Область техники
Изобретение относится к области биотехнологии, а именно к технологии получения биологически активных веществ (БАВ) методами генной инженерии, точнее к методам получения дезоксирибонуклеазы I человека.
Предшествующий уровень техники.
Дезоксирибонуклеаза I (синонимы ДНКаза I, Панкреатическая дезоксирибонуклеаза, Deoxyribonuclease I, DNAse I, шифр международного Классификатора Ферментов ЕС 3.1.21.1) человека - природный внеклеточный фермент, вырабатываемый поджелудочной и слюнными железами. В максимальной концентрации он содержится в ЖКТ, где происходит переваривание ДНК, присутствующей в пище. Низкие концентрации ДНКазы I обнаруживаются в сыворотке здоровых людей. ДНКаза I человека - это гликопротеин, содержащий 260 аминокислот, с молекулярной массой 33000-38000 дальтон. Фермент гомологичен ДНКазе I быка, их аминокислотный состав совпадает на 77%. Пространственная структура ДНКазы быка была определена в работах [Suck, D., С. Oefner, et al. (1984). "Three-dimensional structure of bovine pancreatic DNAse I at 2.5 A resolution." EMBO J 3(10): 2423-2430; Suck, D. and C. Oefner (1986). "Structure of DNAse I at 2.0 A resolution suggests a mechanism for binding to and cutting DNA." Nature 321(6070): 620-625.].
Основным источником ДНКазы I для научных исследований и фармацевтики являлся крупный рогатый скот. Процесс выделения и хроматографической очистки ДНКазы I описан в работах [Funakoshi, A., Y. Tsubota, et al. (1980). "Simple purification and properties of bovine pancreatic deoxyribonuclease I." J Biochem 88(4): 1113-1118.; Paudel, H.K. and T.H. Liao (1986). "Comparison of the three primary structures of deoxyribonuclease isolated from bovine, ovine, and porcine pancreas. Derivation of the amino acid sequence of ovine DNase and revision of the previously published amino acid sequence of bovine DNase." J Biol Chem 261(34): 16012-16017.; Nefsky, В. and A. Bretscher (1989). "Preparation of immobilized monomeric actin and its use in the isolation of protease-free and ribonuclease-free pancreatic deoxyribonuclease I." Eur J Biochem 179(1): 215-219.].
Проблема использования поджелудочной железы быка как исходного сырья для выделения ДНКазы I состоит в том, что в этой ткани содержится сложная смесь протеаз, что затрудняет выделение, и, кроме того, существует вероятность контаминации препарата прионами и вирусами животных.
В исследованиях in vitro было установлено, что ДНКаза может снижать вязкость гнойных секретов легких [Armstrong, J.В. and J.С. White (1950). "Liquefaction of viscous purulent exudates by deoxyribonuclease." Lancet 2(6641): 739-742; Chemick, W.S., G.J. Barbero, et al. (1961). "In-vitro evaluation of effect of enzymes on tracheobronchial secretions from patients withcystic fibrosis." Pediatrics 27: 589-596].
Бычья панкреатическая ДНКаза I (дорназа) была разрешена к клиническому применению в США в 1958 г. как муколитическое средство ингаляционного и парентерального применения, однако из-за многочисленных побочных эффектов вышла из медицинского употребления. Вероятной причиной побочных действий была контаминация препарата другими пищеварительными панкреатическими ферментами быка (он содержал до 2% трипсина и химотрипсина) [Lieberman, J. (1962). "Enzymatic dissolution of pulmonary secretions. An in vitro study of sputum from patients with cystic fibrosis of pancreas." Am J Dis Child 104: 342-348].
Очевидной заменой лекарственных препаратов ДНКазы I быка является рекомбинантная высокоочищенная ДНКаза I человека.
ДНКаза I человека была выделена и частично очищена из поджелудочной железы [Funakoshi, A., Y. Tsubota, et al. (1977). "Purification and properties of human pancreatic deoxyribonuclease I." J Biochem 82(6): 1771-1777], дуоденального сока, сыворотки [Love, J.D. and R.R. Hewitt (1979). "The relationship between human serum and human pancreatic DNAse I." J Biol Chem 254(24): 12588-12594] и мочи [Murai, K., М. Yamanaka, et al. (1978). "Purification and properties of deoxyribonuclease from human urine." Biochim Biophys Acta 517(1): 186-194]. Было установлено, что N-концевой аминокислотой зрелого белка является лейцин [Ito, K., N. Minamiura, et al. (1984). "Human urine DNAse I: immunological identity with human pancreatic DNAse I, and enzymic and proteochemical properties of the enzyme." J Biochem 95(5): 1399-1406].
Ген человеческой ДНКазы был изолирован из библиотеки панкреатической кДНК с использованием олигонуклеотидных зондов. Был выделен клон полноразмерной кДНК, состоящий из 1039 пар оснований. Было выявлено, что он содержит одну длинную открытую рамку считывания, кодирующую полипептид из 260 аминокислот, высокогомологичный бычьей ДНКазе I. При экспрессии данной кДНК в клетках человека А-293 был получен активный фермент [Shak, S., D.J. Capon, et al. (1990). "Recombinant human DNAse I reduces the viscosity of cystic fibrosis sputum." Proc Natl Acad Sci USA 87(23): 9188-9192].
Промышленно пригодная система экспрессии ДНКазы I человека (рчДНКазы), описанная в патенте США 7297526, основана на использовании культивируемых клеток яичника китайского хомячка (СНО-DP7), содержащих множественные копии трансгена рчДНКазы под контролем раннего промотора цитомегаловируса. Данная система позволяет получать рчДНКазу в значительных количествах и используется при промышленном производстве лекарственного препарата рчДНКазы «Дорназа альфа», но в то же время, культивирование клеток СНО требует использования дорогостоящей культуральной среды и сложной системы очистки продукта, включающей стадии вирус-инактивации.
Потенциально более экономичными промышленно пригодными системами экспрессии гена рчДНКазы являются основанные на использовании дрожжей S. cerevisae или P. pastoris либо бактерий Е. coli. Экспрессия гена рчДНКазы в дрожжевых системах экспрессии приводит к появлению N-гликозилированной формы белка (патент США 7118901), экспрессия в Е. coli - к появлению негликозилированной формы. Удельная ферментативная активность рчДНКазы в обоих случаях одинакова. Поскольку структура N-связанных гликанов, продуцируемых дрожжами, значительно отличается от таковой для человека и N-гликаны дрожжей иммуногенны для млекопитающих, экспрессируемая в дрожжевых системах рчДНКаза непригодна для медицинского применения. Таким образом, среди всех систем экспрессии в микроорганизмах, система экспрессии рчДНКазы в клетках Е. coli лучше подходит для получения фармацевтически пригодного продукта.
Существует ряд работ по получению ферментативно активной ДНКазы I быка в бактериальной системе, но возможности повышенной экспрессии гена данного белка ограничены в связи с токсичностью продукта для клеток-продуцентов. В отличие от эукариот, у которых процессы транскрипции и трансляции проходят в разных клеточных компартментах, в клетке Е. coli эти процессы сопряжены. Наличие активной ДНКазы в цитоплазме приводит к деградации геномной ДНК и последующему лизису бактериальной клетки, поэтому повышенная экспрессия ДНКазы неизбежно приводит к генетической нестабильности клона, т.е. отбор суперпродуцентов одновременно является отбором наиболее генетически нестабильных бактерий. Так, в работе [Worrall, A.F. and В.A. Cormolly (1990). "The chemical synthesis of a gene coding for bovine pancreatic DNAse I and its cloning and expression in Escherichia coli" J Biol Chem 265(35): 21889-21895] экспрессировали активную ДНКазу быка в системе E. coli, используя синтетический ген с оптимизированными кодонами и промотор поздних генов фага лямбда pL. Показано, что продукт экспрессии обладал токсичностью для клеток штамма-продуцента, поэтому выход целевого белка лежал в пределах от 100 мкг до 1 мг/л культуры. Активность экспрессированного фермента была сопоставима с активностью нативного фермента быка. Однако значения активности, приведенные авторами (5×108 Ед/г белка), были измерены для не полностью очищенного препарата. В работе [Chen, С.Y., S. С.Lu, et al. (1998). "Cloning, sequencing and expression of a cDNA encoding bovine pancreatic deoxyribonuclease I in Escherichia coli: purification and characterization of the recombinant enzyme." Gene 206(2): 181-184] экспрессировали природную кДНК быка в системе E. coli штамм BL21(DE3)pLysE. Однако уровень экспрессии оставался низким, так как продукт оказался токсичным для бактерий. Клетки лизировались после индукции, фермент обнаруживался как в культуральной среде, так и в клетках. По данным авторов, индуцированная культура давала 3500 Ед/л. Активность выделенного фермента составляла 908 Ед/мг, что сопоставимо с контрольным измерением тем же методом активности природного фермента, выделенного авторами параллельно из тканей быка (938 Ед/мг). Таким образом, расчетная продуктивность данного метода составляет около 4 мг/л культуры.
Несколько больший уровень экспрессии дезоксирибонуклеазы I быка был достигнут при помощи трансформации штамма JM109 клеток E. coli плазмидой pHEL12, несущей ген ДНКазы I под контролем промотора Т7 и последующей инфекции культуры клеток бактериофагом М13/Т7 при одновременной индукции экспрессии целевого гена при помощи ИПТГ [Linardou, H., A.A. Epenetos, et al. (2000). "A recombinant cytotoxic chimera based on mammalian deoxyribonuclease-I." Int J Cancer 86(4): 561-569]. Существенным ограничением данного метода является необходимость инфицирования рабочей культуры бактериофагом, поскольку при промышленной культивации очистка оборудования от бактериофага практически невозможна.
Подробное описание настоящего изобретения
Технической задачей, решаемой авторами, являлось создание технологии получения ферментативно активной рекомбинантной ДНКазы I человека с более высоким выходом.
Технический результат достигался путем создания технологии, включающей в себя новую экспрессионную плазмидную ДНК, кодирующую оптимизированный для экспрессии в бактериальной системе ген ДНКазы I человека, создание штамма продуцента E. coli на ее основе и технологии выделения и модификации ДНКазы I.
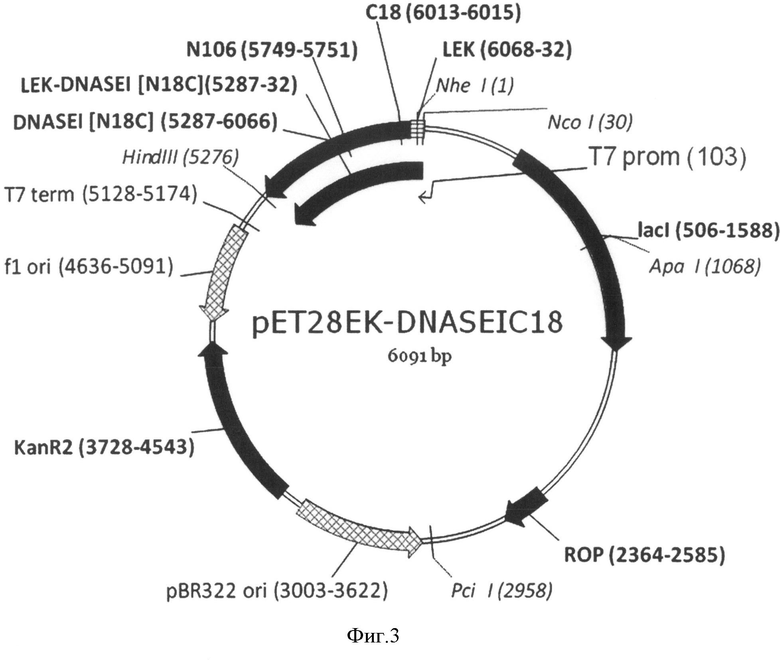
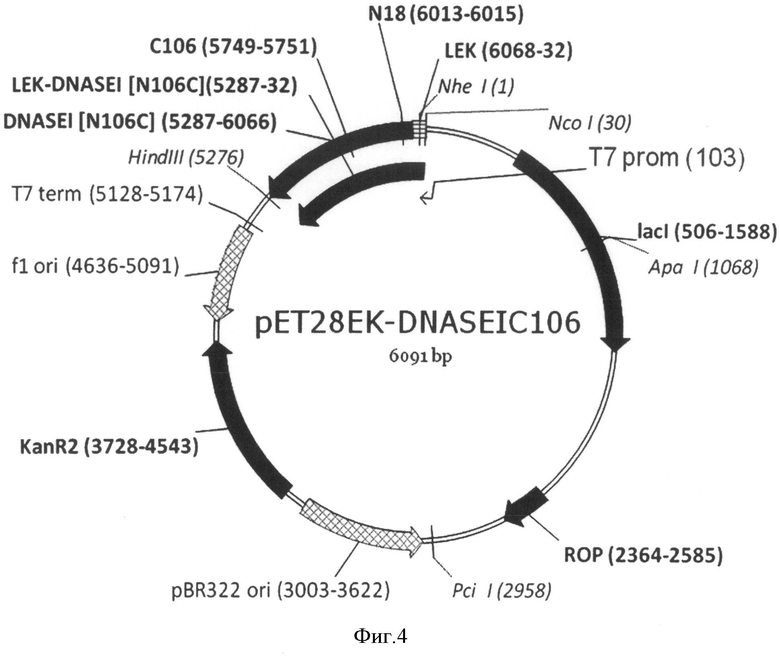
В основе данного решения лежат разработанные авторами экспрессионные плазмиды pET28EK-DNASEI, pET28EK-DNASEIC18 и pET28EK-DNASEIC106, длиной 6091 п.о., содержащие фрагмент ДНК, включающий последовательность, кодирующую синтетический отщепляемый лидерный N-концевой пептид длиной 19 аминокислот, включающий гексагистидиновый кластер и последовательность узнавания энтерокиназы, и слитую с ним в рамке последовательность, кодирующую ДНКазу I человека, либо ее мутеины [N18C] или [N106C], соответственно. Указанный фрагмент содержит оптимальные для E. coli кодоны, позволяющие увеличить уровень экспрессии гетерологичного белка за счет эффективной трансляции всех аминокислот полипептида. Наличие лидерного пептида подавляет ферментативную активность ДНКазы I, что снижает токсичность продукта экспрессии для бактерии, что приводит к значительному увеличению выхода целевого белка.
Целью настоящего изобретения является предоставление экспрессионной плазмиды, содержащей фрагмент ДНК, кодирующий предшественник рекомбинантной ДНКазы I человека или ее мутеина, включающий последовательность, кодирующую отщепляемый N-концевой лидер, включающий гексагистидиновый кластер и последовательность узнавания энтерокиназы, слитую в рамке с последовательностью, кодирующей ДНКазу I человека, под контролем промотора, функционирующего в бактериальной клетке.
Также целью настоящего изобретения является предоставление описанной выше экспрессионной плазмиды, где указанная плазмида выбрана из группы, состоящей из плазмид pET28EK-DNASEI, pET28EK-DNASEIC18 и pET28EK-DNASEIC106.
Также целью настоящего изобретения является предоставление бактерии, принадлежащей к роду Escherichia, трансформированной описанной выше плазмидой, - продуцента предшественника рекомбинантной ДНКазы I человека или ее мутеина.
Также целью настоящего изобретения является предоставление описанной выше бактерии, где указанная бактерия представлена штаммами E. coli BL21[DE3]/pET28EK-DNASEI, E. coli BL21[DE3]/pET28EK-DNASEI18 и E. coli BL21[DE3]/pET28EK-DNASEI106.
Также целью настоящего изобретения является предоставление предшественника рекомбинантной ДНКазы I человека или ее мутеина, содержащего отщепляемый N-концевой лидер, включающий гексагистидиновый кластер, и последовательность узнавания энтерокиназы.
Также целью настоящего изобретения является предоставление описанного выше мутеина ДНКазы I человека, при этом мутеин содержит точечную замену N18C или N106C.
Также целью настоящего изобретения является предоставление способа получения рекомбинантной ДНКазы I человека или ее мутеина, включающий культивирование описанной выше бактерии в питательной среде, выделение телец включения, солюбилизацию белка-предшественника, металлохелатную хроматографию в денатурирующих условиях, рефолдинг белка-предшественника, получение зрелого белка обработкой энтерокиназой и выделение зрелого белка.
Также целью настоящего изобретения является предоставление описанного выше способа, в котором культивируют штамм E. coli BL21[DE3]/pET28EK-DNASEI, E. coli BL21[DE3]/pET28EK-DNASEI18 или E. coli BL21[DE3]/pET28EK-DNASEI106.
Также целью настоящего изобретения является предоставление описанного способа, в котором мутеин содержит точечную замену N18C или N106C.
Также целью настоящего изобретения является предоставление способа получения конъюгатов полиэтиленгликоля и рекомбинантной дезоксирибонуклазы I человека или ее мутеина, включающего предварительное ограниченное восстановление белков, инкубацию с малеимид-полиэтиленгликолем и выделение полученных конъюгатов.
Также целью настоящего изобретения является предоставление описанного способа, в котором мутеин содержит точечную замену N18C или N106C.
Также целью настоящего изобретения является предоставление конъюгата полиэтиленгликоля и рекомбинантной дезоксирибонуклазы I человека или ее мутеина, полученного описанным выше способом.
Подробное описание настоящего изобретения
Для реализации настоящего изобретения главной технической задачей явилось создание способа получения рекомбинантной ДНКазы I человека или ее мутеина, а также их конъюгатов с полиэтиленгликолем, с использованием бактерии, трансформированной экспрессионной плазмидой, содержащей фрагмент ДНК, кодирующий предшественник рекомбинантной ДНКазы I человека или ее мутеина, включающий последовательность, кодирующую отщепляемый N-концевой лидер, включающий гексагистидиновый кластер и последовательность узнавания энтерокиназы, и слитую с ним в рамке последовательность, кодирующую ДНКазу I человека, под контролем промотора, функционирующего в бактериальной клетке.
Термин «экспрессионная плазмида» означает плазмидную ДНК, содержащую все необходимые генетические элементы для экспрессии внедренного в него гена, например, такие как промотор, терминатор. Конкретным примером генетических элементов, необходимых для экспрессии предшественника рекомбинантной ДНКазы I человека в составе экспрессионной кассеты, согласно настоящему изобретению, является, но не ограничивается им, промотор РНК-полимеразы бактериофага Т7.
Фрагментом ДНК, кодирующим предшественник рекомбинантной ДНКазы I человека, согласно настоящему изобретению, является, например, синтетический ген, кодирующий предшественник рекомбинантной ДНКазы I человека или ее мутеина, включающий последовательность, кодирующую отщепляемый N-концевой лидер, включающий гексагистидиновый кластер, и последовательность узнавания энтерокиназы, слитую в рамке с последовательностью, кодирующей ДНКазу I человека. Указанный фрагмент ДНК может быть получен методом ПЦР (см. Пример 1, Фиг.1). Также указанный фрагмент ДНК может быть получен с использованием технологии клонирования фирмы Sloning BioTechnology, описанной в заявке РСТ WO 2005071077.
Чтобы обеспечить эффективную трансляцию клонированного гена в E. coli, предпочтительно, чтобы в последовательности, кодирующей предшественник ДНКазы I человека, все редкие кодоны были заменены синонимичными часто встречающимися кодонами и часто встречающиеся кодоны были распределены в последовательности равномерно, в соответствии с частотностью кодонов экспрессирующихся генов E. coli.
Последовательность гена, кодирующего предшественник рекомбинантной ДНКазы I человека, согласно настоящему изобретению, представлена в Перечне последовательностей под номером SEQ ID NO:1. Аминокислотная последовательность предшественника рекомбинантной ДНКазы I человека согласно настоящему изобретению представлена в Перечне последовательностей под номером SEQ ID NO:2. Аминокислотная последовательность зрелой рекомбинантной ДНКазы I человека представляет собой последовательностей под номером SEQ ID NO:2 без 19 первых аминокислот.
Фрагменты ДНК, которые кодируют по существу тот же белок, могут быть получены, например, путем модификации нуклеотидной последовательности фрагмента ДНК (SEQ ID NO:1), кодирующего предшественник рекомбинантной ДНКазы I человека, например, посредством метода сайт-направленного мутагенеза, так, что один или несколько аминокислотных остатков в определенном сайте будут делегированы, заменены, вставлены или добавлены. Фрагменты ДНК, модифицированные, как описано выше, могут быть получены с помощью традиционных методов обработки с целью получения мутации. Фрагменты ДНК, которые кодируют по существу тот же белок, могут быть получены путем экспрессии фрагментов ДНК, имеющих мутацию, описанную выше, в соответствующей клетке.
ДНКаза I (панкреатическая дезоксирибонуклеаза I) представляет собой эндонуклеазу, гидролизующую как одно-, так и двухцепочечную ДНК с образованием сложной смеси моно- и олигонуклеотидов, содержащих 5'-фосфатные группы.
Термин «мутеин» означает мутантный белок или белок, кодируемый мутантным геном. Предпочтительным мутеином ДНКазы I согласно настоящему изобретению является мутантная ДНКаза I, содержащая замену одной или нескольких аминокислот на цистеин. Наличие дополнительного остатка цистеина в мутеиновых вариантах ДНКазы I человека позволяет производить их направленную модификацию полиэтиленгликолем с получением конъюгатов в ферментативно высокоактивной форме.
Показатели функциональной активности, при которой считается, что полученный белок обладает свойствами ДНКазы I человека, определяются по его способности гидролизовать как одно-, так и двухцепочечные фрагменты ДНК. Так, например, активность ДНКазы I человека можно детектировать методом зимографии как описано в Примере 10. Считается, что вариант белка обладает свойствами предшественника ДНКазы I человека при условии, что активность указанного варианта составляет не ниже 1% активности нативной ДНКазы I человека.
Экспрессионная плазмида согласно настоящему изобретению содержит фрагмент ДНК, кодирующий предшественник рекомбинантной ДНКазы I человека или ее мутеина, включающий последовательность, кодирующую отщепляемый N-концевой лидер, включающий гексагистидиновый кластер, и последовательность узнавания энтерокиназы, слитую в рамке с последовательностью, кодирующей ДНКазу I человека, под контролем промотора, функционирующего в бактериальной клетке.
В качестве рекомбинантной плазмиды согласно настоящему изобретению могут использоваться различные плазмиды, обладающие способностью к экспрессии в клетке-реципиенте, такие как плазмиды pBR322, pMW119, pUC19, pET22b, pET28b и подобные им, но список плазмид не ограничивается ими.
Конкретным вариантом реализации настоящего изобретения являются плазмиды, которые состоят из:
1) фрагмента NheI-NcoI длиной 29 п.о., представляющего собой синтетический адаптер, кодирующий гексагистидиновый кластер;
2) фрагмента NcoI-HindIII вектора рЕТ28а(+) длиной 5246 п.о., содержащего область начала репликации плазмиды pBR322, ген РНК-организующего белка Rop, участок инициации репликации бактериофага f1, последовательность, кодирующую аминогликозид-3'-фосфотрансферазу, промотор РНК-полимеразы бактериофага Т7; участок терминации транскрипции; последовательность, кодирующую репрессор лактозного оперона;
3) фрагмента NheI-HindIII длиной 816 п.о., кодирующего ДНКазу I человека либо ее мутеины [N18C] или [N106C] и слитую в рамке последовательность узнавания энтерокиназой.
Указанные плазмиды содержат уникальные сайты узнавания эндонуклеазами рестрикции: NheI (1), ApaI (1068), PciI (2958), HindIII (5276).
Структуры соответствующих плазмид pET28EK-DNASEI, pET28EK-DNASEIC18, pET28EK-DNASEIC106 приведены на Фиг.2, 3 и 4.
При помощи созданных плазмид можно трансформировать бактериальную клетку, предпочтительно бактерию, принадлежащую к роду Escherichia, восприимчивую к подобной трансформации указанной плазмидой. Выбор конкретной клетки не является критическим, поскольку методология и приемы трансформации хорошо известны специалисту в данной области техники. И хотя в зависимости от вида клетки и условий культивирования полученного трансформанта уровень экспрессии предшественника ДНКазы I человека может варьироваться, факт экспрессии целевого белка будет иметь место при условии успешной трансформации клетки-реципиента.
«Трансформация клетки плазмидой» означает введение плазмиды в клетку с помощью методов, хорошо известных специалисту в данной области техники. Трансформация этой плазмидой приводит к экспрессии гена, кодирующего белок согласно настоящему изобретению, и к синтезу белка в бактериальной клетке. Методы трансформации включают любые стандартные методы, известные специалисту в данной области техники, например метод, описанный в Jac A. Nickoloff, Electroporation Protocols for Microorganisms (Methods in Molecular Biology) // Humana Press; 1st edition (August 15, 1995).
Согласно настоящему изобретению, «бактериальная клетка-продуцент предшественника ДНКазы I человека» означает бактериальную клетку, обладающую способностью к продукции и накоплению предшественника ДНКазы I человека согласно настоящему изобретению, когда бактериальная клетка согласно настоящему изобретению выращивается в указанной питательной среде. Используемый здесь термин «бактериальная клетка-продуцент предшественника ДНКазы I человека» также означает клетку, которая способна накапливать продуцент предшественника ДНКазы I человека в количестве не менее чем 1 мг/л, более предпочтительно, не менее чем 30 мг/л. Указанный предшественник ДНКазы I человека накапливается в указанной клетке предпочтительно в виде телец включения.
Предпочтительно использование бактерии, принадлежащей к роду Escherichia, для трансформации рекомбинантной плазмидой, содержащей фрагмент ДНК, кодирующий предшественник ДНКазы I человека.
Термин "бактерия, принадлежащая к роду Escherichia" может означать, что бактерия относится к роду Escherichia в соответствии с классификацией, известной специалисту в области микробиологии. В качестве примера микроорганизма, принадлежащего к роду Escherichia, может быть упомянута бактерия Escherichia coli (E. coli).
Круг бактерий, принадлежащих к роду Escherichia, не ограничен каким-либо образом, однако, например, бактерии, описанные в книге Neidhardt, F.C. et al. (Escherichia coli and Salmonella typhimurium, American Society for Microbiology, Washington D.C., 1208, Таблица 1), могут быть приведены в качестве примеров.
Конкретным примером штамма-реципиента для получения продуцента предшественника ДНКазы I человека согласно настоящему изобретению является, но не ограничиваются им, штамм Escherichia coli BL21[DE3].
Штамм Escherichia coli BL21[DE3] характеризуется следующими культурально-морфологическими, физиолого-биохимическими признаками и генетическими признаками.
Культурально-морфологические особенности штамма: грамотрицательные палочки образуют нити; на агаризованной среде - беловатые крупные колонии с неровным краем. Активность штамма определяется методом денситометрии электрофореграммы. Штамм хранится в следующих условиях: среда Лурье-Бертрана, 1% глюкозы, 10% глицерина. Штамм размножается в следующих условиях - среда Лурье-Бертрана, 1% глюкозы, канамицина сульфат 30 мкг/мл.
Генетические особенности штамма. Генотип штамма - F- ompT gal dcm lon hsdSB(rB - mB -)λ(DE3 [lacI lacUV5-T7 gene 1 ind1 sam7 nin5]).
Трансформация штамма Escherichia coli BL21[DE3] плазмидами рЕТ28ЕК-DNASEI, pET28EK-DNASEIC18, pET28EK-DNASEIC106 приводит к получению штаммов-продуцентов BL21[DE3]/pET28EK-DNASEI, BL21[DE3]/pET28EK-DNASEI18C, BL21[DE3]/pET28EK-DNASEI106C, соответственно, которые обеспечивали синтез рекомбинантного белка-предшественника ДНКазы I человека в количестве 15-40% от суммарного содержания белка клеток.
Штамм Escherichia coli BL21[DE3]/pET28EK-DNASEI кодирует гибридный белок предшественник LEK-DNASEI, состоящий из аминокислотной последовательности ДНКазы I человека и слитого в рамке N-концевого лидерного пептида длиной 19 аминокислот, содержащего шестигистидиновый кластер и сайт расщепления энтерокиназой, находящийся непосредственно перед первой аминокислотой ДНКазы I человека.
Штамм Escherichia coli BL21[DE3]/pET28EK-DNASEI18C кодирует гибридный белок предшественник LEK-DNASEI[N18C], состоящий из аминокислотной последовательности мутеина ДНКазы I человека [N18C] и слитого в рамке N-концевого лидерного пептида длиной 19 аминокислот, содержащего шестигистидиновый кластер и сайт расщепления энтерокиназой, находящийся непосредственно перед первой аминокислотой мутеина ДНКазы I человека.
Штамм Escherichia coli BL21[DE3]/pET28EK-DNASEI106C кодирует гибридный белок предшественник LEK-DNASEI[N106C], состоящий из аминокислотной последовательности мутеина ДНКазы I человека [N106C] и слитого в рамке N-концевого лидерного пептида длиной 19 аминокислот, содержащего шестигистидиновый кластер и сайт расщепления энтерокиназой, находящийся непосредственно перед первой аминокислотой мутеина ДНКазы I человека.
Способ получения ДНКазы I человека или ее мутеина согласно настоящему изобретению включает культивирование описанной выше бактерии в питательной среде, подходящей для выращивания указанных прокариотических клеток, выделение телец включения, солюбилизацию белка-предшественника, металлохелатную хроматографию в денатурирующих условиях, рефолдинг белка-предшественника, получение зрелого белка обработкой энтерокиназой и выделение указанного зрелого белка рекомбинантной ДНКазы I человека или ее мутеина.
Ковалентная модификация путем присоединения к белковой молекуле полимеров определенной структуры позволяет создавать препараты с пролонгированным действием, то есть преодолеть ряд недостатков препаратов на основе белков, к которым относятся: недостаточная стабильность (in vitro и in vivo), неустойчивость к действию протеаз (особенно пептидов), короткий период полураспада/полувыведения при введении в организм (минуты), иммуногенность и антигенность, наличие выраженных побочных эффектов при длительном клиническом применении [Abuchowski, A.; McCoy, J.R.; Palczuk, N.С.; van Es, Т.; Davis, F.F. (1977), "Effect of covalent attachment of polyethylene glycol on immunogenicity and circulating life of bovine liver catalase". Journal of Biological Chemistry 252 (II): 3582-3586]. Изменение фармакодинамики препарата методом ПЭГилирования используется при производстве ряда разрешенных к клиническому применению препаратов, например, гранулоцитарный колоний стимулирующего фактора (ГКСФ), интерферона бета и др.
Поэтому в рамки настоящего изобретения также включается способ получения конъюгатов полиэтиленгликоля и рекомбинантной ДНКазы I человека или ее мутеина согласно настоящему изобретению, включающий предварительное ограниченное восстановление ДНКазы I человека или ее мутеина согласно настоящему изобретению, инкубацию их с малеимид-полиэтиленгликолем и выделение полученных конъюгатов.
Также в рамки настоящего изобретения включаются конъюгат полиэтиленгликоля и рекомбинантной ДНКазы I человека или ее мутеина, полученный описанным выше способом.
Использование указанного выше способа позволяет проводить синтез и продукцию конъюгатов мутеинов рекомбинантной ДНКазы I человека с полиэтиленгликолем согласно настоящему изобретению с активностью не менее 10% активности нативной ДНКазы I человека или быка.
Особенности плазмид и результаты их практического применения приведены на следующих Фигурах.
Краткое описание Фигур:
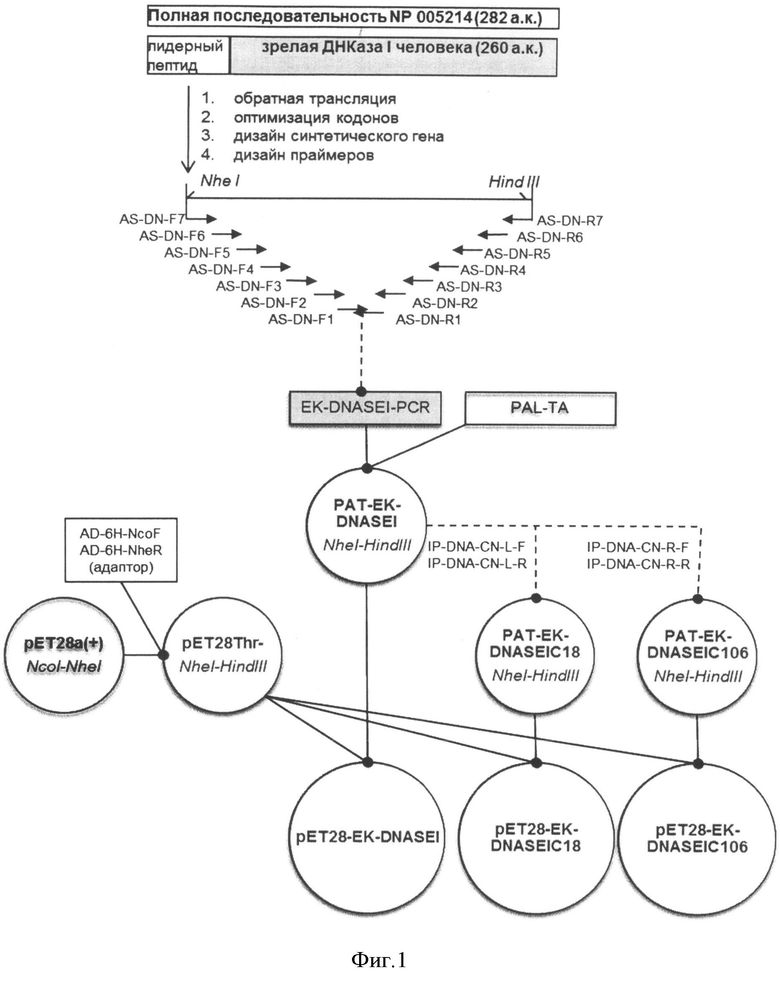
На Фигуре 1 показана схема сборки синтетического гена ДНКазы I человека из олигонуклеотидных праймеров и схема получения экспрессионных плазмид рЕТ28ЕК-DNASEI, pET28EK-DNASEIC18, pET28EK-DNASEIC106.
На Фигуре 2 показана карта экспрессионной плазмиды pET28EK-DNASEI. Используются следующие обозначения: "pBR322ori" область начала репликации плазмиды pBR322; "ROP" - ген РНК-организующего белка Rop; "f1 ori" - участок инициации репликации бактериофага f1; "KanR2" - последовательность, кодирующая аминогликозид-3'-фосфотрансферазу, обеспечивающую устойчивость бактерий к канамицину; "Т7 prom" - промотор РНК-полимеразы бактериофага Т7; "T7term" - участок терминации транскрипции; "lacI" - последовательность, кодирующая репрессор лактозного оперона; "LEK-DNASEI" - открытая рамка считывания (ОРС) полипептида белка-предшественника ДНКазы I человека, включающего отделяемый N-концевой дополнительный пептид "LEK" и зрелую ДНКазу I человека "DNASEI". Стрелками указаны направления транскрипции генов. Курсивом выделены сайты узнавания эндонуклеаз рестрикции, в скобках указаны номера нуклеотидов в точках разрезания. "N18", "N106" - триплеты нуклеотидов, соответствующих сайтам N-гликозилирования природной ДНКазы I человека.
На Фигуре 3 показана карта экспрессионной плазмиды pET28EK-DNASEI18C. Используются обозначения аналогично Фигуре 2, а также: "LEK-DNASEI[N18C]" - открытая рамка считывания (ОРС) полипептида белка-предшественника мутеина ДНКазы I человека [N18C].
На Фигуре 4 показана карта экспрессионной плазмиды pET28EK-DNASEI106C. Используются обозначения аналогично Фигуре 2, а также: "LEK-DNASEI[N106C]" - открытая рамка считывания (ОРС) полипептида белка-предшественника мутеина ДНКазы I человека [N106C].
На Фигуре 5 показана электрофореграмма тотального белка клеток штамма-продуцента BL21[DE3]/pET28EK-DNASEI при индукции. ДСН-ПААГ анализ для культур штамма-продуцента, полученных из трех случайно отобранных колоний. Обозначения "-" и "+" - образцы до индукции и через 2 ч после индукции культур 1 мМ ИПТГ. "М" - маркер молекулярных масс. Молекулярные массы полос маркера указаны в кДа. Положение целевого белка указано стрелкой.
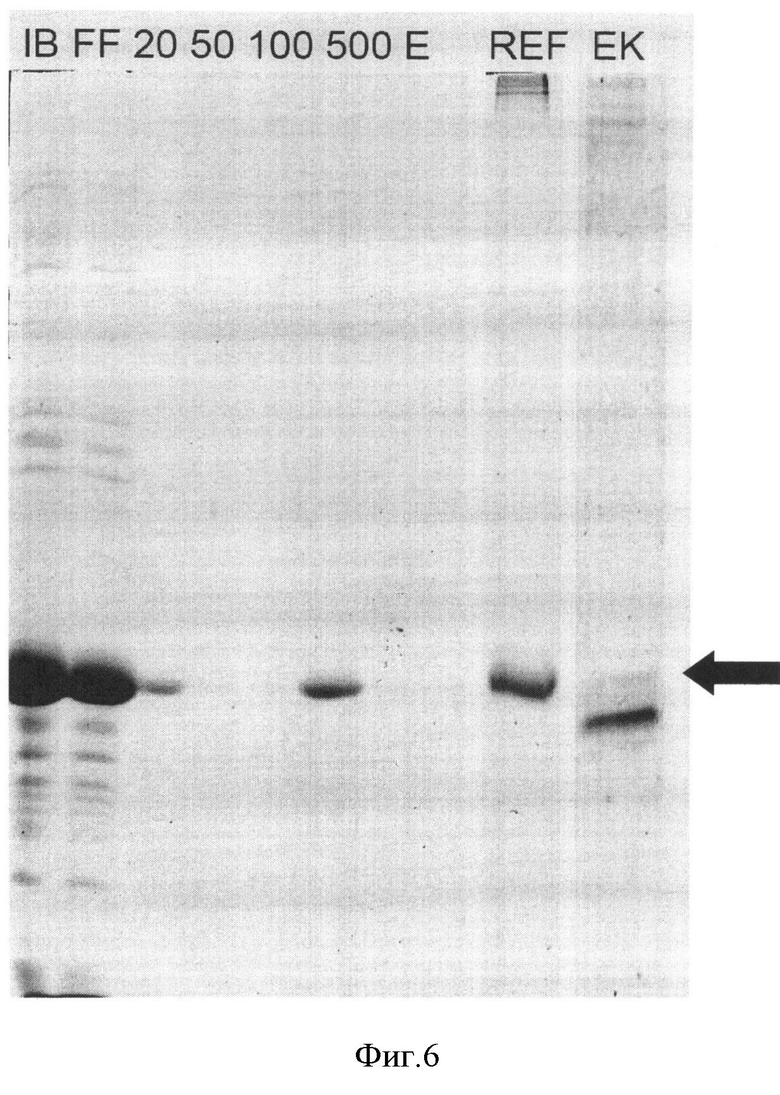
На Фигуре 6 приведены электрофореграммы белковых фракций при очистке, ренатурации и процессинге белка-предшественника рекомбинантной ДНКазы I человека. Обозначения: "IB" - солюбилизированные тельца включения, "FF" - фракция проскока через металлохелатную колонку, "20"-"500" - фракции ступенчатой элюции возрастающими концентрациями имидазола, в мМ; "Е" - фракция элюата при промывке колонки ЭДТА, "REF" - растворимый белок после рефолдинга, "ЕК" - реакционная смесь после 2 ч расщепления энтерокиназой. Дорожки "REF" и "EK" - электрофорез в невосстанавливающих условиях, остальные дорожки - в восстанавливающих. Положение белка-предшественника указано стрелкой.
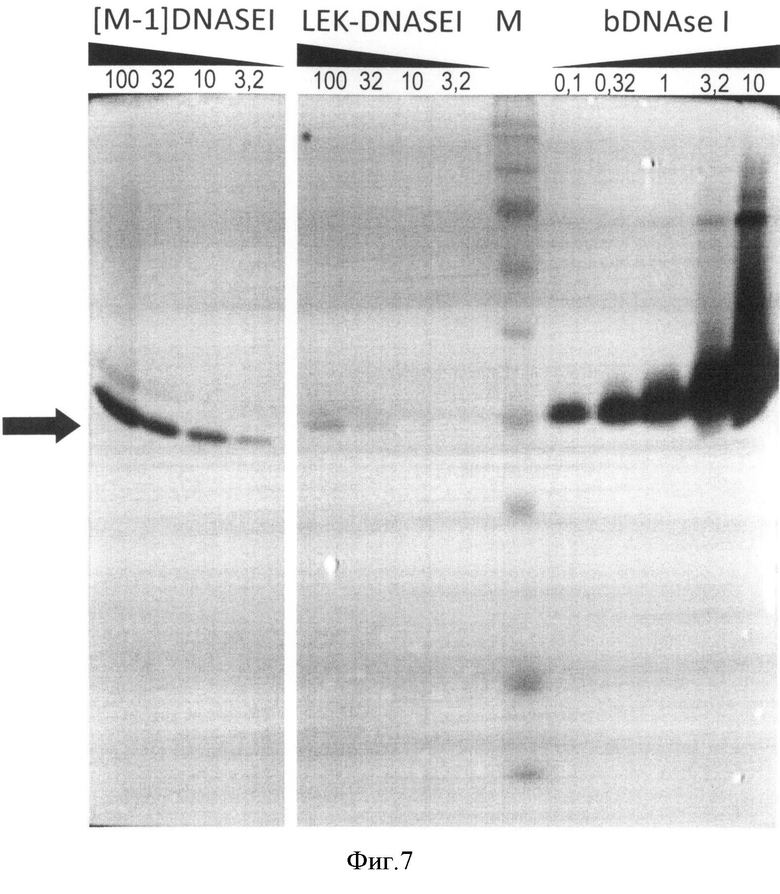
На Фигуре 7 приведена зимограмма белка-предшественника ДНКазы I. Окраска бромистым этидием. Расположение дорожек: "[M-1]DNASEI" - рекомбинантная ДНКаза I человека с дополнительным N-концевым остатком метионина, "LEK-DNASEI" - белок-предшественник ДНКазы I человека, "bDNAse I" - природная ДНКаза I быка, "М" - предокрашенный маркер молекулярных масс. Количество нанесенных на соответствующие дорожки белков указано в нг, положение белка-предшественника ДНКазы I человека указано стрелкой. Ферментативная активность пропорциональна размеру темной зоны на светлом фоне - участку деградации иммобилизованной в полиакриламидном геле двуцепочечной ДНК.
Настоящее изобретение будет более подробно описано ниже со ссылкой на следующие не ограничивающие настоящее изобретение Примеры.
Пример 1. Получение плазмидной ДНК PAT-EK-DNASEI, кодирующей предшественник ДНКазы I человека с отделяемым N-концевым пептидом
Для известной аминокислотной последовательности ДНКазы I человека с добавленным N-концевым лидерным пептидом (SEQ ID NO:3), содержащим последовательность узнавания энтерокиназой, была проведена обратная трансляция в последовательность нуклеотидов ДНК. При этом были использованы кодоны, оптимальные для экспрессии этого гена в E. coli класса В, а также была проведена оптимизация структуры гена по вторичной структуре мРНК, GC составу, обеспечено отсутствие нежелательных регуляторных элементов (например, отсутствие внутренних сайтов связывания рибосом), а также отсутствие протяженных повторов, палиндромов.
Индекс CAI (Codon Adaptation Index), отражающий эффективность экспрессии гена в данном организме, для полученной последовательности составил 0,8, что является хорошим прогностическим показателем для промышленной пригодности полученного на его основе штамма-продуцента. Полученная нуклеотидная последовательность приведена в SEQ ID NO:4.
Синтетический ген, кодирующий полипептид ДНКазы I человека, получали методом полимеразной цепной реакции с использованием синтетических олигонуклеотидов, приведенных в таблице 1, на приборе Терцик МС2 («ДНК-Технология», Россия).
Готовили инкубационную смесь следующего состава: 1х буфер для термостабильной ДНК-полимеразы; 10 пМ каждого праймера; по 2 мМ каждого дезоксирибонуклеотидтрифосфата; 1-2 ед. термостабильной ДНК-полимеразы в объеме 50 мкл. Поверх этой смеси наслаивали 50 мкл легкого минерального масла и вели амплификацию по схеме: 1 цикл - денатурация - 94°C, 3 мин; 25 циклов - денатурация 94°C, 30 сек, отжиг 55°C, 30 сек, достройка 72°C, 45-60 сек; 1 цикл - достройка 72°C, 7 мин.
Продукт ПЦР выделяли из 1% агарозного геля с использованием набора "Wizard SV Gel and PCR Clean-Up System" («Promega», США) по протоколу производителя и лигировали в векторную плазмиду PAL-TA (ЗАО «Евроген», Россия) с использованием ДНК-лигазы фага Т4 и стандартного буферного раствора (Fermentas, Литва). Полученными лигазными смесями трансформировали клетки E. coli штамма DH5α, с генотипом F- φ80lacZΔM15 Δ(lacZYA-argF) U169 recA1 endA1 hsdR17 (rk-, mk+) phoA supE44 λ-thi-1 gyrA96 relA1. Для этого к 200 мкл замороженной суспензии клеток E. coli добавляли 5 мкл лигазной смеси, инкубировали на льду 30 мин, нагревали до 42 С на 45 секунд и инкубировали на льду 5 минут, затем добавляли 800 мкл питательного бульона SOB, инкубировали при 37°C 60 минут, затем переносили суспензию на чашку Петри с твердой агаризованной средой, содержащей ампициллин в концентрации 100 мкг/мл агара и помещали в термостат на 37°С 18 часов. Колонии E. coli, отобранные в результате бело-голубого скрининга, анализировали методом ПЦР с клонов с использованием праймеров к последовательностям реципиентной плазмиды M13rev (SEQ ID NO:19) и M13dir (SEQ ID NO:20). 5 отобранных клонов наращивали в 5 мл питательного бульона 2xYT-Amp и выделяли плазмидную ДНК при помощи набора GeneJET Plasmid Miniprep Kit (Fermentas, Литва) по протоколу производителя. Для полученных плазмид определяли нуклеотидную последовательность области вставки методом ПЦР-секвенирования с использованием стандартных праймеров T7prom (SEQ ID NO:21) и SP6 (SEQ ID NO:22) к последовательности вектора.
Пример 2. Получение плазмид PAT-EK-DNASEIC18 и PAT-EK-DNASEIC106, кодирующих мутеины N18C и N106C ДНКазыI человека с отделяемым N-концевым лидером
Для получения плазмиды PAT-EK-DNASEIC18 проводили сайт-направленный мутагенез плазмиды PAT-EK-DNASEI методом инвертированной ПЦР с использованием праймеров IP-DNA-CN-L-F (SEQ ID NO:23) и IP-DNA-CN-L-R (SEQ ID NO:24).
Для получения плазмиды PAT-EK-DNASEIC106 проводили сайт-направленный мутагенез плазмиды PAT-EK-DNASEI методом инвертированной ПЦР с использованием праймеров IP-DNA-CN-R-F (SEQ ID NO:25) и IP-DNA-CN-R-R (SEQ ID NO:26).
Сайт-направленный мутагенез методом инвертированной ПЦР проводили по [Michael P. Weiner, Tim Gackstetter, Gina L. Costa, John C. Bauer, and Keith A. Kretz. Site-directed Mutagenesis using PCR in Molecular Biology: Current Innovations and Future Trends. Eds. A.M. Griffin and H.G. Griffin. ISBN 1-898486-01-8, 1995. Horizon Scientific Press, PO Box 1, Wymondham, Norfolk, U.K.] с модификациями. Каждый олигонуклеотид фосфорилировали отдельно. Реакцию проводили в буфере трис-HCl, pH 7,5, содержащем 10 мМ MgCl2, 50 мМ дитиотреитола, 1 мМ АТФ и 100 пМ олигонуклеотида, 1 ед. полинуклеотидкиназы фага Т4 (Сибэнзим, Россия) в течение 30 мин при 37°C. После окончания реакции фермент инактивировали при 65°C 10 мин. Затем использовали для проведения инвертированной ПЦР в количестве 20 пМ на реакцию. ПЦР вели с использованием набора "Encyclo PCR kit" (ЗАО Евроген, Россия) по инструкции производителя, по следующей схеме: 1 цикл: 4 мин 94°C, 2 мин 50°C, 2 мин 72°C; затем 11 циклов 1 мин - 94°C, 1 мин - 55°C, 2 мин 72°C. Продукт ПЦР разводили вдвое однократным буфером для эндонуклеазы DpnI, добавляли 10 Ед. DpnI и инкубировали при 37°C 30 мин, затем вносили 2.5 Ед. полимеразы Pfu, переносили на 72°C и инкубировали еще 30 мин. Полученную смесь очищали, используя набор реактивов "Wizard SV Gel and PCR Clean-Up System" («Promega», США) по протоколу производителя. Проводили лигирование очищенного продукта инвертированной ПЦР с использованием ДНК-лигазы фага Т4 и стандартного буферного раствора (Fermentas, Литва) в течение 1 часа при комнатной температуре. Полученной лигазной смесью трансформировали клетки E. coli штамма DH5α. 4 клона E. coli наращивали в 5 мл питательного бульона 2xYT-Amp и выделяли плазмидную ДНК при помощи набора GeneJET Plasmid Miniprep Kit (Fermentas, Литва). Нуклеотидную последовательность полученных плазмид определяли методом ПЦР-секвенирования.
Для секвенирования плазмиды PAT-EK-DNASEIC18 использовали специфический олигонуклеотид SQ-DNA-CN-L-F (SEQ ID NO:27).
Для секвенирования плазмиды PAT-EK-DNASEIC106 использовали специфический олигонуклеотид SQ-DNA-CN-R-F (SEQ ID NO:28).
Пример 3. Получение векторной плазмиды pET28Thr-
Векторная плазмида была получена на основе коммерческого вектора рЕТ28а(+) (Novagen, США), со вставленным коротким ДНК адаптером. Реципиентную плазмиду рЕТ28а (+) обрабатывали последовательно каждой из эндонуклеаз NcoI и NheI, после чего инактивировали ферменты прогреванием 20 мин при 65°C и выделяли из геля набором "Wizard SV Gel and PCR Clean-Up System" («Promega», США) по протоколу производителя, и лигировали с адаптером. Адаптер получали отжигом двух частично комплиментарных олигонуклеотидов AD-6H-NcoF (SEQ ID NO:29) и AD-6H-NheR (SEQ ID NO:30), при отжиге образующих дуплекс с выступающими «липкими» 5'-концами. Для получения адаптера вносили в пробирку по 100 пм каждого олигонуклеотида, нагревали до 95°C и медленно охлаждали до комнатной температуры. Адаптер лигировали с NcoI-NheI фрагментом плазмиды рЕТ28а Т4 ДНК-лигазой (Fermentas), полученными лигазными смесями, трансформировали клетки E. coli штамма DH5α. Колонии E. coli анализировали методом ПЦР с клонов, с использованием олигонуклеотидов AD-6H-NcoF (SEQ ID NO:29) и T7t (SEQ ID NO:31).
Отобранные клоны наращивали в 5 мл 2xYT-Kan и проводили выделение плазмидной ДНК с набором GeneJET Plasmid Miniprep Kit (Fermentas, Литва). Нуклеотидную последовательность области вставки адаптера в полученных плазмидах определяли методом ПЦР-секвенирования с праймера T7t (SEQ ID NO:31).
Пример 4. Получение экспрессионных плазмид pET28EK-DNASEI, pET28EK-DNASEIC18 и pET28EK-DNASEIC106
Реципиентную плазмиду pET28Thr-, полученную как описано в примере 3, обрабатывали эндонуклеазами NheI и HindIII; проводили дефосфорилирование с последующей инактивацией щелочной фосфатазы 10 мин при 65°C. Фрагмент ДНК выделяли из агарозного геля набором Wizard SV Gel and PCR Clean-Up System (Promega, США) по методике производителя.
Донорные плазмиды также обрабатывали эндонуклеазами NheI и HindIII, продукты рестрикции разделяли в 1% агарозном геле и выделяли из геля набором Wizard SV Gel and PCR Clean-Up System (Promega, США).
Для получения экспрессионной плазмиды pET28EK-DNASEI в качестве донорной использовали плазмиду PAT-EK-DNASEI, полученную как описано в примере 1.
Для получения экспрессионной плазмиды pET28EK-DNASEIC18 в качестве донорной использовали плазмиду PAT-EK-DNASEIC18, полученную как описано в примере 2.
Для получения экспрессионной плазмиды pET28EK-DNASEIC106 в качестве донорной использовали плазмиду PAT-EK-DNASEIC106, полученную как описано в примере 2.
Реакцию лигирования очищенных фрагментов донора и акцептора проводили с использованием Т4 ДНК лигазы (Fermentas, Литва) по методике производителя. Лигазной смесью трансформировали клетки Е. coli штамма DH5α, колонии E. coli анализировали методом ПЦР с клонов, с использованием праймеров к последовательности векторов T7t (SEQ ID NO:31) и T7prom (SEQ ID NO:21). Отобранные клоны наращивали в 5 мл 2xYT-Каn и проводили выделение плазмидной ДНК набором GeneJET Plasmid Miniprep Kit (Fermentas, Литва). Нуклеотидную последовательность в области вставки в полученных генетических конструкциях определяли методом ПЦР-секвенирования с использованием стандартных праймеров к последовательности вектора T7t (SEQ ID NO:31) и T7prom (SEQ ID NO:21).
Пример 5. Получение штаммов-продуцентов Е. coli BL21[DE3]/pET28EK-DNASEI, BL21[DE3]/pET28EK-DNASEIC18, BL21[DE3]/pET28EK-DNASEIC106, оценка продуктивности штаммов-продуцентов и локализация целевого белка
Для получения штаммов-продуцентов гибридных белков предшественников ДНКазыI человека и ее мутеинов экспрессионные конструкции, полученные по примеру 5, использовали для трансформации компетентных клеток E. coli BL21[DE3] (с генотипом F-ompT hsdSB (r-m-) gal dcm (DE3)) и проводили отбор клонов, сохраняющих уровень биосинтеза рекомбинантного полипептида не ниже 30-50% от суммарного клеточного белка в течение, по крайней мере, четырех последовательных пассажей.
Для получения штамма E. coli BL21[DE3]/pET28EK-DNASEI - продуцента белка-предшественника ДНКазы I человека клетки штамма E. coli BL21[DE3] трансформировали экспрессионной плазмидой pET28EK-DNASEI.
Для получения штамма E. coli BL21[DE3]/pET28EK-DNASEIC18 - продуцента белка-предшественника мутеина [N18C] ДНКазы I человека клетки штамма E. coli BL21[DE3] трансформировали экспрессионной плазмидой pET28EK-DNASEIC18.
Для получения штамма E. coli BL21[DE3]/pET28EK-DNASEIC106 - продуцента белка-предшественника мутеина [N106C] ДНКазы I человека клетки штамма E. coli BL21[DE3] трансформировали экспрессионной плазмидой pET28EK-DNASEIC106.
Трансформанты E. coli BL21[DE3] высевали на агаризованную среду 2xYT-агар с добавлением канамицина до 30 мкг/мл и глюкозы до 2%, проводили аналитическую экспрессию целевых белков для пяти случайно выбранных клонов типичного фенотипа. Клоны подращивали в питательном бульоне с добавлением канамицина до 30 мкг/мл и раствора глюкозы до 2% в течение 6-7 часов, инокулировали новую порцию питательной среды в соотношении 1:100, растили культуру до достижения оптической плотности 2 О.Е., индуцировали изопропилтио-р-D-галактозидом, и культивировали еще 2 ч. После окончания культивации осадок клеток отделяли центрифугированием, ресуспендировали клетки в растворе 10 мМ Трис-HCl, 2 мМ ЭДТА-Na, 0.1% Тритона-Х100, 10 мкг/мл лизоцима в соотношении 10 мл раствора на 1 г клеточной пасты, выдерживали суспензию 30 мин на льду и проводили разрушение клеток ультразвуковым диспергатором до исчезновения видимой вязкости суспензии. Отбирали образцы для электрофоретического анализа, разделяли в них растворимую и нерастворимую фракцию белков центрифугированием в микроцентрифуге, дополнительно ресуспендировали осадок в том же растворе и осаждали центрифугированием. Результаты электрофоретического анализа тотального белка для штамма BL21[DE3]/pET28EK-DNASEI приведены на Фиг.5, электрофоретическая подвижность и интенсивность полосы целевого белка для мутеинов [N18C] и [N106C] полностью аналогичны. По данным гель-электрофореза белковых фракций все 3 целевых белка практически полностью были локализованы в нерастворимой фракции белков, т.е. находились в форме «телец включения».
Пример 6. Наработка, выделение и очистка в денатурирующих условиях белков-предшественников ДНКазы I человека и ее мутеинов DNASEI[N18C] и DNASEI[N106C]
Получение очищенных денатурированных белков-предшественников ДНКазы I человека и ее мутеинов N18C и N106C из штаммов-продуцентов Е.coli BL21[DE3]/pET28EK-DNASEI; BL21[DE3]/pET28EK-DNASEIC18 и BL21[DE3]/pET28EK-DNASEIC106 проводили по следующей общей схеме: выращивание штаммов-продуцентов, индукция, отделение биомассы, лизис и дезинтеграция клеток в присутствии ЭДТА-Na; отделение телец включения; солюбилизация; металлохелатная хроматография. Для этого штаммы-продуценты высевали из музея петлей истощающим штрихом на чашку Петри с агаризованной средой LB, содержащей 30 мкг/мл канамицина и 2% глюкозы, растили 14 часов при +37°C. Одну отдельную колонию соответствующего штамма переносили в 5 мл жидкой среды LB, содержащей 30 мкг/мл канамицина и 2% глюкозы, и растили на качалке 14 часов при +37°C. Полученными культурами инокулировали 3 колбы по 250 мл среды 2xYT, содержащих 30 мкг/мл канамицина и 0,1% глюкозы, растили на качалке 3,5 часа при +37°C, отбирали образцы бактериальной суспензии для анализа, добавляли ИПТГ до конечной концентрации 1 мМ и растили еще 4 часа.
Отделяли осадки биомассы центрифугированием, осадки клеток ресуспендировали в 20 мл раствора А (50 мМ Трис pH 7,4, 2 мМ ЭДТА), добавляли лизоцим до 10 мкг/мл и Тритон Х100 до 0,1%, инкубировали 30 мин на льду. Проводили разрушение клеток и геномной ДНК при помощи ультразвукового диспергатора пульсами по 10 с до исчезновения повышенной вязкости суспензии. Отделяли осадки центрифугированием 10 мин при 18000 об/мин. Осадки ресуспендировали в 20 мл раствора А, добавляли детергент NP-40 до 1%, суспензии обрабатывали ультразвуковым диспергатором до исчезновения частиц осадка крупнее 1 мм, отделяли тельца включения центрифугированием 10 мин при 20000 об/мин. Полученные осадки телец включения ресуспендировали в растворе А, добавляли NaCl до 500 мМ, суспензию обрабатывали ультразвуковым диспергатором до исчезновения частиц осадка крупнее 1 мм, отделяли осадок центрифугированием 10 мин при 20000 об/мин. Полученные препараты обогащенных телец включения хранили при -70°C. Чистота всех целевых белков в полученных препаратах составляла не менее 80% по данным денситометрии электрофореграмм.
Для проведения солюбилизации целевых белков к навескам приблизительно 50 мг телец включения добавляли раствор Б (8 М мочевины, 50 мМ Трис-HCl, 50 мМ бета-меркаптоэтанола, pH 8.0) в соотношении 10 мл раствора на 1 г телец. Суспензии инкубировали при перемешивании 2 часа при +37°C, отделяли не растворившийся клеточный дебрис центрифугированием 10 мин при 18000 об/мин. Супернатанты наносили на колонки HiTrap Chelating Sepharose (GE Healthcare, США), содержащие хелатированные ионы кобальта и уравновешенные раствором В (6 М мочевины, 50 мМ Трис-HCl, 500 мМ хлорида натрия, pH 7,0). Колонки промывали раствором Г (6 М мочевины, 50 мМ Трис-HCl, 500 мМ хлорида натрия, 50 мМ имидазола pH 7,0) и элюировали целевые белки раствором Д (6 М мочевины, 50 мМ Трис-HCl, 500 мМ хлорида натрия, 50 мМ имидазола pH 7,0). Элюаты концентрировали ультрафильтрацией до конечной концентрации белка 20 мг/мл, обессоливали диафильтрацией и замораживали. Чистота всех целевых белков в полученных препаратах составляла не менее 95% по данным денситометрии электрофореграмм.
Пример 7. Ренатурация белков-предшественников ДНКазы I человека и ее мутеинов [N18C] и [N106C]
Ренатурацию белков проводили при помощи рефолдинга быстрым разбавлением. Дисульфидные связи денатурированных белков-предшественников восстанавливали обработкой 5 мМ дитиотреитола в течение 30 мин при комнатной температуре. Для всех трех белков использовали раствор для рефолдинга одинакового состава, содержащий 50 мМ Tris-HCl pH=8,0, 4 мМ CaCl2, 4 мМ MgSO4, 2 мМ восстановленного глутатиона, 0,4 мМ окисленного глутатиона, 1% ПЭГ 1500, 2 М мочевины. Растворы денатурированных белков-предшественников вводили в растворы для рефолдинга по каплям при постоянном перемешивании, использовали разведение 1:40. Вели рефолдинг 1 ч при комнатной температуре при постоянном перемешивании без доступа кислорода. После этого отделяли выпавший осадок центрифугированием. Выход растворимых ковалентно мономерных белков-предшественников составлял во всех случаях не менее 70% по денситометрии электрофореграммы. Растворы ренатурированных белков концентрировали ультрафильтрацией до конечной концентрации белка 3-4 мг/мл и немедленно отделяли N-концевые пептиды обработкой энтерокиназой.
Пример 8. Отделение N-концевых пептидов от белков-предшественников ДНКазы I человека и ее мутеинов DNASEI[N18C] и DNASEI[N106C].
Отделение N-концевых пептидов проводили при помощи рекомбинантной энтерокиназы ("Sigma", США), используя соотношение фермент:субстрат 1:10 (по массе). Реакцию вели в течение 2 ч при +37°C, используя разбавленные в 2 раза водой растворы белков-предшественников, т.е. при концентрации субстратов 1,5-2 мг/мл. Степень превращения для всех трех белков-предшественников составила около 70% по данным гель-электрофоретического анализа.
Пример 9. Финальная очистка ДНКазы I человека и ее мутеинов DNASEI[N18C] и DNASEI[N106C].
Отделение процессированных белков от белков-предшественников, свободных N-концевых пептидов, энтерокиназы и посторонних примесных белков проводили адсорбцией в объеме, добавляя к реакционным смесям суспензию сорбента Chelating Sepharose FF ("GE Healthcare", США) с иммобилизованными ионами кобальта в соотношении 300 мкл суспензии на 1 мг белка. Реакционные смеси инкубировали с сорбентом 1 ч при комнатной температуре при помешивании. После этого супернатанты отделяли, осадки сорбента промывали 5 объемами раствора 25 мМ Tris-HCl pH8,0, 2 мМ CaCl2, 2 мМ MgSO4, 1 мМ восстановленного глутатиона, 1 М мочевины, объединяли супернатанты, концентрировали их ультрафильтрацией до конечной концентрации общего белка 1,5-2 мг/мл и удаляли восстановленный глютатион диафильтрацией до отрицательной реакции фильтрата с реактивом Эллмана (5,5'-дитиобис-(2-нитробензойной кислотой)). В результате этой процедуры были получены растворы очищенной рекомбинантной ДНКазы I человека и ее мутеинов DNASEI[N18C] и DNASEI[N106C], содержащие не более 5% примесных белков по данным гель-электрофоретического анализа.
Пример 10. Измерение активности рекДНКазы I человека методом зимографии.
Готовили пластину геля для электрофореза по Лэммли. В разделяющий гель перед полимеризацией добавляли ДНК из тимуса крупного рогатого скота до конечной концентрации 10 мкг/мл. Наносили по несколько разведений образцов исследуемых белков в растворе для подготовки образца к электрофорезу по Лэммли, а также аналогично солюбилизированные отмытые тельца включения контрольного белка - ДНКазы I человека с дополнительным N-концевым остатком метионина, и стандарт ферментативной активности - природную ДНКазу I крупного рогатого скота.
По завершении электрофореза отделяли концентрирующий гель, разделяющий гель отмывали в растворе (Тритон Х-100 1%, Трис-HCl pH 7,5, 20 мМ) 3 раза по 10 мин для удаления из геля ЭДТА, додецилсульфата натрия и ренатурации белков в смешанных мицеллах детергентов. Затем гель промывали в растворе (Трис-HCl 20 мМ, NaCl 150 мМ, Твин 20 0,05%, CaCl2 5 мМ, MgCl2 5 мМ, MnSO4 5 мМ) 2 раза по 5 мин. В третью порцию раствора добавляли бромистый этидий до 1 мкг/мл и вели инкубацию геля в течение 30 мин при +37°C. Гель помещали на трансиллюминатор и фотографировали. Продолжали инкубирование на +37°C до проявления видимых зон распада субстрата, но не больше 2-х суток. При денситометрии полученных зимограмм было установлено, что удельная активность белка-предшественника ДНКазы I по отношению к природной ДНКазе I быка составляет не более 0,01%, при этом удельная активность метионил-ДНКазы I составляет около 0,1%, а удельная активность зрелой рекомбинантной ДНКазы I человека - около 1%. Поскольку единственным отличием рекомбинантной ДНКазы от природного белка является отсутствие N-связанных олигосахаридов, было предположено, что ферментативная активность белка может быть восстановлена при конъюгации аминокислот в сайтах N-гликозилирования (позиции 18 и 106) с гидрофильными полимерами.
Пример 11. Направленная конъюгация мутеинов DNASEI[N18C] и DNASEI[N106C] с малеимид-полиэтиленгликолем
Очищенные мутеины DNASEI[N18C] и DNASEI[N106C], содержащие замены остатков аспарагина на остатки цистеина, обрабатывали реактивом ТСЕР (трихлорэтилфосфат) в молярном соотношении 5:1 в течение 10 мин при комнатной температуре для полного восстановления непарного остатка цистеина и добавляли ПЭГ-малеимид 10 кДа ("CreativePEGWorks", США) в молярном соотношении 5:1. Реакционную смесь инкубировали 60 мин при комнатной температуре. Осаждали непрореагировавшие мутеины разбавлением реакционной смеси в 4 раза водой и отделяли осадки центрифугированием. Очистку конъюгатов проводили анионообменной хроматографией на колонках HiTrap Capto DEAE 1 мл ("GE Healthcare", США), уравновешенных раствором 20 мМ Трис-HCl pH 7,0; 5 мМ CaCl2. Супернатанты разбавленных реакционных смесей наносили на колонки немедленно по окончании центрифугирования. Колонки промывали 5 объемами стартового раствора и вели элюцию линейным градиентом концентрации хлорида натрия от 0 до 300 мМ за 20 мл, собирая элюат фракциями по 1 мл. Фракции, содержащие только монозамещенные конъюгаты мутеинов (по данным ДСН-ПААГ анализа), объединяли, концентрировали ультрафильтрацией на мембранах с порогом отсечения 10 кДа, обессоливали диафильтрацией к раствору 20 мМ Трис-HCl, 5 мМ CaCl2 до конечной концентрации белка 1-2 мг/мл и использовали для определения ферментативной активности. Анионообменная хроматография позволяет эффективно отделять конъюгаты от остатков непрореагировавших мутеинов за счет изменения заряда поверхности белка после конъюгации и более ранней элюции конъюгатов.
Пример 12. Определение ферментативной активности в растворе рекомбинантной ДНКазы I человека, мутеинов DNASEI(N18C] и DNASEI[N18C] и их конъюгатов с полиэтиленгликолем 10 кДа
Поскольку ПЭГ-производные мутеинов DNASEI[N18C] и DNASEI[N106C] обладают переменной молекулярной массой и не образуют сфокусированных зон при электрофорезе в ДСН-ПААГ, измерение их ферментативной активности зимографией затруднительно. Для сравнения удельной ферментативной активности конъюгатов, мутеинов и исходной рекомбинантной ДНКазы I человека использовали методику гидролиза сверхскрученной плазмиды в растворе с последующим разделением продуктов реакции в агарозном геле и расчетом степени гидролиза субстрата по денситометрии цифровой фотографии геля.
Гидролиз вели в растворе, содержащем сверхскрученную плазмиду pUC19 в концентрации 30 нг/мкл, 25 мМ Hepes, pH 7.0, 0,1 мкг/мкл бычьего сывороточного альбумина, 4 мМ MgCl2, 4 мМ MnSO4 и/или CaCl2. Реакционные смеси инкубировали ровно 10 мин, реакцию останавливали добавлением Na-ЭДТА до 20 мМ. Было установлено, что удельная ферментативная активность рекомбинантной ДНКазы I человека составляет не более 2% от активности ДНКазы I быка, ферментативная активность мутеинов DNASEI[N18C] и DNASEI[N18C] в данном тесте не выявляется, т.е. составляет менее 10% от удельной активности рекомбинантной ДНКазы I человека. В то же время удельная активность обоих конъюгатов DNASEI[N18C] и DNASEI[N18C] с ПЭГ составляет 10-20% от активности ДНКазы I быка.
Приведенные результаты показали, что включение в последовательность синтезируемого белка лидерного N-концевого пептида, кодируемого оптимальными для Е. coli кодонами снижает токсичность продукта экспрессии для бактерий, что приводит к значительному увеличению выхода целевого белка. N-концевой пептид также позволяет проводить хроматографическую очистку экспрессируемого белка в денатурирующих условиях методом металлохелатной хроматографии, что, в свою очередь, дает возможность получать гомогенный продукт при помощи одной хроматографической стадии. После удаления дополнительного пептида обработкой энтерокиназой и отделения зрелой ДНКазы I повторной металлохелатной хроматографией может быть получен очищенный препарат белка. Полученные указанным способом мутеиновые варианты ДНКазы I человека могут быть превращены в ферментативно высокоактивную форму при направленной конъюгации с полиэтиленгликолем.
Хотя указанное изобретение описано в деталях со ссылкой на Примеры, для специалиста в указанной области техники очевидно, что могут быть совершены различные изменения и произведены эквивалентные замены, и такие изменения и замены не выходят за рамки настоящего изобретения.









Группа изобретений относится к биотехнологии и представляет собой способ получения рекомбинантной ДНКазы I человека или ее мутеина, а также их конъюгатов с полиэтиленгликолем, с использованием бактерии, принадлежащей к роду Escherichia, трансформированной экспрессионной плазмидой, содержащей промотор, функционирующий в бактериальной клетке, фрагмент ДНК, кодирующий гексагистидиновый кластер, фрагмент, кодирующий последовательность узнавания энтерокиназы, слитую в рамке с ДНКазой I человека или ее функционально активным мутеином, содержащим замены аспарагина на цистеин, участок терминации транскрипции, фрагмент вектора рЕТ28а(+), содержащий участок инициации репликации бактериофага fl, последовательность, кодирующую аминогликозид-3'-фосфотрансферазу, область начала репликации плазмиды pBR322, ген РНК-организующего белка Rop, последовательность, кодирующую репрессор лактозного оперона. Предложенное изобретение позволяет получать рекомбинантную ДНКазу I человека или ее мутеин с высоким выходом. 6 н. и 12 з.п. ф-лы, 7 ил., 1 табл., 12 пр.
1. Плазмида для экспрессии в клетках бактерии, принадлежащей к роду Escherichia, неактивного предшественника ДНКазы I человека или ее мутеинов, содержащего отщепляемый N-концевой лидер, включающий гексагистидиновый кластер и последовательность узнавания энтерокиназы, по существу, содержащая:
- промотор, функционирующий в бактериальной клетке;
- фрагмент ДНК, кодирующий гексагистидиновый кластер;
- фрагмент, кодирующий последовательность узнавания энтерокиназы, слитую в рамке с ДНКазой I человека или ее функционально активным мутеином, содержащим замены аспарагина на цистеин;
- участок терминации транскрипции;
- фрагмент вектора рЕТ28а(+), содержащий участок инициации репликации бактериофага fl, последовательность, кодирующую аминогликозид-3'-фосфотрансферазу, область начала репликации плазмиды pBR322, ген РНК-организующего белка Rop, последовательность, кодирующую репрессор лактозного оперона.
2. Плазмида по п.1, отличающаяся тем, что указанным фрагментом ДНК, кодирующим предшественник ДНКазы I, является фрагмент ДНК, представленный в Перечне последовательностей под номером SEQ ID:1, или его вариант.
3. Плазмида по п.1, отличающаяся тем, что указанным промотором является промотор РНК-полимеразы бактериофага Т7.
4. Плазмида по п.1, отличающаяся тем, что указанный мутеин ДНКазы I содержит замену N18C или N106C.
5. Плазмида по п.1, отличающаяся тем, что указанная плазмида выбрана из группы, состоящей из плазмид pET28EK-DNASEI, pET28EK-DNASEIC18 и pET28EK-DNASEIC106.
6. Бактерия, принадлежащая к роду Escherichia, трансформированная плазмидой по п.1, - продуцент неактивного предшественника рекомбинантной ДНКазы I человека или ее мутеина, содержащего замены аспарагина на цистеин.
7. Бактерия по п.6, отличающая тем, что указанный мутеин ДНКазы I содержит замену N18C или N106C.
8. Бактерия по п.6, отличающаяся тем, что указанная бактерия выбрана из группы, включающей бактерии Е. coli BL21[DE3]/pET28EK-DNASEI, E.coli BL21[DE3]/pET28EK-DNASEI18C и Е. coli BL21[DE3]/pET28EK-DNASEI106C.
9. Предшественник рекомбинантной ДНКазы I человека или ее мутеина, содержащий отщепляемый N-концевой лидер, включающий гексагистидиновый кластер и последовательность узнавания энтерокиназы, полученный путем культивирования бактерии по п.6 в питательной среде и предназначенный для получения активной формы ДНКазы 1 человека или ее мутеина.
10. Предшественник рекомбинантной ДНКазы I человека по п.9, содержащий последовательность аминокислот, представленную в Перечне последовательностей под номером SEQ ID NO:2, или ее вариант.
11. Предшественник мутеина ДНКазы I человека по п.9, отличающийся тем, что указанный мутеин содержит замену аспарагина на цистеин N18C или N106C.
12. Способ получения рекомбинантной ДНКазы I человека или ее мутеина, включающий культивирование бактерии по п.6 в питательной среде, выделение телец включения, солюбилизацию белка-предшественника, металлохелатную хроматографию в денатурирующих условиях, рефолдинг белка-предшественника, получение зрелого белка обработкой энтерокиназой и выделение указанного зрелого белка рекомбинантной ДНКазы 1 человека или ее мутеина.
13. Способ по п.12, отличающийся тем, что культивируют штамм Е. coli BL21[DE3]/pET28EK-DNASEI, Е. coli BL21[DE3]/pET28EK-DNASEI18C или Е. coli BL21[DE3]/pET28EK-DNASEI106C.
14. Способ по п.12, отличающийся тем, что указанный мутеин содержит замену N18C или N106C.
15. Способ получения конъюгатов полиэтиленгликоля и рекомбинантного мутеина ДНКазы I человека, полученного способом по п.12, включающий предварительное ограниченное восстановление белка, инкубацию с малеимид-полиэтиленгликолем и выделение полученного конъюгата.
16. Способ по п.15, отличающийся тем, что указанный мутеин содержит точечную замену N18C или N106C.
17. Ферментативно активный конъюгат мутеина рекомбинантной ДНКазы I человека, содержащего замены аспарагина на цистеин, и полиэтиленгликоля, полученный способом по п.15.
18. Ферментативно активный конъюгат по п.17, отличающийся тем, что указанный мутеин содержит точечную замену N18C или N106C.
| RU 2006110371 A, 10.10.2007 | |||
| ПРЕПАРАТ ЧЕЛОВЕЧЕСКОГО ЭНДОСТАТИНА И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2004 |
|
RU2278688C1 |
| МОДИФИЦИРОВАННЫЕ ИНГИБИТОРЫ ПРОТЕИНАЗЫ | 1995 |
|
RU2182598C2 |
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| ФИЗИОЛОГИЧЕСКИ АКТИВНЫЙ ПОЛИПЕПТИДНЫЙ КОНЪЮГАТ, ОБЛАДАЮЩИЙ ПРОЛОНГИРОВАННЫМ ПЕРИОДОМ ПОЛУВЫВЕДЕНИЯ IN VIVO | 2004 |
|
RU2312868C2 |
| LINARDOU H | |||
| ET AL., A recombinant cytotoxic chimera based on mammalian deoxyribonuclease-I, Int J Cancer., 2000, v.86, no.4, pp.561-569 | |||
| US 20040219529 A1, 04.11.2004 | |||
| WO 2005071077 A1, 04.08.2005. | |||

