Настоящее изобретение, рассматриваемое в самом широком смысле, относится к предотвращению формирования и/или стабилизации трехмерных артериальных или венозных тромбов.
В частности, настоящее изобретение относится к применению по меньшей мере одного антитела и/или одного ингибитора для ингибирования активности фактора XII и предотвращению формирования и/или стабилизации тромбов и роста тромбов. Изобретение также относится к фармацевтическому составу и применению фактора XII в качестве антитромботического средства.
Повреждение стенок сосудов немедленно приводит к адгезии и агрегации тромбоцитов, после чего следует активация системы свертывания плазмы и формирование содержащего фибрин тромба, который перекрывает поврежденный участок. Эти события являются критическими для ограничения посттравматической потери крови, но также могут приводить к закупорке пораженных сосудов, что приводит к ишемии и инфаркту жизненно важных органов. В каскадной модели (модели водопада) свертывание крови протекает в виде последовательности реакций, включающих активацию зимогенов ограниченным протеолизом, что в конечном счете приводит к очень быстрому образованию тромбина, который преобразует фибриноген плазмы в фибрин и эффективно активирует тромбоциты. В свою очередь сцепленные с коллагеном или фибрином тромбоциты увеличивают образование фибрина на несколько порядков путем экспонирования прокоагулянта фосфатидилсерина (PS) на своей внешней поверхности, что ускоряет сборку и активацию протеазных комплексов свертывания, и путем прямого взаимодействия между рецепторами тромбоцитов и факторами свертывания.
Для свертывания существуют два сходящихся пути, которые запускаются либо внешними (стенка сосуда) либо внутренними (образованными в крови) компонентами сосудистой системы. "Внешний" путь инициируется комплексом фактора VII (FVII) плазмы и интегрального мембранного белка тканевого фактора (TF), ключевого кофактора свертывания, который отсутствует на поверхности внутренней стороны сосуда, но интенсивно экспрессируется в субэндотелиальном слое сосуда. TF, экспрессируемый в микрополостях кровеносной системы, также может давать вклад в распространение тромба путем поддержки образования тромбина на поверхности активированных тромбоцитов.
"Внутренний" или контактный путь активации инициируется, когда фактор XII (FXII, фактор Хагемана) контактирует с отрицательно заряженными поверхностями в реакции, в которой участвуют высокомолекулярный кининоген и калликреин плазмы крови. FXII может быть активирован макромолекулярными составляющими субэндотелиального матрикса, такими как гликозаминогликаны и коллагены, сульфатиды, нуклеотиды и другие растворимые полианионы, или нефизиологическими материалами, такими как стекло или полимеры. Одним из наиболее эффективных контактных активаторов является каолин, и эта реакция лежит в основе главного клинического теста на свертываемость, (активированное) парциальное тромбопластиновое время (PTT, aPTT). В реакциях, стимулируемых тромбоцитами, активированный FXII затем активирует FXI, a FXIa в свою очередь активирует фактор IX. Независимо от его высокой эффективности при индуцировании свертывания крови in vitro (пато)физиологическая значимость запускаемого фактором FXII внутреннего пути свертывания ставится под сомнения тем фактом, что унаследованный недостаток FXII, а также высокомолекулярного кининогена и калликреина плазмы крови не связан с геморрагическими осложнениями. В сочетании с наблюдениями, выявившими тот факт, что люди и мыши, у которых отсутствуют составляющие внешнего пути, такие как TF, FVII или фактор IX, страдают от острых кровотечений, это приводит к принятой в настоящее время гипотезе, что образование фибрина in vivo инициируется исключительно внешним каскадом (Mackman. N. (2004). Role of tissue factor in hemostasis, thrombosis, and vascular development. Arterioscler. Thromb. Vase. Biol. 24, 1015-1022).
Подобно любому физиологическому механизму, каскад свертывания может быть активирован неверно, что приводит в результате к образованию кровоостанавливающих препятствий в кровеносных сосудах. В результате этого сосуды могут оказаться блокированными, и кровоснабжение дистальных органов может уменьшиться. Этот процесс известен как тромбоэмболия и связан с высокой смертностью. Помимо этого, использование протезирующих устройств, которые находятся в контакте с кровью, является чрезвычайно ограниченным из-за активации каскада коагуляции и образования отложений на поверхности протеза, что часто нарушает его функциональность. Примерами таких протезов являются устройства гемодиализа, кардиопульмональные шунты, сосудистые стенты и постоянные катетеры. При использовании таких устройств для предотвращения осаждения фибрина на поверхность применяются антикоагулянты, такие как гепарин. Однако некоторые пациенты не переносят гепарин, который может вызвать тромбоцитопению (HIT), приводящую к агрегации тромбоцитов и опасному для жизни тромбозу. Помимо этого, все антикоагулянты, используемые в клинике, естественным образом связаны с повышенным риском сильного кровотечения. Таким образом, существует потребность в новых типах антикоагулянтов, которые не связаны с указанными осложнениями и которые могут быть использованы для больных пациентов или для улучшенной терапии, предотвращающей тромбоз без увеличения вероятности возникновения кровотечения.
В этой связи также является очевидной потребность в улучшенных лекарственных средствах для лечения или профилактики тромбозов или сходных расстройств. Таким образом, удовлетворение вышеперечисленных потребностей является задачей, решаемой настоящим изобретением. Уже более пятидесяти лет известно, что недостаток фактора FXII свертывания не связан с увеличением спонтанных или связанных с травмой геморрагических осложнений (Ratnoff, O.D. & Colopy, J.E. (1955) A familial hemorrhagic trait associated with a deficiency of a clot-promoting fraction of plasma. J Clin Invest 34, 602-13). Действительно, хотя они и демонстрируют патологический aPTT (клинический тест на свертываемость, который относится к внутреннему пути коагуляции), люди с дефицитом FXII не страдают от патологических кровотечений даже во время обширного хирургического вмешательства (Colman, R.W. Hemostasis and Thrombosis. Basic principles & clinical practice (eds. Colman R.W., Hirsch. J., Mader V.J., Clowes A.W., & George J.) 103-122 (Lippincott Williams & Wilkins, Philadelphia, 2001). Напротив, дефицит FXII связывают с повышенным риском венозного тромбоза (Kuhli, С, Scharrer, I., Koch, F., Ohrloff, C. & Hattenbach, L.O. (2004) Factor XII deficiency: a thrombophilic risk factor for retinal vein occlusion. Am. J. Ophthalmol. 137, 459-464., Halbmayer, W.M., Mannhalter, C, Feichtinger, C, Rubi, K. & Fischer, M. (1993) Factor XII (Hageman factor) deficiency: a risk factor for development of thromboembolism. Incidence of factor XII deficiency in patients after recurrent venous or arterial thromboembolism and myocardial infarction. Wien. Med. Wochenschr. 143, 43-50). Исследования и истории болезней, подтверждающие это предположение, ссылаются на индексный случай дефицита фактора FXII, случай Джона Хагемана, умершего от легочной эмболии. Гипотеза, что дефицит фактора FXII связан с повышенным риском тромбообразования, ставится под сомнение недавним пересмотром нескольких случаев, в которых сообщается о связи дефицита фактора FXII с тромбозом (Girolami, A., Randi, M.L, Gavasso, S., Lombardi, A.M. & Spiezia, F. (2004) The Occasional Venous Thromboses Seen in Patients with Severe (Homozygous) FXII Deficiency are Probably Due to Associated Risk Factors: A Study of Prevalence in 21 Patients and Review of the Literature. J. Thromb. Thrombolysis 17, 139-143). В большинстве случаев авторы в сочетании с дефицитом фактора FXII идентифицировали сопутствующие врожденные или приобретенные факторы риска тромбообразования, которые могли бы быть ответственными за случаи тромбообразования независимо от FXII. Наиболее обширные эпидемиологические исследования с привлечением хорошо охарактеризованных пациентов (Koster, Т., Rosendaal, F.R., Briet, E. & Vandenbroucke, J.P. (1994) John Hageman′s factor and deep-vein thrombosis: Leiden thrombophilia Study. Br. J. Haematol. 87, 422-424) и семей с дефицитом фактора FXII (Zeerleder, S. et al. (1999) Reevaluation of the incidence of thromboembolic complications in congenital factor XII deficiency-a study on 73 subjects from 14 Swiss families. Thromb. Haemost. 82, 1240-1246) выявили отсутствие корреляции между дефицитом фактора FXII и риском тромбообразования или риском отсутствия образования тромбов.
Неожиданно и вопреки всем сложившимся представлениям специалистов заявители обнаружили, что внутренний путь свертывания крови, связанный с фактором XII, является существенным для формирования тромба in vivo, но не является необходимым для нормального тканеспецифического гемостаза. Эти результаты изменяют давно устоявшуюся концепцию, заключающуюся в том, что свертываемость крови in vivo опосредована исключительно внешним путем, и указывают на фактор XII как играющий ключевую роль в процессе патологического образования тромбов.
Соответственно, первой задачей, решаемой настоящим изобретением, является применение по меньшей мере одного антитела и/или по меньшей мере одного ингибитора для ингибирования фактора XII и предотвращения образования и/или стабилизации трехмерных артериальных или венозных тромбов. Анти-FXII антитело или соответствующий ингибитор может функционировать таким образом, чтобы ингибировать активацию FXII и/или интерферировать с другими частями молекулы FXII, которые в существенной степени вовлечены в активацию FXII.
Вместе с тем фактом, что внутренний путь не является необходимым для гемостаза, это указывает на фактор XII как на новую мишень для эффективной антитромботической терапии. Помимо этого, указанные результаты являются важными для разработки анти-FXII агентов, для контроля других (пато)механизмов, связанных с системой контакта, таких как воспаление, активация комплемента, фибринолиз, ангиогенез и образование кинина.
Таким образом, настоящее изобретение дополнительно предоставляет применение такого антитела и/или ингибитора для лечения или профилактики состояния или расстройства, связанного с образованием артериального тромба, т.е. инсульта или инфаркта миокарда, воспаления, активации комплемента, фибринолиза, ангиогенеза и/или заболеваний, связанных с патологическим образованием кинина, таких как гипотонический шок, отек, включая наследственный ангионевротический отек, бактериальные инфекции, артриты, панкреатиты или суставная подагра.
В частности, применение по меньшей мере одного анти-FXII антитела (например, антитела F1 (MoAb F1, Ravon etal., Blood. 1995 Dec 1; 86 (11): 4134-43)) и/или применение по меньшей мере одного ингибитора протеазы для ингибирования образования тромба, опосредованного FXII, также является объектом настоящего изобретения.
Особенно предпочтительным является ингибитор протеазы, выбранный, например, из следующей группы: ингибитор AT III, ингибитор ангиотензин-превращающего фермента, ингибитор С1, апротинин, ингибитор протеазы альфа-1, антиболевое средство ([(S)-1-карбокси-2-фенилэтил]-карбамоил-L-Arg-L-Val-аргинал), Z-Pro-Pro-альдегид-диметилацетат, DX88 (Dyax Inc., 300 Technology Square, Cambridge, MA 02139, USA; cited in: Williams A. and Baird LG., Transfus Apheresis Sci. 2003 Dec: 29 (3): 255-8), леупептин, ингибиторы пролиловой лигопептидазы, такие как Fmoc-Ala-Pyr-CN, ингибитор трипсина из кукурузы, мутанты бычьего панкреатического ингибитора трипсина, экотин, YAP (антикоагулянтный белок желтоперой камбалы (Yellowfin sole)) и ингибитор-V трипсина Curcurbita maxima, включая изоингибиторы Curcurbita maxima.
Соответственно, настоящее изобретение предоставляет применение такого антитела и/или ингибитора, описанного в настоящем документе, в медицине, а также применение такого антитела и/или ингибитора для изготовления лекарственного средства.
Таким образом, в соответствии другим с аспектом настоящего изобретения предоставляется фармацевтический состав, содержащий по меньшей мере одно антитело и/или один ингибитор, который подходит для ингибирования фактора XII и который предотвращает формирование и/или стабилизацию трехмерных венозных или артериальных тромбов.
В частности, антитело, используемое для фармацевтического состава, представляет собой анти-FXII антитело (например, такое как F1 антитело (MoAb F1, Ravon et al., Blood. 1995 Dec 1;86(11):4134-43)), и ингибитор представляет собой ингибитор протеазы, выбранный, например, без ограничения, из следующей группы: ингибитор AT III, ингибитор ангиотензин-превращающего фермента, ингибитор C1, апротинин, ингибитор протеазы альфа-1, антиболевое средство ([(S)-1-карбокси-2-фенилэтил]-карбамоил-L-Arg-L-Val-аргинал), Z-Pro-Pro-альдегид-диметилацетат, DX88 (Dyax Inc., 300 Technology Square, Cambridge, MA 02139, USA; cited in: Williams A. and Baird LG., Transfus Apheresis Sci. 2003 Dec: 29 (3): 255-8), леупептин, ингибиторы пролилолигопептидазы, такие как Fmoc-Ala-Pyr-CN, ингибитор трипсина из кукурузы, мутанты бычьего панкреатического ингибитора трипсина, экотин, YAP (антикоагулянтный белок желтоперой камбалы (Yellowfin sole)) и ингибитор-V трипсина Curcurbita maxima, включая изоингибиторы Curcurbita maxima.
Антитело также может представлять собой его фрагмент или миметик, сохранивший ингибирующую активность, например аналоги домена ингибитора протеаз типа Куница белка-предшественника амилоида, описанные в патенте US 6,613,890 (см., в частности, колонки 4-8). Другим подходящим ингибитором может быть Hamadarin, раскрытый Harahiko Isawa et al. в The Journal of Biological Chemistry, Vol.277, No.31 (August 2, pp.27651-27658, 2002). Подходящий ингибитор трипсина из кукурузы и способы его получения раскрыты у Zhi-Yuan Chen et al., Applied and Environmental Microbiology, March 1999, p.1320-1324 (см. также ссылку 19, указанную в этом документе). Содержание всех перечисленных выше документов включено в настоящую заявку в полном объеме. В заключение следует особо отметить, что малые молекулы, выделенные, например, с использованием соответствующего ингибирования FXIIa в качестве анализа, на котором основан выбор, также являются частью изобретения, а также соответствующее их применение, описанное выше или ниже. Указанные низкомолекулярные ингибиторы FXIIa могут быть разработаны, исходя из кристаллической структуры FXII. Таким образом, несколько доменов FXII или легкая цепь могут быть рекомбинантно экспрессированы в такой экспрессионной системе, как E.coli, дрожжи, или клетки млекопитающих. Затем белок очищают и кристаллизуют с использованием стандартных процедур, как описано для FXI субстрата FXII (Jin L, et al. (2005) Crystal structures of the FXIa catalytic domain in complex with ecotin mutants reveal substrate-like interactions. J Biol Chem. 280(6):4704-12). В качестве альтернативы могут быть включены низкомолекулярные ингибиторы сериновой протеазы для стабилизации структуры FXII. Такие препараты, содержащие низкомолекулярные ингибиторы белковых мишеней, которые могут быть разработаны, например, с учетом кристаллической структуры этих целевых белков, хорошо известны в данной области техники и включают фармацевтические препараты, которые могут быть, например, введены пациенту системно, например парентерально, перорально или местно.
Термин "парентерально", как он используется в настоящем документе, включает подкожные, внутривенные, внутримышечные, внутриартериальные и интратрахеальные инъекции, инстилляцию, введение в виде спрея и инфузию. Парентеральные составы предпочтительно вводятся внутривенно, либо в форме болюсов, либо в форме постоянной инфузии, либо подкожно, в соответствии и известными процедурами. Предпочтительные жидкие носители, которые хорошо известны для использования при парентеральном введении, включают стерильную воду, физиологический раствор, водную декстрозу, растворы сахаров, этанол, гликоли и масла.
Таблетки и капсулы для перорального введения могут содержать общеизвестные вспомогательные вещества, такие как связующие агенты, наполнители, лубриканты, смачивающие агенты и т.п. Пероральные жидкие составы могут быть в форме водных или масляных суспензий, растворов, эмульсий, сиропов, эликсиров, и т.п. или могут быть представлены в виде сухого продукта, восстанавливаемого водой или другим подходящим носителем для использования. Такие жидкие составы могут содержать общеизвестные добавки, такие как суспендирующие агенты, эмульгаторы, неводные носители и консерванты.
Составы, подходящие для местного применения, могут быть в форме водных или масляных суспензий, растворов, эмульсий, гелей или, предпочтительно, эмульсионных мазей. Составы, подходящие для нанесения в виде спрея, могут быть в форме распыляемой жидкости или сухого порошка.
В соответствии с третьим аспектом настоящего изобретения предоставляется применение фактора XII в качестве антитромботической мишени путем ингибирования фактора XII по меньшей мере одним антителом и/или одним ингибитором и предотвращения, таким образом, образования и/или стабилизации трехмерных тромбов в сосудах.
Сущность настоящего изобретения, обеспечиваемые им преимущества и его дополнительные признаки изложены в нижеследующем подробном описании выполненных экспериментов и их результатов, которое следует изучать совместно с прилагаемыми чертежами, описанными ниже.
При анализе функций внутреннего каскада коагуляции в гемостазе и тромбозе использовали мышей с дефицитом фактора XII. Интравитальная флуоресцентная микроскопия и ультразвуковое измерение параметров потока выявили сильные нарушения в образовании и стабилизации трехмерных тромбов в различных артериальных отделах сосудистой системы. Введение мутантным мышам человеческого фактора XII восстанавливало внутренний путь коагуляции in vitro и формирование артериальных тромбов in vivo. С точки зрения механики процесса, прокоагулянтная активность внутреннего пути в существенной степени опосредована активированными тромбоцитами. Эти результаты указывают на внутренний каскад свертывания крови, инициируемый FXII, как на ключевой элемент в процессе формирования артериальных тромбов, связывающий плазменную коагуляцию с агрегацией тромбоцитов.
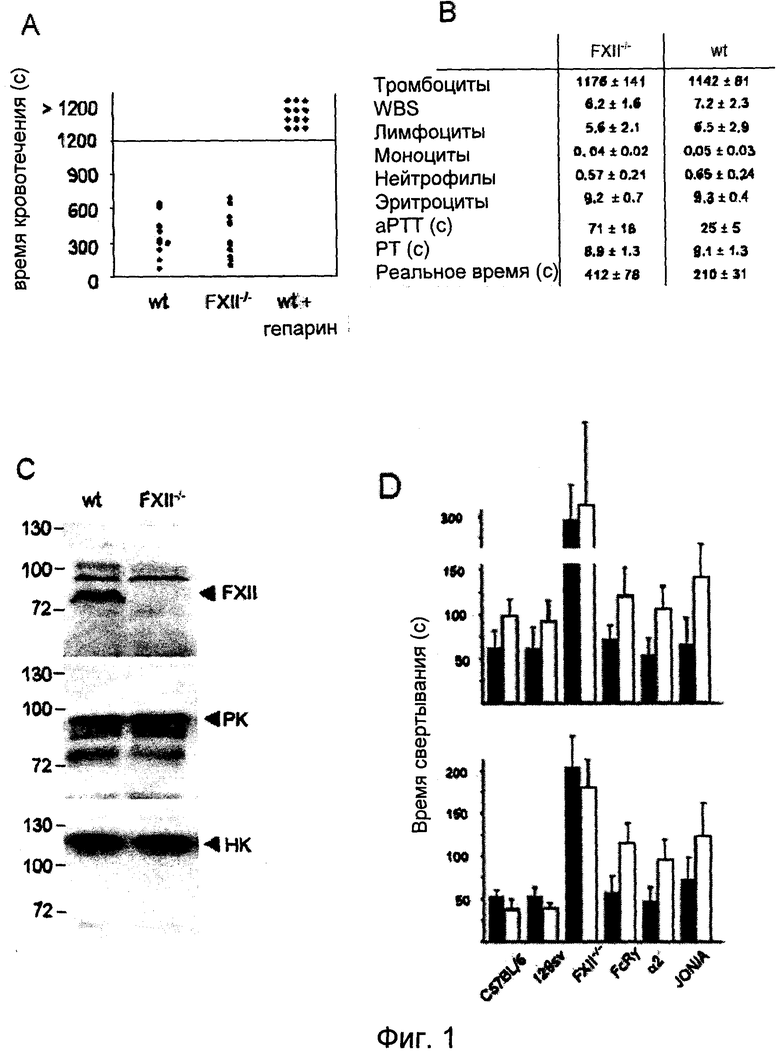
На Фиг.1 представлен анализ свертывания крови у мышей, дефицитных по FXII: (A) Время кровотечения из хвостовой вены у мышей дикого типа (n=12) и FXII-/- мышей (n=11). Каждый символ представляет одну особь. (B) Измерение периферийной крови в тыс./мкл и параметры глобального свертывания для мышей дикого типа и FXII-/- мышей. Расшифровка аббревиатур: количество лейкоцитов (WBC), активированное парциальное тромбопластиновое время (aPTT) и протромбиновое время (PT). Значения представляют собой средние ±SD для 10 мышей каждого генотипа. (C) Белки FXII контактной системы, калликреин плазмы (PK) и высокомолекулярный кининоген (HK) в 0,3 мкл плазмы мышей дикого типа и FXII-/- мышей, определенные методом вестерн-блоттинга с использованием специфических антител. Стандарт молекулярного веса приведен слева. (D) Время свертывания при рекальцификации определяли для плазмы без тромбоцитов (верхняя часть) и обогащенной тромбоцитами плазмы (нижняя часть), полученной от мышей C57BL/6 и 129sv wt, FXII-/-, FcRγ-/- и дефицитных по интегрину α2, после активации каолином (темные столбцы) или коллагеном (светлые столбцы). Эффект JON/A анализировали на плазме C57BL/6 с добавлением 50 мкг/мл антитела. Приводятся средние±STD по 6 экспериментам.
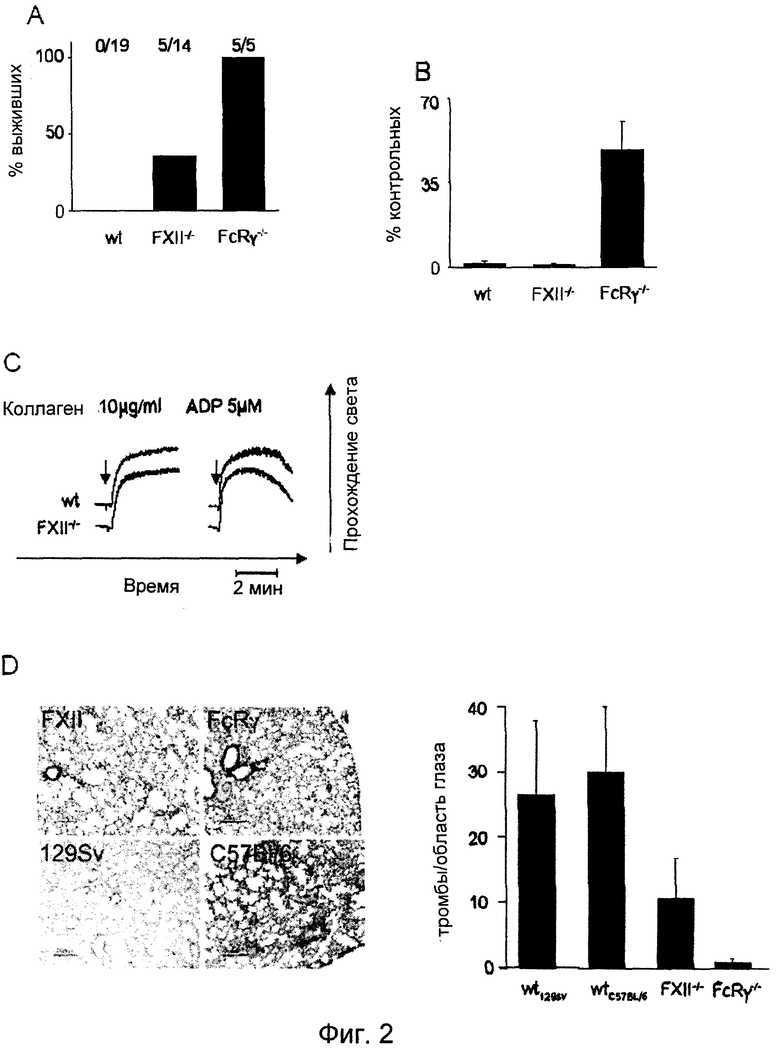
Фиг.2. (A) Наблюдали смертность вследствие тромбоэмболических осложнений, возникших после внутривенной инъекции коллагена (0,8 мг/кг) и эпинефрина (60 мкг/кг). Мыши дикого типа погибали в течение 5 мин. Животные, оставшиеся в живых спустя 30 мин после введения, расценивались как выжившие. (B) Количество тромбоцитов у контрольных (n=19), FXII-/- (n=14) и FcRγ-/- (n=5) мышей спустя 2 мин после инфузии коллагена/эпинефрина. (C) Гепаринизированную обогащенную тромбоцитами плазму, полученную от мышей дикого типа и FXII-/- мышей, стимулировали коллагеном (10 мкг/мл) или ADP (5 мкМ) и регистрировали величину светопроницаемости в стандартном агрегометре. Представленные результаты получены для групп из шести мышей. (D) Окрашенные гематоксилином/эозином срезы легких указанных мышей через 2 мин после инъекции коллагена/эпинефрина. Количество тромбов в поле зрения подсчитывали при увеличении 20×. Столбцы представляют средние ±SDT по 100 полям зрения.
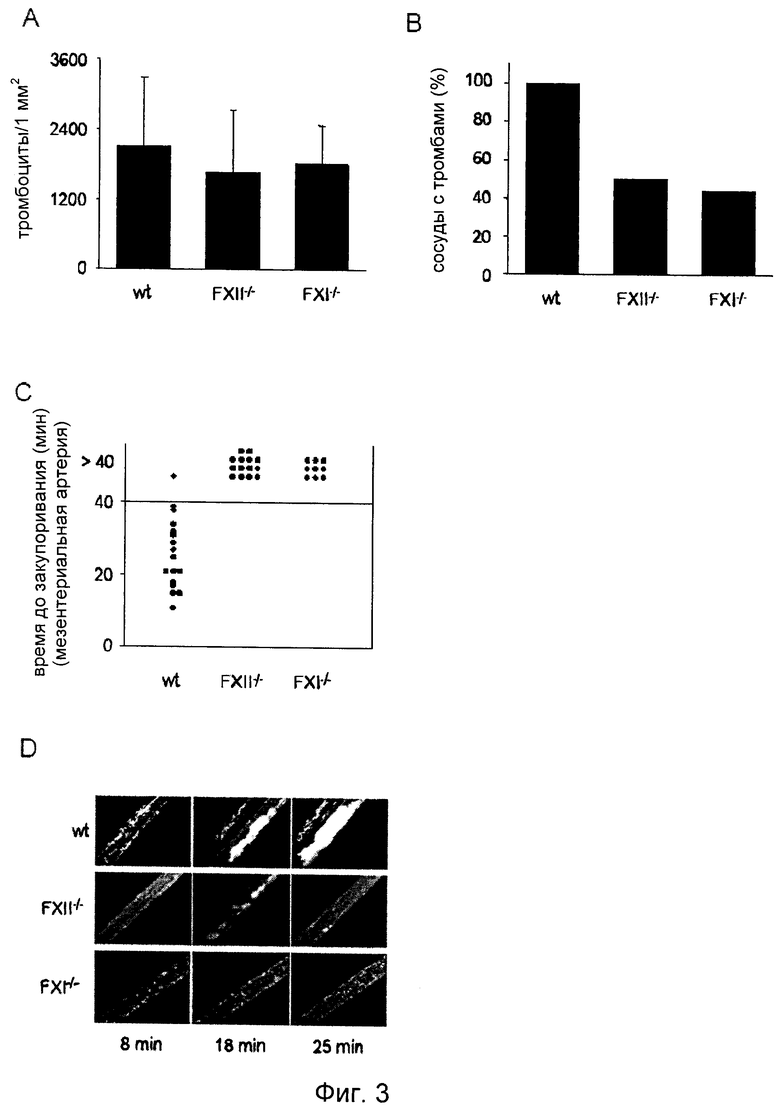
На Фиг.3 показано образование in vivo дефектного тромба у мыши без фактора XII. Образование тромба наблюдали in vivo в мезентериальных артериолах при поражении, вызванном 20% FeCl3. (А) Адгезию единичных тромбоцитов обнаружили у всех линий мышей спустя 5 мин после нанесения повреждения, при этом спустя 7-8 мин после нанесения повреждения наблюдали первые тромбы у мышей дикого типа, в то время как у FXII-/-мышей первые тромбы появлялись спустя 14-35 мин после нанесения повреждения, а у FXI-/- мышей спустя 5-35 мин после нанесения повреждения. (В) Образование тромбов в мезентериальных артериях наблюдали в 100% случаях у мышей дикого типа, но только в 50% случаях у FXII-/- мышей и в 44,4% случаях у FXI-/- мышей. (C) Тромбы, образовавшиеся у мышей дикого типа, закупоривали сосуд в среднем спустя 25 мин после нанесения повреждения, тогда как тромбы, образовавшиеся у мышей, дефицитных по FXII-/- и FXI-/-, не приводили к закупориванию. Каждый символ представляет одну наблюдавшуюся артериолу. (D) Репрезентативные фотографии одного эксперимента.
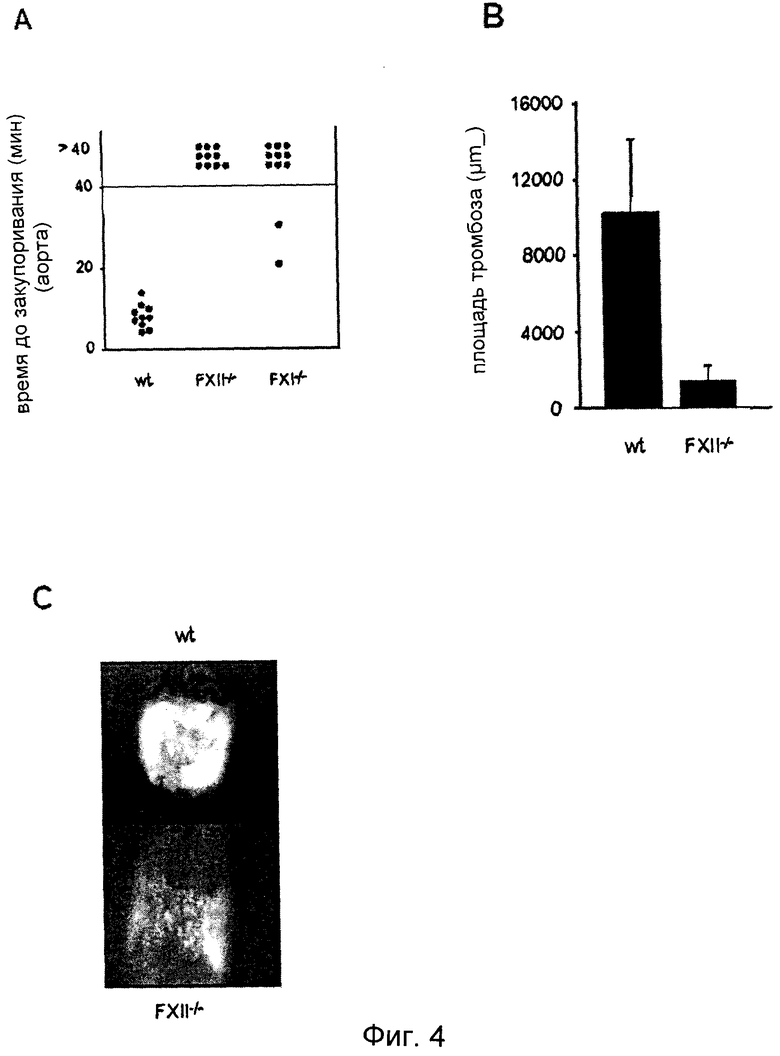
Фиг.4. (A) Мышей дикого типа (n=10), FXII-/- (n=10) и FXI-/- (n=11) анализировали на модели закупоривания артерии. Тромбоз индуцировали в аорте посредством одного плотного сжатия щипцами. Ток крови отслеживали с помощью периваскулярного ультразвукового датчика потока до полного закупоривания. Эксперимент прекращали через 40 мин. Каждый символ представляет одну особь. (B) Механическое повреждение сонной артерии инициировали наложением лигатуры. После удаления нити измеряли в мкм2 площадь тромба у мышей дикого типа (n=10) и FXII-/- (n=10). (C) На фотомикрографиях представлены репрезентативные изображения спустя 2 мин после нанесения повреждения.
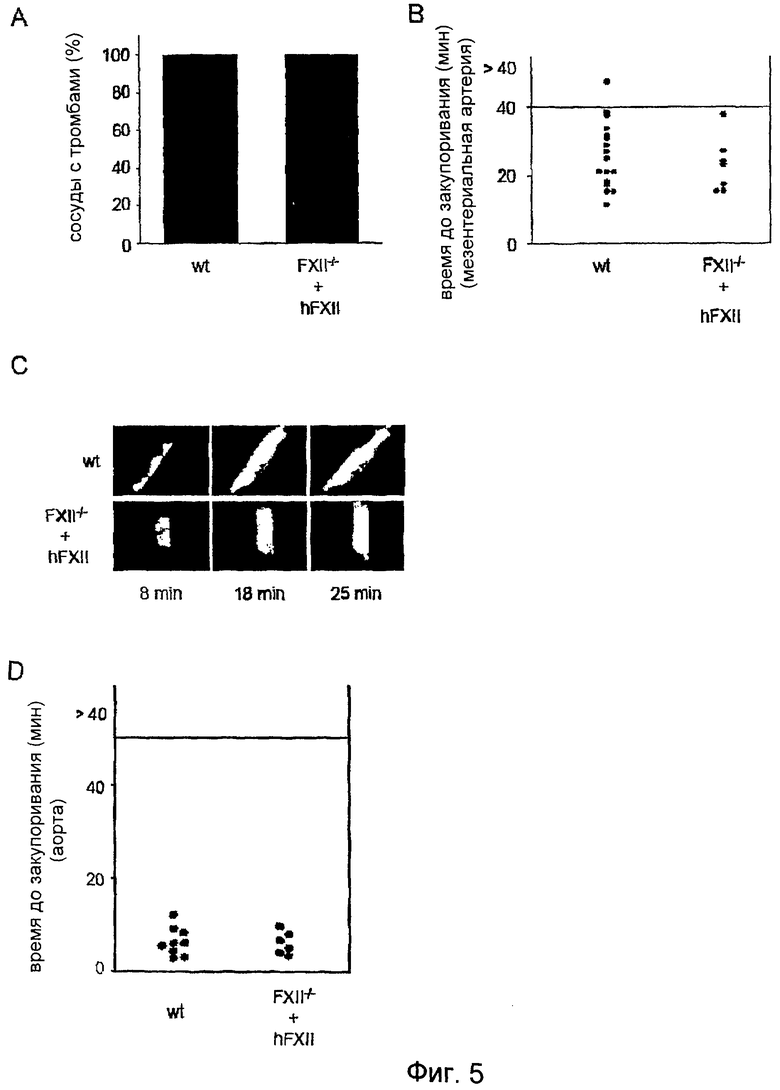
На Фиг.5 показан дефект образования тромба у животных, дефицитных по FXII, которым был введен человеческий FXII. (A) Образование тромба после повреждения, индуцированного FeCl3, наблюдали в мезентериальных артериях в 100% случаях как у мышей дикого типа, так и у FXII-/- мышей, которым был инъецирован человеческий FXII. (B) Формирующиеся тромбы закупоривали сосуд в среднем спустя 25 мин после нанесения повреждения у мышей дикого типа и спустя 22,7 мин после нанесения повреждения у FXII-/- мышей, которым был инъецирован человеческий FXII. Каждый символ представляет одну особь. (C) Представлены репрезентативные фотографии. (D) FXII-/- мыши получали 2 мг/кг hFXII-/-, и в аорте индуцировали тромбоз посредством одного плотного сжатия щипцами. Ток крови отслеживали с помощью периваскулярного ультразвукового датчика потока до полного закупоривания. Эксперимент прекращали через 40 мин. Каждый символ представляет одну особь.
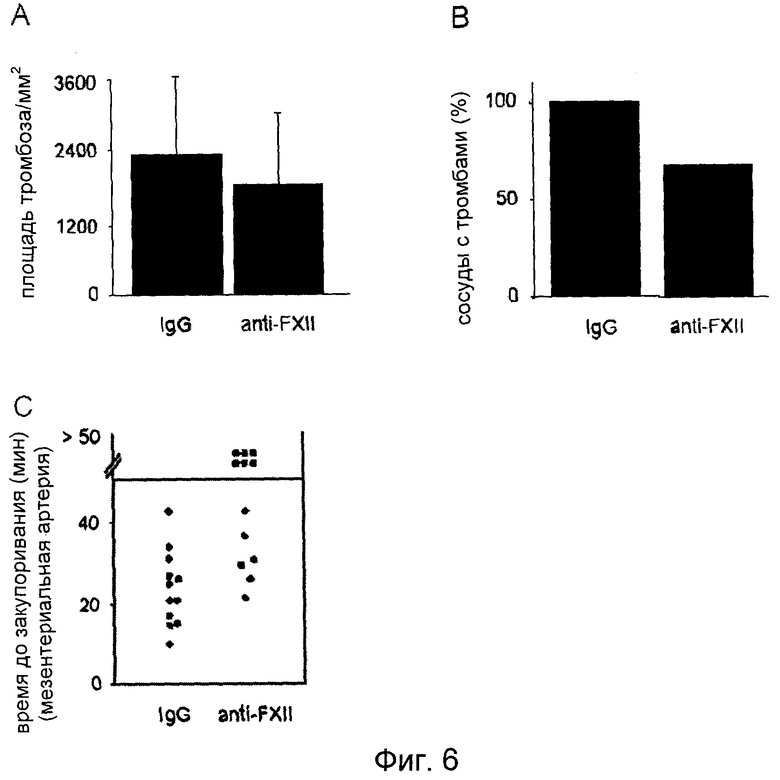
На Фиг.6 показано in vivo ингибированное анти-FXII антителами образование тромба у мыши. Мыши дикого типа получали 2 мг/кг анти-FXII антител или нечеловеческого IgG i.v. Спустя 15 мин проводили мониторинг in vivo образования тромба в мезентериальных артериолах при поражении, вызванном 20% FeCl3. (A) Адгезия единичных тромбоцитов была обнаружена у обоих групп спустя 5 мин после нанесения повреждения. Спустя 7-8 минут наблюдали первые тромбы у мышей контрольной группы, обработанной IgG, тогда как у мышей, обработанных анти-FXII антителами, первые тромбы появились спустя 12-32 минуты после нанесения повреждения. (B) Образование тромбов наблюдали в мезентериальных артериях в 100% случаях у контрольных мышей, но только в 60% случаях у мышей, обработанных анти-FXII антителами. (C) Показано время до полного закупоривания. Каждый символ представляет одну особь.
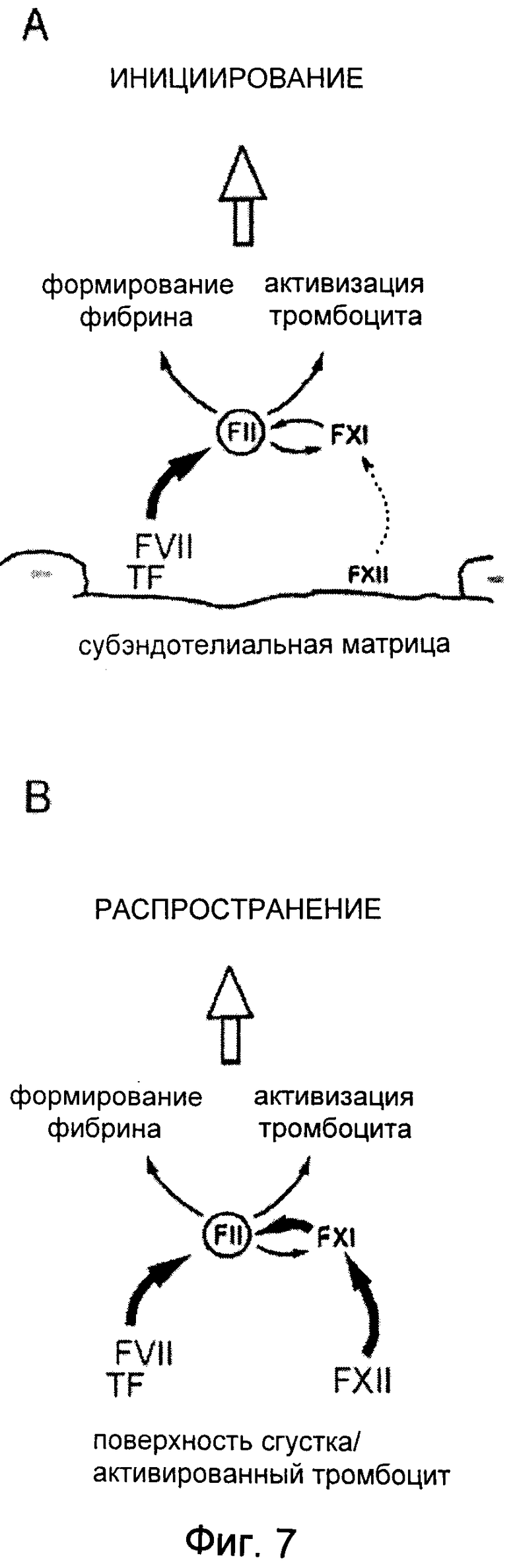
На Фиг.7 показана пересмотренная модель формирования артериального тромба. Сначала на участках очагов повреждений сосудов происходит образование тромбина вследствие экспозиции тканевого фактора (TF) в субэндотелиальном матриксе. TF в комплексе с FVII инициирует внешний путь свертывания крови. На участке повреждения вклад FXII, опосредующего внутренний путь через FXI с образованием тромбина (FII), является незначительным, и им можно пренебречь в случае нормального гемостаза. Соответственно, индивиды с дефицитом FXII не страдают от кровотечений. Образование тромбина инициирует формирование сгустка путем образования фибрина и активации тромбоцитов. Рост тромба: на поверхностях, открытых к растущему тромбу, индуцированный FXII внутренний путь вносит существенный вклад в образование тромбина. Активированный FXII с участием FXI образует дополнительный фибрин. Соответственно, дефицит как по FXII, так и по FXI вызывает серьезное нарушение процесса формирования тромбов.
В настоящем изобретении потенциальный вклад внутреннего пути свертывания при патологическом образовании тромбов in vivo оценивали с помощью моделей артериального тромбоза, основанных на интравитальной микроскопии и измерении параметров потока, с использованием мышей без фактора XII. Несмотря на то что начальная адгезия тромбоцитов на участках повреждений у мутантных животных не меняется, последующее формирование и стабилизация трехмерных тромбов сопровождается серьезными нарушениями. Это нарушение наблюдалось в различных ветвях сосудистой сети и полностью устранялось с помощью экзогенного фактора XII человека. Эти факты указывают на опосредованный фактором XII внутренний путь свертывания крови как на основное звено, связывающее первичный и вторичный гемостаз в пересмотренной модели образования тромбов.
Для анализа функций FXII при свертывании крови in vivo выводили мышей с дефицитом FXII. FXII-/- мыши являлись здоровыми особями, бесплодными и фенотипически неотличимыми от однопометных животных дикого типа. Подробный гистологический и гемостазиологический анализ не выявил корреляций с увеличенным тромбообразованием и кровотечением у FXII-/- мышей, несмотря на увеличенное aPTT, равное 68±17 сек, и время рекальцификации, равное 412±78 сек, плазмы, отобранной из ретроорбитального синуса (дикий тип: 23±4 и 210±31 сек) (Pauer.H.U., et al. (2004). Targeted deletion of murine coagulation factor XII gene-a model for contact phase activation in vivo. Thromb. Haemost. 92, 503-508). Так же как и люди с дефицитом FXII, FXII-/- мыши не подвержены кровотечениям, что подтверждается временем кровотечения при надрезе хвоста, сходным с таковым у животных дикого типа (369.5±201,7 и 355,9±176,1 сек соответственно n=12 на группу, Фиг.1A). Количество клеток периферийной крови у мутантных мышей не отличалось от контроля дикого типа. Следует отметить, что протромбиновое время (PT) у FXII-/- мышей и у мышей дикого типа было схожим (8,9±1,3 и 9,1±1,3 сек), что указывает на то, что недостаток FXII не оказывает негативного влияния на образование фибрина внешней системой свертывания крови (Фиг.1B). Для оценки потенциальных отличий прокоагулянтной активности FXII между человеком и мышью плазму человека с дефицитом FXII (FXII<1%) восстанавливали плазмой мышей дикого типа, или наоборот, и определяли PTT смесей. В каждом случае отмечалась нормализация процесса формирования сгустка, что подтверждает предположение о том, что функция FXII при образовании сгустка является сопоставимой у человека и мыши.
У человека, аналогично случаю дефицита FXII, дефицит белков контактной системы: калликреина плазмы (PK) и высокомолекулярного кининогена (HK), не приводит к повышенному риску кровотечения, несмотря на увеличенное aPTT. Для подтверждения того, что увеличение aPTT у FXII-/- мышей не является следствием дополнительных дефектов белков контактной фазы, был проведен анализ PK и HK плазмы мышей дикого типа и FXII-/-. По данным вестерн-блот анализа уровни HK и PK были эквивалентны у мутантных мышей и мышей дикого типа (Фиг.1C). Функционально, в FXII-/- плазме, подвергнутой воздействию коллагена или каолина, обработка и образование тромбина были серьезно нарушены по сравнению с диким типом.
Свертывание крови и активация тромбоцитов представляют собой комплементарные и взаимозависимые процессы. Тромбоциты взаимодействуют с несколькими факторами свертывания крови и вносят вклад в их активацию, и основной продукт свертывания, тромбин, является мощным активатором тромбоцитов. В этой связи было проведено более подробное изучение вклада тромбоцитов и FXII в формирование сгустка. С этой целью инициировали свертывание, используя либо каолин, который классически активирует FXII, но не оказывает непосредственного воздействия на тромбоциты, либо коллаген, который активирует как FXII, так и тромбоциты, где он взаимодействует со множеством рецепторов, наиболее важными из которых являются интегрин α2β1 и GPVI. В случае присутствия, но не в случае отсутствия тромбоцитов, коллаген очень хорошо подходит для формирования сгустка в плазме дикого типа (Фиг.1D). Напротив, в плазме, содержащей дефектные по активации FcRγ-/- тромбоциты, относительная эффективность коалина и коллагена была похожа на PFP (плазма без тромбоцитов), и схожий эффект наблюдался с PRP (плазма обогащенная тромбоцитами) из интегрина α2-/- мышей. Прокоагулянтная активность тромбоцитов также эффективно запускалась в свертывающейся плазме, и было показано, что фибриновый (фибриногеновый) рецептор αllbβ3 играет ключевую роль в этом процессе, хотя лежащие в его основе механизмы не вполне понятны. В полном соответствии с этими результатами антитело JON/A, блокирующее функции αllbβ3, в существенной степени ингибировало зависящее от тромбоцитов сокращение времени свертывания (Фиг.1D). Вместе эти результаты демонстрируют, что тромбоциты в прокоагулянтном состоянии могут опосредовать индуцированное FXII формирование сгустка.
Для определения, имеет ли индуцированная коллагеном активация FXII функциональные последствия in vivo, на мышах дикого типа и FXII-/- мышах реализовывали модель летальной легочной тромбоэмболии, индуцированной инфузией смеси коллагена (0,8 мг/кг массы тела) и эпинефрина (60 мкг/кг массы тела). Все контрольные мыши (19/19) погибали в течение 5 минут от обширного тромбоза легких и остановки сердца, что сопровождалось более чем 95% уменьшением количества тромбоцитов в кровотоке спустя 2 мин после оказания воздействия (Фиг.2A, 2B). В этих экспериментальных условиях выживало 35,7% (5/14) FXII-/- мышей, хотя количество их периферийных тромбоцитов также уменьшалось, как и у контрольных мышей дикого типа, что указывает на то, что наблюдаемая защита не основана на дефекте активации тромбоцитов. Это предположение было подтверждено исследованиями in vitro, показавшими, что FXII-/- тромбоциты экспрессируют нормальные уровни основных поверхностных гликопротеинов, включая рецепторы коллагена, и что указанные клетки являются нормально активируемыми классическими агонистами, такими как тромбин, аденозиндифосфат (АДФ) или GPVI-специфический агонист, родственный коллагену пептид (измерено активацией экспрессии αllbβ интергина и P-селектина). В полном соответствии с этими результатами, FXII-/- тромбоциты демонстрируют неизмененный агрегационный ответ на коллаген, АДФ (Фиг.2C), PMA или тромбин.
В параллельной серии экспериментов FcRγ-/- мыши подвергались воздействию коллагена/эпинефрина. Эти мыши были полностью защищены от смертельного исхода и количество тромбоцитов не очень сильно уменьшалось спустя 2 минуты после воздействия, что подтверждает обязательность активации тромбоцитов для обеспечения смертельного исхода в этой модели. Эти данные были дополнительно подтверждены результатами гистологических анализов срезов легких, полученных от мышей различных групп. Хотя у мышей дикого типа подавляющее количество сосудов было закупорено, этот эффект был в существенной степени снижен у FXII-/- мышей (выживших и погибших). В полном соответствии с предыдущими публикациями, в легких FcRγ-/- практически не было обнаружено тромбов (Фиг.2D). Эти результаты указывают на то, что in vivo коллаген запускает как активацию тромбоцитов, так и опосредованный FXII внутренний путь свертывания крови, которые в данной модели действуют синергически, формируя легочные тромбы.
Патологическое образование тромбов часто инициируется разрывом или внезапным разрушением атеросклеротической бляшки в артериальной ветви сосудистой сети, что приводит к нефизиологически сильной активации тромбоцитов и прокоагулянтной активности на поверхности субэндотелиальных слоев. Для оценки роли FXII в этих процессах изучали формирование тромбов у мышей дикого типа и FXII-/- мышей, используя различные модели артериальных повреждений. В первой модели индуцировали окислительное повреждение в мезентериальных артериолах (60-100 мкм в диаметре) и исследовали формирование тромбов с помощью in vivo флуоресцентной микроскопии. Мыши дикого типа и FXII-/- мыши получали меченные флуоресцентными метками тромбоциты (1×108) такого же генотипа, и повреждение индуцировали путем топического наложения на 1 минуту фильтровальной бумаги, пропитанной 20% хлоридом железа (FeCl3), что вызывало образование свободных радикалов, ведущее к разрушению эндотелия. Взаимодействие тромбоцитов с поврежденной стенкой сосуда начиналось очень быстро, и спустя пять минут после нанесения повреждения количество надежно прикрепленных тромбоцитов было одинаковым у обеих групп мышей (Фиг. 3A). Однако хотя у мышей дикого типа прикрепившиеся тромбоциты непрерывно мобилизовали дополнительные тромбоциты из кровотока, в результате чего формировались агрегаты, этот процесс был сильно искажен у мутантных мышей. В контрольных сосудах в 100% случаях (17/17) стабильные тромбы диаметром >20 мкм формировались в течение 10 минут после нанесения повреждения и со временем стабильно росли, приводя в конечном счете к полному закупориванию сосудов в 94,1% (16/17) случаях в течение времени наблюдения 40 минут (среднее время закупоривания 25,6±8,9 мин) (Фиг.3). Напротив, у мутантных мышей формирование микроагрегатов или тромбов полностью отсутствовало в 50% (7/14) сосудов. В оставшихся 50% (7/14) сосудов сформировались тромбы, которые, однако, были нестабильными и быстро отделялись от стенки сосуда. Ни в одном из сосудов тромбы с диаметром >20 мкм не оставались прикрепленными к участку поражения дольше 1 минуты. Соответственно, у FXII-/- мышей ни один из сосудов не закупорился за период наблюдения (40 минут). Этот неожиданный результат демонстрирует, что FXII необходим для возникновения и стабилизации обогащенных тромбоцитами тромбов в артериолах, поврежденных FeCl3, и указывает на то, что индуцируемый FXII путь свертывания крови вносит существенный вклад в наблюдаемый тромботический ответ. Это предположение было подтверждено при анализе мышей, дефицитных по FXI, в той же самой модели. Поскольку FXI представляет собой основной субстрат для FXII во "внутреннем" каскаде, у этих мышей также должен наблюдаться похожий дефект образования тромбов. Действительно, практически так же, как и у FXII-/- мышей, по существу нормальная адгезия тромбоцитов на участке повреждения наблюдалась в течение первых трех минут после нанесения повреждения, тогда как образование тромбов было полностью подавлено в сосудах в 55.6% (5/9) случаях. В остальных сосудах сформировавшиеся микроагрегаты и тромбы были нестабильными и непрерывно эмболизировались. В результате ни один из сосудов не закупорился за период наблюдения (40 минут). Эти данные показывают, что мыши, дефицитные по FXI, защищены в модели индуцированного FeCl3 закупоривания сонной артерии.
Известно, что индуцированное FeCl3 формирование артериальных тромбов зависит от тромбоцитов и образования тромбина, но остается неясным, насколько хорошо этот тип повреждения имитирует среду, возникающую в пораженных сосудах, например, при разрыве атеросклеротической бляшки. В этой связи, для исключения возможности того, что массивное индуцированное FeCl3 окислительное поражение создает нефизиологические условия, которые могут искусственно благоприятствовать активации зависящей от FXII контактной фазы, функцию FXII исследовали на хорошо изученной модели артериального тромбоза, в которой повреждение индуцировалось в аорте механически, и кровоток отслеживали с помощью ультразвукового датчика потока. После временного возрастания непосредственно после нанесения повреждения кровоток непрерывно уменьшался в течение нескольких минут у всех тестированных мышей. У всех тестированных мышей дикого типа (10/10) это снижение приводило к полному и необратимому закупориванию сосуда за период времени от 1,6 до 11,1 минут после нанесения повреждения (среднее время закупоривания 5,3±3,0 минут, Фиг.4A). Другую картину наблюдали у FXII-/- мышей, у которых формирование стабильных тромбов было сильно искажено. Хотя у всех животных наблюдали постоянное снижение кровотока в течение первых минут после нанесения повреждения, закупоривание произошло только у 4 из 10 мышей. Более того, закупоривающие тромбы у этих мышей во всех случаях были нестабильными и быстро эмболизировались, так что кровоток восстанавливался за 10-115 сек после закупоривания. Ни один из повторно открывшихся сосудов не закупоривался второй раз. Соответственно, все FXII-/- мыши демонстрировали по существу нормальную скорость кровотока через поврежденный сосуд в конце периода наблюдения (40 минут). Очень похожие результаты были получены на FXI-/- мышах, где у 9 из 11 мышей не сформировались закупоривающие тромбы в течение периода наблюдения (30 минут) (Фиг.5A).
Серьезный дефект формирования артериальных тромбов у FXII-/-мышей был подтвержден на третьей модели, где изучали in vivo мобилизацию тромбоцитов в поврежденной сонной артерии с помощью флуоресцентной микроскопии. От мыши донора получали очищенные тромбоциты, метили их флуоресцентными метками и инъецировали мыши-реципиенту, обладающей таким же генотипом. Повреждение сосудов индуцировали путем наложения сильной лигатуры на сонную артерию, что обязательно вызывает разрушение эндотелиального слоя и часто приводит к разрыву внутренней эластичной оболочки с последующей быстрой адгезией тромбоцитов, запускаемой коллагеном, и формированию тромба на участке повреждения (Gruner et al., Blood 102:12/8/2005 2003). В то время как у животных дикого типа быстро формировались большие стабильные тромбы (площадь тромбов: 102,821±39,344 мкм2; t=5 мин), которые не эмболизировались, у мутантных мышей формировались только небольшие или среднего размера агрегаты, которые часто отрывались от участка повреждения (Фиг.4B, C). Соответственно, у мутантных мышей площадь тромбов была сильно уменьшена (8,120±13,900 мкм2; t=5 мин), хотя первичная адгезия тромбоцитов на стенке сосуда не выглядела дефектной.
Для проверки, является ли тяжелый дефект образования тромбов у FXII-/- мышей результатом отсутствия FXII плазмы или FXII тромбоцитов, либо результатом вторичных, неидентифицированных эффектов дефицита FXII, таких как изменение в сосудистой системе, образование артериальных тромбов изучали на FXII-/- мышах после введения человеческого FXII (2 мкг/г массы тела). Такая обработка нормализовала PTT (27±6 сек) и полностью восстанавливала формирование артериальных тромбов. В поврежденных FeCl3 мезентериальных артериолах в 100% случаев формировались тромбы >20 мкм в течение 10 минут после нанесения повреждения, и все сосуды полностью закупоривались в течение периода наблюдения (Фиг.5A-C). При этом даже прослеживалась тенденция более быстрого закупоривания у восстановленных FXII-/- мышей по сравнению с необработанными контрольными мышами дикого типа (среднее время закупоривания: 22,7±8,2 мин по сравнению с 25,6±8,9 мин). Похожий результат был получен, когда повреждение индуцировали механически на аорте. На всех протестированных сосудах полное и необратимое закупоривание происходило в течение 10 минут после нанесения повреждения (Фиг.5D), что подтверждает тот факт, что недостаток FXII плазмы ответственен за дефект тромбообразования, наблюдаемый у FXII-/- мышей.
Описанные выше исследования продемонстрировали, что FXII играет ключевую роль в формировании артериальных тромбов и может, следовательно, рассматриваться в качестве антитромботической мишени.
Для получения прямого подтверждения этого факта, мышей обрабатывали поликлональными антителами кролика против мышиного FXII или антителами неиммунизированного кролика, и анализировали мобилизацию тромбоцитов и образование тромбов в мезентериальных артериях вслед за нанесением повреждения, индуцированного FeCl3. Как показано на Фиг.6A, адгезия тромбоцитов на участках повреждений была сопоставимой у обеих групп мышей. Однако, хотя в контрольных сосудах в 100% случаях формировались тромбы >20 мкм в течение 10 минут после нанесения повреждения, и все сосуды полностью закупоривались в течение периода наблюдения (Фиг.6B, C), тромбы >20 мкм наблюдались в сосудах только в 67% случаях, и закупоривание сосудов происходило только в 50% случаях животных, обработанных анти-FXII антителами.
В качестве альтернативы, для определения влияния низкомолекулярных ингибиторов FXII, мыши дикого типа подвергались инфузии ингибитором FXII-ингибитором трипсина из кукурузы (CTI, 50 мкг/г массы тела) за 5 минут перед нанесением индуцированного FeCl3 повреждения сонной артерии (Wang et al. (2005) J. Thromb. Heamost. 3: 695-702). Обработка ингибитором увеличивала aPTT (62±11 сек, n=4), но не влияла на кровотечение во время хирургической процедуры. Ни у одного из тестированных животных (0/4) не развились закупоривающие сосуды тромбы в течение 30 минут после применения FeCl3.
Эти результаты демонстрируют, что анти-FXII терапевтические средства, такие как анти-FXII антитела или низкомолекулярные ингибиторы FXII, обеспечивают эффективную защиту от образования артериальных тромбов.
Хотя в течение более чем 50 лет контактная активация FXII рассматривается в качестве стартовой точки внутреннего каскада свертывания крови, этот путь считался нерелевантным для свертывания крови. В настоящем изобретении были использованы три различные in vivo модели для анализа мобилизации тромбоцитов и формирования тромбов на участках повреждения артерий у мышей, дефицитных по FXII, с помощью in situ видеомикроскопии и ультразвукового измерения параметров потока, которые показали невозможность формирования стабильных трехмерных тромбов. В основе этого дефекта лежит недостаток в плазме FXII, а не других компонентов, что было полностью подтверждено инъекцией экзогенного человеческого FXII (Фиг.6), тем самым, также исключая возможность вклада вторичных эффектов дефицита FXII в наблюдаемый фенотип.
Эти результаты являются совершенно неожиданными, поскольку FXII рассматривался как антитромботический, а не протромботический фермент, что основывалось на нескольких сообщениях, демонстрирующих связь дефицита FXII с повышенной вероятностью венозного тромбоза (Kuhli, C, Scharrer. l., Koch. F., Ohrloff. C, and Hattenbach. LO. (2004). Factor XII deficiency: a thrombophilic risk factor for retinal vein occlusion. Am. J. Ophthalmol. 137, 459-464; Halbmayer. W.M., Mannhalter. C, Feichtinger. C, Rubi, K., and Fischer. M. (1993). [Factor XII (Hageman factor) deficiency: a risk factor for development of thromboembolism. Incidence of factor XII deficiency in patients after recurrent venous or arterial thromboembolism and myocardial infarction]. Wien. Med. Wochenschr. 143, 43-50).
Мыши с дефицитом FXII демонстрировали нормальное время коагуляции (Фиг.1) и не показывали никаких признаков спонтанного или повышенного посттравматического (интраоперационного) кровотечения, что подтверждало несущественность FXII для нормального гемостаза. На первый взгляд эти результаты противоречат центральной догме гемостаза, что только те факторы, недостаток которых связан с кровотечением или тромбозом, являются релевантными для свертывания крови. Однако при ближайшем рассмотрении эти данные не противоречат этому утверждению, но вскрывают интересную возможность, заключающуюся в том, что артериальный тромбоз может быть опосредован различными механизмами.
Хотя обсуждавшиеся выше механизмы устойчивого образования тромбов могут быть существенными для формирования гемостатической пробки, данные показывают, что формирование стабильных артериальных тромбов требует дополнительной активации внутреннего пути свертывания крови, по меньшей мере, у мышей. Отсутствуют свидетельства в пользу возможности наличия видоспецифичных различий в функции FXII или субстрата фермента. Все параметры свертывания крови и гемостатический фенотип мутантных мышей соответствуют человеческому дефициту FXII, и все отличия, наблюдавшиеся у животных, были нормализованы путем восстановления человеческим FXII (Фиг.5). Помимо этого, исключена возможность того, что дефект тромбообразования ограничен только конкретной экспериментальной моделью, поскольку он был обнаружен в различных артериальных отделах сосудистой системы независимо от типа повреждения. Определенные трудности может представлять определение того, какой тип поражения наилучшим образом отражает очаг поражения сосуда, образующийся вследствие разрыва атеросклеротической бляшки, что рассматривается в качестве основного триггера острых сердечно-сосудистых синдромов. Атеросклеротические очаги поражения обогащены тромбогенными составляющими и, что особенно важно, TF фибриллярными коллагенами. Было показано, что в процессе атерогенеза усиленный синтез коллагена интимальными гладкомышечными клетками дает существенный вклад в сужение просвета сосуда. Разрыв или разрушение бляшки приводит к контакту коллагеновых фибрилл с текущей кровью, что запускает адгезию и агрегацию тромбоцитов. Помимо этого, они включают активацию FXII, как показано в настоящем документе, для фибриллярного коллагена типа I, который является основным типом коллагена, обнаруженным в стенке сосудов. Однако коллагены скорее всего являются не единственным (пато)физиологическим активатором FXII на участках повреждений. Другие кандидаты могут представлять собой вещества, высвобождаемые из разрушающихся клеток или содержащиеся во внеклеточном матриксе (BKM), включая HSP90 или растворимые и нерастворимые полианионы, например нуклеосомы или гликозаминогликаны.
Среди этих активаторов FXII коллагены, вне всякого сомнения, являются наиболее тромбогенными, поскольку они также эффективно активируют тромбоциты. На участках повреждений тромбоциты прикрепляются к BKM посредством обратимого взаимодействия GPIb-V-IX тромбоцитов с коллаген-связанным vWf, что снижает скорость клеток и тем самым обеспечивает возможность связывания других рецепторов. Среди этих рецепторов основную роль играет GPVI, поскольку он активирует интегрины α2β1 и αllbβ3, которые затем опосредуют стабильную адгезию и участвуют в клеточной активации. Помимо этого, активация тромбоцитов через комплекс GPVI/FcRγ-цепь индуцирует прокоагулирующее состояние клеток, которое характеризуется экспонированием фосфатидилсерина (PS) и продуцированием (несущих PS) мембранных пузырьков и микровезикул. Интегрин α2β1 облегчает этот процесс непосредственно путем сигналов "снаружи-внутрь" и опосредовано путем усиления взаимодействий GPVI-коллаген. Установлено, что содержащие PS мембраны сильно ускоряют две основные реакции процесса свертывания, реакции теназы и протромбиназы. Настоящее изобретение продемонстрировало, что прокоагуляционные тромбоциты облегчают зависящее от FXII свертывание in vitro с помощью механизма, в который вовлечены как комплекс GPVI/FcRγ-цепь, так и α2β1 (Фиг.2). Это может, по меньшей мере отчасти, объяснить, почему мыши с дефицитом α2β1, несмотря на неизмененную адгезию тромбоцитов на участках артериальных повреждений, демонстрировали наличие частичных дефектов при формировании закупоривающих тромбов. Помимо коллагенов, прокоагуляционная активность тромбоцитов эффективно стимулируется свертывающейся плазмой через механизм, зависящий от αllbβ3. В представленных экспериментах блокада αllbβ3 практически полностью ингибировала участие тромбоцитов в FXII-зависимом свертывании, свидетельствуя о том, что хорошо известная антикоагуляционная активность антагонистов αllbβ3 может частично основываться на ингибировании внутреннего пути свертывания, опосредованного FXII. Таким образом, настоящее изобретение указывает на то, что опосредованная FXII контактная система и активация тромбоцитов могут быть взаимозависимыми процессами, которые совместно участвуют в патологическом формировании тромбов.
На основе представленных экспериментальных результатов была предложена модель патологического формирования тромбов, которая схематически представлена на Фиг.7. На участках повреждения сосудов первый слой тромбоцитов вступает в контакт с коллагенами окружающей среды, которая также обогащена TF и фибрином. В этой связи не является удивительным, что адгезия тромбоцитов к поврежденной стенке сосуда не нарушена у FXII-/- мышей, и очень вероятно, что эти клетки являются полностью активированными и находятся в прокоагуляционном состоянии. Однако в растущем тромбе коллагены отсутствуют, и концентрации TF, обеспечиваемые микровезикулами, могут быть ниже по сравнению со стенкой сосуда, и их активность может быть уменьшена под действием TFPI высвобождающегося из активированных тромбоцитов в больших количествах. В таких условиях требуются дополнительные механизмы для поддержания пространственно-временного образования тромбина для активации новых мобилизованных тромбоцитов и инициирования их коагулянтной активности через формирование фибрина. Ярко выраженная неспособность FXII-/- мышей образовывать стабильные тромбы однозначно демонстрирует, что опосредованный FXII внутренний путь свертывания является принципиально важным участником этого процесса. Вместе с наблюдением того, что мыши с низким уровнем TF также демонстрируют нарушенное тромбообразование в артериях, эти результаты указывают на то, что как внешний, так и внутренний пути должны быть в рабочем состоянии и действовать синергически для инициирования формирования трехмерного тромба, который в конечном счете приведет к закупориванию. Напротив, отсутствие кровотечений у FXII-/- мышей указывает на то, что рост тромба в трех измерениях может быть необязательным для перекрывания отверстия в стенке сосуда. Это может объяснить, почему внешний путь, который образует первый тонкий слой фибрина и активированных тромбоцитов, является существенным для сохранения нормального гемостаза. Наши результаты указывают на интересную возможность того, что формирование трехмерных тромбов выполняет функции, отличные от поддержания гемостаза. Они могут включать остановку потока крови в определенных областях травмированной ткани с целью предотвращения распространения патогенов или токсинов потоком крови.
Экспериментальная часть
Животные
Все эксперименты и условия содержания одобрены местным комитетом по содержанию и использованию животных. Классические мутанты мышей без фактора XI (FXI-/-), фактора XII (FXII-/-), интегрина α2 (α2-/-) получали описанным способом (Gailani. D., Lasky. N.M., and Bro-ze. G.J., Jr. (1997). A murine model of factor XI deficiency. Blood Coagul. Fibrinolysis 8, 134-144; Pauer. H.U., Renne. T., Hemmerlein, B., Legler, T., Fritzlar. S., Adham. I., Muller-Esterl. W., Emons. G., Sancken. U., Engel. W., and Burfeind. P. (2004). Targeted deletion of murine coagulation factor XII gene-a model for contact phase activation in vivo. Thromb. Haemost. 92, 503-508; Holtkotter. O., Nieswandt. B., Smyth. N., Muller, W., Hafner. M., Schulte. V., Krieg J., and Eckes. B. (2002). Integrin alpha 2-deficient mice develop normally, are fertile, but display partially defective platelet interaction with collagen. J Biol Chem JID - 2985121R 277, 10789-10794). В качестве контроля использовали C57B/6J мышей (FXI-/-) или Sv129 (FXII-/-). Мышей с дефицитом FcRγ-цепи (Takai, T., Li, M., Sylvestre. D., Clynes, R., and Ravetch. J.V. (1994). FcR gamma chain deletion results in pleiotrophic effector cell defects. Cell 76, 519-529) получали описанным способом (Taconics, Germantown).
Создание анти-FXII антител
Общую клеточную РНК выделяли из печени 129sv wt мышей, и осуществляли синтез FXII-ДНК, используя набор для проведения одноэтапной RT-ПЦР ("one-step RT-PCR Kit") компании Qiagen согласно инструкции производителя. Тяжелую цепь фактора FXII (положения 61-1062, соответствующие остаткам 21-354) модифицировали, используя по 25 пмол каждого 5- и 3-праймеров (ttggatccccaccatggaaagactccaag и ttgaattcgcgcatgaacgaggacag), встраивающих сайты рестрикции BamH I и EcoR I соответственно согласно следующему протоколу: 30 с при 95°C, 60 с при 58°C и 1 мин при 72°C для 30 циклов в термоциклере (Biometra, Gottingen, Germany). Продукт ПЦР клонировали в сайты BamH I и EcoR I вектора экспрессии pGEX-2T (Pharmacia). Затем, после секвенирования, белок экспрессировали в E.coli линии BL21. Экспоненциально растущие бактерии стимулировали 0,5 мМ изопропил-β-D-тиогалактопиранозидом в течение 1 часа, собирали, ресуспендировали в 10 мМ трис-HCl, pH 7,4, содержащем 1 мМ ЭДТА, 200 мМ NaCl, 10 мкг/мл бензамидин гидрохлорида, 10 мкг/мл фенилметилсульфонилфторида и обрабатывали ультразвуком в течение 3 мин 15-секундными импульсами. После центрифугирования при 15000 g в течение 20 мин при 4°C супернатант удаляли и для очистки переносили в колонку с GST-сефарозой (Pharmacia). Элюированный белок имел чистоту >95%, которая была определена анализом окрашенного кумасси SDS-PAGE. Образование поликлональных антител против FXII с GST-тяжелой цепью стимулировали в организме кроликов согласно стандартным процедурам. Антитела выделяли из гипериммунной сыворотки, используя колонки с FXII-тяжелой цепью, слитые с мальтоза-связывающим белком (MBP). Эти слитые белки экспрессировали и очищали, используя систему экспрессии pMAL-c2 и амилозные колонки, аналогично тому, как это описано для GST-слитого конструкта.
Приготовление тромбоцитов
После эфирной анестезии у мышей отбирали кровь из ретроорбитального венозного синуса. Кровь собирали в пробирку, содержащую 20 ед./мл гепарина, и богатую тромбоцитами плазму (prp) получали центрифугированием при 300 g в течение 10 мин при комнатной температуре (RT). Для промывки тромбоцитов, prp центрифугировали при 1000 g в течение 8 мин, осадок дважды ресуспендировали в модифицированном Tyrodes-Hepes буфере (134 мМ NaCl, 0,34 мМ Na2HPO4, 2,9 мМ KCl, 12 мМ NaHCO3, 20 мМ Hepes, 5 мМ глюкозы, 0,35% бычьего сывороточного альбумина, pH 6,6) в присутствии простациклина (0,1 мкг/мл) и апиразы (0,02 ед./мл). Затем тромбоциты ресуспендировали в этом же буфере (pH 7,0, 0,02 ед./мл апиразы), инкубировали при 37°C в течение по меньшей мере 30 мин перед проведением анализа.
Поточная цитометрия
Гепаринизированную цельную кровь разбавляли 1:20 модифицированным Tyrode-HEPES буфером (134 мМ NaCl, 0,34 мМ Na2HPO4, 2,9 мМ KCl, 12 мМ NaHCO3, 20 мМ Hepes [N-2-гидроксиэтилпиперазин-N′-2-этансульфоновая кислота], pH 7,0), содержащим 5 мМ глюкозы, 0,35% бычьего сывороточного альбумина (BSA) и 1 мМ CaCl2. Образцы инкубировали с меченными флуорофором антителами в течение 15 минут при комнатной температуре и сразу анализировали при помощи устройства FACScalibur (Becton Dickinson, Heidelberg, Germany) (Nieswandt. B., Schulte. V., and Bergmeier. W. (2004). Flow-cytometric analysis of mouse platelet function. Methods Mol. Biol. 272, 255-268).
Агрегометрия
Для определения агрегации тромбоцитов измеряли величину светопроницаемости, используя prp (200 мкл с 0,5×106 тромбоцитов/мкл). Величину проницаемости регистрировали в 4-канальном агрегометре Fibrintimer (APACT Laborgerate und Analysensysteme, Hamburg, Germany) в течение 10 мин, выражали в произвольных единицах измерения со 100% проницаемостью, установленной по плазме. Агрегацию тромбоцитов индуцировали добавлением коллагена (10 мкг/мл) и АДФ (5 мкг/мл).
Изучение времени коагуляции
Мышей анестезировали интраперитонеальной инъекцией трибромэтанола (Aldrich) (0,15 мл/10 г веса тела), и скальпелем отрезали 3 мм сегмент кончика хвоста. Следили за кровотечением из хвоста, при этом бережно промокали капельки крови фильтровальной бумагой таким образом, чтобы избежать контакта с поврежденным участком. Момент остановки кровотечения определяли по исчезновению следов крови на бумаге через 15-секундные интервалы. При необходимости, кровотечение останавливали вручную через 20 минут. Где указано, мышей обрабатывали hFXII в количестве 100 мкг/мышь.
Приготовление тромбоцитов для интравитальной микроскопии
Кровь мышей (1 об.) отбирали в 0,5 об. буфера Hepes, содержащего 20 ед./мл гепарина. Кровь центрифугировали при 250 g в течение 10 мин, и богатую тромбоцитами плазму бережно переносили в свежую пробирку. Тромбоциты метили 5-карбоксифлуоресцеин диацетат-N-сукцинимидил эфиром (DCF) и доводили до конечной концентрации 200х106 тромбоцитов/250 мкл (Massberg, S., Sausbier. M., Klatt, P., Bauer. M., Pfeifer A, Siess. W., Fassler. R., Ruth, P., Krombach. F., and Hofmann. F. (1999). Increased adhesion and aggregation of platelets lacking cyclic guanosine S′A-monophosphate kinase I.J Exp Med 189, 1255-1264).
Модель in vivo тромбообразования при FeCl3-индуцированном повреждении
Самцов и самок мышей в возрасте 4-5 недель анестезировали интраперитонеальной инъекцией 2,2,2-трибромэтанола и 2-метил-2-бутанола (Sigma) (0,15 мл/10 г веса тела из расчета 2,5% раствора). Флуоресцентно меченные тромбоциты вводили внутривенно. Мезентерий бережно выводили на поверхность через срединный разрез брюшной стенки. Артериолы (35-50 мкм в диаметре) визуализировали, используя микроскоп Axiovert 200 инвертированный, Carl Zeiss (×10), оборудованный флуоресцентным источником света 100-W HBO и CCD камерой (CV-M300), присоединенной к S-VHS устройству видеозаписи (AG-7355, Panasonic, Matsushita Electric, Japan). После местного нанесения FeCl3 (20%), индуцирующего повреждение сосуда, и обнажения эндотелия состояние артериол отслеживали в течение 40 мин или до полного закупоривания (остановка тока крови в течение >1 мин). Устойчивую адгезию тромбоцитов определяли в виде количества флуоресцентно меченных тромбоцитов, которые осаждались на стенке сосуда в течение 5 минут после повреждения, тромб определяли как агрегат тромбоцитов диаметром больше 20 мкм, время закупоривания сосуда определяли как время, требуемое для остановки крови, текущей в течение по меньшей мере 1 минуты. Во всех экспериментах у мышей выбирали максимум по две артериолы на основе качества экспозиции. Всего было исследовано 17 wt, 14 FXII-/- и 9 FXI-/- артериол.
Интравитальная микроскопия - каротидная артерия
Интравитальную микроскопию поряженной каротидной артерии выполняли по существу, следуя описанию (Massberg. S., Gawaz. M., Gruner. S., Schulte. V., Konrad. I, Zohlnho-fer, D., Heinzmann. U., and Nieswandt. B. (2003). A crucial role of glycoprotein VI for platelet recruitment to the injured arterial wall in vivo. J Exp Med JID - 2985109R 197, 41-49). Излагая вкратце, мышей анестезировали интраперитонеальной инъекцией кетамином/ксилазином (кетамин 100 мг/кг, Parke-Davis, Karlsruhe, Germany; ксилазин 5 мг/кг, Bayer AG, Leverkusen, Germany). Полиэтиленовые катетеры (Portex, Hythe, England) имплантировали в правую яремную вену, и при помощи внутривенной инфузии вводили флуоресцентно меченные тромбоциты (200×106/250 мкл). Повреждение каротидной артерии для обнажения эндотелия индуцировали наложением сильной лигатуры. Перед и после повреждения сосуда флуоресцентно меченные тромбоциты визуализировали in situ при помощи in vivo видеомикроскопии правой общей каротидной артерии, используя микроскоп Zeiss Axiotech (20× водно-иммерсионный объектив, W 20×/0,5, Zeiss, Gottingen, Germany) со 100 Вт HBO ртутной лампой для epi-освещения. Адгезию тромбоцитов и образование тромбов регистрировали в течение 5 мин после индуцирования повреждения, и изображения, записанные на видеопленку, оценивали, используя компьютерную программу для анализа видеоизображений (Visitron, Munich, Germany).
Легочный тромбоэмболизм
Мышей анестезировали интраперитонеальной инъекцией 2,2,2-трибромэтанола и 2-метил-2-бутанола (Aldrich) (0,15 мл/10 г веса тела из расчета 2,5% раствора). От анестезированных мышей получали смесь коллагена (0,8 мг/кг), и в яремную вену инъецировали эпинефрин (60 мкг/кг). У выживших мышей зашивали надрез, и оставляли их выздоравливать. Проводили некроскопию и гистологический анализ легких, фиксированных в 4% формальдегиде, и залитые парафином срезы окрашивали гематоксилином/эозином.
Подсчет тромбоцитов
Подсчет тромбоцитов выполняли методом поточной цитометрии, используя устройство FACScalibur (Becton Dickinson, Heidelberg, Germany). Результаты выражали в виде среднего значения ±S.D или в виде % от контроля (wt, n=19; FXII-/-, n=14 и FcRγ-/-, n=5).
Время закупоривания
Абдоминальную полость анестезированных мышей вскрывали в продольном направлении, и препарировали абдоминальную аорту. Ультразвуковой датчик оценки потока крови размещали вокруг аорты, и индуцировали тромбоз сильным сдавливанием аорты щипцами. Поток крови отслеживали до момента полного закупоривания. Эксперимент завершали вручную через 45 минут. Указано, что непосредственно перед проведением эксперимента внутривенно вводили человеческий фактор XII.
Гистологический анализ
Мышей умерщвляли, быстро вынимали легкие и фиксировали при 4°С в течение 24 часов в забуференном 4% формалине (pH 7,4; Kebo). Ткани обезвоживали и заливали парафином (Histolab Products AB), делали 4-мкм срезы и закрепляли. После удаления парафина ткани окрашивали гематоксилином Майера (Mayers) (Histolab Products AB) и эозином (Surgipath Medical Industries, Inc.).
Электрофорез в SDS-полиакриламидном геле, вестерн-блот анализ и иммуноимпритинг
Плазму (0,3 мкл/дорожка) разделяли 12,5% (вес./об.) электрофорезом в полиакриламидном геле в присутствии 1% (вес./об.) SDS (Laemmli, 1970). Белки переносили на нитроцеллюлозную мембрану в течение 30 мин при 100 мА. Мембраны блокировали буфером PBS, содержащим 4% (вес./об.) порошок сухого молока и 0,05% (вес./об.) твин-20, pH 7,4. Мембраны исследовали, используя 0,5 мкг/мл моноклональных антител к MBK3 (Haaseman J. Immunology 1988). Связанные антитела детектировали вторичными антителами к мышиным IgG, конъюгированными с пероксидазой хрена (разбавление 1:5000) с последующим изучением методом хемилюминесцентного детектирования.
Исследование свертывания
Для определения времени свертывания при рекальцификации 100 мкл мышиной антикоагулированной цитратом плазмы (0,38% цитрата натрия) инкубировали со 100 мкл каждого из: коллагена типа Horm (Nycomed, Munchen, Germany), эллаговой кислоты, сульфата хондроитина (оба фирмы Sigma), каолина или буфера (конечные концентрации 30 мкг/мл), в течение 120 сек при 37°C в коагулометре KC10 "Kugelkoagulometer" (Amelung, Lemgo, Germany). Для тестирования эффекта активации тромбоцитов на FXII-зависимое свертывание крови промытые тромбоциты ресуспендировали в буфере Tyrode, содержащем 4 мМ Са2+ и 5 мМ Са2+-ионофора А23187 (Sigma) в течение 10 мин перед добавлением плазмы, не содержащей тромбоциты. Образование сгустка инициировали рекальцификацией 100 мкл 25 мМ раствора CaCl2, и время до образования сгустка регистрировали измерителем времени свертывания KC4 (Amelung).
Анализ свертывания
Общие и единичные параметры свертывания определяли, используя автоматизированную систему для изучения свертывания крови (BCS, Dade Behring) с реагентами Dade Behring, следуя протоколу, разработанному производителем для образцов крови человека. Основные положения протокола BCS анализа приведены во вложенной инструкции компании Dade Behring, которую можно найти на сайте производителя Dade Behring (http://www.dadebehring.com). D-димеры измеряли методом ELISA от Asserachrom (Roche). Подсчет клеток периферической крови выполняли при помощи устройства Sysmex XE 2100, следуя стандартным протоколам.
Измерение тромбина
Образование тромбина измеряли способом Aronson et al. (Circulation, 1985) с небольшими модификациями. Аликвоты (0,5 мл) плазмы, богатой тромбоцитами или свободной от тромбоцитов, помещали в круглодонные полипропиленовые пробирки, которые покрывали коллагеном типа Horm (100 мкг/мл, 24 ч, 4°C), и добавляли 20 мкл 1 М Ca2+ для инициации коагуляции крови. В лунки микротитровального планшета, содержащие 90 мкл 3,8% цитрата натрия, добавляли образцы (10 мкл) с интервалами 2,5-10 мин в течение 60 мин. Окрашивание проводили через 2 мин путем добавления 50 л 2 ммол/л S-2238 (H-D-Phe-Arg-NH-NO2-HCl, специфического к тромбину субстрата; Chromogenix, Molndal, Sweden) в 1 мол/л трис (pH 8,1). Коэффициент поглощения высвобождаемого окрашенного продукта измеряли спектрофотометрически при длине волны Vmax=405 нм, используя микропланшетный ридер (Easy Reader, EAR 340AT, SLT Lab Instruments GmbH, Vienna, Austria). Для каждой временной точки проводили по три измерения.
Статистическая оценка
Статистический анализ выполняли, используя непараметрический тест Стьюдента.
Хотя здесь по существу проиллюстрированы и описаны только предпочтительные варианты осуществления изобретения, очевидно, что возможны многие модификации и изменения настоящего изобретения в свете приведенной здесь информации и в рамках прилагаемой формулы изобретения без отступления от сущности и предполагаемого объема настоящего изобретения.
Заявленная группа изобретений относится к медицине, в частности к гематологии, и может быть использована для предотвращения формирования и/или стабилизации патологических тромбов. Предложено применение по меньшей мере одного антитела, ингибирующего фактор XII. Предложено также фармацевтическое средство, содержащее по меньшей мере одно антитело, ингибирующее фактор XII. Изобретения обеспечивают возможность предотвращения роста трехмерного внутрипросветного тромба без проявлений влияния на гемостаз. 2 н. и 5 з.п. ф-лы, 7 ил.
1. Применение по меньшей мере одного антитела, ингибирующего фактор XII, для предотвращения нарушения, связанного с формированием венозных или артериальных тромбов, путем предотвращения патологического формирования и/или стабилизации тромбов и, таким образом, предотвращения роста трехмерного внутрипросветного тромба без влияния на гемостаз.
2. Применение по п.1, в котором указанное антитело представляет собой анти-FXII антитело.
3. Применение по п.2, в котором указанное антитело представляет собой ингибитор активации FXII.
4. Применение по любому из пп.1-3 для лечения или профилактики состояний или нарушений, связанных с формированием венозного или артериального тромба, особенно инсульта или инфаркта миокарда.
5. Фармацевтическое средство, содержащее по меньшей мере одно антитело, ингибирующее фактор XII, для предотвращения нарушения, связанного с формированием венозных или артериальных тромбов, путем предотвращения патологического формирования и/или стабилизации тромбов и, таким образом, предотвращения роста трехмерного внутрипросветного тромба.
6. Средство по п.5, в котором указанное антитело представляет собой анти-FXII антитело.
7. Средство по п.5, в котором указанное антитело ингибирует активацию FXII.
| US 4963657 A 16.10.1990 | |||
| ИНГИБИТОРЫ СЕРИНОВЫХ ПРОТЕАЗ | 1997 |
|
RU2172321C2 |
| US 2003109680 A1 12.06.2003 | |||
| Фрикционная дисковая муфта | 1985 |
|
SU1222929A1 |
| ОВАНЕСОВ М.В | |||
| "Влияние факторов внутреннего пути свертывания крови на пространственную динамику роста сгустка" - автореферат диссертации на соискание ученой степени к.б.н., М., 2002 [он-лайн] [Найдено 2011.09.05] найдено из Интернет: | |||

