов с целью их последующего секвенирования. Эта задача не требует рэндомизации разрывов с точностью до 1 нуклеотида: с учетом длины прочтения современных капиллярных или NGS-секвенаторов вполне достаточно, чтобы концы клонов покрывали исследуемую область генома с шагом в одну или несколько сотен нуклеотидных пар. Таким образом, неслучайность первичных разрывов, вносимых панкреатической ДНКазой, обусловленная GC-составом последовательности, степенью ее устойчивости в дуплексе, склонностью к образованию неканонической структуры и прочими факторами, оказывается несущественной для описанных в литературе методов. Однако при оптимизации способности фрагментов генов к экспрессии in vivo существует насущная необходимость получения клонов, отличающихся между собой по длине на одни или несколько кодонов, что повышает требования к рэндомизации точек разрывов. Предложенный нами метод позволяет эффективно решать задачу рэндомизации за счет использования ДНК-полимеразы I, скорость работы которой в качестве 3'-5'-экзонуклеазы практически не зависит от последовательности. Смещая концы получаемых клонов на небольшое, но не фиксированное расстояние от точек первичных разрывов пакреатической ДНКазы, можно достигнуть практически случайного их распределения.
Особенностью большинства описанных методов получения делеций, основанных на использовании нуклеаз, является деградация исходной ДНК в процессе обработки. Таким образом, для получения банка требуется значительное количество исходной ДНК, а полученный банк не может быть размножен. Использование схем с лигированием первичных фрагментов с двуцепочечными адаптерами позволяет решить эту проблему, но приводит к загрязнению банка димерами адаптеров и клонами со вставками незначительного размера. Использование в предложенной схеме ПЦР с симметричным адаптерным праймером, специфичным к константной части случайного праймера, позволяет эффективно решать эту проблему. Несмотря на низкий выход ПЦР с симметричным праймером по сравнению с асимметричным, ее преимуществом является селекция размера клонов, укладывающихся в диапазон размеров от 200 до 700 п.н. (это связано с жесткостью дуплексов ДНК, не позволяющих им образовывать структуры типа «сковорода с ручкой» при таком размере фрагмента). Таким образом, предлагаемая нами схема получения банков при стандартном наборе ферментов наиболее приближена к задаче создания набора клонов, предназначенного для отбора хорошо экспрессирующихся in vivo вариантов, обладающих оптимальными биотехнологическими характеристиками.
Сущность изобретения состоит в возможности устранения принципиального ограничения метода ПНР со случайной затравкой, состоящего в способности случайной затравки образовывать в растворе дуплексы, самопроизвольного размножающиеся при проведении ПЦР в отсутствие специфической ДНК-матрицы.
На первой стадии процедуры получают банк фрагментов исходного гена в форме смеси продуктов ПЦР, последовательно выполняя следующие операции:
(1) в случайные сайты целевого гена, находящегося в форме линейного фрагмента ДНК, вносят однонитевые разрывы с помощью ограниченного гидролиза панкреатической дезоксирибонуклеазой 1 (ДНКазой);
(2) расширяют бреши в сторону 5'-конца с помощью ДНК-полимеразы I Е.coli в отсутствие дезоксирибонуклеотидтрифосфатов;
(3) проводят отжиг синтетических олигонуклеотидов, имеющих на 3'-конце 6-, 11- или 17-членную случайную последовательность, а на 5'-конце - константный участок (20 нуклеотидов), предназначенный для отжига адаптерного праймера («случайные» праймеры); ковалентно присоединяют случайные праймеры, соединившиеся с ДНК-матрицей за счет комплементарных взаимодействий, путем лигирования ДНК-лигазой фага T4;
(4) удаляют избыток не связанного с матричной ДНК случайного праймера путем батч-хроматографии на микропористом стеклянном сорбенте;
(5) проводят реакцию ПЦР с адаптерным праймером («однопраймерная» ПЦР).
Для оптимизации концентрации реагентов, нуждающихся в точных стехиометрических соотношениях с целью подбора желательной средней длины фрагментов делеционных производных, готовят серийные разведения панкреатической дезоксирибонуклеазы 1 и случайного праймера, объединяя продукты реакции, полученные в разных условиях. Для отбора делеционных производных, пригодных для экспрессии in vivo, полученные in vitro фрагменты (очищенные продукты ПЦР) подвергаются клонированию в вектор прямой селекции pQL30, содержащий ген β-галактозидазы E.coli со встроенным полилинкером с мутацией смещения трансляционной рамки считывания. Встройка делеционных производных целевого гена в полилинкер с определенной вероятностью приводит к восстановлению рамки, что позволяет визуально контролировать экспрессию белка в полученных клонах. Существенной особенностью предлагаемого метода является возможность отбора только тех фрагментов исходного гена, которые не только в силу восстановления непрерывности открытой рамки считывания, но и в силу своей последовательности, вторичной и третичной структуры кодируемого продукта обладают способностью к высокоэффективной экспрессии в бактериальных клетках.
1) Гидролиз панкреатической дезоксирибонуклеазой 1 ДНК, содержащей целевой ген, в форме линейного фрагмента в количестве 8-10 мкг разбавляют в объеме 80 мкл в буфере 10 мМ Трис-HCl, pH 7.5, 1 мМ MgCl2, 50 мМ NaCl, 1 мМ восстановленный дитиотрейтол. Полученную смесь делят на четыре аликвоты объемом 20 мкл каждая. В первую пробирку вносят ДНКазу (Promega) с активностью 10-4 ед/мкл, тщательно перемешивают и аликвоту полученного раствора объемом 2 мкл переносят во вторую пробирку. Операцию серийного разбавления ДНКазы с шагом 10 раз повторяют с третьей и четвертой пробирками. Время приготовления серии разбавлений ДНКазы не должно превышать 2 мин.
Подготовленную серию разведений инкубируют 20 мин при 37°C, после чего реакцию останавливают, внося в каждую пробирку по 20 мкл смеси водонасыщенный нейтральный фенол - хлороформ (1:1). Степень деградации ДНК оценивают с помощью электрофореза в 1,8%-агарозном геле. Для дальнейшей работы объединяют и используют образцы с видимой степенью деградации. Для этого объединенную ДНК подвергают фенольной экстракции, осаждают изопропанолом в стандартных условиях и растворяют в минимальном объеме деионизованной воды.
2) Реакция процессивного гидролиза ДНК ДНК-полимеразой 1 из E.coli в направлении 5'→3' конец
К ДНК, полученной на предыдущей стадии, добавляют 4 ед. полимеразы 1 из E.coli (Pharmacia) в соответствующем буфере. Реакцию проводят в течение 1 ч при 37°C и останавливают фенольной экстракцией.
3) Отжиг синтетических олигонуклеотидов
На подготовленную исходную матрицу с внесенными разрывами проводят отжиг «случайных» праймеров:
supl(5'-GGATCCGCAGCATCCGGAGCNNNNNN-3'),
supl-11(5'-GGATCCGCAGCATCCGGAGCNNNNNNNNNNN-3'),
или
supl-17(5'-GGATCCGCAGCATCCGGAGCNNNNNNNNNNNNNNNNN-3'),
имеющих на 3'-конце 6-, 11- или 17-членную случайную последовательность, а на 5'-конце - константный участок (20 нуклеотидов), предназначенный для отжига адаптерного праймера; ковалентно присоединяют случайный праймер, образовавший Уотсон-Криковский дуплекс с ДНК-матрицей, путем лигирования ДНК-лигазой фага T4.
Для этого ДНК, полученную после фенольной экстракции, растворяют в воде и лигазном буфере общим объемом 30 мкл, добавляют 3-5 ед. ДНК-лигазы фага T4. Разделяют смесь на три равные порции по 10 мкл. В первую пробирку вносят 4 пмоль праймеров объеме 1 мкл, перемешав, переносят 2 мкл во вторую пробирку, и затем 2 мкл из второй в третью пробирку.
Лигирование протекает в течение 14 ч при 4°C.
4) Удаление избытка несвязанного случайного праймера путем батч-хроматографии
Избыток несвязанного случайного праймера удаляют путем батч-хроматографии с помощью набора Silica bead DNA gel extraction kit (Fermentas) согласно протоколу производителя.
5) ПЦР с адаптерным праймером
Очищенную лигазную смесь используют в качестве матрицы для проведения ПЦР с адаптерным праймером:
ES1 (5'-GGGGATCCGCAGCATCCGGAGC-3') или
ES3 (5'-GGGGATCCGCAGCATCCG-3').
Проводят 30 циклов ПЦР в объеме 30 мкл, внося в реакционную смесь 4 пмоль адаптерного праймера. Температура отжига праймеров в ходе ПЦР - 60°C. Признаком прохождения реакции является появление набора фрагментов ДНК в диапазоне размеров 150-600 п.н., наблюдаемого при проведении электрофореза в агарозном геле.
6) Клонирование серийных делеций в вектор прямой селекции pQL30
ПЦР-продукт, полученный на предыдущей стадии, очищают с помощью набора Silica bead DNA gel extraction kit (Fermentas) согласно протоколу производителя. Очищенный продукт подвергают воздействию эндонуклеазы рестрикции BamHI при 37°C в течение 2 ч. Тем же ферментом обрабатывается и ДНК вектора pQL30. Лигирование проводят в течение 14 ч при 4°С.Клетки E.coli штамма TG1 трансформируют лигазной смесью и высевают на чашки со средой LB в присутствии 100 мкг/мл ампициллина и X-gal. Для дальнейшей работы отбирают клоны, экспрессирующие активность β-галактозидазы (синяя окраска колоний на среде с X-gal). Учет результатов определения активности β-галактозидазы в колониях трансформантов проводится в срок около 40 ч с момента окончания трансформации, температура культивирования 37°C.
Все отобранные клоны далее анализируют:
- на наличие β-галактозидазной активности в растворимой и нерастворимой клеточной фракции отобранного клона;
- на содержание специфической активности целевого белка в клетках продуцента (обычно методом иммуноскрининга в варианте дот-блоттинга или Вестер-блоттинга).
7) Биосинтез рекомбинантного белка
Полученный продуцент культивируют в течение 14-18 ч при 30°C в жидкой среде (0,5% дрожжевой экстракт, 1% пептон, 0,5% NaCl) с добавлением 100 мкг/мл ампициллина в пробирках объемом 10 мл с 3 мл среды в каждой. Индукцию промотора в клетках продуцента проводят внесением раствора IPTG до конечной концентрации 1 мМ.
8) Определение β-галактозидазной активности в клетках продуцента
Каждую из полученных культур отобранных клонов подвергают дезинтеграции. Для этого клетки, собранные из 600 мкл культуры, суспендируют в 600 мкл буфера, содержащего 25 мМ Трис-HCl, 5 мМ EDTA, панкреатическую РНКазу (1 мг/мл), pH 8.0, добавляют 30 мг стеклянных шариков диаметром 0,5 мм и встряхивают на ручном встряхивателе для пробирок в течение 5 мин на максимальной амплитуде. Обработка ведется при комнатной температуре.
Полученный таким образом грубый клеточный лизат подвергают центрифугированию при 14000 G в течение 15 мин с целью разделения растворимой и нерастворимой клеточных фракций. Нерастворимую клеточную фракцию суспендируют в 600 мкл воды. По 20 мкл растворимой и нерастворимой (в форме суспензии) клеточной фракции от каждой полученной культуры смешивают с 50 мкл субстратной смеси, содержащей 0,02% X-gal, 25 мМ Трис-HCl, pH8,5. Смесь инкубируют при 37°C в течение 2-4 ч, затем производят замер сигнала на планшетном спектрофотометре при λ=620 нм.
9) Анализ выхода целевого белка
Оценку выхода целевого продукта осуществляют при помощи электрофоретического разделения белков по Лэммли с последующим вестерн-блоттингом на нитроцеллюлозной мембране и детекцией целевого белка с использованием антител, специфичных к заданному белку. Для этого из грубого клеточного лизата каждой культуры отбирают по 100 мкл. Лизат подвергают центрифугированию при 14000 G в течение 15 мин и разделяют растворимую и нерастворимую клеточную фракции. Белки растворимой и нерастворимой клеточной фракции солюбилизируют в 30 мкл буфера Лэммли и разделяют в 15% ПААГ в денатурирующих условиях. Затем белки переносят на нитроцеллюлозную мембрану. После окрашивания мембраны красителем Понсо С наблюдают полосу, соответствующую расчетной массе белка (~120 кДа).
Детекция целевого белка на мембране включает в себя: а) блокирование неспецифических связей путем инкубации мембраны с 1% BSA, б) взаимодействие с антителами, специфическими к целевому белку; в) взаимодействие с вторичными антителами, конъюгированными с пероксидазой хрена; г) окрашивание с диаминобензидином (ДАБ) в присутствии 1% пероксида водорода. После окрашивания с ДАБ наблюдают полосу, соответствующую расчетной массе белка (~120 кДа).
Пример 1
Для работы были использованы 27 образцов кДНК гена NS5A-NS5B из HCV, генотип 1b (GenBank accession number JX022751-JX022777) длиной 1670 п.н., наработанные с помощью ПЦР с праймерами 1b_6117_S_L (TCCCCCACGCACTATGTGCC) и 1b_7780_AS_L (CGGTARTGGTCGTCCAGGAC). Образцы перечисленных фрагментов кДНК были подвергнуты вторичной ПЦР-амплификации с целью накопления ДНК целевого гена NS5A-NS5B, объединены в пул и очищены с помощью набора Silica bead DNA gel extraction kit (Fermentas) согласно протоколу производителя.
Очищенный ПЦР-продукт, содержащий целевой ген, в количестве 10 мкг разбавляли в объеме 80 мкл в буфере 10 мМ Трис-HCl, pH 7.5, 1 мМ MgCl2, 50 мМ NaCl, 1 мМ восстановленный дитиотрейтол. Полученную смесь делили на четыре аликвоты объемом 20 мкл каждая. В первую пробирку вносили ДНКазу (MBI Fermentas) с активностью 1×10-4 ед/мкл, тщательно перемешивали и аликвоту полученного раствора объемом 2 мкл переносили во вторую пробирку. Операцию серийного разбавления ДНКазы с шагом 10 раз повторяли с третьей и четвертой пробирками. Время приготовления серии разбавлений ДНКазы не превышало 2 мин.
Подготовленную серию разведении инкубировали 20 мин при 37°C, после чего реакцию останавливали, внося в каждую пробирку по 20 мкл смеси водонасыщенный нейтральный фенол-хлороформ (1:1). Степень деградации ДНК оценивали с помощью электрофореза в 1,8%-агарозном геле. Для дальнейшей работы объединяли и использовали образцы с видимой степенью деградации: концентрация ДНКазы равна 1×10-5-1×10-6 ед/мкл. Объединенную ДНК подвергали фенольной экстракции, осаждали изопропанолом в стандартных условиях и растворяли 10 мкл деионизованной воды.
К 10 мкл каждого образца добавили 1 мкл (4 ед) полимеразы 1 из E.coli в соответствующем буфере и инкубировали в течение 1 ч при 37°C. Реакцию останавливали фенольной экстракцией.
ДНК, полученную после фенольной экстракции, растворяли в воде и лигазном буфере общим объемом 30 мкл, добавляли ДНК-лигазу фага Т4. Разделяли смесь на три равные порции по 10 мкл. В первую пробирку вносили 1 мкл (4пмоль) праймера (supl, supl-11 или supl-17), перемешав, переносили 2 мкл во вторую пробирку, и затем 2 мкл из второй - в третью пробирку. Лигирование протекало в течение 14 ч при 4°C. Затем лигазную смесь использовали в качестве матрицы для проведения ПЦР с праймерами ES1 и ES3. Оценку прохождения реакции оценивали электрофоретически.
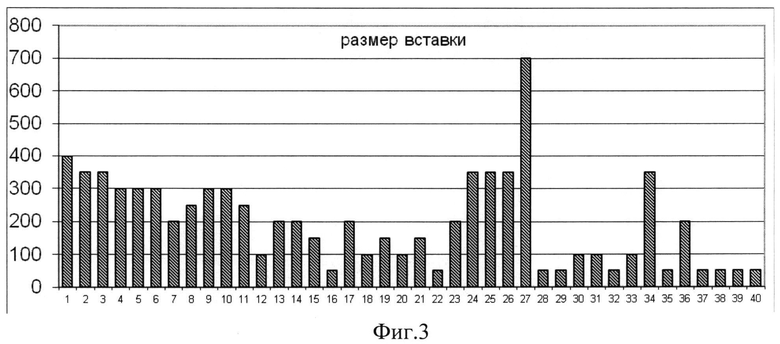
ПЦР-продукт, полученный на предыдущей стадии, очищали с помощью набора Gel extraction kit (MBI Fermentas) согласно протоколу производителя. Очищенный продукт подвергали воздействию эндонуклеазы рестрикции BamHI при 37°C в течение 2 ч. Аналогичным образом обрабатывали плазмидную ДНК вектора pQL30. Лигирование проводили в течение 14 ч при 4°C. Клетки E.coli штамма TG1 трансформировали лигазной смесью и высевали на чашки со средой LB, содержащие 100 мкг/мл ампициллина и 0,05% X-gal. Соотношение синих (с экспрессирующейся вставкой) и белых (без вставки или с неэкспрессирующейся вставкой) колоний составило 63:1460. Из колоний с восстановленной активностью β-галактозидазы была выделена плазмидная ДНК. Наличие соответствующей вставки в клонах по сравнению с нерекомбинантным вектором pQL30 выявлялось с помощью ПЦР со стандартными праймерами pQE-for и pUC-for. В качестве отрицательного контроля использовался нерекомбинантный вектор pQL30. Таким образом, удалось отобрать 40 клонов с размером вставки, визуально превышающим контроль. Размер вставки колебался от 50 до 700 п.н. без существенных отличий между группами.
Все отобранные клоны, представляющие собой производные штамма E.coli TG1, несущие отобранные из банка плазмиды, использовались для наработки рекомбинантных белков. В качестве отрицательного контроля, не содержащего целевой вставки был выбран продуцент полноразмерной β-галактозидазы E.coli (βGal) и ее слитые бифункциональные производные, содержащие гены пектинов из эндоплазматического ретикулума дрожжей Saccharomyces cerevisiae: pLacZ-emp46 и pLacZ-emp47.
Полученные продуценты (включая контрольные культуры) культивировали в течение 14-18 ч при 30°C в жидкой среде (0,5% дрожжевой экстракт, 1% пептон, 0,5% Nad) с добавлением 100 мкг/мл ампициллина в пробирках объемом 10 мл с 3 мл среды в каждой. Индукцию промотора в клетках продуцента проводили добавлением IPTG до 1 мМ. Клетки собирали низкоскоростным центрифугированием и подвергали дезинтеграции, как описано выше.
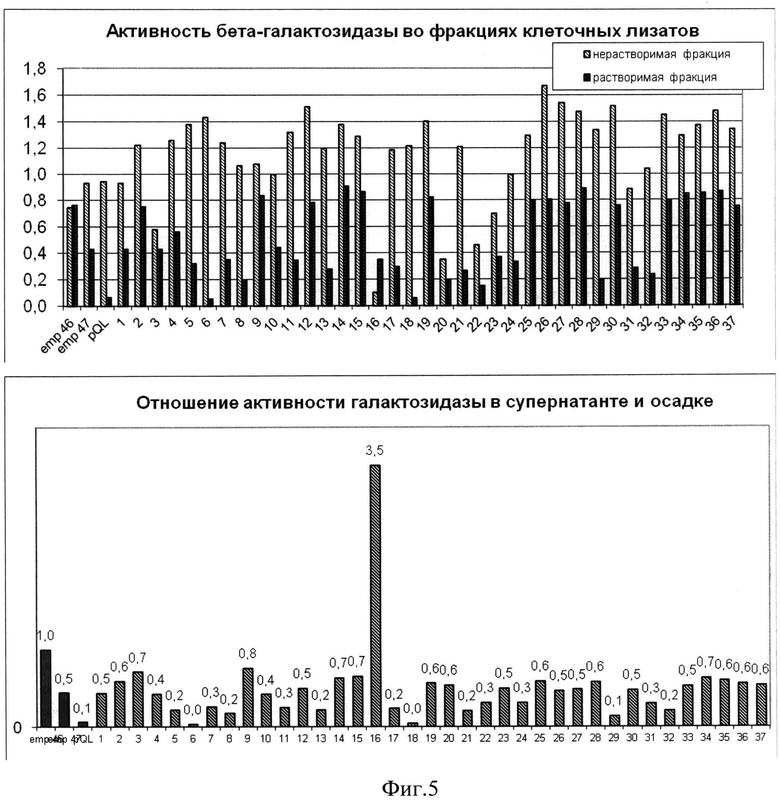
Для количественного определения активности βGal по 100 мкл грубого клеточного лизата разделяли на растворимую и нерастворимую фракцию, приведенные к эквивалентному объему (нерастворимую клеточную фракцию суспендировали в воде до объема 100 мкл). По 20 мкл растворимой и нерастворимой клеточной фракции от каждой полученной культуры смешивали с 50 мкл субстратной смеси, содержащей 0,02% X-gal, 25 мМ Трис-HCl, pH 8,5. В качестве отрицательного контроля использовали клеточный лизат штамма TGl(pQL30). Смесь инкубировали при 37°C в течение 2-4 ч, затем замеряли сигнал на спектрофотометре при длине волны λ=620 нм (фиг.5).
Оценку выхода целевого продукта осуществляли при помощи электрофоретического разделения белков по Лэммли с последующим вестерн-блоттингом на нитроцеллюлозной мембране и детекцией целевого белка с использованием сывороток крови людей, инфицированных гепатитом C. Для этого грубый клеточный лизат центрифугировали при 14000 G в течение 15 мин. Белки растворимой и нерастворимой клеточной фракции солюбилизировали в 30 мкл буфера Лэммли и разделяли в 15% ПААГ в денатурирующих условиях. После переноса белков на нитроцеллюлозную мембрану и обратимого окрашивания перенесенных белков Понсо C наблюдали полосу, соответствующую расчетной массе белка (~130кДа) у клона s3 в растворимой фракции (фиг.6).
Для блокировки неспецифического связывания инкубировали мембрану с перенесенными на нее белками в 1% растворе бычьего сывороточного альбумина, приготовленного на буфере PBS. Затем мембрану с белками инкубировали с пулом сывороток крови 7 пациентов, инфицированных вирусом гепатита С генотипа 1b (общее разведение пулированной сыворотки в буфере PBST составило 1/300), и далее с вторичными антивидовыми антителами к IgG человека, коньюгированными с пероксидазой хрена (в PBST-буфере, разведение антител 1/10000). Окрашивание с ДАБ (до 0,1 мг/мл) проводили в буфере PBS в присутствии 0,01% пероксида водорода. В результате у клона s3 из группы SVR наблюдали полосу, соответствующую расчетной массе белка (~130 кДа), проявляющую ярко выраженную реакцию с антителами пациентов, инфицированных ВГС, но не здоровых доноров (фиг.6).
Краткое описание графических изображений:
Фиг.1. Распределение количества колоний в зависимости от экспрессии гена LacZ: серые столбцы соответствуют колониям белого цвета, полученным на индикаторной среде (βGal-), а черные столбцы - синего цвета (βGal+). Заштрихованные столбцы соответствующие колониям, экспрессирующим активность βGal, и несущим вставку целевого размера (>100 п.н.) по результатам ПЦР.
Фиг.2. На диаграмме показано процентное отношение клонов βGal+ (черный цвет) и клонов βGal+ со вставкой целевого размера (заштрихованные) к общему числу клонов библиотеки.
Фиг.3. На графике показано распределение величины вставки целевого гена внутри банка по результатам ПНР со стандартными праймерами (pQE-for и pUC-for).
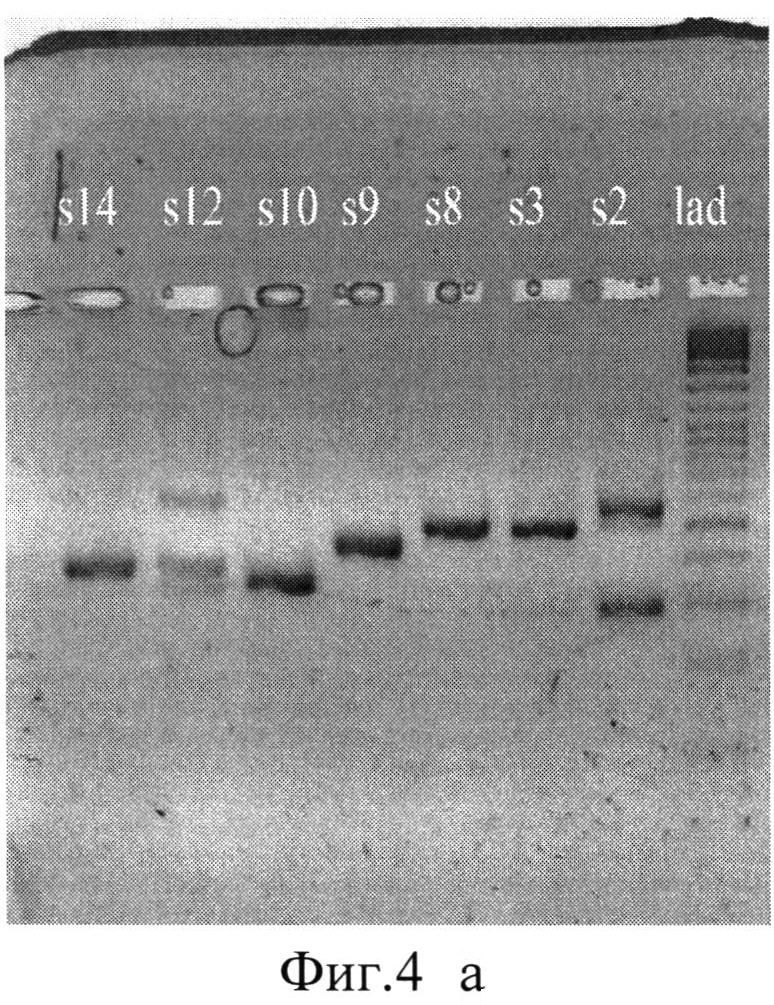
Фиг.4 (а)-(г). Результаты электрофореза в 1,8% агарозном геле очищенных ПНР-продуктов клонов со вставкой.
Фиг.5. Определение удельной активности βGal (нормированной на объем фракции) в растворимой (темные столбцы) и нерастворимой (светлые столбцы) клеточных фракциях. На оси ординат указана величина A620.
Фиг.6. Электрофоретический анализ (а-б) и вестерн-блоттинг (в) белков. Белки разделены в денатурирующем 15% ПААГ, с последующим окрашиванием Понсо C на общий белок, переносом на нитроцеллюлозный фильтр и детекцией целевого белка с использованием сыворотки крови людей, инфицированных гепатитом C.
Источники информации
1. US Patent 4521509, Benkovic, et al, 1985, Method for degrading DNA.
2. US Patent 6265159, Sugino, et al. 2001, Method for producing DNA nested deletions by an in vitro reaction using transposase.
3. J.M. Whitcomb et al. A new PCR based method for the generation of nested deletions, Nucleic Acids Research, 1993, Vol.21, No.17 4143-4146.






Настоящее изобретение относится к области биотехнологии. Предложен способ конструирования библиотек делеционных производных генов на основе ПЦР со случайной затравкой. В исследуемый ген, в форме линейной ДНК вносятся однонитевые разрывы путем обработки панкреатической ДНКазой I в серии разведений. Разрывы расширяют обработкой полимеразой I из Е.coli в отсутствие нуклеотидтрифосфатов. Затем присоединяют к матрице случайную затравку, которая имеет на 3'-конце 6-, 11- или 17-членную случайную последовательность, а на 5'-конце - константный участок (20 нуклеотидов), предназначенный для отжига адаптерного праймера. Далее осуществляют препаративную наработку библиотеки методом ПЦР с симметричным адаптерным праймером, который присоединяют к подготовленной матрице путем обработки T4 ДНК-лигазой, что позволяет эффективно удалять димеры праймеров, образовавшиеся за счет взаимного отжига случайных последовательностей. Полученные in vitro банки делеционых производных в дальнейшем могут быть подвергнуты скринингу с целью отбора вариантов, для дальнейшей их экспрессии in vivo. Способ позволяет получать укороченные варианты практически важных генов с улучшенными биотехнологическими характеристиками. Способ может быть использован для получения библиотек делеционных производных генов для последующего их использования в области биотехнологии, сельского хозяйства и пищевой, фармацевтической, медицине промышленности. 12 ил., 1 пр.
Способ конструирования библиотек делеционных производных генов на основе ПЦР со случайной затравкой, где в исследуемый ген в форме линейной ДНК вносят однонитевые разрывы, присоединяют к матрице случайную затравку, после чего проводят препаративную наработку библиотеки методом ПЦР с симметричным адаптерным праймером, отличающийся тем, что (1) однонитевые разрывы вносят путем обработки панкреатической ДНКазой I в серии разведений; (2) разрывы расширяют обработкой полимеразой I из Е.coli в отсутствие нуклеотидтрифосфатов; (3) затравка имеет на 3'-конце 6-, 11- или 17-членную случайную последовательность, а на 5'-конце - константный участок (20 нуклеотидов), предназначенный для отжига адаптерного праймера; (4) адаптерный праймер присоединяют к подготовленной матрице путем обработки T4 ДНК-лигазой, что позволяет эффективно удалять димеры праймеров, образовавшиеся за счет взаимного отжига случайных последовательностей.
| KING D.A | |||
| et al., “Domain structure and protein interactions of the silent information regulator Sir3 revealed by screening a nested deletion library of protein fragments” (2006) J | |||
| Biol | |||
| Chem., 281, 29, 20107-20119) | |||
| SHEN W | |||
| et al., “A novel method for generating a nested set of unidirectional deletion mutants using mixed |

