Область техники, к которой относится изобретение
Настоящее изобретение относится к химиотерапевтическому средству против рака (упоминаемому далее как "противораковое химиотерапевтическое средство"), а более конкретно к противораковому химиотерапевтическому средству, включающему в комбинации производное соединение холестанола и противораковое средство.
Уровень техники
Множество противораковых средств, используемых в химиотерапии против рака, которая представляет собой один из методов лечения рака, до настоящего времени разрабатывали и классифицировали на основании структуры, механизма действия и т.д. Однако эффективность такого противоракового средства, применяемого в виде единственного средства, является неудовлетворительной. Вместо этого, в последние годы проводили мультилекарственную терапию с применением большого количества противораковых средств преимущественно с точки зрения ослабления неблагоприятных побочных эффектов, и эффективность мультилекарственной терапии была признана.
При таких обстоятельствах необходимы как разработка новой противораковой комбинированной химиотерапии, которая имеет меньшее неблагоприятное побочное действие и более высокую эффективность, чем общепризнанные способы химиотерапии, так и разработка новых химиотерапевтических средств для использования в химиотерапии.
Между тем, ранее было обнаружено, что производное соединение холестанола, у которого сахарная цепь, такая как GlcNAc-Gal-, GlcNAc-Gal-Glc-, Fuc-Gal-, Gal-Glc-, Gal- или GlcNAc-, связана с холестанолом (соединение, у которого насыщена двойная связь в кольце B холестерина), обладает превосходной противоопухолевой активностью (патентные документы с 1 по 4).
Однако не сообщалось о фактах, когда упомянутые выше производные соединения холестанола и еще одно противораковое средство применяют в комбинации.
[Патентный документ 1] JP-A-2000-191685
[Патентный документ 2] JP-A-1999-60592
[Патентный документ 3] WO 2005/007172 (публикация)
[Патентный документ 4] WO 2007/026869 (публикация)
Раскрытие изобретения
Проблемы, которые должны быть решены с помощью изобретения
Таким образом, настоящее изобретение направлено на предоставление противоракового химиотерапевтического средства, которое обладает меньшими побочными эффектами и превосходной эффективностью.
Средства для решения проблем
Принимая во внимание вышеизложенное, авторы настоящего изобретения провели всесторонние исследования и обнаружили, что значительно усиленный противораковый эффект может быть достигнут посредством применения, в комбинации, производного соединения холестанола, представленного формулой (1), или его циклодекстринового соединения включения и известного химиотерапевтического средства (противоракового средства), и, таким образом, комбинированное применение данных фармацевтических средств в противораковой химиотерапии является очень полезным.
Соответственно, настоящее изобретение направлено на следующие пункты с (1) по (14).
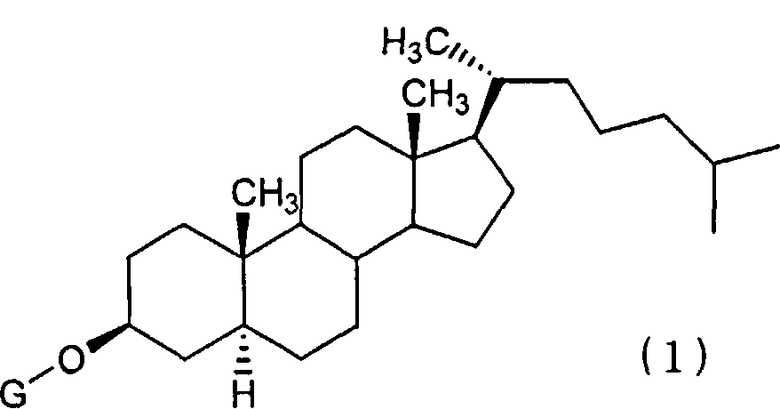
(1) Противораковое химиотерапевтическое средство, содержащее, в комбинации, производное соединение холестанола, представленное формулой (1):

(в которой G представляет GlcNAc-Gal-, GlcNAc-Gal-Glc-, Fuc-Gal-, Gal-Glc-, Gal- или GlcNAc-) или его циклодекстриновое соединение включения и противораковое средство.
(2) Противораковое химиотерапевтическое средство согласно абзацу (1) выше, при этом в формуле (1) G представляет собой GlcNAc-Gal- или GlcNAc-.
(3) Противораковое химиотерапевтическое средство согласно абзацу (1) или (2) выше, при этом противораковым средством является одно или более веществ, выбранных из группы, состоящей из таксанового противоракового средства, противоракового средства платинового комплекса, соединения пеметрекседа и фторурацила.
(4) Противораковое химиотерапевтическое средство согласно абзацу (3) выше, при этом противораковым средством является одно или более веществ, выбранных из группы, состоящей из паклитаксела, доцетаксела, пеметрекседа, 5-FU, цисплатина, оксалиплатина, циклофосфамида и тринотекана.
(5) Противораковое химиотерапевтическое средство согласно абзацу (1) или (2) выше, при этом противораковым средством является одно или более веществ, выбранных из группы, состоящей из таксанового противоракового средства, соединения пеметрекседа и фторурацила.
(6) Противораковое химиотерапевтическое средство согласно абзацу (5) выше, при этом противораковым средством является одно или более веществ, выбранных из группы, состоящей из паклитаксела, доцетаксела, пеметрекседа, 5-FU, циклофосфамида и иринотекана.
(7) Противораковое химиотерапевтическое средство согласно любому из абзацев с (1) по (6) выше, которое представляет собой многокомпонентное средство.
(8) Противораковое химиотерапевтическое средство согласно любому из абзацев с (1) по (6) выше, которое находится в форме набора, включающего лекарственное средство, содержащее в своем составе производное соединение холестанола и лекарственное средство, содержащее в своем составе противораковое средство.
(9) Противораковое химиотерапевтическое средство согласно абзацу (8) выше, при этом лекарственное средство, содержащее в своем составе производное соединение холестанола, представляет собой липосомальную композицию.
(10) Применение в комбинации производного соединения холестанола, представленного формулой (1):
(в которой G представляет GlcNAc-Gal-, GlcNAc-Gal-Glc-, Fuc-Gal-, Gal-Glc-, Gal- или GlcNAc-) или его циклодекстриновое соединение включения и противораковое средство, для получения противоракового химиотерапевтического средства.
(11) Применение согласно абзацу (10) выше, при этом противораковым средством является одно или более веществ, выбранных из группы, состоящей из таксанового противоракового средства, соединения пеметрекседа и фторурацила.
(12) Применение согласно абзацу (11) выше, при этом противораковым средством является одно или более веществ, выбранных из группы, состоящей из паклитаксела, доцетаксела, пеметрекседа, 5-FU, циклофосфамида и иринотекана.
(13) Противораковая химиотерапия, отличающаяся тем, что она включает введение в комбинации производного соединения холестанола, представленного формулой (1):
(в которой G представляет GlcNAc-Gal-, GlcNAc-Gal-Glc-, Fuc-Gal-, Gal-Glc-, Gal- или GlcNAc-) или его циклодекстринового соединения включения и противоракового средства нуждающемуся в этом пациенту.
(14) Противораковая химиотерапия согласно абзацу (13) выше, при этом производное соединение холестанола или его циклодекстриновое соединение включения и противораковое средство вводят нуждающемуся в этом пациенту одновременно или по отдельности с промежутками.
Результаты изобретения
Посредством применения противоракового химиотерапевтического средства и противораковой химиотерапии согласно настоящему изобретению, предупреждение и лечение рака можно осуществлять безопасно и с более высокой эффективностью.
Краткое описание чертежей
Фиг.1-A представляет собой диаграмму, показывающую ингибирующие действия на клеточную пролиферацию противоракового средства (CDDP или L-OHP), GC-CD и противоракового средства + GC-CD на клетках colon26;
Фиг.1-B представляет собой диаграмму, показывающую ингибирующие действия на клеточную пролиферацию противоракового средства (5-FU, PTX, DTX, или CPT), GC-CD и противоракового средства + GC-CD на клетках colon26;
Фиг.1-C представляет собой диаграмму, показывающую ингибирующие действия на клеточную пролиферацию CPA, GC-CD и CPA+GC-CD на клетках colon26;
Фиг.2 представляет собой диаграмму, показывающую ингибирующие действия на клеточную пролиферацию CDDP, GC-CD и CDDP+GC-CD на клетках MKN45, клетках NCIH226 или клетках colo201;
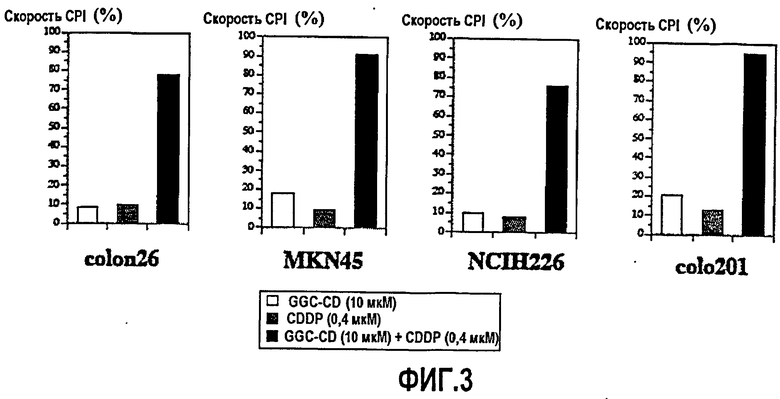
Фиг.3 представляет собой диаграмму, показывающую ингибирующие действия на клеточную пролиферацию CDDP, GGC-CD и CDDP+GGC-CD на клетках colon26, клетках MKN45, клетках NCIH226 или клетках colo201;
Фиг.4 представляет собой диаграмму, показывающую противоопухолевое действие единственного введения CDDP, GC-CD и CDDP+GC-CD против перитонеальной диссеминации, вызванной клетками colon26, интраперитонеально инокулированными мышам;
Фиг.5 представляет собой диаграмму, показывающую противоопухолевое действие множественного введения CDDP, GC-CD и CDDP+GC-CD против перитонеальной диссеминации, вызванной клетками colon26, интраперитонеально инокулированными мышам;
Фиг.6 представляет собой диаграмму, показывающую противоопухолевое действие единственного, отсроченного введения CDDP, GC-CD и CDDP+GC-CD против перитонеальной диссеминации, вызванной клетками colon26, интраперитонеально инокулированными мышам, после подтверждения перитонеальной диссеминации на мезотелии мышей;
Фиг.7 представляет собой диаграмму, показывающую коэффициент выживаемости мышей, которым интраперитонеально инокулировали клетки colon26, при единственном введении CDDP, GC-CD или CDDP+GC-CD (единственном введении CDDP и двойном введении GC-CD соответственно);
Фиг.8 представляет собой диаграмму, показывающую результат ослабления или уменьшения роста опухоли, в которую клетки colon26 подкожно инокулировали мышам, при единственном введении CDDP, GGC-CD или CDDP+GGC-CD; а
Фиг.9 представляет собой диаграмму, показывающую результат подавления метастаза клеток colon26 в легкие, при единственном введении CDDP, GC-CD, GGC-CD, CDDP+GC-CD и CDDP+GGC-CD.
Подробное описание предпочтительных вариантов осуществления
Все специфические производные соединения холестанола, представленные формулой (1) и используемые в настоящем изобретении, являются известными соединениями.
Среди производных соединений холестанола, которые представлены формулой (1) и в которых G представляет собой GlcNAc-Gal-, G предпочтительно представляет собой GlcNAcβ1,3-Galβ- или GlcNAcβ1,4-Galβ-. Среди производных соединений (1) холестанола, в которых G представляет собой GlcNAc-Gal-Glc-, G предпочтительно представляет собой GlcNAcβ1,3-Galβ1,4-Glc-. Среди производных соединений (1) холестанола, в которых G представляет собой Fuc-Gal-, G предпочтительно представляет собой Fucαl,3Gal-. Среди производных соединений (1) холестанола, в которых G представляет собой Gal-Glc-, G предпочтительно представляет собой Galβ1,4Glcβ-. Среди производных соединений (1) холестанола, в которых G представляет собой Gal-, G предпочтительно представляет собой Galβ-. Среди производных соединений (1) холестанола, в которых G представляет собой GlcNAc-, G предпочтительно представляет собой GlcNAcβ-.
Из них вещества, в которых G представляет собой GlcNAc-Gal- и GlcNAc-, являются более предпочтительными, при этом вещества, в которых G представляет собой GlcNAcβ1,4-Galβ- и GlcNAcβ- являются еще более предпочтительными.
Упомянутые выше производные соединения холестанола могут быть получены с помощью способа, например, раскрытого в упомянутых выше патентных документах 1-4, или аналогичного способа.
Производное соединение холестанола, представленное формулой (1), легко образует комплекс включения с циклодекстрином или его производным соединением. Таким образом, производное соединение холестанола, используемое в настоящем изобретении, может представлять собой его циклодекстриновое соединение включения. При образовании подобных соединений включения необходимо принимать в расчет размер гостевой молекулы, подлежащей включению, ван-дер-ваальсово взаимодействие между гостевой молекулой и циклодекстрином и водородную связь между гидроксильными группами циклодекстрина и гостевой молекулой. Вследствие этого нерастворимые гостевые соединения не всегда образуют соответствующие соединения включения. Однако производное соединение холестанола настоящего изобретения может образовывать подходящие комплексы включения с циклодекстрином.
Примеры циклодекстрина, образующего циклодекстриновое соединение включения настоящего изобретения, включают такие циклодекстрины, как α-циклодекстрин, β-циклодекстрин и γ-циклодекстрин; и циклодекстриновые производные соединения, такие как метил-β-циклодекстрин, 2-гидроксипропил-β-циклодекстрин, моноацетил-β-циклодекстрин и 2-гидроксипропил-γ-циклодекстрин. Из них для получения улучшенной растворимости предпочтительным является 2-гидроксипропил-β-циклодекстрин.
Циклодекстриновое соединение включения может быть получено, например, с помощью следующей методики: получают водный раствор циклодекстрина или его производное соединение, имеющее соответствующую концентрацию (например, от 20 до 40%), и к водному раствору добавляют производное соединение холестанола настоящего изобретения, с последующим встряхиванием полученной в результате смеси.
На концентрацию раствора производного соединения холестанола (1) не накладывают никакого особого ограничения, при условии, что производное соединение холестанола может образовывать соединение включения с циклодекстрином. В большинстве случаев, концентрация составляет от приблизительно 1 до приблизительно 50 масс.%, предпочтительно от приблизительно 10 до приблизительно 30 масс.%.
Полученное таким образом циклодекстриновое соединение включения является сильно водорастворимым, и, вследствие этого, эффективно демонстрирует действие гостевой молекулы in vivo. Еще одно преимущество циклодекстринового соединения включения состоит в обеспечении непротиворечивых результатов испытаний in vitro.
В качестве альтернативы, производное соединение холестанола (1) может быть получено в виде липосомальной композиции, посредством чего производное соединение холестанола может быть более эффективно доставлено до участка экспрессии действия. Еще одно преимущество циклодекстринового соединения включения состоит в обеспечении непротиворечивых результатов испытаний in vitro.
Предпочтительно, липосомальная композиция включает производное соединение холестанола настоящего изобретения, мембранный компонент и алифатический или ароматический амин. Содержание производного соединения холестанола в липосомальной композиции составляет предпочтительно от 0,3 до 2,0 моль, более предпочтительно 0,8 до 1,5 моль относительно 1 моль мембранного компонента.
Мембранным компонентом может быть фосфолипид. Конкретные примеры предпочтительно используемых фосфолипидов включают натуральные и синтетические фосфолипиды, такие как фосфатидилхолин, фосфатидилэтаноламин, фосфатидилсерин, фосфатидилинозитол и фосфатидная кислота; их смеси; и модифицированные натуральные фосфолипиды, такие как водный лецитин. Примеры более предпочтительных веществ включают фосфатидилхолин (1α-дипалмитоилфосфатидилхолин (DPPC)).
Алифатический или ароматический амин используют главным образом для положительной зарядки поверхности липидной мембраны. Примеры подобных аминов включают алифатические амины, такие как стеариламин и олеиламин; и ароматические амины, такие как флуоренилэтиламин. Среди них стеариламин используют особенно предпочтительно.
Предпочтительно амин содержится в количестве, равном от 0,04 до 0,15 моль, более предпочтительно от 0,1 до 0,15 моль, относительно 1 моль мембранного компонента (фосфолипида).
В дополнение к упомянутым выше компонентам, при необходимости липосома может дополнительно содержать в своем составе стабилизатор мембранных структур, такой как холестерин, жирная кислота, диацетилфосфат и т.д.
Водным раствором, использующимся для диспергирования мембранного компонента, предпочтительно является вода, физиологический раствор, буфер, водный раствор сахара или их смесь. Может быть использован либо органический, либо неорганический буфер при условии, что буфер обладает буферным действием в условиях концентрации ионов водорода человеческого организма. Примеры подобных буферов включают фосфатный буфер.
Никакое особое ограничение не накладывается на способ получения липосомальной композиции, и могут быть выбраны широко используемые способы. Примеры способа, который может быть использован, включают способы, раскрытые в JP-A-1982-82310, JP-A-1985-12127, JP-A-1985-58915, JP-A-1989-117824, JP-A-1989-167218, JP-A-1992-29925 и JP-A-1997-87168; способ, раскрытый в способах биохимического анализа (1988) 33, p.337; или способ, раскрытый в "Liposome" (опубликованной Nankodo).
Никакое особое ограничение не накладывается на противораковое средство, которое используют в комбинации с производным соединением холестанола, представленного формулой (1) или его циклодекстриновым соединением включения, и могут быть использованы известные противораковые химиотерапевтические средства. Предпочтительно используют стандартные терапевтические средства, которые были приняты в соответствии с таргентной терапией рака.
Конкретные примеры включают алкилирующие агенты, такие как циклофосфамид, ифосфамид, мелфалан (L-PAM), бусульфан и карбохинон; антагонисты метаболизма, такие как 6-меркаптопурин (6-MP), метотрексат (MTX), 5-Фторурацил (5-FU), тегафур, эноцитабин (BHAC) и соединения пеметрекседа (пеметрексед, MTA) и т.д.); карциностатические антибиотики, такие как актиномицин D, даунорубицин, блеомицин, пепломицин, митомицин C, акларубицин и неокарциностатин (NCS); растительные алкалоиды, такие как винкристин, виндезин, винбластин, таксановые противораковые средства (таксотер (доцетаксел) и таксол (паклитаксел, TXL) и т.д.) и иринотекан (CPT-11); и платиновые соединения, такие как цисплатин (CDDP), карбоплатин и оксалиплатин (L-OHP). Данные противораковые средства могут быть использованы по отдельности или в комбинации двух или более веществ.
Как показано в примерах, описанных далее, когда производное соединение холестанола, представленное формулой (1), или его циклодекстриновое соединение включения используют в комбинации с противораковым средством, пролиферация раковых клеток различных типов сильно угнетается по сравнению со случаем введения только одного средства. Вследствие этого данная комбинированная химиотерапия может радикально усиливать терапевтическую эффективность и уменьшение неблагоприятных побочных эффектов, а фармацевтические продукт, содержащий в своем составе данные ингредиенты, представляет собой полезное противораковое химиотерапевтическое средство.
Никакое особое ограничение не накладывается на рак, который можно эффективно лечить посредством введения противоракового химиотерапевтического средства в соответствии с настоящим изобретением. Примеры рака-мишени включают злокачественные опухоли, такие как рак желудка, рак толстой кишки, рак поджелудочной железы, рак матки, рак яичников, рак легких, рак желчного пузыря, рак пищевода, рак печени, рак молочной железы, мезотелиома и рак простаты.
Формой противоракового химиотерапевтического средства настоящего изобретения может являться многокомпонентное средство, в котором упомянутые выше ингредиенты перемешивают в соответствующем соотношении, каждый в эффективном количестве, для образования лекарственной формы однократного применения (тип единой готовой формы), или может являться набор, который состоит из соответствующей лекарственной формы упомянутых выше ингредиентов, каждый из которых образован независимо, причем каждый включает эффективное количество, и который предоставляет возможность введения лекарственных форм одновременно или по отдельности с промежутками (тип двойной готовой формы).
Аналогично общим фармацевтическим готовым формам, никакое особое ограничение не накладывается на лекарственную форму описанной выше готовой формы, и форма может быть любой из твердой формы, такой как таблетка, жидкой формы, такой как инъекционный раствор, сухой порошок, растворяемый перед применением, и т.д.
Никакие особые ограничения не накладываются на способ введения готовой формы, и соответствующий способ может быть определен в зависимости от лекарственной формы средств. Например, инъекционный раствор может быть введен внутривенно, внутримышечно, подкожно, внутрикожно или интраперитонеально, а твердая форма может быть введена перорально или энтерально.
Готовая форма может быть получена с помощью способа, известного в данной области. Также могут быть использованы все фармацевтически приемлемые носители (эксципиенты или разбавители, такие как наполнитель, объемообразующий агент и связующее вещество), используемые в большинстве случаев в данной области.
Например, пероральная твердая форма может быть получена посредством перемешивания ингредиентов лекарственного средства настоящего изобретения с эксципиентом и с необязательным связующим веществом, разрыхлителем, смазочным веществом, красителем, ароматическим средством, отдушкой и т.д. и формирования смеси в таблетки, таблетки с покрытием, гранулы, порошок, капсулы и т.д. с помощью способа, известного в данной области. Данными добавками могут быть добавки, используемые в большинстве случаев в данной области. Примеры эксципиента включают лактозу, сукрозу, натрия хлорид, глюкозу, крахмал, кальция карбонат, каолин, микрокристаллическую целлюлозу и кремниевую кислоту. Примеры связующего вещества включают воду, этанол, пропанол, простой сироп, жидкую глюкозу, жидкий крахмал, жидкий желатин, карбоксиметилцеллюлозу, гидроксипропилцеллюлозу, гидроксипропилкрахмал, метилцеллюлозу, этилцеллюлозу, шеллак, кальция фосфат и поливинилпирролидон. Примеры разрыхлителя включают сухой крахмал, натрия альгинат, агаровый порошок, натрия гидрокарбонат, кальция карбонат, натрия лаурилсульфат, моноглицерилстеарат и лактозу. Примеры смазочных веществ включают очищенный тальк, стеаратные соли, боракс и полиэтиленгликоль. Примеры ароматического средства включают сахарозу, апельсиновую цедру, лимонную кислоту и винную кислоту.
Пероральная жидкая готовая форма может быть получена посредством перемешивания ингредиентов лекарственного средства настоящего изобретения с ароматическим средством, буфером, стабилизатором, отдушкой и т.д. и формирования смеси во внутреннее жидкое средство, сироп, эликсир и т.д. с помощью способа, известного в данной области. Ароматическим средством, используемым при получении, может быть любой из упомянутых выше элементов. Примеры буфера включают натрия цитрат. Примеры стабилизатора включают трагант, гуммиарабик и желатин.
Инъекционные растворы могут быть получены посредством перемешивания ингредиентов лекарственного средства настоящего изобретения с добавками, такими как регулятор pH, буфер, стабилизатор, средство, регулирующее тоничность, и местное анестетическое средство и т.д. и формирования смеси с помощью способа, известного в данной области, для предоставления за счет этого подкожной, внутримышечной и внутривенной инъекционных жидкостей. Примеры регулятора pH и буфера включают натрия цитрат, ЭДТК, тиогликолевую кислоту и тиомолочную кислоту. Примеры местного анестетического средства включают прокаина гидрохлорид и лидокаина гидрохлорид. Примеры средства, регулирующего тоничность включают натрия хлорид и глюкозу.
Суппозитории могут быть получены посредством перемешивания ингредиентов лекарственного средства настоящего изобретения с носителем для готовой формы, известным в данной области, таким как полиэтиленгликоль, ланолин, масло какао и триглицерид жирной кислоты и с необязательным поверхностно-активным веществом, таким как Tween (зарегистрированная торговая марка), и формирования смеси в суппозитории с помощью способа, известного в данной области.
Мази могут быть получены посредством перемешивания ингредиентов лекарственного средства настоящего изобретения с необязательными добавками, используемыми в большинстве случаев в данной области, такими как основа, стабилизатор, увлажняющий компонент и консервант, и формирования смеси в мази с помощью способа, известного в данной области. Примеры основы включают жидкий парафин, медицинский вазелин, осветленный пчелиный воск, октилдодециловый спирт и парафин. Примеры консерванта включают метил-p-гидроксибензоат, этил-p-гидроксибензоат и пропил-p-гидроксибензоат.
Катаплазмы могут быть получены посредством нанесения упомянутых выше мази, крема, геля, пасты и т.д. на используемую в большинстве случаев подложку с помощью обычного способа. Примеры соответствующих подложек включают тканый и нетканый материал, изготовленный из хлопка, штапельного волокна или искусственного волокна, и пленку и пенопласт, изготовленный из мягкого винилхлорида, полиэтилена, полиуретана и т.д.
В большинстве случаев, готовую форму предпочтительно получают для того, чтобы иметь содержание производного соединения холестанола и содержание противоракового средства, равное от 0,0001 до 80 масс.% (в виде эффективного ингредиента).
Когда противораковое химиотерапевтическое средство в соответствии с настоящим изобретением предоставлено в виде набора, набор может быть выполнен с возможностью независимой упаковки соответствующей лекарственной формы, включающей отдельно производное соединение холестанола, представленное формулой (1), или его циклодекстриновое соединение включения и противораковое средство, каждое из которых было получено приведенным выше способом, и использования каждой фармацевтической готовой формы, взятой отдельно из надлежащей соответствующей упаковки перед применением. В качестве альтернативы, каждая фармацевтическая готовая форма может содержаться в упаковке, применяемой каждый раз при комбинированном введении.
Доза противоракового химиотерапевтического средства настоящего изобретения варьирует в зависимости от массы тела, возраста, пола, симптомов у нуждающегося в этом пациента, способа и частоты введения нуждающемуся в этом пациенту и т.д. В большинстве случаев, например, ежедневная доза для взрослого составляет приблизительно от 0,1 до 30 мг/кг в виде производного соединения холестанола (1), предпочтительно от 3 до 10 мг/кг. Доза противоракового средства может попадать в пределы диапазона, установленного относительно средства, или может быть ниже, чем этот диапазон. Никакие особые ограничения не накладываются на частоту введения, и средство может быть введено один раз или несколько раз в день. Предпочтительным является единственное введение в день. При использовании набора, каждая готовая форма, включающая отдельные ингредиенты лекарственного средства, может быть введена одновременно или с перерывами.
Далее, настоящее изобретение будет описано более подробно с помощью примеров, которые не должны трактоваться как ограничение изобретения.
ПРИМЕРЫ
Пример 1
Действие добавления лекарственного средства на подавление пролиферации раковых клеток
Клетки colon26 (полученные из рака толстой кишки мыши) инокулировали на 96-луночный планшет (1×104 клеток/50 мкл, 10%FCS-RPMI среда/лунка) и инкубировали при 37°C на протяжении 16 часов. В каждую лунку добавляли хорошо известное противораковое средство (цисплатин (сокращенно "CDDP"), оксалиплатин (сокращенно "L-OHP"), фторурацил (5-FU), паклитаксел (TXL; сокращенно "PTX"), доцетаксел (TXT; сокращенно "DTX"), иринотекан (CPT-11; сокращенно "CPT") или циклофосфамид (сокращенно "CPA")) и/или циклодекстриновое соединение включения (сокращенно "GC-CD") производного соединения холестанола, в которых G в формуле (1) обозначает GlcNAcβ- (сокращенно "GC"), (многократное разбавление FCS(-)-среда: итоговая концентрация: ≤500 мкм, 50 мкл), с последующим инкубированием при 37°C на протяжении двух дней. GC-CD получали в соответствии со способом, раскрытым в примере 1(2) патентного документа 4. Конкретно, получали 40% водный раствор гидроксипропил-β-циклодекстрина, и к раствору добавляли GC с последующим перемешиванием с встряхиванием (80°C в течение 30 минут) для получения за счет этого GC-CD.
В качестве контроля использовали лунки, в которые была добавлена только FCS(-)-среда. Определение количества жизнеспособных клеток выполняли посредством набора для подсчета клеток (продукт Dojin).
Скорость (%) подавления клеточной пролиферации (CPI) рассчитывали с помощью следующего уравнения. Фиг.1 (фиг.1-A, 1-B и 1-C) показывает результаты.
Скорость CPI (%)=(1- обработанные клетки OD450-650/необработанные клетки OD450-650)×100

Пример 2
Действие подавления пролиферации различных раковых клеток
Повторяли процедуру примера 1 за исключением того, что клетки colon26 заменили на MKN45 (полученные из рака желудочно-кишечного тракта человека), NCIH226 (полученные из рака легких человека) и Colo201 (полученных из рака толстой кишки человека). Скорость CPI (%) определяли аналогичным способом. Фиг.2 показывает результаты.
В примере 2 также использовали циклодекстриновое соединение включения (сокращенно "GGC-CD") производного соединения холестанола, в котором G в формуле (1) обозначает GlcNAcβ1,4-Galβ- (сокращенно "GGC"). GGC-CD получали способом, аналогичным упомянутому выше способу получения GC-CD, за исключением того, что соединение холестанола заменили на GGC. Определяли скорость CPI по отношению раковых клеток. Фиг.3 показывает результаты.
Пример 3
Действие добавления лекарственного средства на подавление пролиферации раковых клеток in vivo
В следующих примерах в качестве испытуемых животных использовали мышей Balb/c (возраст 6 недель, самки).
(1) Клетки colon26 (1×104 клеток/мышь) интраперитонеально инокулировали мышам (0 день). На следующий день после инокуляции (1 день) CDDP и/или GC-CD регулировали физиологическим раствором (изотонический раствор Otsuka) до интересующей концентрации, и мышам интраперитонеально вводили CDDP, GC-CD или CDDP+GC-CD (500 мкл) с последующим выращиванием. На 19 день мышей вскрывали и измеряли массу брыжейки и большого сальника. Контрольной группе (n=10; 10 мышей/группе) вводили только физиологический раствор (500 мкл).
Фиг.4 показывает результаты.
(2) Клетки colon26 (1×104 клеток/мышь) интраперитонеально инокулировали мышам (0 день). На 1 день, 2 день, 3 день, 6 день, 7 день и 8 день CDDP и/или GC-CD регулировали физиологическим раствором (изотонический раствор Otsuka) до интересующей концентрации, и мышам интраперитонеально вводили CDDP, GC-CD или CDDP+GC-CD (500 мкл) с последующим выращиванием. На 21 день мышей вскрывали и измеряли массу брыжейки и большого сальника. Контрольной группе (n=10; 10 мышей/группе) вводили только физиологический раствор (500 мкл).
Фиг.5 показывает результаты.
(3) Клетки colon26 (1×104 клеток/мышь) интраперитонеально инокулировали мышам (0 день). На 7 день, CDDP и/или GC-CD регулировали физиологическим раствором (изотонический раствор Otsuka) до интересующей концентрации, и мышам интраперитонеально вводили CDDP, GC-CD или CDDP+GC-CD (500 мкл) с последующим выращиванием. На 18 день мышей вскрывали и измеряли массу брыжейки и большого сальника. Контрольной группе (n=10; 10 мышей/группе) вводили только физиологический раствор (500 мкл).
Фиг.6 показывает результаты.
Пример 4
Противоопухолевое действие с помощью добавления лекарственного средства
В качестве испытуемых животных использовали мышей Balb/c (возраст 6 недель, самки). Клетки colon26 (1×104 клеток/мышь) интраперитонеально инокулировали мышам (0 день). На 2 день и/или 3 день CDDP и/или GC-CD регулировали физиологическим раствором (изотонический раствор Otsuka) до интересующей концентрации, и мышам интраперитонеально вводили CDDP, GC-CD или CDDP+GC-CD (500 мкл) (см. легенду фиг.7 для графика введения) с последующим выращиванием. Продолжительность выживания (дни) подсчитывали до 43 дня. Контрольной группе (n=10; 10 мыши/группа) вводили только физиологический раствор (500 мкл).
Фиг.7 показывает результаты.
Пример 5
Противоопухолевое действие с помощью добавления лекарственного средства
В качестве испытуемых животных использовали мышей Balb/c (возраст 6 недель, самки). Клетки colon26 (5×104 клеток/мышь) подкожно инокулировали мышам (0 день). После подтверждения, что размер опухоли достиг приблизительно 4 мм (7-10 день после инокуляции), CDDP и/или GGC-CD регулировали физиологическим раствором (изотонический раствор Otsuka) до интересующей концентрации и мышам через хвостовую вену вводили CDDP, GGC-CD или CDDP+GGC-CD (200 мкл) с последующим выращиванием. Отслеживали зависимое от времени изменение размера опухоли до 21 дня и определяли соответствующий объем опухоли. Контрольной группе (n=7; 7 мыши/группа) вводили только физиологический раствор (200 мкл).
Фиг.8 показывает результаты.
Пример 6
Действие подавления ракового метастазирования за счет добавления лекарственного средства
В качестве испытуемых животных использовали мышей Balb/c (возраст 6 недель, самки). Клетки colon26 (5×104 клеток/мышь) интраперитонеально инокулировали мышам (0 день). Непосредственно после инокуляции CDDP и/или GC-CD или GGC-CD регулировали физиологическим раствором (изотонический раствор Otsuka) до интересующей концентрации и мышам через хвостовую вену вводили CDDP, GC-CD (или GGC-CD), или CDDP+GC-CD (или GGC-CD) (200 мкл) с последующим выращиванием. На 14 день мышей разрезали и подсчитывали опухолевые узелки в легких. Контрольной группе (n=10; 10 мыши/группа) вещество не вводили.
Фиг.9 показывает результаты.
Как описано выше в данной заявке, посредством применения в комбинации производного соединения холестанола настоящего изобретения или его циклодекстринового соединения включения и противоракового средства пролиферация различных раковых клеток сильно подавляется, и может быть получен синергичный эффект и/или эффект усиления противоопухолевого действия известного противоракового средства.










Изобретение относится к химико-фармацевтической промышленности и представляет собой противораковое химиотерапевтическое средство, содержащее в комбинации производное холестанола, представленное формулой (1),
где G представляет собой GlcNAc-Gal- или GlcNAc-, или его циклодекстриновое соединение включения и иринотекан. Противораковое химиотерапевтическое средство имеет превосходную эффективность и меньшие побочные действия. 3 н. и 4 з.п. ф-лы, 6 пр., 9 ил.
1. Противораковое химиотерапевтическое средство, содержащее в комбинации производное соединение холестанола, представленное формулой (1):

в которой G представляет GlcNAc-Gal- или GlcNAc-, или его циклодекстриновое соединение включения и противораковое средство, представляющее собой иринотекан.
2. Противораковое химиотерапевтическое средство по п.1, которое представляет собой многокомпонентное средство.
3. Противораковое химиотерапевтическое средство по п.1, которое находится в форме набора, включающего лекарственное средство, содержащее в своем составе производное соединение холестанола и лекарственное средство, содержащее противораковое средство.
4. Противораковое химиотерапевтическое средство по любому из пп.1-3, в котором лекарственное средство, содержащее производное соединение холестанола, представляет собой липосомальную композицию.
5. Применение в комбинации производного соединения холестанола, представленного формулой (1):
в которой G представляет GlcNAc-Gal- или GlcNAc-, или его циклодекстринового соединения включения и противоракового средства, представляющего собой иринотекан, для получения противоракового химиотерапевтического средства.
6. Способ лечения рака, включающий введение в комбинации производного соединения холестанола, представленного формулой (1):

в которой G представляет GlcNAc-Gal- или GlcNAc-, или его циклодекстринового соединения включения и противоракового средства, представляющего собой иринотекан, нуждающемуся в этом пациенту.
7. Способ по п.6, в котором производное соединение холестанола или его циклодекстриновое соединение включения и противораковое средство вводят нуждающемуся в этом пациенту одновременно или по отдельности и с перерывами.
| EP 1921086 A1, 14.05.2008 | |||
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| ПРОТИВООПУХОЛЕВОЕ СРЕДСТВО | 2003 |
|
RU2240793C1 |

