Область техники
В целом, настоящее изобретение относится к области химической промышленности и, в частности, к области фармацевтической промышленности.
В частности, изобретение относится к способу получения компактированных матриц, которые могут представлять собой таблетки или устройства, применимые для высвобождения активных веществ, характеризующиеся замедленным высвобождением, и к компактированным матрицам, полученным таким образом. В частности, изобретение относится к способу получения компактированных матриц, которые могут представлять собой таблетки или устройства для высвобождения активных веществ, который обеспечивает стадию прямого прессования конкретных компонентов и стадию термической обработки.
Уровень техники
На протяжении примерно 40 лет исследования в области фармацевтики были посвящены изучению и разработке новых систем для модифицирования и регулирования высвобождения активных веществ в живых организмах.
Такое модифицирование направлено на замедление высвобождения (замедленное или пролонгированное высвобождение) лекарственных препаратов в организме для снижения частоты введения лекарства и также в ряде случаев на регулирование скорости высвобождения активных веществ (контролируемое высвобождение, КB) с тем, чтобы обеспечить достижение кинетики высвобождения нулевого порядка, т.е. такой кинетики, которая не зависела бы от дозы препарата, содержащейся в лекарственной форме (Extended Release and Targeted Dmg Delivery System, in Remington The Science and Practice of Pharmacy, 21-e издание, глава 47 стр.939-936).
Другие модификации направлены на то, чтобы высвобождение лекарственного препарата происходило в конкретном участке организма при воздействии некоторых стимулирующих высвобождение параметров (рН, температуры, ферментной активности, ионной силы) (Morishita M. et al. J Drug Deliv Sci Technol 16(1): 19-24, 2006) или через заданный период времени или через заранее заданные промежутки времени (отсроченное или пульсирующее высвобождение) (Gazzaniga et al. European Journal of Pharmaceutics и Biopharmaceutics 68(1): 11-18, 2008).
В частности, в формах для перорального введения замедленного высвобождения лекарственного препарата можно добиться путем применения подходящих полимеров, используемых в маленьких количествах в виде покрывающих пленок или в больших количествах для образования матричных систем. В обоих случаях состав пленки или матрицы способен оказывать влияние на высвобождение активных веществ, и, таким образом, во многих случаях скорость высвобождения может быть рассчитана заранее, а затем проверена путем соответствующих исследований растворимости "in vitro" (Kanjickal DG, Lopina ST. Modeling of drug release from polymeric delivery systems - A review. Critical Reviews in Therapeutic Drug Carrier Systems 21(5): 345-386), 2004).
Пленки можно наносить непосредственно на оболочку таблеток или на оболочку гранул или пеллет, которые можно вводить как таковые или после инкапсуляции или превращения в таблетки.
Полимеры, применяемые для создания матриц, регулируют высвобождение лекарственных препаратов вследствие различных скоростей растворения или разрушения таких полимеров или за счет диффузии активного веществ в матрице, которая в случае гидрофильных полимеров может набухать и превращаться в гель и более или менее легко разрушаться (Brazel CS, Peppas NA. Mechanisms of solute and drug transport in relaxing, swella-ble, hydrophilic glassy polymers. Polymer 40(12): 3383-3398, 1999).
Существуют более "интеллектуальные" системы, называемые системами или средствами доставки лекарственного вещества (СДЛВ), которые могут быть получены с применением дорогостоящих и более сложных промышленных способов (Hilt JZ, Peppas NA, International Journal of Pharmaceutics 306(1-2): 15-23, 2005). Прототипом СДЛВ являются так называемые "осмотические" системы, в которых используют полупроницаемые мембраны и осмотическое давление, образующееся внутри этих мембран, для замедления высвобождения лекарственного препарата. Высвобождение лекарственного препарата в растворе или в суспензии происходит благодаря микроотверстиям, создаваемым с помощью лазерного луча на поверхности таблетки, при постоянной скорости в соответствии с кинетикой высвобождения нулевого порядка (US 4160020; WO 03/075894 A1). Недостатком таких систем является возможность в ряде случаев массового высвобождения всей содержащейся дозы, явление, известное как "сброс дозы", с сопутствующими токсичными эффектами для организма в зависимости от вида доставляемого активного вещества. С этой точки зрения, "таблетированные" формы предоставляют больше гарантий, в том числе на стадии контроля качества промышленного производства.
В матричных системах используют недиспергируемые в воде или гидрофобные системы, например этилцеллюлозу, или гидрофильные полимеры, способные набухать в присутствии жидкостей на водной основе, такие как, например, гидроксипропилметилцеллюлоза, которая в зависимости от своей молекулярной массы и степени замещения может также образовывать гели, не очень легко поддающиеся разрушению.
В целом, такие матричные системы получают способом гранулирования или "таблетирования" как для того, чтобы добиться улучшения гомогенизации компонентов и избежать явления расслоения при смешивании порошков и во время их прессования, так и для того, чтобы сделать возможным получение матрицы, эффективно регулирующей высвобождение лекарственного препарата. Такие способы можно проводить во влажных условиях (влажное гранулирование, распылительная сушка) или сухих условиях (сухое гранулирование или вальцевание и экструзия горячего расплава) (Oral Solid Dosage Forms, in Remington The Science and Practice of Pharmacy 21-e издание, глава 45, стр.889-928).
Поскольку промышленные способы, включающие стадию гранулирования, по своей природе считаются экономически нецелесообразными, на предприятиях предпочитают использовать способы прямого прессования, в том числе благодаря наполнителям, разработанным для этой цели и имеющимся на рынке (Gohel MC, Jogani PD. Journal of Pharmacy and Pharmaceutical Sciences 8(1): 76-93, 2005; Goto K, et al. Drug Development and Industrial Pharmacy 25(8)869-878, 1999; Michoel A, et al. Pharmaceutical Development and Technology 7(1)79-87, 2002).
Большинство систем с контролируемым высвобождением, имеющихся на рынке (Colombo et al. Swelling matrices for controlled drug delivery: gel-layer behaviour, mechanisms and optimal performance. Pharm Sci Technol Today 3(6), 2000), регулируют высвобождение лекарственных препаратов с помощью матриц, основанных на применении гидрофильных полимеров (Peppas NA et al. Hydrogels in pharmaceutical formulations. European Journal of Pharmaceutics and Biopharmaceutics 50(1)27-46, 2000).
Как было указано, в общем, системы, эффективно регулирующие высвобождение активных веществ с помощью полимерной матрицы, получают способом влажного гранулирования (ЕР 1681051 A1; US patent 5549913).
Фактически, существует немного примеров контролируемого высвобождения с помощью полимерных матриц, полученных прямым прессованием. Например, М.Е.Pina с коллегами предложил (Pharmaceutical Development and Technology 11(2): 213-228, 2006) способ модифицированного высвобождения ибупрофена, лекарственного препарата, который не очень хорошо растворим в воде, с помощью матрицы, полученной путем прямого прессования, состоящей, главным образом, из гидрофильного полимера, набухающего в водной среде гидроксипропилметилцеллюлозы (ГПМЦ). Пеппас и Сиепманн (Advanced Drug Delivery Reviews 48(2-3): 139-157, 2001) опубликовали полный обзор по моделированию высвобождения лекарственных препаратов из матриц, состоящих из ГПМЦ.
Е.Кроули с коллегами (Е.Crowley.) (International Journal of Pharmaceutics 269(2): 509-522, 2004) предложил способ модифицированного высвобождения гвайфенезина, водорастворимого лекарственного препарата, с помощью матрицы, полученной путем прямого прессования, изготовленной из гидрофобной полиэтилцеллюлозы.
Способы получения матриц для контролируемого высвобождения, которые включают термическую обработку, заслуживают особого внимания, особенно развивающаяся технология экструзии термопластичных полимеров, известная как экструзия горячего расплава (ЭГР), позволяющая получить монолитные матрицы, которые можно использовать для получения гранулята, или непосредственно регулярные геометрические формы, применяемые как "таблетки".
Этот способ включает плавление полимера в присутствии возможных вспомогательных веществ путем нагревания до температуры на 10-60°С выше, чем температура стеклования (Tg) аморфных полимеров или температура плавления полукристаллических полимеров. Такой расплав после приобретения подходящей вязкости продавливают через щель с регулярным сечением, придавая, таким образом, форму указанного сечения, при последующем охлаждении (Repka MA, et al. Drug Development and Industry Pharmacy Part 133(9): 909-926 and Part II 33(10): 1043-1057, 2007).
В известном уровне техники было обнаружено, что в некоторых случаях можно оказывать влияние на высвобождение активных веществ из таблеток, напрямую подвергая таблетки стадии термической обработки.
Омелзукс с соавторами (Omelczuck et al.) (Pharmaceutical Research 10, 542-548, 1993) сообщил, что термическая обработка (при температуре между 40 и 80°С в течение 24 часов) таблеток, содержащих поли(dl-молочную кислоту) (ПМК) и микрокристаллическую целлюлозу, увеличивала время высвобождения теофиллина. На основе представленных кривых растворения сделан вывод, что такое высвобождение происходит согласно сложной кинетике, отличной от кинетики нулевого порядка.
Азарми С. с соавторами (Azarmi S. et al.) (International Journal of Pharmaceutics 246 (2002), 171-177) подтвердил, что таблетки индометацина, полученные путем прямого прессования лекарственного препарата с Eudragit RS РО или RL РО и лактозой в соотношении 3:3:4 и подвергаемые нагреванию при температуре выше 50 или 60°С в течение 2-24 часов, проявляли замедленное высвобождение по сравнению с таблетками, которые не подвергались термической обработке, без заметного изменения предела прочности.
Аналогичные результаты были получены Азарми с соавторами (Azarmi S. et al.) (Pharmaceutical Development и Technology, 10: 233-239, 2005) с таблетками диклофенака натрия (полученными путем прямого прессования диклофенака натрия, Eudragit RS РО или RL РО и лактозы 3:4:3), подвергаемыми нагреванию при 50-70°С в течение 2-24 часов.
Ранее Билла с соавторами (Billa et al.) (Drug Development и Industrial Pharmacy, 24(1), 45-50, 1998) исследовали влияние термической обработки при 60°С на таблетки, содержащие диклофенак натрия/Eudragit NE40D/микрокристаллическую целлюлозу, и отметили достижение замедленного высвобождения, связанного с увеличением предела прочности таблеток.
Данные наблюдений влияния термической обработки таблеток на свойства высвобождения лекарственных препаратов ограничены приведенными выше примерами, т.е. таблетками на основе Eudragit или содержащими ПМК, и результаты, полученные после длительного нагревания, весьма ограничены как в отношении замедления высвобождения, так и полученной кинетики высвобождения.
В области контролируемого высвобождения также известно применение водонерастворимых сшитых полимеров, которые, тем не менее, являются гидрофильными и способными набухать в водной среде (Brazel CS, Peppas NA 1999. Mechanisms of solute and drug transport in relaxing, swellable, hydrophilic glassy polymers. Polymer 40(12): 3383-3398).
К этой категории принадлежит поликарбофил (CAS RN 9003-01-4) (Handbook of Pharmaceutical Excipients, пятое издание. Pharmaceutical Press, стр.539-541, 2006), полимер полиакриловой кислоты, сшитый с дивинилгликолем, который известен своим применением при производстве фармацевтических форм с контролируемым высвобождением, например в форме таблеток, пластинок или пленок, обладающих биоадгезивными свойствами. В качестве примера можно ознакомиться с заявками на патент WO 2005/065685 и WO 01/95888 и патентом США 5102666.
Поликарбофил применяют внутри фармацевтических форм прежде всего благодаря его биоадгезивным свойствам. Робинсон с соавторами (Robinson at al.) (Journal of Pharmaceutical Sciences 89(7): 850-86б, 2000) опубликовали обзор биоадгезивных свойств поликарбофила и других полимеров, применяемых в лекарственных формах. Репка с соавторами (Repka at al.) (Journal of Controlled Release 70(3): 341-351, 2001) исследовали биоадгезивные свойства буккальных пленок, полученных с помощью экструзии горячего расплава, также содержащих поликарбофил.
Также стоит отметить зависящую от рН способность поликарбофила набухать, поглощая воду, с 1000-кратным увеличением своего первоначального объема и 10-кратным увеличением своего первоначального диаметра. Согласно указаниям из монографии US Pharmacopeia 31, относящимся к поликарбофилу, абсорбирующая способность в отношении раствора бикарбоната натрия не должна быть меньше чем 62 г на 1 г сухого полимера.
Благодаря таким характеристикам поликарбофил применяют не только в фармацевтических препаратах, но также в пищевых добавках для лечения кишечных дисфункций, хронического запора, дивертикулита и синдрома раздраженной толстой кишки.
В WO 01/95888 A1 описаны биоадгезивные таблетки с замедленным высвобождением, содержащие активный ингредиент, который подвергается метаболизму под действием 5α-редуктазы, водорастворимый полимер, такой как, например, гидроксипропилметилцеллюлоза, и водонерастворимый, водонабухающий сшитый поликарбоксильный полимер, в частности поликарбофил. Способ приготовления таких таблеток не включает какую-либо стадию нагревания. В WO 2005/065685 предложены биоадгезивные таблетки замедленного высвобождения, содержащие активный ингредиент и полимерную систему, включающую по меньшей мере два полимера, где один полимер представляет собой кислый нерастворимый полимер, а другой полимер представляет собой биоадгезивный полимер; указанная полимерная система может включать, например, этилцеллюлозу, поликарбофил и микрокристаллическую целлюлозу. Согласно этому документу в технологическом процессе для получения таблеток не предусмотрены стадии нагревания.
Краткое описание изобретения
В первом аспекте задача настоящего изобретения состоит в обеспечении компактированной матрицы, содержащей сшитый поликарбоксильный полимер, неразрушаемой и обладающей биоадгезией, способной набухать за счет поглощения воды, образующей неразрушаемый гелеобразный слой, применимой, например, для замедленного высвобождения активных веществ.
Указанную задачу решают путем применения способа получения компактированной биоадгезивной матрицы, включающего следующие стадии:
- приготовление однородной порошковой смеси, содержащей по меньшей мере одну алкилцеллюлозу или одну гидроксиалкилцеллюлозу и водонерастворимый, водонабухающий сшитый поликарбоксильный полимер;
- приготовление из указанной порошковой смеси спрессованных или компактированных заготовок путем прямого прессования или сухого компактирования;
- нагревание спрессованных или компактированных заготовок, полученных таким образом, при температуре в диапазоне 80-250°С в течение 1-60 минут.
В другом аспекте задача изобретения заключается в обеспечении спрессованной заготовки, содержащей вышеуказанную уплотненную биоадгезивную матрицу, способную набухать в воде, для высвобождения активных веществ, характеризующуюся замедленным или контролируемым высвобождением. Указанную задачу решают с помощью способа получения компактированной матрицы, описанного выше, в котором вышеуказанная однородная смесь порошков также содержит по меньшей мере одно активное вещество.
Термин "спрессованная заготовка" предназначен для обозначения не только традиционных таблеток для фармацевтического применения, в частности таблеток для перорального введения, способных высвобождать активные вещества или вещества, которые восстанавливают физиологические состояния, но также других устройств, полученных путем прессования порошка, например уретральных суппозиториев, таблеток и пластинок для вагинального, буккального, назального, стоматологического, отологического, глазного или даже эпидермального применения, способных высвобождать активные вещества или вещества, которые восстанавливают физиологические состояния. Применение таких таблеток и устройств не ограничивается сектором фармацевтической продукции для человека и ветеринарным применением, где под активным веществом понимают лекарственные вещества согласно определению, данному в указанном порядке в директиве ЕС 2004/27 СЕ (ст.1) и 2004/28/СЕ (ст.1), но распространяется и на другие области, такие как медицинские устройства согласно определению, данному в директиве ЕС 93/42/СЕЕ, область продуктов питания согласно определению, данному в ст.2 Правил ЕС (СЕ) No. 178/2002, область пищевых добавок, как определено директивой 2002/46/СЕ, область диетических продуктов и продуктов для детей, как определено директивой ЕС СЕ No. 89/398, область средств защиты растений согласно определению, данному директивой ЕС 91/414/СЕ, область органических и минеральных удобрений согласно определению и классификации Правил ЕС (СЕ) No. 2003/2003, область дезинфицирующих средств и инсектицидов и биоцидов, в общем, согласно определению, данному в директиве ЕС 98/8/СЕ, область моющих средств. Кроме того, радиофармацевтические препараты, радионуклиды и молекулы, меченные радионуклидами, могут содержаться в таких матрицах и высвобождаться из них для диагностических, терапевтических и общих биоцидных целей.
Вышеуказанные порошковые смеси могут также содержать разбавитель. Предпочтительно, когда разбавитель состоит из безводной лактозы (CAS RN 63-42-3) или моногидрата лактозы (CAS RN 64044-51-5) во всех известных аморфных и кристаллических физических формах, также полученных с применением сушки распылением или агломерации, подобно Tabettose® и Pharmatose DCL 15®, и/или микрокристаллической целлюлозы (например, Avicel PH, Emcocel, Tabulose). Можно также применять предварительно полученные смеси лактозы/микрокристаллической целлюлозы, такие как, например, высушенный распылением состав, содержащий 75% моногидрата альфа-лактозы и 25% микрокристаллической целлюлозы (MicroceLac®) или Cellactose®, или другие наполнители, совместно обрабатываемые прямым прессованием, такие как Ludipress, Starlac, Pharmatose DCL 40, Avicel CE 15, Celocal, Proslov.
Вышеуказанную алкилцеллюлозу можно выбрать, например, из группы, содержащей метилцеллюлозу (CAS RN 9004-67-5) и этилцеллюлозу (CAS RN 9004-57-3), а вышеуказанную гидроксиалкилцеллюлозу можно выбрать, например, из группы, содержащей гидроксипропилцеллюлозу (CAS RN 9004-64-2 и RN 78214-41-2), гидроксипропилметилцеллюлозу (CAS RN 9004-65-3), гидроксиэтилцеллюлозу (CAS RN 9004-62-0), гидроксиэтилметилцеллюлозу (CAS RN 9004-42-2).
Также возможно применение, при неполном замещении алкил- или гидроксиалкилцеллюлозы, следующих веществ, в том числе в комбинации друг с другом: кросповидона, повидона (9003-39-8), сополимера винилпирролидона и винилацетата (Kollidon® VA64), ацетатфталата целлюлозы (CAS RN 9004-38-0), фталата гипромеллозы (CAS RN 9050-31-1), поливинилового спирта (CAS RN 9002-89-5), поливинилацетатфталата (CAS RN 34481-48-6), различных циклодекстринов (которые описаны в указанной выше монографии Handbook of Pharmaceutical Excipients, пятое издание. Pharmaceutical Press), различных видов метакрилатных полимеров, также поставляемых под наименованием Eudragit (Rohm GmbH), таких как полимеры, именуемые Е, L, S, RS, RL, PO, NE, RSPM, в различных продуктах, производимых также компаниями Eastman Chemical Company и BASF, триацетата глицерина, триэтилцитрата, ацетилтрибутилцитрата, дибутилсебацината, диэтилфталата, дибутилфталата, диоктилфосфата, полиэтиленгликоля, полиэтиленоксидов (CAS RN 25322-68-3), кальций карбоксиметилцеллюлозы (CAS RN 9050-04-8), натрий карбоксиметилцеллюлозы (CAS RN 9004-32-4), инулина (CAS RN 9005-80-5), хитозана (CAS RN 9012-76-4) и его производных, гуаровой смолы (CAS RN 9000-30-0), ксантановой камеди (11138-66-2) и трагакантовой камеди (CAS RN 900-65-1), карбомера (CAS RN 9003-01-04 и 96827-24-6), каррагенина (как описано в указанной выше монографии Handbook of Pharmaceutical Excipients, пятое издание), альгиновой кислоты (CAS RN 9005-32-7), полоксамера (CAS RN 9003-11-6), алифатических полиэфиров (как описано в указанной выше монографии Handbook of Pharmaceutical Excipients, пятое издание), ацетатбутирата целлюлозы, хитозан лактата, пектина, сополимера полиэтилена и винилацетата, полиэтилена, сополимера поливинилацетата и метакриловой кислоты, карнаубского воска, бутилированного гидроксианизола, аскорбилпальмитата, глицерил пальмитостеарата, гидрогенизированного соевого и касторового масла (Sterotek® К), глицерил моностеарата, d-α-токоферола (витамина Е), витамин Е сукцината, витамина Е и TPGS, метилпарабена, бутилстеарата, стеарилового спирта, монопальмитата сахарозы (сахарного сложного эфира), сложных глицериновых эфиров и эфиров ПЭГ (Gelucire 44/14), полиоксиэтиленалкиловых эфиров, глицерил пальмитостеарата Precirol® ATO 5, минерального масла, касторового масла и наполнителей, используемых для получения шипучих смесей или систем.
Вышеуказанный водонерастворимый сшитый поликарбоксильный полимер, который способен набухать в воде, предпочтительно состоит из поликарбофила (регистрационный номер CAS 9003-01-04).
Температура, при которой нагревают спрессованные заготовки, предпочтительно составляет 90-160°С, а время нагревания обычно составляет 1-30 минут, в частности 1-20 минут. Скорость нагревания, применяемая для нагревания спрессованных заготовок до температуры обработки, может варьировать от 1°С/минут до 50°С/минут.
Прессование порошков, подвергаемых последующей термической обработке, можно осуществлять с применением давлений между 100-500 МПа. Также можно получить спрессованные порошковые заготовки с низким пределом прочности, которые подвергают последующей термической обработке с применением давлений между 5 кПа-100 МПа.
Форма спрессованных заготовок может иметь любую регулярную трехмерную геометрическую конфигурацию, и масса может варьировать в соответствии с необходимостью и применением (человеком или в ветеринарии), превышая 100 г при сельскохозяйственном использовании.
В состав таких спрессованных заготовок можно добавить, при необходимости, все вспомогательные вещества, которые обычно применяют в процессе прессования и которые известны специалистам в данной области техники: скользящие вещества, смазывающие вещества, антиадгезионные агенты, дезинтегрирующие агенты и супердезинтегрирующие агенты, ароматизаторы, подсластители и поглотители.
Предложенные таблетки можно покрыть путем применения классических способов покрытия полимерной пленкой и/или покрытия сухим методом (Pharmaceutical Dosage Forms: Tablets, том 1, 2, 3, издано Н.А.Lieberman, L.Lachman, J.B.Schwartz, Dekker, второе издание, США, 1989) для придания активному веществу устойчивости к действию желудочного сока, растворимости в кишечнике или защиты от неблагоприятных воздействий окружающей среды.
Таблетки, являющиеся предметом настоящего изобретения, можно применять в виде ядра или слоя, содержащего или не содержащего активное вещество, для получения таблеток, известных под названием таблетки с вкладкой, многослойные таблетки и таблетки с ядром (Pharmaceutical Dosage Forms: Tablets, том 1, издано Н.А.Lieberman, L.Lachman, J.B.Schwartz, Dekker, второе издание, США, 1989). Различные дополнительные слои могут иметь качественный состав, идентичный составу, предложенному в настоящем изобретении, и/или различное содержание активных веществ или дополнительного активного вещества или они могут представлять собой различные матрицы, которые уже были описаны или применяются в данной области.
В случае многослойных таблеток матрица согласно изобретению представляет собой по меньшей мере один слой многослойной таблетки.
В случае таблеток с ядром матрица, предложенная в настоящем изобретении, может представлять собой ядро или самый верхний слой, называемый внешним слоем, содержащий или не содержащий активное вещество или содержащий другие активные вещества в каждом слое.
В случае таблеток с вкладкой матрица согласно изобретению может представлять собой как внешний слой, так и вкладку и может содержать активное вещество или несколько активных веществ или не содержать их. Даже если способ согласно настоящему изобретению предпочтительно представляет собой способ прямого прессования, в определенных ситуациях для преодоления некоторых недостатков, которые могли быть вызваны размерами частиц некоторых активных веществ, при приготовлении таблеток или ядер/вкладок или слоев можно по-прежнему применять методы, известные специалистам в данной области техники. К таким известным методам относят влажное гранулирование (влажное гранулирование, гранулирование в псевдоожиженном слое, сушка распылением, замораживание распылением) или сухое гранулирование (сухое гранулирование или вальцевание); или, возможно, способ прессования можно применить к пилюлям, подвергаемым процессу, известному как сферонизация, который позволяет получить гранулы сферической формы и контролируемого размера (Remington, 21-е издание, глава 45, стр.903). Естественно, различные способы гранулирования требуют минимального добавления вспомогательных веществ или наполнителей, необходимых для реализации этих разных способов и известных специалистам в данной области техники. В случае активных веществ, чувствительных к окислению, нагревание можно легко провести в инертной атмосфере, например в атмосфере азота.
Что касается летучих или сублимирующихся активных веществ или в любом случае, при необходимости, термическую обработку можно выполнить в обычной или инертной атмосфере, повышая давление такой атмосферы до величины на 0.5 МПа выше давления окружающей среды.
Стадия охлаждения после нагревания может происходить естественным образом или принудительно, например, путем регулирования охлаждения с помощью вентиляции сухого воздуха или инертного газа (N2, Ar, He) при комнатной температуре или сухого воздуха или инертного газа, охлажденного до температуры ниже комнатной температуры. После нагревания перед упаковкой может потребоваться выдержка в условиях окружающей среды, котороая, в зависимости от выбранного состава, может продолжаться даже 24 часа. В целом, такое время ожидания не влияет на качество продукции, но в любом случае предпочтительно выждать стандартное время, составляющее 5 минут.
Предпочтительный состав порошков для применения в способе согласно изобретению содержит активное вещество, микроцелак, этилцеллюлозу и поликарбофил. Поликарбофил, в общем, составляет 5-35% по массе от общей массы порошковой смеси до прессования и термической обработки, предпочтительно 10-25%.
Этилцеллюлоза и микроцелак, в общем, присутствуют в порошковой смеси в весовом отношении, варьирующем от 1:2 до 2:1, предпочтительно от 0.8:1 до 1,2:1, и составляют 45-95% по массе от общей массы смеси до прессования и термической обработки, предпочтительно 60-80%.
Этилцеллюлоза и поликарбофил, в общем, присутствуют в массовом соотношении, варьирующем от 1:5 до 5:1. Активное вещество содержится в порошковой смеси в количестве, варьирующем от 0,001 ppm (частей на миллион) до 50% по массе от общей массы смеси до прессования и термической обработки. Кроме того, в пределах указанного процентного содержания активное вещество можно смешивать с подходящими вспомогательными веществами для процесса солюбилизации, образующими гидротропические комплексы или комплексы включения; или с веществами, стимулирующими процессы желудочно-кишечного всасывания или в любом случае трансмукозальное всасывание лекарственных препаратов, известными как усилители; или веществами, которые физически или химически стабилизируют активное вещество.
Активное вещество может быть природным, синтетическим или полусинтетическим с фармакологическим действием, подходящим для терапевтического, диагностического или профилактического применения людьми или животными или может представлять собой вещество природного, синтетического или полусинтетического происхождения, биологически, физически или химически активное и подходящее для ухода за растениями (средства защиты растений), в виде удобрения, в виде дезинфицирующего средства и/или дезинфицирующего средства для человека или окружающей среды, или вещество, принадлежащее, в целом, к категории биоцидов, как определено ЕС.
Единственное условие, необходимое для применения активного вещества в способе согласно настоящему изобретению, заключается в том, что оно обладает достаточной термостабильностью в условиях нагревания (температуры, времени), предусмотренных самим способом.
Из спрессованных заготовок согласно настоящему изобретению могут также высвобождаться вещества, обладающие питательной способностью, применяемые в качестве диетических добавок для людей и животных, в том числе как для обычных субъектов, так и субъектов, страдающих от хронических или острых патологических состояний.
Предложенные спрессованные заготовки могут находить применение в качестве косметических продуктов, согласно определению "косметический", действующему в ЕС, если они надлежащим образом скомпонованы с веществами, которые разрешено применять в косметике.
Настоящее изобретение также относится к таблетке с замедленным высвобождением, обладающей свойством биоадгезии и содержащей активное вещество, по меньшей мере одну алкилцеллюлозу или гидроксиалкилцеллюлозу и водонерастворимый, водонабухающий сшитый поликарбоксильный полимер. Предпочтительно указанная таблетка характеризуется контролируемым высвобождением и кинетикой высвобождения активного вещества по существу нулевого порядка в водном растворе при рН 4-8.
Предпочтительно, когда вышеуказанная таблетка также содержит разбавитель, состоящий из безводной лактозы или моногидрата лактозы во всех известных кристаллических и аморфных физических формах, и/или микрокристаллическую целлюлозу. Таблетка также может содержать предварительно приготовленные смеси микрокристаллической целлюлозы/лактозы, известные специалистам в данной области техники как цельный наполнитель, такой как, например, MicroceLac®.
Варьирование соотношений между компонентами, указанное выше, условия прессования порошков или гранулята и условия нагревания, температура и время термической обработки позволяют регулировать скорость, с которой происходит высвобождение активного вещества, подчиняющееся, в целом, кинетике нулевого порядка в диапазоне рН между 4 и 8.
Если активное вещество отсутствует (0%), такая спрессованная и термически обработанная матрица, благодаря своей способности набухать в водной среде и своим биоадгезивным свойствам, все еще может оказывать терапевтическое действие при некоторых желудочно-кишечных дисфункциях или при некоторых патологиях, например при хроническом запоре, дивертикулите, синдроме раздраженной толстой кишки и при всех других патологиях, для лечения которых могут быть полезны описанные характеристики. Такую таблетку можно получить описанным выше способом. Используя свойства указанных матриц, которые после набухания в воде вновь приобретают свою первоначальную форму и размеры благодаря высушиванию, несколько активных веществ, в частности веществ, являющихся термолабильными или трудно получаемыми в твердом состоянии, можно ввести в эту матрицу с помощью имбибиции, т.е. путем пропитывания матриц согласно изобретению, приготовленных без лекарственного препарата, в водных растворах с подходящей концентрацией активного вещества. Через заданное время набухшие таблетки можно извлечь и оставить сушиться на воздухе или с помощью подходящего способа высушивания, путем принудительной вентиляции воздухом или инертным газом и возможного умеренного нагревания, или путем облучения ИК-лампой, или с применением способа лиофилизации для их повторного получения с первоначальной формой и размером.
Краткое описание чертежей
На фиг.1а показана усредненная кривая растворения (n=6) в 0,05 н HCl дилтиазема (ДТЗ), высвобожденного из таблеток, полученных способом согласно настоящему изобретению (пример 1) (Т), по сравнению с таблетками с идентичным составом, но не прошедших термическую обработку (NT). Отрезки отражают 95% доверительные интервалы.
На фиг.1b показаны усредненные кривые растворения (n=6) в фосфатном буфере (рН 7,2) дилтиазема (ДТЗ), высвобожденного из таблеток, полученных способом согласно настоящему изобретению (пример 1) (Т), по сравнению с ДТЗ, высвобожденным из таблеток с идентичным составом, но не прошедших термическую обработку (NT). Отрезки отражают 95% доверительные интервалы.
На фиг.1с показана кривая ДСК (сплошная линия) термически обработанных (150°С в течение 15 минут) таблеток, полученных способом согласно настоящему изобретению (пример 1), по сравнению с кривой ДСК таблеток с идентичным составом, но не прошедших термическую обработку (пунктирная линия).
На фиг.2а показаны усредненные кривые растворения в 0,05 н HCl дилтиазема (ДТЗ), высвобожденного из таблеток, полученных способом согласно настоящему изобретению (пример 2) (Т), по сравнению с ДТЗ, высвобожденным из таблеток с идентичным составом, но не прошедших термическую обработку (NT). Отрезки отражают 95% доверительные интервалы.
На фиг.2b показаны усредненные кривые растворения (n=6) в фосфатном буфере (рН 7,2) дилтиазема (ДТЗ), высвобожденного из таблеток, полученных способом согласно настоящему изобретению (пример 2) (Т), по сравнению с ДТЗ, высвобожденным из таблеток с идентичным составом, но не прошедших термическую обработку (NT). Отрезки отражают 95% доверительные интервалы.
На фиг.2с показана, снизу, кривая ДСК таблеток, полученных согласно настоящему изобретению (пример 2) (сплошная линия), по сравнению с кривой для таблеток с идентичным составом, но термически необработанных. Сверху приведены кривые ДСК для дилтиазема (штриховая линия) и микроцелака (пунктирная линия) по сравнению с кривой ДСК для таблеток с составом, идентичным составу таблеток согласно примеру 2, но термически необработанных (сплошная линия).
На фиг.3а показаны усредненные кривые растворения (n=6) в 0,05 н HCl дилтиазема (ДТЗ), высвобожденного из таблеток, полученных способом согласно настоящему изобретению (пример 3) (Т), по сравнению с ДТЗ, высвобожденным из таблеток с идентичным составом, но не прошедших термическую обработку (NT). Отрезки отражают 95% доверительные интервалы.
На фиг.3b показаны усредненные кривые растворения (n=6) в фосфатном буфере (рН 7,2) дилтиазема (ДТЗ), высвобожденного из таблеток, полученных способом согласно настоящему изобретению (пример 3) (Т), по сравнению с ДТЗ, высвобожденным из таблеток с идентичным составом, но не прошедших термическую обработку (NT). Отрезки отражают 95% доверительные интервалы.
На фиг.3с приведены для сравнения усредненные кривые растворения (n=6) в фосфатном буфере (рН 7,2) дилтиазема (ДТЗ), высвобожденного из 6 таблеток согласно примеру 3 и из 6 таблеток согласно примеру 2.
На фиг.3d приведены для сравнения друг с другом кривые ДСК таблеток согласно примеру 3 (сплошная линия) и таблеток с идентичным составом, но термически необработанных (штриховые линии).
На фиг.4а приведены для сравнения усредненные кривые растворения (n=6) в 0,05 н HCl дилтиазема (ДТЗ), высвобожденного из таблеток согласно изобретению (пример 1) (Т) и из таблеток с идентичным составом, но термически необработанных (NT), только что изготовленных (t=0) и после 47 дней хранения в блистерной упаковке.
На фиг.4b приведены для сравнения усредненные кривые растворения (n=6) в фосфатном буфере (рН 7,2) дилтиазема (ДТЗ), высвобожденного из таблеток согласно изобретению (пример 1) (Т) и из таблеток с идентичным составом, но термически необработанных (NT), только что изготовленных (t=0) и после 47 дней хранения в блистерной упаковке.
На фиг.5а приведены для сравнения усредненные кривые растворения (n=6) в 0.05 N HCl дилтиазема (ДТЗ), высвобожденного из таблеток согласно изобретению (пример 2) (Т) и из таблеток с идентичным составом, но термически необработанных (NT), только что изготовленных (t=0) и после 47 дней хранения в блистерной упаковке.
На фиг.5b приведены для сравнения усредненные кривые растворения в фосфатном буфере (рН 7.2) дилтиазема (ДТЗ), высвобожденного из термически обработанных таблеток согласно изобретению (пример 2) (Т) и из таблеток с идентичным составом, но термически необработанных (NT), только что изготовленных (t=0) и после 47 дней хранения в блистерной упаковке.
На фиг.5с приведены для сравнения усредненные кривые растворения в фосфатном буфере (рН 7.2) дилтиазема (ДТЗ), высвобожденного из термически обработанных таблеток согласно изобретению (пример 1), только что приготовленных (t=0) и после 13 месяцев хранения в блистерной упаковке. Отрезки отражают 95% доверительные интервалы.
На фиг.5d изображена усредненная кривая растворения (n=6) в 0,05 н HCl дилтиазема (ДТЗ), высвобожденного из таблеток партии согласно примеру 2, термически обработанных (Т), хранившихся в блистерной упаковке в течение 32 месяцев. Отрезки отражают 95% доверительные интервалы.
На фиг.5е изображена усредненная кривая растворения (n=6) в фосфатном буфере (рН 7.2) дилтиазема (ДТЗ) из таблеток партии согласно примеру 2, термически обработанных (Т), хранившихся в блистерной упаковке в течение 32 месяцев. Отрезки отражают 95% доверительные интервалы.
На фиг.5f приведены для сравнения усредненные кривые (n=6) доли растворенного дилтиазема (ДТЗ) относительно количества, присутствующего в растворе через 600 минут, в фосфатном буфере (рН 7.2), из разных партий термически обработанных таблеток согласно примеру 2 (Т) и таблеток с идентичным составом, но необработанных (NT), только что изготовленных (t=0) и хранящихся в блистерной упаковке в течение 13 (t=13 месяцев) и 32 месяцев (t=32 месяцев).
На фиг.6 показаны кривые растворения дилтиазема в фосфатном буфере (рН 7,2) из таблеток согласно настоящему изобретению (пример 2), подвергнутых различным способам термической обработки. В частности, на этом графике сравнивается влияние различных способов термической обработки на высвобождение дилтиазема (ДТЗ) в фосфатном буфере (рН 7,2) из таблеток согласно примеру 2. Усредненные кривые растворения (n=6) из таблеток: nt= не обработаны; 150-5=150°С×5 минутная обработка; 90-15=90°С×15 минутная обработка; 150-15=150°С×15 минутная обработка; 130-15=130°С×15 минутная обработка.
На фиг.7 графически показан уровень набухания в процентах (S%) таблеток согласно настоящему изобретению. В частности, на графике приведены для сравнения усредненные кривые набухания (n=3) термически обработанных таблеток согласно примерам 1 и 2. Отрезки отражают значения стандартного отклонения.
На фиг.7а приведены три фотографии, демонстрирующие термически обработанные таблетки согласно настоящему изобретению перед испытанием на растворимость и в конце испытания на растворимость в фосфатном буфере при достижении максимальной степени набухания. Слева направо приведены следующие фотографии: таблетка с первоначальным размером согласно примерам 1 или 2 перед испытанием на растворимость; таблетка согласно примеру 2 при максимальной степени набухания, достигаемой в конце испытания на растворимость при 37°С в фосфатном буфере; таблетка согласно примеру 1 при максимальной степени набухания, достигаемой в конце испытания на растворимость при 37°С в фосфатном буфере.
На фиг.7b приведена увеличенная фотография термически обработанной таблетки согласно настоящему изобретению при максимальной степени набухания, достигаемой в конце испытания на растворимость. В частности, фотография в большом масштабе, которая в неувеличенном виде приведена на фиг.7а, расположена в центре: таблетка согласно примеру 2 при максимальной степени набухания, достигаемой в конце испытания на растворимость при 37°С в фосфатном буфере.
На фиг.7с приведены следующие две фотографии: сверху - фотография трех таблеток согласно изобретению при максимальной степени набухания, внизу - вид в перспективе таблетки согласно изобретению при максимальной степени набухания. В частности, показаны фотографии термически обработанных таблеток согласно примеру 2 при максимальной степени набухания, достигаемой в конце испытания на растворимость при 37°С в фосфатном буфере (рН=7,2).
На фиг.7d представлен график, который иллюстрирует набухание спрессованных матриц, не содержащих активное вещество согласно изобретению. На этом графике, в частности, приведены для сравнения усредненные кривые набухания (n=3) матриц, не содержащих активное вещество и при соотношениях между компонентами согласно примеру 2, полученных двумя различными способами термической обработки (5 или 15 мин при 150°С) или не подвергаемых термической обработке (NT).
На фиг.8 показаны усредненные кривые растворения (n=6) в фосфатном буфере (рН 7,2) гликлазида, высвобожденного из таблеток (Т), полученных способом согласно настоящему изобретению (пример 4) (термически обработанных при 150°С в течение 5 и 15 минут), по сравнению с таблетками с идентичным составом, но не прошедших термическую обработку (NT). Отрезки отражают 95% доверительные интервалы.
На фиг.9 представлена сравнительная характеристика размера термически обработанной (150°С в течение 15 минут) таблетки согласно настоящему изобретению (пример 2), которая хранилась в блистерной упаковке в течение 38 месяцев, и размеров 6 таблеток из этой же партии, которые подвергались испытанию на растворимость в 0,05 н HCl после термической обработки (150°С в течение 15 минут) и хранения в блистерной упаковке в течение 13 месяцев, затем подвергались высушиванию воздухом при комнатной температуре и, наконец, после такого высушивания при комнатной температуре хранились на воздухе на протяжении еще 25 месяцев.
На фиг.10 представлена сравнительная характеристика термически обработанных (150°С в течение 15 минут) таблеток согласно примеру 2 при максимальной степени набухания, достигаемой в конце испытания на растворимость в 0,05 н HCl, и таблетки первоначального размера. В частности, слева показана таблетка согласно примеру 2 при максимальной степени набухания, достигаемой в конце испытания на растворимость в 0,05 н HCl при 37°С. Справа показана таблетка первоначального размера согласно примеру 2.
На фиг.11 представлен график, на котором показаны результаты исследования планарного сжатия (в процентах) порошка поликарбофила, подвергнутого различным способам изотермического нагревания, с применением микроскопа с нагревательным столиком.
На фиг.12 показана сравнительная характеристика кривой ДСК (эндотермический эффект), кривой ТГА (весовой процент относительно температуры) и кривой % планарного сжатия (ПС) относительно температуры порошка поликарбофила, полученной с применением микроскопа с нагревательным столиком (МНС).
На фиг.13 показана микрофотография СЭМ (сканирующего электронного микроскопа) поперечного сечения компактированной порошковой заготовки поликарбофила (не подвергнутой термической обработке). Компактированная заготовка была получена путем приложения давления 750 кПа в течение 15 мин на 100 мг порошка поликарбофила.
На фиг.14 показана микрофотография СЭМ поперечного сечения термически обработанной компактированной заготовки поликарбофила. Компактированная заготовка была получена путем приложения давления 750 кПа в течение 15 мин на 100 мг порошка поликарбофила. Затем компактированную заготовку подвергали нагреванию при 150°С в течение 15 мин в горячем сушильном шкафу.
На фиг.15 показана микрофотография СЭМ этой же пробы, показанной на фиг.14, при большем увеличении.
На фиг.16 показана микрофотография СЭМ поперечного сечения термически обработанной компактированной заготовки этилцеллюлозы/поликарбофила 3:2. Компактированная заготовка была получена путем приложения давления 750 кПа в течение 15 мин на 100 мг порошковой смеси этилцеллюлозы/поликарбофила 3:2. Затем компактированную заготовку подвергали нагреванию при 150°С в течение 15 минут в горячем сушильном шкафу.
На фиг.17 показана микрофотография СЭМ порошка поликарбофила, который не подвергали никакой термической обработке.
На фиг.18 показана микрофотография СЭМ порошка поликарбофила, который подвергали нагреванию при 150°С в течение 15 минут в горячем сушильном шкафу.
Подробное описание изобретения
Настоящее изобретение основано на экспериментальной работе, выполненной с применением смесей различных наполнителей, применяемых для приготовления таблеток, в которых на высвобождение активного вещества могла оказывать влияние энергообработка. Первоначальная задача состояла в получении таблеток, которые, при нагревании в атмосферных условиях, демонстрировали замедление высвобождения, не проявляя разложение компонентов композиции.
Также были предприняты попытки получить композицию, высвобождение активного вещества из которой происходило из тяжело разрушаемой матрицы согласно кинетике нулевого порядка, так чтобы скорость высвобождения со временем не зависела от остаточного количества активного вещества в композиции, что по существу является необходимым требованием для фармацевтических форм с контролируемым высвобождением.
На первой стадии обрабатывали различные композиции, не содержащие лекарственный препарат, и на основе полученного замедления времени распадаемости выбирали наполнители, которые применяли в разрабатываемых таблетках. Затем проводили испытания для оценки устойчивости таких наполнителей к температурам, применяемым для энергообработки.
Затем был выбран модельный лекарственный препарат, который можно было бы использовать для определения параметров высвобождения из таблеток. Выбор пал на дилтиазем гидрохлорид, доступный на рынке как в препаратах с быстрым или стандартным высвобождением, так и в композициях с модифицированным высвобождением.
На основе предварительных испытаний и применяя такую модель лекарственного препарата, были приготовлены таблетки различного состава.
Влияние термической обработки на высвобождение активного вещества из таких таблеток оценивали с помощью испытаний на растворимость, выполненных как в кислой среде, моделирующей среду в желудке, так и в фосфатном буфере, который в некоторой степени имитирует среду кишечника. Модификации, которым подвергали таблетки, исследовали с помощью термоаналитических методов, спектроскопических методов и с применением некоторых физических испытаний, типичных для этого вида фармацевтической формы.
Получение смесей активного вещества с разными наполнителями.
Смешивание компонентов, возможно просеянных, осуществляли в цилиндрических стеклянных контейнерах янтарного цвета с навинчивающейся крышкой, оборудованной тефлоновой пробкой, или в подходящих контейнерах из нержавеющей стали и продолжали в смесителе Turbula® до тех пор, пока смесь компонентов не становилась полностью однородной, в общем следующим способом.
1. Формировали ядро, состоящее из неосновного компонента и равного количества по массе активного вещества.
2. Добавляли все оставшееся активное вещество.
3. Добавляли основные наполнители в весовом количестве, равном весовому количеству порошка, содержащегося в контейнере.
4. Добавляли все количество основных наполнителей.
Для всех смесей каждую порцию порошка перемешивали в течение времени, которое зависело от массы, в целом для самых больших количеств на протяжении максимально 30-40 минут.
Получение таблеток.
Таблетки с массой в диапазоне 150-170 мг приготавливали в кривошипной таблеточной машине, оборудованной вогнутым монопуансоном.
Обработка таблеток и порошков.
Таблетки, подвергаемые обработке, помещали на металлическую основу, при этом каждая таблетка была защищена маленькой металлической сетчатой корзинкой. Обработка проводилась в печи газового хроматографа (HP 5890 серия II) и состояла из нагревания до предварительно установленной температуры обработки и поддержания такой температуры на протяжении предварительно установленного времени. Применяли следующую температурную программу: 0,1 мин при 25°С, достижение конечной температуры при градиенте, равном 30°/мин, и поддержание такой температуры в течение установленного времени, затем принудительное или естественное охлаждение таблеток до комнатной температуры.
После обработки каждой таблетки потерю массы в процентах оценивали, (Δm %), согласно следующему уравнению:
Δm %=(mO-m)/mO*100
где mO представляет собой начальную массу таблетки и m представляет собой массу этой же таблетки после термической обработки.
Обработку порошков для сравнений выполняли в печи газового хроматографа в пирекс-трубках.
Хранение таблеток.
Необработанные и обработанные таблетки хранили при комнатной температуре в поливинилхлоридной блистерной упаковке в течение различных периодов времени: 47 дней, 13 месяцев и 32 месяца. В конце периода консервации оценивали процентное увеличение массы, (Δw %), согласно следующему уравнению:
Δw %=(mC-mO)/mO*100
где mC представляет собой массу таблетки после определенного периода хранения и mO представляет собой исходную массу этой же таблетки. Для обработанных таблеток mO обозначает массу после термической обработки.
Определение твердости таблетки.
Испытание выполняли на обработанных и необработанных таблетках с помощью соответствующего оборудования, рассматривая в качестве достоверных результатов только те, которые были полученные при действительно радиальном разрушении таблетки, а не результаты, обусловленные явлением деформации или разрушением таблетки по диагональным плоскостям. Полученный результат представляет собой радиальный предел прочности и выражается в кп (килопонд = килограмм-сила =9.80665 ньютонов).
Определение количества воды, присутствующей в таблетках.
Исследование выполняли путем титрования Карла Фишера (КФ) с применением подходящего автоматического аппарата (Mettler-Toledo DL38). В качестве агента для титрования использовали Hydranal Composite 5 (Riedel-deHaën), стандартизированный по дигидрату тартрата натрия (Riedel-deHaën). Полученный результат выражали в виде процентного содержания (по массе) воды, содержащейся в 55,0 мг пробе порошка, тщательно взвешенной, отобранной из таблетки, раздробленной в стеклянной ступке. Кроме того, в этом случае исследование выполняли с применением обработанных и необработанных таблеток.
Метод дифференциальной сканирующей калориметрии (ДСК).
Исследования физической стабильности активных веществ и наполнителей, применяемых в композиции предложенных таблеток.
5,0 мг каждого тщательно взвешенного наполнителя/вещества помещали в алюминиевые ампулы, запрессовывали и анализировали с помощью ДСК (Perkin Elmer 7) в потоке азота; анализ также выполняли с применением термически обработанных порошков. Применяемые рабочие условия были следующими: начальная температура (Tstart)=50°С; конечная температура (Tend)=250°С; градиент =10°С/мин.
Контроль таблеток, содержащих активное вещество, методом ДСК.
Таблетки растирали в стеклянной ступке и 5,0 мг тщательно взвешенного порошка, полученного от каждой таблетки, анализировали вышеописанным способом. Кроме того, в этом случае исследование выполняли с применением как необработанных, так и термически обработанных таблеток. Все сканирования выполняли в потоке азота.
Способ определения изменений массы во время нагревания - термогравиметрический анализ (ТГА)
Определение изменений массы активных веществ и наполнителей, порошковых смесей и порошков, полученных при измельчении таблеток, проводили методом ТГА 7 с применением Perkin Elmer, в атмосфере азота, используя те же температуры и градиенты нагревания, которые применяли при термических обработках.
Испытания на распадаемость.
Испытания проводили, используя оборудование в соответствии с монографией Disintegration of Tablets and Capsules of the European Pharmacopoeia, 6-е издание. Температуру применяемой среды, 1 л деионизированной воды, поддерживали при 37±0,1°С. Испытания выполняли с применением 6 таблеток за раз.
Испытания на растворимость.
Испытания на растворимость проводили в устройстве (Distek) в соответствии с монографией Disintegration of Tablets and Capsules of the European Pharmacopoeia, 6-е издание. 1 л среды для растворения, находящейся в стеклянном сосуде, термостатировали до 37±0,1°С и устанавливали скорость вращения лопаток при 50 об/мин. Определение растворенного активного вещества выполняли с применением устройства DAD UV-visible Agilent Technologies 8453, автоматизированного с помощью перистальтического насоса и системы держателей для пробирок "Multicell Transport for Agilent 8453", управляемых связанным программным обеспечением. После каждого снятия показаний среду для растворения помещали обратно в исходный сосуд. Время отбора проб устанавливали через 5 минут в течение первых 20 минут и затем через 10 минут вплоть до 200 минут и потом фиксировали с использованием прогрессии, в зависимости от общего времени исследования. Анализ проводили при аналитической длине волны 236 нм, применяя диафрагму для вычитания фона, установленную между 450 и 600 нм. Определение выполняли путем построения калибровочной кривой в диапазоне концентраций, который учитывает растворение 1% и 100% теоретического содержания активного вещества в таблетках.
Кислая среда для растворения состояла из буфера, полученного путем добавления деионизированной воды к подходящему количеству 37% HCl до объема, необходимого для получения 0,05 н раствора.
Среда для растворения при рН 7,2 состояла из 0,05 М фосфатного буфера, полученного путем растворения в деионизированной воде подходящих количеств гидрофосфата натрия и дигидрофосфата калия и установления рН с помощью подходящих количеств фосфорной кислоты или гидроксида натрия.
Для каждого испытания оценивали профиль растворения 6 таблеток.
ИК-спектрометрия.
ИК-спектры различных веществ и смесей получали с помощью спектрометра Perkin Elmer 1310, приготавливая пробы в KBr-пластинках.
Испытания на адгезию.
Эти измерения выполняли с применением прибора для испытания на растяжение (LLOYD LRX), модифицированного для измерений мукоадгезии (Russo E, Parodi В, Caviglioli G, Cafaggi S, Bignardi et al. J Drug Deliv Sci Technol 14(6): 489-494, 2004).
Для проведения указанных испытаний на плоской поверхности были изготовлены цилиндрические таблетки, масса которых составляла примерно 200 мг, а диаметр примерно 13 мм, прилагая нагрузку, равную 2 тоннам в 1 минуту при применении ручного гидравлического пресса. Такие прессы продают для приготовления пластинок из KBr для ИК-спектрометрии.
Подложка для адгезии состояла из муциновых таблеток (Sigma), масса которых составляла примерно 250 мг, а диаметр примерно 13 мм, изготовленных с помощью пресса для ИК-спектрометрии путем приложения нагрузки 5 тонн в минуту.
Подложку для адгезии прикрепляли к датчику нагрузки; пробу, установленную на термостатируемую основу при 37°С, увлажняли в течение 1 минуты с помощью 200 мкл 0,05 М фосфатного буфера с рН 7,2, температуру которого также поддерживали при 37°С.
Прикладывали предварительную нагрузку 1 Н в течение 2 минут при скорости 10 мм/мин; для оценки адгезии устанавливали растяжение 3 мм при скорости 0,1 мм/с.
Интерпретация полученных кривых.
Из полученных графиков получали следующие параметры:
- максимальная нагрузка [Н];
- работа [Н·мм], полученная как результат интегрирования растяжения × зона нагрузки;
- удельная нагрузка [МПа], полученная из соотношения между максимальной нагрузкой и площадью таблетки (132,73 мм2).
Испытания на прилипание выполняли с применением таблеток, содержащих только наполнители (необработанных и термически обработанных), и с применением таблеток, содержащих активное вещество (необработанных и термически обработанных).
Оценка степени набухания.
Применяемые таблетки погружали в 0,05 М фосфатный буфер с рН 7,2, термостатировали до 37°С и выдерживали при перемешивании с помощью вращающихся лопастей при 50 об/мин. Через определенные промежутки времени (30 или 60 минут) таблетки извлекали из среды, давали стечь жидкости на металлической решетке в течение 30 секунд и взвешивали на аналитических весах.
Степень набухания в процентах, S%, рассчитывали согласно следующему уравнению:
S%=(mt-mo)/mo*100
где mt представляет собой массу таблеток, извлеченных через время t, и mo - начальная масса таблеток.
Это испытание выполняли с применением таблеток, содержащих или не содержащих активные вещества, которые были термически обработаны или не были термически обработаны.
Оценка объема таблеток.
Таблетки погружали в градуированный цилиндр, содержащий известный объем вазелинового масла. Объем таких таблеток оценивали по разнице относительно начального объема жидкости.
Это исследование выполняли с применением обработанных таблеток и этих же таблеток после испытания на растворение.
Чтобы решить вышеупомянутую техническую проблему, исследовали много смесей различных наполнителей, применяя дилтиазем гидрохлорид (ДТЗ) в качестве модели активного вещества.
Некоторые из исследованных наполнителей приведены ниже в таблице 1.
С применением вышеуказанных наполнителей и гидрохлорида дилтиазема приготавливали смеси с тремя, четырьмя и пятью компонентами и изготавливали из них таблетки вышеописанными способами. Затем таблетки подвергали вышеуказанным испытаниям и измерениям.
Было подтверждено, что необходимые результаты в условиях контролируемого высвобождения были получены, когда порошковая смесь состояла по меньшей мере из одного из компонентов, указанных в пункте 1 прилагаемой формулы изобретения.
Дополнительно было изучено влияние термической обработки, предложенной в способе согласно настоящему изобретению, на некоторые компоненты полученной биоадгезивной компактированной матрицы.
На фиг.11 показаны результаты исследования термического сжатия поликарбофила (сжатие относительно времени нагревания). Как можно видеть на этом графике, максимальное планарное сжатие происходит, когда пробу поликарбофила нагревают при 160°С в течение 5 минут.
Влияние планарного сжатия порошка можно оценить с помощью микрофотографий СЭМ, показанных на фиг.17 и 18. На фиг.17 показана гроздь с виноградоподобной морфологией, при этом отдельные виноградины исчезают на фиг.18, где скорее можно различить образования, имеющие непрерывную матрицу (похожие на розу) и меньший суммарный объем: это следствие термической обработки, которой был подвергнут порошок. Более того, на фиг.18 можно наблюдать мостики, связывающие отдельные гранулы, тогда как гранулы на фиг.17 четко отделены друг от друга.
На графике, представленном на фиг.12, три наложенных профиля (ДСК, ТГА и МНС) демонстрируют, что явление сжатия, измеренное как планарное сжатие на фокальной плоской поверхности микроскопа, не связано с какими-либо явлениями разложения поликарбофила, но, возможно, связано с эндотермическими событиями, происходящими в поликарбофиле при температурах выше 50°С и достигающими своего пика с маленькой эндотермической кривой между 128°С и 147°С.
Из сравнения микрофотографий СЭМ, представленных на фиг.13 и 14, хорошо видно, что, когда спрессованную заготовку поликарбофила подвергают термической обработке (150°С в течение 15 минут), его структура резко изменяется: на фиг.14 четко видна трабекулярная матрица, тогда как на фиг.13 не видно ничего подобного.
Трабекулярную матрицу и поры, которые содержатся в такой матрице, можно также увидеть на фотографии с большим увеличением, показанной на фиг.15. С применением смеси этилцеллюлозы и поликарбофила (3:2) было также изучено совместное влияние прессования и нагревания. Как можно видеть на микрофотографии, приведенной на фиг.16, взаимодействие между двумя полимерами вызывает частичную окклюзию двух микрогранул этилцеллюлозы в порах недавно образованной поликарбофильной матрицы.
Ниже приведены, для иллюстрации, а не для ограничения, несколько примеров составов, адаптированных для применения в способе согласно настоящему изобретению, также содержащих наряду с дилтиазем гидрохлоридом (ДТЗ) гликлазид (ГЛЗ).
Пример 1
Следующие компоненты смешивали согласно вышеописанным способам до получения однородного порошка:
Процентные содержания, приведенные в тексте настоящей заявки, если не указано иное, следует понимать как процентные содержания по массе от общей массы порошковой смеси до прессования и до термической обработки.
Из указанной порошковой смеси были приготовлены таблетки путем прямого прессования согласно вышеописанной процедуре.
Часть этих таблеток подвергали термической обработке с помощью вышеописанных способов, выдерживая их при температуре обработки 150°С в течение времени обработки, равного 15 мин. По истечении указанного времени сушильную печь сразу же охлаждали до комнатной температуры путем принудительной вентиляции. Минимальное время выдерживания при комнатной температуре перед упаковкой: 5 минут.
Среднее содержание воды в термически необработанных таблетках (количество проб =20) составляло, согласно методу Карла Фишера, 2.57 мас.% (стандартное отклонение СО=0,09) от массы таблетки и средняя твердость составляла 307,8 Н (31,4 кп; СО=1,2), тогда как содержание воды в термически обработанных таблетках (количество проб =20) составляло 0,88 (СО=0,08) и твердость составляла 405,9 Н (41,4 кп; СО=1,1).
Термически обработанные таблетки (обозначенные как Т) давали кривую растворения в 0,05 N HCL, представленную на фиг.1а, на которой также изображена кривая растворения соответствующих термически необработанных таблеток (обозначенных NT).
На кривых изображены средние значения для шести необработанных таблеток и шести термически обработанных таблеток.
Модификация, вызванная термической обработкой таблеток, хорошо видна на фиг.1а, в отношении высвобождения гидрохлорида дилтиазема в кислой среде. Термическая обработка оказывает влияние на компоненты таблетки, которые образуют матрицу, значительно замедляющую высвобождение лекарственного препарата, которое происходит согласно кинетике нулевого порядка.
На начальной стадии, до достижения 20% высвобождения лекарственного препарата, кривые высвобождения из обеих таблеток выглядят частично совпадающими друг с другом, вероятно, благодаря эффекту разрыва, который предшествует гидратации и загустеванию матриц в таблетках Т. На первом этапе высвобождение из обработанных таблеток в любом случае выглядит немного более быстрым, фактически, через 30 минут матрица высвобождает свыше 11% препарата.
На фиг.1b приведен профиль растворения в фосфатном буфере (рН=7,2) таблеток Т по сравнению с профилем соответствующих таблеток NT.
Здесь также на кривых изображены средние значения для шести необработанных таблеток и шести термически обработанных таблеток.
Влияние обработки на высвобождение ДТЗ в фосфатном буфере даже более очевидно. Матрица, которая образуется после такой обработки, высвобождает лекарственный препарат гораздо более медленно; действительно, между 100 и 850 минутами из матрицы высвобождается примерно 47% введенного лекарственного препарата. t50 (время, за которое высвобождается 50% лекарственного препарата) составляет 4 часа для таблеток NT, тогда как для таблеток Т оно равно примерно 14 часам. Фактически, из таблеток Т через 4 часа высвобождается только 33% введенного лекарственного препарата, и через 850 минут максимальное высвобождение введенного ДТЗ все еще не было достигнуто.
Кроме того, в этом буфере таблетки Т проявляют высвобождение, которое подчиняется кинетике нулевого порядка после короткого начального периода адаптации, связанной с эффектом разрыва.
При значении рН фосфатного буфера матрица, образующаяся в таблетках Т после термической обработки, поглощает водную среду для растворения, набухает и, таким образом, увеличивает свой объем, образуя гелеобразный внешний слой (см. фиг.7а, последний справа), благодаря которому механизм высвобождения лекарственного препарата действует согласно кинетике нулевого порядка. Кроме того, обнаружено, что набухшие матрицы таблеток, которые подвергались процессу нагревания, остаются цельными, т.е. они не подвергаются явлению разрушения во время всего испытания на растворение (см. также набухание), и один раз извлеченные и оставленные сушиться, они вновь приобретают форму и размер таблеток, из которых они были получены, подобно тому, как это имеет место в примере 2 (фиг.9).
Аналогично тому, как это имеет место в примере 2 (фиг.10), в кислой окружающей среде также наблюдается, в меньшей степени, образование гелеобразной кроны.
Сравнение с таблеткой NT позволяет подтвердить, что термическая обработка смеси компонентов приводит к образованию неразрушаемой матрицы, которая высвобождает лекарственный препарат, поддерживая скорость высвобождения постоянной (% высвобожденного вещества/время высвобождения) в течение длительного периода времени. Кроме того, визуально можно наблюдать гелеобразную и полупрозрачную крону - вероятно, отвечающую за регулирование высвобождения лекарственного препарата - которая образуется из этой отдельной матрицы вследствие явления постепенного набухания.
Регулирование скорости высвобождения можно объяснить как явлением набухания матрицы, так и молекулярной диффузией лекарственного препарата, растворенного в водной среде, которая заставляет матрицу набухать, через гелеобразный слой матрицы. Такие явления не наблюдаются в случае таблеток NT, которые в конце испытания на растворение полностью распадались.
Появление модификации, коррелирующей с образованием матрицы, также очевидно из увеличения твердости таблетки, равной 98,04 Н (10 кп).
Превращения, вызванные термической обработкой, в результате которой образуются матрицы, показаны с помощью кривой ДСК, приведенной на фиг.1с.
Пример 2
Следующие компоненты смешивали в соответствии с рабочими процедурами, описанными выше, для получения однородного порошка:
Из этой порошковой смеси были приготовлены таблетки путем прямого прессования согласно вышеописанной процедуре.
Часть этих таблеток подвергали термической обработке вышеописанным способом, выдерживая их при температуре обработки 150°С в течение времени обработки, равного 15 мин. По истечении указанного времени сушильную печь сразу же охлаждали до комнатной температуры путем принудительной вентиляции. Минимальное время выдерживания при комнатной температуре перед упаковкой: 5 минут.
Среднее содержание воды в термически необработанных таблетках (количество проб =20) составляло, согласно методу Карла Фишера, 2,57 мас.% (стандартное отклонение СО=0,08) от массы таблетки и средняя твердость составляла 247,1 Н (25,2 кп) (СО=-1,3), тогда как среднее содержание воды в этих термически обработанных таблеток (количество проб =20) составляло 1,26 (СО=0,05) и твердость составляла 401,0 Н (40,9 кп) (СО=1.1).
Термически обработанные таблетки (Т) давали кривую растворения в 0,05 н HCl, представленную на фиг.2а, на которой также приведена кривая растворения соответствующих термически необработанных таблеток (NT).
На кривых изображены средние значения для шести необработанных таблеток и шести термически обработанных таблеток.
На фиг.2а наблюдается значительное замедление высвобождения ДТЗ, которого можно добиться после термической обработки этих таблеток. Хорошо видно влияние термической обработки на различный количественный состав этого примера - в этом примере содержание поликарбофила удвоено. Такое увеличение содержания поликарбофила в два раза отражается на высвобождении ДТЗ в кислой среде. Наблюдается большее замедление высвобождения, которое не достигает своего максимума даже через 10 часов, тогда как таблетки NT достигают своего максимума через 5 часов. Одновременно начальное высвобождение становится более быстрым по сравнению с начальным высвобождением таблеток NT; можно видеть высвобождение, которое подчиняется кинетике нулевого порядка между 40 и 210 минутами.
На фиг.2b представлена кривая растворения в фосфатном буфере (рН=7,2) шести термически обработанных таблеток Т по сравнению с профилем шести соответствующих необработанных таблеток NT.
На каждой кривой изображено среднее значение для шести таблеток, при этом вертикальные отрезки отражают 95% доверительный интервал.
Что касается предыдущего примера, обработанные таблетки демонстрируют высвобождение, которое подчиняется кинетике нулевого порядка, установившейся после короткого начального периода адаптации. Увеличение содержания поликарбофила обуславливает дополнительное снижение скорости высвобождения по сравнению с предыдущим примером, фактически, через 840 минут растворения из таблеток NT высвобождается 80% введенного лекарственного препарата, тогда как из таблеток Т высвобождается примерно 41%. Из последних через 1400 минут (фиг.3с) высвобождается примерно 66% введенного лекарственного препарата.
Кроме того, в этом случае во время испытания можно наблюдать увеличение объема термически обработанных таблеток наряду с образованием кроны из полупрозрачного гелеобразного вещества, более прозрачной по сравнению с примером 1, которая окружает четко видимое твердое ядро (см. фиг.7а, центральная фотография; 7b, 7с), содержащее все еще не растворенное количество лекарственного препарата.
Образование гелеобразной кроны, меньшей по сравнению с кроной, которая формируется при растворении при рН 7,2, также можно видеть при растворении при кислом рН (фиг.10).
На фиг.9, относящейся к таблеткам, полученным согласно настоящему примеру, продемонстрировано, что, как показано выше, таблетки согласно настоящему изобретению после набухания в воде вновь приобретают свою первоначальную форму и размер после высушивания. Это позволяет распространить применение способа согласно настоящему изобретению также на активные вещества, являющиеся термолабильными или трудно получаемыми в твердом состоянии, которые могут быть введены путем впитывания водных растворов таких веществ спрессованной матрицей, не содержащей активного вещества, полученной с помощью настоящего способа, а затем высушивания пропитанной и набухшей матрицы.
Увеличение твердости таблеток, прошедших термическую обработку, как в примере 1, подтверждает образования матрицы внутри таблеток. В этом примере среднее увеличение твердости больше, чем в предыдущем примере, и составляет 135,3 Н (13,8 кп), и, вероятно, коррелирует с различной консистенцией геля гидратированной матрицы, которая окружает твердое ядро, что отражается на снижении скорости высвобождения, зафиксированном с помощью испытания на растворение.
На фиг.2с показана кривая ДСК, полученная вышеописанными способами, таблеток Т по сравнению с кривой ДСК таблеток NT. В верхней части приведены кривые ДСК физических смесей порошков из MicroceLac (ML) и гидрохлорида дилтиазема (ДТЗ), описанных в настоящем примере.
Превращения, вызванные термической обработкой, в результате которой образуются матрицы, показаны с помощью кривой ДСК, приведенной в нижней части фиг.2с.
Пример 3
Следующие компоненты смешивали согласно рабочим способам, описанным выше, для получения однородного порошка:
Из этой порошковой смеси были приготовлены таблетки путем прямого прессования согласно вышеописанной процедуре.
Часть этих таблеток подвергали термической обработке вышеописанным способом, выдерживая их при температуре обработки 150°С в течение времени обработки, равного 15 мин. По истечении указанного времени сушильную печь сразу же охлаждали до комнатной температуры путем принудительной вентиляции. Минимальное время выдерживания при комнатной температуре перед упаковкой: 5 минут.
Среднее содержание воды в термически необработанных таблетках (количество проб =20) составляло, согласно методу Карла Фишера, 3,14 мас.% (стандартное отклонение СО=0,1) относительно массы таблетки и средняя твердость составляла 236,3 Н (24,1 кп; СО=0.8), тогда как среднее содержание воды в этих термически обработанных таблетках (количество проб =20) составляло 1,26 (СО=0,09) и твердость составляла 259,8 Н (26,5 кп; СО=1,4).
Термически обработанные таблетки (Т) давали кривую растворения в 0,05 н HCl, представленную на фиг.3а, на которой также приведена кривая растворения соответствующих термически необработанных таблеток (NT).
Каждая кривая представляет собой усреденнную кривую растворения по шести таблеткам, при этом вертикальные отрезки отражают значение 95% доверительного интервала.
На фиг.3b представлена кривая растворения в фосфатном буфере (рН=7,2) термически обработанных таблеток (Т) по сравнению с кривой для соответствующих необработанных таблеток (NT). Здесь также каждая кривая представляет собой усредненную кривую по шести необработанным таблеткам и шести термически обработанным таблеткам.
Из приведенных кривых растворения можно видеть, как, даже при увеличении вдвое содержания лекарственного препарата (относительно предыдущих примеров), матрица, являющаяся предметом изобретения, продолжает регулировать высвобождение лекарственного препарата. Влияние термической обработки, как и в предыдущих случаях, более наглядно в фосфатном буфере, чем в HCl. Кинетика высвобождения после начального этапа соответствует нулевому порядку, что подтверждается прямолинейными участками кривых растворения, за исключением необработанных таблеток (NT), кривая которых в фосфатном буфере отклоняется от линейности, вероятно, вследствие явлений разрушения, которые предшествуют полной распадаемости таблеток.
В кислой среде таблетки Т высвобождают лекарственный препарат более быстро в первые 30 минут, затем устанавливается кинетика нулевого порядка, согласно которой высвобождается примерно 50% введенного ДТЗ. Высвобождение из таблеток Т заканчивается через примерно 400 минут, тогда как в таблетках NT высвобождение завершается в пределах 230 минут.
В фосфатном буфере таблетки NT достигают полного высвобождения через примерно 560 минут, тогда как таблетки Т достигают его через 1400 минут.
Кроме того, в этом случае наблюдали набухание обработанных таблеток, которые, после прохождения испытания на растворение в HCl оставались цельными; после испытания в фосфатном буфере таблетки набухают в гораздо большей степени, окружены гелеобразным слоем и способны к восстановлению своей первоначальной формы после извлечения и высушивания.
Крайне интересно сравнить (фиг.3с) профили растворения в фосфатном буфере из этого примера и аналогичный профиль из примера 2. Из параллельности линейных участков двух профилей можно сделать вывод, что матрица из примера 3, образованная составом с тем же соотношением этилцеллюлоза/поликарбофил, как в примере 2, способна регулировать высвобождение ДТЗ с той же скоростью, что и в примере, даже когда содержание лекарственного препарата увеличено вдвое. Конечно, эффект разрыва больше в примере 3.
На фиг.3d показана кривая ДСК, полученная вышеупомянутым способом, таблеток Т по сравнению с кривой ДСК таблеток NT. Превращения, вызванные термической обработкой, в результате которой образуется матрица, воспроизводимо выделяются на кривой ДСК, как показано на фиг.3d.
Оценка устойчивости при хранении.
Таблетки, полученные согласно примерам 1 (фиг.4а и 4b) и 2 (фиг.5а и 5b), после хранения в течение 47 дней в блистерной упаковке исследовали с целью проверки, являлось ли влияние обработки обратимым во времени или нет. Соответствующие кривые растворения в 0,05 н HCl (4а) и в фосфатном буфере (4b) усредненные по шести таблетам, Т и NT, сравнивали при t=0 и при t=47 дней.
Ниже в таблице 2 приведены несколько параметров, полученных при исследовании таблеток согласно примерам 1 и 2, хранившихся в блистерной упаковке.
Как видно из фиг.4 и 5, не существует значительных различий в высвобождении двух видов таблеток после хранения, даже если таблетки повторно приобретали некоторое количество воды, как показано в таблице 2. При исследовании устойчивости, проведенном в течение 32 месяцев с применением таблеток согласно примеру 2, которые хранились в блистерной упаковке при комнатной температуре, содержания воды, измеренные по Карлу Фишеру, составляли 2,1-2,5%, таким образом, на практике восстановление водосодержания при первоначальных значениях подтверждается с течением времени. Это наводит на мысль, что замедление высвобождения после термической обработки обусловлено не потерей матрицей воды, а модификацией физического состояния компонентов, образующих такую матрицу, которая, как оказалось, является необратимой в течение времени хранения. Другие данные исследований, полученные после хранения на протяжении 2-3 лет, подтверждают вышесказанное.
На фиг.5с показана усредненная кривая растворения в фосфатном буфере шести таблеток согласно примеру 1, только что приготовленных и после хранения в блистерной упаковке в течение 13 месяцев при комнатной температуре.
Видно, что обе кривые практически совпадают при наложении.
На фиг.5d показана усредненная кривая растворения в 0,05 н HCl шести таблеток согласно примеру 2 после 32 месяцев хранения в блистерной упаковке и на фиг.5е показана усредненная кривая растворения в фосфатном буфере шести таблеток согласно примеру 2 после такого же периода хранения.
На фиг.5f приведены кривые растворения в виде доли растворенного ДТЗ относительно растворенного количества спустя 600 минут. Такой способ отображения является полезным для того, чтобы подчеркнуть механизмы высвобождения, при этом он не дает информации о скорости высвобождения и замедлении высвобождения активного вещества. На этом графике приведены для сравнения усредненные кривые растворения (n=6) в фосфатном буфере различных партий таблеток согласно примеру 2 и таблеток с идентичным составом, но необработанных термически (NT).
Из этого сравнения видно, что механизм высвобождения лекарственного препарата из матриц различных партий полностью совпадает при наложении и не изменяется даже после хранения в блистерной упаковке в течение 32 месяцев. Этот способ отображения также подчеркивает разный механизм высвобождения из спрессованных матриц согласно настоящему изобретению по сравнению с необработанными таблетками. Кроме того, механизм высвобождения для последних менее воспроизводим.
Сравнивая результаты испытания на твердость, приведенные в таблице 2, с результатами, полученными до хранения, отмечают уменьшение предела прочности таблеток согласно примеру 1 и его увеличение в случае таблеток NT согласно примеру 2. В таблице 3 приведены данные о твердости таблеток согласно примеру 2, хранившихся в течение 32 месяцев.
Исследования термических процессов.
Далее было проведено исследование для выяснения, можно ли также добиться эффекта замедления высвобождения активного вещества, достигаемого путем термической обработки таблеток, заранее подвергая смесь наполнителей или даже одни наполнители такой же обработке с последующим добавлением активного вещества и затем проведением прессования.
Экспериментально было обнаружено, что при нагревании перед прессованием физической смеси всех компонентов согласно примеру 2 при 150°С в течение 15 минут или при таком же нагревании одного наполнителя за раз (например, поликарбофила, этилцеллюлозы или MicroceLac) были получены таблетки, которые после последующего добавления других компонентов не показывали требуемый профиль высвобождения дилтиазема. Нагревание гидрохлорида дилтиазема при 150°С в течение 15 минут, согласно ВЭЖХ, термоаналитическим (ДСК, ТГА и МНС) и спектроскопическим исследованиям, не вызывало никаких физических или химических модификаций этого активного вещества. Гидрохлорид дилтиазема, предварительно обработанный таким образом и используемый для получения таблеток согласно примеру 2, не модифицировал профили растворения из таких матриц и также не модифицировал высвобождение из таблеток NT с идентичным составом.
На основе этих наблюдений сделан вывод, что термическая обработка приводит к созданию матрицы согласно настоящей заявке на патент, только если она проведена с применением таблетки, в которой компоненты, в однородной смеси друг с другом, находятся в тесном контакте друг с другом за счет сил загустевания или пластифицирования, которые возникают во время процесса компактирования или прессования.
Затем способы термической обработки таблеток согласно примеру 2 варьировали: некоторые таблетки обрабатывали в течение 15 минут при 90°С или при 130°С, другие обрабатывали при 150°С в течение 5 минут. Полученные профили растворения, а также, для сравнения, кривая, относящаяся к таблеткам, обработанным способом, описанным в примере 2, и таблеткам NT, приведены на фиг.6.
Из фиг.6 видно, что все таблетки демонстрируют кинетику высвобождения нулевого порядка, даже если таблетки, обработанные при 150°С в течение 5 или 15 минут, проявляют усиленное замедление высвобождения активного вещества по сравнению с таблетками, обработанными при 130°С. Последние таблетки в любом случае демонстрируют замедление высвобождения активного вещества относительно таблеток NT и значительную степень набухания после испытания на растворимость, даже если оба указанных свойства проявляются в меньшей степени, чем свойства, наблюдаемые в случае таблеток, обработанных при 150°С.
Эти данные соответствуют наблюдению, что после испытания на растворимость в фосфатном буфере в таблетках, обработанных при 130° в течение 15 минут, набухание минимально и гелеобразный слой трудно различить, и в таблетках, обработанных при 90°, но в течение только 15 минут, не наблюдается никакая визуальная модификация по сравнению с таблетками NT.
Эти данные указывают, что скорость высвобождения активного вещества можно также корректировать путем выбора подходящей температуры термической обработки и временных параметров.
Испытания на адгезию.
Приготовили таблетки без ДТЗ, содержащие три наполнителя из примера 2 в тех же взаимных массовых соотношениях: испытание на адгезию проводили с применением этих таблеток и с применением таблеток согласно примеру 2 в соответствии с вышеописанной процедурой, выполненной с помощью тензометра. В таблицах 3bis и 4 показано содержание воды в анализируемых таблетках и результаты, полученные в результате этого испытания.
Таблица 4: данные, полученные в результате испытаний на адгезию, выполненных с применением таблеток, не содержащих ДТЗ, и с применением таблеток согласно примеру 2, и соответствующие стандартные отклонения. Указанные результаты представляют собой средние значения по трем измерениям для каждого вида таблеток.
Как можно видеть из данных, представленных в таблице 4, таблетки NT, не содержащие ДТЗ, демонстрируют определенную адгезию к подложке (муцину), имитирующей слизистую оболочку желудочно-кишечного тракта, которое уменьшается после обработки, явление, которое, вероятно, коррелирует со структурными модификациями матриц, образующихся при термической обработке.
Что касается таблеток NT, таблетки, содержащие лекарственный препарат, характеризуются меньшей адгезией, чем таблетки, приготовленные только с наполнителями. Это может быть обусловлено различным составом; фактически, даже если соотношения между компонентами сохраняются постоянными, таблетки без ДТЗ содержат абсолютное количество поликарбофила, большее на 25% по сравнению с примером 2. Результаты, полученные при исследовании обработанных таблеток, показывают уменьшение адгезии в случае обработки при 130°С по сравнению с таблетками NT, содержащими лекарственный препарат, при этом после обработки при 150°С×15 минут адгезионные свойства не уменьшаются.
Испытания на набухание.
Процесс набухания изучали с применением таблеток, полученных согласно примерам 1 и 2, при 37°С в фосфатном буфере при рН 7,2, рН условиях, при которых это явление особенно наглядно. На фиг.7 показана степень набухания в процентах (S%) двух видов таблеток согласно примерам 1 и 2. Полученные кривые описывают среднее набухание трех таблеток по каждому виду, отрезки отражают стандартное отклонение.
В таблице 5 приведены следующие параметры: S%60, S%max, масса и объем таблеток после 24-часового погружения в фосфатный буфер и соответствующие плотности.
Таблица 5: некоторые параметры, оцениваемые с применением таблеток, обработанных согласно примерам 1 и 2; приведенные значения представляют собой среднее по трем таблеткам каждого вида. S%60 представляет собой степень набухания после первого измерения (60 минут), <S%max представляет собой максимальный достигнутый уровень набухания, Tmax - время, за которое достигается максимальное набухание.
Как видно на фиг.7, таблетки, обработанные согласно примеру 2, проявляют более высокие степень и скорость набухания по сравнению с таблетками, обработанными по п.1, как также показано в таблице 5: это свойство, по-видимому, коррелирует с концентрацией поликарбофила в смеси. Следует отметить, что для обоих видов таблеток имеет место увеличение массы, на основе которого рассчитывают S, сохраняющуюся постоянной в течение продолжительного периода, за исключением первых 120 минут.
Таблетки согласно примеру 1 достигают максимального времени, равного 1440 минут (24 часа), после которого они больше не набухают, но, оставаясь погруженными, не распадаются на протяжении до 1830 минут (30,5 часов), тогда как для таблеток согласно примеру 2 это явление не обнаружено, поскольку после достижения максимума через 24 часа (время, в которое было сделано последнее измерение) таблетки проявляют тенденцию к полному разложению между 24 и 30 часами. Эти явления наблюдаются в условиях измерения in vitro, которые включают большой объем водной среды и непрерывное перемешивание.
На фиг.7 представлены профили набухания таблеток согласно примерам 1 и 2 в фосфатном буфере при 37°С. На фиг.7а приведены фотографии таблеток согласно двум примерам, сделанные при их максимальной степени набухания, которые сравниваются с фотографией таблетки при ее первоначальном размере перед испытанием на растворимость.
На фиг.7b показано увеличенное изображение таблетки согласно примеру 2 при ее максимальной степени набухания, на котором хорошо видно твердое центральное ядро.
На фиг.7с представлены фотографии набухших таблеток согласно примеру 2 с различных ракурсов, на которых хорошо видно образование плотной матрицы.
Также измеряли набухание матриц, не содержащих активное вещество. На фиг.7d показаны профили набухания таблеток, содержащих все компоненты согласно примеру 2, за исключением ДТЗ, но в тех же соотношениях, как в указанном примере. Наибольшего набухания достигали матрицы, полученные при самой продолжительной термической обработке, 150°С в течение 15 минут: они достигают максимального значения увеличения массы, равного 1744%, через 5 часов и сопротивляются разрушению, вызванному непрерывным перемешиванием, в течение еще двух часов.
Матрицы, образующиеся при 5-минутном нагревании, достигают максимального значения увеличения массы (1282%) через два часа и сопротивляются разрушении в течение еще 60 минут.
Присутствие лекарственного препарата в матрицах, полученных путем нагревания, замедляет явление набухания вследствие диффузии водной среды в набухающую матрицу. Замедление коррелирует с процессом реверсирования массы, т.е. обратной диффузией активного вещества. Одновременность этих двух явлений может объяснить кинетику высвобождения активного вещества согласно модели нулевого порядка.
Влияние термической обработки на образование матрицы также очевидно из поведения таблеток NT, фиксируемого во время изучения набухания. Действительно, эти таблетки набухают очень мало, только на 80%, в течение первого часа и затем полностью распадаются через 120 минут.
Пример 4
Из этой порошковой смеси были приготовлены таблетки путем прямого прессования согласно вышеописанной процедуре.
Часть этих таблеток подвергали термической обработке вышеописанным способом, удерживая их при температуре обработки 150°С в течение 5 или 15 минут. По истечении указанного времени сушильную печь сразу же охлаждали до комнатной температуры путем принудительной вентиляции. Минимальное время выдерживания при комнатной температуре перед упаковкой: 5 минут.
На фиг.8 представлена усреденная кривая растворения по шести таблеткам для каждого типа обработки (150°С в течение 15 минут и 150°С в течение 5 минут) по сравнению с усредненной кривой растворения таблеток NT в фосфатном буфере при рН 7,2.
Влияние термической обработки на образование матрицы очевидно из регулирования скорости высвобождения гликлазида и наглядно видно из набухания заготовок для образования компактированной гелеобразной кроны.
Ясно, что влияние термической обработки также варьируется в зависимости от вида активного вещества, вследствие различного взаимодействия с компонентами матрицы.
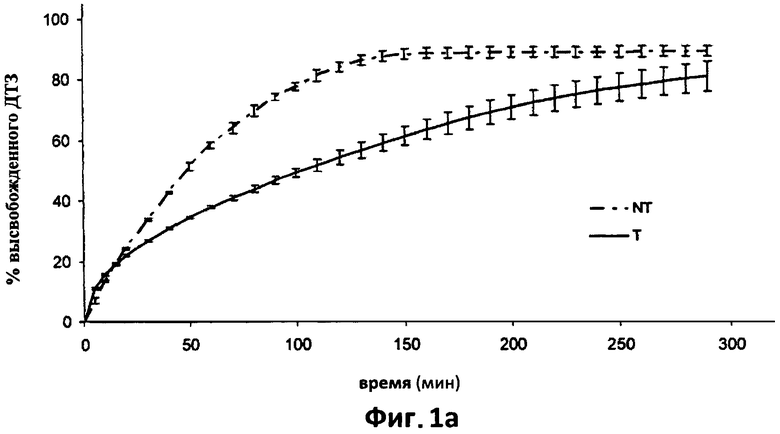
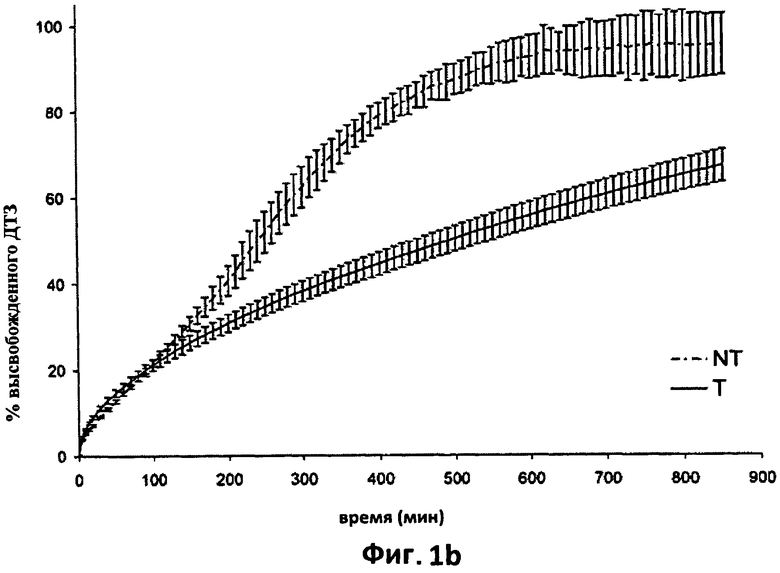
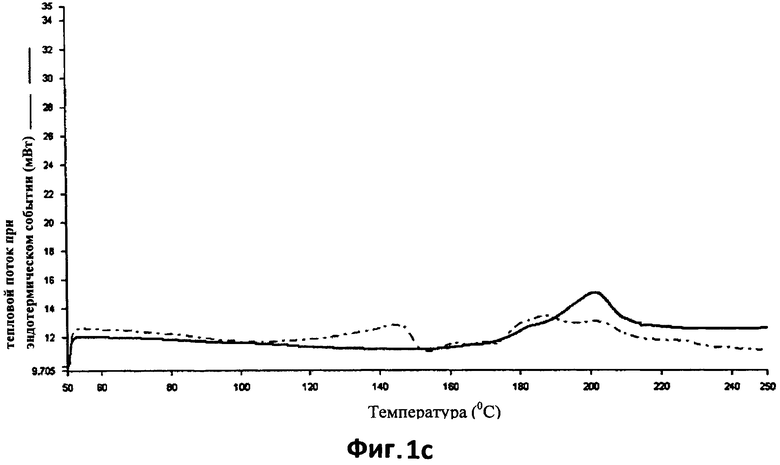

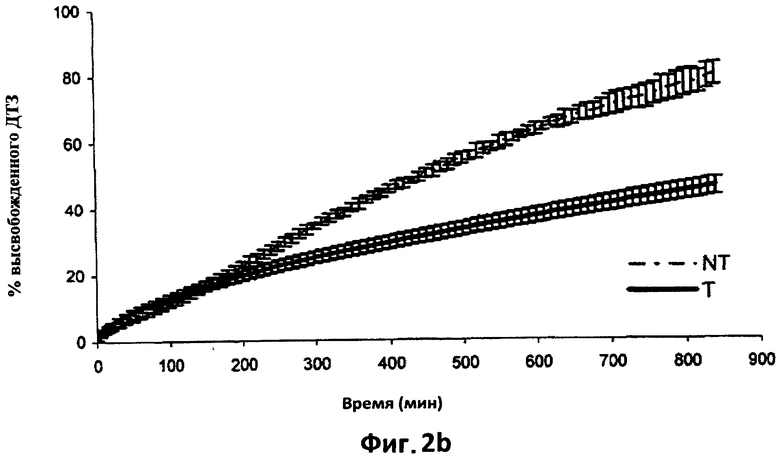

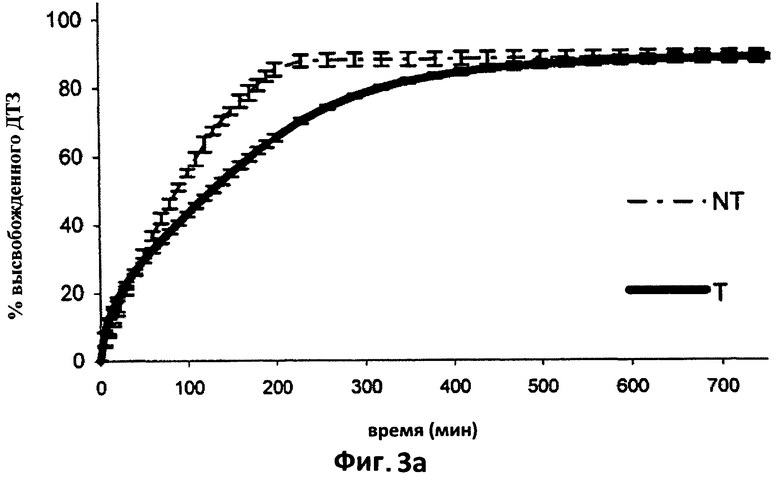

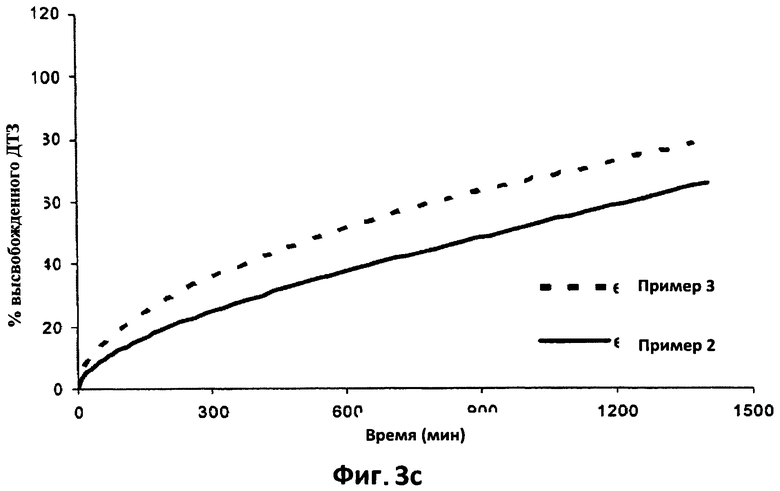

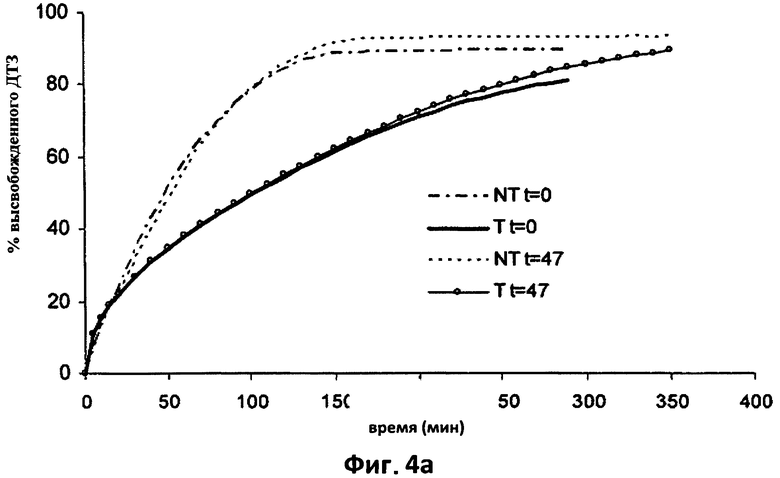
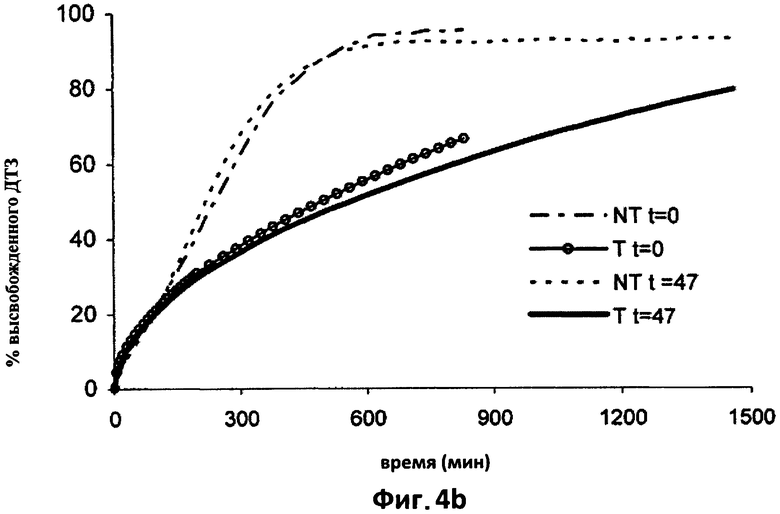

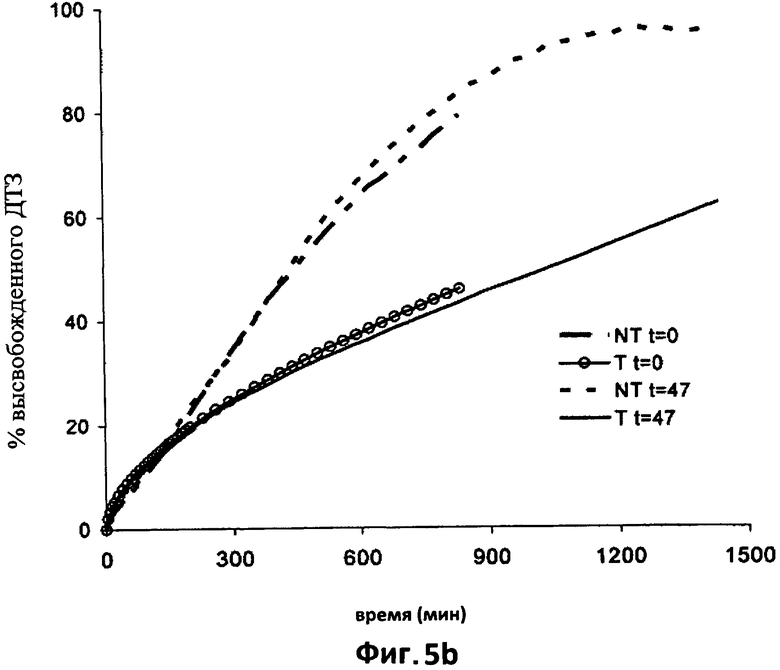
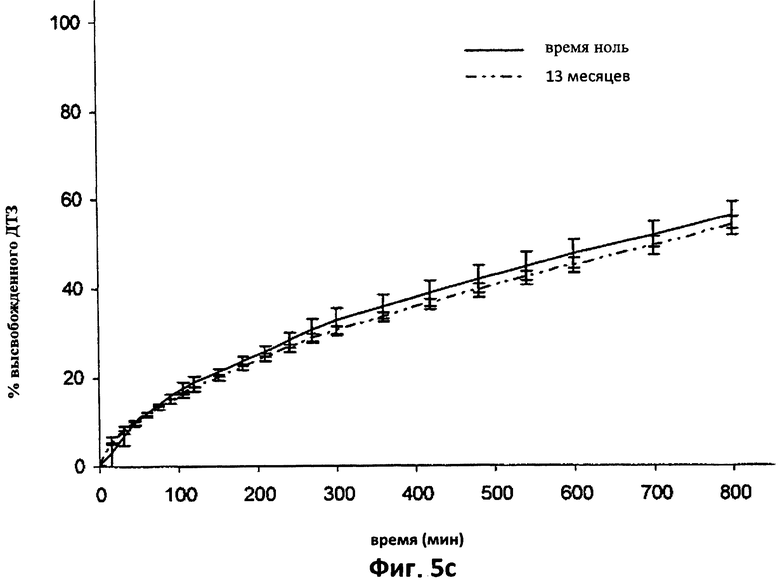
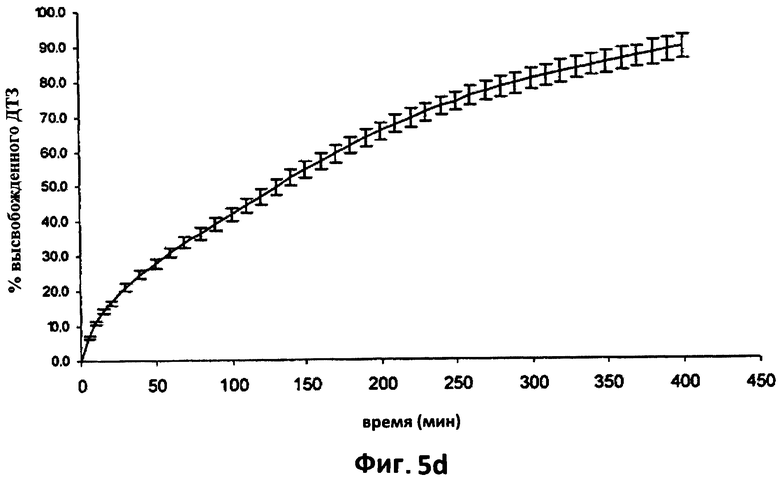
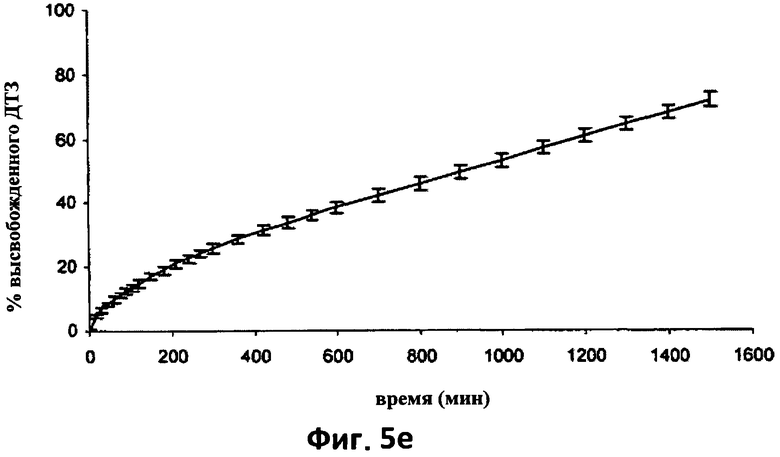
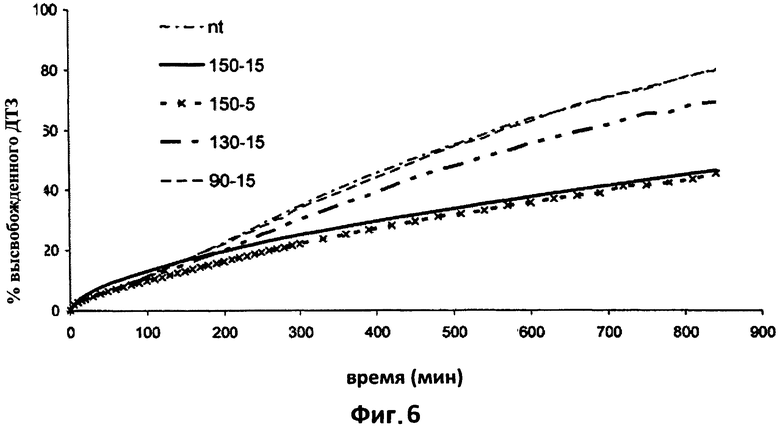
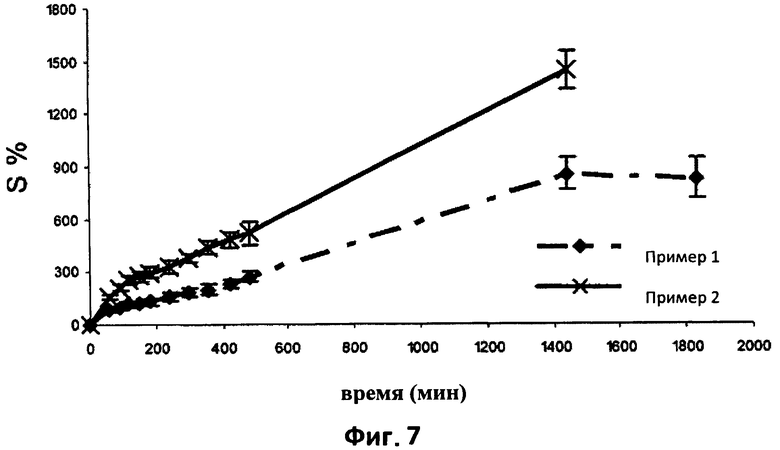



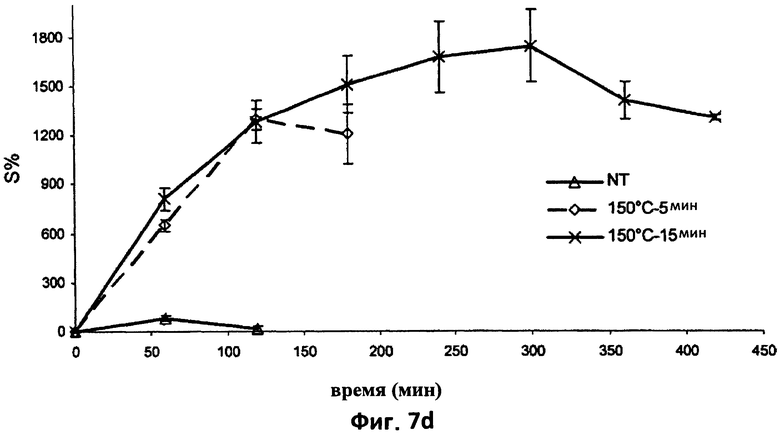

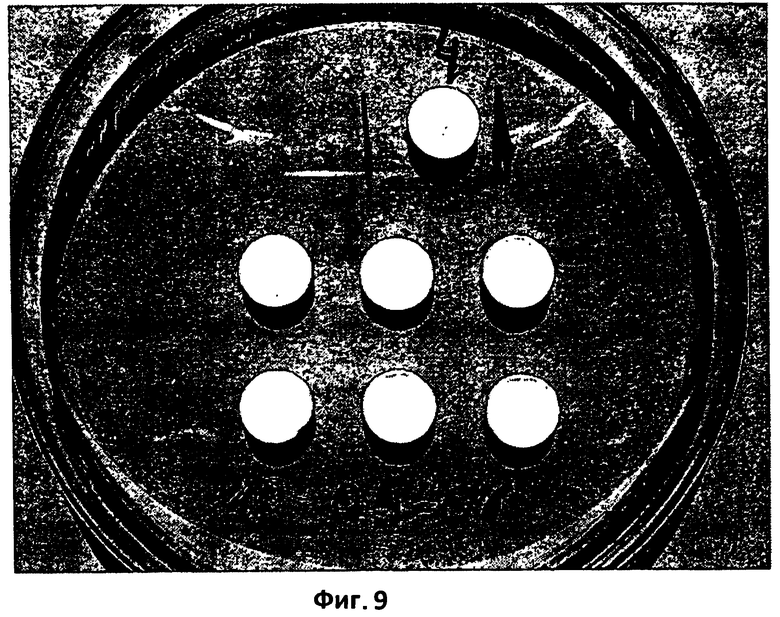
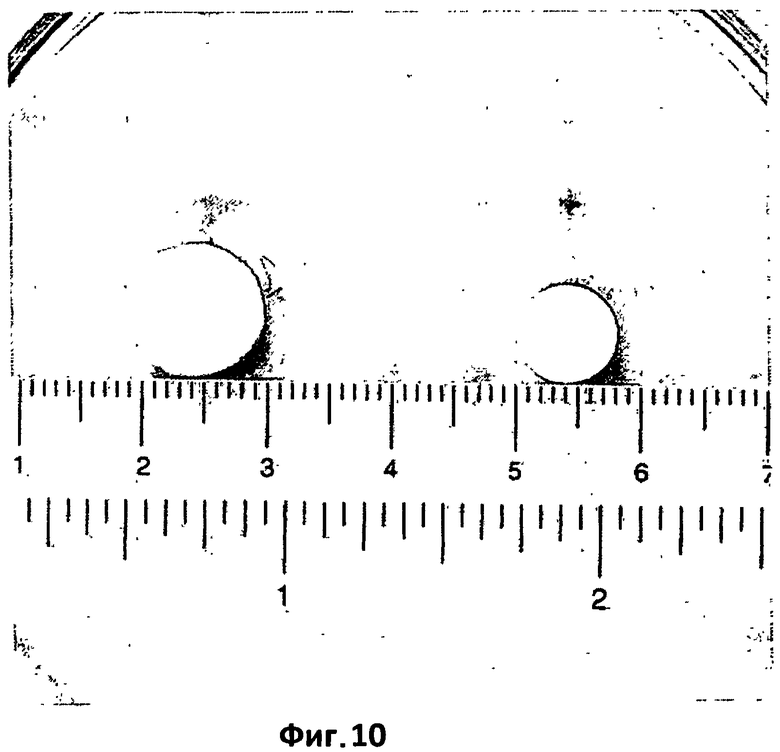

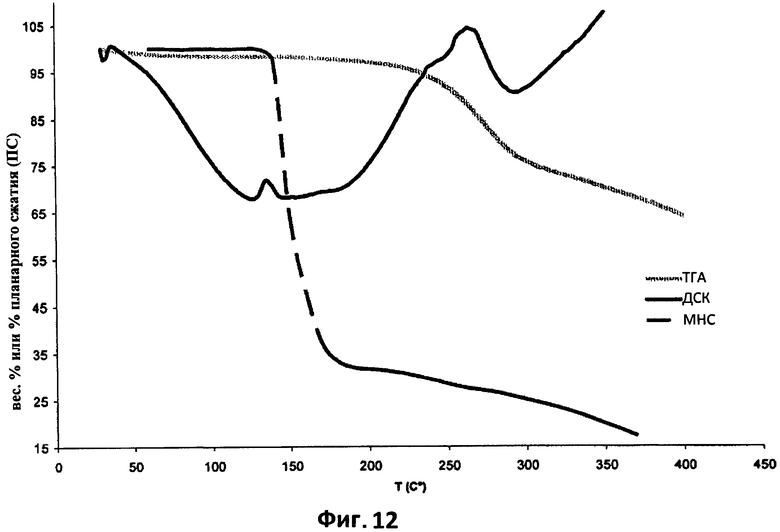


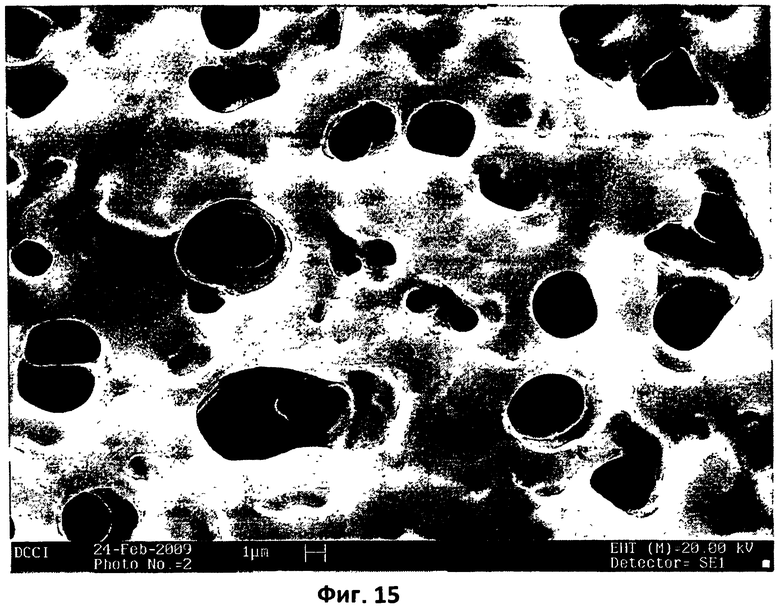
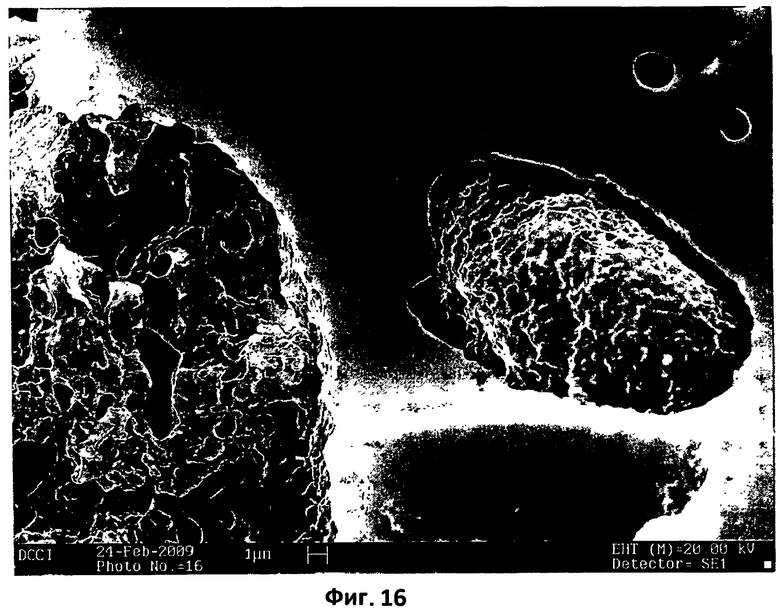


Изобретение относится к медицине. Описан способ получения биоадгезивной компактированной матрицы, который включает приготовление однородной смеси порошков, содержащей по меньшей мере одну алкилцеллюлозу или одну гидроксиалкилцеллюлозу и водонерастворимый, водонабухающий сшитый поликарбоксильный полимер, приготовление из такой порошковой смеси спрессованных заготовок с помощью прямого прессования и нагревание спрессованных заготовок, полученных таким образом, до температуры в диапазоне 80-250°С в течение 1-60 минут. Порошковая смесь может также содержать по меньшей мере одно активное вещество. Спрессованные заготовки, полученные таким образом, характеризуются замедленным высвобождением и проявляют кинетику высвобождения активного вещества по существу нулевого порядка в водном растворе при рН 4-8. 2 н. и 13 з.п. ф-лы, 18 ил., 5 табл., 4 пр.
1. Способ получения биоадгезивной компактированной матрицы для замедленного высвобождения активных веществ, включающий стадии:
- приготовления однородной смеси порошков, содержащей по меньшей мере одну алкилцеллюлозу или одну гидроксиалкилцеллюлозу и водонерастворимый, водонабухающий сшитый поликарбоксильный полимер;
- приготовления спрессованных или компактированных заготовок из указанной порошковой смеси путем прямого прессования;
- нагревания спрессованных или компактированных заготовок, полученных таким образом, при температуре в диапазоне 80-250°С, предпочтительно 90-160°С, в течение 1-60 минут, предпочтительно 1-30 минут.
2. Способ по п.1 для получения биоадгезивных спрессованных заготовок, содержащих по меньшей мере одно активное вещество и выполненных с возможностью замедленного или контролируемого высвобождения по меньшей мере одного указанного активного вещества, причем указанная однородная порошковая смесь также содержит по меньшей мере одно активное вещество.
3. Способ по п.1, отличающийся тем, что указанная однородная порошковая смесь дополнительно содержит разбавитель, выбранный из безводной лактозы или моногидрата лактозы в любой аморфной или кристаллической физической форме, и микрокристаллическую целлюлозу или ее смеси, возможно, в том числе предварительно обработанные, в частности полученный сушкой распылением состав, содержащий 75% моногидрата альфа-лактозы и 25% микрокристаллической целлюлозы (MicroceLak®).
4. Способ по п.1, отличающийся тем, что указанная алкилцеллюлоза выбрана из группы, включающей метилцеллюлозу и этилцеллюлозу, и указанная гидроксиалкилцеллюлоза выбрана из группы, включающей гидроксипропилцеллюлозу, гидроксипропилметилцеллюлозу, гидроксиэтилцеллюлозу, гидроксиэтилметилцеллюлозу.
5. Способ по любому из пп.1-4, отличающийся тем, что указанный водонерастворимый, водонабухающий сшитый поликарбоксильный полимер представляет собой поликарбофил.
6. Способ по любому из пп.1-4, отличающийся тем, что указанная однородная порошковая смесь дополнительно содержит один или более компонентов, выбранных из кросповидона, повидона, сополимера винилпирролидона и винилацетата (Kollidon® VA64), ацетатфталата целлюлозы, фталата гипромеллозы, поливинилового спирта, поливинилацетатфталата, циклодекстрина, метакрилатных полимеров, триацетата глицерина, триэтилцитрата, трибутилцитрата, ацетилтриэтилцитрата, ацетилатрибутилцитрата, дибутилсебацината, диэтилфталата, дибутилфталата, диоктилфосфата, полиэтиленгликоля, полиэтиленоксидов, кальций карбоксиметилцеллюлозы, натрий карбоксиметилцеллюлозы, инулина, хитозана, гуаровой смолы, ксантановой камеди, трагакантовой камеди, карбомера, каррагенина, альгиновой кислоты, полоксамера, алифатических сложных полиэфиров, ацетатбутиратацеллюлозы, хитозанлактата, пектина, сополимера полиэтилена и винилацетата, полиэтилена, сополимера поливинилацетата и метакриловой кислоты, карнаубского воска, бутилированного гидроксианизола, бутилированного гидрокситолуола, аскорбилпальмитата, глицерин пальмитостеарата, гидрогенизированного соевого и касторового масла, глицерин моностеарата, d-α-токоферола (витамина Е), витамин Е сукцината, витамина Е и TPGS, метилпарабена, бутилстеарата, стеарилового спирта, сахароза монопальмитата (сахарного сложного эфира), глицериновых эфиров и эфиров ПЭГ, полиоксиэтиленалкиловых эфиров, глицерин пальмитостеарата, минерального масла, касторового масла.
7. Способ по любому из пп.1-4, отличающийся тем, что указанная однородная порошковая смесь содержит по меньшей мере одно активное вещество, высушенный распылением состав, содержащий 75% моногидрата альфа-лактозы и 25% микрокристаллической целлюлозы (MicroceLac), этилцеллюлозу и поликарбофил.
8. Способ по п.7, отличающийся тем, что поликарбофил составляет 5-35%, предпочтительно 10-25%, по массе от общей массы указанной порошковой смеси.
9. Способ по п.8, отличающийся тем, что этилцеллюлоза и высушенный распылением состав, содержащий 75% моногидрата альфа-лактозы и 25% микрокристаллической целлюлозы (MicroceLac), присутствуют в указанной порошковой смеси в весовом соотношении от 1:2 до 2:1, предпочтительно от 0,8:1 до 1,2:1, и вместе составляют 45-95%, предпочтительно 60-80%, по массе от общей массы указанной порошковой смеси, и этилцеллюлоза присутствует в массовом соотношении к поликарбофилу от 1:5 до 5:1.
10. Способ по п.8 или 9, отличающийся тем, что по меньшей мере одно указанное активное вещество содержится в указанной порошковой смеси в количестве от 0,001 ppm до 50 мас.% от общей массы смеси.
11. Способ по любому из пп.1-4, 8, 9, отличающийся тем, что указанная спрессованная заготовка представляет собой фармацевтическую таблетку, и указанное активное вещество представляет собой фармакологически активное вещество.
12. Биоадгезивная таблетка для замедленного высвобождения активных веществ, содержащая по меньшей мере одну алкилцеллюлозу или одну гидроксиалкилцеллюлозу и водонерастворимый, водонабухающий сшитый поликарбоксильный полимер, предпочтительно представляющий собой поликарбофил, полученная с использованием биоадгезивной компактированной матрицы, полученной в соответствии со способом по п.1.
13. Биоадгезивная таблетка по п.12, дополнительно содержащая по меньшей мере одно активное вещество и выполненная с возможностью замедленного или контролируемого высвобождения по меньшей мере одного указанного активного вещества, полученная в соответствии со способом по п.2.
14. Биоадгезивная таблетка по п.13, характеризующаяся тем, что она проявляет контролируемое высвобождение и кинетику высвобождения активного вещества по существу нулевого порядка в водном растворе при величинах рН в диапазоне 4-8.
15. Таблетка по п.13 или 14, отличающаяся тем, что по меньшей мере одна указанная алкилцеллюлоза представляет собой этилцеллюлозу, и указанный водонерастворимый, водонабухающий сшитый поликарбоксильный полимер представляет собой поликарбофил, дополнительно содержащая разбавитель, выбранный из безводной лактозы или моногидрата лактозы в любой аморфной или кристаллической физической форме, и микрокристаллическую целлюлозу или ее смеси, возможно, также предварительно полученные, в частности высушенный распылением состав, содержащий 75% моногидрата альфа-лактозы и 25% микрокристаллической целлюлозы (MicroceLac).
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| US 5707655 A, 13.01.1998 | |||

