Область техники
Настоящее изобретение относится к соединению, основное структурное звено которого соответствует трициклическому пиразолопиримидину, которое ингибирует действие белка теплового шока 90 (HSP90).
Уровень техники
Белок теплового шока HSP90 представляет собой важнейший внутриклеточный белок-шаперон. Белки-шапероны представляют собой такие белки, которые связываются с различными белками для содействия фолдингу связанных белков. Группу белков, для фолдинга которых требуется HSP90, обычно называют HSP90-зависимыми белками.
Предполагают, что HSP90, также, как и белки со сложной структурой, такие как ко-шапероны, белки-партнеры и иммунофилины, вовлечены в механизм фолдинга HSP90-зависимых белков с помощью HSP90 и что они совместно содействуют фолдингу HSP90-зависимых белков (непатентный документ 1); однако же подробности механизма все еще в достаточной степени не ясны.
Предполагают, что HSP90-зависимые белки образуют комплекс с HSP90, ко-шаперонами и им подобными и затем конформационно меняются до сформированных зрелых белков, и предполагают, что белки подвергаются убиквитированию и деградации протеасомами в том случае, когда они не подверглись нормальному фолдингу с помощью HSP90 и им подобных (непатентные документы с 1 по 4).
В последние годы HSP90 рассматриваются в качестве кандидатов для терапевтических средств для различных заболеваний (например, рака, нейродегенеративных заболеваний, таких как болезнь Альцгеймера, сердечно-сосудистых заболеваний, инфекций, аутоиммунных заболеваний и заболеваний, связанных с апоптозным повреждением клетки) (непатентный документ 2).
В частности, поскольку многие связанные с раком белки, включая молекулярные мишени противораковых средств, представляют собой HSP90-зависимые белки, то предполагается, что ингибиторы HSP90 можно рассматривать в качестве кандидатов для создания противораковых средств. Например, белки со сложной структурой, задействованные в возникновении и развитии рака, такие как Her2, Raf, Akt и теломераза, известны в качестве HSP90-зависимых белков (непатентный документ 1). Предполагают, что эти связанные с раком белки представляют собой измененные белки, возникшие в процессе превращения несформированных белков в сформированные зрелые белки, и они действуют, вызывая злокачественную трансформацию клеток, соответственно, посредством использования HSP90 в качестве белка-шаперона. HSP90 представляет собой белок, присутствующий не только в раковых клетках, но и в нормальных клетках, а также имеются сообщения, что сродство к HSP90-зависимым белкам и АТФ-азная активность, необходимые для проявления его активности в качестве шаперона, являются повышенными в раковых клетках по сравнению с нормальными клетками (непатентные документы с 1 по 3). Вследствие этого ингибиторы HSP90, как предполагается, способны инактивировать связанные с раком белки со сложной структурой одновременно в специфических раковых клетках, и их рассматривают в качестве кандидатов для противораковых средств, которые эффективны и обладают широким спектром противоопухолевого действия.
Гелданамицин, гербимицин, 17-аллиламиногелданамицин (17-AAG) и им подобные представляют собой традиционно известные ингибиторы HSP90 (непатентные документы с 1 по 4). Эти соединения связываются с АТФ-связывающим «карманом» N-конца HSP90 и ингибируют связывание HSP90 с АТФ, с тем, чтобы ингибировать функционирование HSP90 в качестве белка-шаперона. Сообщается о разнообразных соединениях, ингибирующих HSP90, в дополнение к вышеуказанным соединениям (патентный документ 1, патентный документ 2, патентный документ 3, непатентный документ 5 и непатентный документ 6) и также сообщается о производном трициклического пиразолопиримидина (патентный документ 4).
Кроме того, в нескольких публикациях сообщается о запланированных применениях производных трициклического пиразолопиримидина и соединений, имеющих конденсированную кольцевую структуру, у которых также имеется три составляющих гетероциклических кольца, для противораковых целей (патентные документы с 5 по 9 и непатентные документы 7 и 8).
Перечень ссылок
Патентные документы
Патентный документ 1: WO 2005/28434
Патентный документ 2: WO 2008/049105
Патентный документ 3: WO 2008/093075
Патентный документ 4: WO 2008/035629
Патентный документ 5: WO 2004/047755
Патентный документ 6: WO 2006/015263
Патентный документ 7: WO 2005/021568
Патентный документ 8: WO 1998/043991
Патентный документ 9: WO 2008/100447
Непатентные документы:
Непатентный документ 1: Medicinal Research Reviews (2006) Vol. 26, No. 3, 310-338
Непатентный документ 2: TRENDS in Molecular Medicine (2004) Vol. 10, No. 6, 283-290
Непатентный документ 3: British Journal of Pharmacology (2005) 146, 769-780
Непатентный документ 4: TRENDS in Biochemical Sciences (2006) Mar, 31 (3), 164-172
Непатентный документ 5: Journal of Medicinal Chemistry (2005) Vol. 48, No. 13, 4212-4215
Непатентный документ 6: Journal of Medicinal Chemistry (2006) Vol. 49, No. 1, 381-390
Непатентный документ 7: Organic & Biomolecular Chemistry (2003) Vol. 1, No. 23, 4166-4172
Непатентный документ 8: Organic & Biomolecular Chemistry (2006) Vol. 4, No. 9, 1723-1729
Сущность изобретения
Проблема, решаемая изобретением
Несмотря на то, что ингибиторы HSP90 предполагаются для применения в качестве лекарственных средств, а именно, в качестве противораковых средств, но в действительности на настоящий момент нет подобных соединений, которые применялись бы в качестве лекарственных средств.
Способы решения проблемы
В результате широкомасштабных исследований для решения вышеуказанных проблем авторы настоящего изобретения нашли новые соединения, которые ингибируют АТФ-азную активность HSP90 и обладают противоопухолевой активностью, которые представлены формулой (1), как показано ниже, что и составило настоящее изобретение. Кроме того, авторы изобретения осуществили тестирование противоопухолевой активности in vivo, тестирование безопасности, тестирование метаболической стабильности, тестирование ингибирования метаболических ферментов и др. для соединения настоящего изобретения. В результате авторы изобретения обнаружили, что соединение настоящего изобретения имеет различные свойства, необходимые для лекарственных средств.
А именно, настоящее изобретение предоставляет:
[1] Соединение, представленное формулой (1), или его соль:
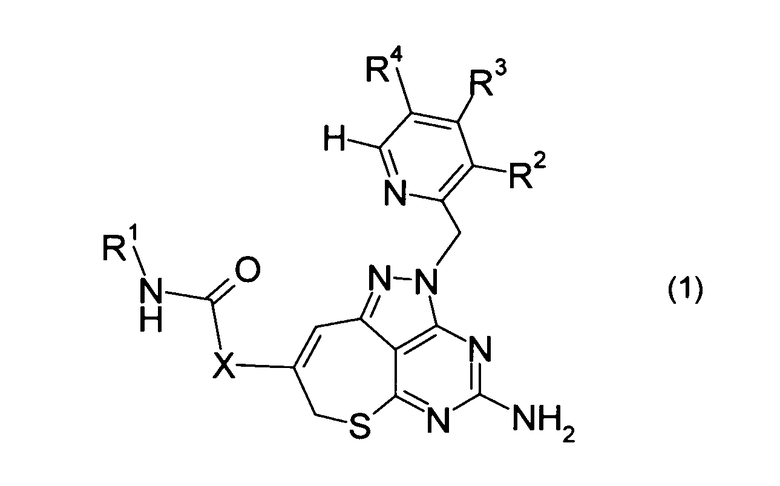
где в формуле (1)
R1 представляет собой С1-С6 алкильную группу, которая может быть замещена от 1 до 3 атомами галогена, или атом водорода,
R2 представляет собой атом галогена,
R3 представляет собой С1-С6 алкильную группу или С1-С6 алкоксильную группу,
R4 представляет собой С1-С6 алкильную группу, которая может быть замещена от 1 до 3 атомами галогена, цианогруппу, атом галогена или атом водорода, и
Х представляет собой одинарную связь или метиленовую группу.
[2] 2-{4-амино-2-[(3-хлор-4-метокси-5-метилпиридин-2-ил)метил]-2,7-дигидро-6-тиа-1,2,3,5-тетраазабензо[cd]азулен-8-ил}-N-метилацетамид.
[3] 2-{4-амино-2-[(3-бром-4-метокси-5-метилпиридин-2-ил)метил]-2,7-дигидро-6-тиа-1,2,3,5-тетраазабензо[cd]азулен-8-ил}-N-метилацетамид.
[4] 2-{4-амино-2-[(3-бром-4-метокси-5-метилпиридин-2-ил)метил]-2,7-дигидро-6-тиа-1,2,3,5-тетраазабензо[cd]азулен-8-ил}ацетамид.
[5] 2-{4-амино-2-[(3-хлор-4-метокси-5-метилпиридин-2-ил)метил]-2,7-дигидро-6-тиа-1,2,3,5-тетраазабензо[cd]азулен-8-ил}-N-(2-фторэтил)ацетамид.
[6] 4-амино-2-[(3-хлор-4-метоксипиридин-2-ил)метил]-N-(2,2-дифторэтил)-2,7-дигидро-6-тиа-1,2,3,5-тетраазабензо[cd]азулен-8-карбоксамид.
[7] Гидробромид соединения в соответствии с каким-либо из примеров с [2] по [6].
[8] Гидрохлорид соединения в соответствии с каким-либо из примеров с [2] по [6].
[9] Метансульфонат соединения в соответствии с каким-либо из примеров с [2] по [6].
[10] Этан-1,2-дисульфонат соединения в соответствии с каким-либо из примеров с [2] по [6].
[11] Ингибитор белков HSP90, включающий в себя соединение в соответствии с каким-либо из примеров с [1] по [10] или его соль.
[12] Средство для ингибирования АТФ-азной активности HSP90, которое включает в себя соединение в соответствии с каким-либо из примеров с [1] по [10] или его соль.
[13] Средство для ингибирования связывания HSP90 с АТФ, которое включает соединение в соответствии с каким-либо из примеров с [1] по [10] или его соль.
[14] Лекарственное средство, которое включает соединение в соответствии с каким-либо из примеров с [1] по [10] или его соль в качестве активного ингредиента.
[15] Противораковое средство, которое включает соединение в соответствии с каким-либо из примеров с [1] по [10] или его соль в качестве активного ингредиента.
[16] Фармацевтическая композиция, которая включает соединение в соответствии с каким-либо из примеров с [1] по [10] или его соль и фармацевтически приемлемый носитель.
[17] Способ лечения рака, который включает введение соединения в соответствии с каким-либо из примеров с [1] по [10] или его соли.
[18] Применение соединения в соответствии с каким-либо из примеров с [1] по [10] или его соли для получения лекарственного средства.
[19] Противораковое средство в соответствии с примером [15], где рак вовлечен в повышенную экспрессию генов HSP90-зависимого(ых) белка(ов).
[20] Способ лечения рака в соответствии с примером [17], где рак вовлечен в повышенную экспрессию генов HSP90-зависимого(ых) белка(ов).
[21] Противораковое средство в соответствии с примером [15], где рак вовлечен в мутацию HSP90-зависимого белка.
[22] Способ лечения рака в соответствии с примером [17], где рак вовлечен в мутацию HSP90-зависимого(ых) белка(ов).
[23] Противораковое средство в соответствии с примером [15], где рак вовлечен в активацию HSP90-зависимого(ых) белка(ов).
[24] Способ лечения рака в соответствии с примером [17], где рак вовлечен в активацию HSP90-зависимого(ых) белка(ов).
[25] Противораковое средство в соответствии с примером [15], где рак вовлечен в активацию внутриклеточного метаболического пути передачи сигналов, с которым связан(ы) HSP90-зависимый(е) белок(ки).
[26] Способ лечения рака в соответствии с примером [17], где рак вовлечен в активацию внутриклеточного метаболического пути передачи сигналов, с которым связан(ы) HSP90-зависимый(е) белок(ки).
[27] Противораковое средство в соответствии с примером [15], где рак зависит от HSP90-зависимого(ых) белка(ов).
[28] Способ лечения рака в соответствии с примером [17], где рак зависит от HSP90-зависимого(ых) белка(ов).
Преимущества изобретения
В соответствии с настоящим изобретением предложено новое производное пиразолопиримидина, обладающее активностью при ингибировании HSP90, которое представлено вышеуказанной формулой (1). Соединение настоящего изобретения может применяться в качестве противоопухолевого средства.
Описание примеров осуществления изобретения
В настоящем изобретении "белок теплового шока 90" или "HSP90" обозначает какого-либо или всех представителей семейства белков теплового шока HSP90, за исключением особо указанных случаев. Семейство HSP90 включает, например, HSP90α, HSP90β, 94 кДа глюкозо-регулируемый белок (GRP94) и связанный с рецептором Hsp75/фактора некроза опухоли белок 1 (TRAP1).
В настоящем изобретении "ингибитор HSP90" обозначает соединение или композицию, которые частично или полностью ингибируют действие HSP90. Примеры ингибитора HSP90 включают соединение или композицию, которые частично или полностью ингибируют экспрессию гена белка теплового шока HSP90, и соединение или композицию, которые частично или полностью ингибируют функционирование HSP90 в качестве белка-шаперона.
В этом документе "функционирование HSP90 в качестве белка-шаперона" обозначает функционирование HSP90 в содействии фолдингу зависимого белка с целью превращения зависимого белка в его функциональную форму или, например, функционирование HSP90 для стабилизации зависимого белка.
Таким образом, конкретные примеры ингибитора HSP90 включают соединение, ингибирующее экспрессию гена белка теплового шока HSP90, соединение, ингибирующее связывание HSP90 с зависимым белком, соединение, ингибирующее связывание HSP90 с ко-шаперонами или иммунофилинами, соединение, ингибирующее связывание HSP90 с АТФ, соединение, ингибирующее АТФ-азную активность HSP90 и соединение, ингибирующее конформационное изменение HSP90. Ингибитор HSP90 может применяться в качестве терапевтического средства при заболевании, вызванном воздействием HSP90.
Примеры HSP90-зависимого белка включают: киназы рецептора ростового фактора, такие как Her2, EGFR, c-Kit, c-Met, KDR, Flt3, IGF-1R и PDGF; внутриклеточные киназы, такие как PDK1, Akt, Raf, S6, Cdk4, Cdk6, Chk1, PLK1, Src, Aurora В, Bcr-Abl, GSK3β и ERK5; стероидные рецепторы, такие как GR, ERα, PR и AR; и другие, такие как HIF-1, Survivin, Tert, Bcl-6 и р53.
В настоящем изобретении примеры "заболеваний, вызванных воздействием HSP90" включают рак, нейродегенеративные заболевания, такие как болезнь Альцгеймера, сердечно-сосудистые заболевания, инфекции, аутоиммунные заболевания и заболевания, связанные с апоптозным повреждением клетки. В настоящем изобретении термины "опухоль" и "рак" могут использоваться взаимозаменяемо. Кроме того, в настоящем изобретении опухоль, злокачественная опухоль, рак, злокачественное новообразование, карцинома, саркома и им подобные в общем могут обозначаться как "опухоль" или "рак".
Отдельные конкретные заместители в формуле (1) настоящего изобретения будут описаны ниже.
R1 представляет собой С1-С6 алкильную группу, которая может быть замещена от 1 до 3 атомами галогена, или атом водорода.
В этом документе "С1-С6 алкильная группа, которая может быть замещена от 1 до 3 атомами галогена" обозначает прямую, разветвленную или циклическую алкильную группу, имеющую от 1 до 6 атомов углерода, которая незамещена или замещена от 1 до 3 атомами галогена. Примеры такой алкильной группы, имеющей от 1 до 6 атомов углерода, включают метильную группу, этильную группу, пропильную группу, изопропильную группу, циклопропильную группу, циклобутильную группу, циклопентильную группу и циклогексилэтильную группу. Примеры атома галогена, являющегося заместителем в такой алкильной группе, включают атом фтора, атом хлора, атом брома и атом йода. Такой заместитель может быть расположен при любом атоме углерода алкильной группы. В том случае, когда алкильная группа имеет несколько заместителей, заместители могут находиться при одном и том же атоме углерода или при разных атомах углерода. Такой заместитель в виде атома галогена для алкильной группы предпочтительно представляет собой атом фтора или атом брома, и более предпочтительно - атом фтора.
R1 предпочтительно представляет собой незамещенную С1-С3 алкильную группу, С1-С3 алкильную группу, имеющую атом фтора в качестве заместителя, или атом водорода, и более предпочтительны метильная группа, этильная группа, фторэтильная группа, дифторэтильная группа или атом водорода.
R2 представляет собой атом галогена. Примеры такого атома галогена включают атом фтора, атом хлора, атом брома и атом йода.
R2 предпочтительно представляет собой атом фтора, атом хлора или атом брома, и более предпочтительно - атом хлора или атом брома.
R3 представляет собой С1-С6 алкильную группу или С1-С6 алкоксильную группу.
В этом документе "С1-С6 алкильная группа" имеет такое же определение, как и в вышеуказанном R2. "С1-С6 алкоксильная группа" обозначает С1-С6 алкоксильную группу, имеющую в своем составе как составную алкильную часть вышеописанную "С1-С6 алкильную группу".
R3 предпочтительно представляет собой С1-С3 алкильную группу или С1-С3 алкоксильную группу, и более предпочтительны метильная группа, этильная группа или метоксигруппа.
R4 представляет собой С1-С6 алкильную группу, которая может быть замещена от 1 до 3 атомами галогена, цианогруппу, атом галогена или атом водорода.
В этом документе "С1-С6 алкильная группа, которая может быть замещена от 1 до 3 атомами галогена" имеет такое же определение как и для вышеуказанного R1.
R4 предпочтительно представляет собой С1-С3 алкильную группу, цианогруппу, атом фтора или атом водорода.
Х предпочтительно представляет собой одинарную связь или метиленовую группу.
Соединение, представленное формулой (1), в соответствии с настоящим изобретением, может представлять собой как стереоизомер, так и оптический изомер, происходящий из-за наличия асимметрического атома углерода. Стереоизомер, оптический изомер и их смесь, все они включаются в настоящее изобретение.
Соединение, представленное формулой (1), в соответствии с настоящим изобретением предпочтительно представляет собой, в частности, любое соединение, выбранное из группы нижеприведенных:
2-{4-амино-2-[(3-хлор-4-метокси-5-метилпиридин-2-ил)метил]-2,7-дигидро-6-тиа-1,2,3,5-тетраазабензо[cd]азулен-8-ил}-N-метилацетамид;
2-{4-амино-2-[(3-бром-4-метокси-5-метилпиридин-2-ил)метил]-2,7-дигидро-6-тиа-1,2,3,5-тетраазабензо[cd]азулен-8-ил}-N-метилацетамид;
2-{4-амино-2-[(3-бром-4-метокси-5-метилпиридин-2-ил)метил]-2,7-дигидро-6-тиа-1,2,3,5-тетраазабензо[cd]азулен-8-ил}ацетамид;
2-{4-амино-2-[(3-хлор-4-метокси-5-метилпиридин-2-ил)метил]-2,7-дигидро-6-тиа-1,2,3,5-тетраазабензо[cd]азулен-8-ил}-N-(2-фторэтил)ацетамид; и
4-амино-2-[(3-хлор-4-метоксипиридин-2-ил)метил]-N-(2,2-дифторэтил)-2,7-дигидро-6-тиа-1,2,3,5-тетраазабензо[cd]азулен-8-карбоксамид.
Соединение, представленное формулой (1), в соответствии с настоящим изобретением может быть превращено в фармакологически приемлемую соль, как это описано. Примеры таких солей включают соли галогеноводородной кислоты, такие как гидрохлоридные или гидройодидные соли; соли неорганической кислоты, такие как нитраты, перхлораты, сульфаты и фосфаты; низшие алкансульфонаты, такие как метансульфонаты, трифторметансульфонаты или этансульфонаты; арилсульфонаты, такие как бензолсульфонаты или п-толуолсульфонаты; соли органической кислоты, такие как формиаты, ацетаты, малаты, фумараты, сукцинаты, цитраты, тартраты, оксалаты или малеаты; и соли аминокислот, такие как орнитинаты, глутаматы или аспартаты. Из них предпочтительными являются соли галогеноводородной кислоты и органической кислоты, и бромат, гидрохлорид, метансульфонат или этан-1,2-дисульфонат представляют собой наиболее предпочтительные.
Соединение, представленное формулой (1), в соответствии с настоящим изобретением, или его соль могут быть представлены в свободной форме или в форме гидрата или сольвата. Оно может быть представлено в свободной форме, или в форме гидратной соли или в подобной форме, что представляется как результат абсорбции влаги из воздуха. Тип сольвата конкретно не ограничен, при условии, что он представляет собой фармакологически приемлемый. А именно, гидрат, этанолат или им подобные представляют собой предпочтительные. Кроме того, в том случае, когда присутствует атом азота в соединении, представленном формулой (1), в соответствии с настоящим изобретением, тогда это может быть форма N-оксида. Такие формы сольвата, гидрата, гидратной соли и N-оксида также включены в объем настоящего изобретения.
В зависимости от типов заместителей или их комбинации соединение, представленное формулой (1), в соответствии с настоящим изобретением может быть представлено в виде изомеров различных типов, включая геометрические изомеры, такие как цис-изомер или транс-изомер, таутомеры или оптические изомеры, такие как d-форма или l-форма. Соединение настоящего изобретения включает в себя все эти изомеры, стереоизомеры и смеси таких изомеров и стереоизомеров, которые смешаны в любых заданных соотношениях, за исключением особо указанных случаев.
Соединение, представленное формулой (1), в соответствии с настоящим изобретением может также включать в себя атомный(е) изотоп(ы) одного или нескольких составляющих его атомов в неопределенном соотношении. Примеры такого атомного изотопа включают дейтерий (2Н), тритий (3Н), йод-125 (125I) и углерод-14 (14С). Эти соединения пригодны к применению в качестве терапевтических или профилактических агентов, или в качестве реагентов, применяемых для исследования, таких как реагенты для количественного анализа, и в качестве диагностических средств, такие как in vivo моделирующие диагностические средства. Вне зависимости от того, являются или не являются они радиоактивными соединениями, все изотопные варианты соединения, представленного формулой (1), включены в объем настоящего изобретения.
Кроме того, настоящее изобретение также включает в качестве активного ингредиента фармацевтической композиции настоящего изобретения соединение, которое превращается в соединение (1), что осуществляется в результате реакции с ферментом, кислотой желудочного сока или им подобными при физиологических условиях in vivo; а именно, соединение, которое превращается в соединение (1) в результате ферментативного окисления, восстановления, гидролиза или им подобных реакций, или "фармакологически приемлемое соединение-пролекарство", которое превращается в соединение (1) в результате гидролиза или подобной реакции под действием кислоты желудочного сока, и др.
Примеры вышеупомянутого пролекарства включают соединения, полученные путем ацилирования, алкилирования или фосфорилирования аминогруппы соединения (1) (например, соединения получены путем превращения вышеупомянутой аминогруппы в эйкозаноил, аланил, пентиламинокарбонил, (5-метил-2-оксо-1,3-диоксолен-4-ил)метоксикарбонил, тетрагидрофуранил, пирролидилметил, пивалоилоксиметил или трет-бутил, и другие соединения).
Пролекарство соединения настоящего изобретения может быть получено из соединения (1) в соответствии с известным способом. Кроме того, пролекарство соединения настоящего изобретения включает в себя пролекарство, которое превращается в соединение (1) при физиологических условиях, как это описано на страницах 163-198 издания "Iyakuhin no Kaihatsu (Development of Medicaments)" Vol. 7, Bunshi Sekkei (Molecular Design), Hirokawa Shoten Со., 1990.
Далее будет описан типичный способ получения соединения, представленного формулой (1). Следует заметить, что подходящие защитные группы могут применяться в каждой реакции, в какой это необходимо, или что желаемое превращение может быть осуществлено посредством обычных химических реакций, широко известных в органической химии. Типы защитных групп и порядок осуществления превращений соответствующих заместителей особым образом не ограничены.
[Основная стадия 1]
Будет описан способ получения соединения, представленного формулой (1), в котором Х представляет собой метиленовую группу.
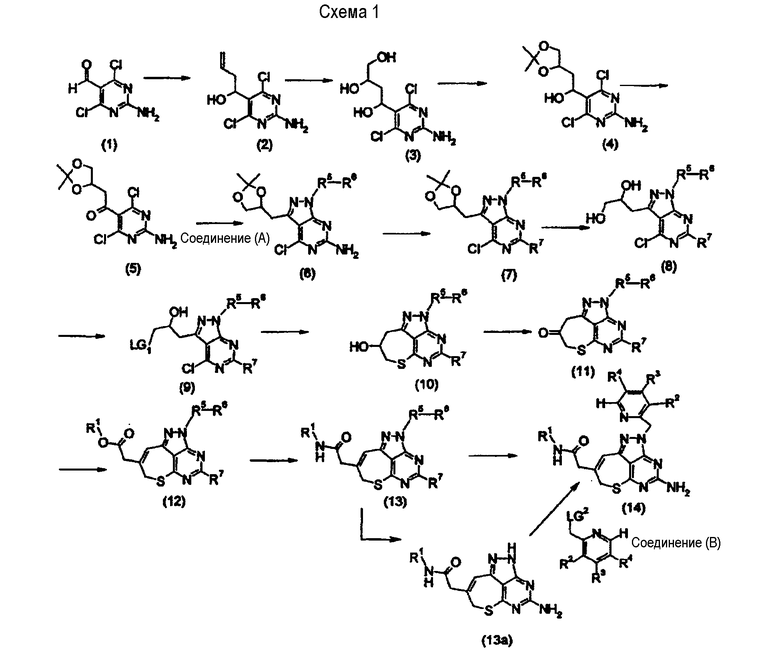
где R1, R2, R3 и R4 представляют собой таковые, как определено выше, соответственно, R5 представляет собой метиленовую группу, которая может быть замещена 1 или 2 алкильными группами, имеющими от 1 до 6 атомов углерода, R6 представляет собой арильную группу, которая может иметь заместитель(ли), или гетероциклическую группу, которая может иметь заместитель(ли), R7 представляет собой аминогруппу, имеющую защитную группу, и каждая из LG1 и LG2 представляет собой уходящую группу. Уходящие группы, LG1 и LG2, включают атом галогена, толуолсульфонилоксигруппу, метансульфонилоксигруппу, трифторметансульфонилоксигруппу и им подобные. Защитные группы для аминогруппы включают бензилоксикарбонильную группу, трет-бутоксикарбонильную группу, бензильную группу и им подобные.
Альдегидное производное (1) (получаемое как коммерчески доступный продукт) обрабатывают аллилбромидом и порошком индия в растворителе, получая соединение (2). Алкеновое производное (2) также может быть получено при использовании альдегидного производного (1) и реагента Гриньяра, такого как аллилмагнийбромид. В качестве растворителя предпочтительным является N,N-диметилформамид, N-метилпирролидон, тетрагидрофуран, диметилсульфоксид или им подобные. Температура реакции находится, предпочтительно, в интервале между -70°С и 100°С, и более предпочтительно - между -20°С и 50°С. Время реакции составляет, предпочтительно, от 1 часа до 48 часов.
Алкеновое производное (2) подвергают 1,2-дигидроксилированию, таким образом получая триольное производное (3). Примеры такого дигидроксилирования алкена включают реакцию с использованием перманганата калия, реакцию присоединения воды в присутствии соли ртути (способ Кучерова-Дениже), реакцию окисления в присутствии осмия с использованием каталитического количества тетраоксида осмия и аминоксида в качестве ко-оксиданта, и реакцию дигидроксилирования с использованием йода (способ Прево или способ Вудворда). Примеры растворителей, применяемых при этих реакциях, включают N,N-диметилформамид, N-метилпирролидон, тетрагидрофуран, диметилсульфоксид, метиленхлорид, ацетон, трет-бутанол, воду и их смеси. Приемлемая температура реакции находится в интервале между 0°С и 100°С, и предпочтительно - между 10°С и 50°С. Время реакции составляет предпочтительно от 1 часа до 72 часов.
Триольное производное (3) может быть превращено в ацетальное производное (4) путем обработки 2,2-диметоксипропаном и каталитическим количеством кислоты в растворителе, таком как N,N-диметилформамид, N-метилпирролидон, тетрагидрофуран или диметилсульфоксид, или путем обработки таким же каталитическим количеством кислоты в ацетоне. Примеры таких каталитических кислот включают кислоты Льюиса, включая п-толуолсульфоновую кислоту, хлористоводородную кислоту, серную кислоту и хлорид цинка в качестве типичных примеров. Температура реакции находится в интервале между 0°С и 100°С, и более предпочтительно - между 10°С и 50°С. Время реакции составляет предпочтительно от 1 часа до 48 часов.
Соединение (4) может быть превращено в кетоновое производное (5) посредством обработки при условиях, соответствующих реакции окисления. Примеры реакции окисления в этом случае включают окисление по реакции Мукаямы, активируемой диметилсульфоксидом, или подобным; окисление по реакции Сверна или реакцию окисления, использующую ее модификацию посредством применения дициклогексилкарбодиимида, трифторуксусного ангидрида, уксусного ангидрида или комплекса триоксид серы-пиридин вместо оксалилхлорида; и реакцию окисления с использованием диоксида марганца. Примеры растворителя, применяемого в данном случае, включают диоксан, тетрагидрофуран и метиленхлорид. Температура реакции находится, предпочтительно, в интервале между -70°С и 50°С. Время реакции составляет, предпочтительно, от 1 часа до 48 часов.
Кетоновое производное (5) может быть превращено в соединение (6) посредством обработки гидразиновым производным (R6-R5-NHNH2) (соединение (А)) в растворителе. Соединение (А) может быть получено способом, описанным в J. Am. Chem. Soc. 1995, 117, 4228-4239, и др. Примеры растворителя, применяемого в этой реакции, включают спирт, метиленхлорид, тетрагидрофуран, диоксан и их смеси. Температура реакции находится, предпочтительно, в интервале между -20°С и 50°С, и более предпочтительно - между 0°С и 30°С. В том случае, когда соединение (А) заменяют его солью, может соответственно применяться эквивалентное количество или избыток основания относительно соли. Пример основания представляет собой триэтиламин. Время реакции составляет, предпочтительно, от 1 часа до 48 часов.
Диольное производное (8) может быть получено путем защиты аминогруппы в 6-положении соединения (6) подходящей защитной группой, такой как бензилоксикарбонильная группа или трет- бутоксикарбонильная группа, чтобы превратить его в соединение (7), и затем обрабатывая ацетальное производное (7) кислотой, такой как серная кислота, хлористоводородная кислота или уксусная кислота. Примеры растворителя, применяемого в этой реакции, включают спирт, воду, диоксан и их смеси. Температура реакции находится, предпочтительно, в интервале между -10°С и 70°С, и более предпочтительно - между 0°С и 40°С. Время реакции составляет предпочтительно от 1 часа до 48 часов.
Соединение (9) может быть получено путем превращения гидроксильной группы соединения (8) в уходящую группу LG1, такую как атом галогена, толуолсульфонилоксильную группу, метансульфонилоксильную группу или трифторметансульфонилоксильную группу, посредством обработки тионилхлоридом, тионилбромидом, толуолсульфонилхлоридом, метансульфонилхлоридом или трифторметансульфонилхлоридом в присутствии основания, например, основываясь на общедоступных сведениях из органической химии. Температура реакции находится, предпочтительно, в интервале между -78°С и 30°С, и более предпочтительно - между
-20°С и 10°С. Время реакции составляет предпочтительно от 1 часа до 48 часов.
Соединение (10) может быть получено путем реакции соединения (9) с бисульфидом натрия в растворителе, таком как N,N-диметилформамид, с последующей обработкой основанием. Альтернативно, соединение (10) также может быть получено путем реакции соединения (9) с тиоацетатом калия в растворителе, таком как N,N-диметилформамид. Примеры основания включают карбонат калия и бикарбонат калия. Предпочтительное основание представляет собой карбонат калия. Приемлемая температура реакции находится в интервале между -30°С и 100°С, и предпочтительно - между -10°С и 70°С.
Примеры способа превращения гидроксильной группы в оксогруппу (превращение соединения (10) в соединение (11)) включают реакцию окисления Мукаямы и реакцию окисления Сверна или реакцию окисления, использующую ее модификацию посредством применения DCC, ангидрида трифторуксусной кислоты, уксусного ангидрида или комплекса триоксид серы-пиридин вместо оксалилхлорида. Эти способы описаны в изданиях The Chemical Society of Japan (ed.), "Jikken Kagaku Koza (Courses in Experimental Chemistry), 4th edition, Vol. 23, Yuki Gosei (Organic Synthesis) V" (Maruzen Со., Ltd., 1992) или им подобных.
Производное в виде сложного эфира уксусной кислоты (12) может быть получено путем реакции гомологизации, включающей в качестве типичного примера реакцию Виттига между неким коммерчески доступным реагентом, выбранным в соответствии с целью получения желаемой группы R1, и кетоновым производным (11). Реакция гомологизации представляет собой достаточно обычную известную реакцию и она может быть описана в издании Jikken Kagaku Koza (Courses in Experimental Chemistry) (4th edition, Vol. 22, издательство The Chemical Society of Japan, Maruzen Со., Ltd.) "Yuki Gosei (Organic Synthesis) I: Tankasuiso, Harogen Kagobutsu (Hydrocarbons, Halogen Compounds)", рр. 57-69.
Производное амида уксусной кислоты (13) может быть получено путем обеспечения направленного действия моноалкиламина на производное сложного эфира уксусной кислоты (12) в спирте, или путем гидролиза производного сложного эфира уксусной кислоты (12) с получением производного уксусной кислоты, и которое затем подвергают реакции конденсации с аминами различных типов (которые можно получить как коммерчески доступные продукты). Реакция гидролиза сложного эфира представляет собой достаточно обычную известную реакцию щелочного гидролиза. Ссылкой на нее является Jikken Kagaku Koza (Courses in Experimental Chemistry) (4th edition, Vol. 22, издательство The Chemical Society of Japan, Maruzen Со., Ltd.) "Yuki Gosei (Organic Synthesis) IV: San, Аминоsan, Peputido (Acids, Amino Acids, Peptides)", рр. 6-11. Способ, как правило применяемый в качестве способа пептидного синтеза, может быть успешно использован в проведении реакции конденсации с аминами. Примеры способа пептидного синтеза включают азидный метод, метод хлорангидридов кислот, метод DCC (дициклогексилкарбодиимидный метод), метод активированных эфиров, карбонилдиимидазольный метод, метод с использованием водорастворимого карбодиимида и метод с использованием диэтилцианофосфата. Эти методы описаны в изданиях М. Bondansky, Y. S. Klausner and М. А. Ondetti, "Peptide Synthesis" (А Wiley-interscience publication, New York, 1976), G. R. Pettit, "Synthetic Peptides" (Elsevier Scientific Publication Company, New York, 1976), The Chemical Society of Japan (ed.), "Jikken Kagaku Koza (Courses in Experimental Chemistry), 4th edition, Vol. 22, Yuki Gosei (Organic Synthesis) IV" (Maruzen Со., Ltd., 1992) или им подобных. Примеры растворителя, применяемого в реакции конденсации с аминами различных типов, включают N,N-диметилформамид, N-метилпирролидон, пиридин, хлороформ, метиленхлорид, тетрагидрофуран, диоксан, ацетонитрил и их смеси. Приемлемая температура реакции находится в интервале между -20°С и 50°С, и предпочтительно - между -10°С и 30°С.
Соединение (13) можно превратить в соединение (13а) посредством кислотной обработки, окислением или гидролизом, и последующую обработку осуществляют в условиях реакции удаления защитной группы, что приемлемо для защитной группы при аминогруппе, имеющей защитную группу (R4), в том случае, когда R6-R5-группа представляет собой такую защитную группу, как 4-метоксибензильная группа. Типичный пример условий проведения реакции удаления защитной группы, приемлемых для защитной группы, будет описан ниже. Например, в том случае, когда аминогруппа, в которую вводится заместитель в виде защитной группы, представляет собой алканоиламиногруппу или ароиламиногруппу, тогда группа может быть превращена в аминогруппу посредством гидролиза с использованием водного раствора гидроксида натрия, гидроксида калия, аммиака или им подобных. В том случае, когда аминогруппа, в которую вводится заместитель в виде защитной группы, представляет собой трет-бутоксикарбониламиногруппу или ди-трет-бутоксикарбониламиногруппу, тогда группа может быть превращена в аминогруппу посредством обработки кислотой, такой как хлористоводородная кислота или трифторуксусная кислота.
Соединение (13а) может быть превращено в соединение (14) посредством обработки производным пиридина (соединение (В)), полученным способом, описанным в J. Med. Chem. 1992, 35, 438-450, и др., в растворителе в присутствии основания. Примеры растворителя включают N,N-диметилформамид, N-метилпирролидон, тетрагидрофуран и диметилсульфоксид. Примеры основания включают гидрид натрия, этоксид натрия, трет-бутоксид калия, гидроксид калия, карбонат калия и карбонат цезия. Приемлемая температура реакции находится в интервале между 0°С и 100°С. Приемлемое время реакции составляет от 1 часа до 48 часов.
С другой стороны, в том случае, когда R6-R5-группа соединения (13) не является защитной группой, но представляет собой интересующий заместитель, тогда соединение (14) может быть получено посредством обработки аминогруппы, имеющей защитную группу (R7), в условиях вышеуказанной реакции удаления защитной группы.
[Основная стадия 2]
Будет описан способ получения соединения, представленного формулой (1), в которой Х представляет собой одинарную связь.
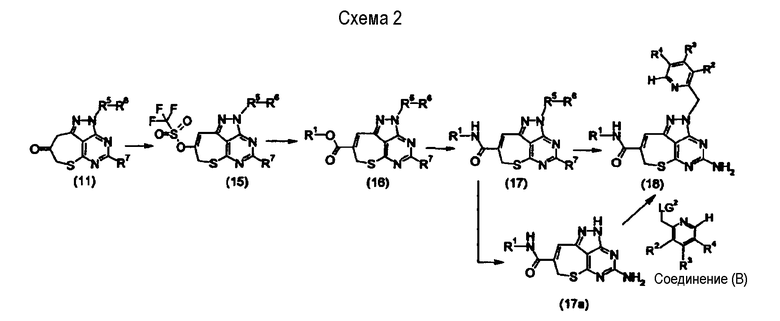
где R1, R2, R3, R4, R5, R6, R7 и LG2 представляют собой такие, как определено выше, соответственно.
Кетоновое производное (11) также может быть превращено в трифлатное соединение (15) посредством обработки трифторметансульфоновым ангидридом в присутствии основания. Предпочтительные примеры растворителя включают метиленхлорид, ацетонитрил, тетрагидрофуран и N,N-диметилформамид. Наиболее предпочтительный растворитель представляет собой метиленхлорид. Предпочтительные примеры основания включают гидрид натрия, гидрид калия, гидроксид натрия, гидроксид калия, карбонат калия, трет-бутоксид натрия, трет-бутоксид калия, пиридин, триэтиламин, DBU и диизопропилэтиламин. Наиболее предпочтительное основание представляет собой триэтиламин. Температура реакции находится, предпочтительно, в интервале между -80°С и 150°С, и более предпочтительно - между -10°С и 40°С.
Трифлатное соединение (15) может быть превращено в форму сложного эфира (16) путем осуществления реакции сочетания с монооксидом углерода в присутствии металлического катализатора и основания в спирте. В качестве такового спирта предпочтительными являются метанол и этанол. Металлический катализатор представляет собой, предпочтительно, палладиевый катализатор. Примеры палладиевого катализатора включают комплекс (1:1) [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий(II)/дихлорметан, дихлорбис(трифенилфосфин)палладий(II) и тетракис(трифенилфосфин)палладий(0). Предпочтительное количество применяемого металлического катализатора составляет от 0,01 до 0,2 молярных эквивалентов относительно трифлатного соединения (15). Предпочтительные примеры основания включают неорганические основания, такие как трифосфат калия, карбонат калия, карбонат натрия и карбонат цезия. Наиболее предпочтительное основание представляет собой трифосфат калия, карбонат натрия или им подобные. Основание предпочтительно применяют в количестве от 1 до 100 молярных эквивалентов относительно трифлатного соединения (15). Примеры добавки включают фосфорорганические соединения, такие как 1,1'-бис(дифенилфосфино)ферроцен (dppf) и трифенилфосфин. Добавку предпочтительно применяют в количестве от 0,05 до 0,2 молярных эквивалентов относительно трифлатного соединения (15). Температура реакции находится, предпочтительно, в интервале между -10°С и температурой кипения растворителя, и наиболее предпочтительно - в интервале между 0°С и 100°С. Время реакции составляет, как правило, приблизительно от 1 часа до 50 часов.
Следующую стадию (превращение соединения (17) в соединение (18)) можно осуществить посредством таких же обработок, как и в способе, показанном на схеме 1 (превращение соединения (13) в соединение (14)).
Так как соединение настоящего изобретения или его соль ингибирует HSP90, то оно может применяться в качестве ингибитора HSP90, в качестве средства для ингибирования АТФ-азной активности HSP90, или в качестве средства для ингибирования связывания HSP90 с АТФ. Следовательно, оно может применяться в качестве лекарственного средства, включающего соединение настоящего изобретения или его соль, и особенно предпочтительно - в качестве противоракового средства.
АТФ-азную активность HSP90 можно проверить посредством анализа АТФ-азы, обычно применяемого специалистом в данной области технике. Например, АТФ-азную активность HSP90 можно определить, используя рекомбинантный белок HSP90 и АТФ в присутствии или в отсутствие тестируемого соединения, как это описано ниже в тестовом примере 1. Альтернативно, для анализа АТФ-азы приемлемо применить, например, способ, описанный в Analytical Biochemistry 327, 176-183 (2004) или в Nature 425, 407-410 (2003).
Ингибировании экспрессии гена белка теплового шока HSP90 можно проверить посредством Нозерн-блоттинга, Вестерн-блоттинга, метода твердофазного иммуноферментного анализа ELISA или им подобными методами, обычно применяемыми специалистом в данной области техники. Например, мРНК выделяют из клеток, культивируемых в присутствии или в отсутствие тестируемого соединения, для последующего осуществления Нозерн-блоттинга. В том случае когда количество мРНК, соответствующей HSP90, во всей мРНК, выделенной из клеток, культивированных в присутствии тестируемого соединения, является пониженным по сравнению с тем количеством этой же мРНК, соответствующей HSP90, выделенной из клеток, культивированных в отсутствие тестированного соединения, тогда тестируемое соединение идентифицируют как соединение, ингибирующее экспрессию гена белка теплового шока HSP90. Альтернативно, можно определить количество белка HSP90 посредством Вестерн-блоттинга, используя способ, описанный, например, в Cancer. Res. 65, 6401-6408 (2005).
Ингибирование связывания HSP90 с зависимым белком можно определить, например, посредством иммунопреципитации и Вестерн-блоттинга, обычно применяемых специалистом в данной области техники. При проведении иммунопреципитации и Вестерн-блоттинга приемлемо использовать способ, описанный, например, в J. Biol. Chem. 277, 10346-10353 (2002).
Ингибирование соединением процесса связывания HSP90 с ко-шаперонами или иммунофилинами можно определить, например, посредством иммунопреципитации и Вестерн-блоттинга, обычно применяемых специалистом в данной области техники. Связывание HSP90 с ко-шаперонами или иммунофилинами можно определить в присутствии или в отсутствие тестируемого соединения посредством способа, описанного, например, в Nature 425, 407-410 (2003).
Ингибирование связывания HSP90 с АТФ можно определить, например, посредством теста на связывание радиоактивно-меченной АТФ с HSP90. Связывание HSP90 с радиоактивно-меченной АТФ возможно определить в присутствии или в отсутствие тестируемого соединения посредством осуществления метода, описанного, например, в J. Biol. Chem. 272, 18608-18613 (1997).
Ингибирование конформационного изменения HSP90 можно определить посредством конформационного анализа, используя, например, бис-АНС (1,1'-бис(4-анилино-5-нафталинсульфоновую кислоту)). Например, может быть осуществлен способ проведения конформационного анализа, описанный в J. Med. Chem. 47, 3865-3873 (2004).
Активность ингибирования клеточного роста можно определить, используя метод тестирования ингибирования роста, который обычно применяет специалист в данной области техники. Активность ингибирования клеточного роста можно определить, например, посредством сравнения уровней клеточного роста (например, для опухолевых клеток) в присутствии или в отсутствие тестируемого соединения, как это описано в нижеприведенном тестовом примере 2.
Уровень роста можно определить, используя тестовую систему для анализа живых клеток. Примеры метода анализа живых клеток включают тест включения [3Н]-тимидина, метод с использованием бромдезоксиуридина (BrdU) и исследование цитотоксичности с помощью МТТ.
Кроме того, in vivo противоопухолевую активность можно определять, используя метод тестирования противоопухолевой активности, обычно применяемый специалистом в данной области техники. Например, различные типы опухолевых клеток трансплантируют мышам, крысам или им подобным и после подтверждения жизнеспособности трансплантированных клеток соединение настоящего изобретения вводили животному посредством перорального введения, внутривенного введения, и др. В дальнейшем, спустя время от нескольких дней до нескольких недель, сравнивали рост опухоли в группе, которой не вводилось средство, с ростом опухоли в той группе, которой вводили соединение, таким образом подтверждали in vivo противоопухолевую активность соединения настоящего изобретения.
Соединение настоящего изобретения можно применять для лечения опухолей или рака, например, такого как рак легкого, рак желудочно-кишечного тракта, рак яичников, рак матки, рак молочной железы, рак печени, рак головы и шеи, рак крови, рак почки, неоплазия яичка, рак простаты, множественная миелома, рак кожи, такой как злокачественная меланома, саркома.
В связи с тем, что соединение настоящего изобретения обладает ингибирующим действием на HSP90, его можно применять при лечении рака, при котором повышена зависимость от HSP90. Такие виды рака, при которых повышена зависимость от HSP90, включают рак, который находится в зависимости от HSP90-зависимого(ых) белка(ов), рак, при котором гены HSP90-зависимого(ых) белка(ов) экспрессируются чрезмерно, рак, при котором HSP90-зависимый(е) белок(ки) претерпевает(ют) мутацию, и им подобные. Более конкретные примеры включают рак, при котором гены Her2, c-Met, Flt3 или им подобные экспрессируются чрезмерно, и рак, при котором c-kit, PDGFR, Raf или им подобные претерпевают мутацию. Впрочем, примеры не ограничиваются только вышеприведенными.
Кроме того, много групп факторов, связанных с раком (RAS-MAPK, PI3K, теломераза, и др.), имеются во внутриклеточном пути передачи сигналов, с которыми связан HSP90-зависимый белок. Если HSP90 ингибируется, то передача сигнала таким факторам тоже ингибируется. И как результат этого, ингибируется активация вышеуказанных внутриклеточных путей передачи сигнала. Таким образом, с этой точки зрения, поскольку соединение настоящего изобретения представляет собой ингибитор HSP90, то предпочтительно его применять для лечения различных типов рака.
Фармацевтическая композиция настоящего изобретения включает соединение настоящего изобретения и фармакологически приемлемый носитель. Она может применяться в виде инъекционных форм различных типов, таких как внутривенная инъекция, внутримышечная инъекция или подкожная инъекция, или ее можно вводить различными способами, такими как пероральное введение или введение через кожу. Фармакологически приемлемый носитель относится к фармакологически приемлемому веществу (например, эксципиенту, разбавителю, вспомогательному веществу, растворителю, и др.), которое связано с транспортировкой соединения настоящего изобретения или композиции, включающей в себя соединение настоящего изобретения, от одних системы или органа к другим системе или органу.
Что касается способа получения готовой лекарственной формы, то выбирают удобную готовую лекарственную форму (например, пероральная готовая лекарственная форма или инъекционная) и общепринятый метод для получения различных типов готовых лекарственных форм может применяться в зависимости от способа введения. Примеры пероральных готовых лекарственных форм включают таблетки, порошки, гранулы, капсулы, пилюли, пастилки, растворы, сиропы, эликсиры, эмульсии и масляные или водные суспензии. В случае перорального введения средство может быть либо в свободной форме, либо в форме соли. Водорастворимую готовую лекарственную форму можно получить путем образования аддукта с кислотой, применяя фармакологически приемлемую кислоту, или путем образования соли щелочного металла, такого как натрий. В том случае, когда готовая лекарственная форма является инъекционной, тогда могут использоваться стабилизатор, консервант, солюбилизатор в готовой лекарственной форме. Инъекция может быть предоставлена в виде лекарственной формы, которую готовят перед применением в виде сохраняемого раствора, который может содержать адъювант или ему подобное, в контейнере, и затем следует превращение ее в твердую лекарственную форму посредством лиофилизации или ей подобных способов. Одна доза может храниться в одном контейнере или многократное число доз может храниться в одном контейнере.
Примеры твердых готовых лекарственных форм включают таблетки, порошки, гранулы, капсулы, пилюли и пастилки. Эти твердые готовые лекарственные формы могут содержать фармацевтически приемлемое вспомогательное вещество совместно с соединением настоящего изобретения. Примеры вспомогательного вещества включают наполнители, объемообразующие агенты, связующие вещества, разрыхлители, солюбилизаторы, увлажняющие вещества и скользящие вещества. Все они могут выборочно смешиваться, как это необходимо для обеспечения получения готовой лекарственной формы.
Примеры жидких готовых лекарственных форм включают растворы, сиропы, эликсиры, эмульсии и суспензии. Эти готовые жидкие лекарственные формы могут содержать фармацевтически приемлемое вспомогательное вещество совместно с соединением настоящего изобретения. Примеры вспомогательного вещества включают суспендирующие агенты и эмульгирующие вещества. Все они могут выборочно смешиваться, как это необходимо для обеспечения получения готовой лекарственной формы.
Соединение настоящего изобретения может применяться для лечения рака у млекопитающих, в частности у людей. Доза и интервал между введением дозы лекарственного препарата могут быть выбраны должным образом на основании заключения врача, в соответствии с локализацией болезни и ростом организма, массой тела, полом или историей болезни пациента. В том случае, когда соединение настоящего изобретения вводят человеку, тогда диапазон доз составляет приблизительно от 0,01 мг/кг массы тела до приблизительно 500 мг/кг массы тела, и предпочтительно, от приблизительно 0,1 мг/кг массы тела до приблизительно 100 мг/кг массы тела в день. В том случае, когда соединение вводят человеку, тогда соединение вводят, предпочтительно, сразу одной дозой или раздельными дозами количеством от двух до четырех в день, и введение предпочтительно повторяют через подходящие интервалы. Дневная доза может превышать вышеуказанную дозу, если это необходимо на основании заключения врача.
Соединение настоящего изобретения может применяться совместно с другими противоопухолевыми средствами. Примеры таковых других противоопухолевых средств включают противоопухолевый антибиотик, противоопухолевый растительный ингредиент, BRM (модификатор биологического отклика), гормон, витамин, противоопухолевое антитело, молекулярно-нацеленное средство и другие противоопухолевые средства.
А именно, примеры алкилирующего средства включают алкилирующие агенты, такие как азотистый иприт, N-оксид азотистого иприта, или хлорамбуцил; азиридоновые алкилирующие средства, такие как карбоквон или тиотепа; эпоксидные алкилирующие средства, как такие дибромманнит или дибромдульцит; нитрозомочевинные алкилирующие средства, такие как кармустин, ломустин, семустин, нимустина гидрохлорид, стрептозоцин, хлорзотоцин или ранимустин; бусульфан; импросульфана тозилат; и Дакарбазин.
Примеры различных типов антиметаболитов включают пуриновые антиметаболиты, такие как 6-меркаптопурин, 6-тиогуанин или тиоинозин; пиримидиновые антиметаболиты, такие как фторурацил, тегафур, тегафур-урацил, кармофур, доксифлуридин, броксуридин, цитарабин или эноцитабин; и антагонисты фолиевой кислоты, такие как метотрексат или триметотрексат.
Примеры противоопухолевых антибиотиков включают антрациклиновые антибиотические противоопухолевые средства, такие как митомицин С, блеомицин, пепломицин, даунорубицин, акларубицин, доксорубицин, пирарубицин, THP-адриамицин, 4'-эпидоксорубицин или эпирубицин; хромомицин А3; и актиномицин D.
Примеры противоопухолевых растительных ингредиентов включают алкалоиды барвинка, такие как виндезин, винкристин или винбластин; таксаны, такие как паклитаксел или доцетаксел; и эпиподофиллотоксины, такие как этопозид или тенипозид.
Примеры модификатора биологического отклика BRM включают фактор некроза опухоли и индометацин.
Примеры гормона включают гидрокортизон, дексаметазон, метилпреднизолон, преднизолон, прастерон, бетаметазон, триамцинолон, оксиметолон, нандролон, метенолон, фосфестрол, этинилэстрадиол, хлормадинон и медроксипрогестерон.
Примеры витамина включают витамин С и витамин А.
Примеры противоопухолевого антитела и молекулярно-нацеленного агента включают трастузумаб, ритуксимаб, цетуксимаб, нимотузумаб, деносумаб, бевацизумаб, инфликсимаб, иматинибмезилат, гефитиниб, эрлотиниб, сунитиниб, лапатиниб и сорафениб.
Примеры других противоопухолевых средств включают цисплатин, карбоплатин, оксалиплатин, тамоксифен, камптотецин, ифосфамид, циклофосфамид, мелфалан, L-аспарагиназу, асеклатон (aseclatone), шизофиллан, пицибанил, прокарбазин, пипоброман, неокарциностатин, гидроксимочевину, убенимекс и крестин.
Настоящее изобретение включает способ предупреждения рака и/или способ лечения рака, который включает введение соединения настоящего изобретения или его соли.
Кроме того, настоящее изобретение также включает применение соединения настоящего изобретения, его соли или его сольвата для получения вышеуказанного лекарственного средства.
Настоящее изобретение будет конкретно описано со ссылкой на показанные ниже примеры; однако настоящее изобретение этим не ограничивается, и их не следует рассматривать в каком-либо смысле как ограничивающие. Реагенты, растворители и исходные вещества, которые особым образом не описаны в этом документе, представляют собой легко доступные из коммерческих источников.
Примеры
(Пример 1)
(1) 1-(2-Амино-4,6-дихлорпиримидин-5-ил)-3-бутен-1-ол
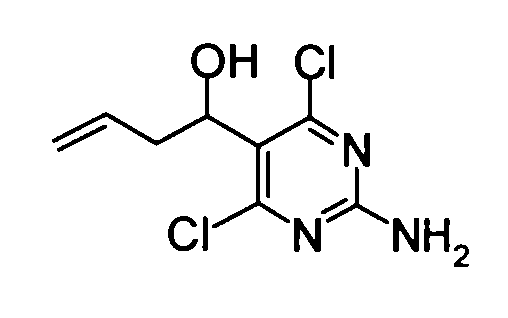
Порошкообразный индий (0,23 г) и порошкообразный цинк (1,31 г) добавляли к смеси, составленной из коммерчески доступных 2-амино-4,6-дихлорпиримидин-5-карбоальдегида (1,92 г) и N,N-диметилформамида (20 мл). Затем йодид натрия (0,15 г) и аллилбромид (1,73 мл) добавляли к смеси при комнатной температуре. Полученную в результате смесь перемешивали в течение 3 часов. Затем реакционную смесь фильтровали через целит, и этилацетат затем добавляли к фильтрату. Полученную в результате смесь последовательно промывали 1н хлористоводородной кислотой и насыщенным солевым раствором в этом порядке. Органический слой сушили над безводным сульфатом натрия и затем концентрировали. Затем гексан добавляли к остатку, и осадок отделяли фильтрованием, таким образом получая указанное в заголовке соединение (1,75 г) в виде твердого вещества.
1Н-ЯМР (ДМСО-D6) δ: 2,55-2,69 (2Н, м), 4,95-5,09 (3Н, м), 5,37 (1Н, д, J=4,1 Гц), 5,67-5,77 (1Н, м), 7,42 (2Н, с).
(2) 1-(2-Амино-4,6-дихлорпиримидин-5-ил)-2-(2,2-диметил-[1,3]диоксолан-4-ил)этан-1-ол
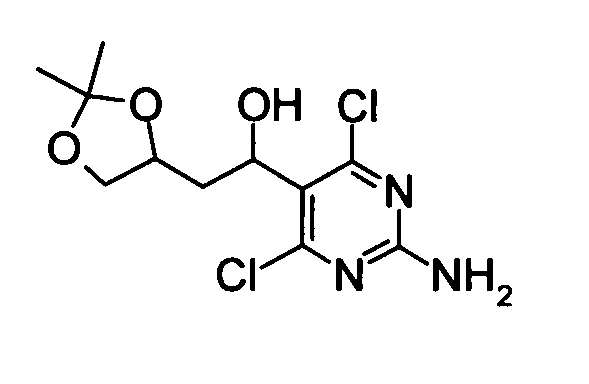
Смесь, составленную из 1-(2-амино-4,6-дихлорпиримидин-5-ил)-3-бутен-1-ола (57,24 г), N-метилморфолин-N-оксида (147,6 г), тетрагидрофурана (500 мл), ацетона (500 мл), воды (500 мл) и тетраоксида осмия (62 мг) перемешивали при комнатной температуре в течение 2 дней. После подтверждения исчезновения веществ добавляли к реакционному раствору насыщенный водный раствор тиосульфата натрия (1 л), и реакционную смесь затем концентрировали приблизительно до объема 1,5 л при пониженном давлении. Остаток насыщали хлоридом натрия, затем следовала экстракция тетрагидрофураном. Органический слой сушили над безводным сульфатом натрия. После фильтрования концентрировали фильтрат при пониженном давлении, и затем растворитель удаляли упариванием. N,N-Диметилформамид (500 мл), 2,2-диметоксипропан (210 мл) и моногидрат п-толуолсульфоновой кислоты (18,61 г) добавляли к полученному в результате остатку. Полученную в результате смесь перемешивали при комнатной температуре в течение 14 часов. Насыщенный раствор бикарбоната натрия (1 л) и воду (1 л) добавляли к реакционной смеси, затем следовала экстракция этилацетатом. Органический слой последовательно промывали водой и насыщенным солевым раствором в этом указанном порядке, и затем его сушили над безводным сульфатом натрия. После фильтрования концентрировали фильтрат приблизительно до объема 100 мл при пониженном давлении. Гексан добавляли к остатку и осадок отделяли фильтрованием, таким образом получая указанное в заголовке соединение (53,88 г) в виде твердого вещества.
1Н-ЯМР (ДМСО-D6) δ: 1,22-1,32 (6Н, м), 1,72-2,23 (2Н, м), 2,50 (1Н, с), 3,50 (1Н, тд, J=14,2, 6,9 Гц), 4,22-3,92 (2Н, м), 5,06-5,36 (2Н, м), 7,43 (2Н, д, J=12,8 Гц).
Масс спектрометрия с ионизацией электрораспылением (ESI-MS) m/z: 308 (М+Н)+.
(3) 1-(2-Амино-4,6-дихлорпиримидин-5-ил)-2-(2,2-диметил-[1,3]диоксолан-4-ил)этан-1-он
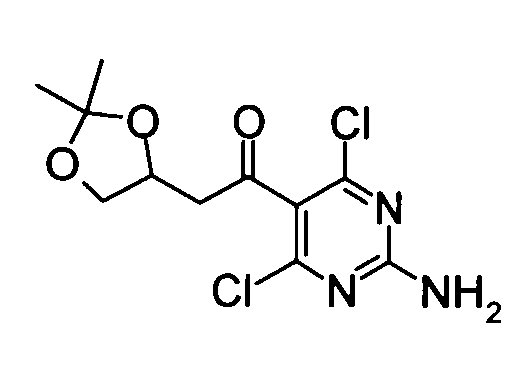
Уксусный ангидрид (149 мл) добавляли по каплям к смеси, составленной из вышеуказанных 1-(2-амино-4,6-дихлорпиримидин-5-ил)-2-(2,2-диметил-[1,3]диоксолан-4-ил)этан-1-ола (74,70 г) и диметилсульфоксида (600 мл) при комнатной температуре в течение 15 минут при охлаждении баней со льдом. Реакционную смесь затем перемешивали при той же температуре, как указано выше, в течение 18 часов. После подтверждения исчезновения веществ реакционный раствор выливали в ледяную воду. Выпавший твердый осадок отделяли фильтрованием, таким образом получая указанное в заголовке соединение (68,26 г).
1Н-ЯМР (CDCl3) δ: 1,37 (3Н, с), 1,42 (3Н, с), 2,98-3,06 (1Н, м), 3,32-3,40 (1Н, м), 3,67-3,72 (1Н, м), 4,25-4,30 (1Н, м), 4,57-4,64 (1Н, м), 5,72 (2Н, с).
ESI-MS m/z: 306 (М+Н)+.
(4) 4-Хлор-3-[(2,2-диметил-1,3-диоксолан-4-ил)метил]-1-(4-метоксибензил)-1H-пиразоло[3,4-d]пиримидин-6-амин
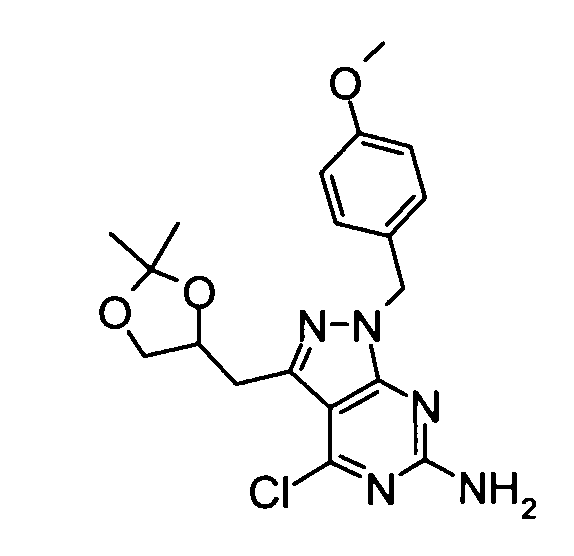
Триэтиламин (83,68 мл) добавляли к смеси, составленной из вышеуказанных 1-(2-амино-4,6-дихлорпиримидин-5-ил)-2-(2,2-диметил-[1,3]диоксолан-4-ил)этан-1-она (61,23 г), гидрохлорида (4-метоксибензил)гидразина (41,50 г), полученного способом, описанным в патенте США № US2003/18197, и дихлорметана (600 мл), в течение 30 минут при охлаждении баней со льдом. Затем по мере того, как температура реакционного раствора постепенно повышалась, его перемешивали в течение 17 часов. Затем к реакционной смеси добавляли 10% водный раствор лимонной кислоты, затем следовала экстракция дихлорметаном. Органический слой сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении. К полученному в результате остатку добавляли 5% водный раствор лимонной кислоты, и затем осадок отделяли фильтрованием. Полученный в результате осадок промывали водой, таким образом получая указанное в заголовке соединение (73,84 г) в виде твердого вещества.
1Н-ЯМР (CDCl3) δ: 1,36 (3Н, с), 1,43 (3Н, с), 3,11 (1Н, дд, J=14,7, 8,1 Гц), 3,43 (1Н, дд, J=14,7, 5,2 Гц), 3,73-3,78 (4Н, м), 4,08 (1Н, дд, J=8,1, 6,0 Гц), 4,54-4,61 (1Н, м), 4,77 (2Н, ушир.с), 5,22 (2Н, с), 6,83 (2Н, д, J=8,5 Гц), 7,24 (2Н, д, J=8,5 Гц).
ESI-MS m/z: 404 (М+Н)+.
(5) Бикарбонат ди-трет-бутил{4-хлор-3-[(2,2-диметил-1,3-диоксолан-4-ил)метил]-1-(4-метоксибензил)-1H-пиразоло[3,4-d]пиримидин-6-ил}имида
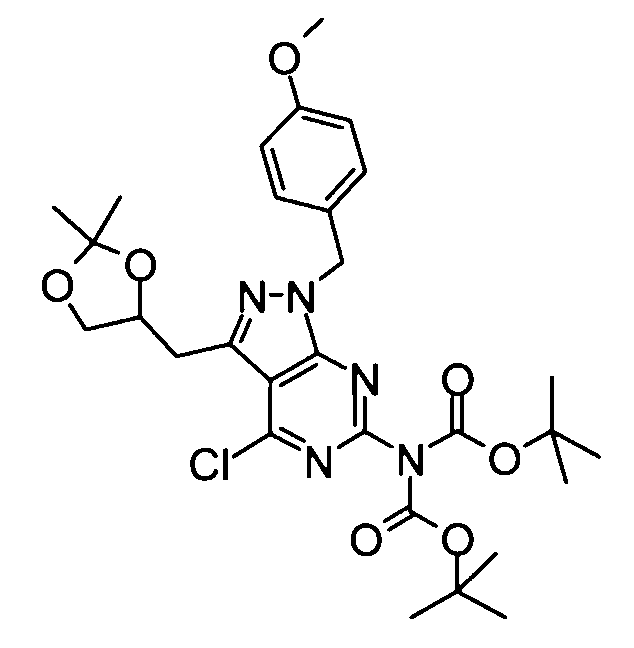
4-Диметиламинопиридин (2,20 г) и ди-трет-бутилбикарбонат (86,59 г) добавляли к смеси, составленной из вышеуказанных 4-хлор-3-[(2,2-диметил-1,3-диоксолан-4-ил)метил]-1-(4-метоксибензил)-1H-пиразоло[3,4-d]пиримидин-6-амина (72,83 г) и тетрагидрофурана (700 мл), и полученную в результате смесь затем перемешивали при комнатной температуре в течение 12 часов. Затем реакционную смесь фильтровали, затем концентрировали фильтрат при пониженном давлении. Полученный в результате остаток очищали с помощью колоночной хроматографии на силикагеле (этилацетат-гексан), таким образом получая указанное в заголовке соединение (70,00 г) в виде аморфного вещества.
1Н-ЯМР (CDCl3) δ: 1,37 (3Н, с), 1,40 (3Н, с), 1,44-1,46 (18Н, м), 3,21-3,29 (1Н, м), 3,48-3,55 (1Н, м), 3,74-3,81 (4Н, м), 4,09-4,15 (1Н, м), 4,58-4,66 (1Н, м), 5,48 (2Н, дд, J=17,3, 15,1 Гц), 6,81 (2Н, д, J=7,8 Гц), 7,27-7,30 (2Н, м).
ESI-MS m/z: 604 (М+Н)+.
(6) Бикарбонат ди-трет-бутил[4-хлор-3-(2,3-дигидроксипропил)-1-(4-метоксибензил)-1H-пиразоло[3,4-d]пиримидин-6-ил]имида
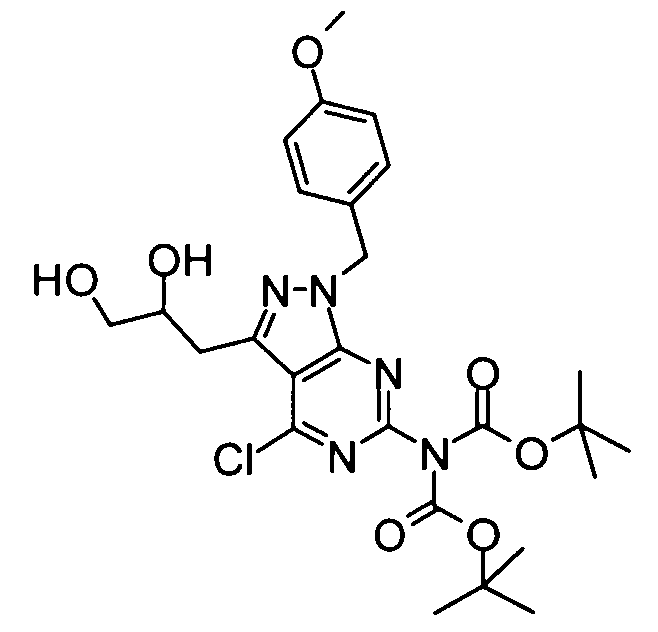
Вышеуказанный бикарбонат ди-трет-бутил{4-хлор-3-[(2,2-диметил-1,3-диоксолан-4-ил)метил]-1-(4-метоксибензил)-1H-пиразоло[3,4-d]пиримидин-6-ил}имида (53,85 г) растворяли в ацетонитриле (500 мл), и затем добавляли к раствору дигидрат хлорида меди(II) (30,39 г). Полученную в результате смесь перемешивали при комнатной температуре в течение 2 часов. Затем насыщенный водный раствор хлорида аммония добавляли к реакционному раствору, затем следовала экстракция этилацетатом. Органический слой промывали насыщенным солевым раствором и затем его сушили над безводным сульфатом натрия, затем концентрировали при пониженном давлении. Полученный в результате остаток очищали с помощью колоночной хроматографии на силикагеле (этилацетат-гексан), таким образом получая указанное в заголовке соединение (37,70 г) в виде аморфного вещества.
1Н-ЯМР (CDCl3) δ: 1,46 (18Н, с), 3,15 (1Н, д, J=3,7 Гц), 3,23-3,33 (2Н, м), 3,62-3,82 (5Н, м), 4,26-4,34 (1Н, м), 5,49 (2Н, т, J=15,9 Гц), 6,82 (2Н, д, J=8,1 Гц), 7,25-7,30 (2Н, м).
ESI-MS m/z: 564 (М+Н)+.
(7) Бикарбонат ди-трет-бутил[8-гидрокси-2-(4-метоксибензил)-2,7,8,9-тетрагидро-6-тиа-1,2,3,5-тетраазабензо[cd]азулен-4-ил]имида
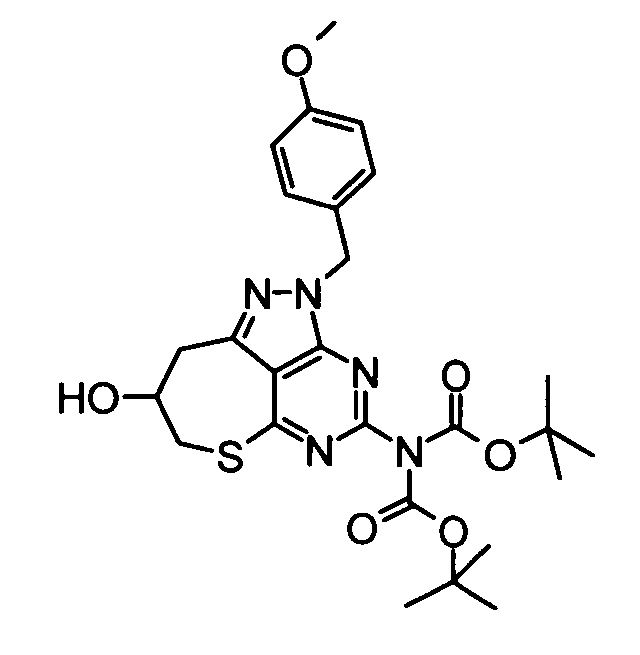
Метансульфонилхлорид (4,23 мл) добавляли по каплям к смеси, составленной из вышеуказанных бикарбоната ди-трет-бутил[4-хлор-3-(2,3-дигидроксипропил)-1-(4-метоксибензил)-1H-пиразоло[3,4-d]пиримидин-6-ил]имида (28,00 г), 2,4,6-коллидина (16,53 мл) и безводного дихлорметана (400 мл), при охлаждении баней со льдом. Полученную в результате смесь затем перемешивали при 4°С в течение 15 часов. Затем к реакционной смеси добавляли 10% водный раствор лимонной кислоты, затем следовала экстракция дихлорметаном. Органический слой сушили над безводным сульфатом натрия и затем его концентрировали при пониженном давлении. Полученный в результате остаток растворяли в N,N-диметилформамиде (300 мл), и затем раствору добавляли моногидрат гидросульфида натрия (5,52 г) к при охлаждении баней со льдом. Затем полученную в результате смесь перемешивали при комнатной температуре в течение 1,5 часов. Затем к реакционной смеси добавляли карбонат калия (10,29 мг) и полученную в результате смесь затем нагревали до 50°С, затем следовал дальнейший процесс перемешивания в течение 5 часов. В дальнейшем к реакционной смеси добавляли этилацетат, и полученную в результате смесь последовательно промывали 10% водным раствором лимонной кислоты и затем насыщенным солевым раствором. Полученный органический раствор сушили над безводным сульфатом натрия и затем его концентрировали при пониженном давлении. Полученный в результате остаток очищали с помощью колоночной хроматографии на силикагеле (этилацетат-гексан), таким образом получая указанное в заголовке соединение (20,59 г) в виде твердого вещества.
1Н-ЯМР (CDCl3) δ: 1,45 (18Н, с), 2,39 (1H, ушир.с), 3,29-3,51 (4Н, м), 4,58 (1Н, ушир.с), 3,76 (3Н, с), 5,42-5,49 (2Н, м), 6,82 (2Н, д, J=8,6 Гц), 7,30 (2Н, д, J=8,6 Гц).
ESI-MS m/z: 544 (М+Н)+.
(8) 4-[Бис(трет-бутоксикарбонил)амино]-2-(4-метоксибензил)-2,7-дигидро-6-тиа-1,2,3,5-тетраазабензо[cd]азулен-8-илацетат.
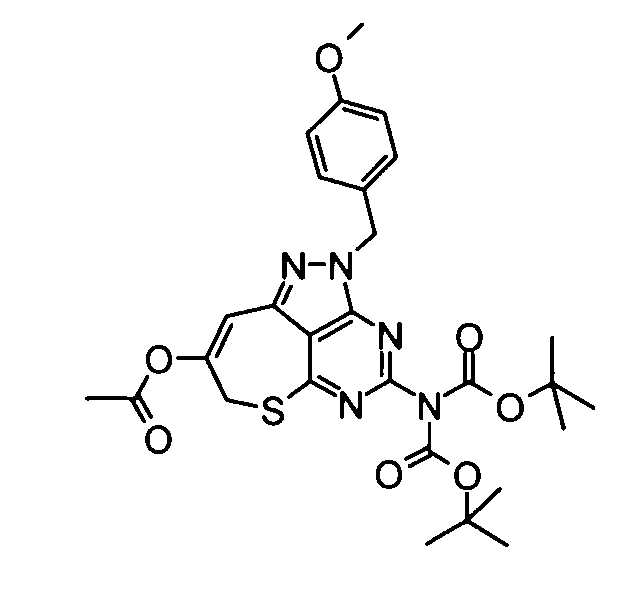
В атмосфере азота уксусный ангидрид (14 мл) добавляли по каплям к смеси, составленной из вышеуказанных бикарбоната ди-трет-бутил[8-гидрокси-2-(4-метоксибензил)-2,7,8,9-тетрагидро-6-тиа-1,2,3,5-тетраазабензо[cd]азулен-4-ил]имида (8,17 г), диметилсульфоксида (74 мл) и пиридина (12 мл) при охлаждении на льду, и полученную в результате смесь затем перемешивали в течение 30 минут. Затем реакционный раствор дополнительно перемешивали при комнатной температуре в течение 15 часов. После подтверждения исчезновения веществ реакционную смесь разбавляли этилацетатом и затем промывали ее насыщенным солевым раствором. Органический слой сушили над безводным сульфатом натрия, затем концентрировали при пониженном давлении. Полученный в результате остаток очищали с помощью колоночной хроматографии на силикагеле (этилацетат-гексан), таким образом получая указанное в заголовке соединение (6,15 г) в виде аморфного вещества.
1Н-ЯМР (CDCl3) δ: 1,44 (18Н, с), 2,26 (3Н, с), 3,77 (3Н, с), 3,88 (2Н, с), 5,50 (2Н, с), 6,68 (1Н, с), 6,83 (2Н, д, J=8,8 Гц), 7,31 (2Н, д, J=8,8 Гц).
ESI-MS m/z: 584 (М+Н)+.
(9) Бикарбонат ди-трет-бутил[2-(4-метоксибензил)-8-оксо-2,7,8,9-тетрагидро-6-тиа-1,2,3,5-тетраазабензо[cd]азулен-4-ил]имида
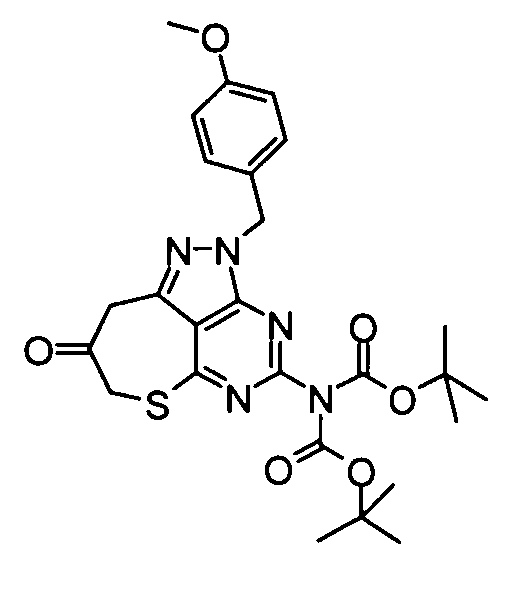
Смесь, составленную из вышеуказанных 4-[бис(трет-бутоксикарбонил)амино]-2-(4-метоксибензил)-2,7-дигидро-6-тиа-1,2,3,5-тетраазабензо[cd]азулен-8-илацетата (6,15 г), метанола (200 мл) и карбоната калия (0,73 г) перемешивали в течение 1,5 часов при охлаждении баней со льдом. После подтверждения исчезновения веществ к реакционной смеси добавляли насыщенный водный раствор хлорида аммония, затем следовала экстракция этилацетатом. Органический слой промывали насыщенным солевым раствором, и затем его сушили над безводным сульфатом натрия, затем концентрировали при пониженном давлении, таким образом получая указанное в заголовке соединение (5,70 г) в виде аморфного вещества.
1Н-ЯМР (CDCl3) δ: 1,46 (18Н, с), 3,84 (2Н, с), 3,77 (3Н, с), 4,23 (2Н, с), 5,48 (2Н, с), 6,83 (2Н, д, J=8,6 Гц), 7,32 (2Н, д, J=8,6 Гц).
ESI-MS m/z: 542 (М+Н)+.
(10) Этил{4-[бис(трет-бутоксикарбонил)амино]-2-(4-метоксибензил)-2,7-дигидро-6-тиа-1,2,3,5-тетраазабензо[cd]азулен-8-ил}ацетат
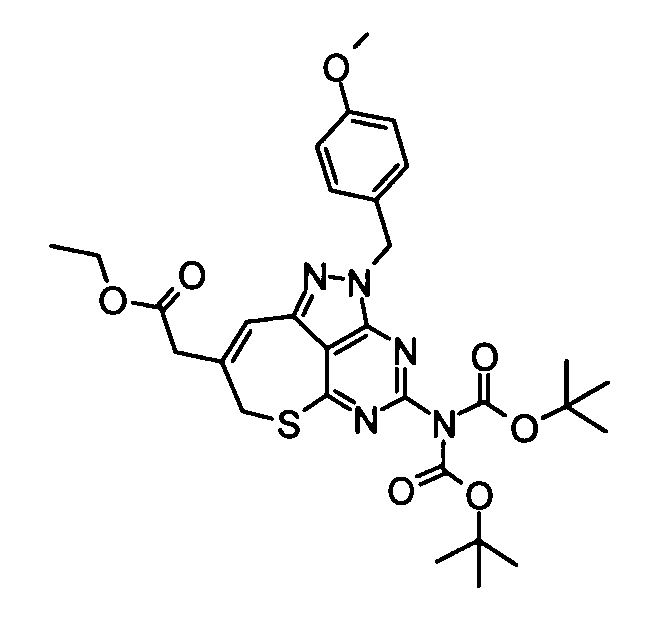
Смесь, составленную из вышеуказанных бикарбоната ди-трет-бутил[2-(4-метоксибензил)-8-oxo-2,7,8,9-тетрагидро-6-тиа-1,2,3,5-тетраазабензо[cd]азулен-4-ил]имида (5,19 г), этил(трифенилфосфанилиден)ацетата (3,51 г) и толуола (300 мл) перемешивали при 65°С в течение 13 часов. Затем реакционную смесь концентрировали при пониженном давлении и остаток затем очищали с помощью колоночной хроматографии на силикагеле (этилацетат-гексан), таким образом получая указанное в заголовке соединение (3,78 г) в виде аморфного вещества.
1Н-ЯМР (CDCl3) δ: 1,29 (3Н, т, J=7,1 Гц), 1,69-1,77 (1Н, м), 2,37-2,40 (1Н, м), 2,46-2,52 (1Н, м), 2,68-2,71 (2Н, м), 4,20 (2Н, кв, J=7,1 Гц), 5,10-5,13 (1Н, м), 5,20 (2Н, ушир.с).
ESI-MS m/z: 612 (М+Н)+
(11) 2-(4-амино-2,7-дигидро-6-тиа-1,2,3,5-тетраазабензо[cd]азулен-8-ил)-N-метилацетамидтрифторацетат
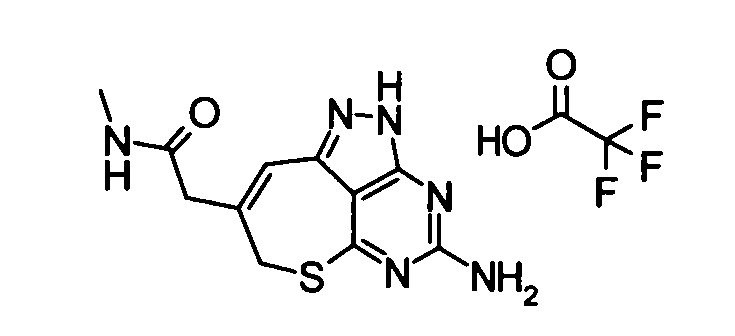
Вышеуказанный этил {4-[бис(трет-бутоксикарбонил)амино]-2-(4-метоксибензил)-2,7-дигидро-6-тиа-1,2,3,5-тетраазабензо[cd]азулен-8-ил}ацетат (2,2 г) растворяли в растворе 40% метиламин/метанол (40 мл), и смешанный раствор затем перемешивали при комнатной температуре в течение 2 часов. Завершение реакции подтверждали с помощью ЖХ-МС, и затем растворитель удаляли упариванием при пониженном давлении. Затем к полученному в результате остатку добавляли анизол (2 мл) и трифторуксусную кислоту (40 мл), и полученную в результате смесь затем перемешивали при 65°С в течение 15 часов. Затем реакционный раствор концентрировали при пониженном давлении, и к остатку добавляли смешанный раствор изопропиловый эфир - диэтиловый эфир. Осадок отделяли фильтрованием, таким образом получая указанное в заголовке соединение (1,53 г) в виде твердого вещества.
ESI-MS m/z: 277 (М+Н)+.
(12) 5-Хлор-4-гидрокси-6-метилникотиновая кислота
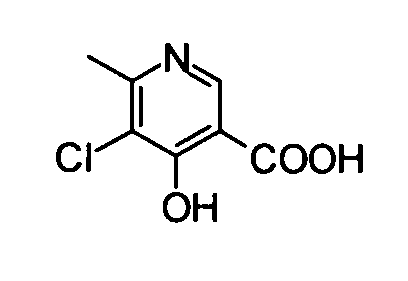
Коммерчески доступную 4-гидрокси-6-метилникотиновую кислоту (300 мг) суспендировали в 3 мл ацетонитрила, и затем к суспензии добавляли N-хлорсукцинимид (380 мг). Полученную в результате смесь перемешивали при комнатной температуре в течение 30 минут. Затем реакционный раствор нагревали до температуры кипения с обратным холодильником в течение 45 минут. После подтверждения исчезновения веществ реакционный раствор охлаждали во льду, и затем осадок отделяли фильтрованием, таким образом получая указанное в заголовке соединение (324 мг) в виде твердого вещества.
1Н-ЯМР (CD3OD) δ: 2,56 (3Н, с), 8,50 (1Н, с).
ESI-MS m/z: 188 (М+Н)+
(13) Метил 4,5-дихлор-6-метилникотинат
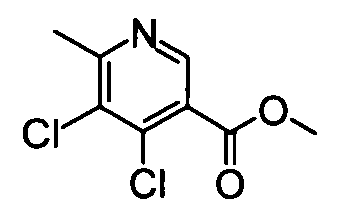
Оксихлорид фосфора (1,13 мл) добавляли к вышеуказанной 5-хлор-4-гидрокси-6-метилникотиновой кислоте (320 мг), и полученную в результате смесь затем нагревали до температуры кипения с обратным холодильником в течение 2 часов. Затем реакционный раствор концентрировали при пониженном давлении, и затем к остатку по каплям добавляли метанол (3 мл) при охлаждении во льду. Полученную в результате смесь перемешивали при комнатной температуре в течение 30 минут, с последующим концентрированием при пониженном давлении. Насыщенный раствор бикарбоната натрия добавляли к остатку при охлаждении во льду, затем следовала экстракция этилацетатом. Органический слой сушили над безводным сульфатом натрия, и затем растворитель удаляли упариванием, таким образом получая продукт неочищенного указанного в заголовке соединения (436 мг) в виде твердого вещества.
ESI-MS m/z: 220 (М+Н)+
(14) Метил-5-хлор-4-метокси-6-метилникотинат
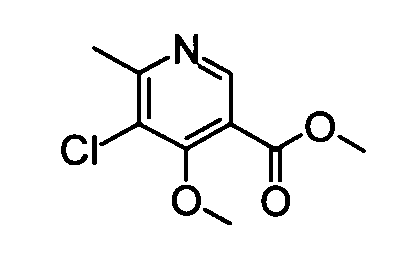
Неочищенный метил-4,5-дихлор-6-метилникотинат (380 мг) растворяли в 3 мл метанола и в токе азота затем добавляли метоксид натрия (120 мг) к раствору при охлаждении во льду. Температура реакционного раствора постепенно повышалась до комнатной температуры, и его перемешивали в течение 18 часов. После подтверждения исчезновения веществ к реакционному раствору добавляли насыщенный водный раствор хлорида аммония при охлаждении во льду, затем следовала экстракция хлороформом. Органический слой сушили над безводным сульфатом натрия, и растворитель затем удаляли упариванием. Остаток очищали с помощью хроматографии на силикагеле (этилацетат-гексан), таким образом получая указанное в заголовке соединение (210 мг) в виде твердого вещества.
1H-ЯМР (CDCl3) δ: 2,67 (3Н, с), 3,95 (4Н, с), 4,00 (3Н, с), 8,76 (1Н, с).
ESI-MS m/z: 216 (М+Н)+
(15) (5-Хлор-4-метокси-6-метилпиридин-3-ил)метанол
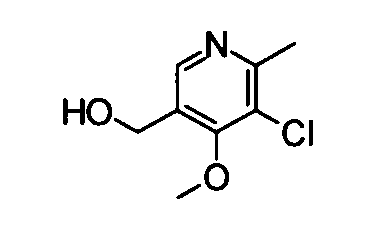
Вышеуказанный метил-5-хлор-4-метокси-6-метилникотинат (1,0 г) растворяли в 30 мл метанола, и затем к раствору добавляли боргидрид натрия (1,75 г). Полученную в результате смесь нагревали до температуры кипения с обратным холодильником в течение 1 часа. Затем насыщенный водный раствор хлорида аммония добавляли к реакционному раствору при охлаждении во льду, затем следовала трехкратная экстракция хлороформом. Органический слой сушили над безводным сульфатом натрия, и растворитель затем удаляли упариванием, таким образом получая указанное в заголовке соединение (0,92 г) в виде маслообразного вещества.
1Н-ЯМР (CDCl3) δ: 2,63 (3Н, с), 4,00 (3Н, с), 4,71 (2Н, ушир.с), 8,33 (1Н, с)
ESI-MS m/z: 188 (М+Н)+
(16) 3-Хлор-5-(хлорметил)-4-метокси-2-метилпиридин
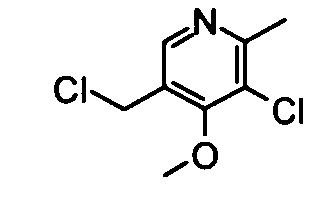
Вышеуказанный (5-хлор-4-метокси-6-метилпиридин-3-ил)метанол (520 мг) растворяли в 20 мл хлороформа, и затем добавляли к раствору тионилхлорид (0,38 мл) при охлаждении во льду. Полученную в результате смесь перемешивали при той же температуре, как указано выше, в течение 3 часов. Затем реакционный раствор концентрировали, и затем добавляли к концентрату этилацетат. Полученную в результате смесь промывали насыщенным раствором бикарбоната натрия, водой и насыщенным солевым раствором в указанном порядке. Органический слой сушили над безводным сульфатом натрия, и растворитель затем удаляли упариванием. Остаток очищали с помощью хроматографии на силикагеле (этилацетат-гексан), таким образом получая указанное в заголовке соединение (550 мг) в виде маслообразного вещества.
1H-ЯМР (CDCl3) δ: 2,64 (3Н, с), 4,05 (3Н, с), 4,61 (2Н, с), 8,35 (1Н, с).
ESI-MS m/z: 206 (М+Н)+
(17) 3-Хлор-4-метокси-2,5-диметилпиридин
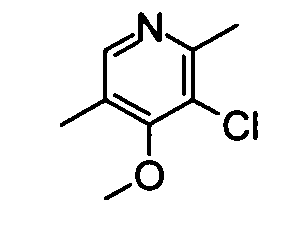
Вышеуказанный 3-хлор-5-(хлорметил)-4-метокси-2-метилпиридин (550 мг) растворяли в 10 мл метанола, и 10% Pd на угле (50 мг) затем добавляли к раствору. Проводили при нормальном давлении каталитическое гидрирование смеси при охлаждении во льду в течение 3 часов. Затем катализатор отделяли фильтрованием, и метанол удаляли упариванием при пониженном давлении. Остаток экстрагировали хлороформом. Органический слой промывали насыщенным раствором бикарбоната натрия и затем его сушили над безводным сульфатом натрия. Растворитель удаляли упариванием и остаток очищали с помощью хроматографии на силикагеле (этилацетат-гексан), таким образом получая указанное в заголовке соединение (365 мг) в виде маслообразного вещества.
1H-ЯМР (CDCl3) δ: 2,25 (3Н, с), 2,59 (3Н, с), 3,89 (3Н, с), 8,16 (1Н, с).
ESI-MS m/z: 172 (М+Н)+
(18) 3-Хлор-4-метокси-2,5-диметилпиридин-1-оксид
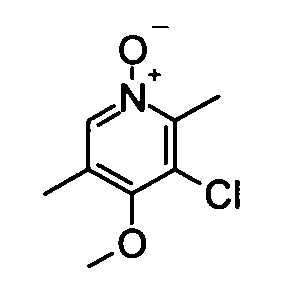
Вышеуказанный 3-хлор-4-метокси-2,5-диметилпиридин (181 мг) растворяли в 5 мл дихлорметана, затем добавляли к раствору пероксид мочевины (169 мг) и фталевый ангидрид (219 мг). Полученную в результате смесь перемешивали при комнатной температуре в течение 2,5 часов. Затем к реакционному раствору добавляли насыщенный водный раствор тиосульфата натрия при охлаждении во льду, и полученную в результате смесь затем разбавляли хлороформом. Водный слой дважды экстрагировали хлороформом. Органический слой сушили над безводным сульфатом натрия и растворитель удаляли упариванием, таким образом получая указанное в заголовке соединение (181 мг) в виде твердого вещества.
1H-ЯМР (CDCl3) δ: 2,24 (3Н, с), 2,62 (3Н, с), 3,87 (3Н, с), 8,07 (1Н, с).
ESI-MS m/z: 188 (М+Н)+
(19) (3-Хлор-4-метокси-5-метилпиридин-2-ил)метанол
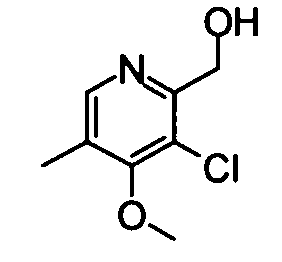
Вышеуказанный 3-хлор-4-метокси-2,5-диметилпиридин-1-оксид (530 мг) суспендировали в 15 мл дихлорметана и затем добавляли к суспензии трифторуксусный ангидрид (0,39 мл) при охлаждении во льду. Полученную в результате смесь перемешивали при комнатной температуре в течение 3 часов. Реакционный раствор разбавляли хлороформом и затем промывали насыщенным раствором бикарбоната натрия. Водный слой экстрагировали хлороформом. Органический слой сушили над безводным сульфатом натрия, и растворитель затем удаляли упариванием, таким образом получая указанное в заголовке соединение (521 мг) в виде маслообразного вещества.
1Н-ЯМР (CDCl3) δ: 2,29 (3Н, с), 3,93 (3Н, с), 4,29 (1Н, ушир.с), 4,72-4,74 (2Н, м), 8,26 (1Н, с).
ESI-MS m/z: 188 (М+Н)+
(20) Гидрохлорид 3-Хлор-2-(хлорметил)-4-метокси-5-метилпиридина
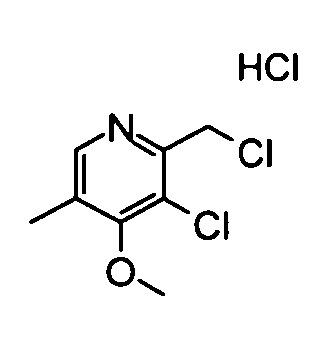
Вышеуказанный (3-хлор-4-метокси-5-метилпиридин-2-ил)метанол (530 мг) растворяли в 20 мл хлороформа, и затем к раствору добавляли по каплям тионилхлорид (1,03 мл) при охлаждении во льду. Полученную в результате смесь перемешивали при комнатной температуре в течение 3 часов. Затем реакционный раствор концентрировали, и затем его промывали смешанным растворителем диэтиловый эфир - гексан, таким образом получая указанное в заголовке соединение (410 мг) в виде твердого вещества.
1H-ЯМР (CDCl3) δ: 2,47 (3Н, с), 4,32 (3Н, с), 5,09 (2Н, с), 8,54 (1Н, с).
ESI-MS m/z: 206 (М+Н)+
(21) 2-{4-Амино-2-[(3-хлор-4-метокси-5-метилпиридин-2-ил)метил]-2,7-дигидро-6-тиа-1,2,3,5-тетраазабензо[cd]азулен-8-ил}-N-метилацетамид
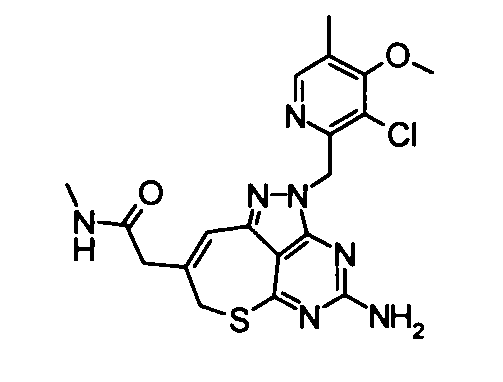
Диметилформамид (1 мл} добавляли к 2-(4-амино-2,7-дигидро-6-тиа-1,2,3,5-тетраазабензо[cd]азулен-8-ил)-N-метилацетамидтрифторацетату (28 мг), гидрохлориду 3-хлор-2-(хлорметил)-4-метокси-5-метилпиридина (36 мг) и карбонату калия (69 мг). Полученную в результате смесь перемешивали при 60°С в течение 2,5 часов. Затем нерастворимое вещество удаляли фильтрованием и растворитель удаляли упариванием в токе азота. Полученный в результате остаток растворяли в диметилсульфоксиде (1 мл) и затем очищали его с помощью препаративной обращенно-фазовой ВЭЖХ. Растворитель удаляли упариванием при пониженном давлении, таким образом получая указанное в заголовке соединение (27,0 мг) в виде твердого вещества.
1Н-ЯМР (CDCl3) δ: 2,24 (4Н, с), 2,82 (3Н, д, J=4,9 Гц), 3,27 (2Н, с), 3,80 (2Н, с), 3,91 (3Н, с), 5,21 (2Н, с), 5,65 (2Н, с), 5,87 (1Н, с), 6,70 (1Н, с), 8,16 (1Н, с).
ESI-MS m/z: 446 (М+Н)+.
(Пример 2)
(1) 5-Бром-2,3-диметил-4-нитропиридин-1-оксид
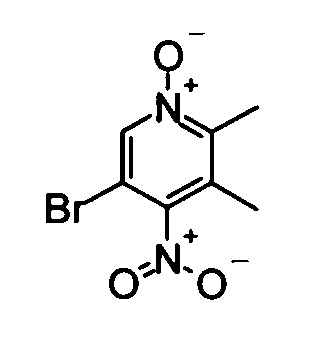
5-Бром-2,3-диметилпиридин-1-оксид (4,61 г), полученный способом, описанным в патенте США № US5250527, растворяли в концентрированной серной кислоте (10 мл). Затем смешанный раствор дымящей азотной кислоты (24 мл) и дымящей серной кислоты (13 мл) добавляли по каплям к вышеуказанному полученному раствору при перемешивании и охлаждении во льду. Полученную в результате смесь перемешивали при той же самой температуре, как указано выше, в течение 30 минут, и затем перемешивали при 90°С в течение 1 часа. После того как реакционный раствор выдержали до охлаждения, его выливали в ледяную воду. Дихлорметан добавляли к реакционному раствору при перемешивании и охлаждении на бане со льдом, и затем смесь нейтрализовали водным раствором аммиака. Реакционный раствор экстрагировали дихлорметаном. Органический слой сушили над безводным сульфатом магния, и затем растворитель удаляли упариванием при пониженном давлении, таким образом получая в качестве продукта неочищенное указанное в заголовке соединение вещество (4,80 г) в виде твердого вещества.
(2) 5-Бром-4-метокси-2,3-диметилпиридин-1-оксид
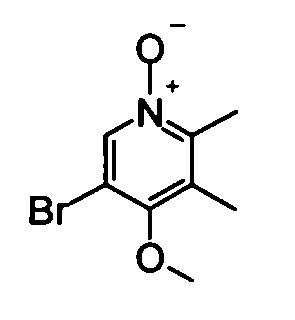
Раствор 1М метоксид натрия - метанол (30 мл) добавляли к метанольному (20 мл) раствору вышеуказанного 5-бром-2,3-диметил-4-нитропиридин-1-оксида (4,80 г) при охлаждении баней со льдом, и полученную в результате смесь перемешивали при комнатной температуре в течение ночи. Затем реакционный раствор концентрировали и остаток растворяли в дихлорметане и воде для разделения жидких фаз. Органический слой сушили над безводным сульфатом натрия и фильтрат концентрировали, таким образом получая в качестве продукта неочищенное указанное в заголовке соединение вещество (4,91 г) в виде твердого вещества.
1Н-ЯМР (CDCl3) δ: 2,30 (3Н, с), 2,47 (3Н, с), 3,84 (3Н, с), 8,36 (1Н, с).
ESI-MS m/z: 232 (М+Н)+
(3) (5-Бром-4-метокси-3-метилпиридин-2-ил)метанол
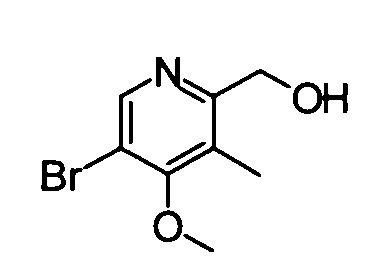
5-Бром-4-метокси-2,3-диметилпиридин-1-оксид (4,91 г) растворяли в дихлорметане (60 мл), и затем к раствору добавляли трифторуксусный ангидрид (20 мл) при комнатной температуре. Полученную в результате смесь перемешивали в течение ночи. Затем реакционный раствор концентрировали и концентрированный раствор затем растворяли в метаноле (50 мл). Полученную в результате смесь перемешивали при 50°С в течение 1 часа. Реакционный раствор концентрировали. Остаток растворяли в дихлорметане и затем промывали насыщенным раствором бикарбоната натрия. Органический слой сушили над безводным сульфатом натрия, и фильтрат затем концентрировали. Остаток очищали с помощью колоночной хроматографии на силикагеле (гексан-этилацетат), таким образом получая указанное в заголовке соединение (3,43 г) в виде твердого вещества.
1Н-ЯМР (CDCl3) δ: 2,17 (3Н, с), 3,89 (3Н, с), 4,62 (2Н, с), 8,50 (1Н, с).
ESI-MS m/z: 232 (М+Н)+
(4) 5-Бром-2-({[трет-бутил(диметил)силил]окси}метил)-4-метокси-3-метилпиридин
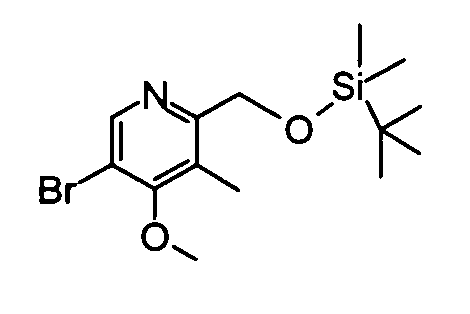
Вышеуказанный (5-бром-4-метокси-3-метилпиридин-2-ил)метанол (500 мг) растворяли в дихлорметане (10 мл) и затем добавляли к раствору трет-бутилдиметилсилилхлорид (487 мг), триэтиламин (0,48 мл) и диметиламинопиридин (30 мг) при охлаждении на бане со льдом. Полученную в результате смесь перемешивали при комнатной температуре в течение ночи. Затем реакционный раствор разбавляли дихлорметаном и затем промывали насыщенным раствором бикарбоната натрия. Органический слой сушили над безводным сульфатом натрия и затем фильтрат концентрировали. Остаток очищали с помощью колоночной хроматографии на силикагеле (гексан-этилацетат), таким образом получая указанное в заголовке соединение (743 мг) в виде маслообразного вещества.
1Н-ЯМР (CDCl3) δ: 0,07 (6Н, с), 0,89 (9Н, с), 2,37 (3Н, с), 3,87 (3Н, с), 4,77 (2Н, с), 8,43 (1Н, с).
(5) 2-({[трет-бутил(диметил)силил]окси}метил)-5-фтор-4-метокси-3-метилпиридин
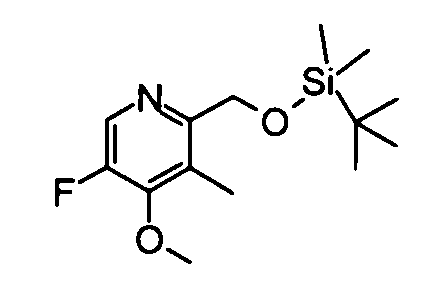
Вышеуказанный 5-бром-2-({[трет-бутил(диметил)силил]окси}метил)-4-метокси-3-метилпиридин (734 мг) растворяли в тетрагидрофуране (30 мл), и затем к раствору добавляли по каплям бутиллитий (1,59 М раствор, 2,42 мл)при -110°С. Полученную в результате смесь перемешивали при той же самой температуре, как указано выше, в течение 1 часа, и раствор N-фторбензолсульфонимида (3,03 г) в тетрагидрофуране (5 мл) затем добавляли по каплям к реакционному раствору при той же самой температуре. Температуру полученной в результате смеси постепенно повышали до -68°С в течение 3 часов, и смесь затем перемешивали при комнатной температуре в течение 30 минут. Затем насыщенный водный раствор хлорида аммония добавляли к реакционному раствору, затем следовала экстракция дихлорметаном. Органический слой сушили над безводным сульфатом натрия и фильтрат затем концентрировали. Остаток очищали с помощью колоночной хроматографии на силикагеле (гексан-этилацетат), таким образом получая указанное в заголовке соединение (353 мг) в виде маслообразного вещества.
1Н-ЯМР (CDCl3) δ: 0,06 (6Н, с), 0,89 (9Н, с), 2,28 (3Н, с), 4,07 (3Н, д, J=3,7 Гц), 4,75 (2Н, с), 8,15 (1Н, д, J=3,7 Гц).
ESI-MS m/z: 286 (М+Н)+.
(6) (5-Фтор-4-метокси-3-метилпиридин-2-ил)метанол
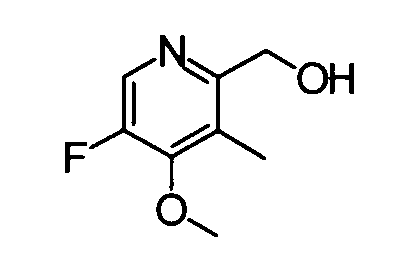
Тетрабутиламмонийфторид (1 М раствор, 1,49 мл) добавляли к раствору вышеуказанного 2-({[трет-бутил(диметил)силил]окси}метил)-5-фтор-4-метокси-3-метилпиридина (353 мг) в тетрагидрофуране (10 мл) при охлаждении на бане со льдом, и полученную в результате смесь перемешивали при комнатной температуре в течение 1 часа. Реакционный раствор разбавляли дихлорметаном и затем промывали водой. Органический слой сушили над безводным сульфатом натрия и фильтрат затем концентрировали. Остаток очищали с помощью колоночной хроматографии на силикагеле (гексан-этилацетат), таким образом получая указанное в заголовке соединение (150 мг) в виде твердого вещества.
1Н-ЯМР (CDCl3) δ: 2,08 (3Н, с), 4,10 (3Н, д, J=4,1 Гц), 4,52 (1Н, с), 4,61 (2Н, с), 8,22 (1Н, д, J=4,1 Гц).
ESI-MS m/z: 172 (М+Н)+.
(7) Гидрохлорид 2-(хлорметил)-5-фтор-4-метокси-3-метилпиридина
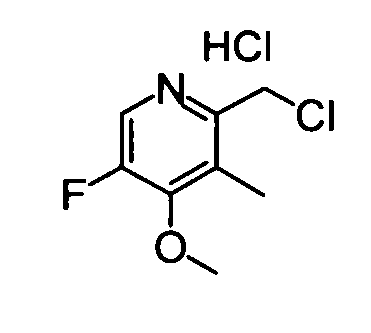
Указанное в заголовке соединение (154 мг) получали в виде твердого вещества таким же синтетическим способом, как в примере 1(20), используя вышеуказанный (5-фтор-4-метокси-3-метилпиридин-2-ил)метанол (135 мг).
ESI-MS m/z: 190 (М+Н)+.
(8) 2-{4-Амино-2-[(5-фтор-4-метокси-3-метилпиридин-2-ил)метил]-2,7-дигидро-6-тиа-1,2,3,5-тетраазабензо-[cd]азулен-8-ил}-N-метилацетамид
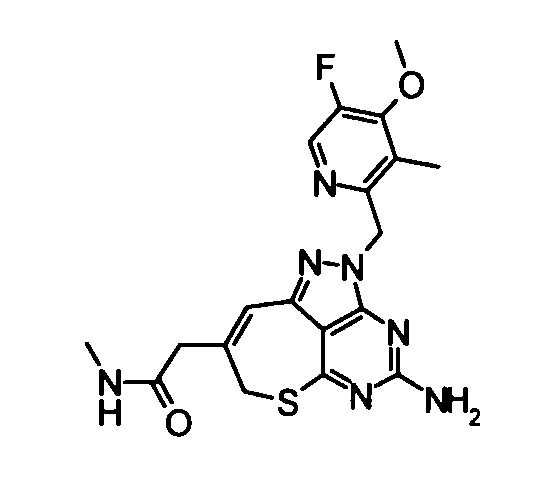
Карбонат калия (85 мг) и гидрохлорид 2-(хлорметил)-5-фтор-4-метокси-3-метилпиридина (52 мг) добавляли к раствору 2-(4-амино-2,7-дигидро-6-тиа-1,2,3,5-тетраазабензо[cd]азулен-8-ил)-N-метилацетамида трифторацетата (60 мг) в N,N-диметилформамиде (5 мл). Полученную в результате смесь перемешивали при 50°С в течение 3,5 часов. Затем нерастворимое вещество удаляли фильтрованием и фильтрат затем концентрировали. Остаток очищали с помощью колоночной хроматографии на силикагеле (хлороформ-метанол), таким образом получая указанное в заголовке соединение (30 мг) в виде твердого вещества.
1Н-ЯМР (CDCl3) δ: 2,27 (3Н, с), 2,83 (3Н, д, J=4,6 Гц), 3,27 (2Н, с), 3,78 (2Н, с), 4,08 (3Н, д, J=4,6 Гц), 5,13 (2Н, с), 5,49 (2Н, с), 5,74 (1Н, ушир.с), 6,70 (1Н, с), 8,19 (1Н, д, J=3,4 Гц).
ESI-MS m/z: 430 (М+Н)+.
Рассчитано в процентах для брутто-формулы C19H20FN7O2S·0,5H2O: С, 52,05; Н, 4,82; N, 22,36; F, 4,33; S, 7,32.
Найдено: С, 52,27; Н, 4,79; N, 22,10; F, 4,44; S, 7,05.
(Пример 3)
(1) 3-Бром-2,5-диметил-4-нитропиридин-1-оксид
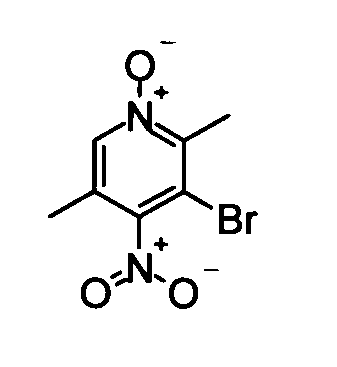
Указанное в заголовке соединение (1,08 г) получали в виде твердого вещества таким же синтетическим способом, как в примере 2(1), используя коммерчески доступный 3-бром-2,5-диметилпиридин-1-оксид (2,35 г).
ESI-MS m/z: 247 (М+Н)+.
(2) 3-Бром-4-метокси-2,5-диметилпиридин-1-оксид
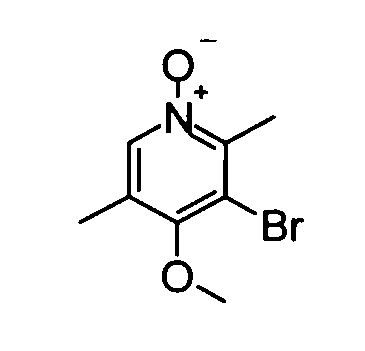
Продукт в виде неочищенного указанного в заголовке соединения (990 мг) получали в виде твердого вещества таким же синтетическим способом, как в примере 2(2), используя вышеуказанный 3-бром-2,5-диметил-4-нитропиридин-1-оксид (1,08 г).
ESI-MS m/z: 234 (М+Н)+.
(3) (3-Бром-4-метокси-5-метилпиридин-2-ил)метанол
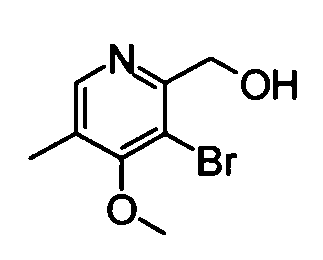
Указанное в заголовке соединение (931 мг) получали в виде твердого вещества таким же синтетическим способом, как в примере 2(3), используя вышеуказанный 3-бром-4-метокси-2,5-диметилпиридин-1-оксид (990 мг).
1Н-ЯМР (CDCl3) δ: 2,33 (3Н, с), 3,90 (3Н, с), 4,38 (1Н, ушир.с), 4,69 (2Н, с), 8,28 (1Н, с).
ESI-MS m/z: 234 (М+Н)+.
(4) Гидрохлорид 3-бром-2-(хлорметил)-4-метокси-5-пиридина
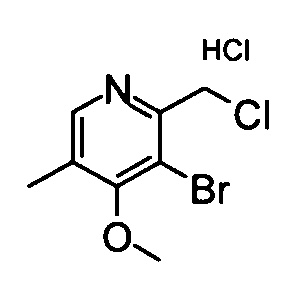
Указанное в заголовке соединение (161 мг) получали в виде твердого вещества таким же синтетическим способом, как в примере 1(20), используя вышеуказанный (3-бром-4-метокси-5-метилпиридин-2-ил)метанол (150 мг).
1Н-ЯМР (CDCl3) δ: 2,50 (3Н, с), 4,21 (3Н, с), 5,18 (2Н, с), 8,47 (1Н, с).
ESI-MS m/z: 250 и 252 (М+Н)+.
(5) 2-{4-Амино-2-[(3-бром-4-метокси-5-метилпиридин-2-ил)метил]-2,7-дигидро-6-тиа-1,2,3,5-тетраазабензо[cd]азулен-8-ил}-N-метилацетамид
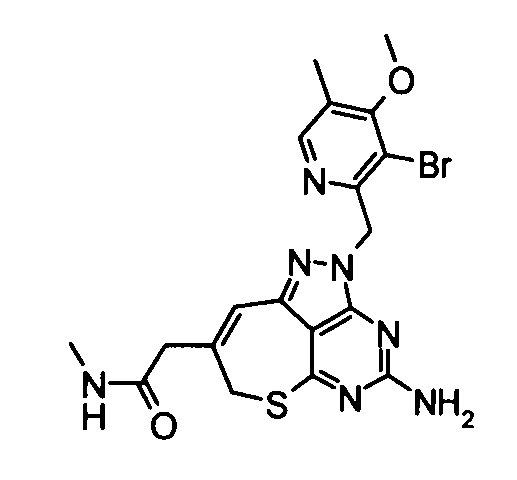
Карбонат калия (160 мг) и гидрохлорид 3-бром-2-(хлорметил)-4-метокси-5-пиридина (166 мг) добавляли к раствору трифторацетата 2-(4-амино-2,7-дигидро-6-тиа-1,2,3,5-тетраазабензо[cd]азулен-8-ил)-N-метилацетамида (150 мг) в N,N-диметилформамиде (5 мл). Полученную в результате смесь перемешивали при 50°С в течение ночи. Затем реакционный раствор разбавляли дихлорметаном и затем промывали водой. Органический слой сушили над безводным сульфатом натрия и фильтрат затем концентрировали. Остаток очищали с помощью колоночной хроматографии на силикагеле (хлороформ-метанол), таким образом получая указанное в заголовке соединение (105 мг) в виде твердого вещества.
1Н-ЯМР (CDCl3) δ: 2,27 (3Н, с), 2,83 (3Н, д, J=4,6 Гц), 3,28 (2Н, с), 3,81 (2Н, с), 3,88 (3Н, с), 5,08-5,14 (2Н, ушир.м), 5,66 (2Н, с), 5,77 (1Н, ушир.с), 6,72 (1Н, с), 8,17 (1Н, с).
ESI-MS m/z: 492 (М+Н)+.
Рассчитано в процентах для брутто-формулы C19H20BrN7O2S·0,25Н2О·0,2 диоксан: С, 46,40; Н, 4,34; N, 19,13; Br, 15,59; S, 6,45.
Найдено: С, 46,21; Н, 4,07; N, 19,16; Br, 15,70; S, 6,27.
(Пример 4)
(1) Трет-бутил[8-(2-амино-2-оксоэтил)-2-(4-метоксибензил)-2,7-дигидро-6-тиа-1,2,3,5-тетраазабензо[cd]азулен-4-ил]карбамат
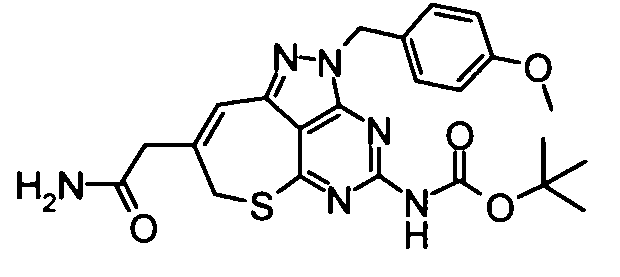
Водный 1н. раствор гидроксида натрия (3 мл) добавляли к этанольному раствору (5 мл) этил {4-[бис(трет-бутоксикарбонил)амино]-2-(4-метоксибензил)-2,7-дигидро-6-тиа-1,2,3,5-тетраазабензо[cd]азулен-8-ил}ацетата (600 мг). Полученную в результате смесь перемешивали при комнатной температуре в течение ночи. Затем добавляли 1н. хлористоводородную кислоту (3 мл) к реакционному раствору и полученный в результате раствор затем концентрировали. Дихлорметан добавляли к к остатку для проведения экстракции. Органический слой сушили над безводным сульфатом натрия и фильтрат затем концентрировали, получая карбоновую кислоту (576 мг). Карбоновую кислоту (576 мг) растворяли в ацетонитриле (10 мл) и затем к раствору добавляли хлорид аммония (157 мг), триэтиламин (0,2 мл) и гидрохлорид 1-этил-3-(3-диметиламинопропил)карбодиимида (282 мг). Полученную в результате смесь перемешивали при комнатной температуре в течение 3 дней. Затем реакционный раствор разбавляли дихлорметаном и туда же затем добавляли воду для разделения жидких фаз. Органический слой промывали насыщенным солевым раствором. Органический слой сушили над безводным сульфатом натрия и затем растворитель удаляли упариванием при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле (хлороформ-метанол), таким образом получая указанное в заголовке соединение (252 мг) в виде твердого вещества.
ESI-MS m/z: 483 (М+Н)+
(2) Трифторацетат 2-(4-амино-2,7-дигидро-6-тиа-1,2,3,5-тетраазабензо[cd]азулен-8-ил)ацетамида
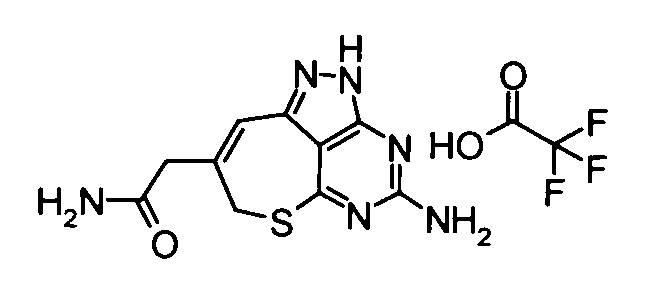
Указанное в заголовке соединение (162 мг) получали в виде твердого вещества таким же синтетическим способом, как в примере 1(11), используя вышеуказанный трет-бутил[8-(2-амино-2-оксоэтил)-2-(4-метоксибензил)-2,7-дигидро-6-тиа-1,2,3,5-тетраазабензо[cd]азулен-4-ил]карбамат (256 мг).
ESI-MS m/z: 263 (М+Н)+.
(3) Мезилат 2-{4-амино-2-[(3-бром-4-метокси-5-метилпиридин-2-ил)метил]-2,7-дигидро-6-тиа-1,2,3,5-тетраазабензо[cd]азулен-8-ил}ацетамида
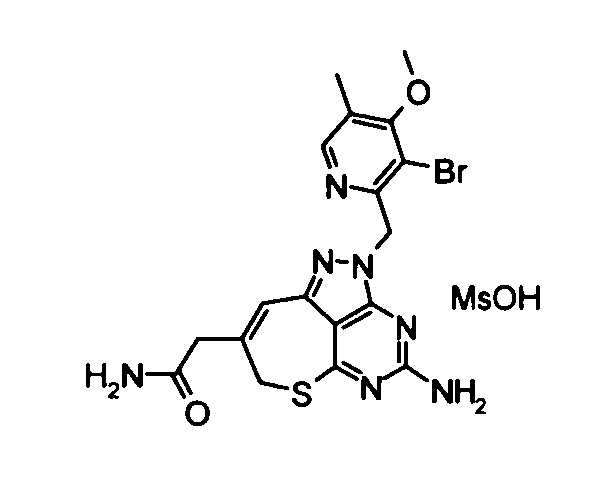
Карбонат калия (110 мг) и гидрохлорид 3-бром-2-(хлорметил)-4-метокси-5-пиридина (114 мг) добавляли к раствору вышеуказанного трифторацетата 2-(4-амино-2,7-дигидро-6-тиа-1,2,3,5-тетраазабензо[cd]азулен-8-ил)ацетамида (100 мг) в N,N-диметилформамиде (5 мл). Полученную в результате смесь перемешивали при 50°С в течение ночи. Затем реакционный раствор разбавляли дихлорметаном и затем промывали водой. Органический слой сушили над безводным сульфатом натрия и фильтрат затем концентрировали. Остаток очищали с помощью колоночной хроматографии на силикагеле (хлороформ-метанол), таким образом получая 2-{4-амино-2-[(3-бром-4-метокси-5-метилпиридин-2-ил)метил]-2,7-дигидро-6-тиа-1,2,3,5-тетраазабензо[cd]азулен-8-ил}ацетамид (90 мг) в виде твердого вещества. Полученное в результате твердое вещество (27 мг) растворяли в диоксане, и водный 0,2 М раствор мезилата (0,283 мл) затем добавляли к раствору при охлаждении баней со льдом, затем следовала лиофилизация, таким образом получали указанное в заголовке соединение (32 мг) в виде твердого вещества.
1Н-ЯМР (CDCl3) δ: 2,30 (3Н, с), 2,84 (3Н, с), 3,35 (2Н, д, J=0,9 Гц), 3,92 (3Н, с), 3,95 (2Н, с), 5,46 (1Н, ушир.с), 5,68 (2Н, с), 5,97 (1Н, ушир.с), 6,75 (1Н, с), 8,19 (1Н, с).
ESI-MS m/z: 476 и 478 (М+Н)+.
(Пример 5)
(1) 2-[4-Амино-2-(4-метоксибензил)-2,7-дигидро-6-тиа-1,2,3,5-тетраазабензо[cd]азулен-8-ил]-N-(2-фторэтил)ацетамид
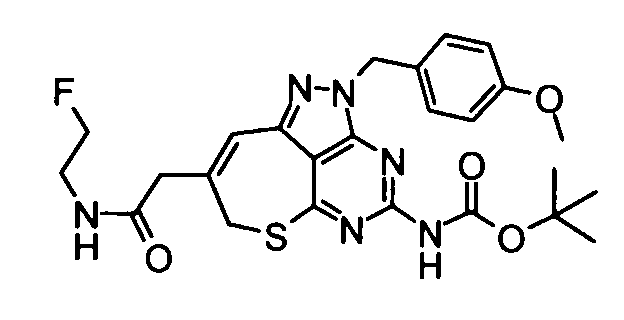
Водный 1н раствор гидроксида натрия (2,7 мл) добавляли к этанольному раствору (10 мл) этил{4-[бис(трет-бутоксикарбонил)амино]-2-(4-метоксибензил)-2,7-дигидро-6-тиа-1,2,3,5-тетраазабензо[cd]азулен-8-ил}ацетата (526 мг), и полученную в результате смесь затем перемешивали при комнатной температуре в течение ночи. Затем растворитель удаляли упариванием, получая натриевую соль карбоновой кислоты. Этот продукт растворяли в N,N-диметилформамиде (10 мл), и затем добавляли к раствору 2-фторэтиламин (171 мг), гидрохлорид 1-этил-3-(3-диметиламинопропил)карбодиимида (247 мг) и 1-гидроксибензотриазол (10 мг) при охлаждении баней со льдом. Полученную в результате смесь перемешивали при комнатной температуре в течение 3 дней. Затем реакционный раствор разбавляли дихлорметаном и туда же затем добавляли воду для осуществления разделения жидких фаз. Органический слой промывали насыщенным солевым раствором. Органический слой сушили над безводным сульфатом натрия и затем растворитель удаляли упариванием при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле (гексан-этилацетат), таким образом получая указанное в заголовке соединение (419 мг) в виде маслообразного вещества.
ESI-MS m/z: 529 (М+Н)+.
(2) Трифторацетат 2-(4-амино-2,7-дигидро-6-тиа-1,2,3,5-тетраазабензо[cd]азулен-8-ил)-N-(2-фторэтил)ацетамида
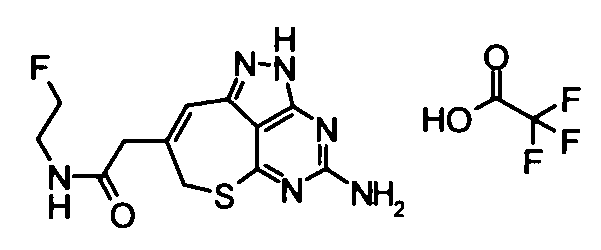
Продукт в виде неочищенного указанного в заголовке соединения (247 мг) получали в виде твердого вещества таким же синтетическим способом, как в примере 1(11), используя вышеуказанный 2-[4-амино-2-(4-метоксибензил)-2,7-дигидро-6-тиа-1,2,3,5-тетраазабензо[cd]азулен-8-ил]-N-(2-фторэтил)ацетамид (418 мг).
ESI-MS m/z: 309 (М+Н)+.
(3) 2-{4-Амино-2-[(3-хлор-4-метокси-5-метилпиридин-2-ил)метил]-2,7-дигидро-6-тиа-1,2,3,5-тетраазабензо[cd]азулен-8-ил}-N-(2-фторэтил)ацетамид
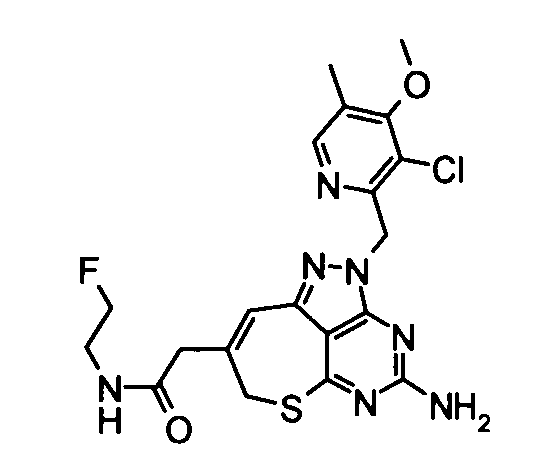
Указанное в заголовке соединение (12 мг) получали в виде твердого вещества таким же синтетическим способом, как в примере 3(5), используя вышеуказанный трифторацетат 2-(4-амино-2,7-дигидро-6-тиа-1,2,3,5-тетраазабензо[cd]азулен-8-ил)-N-(2-фторэтил)ацетамида (60 мг) и гидрохлорид 3-хлор-2-(хлорметил)-4-метокси-5-пиридина (52 мг).
1Н-ЯМР (CDCl3) δ: 2,25 (3Н, с), 3,31 (2Н, с), 3,56 (1Н, дд, J=10,5, 5,0 Гц), 3,63 (1Н, дд, J=10,5, 5,0 Гц), 3,81 (2Н, с), 3,91 (3Н, с), 4,44 (1Н, т, J=4,8 Гц), 4,56 (1Н, т, J=4,8 Гц), 5,11 (2Н, с), 5,65 (2Н, с), 6,09 (1Н, ушир.с), 6,73 (1Н, с), 8,17 (1Н, с).
ESI-MS m/z: 478 (М+Н)+.
Рассчитано в процентах для брутто-формулы C20H21ClFN7O2S·0,75 диоксан: С, 50,80; Н, 5,00; N, 18,02.
Найдено: С, 51,18; Н, 4,86; N, 17,91.
(Пример 6)
(1) 3-Хлор-4-метокси-2-метилпиридин-1-оксид
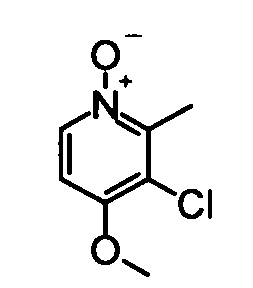
3-Хлор-2-метил-4-нитропиридин-1-оксид (1,00 г), полученный способом, описанным в Po1ish Journal of Chemistry, 1986, 59, 10-12, растворяли в растворе 0,5 М метоксид натрия - метанол (15,9 мл), и полученную в результате смесь затем перемешивали при комнатной температуре в течение ночи. Затем насыщенный водный раствор хлорида аммония добавляли к реакционному раствору, и метанол затем удаляли упариванием при пониженном давлении. Полученный в результате продукт трижды экстрагировали хлороформом. Органический слой сушили над безводным сульфатом магния, и растворитель затем удаляли упариванием при пониженном давлении, таким образом получая указанное в заголовке соединение (897 мг) в виде твердого вещества.
1Н-ЯМР (CDCl3) δ: 2,68 (3Н, с), 3,96 (3Н, с), 6,73 (1Н, д, J=7,3 Гц), 8,20 (1Н, д, J=7,3 Гц).
ESI-MS m/z: 174 (М+Н)+.
(2) (3-Хлор-4-метоксипиридин-2-ил)метилацетат
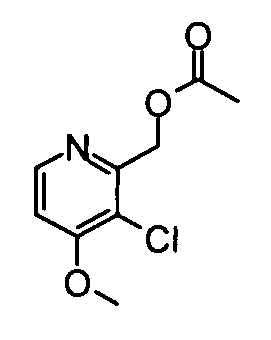
Вышеуказанный 3-хлор-4-метокси-2-метилпиридин-1-оксид (890 мг) растворяли в уксусном ангидриде (12 мл) и полученный в результате раствор затем нагревали до температуры кипения с обратным холодильником в течение 30 минут. Оставляли охлаждаться до комнатной температуры, после чего реакционный раствор концентрировали при пониженном давлении и остаток затем очищали с помощью хроматографии на силикагеле (гексан-этилацетат), таким образом получая указанное в заголовке соединение (576 мг) в виде маслообразного вещества.
1Н-ЯМР (CDCl3) δ: 2,17 (3Н, с), 3,97 (3Н, с), 5,33 (2Н, с), 6,83 (1Н, д, J=5,6 Гц), 8,39 (1Н, д, J=5,6 Гц).
ESI-MS m/z: 216 (М+Н)+.
(3) (3-Хлор-4-метоксипиридин-2-ил) метанол
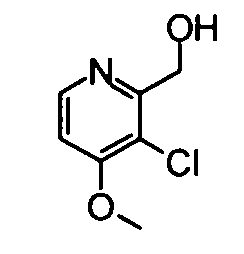
Вышеуказанный (3-хлор-4-метоксипиридин-2-ил)метилацетат (570 мг) растворяли в метаноле (13 мл), и затем добавляли к раствору карбонат калия (731 мг). Полученную в результате смесь перемешивали при 50°С в течение 30 минут. Затем метанол удаляли упариванием при пониженном давлении, и затем к остатку добавляли воду, затем следовала трехкратная экстракция хлороформом. Органический слой сушили над безводным сульфатом магния, и растворитель затем удаляли упариванием при пониженном давлении, таким образом получая указанное в заголовке соединение (437 мг) в виде маслообразного вещества.
1Н-ЯМР (CDCl3) δ: 3,98 (3Н, с), 4,39 (1Н, т, J=4,4 Гц), 4,76 (2Н, д, J=4,4 Гц), 6,84 (1Н, д, J=5,6 Гц), 8,37 (1Н, д, J=5,6 Гц).
ESI-MS m/z: 174 (М+Н)+.
(4) Гидрохлорид 3-хлор-4-метокси-2-(хлорметил)пиридина
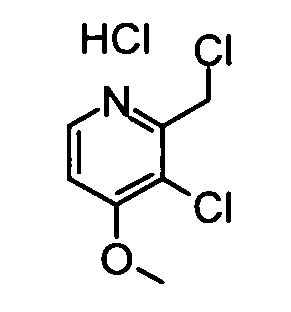
Указанное в заголовке соединение (558 мг) получали в виде твердого вещества таким же синтетическим способом, как в примере 1(20), используя вышеуказанный (3-хлор-4-метоксипиридин-2-ил)метанол (437 мг).
1Н-ЯМР (CDCl3) δ: 4,26 (3Н, с), 5,17 (2Н, с), 7,48 (1Н, д, J=6,8 Гц), 8,77 (1Н, д, J=6,8 Гц).
ESI-MS m/z: 192 (М+Н)+.
(5) 4-Амино-2-[(3-хлор-4-метоксипиридин-2-ил)метил]-N-(2,2-дифторэтил)-2,7-дигидро-6-тиа-1,2,3,5-тетраазабензо[cd]азулен-8-карбоксамид
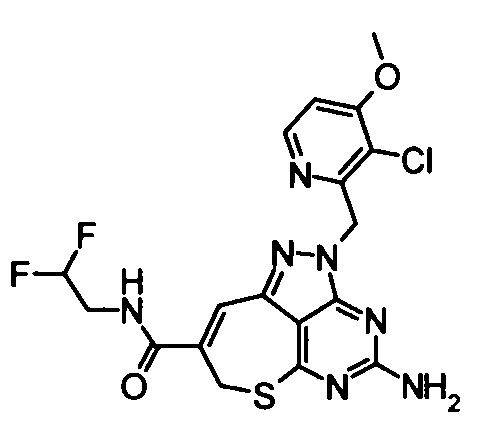
Указанное в заголовке соединение (48 мг) получали в виде твердого вещества таким же синтетическим способом, как в примере 2(8), используя трифторацетат 4-амино-N-(2,2-дифторэтил)-2,7-дигидро-6-тиа-1,2,3,5-тетраазабензо[cd]азулен-8-карбоксамида (50 мг) и 3-хлор-2-(хлорметил)-4-метоксипиридин (35 мг).
1Н-ЯМР (ДМСО-D6) δ: 3,55-3,63 (2Н, м), 3,96 (3Н, с), 4,09 (2Н, с), 5,60 (2Н, с), 5,95-6,19 (1Н, м), 7,06 (2Н, ушир.с), 7,16 (1Н, д, J=5,73 Гц), 7,49 (1Н, с), 8,25 (1Н, д, J=5,73 Гц), 8,82 (1Н, т, J=5,73 Гц).
ESI-MS m/z: 468 (М+Н)+.
Рассчитано в процентах для брутто-формулы C18H16ClF2N7O2S·0,25H2O·0,5 диоксан: С, 46,51; Н, 4,00; N, 18,99; Cl, 6,86; F, 7,36; S, 6,21.
Найдено: С, 46,40; Н, 3,72; N, 19,10; Cl, 6,93; F, 7,29; S, 6,19.
(Пример 7)
Моногидробромид 2-{4-амино-2-[(3-хлор-4-метокси-5-метилпиридин-2-ил)метил]-2,7-дигидро-6-тиа-1,2,3,5-тетраазабензо[cd]азулен-8-ил}-N-метилацетамида
Раствор 20% бромистоводородная кислота - этанол (204 мкл, 0,505 ммоль) добавляли к раствору 2-{4-амино-2-[(3-хлор-4-метокси-5-метилпиридин-2-ил)метил]-2,7-дигидро-6-тиа-1,2,3,5-тетраазабензо[cd]азулен-8-ил}-N-метилацетамид (250 мг, 0,561 ммоль) в ацетонитриле (350 мл) при комнатной температуре. Полученную в результате смесь перемешивали в течение 3 часов, выпавший осадок твердого вещества затем отделяли фильтрованием. Этанол (3 мл) добавляли к полученному в результате твердому веществу, и полученную в результате смесь затем перемешивали в течение 1 дня. Затем твердое вещество отделяли фильтрованием. Твердое вещество затем сушили при пониженном давлении при комнатной температуре в течение 2 дней, таким образом получая указанное в заголовке соединение (128 мг, 0,243 ммоль).
Рассчитано в процентах для брутто-формулы C19H21N7O2SClBr·0,4H2O: С, 42,91; Н, 4,01; О, 7,13; N, 18,57; Cl, 6,68; Br, 15,03; S, 6,08.
Найдено: С, 42,73; Н, 4,11; О, 7,19; N, 18,36; Cl, 6,64; Br, 14,96; S, 6,00.
(Пример 8)
Моногидрохлорид 2-{4-амино-2-[(3-хлор-4-метокси-5-метилпиридин-2-ил)метил]-2,7-дигидро-6-тиа-1,2,3,5-тетраазабензо[cd]азулен-8-ил}-N-метилацетамида
3н хлористоводородную кислоту (0,786 мл, 2,358 ммоль) добавляли к этанольной (30 мл) суспензии вышеуказанного 2-{4-амино-2-[(3-хлор-4-метокси-5-метилпиридин-2-ил)метил]-2,7-дигидро-6-тиа-1,2,3,5-тетраазабензо[cd]азулен-8-ил}-N-метилацетамида (527,52 мг, 1,183 ммоль), при этом все время перемешивая при 25°С. Полученную в результате смесь перемешивали в течение 1 часа. Выпавший осадок твердого вещества отфильтровали, промывали этанолом (6 мл), и затем сушили при пониженном давлении при 40°С в течение 30 минут, таким образом получая указанное в заголовке соединение (531,09 мг, 1,101 ммоль).
Рассчитано в процентах для брутто-формулы C19H21Cl2N7O2S: С, 47,31; Н, 4,39; N, 20,33; О, 6,63; Cl, 14,70; S, 6,65.
Найдено: С, 47,29; Н, 4,40; N, 20,02; О, 6,87; Cl, 14,99; S, 6,83.
(Пример 9)
Монометансульфонат 2-{4-амино-2-[(3-хлор-4-метокси-5-метилпиридин-2-ил)метил]-2,7-дигидро-6-тиа-1,2,3,5-тетраазабензо[cd]азулен-8-ил}-N-метилацетамида
Раствор метансульфоновой кислоты (32 мг, 0,302 ммоль) в диэтиловом эфире (50 мл) добавляли при 5°С к раствору 2-{4-амино-2-[(3-хлор-4-метокси-5-метилпиридин-2-ил)метил]-2,7-дигидро-6-тиа-1,2,3,5-тетраазабензо[cd]азулен-8-ил}-N-метилацетамида (151 мг, 0,339 ммоль) в ацетонитриле (150 мл). Полученную в результате смесь перемешивали при 5°С в течение 25 минут. Затем растворитель удаляли упариванием при пониженном давлении, соответствующее количество диэтилового эфира затем добавляли к остатку, и твердое вещество затем отделяли фильтрованием. Затем этанол (1 мл) добавляли к полученному в результате твердому веществу, и полученную в результате смесь затем перемешивали при комнатной температуре в течение приблизительно 1 дня. Затем твердое вещество отделяли фильтрованием и его затем сушили при пониженном давлении при комнатной температуре в течение 1 дня, таким образом получая указанное в заголовке соединение (145 мг, 0,268 ммоль).
Рассчитано в процентах для брутто-формулы C20H24N7O5S2Cl·0,3H2O: С, 44,71; Н, 4,69; О, 15,45; N, 17,81; Cl, 6,54; S, 11,56.
Найдено: С, 43,88; Н, 4,53; О, 15,49; N, 17,91; Cl, 6,48; S, 11,71.
(Пример 10)
Моноэтан-1,2-дисульфонат 2-{4-амино-2-[(3-хлор-4-метокси-5-метилпиридин-2-ил)метил]-2,7-дигидро-6-тиа-1,2,3,5-тетраазабензо[cd]азулен-8-ил}-N-метилацетамида
Ацетоновый раствор (50 мл) этан-1,2-дисульфоната (70 мг, 0,368 ммоль) добавляли при 5°С к ацетоновому раствору (50 мл) 2-{4-амино-2-[(3-хлор-4-метокси-5-метилпиридин-2-ил)метил]-2,7-дигидро-6-тиа-1,2,3,5-тетраазабензо[cd]азулен-8-ил}-N-метилацетамида (164 мг, 0,368 ммоль). Полученную в результате смесь перемешивали при 5°С в течение 20 минут. Затем реакционный раствор оставляли в состоянии покоя при 5°С в течение 18 часов, и его затем перемешивали при комнатной температуре в течение 7 часов. Затем выпавший осадок твердого вещества отфильтровали, и его затем сушили при пониженном давлении при комнатной температуре в течение приблизительно 1 дня, таким образом получая указанное в заголовке соединение (119 мг, 0,187 ммоль).
Рассчитано в процентах для брутто-формулы C21H26N7O8S3Cl·1,4Н2О: С, 38,06; Н, 4,68; О, 22,29; N, 14,92; Cl, 5,25; S, 13,83.
Найдено: С, 38,14; Н, 4,39; О, 22,74; N, 14,83; Cl, 5,36; S, 14,55.
(Тестовый пример 1: Испытание действия HSP90 на АТФ-азу)
Испытание действия HSP90 на АТФ-азу проводили с использованием рекомбинантного дрожжевого HSP90 (в дальнейшем в этом документе называемом rHsp90). ДНК дрожжевого HSP90 клонировали из библиотеки дрожжевой геномной ДНК в соответствии со стандартным способом. Клонированную ДНК дрожжевого HSP90 включали в плазмиду для экспрессии в Escherichia coli, и плазмиду экспрессировали в Escherichia coli, получая rHsp90.
Тестируемое соединение растворяли в ДМСО до концентрации 10 мМ. Полученный раствор разбавляли ДМСО до двух концентраций, 1 мМ и 0,2 мМ. Каждый из разбавленных растворов далее разбавляли в 10 раз, используя в качестве разбавителя буферный раствор для проведения испытаний (100 мМ Трис, рН 7,4, 20 мМ KCl, 6 мМ MgCl2) (концентрация тестируемого соединения в каждом растворе: 100 мкМ и 20 мкМ; концентрация ДМСО: 10%).
rHsp90 растворяли в ТЭ буферном растворе (20 мМ Трис, рН 7,4, 1 мМ ЭДТА) до концентрации 2,531 мг/мл. Раствор разбавляли буферным раствором для проведения испытаний до концентрации 125 мкг/мл и дозированно распределяли в 96-луночном испытательном планшете в количестве 40 мкл в лунку (конечная концентрация: 100 мкг/мл).
Раствор тестируемого соединения дозированно распределяли в количестве 5 мкл в лунку, и затем растворы в соответствующих лунках смешивали, используя планшетный смеситель. 100 мМ АТФ (Sigma, Catalog No. А-7699) разбавляли буферным раствором для проведения испытаний до 1 мМ и дозированно распределяли в количестве 5 мкл на лунку (конечная концентрация: 100 мкМ). Растворы в соответствующих лунках смешивали, используя планшетный смеситель, и затем испытательный планшет оставляли стоять в термостате при 37°С в течение двух часов.
Реагент BIOMOL GREEN Reagent (BIOMOL, Catalog No. АК-111) дозированно распределяли в количестве 100 мкл на лунку, и реакцию заканчивали. Растворы в соответствующих лунках смешивали, используя планшетный смеситель, и затем 34% цитрат натрия дозированно распределяли в количестве 10 мкл на лунку. Растворы в соответствующих лунках смешивали, используя планшетный смеситель, и затем испытательный планшет оставляли стоять при комнатной температуре в течение 30 минут. Измеряли поглощение при 650 нм для каждой лунки, используя спектрофотометр SpectramaxPLUS (Molecular Devices).
Соотношение поглощения в случае группы с добавлением тестируемого соединения к поглощению в случае группы без тестируемого соединения (Т/С величина) определяли по нижеприведенной расчетной формуле, основываясь на принятом обозначении, что поглощение для лунки, к которой были добавлены тестируемое соединение и rHsp90, было А, поглощение для лунки, к которой был добавлен только rHsp90, было В, и поглощение для лунки, к которой не были добавлены ни тестируемое соединение, ни rHsp90, было С. Расчеты проводили с использованием программного обеспечения Softmax Pro 4.6 (Molecular Devices). Кроме того, рассчитывали степень ингибирования (%), используя нижеприведенную расчетную формулу:
Т/С=(А-С)/(В-С)
Соединения примеров 1-6 проявляли активность ингибирования АТФ-азы на 50% или более при концентрации 2 мкМ.
(Тестовый пример 2: испытание ингибирования клеточного роста)
Испытание ингибирования клеточного роста проводили, используя два типа клеток (линия клеток рака молочной железы человека SK-BR-3 и линия клеток рака легких человека NCI-H460).
Клетки каждого типа суспендировали в питательной среде и высевали в 96-луночный планшет в количестве 2000 клеток/150 мкл/на лунку в случае клеток SK-BR-3 и в количестве 500 клеток/150 мкл/на лунку в случае клеток NCI-H460. Тестируемое соединение растворяли в ДМСО, и этот раствор разбавляли питательной средой, чтобы приготовить раствор тестируемого образца (концентрация ДМСО: 0,5% или менее). На следующий день после высевания к клеткам дополнительно добавляли по 50 мкл ДМСО-содержащей среды, к которой тестируемое соединение не добавляли (в дальнейшем в этом документе называемой разбавленным ДМСО раствором; концентрация ДМСО: 0,5% или менее) или по 50 мкл раствора тестируемого образца. Исследование цитотоксичности с помощью МТТ осуществляли непосредственно по истечении 72 часов после добавления раствора тестируемого образца или разбавленного ДМСО раствора к клеткам. Исследование цитотоксичности с помощью МТТ проводили, как описано ниже.
Раствор 5 мг/мл МТТ (бромид 3-(4,5-диметилтиазол-2-ил)-2,5-дифенилтетразолия) добавляли в количестве 20 мкл на лунку. Затем планшет выдерживали в термостате при 37°С в 5% СО2 в течение четырех часов. Планшет центрифугировали при 1200 об/мин в течение пяти минут, и затем супернатант над клеточной культурой удаляли с помощью всасывающей системы с использованием перекачивающего дозирующего устройства. ДМСО добавляли в количестве 150 мкл на лунку, и образовавшийся формазан растворяли. Планшет перемешивали, используя планшетный смеситель, для равномерного окрашивания соответствующих лунок. Поглощение для каждой лунки измеряли, используя сканирующий спектрофотометр для считывания планшетов, фиксировали значение оптической плотности OD при 540 нм в сравнении с 660 нм.
Т/С (%) для каждой концентрации определяли по нижеприведенной расчетной формуле и строили графическую зависимость доза-результат воздействия, рассчитанный как концентрация ингибирования 50% роста (величина GI50), основываясь на принятом обозначении, что величина оптической плотности OD, измеренная непосредственно после добавления раствора тестируемого образца, была S, величина оптической плотности OD, измеренная спустя 72 часа после добавления раствора тестируемого образца, была Т, и величина оптической плотности OD, измеренная спустя 72 часа после добавления разбавленного раствора ДМСО, была С.
Т/С (%)=(T-S)/(C-S)×100
Полученные результаты показаны ниже.
(Тестовый пример 3: тестирование противоопухолевой активности in vivo)
Тестирование противоопухолевой активности проводили, используя NCI-H1975 (линия клеток немелкоклеточного рака легкого человека).
В день 0 вышеупомянутые клетки трансплантировали подкожно в области правого бока каждого самца бестимусной бесшерстной мыши (4×106 клеток на мышь).
В день 10 замеряли массу тела и диаметр опухоли. Затем мышей в произвольном порядке делили на группы таким образом, чтобы для групп не было статистически значимых различий в величинах массы тела и в определенном объеме опухоли (определенный объем опухоли = наибольший диаметр опухоли × наименьший диаметр опухоли × наименьший диаметр опухоли/2).
В день 11 измеряли диаметр опухоли и затем начинали введение тестируемого соединения.
Тестируемое соединение готовили в концентрации 1,5 мг/мл, и его вводили мышам перорально в дозе 15 мг/10 мл/кг.
Такое введение осуществляли один раз в день в течение периода времени от дня 11 до дня 14, и измеряли диаметр опухоли в течение периода времени от дня 11 до дня 14 и в день 17.
В день 17 опухоль оперативно удаляли и затем измеряли ее массу.
Начальную массу опухоли рассчитывали исходя из объема опухоли, измеренного в День 11, считая удельный вес равным 1.
Возрастание массы опухоли рассчитывали путем вычитания начальной массы опухоли из массы опухоли во влажном состоянии, измеренной в день 17.
Степень ингибирования роста опухоли (GI%=100-Т/С (%)) каждым тестируемым соединением рассчитывали из соотношения (Т/С (%)) возрастания массы опухоли (Т) для группы, которой вводилось тестируемое соединение, к возрастанию массы опухоли (С) для контрольной группы (группа, которой вводился раствор).
Проверка статистической значимости различий между контрольной группой и группой, которой вводилось тестируемое соединение, проводилась в соответствии с критерием множественного сравнения Дуннетта с использованием программы EXSAS ver. 7.5.2.2 (Arm Corp).
Все соединения примеров 1-6 проявляли достоверную активность ингибирования роста опухоли на 50% или более.
(Тестовый пример 4: тестирование ингибирования CYP3A4 с использованием микросом печени человека)
Мидазолам (субстрат; Wako Pure Chemical Industries, Ltd.), 1'-гидроксимидазолам (определяемое целевое соединение; Becton, Dickinson and Company, Japan), Кетоконазол (положительный контроль; Sigma Chemical Со.), НАДФН система регенерации (раствор А и раствор В (Becton, Dickinson and Company, Japan)) использовались в работе.
Соответствующие количества тестируемого соединения, субстрата, определяемого целевого соединения, и соединения положительного контроля взвешивали. Их растворяли в ДМСО и затем разбавляли ацетонитрилом, по необходимости. Готовили 180 мкл реакционного раствора путем смешивания нижеприведенных с (1) по (5), которые были предварительно нагреты до 37°С в течение 10 минут:
(1) Калий-фосфатный буферный раствор (рН 7,4; конечная концентрация: 50 мМ)
(2) Очищенная вода
(3) Мидазолам (конечная концентрация: 2,5 мкМ)
(4) Тестируемое соединение (конечная концентрация: 10 мкМ; отрицательный контроль: нет соединений; положительный контроль: Кетоконазол, конечная концентрация: 0,1 мкМ)
(5) Микросомы печени человека (конечная концентрация: 0,05 мг-P/мл)
Добавляли 20 мкл НАДФН системы регенерации (раствор A/раствор B/очищенная вода=5/1/4) к этому реакционному раствору и реакцию инициировали. Смесь инкубировали в течение 10 минут. Затем отбирали 50 мкл реакционного раствора и добавляли его затем к 200 мкл ацетонитрила, далее следовала остановка реакции. Кроме того, 50 мкл ацетонитрильного раствора, содержащего внутреннее стандартное вещество, добавляли к реакционному раствору.
С той целью, чтобы удалить белки из этого образца, реакционный раствор переносили в 96-луночный собирающий планшет (Waters), используя Captiva (зарегистрированный товарный знак) (GL Sciences). Этот раствор образца обозначали как образец, используемый для анализа посредством жидкостной хроматографии-масс-спектрометрии/масс-спектрометрии (LC-MS/MS).
Отдельно готовили образцы 1'-гидроксимидазолама, имеющие известные концентрации (конечная концентрация: от 0 до 5 мкМ; всего 5 точек). Строили калибровочную кривую, используемую для испытаний, и в образце определяли концентрацию 1'-гидроксимидазолама. Эту реакцию дублировали.
Количество 1'-гидроксимидазолама, образовавшегося в случае отрицательного контроля, принимали за 100%, и процентное количество этого вещества, образовавшееся за счет добавления каждого соединения, затем рассчитывали. Таким образом получали показатель ингибирования CYP3A4.
И в результате получили, что степень ингибирования соединением 1 составляла 15%, степень ингибирования соединением 2 составляла 58%, степень ингибирования соединением 3 составляла 5%, степень ингибирования соединением 4 составляла 43%, степень ингибирования соединением 5 составляла 66%, и степень ингибирования соединением 6 составляла -18%. Положительный контроль, Кетоконазол, показывал степень ингибирования, равную 82%.
(Тестовый пример 5: тестирование противоопухолевой активности in vivo)
Тестирование противоопухолевой активности проводили, используя NCI-Н1650 (линию клеток немелкоклеточного рака легкого человека).
Вышеуказанные клетки трансплантировали подкожно в область правого бока каждого самца бестимусной бесшерстной мыши (1×107 клеток на мышь).
Спустя тридцать девять дней опухоль оперативно удаляли и ее маленький фрагмент, имеющий размер каждой стороны от 2 до 3 мм, формировали. Маленький фрагмент трансплантировали подкожно в область правого бока каждого самца бестимусной бесшерстной мыши (день 0).
В день 14 измеряли массу тела и диаметр опухоли. Затем мышей в произвольном порядке делили на группы таким образом, чтобы для групп не было статистически значимых различий в величинах массы тела и в определенном объеме опухоли (определенный объем опухоли = наибольший диаметр опухоли × наименьший диаметр опухоли × наименьший диаметр опухоли/2).
Затем в день 14 начинали введение тестируемого соединения.
Тестируемое соединение готовили в концентрации 0,65, 0,9 и 1,3 мг/мл, и его вводили мышам перорально в дозе 10 мл/кг.
Такое введение осуществляли один раз в день в течение периода времени от дня 14 до дня 31, и измеряли диаметр опухоли в день 14, день 17, день 21, день 24, день 28 и день 31.
В день 31 опухоль оперативно удаляли и затем измеряли ее массу во влажном состоянии.
Начальную массу опухоли рассчитывали исходя из объема опухоли, измеренного в день 14, считая удельный вес равным 1.
Возрастание массы опухоли рассчитывали путем вычитания начальной массы опухоли из массы опухоли во влажном состоянии, измеренной в день 31.
Степень ингибирования роста опухоли (GI%=100-Т/С (%)) каждым тестируемым соединением рассчитывали из соотношения (Т/С (%)) возрастания массы опухоли (Т) для группы, которой вводилось тестируемое соединение, к возрастанию массы опухоли (С) для контрольной группы (группа, которой вводился раствор).
Проверка статистической значимости различий между контрольной группой и группой, которой вводилось тестируемое соединение, проводилась в соответствии с критерием множественного сравнения Дуннетта с использованием программы EXSAS ver. 7.5.2.2 (Arm Corp).
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНОЕ ПИРАЗОЛОПИРИМИДИНА | 2007 |
|
RU2420530C2 |
| НОВОЕ ПРОИЗВОДНОЕ ЦИКЛИЧЕСКОГО ДИНУКЛЕОТИДА И ЕГО КОНЪЮГАТ АНТИТЕЛО-ЛЕКАРСТВЕННОЕ СРЕДСТВО | 2019 |
|
RU2809547C2 |
| КОНЪЮГАТ АНТИТЕЛО-ЛЕКАРСТВЕННОЕ СРЕДСТВО, СОДЕРЖАЩИЙ НОВОЕ ПРОИЗВОДНОЕ ЦИКЛИЧЕСКОГО ДИНУКЛЕОТИДА | 2021 |
|
RU2836814C1 |
| КЛАСС КОНДЕНСИРОВАННЫХ КОЛЬЦЕВЫХ СОЕДИНЕНИЙ И ИХ ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ | 2021 |
|
RU2831125C1 |
| Новые соединения пиридопиримидинона для модулирования каталитической активности гистонлизиндеметилаз (KDMS) | 2015 |
|
RU2684396C2 |
| АНТАГОНИСТЫ РЕЦЕПТОРА ВАЗОПРЕССИНА V | 2005 |
|
RU2370497C2 |
| БЕНЗОКСЕПИНОВЫЕ ИНГИБИТОРЫ PI3 И СПОСОБЫ ПРИМЕНЕНИЯ | 2010 |
|
RU2557658C2 |
| НОВОЕ ХИНОЛИН-ЗАМЕЩЕННОЕ СОЕДИНЕНИЕ | 2014 |
|
RU2689158C2 |
| НОВОЕ ХИНОЛИН-ЗАМЕЩЕННОЕ СОЕДИНЕНИЕ | 2014 |
|
RU2797117C2 |
| ХИНОЛИНОВЫЕ И ХИНАЗОЛИНОВЫЕ ПРОИЗВОДНЫЕ | 2000 |
|
RU2256654C2 |
Изобретение относится к 2-{4-амино-2-[(3-хлор-4-метокси-5-метилпиридин-2-ил)метил]-2,7-дигидро-6-тиа-1,2,3,5-тетраазабензо[cd]азулен-8-ил}-N-метилацетамиду и его солям, таким как гидробромид, гидрохлорид, метансульфонат, этан-1,2-дисульфонат. Изобретение также относится к лекарственному средству и фармацевтической композиции на основе указанного соединения и его солей для ингибирования АТФ-азной активности HSP90. Технический результат: получено новое соединение и его соли, которые могут найти применение в медицине для лечения рака.
13 н. и 10 з.п. ф-лы, 10 пр.
1. 2-{4-амино-2-[(3-хлор-4-метокси-5-метилпиридин-2-ил)метил]-2,7-дигидро-6-тиа-1,2,3,5-тетраазабензо[cd]азулен-8-ил}-N-метилацетамид.
2. Гидробромид соединения по п.1.
3. Гидрохлорид соединения по п.1.
4. Метансульфонат соединения по п.1.
5. Этан-1,2-дисульфонат соединения по п.1.
6. Ингибитор HSP90, включающий соединение по п.1 или его соль или соединение по любому из пп.2-5.
7. Средство для ингибирования АТФ-азной активности HSP90, которое включает соединение по п.1 или его соль или соединение по любому из пп.2-5.
8. Средство для ингибирования связывания HSP90 с АТФ, которое включает соединение по п.1 или его соль или соединение по любому из пп.2-5.
9. Лекарственное средство для лечения рака, которое включает соединение по п.1 или его соль или соединение по любому из пп.2-5 в качестве активного ингредиента.
10. Противораковое средство, которое включает соединение по п.1 или его соль или соединение по любому из пп.2-5 в качестве активного ингредиента.
11. Фармацевтическая композиция для лечения рака, которая включает соединение по п.1 или его соль или соединение по любому из пп.2-5 и фармацевтически приемлемый носитель.
12. Способ лечения рака, включающий введение соединение по п.1 или его соль или соединение по любому из пп.2-5.
13. Применение соединение по п.1 или его соль или соединение по любому из пп.2-5 для получения лекарственного средства для лечения рака.
14. Противораковое средство по п.10, где рак вовлечен в повышенную экспрессию генов HSP90-зависимого(ых) белка(ов).
15. Способ лечения рака по п.12, где рак вовлечен в повышенную экспрессию генов HSP90-зависимого(ых) белка(ов).
16. Противораковое средство по п.10, где рак вовлечен в мутацию HSP90-зависимого(ых) белка(ов).
17. Способ лечения рака по п.12, где рак вовлечен в мутацию HSP90-зависимого(ых) белка(ов).
18. Противораковое средство по п.10, где рак вовлечен в активацию HSP90-зависимого(ых) белка(ов).
19. Способ лечения рака по п.12, где рак вовлечен в активацию HSP90-зависимого(ых) белка(ов).
20. Противораковое средство по п.10, где рак вовлечен в активацию внутриклеточного метаболического пути передачи сигналов, с которым связан(ы) HSP90-зависимый(е) белок(ки).
21. Способ лечения рака по п.12, где рак вовлечен в активацию внутриклеточного метаболического пути передачи сигналов, с которым связан(ы) HSP90-зависимый(е) белок(ки).
22. Противораковое средство по п.10, где рак зависит от HSP90-зависимого(ых) белка(ов).
23. Способ лечения рака по п.12, где рак зависит от HSP90-зависимого(ых) белка(ов).
| СКЛАДНОЙ ЯЩИК | 1992 |
|
RU2065388C1 |
| WO 2005028434 A2, 31.03.2005 | |||
| ПРОИЗВОДНЫЕ КОНДЕНСИРОВАННЫХ ПОЛИЦИКЛИЧЕСКИХ ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1996 |
|
RU2167877C2 |

