Заявляемое изобретение относится к биотехнологии и химико-фармацевтической промышленности и может быть использовано для создания систем, в частности, липосомальных фармацевтических композиций, осуществляющих направленную транспортировку физиологически активных веществ с целью повышения терапевтической активности лекарственных препаратов, а также обеспечивающих высокую эффективность и стабильность активного вещества в липосомах.
Липосома - самопроизвольно образующиеся в смесях фосфолипидов с водой замкнутые пузырьки, стенка которых состоит из одного или нескольких бислоев фосфолипидов (слоев толщиной в две молекулы), в которые встроены другие вещества, например, белки. Внутри липосом содержится вода или раствор (см. http://ru.wikipedia.org/wiki/%CB%E8%).
Липосомы успешно используются, как носители лекарств, поскольку:
по химическому составу сходны с природными мембранами клеток;
универсальны, что позволяет переносить широкий спектр медицинских химических препаратов;
не вызывают аллергических реакций.
Однако есть ряд трудностей использования липосом в медицине. Во-первых, липосомы поглощаются клетками ретикуло-эндотелиальной системы, причем большее их количество находится в печени, селезенке, костном мозге, лимфатических узлах и кровотоке. Поэтому доставка лекарственных средств с помощью липосом в другие органы и части организма является более сложной задачей. Во-вторых, липопротеины, обмениваясь с липосомами липидами, способствуют разрушению липосом и вытеканию наружу их содержимого. Кроме этого, стоит задача увеличения сроков хранения липосом после их приготовления (см. Кобринский Г. Липосомы в медицине (см. http://n-t.ru/nj/nz/1988/0601.htm)// Наука и жизнь. 1988. №6. (Электронная версия: НиТ. Научные журналы, 2002).
Липосомы, как носители лекарственных средств, широко известны с середины 60-х годов прошлого столетия. За прошедшее время изучены как преимущества, так и недостатки препаратов на основе липосом. К очевидным преимуществам липосом можно отнести биосовместимость, биодеградируемость, способность переносить любые по строению соединения, в том числе, полярные, неполярные, амфифильные, с большой и малой молекулярной массой. Помимо этого, во многих случаях можно достичь адресной доставки лекарственного средства за счет пассивного нацеливания или конъюгации липосомы с биомаркерами.
Помимо указанных недостатков липосом, как лекарственной формы, существуют сложности, связанные с высокой скоростью поглощения липосом макрофагами, что приводит к короткому по длительности лечебному эффекту препарата. Возникают проблемы и со стандартизацией липосомальных форм.
В настоящее время из уровня техники известно несколько различных технологий, позволяющих добиться высокой степени включения лекарственных веществ в структуру липосом и обеспечить их надежное удержание в течение всего срока хранения препарата.
Первым способом является химическая модификация самой молекулы лекарственного препарата с целью повышения ее липофильности. В этом случае модифицированная молекула легко встраивается в липосомальную мембрану, однако, при этом, она является уже совершенно иной субстанцией, требующей проведения дополнительных исследований на биологическую активность (см. Bakker-Woudenberg IA, Storm G, Woodle MC Liposomes in the treatment of infections. J Drug Target (1994) 2:363-371.). Такой подход требует выполнения значительного комплекса работ, проведение полного цикла которых может занимать длительное время, от двух до трех лет.
Второй метод основан на включении лекарственного препарата во внутреннюю водную фазу липосомальной суспензии. Это достигается за счет проведения двойного эмульгирования или повторения нескольких циклов гидратация-заморозка (см. Hiroshi Kikuchi et al. Gene delivery using liposome technology. Journal of Controlled Release 62 (1999) 269-277).
Недостатки известного метода обусловлены проблемами масштабирования, т.к. метод включает в себя выполнение целого ряда нетехнологичных операций, включающих, в том числе, лабораторный синтез. Кроме того, при осуществлении данного способа лекарственное вещество может образовывать комплексные соединения с компонентами мембраны и за счет этого выходить во время хранения липосомальной лекарственной формы во внешнюю среду липосомального раствора.
Известен третий способ, основанный на получении липидной композиции, обладающей повышенным сродством к субстанции. Так как многие лекарственные препараты имеют повышенную кислотность, они могут легко образовывать соли с целым рядом основных соединений, входящих в структуру липосомальной мембраны, при этом они могут эффективно осаждаться на положительно заряженных липосомальных мембранах. Данный подход позволяет добиться практически полного включения лекарственного вещества в структуру липосом без использования сложных и нетехнологичных процедур и без какой-либо модификации самого лекарственного вещества (см. Crommelin DJ Influence of lipid composition and ionic strength on the physical stability ofliposomes. J Pharm Sci (1984) 73:1559-1563.).
Из уровня техники известна фармацевтическая композиция, представляющая собой липосомальную эмульсию, образованную фосфолипидами в виде липосом со средним размером частиц 0,05-0,22 мкм и сульфатированными гликозаминогликанами, при этом в качестве сульфатированных гликозаминогликанов содержит натриевую соль кератансульфата и натриевую соль хондроитинсульфата, причем компоненты находятся в определенном соотношении в мас.%:
при этом сульфатированные гликозаминогликаны находятся как внутри липосом, так и вне липосом, в физиологическом растворе (см. патент РФ на изобретение №2460517 «Фармацевтическая композиция для комплексного лечения заболеваний глазной поверхности у больных с первичной открытоугольной глаукомой», дата подачи 14.07.2011 г., опубликовано 10.09.2012 г.).
Известна биоцидная композиция на липосомальной основе, содержащая четвертичное аммонийное - дидецилдиметиламмоний галогенид в форме клатрата с мочевиной и липид (см. патент РФ на изобретение №2353395 «Биоцидная композиция на липосомальной основе», дата подачи 14.06.2007 г., опубликовано 27.04.2009 г.).
Известна липосомальная композиция, содержащая следующие ингредиенты, вес.ч.: паклитаксел - 2-5; фосфатид - 20-200; холестерин - 2-30; аминокислоты - 0,3-4; лиофилизированный эксципиент - 10-75. При этом способ получения данной композиции включает следующие операции: растворение паклитаксела, фосфатида, холестерина в изопропаноле или этаноле для получения прозрачного раствора, удаление растворителя для образования мембраны, затем добавление водного раствора аминокислот и лиофилизированного эксципиента с последующей гидратацией, обработкой ультразвуком или гомогенизацией для получения липосомального препарата паклитаксела (см. патент РФ на изобретение №2264807 «Липосомальная композиция с паклитакселом для лечения рака и способ ее получения», дата подачи 28.02.2001 г., дата публикации заявки 10.09.2004 г.).
Из уровня техники известен способ получения липосомальной композиции, включающий формирование смеси, содержащей, по крайней мере, один компонент липосомальной мембраны, воду и добавку, диспергирование смеси сводится к тому, что в качестве добавки используют либо субмикропорошок вещества нерастворимого в воде и в органическом растворителе, либо набухающие в воде полимер, либо жидкость, не смешивающуюся с водой и другими компонентами смеси (см. патент РФ на изобретение №2104691 «Способ получения липосомальной композиции (варианты)», дата подачи 20.02.1995 г., дата публикации заявки 27.02.1997 г.).
Известен способ получения липосомальной композиции, включающий формирование смеси липидов с липофильными и гидрофильными добавками, мелкодисперсным порошком и диспергирование, при этом на предварительном этапе подготовки жировой фазы ее обрабатывают аминосодержащими соединениями для перераспределения заряда, а также частично гидратируют липиды, затем вводят в необходимых количествах добавки, причем липосомы формируют на границе раздела двухфазной системы вода/нерастворимые в воде частицы (см. патент на изобретение №2311449 «Способ получения липосомальной композиции», дата подачи 20.12.2005 г., опубликовано 27.11.2007 г).
Кроме того, известен способ получения сухих липосомальных препаратов, включающий в себя смешение фосфолипидов, порошкообразного носителя и биологически активного вещества и дальнейшую обработку полученной смеси, при этом в загрузочный бункер струйной мельницы засыпают композицию, состоящую из биологически активного вещества, фосфолипида из группы, включающей витол и лецитин ПРО, и водорастворимого порошкообразного носителя и подвергают помолу в струйной мельнице при давлении не менее 2,5 атм и расходе газа не менее 100 л/мин, с дальнейшим выделением фракции с размером частиц, меньшим, чем 50 мкм. Как вариант, помолу подвергают смесь фосфолипидов и порошкообразного носителя, а затем добавляют к полученному продукту раствор биологически активного вещества в полярном растворителе, и подвергают суспензию лиофильному высушиванию, обрабатывая ее при необходимости перед сушкой ультразвуком при частоте 40 кГц (см. патент на изобретение №2437649 «Способ получения сухих липосомальных препаратов», дата 03.11.2008 г., опубликовано 27.12.2011 г.).
Наиболее близким техническим решением к заявляемой липосомальной композиции является композиция для доставки лекарственных или диагностических соединений, представляющая собой липосому, имеющую внутреннее пространство, отделенное от среды мембраной, включающей один или несколько липидов, причем внутреннее пространство включает анион, выбранный из полианионизированного моносахарида или полианионизированного дисахарида, и на липосоме создается трансмембранный градиент, эффективный для удерживания веществ внутри липосомы, при этом трансмембранный градиент может представлять собой градиент иона аммония или ион замещенного аммония, в качестве которого может быть использован, например, ион четвертичного аммония, а липид представляет собой фосфатидилхолин или холестерин.
Способ инкапсулирования вещества в липосому включает стадию контактирования композиции с веществом в течение времени, достаточного для того, чтобы указанное вещество стало инкапсулированным в этой липосоме, причем часть вещества, которая становится инкапсулированной в липосомах, составляет, по крайней мере, от 80% до 95%, а вещество является полностью катионным веществом, терапевтическим агентом или детектируемым маркером (см. патент РФ на изобретение №2424792 «Липосомы, используемые для доставки лекарственных средств, дата подачи 02.05.2005 г., опубликовано 27.07.2011 г., заявка РСТ US 2005/15349 20050502).
Недостатки данного изобретения обусловлены невозможностью их использования для получения препаратов на основе сильных органических кислот, так как в этом случае образование солей между лекарственным веществом и четвертичными аммониевыми соединениями приводит к образованию кристаллических кластеров на ее поверхности, что, в свою очередь, снижает стабильность липосомальной суспензии.
Техническим результатом, на достижение которого направлено заявляемое изобретение является повышение степени включения лекарственного (активного) вещества в структуру липосом и увеличение срока хранения липосомального препарата без существенного изменения степени включения лекарственного (активного) вещества.
При этом в качестве лекарственного вещества используют натриевую соль 2-метилтио-6-нитро-1,2-4-триазоло[5,1-с]-1,2,4-триазин-7-она, дигидрат (Триазавирин).
Указанный технический результат достигается тем, что липосомальная фармацевтическая композиция, включающая лекарственное вещество, липиды в виде фосфатидилхолина и холестерина, ионы четвертичного аммония, добавки, согласно изобретению в качестве лекарственного вещества используют натриевую соль 2-метилтио-6-нитро-1,2-4-триазоло[5,1-c]-1,2,4-триазин-7-она, дигидрат, вводимую в липосомы в виде комплексных солей, образованных с четвертичными аммониевыми солями, в качестве которых применяют цетилпиридиний хлорид и гексадецил триметиламмоний хлорид, причем при общем содержании комплексных солей в количестве более 20 мас.% в качестве липидов используют фосфатидилхолин, при содержании менее 20 мас.% - фосфатидилхолин и холестерин, при этом компоненты находятся в определенном соотношении, мас.%:
Фосфатидилхолин представляет собой Эпикурон 200.
В качестве добавок используют антиоксиданты и консерванты, при этом в качестве антиоксидантов применяют смесь токоферолов (Е306) в количестве 0,05-0,1 мас.%, а в качестве консервантов, например, метилпарабен в количестве 0,1-0,3 мас.%.
Указанный результат достигается тем, что способ получения липосомальной фармацевтической композиции, включающий введение лекарственного вещества в липосому, согласно изобретению лекарственное вещество, в качестве которого используют натриевую соль 2-метилтио-6-нитро-1,2-4-триазоло[5,1-с]-1,2,4-триазин-7-она, дигидрат, вводят в липосомы в виде комплексных солей, образованных с четвертичными аммониевыми солями, в качестве которых применяют цетилпиридиний хлорид и гексадецил триметиламмоний хлорид, при этом полученные комплексные соли растворяют в неполярном растворителе и смешивают с липидами и добавками, после чего растворитель удаляют, а полученную в результате липидную пленку охлаждают, высушивают в вакууме и гидратируют до образования суспензии мультиламеллярных везикул, из которой путем ультрафильтрации, проводимой одновременно со стерилизацией при температуре 45-50°C, формируют липосомы.
При этом в качестве липидов используют фосфатидилхолин и холестерин.
Активное лекарственное вещество Триазавирин вводят в состав липидной композиции в виде комплексных солей, образованных с четвертичными аммониевыми солями, обладающими поверхностно-активными и антисептическими свойствами. Благодаря такому способу получения липосомальной суспензии, предотвращается вымывание лекарственного вещества Триазавирина из структуры липосом и повышается его липофильность, при этом комплексные соединения находятся как внутри липосом, так и на ее мембране, а также в физиологическом растворе, что позволяет наиболее эффективно доставлять лекарственное средство.
В данном случае не требуется проведения операций по модификации самого лекарственного вещества, которая заключается во введении алкильных остатков по N-атомам гетероциклического ядра и в результате приводит к изменению структуры активного действующего вещества, а это, в свою очередь, ведет к радикальным изменениям его фармакокинетического профиля. Кроме того, не требуется проведения значительного комплекса дополнительных исследований.
Триазавирин (Н-форма), предназначенный для лечения и профилактики инфекционных заболеваний вирусной природы человека и животных, имеет повышенную кислотность (см. патент на изобретение №2294936 «Натриевая соль 2-метилтио-6-нитро-1,2-4-триазоло[5,1-с]-1,2,4-триазин-7-она, дигидрат, обладающая противовирусной активностью», дата подачи 29.06.2005 г., опубликовано 10.03.2007 г.), что позволяет ему легко образовывать соли с целым рядом основных соединений и, в том числе, эффективно осаждаться на положительно заряженных липосомальных мембранах, что, в свою очередь, позволяет добиваться практически полного включения Тризавирина в структуру липосом без какой-либо его модификации, не применяя при этом каких-либо сложных и нетехнологичных процедур.
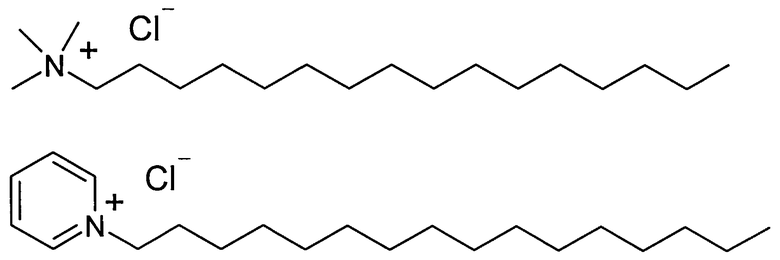
В заявляемом изобретении в качестве четвертичных аммонийных солей были использованы цетилпиридиний хлорид и гексадецил триметиламмоний хлорид, - соли, которые хорошо кристаллизуются из подходящего растворителя, например, спирта или ацетонитрила, а их комплексы с Триазавирином обладают сходными физико-химическими свойствами.
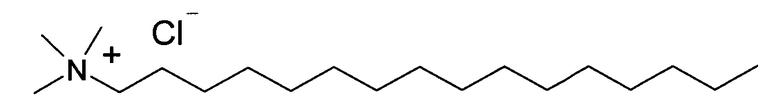
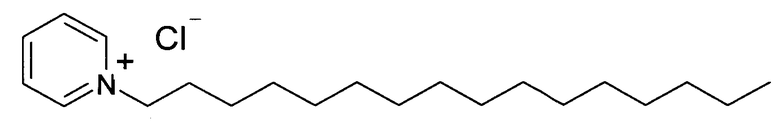
Были получены комплексные соли этих соединений с Триазавирином. Кроме того, были изучены физико-химические свойства данных солей. В отличие от исходного Триазавирина, соли практически нерастворимы в воде, однако хорошо растворимы, например, в хлороформе и триглицеридах. При определенных условиях соли могут образовывать сплошные фазы с компонентами липидных пленок липосом, однако при непродолжительном стоянии могут кристаллизоваться из них. Для преодоления данного недостатка была использована смесь двух различных четвертичных аммониевых солей, имеющая наименьшую склонность к кристаллизации, в частности, была применена эквимолярная смесь вышеуказанных цетилпиридиний хлорида и гексадецил триметиламмоний хлорида. Использование смеси солей позволяет избежать кристаллизации липидной пленки липосом, которая негативно отражается на их стабильности. В то же время положительный заряд на поверхности липосом, равный +19 мВ, позволяет стабилизировать поверхность липидной мембраны и предотвращает слияние липосом во время хранения. При этом дополнительная стерическая стабилизация позволяет значительно замедлить процесс старения липосом.
Для доказательства эффективности использования предлагаемой к защите липосомальной композиции была проведена серия экспериментов по введению солей Триазавирина в структуру липосом. В частности, было проведено сравнение липосом, полученных в соответствии с заявляемым изобретением и синтезированных способом прямой гидратации в присутствии Триазавирина. В результате было обнаружено, что при прямой гидратации липидной пленки степень включения Триазавирина в структуру липосом не превышает 35%, в то время, как при использовании комплексов Триазавирина с четвертичными аммонийными солями она увеличивается до 85%.
Технических решений, совпадающих с совокупностью существенных признаков заявляемого изобретения, не выявлено, что позволяет сделать вывод о соответствии заявляемого изобретения такому условию патентоспособности как «новизна».
Заявляемые существенные признаки, предопределяющие получение указанного технического результата, явным образом не следуют из уровня техники, что позволяет сделать вывод о соответствии заявляемого изобретения такому условию патентоспособности как «изобретательский уровень».
Условие патентоспособности «промышленная применимость» подтверждено на примерах конкретного применения.
Заявляемую липосомальную композицию получают следующим образом.
Активное действующее вещество Триазавирин в виде 5%-ного водного раствора (1 ммоль) по каплям добавляют к 5%-ному раствору смеси солей цетилпиридиний хлорид (0,5 ммоль) и гексадецил триметиламмоний хлорид (0,5 ммоль) и перемешивают до выпадения осадка, который отфильтровывают, а затем сушат при температуре 40-50°C. После этого, полученное комплексное соединение растворяют в 1%-ном растворе хлороформа или метанола и добавляют к нему фосфатидилхолин, например, марки Эпикурон 200, в количестве 75% от суммы твердых веществ в растворе, а также консервант, в качестве которого используют, например, метилпарабен и антиокислитель, в качестве которого используют смесь токоферолов (0.05-0.1%). После этого, растворитель осторожно отгоняют на роторном испарителе, получая липидную пленку липосом, состав которой приведен в Таблице №1.
Формирование пленки осуществляют при повышенной температуре 45-50°C, после чего состав охлаждают до 0°C и сушат в вакууме.
Таким образом, полученная липосомальная пленка содержит от 15 до 25% комплексных солей триазавирина с четвертичными аммониевыми солями и 75-80% липида - фосфатидилхолина. При содержании в пленке солей Триазавирина с четвертичными аммониевыми солями в количестве менее 20 мас.% разница в содержании липидов покрывается за счет добавления холестерина.
Липосомы получают путем последовательного осуществления гидратации липидной пленки в 0,9%-ном растворе хлорида натрия и последующей экструзии через мембрану Anopore с диаметром пор 200 нм, которую проводят при температуре 45-50°C, соответствующей точке плавления липидной пленки. Общее содержание твердых веществ в готовой липосомальной композиции составляет 12,5 г/л, при этом гидратацию пленки проводят непосредственно после ее сушки, так как хранение пленок в течение 2-3 суток может привести к формированию кристаллических дефектов.
Параметры полученных таким способом липосом оценивали с помощью спектроскопии динамического светорассеяния на приборе Malvern Zetasizer Nano SL. Результаты оценки подтвердили, что заявляемая липосомальная композиция, получаемая заявляемым способом, обладает необходимыми для медицинского применения характеристиками, в частности, средний размер липосом составляет 180-190 нм, а индекс полидисперсности 0,10-0,13.
Основные параметры заявляемой липосомальной композиции приведены в Таблице №2.
Осуществление заявляемого изобретения подтверждается примерами конкретного выполнения.
Пример №1.
Получение комплексных солей Триазавирина.
500 мг Триазавирина растворяют в 10 мл воды и добавляют по каплям к 5%-ному водному раствору смеси солей, состоящей из 300 мг цетилпиридиний хлорида и 280 мг гексадецил триметиламмоний хлорида. После непродолжительного перемешивания наблюдается выпадение осадка, который отфильтровывают и сушат при температуре 45-50°C.
Пример №2.
Получение липидной пленки при содержании солей Триазавирина с четвертичными аммониевыми солями менее 20 мас.% (без добавления холестерина).
Навески 250 мг полученного комплекса триазавирина, 750 мг фосфатидилхолина (Эпикурона 200) и 3 мг консерванта, в качестве которого используют метипарабен, растворяют в 75 мл хлороформа. Полученный раствор упаривают на роторном испарителе в круглой колбе объемом 250 мл при средней скорости 50-60 об/мин, вращения ротора и температуре 45-50°C. Полученную пленку быстро охлаждают до 0°C и высушивают в вакууме в течение 6 часов в защищенной от солнечных лучей колбе. После этого готовую пленку хранят в холодильнике при температуре -18°C.
Пример №3.
Получение липидной пленки при содержании солей Триазавирина с четвертичными аммониевыми солями менее 20 мас.% (с добавлением холестерина)
Навески 150 мг полученного комплекса триазавирина, 50 мг холестерина, 800 мг фосфатидилхолина (Эпикурона 200) и 3 мг консерванта, в качестве которого используют метипарабен, и 1 мг смеси токоферолов (Е306) растворяют в 75 мл хлороформа. Полученный раствор упаривают на роторном испарителе в круглой колбе объемом 250 мл при средней скорости 50-60 об/мин, вращения ротора и температуре 45-50°C. Полученную пленку быстро охлаждают до 0°C и высушивают в вакууме в течение 6 часов в защищенной от солнечных лучей колбе. После этого готовую пленку хранят в холодильнике при температуре -18°C.
Пример №4
Гидратация липидной пленки
Липидную пленку после ее получения заливают 50 мл 0,9%-ного раствора хлорида натрия и помещают на перемешивающее устройство типа шейкер. Гидратацию проводят в течение 3 часов при средних оборотах 50-60 об/мин. перемешивающего устройства и температуре 45-50°C. Образовавшуюся суспензию мультиламеллярных везикул центрифугируют для отделения крупных агломератов на оборотах 1500-2000 мин-1.
Пример №5
Формирование липосом.
Полученную суспензию везикул в количестве 20 мл осторожно продавливают через мембранную насадку для ультрафильтрации Anotop 25 с диаметром пор 0,2 мкм при температуре 40°C. Полученный таким способ опалесцирующий коллоидный раствор липосом хранят в защищенном от солнечного света месте при температуре 4°C.
Основные параметры липосомальной композиции (размер липосом и их полидисперсность) не изменялись при хранения в течение 5 месяцев.
| название | год | авторы | номер документа |
|---|---|---|---|
| ЛИПОСОМАЛЬНАЯ КОМПОЗИЦИЯ И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2012 |
|
RU2516893C1 |
| ЛИПОСОМАЛЬНЫЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ СЛАБОКИСЛОТНЫЕ ЛЕКАРСТВЕННЫЕ СРЕДСТВА, И ИХ ПРИМЕНЕНИЕ | 2018 |
|
RU2778886C2 |
| СОЗДАНИЕ ПРОФИЛЯ КОНТРОЛИРУЕМОГО ВЫСВОБОЖДЕНИЯ ЛЕКАРСТВЕННОГО ВЕЩЕСТВА ПРИ ИСПОЛЬЗОВАНИИ ЛИПОСОМНОЙ КОМПОЗИЦИИ В ВОДНЫХ И БЕЗВОДНЫХ РАСТВОРАХ | 2014 |
|
RU2712157C2 |
| СРЕДСТВО ДЛЯ ЛЕЧЕНИЯ ОПУХОЛИ И НАБОР, СОДЕРЖАЩИЙ ЛИПОСОМАЛЬНУЮ КОМПОЗИЦИЮ ГЕМЦИТАБИНА | 2016 |
|
RU2761620C2 |
| УСОВЕРШЕНСТВОВАННЫЕ ЛИПОСОМЫ И ИХ ПРИМЕНЕНИЕ | 2008 |
|
RU2482837C2 |
| СРЕДСТВО ДЛЯ ЛЕЧЕНИЯ ОПУХОЛИ И НАБОР, СОДЕРЖАЩИЙ ЛИПОСОМАЛЬНУЮ КОМПОЗИЦИЮ ГЕМЦИТАБИНА | 2016 |
|
RU2738365C2 |
| Способ получения липидной смеси и липосомальное пероральное противовирусное лечебно-профилактическое средство с использованием указанной липидной смеси | 2020 |
|
RU2746320C1 |
| СРЕДСТВО ДЛЯ ЛЕЧЕНИЯ ОПУХОЛИ И НАБОР, СОДЕРЖАЩИЙ ЛИПОСОМАЛЬНУЮ КОМПОЗИЦИЮ ГЕМЦИТАБИНА | 2016 |
|
RU2768178C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ СЛАБОКИСЛОТНОГО ЛЕКАРСТВЕННОГО СРЕДСТВА И СПОСОБЫ ПРИМЕНЕНИЯ | 2020 |
|
RU2832678C2 |
| ЛИПОСОМАЛЬНОЕ ПЕРОРАЛЬНОЕ ПРОТИВОВИРУСНОЕ ЛЕЧЕБНО-ПРОФИЛАКТИЧЕСКОЕ СРЕДСТВО НА ОСНОВЕ ИНТЕРФЕРОНА-АЛЬФА ЧЕЛОВЕКА | 2008 |
|
RU2361572C1 |
Заявляемое изобретение относится к биотехнологии и химико-фармацевтической промышленности, а именно представляет собой липосомальную фармацевтическую композицию, осуществляющую направленную транспортировку физиологически активных веществ с целью повышения терапевтической активности лекарственных препаратов, а также обеспечивающих высокую эффективность и стабильность активного вещества в липосомах. Благодаря использованию данного изобретения обеспечивается повышение степени включения лекарственного (активного) вещества в структуру липосом и увеличение срока хранения липосомального препарата без существенного изменения степени включения лекарственного (активного) вещества. 2 н. и 5 з.п. ф-лы, 2 табл., 5 пр.
1. Липосомальная фармацевтическая композиция, включающая лекарственное вещество, липиды в виде фосфатидилхолина и холестерина, ионы четвертичного аммония, добавки, согласно изобретению в качестве лекарственного вещества используют натриевую соль 2-метилтио-6-нитро-1,2-4-триазоло[5,1-с]-1,2,4-триазин-7-она, дигидрат, вводимую в липосомы в виде комплексных солей, образованных с четвертичными аммониевыми солями, в качестве которых применяют цетилпиридиний хлорид и гексадецил триметиламмоний хлорид, причем при общем содержании комплексных солей в количестве более 20 мас.% в качестве липидов используют фосфатидилхолин, при содержании менее 20 мас.% - фосфатидилхолин и холестерин, при этом компоненты находятся в определенном соотношении, мас.%:
2. Липосомальная фармацевтическая композиция по п.1, отличающаяся тем, что фосфатидилхолин представляет собой Эпикурон 200.
3. Липосомальная фармацевтическая композиция по п.1, отличающаяся тем, что в качестве добавок используют антиоксиданты и консерванты,
4. Липосомальная фармацевтическая композиция по п.3, отличающаяся тем, что антиоксиданты и консерванты используют при следующем соотношении, мас.%: антиоксиданты - 0,05-0,1; консерванты - 0,1-0,3.
5. Липосомальная фармацевтическая композиция по п.1, отличающаяся тем, что в качестве антиоксидантов применяют смесь токоферолов (Е306), а в качестве консервантов, например, метилпарабен.
6. Способ получения липосомальной фармацевтической композиции по п.1, включающий введение лекарственного вещества в липосому, отличающийся тем, что лекарственное вещество, в качестве которого используют натриевую соль 2-метилтио-6-нитро-1,2-4-триазоло[5,1-c]-1,2,4-триазин-7-она, дигидрат, вводят в липосомы в виде комплексных солей, образованных с четвертичными аммониевыми солями, в качестве которых применяют цетилпиридиний хлорид и гексадецил триметиламмоний хлорид, при этом полученные комплексные соли растворяют в неполярном растворителе и смешивают с липидами и добавками, после чего растворитель удаляют, а полученную в результате липидную пленку охлаждают, высушивают в вакууме и гидратируют до образования суспензии мультиламеллярных везикул, из которой путем ультрафильтрации, проводимой одновременно со стерилизацией при температуре 45-50°C, формируют липосомы.
7. Способ получения липосомальной фармацевтической композиции по п.6, отличающийся тем, что в качестве липидов используют фосфатидилхолин и холестерин.
| ЛИПОСОМЫ, ИСПОЛЬЗУЕМЫЕ ДЛЯ ДОСТАВКИ ЛЕКАРСТВЕННЫХ СРЕДСТВ | 2005 |
|
RU2424792C2 |
| НАТРИЕВАЯ СОЛЬ 2-МЕТИЛТИО-6-НИТРО-1,2-4-ТРИАЗОЛО[5,1-C]-1,2,4-ТРИАЗИН-7(4H)-ОНА, ДИГИДРАТ, ОБЛАДАЮЩАЯ ПРОТИВОВИРУСНОЙ АКТИВНОСТЬЮ | 2005 |
|
RU2294936C1 |
| Karpenko I | |||
| "Antiviral properties, Metabolism, and Pharmocokinetics of a Novel Azolo-1,2,4-Triazine-Derived Inhibitor of Influenza A and B Virus Replication", Antimicrob Agents Chemother, 2010 May; 54(5):2017-2022 | |||

