ОБЛАСТЬ ТЕХНИКИ
[0001]
Согласно настоящему изобретению предложены производные циклических динуклеотидов, обладающие новыми структурами, с активностью агонистов STING, конъюгаты антитела с лекарственным средством, образованные путем конъюгирования новых производных циклических динуклеотидов и антител против целевых клеток друг с другом посредством линкера, и фармацевтическая композиция, содержащая конъюгаты антитела с лекарственным средством, и т.п.
УРОВЕНЬ ТЕХНИКИ
[0002]
STING (стимулятор генов интерферона) представляет собой трансмембранный адапторный белок, локализованный в эндоплазматическом ретикулуме (непатентная публикация 1). STING действует как главная молекула для стимуляции врожденного иммунитета у млекопитающих, и играет роль на первой линии защиты против инвазии патогенов, таких как бактерии и вирусы. Известно, что активация STING вызывается сигналом, образованным, когда множество сенсоров цитоплазматической ДНК обнаруживают экзогенную или эндогенную ДНК. Ожидают, что среди сенсоров цитоплазматической ДНК, cGAS (синтаза циклического ГМФ-АМФ) является важным сенсором ДНК. Когда cGAS обнаруживает ДНК, образуется циклический динуклеотид (2',3'-цГАМФ), и данный 2',3'-цГАМФ непосредственно связывается со STING, чтобы активировать его (непатентная публикация 2). Активированный STING перемещается в аппарат Гольджи и вызывает аутофосфорилирование ТВК1 (Tank-связывающей киназы 1). ТВК-1, активированная аутофосфорилированием, активирует как путь транскрипции IRF3 (регуляторного фактора интерферона 3) (непатентная публикация 3), так и путь транскрипции NFκB (непатентная публикация 4), повышая продукцию воспалительных белков, называемых интерферонами и цитокинами (IFN I типа (интерферон), IL-6 (интерлейкин-6), TNF-α (фактор некроза опухоли-α)). Данные белки приводят в действие систему приобретенного иммунитета, включая Т-клетки, посредством сложных каскадов, которые разрушают патогены и раковые клетки.
[0003]
В недавних исследованиях продемонстрировали, что STING стимулирует не только иммунную защиту хозяина от микроорганизмов, но также противоопухолевый иммунитет. Например, иммуногенные опухоли, трансплантированные мышам с дефицитом STING, быстрее растут, чем опухоли, трансплантированные мышам дикого типа и мышам с дефицитом TRIF (содержащий домен рецептора То11/интерлейкина-1 (IL-1) адапторный белок, индуцирующий интерферон-β). В противоположность мышам с дефицитом TLR (Toll-подобный рецептор), MyD88 (первичный ответ миелоидной дифференцировки 88) и MAVS (митохондриальный противовирусный сигнальный белок), самопроизвольное примирование CD8+ Т-клеток против опухоли также исчезало у мышей с дефицитом STING. Это позволяет предположить, что путь STING, который запускается обнаружением цитоплазматической ДНК, участвует в контроле роста опухоли (непатентная публикация 5). Кроме того, в других исследованиях продемонстрировали, что STING необходим для противоопухолевого действия лучевой терапии (непатентная публикация 6) и терапии антителом против CD47 (непатентная публикация 7). ДНК, происходящая из уничтоженных опухолевых клеток после лечения радиацией или антителом против CD47, перемещается в цитоплазму дендритных клеток, чтобы активировать путь cGAS-STING, а затем вызвать продукцию IFN, чтобы активировать приобретенный иммунитет посредством врожденного иммунитета. Данные исследования позволяют предположить, что перекрестное примирование дендритными клетками, активированными через путь STING, важно, чтобы вызвать приобретенный иммунитет против опухоли.
[0004]
Продемонстрировали, что низкомолекулярный флавоноид DMXAA, который известен как разрушающий сосуды агент, вызывает продукцию IFN I типа в макрофагах и, следовательно, обладает эффективной противоопухолевой активностью в моделях опухоли у мышей (непатентная публикация 8). Ожидали, что DMXAA будет служить в качестве иммунотерапевтического лекарственного средства для немелкоклеточного рака легкого благодаря его превосходному противоопухолевому действию в доклинических испытаниях; тем не менее, DMXAA потерпел неудачу в клинических испытаниях (непатентная публикация 9). В недавнем исследовании выявили, что DMXAA представляет собой агонист, специфичный к STING мыши, и не проявляет межвидовой перекрестной реактивности со STING человека и, следовательно, не способен связываться с ним (непатентная публикация 10). Наконец, обнаружили, что DMXAA неэффективен у людей; тем не менее, исследования в моделях на мышах привели к предположению, что низкомолекулярные лекарственные средства способны усиливать противоопухолевый иммунитет путем эффективного примирования CD8+Т-клеток посредством STING.
[0005]
Продемонстрировали, что когда циклический динуклеотид (ЦДН), другое низкомолекулярное соединение, вводят несущим опухоль мышам, он усиливает противоопухолевый иммунный ответ, опосредованный STING, значительно ингибируя рост опухоли, улучшая показатель выживаемости мышей (непатентная публикация 11). ЦДН классифицируют на бактериальные ЦДН с каноническими двумя 3'-5'-фосфатными связями (циклический-ди-ГМФ, циклический-ди-АМФ, 3',3'-цГАМФ), и ЦДН с неканоническими, смешанными связями с 2'-5'-фосфатной связью (2',3'-цГАМФ), которые продуцирует cGAS млекопитающих. В недавнем исследовании продемонстрировали, что ЦДН со смешанными связями, а не канонические ЦДН, способны универсальным образом активировать различные типы STING (непатентная публикация 12).
[0006]
ЦДН природного типа быстро разлагаются нуклеазами в крови, как большинство молекул нуклеиновых кислот, и, следовательно, их нельзя вводить в исходном виде. По этой причине, были разработаны синтезированные низкомолекулярные соединения, обладающие активностью агонистов STING in vivo (например, патентные публикации 1-26).
[0007]
Агонист STING MIW-815 (альтернативно называемый ADU-S100, ML RR-S2 CDA или ML-RR-CDA-2Na+), который представляет собой противоопухолевый агент, на данный момент проходящий клинические испытания, вводят непосредственно в опухоль. Такой способ непосредственного введения агониста STING в опухоль позволяет введение лекарственного средства только в ограниченную область опухоли и представляет сложность для непосредственного введения лекарственного средства в каждый отдаленный метастаз опухоли, что неблагоприятно ограничивает тип излечимых опухолей. Хотя в непатентной публикации 13 описано, что противоопухолевое действие проявлялось при введении ML RR-S2 CDA, исследование проводили только по внутриопухолевому введению, и противоопухолевое действие при системном введении (например, внутривенном введении) не было продемонстрировано. В непатентной публикации 14 описано, что противоопухолевое действие проявлялось при внутривенном введении агониста STING SB11285 в модели опухоли у мышей, но не уточнено, какая именно структура была у соединения SB11285. В патентной публикации 14 описан конъюгат, содержащий стимулирующее иммунитет соединение, конструкцию антитела и линкер, но не описан какой-либо конкретный пример конъюгата, в котором используется агонист STING в качестве стимулирующего иммунитет соединения. В патентной публикации 26 описан конъюгат, образованный путем конъюгирования ЦДН, обладающего определенной структурой, и антитела друг с другом посредством линкера, но не описано введение конъюгата in vivo в Примерах, и, следовательно, противоопухолевое действие конъюгата не было подтверждено.
Перечень противопоставленных материалов
Патентная литература
[0008]
Патентная публикация 1: WO 2014/099824
Патентная публикация 2: WO 2014/179335
Патентная публикация 3: WO 2014/189805
Патентная публикация 4: WO 2014/189806
Патентная публикация 5: WO 2015/074145
Патентная публикация 6: WO 2015/185565
Патентная публикация 7: WO 2016/096714
Патентная публикация 8: WO 2016/012305
Патентная публикация 9: WO 2016/145102
Патентная публикация 10: WO 2017/027646
Патентная публикация 11: W02017/027645
Патентная публикация 12: WO 2017/075477
Патентная публикация 13: WO 2017/093933
Патентная публикация 14: WO 2017/100305
Патентная публикация 15: WO 2017/123669
Патентная публикация 16: WO 2017/161349
Патентная публикация 17: WO 2017/175147
Патентная публикация 18: WO 2017/175156
Патентная публикация 19: WO 2018/009466
Патентная публикация 20: WO 2018/045204
Патентная публикация 21: WO 2018/060323
Патентная публикация 22: WO 2018/067423
Патентная публикация 23: WO 2018/065360
Патентная публикация 24: WO 2014/093936
Патентная публикация 25: WO 2018/009648
Патентная публикация 26: WO 2018/100558
Непатентная литература
[0009]
Непатентная публикация 1: Nature 2008, 455, 674-678
Непатентная публикация 2: Mol. Cell, 2013, 51, 226-235
Непатентная публикация 3: Science 2015а, 347, ааа2630
Непатентная публикация 4: J. Virol. 2014, 88, 5328-5341
Непатентная публикация 5: Immunity 2014, 41, 830-842
Непатентная публикация 6: Immunity 2014, 41, 843-852
Непатентная публикация 7: Nat. Med. 2015, 21, 1209-1215
Непатентная публикация 8: J. Immonol. 1994, 153, 4684-4693
Непатентная публикация 9: J. Clin. Oncol. 2011, 29, 2965-2971
Непатентная публикация 10: J. Immonol. 2013, 190, 5216-5225
Непатентная публикация 11: Sci. Rep.2016, 6, 19049
Непатентная публикация 12: Mol. Cell, 2015, 59, 891-903
Непатентная публикация 13: Cell Rep.2015, 11, 1018-1030
Непатентная публикация 14: AACR Tumor Immunology and Immunotherapy, 2017, постер A25
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
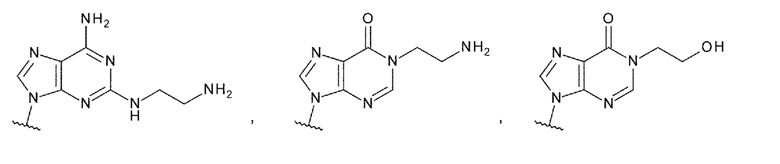
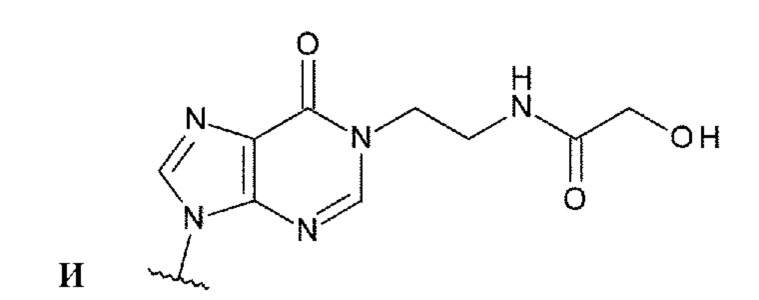
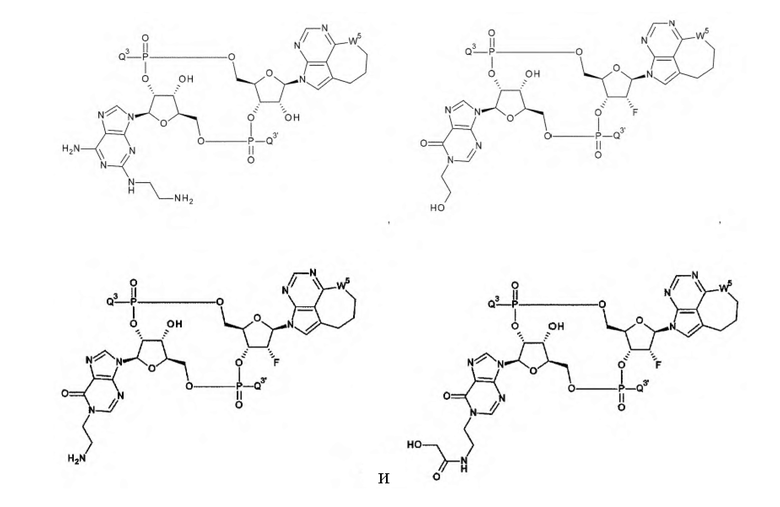
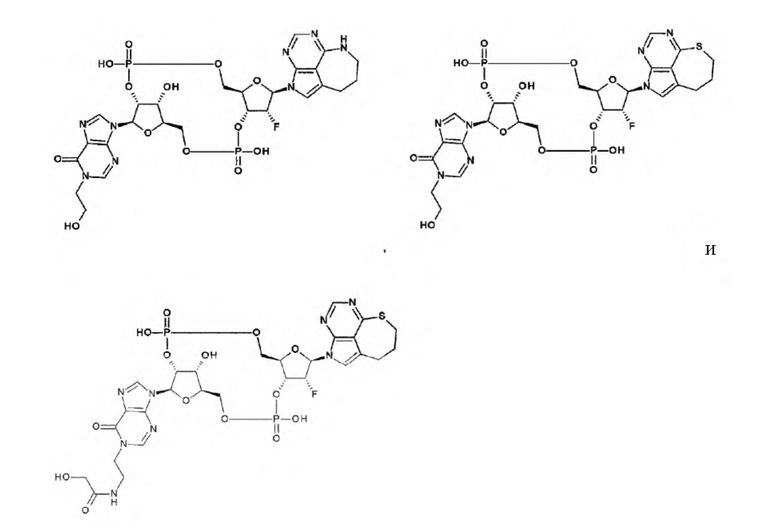
Техническая задача
[0010]
Требуется разработка производных ЦДН с новым остовом, которые обладают активностью агонистов STING и повышают продукцию воспалительных белков, таких как интерфероны и цитокины, чтобы активировать иммунные клетки; и требуются терапевтические агенты и/или способы терапии с применением новых производных ЦДН для лечения заболеваний, связанных с активностью агонистов STING, например, заболеваний, излечимых с помощью активации иммунитета (например, рака). Дополнительно требуются конъюгаты антитела с лекарственным средством, которые образованы путем конъюгирования нового производного ЦДН и антитела против целевых клеток друг с другом посредством линкера и которые позволяют системное введение и способны доставлять агонист STING специфично в целевые клетки и органы (например, очаги опухоли); и терапевтические агенты и/или способы терапии с применением конъюгатов антитела с лекарственным средством для лечения заболеваний, связанных с активностью агонистов STING, например, заболеваний, поддающихся лечению с помощью активации иммунитета (например, рака).
Решение проблемы
[0011]
Для решения описанных выше проблем авторы настоящего изобретения разработали новые производные ЦДН, содержащие слитый трициклический заместитель, и обнаружили, что новые производные ЦДН обладают эффективной активностью агонистов STING и проявляют эффективную противоопухолевую активность. Кроме того, авторы настоящего изобретения разработали конъюгаты антитела с лекарственным средством, образованные путем конъюгирования новых производных ЦДН согласно настоящему изобретению и антитела друг с другом посредством линкера, и обнаружили, что указанные конъюгаты антитела с лекарственным средством при системном введении оказывают противоопухолевое действие на опухоли, экспрессирующие антиген, таким образом, осуществляя настоящее изобретение.
[0012]
В частности, согласно настоящему изобретению предложено следующее.
[1] Конъюгат антитела с лекарственным средством, представленный формулой (II):
[0013]
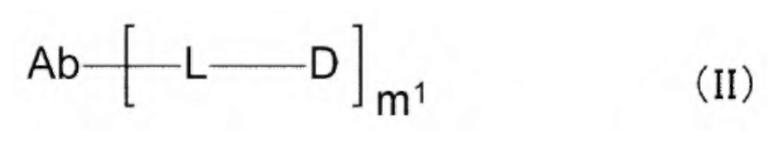
где
m1 находится в диапазоне от 1 до 10;
Ab представляет собой антитело или функциональный фрагмент антитела, где гликан антитела необязательно ремоделирован;
L представляет собой линкер, соединяющий Ab и D;
Ab непосредственно связывается с L через аминокислотный остаток из Ab, или необязательно связывается посредством гликана или ремоделированного гликана Ab с L; и D представляет собой соединение, представленное формулой (I):
[0014]
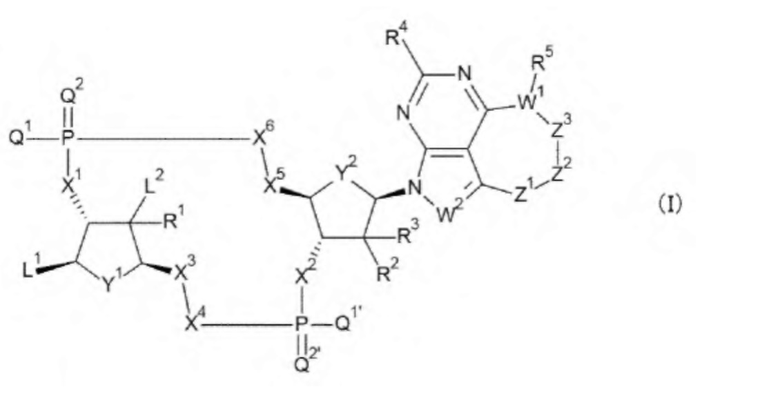
где
L связывается с любой -NH2 или гидроксигруппой, содержащейся в L1 или L2;
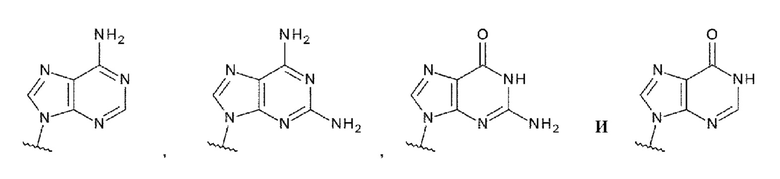
L1 представляет собой группу, выбранную из группы, состоящей из следующих формул:
[0015]
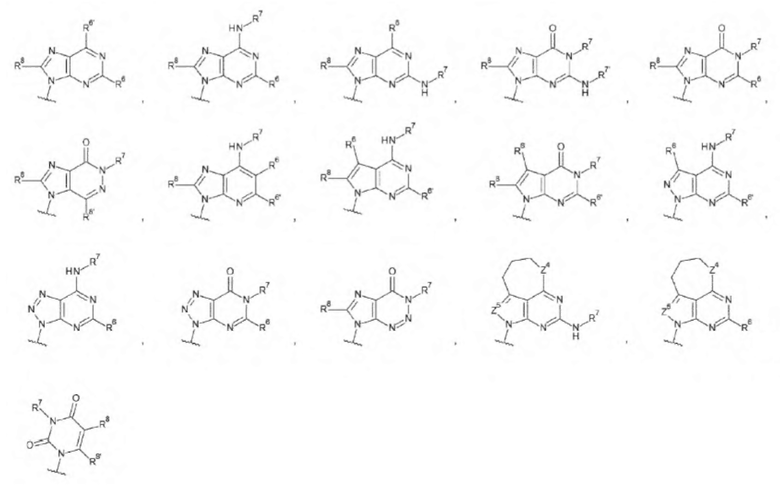
и необязательно содержащую в качестве заместителей в любом положении от одной до трех групп, выбранных из группы, состоящей из гидроксигруппы, -NH2, 2-гидроксиацетиламинометильной группы и 2-[(2-гидроксиацетил)амино]зтильной группы, где
R6 и R6' каждый независимо представляет собой атом водорода, атом галогена, гидроксигруппу, -NH2, С1-С6 алкильную группу, С2-С6 алкенильную группу или С2-С6 алкинильную группу;
R7 и R7' каждый независимо представляет собой атом водорода или С1-С6 алкильную группу, где С1-С6 алкильная группа необязательно содержит в качестве заместителей один или два заместителя, выбранных из группы, состоящей из атома галогена и оксогруппы;
R8 и R8' каждый независимо представляет собой атом водорода или атом галогена;
Z4 представляет собой -CH-, -NH- или атом кислорода; и
Z5 представляет собой атом азота или -СН=,
L2 представляет собой группу, выбранную из (i) и (ii):
(i) при связывании с L, L2 представляет собой -NHR', гидрокси-С1-С6 алкильную группу или амино-С1-С6 алкильную группу, где R' представляет собой атом водорода, С1-С6 алкильную группу, С2-С6 алкенильную группу, С2-С6 алкинильную группу или С3-С6 циклоалкильную группу, и С1-С6 алкильная группа, С2-С6 алкенильная группа или С2-С6 алкинильная группа необязательно содержит в качестве заместителей от одного до шести атомов галогена; и
(ii) при отсутствии связывания с L1, L2 представляет собой атом водорода или атом галогена;
Q1 и Q1' каждый независимо представляет собой гидроксигруппу, тиольную группу или борановую группу (ВН3-);
Q2 и Q2' каждый независимо представляет собой атом кислорода или атом серы;
X1 и X2 каждый независимо представляет собой атом кислорода, атом серы или -СН-;
Y1 и Y2 каждый представляет собой атом кислорода или -СН2-; X3 и X4 представляют собой группу, выбранную из (iii) и (iv):
(iii) когда Y1 представляет собой атом кислорода, Х3-Х4 представляет собой -СН2-О-, -CH2-S-, -СН2-СН2- или -CH2-CF2-; и
(iv) когда Y1 представляет собой -СН2-, Х3-Х4 представляет собой -О-СН2-; X5 и X6 представляют собой группу, выбранную из (v) и (vi):
(v) когда Y2 представляет собой атом кислорода, Х5-Х6 представляет собой -СН2-О-, -CH2-S-, -СН2-СН2- или -CH2-CF2-; и
(vi) когда Y2 представляет собой -СН2-, Х5-Х6 представляет собой -О-СН2-;
R1, R2 и R3 каждый независимо представляет собой атом водорода, атом галогена, -OR, -OC(=O)R', -N3, -NHR, -NR'R'' или -NHC(=O)R', где R' такой, как в определении выше, и R'' представляет собой С1-С6 алкильную группу, С2-С6 алкенильную группу, С2-С6 алкинильную группу или С3-С6 циклоалкильную группу;
W1 представляет собой атом азота, атом кислорода, атом серы или -СН-;
W2 представляет собой атом азота или -СН=;
R4 представляет собой атом водорода, атом галогена или -NH2;
R5 представляет собой группу, выбранную из (vii) - (х):
(vii) где W1 представляет собой атом азота, R5 представляет собой атом водорода, С1-С6 алкильную группу, гидрокси-С1-С6 алкильную группу или амино-С1-С6 алкильную группу;
(viii) где W1 представляет собой атом кислорода, R5 отсутствует;
(ix) где W1 представляет собой атом серы, R5 отсутствует; и
(x) где W1 представляет собой -СН-, R5 представляет собой атом водорода, атом галогена, гидроксигруппу, -NH2 или С1-С6 алкильную группу;
Z1-Z2-Z3 вместе представляют собой -СН2-СН2-СН2-, -CH2-CH2-R''''-, -СН=СН-СН2-, -СН=СХ-СН2-, -СХ=СН-СН2-, -СХ=СХ-СН2-, -С(=O)-СН2-СН2-, -СН2-СН2-С(=O)-, -СН2-СН(СН3)-СН2- или -СН2-СН2-СН(СН3)-, где R''' представляет собой -О- или -СН2-СН2- и X представляет собой атом галогена или группу, представленную любой одной из следующих формул:
[0016]

где
каждая звездочка указывает на связывание с W1, и каждая волнистая линия указывает на связывание с атомом углерода =С-;
[2] Конъюгат антитела с лекарственным средством согласно [1], где W1 представляет собой атом азота;
[3] Конъюгат антитела с лекарственным средством согласно [2], где W1 представляет собой атом азота и R5 представляет собой атом водорода;
[4] Конъюгат антитела с лекарственным средством согласно [1], где W1 представляет собой атом кислорода;
[5] Конъюгат антитела с лекарственным средством согласно [1], где W1 представляет собой атом серы;
[6] Конъюгат антитела с лекарственным средством согласно [1], где W1 представляет собой -СН-;
[7] Конъюгат антитела с лекарственным средством согласно [6], где W1 представляет собой -СН- и R5 представляет собой атом водорода;
[8] Конъюгат антитела с лекарственным средством согласно любому одному из [1]-[7], где Z1, Z2 и Z3 вместе образуют -СН2-СН2-СН2- или -СН=СН-СН2-;
[9] Конъюгат антитела с лекарственным средством согласно любому одному из [1]-[7], где
Z1, Z2 и Z3 вместе образуют -СН2-СН(СН3)-СН2- или -СН2-СН2-СН(СН3)-;
[10] Конъюгат антитела с лекарственным средством согласно любому одному из [1]-[7], где Z1, Z2 и Z3 вместе образуют -CH2-CH2-R''''-, где R''' представляет собой -О- или -СН2-СН2-;
[11] Конъюгат антитела с лекарственным средством согласно любому одному из [1]-[10], где W2 представляет собой -СН=;
[12] Конъюгат антитела с лекарственным средством согласно любому одному из [1]-[10], где W2 представляет собой атом азота;
[13] Конъюгат антитела с лекарственным средством согласно любому одному из [1]-[12], где R4 представляет собой атом водорода;
[14] Конъюгат антитела с лекарственным средством согласно любому одному из [1]-[12], где R4 представляет собой атом фтора;
[15] Конъюгат антитела с лекарственным средством согласно любому одному из [1]-[14], где R8 и R8' в L1 каждый независимо представляет собой атом водорода;
[16] Конъюгат антитела с лекарственным средством согласно любому одному из [1]-[15], где L1 представляет собой группу, выбранную из группы, состоящей из следующих формул:
[0017]
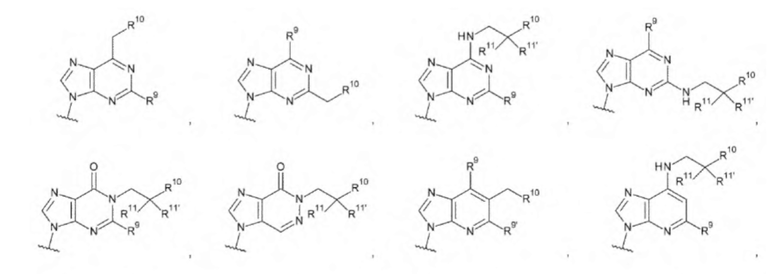
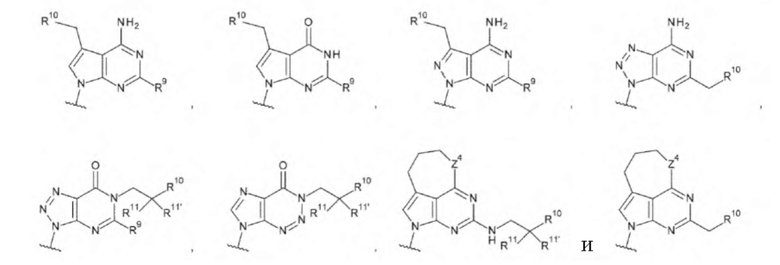
где
R9 и R9' каждый представляет собой атом водорода, атом галогена, гидроксигруппу или -NH2;
R10 представляет собой гидроксигруппу, -NH2, -NHC(=O)CH2OH, CH2NHC(=O)CH2OH, -CH2CH2NHC(=O)CH2OH, гидрокси-С1-С3 алкильную группу или амино-С1-С3 алкильную группу;
R11 и R11' каждый независимо представляет собой атом водорода, атом фтора или метальную группу, или R11 и R11' связаны друг с другом с образованием циклопропана; и
Z4 представляет собой -СН2-, -NH- или атом кислорода;
[17] Конъюгат антитела с лекарственным средством согласно любому одному из [1]-[15], где L1 представляет собой группу, выбранную из группы, состоящей из следующих формул:
[0018]
где
R13 и R13' каждый независимо представляет собой атом водорода, гидроксигруппу или -NH2;
R12 представляет собой гидроксигруппу, -NH2, -CH2OH, -NHC(=O)CH2OH, -CH2NHC(=O)CH2OH или -CH2CH2NHC(=O)CH2OH; и
Z4 такой, как в определении выше;
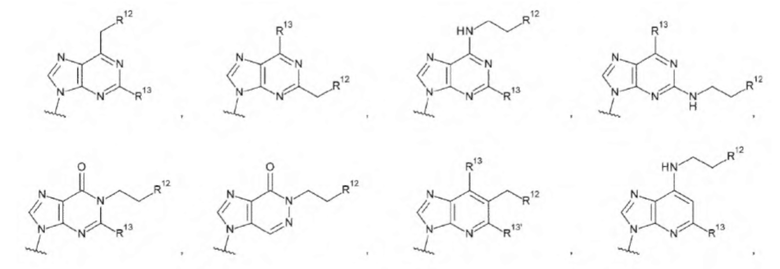
[18] Конъюгат антитела с лекарственным средством согласно любому одному из [1]-[15], где L1 представляет собой группу, выбранную из группы, состоящей из следующих формул:
[0019]
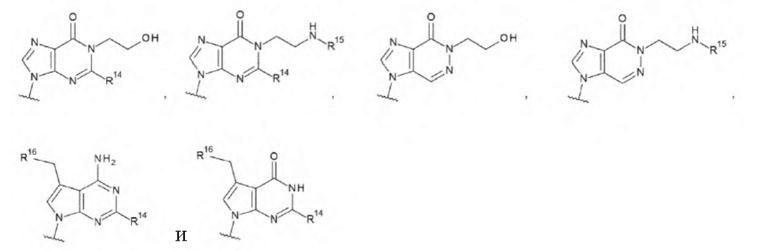
где
R14 представляет собой атом водорода или -NH2;
R15 представляет собой атом водорода или -С(=O)CH2OH; и
R16 представляет собой гидроксигруппу, -NH2, -СН2ОН, -СН2СН2ОН, -CH2NH2 или -CH2CH2NH2;
[19] Конъюгат антитела с лекарственным средством согласно любому одному из [1]-[18], где L2 связывается с L и представляет собой -NH2, -CH2NH2 или -CH2OH;
[20] Конъюгат антитела с лекарственным средством согласно любому одному из [1]-[18], где L2 не связывается с L и представляет собой атом водорода или атом фтора;
[21] Конъюгат антитела с лекарственным средством согласно любому одному из [1]-[20], где Q1 и Q1' каждый независимо представляет собой гидроксигруппу или тиольную группу;
[22] Конъюгат антитела с лекарственным средством согласно любому одному из [1]-[21], где X1 и X2 каждый представляет собой атом кислорода;
[23] Конъюгат антитела с лекарственным средством согласно любому одному из [1]-[22], где Y1 и Y2 каждый представляет собой атом кислорода;
[24] Конъюгат антитела с лекарственным средством согласно любому одному из [1]-[23], где X3 и X4 представляют собой -СН-O-;
[25] Конъюгат антитела с лекарственным средством согласно любому одному из [1]-[24], где X5 и X6 представляют собой -СН-O-;
[26] Конъюгат антитела с лекарственным средством согласно любому одному из [1]-[25], где R1, R2 и R3 каждый независимо представляет собой атом водорода, гидроксигруппу или атом фтора;
[27] Конъюгат антитела с лекарственным средством согласно любому одному из [1]-[26], где D представлен любой одной из следующих двух формул:
[0020]
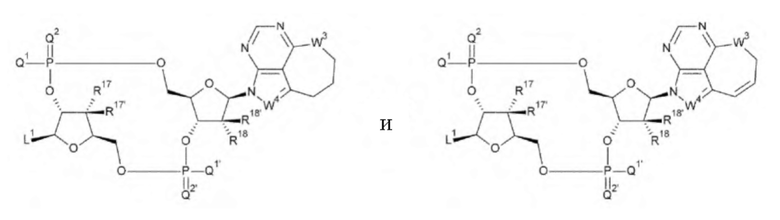
где
L1, Q1, Q1', Q2 и Q2' такие, как в определении выше;
R17, R17', R18 и R18' каждый независимо представляет собой атом водорода, атом галогена, гидроксшруппу или -NH2;
W3 представляет собой -NH-, атом кислорода, атом серы или -СН-; и
W4 представляет собой -СН= или атом азота;
[28] Конъюгат антитела с лекарственным средством согласно [27], где D представлен любой одной из следующих двух формул:
[0021]
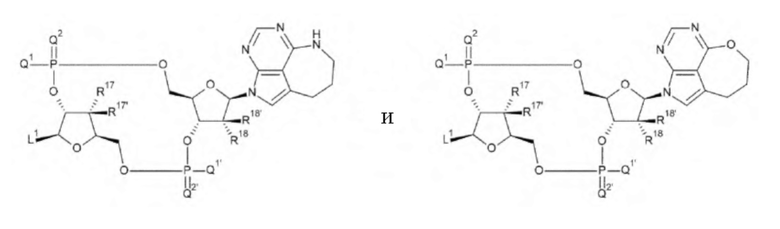
где
L1, Q1, Q1', Q2, Q2', R17, R17', R18 и R18' такие, как в определении выше;
[29] Конъюгат антитела с лекарственным средством согласно [27] или [28], где D представлен любой одной из следующих восьми формул:
[0022]
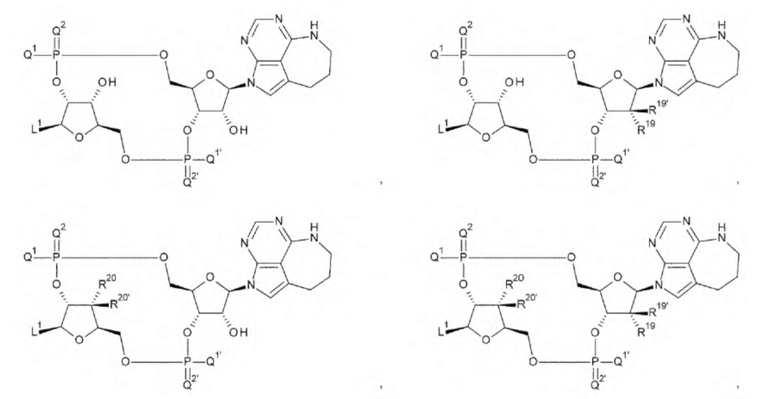
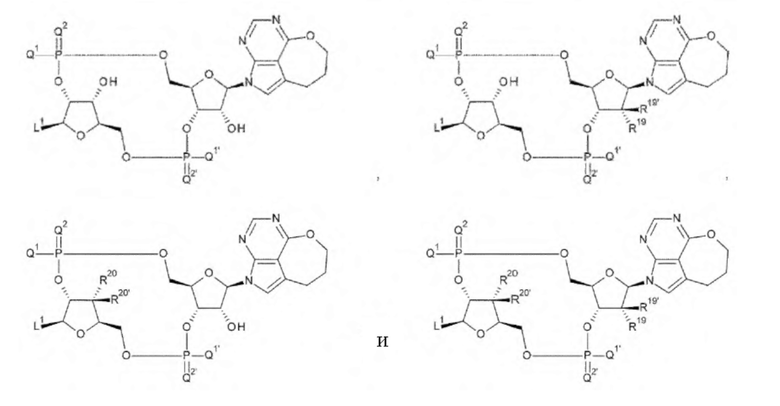
где
L1, Q1, Q1', Q2 и Q2' такие, как в определении выше; и
R19, R19', R20 и R20' каждый независимо представляет собой атом водорода или атом фтора;
[30] Конъюгат антитела с лекарственным средством согласно любому одному из [27]-[29],
где D представлен любой одной из следующих четырех формул:
[0023]
где
L1 такой, как в определении выше;
[31] Конъюгат антитела с лекарственным средством согласно любому одному из [27]-[30], где D представлен любой одной из следующих четырех формул:
[0024]
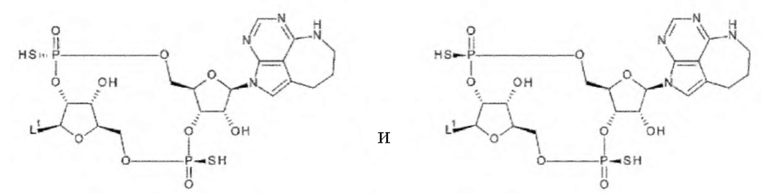
где
L1 такой, как в определении выше;
[32] Конъюгат антитела с лекарственным средством согласно любому одному из [27]-[30], где D представлен любой одной из следующих четырех формул:
[0025]
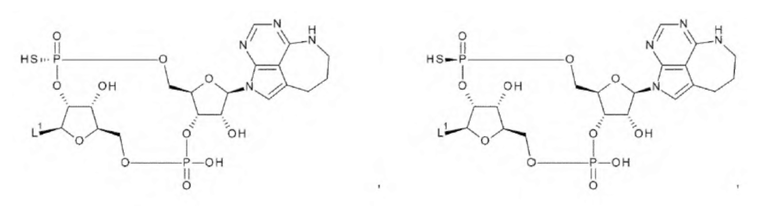
где
L1 такой, как в определении выше;
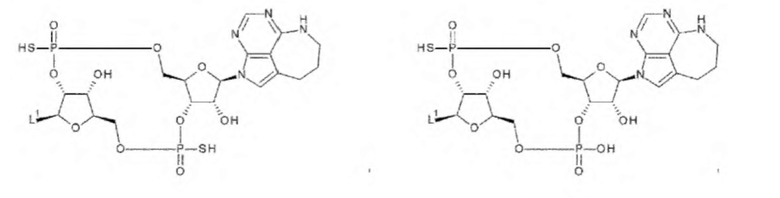
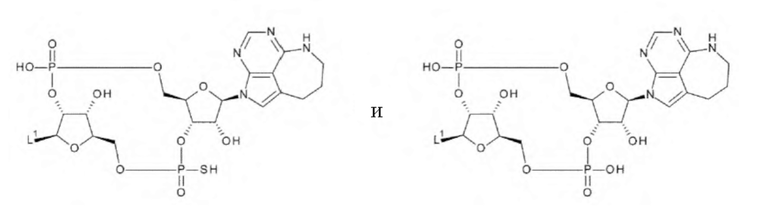
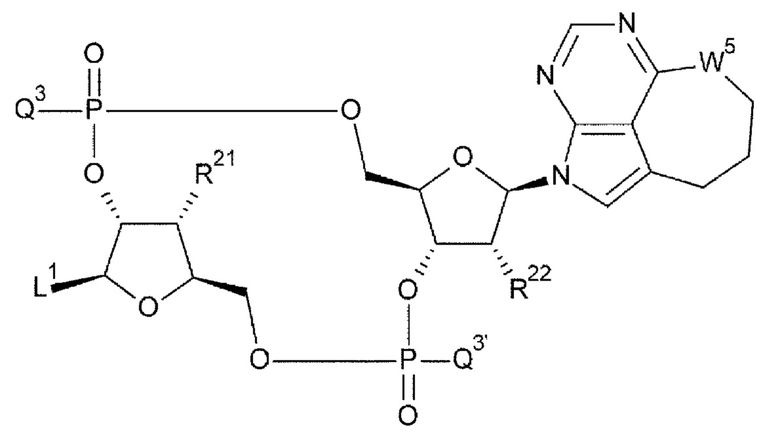
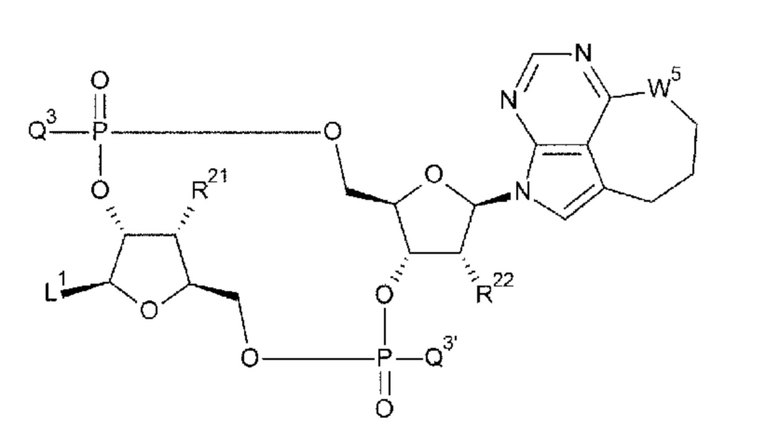
[33] Конъюгат антитела с лекарственным средством согласно любому одному из [1]-[26], где D представлен следующей формулой:
[0026]
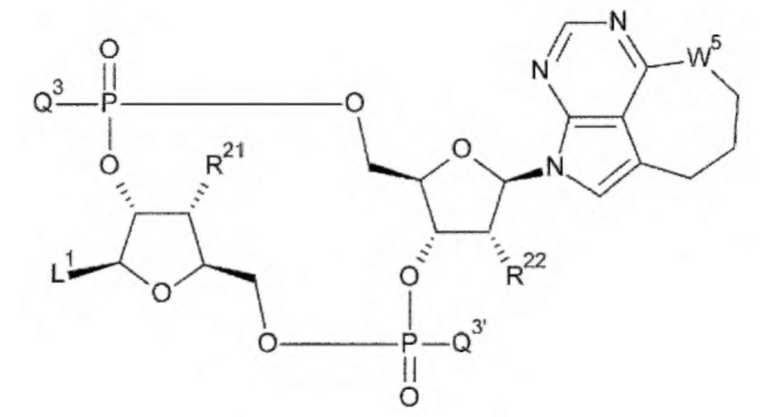
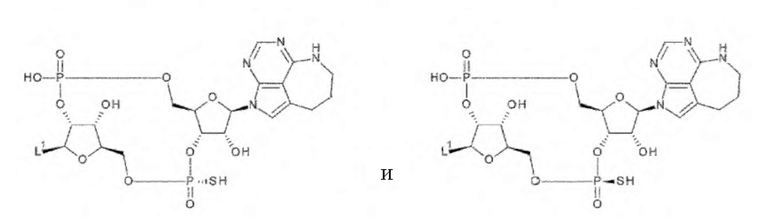
где
L1 такой, как в определении выше;
Q3 и Q3' каждый независимо представляет собой гидроксигруппу или тиольную группу;
R21 и R22 каждый независимо представляет собой гидроксигруппу или атом фтора; и
W3 представляет собой -NH- или атом серы;
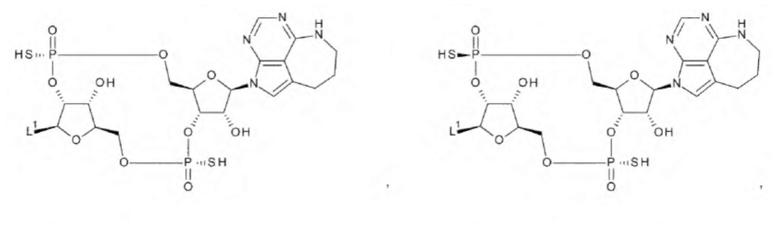
[34] Конъюгат антитела с лекарственным средством согласно [33], где D представлен любой одной из следующих двух формул:
[0027]
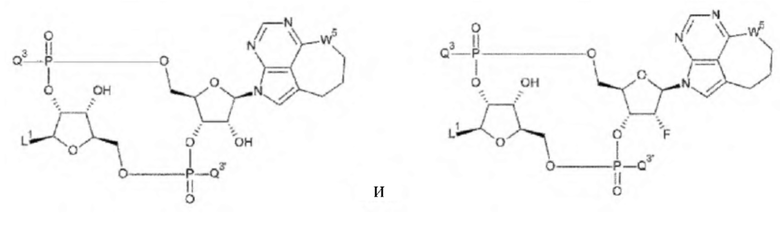
где
L1, Q3, Q3' и W3 такие, как в определении выше:
[35] Конъюгат антитела с лекарственным средством согласно любому одному из [1]-[34], где L1 представлен любой одной из следующих четырех формул:
[0028]
[36] Конъюгат антитела с лекарственным средством согласно любому одному из [1]-[34], где L1 представлен любой одной из следующих четырех формул:
[0029]
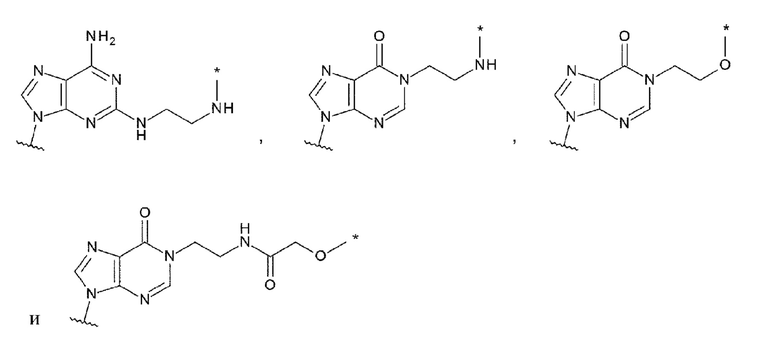
где
каждая звездочка указывает на связывание с L;
[37] Конъюгат антитела с лекарственным средством согласно любому одному из [33], [34] и [36], где D представлен любой одной из следующих четырех формул:
[0030]
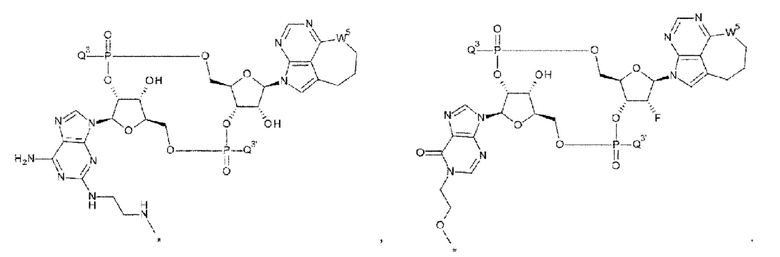
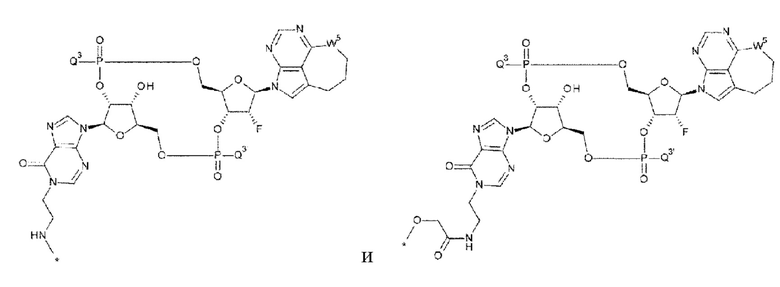
где
каждая звездочка указывает на связывание с L; и
Q3, Q3' и W3 такие, как в определении выше;
[38] Конъюгат антитела с лекарственным средством согласно любому одному из [33], [34], [36] и [37], где D представлен любой одной из следующих четырех формул:
[0031]
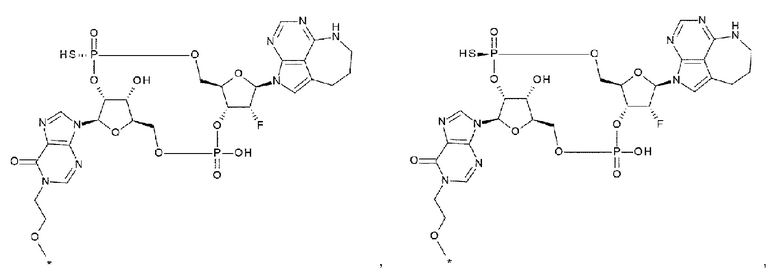
где
каждая звездочка указывает на связывание с L;
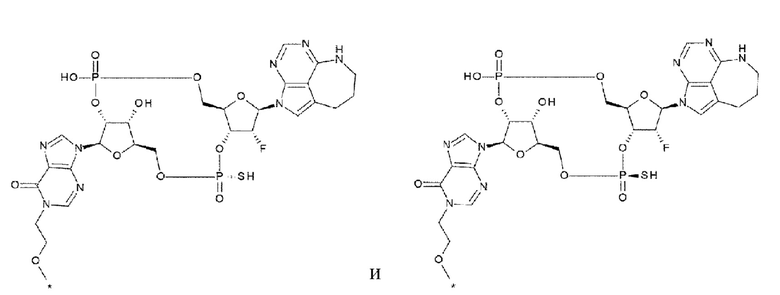
[39] Конъюгат антитела с лекарственным средством согласно любому одному из [33], [34], [36] и [37], где D представлен любой одной из следующих трех формул:
[0032]
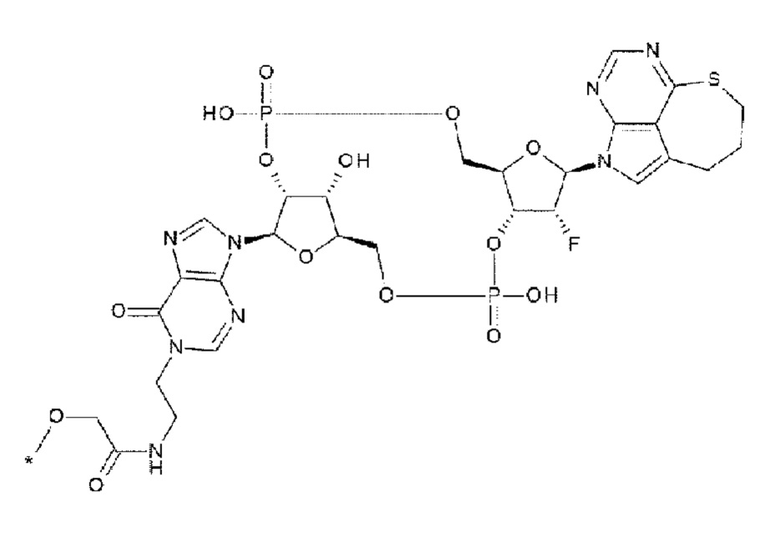
где
каждая звездочка указывает на связывание с L;
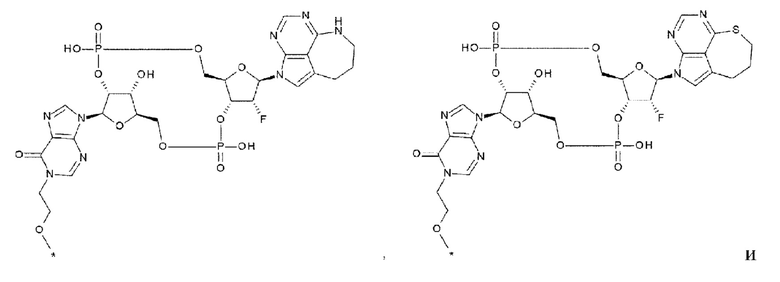
[40] Конъюгат антитела с лекарственным средством согласно любому одному из [33], [34], [36] и [37], где D представлен любой одной из следующих четырех формул:
[0033]
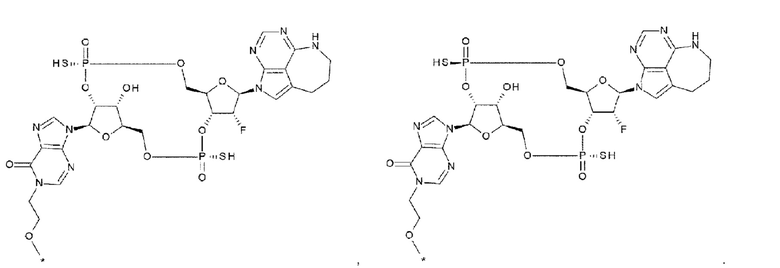
где
каждая звездочка указывает на связывание с L;
[41] Конъюгат антитела с лекарственным средством согласно любому одному из [1]-[40], где линкер L представлен -Lb-La-Lp-Lc-*, где
звездочка указывает на связывание с лекарственным средством D;
Lp представляет собой линкер, состоящий из последовательности аминокислот, расщепляемой в целевой клетке, или отсутствует;
La представляет собой любую одну группу, выбранную из перечисленных далее групп:
-С(=O)-(СН2СН2)n2-С(=O)-,
-С(=O)-(СН2СН2)n2-СН2-С(=O)-,
-C(=O)-(CH2CH2)n2-C(=O)-NH-(CH2CH2)n3-C(=O)-,
-C(=O)-(CH2CH2)n2-C(=O)-NH-(CH2CH2)n3-CH2-C(=O)-,
-C(=O)-(CH2CH2)n2-C(=O)-NH-(CH2CH2O)n3-CH2-C(=O)-,
-(CH2)n4-O-C(=O)-, и
-(CH2)n9-C(=O)-, где
n2 представляет собой целое число от 1 до 3, n3 представляет собой целое число от 1 до 5, п4 представляет собой целое число от 0 до 2 и n9 представляет собой целое число от 2 до 7;
Lb представляет собой спейсер, связывающий La и гликан или ремоделированный гликан Ab, или спейсер, связывающий La и остаток цистеина Ab; и
Lc представляет собой -NH-CH2-, -NH-фенильная группа-СН2-O(С=O)- или -NH-гетероарильная группа-СН2-O(С=O)- или отсутствует;
[42] Конъюгат антитела с лекарственным средством согласно [41], где Lc отсутствует;
[43] Конъюгат антитела с лекарственным средством согласно [41], где Lc представляет собой -NH-CH2-;
[44] Конъюгат антитела с лекарственным средством согласно любому одному из [41]-[43], где Lp представляет собой любой один, выбранный из группы, состоящей из: -GGVA-, -VA-, -GGFG-, -FG-, -GGPI-, -PI-, -GGVCit-, -VCit-, -GGVK-, -VK-, -GGFCit-, -FCit-, -GGFM-, -FM-, -GGLM-, -LM-, -GGICit- и -ICit-;
[45] Конъюгат антитела с лекарственным средством согласно [44], где Lp представляет собой любой один из -GGVA-, -VA-, -GGFG-, -FG-, -GGVCit-, -VCit-, -GGFCit- и -FCit-;
[46] Конъюгат антитела с лекарственным средством согласно любому одному из [41]-[43], где Lp представляет собой любой один из -GGFG-, -GGPI-, -GGVA-, -GGFM-, -GGVCit-, -GGFCit-, -GGICit-, -GGPL-, -GGAQ- и -GGPP-;
[47] Конъюгат антитела с лекарственным средством согласно [46], где Lp представляет собой -GGFG- или -GGPI-;
[48] Конъюгат антитела с лекарственным средством согласно любому одному из [41]-[47], где La представляет собой любой один, выбранный из группы, состоящей из:
-С(=O)-СН2СН2-С(=O)-,
-C(=O)-CH2CH2-C(=O)-NH-(CH2CH2O)3-CH2-C(=O)-,
-C(=O)-CH2CH2-C(=O)-NH-(CH2CH2O)4-CH2-C(=O)- и
-(CH2)5-C(=O);
[49] Конъюгат антитела с лекарственным средством согласно любому одному из [41]-[48], где Lb представлен любой одной из следующих формул:
[0034]
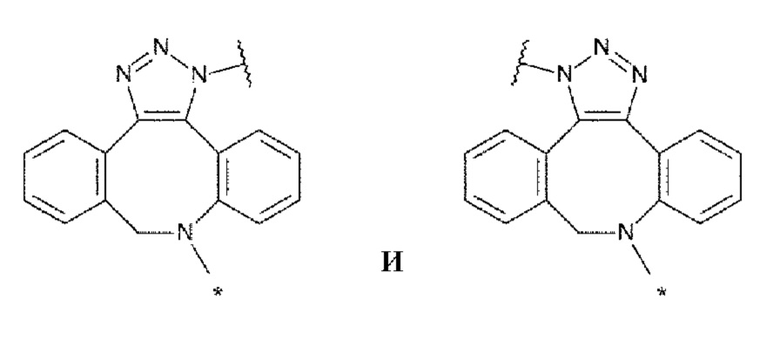
[0035]
, и
[0036]
где, в структурных формулах для Lb, представленных выше,
каждая звездочка указывает на связывание с La, и каждая волнистая линия указывает на связывание с гликаном или ремоделированным гликаном Ab;
[50] Конъюгат антитела с лекарственным средством согласно любому одному из [41]-[48], где Lb представляет собой -(сукпинимид-3-ил-N)-, где -(сукпинимид-3-ил-N)- представляет собой следующую структурную формулу:
[0037]
где звездочка указывает на связывание с La и волнистая линия указывает на связывание с боковой цепью остатка цистеина антитела посредством образования тиоэфира;
[51] Конъюгат антитела с лекарственным средством согласно любому одному из [41] и [46]-[49], где линкер L представлен -Lb-La-Lp-Lc-*, где
звездочка указывает на связывание с лекарственным средством D;
Lp представляет собой -GGFG- или -GGPI-;
La представляет собой -С(=O)-СН2СН2-С(=O)-;
Lb представлен следующей формулой:
[0038]
где, в структурных формулах для Lb, представленных выше,
каждая звездочка указывает на связывание с La, и каждая волнистая линия указывает на связывание с гликаном или ремоделированным гликаном Ab; и
Lc представляет собой -NH-CH2-;
[52] Конъюгат антитела с лекарственным средством согласно любому одному из [1]-[51], где среднее количество конъюгированных молекул лекарственного средства на молекулу антитела в конъюгате антитела с лекарственным средством находится в диапазоне от 1 до 10;
[53] Конъюгат антитела с лекарственным средством согласно [52], где среднее количество конъюгированных молекул лекарственного средства на молекулу антитела в конъюгате антитела с лекарственным средством находится в диапазоне от 1 до 5;
[54] Конъюгат антитела с лекарственным средством согласно любому одному из [1]-[53], где антитело связывается посредством гликана, связанного с Asn297 антитела (N297-гликана), с L;
[55] Конъюгат антитела с лекарственным средством согласно [54], где И297-гликан представляет собой ремоделированный гликан;
[56] Конъюгат антитела с лекарственным средством согласно [54] или [55], где №97-гликан представляет собой N297-(Fuc)MSG1 или N297-(Fuc)SG;
[57] Конъюгат антитела с лекарственным средством согласно любому одному из [1]-[56], где антитело представляет собой антитело против HER2, антитело против HER3, антитело против DLL3, антитело против FAP, антитело против CDH11, антитело против CDH6, антитело против А33, антитело против CanAg, антитело против CD19, антитело против CD20, антитело против CD22, антитело против CD30, антитело против CD33, антитело против CD56, антитело против CD70, антитело против CD98, антитело против TROP2, антитело против СЕА, антитело против Cripto, антитело против EphA2, антитело против G250, антитело против MUC1, антитело против GPNMB, антитело против интегрина, антитело против PSMA, антитело против тенасцина-С, антитело против SLC44A4, антитело против мезотелина, антитело против ENPP3, антитело против CD47, антитело против EGFR, антитело против GPR20 или антитело против DR5;
[58] Конъюгат антитела с лекарственным средством согласно [57], где антитело представляет собой антитело против HER2;
[59] Конъюгат антитела с лекарственным средством согласно [58], где антитело представляет собой антитело, содержащее легкую цепь, состоящую из последовательности аминокислот, представленной в SEQ ID NO: 1, и тяжелую цепь, состоящую из последовательности аминокислот, представленной в SEQ ID NO: 2, или антитело, содержащее легкую цепь, состоящую из последовательности аминокислот, представленной в SEQ ID NO: 1, и тяжелую цепь, состоящую из последовательности аминокислот, представленной в SEQ ID NO: 3;
[60] Конъюгат антитела с лекарственным средством согласно [58], где антитело представляет собой антитело, содержащее легкую цепь, состоящую из последовательности аминокислот, представленной в SEQ ID NO: 28, и тяжелую цепь, состоящую из последовательности аминокислот, представленной в SEQ ID NO: 29, или антитело, содержащее легкую цепь, состоящую из последовательности аминокислот, представленной в SEQ ID NO: 28, и тяжелую цепь, состоящую из последовательности аминокислот, представленной в SEQ ID N0: 30;
[61] Конъюгат антитела с лекарственным средством согласно [57], где антитело представляет собой антитело, содержащее легкую цепь, состоящую из последовательности аминокислот, представленной в SEQ ID NO: 31, и тяжелую цепь, состоящую из последовательности аминокислот, представленной в SEQ ID NO: 32, антитело, содержащее легкую цепь, состоящую из последовательности аминокислот, представленной в SEQ ID NO: 33, и тяжелую цепь, состоящую из последовательности аминокислот, представленной в SEQ ID NO: 34, или антитело, содержащее легкую цепь, состоящую из последовательности аминокислот, представленной в SEQ ID NO: 35, и тяжелую цепь, состоящую из последовательности аминокислот, представленной в SEQ ID NO: 36;
[62] Соединение или фармакологически приемлемая соль соединения, где соединение представлено формулой (Ia):
[0039]
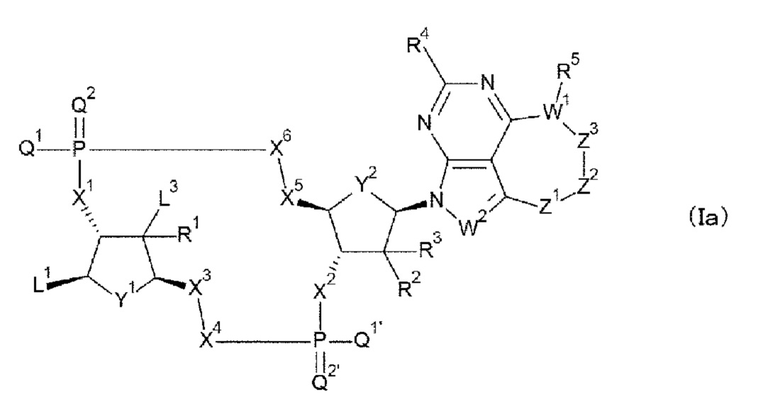
где
L1 представляет собой группу, выбранную из группы, состоящей из следующих формул:
[0040]
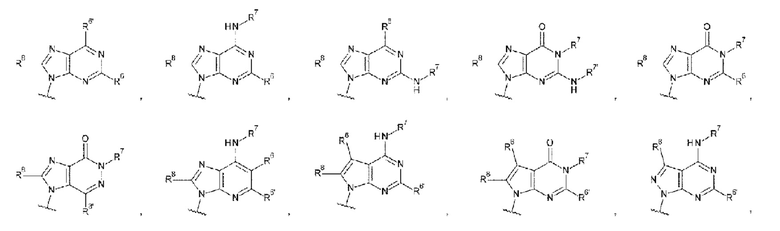
и необязательно содержит в качестве заместителей в любом положении от одной до трех групп, выбранных из группы, состоящей из гидроксигруппы, -NH2, 2-гидроксиацетиламинометильной группы и 2-[(2-гидроксиацетил)амино]этильной группы, где
R6 и R6' каждый независимо представляет собой атом водорода, атом галогена, гидроксигруппу, -NH2, С1-С6 алкильную группу, С2-С6 алкенильную группу или С2-С6 алкинильную группу;
R7 и R7 каждый независимо представляет собой атом водорода или С1-С6 алкильную группу, где С1-С6 алкильная группа необязательно содержит в качестве заместителей один или два заместителя, выбранных из группы, состоящей из атома галогена и оксогруппы;
R8 и R8' каждый независимо представляет собой атом водорода или атом галогена;
Z4 представляет собой -СН-, -NH- или атом кислорода; и
Z5 представляет собой атом азота или -СН=,
L3 представляет собой атом водорода, атом галогена, -NH2, гидрокси-С1-С3 алкильную группу или амино-С1-С3 алкильную группу;
Q1 и Q1' каждый независимо представляет собой гидроксигруппу, тиольную группу или борановую группу (ВН3-);
Q2 и Q2' каждый независимо представляет собой атом кислорода или атом серы;
X1 и X2 каждый независимо представляет собой атом кислорода, атом серы или -СН2-;
Y1 и Y2 каждый представляет собой атом кислорода или -СН2-; X3 и X4 представляют собой группу, выбранную из (iii) и (iv):
(iii) когда Y1 представляет собой атом кислорода, Х3-Х4 представляет собой -СН2-О-, -CH2-S-, -СН2-СН2- или -CH2-CF2-; и
(iv) когда Y1 представляет собой -СН2-, Х3-Х4 представляет собой -O-СН2-; X5 и X6 представляют собой группу, выбранную из (v) и (vi):
(v) когда Y2 представляет собой атом кислорода, Х5-Х6 представляет собой -СН2-О-, -CH2-S-, -СН2-СН2- или -CH2-CF2-; и
(vi) когда Y2 представляет собой -СН2-, Х5-Х6 представляет собой -O-СН2-;
R1, R2 и R3 каждый независимо представляет собой атом водорода, атом галогена, -OR', -OC(=O)R', -N3, -NHR', -NR'R'' или -NHC(=O)R', где R' представляет собой атом водорода, С1-С6 алкильную группу, С2-С6 алкенильную группу, С2-С6 алкинильную группу или С3-С6 циклоалкильную группу, указанная С1-С6 алкильная группа, С2-С6 алкенильная группа или С2-С6 алкинильная группа необязательно содержит в качестве заместителей от одного до шести атомов галогена и R'' представляет собой С1-6 алкильную группу, С2-6 алкенильную группу, С2-6 алкинильную группу или С3-С6 циклоалкильную группу;
W1 представляет собой атом азота, атом кислорода, атом серы или -СН-;
W2 представляет собой атом азота или -СН=;
R4 представляет собой атом водорода, атом галогена или -NH2;
R5 представляет собой группу, выбранную из (vii) - (х):
(vii) где W1 представляет собой атом азота, R5 представляет собой атом водорода, С1-С6 алкильную группу, гидрокси-С1-С6 алкильную группу или амино-С1-С6 алкильную группу;
(viii) где W1 представляет собой атом кислорода, R5 отсутствует;
(ix) где W1 представляет собой атом серы, R5 отсутствует; и
(х) где W1 представляет собой -СН-, R' представляет собой атом водорода, атом галогена, гидроксигруппу, -NH2 или С1-С6 алкильную группу;
Z1-Z2-Z3 вместе представляют собой группу -СН2-СН2-СН2-, -CH2-CH2-R'''-, -СН=СН-СН2-, -СН=СХ-СН2-, -СХ=СН-СН2-, -СХ=СХ-СН2-, -С(=O)-СН2-СН2-, -СН2-СН2-С(=O)-, -СН2-СН(СН3)-СН2- или -СН2-СН2-СН(СНз)-, где R''' представляет собой -О- или -СН2-СН2-и X представляет собой атом галогена или группу, представленную любой одной из следующих формул:
[0041]

где
каждая звездочка указывает на связывание с W1, и каждая волнистая линия указывает на связывание с атомом углерода =С-;
[63] Соединение согласно [62] или фармакологически приемлемая соль указанного соединения, где W1 представляет собой атом азота;
[64] Соединение согласно [63] или фармакологически приемлемая соль указанного соединения, где W1 представляет собой атом азота и R5 представляет собой атом водорода;
[65] Соединение согласно [62] или фармакологически приемлемая соль указанного соединения, где W1 представляет собой атом кислорода;
[66] Соединение согласно [62] или фармакологически приемлемая соль указанного соединения, где W1 представляет собой атом серы;
[67] Соединение согласно [62] или фармакологически приемлемая соль указанного соединения, где W1 представляет собой -СН-;
[68] Соединение согласно [67] или фармакологически приемлемая соль указанного соединения, где W1 представляет собой -СН- и R3 представляет собой атом водорода; [69] Соединение согласно любому одному из [62]-[68] или фармакологически приемлемая соль указанного соединения, где Z1, Z2 и Z3 вместе образуют -СН2-СН2-СН2- или -СН=СН-СН2-;
[70] Соединение согласно любому одному из [62]-[68] или фармакологически приемлемая соль указанного соединения, где Z1, Z2 и Z3 вместе образуют -СН2-СН(СН3)-СН2- или -CH2-СН2-СН(СН3)-;
[71] Соединение согласно любому одному из [62]-[68] или фармакологически приемлемая соль указанного соединения, где Z1, Z2 и Z3 вместе образуют -CH2-CH2-R'''-, где R''' представляет собой -О- или -СН2-СН2-;
[72] Соединение согласно любому одному из [62]-[71] или фармакологически приемлемая соль указанного соединения, где W2 представляет собой -СН-
[73] Соединение согласно любому одному из [62]-[71] или фармакологически приемлемая соль указанного соединения, где W2 представляет собой атом азота;
[74] Соединение согласно любому одному из [62]-[73] или фармакологически приемлемая соль указанного соединения, где R4 представляет собой атом водорода;
[75] Соединение согласно любому одному из [62]-[73] или фармакологически приемлемая соль указанного соединения, где R4 представляет собой атом фтора;
[76] Соединение согласно любому одному из [62]-[75] или фармакологически приемлемая соль указанного соединения, где R8 и R8 в L1 каждый независимо представляет собой атом водорода;
[77] Соединение согласно любому одному из [62]-[76] или фармакологически приемлемая соль указанного соединения, где L1 представляет собой группу, выбранную из группы, состоящей из следующих формул:
[0042]
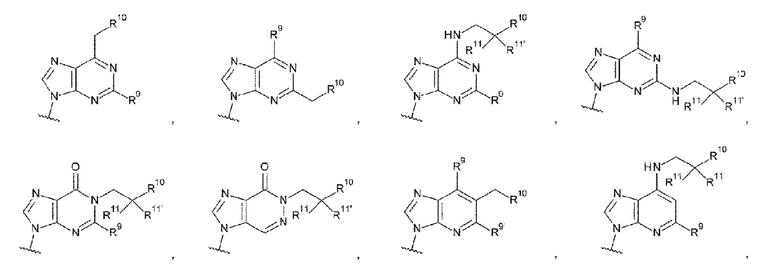
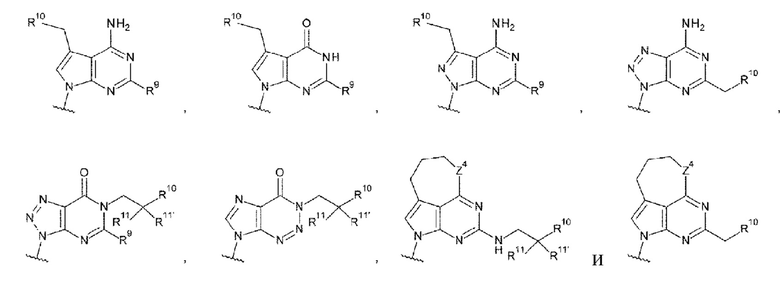
где
R9 и R9' каждый представляет собой атом водорода, атом галогена, гидроксигруппу или -NH2;
R10 представляет собой гидроксигруппу, -NH2, -NHC(=O)CH2OH, CH2NHC(=O)CH2OH, -CH2CH2NHC(=O)CH2OH, гидрокси-С1-С3 алкильную группу или амино-С1-С3 алкильную группу;
R11 и R11' каждый независимо представляет собой атом водорода, атом фтора или метальную группу, или R11 и R11' связаны друг с другом с образованием циклопропана; и
Z4 представляет собой -СН2-, -NH- или атом кислорода;
[78] Соединение согласно любому одному из [62]-[76] или фармакологически приемлемая соль указанного соединения, где L1 представляет собой группу, выбранную из группы, состоящей из следующих формул:
[0043]
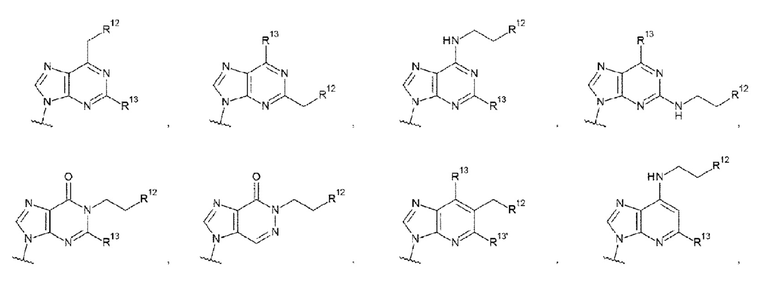
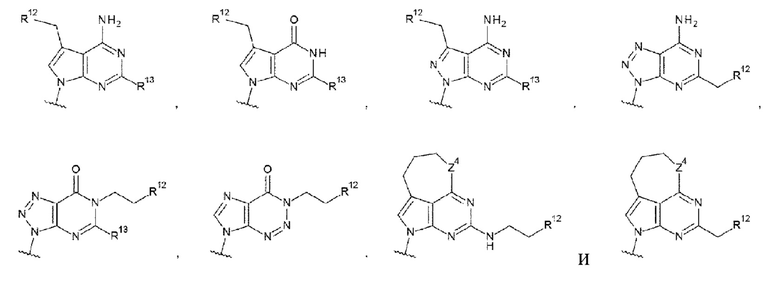
где
R13 и R13' каждый независимо представляет собой атом водорода, гидроксигруппу или -NH2;
R12 представляет собой гидроксигруппу, -NH2, -СН2ОН, -NHC(=O)CH2OH, -CH2NHC(=O)CH2OH или -CH2CH2NHC(=O)CH2OH; и
Z4 такой, как в определении выше;
[79] Соединение согласно любому одному из [62]-[76] или фармакологически приемлемая соль указанного соединения, где L1 представляет собой группу, выбранную из группы, состоящей из следующих формул:
[0044]
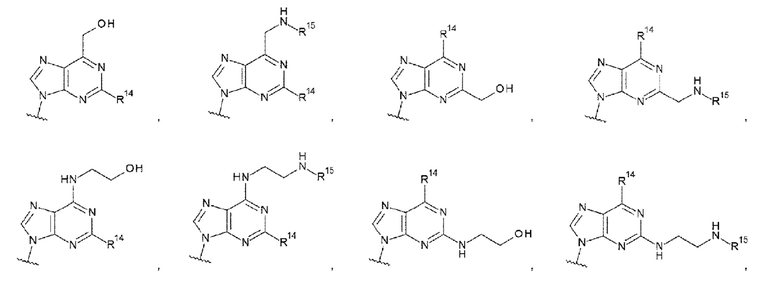
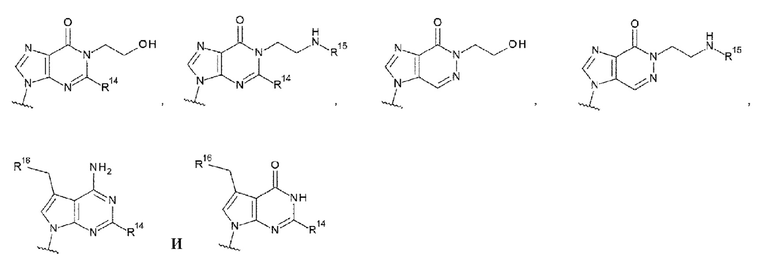
где
R14 представляет собой атом водорода или -NH2;
R15 представляет собой атом водорода или -С(=O)CH2OH; и
R16 представляет собой гидроксигруппу, -NH2, -СН2ОН, -СН2СН2ОН, -CH2NH2 или -CH2CH2NH2;
[80] Соединение согласно любому одному из [62]-[79] или фармакологически приемлемая соль указанного соединения, где L3 представляет собой атом водорода, атом фтора, -NH2, -СН2ОН или -CH2NH2;
[81] Соединение согласно любому одному из [62]-[80] или фармакологически приемлемая соль указанного соединения, где Q1 и Q1' каждый независимо представляет собой гидроксигруппу или тиольную группу;
[82] Соединение согласно любому одному из [62]-[81] или фармакологически приемлемая соль указанного соединения, где X1 и X2' каждый представляет собой атом кислорода;
[83] Соединение согласно любому одному из [62]-[82] или фармакологически приемлемая соль указанного соединения, где Y1 и Y2' каждый представляет собой атом кислорода;
[84] Соединение согласно любому одному из [62]-[83] или фармакологически приемлемая соль указанного соединения, где X3 и X4 представляют собой -СН2-О-;
[85] Соединение согласно любому одному из [62]-[84] или фармакологически приемлемая соль указанного соединения, где X3 и X6 представляют собой -СН2-О-;
[86] Соединение согласно любому одному из [62]-[85] или фармакологически приемлемая соль указанного соединения, где R1, R2 и R3 каждый независимо представляет собой атом водорода, гидроксигруппу или атом фтора;
[87] Соединение согласно любому одному из [62]-[86] или фармакологически приемлемая соль указанного соединения, где соединение представлено любой одной из следующих двух формул:
[0045]
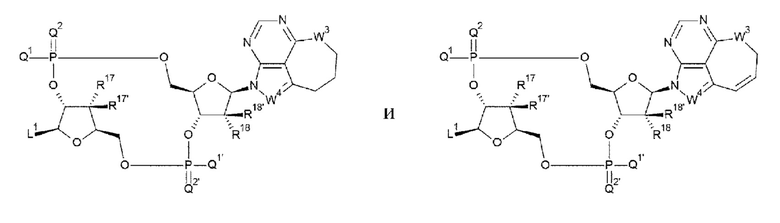
где
L1, Q1, Q1', Q2 и Q2' такие, как в определении выше;
R17, R17', R18 и R18' каждый независимо представляет собой атом водорода, атом галогена, гидроксигруппу или -NH2;
W3 представляет собой -NH-, атом кислорода, атом серы или -СН2-; и
W4 представляет собой -СН= или атом азота;
[88] Соединение согласно [87] или фармакологически приемлемая соль указанного соединения, где соединение представлено любой одной из следующих двух формул:
[0046]
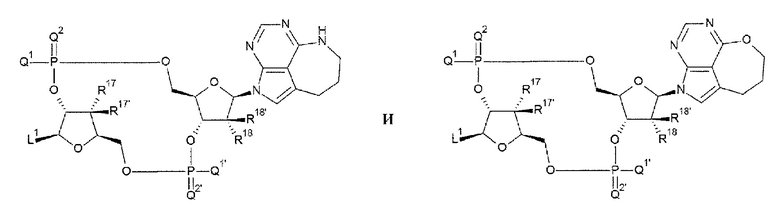
где
L1, Q1, Q1', Q2, Q2', R17, R17', R18 и R18' такие, как в определении выше;
[89] Соединение согласно [87] или [88] или фармакологически приемлемая соль указанного соединения, где соединение представлено любой одной из следующих восьми формул:
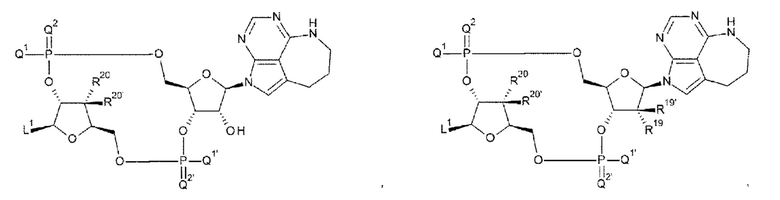
[0047]
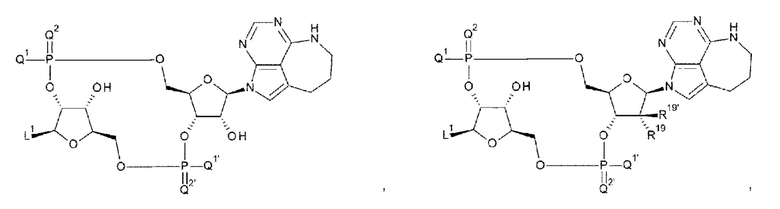
где
L1, Q1, Q1', Q2, и Q2' такие, как в определении выше; и
R19, R19', R20 и R20' каждый независимо представляет собой атом водорода или атом фтора;
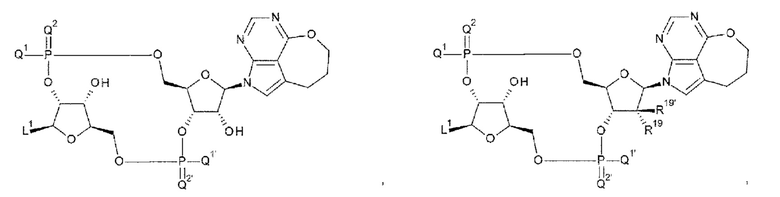
[90] Соединение согласно любому одному из [87]-[89] или фармакологически приемлемая соль указанного соединения, где соединение представлено любой одной из следующих четырех формул:
[0048]
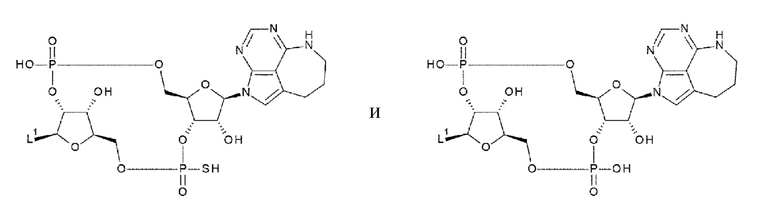
где
L1 такой, как в определении выше;
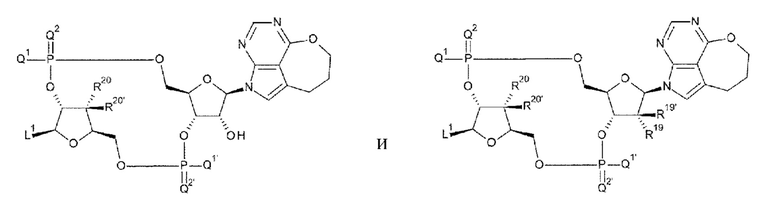
[91] Соединение согласно любому одному из [87]-[90] или фармакологически приемлемая соль указанного соединения, где соединение представлено любой одной из следующих четырех формул:
[0049]
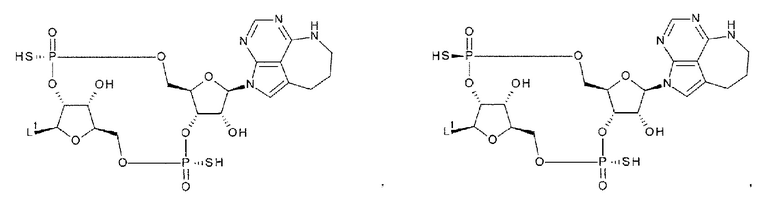
где
L1 такой, как в определении выше;
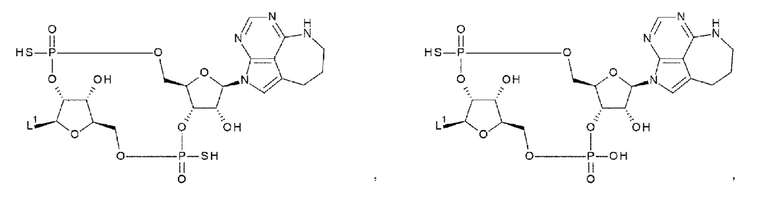
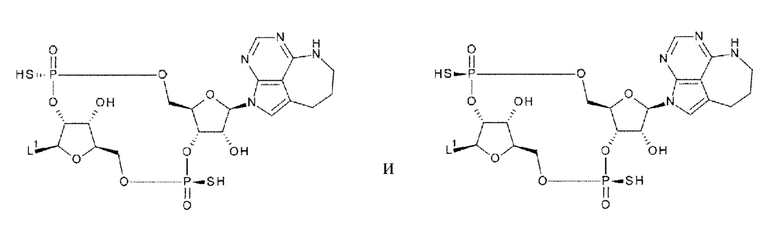
[92] Соединение согласно любому одному из [87]-[90] или фармакологически приемлемая соль указанного соединения, где соединение представлено любой одной из следующих четырех формул:
[0050]
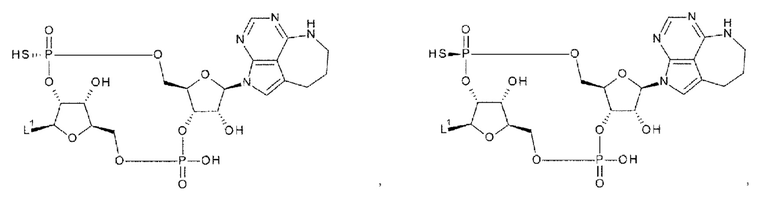
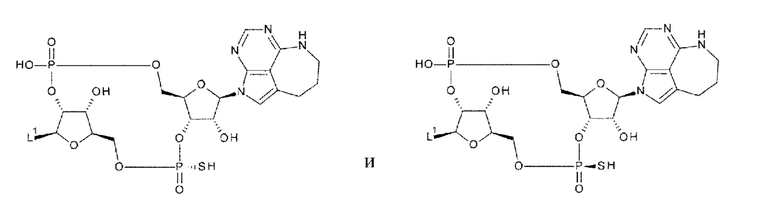
где
L1 такой, как в определении выше;
[93] Соединение согласно любому одному из [62]-[86] или фармакологически приемлемая соль указанного соединения, где соединение представлено следующей формулой:
[0051]
где
L1 такой, как в определении выше;
Q3 и Q3' каждый независимо представляет собой гидроксигруппу или тиольную группу;
R21 и R22 каждый независимо представляет собой гидроксигруппу или атом фтора; и
W3 представляет собой -NH- или атом серы;
[94] Соединение согласно
[93] или фармакологически приемлемая соль указанного соединения, где соединение представлено любой одной из следующих двух формул:
[0052]
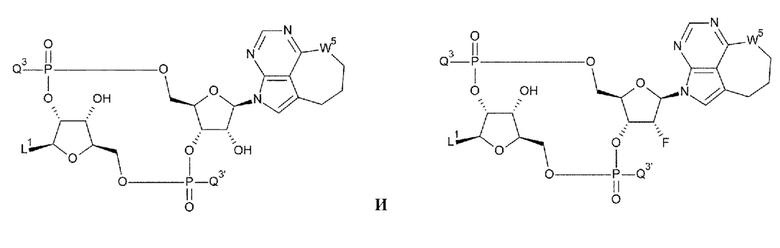
где
L1, Q3, Q3 и W3 такие, как в определении выше;
[95] Соединение согласно любому одному из [62]-[94] или фармакологически приемлемая соль указанного соединения, где L1 представлен любой одной из следующих формул:
[0053]
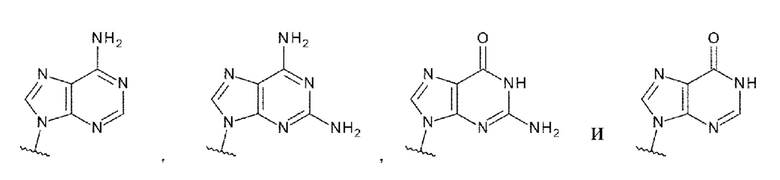
[96] Соединение согласно любому одному из [62]-[94] или фармакологически приемлемая соль указанного соединения, где L1 представлен любой одной из следующих четырех формул:
[0054]
[97] Соединение согласно любому одному из [93], [94] и [96] или фармакологически приемлемая соль указанного соединения, где D представлен любой одной из следующих четырех формул:
[0055]
где
Q3, Q3' и W5 такие, как в определении выше;
[98] Соединение согласно любому одному из [93], [94], [96] и [97] или фармакологически приемлемая соль указанного соединения, где D представлен любой одной из следующих четырех формул:
[0056]

[99] Соединение согласно любому одному из [93], [94], [96] и [97] или фармакологически приемлемая соль указанного соединения, где D представлен любой одной из следующих трех формул:
[0057]
[100] Соединение согласно любому одному из [93], [94], [96] и [97] или фармакологически приемлемая соль указанного соединения, где D представлен любой одной из следующих четырех формул:
[0058]
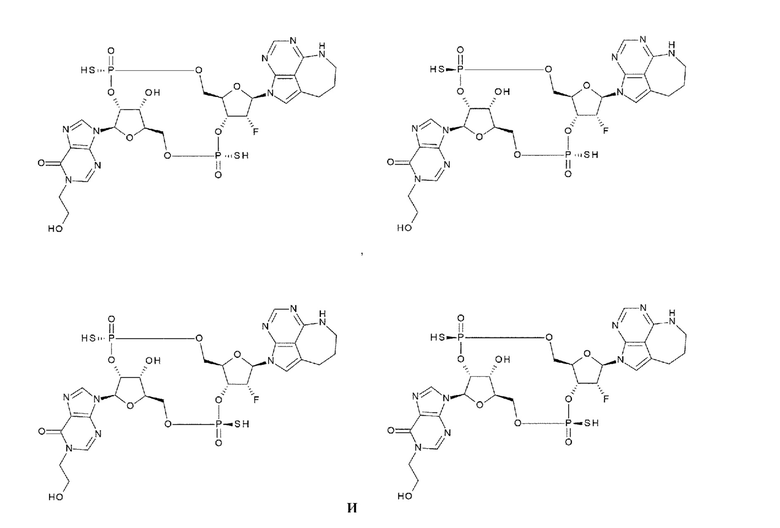
[101] Агонист STTNG, включающий любой один, выбранный из группы, состоящей из конъюгата антитела с лекарственным средством согласно любому одному из [1]-[61] и соединения согласно любому одному из [62]-[100] или фармакологически приемлемой соли указанного соединения;
[102] Фармацевтическая композиция, содержащая любой один, выбранный из группы, состоящей из конъюгата антитела с лекарственным средством согласно любому одному из [1]-[61] и соединения согласно любому одному из [62]-[100] или фармакологически приемлемой соли указанного соединения;
[103] Противоопухолевый агент, включающий любой один, выбранный из группы, состоящей из конъюгата антитела с лекарственным средством согласно любому одному из [1]-[61] и соединения согласно любому одному из [62]-[100] или фармакологически приемлемой соли указанного соединения;
[104] Противоопухолевый агент согласно [103], где опухоль представляет собой рак легких, рак почки, уротелиальный рак, колоректальный рак, рак предстательной железы, мультиформную глиобластому, рак яичников, рак поджелудочной железы, рак молочной железы, меланому, рак печени, рак мочевого пузыря, рак желудка, рак пищевода, рак эндометрия, рак яичка, рак шейки матки, плацентарную хориокарциному, мультиформную глиобластому, опухоль головного мозга, рак головы и шеи, рак щитовидной железы, мезотелиому, стромальную опухоль желудочно-кишечного тракта (GIST), рак желчного пузыря, рак желчного протока, рак надпочечников, плоскоклеточную карциному, лейкоз, злокачественную лимфому, плазмоцитому, миелому или саркому;
[105] Способ лечения рака, указанный способ включает введение любого одного, выбранного из группы, состоящей из конъюгата антитела с лекарственным средством согласно любому одному из [1]-[61], соединения согласно любому одному из [62]-[100] или фармакологически приемлемой соли указанного соединения, агониста STING согласно [101], фармацевтической композиции согласно [102] и противоопухолевого агента согласно [103] или [104];
[106] Способ согласно [105], где рак представляет собой рак легких, рак почки, уротелиальный рак, колоректальный рак, рак предстательной железы, мультиформную глиобластому, рак яичников, рак поджелудочной железы, рак молочной железы, меланому, рак печени, рак мочевого пузыря, рак желудка, рак пищевода, рак эндометрия, рак яичка, рак шейки матки, плацентарную хориокарциному, мультиформную глиобластому, опухоль головного мозга, рак головы и шеи, рак щитовидной железы, мезотелиому, стромальную опухоль желудочно-кишечного тракта (GIST), рак желчного пузыря, рак желчного протока, рак надпочечников, плоскоклеточную карциному, лейкоз, злокачественную лимфому, плазмоцитому, миелому или саркому.
Полезный эффект изобретения.
[0059]
В соответствии с настоящим изобретением предложены новые производные ЦДН. Новые производные ЦДН согласно настоящему изобретению обладают эффективной активностью агонистов STING и проявляют сильную противоопухолевую активность. Кроме того, в соответствии с настоящим изобретением предложены новые конъюгаты антитело-производное ЦДН, которые позволяют системное введение и оказывают противоопухолевое действие на опухоли, экспрессирующие антиген.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
[0060]
[Фигура 1] На фигуре 1 схематически показана форма конъюгата с лекарственным средством, полученная из ремоделированного гликаном SG-типа антитела (молекула (II) на А фигуры 1), и форма конъюгата с лекарственным средством, полученная из ремоделированного гликаном MSG-типа антитела (молекула (II) на В фигуры 1), каждая из которых является формой конъюгата с лекарственным средством согласно настоящему изобретению (молекулой (II)). (а) обозначает лекарственное средство D, (b) обозначает линкер L, (с) обозначает линкер PEG (L(PEG)) и (d) обозначает N297-гликан, где каждый незаштрихованный круг представляет собой NeuAc(Sia), каждый незаштрихованный шестиугольник представляет Man, каждый заштрихованный шестиугольник представляет собой GlcNAc, каждый незаштрихованный ромб представляет Gal и каждый незаштрихованный перевернутый треугольник представляет собой Fuc. Каждый незаштрихованный пятиугольник представляет собой триазольное кольцо, образованное путем реакции между алкином, происходящим из линкера L, и азидной группой, происходящей из линкера PEG. Каждая форма Y представляет собой антитело Ab. Каждый линкер PEG связывается с карбоксильной группой в положении 2 сиаловой кислоты, расположенной на невосстанавливающем конце, посредством амидной связи. Если не указано иное, такой способ иллюстрирования используется повсюду в настоящем описании.
[Фигура 2] На фигуре 2 представлены схематические диаграммы, иллюстрирующие структуры (Fucα1,6)GlcNAc-антитела (молекула (III) на А фигуры 2), ремоделированного гликаном SG-типа антитела (молекула (IV) на В фигуры 2) и ремоделированного гликаном MSG-типа антитела (молекула (IV) на С фигуры 2), каждая из которых представляет собой интермедиат получения формы конъюгата с лекарственным средством согласно настоящему изобретению. На каждой из диаграмм форма Y представляет собой антитело Ab, как на фигуре 1. (е) на А фигуры 2 обозначает N297-гликан, состоящий из дисахарида, в котором положение 1 Fuc и положение 6 GlcNAc связаны посредством α-гликозидной связи. На В и С фигуры 2 (d) обозначает N297-гликан, как на фигуре 1, и (f) обозначает линкер PEG, содержащий азидную группу, где азидная группа, которую подвергнут связыванию с линкером L, показана на конце. Способ связывания каждого линкера PEG, содержащего азидную группу, такой же, как у линкеров PEG на фигуре 1.
[Фигура 3] На фигуре 3 представлены схематические диаграммы, иллюстрирующие этапы получения ремоделированного гликаном SG-типа антитела и ремоделированного гликаном MSG-типа антитела из антитела, полученного в клетках животного. Как показано на фигуре 2, молекулы (III) и (IV) на диаграммах представляют собой (Fucα1,6)GlcNAc-антитело и ремоделированное гликаном SG-типа антитело или ремоделированное гликаном MSG-типа антитело, соответственно. Молекула (V) представляет собой антитело, полученное в клетках животного, и смесь молекул с гетерогенными №97-гликанами. На фигуре 3А показан этап получения гомогенных молекул (Fucα1,6)GlcNAc-антитела (III) путем обработки гетерогенных N297-гликанов (V) гидролазой, такой как EndoS. На фигуре 3В показан этап получения ремоделированного гликаном SG-типа антитела (IV) путем осуществления трансгликозилирования GlcNAc N297-гликана в антителе (III) донорными молекулами гликана SG-типа с применением гликозилтрансферазы, такой как мутантная D233Q/Q303L EndoS. На фигуре 3С показан этап получения ремоделированного гликаном MSG-типа антитела (IV) путем осуществления трансгликозилирования антитела (III) донорными молекулами гликана MSG-типа таким же образом, как на фигуре 3В. Каждая из донорной молекулы гликана SG-типа и донорной молекулы гликана MSG-типа, предназначенных для применения здесь, представляет собой такую молекулу, где сиаловая кислота на ее невосстанавливающем конце модифицирована линкером PEG, содержащим азидную группу, и сиаловая кислота на каждом невосстанавливающем конце ремоделированного N297-гликаном SG-типа антитела или ремоделированного N297-гликаном MSG-типа антитела, которые необходимо получить, модифицирована таким же образом, как показано на фигурах 2В и 2С.
[Фигура 4] На фигуре 4 представлены последовательности аминокислот легкой цепи (SEQ ID NO: 1) и тяжелой цепи (SEQ ID NO: 2) трастузумаба.
[Фигура 5] На фигуре 5 представлены последовательности аминокислот легкой цепи (SEQ ID NO: 1) и тяжелой цепи (SEQ ID NO: 3) модифицированного антитела против HER2.
[Фигура 6] На фигуре 6 представлены последовательности аминокислот (a) STING человека дикого типа, (b) REF-мутированного (R232H) STING человека и (с) HAQ-мутированного (R71H, G230A, R293Q) STING человека.
[Фигура 7] На фигуре 7 продемонстрировано противоопухолевое действие внутриопухолевого введения производных ЦДН. На каждом графике линия с заштрихованными квадратами соответствует группе, которой вводили среду, линия с незаштрихованными квадратами соответствует группе, которой вводили соединение №6а, линия с незаштрихованными перевернутыми треугольниками соответствует группе, которой вводили соединение №8b, и линия с незаштрихованными кругами соответствует группе, которой вводили соединение №9b. На вертикальной оси представлен объем опухоли (мм3), а на горизонтальной оси представлены сутки после трансплантации опухоли.
[Фигура 8] На фигуре 8 продемонстрировано противоопухолевое действие внутривенного введения конъюгата (1) антитела против HER2-ЦДН и конъюгата антитела против LPS-ЦДН (1). На каждом графике линия с заштрихованными квадратами соответствует группе, которой вводили среду, линия с незаштрихованными треугольниками соответствует группе, которой вводили конъюгат (1) антитела против HER2-ЦДН, который был получен путем конъюгирования соединения из примера 8b с модифицированным антителом против HER2, полученным в ссылочном примере 1, и линия с заштрихованными треугольниками соответствует группе, которой вводили конъюгат (1) антитела против LPS-ЦДН, который был получен аналогично путем конъюгирования соединения из примера 8b с модифицированным антителом против LPS, полученным в ссылочном примере 2. На вертикальной оси представлен объем опухоли (мм3), а на горизонтальной оси представлены сутки после трансплантации опухоли.
[Фигура 9] На фигуре 9 продемонстрировано противоопухолевое действие внутривенного введения конъюгатов (2) и (3) антитела против HER2-ЦДН. На диаграмме линия с заштрихованными квадратами соответствует группе, которой вводили среду, линия с незаштрихованными квадратами соответствует группе, которой вводили конъюгат (2) антитела против HER2-ЦДН, и линия с незаштрихованными треугольниками соответствует группе, которой вводили конъюгат (3) антитела против HER2-ЦДН. На вертикальной оси представлен объем опухоли (мм3), а на горизонтальной оси представлены сутки после трансплантации опухоли.
[Фигура 10] На фигуре 10 продемонстрировано противоопухолевое действие внутривенного введения конъюгата (19) антитела против HER2-ЦДН. На диаграмме линия с заштрихованными квадратами соответствует группе, которой вводили среду, и линия с незаштрихованными треугольниками соответствует группе, которой вводили конъюгат (19) антитела против HER2-ЦДН. В конъюгате (19) антитела против HER2-ЦДН лекарственное средство-линкер конъюгировано с антителом путем конъюгирования через цистеин. На вертикальной оси представлен объем опухоли (мм3), а на горизонтальной оси представлены сутки после трансплантации опухоли.
[Фигура 11] На фигуре 11 продемонстрировано противоопухолевое действие внутривенного введения конъюгатов (1) и (9)-(12) антитела против HER2-ЦДН. На диаграмме линия с заштрихованными квадратами соответствует группе, которой вводили среду, линия с незаштрихованными треугольниками соответствует группе, которой вводили конъюгат (9) антитела против HER2-ЦДН, линия с незаштрихованными перевернутыми треугольниками соответствует группе, которой вводили конъюгат (10) антитела против HER2-ЦДН, линия с незаштрихованными ромбами соответствует группе, которой вводили конъюгат (11) антитела против HER2-ЦДН, линия с незаштрихованными кругами соответствует группе, которой вводили конъюгат антитела против HER2-ЦДН (12), и линия с незаштрихованными квадратами соответствует группе, которой вводили конъюгат (1) антитела против HER2-ЦДН. В каждом из конъюгатов (9), (10), (11), (12) и (1) антитела против HER2-ЦДН соединение из примера 8b конъюгировано посредством линкера, где линкеры отличаются друг от друга. На вертикальной оси представлен объем опухоли (мм3), а на горизонтальной оси представлены сутки после трансплантации опухоли.
[Фигура 12] На фигуре 12 продемонстрировано противоопухолевое действие внутривенного введения конъюгата (1) антитела 2 против HER2-ЦДН, антитела 2 против HER2 и соединения №8b. На диаграмме линия с заштрихованными квадратами соответствует группе, которой вводили среду, линия с незаштрихованными треугольниками соответствует группе, которой вводили 60 мкг конъюгата (1) антитела 2 против HER2-ЦДН, линия с незаштрихованными перевернутыми треугольниками соответствует группе, которой вводили 59 мкг антитела 2 против HER2, и линия с незаштрихованными кругами соответствует группе, которой вводили 1,2 мкг соединения №8b. Каждая доза антитела 2 против HER2 и соединения №8b эквивалентна соответствующему компоненту, составляющему конъюгат (1) антитела 2 против HER2-ЦДН. На вертикальной оси представлен объем опухоли (мм3), а на горизонтальной оси представлены сутки после трансплантации опухоли.
[Фигура 13] На фигуре 13 (а) продемонстрировано противоопухолевое действие внутривенного введения конъюгатов (2) и (3) антитела 2 против HER2-ЦДН. На фигуре 13 (b) продемонстрировано противоопухолевое действие внутривенного введения конъюгатов (4), (5), (7) и (8) антитела 2 против HER2-ЦДН. На фигуре 13 (с) продемонстрировано противоопухолевое действие внутривенного введения конъюгата (6) антитела 2 против HER2-ЦДН. На каждом графике линия с заштрихованными квадратами соответствует группе, которой вводили среду, и каждая линия с незаштрихованными символами соответствует группе, в которой оцениваемым субъектам вводили конъюгаты (2)-(8) антитела 2 против HER2-ЦДН. На вертикальной оси представлен объем опухоли (мм3), а на горизонтальной оси представлены сутки после трансплантации опухоли.
[Фигура 14] На фигуре 14 продемонстрировано противоопухолевое действие внутривенного введения конъюгатов (9) и (10) антитела 2 против HER2-ЦДН. На диаграмме линия с заштрихованными квадратами соответствует группе, которой вводили среду, линия с незаштрихованными треугольниками соответствует группе, которой вводили конъюгат (9) антитела 2 против HER2-ЦДН, и линия с незаштрихованными кругами соответствует группе, которой вводили конъюгат (10) антитела 2 против HER2-ЦДН. Конъюгаты (9) и (10) антитела 2 против HER.2-ЦДН представляют собой конъюгаты антитела-ЦДН, в которых используется ремоделированное гликаном MSG-типа антитело со средним количеством конъюгированных молекул лекарственного средства, приблизительно составляющим 2. На вертикальной оси представлен объем опухоли (мм3), а на горизонтальной оси представлены сутки после трансплантации опухоли.
[Фигура 15] На фигуре 15 продемонстрировано противоопухолевое действие внутривенного введения антитела против EphA2 и конъюгата (1) антитела против EphA2-ЦДН. На диаграмме линия с заштрихованными квадратами соответствует группе, которой вводили среду, линия с незаштрихованными кругами соответствует группе, которой вводили антитело против EphA2, и линия с незаштрихованными треугольниками соответствует группе, которой вводили конъюгат (1) антитела против EhpA2-ЦДН. На вертикальной оси представлен объем опухоли (мм3), а на горизонтальной оси представлены сутки после трансплантации опухоли.
[Фигура 16] На фигуре 16 продемонстрировано противоопухолевое действие внутривенного введения антитела против CD33 и конъюгата (1) антитела против CD33-ЦДН. На диаграмме линия с заштрихованными квадратами соответствует группе, которой вводили среду, линия с незаштрихованными кругами соответствует группе, которой вводили антитело против CD33, и линия с незаштрихованными треугольниками соответствует группе, которой вводили конъюгат (1) антитела против CD33-ЦДН. На вертикальной оси представлен объем опухоли (мм3), а на горизонтальной оси представлены сутки после трансплантации опухоли.
[Фигура 17] На фигуре 17 представлены последовательности аминокислот легкой цепи (SEQ ID NO: 28) и тяжелой цепи (SEQ ID NO: 29) пертузумаба.
[Фигура 18] На фигуре 18 представлены последовательности аминокислот легкой цепи (SEQ ID NO: 28) и тяжелой цепи (SEQ ID NO: 30) модифицированного антитела 2 против HER2.
[Фигура 19] На фигуре 19 представлены последовательности аминокислот легкой цепи (SEQ ID NO: 31) и тяжелой цепи (SEQ ID NO: 32) антитела против CD33.
[Фигура 20] На фигуре 20 представлены последовательности аминокислот легкой цепи (SEQ ID NO: 33) и тяжелой цепи (SEQ ID NO: 34) антитела против EphA2.
[Фигура 21] На фигуре 21 представлены последовательности аминокислот легкой цепи (SEQ ID NO: 35) и тяжелой цепи (SEQ ID NO: 36) антитела против CDH6.
[Фигура 22] На фигуре 22 продемонстрировано противоопухолевое действие внутривенного введения конъюгатов (11) и (12) антитела 2 против HER2-ЦДН. На диаграмме линия с заштрихованными квадратами соответствует группе, которой вводили среду, линия с незаштрихованными треугольниками соответствует группе, которой вводили конъюгат (11) антитела 2 против HER2-ЦДН, и линия с незаштрихованными кругами соответствует группе, которой вводили конъюгат (12) антитела 2 против HER2-ЦДН.
ОПИСАНИЕ ВАРИАНТОВ РЕАЛИЗАЦИИ
[0061]
Согласно настоящему изобретению предложены новые производные ЦДН, обладающие активностью агонистов STING, и их конъюгаты антитело-лекарственное средство, и применение любых из них. Новые производные ЦДН согласно настоящему изобретению обладают активностью агонистов STING и активируют иммунные клетки, чтобы вызвать продукцию интерферонов и цитокинов. Новые производные ЦДН согласно настоящему изобретению оказывают противоопухолевое действие посредством активации иммунных клеток. Новые производные ЦДН можно вводить непосредственно в целевую ткань, иммунные функции которой предполагается активировать, или соединить с антителом, способным распознавать и связывать целевые клетки (например, опухолевые клетки или иммунные клетки), посредством любого линкера и вводить системно.
[0062]
STING (стимулятор генов интерферона) представляет собой трансмембранный адапторный белок, локализованный в эндоплазматическом ретикулуме. Известно, что у STING с высокой частотой проявляется врожденный полиморфизм (PLoS One, 21 октября 2013 г., 8(10), е77846). Известны как мутированные формы STING, например, мутация R232H, которая представляет собой мутацию аминокислоты в положении 232 аргинина (R) на гистидин (Н), и мутация HAQ, которая представляет собой мутацию аргинина (R) в положении 71 на гистидин (Н), глицина (G) в положении 230 на аланин (А) и аргинина (R) в положении 293 на глутамин (Q). Известно, что такие полиморфы STING являются причиной различия в интенсивности ответа, вызванного стимуляцией агонистом STING, такого как уровни продукции цитокинов (Genes and Immunity, 2011, 12, 263-269).
Следовательно, проявление активности по отношению к различным типам STING желательно для стабильного действия агониста STING у людей.
[0063]
В данной заявке «рак», «карцинома» и «опухоль» имеют одинаковые значения.
[0064]
В настоящем изобретении «активность активации иммунитета» относится к обуславливанию в некоторой форме активации иммунных клеток, вовлеченных в противоопухолевый иммунитет, таких как моноциты, макрофаги, дендритные клетки, Т-клетки, В-клетки, NK-клетки и нейтрофилы, например, к обуславливанию любых структурных или функциональных изменений в иммунных клетках, включая продукцию цитокинов и хемокинов, повышенную экспрессию иммуностимулирующих маркеров, пониженную экспрессию иммуносупрессивных маркеров, изменение системы внутриклеточной передачи сигналов, такое как фосфорилирование, и изменение экспрессии генов. В объем значения данного термина также входит явление, состоящее в том, что опухолевые клетки вызывают изменение с индукцией противоопухолевого иммунитета, такое как индукция продукции цитокинов и хемокинов, которые индуцируют активацию или миграцию иммунных клеток, повышение чувствительности к иммунным клеткам и т.д.
[0065]
В настоящем изобретении «противоопухолевое действие» относится к индукции уменьшения или регрессии опухоли путем непосредственного или опосредованного влияния лекарственного средства на опухолевые клетки. Противоопухолевым действием называют, например, обуславливание уменьшения количества опухолевых клеток, повреждения опухолевых клеток или регрессии опухоли, например, посредством явления, состоящего в том, что лекарственное средство непосредственно вызывает повреждение опухолевых клеток, что опухолевые клетки активируют противоопухолевый иммунитет посредством стимуляции лекарственным средством, или что лекарственное средство, доставленное в опухолевую клетку, например, высвобождается из клетки и активирует противоопухолевый иммунитет вблизи опухолевой клетки.
[0066]
В настоящем изобретении «цитотоксическая активность» относится к обуславливанию патологического изменения в некоторой форме в клетках, в частности, к обуславливанию, не только непосредственных повреждений, но также любых структурных или функциональных повреждений в клетках, таких как расщепление ДНК, образование нуклеотидных димеров, расщепление хромосом, повреждение митотического аппарата и сниженная активность ферментов.
[0067]
В настоящем изобретении «клетки» включают клетки в животных и культивированные клетки.
[0068]
В данной заявке «атом галогена» относится к атому фтора, атому хлора, атому брома или атому йода.
[0069]
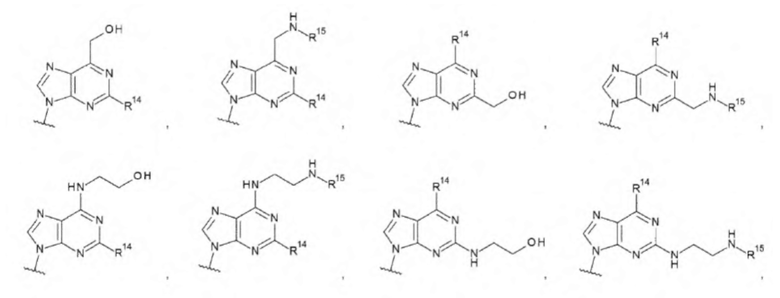
В данной заявке «С1-С6 алкильная группа» относится к линейной или разветвленной алкильной группе, содержащей от одного до шести атомов углерода. «С1-С6 алкильная группа» может содержать циклопропан на алкильной группе, если общее количество атомов углерода не превышает шести. Примеры «С1-С6 алкильной группы» могут включать, но не ограничены следующими структурами:
[0070]
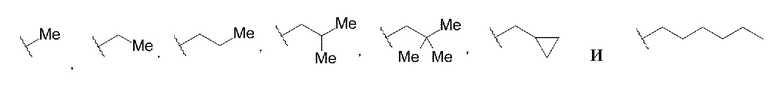
где каждая волнистая линия указывает на положение замены.
[0071]
В данной заявке «С2-С6 алкенильная группа» относится к линейной или разветвленной алкенильной группе, содержащей от двух до шести атомов углерода.
[0072]
В данной заявке «С2-С6 алкинильная группа» относится к линейной или разветвленной алкинильной группе, содержащей от двух до шести атомов углерода.
[0073]
В данной заявке «С3-С6 циклоалкильная группа» относится к насыщенной циклической углеводородной группе, содержащей от трех до шести атомов углерода. «С3-С6 циклоалкильная группа» может содержать в качестве заместителей множество алкильных групп, если общее количество атомов углерода не превышает шести. Примеры «С3-С6 циклоалкильной группы» могут включать, но не ограничены следующими структурами:
[0074]
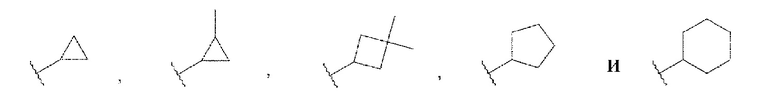
где каждая волнистая линия указывает на положение замены.
[0075]
В данной заявке «гидрокси-С1-С6 алкильная группа» относится к алкильной группе, в которой линейная или разветвленная алкильная группа, содержащая от одного до шести атомов углерода, содержит в качестве заместителей одну или две гидроксигруппы в любом положении. «Гидрокси-С1-С6 алкильная группа» может содержать циклопропан на алкильной группе, если общее количество атомов углерода не превышает шести. Примеры «гидрокси-С1-С6 алкильной группы» могут включать, но не ограничены следующими структурами:
[0076]
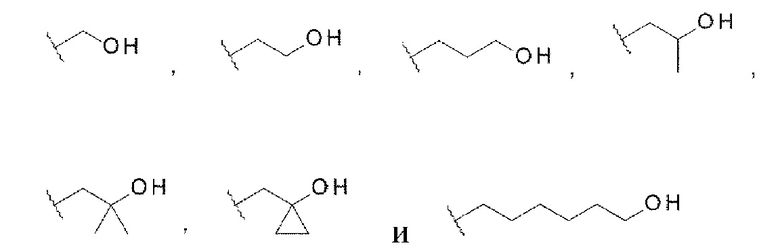
где каждая волнистая линия указывает на положение замены.
[0077]
В настоящем изобретении «амино-С1-С6 алкильная группа» относится к алкильной группе, в которой линейная или разветвленная алкильная группа, содержащая от одного до шести атомов углерода, содержит в качестве заместителей одну или две аминогруппы в любом положении. «Амино-С1-С6 алкильная группа» может содержать циклопропан на алкильной группе, если общее количество атомов углерода не превышает шести. Примеры «амино-С1-С6 алкильной группы» могут включать, но не ограничены следующими структурами:
[0078]
где каждая волнистая линия указывает на положение замены.
[0079]
<1. Новое производное ЦДН>
Новое производное ЦДН согласно настоящему изобретению имеет структуру, представленную формулой (Ia):
[0080]
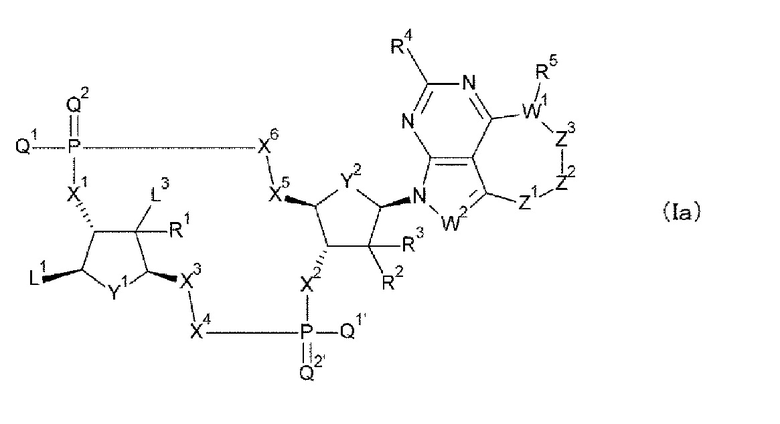
[0081]
L1 представляет собой группу, выбранную из группы, состоящей из следующих формул:
[0082]
и необязательно содержащую в качестве заместителей в любом положении от одной до трех групп, выбранных из группы, состоящей из гидроксигруппы, -NH2, 2-гидроксиацетиламинометильной группы и 2-[(2-гидроксиацетил)амино]этильной группы, где
R6 и R6' каждый независимо представляет собой атом водорода, атом галогена, гидроксигруппу, -NH2, С1-С6 алкильную группу, С2-С6 алкенильную группу или С2-С6 алкинильную группу;
R7 и R7' каждый независимо представляет собой атом водорода или С1-С6 алкильную группу, где С1-С6 алкильная группа необязательно содержит в качестве заместителей один или два заместителя, выбранных из группы, состоящей из атома галогена и оксогруппы;
R8 и R8' каждый независимо представляет собой атом водорода или атом галогена;
Z4 представляет собой -СН2-, -NH- или атом кислорода; и
Z5 представляет собой атом азота или -СН=.
[0083]
L1 представляет собой группу, выбранную из группы, состоящей из следующих формул:
[0084]
и необязательно содержащую в качестве заместителей в любом положении от одной до трех групп, выбранных из группы, состоящей из гидроксигруппы, -NH2, 2-гидроксиацетиламинометильной группы и 2-[(2-гидроксиацетил)амино]этильной группы, где
R6 и R6' каждый независимо представляет собой атом водорода, атом галогена, гидроксигруппу, -NH2, С1-С6 алкильную группу, С2-С6 алкенильную группу или С2-С6 алкинильную группу;
R7 и R7' каждый независимо представляет собой атом водорода или С1-С6 алкильную группу, где С1-С6 алкильная группа необязательно содержит в качестве заместителей один или два заместителя, выбранных из группы, состоящей из атома галогена и оксогруппы;
R8 и R8' каждый независимо представляет собой атом водорода или атом галогена;
Z4 представляет собой -СН2-, -NH- или атом кислорода; и
Z5 представляет собой атом азота или -СН=.
[0085]
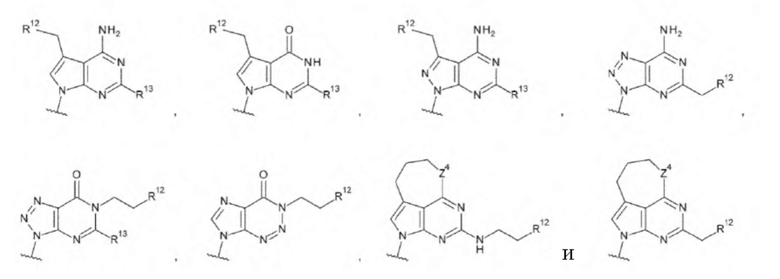
L1 предпочтительно представляет собой группу, выбранную из группы, состоящей из следующих формул:
[0086]
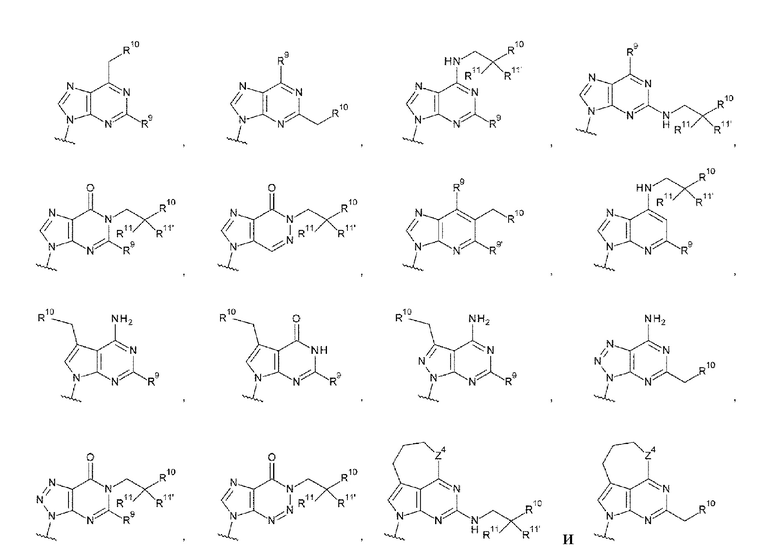
где
R9 и R9' каждый представляет собой атом водорода, атом галогена, гидроксигруппу или -NH2;
R10 представляет собой гидроксигруппу, -NH2, -NHC(=O)CH2OH, CH2NHC(=O)CH2OH, -CH2CH2NHC(=O)CH2OH, гидрокси-С1-С3 алкильную группу или амино-С1-С3 алкильную группу;
R11 и R11' каждый независимо представляет собой атом водорода, атом фтора или метальную группу, или R11 и R11' связаны друг с другом с образованием циклопропана; и
Z4 представляет собой -СН2-, -NH- или атом кислорода.
[0087]
L1 предпочтительно представляет собой группу, выбранную из группы, состоящей из следующих формул:
[0088]
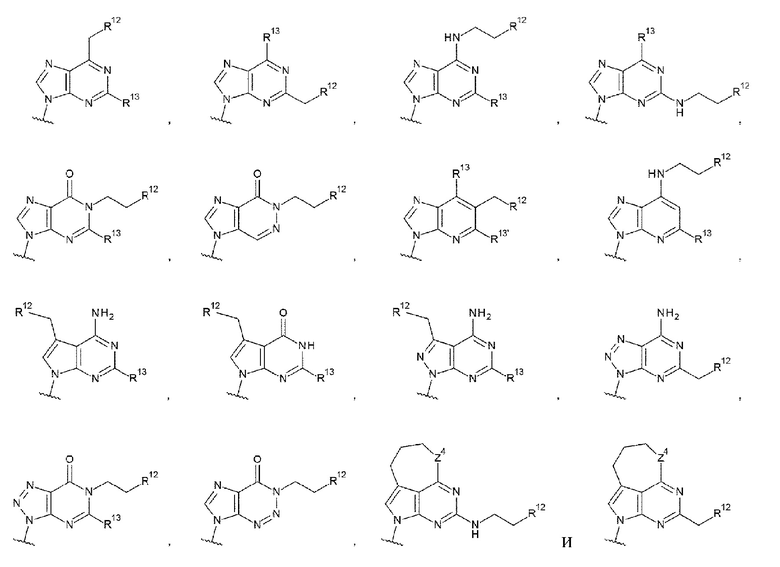
где
R13 и R13' каждый независимо представляет собой атом водорода, гидроксигруппу или -NH2;
R12 представляет собой гидроксигруппу, -NH2, -СН2ОН, -NHC(=O)CH2OH, -CH2NHC(=O)CH2OH или -CH2CH2NHC(=O)CH2OH; и
Z4 такой, как в определении выше.
[0089]
Кроме того, L1 предпочтительно представляет собой группу, выбранную из группы, состоящей из следующих формул:
[0090]
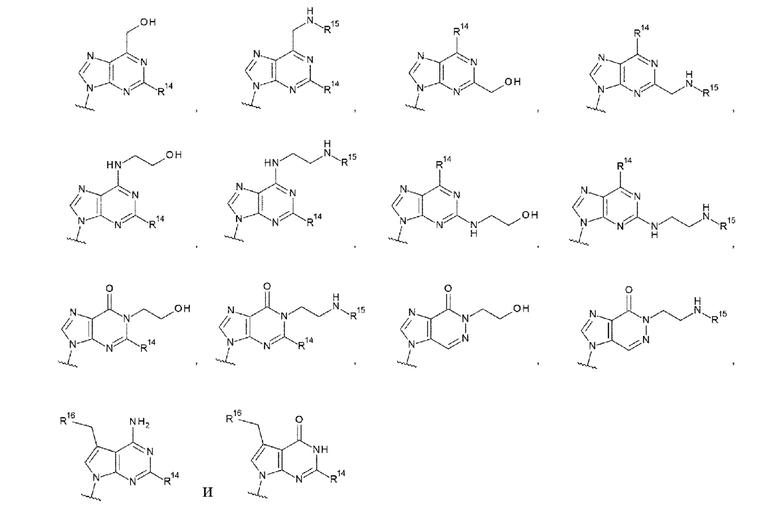
где
R14 представляет собой атом водорода или -NH2;
R15 представляет собой атом водорода или -С(=O)СН2ОН; и
R16 представляет собой гидроксигруппу, -NH2, -СН2ОН, -СН2СН2ОН, -CH2NH2 или -CH2CH2NH2.
[0091]
L1 более предпочтительно представляет собой группу, выбранную из группы, состоящей из следующих формул:
[0092]
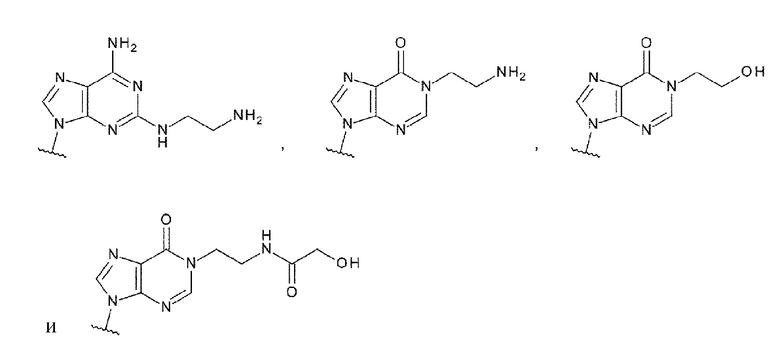
[0093]
L3 выбран из атома водорода, атома галогена, -NH2, гидрокси-С1-С3 алкильной группы и амино-С1-С3 алкильной группы.
[0094]
Q1 и Q1' каждый независимо представляет собой гидроксигруппу, тиольную группу или борановую группу (ВН3-). Q1 предпочтительно представляет собой гидроксигруппу или тиольную группу. Q1' предпочтительно представляет собой гидроксигруппу или тиольную группу. Более предпочтительно, комбинация Q1 и Q1' такая, что Q1 и Q'1 каждый представляет собой тиольную группу, или такая, что Q1 и Q1 каждый представляет собой гидроксигруппу.
[0095]
Q2 и Q2' каждый независимо представляет собой атом кислорода или атом серы. Предпочтительно, Q2 и Q2' каждый представляет собой атом кислорода, или каждый представляет собой атом серы.
[0096]
Комбинация Q1 и Q2 предпочтительно такая, что Q1 представляет собой тиольную группу и Q2 представляет собой атом кислорода, или такая, что Q1 представляет собой тиольную группу и Q2 представляет собой атом серы.
[0097]
Комбинация Q1' и Q2' предпочтительно такая, что Q1' представляет собой тиольную группу и Q2' представляет собой атом кислорода, или такая, что Q1' представляет собой гидроксигруппу и Q2' представляет собой атом кислорода, или такая, что Q1' представляет собой тиольную группу и Q2' представляет собой атом серы.
[0098]
X1 и X2 каждый независимо представляет собой атом кислорода, атом серы или -СН2-. X1 предпочтительно представляет собой атом кислорода. X2 предпочтительно представляет собой атом кислорода. Более предпочтительно, X1 и X2 каждый представляет собой атом кислорода.
[0099]
Y1 и Y2 каждый представляет собой атом кислорода или -CH2-. Y1 предпочтительно представляет собой атом кислорода. Y2 предпочтительно представляет собой атом кислорода. Более предпочтительно, Y1 и Y2 каждый представляет собой атом кислорода.
[0100]
X3 и X4 представляют собой группу, выбранную из (iii) и (iv):
(iii) когда Y1 представляет собой атом кислорода, Х3-Х4 представляет собой -СН2-О-, -CH2-S-, -СН2-СН2- или -CH2-CF2-; и
(iv) когда Y1 представляет собой -СН2-, Х3-Х4 представляет собой -О-СН2-. X3 и X4 предпочтительно представляют собой -СН2-О- в (iii).
[0101]
X5 и X6 представляют собой группу, выбранную из (v) и (vi):
(v) когда Y2 представляет собой атом кислорода, Х5-Х6 представляет собой -СН2-О-, -CH2-S-, -СН2-СН2- или -CH2-CF2-; и
(vi) когда Y2 представляет собой -СН2-, Х5-Х6 представляет собой -О-СН2-. X5 и X6 предпочтительно представляют собой -СН2-О- в (v).
[0102]
R1, R2 и R3 каждый независимо представляет собой атом водорода, атом галогена, -OR', -OC(=O)R', -N3, -NHR, -NR'R'' или -NHC(=O)R', где R' представляет собой атом водорода, С1-С6 алкильную группу, С2-С6 алкенильную группу, С2-С6 алкинильную группу или С3-С6 циклоалкильную группу, указанная С1-С6 алкильная группа, С2-С6 алкенильная группа или С2-С6 алкинильная группа необязательно содержит в качестве заместителей от одного до шести атомов галогена и R'' представляет собой С1-С6 алкильную группу, С2-С6 алкенильную группу, С2-С6 алкинильную группу или С3-С6 циклоалкильную группу.
[0103]
R1 предпочтительно представляет собой атом водорода, гидроксигруппу или атом фтора.
[0104]
R2 предпочтительно представляет собой атом водорода, гидроксигруппу или атом фтора.
[0105]
R3 предпочтительно представляет собой атом водорода, гидроксигруппу или атом фтора.
[0106]
W1 представляет собой атом азота, атом кислорода, атом серы или -СН-.
[0107]
R5 представляет собой группу, выбранную из (vii)-(х):
(vii) где W1 представляет собой атом азота, R5 представляет собой атом водорода, С1-С6 алкильную группу, гидрокси-С1-С6 алкильную группу или амино-С1-С6 алкильную группу;
(viii) где W1 представляет собой атом кислорода, R5 отсутствует;
(ix) где W1 представляет собой атом серы, R5 отсутствует; и
(x) где W1 представляет собой -СН-, R5 представляет собой атом водорода, атом галогена, гидроксигруппу, -NH2 или С1-С6 алкильную группу.
Где W1 представляет собой атом азота, R5 предпочтительно представляет собой атом водорода. Где W1 представляет собой -СН-, R5 предпочтительно представляет собой атом водорода.
[0108]
W2 представляет собой атом азота или -СН=. W2 предпочтительно представляет собой -СН=.
[0109]
R4 представляет собой атом водорода, атом галогена или -NH2. R4 предпочтительно представляет собой атом водорода.
[0110]
Z1-Z2-Z3 вместе представляют собой -СН2-СН2-СН2-, -CH2-CH2-R'''-, -СН=СН-СН2-, -СН=СХ-СН2-, -СХ=СН-СН2-, -СХ=СХ-СН2-, -С(=O)-СН2-СН2-, -СН2-СН2-С(=O)-, -СН2-СН(СН3)-СН2- или -СН2-СН2-СН(СН3)-, где R''' представляет собой -О- или -СН2-СН2- и X представляет собой атом галогена или группу, представленную любой одной из следующих формул:
[0111]

где
каждая звездочка указывает на связывание с W1, и каждая волнистая линия указывает на связывание с атомом углерода =С-.
Z1, Z2 и Z3 предпочтительно вместе образуют -СН2-СН2-СН2-, -СН=СН-СН2-, -СН2-СН(СН3)-СН2-, -СН2-СН2-СН(СН3)- или -CH2-CH2-R'''-, где R''' представляет собой -О- или -СН2-СН2-.
[0112]
Новое производное ЦДН согласно настоящему изобретению предпочтительно имеет структуру, представленную следующей формулой:
[0113]
[0114]
L1 такой, как в определении выше.
[0115]
Q3 и Q3' каждый независимо представляет собой гидроксигруппу или тиольную группу. Предпочтительно, Q3 и Q3' каждый представляет собой тиольную группу.
[0116]
R21 и R22 каждый независимо представляет собой гидроксигруппу или атом фтора. R21 предпочтительно представляет собой гидроксигруппу. R22 предпочтительно представляет собой атом фтора.
[0117]
W3 представляет собой -NH- или атом серы.
[0118]
Способы получения нового производного ЦДН согласно настоящему изобретению описаны далее в разделе
<3. Способы получениям
[0119]
<2. Конъюгат антитела с лекарственным средством>
Новое производное ЦДН согласно настоящему изобретению можно вводить непосредственно в целевую ткань (например, внутриопухолевое введение), или вводить в виде конъюгата антитела с лекарственным средством, в котором производное ЦДН соединено с антителом, способным распознавать и связываться с целевыми клетками (например, опухолевыми клетками или иммунными клетками), посредством любого линкера.
[0120]
Конъюгат антитела с лекарственным средством согласно настоящему изобретению представлен формулой (II):
[0121]
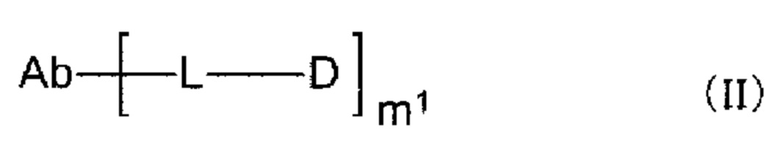
m1 представляет собой количество конъюгированных молекул лекарственного средства на молекулу антитела в конъюгате антитела с лекарственным средством; Ab представляет собой антитело или функциональный фрагмент антитела; L представляет собой линкер, соединяющий Ab и D; D представляет собой описанное выше новое производное ЦДН (в данной заявке, когда используется как часть конъюгата антитела с лекарственным средством, новое производное ЦДН также называют просто «лекарственным средством»).
[0122]
Лекарственное средство D представляет собой соединение, обладающее активностью активировать иммунные клетки, в частности, активностью агониста STING. Когда часть или весь линкер отщепляется в целевой клетке (например, опухолевой клетке или иммунной клетке), лекарственное средство D в исходной структуре высвобождается, чтобы оказать эффект активации иммунной системы. Предполагаемая функция осуществляется посредством повышения чувствительности целевой клетки к иммунным клеткам или активации иммунных клеток посредством целевой клетки. Предполагаемая функция не ограничена конкретной функцией при условии, что это может быть любая функция, относящаяся к активности агонистов STING. Тем не менее, предпочтительно это противоопухолевая активность. То есть, лекарственное средство D, связанное с антителом, нацеленным на опухоль (например, антителом против HER2), посредством любого линкера доставляется в целевые клетки или ткань, где часть или весь линкер отщепляется, и лекарственное средство D оказывает противоопухолевое действие посредством повышения чувствительности целевых клеток к иммунным клеткам или активации иммунных клеток посредством целевых клеток (например, продукции интерферонов или цитокинов).
[0123]
Лекарственное средство D, которое нужно конъюгировать с конъюгатом антитела с лекарственным средством согласно настоящему изобретению, представлено формулой (I):
[0124]
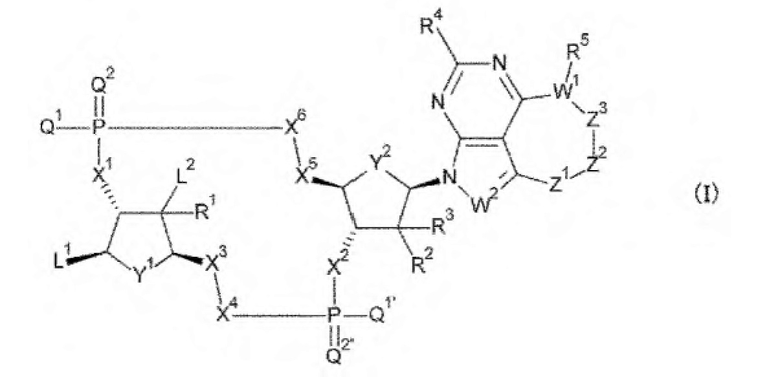
где
L связывается с любой -NH2 или гидроксигруппой, содержащейся в L1 или L2;
L1 такой, как определено в разделе <1. Новое производное ЦДН> выше;
L2 представляет собой группу, выбранную из (i) и (ii):
(i) при связывании с L, L2 представляет собой -NHR', гидрокси-С1-С6 алкильную группу или амино-С1-С6 алкильную группу, где R' представляет собой атом водорода, С1-С6 алкильную группу, С2-С6 алкенильную группу, С2-С6 алкинильную группу или С3-С6 пиклоалкильную группу, и С1-С6 алкильная группа, С2-С6 алкенильная группа или С2-С6 алкинильная группа необязательно содержит в качестве заместителей от одного до шести атомов галогена; и
(ii) при отсутствии связывания с L, L2 представляет собой атом водорода или атом галогена;
Q1, Q1', Q2, Q2', X1, X2, X3, X4, X5, X6, Y1, Y2, R1, R2, R3, R4, R5, W1, W2, Z1, Z2, Z3 такие, как в определении в разделе <1. Новое производное ЦДН> выше.
[0125]
При связывании с L, L2 предпочтительно представляет собой -NH2, -CH2NH2 или - СН2ОН. При отсутствии связывания с L, L2 предпочтительно представляет собой атом водорода или атом фтора.
[0126]
Лекарственное средство D для применения для нового производного ЦДН согласно настоящему изобретению или конъюгата антитела с лекарственным средством согласно настоящему изобретению предпочтительно представлено любой одной из следующих двух формул:
[0127]
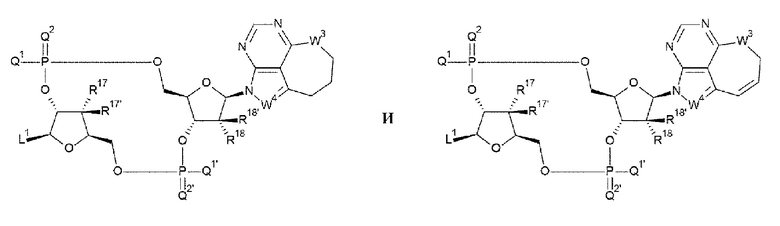
где
L1, Q1, Q1', Q2 и Q2' такие, как в определении выше;
R17, R17', R18 и R18' каждый независимо представляет собой атом водорода, атом галогена, гидроксигруппу или -NH2;
W3 представляет собой -NH-, атом кислорода, атом серы или -СН2-; и
W4 представляет собой -СН= или атом азота.
[0128]
Лекарственное средство D для применения для нового производного ЦДН согласно настоящему изобретению или конъюгата антитела с лекарственным средством согласно настоящему изобретению предпочтительно представлено любой одной из следующих двух формул:
[0129]
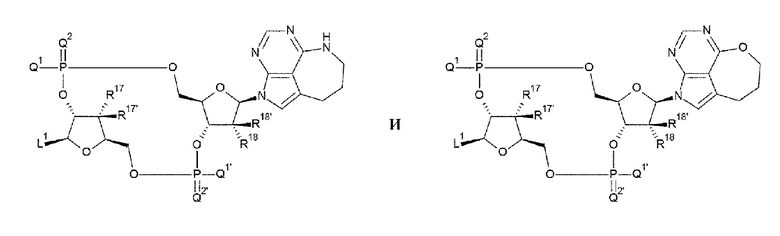
где
L1, Q1, Q1', Q2, Q2', R17, R17', R18 и R18' такие, как в определении выше.
Лекарственное средство D для применения для нового производного ЦДН согласно настоящему изобретению или конъюгата антитела с лекарственным средством согласно настоящему изобретению предпочтительно представлено любой одной из следующих восьми формул:
[0131]
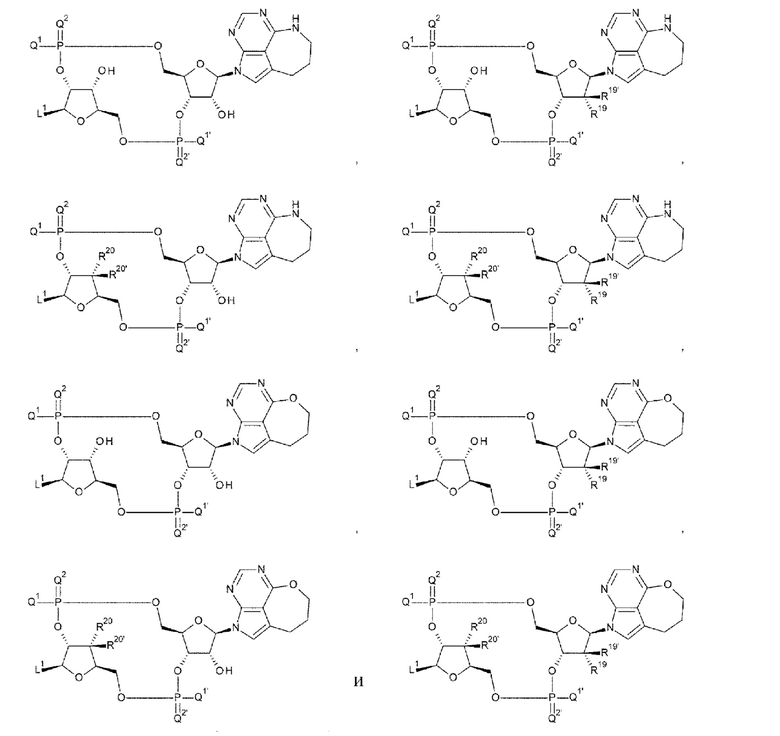
где
L1, Q1, Q1', Q2 и Q2' такие, как в определении выше; и
R19, R19', R20 и R20' каждый независимо представляет собой атом водорода или атом фтора.
Лекарственное средство D для применения для нового производного ЦДН согласно настоящему изобретению или конъюгата антитела с лекарственным средством согласно настоящему изобретению предпочтительно представлено любой одной из следующих четырех формул:
[0132]
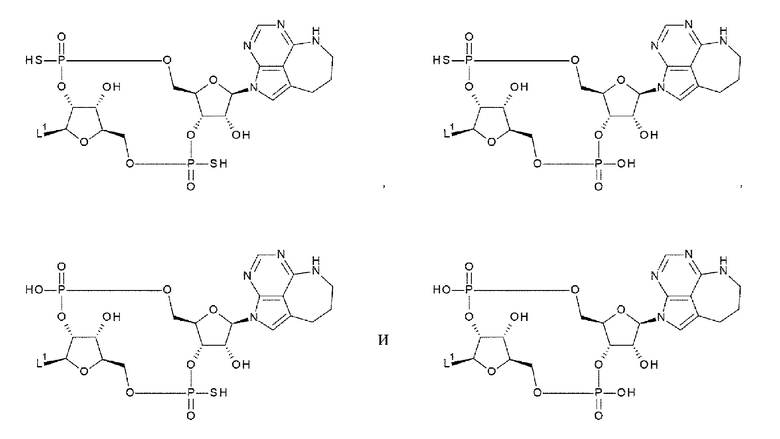
где
L1 такой, как в определении выше.
[0133]
Кроме того, лекарственное средство D для применения для нового производного ЦДН согласно настоящему изобретению или конъюгата антитела с лекарственным средством согласно настоящему изобретению предпочтительно представлено любой одной из следующих четырех формул:
[0134]
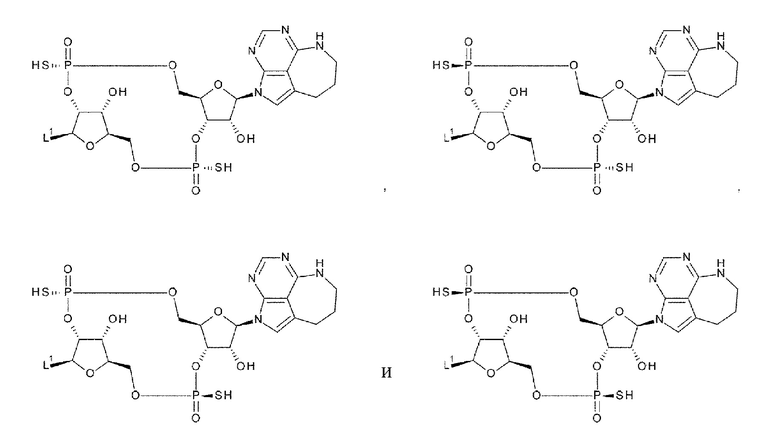
где
L1 такой, как в определении выше.
[0135]
Лекарственное средство D для применения для нового производного ЦДН согласно настоящему изобретению или конъюгата антитела с лекарственным средством согласно настоящему изобретению предпочтительно представлено любой одной из следующих четырех формул:
[0136]
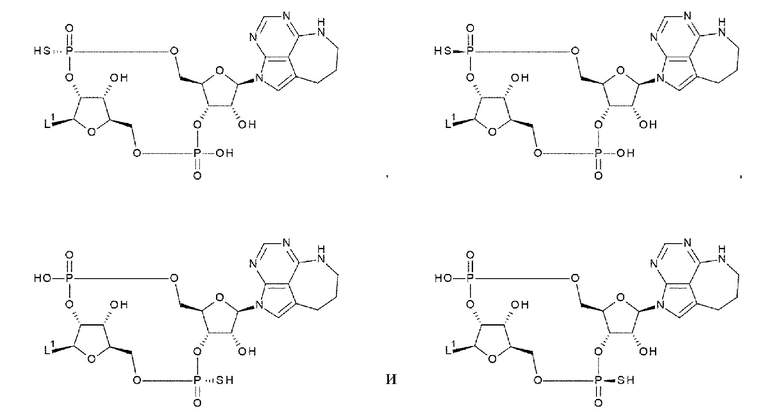
где
[0137]
L1 такой, как в определении выше.
Лекарственное средство D для применения для нового производного ЦДН согласно настоящему изобретению или конъюгата антитела с лекарственным средством согласно настоящему изобретению предпочтительно представлено следующей формулой:
[0138]
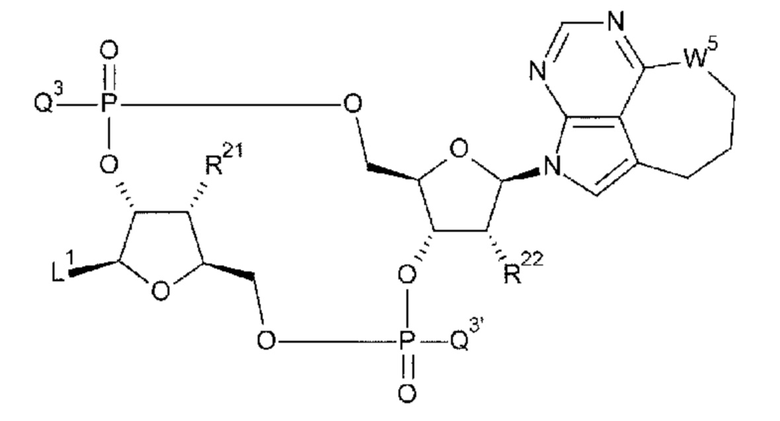
где
L1, Q3, Q3', R21, R22 и W3 такие, как в определении в разделе <1. Новое производное ЦДН> выше.
[0139]
В лекарственном средстве D для применения для нового производного ЦДН согласно настоящему изобретению или конъюгата антитела с лекарственным средством согласно настоящему изобретению, L1 предпочтительно представлен любой одной из следующих формул:
[0140]
[0141]
В лекарственном средстве D для применения для нового производного ЦДН согласно настоящему изобретению или конъюгата антитела с лекарственным средством согласно настоящему изобретению, L1 предпочтительно представлен любой одной из следующих четырех формул:
[0142]
где
каждая звездочка указывает на связывание с L.
[0143]
Лекарственное средство D для применения для нового производного ЦДН согласно настоящему изобретению или конъюгата антитела с лекарственным средством согласно настоящему изобретению предпочтительно представлено любой одной из следующих четырех формул:
[0144]
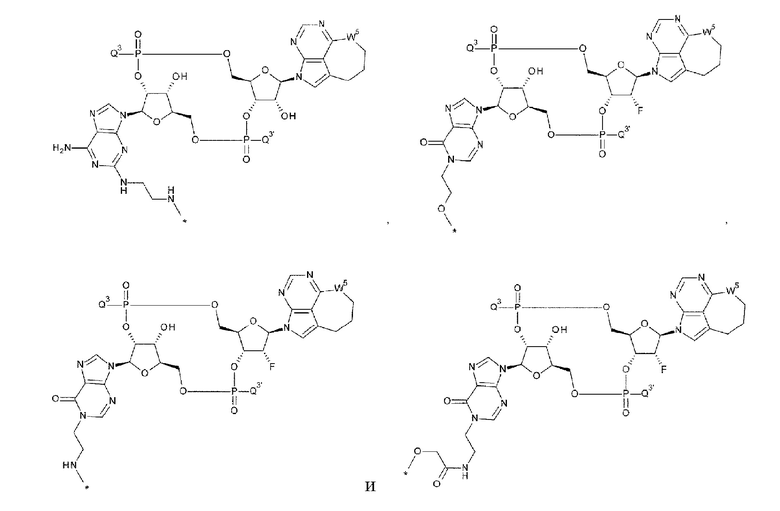
где
каждая звездочка указывает на связывание с L; и
Q3, Q3' и W3 такие, как в определении в разделе <1. Новое производное ЦДН> выше.
[0145]
<2.1. Структура линкера>
Будет описана структура линкера для конъюгирования лекарственного средства с антителом в конъюгате антитела с лекарственным средством согласно настоящему изобретению. Линкер для применения в конъюгате антитела с лекарственным средством согласно настоящему изобретению не ограничен конкретным линкером при условии, что это может быть любой линкер, который специалисты в данной области понимают как линкер, который соединяет антитело и лекарственное средство. Примеры линкера для применения в конъюгате антитела с лекарственным средством согласно настоящему изобретению могут включать, но не ограничены линкерами, описанными в Protein Cell, 2018, 9(1): 33-46, Pharm Res, 2015, 32: 3526-3540, и Int. J. Mol. Sci., 2016, 17, 561. Линкер может представлять собой линкер, который расщепляется in vivo, или линкер, который не расщепляется in vivo, но предпочтительно представляет собой линкер, который расщепляется от vivo.
[0146]
Примеры линкера для применения в конъюгате антитела с лекарственным средством согласно настоящему изобретению могут включать, но не ограничены линкером, который связывает лекарственное средство с гликаном или ремоделированным гликаном в Fc-части антитела (здесь и далее периодически называют «конъюгирование гликаном») (например, как описано в WO 2018/003983), и линкером, который связывает лекарственное средство с любым аминокислотным остатком (например, остатком цистеина или остатком лизина) антитела (например, как описано в WO 2014/057687). Примеры способов, которыми линкер связывает лекарственное средство с любым аминокислотным остатком антитела, могут включать, но не ограничены связыванием с сульфгидрильной группой (SH-группой) цистеина в Ab тиоэфирной связью (в данной заявке периодически называют «конъюгирование цисте ином»), и связыванием с аминогруппой (NH2-группой) лизина в Ab амидной связью (здесь и далее периодически называют «конъюгирование лизином»), и предпочтительно линкер связывается способом конъюгирования цистеином.
[0147]
Предпочтительный линкер L в настоящем изобретении представлен следующей формулой: -Lb-La-Lp-Lc-*
где
звездочка указывает на связывание с любой аминогруппой или гидроксигруппой, содержащейся в L1 или L2 лекарственного средства D.
[0148]
Сначала будет описана Lp.
[0149]
Lp представляет собой линкер, состоящий из последовательности аминокислот, расщепляемой in vivo или в целевой клетке (здесь и далее периодически называют пептидным линкером), или отсутствует.
[0150]
Lp отщепляется, например, под действием фермента, такого как пептидаза и эстераза. Lp представляет собой пептид, состоящий из от двух до семи (предпочтительно от двух до четырех) аминокислот. Lp образует амидную связь на N-конце с карбонильной группой на правом конце La, и образует амидную связь на С-конце с аминогруппой (-NH-) Lc. Амидная связь на С-концевой стороне Lp расщепляется ферментом, таким как пептидаза.
[0151]
Аминокислоты, составляющие Lp, не ограничены конкретными аминокислотами и, например, представляют собой L- или D-аминокислоты, и предпочтительно представляют собой L-аминокислоты. Аминокислоты могут представлять собой не только α-аминокислоты, но могут включать аминокислоту со структурой, например, β-аланина, ε-аминокапроновой кислоты или γ-аминомасляной кислоты, и могут дополнительно включать неприродную аминокислоту, такую как N-метилированная аминокислота. Последовательность аминокислот Lp не ограничена конкретной последовательностью аминокислот, и примеры аминокислот, которые составляют Lp, могут включать глицин (Gly; G), валин (Val; V), аланин (Ala; А), фенилаланин (Phe; F), глутаминовую кислоту (Glu; Е), изолейцин (Не; I), пролин (Pro; Р), цитруллин (Cit), лейцин (Leu; L), метионин (Met; М), серии (Ser; S), лизин (Lys; К), и аспарагиновую кислоту (Asp; D). Предпочтительны среди них глицин (Gly; G), валин (Val; V), аланин (Ala; А), фенилаланин (Phe: F) и цитруллин (Cit). Любые из данных аминокислот могут встречаться множество раз, и Lp имеет последовательность аминокислот, включая свободно выбранные аминокислоты. Паттерн высвобождения лекарственного средства можно контролировать с помощью типа аминокислот.
[0152]
Конкретные примеры Lp могут включать -GGVA-, -VA-, -GGFG-, -FG-, -GGPI-, -PI-, -GGVCit-, -VCit-, -GGVK-, -VK-, -GGFCit-, -FCit-, -GGFM-, -FM-, -GGLM-, -LM-, -GGICit- и -ICit-. Линкер Lp предпочтительно представляет собой -GGVA-, -VA-, -GGFG-, -FG-, -GGVCit-, -VCit-, -GGFCit- или -Fcit-. Линкер Lp более предпочтительно представляет собой -GGVA-, -GGFG или -GGVCit-. Линкер Lp предпочтительно представляет собой -GGFG-или -GGPI-.
[0153]
Затем будет описан La.
[0154]
La представляет собой любой один, выбранный из группы, состоящей из следующих:
-С(=O)-(СН2СН2)n2-С(=O)-,
-С(=O)-(СН2СН2)n2-СН2-С(=O)-,
-C(=O)-(CH2CH2)n2-C(=O)-NH-(CH2CH2)n3-C(=O)-,
-C(=O)-(CH2CH2)n2-C(=O)-NH-(CH2CH2)n3-CH2-C(=O)-,
-C(=O)-(CH2CH2)n2-C(=O)-NH-(CH2CH2O)n3-CH2-C(=O)-,
-(CH2)n4-O-C(=O)-, и -(CH2)n9-C(=O)-
где
n2 представляет собой целое число от 1 до 3 (предпочтительно 1 или 2), n3 представляет собой целое число от 1 до 5 (предпочтительно целое число от 2 до 5, более предпочтительно 3 или 4), n4 представляет собой целое число от 0 до 2 (предпочтительно 0 или 1) и n9 представляет собой целое число от 2 до 7 (предпочтительно целое число от 2 до 5, более предпочтительно 2, 3 или 5).
[0155]
La предпочтительно представляет собой любой один, выбранный из группы, состоящей из следующих:
-С(=O)-СН2СН2-С(=O)-,
-C(=O)-CH2CH2-C(=O)-NH-(CH2CH2O)3-CH2-C(=O)-,
-C(=O)-CH2CH2-C(=O)-NH-(CH2CH2O)4-CH2-C(=O)-,
-C(=O)-(CH2CH2)2-C(=O)-,
-C(=O)-CH2CH2-C(=O)-NH-(CH2CH2)2-C(=O)-,
-C(=O)-CH2CH2-C(=O)-NH-(CH2CH2)2-CH2-C(=O)-,
-CH2-OC(=O),
-OC(=O)-, и
-(CH2)5-C(=O)-.
[0156]
La более предпочтительно представляет собой -С(=O)-СН2СН2-С(=O)-,
-C(=O)-CH2CH2-C(=O)-NH-(CH2CH2O)3-CH2-C(=O)-, или
-(СН2)5-С(=O)-.
[0157]
La еще более предпочтительно представляет собой -С(=O)-СН2СН2-С(=O)-.
[0158]
Далее будет описан Lb.
[0159]
Lb представляет собой спейсер, который будут применять для линкера для конъюгирования гликаном (в данной заявке также называют «спейсером для линкера для конъюгирования гликаном»), или спейсер, который будут применять для конъюгирования цистеином (в данной заявке также называют «спейсер для линкера для конъюгирования цистеином»).
[0160]
<Когда Lb представляет собой «Спейсер для линкера для конъюгирования гликаном»>
Когда Lb представляет собой «спейсер для линкера для конъюгирования гликаном», примеры Lb могут включать, но не ограничены спейсером, представленным следующей формулой:
[0161]
[0162]
или.
[0163]
[0164]
В каждой структурной формуле, представленной выше, каждая звездочка (*) указывает на связывание с -(С=O)- или -CH2- на левом конце La и каждая волнистая линия указывает на связывание с гликаном или ремоделированным гликаном Ab.
[0165]
Когда любой один из Lb-1, Lb-2 и Lb-3 выбран в качестве Lb, сайт триазольного кольца порождает структуры геометрических изомеров, и молекулы Lb содержат любую одну из двух структур или их смесь. Конъюгат антитела с лекарственным средством согласно настоящему изобретению способен связывать множество молекул лекарственного средства с одной молекулой антитела. Когда множество молекул лекарственного средства необходимо связать с одной молекулой антитела, следовательно, присутствует множество молекул Lb (см., например, схематическую диаграмму (1е) конъюгата антитела с лекарственным средством, показанную на схеме Е, описанную далее в разделе <3. Способы получения>). Когда любой один из Lb-1, Lb-2 и Lb-3 выбран в качестве Lb и присутствует множество молекул Lb на молекулу антитела (например, когда m2, который описан далее, представляет собой 1 или 2), сайт триазольного кольца в каждой молекуле Lb порождает структуры геометрических изомеров, и молекулы Lb содержат любую одну из двух структур или их смесь.
[0166]
<Когда Lb представляет собой «Спейсер для линкера для конъюгирования цистеином»>
Когда Lb представляет собой «спейсер для линкера для конъюгирования цисте ином», примеры Lb могут включать, но не ограничены -(сукцинимид-3-ил-N)-. В настоящем изобретении «-(сукцинимид-3-ил-N)-» имеет структуру, представленную следующей формулой:
[0167]
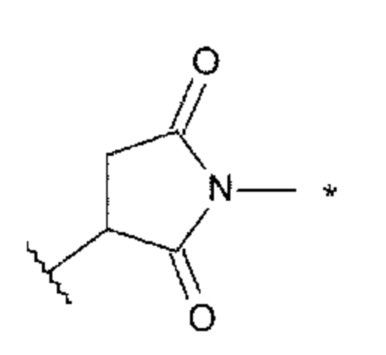
В структурной формуле, представленной выше, звездочка указывает на связывание с La и волнистая линия указывает на связывание с боковой цепью остатка цистеина антитела посредством образования тиоэфира.
[0168]
Далее будет описан Lc.
[0169]
Lc представляет собой -NH-CH2, -NH-фенильная группα-СН2-O(С=O)- или -NH-гетероарильная группα-СН2-O(С=O)- или отсутствует. Здесь фенильная группа предпочтительно представляет собой 1,4-фенильную группу, и гетероарильная группа предпочтительно представляет собой 2,5-пиридильную группу, 3,6-пиридильную группу, 2,5-пиримидильную группу или 2,5-тиенильную группу. Lc предпочтительно представляет собой -NH-CH2- или отсутствует.
[0170]
Более предпочтительный линкер L в настоящем изобретении представляет собой, когда способом связывания лекарственного средства и антитела является «конъюгирование гликаном»,
-ZL1-C(=O)-CH2CH2-C(=O)-GGFG-,
-ZL1-C(=O)-CH2CH2-C(=O)-GGVA-,
-ZL1-C(=O)-CH2CH2-C(=O)-GGVCit-,
-ZL1-C(=O)-CH2CH2-C(=O)-GGFCit-,
-ZL1-C(=O)-CH2CH2-C(=O)-GGICit-,
-ZL1-C(=O)-CH2CH2-C(=O)-GGFM-,
-ZL1-C(=O)-CH2CH2-C(=O)-GGPI-,
-ZL1-C(=O)-CH2CH2-C(=O)-GGLM-,
-ZL1-C(=O)-CH2CH2-C(=O)-FG-,
-ZL1-C(=O)-CH2CH2-C(=O)-VA-,
-ZL1-C(=O)-CH2CH2-C(=O)-GGFG-NH-CH2-,
-ZL1-C(=O)-CH2CH2-C(=O)-GGVA-NH-CH2-,
-ZL1-C(=O)-CH2CH2-C(=O)-GGVCit-NH-CH2-,
-ZLI-C(=O)-CH2CH2-C(=O)-GGFCit-NH-CH2-,
-ZLI-C(=O)-CH2CH2-C(=O)-NH-(CH2CH2O)3-CH2-C(=O)-, или
-ZL1-C(=O)-CH2CH2-C(=O)-NH-(CH2CH2O)4-CH2-C(=O)-,
где ZL1 представляет собой следующую структурную формулу для Lb:
[0171]
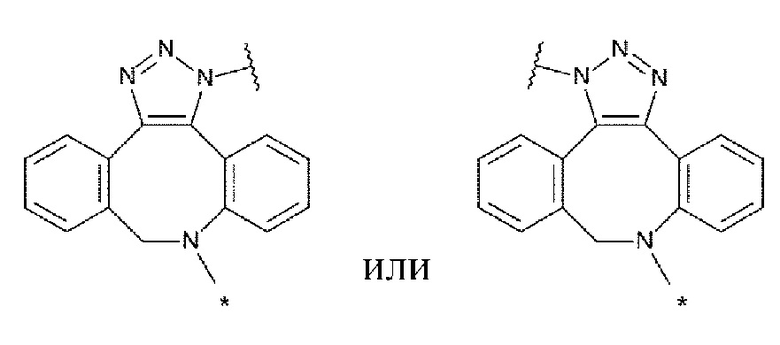
или,
когда способом связывания лекарственного средства и антитела является «конъюгирование цистеином», -ZL2-(CH2)5-C(=O)-GGFG-, -ZL2-(CH2)5-C(=O)-GGVA-, -ZL2-(CH2)5-C(=O)-GGVCit-, -ZL2-(CH2)5-C(=O)-GGFCit-, -ZL2-(CH2)5-C(=O)-GGICit-, -ZL2-(CH2)5-C(=O)-GGFM-, -ZL2-(CH2)5-C(=O)-GGPI-,
-ZL2-(CH2)5-C(=O)-GGLM-,
-ZL2-(CH2)5-C(=O)-FG-,
-ZL2-(CH2)5-C(=O)-VA-,
-ZL2-(CH2)5-C(=O)-GGFG-NH-CH2-,
-ZL2-(CH2)5-C(=O)-GGVA-NH-CH2-,
-ZL2-(CH2)5-C(=O)-GGVCit-NH-CH2-,
-ZL2-(CH2)5-C(=O)-GGFCit-NH-CH2-,
-ZL2-(CH2)5-C(=O)-NH-(CH2CH2O)3-CH2-C(=O)- или
-ZL2-(CH2)5-C(=O)-NH-(CH2CH2O)4-CH2-C(=O)-,
где ZL2 представляет собой -(сукцинимид-3-ил-N)-, представленный следующей структурной формулой для Lb:
[0172]
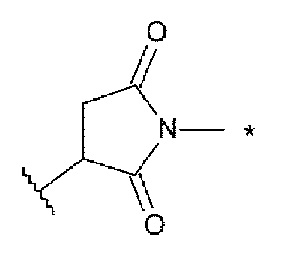
[0173]
Более предпочтительный линкер L в настоящем изобретении такой, что способ связывания лекарственного средства и антитела представляет собой «конъюгирование гликаном» и линкер L представляет собой -ZLI-C(=O)-CH2CH2-C(=O)-GGFG-NH-CH2-, или
-ZL1-C(=O)-CH2CH2-C(=O)-GGPI-NH-CH2-,
где ZL1 представляет собой следующую структурную формулу для Lb:
[0174]
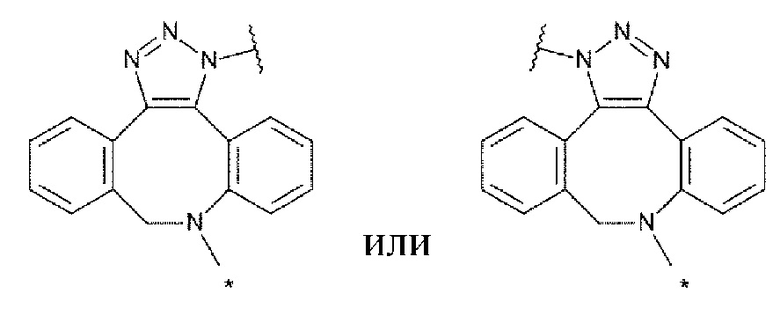
Правый конец в каждом из «предпочтительного линкера L» и «более предпочтительного линкера L» связывается с любым -NH2 или гидроксигруппой, содержащейся в L1 или L2 в формуле (I).
[0176]
<2.2. Антитело и его модификация гликаном>
<2.2.1 Антитело>
В данной заявке «ген» относится к нуклеотидам, включая последовательность нуклеотидов, кодирующую аминокислоты белка, или последовательность нуклеотидов, кодирующую аминокислоты белка, или комплементарную ей цепь, и значение термина «ген» включает, например, полинуклеотид, олигонуклеотид, ДНК, мРНК, кДНК и РНК в качестве последовательности нуклеотидов, включая последовательность нуклеотидов, кодирующую аминокислоты белка, или комплементарную ей цепь.
[0177]
В данной заявке «нуклеотиды», «полинуклеотид» и «последовательность нуклеотидов» имеют те же значения, как и «нуклеиновая кислота», и в объем значения термина «нуклеотиды» или «последовательность нуклеотидов» входят, например, ДНК, РНК, зонд, олигонуклеотид, полинуклеотид и праймер.
[0178]
В данной заявке «полипептид», «пептид» и «белок» используют без всякого различия.
[0179]
В данной заявке «функциональный фрагмент антитела» также называют «антигенсвязывающим фрагментом антитела», и данный термин означает частичный фрагмент антитела с активностью связывания с антигеном, и его примеры могут включать, но не ограничены перечисленными: Fab, F(ab')2, Fv, scFv, диатела, линейные антитела и мультиспецифические антитела, полученные из фрагментов антитела. Кроме того, в объем значения термина «антигенсвязывающий фрагмент антитела» входит Fab', моновалентный фрагмент вариабельной области антитела, полученный путем обработки F(ab')2 в восстанавливающих условиях. Тем не менее, нет ограничения данными молекулами, и пригодна любая молекула, обладающая способностью связывания с антигеном. Такие антигенсвязывающие фрагменты включают не только фрагменты, полученные путем обработки полноразмерной молекулы белка антитела подходящим ферментом, но также белки, полученные в подходящих клетках-хозяевах с применением гена антитела, модифицированного с помощью генной инженерии.
[0180]
Понятие функционального фрагмента согласно настоящему изобретению включает функциональный фрагмент, в котором остался хорошо сохранившийся аспарагин (Asn297), который будет модифицирован N-связанным гликаном, и аминокислоты вблизи Asn297 в Fc-области тяжелой цепи IgG, и который обладает активностью связывания с антигеном.
[0181]
Антитело для применения в конъюгате антитела с лекарственным средством согласно настоящему изобретению относится к иммуноглобулину и представляет собой молекулу, содержащую антигенсвязывающую область, которая иммуноспецифично связывается с антигеном. Антитело согласно настоящему изобретению может относиться к 15 любому классу IgG, IgE, IgM, IgD, IgA и IgY, и предпочтительно представляет собой IgG. Подкласс может быть любым из IgG1, IgG2, IgG3, IgG4, IgA1 и IgA2, и предпочтительно представляет собой IgG1, IgG2 и IgG4 (включая антитела, содержащие мутацию, которая влияет на активности антителозависимой клеточной цитотоксичности (АЗКЦ) и антителозависимого клеточного фагоцитоза (АЗКФ) в Fc-области тяжелой цепи IgG).
[0182]
Если в качестве изотипа антитела согласно настоящему изобретению используют IgG1, эффекторную функцию можно регулировать путем замены части аминокислотных остатков в константной области (см. WO 88/07089, WO 94/28027, WO 94/29351). Примеры мутантов IgG1 могут включать, но не ограничены мутантами с 25 мутацией LALA IgG1 (IgG1-L234A, L235A). L234A, L235A означает замену лейцина на аланин в положениях 234 и 235, соответствующих нумерации EU-индекс (Proceedings of the National Academy of Sciences of United States of America, том 63, №1 (15 мая 1969), стр. 78-85).
[0183]
Антитело можно получить из любого вида, и предпочтительные примеры происхождения могут включать, но не ограничены перечисленными: человека, крысу, мышь и кролика. Если антитело получено из вида, отличного от вида человек, оно предпочтительно представляет собой химерное или гуманизированное антитело с применением хорошо известной методики. Антитело согласно настоящему изобретению может представлять собой поликлональное антитело или моноклональное антитело, и предпочтительно представляет собой моноклональное антитело. Примеры моноклональных антител могут включать, но не ограничены перечисленными: моноклональные антитела, полученные из отличных от человека животных, такие как антитела крысы, антитела мыши и антитела кролика; химерные антитела; гуманизированные антитела; антитела человека; их функциональные фрагменты; и их модифицированные формы.
[0184]
Антитело предпочтительно представляет собой, но не ограничено антителом, нацеленным на опухолевые клетки или иммунные клетки. Антитело более предпочтительно представляет собой антитело, нацеленное на опухолевые клетки.
[0185]
Если используют антитело, нацеленное на опухолевые клетки, предпочтительно, чтобы антитело обладало одним или более свойствами из свойства наличия способности распознавать опухолевые клетки, свойства наличия способности связываться с опухолевыми клетками, свойства наличия способности встраиваться и интернализоваться в опухолевые клетки и свойства вызывать повреждение опухолевых клеток. Лекарственное средство согласно настоящему изобретению для конъюгирования с антителом посредством линкера обладает активностью агонистов STING. Лекарственное средство согласно настоящему изобретению индуцирует интерферон путем активации передачи сигналов регуляторного фактора интерферона-3 (IRF3). Следовательно, если антитело, нацеленное на опухолевые клетки, применяют в конъюгате антитела с лекарственным средством согласно настоящему изобретению, конъюгат антитела с лекарственным средством после введения в организм доставляется в очаг опухоли и внедряется в клетки в опухолях, и часть, представляющая собой линкер, затем отщепляется пептидазой или тому подобным ферментом и часть, представляющая собой лекарственное средство, высвобождается. Предполагается, что высвобожденная часть, представляющая лекарственное средство, активирует противоопухолевый иммунитет и оказывает противоопухолевое действие посредством активности агониста STING.
[0186]
Способность антитела связываться с опухолевыми клетками можно подтвердить, применяя проточную цитометрию. Внедрение антитела в опухолевые клетки можно подтвердить, применяя (1) анализ визуализации антитела, внедренного в клетки, под флуоресцентным микроскопом с помощью вторичного антитела (флуоресцентно меченого), которое связывается с терапевтическим антителом (Cell Death and Differentiation (2008) 15, 751-761), (2) анализ измерения интенсивности флуоресценции, внедренной в клетки со вторичным антителом (флуоресцентно меченым), которое связывается с терапевтическим антителом (Molecular Biology of the Cell, том 15, 5268-5282, декабрь 2004), или (3) анализ Mab-ZAP с применением иммунотоксина, который связывается с терапевтическим антителом, где токсин высвобождается после внедрения в клетки для подавления роста клеток (Bio Techniques 28: 162-165, январь 2000). В качестве иммунотоксина можно применять рекомбинантный комплекс белок-белок каталитического домена дифтерийного токсина и белка G.
[0187]
В настоящем изобретении «высокая способность к интернализации» относится к случаю, когда показатель выживаемости целевых экспрессирующих антиген клеток (например, экспрессирующих HER2 клеток, если используют антитело против HER2) при добавлении интересующего антитела и меченого сапорином антитела IgG против мыши или крысы (представленный как уровень относительно показателя выживаемости клеток без добавления антитела, принятого за 100%) составляет предпочтительно 70% или менее, и более предпочтительно 60% или менее.
[0188]
Если антитело, нацеленное на опухолевые клетки, используют в конъюгате антитела с лекарственным средством согласно настоящему изобретению, предпочтительно, но не обязательно, чтобы само антитело обладало противоопухолевым действием. Предпочтительно, чтобы антитело для применения в конъюгате антитела с лекарственным средством согласно настоящему изобретению обладало свойством интернализации, которое включает миграцию в опухолевые клетки.
[0189]
Противоопухолевая активность лекарственного средства или конъюгата антитела с лекарственным средством относится к цитотоксической активности или направленному против опухолевых клеток действию, или регрессии объема опухоли. Противоопухолевую активность можно подтвердить, применяя любую известную систему оценки in vitro или in vivo.
[0190]
Активность активации иммунитета лекарственного средства и конъюгата антитела с лекарственным средством относится к повышению чувствительности опухолевых клеток к иммунным клеткам или активации иммунных клеток опухолевыми клетками. Активность активации иммунитета можно подтвердить, применяя любую известную систему оценки in vitro или in vivo.
[0191]
Примеры антитела для применения в настоящем изобретении могут включать, но не ограничены перечисленными: антитело против HER2, антитело против HER3, антитело против DLL3, антитело против FAP, антитело против CDH11, антитело против CDH6, антитело против А33, антитело против CanAg, антитело против CD19, антитело против CD20, антитело против CD22, антитело против CD30, антитело против CD33, антитело против CD56, антитело против CD70, антитело против CD98, антитело против TROP2, антитело против СЕА, антитело против Cripto, антитело против EphA2, антитело против G250, антитело против MUC1, антитело против GPNMB, антитело против интегрина, антитело против PSMA, антитело против тенасцина-С, антитело против SLC44A4, антитело против мезотелина, антитело против ENPP3, антитело против CD47, антитело против EGFR, антитело против GPR20 и антитело против DR5. Антитело согласно настоящему изобретению предпочтительно представляет собой антитело против HER2 (например, трастузумаб или пертузумаб), антитело против CDH6, антитело против CD33 или антитело против EphA2 и более предпочтительно антитело против HER2.
[0192]
Антитело согласно настоящему изобретению можно получить, применяя способ, который обычно осуществляют в данной области, который включает иммунизацию животного полипептидным антигеном и сбор и очистку антитела, продуцированного в организме. Происхождение антигена не ограничено людьми, и животных можно иммунизировать антигеном, полученным из отличного от человека животного, такого как мышь и крыса. В данном случае перекрестную реактивность полученного в результате этого антитела, которое связывается с гетерологичным антигеном, с соответствующим антигеном человека можно исследовать, чтобы провести скрининг антител, применимых для лечения заболевания человека.
[0193]
В качестве альтернативы моноклональное антитело можно получить из гибридомы, полученной путем слияния продуцирующей антитело клетки, которая продуцирует антитело против антигена, с клеткой миеломы в соответствии со способом, известным в данной области (например, Kohler и Milstein, Nature (1975) 256, стр. 495-497, Kennett, R. ред., Monoclonal Antibodies, стр. 365-367, Plenum Press, Нью-Йорк (1980)).
[0194]
Антиген можно получить путем генной инженерии, чтобы позволить клеткам-хозяевам продуцировать ген, кодирующий белок антигена.
[0195]
Гуманизированное антитело согласно настоящему изобретению можно получить в соответствии с известным способом (например, Proc. Natl. Acad. Sci. U.S.A., 81, 6851-6855, (1984), Nature (1986) 321, стр. 522-525, WO 90/07861).
[0196]
Например, каждое антитело против HER2 (US 5821337, WO 2004/008099 и т.д.), антитело против CD33 (WO 2014/057687 и т.д.), антитело против CD70 (WO 2004/073656 и т.д.), антитело против EphA2 (WO 2009/028639 и т.д.) и антитело против CDH6 (WO 2018/212136 и т.д.) можно получить в соответствии с известным способом.
[0197]
Например, желательно, чтобы антитело против HER2 согласно настоящему изобретению обладало любым из следующих свойств, но антитело против HER2 не ограничено ими.
(1) Антитело против HER2, обладающее следующими свойствами:
(a) способно специфично связываться с HER2; и
[0198]
(b) обладает активностью интернализоваться в экспрессирующие HER2 клетки путем связывания с HER2.
(2) Антитело согласно (1), способное связываться с внеклеточным доменом HER2.
(3) Антитело согласно (1) или (2), представляющее собой моноклональное антитело.
(4) Антитело согласно любому из пп. (1)-(3), обладающее активностями или активностью антителозависимой клеточной цитотоксичности (АЗКЦ) и/или комплементзависимой цитотоксичности (КЗЦ).
(5) Антитело согласно любому из пп. (1)-(4), представляющее собой моноклональное антитело мыши, химерное моноклональное антитело или гуманизированное моноклональное антитело.
(6) Антитело согласно любому из пп. (1)-(3), в котором константная область тяжелой цепи представляет собой константную область тяжелой цепи IgG1 человека и содержит мутацию, которая вызывает снижение активности АЗКЦ и АЗКФ.
(7) Антитело согласно любому из пп. (1)-(4), представляющее собой гуманизированное моноклональное антитело, содержащее тяжелую цепь, состоящую из последовательности аминокислот, представленной в SEQ ID NO: 2, и легкую цепь, состоящую из последовательности аминокислот, представленной в SEQ ID NO: 1.
(8) Антитело согласно (5), в котором константная область тяжелой цепи представляет собой константную область тяжелой цепи IgG1 человека, и лейцин заменен на аланин в положениях 234 и 235, соответствующих нумерации EU-индекс.
(9) Антитело согласно (8), представляющее собой гуманизированное моноклональное антитело, содержащее тяжелую цепь, состоящую из последовательности аминокислот, представленной в SEQ ID NO: 3, и легкую цепь, состоящую из последовательности аминокислот, представленной в SEQ ID NO: 1.
(10) Антитело согласно любому из пп. (1)-(4), представляющее собой гуманизированное моноклональное антитело, содержащее тяжелую цепь, состоящую из последовательности аминокислот, представленной в SEQ ID NO: 29, и легкую цепь, состоящую из последовательности аминокислот, представленной в SEQ ID NO: 28.
(11) Антитело согласно любому из пп. (1)-(4), представляющее собой гуманизированное моноклональное антитело, содержащее тяжелую цепь, состоящую из последовательности аминокислот, представленной в SEQ ID NO: 30, и легкую цепь, состоящую из последовательности аминокислот, представленной в SEQ ID NO: 28.
(12) Антитело согласно любому из пп. (1)-(11), в котором одна или две аминокислоты удалены на карбоксильном конце тяжелой цепи.
(13) Антитело, полученное с применением способа получения антитела согласно любому из пп. (1)-(12), указанный способ включает этапы: культивирования клетки-хозяина, трансформированной вектором экспрессии, содержащим полинуклеотид, кодирующий антитело; и сбора нацеленного антитела из культуры, полученной на этапе культивирования.
[0199]
<2.2.2 Ремоделирование гликана для антитела>
Недавно сообщали о способе ремоделирования гетерогенных гликанов антитела с помощью ферментативной реакции для внедрения гомогенных гликанов, содержащих функциональную группу (ACS Chem. Biol. 2012, 7, 110-122, ACS Med. Chem. Lett. 2016, 7, 1005-1008). Предприняли попытку применения данной методики ремоделирования гликана для сайт-специфического внедрения лекарственного средства, чтобы синтезировать гомогенный конъюгат антитела с лекарственным средством (ADC) (Bioconjugate Chem. 2015, 26, 2233-2242, Angew. Chem. Int., изд. 2016, 55, 2361-2367, US 2016361436).
[0200]
При ремоделировании гликана сначала используют гидролазу для отщепления гетерогенных гликанов, добавленных в белок (например, антитело), причем только GlcNAc на каждом конце оставляют в неизменном виде, с получением гомогенной белковой молекулы, содержащей добавленный к ней GlcNAc (здесь и далее называют «акцептором»). Затем предоставляют любой отдельно полученный гликан (здесь и далее называют «донором»), и акцептор и донор соединяют друг с другом, применяя гликозилтрансферазу. Таким образом, можно синтезировать гомогенный гликопротеин с любой структурой гликанов.
[0201]
В настоящем изобретении «гликан» относится к структурному звену из двух или более моносахаридов, соединенных друг с другом гликозидными связями. Конкретные моносахариды и гликаны периодически выражают в виде сокращений, таких как «GlcNAc-» и «SG-». Когда любое из данных сокращений используют в структурной формуле, подразумевается, что в представленной аббревиатуре атом кислорода или атом азота, участвующий в гликозидной связи на восстанавливающем конце с другим структурным звеном, не включен в аббревиатуру, обозначающую гликан, если конкретно не указан.
[0202]
В настоящем изобретении каждый моносахарид как основное звено гликана для удобства выражен определением, что в кольцевой структуре положение атома углерода, связанного с атомом кислорода, составляющим кольцо, и непосредственно связанного с гидроксигруппой (или атомом кислорода, вовлеченным в гликозидную связь) представляет собой положение 1 (положение 2 только для сиаловых кислот), если не указано иное. Каждое название соединений в Примерах представлено с учетом всей химической структуры, и данное правило не обязательно применяется.
[0203]
Если гликан в настоящем изобретении выражен в виде обозначения (например, SG, MSG, GlcNAc), то предполагается, что указанное обозначение, если нет других определений, включает атомы углерода, доходящие до восстанавливающего конца, и не включает N или О, вовлеченные в N- или О-гликозидную связь.
[0204]
Конъюгат антитела с лекарственным средством согласно настоящему изобретению представлен следующей формулой:
[0205]
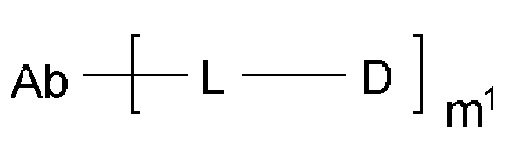
где антитело Ab или функциональный фрагмент антитела связывается боковой цепью своего аминокислотного остатка (например, цистеина, лизина) непосредственно с L, или связывается посредством гликана или ремоделированного гликана Ab с L.
[0206]
Гликаны в Ab согласно настоящему изобретению представляют собой N-связанные гликаны или О-связанные гликаны, и предпочтительно представляют собой N-связанные гликаны.
[0207]
N-связанные гликаны и О-связанные гликаны связываются с боковой цепью аминокислоты антитела посредством N-гликозидной связи и О-гликозидной связи, соответственно.
[0208]
Ab согласно настоящему изобретению представляет собой IgG и предпочтительно представляет собой IgG1, IgG2 или IgG4.
[0209]
IgG содержит хорошо сохранившийся N-связанный гликан на остатке аспарагина в положении 297 Fc-области тяжелой цепи (здесь и далее называют «Asn297 или N297»), и известно, что N-связанный гликан вносит вклад в активность, кинетику и т.д. молекулы антитела (Eon-Duval, А. и др., Biotechnol. Prog. 2012, 28, 608-622, Sanglier-Cianferani, S., Anal. Chem. 2013, 85, 715-736).
[0210]
Последовательность аминокислот в константной области IgG хорошо сохранена, и каждая из аминокислот указана в соответствии с нумерацией EU-индекс в сообщении от Edelman и др. (Proc. Natl. Acad. Sci. U.S.A., 63, 78-85, (1969)). Например, Asn297, к которому добавлен N-связанный гликан в Fc-области, соответствует положению 297, определенному нумерацией EU-индекс, и каждая аминокислота уникально определена путем представления в нумерации EU-индекс, далее если фактическое положение аминокислоты изменилось посредством фрагментации молекулы или делеции участка.
[0211]
В следующей формуле проиллюстрирован случай, когда конъюгат антитела с лекарственным средством согласно настоящему изобретению связывается посредством N297-гликана антитела или функционального фрагмента антитела с L.
[0212]
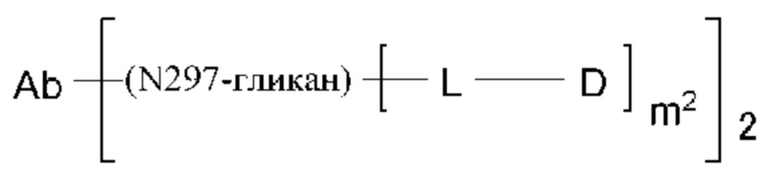
[0213]
Антитело, содержащее ремоделированный гликан, называют ремоделированным гликаном антителом.
[0214]
SGP (α2,6-SGP), аббревиатура для сиалилгликопептида, является типичным N-связанным гликопептидом. SGP можно выделить/очистить из желтка куриного яйца, например, в соответствии со способом, описанным в WO 2011/027868. Очищенные продукты SGP реализуются Tokyo Chemical Industry Co., Ltd., и FUSHIMI Pharmaceutical Co., Ltd. В данной заявке молекула гликана SGP обозначена SG, и гликан, образованный путем удаления одной молекулы GlcNAc на восстанавливающем конце в SG, обозначен SG(10). SG(10) можно получить путем ферментативного гидролиза SGP, например, руководствуясь сообщением Umekawa и др. (Biochim. Biophys. Acta 2010, 1800, 1203-1209). В качестве альтернативы, SG(10) можно приобрести в Tokyo Chemical Industry Co., Ltd. или FUSHIMI Pharmaceutical Co., Ltd.
[0215]
В данной заявке структура гликанов, образованная путем удаления сиаловой кислоты на невосстанавливающем конце только любой одной из разветвленных цепей β-Man в SG(10), обозначена MSG(9), и структура, содержащая сиаловую кислоту только в 1-3 гликане разветвленных цепей, обозначена MSG1, и структура, содержащая сиаловую кислоту только в 1-6 гликане разветвленных цепей, обозначена MSG2.
[0216]
Ремоделированный гликан согласно настоящему изобретению представляет собой N297-(Fuc)SG, N297-(Fuc)MSG1, N297-(Fuc)MSG2 или смесь N297-(Fuc)MSG1 и N297-(Fuc)MSG2, предпочтительно N297-(Fuc)SG, N297-(Fuc)MSG1 или N297-(Fuc)MSG2, и более предпочтительно N297-(Fuc)SG или N297-(Fuc)MSG1.
[0217]
N297-(Fuc)SG представлен следующей структурной формулой или формулой последовательности:
[0218]
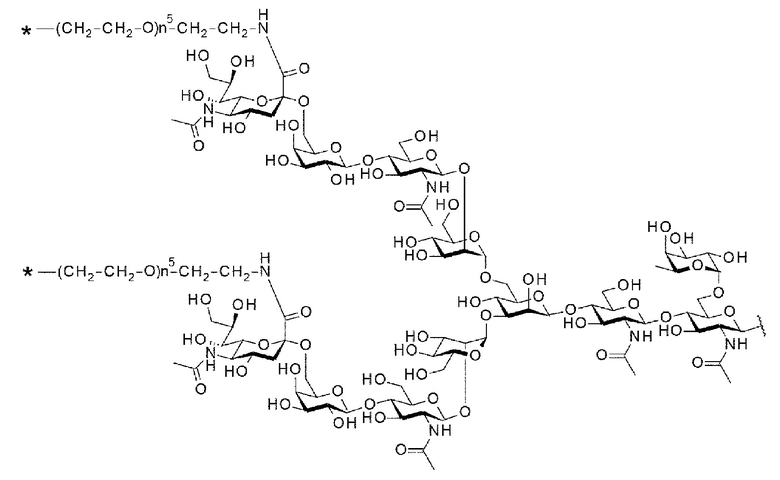
[0219]
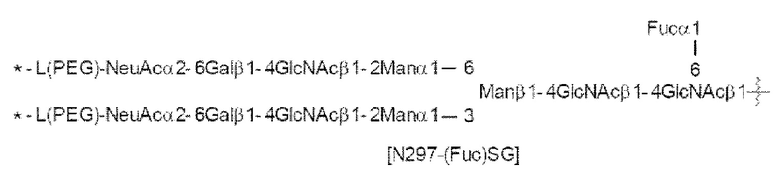
[0220]
В формулах каждая волнистая линия указывает на связывание с Asn297 антитела;
L(PEG) представляет собой -(CH2-CH2-O)n5-CH2-CH2-NH-, где аминогруппа на правом конце указывает на связывание амидной связью с карбоксильной группой в положении 2 сиаловой кислоты на невосстанавливающем конце в каждой из 1-3 цепи и 1-6 цепи разветвленных цепей β-Man в N297-гликане, и каждая звездочка указывает на связывание с линкером L, в частности, атомом азота в положении 1 или 3 1,2,3-триазольного кольца Lb в линкере L; и
n5 представляет собой целое число от 2 до 10, и предпочтительно целое число от 2 до 5.
[0221]
N297-(Fuc)MSG1 представлен следующей структурной формулой или формулой последовательности:
[0222]
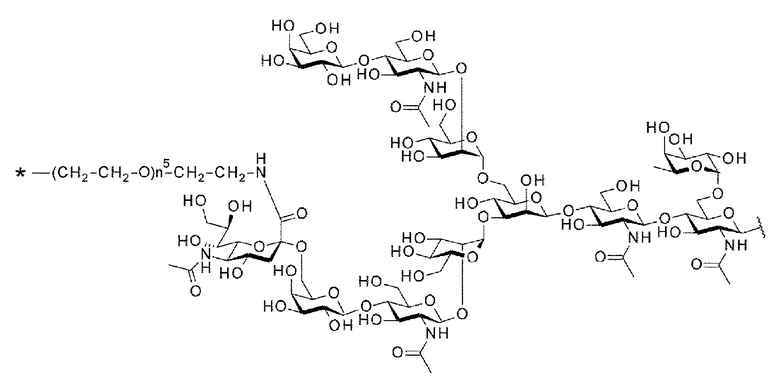
[0223]
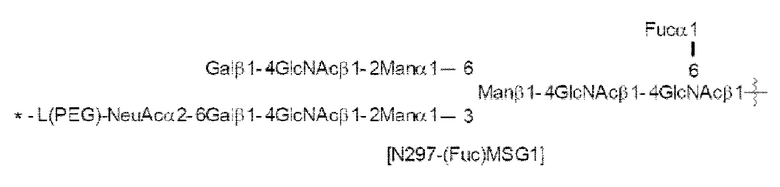
[0224]
В формулах каждая волнистая линия указывает на связывание с Asn297 антитела;
L(PEG) представляет собой -(CH2-CH2-O)n5-CH2-CH2-NH-, где аминогруппа на правом конце указывает на связывание амидной связью с карбоксильной группой в положении 2 сиаловой кислоты на невосстанавливающем конце в 1-3 цепи разветвленных цепей β-Man в N297-гликане;
каждая звездочка указывает на связывание с линкером L, в частности, атомом азота в положении 1 или 3 1,2,3-триазольного кольца Lb в линкере L; и
n5 представляет собой целое число от 2 до 10, и предпочтительно целое число от 2 до 5.
[0225]
N297-(Fuc)MSG2 представлен следующей структурной формулой или формулой последовательности:
[0226]
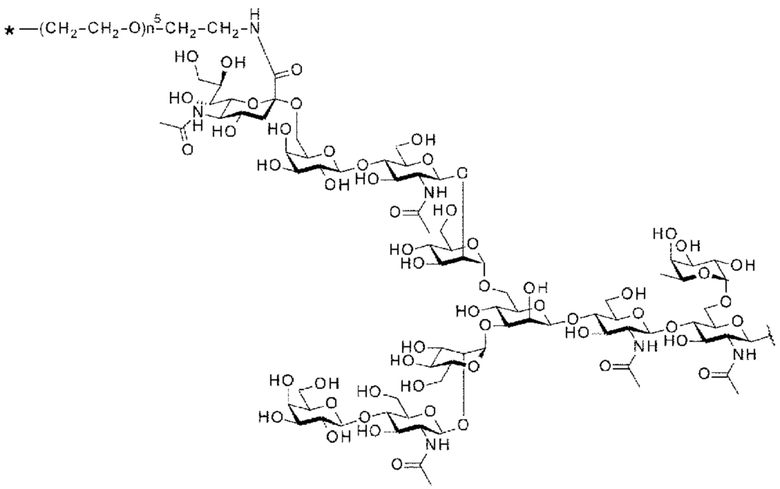
[0227]
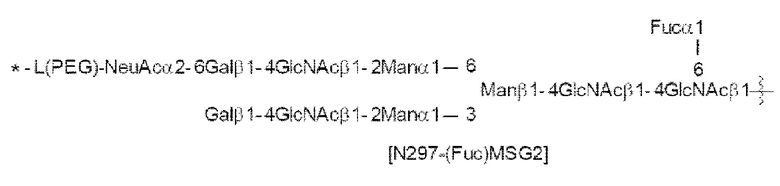
[0228]
В формулах каждая волнистая линия указывает на связывание с Asn297 антитела;
L(PEG) представляет собой -(CH2-CH2-O)n5-CH2-CH2-NH-, где аминогруппа на правом конце указывает на связывание амидной связью с карбоксильной группой в положении 2 сиаловой кислоты на невосстанавливающем конце в 1-6 цепи разветвленных цепей β-Man в N297-гликане, и каждая звездочка указывает на связывание с линкером L, в частности, атомом азота в положении 1 или 3 1,2,3-триазольного кольца Lb в линкере L; и
n5 представляет собой целое число от 2 до 10, и предпочтительно целое число от 2 до 5.
[0229]
Если N297-гликан антитела в конъюгате антитела с лекарственным средством согласно настоящему изобретению представляет собой N297-(Fuc)SG, конъюгат антитела с лекарственным средством представляет собой молекулу, с которой были конъюгированы четыре молекулы линкера L и четыре молекулы лекарственного средства D (m2=2), поскольку антитело представляет собой димер.
Если N297-гликан антитела в конъюгате антитела с лекарственным средством согласно настоящему изобретению представляет собой N297-(Fuc)MSG1, N297-(Fuc)MSG2 или их смесь, то конъюгат антитела с лекарственным средством представляет собой молекулу, с которой были конъюгированы две молекулы линкера L и две молекулы лекарственного средства D (m2=1), поскольку антитело представляет собой димер (см. фигуру 1).
[0230]
N297-гликан предпочтительно представляет собой N297-(Fuc)SG, N297-(Fuc)MSG1 или N297-(Fuc)MSG2 и более предпочтительно представляет собой N297-(Fuc)SG.
[0231]
Если N297-гликан антитела в конъюгате антитела с лекарственным средством согласно настоящему изобретению представляет собой N297-(Fuc)SG, N297-(Fuc)MSG1, или N297-(Fuc)MSG2, можно получить высокогомогенный ADC.
[0232]
<3. Способы получения>
Будут описаны типичные способы получения нового производного ЦДН согласно настоящему изобретению или интермедиатов его получения. Далее в тексте номера соединений, представленные в формулах реакций, используют, чтобы отличить соединения друг от друга. В частности, будут приведены упоминания в форме «соединение формулы (1)», «соединение (1)» и т.д. Соединения с другими номерами будут выражены таким же образом.
[0233]
На схеме А - схеме E далее в тексте заместители R1-R5, L1, L2, W1, W2 и Z1-Z3 синонимичны с описанными выше. Каждый заместитель Ra, Rc, Re и Rg представляет собой боковую цепь природной α-аминокислоты. Их примеры могут включать метальную группу, изопропильную группу, втор-бутильную группу, изобутильную группу и бензильную группу. PRO1 представляет собой защитную группу для первичного спирта. PRO1 предпочтительно представляет собой 4,4'-диметокситритильную группу, 4-метокситритильную группу или тому подобные группы. Каждый PRO2, PRO3, PRO7 и PRO8 представляет собой защитную группу для вторичного спирта. Предпочтительно, каждый PRO2, PRO3, PRO7 и PRO8 представляет собой трет-бутилдиметилсилильную группу, триизопропилсилилоксиметильную группу, бензоильную группу, 2-нитробензильную группу, 4-метокситетрагидропиран-4-ильную группу или тому подобные группы. PRO6 представляет собой защитную группу для карбоновой кислоты. PRO6 предпочтительно представляет собой трет-бутильную группу, бензильную группу или тому подобные группы. Каждый PRO5 и PRO9 представляет собой защитную группу для амина. PRO5 предпочтительно представляет собой трет-бутилоксикарбонильную группу, 9-флуоренилметилоксикарбонильную группу, аллилоксикарбонильную группу, 2,2,2-трихлорэтоксикарбонильную группу, бензилоксикарбонильную группу или тому подобные группы, и PRO9 предпочтительно представляет собой 9-флуоренилметилоксикарбонильную группу или 2-(триметилсилил)этоксикарбонильную группу. PRO4 представляет собой защитную группу для спирта или амина. PRO4 предпочтительно представляет собой трет-бутилдиметилсилильную группу, бензоильную группу или тому подобные группы для спирта, и предпочтительно 2-(триметилсилил)этоксикарбонильную группу, аллилоксикарбонильную группу, трет-бутилоксикарбонильную группу или тому подобные группы для амина. Qa представляет собой атом кислорода или атом серы, и Qb представляет собой гидроксигруппу или тиольную группу. Каждый Qa' и Qb' независимо представляет собой отрицательно заряженный атом кислорода (О-) или атом серы (S-). Rx и Ry каждый независимо представляет собой атом галогена или -O-PRO2. n представляет собой целое число от 1 до 3.
[0234] Схема А
Производное ЦДН согласно настоящему изобретению, представленное в (1), можно получить в соответствии со схемой А, описанной далее.
[0235]
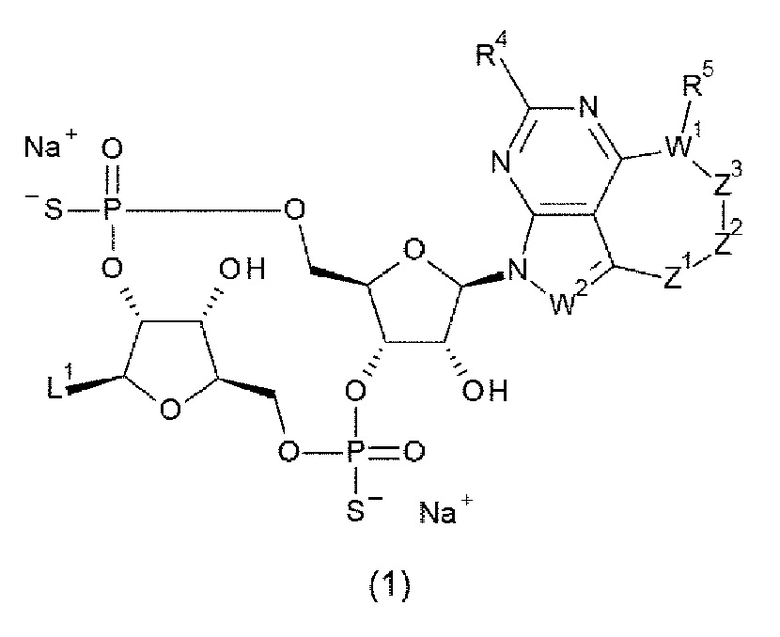
[0236]
Настоящий способ получения представляет собой способ получения соединения, представленного общей формулой (1). Однореакторный синтез применим для этапов А-1-А-5 настоящего способа получения, и это можно осуществить руководствуясь сообщением Gaffhey и др. (Org. Lett. 2010, 12, 3269-3271).
[0237]
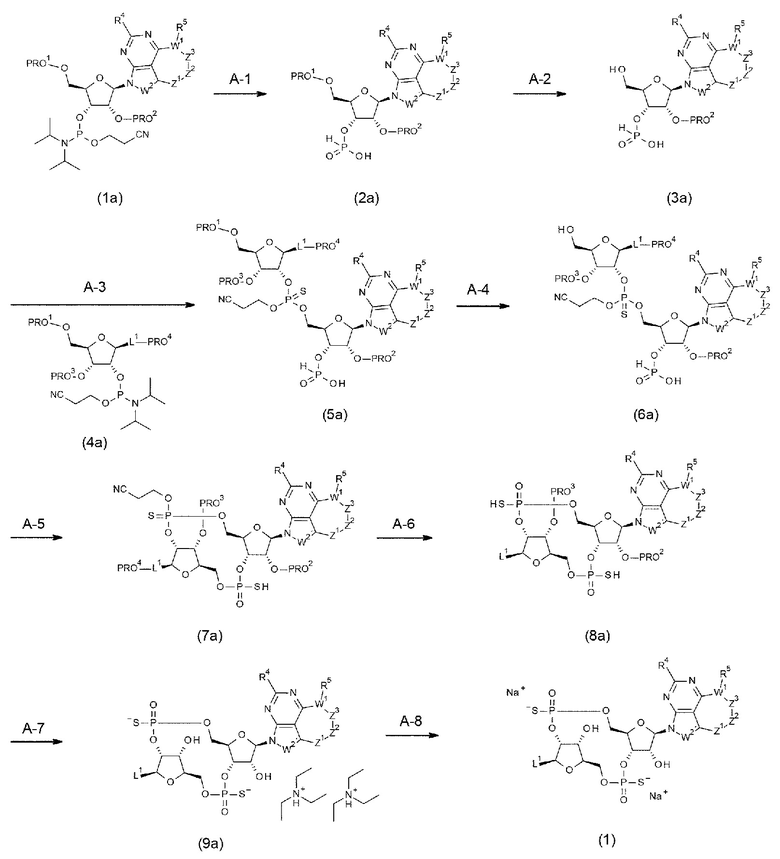
[0238] (Этап А-1)
Данный этап представляет собой этап получения соединения формулы (2а) путем последовательного проведения реакции гидролиза соединения формулы (1а) и удаления цианоэтильной группы из продукта с применением известной методики органической химии.
[0239]
Реакцию гидролиза проводили путем обработки соединения (1а) в растворителе (ацетонитриле, тетрагидрофуране, N,N-диметилформамиде или смешанном растворителе из них) с водой и кислотой (трифторацетат пиридина, 4,5-дицианоимидазол, 1H-тетразол и т.д.) при температуре от -10°С до точки кипения растворителя, используемого для реакции, предпочтительно от 15°С до 35°С. Количество используемых молей воды составляло от 2 молей до избыточного количества молей, предпочтительно от 2 молей до 10 молей, на 1 моль соединения (1а), и количество используемых молей кислоты составляло от 1 моля до избыточного количества молей, предпочтительно от 1 моля до 5 молей, на 1 моль соединения (1а). Время реакции составляло от 1 минуты до 3 часов и предпочтительно от 5 минут до 30 минут. В данную реакционную смесь затем добавляли основание (трет-бутиламин и т.д.), чтобы удалить цианоэтильную группу. Используемое количество молей основания составляло избыточное количество молей, предпочтительно от 30 молей до 50 молей, на 1 моль соединения (1а). Время реакции составляло от 5 минут до 6 часов, и предпочтительно от 15 минут до 1 часа. Реакционную смесь концентрировали при пониженном давлении с получением неочищенного продукта (2а). Неочищенный продукт (2а) можно передать на следующий этап без очистки.
[0240]
(Этап А-2)
Данный этап представляет собой этап получения соединения формулы (3а) путем удаления защитной группы для гидроксигруппы из соединения формулы (2а) с применением известной методики органической химии. До начала реакции данного этапа, неочищенную форму формулы (2а) подвергали азеотропной перегонке от одного до трех раз с ацетонитрилом, по необходимости, для высушивания.
[0241]
Когда PRO1 представлял собой 4,4'-диметокситритильную группу, 4,4'-диметокситритильную группу удаляли путем обработки соединения (2а) в растворителе (дихлорметан, хлороформ, дихлорэтан и т.д.) водой и кислотой (дихлоруксусная кислота, трифторуксусная кислота и т.д.) при температуре от -10°С до точки кипения растворителя, используемого для реакции, предпочтительно от 15°С до 35°С. Количество используемых молей воды составляло избыточное количество молей, предпочтительно от 10 молей до 20 молей, на 1 моль соединения (2а), и кислота была разбавлена растворителем, использованным для реакции, до концентрации от 1% до 50% (об./об.), предпочтительно от 5% до 10% (об./об.), и использовали избыточное количество молей, предпочтительно от 5 молей до 15 молей, разбавленного раствора. Время реакции составляло от 1 минуты до 3 часов, и предпочтительно от 5 минут до 30 минут. Пиридин добавляли в реакционную смесь для гашения. Используемое количество молей пиридина представляло собой количество, достаточное для нейтрализации используемой кислоты, предпочтительно от 2 молей до 10 молей, на 1 моль кислоты. Реакционную смесь концентрировали при пониженном давлении с получением неочищенного продукта (3а). Неочищенный продукт (3а) подвергали азеотропной перегонке от трех до пяти раз с обезвоженным ацетонитрилом. Ацетонитрилу позволяли остаться после последнего азеотропного процесса, и, таким образом, получали от 0,01 M до 1 M раствор в ацетонитриле соединения (3а). Полученный раствор в ацетонитриле непосредственно передавали на следующий этап.
[0242]
(Этап А-3)
Данный этап представляет собой этап получения соединения формулы (5а) путем последовательного проведения реакции сочетания соединения формулы (3а) с соединением формулы (4а) и реакции сульфидирования полученной в результате этого связанной формы с применением известной методики органической химии.
Перед началом реакции данного этапа, соединение (4а) подвергали азеотропной перегонке от трех до пяти раз с обезвоженным ацетонитрилом. Ацетонитрилу позволяли остаться после последнего азеотропного процесса, и, таким образом, получали от 0,01 M до 1 M раствор в ацетонитриле соединения (4а). В данный раствор добавляли осушающий агент (молекулярные сита 3А или молекулярные сита 4А в порошке или гранулах), и раствор хранили до момента применения в атмосфере азота или аргона.
[0243]
Реакцию сочетания проводили путем добавления азеотропно высушенного соединения (4а) в раствор в ацетонитриле соединения (3а) при температуре от 5°С до 35°С.
Время реакции составляло от 1 минуты до 24 часов, и предпочтительно от 5 минут до 6 часов. В данную реакционную смесь затем добавляли сульфидирующий агент (N,N-диметил-N'-(3-сульфанилиден-3Н-1,2,4-дитиазол-5-ил)метанимидамид, 3Н-1,2-бензодитиол-3-он и т.д.) для проведения реакции сульфидирования. Используемое количество молей сульфидирующего агента составляло от 1 моля до 5 молей, предпочтительно от 1 моля до 2 молей, на 1 моль соединения (3а). Время реакции составляло от 5 минут до 24 часов, и предпочтительно от 30 минут до 6 часов. Реакционную смесь концентрировали при пониженном давлении с получением неочищенного продукта (5а). Полученный неочищенный продукт (5а) непосредственно передавали на следующий этап.
[0244]
(Этап А-4)
Данный этап представляет собой этап получения соединения формулы (6а) путем удаления защитной группы для гидроксигруппы из соединения формулы (5а) с применением известной методики органической химии.
[0245]
Когда PRO1 представлял собой 4,4'-диметокситритильную группу, 4,4'-диметокситритильную группу удаляли путем обработки соединения соединения (5а) в растворителе (дихлорметан, хлороформ, дихлорэтан и т.д.) водой и кислотой (дихлоруксусная кислота, трифторуксусная кислота и т.д.) при температуре от -10°С до точки кипения растворителя, используемого для реакции, предпочтительно от 15°С до 35°С. Количество используемых молей воды составляло избыточное количество молей, предпочтительно от 10 до 20 молей, на 1 моль соединения (5а), и кислота была разбавлена растворителем, использованным для реакции, до концентрации от 1% до 50% (об./об.), предпочтительно от 5% до 10% (об./об.), и использовали избыточное количество молей, предпочтительно от 5 молей до 15 молей, разбавленного раствора. Время реакции составляло от 1 минуты до 3 часов, и предпочтительно от 5 минут до 30 минут. Пиридин добавляли в реакционную смесь для гашения. Используемое количество молей пиридина представляло собой количество, достаточное для нейтрализации используемой кислоты, предпочтительно от 10 молей до 200 молей, на 1 моль кислоты. Реакционную смесь концентрировали при пониженном давлении с получением неочищенного продукта (6а). Полученный неочищенный продукт (6а) непосредственно передавали на следующий этап.
[0246]
(Этап А-5)
Данный этап представляет собой этап получения соединения формулы (7а) путем последовательного проведения реакции циклизации и реакции сульфидирования соединения формулы (6а) с применением известной методики органической химии.
Соединение (6а) растворяли в пиридине, и продукт затем концентрировали при пониженном давлении с получением от 0,01 M до 0,5 M раствора пиридина. Реакцию циклизации проводили путем добавления дегидрирующего/конденсирующего агента (2-хлор-5,5-диметил-1,3,2λ5-диоксафосфинан-2-он и т.д.) в данный раствор пиридина при температуре от 5°С до 35°С. Используемое количество молей дегидрирующего/конденсирующего агента составляло от 1 моля до избыточного количества молей, предпочтительно от 3 молей до 5 молей, на 1 моль соединения (6а). Время реакции составляло от 1 минуты до 6 часов, и предпочтительно от 5 минут до 1 часа. Затем проводили реакцию сульфидирования путем добавления воды и сульфидирующего агента (3Н-1,2-бензодитиол-3-он, N,N-диметил-N'-(3-сульфанилиден-3Н-1,2,4-дитиазол-5-ил)метанимидамид и т.д.) в данную реакционную смесь. Используемое количество молей воды составляло избыточное количество молей, предпочтительно от 30 молей до 50 молей, на 1 моль соединения (6а), и используемое количество сульфидирующего агента составляло от 1 моля до 5 молей, предпочтительно от 1 моля до 2 молей, на 1 моль соединения (6а). Время реакции составляло от 5 минут до 12 часов, и предпочтительно от 30 минут до 3 часов. Реакционную смесь добавляли в водный раствор (от 0,1 M до 1 М) гидрокарбоната натрия, и продукт затем перемешивали в течение периода времени от 15 минут до 24 часов для гашения. После того, как реакционную смесь подвергли экстрагированию от одного до пяти раз органическим растворителем (этилацетат, диэтиловый эфир, толуол или смешанный растворитель из них), экстракты объединяли и сушили над безводной солью (безводный сульфат натрия или безводный сульфат магния). Осушающий агент удаляли путем фильтрации и фильтрат концентрировали при пониженном давлении. Полученный в результате этого остаток очищали с помощью колоночной хроматографии на силикагеле [дихлор метан/метанол, этилацетат/метанол, гексан/этилацетат и т.д.], колоночной хроматографии на силикагеле С18 [буфер/ацетонитрил] или их комбинации с получением соединения (7а) в виде смеси двух или более диастереомеров или двух или более чистых диастереомеров. Хотя на данном этапе во множестве случаев получают два диастереомера, дополнительно можно получить один или два диастереомера для некоторых типов сырьевых материалов (1а) и (4а). Даже если полученное соединение (7а) находится в виде смеси множества диастереомеров, смесь можно передать на следующий этап без дополнительной очистки.
[0247]
(Этап А-6)
Данный этап представляет собой этап получения соединения формулы (8а) путем одновременного удаления цианоэтильной группы и всех арильных защитных групп из соединения формулы (7а) с применением известной методики органической химии. Данный этап проводили в автоклаве или в запечатанной пробирке, при необходимости.
[0248]
Когда PRO4 представляла собой бензоильную группу, цианоэтильную группу и бензоильную группу удаляли путем обработки соединения соединения (7а) в растворителе (метанол, этанол, тетрагидрофуран или смешанный растворитель из них) 28% (об./об.) водным аммиаком при температуре от 5°С до точки кипения растворителя, используемого для реакции. Используемое количество молей аммиака составляло избыточное количество молей, предпочтительно от 300 молей до 3000 молей, на 1 моль соединения (7а). Время реакции составляло от 30 минут до 96 часов, и предпочтительно от 2 часов до 48 часов. Реакционную смесь концентрировали, при необходимости, и остаток очищали с помощью препаративной ВЭЖХ [буфер/ацетонитрил, буфер/метанол и т.д.], колоночной хроматографии на силикагеле С18 [буфер/ацетонитрил, буфер/метанол и т.д.] или их комбинации с получением соединения (8а). Даже когда полученное соединение (8а) находится в виде смеси диастереомеров, смесь можно передать на следующий этап без дополнительной очистки. На данном этапе соединение (8а) можно передать на следующий этап без очистки.
[0249]
(Этап А-7)
Данный этап представляет собой этап получения соединения формулы (9а) путем одновременного удаления всех силильных защитных групп из соединения формулы (8а) с применением известной методики органической химии.
Когда каждая из PRO2 и PRO3 представляла собой трет-бутилдиметилсилильную группу, трет-бутилдиметилсилильные группы удаляли путем непосредственной обработки соединения (8а) тригидрофторидом триэтиламина при температуре от 5°С до 100°С, предпочтительно от 35°С до 60°С. Используемое количество молей тригидрофторида триэтиламина составляло избыточное количество молей, предпочтительно от 100 до 200 молей, на 1 моль соединения (8а). Время реакции составляло от 30 минут до 24 часов, и предпочтительно от 2 часов до 12 часов. После охлаждения реакционной смеси до комнатной температуры, охлажденный до температуры льда смешанный от 3:1 до 10:1 (об./об.) раствор 1 M водного раствора гидрокарбоната триэтиламмония и триэтиламина наливали малыми частями в реакционную смесь для гашения. При необходимости, реакционную смесь можно вливать в охлажденный до температуры льда смешанный раствор 1 M водного раствора гидрокарбоната триэтиламмония и триэтиламина. В данном случае, реакционный сосуд промывали ацетонитрилом и водой. Количество молей триэтиламина, которое необходимо использовать, представляет собой количество, достаточное для изменения условий реакционной смеси до слабоосновных, предпочтительно составляет приблизительно 2 моля, на 1 моль тригидрофторида триэтиламина. После того, как компонент органического растворителя реакционной смеси отгоняли при пониженном давлении, оставшийся водный раствор очищали с помощью препаративной ВЭЖХ [буфер/ацетонитрил, буфер/метанол и т.д.], колоночной хроматографии на силикагеле С18 [буфер/ацетонитрил, буфер/метанол и т.д.] или их комбинации с получением соединения (9а) в виде одного диастереомера.
[0250]
(Этап А-8)
Данный этап представляет собой этап получения соединения формулы (1) путем ионного обмена соединения формулы (9а) с применением известной методики органической химии.
Катионообменную смолу (смола ВТ AG (R) 50W-X2, 100 - 200 меш, водородного типа) суспендировали в чистой воде и заполняли ею пустой картридж колонки. Использованное количество катионообменной смолы было в 10 раз - в 50 раз больше по массе, чем количество соединения (9а). После того, как избыточной части чистой воды позволили под действием гравитации стечь вниз, 3 × объемам колонки 1 M водного раствора гидроксида натрия позволили под действием гравитации стечь вниз и 6 × объемам колонки чистой воды затем позволили под действием гравитации стечь вниз. Соединение (9а) растворяли приблизительно в 3 × объемах колонки чистой воды и наполняли им колонку. Если соединение плохо растворимо в чистой воде, можно использовать смесь с небольшим количеством органического растворителя (ацетонитрил, метанол и т.д.). Раствор, которому позволяли под действием гравитации стечь вниз, разделяли и собирали, а затем дополнительно элюировали 6 × объемами колонки чистой воды или т.п., и фракции разделяли и собирали. Фракции, содержащие целевой продукт, объединяли и лиофилизировали с получением соединения (1) в виде одного диастереомера.
[0251]
Схема А
Производное ЦДН согласно настоящему изобретению, представленное в (1'), можно получить в соответствии со схемой А', описанной далее.
[0252]
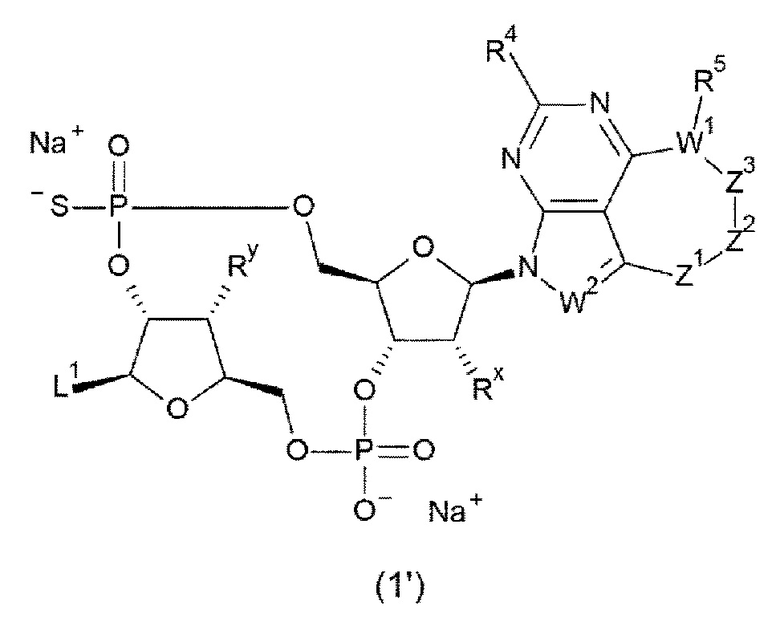
[0253]
Настоящий способ получения представляет собой способ получения соединения, представленного общей формулой (1'), указанный способ представляет собой частично модифицированную форму схемы А. В частности, соединение основной формулы (1') можно получить путем замены этапа А-5 схемы А на этап А'-5, показанный ниже. Если каждый заместитель Rx и Ry представляет собой атом галогена, этап А-7 можно опустить.
[0254]
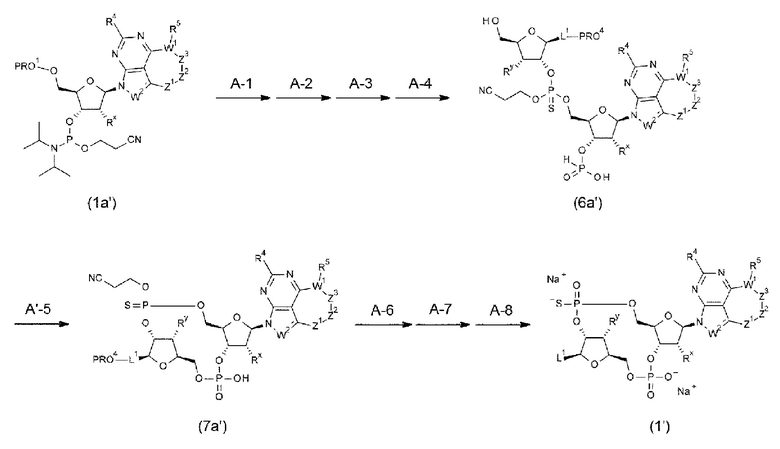
[0255]
(Этап А'-5)
Данный этап представляет собой этап получения соединения формулы (7а') путем последовательного проведения реакции циклизации и реакции окисления соединения формулы (6а') с применением известной методики органической химии.
Соединение (6а') растворяли в пиридине, и продукт затем концентрировали при пониженном давлении с получением от 0,01 M до 0,5 M раствора пиридина. Реакцию циклизации проводили путем добавления дегидрирующего/конденсирующего агента (2-хлор-5,5-диметил-1,3,2λ5-диоксафосфинан-2-он и т.д.) в данный раствор пиридина при температуре от 5°С до 35°С. Используемое количество молей дегидрирующего/конденсирующего агента составляло от 1 моля до избыточного количества молей, предпочтительно от 3 молей до 5 молей, на 1 моль соединения (6а'). Время реакции составляло от 1 минуты до 6 часов, и предпочтительно от 5 минут до 1 часа. Затем проводили реакцию окисления путем добавления воды и окисляющего агента (йод и т.д.) в данную реакционную смесь. Используемое количество молей воды составляло от 0 молей до избыточного количества молей, предпочтительно от 30 молей до 50 молей, на 1 моль соединения (6а'), и используемое количество окисляющего агента составляло от 2 молей до 10 молей, предпочтительно от 3 молей до 5 молей, на 1 моль соединения (6а'). Время реакции составляло от 5 минут до 12 часов, и предпочтительно от 30 минут до 3 часов. Реакционную смесь добавляли в водный раствор (от 0,1 M до 1 М) гидрокарбоната натрия, и продукт перемешивали в течение периода времени от 15 минут до 24 часов для гашения. После того, как реакционную смесь подвергли экстрагированию от одного до пяти раз органическим растворителем (этилацетат, диэтиловый эфир, толуол или смешанный растворитель из них), экстракты объединяли и сушили над безводной солью (безводный сульфат натрия или безводный сульфат магния). Осушающий агент удаляли путем фильтрации и фильтрат концентрировали при пониженном давлении. Полученный в результате этого остаток очищали с помощью колоночной хроматографии на силикагеле [дихлор метан/метанол, этилацетат/метанол, гексан/этилацетат и т.д.], колоночной хроматографии на силикагеле С18 [буфер/ацетонитрил] или их комбинации с получением соединения (7а').
[0256]
Схема А''
Производное ЦДН согласно настоящему изобретению, представленное в (1''), можно получить в соответствии со схемой А'', описанной далее.
[0257]
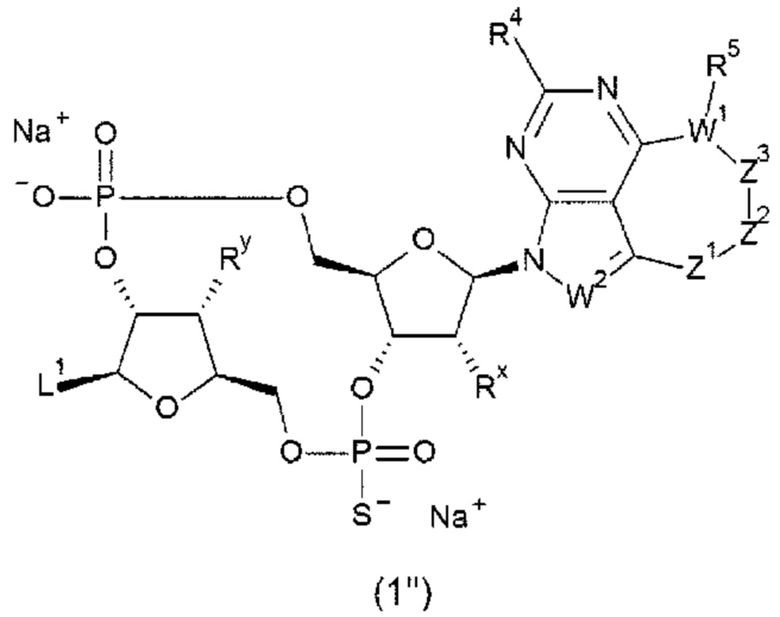
[0258]
Настоящий способ получения представляет собой способ получения соединения, представленного общей формулой (1''), указанный способ представляет собой частично модифицированную форму схемы А. В частности, соединение основной формулы (1'') можно получить путем замены этапа А-3 схемы А на этап А''-3, показанный ниже. Если каждый заместитель Rx и Ry представляет собой атом галогена, этап А-7 можно опустить.
[0259]
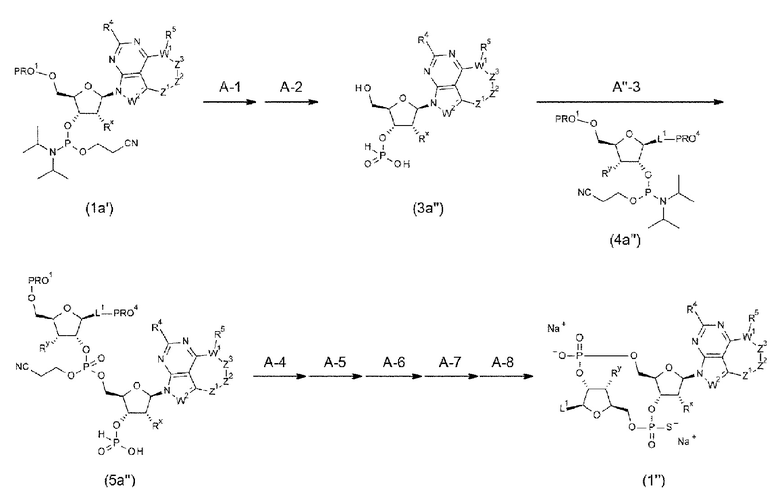
[0260]
(Этап А''-3)
Данный этап представляет собой этап получения соединения формулы (5а'') путем последовательного проведения реакции сочетания соединения формулы (3а'') с соединением формулы (4а'') и реакции окисления полученной в результате этого связанной формы с применением известной методики органической химии.
Перед началом реакции данного этапа, соединение (4а'') подвергали азеотропной перегонке от трех до пяти раз с обезвоженным ацетонитрилом. Ацетонитрилу позволяли остаться после последнего азеотропного процесса, и, таким образом, получали от 0,01 M до 1 M раствора в ацетонитриле соединения (4а''). В данный раствор добавляли осушающий агент (молекулярные сита 3А или молекулярные сита 4А в порошке или гранулах), и раствор хранили до момента применения в атмосфере азота или аргона.
[0261]
Реакцию сочетания проводили путем добавления раствора в ацетонитриле азеотропно высушенного соединения (4а'') в раствор в ацетонитриле соединения (3а'') при температуре от 5°С до 35°С. Время реакции составляло от 1 минуты до 24 часов, и предпочтительно от 5 минут до 6 часов. В данную реакционную смесь затем добавляли окисляющий агент (гидроперекись трет-бутила и т.д.) для проведения реакции окисления.
Используемое количество молей окисляющего агента составляло от 1 моля до 5 молей, предпочтительно от 2 молей до 3 молей, на 1 моль соединения (3а''). Время реакции составляло от 5 минут до 24 часов, и предпочтительно от 30 минут до 6 часов. Насыщенный водный раствор тиосульфата натрия добавляли в реакционную смесь, и продукт перемешивали в течение периода времени от 10 минут до 12 часов для гашения. После того, как реакционную смесь подвергли экстрагированию от одного до пяти раз органическим растворителем (смешанный растворитель из дихлорметана и метанола и т.д.), экстракты объединяли и сушили над безводной солью (безводный сульфат натрия или безводный сульфат магния). Осушающий агент удаляли путем фильтрации и фильтрат концентрировали при пониженном давлении с получением неочищенного продукта (5а''). Полученный неочищенный продукт (5а'') непосредственно передавали на следующий этап.
[0262]
Схема А'''
Производное ЦДН согласно настоящему изобретению, представленное в (1'''), можно получить в соответствии со схемой А''', описанной далее.
[0263]
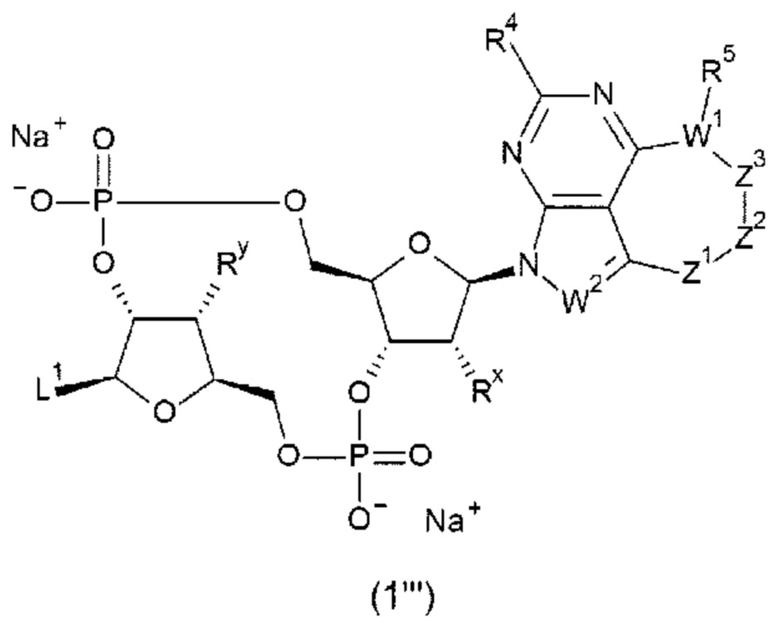
[0264]
Настоящий способ получения представляет собой способ получения соединения, представленного общей формулой (1'''), указанный способ представляет собой частично модифицированную форму схемы А. В частности, соединение основной формулы (1''') можно получить путем замены этапа А-3 и этапа А-5 схемы А, соответственно, на этап А''-3 и этап А'-5. Если каждый заместитель Rx и Ry представляет собой атом галогена, этап А-7 можно опустить.
[0265]
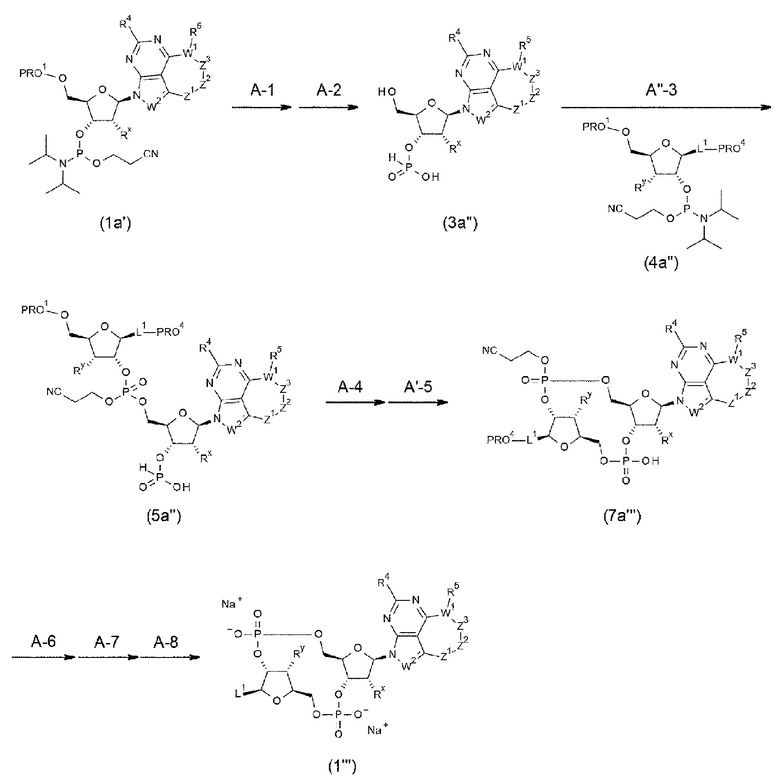
[0266]
Схема В. Предшественник конъюгата (конъюгирование гликаном).
Предшественник конъюгата согласно настоящему изобретению, представленный в (2), можно получить в соответствии со схемой В, описанной далее.
[0267]
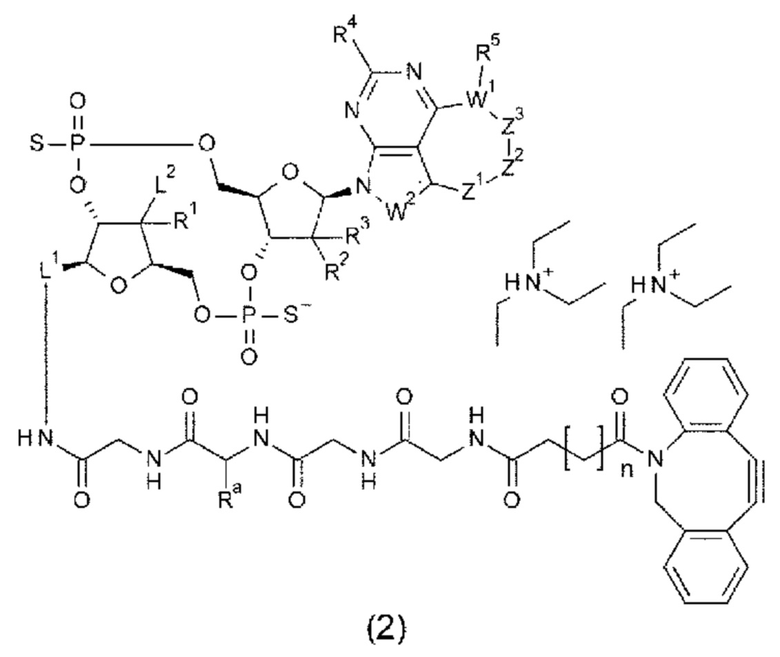
[0268]
Настоящий способ получения представляет собой способ получения предшественника конъюгата (2), где L1 содержит в качестве заместителя -NH2 в любом положении.
[0269]
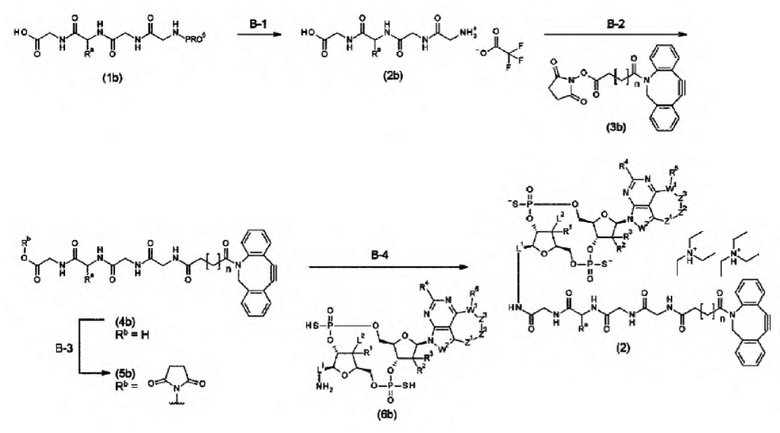
[0270]
(Этап В-1)
Данный этап представляет собой этап получения соединения формулы (2b) путем удаления защитной группы из соединения формулы (1b) с применением известной методики органической химии.
[0271]
Когда PRO5 представлял собой трет-бутилоксикарбонильную группу, защитную группу удаляли путем обработки соединения (1b) в растворителе (дихлорметан, диоксан, ацетонитрил, этилацетат, тетрагидрофуран или смешанный растворитель из них) трифторуксусной кислотой при температуре от -10°С до точки кипения растворителя, используемого для реакции, предпочтительно от 15°С до 35°С. Используемое количество молей трифторуксусной кислоты составляло избыточное количество молей, предпочтительно от 20 молей до 50 молей, на 1 моль соединения (1b). Время реакции составляло от 5 минут до 24 часов, и предпочтительно от 30 минут до 6 часов. Реакционную смесь концентрировали при пониженном давлении, а затем суспендировали в толуоле и полученную суспензию снова концентрировали при пониженном давлении. Данную процедуру повторяли от двух до пяти раз. Растворитель (диэтиловый эфир, диизопр опило вый эфир, гексан, дихлорметан, этилацетат или смешанный растворитель из них) добавляли, чтобы получить взвесь, и твердый продукт затем собирали путем фильтрации с получением неочищенного продукта (2b). Неочищенный продукт (2b) передавали на следующий этап без какой-либо дополнительной очистки.
[0272]
(Этап В-2)
Данный этап представляет собой этап получения соединения формулы (4b) путем осуществления амидирования соединения формулы (2b) соединением формулы (3b) с применением известной методики органической химии.
[0273]
Амидирование проводили путем приведения во взаимодействие соединения (2b) в растворителе (N,N-диметилформамид, N-метилпирролидон, N,N-диметилацетамид, ацетонитрил и т.д.) с основанием (триэтиламин, N,N-диизопропилэтиламин и т.д.) и соединением (3b) при температуре от 5°С до 35°С. Используемое количество молей основания составляло от 1 моля до 5 молей на 1 моль соединения (2b), и используемое количество соединения (3b) составляло от 0,5 молей до 1,5 молей на 1 моль соединения (2b). Время реакции составляло от 10 минут до 72 часов, и предпочтительно от 1 часа до 24 часов. Реакционную смесь вливали в двухслойную смесь органического растворителя (дихлорметан, хлороформ, этилацетат, метанол или смешанный растворитель из них) и воды или кислого водного раствора (от 0,1 до 1 M соляная кислота, водный раствор лимонной кислоты и т.д.), и осуществляли экстрагирование продукта от одного до пяти раз органическим растворителем. Экстракты объединяли и промывали рассолом, а затем сушили над безводной солью (безводный сульфат натрия или безводный сульфат магния). Осушающий агент удаляли путем фильтрации и фильтрат концентрировали при пониженном давлении. В качестве альтернативы, реакционную смесь можно непосредственно концентрировать при пониженном давлении, при этом процесс отделения жидкости опускали, и передать на последующую очистку на колонке с силикагелем. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле [дихлорметан/метанол, этилацетат/метанол и т.д.] с получением соединения (4b). При необходимости, чистоту соединения (4b) можно повысить путем растворения соединения (4b) в хорошем растворителе (этилацетат, ацетонитрил, дихлорметан, метанол, или смешанный растворитель из них), затем повторного преципитирования соединения (4b) с добавлением слабого растворителя (диэтиловый эфир, диизопропиловый эфир, гексан и т.д.), и сбора твердого продукта путем фильтрации.
[0274]
(Этап В-3)
Данный этап представляет собой этап получения соединения формулы (5b) путем проведения этерификации соединения формулы (4b) с применением известной методики органической химии.
Этерификацию проводили путем приведения во взаимодействие соединения (4b) в растворителе (N,N-диметилформамид, N,N-диметилацетамид, N-метилпирролидон, ацетонитрил и т.д.) с N-гидроксисукцинимидом и конденсирующим агентом (1-этил-3-(3-диметиламинопропил)карбодиимида гидрохлорид и т.д.) при температуре от 5°С до 35°С.
Используемое количество молей N-гидроксисукцинимида и используемое количество конденсирующего агента каждое составляло от 1 моля до 3 молей на 1 моль соединения (4b). Время реакции составляло от 30 минут до 72 часов, и предпочтительно от 2 часов до 24 часов. Реакционную смесь разбавляли органическим растворителем (дихлорметан, хлороформ, этилацетат или смешанный растворитель из них), а затем промывали от трех до пяти раз ледяной водой. Органический слой сушили, используя безводную соль (безводный сульфат натрия или безводный сульфат магния). Осушающий агент удаляли путем фильтрации, и фильтрат затем концентрировали при пониженном давлении с получением неочищенного продукта (5b). При необходимости, полученное соединение (5b) можно очистить с помощью колоночной хроматографии на силикагеле С18 [только ацетонитрил]. Чистоту полученного соединения (5b) можно повысить путем растворения соединения (5b) в хорошем растворителе (этилацетат, ацетонитрил, дихлорметан или смешанный растворитель из них), затем повторного преципитирования соединения (5b) с добавлением слабого растворителя (диэтиловый эфир, диизопропиловый эфир, гексан и т.д.), и сбора твердого продукта путем фильтрации.
[0275]
(Этап В-4)
Данный этап представляет собой этап получения соединения формулы (2) путем проведения реакции конденсации соединения формулы (5b) с соединением формулы (6b) с применением известной методики органической химии.
[0276]
Реакцию конденсации проводили путем приведения во взаимодействие соединения (6b) в растворителе (N,N-диметилформамид, N,N-диметилацетамид, N-метилпирролидон, ацетонитрил и т.д.) с основанием (триэтиламин, N,N-диизопропилэтиламин и т.д.) и соединением (5b) при температуре от -10°С до 100°С, предпочтительно от 15°С до 35°С. Используемое количество молей основания составляло от 2 молей до 5 молей на 1 моль соединения (6b), и используемое количество соединения (5b) составляло от 1 моля до 2 молей на 1 моль соединения (6b). Время реакции составляло от 5 минут до 24 часов, и предпочтительно от 1 часа до 6 часов. Бензиламин добавляли в реакционную смесь для гашения. Используемое количество молей бензиламина составляло от 4 молей до 10 молей на 1 моль соединения (6b). Реакционную смесь частично концентрировали при пониженном давлении, при необходимости, и оставшийся раствор очищали с помощью препаративной ВЭЖХ [буфер/ацетонитрил, буфер/метанол и т.д.], колоночной хроматографии на силикагеле С18 [буфер/ацетонитрил, буфер/метанол и т.д.] или их комбинации с получением соединения (2).
[0277]
Схема В'. Предшественник конъюгата (конъюгирование цистенном).
Схема В'
Предшественник конъюгата согласно настоящему изобретению, представленный в (2'), можно получить в соответствии со схемой В', описанной далее.
[0278]
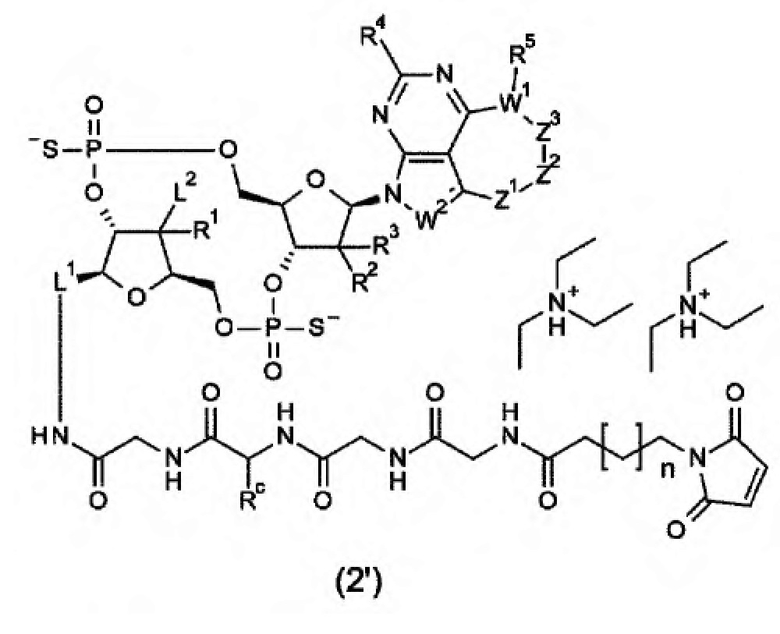
[0279]
Настоящий способ получения представляет собой способ получения предшественника конъюгата (2'), где L1 содержит в качестве заместителя -NH2 в любом положении.
[0280]
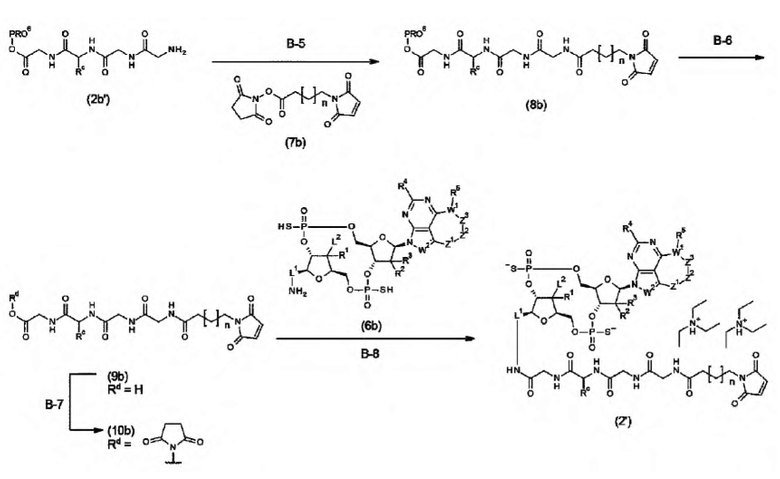
[0281]
(Этап В-5)
Данный этап представляет собой этап получения соединения формулы (8b) путем осуществления амидирования соединения формулы (2b') соединением формулы (7b) с применением известной методики органической химии. Соединение (8b) получали в соответствии с процедурой, описанной на этапе В-2 схемы В, за исключением того, что не используют основание.
[0282]
(Этап В-6)
Данный этап представляет собой этап получения соединения формулы (9b) путем удаления защитной группы из соединения формулы (8b) с применением известной методики органической химии. Соединение (9b) получали в соответствии с процедурой, описанной на этапе В-1 схемы В, за исключением того, что когда PRO6 представляла собой трет-бутильную группу, в процессе очистки использовали колоночную хроматографию на силикагеле [дихлорметан/метанол].
[0283]
(Этап В-7)
Данный этап представляет собой этап получения соединения формулы (10b) путем проведения этерификации соединения формулы (9b) с применением известной методики органической химии. Соединение (10b) получали в соответствии с процедурой, описанной на этапе В-3 схемы В.
[0284]
(Этап В-8)
Данный этап представляет собой этап получения соединения формулы (2') путем проведения реакции конденсации соединения формулы (6b) с соединением формулы (10b) с применением известной методики органической химии. Соединение (2') получали в соответствии с процедурой, описанной на этапе В-4 схемы В.
[0285]
Схема С
Предшественник конъюгата согласно настоящему изобретению, представленный в (3), можно получить в соответствии со схемой С, описанной далее.
[0286]
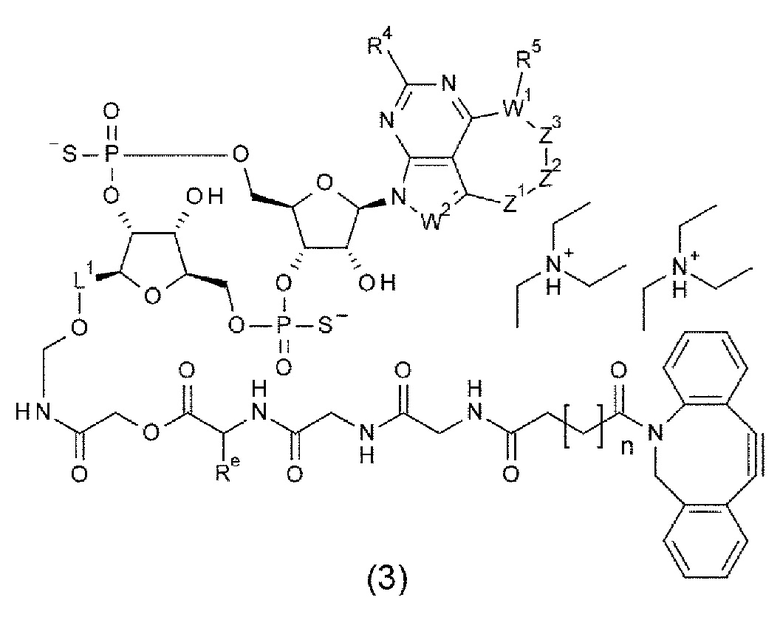
[0287]
Настоящий способ получения представляет собой способ получения предшественника конъюгата (3), где L1 содержит в качестве заместителя гидроксигруппу в любом положении.
[0288]
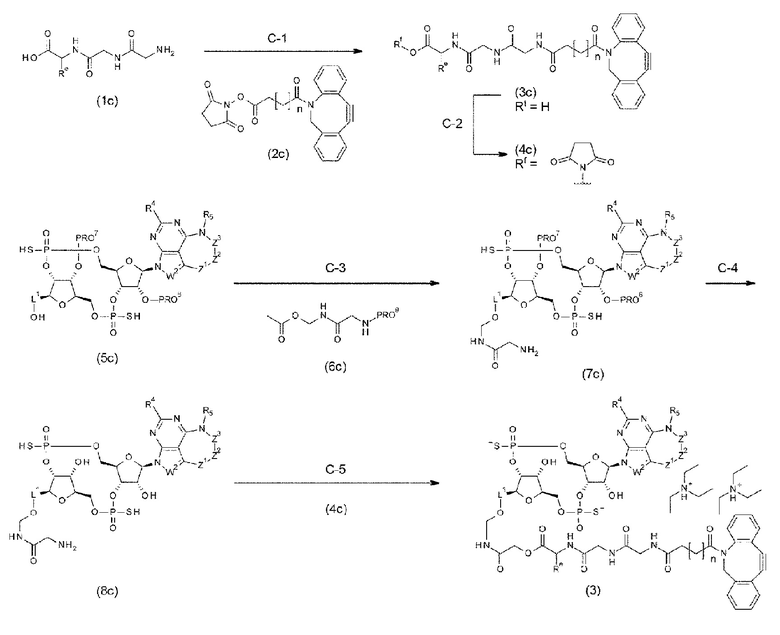
[0289]
(Этап C-1)
Данный этап представляет собой этап получения соединения формулы (3с) путем осуществления амидирования соединения формулы (1с) соединением формулы (2с) с применением известной методики органической химии. Соединение (3с) получали в соответствии с процедурой, описанной на этапе В-2 схемы В.
[0290]
(Этап С-2)
Данный этап представляет собой этап получения соединения формулы (4с) путем проведения этерификации соединения формулы (3с) с применением известной методики органической химии. Соединение (4с) получали в соответствии с процедурой, описанной на этапе В-3 схемы В.
[0291]
(Этап С-3)
Данный этап представляет собой этап получения соединения формулы (7с) путем последовательного проведения реакции сочетания (аминометиленирования) соединения формулы (5с) с соединением формулы (6с) и снятия защитных групп с полученной в результате этого связанной формы с применением известной методики органической химии.
[0292]
Когда PRO9 представляла собой 9-флуоренилметилоксикарбонильную группу, проводили аминометиленирование путем приведения во взаимодействие соединения (5с) в тетрагидрофуране с соединением (6с) и кислотой (п-толуолсульфоновая кислота и т.п.) при температуре от 5°С до 35°С. Используемое количество молей соединения (6с) составляло от 1 моля до 20 молей, предпочтительно от 2 молей до 10 молей, на 1 моль соединения (5с), и используемое количество молей кислоты составляло от 0,05 моля до избыточного количества молей, предпочтительно 0,1 моля до 3 молей, на 1 моль соединения (5с). Время реакции составляло от 30 минут до 72 часов, и предпочтительно от 2 часов до 24 часов. Затем проводили снятие защитных групп путем добавления основания (1,8-диазабицикло[5,4,0]-7-ундецен и т.д.) в реакционную смесь. Когда реакционная смесь содержит суспензию, растворитель (N,N-диметилформамид и т.д.) можно дополнительно добавить, чтобы растворить суспензию, при необходимости, перед проведением реакции. Используемое количество молей основания составляло избыточное количество молей, предпочтительно от 5 молей до 20 молей, на 1 моль соединения (5с). Время реакции составляло от 10 минут до 24 часов, и предпочтительно от 2 часов до 12 часов. Воду добавляли в реакционную смесь, и продукт непосредственно очищали с помощью колоночной хроматографии на силикагеле С18 [буфер/ацетонитрил и т.д.] с получением соединения (7с).
[0293]
(Этап С-4)
Данный этап представляет собой этап получения соединения формулы (8с) путем удаления защитных групп из соединения формулы (7с) с применением известной методики органической химии. Когда PRO7 и PRO8 каждая представляла собой трет бутилдиметилсилильную группу, соединение (8с) получали в соответствии с процедурой, описанной на этапе А-7 схемы А.
[0294]
(Этап С-5)
Данный этап представляет собой этап получения соединения формулы (3) путем проведения реакции конденсации соединения формулы (8с) с соединением формулы (4с) с применением известной методики органической химии. Соединение (3) получали в соответствии с процедурой, описанной на этапе В-4 схемы В.
[0295]
Схема С'
Предшественник конъюгата согласно настоящему изобретению, представленный в (3'), можно получить в соответствии со схемой С', описанной далее.
[0296]
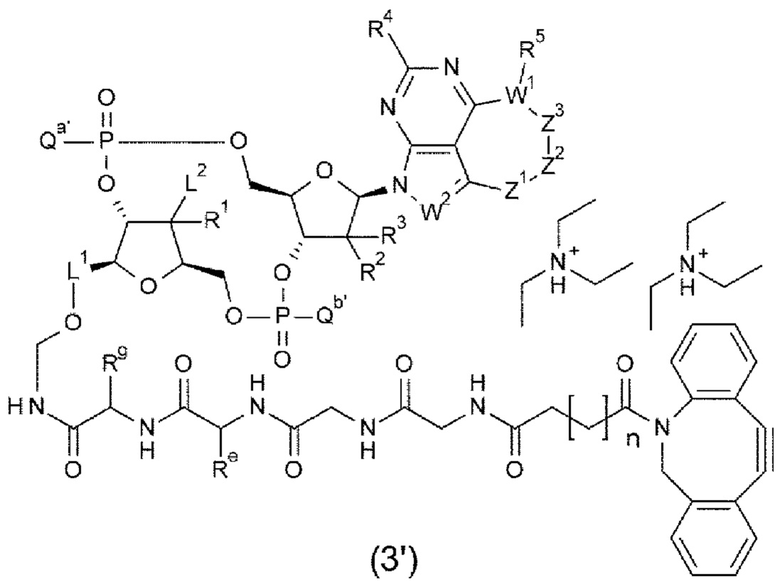
[0297]
Настоящий способ получения представляет собой способ получения предшественника конъюгата (3'), где L1 содержит в качестве заместителя гидроксигруппу в любом положении.
[0298]
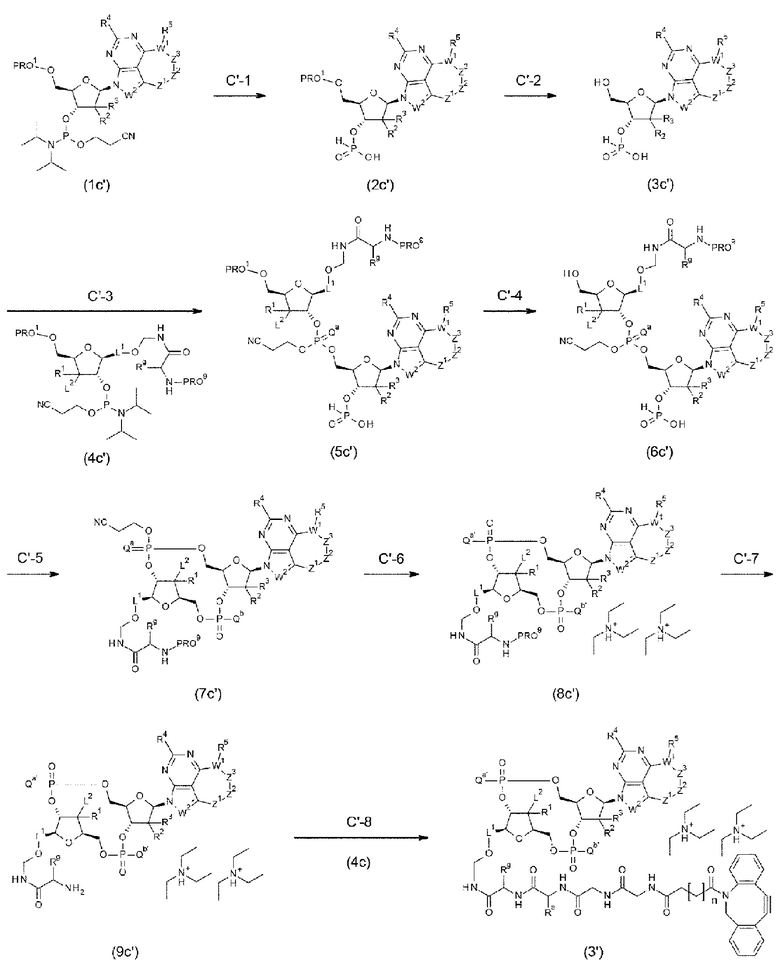
[0299]
(Этап C'-1)
Данный этап представляет собой этап получения соединения формулы (2с') путем последовательного проведения реакции гидролиза соединения формулы (1с') и удаления цианоэтильной группы из продукта с применением известной методики органической химии. Соединение (2с') получали в соответствии с процедурой, описанной на этапе А-1 схемы А.
[0300]
(Этап С'-2)
Данный этап представляет собой этап получения соединения формулы (3с') путем удаления защитной группы для гидроксигруппы из соединения (2с') с применением известной методики органической химии. Соединение (3с') получали в соответствии с процедурой, описанной на этапе А-2 схемы А.
[0301]
(Этап С'-3)
Данный этап представляет собой этап получения соединения формулы (5с') путем последовательного проведения реакции сочетания соединения формулы (3с') с соединением формулы (4с') и реакции сульфидирования или реакции окисления полученной в результате этого связанной формы с применением известной методики органической химии. Соединение (5с') получали в соответствии с процедурой, описанной на этапе А-3 схемы А или этапе А''-3 схемы А''.
[0302]
(Этап С'-4)
Данный этап представляет собой этап получения соединения формулы (6с') путем удаления защитной группы для гидроксигруппы из соединения формулы (5с') с применением известной методики органической химии. Соединение (6с') получали в соответствии с процедурой, описанной на этапе А-4 схемы А.
[0303]
(Этап С'-5)
Данный этап представляет собой этап получения соединения формулы (7с') путем последовательного проведения реакции циклизации соединения формулы (6с') и реакции сульфидирования или реакции окисления продукта с применением известной методики органической химии. Соединение (7с') получали в соответствии с процедурой, описанной на этапе А-5 схемы А или этапе А'-5 схемы А'.
[0304]
(Этап С'-6)
Данный этап представляет собой этап получения соединения формулы (8с') путем одновременного удаления цианоэтильной группы и всех арильных защитных групп из соединения формулы (7с') с применением известной методики органической химии. Соединение (8с') получали в соответствии с процедурой, описанной на этапе А-6 схемы А.
[0305]
(Этап С'-7)
Данный этап представляет собой этап получения соединения формулы (9с') путем одновременного удаления всех силильных защитных групп из соединения формулы (8с') с применением известной методики органической химии. Когда PRO9 представляла собой 2-(триметилсилил)этоксикарбонильную группу, 2-(триметилсилил)этоксикарбонильную группу удаляли путем обработки соединения (8с') раствором в тетрагидрофуране фторида тетрабутиламмония при температуре от 5°С до 100°С, предпочтительно от 35°С до 60°С. Используемое количество молей фторида тетрабутиламмония составляло избыточное количество молей, предпочтительно от 10 до 30 молей, на 1 моль соединения (8с'). Время реакции составляло от 1 часа до 48 часов, и предпочтительно от 4 часов до 24 часов. После того, как реакционную смесь разбавляли добавлением в нее буфера, компонент органического растворителя отгоняли при пониженном давлении, при необходимости. Остаток очищали с помощью препаративной ВЭЖХ [буфер/ацетонитрил, буфер/метанол и т.д.], колоночной хроматографии на силикагеле С18 [буфер/ацетонитрил, буфер/метанол и т.д.] или их комбинации с получением соединения (9с').
[0306]
(Этап С'-8)
Данный этап представляет собой этап получения соединения формулы (3') путем проведения реакции конденсации соединения формулы (9с') с соединением формулы (4с) с применением известной методики органической химии. Соединение (3') получали в соответствии с процедурой, описанной на этапе В-4 схемы В.
[0307]
Схема D. Получение ремоделированного гликаном антитела.
Ремоделированное гликаном антитело можно получить, применяя способ, представленный в следующей формуле, например, руководствуясь способом, описанным в WO 018/003983.
[0308]
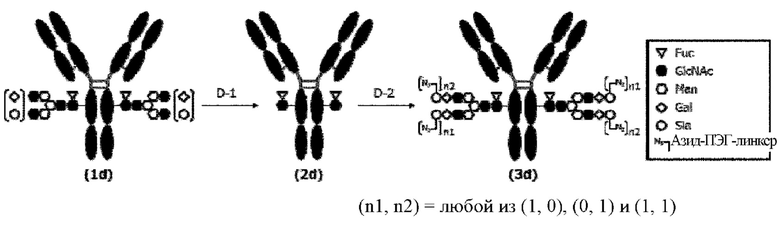
[0309]
(Этап D-1)
Данный этап представляет собой этап получения антитела с укороченным гликаном путем гидролитического расщепления гликозидной связи в GlcNAcβ1-4GlcNAc структуры хитобиозы на восстанавливающем конце N-связанного гликана, связанного с аспарагином в положении 297 последовательности аминокислот нацеленного антитела (N297-связанного гликана), с применением известной ферментативной реакции.
Нацеленное антитело (1d) (10 мг/мл) в буфере (например, фосфатном буфере) подвергали реакции гидролиза гликозидной связи между GlcNAcβ1 и 4GlcNAc в структуре хитобиозы на восстанавливающем конце с применением гидролазы, такой как фермент EndoS дикого типа, при температуре от 0°С до 40°С. Время реакции составляло от 10 минут до 72 часов, и предпочтительно от 1 часа до 6 часов. Используемое количество фермента EndoS дикого типа составляло от 0,1 до 10 мг, предпочтительно от 0,1 до 3 мг, на 100 мг антитела (1d). После завершения реакции продукт очищали с помощью аффинной хроматографии (HiTrap rProtein A FF (5 мл) (производства GE Healthcare)) и/или колонки с гидроксиапатитом (Bio-Scale Mini СНТ, картридж I типа (5 мл) (производства Bio-Rad Laboratories, Inc.)) с получением (Fucα1,6)GlcNAc-антитела (2d).
[0310]
(Этап D-2)
Данный этап представляет собой этап получения ремоделированного гликаном антитела (3d) путем связывания оксазолиновой формы гликана SG-типа или MSG (MSG1, MSG2)-типа (здесь и далее называют «оксазолиновой формой азидгликана»), содержащей линкер PEG, содержащий азидную группу, с (Fucα1,6)GlcNAc-антителом (2d), полученным на этапе D-1, с применением известной ферментативной реакции.
[0311]
Антитело (2d) в буфере (например, фосфатном буфере) подвергали реакции трансгликозилирования путем приведения во взаимодействие с оксазолиновой формой азидгликана в присутствии гликозилтрансферазы, такой как EndoS (D233Q/Q303L), при температуре от 0°С до 40°С. Время реакции составляло от 10 минут до 72 часов, и предпочтительно от 1 часа до 6 часов. Используемое количество фермента EndoS (D233Q/Q303L) составляло от 1 до 10 мг, предпочтительно от 1 до 3 мг, на 100 мг антитела, и используемое количество оксазолиновой формы азидгликана составляло от 2 эквивалентов до избыточного эквивалента, предпочтительно от 4 эквивалентов до 20 эквивалентов. После завершения реакции продукт очищали с помощью аффинной хроматографии (HiTrap rProtein A FF (5 мл) (производства GE Healthcare)) и/или колонки с гидроксиапатитом (Bio-Scale Mini СНТ, картридж I типа (5 мл) (производства Bio-Rad Laboratories, Inc.)) с получением ремоделированного гликаном антитела (3d).
[0312]
При получении ремоделированного гликаном антитела, можно осуществить концентрирование водного раствора антитела, измерение концентрации и замену буфера в соответствии с Общими процедурами от А до С, описанными далее.
Оксазолиновую форму азидгликана SG-типа синтезировали, руководствуясь способом, описанным в WO2018/003983. В качестве примера, способ синтеза [N3-PEG(3)]2-SG(10)-Ox (соединение 1-10, описанное в WO2018/003983) представлен в следующей формуле.
[0313]
[0314]
Аналогично, оксазолиновую форму азидгликана MSG-типа синтезировали, руководствуясь способом, описанным в WO2018/003983. В качестве примера, способ синтеза [N3-PEG(3)]-MSG1(9)-Ox (соединение 1-11, описанное в WO 2018/003983) представлен в следующей формуле.
[0315]
[0316]
Схема Е. Конъюгирование антитела и лекарственного средства (конъюгирование гликаном 1).
[0317]
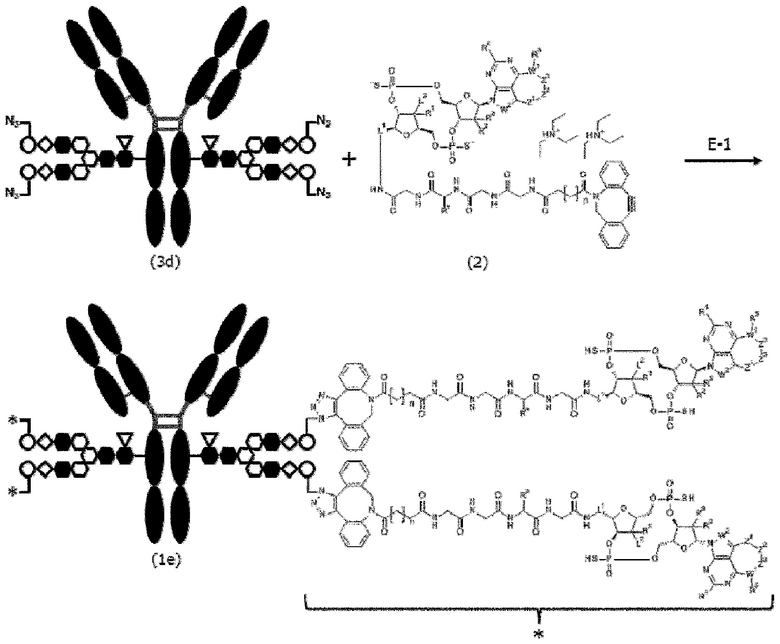
[0318]
В формуле две звездочки (*) на левой стороне конъюгата антитела с лекарственным средством (1е) обозначают молекулу лекарственное средство-линкер, обозначенную звездочкой на правой стороне.
Настоящий способ получения представляет собой способ получения конъюгата антитела с лекарственным средством (1е) путем связывания ремоделированного гликаном антитела (3d), полученного на этапе D-2 схемы D, и предшественника конъюгата (2), полученного на этапе В-4 схемы В, посредством реакции SPAAC (промотируемой напряжением реакции азид-алкинового циклоприсоединения, J. Am. Chem. Soc. 2004, 126, 15046-15047).
[0319]
(Этап E-1)
Реакцию SPAAC проводили путем смешивания буферного раствора (фосфатного буфера, ацетатного буфера, боратного буфера и т.д.) ремоделированного гликаном антитела (3d) и раствора предшественника конъюгата (2), растворенного в подходящем растворителе (диметилсульфоксид, N,N-диметилформамид, N,N-диметилацетамид, N-метилпирролидон, пропиленгликоль или смешанный растворитель из них). Количество молей предшественника конъюгата (2) составляет от 2 молей до избыточного количества молей, предпочтительно от 4 молей до 30 молей, на 1 моль ремоделированного гликаном антитела (3d), и отношение органического растворителя к буферному раствору антитела предпочтительно составляет от 1% до 200% (об./об.). Температура реакции составляла от 0°С до 37°С, и предпочтительно от 15°С до 25°С, и время реакции составляло от 1 часа до 150 часов, и предпочтительно от 6 часов до 72 часов. рН реакционной смеси предпочтительно составлял от 5 до 9. Реакционную смесь очищали в соответствии со способом, описанным далее в Общей процедуре D, с получением конъюгата антитела с лекарственным средством (1е).
[0320]
Схема Е'. Конъюгирование антитела и лекарственного средства (конъюгирование цистеином).
Конъюгат антитела с лекарственным средством согласно настоящему изобретению с конъюгированием через цистеин можно получить, например, руководствуясь способом, описанным в WO2014/057687, с применением нацеленного антитела, полученного, например, в соответствии со ссылочным примером 1, и предшественника конъюгата (2'), содержащего малеимидную группу, полученного на этапе В-8 схемы В'.
[0321]
Схема Е''. Конъюгирование антитела и лекарственного средства (конъюгирование гликаном 2).
При замене предшественника конъюгата (2) на предшественник конъюгата (3'), полученный на этапе С'-8 схемы С', получали конъюгат антитела с лекарственным средством (1е''), показанный в следующей формуле.
[0322]
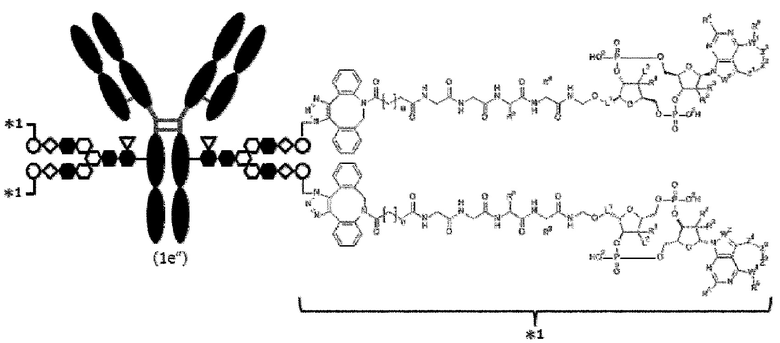
[0323]
В формуле две звездочки (*1) на левой стороне конъюгата антитела с лекарственным средством (1е'') обозначают молекулу лекарственное средство-линкер, обозначенную звездочкой на правой стороне.
[0324]
Конъюгаты антитела с лекарственным средством можно отличить друг от друга путем замены буфера, очистки, измерения концентрации антитела и измерения среднего количества конъюгированных молекул лекарственного средства на молекулу антитела в соответствии с Общими процедурами D - G, описанными далее.
[0325]
Общая процедура А. Концентрирование водного раствора антитела.
Раствор антитела или конъюгата антитела с лекарственным средством помещали в ультрацентрифужное фильтрующее устройство Amicon (R) (50000 NMWL, Merck Millipore Ltd.), и раствор антитела или конъюгата антитела с лекарственным средством концентрировали посредством процедуры центрифугирования (центрифугирование при 2000 g - 4000 g в течение от 5 до 20 минут) с использованием центрифуги (Allegra X-15R, Beckman Coulter, Inc.).
[0326]
Общая процедура В. Измерение концентрации антитела.
Измерение концентрации антитела проводили, применяя УФ-измерительное устройство (Nanodrop 1000, Thermo Fisher Scientific), в соответствии со способом, рекомендованным производителем. При измерении использовали коэффициенты поглощения на 280 им, различные среди антител (1,3 мл мг-1 см-1 - 1,8 мл мг-1 см-1).
[0327]
Общая процедура С. Замена буфера для антитела.
Буфер (фосфатно-солевой буферный раствор (рН 6,0), фосфатный буфер (рН 6,0) и т.д.) добавляли в водный раствор антитела, и продукт концентрировали в соответствии со способом, описанным в Общей процедуре А. Данную процедуру проводили несколько раз, и концентрацию антитела затем измеряли в соответствии со способом, описанным в Общей процедуре В. В данный буферный раствор антитела подходящим образом добавляли буфер (фосфатно-солевой буферный раствор (рН 6,0), фосфатный буфер (рН 6,0) и т.д.) с получением буферного раствора антитела с заданной концентрацией (например, приблизительно 10 мг/мл).
[0328]
Общая процедура D. Очистка конъюгата антитела с лекарственным средством (гель-фильтрационная хроматография).
Колонку NAP (NAP-5, NAP-10, NAP-25 (производства GE Healthcare)) уравновешивали ацетатным буфером (10 мМ ацетатный буфер, 5% сорбит, рН 5,5; в данной заявке называют ABS) или другим подходящим буфером. Данную колонку NAP наполняли реакционной смесью конъюгата антитела с лекарственным средством, и буферу в количестве, рекомендованном производителем, позволяли под действием гравитации стечь вниз, чтобы отделить и собрать фракцию антитела. Колонку NAP снова наполняли данной фракцией, и буферу в количестве, рекомендованном производителем, позволяли под действием гравитации стечь вниз, чтобы отделить и собрать фракцию антитела. Данную процедуру повторяли всего два или три раза с получением конъюгата антитела с лекарственным средством, при этом не связавшееся лекарственное средство-линкер, диметилсульфоксид и пропиленгликоль удаляли. При необходимости, концентрацию раствора конъюгата антитела с лекарственным средством корректировали с помощью Общих процедур А и С.
[0329]
Общая процедура E. Измерение концентрации антитела и среднего количества конъюгированных молекул лекарственного средства на молекулу антитела в конъюгате антитела с лекарственным средством (УФ-способ).
Концентрацию конъюгированного лекарственного средства в конъюгате антитела с лекарственным средством можно рассчитать путем измерения поглощения водного раствора конъюгата антитела с лекарственным средством на двух длинах волн 280 нм и 260 нм (иногда использовали длину волны, отличную от 260 нм) с применением абсорбциометра (спектрометра УФ/ВИД Lambda 25, PerkinElmer, Inc.), а затем проведения расчета, показанного ниже. Суммарное поглощение на любой длине волны равно сумме поглощений всех поглощающих свет химических соединений, присутствующих в системе (аддитивность поглощения), и, следовательно, с допущением, что молярные коэффициенты поглощения антитела и лекарственного средства неизменны до и после конъюгирования антитела и лекарственного средства, концентрация антитела и концентрация лекарственного средства в конъюгате антитела с лекарственным средством выражены следующими выражениями:
A280 = AD,280 + AA,280 = εD,280CD + εA,280CA Выражение (I)
А260 = AD,260 + AA,260 = εD,260CD + εA,260CA Выражение (II)
где A280 обозначает поглощение водного раствора конъюгата антитела с лекарственным средством на 280 нм, А260 обозначает поглощение водного раствора конъюгата антитела с лекарственным средством на 260 нм, AA,280 обозначает поглощение антитела на 280 нм, AA,260 обозначает поглощение антитела на 260 нм, AD,280 обозначает поглощение предшественника конъюгата на 280 нм, AD,260 обозначает поглощение предшественника конъюгата на 260 нм, εA,280 обозначает молярный коэффициент поглощения антитела на 280 нм, εA,260 обозначает молярный коэффициент поглощения антитела на 260 нм, εD,280 обозначает молярный коэффициент поглощения предшественника конъюгата на 280 нм, εD,260 обозначает молярный коэффициент поглощения предшественника конъюгата на 260 нм, CA обозначает концентрацию антитела в конъюгате антитела с лекарственным средством и CD обозначает концентрацию лекарственного средства в конъюгате антитела с лекарственным средством. Для εA,280, εA,260, εD,280 и εD,260 выше использовали значения, полученные заранее (оценки на основании расчета или измеренных значений). Например, εA,280 можно оценить по последовательности аминокислот антитела, применяя известный способ расчета (Protein Science, 1995, том 4, 2411-2423). Для εA,260 использовали значения, рассчитанные по измеренному значению, полученному путем УФ-измерения антитела и оценки εA,280. В Примерах εA,280 = 215380 и εA,260 = 110117 использовали в качестве молярных коэффициентов поглощения модифицированного антитела против HER2. εA,280 = 227300 и εA,260 = 110710 использовали в качестве молярных коэффициентов поглощения модифицированного антитела против LPS. εD,280 и εD,260 можно получить на основании закона Ламберта-Бера (поглощение = молярность × молярный коэффициент поглощения × оптическая длина пути в кювете) путем измерения поглощения раствора, в котором предшественник конъюгата, который предполагалось использовать, растворяли при определенной молярности. Молярный коэффициент поглощения предшественника конъюгата в Примерах получали каждый раз при УФ-измерении. CA и CD можно определить путем измерения А280 и А260 водного раствора конъюгата антитела с лекарственным средством и их подстановки в выражения (I) и (II), чтобы решить систему уравнений. Кроме того, среднее количество конъюгированных молекул лекарственного средства на молекулу антитела можно определить путем деления CD на CA.
[0330]
Общая процедура F. Измерение концентрации антитела и среднего количества конъюгированных молекул лекарственного средства на молекулу антитела в конъюгате антитела с лекарственным средством (обращенно-фазовая высокоэффективная жидкостная хроматография: ОФ-ВЭЖХ)
Дополнительно к Общей процедуре Е, описанной выше, анализ методом высокоэффективной жидкостной хроматографии с применением следующего способа позволяет определить концентрацию антитела и среднее количество конъюгированных молекул лекарственного средства на молекулу антитела в конъюгате антитела с лекарственным средством.
[0331]
[F-1. Подготовка образца для анализа ВЭЖХ (восстановление конъюгата антитела с лекарственным средством)]
Раствор конъюгата антитела с лекарственным средством (приблизительно 1 мг/мл, 60 мкл) смешивали с водным раствором дитиотреитола (ДТТ) (100 мМ, 15 мкл). Смесь инкубировали при 37°С в течение 30 минут, чтобы расщепить дисульфидную связь между L-цепью и H-цепью конъюгата антитела с лекарственным средством. Данную реакционную смесь непосредственно использовали для анализа ВЭЖХ.
[0332]
[F-2. Анализ ВЭЖХ]
Типичные условия анализа описаны далее.
Система ВЭЖХ: система ВЭЖХ Agilent 1290 (Agilent Technologies)
Детектор: ультрафиолетовый абсорбциометр (длина волны измерения: 280 нм)
Колонка: Acquity ВЕН Phenyl (2,1 × 50 мм, 1,7 мкм, производства Waters Corporation)
Температура колонки: 75°С
Скорость потока: 0,8 мл/мин
Впрыскиваемый объем образца: 10 мкл
Подвижная фаза А: водный раствор 0,1% трифторуксусной кислоты (ТФУ)/15% изопропилового спирта
Подвижная фаза В: раствор в ацетонитриле 0,075% ТФУ/15% изопропилового спирта
Программа градиента (подвижная фаза В): 14% - 36% (0 мин - 15 мин), 36% - 80% (15-17 мин), 80% - 14% (17 мин - 17,1 мин), 14% - 14% (17,1 мин - 23 мин)
[F-3. Анализ результатов]
[F-3-1] По сравнению с L-цепью (L0) и H-цепью (Н0) антитела без какой-либо конъюгированной молекулы лекарственного средства, H-цепь с конъюгированной(-ыми) молекулой(-ами) лекарственного средства (H-цепь с одной конъюгированной молекулой лекарственного средства: H1, H-цепь с двумя конъюгированными молекулами лекарственного средства: Н2) обладает гидрофобностью, которая возрастает пропорционально количеству конъюгированных молекул лекарственного средства, что приводит к более длительному времени удерживания и, следовательно, L0, Н0, H1 и Н2 элюируются, по существу, в данном порядке. Путем сравнения времени удерживания каждой из L0 и Н0, каждый обнаруженный пик можно отнести к любой одной из L0H0, H1 и Н2.
[0333]
[F-3-2] Поскольку каждое лекарственное средство-линкер поглощает УФ, значения площади пика корректировали, используя следующее выражение с молярными коэффициентами поглощения H-цепи и лекарственного средства-линкера, соответствующими количеству конъюгированных молекул лекарственное средство-линкер.
[0334]
[Выражение 1]
Скорректированная площадь пика H-цепи (HPAi) = площадь пика × (молярный коэффициент поглощения Н-цепи/(молярный коэффициент поглощения Н-цепи + количество конъюгированных молекул лекарственного средства × молярный коэффициент поглощения лекарственного средства-линкера))
[0335]
Здесь использовали оценки, рассчитанные с применением известного способа расчета, описанного в Общей процедуре Е, для молярных коэффициентов поглощения (280 нм) L-цепи и H-цепи для каждого антитела. Для модифицированного антитела против HER2, использовали 26213 и 81478 в качестве молярного коэффициента поглощения L-цепи и молярного коэффициента поглощения H-цепи, соответственно. Для модифицированного антитела против LPS, аналогично, использовали 27703 и 85948 в качестве молярного коэффициента поглощения L-цепи и молярного коэффициента поглощения Н-цепи, соответственно. Для молярного коэффициента поглощения (280 нм) лекарственного средства-линкера, измеренное значение для предшественника конъюгата использовали в случае конъюгирования посредством реакции SPAAC, и, в случае конъюгирования цистеином, использовали измеренное значение для соединения, в котором малеимидную группу превратили в тиоэфир сукцинимида посредством реакции предшественника конъюгата с меркаптоэтанолом или N-ацетилцистеином.
[0336]
[F-3-3] Отношение площадей пиков (%) каждой цепи к суммарным скорректированным значениям площадей пиков рассчитывали, используя следующее выражение.
[0337]
[Выражение 2]
[0338]
[F-3-4] Среднее количество конъюгированных молекул лекарственного средства на молекулу антитела (DAR) в конъюгате антитела с лекарственным средством рассчитывали, используя следующее выражение.
[0339]
[Выражение 3]

[0340]
[F-3-5] Концентрацию антитела в конъюгате антитела с лекарственным средством рассчитывали, используя следующее выражение.
[0341]
[Выражение 4]
Концентрация антитела (СА) (мг/мл) = (поглощение комплекса конъюгата антитела с лекарственным средством × степень разведения × молекулярная масса антитела)/(молярный коэффициент поглощения антитела + среднее количество конъюгированных молекул лекарственного средства × молярный коэффициент поглощения лекарственного средства-линкера)
[0342]
Здесь измеренное значение для водного раствора конъюгата антитела с лекарственным средством использовали в качестве поглощения (280 нм) комплекса антитело-лекарственное средство. Степень разведения указывает на то, во сколько раз разбавляли водный раствор конъюгата антитела с лекарственным средством при измерении поглощения, и обычно использовали разведение в четыре раза. Для молярного коэффициента поглощения (280 нм) антитела использовали оценку, рассчитанную с использованием известного способа расчета, описанного в Общей процедуре Е. Для среднего количества конъюгированных молекул лекарственного средства использовали значение, полученное в [F-3-4]. Для молярного коэффициента поглощения (280 нм) лекарственного средства-линкера использовали измеренное значение для предшественника конъюгата в случае конъюгирования посредством реакции SPAAC и, в случае конъюгирования цистеином, использовали измеренное значение для соединения, в котором малеимидную группу превратили в тиоэфир сукцинимида посредством реакции лекарственного средства-линкера с меркаптоэтанолом или N-ацетилцистеином.
[0343]
Общая процедура G: Измерение концентрации антитела и среднего количества конъюгированных молекул лекарственного средства на молекулу антитела в конъюгате антитела с лекарственным средством (высокоэффективная жидкостная хроматография с гидрофобным взаимодействием: ГВ-ВЭЖХ)
Дополнительно к Общим процедурам E и F, описанным выше, анализ методом высокоэффективной жидкостной хроматографии с применением следующего способа позволяет определить концентрацию антитела и среднее количество конъюгированных молекул лекарственного средства на молекулу антитела в конъюгате антитела с лекарственным средством.
[0344]
[G-1. Получение образца для анализа ВЭЖХ]
Раствор конъюгата антитела с лекарственным средством (приблизительно 1 мг/мл, 60 мкл) непосредственно использовали в анализе ВЭЖХ.
[0345]
[G-2. Анализ ВЭЖХ]
Типичными условиями анализа являются следующие два.
Система ВЭЖХ: SHIMADZU СВМ-20А (Shimadzu Corporation)
Детектор: ультрафиолетовый абсорбциометр (длина волны измерения: 280 нм)
Колонка: TSK-gel Butyl-NPR (4,6 × 100 мм, 2,5 мкм, производства Tosoh Corporation)
Температура колонки: постоянная температура приблизительно 25°С
Подвижная фаза А: содержащий 1,5 M сульфат аммония 25 мМ фосфатный буфер (рН=7,0)
Подвижная фаза В: 25 мМ фосфатный буфер (рН=7,0)/изопропиловый спирт (3:1)
Скорость потока: 0,8 мл/мин
Впрыскиваемый объем образца: 15 мкл
Программа градиента (подвижная фаза В): 10% - 15% (0 мин - 5 мин), 15% - 65% (5 мин - 20 мин)
или
Система ВЭЖХ: SHIMADZU СВМ-20А (Shimadzu Corporation)
Детектор: ультрафиолетовый абсорбциометр (длина волны измерения: 280 нм)
Колонка: PolyPROPYL А (4,6 × 100 мм, 3 мкм, 1500 ангстрем, производства PolyLC Inc.)
Температура колонки: постоянная температура приблизительно 40°С
Подвижная фаза А: содержащий 1,5 M сульфат аммония 20 мМ фосфатный буфер (рН=7,4)
Подвижная фаза В: 20 мМ фосфатный буфер (рН=7,4)
Скорость потока: 0,8 мл/мин
Впрыскиваемый объем образца: 15 мкл
Программа градиента (подвижная фаза В): 40% - 80% (0 мин -20 мин)
[0346]
[G-3. Анализ результатов]
[G-3-1] Гидрофобность возрастает пропорционально количеству молекул лекарственного средства, конъюгированных с молекулой антитела, и дается более длительное время удерживания, и, следовательно, в случае конъюгирования посредством реакции SPAAC, продукты с DAR=0, DAR=2 и DAR=4 элюируются, по существу, в следующем порядке. Путем сравнения времени удерживания продукта с DAR=0, каждый детектированный пик можно отнести к любому одному из продуктов с DAR=2 и с DAR=4. Для некоторых типов антител или лекарственных средств-линкеров можно детектировать пики для продукта с DAR=1 и с DAR=3. DAR для детектированного пика оценивают в некоторых случаях путем фракционирования пика посредством ГВ-ВЭЖХ, а затем измерения масс-спектра.
[G-3-2] Поскольку каждое лекарственное средство-линкер поглощает УФ, значения площади пика корректировали, используя следующее выражение с молярными коэффициентами поглощения антитела и лекарственного средства-линкера, соответствующими количеству конъюгированных молекул лекарственное средство-линкер.
[0347]
[Выражение 5]
Скорректированная площадь пика антитела (WPAi) = площадь пика × (молярный коэффициент поглощения антитела/(молярный коэффициент поглощения антитела + количество конъюгированных молекул лекарственного средства × молярный коэффициент поглощения лекарственного средства-линкера))
[0348]
Здесь использовали оценки, рассчитанные с применением известного способа расчета, описанного в Общей процедуре Е, для определения молярного коэффициента поглощения (280 нм) антитела. Для определения молярного коэффициента поглощения (280 нм) лекарственного средства-линкера использовали измеренное значение для предшественника конъюгата.
[0349]
[G-3-3] Отношение площадей пиков (%) антитела к суммарным корректированным значениям площадей пиков рассчитывали, используя следующее выражение.
[0350]
[Выражение 6]
[0351]
[G-3-4] Среднее количество конъюгированных молекул лекарственного средства на молекулу антитела в конъюгате антитела с лекарственным средством рассчитывали, используя следующее выражение.
[0352]
[Выражение 7]

[0353]
[G-3-5] Концентрацию антитела в конъюгате антитела с лекарственным средством рассчитывали, используя выражение, представленное в [F-3-5]. Затем использовали значение, полученное в [G-3-4], в качестве среднего количества конъюгированных молекул лекарственного средства.
[0354]
Могут присутствовать стереоизомеры, оптические изомеры благодаря асимметричному атому углерода, геометрические изомеры, таутомеры или оптические изомеры, такие как d-формы, l-формы, и атропоизомеры нового производного ЦДН и конъюгата антитела с лекарственным средством согласно настоящему изобретению, и интермедиата получения любого из них. Все данные изомеры, оптические изомеры и их смеси входят в объем настоящего изобретения.
[0355]
Количество конъюгированных молекул лекарственного средства на молекулу антитела является важным фактором, оказывающим влияние на эффективность и безопасность конъюгата антитела с лекарственным средством согласно настоящему изобретению. Конъюгаты антитела с лекарственным средством получают при условиях реакции, таких как количества сырьевых материалов и реагентов для взаимодействия, предусмотренных для получения постоянного количества конъюгированных молекул лекарственного средства; тем не менее, в противоположность химической реакции низкомолекулярных соединений, обычно получают смесь с различными количествами конъюгированных молекул лекарственного средства. Количества конъюгированных молекул лекарственного средства на молекулу антитела определены как среднее значение, а именно, среднее количество конъюгированных молекул лекарственного средства (DAR). Количество молекул производного циклического динуклеотида, конъюгированных с молекулой антитела, можно контролировать, и можно конъюгировать от 1 до 10 молекул производного циклического динуклеотида в контексте среднего количества конъюгированных молекул лекарственного средства на молекулу антитела, но предпочтительно указанное количество составляет от одной до восьми и, более предпочтительно, от одной до пяти молекул.
[0356]
Когда антитело Ab связывается посредством ремоделированного гликана антитела Ab с L в конъюгате антитела с лекарственным средством согласно настоящему изобретению, количество конъюгированных молекул лекарственного средства на молекулу антитела в конъюгате антитела с лекарственным средством, m2, представляет собой целое число, равное 1 или 2. Когда гликан представляет собой N297-гликан и гликан представляет собой N297-(Fuc)SG, m2 представляет собой 2 и DAR находится в диапазоне от 3 до 5 (предпочтительно, в диапазоне от 3,2 до 4,8, более предпочтительно, в диапазоне от 3,5 до 4,2). Когда N297-гликан представляет собой N297-(Fuc)MSG1, N297-(Fuc)MSG2 или смесь N297-(Fuc)MSG1 и N297-(Fuc)MSG2, m2 представляет собой 1 и DAR находится в диапазоне от 1 до 3 (предпочтительно, в диапазоне от 1,0 до 2,5, более предпочтительно, в диапазоне от 1,2 до 2,2).
[0357]
Специалисты в данной области могут разработать реакцию для конъюгирования необходимого количества молекул лекарственного средства с каждой молекулой антитела на основании описания в Примерах в настоящей заявке, и получить антитело с контролируемым количеством конъюгированных молекул производного циклического динуклеотида.
[0358]
Производное ЦДН и конъюгат антитела с лекарственным средством согласно настоящему изобретению, и интермедиат получения любого из них, может поглощать влагу, позволять адгезию поглощенной воды или становиться гидратом, когда его оставляют в атмосфере или перекристаллизуют. Такие соединения и соли, содержащие воду, также входят в объем настоящего изобретения.
[0359]
Каждое производное ЦДН и конъюгат антитела с лекарственным средством согласно настоящему изобретению, и интермедиат получения любого из них, можно превратить в фармацевтически приемлемую соль, при необходимости, когда они содержат основную группу, такую как аминогруппа. Примеры таких солей могут включать соли галогеноводородов, такие как гидрохлориды и гидройодиды; соли неорганических кислот, такие как нитраты, перхлораты, сульфаты и фосфаты; низшие алкансульфонаты, такие как метансульфонаты, трифторметансульфонаты и этансульфонаты; арилсульфонаты, такие как бензолсульфонаты и п-толуолсульфонаты; соли органических кислот, такие как форматы, ацетаты, малаты, фумараты, сукцинаты, цитраты, тартраты, оксалаты и малеаты; и соли аминокислот, такие как орнитинаты, глутаматы и аспартаты.
[0360]
Так как производное ЦДН или конъюгат антитела с лекарственным средством согласно настоящему изобретению содержит фосфатную группу и/или тиофосфатную группу в структуре, как правило, может образоваться соль присоединения основания. Когда интермедиат получения содержит кислую группу, такую как карбоксигруппа, как правило, может аналогично образоваться соль присоединения основания. Примеры фармацевтически приемлемых солей могут включать соли щелочных металлов, такие как соли натрия, соли калия и соли лития; соли щелочноземельных металлов, такие как соли кальция и соли магния; неорганические соли, такие как соли аммония; и соли органического амина, такие как соли дибензиламина, соли морфолина, соли алкилового эфира фенилглицина, соли этилендиамина, соли N-метилглюкамина, соли диэтиламина, соли триэтиламина, соли циклогексиламина, соли дициклогексил амина, соли N,N'-дибензилэтилендиамина, соли диэтаноламина, соли N-бензил-N-(2-фенилэтокси)амина, соли пиперазина, соли тетраметиламмония и соли трис(гидроксиметил)аминометана.
[0361]
Каждое производное ЦДН и конъюгат антитела с лекарственным средством согласно настоящему изобретению, и интермедиат получения любого из них, может присутствовать в виде гидрата, например, образованного путем поглощения влаги из воздуха. Сольват согласно настоящему изобретению не ограничен конкретным сольватом при условии, что он может представлять собой любой фармацевтически приемлемый сольват. В частности, предпочтительны гидраты, этанолсольваты, 2-пропанолсольваты и т.д. Каждое производное ЦДН и конъюгат антитела с лекарственным средством согласно настоящему изобретению, и интермедиат получения любого из них, может находиться в форме N-оксида, когда атом азота присутствует в нем. Данные сольваты и формы N-оксида входят в объем настоящего изобретения. Каждое производное ЦДН и конъюгат антитела с лекарственным средством согласно настоящему изобретению, и интермедиат получения любого из них, может находиться в форме сульфоксида, когда атом серы присутствует в нем. Данные сольваты и сульфоксидные формы входят в объем настоящего изобретения.
[0362]
В объем настоящего изобретения входят соединения, меченые различными радиоактивными или нерадиоактивными изотопами. Каждое производное ЦДН и конъюгат антитела с лекарственным средством согласно настоящему изобретению, и интермедиат получения любого из них, может содержать один или более составляющих атомов с неприродными соотношениями атомных изотопов. Примеры атомных изотопов могут включать дейтерий (2Н), тритий (3Н), йод-125 (125I) и углерод-14 (14С). Соединение согласно настоящему изобретению можно пометить радиоактивной меткой с радиоактивным изотопом, таким как тритий (3Н), йод-125 (125I) и углерод-14 (14С). Меченое радиоактивной меткой соединение полезно в качестве терапевтического или профилактического агента, реагента для исследования, такого как реагент для анализа, и диагностического агента, такого как диагностический агент для визуализации in vivo. Все изотопные варианты конъюгата антитела с лекарственным средством согласно настоящему изобретению входят в объем настоящего изобретения, независимо от того, радиоактивные они или нет.
[0363]
<4. Лекарственное средство>
Производное ЦДН или конъюгат антитела с лекарственным средством согласно настоящему изобретению проявляет противоопухолевую иммунную активность или цитотоксическую активность по отношению к раковым клеткам, и, следовательно, его можно применять в качестве лекарственного средства, в частности, терапевтического агента и/или профилактического агента для лечения рака, или противоопухолевого агента.
[0364]
Примеры типов рака, при которых можно применять производное ЦДН или конъюгат антитела с лекарственным средством согласно настоящему изобретению, могут включать рак легких (немелкоклеточный рак легких, мелкоклеточный рак легких и т.д.), рак почки, уротелиальный рак, колоректальный рак, рак предстательной железы, мультиформную глиобластому, рак яичников (опухоль поверхностного эпителия, стромальную опухоль, герминогенную опухоль и т.д.), рак поджелудочной железы, рак молочной железы, меланому, рак печени, рак мочевого пузыря, рак желудка, рак пищевода, рак эндометрия, рак яичка (семиному, несеминому), рак шейки матки, плацентарную хориокарциному, мультиформную глиобластому, опухоль головного мозга, рак головы и шеи, рак щитовидной железы, мезотелиому, стромальную опухоль желудочно-кишечного тракта (GIST), рак желчного пузыря, рак желчного протока, рак надпочечников, плоскоклеточную карциному, лейкоз, злокачественную лимфому, плазмоцитому, миелому и саркому. Тем не менее, применения конъюгата антитела с лекарственным средством не ограничены перечисленными, при условии, что раковые клетки как терапевтическая мишень экспрессируют белок, который распознают антитела в конъюгате антитела с лекарственным средством.
[0365]
Производное ЦДН или конъюгат антитела с лекарственным средством согласно настоящему изобретению предпочтительно можно вводить млекопитающим, и более предпочтительно его вводят человеку.
[0366]
Вещества для применения в фармацевтической композиции, содержащей производное ЦДН или конъюгат антитела с лекарственным средством согласно настоящему изобретению, можно подходящим образом выбрать для применения из вспомогательных веществ для получения состава и тому подобных веществ, которые, как правило, используют в данной области, с учетом дозы или концентрации введения.
[0367]
Производное ЦДН или конъюгат антитела с лекарственным средством согласно настоящему изобретению можно вводить в виде фармацевтической композиции, содержащей один или более фармацевтически совместимых компонентов. Например, фармацевтическая композиция обычно содержит один или более фармацевтических носителей (например, стерилизованную жидкость (включая воду и масло (нефть и масло животного происхождения, растительного происхождения или синтетического происхождения (такое как арахисовое масло, соевое масло, минеральное масло и кунжутное масло)))). Вода представляет собой более типичный носитель, когда фармацевтическую композицию вводят внутривенно. Солевой раствор, водный раствор декстрозы и водный раствор глицерина также можно применять в качестве жидкого носителя, в частности, для раствора для инъекций. Подходящие фармацевтические вспомогательные вещества известны в данной области. При необходимости, композиция выше также может содержать следовое количество увлажняющего агента, эмульгирующего агента или рН-буферного вещества. Примеры подходящих фармацевтических носителей описаны в «Remington's Pharmaceutical Sciences», E.W. Martin. Включение их в состав зависит от способа введения.
[0368]
Известны различные системы доставки, и их можно применять для введения производного ЦДН или конъюгата антитела с лекарственным средством согласно настоящему изобретению. Примеры способа введения могут включать, но не ограничены перечисленными: внутрикожный, внутримышечный, интраперитонеальный, внутривенный и подкожный пути. Введение можно осуществить, например, путем инъекции или болюсной инъекции. В конкретном предпочтительном варианте реализации введение производного ЦДН или конъюгата антитела с лекарственным средством осуществляют путем инъекции. Парентеральное введение представляет собой предпочтительный путь введения.
[0369]
В типичном варианте реализации фармацевтическую композицию, содержащую описанный выше конъюгат антитела с лекарственным средством, составляют в виде фармацевтической композиции, подходящей для внутривенного введения людям, в соответствии с обычными процедурами. Композиция для внутривенного введения обычно представляет собой раствор в стерильном и изотоническом водном буфере. При необходимости, лекарственное средство может содержать солюбилизирующий агент и местное анестезирующее средство, чтобы облегчить боль в месте инъекции (например, лигнокаин). Как правило, компоненты выше предложены либо отдельно в виде высушенного лиофилизированного порошка, либо в виде безводного концентрата в плотно запечатанном контейнере, таком как ампула или саше, с указанием количества активного агента, либо в виде смеси в единичной лекарственной форме. Если фармацевтическая композиция предназначена для введения путем инфузии, ее можно вводить из инфузионного флакона, содержащего воду или солевой раствор степени чистоты стерильного фармацевтического средства. Если фармацевтическую композицию вводят путем инъекции, может быть предложена ампула, содержащая стерильную воду или солевой раствор для инъекции, так что упомянутые выше компоненты смешивают друг с другом перед введением. Фармацевтическая композиция может быть предложена в виде раствора.
[0370]
Фармацевтическая композиция согласно настоящему изобретению может представлять собой фармацевтическую композицию, содержащую только производное ЦДН или конъюгат антитела с лекарственным средством согласно настоящему изобретению, или фармацевтическую композицию, содержащую производное ЦДН или конъюгат антитела с лекарственным средством и по меньшей мере один агент для лечения рака, отличный от производного ЦДН или конъюгата антитела с лекарственным средством. Производное ЦДН или конъюгат антитела с лекарственным средством согласно настоящему изобретению можно вводить в комбинации с другими агентами для лечения рака, обеспечивая улучшенное противораковое действие. Другие противораковые агенты для применения для таких целей можно вводить индивиду одновременно, отдельно или последовательно с введением производного ЦДН или конъюгата антитела с лекарственным средством, и можно вводить с различными интервалами между введениями. Примеры таких агентов для лечения рака могут включать абраксан, карбоплатин, цисплатин, гемцитабин, иринотекан (СРТ-11), паклитаксел, пеметрексед, сорафениб, винбластин, агенты, описанные в международной публикации № WO2003/038043, аналоги LH-RH (лейпрорелин, гозерелин и т.д.), фосфат эстрамустина, антагонисты эстрогена (тамоксифен, ралоксифен и т.д.), ингибиторы ароматазы (анастрозол, летрозол, эксеместан и т.д.) и ингибиторы контрольных точек иммунного ответа (ниволумаб, ипилимумаб и т.д.), но не ограничены ими, и приемлем любой агент, обладающий противоопухолевой активностью.
[0371]
Описанную фармацевтическую композицию можно включить в лиофилизированный состав или жидкий состав, в виде состава, обладающего выбранной композицией и необходимой чистотой. При включении в лиофилизированный состав, фармацевтическую композицию можно включить в состав формы, содержащей подходящие для состава вспомогательные вещества, которые применяют в данной области. Также для получения жидкости, фармацевтическую композицию можно включить в состав жидкой формы, содержащей различные вспомогательные вещества для состава, которые применяют в данной области.
[0372]
Компоненты и концентрация фармацевтической композиции могут изменяться в зависимости от способов введения; тем не менее, конъюгат антитела с лекарственным средством, содержащийся в фармацевтической композиции согласно настоящему изобретению, может оказывать фармацевтическое действие даже в меньшей дозе, так как аффинность конъюгата антитела с лекарственным средством к антигену выше, то есть, так как аффинность конъюгата антитела с лекарственным средством в контексте константы диссоциации (значения Kd) от антигена выше (меньшее значение Kd). Таким образом, при определении дозы конъюгата антитела с лекарственным средством, дозу можно установить с учетом условия, относящегося к аффинности конъюгата антитела с лекарственным средством к антигену. Когда производное ЦДН или конъюгат антитела с лекарственным средством согласно настоящему изобретению вводят человеку, например, приблизительно от 0,001 до 100 мг/кг можно вводить однократно или несколькими частями с интервалами от 1 до 180 суток.
[0373]
Далее настоящее изобретение будет описано со ссылкой на Примеры; тем не менее, настоящее изобретение не ограничено Примерами.
Примеры
[0374]
В примерах ниже комнатная температура относится к температуре от 15°С до 35°С. Используемый обезвоженный ацетонитрил представлял собой ацетонитрил (обезвоженный) -Super-, реализуемый KANTO CHEMICAL CO., INC., или ацетонитрил (сверхобезвоженный), реализуемый Wako Pure Chemical Industries, Ltd. Используемый пиридин представлял собой пиридин (обезвоженный) -Super-, реализуемый KANTO CHEMICAL CO., INC. Хроматографию на силикагеле проводили, применяя Biotage SNAP Ultra (производства Biotage), Chromatorex Q-Pack SI (производства FUJI SILYSIA CHEMICAL LTD.) или Purif-Pack-Ex SI (производства Shoko Science Co., Ltd.). Колоночную хроматографию на силикагеле DIOL проводили, применяя Chromatorex Q-pack DIOL (производства FUJI SILYSIA CHEMICAL LTD.). Колоночную хроматографию на силикагеле С18 проводили, применяя Biotage SNAP Ultra C18 (производства Biotage). Колоночную хроматографию на аминосиликагеле проводили, применяя Biotage SNAP Isolute NH2 (производства Biotage). Препаративную ВЭЖХ проводили, применяя систему ВЭЖХ SHIMADZU SPD-M10A (Shimadzu Corporation) или тому подобные системы. В качестве препаративной колонки использовали Kinetex (5 мкм, С18, 100 ангстрем, 250 × 30,0 мм, производства Phenomenex) или Kinetex (5 мкм, С18, 100 ангстрем, 250 × 21,2 мм, производства Phenomenex).
[0375]
Для измерения спектральных данных использовали следующие устройства. Измерения для 1H-ЯМР спектров проводили, используя JEOL ECS-400 (400 МГц), Varian 400-MR (400 МГц) или Varian Unity Inova 500 (500 МГц). Измерение для 31Р-ЯМР спектров проводили, применяя JEOL ECS-400 (160 МГц). Измерение для масс-спектров проводили, применяя систему ЖХ/МС Agilent 6130 Quadrupole (Agilent Technologies). Измерение ЖХ/МС проводили при следующих условиях [колонка: Develosil Combi-RP, 5 мкм, 50 × 2,0 мм (производства Nomura Chemical Co., Ltd.), подвижная фаза: 0,1% об. муравьиной кислоты в ацетонитриле/0,1% об. муравьиной кислоты в дистиллированной воде, 0,1% об. муравьиной кислоты в ацетонитриле: 2% - 100% (0 мин - 5 мин или 0 мин - 10 мин)].
[0376]
Пример 1. Синтез ЦДН1
(5R,7R,8R,12aR,14R,15R,15aS,16R)-7-(6-амино-9H-пурин-9-ил)-15,16-дигидрокси-2,10-бис(сульфанил)-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-дион
[0377]
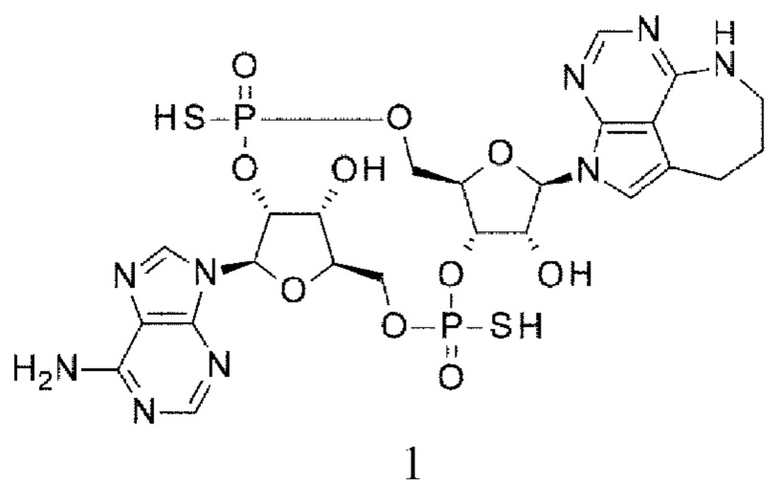
1а (Диастереомер 1)
1b (Диастереомер 2)
1с (Диастереомер 3)
[0378]
[Схема синтеза]
[0379]
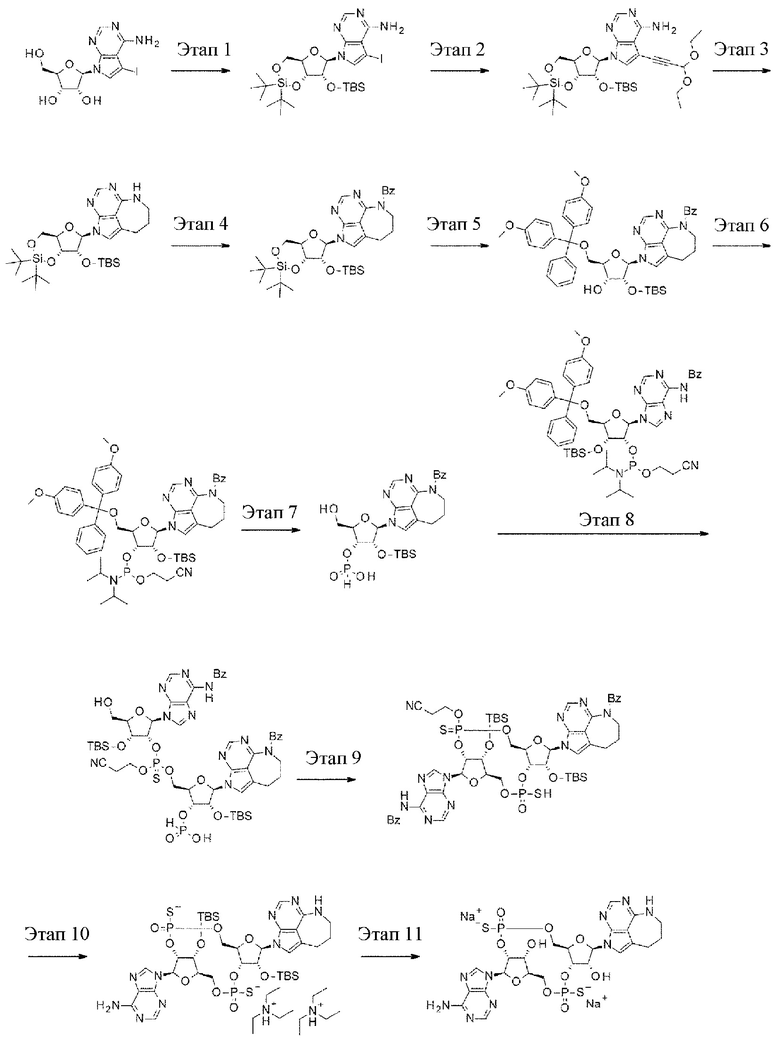
[0380]
(Этап 1)
7-{2-О-[трет-бутил(диметил)силил]-3,5-О-(ди-трет-бутилсилилиден)-β-D-рибофуранозил}-5-йод-7Н-пирроло[2,3-d]пиримидин-4-амин
[0381]
К раствору 5-йодтуберцидина (1,0 г) как соединения, известного в литературе (Tetrahedron 2007, 63, 9850-9861), в N,N-диметилформамиде (10 мл) медленно добавляли по каплям ди-трет-бутилсилил-бис(трифторметансульфонат) (1,24 мл) при 0°С, и реакционную смесь затем перемешивали при той же температуре в течение 30 минут. Имидазол (868 мг) добавляли туда при 0°С, температуру затем повышали до комнатной температуры и реакционную смесь перемешивали в течение 30 минут. При комнатной температуре туда добавляли трет-бутилдиметилхлорсилан, и реакционную смесь перемешивали при той же температуре в течение ночи. Насыщенный водный раствор гидрокарбоната натрия добавляли в реакционную смесь, чтобы погасить реакцию, и продукт затем осуществляли экстрагирование этилацетатом. Органический слой промывали рассолом, а затем сушили над безводным сульфатом натрия. Осушающий агент удаляли путем фильтрации и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле [гексан/этилацетат] с получением указанного в заголовке соединения (910 мг).
МС (ИЭР) m/z: 647 (М+Н)+.
1H-ЯМР (CDCl3) δ: 8,25 (1H, с), 7,03 (1H, с), 6,10 (1H, с), 5,63 (2Н, уш. с), 4,49-4,44 (2Н, м), 4,26 (1Н, дд, J=9,7, 4,8 Гц), 4,17 (1H, м), 4,00 (1H, т, J=9,7 Гц), 1,09 (9Н, с), 1,04 (9Н, с), 0,91 (9Н, с), 0,13 (3Н, с), 0,11 (3Н, с).
[0382]
(Этап 2)
7-{2-O-[трет-бутил(диметил)силил]-3,5-O-(ди-трет-бутилсилилиден)-β-D-рибофуранозил}-5-(3,3-диэтоксипроп-1-ин-1-ил)-7H-пирроло[2,3-d]пиримидин-4-амин
В смешанный раствор соединения, полученного на этапе 1 (910 мг), в N,N-диметилформамиде (3,0 мл)/тетрагидрофуране (9,0 мл) добавляли пропаргилальдегид-диметилацеталь (1,01 мл), триэтиламин (0,392 мл), тетракис(трифенилфосфин)палладий(0) (163 мг) и йодид меди(I) (53,6 мг) в данном порядке, и реакционную смесь перемешивали при 40°С в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия и этилацетата добавляли в реакционную смесь, которую осуществляли экстрагирование этилацетатом. Органический слой промывали рассолом, а затем сушили над безводным сульфатом натрия. Осушающий агент удаляли путем фильтрации и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле [гексан/этилацетат] с получением указанного в заголовке соединения (878 мг).
МС (ИЭР) m/z: 647 (М+Н)+.
1Н-ЯМР (CDCl3) δ: 8,27 (1H, с), 7,17 (1H, с), 6,09 (1H, с), 5,56 (2Н, уш. с), 5,50 (1H, с), 4,48 (1H, дд, J=9,1, 4,9 Гц), 4,42 (1Н, д, J=4,9 Гц), 4,25 (1H, дд, J=9,4, 4,6 Гц), 4,17 (1H, м), 4,00 (1H, т, J=9,7 Гц), 3,85-3,77 (2Н, м), 3,66 (2Н, м), 1,28 (6Н, т, J=7,3 Гц), 1,08 (9Н, с), 1,04 (9Н, с), 0,91 (9Н, с), 0,13 (3Н, с), 0,11 (3Н, с).
[0383]
(Этап 3)
2-{2-O-[трет-бутил(диметил)силил]-3,5-O-(ди-трет-бутилсилилиден)-β-D-рибофуранозил}-6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен
К раствору соединения, полученного на этапе 2 (878 мг), в этаноле (8,8 мл), добавляли 10% палладий/углерод (М), влажный (500 мг), и реакционную смесь перемешивали в атмосфере водорода при комнатной температуре в течение 9 часов. Катализатор удаляли путем фильтрации, а затем промывали дихлорметаном и фильтрат концентрировали при пониженном давлении. К раствору остатка в уксусной кислоте (8,8 мл) добавляли 10% палладий/углерод (М), влажный (500 мг), и реакционную смесь перемешивали в атмосфере водорода при 40°С в течение 2 суток. Катализатор удаляли путем фильтрации, а затем промывали дихлорметаном и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле [гексан/этилацетат/0,1% триэтиламин] с получением указанного в заголовке соединения (603 мг).
МС (ИЭР) m/z: 561 (М+Н)+.
1H-ЯМР (CDCl3) δ: 8,47 (1H, уш. с), 8,07 (1H, с), 6,70 (1H, с), 6,14 (1Н, с), 4,47-4,43 (2Н, м), 4,29 (1H, дд, J=9,1, 4,8 Гц), 4,15 (1Н, м), 3,99 (1H, т, J=9,7 Гц), 3,55 (2Н, м), 2,89 (2Н, т, J=5,4 Гц), 2,04 (2Н, м), 1,09 (9Н, с), 1,04 (9Н, с), 0,90 (9Н, с), 0,10 (3Н, с), 0,10 (3Н, с).
[0384]
(Этап 4)
6-бензоил-2-{2-O-[трет-бутил(диметил)силил]-3,5-O-(ди-трет-бутилсилилиден)-β-D-рибофуранозил}-6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен
К раствору соединения, полученного на этапе 3 (2,17 г), в дихлорметане (21,7 мл), добавляли пиридин (1,56 мл), N,N-диметиламинопиридин (94,5 мг) и бензоилхлорид (0,898 мл) в данном порядке при комнатной температуре, и реакционную смесь перемешивали при 50°С в течение 15 часов. Насыщенный водный раствор гидрокарбоната натрия добавляли в реакционную смесь, чтобы погасить реакцию. После экстрагирования дихлорметаном органический слой сушили над безводным сульфатом натрия. Осушающий агент удаляли путем фильтрации и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле [гексан/этилацетат/0,1% триэтиламин] с получением указанного в заголовке соединения (1,91 г).
МС (ИЭР) m/z: 665 (М+Н)+.
1H-ЯМР (CDCl3) δ: 8,08 (1H, с), 7,37-7,33 (3Н, м), 7,23 (2Н, т, J=7,6 Гц), 6,97 (1H, с), 6,21 (1H, с), 4,50-4,46 (2Н, м), 4,37-4,30 (2Н, м), 4,28-4,09 (2Н, м), 4,02 (1Н, т, J=10,0 Гц), 3,03 (2Н, т, J=6,3 Гц), 2,29-2,17 (2Н, м), 1,10 (9Н, с), 1,05 (9Н, с), 0,90 (9Н, с), 0,10 (6Н, с).
[0385]
(Этап 5)
6-бензоил-2-{5-O-[бис(4-метоксифенил)(фенил)метил]-2-O-[трет-бутил(диметил)силил]-β-D-рибофуранозил}-6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен
К раствору соединения, полученного на этапе 4 (1,91 г), в дихлорметане (15 мл), добавляли смесь фтороводорода-пиридина (0,30 мл) и пиридина (1,88 мл), полученную при 0°С, и реакционную смесь перемешивали при 0°С в течение 2 часов. Насыщенный водный раствор гидрокарбоната натрия добавляли в реакционную смесь, чтобы погасить реакцию. После того, как реакционную смесь подвергли экстрагированию дихлорметаном, органический слой сушили над безводным сульфатом натрия. Осушающий агент удаляли путем фильтрации и фильтрат концентрировали при пониженном давлении. Остаток растворяли в пиридине (15 мл), добавляли туда 4,4'-диметокситритилхлорид (1,17 г), и реакционную смесь перемешивали при 0°С в течение 12 часов. Метанол добавляли туда, реакционную смесь перемешивали в течение 30 минут, и насыщенный водный раствор гидрокарбоната натрия затем добавляли туда, чтобы погасить реакцию. После того, как реакционную смесь подвергли экстрагированию дихлорметаном, органический слой сушили над безводным сульфатом натрия. Осушающий агент удаляли путем фильтрации и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле [гексан/этилацетат/0,1% триэтиламин] с получением указанного в заголовке соединения (1,98 г).
МС (ИЭР) m/z: 827 (М+Н)+.
1H-ЯМР (CDCl3) δ: 8,07 (1H, с), 7,47 (2Н, м), 7,37-7,19 (13Н, м), 6,84 (4Н, м), 6,37 (1H, д, J=5,5 Гц), 4,75 (1Н, т, J=5,2 Гц), 4,38-4,20 (4Н, м), 3,80 (6Н, с), 3,53 (1H, дд, J=10,7, 2,8 Гц), 3,40 (1H, дд, J=11,0, 3,1 Гц), 2,83 (1H, д, J=3,7 Гц), 2,78 (2Н, т, J=6,4 Гц), 2,17 (2Н, м), 0,81 (9Н, с), -0,03 (3Н, с), -0,21 (3Н, с).
[0386]
(Этап 6)
6-бензоил-2-(5-O-[бис(4-метоксифенил)(фенил)метил]-2-O-[трет-бутил(диметил)силил]-3-O-{(2-цианоэтокси)[ди(пропан-2-ил)амино]фосфанил}-β-D-рибофуранозил)-6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен
К раствору соединения, полученного на этапе 5 (1,98 г), в дихлорметане (23,9 мл) добавляли N,N-диизопропилэтиламин (1,02 мл) и 2-цианоэтил-N,N-диизопропилхлорфосфорамидит (1,07 мл), и реакционную смесь перемешивали при комнатной температуре в течение 15 часов. Насыщенный водный раствор гидрокарбоната натрия добавляли в реакционную смесь, чтобы погасить реакцию. После того, как реакционную смесь подвергли экстрагированию дихлорметаном, органический слой сушили над безводным сульфатом натрия. Осушающий агент удаляли путем фильтрации и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле [гексан/этилацетат] с получением указанного в заголовке соединения (2,06 г) в виде смеси диастереомеров у атома фосфора (соотношение диастереомеров = 7:3).
MC (ИЭР) m/z: 1027 (М+Н)+.
1Н-ЯМР (CDCl3) δ: 8,06 (0,3Н, с), 8,04 (0,7Н, с), 7,50-7,16 (15Н, м), 6,85-6,79 (4Н, м), 6,35 (0,7Н, д, J=6,7 Гц), 6,31 (0,3Н, д, J=6,1 Гц), 4,84 (0,7Н, дд, J=7,0, 4,6 Гц), 4,78 (0,3Н, т, J=5,8 Гц), 4,43-4,17 (4Н, м), 4,04-3,85 (1,3Н, м), 3,80-3,76 (6Н, м), 3,69-3,43 (3Н, м), 3,50 (0,7Н, дд, J=10,6, 3,3 Гц), 3,33-3,26 (1H, м), 2,87-2,76 (2Н, м), 2,74-2,60 (1,4Н, м), 2,31 (0,6Н, т, J=6,7 Гц), 2,23-2,11 (2Н, м), 1,21-1,13 (7,8Н, м), 1,04 (4,2Н, д, 3=6,7 Гц), 0,73 (2,7Н, с), 0,72 (6,3Н, с), -0,03 (0,9Н, с), -0,06 (2,1Н, с), -0,24 (3Н, с).
[0387]
(Этап 7)
6-бензоил-2-{2-O-[трет-бутил(диметил)силил]-3-О-[гидрокси(оксо)-λ5-фосфанил]-β-D-рибофуранозил}-6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен
К раствору соединения, полученного на этапе 6 (1,37 г), в ацетонитриле (6,67 мл) добавляли воду (48 мкл) и пиридиновую соль трифторуксусной кислоты (335 мг), и реакционную смесь перемешивали при комнатной температуре в течение 15 минут. В реакционную смесь добавляли трет-бутиламин (6,67 мл), и реакционную смесь перемешивали при комнатной температуре в течение 15 минут. После концентрирования реакционной смеси при пониженном давлении остаток дважды подвергали азеотропной перегонке с ацетонитрилом (5 мл). Воду (0,240 мл) добавляли в раствор остатка в дихлорметане (16,7 мл), в который добавляли раствор дихлоруксусной кислоты (0,953 мл) в дихлорметане (16,7 мл), и реакционную смесь перемешивали при комнатной температуре в течение 15 минут. Пиридин (1,82 мл) добавляли туда, чтобы погасить реакцию, и реакционную смесь затем концентрировали при пониженном давлении. Остаток подвергали трехкратной азеотропной перегонке с обезвоженным ацетонитрилом (10 мл), при этом приблизительно 5 мл ацетонитрила позволяли остаться после последней процедуры. Полученный в результате этого раствор в ацетонитриле указанного в заголовке соединения непосредственно использовали для следующей реакции.
[0388]
(Этап 8)
Доступный для приобретения (ChemGenes Corporation) N-бензоил-5'-O-[бис(4-метоксифенил)(фенил)метил]-3'-O-[трет-бутил(диметил)силил]-2'-O-{(2-цианоэтокси)[ди(пропан-2-ил)амино]фосфанил}аденозин (1,31 г) подвергали трехкратной азеотропной перегонке с обезвоженным ацетонитрилом (10 мл), при этом приблизительно 5 мл ацетонитрила позволяли остаться после последней процедуры, и добавляли туда молекулярные сита 3А, 1/16 (5 гранулоподобных частиц). Данный раствор в ацетонитриле добавляли в раствор, синтезированный на этапе 7, и реакционную смесь перемешивали в атмосфере азота при комнатной температуре в течение 20 минут. В реакционную смесь добавляли N,N-диметил-N'-(3-сульфанилиден-3Н-1,2,4-дитиазол-5-ил)метанимидамид (300 мг), и реакционную смесь перемешивали при комнатной температуре в течение 30 минут, а затем концентрировали при пониженном давлении. Добавляли воду (0,240 мл) в раствор остатка в дихлорметане (19,0 мл), добавляли туда раствор дихлоруксусной кислоты (1,20 мл) в дихлорметане (19,0 мл), и реакционную смесь перемешивали при комнатной температуре в течение 15 минут. После добавления туда пиридина (13,2 мл), чтобы погасить реакцию, продукт концентрировали при пониженном давлении. Полученный в результате этого неочищенный продукт непосредственно использовали для следующей реакции.
[0389]
(Этап 9)
N-{9-[(5R,7R,8R,12aR,14R,15R,15aR,16R)-14-(6-бензоил-6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)-15,16-бис{[трет-бутил(диметил)силил]окси}-10-(2-цианоэтокси)-2-оксо-2-сульфанил-10-сульфанилиденоктагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-7-ил]-9Н-пурин-6-ил}бензамид
После того, как раствор неочищенного продукта, полученный на этапе 8, в пиридине (39,6 мл) концентрировали до приблизительно 25 мл, добавляли туда 2-хлор-5,5-диметил-1,3,2λ5-диоксафосфинан-2-он (908 мг) и реакционную смесь перемешивали при комнатной температуре в течение 30 минут. Воду (0,84 мл) и 3Н-1,2-бензодитиол-3-он (336 мг) добавляли туда, и реакционную смесь перемешивали при комнатной температуре в течение 15 минут. Реакционную смесь вливали в водный раствор (180 мл) гидрокарбоната натрия (5,25 г), и продукт перемешивали при комнатной температуре в течение 30 минут, а затем осуществляли экстрагирование этилацетатом. После высушивания органического слоя над безводным сульфатом натрия осушающий агент удаляли путем фильтрации и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле [гексан/этилацетат/метанол] с получением указанного в заголовке соединения (507 мг) в виде смеси диастереомеров у атома фосфора.
МС (ИЭР) m/z: 1219 (М+Н)+.
[0390]
(Этап 10)
Бис(N,N-диэтилэтанаминия)(5R,7R,8R,12aR,14R,15R,15aR,16R)-7-(6-амино-9H-пурин-9-ил)-15,16-бис{[трет-бутил(диметил)силил]окси}-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
К раствору соединения, полученного на этапе 9 (507 мг), в метаноле (5 мл) добавляли 28% водный аммиак (5 мл), и реакционную смесь перемешивали при комнатной температуре в течение 14 часов. После концентрирования реакционной смеси остаток очищали с помощью колоночной хроматографии на силикагеле С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил] с получением указанного в заголовке соединения (301 мг) в виде смеси диастереомеров у атома фосфора.
МС (ИЭР) m/z: 958 (М+Н)+.
[0391]
(Этап 11)
Динатрия(5R,7R,8R,12aR,14R,15R,15aS,16R)-7-(6-амино-9H-пурин-9-ил)-15,16-дигидрокси-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
Тригидрофторид триэтиламина (3,84 мл) добавляли к соединению, полученному на этапе 10 (301 мг), и реакционную смесь перемешивали при 45°С в течение 3 часов. В реакционную смесь добавляли охлажденную до температуры льда смесь 1 M водного раствора гидрокарбоната триэтиламмония (20 мл) и триэтиламина (4 мл) при комнатной температуре. После концентрирования реакционной смеси при пониженном давлении реакционную смесь очищали с помощью колоночной хроматографии на силикагеле С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил] и препаративной ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 0% - 25% (0 мин - 40 мин)], чтобы разделить диастереомеры у атома фосфора. Полученное в результате этого соединение (соль триэтиламина) превращали в натриевую соль с помощью следующей процедуры.
[0392]
[Превращение в натриевую соль]
Смолу ВТ AG (R) 50W-X2 (чистую для биотехнологии, 100-200 меш, водородная форма) (500 мг) суспендировали в чистой воде и заполняли ею пустую колонку. После того, как избыточной части чистой воды позволили под действием гравитации стечь вниз, 1 M водному раствору гидроксида натрия (5 мл) и чистой воды (10 мл) позволяли под действием гравитации стечь вниз в данном порядке. Полученное выше соединение растворяли в чистой воде (5 мл) и заполняли им колонку. Раствор, которому позволяли под действием гравитации стечь вниз, разделяли, а затем дополнительно элюировали чистой водой (10 мл). Фракции, содержащие целевой продукт, объединяли и лиофилизировали с получением диастереомера 1 (83,4 мг), диастереомера 2 (44,8 мг) и диастереомера 3 (13,1 мг) указанного в заголовке соединения (время удерживания при ВЭЖХ: диастереомер 1 > 2, 3).
Диастереомер 1
МС (ИЭР) m/z: 730 (М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,74 (1Н, с), 8,17 (1H, с), 8,02 (1Н, с), 7,10 (1H, с), 6,34 (1H, д, J=8,5 Гц), 6,30 (1H, д, J=4,8 Гц), 5,41-5,34 (1H, м), 5,19-5,13 (1H, м), 4,85 (1H, д, J=3,6 Гц), 4,79 (1Н, т, J=4,5 Гц), 4,52-4,41 (2Н, м), 4,40-4,31 (2Н, м), 4,07-3,97 (2Н, м), 3,52-3,47 (2Н, м), 2,90-2,76 (2Н, м), 2,05-1,95 (2Н, м).
31Р-ЯМР (CD3OD) δ: 57,9 (с), 54,5 (с).
Диастереомер 2
МС (ИЭР) m/z: 730 (М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,82 (1H, с), 8,17 (1H, с), 8,02 (1H, с), 7,13 (1H, с), 6,35 (1H, д, J=2,4 Гц), 6,33 (1H, с), 5,50-5,43 (2Н, м), 4,80 (1H, дд, J=6,7, 4,2 Гц), 4,52-4,28 (5Н, м), 4,02 (1H, д, J=12,1 Гц), 3,93-3,86 (1Н, м), 3,54-3,47 (2Н, м), 2,95-2,88 (2Н, м), 2,05-1,97 (2Н, м).
31Р-ЯМР (CD3OD) δ: 63,0 (с), 60,2 (с).
Диастереомер 3
МС (ИЭР) m/z: 730 (М+Н)+.
1H-ЯМР (CD3OD) δ: 9,16 (1H, с), 8,17 (1H, с), 8,02 (1H, с), 7,12 (1H, с), 6,35 (1H, д, J=8,5 Гц), 6,29 (1H, д, J=6,7 Гц), 5,63-5,56 (1Н, м), 5,54-5,46 (1H, м), 4,79 (1Н, дд, J=6,7, 4,8 Гц), 4,53-4,43 (2Н, м), 4,36-4,28 (2Н, м), 4,26-4,19 (1H, м), 4,16-4,09 (1H, м), 3,93-3,86 (1H, м), 3,52-3,47 (2Н, м), 2,92-2,87 (2Н, м), 2,04-1,95 (2Н, м).
31Р-ЯМР (CD3OD) δ: 62,8 (с), 58,7 (с).
[0393]
Пример 2. Синтез ЦДН2
(5R,7R,8R,12aR,14R,15R,15aS,16R)-7-(6-амино-9Н-пурин-9-ил)-15,16-дигидрокси-2,10-бис(сульфанил)-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-дион
(Диастереомер 4 соединения 1, описанного в примере 1)
[0394]
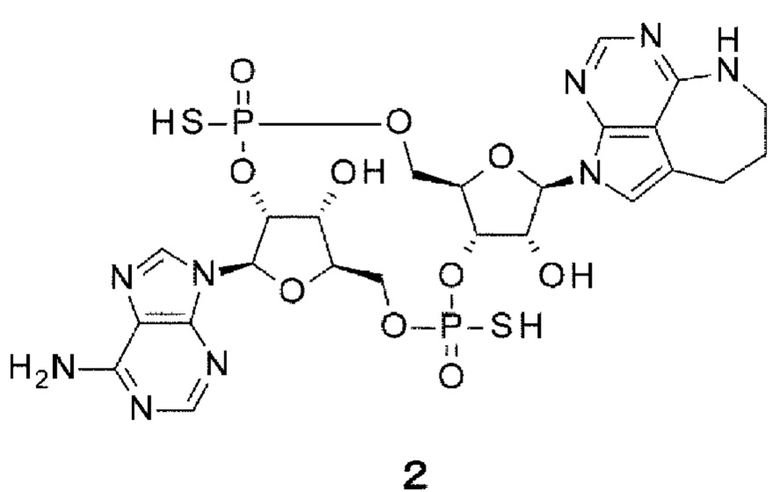
2а (Диастереомер 4 соединения 1)
[0395]
[Схема синтеза]
[0396]
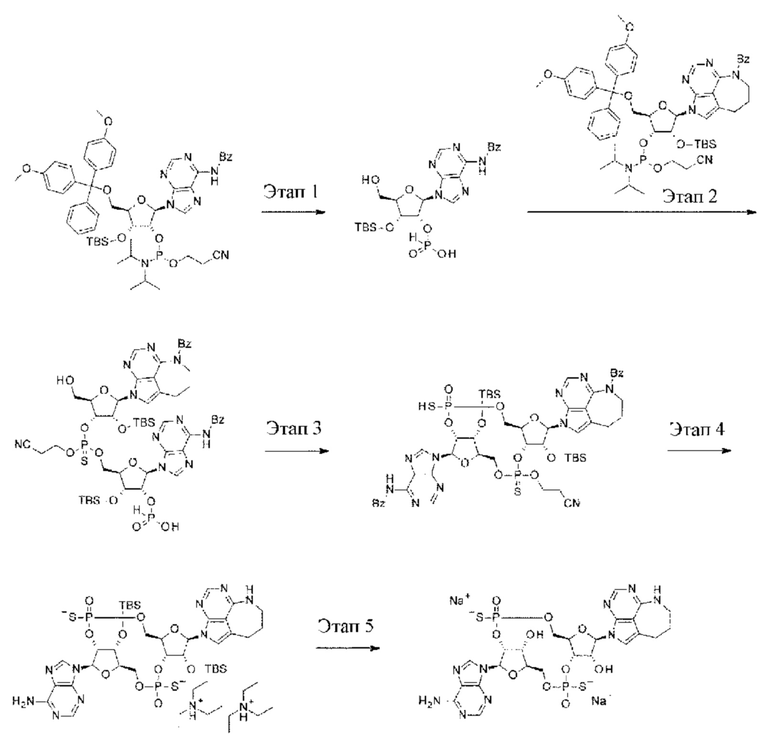
[0397] (Этап 1)
N-бензоил-3'-O-[трет-бутил(диметил)силил]-2'-O-[гидрокси(оксо)-λ5-фосфанил]аденозин
С применением доступного для приобретения (ChemGenes Corporation) N-бензоил-5'-O-[бис(4-метоксифенил)(фенил)метил]-3'-O-[трет-бутил(диметил)силил]-2'-O-{(2-цианоэтокси)[ди(пропан-2-ил)амино]фосфанил}аденозина (962 мг) проводили реакцию таким же образом, как на этапе 7 примера 1, с получением раствора в ацетонитриле указанного в заголовке соединения. Данный раствор в ацетонитриле непосредственно использовали для следующей реакции.
[0398]
(Этап 2)
С применением соединения, полученного на этапе 1, и соединения, полученного на этапе 6 примера 1 (1,00 г), проводили реакцию таким же образом, как на этапе 8 примера 1. Полученный в результате этого неочищенный продукт непосредственно использовали для следующей реакции.
[0399] (Этап 3)
N-{9-[(5R,7R,8R,12aR,14R,15R,15aR,16R)-14-(6-бензоил-6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)-15,16-бис{[трет-бутил(диметил)силил]окси}-2-(2-цианоэтокси)-10-оксо-10-сульфанил-2-сульфанилиденоктагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10] пентаоксадифосфациклотетрадецин-7-ил]-9Н-пурин-6-ил}бензамид
С применением неочищенного продукта, полученного на этапе 2, проводили реакцию таким же образом, как на этапе 9 примера 1, с получением указанного в заголовке соединения (367 мг) в виде смеси диастереомеров у атома фосфора.
МС (ИЭР) m/z: 1219(М+Н)+.
[0400] (Этап 4)
Бис(N,N-диэтилэтанаминия) (5R,7R,8R,12aR,14R,15R,15aR,16R)-7-(6-амино-9H-пурин-9-ил)-15,16-бис{[трет-бутил(диметил)силил]окси}-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2R5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
С применением соединения, полученного на этапе 3 (367 мг), проводили реакцию таким же образом, как на этапе 10 примера 1, с получением диастереомера 1 (115 мг: с примесями) и диастереомера 4(101 мг: с примесями) указанного в заголовке соединения.
Диастереомер 1 (менее полярный)
МС (ИЭР) m/z: 958(М+Н)+.
Диастереомер 4 (более полярный)
МС (ИЭР) m/z: 958(М+Н)+.
[0401]
(Этап 5)
Динатрия (5R,7R,8R,12aR,14R,15R,15aS,16R)-7-(6-амино-9H-пурин-9-ил)-15,16-дигидрокси-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
(Диастереомер 4 соединения 1)
С применением соединения, полученного на этапе 4 (диастереомер 4) (101 мг: с примесями), проводили реакцию таким же образом, как на этапе 11 примера 1, и затем проводили очистку при следующих [Условиях очистки] с получением указанного в заголовке соединения в виде соли триэтиламина.
[Условия очистки] Хроматография на колонке с силикагелем С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил], препаративная ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 5% - 25% (0 мин - 40 мин)], и препаративная ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/метанол, метанол: 5% - 100% (0 мин - 40 мин)].
Полученную соль триэтиламина подвергали солевому обмену таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением указанного в заголовке соединения (28,5 мг).
МС (ИЭР) m/z: 730(М+Н)+.
1H-ЯМР (CD3OD) δ: 9,11 (1H, с), 8,19 (1H, с), 8,02 (1H, с), 7,08 (1H, с), 6,35 (1H, д, J=8,5 Гц), 6,27 (1H, д, J=4,8 Гц), 5,43-5,36 (1Н, м), 5,29-5,21 (1H, м), 4,95-4,88 (1H, м), 4,80 (1Н, дд, J=4,5, 2,3 Гц), 4,50-4,43 (1H, м), 4,42-4,33 (2Н, м), 4,30-4,22 (1H, м), 4,20-4,03 (2Н, м), 3,52-3,46 (2Н, м), 2,85-2,66 (2Н, м), 2,05-1,90 (2Н, м).
31Р-ЯМР (CD3OD) δ: 58,1 (с), 54,1 (с).
[0402]
Пример 3. Синтез ЦДНЗ
(5R,7R,8R,12aR,14R,15R,15aS,16R)-7-(6-амино-9H-пурин-9-ил)-2,10,15,16-тетрагидрокси-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-дион
[0403]
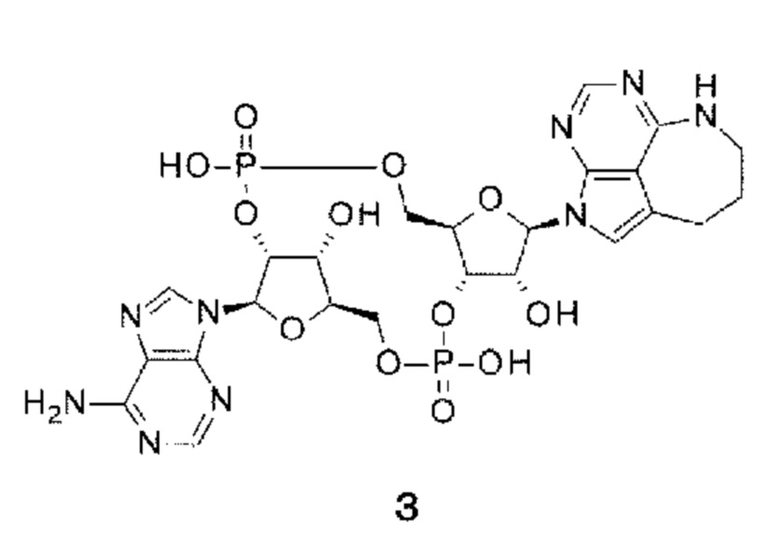
[0404]
[Схема синтеза]
[0405]
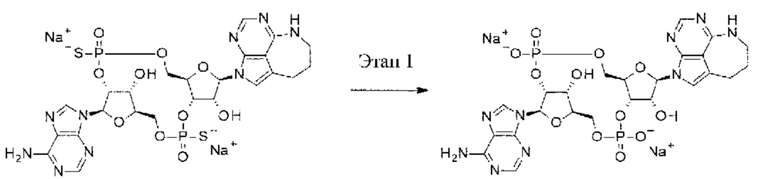
[0406]
(Этап 1)
Динатрия (5R,7R,8R,12aR,14R,15R,15aS,16R)-7-(6-амино-9H-пурин-9-ил)-15,16-дигидрокси-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(олат)
К раствору соединения, полученного на этапе 11 примера 1 (диастереомер 1) (30,0 мг), в ацетоне (0,5 мл)/воде (0,2 мл) добавляли триэтиламин (0,27 мл) и йодметан (60 мкл), и реакционную смесь перемешивали в течение 1 дня. После концентрирования реакционной смеси при пониженном давлении, реакционную смесь очищали с помощью препаративной ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 0% - 20% (0 мин - 40 мин)]. Полученное соединение подвергали солевому обмену таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением указанного в заголовке соединения (21,2 мг).
МС (ПЭР) m/z: 698(М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,55 (1Н, с), 8,18 (1Н, с), 8,01 (1H, с), 7,32 (1H, с), 6,26 (1H, с), 6,13 (1H, с), 5,00-4,85 (2Н, м), 4,68-4,64 (1H, м), 4,48-4,23 (5Н, м), 4,15-4,04 (2Н, м), 3,49-3,39 (2Н, м), 2,90-2,66 (2Н, м), 1,98-1,83 (2Н, м).
31Р-ЯМР (CD3OD) δ: -0,22 (с).
[0407]
Пример 4. Синтез ЦДН4
(5R,7R,8R,12aR,14R,15R,15aS,16R)-7-(2-амино-6-оксо-1,6-дигидро-9Н-пурин-9-ил)-15,16-дигидрокси-2,10-бис(сульфанил)-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-дион
[0408]
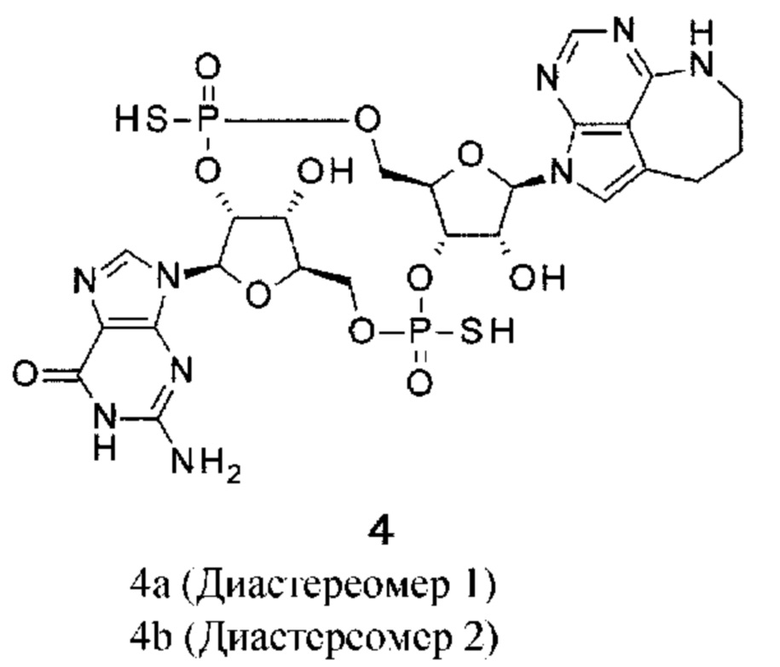
[0409]
[Схема синтеза]
[0410]
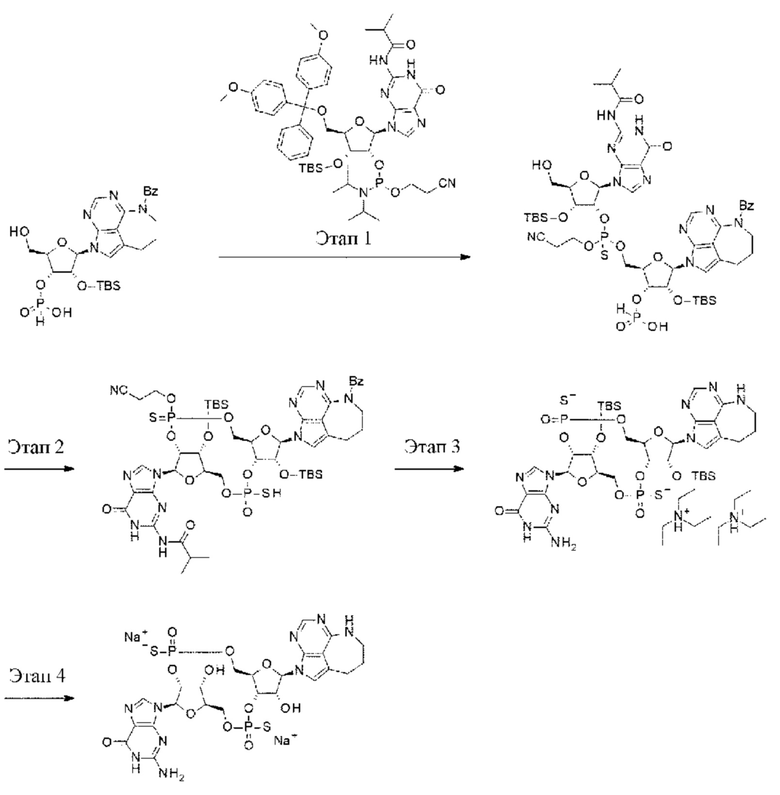
[0411]
(Этап 1)
Реакцию этапа 7 примера 1 проводили в следующем масштабе (сырьевой материал: 1,01 г). С применением раствора в ацетонитриле полученного соединения и доступного для приобретения (Wuhu Nuowei Chemistry Co., Ltd.) 5'-O-[бис(4-метоксифенил)(фенил)метил]-3'-О-[трет-бутил(диметил)силил]-2'-O-{(2-цианоэтокси)[ди(пропан-2-ил)амино]фосфанил}-N-(2-метилпропаноил)гуанозина (954 мг) проводили реакцию таким же образом, как на этапе 8 примера 1. Полученный в результате этого неочищенный продукт непосредственно использовали для следующей реакции.
[0412]
(Этап 2)
N-{9-[(5R,7R,8R,12aR,14R,15R,15aR,16R)-14-(6-бензоил-6,7,8,9-тетрагидро-2H-2,3,5,6-тетраазабензо[cd]азулен-2-ил)-15,16-бис{[трет-бутил(диметил)силил]окси}-10-(2-цианоэтокси)-2-оксо-2-сульфанил-10-сульфанилиденоктагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-7-ил]-6-оксо-6,9-дигидро-1Н-пурин-2-ил}-2-метилпропанамид
С применением неочищенного продукта, полученного на этапе 1, проводили реакцию таким же образом, как на этапе 9 примера 1 с получением указанного в заголовке соединения (357 мг) в виде смеси диастереомеров у атома фосфора.
MC (ИЭР) m/z: 1201(М+Н)+.
[0413]
(Этап 3)
Бис(N,N-диэтилэтанаминия) (5R,7R,8R,12aR,14R,15R,15aR,16R)-7-(2-амино-6-оксо-1,6-дигидро-9Н-пурин-9-ил)-15,16-бис{[трет-бутил(диметил)силил]окси}-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
С применением соединения, полученного на этапе 3 (357 мг), проводили реакцию таким же образом, как на этапе 10 примера 1 с получением указанного в заголовке соединения (241 мг) в виде смеси диастереомеров у атома фосфора.
МС (ИЭР) m/z: 974(М+Н)+.
[0414]
(Этап 4)
Динатрия (5R,7R,8R,12aR,14R,15R,15aS,16R)-7-(2-амино-6-оксо-1,6-дигидро-9H-пурин-9-ил)-15,16-дигидрокси-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
С применением соединения, полученного на этапе 3 (241 мг), проводили реакцию таким же образом, как на этапе 11 примера 1, и диастереомеры у атома фосфора разделяли при следующих [Условиях очистки] с получением двух диастереомеров указанного в заголовке соединения в виде солей триэтиламина.
[Условия очистки] Хроматография на колонке с силикагелем С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил] и препаративная ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 5% - 20% (0 мин - 40 мин)].
Каждую из полученных солей триэтиламина подвергали солевому обмену таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением диастереомера 1 (56,7 мг) и диастереомера 2 (25,9 мг) указанного в заголовке соединения (время удерживания при ВЭЖХ: диастереомер 1>2).
Диастереомер 1 (менее полярный)
МС (ИЭР) m/z: 746(М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,03 (1H, с), 8,00 (1Н, с), 7,11 (1H, с), 6,27 (1H, д, J=3,0 Гц), 5,99 (1H, д, J=8,5 Гц), 5,67-5,61 (1H, м), 5,27-5,21 (1H, м), 4,85 (1H, д, J=3,6 Гц), 4,73 (1H, дд, J=3,9, 2,0 Гц), 4,48-4,39 (2Н, м), 4,38-4,30 (2Н, м), 4,18-4,08 (2Н, м), 3,51-3,45 (2Н, м), 2,80-2,71 (1H, м), 2,63-2,53 (1H, м), 2,02-1,84 (2Н, м).
31Р-ЯМР (CD3OD) δ: 57,6 (с), 53,5 (с).
Диастереомер 2 (более полярный)
МС (ИЭР) m/z: 746(М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,20 (1H, с), 8,01 (1H, с), 7,19 (1H, с), 6,32 (1H, д, J=6,0 Гц), 6,05 (1H, д, J=8,5 Гц), 5,67-5,53 (1Н, м), 5,47-5,40 (1H, м), 4,77-4,71 (1H, м), 4,51-4,46 (1H, м), 4,45-4,30 (3Н, м), 4,28-4,25 (1H, м), 4,19-4,08 (1H, м), 3,96-3,89 (1H, м), 3,53-3,46 (2Н, м), 2,92-2,79 (2Н, м), 2,05-1,93 (2Н, м).
31Р-ЯМР (CD3OD) δ: 61,7 (с), 59,5 (с).
[0415]
Пример 5. Синтез ЦДН5
(5R,7R,8R,12aR,14R,15R,15aS,16R)-7-[1-(2-аминоэтил)-6-оксо-1,6-дигидро-9Н-пурин-9-ил]-15,16-дигидрокси-2,10-бис(сульфанил)-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-дион
[0416]
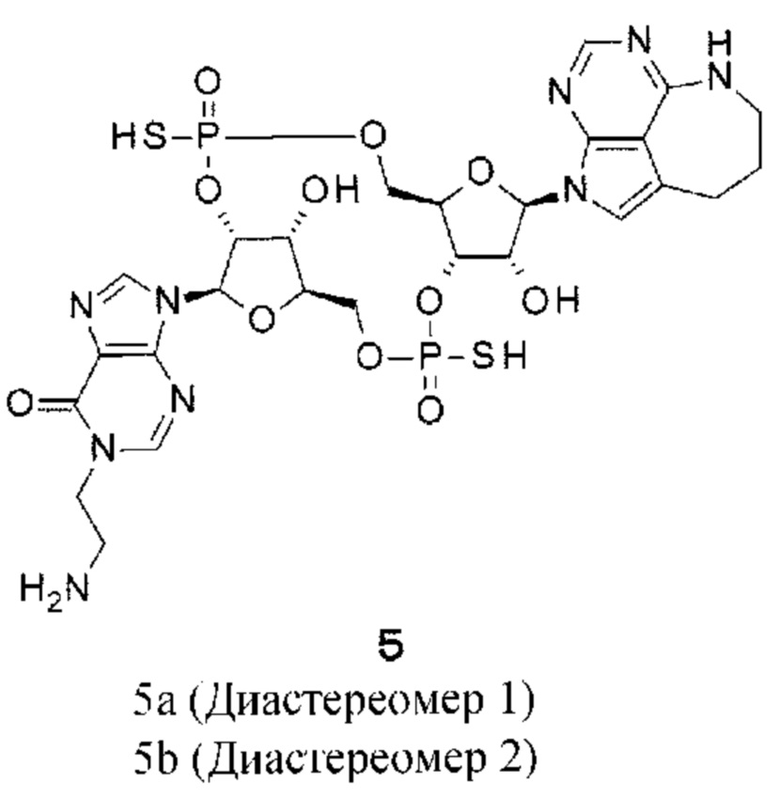
[0417]
[Схема синтеза]
[0418]
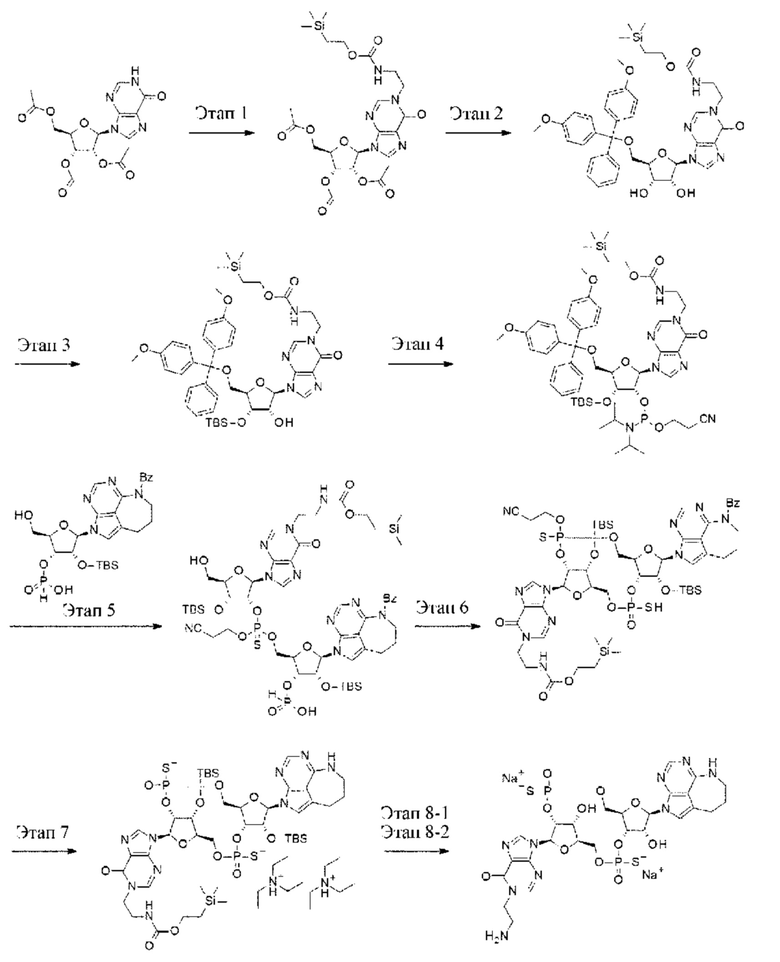
[0419]
(Этап 1)
2',3',5'-три-O-ацетил-1-[2-({[2-(триметилсилил)этокси]карбонил}амино)этил]инозин
В суспензию 2',3',5'-три-O-ацетилинозина (5,00 г) в тетрагидрофуране (90 мл) добавляли 2-(триметилсилил)этил-(2-гидроксиэтил)карбамат (3,12 г) и трифенилфосфин (3,99 г), и добавляли туда раствор дипропан-2-ил-(E)-диазен-1,2-дикарбоксилата (3,05 мл) в тетрагидрофуране (10 мл), и реакционную смесь перемешивали при комнатной температуре в течение 15 часов. После концентрирования реакционной смеси при пониженном давлении остаток очищали с помощью колоночной хроматографии на силикагеле [гексан/этилацетат/метанол] с получением указанного в заголовке соединения (3,01 г).
МС (ИЭР) m/z: 582(М+Н)+.
1Н-ЯМР (CDCl3) δ: 7,96 (1H, с), 7,93 (1Н, с), 6,10 (1H, д, J=4,8 Гц), 5,86 (1H, т, J=5,4 Гц), 5,58 (1H, т, J=5,l Гц), 4,96 (1H, т, J=7,3 Гц), 4,47-4,40 (2Н, м), 4,36 (1Н, дд, J=13,0, 5,1 Гц), 4,24 (2Н, т, J=5,4 Гц), 4,15 (2Н, т, J=8,8 Гц), 3,55 (2Н, к, J=6,0 Гц), 2,15 (3Н, с), 2,13 (3Н, с), 2,10 (3Н, с), 0,97 (2Н, т, J=8,8 Гц), 0,03 (9Н, с).
[0420]
(Этап 2)
5'-O-[бис(4-метоксифенил)(фенил)метил]-1-[2-({[2-(триметилсилил)этокси]карбонил}амино)этил]инозин
К раствору соединения, полученного на этапе 1 (3,01 г), в тетрагидрофуране (15 мл)-метаноле (15 мл) добавляли карбонат калия (100 мг), и реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Уксусную кислоту (83 мкл) добавляли в реакционную смесь, которую концентрировали при пониженном давлении, и остаток подвергали азеотропной перегонке с пиридином. Продукт снова растворяли в пиридине (30 мл), в который добавляли 4,4'-диметокситритилхлорид (2,10 г) при 0°С, и реакционную смесь перемешивали в течение 30 минут, а затем хранили в холодильной камере в течение ночи. Метанол (1 мл) добавляли в реакционную смесь, которую перемешивали в течение 30 минут, а затем концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле [гексан/этилацетат/метанол/0,1% триэтиламин] с получением указанного в заголовке соединения (3,61 г).
МС (ИЭР) m/z: 758(М+Н)+.
1Н-ЯМР (CDCl3) δ: 7,92 (1H, с), 7,76 (1H, с), 7,37 (2Н, д, J=7,3 Гц), 7,29-7,14 (7Н, м), 6,78 (4Н, д, J=8,5 Гц), 5,94 (1H, д, J=5,4 Гц), 5,63 (1H, уш. с), 4,81-4,74 (1H, м), 4,46-4,41 (1H, м), 4,36-4,31 (1H, м), 4,19-4,05 (4Н, м), 3,76 (6Н, с), 3,52-3,44 (2Н, м), 3,44-3,31 (2Н, м), 0,99-0,91 (2Н, м), 0,02 (9Н, с) (показаны только наблюдаемые пики).
[0421]
(Этап 3)
5'-O-[бис(4-метоксифенил)(фенил)метил]-3'-O-[трет-бутил(диметил)силил]-1-[2-({[2-(триметилсилил)этокси]карбонил}амино)этил]инозин
К раствору соединения, полученного на этапе 2 (3,61 г), в дихлорметане (18 мл) добавляли имидазол (811 мг) и трет-бутил(хлор)диметилсилан (861 мг), и реакционную смесь перемешивали при комнатной температуре в течение 17 часов. Насыщенный водный раствор гидрокарбоната натрия добавляли в реакционную смесь, которую осуществляли экстрагирование дихлорметаном. После высушивания органического слоя над безводным сульфатом натрия осушающий агент удаляли путем фильтрации и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле [гексан/этилацетат/0,1% триэтиламин] с получением указанного в заголовке соединения (1,61 г) и 5'-O-[бис(4-метоксифенил)(фенил)метил]-2'-O-[трет-бутил(диметил)силил]-1-[2-({[2-(триметилсилил)этокси]карбонил}амино)этил]инозина (1,31 г) в качестве региоизомера указанного в заголовке соединения.
МС (ИЭР) m/z: 872(М+Н)+.
1Н-ЯМР (CDCl3) δ: 7,98 (1H, с), 7,85 (1H, с), 7,39 (2Н, д, J=7,9 Гц), 7,32-7,15 (7Н, м), 6,78 (4Н, д, J=9,1 Гц), 5,93 (1H, д, J=4,8 Гц), 5,22-5,11 (1H, м), 4,60 (1Н, к, 3=5,6 Гц), 4,47 (1H, т, J=4,2 Гц), 4,28-4,08 (5Н, м), 3,77 (6Н, с), 3,59-3,49 (2Н, м), 3,45 (1H, дд, J=10,3, 3,0 Гц), 3,26 (1H, дд, J=10,3, 3,9 Гц), 3,15-3,08 (1H, м), 0,95 (2Н, т, J=8,5 Гц), 0,88 (9Н, с), 0,07 (3Н, с), 0,02 (9Н, с), 0,00 (3Н, с).
Региоизомер (2'-O-TBS-форма)
МС (ИЭР) m/z: 872(М+Н)+.
1H-ЯМР (CDCl3) δ: 7,99 (1Н, с), 7,82 (1Н, с), 7,46-7,41 (2Н, м), 7,35-7,19 (7Н, м), 6,84-6,78 (4Н, м), 5,98 (1H, д, J=5,4 Гц), 5,06-4,96 (1H, м), 4,84 (1H, т, J=5,4 Гц), 4,34-4,08 (6Н, м), 3,78 (6Н, с), 3,54 (2Н, к, J=5,8 Гц), 3,48 (1H, дд, J=10,6, 2,7 Гц), 3,39 (1Н, дд, J=10,6, 3,9 Гц), 2,71 (1H, д, J=4,2 Гц), 0,99-0,91 (2Н, м), 0,85 (9Н, с), 0,03 (9Н, с), 0,02 (3Н, с), -0,12 (3Н, с).
[0422]
(Этап 4)
5'-O-[бис(4-метоксифенил)(фенил)метил]-3'-O-[трет-бугил(диметил)силил]-2'-O-{(2-цианоэтокси)[ди(пропан-2-ил)амино]фосфанил}-1-[2-({[2-(триметилсилил)этокси]карбонил}амино)этил]инозин
К раствору соединения, полученного на этапе 3 (1,61 г), в дихлорметане (18,5 мл) добавляли 4,5-дицианоимидазол (240 мг) и 2-цианоэтил-N,N,N',N'-тетраизопропилфосфородиамидит (0,703 мл), и реакционную смесь перемешивали при комнатной температуре в течение 15 часов. Насыщенный водный раствор гидрокарбоната натрия добавляли в реакционную смесь, чтобы погасить реакцию. После того, как реакционную смесь подвергли экстрагированию дихлорметаном, органический слой сушили над безводным сульфатом натрия. Осушающий агент удаляли путем фильтрации и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле DIOL [гексан/этилацетат] с получением указанного в заголовке соединения (1,95 г) в виде смеси диастереомеров у атома фосфора (соотношение диастереомеров = 61:39).
МС (ИЭР) m/z: 1072(М+Н)+.
1Н-ЯМР (CDCl3) δ: 8,04 (0,39Н, с), 7,99 (0,61Н, с), 7,83 (0,39Н, с), 7,82 (0,61Н, с), 7,42 (2Н, д, J=7,3 Гц), 7,35-7,15 (7Н, м), 6,85-6,77 (4Н, м), 6,15 (0,61H, д, J=6,0 Гц), 6,09 (0,39Н, д, J=4,8 Гц), 5,34-5,24 (0,61Н, м), 5,12-5,03 (0,39Н, м), 4,86-4,76 (0,39Н, м), 4,72-4,62 (0,61Н, м), 4,47-4,42 (0,39Н, м), 4,42-4,36 (0,69Н, м), 4,31-4,05 (6Н, м), 3,78 (6Н, с), 3,78-3,65 (1H, м), 3,61-3,39 (7Н, м), 3,35 (0,61Н, дд, J=10,6, 3,9 Гц), 3,28 (0,39Н, дд, J=10,9, 4,2 Гц), 2,49 (0,78Н, т, J=6,0 Гц), 2,29 (1,22Н, т, J=5,7 Гц), 1,30-0,94 (12Н, м), 0,85 (5,49Н, с), 0,84 (3,51Н, с), 0,09 (1,17Н, с), 0,08 (1,83Н, с), 0,03 (9Н, с), 0,02 (1,83Н, с), 0,00 (1,17Н, с).
[0423]
(Этап 5)
Ту же реакцию, что и на этапе 7 примера 1, проводили в следующем масштабе (сырьевой материал: 910 мг). С применением раствора в ацетонитриле полученного соединения и полученного на этапе 4 соединения (950 мг) проводили реакцию таким же образом, как на этапе 8 примера 1. Полученный в результате этого неочищенный продукт непосредственно использовали для следующей реакции.
[0424]
(Этап 6)
2-(триметилсилил)этил-(2-{9-[(5R,7R,8R,12aR,14R,15R,15aR,16R)-14-(6-бензоил-6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)-15,16-бис{[трет-бутил(диметил)силил]окси}-10-(2-цианоэтокси)-2-оксо-2-сульфанил-10-сульфанилиденоктагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-7-ил]-6-оксо-6,9-дигидро-1H-пурин-1-ил}этил)карбамат
С применением неочищенного продукта, полученного на этапе 5, проводили реакцию таким же образом, как на этапе 9 примера 1, с получением указанного в заголовке соединения (602 мг) в виде смеси диастереомеров у атома фосфора.
МС (ИЭР) m/z: 1303(М+Н)+.
[0425]
(Этап 7)
Бис(N,N-диэтилэтанаминия) (5R,7R,8R,12aR,14R,15R,15aR,16R)-15,16-бис{[трет-бутил(диметил)силил]окси}-2,10-диоксо-7-{6-оксо-1-[2-({[2-(триметилсилил)этокси]карбонил}амино)этил]-1,6-дигидро-9Н-пурин-9-ил}-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
С применением соединения, полученного на этапе 6 (602 мг), проводили реакцию таким же образом, как на этапе 10 примера 1, с получением диастереомера 1 (205 мг: с примесями) и диастереомера 2 (244 мг: с примесями) указанного в заголовке соединения.
Диастереомер 1 (менее полярный)
МС (ИЭР) m/z: 1146(М+Н)+.
Диастереомер 2 (более полярный)
МС (ИЭР) m/z: 1146(М+Н)+.
[0426]
(Этап 8-1)
Динатрия (5R,7R,8R,12aR,14R,15R,15aS,16R)-7-[1-(2-аминоэтил)-6-оксо-1,6-дигидро-9Н-пурин-9-ил]-15,16-дигидрокси-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
(Диастереомер 1)
Тригидрофторид триэтиламина (1,31 мл) добавляли к соединению, полученному на этапе 7 (диастереомер 1) (145 мг: с примесями), и реакционную смесь перемешивали при 45°С в течение 3 часов. В реакционную смесь добавляли охлажденную до температуры льда смесь 1 М раствора гидрокарбоната триэтиламмония (10 мл) и триэтиламина (2 мл). Реакционную смесь концентрировали при пониженном давлении, а затем очищали с помощью колоночной хроматографии на силикагеле С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил]. К раствору соединения, полученного в тетрагидрофуране (4 мл), добавляли раствор в тетрагидрофуране фторида тетрабутиламмония (приблизительно 1 М, 2 мл), и реакционную смесь перемешивали при комнатной температуре в течение 39 часов. В реакционную смесь добавляли 10 мМ водный раствор ацетата триэтил аммония (4 мл), и реакционную смесь концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил] и препаративной ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 5% - 50% (0 мин - 40 мин)]. Замену соли осуществляли таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, за исключением того, что при замене соли для полученного соединения (соли триэтиламина) применяли смешанный раствор ацетонитрила-метанола-чистой воды (1:1:1) в качестве растворителя и элюента, чтобы растворить соединение, с получением указанного в заголовке соединения (72,5 мг).
МС (ИЭР) m/z: 774(М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,60 (1H, с), 8,15 (1H, с), 8,02 (1H, с), 7,11 (1H, с), 6,26 (1H, д, J=4,8 Гц), 6,24 (1H, т, J=5,l Гц), 5,47 (1Н, дт, J=8,2, 4,2 Гц), 5,23-5,17 (1H, м), 4,77-4,73 (2Н, м), 4,52-4,44 (2Н, м), 4,36-4,19 (3Н, м), 4,14-4,02 (3Н, м), 3,48 (2Н, т, J=4,8 Гц), 3,31-3,26 (2Н, м), 2,90-2,74 (2Н, м), 2,01-1,93 (2Н, м).
31Р-ЯМР (CD3OD) δ: 57,7 (с), 54,7 (с).
[0427]
(Этап 8-2)
Динатрия (5R,7R,8R,12aR,14R,15R,15aS,16R)-7-[1-(2-аминоэтил)-6-оксо-1,6-дигидро-9Н-пурин-9-ил]-15,16-дигидрокси-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
(Диастереомер 2)
С применением соединения, полученного на этапе 7 (диастереомер 2) (133 мг: с примесями), проводили реакцию и замену соли таким же образом, как на этапе 8-1, с получением указанного в заголовке соединения (55,4 мг).
МС (ИЭР) m/z: 774(М+Н)+.
1H-ЯМР (CD3OD) δ: 8,72 (1H, с), 8,25 (1H, с), 8,02 (1H, с), 7,11 (1H, с), 6,31 (1H, д, J=6,7 Гц), 6,28 (1H, д, J=8,5 Гц), 5,47-5,38 (2Н, м), 4,77 (1H, дд, J=6,7, 4,2 Гц), 4,48 (1H, д, J=4,2 Гц), 4,46-4,37 (2Н, м), 4,37-4,29 (3Н, м), 4,27-4,18 (1H, м), 4,08-4,02 (1H, м), 3,92-3,85 (1H, м), 3,53-3,46 (2Н, м), 3,28-3,23 (2Н, м), 2,93-2,86 (2Н, м), 2,04-1,96 (2Н, м).
31Р-ЯМР (CD3OD) δ: 62,6 (с), 60,0 (с).
[0428]
Пример 6.
Синтез ЦДН6
(5R,7R,8R,12aR,14R,15R,15aS,16R)-15,16-дигидрокси-7-[1-(2-гидроксиэтил)-6-оксо-1,6-дигидро-9Н-пурин-9-ил]-2,10-бис(сульфанил)-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-дион
[0429]
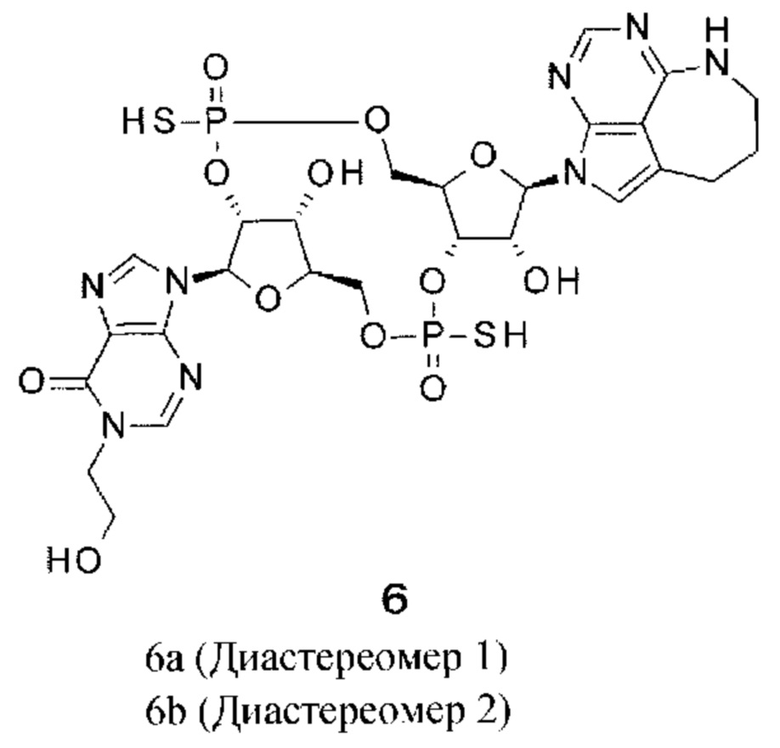
[0430]
[Схема синтеза]
[0431]
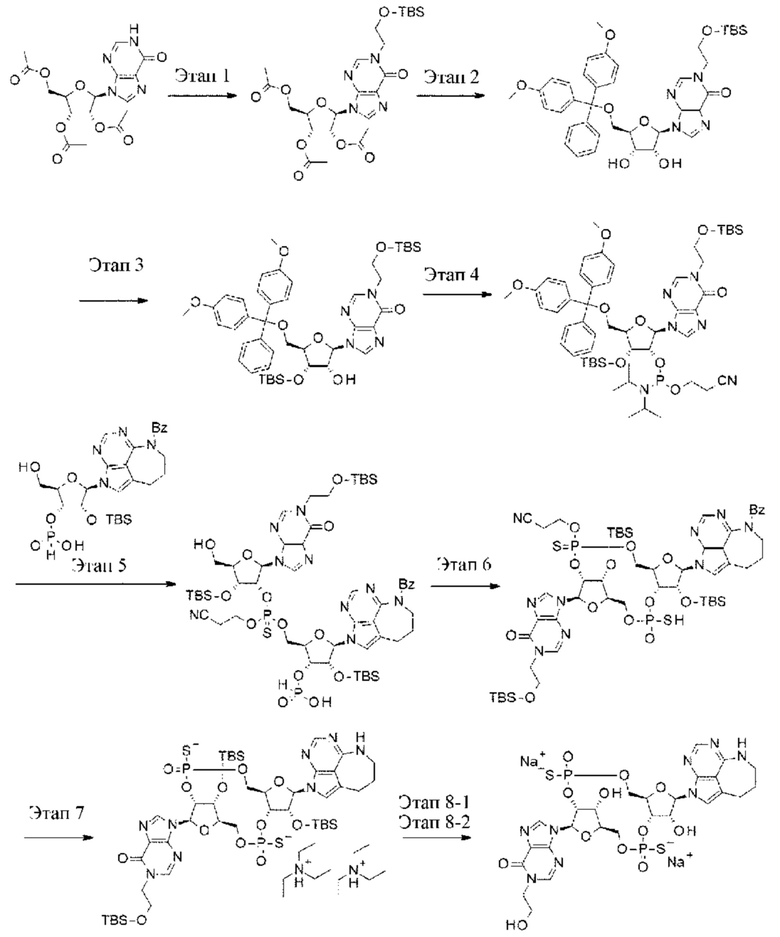
[0432]
(Этап 1)
2',3',5'-три-O-ацетил-1-(2-{[трет-бутил(диметил)силил]окси}этил)инозин
В суспензию доступного для приобретения (Ark Pharm, Inc.) 2',3',5'-три-O-ацетилинозина (10,0 г) в тетрагидрофуране (100 мл) добавляли 2-{[трет-бутил(диметил)силил]окси}этан-1-ол (5,37 г) и трифенилфосфин (7,69 г), и затем добавляли туда дипропан-2-ил-(Е)-диазен-1,2-дикарбоксилат (6,10 мл), и реакционную смесь перемешивали при комнатной температуре в течение 6 часов. После концентрирования реакционной смеси при пониженном давлении, остаток очищали с помощью колоночной хроматографии на силикагеле [гексан/этилацетат/дихлорметан] с получением указанного в заголовке соединения в виде смеси (10,6 г) с оксидом трифенилфосфина.
МС (ИЭР) m/z: 553(М+Н)+.
1Н-ЯМР (CDCl3) δ: 8,05 (1H, с), 7,92 (1Н, с), 6,12 (1H, д, J=5,4 Гц), 5,86 (1H, т, J=5,4 Гц), 5,59 (1H, дд, J=5,4, 4,2 Гц), 4,47-4,41 (2Н, м), 4,38-4,31 (1H, м), 4,22-4,17 (2Н, м), 3,89 (2Н, т, J=4,8 Гц), 2,15 (3Н, с), 2,14 (3Н, с), 2,08 (3Н, с), 0,83 (9Н, с), -0,06 (3Н, с), -0,06 (3Н, с).
[0433]
(Этап 2)
5'-O-[бис(4-метоксифенил)(фенил)метил]-1-(2-{[трет-бутил(диметил)силил]окси}этил)инозин
С применением соединения, полученного на этапе 1 (10,6 г), проводили реакцию таким же образом, как на этапе 2 примера 5, с получением указанного в заголовке соединения в виде смеси (7,21 г) с оксидом трифенилфосфина.
МС (ИЭР) m/z: 729(М+Н)+.
1Н-ЯМР (CDCl3) δ: 8,01 (1H, с), 7,97 (1H, с), 7,35-7,30 (2Н, м), 7,25-7,17 (7Н, м), 6,81-6,76 (4Н, м), 5,95 (1H, д, J=5,4 Гц), 5,13 (1H, уш. с), 4,68-4,61 (1H, м), 4,43-4,36 (2Н, м), 4,31-4,23 (1H, м), 4,15-4,08 (1H, м), 3,89 (2Н, т, J=4,5 Гц), 3,77 (6Н, с), 3,42 (1H, дд, J=10,3, 3,6 Гц), 3,34 (1H, дд, J=10,3, 3,6 Гц), 3,10 (1H, уш. с), 0,83 (9Н, с), -0,06 (3Н, с), -0,07 (3Н, с).
[0434]
(Этап 3)
5'-O-[бис(4-метоксифенил)(фенил)метил]-3'-O-[трет-бутил(диметил)силил]-1-(2-{[трет-бутил(диметил)силил]окси}этил)инозин
С применением соединения, полученного на этапе 2 (7,21 г), проводили реакцию таким же образом, как на этапе 3 примера 5, с получением указанного в заголовке соединения (2,17 г) и 5'-O-[бис(4-метоксифенил)(фенил)метил]-2'-O-[трет-бутил(диметил)силил]-1-(2-{[трет-бутил(диметил)силил]окси}этил)инозина (2,55 г) в качестве региоизомера указанного в заголовке соединения.
МС (ИЭР) m/z: 843(М+Н)+.
1Н-ЯМР (CDCl3) δ: 7,99 (1Н, с), 7,97 (1Н, с), 7,43-7,39 (2Н, м), 7,33-7,19 (7Н, м), 6,83-6,77 (4Н, м), 5,96 (1H, д, J=4,2 Гц), 4,56-4,50 (2Н, м), 4,33-4,25 (1H, м), 4,19-4,02 (2Н, м), 3,89 (2Н, т, J=4,8 Гц), 3,78 (6Н, с), 3,45 (1Н, дд, J=10,9, 4,2 Гц), 3,27 (1H, дд, J=10,9, 4,2 Гц), 3,03 (1H, д, J=6,0 Гц), 0,88 (9Н, с), 0,82 (9Н, с), 0,07 (3Н, с), -0,01 (3Н, с), -0,07 (3Н, с), -0,07 (3Н, с).
Региоизомер (2'-O-TBS-форма)
МС (ИЭР) m/z: 843(М+Н)+.
1Н-ЯМР (CDCl3) δ: 7,98 (1H, с), 7,94 (1H, с), 7,46-7,42 (2Н, м), 7,35-7,20 (7Н, м), 6,85-6,79 (4Н, м), 5,99 (1H, д, J=5,4 Гц), 4,83 (1Н, т, J=5,l Гц), 4,33-4,29 (1H, м), 4,27-4,24 (1H, м), 4,24-4,12 (2Н, м), 3,90 (2Н, т, J=4,5 Гц), 3,79 (3Н, с), 3,78 (3Н, с), 3,48 (1H, дд, J=10,3, 3,0 Гц), 3,40 (1H, дд, J=10,3, 3,0 Гц), 2,71 (1H, д, J=3,6 Гц), 0,86 (9Н, с), 0,83 (9Н, с), 0,01 (3Н, с), -0,07 (3Н, с), -0,07 (3Н, с), -0,11 (3H, с).
[0435]
(Этап 4)
5'-O-[бис(4-метоксифенил)(фенил)метил]-3'-O-[трет-бутил(диметил)силил]-1-(2-{[трет-бутил(диметил)силил]окси}этил)-2'-O-{(2-цианоэтокси)[ди(пропан-2-ил)амино]фосфанил}инозин
С применением соединения, полученного на этапе 3 (2,17 г), проводили реакцию таким же образом, как на этапе 4 примера 5, с получением указанного в заголовке соединения (2,65 г) в виде смеси диастереомеров у атома фосфора.
МС (ИЭР) m/z: 1043(М+Н)+.
1Н-ЯМР (CDCl3) δ: 8,03 (0,53Н, с), 8,01 (0,47Н, с), 7,97 (0,53Н, с), 7,93 (0,47Н, с), 7,45-7,41 (2Н, м), 7,35-7,19 (7Н, м), 6,83-6,78 (4Н, м), 6,17 (0,53Н, д, J=4,2 Гц), 6,05 (0,47Н, д, J=4,2 Гц), 4,87-4,80 (0,47Н, м), 4,64-4,58 (0,53Н, м), 4,46-4,40 (1H, м), 4,30-4,05 (3H, м), 3,92-3,87 (2Н, м), 3,78 (6Н, с), 3,86-3,40 (5Н, м), 3,33-3,24 (1H, м), 2,54 (0,94Н, т, J=6,0 Гц), 2,43 (1,06Н, т, J=6,7 Гц), 1,16-1,09 (9Н, м), 1,01-0,97 (3H, м), 0,83 (4,23Н, с), 0,83 (4,77Н, с), 0,82 (9Н, с), 0,07 (1,41Н, с), 0,04 (1,59Н, с), -0,02 (3H, с), -0,07 (1,41Н, с), -0,08 (1,59Н, с), -0,08 (3H, с).
[0436]
(Этап 5)
Ту же реакцию, что и на этапе 7 примера 1, проводили в следующем масштабе (сырьевой материал: 935 мг). С применением раствора в ацетонитриле полученного соединения и полученного на этапе 4 соединения (950 мг) проводили реакцию таким же образом, как на этапе 8 примера 1. Полученный в результате этого неочищенный продукт непосредственно использовали для следующей реакции.
[0437]
(Этап 6)
3-({(5R,7R,8R,12aR,14R,15R,15aR,16R)-14-(6-бензоил-6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)-15,16-бис{[трет-бутил(диметил)силил]окси}-7-[1-(2-{[трет-бутил(диметил)силил]окси}этил)-6-оксо-1,6-дигидро-9Н-пурин-9-ил]-2-оксо-2-сульфанил-10-сульфанилиденоктагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-10-ил}окси)пропаннитрил
С применением неочищенного продукта, полученного на этапе 5, проводили реакцию таким же образом, как на этапе 9 примера 1, с получением указанного в заголовке соединения (494 мг) в виде смеси диастереомеров у атома фосфора.
МС (ИЭР) m/z: 1274(М+Н)+.
[0438]
(Этап 7)
Бис(N,N-диэтилэтанаминия) (5R,7R,8R,12aR,14R,15R,15aR,16R)-15,16-бис{[трет-бутил(диметил)силил]окси}-7-[1-(2-{[трет-бутил(диметил)силил]окси}этил)-6-оксо-1,6-дигидро-9Н-пурин-9-ил]-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
С применением соединения, полученного на этапе 6 (494 мг), проводили реакцию таким же образом, как на этапе 10 примера 1, с получением диастереомера 1 (88,5 мг: с примесями) и диастереомера 2 (70,7 мг: с примесями) указанного в заголовке соединения.
Диастереомер 1 (менее полярный)
МС (ИЭР) m/z: 1003(M-C6H15Si+2H)+.
Диастереомер 2 (более полярный)
МС (ИЭР) m/z: 1003(M-C6H15Si+2H)+.
[0439]
(Этап 8-1)
Динатрия (5R,7R,8R,12aR,14R,15R,15aS,16R)-15,16-дигидрокси-7-[1-(2-гидроксиэтил)-6-оксо-1,6-дигидро-9Н-пурин-9-ил]-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
(Диастереомер 1)
С применением соединения, полученного на этапе 7 (диастереомер 1) (88,5 мг: с примесями), проводили реакцию таким же образом, как на этапе 11 примера 1, и затем проводили очистку при следующих [Условиях очистки] с получением указанного в заголовке соединения в виде соли триэтиламина.
[Условия очистки] Хроматография на колонке с силикагелем С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил] и препаративная ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 5% - 30% (0 мин - 40 мин)].
Полученную соль триэтиламина подвергали солевому обмену таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением указанного в заголовке соединения (25,7 мг).
МС (ИЭР) m/z: 775(М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,63 (1H, с), 8,22 (1H, с), 8,02 (1H, с), 7,11 (1H, с), 6,30-6,24 (2Н, м), 5,46-5,37 (1H, м), 5,23-5,15 (1Н, м), 4,83-4,79 (1H, м), 4,78-4,74 (1H, м), 4,53-4,42 (2Н, м), 4,35-4,16 (3H, м), 4,16-3,97 (3H, м), 3,83-3,78 (2Н, м), 3,52-3,47 (2Н, м), 2,88-2,81 (2Н, м), 2,03-1,95 (2Н, м).
31Р-ЯМР (CD3OD) δ: 57,8 (с), 54,4 (с).
[0440]
(Этап 8-2)
Динатрия (5R,7R,8R,12aR,14R,15R,15aS,16R)-15,16-дигидрокси-7-[l-(2-гидроксиэтил)-6-оксо-1,6-дигидро-9Н-пурин-9-ил]-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
(Диастереомер 2)
С применением соединения, полученного на этапе 7 (диастереомер 2) (70,7 мг: с примесями), проводили реакцию таким же образом, как на этапе 11 примера 1, и затем проводили очистку при следующих [Условиях очистки] с получением указанного в заголовке соединения в виде соли триэтиламина.
[Условия очистки] Хроматография на колонке с силикагелем С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил], препаративная ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 5% - 25% (0 мин - 40 мин)], и препаративная ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/метанол, метанол: 15% - 70% (0 мин - 40 мин)].
Полученную соль триэтиламина подвергали солевому обмену таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением указанного в заголовке соединения (17,8 мг).
МС (ИЭР) m/z: 775(М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,72 (1H, с), 8,23 (1Н, с), 8,02 (1Н, с), 7,11 (1H, с), 6,30 (2Н, дд, J=13,6, 7,6 Гц), 5,48-5,39 (2Н, м), 4,78 (1H, дд, J=6,7, 4,2 Гц), 4,51-4,28 (5Н, м), 4,26-4,13 (2Н, м), 4,06-4,00 (1H, м), 3,93-3,86 (1Н, м), 3,85-3,80 (2Н, м), 3,52-3,47 (2Н, м), 2,94-2,88 (2Н, м), 2,05-1,97 (2Н, м).
31Р-ЯМР (CD3OD) δ: 62,9 (с), 60,0 (с).
[0441]
Пример 7. Синтез ЦДН7
N-(2-{9-[(5R,7R,8R,12aR,14R,15R,15aS,16R)-15,16-дигидрокси-2,10-диоксо-2,10-бис(сульфанил)-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-7-ил]-6-оксо-6,9-дигидро-1Н-пурин-1-ил}этил)-2-гидрокеиацетамид
[0442]
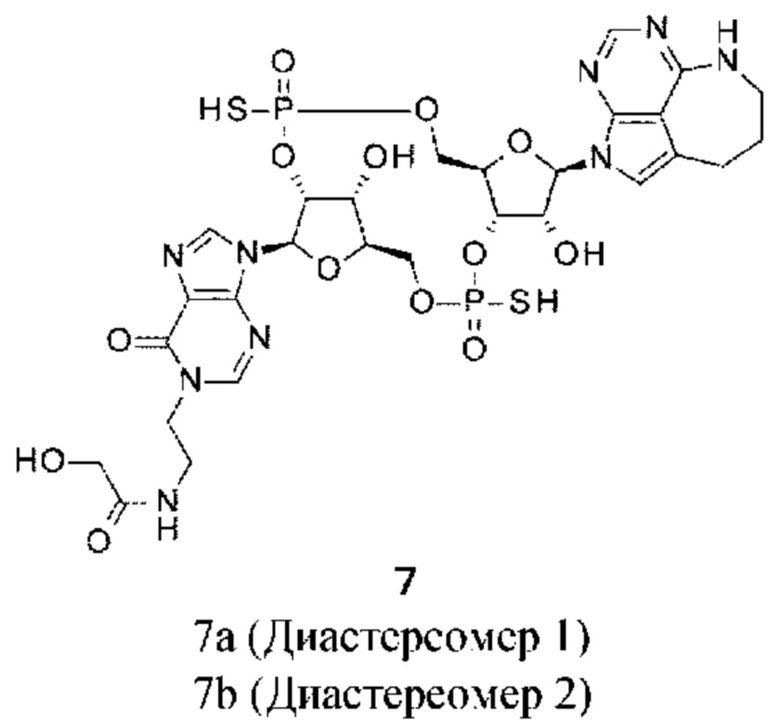
[0443]
[Схема синтеза]
[0444]
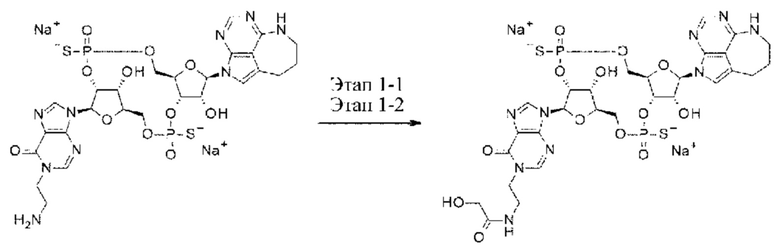
[0445]
(Этап 1-1)
Динатрия (5R,7R,8R,12aR,14R,15R,15aS,16R)-15,16-дигидрокси-7-{l-[2-(2-гидроксиацетамид)этил]-6-оксо-1,6-дигидро-9Н-пурин-9-ил}-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
(Диастереомер 1)
К раствору соединения, полученного на этапе 8-1 примера 5 (10,0 мг), в N,N-диметилформамиде (0,5 мл), добавляли триэтиламин (8 мкл) и 1-[(гидроксиацетил)окси]-2,5-дион (5,3 мг), и реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь разбавляли 10 мМ водным раствором ацетата триэтиламмония и очищали с помощью колоночной хроматографии на силикагеле С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил] и препаративной ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 0% - 30% (0 мин - 40 мин)]. Полученное соединение (соль триэтиламина) подвергали солевому обмену таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением указанного в заголовке соединения (10,5 мг).
МС (ИЭР) m/z: 832(М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,57 (1H, с), 8,04 (1H, с), 8,03 (1H, с), 7,13 (1H, с), 6,26 (1H, д, J=4,2 Гц), 6,24-6,19 (1H, м), 5,57-5,49 (1H, м), 5,26-5,18 (1Н, м), 4,80 (1H, д, J=3,6 Гц), 4,76 (1Н, т, J=4,5 Гц), 4,51-4,41 (2Н, м), 4,35-4,17(3H, м), 4,11-3,95 (3H, м), 3,91 (2Н, с), 3,62-3,55 (2Н, м), 3,52-3,45 (2Н, м), 2,89-2,65 (2Н, м), 2,02-1,91 (2Н, м).
31Р-ЯМР (CD3OD) δ: 57,6 (с), 54,3 (с).
[0446]
(Этап 1-2)
Динатрия (5R,7R,8R,12aR,14R,15R,15aS,16R)-15,16-дигидрокси-7-{1-[2-(2-гидроксиацетамид)этил]-6-оксо-1,6-дигидро-9Н-пурин-9-ил}-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
(Диастереомер 2)
С применением соединения, полученного на этапе 8-2 (30,0 мг) примера 5, проводили реакцию таким же образом, как на этапе 1-1, и затем проводили очистку при следующих [Условиях очистки] с получением указанного в заголовке соединения в виде соли триэтиламина.
[Условия очистки] Хроматография на колонке с силикагелем С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил] и препаративная ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 5% - 30% (0 мин - 40 мин)].
[0447]
Полученную соль триэтиламина подвергали солевому обмену таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением указанного в заголовке соединения (23,6 мг).
МС (ИЭР) m/z: 832(М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,69 (1Н, с), 8,14 (1Н, с), 8,02 (1Н, с), 7,11 (1Н, с), 6,31 (1Н, д, J=6,7 Гц), 6,26 (1Н, д, J=7,9 Гц), 5,49-5,40 (2Н, м), 4,77 (1Н, дд, J=6,7, 4,8 Гц), 4,48 (1Н, д, J=4,2 Гц), 4,46-4,28 (4Н, м), 4,22 (2Н, т, J=5,4 Гц), 4,06-4,00 (1Н, м), 3,94 (2Н, с), 3,92-3,86 (1Н, м), 3,70-3,55 (2Н, м), 3,52-3,47 (2Н, м), 2,92-2,86 (2Н, м), 2,04-1,96 (2Н, м).
31Р-ЯМР (CD3OD) δ: 62,7 (с), 59,9 (с).
[0448]
Пример 8. Синтез ЦДН8
(5R,7R,8R,12aR,14R,15R,15aS,16R)-7-{6-амино-2-[(2-аминоэтил)амино]-9H-пурин-9-ил}-15,16-дигидрокси-2,10-бис(сульфанил)-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагадро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-дион
[0449]
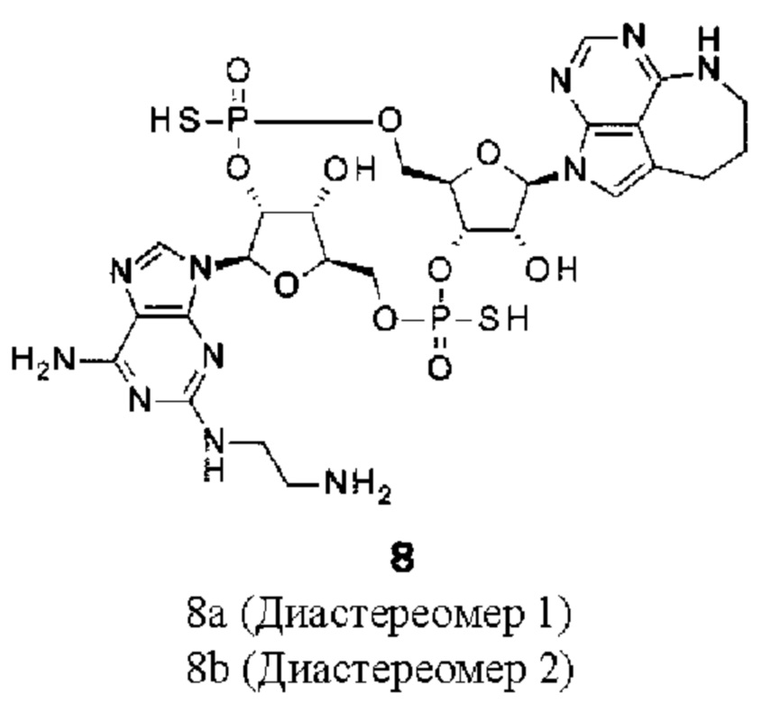
[0450]
[Схема синтеза]
[0451]
[0452]
(Этап 1)
5'-O-[бис(4-метоксифенил)(фенил)метил]-2',3'-бис-O-[трет-бутил(диметил)силил]-2-хлораденозин
К раствору 5'-O-[бис(4-метоксифенил)(фенил)метил]-2-хлораденозина (29,1 г) как соединения, известного в литературе (J. Med. Chem. 1989, 32, 1135-1140), в N,N-диметилформамиде (145 мл), добавляли имидазол (16,4 г) и трет-бутилдиметилхлорсилан (18,2 г), и реакционную смесь перемешивали при комнатной температуре в течение 18 часов. После того, как в реакционную смесь добавляли воду, чтобы погасить реакцию, осуществляли экстрагирование продукта этилацетатом. Органический слой промывали рассолом, а затем сушили над безводным сульфатом натрия. Осушающий агент удаляли путем фильтрации и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле [гексан/этилацетат/0,1% триэтиламин] с получением указанного в заголовке соединения (34,9 г).
МС (ИЭР) m/z: 832(М+Н)+.
1H-ЯМР (CDCl3) δ: 8,02 (1H, с), 7,47-7,42 (2Н, м), 7,36-7,32 (4Н, м), 7,31-7,18 (3H, м), 6,84-6,79 (4Н, м), 5,90 (1H, д, J=4,8 Гц), 5,72 (2Н, уш. с), 4,74 (1H, дд, J=4,5, 2,3 Гц), 4,25 (1H, дд, J=4,2, 2,3 Гц), 4,21 (1H, к, J=4,2 Гц), 3,78 (6Н, с), 3,58 (1H, дд, J=10,9, 4,2 Гц), 3,33 (1Н, дд, J=10,9, 4,2 Гц), 0,84 (9Н, с), 0,82 (9Н, с), 0,04 (3H, с), -0,01 (3H, с), -0,02 (3H, с), -0,17 (3H, с).
[0453]
(Этап 2)
N-ацетил-5'-O-[бис(4-метоксифенил)(фенил)метил]-2',3'-бис-O-[трет-бутил(диметил)силил]-2-хлораденозин
К раствору соединения, полученного на этапе 1 (34,9 г), в пиридине (210 мл) добавляли уксусный ангидрид (140 мл) и 4-диметиламинопиридин (515 мг), и реакционную смесь перемешивали в атмосфере азота при комнатной температуре в течение 21 часа. После разбавления реакционной смеси дихлорметаном (100 мл) добавляли туда насыщенный водный раствор гидрокарбоната натрия, и реакционную смесь осуществляли экстрагирование дихлорметаном. После высушивания органического слоя над безводным сульфатом натрия осушающий агент удаляли путем фильтрации и фильтрат концентрировали при пониженном давлении. Дихлорметан (210 мл) и морфолин (7,30 мл) добавляли к остатку, и продукт перемешивали при комнатной температуре в течение 1 часа. Насыщенный водный раствор хлорида аммония добавляли в реакционную смесь, которую осуществляли экстрагирование дихлорметаном. После высушивания органического слоя над безводным сульфатом натрия осушающий агент удаляли путем фильтрации и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле [гексан/этилацетат/0,1% триэтиламин] с получением указанного в заголовке соединения (45,4 г: с примесями). Полученное соединение непосредственно использовали для следующей реакции без дополнительной очистки.
1Н-ЯМР (CDCl3) δ: 8,95 (1H, уш. с), 8,22 (1H, с), 7,45-7,42 (2Н, м), 7,34-7,20 (7Н, м), 6,82 (4Н, дк, 3=9,4, 2,7 Гц), 5,96 (1H, д, J=4,8 Гц), 4,72-4,69 (1H, м), 4,23 (2Н, уш. с), 3,79 (6Н, с), 3,59 (1H, дд, J=10,9, 3,6 Гц), 3,35 (1H, дд, J=10,3, 3,6 Гц), 2,73 (3H, с), 0,83 (9Н, с), 0,82 (9Н, с), 0,04 (3H, с), 0,00 (3H, с), -0,03 (3H, с), -0,18 (3H, с).
[0454]
(Этап 3)
N-ацетил-5'-O-[бис(4-метоксифенил)(фенил)метил]-2-хлораденозин
К раствору соединения, полученного на этапе 2 (45,4 г: с примесями), в тетрагидрофуране (200 мл) добавляли раствор в тетрагидрофуране фторида тетрабутиламмония (приблизительно 1,0 М, 100 мл), и реакционную смесь перемешивали в атмосфере азота при комнатной температуре в течение 3 часов. Насыщенный водный раствор хлорида аммония добавляли в реакционную смесь, которую осуществляли экстрагирование дихлорметаном. После высушивания органического слоя над безводным сульфатом натрия осушающий агент удаляли путем фильтрации и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле [дихлорметан/ацетон/0,1% триэтиламин] с получением указанного в заголовке соединения (23,0 г).
1Н-ЯМР (CDCl3) δ: 8,71 (1H, уш. с), 8,16 (1H, с), 7,28-7,16 (9Н, м), 6,78-6,73 (4Н, м), 5,99 (1H, д, J=5,4 Гц), 4,86 (1H, т, J=5,l Гц), 4,49 (1Н, дд, J=5,l, 2,7 Гц), 4,39 (1H, к, J=3,2 Гц), 3,78 (3H, с), 3,77 (3H, с), 3,42 (1Н, дд, J=10,9, 3,6 Гц), 3,35 (1H, дд, J=10,6, 3,3 Гц), 2,66 (3H, с).
[0455]
(Этап 4)
N-ацетил-5'-O-[бис(4-метоксифенил)(фенил)метил]-3'-O-[трет-бутил(диметил)силил]-2-хлораденозин
К раствору соединения, полученного на этапе 3 (23,0 г), в N,N-диметилформамиде (178 мл) добавляли имидазол (5,94 г) и трет-бутилдиметилхлорсилан (6,44 г), и реакционную смесь перемешивали в атмосфере азота при комнатной температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия добавляли в реакционную смесь, которую осуществляли экстрагирование дихлорметаном. После высушивания органического слоя над безводным сульфатом натрия осушающий агент удаляли путем фильтрации и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле [гексан/этилацетат/0,1% триэтиламин] с получением указанного в заголовке соединения (9,01 г).
1Н-ЯМР (CDCl3) δ: 8,42 (1H, уш. с), 8,14 (1H, с), 7,38-7,35 (2Н, м), 7,29-7,18 (7Н, м), 6,80-6,76 (4Н, м), 5,99 (1H, д, J=4,2 Гц), 4,71-4,66 (2Н, м), 4,17-4,14 (1H, м), 3,78 (6Н, с), 3,49 (1H, дд, J=10,6, 3,3 Гц), 3,29 (1H, дд, J=10,9, 4,2 Гц), 3,02 (1Н, д, J=5,4 Гц), 2,67 (3H, с), 0,89 (9Н, с), 0,11 (3H, с), 0,02 (3H, с).
[0456]
(Этап 5)
N-ацетил-5'-O-[бис(4-метоксифенил)(фенил)метил]-3'-O-[трет-бутил(диметил)силил]-2-хлор-2'-O-{(2-цианоэтокси)[ди(пропан-2-ил)амино]фосфанил}аденозин
С применением соединения, полученного на этапе 4 (9,01 г), проводили реакцию таким же образом, как на этапе 6 примера 1, с получением указанного в заголовке соединения (10,6 г) в виде смеси диастереомеров у атома фосфора (соотношение диастереомеров = 65:35).
1Н-ЯМР (CDCl3) δ: 8,40 (1H, уш. с), 8,31 (0,35Н, с), 8,25 (0,65Н, с), 7,38 (2Н, д, J=7,3 Гц), 7,29-7,19 (7Н, м), 6,80 (4Н, дд, 3=9,1, 2,4 Гц), 6,27 (0,35Н, д, J=3,0 Гц), 6,13 (0,65Н, д, J=3,6 Гц), 4,88-4,83 (0,65Н, м), 4,69-4,65 (0,35Н, м), 4,56 (1H, т, J=4,8 Гц), 4,23-4,17 (1Н, м), 3,93-3,78 (1H, м), 3,78 (6Н, с), 3,64-3,54 (4Н, м), 3,33-3,28 (1H, м), 2,67 (3H, с), 2,56 (1,3H, т, J=6,3 Гц), 2,52 (0,7Н, т, J=6,3 Гц), 1,16 (2,1Н, д, J=7,3 Гц), 1,14 (3,9Н, д, J=6,0 Гц), 1,12 (3,9Н, д, J=6,0 Гц), 1,02 (2,1Н, д, J=6,7 Гц), 0,83 (5,9Н, с), 0,82 (3,1Н, с), 0,10 (1,9Н, с), 0,07 (1,1Н, с), 0,01Н (3H, с).
[0457]
(Этап 6)
N,N-диэтилэтанаминия (5R,7R,8R,12aR,14R,15R,15aR,16R)-7-(6-ацетамидо-2-хлор-9Н-пурин-9-ил)-14-(6-бензоил-6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)-15,16-бис{[трет-бутил(диметил)силил]окси}-10-(2-цианоэтокси)-2-оксо-10-сульфанилиденоктагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2-тиолят
Ту же реакцию, что и на этапе 7 примера 1 проводили в следующем масштабе (сырьевой материал: 2,06 г). С применением раствора в ацетонитриле полученного соединения и соединения, полученного на этапе 5 (1,98 г), проводили реакцию таким же образом, как на этапе 8 примера 1 и этапе 9 примера 1, с получением диастереомера 1 (180 мг) и диастереомера 2 (167 мг: с примесями) указанного в заголовке соединения.
Диастереомер 1 (менее полярный)
МС (ИЭР) m/z: 1191(М+Н)+.
1Н-ЯМР (CD3OD) δ: 9,10 (1H, с), 8,00 (1H, с), 7,40-7,36 (2Н, м), 7,30-7,23 (4Н, м), 6,38 (1H, д, J=8,5 Гц), 6,35 (1H, д, J=3,0 Гц), 5,67-5,60 (1H, м), 5,09-5,04 (1H, м), 4,72 (1H, д, J=3,6 Гц), 4,50-4,29 (8Н, м), 4,07 (1H, дд, J=12,4, 4,5 Гц), 3,91-3,83 (1H, м), 3,54-3,45 (1H, м), 3,17 (6Н, к, J=7,3 Гц), 3,08 (2Н, т, J=6,0 Гц), 2,45-2,42 (2Н, м), 2,42 (3H, с), 2,28-2,23 (2Н, м), 1,29 (9Н, т, J=7,3 Гц), 1,01 (9Н, с), 0,90 (9Н, с), 0,29 (3H, с), 0,28 (3H, с), 0,25 (3H, с), 0,10 (3H, с).
Диастереомер 2 (более полярный)
МС (ИЭР) m/z: 1191(М+Н)+.
[0458]
(Этап 7-1)
(5R,7R,8R,12aR,l4R,15R,15aR,16R)-7-{6-амино-2-[(2-аминоэтил)амино]-9Н-пурин-9-ил}-15,16-бис{[трет-бутил(диметил)силил]окси}-2,10-бис(сульфанил)-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-дион
К раствору соединения, полученного на этапе 6 (диастереомер 1) (49,6 мг), в метаноле (1,28 мл) добавляли этилендиамин (256 мкл) и реакционную смесь перемешивали при 60°С в течение 2 часов, а затем проводили реакцию, применяя микроволновой реактор, при 120°С в течение 2 часов. Продукт очищали с помощью препаративной ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 40% - 70% (0 мин - 30 мин)] с получением указанного в заголовке соединения (37,2 мг).
МС (ИЭР) m/z: 1016(М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,27 (1H, с), 7,99 (1Н, с), 7,16 (1H, с), 6,25 (1H, д, J=4,2 Гц), 6,09 (1H, д, J=8,5 Гц), 5,41-5,35 (1H, м), 5,12-5,08 (1H, м), 4,86-4,80 (2Н, м), 4,69 (1H, т, J=4,5 Гц), 4,51-4,45 (1H, м), 4,29-4,23 (2Н, м), 4,11-4,03 (2Н, м), 3,51-3,44 (4Н, м), 3,13-3,04 (2Н, м), 2,80-2,76 (2Н, м), 2,02-1,92 (2Н, м), 0,99 (9Н, с), 0,84 (9Н, с), 0,32 (3H, с), 0,29 (3H, с), 0,24 (3H, с), 0,07 (3H, с).
[0459]
(Этап 7-2)
Бис(N,N-диэтилэтанаминия) (5R,7R,8R,12aR,14R,15R,15aR,16R)-7-{6-амино-2-[(2-аминоэтил)амино]-9Н-пурин-9-ил}-15,16-бис{[трет-бутил(диметил)силил]окси}-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
К раствору соединения, полученного на этапе 6 (диастереомер 2) (50,0 мг: с примесями), в метаноле (1,29 мл) добавляли этилендиамин (25,8 мкл) и реакционную смесь перемешивали при 60°С в течение 2 часов, а затем проводили реакцию, применяя микроволновой реактор, при 120°С в течение 2 часов. Продукт очищали с помощью препаративной ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 30% - 50% (0 мин - 30 мин)] с получением указанного в заголовке соединения (23,7 мг).
МС (ИЭР) m/z: 1016(М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,16 (1H, с), 8,00 (1H, с), 7,08 (1H, с), 6,33 (1H, д, J=7,3 Гц), 6,13 (1H, д, J=8,5 Гц), 5,51-5,48 (1H, м), 5,30 (1H, т, J=4,8 Гц), 5,13-5,06 (1Н, м), 4,95 (1Н, д, J=4,2 Гц), 4,66-4,55 (2Н, м), 4,24 (1H, с), 4,08 (1H, дд, J=12,4, 4,5 Гц), 3,89-3,83 (1Н, м), 3,69-3,61 (1H, м), 3,50-3,33 (4Н, м), 3,12-3,01 (14Н, м), 2,89 (2Н, т, J=5,4 Гц), 2,03-1,96 (2Н, м), 1,25 (18Н, т, J=7,3 Гц), 0,99 (9Н, с), 0,74 (9Н, с), 0,27 (6Н, с), 0,18 (3H, с), -0,08 (3H, с).
[0460]
(Этап 8-1)
Динатрия (5R,7R,8R,12aR,14R,15R,15aS,16R)-7-{6-амино-2-[(2-аминоэтил)амино]-9Н-пурин-9-ил}-15,16-дигидрокси-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
(Диастереомер 1)
С применением соединения, полученного на этапе 7-1 (37,2 мг), проводили реакцию таким же образом, как на этапе 11 примера 1, и продукт затем очищали с помощью препаративной ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 2% - 20% (0 мин - 30 мин)] с получением указанного в заголовке соединения в виде соли триэтиламина.
[0461]
Полученную соль триэтиламина подвергали солевому обмену таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением указанного в заголовке соединения (16,5 мг).
МС (ИЭР) m/z: 788(М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,21 (1H, уш. с), 8,01 (1H, с), 7,07 (1H, с), 6,27 (1H, д, J=3,6 Гц), 6,10 (1H, д, J=8,5 Гц), 5,51-5,41 (1H, м), 5,16-5,11 (1H, м), 4,83 (1H, д, J=3,6 Гц), 4,73 (1H, т, J=4,2 Гц), 4,50-4,45 (2Н, м), 4,35-4,29 (2Н, м), 4,16-4,04 (2Н, м), 3,50-3,42 (4Н, м), 3,17-3,05 (2Н, м), 2,82-2,66 (2Н, м), 2,04-1,92 (2Н, м).
31Р-ЯМР (CD3OD) δ: 57,9 (с), 54,2 (с).
[0462]
(Этап 8-2)
Динатрия (5R,7R,8R,12aR,14R,15R,15aS,16R)-7-{6-амино-2-[(2-аминоэтил)амино]-9Н-пурин-9-ил}-15,16-дигидрокси-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
(Диастереомер 2)
С применением соединения, полученного на этапе 7-2 (23,7 мг), проводили реакцию таким же образом, как на этапе 11 примера 1, и продукт затем очищали с помощью препаративной ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 2% - 20% (0 мин - 30 мин)] с получением указанного в заголовке соединения в виде соли триэтиламина.
[0463]
Полученную соль триэтиламина подвергали солевому обмену таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением указанного в заголовке соединения (14,9 мг).
МС (ИЭР) m/z: 788(М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,27 (1Н, уш. с), 8,02 (1Н, с), 7,15 (1Н, с), 6,31 (1Н, д, J=6,0 Гц), 6,12 (1Н, д, J=8,5 Гц), 5,45-5,33 (2Н, м), 4,75 (1Н, дд, J=5,7, 4,5 Гц), 4,50 (1Н, д, J=4,2 Гц), 4,47-4,30 (4Н, м), 4,17-4,13 (1Н, уш. м), 3,94-3,89 (1Н, м), 3,69-3,59 (1Н, уш. м), 3,51-3,44 (3H, м), 3,21-3,07 (2Н, м), 2,88-2,85 (2Н, м), 2,03-1,97 (2Н, м).
31Р-ЯМР (CD3OD) δ: 62,2 (с), 59,8 (с).
[0464]
Пример 9. Синтез ЦДН9.
(5R,7R,8R,12aR,14R,15R,15aS,16R)-7-{6-амино-2-[(2-гидроксиэтил)амино]-9H-пурин-9-ил}-15,16-дигидрокси-2,10-бис(сульфанил)-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ,5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-дион
[0465]
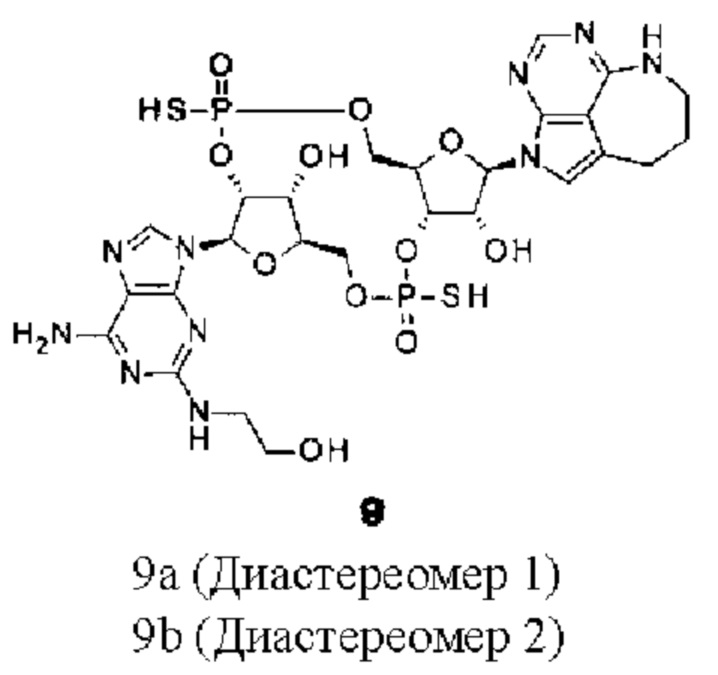
[0466]
[Схема синтеза]
[0467]
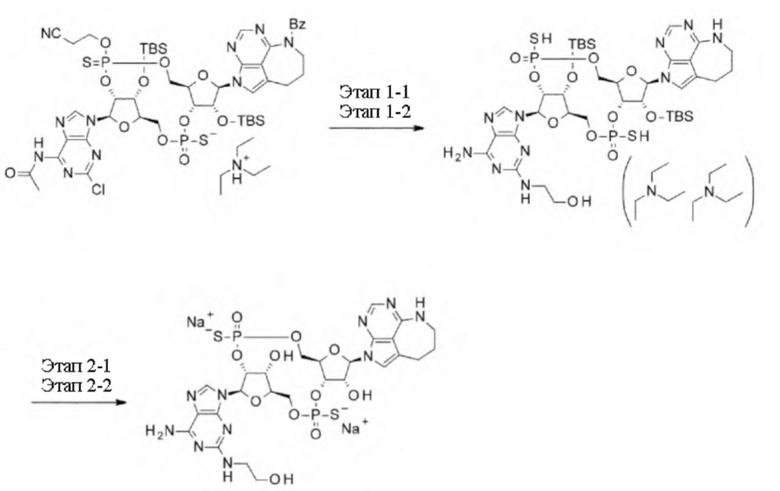
[0468]
(Этап 1-1)
(5R,7R,8R,12aR,14R,15R,15aR,16R)-7-{6-амино-2-[(2-гидроксиэтил)амино]-9H-пурин-9-ил}-15,16-бис{[трет-бутил(диметил)силил]окси}-2,10-бис(сульфанил)-14-(6,7,8,9-тетрагадро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагадро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-дион
К раствору соединения, полученного на этапе 6 примера 8 (диастереомер 1) (50,1 мг), в метаноле (1,29 мл) добавляли 2-аминоэтанол (258 мкл), и реакционную смесь перемешивали при 60°С в течение 2 часов, а затем проводили реакцию, применяя микроволновой реактор, при 120°С в течение 2 часов. Продукт очищали с помощью препаративной ВЭЖХ [10 мМ раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 20% - 60% (0 мин - 30 мин)] с получением указанного в заголовке соединения (39,4 мг) в виде смеси, содержащей соединение, полученное из этаноламина.
МС (ИЭР) m/z: 1017(М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,32 (1Н, с), 8,01 (1Н, с), 7,16 (1Н, с), 6,28 (1Н, д, J=5,4 Гц), 6,13 (1Н, д, J=9,l Гц), 5,44-5,38 (1Н, м), 5,19-5,14 (1Н, м), 4,98-4,83 (2Н, м), 4,78-4,75 (1Н, м), 4,45-4,39 (1Н, м), 4,28-4,22 (1Н, м), 4,18 (1Н, с), 4,13-4,07 (1Н, м), 4,04-3,99 (1Н, м), 3,67 (2Н, т, J=5,4 Гц), 3,51-3,42 (4Н, м), 2,86 (2Н, т, J=5,4 Гц), 2,04-1,98 (2Н, м), 0,98 (9Н, с), 0,82 (9Н, с), 0,31 (3H, с), 0,27 (3H, с), 0,22 (3H, с), 0,05 (3H, с).
[0469]
(Этап 1-2)
Бис(ад-диэтилэтанаминия) (5R,7R,8R,12aR,14R,15R,15aR,16R)-7-{6-амино-2-[(2-гидроксиэтил)амино]-9Н-пурин-9-ил}-15,16-бис{[трет-бутил(диметил)силил]окси}-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
К раствору соединения, полученного на этапе 6 примера 8 (диастереомер 2) (49,3 мг: с примесями), в метаноле (1,27 мл) добавляли 2-аминоэтанол (254 мкл), и реакционную смесь перемешивали при 60°С в течение 2 часов, а затем проводили реакцию, применяя микроволновой реактор, при 120°С в течение 3 часов. Продукт очищали с помощью препаративной ВЭЖХ [10 мМ раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 20% - 60% (0 мин - 30 мин)] с получением указанного в заголовке соединения (26,1 мг).
МС (ИЭР) m/z: 1017(М+Н)+.
1H-ЯМР (CD3OD) δ: 8,25 (1H, с), 8,01 (1Н, с), 7,10 (1H, с), 6,36 (1H, д, J=7,3 Гц), 6,17 (1H, д, J=7,9 Гц), 5,59-5,53 (1H, м), 5,41 (1H, т, J=4,5 Гц), 5,21-5,14 (1H, м), 5,02-4,95 (2Н, м), 4,70-4,61 (2Н, м), 4,18 (1H, с), 4,03 (1H, дд, J=12,l, 4,8 Гц), 3,91-3,86 (1Н, м), 3,75-3,69 (2Н, м), 3,52-3,43 (4Н, м), 3,14 (12Н, к, J=7,3 Гц), 2,93-2,91 (2Н, м), 2,04-1,99 (2Н, м), 1,28 (18Н, т, J=7,6 Гц), 0,99 (9Н, с), 0,75 (9Н, с), 0,27 (6Н, с), 0,21 (3H, с), -0,05 (3H, с).
[0470]
(Этап 2-1)
Динатрия (5R,7R,8R,12aR,14R,15R,15aS,16R)-7-{6-амино-2-[(2-гидроксиэтил)амино]-9H-пурин-9-ил}-15,16-дигидрокси-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
(Диастереомер 1)
С применением смеси, полученной на этапе 1-1 (39,4 мг), проводили реакцию таким же образом, как на этапе 11 примера 1, и затем проводили очистку при следующих [Условиях очистки] с получением указанного в заголовке соединения в виде соли триэтиламина.
[Условия очистки] препаративная ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 2% - 20% (0 мин - 30 мин)].
Полученную соль триэтиламина подвергали солевому обмену таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением указанного в заголовке соединения (16,8 мг).
МС (ИЭР) m/z: 789(М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,25 (1H, с), 8,02 (1Н, с), 7,10 (1H, с), 6,30 (1H, д, J=3,6 Гц), 6,15 (1H, д, J=8,5 Гц), 5,49-5,42 (1H, м), 5,21-5,16 (1H, м), 4,87-4,85 (1H, м), 4,77 (1H, т, J=4,2 Гц), 4,50-4,35 (3H, м), 4,31 (1H, с), 4,12-4,10 (2Н, м), 3,64 (2Н, т, J=5,4 Гц), 3,51-3,38 (4Н, м), 2,85-2,70 (2Н, м), 2,02-1,94 (2Н, м).
31Р-ЯМР (CD3OD) δ: 57,8 (с), 53,9 (с).
[0471]
(Этап 2-2)
Динатрия (5R,7R,8R,12aR,14R,15R,15aR,16R)-7-{6-амино-2-[(2-гидроксиэтил)амино]-9Н-пурин-9-ил}-15,16-дигидрокси-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
(Диастереомер 2)
С применением соединения, полученного на этапе 1-2 (26,1 мг), проводили реакцию таким же образом, как на этапе 11 примера 1, и затем проводили очистку при следующих [Условиях очистки] с получением указанного в заголовке соединения в виде соли триэтиламина.
[Условия очистки] препаративная ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 2% - 20% (0 мин - 30 мин)].
Полученную соль триэтиламина подвергали солевому обмену таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением указанного в заголовке соединения (18,6 мг).
МС (ИЭР) m/z: 789(М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,35 (1Н, с), 8,03 (1Н, с), 7,17 (1Н, с), 6,33 (1Н, д, J=6,0 Гц), 6,16 (1Н, д, J=8,5 Гц), 5,49-5,41 (2Н, м), 4,80 (1Н, т, J=5,4 Гц), 4,51-4,26 (5Н, м), 4,07 (1Н, д, J=12,7 Гц), 3,94-3,89 (1Н, м), 3,67 (2Н, 5, J=5,7 Гц), 3,53-3,39 (4Н, м), 2,89 (2Н, т, J=5,4 Гц), 2,03-1,99 (2Н, м).
31Р-ЯМР (CD3OD) δ: 62,8 (с), 60,3 (с).
[0472]
Пример 10. Синтез ЦДН10.
(5R,7R,8R,12aR,14R,15R,15aS,16R)-7-{6-амино-2-[(2-амино-2-метилпропил)амино]-9H-пурин-9-ил}-15,16-дигадрокси-2,10-бис(сульфанил)-14-(6,7,8,9-тетрагадро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагадро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-дион
[0473]
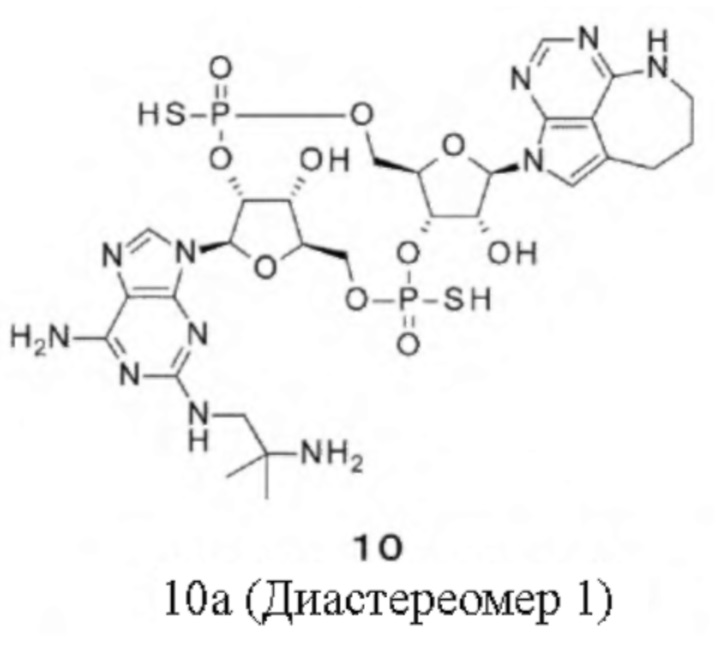
[0474]
[Схема синтеза]
[0475]
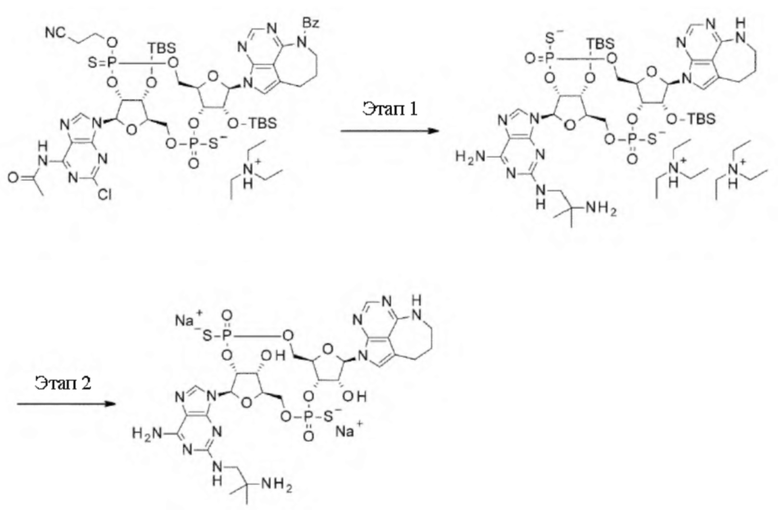
[0476]
(Этап 1)
Бис(N,N-диэтилэтанаминия) (5R,7R,8R,12aR,14R,15R,15aR,16R)-7-{6-амино-2-[(2-амино-2-метилпропил)амино]-9Н-пурин-9-ил}-15,16-бис{[трет-бутал(диметил)силил]окси}-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
К раствору соединения, полученного на этапе 6 примера 8 (диастереомер 1) (41,0 мг), в метаноле (1,10 мл) добавляли 1,2-диамино-2-метилпропан (210 мкл), и реакционную смесь перемешивали при 60°С в течение 2 часов, а затем проводили реакцию, применяя микроволновой реактор, при 120°С в течение 6 часов. Продукт просто очищали с помощью препаративной ВЭЖХ [10 мМ раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 20% - 60% (0 мин - 30 мин)] с получением указанного в заголовке соединения (16,3 мг: с примесями). Полученное соединение непосредственно использовали для следующей реакции без дополнительной очистки.
МС (ИЭР) m/z: 1044(М+Н)+.
[0477]
(Этап 2)
Динатрия (5R,7R,8R,12aR,14R,15R,15aS,16R)-7-{6-амино-2-[(2-амино-2-метилпропил)амино]-9Н-пурин-9-ил}-15,16-дигидрокси-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
С применением соединения, полученного на этапе 1 (16,3 мг: с примесями), проводили реакцию таким же образом, как на этапе 11 примера 1, и затем проводили очистку при следующих [Условиях очистки] с получением указанного в заголовке соединения в виде соли триэтиламина.
[Условия очистки] препаративная ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 5% - 25% (0 мин - 30 мин)].
Полученную соль триэтиламина подвергали солевому обмену таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением указанного в заголовке соединения (6,4 мг).
МС (ИЭР) m/z: 816(М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,28 (1H, уш. с), 8,01 (1H, с), 7,05 (1H, с), 6,27 (1H, д, J=4,2 Гц), 6,15 (1H, д, J=7,9 Гц), 5,41-5,27 (1H, м), 5,13-5,08 (1H, м), 4,84 (1H, д, J=3,6 Гц), 4,73 (1H, т, J=4,5 Гц), 4,50-4,44 (2Н, м), 4,36-4,31 (2Н, м), 4,16-4,00 (2Н, м), 3,49 (2Н, дд, J=6,3, 3,3 Гц), 3,31-3,25 (2Н, м), 2,84-2,70 (2Н, м), 2,03-1,91 (2Н, м), 1,34 (3H, с), 1,30 (3H, с).
31Р-ЯМР (CD3OD) δ: 57,9 (с), 54,5 (с).
[0478]
Пример 11. Синтез ЦДН11.
(5R,7R,8R,12aR,14R,15R,15aS,16R)-7-[6-амино-2-(аминометил)-9H-пурин-9-ил]-15,16-дигидрокси-2,10-бис(сульфанил)-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-дион
[0479]
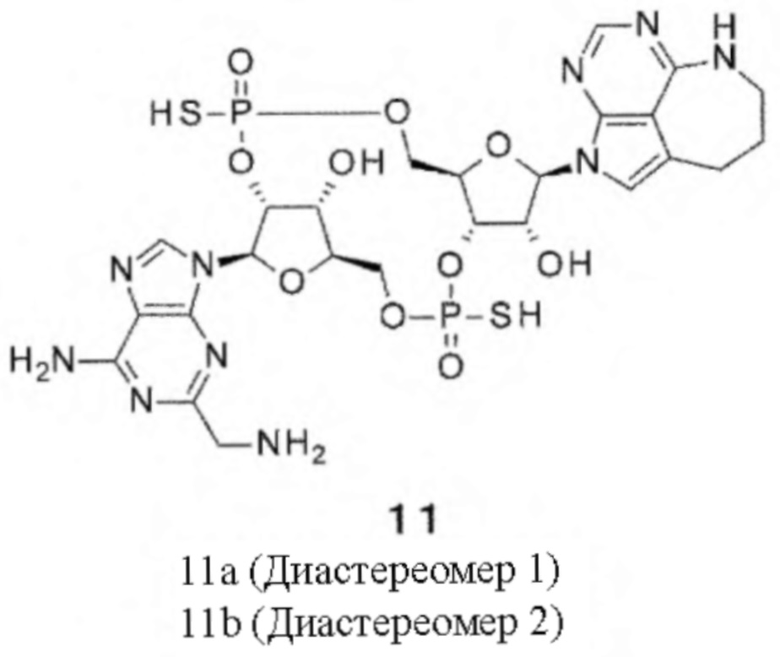
[0480]
[Схема синтеза]
[0481]
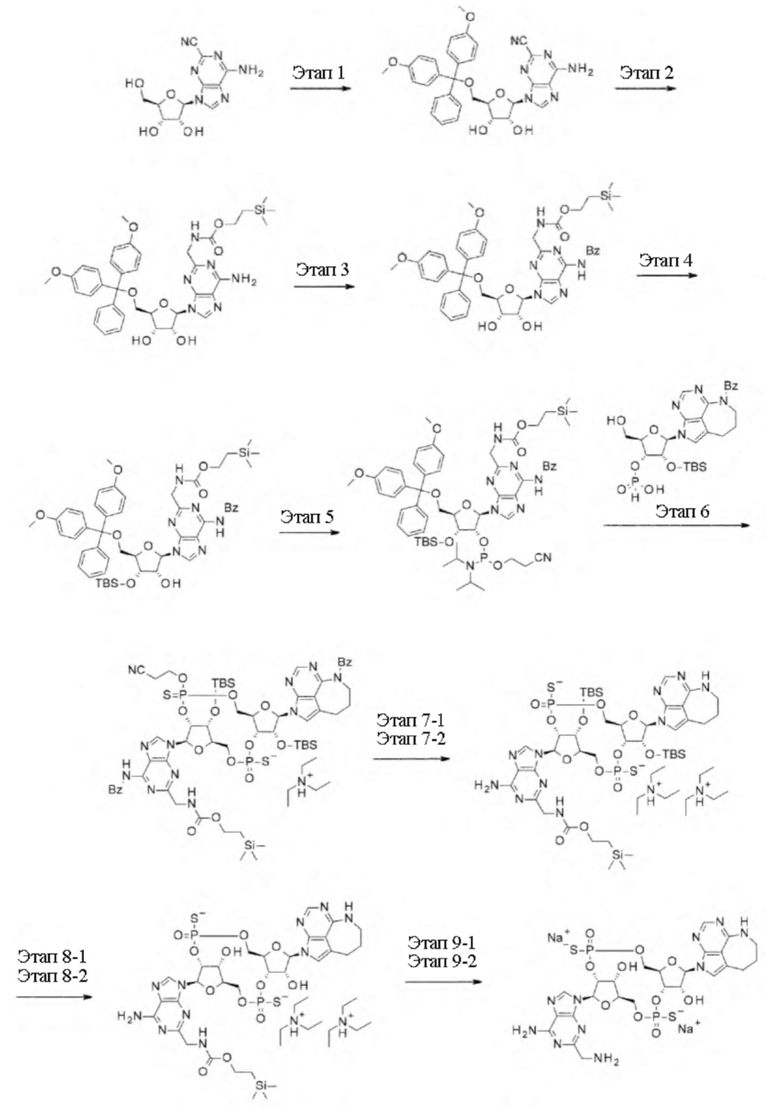
[0482] (Этап 1)
5'-O-[бис(4-метоксифенил)(фенил)метил]-2-цианоаденозин.
К раствору 2-цианоаденозина (440 мг) как соединения, известного в литературе (J. Am. Chem. Soc. 1989, 111, 8502-8504), в пиридине (8,00 мл) добавляли 4,4'-диметокситритилхлорид (642 мг), и реакционную смесь перемешивали в атмосфере азота при комнатной температуре в течение 4 часов. После добавления метанола (10 мл) в реакционную смесь, чтобы погасить реакцию, продукт концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле [гексан/этилацетат/метанол/0,1% триэтиламин] с получением указанного в заголовке соединения (528 мг).
1H-ЯМР (CDCl3) δ: 8,21 (1H, с), 7,31-7,17 (9Н, м), 6,79-6,70 (4Н, м), 5,99 (1H, д, J=5,4 Гц), 5,86 (2Н, уш. с), 4,86 (1Н, к, J=4,6 Гц), 4,65 (1H, т, J=3,6 Гц), 4,48-4,45 (1H, м), 4,41 (1H, к, J=3,0 Гц), 3,79 (6Н, с), 3,46 (1H, дд, J=10,9, 3,6 Гц), 3,34 (1H, дд, J=10,6, 3,3 Гц), 2,93 (1H, д, J=2,4 Гц).
[0483]
(Этап 2)
5'-O-[бис(4-метоксифенил)(фенил)метил]-2-[({[2-(триметилсилил)этокси]карбонил}амино)метил]аденозин
К раствору соединения, полученного на этапе 1 (14,3 г), в тетрагидрофуране (500 мл) добавляли раствор в тетрагидрофуране алюмогидрида лития (приблизительно 2,5 М, 29,0 мл) и реакционную смесь перемешивали в атмосфере азота при 40°С в течение 2 часов. Реакционную смесь охлаждали до температуры льда, добавляли в нее насыщенный водный раствор гидрокарбоната натрия (450 мл), и реакционную смесь перемешивали в течение 10 минут, а затем добавляли туда 1-({[2-(триметилсилил)этокси]карбонил}окси)пирролидин-2,5-дион (25,0 г), чтобы реакция прошла при комнатной температуре в течение 18 часов. Добавляли туда насыщенный водный раствор соли Рошеля, и реакционную смесь перемешивали в течение 2,5 часов, а затем осуществляли экстрагирование смесью дихлорметан/метанол. После высушивания органического слоя над безводным сульфатом натрия осушающий агент удаляли путем фильтрации и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле [гексан/этилацетат/0,1% триэтиламин] с получением указанного в заголовке соединения (10,8 г).
1H-ЯМР (CDCl3) δ: 8,01 (1H, с), 7,26-7,15 (9Н, м), 6,75-6,71 (4Н, м), 6,37 (1H, уш. с), 5,93 (1H, д, J=6,0 Гц), 5,67 (2Н, уш. с), 5,59 (1H, уш. с), 4,77-4,74 (1H, м), 4,46-4,37 (4Н, м), 4,21 (2Н, т, J=8,5 Гц), 3,76 (3H, с), 3,76 (3H, с), 3,42 (1H, дд, J=10,6, 3,3 Гц), 3,25 (1H, дд, J=10,6, 3,3 Гц), 3,16 (1H, уш. с), 1,03 (2Н, т, J=8,5 Гц), 0,05 (9Н, с).
[0484]
(Этап 3)
N-бензоил-5'-O-[бис(4-метоксифенил)(фенил)метил]-2-[({[2-(триметилсилил)этокси]карбонил}амино)метил]аденозин
К раствору соединения, полученного на этапе 2 (10,8 г), в пиридине (70,0 мл) добавляли хлортриметилсилан (15,0 мл), и реакционную смесь перемешивали в атмосфере азота при комнатной температуре в течение 2 часов. Бензоилхлорид (8,44 мл) добавляли в реакционную смесь и дополнительно перемешивали ее в течение 2 часов. Реакционную смесь охлаждали до 0°С и перемешивали в течение 10 минут с добавлением воды (21,0 мл), а затем дополнительно перемешивали при той же температуре в течение 20 минут с добавлением 28% водного аммиака (31,4 мл). Температуру повышали до комнатной температуры и реакционную смесь дополнительно перемешивали в течение 3 часов, а затем концентрировали при пониженном давлении. Остаток суспендировали в этилацетате, и твердый продукт удаляли путем фильтрации. Фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле [гексан/этилацетат/метанол/0,1% триэтиламин] с получением указанного в заголовке соединения (9,47 г).
МС (ИЭР) m/z: 847(М+Н)+.
1H-ЯМР (CDCl3) δ: 9,54 (1H, уш. с), 8,17 (2Н, д, J=6,7 Гц), 7,91 (1Н, уш. с), 7,66-7,52 (3H, м), 7,35-7,10 (9Н, м), 6,75 (4Н, д, J=8,5 Гц), 6,45 (1Н, уш. с), 6,23 (1Н, уш. с), 6,03 (1Н, д, J=6,7 Гц), 4,70-4,65 (2Н, м), 4,45-4,19 (5Н, м), 3,73 (6Н, с), 3,38-3,32 (2Н, м), 2,65 (1H, уш. с), 1,05 (2Н, т, J=8,8 Гц), 0,00 (9Н, с).
[0485]
(Этап 4)
N-бензоил-5'-O-[бис(4-метоксифенил)(фенил)метил]-3'-O-[трет-бутил(диметил)силил]-2-[({[2-(триметилсилил)этокси]карбонил}амино)метил]аденозин
С применением соединения, полученного на этапе 3 (9,47 г), проводили реакцию таким же образом, как на этапе 4 примера 8, с получением указанного в заголовке соединения (3,13 г).
МС (ИЭР)m/z: 961(М+Н)+.
1H-ЯМР (CDCl3) δ: 8,87 (1H, уш. с), 8,24 (1H, уш. с), 8,02 (2Н, д, J=7,3 Гц), 7,64-7,51 (3H, м), 7,40-7,18 (9Н, м), 6,81-6,77 (4Н, м), 6,08 (1Н, д, J=4,8 Гц), 5,85 (1H, уш. с), 4,70-4,52 (4Н, м), 4,23-4,17 (3H, м), 3,77 (6Н, с), 3,50 (1H, дд, J=10,9, 3,0 Гц), 3,29 (1H, дд, J=10,9, 4,2 Гц), 3,21 (1H, д, J=6,0 Гц), 1,06-1,02 (2Н, м), 0,89 (9Н, с), 0,09 (3H, с), 0,05 (9Н, с), 0,01 (3H, с).
[0486]
(Этап 5)
N-бензоил-5'-O-[бис(4-метоксифенил)(фенил)метил]-3'-O-[трет-бутил(диметил)силил]-2'-O-{(2-цианоэтокси)[ди(пропан-2-ил)амино]фосфанил}-2-[({[2-(триметилсилил)этокси]карбонил}амино)метил]аденозин
К раствору соединения, полученного на этапе 4 (1,49 г), в дихлорметане (15,5 мл) добавляли N,N-диизопропилэтиламин (1,58 мл) и 2-цианоэтил-N,N-диизопропилхлорфосфорамидит (1,04 мл), и реакционную смесь перемешивали в атмосфере азота при комнатной температуре в течение 2 часов. Реакционную смесь концентрировали при пониженном давлении, и остаток очищали с помощью колоночной хроматографии на силикагеле [гексан/этилацетат/0,1% триэтиламин] и хроматографии на колонке с силикагелем С18 [ацетонитрил: 100%] с получением указанного в заголовке соединения (1,39 г) в виде смеси диастереомеров у атома фосфора (соотношение диастереомеров =6:4).
1Н-ЯМР (CDCl3) δ 8,84 (1H, с), 8,33 (0,6Н, с), 8,28 (0,4Н, с), 8,02-7,99 (2Н, м), 7,64-7,59 (1H, м), 7,55-7,51 (2Н, м), 7,42-7,20 (9Н, м), 6,82-6,79 (4Н, м), 6,30 (0,4Н, д, J=4,2 Гц), 6,25 (0,6Н, д, J=4,2 Гц), 5,95-5,88 (1H, м), 4,89-4,77 (1H, м), 4,60-4,58 (2Н, м), 4,51-4,45 (1H, м), 4,25-4,18 (3H, м), 3,86-3,46 (5Н, м), 3,78 (6Н, с), 3,35-3,29 (1H, м), 2,53 (1,2Н, т, J=6,3 Гц), 2,38 (0,8Н, т, J=6,3 Гц), 1,16-0,98 (14Н, м), 0,85 (3,6Н, с), 0,84 (5,4Н, с), 0,10 (1,8Н, с), 0,08 (1,2Н, с), 0,05 (9Н, с), 0,01 (1,2Н, с). -0,01 (1,8Н, с).
[0487]
(Этап 6)
N,N-диэтилэтанаминия (5R,7R,8R,12aR,14R,15R,15aR,16R)-7-{6-бензамидо-2-[({[2-(триметилсилил)этокси]карбонил}амино)метил]-9Н-пурин-9-ил}-14-(6-бензоил-6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)-15,16-бис{[трет-бутил(диметил)силил]окси}-10-(2-цианоэтокси)-2-оксо-10-сульфанилиденоктагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2-тиолят
Ту же реакцию, что и на этапе 7 примера 1, проводили в следующем масштабе (сырьевой материал: 1,94 г). С применением раствора в ацетонитриле полученного соединения и соединения, полученного на этапе 5 (2,19 г), проводили реакцию таким же образом, как на этапе 8 примера 1 и этапе 9 примера 1, с получением диастереомера 1 (138 мг) и диастереомера 2 (82,8 мг) указанного в заголовке соединения.
Диастереомер 1 (менее полярный)
1H-ЯМР (CD3OD) δ: 9,08 (1H, с), 8,11 (2Н, д, J=7,3 Гц), 7,98 (1Н, с), 7,67 (1H, т, J=7,6 Гц), 7,57 (2Н, т, J=7,9 Гц), 7,37 (2Н, д, J=7,9 Гц), 7,28-7,22 (4Н, м), 6,54 (1H, д, J=8,5 Гц), 6,36 (1H, д, J=l,8 Гц), 5,66-5,59 (1H, м), 5,07-5,02 (1Н, м), 4,85-4,83 (1H, м), 4,72 (1H, д, J=3,6 Гц), 4,58-4,08 (12Н, м), 3,88-3,78 (1Н, м), 3,49-3,38 (1H, м), 3,21 (6Н, к, J=7,3 Гц), 3,05-3,00 (2Н, м), 2,49-2,40 (1H, м), 2,34-2,26 (1H, м), 2,09-2,03 (2Н, м), 1,31 (9Н, т, J=7,6 Гц), 1,21-1,09 (2Н, м), 1.02 (9Н, с), 0,91 (9Н, с), 0,30 (3H, с), 0,29 (3H, с), 0,26 (3H, с), 0,12 (3H, с), 0,05 (9Н, с).
Диастереомер 2 (более полярный)
MC(ИЭР)m/z: 1392(М+Н)+.
1H-ЯМР (CD3OD) δ: 8,91 (1H, с), 8,10-8,07 (2Н, м), 7,94 (1Н, с), 7,67-7,63 (1Н, м), 7,59-7,54 (2Н, м), 7,41-7,20 (6Н, м), 6,54 (1Н, д, J=8,5 Гц), 6,22 (1Н, д, J=5,4 Гц), 5,36-5,30 (1H, м), 3,20 (6Н, к, J=7,3 Гц), 3,04-3,00 (2Н, м), 2,85-2,75 (2Н, м), 2,23-2,13 (2Н, м), 1,30 (9Н, т, J=7,3 Гц), 1.03 (9Н, с), 0,79 (9Н, с), 0,05 (9Н, с) (показаны только наблюдаемые пики).
[0488]
(Этап 7-1)
Бис(N,N-диэтилэтанаминия) (5R,7R,8R,12aR,14R,15R,15aR,16R)-7-{6-амино-2-[({[2-(триметилсилил)этокси]карбонил}амино)метил]-9Н-пурин-9-ил}-15,16-бис{[трет-бутил(диметил)силил]окси}-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
С применением соединения, полученного на этапе 6 (диастереомер 1) (42,3 мг), проводили реакцию таким же образом, как на этапе 10 примера 1, с получением указанного в заголовке соединения (25,9 мг).
МС (ИЭР) m/z: 1131(М+Н)+.
1H-ЯМР (CD3OD) δ: 8,72 (1H, с), 8,00 (1Н, с), 7,26 (1H, с), 6,35 (1H, д, J=9,l Гц), 6,26 (1H, д, J=4,8 Гц), 5,42-5,36 (1H, м), 5,20-5,15 (1H, м), 4,91-4,87 (2Н, м), 4,80-4,78 (1H, м), 4,43 (1H, т, J=11,2 Гц), 4,36-4,28 (3H, м), 4,20-4,15 (3H, м), 4,09-3,99 (2Н, м), 3,51 (2Н, д, J=6,7 Гц), 3,13 (12Н, к, J=7,3 Гц), 2,85 (2Н, уш. с), 2,01-1,97 (2Н, м), 1,25 (18Н, т, J=7,3 Гц), 1,07-1,00 (2Н, м), 1,00 (9Н, с), 0,82 (9Н, с), 0,32 (3H, с), 0,28 (3H, с), 0,25 (3H, с), 0,07 (12Н, с).
[0489]
(Этап 7-2)
Бис(N,N-диэтилэтанаминия) (5R,7R, 8R,12aR,14R,15R,15aR,16R)-7-{6-амино-2-[({[2-(триметилсилил)этокси]карбонил}амино)метил]-9Н-пурин-9-ил}-15,16-бис{[трет-бутил(диметил)силил]окси}-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
С применением соединения, полученного на этапе 6 (диастереомер 2) (82,8 мг), проводили реакцию таким же образом, как на этапе 10 примера 1, с получением указанного в заголовке соединения (45,2 мг).
1Н-ЯМР (CD3OD) δ: 8,61 (1H, с), 8,01 (1H, с), 7,08 (1H, с), 6,34 (2Н, т, J=7,9 Гц), 5,49 (1H, дд, J=10,6, 4,5 Гц), 5,42 (1Н, т, J=5,l Гц), 5,24-5,17 (1Н, м), 5,00-4,95 (2Н, м), 4,69-4,57 (2Н, м), 4,36 (2Н, т, J=17,8 Гц), 4,22-4,15 (3H, м), 4,05 (1H, дд, =12,4, 5,1 Гц), 3,90-3,85 (1Н, м), 3,51 (2Н, д, J=9,1 Гц), 3,17 (12Н, к, J=7,3 Гц), 2,92 (2Н, т, J=5,4 Гц), 2,04-1,99 (2Н, м), 1,29 (18Н, т, J=7,3 Гц), 1,07-0,98 (2Н, м), 1,00 (9Н, с), 0,74 (9Н, с), 0,28 (3H, с), 0,28 (3H, с), 0,21 (3H, с), 0,07 (9Н, с), -0,06 (3H, с).
[0490]
(Этап 8-1)
Бис(N,N-диэтилэтанаминия) (5R,7R,8R,12aR,14R,15R,15aS,16R)-7-{6-амино-2-[({[2-(триметилсилил)этокси]карбонил}амино)метил]-9Н-пурин-9-ил}-15,16-дигидрокси-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
Тригидрофторид триэтиламина (700 мкл) добавляли к соединению (25,9 мг), полученному на этапе 7-1, и реакционную смесь перемешивали при 55°С в течение 2 часов. Охлажденную до температуры льда смесь 1 М водного раствора карбоната триэтиламмония (3,5 мл) и триэтиламина (1,10 мл) добавляли в реакционную смесь, которую очищали с помощью препаративной ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 10% - 40% (0 мин - 30 мин)] с получением указанного в заголовке соединения (19,1 мг).
МС (ИЭР) m/z: 903(М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,71 (1H, с), 8,03 (1Н, с), 7,10 (1H, с), 6,37 (1H, д, J=7,9 Гц), 6,28 (1H, д, J=4,2 Гц), 5,38-5,33 (1H, м), 5,18-5,13 (1Н, м), 4,84-4,80 (2Н, м), 4,50-4,40 (2Н, м), 4,35-4,40 (4Н, м), 4,21-4,16 (2Н, м), 4,07-4,00 (2Н, м), 3,51-3,49 (2Н, м), 3,07 (12Н, к, J=7,3 Гц), 2,85 (2Н, т, J=5,4 Гц), 2,02-1,97 (2Н, м), 1,23 (18Н, т, J=7,3 Гц), 1,04 (2Н, т, J=8,2 Гц), 0,07 (9Н, с).
[0491]
(Этап 8-2)
Бис(N,N-диэтилэтанаминия) (5R,7R,8R,12aR,14R,15R,15aS,16R)-7-{6-амино-2-[({[2-(триметилсилил)этокси]карбонил}амино)метил]-9Н-пурин-9-ил}-15,16-дигидрокси-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
С применением соединения, полученного на этапе 7-2 (45,2 мг), проводили реакцию таким же образом, как на этапе 8-1, с получением указанного в заголовке соединения (37,1 мг).
МС (ИЭР) m/z: 903(М+Н)+.
1H-ЯМР (CD3OD) δ: 8,75 (1H, с), 8,02 (1Н, с), 7,13 (1H, с), 6,36 (1H, д, J=9,l Гц), 6,33 (1H, д, J=6,7 Гц), 5,51-5,42 (2Н, м), 4,81 (1H, дд, J=6,7, 4,8 Гц), 4,51-4,28 (7Н, м), 4,18 (2Н, дт, J=8,3, 2,6 Гц), 4,02 (1H, д, J=12,7 Гц), 3,92-3,87 (1H, м), 3,51-3,47 (2Н, м), 3,13 (12Н, к, J=7,3 Гц), 2,93-2,90 (2Н, м), 2,04-1,98 (2Н, м), 1,27 (18Н, т, J=7,3 Гц), 1,06-0,99 (2Н, м), 0,06 (9Н, с).
[0492]
(Этап 9-1)
Динатрия (5R,7R,8R,12aR,14R,15R,15aS,16R)-7-[6-амино-2-(амино[({[2триметилсилил)этокси]карбомил]амино)метил]-9H-пурин-9-ил}-15,16-дигидрокси-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
(Диастереомер 1)
К раствору соединения, полученного на этапе 8-1 (19,1 мг), в тетрагидрофуране (576 мкл) добавляли раствор в тетрагидрофуране фторида тетрабутиламмония (приблизительно 1 М, 288 мкл) и реакционную смесь перемешивали в атмосфере азота при комнатной температуре в течение ночи, и затем проводили очистку при следующих [Условиях очистки] с получением указанного в заголовке соединения в виде соли триэтиламина. [Условия очистки] препаративная ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 0%-30% (0 мин - 40 мин)] и Sep-Pak (R) С18 [вода/ацетонитрил].
Полученную соль триэтиламина подвергали солевому обмену таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением указанного в заголовке соединения (8,4 мг).
МС (ИЭР) m/z: 759(М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,48 (1H, с), 8,03 (1Н, с), 7,04 (1H, с), 6,27 (1H, д, J=3,6 Гц), 6,24 (1H, д, J=8,5 Гц), 5,98-5,93 (1H, м), 5,04-4,99 (1Н, м), 4,81-4,79 (2Н, м), 4,45-4,39 (2Н, м), 4,31-4,27 (2Н, м), 4,12-3,99 (4Н, м), 3,54-3,44 (2Н, м), 2,88-2,85 (2Н, м), 2,02-1,97 (2Н, м).
31Р-ЯМР (CD3OD) δ: 57,6, 55,5.
[0493]
(Этап 9-2)
Динатрия (5R,7R,8R,12aR,14R,15R,15aS,16R)-7-[6-амино-2-(аминометил)-9Н-пурин-9-ил]-15,16-дигидрокси-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
(Диастереомер 2)
С применением соединения, полученного на этапе 8-2 (37,1 мг), проводили реакцию таким же образом, как на этапе 9-1, с получением указанного в заголовке соединения (12,8 мг).
МС (ИЭР) m/z: 759(М+Н)+.
1H-ЯМР (CD3OD)δ: 8,61 (1H, с), 8,02 (1Н, с), 7,13 (1H, с), 6,31 (1H, д, J=6,0 Гц), 6,28 (1H, д, J=8,5 Гц), 5,61-5,55 (1H, м), 5,38-5,35 (1H, м), 4,80 (1H, т, J=5,l Гц), 4,54 (1H, д, J=4,2 Гц), 4,48-4,28 (4Н, м), 4,13 (2Н, с), 4,08-4,04 (1H, м), 3,94-3,90 (1H, м), 3,52-3,49 (2Н, м), 2,90-2,88 (2Н, м), 2,03-1,98 (2Н, м).
31Р-ЯМР (CD3OD) δ: 62,2 (с), 60,0 (с).
[0494]
Пример 12. Синтез ЦДН12.
(5R,7R,8R,12aR,14R,15R,15aS,16R)-7-[6-амино-2-(гидроксиметил)-9H-пурин-9-ил]-15,16-дигидрокси-2,10-бис(сульфанил)-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-дион
[0495]
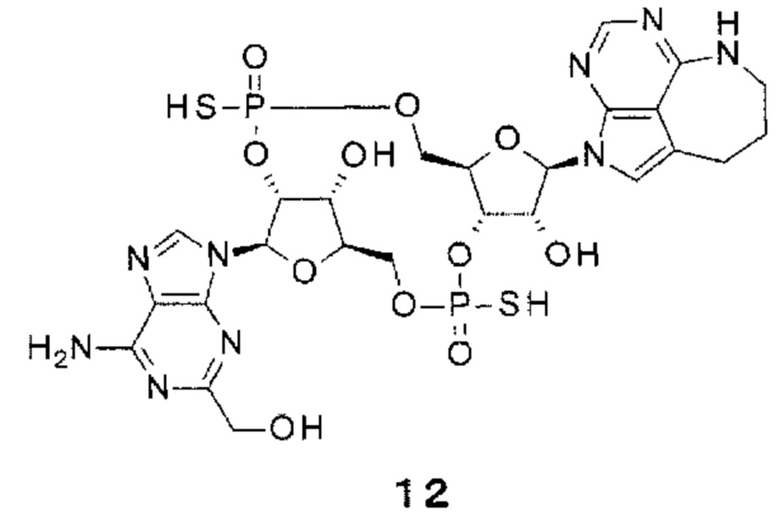
12а (Диастереомер 1)
12b (Диастереомер 2)
[0496]
[Схема синтеза]
[0497]
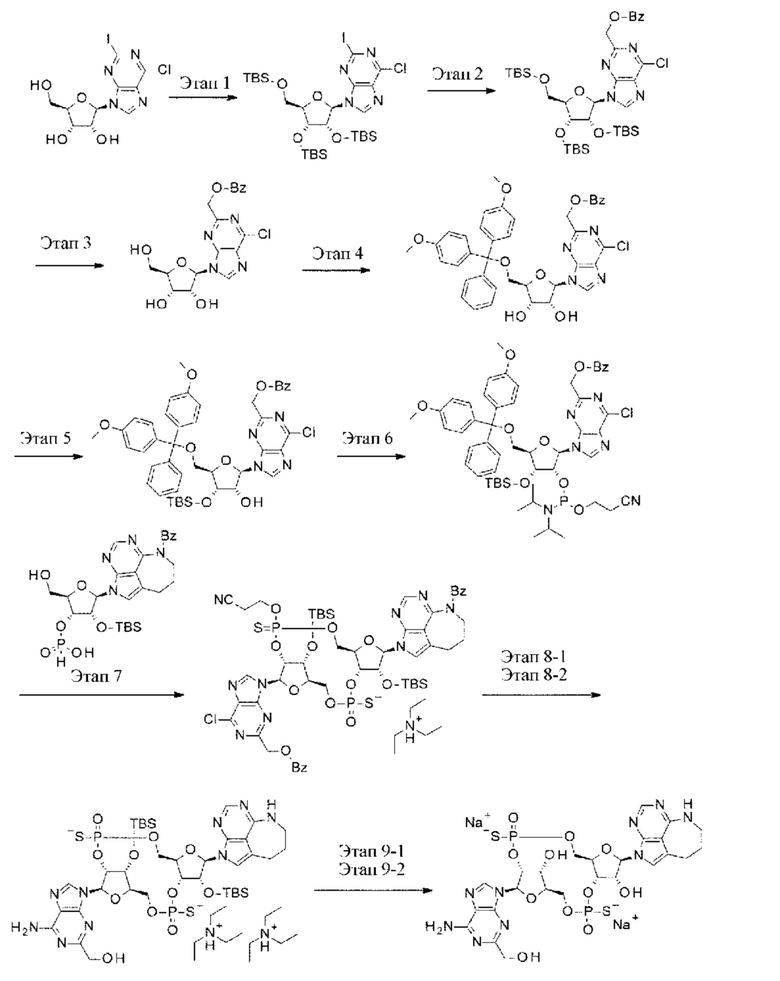
[0498] (Этап 1)
6-хлор-2-йод-9-{2,3,5-трис-O-[трет-бутил(диметил)силил]-β-D-рибофуранозил}-9Н-пурин
К раствору доступного для приобретения (Amadis Chemical Company Limited) 6-хлор-2-йод-9-β-D-рибофуранозил-9Н-пурина (9,65 г) в диметиловом эфире этиленгликоля (120 мл) добавляли N,N-диизопропилэтиламин (40,7 мл) и трет-бутилдиметилсилилтрифторметансульфонат (26,9 мл) при 0°С, и температуру повышали до комнатной температуры в атмосфере азота и реакционную смесь перемешивали в течение 19 часов. Реакционную смесь охлаждали до 0°С, и насыщенный водный раствор гидрокарбоната натрия добавляли в реакционную смесь, чтобы погасить реакцию, и реакционную смесь затем осуществляли экстрагирование дихлорметаном. После высушивания органического слоя над безводным сульфатом натрия осушающий агент удаляли путем фильтрации и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле [гексан/этилацетат] с получением указанного в заголовке соединения (13,7 г).
1H-ЯМР (CDCl3) δ: 8,48 (1H, с), 6,02 (1H, д, J=4,2 Гц), 4,54 (1H, т, J=4,5 Гц), 4,29 (1H, т, J=4,5 Гц), 4,18-4,15 (1H, м), 4,04 (1H, дд, J=11,5, 4,2 Гц), 3,80 (1H, дд, J=11,5, 2,4 Гц), 0,96 (9Н, с), 0,93 (9Н, с), 0,84 (9Н, с), 0,17 (3H, с), 0,16 (3H, с), 0,10 (3H, с), 0,09 (3H, с), 0,01 (3H, с), -0,16 (3H, с).
[0499]
(Этап 2)
2-[(бензоилокси)метил]-6-хлор-9-{2,3,5-трис-O-[трет-бутил(диметил)силил]-β-D-рибофуранозил}-9Н-пурин
К раствору соединения, полученного на этапе 1 (13,7 г), в тетрагидрофуране (121 мл) добавляли тетракис(трифенилфосфин)палладий(0) (2,10 г) и (бензилоксиметил)цинк йодид, полученный способом, описанным ниже (приблизительно 0,9 М, 30,2 мл), в атмосфере азота и реакционную смесь перемешивали при комнатной температуре в течение 20 часов. После того, как в реакционную смесь добавляли насыщенный водный раствор хлорида аммония, чтобы погасить реакцию, осуществляли экстрагирование продукта дихлорметаном. После высушивания органического слоя над безводным сульфатом натрия осушающий агент удаляли путем фильтрации и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле [гексан/этилацетат] с получением указанного в заголовке соединения (7,29 г).
[Получение йодида (бензилоксиметил)цинка]
После того, как суспензию порошка цинка (5,99 г) в тетрагидрофуране (17,1 мл) обрабатывали ультразвуком в атмосфере азота, раствор йодметилбензоата (12,0 г) в тетрагидрофуране (21,3 мл) добавляли туда при 10-15°С, и реакционную смесь перемешивали при той же температуре в течение 1,5 часов с получением раствора в тетрагидрофуране йодида (бензилоксиметил)цинка (приблизительно 0,9 М, 38,4 мл).
МС (ИЭР) m/z: 763(М+Н)+.
1Н-ЯМР (CDCl3) δ: 8,58 (1H, с), 8,13 (1H, дд, J=8,5, 1,2 Гц), 7,60-7,56 (1H, м), 7,47-7,43 (2Н, м), 6,09 (1H, д, J=4,8 Гц), 5,60 (1H, д, J=13,9 Гц), 5,56 (1H, д, J=13,9 Гц), 4,48 (1H, т, J=4,5 Гц), 4,27 (1H, т, J=4,2 Гц), 4,15-4,11 (1H, м), 4,03 (1H, дд, J=11,5, 3,0 Гц), 3,80 (1Н, дд, J=11,5, 2,4 Гц), 0,96 (9Н, с), 0,90 (9Н, с), 0,76 (9Н, с), 0,16 (3H, с), 0,15 (3H, с), 0,08 (3H, с), 0,06 (3H, с), -0,07 (3H, с), -0,27 (3H, с).
[0500]
(Этап 3)
2-[(бензоилокси)метил]-6-хлор-9-β-D-рибофуранозил-9Н-пурин
К раствору соединения, полученного на этапе 2 (7,29 г), в тетрагидрофуране (47,7 мл) добавляли раствор в тетрагидрофуране фторида тетрабутиламмония (приблизительно 1 М, 38 мл) в атмосфере азота при 0°С, и реакционную смесь перемешивали при той же температуре в течение 2,5 часов. После того, как в реакционную смесь добавляли насыщенный водный раствор хлорида аммония, чтобы погасить реакцию, осуществляли экстрагирование продукта дихлорметаном. После высушивания органического слоя над безводным сульфатом натрия осушающий агент удаляли путем фильтрации и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле [гексан/этилацетат] с получением указанного в заголовке соединения (3,69 г).
1H-ЯМР (CDCl3) δ: 8,23 (1H, с), 8,15 (2Н, дд, J=8,5, 1,2 Гц), 7,63-7,59 (1Н, м), 7,48 (2Н, т, J=7,9 Гц), 5,89 (1H, д, J=6,0 Гц), 5,61 (1H, д, J=13,9 Гц), 5,56 (1Н, д, J=14,5 Гц), 4,90 (1H, к, J=5,6 Гц), 4,46-4,43 (1H, м), 4,28 (1H, к, J=2,2 Гц), 4,02 (1H, дд, J=10,0, 2,7 Гц), 3,84-3,79 (1H, м), 3,71-3,65 (1H, м), 3,56-3,53 (1H, м), 2,70 (1Н, д, J=2,4 Гц).
[0501]
(Этап 4)
2-[(бензоилокси)метил]-9-{5-O-[бис(4-метоксифенил)(фенил)метил]-β-D-рибофуранозил}-6-хлор-9Н-пурин
К раствору соединения, полученного на этапе 3 (2,36 г), в пиридине (56 мл) добавляли 4,4'-диметокситритилхлорид (2,30 г), и реакционную смесь перемешивали в атмосфере азота при комнатной температуре в течение 17 часов. Этанол (20 мл) добавляли в реакционную смесь, которую дополнительно перемешивали в течение приблизительно 10 минут, а затем концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле [гексан/этилацетат] с получением указанного в заголовке соединения (1,41 г).
МС (ИЭР) m/z: 745(M+Na)+.
1Н-ЯМР (CDCl3) δ: 8,32 (1H, с), 8,14-8,11 (2Н, м), 7,64-7,59 (1H, м), 7,49-7,44 (2Н, м), 7,23-7,12 (9Н, м), 6,72 (4Н, д, J=7,9 Гц), 5,94 (1H, д, J=5,4 Гц), 5,64 (1H, д, J=15,1 Гц), 5,59 (1H, д, J=14,5 Гц), 4,83-4,77 (2Н, м), 4,37-4,33 (2Н, м), 3,77 (6Н, с), 3,35 (1Н, дд, J=10,6, 3,3 Гц), 3,28 (1H, дд, J=10,9, 3,6 Гц), 2,64 (1Н, с).
[0502]
(Этап 5)
2-[(бензоилокси)метил]-9-{5-O-[бис(4-метоксифенил)(фенил)метил]-3-O-[трет-бутил(диметил)силил]-β-D-рибофуранозил}-6-хлор-9Н-пурин
К раствору соединения, полученного на этапе 4 (2,61 г), в диметиловом эфире этиленгликоля (72,0 мл) добавляли N,N-диизопропилэтиламин (1,89 мл) и трет-бутилдиметилсилилтрифторметансульфонат (1,24 мл), и реакционную смесь перемешивали в атмосфере азота при комнатной температуре в течение 1,5 часов. Насыщенный водный раствор гидрокарбоната натрия добавляли в реакционную смесь, чтобы погасить реакцию, и продукт затем осуществляли экстрагирование дихлорметаном. После высушивания органического слоя над безводным сульфатом натрия осушающий агент удаляли путем фильтрации и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле [гексан/этилацетат/0,1% триэтиламин] с получением указанного в заголовке соединения (1,10 г).
1H-ЯМР (CDCl3) δ: 8,35 (1H, с), 8,12-8,10 (2Н, м), 7,60-7,56 (1H, м), 7,46-7,42 (2Н, м), 7,37-7,35 (2Н, м), 7,28-7,20 (7Н, м), 6,81-6,75 (4Н, м), 6,00 (1H, д, J=4,8 Гц), 5,50 (2Н, с), 4,69 (1H, к, J=5,6 Гц), 4,39 (1H, дд, J=5,l, 3,9 Гц), 4,16 (1H, к, J=3,8 Гц), 3,77 (6Н, с), 3,45 (1H, дд, J=10,6, 3,3 Гц), 3,31 (1H, дд, J=10,9, 4,2 Гц), 3,06 (1H, д, J=6,7 Гц), 0,86 (9Н, с), 0,04 (3H, с), -0,02 (3H, с).
[0503]
(Этап 6)
2-[(бензоилокси)метил]-9-(5-O-[бис(4-метоксифенил)(фенил)метил]-3-O-[трет-бутил(диметил)силил]-2-O-{(2-цианоэтокси)[ди(пропан-2-ил)амино]фосфанил}-β-D-рибофуранозил)-6-хлор-9Н-пурин
С применением соединения, полученного на этапе 5 (511 мг), проводили реакцию таким же образом, как на этапе 6 примера 1, с получением указанного в заголовке соединения (569 мг) в виде смеси диастереомеров у атома фосфора (соотношение диастереомеров =6:4).
1H-ЯМР (CDCl3) δ: 8,43 (0,6Н, с), 8,39 (0,4Н, с), 8,10 (2Н, д, J=7,9 Гц), 7,58 (1Н, т, J=7,6 Гц), 7,47-7,38 (4Н, м), 7,31-7,18 (7Н, м), 6,82-6,78 (4Н, м), 6,25 (0,4Н, д, J=4,8 Гц), 6,21 (0,6Н, д, J=4,8 Гц), 5,49 (1H, д, J=13,9 Гц), 5,45 (1H, д, 13,9 Гц), 4,98-4,93 (0,6Н, м), 4,83-4,78 (0,4Н, м), 4,45 (0,6Н, т, J=4,2 Гц), 4,36 (0,4Н, т, J=4,2 Гц), 4,21-4,17 (1H, м), 3,78 (6Н, с), 3,76-3,32 (6Н, м), 2,46 (1,2Н, т, J=6,3 Гц), 2,30 (0,8Н, т, J=6,3 Гц), 1,28-0,86 (12Н, м), 0,825 (5,4Н, с), 0,815 (3,6Н, с), 0,08 (1,8Н, с), 0,04 (1,2Н, с), -0,01 (1,8Н, с), -0,02 (1,2Н, с).
[0504]
(Этап 7)
N,N-диэтилэтанаминия (5R,7R,8R,12aR,14R,15R,15aR,16R)-7-{2-[(бензоилокси)метил]-6-хлор-9Н-пурин-9-ил}-14-(6-бензоил-6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)-15,16-бис{[трет-бутил(диметил)силил]окси}-10-(2-цианоэтокси)-2-оксо-10-сульфанилиденоктагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2-тиолят
Ту же реакцию, что и на этапе 7 примера 1, проводили в следующем масштабе (сырьевой материал: 2,72 г). С применением раствора в ацетонитриле полученного соединения и соединения, полученного на этапе 6 (3,03 г), проводили реакцию таким же образом, как на этапе 8 примера 1 и этапе 9 примера 1, с получением диастереомера 1 (351 мг) и диастереомера 2 (351 мг) указанного в заголовке соединения.
Диастереомер 1 (менее полярный)
МС (ИЭР) m/z: 1268(М+Н)+.
1Н-ЯМР (CD3OD) δ: 9,30 (1H, с), 8,14 (2Н, д, J=7,3 Гц), 7,99 (1H, с), 7,64 (1H, т, J=7,3 Гц), 7,52 (2Н, т, J=7,9 Гц), 7,40-7,36 (2Н, м), 7,30-7,23 (4Н, м), 6,43 (1H, д, J=8,5 Гц), 6,37 (1H, д, J=3,0 Гц), 5,62-5,56 (1H, м), 5,62 (2Н, с), 5,06-5,01 (1Н, м), 4,83 (1H, дд, J=4,5, 2,7 Гц), 4,69 (1H, д, J=4,2 Гц), 4,49-4,28 (7Н, м), 4,08 (1H, дд, J=12,l, 4,8 Гц), 3,81-3,70 (1H, м), 3,46-3,38 (1H, м), 3,17-3,10 (8Н, м), 2,29-2,23 (4Н, м), 1,28 (9Н, т, J=7,3 Гц), 0,91 (9Н, с), 0,90 (9Н, с), 0,29 (3H, с), 0,20 (3H, с), 0,16 (3H, с), 0,11 (3H, с).
Диастереомер 2 (более полярный)
МС (ИЭР) m/z: 1268(М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,97 (1H, с), 8,14 (2Н, д, J=8,5 Гц), 7,94 (1H, с), 7,68-7,63 (1H, м), 7,55-7,50 (2Н, м), 7,39-7,36 (2Н, м), 7,26-7,21 (4Н, м), 6,41 (1H, д, J=7,9 Гц), 6,21 (1H, д, J=5,4 Гц), 5,64 (1H, д, J=15,l Гц), 5,57 (1H, д, J=15,l Гц), 5,25-5,18 (2Н, м), 5,13-5,10 (1H, м), 5,04-5,01 (1H, м), 4,94-4,78 (3H, м), 4,51 (1Н, т, J=10,9 Гц), 4,33-4,06 (6Н, м), 3,15 (6Н, к, J=7,3 Гц), 3,08-2,96 (2Н, м), 2,84-2,71 (2Н, м), 2,25-2,19 (2Н, м), 1,28 (9Н, т, J=7,3 Гц), 0,91 (9Н, с), 0,79 (9Н, с), 0,19 (6Н, с), 0,14 (3H, с)-0,07 (3H, с).
[0505]
(Этап 8-1)
Бис(N,N-диэтилэтанаминия) (5R,7R,8R,12aR,14R,15R,15aR,16R)-7-[6-амино-2-(гидроксиметил)-9Н-пурин-9-ил]-15,16-бис{[трет-бутил(диметил)силил]окси}-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
К раствору соединения, полученного на этапе 7 (диастереомер 1) (37,3 мг), в метаноле (0,500 мл) добавляли 28% водный раствор аммиака (0,500 мл) и реакционную смесь перемешивали в запечатанной пробирке при 60°С в течение 3 часов. Реакционную смесь непосредственно очищали с помощью препаративной ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 35% - 55% (0 мин - 30 мин)] с получением указанного в заголовке соединения (22,0 мг: с примесями).
МС (ИЭР) m/z: 988(М+Н)+.
[0506]
(Этап 8-2)
Бис(N,N-диэтилэтанаминия) (5R,7R,8R,12aR,14R,15R,15aR,16R)-7-[6-амино-2-(гидроксиметил)-9Н-пурин-9-ил]-15,16-бис{[трет-бутил(диметил)силил]окси}-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
К раствору соединения, полученного на этапе 7 (диастереомер 2) (37,7 мг), в тетрагидрофуране (0,500 мл) добавляли 28% водный раствор аммиака (0,500 мл) и реакционную смесь перемешивали в запечатанной пробирке при 60°С в течение 3 часов. Туда дополнительно добавляли 28% водный раствор аммиака (0,500 мл), и реакционную смесь дополнительно перемешивали в течение 3 часов. Туда дополнительно добавляли 28% водный раствор аммиака (0,500 мл), и реакционную смесь дополнительно перемешивали в течение ночи. Реакционную смесь непосредственно очищали с помощью препаративной ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 20% -50% (0 мин - 30 мин)] с получением указанного в заголовке соединения (19,3 мг).
МС (ИЭР) m/z: 988(М+Н)+.
1H-ЯМР (CD3OD) δ: 8,64 (1H, с), 8,02 (1H, с), 7,09 (1H, с), 6,35 (2Н, д, J=7,9 Гц), 5,48-5,41 (2Н, м), 5,26-5,19 (1H, м), 5,00-4,95 (2Н, м), 4,70-4,53 (4Н, м), 4,22 (1H, с), 4,06 (1Н, дд, J=12,l, 4,8 Гц), 3,91-3,86 (1H, м), 3,53-3,48 (2Н, м), 3,18 (12Н, к, J=7,3 Гц), 2,92 (2Н, т, J=5,4 Гц), 2,04-1,99 (2Н, м), 1,29 (18Н, т, J=7,3 Гц), 1,00 (9Н, с), 0,74 (9Н, с), 0,28 (6Н, с), 0,21 (3H, с), -0,06 (3H, с).
[0507]
(Этап 9-1)
Динатрия (5R,7R,8R42aR,14R,15R,15aS,16R)-7-[6-амино-2-(гидроксиметил)-9H-пурин-9-ил]-15,16-дигидрокси-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
С применением соединения, полученного на этапе 8-1 (22,0 мг: с примесями), проводили реакцию таким же образом, как на этапе 11 примера 1, и затем проводили очистку при следующих [Условиях очистки] с получением указанного в заголовке соединения в виде соли триэтиламина.
[Условия очистки] препаративная ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 2%-30% (0 мин - 30 мин)].
Полученную соль триэтиламина подвергали солевому обмену таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением указанного в заголовке соединения (8,0 мг).
МС (ИЭР) m/z: 760(М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,70 (1H, с), 8,03 (1Н, с), 7,14 (1H, с), 6,39 (1H, д, J=8,5 Гц), 6,29 (1H, д, J=3,6 Гц), 5,40-5,35 (1H, м), 5,15 (1H, дт, J=9,1, 3,8 Гц), 4,84 (1H, д, J=3,6 Гц), 4,80 (1H, т, J=4,5 Гц), 4,57 (2Н, с), 4,51-4,43 (2Н, м), 4,38-4,31 (2Н, м), 4,09-4,00 (2Н, м), 3,52-3,48 (2Н, м), 2,89-2,76 (2Н, м), 2,01-1,97 (2Н, м).
31Р-ЯМР (CD3OD) δ: 58,1 (с), 54,4 (с).
[0508]
(Этап 9-2)
Динатрия (5R,7R,8R,12aR,14R,15R,15aS,16R)-7-[6-амино-2-(гидроксиметил)-9Н-пурин-9-ил]-15,16-дигидрокси-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
С применением соединения, полученного на этапе 8-2 (19,3 мг), проводили реакцию таким же образом, как на этапе 11 примера 1, и затем проводили очистку при следующих [Условиях очистки] с получением указанного в заголовке соединения в виде соли триэтиламина.
[Условия очистки] препаративная ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 2%-30% (0 мин - 30 мин)].
Полученную соль триэтиламина подвергали солевому обмену таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением указанного в заголовке соединения (6,5 мг).
МС (ИЭР) m/z: 760(М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,76 (1H, с), 8,05 (1H, с), 7,18 (1H, с), 6,38 (1H, д, J=8,5 Гц), 6,33 (1H, д,.1=6,7 Гц), 5,49-5,43 (2Н, м), 4,81 (1Н, дд, J=6,3, 4,5 Гц), 4,58 (2Н, с), 4,51-4,29 (5Н, м), 4,04 (1Н, д, J=12,l Гц), 3,93-3,88 (1H, м), 3,54-3,52 (2Н, м), 2,92 (2Н, т, J=5,4 Гц), 2,05-2,00 (2Н, м).
31Р-ЯМР (CD3OD) δ: 63,0 (с), 60,3 (с).
[0509]
Пример 13. Синтез ЦДН13.
(5R,7R,8R,12aR,14R,15R,15aS,16R)-7-{6-[(2-аминоэтил)амино]-9Н-пурин-9-ил}-15,16-дигидрокси-2,10-бис(сульфанил)-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-дион
[0510]
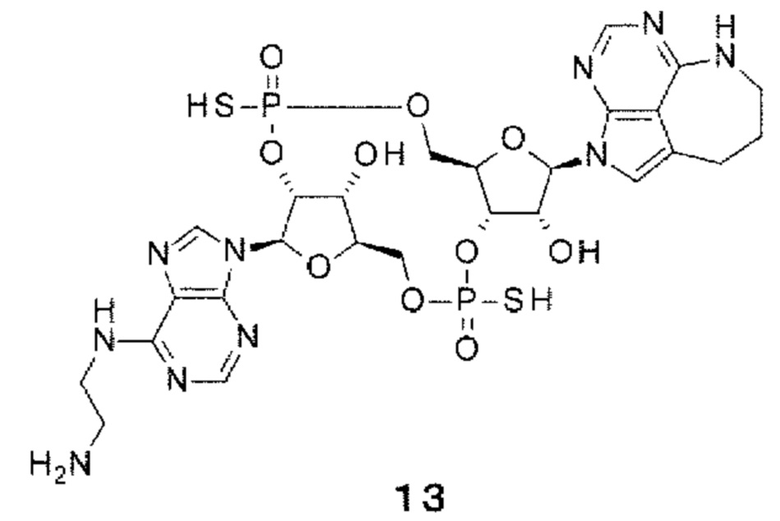
13а (Диастереомер 1)
13b (Диастереомер 2)
[0511]
[Схема синтеза]
[0512]

[0513]
(Этап 1)
9-{5-O-[бис(4-метоксифенил)(фенил)метил]-3-О-[трет-бутил(диметил)силил]-β-D-рибофуранозил}-6-хлор-9Н-пурин
С применением 9-{5-O-[бис(4-метоксифенил)(фенил)метил]-β-D-рибофуранозил}-6-хлор-9Н-пурина (15,3 г) как соединения, известного в литературе (J. Org. Chem. 2000, 65, 5104-5113), проводили реакцию таким же образом, как на этапе 3 примера 5, с получением указанного в заголовке соединения (8,44 г) и 9-{5-0-[бис(4-метоксифенил)(фенил)метил]-2-O-[трет-бутил(диметил)силил]-β-D-рибофуранозил}-6-хлор-9Н-пурина (5,77 г) в качестве региоизомера указанного в заголовке соединения.
МС (ИЭР) m/z: 703(М+Н)+.
1Н-ЯМР (CDCl3) δ: 8,71 (1H, с), 8,37 (1H, с), 7,40-7,37 (2Н, м), 7,31-7,19 (7Н, м), 6,82-6,78 (4Н, м), 6,06 (1Н, д, J=4,9 Гц), 4,79-4,74 (1H, м), 4,59 (1Н, дд, J=4,9, 3,9 Гц), 4,20 (1Н, дд, J=3,9, 1,9 Гц), 3,79 (3H, с), 3,78 (3H, с), 3,52 (1H, дд, J=10,7, 3,4 Гц), 3,29 (1Н, дд, J=10,7, 3,9 Гц), 3,08 (1H, д, J=6,8 Гц), 0,90 (9Н, с), 0,10 (3H, с), 0,03 (3H, с).
Региоизомер (2'-O-TBS-форма)
МС (ИЭР) m/z: 703(М+Н)+.
1Н-ЯМР (CDCl3) δ: 8,67 (1H, с), 8,36 (1H, с), 7,46-7,42 (2Н, м), 7,36-7,20 (7Н, м), 6,84-6,80 (4Н, м), 6,11 (1H, д, J=5,4 Гц), 5,00-4,97 (1Н, м), 4,40-4,35 (1H, м), 4,31-4,28 (1H, м), 3,79 (3H, с), 3,79 (3H, с), 3,52 (1H, дд, J=10,7, 2,9 Гц), 3,42 (1H, дд, J=10,7, 3,9 Гц), 2,68 (1H, д, J=3,9 Гц), 0,84 (9Н, с), 0,00 (3H, с), -0,16 (3H, с).
[0514]
(Этап 2)
9-(5-O-[бис(4-метоксифенил)(фенил)метил]-3-O-[трет-бутил(диметил)силил]-2-O-{(2-цианоэтокси)[ди(пропан-2-ил)амино]фосфанил}-β-D-рибофуранозил)-6-хлор-9Н-пурин
С применением соединения, полученного на этапе 1 (5,39 г), проводили реакцию таким же образом, как на этапе 4 примера 5, с получением указанного в заголовке соединения (5,78 г) в виде смеси диастереомеров у атома фосфора (соотношение диастереомеров =1:1).
МС (ИЭР) m/z: 903(М+Н)+.
1Н-ЯМР (CDCl3) δ: 8,69 (0,5Н, с), 8,67 (0,5Н, с), 8,43 (0,5Н, с), 8,41 (0,5Н, с), 7,43-7,37 (2Н, м), 7,32-7,19 (7Н, м), 6,83-6,78 (4Н, м), 6,30 (0,5Н, д, J=4,4 Гц), 6,21 (0,5Н, д, J=4,9 Гц), 5,06-5,00 (0,5Н, м), 4,86-4,80 (0,5Н, м), 4,56-4,50 (1H, м), 4,27-4,20 (1H, м), 3,79 (6Н, с), 3,75-3,62 (1H, м), 3,57-3,46 (4Н, м), 3,30 (1H, дт, J=10,7, 3,9 Гц), 2,50 (1H, т, J=6,3 Гц), 2,37 (1H, т, J=6,6 Гц), 1,13-1,06 (9Н, м), 0,90 (1,5Н, с), 0,89 (1,5Н, с), 0,86 (4,5Н, с), 0,85 (4,5Н, с), 0,12 (1,5Н, с), 0,08 (1,5Н, с), 0,02 (1,5Н, с), 0,02 (1,5Н, с).
[0515]
(Этап 3)
Ту же реакцию, что и на этапе 7 примера 1 проводили в следующем масштабе (сырьевой материал: 1,84 г). С применением раствора в ацетонитриле полученного соединения и соединения, полученного на этапе 2 (1,62 г), проводили реакцию таким же образом, как на этапе 8 примера 1. Полученный в результате этого неочищенный продукт непосредственно использовали для следующей реакции.
[0516]
(Этап 4)
3-{[(5R,7R,8R,12aR,14R,15R,15aR,16R)-14-(6-бензоил-6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)-15,16-бис{[трет-бутил(диметил)силил]окси}-7-(6-хлор-9Н-пурин-9-ил)-2-оксо-2-сульфанил-10-сульфанилиденоктагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10] пентаоксадифосфациклотетрадецин-10-ил]окси}пропаннитрил
С применением неочищенного продукта, полученного на этапе 3, проводили реакцию таким же образом, как на этапе 9 примера 1, с получением указанного в заголовке соединения (626 мг) в виде смеси диастереомеров у атома фосфора.
МС (ИЭР) m/z: 1134(М+Н)+.
[0517]
(Этап 5)
Бис(N,N-диэтилэтанаминия) (5R,7R,8R,12aR,14R,15R,15aR,16R)-7-{6-[(2-аминоэтил)амино]-9Н-пурин-9-ил}-15,16-бис{[трет-бутил(диметил)силил]окси}-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
К раствору соединения, полученного на этапе 4 (299 мг: смесь диастереомеров), в этаноле (10 мл) добавляли этилендиамин (0,352 мл) и триэтиламин (0,735 мл), и реакционную смесь перемешивали при 60°С в течение 15 часов. После концентрирования реакционной смеси при пониженном давлении, остаток очищали с помощью колоночной хроматографии на силикагеле С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил] с получением диастереомера 1 (122 мг: с примесями) и диастереомера 2(111 мг: с примесями) указанного в заголовке соединения. Диастереомер 1 (менее полярный)
МС (ИЭР) m/z: 1001(М+Н)+.
Диастереомер 2 (более полярный)
МС (ИЭР) m/z: 1001(М+Н)+.
[0518]
(Этап 6-1)
Динатрия (5R,7R,8R,12aR,14R,15R,15aS,16R)-7-{6-[(2-аминоэтил)амино]-9Н-пурин-9-ил}-15,16-дигидрокси-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
(Диастереомер 1)
С применением соединения, полученного на этапе 5 (диастереомер 1) (122 мг: с примесями), проводили реакцию таким же образом, как на этапе 11 примера 1, и затем проводили очистку при следующих [Условиях очистки] с получением указанного в заголовке соединения в виде соли триэтиламина.
[Условия очистки] Хроматография на колонке с силикагелем С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил] и препаративная ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 5%-30% (0 мин - 40 мин)].
Полученную соль триэтиламина подвергали солевому обмену таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением указанного в заголовке соединения (29,6 мг).
МС (ИЭР) m/z: 773(М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,77 (1H, с), 8,27 (1H, с), 8,03 (1Н, с), 7,10 (1H, с), 6,35 (1H, д, J=8,5 Гц), 6,27 (1H, д, J=4,8 Гц), 5,41 (1H, ддд, J=7,9, 4,2, 2,1 Гц), 5,21-5,14 (1Н, м), 4,84-4,77 (2Н, м), 4,49-4,38 (2Н, м), 4,35-4,26 (2Н, м), 4,09-3,99 (2Н, м), 3,92-3,80 (2Н, м), 3,51-3,45 (2Н, м), 3,22 (2Н, т, J=6,0 Гц), 2,89-2,81 (2Н, м), 2,02-1,94 (2Н, м).
31Р-ЯМР (CD3OD) δ: 57,8 (с), 55,0 (с).
[0519]
(Этап 6-2)
Динатрия (5R,7R,8R,12aR,14R,15R,15aS,16R)-7-{6-[(2-аминоэтил)амино]-9Н-пурин-9-ил}-15,16-дигидрокси-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
(Диастереомер 2)
С применением соединения, полученного на этапе 5 (диастереомер 2) (119 мг: с примесями), проводили реакцию таким же образом, как на этапе 11 примера 1, и затем проводили очистку при следующих [Условиях очистки] с получением указанного в заголовке соединения в виде соли триэтиламина.
[Условия очистки] Хроматография на колонке с силикагелем С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил] и препаративная ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 5%-25% (0 мин - 40 мин)].
Полученную соль триэтиламина подвергали солевому обмену таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением указанного в заголовке соединения (15,6 мг).
МС (ИЭР) m/z: 773(М+Н)+.
1H-ЯМР (CD3OD) δ: 8,83 (1H, с), 8,27 (1H, с), 8,02 (1Н, с), 7,10 (1H, с), 6,35 (1H, д, J=7,9 Гц), 6,32 (1H, д, J=6,7 Гц), 5,55-5,43 (2Н, м), 4,81 (1H, дд, J=7,0, 4,5 Гц), 4,52-4,29 (5Н, м), 4,06-4,00 (1H, м), 3,93-3,80 (3H, м), 3,52-3,47 (2Н, м), 3,21 (2Н, т, J=5,7 Гц), 2,94-2,88 (2Н, м), 2,05-1,96 (2Н, м).
31Р-ЯМР (CD3OD) δ: 63,1 (с), 60,1 (с).
[0520]
Пример 14. Синтез ЦДН14.
(5R,7R,8R,12aR,14R,15R,15aS,16R)-15,16-дигидрокси-7-{6-[(2-гидроксиэтил)амино]-9Н-пурин-9-ил}-2,10-бис(сульфанил)-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-дион
[0521]
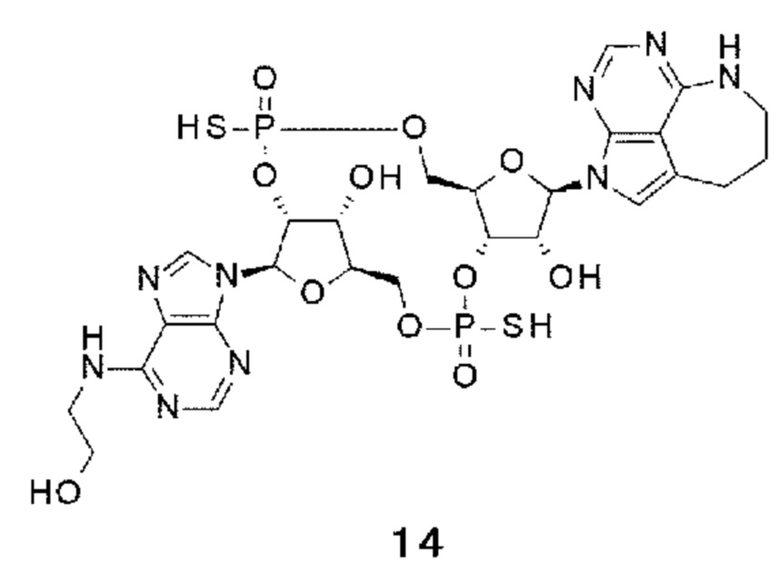
14а (Диастереомер 1)
14b (Диастереомер 2)
[0522]
[Схема синтеза]
[0523]
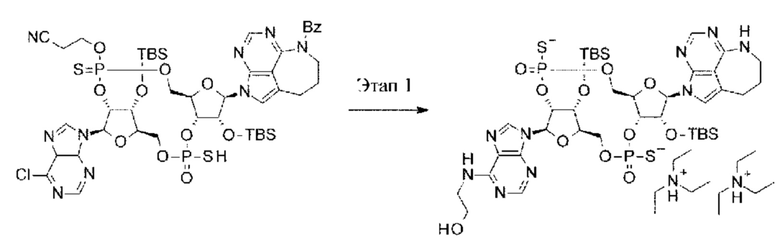
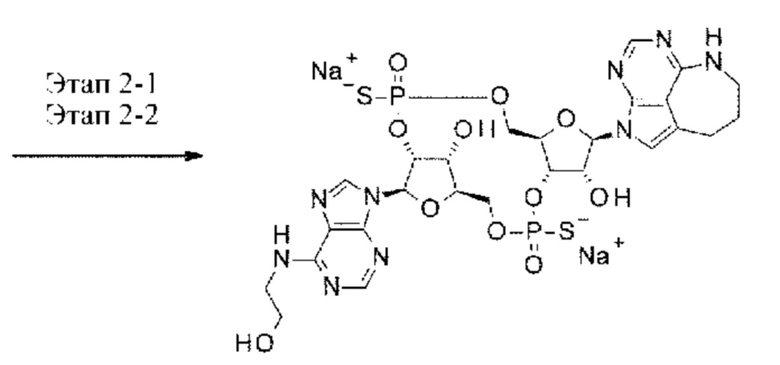
[0524]
(Этап 1)
Бис(N,N-диэтилэтанаминия) (5R,7R,8R,12aR,14R,15R,15aR,16R)-15,16-бис{[трет-бутил(диметил)силил]окси}-7-{6-[(2-гидроксиэтил)амино]-9Н-пурин-9-ил}-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
К раствору соединения, полученного на этапе 4 примера 13 (313 мг), в этаноле (10 мл) добавляли 2-аминоэтанол (0,330 мл) и триэтиламин (0,769 мл), и реакционную смесь перемешивали при 60°С в течение 15 часов. После концентрирования реакционной смеси при пониженном давлении остаток очищали с помощью колоночной хроматографии на силикагеле С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил] с получением диастереомера 1(111 мг: с примесями) и диастереомера 2 (102 мг: с примесями) указанного в заголовке соединения.
Диастереомер 1 (менее полярный)
МС (ИЭР) m/z: 1002(М+Н)+.
Диастереомер 2 (более полярный)
МС (ИЭР) m/z: 1002(М+Н)+.
[0525]
(Этап 2-1)
Динатрия (5R,7R,8R,12aR,14R,15R,15aS,16R)-15,16-дигидрокси-7-{6-[(2-гидроксиэтил)амино]-9Н-пурин-9-ил}-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
(Диастереомер 1)
С применением соединения, полученного на этапе 1 (диастереомер 1) (111 мг: с примесями), проводили реакцию таким же образом, как на этапе 11 примера 1, и затем проводили очистку при следующих [Условиях очистки] с получением указанного в заголовке соединения в виде соли триэтиламина.
[Условия очистки] Хроматография на колонке с силикагелем С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил] и препаративная ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 5% - 30% (0 мин - 40 мин)].
Полученную соль триэтиламина подвергали солевому обмену таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением указанного в заголовке соединения (47,1 мг).
МС (ИЭР) m/z: 774(М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,70 (1H, с), 8,22 (1H, с), 8,03 (1H, с), 7,10 (1H, с), 6,34 (1H, д, J=8,5 Гц), 6,29 (1H, д, J=4,8 Гц), 5,41-5,34 (1H, м), 5,19-5,13 (1H, м), 4,84 (1Н, д, J=4,2 Гц), 4,79 (1H, дд, J=4,8, 2,4 Гц), 4,52-4,41 (2Н, м), 4,39-4,31 (2Н, м), 4,07-3,96 (2Н, м), 3,81-3,66 (4Н, м), 3,52-3,47 (2Н, м), 2,90-2,77 (2Н, м), 2,03-1,95 (2Н, м).
31Р-ЯМР (CD3OD) δ: 57,9 (с), 54,4 (с).
[0526]
(Этап 2-2)
Динатрия (5R,7R,8R,12aR,14R,15R,15aS,16R)-15,16-дигидрокси-7-{6-[(2-гидроксиэтил)амино]-9Н-пурин-9-ил}-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
(Диастереомер 2)
С применением соединения, полученного на этапе 1 (диастереомер 2) (102 мг: с примесями), проводили реакцию таким же образом, как на этапе 11 примера 1, и затем проводили очистку при следующих [Условиях очистки] с получением указанного в заголовке соединения в виде соли триэтиламина.
[Условия очистки] Хроматография на колонке с силикагелем С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил] и препаративная ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 5%-25% (0 мин - 40 мин)].
Полученную соль триэтиламина подвергали солевому обмену таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением указанного в заголовке соединения (27,1 мг).
МС (ИЭР) m/z: 774(М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,78 (1H, с), 8,22 (1H, с), 8,02 (1H, с), 7,12 (1H, с), 6,34 (1H, д, J=1,8 Гц), 6,32 (1H, с), 5,52-5,42 (2Н, м), 4,80 (1H, дд, J=6,7, 4,8 Гц), 4,50-4,28 (5Н, м), 4,05-3,98 (1H, м), 3,93-3,86 (1H, м), 3,81-3,68 (4Н, м), 3,53-3,47 (2Н, м), 2,95-2,88 (2Н, м), 2,05-1,98 (2Н, м).
31Р-ЯМР (CD3OD) δ: 63,0 (с), 60,2 (с).
[0527]
Пример 15. Синтез ЦДН15.
N-[2-({9-[(5R,7R,8R,12aR,14R,15R,15aS,16R)-15,16-дигидрокси-2,10-диоксо-2,10-бис(сульфанил)-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-7-ил]-9Н-пурин-6-ил}амино)этил]-2-гидроксиацетамид
[0528]
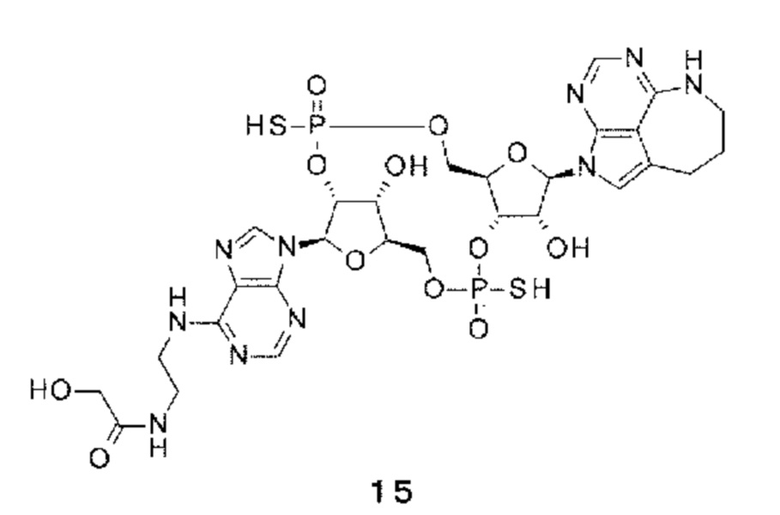
15 а (Диастереомер 1)
[0529]
[Схема синтеза]
[0530]
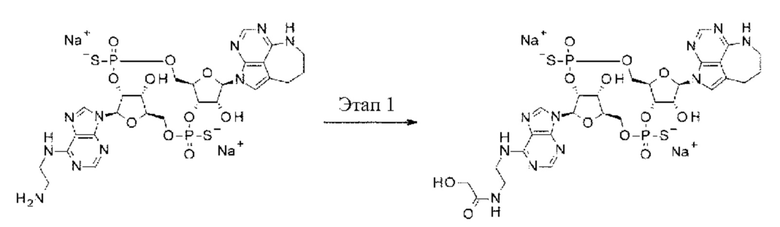
[0531]
(Этап 1)
Динатрия (5R,7R,8R,12aR,14R,15R,15aS,16R)-15,16-дигидрокси-7-(6-{[2-(2-гидроксиацетамид)этил]амино}-9Н-пурин-9-ил)-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
С применением соединения, полученного на этапе 6-2 примера 13 (10,0 мг), проводили реакцию таким же образом, как на этапе 1-1 примера 7, и затем проводили очистку при следующих [Условиях очистки] с получением указанного в заголовке соединения в виде соли триэтиламина.
[Условия очистки] Хроматография на колонке с силикагелем С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил], препаративная ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 5%-35% (0 мин - 40 мин)].
Полученную соль триэтиламина подвергали солевому обмену таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением указанного в заголовке соединения (6,6 мг).
МС (ИЭР) m/z: 831(М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,78 (1H, с), 8,24 (1H, с), 8,02 (1Н, с), 7,12 (1H, с), 6,33 (2Н, д, J=6,7 Гц), 5,52-5,42 (2Н, м), 4,80 (1H, дд, J=6,7, 4,2 Гц), 4,50-4,27 (5Н, м), 4,04-3,98 (1H, м), 3,95 (2Н, с), 3,93-3,86 (1H, м), 3,83-3,73 (2Н, м), 3,58-3,46 (4Н, м), 2,95-2,88 (2Н, м), 2,05-1,98 (2Н, м).
31Р-ЯМР (CD3OD) δ: 63,1 (с), 60,4 (с).
[0532]
Пример 16. Синтез ЦДН16.
(5R,7R,8R,12aR,14R,15R,15aS,16R)-7-{2-амино-6-[(2-гидроксиэтил)амино]-9Н-пурин-9-ил}-15,16-дигидрокси-2,10-бис(сульфанил)-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-дион
[0533]
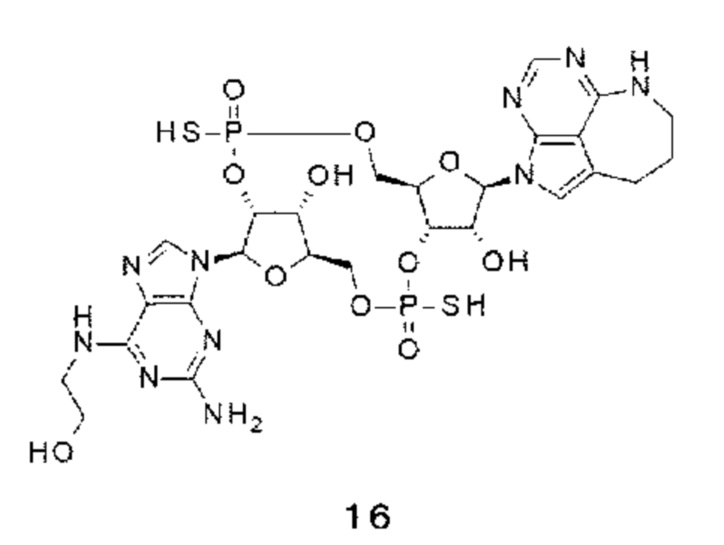
16а (Диастереомер 1)
16b (Диастереомер 2)
[0534]
[Схема синтеза]
[0535]
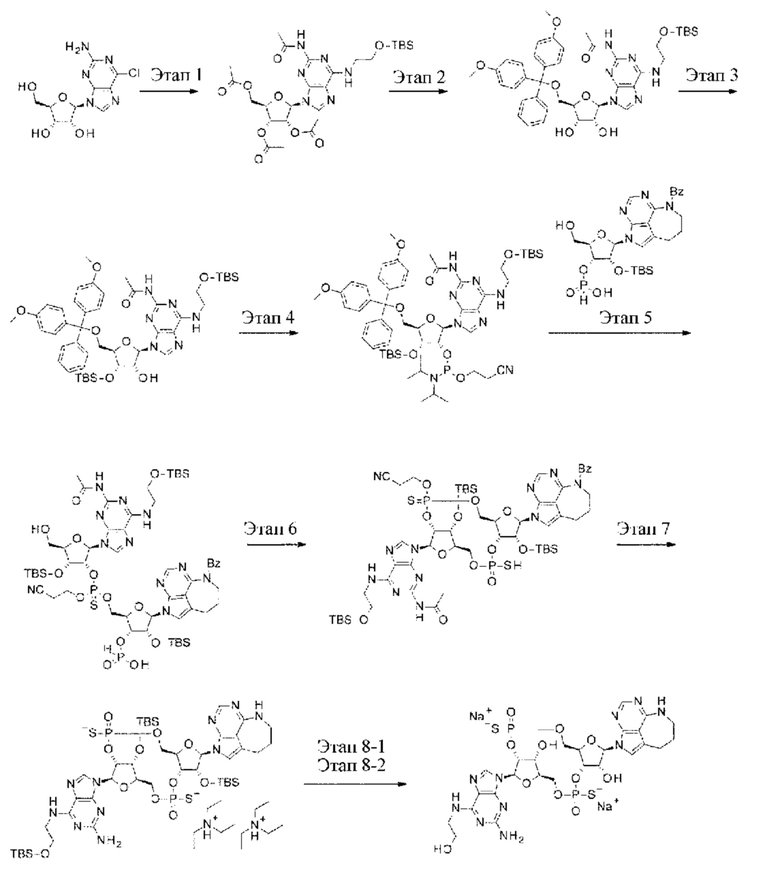
[0536]
(Этап 1)
2-ацетамидо-2',3',5'-три-O-ацетил-N-(2-{[трет-бутил(диметил)силил]окси}этил)аденозин
К раствору доступного для приобретения (Tokyo Chemical Industry Co., Ltd.) 6-хлор-9-β-D-рибофуранозил-9Н-пурин-2-амина (5,00 г) в этаноле (30 мл) добавляли 2-{[трет-бутил(диметил)силил]окси}этан-1-амин (3,49 г) и N,N-диизопропилэтиламин (4,33 мл), и реакционную смесь перемешивали при 80°С в течение 65 часов. После концентрирования реакционной смеси при пониженном давлении к остатку добавляли пиридин (15 мл) и уксусный ангидрид (15 мл), и реакционную смесь перемешивали при 70°С в течение 4 часов.
После концентрирования реакционной смеси при пониженном давлении, насыщенный водный раствор гидрокарбоната натрия добавляли в реакционную смесь, которую осуществляли экстрагирование этилацетатом. После высушивания органического слоя над безводным сульфатом натрия осушающий агент удаляли путем фильтрации и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле [гексан/этилацетат] с получением указанного в заголовке соединения (8,91 г).
МС (ИЭР) m/z: 609(М+Н)+.
1Н-ЯМР (CDCl3) δ: 7,84 (1H, с), 7,77 (1H, с), 6,14 (1H, уш. с), 6,03 (1H, д, J=4,8 Гц), 5,92 (1H, т, J=5,l Гц), 5,72 (1H, т, J=5,l Гц), 4,50-4,40 (2Н, м), 4,35 (1Н, дд, J=11,8, 4,5 Гц), 3,83 (2Н, т, J=5,l Гц), 3,76-3,63 (2Н, м), 2,54 (3H, с), 2,14 (3H, с), 2,10 (6Н, с), 0,91 (9Н, с), 0,07 (6Н, с).
[0537]
(Этап 2)
2-ацетамидо-5'-O-[бис(4-метоксифенил)(фенил)метил]-N-(2-{[трет-бутил(диметил)силил]окси}этил)аденозин
К раствору соединения, полученного на этапе 1 (4,00 г), в дихлорметане (40 мл) добавляли раствор в метаноле метоксида натрия (1,0 М, 6,64 мл), и реакционную смесь перемешивали при 0°С в течение 1 часа. После того как уксусную кислоту (0,413 мл) и пиридин (0,5 мл) добавляли в реакционную смесь, чтобы погасить реакцию, реакционную смесь концентрировали при пониженном давлении. Пиридин добавляли к остатку, и продукт затем частично концентрировали при пониженном давлении с получением раствора пиридина (приблизительно 20 мл). В данный раствор добавляли 4,4'-диметокситритилхлорид (4,68 г) при 0°С, и реакционную смесь перемешивали при той же температуре в течение 30 минут, а затем хранили при 4°С в течение ночи. Метанол (2 мл) добавляли в реакционную смесь, которую перемешивали в течение 30 минут, а затем концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле [гексан/этилацетат/метанол/0,1% триэтиламин] с получением указанного в заголовке соединения (4,41 г).
МС (ИЭР) m/z: 785(М+Н)+.
1Н-ЯМР (CDCl3) δ: 7,97 (1H, с), 7,90 (1H, с), 7,21-7,08 (9Н, м), 6,73-6,67 (4Н, м), 6,14 (1H, с), 5,88 (1H, д, J=6,7 Гц), 4,91-4,85 (1H, м), 4,46 (1H, т, J=3,0 Гц), 4,32 (1H, д, J=5,4 Гц), 3,86-3,80 (2Н, м), 3,76 (3H, с), 3,76 (3H, с), 3,70-3,66 (1Н, м), 3,42-3,34 (2Н, м), 3,13 (1H, дд, J=10,6, 2,7 Гц), 2,24 (3H, с), 0,90 (9Н, с), 0,07 (3H, с), 0,07 (3H, с) (показаны только наблюдаемые пики).
[0538]
(Этап 3)
2-ацетамидо-5'-O-[бис(4-метоксифенил)(фенил)метил]-3'-O-[трет-бутил(диметил)силил]-N-(2-{[трет-бутил(диметил)силил] окси}этил)аденозин
С применением соединения, полученного на этапе 2 (4,41 г), проводили реакцию таким же образом, как на этапе 3 примера 5, с получением указанного в заголовке соединения (1,75 г) и 2-ацетамидо-5'-O-[бис(4-метоксифенил)(фенил)метил]-2'-O-[трет-бутил(диметил)силил]-N-(2-{[трет-бутил(диметил)силил]окси}этил)аденозина (1,31 г) в качестве региоизомера указанного в заголовке соединения.
МС (ИЭР) m/z: 899(М+Н)+.
1Н-ЯМР (CDCl3) δ: 7,89 (1H, с), 7,69 (1H, уш. с), 7,39-7,34 (2Н, м), 7,29-7,18 (7Н, м), 6,80-6,75 (4Н, м), 6,11 (1H, уш. с), 5,89 (1H, д, J=5,4 Гц), 4,65 (1H, дд, J=5,4, 2,7 Гц), 4,43 (1H, дд, J=5,l, 3,3 Гц), 4,19-4,15 (1H, м), 3,83 (2Н, дд, J=5,4, 2,7 Гц), 3,77 (6Н, с), 3,74-3,63 (2Н, м), 3,40 (1H, дд, J=10,9, 3,6 Гц), 3,22 (1H, дд, J=10,9, 3,9 Гц), 2,43 (3H, с), 0,90 (9Н, с), 0,88 (9Н, с), 0,10 (3H, с), 0,06 (6Н, с), 0,03 (3H, с) (показаны только наблюдаемые пики).
Региоизомер (2'-O-TBS-форма)
МС (ИЭР) m/z: 899(М+Н)+.
1Н-ЯМР (CDCl3) δ: 7,84 (1H, с), 7,52 (1H, уш. с), 7,48-7,44 (2Н, м), 7,37-7,32 (4Н, м), 7,29-7,18 (3H, м), 6,83-6,78 (4Н, м), 6,12 (1H, уш. с), 5,89 (1H, д, J=6,0 Гц), 4,96-4,89 (1H, м), 4,32-4,27 (1H, м), 4,24 (1H, дд, J=3,2, 1,6 Гц), 3,84 (2Н, дд, J=5,4, 2,7 Гц), 3,78 (6Н, с), 3,75-3,65 (2Н, м), 3,48 (1H, дд, J=10,6, 2,7 Гц), 3,35 (1H, дд, J=10,6, 3,6 Гц), 2,72 (1Н, д, J=3,0 Гц), 2,37 (3H, с), 0,91 (9Н, с), 0,84 (9Н, с), 0,07 (6Н, с), -0,01 (3H, с), -0,17 (3H, с).
[0539]
(Этап 4)
2-ацетамидо-5'-O-[бис(4-метоксифенил)(фенил)метил]-3'-O-[трет-бутил(диметил)силил]-N-(2-{[трет-бутил(диметил)силил]окси}этил)-2'-O-{(2-цианоэтокси)[ди(пропан-2-ил)амино]фосфанил}аденозин
С применением соединения, полученного на этапе 3 (1,75 г), проводили реакцию таким же образом, как на этапе 4 примера 5, с получением указанного в заголовке соединения (2,08 г) в виде смеси диастереомеров у атома фосфора (соотношение диастереомеров=6:4).
МС (ИЭР) m/z: 1099(М+Н)+.
1Н-ЯМР (CDCl3) δ: 7,90 (0,6Н, с), 7,88 (0,4Н, с), 7,61 (1H, д, J=7,3 Гц), 7,45-7,18 (9Н, м), 6,81 (4Н, м), 6,10 (0,6Н, д, J=5,4 Гц), 6,09 (1H, уш. с), 6,06 (0,4Н, д, J=4,8 Гц), 4,95-4,85 (0,6Н, м), 4,76-4,69 (0,4Н, с), 4,45-4,41 (0,6Н, м), 4,40-4,36 (0,4Н, м), 4,19-4,13 (1H, м), 3,86-3,80 (2Н, м), 3,78 (6Н, с), 3,75-3,41 (8Н, м), 3,32-3,22 (1H, м), 2,55-2,45 (3H, м), 2,36-2,30 (1H, м), 1,30-1,10 (9Н, м), 0,92 (1,2Н, д, J=6,7 Гц), 0,90 (9Н, с), 0,85 (9Н, с), 0,76 (1,8Н, д, J=6,7 Гц), 0,10 (1,8Н, с), 0,07 (1,2Н, с), 0,06 (6Н, с), 0,00 (3H, с).
[0540]
(Этап 5)
Ту же реакцию, что и на этапе 7 примера 1, проводили в следующем масштабе (сырьевой материал: 981 мг). С применением раствора в ацетонитриле полученного соединения и полученного на этапе 4 соединения (1,05 г), проводили реакцию таким же образом, как на этапе 8 примера 1. Полученный в результате этого неочищенный продукт непосредственно использовали для следующей реакции.
[0541]
(Этап 6)
N-{9-[(5R,7R,8R,12aR,14R,15R,15aR,16R)-14-(6-бензоил-6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)-15,16-бис{[трет-бутил(диметил)силил]окси}-10-(2-цианоэтокси)-2-оксо-2-сульфанил-10-сульфанилиденоктагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-7-ил]-6-[(2-{[трет-бутил(диметил)силил]окси}этил)амино]-9Н-пурин-2-ил}ацетамид
С применением неочищенного продукта, полученного на этапе 5, проводили реакцию таким же образом, как на этапе 9 примера 1, с получением указанного в заголовке соединения (413 мг) в виде смеси диастереомеров у атома фосфора.
МС (ИЭР) m/z: 1330(М+Н)+.
[0542]
Этап 7)
Бис(N,N-диэтилэтанаминия) (5R,7R,8R,12aR,14R,15R,15aR,16R)-7-{2-амино-6-[(2-{[трет-бутил(диметил)силил]окси}этил)амино]-9Н-пурин-9-ил}-15,16-бис{[трет-бутил(диметил)силил]окси}-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
Соединение, полученное на этапе 6 (413 мг), растворяли в метаноле (5 мл) и 28% водном аммиаке (5 мл), и реакционную смесь перемешивали при комнатной температуре в течение 63 часов. После концентрирования реакционной смеси при пониженном давлении остаток просто очищали с помощью колоночной хроматографии на силикагеле С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил], и менее полярный диастереомер 1 и более полярный диастереомер 2 отделяли друг от друга. Каждый диастереомер снова растворяли в метаноле (5 мл) и 28% водном аммиаке (5 мл), и каждую реакционную смесь перемешивали при 100°С в течение 2 суток. После того, как каждую реакционную смесь концентрировали при пониженном давлении, каждый остаток очищали с помощью колоночной хроматографии на силикагеле С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил] с получением диастереомера 1 (70,4 мг: с примесями) и диастереомера 2 (65,1 мг: с примесями) указанного в заголовке соединения.
Диастереомер 1 (менее полярный)
МС (ИЭР) m/z: 1131(М+Н)+.
Диастереомер 2 (более полярный)
МС (ИЭР) m/z: 1131(М+Н)+.
[0543]
(Этап 8-1)
Динатрия (5R,7R,8R,12aR,14R,15R,15aS,16R)-7-{2-амино-6-[(2-гидроксиэтил)амино]-9Н-пурин-9-ил}-15,16-дигидрокси-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
(Диастереомер 1)
С применением соединения, полученного на этапе 7 (диастереомер 1) (70,4 мг: с примесями), проводили реакцию таким же образом, как на этапе 11 примера 1, и затем проводили очистку при следующих [Условиях очистки] с получением указанного в заголовке соединения в виде соли триэтиламина.
[Условия очистки] Хроматография на колонке с силикагелем С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил] и препаративная ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 5%-30% (0 мин - 40 мин)].
Полученную соль триэтиламина подвергали солевому обмену таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением указанного в заголовке соединения (20,8 мг).
МС (ИЭР) m/z: 789(М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,24 (1H, с), 8,02 (1Н, с), 7,09 (1H, с), 6,29 (1H, д, J=4,2 Гц), 6,12 (1H, д, J=8,5 Гц), 5,46-5,39 (1H, м), 5,22-5,15 (1H, м), 4,84 (1H, д, J=3,6 Гц), 4,80 (1H, т, J=4,5 Гц), 4,50-4,32 (3H, м), 4,32-4,28 (1H, м), 4,12-3,98 (2Н, м), 3,74 (2Н, т, J=5,4 Гц), 3,68-3,60 (2Н, м), 3,48 (2Н, т, J=5,4 Гц), 2,87-2,70 (2Н, м), 2,01-1,93 (2Н, м).
31Р-ЯМР (CD3OD) δ: 57,8 (с), 54,1 (с).
[0544]
(Этап 8-2)
Динатрия (5R,7R,8R,12aR,14R,15R,15aS,16R)-7-{2-амино-6-[(2-гидроксиэтил)амино]-9Н-пурин-9-ил}-15,16-дигидрокси-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
(Диастереомер 2)
С применением соединения, полученного на этапе 7 (диастереомер 2) (65,1 мг: с примесями), проводили реакцию таким же образом, как на этапе 11 примера 1, и затем проводили очистку при следующих [Условиях очистки] с получением указанного в заголовке соединения в виде соли триэтиламина.
[Условия очистки] Хроматография на колонке с силикагелем С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил], препаративная ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 5%-30% (0 мин - 40 мин)], и препаративная ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/метанол, метанол: 0%-60% (0 мин - 40 мин)].
Полученную соль триэтиламина подвергали солевому обмену таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением указанного в заголовке соединения (7,9 мг).
МС (ИЭР) m/z: 789(М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,39 (1H, с), 8,02 (1H, с), 7,13 (1H, с), 6,33 (1H, д, J=6,7 Гц), 6,13 (1H, д, J=8,5 Гц), 5,51-5,40 (2Н, м), 4,83-4,78 (1Н, м), 4,51-4,29 (4Н, м), 4,28-4,23 (1H, м), 4,07-4,00 (1H, м), 3,94-3,87 (1H, м), 3,75 (2Н, т, J=5,7 Гц), 3,69-3,62 (2Н, м), 3,53-3,47 (2Н, м), 2,91 (2Н, т, J=5,7 Гц), 2,05-1,97 (2Н, м).
31Р-ЯМР (CD3OD) δ: 62,9 (с), 60,3 (с).
[0545]
Пример 17. Синтез ЦДН17.
(5R,7R,8R,12aR,14R,15R,15aS,16R)-15,16-дигидрокси-7-[6-(гидроксиметил)-9Н-пурин-9-ил]-2,10-бис(сульфанил)-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-дион
[0546]
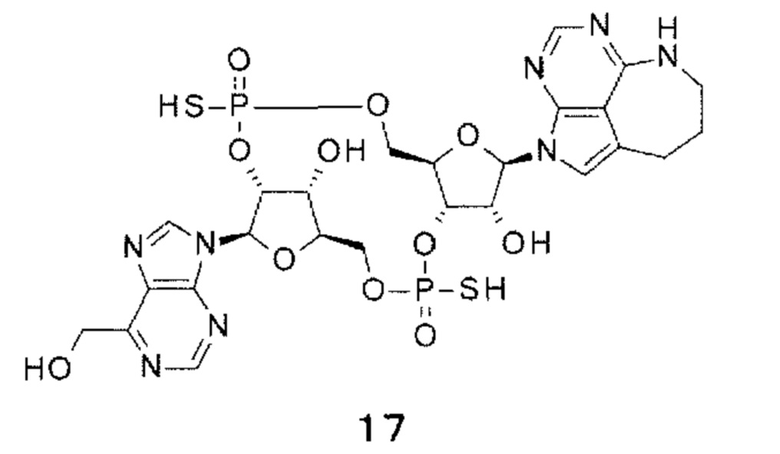
17а (Диастереомер 1)
17b (Диастереомер 2)
[0547]
[Схема синтеза]
[0548]
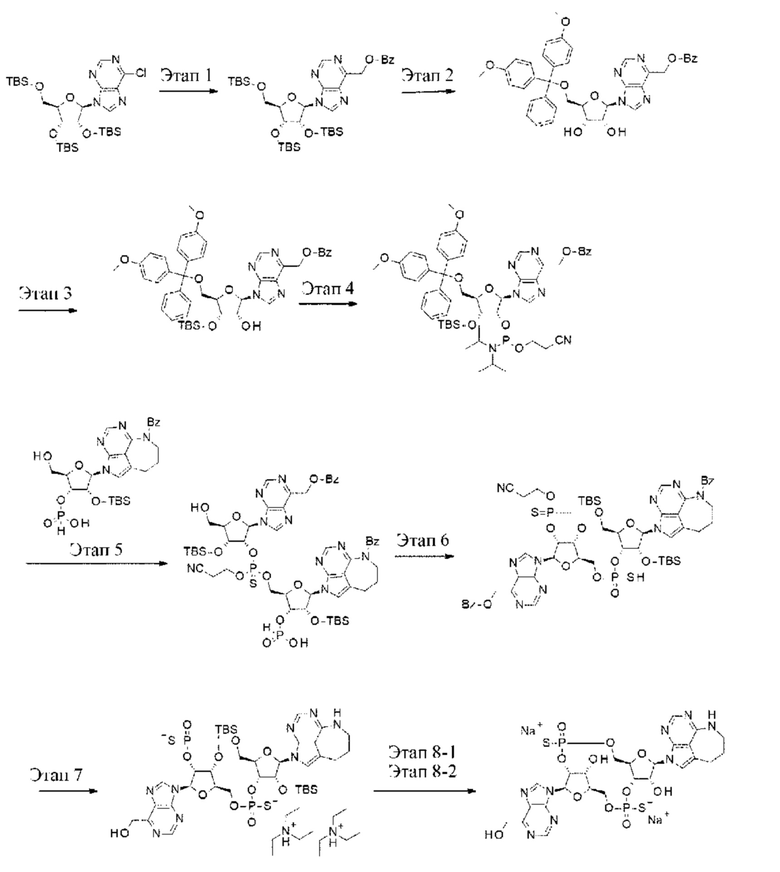
[0549]
(Этап 1)
6-[(бензоилокси)метил]-9-{2,3,5-трис-O-[трет-бутил(диметил)силил]-β-D-рибофуранозил}-9Н-пурин
С применением 6-хлор-9-{2,3,5-трис-O-[трет-бутил(диметил)силил]-β-D-рибофуранозил}-9Н-пурина (10,7 г) как соединения, известного в литературе (J. Org. Chem. 1997, 62, 6833-6841), проводили реакцию таким же образом, как на этапе 2 примера 12, с получением указанного в заголовке соединения (10,4 г).
МС (ИЭР) m/z: 729(М+Н)+.
1Н-ЯМР (CDCl3) δ: 8,94 (1Н, с), 8,50 (1H, с), 8,18-8,13 (2H, м), 7,60-7,54 (1Н, м), 7,48-7,41 (2Н, м), 6,13 (1H, д, J=4,2 Гц), 5,88 (2Н, с), 4,63 (1H, дд, J=4,5, 2,1 Гц), 4,34 (1H, дд, J=4,2, 2,1 Гц), 4,17-4,14 (1H, м), 4,04 (1Н, дд, J=11,5, 3,6 Гц), 3,80 (1H, дд, J=11,5, 2,4 Гц), 0,94 (9Н, с), 0,93 (9Н, с), 0,80 (9Н, с), 0,14 (3H, с), 0,13 (3H, с), 0,11 (3H, с), 0,10 (3H, с), -0,02 (3H, с), -0,20 (3H, с).
[0550]
(Этап 2)
6-[(бензоилокси)метил]-9-{5-O-[бис(4-метоксифенил)(фенил)метил]-β-D-рибофуранозил}-9Н-пурин
К раствору соединения, полученного на этапе 1 (10,3 г), в тетрагидрофуране (50 мл) добавляли раствор в тетрагидрофуране фторида тетрабутиламмония (приблизительно 1 М, 49,4 мл) и реакционную смесь перемешивали при 0°С в течение 4 часов. Уксусную кислоту (2,83 мл) добавляли в реакционную смесь, которую затем концентрировали при пониженном давлении. Остаток частично очищали с помощью колоночной хроматографии на силикагеле [дихлорметан/метанол]. К раствору неочищенного продукта в пиридине (50 мл) добавляли 4,4'-диметокситритилхлорид (10,1 г) при 0°С, и реакционную смесь перемешивали при 4°С в течение ночи. Метанол (2 мл) добавляли в реакционную смесь, которую перемешивали в течение 1 часа. Насыщенный водный раствор гидрокарбоната натрия добавляли в реакционную смесь, которую осуществляли экстрагирование дихлорметаном. После высушивания органического слоя над безводным сульфатом натрия осушающий агент удаляли путем фильтрации и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле [гексан/этилацетат/0,1% триэтиламин] с получением указанного в заголовке соединения (4,62 г).
МС (ИЭР) m/z: 689(М+Н)+.
1Н-ЯМР (CDCl3) δ: 8,91 (1H, с), 8,33 (1H, с), 8,15-8,11 (2Н, м), 7,60-7,54 (1H, м), 7,46-7,40 (2Н, м), 7,27-7,12 (9Н, м), 6,75-6,70 (4Н, м), 6,04 (1H, д, J=6,0 Гц), 5,88 (2Н, с), 5,43 (1Н, уш. с), 4,90-4,84 (1H, м), 4,48-4,41 (2Н, м), 3,76 (6Н, с), 3,44 (1Н, дд, J=10,6, 3,3 Гц), 3,30 (1H, дд, J=10,6, 3,3 Гц), 3,09 (1H, уш. с).
[0551]
(Этап 3)
6-[(бензоилокси)метил]-9-{5-O-[бис(4-метоксифенил)(фенил)метил]-3-O-[трет-бутил(диметил)силил]-β-D-рибофуранозил}-9Н-пурин
С применением соединения, полученного на этапе 2 (4,53 г), проводили реакцию таким же образом, как на этапе 3 примера 5, с получением указанного в заголовке соединения (2,09 г) и 6-[(бензоилокси)метил]-9-{5-O-[бис(4-метоксифенил)(фенил)метил]-2-O-[трет-бутил(диметил)силил]-β-D-рибофуранозил}-9Н-пурина (1,81 г) в качестве региоизомера указанного в заголовке соединения.
МС (ИЭР) m/z: 803(М+Н)+.
1Н-ЯМР (CDCl3) δ: 8,92 (1Н, с), 8,32 (1Н, с), 8,16-8,12 (2Н, м), 7,60-7,54 (1Н, м), 7,47-7,36 (4Н, м), 7,31-7,17 (7Н, м), 6,81-6,76 (4Н, м), 6,07 (1H, д, J=4,8 Гц), 5,87 (2Н, с), 4,81-4,74 (1H, м), 4,62-4,57 (1H, м), 4,22-4,17 (1H, м), 3,77 (6Н, с), 3,52 (1H, дд, J=10,9, 3,6 Гц), 3,26 (1H, дд, J=10,9, 3,6 Гц), 3,12 (1H, д, J=6,7 Гц), 0,89 (9Н, с), 0,09 (3H, с), 0,02 (3H, с).
Региоизомер (2'-O-TBS-форма)
МС (ИЭР) m/z: 803(М+Н)+.
1Н-ЯМР (CDCl3) δ: 8,87 (1Н, с), 8,31 (1Н, с), 8,17-8,13 (2Н, м), 7,60-7,55 (1Н, м), 7,48-7,42 (4Н, м), 7,35-7,31 (4Н, м), 7,28-7,17 (3H, м), 6,83-6,78 (4Н, м), 6,12 (1H, д, J=5,4 Гц), 5,88 (2Н, с), 4,99 (1H, дд, J=5,l, 2,6 Гц), 4,40-4,34 (1H, м), 4,28 (1H, дд, J=3,6, 2,0 Гц), 3,77 (6Н, с), 3,53 (1H, дд, J=10,9, 3,0 Гц), 3,40 (1H, дд, J=10,9, 3,9 Гц), 2,68 (1H, д, J=4,8 Гц), 0,84 (9Н, с), 0,00 (3H, с), -0,14 (3H, с).
[0552]
(Этап 4)
6-[(бензоилокси)метил]-9-(5-O-[бис(4-метоксифенил)(фенил)метил]-3-O-[трет-бутил(диметил)силил]-2-O-{(2-цианоэтокси)[ди(пропан-2-ил)амино]фосфанил}-β-D-рибофуранозил)-9Н-пурин
С применением соединения, полученного на этапе 3 (2,04 г), проводили реакцию таким же образом, как на этапе 4 примера 5, с получением указанного в заголовке соединения (2,37 г) в виде смеси диастереомеров у атома фосфора (соотношение диастереомеров=56:44).
MC (ИЭР)m/z: 1003(М+Н)+.
1Н-ЯМР (CDCl3) δ: 8,88 (0,44Н, с), 8,87 (0,56Н, с), 8,37 (0,44Н, с), 8,35 (0,56Н, с), 8,16-8,11 (2Н, м), 7,60-7,54 (1H, м), 7,47-7,38 (4Н, м), 7,33-7,16 (7Н, м), 6,83-6,76 (4Н, м), 6,31 (0,44Н, д, J=4,8 Гц), 6,21 (0,56Н, д, J=4,8 Гц), 5,87 (0,88Н, с), 5,86 (1,12Н, с), 5,08-5,01 (0,56Н, м), 4,86-4,80 (0,44Н, м), 4,56-4,50 (1H, м), 4,27-4,20 (1H, м), 3,85-3,54 (2Н, м), 3,78 (6Н, с), 3,54-3,44 (3H, м), 3,34-3,25 (1H, м), 2,51 (1,12Н, т, J=6,3 Гц), 2,34 (0,88Н, т, J=6,3 Гц), 1,14-1,05 (9Н, м), 0,90 (3H, д, J=6,7 Гц), 0,85 (5,04Н, с), 0,85 (3,96Н, с), 0,11 (1,68Н, с), 0,08 (1,32Н, с), 0,01 (3H, с).
[0553]
(Этап 5)
Ту же реакцию, что и на этапе 7 примера 1, проводили в следующем масштабе (сырьевой материал: 783 мг). С применением раствора в ацетонитриле полученного соединения и полученного на этапе 4 соединения (765 мг), проводили реакцию таким же образом, как на этапе 8 примера 1. Полученный в результате этого неочищенный продукт непосредственно использовали для следующей реакции.
[0554]
(Этап 6)
{9-[(5R,7R,8R,12aR,14R,15R,15aR,16R)-14-(6-бензоил-6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)-15,16-бис{[трет-бутил(диметил)силил]окси}-10-(2-цианоэтокси)-2-оксо-2-сульфанил-10-сульфанилиденоктагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10] пентаоксадифосфациклотетрадецин-7-ил]-9Н-пурин-6-ил}метилбензоат
С применением неочищенного продукта, полученного на этапе 5, проводили реакцию таким же образом, как на этапе 9 примера 1, с получением указанного в заголовке соединения (345 мг) в виде смеси диастереомеров у атома фосфора. MC (ИЭР) m/z: 1234(М+Н)+.
[0555]
(Этап 7)
Бис(N,N-диэтилэтанаминия) (5R,7R,8R,12aR,14R,15R,15aR,16R)-15,16-бис{[трет-бутил(диметил)силил]окси}-7-[6-(гидроксиметил)-9Н-пурин-9-ил]-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
С применением соединения, полученного на этапе 6 (345 мг), проводили реакцию таким же образом, как на этапе 10 примера 1, с получением диастереомера 1 (101 мг: с примесями) и диастереомера 2 (89,9 мг: с примесями) указанного в заголовке соединения.
Диастереомер 1 (менее полярный)
МС (ИЭР) m/z: 973(М+Н)+.
Диастереомер 2 (более полярный)
МС (ИЭР) m/z: 973(М+Н)+.
[0556]
(Этап 8-1)
Динатрия (5R,7R,8R,12aR,14R,15R,15aS,16R)-15,16-дигидрокси-7-[6-(гидроксиметил)-9Н-пурин-9-ил]-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
(Диастереомер 1)
С применением соединения, полученного на этапе 7 (диастереомер 1) (101 мг: с примесями), проводили реакцию таким же образом, как на этапе 11 примера 1, и затем проводили очистку при следующих [Условиях очистки] с получением указанного в заголовке соединения в виде соли триэтиламина.
[Условия очистки] Хроматография на колонке с силикагелем С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил], препаративная ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 5%-30% (0 мин-40 мин)].
Полученную соль триэтиламина подвергали солевому обмену таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением указанного в заголовке соединения (59,2 мг).
МС (ИЭР) m/z: 745(М+Н)+.
1H-ЯМР (CD3OD) δ: 9,07 (1H, с), 8,88 (1Н, с), 8,03 (1Н, с), 7,10 (1Н, с), 6,48 (1H, д, J=8,5 Гц), 6,29 (1H, д, J=4,8 Гц), 5,46 (1H, дт, J=7,9, 4,2 Гц), 5,21-5,15 (1H, м), 5,10 (2Н, с), 4,84-4,80 (1H, м), 4,79 (1H, т, J=4,5 Гц), 4,51-4,42 (2Н, м), 4,37-4,34 (1H, м), 4,33-4,24 (1H, м), 4,09-3,97 (2Н, м), 3,52-3,47 (2Н, м), 2,89-2,82 (2Н, м), 2,03-1,95 (2Н, м).
31Р-ЯМР (CD3OD) δ: 57,9 (с), 54,8 (с).
[0557]
(Этап 8-2)
Динатрия (5R,7R,8R,12aR,14R,15R,15aS,16R)-15,l6-дигидрокси-7-[6-(гидроксиметил)-9Н-пурин-9-ил]-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
(Диастереомер 2)
С применением соединения, полученного на этапе 7 (диастереомер 2) (88,5 мг: с примесями), проводили реакцию таким же образом, как на этапе 11 примера 1, и затем проводили очистку при следующих [Условиях очистки] с получением указанного в заголовке соединения в виде соли триэтиламина.
[Условия очистки] Хроматография на колонке с силикагелем С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил], препаративная ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 5%-30% (0 мин-40 мин)], и препаративная ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/метанол, метанол: 10%-70% (0 мин-40 мин)].
Полученную соль триэтиламина подвергали солевому обмену таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением указанного в заголовке соединения (27,7 мг).
МС (ИЭР) m/z: 745(М+Н)+.
1Н-ЯМР (CD3OD) δ: 9,16 (1H, с), 8,87 (1H, с), 8,03 (1H, с), 7,11 (1H, с), 6,51 (1H, д, J=8,5 Гц), 6,33 (1Н, д,.1=6,7 Гц), 5,58-5,45 (2Н, м), 5,12-5,09 (2Н, м), 4,81-4,76 (1Н, м), 4,53-4,49 (1Н, м), 4,48-4,36 (2Н, м), 4,34 (2Н, с), 4,09-4,02 (1H, м), 3,92-3,85 (1Н, м), 3,53-3,47 (2Н, м), 2,94-2,87 (2Н, м), 2,05-1,97 (2Н, м).
31Р-ЯМР (CD3OD) δ: 63,1 (с), 60,1 (с).
[0558]
Пример 18. Синтез ЦДН18.
(5R,7R,8R,12aR,14R,15R,15aS,16R)-7-[4-амино-5-(3-аминопропил)-7H-пирроло[2,3-(1]пиримидин-7-ил]-15,16-дигидрокси-2,10-бис(сульфанил)-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2H,10H,12H-5,8-метано-2λ5,10λ5,-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-дион
[0559]
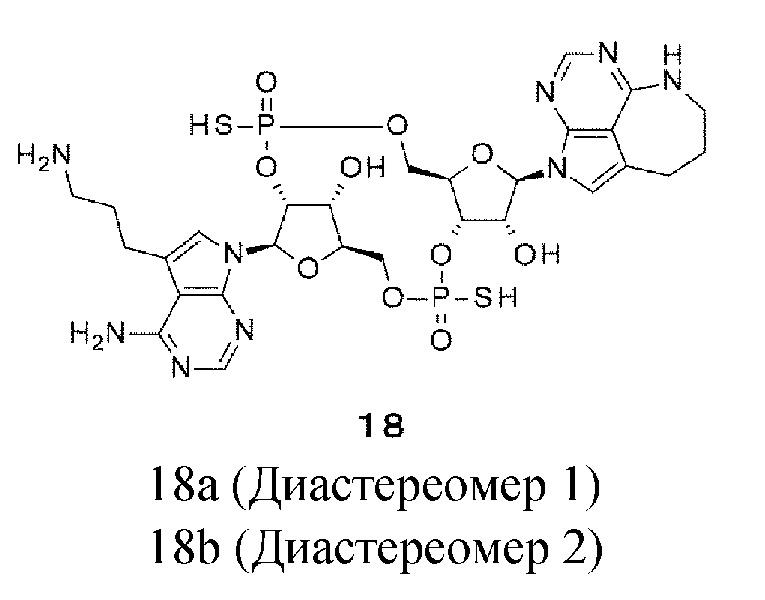
18а (Диастереомер 1)
18b (Диастереомер 2)
[0560]
[Схема синтеза]
[0561]
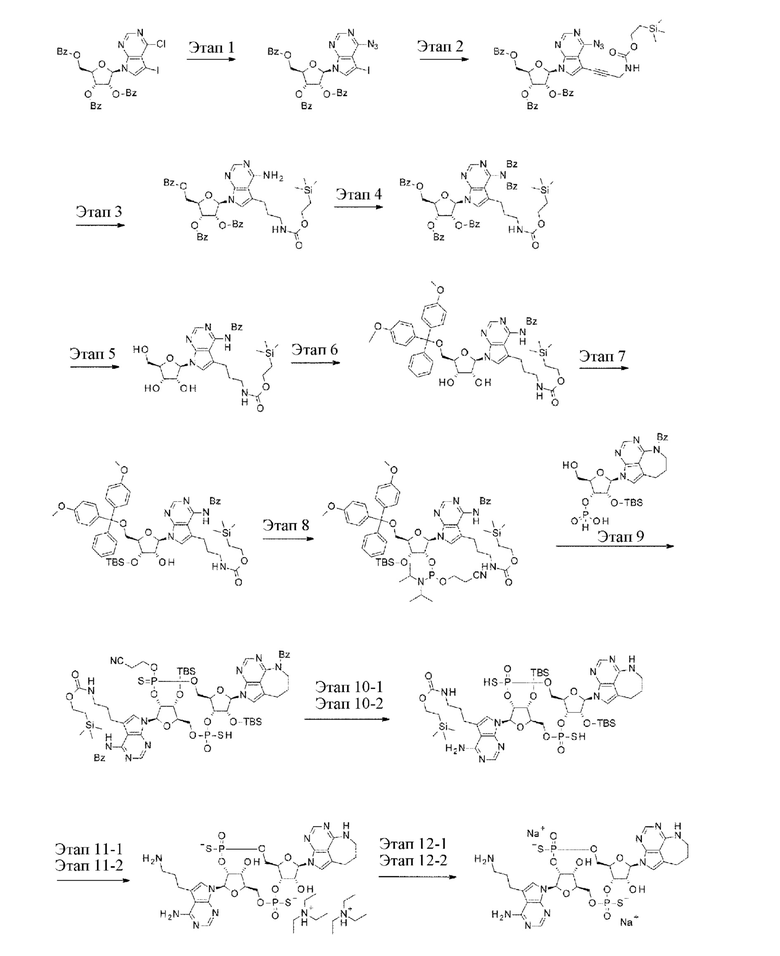
[0562]
(Этап 1)
4-азидо-5-йод-7-(2,3,5-три-O-бензоил-β-D-рибофуранозил)-7Н-пирроло[2,3-d]пиримидин
К раствору 4-хлор-5-йод-7-(2,3,5-три-O-бензоил-β-D-рибофуранозил)--7Н-пирроло[2,3-d]пиримидина (17,96 г) как соединения, известного в литературе (Synth. Commun. 2012, 42, 358-374), в N,N-диметилформамиде (90 мл) добавляли азид тетрабутиламмония (10,1 г), и реакционную смесь перемешивали при 80°С в течение 30 минут. Температуру реакционной смеси возвращали к комнатной температуре, и насыщенный водный раствор гидрокарбоната натрия и этилацетата добавляли в реакционную смесь, которую осуществляли экстрагирование этилацетатом. Органический слой промывали рассолом, а затем сушили над безводным сульфатом натрия. Осушающий агент удаляли путем фильтрации и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле [гексан/этилацетат] с получением указанного в заголовке соединения (17,66 г).
1H-ЯМР (CDCl3) δ: 9,18 (1H, с), 8,13-8,09 (2Н, м), 8,03-7,99 (2Н, м), 7,95-7,90 (2Н, м), 7,64-7,34 (10Н, м), 6,71 (1H, д, J=5,l Гц), 6,20 (1H, т, J=5,5 Гц), 6,11 (1H, т, J=5,3 Гц), 4,96 (1H, дд, J=12.2. 3,2 Гц), 4,88-4,84 (1H, м), 4,70 (1H, дд, J=12.2. 3,2 Гц).
[0563]
(Этап 2)
4-азидо-7-(2,3,5-три-O-бензоил-β-D-рибофуранозил)-5-[3-({[2-(триметилсилил)этокси]карбонил}амино)проп-1-ин-1-ил]-7Н-пирроло[2,3-d]пиримидин
В смешанный раствор соединения, полученного на этапе 1 (9,64 г), в N,N-диметилформамиде (30 мл)/тетрагидрофуране (100 мл) добавляли 2-(триметилсилил)этилпроп-2-ин-1-илкарбамат (6,58 г), триэтиламин (4,57 мл), тетракис(трифенилфосфин)палладий(0) (763 мг) и йодид меди(1) (251 мг) в данном порядке, и реакционную смесь перемешивали при комнатной температуре в течение ночи. Воду добавляли в реакционную смесь, которую осуществляли экстрагирование этилацетатом. Органический слой промывали рассолом, а затем сушили над безводным сульфатом натрия. Осушающий агент удаляли путем фильтрации и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле [гексан/этилацетат] с получением указанного в заголовке соединения (7,20 г).
1H-ЯМР (CDCl3) δ: 9,16 (1H, с), 8,14-8,08 (2Н, м), 8,03-7,99 (2Н, м), 7,94-7,90 (2Н, м), 7,66 (1H, с), 7,65-7,35 (9Н, м), 6,66 (1Н, д, J=5,1 Гц), 6,22 (1H, т, J=5,5 Гц), 6,10 (1H, т, J=5,3 Гц), 5,07 (1H, уш. с), 4,95 (1H, дд, J=12,3, 2,9 Гц), 4,88-4,84 (1H, м), 4,70 (1H, дд, J=12,3, 3,7 Гц), 4,29 (2Н, д, J=5,5 Гц), 4,21 (2Н, т, J=8,6 Гц), 1,02 (2Н, т, J=8,6 Гц), 0,05 (9Н, с).
[0564]
(Этап 3)
7-(2,3,5-три-O-бензоил-β-D-рибофуранозил)-5-[3-({[2-(триметилсилил)этокси]карбонил}амино)пропил]-7Н-пирроло[2,3-(1]пиримидин-4-амин
В смешанный раствор соединения, полученного на этапе 2 (7,20 г), в метаноле (70 мл)/тетрагидрофуране (70 мл) добавляли 20% гидроксид палладия на углероде (2 г), и реакционную смесь перемешивали в атмосфере водорода при комнатной температуре в течение 2 часов. После удаления катализатора путем фильтрации, фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле [гексан/этилацетат] с получением указанного в заголовке соединения (5,45 г).
1H-ЯМР (CDCl3) δ: 8,26 (1H, с), 8,19-8,15 (2Н, м), 8,02-7,97 (2Н, м), 7,96-7,92 (2Н, м), 7,65-7,48 (5Н, м), 7,43-7,32 (4Н, м), 6,82 (1Н, с), 6,73 (1H, д, J=6,3 Гц), 6,17 (1H, т, J=5,9 Гц), 6,11 (1H, дд, J=5,9, 3,9 Гц), 5,23 (2Н, с), 4,90 (1H, дд, J=12.2. 3,4 Гц), 4,77-4,70 (2Н, м), 4,63 (1H, дд, J=12.2. 3,4 Гц), 4,15 (2Н, т, J=8,6 Гц), 3,20-3,10 (2Н, м), 2,62 (2Н, т, J=7,6 Гц), 1,72-1,62 (2Н, м), 1,01-0,94 (2Н, м), 0,04 (9Н, с).
[0565]
(Этап 4)
N,N-дибензоил-7-(2,3,5-три-O-бензоил-β-D-рибофуранозил)-5-[3-({[2-(триметилсилил)этокси]карбонил}амино)пропил]-7Н-пирроло[2,3-d]пиримидин-4-амин
К раствору соединения, полученного на этапе 3 (5,45 г), в дихлорметане (60 мл) добавляли пиридин (1,69 мл) и бензоилхлорид (2,01 мл) в данном порядке при 0°С, и реакционную смесь перемешивали при 0°С в течение 5 минут, дополнительно добавляли в нее триэтиламин (2,91 мл), и реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Насыщенный водный раствор гидрокарбоната натрия и дихлорметана добавляли в реакционную смесь, которую осуществляли экстрагирование дихлорметаном. После высушивания органического слоя над безводным сульфатом натрия осушающий агент удаляли путем фильтрации и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле [гексан/этилацетат], а затем измельчали в порошок с этилацетатом и гексаном. Полученный в результате этого твердый продукт собирали путем фильтрации с получением указанного в заголовке соединения (6,16 г).
1H-ЯМР (CDCl3) δ: 8,54 (1H, с), 8,20-8,16 (2Н, м), 8,02-7,98 (2Н, м), 7,96-7,92 (2Н, м), 7,82-7,76 (4Н, м), 7,65-7,32 (15Н, м), 7,18 (1H, уш. с), 6,82 (1H, д, J=5,9 Гц), 6,17 (1H, т, J=6,l Гц), 6,13 (1H, дд, J=5,9, 3,5 Гц), 4,94 (1Н, дд, J=12.2. 3,2 Гц), 4,79-4,75 (1Н, м), 4,65 (1H, дд, 3=12.2. 3,2 Гц), 4,41 (1H, уш. с), 4,08 (2Н, т, J=8,6 Гц), 2,95-2,87 (2Н, м), 2,58-2,50 (2Н, м), 1,54-1,52 (2Н, м), 0,97-0,91 (2Н, м), 0,02 (9Н, с).
[0566]
(Этап 5)
N-бензоил-7-бензоил-β-D-рибофуранозил-5-[3-({[2-(триметилсилил)этокси]карбонил}амино)пропил]-7Н-пирроло[2,3-(1]пиримидин-4-амин
К раствору соединения, полученного на этапе 4 (6,16 г), в тетрагидрофуране (250 мл) добавляли по каплям раствор в метаноле метоксида натрия (1,0 М, 12,0 мл) при 0°С, и реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Уксусную кислоту (1,07 мл) добавляли в реакционную смесь при 0°С, и реакционную смесь затем концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле [этилацетат/метанол], а затем измельчали в порошок с этилацетатом и гексаном. Полученный в результате этого твердый продукт собирали путем фильтрации с получением указанного в заголовке соединения (3,35 г).
1H-ЯМР (CD3OD) δ: 8,62 (1H, с), 8,04 (2Н, д, J=7,0 Гц), 7,69-7,62 (1H, м), 7,60-7,54 (3H, м), 6,26 (1H, д, J=6,3 Гц), 4,59 (1H, т, J=5,7 Гц), 4,31 (1H, дд, J=5,1, 3,5 Гц), 4,12-4,01 (3H, м), 3,86 (1H, дд, J=12,3, 2,9 Гц), 3,76 (1H, дд, J=12,1, 3,5 Гц), 3,01 (2Н, т, J=6,7 Гц), 2,76 (2Н, т, J=7,4 Гц), 1,86-1,76 (2Н, м), 0,93 (2Н, т, J=8,2 Гц), 0,03 (9Н, с).
[0567]
(Этап 6)
N-бензоил-7-{5-O-[бис(4-метоксифенил)(фенил)метил]-бензоил-β-D-рибофуранозил}-5-[3-({[2-(триметилсилил)этокси]карбонил}амино)пропил]-7Н-пирроло[2,3-d]пиримидин-4-амин
Полученное на этапе 5 соединение (3,35 г) подвергали азеотропной перегонке с пиридином, и 4,4'-диметокситритилхлорид (2,38 г) добавляли в раствор остатка в безводном пиридине (50 мл) при 0°С, и реакционную смесь перемешивали при комнатной температуре в течение ночи. После этого в реакционную смесь добавляли этанол (5 мл), чтобы погасить реакцию, и продукт затем концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле [гексан/этилацетат] с получением указанного в заголовке соединения (4,34 г).
1Н-ЯМР (CDCl3) δ: 8,51 (1H, уш. с), 8,30 (1H, уш. с), 8,05-7,90 (1H, м), 7,56-7,42 (3H, м), 7,39-7,31 (2Н, м), 7,29-7,16 (10Н, м), 6,81-6,75 (4Н, м), 6,22 (1H, д, J=5,9 Гц), 4,91 (1H, уш. с), 4,76-4,67 (2Н, м), 4,45-4,41 (1H, м), 4,38-4,29 (1H, м), 4,05-3,95 (2Н, м), 3,77 (6Н, с), 3,50 (1H, дд, J=10,6, 3,1 Гц), 3,37-3,27 (1H, м), 3,19-2,94 (4Н, м), 2,66 (1H, уш. с), 1,82 (1H, уш. с), 0,93-0,76 (2Н, м), -0,01 (9Н, с).
[0568]
(Этап 7)
Н-бензоил-7-{5-O-[бис(4-метоксифенил)(фенил)метил]-3-O-[трет-бутил(диметил)силил]-бензоил-β-D-рибофуранозил}-5-[3-({[2-(триметилсилил)этокси]карбонил}амино)пропил]-7Н-пирроло[2,3-d]пиримидин-4-амин (3'-O-TBS-форма)
С применением соединения, полученного на этапе 6 (4,34 г), проводили реакцию таким же образом, как на этапе 4 примера 8, с получением указанного в заголовке соединения (1,51 г) и К-бензоил-7-{5-O-[бис(4-метоксифенил)(фенил)метил]-2-O-[трет-бутил(диметил)силил]-бензоил-β-D-рибофуранозил}-5-[3-({[2-(триметилсилил)этокси]карбонил}амино)пропил]-7H-пирроло[2,3-d]пиримидин-4-амина (2'-O-TBS-форма) (2,01 г) в качестве региоизомера указанного в заголовке соединения. (3'-OTBS-форма) (более полярная)
1Н-ЯМР (CDCl3) δ: 8,38-8,26 (2Н, м), 8,10-8,03 (1H, м), 7,58-7,40 (5Н, м), 7,36-7,20 (10Н, м), 6,86-6,77 (4Н, м), 6,30-6,22 (1Н, м), 5,02-4,89 (1H, м), 4,63-4,45 (2Н, м), 4,17-4,10 (1H, м), 4,04-3,92 (2Н, м), 3,79 (3H, с), 3,79 (3H, с), 3,59-3,52 (1H, м), 3,32-3,23 (1H, м), 3,17-2,80 (4Н, м), 1,86-1,64 (2Н, м), 0,89 (9Н, с), 0,86-0,72 (2Н, м), 0,09 (3H, с), 0,01 (3H, с), -0,01 (9Н, с). (2'-OTBS-форма) (менее полярная)
1H-ЯМР (CDCl3) δ: 8,39-8,25 (2Н, м), 8,04 (1H, с), 7,57-7,42 (5Н, м), 7,37-7,18 (10Н, м), 6,87-6,80 (4Н, м), 6,33 (1H, д, J=5,l Гц), 5,01 (1H, уш. с), 4,78 (1H, т, J=5,7 Гц), 4,40-4,33 (1H, м), 4,29-4,25 (1H, м), 4,05-3,92 (2Н, м), 3,80 (3H, с), 3,79 (3H, с), 3,59-3,51 (1H, м), 3,44-3,37 (1H, м), 3,16-3,03 (1Н, м), 2,99-2,81 (3H, м), 1,74-1,61 (2Н, м), 0,83 (9Н, с), 0,80-0,70 (2Н, м), 0,00-0,03 (12Н, м), -0,20 (3H, с).
[0569]
(Этап 8)
N-бензоил-7-(5-O-[бис(4-метоксифенил)(фенил)метил]-3-O-[трет-бутил(диметил)силил]-2-O-{(2-цианоэтокси)[ди(пропан-2-ил)амино]фосфанил}-бензоил-β-D-рибофуранозил)-5-[3-({[2-(триметилсилил)этокси]карбонил}амино)пропил]-7Н-пирроло[2,3-d]пиримидин-4-амин
С применением соединения, полученного на этапе 7 (3'-OTBS-форма) (1,51 г), проводили реакцию таким же образом, как на этапе 6 примера 1, с получением диастереомера 1 (0,77 г) и диастереомера 2 (0,48 г) указанного в заголовке соединения, в виде диастереомеров у атома фосфора.
Диастереомер 1 (менее полярный)
1Н-ЯМР (CDCl3) δ: 8,36-8,27 (2Н, м), 8,02 (1H, уш. с), 7,57-7,40 (5Н, м), 7,37-7,21 (10Н, м), 6,87-6,77 (4Н, м), 6,39 (1Н, д, J=5,l Гц), 4,98 (1H, уш. с), 4,85-4,73 (1Н, м), 4,56-4,47 (1H, м), 4,20-4,15 (1H, м), 4,05-3,92 (2Н, м), 3,88-3,67 (8Н, м), 3,63-3,47 (3H, м), 3,33-3,23 (1H, м), 3,15-2,82 (3H, м), 2,60-2,45 (2Н, м), 1,83-1,64 (2Н, м), 1,10 (6Н, д, J=6,7 Гц), 0,92 (6Н, д, J=6,7 Гц), 0,87 (9Н, с), 0,81-0,71 (2Н, м), 0,12 (3H, с), 0,03 (3H, с), -0,01 (9Н, с).
Диастереомер 2 (более полярный)
1H-ЯМР (CDCl3) δ: 8,36-8,28 (2Н, м), 8,05 (1H, уш. с), 7,57-7,38 (5Н, м), 7,36-7,19 (10Н, м), 6,84-6,79 (4Н, м), 6,48-6,40 (1H, м), 5,02 (1H, уш. с), 4,65-4,42 (2Н, м), 4,19-4,15 (1H, м), 4,06-3,90 (2Н, м), 3,80-3,48 (11Н, м), 3,32-3,24 (1H, м), 3,17-3,03 (2Н, м), 2,99-2,86 (1Н, м), 2,51-2,40 (2Н, м), 1,86-1,69 (2Н, м), 1,18-1,02 (12Н, м), 0,85 (9Н, с), 0,81-0,71 (2Н, м), 0,08 (3H, с), 0,02 (3H, с), -0,01 (9Н, с).
[0570]
[(Этап 9)
2-(триметилсилил)этил(3-{4-бензамидо-7-[(5R,7R,8R,12aR,14R,15R,15aR,16R)-14-(6-бензоил-6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)-15,16-бис{[трет-бутил(диметил)силил]окси}-10-(2-цианоэтокси)-2-оксо-2-сульфанил-10-сульфанилиденоктагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациютотетрадецин-7-ил]-7Н-пирроло[2,3-d]пиримидин-5-ил}пропил)карбамат
Ту же реакцию, что и на этапе 7 примера 1, проводили в следующем масштабе (сырьевой материал: 1,08 г). С применением раствора в ацетонитриле полученного соединения и соединения, полученного на этапе 8 (1,25 г: смесь диастереомеров), проводили реакцию таким же образом, как на этапе 8 примера 1 и этапе 9 примера 1, с получением указанного в заголовке соединения в виде смеси диастереомеров у атома фосфора. Диастереомеры у атома фосфора разделяли с помощью препаративной ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 10%-50% (0 мин-35 мин)] с получением диастереомера 1 (0,19 г) и диастереомера 2 (0,043 г) указанного в заголовке соединения.
Диастереомер 1 (менее полярный)
МС (ИЭР) m/z: 1419 (М+Н)+.
Диастереомер 2 (более полярный)
МС (ИЭР) m/z: 1419 (М+Н)+.
[0571]
(Этап 10-1)
2-(триметилсилил)этил(3-{4-амино-7-[(5R,7R,8R,12aR,14R,15R,15aR,16R)-15,16-бис{[трет-бутил(диметил)силил]окси}-2,10-диоксо-2,10-бис(сульфанил)-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[с(1]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10] пентаоксадифосфациклотетрадецин-7-ил]-7Н-пирроло[2,3-d]пиримидин-5-ил}пропил)карбамат
С применением соединения, полученного на этапе 9 (диастереомер 1) (0,19 г), проводили реакцию таким же образом, как на этапе 8-1 примера 12. Продукт очищали с помощью препаративной ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 40%-70% (0 мин-35 мин)] с получением указанного в заголовке соединения (58 мг). МС (ИЭР) m/z: 1158(М+Н)+.
[0572]
(Этап 10-2)
2-(триметилсилил)этил(3-{4-амино-7-[(5R,7R,8R,12aR,14R,15R,15aR,16R)-15,16-бис{[трет-бутил(диметил)силил]окси}-2,10-диоксо-2,10-бис(сульфанил)-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10] пентаоксадифосфациклотетрадецин-7-ил]-7Н-пирроло[2,3-d]пиримидин-5-ил}пропил)карбамат
С применением соединения, полученного на этапе 9 (диастереомер 2) (43 мг), проводили реакцию таким же образом, как на этапе 8-1 примера 12. Продукт очищали с помощью препаративной ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 30%-60% (0 мин-35 мин)] с получением указанного в заголовке соединения (15 мг). МС (ИЭР) m/z: 1158 (М+Н)+.
[0573]
(Этап 11-1)
Бис(N,N-диэтилэтанаминия)(5R,7R,8R,12aR,14R,15R,15aS,16R)-7-[4-амино-5-(3-аминопропил)-7Н-пирроло[2,3-d]пиримидин-7-ил]-15,16-дигидрокси-2,10-диоксо-14-(6,7,8,9-тетрагидро-2H-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2H,10H,12H-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
К раствору соединения, полученного на этапе 10-1 (58 мг), в тетрагидрофуране (1 мл) добавляли раствор в тетрагидрофуране фторида тетрабутиламмония (приблизительно 1 М, 5 мл), и реакционную смесь перемешивали при комнатной температуре в течение ночи. Так как реакция еще не завершилась, реакционную смесь дополнительно перемешивали при 40°С в течение 3 часов. Реакционную смесь концентрировали при пониженном давлении, а затем очищали с помощью препаративной ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 10%-40% (0 мин-35 мин)]. Продукт дополнительно очищали с помощью Sep-Pak (R) С18 [0,1% водный раствор триэтиламина/ацетонитрил, ацетонитрил: 0%-30%] с получением указанного в заголовке соединения (25 мг).
МС (ИЭР) m/z: 786(М+Н)+.
[0574]
(Этап 11-2)
Бис(N,N-диэтилэтанаминия) (5R,7R,8R,12aR,14R,15R,15aS,16R)-7-[4-амино-5-(3-аминопропил)-7Н-пирроло[2,3-d]пиримидин-7-ил]-15,16-дигидрокси-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
С применением соединения, полученного на этапе 10-2 (15 мг), проводили реакцию таким же образом, как на этапе 11-1 с получением указанного в заголовке соединения (7,2 мг).
МС (ПЭР) m/z: 786(М+Н)+.
[0575]
(Этап 12-1)
Динатрия (5R,7R,8R,12aR,14R,15R,15aS,16R)-7-[4-амино-5-(3-аминопропил)-7H-пирроло[2,3-d]пиримидин-7-ил]-15,16-дигидрокси-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ+-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
(Диастереомер 1)
С применением соединения, полученного на этапе 11-1 (25 мг), замену соли осуществляли таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением указанного в заголовке соединения (17 мг).
МС (ИЭР) m/z: 786(М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,028 (1H, с), 8,025 (1H, с), 7,71 (1H, с), 7,09 (1H, с), 6,51 (1H, д, J=8,2 Гц), 6,26 (1H, д, J=5,l Гц), 5,38-5,29 (1H, м), 5,17-5,10 (1H, м), 4,85-4,80 (1Н, м), 4,75 (1Н, д, J=3,9 Гц), 4,44-4,37 (1H, м), 4,33-4,23 (3H, м), 4,09-3,97 (2Н, м), 3,52-3,46 (2Н, м), 3,00-2,72 (6Н, м), 2,13-2,02 (2Н, м), 2,02-1,93 (2Н, м).
31Р-ЯМР (CD3OD) δ: 56,65 (с), 55,31 (с).
[0576]
(Этап 12-2)
Динатрия (5R,7R,8R,12aR,14R,15R,15aS,16R)-7-[4-амино-5-(3-аминопропил)-7Н-пирроло[2,3-d]пиримидин-7-ил]-15,16-дигидрокси-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
(Диастереомер 2)
С применением соединения, полученного на этапе 11-2 (7,2 мг), замену соли осуществляли таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением указанного в заголовке соединения (6,0 мг). МС (ИЭР) m/z: 786 (М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,02 (1H, с), 8,01 (1H, с), 7,75 (1Н, с), 7,07 (1H, с), 6,52 (1H, д, J=8,6 Гц), 6,29 (1H, д, J=7,4 Гц), 5,61-5,55 (1H, м), 5,47-5,37 (1H, м), 4,86-4,83 (1H, м), 4,56-4,43 (2Н, м), 4,31-4,21 (3H, м), 4,06-3,99 (1H, м), 3,91-3,84 (1H, м), 3,54-3,46 (3H, м), 3,00-2,87 (5Н, м), 2,29-2,07 (2Н, м), 2,06-1,95 (2Н, м).
31Р-ЯМР (CD3OD) δ: 63,08 (с), 58,68 (с).
[0577]
Пример 19. Синтез ЦДН19.
(5R,7R,8R,12aR,14R,15R,15aS,16R)-7-(6-амино-9H-пурин-9-ил)-15,16-дигидрокси-2,10-бис(сульфанил)-14-(6,7,8,9-тетрагидро-2Н-1,2,3,5,6-пентаазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-дион
[0578]
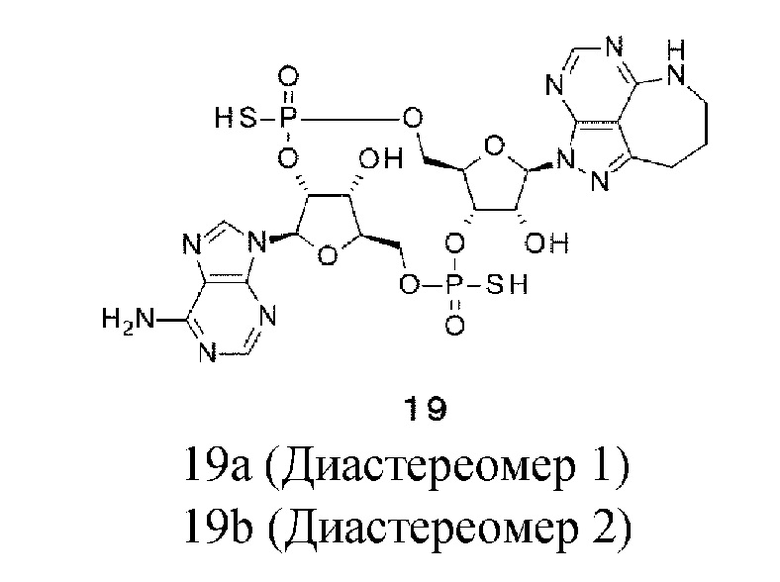
19а (Диастереомер 1)
19b (Диастереомер 2)
[0579]
[Схема синтеза]
[0580]
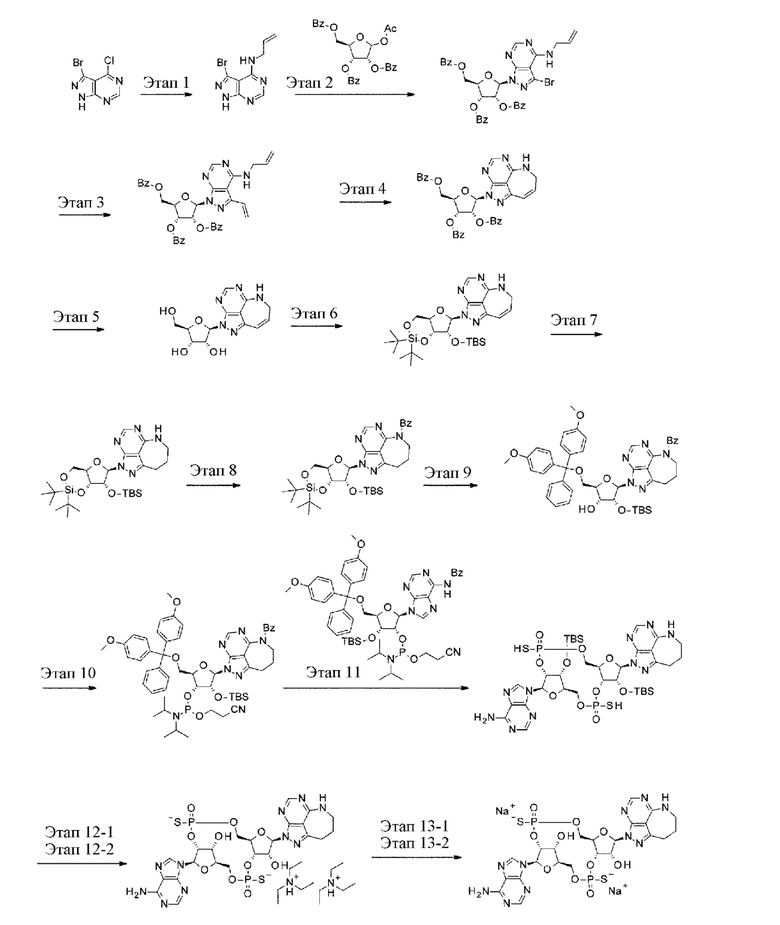
[0581]
(Этап 1)
3-бром-N-(проп-2-ен-1-ил)-1Н-пиразоло[3,4-d]пиримидин-4-амин
Доступный для приобретения (BePharm Ltd.) 3-бром-4-хлор-1Н-пиразоло[3,4-d]пиримидин (8,50 г) суспендировали в диоксане (140 мл), в который добавляли N,N,-диизопропилэтиламин (12,5 мл) и аллиламин (11 мл) при комнатной температуре, и реакционную смесь перемешивали при той же температуре в течение 70 часов. Реакционную смесь концентрировали при пониженном давлении, и из остатка делали взвесь с помощью дихлорметана и этилацетата, из которой затем собирали твердый продукт путем фильтрации (твердый продукт 1). После того, как фильтрат концентрировали при пониженном давлении, ту же процедуру повторяли дважды с получением твердого продукта 2 и твердого продукта 3. Конечный фильтрат очищали с помощью колонки с силикагелем [гексан/этилацетат] с получением твердого продукта 4. Кроме того, твердый продукт 1 очищали с помощью колонки с силикагелем [гексан/этилацетат] с получением твердого продукта 5. Твердый продукт 4 и твердый продукт 5 объединяли с получением указанного в заголовке соединения (4,48 г). Твердый продукт 2 и твердый продукт 3 объединяли с получением указанного в заголовке соединения с примесями (3,78 г).
МС (ИЭР) m/z: 254(М+Н)+.
1Н-ЯМР (ДМСО-d6) δ: 13,82 (1Н, с), 8,26 (1H, с), 7,23 (1H, т, J=5,9 Гц), 5,96 (1H, м), 5,18 (1H, дд, J=17,1, 1,5 Гц), 5,10 (1H, дд, J=10,3, 1,5 Гц), 4,19 (2Н, м).
[0582]
(Этап 2)
3-бром-N-(проп-2-ен-1-ил)-1-(2,3,5-три-O-бензоил-бензоил-β-D-рибофуранозил)-1Н-пиразоло[3,4-d]пиримидин-4-амин
Полученное на этапе 1 соединение (1,00 г) и доступную для приобретения (Ark Pharm) 1-O-ацетил-2,3,5-три-O-бензоил-β-D-рибофуранозу (2,58 г) суспендировали в нитрометане (50 мл), а затем растворяли путем нагревания. Комплекс трифторид бора-диэтиловый эфир (0,63 мл) добавляли туда при нагревании до кипения с обратным холодильником, и реакционную смесь перемешивали при той же температуре в течение 1 часа. Туда дополнительно добавляли 1-O-ацетил-2,3,5-три-O-бензоил-β-D-рибофуранозу (0,40 г) и комплекс трифторид бора-диэтиловый эфир (0,10 мл), и реакционную смесь дополнительно перемешивали в течение 3 часов. После концентрирования реакционной смеси при пониженном давлении остаток очищали с помощью колоночной хроматографии на силикагеле [гексан/этилацетат] с получением указанного в заголовке соединения (1,80 г).
МС (ИЭР) m/z: 698(М+Н)+.
1Н-ЯМР (CDCl3) δ: 8,40 (1H, с), 8,13 (2Н, м), 7,96 (4Н, м), 7,59-7,32 (9Н, м), 6,79 (1H, д, J=3,4 Гц), 6,39 (1H, дд, J=5,l, 3,7 Гц), 6,25 (1H, т, J=5,6 Гц), 6,17 (1H, т, J=5,6 Гц), 6,00 (1H, м), 5,31 (1H, дд, J=17,l, 1,0 Гц), 5,24 (1H, дд, J=10,5, 1,2 Гц), 4,81 (1H, м), 4,75 (1H, дд, J=12,0, 3,7 Гц), 4,63 (1H, дд, J=12.2. 4,4 Гц), 4,30 (2Н, м).
[0583]
(Этап 3)
3-этенил-N-(проп-2-ен-1-ил)-1-(2,3,5-три-O-бензоил-бензоил-β-D-рибофуранозил)-1Н-пиразоло[3,4-d]пиримидин-4-амин
Раствор соединения, полученного на этапе 2 (11,65 г), в толуоле (120 мл) обрабатывали ультразвуком для дегазации при пониженном давлении. Трибутилвинилтин (12,8 мл) и тетракис(трифенилфосфин)палладий(0) (2,73 г) добавляли в реакционную смесь в атмосфере азота и реакционную смесь грели до кипения с обратным холодильником в течение 2 часов. После концентрирования реакционной смеси при пониженном давлении остаток очищали с помощью колоночной хроматографии на силикагеле [дихлорметан/этилацетат] с получением указанного в заголовке соединения (10,07 г).
МС (ИЭР) m/z: 646(М+Н)+.
1Н-ЯМР (CDCl3) δ: 8,42 (1H, с), 8,09 (2Н, м), 7,99 (2Н, м), 7,95 (2Н, м), 7,59-7,50 (3H, м), 7,42-7,33 (6Н, м), 6,90-6,84 (2Н, м), 6,43 (1H, дд, J=5,4, 3,4 Гц), 6,32 (1H, т, J=5,9 Гц), 6,00 (1H, м), 5,94 (1H, дд, J=17,6, 1,0 Гц), 5,67 (1Н, дд, J=11,2, 1,0 Гц), 5,56 (1Н, т, J=5,6 Гц), 5,27 (1H, дд, J=17,1, 1,0 Гц), 5,22 (1H, дд, J=10,3, 1,0 Гц), 4,82 (1Н, м), 4,77 (1Н, дд, J=12.2. 3,9 Гц), 4,62 (1H, дд, J=11,7, 4,9 Гц), 4,29 (2Н, м).
[0584]
(Этап 4)
2-(2,3,5-три-O-бензоил-бензоил-β-D-рибофуранозил)-6,7-дигидро-2Н-1,2,3,5,6-пентаазабензо [cd] азулен
Полученное на этапе 3 соединение (9,0 г) дважды подвергали азеотропной перегонке с бензолом. К раствору остатка в дихлорметане (480 мл) добавляли (+)-10-камфорсульфоновую кислоту (4,2 г) и бензилиден[1,3-бис(2,4,6-триметилфенил)имидазолидин-2-илиден]дихлорид(трициклогексил-λ5-фосфанил)рутений (катализатор Граббса второго поколения) (360 мг), и реакционную смесь грели до кипения с обратным холодильником в течение 3 часов. Дополнительно добавляли туда катализатор Граббса второго поколения (360 мг), и реакционную смесь дополнительно грели до кипения с обратным холодильником в течение 1 часа. Реакционную смесь охлаждали до комнатной температуры, промывали насыщенным водным раствором гидрокарбоната натрия и рассолом в данном порядке, а затем сушили над безводным сульфатом натрия. Осушающий агент удаляли путем фильтрации и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле [гексан/этилацетат] с получением указанного в заголовке соединения (6,39 г).
МС (ИЭР) m/z: 618(М+Н)+.
1Н-ЯМР (CDCl3) δ: 8,39 (1H, с), 8,11 (2Н, м), 7,99 (2Н, м), 7,96 (2Н, м), 7,57-7,52 (3H, м), 7,42-7,35 (6Н, м), 6,83 (1H, д, J=2,9 Гц), 6,76 (1H, д, J=10,7 Гц), 6,43 (1H, дд, J=5,1, 3,2 Гц), 6,33 (1H, т, J=5,9 Гц), 6,12 (1H, м), 5,61 (1H, уш. с), 4,83 (1H, м), 4,78 (1Н, дд, J=12.2. 3,9 Гц), 4,63 (1H, дд, J=12,0, 4,6 Гц), 4,16 (2Н, м).
[0585]
(Этап 5)
2-бензоил-β-D-рибофуранозил-6,7-дигидро-2Н-1,2,3,5,6-пентаазабензо[cd]азулен
В смешанный раствор соединения, полученного на этапе 4 (620 мг), в метаноле (10 мл)/тетрагидрофуране (5,0 мл) добавляли раствор в метаноле метоксида натрия (1,0 М, 0,10 мл) при комнатной температуре, и реакционную смесь перемешивали при той же температуре в течение 18 часов. Реакционную смесь нейтрализовали 1 N соляной кислотой, а затем концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле [дихлорметан/метанол] с получением указанного в заголовке соединения (279 мг).
МС (ИЭР) m/z: 306(М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,23 (1H, с), 6,79 (1H, м), 6,22-6,17 (2Н, м), 4,74 (1H, т, J=5,1 Гц), 4,42 (1H, т, J=4,6 Гц), 4,14 (2Н, дд, J=5,9, 1,5 Гц), 4,11 (1H, к, J=4,1 Гц), 3,81 (1H, дд, J=12,4, 3,2 Гц), 3,68 (1Н, дд, J=12,4,4,6 Гц).
[0586]
(Этап 6)
2-{2-O-[трет-бутил(диметил)силил]-3,5-O-(ди-трет-бутилсилилиден)-бензоил-β-D-рибофуранозил}-6,7-дигидро-2Н-1,2,3,5,6-пентаазабензо[cd]азулен
С применением соединения, полученного на этапе 5 (2,81 г), проводили реакцию таким же образом, как на этапе 1 примера 1, с получением указанного в заголовке соединения (4,72 г).
1H-ЯМР (CDCl3) δ: 8,38 (1H, с), 6,85 (1H, дт, J=11,0, 1,4 Гц), 6,28 (1H, с), 6,16-6,08 (1H, м), 5,78 (1H, уш. с), 4,71-4,63 (2Н, м), 4,39 (1Н, дд, J=9,0, 5,1 Гц), 4,22-4,10 (3H, м), 3,96 (1H, дд, J=10,6, 9,0 Гц), 1,11 (9Н, с), 1,05 (9Н, с), 0,90 (9Н, с), 0,11 (3H, с), 0,09 (3H, с).
[0587]
(Этап 7)
2-{2-O-[трет-бутил(диметил)силил]-3,5-O-(ди-трет-бутилсилилиден)-бензоил-β-D-рибофуранозил}-6,7,8,9-тетрагидро-2Н-1,2,3,5,6-пентаазабензо[cd]азулен
К раствору соединения, полученного на этапе 6 (2,12 г), в тетрагидрофуране (20 мл) добавляли уксусную кислоту (три капли с помощью пипетки Пастера) и 10% палладий-углерод (AD), влажный (0,82 г), и реакционную смесь перемешивали в атмосфере водорода при комнатной температуре в течение 3 часов. Катализатор удаляли путем фильтрации, а затем промывали тетрагидрофураном и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле [гексан/этилацетат] с получением указанного в заголовке соединения (2,09 г).
1H-ЯМР (CDCl3) δ: 8,29 (1H, с), 6,33 (1H, уш. с), 6,27 (1H, с), 4,70 (1H, д, J=4,7 Гц), 4,62 (1H, дд, J=9,6, 4,9 Гц), 4,39 (1H, дд, J=9,0, 5,1 Гц), 4,21-4,08 (1Н, м), 3,95 (1Н, дд, J=10,4, 9,2 Гц), 3,65-3,58 (2Н, м), 3,15-2,99 (2Н, м), 2,22-2,10 (2Н, м), 1,11 (9Н, с), 1,05 (9Н, с), 0,90 (9Н, с), 0,10 (3H, с), 0,09 (3H, с).
[0588]
(Этап 8)
6-бензоил-2-{2-O-[трет-бутил(диметил)силил]-3,5-O-(ди-трет-бутилсилилиден)-бензоил-β-D-рибофуранозил}-6,7,8,9-тетрагидро-2Н-1,2,3,5,6-пентаазабензо[cd]азулен
С применением соединения, полученного на этапе 7 (2,09 г), проводили реакцию таким же образом, как на этапе 4 примера 1, с получением указанного в заголовке соединения (2,23 г).
1H-ЯМР (CDCl3) δ: 8,19 (1H, с), 7,47-7,40 (3H, м), 7,33-7,28 (2Н, м), 6,34 (1H, с), 4,72 (1Н, д, J=4,7 Гц), 4,66 (1H, дд, J=9,4, 4,7 Гц), 4,49 (1H, дд, J=14,7, 8,0 Гц), 4,40 (1H, дд, J=9,0, 5,1 Гц), 4,24-4,09 (2Н, м), 3,96 (1H, дд, J=10,6, 9,0 Гц), 3,26-3,11 (2Н, м), 2,45-2,23 (2Н, м), 1,12 (9Н, с), 1,05 (9Н, с), 0,90 (9Н, с), 0,12 (3H, с), 0,10 (3H, с).
[0589]
(Этап 9)
6-бензоил-2-{5-O-[бис(4-метоксифенил)(фенил)метил]-2-O-[трет-бутил(диметил)силил]-β-D-рибофуранозил}-6,7,8,9-тетрагидро-2Н-1,2,3,5,6-пентаазабензо[cd]азулен
С применением соединения, полученного на этапе 8 (2,23 г), проводили реакцию таким же образом, как на этапе 5 примера 1, с получением указанного в заголовке соединения (2,69 г).
1H-ЯМР (CDCl3) δ: 8,22 (1H, с), 7,55-7,51 (2Н, м), 7,46-7,36 (7Н, м), 7,32-7,26 (2Н, м), 7,26-7,16 (3H, м), 6,80-6,73 (4Н, м), 6,43 (1H, д, J=5,5 Гц), 5,32 (1H, т, J=5,5 Гц), 4,43 (1H, дд, J=14,3, 8,0 Гц), 4,34-4,29 (1H, м), 4,25-4,13 (2Н, м), 3,78 (3H, с), 3,77 (3H, с), 3,46 (1H, дд, J=10,4, 3,3 Гц), 3,17-3,05 (3H, м), 2,79 (1H, д, J=3,5 Гц), 2,40-2,19 (2Н, м), 0,82 (9Н, с), 0,03 (3H, с), -0,13 (3H, с).
[0590]
(Этап 10)
6-бензоил-2-(5-O-[бис(4-метоксифенил)(фенил)метил]-2-O-[трет-бутил(диметил)силил]-3-O-{(2-цианоэтокси)[ди(пропан-2-ил)амино]фосфанил}-бензоил-β-D-рибофуранозил)-6,7,8,9-тетрагидро-2Н-1,2,3,5,6-пентаазабензо[cd]азулен
С применением соединения, полученного на этапе 9 (2,69 г), проводили реакцию таким же образом, как на этапе 4 примера 5, с получением указанного в заголовке соединения (2,93 г) в виде смеси диастереомеров у атома фосфора (соотношение диастереомеров = 6:4).
1Н-ЯМР (CDCl3) δ: 8,22 (0,4Н, с), 8,21 (0,6Н, с), 7,56-7,50 (2Н, м), 7,44-7,37 (7Н, м), 7,28-7,18 (5Н, м), 6,81-6,73 (4Н, м), 6,44 (0,6Н, д, J=6,3 Гц), 6,40 (0,4Н, д, J=6,3 Гц), 5,34-5,28 (1H, м), 4,47-4,35 (2,4Н, м), 4,32-4,26 (0,6Н, м), 4,25-4,16 (1Н, м), 4,03-3,84 (1H, м), 3,80-3,73 (6Н, м), 3,69-3,44 (4Н, м), 3,18-3,00 (3H, м), 2,73-2,59 (1H, м), 2,40-2,19 (3H, м), 1,22-1,14 (8,4Н, м), 1,01 (3,6Н, д, J=6,7 Гц), 0,76 (3,6Н, с), 0,74 (5,4Н, с), 0,02 (1,2Н, с), 0,01 (1,8Н, с), -0,12 (1,8Н, с), -0,15 (1,2Н, с).
[0591]
(Этап 11)
(5R,7R,8R,12aR,14R,15R,15aR,16R)-7-(6-амино-9H-пурин-9-ил)-15,16-бис{[трет-бутил(диметил)силил]окси}-2,10-бис(сульфанил)-14-(6,7,8,9-тетрагидро-2Н-1,2,3,5,6-пентаазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-дион
С применением соединения, полученного на этапе 10 (1,04 г), проводили реакцию таким же образом, как на этапе 7 примера 1, с получением раствора в ацетонитриле 6-бензоил-2-{2-O-[трет-бутил(диметил)силил]-3-O-(дигидроксифосфанил)-β-D-рибофуранозил}-6,7,8,9-тетрагидро-2Н-1,2,3,5,6-пентаазабензо[cd]азулена. С применением данного раствора в ацетонитриле и доступного для приобретения (Cool Pharm Ltd.) N-бензоил-5'-O-[бис(4-метоксифенил)(фенил)метил]-3'-O-[трет-бутил(диметил)силил]-2'-O-{(2-цианоэтокси)[ди(пропан-2-ил)амино]фосфанил}аденозина (1,20 г) проводили реакцию таким же образом, как на этапе 8 примера 1, этапе 9 примера 1 и этапе 10 примера 1 с получением указанного в заголовке соединения в виде смеси диастереомеров у атома фосфора. Диастереомеры у атома фосфора разделяли с помощью препаративной ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 25-50% (0 мин -35 мин)] с получением диастереомера 1 (15 мг) и диастереомера 2 (55 мг) указанного в заголовке соединения (время удерживания при ВЭЖХ: диастереомер 1>2).
Диастереомер 1 (менее полярный)
МС (ИЭР) m/z: 959(М+Н)+.
Диастереомер 2 (более полярный)
МС (ИЭР) m/z: 959(М+Н)+.
[0592]
(Этап 12-1)
Бис(N,N-диэтилэтанаминия) (5R,7R, 8R,12aR,14R,15R,15aS,16R)-7-(6-амино-9Н-пурин-9-ил)-15,16-дигидрокси-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-1,2,3,5,6-пентаазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
Тригидрофторид триэтиламина (1 мл) добавляли к соединению, полученному на этапе 11 (диастереомер 1) (20 мг), и реакционную смесь перемешивали при 45°С в течение 2 часов. Реакционную смесь добавляли в охлажденный до температуры льда смешанный раствор 1 М водного раствора гидрокарбоната триэтиламмония (3 мл) и триэтиламина (1 мл), чтобы погасить реакцию. Продукт очищали с помощью Sep-Pak (R) С18 [0,1% триэтиламин вода/ацетонитрил, ацетонитрил: 0%-17%] с получением указанного в заголовке соединения (15 мг).
МС (ИЭР) m/z: 731(М+Н)+.
[0593]
(Этап 12-2)
Бис(N,N-диэтилэтанаминия)(5R,7R,8R,12aR,14R,15R,15aS,16R)-7-(6-амино-9H-пурин-9-ил)-15,16-дигидрокси-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-1,2,3,5,6-пентаазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
С применением соединения, полученного на этапе 11 (диастереомер 2) (43 мг), проводили реакцию таким же образом, как на этапе 12-1, с получением указанного в заголовке соединения (34 мг).
МС (ИЭР) m/z: 731(М+Н)+.
[0594]
(Этап 13-1)
Динатрия (5R,7R,8R,12aR,14R,15R,15aS,16R)-7-(6-амино-9Н-пурин-9-ил)-15,16-дигидрокси-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-1,2,3,5,6-пентаазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
(Диастереомер 1)
С применением соединения, полученного на этапе 12-1 (15 мг), замену соли осуществляли таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением смеси диастереомеров указанного в заголовке соединения (12 мг).
МС (ИЭР) m/z: 731(М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,79 (1H, с), 8,18 (1H, с), 8,14 (1H, с), 6,37-6,34 (2Н, м), 5,50 (1H, дд, J=10,8, 5,3 Гц), 5,40-5,33 (1Н, м), 5,06 (1H, дд, J=4,5, 2,9 Гц), 4,94 (1H, д, J=3,5 Гц), 4,67-4,30 (4Н, м), 4,03 (1H, ддд, J=12,5, 6,1, 2,2 Гц), 3,93 (1H, дт, J=18,l, 6,1 Гц), 3,58 (2Н, д, J=7,4 Гц), 3.02 (2Н, т, J=5,9 Гц), 2,18-2,04 (2Н, м).
31Р-ЯМР (CD3OD) δ: 56,5 (с), 54,2 (с).
[0595]
(Этап 13-2)
Динатрия (5R,7R,8R,12aR,14R,15R,15aS,16R)-7-(6-амино-9H-пурин-9-ил)-15,16-дигидрокси-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-1,2,3,5,6-пентаазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
(Диастереомер 2)
С применением соединения, полученного на этапе 12-2 (34 мг), замену соли осуществляли таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением смеси диастереомеров указанного в заголовке соединения (28 мг: соотношение диастереомеров = 4:1).
МС (ИЭР) m/z: 731(М+Н)+.
1Н-ЯМР (CD3OD) δ: 9,16 (0,2Н, с), 8,85 (0,8Н, с), 8,19 (0,2Н, с), 8,17 (0,8Н, с), 8,14 (0,8Н, с), 8,13 (0,2Н, с), 6,38-6,30 (2Н, м), 5,67-5,61 (0,8Н, м), 5,57-5,36 (1,2Н, м), 5,22 (0,8Н, дд, J=5,9, 4.3 Гц), 5,10 (0,2Н, т, J=4,5 Гц), 4,64-4,26 (4,8Н, м), 4,16-4,10 (0,2Н, м), 4,05-3,98 (1H, м), 3,86-3,79 (1H, м), 3,62-3,54 (2Н, м), 3,07 (1,6Н, т, J=5,7 Гц), 2,98 (0,4Н, т, J=5,9 Гц), 2,17-2,04 (2Н, м).
31Р-ЯМР (CD3OD) δ: 63,5 (с), 63,4 (с), 60,0 (с), 59,9 (с).
[0596]
Пример 20. Синтез ЦДН20.
(5R,7R,8R,12aR,14R,15R,15aS,16R)-7-(6-амино-9H-пурин-9-ил)-14-(8,9-дигидро-6-окса-2,3,5-триазабензо[cd]азулен-2(7Н)-ил)-15,16-дигидрокси-2,10-бис(сульфанил)октагидро-2Н, 10Н, 12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-дион
[0597]
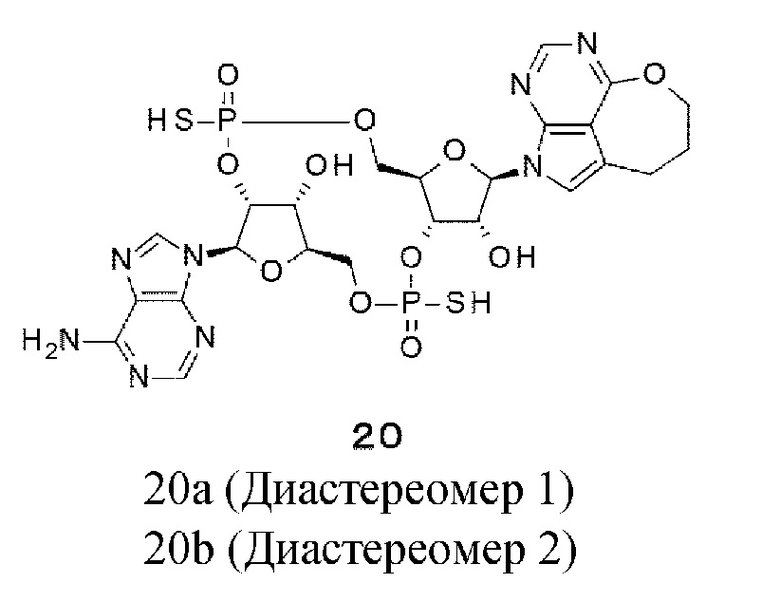
20а (Диастереомер 1)
20b (Диастереомер 2)
[0598]
[Схема синтеза]
[0599]
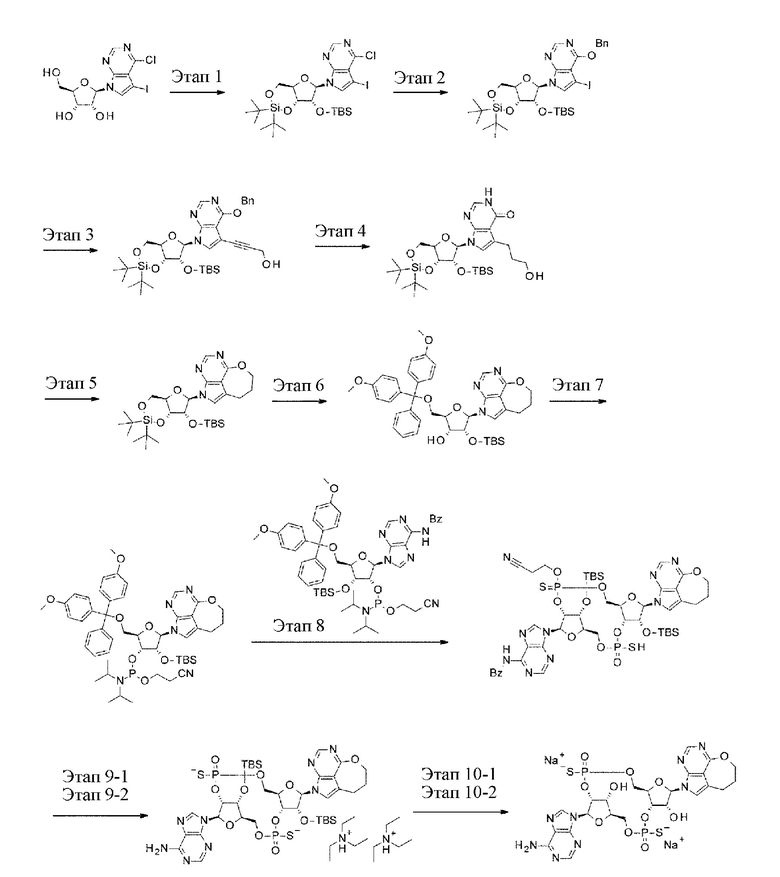
[0600] (Этап 1)
7-{2-O-[трет-бутил(диметил)силил]-3,5-O-(ди-трет-бутилсилилиден)-бензоил-β-D-рибофуранозил}-4-хлор-5-йод-7Н-пирроло[2,3-d]пиримидин
С применением 4-хлор-5-йод-7-р-Б-рибофуранозил-7Н-пирроло[2,3-d]пиримидина (3,15 г) как соединения, известного в литературе (J. Med. Chem. 2008, 51, 3934-3945), проводили реакцию таким же образом, как на этапе 1 примера 1, с получением указанного в заголовке соединения (2,97 г).
1Н-ЯМР (CDCl3) δ: 8,62 (1H, с), 7,38 (1H, с), 6,18 (1Н, с), 4,53-4,47 (1Н, м), 4,44 (1H, д, J=3,9 Гц), 4,24-4,18 (2Н, м), 4,06-3,98 (1Н, м), 1,09 (9Н, с), 1,05 (9Н, с), 0,92 (9Н, с), 0,13 (3H, с), 0,12 (3H, с).
[0601]
(Этап 2)
4-(бензилокси)-7-{2-O-[трет-бутил(диметил)силил]-3,5-O-(ди-трет-бутилсилилиден)-β-D-рибофуранозил}-5-йод-7Н-пирроло[2,3-d]пиримидин
К раствору соединения, полученного на этапе 1 (4,97 г), и бензилового спирта (1,0 мл) в тетрагидрофуране (50 мл) добавляли гидрид натрия (содержащий 37% минеральное масло) (426 мг) при охлаждении льдом, температуру повышали до комнатной температуры и реакционную смесь перемешивали в течение ночи. Насыщенный водный раствор хлорида аммония добавляли в реакционную смесь при охлаждении льдом, чтобы погасить реакцию. После того, как реакционную смесь подвергли экстрагированию этилацетатом, органический слой промывали водой и рассолом и сушили над безводным сульфатом натрия. Осушающий агент удаляли путем фильтрации и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле [гексан/этилацетат] с получением указанного в заголовке соединения (3,38 г).
1Н-ЯМР (CDCl3) δ: 8,43 (1H, с), 7,62-7,57 (2Н, м), 7,43-7,37 (2Н, м), 7,36-7,30 (1H, м), 7,13 (1H, с), 6,15 (1Н, с), 5,65 (1H, д, J=13,7 Гц), 5,62 (1H, д, J=13,7 Гц), 4,50-4,43 (2Н, м), 4,27 (1H, дд, J=9,2, 4,9 Гц), 4,21-4,12 (1H, м), 4,01 (1H, дд, J=10,4, 9,2 Гц), 1,09 (9Н, с), 1,04 (9Н, с), 0,91 (9Н, с), 0,12 (3H, с), 0,11 (3H, с).
[0602]
(Этап 3)
4-(бензилокси)-7-{2-O-[трет-бутил(диметил)силил]-3,5-O-(ди-трет-бутилсилилиден)-β-D-рибофуранозил}-5-(3-гидроксипроп-1-ин-1-ил)-7Н-пирроло[2,3-d]пиримидин
С применением соединения, полученного на этапе 2 (3,38 г), и 2-пропин-1-ола (1,35 мл) проводили реакцию таким же образом, как на этапе 2 примера 1, за исключением того, что температуру реакции устанавливали на уровне комнатной температуры, с получением указанного в заголовке соединения (2,22 г).
1Н-ЯМР (CDCl3) δ: 8,46 (1H, с), 7,59-7,55 (2Н, м), 7,43-7,33 (3H, м), 7,22 (1H, с), 6,16 (1H, с), 5,60 (1H, д, J=12,7 Гц), 5,57 (1H, д, J=12,7 Гц), 4,51-4,41 (4Н, м), 4,27 (1Н, дд, J=9,5, 5,0 Гц), 4,19 (1H, дт, J=9,9, 5,0 Гц), 4,01 (1Н, дд, J=9,9, 9,5 Гц), 1,52 (1H, т, J=6,3 Гц), 1,08 (9Н, с), 1,04 (9Н, с), 0,91 (9Н, с), 0,13 (3H, с), 0,11 (3H, с).
[0603]
(Этап 4)
7-{2-O-[трет-бутил(диметил)силил]-3,5-O-(ди-трет-бутилсилилиден)-бензоил-β-D-рибофуранозил}-5-(3-гидроксипропил)-3,7-дигидро-4Н-пирроло[2,3-d]пиримидин-4-он
С применением соединения, полученного на этапе 3 (3,38 г), проводили реакцию таким же образом, как на этапе 7 примера 19, за исключением того, что растворитель реакции заменяли на смесь метанола (20 мл)/тетрагидрофурана (20 мл) с получением указанного в заголовке соединения (0,92 г).
1Н-ЯМР (CDCl3) δ: 11,36 (1H, уш. с), 7,86 (1H, с), 6,69 (1H, с), 6,10 (1H, с), 4,48 (1Н, дд, J=9,2, 4,9 Гц), 4,37 (1H, д, J=5,1 Гц), 4,23 (1H, дд, J=9,3, 4,8 Гц), 4,16 (1H, дт, J=9,8, 4,8 Гц), 4,02 (1H, дд, J=9,8, 9,3 Гц), 3,84 (1H, т, J=6,3 Гц), 3,58 (2Н, дд, J=11,9, 6,1 Гц), 3,03-2,90 (2Н, м), 1,86 (2Н, с), 1,09 (9Н, с), 1,04 (9Н, с), 0,90 (9Н, с), 0,10 (6Н, с).
[0604]
(Этап 5)
2-{2-O-[трет-бутил(диметил)силил]-3,5-O-(ди-трет-бутилсилилиден)-β-D-рибофуранозил}-2,7,8,9-тетрагидро-6-окса-2,3,5-триазабензо[cd]азулен
К раствору соединения, полученного на этапе 4 (0,92 г), в тетрагидрофуране (32 мл) добавляли трифенилфосфин (0,62 г) и диизопропилазодикарбоксилат (0,47 мл) при охлаждении льдом, температуру повышали до комнатной температуры и реакционную смесь перемешивали в течение 1 часа. После концентрирования реакционной смеси при пониженном давлении остаток очищали с помощью колоночной хроматографии на силикагеле [гексан/этилацетат] с получением указанного в заголовке соединения (0,77 г).
1Н-ЯМР (CDCl3) δ: 8,44 (1H, с), 6,85 (1H, с), 6,19 (1H, с), 4,56-4,51 (2Н, м), 4,50-4,44 (2Н, м), 4,35 (1H, дд, J=9,5, 5,0 Гц), 4,17 (1H, дт, J=10,0, 5,0 Гц), 4,00 (1H, дд, J=10,0, 9,5 Гц), 2,98-2,92 (2Н, м), 2,28-2,20 (2Н, м), 1,10 (9Н, с), 1,05 (9Н, с), 0,91 (9Н, с), 0,12 (3H, с), 0,11 (3H, с).
[0605]
(Этап 6)
2-{5-O-[бис(4-метоксифенил)(фенил)метил]-2-O-[трет-бутил(диметил)силил]-β-D-рибофуранозил}-2,7,8,9-тетрагидро-6-окса-2,3,5-триазабензо[cd]азулен
С применением соединения, полученного на этапе 5 (0,95 г), проводили реакцию таким же образом, как на этапе 5 примера 1, с получением указанного в заголовке соединения (1,13 г).
1Н-ЯМР (CDCl3) δ: 8,42 (1H, с), 7,48-7,43 (2Н, м), 7,37-7,21 (7Н, м), 7,19 (1H, с), 6,85-6,79 (4Н, м), 6,35 (1H, д, J=5,4 Гц), 4,71 (1H, т, J=5,4 Гц), 4,55-4,48 (2Н, м), 4,35 (1H, дд, J=8,2, 4,3 Гц), 4,22 (1H, к, J=3,0 Гц), 3,79 (3H, с), 3,79 (3H, с), 3,53 (1H, дд, J=10,6, 3,1 Гц), 3,37 (1H, дд, J=10,6, 3,1 Гц), 2,80 (1H, д, J=4,3 Гц), 2,71 (2Н, т, J=5,5 Гц), 2,23-2,15 (2Н, м), 0,83 (9Н, с), -0,04 (3H, с), -0,16 (3H, с).
[0606]
(Этап 7)
2-(5-O-[бис(4-метоксифенил)(фенил)метил]-2-O-[трет-бутил(диметил)силил]-3-O-{(2-цианоэтокси)[ди(пропан-2-ил)амино]фосфанил}-бензоил-β-D-рибофуранозил)-2,7,8,9-тетрагидро-6-окса-2,3,5-триазабензо[cd]азулен
С применением соединения, полученного на этапе 6 (1,13 г), проводили реакцию таким же образом, как на этапе 4 примера 5, за исключением того, что для очистки применяли колоночную хроматографию на силикагеле [гексан/этилацетат/0,1% триэтиламин], с получением указанного в заголовке соединения (1,00 г) в виде смеси диастереомеров у атома фосфора (соотношение диастереомеров = 6:4).
1H-ЯМР (CDCl3) δ: 8,41 (0,4Н, с), 8,39 (0,6Н, с), 7,51-7,43 (2Н, м), 7,39-7,17 (8Н, м), 6,85-6,79 (4Н, м), 6,34 (0,6Н, д, J=6,7 Гц), 6,30 (0,4Н, д, J=5,9 Гц), 4,82 (0,6Н, дд, J=6,7, 4,7 Гц), 4,76 (0,4Н, дд, J=5,9, 4,7 Гц), 4,55-4,47 (2Н, м), 4,43-4,35 (1,2Н, м), 4,30-4,25 (0,8Н, м), 4,05-3,85 (1H, м), 3,81-3,76 (6Н, м), 3,70-3,47 (4Н, м), 3,32-3,24 (1H, м), 2,79-2,64 (3,2Н, м), 2,31 (0,8Н, т, 3=6,5 Гц), 2,23-2,14 (2Н, м), 1,20-1,15 (8,4Н, м), 1,03 (3,6Н, д, J=7,0 Гц), 0,75 (3,6Н, с), 0,73 (5,4Н, с), -0,03 (1,2Н, с), -0,08 (1,8Н, с), -0,20 (1,2Н, с), -0,22 (1,8Н, с).
[0607]
(Этап 8)
N-{9-[(5R,7R,8R,12aR,14R,15R,15aR,16R)-15,16-бис{[трет-бутил(диметил)силил]окси}-10-(2-цианоэтокси)-14-(8,9-дигидро-6-окса-2,3,5-триазабензо[cd]азулен-2(7Н)-ил)-2-оксо-2-сульфанил-10-сульфанилиденоктагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациютотетрадецин-7-ил]-9Н-пурин-6-ил}бензамид
С применением соединения, полученного на этапе 7 (1,00 г), проводили реакцию таким же образом, как на этапе 7 примера 1, с получением раствора в ацетонитриле 2-{2-O-[трет-бутил(диметил)силил]-3-O-(дигидроксифосфанил)-бензоил-β-D-рибофуранозил}-2,7,8,9-тетрагидро-6-окса-2,3,5-триазабензо[cd]азулена. С применением данного раствора в ацетонитриле и доступного для приобретения (Cool Pharm Ltd.) N-бензоил-5'-O-[бис(4-метоксифенил)(фенил)метил]-3'-O-[трет-бутил(диметил)силил]-2'-O-{(2-цианоэтокси)[ди(пропан-2-ил)амино]фосфанил}аденозина (1,28 г) проводили реакцию таким же образом, как на этапе 8 примера 1 и этапе 9 примера 1, с получением смеси, содержащей диастереомер 1 указанного в заголовке соединения, и смеси, содержащей диастереомер 2 указанного в заголовке соединения.
Диастереомер 1 (менее полярный)
МС (ИЭР) m/z: 1116(М+Н)+.
Диастереомер 2 (более полярный)
МС (ИЭР) m/z: 1116(М+Н)+.
[0608] (Этап 9-1)
Бис(N,N-диэтилэтанаминия)(5R,7R,8R,12aR,14R,15R,15aR,16R)-7-(6-амино-9H-пурин-9-ил)-15,16-бис{[трет-бутил(диметил)силил]окси}-14-(8,9-дигидро-6-окса-2,3,5-триазабензо[cd]азулен-2(7H)-ил)-2,10-диоксооктагидро-2H,10H,12H-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
С применением всего количества полученного на этапе 8 соединения (смеси, содержащей диастереомер 1) проводили реакцию таким же образом, как на этапе 10 примера 1, и затем проводили очистку при следующих [Условиях очистки] с получением указанного в заголовке соединения (73 мг) в виде соли триэтиламина.
[Условия очистки] ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 30%-60% (0 мин-35 мин)].
МС (ИЭР) m/z: 959(М+Н)+.
[0609]
(Этап 9-2)
Бис(N,N-диэтилэтанаминия)(5R,7R,8R,12aR,14R,15R,15aR,16R)-7-(6-амино-9H-пурин-9-ил)-15,16-бис{[трет-бутил(диметил)силил]окси}-14-(8,9-дигидро-6-окса-2,3,5-триазабензо[св]азулен-2(7Н)-ил)-2,10-диоксооктагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
С применением всего количества полученного на этапе 8 соединения (смеси, содержащей диастереомер 2) проводили реакцию таким же образом, как на этапе 10 примера 1, и затем проводили очистку при следующих [Условиях очистки] с получением указанного в заголовке соединения (58 мг) в виде соли триэтиламина.
[Условия очистки] ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 25%-50% (0 мин-35 мин)].
МС (ИЭР) m/z: 959(М+Н)+.
[0610]
(Этап 10-1)
Динатрия (5R,7R,8R,12aR,14R,15R,15aS,16R)-7-(6-амино-9H-пурин-9-ил)-14-(8,9-дигидро-6-окса-2,3,5-триазабензо[cd]азулен-2(7Н)-ил)-15,16-дигидрокси-2,10-диоксооктагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
(Диастереомер 1)
С применением соединения, полученного на этапе 9-1 (68 мг), проводили реакцию таким же образом, как на этапе 12-1 примера 19, а затем проводили замену соли таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением указанного в заголовке соединения (42 мг).
МС (ИЭР) m/z: 731(М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,73 (1H, с), 8,31 (1H, с), 8,17 (1Н, с), 7,36 (1H, с), 6,38 (1H, д, J=4,7 Гц), 6,34 (1H, д, J=8,2 Гц), 5,42-5,34 (1H, м), 5,24-5,16 (1H, м), 4,86-4,81 (2Н, м), 4,64-4,29 (6Н, м), 4,12-4,01 (2Н, м), 2,98-2,81 (2Н, м), 2,28-2,13 (2Н, м).
31Р-ЯМР (CD3OD) δ: 58,2 (с), 54,4 (с).
[0611]
(Этап 10-2)
Динатрия (5R,7R,8R,12aR,14R,15R,15aS,16R)-7-(6-амино-9Н-пурин-9-ил)-14-(8,9-дигидро-6-окса-2,3,5-триазабензо[cd]азулен-2(7Н)-ил)-15,16-дигидрокси-2,10-диоксооктагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
(Диастереомер 2)
С применением соединения, полученного на этапе 9-2 (58 мг), проводили реакцию таким же образом, как на этапе 12-1 примера 19, а затем проводили замену соли таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением указанного в заголовке соединения (35 мг).
МС (ИЭР) m/z: 731(М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,81 (1H, с), 8,31 (1H, с), 8,17 (1H, с), 7,38 (1H, с), 6,41 (1H, д, J=6,7 Гц), 6,34 (1H, д, J=8,6 Гц), 5,56-5,41 (2Н, м), 4,87 (1Н, м), 4,64-4,27 (7Н, м), 4,06-3,99 (1H, м), 3,92-3,88 (1H, м), 2,99 (2Н, т, J=5,5 Гц), 2,28-2,19 (2Н, м).
31Р-ЯМР (CD3OD) δ: 63,1 (с), 60,5 (с).
[0612]
Пример 21. Синтез лекарственного средства-линкера 1
[Схема синтеза]
[0613]
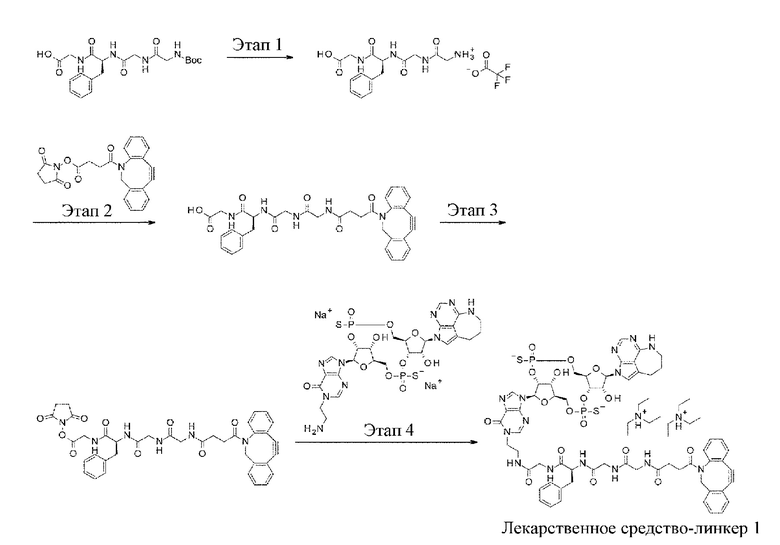
[0614]
(Этап 1)
N-(азаниумилацетил)глицил-L-фенилаланилглицинатрифторацетат
К раствору доступного для приобретения (Bachem Holding AG) N-(трет-бутоксикарбонил)глицилглицил-L-фенилаланилглицина (3,00 г) в дихлорметане (30 мл) добавляли трифторуксусную кислоту (15 мл) при комнатной температуре и реакционную смесь перемешивали при той же температуре в течение 3 часов. Реакционную смесь концентрировали при пониженном давлении, а затем суспендировали в толуоле и снова концентрировали при пониженном давлении. Данную процедуру концентрирования дополнительно повторяли дважды. Из остатка делали взвесь с диэтиловым эфиром (100 мл), а затем собирали путем фильтрации с получением неочищенной формы указанного в заголовке соединения (3,27 г). МС (ИЭР) m/z: 337(М+Н)+.
1Н-ЯМР (ДМСО-d6) δ: 12,60 (1H, уш. с), 8,48 (1H, т, J=5,6 Гц), 8,44 (1H, т, J=5,9 Гц), 8,31 (1Н, д, J=8,8 Гц), 7,97 (3H, уш. с), 7,28-7,16 (5Н, м), 4,58 (1H, м), 3,87 (1H, дд, J=16,8, 5,6 Гц), 3,78 (2Н, д, J=5,9 Гц), 3,67 (1H, дд, J=17,l, 5,4 Гц), 3,56 (2Н, уш. д, J=4,4 Гц), 3,05 (1H, дд, J=13,7, 3,9 Гц), 2,74 (1H, дд, J=13,7, 10,3 Гц).
[0615]
(Этап 2)
N-[4-(11,12-дидегидродибензо[b,f]азоцин-5(6Н)-ил)-4-оксобутаноил]глицилглицил-L-фенилаланилглицин
К раствору соединения, полученного на этапе 1 (2,09 г), в N,N-диметилформамиде (46,4 мл) добавляли триэтиламин (0,804 мл) и 1-{[4-(11,12-дидегидродибензо[b,f]азоцин-5(6Н)-ил)-4-оксобутаноил]окси}пирролидин-2,5-дион (1,87 г) и реакционную смесь перемешивали при комнатной температуре в течение 21 часа. Реакционную смесь концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле [дихлорметан/метанол]. В раствор в дихлорметане полученного в результате этого соединения добавляли диэтиловый эфир, чтобы получить взвесь, и соединение собирали путем фильтрации с получением указанного в заголовке соединения (2,10 г).
МС (ИЭР) m/z: 624(М+Н)+.
1H-ЯМР (ДМСО-d6) δ: 8,20-7,91 (4Н, м), 7,68-7,13 (13Н, м), 4,98 (1H, дд, J=13,9, 3,2 Гц), 4,51-4,46 (1H, м), 3,73-3,47 (7Н, м), 3,00 (1H, дд, J=13,9, 4,1 Гц), 2,73 (1H, т, J=11,7 Гц), 2,67-2,57 (1H, м), 2,29-2,22 (1H, м), 2,06-2,01 (1H, м), 1,80-1,73 (1H, м) (показаны только наблюдаемые пики).
[0616]
(Этап 3)
2,5-диоксопирролидин-1-ил-Т-[4-(11,12-дидегидродибензо[b,f]азоцин-5(6Н)-ил)-4-оксобутаноил]глицилглицил-L-фенилаланилглицинат
К раствору соединения, полученного на этапе 2 (2,10 г), в N,N-диметилформамиде (33,7 мл) добавляли N-гидроксисукцинимид (426 мг) и 1-этил-3-(3-диметиламинопропил)-карбодиимида гидрохлорид (710 мг), и реакционную смесь перемешивали в атмосфере азота при комнатной температуре в течение 16 часов. Реакционную смесь разбавляли дихлорметаном, а затем промывали три раза ледяной водой и сушили над безводным сульфатом натрия. Осушающий агент удаляли путем фильтрации и фильтрат концентрировали при пониженном давлении. Этилацетат добавляли в масляный остаток, чтобы преципитировать твердый продукт. Растворитель отгоняли при пониженном давлении, и диэтиловый эфир добавляли в полученный в результате этого твердый продукт, чтобы получить взвесь, и твердый продукт собирали путем фильтрации с получением указанного в заголовке соединения (2,18 г).
1H-ЯМР (ДМСО-d6) δ: 8,74-8,69 (1Н, м), 8,16-8,08 (2Н, м), 8,00-7,93 (1H, м), 7,71-7,15 (13Н, м), 5,00 (1H, дд, J=13,9, 3,0 Гц), 4,55-4,49 (1H, м), 4,27 (2Н, т, J=6,0 Гц), 3,77-3,68 (1H, м), 3,64-3,50 (4Н, м), 3,02 (1Н, дд, J=13,9, 4,2 Гц), 2,82-2,73 (5Н, м), 2,69-2,58 (1Н, м), 2,33-2,24 (1H, м), 2,10-2,02 (1Н, м), 1,83-1,75 (1H, м).
[0617]
(Этап 4)
Бис(N,N-диэтилэтанаминия) N-[4-(11,12-дидегидродибензо[b,f]азоцин-5(6Н)-ил)-4-оксобутаноил]глицилглицил-L-фенилаланил-N-(2-{9-[(5R,7R,8R,12aR,14R,15R,15aS,16R)-15,16-дигидрокси-2,10-диоксо-2,10-дисульфид-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-7-ил]-6-оксо-6,9-дигидро-1Н-пурин-1-ил}этил)глицинамид
(Лекарственное средство-линкер 1)
К раствору соединения, полученного на этапе 8-2 примера 5 (10,0 мг), в N,N-диметилформамиде (1 мл) добавляли триэтиламин (8 мкл) и полученное на этапе 3 соединение (17,6 мг), и реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Бензиламин (3 мкл) добавляли в реакционную смесь, которую перемешивали при комнатной температуре в течение 1 часа. В реакционную смесь добавляли 10 мМ водный раствор ацетата триэтиламмония и метанол, и реакционную смесь очищали с помощью колоночной хроматографии на силикагеле С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил] и препаративной ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 5%-50% (0 мин-40 мин)] с получением указанного в заголовке соединения (10,9 мг).
МС (ИЭР) m/z: 1379(М+Н)+.
1H-ЯМР (CD3OD) δ: 8,72 (1H, д, J=10,0 Гц), 8,15 (1H, д, J=10,0 Гц), 8,02 (1Н, с), 7,63-7,50 (2Н, м), 7,42-7,37 (3H, м), 7,32-7,13 (8Н, м), 7,12 (1H, с), 6,31 (1H, д, J=6,7 Гц), 6,25 (1H, д, J=8,5 Гц), 5,51-5,40 (2Н, м), 5,09-4,99 (1Н, м), 4,85-4,77 (1H, м), 4,53-4,42 (2Н, м), 4,42-4,15 (5Н, м), 4,04-3,96 (1H, м), 3,92-3,46 (12Н, м), 3,18 (12Н, к, J=7,3 Гц), 3,16-2,73 (5Н, м), 2,40-2,23 (2Н, м), 2,06-1,94 (4Н, м), 1,29 (18Н, т, J=7,3 Гц).
[0618]
Пример 22. Синтез лекарственного средства-линкера 2.
[Схема синтеза]
[0619]
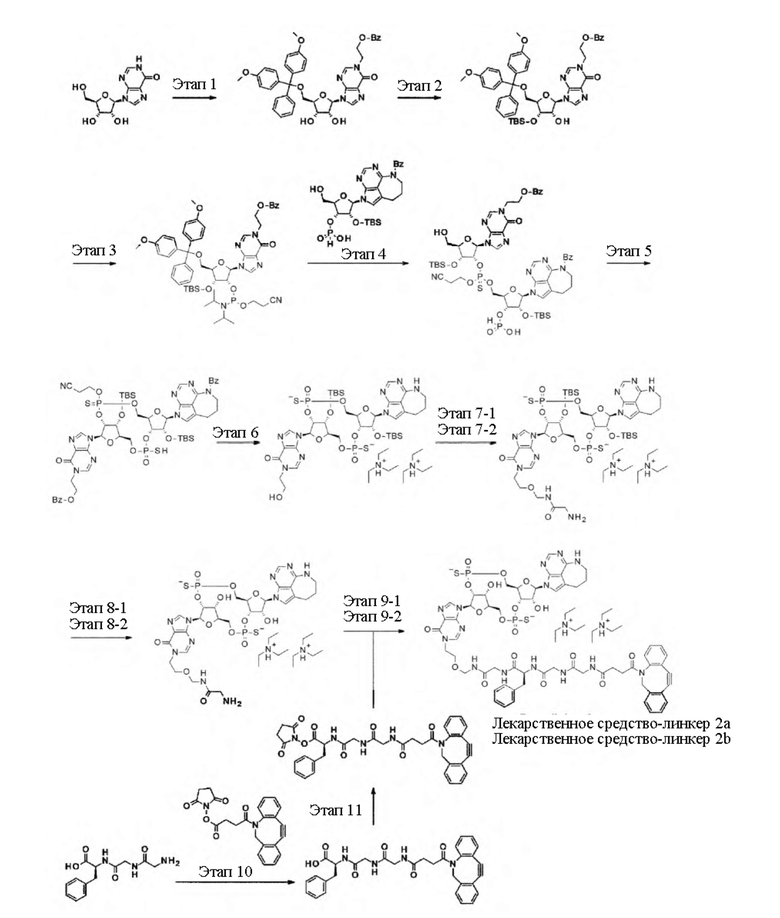
[0620]
(Этап 1)
1-[2-(бензоилокси)этил]-5'-O-[бис(4-метоксифенил)(фенил)метил]инозин
К раствору инозина (10,0 г) в пиридине (50 мл) и N,N-диметилацетамиде (50 мл) добавляли 4,4'-диметокситритилхлорид (15,2 г) при 0°С, и реакционную смесь затем перемешивали при 4°С в течение 64 часов. Метанол (2 мл) добавляли в реакционную смесь, которую перемешивали в течение 10 минут, а затем концентрировали до приблизительно 50 мл. К остатку добавляли 2-бромэтилбензоат (7,02 мл) и 2,3,4,6,7,8,9,10-октагидропиримидо[1,2-а]азепин (13,9 мл), и реакционную смесь перемешивали при комнатной температуре в течение 1 дня. Насыщенный водный раствор гидрокарбоната натрия и воды добавляли в реакционную смесь, которую осуществляли экстрагирование этилацетатом. Органический слой промывали рассолом, а затем сушили над безводным сульфатом натрия. Осушающий агент удаляли путем фильтрации и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле [гексан/этилацетат/метанол/0,1% триэтиламин] с получением указанного в заголовке соединения (15,2 г).
МС (ИЭР) m/z: 719(М+Н)+.
1Н-ЯМР (CDCl3) δ: 8,00 (1H, с), 7,98 (1Н, с), 7,98-7,94 (2Н, м), 7,62-7,15 (12Н, м), 6,80-6,75 (4Н, м), 5,95 (1H, д, Т=5,4 Гц), 4,82-4,79 (1Н, м), 4,72-4,64 (3H, м), 4,55-4,34 (5Н, м), 3,77 (6Н, с), 3,43 (1H, дд, 10,6, 3,9 Гц), 3,34 (1H, дд, J=10,6, 3,6 Гц).
[0621]
(Этап 2)
1-[2-(бензоилокси)этил]-5'-O-[бис(4-метоксифенил)(фенил)метил]-3'-O-[трет-бутил(диметил)силил]инозин
С применением соединения, полученного на этапе 1 (3,01 г), проводили реакцию таким же образом, как на этапе 3 примера 5, с получением указанного в заголовке соединения (1,20 г) и 1-[2-(бензоилокси)этил]-5'-O-[бис(4-метоксифенил)(фенил)метил]-2'-O-[трет-бутил(диметил)силил]инозина (1,22 г) в качестве региоизомера указанного в заголовке соединения.
МС (ИЭР) m/z: 833(М+Н)+.
1Н-ЯМР (CDCl3) δ: 8,03 (1H, с), 7,98-7,96 (1H, м), 7,96 (1H, с), 7,96-7,94 (1H, м), 7,59-7,52 (1H, м), 7,44-7,38 (4Н, м), 7,32-7,15 (7Н, м), 6,83-6,77 (4Н, м), 5,94 (1Н, д, J=4,8 Гц), 4,69-4,63 (2Н, м), 4,59-4,35 (4Н, м), 4,16 (1Н, дд, J=3,8, 1,9 Гц), 3,77 (6Н, д, J=l,8 Гц), 3,47 (1H, дд, J=10,9, 3,0 Гц), 3,27 (1H, дд, J=10,9, 4,2 Гц), 3,00 (1H, д, J=6,7 Гц), 0,87 (9Н, с), 0,06 (3H, с), -0,01 (3H, с).
(2'-O-TBS-форма)
МС (ИЭР) m/z: 833(М+Н)+.
1H-ЯМР (CDCl3) δ: 8,01 (1Н, с), 7,97-7,93 (2Н, м), 7,91 (1H, с), 7,59-7,53 (1Н, м), 7,45-7,38 (4Н, м), 7,35-7,17 (7Н, м), 6,83-6,77 (4Н, м), 5,97 (1H, д, J=6,0 Гц), 4,84 (1H, т, J=5,4 Гц), 4,71-4,60 (2Н, м), 4,52-4,37 (2Н, м), 4,33-4,28 (1H, м), 4,28-4,24 (1H, м), 3,78 (3H, с), 3,77 (3H, с), 3,47 (1H, дд, J=10,9, 3,0 Гц), 3,38 (1H, дд, J=10,9, 3,6 Гц), 2,71 (1H, д, J=3,0 Гц), 0,80 (9Н, с), -0,03 (3H, с), -0,19 (3H, с).
[0622]
(Этап 3)
1-[2-(бензоилокси)этил]-5'-O-[бис(4-метоксифенил)(фенил)метил]-3'-O-[трет-бутил(диметил)силил]-2'-O-{(2-цианоэтокси)[ди(пропан-2-ил)амино]фосфанил}инозин
С применением соединения, полученного на этапе 2 (1,20 г), проводили реакцию таким же образом, как на этапе 4 примера 5, с получением указанного в заголовке соединения (1,41 г) в виде смеси диастереомеров у атома фосфора (соотношение диастереомеров = 0,55:0,45).
МС (ИЭР) m/z: 1033(М+Н)+.
1H-ЯМР (CDCl3) δ: 8,05 (0,45Н, с), 8,04 (0,55Н, с), 7,99-7,95 (2Н, м), 7,95 (0,55Н, с), 7,92 (0,45Н, с), 7,59-7,53 (1H, м), 7,45-7,39 (4Н, м), 7,35-7,10 (7Н, м), 6,83-6,78 (4Н, м), 6,15 (0,55Н, д, J=5,4 Гц), 6,08 (0,45Н, д, J=6,0 Гц), 4,86-4,49 (3H, м), 4,49-4,35 (3H, м), 4,25-4,10 (1H, м), 3,78 (6Н, с), 3,72-3,41 (5Н, м), 3,35-3,25 (1H, м), 2,47 (1H, т, J=6,7 Гц), 2,32 (1H, т, 3=6,3 Гц), 1,33-1,24 (6Н, м), 1,13-1,03 (6Н, м), 0,84 (4,05Н, с), 0,84 (4,95Н, с), 0,08 (1,35Н, с), 0,05 (1,65Н, с), 0,00 (1,35Н, с), -0,01 (1,65Н, с).
[0623]
(Этап 4)
Ту же реакцию, что и на этапе 7 примера 1, проводили в следующем масштабе (сырьевой материал: 1,40 г). С применением раствора в ацетонитриле полученного соединения и соединения, полученного на этапе 3 (1,41 г), проводили реакцию таким же образом, как на этапе 8 примера 1. Полученный в результате этого неочищенный продукт непосредственно использовали для следующей реакции.
[0624]
(Этап 5)
2-{9-[(5R,7R,8R,12aR,14R,15R,15aR,16R)-14-(6-бензоил-6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)-15,16-бис{[трет-бутил(диметил)силил]окси}-10-(2-цианоэтокси)-2-оксо-2-сульфанил-10-сульфанилиденоктагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-7-ил]-6-оксо-6,9-дигидро-1H-пурин-1-ил}этилбензоат
С применением неочищенного продукта, полученного на этапе 4, проводили реакцию таким же образом, как на этапе 9 примера 1, с получением указанного в заголовке соединения (778 мг) в виде смеси диастереомеров у атома фосфора.
МС (ИЭР) m/z: 1264(М+Н)+.
[0625]
(Этап 6)
Бис(N,N-диэтилэтанаминия) (5R,7R,8R,12aR,14R,15R,15aR,16R)-15,16-бис {[трет-бутил(диметил)силил]окси}-7-[1-(2-гидроксиэтил)-6-оксо-1,6-дигидро-9Н-пурин-9-ил]-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
С применением соединения, полученного на этапе 5 (778 мг), проводили реакцию таким же образом, как на этапе 10 примера 1, с получением диастереомера 1 (255 мг) и диастереомера 2 (с примесями) указанного в заголовке соединения. Диастереомер 2 снова очищали с помощью препаративной ВЭЖХ [вода/ацетонитрил с 0,2% триэтиламина, ацетонитрил с 0,2% триэтиламина: 5%-50% (0 мин-40 мин)] с получением диастереомера 2 (94,6 мг) указанного в заголовке соединения.
Диастереомер 1 (менее полярный)
МС (ИЭР) m/z: 1003(М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,66 (1H, с), 8,21 (1H, с), 8,04 (1Н, с), 7,33 (1H, с), 6,27 (1H, д, J=5,1 Гц), 6.25 (1H, д, 3=3,6 Гц), 5,39-5,29 (1H, м), 5,18-5,11 (1H, м), 4,85-4,81 (1H, м), 4,79-4,74 (1H, м), 4,71-4,66 (1H, м), 4,50-4,42 (1Н, м), 4,36-4,21 (2Н, м), 4,09-3,98 (2Н, м), 3,85-3,78 (2Н, м), 3,78-3,69 (2Н, м), 3,55-3,46 (2Н, м), 3,17 (12Н, к, J=7,3 Гц), 2,98-2,75 (2Н, м), 2,05-1,88 (2Н, м), 1,28 (18Н, т, J=7,3 Гц), 0,98 (9Н, с), 0,85 (9Н, с), 0,31 (3H, с), 0,27 (3H, с), 0,25 (3H, с), 0,09 (3H, с).
Диастереомер 2 (более полярный)
МС (ИЭР) m/z: 1003(М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,50 (1H, с), 8,22 (1H, с), 8,07 (1H, с), 7,20 (1H, с), 6,33 (1H, д, J=7,3 Гц), 6.26 (1H, д, 3=9,1 Гц), 5,59-5,44 (1H, м), 5,38-5,32 (1H, м), 5,21-5,11 (1H, м), 4,99-4,89 (2Н, м), 4,68-4,54 (2Н, м), 4,25-4,12 (3H, м), 4,09-4,03 (1H, м), 3,90-3,80 (3H, м), 3,59-3,51 (2Н, м), 3,20 (12Н, к, J=7,3 Гц), 2,96-2,89 (2Н, м), 2,07-1,98 (2Н, м), 1,30 (18Н, т, J=7,3 Гц), 0,99 (9Н, с), 0,74 (9Н, с), 0,27 (3H, с), 0,27 (3H, с), 0,20 (3H, с), -0,05 (3H, с).
[0626]
(Этап 7-1)
Бис(N,N-диэтилэтанаминия) (5R,7R,8R,12aR,14R,15R,15aR,16R)-15,16-бис {[трет-бутил(диметил)силил]окси}-7-(1-{2-[(глициламино)метокси]этил}-6-оксо-1,6-дигидро-9Н-пурин-9-ил)-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
(Диастереомер 1)
К раствору соединения, полученного на этапе 6 (диастереомер 1) (30 мг), в тетрагидрофуране (0,5 мл) добавляли [(N-{[(9Н-флуорен-9-ил)метокси]карбонил}глицил)амино]метилацетат (91,7 мг) и моногидрат п-толуолсульфоновой кислоты (11,8 мг), и реакционную смесь перемешивали при комнатной температуре в течение 6 часов. В реакционную смесь добавляли N,N-диметилформамид (0,5 мл) и 1,8-диазабицикло[5,4,0]-7-ундецен(56 мкл), и реакционную смесь перемешивали при комнатной температуре в течение 3 часов. В реакционную смесь добавляли 10 мМ водный раствор ацетата триэтиламмония, и реакционную смесь очищали с помощью колоночной хроматографии на силикагеле С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил] с получением указанного в заголовке соединения (25,6 мг), содержащего сырьевой материал в качестве примеси.
MC (ИЭР) m/z: 1089(М+Н)+.
[0627] (Этап 7-2)
Бис(N,N-диэтилэтанаминия) (5R,7R,8R,12aR,14R,15R,15aR,16R)-15,16-бис{[трет-бутил(диметил)силил]окси}-7-(1-{2-[(глициламино)метокси]этил}-6-оксо-1,6-дигидро-9Н-пурин-9-ил)-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1] [1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят) (Диастереомер 2)
С применением соединения, полученного на этапе 6 (диастереомер 2) (84,6 мг), проводили реакцию таким же образом, как на этапе 7-1, с получением указанного в заголовке соединения (70,9 мг), содержащего сырьевой материал в качестве примеси. MC (ИЭР) m/z: 1089 (М+Н)+.
[0628] (Этап 8-1)
Бис(N,N-диэтилэтанаминия) (5R,7R,8R,12aR,14R,15R,15aS,16R)-7-(1-{2-[(глициламино)метокси]этил}-6-оксо-1,6-дигидро-9H-пурин-9-ил)-15,16-дигидроксио-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят), диастереомер 1
К соединению, полученному на этапе 7-1 (25,6 мг), добавляли тригидрофторид триэтиламина (2 мл), и реакционную смесь перемешивали при 45°С в течение 3 часов. В реакционную смесь добавляли охлажденную до температуры льда смесь 1 М раствора гидрокарбоната триэтиламмония (10 мл) и триэтиламина (2 мл) при комнатной температуре. Реакционную смесь концентрировали при пониженном давлении, а затем очищали с помощью колоночной хроматографии на силикагеле С18 (10 мМ водный раствор ацетата триэтиламмония/ацетонитрил) с получением указанного в заголовке соединения (16,6 мг: с примесью, происходящей из сырьевого материала с этапа 7-1).
МС (ИЭР) m/z: 861 (М+Н)+.
[0629] (Этап 8-2)
Бис(N,N-диэтилэтанаминия) (5R,7R,8R,12aR,14R,15R,15aS,16R)-7-(1-{2-[(глициламино)метокси]этил}-6-оксо-1,6-дигидро-9Н-пурин-9-ил)-15,16-дигидрокси-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят) (Диастереомер 2)
С применением соединения, полученного на этапе 7-2 (70,9 мг), проводили реакцию таким же образом, как на этапе 8-1, с получением указанного в заголовке соединения (51,7 мг: с примесью, происходящей из сырьевого материала с этапа 7-2).
МС (ИЭР) m/z: 861 (М+Н)+.
[0630] (Этап 9-1)
Бис(N,N-диэтилэтанаминия) N-[4-(11,12-дидегидродибензо[b,f]азоцин-5(6Н)-ил)-4-оксобутаноил]глицилглицил-L-фенилаланил-N-[(2-{9-[(5R,7R,8R,12aR,14R,15R,15aS,16R)-15,16-дигидрокси-2,10-диоксо-2,10-дисульфид-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1] [1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-7-ил]-6-оксо-6,9-дигидро-1Н-пурин-1-ил}этокси)метил]глицинамид (Лекарственное средство-линкер 2а: диастереомер 1)
К раствору соединения, полученного на этапе 8-1 (16,6 мг), в N,N-диметилформамиде (0,5 мл) добавляли триэтиламин (6 мкл) и соединение, полученное на этапе 11, описанное далее (15,5 мг), и реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Бензиламин (3 мкл) добавляли в реакционную смесь, которую перемешивали при комнатной температуре в течение 1 часа. Туда добавляли 10 мМ водный раствор ацетата триэтиламмония и метанол, и реакционную смесь очищали с помощью колоночной хроматографии на силикагеле С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил] и препаративной ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 10% - 45% (0 мин - 30 мин)] с получением указанного в заголовке соединения (5,1 мг).
MC (ИЭР) m/z: 1409 (М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,66-8,60 (1H, м), 8,17 (1H, с), 8,02 (1H, с), 7,65-7,48 (2Н, м), 7,43-7,36 (3Н, м), 7,31-7,13 (8Н, м), 7,11 (1Н, с), 6,30-6,21 (2Н, м), 5,46-5,37 (1H, м), 5,23-5,16 (1H, м), 5,08-4,99 (1H, м), 4,86-4,81 (1Н, м), 4,80-4,75 (1H, м), 4,70-4,40 (7Н, м), 4,40-4,20 (3Н, м), 4,10-3,97 (3Н, м), 3,86-3,58 (8Н, м), 3,51-3,43 (3Н, м), 3,18 (12Н, к, J=7,3 Гц), 3,01-2,93 (1H, м), 2,85-2,72 (3Н, м), 2,37-2,15 (2Н, м), 2,01-1,93 (2Н, м), 1,29 (18Н, т, J=7,3 Гц) (показаны только наблюдаемые пики).
[0631] (Этап 9-2)
Бис(N,N-диэтилэтанаминия) N-[4-(11,12-дидегидродибензо[b,f]азоцин-5(6Н)-ил)-4-оксобутаноил]глицилглицил-L-фенилаланил-N-[(2-{9-[(5R,7R,8R,12aR,14R,15R,15aS,16R)-15,16-дигидрокси-2,10-диоксо-2,10-дисульфид-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[сс1]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1] [1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-7-ил]-6-оксо-6,9-дигидро-1Н-пурин-1-ил}этокси)метил]глицинамид (Лекарственное средство-линкер 2b: диастереомер 2)
С применением соединения, полученного на этапе 8-2 (51,7 мг), проводили реакцию таким же образом, как на этапе 9-1, а затем проводили очистку при следующих [Условиях очистки] с получением указанного в заголовке соединения (33,7 мг).
[Условия очистки] Хроматография на колонке с силикагелем С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил], препаративная ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 10% - 50% (0 мин - 30 мин)]. MC(ИЭР)m/z: 1409(М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,73 (1H, д, J=6,7 Гц), 8,19 (1H, д, J=3,0 Гц), 8,02 (1Н, с), 7,66-7,50 (2Н, м), 7,43-7,37 (3Н, м), 7,33-7,13 (8Н, м), 7,11 (1Н, с), 6,33-6,23 (2Н, м), 5,51-5,38 (2Н, м), 5,04 (1H, т, J=13,6 Гц), 4,83-4,77 (1Н, м), 4,64-4,55 (2Н, м), 4,52-4,26 (6Н, м), 4,25-3,97 (2Н, м), 3,93-3,45 (13Н, м), 3,19 (12Н, к, J=7,3 Гц), 3,17-3,11 (1H, м), 3,02-2,92 (1H, м), 2,91-2,73 (3Н, м), 2,40-2,24 (2Н, м), 2,07-1,95 (3Н, м), 1,30 (18Н, т, J=7,3 Гц).
[0632] (Этап 10)
N-[4-(11,12-дидегидродибензо[b,f]азоцин-5(6Н)-ил)-4-оксобутаноил]глицилглицил-L-фенилаланин
К раствору доступной для приобретения (Bachem Holding AG) (2S)-2-[[2-[(2-аминоацетил)амино]ацетил]амино]-3-фенилпропионовой кислоты (2,86 г) в N,N-диметилформамиде (51,2 мл) добавляли триэтиламин (2,56 мл) и доступный для приобретения (Click Chemistry Tools) 1-{[4-(11,12-дидегидродибензо[b,f]азоцин-5(6Н)-ил)-4-оксобутаноил]окси}пирролидин-2,5-дион (3,69 г), и реакционную смесь перемешивали при комнатной температуре в течение 24 часов. Водный раствор (500 мл) моногидрата лимонной кислоты (24,0 г) добавляли в реакционную смесь, которую осуществляли экстрагирование этилацетатом. После высушивания органического слоя над безводным сульфатом натрия осушающий агент удаляли путем фильтрации и фильтрат концентрировали при пониженном давлении. Остаток растворяли в смешанном растворе этилацетата/ацетонитрила, а затем преципитировали диизопропиловым эфиром и собирали путем фильтрации с получением указанного в заголовке соединения (4,30 г).
1H-ЯМР (ДМСО-d6) δ: 12,8 (1H, уш. с), 8,15-7,95 (3Н, м), 7,68-7,17 (13Н, м), 5,01 (1H, д, J=14,2 Гц), 4,41-4,37 (1H, м), 3,74-3,57 (5Н, м), 3,05-3,01 (1H, м), 2,87 (1H, дд, J=14,2, 9,3 Гц), 2,68-2,59 (1H, м), 2,32-2,25 (1H, м), 2,09-2,03 (1H, м), 1,82-1,76 (1H, м).
[0633] (Этап 11)
2,5-диоксопирролидин-1-ил-N-[4-(11,12-дидегидродибензо[b,f]азоцин-5(6Н)-ил)-4-оксобутаноил]глицилглицил-L-фенилаланинат
К раствору соединения, полученного на этапе 10 (2,10 г), в N,N-диметилформамиде (75,9 мл) добавляли N-гидроксисукцинимид (961 мг) и 1-этил-3-(3-диметиламинопропил)карбодиимида гидрохлорид (1,60 г) и реакционную смесь перемешивали в атмосфере азота при комнатной температуре в течение 21 часа. Реакционную смесь разбавляли дихлорметаном, промывали три раза ледяной водой, а затем сушили над безводным сульфатом натрия. Осушающий агент удаляли путем фильтрации и фильтрат концентрировали при пониженном давлении. Толуол добавляли к остатку, и продукт снова концентрировали при пониженном давлении. Остаток растворяли в ацетонитриле, и продукт очищали с помощью колоночной хроматографии на силикагеле С18 [ацетонитрил: 100%]. Фракцию, содержащую целевой продукт, концентрировали при пониженном давлении, и после этого диизопропиловый эфир добавляли к остатку, чтобы получить взвесь. Полученный твердый продукт собирали путем фильтрации с получением указанного в заголовке соединения (2,59 г).
1Н-ЯМР (ДМСО-d6) δ: 8,58-8,51 (1Н,м), 8,17-8,00 (2Н, м), 7,66-7,20 (13Н,м), 5,02-4,98 (1Н, м), 4,90-4,85 (1Н, м), 3,78-3,57 (5Н, м), 3,24-3,19 (1Н, м), 3,06-3,00 (1Н, м), 2,82 (4Н, уш. с), 2,67-2,58 (1Н, м), 2,32-2,23 (1Н, м), 2,09-2,02 (1Н, м), 1,82-1,75 (1Н, м).
[0634]
Пример 23. Синтез лекарственного средства-линкера 3.
[Схема синтеза]
[0635]
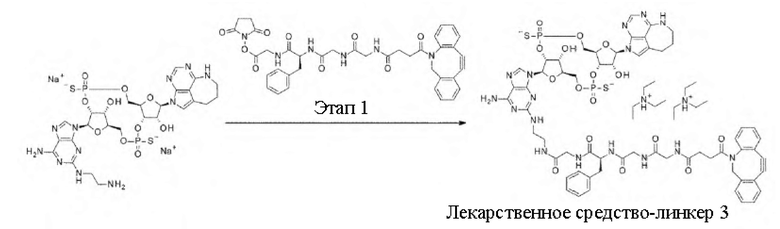
[0636] (Этап 1)
Бис(N,N-диэтилэтанаминия) N-[4-(11,12-дидегидродибензо[b,f]азоцин-5(6Н)-ил)-4-оксобутаноил]глицилглицил-L-фенилаланил-N-[2-({6-амино-9-[(5R,7R,8R,12aR,14R,15R,15aS,16R)-15,16-дигидрокси-2,10-диоксо-2,10-дисульфид-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-7-ил]-9Н-пурин-2-ил}амино)этил]глицинамид
(Лекарственное средство-линкер 3)
К раствору соединения, полученного на этапе 8-2 примера 8 (4,8 мг), в N,N-диметилформамиде (0,29 мл) добавляли триэтиламин (1,9 мкл) и соединение, полученное на этапе 3 примера 21 (5,2 мг), и реакционную смесь перемешивали в атмосфере азота при комнатной температуре в течение 1,5 часов. После добавления туда бензиламина (3,2 мкл), чтобы погасить реакцию, продукт очищали с помощью препаративной ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 20% - 40% (0 мин - 30 мин)] с получением указанного в заголовке соединения (8,0 мг). MC(ИЭР)m/z: 1393(М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,35 (1H, уш. с), 8,02 (1H, с), 7,61-7,15 (14Н, м), 6,33 (1H, дд, J=6,0, 3,0 Гц), 6,15-6,09 (1H, м), 5,49-5,37 (2Н, м), 5,05 (1H, дд, J=13,9, 12,1 Гц), 4,85-4,79 (1H, м), 4,53-4,21 (6Н, м), 4,06-3,61 (8Н, м), 3,51-3,13 (6Н, м), 3,16 (12Н, к, J=7,3 Гц), 3,06-2,65 (7Н, м), 2,35-1,94 (4Н, м), 1,28 (18Н, т, J=7,6 Гц).
[0637]
Пример 24. Синтез лекарственного средства-линкера 4.
[Схема синтеза]
[0638]
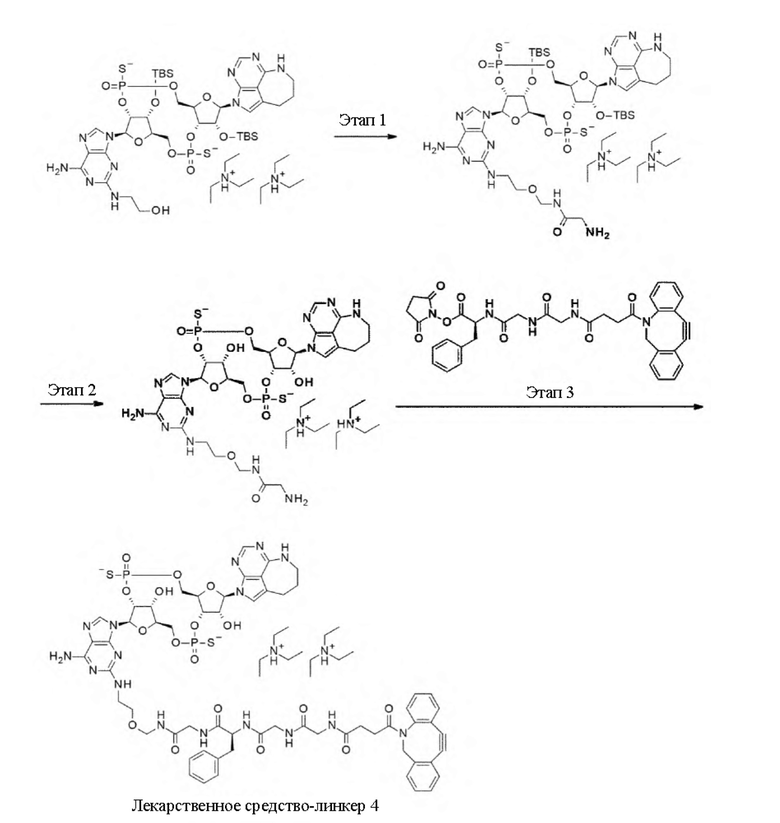
[0639] (Этап 1)
Бис(N,N-диэтилэтанаминия) (5R,7R,8R,12aR,14R,15R,15aR,16R)-7-[6-амино-2-({2-[(глициламино)метокси]этил}амино)-9Н-пурин-9-ил]-15,16-бис{[трет-бутил(диметил)силил]окси}-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабешо[cd]азулен-2-ил)октагадро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
С применением соединения, полученного на этапе 1-2 примера 9 (17,6 мг), проводили реакцию таким же образом, как на этапе 7-1 примера 22, с получением указанного в заголовке соединения (8,3 мг). МС (ИЭР) m/z: 1103(М+Н)+.
[0640] (Этап 2)
Бис(N,N-диэтилэтанаминия) (5R,7R,8R,12aR,14R,15R,15aS,16R)-7-[6-амино-2-({2-[(глициламино)метокси]этил}амино)-9Н-пурин-9-ил]-15,16-дигидрокси-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[сс1]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
С применением соединения, полученного на этапе 1 (10,7 мг), проводили реакцию таким же образом, как на этапе 8-1 примера 22, с получением указанного в заголовке соединения (7,6 мг).
МС (ИЭР) m/z: 875(М+Н)+.
(Этап 3)
Бис(N,N-диэтилэтанаминия) N-[4-(11,12-дидегидродибензо[b,f]азоцин-5(6Н)-ил)-4-оксобутаноил]глицилглицил-L-фенилаланил-N-{[2-({6-амино-9-[(5R,7R,8R,12aR,14R,15R,15aS,16R)-15,16-дигидрокси-2,10-диоксо-2,10-дисульфид-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-7-ил]-9Н-пурин-2-ил}амино)этокси]метил}глицинамид
(Лекарственное средство-линкер 4)
С применением соединения, полученного на этапе 2 (7,6 мг), проводили реакцию таким же образом, как на этапе 9-1 примера 22. Очистку проводили при следующих [Условиях очистки] с получением указанного в заголовке соединения (7,6 мг) в виде соли триэтиламина.
[Условия очистки] препаративная ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 20% - 40% (0 мин - 30 мин)].
MC (ИЭР) m/z: 1423(M+H)+.
1Н-ЯМР (CD3OD) δ: 8,37 (1Н, уш. с), 8,01 (1Н, д, J=2,4 Гц), 7,63-7,11 (14Н, м), 6,33 (1Н, д, J=6,7 Гц), 6,17 (1Н, д, J=7,3 Гц), 5,51-5,36 (2Н, м), 5,09-5,03 (1Н, м), 4,84-4,80 (1Н, м), 4,63-4,25 (8Н, м), 4,07-3,58 (9Н, м), 3,50-3,41 (4Н, м), 3,28-2,72 (8Н, м), 3,18 (12Н, к, J=7,3 Гц), 2,45-1,96 (4Н, м), 1,29 (18Н, т, J=7,3 Гц).
[0641]
Пример 25. Синтез ремоделированного гликаном антитела 1.
Синтез модифицированного антитела против HER2-[SG-(N3)2]2
[Схема синтеза]
[0642]
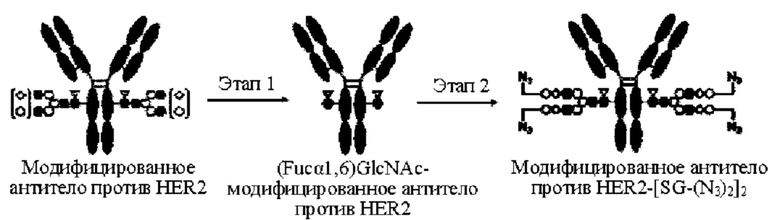
[0643]
(Этап 1)
Получение (Fuca1,6)ClcNAc-модифицированного антитела против HER2.
В фосфатно-солевой буферный раствор модифицированного антитела против HER2, полученный в соответствии со ссылочным примером 1 (20 мл, 12,6 мг/мл, рН 6,0), добавляли фосфатно-солевой буферный раствор EndoS дикого типа (0,147 мл, 7,70 мг/мл, рН 6,0), и реакционную смесь встряхивали при 37°С в течение 2 часов и 15 минут. Степень прохождения реакции проверяли, применяя станцию для электрофореза Experion (производства Bio-Rad Laboratories, Inc.). После завершения реакции проводили очистку с помощью аффинной хроматографии и очистку с помощью колоночной хроматографии на гидроксиапатите в соответствии со следующими способами. (1) Очистка с помощью аффинной хроматографии. Устройство для очистки: АКТА avant 25 (производства GE Healthcare) Колонка: HiTrap rProtein A FF (5 мл) (производства GE Healthcare) Скорость потока: 5 мл/мин (1,25 мл/мин при загрузке)
Реакционную смесь, полученную как описано выше, очищали двумя отдельными процедурами. При подсоединении к колонке реакционную смесь добавляли в колонку, и 2 объема колонки буфера для связывания (20 мМ фосфатного буфера (рН 6,0)) пропускали при 1,25 мл/мин и 5 объемов колонки указанного буфера дополнительно пропускали при 5 мл/мин. При промежуточной промывке пропускали 15 объемов колонки раствора для промывки (20 мМ фосфатный буфер (рН 7,0), 0,5 М раствор хлорида натрия). При элюировании пропускали 6 объемов колонки элюирующего буфера (ImmunoPure IgG Elution buffer, производства Pierce). Элюат незамедлительно нейтрализовали 1 М Трис-буфером (рН 9,0). Во фракциях, содержащих целевой продукт, осуществляли замену буфера на 5 мМ раствор фосфатного буфера/50 мМ 2-морфолиноэтансульфоновой кислоты (MES) (рН 6,8) в соответствии со способом, описанным в Общей процедуре С. Концентрацию антитела в полученном буферном растворе измеряли в соответствии со способом, описанным в Общей процедуре В, таким образом, получая частично очищенный раствор указанного в заголовке антитела (26,57 мг/мл, 9,0 мл). (2) Очистка с помощью хроматографии на гидроксиапатите Устройство очистки: АКТА avant 25 (производства GE Healthcare) Колонка: Bio-Scale Mini СНТ, картридж I типа (5 мл) (производства Bio-Rad Laboratories, Inc.) Скорость потока: 5 мл/мин (1,25 мл/мин при загрузке)
Раствор, полученный в (1), добавляли в колонку и 2 объема колонки раствора А (5 мМ фосфатный буфер, 50 мМ раствор MES (рН 6,8)) пропускали при 1,25 мл/мин и 3 объема колонки того же раствора дополнительно пропускали при 5 мл/мин. После этого проводили элюирование раствором А и раствором В (5 мМ фосфатный буфер/50 мМ раствор MES (рН 6,8), 2 М раствор хлорида натрия). Условия элюирования были следующими: раствор А:раствор В = от 100:0 до 0:100 (5 объемов колонки). Далее, пропускали 5 объемов колонки раствора для промывки (500 мМ фосфатный буфер (рН 6,5)). Во фракциях, содержащих целевой продукт, осуществляли замену буфера на 20 мМ фосфатный буфер (рН 6,0) в соответствии со способом, описанным в Общей процедуре С. Концентрацию антитела в полученном буферном растворе измеряли в соответствии со способом, описанным в Общей процедуре В, таким образом, получая раствор указанного в заголовке антитела (17,29 мг/мл, приблизительно 13 мл).
[0644] (Этап 2)
Получение модифицированного антитела против HER2-[SG-(N3)2]2
К антителу, полученному на этапе 1, в 20 мМ фосфатном буферном растворе (17,29 мг/мл, 13 мл, рН 6,0) добавляли [N3-PEG(3)]2-SG(10)Ox (соединение 1-10 в WO 2018/003983) (52 мг) в 20 мМ фосфатном буферном растворе (рН 6,0) (3,0 мл + 1,0 мл для промывки) и фосфатно-солевой буферный раствор EndoS (D233Q/Q303L) (0,698 мл, 5,8 мг/мл, рН 6,0), и реакционную смесь встряхивали при 30°С в течение 4 часов. Реакционную смесь хранили при -80°С в течение 15 часов, а затем размораживали при 30°С, и дополнительно добавляли [N3-PEG(3)]2-SG(10)Ox (7,4 мг) и фосфатно-солевой буферный раствор EndoS (D233Q/Q303L) (0,155 мл, 5,8 мг/мл, рН 6,0) в реакционную смесь, которую встряхивали при 30°С в течение 2 часов. Степень прохождения реакции проверяли, применяя станцию для электрофореза Experion (производства Bio-Rad Laboratories, Inc.). После завершения реакции проводили очистку с помощью аффинной хроматографии и очистку с помощью хроматографии на гидроксиапатите, как на этапе 1. Фракции, содержащие целевой продукт (всего семь фракций), разделяли на четыре первые фракции и три последние фракции, и в каждой фракции осуществляли замену буфера на фосфатно-солевой буферный раствор (рН 6,0) в соответствии со способом, описанным в Общей процедуре С.Концентрацию антитела в каждом полученном буферном растворе измеряли в соответствии со способом, описанным в Общей процедуре В, таким образом, получая раствор указанного в заголовке антитела (четыре первые фракции: 14,99 мг/мл, 10 мл) и раствор указанного в заголовке антитела (три последние фракции: 10,97 мг/мл, 6,2 мл).
[0645]
Пример 26. Синтез ремоделированного гликаном антитела 2.
Получение модифицированного антитела против LPS-[SG-(N3)2]2
[Схема синтеза]
[0646]
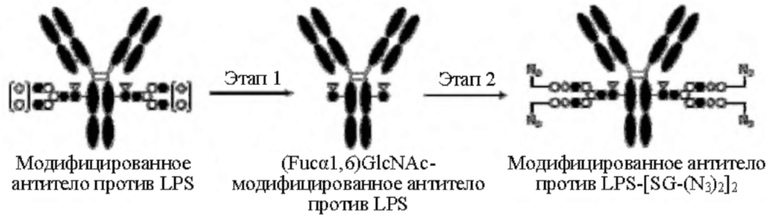
[0647] (Этап 1)
Получение (Fuca1,6)ClcNAc-модифицированного антитела против LPS.
С применением фосфатно-солевого буферного раствора модифицированного антитела против LPS, полученного в соответствии со ссылочным примером 2 (8,5 мл, 10,96 мг/мл, рН 6,0), проводили те же процедуры, как на этапе 1 примера 25, с получением указанного в заголовке антитела в 20 мМ фосфатном буферном растворе (11,70 мг/мл, 7,5 мл, рН 6,0).
[0648] (Этап 2)
Получение модифицированного антитела против LPS-[SG-(N3)2]2
С применением антитела в 20 мМ фосфатном буферном растворе, полученного на этапе 1 (11,70 мг/мл, 7,5 мл, рН 6,0), и [N3-PEG(3)]2-SG(10)Ox (20,3 мг), проводили те же процедуры, как на этапе 2 примера 25, с получением фосфатно-солевого буферного раствора указанного в заголовке антитела (10,55 мг/мл, 7,5 мл, рН 6,0).
[0649]
Пример 27. Синтез конъюгата 1 антитела с лекарственным средством (синтез конъюгата 1 антитела против HER2-ЦДН).
Фосфатно-солевой буферный раствор ремоделированного гликаном антитела 1 (рН 6,0) (10,97 мг/мл, 0,500 мл) разбавляли пропиленгликолем (0,250 мл). В данный раствор добавляли смесь раствора в диметилсульфоксиде лекарственного средства-линкера 3 (10 мМ, 0,0907 мл, 24 эквивалента на молекулу антитела) и пропиленгликоля (0,159 мл), и проводили реакцию полученной смеси во вращающемся устройстве для пробирок (MTR-103, AS ONE Corporation) при комнатной температуре в течение 2 суток. Реакционную смесь очищали в соответствии со способом, описанным в Общей процедуре D, с получением раствора конъюгата нацеленного антитела с лекарственным средством в ABS (10 мМ ацетатный буфер, 5% сорбит, рН 5,5) (3,5 мл).
Анализ проводили в соответствии со способами, описанными в Общих процедурах Е и G, с получением следующих результатов. Концентрация антитела: 0,97 мг/мл Выход антитела: 3,41 мг (62%)
Среднее количество конъюгированных молекул лекарственного средства: 3,6
[0650]
Пример 28. Синтез конъюгата 2 антитела с лекарственным средством (синтез конъюгата 2 антитела против HER2-ЦДН).
Фосфатно-солевой буферный раствор ремоделированного гликаном антитела 1 (рН 6,0) (10,97 мг/мл, 0,500 мл) разбавляли пропиленгликолем (0,250 мл). В данный раствор добавляли смесь раствора в диметилсульфоксиде лекарственного средства-линкера 4 (10 мМ, 0,0907 мл, 24 эквивалента на молекулу антитела) и пропиленгликоля (0,159 мл) и проводили реакцию полученной смеси во вращающемся устройстве для пробирок (MTR-103, AS ONE Corporation) при комнатной температуре в течение 2 суток. Реакционную смесь очищали в соответствии со способом, описанным в Общей процедуре D, с получением раствора конъюгата нацеленного антитела с лекарственным средством в ABS (3,5 мл).
Анализ проводили в соответствии со способами, описанными в Общих процедурах Е и G, с получением следующих результатов.
Концентрация антитела: 1,08 мг/мл
Выход антитела: 3,78 мг (69%)
Среднее количество конъюгированных молекул лекарственного средства: 3,2
[0651]
Пример 29. Синтез конъюгата 3 антитела с лекарственным средством (синтез конъюгата 3 антитела против HER2-ЦДН).
Фосфатно-солевой буферный раствор ремоделированного гликаном антитела 1 (рН 6,0) (10,97 мг/мл, 0,500 мл) разбавляли пропиленгликолем (0,250 мл). В данный раствор добавляли смесь раствора в диметилсульфоксиде лекарственного средства-линкер а 2а (10 мМ, 0,0907 мл, 24 эквивалента на молекулу антитела) и пропиленгликоля (0,159 мл), и проводили реакцию полученной смеси во вращающемся устройстве для пробирок (MTR-103, AS ONE Corporation) при комнатной температуре в течение 47 часов. Реакционную смесь очищали в соответствии со способом, описанным в Общей процедуре D, с получением раствора конъюгата нацеленного антитела с лекарственным средством в ABS (3,5 мл).
Анализ проводили в соответствии со способами, описанными в Общих процедурах Е и G, с получением следующих результатов.
Концентрация антитела: 0,91 мг/мл
Выход антитела: 3,17 мг (58%)
Среднее количество конъюгированных молекул лекарственного средства: 3,6
[0652]
Пример 30. Синтез конъюгата 4 антитела с лекарственным средством (синтез конъюгата 1 антитела против LPS-ЦДН).
Фосфатно-солевой буферный раствор ремоделированного гликаном антитела 2 (рН 6,0) (10,55 мг/мл, 1,00 мл) разбавляли пропиленгликолем (0,500 мл). В данный раствор добавляли смесь раствора в диметилсульфоксиде лекарственного средства-линкера 3 (10 мМ, 0,174 мл, 24 эквивалента на молекулу антитела) и пропиленгликоля (0,326 мл) и проводили реакцию полученной смеси во вращающемся устройстве для пробирок (MTR-103, AS ONE Corporation) при комнатной температуре в течение 2 суток. Реакционную смесь очищали в соответствии со способом, описанным в Общей процедуре D, с получением раствора конъюгата нацеленного антитела с лекарственным средством в ABS (6,5 мл).
Анализ проводили в соответствии со способами, описанными в Общих процедурах Е и G, с получением следующих результатов.
Концентрация антитела: 1,11 мг/мл
Выход антитела: 7,23 мг (69%)
Среднее количество конъюгированных молекул лекарственного средства: 3,9
[0653]
Пример 31. Синтез ЦДН21.
(5R,7R,8R,12aR,14R,15R,15а8,16R)-7-(6-амино-9Н-пурин-9-ил)-15,16-дигидрокси-2,10-бис(сульфанил)-14-(7,8,9,10-тетрагидро-2Н-6-окса-2,3,5-триазациклоокта[1,2,3-cd]инден-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-дион
[0654]
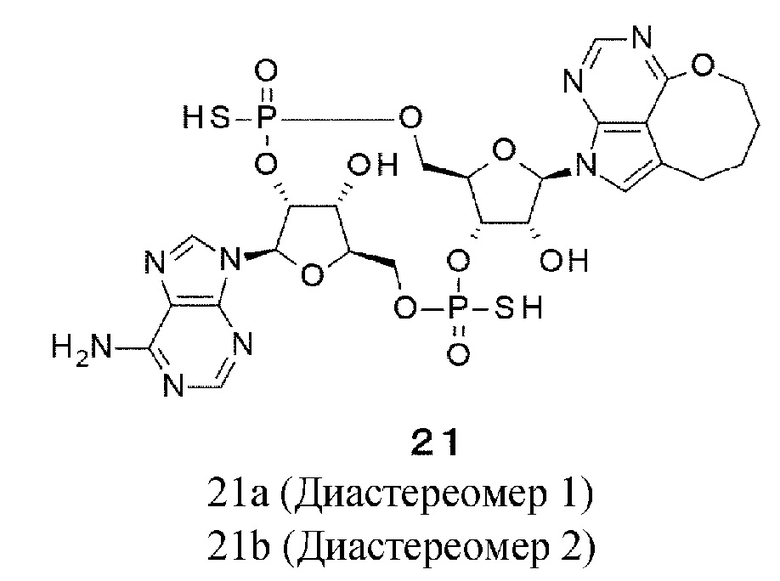
[0655]
[Схема синтеза]
[0656]
[0657]
(Этап 1)
4-(бензилокси)-7-{2-O-[трет-бутил(диметил)силил]-3,5-O-(ди-трет-бутилсилилиден)-β-D-рибофуранозил}-5-(4-гидроксибут-1-ин-1-ил)-7Н-пирроло[2,3-d]пиримидин
С применением соединения, полученного на этапе 2 примера 20 (3,37 г), и 3-бутин-1-ола (1,72 мл) проводили реакцию таким же образом, как на этапе 2 примера 1, за исключением того, что температуру реакции устанавливали на уровне комнатной температуры, с получением указанного в заголовке соединения (2,74 г).
1Н-ЯМР (CDCl3) δ: 8,44 (1H, с), 7,58-7,53 (2Н, м), 7,42-7,31 (3Н, м), 7,16 (1H, с), 6,16 (1H, с), 5,61 (2Н, дд, J=14,9, 12,5 Гц), 4,47 (1Н, дд, J=9,2, 4,9 Гц), 4,42 (1Н, д, J=4,7 Гц), 4,26 (1Н, дд, J=9,4, 4,7 Гц), 4,17 (1H, тд, J=9,9, 4,8 Гц), 4,01 (1Н, т, J=9,6 Гц), 3,71-3,64 (2Н, м), 2,65 (2Н, т, J=6,l Гц), 1,87 (1H, т, J=6,5 Гц), 1,08 (9Н, с), 1,04 (9Н, с), 0,91 (9Н, с), 0,11 (3Н, с), 0,10 (3Н, с).
[0658] (Этап 2)
7-{2-O-[трет-бутил(диметил)силил]-3,5-O-(ди-трет-бутилсилилиден)-β-D-рибофуранозил}-5-(4-гидроксибутил)-3,7-дигидро-4Н-пирроло[2,3-d]пиримидин-4-он
С применением соединения, полученного на этапе 1 (2,74 г), проводили реакцию таким же образом, как на этапе 7 примера 19, за исключением того, что растворитель реакции заменяли на смешанный растворитель метанол (30 мл)/тетрагидрофуран (30 мл), с получением указанного в заголовке соединения (2,21 г).
1H-ЯМР (CDCl3) δ: 11,55 (1H, уш. с), 7,90 (1H, с), 6,63 (1H, с), 6,09 (1H, с), 4,47 (1Н, дд, J=9,0, 5,1 Гц), 4,38 (1H, д, J=4,7 Гц), 4,24 (1H, дд, J=9,4, 4,7 Гц), 4,19-4,11 (1H, м), 4,01 (1H, т, J=9,8 Гц), 3,82-3,74 (2Н, м), 2,88 (1H, уш. с), 2,83-2,74 (2Н, м), 1,85-1,75 (2Н, м), 1,71-1,62 (2Н, м), 1,09 (9Н, с), 1,04 (9Н, с), 0,90 (9Н, с), 0,102 (3Н, с), 0,099 (3Н, с).
[0659] (Этап 3)
2-{2-О-[трет-бутил(диметил)силил]-3,5-О-(ди-трет-бутилсилилиден)-β-В-рибофуранозил}-7,8,9,10-тетрагидро-2Н-6-окса-2,3,5-триазациклоокта[1,2,3-cd]инден
С применением соединения, полученного на этапе 2 (1,89 г), проводили реакцию таким же образом, как на этапе 5 примера 20, с получением указанного в заголовке соединения (1,40 г).
1H-ЯМР (CDCl3) δ: 8,41 (1H, с), 6,84 (1H, с), 6,21 (1Н, с), 4,54-4,43 (4Н, м), 4,31 (1Н, дд, J=9,6, 4,9 Гц), 4,17 (1H, тд, J=10,0, 5,1 Гц), 4,00 (1H, дд, J=10,4, 9,2 Гц), 2,87-2,73 (2Н, м), 2,05-1,87 (4Н, м), 1,10 (9Н, с), 1,05 (9Н, с), 0,91 (9Н, с), 0,12 (3Н, с), 0,10 (3Н, с).
[0660] (Этап 4)
2-{5-O-[бис(4-метоксифенил)(фенил)метил]-2-O-[трет-бутил(диметил)силил]-β-D-рибофуранозил}-7,8,9,10-тетрагидро-2Н-6-окса-2,3,5-триазациклоокта[1,2,3-с(1]инден
С применением соединения, полученного на этапе 3 (1,61 г), проводили реакцию таким же образом, как на этапе 5 примера 1, с получением указанного в заголовке соединения (1,95 г).
1Н-ЯМР (CDCl3) δ: 8,40 (1H, с), 7,49-7,43 (2Н, м), 7,38-7,20 (8Н, м), 6,86-6,78 (4Н, м), 6,38 (1H, д, J=5,5 Гц), 4,70 (1Н, т, J=5,3 Гц), 4,54-4,45 (2Н, м), 4,38-4,31 (1Н, м), 4,26-4,18 (1H, м), 3,79 (3Н, с), 3,79 (3Н, с), 3,53 (1H, дд, J=10,6, 2,3 Гц), 3,37 (1H, дд, J=10,6, 3,1 Гц), 2,81 (1H, д, J=3,9 Гц), 2,56-2,45 (2Н, м), 2,00-1,91 (2Н, м), 1,86-1,76 (2Н, м), 0,82 (9Н, с), -0,04 (3Н, с), -0,17(3Н, с).
[0661] (Этап 5)
2-(5-О-[бис(4-метоксифенил)(фенил)метил]-2-О-[трет-бутил(диметил)силил]-3-О-{(2-цианоэтокси)[ди(пропан-2-ил)амино]фосфанил}-β-D-рибофуранозил)-7,8,9,10-тетрагидро-2Н-6-окса-2,3,5-триазациклоокта[1,2,3-cd]инден
С применением соединения, полученного на этапе 4 (1,95 г), процессы проводили таким же образом, как на этапе 4 примера 5, с получением указанного в заголовке соединения (2,20 г) в виде смеси диастереомеров у атома фосфора (соотношение диастереомеров=6:4).
1H-ЯМР (CDCl3) δ: 8,39 (0,4Н, с), 8,37 (0,6Н, с), 7,51-7,45 (2Н, м), 7,40-7,20 (8Н, м), 6,86-6,78 (4Н, м), 6,37 (0,6Н, д, J=6,7 Гц), 6,32 (0,4Н, д, J=6,3 Гц), 4,83-4,78 (0,6Н, м), 4,77-4,70 (0,4Н, м), 4,56-4,44 (2Н, м), 4,42-4,33 (1,4Н, м), 4,29-4,24 (0,6Н, м), 4,07-3,86 (1Н, м), 3,82-3,75 (6Н, м), 3,70-3,46 (4Н, м), 3,32-3,24 (1Н, м), 2,76-2,65 (1Н, м), 2,63-2,49 (2Н, м), 2,32 (1H, т, J=6,7 Гц), 2,01-1,90 (2Н, м), 1,87-1,74 (2Н, м), 1,23-1,12 (8,4Н, м), 1,03 (3,6Н, д, J=6,7 Гц), 0,74 (3,6Н, с), 0,72 (5,4Н, с), -0,04 (1,2Н, с), -0,08 (1,8Н, с), -0,21 (1,2Н, с), -0,23 (1,8Н, с).
[0662] (Этап 6)
(5R,7R,8R,12aR,14R,15R,15aR,16R)-7-(6-амино-9H-пурин-9-ил)-15,16-бис{[трет-бутил(диметил)силил]окси}-2,10-бис(сульфанил)-14-(7,8,9,10-тетрагидро-2Н-6-окса-2,3,5-триазациклоокта[1,2,3-cd]инден-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-дион
С применением соединения, полученного на этапе 5 (1,18 г), проводили реакцию таким же образом, как на этапе 7 примера 1, с получением раствора в ацетонитриле 2-{2-O-[трет-бутил(диметил)силил]-3-O-[гидрокси(оксо)-λ5-фосфанил]-β-D-рибофуранозил}-7,8,9,10-тетрагидро-2Н-6-окса-2,3,5-триазациклоокта[1,2,3-d]индена. С применением данного раствора в ацетонитриле и доступного для приобретения (Cool Pharm Ltd.) N-бензоил-5'-O-[бис(4-метоксифенил)(фенил)метил]-3'-O-[трет-бутил(диметил)силил]-2'-O-{(2-цианоэтокси)[ди(пропан-2-ил)амино]фосфанил}аденозина (1,49 г) проводили реакцию таким же образом, как на этапах 8, 9 и 10 примера 1, с получением указанного в заголовке соединения в виде смеси диастереомеров у атома фосфора. Данную смесь очищали с помощью ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 25% - 60% (0 мин - 35 мин)] с получением диастереомера 1 (50 мг) и диастереомера 2 (34 мг) указанного в заголовке соединения.
Диастереомер 1 (менее полярный)
МС (ИЭР) m/z: 973(М+Н)+.
Диастереомер 2 (более полярный)
МС (ИЭР) m/z: 973(М+Н)+.
[0663] (Этап 7-1)
Динатрия (5R,7R,8R,12aR,14R,15R,15aS,16R)-7-(6-амино-9H-пурин-9-ил)-15,16-дигидрокси-2,10-диоксо-14-(7,8,9,10-тетрагидро-2Н-6-окса-2,3,5-триазациклоокта[1,2,3-cd]инден-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят) (Диастереомер 1)
С применением соединения, полученного на этапе 6 (диастереомер 1) (50 мг), проводили реакцию таким же образом, как на этапе 11 примера 1, и продукт затем очищали с помощью Sep-Pak (R) С18 [0,1% водный раствор триэтиламина/ацетонитрил = 5:1] с получением соли триэтиламина указанного в заголовке соединения.
Полученную соль триэтиламина подвергали солевому обмену таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением указанного в заголовке соединения (33 мг). МС (ИЭР) m/z: 745(М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,73 (1H, с), 8,30 (1H, с), 8,17 (1H, с), 7,40 (1H, с), 6,39 (1H, д, J=4,3 Гц), 6,34 (1H, д, Т=8,6 Гц), 5,40-5,36 (1H, м), 5,22-5,17 (1Н, м), 4,87-4,84 (1H, м), 4,80 (1H, т, J=4,5 Гц), 4,64-4,31 (6Н, м), 4,11-4,03 (2Н, м), 2,82 (1Н, дд, J=16,4, 8,6 Гц), 2,68 (1H, дд, J=16,2, 8,8 Гц), 2,06-1,71 (4Н, м).
31Р-ЯМР (CD3OD) δ: 58,1 (с), 54,2 (с).
[0664] (Этап 7-2)
Динатрия (5R,7R,8R,12aR,14R,15R,15aS,16R)-7-(6-амино-9Н-пурин-9-ил)-15,16-дигидрокси-2,10-диоксо-14-(7,8,9,10-тетрагвдро-2Н-6-окса-2,3,5-триазациклоокта[1,2,3-cd]инден-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят) (Диастереомер 2)
С применением соединения, полученного на этапе 6 (диастереомер 2) (34 мг), проводили реакцию таким же образом, как на этапе 11 примера 1, и продукт затем очищали с помощью Sep-Pak (R) С18 [0,1% водный раствор триэтиламина/ацетонитрил = 5:1] с получением соли триэтиламина указанного в заголовке соединения.
Полученную соль триэтиламина подвергали солевому обмену таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением указанного в заголовке соединения (21 мг). МС (ПЭР) m/z: 745 (М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,81 (1H, с), 8,30 (1H, с), 8,17 (1Н, с), 7,40 (1H, с), 6,43 (1H, д, J=6,7 Гц), 6,34 (1H, д, J=8,6 Гц), 5,55-5,42 (2Н, м), 4,87-4,84 (1H, м), 4,59-4,28 (7Н, м), 4,06-3,99 (1H, м), 3,94-3,86 (1H, м), 2,96-2,81 (2Н, м), 2,07-1,94 (2Н, м), 1,93-1,80 (2Н, м).
31Р-ЯМР (CD3OD) δ: 63,1 (с), 60,5 (с).
[0665]
Пример 32. Синтез ЦДН22.
(5R,7R,8R,12aR,14R,15R,15aS,16R)-7-(6-амино-9H-пурин-9ил)-15,16-дигидрокси-14-[(7S)-7-метил-8,9-дигидро-6-окса-2,3,5-триазабензо[cd]азулен-2(7Н)-ил]-2,10-бис(сульфанил)октагидро-2Н,10Н, 12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1] [1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-дион
[0666]
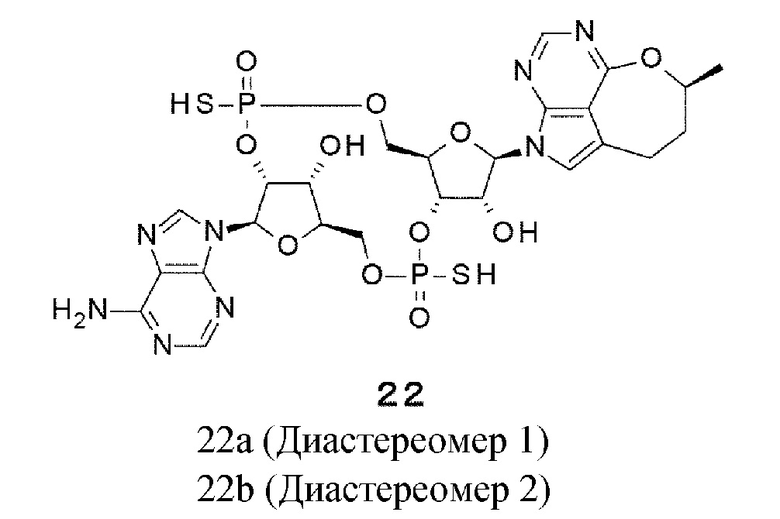
[0667]
[Схема синтеза]
[0668]
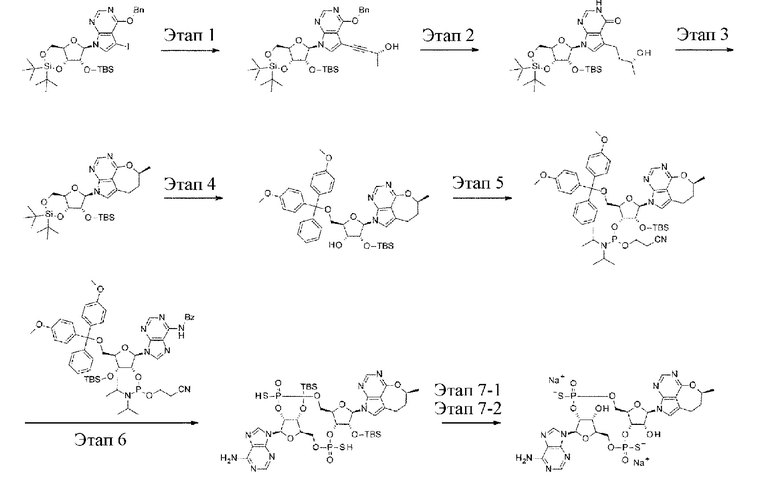
[0669] (Этап 1)
4-(бензилокси)-7-{2-O-[трет-бутил(диметил)силил]-3,5-О-(ди-трет-бутилсилилиден)-β-D-рибофуранозил}-5-[(3R)-3-гидроксибут-1-ин-1-ил]-7Н-пирроло[2,3-d]пиримидин
С применением соединения, полученного на этапе 2 примера 20 (2,54 г), и (R)-(+)-3-бутин-2-ола (1,36 мл) проводили реакцию таким же образом, как на этапе 2 примера 1, за исключением того, что температуру реакции устанавливали на уровне комнатной температуры, с получением указанного в заголовке соединения (1,85 г).
1H-ЯМР (CDCl3) δ: 8,46 (1H, с), 7,57 (2Н, д, J=7,0 Гц), 7,44-7,32 (3Н, м), 7,20 (1H, с), 6,16 (1H, с), 5,58 (1H, д, J=12,9 Гц), 5,55 (1H, д, J=13,7 Гц), 4,71-4,63 (1H, м), 4,48 (1H, дд, J=9,2, 4,9 Гц), 4,42 (1H, д, J=4,7 Гц), 4,26 (1H, дд, J=9,4, 4,7 Гц), 4,23-4,14 (1H, м), 4,01 (1H, т, J=9,6 Гц), 1,70 (1H, д, J=5,5 Гц), 1,42 (3Н, д, J=6,7 Гц), 1,09 (9Н, с), 1,04 (9Н, с), 0,91 (9Н, с), 0,12 (3Н, с), 0,11(3Н, с).
[0670]
(Этап 2)
7-{2-О-[трет-бутил(диметил)силил]-3,5-О-(ди-трет-бутилсилилиден)-β-D-рибофуранозил}-5-[(3R)-3-гидроксибутил]-3,7-дигидро-4Н-пирроло[2,3-d]пиримидин-4-он
С применением соединения, полученного на этапе 1 (1,85 г), проводили реакцию таким же образом, как на этапе 7 примера 19, за исключением того, что растворитель реакции заменяли на смешанный растворитель метанол (15 мл)/тетрагидрофуран (15 мл), с получением указанного в заголовке соединения (1,34 г).
1H-ЯМР (CDCl3) δ: 11,81 (1H, с), 7,89 (1H, с), 6,68 (1H, с), 6,12 (1H, с), 4,49 (1H, дд, J=9,2, 4.5 Гц), 4,34 (1H, д, J=4,3 Гц), 4,27-4,14 (3Н, м), 4,03 (1H, т, J=9,6 Гц), 3,77-3,65 (1H, м), 3,17-3.06 (1H, м), 2,85-2,74 (1H, м), 1,84-1,73 (1H, м), 1,71-1,61 (1Н, м), 1,15 (3Н, д, J=6,3 Гц), 1,09 (9Н, с), 1,04 (9Н, с), 0,90 (9Н, с), 0,10 (6Н, с).
[0671] (Этап 3)
(7S)-2-{2-O-[трет-бутил(диметил)силил]-3,5-O-(ди-трет-бутилсилилиден)-β-D-рибофуранозил}-7-метил-2,7,8,9-тетрагидро-6-окса-2,3,5-триазабензо[cd]азулен
С применением соединения, полученного на этапе 2 (1,34 г), проводили реакцию таким же образом, как на этапе 5 примера 20, с получением указанного в заголовке соединения (0,70 г).
1H-ЯМР (CDCl3) δ: 8,43 (1H, с), 6,82 (1H, с), 6,19 (1H, с), 4,58-4,43 (3Н, м), 4,33 (1H, дд, J=9,6, 5,0 Гц), 4,17 (1H, тд, J=10,0, 5,0 Гц), 4,00 (1H, дд, J=10,4, 9,2 Гц), 3,06-2,97 (1H, м), 2,89-2,79 (1H, м), 2,23-2,08 (2Н, м), 1,60 (3Н, д, J=6,3 Гц), 1,10 (9Н, с), 1,05 (9Н, с), 0,91 (9Н, с), 0,12 (3Н, с), 0,11(3Н, с).
[0672] (Этап 4)
(7S)-2-{5-O-[бис(4-метоксифенил)(фенил)метил]-2-O-[трет-бутил(диметил)силил]-β-D-рибофуранозил}-7-метил-2,7,8,9-тетрагидро-6-окса-2,3,5-триазабензо[cd]азулен
С применением соединения, полученного на этапе 3 (0,70 г), проводили реакцию таким же образом, как на этапе 5 примера 1, с получением указанного в заголовке соединения (0,82 г).
1Н-ЯМР (CDCl3) δ: 8,41 (1H, с), 7,49-7,43 (2Н, м), 7,37-7,20 (7Н, м), 7,16 (1H, с), 6,85-6,78 (4Н, м), 6,35 (1H, д, J=5,5 Гц), 4,72 (1H, т, J=5,3 Гц), 4,57-4,47 (1H, м), 4,36 (1H, дд, J=9,0, 3,9 Гц), 4,22 (1H, к, J=3,l Гц), 3,79 (3Н, с), 3,79 (3Н, с), 3,52 (1H, дд, J=10,6, 2,7 Гц), 3,37 (1H, дд, J=10,6, 3,1 Гц), 2,81 (1H, д, J=3,9 Гц), 2,78-2,58 (2Н, м), 2,16-2,07 (2Н, м), 1,59 (3Н, д, J=6,7 Гц), 0,83 (9Н, с), -0,03 (3Н, с), -0,15 (3Н, с).
[0673] (Этап 5)
(7S)-2-(5-O-[бис(4-метоксифенил)(фенил)метил]-2-O-[трет-бутил(диметил)силил]-3-O-{(2-цианоэтокси)[ди(пропан-2-ил)амино]фосфанил}-β-D-рибофуранозил)-7-метил-2,7,8,9-тетрагидро-6-окса-2,3,5-триазабензо[cd]азулен
С применением соединения, полученного на этапе 4 (0,82 г), процессы проводили таким же образом, как на этапе 4 примера 5, с получением указанного в заголовке соединения (0,85 г) в виде смеси диастереомеров у атома фосфора (соотношение диастереомеров = 6:4).
1H-ЯМР (CDCl3) δ: 8,40 (0,4Н, с), 8,38 (0,6Н, с), 7,50-7,43 (2Н, м), 7,39-7,16 (8Н, м), 6,86-6,78 (4Н, м), 6,34 (0,6Н, д, J=6,7 Гц), 6,30 (0,4Н, д, J=5,9 Гц), 4,86-4,80 (0,6Н, м), 4,79-4,74 (0,4Н, м), 4,55-4,46 (1Н, м), 4,44-4,35 (1,4Н, м), 4,29-4,24 (0,6Н, м), 4,05-3,85 (1Н, м), 3,82-3,75 (6Н, м), 3,69-3,47(4Н, м), 3,31-3,24(1Н, м), 2,82-2,61 (3Н, м), 2,31 (1H, т, J=6,7 Гц), 2,17-2,07(2Н, м), 1,58-1,55 (3Н, м), 1,22-1,13 (8,4Н, м), 1,03 (3,6Н, д, J=6,7 Гц), 0,75 (3,6Н, с), 0,74 (5,4Н, с), -0,03 (1,2Н, с), -0,07 (1,8Н, с), -0,19 (1,2Н, с), -0,21 (1,8Н, с).
(Этап 6)
(5R,7R,8R,12aR,14R,15R,15aR,16R)-7-(6-амино-9Н-пурин-9-ил)-15,16-бис{[трет-бутил(диметил)силил]окси}-14-[(7S)-7-метил-8,9-дигидро-6-окса-2,3,5-триазабешо[cd]азулен-2(7Н)-ил]-2,10-бис(сульфанил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10] пентаоксадифосфациклотетрадецин-2,10-дион
С применением соединения, полученного на этапе 5 (0,85 г), проводили реакцию таким же образом, как на этапе 7 примера 1, с получением раствора в ацетонитриле (7S)-2-{2-O-[трет-бутил(диметил)силил]-3-O-[гидрокси(оксо)-λ5-фосфанил]-β-D-рибофуранозил}-7-метил-2,7,8,9-тетрагидро-6-окса-2,3,5-триазабензо[cd]азулена. С применением данного раствора в ацетонитриле и доступного для приобретения (Cool Pharm Ltd.) N-бензоил-5'-O-[бис(4-метоксифенил)(фенил)метил]-3'-O-[трет-бутил(диметил)силил]-2'-O-{(2-цианоэтокси)[ди(пропан-2-ил)амино]фосфанил}аденозина (1,12 г) проводили реакцию таким же образом, как на этапах 8, 9 и 10 примера 1, с получением указанного в заголовке соединения в виде смеси диастереомеров у атома фосфора. Данную смесь очищали с помощью ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 25% - 60% (0 мин - 35 мин)] с получением диастереомера 1 (95 мг) и диастереомера 2 (44 мг) указанного в заголовке соединения (время удерживания при ВЭЖХ: диастереомер 1>2). Диастереомер 1 (менее полярный) МС (ИЭР) m/z: 973(М+Н)+. Диастереомер 2 (более полярный) МС (ИЭР) m/z: 973(М+Н)+.
[0675] (Этап 7-1)
Динатрия (5R,7R,8R,12aR,14R,15R,15aS,16R)-7-(6-амино-9Н-пурин-9-ил)-15,16-дигидрокси-14-[(7S)-7-метил-8,9-дигидро-6-окса-2,3,5-триазабензо[cd]азулен-2(7H)-ил]-2,10-диоксооктагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят) (Диастереомер 1)
С применением соединения, полученного на этапе 6 (диастереомер 1) (95 мг), проводили реакцию таким же образом, как на этапе 11 примера 1, и продукт затем очищали с помощью Sep-Pak (R) С18 [0,1% водный раствор триэтиламина/ацетонитрил = 5:1] с получением соли триэтиламина указанного в заголовке соединения.
Полученную соль триэтиламина подвергали солевому обмену таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением указанного в заголовке соединения (57 мг). МС (ИЭР) m/z: 745(М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,72 (1H, с), 8,30 (1H, с), 8,17 (1H, с), 7,35 (1H, с), 6,37 (1H, д, J=4,3 Гц), 6,34 (1H, д, J=8,2 Гц), 5,40-5,35 (1H, м), 5,22-5,17 (1H, м), 4,85-4,81 (2Н, м), 4,66-4,58 (1H, м), 4,53-4,40 (2Н, м), 4,39-4,30 (2Н, м), 4,12-4,01 (2Н, м), 3,02-2,93 (1H, м), 2,80-2,68 (1H, м), 2,23-2,14 (1H, м), 2,12-2,00 (1H, м), 1,57 (3Н, д, J=6,3 Гц).
31Р-ЯМР (CD3OD) δ: 58,1 (с), 54,3 (с).
[0676] (Этап 7-2)
Динатрия (5R,7R,8R,12aR,14R,15R,15aS,16R)-7-(6-амино-9H-пурин-9-ил)-15,16-дигидрокси-14-[(7S)-7-метил-8,9-дигидро-6-окса-2,3,5-триазабензо[cd]азулен-2(7Н)-ил]-2,10-диоксооктагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят) (Диастереомер 2)
С применением соединения, полученного на этапе 6 (диастереомер 2) (44 мг), проводили реакцию таким же образом, как на этапе 11 примера 1, и продукт затем очищали с помощью Sep-Pak (R) С18 [0,1% водный раствор триэтиламина/ацетонитрил = 5:1] с получением соли триэтиламина указанного в заголовке соединения.
Полученную соль триэтиламина подвергали солевому обмену таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением указанного в заголовке соединения (30 мг). МС (ИЭР) m/z: 745(М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,81 (1H, с), 8,30 (1H, с), 8,17 (1H, с), 7,36 (1H, с), 6,41 (1H, д, J=6,7 Гц), 6,34 (1Н, д,.1=8,6 Гц), 5,55-5,42 (2Н, м), 4,87-4,84 (1Н, м), 4,65-4,57 (1Н, м), 4,55-4,28 (5Н, м), 4,06-3,99 (1H, м), 3,93-3,86 (1H, м), 3,11-3,01 (1H, м), 2,95-2,83 (1H, м), 2,29-2,19 (1H, м), 2,16-2,03 (1H, м), 1,57 (3Н, д, J=6,3 Гц). 31Р-ЯМР (CD3OD) δ: 63,7 (с), 61,2 (с).
[0677]
Пример 33. Синтез ЦДН23.
(5R,7R,8R,12aR,14R,15R,15aS,16R)-7-(6-амино-9Н-пурин-9-ил)-14-(8,9-дигидро-6-тиа-2,3,5-триазабензо[cd]азулен-2(7Н)-ил)-15,16-дигидрокси-2,10-бис(сульфанил)октагидро-2Н,10Н, 12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-дион
[0678]
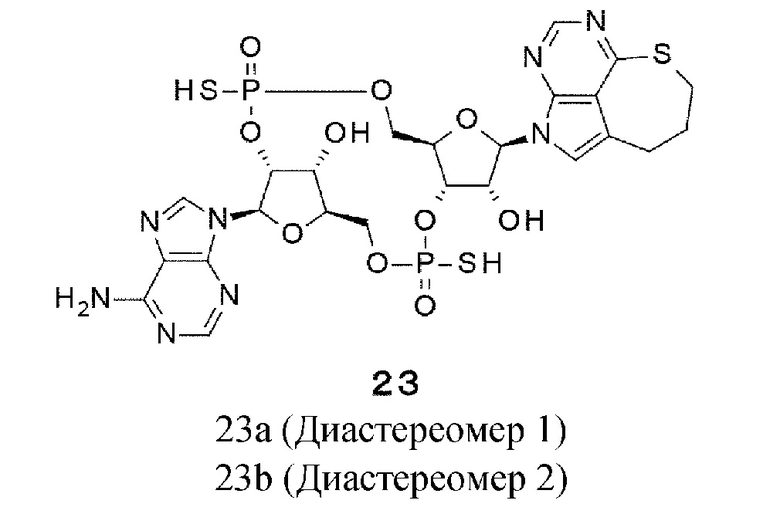
[0679]
[Схема синтеза]
[0680]
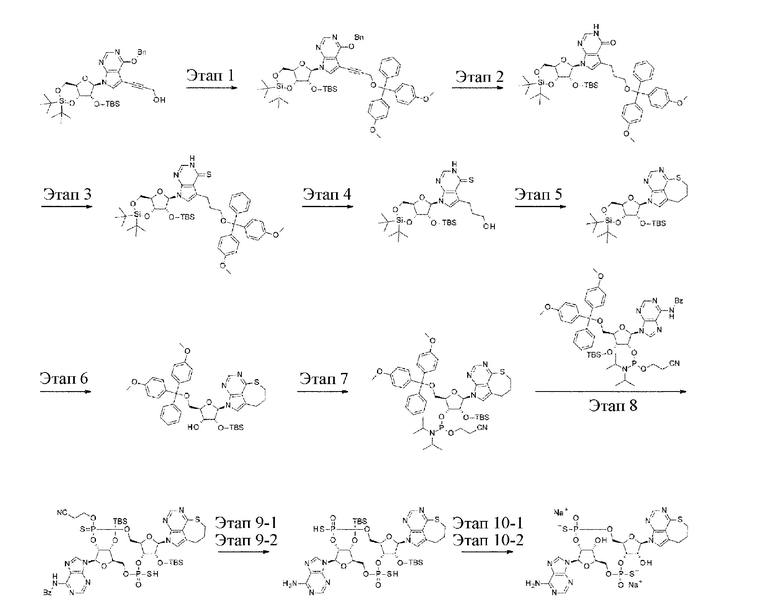
[0681]
(Этап 1)
4-(бензилокси)-5-{3-[бис(4-метоксифенил)(фенил)метокси]проп-1-ин-1-ил}-7-{2-O-[трет-бутил(диметил)силил]-3,5-O-(ди-трет-бутилсилилиден)-β-D-рибофуранозил}-7Н-пирроло[2,3-d]пиримидин
С применением соединения, полученного на этапе 3 примера 20 (4,17 г), проводили реакцию таким же образом, как на этапе 1 примера 11, за исключением того, что растворитель реакции заменяли на смешанный растворитель дихлорметан (40 мл)/пиридин (40 мл), с получением указанного в заголовке соединения (5,70 г).
1Н-ЯМР (CDCl3) δ: 8,44 (1Н, с), 7,53-7,47 (4Н, м), 7,41-7,35 (4Н, м), 7,33-7,11 (7Н, м), 6,86-6,79 (4Н, м), 6,17 (1H, с), 5,61 (1Н, д, J=13,7 Гц), 5,58 (1Н, д, J=13,7 Гц), 4,49 (1Н, дд, J=9,0, 5,0 Гц), 4,44 (1H, д, J=4,8 Гц), 4,29 (1Н, дд, J=9,6, 5,0 Гц), 4,23-4,15 (1H, м), 4,04 (1H, т, J=9,8 Гц), 3,98 (2Н, с), 3,78 (6Н, с), 1,10 (9Н, с), 1,05 (9Н, с), 0,91 (9Н, с), 0,12 (3Н, с), 0,11 (3Н, с).
[0682]
(Этап 2)
5-{3-[бис(4-метоксифенил)(фенил)метокси]пропил}-7-{2-O-[трет-бутил(диметил)силил]-3,5-O-(ди-трет-бутилсилилиден)-β-D-рибофуранозил}-3,7-дигидро-4Н-пирроло[2,3-d]пиримидин-4-он
В смешанный раствор соединения, полученного на этапе 1 (5,70 г), в метаноле (100 мл)/тетрагидрофуране (50 мл) добавляли формиат аммония (3,71 г) и 10% палладий-углерод (AD), влажный (2 г), и реакционную смесь перемешивали при комнатной температуре в течение 3 часов. После удаления катализатора путем фильтрации, фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле [гексан/этилацетат] с получением указанного в заголовке соединения (4,56 г).
1H-ЯМР (CDCl3) δ: 11,61 (1H, уш. с), 7,75 (1H, с), 7,48-7,44 (2Н, м), 7,37-7,14 (7Н, м), 6,84-6,79 (4Н, м), 6,57 (1H, с), 6,05 (1H, с), 4,45 (1H, дд, J=9,2, 4,9 Гц), 4,35 (1H, д, J=5,l Гц), 4,23 (1H, дд, J=9,6, 4,9 Гц), 4,16-4,10 (1H, м), 3,97 (1H, т, Г=9,8 Гц), 3,78 (6Н, с), 3,19-3,08 (2Н, м), 2,90 (2Н, т, J=7,8 Гц), 2,07-1,98 (2Н, м), 1,09 (9Н, с), 1,04 (9Н, с), 0,89 (9Н, с), 0,09 (3Н, с), 0,08 (3Н, с).
[0683] (Этап 3)
5-{3-[бис(4-метоксифенил)(фенил)метокси]пропил}-7-{2-О-[трет-бутил(диметил)силил]-3,5-О-(ди-трет-бутилсилилиден)-β-D-рибофуранозил}-3,7-дигидро-4Н-пирроло[2,3-d]пиримидин-4-тион
К раствору соединения, полученного на этапе 2 (1,01 г), в дихлорметане (10 мл) добавляли пиридин (0,461 мл), и добавляли туда по каплям трифторметансульфоновый ангидрид (0,385 мл) при охлаждении льдом, и реакционную смесь перемешивали в течение 30 минут. Суспензию моногидросульфида натрия н-гидрата (2,54 г) в N,N-диметилформамиде (25 мл) добавляли в реакционную смесь при той же температуре, и реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Насыщенный водный раствор гидрокарбоната натрия и этилацетата добавляли в реакционную смесь, которую фильтровали через целит, а затем осуществляли экстрагирование этилацетатом. Органический слой промывали рассолом и сушили над безводным сульфатом натрия. Осушающий агент удаляли путем фильтрации и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле [гексан/этилацетат] с получением указанного в заголовке соединения (0,51 г).
1H-ЯМР (CDCl3) δ: 10,83 (1H, с), 7,83 (1H, с), 7,50-7,44 (2Н, м), 7,39-7,14 (7Н, м), 6,87-6,79 (4Н, м), 6,72 (1H, с), 6,07 (1H, с), 4,48-4,42 (1H, м), 4,31 (1H, д, J=4,3 Гц), 4,21-4,09 (2Н, м), 3,99-3,92 (1H, м), 3,79 (6Н, с), 3,19-3,09 (4Н, м), 2,09-1,98 (2Н, м), 1,08 (9Н, с), 1,04 (9Н, с), 0,90 (9Н, с), 0,09 (3Н, с), 0,09 (3Н, с).
[0684] (Этап 4)
7-{2-О-[трет-бутил(диметил)силил]-3,5-О-(ди-трет-бутилсилилиден)-β-D-рибофуранозил}-5-(3-гидроксипропил)-3,7-дигидро-4Н-пирроло[2,3-(1]пиримидин-4-тион
К раствору соединения, полученного на этапе 3 (2,79 г), в дихлорметане (80 мл) добавляли дистиллированную воду (4 мл), и добавляли туда по каплям дихлоруксусную кислоту (1,28 мл) при охлаждении льдом, и реакционную смесь перемешивали в течение 30 минут. Пиридин (2,50 мл) добавляли в реакционную смесь при той же температуре, и реакционную смесь затем концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле [гексан/этилацетат] с получением указанного в заголовке соединения (0,96 г).
1H-ЯМР (CDCl3) δ: 11,16 (1H, с), 7,89 (1H, с), 6,83 (1H, с), 6,11 (1H, с), 4,51-4,45 (1H, м), 4,36-4,29 (1H, м), 4,22-4,13 (2Н, м), 4,06-3,97 (1Н, м), 3,68 (2Н, т, J=6,l Гц), 3,27-3,09 (2Н, м), 2,25-2,13 (1H, уш. м), 2,00-1,93 (2Н, м), 1,08 (9Н, с), 1,04 (9Н, с), 0,90 (9Н, с), 0,10 (6Н, с).
[0685] (Этап 5)
2-{2-О-[трет-бутил(диметил)силил]-3,5-О-(ди-трет-бутилсилилиден)-β-D-рибофуранозил}-2,7,8,9-тетрагидро-6-тиа-2,3,5-триазабензо[cd]азулен
С применением соединения, полученного на этапе 4 (0,35 г), проводили реакцию таким же образом, как на этапе 5 примера 20, с получением указанного в заголовке соединения (0,25 г).
1H-ЯМР (CDCl3) δ: 8,53 (1H, с), 6,92 (1H, с), 6,21 (1Н, с), 4,50-4,44 (2Н, м), 4,30 (1Н, дд, J=9,6, 4,9 Гц), 4,17 (1H, тд, J=10,0, 5,0 Гц), 4,00 (1H, дд, J=10,4, 9,2 Гц), 3,18-3,12 (2Н, м), 3,06-3,00 (2Н, м), 2,39-2,30 (2Н, м), 1,09 (9Н, с), 1,05 (9Н, с), 0,91 (9Н, с), 0,12 (3Н, с), 0,11 (3Н, с).
[0686] (Этап 6)
2-{5-О-[бис(4-метоксифенил)(фенил)метил]-2-О-[трет-бутил(диметил)силил]-р-О-рибофуранозил}-2,7,8,9-тетрагидро-6-тиа-2,3,5-триазабензо[cd]азулен
С применением соединения, полученного на этапе 5 (0,41 г), проводили реакцию таким же образом, как на этапе 5 примера 1, с получением указанного в заголовке соединения (0,46 г).
1Н-ЯМР (CDCl3) δ: 8,52 (1H, с), 7,48-7,42 (2Н, м), 7,37-7,20 (8Н, м), 6,85-6,78 (4Н, м), 6,36 (1H, д, J=5,l Гц), 4,70 (1Н, т, J=5,1 Гц), 4,37 (1Н, дд, J=8,8, 4,1 Гц), 4,23-4,19 (1H, м), 3,79 (3Н, с), 3,79 (3Н, с), 3,53 (1H, дд, J=10,6, 2,7 Гц), 3,38 (1H, дд, J=10,6, 3,1 Гц), 3,16-3,09 (2Н, м), 2,78 (1H, д, J=4,l Гц), 2,76-2,70 (2Н, м), 2,30-2,22 (2Н, м), 0,83 (9Н, с), -0,03 (3Н, с), -0,14 (3Н, с).
[0687] (Этап 7)
2-(5-О-[бис(4-метоксифенил)(фенил)метил]-2-О-[трет-бутил(диметил)силил]-3-О-{(2-цианоэтокси)[ди(пропан-2-ил)амино]фосфанил}-р-О-рибофуранозил)-2,7,8,9-тетрагидро-6-тиа-2,3,5-триазабензо[cd]азулен
С применением соединения, полученного на этапе 6 (0,46 г), процессы проводили таким же образом, как на этапе 4 примера 5, с получением указанного в заголовке соединения (0,48 г) в виде смеси диастереомеров у атома фосфора (соотношение диастереомеров=6:4).
1H-ЯМР (CDCl3) δ: 8,51 (0,4Н, с), 8,49 (0,6Н, с), 7,50-7,43 (2Н, м), 7,38-7,23 (8Н, м), 6,86-6,78 (4Н, м), 6,36 (0,6Н, д, J=6,7 Гц), 6,32 (0,4Н, д, J=5,5 Гц), 4,83-4,78 (0,6Н, м), 4,76-4,71 (0,4Н, м), 4,45-4,35 (1,4Н, м), 4,29-4,24 (0,6Н, м), 4,04-3,84 (1H, м), 3,82-3,75 (6Н, м), 3,70-3,48 (4Н, м), 3,32-3,25 (1H, м), 3,16-3,09 (2Н, м), 2,84-2,73 (2Н, м), 2,73-2,61 (1H, м), 2,36-2,20 (3Н, м), 1,22-1,13 (8,4Н, м), 1,03 (3,6Н, д, J=7,0 Гц), 0,76 (3,6Н, с), 0,75 (5,4Н, с), -0,03 (1,2Н, с), -0,07 (1,8Н, с), -0,18 (1,2Н, с), -0,20 (1,8Н, с).
[0688] (Этап 8)
N-{9-[(5R,7R,8R,12aR,14R,15R,15aR,16R)-15,16-бис{[трет-бутил(диметил)силил]окси}-10-(2-цианоэтокси)-14-(8,9-дигидро-6-тиа-2,3,5-триазабензо[сс1]азулен-2(7Н)-ил)-2-оксо-2-сульфанил-10-сульфанилиденоктагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-7-ил]-9Н-пурин-6-ил}бензамид
С применением соединения, полученного на этапе 7 (0,48 г), проводили реакцию таким же образом, как на этапе 7 примера 1, с получением раствора в ацетонитриле 2-{2-О-[трет-бутил(диметил)силил]-3-О-[гидрокси(оксо)-λ5-фосфанил]-β-D-рибофуранозил}-2,7,8,9-тетрагидро-6-тиа-2,3,5-триазабензо[cd]азулена. С применением данного раствора в ацетонитриле и доступного для приобретения (Cool Pharm Ltd.) N-бензоил-5'-О-[бис(4-метоксифенил)(фенил)метил]-3'-О-[трет-бутил(диметил)силил]-2'-О-{(2-цианоэтокси)[ди(пропан-2-ил)амино]фосфанил}аденозина (0,61 г) проводили реакцию таким же образом, как на этапах 8 и 9 примера 1, с получением указанного в заголовке соединения в виде смеси диастереомеров у атома фосфора. Данную смесь очищали с помощью ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 40% - 90% (0 мин - 35 мин)] с получением диастереомера 1 (40 мг) и диастереомера 2 (18 мг) указанного в заголовке соединения. Диастереомер 1 (менее полярный) МС (ИЭР) m/z: 1132(М+Н)+. Диастереомер 2 (более полярный) МС (ИЭР) m/z: 1132(М+Н)+.
[0689] (Этап 9-1)
(5R,7R,8R,12aR,14R,15R,15aR,16R)-7-(6-амино-9H-пурин-9-ил)-15,16-бис{[трет-бутил(диметил)силил]окси}-14-(8,9-дигидро-6-тиа-2,3,5-триазабензо[cd]азулен-2(7Н)-ил)-2,10-бис(сульфанил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-дион
С применением соединения, полученного на этапе 8 (диастереомер 1) (40 мг), проводили реакцию таким же образом, как на этапе 10 примера 1, и продукт затем очищали с помощью препаративной ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 30% - 60% (0 мин - 35 мин)] с получением указанного в заголовке соединения (35 мг). МС (ИЭР) m/z: 975(М+Н)+.
[0690] (Этап 9-2)
(5R,7R,8R,12aR,14R,15R,15aR,16R)-7-(6-амино-9H-пурин-9-ил)-15,16-бис{[трет-бутил(диметил)силил]окси}-14-(8,9-дигидро-6-тиа-2,3,5-триазабензо[cd]азулен-2(7Н)-ил)-2,10-бис(сульфанил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,1λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-дион
С применением соединения, полученного на этапе 8 (диастереомер 2) (18 мг), проводили реакцию таким же образом, как на этапе 10 примера 1, и продукт затем очищали с помощью препаративной ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 25% - 50% (0 мин - 35 мин)] с получением указанного в заголовке соединения (15 мг). МС (ИЭР) m/z: 975(М+Н)+.
[0691] (Этап 10-1)
Динатрия (5R,7R,8R,12aR,14R,15R,15aS,16R)-7-(6-амино-9H-пурин-9-ил)-14-(8,9-дигидро-6-тиа-2,3,5-триазабензо [cd]азулен-2(7Н)-ил)-15,16-дигидрокси-2,10-диоксооктагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят) (Диастереомер 1)
С применением соединения, полученного на этапе 9-1 (35 мг), проводили реакцию таким же образом, как на этапе 11 примера 1, и продукт очищали с помощью Sep-Pak (R) С18 [0,1% водный раствор триэтиламина/ацетонитрил = 5:1] с получением соли триэтиламина указанного в заголовке соединения.
Полученную соль триэтиламина подвергали солевому обмену таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением указанного в заголовке соединения (18 мг). МС (ИЭР) m/z: 747(М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,73 (1H, с), 8,41 (1H, с), 8,17 (1H, с), 7,48 (1H, с), 6,40 (1H, д, J=4,3 Гц), 6,34 (1H, д, J=8,6 Гц), 5,40-5,35 (1H, м), 5,23-5,17 (1H, м), 4,86-4,79 (2Н, м), 4,54-4,41 (2Н, м), 4,39-4,31 (2Н, м), 4,12-4,01 (2Н, м), 3,23-3,16 (2Н, м), 3,05-2,86 (2Н, м), 2,36-2,20 (2Н, м). 31Р-ЯМР (CD3OD) δ: 58,1 (с), 54,2 (с).
[0692] (Этап 10-2)
Динатрия (5R,7R,8R,12aR,14R,15R,15aS,16R)-7-(6-амино-9Н-пурин-9-ил)-14-(8,9-дигидро-6-тиа-2,3,5-триазабензо[cd]азулен-2(7Н)-ил)-15,16-дигидрокси-2,10-диоксооктагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят) (Диастереомер 2)
С применением соединения, полученного на этапе 9-2 (15 мг), проводили реакцию таким же образом, как на этапе 11 примера 1, и продукт очищали с помощью Sep-Pak (R) С18 [0,1% водный раствор триэтиламина/ацетонитрил = 5:1] с получением соли триэтиламина указанного в заголовке соединения.
Полученную соль триэтиламина подвергали солевому обмену таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением указанного в заголовке соединения (8,1 мг). МС (ИЭР) m/z: 747(М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,81 (1H, с), 8,41 (1H, с), 8,17 (1H, с), 7,49 (1H, с), 6,43 (1H, д, J=6,7 Гц), 6,34 (1Н, д, J=8,2 Гц), 5,55-5,41 (2Н, м), 4,87-4,82 (1Н, м), 4,57-4,27 (5Н, м), 4,07-3,99 (1Н, м), 3,94-3,86 (1H, м), 3,24-3,16 (2Н, м), 3,12-3,03 (2Н, м), 2,39-2,25 (2Н, м). 31Р-ЯМР (CD3OD) δ: 63,0 (с), 60,5 (с).
[0693]
Пример 34. Синтез ЦДН24.
1-[(5R,7R,8R,12aR,14R,15R,15а8,16R)-15,16-дигидрокси-2,10-диоксо-2,10-бис(сульфанил)-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабешо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-7-ил]пиримидин-2,4(1Н,3Н)-дион
[0694]
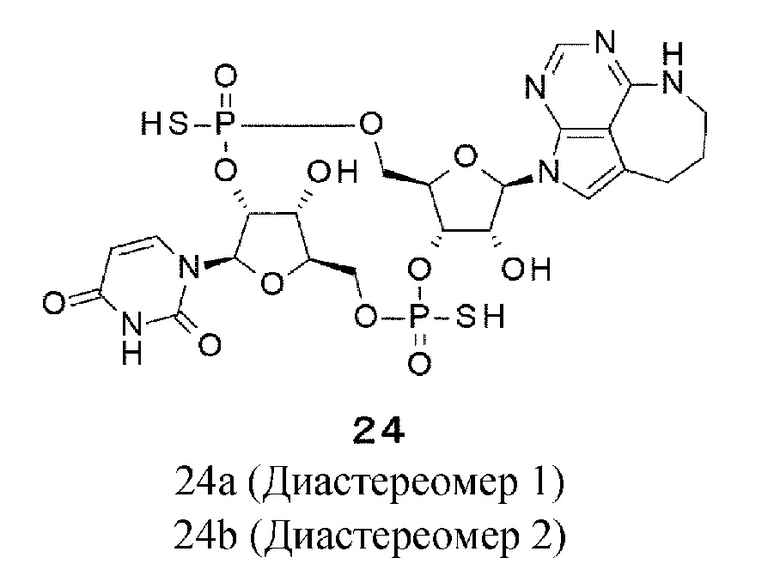
[0695]
[Схема синтеза]
[0696]
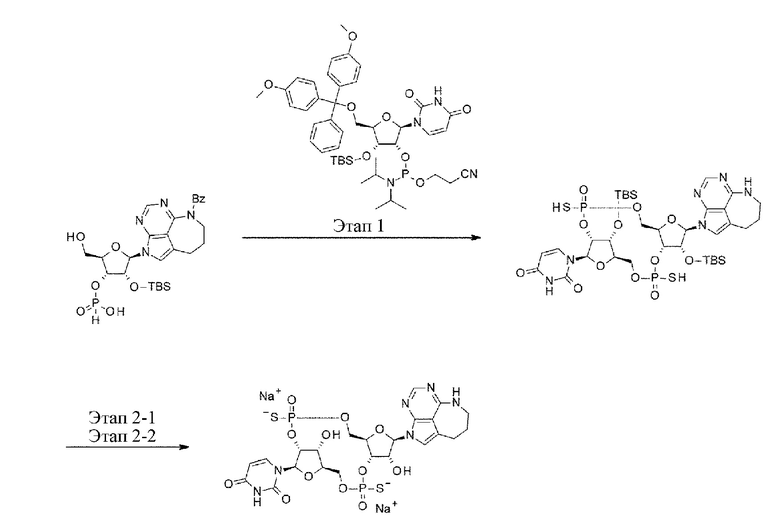
[0697]
(Этап 1)
1-[(5R,7R,8R,12aR,14R,15R,15aR,16R)-15,16-бис{[трет-бутил(диметил)силил]окси}-2,10-диоксо-2,10-бис(сульфанил)-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-7-ил]пиримидин-2,4(1Н,3Н)-дион
Ту же реакцию, что и на этапе 7 примера 1, проводили в следующем масштабе (сырьевой материал: 1,01 г). С применением раствора в ацетонитриле полученного соединения и доступного для приобретения (Angene International Limited) 5'-О-[бис(4-метоксифенил)(фенил)метил]-3'-О-[трет-бутил(диметил)силил]-2'-О-{(2-цианоэтокси)[ди(пропан-2-ил)амино]фосфанил}уридина (1,03 г) проводили реакцию таким же образом, как на этапах 8, 9 и 10 примера 1, с получением указанного в заголовке соединения в виде смеси диастереомеров у атома фосфора. Данную смесь очищали с помощью ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 25% - 60% (0 мин - 35 мин)] с получением диастереомера 1 (50 мг) и диастереомера 2 (23 мг) указанного в заголовке соединения. Диастереомер 1 (менее полярный) МС (ИЭР) m/z: 935(М+Н)+. Диастереомер 2 (более полярный) МС (ИЭР) m/z: 935(М+Н)+.
[0698] (Этап 2-1)
Динатрия (5R,7R,8R,12aR,14R,15R,15aS,16R)-7-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-15,16-дигидрокси-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят) (Диастереомер 1)
С применением соединения, полученного на этапе 1 (диастереомер 1) (50 мг), проводили реакцию таким же образом, как на этапе 11 примера 1, и продукт затем очищали с помощью Sep-Pak (R) С18 [0,1% водный раствор триэтиламина/ацетонитрил = 5:1] с получением соли триэтиламина указанного в заголовке соединения.
Полученную соль триэтиламина подвергали солевому обмену таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением указанного в заголовке соединения (30 мг). МС (ИЭР) m/z: 707(М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,09 (1H, д, J=8,2 Гц), 8,03 (1H, с), 7,16 (1H, с), 6,32 (1H, д, J=8,6 Гц), 6,28 (1Н, д, J=4,3 Гц), 5,82 (1H, д, J=8,2 Гц), 5,08-5,01 (1H, м), 4,93-4,84 (1H, м), 4,73 (1Н, т, J=4,5 Гц), 4,68 (1H, д, J=3,9 Гц), 4,48-4,38 (2Н, м), 4,33-4,24 (1H, м), 4,23 (1H, д, J=2,3 Гц), 4,09-3,99 (2Н, м), 3,56-3,46 (2Н, м), 2,96-2,83 (2Н, м), 2,07-1,95 (2Н, м).
31Р-ЯМР (CD3OD) δ: 58,3 (с), 54,6 (с).
[0699]
(Этап 2-2)
Динатрия (5R,7R,8R,12aR,14R,15R,15aS,16R)-7-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-15,16-дигидрокси-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[с(1]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
(Диастереомер 2)
С применением соединения, полученного на этапе 1 (диастереомер 2) (23 мг), проводили реакцию таким же образом, как на этапе 11 примера 1, и продукт затем очищали с помощью Sep-Pak (R) С18 [0,1% водный раствор триэтиламина/ацетонитрил = 5:1] с получением соли триэтиламина указанного в заголовке соединения.
Полученную соль триэтиламина подвергали солевому обмену таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением указанного в заголовке соединения (11 мг). МС (ИЭР) m/z: 707(М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,08 (1H, д, J=7,8 Гц), 8,01 (1H, с), 7,16 (1H, с), 6,35 (1H, д, J=8,6 Гц), 6,31 (1H, д, J=6,7 Гц), 5,85 (1H, д, J=7,8 Гц), 5,38-5,33 (1Н, м), 5,04-4,96 (1H, м), 4,75 (1H, дд, J=6,5, 4,5 Гц), 4,50-4,34 (3Н, м), 4,33-4,26 (1H, м), 4,22-4,17 (1H, м), 4,04-3,97 (1H, м), 3,91-3,84 (1H, м), 3,53-3,46 (2Н, м), 2,97-2,87 (2Н, м), 2,05-1,95 (2Н, м). 31Р-ЯМР (CD3OD) S: 63,2 (с), 60,2 (с).
[0700]
Пример 35. Синтез ЦДН25.
(5R,7R,8R,12aR,14R,15R,15aS,16R)-15,16-дигидрокси-7-(6-оксо-1,6-дигидро-9Н-пурин-9-ил)-2,10-бис(сульфанил)-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-дион
[0701]
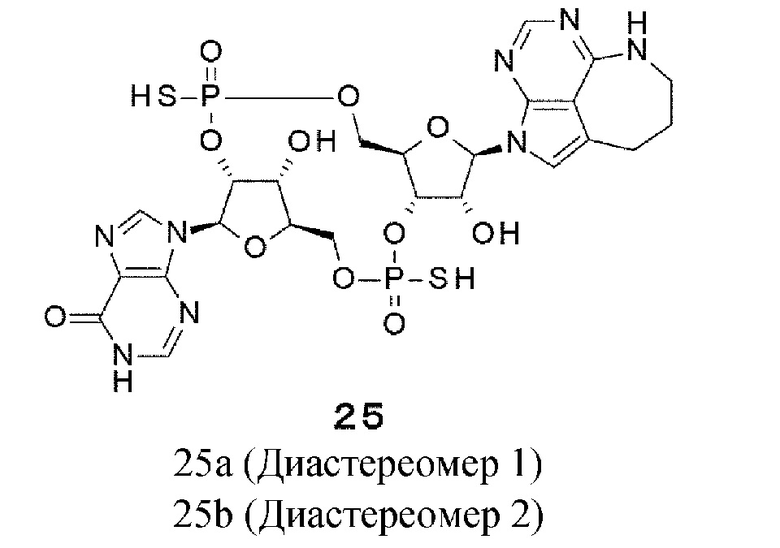
[Схема синтеза] [0703]
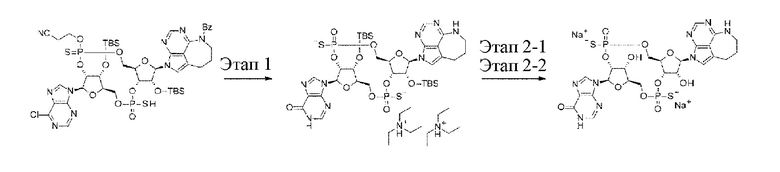
[0704] (Этап 1)
Бис(N,N-диэтилэтанаминия) (5R,7R,8R,12aR,14R,15R,15aR,16R)-15,16-бис{[трет-бутил(диметил)силил]окси}-2,10-диоксо-7-(6-оксо-1,6-дигидро-9Н-пурин-9-ил)-14-(6,7,8,9-тетрагидро-2Н-23,5,6-тетраазабензо[се]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
К раствору соединения (313 мг), полученного на этапе 4 примера 13, в тетрагидрофуране (5,0 мл) добавляли N-[(Е)-(пиридин-2-ил)метилиден]гидроксиламин (337 мг) и N,N,N',N'-тетраметилгуанидин (0,346 мл), и реакционную смесь перемешивали при комнатной температуре в течение 1 дня. Метанол (5,0 мл) и 28% водного аммиака (5,0 мл) добавляли в реакционную смесь, которую перемешивали при 50°С в течение 5 часов. После концентрирования реакционной смеси при пониженном давлении остаток очищали с помощью колоночной хроматографии на силикагеле С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил] с получением диастереомера 1 (106 мг: с примесями) и диастереомера 2 (105 мг: с примесями) указанного в заголовке соединения. Диастереомер 1 (менее полярный) МС (ИЭР) m/z: 959(М+Н)+. Диастереомер 2 (более полярный) МС (ИЭР) m/z: 959(М+Н)+.
[0705] (Этап 2-1)
Динатрия (5R,7R,8R,12aR,14R,15R,15aS,16R)-15,16-дигидрокси-2,10-диоксо-7-(6-оксо-l,6-дигидро-9Н-пурин-9-ил)-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят) (Диастереомер 1)
С применением соединения (диастереомера 1) (106 мг: с примесями), полученного на описанном выше этапе 1, проводили реакцию таким же образом, как на этапе 11 примера 1, и очистку проводили при следующих [Условиях очистки] с получением указанного в заголовке соединения в виде соли триэтиламина.
[Условия очистки] Хроматография на колонке с силикагелем С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил] и препаративная ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/раствор ацетонитрила-метанола (1:1), раствор ацетонитрила-метанола (1:1): 7% - 50% (0 мин - 40 мин)].
Полученную соль триэтиламина подвергали солевому обмену таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением указанного в заголовке соединения (43,4 мг). МС (ИЭР) m/z: 731(М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,66 (1H, с), 8,02 (2Н, с), 7,09 (1H, с), 6,30 (1H, д, J=6,8 Гц), 6,28 (1H, д, J=4,8 Гц), 5,45-5,38 (1H, м), 5,20-5,13 (1Н, м), 4,82 (1Н, д, J=4,2 Гц), 4,77 (1H, т, J=4,5 Гц), 4,52-4,41 (2Н, м), 4,36-4,27 (2Н, м), 4,08-3,97 (2Н, м), 3,53-3,46 (2Н, м), 2,88-2,80 (2Н, м), 2,04-1,95 (2Н, м).
31Р-ЯМР (CD3OD) δ: 57,7 (с), 54,6 (с).
[0706]
(Этап 2-2)
Динатрия (5R,7R,8R,12aR,14R,15R,15aS,16R)-15,16-дигидрокси-2,10-диоксо-7-(6-оксо-l,6-дигидро-9Н-пурин-9-ил)-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят) (Диастереомер 2)
С применением соединения (диастереомера 2) (105 мг: с примесями), полученного на описанном выше этапе 1, проводили реакцию таким же образом, как на этапе 11 примера 1, и очистку проводили при следующих [Условиях очистки] с получением указанного в заголовке соединения в виде соли триэтиламина.
[Условия очистки] Хроматография на колонке с силикагелем С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил] и препаративная ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/раствор ацетонитрила-метанола (1:1), раствор ацетонитрила-метанола (1:1): 7% - 45% (0 мин - 40 мин)].
Полученную соль триэтиламина подвергали солевому обмену таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением указанного в заголовке соединения (21,5 мг). МС (ИЭР) m/z: 731(М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,72 (1H, с), 8,03 (1H, с), 8,02 (1Н, с), 7,11 (1H, с), 6,32 (1H, д, J=6,0 Гц), 6,30 (1H, д, J=8,0 Гц), 5,49-5,40 (2Н, м), 4,77 (1Н, дд, J=6,7, 4,2 Гц), 4,49 (1H, д, J=4,5 Гц), 4,47-4,29 (4Н, м), 4,07-4,01 (1H, м), 3,93-3,86 (1Н, м), 3,52-3,47 (2Н, м), 2,90 (2Н, т, J=5,4 Гц), 2,05-1,97 (2Н, м).
31Р-ЯМР (CD3OD) δ: 62,9 (с), 60,0 (с) [0707]
Пример 36. Синтез ЦДН26.
(5R,7R,8R,12aR,14R,15R,15aS,16R)-15,16-дигидрокси-7-[1-(3-гидроксипропил)-6-оксо-1,6-дигидро-9Н-пурин-9-ил]-2,10-бис(сульфанил)-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[с(1]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-дион
[0708]
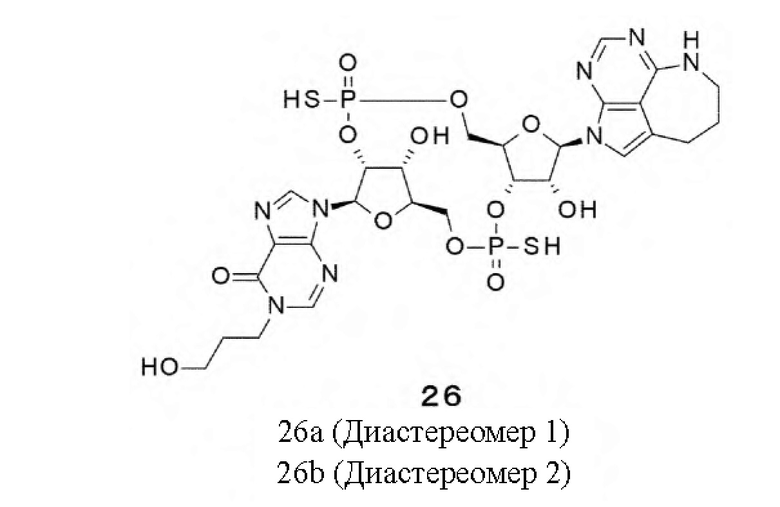
[0709]
[Схема синтеза] [0710]
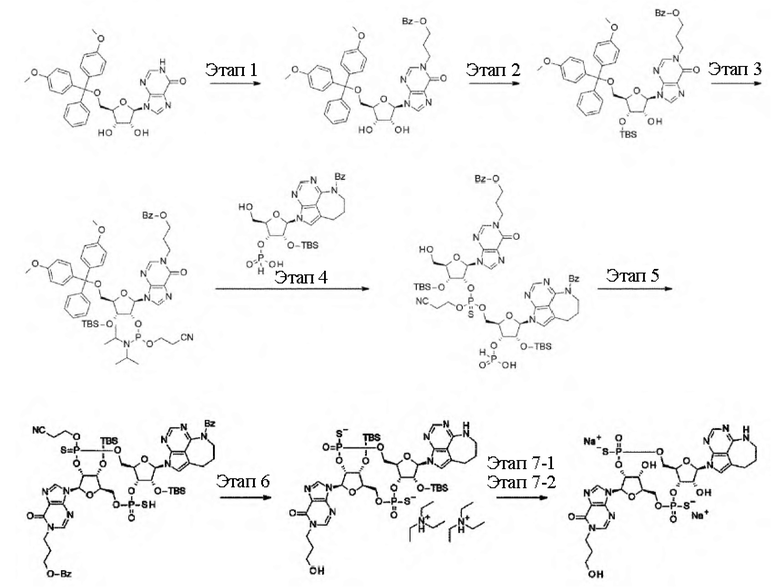
[0711] (Этап 1)
1-[3-(бензоилокси)пропил]-5'-О-[бис(4-метоксифенил)(фенил)метил]инозин
К раствору 3-бромпропан-1-ола (1,14 мл) в тетрагидрофуране (25 мл) добавляли триэтиламин (1,83 мл) и бензоилхлорид (1,43 мл), и реакционную смесь перемешивали при комнатной температуре в течение 6 часов. Реакционную смесь фильтровали и промывали тетрагидрофураном и фильтрат концентрировали при пониженном давлении. К раствору остатка в обезвоженном N,N-диметилацетамиде (25 мл) добавляли доступный для приобретения (Aamdis Chemical) 5'-O-[бис(4-метоксифенил)(фенил)метил]инозин (5,0 г) и 1,8-диазабицикло[5,4,0]-7-ундецен (2,75 мл), и реакционную смесь перемешивали при комнатной температуре в течение 3 суток. Воду добавляли в реакционную смесь, которую осуществляли экстрагирование этилацетатом. Органический слой промывали рассолом, а затем сушили над безводным сульфатом натрия. Осушающий агент удаляли путем фильтрации и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле [гексан/этил ацетат/метанол] с получением указанного в заголовке соединения (4,41 г).
МС (ИЭР) m/z: 733(М+Н)+.
1H-ЯМР (CDCl3) δ: 8,00-7,98 (1H, м), 7,98-7,96 (1Н, м), 7,97 (1Н, с), 7,96 (1Н, с), 7,58-7,52 (1H, м), 7,46-7,39 (2Н, м), 7,35-7,30 (2Н, м), 7,26-7,16 (7Н, м), 6,81-6,75 (4Н, м), 5,87 (1H, д, J=6,0 Гц), 4,85 (1H, д, J=3,6 Гц), 4,67-4,62 (1H, м), 4,43-4,34 (3H, м), 4,30-4,16 (2Н, м), 3,78-3,75 (1H, м), 3,77 (6Н, с), 3,42 (1Н, дд, J=10,3, 3,6 Гц), 3,33 (1H, дд, J=10,3, 3,6 Гц), 3,02 (1H, д, J=2,4 Гц), 2,32 (2Н, дд, J=11,8, 5,7 Гц).
[0712]
(Этап 2)
1-[3-(бегооилокси)пропил]-5'-O-[бис(4-метоксифенил)(фенил)метил]-3'-O-[трет-бутил(диметил)силил]инозин
С применением соединения (4,41 г), полученного на описанном выше этапе 1, проводили реакцию таким же образом, как на этапе 3 примера 5, с получением указанного в заголовке соединения (1,60 г) и 1-[3-(бензоилокси)пропил]-5'-O-[бис(4-метоксифенил)(фенил)метил]-2'-O-[трет-бутил(диметил)силил]инозина (1,70 г) в качестве региоизомера указанного в заголовке соединения.
МС (ИЭР) m/z: 847(М+Н)+.
1H-ЯМР (CDCl3) δ: 8,01 (1H, д, J=l,2 Гц), 8,00 (1Н, с), 7,99 (1H, д, J=1,2 Гц), 7,93 (1Н, с), 7,60-7,52 (1H, м), 7,46-7,38 (4Н, м), 7,33-7,16 (7Н, м), 6,83-6,77 (4Н, м), 5,90 (1H, д, J=4,8 Гц), 4,58-4,52 (1H, м), 4,50-4,46 (1H, м), 4,39 (2H, т, J=6,0 Гц), 4,28-4,12 (3H, м), 3,78 (3H, с), 3,77 (3H, с), 3,46 (1Н, дд, J=10,6, 3,9 Гц), 3,26 (1H, дд, J=10,6, 3,9 Гц), 2,99 (1Н, д, J=6,7 Гц), 2,30 (2Н, дд, J=13,3, 6,0 Гц), 0,88 (9Н, с), 0,07 (3H, с), 0,00 (3H, с).
Региоизомер (2'-О-TBS-форма)
МС (ИЭР) m/z: 847(М+Н)+.
1Н-ЯМР (CDCl3) δ: 8,01 (1H, д, J=1,8 Гц), 7,99 (1Н, д, J=1,8 Гц), 7,98 (1H, с), 7,87 (1Н, с), 7,59-7,54 (1H, м), 7,47-7,40 (4Н, м), 7,36-7,17 (7Н, м), 6,84-6,78 (4Н, м), 5,94 (1H, д, J=5,4 Гц), 4,84 (1H, т, J=5,4 Гц), 4,41-4,35 (2Н, м), 4,33-4,28 (1H, м), 4,28-4,19 (3H, м), 3,78 (3H, с), 3,78 (3H, с), 3,48 (1H, дд, J=10,9, 3,0 Гц), 3,37 (1H, дд, J=10,9, 3,0 Гц), 2,68 (1H, д, J=3,6 Гц), 2,34-2,26 (2Н, м), 0,84 (9Н, с), 0,01 (3H, с), -0,13 (3H, с).
[0713]
(Этап 3)
1-[3-(бензоилокси)пропил]-5'-O-[бис(4-метоксифенил)(фенил)метил]-3'-O-[трет-бутил(диметил)силил]-2'-O-{(2-цианоэтокси)[ди(пропан-2-ил)амино]фосфанил}инозин
С применением соединения (1,60 г), полученного на описанном выше этапе 2, проводили реакцию таким же образом, как на этапе 4 примера 5, с получением указанного в заголовке соединения (1,91 г) в виде смеси диастереомеров у атома фосфора (соотношение диастереомеров=67:33). МС (ИЭР) m/z: 1047(М+Н)+.
1H-ЯМР (CDCl3) δ: 8,04-8,00 (0,33Н, м), 8,03 (1H, с), 8,02 (1H, с), 7,91 (0,67Н, д, J=14,5 Гц), 7,60-7,53 (1H, м), 7,49-7,40 (4Н, м), 7,35-7,17 (7Н, м), 6,84-6,78 (4Н, м), 6,14 (0,67Н, д, J=5,1 Гц), 6,06 (0,33Н, д, J=6,0 Гц), 4,86-4,78 (0,33Н, м), 4,68-4,61 (0,67Н, м), 4,44-4,35 (2Н, м), 4,29-4,09 (4Н, м), 3,78 (6Н, с), 3,65-3,42 (6,33Н, м), 3,34-3,24 (0,67Н, м), 2,76 (1,34Н, т, J=6,6 Гц), 2,50 (0,66Н, т, J=6,6 Гц), 2,38 (1,34Н, т, J=6,6 Гц), 2,30 (0,66Н, т, J=6,6 Гц), 1,30-1,24 (6Н, м), 1,15-1,07 (4,02Н, м), 0,95 (1,98Н, д, J=6,6 Гц), 0,84 (9Н, с), 0,09 (0,99Н, с), 0,05 (2,01Н, с), 0,00 (3H, с).
[0714]
(Этап 4)
Ту же реакцию, что и на этапе 7 примера 1, проводили в следующем масштабе (сырьевой материал: 981 мг). С применением раствора в ацетонитриле полученного соединения и соединения (1,03 г), полученного на описанном выше этапе 3, проводили реакцию таким же образом, как на этапе 8 примера 1, и полученный в результате этого неочищенный продукт непосредственно использовали для следующей реакции.
[0715]
(Этап 5)
3-{9-[(5R,7R,8R,12aR,14R,15R,15aR,16R)-14-(6-бензоил-6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)-15,16-бис{[трет-бутил(диметил)силил]окси}-10-(2-цианоэтокси)-2-оксо-2-сульфанил-10-сульфанилиденоктагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-7-ил]-6-оксо-6,9-дигидро-1H-пурин-1-ил}пропилбензоат
С применением неочищенного продукта, полученного на описанном выше этапе 4, проводили реакцию таким же образом, как на этапе 9 примера 1, с получением указанного в заголовке соединения (774 мг) в виде смеси диастереомеров у атома фосфора.
MC(ИЭР)m/z: 1278(М+Н)+.
[0716]
(Этап 6)
Бис(N,N-диэтилэтанаминия) (5R, 7R, 8R, 12aR, 14R, 15R, 15aR, 16R)-15,16-бис{[трет-бутил(диметил)силил]окси}-7-[1-(3-гидроксипропил)-6-оксо-1,6-дигидро-9Н-пурин-9-ил]-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
С применением соединения (774 мг), полученного на описанном выше этапе 5, проводили реакцию таким же образом, как на этапе 10 примера 1, и продукт очищали с помощью колоночной хроматографии на силикагеле С18 [0,2% водный раствор триэтиламина/ацетонитрил] с получением диастереомера 1 (101 мг: с примесями) и диастереомера 2 (90,8 мг: с примесями) указанного в заголовке соединения.
Диастереомер 1 (менее полярный)
MC(ИЭР)m/z: 1017(M+H)+.
Диастереомер 2 (более полярный)
МС(ИЭР) m/z: 1017(М+Н)+.
[0717]
(Этап 7-1)
Динатрия (5R,7R,8R,12aR,14R,15R,15aS,16R)-15,16-дигидрокси-7-[1-(3-гидроксипропил)-6-оксо-1,6-дигидро-9Н-пурин-9-ил]-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
(Диастереомер 1)
С применением соединения (диастереомера 1) (101 мг: с примесями), полученного на описанном выше этапе 6, проводили реакцию таким же образом, как на этапе 11 примера 1, и очистку проводили при следующих [Условиях очистки] с получением указанного в заголовке соединения в виде соли триэтиламина.
[Условия очистки] Хроматография на колонке с силикагелем С18 [10 мМ водный раствор ацетата триэтиламмония/ацето нитрил].
Полученную соль триэтиламина подвергали солевому обмену таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением указанного в заголовке соединения (48,8 мг).
МС (ИЭР) m/z: 789(М+Н)+.
1H-ЯМР (CD3OD) δ: 8,65 (1H, с), 8,28 (1H, с), 8,03 (1H, с), 7,09 (1H, с), 6,28 (1H, с), 6,27 (1H, д, J=4,8 Гц), 5,46-5,38 (1H, м), 5,21-5,13 (1H, м), 4,83-4,80 (1H, м), 4,79-4,75 (1H, м), 4,52-4,39 (2Н, м), 4,36-4,28 (2Н, м), 4,26-4,17 (1H, м), 4,17-4,08 (1H, м), 4,08-3,97 (2Н, м), 3,59 (2Н, т, J=5,7 Гц), 3,49 (2Н, т, J=4,8 Гц), 2,91-2,74 (2Н, м), 2,02-1,92 (4Н, м).
31Р-ЯМР (CD3OD) δ: 57,6 (с), 54,6 (с).
[0718]
(Этап 7-2)
Динатрия (5R,7R,8R,12aR,14R,15R,15aS,16R)-15,16-дигидрокси-7-[1-(3-гидроксипропил)-6-оксо-1,6-дигидро-9Н-пурин-9-ил]-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
(Диастереомер 2)
С применением соединения (диастереомера 2) (90,8 мг: с примесями), полученного на описанном выше этапе 6, проводили реакцию таким же образом, как на этапе 11 примера 1, и очистку проводили при следующих [Условиях очистки] с получением указанного в заголовке соединения в виде соли триэтиламина.
[Условия очистки] Хроматография на колонке с силикагелем С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил] и препаративная ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 3% - 20% (0 мин - 40 мин)].
Полученную соль триэтиламина подвергали солевому обмену таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением указанного в заголовке соединения (22,3 мг).
МС (ИЭР) m/z: 789(М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,71 (1H, с), 8,29 (1H, с), 8,02 (1H, с), 7,11 (1H, с), 6,32 (1H, д, J=6,7 Гц), 6,28 (1H, д, J=8,5 Гц), 5,48-5,38 (2Н, м), 4,80-4,74 (1H, м), 4,51-4,47 (1H, м), 4,47-4,28 (4Н, м), 4,27-4,14 (2Н, м), 4,07-4,01 (1H, м), 3,92-3,86 (1H, м), 3,60 (2Н, т, J=6,0 Гц), 3,53-3,47 (2Н, м), 2,93-2,87 (2Н, м), 2,06-1,94 (4Н, м).
31Р-ЯМР (CD3OD) δ: 62,8 (с), 59,9 (с).
[0719]
Пример 37. Синтез ЦДН27.
(5R,7R,8R,12aR,14R,15R,15aS,16R)-7-[2-амино-1-(2-гидроксиэтил)-6-оксо-1,6-дигидро-9Н-пурин-9-ил]-15,16-дигидрокси-2,10-бис(сульфанил)-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-дион
[0720]
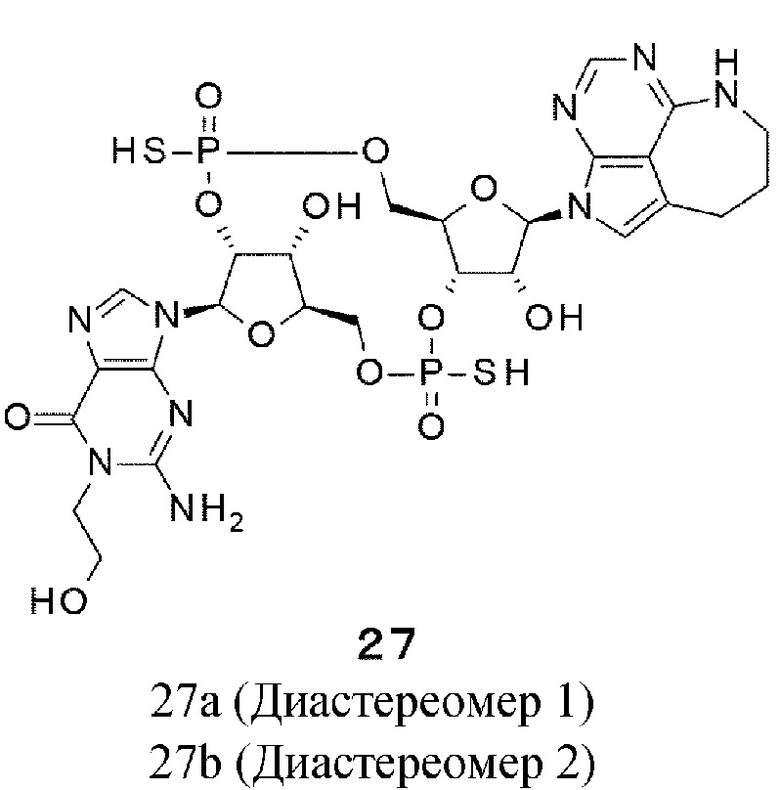
[0721]
[Схема синтеза]
[0722]
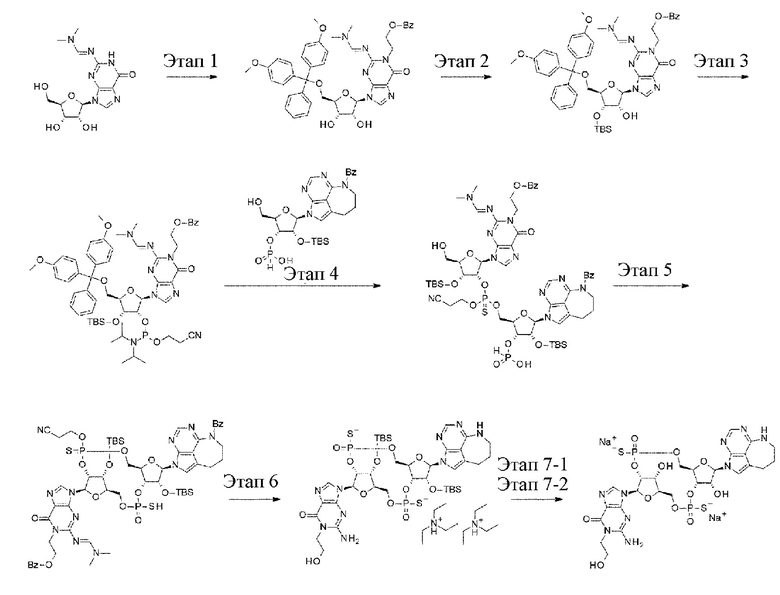
[0723]
(Этап 1)
1-[2-(бензоилокси)этил]-5'-О-[бис(4-метоксифенил)(фенил)метил]-N-[(диметиламино)метилиден]гуанозин
В смешанный раствор N-[(диметиламино)метилиден]гуанозина (10,0 г) как соединения, известного в литературе (Journal of Organic Chemistry, 1994, 59, 7243-7248), в N,N-диметилацетамиде (50 мл)-пиридине (50 мл) добавляли 4,4'-диметокситритилхлорид (10,5 г) при 0°С, и реакционную смесь перемешивали при 4°С в течение 16 часов. В реакционную смесь добавляли 2-бромоэтилбензоат (6,54 мл) и 1,8-диазабицикло[5,4,0]-7-ундецен (11,0 мл), и реакционную смесь перемешивали при комнатной температуре в течение 2 суток. Насыщенный водный раствор гидрокарбоната натрия и воды добавляли в реакционную смесь, которую осуществляли экстрагирование этилацетатом. Органический слой промывали рассолом и сушили над безводным сульфатом натрия. Осушающий агент удаляли путем фильтрации и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле [гексан/этилацетат/метанол] с получением указанного в заголовке соединения в виде смеси с оксидом трифенилфосфина (16,7 г).
МС (ИЭР) m/z: 789(М+Н)+.
1H-ЯМР (CDCl3) δ: 8,17 (1H, с), 7,93-7,89 (2Н, м), 7,55 (1H, с), 7,54-7,48 (1H, м), 7,42-7,36 (4Н, м), 7,31-7,26 (4Н, м), 7,25-7,19 (2Н, м), 7,16-7,11 (1H, м), 6,82-6,76 (4Н, м), 5,96 (1H, д, J=6,7 Гц), 4,79-4,70 (1H, м), 4,70-4,61 (2Н, м), 4,60-4,52 (1H, м), 4,52-4,45 (1H, м), 4,41-4,38 (1H, м), 4,34-4,30 (1H, м), 3,75 (3H, с), 3,75 (3H, с), 3,39-3,36 (2Н, м), 2,89 (3H, с), 2,80 (3H, с).
[0724]
(Этап 2)
1-[2-(бензоилокси)этил]-5'-O-[бис(4-метоксифенил)(фенил)метил]-3'-O-[трет-бутил(диметил)силил]-N-[(диметиламино)метилиден]гуанозин
С применением соединения (15,7 г), полученного на описанном выше этапе 1, проводили реакцию таким же образом, как на этапе 3 примера 5, с получением указанного в заголовке соединения (4,82 г) и 1-[2-(бензоилокси)этил]-5'-O-[бис(4-метоксифенил)(фенил)метил]-2'-O-[трет-бутил(диметил)силил]-N-[(диметиламино)метилиден]гуанозина (6,01 г) в качестве региоизомера указанного в заголовке соединения.
МС (ИЭР) m/z: 903(М+Н)+.
1Н-ЯМР (CDCl3) 5: 8,24 (1H, с), 7,99-7,95 (2Н, м), 7,85 (1H, с), 7,56-7,50 (1H, м), 7,44-7,38 (4Н, м), 7,34-7,27 (6Н, м), 7,24-7,18 (1H, м), 6,84-6,79 (4Н, м), 5,98 (1Н, д, J=4,2 Гц), 4,87-4,77 (2Н, м), 4,72-4,61 (2Н, м), 4,41-4,36 (2Н, м), 4,16-4,09 (1H, м), 3,78 (3H, с), 3,78 (3H, с), 3,45 (1H, дд, J=10,6, 3,9 Гц), 3,25 (1H, дд, J=10,6, 3,9 Гц), 3,05 (1H, д, J=5,4 Гц), 2,91 (3H, с), 2,78 (3H, с), 0,86 (9Н, с), 0,05 (3H, с), -0,04 (3H, с).
Региоизомер (2'-O-TBS-форма)
МС (ИЭР) m/z: 903(М+Н)+.
1H-ЯМР (CDCl3) δ: 8,23 (1H, с), 7,97-7,91 (2Н, м), 7,82 (1H, с), 7,56-7,50 (1H, м), 7,45-7,36 (4Н, м), 7,35-7,26 (6Н, м), 7,25-7,19 (1H, м), 6,85-6,79 (4Н, м), 5,98 (1Н, д, J=5,4 Гц), 4,88-4,77 (2Н, м), 4,73-4,62 (3H, м), 4,32-4,27 (1H, м), 4,23-4,19 (1H, м), 3,79 (3H, с), 3,79 (3H, с), 3,47 (1H, дд, J=10,9, 3,6 Гц), 3,37 (1H, дд, J=10,9, 3,6 Гц), 2,90 (3H, с), 2,76 (1H, д, J=2,5 Гц), 2,75 (3H, с), 0,84 (9Н, с), 0,02 (3H, с), -0,15 (3H, с).
[0725]
(Этап 3)
1-[2-(бензоилокси)этил]-5'-O-[бис(4-метоксифенил)(фенил)метил]-3'-O-[трет-бутил(диметил)силил]-2'-O-{(2-цианоэтокси)[ди(пропан-2-ил)амино]фосфанил}-N-[(диметиламино)метилиден]гуанозин
С применением соединения (4,81 г), полученного на описанном выше этапе 2, проводили реакцию таким же образом, как на этапе 4 примера 5, с получением указанного в заголовке соединения (5,41 г) в виде смеси диастереомеров у атома фосфора (соотношение диастереомеров=7:3).
МС (ИЭР) m/z: 1103(М+Н)+.
1H-ЯМР (CDCl3) δ: 8,33 (0,7Н, с), 8,31 (0,3Н, с), 7,98-7,94 (2Н, м), 7,87 (0,3Н, с), 7,80 (0,7Н, с), 7,56-7,50 (1H, м), 7,46-7,37 (4Н, м), 7,35-7,27 (6Н, м), 7,24-7,18 (1H, м), 6,85-6,80 (4Н, м), 6,14 (0,7Н, д, J=6,0 Гц), 6,13 (0,3Н, д, J=5,4 Гц), 4,92-4,59 (4Н, м), 4,55-4,48 (0,7Н, м), 4,33-4,29 (0,3Н, м), 4,25-4,21 (0,6Н, м), 4,17-4,13 (1,4Н, м), 3,79 (3H, с), 3,79 (3H, с), 3,60-3,38 (5Н, м), 3,33-3,23 (1H, м), 2,93 (2,1Н, с), 2,92 (0,9Н, с), 2,82 (2,1Н, с), 2,81 (0,9Н, с), 2,47-2,42 (0,6Н, м), 2,34-2,27 (1,4Н, м), 1,09 (4,2Н, д, J=6,7 Гц), 1,07 (1,8Н, д, J=7,3 Гц), 1,05 (4,2Н, д, J=6,7 Гц), 0,91 (1,8Н, д, J=7,3 Гц), 0,84 (9Н, с), 0,07 (0,9Н, с), 0,04 (2,1Н, с), -0,02 (3H, с).
[0726]
(Этап 4)
Ту же реакцию, что и на этапе 7 примера 1, проводили в следующем масштабе (сырьевой материал: 1,68 г). С применением раствора в ацетонитриле полученного соединения и соединения (1,81 г), полученного на описанном выше этапе 3, проводили реакцию таким же образом, как на этапе 8 примера 1, и полученный в результате этого неочищенный продукт непосредственно использовали для следующей реакции.
[0727]
(Этап 5)
2-(9-[(5R,7R,8R,12aR,14R,15R,15aR,16R)-14-(6-бензоил-6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)-15,16-бис{[трет-бутил(диметил)силил]окси}-10-(2-цианоэтокси)-2-оксо-2-сульфанил-10-сульфанилиденоктагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-7-ил]-2-{(E)-[(диметиламино)метилиден]амино}-6-оксо-6,9-дигидро-1Н-пурин-1-ил)этилбензоат
С применением неочищенного продукта, полученного на описанном выше этапе 4, проводили реакцию таким же образом, как на этапе 9 примера 1, с получением указанного в заголовке соединения (1,15 г) в виде смеси диастереомеров у атома фосфора. МС (ИЭР) m/z: 1334(М+Н)+.
[0728]
(Этап 6)
Бис(N,N-диэтилэтанаминия) (5R,7R,8R,12aR,14R,15R,15aR,16R)-7-[2-амино-1-(2-гидроксиэтил)-6-оксо-1,6-дигидро-9Н-пурин-9-ил]-15,16-бис{[трет-бутил(диметил)силил]окси}-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
С применением соединения (1,15 г), полученного на описанном выше этапе 5, проводили реакцию таким же образом, как на этапе 10 примера 1, с получением диастереомера 1 (134 мг: с примесями) и диастереомера 2 (127 мг: с примесями) указанного в заголовке соединения.
Диастереомер 1 (менее полярный)
MC(ИЭР)m/z: 1018(М+Н)+.
Диастереомер 2 (более полярный)
MC(ИЭР)m/z: 1018(М+Н)+.
[0729]
(Этап 7-1)
Динатрия (5R,7R,8R,12aR,14R,15R,15aS,16R)-7-[2-амино-1-(2-гидроксиэтил)-6-оксо-1,6-дигидро-9Н-пурин-9-ил]-15,16-дигидрокси-2Д0-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[с(1]азулен-2-ил)октагидро-2Н,10H,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
(Диастереомер 1)
С применением соединения (диастереомера 1) (134 мг: с примесями), полученного на описанном выше этапе 6, проводили реакцию таким же образом, как на этапе 11 примера 1, а затем проводили очистку при следующих [Условиях очистки] с получением указанного в заголовке соединения в виде соли триэтиламина.
[Условия очистки] Хроматография на колонке с силикагелем С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил] и препаративная ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 5% - 25% (0 мин - 40 мин)].
Полученную соль триэтиламина подвергали солевому обмену таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением указанного в заголовке соединения (36,0 мг).
МС (ПЭР) m/z: 790(М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,01 (1H, с), 7,99 (1Н, с), 7,17 (1H, с), 6,23 (1H, д, J=2,4 Гц), 5,96 (1H, д, J=8,5 Гц), 5,67-5,58 (1H, м), 5,29-5,22 (1Н, м), 4,95-4,85 (1H, м), 4,83-4,79 (1H, м), 4,48-4,40 (2Н, м), 4,39-4,31 (2Н, м), 4,22-4,09 (3H, м), 3,73-3,66 (2Н, м), 3,56-3,50 (1H, м), 3,50-3,44 (2Н, м), 2,80-2,68 (1Н, м), 2,48-2,35 (1H, м), 2,00-1,88 (1Н, м), 1,87-1,77 (1Н, м).
31Р-ЯМР (CD3OD) δ: 57,6 (с), 53,1 (с).
[0730]
(Этап 7-2)
Динатрия (5R,7R,8R,12aR,14R,15R,15aS,16R)-7-[2-амино-1-(2-гидроксиэтил)-6-оксо-1,6-дигидро-9Н-пурин-9-ил]-15,16-дигидрокси-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
(Диастереомер 2)
С применением соединения (диастереомера 2) (127 мг: с примесями), полученного на описанном выше этапе 6, проводили реакцию таким же образом, как на этапе 11 примера 1, а затем проводили очистку при следующих [Условиях очистки] с получением указанного в заголовке соединения в виде соли триэтиламина.
[Условия очистки] Хроматография на колонке с силикагелем С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил] и препаративная ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 3% - 20% (0 мин - 40 мин)].
Полученную соль триэтиламина подвергали солевому обмену таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением указанного в заголовке соединения (20,1 мг).
МС (ПЭР) m/z: 790(М+Н)+.
1H-ЯМР (CD3OD) δ: 8,20 (1H, с), 8,02 (1H, с), 7,19 (1H, с), 6,31 (1H, д, J=6,0 Гц), 6,05 (1H, д, J=8,5 Гц), 5,62-5,52 (1Н, м), 5,47-5,40 (1H, м), 4,80-4,75 (1H, м), 4,51-4,47 (1H, м), 4,47-4,21 (5Н, м), 4,16-4,09 (1H, м), 3,99-3,89 (2Н, м), 3,84-3,78 (2Н, м), 3,52-3,46 (2Н, м), 2,93-2,83 (1H, м), 2,82-2,72 (1Н, м), 2,03-1,92 (2Н, м).
31Р-ЯМР (CD3OD) δ: 61,6 (с), 59,6 (с).
[0731]
Пример 38. Синтез ЦДН28.
N-[2-({6-амино-9-[(5R,7R,8R,12aR,14R,15R,15aS,16R)-15,16-дигидрокси-2,10-диоксо-2,10-бис(сульфанил)-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-7-ил]-9Н-пурин-2-ил}амино)этил]-2-гидроксиацетамид
[0732]
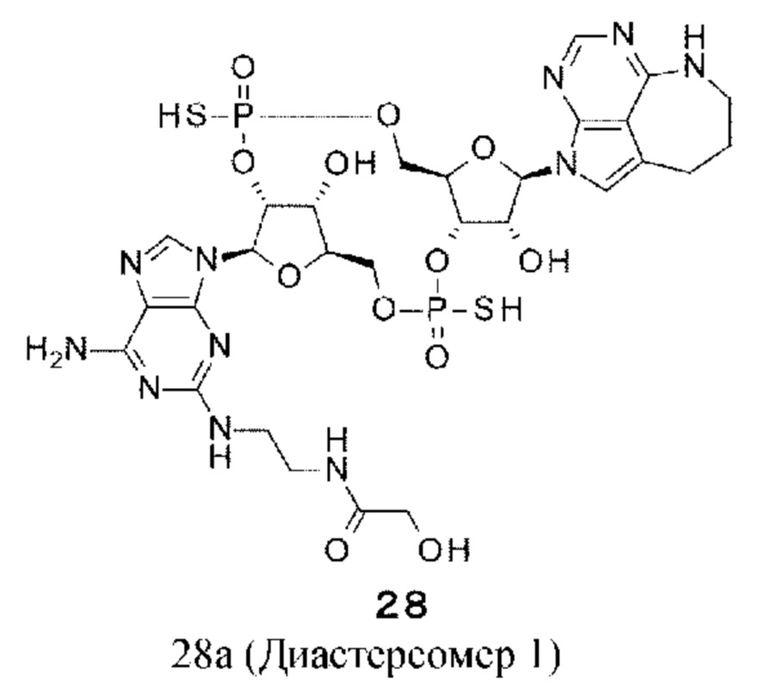
[0733]
[Схема синтеза]
[0734]
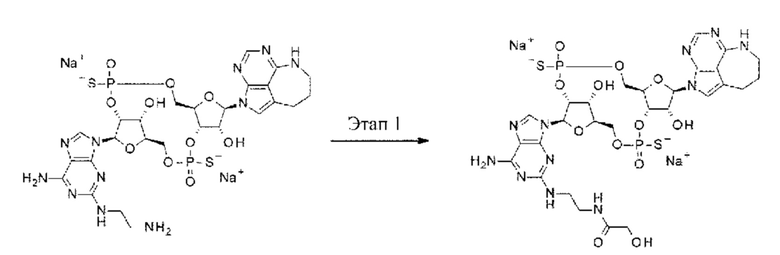
[0735]
(Этап 1)
Динатрия (5R,7R,8R,12aR,14R,15R,15aS,16R)-7-(6-амино-2-{[2-(2-гидроксиацетамид)этил]амино}-9Н-пурин-9-ил)-15,16-дигидрокси-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабегоо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
С применением соединения (6,6 мг), полученного на этапе 8-2 примера 8, проводили реакцию таким же образом, как на этапе 1-1 примера 7, и затем проводили очистку при следующих [Условиях очистки] с получением указанного в заголовке соединения в виде соли триэтиламина.
[Условия очистки] препаративная ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 2% - 30% (0 мин - 30 мин)].
Полученную соль триэтиламина подвергали солевому обмену таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением указанного в заголовке соединения (2,9 мг).
МС (ИЭР) m/z: 846(М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,33 (1H, с), 8,02 (1H, с), 7,16 (1H, с), 6,33 (1H, д, J=6,0 Гц), 6,18 (1H, д, J=8,5 Гц), 5,53-5,45 (2Н, м), 4,79 (1Н, т, J=5,1 Гц), 4,50-4,25 (5Н, м), 4,10 (1Н, д, J=11,5 Гц), 3,97 (2Н, с), 3,94-3,89 (1H, м), 3,53-3,39 (6Н, м), 2,88 (2Н, т, J=5,7 Гц), 2,04-1,98 (2Н, м). 31Р-ЯМР (CD3OD) δ: 62,6, 60,1.
[0736]
Пример 39. Синтез ЦДН29.
N-({6-амино-9-[(5R,7R,8R,12aR,14R,15R,15aS,16R)-15,16-дигидрокси-2,10-диоксо-2,10-бис(сульфанил)-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-7-ил]-9Н-пурин-2-ил}метил)-2-гидроксиацетамид
[0737]
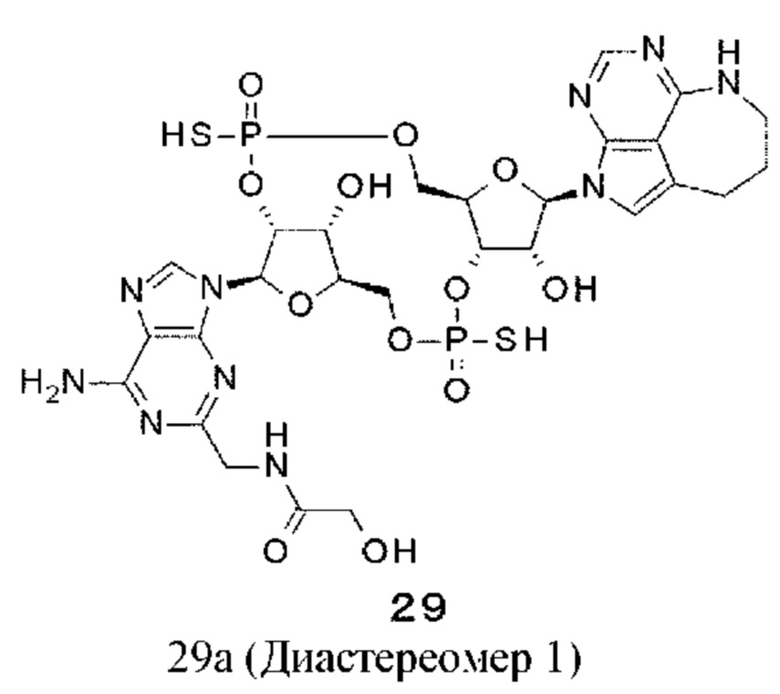
[0738]
[Схема синтеза]
[0739]
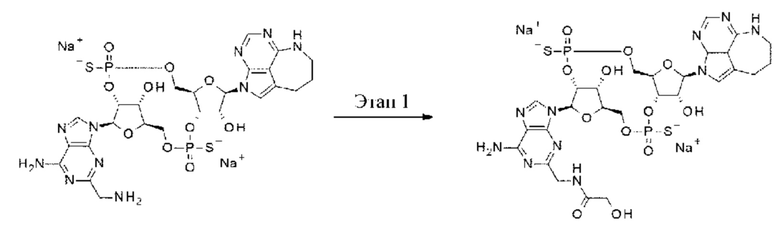
[0740]
(Этап 1)
Динатрия (5R,7R,8R,12aR,14R,15R,15aS,16R)-7-{6-амино-2-[(2-гидроксиацетамид)метил]-9Н-пурин-9-ил}-15,16-дигидрокси-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
С применением соединения (4,6 мг), полученного на этапе 9-2 примера 11, проводили реакцию таким же образом, как на этапе 1-1 примера 7, и продукт очищали с помощью препаративной ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 2% - 30% (0 мин - 30 мин)] с получением указанного в заголовке соединения в виде соли триэтиламина.
[0741]
Полученную соль триэтиламина подвергали солевому обмену таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением указанного в заголовке соединения (4,0 мг).
МС (ИЭР) m/z: 817(М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,75 (1H, с), 8,02 (1H, с), 7,12 (1H, с), 6,36 (1H, д, J=8,5 Гц), 6,33 (1H, д, J=6,7 Гц), 5,49-5,41 (2Н, м), 4,80 (1H, дд, J=6,7, 4,8 Гц), 4,50-4,29 (7Н, м), 4,07 (2Н, с), 4,05-4,01 (1Н, м), 3,92-3,87 (1H, м), 3,51-3,49 (2Н, м), 2,93-2,90 (2Н, м), 2,03-1,99 (2Н, м).
31Р-ЯМР (CD3OD) δ: 63,2, 60,3.
[0742]
Пример 40. Синтез ЦДН30.
(5R,7R,8R,12aR,14R,15R,15aS,16R)-7-(6-амино-2-{[(1-аминоциклопропил)метил]амино}-9H-пурин-9-ил)-15,16-дигидрокси-2,10-бис(сульфанил)-14-(6,7,8,9-тетрагидро-2H-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-дион
[0743]
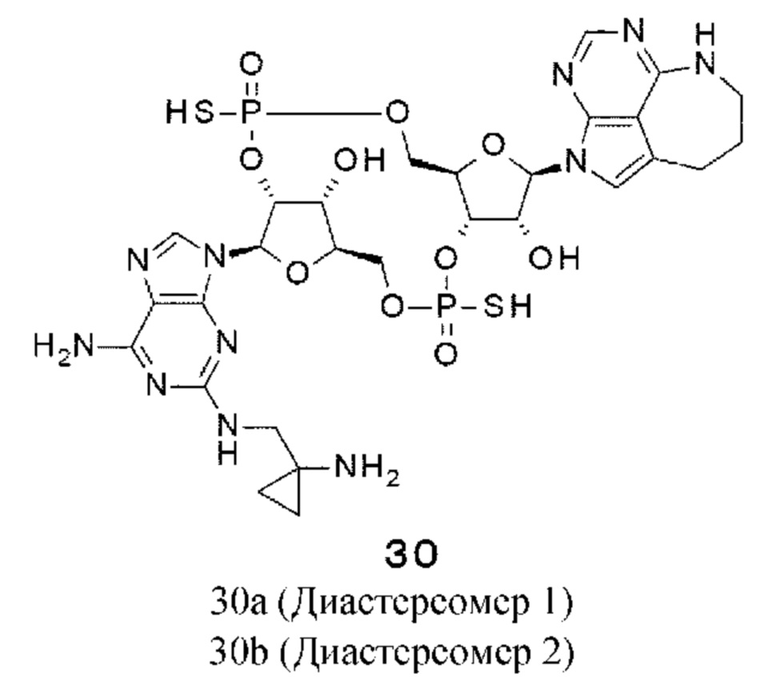
[0744]
[Схема синтеза]
[0745]
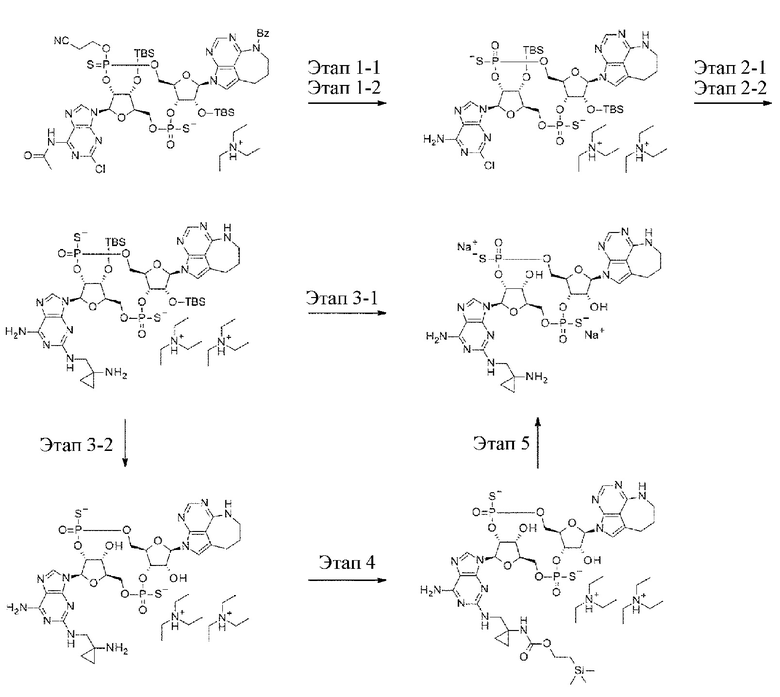
[0746]
(Этап 1-1)
Бис(N,N-диэтилэтанаминия) (5R,7R,8R,12aR,14R,15R,15aR,16R)-7-(6-амино-2-хлор-9H-пурин-9-ил)-15,16-бис{[трет-бутил(диметил)силил]окси}-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
С применением соединения (диастереомера 1) (590 мг), полученного на этапе 6 примера 8, проводили реакцию таким же образом, как на этапе 10 примера 1, с получением указанного в заголовке соединения (420 мг).
МС (ИЭР) m/z: 992(М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,76 (1Н, с), 7,97 (1Н, с), 7,30 (1Н, с), 6,23 (1Н, д, J=4,8 Гц), 6,22 (1Н, д, J=7,9 Гц), 5,43-5,37 (1Н, м), 5,19-5,15 (1H, м), 4,85-4,77 (3H, м), 4,44-4,31 (2Н, м), 4,22 (1Н, уш. с), 4,08-4,00 (2Н, м), 3,52-3,48 (2Н, м), 3,14 (12Н, к, J=7,3 Гц), 2,84-2,81 (2Н, м), 2,02-1,92 (2Н, м), 1,26 (18Н, т, J=7,3 Гц), 1,00 (9Н, с), 0,85 (9Н, с), 0,33 (3H, с), 0,28 (3H, с), 0,27 (3H, с), 0,10 (3H, с).
[0747]
(Этап 1-2)
Бис(N,N-диэтилэтанаминия) (5R,7R,8R,12aR,14R,15R,15aR,16R)-7-(6-амино-2-хлор-9H-пурин-9-ил)-15,16-бис{[трет-бутил(диметил)силил]окси}-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
С применением соединения (диастереомера 2) (710 мг), полученного на этапе 6 примера 8, проводили реакцию таким же образом, как на этапе 10 примера 1, с получением указанного в заголовке соединения (452 мг).
МС (ИЭР) m/z: 992(М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,55 (1H, с), 8,01 (1Н, с), 7,07 (1H, с), 6,34 (1H, д, J=7,3 Гц), 6,20 (1H, д, J=8,5 Гц), 5,52-5,44 (1H, м), 5,38-5,36 (1H, м), 5,17-5,10 (1H, м), 4,98-4,95 (2Н, м), 4,67-4,57 (2Н, м), 4,25 (1H, уш. с), 4,11-4,07 (1H, м), 3,89-3,84 (1H, м), 3,52-3,49 (2Н, м), 3,18 (12Н, к, 7,3 Гц), 2,93-2,91 (2Н, м), 2,03-1,99 (2Н, м), 1,30 (18Н, т, J=7,3 Гц), 1,00 (9Н, с), 0,74 (9Н, с), 0,27 (6Н, с), 0,20 (3H, с), -0,28 (3H, с).
[0748]
(Этап 2-1)
Бис(N,N-диэтилэтанаминия)(5R,7R,8R,12aR,14R,15R,15aR,16R)-7-(6-амино-2-{[(1-аминоциклопропил)метил]амино}-9Н-пурин-9-ил)-15,16-бис{[трет-бутил(диметил)силил]окси}-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
К раствору 1-(аминометил)циклопропан-1-амина⋅2HCl (309 мг) в метаноле (40 мл) добавляли МП-карбонатную смолу (5,45 г), и реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Смолу удаляли путем фильтрации и фильтрат концентрировали при пониженном давлении. Раствор остатка в метаноле (0,837 мл) добавляли к соединению (30,0 мг), полученному на описанном выше этапе 1-1, и проводили реакцию продукта с помощью микроволнового реактора при 120°С в течение 4 часов. После концентрирования реакционной смеси при пониженном давлении остаток очищали с помощью препаративной ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 30% - 60% (0 мин - 30 мин)] с получением смеси, содержащей указанное в заголовке соединение. Полученную смесь непосредственно использовали для следующего этапа.
MC(ИЭР)m/z: 1042(М+Н)+.
[0749]
(Этап 2-2)
Бис(N,N-диэтилэтанаминия) (5R,7R,8R,12aR,14R,15R,15aR,16R)-7-(6-амино-2-{[(1-аминоциклопропил)метил]амино}-9Н-пурин-9-ил)-15,16-бис{[трет-бутил(диметил)силил]окси}-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[с(1]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
С применением соединения (58,6 мг), полученного на описанном выше этапе 1-2, смесь, содержащую указанное в заголовке соединение, получали таким же образом, как на описанном выше этапе 2-1. Полученную смесь непосредственно использовали для следующей реакции.
MC(ИЭР)m/z: 1042(М+Н)+.
[0750]
(Этап 3-1)
Динатрия (5R,7R,8R,12aR,14R,15R,15aS,16R)-7-(6-амино-2-{[(1-аминоциклопропил)метил]амино}-9Н-пурин-9-ил)-15,16-дигидрокси-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1] [1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
(Диастереомер 1)
С применением смеси, полученной на описанном выше этапе 2-1, проводили реакцию таким же образом, как на этапе 11 примера 1, и продукт затем очищали с помощью препаративной ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 2% - 20% (0 мин - 30 мин)] с получением указанного в заголовке соединения в виде соли триэтиламина.
Полученную соль триэтиламина подвергали солевому обмену таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением указанного в заголовке соединения (2,0 мг).
МС (ИЭР) m/z: 812(M-2Na+1H)-.
1Н-ЯМР (CD3OD) δ: 8,25 (1H, с), 8,01 (1H, с), 7,04 (1H, с), 6,28 (1H, д, J=4,2 Гц), 6,12 (1H, д, J=7,9 Гц), 5,43-5,35 (1H, м), 5,15-5,11 (1H, м), 4,75 (1H, т, J=4,2 Гц), 4,65-4,56 (1H, м), 4,49-4,43 (2Н, м), 4,37-4,31 (2Н, м), 4,15-4,01 (2Н, м), 3,73-3,59 (1Н, м), 3,50-3,47 (2Н, м), 3,22-3,15 (1H, м), 2,85-2,68 (2Н, м), 2,00-1,93 (2Н, м), 0,87-0,80 (4Н, м).
31Р-ЯМР (CD3OD) δ: 57,7 (с), 54,3 (с).
[0751]
(Этап 3-2)
Бис(N,N-диэтилэтанаминия) (5R,7R,8R,12aR,14R,15R,15aS,16R)-7-(6-амино-2-{[(1-аминоциклопропил)метил]амино}-9Н-пурин-9-ил)-15,16-дигидрокси-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
С применением смеси, полученной на описанном выше этапе 2-2, проводили реакцию таким же образом, как на этапе 11 примера 1, с получением смеси, содержащей указанное в заголовке соединение. Полученную смесь непосредственно использовали для следующей реакции.
[0752]
(Этап 4)
Бис(N,N-диэтилэтанаминия) (5R,7R,8R,12aR,14R,15R,15aS,16R)-7- [6-амино-2-({[1-({[2-(триметилсилил)этокси]карбонил}амино)циклопропил]метил}амино)-9Н-пурин-9-ил]-15,16-дигидрокси-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
К раствору смеси, полученной на описанном выше этапе 3-2, в N,N-диметилформамиде (1,0 мл) добавляли триэтиламин (40,8 мкл) и 2,5-диоксопирролидин-1-ил(2-(триметилсилил)этил)карбонат (39,5 мг), и реакционную смесь перемешивали при комнатной температуре в течение 1 часа. После гашения путем добавления воды в реакционную смесь продукт очищали с помощью препаративной ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 10% - 50% (0 мин - 30 мин)] с получением указанного в заголовке соединения (11,0 мг).
МС (ИЭР) m/z: 958(М+Н)+.
1H-ЯМР (CD3OD) δ: 8,40 (1H, с), 8,03 (1H, с), 7,12 (1H, с), 6,32 (1H, д, J=6,7 Гц), 6,18 (1H, д, Г=8,5 Гц), 5,53-5,49 (1H, м), 5,46-5,39 (1H, м), 4,83 (1H, дд, J=6,3, 4,5 Гц), 4,53-4,30 (4Н, м), 4,26-4,24 (1H, м), 4,13-4,08 (2Н, м), 4,04-4,00 (1H, м), 3,93-3,88 (1H, м), 3,60 (1H, д, J=13,9 Гц), 3,52-3,49 (2Н, м), 3,40 (1H, д, J=13,9 Гц), 3,04 (12Н, к, 7,3 Гц), 2,93-2,90 (2Н, м), 2,04-1,99 (2Н, м), 1,23 (18Н, т, J=7,3 Гц), 0,96-0,92 (2Н, м), 0,84-0,80 (2Н, м), 0,76-0,73 (2Н, м), 0,02 (9Н, с).
[0753]
(Этап 5)
Динатрия (5R,7R,8R,12aR,14R,15R,15aS,16R)-7-(6-амино-2-{[(1-аминоциклопропил)метил]амино}-9Н-пурин-9-ил)-15,16-дигидрокси-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
(Диастереомер 2)
К раствору соединения (11,0 мг), полученного на описанном выше этапе 4, в тетрагидрофуране (474 мкл) добавляли раствор в тетрагидрофуране фторида тетрабутиламмония (приблизительно 1 М, 237 мкл) и реакционную смесь перемешивали в атмосфере азота при 40°С в течение 3 часов. После гашения путем добавления 10 мМ водного раствора ацетата триэтиламмония в реакционную смесь, продукт очищали с помощью препаративной ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 2% - 30% (0 мин - 30 мин)] и Sep-Pak (R) С18 [вода/ацетонитрил/0,1% триэтиламин] с получением указанного в заголовке соединения в виде соли триэтиламина.
Полученную соль триэтиламина подвергали солевому обмену таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением указанного в заголовке соединения (4,1 мг).
МС (ИЭР) m/z: 812(M-2Na+1H)-.
1Н-ЯМР (CD3OD) δ: 8,34 (1H, с), 8,01 (1H, с), 7,14 (1H, с), 6,32 (1H, д, J=6,0 Гц), 6,16 (1H, д, J=8,5 Гц), 5,43-5,37 (2Н, м), 4,78 (1H, т, J=5,4 Гц), 4,50-4,28 (5Н, м), 4,12-4,08 (1H, м), 3,95-3,89 (1Н, м), 3,69-3,64 (1Н, м), 3,51-3,48 (2Н, м), 3,26 (1Н, д, J=14,5), 2,89-2,86 (2Н, м), 2,03-1,98 (2Н, м), 0,83-0,79 (4Н, м).
31Р-ЯМР (CD3OD) δ: 62,3 (с), 60,0 (с).
[0754]
Пример 41. Синтез ЦДН31.
(5R,7R,8R,12aR,14R,15R,15aS,16R)-15,16-дигидрокси-7-[2-(гидроксиметил)-6-(метиламино)-9Н-пурин-9-ил]-2,10-бис(сульфанил)-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-дион
[0755]
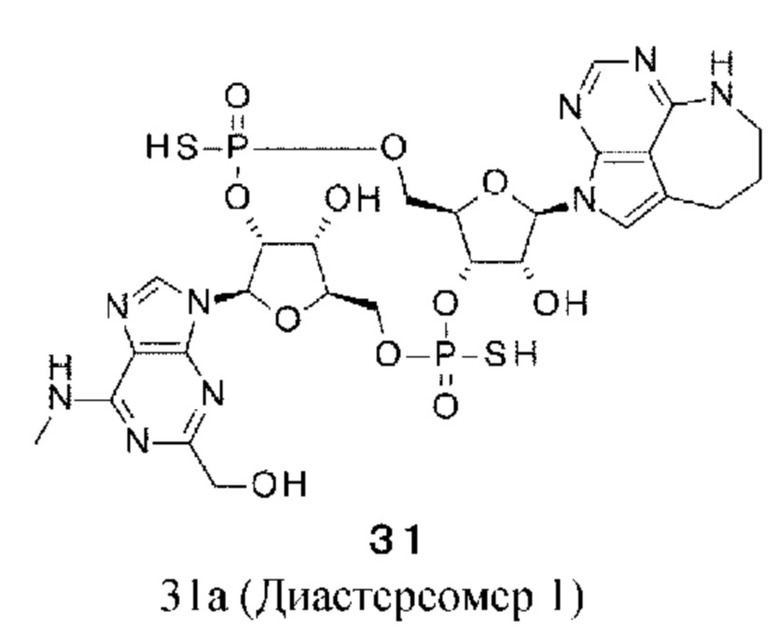
[0756]
[Схема синтеза]
[0757]
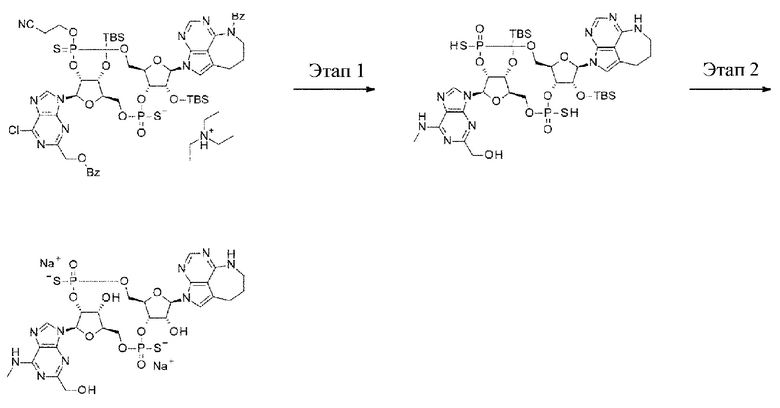
[0758]
(Этап 1)
(5R,7R,8R,12aR,14R,15R,15aR,16R)-15,16-бис{[трет-бутил(диметил)силил]окси}-7-[2-(гидроксиметил)-6-(метиламино)-9Н-пурин-9-ил]-2,10-бис(сульфанил)-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-дион
К раствору диастереомера 2 (более полярного), полученного на этапе 7 примера 12 (56,5 мг), в метаноле (1,00 мл) добавляли 40% водный раствор метиламина (1,00 мл), и реакционную смесь перемешивали в запечатанной пробирке при 60°С в течение 3 часов. Реакционную смесь концентрировали при пониженном давлении с получением смеси, содержащей указанное в заголовке соединение. Полученную смесь непосредственно использовали для следующей реакции.
МС (ИЭР) m/z: 1002(М+Н)+.
[0759]
(Этап 2)
Динатрия (5R,7R,8R,12aR,14R,15R,15aS,16R)-15,16-дигидрокси-7-[2-(гидроксиметил)-6-(метиламино)-9Н-пурин-9-ил]-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
С применением смеси, полученной на описанном выше этапе 1, проводили реакцию таким же образом, как на этапе 11 примера 1, и продукт затем очищали с помощью препаративной ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 2% - 20% (0 мин - 30 мин)] с получением указанного в заголовке соединения в виде соли триэтиламина.
Полученную соль триэтиламина подвергали солевому обмену таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением указанного в заголовке соединения (16,0 мг).
МС (ИЭР) m/z: 774(М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,70 (1H, с), 8,02 (1H, с), 7,13 (1H, с), 6,38 (1H, д, J=9,1 Гц), 6,33 (1H, д, J=7,3 Гц), 5,49-5,42 (2Н, м), 4,80 (1H, дд, J=6,7, 4,2 Гц), 4,60 (2Н, с), 4,50-4,28 (5Н, м), 4,05-4,00 (1H, м), 3,92-3,87 (1H, м), 3,52-3,49 (2Н, м), 3,14 (3H, уш. с), 2,91 (2Н, т, J=5,4 Гц), 2,04-1,99 (2Н, м).
31Р-ЯМР (CD3OD) δ: 63,1 (с), 60,3 (с).
[0760]
Пример 42. Синтез ЦДН32.
(5R,7R,8R,12aR,14R,15aS,16R)-16-гидрокси-7-[1-(2-гидроксиэтил)-6-оксо-1,6-дигидро-9Н-пурин-9-ил]-2,10-бис(сульфанил)-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-дион
[0761]
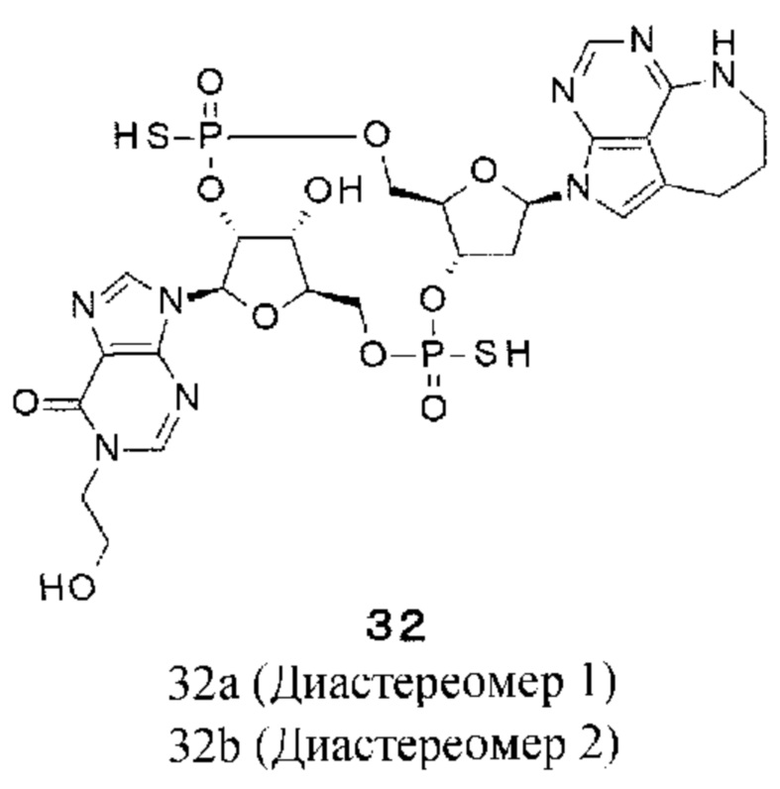
[0762]
[Схема синтеза]
[0763]
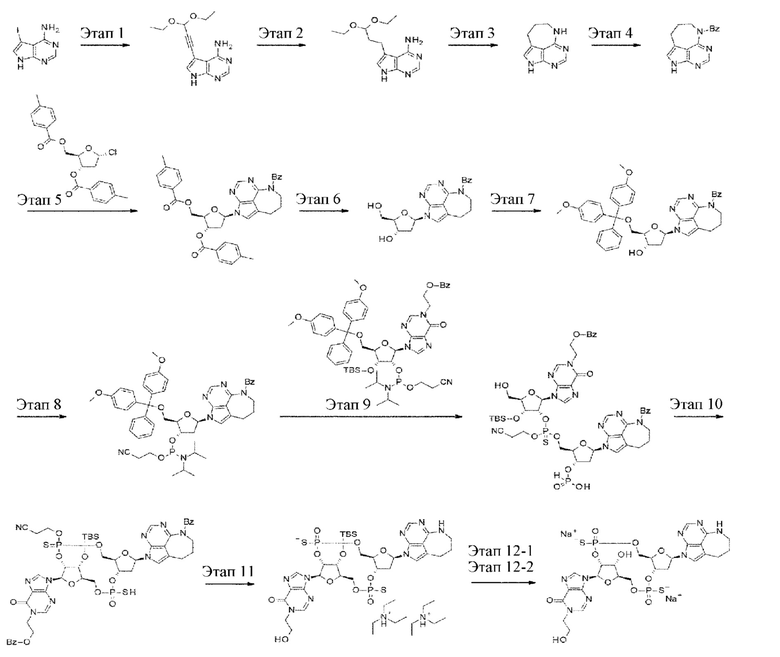
[0764]
(Этап 1)
5-(3,3-диэтоксипроп-1-ин-1-ил)-7Н-пирроло[2,3-с1]пиримидин-4-амин
К раствору доступного для приобретения (PharmaBlock Sciences (Nanjing), Inc.) 5-йод-7Н-пирроло[2,3-с1]пиримидин-4-амина (22 г) в N,N-диметилформамиде (70 мл) добавляли йодид меди (1,61 г), дихлорид бис(трифенилфосфин)палладия (5,94 г) и триэтиламин (35 мл). Пропаргилальдегиддиэтилацеталь (22 мл) добавляли туда в течение 2 часов, и реакционную смесь перемешивали при комнатной температуре в течение ночи. Хлороформ (350 мл) добавляли в реакционную смесь, которую промывали дважды водой. После высушивания органического слоя сульфатом магния, осушающий агент удаляли путем фильтрации и фильтрат концентрировали при пониженном давлении. Этилацетат (350 мл) добавляли к остатку и продукт перемешивали в течение ночи. Твердый преципитированный продукт собирали путем фильтрации с получением указанного в заголовке соединения (9,60 г).
МС (ИЭР) m/z: 261(М+Н)+.
1H-ЯМР (ДМСО-d6) δ: 12,04 (1H, уш. с), 8,11 (1H, уш. с), 7,57 (1Н, с), 6,56 (2Н, уш. с), 5,59 (1H, с), 3,68 (2Н, м), 3,57 (2Н, м), 1,17 (6Н, т, J=7,3 Гц).
[0765]
(Этап 2)
5-(3,3-диэтоксипропил)-7Н-пирроло[2,3-d]пиримидин-4-амин
В смешанный раствор соединения (17,9 г), полученного на описанном выше этапе 1, в тетрагидрофуране (160 мл)/этаноле (80 мл) добавляли 10% палладий-углерод (М) влажный (25,0 г), и реакционную смесь перемешивали в атмосфере водорода при комнатной температуре в течение ночи. Катализатор удаляли путем фильтрации с помощью целита и фильтрат концентрировали при пониженном давлении с получением неочищенной формы указанного в заголовке соединения (17,0 г).
МС (ИЭР) m/z: 265(М+Н)+.
[0766]
(Этап 3)
6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен
Соединение (50,21 г), полученное на описанном выше этапе 2, растворяли в 90% водном растворе уксусной кислоты (344 мл), и реакционную смесь перемешивали при 50°С в течение ночи. После подтверждения исчезновения сырьевого материала, палладий-углерод (М), влажный (60 г), добавляли в реакционную смесь, которую перемешивали в атмосфере водорода при 40°С в течение ночи. Катализатор удаляли путем фильтрации с помощью целита и фильтрат концентрировали при пониженном давлении. Насыщенный водный раствор гидрокарбоната натрия (350 мл) добавляли к остатку, и продукт подвергали семь раз экстрагированию хлороформом/метанолом (9:1). Органический слой концентрировали при пониженном давлении, и остаток затем очищали с помощью колоночной хроматографии на силикагеле [хлороформ/метанол] с получением указанного в заголовке соединения (18,47 г).
МС (ПЭР) m/z: 175(М+Н)+.
1Н-ЯМР (ДМСО-d6) δ: 11,26 (1H, уш. с), 7,97 (1H, с), 7,37 (1H, уш. с), 6,86 (1H, уш. с), 3,35 (2Н, м), 2,80 (2Н, т, J=5,4 Гц), 1,88 (2Н, м).
[0767]
(Этап 4)
Фенил(2,7,8,9-тетрагидро-6Н-2,3,5,6-тетраазабензо[cd]азулен-6-ил)метанон
В суспензию соединения (8,47 г), полученного на описанном выше этапе 3, в дихлорметане (120 мл) добавляли обезвоженный пиридин (39,2 мл), N,N-диметиламинопиридин (2,38 г) и бензоилхлорид (22,6 мл) в данном порядке, и реакционную смесь перемешивали при комнатной температуре в течение 1 часа. После концентрирования реакционной смеси при пониженном давлении добавляли к остатку хлороформ (150 мл), метанол (60 мл) и триэтиламин (50 мл) и продукт перемешивали при комнатной температуре в течение 3 часов. Реакционную смесь вливали в двухслойную смесь хлороформа и воды и осуществляли экстрагирование хлороформом. Органический слой промывали дважды 25% (масса/объем) водным раствором гидросульфата калия и сушили над безводным сульфатом магния. Осушающий агент удаляли путем фильтрации и фильтрат концентрировали при пониженном давлении. Этилацетат (50 мл) и гексан (125 мл) добавляли в данном порядке к остатку, чтобы получить взвесь, которую затем перемешивали в течение 1 часа. Твердый преципитированный продукт собирали путем фильтрации с получением указанного в заголовке соединения (9,89 г).
МС (ИЭР) m/z: 279(М+Н)+.
1H-ЯМР (CDCl3) δ: 10,07 (1H, уш. с), 8,11 (1H, с), 7,39-7,21 (5Н, м), 7,12 (1Н, с), 4,32 (2Н, м), 3,06 (2Н, м), 2,26 (2Н, м).
[0768]
(Этап 5)
6-бензоил-2-[2-дезокси-3,5-бис-O-(4-метилбензоил)-β-D-эритропентофуранозил]-6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен
В суспензию соединения (7,10 г), полученного на описанном выше этапе 4, в ацетонитриле (80 мл) добавляли порошкованный гидроксид калия (2,9 г) и трис[2-(2-метоксиэтокси)этил]амин (0,41 мл) в атмосфере азота и реакционную смесь перемешивали при комнатной температуре в течение 15 минут. При охлаждении льдом добавляли туда 2-дезокси-3,5-бис-O-(4-метилбензоил)-α-D-эритропентофуранозилхлорид (10,12 г) как соединение, известное в литературе (Synlett 2004(2): 335-337), и ацетонитрил (60 мл), температуру повышали до комнатной температуры и реакционную смесь перемешивали в течение ночи. Насыщенный водный раствор хлорида аммония добавляли в реакционную смесь, чтобы погасить реакцию. Твердый преципитированный продукт собирали путем фильтрации, а затем промывали водой с получением указанного в заголовке соединения (9,70 г).
МС (ИЭР) m/z: 631(М+Н)+.
1Н-ЯМР (CDCl3) δ: 8,10 (1H, с), 8,00-7,96 (4Н, м), 7,37-7,22 (9Н, м), 7,10 (1H, с), 6,88 (1H, дд, J=8,5, 5,4 Гц), 5,76 (1H, м), 4,77 (1H, дд, J=11,8, 3,9 Гц), 4,65-4,57 (2Н, м), 4,32 (1H, м), 4,20 (1H, м), 2,90-2,78 (3H, м), 2,73 (1H, ддд, J=2,1, 5,7, 8,5 Гц), 2,44 (6Н, с), 2,19 (2Н, м).
[0769]
(Этап 6)
6-бензоил-2-(2-дезокси-β-D-эритропентофуранозил)-6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен
В смешанный раствор соединения (8,69 г), полученного на описанном выше этапе 5, в метаноле (45 мл)-тетрагидрофуране (135 мл) добавляли по каплям 2 N водный раствор гидроксида натрия (27,6 мл) при -10°С в течение 20 минут. После перемешивания при той же температуре в течение 2 часов добавляли туда 1 N соляную кислоту (58 мл), чтобы погасить реакцию. После концентрирования реакционной смеси при пониженном давлении остаток очищали с помощью колоночной хроматографии на силикагеле [хлороформ/метанол] с получением указанного в заголовке соединения (4,09 г).
МС (ИЭР) m/z: 395(М+Н)+.
1Н-ЯМР (CDCl3) δ: 8,04 (1H, с), 7,39-7,23 (5Н, м), 7,05 (1H, с), 6,27 (1H, дд, J=9,7, 5,4 Гц), 6,00 (1H, д, J=10,9 Гц), 4,77 (1Н, д, J=4,8 Гц), 4,41 (1H, дд, J=14,5, 7,9 Гц), 4,19 (1H, с), 4,15 (1H, дд, J=14,5, 7,9 Гц), 3,94 (1H, д, J=12,7 Гц), 3,74 (1H, м), 3,16-2,95 (3H, м), 2,30-2,14 (3H, м), 1,96 (1H, с).
[0770]
(Этап 7)
6-бензоил-2-{5-O-[бис(4-метоксифенил)(фенил)метил]-2-дезокси-β-D-эритропентофуранозил}-6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен
С применением соединения (4,55 г), полученного на описанном выше этапе 6, проводили реакцию таким же образом, как на этапе 1 примера 11, с получением указанного в заголовке соединения (6,38 г).
МС (ИЭР) m/z: 697(М+Н)+.
1Н-ЯМР (CDCl3) δ: 8,08 (1H, с), 7,43 (2Н, д, J=7,9 Гц), 7,36-7,19 (13Н, м), 6,84-6,76 (5Н, м), 4,66 (1H, уш. с), 4,31 (1H, м), 4,21 (1H, м), 4,07 (1H, м), 3,79 (6Н, с), 3,44 (1Н, дд, J=10,0, 3,9 Гц), 3,38 (1H, дд, J=10,3, 4,8 Гц), 2,85 (2Н, т, J=6,3 Гц), 2,66 (1H, м), 2,47 (1H, ддд, J=4,1, 6,5, 13,9 Гц), 2,18 (2Н, м) (показаны только наблюдаемые пики).
[0771]
(Этап 8)
6-бензоил-2-(5-O-[бис(4-метоксифенил)(фенил)метил]-3-O-{(2-цианоэтокси)[ди(пропан-2-ил)амино]фосфанил}-2-дезокси-β-D-эритропентофуранозил)-6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо [cd]азулен
С применением соединения (6,37 г), полученного на описанном выше этапе 7, проводили реакцию таким же образом, как на этапе 6 примера 1, с получением указанного в заголовке соединения (6,05 г) в виде смеси диастереомеров (соотношение диастереомеров=1:1).
1H-ЯМР (CDCl3) δ: 8,09 (0,5Н, с), 8,08 (0,5Н, с), 7,46-7,41 (2Н, м), 7,35-7,19 (13Н, м), 6,84-6,75 (5Н, м), 4,82-4,76 (1H, м), 4,35-4,19 (3H, м), 3,89-3,54 (4Н, м), 3,79 (1,5Н, с), 3,79 (1,5Н, с), 3,78 (1,5Н, с), 3,78 (1,5Н, с), 3,42 (1H, тд, J=9,8, 3,8 Гц), 3,38-3,30 (1H, м), 2,81 (2Н, т, J=6,3 Гц), 2,74-2,66 (1H, м), 2,63-2,49 (1H, м), 2,62 (1H, т, J=6,0 Гц), 2,45 (1H, т, J=6,3 Гц), 2,21-2,14 (2Н, м), 1,20-1,17 (9Н, м), 1,11 (3H, д, J=6,7 Гц).
[0772]
(Этап 9)
С применением соединения (868 мг), полученного на описанном выше этапе 8, проводили реакцию таким же образом, как на этапе 7 примера 1, с получением раствора в ацетонитриле 6-бензоил-2-{2-дезокси-3-O-[гидрокси(оксо)-λ5-фосфанил]-β-D-эритропентофуранозил}-6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулена. С применением данного раствора в ацетонитриле и соединения (1,00 г), полученного на этапе 3 примера 22, проводили реакцию таким же образом, как на этапе 8 примера 1, и полученный в результате этого неочищенный продукт непосредственно использовали для следующей реакции.
[0773]
(Этап 10)
2-{9-[(5R,7R,8R,12aR,14R,15aS,16R)-14-(6-бензоил-6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)-16-{[трет-бутил(диметил)силил]окси}-10-(2-цианоэтокси)-2-оксо-2-сульфанил-10-сульфанилиденоктагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-7-ил]-6-оксо-6,9-дигидро-1Н-пурин-1-ил}этилбензоат
С применением неочищенного продукта, полученного на этапе 9, проводили реакцию таким же образом, как на описанном выше этапе 9 примера 1, с получением указанного в заголовке соединения (702 мг) в виде смеси диастереомеров у атома фосфора.
МС (ИЭР) m/z: 1134(М+Н)+.
[0774]
(Этап 11)
Бис(N,N-диэтилэтанаминия) (5R,7R,8R,12aR,14R,15aS,16R)-16-{[трет-бутил(диметил)силил]окси}-7-[1-(2-гидроксиэтил)-6-оксо-1,6-дигидро-9Н-пурин-9-ил]-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1]1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
С применением соединения (702 мг), полученного на описанном выше этапе 10, проводили реакцию таким же образом, как на этапе 10 примера 1, с получением диастереомера 1(119 мг: с примесями) и диастереомера 2(113 мг: с примесями) указанного в заголовке соединения. Диастереомер 1 (менее полярный) МС (ИЭР) m/z: 873(М+Н)+.
Диастереомер 2 (более полярный)
МС (ИЭР) m/z: 873(М+Н)+.
[0775]
(Этап 12-1)
Динатрия (5R,7R,8R,12aR,14R,15aS,16R)-16-гидрокси-7-[1-(2-гидроксиэтил)-6-оксо-1,6-дигидро-9Н-пурин-9-ил]-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
(Диастереомер 1)
С применением соединения (диастереомера 1) (119 мг: с примесями), полученного на описанном выше этапе 11, проводили реакцию таким же образом, как на этапе 11 примера 1, а затем проводили очистку при следующих [Условиях очистки] с получением указанного в заголовке соединения в виде соли триэтиламина.
[Условия очистки] Хроматография на колонке с силикагелем С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил] и препаративная ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 5% - 30% (0 мин - 40 мин)].
Полученную соль триэтиламина подвергали солевому обмену таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением указанного в заголовке соединения (51,8 мг).
МС (ПЭР) m/z: 759(М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,69 (1H, с), 8,24 (1H, с), 8,02 (1H, с), 7,09 (1H, с), 6,68 (1H, т, J=7,0 Гц), 6,29 (1H, д, J=8,5 Гц), 5,36-5,27 (2Н, м), 4,75-4,71 (1H, м), 4,41-4,30 (3H, м), 4,28-4,12 (3H, м), 4,06-4,00 (1H, м), 3,94-3,84 (1H, м), 3,82 (2Н, т, J=4,8 Гц), 3,52-3,47 (2Н, м), 2,91-2,69 (4Н, м), 2,04-1,96 (2Н, м).
31Р-ЯМР (CD3OD) 5: 57,3 (с), 54,9 (с).
[0776]
(Этап 12-2)
Динатрия (5R,7R,8R,12aR,14R,15aS,16R)-16-гидрокси-7-[1-(2-гидроксиэтил)-6-оксо-1,6-дигидро-9Н-пурин-9-ил]-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[с(1]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
(Диастереомер 2)
С применением соединения (диастереомера 2) (113 мг: с примесями), полученного на описанном выше этапе 11, проводили реакцию таким же образом, как на этапе 11 примера 1, а затем проводили очистку при следующих [Условиях очистки] с получением указанного в заголовке соединения в виде соли триэтиламина.
[Условия очистки] Хроматография на колонке с силикагелем С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил] и препаративная ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 5% - 25% (0 мин - 40 мин)].
Полученную соль триэтиламина подвергали солевому обмену таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением указанного в заголовке соединения (65,4 мг).
МС (ИЭР) m/z: 759(М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,78 (1H, с), 8,24 (1H, с), 8,02 (1H, с), 7,08 (1H, с), 6,73 (1H, дд, J=9,1, 5,4 Гц), 6,29 (1H, д, J=8,5 Гц), 5,59-5,52 (1H, м), 5,45-5,37 (1H, м), 4,45 (1H, д, J=4,2 Гц), 4,40-4,16 (6Н, м), 4,01 (1H, д, J=12,7 Гц), 3,87-3,75 (3H, м), 3,53-3,46 (2Н, м), 2,93-2,88 (2Н, м), 2,87-2,65 (2Н, м), 2,06-1,96 (2Н, м).
31Р-ЯМР (CD3OD) δ: 63,1 (с), 57,3 (с).
[0777]
Пример 43. Синтез ЦДН33.
(5R,7R,8R,12aR,14R,15S,15aR,16R)-15-фтор-16-гидрокси-7-[1-(2-гидроксиэтил)-6-оксо-1,6-дигидро-9Н-пурин-9-ил]-2,10-бис(сульфанил)-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-дион
[0778]
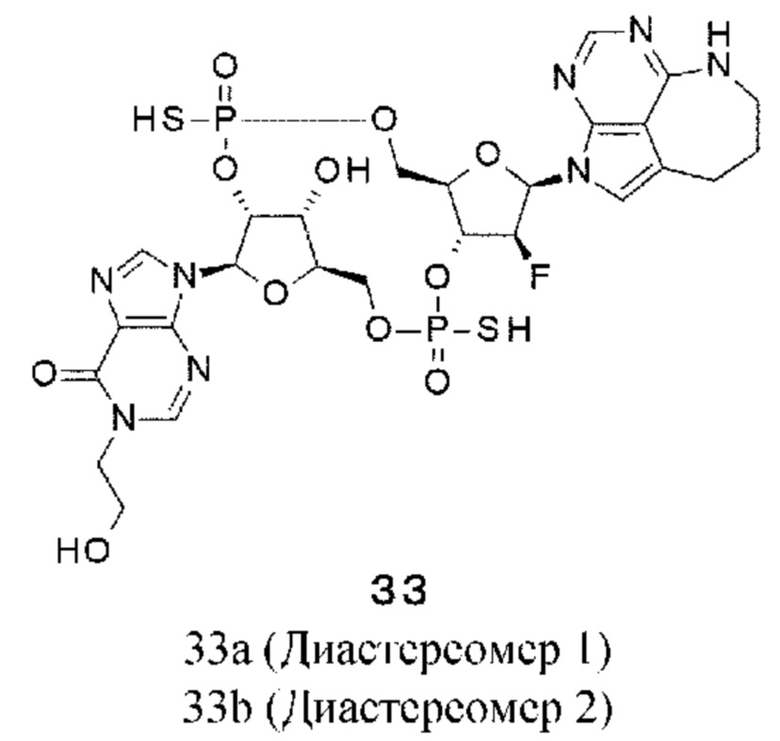
[0779]
[Схема синтеза]
[0780]
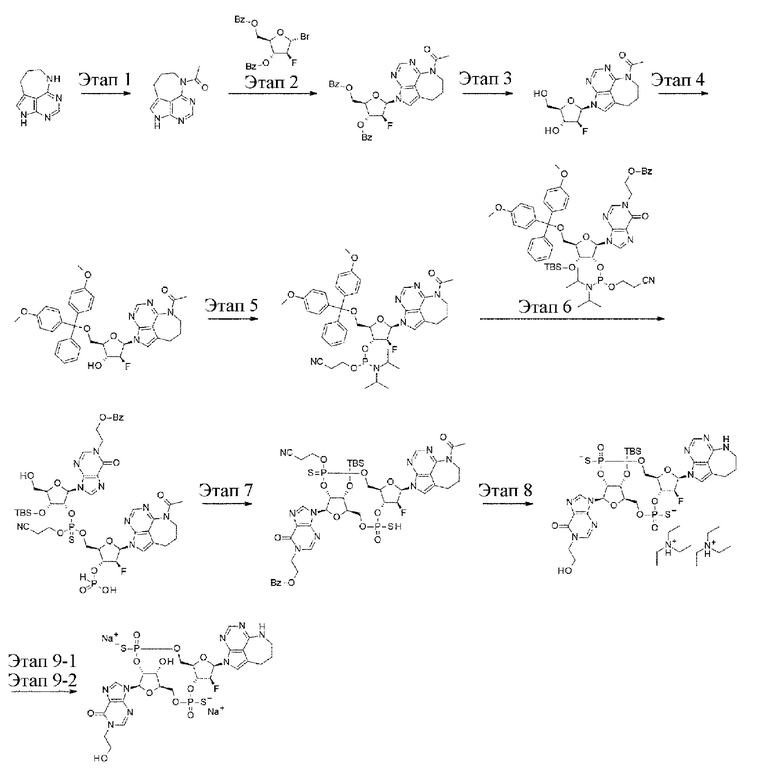
[0781]
(Этап 1)
1-(2,7,8,9-тетрагидро-6Н-2,3,5,6-тетраазабензо[cd]азулен-6-ил)этан-1-он
Соединение (6,88 г), полученное на этапе 3 примера 42, растворяли в уксусном ангидриде (48 мл) и реакционную смесь перемешивали при 90°С. Реакционную смесь концентрировали при пониженном давлении и остаток растворяли в смеси хлороформ (100 мл)/метанол (50 мл)/триэтиламин (30 мл). После перемешивания при комнатной температуре в течение 4 часов реакционную смесь вливали в двухслойную смесь хлороформа и воды и осуществляли экстрагирование хлороформом. После высушивания органического слоя над сульфатом магния осушающий агент удаляли путем фильтрации и фильтрат концентрировали при пониженном давлении. Гексан/этилацетат (1:2) добавляли к остатку, чтобы получить взвесь, и твердый продукт затем собирали путем фильтрации с получением указанного в заголовке соединения (7,18 г).
МС (ИЭР) m/z: 217(М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,47 (1H, с), 7,21 (1H, с), 4,11 (2Н, д, J=8,5 Гц), 2,97 (2Н, т, J=6,3 Гц), 2,46 (3H, с), 2,06 (2Н, м).
[0782]
(Этап 2)
6-ацетил-2-(3,5-ди-O-бензоил-2-дезокси-2-фтор-β-D-арабинофуранозил)-6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен
С применением соединения (6,00 г), полученного на описанном выше этапе 1, и доступного для приобретения (Carbosynth Limited) [(2R,3R,4S,5R)-3-бензоилокси-5-бром-4-фтор-тетрагидрофуран-2-ил]метилбензоата (14,1 г) проводили реакцию таким же образом, как на этапе 5 примера 42, с получением указанного в заголовке соединения (11,45 г).
МС (ИЭР) m/z: 559(М+Н)+.
1Н-ЯМР (CDCl3) δ: 8,58 (1H, с), 8,12 (4Н, т, J=7,3 Гц), 7,69-7,44 (6Н, м), 7,28 (1H, д, J=2,4 Гц), 6,92 (1H, дд, J=23,3, 2,7 Гц), 5,77 (1H, дд, J=17,5, 3,0 Гц), 5,34 (1H, дд, J=50,2, 3,0 Гц), 4,83 (2Н, дд, J=11,8, 4,5 Гц), 4,76 (2Н, дд, J=11,8, 5,1 Гц), 4,56 (1H, м), 4,14 (2Н, м), 2,90 (2Н, т, J=6,7 Гц), 2,07 (3H, с).
[0783]
(Этап 3)
6-ацетил-2-(2-дезокси-2-фтор-β-D-арабинофуранозил)-6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен
В смешанный раствор соединения (8,70 г), полученного на описанном выше этапе 2, в метаноле (58 мл)/тетрагидрофуране (117 мл) добавляли по каплям 2 N водный раствор гидроксида натрия (32 мл) при -20°С в течение 12 минут. После перемешивания при той же температуре в течение 3 часов добавляли туда 1 N соляную кислоту (66 мл), чтобы погасить реакцию. Реакционную смесь концентрировали при пониженном давлении и остаток затем очищали с помощью колоночной хроматографии на силикагеле [хлороформ/метанол] с получением указанного в заголовке соединения (3,27 г).
МС (ИЭР) m/z: 351(М+Н)+.
1Н-ЯМР (CDCl3) δ: 8,54 (1H, с), 7,20 (1H, д, J=1,8 Гц), 6,68 (1H, дд, J=17,5, 4,2 Гц), 5,16 (1H, ддд, J=52,0, 2,4, 1,2 Гц), 4,75 (1H, ддд, J=19,2, 2,6, 1,3 Гц), 4,16 (1H, м), 4,08 (2Н, м), 3,99 (1H, дд, J=12,1, 3,6 Гц), 3,92 (1H, дд, J=12,1, 4,2 Гц), 2,93 (2Н, м), 2,52 (3H, с), 2,07 (2Н, м) (показаны только наблюдаемые пики).
[0784]
(Этап 4)
6-ацетил-2-{5-O-[бис(4-метоксифенил)(фенил)метил]-2-дезокси-2-фтор-β-D-арабинофуранозил}-6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен
С применением соединения (3,95 г), полученного на описанном выше этапе 3 (3,95 г), проводили реакцию таким же образом, как на этапе 1 примера 11, с получением указанного в заголовке соединения (5,66 г).
МС (ИЭР) m/z: 653(М+Н)+.
1Н-ЯМР (CDCl3) δ: 8,56 (1H, с), 7,50-7,20 (ЮН, м), 6,85-6,78 (5Н, м), 5,07 (1H, дт, J=51,8, 3,2 Гц), 4,59 (1H, уш. д, J=18,1 Гц), 4,21-4,02 (3H, м), 3,80 (3H, с), 3,79 (3H, с), 3,50 (1H, дд, J=10,3, 5,4 Гц), 3,44 (1H, дд, J=10,0, 5,1 Гц), 2,88 (2Н, м), 2,54 (3H, с), 2,42 (1H, уш. с), 2,07 (2Н, м).
[0785]
(Этап 5)
6-ацетил-2-(5-O-[бис(4-метоксифенил)(фенил)метил]-3-O-{(2-цианоэтокси)[ди(пропан-2-ил)амино]фосфанил}-2-дезокси-2-фтор-β-D-арабинофуранозил)-6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен
С применением соединения (5,68 г), полученного на описанном выше этапе 4, проводили реакцию таким же образом, как на этапе 6 примера 1, с получением указанного в заголовке соединения (5,08 г) в виде смеси диастереомеров у атома фосфора (соотношение диастереомеров=1:1).
МС (ИЭР) m/z: 853(М+Н)+.
1Н-ЯМР (CDCl3) δ: 8,58 (0,5Н, с), 8,57 (0,5Н, с), 7,51-7,20 (10Н, м), 6,85-6,76 (5Н, м), 5,24-5,02 (1H, м), 4,76-4,60 (1H, м), 4,21-4,05 (3H, м), 3,91-3,74 (1H, м), 3,80 (1,5Н, с), 3,79 (1,5Н, с), 3,79 (1,5Н, с), 3,79 (1,5Н, с), 3,69-3,55 (3H, м), 3,49-3,37 (2Н, м), 2,95-2,80 (2Н, м), 2,61 (1H, т, J=6,3 Гц), 2,54 (3H, с), 2,43 (1Н, т, J=6,7 Гц), 2,11-2,03 (2Н, м), 1,21-1,17 (9Н, м), 1,11 (3H, д, J=7,3 Гц).
[0786]
(Этап 6)
С применением соединения (1,00 г), полученного на описанном выше этапе 5, проводили реакцию таким же образом, как на этапе 7 примера 1, с получением раствора в ацетонитриле 6-ацетил-2-{2-дезокси-2-фтор-3-O-[гидрокси(оксо)-λ5-фосфанил]-β-D-арабинофуранозил}-6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[сс1]азулена. С применением данного раствора в ацетонитриле и соединения (1,21 г), полученного на этапе 3 примера 22, проводили реакцию таким же образом, как на этапе 8 примера 1, и полученный в результате этого неочищенный продукт непосредственно использовали для следующей реакции.
[0787]
(Этап 7)
2-{9-[(5R,7R,8R,12aR,14R,15S,15aR,16R)-14-(6-ацетил-6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)-16-{[трет-бутил(диметил)силил]окси}-10-(2-цианоэтокси)-15-фтор-2-оксо-2-сульфанил-10-сульфанилиденоктагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-7-ил]-6-оксо-6,9-дигидро-1Н-пурин-1-ил}этилбензоат
С применением неочищенного продукта, полученного на описанном выше этапе 6, проводили реакцию таким же образом, как на этапе 9 примера 1, с получением указанного в заголовке соединения (757 мг) в виде смеси диастереомеров у атома фосфора.
MC (ИЭР) m/z: 1090(М+Н)+.
[0788]
(Этап 8)
Бис(N,N-диэтилэтанаминия) (5R,7R,8R,12aR,14R,15S,15aR,16R)-16-{[трет-бутил(диметил)силил]окси}-15-фтор-7-[1-(2-гидроксиэтил)-6-оксо-1,6-дигидро-9Н-пурин-9-ил]-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
С применением соединения (757 мг), полученного на описанном выше этапе 7, проводили реакцию таким же образом, как на этапе 10 примера 1, с получением диастереомера 1 (113 мг: с примесями) и диастереомера 2 (108 мг: с примесями) указанного в заголовке соединения. Диастереомер 1 (менее полярный) МС (ИЭР) m/z: 891(М+Н)+.
Диастереомер 2 (более полярный)
МС (ИЭР) m/z: 891(М+Н)+.
[0789]
(Этап 9-1)
Динатрия (5R,7R,8R,12aR,14R,15S,15aR,16R)-15-фтор-16-гидрокси-7-[1-(2-гидроксиэтил)-6-оксо-1,6-дигидро-9Н-пурин-9-ил]-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
(Диастереомер 1)
С применением соединения (диастереомера 1) (113 мг: с примесями), полученного на описанном выше этапе 8, проводили реакцию таким же образом, как на этапе 11 примера 1, а затем проводили очистку при следующих [Условиях очистки] с получением указанного в заголовке соединения в виде соли триэтиламина.
[Условия очистки] Хроматография на колонке с силикагелем С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил] и препаративная ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 5% - 30% (0 мин - 30 мин)].
Полученную соль триэтиламина подвергали солевому обмену таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением указанного в заголовке соединения (28,8 мг).
МС (ИЭР) m/z: 777(М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,67 (1H, с), 8,24 (1Н, с), 8,04 (1Н, с), 7,05 (1H, с), 6,73 (1Н, дд, J=23,9, 2,7 Гц), 6,28 (1H, д, J=8,5 Гц), 5,43-5,24 (3H, м), 4,77-4,72 (1Н, м), 4,51-4,32 (4Н, м), 4,26-4,12 (2Н, м), 4,06-3,91 (2Н, м), 3,82 (2Н, т, J=5,1 Гц), 3,50 (2Н, т, J=5,1 Гц), 2,92-2,85 (2Н, м), 2,06-1,97 (2Н, м).
31Р-ЯМР (CD3OD) δ: 57,8 (с), 54,7 (с).
[0790]
(Этап 9-2)
Динатрия (5R,7R,8R,12aR,14R,15S,15aR,16R)-15-фтор-16-гидрокси-7-[1-(2-гидроксиэтил)-6-оксо-1,6-дигидро-9Н-пурин-9-ил]-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
(Диастереомер 2)
С применением соединения (диастереомера 2) (108 мг: с примесями), полученного на описанном выше этапе 8, проводили реакцию таким же образом, как на этапе 11 примера 1, а затем проводили очистку при следующих [Условиях очистки] с получением указанного в заголовке соединения в виде соли триэтиламина.
[Условия очистки] Хроматография на колонке с силикагелем С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил] и препаративная ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 5% - 20% (0 мин - 30 мин)].
Полученную соль триэтиламина подвергали солевому обмену таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением указанного в заголовке соединения (3,2 мг). МС (ИЭР) m/z: 777(М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,76 (1H, с), 8,23 (1Н, с), 8,04 (1Н, с), 7,06 (1H, с), 6,73 (1Н, дд, Г=24,5, 2,1 Гц), 6,28 (1H, д, J=8,5 Гц), 5,53-5,45 (1H, м), 5,43-5,27 (2Н, м), 4,62-4,49 (1H, м), 4,45-4,41 (1H, м), 4,40-4,27 (2Н, м), 4,27-4,15 (3H, м), 4,03-3,92 (2Н, м), 3,87-3,78 (2Н, м), 3,51 (2Н, т, J=5,6 Гц), 2,90 (2Н, т, J=5,6 Гц), 2,08-1,96 (2Н, м).
31Р-ЯМР (CD3OD) δ: 63,0 (с), 57,8 (с).
[0791]
Пример 44. Синтез ЦДН34.
(5R,7R,8R,12aR,14R,15R,15aR,16R)-15-фтор-16-гидрокси-7-[1-(2-гидроксиэтил)-6-оксо-1,6-дигидро-9Н-пурин-9-ил]-2,10-бис(сульфанил)-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-дион
[0792]
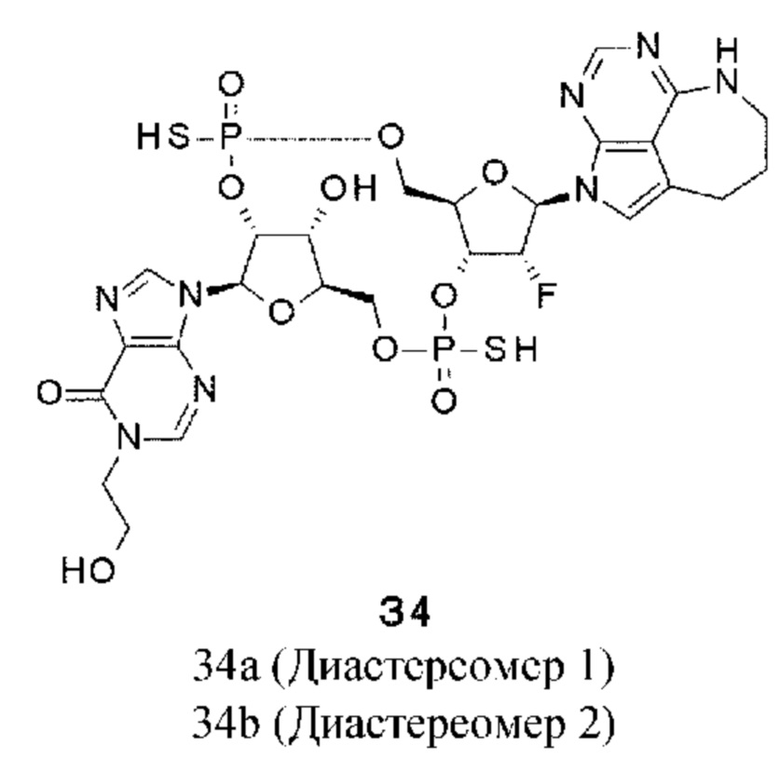
[0793]
[Схема синтеза]
[0794]
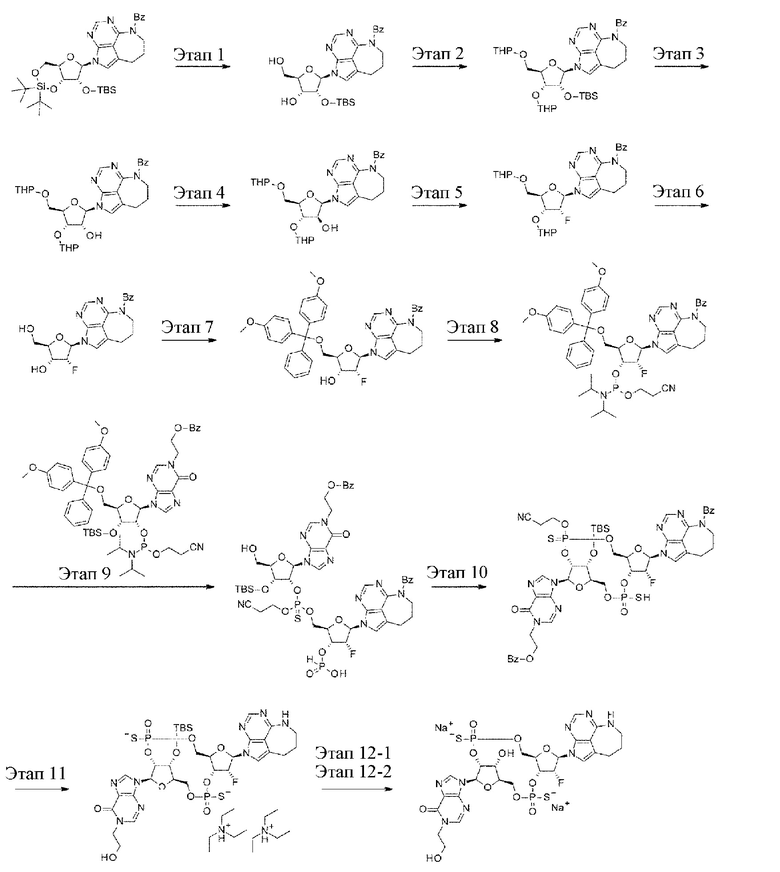
[0795] (Этап 1)
6-бензоил-2-{2-O-[трет-бутил(диметил)силил]-β-D-рибофуранозил}-6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен
В смешанный раствор соединения (35,80 г), полученного на этапе 4 примера 1, в дихлорметане (322 мл)/пиридине (35 мл) добавляли раствор фтороводорода/пиридина (6,33 г) в дихлорметане (36 мл) при охлаждении льдом в течение 5 минут, и реакционную смесь перемешивали при той же температуре в течение 3 часов. Насыщенный водный раствор гидрокарбоната натрия (268 мл) и рассола (143 мл) добавляли в реакционную смесь в данном порядке, чтобы погасить реакцию, и осуществляли экстрагирование продукта этилацетатом. После высушивания органического слоя над безводным сульфатом натрия осушающий агент удаляли путем фильтрации и фильтрат концентрировали при пониженном давлении. Гексан/этилацетат (1:1) (108 мл) добавляли к остатку, чтобы получить взвесь, которую затем перемешивали при 50°С в течение 30 минут, и дополнительно добавляли туда гексан (161 мл) и продукт дополнительно перемешивали в течение 2 часов. Твердый преципитированный продукт собирали путем фильтрации и промывали гексаном/этилацетатом (4:1) (143 мл) с получением указанного в заголовке соединения (26,81 г).
МС (ПЭР) m/z: 525(М+Н)+.
1Н-ЯМР (ДМСО-d6) δ: 7,98 (1H, с), 7,65 (1H, с), 7,39 (1Н, м), 7,26-7,20 (4Н, м), 6,19 (1H, д, J=6,5 Гц), 5,15 (1H, т, J=5,6 Гц), 5,00 (1H, д, J=4,8 Гц)4,48 (1H, т, J=5,6 Гц), 4,27 (1H, м), 4,11-4,02 (2Н, м), 3,97 (1H, м), 3,67-3,57 (2Н, м), 2,99 (2Н, м), 2,23-2,07 (2Н, м), 0,68 (9Н, с), -0,11 (3H, с), -0,26 (3H, с).
[0796]
(Этап 2)
6-бензоил-2-{2-O-[трет-бутил(диметил)силил]-3,5-бис-O-(оксан-2-ил)-β-D-рибофуранозил}-6,7,8,9-тетрагидро-2H-2,3,5,6-тетраазабензо[cd]азулен
К раствору соединения (19,93 г), полученного на описанном выше этапе 1, и 3,4-дигидро-2Н-пирана (35 мл) в N,N-диметилформамиде (200 мл) добавляли моногидрат п-толуолсульфоновой кислоты (7,25 г) при охлаждении льдом, и реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Насыщенный водный раствор гидрокарбоната натрия добавляли в реакционную смесь при охлаждении льдом, чтобы погасить реакцию, и реакционную смесь осуществляли экстрагирование этилацетатом. Органический слой промывали водой и рассолом в данном порядке и сушили над безводным сульфатом натрия. Осушающий агент удаляли путем фильтрации и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле [гексан/этилацетат] с получением указанного в заголовке соединения (24,73 г).
1Н-ЯМР (CDCl3) δ: 8,10-8,07 (1H, м), 7,59-7,35 (1H, м), 7,35-7,27 (3Н, м), 7,25-7,17 (2Н, м), 6,44-6,36 (1H, м), 4,90-3,36 (13Н, м), 3,06-2,96 (2Н, м), 2,31-2,15 (2Н, м), 2,01-1,43 (12Н, м), 0,84-0,73 (9Н, м), 0,04-(-0,35) (6Н, м).
[0797]
(Этап 3)
6-бензоил-2-[3,5-бис-O-(оксан-2-ил)-β-D-рибофуранозил]-6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен
К раствору соединения (24,73 г), полученного на описанном выше этапе 2, и уксусной кислоты (3,1 мл) в тетрагидрофуране (250 мл) добавляли раствор в тетрагидрофуране фторида тетрабутиламмония (приблизительно 1 М, 55 мл) при охлаждении льдом, и реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь концентрировали при пониженном давлении, этилацетат добавляли к остатку и продукт промывали водой и рассолом в данном порядке. После высушивания органического слоя над безводным сульфатом натрия осушающий агент удаляли путем фильтрации и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле [гексан/этилацетат] с получением указанного в заголовке соединения (18,74 г).
1H-ЯМР (CDCl3) δ: 8,12-8,09 (1H, м), 7,49-7,30 (4Н, м), 7,28-7,20 (2Н, м), 6,41-6,30 (1H, м), 4,83-4,18 (7Н, м), 4,12-3,50 (7Н, м), 3,06-2,97 (2Н, м), 2,31-2,17 (2Н, м), 1,96-1,47 (12Н, м).
[0798]
(Этап 4)
6-бензоил-2-[3,5-бис-O-(оксан-2-ил)-β-D-арабинофуранозил]-6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен
К раствору соединения (18,74 г), полученного на описанном выше этапе 3, и пиридина (13,1 мл) в дихлорметане (300 мл) добавляли по каплям трифторметансульфоновый ангидрид (11 мл) при охлаждении льдом, и реакционную смесь перемешивали в течение 10 минут. Рассол добавляли в реакционную смесь, чтобы погасить реакцию, осуществляли экстрагирование продукта дихлорметаном и органический слой сушили над безводным сульфатом натрия. Осушающий агент удаляли путем фильтрации и фильтрат концентрировали при пониженном давлении. Остаток растворяли в тетрагидрофуране (300 мл), добавляли по каплям раствор нитрита тетрабутиламмония (28,34 г) в тетрагидрофуране (150 мл) при охлаждении льдом и реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь концентрировали при пониженном давлении, этилацетат добавляли к остатку и продукт промывали водой и рассолом в данном порядке. После высушивания органического слоя над безводным сульфатом натрия осушающий агент удаляли путем фильтрации и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле [гексан/этилацетат] с получением указанного в заголовке соединения (10,46 г).
1H-ЯМР (CDCl3) δ: 8,13-8,06 (1H, м), 7,63-7,30 (4Н, м), 7,29-7,18 (2Н, м), 6,79-6,55 (1H, м), 4,93-3,45 (14Н, м), 3,11-2,95 (2Н, м), 2,32-2,14 (2Н, м), 1,98-1,44 (12Н, м).
[0799]
(Этап 5)
6-бензоил-2-[2-дезокси-2-фтор-3,5-бис-O-(оксан-2-ил)-β-D-рибофуранозил]-6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен
К раствору соединения (10,46 г), полученного на описанном выше этапе 4, и пиридина (7,3 мл) в дихлорметане (200 мл) добавляли по каплям трифторметансульфоновый ангидрид (6,1 мл) при охлаждении льдом и реакционную смесь перемешивали в течение 10 минут.Рассол добавляли в реакционную смесь, чтобы погасить реакцию, осуществляли экстрагирование продукта дихлорметаном и органический слой сушили над безводным сульфатом натрия. Осушающий агент удаляли путем фильтрации и фильтрат концентрировали при пониженном давлении. Остаток растворяли в тетрагидрофуране (200 мл), добавляли туда раствор в тетрагидрофуране фторида тетрабутил аммония (приблизительно 1 М, 150 мл) и продукт перемешивали при той же температуре в течение 3 часов. Насыщенный водный раствор хлорида аммония добавляли в реакционную смесь, которую осуществляли экстрагирование этилацетатом. Органический слой промывали рассолом и сушили над безводным сульфатом натрия, осушающий агент затем удаляли путем фильтрации и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле [гексан/этилацетат] с получением указанного в заголовке соединения (7,65 г).
1H-ЯМР (CDCl3) δ: 8,13-8,08 (1H, м), 7,53-7,31 (4Н, м), 7,26-7,22 (2Н, м), 6,68-6,53 (1H, м), 5,42-5,08 (1H, м), 4,93-4,18 (6Н, м), 4,10-3,76 (3Н, м), 3,71-3,47 (3Н, м), 3,06-2,96 (2Н, м), 2,29-2,18 (2Н, м), 1,96-1,47 (12Н, м).
[0800]
(Этап 6)
6-бензоил-2-(2-дезокси-2-фтор-β-D-рибофуранозил)-6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен
К раствору соединения (7,65 г), полученного на описанном выше этапе 5, в этаноле (150 мл) добавляли п-толуолсульфонат пиридиния (6,62 г) и реакционную смесь перемешивали при 50°С в течение 3 часов. Реакционную смесь концентрировали при пониженном давлении, этилацетат добавляли к остатку и продукт промывали насыщенным водным раствором гидрокарбоната натрия и рассолом в данном порядке. После высушивания органического слоя над безводным сульфатом натрия осушающий агент удаляли путем фильтрации и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле [гексан/этилацетат] с получением указанного в заголовке соединения (3,55 г).
1H-ЯМР (CDCl3) δ: 8,05 (1H, с), 7,41-7,35 (3Н, м), 7,30-7,24 (2Н, м), 7,06 (1H, с), 6,07-6,00 (2Н, м), 5,85 (1Н, ддд, J=52,8, 6,7, 4,7 Гц), 4,66 (1H, д, J=3,9 Гц), 4,42-4,31 (2Н, м), 4,20 (1Н, м), 3,93 (1Н, дд, J=12,9, 1,6 Гц), 3,74 (1H, тд, J=12,3, 1,6 Гц), 3,12-2,96 (2Н, м), 2,51 (1Н, с), 2,33-2,15 (2Н, м).
[0801]
(Этап 7)
6-бензоил-2-{5-O-[бис(4-метоксифенил)(фенил)метил]-2-дезокси-2-фтор-β-D-рибофуранозил}-6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен
С применением соединения (3,55 г), полученного на описанном выше этапе 6, проводили реакцию таким же образом, как на этапе 1 примера 11, с получением указанного в заголовке соединения (5,77 г).
1Н-ЯМР (CDCl3) δ: 8,09 (1Н, с), 7,45-7,41 (2Н, м), 7,36-7,17 (13Н, м), 6,85-6,79 (4Н, м), 6,53 (1H, дд, J=17,2, 2,3 Гц), 5,40 (1Н, ддд, J=53,2, 4,8, 2,3 Гц), 4,83-4,72 (1H, м), 4,32-4,21 (2Н, м), 4,19-4,14 (1H, м), 3,79 (3Н, с), 3,79 (3Н, с), 3,59 (1H, дд, J=11,0, 2,7 Гц), 3,45 (1H, дд, J=11,0, 3,5 Гц), 2,79 (2Н, т, J=6,3 Гц), 2,45 (1H, с), 2,24-2,11 (2Н, м).
[0802]
(Этап 8)
6-бензоил-2-(5-O-[бис(4-метоксифенил)(фенил)метил]-3-O-{(2-цианоэтокси)[ди(пропан-2-ил)амино]фосфанил}-2-дезокси-2-фтор-β-D-рибофуранозил)-6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен
С применением соединения (5,77 г), полученного на описанном выше этапе 7, проводили реакцию таким же образом, как на этапе 6 примера 1, с получением указанного в заголовке соединения (5,95 г) в виде смеси диастереомеров у атома фосфора (соотношение диастереомеров=1:1).
1H-ЯМР (CDCl3) δ: 8,10 (0,5Н, с), 8,09 (0,5Н, с), 7,45-7,12 (15Н, м), 6,84-6,75 (4Н, м), 6,57-6,46 (1H, м), 5,61-5,33 (1H, м), 5,07-4,83 (1H, м), 4,34-4,18 (3Н, м), 3,93-3,72 (7Н, м), 3,69-3,49 (4Н, м), 3,38-3,27 (1H, м), 2,87-2,68 (2Н, м), 2,61 (1H, тд, J=6,3, 1,6 Гц), 2,40 (1H, тд, J=6,4, 2,1 Гц), 2,21-2,12 (2Н, м), 1,21-1,13 (9Н, м), 1,03 (3Н, д, J=6,7 Гц).
[0803]
(Этап 9)
С применением соединения (1,02 г), полученного на описанном выше этапе 8, проводили реакцию таким же образом, как на этапе 7 примера 1, с получением раствора в ацетонитриле 6-бензоил-2-{2-дезокси-2-фтор-3-O-[гидрокси(оксо)-λ5-фосфанил]-β-D-рибофуранозил}-6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулена. С применением полученного раствора в ацетонитриле и соединения (1,15 г), полученного на этапе 3 примера 22, проводили реакцию таким же образом, как на этапе 8 примера 1, и полученный в результате этого неочищенный продукт непосредственно использовали для следующей реакции.
[0804]
(Этап 10)
2-{9-[(5R,7R,8R,12aR,14R,15R,15aR,16R)-14-(6-бензоил-6,7,8,9-тетрагидро-2H-2,3,5,6-тетраазабензо[cd]азулен-2-ил)-16-{[трет-бутил(диметил)силил]окси}-10-(2-цианоэтокси)-15-фтор-2-оксо-2-сульфанил-10-сульфанилиденоктагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-7-ил]-6-оксо-6,9-дигидро-1H-пурин-1-ил}этилбензоат
С применением неочищенного продукта, полученного на описанном выше этапе 9, проводили реакцию таким же образом, как на этапе 9 примера 1, с получением указанного в заголовке соединения (818 мг: с примесями) в виде смеси диастереомеров у атома фосфора.
МС (ИЭР) m/z: 1152(М+Н)+.
[0805]
(Этап 11)
Бис(N,N-диэтилэтанаминия) (5R,7R,8R,12aR,14R,15R,15aR,16R)-16-{[трет-бутил(диметил)силил]окси}-15-фтор-7-[1-(2-гидроксиэтил)-6-оксо-1,6-дигидро-9Н-пурин-9-ил]-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
С применением соединения (818 мг), полученного на описанном выше этапе 10, проводили реакцию таким же образом, как на этапе 10 примера 1, с получением диастереомера 1 (107 мг: с примесями) и диастереомера 2 (101 мг: с примесями) указанного в заголовке соединения.
Диастереомер 1 (менее полярный)
МС (ИЭР) m/z: 891 (М+Н)+.
Диастереомер 2 (более полярный)
МС (ИЭР) m/z: 891 (М+Н)+.
[0806]
(Этап 12-1)
Динатрия (5R,7R,8R,12aR,14R,15R,15aR,16R)-15-фтор-16-гидрокси-7-[1-(2-гидроксиэтил)-6-оксо-1,6-дигидро-9Н-пурин-9-ил]-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10H,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
(Диастереомер 1)
С применением соединения (диастереомера 1) (107 мг: с примесями), полученного на описанном выше этапе 11, проводили реакцию таким же образом, как на этапе 11 примера 1, а затем проводили очистку при следующих [Условиях очистки] с получением указанного в заголовке соединения в виде соли триэтиламина.
[Условия очистки] Хроматография на колонке с силикагелем С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил] и препаративная ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 5% - 30% (0 мин - 30 мин)].
Полученную соль триэтиламина подвергали солевому обмену таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением указанного в заголовке соединения (29,1 мг).
МС (ИЭР) m/z: 777 (М+Н)+.
1H-ЯМР (CD3OD) δ: 8,58 (1H, м), 8,11 (1H, м), 8,03 (1H, с), 7,11 (1H, с), 6,47 (1H, д, J=17,5 Гц), 6,26 (1H, д, J=8,5 Гц), 5,53-5,36 (2Н, м), 5,29-5,17 (1H, м), 4,77 (1H, д, J=4,2 Гц), 4,54-4,46 (1H, м), 4,44-4,38 (1H, м), 4,35-4,32 (1H, м), 4,30-4,25 (2Н, м), 4,25-4,16 (1H, м), 4,06-3,99 (1H, м), 3,96-3,85 (1H, м), 3,82-3,71 (2Н, м), 3,54-3,42 (2Н, м), 2,77-2,68 (1H, м), 2,66-2,55 (1H, м), 2,02-1,81 (2Н, м).
31Р-ЯМР (CD3OD) δ: 57,5 (с), 53,0 (с).
[0807]
(Этап 12-2)
Динатрия (5R,7R,8R,12aR,14R,15R,15aR,16R)-15-фтор-16-гидрокси-7-[1-(2-гидроксиэтил)-6-оксо-1,6-дигидро-9Н-пурин-9-ил]-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
(Диастереомер 2)
С применением соединения (диастереомера 2) (101 мг: с примесями), полученного на описанном выше этапе 11, проводили реакцию таким же образом, как на этапе 11 примера 1, а затем проводили очистку при следующих [Условиях очистки] с получением указанного в заголовке соединения в виде соли триэтиламина.
[Условия очистки] Хроматография на колонке с силикагелем С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил] и препаративная ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 5% - 20% (0 мин - 30 мин)].
Полученную соль триэтиламина подвергали солевому обмену таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением указанного в заголовке соединения (11,2 мг).
МС (ПЭР) m/z: 777 (М+Н)+.
1H-ЯМР (CD3OD) δ: 8,61 (1H, м), 8,16 (1H, м), 8,02 (1H, м), 7,36 (1H, с), 6,49 (1H, дд, J=16,0, 2,1 Гц), 6,28 (1H, д, J=8,5 Гц), 5,56-5,33 (3Н, м), 4,58-4,49 (2Н, м), 4,45-4,37 (2Н, м), 4,31-4,27 (1H, м), 4,25-4,16 (1H, м), 4,10-3,98 (3Н, м), 3,80 (2Н, т, J=5,1 Гц), 3,48 (2Н, дд, 3=6,7, 3,6 Гц), 2,90-2,72 (2Н, м), 2,00-1,90 (2Н, м).
31Р-ЯМР (CD3OD) δ: 59,5 (с), 57,7 (с).
[0808]
Пример 45. Синтез ЦДН35.
(5R,7R,8R,12aR,14R,15R,15aR,16R)-7-[1-(2-аминоэтил)-6-оксо-1,6-дигидро-9Н-пурин-9-ил]-15-фтор-16-гидрокси-2,10-бис(сульфанил)-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-дион
[0809]
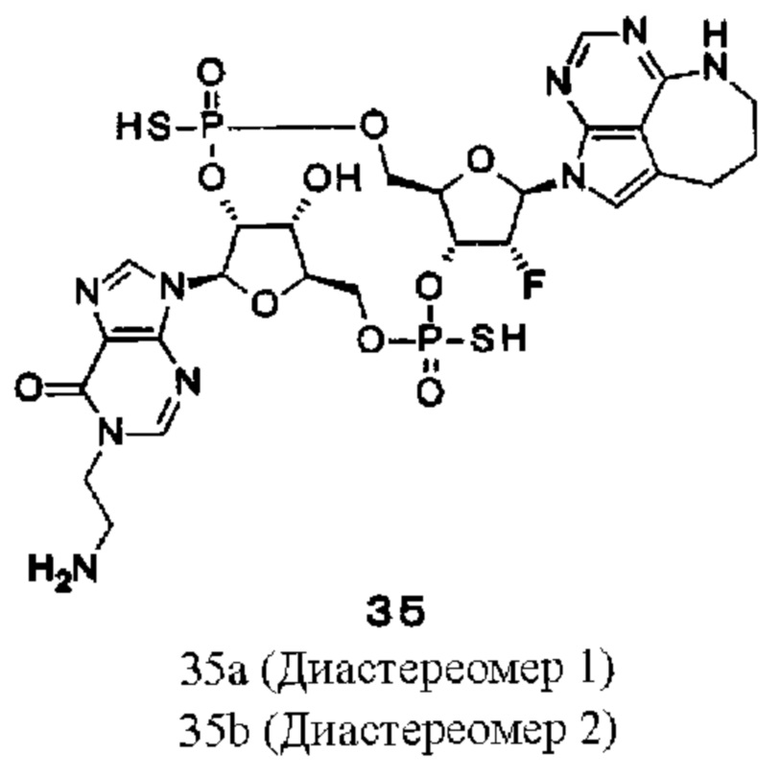
[0810]
[Схема синтеза]
[0811]
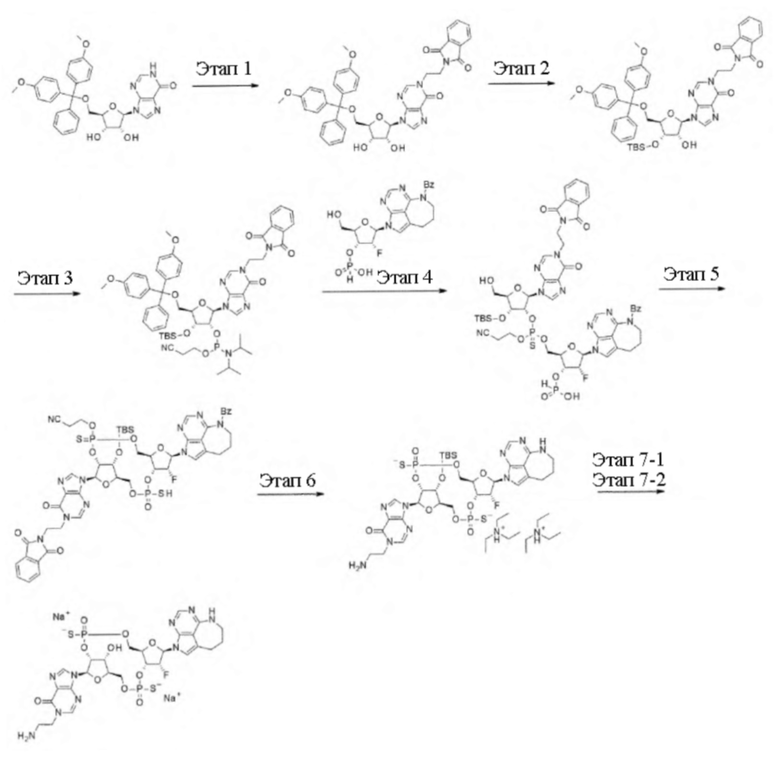
[0812]
(Этап 1)
5'-O-[бис(4-метоксифенил)(фенил)метил]-1-[2-(1,3-диоксо-1,3-дигидро-2Н-изоиндол-2-ил)этил]инозин
В суспензию доступного для приобретения (Aamdis Chemical) 5'-O-[бис(4-метоксифенил)(фенил)метил]инозина (13,0 г) в N,N-диметилацетамиде (60 мл) добавляли N-(2-бромоэтил)фталимид (7,02 г) и 1,8-диазабицикло[5,4,0]-7-ундецен (4,1 мл) и реакционную смесь перемешивали при комнатной температуре в течение ночи. Туда дополнительно добавляли N-(2-бромоэтил)фталимид (1,75 г) и 1,8-диазабицикло[5,4,0]-7-ундецен (1,1 мл) и реакционную смесь дополнительно перемешивали в течение 1 дня. В реакционную смесь добавляли воду, чтобы погасить реакцию, и осуществляли экстрагирование продукта дихлорметаном. После высушивания органического слоя над безводным сульфатом натрия осушающий агент удаляли путем фильтрации и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле [этилацетат/метанол] с получением указанного в заголовке соединения (12,4 г).
1H-ЯМР (CDCl3) δ: 7,83 (1H, с), 7,76-7,67 (4Н, м), 7,64 (1H, с), 7,35-7,33 (2Н, м), 7,25-7,11 (7Н, м), 6,74-6,70 (4Н, м), 5,93 (1H, д, J=5,1 Гц), 5,68 (1H, д, J=3,9 Гц), 4,71 (1H, к, J=4,8 Гц), 4,43 (1H, м), 4,37-4,18 (3Н, м), 4,10-4,06 (2Н, м), 3,730 (3Н, с), 3,728 (3Н, с), 3,51 (1H, м), 3,36 (1H, дд, J=10,6, 3,9 Гц), 3,32 (1H, дд, J=11,0, 5,5 Гц).
[0813]
(Этап 2)
5'-O-[бис(4-метоксифенил)(фенил)метил]-3'-O-[трет-бутил(диметил)силил]-1-[2-(1,3-диоксо-1,3-дигидро-2Н-изоиндол-2-ил)этил]инозин
С применением соединения (12,4 г), полученного на описанном выше этапе 1, проводили реакцию таким же образом, как на этапе 3 примера 5, с получением указанного в заголовке соединения (4,18 г) и 5'-O-[бис(4-метоксифенил)(фенил)метил]-2'-O-[трет-бутил(диметил)силил]-1-[2-(1,3-диоксо-1,3-дигидро-2Н-изоиндол-2-ил)этил]инозина (6,31 г) в качестве региоизомера указанного в заголовке соединения.
1H-ЯМР (CDCl3) δ: 8,00 (1H, с), 7,82-7,77 (2Н, м), 7,74 (1H, с), 7,72-7,67 (2Н, м), 7,41-7,39 (2Н, м), 7,32-7,19 (7Н, м), 6,83-6,78 (4Н, м), 5,90 (1H, д, J=5,1 Гц), 4,53-4,41 (3Н, м), 4,32-4,25 (1H, м), 4,19-4,11 (3Н, м), 3,79 (3Н, с), 3,78 (3Н, с), 3,46 (1H, дд, J=10,6, 3,1 Гц), 3,24 (1H, дд, J=10,8, 4,1 Гц), 2,98 (1Н, д, J=6,7 Гц), 0,85 (9Н, с), 0,04 (3Н, с), -0,03 (3Н, с).
Региоизомер (2'-O-TBS-форма)
1H-ЯМР (CDCl3) δ: 7,97 (1Н, с), 7,82-7,78 (2Н, м), 7,73-7,69 (2Н, м), 7,66 (1H, с), 7,44-7,41 (2Н, м), 7,33-7,18 (7Н, м), 6,81 (4Н, д, J=7,8 Гц), 5,91 (1H, д, J=5,9 Гц), 4,82 (1H, т, J=5,5 Гц), 4,43 (1H, м), 4,34-4,23 (3Н, м), 4,18-4,08 (2Н, м), 3,79 (6Н, с), 3,46 (1H, дд, J=10,6, 2,7 Гц), 3,36 (1H, дд, J=10,6, 3,5 Гц), 2,70 (1H, д, J=3,1 Гц), 0,83 (9Н, с), -0,04 (3Н, с), -0,19 (3Н, с).
[0814]
(Этап 3)
5'-O-[бис(4-метоксифенил)(фенил)метил]-3'-O-[трет-бутил(диметил)силил]-2'-O-{(2-цианоэтокси)[ди(пропан-2-ил)амино]фосфанил}-1-[2-(1,3-диоксо-1,3-дигидро-2Н-изоиндол-2-ил)этил]инозин
С применением соединения (8,89 г), полученного на описанном выше этапе 2, проводили реакцию таким же образом, как на этапе 6 примера 1, с получением указанного в заголовке соединения (9,45 г) в виде смеси диастереомеров у атома фосфора (соотношение диастереомеров=1:1).
1H-ЯМР (CDCl3) δ: 8,01 (0,5Н, с), 8,00 (0,5Н, с), 7,82-7,77 (2Н, м), 7,74 (0,5Н, с), 7,72-7,67 (2,5Н, м), 7,42 (2Н, д, J=7,8 Гц), 7,33-7,18 (7Н, м), 6,81 (4Н, д, J=8,6 Гц), 6,10 (0,5Н, д, J=5,5 Гц), 6,04 (0,5Н, д, J=5,1 Гц), 4,75 (0,5Н, м), 4,60 (0,5Н, м), 4,49-4,41 (1Н, м), 4,38-4,23 (2Н, м), 4,22-4,05 (3Н, м), 3,79 (6Н, с), 3,78-3,65 (1H, м), 3,62-3,39 (4Н, м), 3,33-3,23 (1Н, м), 2,49 (1H, т, J=6,3 Гц), 2,34 (1Н, т, J=6,7 Гц), 1,12-1,08 (9Н, м), 0,91 (3Н, д, J=7,0 Гц), 0,82 (9Н, с), 0,06 (1,5Н, с), 0,03 (1,5Н, с), -0,03 (3Н, с).
[0815]
(Этап 4)
С применением соединения (1,30 г), полученного на этапе 8 примера 44, проводили реакцию таким же образом, как на этапе 7 примера 1, с получением раствора в ацетонитриле 6-бензоил-2-{2-дезокси-2-фтор-3-O-[гидрокси(оксо)-λ5-фосфанил]-β-D-рибофуранозил}-6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулена. С применением полученного раствора в ацетонитриле и соединения (1,50 г), полученного на описанном выше этапе 3, проводили реакцию таким же образом, как на этапе 8 примера 1, и полученный в результате этого неочищенный продукт непосредственно использовали для следующей реакции.
[0816]
(Этап 5)
3-{[(5R,7R,8R,12aR,14R,15R,15aR,16R)-14-(6-бензоил-6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)-16-{[трет-бутил(диметил)силил]окси}-7-{1-[2-(1,3-диоксо-1,3-дигидро-2Н-изоиндол-2-ил)этил]-6-оксо-1,6-дигидро-9Н-пурин-9-ил}-15-фтор-2-оксо-2-сульфанил-10-сульфанилиденоктагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-10-ил]окси}пропаннитрил
С применением неочищенного продукта, полученного на описанном выше этапе 4, проводили реакцию таким же образом, как на этапе 9 примера 1, с получением указанного в заголовке соединения (828 мг) в виде смеси диастереомеров у атома фосфора.
МС (ИЭР) m/z: 1177 (М+Н)+.
[0817]
(Этап 6)
Бис(N,N-диэтилэтанаминия)(5R,7R,8R,12aR,14R,15R,15aR,16R)-7-[1-(2-аминоэтил)-6-оксо-1,6-дигидро-9Н-пурин-9-ил]-16-{[трет-бутил(диметил)силил]окси}-15-фтор-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
В смешанный раствор соединения (828 мг), полученного на описанном выше этапе 5, в этаноле (5,0 мл)/тетрагидрофуране (5,0 мл) добавляли моногидрат гидразина (0,342 мл) и реакционную смесь перемешивали при 50°С в течение 6 часов. После концентрирования реакционной смеси при пониженном давлении остаток очищали с помощью колоночной хроматографии на силикагеле С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил] с получением диастереомера 1 (90,9 мг: с примесями) и диастереомера 2 (91,1 мг: с примесями) указанного в заголовке соединения. Диастереомер 1 (менее полярный)
МС (ИЭР) m/z: 890 (М+Н)+.
Диастереомер 2 (более полярный)
МС (ИЭР) m/z: 890 (М+Н)+.
[0818]
(Этап 7-1)
Динатрия (5R,7R,8R,12aR,14R,15R,15aR,16R)-7-[1-(2-аминоэтил)-6-оксо-1,6-дигидро-9Н-пурин-9-ил]-15-фтор-16-гидрокси-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят) (Диастереомер 1)
С применением соединения (90,9 мг: с примесями), полученного на описанном выше этапе 6 (диастереомер 1), проводили реакцию таким же образом, как на этапе 11 примера 1, а затем проводили очистку при следующих [Условиях очистки] с получением указанного в заголовке соединения в виде соли триэтиламина.
[Условия очистки] Хроматография на колонке с силикагелем С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил] и препаративная ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/раствор ацетонитрила-метанола (1:1), раствор ацетонитрила-метанола (1:1): 10% - 50% (0 мин - 40 мин)].
Полученную соль триэтиламина подвергали солевому обмену таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением указанного в заголовке соединения (20,2 мг).
МС (ИЭР) m/z: 776 (М+Н)+.
1H-ЯМР (CD3OD) δ: 8,54 (1H, с), 8,03 (1H, с), 7,99 (1H, с), 7,13 (1H, с), 6,44 (1H, д, J=18,1 Гц), 6,22 (1H, д, J=7,9 Гц), 5,56-5,38 (2Н, м), 5,33-5,21 (1H, м), 4,72 (1H, д, J=4,2 Гц), 4,58-4,49 (1H, м), 4,41-4,24 (4Н, м), 4,24-4,17 (1H, м), 4,05-3,98 (1H, м), 3,86-3,76 (1H, м), 3,50-3,42 (2Н, м), 3,27-3,16 (2Н, м), 2,78-2,68 (1H, м), 2,59-2,49 (1H, м), 1,98-1,80 (2Н, м).
31Р-ЯМР (CD3OD) δ: 57,5 (с), 53,1 (с).
[0819]
(Этап 7-2)
Динатрия (5R,7R,8R,12aR,14R,15R,15aR,16R)-7-[1-(2-аминоэтил)-6-оксо-1,6-дигидро-9Н-пурин-9-ил]-15-фтор-16-гидрокси-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
(Диастереомер 2)
С применением соединения (диастереомера 2) (91,1 мг: с примесями), полученного на описанном выше этапе 6, проводили реакцию таким же образом, как на этапе 11 примера 1, а затем проводили очистку при следующих [Условиях очистки] с получением указанного в заголовке соединения в виде соли триэтиламина.
[Условия очистки] Хроматография на колонке с силикагелем С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил] и препаративная ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/раствор ацетонитрила-метанола (1:1), раствор ацетонитрила-метанола (1:1): 10% - 45% (0 мин - 40 мин)].
Полученную соль триэтиламина подвергали солевому обмену таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением указанного в заголовке соединения (26,3 мг).
МС (ИЭР) m/z: 776 (М+Н)+.
1H-ЯМР (CD3OD) δ: 8,56 (1H, с), 8,08 (1Н, с), 8,02 (1H, с), 7,37 (1Н, с), 6,48 (1H, д, J=16,2 Гц), 6,23 (1H, д, J=7,9 Гц), 5,58-5,32 (3Н, м), 4,65-4,27 (6Н, м), 4,07-3,96 (3Н, м), 3,48-3,42 (2Н, м), 3,38-3,23 (2Н, м), 2,85-2,75 (1Н, м), 2,69-2,59 (1H, м), 1,99-1,81 (2Н, м).
31Р-ЯМР (CD3OD) δ: 59,0 (с), 57,6 (с).
[0820]
Пример 46. Синтез ЦДН36.
N-(2-{9-[(5R,7R,8R,12aR,14R,15R,15aR,16R)-15-фтор-16-гидрокси-2,10-диоксо-2,10-бис(сульфанил)-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-7-ил]-6-оксо-6,9-дигидро-1Н-пурин-1-ил}этил)-2-гидроксиацетамид
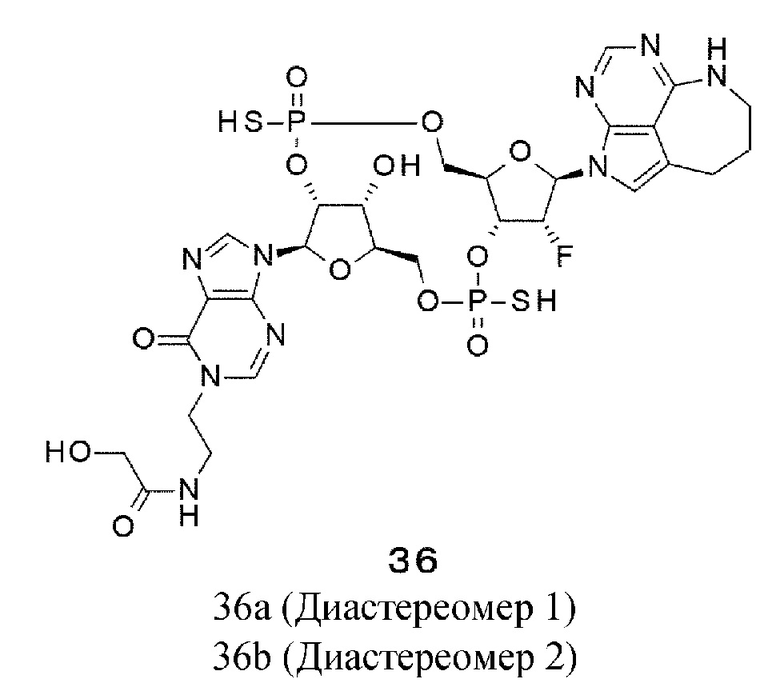
[0822]
[Схема синтеза]
[0823]
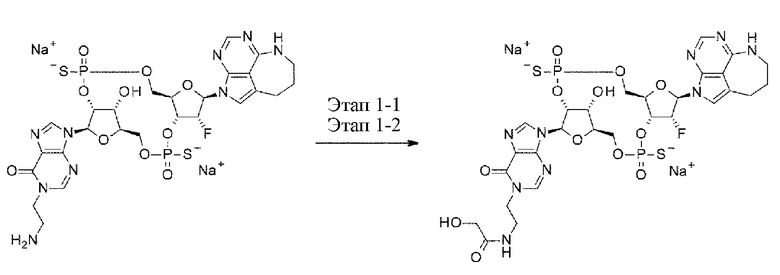
[0824]
(Этап 1-1)
Динатрия (5R,7R,8S,12aR,14R,15R,15aS,16R)-16-фтор-15-гидрокси-7-{1-[2-(2-гидроксиацетамид)этил]-6-оксо-1,6-дигидро-9Н-пурин-9-ил}-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят) (Диастереомер 1)
С применением соединения (15,0 мг), полученного на этапе 7-1 примера 45, проводили реакцию таким же образом, как на этапе 1-1 примера 7, а затем проводили очистку при следующих [Условиях очистки] с получением указанного в заголовке соединения в виде соли триэтиламина.
[Условия очистки] Хроматография на колонке с силикагелем С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил] и препаративная ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/раствор ацетонитрила-метанола (1:1), раствор ацетонитрила-метанола (1:1): 10% - 45% (0 мин - 40 мин)].
Полученную соль триэтиламина подвергали солевому обмену таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением указанного в заголовке соединения (10,3 мг).
МС (ИЭР) m/z: 834 (М+Н)+.
1H-ЯМР (CD3OD) δ: 8,46 (1H, уш. м), 8,04 (1H, с), 7,82 (1H, уш. м), 7,15 (1H, уш. м), 6,43 (1H, д, J=16,9 Гц), 6,17 (1H, дд, J=9,1, 4,5 Гц), 5,70-5,24 (3Н, м), 4,81-4,75 (1H, м), 4,52-4,44 (1H, м), 4,43-4,26 (4Н, м), 4,24-3,94 (2Н, м), 3,89-3,84 (2Н, м), 3,72-3,37 (5Н, м), 2,75-2,65 (1H, м), 2,48-2,32 (1H, м), 1,98-1,76 (2Н, м).
31Р-ЯМР (CD3OD) δ: 57,0 (с), 53,0 (с).
[0825]
(Этап 1-2)
Динатрия (5R,7R,8S,12aR,14R,15R,15aS,16R)-16-фтор-15-гидрокси-7-{1-[2-(2-гидроксиацетамид)этил]-6-оксо-1,6-дигидро-9Н-пурин-9-ил}-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят) (Диастереомер 2)
С применением соединения (15,0 мг), полученного на этапе 7-2 примера 45, проводили реакцию таким же образом, как на этапе 1-1 примера 7, а затем проводили очистку при следующих [Условиях очистки] с получением указанного в заголовке соединения в виде соли триэтиламина.
[Условия очистки] Хроматография на колонке с силикагелем С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил] и препаративная ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/раствор ацетонитрила-метанола (1:1), раствор ацетонитрила-метанола (1:1): 10% - 45% (0 мин - 40 мин)].
Полученную соль триэтиламина подвергали солевому обмену таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением указанного в заголовке соединения (10,6 мг).
МС (ИЭР) m/z: 834 (М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,54 (1Н, уш. с), 8,03 (1H, с), 7,97 (1H, уш. с), 7,35 (1Н, уш. с), 6,48 (1H, д, J=15,7 Гц), 6,23 (1Н, д, J=8,5 Гц), 5,65-5,39 (3Н, м), 4,57-4,47 (2Н, м), 4,46-4,36 (2Н, м), 4,31-4,19 (2Н, м), 4,07-3,96 (2Н, м), 3,95-3,78 (3Н, м), 3,67-3,43 (4Н, м), 2,85-2,75 (1H, м), 2,71-2,59 (1H, м), 2,00-1,84 (2Н, м).
31Р-ЯМР (CD3OD) δ: 59,1 (с), 57,5 (с).
[0826]
Пример 47. Синтез ЦДН37.
(5R,7R,8R,12aR,14R,15R,15aR,16R)-7-{6-амино-2-[(2-аминоэтил)амино]-9Н-пурин-9-ил}-15-фтор-16-гидрокси-2,10-бис(сульфанил)-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-дион
[0827]
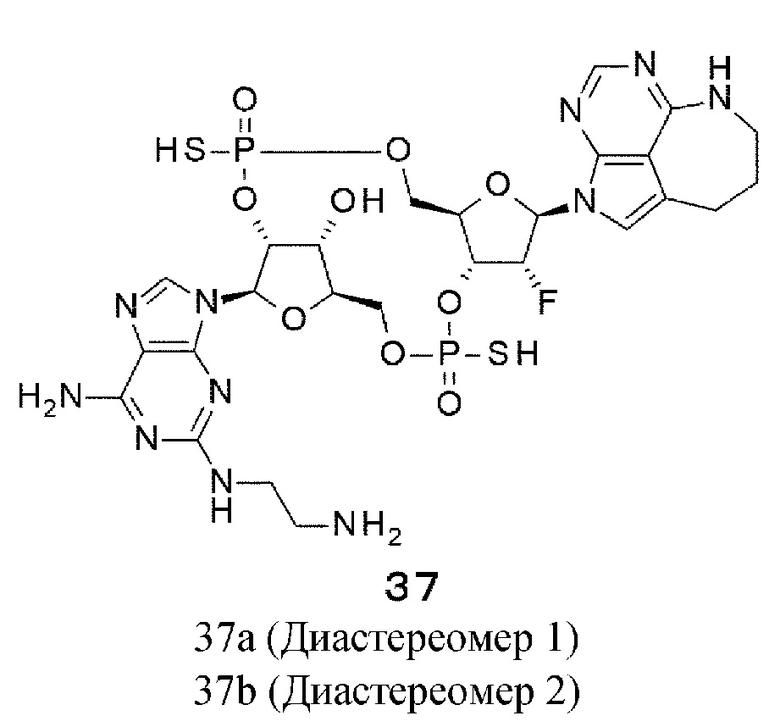
[0828]
[Схема синтеза]
[0829]
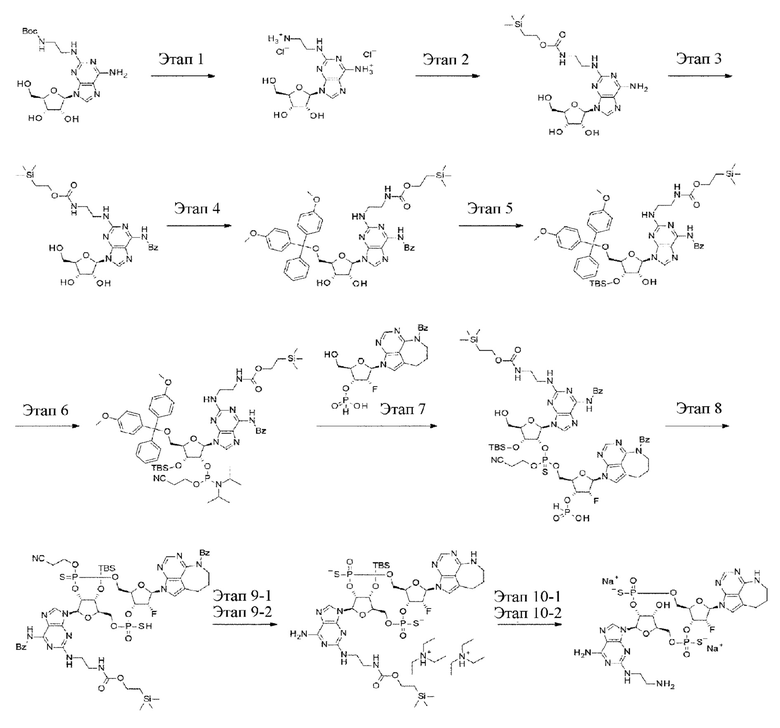
[0830] (Этап 1)
2-[(2-азаниумилэтил)амино] аденозина дихлорид
К раствору 2-({2-[(трет-бутоксикарбонил)амино]этил}амино)аденозина (28,7 г) как соединения, известного в литературе (WO 2012/159072), в метаноле (240 мл) добавляли раствор в диоксане хлорида водорода (приблизительно 4 М, 240 мл) и реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь концентрировали до приблизительно 50 мл при пониженном давлении, и твердый преципитированный продукт затем суспендировали в диэтиловом эфире и собирали путем фильтрации с получением указанного в заголовке соединения (28,8 г).
1H-ЯМР (CD3OD) δ: 8,32 (1H, с), 5,98 (1Н, д, J=6,0 Гц), 4,51 (1H, т, J=5,4 Гц), 4,30 (1H, дд, J=5,1, 3,3 Гц), 4,14-4,11 (1H, м), 3,86-3,72 (4Н, м), 3,23 (2Н, т, J=6,0 Гц).
[0831]
(Этап 2)
2-{[2-({[2-(триметилсилил)этокси]карбонил}амино)этил]амино}аденозин
В смесь соединения (28,8 г), полученного на описанном выше этапе 1, в тетрагидрофуране (330 мл)/воде (70 мл) добавляли триэтиламин (37 мл) и 1-({[2-(триметилсилил)этокси]карбонил}окси)пирролидин-2,5-дион (18,4 г) и реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Реакционную смесь концентрировали при пониженном давлении и подвергали азеотропной перегонке с толуолом. Остаток суспендировали в толуоле и твердый продукт собирали путем фильтрации. Полученный твердый продукт растворяли в дихлорметане и метаноле и продукт концентрировали при пониженном давлении. Тетрагидрофуран добавляли к остатку, твердый преципитированный продукт удаляли путем фильтрации и фильтрат концентрировали при пониженном давлении. Остаток растворяли в дихлорметане и метаноле, и продукт концентрировали при пониженном давлении. Твердый преципитированный продукт суспендировали в дихлорметане и собирали путем фильтрации с получением указанного в заголовке соединения (22,5 г).
1H-ЯМР (CD3OD) δ: 7,92 (1H, с), 5,83 (1H, д, J=6,0 Гц), 4,77 (1H, т, J=5,4 Гц), 4,33 (1H, дд, J=4,8, 3,0 Гц), 4,14-4,10 (3Н, м), 3,87 (1H, дд, J=12,4, 2,7 Гц), 3,73 (1H, дд, J=12,1, 3,0 Гц), 3,50-3,42 (2Н, м), 3,32-3,29 (2Н, м), 0,98-0,86 (2Н, м), 0,03 (9Н, с).
[0832]
(Этап 3)
Н-бензоил-2-{[2-({[2-(триметилсилил)этокси]карбонил}амино)этил]амино}аденозин
С применением соединения (24,7 г), полученного на описанном выше этапе 2, проводили реакцию таким же образом, как на этапе 3 примера 11, с получением указанного в заголовке соединения (22,1 г).
1H-ЯМР (CD3OD) δ: 8,19 (1H, с), 8,05 (2Н, д, J=7,9 Гц), 7,63 (1H, т, J=7,6 Гц), 7,54 (2Н, т, J=7,6 Гц), 5,95 (1H, д, J=5,4 Гц), 4,75 (1H, т, J=5,4 Гц), 4,36 (1H, т, J=4,5 Гц), 4,11-4,07 (3Н, м), 3,86 (1H, дд, J=12,4, 3,3 Гц), 3,75 (1H, дд, J=12,1, 3,6 Гц), 3,59-3,47 (2Н, м), 3,36-3,33 (2Н, м), 0,92 (2Н, т, J=8,2 Гц), 0,00 (9Н, с).
[0833]
(Этап 4)
N-бензоил-5'-O-[бис(4-метоксифенил)(фенил)метил]-2-{[2-({[2-(триметилсилил)этокси]карбонил}амино)этил]амино}аденозин
С применением соединения (22,1 г), полученного на описанном выше этапе 3, проводили реакцию таким же образом, как на этапе 1 примера 11, с получением указанного в заголовке соединения (29,5 г).
1H-ЯМР (ДМСО-d6) δ: 10,7 (1H, с), 8,09 (1H, с), 8,01-7,99 (2Н, м), 7,64-7,60 (1H, м), 7,52 (2Н, т, J=7,6 Гц), 7,36-7,34 (2Н, м), 7,26-7,17 (7Н, м), 7,05-7,02 (1H, м), 6,95-6,92 (1H, м), 6,85-6,80 (4Н, м), 5,88 (1H, д, J=4,8 Гц), 5,53 (1H, д, J=5,4 Гц), 5,20 (1H, д, J=5,4 Гц), 4,72-4,63 (1H, м), 4,34-4,26 (1H, м), 4,05-3,99 (3Н, м), 3,72-3,71 (6Н, м), 3,31-3,10 (6Н, м), 0,89 (2Н, т, J=8,5 Гц), 0,00 (9Н, с).
[0834]
(Этап 5)
N-бензоил-5'-O-[бис(4-метоксифенил)(фенил)метил]-3'-O-[трет-бутил(диметил)силил]-2-{[2-({[2-(триметилсилил)этокси]карбонил}амино)этил]амино}аденозин
С применением соединения (29,5 г), полученного на описанном выше этапе 4, проводили реакцию таким же образом, как на этапе 3 примера 5, с получением указанного в заголовке соединения (6,85 г).
1H-ЯМР (ДМСО-d6, 90°С) δ: 10,3 (1H, уш. с), 8,02 (1H, с), 7,99 (2Н, д, J=7,3 Гц), 7,60 (1H, т, J=7,3 Гц), 7,51 (2Н, т, J=7,6 Гц), 7,37 (2Н, д, J=7,3 Гц), 7,28-7,20 (7Н, м), 6,85-6,82 (4Н, м), 6,69-6,53 (2Н, м), 5,86 (1H, д, J=5,4 Гц), 5,07-5,04 (1H, м), 4,80-4,74 (1H, м), 4,36-4,33 (1H, м), 4,08-3,99 (3Н, м), 3,74 (6Н, с), 3,37-3,15 (6Н, м), 0,89 (2Н, т, J=7,9 Гц), 0,86 (9Н, с), 0,09 (3Н, с), 0,04 (3Н, с), 0,01 (9Н, с).
[0835]
(Этап 6)
N-бензоил-5'-O-[бис(4-метоксифенил)(фенил)метил]-3'-O-[трет-бутил(диметил)силил]-2'-O-{(2-цианоэтокси)[ди(пропан-2-ил)амино]фосфанил}-2-{[2-({[2-(триметилсилил)этокси]карбонил}амино)этил]амино}аденозин
С применением соединения (6,22 г), полученного на описанном выше этапе 5, проводили реакцию таким же образом, как на этапе 6 примера 1, с получением указанного в заголовке соединения (6,18 г) в виде смеси диастереомеров у атома фосфора (соотношение диастереомеров=1:1).
1H-ЯМР (CD3Cl) δ: 8,82 (1H, с), 8,00-7,97 (2Н, м), 7,88 (0,5Н, с), 7,85 (0,5Н, с), 7,59 (1H, т, J=7,0 Гц), 7,51 (2Н, т, J=7,6 Гц), 7,43 (2Н, д, J=7,3 Гц), 7,33-7,20 (7Н, м), 6,81 (4Н, д, J=8,5 Гц), 6,08-6,01 (1H, м), 5,19-4,88 (2Н, м), 4,51-4,42 (1H, м), 4,20-4,08 (3Н, м), 3,82-3,24 (11Н, м), 3,78 (6Н, с), 2,52 (1H, 5, J=6,3 Гц), 2,38-2,34 (1H, м), 1,14-1,09 (9Н, м), 0,90-0,85 (2Н, м), 0,87 (9Н, с), 0,12 (1,5Н, с), 0,09 (1,5Н, с), 0,03 (3Н, с), -0,01 (9Н, с).
[0836]
(Этап 7)
С применением соединения (1,03 г), полученного на этапе 8 примера 44, проводили реакцию таким же образом, как на этапе 7 примера 1, с получением раствора в ацетонитриле 6-бензоил-2-{2-дезокси-2-фтор-3-O-[гидрокси(оксо)-λ5-фосфанил]-β-D-рибофуранозил}-6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулена. С применением полученного раствора в ацетонитриле и соединения (999 мг), полученного на описанном выше этапе 6, проводили реакцию таким же образом, как на этапе 8 примера 1, и полученный в результате этого неочищенный продукт непосредственно использовали для следующей реакции.
[0837]
(Этап 8)
2-(триметилсилил)этил[2-({6-бензамидо-9-[(5R,7R,8R,12aR,14R,15R,15aR,16R)-14-(6-бензоил-6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)-16-{[трет-бутил(диметил)силил]окси}-10-(2-цианоэтокси)-15-фтор-2-оксо-2-сульфанил-10-сульфанилиденоктагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-7-ил]-9Н-пурин-2-ил}амино)этил] карбамат
С применением смеси, полученной на описанном выше этапе 7, проводили реакцию таким же образом, как на этапе 9 примера 1, с получением диастереомера 1 (370 мг: с примесями) и диастереомера 2 (201 мг: с примесями) указанного в заголовке соединения. Диастереомер 1 (менее полярный)
MC (ИЭР) m/z: 1307 (М-Н)-.
Диастереомер 2 (более полярный)
MC (ИЭР) m/z: 1307 (М-Н)-.
[0838]
(Этап 9-1)
Бис(N,N-диэтилэтанаминия)(5R,7R,8R,12aR,14R,15R,15aR,16R)-7-(6-амино-2-{[2-({[2-(триметилсилил)этокси]карбонил}амино)этил]амино}-9Н-пурин-9-ил)-16-{[трет-бутил(диметил)силил]окси}-15-фтор-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
С применением соединения (370 мг: с примесями), полученного на описанном выше этапе 8 (диастереомер 1), проводили реакцию таким же образом, как на этапе 10 примера 1, с получением указанного в заголовке соединения (134 мг: с примесями).
МС (ИЭР) m/z: 1048 (М+Н)+.
[0839]
(Этап 9-2)
Бис(N,N-диэтилэтанаминия)(5R,7R,8R,12aR,14R,15R,15aR,16R)-7-(6-амино-2-{[2-({[2-(триметилсилил)этокси]карбонил}амино)этил]амино}-9Н-пурин-9-ил)-16-{[трет-бутил(диметил)силил]окси}-15-фтор-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
С применением соединения (диастереомера 2) (201 мг: с примесями), полученного на описанном выше этапе 8, проводили реакцию таким же образом, как на этапе 10 примера 1, с получением указанного в заголовке соединения (49,1 мг: с примесями).
MC (ИЭР) m/z: 1048 (М+Н)+.
[0840]
(Этап 10-1)
Динатрия (5R,7R,8R,12aR,14R,15R,15aR,16R)-7-{6-амино-2-[(2-аминоэтил)амино]-9H-пурин-9-ил}-15-фтор-16-гидрокси-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят) (Диастереомер 1)
С применением соединения (134 мг), полученного на описанном выше этапе 9-1, проводили реакцию таким же образом, как на этапе 5 примера 40, и продукт затем очищали с помощью колоночной хроматографии на силикагеле С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил] с получением указанного в заголовке соединения в виде соли триэтиламина.
Полученную соль триэтиламина подвергали солевому обмену таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением указанного в заголовке соединения (22 мг).
МС (ИЭР) m/z: 790 (М+Н)+.
1H-ЯМР (CD3OD) δ: 8,04 (1H, уш. с), 8,01 (1Н, с), 7,06 (1H, с), 6,43 (1H, д, J=16,9 Гц), 6,00 (1H, д, J=7,3 Гц), 5,78-5,55 (1H, м), 5,39 (1H, дд, J=51,7, 3,9 Гц), 5,31-5,18 (1H, м), 4,80 (1H, д, J=3,6 Гц), 4,50-4,44 (1H, м), 4,40-4,32 (4Н, м), 4,14-4,10 (1H, м), 3,52-3,40 (2Н, м), 3,35-3,23 (2Н, м), 3,06-2,90 (2Н, м), 2,63-2,53 (1H, м), 2,40-2,20 (1H, м), 1,99-1,76 (2Н, м).
31Р-ЯМР (CD3OD) δ: 57,2 (с), 52,6 (с).
[0841] (Этап 10-2)
Динатрия (5R,7R,8R,12aR,14R,15R,15aR,16R)-7-{6-амино-2-[(2-аминоэтил)амино]-9Н-пурин-9-ил}-15-фтор-16-гидрокси-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят) (Диастереомер 2)
С применением соединения (49,1 мг: с примесями), полученного на описанном выше этапе 9-2, проводили реакцию таким же образом, как на этапе 5 примера 40, и продукт затем очищали с помощью колоночной хроматографии насиликагеле С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил] с получением указанного в заголовке соединения в виде соли триэтиламина.
Полученную соль триэтиламина подвергали солевому обмену таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением указанного в заголовке соединения (21 мг).
МС (ИЭР) m/z: 790 (М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,02 (1H, уш. с), 8,01 (1H, с), 7,40 (1H, с), 6,48 (1H, д, J=16,3 Гц), 6,01 (1Н, д, J=7,9 Гц), 5,80-5,63 (1H, м), 5,45-5,28 (2Н, м), 4,54-4,48 (2Н, м), 4,41-4,36 (2Н, м), 4,28-4,20 (2Н, м), 4,08-4,03 (1H, м), 3,54-3,41 (2Н, м), 3,40-2,52 (6Н, м), 2,03-1,84 (2Н, м).
31Р-ЯМР (CD3OD) δ: 57,8 (с).
[0842]
Пример 48. Синтез ЦДН38.
(5S,7R,8R,12aR,14R,15R,15aS)-15-гидрокси-7-[1-(2-гидроксиэтил)-6-оксо-1,6-дигидро-9Н-пурин-9-ил]-2,10-бис(сульфанил)-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-дион
[0843]
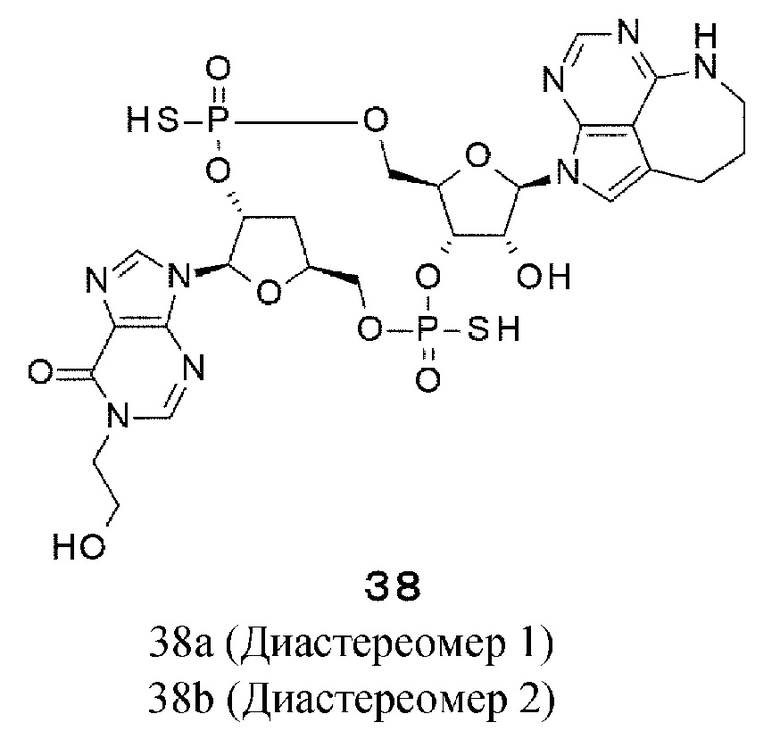
[0844]
[Схема синтеза]
[0845]
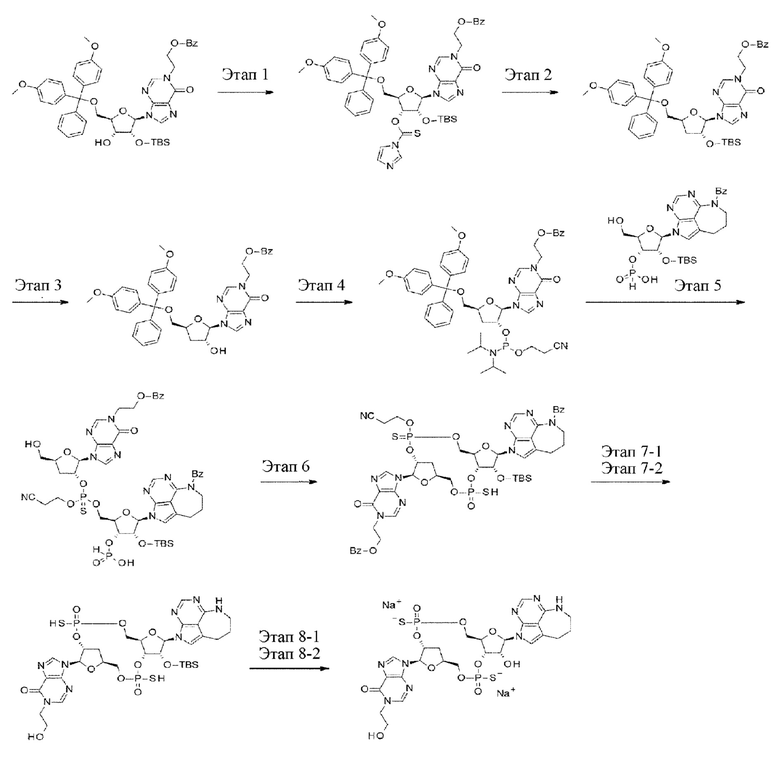
[0846]
(Этап 1)
1-[2-(бензоилокси)этил]-5'-O-[бис(4-метоксифенил)(фенил)метил]-2'-O-[трет-бутил(диметил)силил]-3'-O-(1Н-имидазол-1-карботиоил)инозин
К раствору соединения (2'-O-TBS-форма) (1,45 г), полученного на этапе 2 примера 22, в N,N-диметилформамиде (8,7 мл) добавляли N,N-диметил-4-аминопиридин (213 мг) и ди(1Н-имидазол-1-ил)метантион (1,55 г) при комнатной температуре и реакционную смесь перемешивали в течение 20 часов. Реакционную смесь разбавляли этилацетатом и промывали насыщенным водным раствором гидрокарбоната натрия и рассолом в данном порядке. После высушивания органического слоя над безводным сульфатом натрия осушающий агент удаляли путем фильтрации и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле [гексан/этилацетат] с получением указанного в заголовке соединения (1,58 г).
МС (ИЭР) m/z: 943 (М+Н)+.
1H-ЯМР (CDCl3) δ: 8,38 (1H, с), 8,05 (1H, с), 8,02-7,94 (3Н, м), 7,93 (1H, с), 7,66-7,65 (1H, м), 7,60-7,52 (1H, м), 7,46-7,18 (10Н, м), 7,08 (1H, с), 6,83-6,80 (4Н, м), 6,07 (1H, дд, J=5,4, 3,0 Гц), 6,02 (1H, д, J=6,0 Гц), 5,20 (1Н, дд, 3=6,7, 5,4 Гц), 4,72-4,63 (2Н, м), 4,55-4,38 (3Н, м), 3,78 (3Н, с), 3,77 (3Н, с), 3,57-3,56 (2Н, м), 0,65 (9Н, с), -0,11 (3Н, с), -0,26 (3Н, с).
[0847] (Этап 2)
1-[2-(бензоилокси)этил]-5'-O-[бис(4-метоксифенил)(фенил)метил]-2'-O-[трет-бутил(диметил)силил]-3'-дезоксиинозин
К раствору соединения (1,69 г), полученного на описанном выше этапе 1, в бензоле (10 мл) добавляли гидрид трибутилтина (1,42 мл) и 2,2'-азобис(изобутиронитрил) (29,4 мг) при комнатной температуре и реакционную смесь перемешивали при 80°С в течение 4 часов. Реакционную смесь концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле [гексан/этилацетат] с получением указанного в заголовке соединения (1,12 г).
МС (ИЭР) m/z: 817 (М+Н)+.
1H-ЯМР (CDCl3) δ: 7,99-7,96 (4Н, м), 7,60-7,55 (1H, м), 7,46-7,19 (11Н, м), 6,83-6,81 (4Н, м), 5,94 (1H, д, 3=1,2 Гц), 4,68-4,60 (4Н, м), 4,52-4,39 (2Н, м), 3,79 (6Н, с), 3,42-3,34 (2Н, м), 2,15-2,08 (1H, м), 1,94-1,89 (1H, м), 0,86 (9Н, с), 0,06 (3Н, с), 0,04 (3Н, с).
[0848] (Этап 3)
1-[2-(бензоилокси)этил]-5'-O-[бис(4-метоксифенил)(фенил)метил]-3'-дезоксиинозин
К раствору соединения (1,75 г), полученного на описанном выше этапе 2, в тетрагидрофуране (11 мл) добавляли раствор в тетрагидрофуране фторида тетрабутиламмония (приблизительно 1 М, 2,6 мл) при 0°С и реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Реакционную смесь концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле [гексан/этилацетат] с получением указанного в заголовке соединения (1,29 г).
МС (ИЭР) m/z: 703 (М+Н)+.
1H-ЯМР (CDCl3) δ: 7,99-7,95 (4Н, м), 7,60-7,55 (1H, м), 7,45-7,37 (4Н, м), 7,29-7,17 (7Н, м), 6,81-6,78 (4Н, м), 5,88 (1Н, д, J=3,0 Гц), 4,76-4,63 (4Н, м), 4,52-4,40 (2Н, м), 3,78 (6Н, с), 3,36 (1H, дд, J=10,3, 3,0 Гц), 3,29 (1H, дд, J=10,3, 4,8 Гц), 2,30-2,23 (1H, м), 2,18-2,12 (1H, м) (показаны только наблюдаемые пики).
[0849]
(Этап 4)
1-[2-(бензоилокси)этил]-5'-O-[бис(4-метоксифенил)(фенил)метил]-2'-O-{(2-цианоэтокси)[ди(пропан-2-ил)амино]фосфанил}-3'-дезоксиинозин
С применением соединения (1,28 г), полученного на описанном выше этапе 3, проводили реакцию таким же образом, как на этапе 4 примера 5, с получением указанного в заголовке соединения (1,47 г) в виде смеси диастереомеров у атома фосфора (соотношение диастереомеров=1:1).
МС (ИЭР) m/z: 820 (M-C6H14N+OH+H)+.
1H-ЯМР (CDCl3) δ: 7,99-7,94 (4Н, м), 7,59-7,56 (1H, м), 7,46-7,42 (4Н, м), 7,33-7,18 (7Н, м), 6,81 (4Н, д, J=8,5 Гц), 6,13 (0,5Н, уш. с), 6,07 (0,5Н, уш. с), 4,84-4,39 (6Н, м), 3,84-3,52 (10Н, м), 3,42-3,34 (2Н, м), 2,59 (1H, т, J=6,3 Гц), 2,52 (1H, т, J=6,3 Гц), 2,30-2,07 (2Н, м), 1,18-1,07 (12Н, м).
[0850]
(Этап 5)
Ту же реакцию, что и на этапе 7 примера 1, проводили в следующем масштабе (сырьевой материал: 836 мг). С применением раствора в ацетонитриле полученного соединения и соединения (735 мг), полученного на описанном выше этапе 4, проводили реакцию таким же образом, как на этапе 8 примера 1, и полученный в результате этого неочищенный продукт непосредственно использовали для следующей реакции.
[0851]
(Этап 6)
2-{9-[(5S,7R,8R,12aR,14R,15R,15aR)-14-(6-бензоил-6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)-15-{[трет-бутил(диметил)силил]окси}-10-(2-цианоэтокси)-2-оксо-2-сульфанил-10-сульфанилиденоктагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-7-ил]-6-оксо-6,9-дигидро-1Н-пурин-1-ил}этилбензоат
С применением неочищенного продукта, полученного на описанном выше этапе 5, проводили реакцию таким же образом, как на этапе 9 примера 1, с получением диастереомера 1 (67,6 мг) и диастереомера 2 (91,6 мг) указанного в заголовке соединения (время удерживания при ВЭЖХ: диастереомер 1>2).
Диастереомер 1 (менее полярный)
МС (ИЭР) m/z: 1134 (М+Н)+.
Диастереомер 2 (более полярный)
МС (ИЭР) m/z: 1134 (М+Н)+.
[0852]
(Этап 7-1)
(5S,7R,8R,12aR,14R,15R,15aR)-15-{[трет-бутил(диметил)силил]окси}-7-[1-(2-гидроксиэтил)-6-оксо-1,6-дигидро-9Н-пурин-9-ил]-2,10-бис(сульфанил)-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[с(1]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-дион
С применением соединения (диастереомера 1) (67,6 мг), полученного на описанном выше этапе 6, проводили реакцию таким же образом, как на этапе 10 примера 1, и продукт очищали с помощью препаративной ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 0% - 50% (0 мин - 30 мин)] с получением указанного в заголовке соединения (34,9 мг).
МС (ИЭР) m/z: 873(М+Н)+.
[0853]
(Этап 7-2)
(5S,7R,8R,12aR,14R,15R,15aR)-15-{[трет-бутил(диметил)силил]окси}-7-[1-(2-гидроксиэтил)-6-оксо-1,6-дигидро-9Н-пурин-9-ил]-2,10-бис(сульфанил)-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10] пентаоксадифосфациклотетрадецин-2,10-дион
С применением соединения (диастереомера 2) (91,6 мг), полученного на описанном выше этапе 6, проводили реакцию таким же образом, как на этапе 10 примера 1, и продукт очищали с помощью препаративной ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 0% - 50% (0 мин - 30 мин)] с получением указанного в заголовке соединения (44,2 мг).
МС (ИЭР) m/z: 873 (М+Н)+.
[0854]
(Этап 8-1)
Динатрия (5S,7R,8R,12aR,14R,15R,15aS)-15-гидрокси-7-[1-(2-гидроксиэтил)-6-оксо-1,6-дигидро-9Н-пурин-9-ил]-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
(Диастереомер 1)
С применением соединения (34,9 мг), полученного на описанном выше этапе 7-1, проводили реакцию таким же образом, как на этапе 11 примера 1, и продукт затем очищали с помощью Sep-Pak (R) С18 [0,1% водный раствор триэтиламина/ацетонитрил] с получением указанного в заголовке соединения в виде соли триэтиламина.
Полученную соль триэтиламина подвергали солевому обмену таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением указанного в заголовке соединения (21,9 мг).
МС (ИЭР) m/z: 759 (М+Н)+.
1H-ЯМР (CD3OD) δ: 8,34 (1H, уш. с), 8,23 (1H, с), 8,02 (1H, с), 7,24 (1H, уш. с), 6,28 (1H, д, J=3,6 Гц), 6,15 (1H, д, J=3,0 Гц), 5,37-5,32 (1H, м), 4,99-4,93 (1Н, м), 4,74-4,58 (3Н, м), 4,36-4,29 (2Н, м), 4,24-4,13 (3Н, м), 4,00-3,91 (1H, м), 3,82 (2Н, т, J=4,8 Гц), 3,51-3,49 (2Н, м), 3,00-2,90 (3Н, м), 2,57-2,51 (1H, м), 2,02-1,97 (2Н, м).
31Р-ЯМР (CD3OD) δ: 59,7 (с), 56,2 (с).
[0855]
(Этап 8-2)
Динатрия (5S,7R,8R,12aR,14R,15R,15aS)-15-гидрокси-7-[1-(2-гидроксиэтил)-6-оксо-1,6-дигидро-9Н-пурин-9-ил]-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
(Диастереомер 2)
С применением соединения (44,2 мг), полученного на описанном выше этапе 7-2, проводили реакцию таким же образом, как на этапе 11 примера 1, и продукт затем очищали с помощью Sep-Pak (R) С18 [0,1% водный раствор триэтиламина/ацетонитрил] с получением указанного в заголовке соединения в виде соли триэтиламина.
Полученную соль триэтиламина подвергали солевому обмену таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением указанного в заголовке соединения (26,5 мг).
МС (ИЭР) m/z: 759 (М+Н)+.
1H-ЯМР (CD3OD) δ: 8,48-8,46 (1H, уш. м), 8,24 (1H, с), 8,03 (1H, с), 7,41-7,38 (1H, уш. м), 6,32 (1H, д, J=4,8 Гц), 6,15 (1H, д, J=4,2 Гц), 5,46-5,41 (1Н, м), 5,21-5,17 (1Н, м), 4,68-4,65 (1H, м), 4,59-4,54 (2Н, м), 4,50-4,44 (1H, м), 4,35-4,32 (1H, м), 4,21-4,18 (2Н, м), 4,02-3,92 (2Н, м), 3,84-3,81 (2Н, м), 3,52-3,50 (2Н, м), 2,96-2,83 (3Н, м), 2,54-2,48 (1H, м), 2,03-1,98 (2Н, м).
31Р-ЯМР (CD3OD) δ: 60,3 (с), 59,1 (с).
[0856]
Пример 49. Синтез ЦДН39.
(5R,7R,8R,12aR,14R,15R,15aS,16R)-16-фтор-15-гидрокси-7-[1-(2-гидроксиэтил)-6-оксо-1,6-дигидро-9Н-пурин-9-ил]-2,10-бис(сульфанил)-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-дион
[0857]
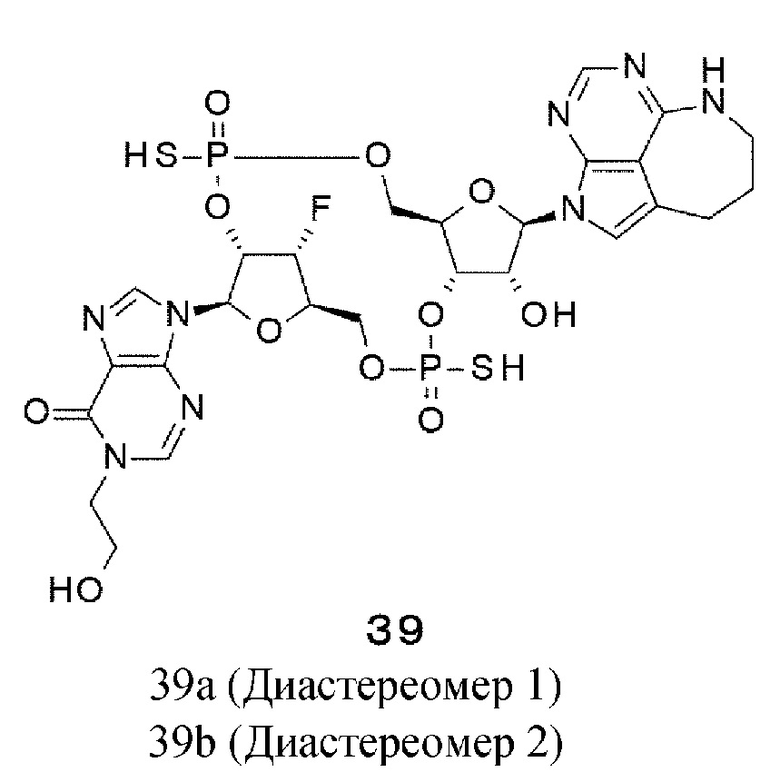
[0858]
[Схема синтеза]
[0859]
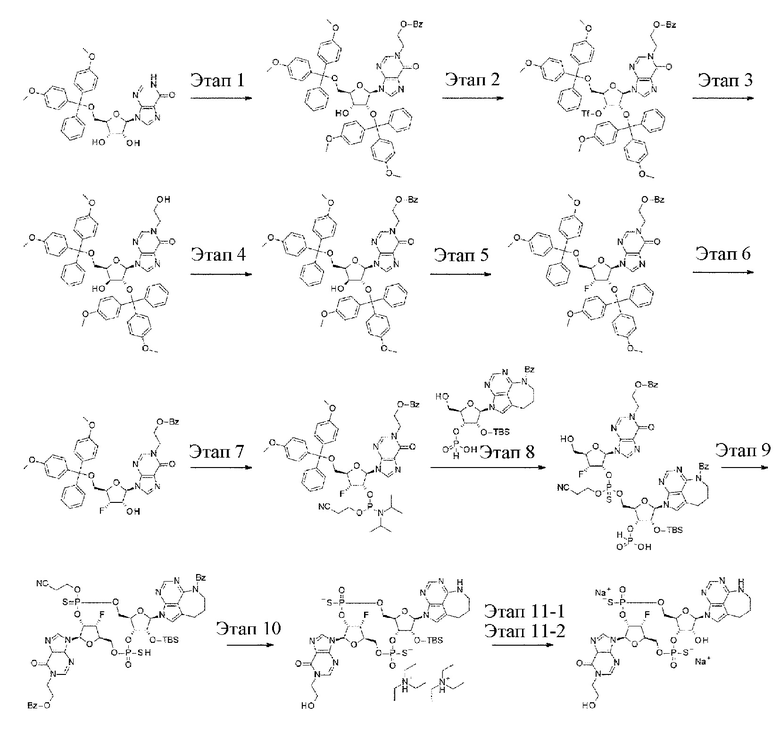
[0860]
(Этап 1)
1-[2-(бензоилокси)этил]-2',5'-бис-О-[бис(4-метоксифенил)(фенил)метил]инозин
В смешанный раствор доступного для приобретения (Amadis Chemical Company Limited) 5'-O-[бис(4-метоксифенил)(фенил)метил]инозина (20,0 г) в пиридине (50 мл)/N,N-диметилацетамиде (70,1 мл) добавляли 4,4'-диметокситритилхлорид (12,5 г), 2-бромоэтилбензоат (6,60 мл) и 1,8-диазабицикло[5,4,0]-7-ундецен (13,1 мл), и реакционную смесь перемешивали при комнатной температуре в течение 20 часов. Насыщенный водный раствор гидрокарбоната натрия и воды добавляли в реакционную смесь, чтобы погасить реакцию, и продукт затем осуществляли экстрагирование этилацетатом. Органический слой промывали рассолом и сушили над безводным сульфатом натрия. Осушающий агент удаляли путем фильтрации и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле [гексан/этилацетат/0,1% триэтиламин] с получением указанного в заголовке соединения (24,1 г: с примесями).
МС (ИЭР) m/z: 1121 (М+Н)+.
[0861]
(Этап 2)
1-[2-(бензоилокси)этил]-2',5'-бис-O-[бис(4-метоксифенил)(фенил)метил]-3'-O-(трифторметансульфонил)инозин
К раствору соединения (24,1 г), полученного на описанном выше этапе 1, в дихлорметане (120 мл) добавляли пиридин (19,0 мл), и медленно добавляли туда по каплям трифторметансульфоновый ангидрид (5,96 мл) при 0°С, и реакционную смесь перемешивали при той же температуре в течение 1 часа. Насыщенный водный раствор гидрокарбоната натрия и воды добавляли в реакционную смесь, чтобы погасить реакцию, и продукт затем осуществляли экстрагирование дихлорметаном, и органический слой сушили над безводным сульфатом натрия. Осушающий агент удаляли путем фильтрации и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле [гексан/этилацетат/0,1% триэтиламин] с получением указанного в заголовке соединения (19,7 г: с примесями).
МС (ИЭР) m/z: 1153 (М+Н)+.
[0862]
(Этап 3)
9-{2,5-бис-O-[бис(4-метоксифенил)(фенил)метил]-β-D-ксилофуранозил}-1-(2-гидроксиэтил)-1,9-дигидро-6Н-пурин-6-он
К раствору соединения (19,7 г), полученного на описанном выше этапе 2, в N,N-диметилформамиде (85,4 мл) добавляли ацетат цезия (8,20 г) и реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Метанол (85,4 мл) и карбонат калия (4,72 г) добавляли в реакционную смесь, которую перемешивали при комнатной температуре в течение 2 часов. В реакционную смесь добавляли воду, чтобы погасить реакцию, и продукт затем осуществляли экстрагирование этилацетатом. Органический слой промывали рассолом и сушили над безводным сульфатом натрия.
Осушающий агент удаляли путем фильтрации и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле [гексан/этилацетат/метанол/0,1% триэтиламин] с получением указанного в заголовке соединения (12,2 г: с примесями).
МС (ИЭР) m/z: 917 (М+Н)+.
[0863]
(Этап 4)
1-[2-(бензоилокси)этил]-9-{2,5-бис-O-[бис(4-метоксифенил)(фенил)метил]-β-D-ксилофуранозил}-1,9-дигидро-6Н-пурин-6-он
К раствору соединения (12,2 г), полученного на описанном выше этапе 3, в дихлорметане (48,8 мл) добавляли пиридин (1,61 мл) и бензойный ангидрид (3,16 г), и реакционную смесь перемешивали при комнатной температуре в течение 64 часов. Насыщенный водный раствор гидрокарбоната натрия и воды добавляли в реакционную смесь, чтобы погасить реакцию, и осуществляли экстрагирование продукта этилацетатом. Органический слой промывали рассолом, а затем сушили над безводным сульфатом натрия. Осушающий агент удаляли путем фильтрации и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле [гексан/этилацетат/метанол/0,1% триэтиламин] с получением указанного в заголовке соединения (7,36 г: с примесями).
МС (ИЭР) m/z: 1021 (М+Н)+.
[0864]
(Этап 5)
1-[2-(бензоилокси)этил]-2',5'-бис-O-[бис(4-метоксифенил)(фенил)метил]-3'-дезокси-3'-фторинозин
К раствору соединения (7,36 г), полученного на описанном выше этапе 4, в дихлорметане (37,0 мл) добавляли 2,6-лутидин (3,34 мл), в который затем добавляли трифторид N,N-диэтиламиносеры (1,42 г) при -78°С и температуру постепенно повышали до комнатной температуры в атмосфере азота, и после этого реакционную смесь перемешивали в течение ночи. Насыщенный водный раствор гидрокарбоната натрия медленно добавляли в реакционную смесь, чтобы погасить реакцию, добавляли туда воду и осуществляли экстрагирование продукта этилацетатом. Органический слой промывали рассолом, а затем сушили над безводным сульфатом натрия. Осушающий агент удаляли путем фильтрации и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле [гексан/этилацетат 0,1% триэтиламин] с получением указанного в заголовке соединения (6,89 г: с примесями).
МС (ИЭР) m/z: 1023 (М+Н)+.
[0865]
(Этап 6)
1-[2-(бензоилокси)этил]-5'-O-[бис(4-метоксифенил)(фенил)метил]-3'-дезокси-3'-фторинозин
К раствору соединения (6,89 г), полученного на описанном выше этапе 5, в дихлорметане (25,0 мл) добавляли воду (0,303 мл) и дихлоруксусную кислоту (1,39 мл) и реакционную смесь перемешивали при комнатной температуре в течение 63 часов. Пиридин (2,0 мл) добавляли в реакционную смесь, которую концентрировали при пониженном давлении. Остаток частично очищали с помощью колоночной хроматографии на силикагеле [гексан/этилацетат/метанол] и колоночной хроматографии на силикагеле DIOL [гексан/этилацетат]. К раствору соединения (25,0 мл), полученному в пиридине, добавляли 4,4'-диметокситритилхлорид (1,83 г) при 0°С, и реакционную смесь перемешивали при 4°С в течение 21 часа. Метанол (1,0 мл) добавляли в реакционную смесь, которую перемешивали при 4°С в течение 1 часа, а затем концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле [гексан/этилацетат/метанол/0,1% триэтиламин] с получением указанного в заголовке соединения (1,33 г).
МС (ИЭР) m/z: 721 (М+Н)+.
1H-ЯМР (CDCl3) δ: 7,99 (1H, с), 7,95 (1H, д, J=1,2 Гц), 7,93 (1H, с), 7,93 (1H, д, J=1,5 Гц), 7,58-7,51 (1H, м), 7,43-7,37 (2Н, м), 7,35-7,30 (2Н, м), 7,26-7,13 (7Н, м), 6,82-6,75 (4Н, м), 5,98 (1H, д, J=7,3 Гц), 5,13 (1H, дд, J=54,7, 4,1 Гц), 5,05-4,92 (1H, м), 4,63 (2Н, т, J=5,1 Гц), 4,56-4,25 (4Н, м), 3,76 (3Н, с), 3,76 (3Н, с), 3,45 (1H, дд, J=10,9, 3,6 Гц), 3,34 (1H, дд, J=10,9, 3,6 Гц).
[0866]
(Этап 7)
1-[2-(бензоилокси)этил]-5'-O-[бис(4-метоксифенил)(фенил)метил]-2'-O-{(2-цианоэтокси)[ди(пропан-2-ил)амино]фосфанил}-3'-дезокси-3'-фторинозин
С применением соединения (1,33 г), полученного на описанном выше этапе 6, проводили реакцию таким же образом, как на этапе 4 примера 5, с получением указанного в заголовке соединения (1,49 г) в виде смеси диастереомеров у атома фосфора (соотношение диастереомеров=1:1).
МС (ИЭР) m/z: 921 (М+Н)+.
1H-ЯМР (CDCl3) δ: 7,99 (0,5Н, с), 7,98-7,93 (3Н, м), 7,88 (0,5Н, с), 7,58-7,52 (1H, м), 7,45-7,37 (4Н, м), 7,33-7,18 (7Н, м), 6,85-6,77 (4Н, м), 6,14 (0,5Н, д, J=7,3 Гц), 6,10 (0,5Н, д, J=7,9 Гц), 5,27-5,01 (2Н, м), 4,70-4,61 (2Н, м), 4,57-4,30 (3Н, м), 3,78 (3Н, с), 3,77 (3Н, с), 3,62-3,33 (6Н, м), 2,56 (1Н, т, J=6,3 Гц), 2,32 (1Н, т, J=6,3 Гц), 1,12 (6Н, д, J=6,3 Гц), 1,04 (3Н, д, J=6,3 Гц), 0,76 (3Н, д, J=6,3 Гц).
[0867]
(Этап 8)
Ту же реакцию, что и на этапе 7 примера 1, проводили в следующем масштабе (сырьевой материал: 948 мг). С применением раствора в ацетонитриле полученного соединения и соединения (850 мг), полученного на описанном выше этапе 7, проводили реакцию таким же образом, как на этапе 8 примера 1, и полученный в результате этого неочищенный продукт непосредственно использовали для следующей реакции.
[0868]
(Этап 9)
2-{9-[(5R,7R,8S,12aR,14R,15R,15aR,16R)-14-(6-бензоил-6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)-15-{[трет-бутил(диметил)силил]окси}-10-(2-цианоэтокси)-16-фтор-2-оксо-2-сульфанил-10-сульфанилиденоктагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-7-ил]-6-оксо-6,9-дигидро-1H-пурин-1-ил}этилбензоат
С применением неочищенного продукта, полученного на описанном выше этапе 8, проводили реакцию таким же образом, как на этапе 9 примера 1, с получением указанного в заголовке соединения (709 мг) в виде смеси диастереомеров у атома фосфора.
МС (ИЭР) m/z: 1152 (М+Н)+.
[0869]
(Этап 10)
Бис(N,N-диэтилэтанаминия) (5R,7R,8S,12aR,14R,15R,15aR,16R)-15-{[трет-бутил(диметил)силил]окси}-16-фтор-7-[1-(2-гидроксиэтил)-6-оксо-1,6-дигидро-9Н-пурин-9-ил]-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфацикл отетрадецин-2,10-бис(тиолят)
С применением соединения (709 мг), полученного на описанном выше этапе 9, проводили реакцию таким же образом, как на этапе 10 примера 1, с получением диастереомера 1 (108 мг: с примесями) и диастереомера 2 (102 мг: с примесями) указанного в заголовке соединения. Диастереомер 1 (менее полярный)
МС (ИЭР) m/z: 891 (М+Н)+.
Диастереомер 2 (более полярный)
МС (ИЭР) m/z: 891 (М+Н)+.
[0870]
(Этап 11-1)
Динатрия (5R,7R,8S,12aR,14R,15R,15aS,16R)-16-фтор-15-гидрокси-7-[1-(2-гидроксиэтил)-6-оксо-1,6-дигидро-9Н-пурин-9-ил]-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентао ксадифосфацикл отетрадецин-2,10-бис(тиолят)
(Диастереомер 1)
С применением соединения (диастереомера 1) (108 мг: с примесями), полученного на описанном выше этапе 10, проводили реакцию таким же образом, как на этапе 11 примера 1, а затем проводили очистку при следующих [Условиях очистки] с получением указанного в заголовке соединения в виде соли триэтиламина.
[Условия очистки] Хроматография на колонке с силикагелем С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил] и препаративная ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 3% - 30% (0 мин - 40 мин)].
Полученную соль триэтиламина подвергали солевому обмену таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением указанного в заголовке соединения (40,1 мг).
МС (ИЭР) m/z: 777 (М+Н)+.
1H-ЯМР (CD3OD) δ: 8,66 (1H, м), 8,26 (1Н, с), 8,02 (1Н, с), 7,09 (1H, с), 6,29-6,24 (2Н, м), 5,68-5,45 (2Н, м), 5,26-5,18 (1H, м), 4,78-4,73 (1H, м), 4,62-4,42 (3Н, м), 4,26-3,98 (5Н, м), 3,85-3,78 (2Н, м), 3,53-3,47 (2Н, м), 2,91-2,84 (2Н, м), 2,04-1,96 (2Н, м).
31Р-ЯМР (CD3OD) δ: 58,0 (с), 56,9 (с).
[0871]
(Этап 11-2)
Динатрия (5R,7R,8S,12aR,14R,15R,15aS,16R)-16-фтор-15-гидрокси-7-[1-(2-гидроксиэтил)-6-оксо-1,6-дигидро-9Н-пурин-9-ил]-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят) (Диастереомер 2)
С применением соединения (диастереомера 2) (102 мг: с примесями), полученного на описанном выше этапе 10, проводили реакцию таким же образом, как на этапе 11 примера 1, а затем проводили очистку при следующих [Условиях очистки] с получением указанного в заголовке соединения в виде соли триэтиламина.
[Условия очистки] Хроматография на колонке с силикагелем С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил] и препаративная ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 3% - 25% (0 мин - 40 мин)].
Полученную соль триэтиламина подвергали солевому обмену таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением указанного в заголовке соединения (26,7 мг).
МС (ИЭР) m/z: 777(М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,73 (1H, м), 8,25 (1H, с), 8,02 (1H, с), 7,10 (1H, уш. с), 6,33 (1H, д, J=6,7 Гц), 6,28 (1Н, д, J=9,1 Гц), 5,65-5,50 (1Н, м), 5,49-5,43 (1Н, м), 5,31 (1Н, дд, J=54,4, 3,6 Гц), 4,79 (1Н, дд, J=6,3, 4,5 Гц), 4,62-4,34 (4Н, м), 4,27-4,14 (2Н, м), 4,08-4,01 (1H, м), 3,93-3,87 (1H, м), 3,86-3,80 (2Н, м), 3,53-3,47 (2Н, м), 2,95-2,89 (2Н, м), 2,06-1,97 (2Н, м).
31Р-ЯМР (CD3OD) δ: 63,1 (с), 59,7 (с).
[0872]
Пример 50. Синтез ЦДН40.
(5R,7R,8S,12aR,14R,15R,15aS,16R)-7-[1-(2-аминоэтил)-6-оксо-1,6-дигидро-9Н-пурин-9-ил]-16-фтор-15-гидрокси-2,10-бис(сульфанил)-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-дион
[0873]
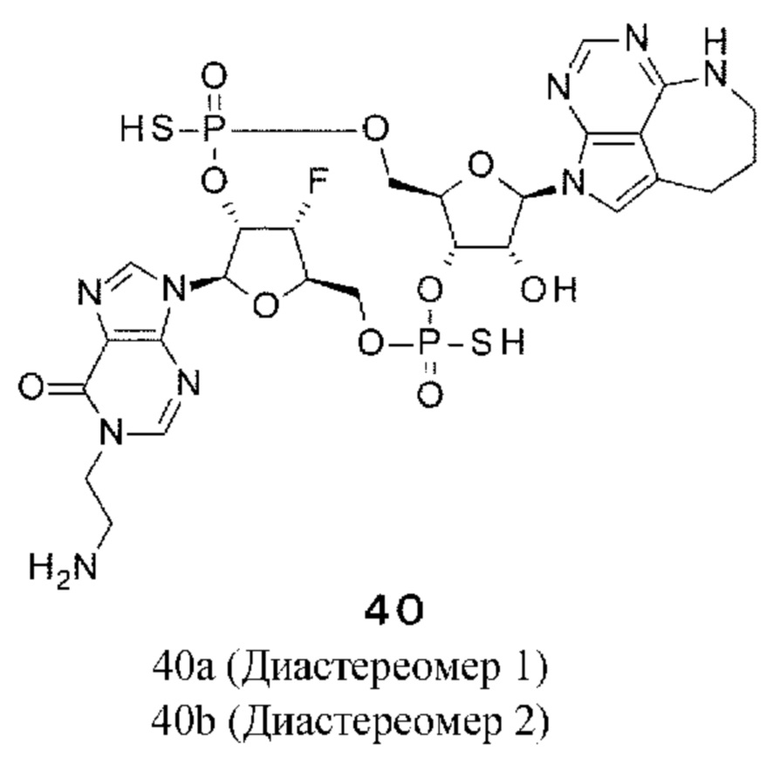
[0874]
[Схема синтеза]
[0875]
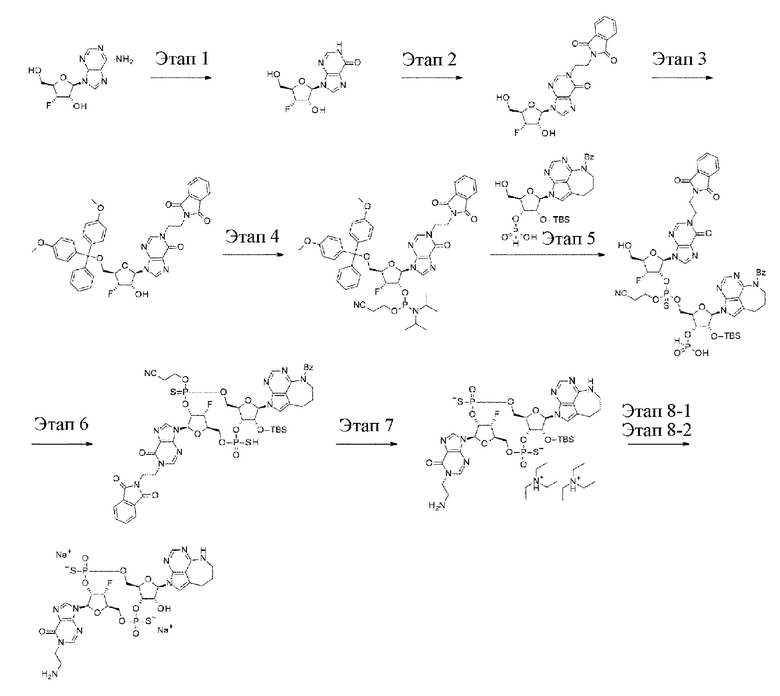
[0876]
(Этап 1)
3'-дезокси-3'-фторинозин
К раствору доступного для приобретения (Angene International Limited) 3'-дезокси-3'-фтораденозина (2,38 г) в уксусной кислоте (120 мл) добавляли малыми частями водный раствор (48 мл) нитрита натрия (6,10 г), и реакционную смесь перемешивали при комнатной температуре в течение 43 часов. Реакционную смесь концентрировали при пониженном давлении и дважды подвергали азеотропной перегонке с толуолом. Остаток очищали с помощью колоночной хроматографии на силикагеле [дихлорметан/метанол] с получением указанного в заголовке соединения (3,76 г).
МС (ИЭР) m/z: 271(М+Н)+.
1H-ЯМР (CD3OD) δ: 8,30 (1H, с), 8,05 (1H, с), 6,03 (1H, д, J=7,8 Гц), 5,08 (1H, дд, J=54,4, 4,1 Гц), 4,89-4,85 (1H, м), 4,38 (1H, дт, J=26,8, 3,2 Гц), 3,82-3,73 (2Н, м).
(Этап 2)
3'-дезокси-1-[2-(1,3-Диоксо-1,3-дигидро-2Н-изоиндол-2-ил)этил]-3'-фторинозин
К раствору соединения (3,76 г), полученного на описанном выше этапе 1, в N,N-диметилацетамиде (37,6 мл) добавляли 2-(2-бромоэтил)-1Н-изоиндол-1,3(2Н)-дион (6,60 мл) и 1,8-диазабицикло[5,4,0]-7-ундецен (3,12 мл), и реакционную смесь перемешивали при комнатной температуре в течение 3 суток. Реакционную смесь концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле [гексан/этилацетат/метанол] с получением указанного в заголовке соединения (3,51 г: с примесями).
МС (ИЭР) m/z: 444(М+Н)+.
1H-ЯМР (CDCl3) δ: 7,87 (1H, с), 7,84 (1Н, с), 7,81 (2Н, дд, J=5,4, 3,0 Гц), 7,73 (2Н, дд, J=5,4, 3,0 Гц), 5,83 (1H, д, J=7,9 Гц), 5,28 (1H, дд, J=11,5, 2,4 Гц), 5,18 (1H, дд, J=55,0, 4,2 Гц), 5,01-4,87 (1H, м), 4,49 (1H, д, J=28,4 Гц), 4,43-4,28 (2Н, м), 4,19-4,08 (2Н, м), 3,95 (1H, д, J=8,2 Гц), 3,91-3,84 (1H, м), 3,76 (1Н, д, J=13,3 Гц).
[0878]
(Этап 3)
5'-O-[бис(4-метоксифенил)(фенил)метил]-3'-дезокси-1-[2-(1,3-диоксо-1,3-дигидро-2Н-изоиндол-2-ил)этил]-3'-фторинозин
С применением соединения (3,51 г), полученного на описанном выше этапе 2, проводили реакцию таким же образом, как на этапе 1 примера 11, с получением указанного в заголовке соединения (4,05 г).
МС (ИЭР) m/z: 746 (М+Н)+.
1H-ЯМР (CDCl3) δ: 7,95 (1H, с), 7,79 (2Н, дд, J=5,4, 3,0 Гц), 7,73 (1H, с), 7,70 (2Н, дд, J=5,4, 3,0 Гц), 7,34-7,29 (2Н, м), 7,25-7,18 (7Н, м), 6,81-6,76 (4Н, м), 5,92 (1H, д, J=7,3 Гц), 5,12 (1H, дд, J=54,4, 4,2 Гц), 4,97-4,85 (1H, м), 4,55-4,40 (2Н, м), 4,30-4,22 (1H, м), 4,21-4,01 (2Н, м), 3,77 (6Н, с), 3,73 (1H, д, J=10,3 Гц), 3,43 (1H, дд, J=10,9, 3,6 Гц), 3,32 (1Н, дд, J=10,9, 3,6 Гц).
[0879]
(Этап 4)
5'-O-[бис(4-метоксифенил)(фенил)метил]-2'-O-{(2-цианоэтокси)[ди(пропан-2-ил)амино]фосфанил}-3'-дезокси-1-[2-(1,3-Диоксо-1,3-дигидро-2Н-изоиндол-2-ил)этил]-3'-фторинозин
С применением соединения (4,05 г), полученного на описанном выше этапе 3, проводили реакцию таким же образом, как на этапе 4 примера 5, с получением указанного в заголовке соединения (4,35 г) в виде смеси диастереомеров у атома фосфора (соотношение диастереомеров=1:1).
МС (ИЭР) m/z: 946 (М+Н)+.
1H-ЯМР (CDCl3) δ: 7,95 (0,5Н, с), 7,94 (0,5Н, с), 7,84-7,78 (2Н, м), 7,73-7,68 (2Н, м), 7,65 (1H, с), 7,43-7,41 (2Н, м), 7,32-7,15 (7Н, м), 6,84-6,77 (4Н, м), 6,09 (0,5Н, д, J=7,9 Гц), 6,05 (0,5Н, д, J=7,9 Гц), 5,31-5,16 (1H, м), 5,15-4,98 (1H, м), 4,51-4,21 (3Н, м), 4,20-4,05 (2Н, м), 3,793 (1,5Н, с), 3,789 (3Н, с), 3,784 (1,5Н, с), 3,66-3,55 (2Н, м), 3,50-3,30 (4Н, м), 2,56 (1H, т, J=6,3 Гц), 2,41 (1H, т, J=6,3 Гц), 1,16 (3Н, д, J=7,3 Гц), 1,14 (3Н, д, J=7,3 Гц), 1,10 (3Н, д, J=7,3 Гц), 0,83 (3Н, д, J=7,3 Гц).
[0880]
(Этап 5)
Ту же реакцию, что и на этапе 7 примера 1, проводили в следующем масштабе (сырьевой материал: 1,47 г). С применением раствора в ацетонитриле полученного соединения и соединения (1,35 г), полученного на описанном выше этапе 4, проводили реакцию таким же образом, как на этапе 8 примера 1, и полученный в результате этого неочищенный продукт непосредственно использовали для следующей реакции.
[0881]
(Этап 6)
3-{[(5R,7R,8S,12aR,14R,15R,15aR,16R)-14-(6-бензоил-6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)-15-{[трет-бутил(диметил)силил]окси}-7-{1-[2-(1,3-диоксо-1,3-дигидро-2Н-изоиндол-2-ил)этил]-6-оксо-1,6-дигидро-9Н-пурин-9-ил}-16-фтор-2-оксо-2-сульфанил-10-сульфанилиденоктагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-10-ил]окси}пропаннитрил
С применением неочищенного продукта, полученного на описанном выше этапе 5, проводили реакцию таким же образом, как на этапе 9 примера 1, с получением указанного в заголовке соединения (1,25 г) в виде смеси диастереомеров у атома фосфора.
МС (ИЭР) m/z: 1177(М+Н)+.
[0882]
(Этап 7)
Бис(N,N-диэтилэтанаминия) (5R,7R,8S,12aR,14R,15R,15aR,16R)-7-[1-(2-аминоэтил)-6-оксо-1,6-дигидро-9Н-пурин-9-ил]-15-{[трет-бутил(диметил)силил]окси}-16-фтор-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
В смешанный раствор соединения (1,25 г), полученного на описанном выше этапе 6, в этаноле (7,5 мл)/тетрагидрофуране (7,5 мл) добавляли моногидрат гидразина (0,599 мл), и реакционную смесь перемешивали при 50°С в течение 6 часов. После концентрирования реакционной смеси при пониженном давлении остаток очищали с помощью колоночной хроматографии на силикагеле С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил] с получением диастереомера 1 (145 мг: с примесями) и диастереомера 2 (198 мг: с примесями) указанного в заголовке соединения (время удерживания при ВЭЖХ: диастереомер 1>2).
Диастереомер 1 (менее полярный)
МС (ИЭР) m/z: 890(М+Н)+.
Диастереомер 2 (более полярный)
МС (ИЭР) m/z: 890(М+Н)+.
[0883]
(Этап 8-1)
Динатрия (5R,7R,8S,12aR,14R,15R,15aS,16R)-7-[1-(2-аминоэтил)-6-оксо-1,6-дигидро-9Н-пурин-9-ил]-16-фтор-15-гидрокси-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
(Диастереомер 1)
С применением соединения (диастереомера 1) (145 мг: с примесями), полученного на описанном выше этапе 7, проводили реакцию таким же образом, как на этапе 11 примера 1, а затем проводили очистку при следующих [Условиях очистки] с получением указанного в заголовке соединения в виде соли триэтиламина.
[Условия очистки] Хроматография на колонке с силикагелем С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил] и препаративная ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/раствор ацетонитрила-метанола (1:1), раствор ацетонитрила-метанола (1:1): 5% - 50% (0 мин - 40 мин)].
Полученную соль триэтиламина подвергали солевому обмену таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением указанного в заголовке соединения (70,7 мг).
МС (ИЭР) m/z: 776(М+Н)+.
1H-ЯМР (CD3OD) δ: 8,64 (1H, с), 8,23 (1H, с), 8,02 (1H, с), 7,07 (1H, с), 6,27 (1H, д, J=4,5 Гц), 6,25 (1H, д, J=6,7 Гц), 5,67-5,43 (2Н, м), 5,23-5,16 (1H, м), 4,76 (1Н, т, J=5,l Гц), 4,63-4,45 (3H, м), 4,31-4,03 (5Н, м), 3,51-3,46 (2Н, м), 3,31-3,26 (2Н, м), 2,90-2,83 (2Н, м), 2,03-1,94 (2Н, м).
31Р-ЯМР (CD3OD) δ: 57,9 (с), 56,9 (с).
[0884]
(Этап 8-2)
Динатрия (5R,7R,8S,12aR,14R,15R,15aS,16R)-7-[1-(2-аминоэтил)-6-оксо-1,6-дигидро-9Н-пурин-9-ил]-16-фтор-15-гидрокси-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
(Диастереомер 2)
С применением соединения (диастереомера 2) (198 мг: с примесями), полученного на описанном выше этапе 7, проводили реакцию таким же образом, как на этапе 11 примера 1, а затем проводили очистку при следующих [Условиях очистки] с получением указанного в заголовке соединения в виде соли триэтиламина.
[Условия очистки] Хроматография на колонке с силикагелем С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил] и препаративная ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/раствор ацетонитрила-метанола (1:1), раствор ацетонитрила-метанола (1:1): 5% - 50% (0 мин - 40 мин)].
Полученную соль триэтиламина подвергали солевому обмену таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением указанного в заголовке соединения (69,7 мг).
МС (ИЭР) m/z: 776(М+Н)+.
1H-ЯМР (CD3OD) δ: 8,73 (1H, с), 8,27 (1H, с), 8,02 (1H, с), 7,09 (1H, с), 6,31 (1H, д, J=6,7 Гц), 6,26 (1Н, д, J=8,5 Гц), 5,62-5,47 (1Н, м), 5,47-5,41 (1H, м), 5,30 (1H, дд, J=53,8, 3,6 Гц), 4,78 (1H, дд, J=6,7, 4,2 Гц), 4,58 (1H, д, J=26,0 Гц), 4,51-4,41 (2Н, м), 4,40-4,23 (3H, м), 4,11-4,05 (1H, м), 3,93-3,86 (1H, м), 3,53-3,46 (2Н, м), 3,36-3,28 (2Н, м), 2,90 (2Н, т, J=5,7 Гц), 2,05-1,96 (2Н, м).
31Р-ЯМР (CD3OD) δ: 62,8 (с), 59,4 (с).
[0885]
Пример 51. Синтез ЦДН41.
N-(2-{9-[(5R,7R,8S,12aR,14R,15R,15aS,16R)-16-фтор-15-гидрокси-2,10-диоксо-2,10-бис(сульфанил)-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-7-ил]-6-оксо-6,9-дигидро-1Н-пурин-1-ил}этил)-2-гидроксиацетамид
[0886]
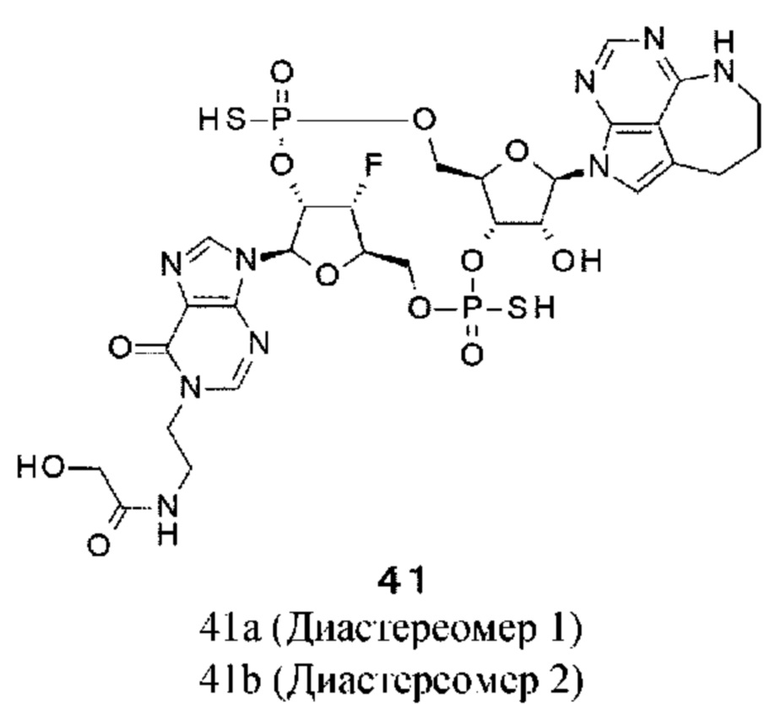
[0887]
[Схема синтеза]
[0888]
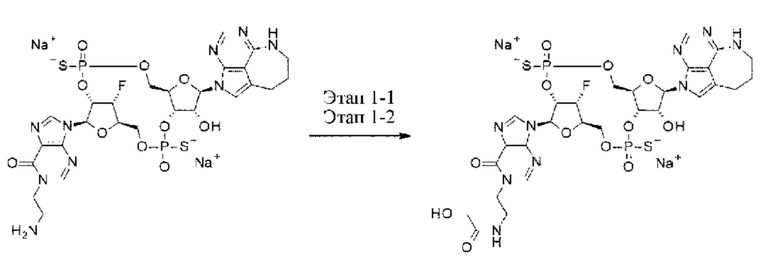
[0889]
(Этап 1-1)
Динатрия (5R,7R,8S,12aR,14R,15R,15aS,16R)-16-фтор-15-гидрокси-7-{l-[2-(2-гидроксиацетамид)этил]-6-оксо-1,6-дигидро-9Н-пурин-9-ил}-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
(Диастереомер 1)
С применением соединения (25,0 мг), полученного на этапе 8-1 примера 50, проводили реакцию таким же образом, как на этапе 1-1 примера 7, а затем проводили очистку при следующих [Условиях очистки] с получением указанного в заголовке соединения в виде соли триэтиламина.
[Условия очистки] Хроматография на колонке с силикагелем С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил] и препаративная ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 5% - 30% (0 мин - 40 мин)].
Полученную соль триэтиламина подвергали солевому обмену таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением указанного в заголовке соединения (18,6 мг).
МС (ИЭР) m/z: 834(М+Н)+.
1H-ЯМР (CD3OD) δ: 8,60 (1H, м), 8,12 (1H, м), 8,02 (1H, с), 7,11 (1H, уш. с), 6,27 (1H, д, J=4,9 Гц), 6,23 (1H, д, J=9,l Гц), 5,75-5,58 (1H, м), 5,54 (1Н, дд, J=53,5, 3,0 Гц), 5,28-5,20 (1H, м), 4,75 (1Н, т, 3=5,2 Гц), 4,62-4,52 (1Н, м), 4,52-4,42 (2Н, м), 4,29-4,01 (5Н, м), 3,92 (2Н, с), 3,64-3,58 (2Н, м), 3,53-3,47 (2Н, м), 2,93-2,76 (2Н, м), 2,06-1,93 (2Н, м).
31Р-ЯМР (CD3OD) δ: 57,9 (с), 56,7 (с).
[0890]
(Этап 1-2)
Динатрия (5R,7R,8S,12aR,14R,15R,15aS,16R)-16-фтор-15-гидрокси-7-{1-[2-(2-гидроксиацетамид)этил]-6-оксо-1,6-дигидро-9Н-пурин-9-ил}-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
(Диастереомер 2)
С применением соединения (диастереомера 2) (15,0 мг), полученного на этапе 8-2 примера 50, проводили реакцию таким же образом, как на этапе 1-1 примера 7, а затем проводили очистку при следующих [Условиях очистки] с получением указанного в заголовке соединения в виде соли триэтиламина.
[Условия очистки] Хроматография на колонке с силикагелем С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил] и препаративная ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 5% - 30% (0 мин - 40 мин)].
Полученную соль триэтиламина подвергали солевому обмену таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением указанного в заголовке соединения (7,4 мг).
МС (ИЭР) m/z: 834(М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,69 (1H, м), 8,16 (1H, с), 8,02 (1H, с), 7,11 (1H, с), 6,32 (1H, д, J=6,7 Гц), 6,26 (1H, д, J=8,6 Гц), 5,67-5,51 (1H, м), 5,48-5,43 (1H, м), 5,29 (1H, дд, J=54,0, 3,7 Гц), 4,77 (1H, дд, J=6,4, 4,6 Гц), 4,62-4,33 (2Н, м), 4,25-4,17 (2Н, м), 4,08-4,01 (1H, м), 3,94 (2Н, с), 3,93-3,85 (1H, м), 3,70-3,56 (2Н, м), 3,52-3,46 (2Н, м), 2,93-2,86 (2Н, м), 2,04-1,97 (4Н, м).
31Р-ЯМР (CD3OD) δ: 62,8 (с), 59,5 (с).
[0891]
Пример 52. Синтез ЦДН42.
(5R,7R,8S,12aR,14R,15R,15aS,16R)-7-{6-амино-2-[(2-аминоэтил)амино]-9H-пурин-9-ил}-16-фтор-15-гидрокси-2,10-бис(сульфанил)-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-дион
[0892]
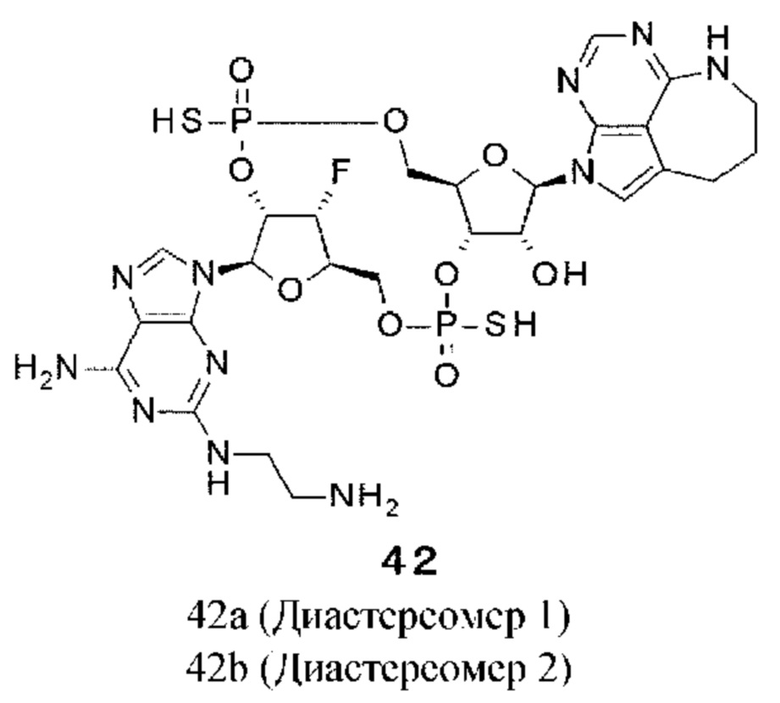
[0893]
[Схема синтеза]
[0894]
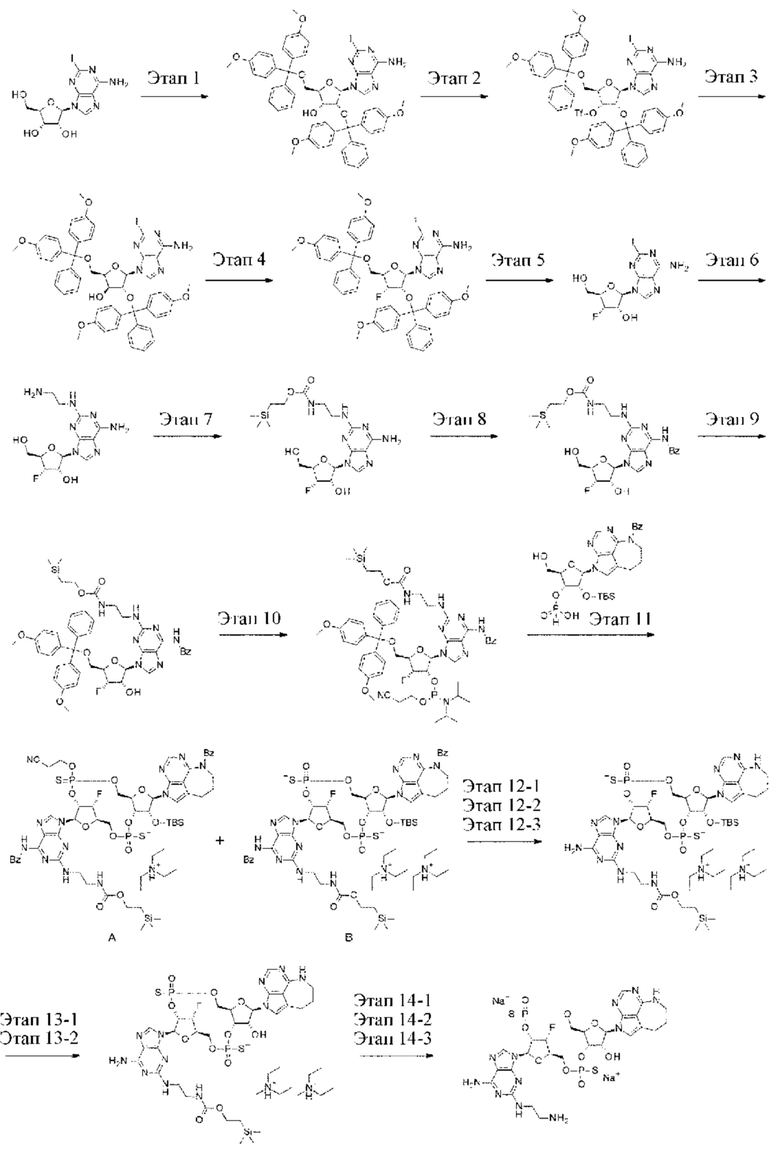
[0895]
(Этап 1)
2',5'-бис-О-[бис(4-метоксифенил)(фенил)метил]-2-йодаденозин
Доступный для приобретения (Amadis Chemical Company Limited) 2-йодаденозин (1,35 г) подвергали трехкратной азеотропной перегонке с пиридином. К раствору остатка в обезвоженном пиридине (17,0 мл) добавляли 4,4'-диметокситритилхлорид (2,35 г) и реакционную смесь перемешивали в атмосфере азота при комнатной температуре в течение ночи. Метанол (5,00 мл) добавляли в реакционную смесь, чтобы погасить реакцию, и продукт затем концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле [гексан/этилацетат/0,1% триэтиламин] с получением указанного в заголовке соединения (1,96 г: с примесями).
МС (ИЭР) m/z: 998(М+Н)+.
1H-ЯМР (CDCl3) δ: 7,88 (1H, с), 7,29-7,12 (18Н, м), 6,73-6,60 (8Н, м), 6,31 (1H, д, J=7,9 Гц), 5,69 (2Н, уш. с), 5,13 (1H, дд, J=7,6, 4,5 Гц), 4,07 (1H, т, J=3,3 Гц), 3,77 (6Н, с), 3,76 (3H, с), 3,74 (3H, с), 3,14 (2Н, д, J=3,6 Гц), 2,87 (1H, д, J=4,2 Гц), 2,28 (1H, с).
[0896]
(Этап 2)
2',5'-бис-O-[бис(4-метоксифенил)(фенил)метил]-2-йод-3'-O-(трифторметансульфонил)аденозин
Соединение (100 мг), полученное на описанном выше этапе 1, подвергали трехкратной азеотропной перегонке с толуолом. К раствору остатка в обезвоженном дихлорметане (1,0 мл) добавляли пиридин (0,20 мл) и трифторметансульфоновый ангидрид (27,0 мкл) в атмосфере азота при 0°С и реакционную смесь перемешивали при той же температуре в течение 80 минут. Дополнительно добавляли туда трифторметансульфоновый ангидрид (27,0 мкл) и реакционную смесь дополнительно перемешивали в течение 80 минут. Насыщенный водный раствор гидрокарбоната натрия добавляли в реакционную смесь, чтобы погасить реакцию, и продукт затем осуществляли экстрагирование дихлорметаном. После высушивания органического слоя над безводным сульфатом натрия осушающий агент удаляли путем фильтрации и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле [гексан/этилацетат/0,1% триэтиламин] с получением указанного в заголовке соединения (57,2 мг).
МС (ИЭР) m/z: 1130(М+Н)+.
1Н-ЯМР (CDCl3) δ: 7,73 (1H, с), 7,32-7,09 (18Н, м), 6,79-6,70 (6Н, м), 6,60-6,56 (2Н, м), 6,19 (1H, дд, J=8,2, 3,9 Гц), 6,01 (1Н, д, J=7,9 Гц), 5,59 (2Н, уш. с), 4,05 (1Н, д, J=3,6 Гц), 3,98 (1H, т, J=6,7 Гц), 3,78 (3H, с), 3,77 (3H, с), 3,71 (3H, с), 3,70 (3H, с), 3,39 (1H, дд, J=10,9, 6,0 Гц), 3,17 (1H, дд, J=10,6, 7,0 Гц).
[0897]
(Этап 3)
9-{2,5-бис-O-[бис(4-метоксифенил)(фенил)метил]-β-D-ксилофуранозил}-2-йод-9Н-пурин-6-амин
К раствору соединения (2,76 г), полученного на описанном выше этапе 2, в N,N-диметилформамиде (24,4 мл) добавляли ацетат цезия (1,20 г) и реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Метанол (24,4 мл) и карбонат калия (675 мг) добавляли в реакционную смесь, которую дополнительно перемешивали в течение 2 часов. В реакционную смесь добавляли воду, чтобы преципитировать твердый продукт, и компонент метанол затем отгоняли при пониженном давлении. Полученный в результате этого твердый продукт собирали путем фильтрации с получением указанного в заголовке соединения (2,38 г).
1Н-ЯМР (CDCl3) δ: 8,02 (1Н, с), 7,42-7,13 (18Н, м), 6,79-6,72 (8Н, м), 5,68 (2Н, уш. с), 5,55 (1H, д, J=l,2 Гц), 5,49 (1H, д, J=9,l Гц), 4,48 (1Н, с), 4,41-4,37 (1H, м), 4,06 (1H, дд, J=9,l, 3,6 Гц), 3,77 (9Н, с), 3,76 (3H, с), 3,56-3,48 (2Н, м).
[0898]
(Этап 4)
2',5'-бис-O-[бис(4-метоксифенил)(фенил)метил]-3'-дезокси-3'-фтор-2-йодаденозин
К раствору соединения (2,54 г), полученного на описанном выше этапе 3, в дихлорметане (17,0 мл) добавляли пиридин (1,13 мл) и добавляли туда трифторид N,N-диэтиламиносеры (0,397 мл) при охлаждении льдом, и реакционную смесь перемешивали в атмосфере азота при комнатной температуре в течение 40 минут. Насыщенный водный раствор гидрокарбоната натрия добавляли в реакционную смесь, чтобы погасить реакцию, и продукт затем осуществляли экстрагирование дихлорметаном. После высушивания органического слоя над безводным сульфатом натрия осушающий агент удаляли путем фильтрации и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле [гексан/этилацетат/0,1% триэтиламин] с получением указанного в заголовке соединения (2,02 г: с примесями).
МС (ИЭР) m/z: 1000(М+Н)+.
[0899]
(Этап 5)
3'-дезокси-3'-фтор-2-йодаденозин
К раствору смеси (2,02 г), полученной на описанном выше этапе 4, в дихлорметане (20,2 мл) добавляли воду (0,364 мл) и дихлоруксусную кислоту (0,996 мл) при охлаждении льдом, и реакционную смесь перемешивали при комнатной температуре в течение 15 минут. Пиридин (1,95 мл) добавляли в реакционную смесь, чтобы погасить реакцию, и продукт концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле [гексан/этилацетат] с получением указанного в заголовке соединения (1,34 г: с примесями).
1Н-ЯМР (CD3OD) δ: 5,94 (1H, д, J=7,9 Гц), 5,09 (1H, дд, J=54,7, 4,3 Гц), 4,95-4,89 (1H, м), 4,40 (1H, д, J=27,3 Гц), 3,87-3,76 (2Н, м) (показаны только наблюдаемые пики).
[0900]
(Этап 6)
2-[(2-аминоэтил)амино]-3'-дезокси-3'-фтораденозин
Этилендиамин (1,35 мл) добавляли к соединению (1,34 г), полученному на описанном выше этапе 5, и реакционную смесь перемешивали при 110°С в течение 1 часа. Реакционную смесь концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил] с получением указанного в заголовке соединения (543 мг: с примесями).
1Н-ЯМР (CD3OD) δ: 7,94 (1H, с), 5,88 (1, d, J=7,9 Гц), 5,09 (1H, дд, J=54,7, 4,5 Гц), 4,95 (1H, дк, J=25,l, 4,2 Гц), 4,38 (1H, дт, J=27,6, 2,7 Гц), 3,84-3,76 (2Н, м), 3,56-3,53 (2Н, м), 3,01 (2Н, т, J=5,7 Гц).
[0901]
(Этап 7)
3'-дезокси-3'-фтор-2-{[2-({[2-(триметилсилил)этокси]карбонил}амино)этил]амино}аденозин
К раствору соединения (543 мг), полученного на описанном выше этапе 6, в тетрагидрофуране (10 мл) добавляли 1-({[2-(триметилсилил)этокси]карбонил}окси)пирролидин-2,5-дион (961 мг), N,N-диметилформамид (5,0 мл) и метанол (5,0 мл) и реакционную смесь перемешивали при комнатной температуре в течение 1,5 часов. Туда дополнительно добавляли 1-({[2-(триметилсилил)этокси]карбонил}окси)пирролидин-2,5-дион (800 мг) и реакционную смесь дополнительно перемешивали в течение 40 минут. После концентрирования реакционной смеси при пониженном давлении остаток очищали с помощью колоночной хроматографии на силикагеле [гексан/этилацетат] с получением указанного в заголовке соединения (483 мг).
1H-ЯМР (CD3OD) δ: 7,87 (1H, с), 5,83 (1H, д, J=7,9 Гц), 5,15-4,96 (2Н, м), 4,35 (1H, дт, J=27,4, 2,7 Гц), 4,11-4,04 (2Н, м), 3,84-3,73 (2Н, м), 3,48-3,38 (2Н, м), 2,96-2,83 (2Н, м), 0,95-0,83 (2Н, м), 0,00 (9Н, с).
[0902]
(Этап 8)
N-бензоил-3'-дезокси-3'-фтор-2-{[2-({[2-(триметилсилил)этокси]карбонил}амино)этил]амино}аденозин
С применением соединения (483 мг), полученного на описанном выше этапе 7, проводили реакцию таким же образом, как на этапе 3 примера 11, с получением указанного в заголовке соединения (374 мг).
1H-ЯМР (CDCl3) δ: 9,16-9,03 (1Н, м), 8,01 (2Н, д, J=7,3 Гц), 7,63-7,33 (4Н, м), 5,85-5,60 (2Н, м), 5,21-4,91 (2Н, м), 4,45 (1H, д, J=27,8 Гц), 4,19-4,06 (2Н, м), 3,92 (1Н, д, J=12,7 Гц), 3,77-3,34 (6Н, м), 1,05-0,87 (2Н, м), -0,02 (9Н, с) (показаны только наблюдаемые пики).
[0903]
(Этап 9)
N-бензоил-5'-O-[бис(4-метоксифенил)(фенил)метил]-3'-дезокси-3'-фтор-2-{[2-({[2-(триметилсилил)этокси]карбонил}амино)этил]амино}аденозин
С применением соединения (374 мг), полученного на описанном выше этапе 8, проводили реакцию таким же образом, как на этапе 1 примера 11, с получением указанного в заголовке соединения (398 мг).
МС (ИЭР) m/z: 878(М+Н)+.
1Н-ЯМР (CDCl3) δ: 8,94 (1H, уш. с), 8,01-7,98 (2Н, м), 7,88-7,81 (1H, м), 7,62-7,49 (3H, м), 7,40-7,20 (9Н, м), 6,83-6,78 (4Н, м), 6,12-5,48 (2Н, м), 5,22-4,98 (2Н, м), 4,52 (1H, д, J=27,8 Гц), 4,24-4,10 (2Н, м), 3,77 (6Н, с), 3,65-3,29 (8Н, м), 1,04-0,88 (2Н, м), 0,01 (9Н, уш. с).
[0904]
(Этап 10)
N-бензоил-5'-O-[бис(4-метоксифенил)(фенил)метил]-2'-O-{(2-цианоэтокси)[ди(пропан-2-ил)амино]фосфанил}-3'-дезокси-3'-фтор-2-{[2-({[2-(триметилсилил)этокси]карбонил}амино)этил]амино}аденозин
С применением соединения (398 мг), полученного на описанном выше этапе 9, проводили реакцию таким же образом, как на этапе 6 примера 1, с получением указанного в заголовке соединения (434 мг) в виде смеси диастереомеров у атома фосфора (соотношение диастереомеров = 1:1).
1Н-ЯМР (CDCl3) δ: 8,79-8,75 (1H, м), 8,00-7,97 (2Н, м), 7,83 (0,5Н, с), 7,77 (0,5Н, с), 7,61-7,21 (12Н, м), 6,84-6,79 (4Н, м), 6,07-5,96 (1H, м), 5,58-5,10 (3H, м), 4,50-4,40 (1H, м), 4,11-4,06 (2Н, м), 3,87-3,80 (1H, м), 3,79 (3H, с), 3,78 (3H, с), 3,65-3,20 (10Н, м), 2,63-2,59 (1H, м), 2,39 (1H, т, J=6,3 Гц), 1,17 (3H, д, J=6,7 Гц), 1,15 (3H, д, J=6,7 Гц), 1,10 (3H, д, J=6,7 Гц), 0,92-0,86 (2Н, м), 0,81 (3H, д, J=6,7 Гц), -0,01 (9Н, с).
[0905]
(Этап 11)
Соединение А:
N,N-диэтилэтанаминия (5R,7R,8S,12aR,14R,15R,15aR,16R)-7-(6-бензамидо-2-{[2-({[2-(триметилсилил)этокси]карбонил}амино)этил]амино}-9Н-пурин-9-ил)-14-(6-бензоил-6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)-15-{[трет-бутил(диметил)силил]окси}-10-(2-цианоэтокси)-16-фтор-2-оксо-10-сульфанилиденоктагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1]1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2-тиолят
Соединение В:
Бис(N,N-диэтилэтанаминия) (5R,7R,8S,12aR,14R,15R,15aR,16R)-7-(6-бензамидо-2-{[2-({[2-(триметилсилил)этокси]карбонил}амино)этил]амино}-9Н-пурин-9-ил)-14-(6-бензоил-6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)-15-{[трет-бутил(диметил)силил]окси}-16-фтор-2,10-диоксооктагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
Ту же реакцию, что и на этапе 7 примера 1, проводили в следующем масштабе (сырьевой материал: 502 мг). С применением раствора в ацетонитриле полученного соединения и соединения (434 мг), полученного на описанном выше этапе 10, проводили реакцию таким же образом, как на этапе 8 примера 1 и этапе 9 примера 1, с получением диастереомера 1 (43,9 мг) и диастереомера 2 (27,0 мг) указанного в заголовке соединения А, и диастереомера 1 (55,0 мг) и диастереомера 2 (121 мг) указанного в заголовке соединения В (каждый с примесями).
Диастереомер 1 соединения А (менее полярный)
MC(ИЭР) m/z: 1309(М+Н)+.
Диастереомер 2 соединения А (более полярный)
MC(ИЭР) m/z: 1309(М+Н)+.
Диастереомер 1 соединения В (менее полярный)
МС(ИЭР) m/z: 1256(М+Н)+.
Диастереомер 2 соединения В (более полярный)
MC(ИЭР) m/z: 1256(М+Н)+.
[0906]
(Этап 12-1)
Бис(N,N-диэтилэтанаминия) (5R,7R,8S,12aR,14R,15R,15aR,16R)-7-(6-амино-2-{[2-({[2-(триметилсилил)этокси]карбонил}амино)этил]амино}-9Н-пурин-9-ил)-15-{[трет-бутил(диметил)силил]окси}-16-фтор-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
С применением соединения А (диастереомер 1) (43,9 мг), полученного на описанном выше этапе 11, проводили реакцию таким же образом, как на этапе 10 примера 1, с получением указанного в заголовке соединения.
МС (ИЭР) m/z: 1048(М+Н)+.
[0907]
(Этап 124)
Бис(N,N-диэтилэтанаминия) (5R,7R,8S,12aR,14R,15R,15aR,16R)-7-(6-амино-2-{[2-({[2-(триметилсилил)этокси]карбонил}амино)этил]амино}-9Н-пурин-9-ил)-15-{[трет-бутил(диметил)силил]окси}-16-фтор-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
С применением соединения В (диастереомер 1) (55,0 мг), полученного на описанном выше этапе 11, проводили реакцию таким же образом, как на этапе 10 примера 1, с получением указанного в заголовке соединения.
МС (ИЭР) m/z: 1048(М+Н)+.
[0908]
(Этап 12-2)
Бис(N,N-диэтилэтанаминия) (5R,7R,8S,12aR,14R,15R,15aR,16R)-7-(6-амино-2-{[2-({[2-(триметилсилил)этокси]карбонил}амино)этил]амино}-9Н-пурин-9-ил)-15-{[трет-бутил(диметил)силил]окси}-16-фтор-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
С применением соединения А (диастереомер 2) (27,0 мг: с примесями), полученного на описанном выше этапе 11, проводили реакцию таким же образом, как на этапе 10 примера 1, с получением указанного в заголовке соединения.
МС (ИЭР) m/z: 1048(М+Н)+.
[0909]
(Этап 13-1)
Бис(N,N-диэтилэтанаминия) (5R,7R,8S,12aR,14R,15R,15aS,16R)-7-(6-амино-2-{[2-({[2-(триметилсилил)этокси]карбонил}амино)этил]амино}-9Н-пурин-9-ил)-16-фтор-15-гидрокси-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2НД0НД2Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
Полученное на описанном выше этапе 12-1 соединение и полученное на этапе 12'-1 соединение объединяли и проводили реакцию таким же образом, как на этапе 11 примера 1, с получением указанного в заголовке соединения (36,4 мг).
МС (ИЭР) m/z: 934(М+Н)+.
[0910]
(Этап 13-2)
Бис(N,N-диэтилэтанаминия) (5R,7R,8S,12aR,14R,15R,15aS,16R)-7-(6-амино-2-{[2-({[2-(триметилсилил)этокси]карбонил}амино)этил]амино}-9Н-пурин-9-ил)-16-фтор-15-гидрокси-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
С применением соединения, полученного на описанном выше этапе 12-2, проводили реакцию таким же образом, как на этапе 11 примера 1, с получением указанного в заголовке соединения (12,4 мг).
МС (ИЭР) m/z: 934(М+Н)+.
[0911]
(Этап 14-1)
Динатрия (5R,7R,8S,12aR,14R,15R,15aS,16R)-7-{6-амино-2-[(2-аминоэтил)амино]-9Н-пурин-9-ил}-16-фтор-15-гидрокси-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
(Диастереомер 1)
С применением соединения (36,4 мг), полученного на описанном выше этапе 13-1, проводили реакцию таким же образом, как на этапе 5 примера 40, а затем проводили очистку при следующих [Условиях очистки] с получением указанного в заголовке соединения в виде соли триэтиламина.
[Условия очистки] препаративная ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 2% - 30% (0 мин - 40 мин)] и Sep-Pak (R) С18 [вода/ацетонитрил/0,1% триэтиламин].
Полученную соль триэтиламина подвергали солевому обмену таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением указанного в заголовке соединения (21,0 мг).
МС (ИЭР) m/z: 790(М+Н)+.
1H-ЯМР (CD3OD) δ: 8,26 (1H, уш. с), 8,01 (1H, с), 7,04 (1H, с), 6,27 (1Н, д, J=4,8 Гц), 6,13 (1H, д, J=7,9 Гц), 5,69-5,50 (1H, м), 5,57 (1H, дд, J=53,5, 2,7 Гц), 5,14-5,10 (1H, м), 4,73 (1H, т, J=4,8 Гц), 4,62-4,53 (1H, м), 4,50-4,44 (2Н, м), 4,24-4,01 (3H, м), 3,67-3,58 (1H, м), 3,50-3,44 (3H, м), 3,20-3,04 (2Н, м), 2,83-2,81 (2Н, м), 2,04-1,94 (2Н, м).
31Р-ЯМР (CD3OD) δ: 58,4 (с), 56,5 (с).
[0912]
(Этап 14-2)
Бис(N,N,N-трибутилбутан-1-аминия) (5R,7R,8S,12aR,14R,15R,15aS,16R)-7-{6-амино-2-[(2-аминоэтил)амино]-9Н-пурин-9-ил}-16-фтор-15-гидрокси-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
С применением соединения (29,5 мг), полученного на описанном выше этапе 13-2, проводили реакцию таким же образом, как на этапе 5 примера 40, а затем проводили очистку при следующих [Условиях очистки] с получением указанного в заголовке соединения (24,2 мг: с примесями).
[Условия очистки] препаративная ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 2% - 30% (0 мин - 30 мин)], Sep-Pak (R) С18 [вода/ацетонитрил/0,1% триэтиламин] и препаративная ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 10% - 25% (0 мин - 30 мин)].
1Н-ЯМР (CD3OD) δ: 8,27 (1H, с), 8,02 (1H, с), 7,13 (1H, с), 6,32 (1H, д, J=6,0 Гц), 6,15 (1H, д, J=7,9 Гц), 5,63-5,51 (1H, м), 5,39-5,35 (1H, м), 5,30 (1H, дд, J=54,4, 3,6 Гц), 4,77 (1H, т, J=5,l Гц), 4,60-4,34 (4Н, м), 4,19-4,13 (1H, м), 3,92-3,88 (1H, м), 3,70-3,62 (1H, м), 3,51-3,45 (3H, м), 3,26-3,07 (18Н, м), 2,98-2,87 (2Н, м), 2,03-1,99 (2Н, м), 1,70-1,62 (16Н, м), 1,47-1,37 (16Н, м), 1,03 (24Н, т, J=7,3 Гц).
[0913]
(Этап 14-2')
Динатрия (5R,7R,8S,12aR,14R,15R,15aS,16R)-7-{6-амино-2-[(2-аминоэтил)амино]-9Н-пурин-9-ил}-16-фтор-15-гидрокси-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
(Диастереомер 2)
Соединение (19,0 мг: с примесями), полученное на описанном выше этапе 14-2, очищали с помощью препаративной ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 10% - 25% (0 мин - 30 мин)].
Полученное в результате этого соединение подвергали солевому обмену таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением указанного в заголовке соединения (8,4 мг).
МС (ПЭР) m/z: 790(М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,27 (1H, с), 8,01 (1H, с), 7,12 (1H, с), 6,31 (1H, д, J=6,0 Гц), 6,15 (1H, д, J=8,5 Гц), 5,65-5,49 (1H, м), 5,38-5,34 (1H, м), 5,30 (1H, дд, J=55,0, 3,0 Гц), 4,77 (1Н, дд, J=5,7, 4,5 Гц), 4,60-4,34 (4Н, м), 4,19-4,13 (1H, м), 3,92-3,88 (1H, м), 3,68-3,61 (1H, м), 3,51-3,45 (3H, м), 3,22-3,07 (2Н, м), 2,89-2,87 (2Н, м), 2,03-1,98 (2Н, м).
31Р-ЯМР (CD3OD) δ: 62,6 (с), 59,5 (с).
[0914]
Пример 53. Синтез ЦДН43.
(5S,7R,8R,12aR,14R,15R,15aS,16R)-16-амино-7-(2-амино-6-оксо-1,6-дигидро-9Н-пурин-9-ил)-15-гидрокси-2,10-бис(сульфанил)-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-дион
[0915]
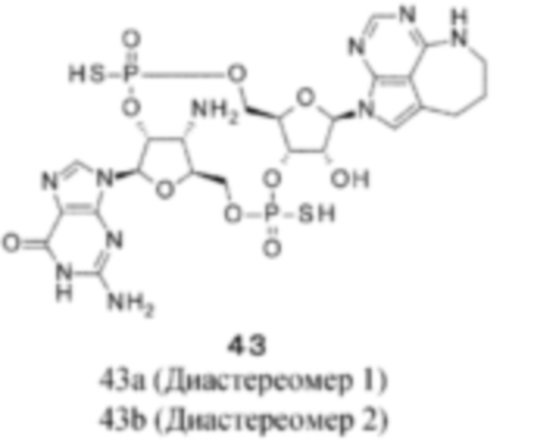
[0916]
[Схема синтеза]
[0917]
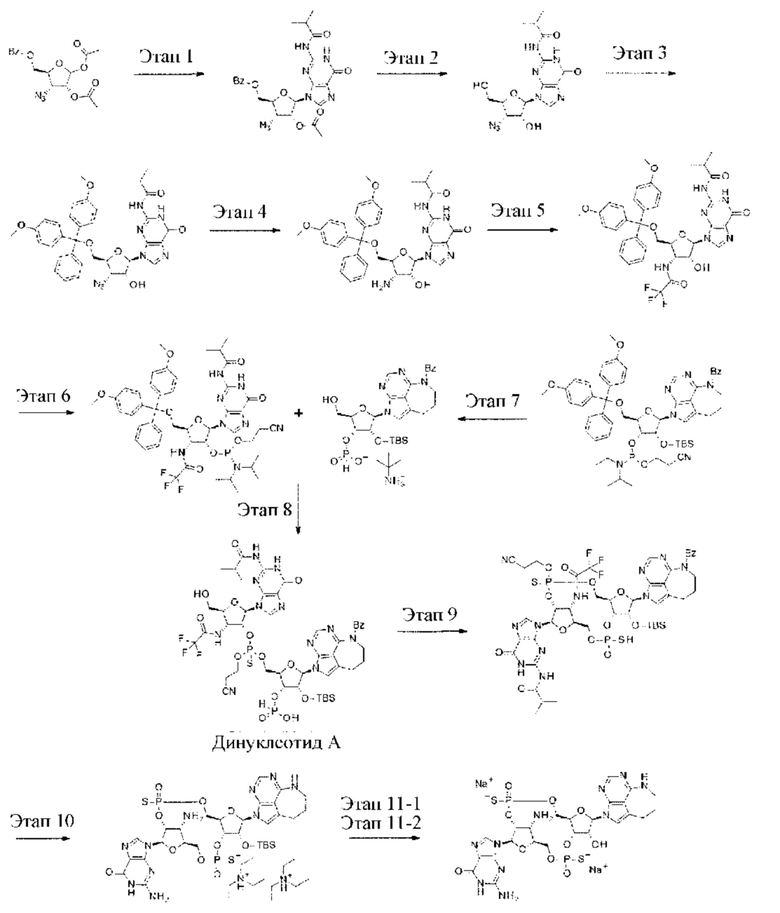
[0918]
(Этап 1)
2'-O-ацетил-3'-азидо-5'-O-бензоил-3'-дезокси-N-(2-метилпропаноил)гуанозин
К раствору 1,2-ди-O-ацетил-3-азидо-5-O-бензоил-3-дезокси-O-рибофуранозы (4,0 г) как соединения, известного в литературе (Reel. Trav. Chim. Pay-Bas 1986, 105, 85-91), в ацетонитриле (60 мл) добавляли N2-изобутирилгуанин (3,65 г) и N,O-бис(триметилсилил)ацетамид (8,08 мл) при комнатной температуре и реакционную смесь перемешивали при 70°С в течение 3 часов. Триметилсилилтрифторметансульфонат (2,98 мл) добавляли туда при 70°С, и реакционную смесь перемешивали при той же температуре в течение 1 дня. После охлаждения реакционной смеси добавляли туда насыщенный водный раствор гидрокарбоната натрия, чтобы погасить реакцию, и осуществляли экстрагирование продукта этилацетатом. Органический слой промывали рассолом и сушили над безводным сульфатом натрия. Осушающий агент удаляли путем фильтрации и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле [гексан/этилацетат] с получением указанного в заголовке соединения (4,82 г).
1Н-ЯМР (CDCl3) δ: 12,2 (1H, с), 9,42 (1H, с), 8,02 (2Н, м), 7,71 (1H, с), 7,63 (1H, м), 7, 48 (2Н, м), 5,97 (1H, д, J=3,9 Гц), 5,87 (1Н, дд, J=5,5, 3,9 Гц), 5,10 (1H, дд, J=11,7, 5,5 Гц), 4,93 (1H, т, J=5,9 Гц), 4,66 (1H, дд, J=11,7, 5,5 Гц), 4,14 (1Н, к, J=7,2 Гц), 2,77 (1H, м), 2,21 (3H, с), 1,31 (6Н, м).
[0919]
(Этап 2)
3'-азидо-3'-дезокси-N-(2-метилпропаноил)гуанозин
В смешанный раствор соединения (5,48 г), полученного на описанном выше этапе 1, в тетрагидрофуране (64 мл)/метаноле (32 мл) добавляли 5 М гидроксид натрия (17 мл) при 0°С и реакционную смесь перемешивали в течение 15 минут. Уксусную кислоту (5,08 мл) добавляли в реакционную смесь, чтобы погасить реакцию, и реакционную смесь концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле [дихлорметан/метанол] с получением указанного в заголовке соединения (3,8 г).
1Н-ЯМР (CD3OD) δ: 8,29 (1H, с), 5,97 (1H, д, J=5,7 Гц), 4,86 (1H, т, J=5,3 Гц), 4,29 (1H, т, J=5,3 Гц), 4,10 (1Н, дт, J=5,9, 2,4 Гц), 3,87 (1H, дд, J=12,l, 3,1 Гц), 3,76 (1Н, дд, J=12,3, 3,3 Гц), 2,74 (1H, м), 1,24 (6Н, д, J=6,7 Гц).
[0920]
(Этап 3)
3'-азидо-5'-O-[бис(4-метоксифенил)(фенил)метил]-3'-дезокси-N-(2-метилпропаноил)гуанозин
С применением соединения (3,8 г), полученного на описанном выше этапе 2, проводили реакцию таким же образом, как на этапе 1 примера 11, с получением указанного в заголовке соединения (5,78 г).
1H-ЯМР (CDCl3) δ: 11,9 (1H, с), 7,68 (2Н, д, J=7,4 Гц), 7,60 (1Н, с), 7,57 (2Н, д, J-8,6 Гц), 7,51 (2Н, д, J=8,6 Гц), 7,39-7,18 (3H, м), 7,02 (1H, д, J=4,7 Гц), 6,95 (2Н, д, J=9,0 Гц), 6,89 (2Н, д, J=9,0 Гц), 5,84 (1H, м), 5,63 (1H, д, J=7,8 Гц), 4,58 (1H, дд, J=6,7, 2,3 Гц), 3,99 (1H, м), 3,83 (3H, с), 3,81 (3H, с), 3,63 (1H, дд, J=11,0, 1,6 Гц), 2,92 (1H, дд, J=10,8, 2,5 Гц), 0,91 (1H, м), 69 (3H, д, J=6,7 Гц), 0,21 (3H, д, J=7,0 Гц).
[0921]
(Этап 4)
3'-амино-5'-O-[бис(4-метоксифенил)(фенил)метил]-3'-дезокси-N-(2-метилпропаноил)гуанозин
К раствору соединения (5,17 г), полученного на описанном выше этапе 3, в метаноле (60 мл) добавляли трифенилфосфин (3,98 г) и реакционную смесь перемешивали при комнатной температуре в течение 4 часов. Реакционную смесь концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле [гексан/этилацетат] с получением указанного в заголовке соединения (2,22 г).
1Н-ЯМР (CDCl3) δ: 7,77 (2Н, д, J=6,7 Гц), 7,49-7,17 (8Н, м), 6,86-6,77 (4Н, м), 5,93 (1H, д, J=2,7 Гц), 5,85 (1H, д, J=3,5 Гц), 5,02 (1Н, дд, J=6,7, 2,7 Гц), 4,74 (1Н, м), 4,32 (1H, м), 4,06 (1H, м), 3,79 (3H, с), 3,78 (3H, с), 3,72 (1H, т, J=5,9 Гц), 3,49 (1H, дд, J=10,4, 3,3 Гц), 3,33 (1Н, дд, J=10,4, 3,7 Гц), 2,26 (1H, м), 1,21-0,95 (6Н, м).
[0922]
(Этап 5)
5'-O-[бис(4-метоксифенил)(фенил)метил]-3'-дезокси-N-(2-метилпропаноил)-3'-(2,2,2-трифторацетамид)гуанозин
К раствору соединения (2,22 г), полученного на описанном выше этапе 4, в N,N-диметилформамиде (20 мл) добавляли этилтрифторацетат (4,0 мл) и реакционную смесь перемешивали при 50°С в течение 2 суток. После охлаждения реакционной смеси добавляли туда насыщенный водный раствор гидрокарбоната натрия, чтобы погасить реакцию, и осуществляли экстрагирование продукта этилацетатом. Органический слой промывали рассолом, а затем сушили над безводным сульфатом натрия. Осушающий агент удаляли путем фильтрации и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле [дихлорметан/метанол] с получением указанного в заголовке соединения (2,06 г).
МС (ИЭР) m/z: 751(М+Н)+.
1Н-ЯМР (CDCl3) δ: 12,0 (1H, уш. с), 7,81 (1H, уш. с), 7,65 (1H, с), 7,59 (2Н, д, J=7,4 Гц), 7,47 (2Н, д, J=9,0 Гц), 7,43 (2Н, д, J=8,6 Гц), 7,27-7,18 (3H, м), 6,87 (2Н, д, J=9,0 Гц), 6,82 (2Н, д, J=9,0 Гц), 5,72 (1H, д, J=6,3 Гц), 5,67 (1Н, м), 5,02 (1Н, м), 4,33 (1H, м), 3,80 (3H, с), 3,78 (3H, с), 3,60 (1H, дд, J=10,6, 1,6 Гц), 3,29 (1Н, дд, J=10,6, 2,7 Гц), 1,36 (1H, м), 0,82 (3H, д, J=7,0 Гц), 0,44 (3H, д, J=6,7 Гц).
[0923]
(Этап 6)
5'-O-[бис(4-метоксифенил)(фенил)метил]-2'-O-{(2-цианоэтокси)[ди(пропан-2-ил)амино]фосфанил}-3'-дезокси-N-(2-метилпропаноил)-3'-(2,2,2-трифторацетамид)гуанозин
С применением соединения (1,0 г), полученного на описанном выше этапе 5, проводили реакцию таким же образом, как на этапе 4 примера 5, с получением указанного в заголовке соединения (780 мг) в виде смеси диастереомеров у атома фосфора.
[0924]
(Этап 7)
2-метилпропан-2-аминия6-бензоил-2-{2-O-[трет-бутил(диметил)силил]-3-O-[оксид(оксо)-λ5-фосфанил]-β-D-рибофуранозил}-6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен
К раствору соединения (5,0 г), полученного на описанном выше этапе 6 примера 1, в ацетонитриле (30 мл) добавляли воду (0,18 мл) и пиридиновую соль трифторуксусной кислоты (1,2 г) и реакционную смесь перемешивали при комнатной температуре в течение 30 минут. В реакционную смесь добавляли трет-бутиламин (30 мл) и реакционную смесь перемешивали при комнатной температуре в течение 30 минут. После концентрирования реакционной смеси при пониженном давлении остаток дважды подвергали азеотропной перегонке с ацетонитрилом. К раствору остатка в дихлорметане (50 мл) добавляли воду (0,88 мл) и раствор дихлоруксусной кислоты (3,2 мл) в дихлорметане (50 мл) в данном порядке и реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Метанол (5 мл) и пиридин (6,3 мл) добавляли туда, чтобы погасить реакцию, и реакционную смесь концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле DIOL [гексан/этил ацетат → дихлорметан/метанол] с получением указанного в заголовке соединения (3,0 г).
МС (ИЭР)m/z: 589(М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,01 (1H, с), 7,65 (1H, с), 7,44 (1H, м), 7,34-7,26 (4Н, м), 7,02 (1H, д, J=639,3 Гц), 6,24 (1H, с), 4,89 (1H, м), 4,81 (1Н, ддд, J=10,6, 5,1, 1,6 Гц), 4,43-4,27 (3H, м), 3,90 (2Н, м), 3,14 (2Н, м), 2,30 (2Н, м), 1,42 (9Н, с), 0,80 (9Н, с), -0,27 (6Н, с).
[0925]
(Этап 8)
Динуклеотид А
В смешанный раствор соединения (543 мг), полученного на описанном выше этапе 7, в дихлорметане (6,0 мл)/ацетонитриле (6,0 мл) добавляли молекулярные сита 3А, 1/16 (500 мг), и 4,5-дицианоимидазол (126 мг), и реакционную смесь перемешивали при комнатной температуре в течение 15 минут. Соединение (780 мг), полученное на описанном выше этапе 6, добавляли в реакционную смесь, которую перемешивали в течение 5 часов, и добавляли туда N,N-диметил-N'-(3-сульфанилиден-3H-1,2,4-дитиазол-5-ил)метанимидамид (219 мг) и реакционную смесь дополнительно перемешивали в течение 2 часов. Молекулярные сита 3А удаляли из реакционной смеси путем фильтрации и насыщенный водный раствор гидрокарбоната натрия добавляли в фильтрат, который осуществляли экстрагирование этилацетатом. После высушивания органического слоя над безводным сульфатом натрия осушающий агент удаляли путем фильтрации и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле [гексан/этилацетат]. К раствору полученного в результате этого соединения (740 мг) в дихлорметане (6,0 мл) добавляли воду (0,086 мл) и раствор дихлоруксусной кислоты (0,158 мл) в дихлорметане (6,0 мл) в данном порядке и реакционную смесь перемешивали при комнатной температуре в течение 1,5 часов. Добавляли туда пиридин (0,31 мл), чтобы погасить реакцию, и реакционную смесь затем концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле [дихлорметан/метанол] с получением указанного в заголовке соединения (520 мг) в виде смеси диастереомеров у атома фосфора.
МС (ИЭР) m/z: 1168(М+Н)+.
[0926]
(Этап 9)
N-{9-[(5S,7R,8R,12aR,14R,15R,15aR,16R)-14-(6-бензоил-6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)-15-{[трет-бутил(диметил)силил]окси}-10-(2-цианоэтокси)-2-оксо-2-сульфанил-10-сульфанилиден-16-(2,2,2-трифторацетамид)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-7-ил]-6-оксо-6,9-дигидро-1Н-пурин-2-ил}-2-метилпропанамид
С применением соединения (270 мг), полученного на описанном выше этапе 8, проводили реакцию таким же образом, как на этапе 9 примера 1, с получением указанного в заголовке соединения (110 мг) в виде смеси диастереомеров у атома фосфора.
МС (ИЭР) m/z: 1182(М+Н)+.
[0927]
(Этап 10)
Бис(N,N-диэтилэтанаминий) (5S,7R,8R,12aR,14R,15R,15aR,16R)-16-амино-7-(2-амино-6-оксо-1,6-дигидро-9Н-пурин-9-ил)-15-{[трет-бутил(диметил)силил]окси}-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
С применением соединения (110 мг), полученного на описанном выше этапе 9, проводили реакцию таким же образом, как на этапе 10 примера 1, с получением диастереомера 1 (34,2 мг) и диастереомера 2 (19,3 мг) указанного в заголовке соединения.
Диастереомер 1 (менее полярный)
MC (ИЭР) m/z: 859(М+Н)+.
Диастереомер 2 (более полярный)
MC (ИЭР) m/z: 859(М+Н)+.
[0928]
(Этап 11-1)
Динатрия (5S,7R,8R,12aR,14R,15R,15aS,16R)-16-амино-7-(2-амино-6-оксо-1,6-дигидро-9Н-пурин-9-ил)-15-гидрокси-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
(Диастереомер 1)
С применением соединения (34,2 мг), полученного на описанном выше этапе 10 (диастереомер 1), проводили реакцию таким же образом, как на этапе 11 примера 1, и продукт затем очищали с помощью колоночной хроматографии на силикагеле С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил] с получением указанного в заголовке соединения в виде соли триэтиламина.
Полученную соль триэтиламина подвергали солевому обмену таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением указанного в заголовке соединения (8,9 мг).
МС (ИЭР) m/z: 745(М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,01 (1H, с), 7,93 (1H, с), 7,01 (1H, с), 6,20 (1H, д, J=3,9 Гц), 6,01 (1H, д, J=8,6 Гц), 5,65 (1Н, м), 5,19 (1Н, дт, J=9,5, 4,0 Гц), 4,68 (1H, т, J=4,3 Гц), 4,40-4,28 (2Н, м), 4,23 (1H, м), 4,17 (1Н, м), 4,11-4,01 (3H, м), 3,41 (2Н, м), 2,75-2,53 (2Н, м), 1,98-1,78 (2Н, м).
31Р-ЯМР (CD3OD) δ: 58,0 (с), 54,3 (с).
[0929]
(Этап 11-2)
Динатрия (5S,7R,8R,12aR,14R,15R,15aS,16R)-16-амино-7-(2-амино-6-оксо-l,6-дигидро-9H-пурин-9-ил)-15-гидрокси-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
(Диастереомер 2)
С применением соединения (диастереомера 2) (19,3 мг), полученного на описанном выше этапе 10, проводили реакцию таким же образом, как на этапе 11 примера 1, и продукт затем очищали с помощью колоночной хроматографии на силикагеле С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил] с получением указанного в заголовке соединения в виде соли триэтиламина.
Полученную соль триэтиламина подвергали солевому обмену таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением указанного в заголовке соединения (5,6 мг).
МС (ИЭР) m/z: 745(М+Н)+.
1H-ЯМР (ДМСО-d6) δ: 10,7 (1H, с), 8,40 (2Н, уш. с), 8,09 (1Н, с), 8,03 (1H, с), 7,74 (1H, уш. с), 7,22 (1H, с), 6,95 (1H, уш. с), 6,53 (2Н, уш. с), 61,6 (2Н, т, J=8,2 Гц), 5,57 (1H, к, J=8,0 Гц), 5,25 (1H, дд, J=7,8, 4,3 Гц), 4,61 (1H, дд, J=7,6, 4,5 Гц), 4,38 (1H, с), 4,27-4,11 (3H, м), 3,85-3,71 (3H, м), 3,35 (2Н, м), 2,80 (2Н, м), 1,93 (2Н, м).
31Р-ЯМР (ДМСО-d6) δ: 60,3 (с), 58,3 (с).
[0930]
Пример 54. Синтез ЦДН44.
(5R,7R,8S,12aR,14R,15R,15aR,16R)-15,16-дифтор-7-[1-(2-гидроксиэтил)-6-оксо-1,6-дигидро-9Н-пурин-9-ил]-2,10-бис(сульфанил)-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-дион
[0931]
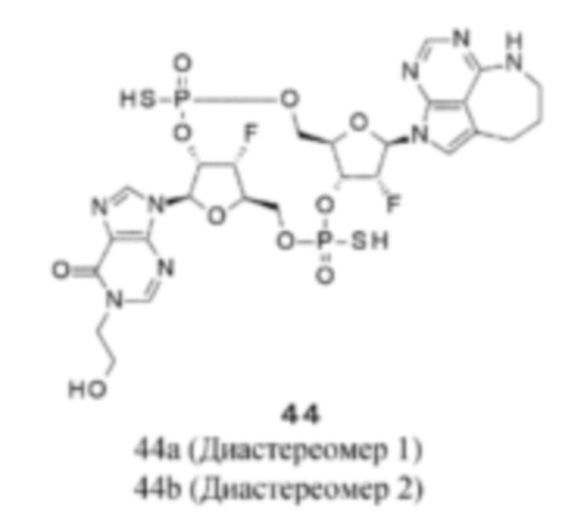
[0932]
[Схема синтеза]
[0933]
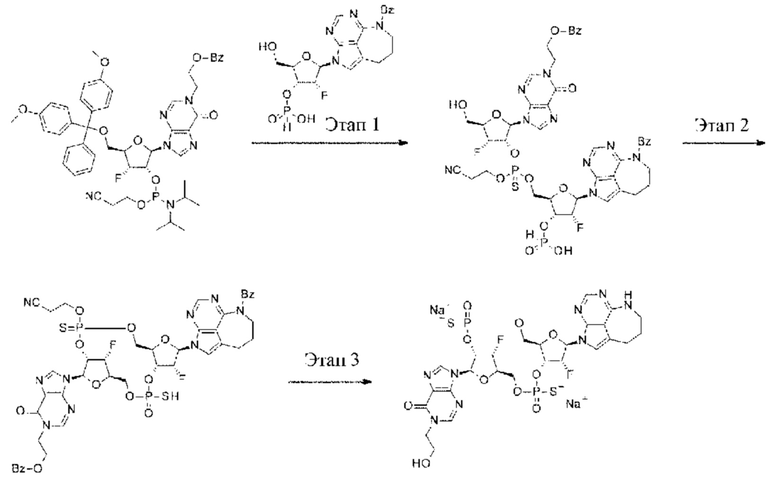
[0934]
(Этап 1)
С применением соединения (636 мг), полученного на этапе 8 примера 44, проводили реакцию таким же образом, как на этапе 7 примера 1, с получением раствора в ацетонитриле 6-бензоил-2-{2-дезокси-2-фтор-3-O-[гидрокси(оксо)-λ5-фосфанил]-β-D-рибофуранозил}-6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулена. С применением полученного раствора в ацетонитриле и полученного на этапе 7 соединения примера 49 (640 мг) проводили реакцию таким же образом, как на этапе 8 примера 1, и полученный в результате этого неочищенный продукт непосредственно использовали для следующей реакции.
[0935]
(Этап 2)
2-{9-[(5R,7R,8S,12aR,14R,15R,15aR,16R)-14-(6-бензоил-6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)-10-(2-цианоэтокси)-15,16-дифтор-2-оксо-2-сульфанил-10-сульфанилиденоктагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-7-ил]-6-оксо-6,9-дигидро-1Н-пурин-1-ил}этилбензоат
С применением неочищенного продукта, полученного на описанном выше этапе 1, проводили реакцию таким же образом, как на этапе 9 примера 1, с получением указанного в заголовке соединения (228 мг: с примесями) в виде смеси диастереомеров у атома фосфора.
МС (ИЭР) m/z: 1040(М+Н)+.
[0936]
(Этап 3)
Динатрия (5R,7R,8S,12aR,14R,15R,15aR,16R)-15,16-дифтор-7-[1-(2-гидроксиэтил)-6-оксо-1,6-дигидро-9Н-пурин-9-ил]-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
Соединение (228 мг), полученное на описанном выше этапе 2, растворяли в метаноле (5 мл) и 28% водном растворе аммиака (5 мл), и реакционную смесь перемешивали при 60°С в течение 12 часов. Реакционную смесь концентрировали при пониженном давлении, а затем очищали с помощью колоночной хроматографии на силикагеле С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил], препаративной ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 5% - 30% (0 мин - 40 мин)] и препаративной ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/раствор ацетонитрила-метанола (1:1), раствор ацетонитрила-метанола (1:1): 7% - 50% (0 мин - 40 мин)] в данном порядке с получением диастереомера 1 и диастереомера 2 указанного в заголовке соединения в виде солей триэтиламина (время удерживания при ВЭЖХ: диастереомер 1>2).
Каждую из полученных солей триэтиламина подвергали солевому обмену таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением диастереомера 1 (12,5 мг) и диастереомера 2 (15,8 мг) указанного в заголовке соединения.
Диастереомер 1 (менее полярный)
МС (ПЭР) m/z: 779(М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,63 (1H, с), 8,16 (1H, с), 8,03 (1H, с), 7,08 (1H, с), 6,47 (1H, дд, J=17,5, 1,8 Гц), 6,27 (1H, д, J=7,9 Гц), 5,64-5,36 (3H, м), 5,31-5,20 (1Н, м), 4,62-4,50 (2Н, м), 4,44-4,39 (1H, м), 4,32-4,12 (3H, м), 4,10-4,03 (1H, м), 3,99-3,91 (1H, м), 3,83-3,72 (2Н, м), 3,52-3,46 (2Н, м), 2,78-2,72 (2Н, м), 2,04-1,85 (2Н, м).
31Р-ЯМР (CD3OD) δ: 57,5 (с), 55,0 (с).
Диастереомер 2 (более полярный)
МС (ПЭР) m/z: 779(М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,69 (1H, с), 8,21 (1Н, с), 8,03 (1Н, с), 7,25 (1H, с), 6,49 (1Н, дд, Г=15,1, 3,0 Гц), 6,28 (1H, д, J=9,l Гц), 5,64-5,28 (4Н, м), 4,61-4,39 (4Н, м), 4,26-4,17 (1H, м), 4,13-3,95 (3H, м), 3,84-3,77 (2Н, м), 3,53-3,45 (2Н, м), 2,92-2,77 (2Н, м), 2,02-1,92 (2Н, м).
31Р-ЯМР (CD3OD) δ: 60,7 (с), 57,4 (с).
[0937]
Пример 55. Синтез ЦДН45.
(5R,7R,8S,12aR,14R,15R,15aR,16R)-7-{6-амино-2-[(2-аминоэтил)амино]-9Н-пурин-9-ил}-15,16-дифтор-2,10-бис(сульфанил)-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-дион
[0938]
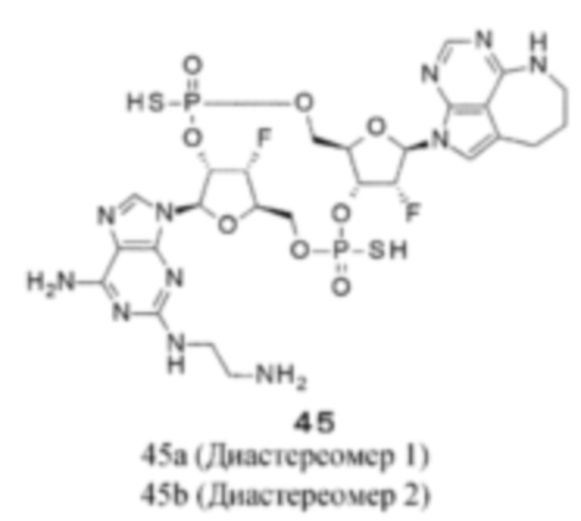
[0939]
[Схема синтеза]
[0940]
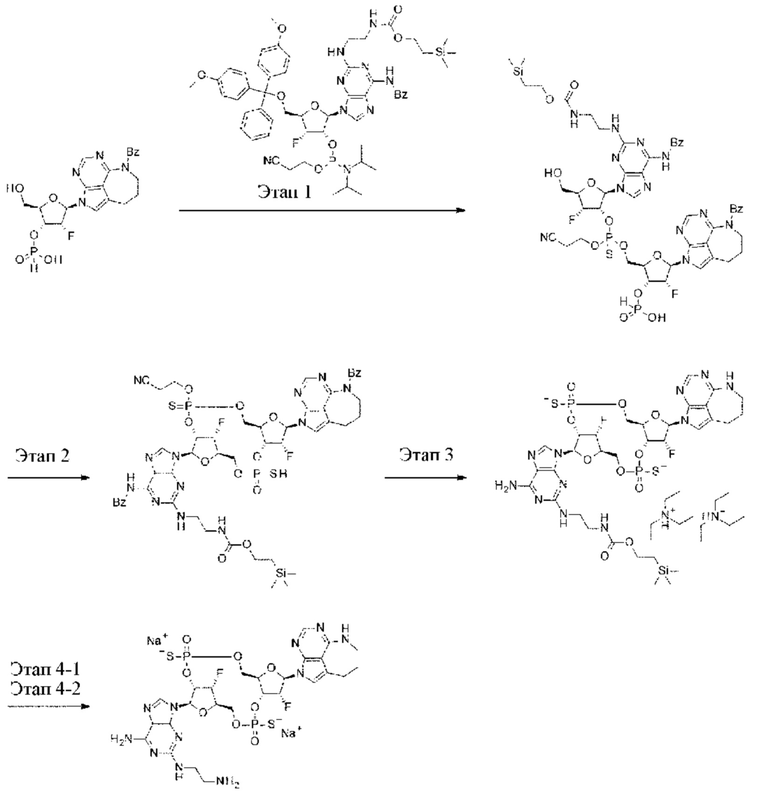
[0941]
(Этап 1)
С применением соединения (696 мг), полученного на этапе 8 примера 44, проводили реакцию таким же образом, как на этапе 7 примера 1, с получением раствора в ацетонитриле 6-бензоил-2-{2-дезокси-2-фтор-3-O-[гидрокси(оксо)-λ5-фосфанил]-β-D-рибофуранозил}-6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулена. С применением полученного раствора в ацетонитриле и соединения (738 мг), полученного на этапе 10 примера 52, проводили реакцию таким же образом, как на этапе 8 примера 1, и полученный в результате этого неочищенный продукт непосредственно использовали для следующей реакции.
[0942]
(Этап 2)
2-(триметилсилил)этил[2-({6-бензамидо-9-[(5R,7R,8S,12aR,14R,15R,15aR,16R)-14-(6-бензоил-6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)-10-(2-цианоэтокси)-15,16-дифтор-2-оксо-2-сульфанил-10-сульфанилиденоктагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-7-ил]-9Н-пурин-2-ил}амино)этил]карбамат
С применением смеси, полученной на описанном выше этапе 1, проводили реакцию таким же образом, как на этапе 9 примера 1, с получением смеси, содержащей указанное в заголовке соединение (1,31 г). Полученную смесь непосредственно использовали для следующей реакции.
МС (ИЭР) m/z: 1195(М-Н)-.
[0943]
(Этап 3)
Бис(N,N-диэтилэтанаминия) (5R,7R,8S,12aR,14R,15R,15aR,l6R)-7-(6-амино-2-{[2-({[2-(триметилсилил)этокси]карбонил}амино)этил]амино}-9Н-пурин-9-ил)-15,16-дифтор-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
С применением смеси (1,31 г), полученной на описанном выше этапе 2, проводили реакцию таким же образом, как на этапе 10 примера 1, с получением диастереомера 1 (249 мг: с примесями) и диастереомера 2 (344 мг: с примесями) указанного в заголовке соединения.
Диастереомер 1 (менее полярный)
МС (ИЭР) m/z: 936(М+Н)+.
Диастереомер 2 (более полярный)
МС (ИЭР) m/z: 936(М+Н)+.
[0944]
(Этап 4-1)
Динатрия (5R,7R,8S,12aR,14R,15R,15а,16R)-7-{6-амино-2-[(2-аминоэтил)амино]-9Н-пурин-9-ил}-15,16-дифтор-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
(Диастереомер 1)
С применением соединения (диастереомера 1) (249 мг: с примесями), полученного на описанном выше этапе 3, проводили реакцию таким же образом, как на этапе 5 примера 40, а затем проводили очистку при следующих [Условиях очистки] с получением указанного в заголовке соединения в виде соли триэтиламина.
[Условия очистки] препаративная ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 10% - 40% (0 мин - 30 мин)], и препаративная ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 0% -30% (0 мин - 30 мин)].
Полученную соль триэтиламина подвергали солевому обмену таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением указанного в заголовке соединения (9,2 мг).
МС (ИЭР) m/z: 792(М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,17 (1H, уш. с), 8,01 (1Н, с), 7,01 (1H, с), 6,45 (1H, д, J=17,5 Гц), 6,07 (1H, д, J=8,5 Гц), 5,84-5,64 (1H, м), 5,61 (1H, дд, J=53,5, 3,3 Гц), 5,40 (1H, дд, J=52,0, 4,2 Гц), 5,30-5,18 (1H, м), 4,59-4,17 (6Н, м), 3,54-3,42 (2Н, м), 3,39-3,31 (2Н, м), 3,05-2,98 (2Н, м), 2,69-2,51 (2Н, м), 2,02-1,83 (2Н, м).
31Р-ЯМР (CD3OD) δ: 57,3 (с), 54,8 (с).
[0945]
(Этап 4-2)
Динатрия (5R,7R,8S,12aR,14R,15R,15aR,16R)-7-{6-амино-2-[(2-аминоэтил)амино]-9Н-пурин-9-ил}-15,16-дифтор-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
(Диастереомер 2)
С применением соединения (диастереомера 2) (344 мг: с примесями), полученного на описанном выше этапе 3, проводили реакцию таким же образом, как на этапе 5 примера 40, а затем проводили очистку при следующих [Условиях очистки] с получением указанного в заголовке соединения в виде соли триэтиламина.
[Условия очистки] препаративная ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 10% - 40% (0 мин - 30 мин)] и препаративная ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 10% -30% (0 мин-30 мин)].
Полученную соль триэтиламина подвергали солевому обмену таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением указанного в заголовке соединения (4,7 мг).
МС (ИЭР) m/z: 792(М+Н)+.
1H-ЯМР (CD3OD) δ: 8,05 (1H, уш. с), 8,01 (1H, с), 7,35 (1H, с), 6,50 (1H, д, J=16,3 Гц), 6,06 (1H, д, J=8,5 Гц), 5,98-5,75 (1H, м), 5,48-5,28 (3H, м), 4,59-4,28 (3H, м), 4,29-4,22 (1H, м), 4,04-3,98 (1H, м), 3,56-3,42 (2Н, м), 3,36-2,61 (6Н, м), 2,05-1,86 (2Н, м).
31Р-ЯМР (CD3OD) δ: 58,4 (уш. с), 57,6 (с).
[0946]
Пример 56. Синтез ЦДН46.
(5R,7R,8R,12aR,14R,15S,15aR,16R)-7-(6-амино-9H-пурин-9-ил)-l5,16-дигидрокси-2,10-бис(сульфанил)-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)декагидро-2Н,10Н-5,8-метано-2λ5,10λ5-циклопента[1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-дион
[0947]
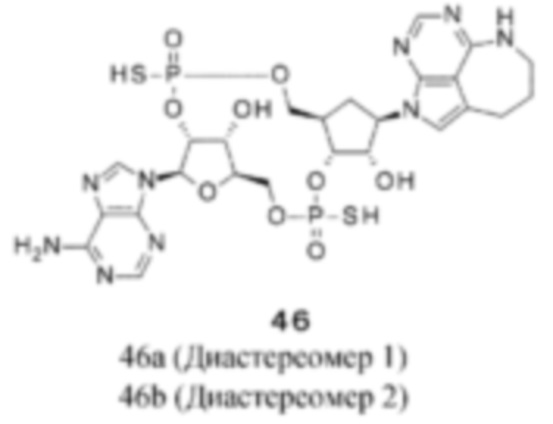
[0948]
[Схема синтеза]
[0949]
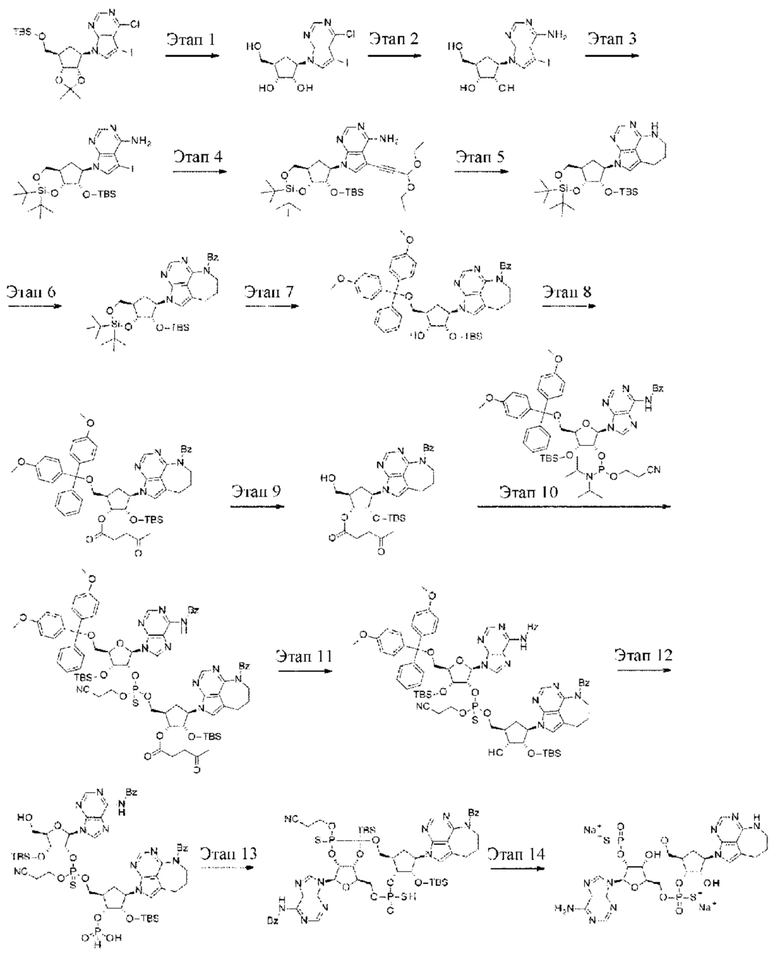
[0950]
(Этап 1)
(1R,2S,3R,5R)-3-(4-хлор-5-йод-7Н-пирроло[2,3-d]пиримидин-7-ил)-5-(гидроксиметил)циклопентан-1,2-диол
В 7-[(3aS,4R,6R,6aR)-6-({[трет-бутил(диметил)силил]окси}метил)-2,2-диметилтетрагидро-2Н,3аН-циклопента[d][1,3]диоксол-4-ил]-4-хлор-5-йод-7Н-пирроло[2,3-d]пиримидин (6,0 г) как соединение, известное в литературе (WO 2015/199136), добавляли трифторуксусную кислоту (36 мл) и воду (12 мл) в данном порядке и реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Реакционную смесь концентрировали при пониженном давлении с получением неочищенной формы указанного в заголовке соединения. Полученный неочищенный продукт непосредственно использовали для следующей реакции.
[0951]
(Этап 2)
(1R,2S,3R,5R)-3-(4-амино-5-йод-7Н-пирроло[2,3-d]пиримидин-7-ил)-5-(гидроксиметил)циклопентан-1,2-диол
К раствору соединения (4,4 г), полученного на описанном выше этапе 1, в 1,4-диоксане (40 мл) добавляли 28% водный аммиак (40 мл) и реакционную смесь перемешивали при 90°С в течение 72 часов. После концентрирования реакционной смеси при пониженном давлении остаток очищали с помощью колоночной хроматографии на силикагеле С18 [вода/метанол] с получением указанного в заголовке соединения (3,4 г).
1Н-ЯМР (CD3OD) δ: 8,07 (1H, с), 7,56 (1H, с), 6,57 (1Н, уш. с), 4,89 (1Н, дд, J=19,2, 8,6 Гц), 4,79 (1H, д, 6,7 Гц), 4,70 (1H, т, J=5,3 Гц), 4,59 (1H, д, J=4,3 Гц), 4,15 (1H, м), 3,78 (1H, м), 3,45 (2Н, м), 2,14 (1H, м), 2,03 (1Н, м), 1,48 (1H, м).
[0952]
(Этап 3)
7-[(4aR,6R,7S,7aR)-2,2-ди-трет-бутил-7-{[трет-бутил(диметил)силил]окси}гексагидро-2H-циклопента[d][1,3,2]диоксасилин-6-ил]-5-йод-7Н-пирроло[2,3-d]пиримидин-4-амин
С применением соединения (3,3 г), полученного на описанном выше этапе 2, проводили реакцию таким же образом, как на этапе 1 примера 1, с получением указанного в заголовке соединения (4,6 г).
1Н-ЯМР (CDCl3) δ: 8,27 (1Н, с), 7,00 (1H, с), 5,69 (1H, уш. с), 4,92 (1H, дт, J=9,0, 1,2 Гц), 4,32 (3H, м), 3,98 (1H, т, J=10,8 Гц), 2,58 (1H, м), 2,26 (1H, м), 1,74 (1H, уш. с), 1,49 (1Н, дт, J=12,5, 8,6 Гц), 1,13 (9Н, с), 1,09 (9Н, с), 0,86 (9Н, с), 0,06 (3H, с), 0,00 (3H, с).
[0953]
(Этап 4)
7-[(4aR,6R,7S,7aR)-2,2-ди-трет-бутил-7-{[трет-бутил(диметил)силил]окси}гексагидро-2Н-циклопента[d][1,3,2]диоксасилин-6-ил]-5-(3,3-диэтоксипроп-1-ин-1-ил)-7Н-пирроло[2,3-d]пиримидин-4-амин
С применением соединения (4,6 г), полученного на описанном выше этапе 3, проводили реакцию таким же образом, как на этапе 2 примера 1, с получением указанного в заголовке соединения (3,6 г).
1Н-ЯМР (CDCl3) δ: 8,30 (1H, с), 7,15 (1H, с), 5,61 (1H, уш. с), 5,55 (1H, с), 4,90 (1H, дт, J=9,0, 1,2 Гц), 4,32 (3H, м), 3,97 (1H, т, J=10,8 Гц), 3,86 (2Н, м), 3,71 (2Н, м), 2,58 (1H, м), 2,28 (1H, м), 1,68 (1H, с), 1,48 (1Н, м), 1,32 (6Н, т, J=7,0 Гц), 1,13 (9Н, с), 1,09 (9Н, с), 0,86 (9Н, с), 0,05 (3H, с), 0,04 (3H, с).
[0954]
(Этап 5)
2-[(4aR,6R,7S,7aR)-2,2-ди-трет-бутил-7-{[трет-бутил(диметил)силил]окси}гексагидро-2Н-циклопента[d][1,3,2]диоксасилин-6-ил]-6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен
С применением соединения (3,60 г), полученного на описанном выше этапе 4, проводили реакцию таким же образом, как на этапе 3 примера 1, с получением указанного в заголовке соединения (2,44 г).
МС (ИЭР) m/z: 589(М+Н)+.
1Н-ЯМР (CDCl3) δ: 8,18 (1H, с), 6,66 (1H, с), 6,55 (1Н, уш. с), 4,94 (1H, м), 4,36-4,25 (3H, м), 3,97 (1H, т, J=10,8 Гц), 3,57 (2Н, м), 2,93 (2Н, т, J=5,5 Гц), 2,58 (1Н, м), 2,27 (1Н, м), 2,09 (1H, м), 1,46 (1Н, дт, J=12,5, 8,6 Гц), 1,29 (1Н, м), 1,12 (9Н, с), 1,08 (9Н, с), 0,86 (9Н, с), 0,04 (3H, с), 0,03 (3H, с).
[0955]
(Этап 6)
{2-[(4aR,6R,7S,7aR)-2,2-ди-трет-бутил-7-{[трет-бутил(диметил)силил]окси}гексагидро-2Н-циклопента[d][1,3,2]диоксасилин-6-ил]-2,7,8,9-тетрагидро-6Н-2,3,5,6-тетраазабензо[cd]азулен-6-ил}(фенил)метанон
С применением соединения (2,44 г), полученного на описанном выше этапе 5, проводили реакцию таким же образом, как на этапе 4 примера 1, с получением указанного в заголовке соединения (2,01 г).
МС (ИЭР) m/z: 663(М+Н)+.
1Н-ЯМР (CDCl3) δ: 8,12 (1H, с), 7,42-7,25 (5Н, м), 6,97 (1H, с), 5,04 (1H, т, J=9,0 Гц), 4,44 (1H, м), 4,38 (1H, дд, J=10,0, 4,9 Гц), 4,33-4,23 (3H, м), 4,00 (1H, т, J=10,8 Гц), 3,06 (2Н, м), 2,60 (1H, м), 2,29 (3H, м), 1,57 (1H, м), 1,14 (9Н, с), 1,10 (9Н, с), 0,84 (9Н, с), 0,06 (3H, с), 0,05 (3H, с).
[0956]
(Этап 7)
{2-[(1R,2S,3R,4R)-4-{[бис(4-метоксифенил)(фенил)метокси]метил}-2-{[трет-бутил(диметил)силил]окси}-3-гидроксициклопентил]-2,7,8,9-тетрагидро-6Н-2,3,5,6-тетраазабензо[cd]азулен-6-ил}(фенил)метанон
С применением соединения (2,01 г), полученного на описанном выше этапе 6, проводили реакцию таким же образом, как на этапе 5 примера 1, с получением указанного в заголовке соединения (2,13 г).
1Н-ЯМР (CDCl3) δ: 8,03 (1H, с), 7,49 (2Н, м), 7,38-7,19 (13Н, м), 7,02 (1H, с), 6,83 (4Н, м), 5,07 (1H, м), 4,60 (1H, дд, J=8,6, 5,1 Гц), 4,29 (2Н, м), 3,99 (1Н, м), 3,79 (6Н, с), 3,33 (1H, дд, J=9,4, 3,9 Гц), 3,22 (1H, дд, J=9,2, 4,1 Гц), 2,97 (2Н, т, J=6,5 Гц), 2,68 (1Н, д, J=1,6 Гц), 2,37-2,18 (5Н, м), 0,73 (9Н, с), -0,18 (3H, с), -0,47 (3H, с).
[0957]
(Этап 8)
(1R,2S,3R,5R)-3-(6-бензоил-6,7,8,9-тетрагидро-2H-2,3,5,6-тетраазабензо[cd]азулен-2-ил)-5-{[бис(4-метоксифенил)(фенил)метокси]метил}-2-{[трет-бутил(диметил)силил]окси}циклопентил-4-оксопентаноат
К раствору левулиновой кислоты (2,96 г) в тетрогидрофуране (20 мл) добавляли N,N-дициклогексилкарбодиимид (2,63 г) и реакционную смесь перемешивали при комнатной температуре в течение 12 часов. После удаления преципитата путем фильтрации, фильтрат концентрировали при пониженном давлении. К раствору остатка в дихлорметане (20 мл) добавляли соединение (2,10 г), полученное на описанном выше этапе 7, и 4-диметиламинопиридин (155 мг) и реакционную смесь перемешивали при комнатной температуре в течение 1 часа. После добавления насыщенного водного раствора гидрокарбоната натрия в реакционную смесь, чтобы погасить реакцию, осуществляли экстрагирование продукта этилацетатом, и органический слой сушили над безводным сульфатом натрия. Осушающий агент удаляли путем фильтрации и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле [гексан/этилацетат] с получением указанного в заголовке соединения (2,35 г).
1H-ЯМР (CDCl3) δ: 8,02 (1H, с), 7,49 (2Н, м), 7,40-7,19 (13Н, м), 7,04 (1H, с), 6,84 (4Н, м), 5,26 (1H, дд, J=5,l, 2,0 Гц), 5,06 (1Н, к, J=9,0 Гц), 4,61 (1H, дд, J=8,8, 4,9 Гц), 4,28 (2Н, м), 3,79 (6Н, с), 3,39 (1H, дд, J=9,2, 3,7 Гц), 3,19 (1H, дд, J=9,4, 3,9 Гц), 3,00-1,85 (14Н, м), 0,64 (9Н, с), -0,13 (3H, с), -0,45 (3H, с).
[0958]
(Этап 9)
(1H,2S,3R,5R)-3-(6-бензоил-6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)-2-{[трет-бутил(диметил)силил]окси}-5-(гидроксиметил)циклопентил-4-оксопентаноат
К раствору соединения (2,35 г), полученного на описанном выше этапе 8, в дихлорметане (25 мл) добавляли воду (0,24 мл) и раствор дихлоруксусной кислоты (1,05 мл) в дихлорметане (25 мл) в данном порядке, и реакционную смесь перемешивали при комнатной температуре в течение 1 часа. После добавления в реакционную смесь метанола (1,0 мл) и насыщенного водного раствора гидрокарбоната натрия, чтобы погасить реакцию, осуществляли экстрагирование продукта этилацетатом и органический слой сушили над безводным сульфатом натрия. Осушающий агент удаляли путем фильтрации и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле [гексан/этилацетат] с получением указанного в заголовке соединения (1Д 5 г).
МС (ИЭР) m/z: 621(М+Н)+.
1Н-ЯМР (CDCl3) δ: 8,05 (1H, с), 7,38-7,17 (5Н, м), 7,04 (1H, с), 5,19 (1Н, дд, J=4,5, 1,4 Гц), 4,82-4,69 (2Н, м), 4,47 (1H, дд, J=14,5, 7,4 Гц), 4,31 (1H, д, J=7,8 Гц), 4,15-4,08 (1H, м), 3,83 (1H, м), 3,75 (1H, д, J=10,8 Гц), 3,11-2,95 (2Н, м), 2,90-2,51 (4Н, м), 2,47-2,32 (2Н, м), 2,32-2,11 (3H, м), 2,22 (3H, с), 0,68 (9Н, с), -0,16 (3H, с), -0,50 (3H, с).
[0959]
(Этап 10)
N-бензоил-2'-O-[({(1H,2R,3S,4R)-4-(6-бензоил-6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)-3-{[трет-бутил(диметил)силил]окси}-2-[(4-оксопентаноил)окси]циклопентил}метокси)(2-цианоэтокси)фосфоротиоил]-5'-O-[бис(4-метоксифенил)(фенил)метил]-3'-O-[трет-бутил(диметил)силил]аденозин
К раствору соединения (550 мг), полученного на описанном выше этапе 9, в ацетонитриле (12 мл) добавляли молекулярные сита 3А, 1/16 (500 мг), и N-бензоил-5'-O-[бис(4-метоксифенил)(фенил)метил]-3'-O-[трет-бутил(диметил)силил]-2'-O-{(2-цианоэтокси)[ди(пропан-2-ил)амино]фосфанил}аденозин (1,23 г) и реакционную смесь перемешивали при комнатной температуре в течение 15 минут. В реакционную смесь добавляли 4,5-дицианоимидазол и реакционную смесь перемешивали в течение 1 часа, и после этого добавляли туда 1,1-диоксид 3H-1,2-бензотиол-3-она (355 мг), и реакционную смесь дополнительно перемешивали в течение 1 часа. Молекулярные сита 3А удаляли из реакционной смеси путем фильтрации и насыщенный водный раствор гидрокарбоната натрия добавляли в фильтрат, который осуществляли экстрагирование этилацетатом. После высушивания органического слоя над безводным сульфатом натрия осушающий агент удаляли путем фильтрации и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле [гексан/этилацетат] с получением указанного в заголовке соединения (1,17 г) в виде смеси диастереомеров у атома фосфора.
МС (ИЭР) m/z: 1539(М+Н)+.
[0960]
(Этап 11)
N-бензоил-2'-O-[{[(1R,2R,3S,4R)-4-(6-бензоил-6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)-3-{[трет-бутил(диметил)силил]окси}-2-гидроксициклопентил]метокси}(2-цианоэтокси)фосфоротиоил]-5'-O-[бис(4-метоксифенил)(фенил)метил]-3'-O-[трет-бутил(диметил)силил]аденозин
К раствору соединения (1,22 г), полученного на описанном выше этапе 10, в ацетонитриле (2,0 мл) добавляли смешанный раствор моногидрата гидразина (0,25 мл) в уксусной кислоте (5,0 мл)/пиридине (7,5 мл) и реакционную смесь перемешивали при комнатной температуре в течение 30 минут. После того, как в реакционную смесь добавляли воду, чтобы погасить реакцию, осуществляли экстрагирование продукта этилацетатом и органический слой сушили над безводным сульфатом натрия. Осушающий агент удаляли путем фильтрации и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле [гексан/этилацетат] с получением указанного в заголовке соединения (770 мг) в виде смеси диастереомеров у атома фосфора.
МС (ИЭР) m/z: 1442(М+Н)+.
[0961]
(Этап 12)
N-бензоил-2'-O-[{[(1R,2R,3S,4R)-4-(6-бензоил-6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)-3-{[трет-бутил(диметил)силил]окси}-2-{[гидрокси(оксо)-λ5-фосфанил]окси}циклопентил]метокси}(2-цианоэтокси)фосфоротиоил]-3'-O-[трет-бутил(диметил)силил]аденозин
К раствору соединения (770 мг), полученного на описанном выше этапе 11, в пиридине (8,0 мл) добавляли дифенилфосфит (0,61 мл) и реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Воду (20 мл), ацетонитрил (8,0 мл) и водный раствор ацетата триэтиламмония (2 М, 1,6 мл) добавляли в реакционную смесь, которую перемешивали в течение 1 часа, и реакционную смесь затем концентрировали при пониженном давлении. К раствору остатка в дихлорметане (5,0 мл) добавляли воду (0,096 мл) и раствор дихлоруксусной кислоты (0,22 мл) в дихлорметане (5,0 мл) в данном порядке, и реакционную смесь перемешивали в течение 30 минут. Пиридин (0,43 мл) добавляли туда, чтобы погасить реакцию, и реакционную смесь затем концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле [гексан/этилацетат] с получением указанного в заголовке соединения (400 мг) в виде смеси диастереомеров у атома фосфора.
MC(H3P)m/z: 1203(М+Н)+.
[0962] (Этап 13)
N-{9-[(5R,7R,8R,12aR,14R,15S,15aR,16R)-14-(6-бензоил-6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)-15,16-бис{[трет-бутил(диметил)силил]окси}-10-(2-цианоэтокси)-2-оксо-2-сульфанил-10-сульфанилидендекагидро-2Н,10Н-5,8-метано-2λ5,10λ5-циклопента[l][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-7-ил]-9Н-пурин-6-ил}бензамид
С применением соединения (400 мг), полученного на описанном выше этапе 12, проводили реакцию таким же образом, как на этапе 9 примера 1, с получением указанного в заголовке соединения (300 мг) в виде смеси диастереомеров у атома фосфора.
MC(ИЭР)m/z: 1217(М+Н)+.
[0963] (Этап 14)
Динатрия (5R,7R,8R,12aR,14R,15S,15aR,16R)-7-(6-амино-9H-пурин-9-ил)-15,16-дигидрокси-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)декагидро-2Н,10Н-5,8-метано-2λ5,10λ5-циклопента[1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
К раствору соединения (300 мг), полученного на описанном выше этапе 13, в метаноле (10 мл) добавляли 28% водный аммиак (10 мл) и реакционную смесь перемешивали при комнатной температуре в течение 12 часов. После концентрирования реакционной смеси при пониженном давлении добавляли к остатку тригидрофторидтриэтиламина (3,0 мл) и продукт перемешивали при 45°С в течение 2 часов. Реакционную смесь добавляли по каплям в охлажденный до температуры льда смешанный раствор 1 Μ раствора гидрокарбоната триэтиламмония (18 мл)/триэтиламина (6,0 мл), чтобы погасить реакцию. Реакционную смесь концентрировали при пониженном давлении, а затем очищали с помощью колоночной хроматографии на силикагеле С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил] с получением диастереомера 1 и диастереомера 2 указанного в заголовке соединения в виде солей триэтиламина.
Каждую из полученных солей триэтиламина подвергали солевому обмену таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением диастереомера 1 (45,6 мг) и диастереомера 2 (12,6 мг) указанного в заголовке соединения.
Диастереомер 1 (менее полярный)
МС (ИЭР) m/z: 728(М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,69 (1Н, с), 8,10 (1Н, с), 7,93 (1Н, с), 7,10 (1Н, с), 6,29 (1Н, д, J=8,2 Гц), 5,38 (1Н, дк, J=9,7,2,7 Гц), 4,99 (2Н, м), 4,79 (1Н, д, J=3,9 Гц), 4,61 (1H, дд, J=7,2, 4,9 Гц), 4,36 (1H, м), 4,29 (1H, м), 4,19 (1H, м), 3,99 (1H, м), 3,89 (1Н, к, J=9,9 Гц), 3,47 (2Н, т, J=4,9 Гц), 2,80 (2Н, к, J=5,7 Гц), 2,72 (1H, м), 2,37 (1H, дт, J=16,4, 6,7 Гц), 1,94 (2Н, м), 1,61 (1H, дт, J=16,4, 6,7 Гц).
31Р-ЯМР (CD3OD) δ: 58,3 (с), 54,0 (с).
Диастереомер 2 (более полярный)
МС (ИЭР) m/z: 728(М+Н)+.
1H-ЯМР (CD3OD) δ: 8,74 (1H, с), 8,10 (1H, с), 7,95 (1H, с), 7,09 (1H, с), 6,26 (1H, д, J=8,6 Гц), 5,44 (1H, м), 5,24 (1H, м), 5,06 (1Н, к, J=9,3 Гц), 4,59 (1Н, дд, J=9,4, 4,3 Гц), 4,51 (1Н, д, J=4,3 Гц), 4,34-4,21 (3Н, м), 3,94 (1H, м), 3,74 (1H, м), 3,47 (2Н, м), 2,85 (2Н, т, J=5,5 Гц), 2,54 (1H, м), 2,45 (1H, дт, J=17,l, 6,7 Гц), 1,95 (2Н, м), 1,38 (1H, ддд, J=14,6, 8,3, 5,0 Гц).
31Р-ЯМР (CD3OD) δ: 62,7 (с), 60,1 (с).
[0964]
Пример 57. Синтез ЦДН47.
(5R,7R,8R,12aR,14R,15R,15aS,16R)-7-(6-амино-9H-пурин-9-ил)-15,16-дигидрокси-2,10-бис(сульфанил)-10-сульфанилиден-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро)-2Η,10Η,12Η-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2-он
[0965]
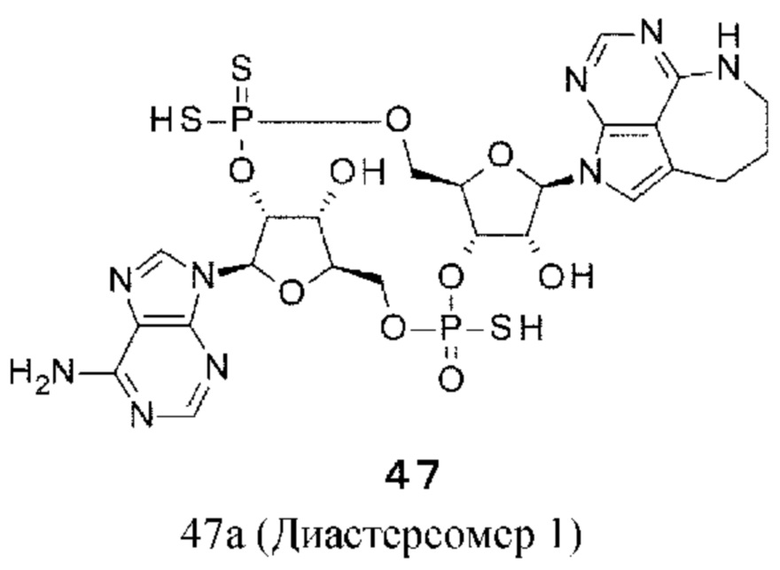
[0966]
[Схема синтеза]
[0967]
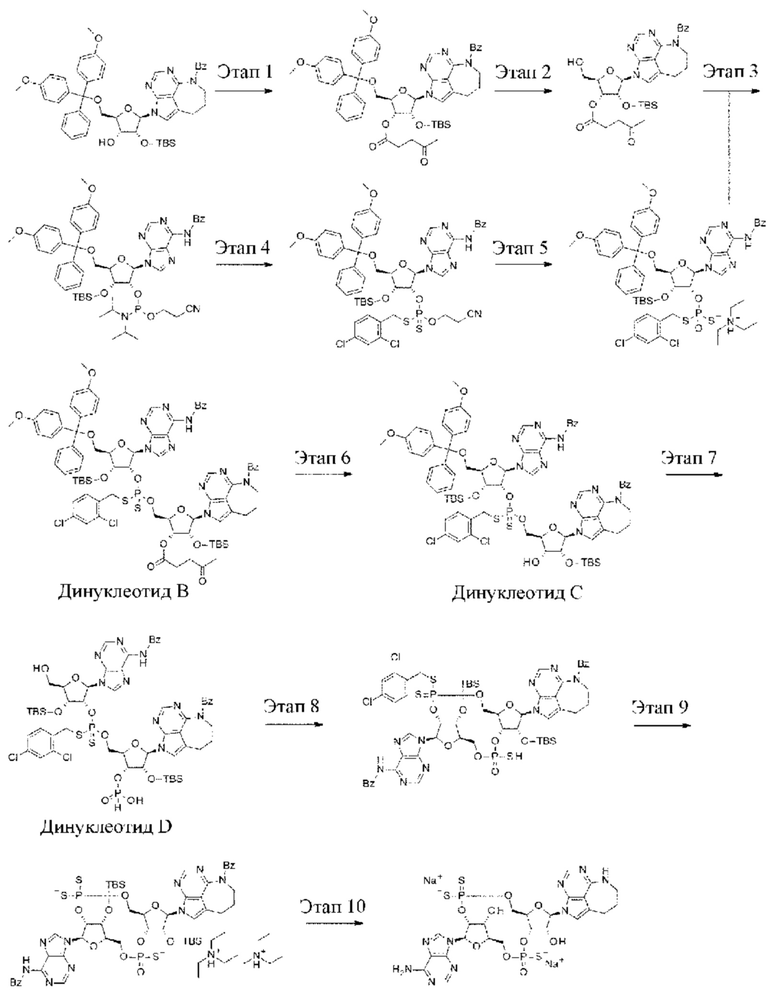
[0968]
(Этап 1)
6-бензоил-2-{5-O-[бис(4-метоксифенил)(фенил)метил]-2-O-[трет-бутил(диметил)силил]-3-O-(4-оксопентаноил)-β-D-рибофуранозил}-6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен
С применением соединения, полученного на этапе 5 примера 1, проводили реакцию таким же образом, как на этапе 8 примера 56, с получением указанного в заголовке соединения (2,8 г).
1H-ЯМР (CDCl3) δ: 8,08 (1H, с), 7,48-7,19 (15Н, м), 6,37 (4Н, д, J=6,7 Гц), 5,48 (1H, дд, J=5,l, 2,3 Гц), 4,83 (1H, дд, J=6,7, 5,1 Гц), 4,37-4,00 (3Н, м), 3,80 (3Н, с), 3,79 (3Н, с), 3,69 (1H, м), 3,54 (1H, дд, J=10,6, 2,7 Гц), 3,39 (1H, дд, J=11,0, 2,7 Гц), 2,90-2,57 (6Н, м), 2,20 (3Н, с), 2,20-2,11 (2Н, м), 0,69 (9Н, с), -0,03 (3Н, с), -0,29 (3Н, с).
[0969]
(Этап 2)
6-бензоил-2-{2-O-[трет-бутил(диметил)силил]-3-O-(4-оксопентаноил)-β-D-рибофуранозил}-6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен
С применением соединения (2,8 г), полученного на описанном выше этапе 1, проводили реакцию таким же образом, как на этапе 9 примера 56, с получением указанного в заголовке соединения (1,74 г).
1H-ЯМР (CDCl3) δ: 8,06 (1Н, с), 7,38-7,20 (5Н, м), 7,04 (1H, с), 6,32 (1H, дд, J=12,l, 1,6 Гц), 5,64 (1H, д, J=7,8 Гц), 5,47 (1H, д, J=5,1 Гц), 5,16 (1H, дд, J=7,8, 5,1 Гц), 4,40 (1H, м), 4,29 (1H, с), 4,17 (1H, м), 3,91 (1H, м), 3,75 (1H, м), 3,02 (2Н, м), 2,90-2,60 (4Н, м), 2,30-2,14 (2Н, м), 2, 22 (3Н, с), 0,68 (9Н, с), -0,15 (3Н, с), -0,46 (3Н, с).
[0970]
(Этап 3)
N-бензоил-5'-O-[бис(4-метоксифенил)(фенил)метил]-3'-O-[трет-бутил(диметил)силил]-2'-O-[(2-цианоэтокси){[(2,4-дихлорфенил)метил]сульфанил}фосфоротиоил]аденозин
К раствору доступного для приобретения (ChemGenes Corporation) N-бензоил-5'-O-[бис(4-метоксифенил)(фенил)метил]-3'-O-[трет-бутил(диметил)силил]-2'-O-{(2-цианоэтокси)[ди(пропан-2-ил)амино]фосфанил}аденозина (3,73 г) в ацетонитриле (30 мл) добавляли молекулярные сита 3А, 1/16 (1,0 г), и 2,4-дихлорбензилмеркаптан (1,8 мл), и реакционную смесь перемешивали при комнатной температуре в течение 10 минут. Перхлорат имидазола (3,18 г) добавляли в реакционную смесь, которую перемешивали в течение 2,5 часов, а затем добавляли туда серу (242 мг), и реакционную смесь дополнительно перемешивали в течение 1 часа. Молекулярные сита 3А удаляли из реакционной смеси путем фильтрации, и насыщенный водный раствор гидрокарбоната натрия добавляли в фильтрат, который осуществляли экстрагирование этилацетатом. После высушивания органического слоя над безводным сульфатом натрия осушающий агент удаляли путем фильтрации и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле [гексан/этилацетат] с получением указанного в заголовке соединения (2,73 г) в виде смеси диастереомеров у атома фосфора.
МС (ИЭР) m/z: 1111(М+Н)+.
1H-ЯМР (CDCl3) δ: 8,89 (1H, с), 8,74 (1H, д, J=1,8 Гц), 8,32 (0,5Н, с), 8,26 (0,5Н, с), 8,02 (2Н, м), 7,64-7,61 (15Н, м), 6,82 (4Н, д, J=9,0), 6,45 (0,5Н, д, J=6,7 Гц), 6,38 (0,5Н, д, 3=6,3 Гц), 5,88 (0,5Н, ддд, J=14,2, 6,6, 4,8 Гц), 5,74 (0,5Н, ддд, J=14,4, 6,4, 4,8 Гц), 4,75 (0,5Н, дд, J=4,7, 2,7 Гц), 4,64 (0,5Н, дд, J=4,7, 2,3 Гц), 4,22-3,82 (5Н, м), 3,78 (6Н, с), 3,58-3,53 (2Н, м), 3,34-3,29 (2Н, м), 2, 57 (1H, т, J=6,3 Гц), 2,48 (1H, т, J=6,3 Гц), 0,90 (4,5Н, с), 0,88 (4,5Н, с), 0,16 (1,5Н, с), 0,11 (1,5Н, с), 0,07 (1,5Н, с), 0,04 (1,5Н, с).
[0971]
(Этап 4)
Ν,Ν-диэтилэтанаминия N-бензоил-5'-O-[бис(4-метоксифенил)(фенил)метил]-3'-O-[трет-бутил(диметил)силил]-2'-O-[{[(2,4-дихлорфенил)метил]сульфанил}(сульфид)фосфорил]аденозин
К раствору соединения (2,66 г), полученного на описанном выше этапе 3, в ацетонитриле (30 мл) добавляли триэтиламин (30 мл) и реакционную смесь перемешивали при комнатной температуре в течение 4 часов. Реакционную смесь концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле DIOL [гексан/этилацетат] с получением указанного в заголовке соединения (2,5 г) в виде смеси диастереомеров у атома фосфора.
МС (ИЭР) m/z: 1058(М+Н)+.
[0972]
(Этап 5)
Динуклеотид В
К раствору соединения (2,5 г), полученного на описанном выше этапе 4, в дихлорметане (10 мл) добавляли молекулярные сита 4А, 1/16 (1,0 г), соединение (930 мг), полученное на описанном выше этапе 2 и 1-метилимидазол (1,18 мл) в данном порядке, и реакционную смесь перемешивали при комнатной температуре в течение 20 минут. В реакционную смесь добавляли 2,4,6-триизопропилбензолсульфонилхлорид (905 мг), и реакционную смесь дополнительно перемешивали в течение 4 часов. Молекулярные сита 4А удаляли из реакционной смеси путем фильтрации, и насыщенный водный раствор гидрокарбоната натрия добавляли в фильтрат, который осуществляли экстрагирование этилацетатом. После высушивания органического слоя над безводным сульфатом натрия осушающий агент удаляли путем фильтрации и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле [гексан/этилацетат] с получением указанного в заголовке соединения (1,04 г) в виде смеси диастереомеров у атома фосфора.
MC(ИЭР)m/z: 1662(М+Н)+.
[0973]
(Этап 6)
Динуклеотид С
С применением соединения (1,04 г), полученного на описанном выше этапе 5, проводили реакцию таким же образом, как на этапе 11 примера 56, с получением указанного в заголовке соединения (820 мг) в виде смеси диастереомеров у атома фосфора.
MC(H3P)m/z: 1564(М+Н)+.
[0974]
(Этап 7)
Динуклеотид D
С применением соединения (820 мг), полученного на описанном выше этапе 6, проводили реакцию таким же образом, как на этапе 12 примера 56, с получением указанного в заголовке соединения (440 мг) в виде смеси диастереомеров у атома фосфора.
[0975]
(Этап 8)
N-{9-[(5R,7R,8R,12aR,14R,15R,15aR,16R)-14-(6-бензоил-6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)-15,16-бис{[трет-бутил(диметил)силил]окси}-10-{[(2,4дихлорфенил)метил]сульфанил}-2-оксо-2-сульфанил-10-сульфанилиденоктагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-7-ил]-9Н-пурин-6-ил}бензамид
С применением соединения (440 мг), полученного на описанном выше этапе 7, проводили реакцию таким же образом, как на этапе 9 примера 1, с получением указанного в заголовке соединения (360 мг).
МС (ИЭР) m/z: 1340(М+Н)+.
[0976]
(Этап 9)
Бис(N,N-диэтилэтанаминия) (5R,7R,8R,12aR,14R,15R,15aR,16R)-7-(6-6ензамидо-9H-пурин-9-ил)-14-(6-бензоил-6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)-15,16-бис{[трет-бутил(диметил)силил]окси}-2-оксо-10-сульфанилиденоктагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
В раствор в диметилсульфоксиде соединения (360 мг), полученного на описанном выше этапе 8, добавляли 1-додекантиол (434 мг) и 1,8-диазабицикло[5,4,0]-7-ундецен (0,40 мл) и реакционную смесь перемешивали при комнатной температуре в течение 6 часов. Реакционную смесь непосредственно очищали с помощью колоночной хроматографии на силикагеле С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил] с получением указанного в заголовке соединения (130 мг).
МС (ИЭР) m/z: 1182(М+Н)+.
[0977]
(Этап 10)
Динатрия (2S,5R,7R,8R,12aR,14R,15R,15aS,16R)-7-(6-амино-9H-пурин-9-ил)-15,16-дигидрокси-2-оксо-10-сульфанилиден-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
С применением соединения (130 мг), полученного на описанном выше этапе 9, проводили реакцию таким же образом, как на этапе 14 примера 56, и продукт затем очищали с помощью колоночной хроматографии на силикагеле С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил] с получением указанного в заголовке соединения в виде соли триэтиламина.
Полученную соль триэтиламина подвергали солевому обмену таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением указанного в заголовке соединения (62,8 мг).
МС (ИЭР) m/z: 746(М+Н)+.
1H-ЯМР (CD3OD) δ: 8,70 (1Н, с), 8,07 (1Н, с), 8,03 (1Н, с), 7,25 (1Н, с), 6,26 (1Н, д, J=8,6 Гц), 6,21 (1Н, д, J=5,9 Гц), 5,52 (1Н, дк, J=14,1, 5,2 Гц), 5,36 (1Н, м), 4,70 (1Н, дд, J=5,7, 4,5 Гц), 4,42 (1Н, д, J=4,3 Гц), 4,37-4,26 (3Н, м), 4,21 (1Н, м), 3,96 (1Н, м), 3,84 (1Н, м), 3,50 (2Н, м), 2,81 (2Н,м), 1,93 (2Н,м).
31Р-ЯМР (CD3OD) δ: 119,8 (с), 59,4 (с).
[0978]
Пример 58. Синтез ЦДН48.
(5R,7R,8R,12aR,14R,15R,15aR,16R)-14-(8,9-дигидро-6-тиа-2,3,5-триазабензо[cd] азулен-2(7Н)-ил)-15-фтор-16-гидрокси-7-[1-(2-гадроксиэтил)-6-оксо-1,6-дигидро-9Н-пурин-9-ил]-2,10-бис(сульфанил)октагидро-2Н,10Н,12H-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-дион
[0979]
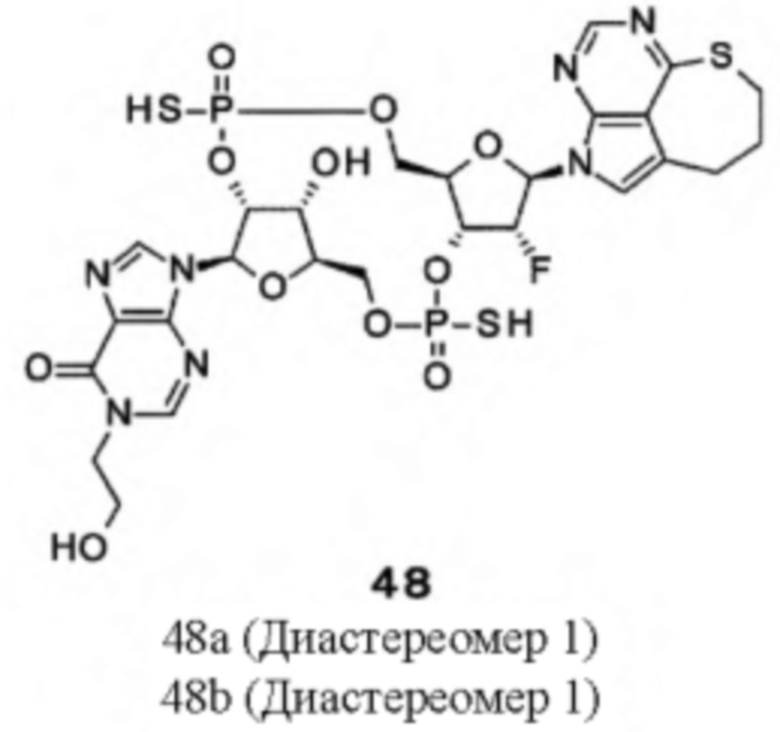
[0980]
[Схема синтеза]
[0981]
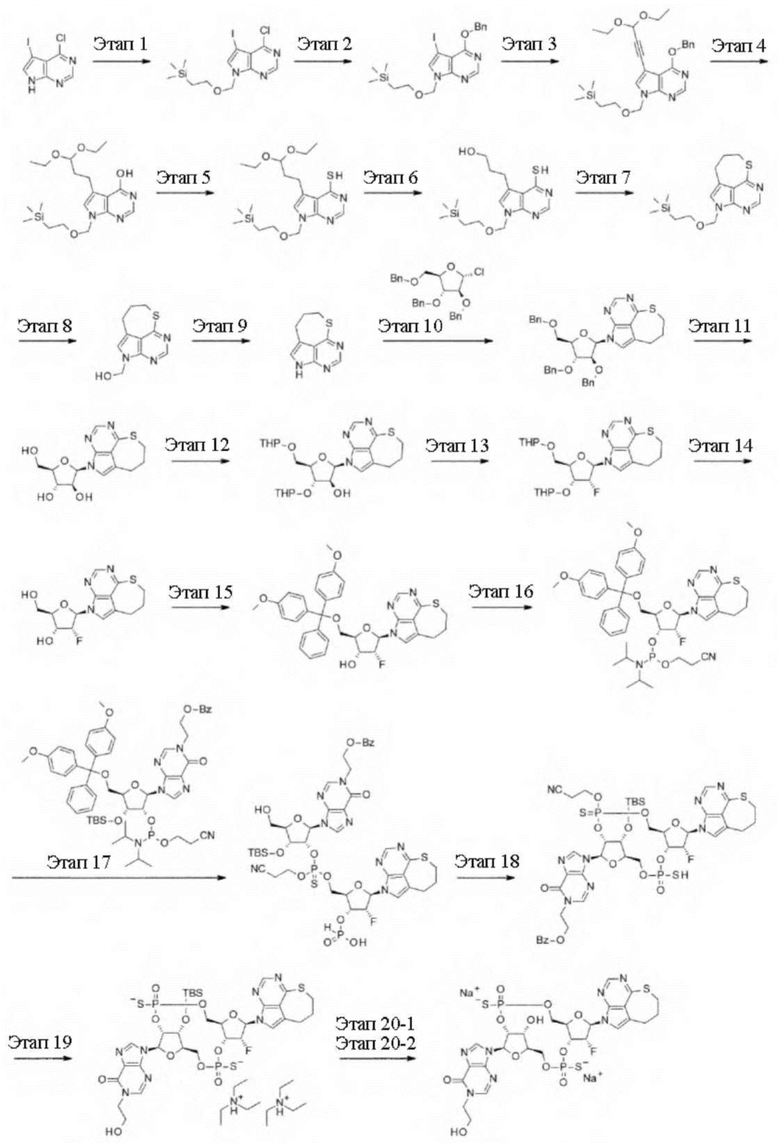
[0982]
(Этап 1)
4-хлор-5-йод-7-{[2-(триметилсилил)этокси]метил}-7Н-пирроло[2,3-d]пиримидин
К раствору доступного для приобретения (PharmaBlock Sciences (Nanjing), Inc.) 4-хлор-5-йод-7Н-пирроло[2,3-d]пиримидина (73,8 г) в Ν,Ν-диметилформамиде (10 мл) добавляли гидрид натрия (содержащий 45% минеральное масло) (13,3 г) при охлаждении льдом, и реакционную смесь перемешивали в течение 40 минут при повышении температуры до комнатной температуры. Реакционную смесь снова охлаждали до температуры льда, добавляли туда [2-(хлорметокси)этил](триметил)силан (51,0 мл) в течение 10 минут и реакционную смесь затем перемешивали при той же температуре в течение 30 минут. В по большей части отвердевшую реакционную смесь добавляли воду (260 мл) малыми частями, чтобы погасить реакцию. Твердый продукт собирали путем фильтрации, промывали водой (1500 мл) и гексаном (600 мл) и сушили при пониженном давлении при 40°С с получением указанного в заголовке соединения (97,63 г).
МС (ИЭР) m/z: 410(М+Н)+.
1H-ЯМР (CDCl3) δ: 8,64 (1H, с), 7,54 (1Н, с), 5,61 (2Н, с), 3,52 (2Н, т, J=8,3 Гц), 0,92 (2Н, т, J=8,3 Гц), -0,04 (9Н, с).
[0983]
(Этап 2)
4-(бензилокси)-5-йод-7-{[2-(триметилсилил)этокси]метил}-7Н-пирроло[2,3-d]пиримидин
К раствору бензилового спирта (27 мл) в Ν,Ν-диметилформамиде (170 мл) добавляли гидрид натрия (содержащий 45% минеральное масло) (12 г) при охлаждении льдом и реакционную смесь перемешивали в течение 40 минут при повышении температуры до комнатной температуры. Реакционную смесь снова охлаждали до температуры льда, суспензию соединения (97,63 г), полученного на описанном выше этапе 1, в Ν,Ν-диметилформамиде (360 мл) добавляли туда в течение 40 минут, и реакционную смесь перемешивали при той же температуре в течение 35 минут. Ледяную стружку и насыщенный водный раствор хлорида аммония добавляли в реакционную смесь, чтобы погасить реакцию. Реакционную смесь вливали в двухслойную смесь насыщенного водного раствора хлорида аммония и этилацетата, и осуществляли экстрагирование этилацетатом:толуолом (9:1). Органический слой промывали дважды водой и дважды рассолом, а затем сушили над безводным сульфатом натрия. Осушающий агент удаляли путем фильтрации и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле [гексан/этилацетат] с получением указанного в заголовке соединения (107,7 г).
МС (ИЭР) m/z: 482(М+Н)+.
1H-ЯМР (CDCl3) δ: 8,47 (1Н, с), 7,61 (2Н, д, J=7,3 Гц), 7,41 (2Н, т, J=7,6 Гц), 7,36-7,30 (1H, м), 7,30 (1H, с), 5,65 (2Н, с), 5,57 (2Н, с), 3,52 (2Н, т, J=8,3 Гц), 0,91 (2Н, т, J=8,3 Гц), -0,05 (9Н, с).
[0984]
(Этап 3)
4-(бензилокси)-5-(3,3-диэтоксипроп-1-ин-1-ил)-7-{[2-(триметилсилил)этокси]метил}-7Н-пирроло[2,3-d]пиримидин
В смешанный раствор соединения (113,4 г), полученного на описанном выше этапе 2, в ацетонитриле (1000 мл)/триэтиламине (98 мл) добавляли йодид меди (4,49 г), тетракистрифенилфосфинпалладий(0) (8,17 г) и 3,3-диэтоксипроп-1-ин (104 мл) в атмосфере азота при комнатной температуре, и реакционную смесь перемешивали при той же температуре в течение 4,5 часов. После концентрирования реакционной смеси при пониженном давлении к остатку добавляли этилацетат и гексан и твердый преципитированный продукт удаляли путем фильтрации. Твердый продукт промывали смесью этилацетата:гексана (1:1) и фильтрат затем концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле [гексан/этилацетат] с получением указанного в заголовке соединения (145,5 г: с примесями).
МС (ИЭР) m/z: 482(М+Н)+.
[0985]
(Этап 4)
5-(3,3-диэтоксипропил)-7-{[2-(триметилсилил)этокси]метил}-7Н-пирроло[2,3-d]пиримидин-4-ол
К раствору соединения (145,5 г), полученного на описанном выше этапе 3, в этаноле (900 мл) добавляли катализатор 10% палладий/углерод (М), влажный (50,2 г), и реакционную смесь перемешивали в атмосфере водорода при комнатной температуре в течение 5 часов. Дихлорметан (500 мл) добавляли в реакционную смесь, катализатор удаляли путем фильтрации с помощью целита и фильтрат концентрировали при пониженном давлении. Остаток очищали дважды с помощью колоночной хроматографии на силикагеле [гексан/этилацетат] с получением указанного в заголовке соединения (59,6 г).
МС (ИЭР)m/z: 418(M+Na)+, 394[M-H]-.
1Н-ЯМР (CDCl3) δ: 11,23 (1H, уш. с), 7,85 (1H, с), 6,79 (1H, с), 5,47 (2Н, с), 4,58 (1H, т, J=5,9 Гц), 3,69 (2Н, м), 3,57-3,49 (4Н, м), 2,90 (2Н, т, J=7,8 Гц), 2,07 (2Н, м), 1,23 (6Н, т, J=7,l Гц), 0,91 (2Н, т, J=8,1 Гц), -0,04 (9Н, с).
[0986]
(Этап 5)
5-(3,3-диэтоксипропил)-7-{[2-(триметилсилил)этокси]метил}-7Н-пирроло[2,3-d]пиримидин-4-тиол
К раствору соединения (59,6 г), полученного на описанном выше этапе 4, в обезвоженном дихлорметане (300 мл) добавляли 2,6-лутидин (42 мл) в атмосфере азота. Трифторметансульфоновый ангидрид (31 мл) добавляли туда по каплям при -20°С в течение 20 минут и реакционную смесь перемешивали при той же температуре в течение 20 минут. Туда добавляли Ν,Ν-диметилформамид (500 мл) и моногидросульфида натрия гидрат (33,5 г) при охлаждении льдом, температуру повышали до комнатной температуры и реакционную смесь затем перемешивали в течение 2,5 часов. Реакционную смесь концентрировали при пониженном давлении, чтобы отогнать компоненты с низкой точкой кипения. Остаток вливали в двухслойную смесь этилацетата и охлажденного до температуры льда насыщенного водного раствора хлорида аммония, и осуществляли экстрагирование смесью этил ацетат: толуол (9:1). Органический слой однократно промывали насыщенным водным раствором хлорида аммония и два раза рассолом, а затем сушили над безводным сульфатом натрия. Осушающий агент удаляли путем фильтрации и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле [гексан/этилацетат] с получением смеси указанного в заголовке соединения и 2,6-лутидина. Полученную смесь вливали в двухслойную смесь этилацетата и 1 N соляной кислоты и дважды осуществляли экстрагирование этилацетатом. Органический слой промывали три раза рассолом, а затем сушили над безводным сульфатом натрия. Осушающий агент удаляли путем фильтрации и фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (57,6 г).
МС (ИЭР) m/z: 410[М-Н]-.
1Н-ЯМР (CDCl3) δ: 11,69 (1H, уш. с), 7,90 (1H, с), 6,96 (1H, с), 5,49 (2Н, с), 4,61 (1H, т, J=5,9 Гц), 3,71 (2Н, м), 3,55 (2Н, м), 3,49 (2Н, т, J=8,1 Гц), 3,14 (2Н, т, J=7,8 Гц), 2,08 (2Н, м), 1,23 (6Н, т, J=7,1 Гц), 0,90 (2Н, т, J=8,3 Гц), -0,04 (9Н, с).
[0987]
(Этап 6)
3-(4-сульфанил-7-{[2-(триметилсилил)этокси]метил}-7Н-пирроло[2,3-d]пиримидин-5-ил)пропан-1-ол
Соединение (31,62 г), полученное на описанном выше этапе 5, растворяли в 80% водном растворе уксусной кислоты (300 мл), и реакционную смесь перемешивали при комнатной температуре в течение 30 минут. После подтверждения исчезновения сырьевого материала реакционную смесь охлаждали до температуры льда, аккуратно добавляли туда малыми частями борогидрид натрия (1,45 г) и реакционную смесь перемешивали при той же температуре в течение 30 минут. Затем добавляли туда триацетоксиборогидрид натрия (24,4 г) в течение 15 минут и реакционную смесь перемешивали при той же температуре в течение 1,5 часов. Реакционную смесь концентрировали до приблизительно 1/5 исходного объема при пониженном давлении. Гидрокарбонат натрия (твердый) аккуратно добавляли к остатку для нейтрализации до некоторой степени, реакционную смесь осуществляли экстрагирование этилацетатом. Органический слой промывали насыщенным гидрокарбонатом натрия и рассолом в данном порядке и сушили над безводным сульфатом натрия. Осушающий агент удаляли путем фильтрации и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле [гексан/этилацетат] с получением указанного в заголовке соединения (17,93 г).
МС (ИЭР) m/z: 340[М+Н]+.
1H-ЯМР (CDCl3) δ: 11,92 (1H, уш. с), 7,95 (1H, с), 7,01 (1H, с), 5,51 (2Н, с), 3,70 (2Н, т, J=5,9 Гц), 3,50 (2Н, т, J=8,1 Гц), 3,23 (2Н, т, J=7,3 Гц), 2,33 (1H, уш. с), 1,99 (2Н, м), 0,91 (2Н, т, J=8,3 Гц), -0,04 (9Н, с).
[0988]
(Этап 7)
2-{[2-(триметилсилил)этокси]метил}-2,7,8,9-тетрагидро-6-тиа-2,3,5-триазабензо[с(1]азулен
К раствору соединения (31,31 г), полученного на описанном выше этапе 6, в обезвоженном тетрагидрофуране (600 мл) добавляли трифенилфосфин (25,4 г) и диизопропилазодикарбоксилат (21,8 г) в атмосфере азота при 0°С и реакционную смесь перемешивали при той же температуре в течение 1 часа. После концентрирования реакционной смеси при пониженном давлении остаток очищали с помощью колоночной хроматографии на силикагеле [дихлорметан/этилацетат] и колоночной хроматографии на силикагеле [гексан/этилацетат] в данном порядке с получением указанного в заголовке соединения (35,93 г: с примесями).
МС (ИЭР) m/z: 322[М+Н]+.
1Н-ЯМР (CDCl3) δ: 8,57 (1H, с), 7,08 (1H, с), 5,58 (2Н, с), 3,52 (2Н, т, J=8,3 Гц), 3,17 (2Н, м), 3,06 (2Н, т, J=5,6 Гц), 2,36 (2Н, м), 0,92 (2Н, т, J=8,3 Гц), -0,05 (9Н, с).
[0989]
(Этап 8)
(8,9-дигидро-6-тиа-2,3,5-триазабензо[cd]азулен-2(7Н)-ил)метанол
К раствору соединения (35,93 г), полученного на описанном выше этапе 7, в дихлорметане (150 мл) добавляли трифторуксусную кислоту (150 мл) при комнатной температуре и реакционную смесь перемешивали при той же температуре в течение 1,5 часов. Реакционную смесь концентрировали при пониженном давлении, а затем подвергали азеотропной перегонке четыре раз с толуолом. Смесь дихлорметан:гексан (1:2) добавляли к остатку, и твердый преципитированный продукт собирали путем фильтрации (твердый продукт 1). После того, как фильтрат концентрировали при пониженном давлении, остаток очищали с помощью колоночной хроматографии на силикагеле [гексан/этилацетат → этилацетат/метанол] с получением твердого продукта 2. Твердый продукт 1 и твердый продукт 2 объединяли с получением указанного в заголовке соединения (20,13 г).
МС (ИЭР) m/z: 222[М+Н]+.
1H-ЯМР (CDCl3) δ: 8,60 (1H, с), 7,19 (1H, с), 5,71 (2Н, с), 3,21 (2Н, м), 3,07 (2Н, м), 2,38 (2Н, м) (показаны только наблюдаемые пики).
[0990]
(Этап 9)
2,7,8,9-тетрагидро-6-тиа-2,3,5-триазабензо[cd]азулен
В суспензию соединения (20,13 г), полученного на описанном выше этапе 8, в метаноле (250 мл) добавляли 28% водный раствор аммиака (150 мл) и реакционную смесь перемешивали при комнатной температуре в течение 1,5 часов. Реакционную смесь концентрировали до приблизительно половины объема при пониженном давлении. Твердый преципитированный продукт собирали путем фильтрации и промывали этанолом с получением твердого продукта 1. Фильтрат концентрировали при пониженном давлении, и твердый продукт 2 получали с помощью такой же процедуры. Фильтрат наносили на силикагель, а затем очищали с помощью колоночной хроматографии на силикагеле [дихлорметан/метанол]. Фракции, содержащие целевой продукт, концентрировали при пониженном давлении, взвесь промывали этанолом и твердый продукт затем собирали путем фильтрации (твердый продукт 3). Твердый продукт 1, твердый продукт 2 и твердый продукт 3 объединяли с получением указанного в заголовке соединения (12,36 г).
МС (ИЭР) m/z: 192[М+Н]+.
1H-ЯМР (CDCl3) δ: 10,53 (1H, уш. с), 8,57 (1H, с), 7,10 (1H, с), 3,18 (2Н, м), 3,08 (2Н, т, J=5,6 Гц), 2,37 (2Н, м).
[0991]
(Этап 10)
2-(2,3,5-три-O-бензил-β-D-арабинофуранозил)-2,7,8,9-тетрагидро-6-тиа-2,3,5-триазабензо[cd] азулен
В суспензию соединения (13,47 г), полученного на описанном выше этапе 9, в обезвоженном ацетонитриле (350 мл) добавляли порошкованный гидроксид калия (10,3 г) и трис[2-(2-метоксиэтокси)этил]амин (1,13 мл) в атмосфере азота и реакционную смесь перемешивали при комнатной температуре в течение 1,5 часов. Раствор хлорида 2,3,5-три-О-бензил-α-D-арабинофуранозила (40,2 г) как соединения, известного в литературе (J. Med. Chem. 1976, 19, 6, 814-816), в ацетонитриле (100 мл) добавляли туда малыми частями при охлаждении льдом, и температуру повышали до комнатной температуры и реакционную смесь перемешивали в течение 4 часов. Не растворившееся вещество удаляли путем фильтрации и промывали ацетонитрилом. Фильтрат концентрировали при пониженном давлении и остаток очищали дважды с помощью колоночной хроматографии на силикагеле [гексан/этилацетат] с получением указанного в заголовке соединения (26,19 г).
МС (ИЭР) m/z: 594[М+Н]+.
1H-ЯМР (CDCl3) δ: 8,51 (1H, с), 7,37-7,17 (14Н, м), 6,86 (2Н, м), 6,82 (1H, д, J=4,9 Гц), 4,68 (1H, д, J=11,7 Гц), 4,59 (1H, д, J=11,7 Гц), 4,54 (1H, д, J=13,2 Гц), 4,52 (1H, д, J=11,7 Гц), 4,36-4,33 (2Н, м), 4,22 (1H, д, J=11,7 Гц), 4,14-4,08 (2Н, м), 3,77 (1H, дд, J=10,7, 3,9 Гц), 3,72 (1H, дд, J=10,5, 4,1 Гц), 3,13 (2Н, м), 2,81 (2Н, м), 2,27 (2Н, м).
[0992]
(Этап 11)
2-β-D-арабинофуранозил-2,7,8,9-тетрагидро-6-тиа-2,3,5-триазабензо[cd]азулен
К раствору соединения (26,19 г), полученного на описанном выше этапе 10, в обезвоженном дихлорметане (300 мл) добавляли раствор в дихлорметане трихлорида бора (1 М, 200 мл) в атмосфере азота при -78°С, и реакционную смесь перемешивали при той же температуре в течение 2 часов, и температуру затем повышали до 0°С и реакционную смесь дополнительно перемешивали в течение 4 часов. Реакционную смесь снова охлаждали до -78°С, добавляли туда раствор метанола (80 мл) в дихлорметане (160 мл) и реакционную смесь перемешивали в течение 30 минут при повышении температуры до комнатной температуры. Реакционную смесь концентрировали при пониженном давлении и дважды подвергали азеотропной перегонке с этанолом. Этанол (200 мл) и диэтиловый эфир (100 мл) добавляли к остатку, чтобы получить взвесь, и твердый продукт собирали путем фильтрации (твердый продукт 1). Фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле [дихлорметан/метанол]. Фракции, содержащие целевой продукт, концентрировали при пониженном давлении, добавляли туда этанол, чтобы получить взвесь, и твердый продукт собирали путем фильтрации (твердый продукт 2). Твердый продукт 1 и твердый продукт 2 объединяли с получением указанного в заголовке соединения (13,2 г).
МС (ИЭР) m/z: 324[М+Н]+.
1H-ЯМР (CD3OD) δ: 8,73 (1H, с), 7,96 (1H, с), 6,70 (1H, д, J=4,9 Гц), 4,32 (1H, т, J=4,6 Гц), 4,25 (1H, т, J=4,6 Гц), 3,97 (1H, м), 3,90 (1Н, дд, J=12,0, 3,2 Гц), 3,85 (1H, дд, J=12,0, 4,6 Гц), 3,53 (2Н, м), 3,17 (2Н, м), 2,43 (2Н, м).
[0993]
(Этап 12)
2-[3,5-бис-O-(оксан-2-ил)-β-D-арабинофуранозил]-2,7,8,9-тетрагидро-6-тиа-2,3,5-триазабензо[cd]азулен
К раствору соединения (15,35 г), полученного на описанном выше этапе 11, в обезвоженном диметилсульфоксиде (160 мл) добавляли 3,4-дигидро-2Н-пиран (17,2 мл) и моногидрат п-толуолсульфоновой кислоты (9,02 г) при 0°С и реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Туда дополнительно добавляли 3,4-дигидро-2Н-пиран (8,6 мл) и реакционную смесь перемешивали в течение 45 минут, и незамедлительно после этого добавляли туда триэтиламин (13 мл), чтобы погасить реакцию. Реакционную смесь вливали в двухслойную смесь этилацетата и насыщенного водного раствора гидрокарбоната натрия и осуществляли экстрагирование этилацетатом. Органический слой однократно промывали водой и дважды промывали рассолом, а затем сушили над безводным сульфатом натрия. Осушающий агент удаляли путем фильтрации и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле [гексан/этилацетат] с получением указанного в заголовке соединения (10,81 г) в виде смеси четырех диастереомеров.
МС (ИЭР) m/z: 492[М+Н]+.
1Н-ЯМР (CDCl3) δ: 8,548 (0,2Н, с), 8,546 (0,3Н, с), 8,54 (0,3Н, с), 8,53 (0,2Н, с), 7,54 (0,2Н, с), 7,53 (0,3Н, с), 7,51 (0,2Н, с), 7,44 (0,3Н, с), 6,75 (0,2Н, д, J=5,4 Гц), 6,71 (0,2Н, д, J=5,9 Гц), 6,57 (0,3Н, д, J=5,9 Гц), 6,56 (0,3Н, д, J=5,9 Гц), 4,87-4,69 (2Н, м), 4,55-3,54 (10Н, м), 3,18-3,12 (2Н, м), 3,10-2,96 (2Н, м), 2,40-2,30 (2Н, м), 1,92-1,51 (12Н, м).
[0994]
(Этап 13)
2-[2-дезокси-2-фтор-3,5-бис-O-(оксан-2-ил)-β-D-рибофуранозил]-2,7,8,9-тетрагидро-6-тиа-2,3,5-триазабензо[cd]азулен
К раствору соединения (10,81 г), полученного на описанном выше этапе 12, в обезвоженном дихлорметане (150 мл) добавляли пиридин (5,3 мл) и трифторметансульфоновый ангидрид (5,6 мл) в атмосфере азота при 0°С, и реакционную смесь перемешивали при той же температуре в течение 1 часа. После добавления ледяной стружки в реакционную смесь, чтобы погасить реакцию, реакционную смесь вливали в двухслойную смесь этилацетата и насыщенного водного раствора гидрокарбоната натрия, и осуществляли экстрагирование этилацетатом. Органический слой промывали дважды рассолом и сушили над безводным сульфатом натрия. Осушающий агент удаляли путем фильтрации и фильтрат концентрировали при пониженном давлении с получением неочищенной формы трифлата в аморфной форме. Полученную неочищенную форму трифлата растворяли в обезвоженном тетрагидрофуране (150 мл), в который добавляли малыми частями раствор в тетрагидрофуране фторида тетрабутиламмония (приблизительно 1 М, 154 мл) при охлаждении льдом, и реакционную смесь перемешивали при той же температуре в течение ночи. Насыщенный водный раствор хлорида аммония добавляли в реакционную смесь, чтобы погасить реакцию. Реакционную смесь концентрировали до приблизительно половины объема при пониженном давлении. Остаток вливали в двухслойную смесь этилацетата и насыщенного водного раствора хлорида аммония и осуществляли экстрагирование этилацетатом. Органический слой однократно промывали насыщенным водным раствором хлорида аммония и дважды промывали рассолом. Водный слой снова осуществляли экстрагирование этилацетатом и экстракт промывали рассолом. Органические слои объединяли и сушили над безводным сульфатом натрия. Осушающий агент удаляли путем фильтрации и фильтрат концентрировали при пониженном давлении с получением неочищенной формы указанного в заголовке соединения (40,37 г).
МС (ИЭР) m/z: 494[М+Н]+.
[0995]
(Этап 14)
2-(2-дезокси-2-фтор-β-D-рибофуранозил)-2,7,8,9-тетрагидро-6-тиа-2,3,5-триазабензо[cd]азулен
К раствору соединения (40,37 г), полученного на описанном выше этапе 13, в метаноле (400 мл) добавляли моногидрат п-толуолсульфоновой кислоты (2,09 г), и реакционную смесь перемешивали при 60°С в течение 4 часов. Триэтиламин (16 мл) добавляли в реакционную смесь, чтобы погасить реакцию. Реакционную смесь концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле [гексан/этилацетат → этилацетат/метанол]. Фракции, содержащие целевой продукт, концентрировали при пониженном давлении до тех пор, пока фракции не стали взвесью, и твердый продукт собирали путем фильтрации. Полученный твердый продукт промывали гексаном/этилацетатом (1:1) с получением твердого продукта 1. Фильтрат концентрировали при пониженном давлении, и остаток очищали с помощью колоночной хроматографии на силикагеле [дихлорметан/метанол] с получением твердого продукта 2. Твердый продукт 1 и твердый продукт 2 объединяли с получением указанного в заголовке соединения (5,32 г).
МС (ИЭР) m/z: 326[М+Н]+.
1Н-ЯМР (CDCl3) δ: 8,51 (1H, с), 7,01 (1H, с), 6,00 (1Н, дд, J=13,7, 6,3 Гц), 5,95 (1H, дд, J=11,7, 2,0 Гц), 5,87 (1H, ддд, J=52,7, 6,3, 4,9 Гц), 4,69 (1Н, м), 4,32 (1H, уш. с), 3,96 (1H, д, J=12,7 Гц), 3,77 (1Н, м), 3,17 (2Н, м), 3,04 (2Н, м), 2,41-2,31 (3Н, м).
[0996]
(Этап 15)
2-{5-O-[бис(4-метоксифенил)(фенил)метил]-2-дезокси-2-фтор-β-D-рибофуранозил}-2,7,8,9-тетрагидро-6-тиа-2,3,5-триазабензо[cd]азулен
С применением соединения (5,32 г), полученного на описанном выше этапе 14, проводили реакцию таким же образом, как на этапе 1 примера 11, с получением указанного в заголовке соединения (10,1 г).
МС (ИЭР) m/z: 628[М+Н]+.
1Н-ЯМР (CDCl3) δ: 8,54 (1H, с), 7,42 (2Н, д, J=7,3 Гц), 7,32-7,21 (8Н, м), 6,81 (4Н, м), 6,52 (1H, дд, J=17,3, 2,2 Гц), 5,37 (1H, ддд, J=53,3, 4,4, 2,4 Гц), 4,76 (1H, м), 4,16 (1H, м), 3,789 (3Н, с), 3,786 (3Н, с), 3,59 (1Н, дд, J=10,7, 2,4 Гц), 3,44 (1Н, дд, J=10,7, 3,4 Гц), 3,12 (2Н, м), 2,76 (2Н, т, J=5,6 Гц), 2,27 (2Н, м), 2,18 (1Н, дд, J=7,8, 2,9 Гц).
[0997]
(Этап 16)
2-(5-O-[бис(4-метоксифенил)(фенил)метил]-3-O-{(2-цианоэтокси)[ди(пропан-2-ил)амино]фосфанил}-2-дезокси-2-фтор-β-D-рибофуранозил)-2,7,8,9-тетрагидро-6-тиа-2,3,5-триазабензо[cd]азулен
С применением соединения (10,1 г), полученного на описанном выше этапе 15, проводили реакцию таким же образом, как на этапе 6 примера 1, с получением указанного в заголовке соединения (12,6 г) в виде смеси диастереомеров у атома фосфора (соотношение диастереомеров=1:1).
1H-ЯМР (CDCl3) δ: 8,53 (1Н, с), 7,40 (2Н, м), 7,34-7,17 (8Н, м), 6,84-6,74 (4Н, м), 6,53 (0,5Н, дд, J=17,3, 2,2 Гц), 6,48 (0,5Н, дд, J=17,6, 1,5 Гц), 5,50-5,31 (1Н, м), 4,99 (0,5Н, м), 4,85 (0,5Н, м), 4,31-4,26 (1H, м), 3,93-3,76 (1H, м), 3,792 (1,5Н, с), 3,789 (1,5Н, с), 3,779 (1,5Н, с), 3,776 (1,5Н, с), 3,67-3,51 (4Н, м), 3,34-3,30 (1Н, м), 3,13-3,10 (2Н, м), 2,76-2,69 (2Н, м), 2,61 (1H, тд, J=6,3, 2,4 Гц), 2,39 (1H, м), 2,28-2,21 (2Н, м), 1,19-1,15 (9Н, м), 1,03 (3Н, д, J=6,8 Гц).
[0998]
(Этап 17)
С применением соединения (740 мг), полученного на описанном выше этапе 16, проводили реакцию таким же образом, как на этапе 7 примера 1, с получением раствора в ацетонитриле 2-{2-дезокси-2-фтор-3-O-[гидрокси(оксо)-λ5-фосфанил]-β-D-рибофуранозил}-2,7,8,9-тетрагидро-6-тиа-2,3,5-триазабензо[cd]азулена. С применением полученного раствора в ацетонитриле и полученного на этапе 3 примера 22 соединения (924 мг), проводили реакцию таким же образом, как на этапе 8 примера 1, и полученный в результате этого неочищенный продукт непосредственно использовали для следующей реакции.
[0999]
(Этап 18)
2-{9-[(5R,7R,8R,12aR,14R,15R,15aR,16R)-16-{[трет-бутил(диметил)силил]окси}-10-(2-цианоэтокси)-14-(8,9-дигидро-6-тиа-2,3,5-триазабензо[cd]азулен-2(7Н)-ил)-15-фтор-2-оксо-2-сульфанил-10-сульфанилиденоктагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-7-ил]-6-оксо-6,9-дигидро-1Н-пурин-1-ил}этилбензоат
С применением неочищенного продукта, полученного на описанном выше этапе 17, проводили реакцию таким же образом, как на этапе 9 примера 1, с получением указанного в заголовке соединения (502 мг: с примесями) в виде смеси диастереомеров у атома фосфора.
МС (ИЭР) m/z: 1065(М+Н)+.
[1000]
(Этап 19)
Бис(N,N-диэтилэтанаминия) (5R,7R,8R,12aR,14R,15R,15aR,16R)-16-{[трет-бутил(диметил)силил]окси}-14-(8,9-дигидро-6-тиа-2,3,5-триазабензо[cd]азулен-2(7Н)-ил)-15-фтор-7-[1-(2-гидроксиэтил)-6-оксо-1,6-дигидро-9Н-пурин-9-ил]-2,10-диоксооктагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
С применением соединения (502 мг), полученного на описанном выше этапе 18, проводили реакцию таким же образом, как на этапе 10 примера 1, с получением диастереомера 1 (67,8 мг: с примесями) и диастереомера 2 (69,5 мг: с примесями) указанного в заголовке соединения. Диастереомер 1 (менее полярный)
МС (ИЭР) m/z: 908(М+Н)+.
Диастереомер 2 (более полярный)
МС (ИЭР) m/z: 908(М+Н)+.
[1001]
(Этап 20-1)
Динатрия (5R,7R,8R,12aR,14R,15R,15aR,16R)-14-(8,9-дигидро-6-тиа-2,3,5-триазабензо[cd]азулен-2(7Н)-ил)-15-фтор-16-гидрокси-7-[1-(2-гидроксиэтил)-6-оксо-1,6-дигидро-9H-пурин-9-ил]-2,10-диоксооктагидро-2H,10H,12H-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
(Диастереомер 1)
С применением соединения, полученного на этапе 19 выше (диастереомер 1) (67,8 мг: с примесями), проводили реакцию таким же образом, как на этапе 11 примера 1, а затем проводили очистку при следующих [Условиях очистки] с получением указанного в заголовке соединения в виде соли триэтиламина.
[Условия очистки] Хроматография на колонке с силикагелем С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил], препаративная ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/раствор ацетонитрила-метанола (1:1), раствор ацетонитрила-метанола (1:1): 10% - 45% (0 мин - 40 мин)] и препаративная ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/метанол, метанол: 25% - 75% (0 мин - 40 мин)].
Полученную соль триэтиламина подвергали солевому обмену таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением указанного в заголовке соединения (21,2 мг).
МС (ИЭР) m/z: 794(М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,55 (1Н, уш. с), 8,41 (1Н, с), 8,09 (1Н, уш. с), 7,51 (1Н, с), 6,58 (1Н, д, J=16,9 Гц), 6,25 (1Н, д, J=7,9 Гц), 5,55-5,34 (2Н, м), 5,30-5,17 (1Н, м), 4,74 (1Н, д, J=4,2 Гц), 4,55-4,47 (1Н, м), 4,46-4,40 (1Н, м), 4,37-4,30 (2Н, м), 4,28-4,16 (2Н, м), 4,05-3,99 (1Н, м), 3,90-3,70 (3Н, м), 3,28-3,20 (1Н, м), 3,18-3,10 (1Н, м), 2,91-2,82 (1Н, м), 2,76-2,64 (1Н, м), 2,33-2,22 (1Н, м), 2,21-2,09 (1Н, м).
31Р-ЯМР (CD3OD) δ: 57,6 (с), 52,7 (с).
[1002]
(Этап 20-2)
Динатрия (5R,7R,8R,12aR,14R,15R,15aR,16R)-14-(8,9-дигидро-6-тиа-2,3,5-триазабензо[cd]азулен-2(7Н)-ил)-15-фтор-16-гидрокси-7-[1-(2-гидроксиэтил)-6-оксо-1,6-дигидро-9Н-пурин-9-ил]-2,10-диоксооктагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
(Диастереомер 2)
С применением соединения, полученного на этапе 19 выше (диастереомер 2) (69,5 мг: с примесями), проводили реакцию таким же образом, как на этапе 11 примера 1, а затем проводили очистку при следующих [Условиях очистки] с получением указанного в заголовке соединения в виде соли триэтиламина.
[Условия очистки] Хроматография на колонке с силикагелем С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил], препаративная ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/раствор ацетонитрила-метанола (1:1), раствор ацетонитрила-метанола (1:1): 10% - 45% (0 мин - 40 мин)], и препаративная ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/метанол, метанол: 25% - 75% (0 мин - 40 мин)].
Полученную соль триэтиламина подвергали солевому обмену таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением указанного в заголовке соединения (14,9 мг).
МС (ИЭР) m/z: 794(М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,60 (1H, с), 8,41 (1H, с), 8,16 (1H, с), 7,72 (1H, с), 6,60 (1H, д, J=15,7 Гц), 6,28 (1H, д, J=7,9 Гц), 5,61-5,33 (3Н, м), 4,59-4,49 (2Н, м), 4,48-4,39 (2Н, м), 4,34-4,27 (1H, м), 4,25-4,16 (1H, м), 4,11-3,99 (3Н, м), 3,86-3,75 (2Н, м), 3,25-3,11 (2Н, м), 3,05-2,90 (2Н, м), 2,35-2,17 (2Н, м).
31Р-ЯМР (CD3OD) δ: 59,1 (с), 57,9 (с).
[1003]
Пример 59. Синтез ЦДН49.
(5R,7R,8R,12aR,14R,15R,15aR,16R)-7-[1-(2-аминоэтил)-6-оксо-1,6-дигидро-9Н-пурин-9-ил]-14-(8,9-дигидро-6-тиа-2,3,5-триазабензо[cd]азулен-2(7Н)-ил)-15-фтор-16-гидрокси-2,10-бис(сульфанил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-дион
[1004]
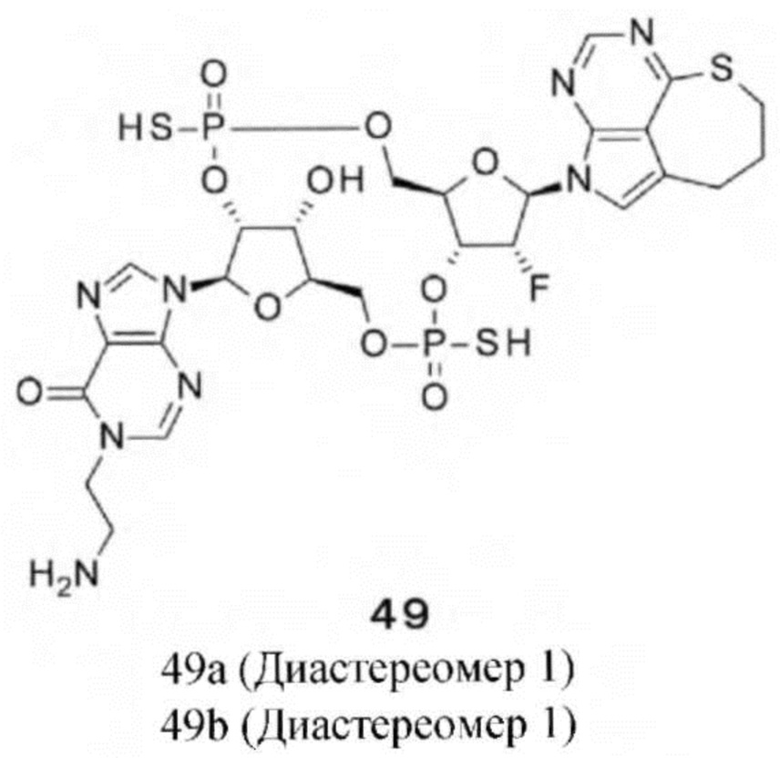
[1005]
[Схема синтеза]
[1006]
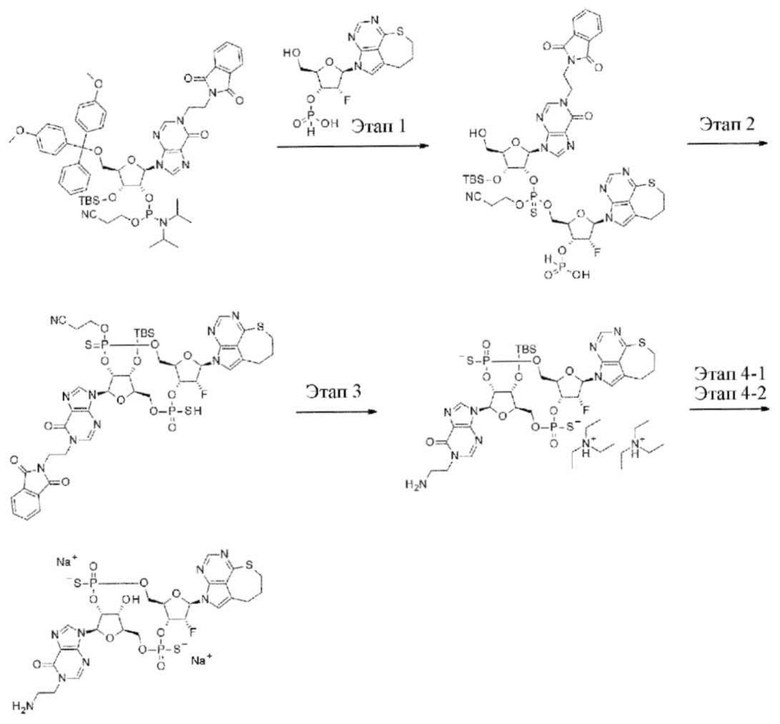
[1007]
(Этап 1)
С применением соединения, полученного на этапе 16 примера 58 (1,80 г), проводили реакцию таким же образом, как на этапе 7 примера 1, с получением раствора в ацетонитриле 2-{2-дезокси-2-фтор-3-O-[гидрокси(оксо)-λ5-фосфанил]-β-D-рибофуранозил}-2,7,8,9-тетрагидро-6-тиа-2,3,5-триазабензо[cd]азулена. С применением полученного раствора в ацетонитриле и полученного на этапе 3 примера 45 соединения (2,30 г) проводили реакцию таким же образом, как на этапе 8 примера 1, и полученный в результате этого неочищенный продукт непосредственно использовали для следующей реакции.
[1008]
(Этап 2)
3-{[(5R,7R,8R,12aR,14R,15R,15aR,16R)-16-{[трет-бутил(диметил)силил]окси}-14-(8,9-дигидро-6-тиа-2,3,5-триазабензо[cd]азулен-2(7Н)-ил)-7-{1-[2-(1,3-диоксо-1,3-дигидро-2Н-изоиндол-2-ил)этил]-6-оксо-1,6-дигидро-9Н-пурин-9-ил}-15-фтор-2-оксо-2-сульфанил-10-сульфанилиденоктагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-10-ил]окси}пропаннитрил
С применением неочищенного продукта, полученного на этапе 1 выше, проводили реакцию таким же образом, как на этапе 9 примера 1, с получением указанного в заголовке соединения (1,22 г) в виде смеси диастереомеров у атома фосфора.
МС (ИЭР) m/z: 1090(М+Н) +.
[1009]
(Этап 3)
Бис(N,N-диэтилэтанаминия) (5R,7R,8R,12aR,14R,15R,15aR,16R)-7-[1-(2-аминоэтил)-6-оксо-1,6-дигидро-9Н-пурин-9-ил]-16-{[трет-бутил(диметил)силил]окси}-14-(8,9-дигидро-6-тиа-2,3,5-триазабензо[cd]азулен-2(7Н)-ил)-15-фтор-2,10-диоксооктагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
С применением соединения, полученного на этапе 2 выше (1,22 г), проводили реакцию таким же образом, как на этапе 6 примера 45, и продукт затем очищали с помощью колоночной хроматографии на силикагеле С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил] с получением диастереомера 1 (108 мг: с примесями) и диастереомера 2(111 мг: с примесями) указанного в заголовке соединения.
Диастереомер 1 (менее полярный)
МС (ИЭР) m/z: 907(М+Н)+.
Диастереомер 2 (более полярный)
МС (ИЭР) m/z: 907(М+Н)+.
[1010]
(Этап 4-1)
Динатрия (5R,7R,8R,12aR,14R,15R,15aR,16R)-7-[1-(2-аминоэтил)-6-оксо-1,6-дигидро-9Н-пурин-9-ил]-14-(8,9-дигидро-6-тиа-2,3,5-триазабензо[cd]азулен-2(7Н)-ил)-15-фтор-16-гидрокси-2,10-диоксооктагидро-2H,10H,12H-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
(Диастереомер 1)
С применением соединения, полученного на этапе 3 выше (диастереомер 1) (108 мг: с примесями), проводили реакцию таким же образом, как на этапе 11 примера 1, а затем проводили очистку при следующих [Условиях очистки] с получением указанного в заголовке соединения в виде соли триэтиламина.
[Условия очистки] Хроматография на колонке с силикагелем С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил] и препаративная ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/раствор ацетонитрила-метанола (1:1), раствор ацетонитрила-метанола (1:1): 10% - 50% (0 мин - 40 мин)].
Полученную соль триэтиламина подвергали солевому обмену таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением указанного в заголовке соединения (44,4 мг).
МС (ИЭР) m/z: 793(М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,50 (1Н, с), 8,42 (1Н, с), 7,92 (1Н, с), 7,56 (1Н, с), 6,56 (1Н, д, J=16,3 Гц), 6,21 (1Н, д, J=6,0 Гц), 5,57-5,40 (2Н, м), 5,35-5,22 (1Н, м), 4,73-4,67 (1Н, м), 4,58-4,49 (1Н, м), 4,45-4,26 (4Н, м), 4,24-4,15 (1Н, м), 4,05-3,96 (1Н, м), 3,78-3,51 (1Н, м), 3,26-3,06 (4Н, м), 2,93-2,82 (1Н, м), 2,70-2,51 (1Н, м), 2,29-2,07 (2Н, м).
31Р-ЯМР (CD3OD) δ: 57,5 (с), 52,9 (с).
[1011]
(Этап 4-2)
Динатрия (5R,7R,8R,12aR,14R,15R,15aR,16R)-7-[1-(2-аминоэтил)-6-оксо-1,6-дигидро-9Н-пурин-9-ил]-14-(8,9-дигидро-6-тиа-2,3,5-триазабензо[cd]азулен-2(7Н)-ил)-15-фтор-16-гидрокси-2,10-диоксооктагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
(Диастереомер 2)
С применением С применением соединения, полученного на этапе 3 (диастереомер 2) (111 мг: с примесями), проводили реакцию таким же образом, как на этапе 11 примера 1, а затем проводили очистку при следующих [Условиях очистки] с получением указанного в заголовке соединения в виде соли триэтиламина.
[Условия очистки] Хроматография на колонке с силикагелем С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил], препаративная ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 5% - 25% (0 мин - 40 мин)] и препаративная ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/метанол, метанол: 20% - 60% (0 мин - 40 мин)].
Полученную соль триэтиламина подвергали солевому обмену таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением указанного в заголовке соединения (40,6 мг).
МС (ИЭР) m/z: 793(М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,57 (1Н, с), 8,41 (1Н, с), 8,13 (1Н, с), 7,72 (1Н, с), 6,59 (1Н, дд, J=15,7, 1,8 Гц), 6,26 (1H, д, J=8,5 Гц), 5,61-5,34 (3Н, м), 4,57-4,48 (2Н, м), 4,48-4,38 (2Н, м), 4,38-4,28 (2Н, м), 4,08-3,98 (3Н, м), 3,29-3,21 (2Н, м), 3,20-3,12 (2Н, м), 3,02-2,92 (1H, м), 2,92-2,81 (1H, м), 2,29-2,15 (2Н, м).
31Р-ЯМР (CD3OD) δ: 58,7 (с), 57,8 (с).
[1012]
Пример 60. Синтез ЦДН50.
N-(2-{9-[(5R,7R,8R,12aR,14R,15R,15aR,16R)-14-(8,9-дигидро-6-тиа-2,3,5-триазабензо[cd]азулен-2(7Н)-ил)-15-фтор-16-гидрокси-2,10-диоксо-2,10-бис(сульфанил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-7-ил]-6-оксо-6,9-дигидро-1Н-пурин-1-ил}этил)-2-гидроксиацетамид
[1013]
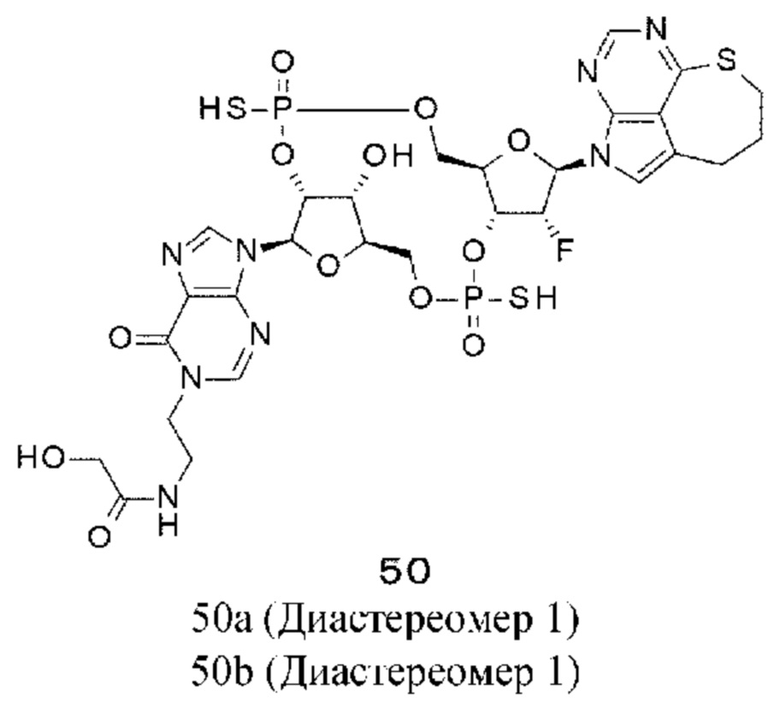
[1014]
[Схема синтеза]
[1015]
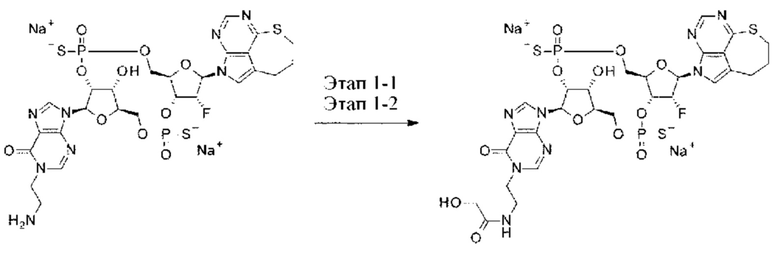
[1016]
(Этап 1-1)
Динатрия (5R,7R,8R,12aR,14R,15R,15aR,16R)-14-(8,9-дигидро-6-тиа-2,3,5-триазабензо[cd]азулен-2(7Н)-ил)-15-фтор-16-гидрокси-7-{1-[2-(2-гидроксиацетамид)этил]-6-оксо-1,6-дигидро-9Н-пурин-9-ил}-2,10-диоксооктагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
(Диастереомер 1)
С применением соединения, полученного на этапе 4-1 примера 59 (20,0 мг), проводили реакцию таким же образом, как на этапе 1-1 примера 7, а затем проводили очистку при следующих [Условиях очистки] с получением указанного в заголовке соединения в виде соли триэтиламина.
[Условия очистки] Хроматография на колонке с силикагелем С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил] и препаративная ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 10% - 30% (0 мин - 40 мин)].
Полученную соль триэтиламина подвергали солевому обмену таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением указанного в заголовке соединения (15,6 мг).
МС (ИЭР) m/z: 851(М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,43 (1Н, с), 8,40 (1Н, уш. с), 7,66 (1Н, уш. с), 7,58 (1Н, с), 6,53 (1Н, д, J=16,3 Гц), 6,14 (1Н, д, J=8,5 Гц), 5,73-5,64 (1Н, м), 5,59-5,42 (1Н, м), 5,42-5,29 (1Н, м), 4,80-4,74 (1Н, м), 4,53-4,26 (5Н, м), 4,21-4,12 (1Н, м), 3,99-3,92 (1Н, м), 3,83 (2Н, с), 3,66-3,56 (1Н, м), 3,43-3,26 (2Н, м), 3,23-3,06 (2Н, м), 2,89-2,79 (1Н, м), 2,49-2,33 (1Н, м), 2,27-2,15 (1Н, м), 2,15-2,02 (1Н, м).
31Р-ЯМР (CD3OD) δ: 57,0 (с), 52,6 (с).
[1017]
(Этап 1-2)
Динатрия (5R,7R,8R,12aR,14R,15R,15aR,16R)-14-(8,9-дигидро-6-тиа-2,3,5-триазабензо[cd]азулен-2(7Н)-ил)-15-фтор-16-гидрокси-7-{1-[2-(2-гидроксиацетамид)этил]-6-оксо-1,6-дигидро-9Н-пурин-9-ил}-2,10-диоксооктагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
(Диастереомер 2)
С применением соединения, полученного на этапе 4-2 примера 59 (10,0 мг), проводили реакцию таким же образом, как на этапе 1-1 примера 7, а затем проводили очистку при следующих [Условиях очистки] с получением указанного в заголовке соединения в виде соли триэтиламина.
[Условия очистки] Хроматография на колонке с силикагелем С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил] и препаративная ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 7% - 25% (0 мин - 40 мин)].
Полученную соль триэтиламина подвергали солевому обмену таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением указанного в заголовке соединения (6,6 мг).
МС (ИЭР) m/z: 851(М+Н)+.
1H-ЯМР (CD3OD) δ: 8,46 (1H, с), 8,42 (1H, с), 7,84 (1H, с), 7,78 (1H, с), 6,59 (1H, д, J=15,1 Гц), 6,20 (1H, д, J=7,9 Гц), 5,69-5,38 (3Н, м), 4,60-4,50 (2Н, м), 4,48-4,38 (2Н, м), 4,31-4,20 (2Н, м), 4,10-3,93 (2Н, м), 3,87 (2Н, с), 3,73-3,57 (2Н, м), 3,52-3,41 (1H, м), 3,25-3,10 (2Н, м), 3,01-2,90 (1H, м), 2,83-2,71 (1H, м), 2,30-2,11 (2Н, м).
31Р-ЯМР (CD3OD) δ: 58,2 (с), 57,6 (с).
[1018]
Пример 61. Синтез ЦДН51.
(5R,7R,8R,12aR,14R,15R,15aR,16R)-7-{6-амино-2-[(2-аминоэтил)амино]-9Н-пурин-9-ил}-14-(8,9-дигидро-6-тиа-2,3,5-триазабензо[cd]азулен-2(7Н)-ил)-15-фтор-16-гидрокси-2,10-бис(сульфанил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-дион
[1019]
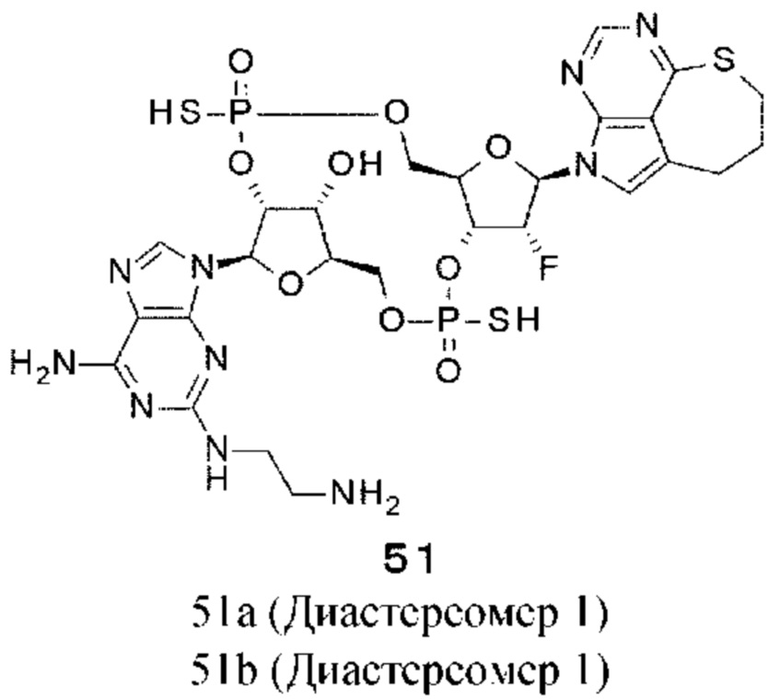
[1020]
[Схема синтеза]
[1021]
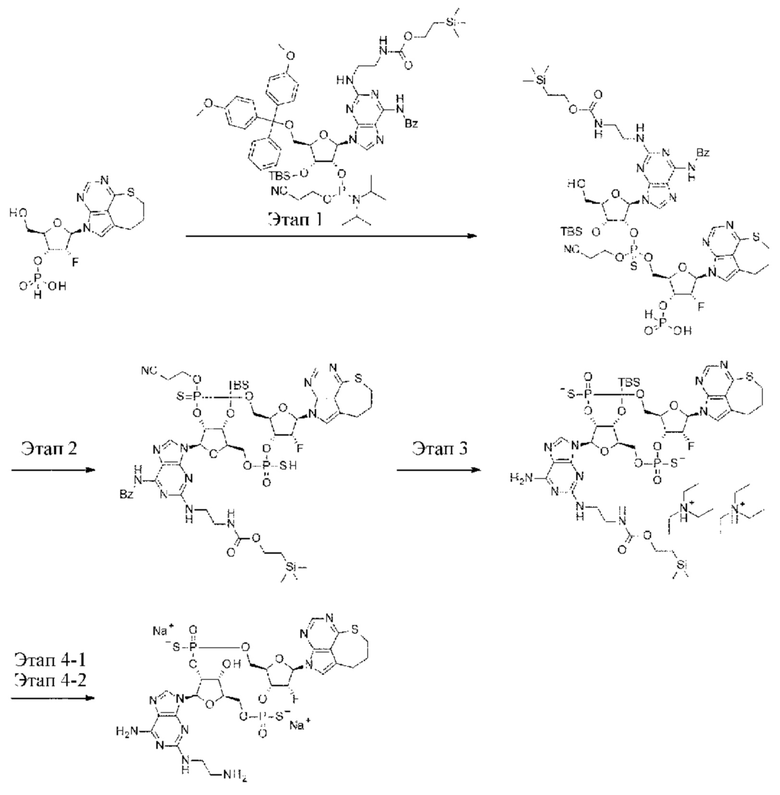
[1022]
(Этап 1)
С применением соединения, полученного на этапе 16 примера 58 (1,80 г), проводили реакцию таким же образом, как на этапе 7 примера 1, с получением раствора в ацетонитриле 2-{2-дезокси-2-фтор-3-O-[гидрокси(оксо)-λ5-фосфанил]-β-D-рибофуранозил}-2,7,8,9-тетрагидро-6-тиа-2,3,5-триазабензо[cd]азулена. С применением полученного раствора в ацетонитриле и полученного на этапе 6 примера 47 соединения (3,10 г) проводили реакцию таким же образом, как на этапе 8 примера 1, и полученный в результате этого неочищенный продукт непосредственно использовали для следующей реакции.
[1023]
(Этап 2)
2-(триметилсилил)этил[2-({6-бензамидо-9-[(5R,7R,8R,12aR,14R,15R,15aR,16R)-16-{[трет-бутил(диметил)силил]окси}-10-(2-цианоэтокси)-14-(8,9-дигидро-6-тиа-2,3,5-триазабензо[cd]азулен-2(7Н)-ил)-15-фтор-2-оксо-2-сульфанил-10-сульфанилиденоктагидро-2H,10H,12H-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-7-ил]-9Н-пурин-2-ил}амино)этил]карбамат
С применением неочищенного продукта, полученного на этапе 1 выше, проводили реакцию таким же образом, как на этапе 9 примера 1, с получением указанного в заголовке соединения (1,83 г: с примесями).
МС (ИЭР) m/z: 1222(М+Н)+.
[1024]
(Этап 3)
Бис(N,N-диэтилэтанаминия) (5R,7R,8R,12aR,14R,15R,15aR,16R)-7-(6-амино-2-{[2-({[2-(триметилсилил)этокси]карбонил}амино)этил]амино}-9Н-пурин-9-ил)-16-{[трет-бутил(диметил)силил]окси}-14-(8,9-дигидро-6-тиа-2,3,5-триазабензо[cd]азулен-2(7Н)-ил)-15-фтор-2,10-диоксооктагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
С применением смеси, полученной на этапе 2 выше (1,83 г), проводили реакцию таким же образом, как на этапе 10 примера 1, с получением диастереомера 1 (151 мг: с примесями) и диастереомера 2 (103 мг: с примесями) указанного в заголовке соединения.
Диастереомер 1 (менее полярный)
МС (ИЭР) m/z: 1065(М+Н)+.
Диастереомер 2 (более полярный)
МС (ИЭР) m/z: 1065(М+Н)+.
[1025]
(Этап 4-1)
Динатрия (5R,7R,8R,12aR,14R,15R,15aR,16R)-7-{6-амино-2-[(2-аминоэтил)амино]-9Н-пурин-9-ил}-14-(8,9-дигидро-6-тиа-2,3,5-триазабензо[cd]азулен-2(7Н)-ил)-15-фтор-16-гидрокси-2,10-диоксооктагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
(Диастереомер 1)
С применением соединения, полученного на этапе 3 выше (диастереомер 1) (151 мг: с примесями), проводили реакцию таким же образом, как на этапе 5 примера 40, а затем проводили очистку при следующих [Условиях очистки] с получением указанного в заголовке соединения в виде соли триэтиламина.
[Условия очистки] Хроматография на колонке с силикагелем С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил] и препаративная ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 2% - 30% (0 мин - 30 мин)].
Полученную соль триэтиламина подвергали солевому обмену таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением указанного в заголовке соединения (10,6 мг).
МС (ИЭР) m/z: 807(М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,39 (1H, с), 7,97 (1H, уш. с), 7,55 (1H, с), 6,51 (1Н, д, J=16,3 Гц), 6,00-5,92 (1H, м), 5,83-5,65 (1H, м), 5,44 (1H, дд, J=52,0, 3,6 Гц), 5,34-5,20 (1H, м), 4,77 (1H, д, J=3,6 Гц), 4,49-4,31 (5Н, м), 4,11-4,07 (1H, м), 3,28-2,72 (8Н, м), 2,29-1,99 (2Н, м).
31Р-ЯМР (CD3OD) δ: 57,3 (с), 52,3 (с).
[1026]
(Этап 4-2)
Динатрия (5R,7R,8R,12aR,14R,15R,15aR,16R)-7-{6-амино-2-[(2-аминоэтил)амино]-9Н-пурин-9-ил}-14-(8,9-дигидро-6-тиа-2,3,5-триазабензо[cd]азулен-2(7Н)-ил)-15-фтор-16-гидрокси-2,10-диоксооктагидро-2H,10H,12H-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
(Диастереомер 2)
С применением соединения, полученного на этапе 3 выше (диастереомер 2) (103 мг: с примесями), проводили реакцию таким же образом, как на этапе 5 примера 40, а затем проводили очистку при следующих [Условиях очистки] с получением указанного в заголовке соединения в виде соли триэтиламина.
[Условия очистки] Хроматография на колонке с силикагелем С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил] и препаративная ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 10% - 30% (0 мин - 30 мин)].
Полученную соль триэтиламина подвергали солевому обмену таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением указанного в заголовке соединения (12,1 мг).
МС (ИЭР) m/z: 807(М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,39 (1Н, с), 7,95 (1Н, уш. с), 7,82 (1Н, с), 6,58 (1Н, д, J=15,l Гц), 6,00-5,95 (1Н, м), 5,90-5,71 (1Н, м), 5,40 (1Н, дд, J=51,7, 3,3 Гц), 5,38-5,25 (1Н, м), 4,53-4,39 (4Н, м), 4,28-4,18 (2Н, м), 4,10-4,05 (1Н, м), 3,27-2,54 (8Н, м), 2,34-2,10 (2Н, м).
31Р-ЯМР (CD3OD) δ: 57,9 (с), 57,3 (с).
[1027]
Пример 62. Синтез ЦДН52.
(5R,7R,8R,12aR,14R,15R,15aR,16R)-15-фтор-2,16-дигидрокси-7-[1-(2-гидроксиэтил)-6-оксо-1,6-дигидро-9Н-пурин-9-ил]-10-сульфанил-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-дион
[1028]
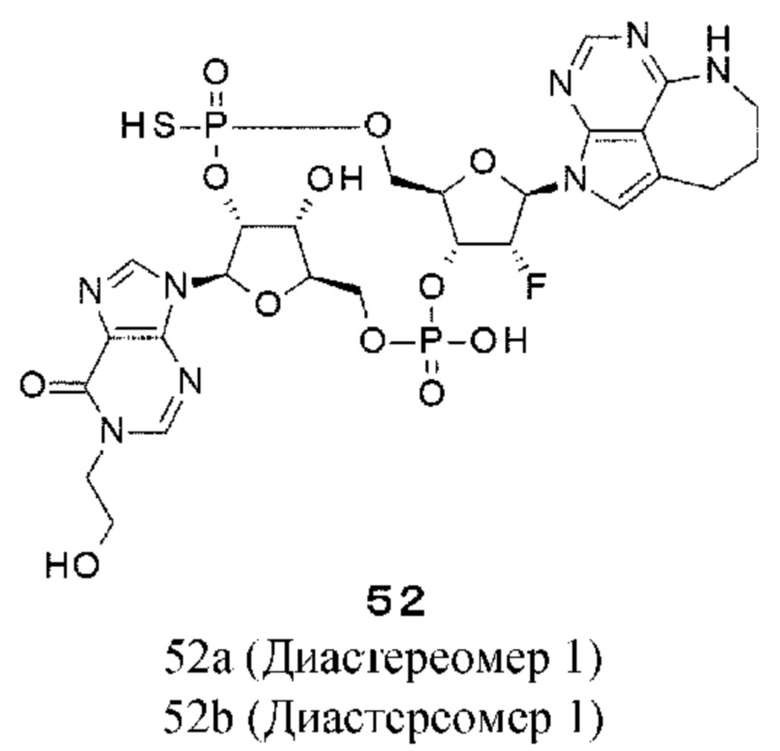
[1029]
[Схема синтеза]
[1030]
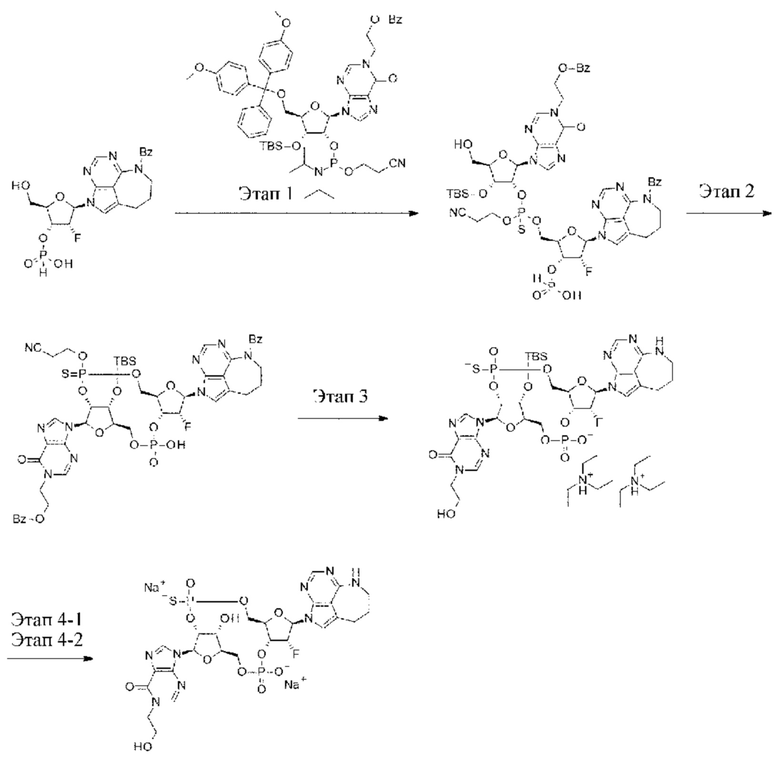
[1031]
(Этап 1)
С применением соединения, полученного на этапе 8 примера 44 (1,00 г), проводили реакцию таким же образом, как на этапе 7 примера 1, с получением раствора в ацетонитриле 6-бензоил-2-{2-дезокси-2-фтор-3-0-[гидрокси(оксо)-λ5,-фосфанил]-β-D-рибофуранозил}-6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулена. С применением полученного раствора в ацетонитриле и полученного на этапе 3 примера 22 соединения (1,13 г), проводили реакцию таким же образом, как на этапе 8 примера 1, и полученный в результате этого неочищенный продукт непосредственно использовали для следующей реакции.
[1032]
(Этап 2)
2-{9-[(5R,7R,8R,12aR,14R,15R,15aR,16R)-14-(6-бензоил-6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)-16-{[трет-бутил(диметил)силил]окси}-10-(2-цианоэтокси)-15-фтор-2-гидрокси-2-оксо-10-сульфанилиденоктагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-7-ил]-6-оксо-6,9-дигидро-1Н-пурин-1-ил}этилбензоат
Раствор неочищенного продукта, полученного на этапе 1 выше, в пиридине (32,5 мл) концентрировали до приблизительно 25 мл, а затем добавляли туда 2-хлор-5,5-диметил-1,3,2λ5-диоксафосфинан-2-он (945 мг), и реакционную смесь перемешивали при комнатной температуре в течение 30 минут. Йод (1,11 г) добавляли в реакционную смесь, которую перемешивали в течение 1 часа. Реакционную смесь вливали в водный раствор (150 мл) гидрокарбоната натрия (4,30 г) и продукт перемешивали в течение 30 минут, а затем осуществляли экстрагирование этилацетатом. После высушивания органического слоя над безводным сульфатом натрия осушающий агент удаляли путем фильтрации и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле [гексан/этилацетат/метанол] с получением указанного в заголовке соединения (671 мг: с примесями) в виде смеси диастереомеров у атома фосфора.
МС (ИЭР) m/z: 1136(М+Н)+.
[1033]
(Этап 3)
Бис(N,N-диэтилэтанаминия) (5R,7R,8R,12aR,14R,15R,15aR,16R)-16-{[трет-бутил(диметил)силил]окси}-15-фтор-7-[1-(2-гидроксиэтил)-6-оксо-1,6-дигидро-9Н-пурин-9-ил]-2,10-диоксо-10-сульфид-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2-олат
С применением соединения, полученного на этапе 2 выше (671 мг), проводили реакцию таким же образом, как на этапе 10 примера 1, с получением диастереомера 1 (55,6 мг: с примесями) и диастереомера 2 (65,7 мг: с примесями) указанного в заголовке соединения.
Диастереомер 1 (менее полярный)
МС (ИЭР) m/z: 875(М+Н)+.
Диастереомер 2 (более полярный)
МС (ИЭР) m/z: 875(М+Н)+.
[1034]
(Этап 4-1)
Динатрия (5R,7R,8R,12aR,14R,15R,15aR,16R)-15-фтор-16-гидрокси-7-[1-(2-гидроксиэтил)-6-оксо-1,6-дигидро-9Н-пурин-9-ил]-2,10-диоксо-10-сульфид-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2-олат
(Диастереомер 1)
С применением соединения, полученного на этапе 3 выше (диастереомер 1) (55,6 мг: с примесями), проводили реакцию таким же образом, как на этапе 11 примера 1, а затем проводили очистку при следующих [Условиях очистки] с получением указанного в заголовке соединения в виде соли триэтиламина.
[Условия очистки] Хроматография на колонке с силикагелем С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил] и препаративная ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 5% - 25% (0 мин - 30 мин)].
Полученную соль триэтиламина подвергали солевому обмену таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением указанного в заголовке соединения (11,3 мг).
МС (ИЭР) m/z: 761(М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,55 (1H, м), 8,15 (1H, м), 8,03 (1H, с), 7,14 (1H, д, J=4,8 Гц), 6,46 (1H, д, J=18,1 Гц), 6,28 (1H, д, J=7,9 Гц), 5,50-5,29 (2Н, м), 5,16-5,04 (1H, м), 4,74-4,69 (1H, м), 4,40-4,18 (6Н, м), 4,13-4,07 (1H, м), 4,02-3,91 (1H, м), 3,84-3,74 (2Н, м), 3,53-3,43 (2Н, м), 2,81-2,63 (2Н, м), 2,02-1,85 (2Н, м).
31Р-ЯМР (CD3OD) δ: 53,4 (с), -0,86 (с).
[1035]
(Этап 4-2)
Динатрия (5R,7R,8R,12aR,14R,15R,15aR,16R)-15-фтор-16-гидрокси-7-[1-(2-гидроксиэтил)-6-оксо-1,6-дигидро-9Н-пурин-9-ил]-2,10-диоксо-10-сульфид-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2-олат
(Диастереомер 2)
С применением соединения, полученного на этапе 3 выше (диастереомер 2) (65,7 мг: с примесями), проводили реакцию таким же образом, как на этапе 11 примера 1, а затем проводили очистку при следующих [Условиях очистки] с получением указанного в заголовке соединения в виде соли триэтиламина.
[Условия очистки] Хроматография на колонке с силикагелем С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил] и препаративная ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 3% - 20% (0 мин - 30 мин)].
Полученную соль триэтиламина подвергали солевому обмену таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением указанного в заголовке соединения (23,5 мг).
МС (ИЭР) m/z: 761(М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,58 (1H, д, J=3,0 Гц), 8,19 (1Н, д, J=2,4 Гц), 8,03 (1H, с), 7,40 (1H, с), 6,49 (1H, дд, J=16,3, 1,8 Гц), 6,29 (1H, д, J=8,5 Гц), 5,51-5,22 (3Н, м), 4,59-4,55 (1H, м), 4,43-4,17 (5Н, м), 4,14-4,01 (3Н, м), 3,85-3,78 (2Н, м), 3,51-3,44 (2Н, м), 2,90-2,75 (2Н, м), 1,99-1,90 (2Н, м).
31Р-ЯМР (CD3OD) δ: 59,7 (с), -0,75 (с).
[1036]
Пример 63. Синтез ЦДН53.
(5R,7R,8R,12aR,14R,15R,15aR,16R)-15-фтор-10,16-дигидрокси-7-[1-(2-гидроксиэтил)-6-оксо-1,6-дигидро-9Н-пурин-9-ил]-2-сульфанил-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-дион
[1037]
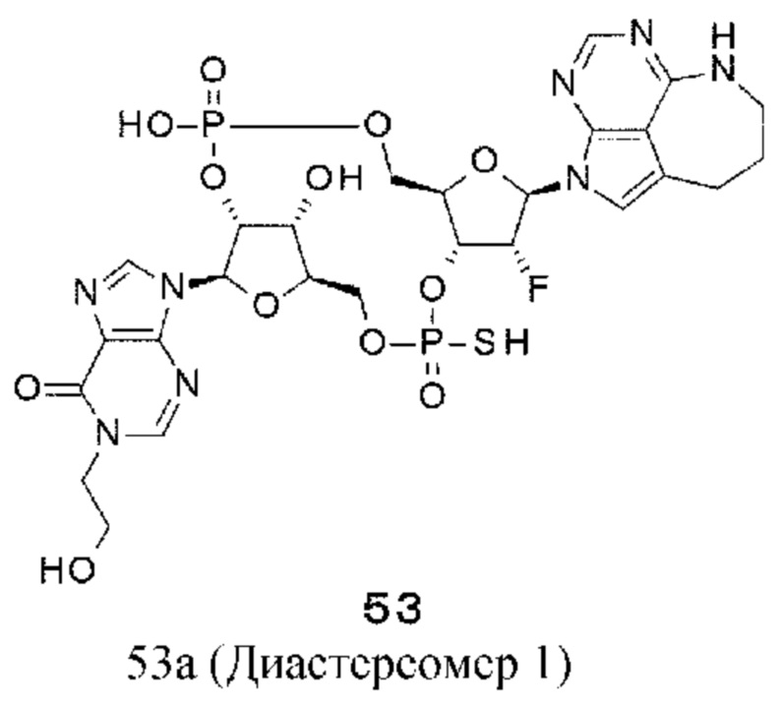
[1038]
[Схема синтеза]
[1039]
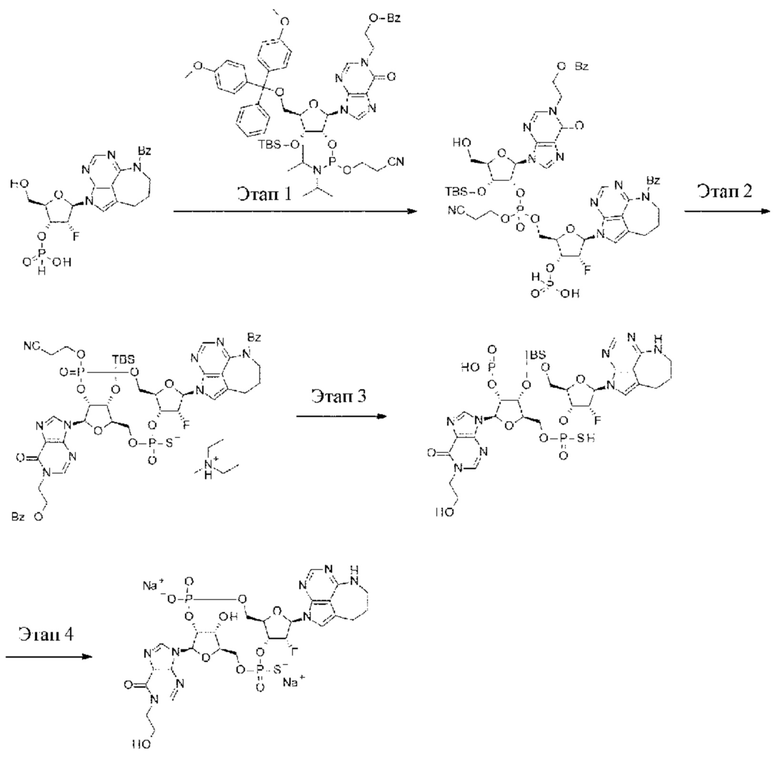
[1040]
(Этап 1)
С применением соединения, полученного на этапе 8 примера 44 (1,02 г), проводили реакцию таким же образом, как на этапе 7 примера 1, с получением раствора в ацетонитриле 6-бензоил-2-{2-дезокси-2-фтор-3-O-[гидрокси(оксо)-λ5-фосфанил]-β-D-рибофуранозил}-6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулена (раствор в ацетонитриле А). Полученное на этапе 3 примера 22 соединение (1,23 г) азеотропно обезвоживали три раза обезвоженным ацетонитрилом (10 мл). После последнего процесса позволили остаться приблизительно 7 мл ацетонитрила и добавляли туда молекулярные сита 3А, 1/16 (5 гранулоподобных частиц) (раствор в ацетонитриле В). Раствор в ацетонитриле А и раствор в ацетонитриле В смешивали друг с другом и смесь перемешивали в атмосфере азота при комнатной температуре в течение 15 минут. Раствор в декане трет-бутилгидропероксида (5,5 М, 0,50 мл) добавляли в реакционную смесь, которую перемешивали в течение 40 минут, и охлаждали льдом реакционную смесь, в которую добавляли водный раствор (1,1 мл) пентагидрата тиосульфата натрия (826 мг), и реакционную смесь перемешивали в течение 10 минут. Воду (20 мл) добавляли в реакционную смесь, которую осуществляли экстрагирование смесью дихлорметан-метанол. После высушивания органического слоя над безводным сульфатом натрия осушающий агент удаляли путем фильтрации и фильтрат концентрировали при пониженном давлении. К раствору остатка в дихлорметане (15,9 мл) добавляли воду (0,200 мл) и раствор дихлоруксусной кислоты (1,00 мл) в дихлорметане (15,9 мл) в данном порядке, и реакционную смесь перемешивали при комнатной температуре в течение 15 минут. После добавления пиридина (11,0 мл) в реакционную смесь, чтобы погасить реакцию, реакционную смесь концентрировали при пониженном давлении. Полученный в результате этого неочищенный продукт непосредственно использовали для следующей реакции.
[1041]
(Этап 2)
Ν,Ν-диэтилэтанаминия (5R,7R,8R,12aR,14R,15R,15aR,16R)-7-{1-[2-(бензоилокси)этил]-6-оксо-1,6-дигидро-9Н-пурин-9-ил}-14-(6-бензоил-6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)-16-{[трет-бутил(диметил)силил]окси}-10-(2-цианоэтокси)-15-фтор-2,10-диоксооктагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2-тиолят
Неочищенный продукт, полученный на этапе 1, приводили в реакцию таким же образом, как на этапе 9 примера 1, с получением указанного в заголовке соединения (122 мг: с примесями).
МС (ИЭР) m/z: 1136(М+Н)+.
[1042]
(Этап 3)
(5R,7R,8R,12aR,14R,15R,15aR,16R)-16-{[трет-бутил(диметил)силил]окси}-15-фтор-10-гидрокси-7-[1-(2-гидроксиэтил)-6-оксо-1,6-дигидро-9Н-пурин-9-ил]-2-сульфанил-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-дион
С применением соединения, полученного на этапе 2 выше (122 мг, с примесями), проводили реакцию таким же образом, как на этапе 10 примера 1, и полученный в результате этого неочищенный продукт непосредственно использовали для следующей реакции.
МС (ИЭР) m/z: 875(М+Н)+.
[1043]
(Этап 4)
Динатрия (5R,7R,8R,12aR,14R,15R,15aR,16R)-15-фтор-16-гидрокси-7-[1-(2-гидроксиэтил)-6-оксо-1,6-дигидро-9Н-пурин-9-ил]-2,10-диоксо-2-сульфид-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-10-олат
С применением неочищенного продукта, полученного на этапе 3 выше, проводили реакцию таким же образом, как на этапе 11 примера 1, и продукт затем очищали с помощью препаративной ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 2% - 30% (0 мин - 30 мин)] с получением указанного в заголовке соединения в виде соли триэтиламина. Полученную соль триэтиламина подвергали солевому обмену таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением указанного в заголовке соединения (30 мг).
МС (ИЭР) m/z: 761(М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,58 (1Н, с), 8,12 (1Н, с), 8,02 (1Н, с), 7,13 (1Н, с), 6,47 (1Н, д, J=18,1 Гц), 6,27 (1Н, д, J=8,5 Гц), 5,41 (1Н, дд, J=51,7, 3,9 Гц), 5,29-5,16 (2Н, м), 4,58-4,52 (2Н, м), 4,36-4,17 (5Н, м), 4,05-3,90 (2Н, м), 3,82-3,71 (2Н, м), 3,52-3,43 (2Н, м), 2,76-2,63 (2Н, м), 2,02-1,85 (2Н, м).
31Р-ЯМР (CD3OD) δ: 58,0 (с), -0,97 (с).
[1044]
Пример 64. Синтез ЦДН54.
(5R,7R,8R,12aR,14R,15R,15aR,16R)-15-фтор-2,10,16-тригидрокси-7-[1-(2-гидроксиэтил)-6-оксо-1,6-дигидро-9H-пурин-9-ил]-14-(6,7,8,9-тетрагидро-2H-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-дион
[1045]
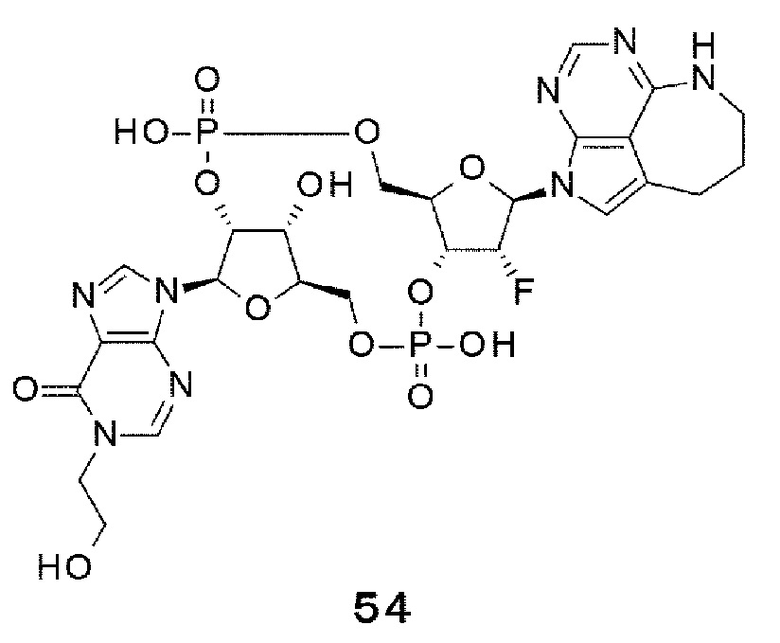
[1046]
[Схема синтеза]
[1047]
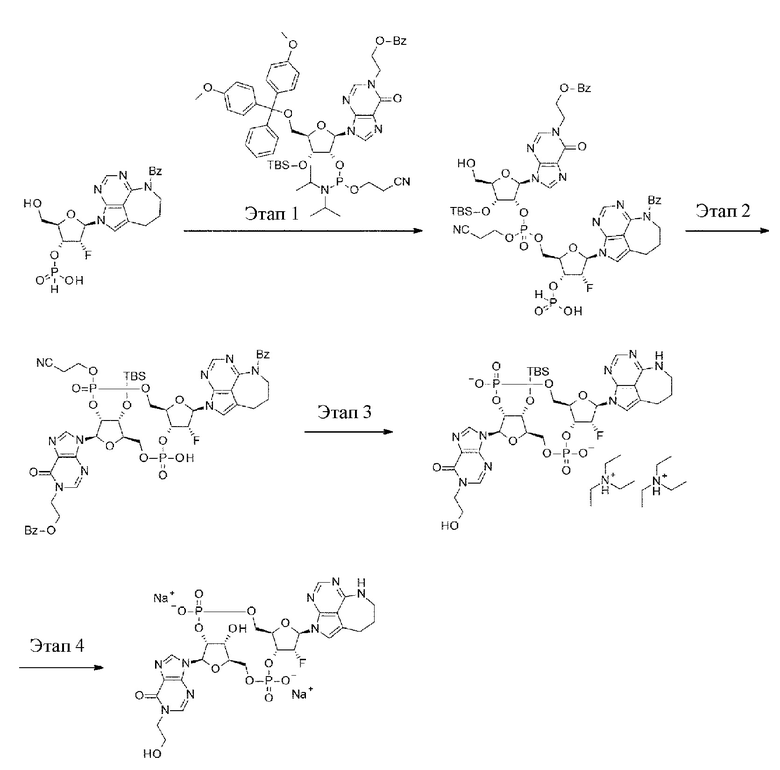
[1048]
(Этап 1)
С применением соединения, полученного на этапе 8 примера 44 (1,00 г), и полученного на этапе 3 примера 22 соединения (1,13 г) проводили реакцию таким же образом, как на этапе 1 примера 63, и полученный в результате этого неочищенный продукт непосредственно использовали для следующей реакции.
[1049]
(Этап 2)
2-{9-[(5R,7R,8R,12aR,14R,15R,15aR,16R)-14-(6-бензоил-6,7,8,9-тетрагидро-2H-2,3,5,6-тетраазабензо[cd]азулен-2-ил)-16-{[трет-бутил(диметил)силил]окси}-10-(2-цианоэтокси)-15-фтор-2-гидрокси-2,10-диоксооктагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-7-ил]-6-оксо-6,9-дигидро-1Н-пурин-1-ил}этилбензоат
С применением неочищенного продукта, полученного на этапе 1 выше, проводили реакцию таким же образом, как на этапе 2 примера 62, с получением указанного в заголовке соединения (602 мг: с примесями) в виде смеси диастереомеров у атома фосфора.
МС (ИЭР) m/z: 1120 (М+Н)+.
[1050]
(Этап 3)
Бис(N,N-диэтилэтанаминия) (5R,7R,8R,12aR,14R,15R,15aR,16R)-16-{[трет-бутил(диметил)силил]окси}-15-фтор-7-[1-(2-гидроксиэтил)-6-оксо-1,6-дигидро-9Н-пурин-9-ил]-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2H,10H,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(олат)
С применением соединения, полученного на этапе 2 выше (602 мг), проводили реакцию таким же образом, как на этапе 10 примера 1, с получением указанного в заголовке соединения (90,8 мг: с примесями).
МС (ИЭР) m/z: 859 (М+Н)+.
[1051]
(Этап 4)
Динатрия (5R,7R,8R,12aR,14R,15R,15aR,16R)-15-фтор-16-гидрокси-7-[1-(2-гидроксиэтил)-6-оксо-1,6-дигидро-9Н-пурин-9-ил]-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(олат)
С применением соединения, полученного на этапе 3 выше (90,8 мг: с примесями), проводили реакцию таким же образом, как на этапе 11 примера 1, а затем проводили очистку при следующих [Условиях очистки] с получением указанного в заголовке соединения в виде соли триэтиламина.
[Условия очистки] Хроматография на колонке с силикагелем С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил] и препаративная ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 1%-20% (0 мин - 40 мин)].
Полученную соль триэтиламина подвергали солевому обмену таким же образом, как в [Превращении в натриевую соль], описанном на этапе 11 примера 1, с получением указанного в заголовке соединения (40,4 мг).
МС (ИЭР) m/z: 745 (М+Н)+.
1H-ЯМР (CD3OD) δ: 8,56 (1Н, с), 8,19 (1Н, с), 8,03 (1Н, с), 7,10 (1Н, с), 6,46 (1Н, д, J=18,7 Гц), 6,31 (1Н, д, J=8,5 Гц), 5,51-5,32 (1Н, м), 5,24-5,01 (2Н, м), 4,61-4,56 (1Н, м), 4,41-4,21 (5Н, м), 4,21-3,97 (3Н, м), 3,87-3,75 (2Н, м), 3,54-3,41 (2Н, м), 2,83-2,67 (2Н, м), 2,02-1,85 (2Н, м).
31Р-ЯМР (CD3OD) δ: -0,59 (с), -0,81 (с).
[1052]
Пример 65. Синтез лекарственного средства-линкера 5.
[Схема синтеза]
[1053]
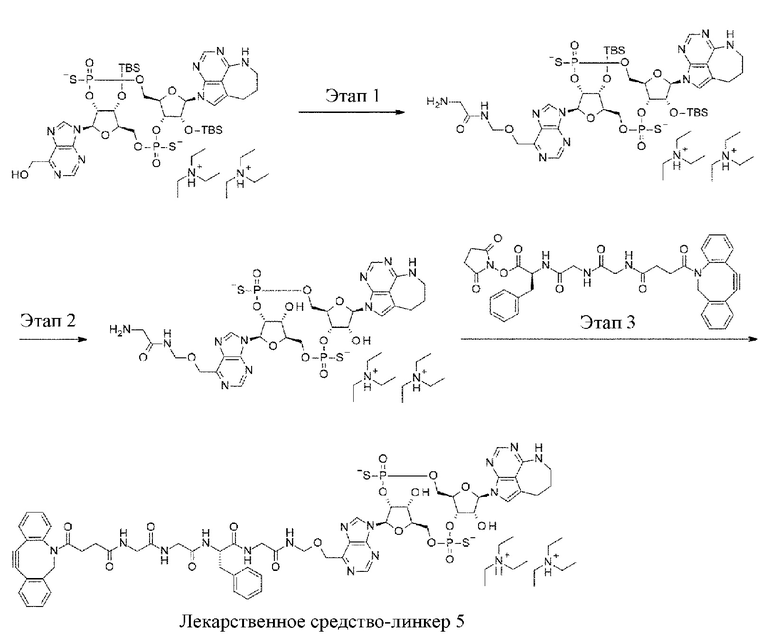
[1054]
(Этап 1)
Бис(N,N-диэтилэтанаминия) (5R,7R,8R,12aR,14R,15R,15aR,16R)-15,16-бис{[трет-бутил(диметил)силил]окси}-7-(6-{[(глициламино)метокси]метил}-9Н-пурин-9-ил)-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
С применением соединения, полученного на этапе 7 примера 17 (диастереомер 2) (80,6 мг), проводили реакцию таким же образом, как на этапе 7-1 примера 22, с получением указанного в заголовке соединения (66,1 мг: с примесями).
МС (ИЭР) m/z: 1059 (М+Н)+.
[1055]
(Этап 2)
Бис(N,N-диэтилэтанаминия) (5R,7R,8R,12aR,14R,15R,15aS,16R)-7-(6-{[(глициламино)метокси]метил}-9Н-пурин-9-ил)-15,16-дигидрокси-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
С применением соединения, полученного на этапе 1 выше (66,1 мг), проводили реакцию таким же образом, как на этапе 8-1 примера 22, с получением указанного в заголовке соединения (40,4 мг: с примесями).
МС (ИЭР)m/z: 831 (М+Н)+.
[1056]
(Этап 3)
Бис(N,N-диэтилэтанаминия) N-[4-(11,12-дидегидродибензо[b,f]азоцин-5(6Н)-ил)-4-оксобутаноил]глицилглицил-L-фенилаланил-N-[({9-[(5R,7R,8R,12aR,14R,15R,15aS,16R)-15,16-дигидрокси-2,10-диоксо-2,10-дисульфид-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-7-ил]-9Н-пурин-6-ил}метокси)метил]глицинамид
(Лекарственное средство-линкер 5)
С применением соединения, полученного на этапе 2 выше (40,4 мг), проводили реакцию таким же образом, как на этапе 9-1 примера 22, и продукт затем очищали с помощью колоночной хроматографии на силикагеле С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил] и препаративной ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 10%-45% (0 мин - 30 мин)] с получением указанного в заголовке соединения (5,0 мг).
MC (ИЭР) m/z: 1379 (М+Н)+.
1Н-ЯМР (CD3OD) δ: 9,17 (1Н, с), 8,83 (1Н, д, J=4,8 Гц), 8,01 (1Н, д, J=3,6 Гц), 7,63-7,45 (2Н, м), 7,40-7,34 (3Н, м), 7,29-7,12 (8Н, м), 7,09 (1Н, с), 6,47 (1Н, д, J=8,5 Гц), 6,31 (1Н, д, J=6,7 Гц), 5,56-5,47 (2Н, м), 5,05-4,91 (4Н, м), 4,88-4,73 (3Н, м), 4,57-4,30 (5Н, м), 4,09-4,01 (1Н, м), 4,00-3,55 (9Н, м), 3,52-3,45 (2Н, м), 3,18 (12Н, к, J=7,3 Гц), 3,02-2,94 (1Н, м), 2,90-2,71 (3Н, м), 2,35-2,20 (2Н, м), 2,04-1,92 (3Н, м), 1,27 (18Н, т, J=7,3 Гц).
[1057]
Пример 66. Синтез лекарственного средства-линкера 6.
[Схема синтеза]
[1058]
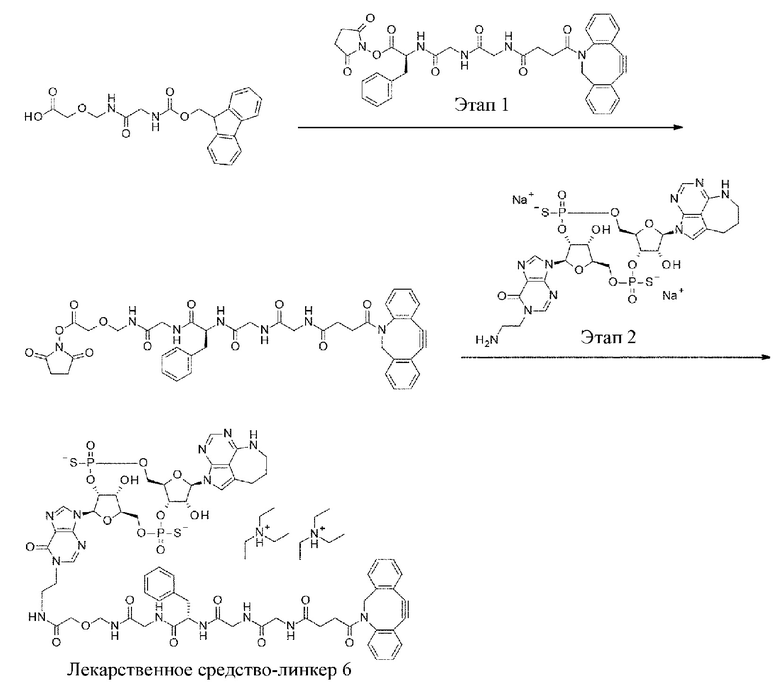
[1059]
(Этап 1)
N-[4-(11,12-дидегидродибензо[b,f]азоцин-5(6Н)-ил)-4-оксобутаноил]глицилглицил-L-фенилаланил-N-({2-[(2,5-диоксопирролидин-1-ил)окси]-2-оксоэтокси}метил)глицинамид
К раствору {[(N-{[(9Н-флуорен-9-ил)метокси]карбонил}глицил)амино]метокси}уксусной кислоты (955 мг) как соединения, известного в литературе (WO 2014/057687), в N,N-диметилформамиде (8,0 мл) добавляли 1,8-диазабицикло[5,4,0]-7-ундецен (0,74 мл) и реакционную смесь перемешивали при комнатной температуре в течение 1 часа (реакционная смесь А). К раствору соединения, полученного на этапе 10 примера 22 (938 мг), в N,N-диметилформамиде (8,0 мл) добавляли N-гидроксисукцинимид (229 мг) и гидрохлорид 1-этил-3-(3-диметиламинопропил)-карбодиимида (380 мг) и реакционную смесь перемешивали при комнатной температуре в течение 50 минут (реакционная смесь В). Реакционную смесь А добавляли в реакционную смесь В и продукт перемешивали при комнатной температуре в течение 1 часа. Дихлорметан (50 мл) и 10% водный раствор лимонной кислоты (10 мл) добавляли в реакционную смесь, которую осуществляли экстрагирование дихлорметаном. Органический слой промывали рассолом, а затем сушили над безводным сульфатом натрия. Осушающий агент удаляли путем фильтрации и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле [хлороформ/(нижний слой хлороформа/метанол/вода = 7:3:1)]. К раствору соединения, полученного в N,N-диметилформамиде (8,0 мл), добавляли N-гидроксисукцинимид (229 мг) и 1-этил-3-(3-диметиламинопропил)-карбодиимида гидрохлорид (380 мг), и реакционную смесь перемешивали при комнатной температуре в течение 30 минут. Дихлорметан (100 мл) и воду (25 мл) добавляли в реакционную смесь, которую осуществляли экстрагирование дихлорметаном. Органический слой промывали рассолом, а затем сушили над безводным сульфатом натрия. Осушающий агент удаляли путем фильтрации и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле [хлороформ/метанол]. Фракции, содержащие целевой продукт, концентрировали при пониженном давлении, и диэтиловый эфир добавляли к остатку, чтобы получить взвесь. Полученный твердый продукт собирали путем фильтрации с получением указанного в заголовке соединения (412 мг).
1H-ЯМР (ДМСО-d6) δ: 8,72 (1Н, м), 8,32 (1Н, м), 8,17-7,96 (3Н, м), 7,71-7,15 (13Н, м), 5,01 (1Н, д, J=13,9 Гц), 4,70-4,48 (5Н, м), 3,81-3,51 (7Н, м), 3,05 (1Н, дд, J=14,2, 3,9 Гц), 2,83 (4Н, с), 2,80 (1Н, м), 2,64 (1Н, м), 2,28 (1Н, м), 2,07 (1Н, м), 1,79 (1Н, м).
[1060]
(Этап 2)
Бис(N,N-диэтилэтанаминия) N-[4-(11,12-дидегидродибензо[b,f]азоцин-5(6Н)-ил)-4-оксобутаноил]глицилглицил-L-фенилаланил-N-({2-[(2-{9-[(5R,7R,8R,12aR,14R,15R,15aS,16R)-15,16-дигидрокси-2,10-диоксо-2,10-дисульфид-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-7-ил]-6-оксо-6,9-дигидро-1Н-пурин-1-ил}этил)амино]-2-оксоэтокси}метил)глицинамид
(Лекарственное средство-линкер 6)
С применением соединения, полученного на этапе 8-2 примера 5 (20,0 мг), и полученного на этапе 1 выше соединения (23,7 мг) проводили реакцию таким же образом, как на этапе 4 примера 21, и продукт затем очищали с помощью колоночной хроматографии на силикагеле С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил], препаративной ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 20%-45% (0 мин - 40 мин)] и препаративной ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/метанол, метанол: 30%-80% (0 мин - 40 мин)] с получением указанного в заголовке соединения (14,1 мг).
MC (ИЭР) m/z: 1466 (М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,74-8,69 (1Н, м), 8,17-8,12 (1Н, м), 8,04-7,96 (1Н, м), 7,65-7,13 (13Н, м), 7,13-7,04 (1Н, м), 6,33-6,23 (2Н, м), 5,50-5,31 (2Н, м), 5,07-4,94 (2Н, м), 4,85-4,76 (1Н, м), 4,71-4,26 (10Н, м), 4,18-3,53 (13Н, м), 3,53-3,33 (3Н, м), 3,19 (12Н, к, J=7,3 Гц), 3,05-2,92 (1Н, м), 2,91-2,70 (3Н, м), 2,40-2,20 (2Н, м), 2,07-1,87 (2Н, м), 1,29 (18Н, т, J=7,3 Гц).
[1061]
Пример 67. Синтез лекарственного средства-линкера 7.
[Схема синтеза]
[1062]
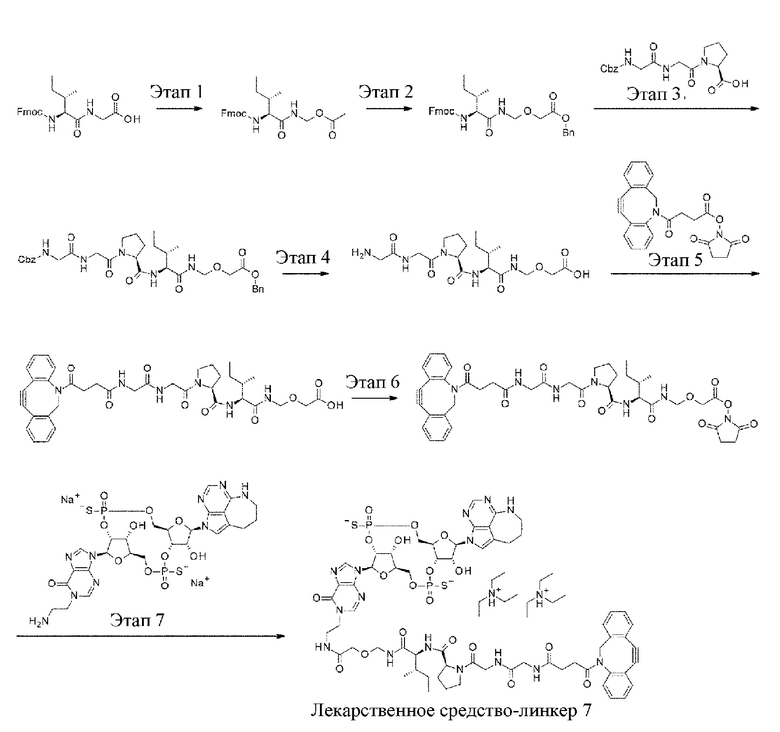
[1063]
(Этап 1)
[(N-{[(9Н-флуорен-9-ил)метокси]карбонил}-L-изолейцил)амино]метилацетат
В смесь доступного для приобретения (Iris Biotech GmbH) N-{[(9Н-флуорен-9-ил)метокси]карбонил}-L-изолейцилглицина (2,50 г) в тетрагидрофуране (45 мл) - толуоле (15 мл) добавляли пиридин (0,588 мл) и тетраацетат свинца (3,24 г) при комнатной температуре, и реакционную смесь перемешивали при 65°С в течение 3 часов. Реакционную смесь разбавляли этилацетатом и промывали водой и рассолом в данном порядке. После высушивания органического слоя над безводным сульфатом натрия осушающий агент удаляли путем фильтрации и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле [гексан/этилацетат] с получением указанного в заголовке соединения (2,06 г)
МС (ИЭР) m/z: 447 (M+Na)+.
1Н-ЯМР (CDCl3) δ: 7,77 (2Н, д, J=7,9 Гц), 7,58 (2Н, д, J=7,3 Гц), 7,40 (2Н, т, J=7,3 Гц), 7,32 (2Н, тд, J=7,6, 1,2 Гц), 6,97-6,92 (1Н, уш. м), 5,32-5,20 (3Н, м), 4,47-4,37 (2Н, м), 4,21 (1Н, т, J=6,7 Гц), 4,05-4,01 (1Н, м), 2,04 (3Н, с), 1,92-1,84 (1Н, уш. м), 1,51-1,41 (1Н, уш. м), 1,17-1,07 (1Н, уш. м), 0,93-0,89 (6Н, м).
[1064]
(Этап 2)
Бензил {[(N-{[(9Н-флуорен-9-ил)метокси]карбонил}-L-изолейцил)амино]метокси}ацетат
В суспензию полученного на этапе 1 выше соединения (2,06 г) в тетрагидрофуране (48 мл) добавляли бензилгликолят (1,38 мл) и моногидрат п-толуолсульфоновой кислоты (92,3 мг) при 0°С и реакционную смесь перемешивали при комнатной температуре в течение 4 часов. Реакционную смесь разбавляли этилацетатом и промывали насыщенным раствором гидрокарбоната натрия и водный слой затем осуществляли экстрагирование этилацетатом. Органический слой промывали рассолом и сушили над безводным сульфатом натрия. Осушающий агент удаляли путем фильтрации и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле [гексан/этилацетат] с получением указанного в заголовке соединения (1,72 г).
МС (ИЭР) m/z: 553 (M+Na)+.
1Н-ЯМР (CDCl3) δ: 7,76 (2Н, д, J=7,9 Гц), 7,58 (2Н, д, J=7,3 Гц), 7,42-7,29 (9Н, м), 6,74-6,69 (1Н, уш. м), 5,24-5,21 (1Н, уш. м), 5,17 (2Н, с), 4,86 (2Н, д, J=6,7 Гц), 4,48-4,39 (2Н, м), 4,23-4,19 (3Н, м), 4,04-4,00 (1Н, м), 1,95-1,87 (1Н, уш. м), 1,50-1,41 (1Н, уш. м), 1,15-1,07 (1Н, уш. м), 0,93-0,89 (6Н, м).
[1065]
(Этап 3)
N-[(бензилокси)карбонил]глицилглицил-L-пролил-N-{[2-(бензилокси)-2-оксоэтокси]метил}-L-изолейцинамид
В суспензию полученного на этапе 2 выше соединения (1,72 г) в ацетонитриле (40 мл) добавляли 1,8-диазабицикло[5,4,0]-7-ундецен (0,290 мл) при комнатной температуре и реакционную смесь перемешивали в течение 1 часа (реакционная смесь А). В суспензию доступного для приобретения N-[(бензилокси)карбонил]глицилглицил-L-пролина (1,41 г) в ацетонитриле (20 мл) добавляли 3Н-[1,2,3]триазоло[4,5-b]пиридин-3-ол (529 мг), гидрохлорид 1-этил-3-(3-диметиламинопропил)-карбодиимида (746 мг) и N,N-диизопропилэтиламин (0,678 мл) при комнатной температуре, и реакционную смесь перемешивали в течение 1 часа. Данную реакционную смесь добавляли в реакционную смесь А выше при комнатной температуре и продукт перемешивали в течение 6 часов, а затем концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле [хлороформ/метанол] с получением указанного в заголовке соединения (1,56 г).
МС (ИЭР) m/z: 676 (M+Na)+.
1H-ЯМР (CD3OD) δ: 7,37-7,27 (10Н, м), 5,17 (2Н, с), 5,09 (2Н, с), 4,82-4,70 (2Н, м), 4,56-4,44 (1Н, м), 4,17-4,14 (3Н, м), 4,07-3,96 (2Н, м), 3,83-3,82 (2Н, м), 3,64-3,48 (2Н, м), 2,34-1,78 (5Н, м), 1,61-1,51 (1Н, м), 1,25-1,13 (1Н, м), 0,95-0,87 (6Н, м).
[1066]
(Этап 4)
Глицилглицил-L-пролил-Н-[(карбоксиметокси)метил]-L-изолейцинамид
В смесь соединения, полученного на этапе 3 выше (1,56 г), в метаноле (8 мл)-тетрагидрофуране (24 мл)-дихлорметане (8 мл) добавляли 10% палладий-углерод (М), влажный (1,4 г), и реакционную смесь перемешивали в атмосфере водорода при комнатной температуре в течение 23 часов. Смесь метанол/тетрагидрофуран (1:1) (50 мл) добавляли в реакционную смесь, которую фильтровали с помощью целита. Целит промывали смесью метанола/тетрагидрофурана (1:1). После того, как фильтрат концентрировали при пониженном давлении, к остатку добавляли метанол (20 мл), тетрагидрофуран (20 мл) и 10% палладий-углерод (М), влажный (1,0 г), и реакционную смесь перемешивали в атмосфере водорода при комнатной температуре в течение 3 часов. Реакционную смесь фильтровали с помощью целита и целит затем промывали метанолом/тетрагидрофураном (1:1). Фильтрат концентрировали при пониженном давлении с получением неочищенной формы указанного в заголовке соединения (1,02 г).
МС (ИЭР) m/z: 430 (М+Н)+.
(Этап 5)
N-[4-(11,12-дидегидродибензо[b,f]азоцин-5(6Н)-ил)-4-оксобутаноил]глицилглицил-L-пролил-N-[(карбоксиметокси)метил]-L-изолейцинамид
К раствору соединения, полученного на этапе 4 выше (1,02 г), в N,N-диметилформамиде (24 мл) добавляли 1-{[4-(11,12-дидегидродибензо[b,f]азоцин-5(6Н)-ил)-4-оксобутаноил]окси}пирролидин-2,5-дион (956 мг) и N,N-диизопропилэтиламин (0,496 мл) и реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле [хлороформ/метанол]. Фракции, содержащие целевой продукт, концентрировали при пониженном давлении, и этилацетат добавляли к остатку, чтобы отвердить. Полученный твердый продукт собирали путем фильтрации с получением указанного в заголовке соединения (1,06 г).
МС (ИЭР) m/z: 739 (M+Na)+, 715 (M-H)-.
1Н-ЯМР (CD3OD) δ: 7,64-7,59 (2Н, м), 7,48-7,44 (3Н, м), 7,38-7,23 (3Н, м), 5,15-5,12 (1Н, м), 4,74-4,70 (2Н, м), 4,64-4,48 (1Н, м), 4,25-4,18 (1Н, м), 4,11-3,96 (3Н, м), 3,91-3,85 (1Н, м), 3,78-3,57 (5Н, м), 2,84-2,75 (1Н, м), 2,39-2,16 (3Н, уш. м), 2,08-1,84 (5Н, уш. м), 1,61-1,52 (1Н, уш. м), 1,23-1,15 (1Н, уш. м), 0,96-0,87 (6Н, м).
[1068]
(Этап 6)
N-[4-(11,12-дидегидродибензо[b,f]азоцин-5(6Н)-ил)-4-оксобутаноил]глицилглицил-L-пролил-N-({2-[(2,5-диоксопирролидин-1-ил)окси]-2-оксоэтокси}метил)-L-изолейцинамид
В суспензию полученного на этапе 5 выше соединения (1,06 г) в N,N-диметилформамиде (16 мл) добавляли N-гидроксисукцинимид (187 мг) и гидрохлорид 1-этил-3-(3-диметиламинопропил)-карбодиимида (312 мг) и реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Реакционную смесь разбавляли хлороформом и промывали три раза водой. После высушивания органического слоя над безводным сульфатом натрия осушающий агент удаляли путем фильтрации и фильтрат концентрировали при пониженном давлении. Этилацетат добавляли к остатку, и полученный твердый продукт собирали путем фильтрации. В твердый продукт, собранный путем фильтрации, добавляли диэтиловый эфир, чтобы получить взвесь, и твердый продукт затем собирали путем фильтрации с получением указанного в заголовке соединения (767 мг).
МС (ИЭР) m/z: 836 (M+Na)+.
1Н-ЯМР (CD3OD) δ: 7,68-7,56 (2Н, м), 7,48-7,44 (3Н, м), 7,38-7,23 (3Н, м), 5,15-5,12 (1Н, м), 4,83-4,70 (2Н, м), 4,64-4,50 (3Н, м), 4,24-3,55 (8Н, м), 2,83-2,76 (5Н, м), 2,38-2,18 (3Н, м), 2,11-1,84 (5Н, уш. м), 1,60-1,52 (1Н, м), 1,25-1,13 (1Н, м), 0,98-0,87 (6Н, м).
[1069]
(Этап 7)
Бис(N,N-диэтилэтанаминия) N-[4-(11,12-дидегидродибензо[b,f]азоцин-5(6Н)-ил)-4-оксобутаноил]глицилглицил-L-пролил-N-({2-[(2-{9-[(5R,7R,8R,12aR,14R,15R,15aS,16R)-15,16-дигидрокси-2,10-диоксо-2,10-дисульфид-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-7-ил]-6-оксо-6,9-дигидро-1Н-пурин-1-ил}этил)амино]-2-оксоэтокси}метил)-L-изолейцинамид
(Лекарственное средство-линкер 7)
С применением соединения, полученного на этапе 8-2 примера 5 (20,0 мг), и полученного на этапе 6 выше соединения (20,9 мг) проводили реакцию таким же образом, как на этапе 4 примера 21, и продукт затем очищали с помощью колоночной хроматографии на силикагеле С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил], препаративной ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 20%-45% (0 мин - 40 мин)] и препаративной ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/метанол, метанол: 30%-80% (0 мин - 40 мин)] с получением указанного в заголовке соединения (11,9 мг).
МС (ИЭР) m/z: 1472 (М+Н)+
1Н-ЯМР (CD3OD) δ: 8,71 (1Н, уш. с), 8,19-8,13 (1Н, м), 8,02 (1Н, с), 7,64-7,56 (2Н, м), 7,48-7,41 (3Н, м), 7,37-7,08 (4Н, м), 6,34-6,23 (2Н, м), 5,48-5,36 (2Н, м), 5,16-5,08 (1Н, м), 4,98-4,55 (6Н, м), 4,52-4,15 (7Н, м), 4,07-3,81 (6Н, м), 3,80-3,45 (8Н, м), 3,19 (12Н, к, J=7,3 Гц), 2,93-2,85 (2Н, м), 2,85-2,71 (1Н, м), 2,45-2,30 (1Н, м), 2,30-2,11 (2Н, м), 2,08-1,83 (6Н, м), 1,66-1,45 (1Н, м), 1,29 (18Н, т, J=7,3 Гц), 1,28-1,10 (1Н, м), 1,01-0,85 (7Н, м).
[1070]
Пример 68. Синтез лекарственного средства-линкера 8.
[Схема синтеза]
[1071]
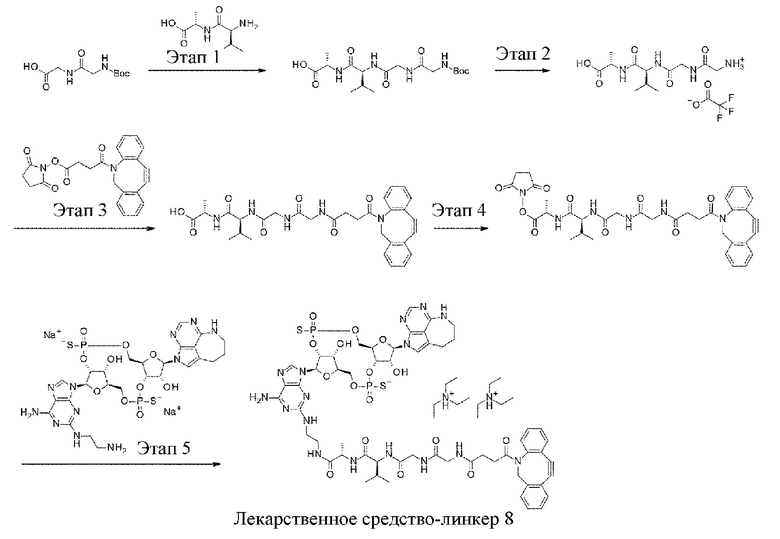
[1072]
(Этап 1)
N-(трет-бутоксикарбонил)глицилглицил-L-валил-L-аланин
К раствору доступного для приобретения (Chemfun Medical Technology (Shanghai) Co., Ltd.) N-(трет-бутоксикарбонил)глицилглицина (5,00 г) и N-гидроксисукцинимида (2,97 г) в N,N-диметилформамиде (50 мл) добавляли гидрохлорид 1-этил-3-(3-диметиламинопропил)-карбодиимида (4,95 г) в атмосфере азота при 0°С, и температуру повышали до комнатной температуры и реакционную смесь перемешивали в течение 1 часа. Триэтиламин (3,6 мл) и доступный для приобретения (KOKUSAN CHEMICAL Co., Ltd.) L-валил-L-аланин (4,05 г) добавляли в реакционную смесь, которую перемешивали при той же температуре в течение 6 часов. Реакционную смесь концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле [дихлорметан/метанол]. Фракции, содержащие целевой продукт, концентрировали при пониженном давлении и добавляли туда диэтиловый эфир, чтобы получить взвесь. Полученный твердый продукт собирали путем фильтрации с получением указанного в заголовке соединения (4,99 г).
МС (ИЭР) m/z: 401 (М-Н)+.
1Н-ЯМР (ДМСО-d6) δ: 12,43 (1Н, уш.), 8,27 (1Н, д, J=6,8 Гц), 7,92 (1Н, т, J=5,4 Гц), 7,79 (1Н, д, J=9,3 Гц), 6,99 (1Н, т, J=5,9 Гц), 4,19 (1Н, т, J=8,1 Гц), 4,13 (1Н, м), 3,73 (2Н, д, J=5,4 Гц), 3,52 (2Н, д, J=5,9 Гц), 1,92 (1Н, дд, м), 1,35 (9Н, с), 1,24 (3Н, д, J=7,3 Гц), 0,85 (3Н, д, J=6,3 Гц), 0,79 (3Н, д, J=6,8 Гц).
[1073]
(Этап 2)
Трифторацетат N-(азаниумилацетил)глицил-L-валил-L-аланина.
С применением соединения, полученного на этапе 1 выше (1,00 г), проводили реакцию таким же образом, как на этапе 1 примера 21, с получением неочищенной формы указанного в заголовке соединения (1,10 г).
МС (ИЭР) m/z: 301 (М-Н)-.
1Н-ЯМР (ДМСО-d6) δ: 12,49 (1Н, уш.), 8,54 (1Н, т, J=5,6 Гц), 8,34 (1Н, д, J=6,8 Гц), 8,03 (1Н, д, J=9,3 Гц), 8,00 (3Н, уш. с), 4,24 (1Н, дд, J=8,8, 6,8 Гц), 4,17 (1Н, м), 3,93-3,84 (2Н, м), 3,62-3,58 (2Н, м), 1,96 (1Н, м), 1,27 (3Н, д, J=7,3 Гц), 0,89 (3Н, д, J=6,8 Гц), 0,84 (3Н, д, J=6,8 Гц).
[1074]
(Этап 3)
N-[4-(11,12-дидегидродибензо[b,f]азоцин-5(6Н)-ил)-4-оксобутаноил]глицилглицил-L-валил-L-аланин
С применением соединения, полученного на этапе 2 выше (861 мг), проводили реакцию таким же образом, как на этапе 2 примера 21, с получением указанного в заголовке соединения (737 мг).
МС (ИЭР) m/z: 590 (М+Н)+.
1Н-ЯМР (ДМСО-d6) δ: 12,46 (1Н, с), 8,28 (1Н, т, J=8,1 Гц), 8,18 (1Н, т, J=5,9 Гц), 8,13 (0,5Н, т, J=5,6 Гц), 8,04 (0,5Н, т, J=5,9 Гц), 7,99 (0,5Н, т, J=5,9 Гц), 7,73 (0,5Н, д, J=1,5 Гц), 7,67 (1Н, м), 7,61 (1Н, м), 7,53-7,44 (3Н, м), 7,40-7,28 (3Н, м), 5,03 (0,5Н, д, J=6,3 Гц), 5,00 (0,5Н, д, J=6,3 Гц), 4,21 (1Н, дд, J=9,0, 7,1 Гц), 4,18-4,08 (1Н, м), 3,78-3,67 (2Н, м), 3,65-3,55 (3Н, м), 2,72-2,59 (1Н, м), 2,28 (1Н, д, J=7,8 Гц), 2,09-2,03 (1Н, м), 1,99-1,91 (1Н, м), 1,79 (1Н, м), 1,25 (1,5Н, д, J=5,4 Гц), 1,24 (1,5Н, д, J=5,4 Гц), 0,87 (3Н, д, J=6,8 Гц), 0,82-0,79 (3Н, м).
[1075]
(Этап 4)
2,5-диоксопирролидин-1-ил-N-[4-(11,12-дидегидродибензо[b,f]азоцин-5(6Н)-ил)-4-оксобутаноил]глицилглицил-L-валил-L-аланинат
С применением соединения, полученного на этапе 3 выше (260 мг), проводили реакцию таким же образом, как на этапе 3 примера 21, с получением указанного в заголовке соединения (84 мг).
МС (ИЭР) m/z: 687 (М+Н)+.
1Н-ЯМР (ДМСО-d6) δ: 8,73 (1Н, т, J=7,0 Гц), 8,18 (0,5Н, т, J=5,7 Гц), 8,12 (0,5Н, т, J=5,7 Гц), 8,04 (0,5Н, т, J=5,7 Гц), 7,98 (0,5Н, т, J=5,7 Гц), 7,84-7,60 (3Н, м), 7,52-7,44 (3Н, м), 7,40-7,28 (3Н, м), 5,03 (0,5Н, д, J=6,0 Гц), 5,00 (0,5Н, д, J=6,0 Гц), 4,69-4,58 (1Н, м), 4,22 (1Н, т, J=7,6 Гц), 3,80-3,54 (5Н, м), 2,80 (4Н, с), 2,73-2,59 (1Н, м), 2,34-2,24 (1Н, м), 2,11-2,02 (1Н, м), 1,99-1,91 (1Н, м), 1,79 (1Н, м), 1,45 (1,5Н, д, J=3,6 Гц), 1,43 (1,5Н, д, J=3,6 Гц), 0,85 (3Н, д, J=6,7 Гц), 0,83-0,79 (3Н, м).
[1076]
(Этап 5)
Бис(N,N-диэтилэтанаминия) N-[4-(11,12-дидегидродибензо[b,f]азоцин-5(6Н)-ил)-4-оксобутаноил]глицилглицил-L-валил-N-[2-({6-амино-9-[(5R,7R,8R,12aR,14R,15R,15aS,16R)-15,16-дигидрокси-2,10-диоксо-2,10-дисульфид-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-7-ил]-9Н-пурин-2-ил}амино)этил]-L-аланинамид
(Лекарственное средство-линкер 8)
С применением соединения, полученного на этапе 8-2 примера 8 (5,3 мг), и полученного на этапе 4 выше соединения (4,5 мг) проводили реакцию таким же образом, как на этапе 1 примера 23, и продукт затем очищали с помощью препаративной ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 25%-35% (0 мин - 30 мин)] с получением указанного в заголовке соединения (5,1 мг).
МС (ИЭР) m/z: 1359 (М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,37 (1Н, уш. с), 8,02 (1Н, с), 7,66-7,13 (9Н, м), 6,33 (1Н, д, J=6,7 Гц), 6,13 (1Н, д, J=9,1 Гц), 5,49-5,42 (2Н, м), 5,13-5,04 (1Н, м), 4,83-4,80 (1Н, м), 4,51-3,63 (14Н, м), 3,51-3,37 (4Н, м), 3,14 (12Н, к, J=7,3 Гц), 2,90-2,72 (5Н, м), 2,36-1,96 (6Н, м), 1,34-1,25 (21Н, м), 0,98-0,86 (6Н, м).
[1077]
Пример 69. Синтез лекарственного средства-линкера 9.
[Схема синтеза]
[1078]
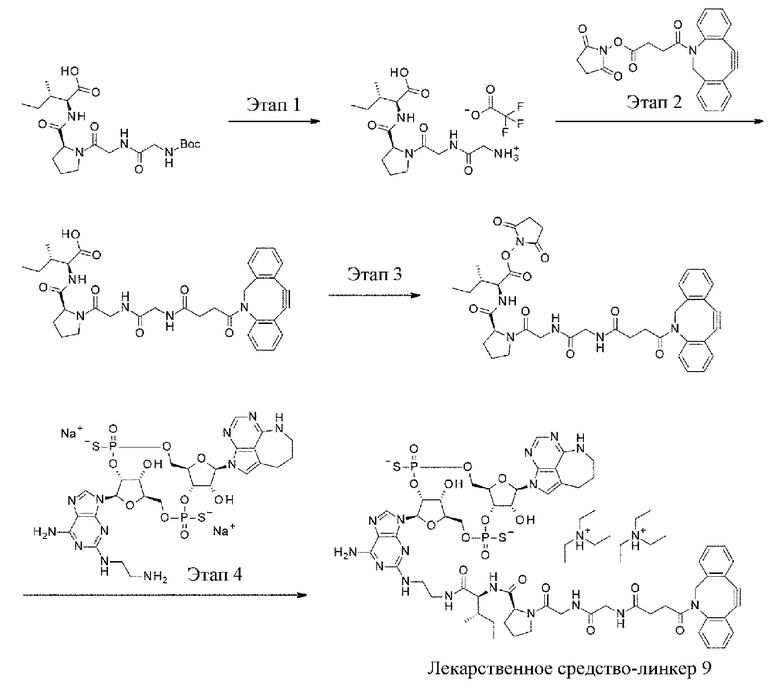
[1079]
(Этап 1)
Трифторацетат N-(азаниумилацетил)глицил-L-пролил-L-изолейцина
С применением доступного для приобретения (Hangzhou Peptide Biochem Co., Ltd.) N-(трет-бутоксикарбонил)глицилглицил-L-пролил-L-изолейцина (1,00 г) проводили реакцию таким же образом, как на этапе 1 примера 21, с получением неочищенной формы указанного в заголовке соединения (1,02 г).
МС (ИЭР) m/z: 343 (М+Н)+.
1Н-ЯМР (ДМСО-d6) δ: 12,61 (1Н, с), 8,51 (0,7Н, т, J=5,1 Гц), 8,48 (0,3Н, т, J=4,9 Гц), 8,35 (0,3Н, д, J=8,8 Гц), 8,05 (0,7Н, д, J=8,3 Гц), 7,99 (3Н, уш. с), 4,55 (0,3Н, дд, J=8,3, 2,4 Гц), 4,46 (0,7Н, дд, J=8,8, 2,9 Гц), 4,23 (0,3Н, дд, J=8,5, 5,6 Гц), 4,15 (0,7Н, дд, J=8,3, 5,9 Гц), 4,06 (0,7Н, дд, J=17,6, 5,4 Гц), 3,99-3,95 (1Н, м), 3,63-3,36 (4,3Н, м), 2,28-2,20 (0,3Н, м), 2,07-2,00 (0,7Н, м), 1,96-1,74 (4Н, м), 1,46-1,36 (1Н, м), 1,26-1,13 (1Н, м), 0,89-0,83 (6Н, м).
[1080]
(Этап 2)
N-[4-(11,12-дидегидродибензо[b,f]азоцин-5(6Н)-ил)-4-оксобутаноил]глицилглицил-L-пролил-L-изолейцин
С применением соединения, полученного на этапе 1 выше (1,02 г), проводили реакцию таким же образом, как на этапе 2 примера 21, с получением указанного в заголовке соединения (993 мг).
МС (ИЭР) m/z: 630 (М+Н)+.
1Н-ЯМР (ДМСО-d6) δ: 12,57 (1Н, с), 8,30 (0,3Н, дд, J=8,5, 3,6 Гц), 8,14-8,06 (1Н, м), 8,00 (0,7Н, д, J=7,9 Гц), 7,88-7,80 (1Н, м), 7,70-7,66 (1Н, м), 7,63-7,60 (1Н, м), 7,53-7,27 (6Н, м), 5,04 (0,7Н, д, J=14,5 Гц), 5,02 (0,3Н, д, J=14,5 Гц), 4,56 (0,3Н, дд, J=8,5, 2,4 Гц), 4,47-4,43 (0,7Н, м), 4,20 (0,3Н, дд, J=8,2, 5,7 Гц), 4,12 (0,7Н, м), 3,99-3,36 (7Н, м), 2,68-2,58 (1Н, м), 2,34-2,18 (1,3Н, м), 2,09-1,74 (6,7Н, м), 1,44-1,34 (1Н, м), 1,25-1,12 (1Н, м), 0,87-0,80 (6Н, м).
[1081]
(Этап 3)
2,5-диоксопирролидин-1-ил-N-[4-(11,12-дидегидродибензо[b,f]азоцин-5(6Н)-ил)-4-оксобутаноил]глицилглицил-L-пролил-L-изолейцинат
С применением соединения, полученного на этапе 2 выше (300 мг), проводили реакцию таким же образом, как на этапе 3 примера 21, с получением указанного в заголовке соединения (287 мг).
МС (ИЭР) m/z: 727 (М+Н)+.
1H-ЯМР (ДМСО-d6) δ: 8,77-8,67 (0,3Н, м), 8,50 (0,5Н, д, J=8,3 Гц), 8,31 (0,2Н, м), 8,10-8,04 (1Н, м), 7,88-7,79 (1Н, м), 7,68-7,57 (2Н, м), 7,49-7,26 (6Н, м), 5,01 (1Н, д, J=14,2 Гц), 4,80-4,42 (2Н, м), 3,97-3,84 (1Н, м), 3,79-3,34 (6Н, м), 2,78 (4Н, с), 2,64-2,57 (1Н, м), 2,30-1,74 (8Н, м), 1,56-1,17 (2Н, м), 1,00-0,80 (6Н, м).
[1082]
(Этап 4)
Бис(N,N-диэтилэтанаминия) N-[4-(11,12-дидегидродибензо[b,f]азоцин-5(6Н)-ил)-4-оксобутаноил]глицилглицил-L-пролил-N-[2-({6-амино-9-[(5R,7R,8R,12aR,14R,15R,15aS,16R)-15,16-дигидрокси-2,10-диоксо-2,10-дисульфид-14-(6,7,8,9-тетрагидро-2H-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2H,10H,12H-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-7-ил]-9Н-пурин-2-ил}амино)этил]-L-изолейцинамид
(Лекарственное средство-линкер 9)
С применением соединения, полученного на этапе 8-2 примера 8 (51,8 мг), и полученного на этапе 3 выше соединения (36,9 мг) проводили реакцию таким же образом, как на этапе 1 примера 23, и продукт затем очищали с помощью препаративной ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 20%-35% (0 мин - 30 мин)] с получением указанного в заголовке соединения (62,5 мг).
МС (ИЭР) m/z: 1397 (М-Н)-.
[1083]
Пример 70. Синтез лекарственного средства-линкера 10.
[Схема синтеза]
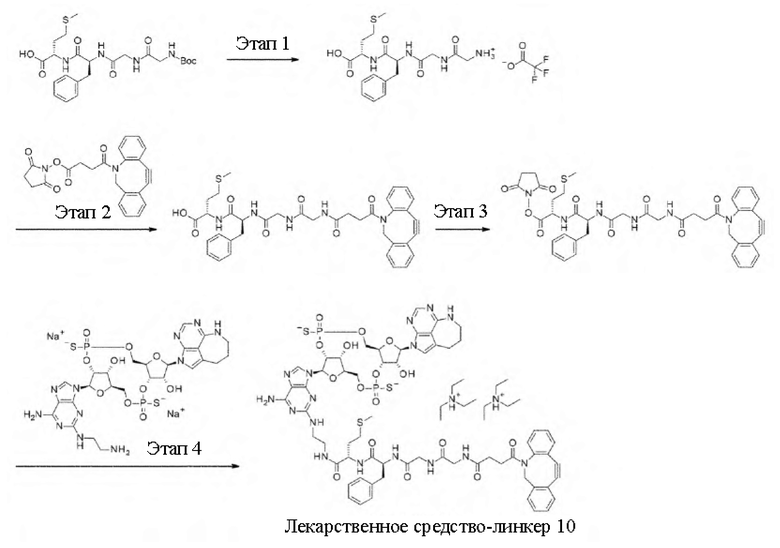
[1085]
(Этап 1)
Трифторацетат N-(азаниумилщетил)глицин-L-фенилаланил-L-метионина
С применением доступного для приобретения (Hangzhou Peptide Biochem Co., Ltd.) N-(трет-бутоксикарбонил)глицилглицил-L-фенилаланил-L-метионина (1,00 г) проводили реакцию таким же образом, как на этапе 1 примера 21, с получением неочищенной формы указанного в заголовке соединения (1,19 г).
МС (ИЭР) m/z: 411 (М+Н)+.
1Н-ЯМР (ДМСО-d6) δ: 8,48 (1Н, т, J=5,4 Гц), 8,41 (1Н, д, J=7,8 Гц), 8,27 (1Н, д, J=8,3 Гц), 7,97 (3Н, м), 7,28-7,18 (5Н, м), 4,58 (1Н, м), 4,32 (1Н, м), 3,85 (1Н, дд, J=17,1, 5,9 Гц), 3,68 (1Н, дд, J=16,6, 5,4 Гц), 3,56 (2Н, м), 3,04 (1Н, дд, J=13,9, 3,7 Гц), 2,74 (1Н, дд, J=13,7, 10,3 Гц), 2,47 (1Н, м), 2,05 (3Н, с), 2,00 (1Н, м), 1,88 (1Н, м), 1,48 (1Н, д, J=10,3 Гц) (показаны только наблюдаемые пики).
[1086]
(Этап 2)
N-[4-(11,12-дидегидродибензо[b,f]азоцин-5(6Н)-ил)-4-оксобутаноил]глицилглицил-L-фенилаланил-L-метионин
С применением соединения, полученного на этапе 1 выше (1,03 г), проводили реакцию таким же образом, как на этапе 2 примера 21, с получением указанного в заголовке соединения (652 мг).
МС (ИЭР) m/z: 698 (М+Н)+.
1Н-ЯМР (ДМСО-d6) δ: 12,67 (1Н, уш. с), 8,28 (0,7Н, д, J=7,9 Гц), 8,25 (0,3Н, д, J=7,9 Гц), 8,17 (0,7Н, т, J=5,7 Гц), 8,10 (0,3Н, т, J=5,7 Гц), 8,04-7,93 (2Н, м), 7,73-7,15 (13Н, м), 5,00 (1Н, д, J=13,9 Гц), 4,56-4,49 (1Н, м), 4,35-4,28 (1Н, м), 3,74-3,49 (5Н, м), 3,03 (1Н, дд, J=13,9, 3,6 Гц), 2,79-2,22 (5Н, м), 2,11-1,76 (4Н, м), 2,02 (2,1Н, с), 2,02 (0,9Н, с).
[1087]
(Этап 3)
2,5-диоксопирролидин-1-ил-N-[4-(11,12-дидегидродибензо[b,f]азоцин-5(6Н)-ил)-4-оксобутаноил]глицилглицил-L-фенилаланил-L-метионинат
С применением соединения, полученного на этапе 2 выше (201 мг), проводили реакцию таким же образом, как на этапе 3 примера 21, с получением указанного в заголовке соединения (95 мг).
МС (ИЭР) m/z: 795 (М+Н)+.
1Н-ЯМР (ДМСО-d6) δ: 8,84-8,59 (1Н, м), 8,17-7,96 (3Н, м), 7,72-7,15 (13Н, м), 5,01 (0,55Н, д, J=14,5 Гц), 5,00 (0,45Н, д, J=13,9 Гц), 4,86-4,74 (1Н, м), 4,56-4,48 (1Н, м), 3,75-3,50 (5Н, м), 3,05-2,52 (6Н, м), 2,82 (4Н, уш. с), 2,33-1,73 (4Н, м), 2,05 (3Н, с).
[1088]
(Этап 4)
Бис(N,N-диэтилэтанаминия) N-[4-(11,12-дидегидродибензо[b,f]азоцин-5(6Н)-ил)-4-оксобутаноил]глицилглицил-L-фенилаланил-N-[2-({6-амино-9-[(5R,7R,8R,12aR,14R,15R,15aS,16R)-15,16-дигидрокси-2,10-диоксо-2,10-дисульфид-14-(6,7,8,9-тетрагидро-2H-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2H,10H,12H-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-7-ил]-9Н-пурин-2-ил}амино)этил]-L-метионинамид
(Лекарственное средство-линкер 10)
С применением соединения, полученного на этапе 8-2 примера 8 (6,0 мг), и полученного на этапе 3 выше соединения (5,5 мг) проводили реакцию таким же образом, как на этапе 1 примера 23, и продукт затем очищали с помощью препаративной ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 20%-40% (0 мин - 30 мин)] с получением указанного в заголовке соединения (2,0 мг).
МС (ИЭР) m/z: 1467 (М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,23 (1Н, уш. с), 7,93 (1Н, с), 7,54-7,07 (14Н, м), 6,23 (1Н, д, J=6,7 Гц), 6,05-5,98 (1Н, м), 5,41-5,31 (2Н, м), 5,00-4,93 (1Н, м), 4,73-4,69 (1Н, м), 4,41-3,52 (14Н, м), 3,44-2,62 (12Н, м), 3,08 (12Н, к, J=7,3 Гц), 2,38-2,13 (3Н, м), 2,02-1,80 (8Н, м), 1,19 (18Н, т, J=7,3 Гц).
[1089]
Пример 71. Синтез лекарственного средства-линкера 11.
[Схема синтеза]
[1090]
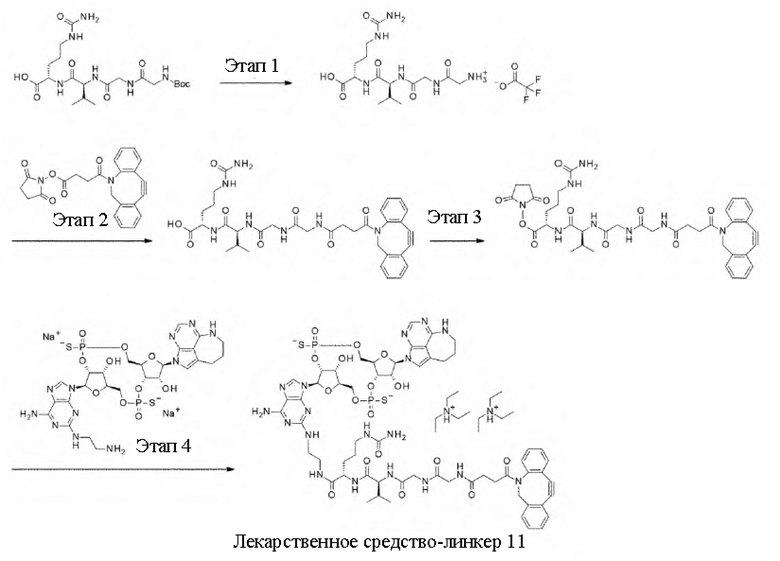
[1091]
(Этап 1)
Трифторацетат N-(азаниумилщетил)глицил-L-валил-N5-карбамоил-L-орнитина.
С применением доступного для приобретения (Hangzhou Peptide Biochem Co., Ltd.) N-(трет-бутоксикарбонил)глицилглицил-L-валил-N5-карбамоил-L-орнитина (1,00 г), проводили реакцию таким же образом, как на этапе 1 примера 21, с получением неочищенной формы указанного в заголовке соединения (1,02 г).
МС (ИЭР) m/z: 389 (М+Н)+.
1Н-ЯМР (ДМСО-d6) δ: 12,52 (1Н, уш. с), 8,55 (1Н, т, 3=5,4 Гц), 8,29 (1Н, д, J=7,3 Гц), 8,03-7,96 (4Н, м), 5,97 (1Н, уш. с), 5,40 (1Н, уш. с), 4,26 (1Н, т, J=7,8 Гц), 4,11 (1Н, м), 3,93-3,85 (2Н, м), 3,60 (2Н, д, J=5,9 Гц), 2,95 (2Н, уш. с), 1,97 (1Н, м), 1,69 (1Н, м), 1,56 (1Н, м), 1,39 (2Н, м), 0,88 (3Н, д, J=6,8 Гц), 0,84 (3Н, д, J=6,8 Гц) (показаны только наблюдаемые пики).
[1092]
(Этап 2)
N-[4-(11,12-дидегидродибензо[b,f]азоцин-5(6Н)-ил)-4-оксобутаноил]глицилглицил-L-валил-N5-карбамоил-L-орнитин
С применением соединения, полученного на этапе 1 выше (1,02 г), проводили реакцию таким же образом, как на этапе 2 примера 21, с получением указанного в заголовке соединения (795 мг).
МС (ИЭР) m/z: 676 (М+Н)+.
1Н-ЯМР (ДМСО-d6) δ: 12,49 (1Н, с), 8,28-8,22 (1Н, м), 8,18 (0,5Н, т, J=6,0 Гц), 8,13 (0,5Н, т, J=5,7 Гц), 8,06 (0,5Н, т, J=5,7 Гц), 8,01 (0,5Н, т, J=6,0 Гц), 7,75-7,71 (1Н, м), 7,69-7,65 (1Н, м), 7,62-7,59 (1Н, м), 7,52-7,43 (3Н, м), 7,40-7,28 (3Н, м), 5,93 (1Н, т, J=5,7 Гц), 5,38 (2Н, уш. с), 5,02 (0,5Н, д, J=13,9 Гц), 5,01 (0,5Н, д, J=13,9 Гц), 4,24 (1Н, дд, J=8,8, 7,0 Гц), 4,10 (1Н, м), 3,79-3,54 (5Н, м), 2,93 (2Н, к, J=6,4 Гц), 2,73-2,60 (1Н, м), 2,29 (1Н, м), 2,09-1,91 (2Н, м), 1,79 (1Н, м), 1,72-1,63 (1Н, м), 1,60-1,50 (1Н, м), 1,43-1,33 (2Н, м), 0,86 (3Н, д, J=6,7 Гц), 0,81 (1,5Н, д, J=6,7 Гц), 0,80 (1,5Н, д, J=6,7 Гц).
[1093]
(Этап 3)
2,5-диоксопирролидин-1-ил-N-[4-(11,12-дидегидродибензо[b,f]азоцин-5(6Н)-ил)-4-оксобутаноил]глицилглицил-L-валил-N5-карбамоил-L-орнитинат
С применением соединения, полученного на этапе 2 выше (300 мг), проводили реакцию таким же образом, как на этапе 3 примера 21, с получением указанного в заголовке соединения (177 мг).
МС (ИЭР) m/z: 773 (М+Н)+.
1Н-ЯМР (ДМСО-d6) δ: 8,75 (1Н, т, J=6,1 Гц), 8,20 (0,5Н, т, J=6,1 Гц), 8,15 (0,5Н, т, J=5,6 Гц), 8,06 (0,5Н, т, J=5,9 Гц), 8,02 (0,5Н, т, J=5,9 Гц), 7,83 (0,5Н, д, J=8,8 Гц), 7,77 (0,5Н, д, J=8,8 Гц), 7,72 (0,5Н, д, J=7,8 Гц), 7,67 (0,5Н, д, J=6,3 Гц), 7,62-7,59 (1Н, м), 7,52-7,44 (3Н, м), 7,39-7,29 (3Н, м), 6,03 (1Н, уш. с), 5,43 (2Н, уш. с), 5,03 (0,5Н, д, J=13,7 Гц), 5,01 (0,5Н, д, J=14,2 Гц), 4,59-4,54 (1Н, м), 4,25 (1Н, м), 3,79-3,68 (5Н, м), 2,97 (2Н, к, J=6,5 Гц), 2,80 (4Н, с), 2,71-2,60 (1Н, м), 2,28 (1Н, м), 2,10-2,03 (1Н, м), 2,00-1,93 (1Н, м), 1,88-1,72 (3Н, м), 1,54-1,49 (2Н, м), 0,85 (3Н, д, J=5,4 Гц), 0,81 (1,5Н, д, J=4,9 Гц), 0,80 (1,5Н, д, J=6,8 Гц).
[1094]
(Этап 4)
Бис(N,N-диэтилэтанаминия) N-[4-(11,12-дидегидродибензо[b,f]азоцин-5(6Н)-ил)-4-оксобутаноил]глицилглицил-L-валил-N-[2-({6-амино-9-[(5R,7R,8R,12aR,14R,15R,15aS,16R)-15,16-дигидрокси-2,10-диоксо-2,10-дисульфид-14-(6,7,8,9-тетрагидро-2H-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2H,10H,12H-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-7-ил]-9Н-пурин-2-ил}амино)этил]-N5-карбамоил-L-орнитинамид
(Лекарственное средство-линкер 11)
С применением соединения, полученного на этапе 8-2 примера 8 (5,0 мг), и полученного на этапе 3 выше соединения (3,8 мг) проводили реакцию таким же образом, как на этапе 1 примера 23, и продукт затем очищали с помощью препаративной ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 20%-35% (0 мин - 30 мин)] с получением указанного в заголовке соединения (3,0 мг).
МС (ИЭР) m/z: 1443 (М-Н)-.
[1095]
Пример 72. Синтез лекарственного средства-линкера 12.
[Схема синтеза]
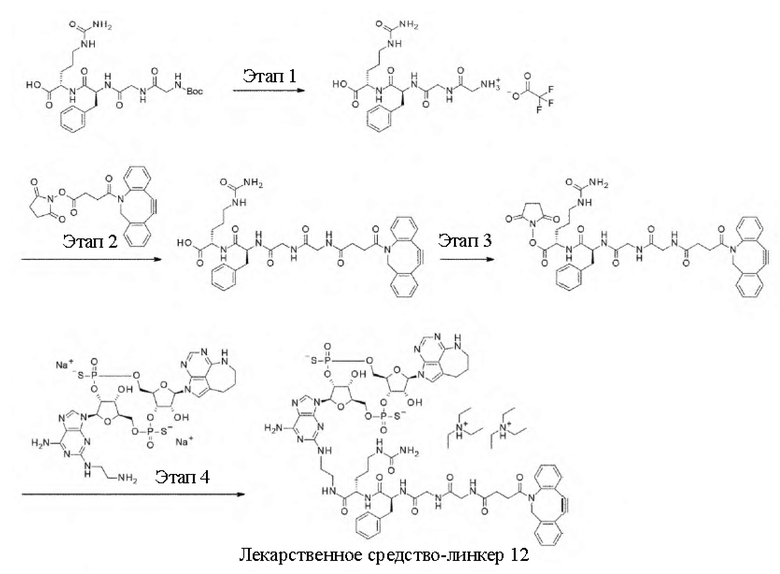
[1097]
(Этап 1)
Трифторацетат N-(азаниумилацетил)глтцин-L-фенилаланил-N5-карбамоил-L-орнитина
С применением доступного для приобретения (Hangzhou Peptide Biochem Co., Ltd.) N-(трет-бутоксикарбонил)глицилглицил-L-фенилаланил-N5-карбамоил-L-орнитина (1,00 г) проводили реакцию таким же образом, как на этапе 1 примера 21, с получением неочищенной формы указанного в заголовке соединения (1,05 г).
МС (ИЭР) m/z: 437 (М+Н)+.
1Н-ЯМР (ДМСО-d6) δ: 12,63 (1Н, уш. с), 8,49 (1Н, т, J=5,4 Гц), 8,40 (1Н, д, J=7,8 Гц), 8,26 (1Н, д, J=8,3 Гц), 7,97 (3Н, уш. с), 7,28-7,17 (5Н, м), 6,00 (1Н, уш. с), 5,41 (2Н, уш. с), 4,60 (1Н, м), 4,17 (1Н, м), 3,85 (1Н, дд, J=16,6, 5,4 Гц), 3,67 (1Н, дд, J=16,6, 5,4 Гц), 3,56 (2Н, д, J=5,9 Гц), 3,03 (1Н, дд, J=13,9, 3,7 Гц), 2,96 (2Н, уш. с), 2,73 (1Н, дд, J=13,9, 10,5 Гц), 1,73 (1Н, м), 1,58 (1Н, м), 1,40 (2Н, м).
[1098]
(Этап 2)
N-[4-(11,12-дидегидродибензо[b,f]азоцин-5(6Н)-ил)-4-оксобутаноил]глицилглицил-L-фенилаланил-N5-карбамоил-L-орнитин
С применением соединения, полученного на этапе 1 выше (1,05 г), проводили реакцию таким же образом, как на этапе 2 примера 21, с получением указанного в заголовке соединения (550 мг).
МС (ИЭР) m/z: 724 (М+Н)+.
1Н-ЯМР (ДМСО-d6) δ: 12,60 (1Н, с), 8,29 (1Н, т, J=9,1 Гц), 8,17 (0,5Н, т, J=5,7 Гц), 8,10 (0,5Н, т, J=5,7 Гц), 8,04-7,94 (2Н, м), 7,72-7,14 (13Н, м), 5,94 (1Н, уш.), 5,39 (2Н, с), 5,01 (0,5Н, д, J=13,9 Гц), 5,00 (0,5Н, д, J=14,5 Гц), 4,56 (1Н, м), 4,15 (1Н, м), 3,74-3,49 (5Н, м), 3,04-2,93 (3Н, м), 2,77-2,58 (2Н, м), 2,28 (1Н, м), 2,05 (1Н, м), 1,83-1,70 (2Н, м), 1,62-1,52 (1Н, м), 1,46-1,32 (2Н, м).
[1099]
(Этап 3)
2,5-диоксопирролидин-1-ил-N-[4-(11,12-дидегидродибензо[b,f]азоцин-5(6Н)-ил)-4-оксобутаноил]глицилглицил-L-фенилаланил-N5-карбамоил-L-орнитинат
С применением соединения, полученного на этапе 2 выше (225 мг), проводили реакцию таким же образом, как на этапе 3 примера 21, с получением указанного в заголовке соединения (226 мг).
МС (ИЭР) m/z: 821 (М+Н)+.
1Н-ЯМР (ДМСО-d6) δ: 8,80 (1Н, т, J=7,6 Гц), 8,19-7,96 (3Н, м), 7,72-7,15 (13Н, м), 6,03-5,89 (1Н, уш.), 5,43 (2Н, уш. с), 5,01 (0,5Н, д, J=14,0 Гц), 4,99 (0,5Н, д, J=14,0 Гц), 4,65-4,53 (2Н, м), 3,74-3,50 (5Н, м), 3,03-2,95 (3Н, м), 2,81 (4Н, с), 2,78-2,58 (2Н, м), 2,28 (1Н, м), 2,07 (1Н, м), 1,91-1,73 (3Н, м), 1,56-1,49 (2Н, м).
[1100]
(Этап 4)
Бис(N,N-диэтилэтанаминия) N-[4-(11,12-дидегидродибензо[b,f]азоцин-5(6Н)-ил)-4-оксобутаноил]глицилглицил-L-фенилаланил-N-[2-({6-амино-9-[(5R,7R,8R,12aR,14R,15R,15aS,16R)-15,16-дигидрокси-2,10-диоксо-2,10-дисульфид-14-(6,7,8,9-тетрагидро-2H-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2H,10H,12H-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-7-ил]-9Н-пурин-2-ил}амино)этил]-N5-карбамоил-L-орнитинамид
(Лекарственное средство-линкер 12)
С применением соединения, полученного на этапе 8-2 примера 8 (5,1 мг), и полученного на этапе 3 выше соединения (4,0 мг) проводили реакцию таким же образом, как на этапе 1 примера 23, и продукт затем очищали с помощью препаративной ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 20%-35% (0 мин - 30 мин)] с получением указанного в заголовке соединения (4,6 мг).
МС (ИЭР) m/z: 1493 (М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,31 (1Н, уш. с), 8,02 (1Н, с), 7,62-7,14 (14Н, м), 6,32 (1Н, д, J=6,0 Гц), 6,11 (1Н, уш. д, J=5,4 Гц), 5,55-5,41 (2Н, м), 5,06 (1Н, дд, J=13,9, 11,5 Гц), 4,86-2,73 (28Н, м), 3,19 (12Н, к, J=7,5 Гц), 2,39-2,16 (2Н, м), 2,04-1,94 (3Н, м), 1,84-1,74 (1Н, м), 1,73-1,62 (1Н, м), 1,51-1,37 (2Н, м), 1,29 (18Н, т, J=7,3 Гц).
[1101]
Пример 73. Синтез лекарственного средства-линкера 13.
[Схема синтеза]
[1102]
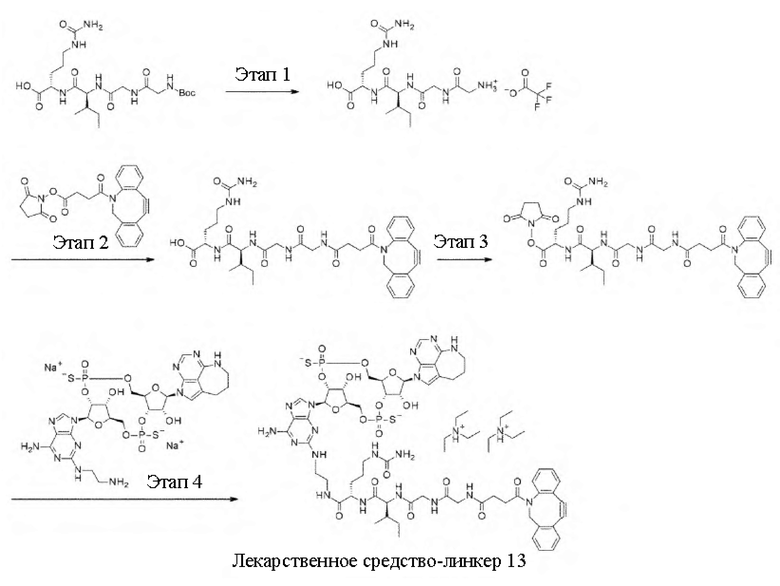
[1103]
(Этап 1)
Трифторацетат N-(азаниумилацетил)глицил-L-изолейцил-N5-карбамоил-L-орнитина
С применением доступного для приобретения (Hangzhou Peptide Biochem Co., Ltd.) N-(трет-бутоксикарбонил)глицилглицил-L-изолейцил-N5-карбамоил-L-орнитина (1,00 г) проводили реакцию таким же образом, как на этапе 1 примера 21, с получением неочищенной формы указанного в заголовке соединения (1,09 г).
МС (ИЭР) m/z: 403 (М+Н)+.
1Н-ЯМР (ДМСО-d6) δ: 8,54 (1Н, т, J=5,4 Гц), 8,30 (1Н, д, J=7,3 Гц), 8,03 (1Н, д, J=9,3 Гц), 7,99 (3Н, уш. с), 5,99 (1Н, уш. с), 4,27 (1Н, т, J=8,l Гц), 4,11 (1Н, м), 3,92-3,83 (2Н, м), 3,60 (2Н, м), 2,95 (2Н, м), 1,71 (2Н, м), 1,56 (1Н, м), 1,48-1,33 (3Н, м), 1,08 (1Н, м), 0,86 (3Н, д, J=6,8 Гц), 0,81 (3Н, т, J=7,3 Гц) (показаны только наблюдаемые пики).
[1104]
(Этап 2)
N-[4-(11,12-дидегидродибензо[b,f]азоцин-5(6Н)-ил)-4-оксобутаноил]глицилглицил-L-изолейцил-N5-карбамоил-L-орнитин
С применением соединения, полученного на этапе 1 выше (1,08 г), проводили реакцию таким же образом, как на этапе 2 примера 21, с получением указанного в заголовке соединения (524 мг).
МС (ИЭР) m/z: 690 (М+Н)+.
1Н-ЯМР (ДМСО-d6) δ: 12,47 (1Н, уш. с), 8,25 (1Н, т, J=6,0 Гц), 8,17 (0,5Н, т, J=5,7 Гц), 8,12 (0,5Н, т, J=6,0 Гц), 8,04 (0,5Н, т, J=6,0 Гц), 7,99 (0,5Н, т, J=6,0 Гц), 1,16-1,66 (2Н, м), 7,62-7,59 (1Н, м), 7,52-7,29 (6Н, м), 5,92 (1Н, т, J=5,4 Гц), 5,38 (2Н, с), 5,02 (0,5Н, д, J=14,5 Гц), 5,01 (0,5Н, д, J=13,9 Гц), 4,25 (1Н, т, J=8,2 Гц), 4,09 (1Н, м), 3,78-3,54 (5Н, м), 2,93 (2Н, к, J=6,4 Гц), 2,73-2,59 (1Н, м), 2,33-2,24 (1Н, м), 2,10-2,02 (1Н, м), 1,79 (1Н, ддд, J=16,6, 7,3, 5,7 Гц), 1,74-1,63 (2Н, м), 1,60-1,50 (1Н, м), 1,45-1,33 (3Н, м), 1,09-1,00 (1Н, м), 0,84 (3Н, д, J=6,7 Гц), 0,79 (1,5Н, т, J=6,7 Гц), 0,77 (1,5Н, т, J=7,0 Гц).
[1105]
(Этап 3)
2,5-диоксопирролидин-1-ил-N-[4-(11,12-дидегидродибензо[b,f]азоцин-5(6Н)-ил)-4-оксобутаноил]глицилглицил-L-изолейцил-N5-карбамоил-L-орнитинат
С применением соединения, полученного на этапе 2 выше (250 мг), проводили реакцию таким же образом, как на этапе 3 примера 21, с получением указанного в заголовке соединения (224 мг).
МС (ИЭР) m/z: 787 (М+Н)+.
1Н-ЯМР (ДМСО-d6) δ: 8,75 (1Н, т, J=6,4 Гц), 8,19 (0,5Н, т, J=5,8 Гц), 8,14 (0,5Н, т, J=5,8 Гц), 8,06 (0,5Н, т, J=5,8 Гц), 8,01 (0,5Н, т, J=5,8 Гц), 7,89-7,65 (2Н, м), 7,63-7,58 (1Н, м), 7,53-7,27 (6Н, м), 5,96 (1Н, м), 5,42 (2Н, уш. с), 5,02 (0,5Н, д, J=14,0 Гц), 5,01 (0,5Н, д, J=14,0 Гц), 4,64-4,51 (1Н, м), 4,26 (1Н, т, J=8,2 Гц), 3,79-3,55 (5Н, м), 3,01-2,93 (2Н, м), 2,80 (4Н, уш. с), 2,73-2,58 (1Н, м), 2,28 (1Н, м), 2,12-2,02 (1Н, м), 1,87-1,68 (4Н, м), 1,54-1,38 (3Н, м), 1,11-1,02 (1Н, м), 0,83 (3Н, д, J=6,7 Гц), 0,78 (1,5Н, т, J=6,7 Гц), 0,76 (1,5Н, т, J=6,7 Гц).
[1106]
(Этап 4)
Бис(N,N-диэтилэтанаминия) N-[4-(11,12-дидегидродибензо[b,f]азоцин-5(6Н)-ил)-4-оксобутаноил]глицилглицил-L-изолейцил-N-[2-({6-амино-9-[(5R,7R,8R,12aR,14R,15R,15aS,16R)-15,16-дигидрокси-2,10-диоксо-2,10-дисульфид-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2H,10H,12H-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-7-ил]-9Н-пурин-2-ил}амино)этил]-N5-карбамоил-L-орнитинамид
(Лекарственное средство-линкер 13)
С применением соединения, полученного на этапе 8-2 примера 8 (5,4 мг), и полученного на этапе 3 выше соединения (4,0 мг) проводили реакцию таким же образом, как на этапе 1 примера 23, и продукт затем очищали с помощью препаративной ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 20%-35% (0 мин - 30 мин)] с получением указанного в заголовке соединения (4,7 мг).
МС (ИЭР) m/z: 1457 (М-Н)-.
[1107]
Пример 74. Синтез лекарственного средства-линкера 14.
[Схема синтеза]
[1108]
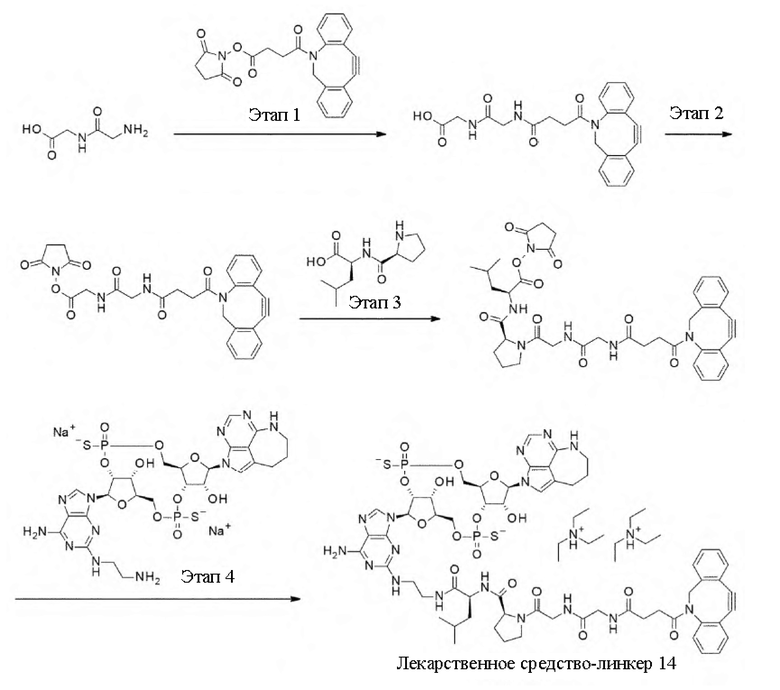
[1109]
(Этап 1)
N-[4-(11,12-дидегидродибензо[b,f]азоцин-5(6Н)-ил)-4-оксобутаноил]глицилглицин
С применением доступного для приобретения (Tokyo Chemical Industry Co., Ltd.) глицилглицина (0,61 г) проводили реакцию таким же образом, как на этапе 2 примера 21, с получением указанного в заголовке соединения (1,08 г).
МС (ИЭР) m/z: 420 (М+Н)+.
1Н-ЯМР (ДМСО-d6) δ: 12,57 (1Н, с), 8,12 (1Н, т, J=6,0 Гц), 8,07 (1Н, т, J=5,7 Гц), 7,69-7,67 (1Н, м), 7,62-7,60 (1Н, м), 7,52-7,29 (6Н, м), 5,03 (1Н, д, J=13,9 Гц), 3,73 (2Н, д, J=6,0 Гц), 3,70-3,55 (3Н, м), 2,64 (1Н, м), 2,28 (1Н, м), 2,06 (1Н, ддд, J=15,3, 7,7, 5,6 Гц), 1,79 (1Н, ддд, J=16,5, 7,4, 5,6 Гц).
[1110]
(Этап 2)
2,5-диоксопирролидин-1-ил-N-[4-(11,12-дидегидродибензо[b,f]азоцин-5(6Н)-ил)-4-оксобутаноил]глицилглицинат
С применением соединения, полученного на этапе 1 выше (1,07 г), проводили реакцию таким же образом, как на этапе 3 примера 21, с получением указанного в заголовке соединения (942 мг).
МС (ИЭР) m/z: 517 (М+Н)+.
1Н-ЯМР (ДМСО-d6) δ: 8,40 (1Н, т, J=6,0 Гц), 8,19 (1Н, т, J=6,0 Гц), 7,69-7,67 (1Н, м), 7,64-7,62 (1Н, м), 7,53-7,29 (6Н, м), 5,05 (1Н, д, J=14,5 Гц), 4,26 (1Н, дд, J=18,1, 6,0 Гц), 4,19 (1Н, дд, J=18,1, 6,0 Гц), 3,72-3,59 (3Н, м), 2,81 (4Н, уш. с), 2,68-2,59 (1Н, м), 2,33-2,24 (1Н, м), 2,07 (1Н, ддд, J=15,4, 7,6, 5,7 Гц), 1,80 (1Н, ддд, J=16,3, 7,3, 5,4 Гц).
[1111]
(Этап 3)
2,5-диоксопирролидин-1-ил-N-[4-(11,12-дидегидродибензо[b,f]азоцин-5(6Н)-ил)-4-оксобутаноил]глицилглицил-L-пролил-L-лейцинат
В суспензию полученного на этапе 2 выше соединения (250 мг) и доступного для приобретения (KOKUSAN CHEMICAL Co., Ltd.) и L-пролил-L-лейцина (166 мг) в N,N-диметилформамиде (5,0 мл) добавляли N,N-диизопропилэтиламин (0,25 мл), и реакционную смесь перемешивали при комнатной температуре в течение 2,5 часов. Туда, дополнительно добавляли L-пролил-L-лейцин (833 мг) и реакционную смесь перемешивали в течение ночи. После концентрирования реакционной смеси при пониженном давлении к остатку добавляли хлороформ (50 мл) и 10% водный раствор лимонной кислоты (10 мл) и осуществляли экстрагирование продукта хлороформом. Органический слой концентрировали при пониженном давлении и остаток частично очищали с помощью колоночной хроматографии на силикагеле [хлороформ/нижний слой (хлороформ:метанол:вода = 7:3:1)]. К раствору неочищенного продукта в N,N-диметилформамиде (5,0 мл) добавляли N-гидроксисукцинимид (67 мг) и 1-этил-3-(3-диметиламинопропил)-карбодиимида гидрохлорид (111 мг) и реакционную смесь перемешивали при комнатной температуре в течение 1,5 часов. Реакционную смесь концентрировали при пониженном давлении, хлороформ (40 мл) и воду (15 мл) добавляли к остатку и осуществляли экстрагирование продукта хлороформом. После высушивания органического слоя над безводным сульфатом натрия осушающий агент удаляли путем фильтрации и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле [хлороформ/метанол], [этилацетат/метанол] в данном порядке с получением указанного в заголовке соединения (127 мг).
МС (ИЭР) m/z: 727 (М+Н)+.
1H-ЯМР (ДМСО-d6) δ: 8,85 (0,3Н, м), 8,56 (0,5Н, д, J=7,3 Гц), 8,33 (0,2Н, м), 8,09 (1Н, м), 7,91-7,79 (1Н, м), 7,70-7,28 (8Н, м), 5,04 (0,7Н, д, J=14,5), 5,03 (0,3Н, д, J=13,9), 4,71-4,29 (2Н, м), 4,01-3,30 (7Н, м), 2,79 (4Н, уш. с), 2,63 (1Н, м), 2,33-1,64 (10Н, м), 0,93-0,83 (6Н, м).
[1112]
(Этап 4)
Бис(N,N-диэтилэтанаминия) N-[4-(11,12-дидегидродибензо[b,f]азоцин-5(6Н)-ил)-4-оксобутаноил]глицилглицил-L-пролил-N-[2-({6-амино-9-[(5R,7R,8R,12aR,14R,15R,15aS,16R)-15,16-дигидрокси-2,10-диоксо-2,10-дисульфид-14-(6,7,8,9-тетрагидро-2H-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2H,10H,12H-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-7-ил]-9Н-пурин-2-ил}амино)этил]-L-изолейцинамид
(Лекарственное средство-линкер 14)
С применением соединения, полученного на этапе 8-2 примера 8 (6,9 мг), и полученного на этапе 3 выше соединения (4,7 мг) проводили реакцию таким же образом, как на этапе 1 примера 23, и продукт затем очищали с помощью препаративной ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 20%-40% (0 мин - 30 мин)] с получением указанного в заголовке соединения (7,2 мг).
МС (ИЭР) m/z: 1397 (М-Н)-.
[1113]
Пример 75. Синтез лекарственного средства-линкера 15.
[Схема синтеза]
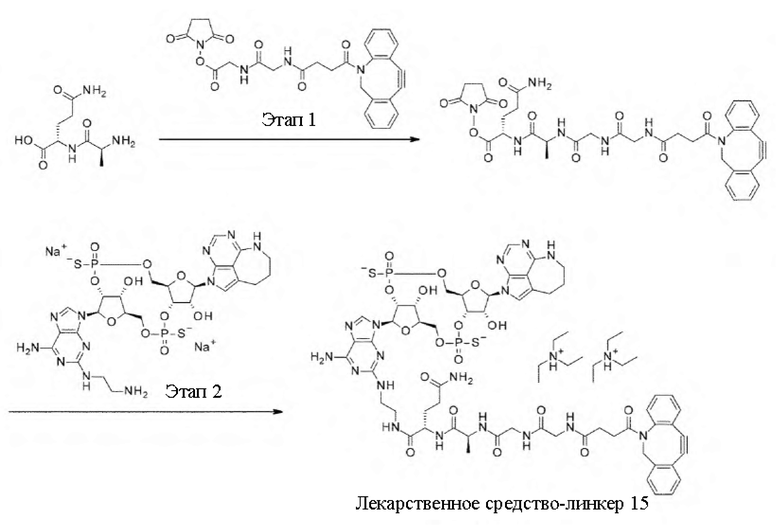
[1115]
(Этап 1)
2,5-диоксопирролидин-1-ил-N-[4-(11,12-дидегадродибензо[b,f]азоцин-5(6Н)-ил)-4-оксобутаноил]глицилглицил-L-аланил-L-глутаминат
С применением доступного для приобретения (Wako Pure Chemical Industries, Ltd.) L-аланил-L-глутамина (263 мг) и полученного на этапе 2 примера 74 соединения (250 мг) проводили реакцию таким же образом, как на этапе 3 примера 74, с получением указанного в заголовке соединения (90 мг).
МС (ИЭР) m/z: 716 (М+Н)+.
1Н-ЯМР (ДМСО-d6) δ: 8,59 (0,8Н, д, J=7,3 Гц), 8,48 (0,2Н, м), 8,20-7,92 (3Н, м), 7,70-7,25 (9Н, м), 6,85 (1Н, уш. с), 5,02 (1Н, д, J=13,9 Гц), 4,62 (1Н, м), 4,33 (1Н, м), 3,75-3,56 (5Н, м), 2,80 (4Н, уш. с), 2,64 (1Н, м), 2,33-2,21 (3Н, м), 2,15-2,03 (2Н, м), 1,99-1,87 (1Н, м), 1,83-1,76 (1Н, м), 1,24-1,17 (3Н, м).
[1116]
(Этап 2)
Бис(N,N-диэтилэтанаминий)N-[4-(11,12-дидегидродибензо[b,f]азоцин-5(6Н)-ил)-4-оксобутаноил]глицилглицил-L-аланил-N1-[2-({6-амино-9-[(5R,7R,8R,12aR,14R,15R,15aS,16R)-15,16-дигидрокси-2,10-диоксо-2,10-дисульфид-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-7-ил]-9Н-пурин-2-ил}амино)этил]-L-глутамамид
(Лекарственное средство-линкер 15)
С применением соединения, полученного на этапе 8-2 примера 8 (6,9 мг), и полученного на этапе 1 выше соединения (4,7 мг) проводили реакцию таким же образом, как на этапе 1 примера 23, и продукт затем очищали с помощью препаративной ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 20%-40% (0 мин - 30 мин)] с получением указанного в заголовке соединения (3,9 мг).
МС (ИЭР) m/z: 1388 (М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,38-8,35 (1Н, м), 8,02 (1Н, с), 7,66-7,13 (9Н, м), 6,33 (1Н, д, J=6,7 Гц), 6,14-6,10 (1Н, м), 5,51-5,42 (2Н, м), 5,14-5,06 (1Н, м), 4,84-4,79 (1Н, м), 4,53-3,64 (14Н, м), 3,56-3,11 (6Н, м), 3,15 (12Н, к, J=7,5 Гц), 2,92-2,69 (4Н, м), 2,42-1,93 (8Н, м), 1,45-1,24 (3Н, м), 1,27 (18 Н, т, J=7,3 Гц).
[1117]
Пример 76. Синтез лекарственного средства-линкера 16.
[Схема синтеза]
[1118]
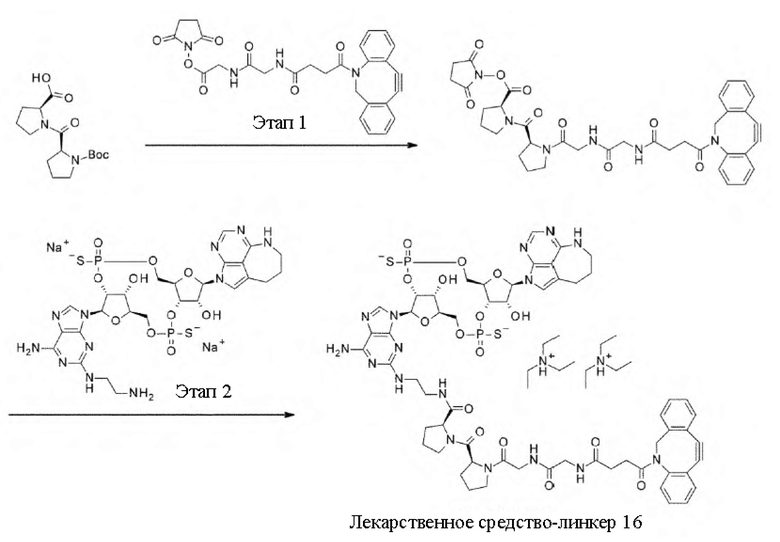
[1119]
(Этап 1)
2,5-диоксопирролидин-1-ил-N-[4-(11,12-дидегидродибензо[b,f]азоцин-5(6Н)-ил)-4-оксобутаноил]глицилглицил-L-пролил-L-пролинат
С применением доступного для приобретения (Cool Pharm Ltd.) 1-(трет-бутоксикарбонил)-L-пролил-L-пролина (777 мг) проводили реакцию таким же образом, как на этапе 1 примера 21, с получением неочищенной формы трифторацетата 1-[(2S)-пирролидин-1-ий-2-карбонил]-L-пролина. С применением данного неочищенного продукта и полученного на этапе 2 примера 74 соединения (428 мг) проводили реакцию таким же образом, как на этапе 3 примера 74, с получением указанного в заголовке соединения (238 мг).
МС (ИЭР) m/z: 711 (М+Н)+.
1Н-ЯМР (ДМСО-d6) δ: 8,12-8,08 (1Н, м), 7,90 (0,2Н, с), 7,82 (0,8Н, д, J=4,8 Гц), 7,70-7,61 (2Н, м), 7,52-7,29 (6Н, м), 5,07-5,01 (1Н, м), 4,88-4,86 (0,2Н, м), 4,77-4,74 (0,2Н, м), 4,69-4,62 (0,8Н, м), 4,60-4,56 (0,8Н, м), 4,02-3,38 (9Н, м), 2,79 (4Н, уш. с), 2,68-2,58 (1Н, м), 2,37-2,24 (2Н, м), 2,16-1,73 (9Н, м).
[1120]
(Этап 2)
Бис(N,N-диэтилэтанаминия) N-[4-(11,12-дидегидродибензо[b,f]азоцин-5(6Н)-ил)-4-оксобутаноил]глицилглицил-L-пролил-N-[2-({6-амино-9-[(5R,7R,8R,12aR,14R,15R,15aS,16R)-15,16-дигидрокси-2,10-диоксо-2,10-дисульфид-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-7-ил]-9Н-пурин-2-ил}амино)этил]-L-пролинамид
(Лекарственное средство-линкер 16)
С применением соединения, полученного на этапе 8-2 примера 8 (6,9 мг), и полученного на этапе 1 выше соединения (4,6 мг) проводили реакцию таким же образом, как на этапе 1 примера 23, и продукт затем очищали с помощью препаративной ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 20%-40% (0 мин - 30 мин)] с получением указанного в заголовке соединения (5,8 мг).
МС (ИЭР) m/z: 1383 (М+Н)+.
[1121]
Пример 77. Синтез лекарственного средства-линкера 17.
[Схема синтеза]
[1122]
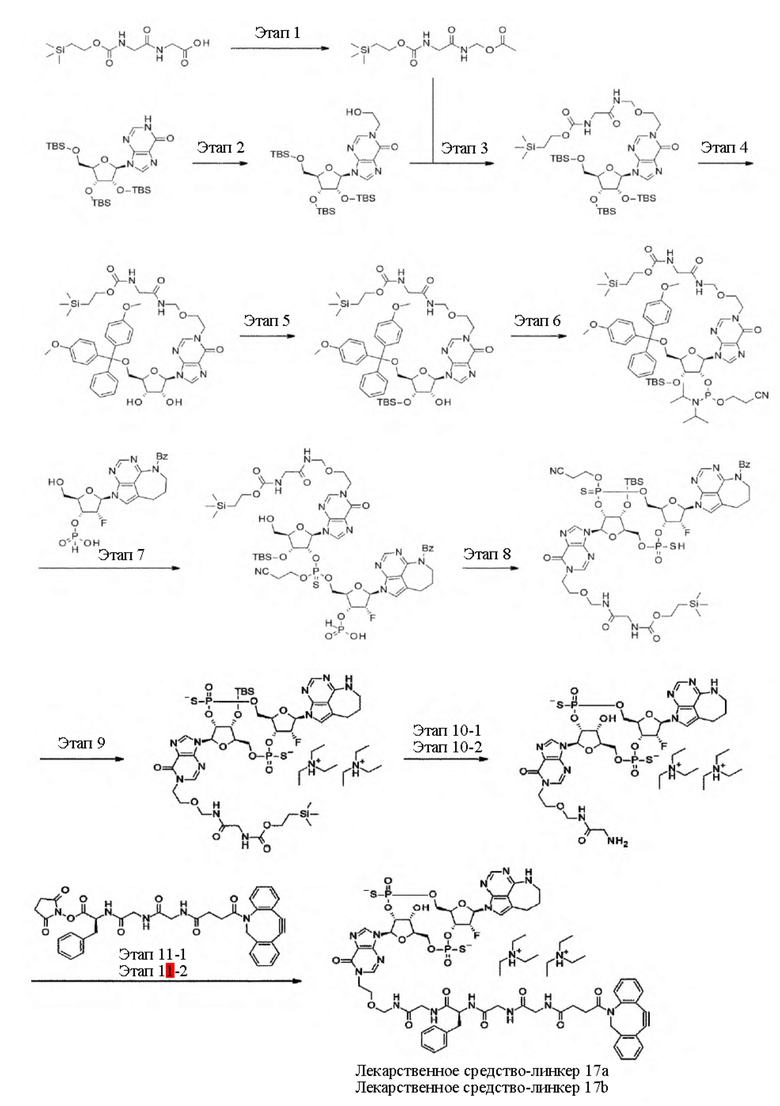
[1123]
(Этап 1)
[(N-{[2-(триметилсилил)этокси]карбонил}глицил)амино]метилацетат
С применением доступного для приобретения (Sundia) N-{[2-(триметилсилил)этокси]карбонил}глицилглицина (9,32 г) проводили реакцию таким же образом, как на этапе 1 примера 67, с получением указанного в заголовке соединения (8,24 г).
1Н-ЯМР (CDCl3) δ: 7,14 (1Н, уш. с), 5,27 (2Н, д, J=7,3 Гц), 5,20 (1Н, уш. с), 4,22-4,16 (2Н, м), 3,88 (2Н, д, J=6,0 Гц), 2,08 (3Н, с), 1,04-0,97 (2Н, м), 0,05 (9Н, с).
[1124]
(Этап 2)
2',3',5'-трис-O-[трет-бутил(диметил)силил]-1-(2-гидроксиэтил)инозин
В смешанный раствор 2',3',5'-трис-O-[трет-бутил(диметил)силил]инозина (31,3 г) как соединения, известного в литературе (Chem. Pharm. Bull. 1987, 35 (1), 72-79), в тетрагидрофуране (75 мл)-N,N-диметилацетамиде (75 мл) добавляли, 2-бромоэтанол (4,82 мл) и 1,8-диазабицикло[5,4,0]-7-ундецен (7,65 мл), и реакционную смесь перемешивали при комнатной температуре в течение 23 часов. Воду и этилацетат добавляли в реакционную смесь, которую осуществляли экстрагирование этилацетатом. Органический слой промывали рассолом и сушили над безводным сульфатом натрия, и осушающий агент затем удаляли путем фильтрации и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле [гексан/этилацетат] с получением указанного в заголовке соединения (29,4 г).
МС (ИЭР) m/z: 655 (М+Н)+.
1Н-ЯМР (CDCl3) δ: 8,16 (1Н, с), 7,99 (1Н, д, J=2,4 Гц), 5,97 (1Н, д, J=4,2 Гц), 4,40-4,25 (3Н, м), 4,18-4,06 (3Н, м), 4,03-3,92 (2Н, м), 3,79 (1Н, дд, J=11,5, 2,4 Гц), 3,08-2,83 (1Н, уш. м), 0,96 (9Н, с), 0,92 (9Н, с), 0,82 (9Н, с), 0,15 (3Н, с), 0,14 (3Н, с), 0,09 (3Н, с), 0,08 (3Н, с), -0,02 (3Н, с), -0,15 (3Н, с).
[1125]
(Этап 3)
2',3',5'-трис-О-[трет-бутил(диметил)силил]-1-(2-{[(N-{[2-(триметилсилил)этокси]карбонил}глицил)амино]метокси}этил)инозин
К раствору соединения, полученного на этапе 2 выше (15,6 г), в толуоле (46,8 мл) добавляли полученное на этапе 1 выше соединение (10,4 г) и пиридин (9,63 мл), и реакционную смесь перемешивали при 110°С в течение 12 часов. Полученное на этапе 1 выше соединение (3,46 г) дополнительно добавляли в реакционную смесь, которую перемешивали при 110°С в течение 1 дня. Насыщенный водный раствор гидрокарбоната натрия и дихлорметана добавляли в реакционную смесь, которую осуществляли экстрагирование дихлорметаном. После высушивания органического слоя над безводным сульфатом натрия осушающий агент удаляли путем фильтрации и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле [гексан/этилацетат] с получением указанного в заголовке соединения (20,6 г: с примесями).
МС (ИЭР) m/z: 885 (М+Н)+.
[1126]
(Этап 4)
5'-O-[бис(4-метоксифенил)(фенил)метил]-1-(2-{[(N-{[2-(триметилсилил)этокси]карбонил}глицил)амино]метокси}этил)инозин
К раствору соединения, полученного на этапе 3 выше (20,6 г), в тетрагидрофуране (50 мл) добавляли тригидрофторид триэтиламина (10 мл) и реакционную смесь перемешивали при комнатной температуре в течение 17 часов. В реакционную смесь медленно добавляли смесь 1 М раствора гидрокарбоната триэтиламмония (50 мл) и триэтиламина (10 мл) при охлаждении льдом, и реакционную смесь затем концентрировали при пониженном давлении. Остаток частично очищали с помощью колоночной хроматографии на силикагеле С18 [вода/ацетонитрил], а затем лиофилизировали. Полученную неочищенную форму подвергали азеотропной перегонке с пиридином, и 4,4'-диметокситритилхлорид (4,73 г) добавляли в раствор остатка в пиридине (50 мл) при 0°С, и реакционную смесь перемешивали при 4°С в течение 17 часов. Метанол (2 мл) добавляли в реакционную смесь, которую перемешивали при комнатной температуре в течение 15 минут, а затем концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле [гексан/этилацетат/метанол/0,1% триэтиламин] с получением указанного в заголовке соединения (9,18 г: с примесями).
МС (ИЭР) m/z: 845 (М+Н)+.
1Н-ЯМР (CDCl3) δ: 7,94 (1Н, с), 7,88 (1Н, с), 7,65 (1Н, уш. с), 7,41-7,36 (2Н, м), 7,32-7,15 (7Н, м), 6,83-6,76 (4Н, м), 5,96 (1Н, д, J=6,1 Гц), 5,73-5,65 (2Н, м), 4,87-4,80 (1Н, м), 4,76-4,61 (2Н, м), 4,44-4,39 (1Н, м), 4,35-4,30 (1Н, м), 4,22-4,05 (4Н, м), 3,83-3,73 (2Н, м), 3,77 (6Н, с), 3,72-3,67 (2Н, м), 3,48-3,32 (3Н, м), 0,99-0,91 (2Н, м), 0,02 (9Н, с).
[1127]
(Этап 5)
5'-O-[бис(4-метоксифенил)(фенил)метил]-3'-O-[трет-бутил(диметил)силил]-1-(2-{[(N-{[2-(триметилсилил)этокси]карбонил}глицил)амино]метокси}этил)инозин
С применением соединения, полученного на этапе 4 выше (5,96 г), проводили реакцию таким же образом, как на этапе 3 примера 5, с получением указанного в заголовке соединения (2,33 г) и 5'-O-[бис(4-метоксифенил)(фенил)метил]-2'-O-[трет-бутил(диметил)силил]-1-(2-{[(N-{[2-(триметилсилил)этокси]карбонил}глицил)амино]метокси}этил)инозина (2,45 г) в качестве региоизомера указанного в заголовке соединения.
МС (ИЭР) m/z: 959 (М+Н)+.
1Н-ЯМР (CDCl3) δ: 7,99 (1Н, с), 7,93 (1Н, с), 7,45-7,39 (2Н, м), 7,35-7,18 (7Н, м), 7,05 (1Н, уш. с), 6,84-6,77 (4Н, м), 5,92 (1Н, д, J=5,4 Гц), 5,46 (1Н, уш. с), 4,71-4,61 (3Н, м), 4,54-4,51 (1Н, м), 4,22-4,10 (5Н, м), 3,81-3,76 (2Н, м), 3,78 (3Н, с), 3,78 (3Н, с), 3,74 (2Н, д, J=6,0 Гц), 3,48 (1Н, дд, J=10,9, 4,2 Гц), 3,26 (1Н, дд, J=10,9, 4,2 Гц), 3,16 (1Н, д, J=6,7 Гц), 1,00-0,93 (2Н, м), 0,89 (9Н, с), 0,09 (3Н, с), 0,02 (9Н, с), 0,02 (3Н, с).
Региоизомер (2'-O-TBS-форма)
МС (ИЭР) m/z: 959 (М+Н)+.
1Н-ЯМР (CDCl3) δ: 7,99 (1Н, с), 7,91 (1Н, с), 7,48-7,42 (2Н, м), 7,37-7,18 (8Н, м), 6,85-6,78 (4Н, м), 5,96 (1Н, д, J=5,4 Гц), 5,63 (1Н, уш. с), 4,88 (1Н, т, J=5,l Гц), 4,66 (2Н, д, J=6,7 Гц), 4,36-4,32 (1Н, м), 4,27-4,19 (2Н, м), 4,18-4,10 (3Н, м), 3,81-3,74 (4Н, м), 3,78 (3Н, с), 3,78 (3Н, с), 3,50 (1Н, дд, J=10,9, 3,6 Гц), 3,38 (1Н, дд, J=10,9, 3,6 Гц), 2,73 (1Н, д, J=4,2 Гц), 0,97-0,90 (2Н, м), 0,86 (9Н, с), 0,02 (3Н, с), 0,01 (9Н, с), - 0,09 (3Н, с).
[1128]
(Этап 6)
5'-O-[бис(4-метоксифенил)(фенил)метил]-3'-O-[трет-бутил(диметил)силил]-2'-O-{(2-цианоэтокси)[ди(пропан-2-ил)амино]фосфанил}-1-(2-{[(N-{[2-(триметилсилил)этокси]карбонил}глицил)амино]метокси}этил)инозин
С применением соединения, полученного на этапе 5 выше (2,33 г), проводили реакцию таким же образом, как на этапе 4 примера 5, с получением указанного в заголовке соединения (2,72 г) в виде смеси диастереомеров у атома фосфора (соотношение диастереомеров = 6:4).
МС (ИЭР) m/z: 1159 (M+H)+.
1H-Ямр (CDCl3) δ: 8,03 (0,4Н, с), 8,02 (0,6Н, с), 7,95 (0,6Н, с), 7,92 (0,4Н, с), 7,46-7,40 (2Н, м), 7,35-7,17 (7Н, м), 6,88 (1Н, уш. с), 6,84-6,78 (4Н, м), 6,15 (0,6Н, д, J=4,2 Гц), 6,10 (0,4Н, д, J=4,8 Гц), 5,34 (1Н, уш. с), 4,86-4,61 (3Н, м), 4,48-4,42 (1Н, м), 4,29-4,09 (5Н, м), 3,83-3,44 (9Н, м), 3,79 (3Н, с), 3,78 (3Н, с), 3,32-3,23 (1Н, м), 2,58-2,49 (1Н, м), 2,44-2,38 (1Н, м), 1,15 (3,6Н, д, J=6,7 Гц), 1,11 (6Н, д, J=6,7 Гц), 1,04-0,92 (2Н, м), 0,97 (2,4Н, д, J=6,7 Гц), 0,85 (3,6Н, с), 0,84 (5,4Н, с), 0,09 (1,2Н, с), 0,06 (1,8Н, с), 0,03 (9Н, с), 0,00 (3Н, с).
[1129]
(Этап 7)
С применением соединения, полученного на этапе 8 примера 44 (2,15 г), проводили реакцию таким же образом, как на этапе 7 примера 1, с получением раствора в ацетонитриле 6-бензоил-2-{2-дезокси-2-фтор-3-О-[гидрокси(оксо)-λ5-фосфанил]-β-D-рибофуранозил}-6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулена. С применением полученного раствора в ацетонитриле и полученного на этапе 6 выше соединения (2,72 г), проводили реакцию таким же образом, как на этапе 8 примера 1, и полученный в результате этого неочищенный продукт непосредственно использовали для следующей реакции.
[1130]
(Этап 8)
2-(триметилсилил)этил-(2-{[(2-{9-[(5R,7R,8R,12aR,14R,15R,15aR,16R)-14-(6-бензоил-6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)-16-{[трет-бутил(диметил)силил]окси}-10-(2-цианоэтокси)-15-фтор-2-оксо-2-сульфанил-10-сульфанилиденоктагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-7-ил]-6-оксо-6,9-дигидро-1Н-пурин-1-ил}этокси)метил]амино}-2-оксоэтил)карбамат
С применением неочищенного продукта, полученного на этапе 7 выше, проводили реакцию таким же образом, как на этапе 9 примера 1, с получением указанного в заголовке соединения (1,47 г: с примесями) в виде смеси диастереомеров у атома фосфора.
МС (ИЭР) m/z: 1278(M+H)+.
[1131]
(Этап 9)
Бис(N,N-диэтилэтанаминия) (5R,7R,8R,12aR,14R,15R,15aR,16R)-16-{[трет-бутил(диметил)силил]окси}-15-фтор-2,10-диоксо-7-[6-оксо-1-(2-{[(N-{[2-(триметилсилил)этокси]карбонил}глицил)амино]метокси}этил)-1,6-дигидро-9Н-пурин-9-ил]-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2H,10H,12H-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
В смешанный раствор соединения, полученного на этапе 8 выше (1,47 г), в метаноле (10 мл)/тетрагидрофуране (10 мл) добавляли 28% водный аммиак (10 мл) и реакционную смесь перемешивали при 50°С в течение 6 часов. После концентрирования реакционной смеси при пониженном давлении остаток очищали с помощью колоночной хроматографии на силикагеле С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил] с получением диастереомера 1 (204 мг: с примесями) и диастереомера 2 (205 мг: с примесями) указанного в заголовке соединения.
Диастереомер 1 (менее полярный)
МС (ИЭР) m/z: 1121 (М+Н)+.
Диастереомер 2 (более полярный)
МС (ИЭР) m/z: 1121 (М+Н)+.
[1132]
(Этап 10-1)
Бис(N,N-диэтилэтанаминия) (5R,7R,8R,12aR,14R,15R.15aR,16R)-15-фтор-7-(1-{2-[(глициламино)метокси]этил}-6-оксо-1,6-дигидро-9Н-пурин-9-ил)-16-гидрокси-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2H,10H,12H-5,8-метано-2λ5,10λ5-фуро[3,2-1] [1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
С применением соединения, полученного на этапе 9 выше (диастереомер 1) (204 мг). проводили реакцию таким же образом, как на этапе 9-1 примера 11, и продукт затем очищали с помощью колоночной хроматографии на силикагеле С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил] и препаративной ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/раствор ацетонитр ила-метанола (1:1), раствор ацетонитрила-метанола (1:1): 10% - 50% (0 мин - 40 мин)] с получением указанного в заголовке соединения (40,7 мг: с примесями).
МС (ИЭР) m/z: 863(M+H)+.
[1133]
(Этап 10-2)
Бис(N,N-диэтилэтанаминия) (5R,7R,8R,12aR,14R,15R,15aR,16R)-15-фтор-7-(1-{2-[(глициламино)метокси]этил}-6-оксо-1,6-дигидро-9Н-пурин-9-ил)-16-гидрокси-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2H,10H,12H-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(тиолят)
С применением соединения, полученного на этапе 9 выше (диастереомер 2) (205 мг), проводили реакцию таким же образом, как на этапе 9-1 примера 11, и продукт затем очищали с помощью колоночной хроматографии на силикагеле С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил] и препаративной ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/раствор ацетонитр ила-метанола (1:1), раствор ацетонитрила-метанола (1:1): 10% - 50% (0 мин - 40 мин)] с получением указанного в заголовке соединения (50,8 мг: с примесями).
МС (ИЭР) m/z: 863(M+H)+.
[1134]
(Этап 11-1)
Бис(N,N-диэтилэтанаминия) N-[4-(11,12-дидегидродибензо[b,f]азоцин-5(6Н)-ил)-4-оксобутаноил]глицилглицил-L-фенилаланил-N-[(2-{9-[(5R,7R,8R,12aR,14R,15R,15aR,16R)-15-фтор-16-гидрокси-2,10-диоксо-2,10-дисульфид-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-7-ил]-6-оксо-6,9-дигидро-1Н-пурин-1-ил}этокси)метил]глицинамид (Лекарственное средство-линкер 17а: диастереомер 1)
С применением соединения, полученного на этапе 10-1 выше (40,7 мг), проводили реакцию таким же образом, как на этапе 9-1 примера 22, и продукт затем очищали с помощью колоночной хроматографии на силикагеле С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил]. препаративной ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил. ацетонитрил: 20% - 45% (0 мин - 40 мин)] и препаративной ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/метанол, метанол: 40% - 90% (0 мин - 40 мин)] с получением указанного в заголовке соединения (25,1 мг).
МС (ИЭР) m/z: 1411 (М+Н)+.
1H-ЯМР (CD3OD) δ: 8,58 (1Н, с), 8,09 (1Н, с), 8,04 (1Н, с), 7,57-7,49 (2Н, м), 7,43-7,34 (3Н, м), 7,32-7,08 (9Н, м), 6,47 (1Н, д, J=16,9 Гц), 6,23 (1Н, д, J=7,9 Гц), 5,56-5,37 (2Н, м), 5,31-5,17 (1Н, м), 5,03 (1Н, д, J=13.9 Гц), 4,79 (1Н, д, J=4.2 Гц), 4,64-4,38 (6Н, м), 4,36-4,21 (4Н, м), 4,05-3,60 (10Н, м), 3,53-3,42 (3Н, м), 3,18 (12Н, к, J=7.3 Гц), 3,01-2,92 (1Н, м), 2,86-2,73 (1Н, м), 2,70-2,54 (2Н, м), 2,37-2,16 (2Н, м), 2,06-1,77 (3Н, м), 1,28 (18Н, т, J=7,3 Гц).
[1135]
(Этап 11-2)
Бис(N,N-диэтилэтанаминия) N-[4-(11,12-дидегидродибензо[b,f]азоцин-5(6Н)-ил)-4-оксобутаноил]глицилглицил-L-фенилаланил-N-[(2-{9-[(5R,7R,8R,12aR,14R,15R,15aR,16R)-15-фтор-16-гидрокси-2,10-диоксо-2,10-дисульфид-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-7-ил]-6-оксо-6,9-дигидро-1Н-пурин-1 -ил} этокси)метил]глицинамид (Лекарственное средство-линкер 17b: диастереомер 2)
С применением соединения, полученного на этапе 10-2 выше (50,8 мг), проводили реакцию таким же образом, как на этапе 9-1 примера 22, и продукт затем очищали с помощью колоночной хроматографии на силикагеле С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил], препаративной ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 25% - 45% (0 мин - 40 мин)] и препаративной ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/метанол, метанол: 45% - 90% (0 мин - 40 мин)] с получением указанного в заголовке соединения (23,4 мг).
МС (ИЭР) m/z: 1411 (М+Н)+.
1H-ЯМР (CD3OD) δ: 8,67 (1Н, с), 8,14 (1Н, с), 8,02 (1Н, с), 7,67-7,50 (2Н, м), 7,43-7,36 (3Н, м), 7,34-7,12 (9Н, м), 6,48 (1Н, д, J=15,1 Гц), 6,26 (1Н, т, J=8,8 Гц), 5,60-5,31 (3Н, м), 5,09-5,00 (1Н, м), 4,61-4,22 (9Н. м), 4.11-3,59 (13Н, м), 3,50-3,44 (2Н, м), 3.18 (12Н. к. J=7.3 Гц), 3,04-2,93 (1Н, м), 2,87-2,74 (3Н, м), 2,39-2,22 (2Н, м), 2,06-1,85 (3Н, м), 1,28 (18Н, т, J=7,3 Гц).
[1136]
Пример 78. Синтез лекарственного средства-линкера 18.
[Схема синтеза]
[1137]
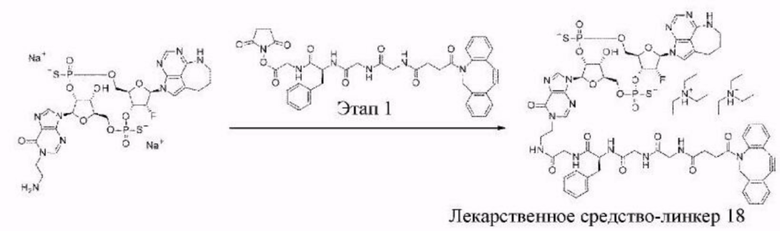
[1138]
(Этап 1)
Бис(N,N-диэтилэтанаминия) N-[4-(11,12-дидегидродибензо[b,f]азоцин-5(6Н)-ил)-4-оксобутаноил]глицилглицил-L-фенилаланил-N-(2-{9-[(5R,7R,8R,12aR,14R,15R,15aR,16R)-15-фтор-16-гидрокси-2,10-диоксо-2,10-дисульфид-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-7-ил]-6-оксо-6,9-дигидро-1Н-пурин-1-ил}этил)глицинамид (Лекарственное средство-линкер 18)
С применением соединения, полученного на этапе 7-2 примера 45 (10,0 мг), и полученного на этапе 3 примера 21 соединения (8,8 мг) проводили реакцию таким же образом, как на этапе 4 примера 21 и продукт затем очищали с помощью колоночной хроматографии на силикагеле С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил], препаративной ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 25% - 50% (0 мин - 40 мин)] и препаративной ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/метанол, метанол: 45% - 90% (0 мин - 40 мин)] с получением указанного в заголовке соединения (10,9 мг).
MC (ИЭР) m/z: 1381 (М+Н)+.
1H-ЯМР (CD3OD) δ: 8,61 (1Н, с), 8,03 (1Н, с), 8,02 (1Н, с), 7,64-7,49 (2Н, м), 7,44-7,37 (3Н, м), 7,35-7,11 (9Н, м), 6,48 (1Н, дд, J=15,1, 1,8 Гц), 6,24 (1Н, д, J=8,5 Гц), 5,62-5,39 (3Н, м), 5,03 (1Н, дд, J=18,4, 14,2 Гц), 4,55-4,37 (5Н, м), 4,29-4,18 (2Н, м), 4,04-3,91 (3Н, м), 3,87-3,52 (8Н, м), 3,50-3,41 (3Н, м), 3,19 (12Н, к, J=7,3 Гц), 3,18-3,10 (1Н, м), 3,02-2,92 (1Н, м), 2,86-2,66 (3Н, м), 2,40-2,21 (2Н, м), 2,06-1,84 (3Н, м), 1,29 (18Н, т, J=7,3 Гц).
[1139]
Пример 79. Синтез лекарственного средства-линкера 19.
[Схема синтеза]
[1140]
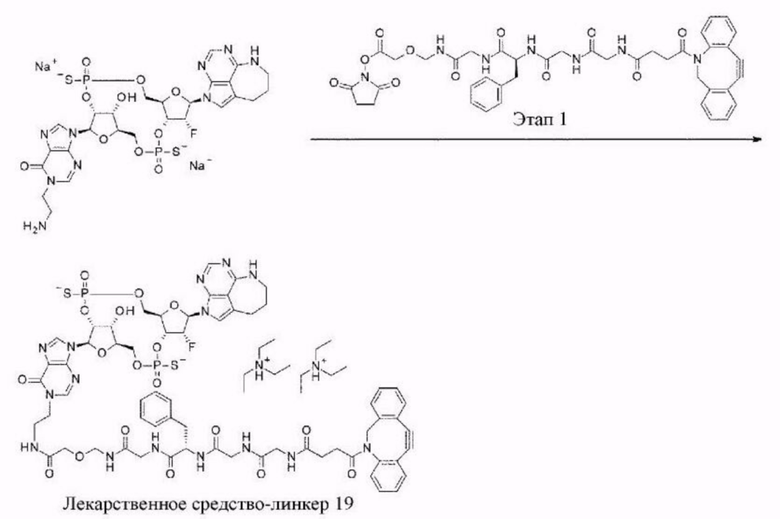
[1141]
(Этап 1)
Бис(N,N-диэтилэтанаминия) N-[4-(11,12-дидегидродибензо[b,f]азоцин-5(6H)-ил)-4-оксобутаноил]глицилглицил-L-фенилаланил-N-({2-[(2-{9-[(5R,7R,8R,12aR,14R,15R,15aR,16R)-15-фтор-16-гидрокси-2,10-диоксо-2,10-дисульфид-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-7-ил]-6-оксо-6,9-дигидро-1Н-пурин-1-ил}этил)амино]-2-оксоэтокси}метил)глицинамид (Лекарственное средство-линкер 19)
С применением соединения, полученного на этапе 7-2 примера 45 (20,0 мг), и полученного на этапе 1 примера 66 соединения (19,8 мг) проводили реакцию таким же образом, как на этапе 4 примера 21, и продукт затем очищали с помощью колоночной хроматографии на силикагеле С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил], препаративной ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацето нитрил: 20% - 45% (0 мин - 40 мин)] и препаративной ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/метанол, метанол: 30% - 80% (0 мин - 40 мин)] с получением указанного в заголовке соединения (22,1 мг).
МС (ИЭР) m/z: 1468(M+H)+.
1H-ЯМР (CD3OD) δ: 8,66 (1Н, уш. с), 8,10 (1Н, уш. с), 8,04-7,96 (1Н, м), 7,66-7,35 (5Н, м), 7,33-7,12 (9Н, м), 6,52-6,41 (1Н, м), 6,28-6,22 (1Н, м), 5,64-5,31 (3Н, м), 5,07-4,99 (1Н, м), 4,70-4,20 (9Н, м), 4,11-3,51 (14Н, м), 3,50-3,30 (3Н, м), 3,27-3,10 (1Н, м), 3,19 (12Н, к, J=7,3 Гц), 3,07-2,93 (1Н, м), 2,89-2,65 (3Н, м), 2,41-2,18 (2Н, м), 2,05-1,82 (2Н, м), 1,29 (18Н, т, J=7,3 Гц).
[1142]
Пример 80. Синтез лекарственного средства-линкера 20.
[Схема синтеза]
[1143]
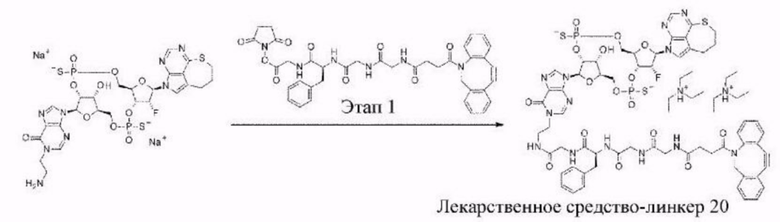
[1144]
(Этап 1)
Бис(N,N-диэтилэтанаминия) N-[4-(11,12-дидегидродибензо[b,f]азоцин-5(6Н)-ил)-4-оксобутаноил]глицилглицил-L-фенилаланил-N-(2-{9-[(5R,7R,8R,12aR,14R,15R,15aR,16R)-14-(8,9-дигидро-6-тиа-2,3,5-триазабензо[cd]азулен-2(7Н)-ил)-15-фтор-16-гидрокси-2,10-диоксульфидоктагидро-2H,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,21][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-7-ил]-6-оксо-6,9-дигидро-1Н-пурин-1 -ил}этил)глицинамид (Лекарственное средство-линкер 20)
С применением соединения, полученного на этапе 4-2 примера 59 (25,0 мг), и полученного на этапе 3 примера 21 соединения (25,8 мг) проводили реакцию таким же образом, как на этапе 4 примера 21, и продукт затем очищали с помощью колоночной хроматографии на силикагеле С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил], препаративной ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 30% - 50% (0 мин - 40 мин)] и препаративной ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/метанол, метанол: 50% - 90% (0 мин - 40 мин)] с получением указанного в заголовке соединения (33,1 мг).
МС (ИЭР)т/г: 1398(M+H)+.
1Н-ЯМР (CD3OD) δ: 8,58 (1Н, уш. с), 8,39 (1Н, д, J=8,5 Гц), 8,00 (1Н, уш. с), 7,71 (1Н, уш. с), 7,64-7,49 (2Н, м), 7,44-7,37 (3Н, м), 7,32-7,10 (8Н, м), 6,59 (1Н, д, J=15,1 Гц), 6,23 (1Н, д, J=8,5 Гц), 5,65-5,36 (3Н, м), 5,03 (1Н, дд, J=16,6, 14,2 Гц), 4,57-4,38 (5Н, м), 4,30-4,17 (2Н, м), 4,07-3,95 (2Н, м), 3,94-3,50 (9Н, м), 3,50-3,35 (1Н, м), 3,19 (12Н, к, J=7,3 Гц), 3,18-3,07 (3Н, м), 3,04-2,73 (4Н, м), 2,40-2,11 (4Н, м), 2,05-1,92 (1Н, м), 1,29 (18Н, т, J=7,3 Гц).
[1145]
Пример 81. Синтез лекарственного средства-линкера 21.
[Схема синтеза]
[1146]
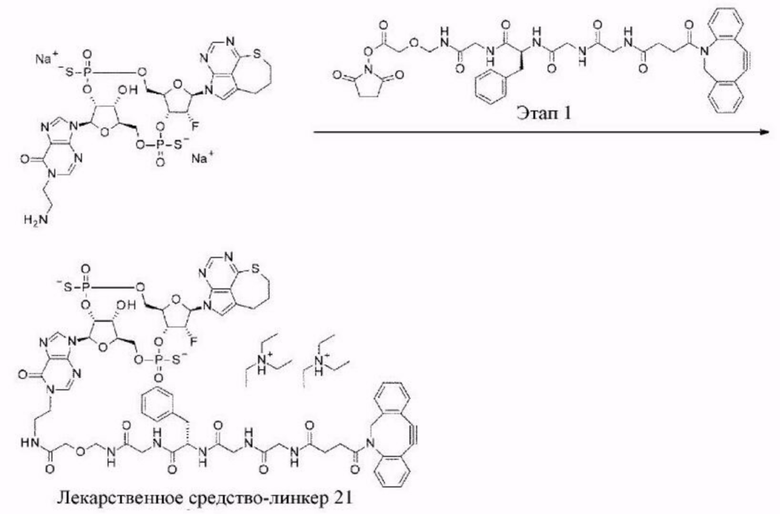
[1147]
(Этап 1)
Бис(N,N-диэтилэтанаминия) N-[4-(11,12-дидегидродибензо[b,f]азоцин-5(6Н)-ил)-4-оксобутаноил]глицилглицил-L-фенилаланил-N-({2-[(2-{9-[(5R,7R,8R,12aR,14R,15R,15aR,16R)-14-(8,9-дигидро-6-тиа-2,3,5-триазабензо[cd]азулен-2(7Н)-ил)-15-фтор-16-гидрокси-2,10-диоксо-2,10-дисульфидоктагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-7-ил]-6-оксо-6,9-дигидро-1Н-пурин-1-ил}этил)амино]-2-оксоэтокси}метил)глицинамид (Лекарственное средство-линкер 21)
С применением соединения, полученного на этапе 4-2 примера 59 (15,0 мг), и полученного на этапе 1 примера 66 соединения (17,4 мг) проводили реакцию таким же образом, как на этапе 4 примера 21, и продукт затем очищали с помощью колоночной хроматографии на силикагеле С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил], препаративной ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 25% - 50% (0 мин - 40 мин)] и препаративной ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/метанол, метанол: 40% - 90% (0 мин - 40 мин)] с получением указанного в заголовке соединения (21,8 мг).
МС (ИЭР) m/z: 1485(M+H)+.
1Н-ЯМР (CD3OD) δ: 8,61 (1Н, уш. с), 8,41-8,34 (1Н, м), 8,10-8,05 (1Н, м), 7,72-7,37 (6Н, м), 7,32-7,12 (8Н, м), 6,63-6,50 (1Н, м), 6,27-6,22 (1Н, м), 5,65-5,31 (3Н, м), 5,07-4,94 (1Н, м), 4,68-4,20 (10Н, м), 4,11-3,53 (14Н, м), 3,26-3,08 (2Н, м), 3,19 (12Н, к, J=7,3 Гц), 3,06-2,67 (4Н, м), 2,40-2,11 (4Н, м), 2,05-1,88 (1Н, м), 1,29 (18Н, т, J=7,3 Гц).
[1148]
Пример 82. Синтез лекарственного средства-линкера 22.
[Схема синтеза]
[1149]
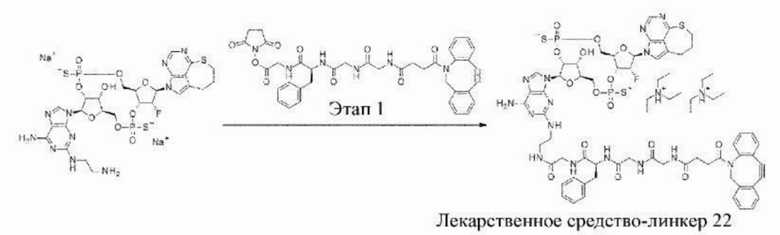
[1150]
(Этап 1)
Бис(N,N-диэтилэтанаминия) N-[4-(11,12-дидегидродибензо[b,f]азоцин-5(6Н)-ил)-4-оксобутаноил]глицилглицил-L-фенилаланил-N-[2-({6-амино-9-[(5R,7R,8R,12aR,14R,15R,15aR,16R)-14-(8,9-дигидро-6-тиа-2,3,5-триазабензо[cd]азулен-2(7Н)-ил)-15-фтор-16-гидрокси-2,10-диоксо-2,10-дисульфидоктагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-7-ил]-9Н-пурин-2-ил} амино)этил] глицинамид (Лекарственное средство-линкер 22)
С применением соединения, полученного на этапе 4-2 примера 61 (4,4 мг), и полученного на этапе 3 примера 21 соединения (3,5 мг) проводили реакцию таким же образом, как на этапе 4 примера 21, и продукт затем очищали с помощью препаративной ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 25% -45% (0 мин - 30 мин)] с получением указанного в заголовке соединения (6,4 мг).
MC (ИЭР) m/z: 1412 (М+Н)+.
1H-ЯМР (CD3OD) δ: 8,37 (1Н, д, J=1,2 Гц), 8,10-7,98 (1Н, м), 7,75 (1Н, с), 7,62-7,13 (13Н, м), 6,59 (1Н, д, J=16,3 Гц), 6,01 (1Н, уш. с), 5,82-5,62 (1Н, м), 5,54-5,33 (2Н, м), 5,04 (1Н, д, J=14,5 Гц), 4,57-3,48 (14Н, м), 3,35-2,65 (12Н, м), 3,17 (12Н, к, J=7,3 Гц), 2,35-2,11 (4Н, м), 2,00-1,92 (1Н, м), 1,28 (18Н, т, J=7,3 Гц).
[1151]
Пример 83. Синтез лекарственного средства-линкера 23.
[Схема синтеза]
[1152]
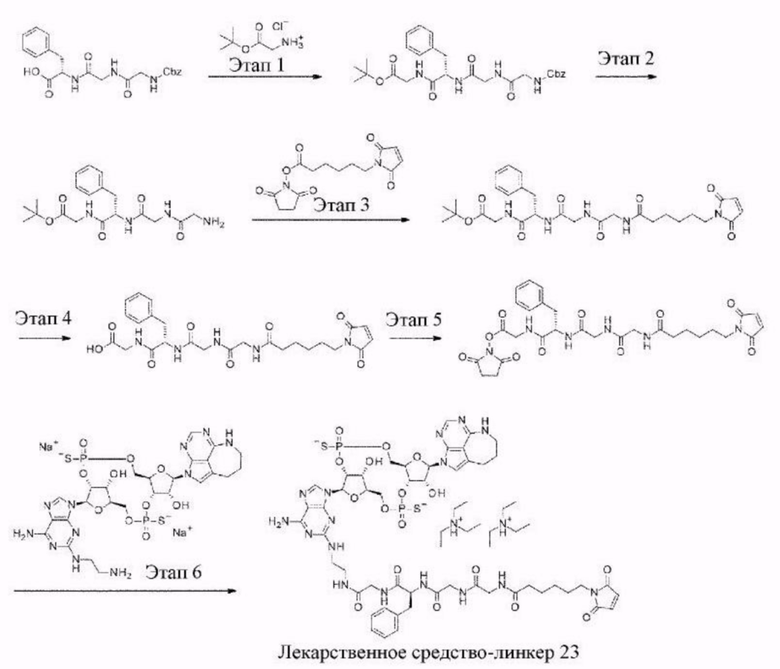
[1153]
(Этап 1)
Трет-бутил N-[(бензилокси)карбонил]глицилглицил-L-фенилаланилглицинат
К раствору доступного для приобретения (Bachem Holding AG) N-[(бензилокси)карбонил]глицилглицил-L-фенилаланина (5,00 г) и гидрохлорида трет-бутилового эфира глицина (2,03 г) в N,N-диметилформамиде (50 мл) добавляли N,N-диизопропилэтиламин (4,11 мл) и 1-этил-3-(3-диметиламинопропил)-карбодиимида гидрохлорид (3,01 г) при охлаждении льдом, и реакционную смесь перемешивали при повышении температуры до комнатной температуры в течение ночи. Реакционную смесь вливали в двухслойную смесь хлороформа и насыщенного водного раствора гидрокарбоната натрия и осуществляли экстрагирование хлороформом. Органический слой промывали водой и рассолом, а затем сушили над безводным сульфатом натрия. Осушающий агент удаляли путем фильтрации и фильтрат концентрировали при пониженном давлении и дважды подвергали азеотропной перегонке с толуолом. Остаток очищали с помощью колоночной хроматографии на силикагеле [дихлорметан/метанол] с получением указанного в заголовке соединения (4,51 г).
МС (ИЭР) m/z: 527(M+H)+.
1H-ЯМР (ДМСО-d6) δ: 8,40 (1Н, т, J=5,7 Гц), 8,15 (1Н, д, J=9,1 Гц), 8,00 (1Н, т, J=5,7 Гц), 7,51 (1Н, т, J=6,0 Гц), 7,38-7,16 (10Н, м), 5,03 (2Н, с), 4,52 (1Н, м), 3,80-3,54 (6Н, м), 3,05 (1Н, дд, J=13,9, 3,6 Гц), 2,76 (1Н, дд, J=14,2, 10,6 Гц), 1,41 (9Н, с).
[1154]
(Этап 2)
Трет-бутилглицилглицил-L-фенилаланилглицинат
В смесь соединения, полученного на этапе 1 выше (4,51 г), в метаноле (20 мл)-дихлорметане (80 мл) добавляли 10% палладий-углерод (М), влажный (750 мг), и реакционную смесь перемешивали в атмосфере водорода при комнатной температуре в течение ночи. Туда дополнительно добавляли 10% палладий-углерод (М), влажный (1,5 г), и реакционную смесь перемешивали в атмосфере водорода при комнатной температуре в течение ночи. Реакционную смесь фильтровали с помощью целита и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на аминосиликагеле [дихлор метан/метанол] с получением указанного в заголовке соединения (3,18 г).
МС (ИЭР) m/z: 393(M+H)+.
1H-ЯМР (ДМСО-d6) δ: 8,42 (1Н, т, J=6,1 Гц), 8,20 (1Н, д, J=8,8 Гц), 8,00 (1Н, уш. с), 7,28-7,16 (5Н, м), 4,53 (1Н, м), 3,78-3,73 (3Н, м), 3,61 (1Н, уш. д, J=16,1 Гц), 3,06 (3H, дд, J=15,4, 5,6 Гц), 2,76 (1Н, дд, J=14,2, 10,3 Гц), 1,88 (2Н, уш.), 1,41 (9Н, с).
[1155]
(Этап 3)
Трет-бутил-N-[6-(2,5-диоксо-2,5-дигидро-1Н-пиррол-1-ил)гексаноил]глицилглицил-L-фенилаланилглицинат
Полученное на этапе 2 выше соединение (2,65 г) и доступный для приобретения (Tokyo Chemical Industry Co., Ltd.) N-сукцинимидил-6-малеимидгексаноат (2,20 г) растворяли в N,N-диметилформамиде (20 мл), и реакционную смесь перемешивали при комнатной температуре в течение 7 часов. После концентрирования реакционной смеси при пониженном давлении остаток вливали в двухслойную смесь дихлорметана и воды и осуществляли экстрагирование дихлорметаном. Органический слой промывали три раза водой и один раз рассолом, а затем сушили над безводным сульфатом натрия. Осушающий агент удаляли путем фильтрации и фильтрат концентрировали при пониженном давлении.
Остаток очищали с помощью колоночной хроматографии на силикагеле [дихлор метан/метанол] с получением указанного в заголовке соединения (3,52 г).
МС (ИЭР) m/z: 586(M+H)+.
1H-ЯМР (CDCl3) δ: 7,28-7,17 (5Н, м), 7,09 (1Н, т, J=5,1 Гц), 6,96 (1Н, т, J=5,1 Гц), 6,91 (1Н, д, J=8,3 Гц), 6,68 (2Н, с), 6,52 (1Н, т, J=4,9 Гц), 4,86 (1Н, к, J=7,3 Гц), 4,02-3,88 (5Н, м), 3,82 (1Н, дд, J=18,1, 4,9 Гц), 3,51 (2Н, т, J=7,1 Гц), 3,15 (1Н, дд, J=14,2, 6,3 Гц), 3,04 (1Н, дд, J=13,7, 7,3 Гц), 2,25 (2Н, т, J=7,6 Гц), 1,70-1,57 (4Н, м), 1,45 (9Н, с), 1,32 (2Н, с).
[1156]
(Этап 4)
N-[6-(2,5-диоксо-2,5-дигидро-1Н-пиррол-1-ил)гексаноил]глицилглицил-L-фенилаланилглицин
К раствору соединения, полученного на этапе 3 выше (1,00 г), в дихлорметане (9,0 мл) добавляли трифторуксусную кислоту (4,5 мл) при 0°С и реакционную смесь перемешивали при комнатной температуре в течение 2,5 часов. Реакционную смесь концентрировали при пониженном давлении и дважды подвергали азеотропной перегонке с толуолом. Остаток очищали с помощью колоночной хроматографии на силикагеле [дихлор метан/метанол] с получением указанного в заголовке соединения (534 мг).
МС (ИЭР) m/z: 530(M+H)+.
1H-ЯМР (ДМСО-d6) δ: 12,59 (1Н, уш.), 8,34 (1Н, т, J=5,9 Гц), 8,10 (1Н, д, J=8,8 Гц), 8,07 (1Н, т, J=5,9 Гц), 7,99 (1Н, т, J=5,6 Гц), 7,27-7,16 (5Н, м), 7,00 (2Н, с), 4,52 (1Н, м), 3,76 (2Н, д, J-5,9 Гц), 3,74 (1Н, м), 3,66 (2Н, д, J=5,4 Гц), 3,57 (1Н, дд, J=16,8, 5,6 Гц), 3,36 (2Н, т, J=7,1 Гц), 3,04 (1Н, дд, J=13,9, 4,1 Гц), 2,77 (1Н, дд, J=13,9, 10,0 Гц), 2,10 (2Н, т, J=7,3 Гц), 1,51-1,43 (4Н,м), 1,19(2Н,м).
[1157]
(Этап 5)
2,5-диоксопирролидин-1-ил-N-[6-(2,5-диоксо-2,5-дигидро-1Н-пиррол-1-ил)гексаноил]глицилглицил-L-фенилаланилглицинат
С применением соединения, полученного на этапе 4 выше (462 мг), проводили реакцию таким же образом, как на этапе 3 примера 21, с получением указанного в заголовке соединения (258 мг).
МС (ИЭР) m/z: 627(M+H)+.
1H-ЯМР (ДМСО-d6) δ: 8,70 (0,85Н, т, J=6,1 Гц), 8,44 (0,15Н, т, J=5,9 Гц), 8,17 (0,85Н, д, J=8,8 Гц), 8,11 (0,15Н, д, J=8,8 Гц), 8,06 (1Н, т, J=5,6 Гц), 7,99-7,95 (1Н, м), 7,27-7,16 (5Н, м), 6,99 (2Н, с), 4,53 (1Н, м), 4,28 (1,7Н, д, J=5,9 Гц), 3,85 (0,3Н, д, J=5,9 Гц), 3,74 (1Н, дд, J=17,1, 5,9 Гц), 3,66 (2Н, д, J=5,9 Гц), 3,58 (1Н, дд, J=16,8, 5,6 Гц), 3,36 (2Н, т, J=7,1 Гц), 3,04 (1Н, дд, J=13,9, 4,1 Гц), 2,81 (4Н, уш. с), 2,77 (1Н, дд, J=10,0, 3,7 Гц), 2,10 (2Н, т, J=7,3 Гц), 1,51-1,44 (4Н, м), 1,20 (2Н, д, J=7,3 Гц).
[1158]
(Этап 6)
Бис(N,N-диэтилэтанаминия) N-[6-(2,5-диоксо-2,5-дигидро-1Н-пиррол-1-ил)гексаноил]глицилглицил-L-фенилаланил-N-[2-({6-амино-9-[(5R,7R,8R, 12aR, 14R, 15R, 15aS, 16R)-15,16-дигидрокси-2,10-диоксо-2,10-дисульфид-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-7-ил]-9Н-пурин-2-ил}амино)этил]глицинамид (Лекарственное средство-линкер 23)
С применением соединения, полученного на этапе 8-2 примера 8 (9,5 мг), и полученного на этапе 5 выше соединения (7,5 мг) проводили реакцию таким же образом, как на этапе 1 примера 23, и продукт затем очищали с помощью препаративной ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 10% - 30% (0 мин - 30 мин)] с получением указанного в заголовке соединения (7,8 мг).
МС (ИЭР) m/z: 1299(M+H)+.
1H-ЯМР (CD3OD) δ: 8,35 (1Н, с), 8,02 (1Н, с), 7,29-7,15 (6Н, м), 6,76 (2Н, с), 6,32 (1Н, д, J=6,7 Гц), 6,11 (1Н, д, 8,5 Гц), 5,48-5,36 (2Н, м), 4,82-4,79 (1Н, м), 4,50-4,25 (6Н, м), 4,06-3,81 (7Н, м), 3,61-3,37 (9Н, м), 3,17-2,89 (4Н, м), 3,14 (12Н, к, J=7,3 Гц), 2,21-1,96 (4Н, м), 1,58-1,48 (4Н, м), 1,30-1,21 (2Н, м), 1,27 (18Н, т, J=7,3 Гц).
[1159]
Пример 84. Синтез ремоделированного гликаном антитела 3.
Получение антитела 2 против HER2-[SG-(N3)2]2
[Схема синтеза]
[1160]
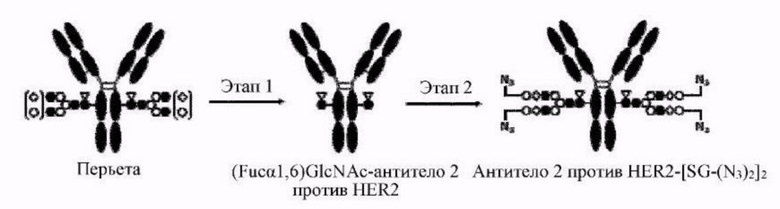
[1161]
(Этап 1)
Получение (Fucα1,6)GlcNAc-антитела 2 против HER2
В доступной для приобретения капельнице для внутривенного введения Перьеты (R) 420 мг/14 мл (Chugai Pharmaceutical Co., Ltd.) (3,5 мл) осуществляли замену буфера в соответствии с Общей процедурой С с получением раствора в фосфатно-солевом буфере (6,0 мл, 15,39 мг/мл, рН 6,0). Те же процедуры, как на этапе 1 примера 25, проводили с получением раствора в 20 мМ фосфатном буфере указанного в заголовке антитела (12,96 мг/мл, 7,5 мл, рН 6,0).
[1162]
(Этап 2)
Получение антитела 2 против HER2-[SG-(N3)2]2
С применением 20 мМ фосфатного буферного раствора антитела, полученного на этапе 1 выше (12,96 мг/мл, 7,5 мл, рН 6,0), и [N3-PEG(3)]2-SG(10)Ox (22,5 мг) проводили те же процедуры, как на этапе 2 примера 25, с получением фосфатно-солевого буферного раствора указанного в заголовке антитела (10,21 мг/мл, 9,0 мл, рН 6,0).
[1163]
Пример 85. Синтез ремоделированного гликаном антитела 4.
Получение модифицированного антитела 2 против HER2-[SG-(N3)2]2
[Схема синтеза]
[1164]
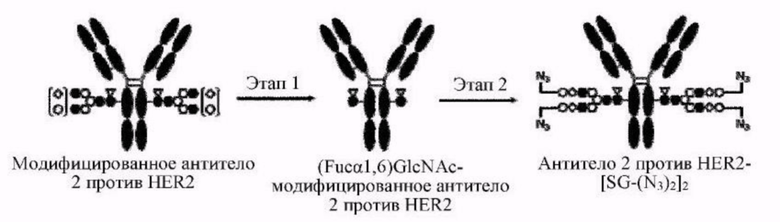
[1165]
(Этап 1)
Получение (Fucα1,6)GlcHAc-модифицированного антитела 2 против HER2.
С применением фосфатно-солевого буферного раствора модифицированного антитела 2 против HER2, полученного в соответствии со ссылочным примером 5 (24 мл, 12,25 мг/мл, рН 6,0), проводили те же процедуры, как на этапе 1 примера 25, с получением раствора в 20 мМ фосфатном буфере указанного в заголовке антитела (20,86 мг/мл, 12,5 мл, рН 6,0).
[1166]
(Этап 2)
Получение модифицированного антитела 2 против HER2-[SG-(N3)2]2
С применением 20 мМ фосфатного буферного раствора антитела, полученного на этапе 1 выше (20,86 мг/мл, 12,5 мл, рН 6,0), и [N3-PEG(3)]2-SG(10)Ox (52 мг) проводили те же процедуры, как на этапе 2 примера 25, с получением фосфатно-солевого буферного раствора указанного в заголовке антитела (10,87 мг/мл, 22 мл, рН 6,0).
[1167]
Пример 86. Синтез ремоделированного гликаном антитела 5.
Получение антитела против CD33-[SG-(N3)2]2
[Схема синтеза]
[1168]

[1169]
(Этап 1)
Получение (Fucα1,6)GlcHAc-антитела против CD33.
С применением фосфатно-солевого буферного раствора антитела против CD33, полученного в соответствии со ссылочным примером 6 (9,0 мл, 11,56 мг/мл, рН 6,0), проводили те же процедуры, как на этапе 1 примера 25, с получением раствора в 20 мМ фосфатном бу4)ере указанного в заголовке антитела (11,62 мг/мл, 8 мл, рН 6,0).
[1170]
(Этап 2)
Получение антитела против CD33-[SG-(N3)2]2
С применением 20 мМ фосфатного буферного раствора антитела, полученного на этапе 1 выше (11,62 мг/мл, 8 мл, рН 6,0), и [N3-PEG(3)]2-SG(10)Ox (21,5 мг) проводили те же процедуры, как описанные на этапе 2 примера 25, с получением фосфатно-солевого буферного раствора указанного в заголовке антитела (10,01 мг/мл, 8 мл, рН 6,0).
[1171]
Пример 87. Синтез ремоделированного гликаном антитела 6.
Получение антитела против EphA2-[SG-(N3)2]2
[Схема синтеза]
[1172]
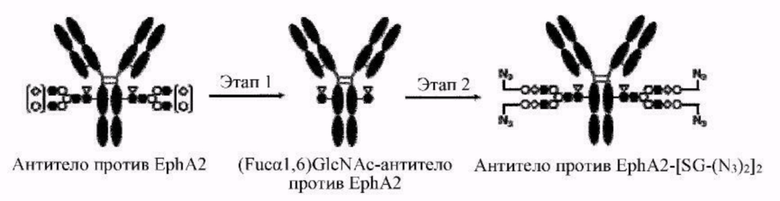
[1173]
(Этап 1)
Получение (Fucα1,6)GlcNAc-антитела против EphA2.
С применением фосфатно-солевого буферного раствора антитела против EphA2, полученного в соответствии со ссылочным примером 7 (8,0 мл, 12,83 мг/мл, рН 6,0), проводили те же процедуры, как на этапе 1 примера 25, с получением раствора в 20 мМ фосфатном буфере указанного в заголовке антитела (13,51 мг/мл, 7 мл, рН 6,0).
[1174]
(Этап 2)
Получение антитела против EphA2-[SG-(N3)2]2
С применением 20 мМ фосфатного буферного раствора антитела, полученного на этапе 1 выше (13,51 мг/мл, 7 мл, рН 6,0), и [N3-PEG(3)]2-SG(10)Ox (21,7 мг) проводили те же процедуры, как на этапе 2 примера 25, с получением фосфатно-солевого буферного раствора указанного в заголовке антитела (8,91 мг/мл, 7,5 мл, рН 6,0).
[1175]
Пример 88. Синтез ремоделированного гликаном антитела 7.
Получение антитела против CDH6-[SG-(N.3)2]2
[Схема синтеза]
[1176]
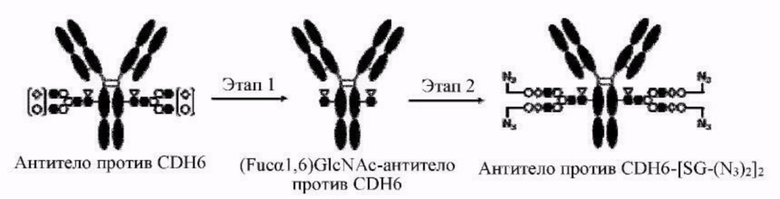
[1177]
(Этап 1)
Получение (Fucα1,6)GlcNAc-антитела против CDH6.
Осуществляли замену буфера HBSor (25 мМ гистидин/5% сорбит, рН=6,0) (5,0 мл, 20,0 мг/мл) антитела против CDH6, полученного в соответствии со ссылочным примером 8, на фосфатно-солевой буферный раствор (рН=6,0) в соответствии с Общей процедурой С, и те же процедуры, как на этапе 1 примера 25, затем проводили с получением раствора в 20 мМ фосфатном буфере указанного в заголовке антитела (9,58 мг/мл, 9,0 мл, рН 6,0).
[1178]
(Этап 2)
Получение антитела против CDH6-[SG-(N3)2]2
С применением 20 мМ фосфатного буферного раствора антитела, полученного на этапе 1 выше (9,58 мг/мл, 9,0 мл, рН 6,0), и [N3-PEG(3)]2-SG(10)Ox (24,5 мг) проводили те же процедуры, как на этапе 2 примера 25, с получением фосфатно-солевого буферного раствора указанного в заголовке антитела (10,69 мг/мл, 7,5 мл, рН 6,0).
[1179]
Пример 89. Синтез ремоделированного гликаном антитела 8.
Получение модифицированного антитела 2 против HER2-[MSG1-(N3)]2
[Схема синтеза]
[1180]
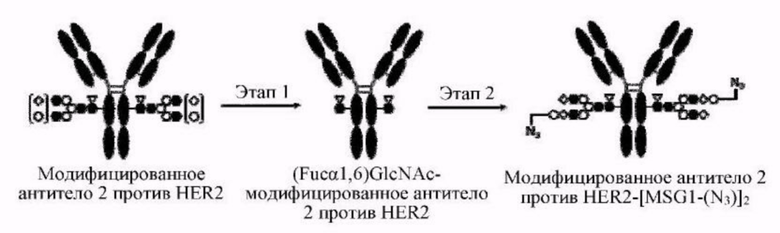
[1181]
(Этап 1)
Получение (Fucα1,6)GlcNAc-модифицированного антитела 2 против HER2.
Ту же реакцию, что и на этапе 1 примера 85, проводили с буферным раствором сырьевого антитела (10 мл, 12,25 мг/мл, рН 6,0) с получением раствора в 20 мМ фосфатном буфере указанного в заголовке антитела (14,84 мг/мл, 7,5 мл, рН 6,0).
[1182]
(Этап 2)
Получение модифицированного антитела 2 против HER2-[MSG1-(N3)]2
С применением 20 мМ фосфатного буферного раствора антитела, полученного на этапе 1 выше (14,84 мг/мл, 7,5 мл, рН 6,0), и [N3-PEG(3)]-MSG1(9)-Ox (соединение 1-11 в WO 2018/003983) (19 мг) проводили те же процедуры, как на этапе 2 примера 25, с получением фосфатно-солевого буферного раствора указанного в заголовке антитела (10,48 мг/мл, 9,25 мл, рН 6,0).
[1183]
Пример 90. Синтез конъюгата 5 антитела с лекарственным средством (синтез конъюгата 4 антитела против HER2-ЦДН)
Фосфатно-солевой буферный раствор ремоделированного гликаном антитела 1 (рН 6,0) (10,97 мг/мл, 0,500 мл) разбавляли пропиленгликолем (0,250 мл). В данный раствор добавляли смесь раствора в диметилсульфоксиде лекарственного средства-линкера 1 (10 мМ, 0,091 мл, 24 эквивалента на молекулу антитела) и пропиленгликоля (0,159 мл) и проводили реакцию полученной смеси во вращающемся устройстве для пробирок (MTR-103, AS ONE Corporation) при комнатной температуре в течение 3 суток. Реакционную смесь очищали в соответствии со способом, описанным в Общей процедуре D, с получением раствора конъюгата нацеленного антитела с лекарственным средством в ABS (3,5 мл).
Анализ проводили в соответствии со способами, описанными в Общих процедурах Е и G, с получением следующих результатов.
Концентрация антитела: 0,49 мг/мл
Выход антитела: 1,71 мг (31%)
Среднее количество конъюгированных молекул лекарственного средства: 3,6
[1184]
Пример 91. Синтез конъюгата 6 антитела с лекарственным средством (синтез конъюгата 5 антитела против НЕР2-ЦДН).
Фосфатно-солевой буферный раствор ремоделированного гликаном антитела 1 (рН 6,0) (10,97 мг/мл, 0,500 мл) разбавляли пропиленгликолем (0,250 мл). В данный раствор добавляли смесь раствора в диметилсульфоксиде лекарственного средства-линкера 2а (10 мМ, 0,091 мл, 24 эквивалента на молекулу антитела) и пропиленгликоля (0,159 мл) и проводили реакцию полученной смеси во вращающемся устройстве для пробирок (MTR-103, AS ONE Corporation) при комнатной температуре в течение 2 суток. Реакционную смесь очищали в соответствии со способом, описанным в Общей процедуре D, с получением раствора конъюгата нацеленного антитела с лекарственным средством в ABS (3,5 мл).
Анализ проводили в соответствии со способами, описанными в Общих процедурах Е и G, с получением следующих результатов. Концентрация антитела: 0,91 мг/мл Выход антитела: 3,17 мг (58%)
Среднее количество конъюгированных молекул лекарственного средства: 3,6
[1185]
Пример 92. Синтез конъюгата 7 антитела с лекарственным средством (синтез конъюгата 6 антитела против НЕР2-ЦДН).
Фосфатно-солевой буферный раствор ремоделированного гликаном антитела 1 (рН 6,0) (10,70 мг/мл, 4,00 мл) разбавляли пропиленгликолем (2,00 мл). В данный раствор добавляли смесь раствора в диметилсульфоксиде лекарственного средства-линкера 2b (10 мМ, 0,707 мл, 24 эквивалента на молекулу антитела) и пропиленгликоля (1,293 мл) и проводили реакцию полученной смеси во вращающемся устройстве для пробирок (MTR-103, AS ONE Corporation) при комнатной температуре в течение 3 суток. Реакционную смесь очищали в соответствии со способом, описанным в Общей процедуре D, с получением раствора конъюгата нацеленного антитела с лекарственным средством в ABS (24,5 мл).
Анализ проводили в соответствии со способами, описанными в Общих процедурах Е и G, с получением следующих результатов. Концентрация антитела: 0,96 мг/мл Выход антитела: 23,57 мг (59%)
Среднее количество конъюгированных молекул лекарственного средства: 3,7
[1186]
Пример 93. Синтез конъюгата 8 антитела с лекарственным средством (синтез конъюгата 7 антитела против НЕР2-ЦДН).
Фосфатно-солевой буферный раствор ремоделированного гликаном антитела 1 (рН 6,0) (10,70 мг/мл, 4,00 мл) разбавляли пропиленгликолем (2,00 мл). В данный раствор добавляли смесь раствора в диметилсульфоксиде лекарственного средства-линкера 5 (10 мМ, 0,707 мл, 24 эквивалента на молекулу антитела) и пропиленгликоля (1,293 мл) и проводили реакцию полученной смеси во вращающемся устройстве для пробирок (MTR-103, AS ONE Corporation) при комнатной температуре в течение 2 суток. Реакционную смесь очищали в соответствии со способом, описанным в Общей процедуре D, с получением раствора конъюгата нацеленного антитела с лекарственным средством в ABS (24,5 мл).
Анализ проводили в соответствии со способами, описанными в Общих процедурах Е и G, с получением следующих результатов. Концентрация антитела: 1,10 мг/мл Выход антитела: 26,86 мг (67%)
Среднее количество конъюгированных молекул лекарственного средства: 3,7.
[1187]
Пример 94. Синтез конъюгата 9 антитела с лекарственным средством (синтез конъюгата 8 антитела против HER2-ЦДН).
Фосфатно-солевой буферный раствор ремоделированного гликаном антитела 1 (рН 6,0) (10,70 мг/мл, 4,00 мл) разбавляли пропиленгликолем (2,00 мл). В данный раствор добавляли смесь раствора в диметилсульфоксиде лекарственного средства-линкера 6 (10 мМ, 0,707 мл, 24 эквивалента на молекулу антитела) и пропиленгликоля (1,293 мл) и проводили реакцию полученной смеси во вращающемся устройстве для пробирок (MTR-103, AS ONE Corporation) при комнатной температуре в течение 2 суток. Реакционную смесь очищали в соответствии со способом, описанным в Общей процедуре D, с получением раствора конъюгата нацеленного антитела с лекарственным средством в ABS (24,5 мл).
Анализ проводили в соответствии со способами, описанными в Общих процедурах Е и G, с получением следующих результатов. Концентрация антитела: 1,34 мг/мл Выход антитела: 32,92 мг (82%)
Среднее количество конъюгированных молекул лекарственного средства: 3,7.
[1188]
Пример 95. Синтез конъюгата 10 антитела с лекарственным средством (синтез конъюгата 2 антитела против LPS-ЦДН).
Фосфатно-солевой буферный раствор ремоделированного гликаном антитела 2 (рН 6,0) (10,55 мг/мл, 0,500 мл) разбавляли пропиленгликолем (0,250 мл). В данный раствор добавляли смесь раствора в диметилсульфоксиде лекарственного средства-линкера 2а (10 мМ, 0,087 мл, 24 эквивалента на молекулу антитела) и пропиленгликоля (0,163 мл) и проводили реакцию полученной смеси во вращающемся устройстве для пробирок (MTR-103, AS ONE Corporation) при комнатной температуре в течение 2 суток. Реакционную смесь очищали в соответствии со способом, описанным в Общей процедуре D, с получением раствора конъюгата нацеленного антитела с лекарственным средством в ABS (3,5 мл).
Анализ проводили в соответствии со способами, описанными в Общих процедурах Е и G, с получением следующих результатов. Концентрация антитела: 0,88 мг/мл. Выход антитела: 3,08 мг (62%).
Среднее количество конъюгированных молекул лекарственного средства: 3,6.
[1189]
Пример 96. Синтез конъюгата 11 антитела с лекарственным средством (синтез конъюгата 1 антитела против EphA2-ЦДН).
Фосфатно-солевой буферный раствор ремоделированного гликаном антитела 6 (рН 6,0) (8,91 мг/мл, 0,500 мл) разбавляли пропиленгликолем (0,250 мл). В данный раствор добавляли смесь раствора в диметилсульфоксиде лекарственного средства-линкера 2b (10 мМ, 0,073 мл, 24 эквивалента на молекулу антитела) и пропиленгликоля (0,177 мл) и проводили реакцию полученной смеси во вращающемся устройстве для пробирок (MTR-103, AS ONE Corporation) при комнатной температуре в течение 2 суток. Реакционную смесь очищали в соответствии со способом, описанным в Общей процедуре D, с получением раствора конъюгата нацеленного антитела с лекарственным средством в ABS (3,5 мл).
Анализ проводили в соответствии со способами, описанными в Общих процедурах Е и F, с получением следующих результатов. Концентрация антитела: 0,60 мг/мл. Выход антитела: 2,10 мг (47%).
Среднее количество конъюгированных молекул лекарственного средства: 3,8.
[1190]
Пример 97. Синтез конъюгата 12 антитела с лекарственным средством (синтез конъюгата 1 антитела против CD33-ЦДН).
Фосфатно-солевой буферный раствор ремоделированного гликаном антитела 5 (рН 6,0) (10,01 мг/мл, 0,500 мл) разбавляли пропиленгликолем (0,250 мл). В данный раствор добавляли смесь раствора в диметилсульфоксиде лекарственного средства-линкера 2b (10 мМ, 0,083 мл, 24 эквивалента на молекулу антитела) и пропиленгликоля (0,167 мл) и проводили реакцию полученной смеси во вращающемся устройстве для пробирок (MTR-103, AS ONE Corporation) при комнатной температуре в течение 2 суток. Реакционную смесь очищали в соответствии со способом, описанным в Общей процедуре D, с получением раствора конъюгата нацеленного антитела с лекарственным средством в ABS (3,5 мл).
Анализ проводили в соответствии со способами, описанными в Общих процедурах Е и G, с получением следующих результатов. Концентрация антитела: 0,73 мг/мл. Выход антитела: 2,57 мг (51%).
Среднее количество конъюгированных молекул лекарственного средства: 3,9.
[1191]
Пример 98. Синтез конъюгата 13 антитела с лекарственным средством (синтез конъюгата 1 антитела против CDH6-ЦДН).
Фосфатно-солевой буферный раствор ремоделированного гликаном антитела 7 (рН 6,0) (10,69 мг/мл, 0,500 мл) разбавляли пропиленгликолем (0,250 мл). В данный раствор добавляли смесь раствора в диметилсульфоксиде лекарственного средства-линкера 2b (10 мМ, 0,088 мл, 24 эквивалента на молекулу антитела) и пропиленгликоля (0,162 мл) и проводили реакцию полученной смеси во вращающемся устройстве для пробирок (MTR-103, AS ONE Corporation) при комнатной температуре в течение 3 суток. Реакционную смесь очищали в соответствии со способом, описанным в Общей процедуре D, с получением раствора конъюгата нацеленного антитела с лекарственным средством в ABS (3,5 мл).
Анализ проводили в соответствии со способами, описанными в Общих процедурах Е и G, с получением следующих результатов. Концентрация антитела: 0,96 мг/мл. Выход антитела: 3,37 мг (63%).
Среднее количество конъюгированных молекул лекарственного средства: 3,8.
[1192]
Пример 99. Синтез конъюгата 14 антитела с лекарственным средством (синтез конъюгата 1 антитела 2 против HER2-ЦДН).
Фосфатно-солевой буферный раствор ремоделированного гликаном антитела 3 (рН 6,0) (10,21 мг/мл, 1,50 мл) разбавляли пропиленгликолем (0,750 мл). В данный раствор добавляли смесь раствора в диметилсульфоксиде лекарственного средства-линкера 3 (10 мМ, 0,253 мл, 24 эквивалента на молекулу антитела) и пропиленгликоля (0,497 мл) и проводили реакцию полученной смеси во вращающемся устройстве для пробирок (MTR-103, AS ONE Corporation) при комнатной температуре в течение 2 суток. Реакционную смесь очищали в соответствии со способом, описанным в Общей процедуре D, с получением раствора конъюгата нацеленного антитела с лекарственным средством в ABS (9,5 мл).
Анализ проводили в соответствии со способами, описанными в Общих процедурах Е и G, с получением следующих результатов. Концентрация антитела: 1,02 мг/мл. Выход антитела: 9,73 мг (64%).
Среднее количество конъюгированных молекул лекарственного средства: 3,7.
[1193]
Пример 100. Синтез конъюгата 15 антитела с лекарственным средством (синтез конъюгата 9 антитела против HER2-ЦДН).
Фосфатно-солевой буферный раствор ремоделированного гликаном антитела 1 (рН 6,0) (10,70 мг/мл, 0,500 мл) разбавляли пропиленгликолем (0,250 мл). В данный раствор добавляли смесь раствора в диметилсульфоксиде лекарственного средства-линкера 8 (10 мМ, 0,088 мл, 24 эквивалента на молекулу антитела) и пропиленгликоля (0,162 мл) и проводили реакцию полученной смеси во вращающемся устройстве для пробирок (MTR-103, AS ONE Corporation) при комнатной температуре в течение 2 суток. Реакционную смесь очищали в соответствии со способом, описанным в Общей процедуре D, с получением раствора конъюгата нацеленного антитела с лекарственным средством в ABS (3,5 мл).
Анализ проводили в соответствии со способами, описанными в Общих процедурах Е и G, с получением следующих результатов. Концентрация антитела: 0,93 мг/мл. Выход антитела: 3,24 мг (61%).
Среднее количество конъюгированных молекул лекарственного средства: 3,6.
[1194]
Пример 101. Синтез конъюгата 16 антитела с лекарственным средством (синтез конъюгата 10 антитела против HER2-ЦДН).
Фосфатно-солевой буферный раствор ремоделированного гликаном антитела 1 (рН 6,0) (10,35 мг/мл, 13,50 мл) разбавляли пропиленгликолем (6,750 мл). В данный раствор добавляли смесь раствора в диметилсульфоксиде лекарственного средства-линкера 9 (10 мМ, 2,310 мл, 24 эквивалента на молекулу антитела) и пропиленгликоля (4,440 мл) и проводили реакцию полученной смеси во вращающемся устройстве для пробирок (MTR-103, AS ONE Corporation) при комнатной температуре в течение 2 суток. Реакционную смесь очищали в соответствии со способом, описанным в Общей процедуре D, с получением раствора конъюгата нацеленного антитела с лекарственным средством в ABS (74,3 мл).
Анализ проводили в соответствии со способами, описанными в Общих процедурах Е и G, с получением следующих результатов. Концентрация антитела: 1,36 мг/мл. Выход антитела: 100,8 мг (72%).
Среднее количество конъюгированных молекул лекарственного средства: 3,7.
[1195]
Пример 102. Синтез конъюгата 17 антитела с лекарственным средством (синтез конъюгата 11 антитела против HER2-ЦДН).
Фосфатно-солевой буферный раствор ремоделированного гликаном антитела 1 (рН 6,0) (10,70 мг/мл, 0,500 мл) разбавляли пропиленгликолем (0,250 мл). В данный раствор добавляли смесь раствора в диметилсульфоксиде лекарственного средства-линкера 10 (10 мМ, 0,088 мл, 24 эквивалента на молекулу антитела) и пропиленгликоля (0,162 мл) и проводили реакцию полученной смеси во вращающемся устройстве для пробирок (MTR-103, AS ONE Corporation) при комнатной температуре в течение 3 суток. Реакционную смесь очищали в соответствии со способом, описанным в Общей процедуре D, с получением раствора конъюгата нацеленного антитела с лекарственным средством в ABS (3,5 мл).
Анализ проводили в соответствии со способами, описанными в Общих процедурах Е и G, с получением следующих результатов. Концентрация антитела: 0,63 мг/мл. Выход антитела: 2,22 мг (42%).
Среднее количество конъюгированных молекул лекарственного средства: 3,5.
[1196]
Пример 103. Синтез конъюгата 18 антитела с лекарственным средством (синтез конъюгата 12 антитела против HER2-ЦДН).
Фосфатно-солевой буферный раствор ремоделированного гликаном антитела 1 (рН 6,0) (10,70 мг/мл, 0,500 мл) разбавляли пропиленгликолем (0,250 мл). В данный раствор добавляли смесь раствора в диметилсульфоксиде лекарственного средства-линкера 11 (10 мМ, 0,088 мл, 24 эквивалента на молекулу антитела) и пропиленгликоля (0,162 мл) и проводили реакцию полученной смеси во вращающемся устройстве для пробирок (MTR-103, AS ONE Corporation) при комнатной температуре в течение 3 суток. Реакционную смесь очищали в соответствии со способом, описанным в Общей процедуре D, с получением раствора конъюгата нацеленного антитела с лекарственным средством в ABS (3,5 мл).
Анализ проводили в соответствии со способами, описанными в Общих процедурах Е и G, с получением следующих результатов. Концентрация антитела: 0,94 мг/мл. Выход антитела: 3,29 мг (62%).
Среднее количество конъюгированных молекул лекарственного средства: 3,6.
[1197]
Пример 104. Синтез конъюгата 19 антитела с лекарственным средством (синтез конъюгата 13 антитела против HER2-ЦДН).
Фосфатно-солевой буферный раствор ремоделированного гликаном антитела 1 (рН 6,0) (10,70 мг/мл, 0,500 мл) разбавляли пропиленгликолем (0,250 мл). В данный раствор добавляли смесь раствора в диметилсульфоксиде лекарственного средства-линкера 12 (10 мМ, 0,088 мл, 24 эквивалента на молекулу антитела) и пропиленгликоля (0,162 мл) и проводили реакцию полученной смеси во вращающемся устройстве для пробирок (MTR-103, AS ONE Corporation) при комнатной температуре в течение 3 суток. Реакционную смесь очищали в соответствии со способом, описанным в Общей процедуре D, с получением раствора конъюгата нацеленного антитела с лекарственным средством в ABS (3,5 мл).
Анализ проводили в соответствии со способами, описанными в Общих процедурах Е и G, с получением следующих результатов. Концентрация антитела: 0,92 мг/мл. Выход антитела: 3,20 мг (60%).
Среднее количество конъюгированных молекул лекарственного средства: 3,5.
[1198]
Пример 105. Синтез конъюгата 20 антитела с лекарственным средством (синтез конъюгата 14 антитела против HER2-ЦДН).
Фосфатно-солевой буферный раствор ремоделированного гликаном антитела 1 (рН 6,0) (10,70 мг/мл, 0,500 мл) разбавляли пропиленгликолем (0,250 мл). В данный раствор добавляли смесь раствора в диметилсульфоксиде лекарственного средства-линкера 13 (10 мМ, 0,088 мл, 24 эквивалента на молекулу антитела) и пропиленгликоля (0,162 мл) и проводили реакцию полученной смеси во вращающемся устройстве для пробирок (MTR-103, AS ONE Corporation) при комнатной температуре в течение 3 суток. Реакционную смесь очищали в соответствии со способом, описанным в Общей процедуре D, с получением раствора конъюгата нацеленного антитела с лекарственным средством в ABS (3,5 мл).
Анализ проводили в соответствии со способами, описанными в Общих процедурах Е и G, с получением следующих результатов. Концентрация антитела: 1,01 мг/мл. Выход антитела: 3,52 мг (66%).
Среднее количество конъюгированных молекул лекарственного средства: 3,5.
[1199]
Пример 106. Синтез конъюгата 21 антитела с лекарственным средством (синтез конъюгата 15 антитела против HER2-ЦДН).
Фосфатно-солевой буферный раствор ремоделированного гликаном антитела 1 (рН 6,0) (10,70 мг/мл, 0,500 мл) разбавляли пропиленгликолем (0,250 мл). В данный раствор добавляли смесь раствора в диметилсульфоксиде лекарственного средства-линкера 14 (10 мМ, 0,088 мл, 24 эквивалента на молекулу антитела) и пропиленгликоля (0,162 мл) и проводили реакцию полученной смеси во вращающемся устройстве для пробирок (MTR-103, AS ONE Corporation) при комнатной температуре в течение 2 суток. Реакционную смесь очищали в соответствии со способом, описанным в Общей процедуре D, с получением раствора конъюгата нацеленного антитела с лекарственным средством в ABS (3,5 мл).
Анализ проводили в соответствии со способами, описанными в Общих процедурах Е и G, с получением следующих результатов. Концентрация антитела: 0,78 мг/мл. Выход антитела: 2,74 мг (51%).
Среднее количество конъюгированных молекул лекарственного средства: 3,6.
[1200]
Пример 107. Синтез конъюгата 22 антитела с лекарственным средством (синтез конъюгата 16 антитела против HER2-ЦДН).
Фосфатно-солевой буферный раствор ремоделированного гликаном антитела 1 (рН 6,0) (10,70 мг/мл, 0,500 мл) разбавляли пропиленгликолем (0,250 мл). В данный раствор добавляли смесь раствора в диметилсульфоксиде лекарственного средства-линкера 15 (10 мМ, 0,088 мл, 24 эквивалента на молекулу антитела) и пропиленгликоля (0,162 мл) и проводили реакцию полученной смеси во вращающемся устройстве для пробирок (MTR-103, AS ONE Corporation) при комнатной температуре в течение 2 суток. Реакционную смесь очищали в соответствии со способом, описанным в Общей процедуре D, с получением раствора конъюгата нацеленного антитела с лекарственным средством в ABS (3,5 мл).
Анализ проводили в соответствии со способами, описанными в Общих процедурах Е и G, с получением следующих результатов. Концентрация антитела: 1,32 мг/мл. Выход антитела: 4,61 мг (86%).
Среднее количество конъюгированных молекул лекарственного средства: 3,6.
[1201]
Пример 108. Синтез конъюгата23 антитела с лекарственным средством (синтез конъюгата 17 антитела против HER2-ЦДН).
Фосфатно-солевой буферный раствор ремоделированного гликаном антитела 1 (рН 6,0) (10,70 мг/мл, 0,500 мл) разбавляли пропиленгликолем (0,250 мл). В данный раствор добавляли смесь раствора в диметилсульфоксиде лекарственного средства-линкера 16 (10 мМ, 0,088 мл, 24 эквивалента на молекулу антитела) и пропиленгликоля (0,162 мл) и проводили реакцию полученной смеси во вращающемся устройстве для пробирок (MTR-103, AS ONE Corporation) при комнатной температуре в течение 2 суток. Реакционную смесь очищали в соответствии со способом, описанным в Общей процедуре D, с получением раствора конъюгата нацеленного антитела с лекарственным средством в ABS (3,5 мл).
Анализ проводили в соответствии со способами, описанными в Общих процедурах Е и G, с получением следующих результатов. Концентрация антитела: 0,93 мг/мл. Выход антитела: 3,25 мг (61%).
Среднее количество конъюгированных молекул лекарственного средства: 3,6.
[1202]
Пример 109. Синтез конъюгата 24 антитела с лекарственным средством (синтез конъюгата 18 антитела против HER2-ЦДН).
Фосфатно-солевой буферный раствор ремоделированного гликаном антитела 1 (рН 6,0) (10,70 мг/мл, 4,00 мл) разбавляли пропиленгликолем (2,00 мл). В данный раствор добавляли смесь раствора в диметилсульфоксиде лекарственного средства-линкера 7 (10 мМ, 0,707 мл, 24 эквивалента на молекулу антитела) и пропиленгликоля (1,293 мл) и проводили реакцию полученной смеси во вращающемся устройстве для пробирок (MTR-103, AS ONE Corporation) при комнатной температуре в течение 3 суток. Реакционную смесь очищали в соответствии со способом, описанным в Общей процедуре D, с получением раствора конъюгата нацеленного антитела с лекарственным средством в ABS (24,5 мл).
Анализ проводили в соответствии со способами, описанными в Общих процедурах Е и G, с получением следующих результатов. Концентрация антитела: 1,33 мг/мл. Выход антитела: 32,64 мг (82%).
Среднее количество конъюгированных молекул лекарственного средства: 3,7.
[1203]
Пример 110. Синтез конъюгата 25 антитела с лекарственным средством (синтез конъюгата 2 антитела 2 против HER2-ЦДН).
Фосфатно-солевой буферный раствор ремоделированного гликаном антитела 4 (рН 6,0) (10,87 мг/мл, 8,00 мл) разбавляли пропиленгликолем (4,00 мл). В данный раствор добавляли смесь раствора в диметилсульфоксиде лекарственного средства-линкера 17а (10 мМ, 1,079 мл, 18 эквивалентов на молекулу антитела) и пропиленгликоля (2,921 мл) и проводили реакцию полученной смеси во вращающемся устройстве для пробирок (MTR-103, AS ONE Corporation) при комнатной температуре в течение 3 суток. Реакционную смесь очищали в соответствии со способом, описанным в Общей процедуре D, с получением раствора конъюгата нацеленного антитела с лекарственным средством в ABS (44,5 мл).
Анализ проводили в соответствии со способами, описанными в Общих процедурах Е и G, с получением следующих результатов. Концентрация антитела: 1,16 мг/мл. Выход антитела: 51,5 мг (59%).
Среднее количество конъюгированных молекул лекарственного средства: 3,8.
[1204]
Пример 111. Синтез конъюгата 26 антитела с лекарственным средством (синтез конъюгата 3 антитела 2 против HER2-ЦДН).
Фосфатно-солевой буферный раствор ремоделированного гликаном антитела 4 (рН 6,0) (10,87 мг/мл, 8,00 мл) разбавляли пропиленгликолем (4,00 мл). В данный раствор добавляли смесь раствора в диметилсульфоксиде лекарственного средства-линкера 17b (10 мМ, 1,079 мл, 18 эквивалентов на молекулу антитела) и пропиленгликоля (2,921 мл) и проводили реакцию полученной смеси во вращающемся устройстве для пробирок (MTR-103, AS ONE Corporation) при комнатной температуре в течение 3 суток. Реакционную смесь очищали в соответствии со способом, описанным в Общей процедуре D, с получением раствора конъюгата нацеленного антитела с лекарственным средством в ABS (44,5 мл).
Анализ проводили в соответствии со способами, описанными в Общих процедурах Е и G, с получением следующих результатов. Концентрация антитела: 1,24 мг/мл. Выход антитела: 54,99 мг (63%).
Среднее количество конъюгированных молекул лекарственного средства: 3,9.
[1205]
Пример 112. Синтез конъюгата 27 антитела с лекарственным средством (синтез конъюгата 4 антитела 2 против HER2-ЦДН).
Фосфатно-солевой буферный раствор ремоделированного гликаном антитела 4 (рН 6,0) (10,89 мг/мл, 1,00 мл) разбавляли пропиленгликолем (0,500 мл). В данный раствор добавляли смесь раствора в диметилсульфоксиде лекарственного средства-линкера 19 (10 мМ, 0,180 мл, 24 эквивалента на молекулу антитела) и пропиленгликоля (0,320 мл) и проводили реакцию полученной смеси во вращающемся устройстве для пробирок (MTR-103, AS ONE Corporation) при комнатной температуре в течение 3 суток. Реакционную смесь очищали в соответствии со способом, описанным в Общей процедуре D, с получением раствора конъюгата нацеленного антитела с лекарственным средством в ABS (7,0 мл).
Анализ проводили в соответствии со способами, описанными в Общих процедурах Е и G, с получением следующих результатов. Концентрация антитела: 0,79 мг/мл. Выход антитела: 5,53 мг (51%).
Среднее количество конъюгированных молекул лекарственного средства: 3,9.
[1206]
Пример 113. Синтез конъюгата 28 антитела с лекарственным средством (синтез конъюгата 5 антитела 2 против HER2-ЦДН).
Фосфатно-солевой буферный раствор ремоделированного гликаном антитела 4 (рН 6,0) (10,89 мг/мл, 1,00 мл) разбавляли пропиленгликолем (0,500 мл). В данный раствор добавляли смесь раствора в диметилсульфоксиде лекарственного средства-линкера 21 (10 мМ, 0,180 мл, 24 эквивалента на молекулу антитела) и пропиленгликоля (0,320 мл) и проводили реакцию полученной смеси во вращающемся устройстве для пробирок (MTR-103, AS ONE Corporation) при комнатной температуре в течение 3 суток. Реакционную смесь очищали в соответствии со способом, описанным в Общей процедуре D, с получением раствора конъюгата нацеленного антитела с лекарственным средством в ABS (7,0 мл).
Анализ проводили в соответствии со способами, описанными в Общих процедурах Е и G, с получением следующих результатов. Концентрация антитела: 0,84 мг/мл. Выход антитела: 5,91 мг (54%).
Среднее количество конъюгированных молекул лекарственного средства: 3,9.
[1207]
Пример 114. Синтез конъюгата 29 антитела с лекарственным средством (синтез конъюгата 6 антитела 2 против HER2-ЦДН).
Фосфатно-солевой буферный раствор ремоделированного гликаном антитела 4 (рН 6,0) (10,89 мг/мл, 1,00 мл) разбавляли пропиленгликолем (0,500 мл). В данный раствор добавляли смесь раствора в диметилсульфоксиде лекарственного средства-линкера 22 (10 мМ, 0,180 мл, 24 эквивалента на молекулу антитела) и пропиленгликоля (0,320 мл) и проводили реакцию полученной смеси во вращающемся устройстве для пробирок (MTR-103, AS ONE Corporation) при комнатной температуре в течение 2 суток. Реакционную смесь очищали в соответствии со способом, описанным в Общей процедуре D, с получением раствора конъюгата нацеленного антитела с лекарственным средством в ABS (5,0 мл).
Анализ проводили в соответствии со способами, описанными в Общих процедурах Е и G, с получением следующих результатов. Концентрация антитела: 0,92 мг/мл. Выход антитела: 4,60 мг (42%).
Среднее количество конъюгированных молекул лекарственного средства: 3,5.
[1208]
Пример 115. Синтез конъюгата 30 антитела с лекарственным средством (синтез конъюгата 7 антитела 2 против HER2-ЦДН).
Фосфатно-солевой буферный раствор ремоделированного гликаном антитела 4 (рН 6,0) (10,89 мг/мл, 1,00 мл) разбавляли пропиленгликолем (0,500 мл). В данный раствор добавляли смесь раствора в диметилсульфоксиде лекарственного средства-линкера 18 (10 мМ, 0,180 мл, 24 эквивалента на молекулу антитела) и пропиленгликоля (0,320 мл) и проводили реакцию полученной смеси во вращающемся устройстве для пробирок (MTR-103, AS ONE Corporation) при комнатной температуре в течение 2 суток. Реакционную смесь очищали в соответствии со способом, описанным в Общей процедуре D, с получением раствора конъюгата нацеленного антитела с лекарственным средством в ABS (7,0 мл).
Анализ проводили в соответствии со способами, описанными в Общих процедурах Е и G, с получением следующих результатов. Концентрация антитела: 1,08 мг/мл. Выход антитела: 7,53 мг (69%).
Среднее количество конъюгированных молекул лекарственного средства: 3,9.
[1209]
Пример 116. Синтез конъюгата 31 антитела с лекарственным средством (синтез конъюгата 8 антитела 2 против HER2-ЦДН).
Фосфатно-солевой буферный раствор ремоделированного гликаном антитела 4 (рН 6,0) (10,89 мг/мл, 1,00 мл) разбавляли пропиленгликолем (0,500 мл). В данный раствор добавляли смесь раствора в диметилсульфоксиде лекарственного средства-линкера 20 (10 мМ, 0,180 мл, 24 эквивалента на молекулу антитела) и пропиленгликоля (0,320 мл) и проводили реакцию полученной смеси во вращающемся устройстве для пробирок (MTR-103, AS ONE Corporation) при комнатной температуре в течение 2 суток. Реакционную смесь очищали в соответствии со способом, описанным в Общей процедуре D, с получением раствора конъюгата нацеленного антитела с лекарственным средством в ABS (7,0 мл).
Анализ проводили в соответствии со способами, описанными в Общих процедурах Е и G, с получением следующих результатов. Концентрация антитела: 1,09 мг/мл. Выход антитела: 7,62 мг (70%).
Среднее количество конъюгированных молекул лекарственного средства: 3,8.
[1210]
Пример 117. Синтез конъюгата 32 антитела с лекарственным средством (синтез конъюгата 9 антитела 2 против HER2-ЦДН).
Фосфатно-солевой буферный раствор ремоделированного гликаном антитела 8 (рН 6,0) (10,48 мг/мл, 1,00 мл) разбавляли пропиленгликолем (0,500 мл). В данный раствор добавляли смесь раствора в диметилсульфоксиде лекарственного средства-линкера 17а (10 мМ, 0,087 мл, 12 эквивалентов на молекулу антитела) и пропиленгликоля (0,413 мл) и проводили реакцию полученной смеси во вращающемся устройстве для пробирок (MTR-103, AS ONE Corporation) при комнатной температуре в течение 2 суток. Реакционную смесь очищали в соответствии со способом, описанным в Общей процедуре D, с получением раствора конъюгата нацеленного антитела с лекарственным средством в ABS (7,0 мл).
Анализ проводили в соответствии со способами, описанными в Общих процедурах Е и G, с получением следующих результатов. Концентрация антитела: 0,77 мг/мл. Выход антитела: 5,36 мг (51%).
Среднее количество конъюгированных молекул лекарственного средства: 1,8.
[1211]
Пример 118. Синтез конъюгата 33 антитела с лекарственным средством (синтез конъюгата 10 антитела 2 против HER2-ЦДН).
Фосфатно-солевой буферный раствор ремоделированного гликаном антитела 8 (рН 6,0) (10,48 мг/мл, 1,00 мл) разбавляли пропиленгликолем (0,500 мл). В данный раствор добавляли смесь раствора в диметилсульфоксиде лекарственного средства-линкера 17b (10 мМ, 0,087 мл, 12 эквивалентов на молекулу антитела) и пропиленгликоля (0,413 мл) и проводили реакцию полученной смеси во вращающемся устройстве для пробирок (MTR-103, AS ONE Corporation) при комнатной температуре в течение 2 суток. Реакционную смесь очищали в соответствии со способом, описанным в Общей процедуре D, с получением раствора конъюгата нацеленного антитела с лекарственным средством в ABS (7,0 мл).
Анализ проводили в соответствии со способами, описанными в Общих процедурах Е и G, с получением следующих результатов. Концентрация антитела: 0,91 мг/мл. Выход антитела: 6,34 мг (61%).
Среднее количество конъюгированных молекул лекарственного средства: 1,8.
[1212]
Пример 119. Синтез конъюгата 34 антитела с лекарственным средством (синтез конъюгата 19 антитела против HER2-ЦДН).
В фосфатном буферном растворе антитела против HER2, полученного в соответствии со ссылочным примером 1, осуществляли замену буфера в соответствии с Общей процедурой С с получением раствора антитела в фосфатно-солевом буферном растворе/5 мМ ЭДТА (7,69 мг/мл). В данный раствор антитела (0,65 мл) добавляли водный раствор гидрохлорида трис(2-карбоксиэтил)фосфина (10 мМ, 20,7 мкл) и водный раствор вторичного кислого фосфата калия (1 М, 9,8 мкл). После подтверждения, что рН реакционной смеси находился в пределах 7,4±1,0, реакционную смесь перемешивали при 37°С в течение 2 часов. Добавляли туда раствор в ДМСО лекарственного средства-линкера 23 (10 мМ, 0,0689 мл) и проводили реакцию полученной смеси во вращающемся устройстве для пробирок (MTR-103, AS ONE Corporation) при комнатной температуре в течение 1 часа. Водный раствор N-ацетил-L-цистеина (100 мМ, 6,9 мкл) добавляли в реакционную смесь, чтобы погасить реакцию. В реакционной смеси осуществляли замену буфера в соответствии со способом, описанным в Общей процедуре С, с получением раствора в ABS конъюгата нацеленного антитела с лекарственным средством (1,6 мл).
Анализ проводили в соответствии со способами, описанными в Общих процедурах Е и G, с получением следующих результатов. Концентрация антитела: 1,46 мг/мл. Выход антитела: 2,34 мг (47%).
Среднее количество конъюгированных молекул лекарственного средства: 6,5.
[1213]
Пример 120. Синтез лекарственного средства-линкера 24.
[Схема синтеза]
[1214]
[1215]
(Этап 1)
С применением соединения, полученного на этапе 8 примера 44 (1,55 г), проводили реакцию таким же образом, как на этапе 7 примера 1, с получением раствора в ацетонитриле 6-бензоил-2-{2-дезокси-2-фтор-3-О-[гидрокси(оксо)-λ5-фосфанил]-β-D-рибофуранозил}-6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулена. С применением полученного раствора в ацетонитриле и полученного на этапе 6 примера 77 соединения (1,96 г) проводили реакцию таким же образом, как на этапе 8 примера 1, и полученный в результате этого неочищенный продукт непосредственно использовали для следующей реакции.
[1216]
(Этап 2)
2-(триметилсилил)этил-(2-{[(2-{9-[(5R,7R,8R,12aR,14R,15R,15aR,16R)-14-(6-бензоил-6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)-16-{[трет-бутил(диметил)силил]окси}-10-(2-цианоэтокси)-15-фтор-2-гидрокси-2-оксо-10-сульфанилиденоктагидро-2H,10H,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-7-ил]-6-оксо-6,9-дигидро-1Н-пурин-1-ил}этокси)метил]амино}-2-оксоэтил)карбамат
С применением неочищенного продукта, полученного на этапе 1 выше, проводили реакцию таким же образом, как на этапе 2 примера 62, с получением указанного в заголовке соединения (1,07 г: с примесями) в виде смеси диастереомеров у атома фосфора.
МС (ИЭР) m/z: 1262(M+H)+.
[1217]
(Этап 3)
Бис(N,N-диэтилэтанаминия) (5R,7R,8R,12aR,14R,15R,16R)-16-{[трет-бутил(диметил)силил]окси}-15-фтор-2,10-диоксо-7-[6-оксо-1-(2-{[(М-{[2-(триметилсилил)этокси]карбонил}глицил)амино]метокси}этил)-1,6-дигидро-9Н-пурин-9-ил]-10-сульфид-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2H,10H,12H-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2-олат
С применением соединения (1,07 г), полученного на этапе 2 выше, проводили реакцию таким же образом, как на этапе 10 примера 1, и продукт затем очищали с помощью колоночной хроматографии на силикагеле С 18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил] и препаративной ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил-метанол (1:1), ацетонитрил-метанол (1:1): 25% - 90% (0 мин - 40 мин)] с получением диастереомера 1 (67,8 мг: с примесями) и диастереомера 2 (56,6 мг: с примесями) указанного в заголовке соединения.
Диастереомер 1 (менее полярный)
МС (ИЭР) m/z: 1105 (М+Н)+.
Диастереомер 2 (более полярный)
МС (ИЭР) m/z: 1105 (M+H)+.
[1218]
(Этап 4-1)
Бис(N,N-диэтилэтанаминия) (5R,7R,8R,12aR,14R,15R,16R)-15-фтор-7-(l-{2-[(глициламино)метокси]этил}-6-оксо-1,6-дигидро-9Н-пурин-9-ил)-16-гидрокси-2,10-диоксо-10-сульфид-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2-олат
С применением соединения; полученного на этапе 3 выше (диастереомер 1) (67,8 мг), проводили реакцию таким же образом, как на этапе 9-1 примера 11, и продукт затем очищали с помощью колоночной хромато графин на силикагеле С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил] и препаративной ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацето нитрил-метанол (1:1), ацетонитрил-метанол (1:1): 10% - 60% (0 мин - 30 мин)] с получением указанного в заголовке соединения (33,7 мг: с примесями).
МС (ИЭР) m/z: 847(M+H)+.
[1219]
(Этап 4-2)
Бис(N,N-диэтилэтанаминия) (5R,7R,8R,12aR,14R,15R,16R)-15-фтор-7-(l-{2-[(глициламино)метокси]этил}-6-оксо-1,6-дигидро-9Н-пурин-9-ил)-16-гидрокси-2,10-диоксо-10-сульфид-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2-олат
С применением соединения, полученного на этапе 3 выше (диастереомер 2) (56,6 мг), проводили реакцию таким же образом, как на этапе 9-1 примера 11, и продукт затем очищали с помощью колоночной хроматографии на силикагеле С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил] и препаративной ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил-метанол (1:1), ацетонитрил-метанол (1:1): 10%-60% (0 мин - 30 мин)] с получением указанного в заголовке соединения (32,6 мг: с примесями).
МС (ИЭР) m/z: 847(М+Н)+.
[1220]
(Этап 5-1)
Бис(N,N-диэтилэтанаминия) N-[4-(11,12-дидегидродибензо[b,f]азоцин-5(6Н)-ил)-4-оксобутаноил]глицилглицил-L-фенилаланил-N-[(2-{9-[(5R,7R,8R,12aR,14R,15R,15aR,16R)-15-фтор-16-гидрокси-2-оксид-2,10-диоксо-10-сульфид-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-7-ил]-6-оксо-6,9-дигидро-1Н-пурин-1-ил}этокси)метил]глицинамид (Диастереомер 1)
С применением соединения, полученного на этапе 4-1 выше (33,7 мг), и полученного на этапе 11 примера 22 соединения (21,3 мг) проводили реакцию таким же образом, как на этапе 9-1 примера 22, и очистку проводили при следующих [Условиях очистки] с получением указанного в заголовке соединения (11,2 мг).
[Условия очистки] Хроматография на колонке с силикагелем С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил] и препаративная ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/метанол, метанол: 40%-90% (0 мин - 40 мин)].
МС (ИЭР) m/z: 1395(М+Н)+.
1H-ЯМР (CD3OD) δ: 8,57-8,54 (1H, м), 8,16-8,10 (1H, м), 8,04 (1H, с), 7,64-7,49 (2Н, м), 7,44-7,34 (3Н, м), 7,33-7,11 (9Н, м), 6,46 (1H, д, J=18,1 Гц), 6,26 (1H, д, J=8,5 Гц), 5,54-5,30 (2Н, м), 5,18-5,00 (2Н, м), 4,75-4,71 (1H, м), 4,65-4,20 (10Н, м), 4,13-3,93 (2Н, м), 3,88-3,60 (8Н, м), 3,55-3,40 (2Н, м), 3,26-3,15 (1H, м), 3,18 (12Н, к, J=7,3 Гц), 3,02-2,91 (1Н, м), 2,87-2,61 (3Н, м), 2,38-2,18 (2Н, м), 2,06-1,80 (3Н, м), 1,28 (18Н, т, J=7,3 Гц).
[1221]
(Этап 5-2)
Бис(N,N-диэтилэтанаминия) N-[4-(11,12-дидегидродибензо[b,f]азоцин-5(6Н)-ил)-4-оксобутаноил]глицилглицил-L-фенилаланил-Х-[(2-{9-[(5R,7R,8R,12aR,14R,15R,15aR,16R)-15-фтор-16-гидрокси-2-оксид-2,10-диоксо-10-сульфид-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-7-ил]-6-оксо-6,9-дигидро-1Н-пурин-1-ил}этокси)метил]глицинамид
(Диастереомер 2)
С применением соединения, полученного на этапе 4-2 выше (32,6 мг), и полученного на этапе 11 примера 22 соединения (20,6 мг) проводили реакцию таким же образом, как на этапе 9-1 примера 22, и очистку проводили при следующих [Условиях очистки] с получением указанного в заголовке соединения (16,7 мг).
[Условия очистки] Хроматография на колонке с силикагелем С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил], препаративная ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/метанол, метанол: 40%-90% (0 мин - 40 мин)] и препаративная ВЭЖХ [100 мМ гексафтор-2-пропанол, 8 мМ водный раствор триэтиламина/ацетонитрил, ацетонитрил: 10%-45% (0 мин - 40 мин)].
MC(H3P)m/z: 1395(М+Н)+.
1H-ЯМР (CD3OD) δ: 8,58 (1H, с), 8,17-8,13 (1H, м), 8,02 (1H, с), 7,64-7,50 (2Н, м), 7,42-7,35 (4Н, м), 7,32-7,12 (8Н, м), 6,49 (1Н, д, J=16,3 Гц), 6,27 (1Н, дд, J=8,2, 6,3 Гц), 5,50-5,21 (3Н, м), 5,08-5,00 (1H, м), 4,66-4,23 (9Н, м), 4,14-3,98 (3Н, м), 3,91-3,52 (9Н, м), 3,48-3,41 (2Н, м), 3,23-3,12 (1H, м), 3,18 (12Н, к, J=7,3 Гц), 3,03-2,94 (1H, м), 2,85-2,75 (3Н, м), 2,40-2,23 (2Н, м), 2,06-1,87 (3Н, м), 1,28 (18Н, т, J=7,3 Гц).
[1222]
Пример 121. Синтез лекарственного средства-линкера 25.
[Схема синтеза]
[1223]
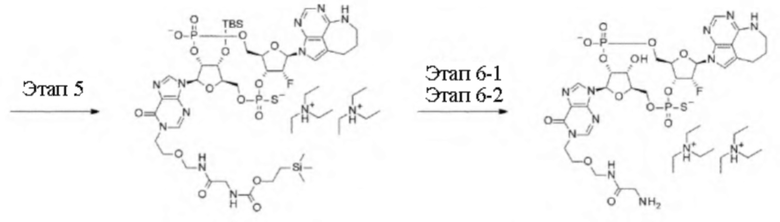
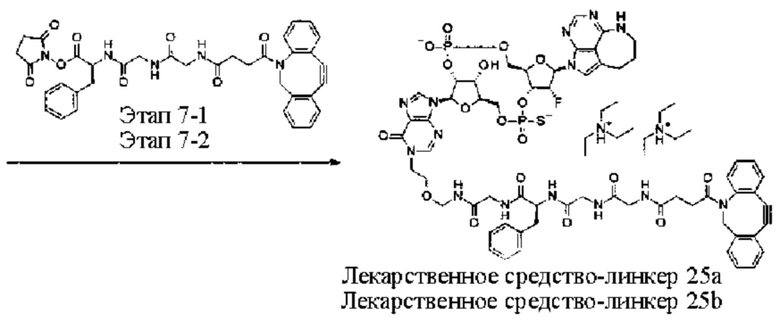
[1224]
(Этап 1)
N,N-диэтилэтанаминия 6-бензоил-2-{5-O-[бис(4-метоксифенил)(фенил)метил]-2-дезокси-2-фтор-3-O-[оксид(оксо)-λ5-фосфанил]-β-В-рибофуранозил}-6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен
К раствору соединения, полученного на этапе 7 примера 44 (1,49 г), в пиридине (10,4 мл) добавляли дифенилфосфит (599 мкл) при охлаждении льдом, и температуру повышали до комнатной температуры и реакционную смесь перемешивали в течение 30 минут. Туда дополнительно добавляли дифенилфосфит (200 мкл) и реакционную смесь дополнительно перемешивали в течение 2 часов. Воду (1,5 мл) добавляли в реакционную смесь при охлаждении льдом и реакционную смесь перемешивали при комнатной температуре в течение 30 минут, а затем концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле [этилацетат/метанол/0,1% триэтиламин] с получением указанного в заголовке соединения (1,41 г).
МС (ИЭР) m/z: 779(М+Н)+.
1H-ЯМР (CD3OD) δ: 8,01 (1H, с), 7,54 (1H, с), 7,42-7,39 (2Н, м), 7,32-7,19 (10Н, м), 7,14-7,09 (2Н, с), 6,84 (1H, дд, J=628,7, 1,8 Гц), 6,83-6,79 (4Н, м), 6,53 (1H, дд, J=17,5, 1,8 Гц), 5,54 (1H, ддд, J=52,l, 4,0, 2,0 Гц), 5,37-5,27 (1H, м), 4,31-4,21 (3Н, м), 3,55 (1Н, дд, J=11,2, 2,1 Гц), 3,38 (1H, дд, J=10,9, 3,0 Гц), 3,34 (6Н, с), 3,19 (6Н, к, J=7,5 Гц), 2,84-2,69 (2Н, м), 2,18-2,12 (2Н, м), 1,29 (9Н, т, J=7,3 Гц).
[1225]
(Этап 2)
6-бензоил-2-{2-дезокси-2-фтор-3-0-[гидрокси(оксо)-λ5-фосфанил]-β-В-рибофуранозил}-6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен
К раствору соединения, полученного на этапе 1 выше (1,41 г), в дихлорметане (20,0 мл) добавляли воду (289 мкл) и раствор дихлоруксусной кислоты (1,14 мл) в дихлорметане (20,0 мл) и реакционную смесь перемешивали при комнатной температуре в течение 10 минут. Пиридин (2,19 мл) добавляли в реакционную смесь, чтобы погасить реакцию, и реакционную смесь затем концентрировали при пониженном давлении. Пиридиновую соль трифторуксусной кислоты (402 мг) добавляли к остатку и продукт подвергали трехкратной азеотропной перегонке с обезвоженным ацетонитрилом (15 мл), причем приблизительно 10 мл ацетонитрила позволяли остаться после последней процедуры. Полученный раствор в ацетонитриле непосредственно использовали для следующей реакции.
МС (ИЭР) m/z: 477(М+Н)+.
[1226]
(Этап 3)
С применением соединения, полученного на этапе 6 примера 77 (2,15 г), и раствора в ацетонитриле полученного на этапе 2 выше соединения проводили реакцию таким же образом, как на этапе 1 примера 63, и полученный в результате этого неочищенный продукт непосредственно использовали для следующей реакции. [1227] (Этап 4)
2-(триметилсилил)этил-(2-{ [(2- {9-[(5R,7R,8R, 12aR, 14R, 15R, 15aR, 16R)- 14-(6-бензоил-6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)-16-{[трет-бутил(диметил)силил]окси}-10-(2-цианоэтокси)-15-фтор-2,10-диоксо-2-сульфанилоктагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-7-ил]-6-оксо-6,9-дигидро-1Н-пурин-1-ил}этокси)метил]амино}-2-оксоэтил)карбамат
С применением неочищенного продукта, полученного на этапе 3 выше, проводили реакцию таким же образом, как на этапе 9 примера 1, с получением указанного в заголовке соединения (1,00 г: с примесями). MC(ИЭР) m/z: 1262(М+Н)+.
[1228]
(Этап 5)
Бис(N,N-диэтилэтанаминия) (5R,7R,8R,12aR,14R,15R,15aR,16R)-16-{[трет-бутил(диметил)силил]окси}-15-фтор-2,10-диоксо-7-[6-оксо-1-(2-{[(N-{[2-(триметилсилил)этокси]карбонил}глицил)амино]метокси}этил)-1,6-дигидро-9Н-пурин-9-ил]-2-сульфид-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-10-олат
С применением соединения, полученного на этапе 4 выше (1,00 г), проводили реакцию таким же образом, как на этапе 10 примера 1, с получением диастереомера 1 (191 мг: с примесями) и диастереомера 2 (369 мг: с примесями) указанного в заголовке соединения.
Диастереомер 1 (менее полярный)
МС (ИЭР) m/z: 1105(М+Н)+.
Диастереомер 2 (более полярный)
МС (ИЭР) m/z: 1105(М+Н)+.
[1229]
(Этап 6-1)
Бис(N,N-диэтилэтанаминия) (5R,7R,8R,12aR,14R,15R,15aR,16R)-15-фтор-7-(1-{2-[(глициламино)метокси]этил}-6-оксо-1,6-дигидро-9Н-пурин-9-ил)-16-гидрокси-2,10-диоксо-2-сульфид-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-10-олат
С применением соединения, полученного на этапе 5 выше (диастереомер 1) (191 мг), проводили реакцию таким же образом, как на этапе 5 примера 40, с получением указанного в заголовке соединения (21,9 мг: с примесями).
МС (ИЭР) m/z: 847(М+Н)+.
1H-ЯМР (CD3OD) δ: 8,39 (1H, с), 8,03 (1H, с), 7,71 (1H, с), 7,24 (1H, с), 6,45 (1H, д, J=17,5 Гц), 6,15 (1H, д, J=8,5 Гц), 5,79-5,65 (2Н, м), 5,46-5,36 (1H, м), 4,63-4,23 (9Н, м), 4,01-3,95 (1H, м), 3,65-3,57 (2Н, м), 3,52-3,37 (5Н, м), 3,15 (12Н, к, J=7,3 Гц), 2,76-2,68 (1H, м), 2,39-2,30 (1H, м), 1,92-1,82 (2Н, м), 1,28 (18Н, т, J=7,6 Гц).
[1230]
(Этап 6-2)
Бис(N,N-диэтилэтанаминия) (5R,7R,8R,12aR,14R,15R,15aR,16R)-15-фтор-7-(1-{2-[(глициламино)метокси]этил}-6-оксо-1,6-дигидро-9Н-пурин-9-ил)-16-гидрокси-2,10-диоксо-2-сульфид-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-10-олат
С применением соединения, полученного на этапе 5 выше (диастереомер 2) (369 мг), проводили реакцию таким же образом, как на этапе 5 примера 40, с получением указанного в заголовке соединения (137 мг: с примесями).
МС (ИЭР) m/z: 847(М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,96 (1H, с), 8,14 (1Н, с), 8,02 (1H, с), 7,04 (1Н, с), 6,46 (1H, д, J=19,3 Гц), 6,28 (1H, д, J=8,5 Гц), 5,61 (1H, дд, J=52,0, 4,2 Гц), 5,38-5,23 (2Н, м), 4,69-4,63 (2Н, м), 4,56 (1Н, д, J=10,3 Гц), 4,39-4,30 (4Н, м), 4,26-4,17 (3Н, м), 3,97-3,91 (1H, м), 3,84-3,72 (2Н, м), 3,52-3,46 (2Н, м), 3,32-3,25 (2Н, м), 2,97 (12Н, к, J=7,3 Гц), 2,74-2,63 (2Н, м), 2,03-1,84 (2Н, м), 1,21 (18Н, т, J=7,3 Гц).
[1231]
(Этап 7-1)
Бис(N,N-диэтилэтанаминия) N-[4-(11,12-дидегидродибензо[b,f]азоцин-5(6Н)-ил)-4-оксобутаноил]глицилглицил-L-фенилаланил-М-[(2-{9-[(5R,7R,8R,12aR,14R,15R,15aR,16R)-15-фтор-16-гидрокси-10-оксид-2,10-диоксо-2-сульфид-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-7-ил]-6-оксо-6,9-дигидро-1Н-пурин-1-ил}этокси)метил]глицинамид
(Лекарственное средство-линкер 25а: диастереомер 1)
С применением соединения, полученного на этапе 6-1 выше (21,9 мг), проводили реакцию таким же образом, как на этапе 9-1 примера 22, а затем проводили очистку при следующих [Условиях очистки] с получением указанного в заголовке соединения (26,3 мг). [Условия очистки] препаративная ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 30%-50% (0 мин - 30 мин)] и Sep-Pak (R) С18 [вода/ацетонитрил/0,1%триэтиламин].
MC(ИЭР) m/z: 1395(М+Н)+.
1H-ЯМР (CD3OD) δ: 8,58 (1H, д, J=4,2 Гц), 8,08-8,02 (2Н, м), 7,64-7,13 (14Н, м), 6,46 (1H, д, J=18,1 Гц), 6,26-6,21 (1H, м), 5,45 (1H, д, J=53,2 Гц), 5,35-5,18 (2Н, м), 5,07-5,01 (1H, м), 4,62-4,13 (10Н, м), 4,09-3,90 (2Н, м), 3,87-3,43 (11Н, м), 3,21-3,09 (1H, м), 3,18 (12Н, к, J=7,3 Гц), 3,00-2,89 (1H, м), 2,83-2,52 (3Н, м), 2,37-2,21 (2Н, м), 2,03-1,79 (3Н, м), 1,28 (18Н, т, J=7,3 Гц).
[1232]
(Этап 7-2)
Бис(N,N-диэтилэтанаминия) N-[4-(11,12-дидегидродибензо[b,f]азоцин-5(6Н)-ил)-4-оксобутаноил]глицилглицил-L-фенилаланил-М-[(2-{9-[(5R,7R,8R,12aR,14R,15R,15aR,16R)-15-фтор-16-гидрокси-10-оксид-2,10-диоксо-2-сульфид-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-7-ил]-6-оксо-6,9-дигидро-1Н-пурин-1-ил}этокси)метил]глицинамид
(Лекарственное средство-линкер 25b: диастереомер 2)
С применением соединения, полученного на этапе 6-2 выше (47,5 мг), проводили реакцию таким же образом, как на этапе 9-1 примера 22, а затем проводили очистку при следующих [Условиях очистки] с получением указанного в заголовке соединения (15,1 мг). [Условия очистки] препаративная ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил, ацетонитрил: 30%-50% (0 мин - 30 мин)] и Sep-Pak (R) С18 [в од а/ацето нитрил/0,1% триэтиламин].
МС (ИЭР) m/z: 1395(М+Н)+.
1Н-ЯМР (CD3OD) δ: 8,99 (1H, д, J=3,6 Гц), 8,14 (1H, д, J=l,8 Гц), 8,02-8,00 (1H, м), 7,64-7,13 (13Н, м), 7,01-6,98 (1H, м), 6,47 (1H, дд, J=19,0, 2,7 Гц), 6,27 (1H, дд, J=8,5, 4,2 Гц), 5,67-5,52 (1H, м), 5,37-5,16 (2Н, м), 5,08-5,01 (1H, м), 4,67-4,14 (11Н, м), 4,10-3,61 (10Н, м), 3,47-3,42 (2Н, м), 3,17-3,10 (1Н, м), 3,15 (12Н, к, J=7,5 Гц), 3,00-2,92 (1H, м), 2,84-2,75 (1Н, м), 2,63-2,54 (2Н, м), 2,32-2,20 (2Н, м), 2,03-1,79 (3Н, м), 1,28 (18Н, т, J=7,3 Гц).
[1233]
Пример 122. Синтез лекарственного средства-линкера 26.
[Схема синтеза]
[1234]
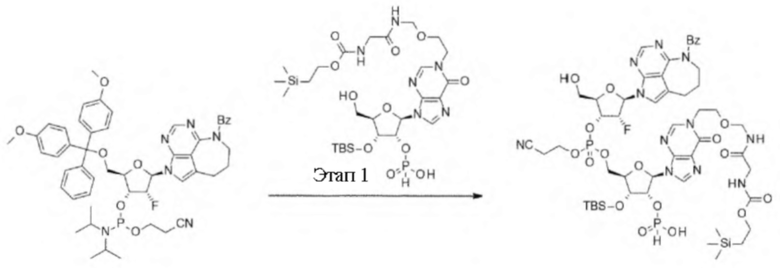
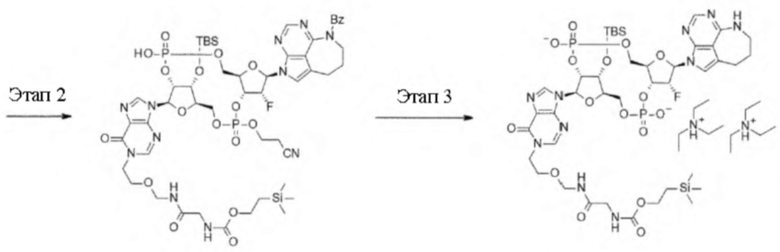
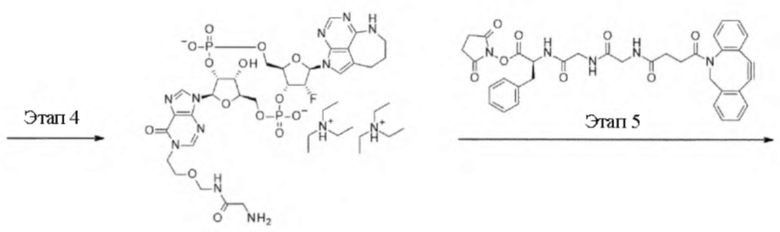
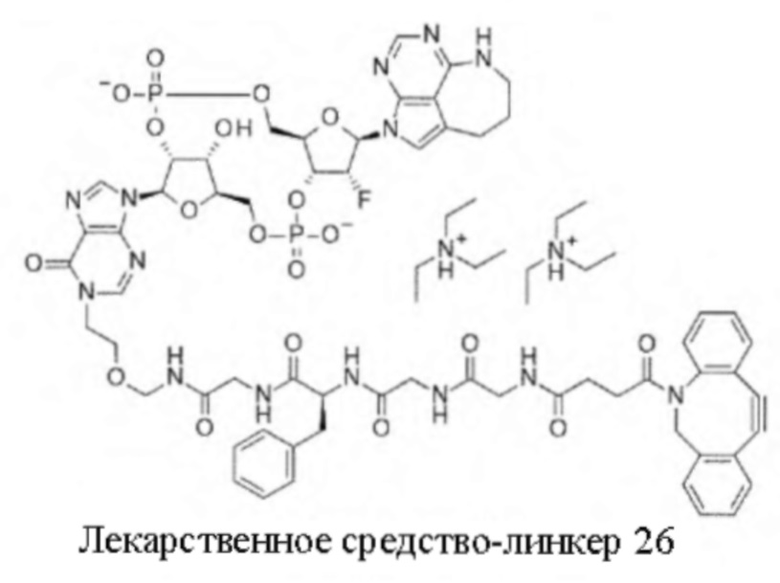
[1235]
(Этап 1)
С применением соединения, полученного на этапе 6 примера 77 (1,26 г), проводили реакцию таким же образом, как на этапе 7 примера 1, с получением раствора в ацетонитриле 3'O-[трет-бутил(дпметил)силил]-2'-O-[гидрокси(оксо)-λ5-фосфанил]-1-(2-{[(N-{[2-(триметилсилил)этокси]карбонил}глицил)амино]метокси}этил)инозина. С применением полученного раствора в ацетонитриле и полученного на этапе 8 примера 44 соединения (1,00 г) проводили реакцию таким же образом, как на этапе 1 примера 63, и полученный в результате этого неочищенный продукт непосредственно использовали для следующей реакции.
[1236]
(Этап 2)
2-(триметилсилил)этил-(2-{[(2-{9-[(5R,7R,8R,12aR,14R,15R,15aR,16R)-14-(6-бензоил-6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)-16-{[трет-бутил(диметил)силил]окси}-10-(2-цианоэтокси)-15-фтор-2-гидрокси-2,10-диоксооктагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-7-ил]-6-оксо-6,9-дигидро-1Н-пурин-1-ил}этокси)метил]амино}-2-оксоэтил)карбамат
С применением неочищенного продукта, полученного на этапе 1 выше, проводили реакцию таким же образом, как на этапе 2 примера 62, с получением указанного в заголовке соединения (678 мг: с примесями).
МС (ИЭР) m/z: 1246(М+Н)+.
[1237]
(Этап 3)
2-(триметилсилил)этил-(2-{[(2-{9-[(5R,7R,8R,12aR,14R,15R,15aR,16R)-14-(6-бензоил-6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)-16-{[трет-бутил(диметил)силил]окси}-10-(2-цианоэтокси)-15-фтор-2-гидрокси-2,10-диоксооктагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-7-ил]-6-оксо-6,9-дигидро-1Н-пурин-1-ил}этокси)метил]амино}-2-оксоэтил)карбамат
С применением соединения, полученного на этапе 2 выше (678 мг), проводили реакцию таким же образом, как на этапе 10 примера 1, и продукт затем очищали с помощью колоночной хроматографии на силикагеле С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил] и препаративной ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/раствор ацетонитрила-метанола (1:1), раствор ацетонитрила-метанола (1:1): 25%-90% (0 мин - 30 мин)] с получением указанного в заголовке соединения (99,6 мг: с примесями).
MC(H3P)m/z: 1089(М+Н)+.
[1238]
(Этап 4)
Бис(N,N-диэтилэтанаминия) (5R,7R,8R,12aR,14R,15R,16R)-15-фтор-7-(1-{2-[(глициламино)метокси]этил}-6-оксо-1,6-дигидро-9Н-пурин-9-ил)-16-гидрокси-2,10-диоксо-14-(6,7,8,9-тетрагидро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1] [1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-2,10-бис(олат)
С применением соединения, полученного на этапе 3 выше (99,6 мг), проводили реакцию таким же образом, как на этапе 9-1 примера 11, и продукт затем очищали с помощью колоночной хроматографии на силикагеле С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил] и препаративной ВЭЖХ [10 мМ водный раствор ацетата триэтиламмония/раствор ацетонитрила-метанола (1:1), раствор ацетонитрила-метанола (1:1): 10%-60% (0 мин - 30 мин)] с получением указанного в заголовке соединения (60,8 мг: с примесями).
МС (ИЭР) m/z: 831(М+Н)+.
[1239]
(Этап 5)
Бис(N,N-диэтилэтанаминия) N-[4-(11,12-дидегидродибензо[b,f]азоцин-5(6Н)-ил)-4-оксобутаноил]глицилглицил-L-фенилаланил-М-[(2-{9-[(5R,7R,8R,12aR,14R,15R,15aR,16R)-15-фтор-16-гидрокси-2,10-диоксид-2,10-диоксо-14-(6,7,8,9-тетрагвдро-2Н-2,3,5,6-тетраазабензо[cd]азулен-2-ил)октагидро-2Н,10Н,12Н-5,8-метано-2λ5,10λ5-фуро[3,2-1][1,3,6,9,11,2,10]пентаоксадифосфациклотетрадецин-7-ил]-6-оксо-6,9-дигидро-1Н-пурин-1-ил}этокси)метил]глицинамид
С применением соединения, полученного на этапе 4 выше (60,8 мг), и полученного на этапе 11 примера 22 соединения (21,3 мг) проводили реакцию таким же образом, как на этапе 9-1 примера 22, и очистку проводили при следующих [Условиях очистки] с получением указанного в заголовке соединения (30,5 мг).
[Условия очистки] Хроматография на колонке с силикагелем С18 [10 мМ водный раствор ацетата триэтиламмония/ацетонитрил] и препаративная ВЭЖХ [100 мМ гексафтор-2-пропанол, 8 мМ водный раствор триэтиламина/ацетонитрил, ацетонитрил: 10%-45% (0 мин - 40 мин)].
МС (ИЭР) m/z: 1379(М+Н)+.
1H-ЯМР (CD3OD) δ: 8,56 (1H, с), 8,16-8,10 (1H, м), 8,05 (1H, с), 7,63-7,49 (2Н, м), 7,45-7,35 (3Н, м), 7,33-7,12 (9Н, м), 6,45 (1H, д, J=18,3 Гц), 6,27 (1H, д, J=9,8 Гц), 5,55-4,99 (4Н, м), 4,65-4,42 (5Н, м), 4,38-4,01 (8Н, м), 3,89-3,60 (9Н, м), 3,52-3,42 (2Н, м), 3,18 (12Н, к, J=7,3 Гц), 3,00-2,91 (1H, м), 2,87-2,60 (3Н, м), 2,38-2,19 (2Н, м), 2,04-1,81 (3Н, м), 1,28 (18Н, т, J=7,3 Гц).
[1240]
Пример 123. Синтез конъюгата35 антитела с лекарственным средством (синтез конъюгата 11 антитела 2 против HER2-ЦДН)
Фосфатно-солевой буферный раствор ремоделированного гликаном антитела 4 (рН 6,0) (10,87 мг/мл, 1,00 мл) разбавляли пропиленгликолем (0,500 мл). В данный раствор добавляли смесь раствора в диметилсульфоксиде лекарственного средства-линкера 24а (10 мМ, 0,135 мл, 18 эквивалентов на молекулу антитела) и пропиленгликоля (0,365 мл) и проводили реакцию полученной смеси во вращающемся устройстве для пробирок (MTR-103, AS ONE Corporation) при комнатной температуре в течение 2 суток. Реакционную смесь очищали в соответствии со способом, описанным в Общей процедуре D, с получением раствора конъюгата нацеленного антитела с лекарственным средством в ABS (7,0 мл).
Анализ проводили в соответствии со способами, описанными в Общих процедурах Е и F, с получением следующих результатов.
Концентрация антитела: 1,08 мг/мл.
Выход антитела: 7,54 мг (69%).
Среднее количество конъюгированных молекул лекарственного средства: 3,8.
[1241]
Пример 124. Синтез конъюгата 36 антитела с лекарственным средством (синтез конъюгата 12 антитела 2 против HER2-ЦДН).
Фосфатно-солевой буферный раствор ремоделированного гликаном антитела 4 (рН 6,0) (10,87 мг/мл, 1,00 мл) разбавляли пропиленгликолем (0,500 мл). В данный раствор добавляли смесь раствора в диметилсульфоксиде лекарственного средства-линкера 24b (10 мМ, 0,135 мл, 18 эквивалентов на молекулу антитела) и пропиленгликоля (0,365 мл) и проводили реакцию полученной смеси во вращающемся устройстве для пробирок (MTR-103, AS ONE Corporation) при комнатной температуре в течение 2 суток. Реакционную смесь очищали в соответствии со способом, описанным в Общей процедуре D, с получением раствора конъюгата нацеленного антитела с лекарственным средством в ABS (7,0 мл).
Анализ проводили в соответствии со способами, описанными в Общих процедурах Е и F, с получением следующих результатов.
Концентрация антитела: 1,12 мг/мл.
Выход антитела: 7,85 мг (72%).
Среднее количество конъюгированных молекул лекарственного средства: 3,8.
[1242]
Пример 125. Синтез конъюгата 37 антитела с лекарственным средством (синтез конъюгата 13 антитела 2 против HER2-ЦДН).
Фосфатно-солевой буферный раствор ремоделированного гликаном антитела 4 (рН 6,0) (10,87 мг/мл, 2,00 мл) разбавляли пропиленгликолем (1,00 мл). В данный раствор добавляли смесь раствора в диметилсульфоксиде лекарственного средства-линкера 26 (10 мМ, 0,270 мл, 18 эквивалентов на молекулу антитела) и пропиленгликоля (0,730 мл) и проводили реакцию полученной смеси во вращающемся устройстве для пробирок (MTR-103, AS ONE Corporation) при комнатной температуре в течение 2 суток. Реакционную смесь очищали в соответствии со способом, описанным в Общей процедуре D, с получением раствора конъюгата нацеленного антитела с лекарственным средством в ABS (14,0 мл).
Анализ проводили в соответствии со способами, описанными в Общих процедурах Е и F, с получением следующих результатов.
Концентрация антитела: 1,05 мг/мл.
Выход антитела: 14,71 мг (68%).
Среднее количество конъюгированных молекул лекарственного средства: 3,8.
[1243]
Пример 126. Синтез конъюгата 38 антитела с лекарственным средством (синтез конъюгата 14 антитела 2 против HER2-ЦДН).
Фосфатно-солевой буферный раствор ремоделированного гликаном антитела 4 (рН 6,0) (10,89 мг/мл, 3,00 мл) разбавляли пропиленгликолем (1,50 мл). В данный раствор добавляли смесь раствора в диметилсульфоксиде лекарственного средства-линкера 25а (10 мМ, 0,540 мл, 24 эквивалента на молекулу антитела) и пропиленгликоля (0,960 мл) и проводили реакцию полученной смеси во вращающемся устройстве для пробирок (MTR-103, AS ONE Corporation) при комнатной температуре в течение 3 суток. Реакционную смесь очищали в соответствии со способом, описанным в Общей процедуре D, с получением раствора конъюгата нацеленного антитела с лекарственным средством в ABS (19 мл).
Анализ проводили в соответствии со способами, описанными в Общих процедурах Е и G, с получением следующих результатов.
Концентрация антитела: 1,38 мг/мл.
Выход антитела: 26,28 мг (80%).
Среднее количество конъюгированных молекул лекарственного средства: 3,7.
[1244]
Пример 127. Синтез конъюгата 39 антитела с лекарственным средством (синтез конъюгата 15 антитела 2 против HER2-ЦДН).
Фосфатно-солевой буферный раствор ремоделированного гликаном антитела 4 (рН 6,0) (10,89 мг/мл, 0,500 мл) разбавляли пропиленгликолем (0,250 мл). В данный раствор добавляли смесь раствора в диметилсульфоксиде лекарственного средства-линкера 25b (10 мМ, 0,090 мл, 24 эквивалента на молекулу антитела) и пропиленгликоля (0,160 мл) и проводили реакцию полученной смеси во вращающемся устройстве для пробирок (MTR-103, AS ONE Corporation) при комнатной температуре в течение 2 суток. Реакционную смесь очищали в соответствии со способом, описанным в Общей процедуре D, с получением раствора конъюгата нацеленного антитела с лекарственным средством в ABS (3,5 мл).
Анализ проводили в соответствии со способами, описанными в Общих процедурах Е и G, с получением следующих результатов.
Концентрация антитела: 0,93 мг/мл.
Выход антитела: 3,25 мг (60%).
Среднее количество конъюгированных молекул лекарственного средства: 3,5.
[1245]
(Ссылочный пример 1: получение антитела против HER2).
В данной заявке «трастузумаб», который также называют герцептином (R), huMAb4D5-8 или rhuMAb4D5-8, представляет собой гуманизированное антитело IgG1, содержащее легкую цепь, состоящую из последовательности аминокислот, представленной в SEQ ID NO: 1, и тяжелую цепь, состоящую из последовательности аминокислот, представленной в SEQ ID NO: 2. На US 5821337 ссылались ради последовательности аминокислот.На фигуре 4 представлены последовательности аминокислот легкой цепи (SEQ ID NO: 1) и тяжелой цепи (SEQ ID NO: 2) трастузумаба.
[1246]
На основе антитела против HER2, используемого в данной заявке, антитело IgG1 из модифицированного в константной области трастузумаба (здесь и далее также называют модифицированным антителом против HER2) было разработано и произведено путем осуществления мутации лейцина (L) на аланин (А) в положениях 234 и 235, соответствующих нумерации EU-индекс, в последовательности аминокислот тяжелой цепи трастузумаба (в данной заявке также называют мутацией LALA). На фигуре 5 представлены последовательности аминокислот легкой цепи (SEQ ID NO: 1) и тяжелой цепи (SEQ ID NO: 3) модифицированного антитела против HER2.
[1247]
(Ссылочный пример 2: получение антитела против LPS).
Антитело против LPS получали, руководствуясь WO 2015/046505. Изотип антитела против LPS, используемый в Примерах, представляет собой IgG1, и антитело против LPS содержит мутацию LALA (здесь и далее также называют модифицированным антителом против LPS). В SEQ ID NO: 26 и SEQ ID NO: 27, соответственно, представлены последовательности аминокислот легкой цепи и тяжелой цепи модифицированного антитела против LPS, используемого в Примерах.
[1248]
(Ссылочный пример 3. Синтез ML-RR-CDA⋅2Na+)
ML-RR-CDA⋅2Na+, используемое в данной заявке в качестве эталонного соединения, синтезировали в соответствии со способом, описанным в Патентной публикации 3 (WO 2014/189805).
[1249]
(Ссылочный пример 4. Синтез 2',3'-цГАМФ)
С применением cGAS, 2',3'-цГАМФ, используемое в данной заявке в качестве эталонного соединения, ферментативно синтезировали из АТФ и ГТФ. Получение cGAS и ферментативную реакцию осуществляли, применяя способ, описанный в публикации (Immunity, 2013, 39, 1019-1031, Cell Rep.2014, 6, 421-430), с подходящей модификацией. Очистку проводили с помощью колоночной хроматографии, применяя слабоосновную анионообменную смолу (DIAION WA10) и синтетический адсорбент (SEPABEADS SP207SS).
[1250]
(Ссылочный пример 5: получение антитела 2 против HER2).
«Пертузумаб», который также называют Перьета (R), представляет собой гуманизированное антитело IgG1, содержащее легкую цепь, состоящую из последовательности аминокислот, представленной в SEQ ID NO: 28, и тяжелую цепь, состоящую из последовательности аминокислот, представленной в SEQ ID NO: 29. На WO 2004/008099 ссылались ради последовательности аминокислот. В данной заявке пертузумаб также называют антителом 2 против HER2. На фигуре 17 представлены последовательности аминокислот легкой цепи (SEQ ID NO: 28) и тяжелой цепи (SEQ ID NO: 29) пертузумаба.
[1251]
Как описано в данной заявке, было разработано и получено антитело 2 против HER2, содержащее не только мутацию LALA, но также константную область аллотипа Glm3 с мутацией лизина (К) на аргинин (R) в положении 214, соответствующем нумерации EU-индекс, в последовательности аминокислот тяжелой цепи (в данной заявке также называют модифицированным антителом 2 против HER2). На фигуре 18 представлены последовательности аминокислот легкой цепи (SEQ ID NO: 28) и тяжелой цепи (SEQ ID NO: 30) модифицированного антитела 2 против HER2, используемого в Примерах.
[1252]
(Ссылочный пример 6: получение антитела против CD33)
Антитело против CD33 получали, руководствуясь WO 2014/057687. Изотип антитела против CD33, используемый в Примерах, представляет собой IgG1, и антитело против CD33 содержит мутацию LALA. На фигуре 19 представлены последовательности аминокислот легкой цепи (SEQ ID NO: 31) и тяжелой цепи (SEQ ID NO: 32) антитела против CD33, используемого в Примерах.
[1253]
(Ссылочный пример 7: получение антитела против EphA2)
Антитело против EphA2 получали, руководствуясь WO 2009/028639. Изотип антитела против EphA2, используемый в Примерах, представляет собой IgG1. На фигуре 20 представлены последовательности аминокислот легкой цепи (SEQ ID NO: 33) и тяжелой цепи (SEQ ID NO: 34) антитела против EphA2, используемого в Примерах.
[1254]
(Ссылочный пример 8: получение антитела против CDH6)
Антитело против CDH6 получали, руководствуясь WO 2018/212136. Изотип антитела против CDH6, используемый в Примерах, представляет собой IgG1, и антитело против CDH6 содержит не только мутацию LALA, но также мутацию пролина (Р) на глицин (G) в положении 329, определенном нумерацией EU-индекс, в последовательности аминокислот тяжелой цепи. На фигуре 21 представлены последовательности аминокислот легкой цепи (SEQ ID NO: 35) и тяжелой цепи (SEQ ID NO: 36) антитела против CDH6, используемого в Примерах.
[1255]
(Пример тестирования 1) Оценка активности агонистов STING с помощью репортерных клеток.
<Анализ репортерного гена>
Активность человеческих агонистов STING оценивали, применяя клетки THPl-Dual (ТМ) (HAQ-мутированные) (InvivoGen, Калифорния, США), с помощью которых можно подтвердить активацию сигнального пути регуляторного фактора интерферона-3 (IRF3), который является нижележащим по отношению к сигнальному пути STING. Активность мышиных агонистов STING оценивали, применяя клетки RAW-Dual (ТМ) (InvivoGen).
[1256]
Анализ проводили, как описано далее. Сначала тестируемое соединение, разбавленное в ФБР, разделяли на аликвоты в прозрачном 96-луночном планшете (Corning Incorporated, Нью-Йорк, США) по 20 мкл/лунку. Затем добавляли репортерные клетки, суспендированные в аналитическом буфере (среде RPMI1640 или среде DMEM, содержащей 10% бычий сывороточный альбумин), по 180 мкл/лунку (1 × 105 клеток/лунку), чтобы начать стимуляцию. После культивирования клеток в окружающей среде при 37°С и 5% СО2 в течение 24 часов, продукт центрифугировали, чтобы собрать супернатант.В белый 384-луночный планшет добавляли 6 мкл собранного супернатанта, и добавляли туда 15 мкл раствора QUANTI-Luc (InvivoGen). После того, как продукт хорошо смешали, измеряли испускание, применяя спектрофотометр для прочтения планшетов (PerkinElmer, Inc., Массачусетс, США). Максимальное подсчитанное количество для клеток, обработанных от 1,37 до 100 мкМ ML-RR-CDA⋅2Na+ (Соединения 21 в WO 2014/189805), приняли за 100% и количество для клеток, обработанных ФБР, приняли за 0%, и концентрацию, необходимую для получения для тестируемого соединения количества 50%, рассчитывали как значение ЕС50 (мкМ), применяя GraphPad Prism (GraphPad Software, Калифорния, США). В таблице 1 показаны результаты тестирования активности человеческих агонистов STING.
[1257]
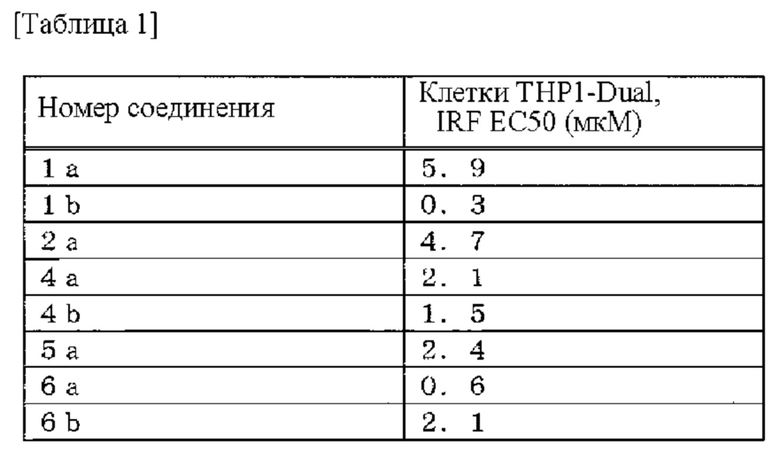
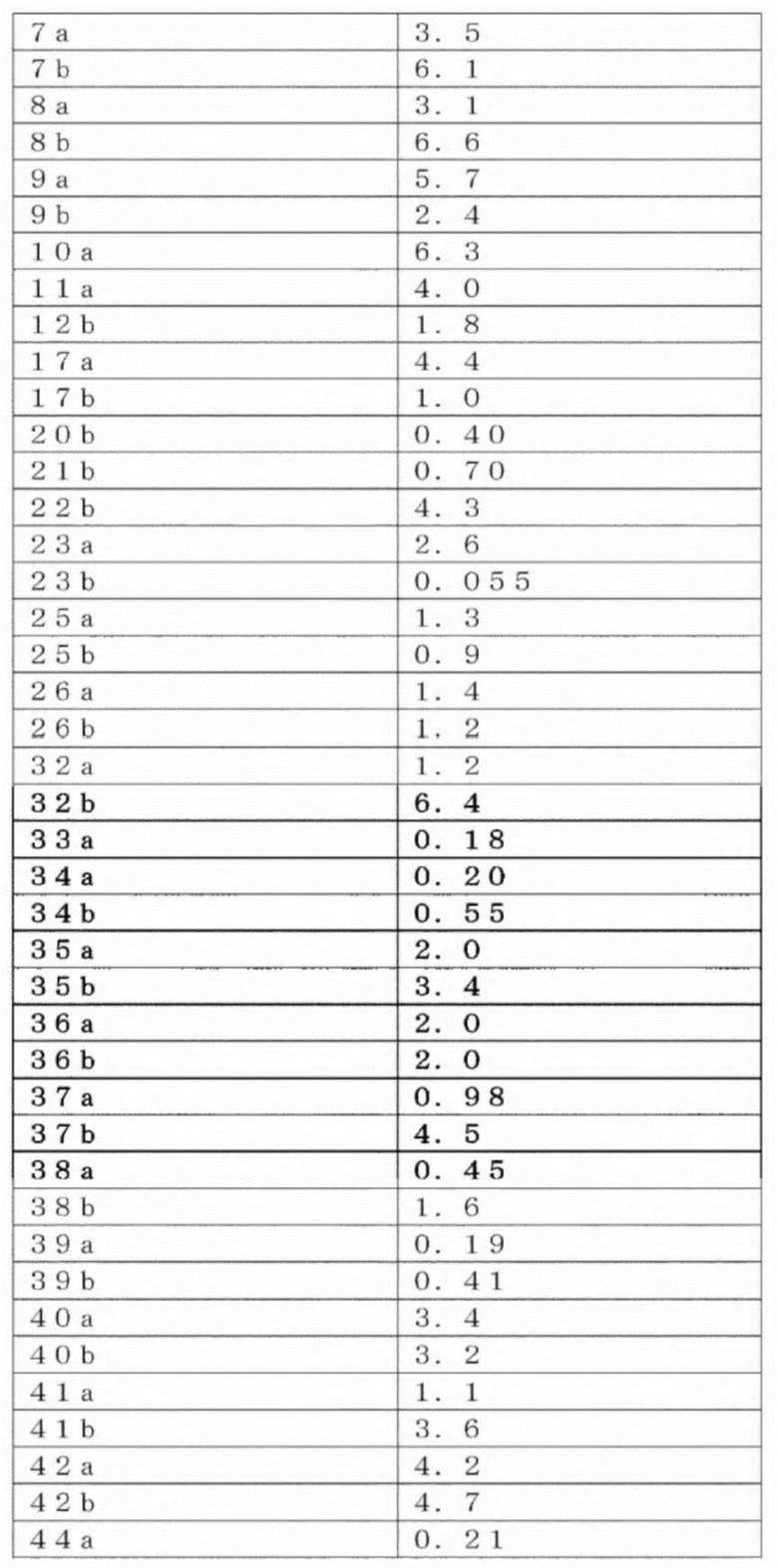
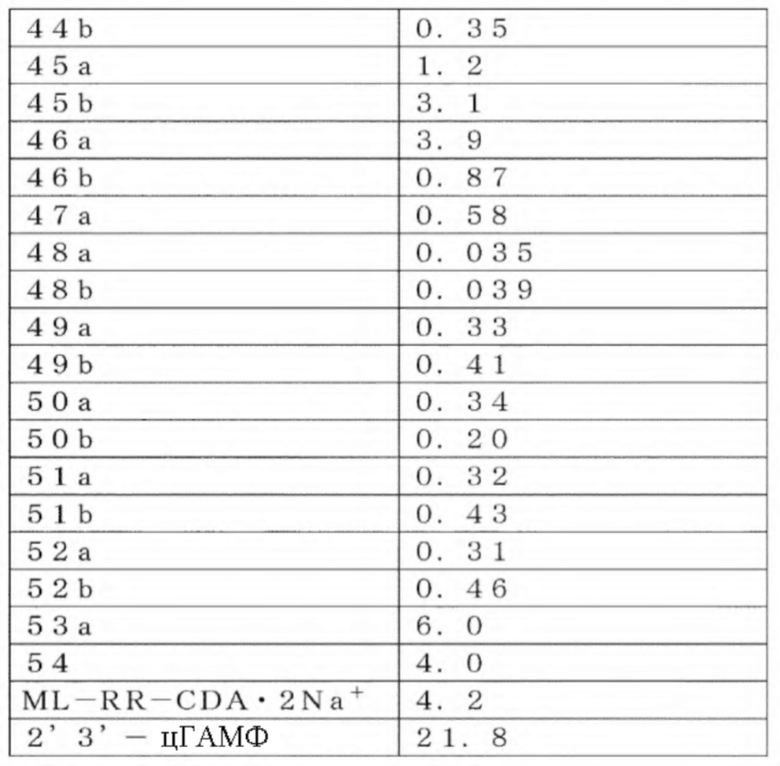
[1258]
Полученные результаты показали, что соединения согласно настоящему изобретению обладают агонистической активностью против STING человека. Кроме того, подтвердили, что соединения согласно настоящему изобретению обладают агонистической активностью, сравнимой с таковой или выше, чем таковая у существующих ЦДН против STING мыши.
[1259]
(Пример тестирования 2) Анализ теплового сдвига белков, проведенный с рекомбинантным белком с доменом, связывающим С-конец STING.
[1260]
(i) Конструирование плазмид экспрессии.
[1261]
<Конструирование плазмиды экспрессии ТМЕМ173 человека>.
Для плазмиды для экспрессии STING человека (здесь и далее периодически называют ТМЕМ173 человека) в клетках млекопитающих приобретали клон кДНК ТМЕМ173 человека (номер доступа NM_198282.3, плазмида экспрессии Н232 (REF)-мутированного STING) (GeneCopoeia, Inc., Мэриленд, США) с мутацией аргинина (R) на гистидин (Н) в положении 232 (здесь и далее называют мутацией Н232 или мутацией REF). В SEQ ID NO: 4 и SEQ ID NO: 5, соответственно, показаны последовательность аминокислот H232(REF)-мутированного STING человека и его последовательность нуклеотидов. Кроме того, плазмиду экспрессии STING дикого типа и плазмиду экспрессии мутированного STING получали посредством сайт-специфического мутагенеза на основе способа обратной ПЦР, используя в качестве матрицы плазмиду экспрессии Н232-мутированного STING. В частности, ПЦР сначала проводили, используя два праймера (5'- (WT) fwd) (SEQ ID NO: 12) и 5'-
(WT) fwd) (SEQ ID NO: 12) и 5'- (WT) rev)) (SEQ ID NO: 13) и набор для мутагенеза KOD-Plus (SMK-101) (TOYOBO CO., LTD.), и подтверждали успешное конструирование целевой плазмиды экспрессии STING дикого типа (R232) посредством секвенирования ДНК. В SEQ ID NO: 6 и SEQ ID NO: 7, соответственно, показана последовательность аминокислот STING дикого типа человека и его последовательность нуклеотидов.
(WT) rev)) (SEQ ID NO: 13) и набор для мутагенеза KOD-Plus (SMK-101) (TOYOBO CO., LTD.), и подтверждали успешное конструирование целевой плазмиды экспрессии STING дикого типа (R232) посредством секвенирования ДНК. В SEQ ID NO: 6 и SEQ ID NO: 7, соответственно, показана последовательность аминокислот STING дикого типа человека и его последовательность нуклеотидов.
[1262]
Далее получали HAQ (R71H, G230A и R293Q)- мутированную форму таким же образом, как для плазмиды экспрессии STING дикого типа. В частности, проводили ПЦР, используя два праймера  (H232R/G230A fwd) (SEQ ID NO: 14) и
(H232R/G230A fwd) (SEQ ID NO: 14) и  (H232R/G230Arev) (SEQ ID NO: 15)) и набор для мутагенеза с плазмидой экспрессии Н232-мутированного STING в качестве матрицы. Плазмиду для G230A-мутированного STING получали путем введения мутации одновременно в два положения. Кроме того, проводили ПЦР, используя два праймера
(H232R/G230Arev) (SEQ ID NO: 15)) и набор для мутагенеза с плазмидой экспрессии Н232-мутированного STING в качестве матрицы. Плазмиду для G230A-мутированного STING получали путем введения мутации одновременно в два положения. Кроме того, проводили ПЦР, используя два праймера 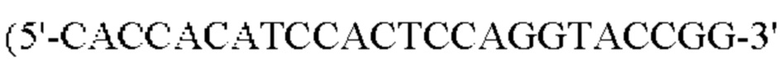 (R71H fwd) (SEQ ID NO: 16) и
(R71H fwd) (SEQ ID NO: 16) и 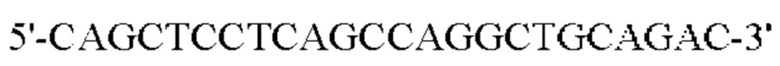 (R71H rev) (SEQ ID NO: 17)) и набор для мутагенеза с плазмидой экспрессии G230A-мутированного STING в качестве матрицы с получением плазмиды экспрессии R71H/G230A-мутированного STING.
(R71H rev) (SEQ ID NO: 17)) и набор для мутагенеза с плазмидой экспрессии G230A-мутированного STING в качестве матрицы с получением плазмиды экспрессии R71H/G230A-мутированного STING.
[1263]
Затем проводили ПЦР, используя два праймера (5'-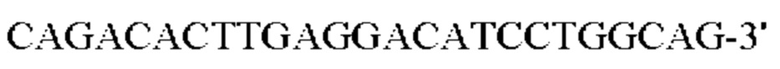 (R293Q fwd) (SEQ ID NO: 18) и 5'-
(R293Q fwd) (SEQ ID NO: 18) и 5'-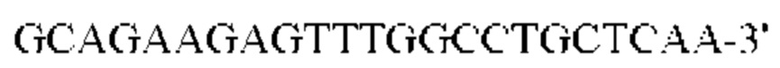 (R293Q rev) (SEQ ID NO: 19)) и набор для мутагенеза с плазмидой экспрессии R71H/G230A-мутированного STING в качестве матрицы с получением плазмиды экспрессии HAQ (R71H/G230A/R293Q)-мутированной формы. В SEQ ID NO: 8 и SEQ ID NO: 9, соответственно, показаны последовательность аминокислот HAQ-мутированного STING человека и его последовательность нуклеотидов.
(R293Q rev) (SEQ ID NO: 19)) и набор для мутагенеза с плазмидой экспрессии R71H/G230A-мутированного STING в качестве матрицы с получением плазмиды экспрессии HAQ (R71H/G230A/R293Q)-мутированной формы. В SEQ ID NO: 8 и SEQ ID NO: 9, соответственно, показаны последовательность аминокислот HAQ-мутированного STING человека и его последовательность нуклеотидов.
[1264]
На фигуре 6 представлены последовательности аминокислот STING дикого типа человека, STING REF-типа и STING HAQ-типа.
[1265]
<Конструирование плазмиды экспрессии рекомбинантного белка с доменом, связывающим С-конец STING, ит.д.>
кДНК белка С-концевого связывающего домена STING человека (АК 139-342) (запись в базе данных UniProt Q86WV6) получали из плазмиды экспрессии каждого полноразмерного клона кДНК ТМЕМ173 человека (дикого типа, Н232-мутированного и HAQ-мутированного) посредством ПЦР с двумя праймерами (5'-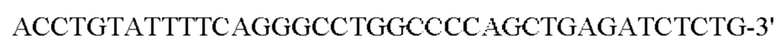 (hST Fw_v2) (SEQ ID NO: 20) и
(hST Fw_v2) (SEQ ID NO: 20) и 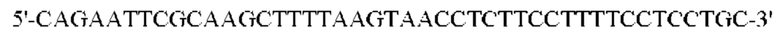 (hST Rv_V3) (SEQ ID NO: 21)). Каждый продукт ПЦР встраивали в pET15b, вектор для экспрессии в Escherichia coli, с применением набора для клонирования In-Fusion HD (Takara Bio Inc.), так что метку 6xHis, состоящую из шести нуклеотидов гистидина, авидиновую метку и сайт расщепления протеазой TEV добавляли на N-конец, чтобы сконструировать плазмиды экспрессии pET15b-HisAviTEV-hSTING (139-342) дикого типа человека, pET15b-HisAviTEV-hSTING (139-342) REF-мутированной формы человека и pET15b-HisAviTEV-hSTING (139-342) AQ-мутированной формы человека.
(hST Rv_V3) (SEQ ID NO: 21)). Каждый продукт ПЦР встраивали в pET15b, вектор для экспрессии в Escherichia coli, с применением набора для клонирования In-Fusion HD (Takara Bio Inc.), так что метку 6xHis, состоящую из шести нуклеотидов гистидина, авидиновую метку и сайт расщепления протеазой TEV добавляли на N-конец, чтобы сконструировать плазмиды экспрессии pET15b-HisAviTEV-hSTING (139-342) дикого типа человека, pET15b-HisAviTEV-hSTING (139-342) REF-мутированной формы человека и pET15b-HisAviTEV-hSTING (139-342) AQ-мутированной формы человека.
[1266]
Для кДНК для экспрессии белка С-концевого связывающего домена STLNG мыши (АК 138-341) (запись в базе данных UniProt Q3TBT3), искусственно синтезировали в Eurofins Genomics К. К. и использовали кДНК, соответствующую аминокислотам в положениях 138 - 341 в последовательности кДНК ТМЕМ173 мыши. В SEQ ID NO: 10 и SEQ ID NO: 11, соответственно, показаны последовательность аминокислот STING мыши и его последовательность нуклеотидов. Синтезированную кДНК встраивали в pET15b, вектор экспрессии в Escherichia coli, с применением набора для клонирования In-Fusion HD, так что метку 6xHis, состоящую из шести нуклеотидов гистидина, авидиновую метку и сайт расщепления протеазой TEV добавляли на N-конец, чтобы сконструировать плазмиду экспрессии формы pET15b-HisAviTEV-mSTING (138-341) дикого типа мыши.
[1267]
Искусственно синтезированную кДНК BirA Е. coli (запись в базе данных UniProt Р06709) встраивали в вектор pCDF Duet-1, чтобы сконструировать плазмиду экспрессии pCDF Duet-1 BirA (1-321).
[1268]
(ii) Способ получения белка С-концевого связывающего домена STING.
Каждой из полученных таким образом плазмид экспрессии pET15b-HisAviTEV-hSTING (139-342) (белок С-концевого связывающего домена STING дикого типа человека, REF-мутированный белок человека и AQ-мутированный белок человека) и плазмидой экспрессии pET15b-HisAviTEV-mSTING (138-341) (белок С-концевого связывающего домена STING дикого типа мыши) трансформировали компетентные клетки Rosetta 2 Е. coli (DE3) (Merck Millipore, Массачусетс, США) одновременно с плазмидой экспрессии pCDF_Duet-l BirA (1-321) для получения экспрессирующих HisAviTEV-STING штаммов. Каждый из данных экспрессионных штаммов добавляли в среду ТВ, содержащую 100 мкг/мл ампициллина, 50 мкг/мл стрептомицина и 30 мкг/мл канамицина, культивировали при 37°С, затем приводили в контакт со 100 мкМ IPTG для индуцируемой экспрессии и дополнительно культивировали при 16°С.
[1269]
Культуральный раствор центрифугировали и полученные в результате этого бактериальные клетки суспендировали в 50 мМ HEPES, рН 8,0, 500 мМ NaCl, 20 мМ имидазол, 1 мМ ДТТ, 5% (масса/объем) глицерин, полная, без ЭДТА, а затем подвергали замораживанию/размораживанию. После добавления лизоцима и ДНКазы I выделяли белки путем разрушения ультразвуком, и осуществляли центрифугирование и собирали полученный в результате этого супернатант.Полученный супернатант очищали, применяя систему для хроматографии АКТА express (GE Healthcare, Иллинойс, США) с колонкой HisTrap FF (GE Healthcare), и элюировали буфером (20 мМ HEPES, рН 7,5, 120 мМ NaCl, 20% глицерин, 0,8 мМ ДТТ) через колонку Superdex 200 16/60 (GE Healthcare). Фракции, содержащие белки с целевой молекулярной массой, собирали с помощью эксклюзионной хроматографии (ЭХ) как белок His-Avi-TEV-hSTING (139-342) человека дикого типа, REF-мутированный белок HisAviTEV-hSTING (139-342) человека, AQ-мутированный белок HisAviTEV-hSTING (139-342) человека и белок His-Avi-TEV-mSTING (138-341) мыши дикого типа. Концентрации белка измеряли, применяя NanoDrop2000 (Thermo Fisher Scientific, Массачусетс, США), и белки криоконсервировали при -80°С до момента применения.
[1270]
В SEQ ID NO: 22, SEQ ID NO: 23, SEQ ID NO: 24 и SEQ ID NO: 25, соответственно, представлены последовательности аминокислот белка HisAviTEV-hSTING (139-342) человека дикого типа, REF-мутированного белка HisAviTEV-hSTING (139-342) человека, AQ-модифицированного белка His AviTEV-hS TING (139-342) человека и белка His-Avi-TEV-mSTING (138-341) мыши дикого типа.
[1271]
(iii) Тестирование связывания STING
Способность связывания каждого соединения с белками С-концевого связывающего домена STING оценивали с помощью анализа теплового сдвига белков, в котором использовали в качестве показателя повышение температуры термической денатурации белка.
[1272]
В частности, смешивали 3 мкл тестируемого соединения (конечная концентрация: 0,5 мМ), 3 мкл оранжевого красителя для белкового геля SYPRO (Thermo Fisher Scientific) (конечная концентрация: 20-кратная) и 6 мкл белка STING с применением аналитического буфера (20 мМ трис-HCl, рН 7,5, 120 мМ NaCl) в 384-луночном планшете для ПЦР в реальном времени, и смешивали с помощью встряхивателя для планшетов. С помощью системы для ПЦР в реальном времени (Thermo Fisher Scientific) температуру повышали от 25°С до 95°С со скоростью 0,03°С/сек и температуру термической денатурации белка измеряли, используя в качестве показателя флуоресценцию, излученную SYPRO Orange. По полученному измерению определяли Tm (среднюю точку конформационного перехода с разворачиванием) (°С) как температуру, при которой уровень повышения интенсивности флуоресценции был максимальным, применяя аналитическое программное обеспечение Protein Thermal Shift (Thermo Fisher Scientific). Значение Tm для лунки без соединения вычитали из значения Tm для каждого тестируемого соединения, чтобы рассчитать сдвиг Tm, вызванный соединением, в виде ΔTm (°С). В таблице 2 показаны результаты тестирования связывания с белками STING.
[1273]
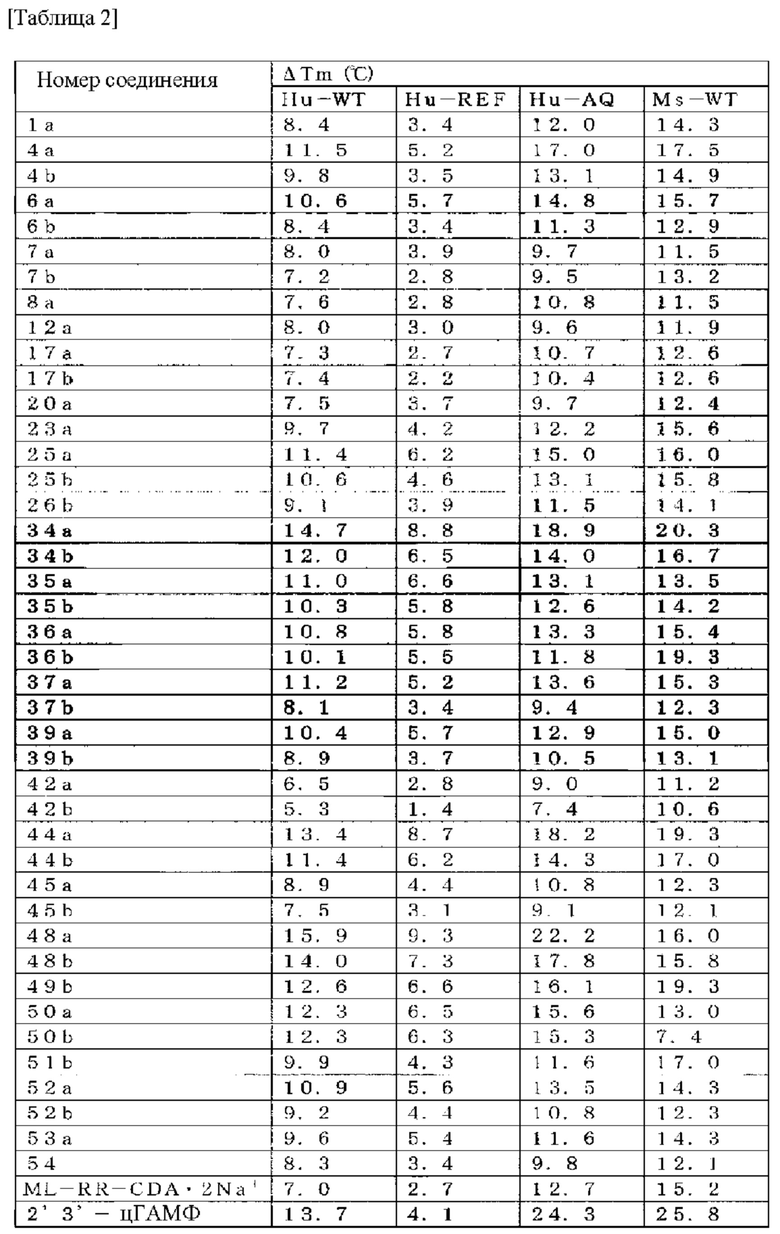
[1274]
Полученные результаты показали, что соединения согласно настоящему изобретению обладают активностью связывания с человеческим STING дикого типа и мутированным STING, и STING мыши дикого типа.
[1275]
(Пример тестирования 3) Тестирование противоопухолевого действия (1)
Мыши: Перед использованием для проведения эксперимента, самкам мышей BALB/c (BALB/cAnNCrlCrlj) (Charles River Laboratories Japan, Inc.) в возрасте 5 недель позволяли привыкнуть к условиям без специфических патогенных факторов (СПФ, SPF) в течение 4 суток или дольше.
[1276]
Измерение, расчетная формула. Во всех исследованиях большую ось и малую ось опухоли измеряли два или три раза в неделю, применяя штангенциркуль с электронным отсчетом (CD15-CX, Mitutoyo Corporation), чтобы рассчитать объем опухоли (мм3). Расчетная формула представлена далее.
Объем опухоли (мм3)=0,5 × большая ось (мм) × [малая ось (мм)]2
Каждое тестируемое соединение разбавляли физиологическим солевым раствором (Otsuka Pharmaceutical Factory, Inc.) для применения. При его введении вводили внутрь опухоли 50 мкл.
[1277]
Использовали клетки линии колоректального рака мыши CT26.WT (CRL2638), приобретенные из Американской коллекции типовых культур. Клетки CT26.WT суспендировали в физиологическом солевом растворе, 1,0 × 106 клеток трансплантировали подкожно в правые подмышечные впадины каждой мыши BALB/c (сутки 0) и на 7 сутки после этого мышей произвольно делили на группы. Каждое тестируемое соединение в дозе 10 мкг вводили внутрь опухоли на 7, 9 и 11 сутки, всего три раза. Группу, которой вводили физиологический солевой раствор, использовали в качестве группы, которой вводили среду. Количество мышей в каждой группе составляло пять или шесть.
Результаты представлены на фигурах 7а и 7b. На каждом графике линия с заштрихованными квадратами соответствует группе, которой вводили среду, линия с незаштрихованными квадратами - группе, которой вводили соединение №6а, линия с незаштрихованными перевернутыми треугольниками - группе, которой вводили соединение №8b, и линия с незаштрихованными кругами - группе, которой вводили соединение №9b. На вертикальной оси представлен объем опухоли (мм3), а на горизонтальной оси представлены сутки после трансплантации опухоли. Рост опухоли прогрессировал в группе, которой вводили среду. В противоположность этому, рост опухоли значительно подавлялся в группах, которым вводили соединение.
[1278]
Полученные результаты подтвердили противоопухолевое действие внутриопухолевого введения производных циклических динуклеотидов.
[1279]
(Пример тестирования 4) Тестирование противоопухолевого действия (2)
Ген HER2 человека внедряли в линию клеток колоректального рака мыши СТ26.WT (CRL2638), приобретенную из Американской коллекции типовых культур, с получением клеток CT26.WT-hHER2. В частности, кДНК амплифицировали, применяя плазмиду, содержащую кДНК HER2 человека (клон ID IOH82145; Thermo Fisher Scientific), и встраивали в ретровирусный вектор pQCXIN (Takara Bio Inc.), применяя набор для клонирования In-Fusion HD (Clontech Laboratories, Inc.). Ретровирусным вектором pQCXIN со встроенным HER2 человека трансфицировали линию клеток EcoPack2-293 (Takara Bio Inc.) с помощью липофектамина 3000 (Thermo Fisher Scientific), собирали супернатант, который содержал вирус, и клетки CT26.WT инфицировали указанным вирусом. Клетки поддерживали в среде с 250 мкг/мл генетицина (Thermo Fisher Scientific).
[1280]
Каждый конъюгат антитело-ЦДН разбавляли ацетатным буфером (10 мМ ацетатный буфер, 5% сорбит, рН 5,5) (NACALAI TESQUE, INC.) для применения. При его введении 200 мкл вводили в хвостовую вену.
[1281]
Клетки CT26.WT-hHER2 суспендировали в физиологическом солевом растворе, 5,0 × 106 клеток трансплантировали подкожно в правые подмышечные впадины каждой мыши BALB/c (сутки 0) и на 7 сутки после этого мышей произвольно делили на группы. Каждое антитело-ЦДН конъюгат в дозе 30 мкг вводили в хвостовую вену на 7 сутки, всего один раз. Группу, которой вводили ацетатный буфер, использовали в качестве группы, которой вводили среду. Количество мышей в каждой группе составляло восемь.
[1282]
Результаты представлены на фигуре 8. На каждом графике линия с заштрихованными квадратами соответствует группе, которой вводили среду, линия с незаштрихованными треугольниками - группе, которой вводили конъюгат (1) антитела против HER2-ЦДН, который был получен путем конъюгирования соединения из примера 8b с модифицированным антителом против HER2, полученным в ссылочном примере 1, и линия с заштрихованными треугольниками - группе, которой вводили конъюгат (1) антитела против LPS-ЦДН, который был получен аналогично путем конъюгирования соединения из примера 8b с модифицированным антителом против LPS, полученным в ссылочном примере 2. На вертикальной оси представлен объем опухоли (мм3), а на горизонтальной оси представлены сутки после трансплантации опухоли. Рост опухоли прогрессировал в группе, которой вводили среду, и группе, которой вводили конъюгат (1) антитела против LPS-ЦДН, который не связывался с HER2. В противоположность этому, рост опухоли значительно подавлялся в группе, которой вводили конъюгат (1) антитела против HER2- ЦДН.
[1283]
Полученные результаты подтверждают зависимое от мишени антитела противоопухолевое действие внутривенного введения конъюгата (1) антитела против HER2- ЦДН.
[1284]
(Пример тестирования 5) Тестирование противоопухолевого действия (3).
Клетки CT26.WT-hHER2 суспендировали в физиологическом солевом растворе, 5,0 × 106 клеток трансплантировали подкожно в правые подмышечные впадины каждой мыши BALB/c (сутки 0), и на 6 сутки после этого мышей произвольно делили на группы. Каждый конъюгат антитело-ЦДН в дозе 30 мкг вводили в хвостовую вену на 6 сутки, всего один раз. Группу, которой вводили ацетатный буфер, использовали в качестве группы, которой вводили среду. Количество мышей в каждой группе составляло шесть или восемь.
[1285]
Результаты представлены на фигуре 9. На диаграмме линия с заштрихованными квадратами соответствует группе, которой вводили среду, линия с незаштрихованными квадратами - группе, которой вводили конъюгат (2) антитела против HER2-ЦДН, и линия с незаштрихованными треугольниками - группе, которой вводили конъюгат (3) антитела против HER2-ЦДН. На вертикальной оси представлен объем опухоли (мм3), а на горизонтальной оси представлены сутки после трансплантации опухоли. Рост опухоли прогрессировал в группе, которой вводили среду. В противоположность этому, рост опухоли значительно подавлялся в группах, которым вводили конъюгаты (2) и (3) антитела против HER2-ЦДН.
[1286]
Полученные результаты подтверждают эффективное противоопухолевое действие конъюгатов антитела против HER2-ЦДН.
[1287]
(Пример тестирования 6) Тестирование противоопухолевого действия (4).
Клетки CT26.WT-hHER2 суспендировали в физиологическом солевом растворе, 5,0 × 106 клеток трансплантировали подкожно в правые подмышечные впадины каждой мыши BALB/c (сутки 0) и на 7 сутки после этого мышей произвольно делили на группы. Конъюгат (19) антитело-ЦДН в дозе 30 мкг вводили в хвостовую вену на 7 сутки, всего один раз. Группу, которой вводили ацетатный буфер, использовали в качестве группы, которой вводили среду. Количество мышей в каждой группе составляло шесть.
Результаты представлены на фигуре 10. На диаграмме линия с заштрихованными квадратами соответствует группе, которой вводили среду, а линия с незаштрихованными треугольниками - группе, которой вводили конъюгат (19) антитела против HER2-ЦДН. в конъюгате (19) антитела против HER2-ЦДН лекарственное средство-линкер конъюгировано с антителом путем конъюгирования через цистеин. На вертикальной оси представлен объем опухоли (мм3), а на горизонтальной оси представлены сутки после трансплантации опухоли. Рост опухоли прогрессировал в группе, которой вводили среду. В противоположность этому, рост опухоли значительно подавлялся в группе, которой вводили конъюгат (19) антитела против HER2-ЦДН.
[1288]
Полученные результаты подтверждают эффективное противоопухолевое действие конъюгата антитела против HER2-ЦДН с конъюгированием через цистеин антитела и лекарственного средства-линкера.
[1289]
(Пример тестирования 7) Тестирование противоопухолевого действия (5).
Клетки CT26.WT-hHER2 суспендировали в физиологическом солевом растворе, 5,0 × 106 клеток трансплантировали подкожно в правые подмышечные впадины каждой мыши BALB/c (сутки 0), и на 6 сутки после этого мышей произвольно делили на группы. Каждый конъюгат антитело-ЦДН в дозе 30 мкг вводили в хвостовую вену на 6 сутки, всего один раз. Группу, которой вводили ацетатный буфер, использовали в качестве группы, которой вводили среду. Количество мышей в каждой группе составляло пять.
[1290]
Результаты представлены на фигуре 11. На диаграмме линия с заштрихованными квадратами соответствует группе, которой вводили среду, линия с незаштрихованными треугольниками - группе, которой вводили конъюгат (9) антитела против HER2- ЦДН, линия с незаштрихованными перевернутыми треугольниками - группе, которой вводили конъюгат (10) антитела против HER2- ЦДН, линия с незаштрихованными ромбами - группе, которой вводили конъюгат (11) антитела против HER2- ЦДН, линия с незаштрихованными кругами - группе, которой вводили конъюгат (12) антитела против HER2- ЦДН, и линия с незаштрихованными квадратами - группе, которой вводили конъюгат (1) антитела против НЕК2-ЦДН. В каждом из конъюгатов (9), (10), (11), (12) и (1) антитела против HER2- ЦДН соединение из примера 8b конъюгировано посредством линкера, где линкеры отличаются друг от друга. На вертикальной оси представлен объем опухоли (мм3), а на горизонтальной оси представлены сутки после трансплантации опухоли. Рост опухоли прогрессировал в группе, которой вводили среду. В противоположность этому, рост опухоли значительно подавлялся в группах, которым вводили конъюгаты (9), (10), (11) и (12) антитела против HER2- ЦДН, как и в случае группы, которой вводили конъюгат (1) антитела против HER2-ЦДН.
[1291]
Полученные результаты подтвердили, что конъюгаты антитела против HER2-ЦДН оказывают противоопухолевое действие даже с различными линкерами.
[1292]
(Пример тестирования 8) Тестирование противоопухолевого действия (6).
Клетки CT26.WT-hHER2 суспендировали в физиологическом солевом растворе, 5,0 × 106 клеток трансплантировали подкожно в правые подмышечные впадины каждой мыши BALB/c (сутки 0) и на 7 сутки после этого мышей произвольно делили на группы. Каждый образец для введения вводили в хвостовую вену на 7 сутки, всего один раз. Группу, которой вводили ацетатный буфер, использовали в качестве группы, которой вводили среду. Количество мышей в каждой группе составляло пять.
[1293]
Результаты представлены в таблице 12. На диаграмме линия с заштрихованными квадратами соответствует группе, которой вводили среду, линия с незаштрихованными треугольниками - группе, которой вводили 60 мкг конъюгата (1) антитела 2 против HER2-ЦДН, линия с незаштрихованными перевернутыми треугольниками - группе, которой вводили 59 мкг антитела 2 против HER2, и линия с незаштрихованными кругами - группе, которой вводили 1,2 мкг соединения №8b. Каждая из доз антитела 2 против HER2 и соединения №8b эквивалентна соответствующему компоненту, составляющему конъюгат (1) антитела 2 против HER2-ЦДН. На вертикальной оси представлен объем опухоли (мм3), а на горизонтальной оси представлены сутки после трансплантации опухоли. Рост опухоли прогрессировал в группе, которой вводили среду. В противоположность этому, рост опухоли значительно подавлялся в группе, которой вводили конъюгат (1) антитела 2 против HER2-ЦДН. Тем не менее, рост опухоли не подавлялся в каждой группе, которой вводили эквивалент антитела 2 против HER2 или соединения №8b.
[1294]
Полученные результаты подтвердили, что конъюгат (1) антитела 2 против HER2-ЦДН оказывает противоопухолевое действие, и что эквивалент антитела 2 против HER2 и эквивалент соединения №8b не оказывают противоопухолевое действие, когда их вводят в хвостовую вену.
[1295]
(Пример тестирования 9) Тестирование противоопухолевого действия (7).
Клетки CT26.WT-hHER2 суспендировали в физиологическом солевом растворе, 5,0 × 106 клеток трансплантировали подкожно в правые подмышечные впадины каждой мыши BALB/c (сутки 0) и на 7 сутки после этого мышей произвольно делили на группы. Каждый конъюгат антитело-ЦДН в дозе 30 мкг вводили в хвостовую вену на 7 сутки, всего один раз. Группу, которой вводили ацетатный буфер, использовали в качестве группы, которой вводили среду. Количество мышей в каждой группе составляло пять или шесть.
[1296]
Результаты представлены на фигурах 13(a)-13(c). На каждом графике линия с заштрихованными квадратами соответствует группе, которой вводили среду, и каждая линия с незаштрихованными символами соответствует группе, в которой оцениваемым субъектам вводили конъюгаты (2), (3), (4), (5), (6), (7) и (8) антитела 2 против HER2-ЦДН. На вертикальной оси представлен объем опухоли (мм3), а на горизонтальной оси представлены сутки после трансплантации опухоли. Рост опухоли прогрессировал в группе, которой вводили среду. В противоположность этому, рост опухоли значительно подавлялся в группах, которым вводили конъюгаты (2), (3), (4), (5), (6), (7) и (8) антитела 2 против HER2-ЦДН.
[1297]
Полученные результаты подтверждают эффективное противоопухолевое действие конъюгатов антитела 2 против НЕК2-ЦДН.
[1298]
(Пример тестирования 10) Тестирование противоопухолевого действия (8).
Клетки CT26.WT-hHER2 суспендировали в физиологическом солевом растворе, 5,0 × 106 клеток трансплантировали подкожно в правые подмышечные впадины каждой мыши BALB/c (сутки 0) и на 7 сутки после этого мышей произвольно делили на группы. Каждый конъюгат антитело-ЦДН в дозе 60 мкг вводили в хвостовую вену на 7 сутки, всего один раз. Группу, которой вводили ацетатный буфер, использовали в качестве группы, которой вводили среду. Количество мышей в каждой группе составляло пять.
[1299]
Результаты представлены на фигуре 14. На диаграмме линия с заштрихованными квадратами соответствует группе, которой вводили среду, линия с незаштрихованными треугольниками - группе, которой вводили конъюгат (9) антитела 2 против НЕК2-ЦДН, и линия с незаштрихованными кругами - группе, которой вводили конъюгат (10) антитела 2 против НЕК2-ЦДН. Конъюгаты (9) и (10) антитела 2 против НЕК2-ЦДН представляют собой конъюгаты антитела-ЦДН, в которых используется ремоделированное гликаном MSG-типа антитело со средним количеством конъюгированных молекул лекарственного средства, приблизительно составляющим 2. На вертикальной оси представлен объем опухоли (мм3), а на горизонтальной оси представлены сутки после трансплантации опухоли. Рост опухоли прогрессировал в группе, которой вводили среду. В противоположность этому, рост опухоли значительно подавлялся в группах, которым вводили конъюгаты (9) и (10) антитела 2 против HER2-ЦДН.
[1300]
Полученные результаты подтверждают эффективное противоопухолевое действие конъюгатов антитела 2 против HER2-ЦДН со средним количеством конъюгированных молекул лекарственного средства, приблизительно составляющим 2.
[1301]
(Пример тестирования 11) Тестирование противоопухолевого действия (9).
Ген EphA2 человека внедряли в линию клеток колоректального рака мыши СТ26.WT (CRL2638), приобретенную из Американской коллекции типовых культур, с получением клеток CT26.WT-hEphA2. В частности, кДНК амплифицировали, применяя pDONR221, содержащий кДНК EphA2 человека (Thermo Fisher Scientific), и встраивали в ретровирусный вектор pLNCX (Takara Bio Inc.), применяя систему для превращения векторов Gateway (Thermo Fisher Scientific). Ретровирусным вектором pLNCX со встроенным EphA2 человека трансфицировали линию клеток EcoPack2-293 (Takara Bio Inc.) с помощью липофектамина 2000 (Thermo Fisher Scientific), собирали супернатант, который содержал вирус, и клетки CT26.WT инфицировали указанным вирусом. Клетки поддерживали в среде с 500 мкг/мл генетицина (Thermo Fisher Scientific).
[1302]
Клетки CT26.WT-hEphA2 суспендировали в фосфатном буфере, 1,9 × 106 клеток трансплантировали подкожно в правые подмышечные впадины каждой мыши BALB/c (сутки 0) и на 7 сутки после этого мышей произвольно делили на группы. Каждый из антитела против EphA2 и конъюгата (1) антитела против EphA2-ЦДН в дозе 60 мкг вводили в хвостовую вену на 7 сутки, всего один раз. Группу, которой вводили ацетатный буфер, использовали в качестве группы, которой вводили среду. Количество мышей в каждой группе составляло восемь.
[1303]
Результаты представлены на фигуре 15. На диаграмме линия с заштрихованными квадратами соответствует группе, которой вводили среду, линия с незаштрихованными кругами - группе, которой вводили антитело против EphA2, и линия с незаштрихованными треугольниками - группе, которой вводили конъюгат (1) антитела против EphA2-ЦДН. На вертикальной оси представлен объем опухоли (мм3), а на горизонтальной оси представлены сутки после трансплантации опухоли. Рост опухоли прогрессировал в группе, которой вводили среду. Рост опухоли не подавлялся в группе, которой вводили антитело против EphA2. В противоположность этому, рост опухоли значительно подавлялся в группе, которой вводили конъюгат (1) антитела против EphA2-ЦДН.
[1304]
Полученные результаты, с применением модели, в которой антитело против EphA2 не оказывает противоопухолевого действия, подтвердили эффективное противоопухолевое действие конъюгата антитела против EphA2-ЦДН.
[1305]
(Пример тестирования 12) Тестирование противоопухолевого действия (10).
Ген CD33 человека внедряли в линию лимфоидных клеток мыши P388D1 (CCL-46), приобретенную из Американской коллекции типовых культур, для получения клеток P388Dl-hCD33. В частности, получали лентивирусный вектор pLVSIN (Takara Bio Inc.) со встроенной в него кДНК CD33 человека, и трансфицировали им линию клеток Lenti-X293T (Takara Bio Inc.), применяя смесь Lentiviral High Titer Packaging Mix (Takara Bio Inc.), собирали супернатант, который содержал вирус, и клетки P388D1 инфицировали указанным вирусом. Клетки поддерживали в среде с 2 мкг/мл пуромицина (Thermo Fisher Scientific).
[1306]
Приобретали самок мышей DBA/2 (DBA/2NCrl) (Charles River Laboratories Japan, Inc.) в возрасте четырех недель и позволяли им привыкнуть к условиям СПФ в течение 5 суток перед использованием для эксперимента.
[1307]
Клетки P388D1-hCD33 суспендировали в фосфатном буфере, 1,0 × 106 клеток трансплантировали подкожно в правые подмышечные впадины каждой мыши DBA/2 (сутки 0) и на 4 сутки после этого мышей произвольно делили на группы. Каждый из антитела против CD33 и конъюгата (1) антитела против CD33-ЦДН в дозе 60 мкг вводили в хвостовую вену на 4 сутки, всего однократно. Группу, которой вводили ацетатный буфер, использовали в качестве группы, которой вводили среду. Количество мышей в каждой группе составляло 10.
[1308]
Результаты представлены на фигуре 16. На диаграмме линия с заштрихованными квадратами соответствует группе, которой вводили среду, линия с незаштрихованными кругами - группе, которой вводили антитела против CD33, и линия с незаштрихованными треугольниками - группе, которой вводили конъюгат (1) антитела против CD33-ЦДН. На вертикальной оси представлен объем опухоли (мм3), а на горизонтальной оси представлены сутки после трансплантации опухоли. Рост опухоли прогрессировал в группе, которой вводили среду. Рост опухоли не подавлялся в группе, которой вводили антитело против CD33. В противоположность этому, рост опухоли значительно подавлялся в группе, которой вводили конъюгат (1) антитела против CD33-ЦДН.
[1309]
Полученные результаты, с применением модели, в которой антитело против CD33 не оказывает противоопухолевого действия, подтвердили эффективное противоопухолевое действие конъюгата антитела против CD33-ЦДН.
[1310]
(Пример тестирования 13) Тестирование противоопухолевого действия (11).
Клетки CT26.WT-hHER2 суспендировали в физиологическом солевом растворе, 5,0 х 106 клеток трансплантировали подкожно в правые подмышечные впадины каждой мыши BALB/c (сутки 0) и на 7 сутки после этого мышей произвольно делили на группы. Каждый конъюгат антитело-ЦДН в дозе 60 мкг вводили в хвостовую вену на 7 сутки, всего один раз. Группу, которой вводили ацетатный буфер, использовали в качестве группы, которой вводили среду. Количество мышей в каждой группе составляло шесть.
[1311]
Результаты представлены на фигуре 22. На диаграмме линия с заштрихованными квадратами соответствует группе, которой вводили среду, линия с незаштрихованными треугольниками - группе, которой вводили конъюгат (11) антитела 2 против HER2-ЦДН, и линия с незаштрихованными кругами - группе, которой вводили конъюгат (12) антитела 2 против HER2-ЦДН. Каждое соединение №52а и соединение №52b, конъюгированное в конъюгатах (11) и (12) антитела 2 против HER/2-ЦДН, соответственно, представляет собой ЦДН, содержащий фосфатную группу. На вертикальной оси представлен объем опухоли (мм3), а на горизонтальной оси представлены сутки после трансплантации опухоли. Рост опухоли прогрессировал в группе, которой вводили среду. В противоположность этому, рост опухоли значительно подавлялся в группах, которым вводили конъюгаты (11) и (12) антитела 2 против HER2-ЦДН.
[1312]
Полученные результаты подтвердили эффективное противоопухолевое действие конъюгатов (11) и (12) антитела 2 против HER2-ЦДН, в которых с ЦДН конъюгирована фосфатная группа.
Промышленная применимость.
[1313]
В соответствии с настоящим изобретением предложены новые производные ЦДН, которые обладают эффективной активностью агонистов STING и проявляют эффективное противоопухолевое действие. Кроме того, в соответствии с настоящим изобретением предложены конъюгаты антитела с лекарственным средством, содержащие новые производные ЦДН. Они полезны в качестве терапевтических агентов для лечения заболеваний, связанных с активностью агонистов STING (например, рака).
Текст перечня последовательностей в нестандартном формате.
[1314]
SEQ ID NO: 1: Последовательность аминокислот легкой цепи трастузумаба
SEQ ID NO: 2: Последовательность аминокислот тяжелой цепи трастузумаба
SEQ ID NO: 3: Последовательность аминокислот тяжелой цепи модифицированного антитела против HER2
SEQ ID NO: 4: Последовательность аминокислот Н232(REF)-мутированного STING человека
SEQ ID NO: 5: Последовательность нуклеиновой кислоты Н232(REF)-мутированного STING человека
SEQ ID NO: 6: Последовательность аминокислот STING дикого типа человека
SEQ ID NO: 7: Последовательность нуклеиновой кислоты STING дикого типа человека
SEQ ID NO: 8: Последовательность аминокислот HAQ-мутированного STING человека
SEQ ID NO: 9: Последовательность нуклеиновой кислоты HAQ-мутированного STING человека
SEQ ID NO: 10: Последовательность аминокислот STING мыши
SEQ ID NO: 11: Последовательность нуклеиновой кислоты STING мыши
SEQ ID NO: 12-21: Последовательности праймеров
SEQ ID NO: 22: Последовательность аминокислот белка HisAviTEV-hSTING (139-342) человека дикого типа
SEQ ID NO: 23: Последовательность аминокислот REF-мутированного белка HisAviTEV-hSTING (139-342) человека
SEQ ID NO: 24: Последовательность аминокислот AQ-мутированного белка HisAviTEV-hSTING (139-342) человека
SEQ ID NO: 25: Последовательность аминокислот белка His-Avi-TEV-mSTING (138-341) мыши дикого типа
SEQ ID NO: 26: Последовательность аминокислот легкой цепи модифицированного антитела против LPS
SEQ ID NO: 27: Последовательность аминокислот тяжелой цепи модифицированного антитела против LPS
SEQ ID NO: 28: Последовательность аминокислот легкой цепи пертузумаба
SEQ ID NO: 29: Последовательность аминокислот тяжелой цепи пертузумаба
SEQ ID NO: 30: Последовательность аминокислот тяжелой цепи модифицированного антитела 2 против HER2
SEQ ID NO: 31: Последовательность аминокислот легкой цепи антитела против CD33 SEQ ID NO: 32: Последовательность аминокислот тяжелой цепи антитела против CD33 SEQ ID NO: 33: Последовательность аминокислот легкой цепи антитела против EphA2 SEQ ID NO: 34: Последовательность аминокислот тяжелой цепи антитела против EphA2 SEQ ID NO: 35: Последовательность аминокислот легкой цепи антитела против CDH6 SEQ ID NO: 36: Последовательность аминокислот тяжелой цепи антитела против CDH6
--->
ПЕРЕЧЕНЬ ПОСЛЕДОВАТЕЛЬНОСТЕЙ
<110> DAIICHI SANKYO COMPANY, LIMITED
<120> НОВОЕ ПРОИЗВОДНОЕ ЦИКЛИЧЕСКОГО ДИНУКЛЕОТИДА И ЕГО КОНЪЮГАТ
АНТИТЕЛО-ЛЕКАРСТВЕННОЕ СРЕДСТВО
<130> SAP-855-PCT
<141> 2019-09-06
<150> JP 2018167369
<151> 2018-09-06
<160> 36
<170> PatentIn версии 3.5
<210> 1
<211> 214
<212> ПРТ
<213> Искусственная последовательность
<220>
<223> Легкая цепь трастузумаба
<400> 1
Asp Ile Gln Met Thr Gln Ser Pro Ser Ser Leu Ser Ala Ser Val Gly
1 5 10 15
Asp Arg Val Thr Ile Thr Cys Arg Ala Ser Gln Asp Val Asn Thr Ala
20 25 30
Val Ala Trp Tyr Gln Gln Lys Pro Gly Lys Ala Pro Lys Leu Leu Ile
35 40 45
Tyr Ser Ala Ser Phe Leu Tyr Ser Gly Val Pro Ser Arg Phe Ser Gly
50 55 60
Ser Arg Ser Gly Thr Asp Phe Thr Leu Thr Ile Ser Ser Leu Gln Pro
65 70 75 80
Glu Asp Phe Ala Thr Tyr Tyr Cys Gln Gln His Tyr Thr Thr Pro Pro
85 90 95
Thr Phe Gly Gln Gly Thr Lys Val Glu Ile Lys Arg Thr Val Ala Ala
100 105 110
Pro Ser Val Phe Ile Phe Pro Pro Ser Asp Glu Gln Leu Lys Ser Gly
115 120 125
Thr Ala Ser Val Val Cys Leu Leu Asn Asn Phe Tyr Pro Arg Glu Ala
130 135 140
Lys Val Gln Trp Lys Val Asp Asn Ala Leu Gln Ser Gly Asn Ser Gln
145 150 155 160
Glu Ser Val Thr Glu Gln Asp Ser Lys Asp Ser Thr Tyr Ser Leu Ser
165 170 175
Ser Thr Leu Thr Leu Ser Lys Ala Asp Tyr Glu Lys His Lys Val Tyr
180 185 190
Ala Cys Glu Val Thr His Gln Gly Leu Ser Ser Pro Val Thr Lys Ser
195 200 205
Phe Asn Arg Gly Glu Cys
210
<210> 2
<211> 450
<212> ПРТ
<213> Искусственная последовательность
<220>
<223> Тяжелая цепь трастузумаба
<400> 2
Glu Val Gln Leu Val Glu Ser Gly Gly Gly Leu Val Gln Pro Gly Gly
1 5 10 15
Ser Leu Arg Leu Ser Cys Ala Ala Ser Gly Phe Asn Ile Lys Asp Thr
20 25 30
Tyr Ile His Trp Val Arg Gln Ala Pro Gly Lys Gly Leu Glu Trp Val
35 40 45
Ala Arg Ile Tyr Pro Thr Asn Gly Tyr Thr Arg Tyr Ala Asp Ser Val
50 55 60
Lys Gly Arg Phe Thr Ile Ser Ala Asp Thr Ser Lys Asn Thr Ala Tyr
65 70 75 80
Leu Gln Met Asn Ser Leu Arg Ala Glu Asp Thr Ala Val Tyr Tyr Cys
85 90 95
Ser Arg Trp Gly Gly Asp Gly Phe Tyr Ala Met Asp Tyr Trp Gly Gln
100 105 110
Gly Thr Leu Val Thr Val Ser Ser Ala Ser Thr Lys Gly Pro Ser Val
115 120 125
Phe Pro Leu Ala Pro Ser Ser Lys Ser Thr Ser Gly Gly Thr Ala Ala
130 135 140
Leu Gly Cys Leu Val Lys Asp Tyr Phe Pro Glu Pro Val Thr Val Ser
145 150 155 160
Trp Asn Ser Gly Ala Leu Thr Ser Gly Val His Thr Phe Pro Ala Val
165 170 175
Leu Gln Ser Ser Gly Leu Tyr Ser Leu Ser Ser Val Val Thr Val Pro
180 185 190
Ser Ser Ser Leu Gly Thr Gln Thr Tyr Ile Cys Asn Val Asn His Lys
195 200 205
Pro Ser Asn Thr Lys Val Asp Lys Lys Val Glu Pro Lys Ser Cys Asp
210 215 220
Lys Thr His Thr Cys Pro Pro Cys Pro Ala Pro Glu Leu Leu Gly Gly
225 230 235 240
Pro Ser Val Phe Leu Phe Pro Pro Lys Pro Lys Asp Thr Leu Met Ile
245 250 255
Ser Arg Thr Pro Glu Val Thr Cys Val Val Val Asp Val Ser His Glu
260 265 270
Asp Pro Glu Val Lys Phe Asn Trp Tyr Val Asp Gly Val Glu Val His
275 280 285
Asn Ala Lys Thr Lys Pro Arg Glu Glu Gln Tyr Asn Ser Thr Tyr Arg
290 295 300
Val Val Ser Val Leu Thr Val Leu His Gln Asp Trp Leu Asn Gly Lys
305 310 315 320
Glu Tyr Lys Cys Lys Val Ser Asn Lys Ala Leu Pro Ala Pro Ile Glu
325 330 335
Lys Thr Ile Ser Lys Ala Lys Gly Gln Pro Arg Glu Pro Gln Val Tyr
340 345 350
Thr Leu Pro Pro Ser Arg Glu Glu Met Thr Lys Asn Gln Val Ser Leu
355 360 365
Thr Cys Leu Val Lys Gly Phe Tyr Pro Ser Asp Ile Ala Val Glu Trp
370 375 380
Glu Ser Asn Gly Gln Pro Glu Asn Asn Tyr Lys Thr Thr Pro Pro Val
385 390 395 400
Leu Asp Ser Asp Gly Ser Phe Phe Leu Tyr Ser Lys Leu Thr Val Asp
405 410 415
Lys Ser Arg Trp Gln Gln Gly Asn Val Phe Ser Cys Ser Val Met His
420 425 430
Glu Ala Leu His Asn His Tyr Thr Gln Lys Ser Leu Ser Leu Ser Pro
435 440 445
Gly Lys
450
<210> 3
<211> 450
<212> ПРТ
<213> Искусственная последовательность
<220>
<223> Тяжелая цепь модифицированного антитела против HER2
<400> 3
Glu Val Gln Leu Val Glu Ser Gly Gly Gly Leu Val Gln Pro Gly Gly
1 5 10 15
Ser Leu Arg Leu Ser Cys Ala Ala Ser Gly Phe Asn Ile Lys Asp Thr
20 25 30
Tyr Ile His Trp Val Arg Gln Ala Pro Gly Lys Gly Leu Glu Trp Val
35 40 45
Ala Arg Ile Tyr Pro Thr Asn Gly Tyr Thr Arg Tyr Ala Asp Ser Val
50 55 60
Lys Gly Arg Phe Thr Ile Ser Ala Asp Thr Ser Lys Asn Thr Ala Tyr
65 70 75 80
Leu Gln Met Asn Ser Leu Arg Ala Glu Asp Thr Ala Val Tyr Tyr Cys
85 90 95
Ser Arg Trp Gly Gly Asp Gly Phe Tyr Ala Met Asp Tyr Trp Gly Gln
100 105 110
Gly Thr Leu Val Thr Val Ser Ser Ala Ser Thr Lys Gly Pro Ser Val
115 120 125
Phe Pro Leu Ala Pro Ser Ser Lys Ser Thr Ser Gly Gly Thr Ala Ala
130 135 140
Leu Gly Cys Leu Val Lys Asp Tyr Phe Pro Glu Pro Val Thr Val Ser
145 150 155 160
Trp Asn Ser Gly Ala Leu Thr Ser Gly Val His Thr Phe Pro Ala Val
165 170 175
Leu Gln Ser Ser Gly Leu Tyr Ser Leu Ser Ser Val Val Thr Val Pro
180 185 190
Ser Ser Ser Leu Gly Thr Gln Thr Tyr Ile Cys Asn Val Asn His Lys
195 200 205
Pro Ser Asn Thr Lys Val Asp Lys Lys Val Glu Pro Lys Ser Cys Asp
210 215 220
Lys Thr His Thr Cys Pro Pro Cys Pro Ala Pro Glu Ala Ala Gly Gly
225 230 235 240
Pro Ser Val Phe Leu Phe Pro Pro Lys Pro Lys Asp Thr Leu Met Ile
245 250 255
Ser Arg Thr Pro Glu Val Thr Cys Val Val Val Asp Val Ser His Glu
260 265 270
Asp Pro Glu Val Lys Phe Asn Trp Tyr Val Asp Gly Val Glu Val His
275 280 285
Asn Ala Lys Thr Lys Pro Arg Glu Glu Gln Tyr Asn Ser Thr Tyr Arg
290 295 300
Val Val Ser Val Leu Thr Val Leu His Gln Asp Trp Leu Asn Gly Lys
305 310 315 320
Glu Tyr Lys Cys Lys Val Ser Asn Lys Ala Leu Pro Ala Pro Ile Glu
325 330 335
Lys Thr Ile Ser Lys Ala Lys Gly Gln Pro Arg Glu Pro Gln Val Tyr
340 345 350
Thr Leu Pro Pro Ser Arg Glu Glu Met Thr Lys Asn Gln Val Ser Leu
355 360 365
Thr Cys Leu Val Lys Gly Phe Tyr Pro Ser Asp Ile Ala Val Glu Trp
370 375 380
Glu Ser Asn Gly Gln Pro Glu Asn Asn Tyr Lys Thr Thr Pro Pro Val
385 390 395 400
Leu Asp Ser Asp Gly Ser Phe Phe Leu Tyr Ser Lys Leu Thr Val Asp
405 410 415
Lys Ser Arg Trp Gln Gln Gly Asn Val Phe Ser Cys Ser Val Met His
420 425 430
Glu Ala Leu His Asn His Tyr Thr Gln Lys Ser Leu Ser Leu Ser Pro
435 440 445
Gly Lys
450
<210> 4
<211> 379
<212> ПРТ
<213> Homo sapiens
<400> 4
Met Pro His Ser Ser Leu His Pro Ser Ile Pro Cys Pro Arg Gly His
1 5 10 15
Gly Ala Gln Lys Ala Ala Leu Val Leu Leu Ser Ala Cys Leu Val Thr
20 25 30
Leu Trp Gly Leu Gly Glu Pro Pro Glu His Thr Leu Arg Tyr Leu Val
35 40 45
Leu His Leu Ala Ser Leu Gln Leu Gly Leu Leu Leu Asn Gly Val Cys
50 55 60
Ser Leu Ala Glu Glu Leu Arg His Ile His Ser Arg Tyr Arg Gly Ser
65 70 75 80
Tyr Trp Arg Thr Val Arg Ala Cys Leu Gly Cys Pro Leu Arg Arg Gly
85 90 95
Ala Leu Leu Leu Leu Ser Ile Tyr Phe Tyr Tyr Ser Leu Pro Asn Ala
100 105 110
Val Gly Pro Pro Phe Thr Trp Met Leu Ala Leu Leu Gly Leu Ser Gln
115 120 125
Ala Leu Asn Ile Leu Leu Gly Leu Lys Gly Leu Ala Pro Ala Glu Ile
130 135 140
Ser Ala Val Cys Glu Lys Gly Asn Phe Asn Val Ala His Gly Leu Ala
145 150 155 160
Trp Ser Tyr Tyr Ile Gly Tyr Leu Arg Leu Ile Leu Pro Glu Leu Gln
165 170 175
Ala Arg Ile Arg Thr Tyr Asn Gln His Tyr Asn Asn Leu Leu Arg Gly
180 185 190
Ala Val Ser Gln Arg Leu Tyr Ile Leu Leu Pro Leu Asp Cys Gly Val
195 200 205
Pro Asp Asn Leu Ser Met Ala Asp Pro Asn Ile Arg Phe Leu Asp Lys
210 215 220
Leu Pro Gln Gln Thr Gly Asp Arg Ala Gly Ile Lys Asp Arg Val Tyr
225 230 235 240
Ser Asn Ser Ile Tyr Glu Leu Leu Glu Asn Gly Gln Arg Ala Gly Thr
245 250 255
Cys Val Leu Glu Tyr Ala Thr Pro Leu Gln Thr Leu Phe Ala Met Ser
260 265 270
Gln Tyr Ser Gln Ala Gly Phe Ser Arg Glu Asp Arg Leu Glu Gln Ala
275 280 285
Lys Leu Phe Cys Arg Thr Leu Glu Asp Ile Leu Ala Asp Ala Pro Glu
290 295 300
Ser Gln Asn Asn Cys Arg Leu Ile Ala Tyr Gln Glu Pro Ala Asp Asp
305 310 315 320
Ser Ser Phe Ser Leu Ser Gln Glu Val Leu Arg His Leu Arg Gln Glu
325 330 335
Glu Lys Glu Glu Val Thr Val Gly Ser Leu Lys Thr Ser Ala Val Pro
340 345 350
Ser Thr Ser Thr Met Ser Gln Glu Pro Glu Leu Leu Ile Ser Gly Met
355 360 365
Glu Lys Pro Leu Pro Leu Arg Thr Asp Phe Ser
370 375
<210> 5
<211> 1140
<212> ДНК
<213> Homo sapiens
<400> 5
atgccccact ccagcctgca tccatccatc ccgtgtccca ggggtcacgg ggcccagaag 60
gcagccttgg ttctgctgag tgcctgcctg gtgacccttt gggggctagg agagccacca 120
gagcacactc tccggtacct ggtgctccac ctagcctccc tgcagctggg actgctgtta 180
aacggggtct gcagcctggc tgaggagctg cgccacatcc actccaggta ccggggcagc 240
tactggagga ctgtgcgggc ctgcctgggc tgccccctcc gccgtggggc cctgttgctg 300
ctgtccatct atttctacta ctccctccca aatgcggtcg gcccgccctt cacttggatg 360
cttgccctcc tgggcctctc gcaggcactg aacatcctcc tgggcctcaa gggcctggcc 420
ccagctgaga tctctgcagt gtgtgaaaaa gggaatttca acgtggccca tgggctggca 480
tggtcatatt acatcggata tctgcggctg atcctgccag agctccaggc ccggattcga 540
acttacaatc agcattacaa caacctgcta cggggtgcag tgagccagcg gctgtatatt 600
ctcctcccat tggactgtgg ggtgcctgat aacctgagta tggctgaccc caacattcgc 660
ttcctggata aactgcccca gcagaccggt gaccatgctg gcatcaagga tcgggtttac 720
agcaacagca tctatgagct tctggagaac gggcagcggg cgggcacctg tgtcctggag 780
tacgccaccc ccttgcagac tttgtttgcc atgtcacaat acagtcaagc tggctttagc 840
cgggaggata ggcttgagca ggccaaactc ttctgccgga cacttgagga catcctggca 900
gatgcccctg agtctcagaa caactgccgc ctcattgcct accaggaacc tgcagatgac 960
agcagcttct cgctgtccca ggaggttctc cggcacctgc ggcaggagga aaaggaagag
1020
gttactgtgg gcagcttgaa gacctcagcg gtgcccagta cctccacgat gtcccaagag
1080
cctgagctcc tcatcagtgg aatggaaaag cccctccctc tccgcacgga tttctcttag
1140
<210> 6
<211> 379
<212> ПРТ
<213> Homo sapiens
<400> 6
Met Pro His Ser Ser Leu His Pro Ser Ile Pro Cys Pro Arg Gly His
1 5 10 15
Gly Ala Gln Lys Ala Ala Leu Val Leu Leu Ser Ala Cys Leu Val Thr
20 25 30
Leu Trp Gly Leu Gly Glu Pro Pro Glu His Thr Leu Arg Tyr Leu Val
35 40 45
Leu His Leu Ala Ser Leu Gln Leu Gly Leu Leu Leu Asn Gly Val Cys
50 55 60
Ser Leu Ala Glu Glu Leu Arg His Ile His Ser Arg Tyr Arg Gly Ser
65 70 75 80
Tyr Trp Arg Thr Val Arg Ala Cys Leu Gly Cys Pro Leu Arg Arg Gly
85 90 95
Ala Leu Leu Leu Leu Ser Ile Tyr Phe Tyr Tyr Ser Leu Pro Asn Ala
100 105 110
Val Gly Pro Pro Phe Thr Trp Met Leu Ala Leu Leu Gly Leu Ser Gln
115 120 125
Ala Leu Asn Ile Leu Leu Gly Leu Lys Gly Leu Ala Pro Ala Glu Ile
130 135 140
Ser Ala Val Cys Glu Lys Gly Asn Phe Asn Val Ala His Gly Leu Ala
145 150 155 160
Trp Ser Tyr Tyr Ile Gly Tyr Leu Arg Leu Ile Leu Pro Glu Leu Gln
165 170 175
Ala Arg Ile Arg Thr Tyr Asn Gln His Tyr Asn Asn Leu Leu Arg Gly
180 185 190
Ala Val Ser Gln Arg Leu Tyr Ile Leu Leu Pro Leu Asp Cys Gly Val
195 200 205
Pro Asp Asn Leu Ser Met Ala Asp Pro Asn Ile Arg Phe Leu Asp Lys
210 215 220
Leu Pro Gln Gln Thr Gly Asp His Ala Gly Ile Lys Asp Arg Val Tyr
225 230 235 240
Ser Asn Ser Ile Tyr Glu Leu Leu Glu Asn Gly Gln Arg Ala Gly Thr
245 250 255
Cys Val Leu Glu Tyr Ala Thr Pro Leu Gln Thr Leu Phe Ala Met Ser
260 265 270
Gln Tyr Ser Gln Ala Gly Phe Ser Arg Glu Asp Arg Leu Glu Gln Ala
275 280 285
Lys Leu Phe Cys Arg Thr Leu Glu Asp Ile Leu Ala Asp Ala Pro Glu
290 295 300
Ser Gln Asn Asn Cys Arg Leu Ile Ala Tyr Gln Glu Pro Ala Asp Asp
305 310 315 320
Ser Ser Phe Ser Leu Ser Gln Glu Val Leu Arg His Leu Arg Gln Glu
325 330 335
Glu Lys Glu Glu Val Thr Val Gly Ser Leu Lys Thr Ser Ala Val Pro
340 345 350
Ser Thr Ser Thr Met Ser Gln Glu Pro Glu Leu Leu Ile Ser Gly Met
355 360 365
Glu Lys Pro Leu Pro Leu Arg Thr Asp Phe Ser
370 375
<210> 7
<211> 1140
<212> ДНК
<213> Homo sapiens
<400> 7
atgccccact ccagcctgca tccatccatc ccgtgtccca ggggtcacgg ggcccagaag 60
gcagccttgg ttctgctgag tgcctgcctg gtgacccttt gggggctagg agagccacca 120
gagcacactc tccggtacct ggtgctccac ctagcctccc tgcagctggg actgctgtta 180
aacggggtct gcagcctggc tgaggagctg cgccacatcc actccaggta ccggggcagc 240
tactggagga ctgtgcgggc ctgcctgggc tgccccctcc gccgtggggc cctgttgctg 300
ctgtccatct atttctacta ctccctccca aatgcggtcg gcccgccctt cacttggatg 360
cttgccctcc tgggcctctc gcaggcactg aacatcctcc tgggcctcaa gggcctggcc 420
ccagctgaga tctctgcagt gtgtgaaaaa gggaatttca acgtggccca tgggctggca 480
tggtcatatt acatcggata tctgcggctg atcctgccag agctccaggc ccggattcga 540
acttacaatc agcattacaa caacctgcta cggggtgcag tgagccagcg gctgtatatt 600
ctcctcccat tggactgtgg ggtgcctgat aacctgagta tggctgaccc caacattcgc 660
ttcctggata aactgcccca gcagaccggt gaccgtgctg gcatcaagga tcgggtttac 720
agcaacagca tctatgagct tctggagaac gggcagcggg cgggcacctg tgtcctggag 780
tacgccaccc ccttgcagac tttgtttgcc atgtcacaat acagtcaagc tggctttagc 840
cgggaggata ggcttgagca ggccaaactc ttctgccgga cacttgagga catcctggca 900
gatgcccctg agtctcagaa caactgccgc ctcattgcct accaggaacc tgcagatgac 960
agcagcttct cgctgtccca ggaggttctc cggcacctgc ggcaggagga aaaggaagag
1020
gttactgtgg gcagcttgaa gacctcagcg gtgcccagta cctccacgat gtcccaagag
1080
cctgagctcc tcatcagtgg aatggaaaag cccctccctc tccgcacgga tttctcttag
1140
<210> 8
<211> 379
<212> ПРТ
<213> Homo sapiens
<400> 8
Met Pro His Ser Ser Leu His Pro Ser Ile Pro Cys Pro Arg Gly His
1 5 10 15
Gly Ala Gln Lys Ala Ala Leu Val Leu Leu Ser Ala Cys Leu Val Thr
20 25 30
Leu Trp Gly Leu Gly Glu Pro Pro Glu His Thr Leu Arg Tyr Leu Val
35 40 45
Leu His Leu Ala Ser Leu Gln Leu Gly Leu Leu Leu Asn Gly Val Cys
50 55 60
Ser Leu Ala Glu Glu Leu His His Ile His Ser Arg Tyr Arg Gly Ser
65 70 75 80
Tyr Trp Arg Thr Val Arg Ala Cys Leu Gly Cys Pro Leu Arg Arg Gly
85 90 95
Ala Leu Leu Leu Leu Ser Ile Tyr Phe Tyr Tyr Ser Leu Pro Asn Ala
100 105 110
Val Gly Pro Pro Phe Thr Trp Met Leu Ala Leu Leu Gly Leu Ser Gln
115 120 125
Ala Leu Asn Ile Leu Leu Gly Leu Lys Gly Leu Ala Pro Ala Glu Ile
130 135 140
Ser Ala Val Cys Glu Lys Gly Asn Phe Asn Val Ala His Gly Leu Ala
145 150 155 160
Trp Ser Tyr Tyr Ile Gly Tyr Leu Arg Leu Ile Leu Pro Glu Leu Gln
165 170 175
Ala Arg Ile Arg Thr Tyr Asn Gln His Tyr Asn Asn Leu Leu Arg Gly
180 185 190
Ala Val Ser Gln Arg Leu Tyr Ile Leu Leu Pro Leu Asp Cys Gly Val
195 200 205
Pro Asp Asn Leu Ser Met Ala Asp Pro Asn Ile Arg Phe Leu Asp Lys
210 215 220
Leu Pro Gln Gln Thr Ala Asp Arg Ala Gly Ile Lys Asp Arg Val Tyr
225 230 235 240
Ser Asn Ser Ile Tyr Glu Leu Leu Glu Asn Gly Gln Arg Ala Gly Thr
245 250 255
Cys Val Leu Glu Tyr Ala Thr Pro Leu Gln Thr Leu Phe Ala Met Ser
260 265 270
Gln Tyr Ser Gln Ala Gly Phe Ser Arg Glu Asp Arg Leu Glu Gln Ala
275 280 285
Lys Leu Phe Cys Gln Thr Leu Glu Asp Ile Leu Ala Asp Ala Pro Glu
290 295 300
Ser Gln Asn Asn Cys Arg Leu Ile Ala Tyr Gln Glu Pro Ala Asp Asp
305 310 315 320
Ser Ser Phe Ser Leu Ser Gln Glu Val Leu Arg His Leu Arg Gln Glu
325 330 335
Glu Lys Glu Glu Val Thr Val Gly Ser Leu Lys Thr Ser Ala Val Pro
340 345 350
Ser Thr Ser Thr Met Ser Gln Glu Pro Glu Leu Leu Ile Ser Gly Met
355 360 365
Glu Lys Pro Leu Pro Leu Arg Thr Asp Phe Ser
370 375
<210> 9
<211> 1140
<212> ДНК
<213> Homo sapiens
<400> 9
atgccccact ccagcctgca tccatccatc ccgtgtccca ggggtcacgg ggcccagaag 60
gcagccttgg ttctgctgag tgcctgcctg gtgacccttt gggggctagg agagccacca 120
gagcacactc tccggtacct ggtgctccac ctagcctccc tgcagctggg actgctgtta 180
aacggggtct gcagcctggc tgaggagctg caccacatcc actccaggta ccggggcagc 240
tactggagga ctgtgcgggc ctgcctgggc tgccccctcc gccgtggggc cctgttgctg 300
ctgtccatct atttctacta ctccctccca aatgcggtcg gcccgccctt cacttggatg 360
cttgccctcc tgggcctctc gcaggcactg aacatcctcc tgggcctcaa gggcctggcc 420
ccagctgaga tctctgcagt gtgtgaaaaa gggaatttca acgtggccca tgggctggca 480
tggtcatatt acatcggata tctgcggctg atcctgccag agctccaggc ccggattcga 540
acttacaatc agcattacaa caacctgcta cggggtgcag tgagccagcg gctgtatatt 600
ctcctcccat tggactgtgg ggtgcctgat aacctgagta tggctgaccc caacattcgc 660
ttcctggata aactgcccca gcagaccgct gaccgtgctg gcatcaagga tcgggtttac 720
agcaacagca tctatgagct tctggagaac gggcagcggg cgggcacctg tgtcctggag 780
tacgccaccc ccttgcagac tttgtttgcc atgtcacaat acagtcaagc tggctttagc 840
cgggaggata ggcttgagca ggccaaactc ttctgccaga cacttgagga catcctggca 900
gatgcccctg agtctcagaa caactgccgc ctcattgcct accaggaacc tgcagatgac 960
agcagcttct cgctgtccca ggaggttctc cggcacctgc ggcaggagga aaaggaagag
1020
gttactgtgg gcagcttgaa gacctcagcg gtgcccagta cctccacgat gtcccaagag
1080
cctgagctcc tcatcagtgg aatggaaaag cccctccctc tccgcacgga tttctcttag
1140
<210> 10
<211> 378
<212> ПРТ
<213> Mus musculus
<400> 10
Met Pro Tyr Ser Asn Leu His Pro Ala Ile Pro Arg Pro Arg Gly His
1 5 10 15
Arg Ser Lys Tyr Val Ala Leu Ile Phe Leu Val Ala Ser Leu Met Ile
20 25 30
Leu Trp Val Ala Lys Asp Pro Pro Asn His Thr Leu Lys Tyr Leu Ala
35 40 45
Leu His Leu Ala Ser His Glu Leu Gly Leu Leu Leu Lys Asn Leu Cys
50 55 60
Cys Leu Ala Glu Glu Leu Cys His Val Gln Ser Arg Tyr Gln Gly Ser
65 70 75 80
Tyr Trp Lys Ala Val Arg Ala Cys Leu Gly Cys Pro Ile His Cys Met
85 90 95
Ala Met Ile Leu Leu Ser Ser Tyr Phe Tyr Phe Leu Gln Asn Thr Ala
100 105 110
Asp Ile Tyr Leu Ser Trp Met Phe Gly Leu Leu Val Leu Tyr Lys Ser
115 120 125
Leu Ser Met Leu Leu Gly Leu Gln Ser Leu Thr Pro Ala Glu Val Ser
130 135 140
Ala Val Cys Glu Glu Lys Lys Leu Asn Val Ala His Gly Leu Ala Trp
145 150 155 160
Ser Tyr Tyr Ile Gly Tyr Leu Arg Leu Ile Leu Pro Gly Leu Gln Ala
165 170 175
Arg Ile Arg Met Phe Asn Gln Leu His Asn Asn Met Leu Ser Gly Ala
180 185 190
Gly Ser Arg Arg Leu Tyr Ile Leu Phe Pro Leu Asp Cys Gly Val Pro
195 200 205
Asp Asn Leu Ser Val Val Asp Pro Asn Ile Arg Phe Arg Asp Met Leu
210 215 220
Pro Gln Gln Asn Ile Asp Arg Ala Gly Ile Lys Asn Arg Val Tyr Ser
225 230 235 240
Asn Ser Val Tyr Glu Ile Leu Glu Asn Gly Gln Pro Ala Gly Val Cys
245 250 255
Ile Leu Glu Tyr Ala Thr Pro Leu Gln Thr Leu Phe Ala Met Ser Gln
260 265 270
Asp Ala Lys Ala Gly Phe Ser Arg Glu Asp Arg Leu Glu Gln Ala Lys
275 280 285
Leu Phe Cys Arg Thr Leu Glu Glu Ile Leu Glu Asp Val Pro Glu Ser
290 295 300
Arg Asn Asn Cys Arg Leu Ile Val Tyr Gln Glu Pro Thr Asp Gly Asn
305 310 315 320
Ser Phe Ser Leu Ser Gln Glu Val Leu Arg His Ile Arg Gln Glu Glu
325 330 335
Lys Glu Glu Val Thr Met Asn Ala Pro Met Thr Ser Val Ala Pro Pro
340 345 350
Pro Ser Val Leu Ser Gln Glu Pro Arg Leu Leu Ile Ser Gly Met Asp
355 360 365
Gln Pro Leu Pro Leu Arg Thr Asp Leu Ile
370 375
<210> 11
<211> 1137
<212> ДНК
<213> Mus musculus
<400> 11
atgccatact ccaacctgca tccagccatc ccacggccca gaggtcaccg ctccaaatat 60
gtagccctca tctttctggt ggccagcctg atgatccttt gggtggcaaa ggatccacca 120
aatcacactc tgaagtacct agcacttcac ctagcctcgc acgaacttgg actactgttg 180
aaaaacctct gctgtctggc tgaagagctg tgccatgtcc agtccaggta ccagggcagc 240
tactggaagg ctgtgcgcgc ctgcctggga tgccccatcc actgtatggc tatgattcta 300
ctatcgtctt atttctattt cctccaaaac actgctgaca tatacctcag ttggatgttt 360
ggccttctgg tcctctataa gtccctaagc atgctcctgg gccttcagag cttgactcca 420
gcggaagtct ctgcagtctg tgaagaaaag aagttaaatg ttgcccacgg gctggcctgg 480
tcatactaca ttgggtactt gcggttgatc ttaccagggc tccaggcccg gatccgaatg 540
ttcaatcagc tacataacaa catgctcagt ggtgcaggga gccgaagact gtacatcctc 600
tttccattgg actgtggggt gcctgacaac ctgagtgtag ttgaccccaa cattcgattc 660
cgagatatgc tgccccagca aaacatcgac cgtgctggca tcaagaatcg ggtttattcc 720
aacagcgtct acgagattct ggagaacgga cagccagcag gcgtctgtat cctggagtac 780
gccaccccct tgcagaccct gtttgccatg tcacaggatg ccaaagctgg cttcagtcgg 840
gaggatcggc ttgagcaggc taaactcttc tgccggacac ttgaggaaat cctggaagat 900
gtccccgagt ctcgaaataa ctgccgcctc attgtctacc aagaacccac agacggaaac 960
agtttctcac tgtctcagga ggtgctccgg cacattcgtc aggaagaaaa ggaggaggtt
1020
accatgaatg cccccatgac ctcagtggca cctcctccct ccgtactgtc ccaagagcca
1080
agactcctca tcagtggtat ggatcagcct ctcccactcc gcactgacct catctga 1137
<210> 12
<211> 27
<212> ДНК
<213> Искусственная последовательность
<220>
<223> праймер
<400> 12
cgtgctggca tcaaggatcg ggtttac 27
<210> 13
<211> 27
<212> ДНК
<213> Искусственная последовательность
<220>
<223> праймер
<400> 13
gtcaccggtc tgctggggca gtttatc 27
<210> 14
<211> 33
<212> ДНК
<213> Искусственная последовательность
<220>
<223> праймер
<400> 14
gctgaccgtg ctggcatcaa ggatcgggtt tac 33
<210> 15
<211> 25
<212> ДНК
<213> Искусственная последовательность
<220>
<223> праймер
<400> 15
ggtctgctgg ggcagtttat ccagg 25
<210> 16
<211> 24
<212> ДНК
<213> Искусственная последовательность
<220>
<223> праймер
<400> 16
caccacatcc actccaggta ccgg 24
<210> 17
<211> 24
<212> ДНК
<213> Искусственная последовательность
<220>
<223> праймер
<400> 17
cagctcctca gccaggctgc agac 24
<210> 18
<211> 25
<212> ДНК
<213> Искусственная последовательность
<220>
<223> праймер
<400> 18
cagacacttg aggacatcct ggcag 25
<210> 19
<211> 23
<212> ДНК
<213> Искусственная последовательность
<220>
<223> праймер
<400> 19
gcagaagagt ttggcctgct caa 23
<210> 20
<211> 39
<212> ДНК
<213> Искусственная последовательность
<220>
<223> праймер
<400> 20
acctgtattt tcagggcctg gccccagctg agatctctg 39
<210> 21
<211> 44
<212> ДНК
<213> Искусственная последовательность
<220>
<223> праймер
<400> 21
cagaattcgc aagcttttaa gtaacctctt ccttttcctc ctgc 44
<210> 22
<211> 243
<212> ПРТ
<213> Искусственная последовательность
<220>
<223> HisAviTEV-hSTING (139-342) человека дикого типа
<400> 22
Met Gly Ser Ser His His His His His His Ser Ser Gly Ser Gly Leu
1 5 10 15
Asn Asp Ile Phe Glu Ala Gln Lys Ile Glu Trp His Glu Gly Gly Ser
20 25 30
Glu Asn Leu Tyr Phe Gln Gly Leu Ala Pro Ala Glu Ile Ser Ala Val
35 40 45
Cys Glu Lys Gly Asn Phe Asn Val Ala His Gly Leu Ala Trp Ser Tyr
50 55 60
Tyr Ile Gly Tyr Leu Arg Leu Ile Leu Pro Glu Leu Gln Ala Arg Ile
65 70 75 80
Arg Thr Tyr Asn Gln His Tyr Asn Asn Leu Leu Arg Gly Ala Val Ser
85 90 95
Gln Arg Leu Tyr Ile Leu Leu Pro Leu Asp Cys Gly Val Pro Asp Asn
100 105 110
Leu Ser Met Ala Asp Pro Asn Ile Arg Phe Leu Asp Lys Leu Pro Gln
115 120 125
Gln Thr Gly Asp Arg Ala Gly Ile Lys Asp Arg Val Tyr Ser Asn Ser
130 135 140
Ile Tyr Glu Leu Leu Glu Asn Gly Gln Arg Ala Gly Thr Cys Val Leu
145 150 155 160
Glu Tyr Ala Thr Pro Leu Gln Thr Leu Phe Ala Met Ser Gln Tyr Ser
165 170 175
Gln Ala Gly Phe Ser Arg Glu Asp Arg Leu Glu Gln Ala Lys Leu Phe
180 185 190
Cys Arg Thr Leu Glu Asp Ile Leu Ala Asp Ala Pro Glu Ser Gln Asn
195 200 205
Asn Cys Arg Leu Ile Ala Tyr Gln Glu Pro Ala Asp Asp Ser Ser Phe
210 215 220
Ser Leu Ser Gln Glu Val Leu Arg His Leu Arg Gln Glu Glu Lys Glu
225 230 235 240
Glu Val Thr
<210> 23
<211> 243
<212> ПРТ
<213> Искусственная последовательность
<220>
<223> REF-мутант HisAviTEV-hSTING (139-342) человека
<400> 23
Met Gly Ser Ser His His His His His His Ser Ser Gly Ser Gly Leu
1 5 10 15
Asn Asp Ile Phe Glu Ala Gln Lys Ile Glu Trp His Glu Gly Gly Ser
20 25 30
Glu Asn Leu Tyr Phe Gln Gly Leu Ala Pro Ala Glu Ile Ser Ala Val
35 40 45
Cys Glu Lys Gly Asn Phe Asn Val Ala His Gly Leu Ala Trp Ser Tyr
50 55 60
Tyr Ile Gly Tyr Leu Arg Leu Ile Leu Pro Glu Leu Gln Ala Arg Ile
65 70 75 80
Arg Thr Tyr Asn Gln His Tyr Asn Asn Leu Leu Arg Gly Ala Val Ser
85 90 95
Gln Arg Leu Tyr Ile Leu Leu Pro Leu Asp Cys Gly Val Pro Asp Asn
100 105 110
Leu Ser Met Ala Asp Pro Asn Ile Arg Phe Leu Asp Lys Leu Pro Gln
115 120 125
Gln Thr Gly Asp His Ala Gly Ile Lys Asp Arg Val Tyr Ser Asn Ser
130 135 140
Ile Tyr Glu Leu Leu Glu Asn Gly Gln Arg Ala Gly Thr Cys Val Leu
145 150 155 160
Glu Tyr Ala Thr Pro Leu Gln Thr Leu Phe Ala Met Ser Gln Tyr Ser
165 170 175
Gln Ala Gly Phe Ser Arg Glu Asp Arg Leu Glu Gln Ala Lys Leu Phe
180 185 190
Cys Arg Thr Leu Glu Asp Ile Leu Ala Asp Ala Pro Glu Ser Gln Asn
195 200 205
Asn Cys Arg Leu Ile Ala Tyr Gln Glu Pro Ala Asp Asp Ser Ser Phe
210 215 220
Ser Leu Ser Gln Glu Val Leu Arg His Leu Arg Gln Glu Glu Lys Glu
225 230 235 240
Glu Val Thr
<210> 24
<211> 243
<212> ПРТ
<213> Искусственная последовательность
<220>
<223> AQ-мутант HisAviTEV-hSTING (139-342) человека
<400> 24
Met Gly Ser Ser His His His His His His Ser Ser Gly Ser Gly Leu
1 5 10 15
Asn Asp Ile Phe Glu Ala Gln Lys Ile Glu Trp His Glu Gly Gly Ser
20 25 30
Glu Asn Leu Tyr Phe Gln Gly Leu Ala Pro Ala Glu Ile Ser Ala Val
35 40 45
Cys Glu Lys Gly Asn Phe Asn Val Ala His Gly Leu Ala Trp Ser Tyr
50 55 60
Tyr Ile Gly Tyr Leu Arg Leu Ile Leu Pro Glu Leu Gln Ala Arg Ile
65 70 75 80
Arg Thr Tyr Asn Gln His Tyr Asn Asn Leu Leu Arg Gly Ala Val Ser
85 90 95
Gln Arg Leu Tyr Ile Leu Leu Pro Leu Asp Cys Gly Val Pro Asp Asn
100 105 110
Leu Ser Met Ala Asp Pro Asn Ile Arg Phe Leu Asp Lys Leu Pro Gln
115 120 125
Gln Thr Ala Asp Arg Ala Gly Ile Lys Asp Arg Val Tyr Ser Asn Ser
130 135 140
Ile Tyr Glu Leu Leu Glu Asn Gly Gln Arg Ala Gly Thr Cys Val Leu
145 150 155 160
Glu Tyr Ala Thr Pro Leu Gln Thr Leu Phe Ala Met Ser Gln Tyr Ser
165 170 175
Gln Ala Gly Phe Ser Arg Glu Asp Arg Leu Glu Gln Ala Lys Leu Phe
180 185 190
Cys Gln Thr Leu Glu Asp Ile Leu Ala Asp Ala Pro Glu Ser Gln Asn
195 200 205
Asn Cys Arg Leu Ile Ala Tyr Gln Glu Pro Ala Asp Asp Ser Ser Phe
210 215 220
Ser Leu Ser Gln Glu Val Leu Arg His Leu Arg Gln Glu Glu Lys Glu
225 230 235 240
Glu Val Thr
<210> 25
<211> 243
<212> ПРТ
<213> Искусственная последовательность
<220>
<223> HisAviTEV-hSTING (139-342) мыши дикого типа
<400> 25
Met Gly Ser Ser His His His His His His Ser Ser Gly Ser Gly Leu
1 5 10 15
Asn Asp Ile Phe Glu Ala Gln Lys Ile Glu Trp His Glu Gly Gly Ser
20 25 30
Glu Asn Leu Tyr Phe Gln Gly Leu Thr Pro Ala Glu Val Ser Ala Val
35 40 45
Cys Glu Glu Lys Lys Leu Asn Val Ala His Gly Leu Ala Trp Ser Tyr
50 55 60
Tyr Ile Gly Tyr Leu Arg Leu Ile Leu Pro Gly Leu Gln Ala Arg Ile
65 70 75 80
Arg Met Phe Asn Gln Leu His Asn Asn Met Leu Ser Gly Ala Gly Ser
85 90 95
Arg Arg Leu Tyr Ile Leu Phe Pro Leu Asp Cys Gly Val Pro Asp Asn
100 105 110
Leu Ser Val Val Asp Pro Asn Ile Arg Phe Arg Asp Met Leu Pro Gln
115 120 125
Gln Asn Ile Asp Arg Ala Gly Ile Lys Asn Arg Val Tyr Ser Asn Ser
130 135 140
Val Tyr Glu Ile Leu Glu Asn Gly Gln Pro Ala Gly Val Cys Ile Leu
145 150 155 160
Glu Tyr Ala Thr Pro Leu Gln Thr Leu Phe Ala Met Ser Gln Asp Ala
165 170 175
Lys Ala Gly Phe Ser Arg Glu Asp Arg Leu Glu Gln Ala Lys Leu Phe
180 185 190
Cys Arg Thr Leu Glu Glu Ile Leu Glu Asp Val Pro Glu Ser Arg Asn
195 200 205
Asn Cys Arg Leu Ile Val Tyr Gln Glu Pro Thr Asp Gly Asn Ser Phe
210 215 220
Ser Leu Ser Gln Glu Val Leu Arg His Ile Arg Gln Glu Glu Lys Glu
225 230 235 240
Glu Val Thr
<210> 26
<211> 214
<212> ПРТ
<213> Искусственная последовательность
<220>
<223> Легкая цепь модифицированного антитела против LPS
<400> 26
Asn Ile Val Met Thr Gln Ser Pro Asp Ser Leu Ala Val Ser Leu Gly
1 5 10 15
Glu Arg Ala Thr Ile Ser Cys Lys Ala Ser Glu Asn Val Gly Asn Ser
20 25 30
Val Ser Trp Tyr Gln Gln Lys Pro Gly Gln Ser Pro Lys Pro Leu Ile
35 40 45
Tyr Gly Ala Ser Asn Arg Tyr Thr Gly Val Pro Asp Arg Phe Ser Gly
50 55 60
Ser Gly Ser Ala Thr Asp Phe Thr Leu Thr Ile Ser Ser Leu Gln Ala
65 70 75 80
Glu Asp Val Ala Val Tyr His Cys Gly Gln Ser Tyr Ser Tyr Pro Tyr
85 90 95
Thr Phe Gly Gly Gly Thr Lys Val Glu Ile Lys Arg Thr Val Ala Ala
100 105 110
Pro Ser Val Phe Ile Phe Pro Pro Ser Asp Glu Gln Leu Lys Ser Gly
115 120 125
Thr Ala Ser Val Val Cys Leu Leu Asn Asn Phe Tyr Pro Arg Glu Ala
130 135 140
Lys Val Gln Trp Lys Val Asp Asn Ala Leu Gln Ser Gly Asn Ser Gln
145 150 155 160
Glu Ser Val Thr Glu Gln Asp Ser Lys Asp Ser Thr Tyr Ser Leu Ser
165 170 175
Ser Thr Leu Thr Leu Ser Lys Ala Asp Tyr Glu Lys His Lys Val Tyr
180 185 190
Ala Cys Glu Val Thr His Gln Gly Leu Ser Ser Pro Val Thr Lys Ser
195 200 205
Phe Asn Arg Gly Glu Cys
210
<210> 27
<211> 451
<212> ПРТ
<213> Искусственная последовательность
<220>
<223> Тяжелая цепь модифицированного антитела против LPS
<400> 27
Gln Val Gln Leu Val Gln Ser Gly Ala Glu Val Lys Lys Pro Gly Ala
1 5 10 15
Ser Val Lys Val Ser Cys Lys Ala Ser Gly Tyr Thr Phe Thr Thr Tyr
20 25 30
Trp Ile Asn Trp Val Arg Gln Ala Pro Gly Gln Gly Leu Glu Trp Met
35 40 45
Gly Asn Ile Tyr Pro Gly Thr Arg Ser Ser Asn Tyr Asn Glu Lys Phe
50 55 60
Lys Asn Arg Val Thr Leu Thr Val Asp Thr Ser Thr Ser Thr Ala Tyr
65 70 75 80
Met Glu Leu Ser Ser Leu Arg Ser Glu Asp Thr Ala Val Tyr Tyr Cys
85 90 95
Thr Arg Val Tyr Tyr Asp His Val Gly Tyr Tyr Phe Asp Tyr Trp Gly
100 105 110
Gln Gly Thr Leu Val Thr Val Ser Ser Ala Ser Thr Lys Gly Pro Ser
115 120 125
Val Phe Pro Leu Ala Pro Ser Ser Lys Ser Thr Ser Gly Gly Thr Ala
130 135 140
Ala Leu Gly Cys Leu Val Lys Asp Tyr Phe Pro Glu Pro Val Thr Val
145 150 155 160
Ser Trp Asn Ser Gly Ala Leu Thr Ser Gly Val His Thr Phe Pro Ala
165 170 175
Val Leu Gln Ser Ser Gly Leu Tyr Ser Leu Ser Ser Val Val Thr Val
180 185 190
Pro Ser Ser Ser Leu Gly Thr Gln Thr Tyr Ile Cys Asn Val Asn His
195 200 205
Lys Pro Ser Asn Thr Lys Val Asp Lys Arg Val Glu Pro Lys Ser Cys
210 215 220
Asp Lys Thr His Thr Cys Pro Pro Cys Pro Ala Pro Glu Ala Ala Gly
225 230 235 240
Gly Pro Ser Val Phe Leu Phe Pro Pro Lys Pro Lys Asp Thr Leu Met
245 250 255
Ile Ser Arg Thr Pro Glu Val Thr Cys Val Val Val Asp Val Ser His
260 265 270
Glu Asp Pro Glu Val Lys Phe Asn Trp Tyr Val Asp Gly Val Glu Val
275 280 285
His Asn Ala Lys Thr Lys Pro Arg Glu Glu Gln Tyr Asn Ser Thr Tyr
290 295 300
Arg Val Val Ser Val Leu Thr Val Leu His Gln Asp Trp Leu Asn Gly
305 310 315 320
Lys Glu Tyr Lys Cys Lys Val Ser Asn Lys Ala Leu Pro Ala Pro Ile
325 330 335
Glu Lys Thr Ile Ser Lys Ala Lys Gly Gln Pro Arg Glu Pro Gln Val
340 345 350
Tyr Thr Leu Pro Pro Ser Arg Glu Glu Met Thr Lys Asn Gln Val Ser
355 360 365
Leu Thr Cys Leu Val Lys Gly Phe Tyr Pro Ser Asp Ile Ala Val Glu
370 375 380
Trp Glu Ser Asn Gly Gln Pro Glu Asn Asn Tyr Lys Thr Thr Pro Pro
385 390 395 400
Val Leu Asp Ser Asp Gly Ser Phe Phe Leu Tyr Ser Lys Leu Thr Val
405 410 415
Asp Lys Ser Arg Trp Gln Gln Gly Asn Val Phe Ser Cys Ser Val Met
420 425 430
His Glu Ala Leu His Asn His Tyr Thr Gln Lys Ser Leu Ser Leu Ser
435 440 445
Pro Gly Lys
450
<210> 28
<211> 214
<212> ПРТ
<213> Искусственная последовательность
<220>
<223> Легкая цепь пертузумаба
<400> 28
Asp Ile Gln Met Thr Gln Ser Pro Ser Ser Leu Ser Ala Ser Val Gly
1 5 10 15
Asp Arg Val Thr Ile Thr Cys Lys Ala Ser Gln Asp Val Ser Ile Gly
20 25 30
Val Ala Trp Tyr Gln Gln Lys Pro Gly Lys Ala Pro Lys Leu Leu Ile
35 40 45
Tyr Ser Ala Ser Tyr Arg Tyr Thr Gly Val Pro Ser Arg Phe Ser Gly
50 55 60
Ser Gly Ser Gly Thr Asp Phe Thr Leu Thr Ile Ser Ser Leu Gln Pro
65 70 75 80
Glu Asp Phe Ala Thr Tyr Tyr Cys Gln Gln Tyr Tyr Ile Tyr Pro Tyr
85 90 95
Thr Phe Gly Gln Gly Thr Lys Val Glu Ile Lys Arg Thr Val Ala Ala
100 105 110
Pro Ser Val Phe Ile Phe Pro Pro Ser Asp Glu Gln Leu Lys Ser Gly
115 120 125
Thr Ala Ser Val Val Cys Leu Leu Asn Asn Phe Tyr Pro Arg Glu Ala
130 135 140
Lys Val Gln Trp Lys Val Asp Asn Ala Leu Gln Ser Gly Asn Ser Gln
145 150 155 160
Glu Ser Val Thr Glu Gln Asp Ser Lys Asp Ser Thr Tyr Ser Leu Ser
165 170 175
Ser Thr Leu Thr Leu Ser Lys Ala Asp Tyr Glu Lys His Lys Val Tyr
180 185 190
Ala Cys Glu Val Thr His Gln Gly Leu Ser Ser Pro Val Thr Lys Ser
195 200 205
Phe Asn Arg Gly Glu Cys
210
<210> 29
<211> 449
<212> ПРТ
<213> Искусственная последовательность
<220>
<223> Тяжелая цепь пертузумаба
<400> 29
Glu Val Gln Leu Val Glu Ser Gly Gly Gly Leu Val Gln Pro Gly Gly
1 5 10 15
Ser Leu Arg Leu Ser Cys Ala Ala Ser Gly Phe Thr Phe Thr Asp Tyr
20 25 30
Thr Met Asp Trp Val Arg Gln Ala Pro Gly Lys Gly Leu Glu Trp Val
35 40 45
Ala Asp Val Asn Pro Asn Ser Gly Gly Ser Ile Tyr Asn Gln Arg Phe
50 55 60
Lys Gly Arg Phe Thr Leu Ser Val Asp Arg Ser Lys Asn Thr Leu Tyr
65 70 75 80
Leu Gln Met Asn Ser Leu Arg Ala Glu Asp Thr Ala Val Tyr Tyr Cys
85 90 95
Ala Arg Asn Leu Gly Pro Ser Phe Tyr Phe Asp Tyr Trp Gly Gln Gly
100 105 110
Thr Leu Val Thr Val Ser Ser Ala Ser Thr Lys Gly Pro Ser Val Phe
115 120 125
Pro Leu Ala Pro Ser Ser Lys Ser Thr Ser Gly Gly Thr Ala Ala Leu
130 135 140
Gly Cys Leu Val Lys Asp Tyr Phe Pro Glu Pro Val Thr Val Ser Trp
145 150 155 160
Asn Ser Gly Ala Leu Thr Ser Gly Val His Thr Phe Pro Ala Val Leu
165 170 175
Gln Ser Ser Gly Leu Tyr Ser Leu Ser Ser Val Val Thr Val Pro Ser
180 185 190
Ser Ser Leu Gly Thr Gln Thr Tyr Ile Cys Asn Val Asn His Lys Pro
195 200 205
Ser Asn Thr Lys Val Asp Lys Lys Val Glu Pro Lys Ser Cys Asp Lys
210 215 220
Thr His Thr Cys Pro Pro Cys Pro Ala Pro Glu Leu Leu Gly Gly Pro
225 230 235 240
Ser Val Phe Leu Phe Pro Pro Lys Pro Lys Asp Thr Leu Met Ile Ser
245 250 255
Arg Thr Pro Glu Val Thr Cys Val Val Val Asp Val Ser His Glu Asp
260 265 270
Pro Glu Val Lys Phe Asn Trp Tyr Val Asp Gly Val Glu Val His Asn
275 280 285
Ala Lys Thr Lys Pro Arg Glu Glu Gln Tyr Asn Ser Thr Tyr Arg Val
290 295 300
Val Ser Val Leu Thr Val Leu His Gln Asp Trp Leu Asn Gly Lys Glu
305 310 315 320
Tyr Lys Cys Lys Val Ser Asn Lys Ala Leu Pro Ala Pro Ile Glu Lys
325 330 335
Thr Ile Ser Lys Ala Lys Gly Gln Pro Arg Glu Pro Gln Val Tyr Thr
340 345 350
Leu Pro Pro Ser Arg Glu Glu Met Thr Lys Asn Gln Val Ser Leu Thr
355 360 365
Cys Leu Val Lys Gly Phe Tyr Pro Ser Asp Ile Ala Val Glu Trp Glu
370 375 380
Ser Asn Gly Gln Pro Glu Asn Asn Tyr Lys Thr Thr Pro Pro Val Leu
385 390 395 400
Asp Ser Asp Gly Ser Phe Phe Leu Tyr Ser Lys Leu Thr Val Asp Lys
405 410 415
Ser Arg Trp Gln Gln Gly Asn Val Phe Ser Cys Ser Val Met His Glu
420 425 430
Ala Leu His Asn His Tyr Thr Gln Lys Ser Leu Ser Leu Ser Pro Gly
435 440 445
Lys
<210> 30
<211> 449
<212> ПРТ
<213> Искусственная последовательность
<220>
<223> Тяжелая цепь модифицированного антитела 2 против HER2
<400> 30
Glu Val Gln Leu Val Glu Ser Gly Gly Gly Leu Val Gln Pro Gly Gly
1 5 10 15
Ser Leu Arg Leu Ser Cys Ala Ala Ser Gly Phe Thr Phe Thr Asp Tyr
20 25 30
Thr Met Asp Trp Val Arg Gln Ala Pro Gly Lys Gly Leu Glu Trp Val
35 40 45
Ala Asp Val Asn Pro Asn Ser Gly Gly Ser Ile Tyr Asn Gln Arg Phe
50 55 60
Lys Gly Arg Phe Thr Leu Ser Val Asp Arg Ser Lys Asn Thr Leu Tyr
65 70 75 80
Leu Gln Met Asn Ser Leu Arg Ala Glu Asp Thr Ala Val Tyr Tyr Cys
85 90 95
Ala Arg Asn Leu Gly Pro Ser Phe Tyr Phe Asp Tyr Trp Gly Gln Gly
100 105 110
Thr Leu Val Thr Val Ser Ser Ala Ser Thr Lys Gly Pro Ser Val Phe
115 120 125
Pro Leu Ala Pro Ser Ser Lys Ser Thr Ser Gly Gly Thr Ala Ala Leu
130 135 140
Gly Cys Leu Val Lys Asp Tyr Phe Pro Glu Pro Val Thr Val Ser Trp
145 150 155 160
Asn Ser Gly Ala Leu Thr Ser Gly Val His Thr Phe Pro Ala Val Leu
165 170 175
Gln Ser Ser Gly Leu Tyr Ser Leu Ser Ser Val Val Thr Val Pro Ser
180 185 190
Ser Ser Leu Gly Thr Gln Thr Tyr Ile Cys Asn Val Asn His Lys Pro
195 200 205
Ser Asn Thr Lys Val Asp Lys Arg Val Glu Pro Lys Ser Cys Asp Lys
210 215 220
Thr His Thr Cys Pro Pro Cys Pro Ala Pro Glu Ala Ala Gly Gly Pro
225 230 235 240
Ser Val Phe Leu Phe Pro Pro Lys Pro Lys Asp Thr Leu Met Ile Ser
245 250 255
Arg Thr Pro Glu Val Thr Cys Val Val Val Asp Val Ser His Glu Asp
260 265 270
Pro Glu Val Lys Phe Asn Trp Tyr Val Asp Gly Val Glu Val His Asn
275 280 285
Ala Lys Thr Lys Pro Arg Glu Glu Gln Tyr Asn Ser Thr Tyr Arg Val
290 295 300
Val Ser Val Leu Thr Val Leu His Gln Asp Trp Leu Asn Gly Lys Glu
305 310 315 320
Tyr Lys Cys Lys Val Ser Asn Lys Ala Leu Pro Ala Pro Ile Glu Lys
325 330 335
Thr Ile Ser Lys Ala Lys Gly Gln Pro Arg Glu Pro Gln Val Tyr Thr
340 345 350
Leu Pro Pro Ser Arg Glu Glu Met Thr Lys Asn Gln Val Ser Leu Thr
355 360 365
Cys Leu Val Lys Gly Phe Tyr Pro Ser Asp Ile Ala Val Glu Trp Glu
370 375 380
Ser Asn Gly Gln Pro Glu Asn Asn Tyr Lys Thr Thr Pro Pro Val Leu
385 390 395 400
Asp Ser Asp Gly Ser Phe Phe Leu Tyr Ser Lys Leu Thr Val Asp Lys
405 410 415
Ser Arg Trp Gln Gln Gly Asn Val Phe Ser Cys Ser Val Met His Glu
420 425 430
Ala Leu His Asn His Tyr Thr Gln Lys Ser Leu Ser Leu Ser Pro Gly
435 440 445
Lys
<210> 31
<211> 218
<212> ПРТ
<213> Искусственная последовательность
<220>
<223> Легкая цепь антитела против CD33
<400> 31
Asp Ile Gln Leu Thr Gln Ser Pro Ser Thr Leu Ser Ala Ser Val Gly
1 5 10 15
Asp Arg Val Thr Ile Thr Cys Arg Ala Ser Glu Ser Leu Asp Asn Tyr
20 25 30
Gly Ile Arg Phe Leu Thr Trp Phe Gln Gln Lys Pro Gly Lys Ala Pro
35 40 45
Lys Leu Leu Met Tyr Ala Ala Ser Asn Gln Gly Ser Gly Val Pro Ser
50 55 60
Arg Phe Ser Gly Ser Gly Ser Gly Thr Glu Phe Thr Leu Thr Ile Ser
65 70 75 80
Ser Leu Gln Pro Asp Asp Phe Ala Thr Tyr Tyr Cys Gln Gln Thr Lys
85 90 95
Glu Val Pro Trp Ser Phe Gly Gln Gly Thr Lys Val Glu Val Lys Arg
100 105 110
Thr Val Ala Ala Pro Ser Val Phe Ile Phe Pro Pro Ser Asp Glu Gln
115 120 125
Leu Lys Ser Gly Thr Ala Ser Val Val Cys Leu Leu Asn Asn Phe Tyr
130 135 140
Pro Arg Glu Ala Lys Val Gln Trp Lys Val Asp Asn Ala Leu Gln Ser
145 150 155 160
Gly Asn Ser Gln Glu Ser Val Thr Glu Gln Asp Ser Lys Asp Ser Thr
165 170 175
Tyr Ser Leu Ser Ser Thr Leu Thr Leu Ser Lys Ala Asp Tyr Glu Lys
180 185 190
His Lys Val Tyr Ala Cys Glu Val Thr His Gln Gly Leu Ser Ser Pro
195 200 205
Val Thr Lys Ser Phe Asn Arg Gly Glu Cys
210 215
<210> 32
<211> 446
<212> ПРТ
<213> Искусственная последовательность
<220>
<223> Тяжелая цепь антитела против CD33
<400> 32
Glu Val Gln Leu Val Gln Ser Gly Ala Glu Val Lys Lys Pro Gly Ser
1 5 10 15
Ser Val Lys Val Ser Cys Lys Ala Ser Gly Tyr Thr Ile Thr Asp Ser
20 25 30
Asn Ile His Trp Val Arg Gln Ala Pro Gly Gln Ser Leu Glu Trp Ile
35 40 45
Gly Tyr Ile Tyr Pro Tyr Asn Gly Gly Thr Asp Tyr Asn Gln Lys Phe
50 55 60
Lys Asn Arg Ala Thr Leu Thr Val Asp Asn Pro Thr Asn Thr Ala Tyr
65 70 75 80
Met Glu Leu Ser Ser Leu Arg Ser Glu Asp Thr Ala Phe Tyr Tyr Cys
85 90 95
Val Asn Gly Asn Pro Trp Leu Ala Tyr Trp Gly Gln Gly Thr Leu Val
100 105 110
Thr Val Ser Ser Ala Ser Thr Lys Gly Pro Ser Val Phe Pro Leu Ala
115 120 125
Pro Ser Ser Lys Ser Thr Ser Gly Gly Thr Ala Ala Leu Gly Cys Leu
130 135 140
Val Lys Asp Tyr Phe Pro Glu Pro Val Thr Val Ser Trp Asn Ser Gly
145 150 155 160
Ala Leu Thr Ser Gly Val His Thr Phe Pro Ala Val Leu Gln Ser Ser
165 170 175
Gly Leu Tyr Ser Leu Ser Ser Val Val Thr Val Pro Ser Ser Ser Leu
180 185 190
Gly Thr Gln Thr Tyr Ile Cys Asn Val Asn His Lys Pro Ser Asn Thr
195 200 205
Lys Val Asp Lys Arg Val Glu Pro Lys Ser Cys Asp Lys Thr His Thr
210 215 220
Cys Pro Pro Cys Pro Ala Pro Glu Ala Ala Gly Gly Pro Ser Val Phe
225 230 235 240
Leu Phe Pro Pro Lys Pro Lys Asp Thr Leu Met Ile Ser Arg Thr Pro
245 250 255
Glu Val Thr Cys Val Val Val Asp Val Ser His Glu Asp Pro Glu Val
260 265 270
Lys Phe Asn Trp Tyr Val Asp Gly Val Glu Val His Asn Ala Lys Thr
275 280 285
Lys Pro Arg Glu Glu Gln Tyr Asn Ser Thr Tyr Arg Val Val Ser Val
290 295 300
Leu Thr Val Leu His Gln Asp Trp Leu Asn Gly Lys Glu Tyr Lys Cys
305 310 315 320
Lys Val Ser Asn Lys Ala Leu Pro Ala Pro Ile Glu Lys Thr Ile Ser
325 330 335
Lys Ala Lys Gly Gln Pro Arg Glu Pro Gln Val Tyr Thr Leu Pro Pro
340 345 350
Ser Arg Glu Glu Met Thr Lys Asn Gln Val Ser Leu Thr Cys Leu Val
355 360 365
Lys Gly Phe Tyr Pro Ser Asp Ile Ala Val Glu Trp Glu Ser Asn Gly
370 375 380
Gln Pro Glu Asn Asn Tyr Lys Thr Thr Pro Pro Val Leu Asp Ser Asp
385 390 395 400
Gly Ser Phe Phe Leu Tyr Ser Lys Leu Thr Val Asp Lys Ser Arg Trp
405 410 415
Gln Gln Gly Asn Val Phe Ser Cys Ser Val Met His Glu Ala Leu His
420 425 430
Asn His Tyr Thr Gln Lys Ser Leu Ser Leu Ser Pro Gly Lys
435 440 445
<210> 33
<211> 219
<212> ПРТ
<213> Искусственная последовательность
<220>
<223> Легкая цепь антитела против EphA2
<400> 33
Asp Ile Val Met Thr Gln Ser Pro Leu Ser Leu Pro Val Thr Pro Gly
1 5 10 15
Glu Pro Ala Ser Ile Ser Cys Arg Ser Ser Gln Ser Ile Val His Ser
20 25 30
Ser Gly Ile Thr Tyr Leu Glu Trp Tyr Leu Gln Lys Pro Gly Gln Ser
35 40 45
Pro Gln Leu Leu Ile Tyr Lys Val Ser Asn Arg Phe Ser Gly Val Pro
50 55 60
Asp Arg Phe Ser Gly Ser Gly Ser Gly Thr Asp Phe Thr Leu Lys Ile
65 70 75 80
Ser Arg Val Glu Ala Glu Asp Val Gly Val Tyr Tyr Cys Phe Gln Gly
85 90 95
Ser His Val Pro Tyr Thr Phe Gly Gln Gly Thr Lys Val Glu Ile Lys
100 105 110
Arg Thr Val Ala Ala Pro Ser Val Phe Ile Phe Pro Pro Ser Asp Glu
115 120 125
Gln Leu Lys Ser Gly Thr Ala Ser Val Val Cys Leu Leu Asn Asn Phe
130 135 140
Tyr Pro Arg Glu Ala Lys Val Gln Trp Lys Val Asp Asn Ala Leu Gln
145 150 155 160
Ser Gly Asn Ser Gln Glu Ser Val Thr Glu Gln Asp Ser Lys Asp Ser
165 170 175
Thr Tyr Ser Leu Ser Ser Thr Leu Thr Leu Ser Lys Ala Asp Tyr Glu
180 185 190
Lys His Lys Val Tyr Ala Cys Glu Val Thr His Gln Gly Leu Ser Ser
195 200 205
Pro Val Thr Lys Ser Phe Asn Arg Gly Glu Cys
210 215
<210> 34
<211> 449
<212> ПРТ
<213> Искусственная последовательность
<220>
<223> Тяжелая цепь антитела против EphA2
<400> 34
Gln Val Gln Leu Val Gln Ser Gly Ala Glu Val Lys Lys Pro Gly Ala
1 5 10 15
Ser Val Lys Val Ser Cys Lys Ala Ser Gly Tyr Thr Phe Ile Asp Tyr
20 25 30
Ser Met His Trp Val Arg Gln Ala Pro Gly Gln Gly Leu Glu Trp Met
35 40 45
Gly Trp Ile Asn Thr Tyr Thr Gly Glu Pro Thr Tyr Ser Asp Asp Phe
50 55 60
Lys Gly Arg Val Thr Ile Thr Ala Asp Thr Ser Thr Ser Thr Ala Tyr
65 70 75 80
Leu Glu Leu Ser Ser Leu Arg Ser Glu Asp Thr Ala Val Tyr Tyr Cys
85 90 95
Ala Thr Tyr Tyr Arg Tyr Glu Arg Asp Phe Asp Tyr Trp Gly Gln Gly
100 105 110
Thr Leu Val Thr Val Ser Ser Ala Ser Thr Lys Gly Pro Ser Val Phe
115 120 125
Pro Leu Ala Pro Ser Ser Lys Ser Thr Ser Gly Gly Thr Ala Ala Leu
130 135 140
Gly Cys Leu Val Lys Asp Tyr Phe Pro Glu Pro Val Thr Val Ser Trp
145 150 155 160
Asn Ser Gly Ala Leu Thr Ser Gly Val His Thr Phe Pro Ala Val Leu
165 170 175
Gln Ser Ser Gly Leu Tyr Ser Leu Ser Ser Val Val Thr Val Pro Ser
180 185 190
Ser Ser Leu Gly Thr Gln Thr Tyr Ile Cys Asn Val Asn His Lys Pro
195 200 205
Ser Asn Thr Lys Val Asp Lys Arg Val Glu Pro Lys Ser Cys Asp Lys
210 215 220
Thr His Thr Cys Pro Pro Cys Pro Ala Pro Glu Leu Leu Gly Gly Pro
225 230 235 240
Ser Val Phe Leu Phe Pro Pro Lys Pro Lys Asp Thr Leu Met Ile Ser
245 250 255
Arg Thr Pro Glu Val Thr Cys Val Val Val Asp Val Ser His Glu Asp
260 265 270
Pro Glu Val Lys Phe Asn Trp Tyr Val Asp Gly Val Glu Val His Asn
275 280 285
Ala Lys Thr Lys Pro Arg Glu Glu Gln Tyr Asn Ser Thr Tyr Arg Val
290 295 300
Val Ser Val Leu Thr Val Leu His Gln Asp Trp Leu Asn Gly Lys Glu
305 310 315 320
Tyr Lys Cys Lys Val Ser Asn Lys Ala Leu Pro Ala Pro Ile Glu Lys
325 330 335
Thr Ile Ser Lys Ala Lys Gly Gln Pro Arg Glu Pro Gln Val Tyr Thr
340 345 350
Leu Pro Pro Ser Arg Glu Glu Met Thr Lys Asn Gln Val Ser Leu Thr
355 360 365
Cys Leu Val Lys Gly Phe Tyr Pro Ser Asp Ile Ala Val Glu Trp Glu
370 375 380
Ser Asn Gly Gln Pro Glu Asn Asn Tyr Lys Thr Thr Pro Pro Val Leu
385 390 395 400
Asp Ser Asp Gly Ser Phe Phe Leu Tyr Ser Lys Leu Thr Val Asp Lys
405 410 415
Ser Arg Trp Gln Gln Gly Asn Val Phe Ser Cys Ser Val Met His Glu
420 425 430
Ala Leu His Asn His Tyr Thr Gln Lys Ser Leu Ser Leu Ser Pro Gly
435 440 445
Lys
<210> 35
<211> 213
<212> ПРТ
<213> Искусственная последовательность
<220>
<223> Легкая цепь антитела против CDH6
<400> 35
Asp Ile Gln Met Thr Gln Ser Pro Ser Ser Leu Ser Ala Ser Val Gly
1 5 10 15
Asp Arg Val Thr Ile Thr Cys Lys Ala Ser Gln Asn Ile Tyr Lys Asn
20 25 30
Leu Ala Trp Tyr Gln Gln Lys Pro Gly Lys Ala Pro Lys Leu Leu Ile
35 40 45
Tyr Asp Ala Asn Thr Leu Gln Thr Gly Val Pro Ser Arg Phe Ser Gly
50 55 60
Ser Gly Ser Gly Ser Asp Phe Thr Leu Thr Ile Ser Ser Leu Gln Pro
65 70 75 80
Glu Asp Phe Ala Thr Tyr Phe Cys Gln Gln Tyr Tyr Ser Gly Trp Ala
85 90 95
Phe Gly Gln Gly Thr Lys Val Glu Ile Lys Arg Thr Val Ala Ala Pro
100 105 110
Ser Val Phe Ile Phe Pro Pro Ser Asp Glu Gln Leu Lys Ser Gly Thr
115 120 125
Ala Ser Val Val Cys Leu Leu Asn Asn Phe Tyr Pro Arg Glu Ala Lys
130 135 140
Val Gln Trp Lys Val Asp Asn Ala Leu Gln Ser Gly Asn Ser Gln Glu
145 150 155 160
Ser Val Thr Glu Gln Asp Ser Lys Asp Ser Thr Tyr Ser Leu Ser Ser
165 170 175
Thr Leu Thr Leu Ser Lys Ala Asp Tyr Glu Lys His Lys Val Tyr Ala
180 185 190
Cys Glu Val Thr His Gln Gly Leu Ser Ser Pro Val Thr Lys Ser Phe
195 200 205
Asn Arg Gly Glu Cys
210
<210> 36
<211> 452
<212> ПРТ
<213> Искусственная последовательность
<220>
<223> Тяжелая цепь антитела против CDH6
<400> 36
Glu Val Gln Leu Val Gln Ser Gly Ala Glu Val Lys Lys Pro Gly Ala
1 5 10 15
Ser Val Lys Val Ser Cys Lys Ala Ser Gly Tyr Thr Phe Thr Arg Asn
20 25 30
Phe Met His Trp Val Arg Gln Ala Pro Gly Gln Gly Leu Glu Trp Met
35 40 45
Gly Trp Ile Tyr Pro Gly Asp Gly Glu Thr Glu Tyr Ala Gln Lys Phe
50 55 60
Gln Gly Arg Val Thr Ile Thr Ala Asp Thr Ser Thr Ser Thr Ala Tyr
65 70 75 80
Met Glu Leu Ser Ser Leu Arg Ser Glu Asp Thr Ala Val Tyr Tyr Cys
85 90 95
Ala Arg Gly Val Tyr Gly Gly Phe Ala Gly Gly Tyr Phe Asp Phe Trp
100 105 110
Gly Gln Gly Thr Leu Val Thr Val Ser Ser Ala Ser Thr Lys Gly Pro
115 120 125
Ser Val Phe Pro Leu Ala Pro Ser Ser Lys Ser Thr Ser Gly Gly Thr
130 135 140
Ala Ala Leu Gly Cys Leu Val Lys Asp Tyr Phe Pro Glu Pro Val Thr
145 150 155 160
Val Ser Trp Asn Ser Gly Ala Leu Thr Ser Gly Val His Thr Phe Pro
165 170 175
Ala Val Leu Gln Ser Ser Gly Leu Tyr Ser Leu Ser Ser Val Val Thr
180 185 190
Val Pro Ser Ser Ser Leu Gly Thr Gln Thr Tyr Ile Cys Asn Val Asn
195 200 205
His Lys Pro Ser Asn Thr Lys Val Asp Lys Arg Val Glu Pro Lys Ser
210 215 220
Cys Asp Lys Thr His Thr Cys Pro Pro Cys Pro Ala Pro Glu Ala Ala
225 230 235 240
Gly Gly Pro Ser Val Phe Leu Phe Pro Pro Lys Pro Lys Asp Thr Leu
245 250 255
Met Ile Ser Arg Thr Pro Glu Val Thr Cys Val Val Val Asp Val Ser
260 265 270
His Glu Asp Pro Glu Val Lys Phe Asn Trp Tyr Val Asp Gly Val Glu
275 280 285
Val His Asn Ala Lys Thr Lys Pro Arg Glu Glu Gln Tyr Asn Ser Thr
290 295 300
Tyr Arg Val Val Ser Val Leu Thr Val Leu His Gln Asp Trp Leu Asn
305 310 315 320
Gly Lys Glu Tyr Lys Cys Lys Val Ser Asn Lys Ala Leu Gly Ala Pro
325 330 335
Ile Glu Lys Thr Ile Ser Lys Ala Lys Gly Gln Pro Arg Glu Pro Gln
340 345 350
Val Tyr Thr Leu Pro Pro Ser Arg Glu Glu Met Thr Lys Asn Gln Val
355 360 365
Ser Leu Thr Cys Leu Val Lys Gly Phe Tyr Pro Ser Asp Ile Ala Val
370 375 380
Glu Trp Glu Ser Asn Gly Gln Pro Glu Asn Asn Tyr Lys Thr Thr Pro
385 390 395 400
Pro Val Leu Asp Ser Asp Gly Ser Phe Phe Leu Tyr Ser Lys Leu Thr
405 410 415
Val Asp Lys Ser Arg Trp Gln Gln Gly Asn Val Phe Ser Cys Ser Val
420 425 430
Met His Glu Ala Leu His Asn His Tyr Thr Gln Lys Ser Leu Ser Leu
435 440 445
Ser Pro Gly Lys
450
<---
| название | год | авторы | номер документа |
|---|---|---|---|
| КОНЪЮГАТ АНТИТЕЛО-ЛЕКАРСТВЕННОЕ СРЕДСТВО, СОДЕРЖАЩИЙ НОВОЕ ПРОИЗВОДНОЕ ЦИКЛИЧЕСКОГО ДИНУКЛЕОТИДА | 2021 |
|
RU2836814C1 |
| КОНЪЮГАТ ЛИГАНД-ЛЕКАРСТВЕННОЕ СРЕДСТВО АНАЛОГА ЭКЗАТЕКАНА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ | 2019 |
|
RU2793316C2 |
| КОНЪЮГАТ АНТИТЕЛО-ПРОИЗВОДНОЕ ПИРРОЛОБЕНЗОДИАЗЕПИНА | 2018 |
|
RU2820928C2 |
| СПОСОБ ПОЛУЧЕНИЯ КОНЪЮГАТА АНТИТЕЛО-ЛЕКАРСТВЕННОЕ СРЕДСТВО | 2021 |
|
RU2841377C1 |
| СОЕДИНЕНИЯ АЗАЛАКТАМА В КАЧЕСТВЕ ИНГИБИТОРОВ HPK1 | 2021 |
|
RU2819642C1 |
| ДИГИДРОНАФТИРИДИНЫ И РОДСТВЕННЫЕ СОЕДИНЕНИЯ, ПОДХОДЯЩИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗ ДЛЯ ЛЕЧЕНИЯ ПРОЛИФЕРАТИВНЫХ ЗАБОЛЕВАНИЙ | 2018 |
|
RU2804468C2 |
| ЛИНКЕРЫ НА ОСНОВЕ СУЛЬФОМАЛЕИМИДА И СООТВЕТСТВУЮЩИЕ КОНЪЮГАТЫ | 2019 |
|
RU2815199C2 |
| КОНЪЮГАТЫ АНТИТЕЛА И ЛЕКАРСТВЕННОГО СРЕДСТВА (ADC) И КОНЪЮГАТЫ АНТИТЕЛА И ПРОЛЕКАРСТВА (APDC), СОДЕРЖАЩИЕ ФЕРМЕНТАТИВНО РАСЩЕПЛЯЕМЫЕ ГРУППЫ | 2016 |
|
RU2751512C2 |
| КОНЪЮГАТ АНТИТЕЛО К B7H3-АНАЛОГ ЭКЗАТЕКАНА И ЕГО ПРИМЕНЕНИЕ В МЕДИЦИНЕ | 2019 |
|
RU2785664C2 |
| Соединения бензотиа(ди)азепина и их применение в качестве модуляторов желчных кислот | 2021 |
|
RU2841263C1 |
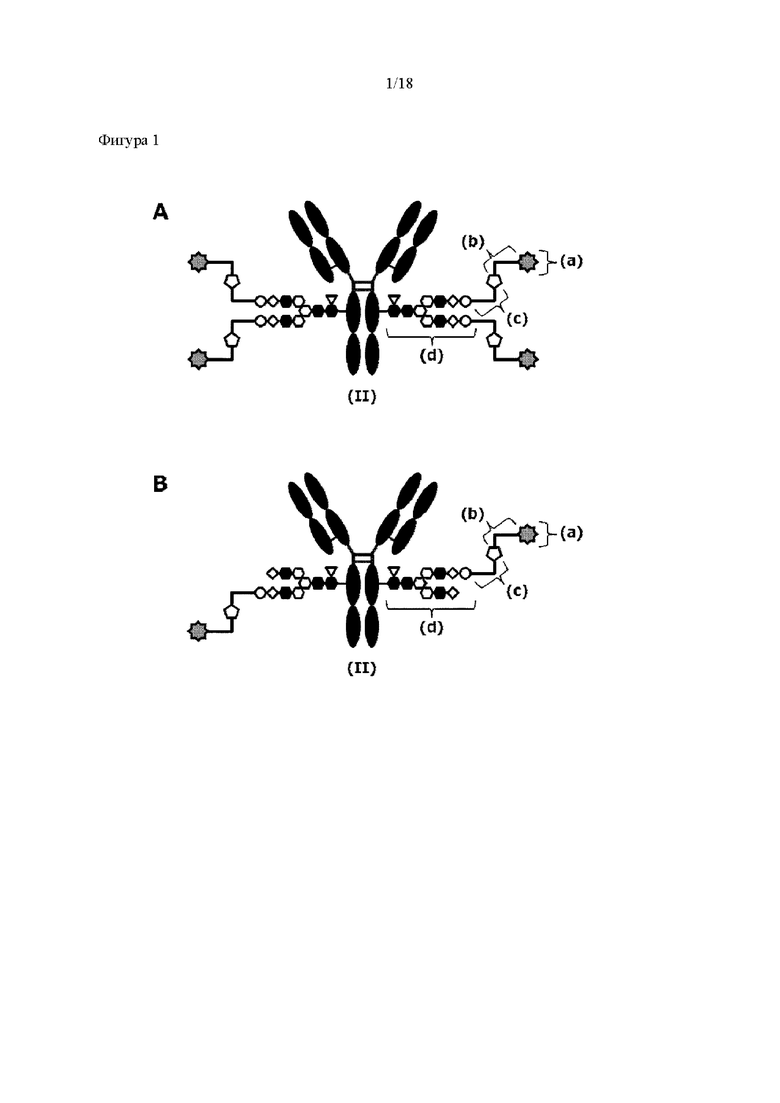
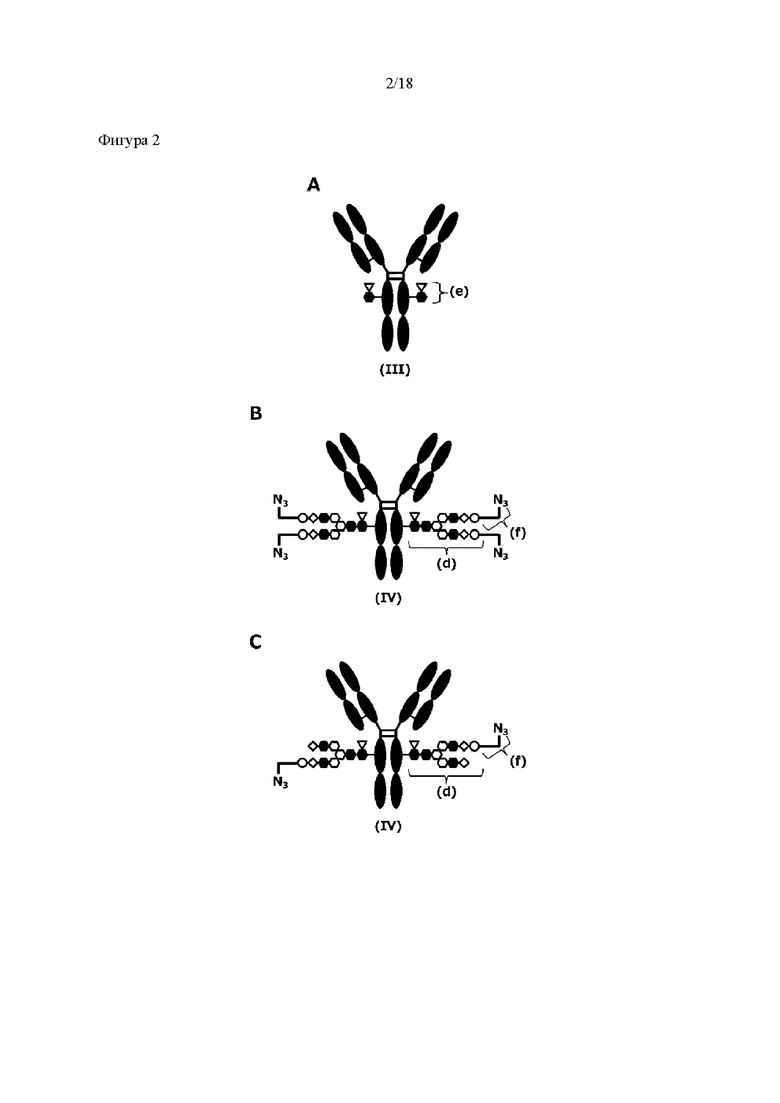
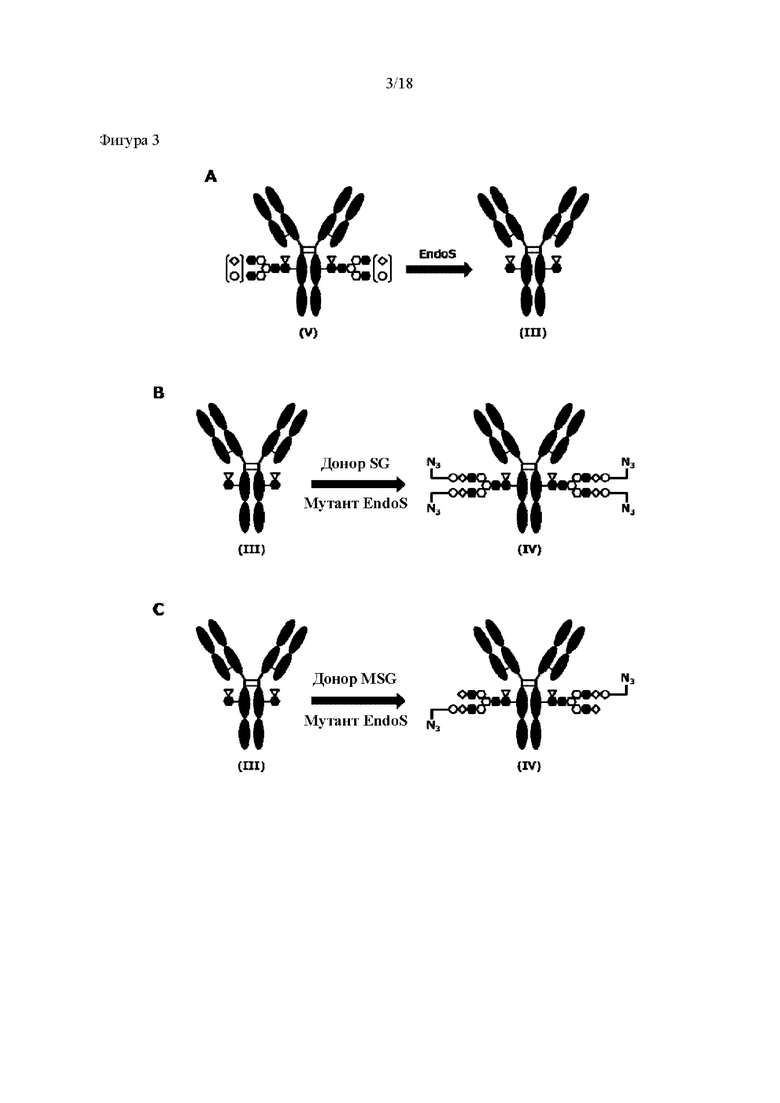



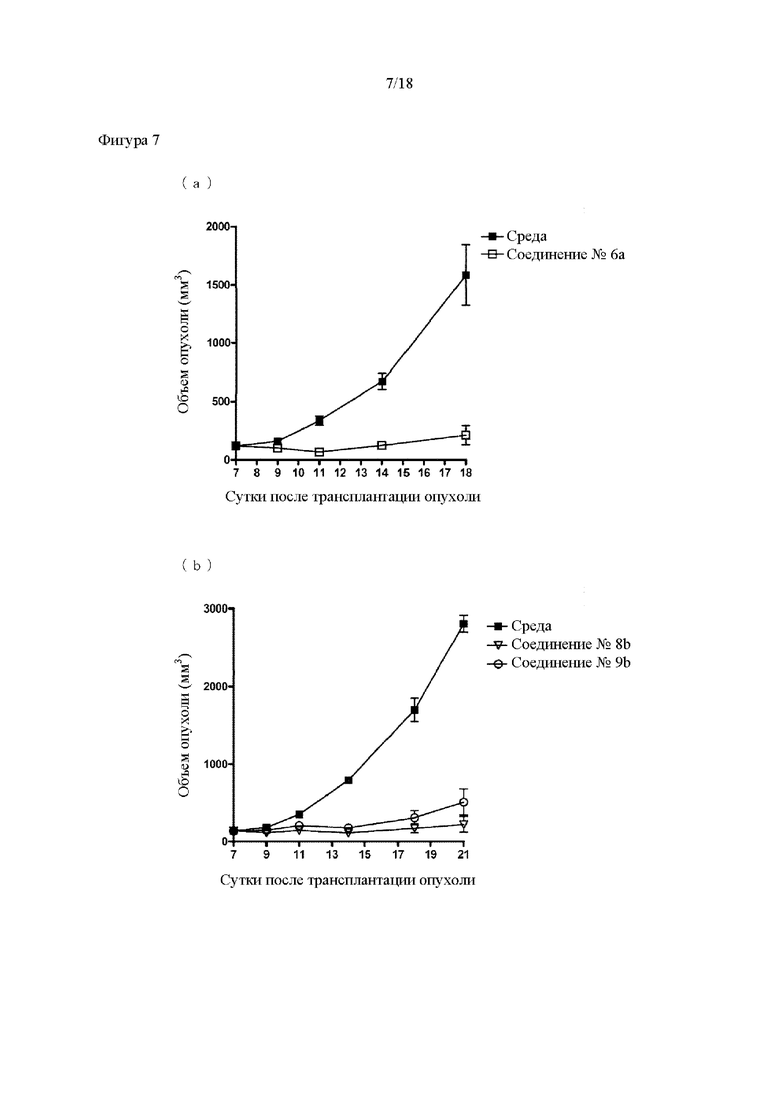
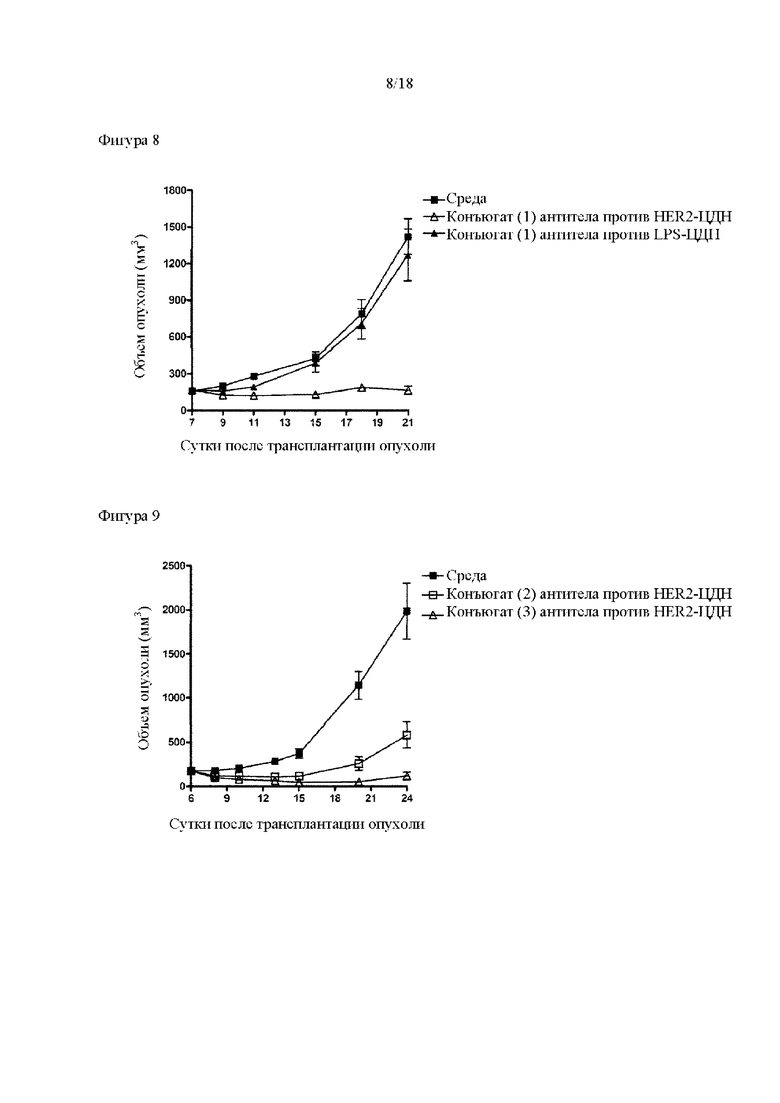
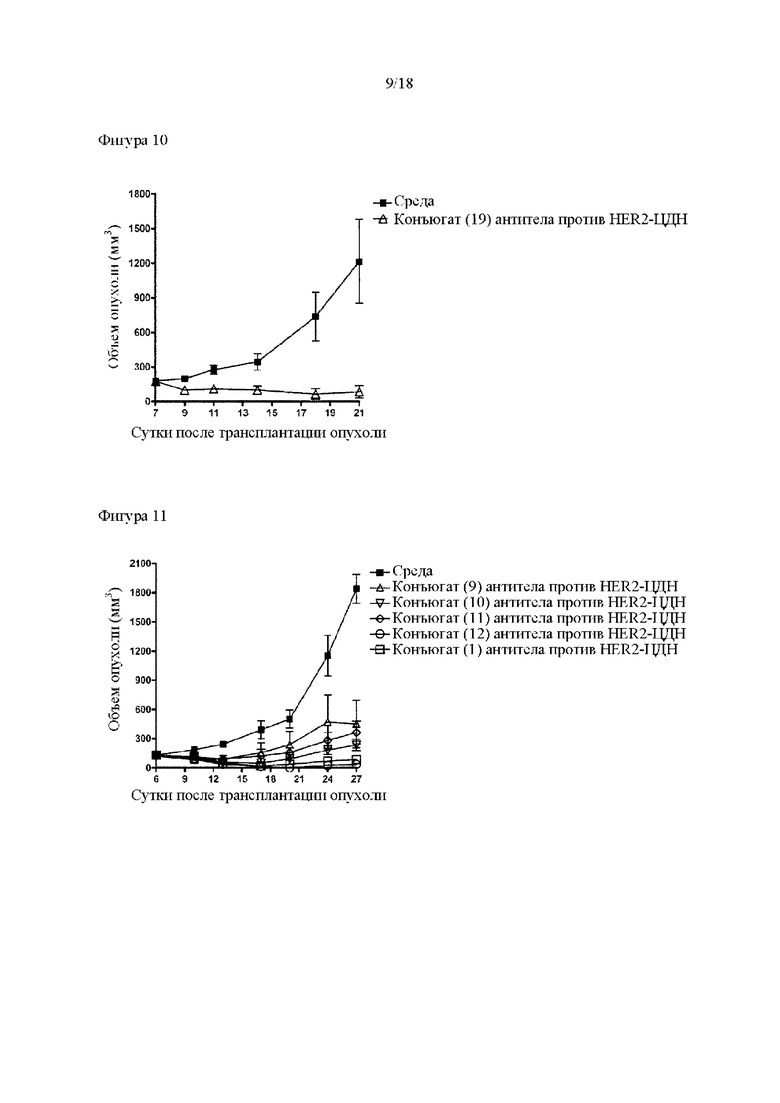
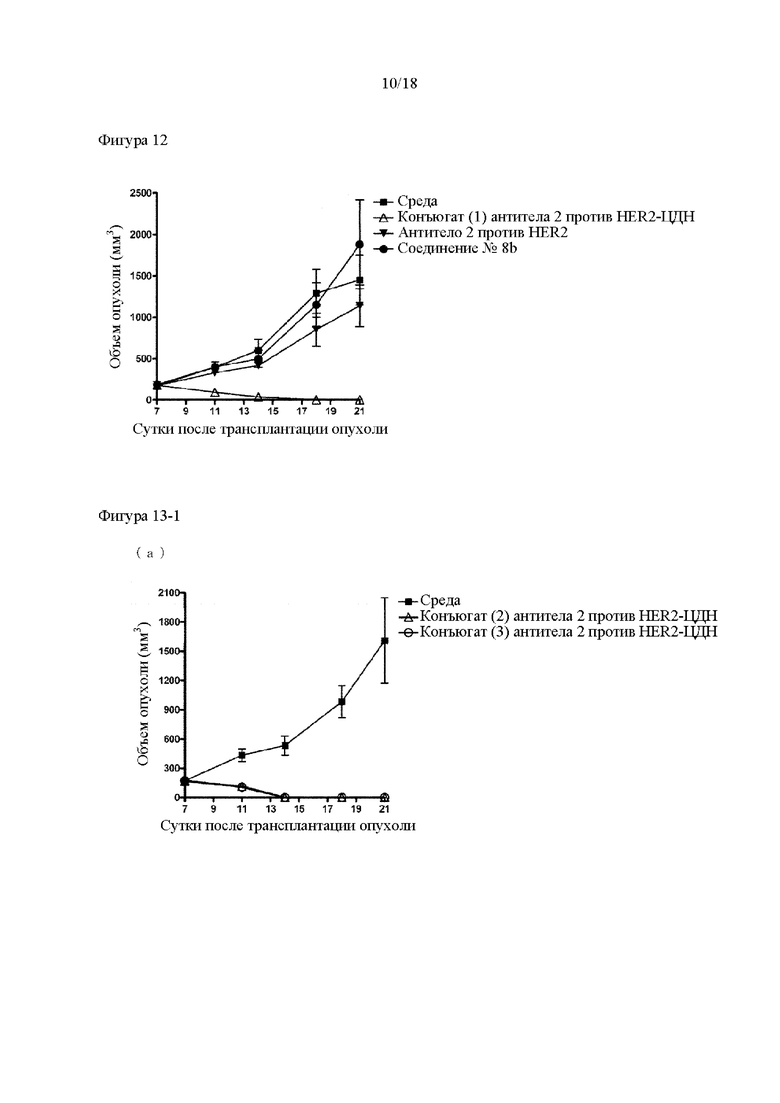
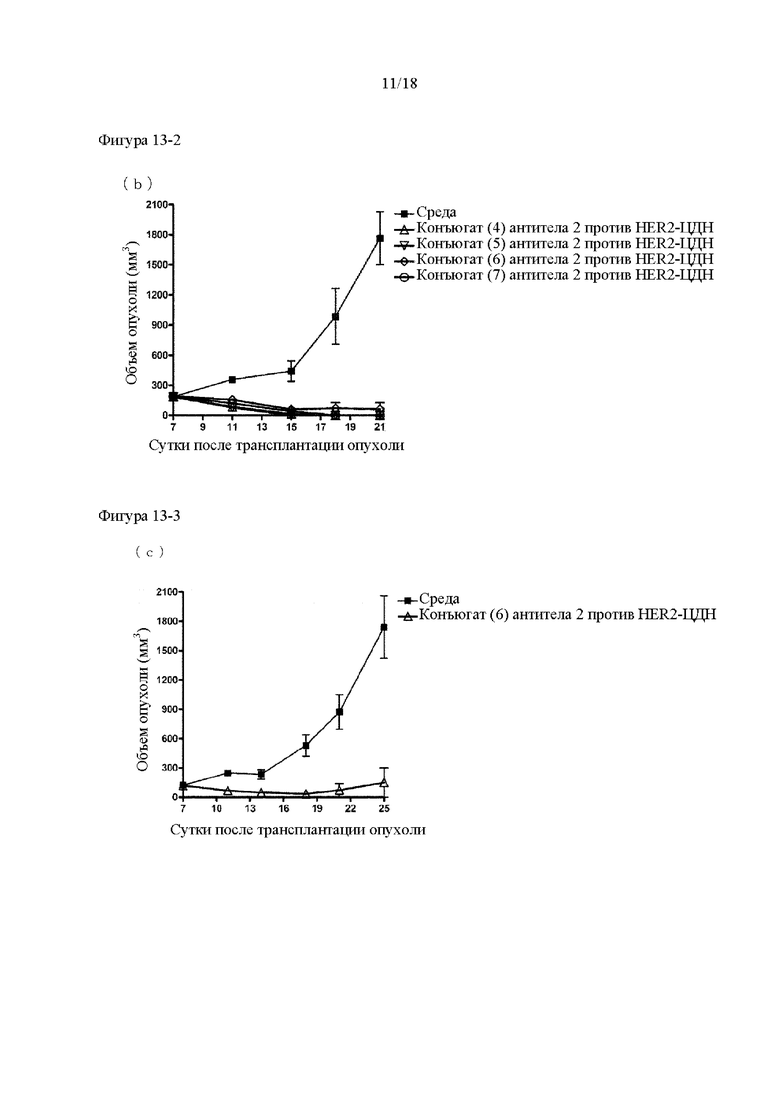
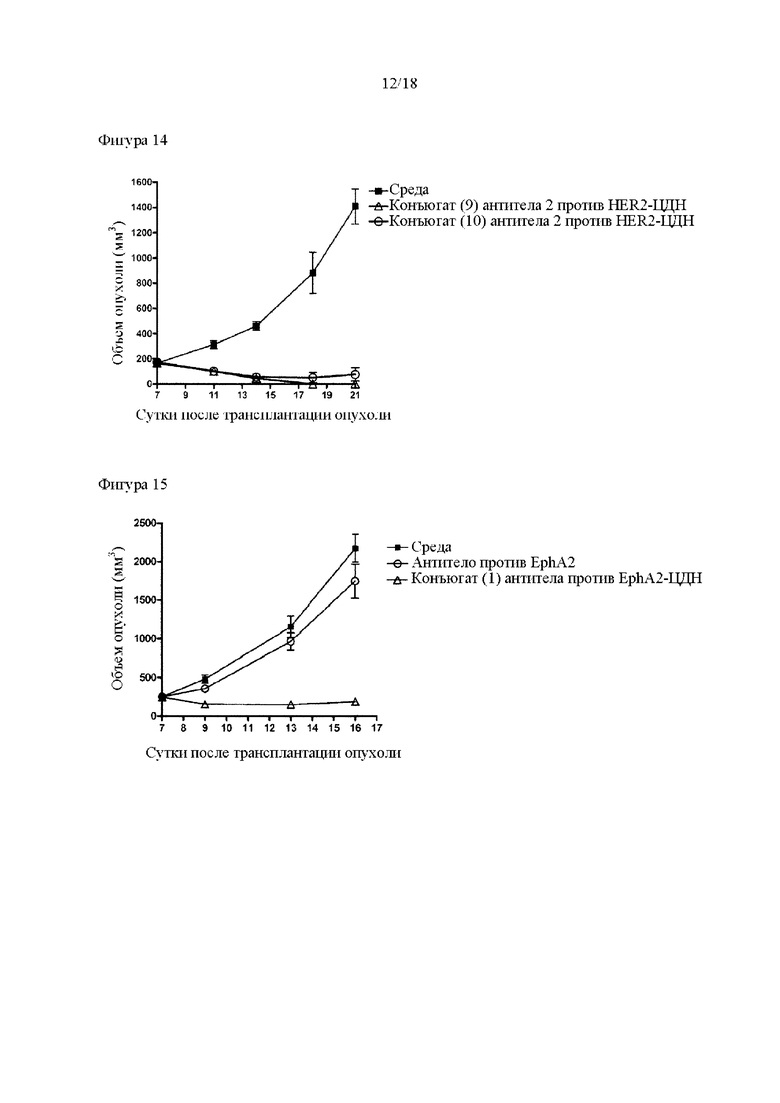
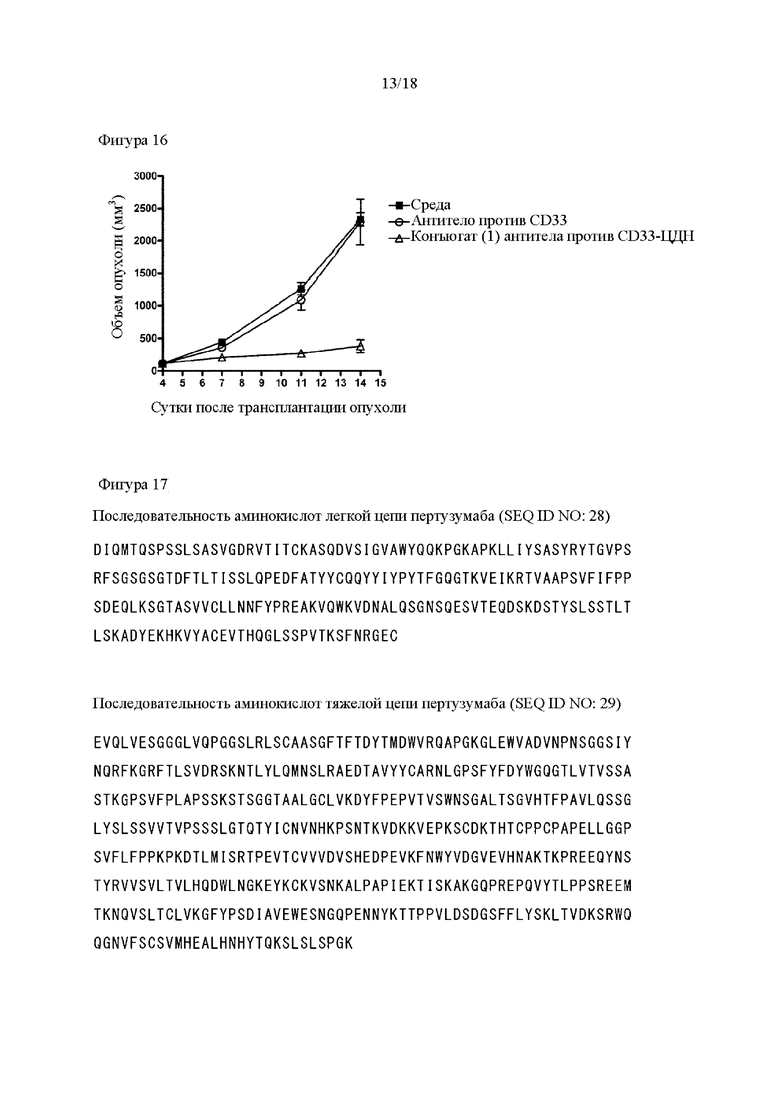




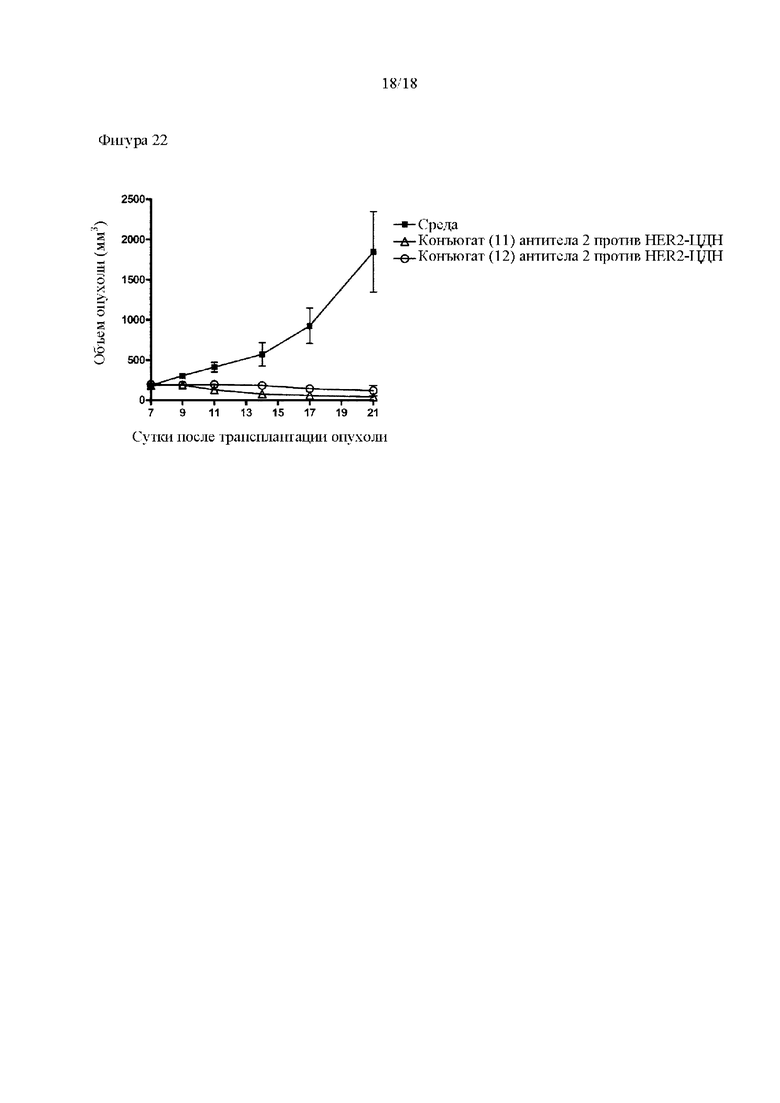
Изобретение относится к новым производным циклических динуклеотидов (ЦДН) с активностью агонистов STING, конъюгатам антитела с лекарственным средством, образованным путем конъюгирования новых производных циклических динуклеотидов и антител против целевых клеток друг с другом посредством линкера, фармацевтической композиции, содержащей конъюгаты антитела с лекарственным средством, и способу лечения рака, связанного с активностью агониста STING. 6 н. и 76 з.п. ф-лы, 22 ил., 2 табл., 127 пр.
1. Конъюгат антитела с лекарственным средством, представленный формулой (II)
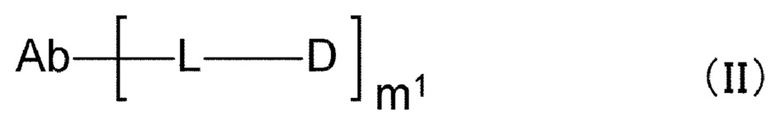 ,
,
где m1 находится в диапазоне от 1 до 10;
Ab представляет собой антитело или функциональный фрагмент антитела, где гликан антитела необязательно ремоделирован;
L представляет собой линкер, соединяющий Ab и D;
Ab связывается непосредственно с L через аминокислотный остаток из Ab или необязательно связывается посредством гликана или ремоделированного гликана Ab с L; и
D представляет собой соединение, представленное формулой (I)
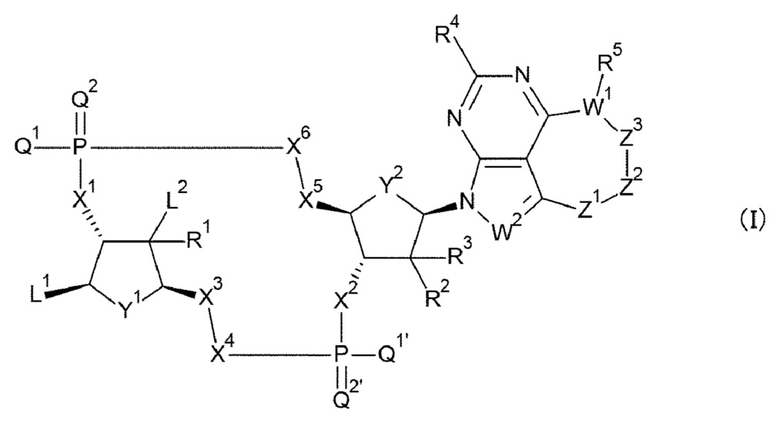 ,
,
где L связывается с любой -NH2 или гидроксигруппой, содержащейся в L1;
L1 представляет собой группу, выбранную из группы, состоящей из следующих формул:
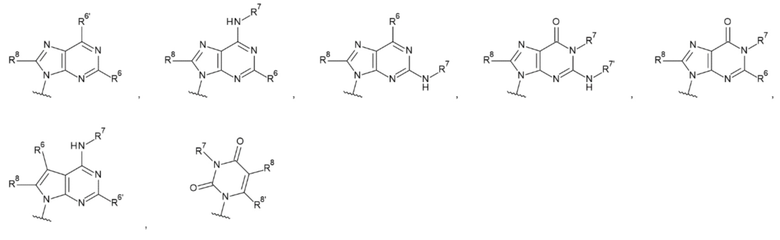
и необязательно содержащую в качестве заместителей в любом положении от одной до трех групп, выбранных из группы, состоящей из гидроксигруппы, -NH2, 2-гидроксиацетиламинометильной группы и 2-[(2-гидроксиацетил)амино]этильной группы,
где R6 и R6' каждый независимо представляет собой атом водорода, -NH2 или C1-C6 алкильную группу;
R7 и R7' каждый независимо представляет собой атом водорода или C1-C6 алкильную группу, где C1-C6 алкильная группа необязательно содержит в качестве заместителей один или два заместителя, выбранных из оксогруппы;
R8 и R8' представляют собой атом водорода;
L2 представляет собой атом водорода;
Q1 и Q1' каждый независимо представляет собой гидроксигруппу или тиольную группу;
Q2 и Q2' каждый независимо представляет собой атом кислорода или атом серы;
X1 и X2 представляют собой атом кислорода;
Y1 представляет собой атом кислорода;
Y2 представляет собой атом кислорода или -CH2-;
X3 и X4 представляют собой группу, выбранную из (iii):
(iii) когда Y1 представляет собой атом кислорода, X3-X4 представляет собой -CH2-O-; и
X5 и X6 представляют собой группу, выбранную из (v) и (vi):
(v) когда Y2 представляет собой атом кислорода, X5-X6 представляет собой -CH2-O-; и
(vi) когда Y2 представляет собой -CH2-, X5-X6 представляет собой -O-CH2-;
R1, R2 и R3 каждый независимо представляет собой атом водорода, атом галогена, -OR' или -NHR', где R' представляет собой атом водорода;
W1 представляет собой атом азота, атом кислорода или атом серы;
W2 представляет собой атом азота или -CH=;
R4 представляет собой атом водорода;
R5 представляет собой группу, выбранную из (vii) - (ix):
(vii) где W1 представляет собой атом азота, R5 представляет собой атом водорода;
(viii) где W1 представляет собой атом кислорода, R5 отсутствует;
(ix) где W1 представляет собой атом серы, R5 отсутствует; и
Z1-Z2-Z3, взятые вместе, представляют собой -CH2-CH2-CH2-, -CH2-CH2-R'''- или -CH2-CH2-CH(CH3)-, где R''' представляет собой -CH2-CH2-.
2. Конъюгат антитела с лекарственным средством по п. 1, отличающийся тем, что W1 представляет собой атом азота.
3. Конъюгат антитела с лекарственным средством по п. 2, отличающийся тем, что W1 представляет собой атом азота и R5 представляет собой атом водорода.
4. Конъюгат антитела с лекарственным средством по п. 1, отличающийся тем, что W1 представляет собой атом кислорода.
5. Конъюгат антитела с лекарственным средством по п. 1, отличающийся тем, что W1 представляет собой атом серы.
6. Конъюгат антитела с лекарственным средством по любому из пп. 1-5, отличающийся тем, что Z1, Z2 и Z3 вместе образуют -CH2-CH2-CH2-.
7. Конъюгат антитела с лекарственным средством по любому из пп. 1-5, отличающийся тем, что Z1, Z2 и Z3 вместе образуют -CH2-CH2-CH(CH3)-.
8. Конъюгат антитела с лекарственным средством по любому из пп. 1-5, отличающийся тем, что Z1, Z2 и Z3 вместе образуют -CH2-CH2-R'''-, где R''' представляет собой -CH2-CH2-.
9. Конъюгат антитела с лекарственным средством по любому из пп. 1-8, отличающийся тем, что W2 представляет собой -CH=.
10. Конъюгат антитела с лекарственным средством по любому из пп. 1-8, отличающийся тем, что W2 представляет собой атом азота.
11. Конъюгат антитела с лекарственным средством по любому из пп. 1-10, отличающийся тем, что L1 представляет собой группу, выбранную из группы, состоящей из следующих формул:
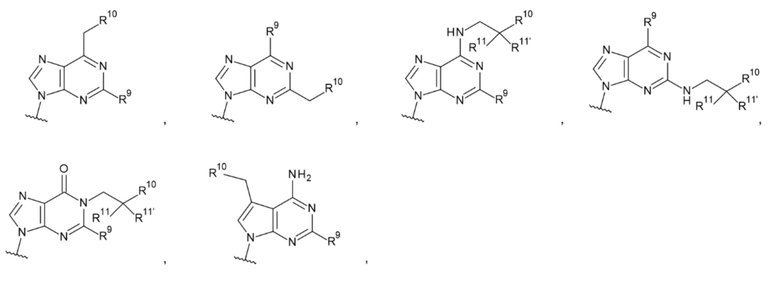
где R9 представляет собой атом водорода или -NH2;
R10 представляет собой гидроксигруппу, -NH2, -NHC(=O)CH2OH, -CH2NHC(=O)CH2OH, -CH2CH2NHC(=O)CH2OH, гидрокси-C1-C3 алкильную группу или амино-C1-C3 алкильную группу; и
R11 и R11' каждый независимо представляет собой атом водорода или метильную группу.
12. Конъюгат антитела с лекарственным средством по любому из пп. 1-10, отличающийся тем, что L1 представляет собой группу, выбранную из группы, состоящей из следующих формул:
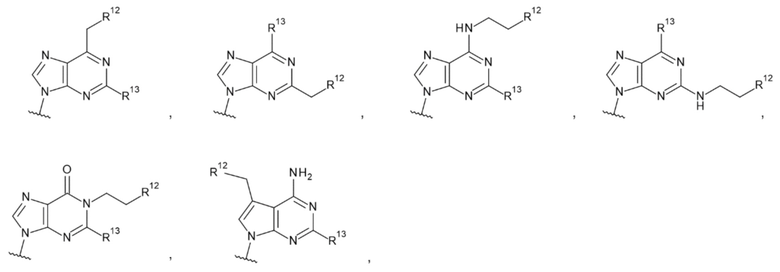
где R13 представляет собой атом водорода или -NH2; и
R12 представляет собой гидроксигруппу, -NH2, -CH2OH, -NHC(=O)CH2OH, -CH2NHC(=O)CH2OH или -CH2CH2NHC(=O)CH2OH.
13. Конъюгат антитела с лекарственным средством по любому из пп. 1-10, отличающийся тем, что L1 представляет собой группу, выбранную из группы, состоящей из следующих формул:
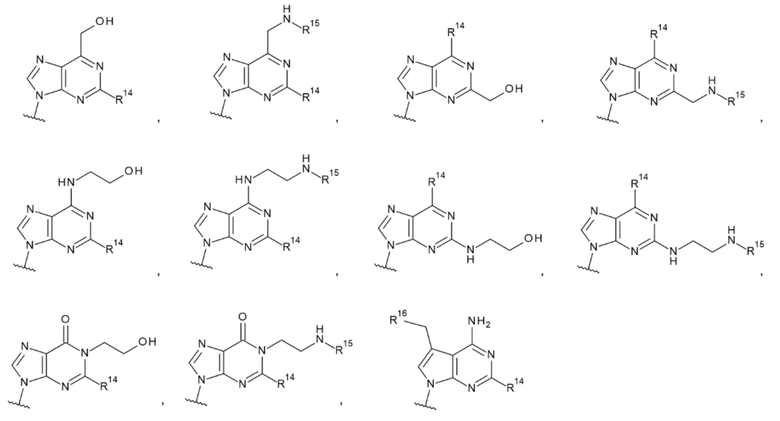
где R14 представляет собой атом водорода или -NH2;
R15 представляет собой атом водорода или -C(=O)CH2OH; и
R16 представляет собой гидроксигруппу, -NH2, -CH2OH, -CH2CH2OH, -CH2NH2 или -CH2CH2NH2.
14. Конъюгат антитела с лекарственным средством по любому из пп. 1-13, отличающийся тем, что Y1 и Y2 каждый представляет собой атом кислорода.
15. Конъюгат антитела с лекарственным средством по любому из пп. 1-14, отличающийся тем, что X5 и X6 представляют собой -CH2-O-.
16. Конъюгат антитела с лекарственным средством по любому из пп. 1-15, отличающийся тем, что R1, R2 и R3 каждый независимо представляет собой атом водорода, гидроксигруппу или атом фтора.
17. Конъюгат антитела с лекарственным средством по любому из пп. 1-16, отличающийся тем, что D представлен любой одной из следующих двух формул:
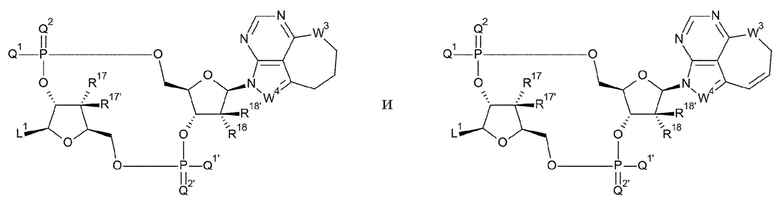 ,
,
где L1, Q1, Q1', Q2 и Q2' такие, как определено выше;
R17, R17', R18 и R18' каждый независимо представляет собой атом водорода, атом галогена, гидроксигруппу или -NH2;
W3 представляет собой -NH-, атом кислорода, атом серы; и
W4 представляет собой -CH= или атом азота.
18. Конъюгат антитела с лекарственным средством по п. 17, отличающийся тем, что D представлен любой одной из следующих двух формул:
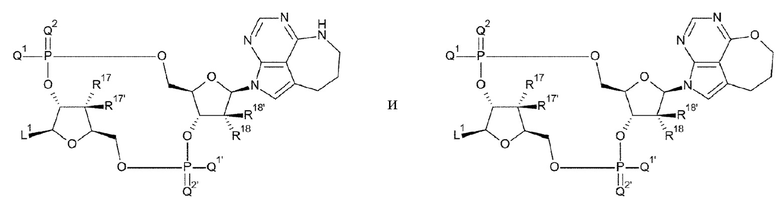 ,
,
где L1, Q1, Q1', Q2, Q2', R17, R17', R18 и R18' такие, как определено выше.
19. Конъюгат антитела с лекарственным средством по п. 17 или 18, отличающийся тем, что D представлен любой одной из следующих восьми формул:
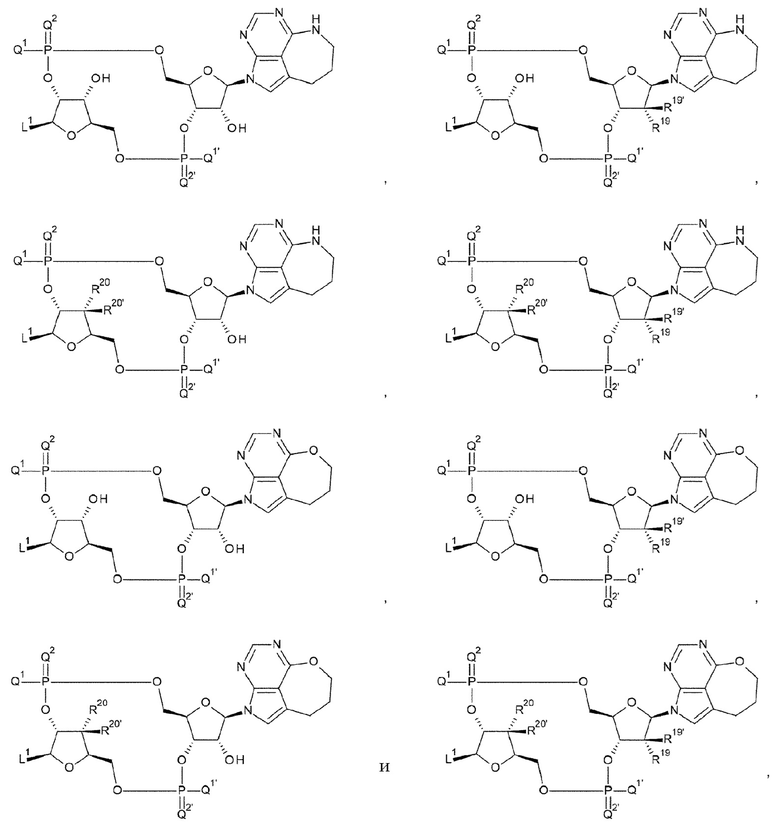
где L1, Q1, Q1', Q2 и Q2' такие, как определено выше; и
R19, R19', R20 и R20' каждый независимо представляет собой атом водорода или атом фтора.
20. Конъюгат антитела с лекарственным средством по любому из пп. 17-19, отличающийся тем, что D представлен любой одной из следующих четырех формул:
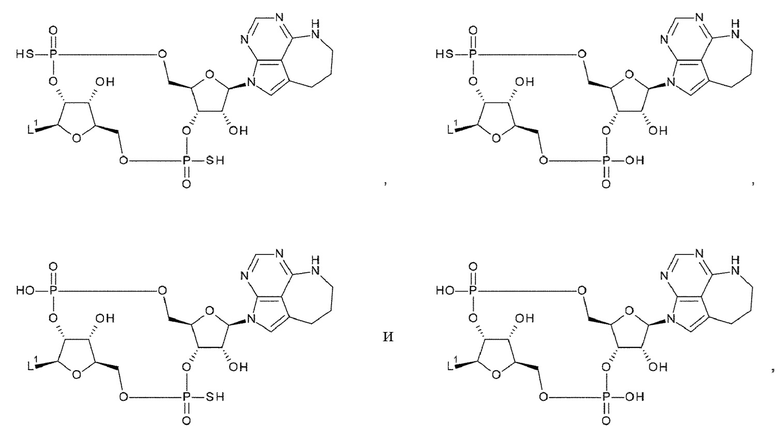
где L1 является таким, как определено выше.
21. Конъюгат антитела с лекарственным средством по любому из пп. 17-20, отличающийся тем, что D представлен любой одной из следующих четырех формул:
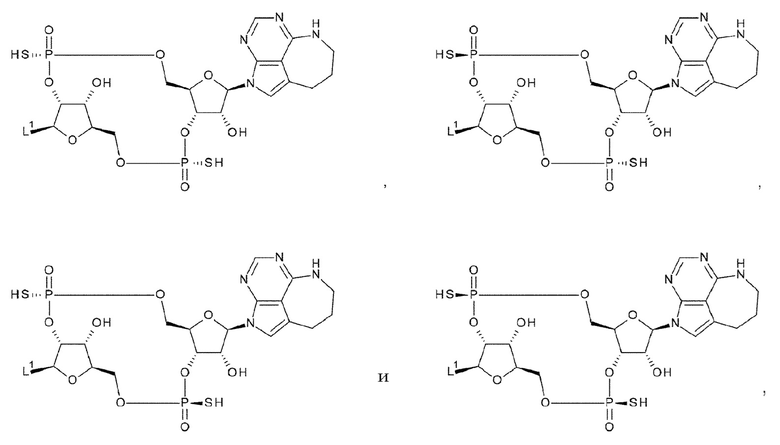
где L1 является таким, как определено выше.
22. Конъюгат антитела с лекарственным средством по любому из пп. 17-20, отличающийся тем, что D представлен любой одной из следующих четырех формул:
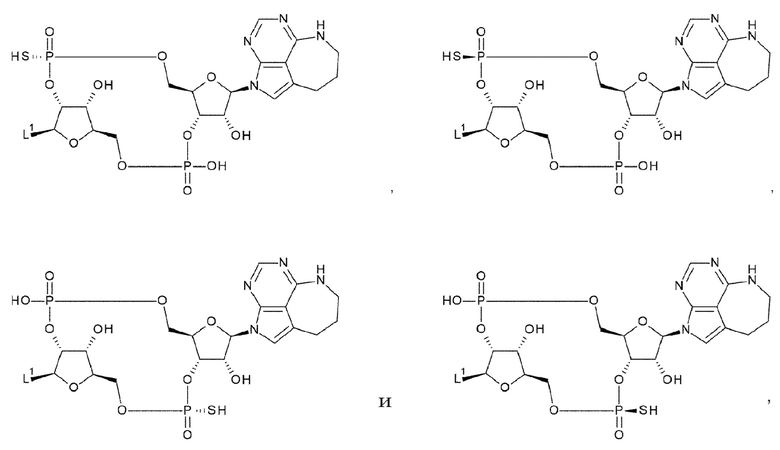
где L1 является таким, как определено выше.
23. Конъюгат антитела с лекарственным средством по любому из пп. 1-16, отличающийся тем, что D представлен следующей формулой:
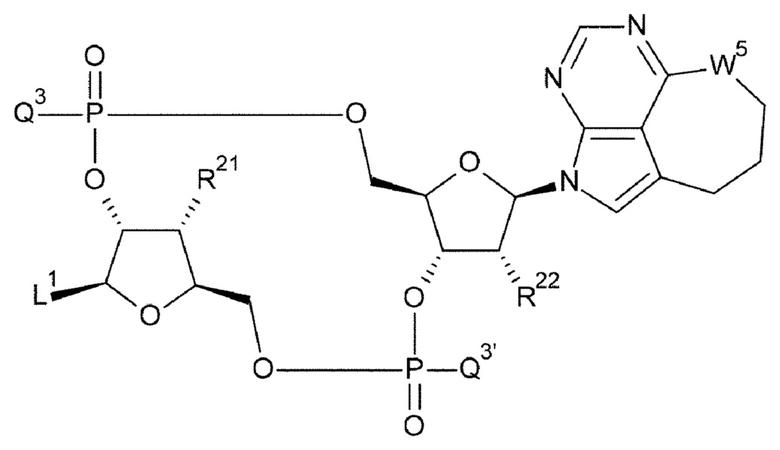 ,
,
где L1 является таким, как определено выше;
Q3 и Q3' каждый независимо представляет собой гидроксигруппу или тиольную группу;
R21 и R22 каждый независимо представляет собой гидроксигруппу или атом фтора; и
W5 представляет собой -NH- или атом серы.
24. Конъюгат антитела с лекарственным средством по п. 23, отличающийся тем, что D представлен любой одной из следующих двух формул:
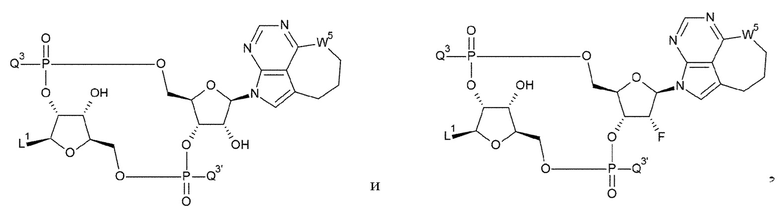
где L1, Q3, Q3' и W5 являются такими, как определено выше.
25. Конъюгат антитела с лекарственным средством по любому из пп. 1-24, отличающийся тем, что L1 представлен любой одной из следующих четырех формул:
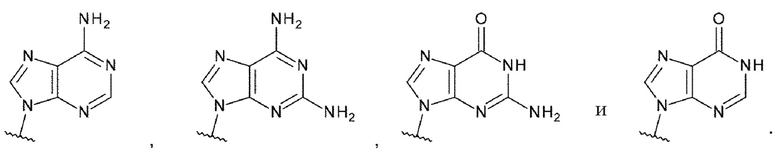
26. Конъюгат антитела с лекарственным средством по любому из пп. 1-24, отличающийся тем, что L1 представлен любой одной из следующих четырех формул:
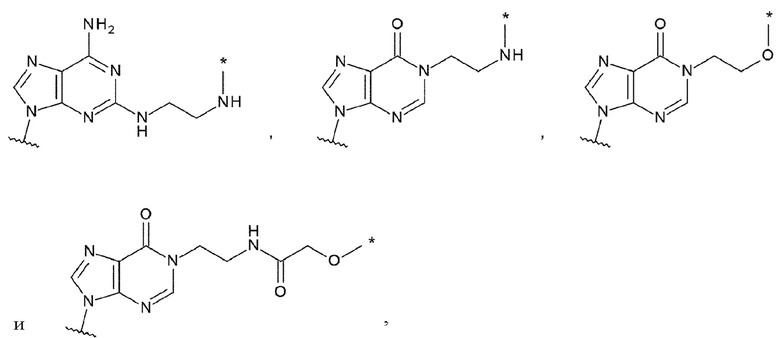
где каждая звездочка указывает на связывание с L.
27. Конъюгат антитела с лекарственным средством по любому из пп. 23, 24 и 26, отличающийся тем, что D представлен любой одной из следующих четырех формул:
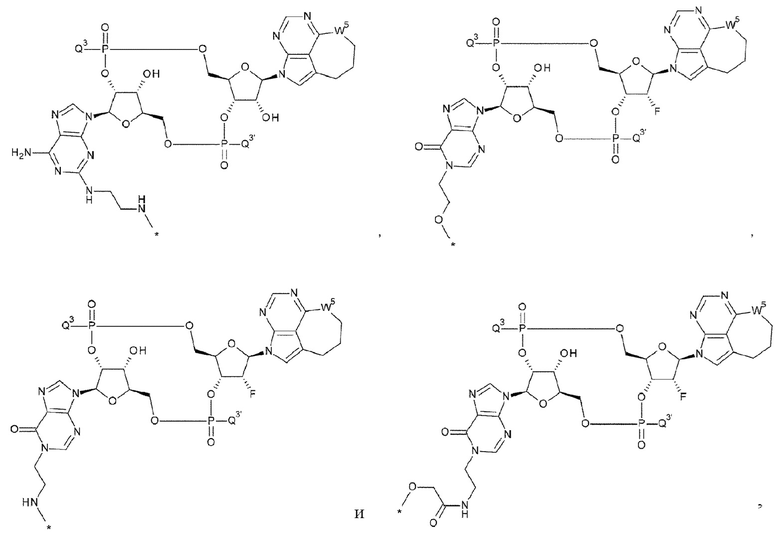
где каждая звездочка указывает на связывание с L; и
Q3, Q3' и W5 являются такими, как определено выше.
28. Конъюгат антитела с лекарственным средством по любому из пп. 23, 24, 26 и 27, отличающийся тем, что D представлен любой одной из следующих четырех формул:
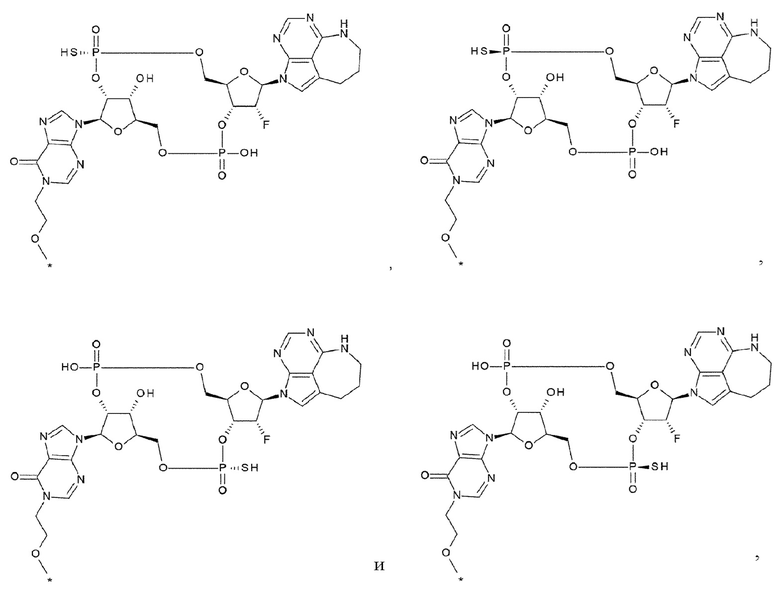
где каждая звездочка указывает на связывание с L.
29. Конъюгат антитела с лекарственным средством по любому из пп. 23, 24, 26 и 27, отличающийся тем, что D представлен любой одной из следующих трех формул:
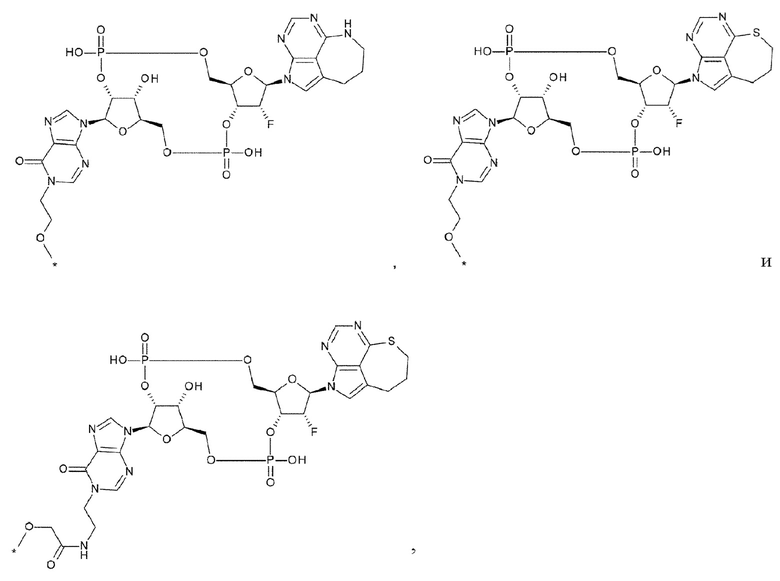
где каждая звездочка указывает на связывание с L.
30. Конъюгат антитела с лекарственным средством по любому из пп. 23, 24, 26 и 27, отличающийся тем, что D представлен любой одной из следующих четырех формул:
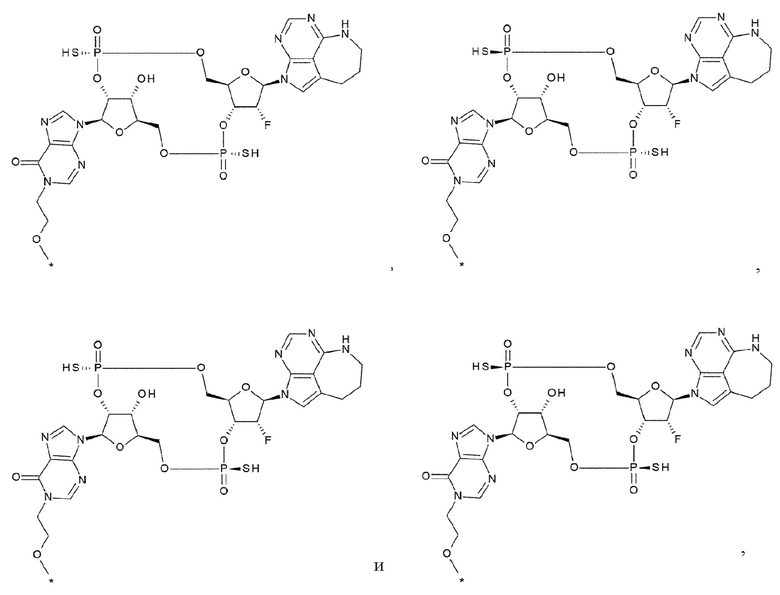
где каждая звездочка указывает на связывание с L.
31. Конъюгат антитела с лекарственным средством по любому из пп. 1-30, отличающийся тем, что линкер L представлен -Lb-La-Lp-Lc-*,
где звездочка указывает на связывание с лекарственным средством D;
Lp представляет собой линкер, состоящий из последовательности аминокислот, расщепляемой в целевой клетке, или отсутствует;
La представляет собой любую одну группу, выбранную из следующих групп:
-C(=O)-(CH2CH2)n2-C(=O)-,
-C(=O)-(CH2CH2)n2-CH2-C(=O)-,
-C(=O)-(CH2CH2)n2-C(=O)-NH-(CH2CH2)n3-C(=O)-,
-C(=O)-(CH2CH2)n2-C(=O)-NH-(CH2CH2)n3-CH2-C(=O)-,
-C(=O)-(CH2CH2)n2-C(=O)-NH-(CH2CH2O)n3-CH2-C(=O)-,
-(CH2)n4-O-C(=O)- и
-(CH2)n9-C(=O)-,
где n2 представляет собой целое число от 1 до 3, n3 представляет собой целое число от 1 до 5, n4 представляет собой целое число от 0 до 2 и n9 представляет собой целое число от 2 до 7;
Lb представляет собой спейсер, связывающий La и гликан или ремоделированный гликан Ab, или спейсер, связывающий La и остаток цистеина Ab; и
Lc представляет собой -NH-CH2-, -NH-фенильная группа-CH2-O(C=O)- или -NH-гетероарильная группа-CH2-O(C=O)- или отсутствует.
32. Конъюгат антитела с лекарственным средством по п. 31, отличающийся тем, что Lc отсутствует.
33. Конъюгат антитела с лекарственным средством по п. 31, отличающийся тем, что Lc представляет собой -NH-CH2-.
34. Конъюгат антитела с лекарственным средством по любому из пп. 31-33, отличающийся тем, что Lp представляет собой любой один, выбранный из группы, состоящей из:
-GGVA-, -VA-, -GGFG-, -FG-, -GGPI-, -PI-, -GGVCit-, -VCit-, -GGVK-, -VK-, -GGFCit-, -FCit-, -GGFM-, -FM-, -GGLM-, -LM-, -GGICit- и -ICit-.
35. Конъюгат антитела с лекарственным средством по п. 34, отличающийся тем, что Lp представляет собой любой один из -GGVA-, -VA-, -GGFG-, -FG-, -GGVCit-, -VCit-, -GGFCit- и -FCit-.
36. Конъюгат антитела с лекарственным средством по любому из пп. 31-33, отличающийся тем, что Lp представляет собой любой один из -GGFG-, -GGPI-, -GGVA-, -GGFM-, -GGVCit-, -GGFCit-, -GGICit-, -GGPL-, -GGAQ- и -GGPP-.
37. Конъюгат антитела с лекарственным средством по п. 36, отличающийся тем, что Lp представляет собой -GGFG- или -GGPI-.
38. Конъюгат антитела с лекарственным средством по любому из пп. 31-37, отличающийся тем, что La представляет собой любой один, выбранный из группы, состоящей из:
-C(=O)-CH2CH2-C(=O)-,
-C(=O)-CH2CH2-C(=O)-NH-(CH2CH2O)3-CH2-C(=O)-,
-C(=O)-CH2CH2-C(=O)-NH-(CH2CH2O)4-CH2-C(=O)- и
-(CH2)5-C(=O)-.
39. Конъюгат антитела с лекарственным средством по любому из пп. 31-38, отличающийся тем, что Lb представлен любой одной из следующих формул:
 и
и
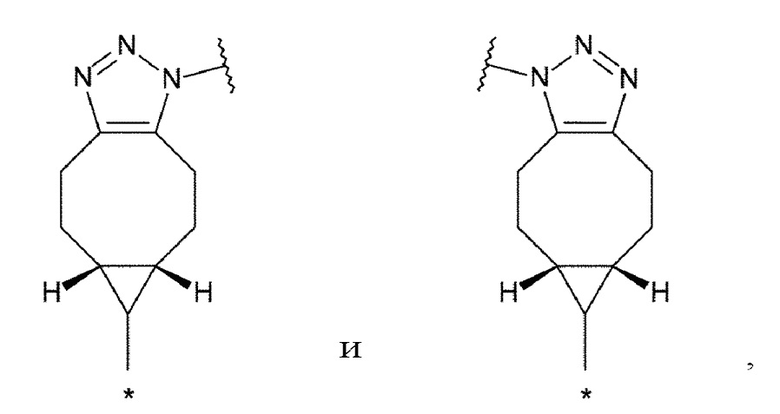
где в структурных формулах для Lb, представленных выше,
каждая звездочка указывает на связывание с La и каждая волнистая линия указывает на связывание с гликаном или ремоделированным гликаном Ab.
40. Конъюгат антитела с лекарственным средством по любому из пп. 31-38, отличающийся тем, что Lb представляет собой -(сукцинимид-3-ил-N)-, где -(сукцинимид-3-ил-N)- представляет собой следующую структурную формулу:
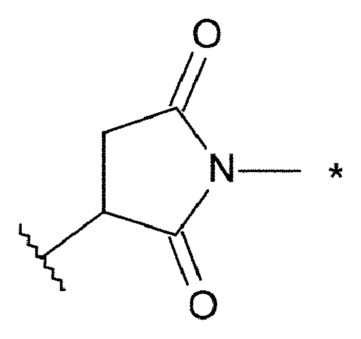 ,
,
где звездочка указывает на связывание с La и волнистая линия указывает на связывание с боковой цепью остатка цистеина антитела посредством образования тиоэфира.
41. Конъюгат антитела с лекарственным средством по любому из пп. 31 и 36-39, отличающийся тем, что линкер L представлен -Lb-La-Lp-Lc-*,
где звездочка указывает на связывание с лекарственным средством D;
Lp представляет собой -GGFG- или -GGPI-;
La представляет собой -C(=O)-CH2CH2-C(=O)-;
Lb представлен следующей формулой:
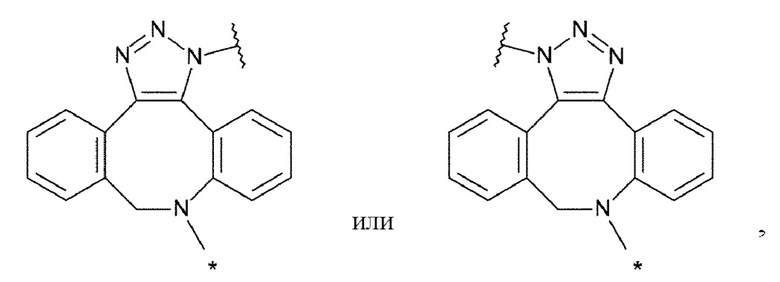
где в структурных формулах для Lb, представленных выше,
каждая звездочка указывает на связывание с La и каждая волнистая линия указывает на связывание с гликаном или ремоделированным гликаном Ab; и
Lc представляет собой -NH-CH2-.
42. Конъюгат антитела с лекарственным средством по любому из пп. 1-41, отличающийся тем, что среднее количество конъюгированных молекул лекарственного средства на молекулу антитела в конъюгате антитела с лекарственным средством находится в диапазоне от 1 до 10.
43. Конъюгат антитела с лекарственным средством по п. 42, отличающийся тем, что среднее количество конъюгированных молекул лекарственного средства на молекулу антитела в конъюгате антитела с лекарственным средством находится в диапазоне от 1 до 5.
44. Конъюгат антитела с лекарственным средством по любому из пп. 1-39 и 41-43 отличающийся тем, что антитело связывается посредством гликана, связанного с Asn297 антитела (N297-гликана), с L.
45. Конъюгат антитела с лекарственным средством по п. 44, отличающийся тем, что N297-гликан представляет собой ремоделированный гликан.
46. Конъюгат антитела с лекарственным средством по п. 44 или 45, отличающийся тем, что N297-гликан представляет собой N297-(Fuc)MSG1 или N297-(Fuc)SG.
47. Конъюгат антитела с лекарственным средством по любому из пп. 1-46, отличающийся тем, что антитело или антигенсвязывающий фрагмент антитела представляет собой антитело против HER2, антитело против HER3, антитело против DLL3, антитело против FAP, антитело против CDH11, антитело против CDH6, антитело против A33, антитело против CanAg, антитело против CD19, антитело против CD20, антитело против CD22, антитело против CD30, антитело против CD33, антитело против CD56, антитело против CD70, антитело против CD98, антитело против TROP2, антитело против CEA, антитело против Cripto, антитело против EphA2, антитело против G250, антитело против MUC1, антитело против GPNMB, антитело против интегрина, антитело против PSMA, антитело против тенасцина-C, антитело против SLC44A4, антитело против мезотелина, антитело против ENPP3, антитело против CD47, антитело против EGFR, антитело против GPR20 или антитело против DR5 или антигенсвязывающий фрагмент указанного антитела.
48. Конъюгат антитела с лекарственным средством по любому из пп. 1-47, отличающийся тем, что антитело или антигенсвязывающий фрагмент антитела представляет собой любое, выбранное из группы, состоящей из IgG, представляющего собой IgG1, IgG2, IgG3 или IgG4, IgE, IgM, IgD, IgA, представляющего собой IgA1 или IgA2, и IgY.
49. Конъюгат антитела с лекарственным средством по п. 48, отличающийся тем, что антитело или антигенсвязывающий фрагмент антитела представляет собой IgG1.
50. Конъюгат антитела с лекарственным средством по п. 49, отличающийся тем, что часть аминокислотных остатков в константной области заменены.
51. Конъюгат антитела с лекарственным средством по п. 50, отличающийся тем, что замена включает замену лейцина аланином в положениях 234 и 235, соответствующих нумерации EU-индекс.
52. Конъюгат антитела с лекарственным средством по любому из пп. 1-51, отличающийся тем, что антитело или антигенсвязывающий фрагмент антитела представляет собой белок, полученный в клетках-хозяевах с применением генетически модифицированного гена антитела.
53. Конъюгат антитела с лекарственным средством по п. 47, отличающийся тем, что антитело или антигенсвязывающий фрагмент антитела представляет собой антитело против HER2 или антигенсвязывающий фрагмент указанного антитела.
54. Конъюгат антитела с лекарственным средством по п. 53, отличающийся тем, что антитело или антигенсвязывающий фрагмент антитела представляет собой одно из следующих (i)-(iii):
(i) антитело, содержащее легкую цепь, состоящую из последовательности аминокислот, представленной в SEQ ID NO: 1, и тяжелую цепь, состоящую из последовательности аминокислот, представленной в SEQ ID NO: 2, или антигенсвязывающий фрагмент указанного антитела, или антитело, содержащее легкую цепь, состоящую из последовательности аминокислот, представленной в SEQ ID NO: 1, и тяжелую цепь, состоящую из последовательности аминокислот, представленной в SEQ ID NO: 3, или антигенсвязывающий фрагмент указанного антитела;
(ii) антитело или антигенсвязывающий фрагмент антитела, как определено в (i), причем часть аминокислотных остатков в константной области заменена; и
(iii) белок, полученный в клетках-хозяевах с применением гена (a), модифицированного методами генной инженерии из гена (b) антитела или антигенсвязывающего фрагмента антитела, как определено в (i).
55. Конъюгат антитела с лекарственным средством по п. 53, отличающийся тем, что антитело или антигенсвязывающий фрагмент антитела представляет собой одно из следующих (i)-(iii):
(i) антитело, содержащее легкую цепь, состоящую из последовательности аминокислот, представленной в SEQ ID NO: 28, и тяжелую цепь, состоящую из последовательности аминокислот, представленной в SEQ ID NO: 29, или антигенсвязывающий фрагмент указанного антитела, или антитело, содержащее легкую цепь, состоящую из последовательности аминокислот, представленной в SEQ ID NO: 28, и тяжелую цепь, состоящую из последовательности аминокислот, представленной в SEQ ID NO: 30, или антигенсвязывающий фрагмент указанного антитела;
(ii) антитело или антигенсвязывающий фрагмент антитела, как определено в (i), причем часть аминокислотных остатков в константной области заменена; и
(iii) белок, полученный в клетках-хозяевах с применением гена (a), модифицированного методами генной инженерии из гена (b) антитела или антигенсвязывающего фрагмента антитела, как определено в (i).
56. Конъюгат антитела с лекарственным средством по пп. 54 или 55, отличающийся тем, что замена включает замену лейцина аланином в положениях 234 и 235, определенных согласно нумерации EU-индекс.
57. Конъюгат антитела с лекарственным средством по любому из пп. 53-56, отличающийся тем, что одна или две аминокислоты удалены на карбоксильном конце тяжелой цепи антитела или антигенсвязывающего фрагмента антитела.
58. Конъюгат антитела с лекарственным средством по п. 47, отличающийся тем, что антитело представляет собой антитело, содержащее легкую цепь, состоящую из последовательности аминокислот, представленной в SEQ ID NO: 31, и тяжелую цепь, состоящую из последовательности аминокислот, представленной в SEQ ID NO: 32, антитело, содержащее легкую цепь, состоящую из последовательности аминокислот, представленной в SEQ ID NO: 33, и тяжелую цепь, состоящую из последовательности аминокислот, представленной в SEQ ID NO: 34, или антитело, содержащее легкую цепь, состоящую из последовательности аминокислот, представленной в SEQ ID NO: 35, и тяжелую цепь, состоящую из последовательности аминокислот, представленной в SEQ ID NO: 36.
59. Конъюгат антитела с лекарственным средством по любому из пп. 1-58, отличающийся тем, что указанный конъюгат антитела с лекарственным средством обладает активностью агониста STING.
60. Фармацевтическая композиция для лечения заболеваний, связанных с активностью агониста STING, содержащая эффективное количество конъюгата антитела с лекарственным средством по любому из пп. 1-58 и один или более фармацевтически приемлемых вспомогательных веществ.
61. Конъюгат антитела с лекарственным средством по любому из пп. 1-58, обладающий противоопухолевой активностью.
62. Конъюгат антитела с лекарственным средством по п. 61, отличающийся тем, что опухоль представляет собой рак легких, рак почки, уротелиальный рак, колоректальный рак, рак предстательной железы, мультиформную глиобластому, рак яичников, рак поджелудочной железы, рак молочной железы, меланому, рак печени, рак мочевого пузыря, рак желудка, рак пищевода, рак эндометрия, рак яичка, рак шейки матки, плацентарную хориокарциному, мультиформную глиобластому, опухоль головного мозга, рак головы и шеи, рак щитовидной железы, мезотелиому, стромальную опухоль желудочно-кишечного тракта (GIST), рак желчного пузыря, рак желчного протока, рак надпочечников, плоскоклеточную карциному, лейкоз, злокачественную лимфому, плазмоцитому, миелому или саркому.
63. Способ лечения рака, связанного с активностью агониста STING, где указанный способ включает введение эффективного количества конъюгата антитела с лекарственным средством по любому из пп. 1-59, 61-62 или фармацевтической композиции по п. 60.
64. Способ по п. 63, отличающийся тем, что рак представляет собой рак легких, рак почки, уротелиальный рак, колоректальный рак, рак предстательной железы, мультиформную глиобластому, рак яичников, рак поджелудочной железы, рак молочной железы, меланому, рак печени, рак мочевого пузыря, рак желудка, рак пищевода, рак эндометрия, рак яичка, рак шейки матки, плацентарную хориокарциному, мультиформную глиобластому, опухоль головного мозга, рак головы и шеи, рак щитовидной железы, мезотелиому, стромальную опухоль желудочно-кишечного тракта (GIST), рак желчного пузыря, рак желчного протока, рак надпочечников, плоскоклеточную карциному, лейкоз, злокачественную лимфому, плазмоцитому, миелому или саркому.
65. Конъюгат антитела с лекарственным средством, представленный формулой (II)
,
где m1 находится в диапазоне от 3 до 5;
Ab представляет собой антитело против HER2, где гликан антитела ремоделирован;
L представляет собой линкер, соединяющий Ab и D;
где линкер L представлен -Lb-La-Lp-Lc-*,
где звездочка указывает на связывание с лекарственным средством D;
Lp представляет собой -GGFG-;
La представляет собой -C(=O)-CH2CH2-C(=O)-;
Lb представлен следующей формулой:
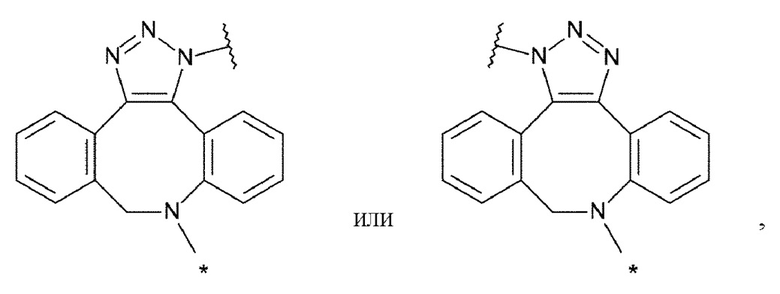
где в структурных формулах для Lb, представленных выше,
каждая звездочка указывает на связывание с La и каждая волнистая линия указывает на связывание с ремоделированным гликаном Ab; и
Lc представляет собой -NH-CH2-;
Ab связывается посредством гликана, связанного с Asn297 антитела (N297-(Fuc)SG), с L; и
D представляет собой соединение, представленное формулой (I)
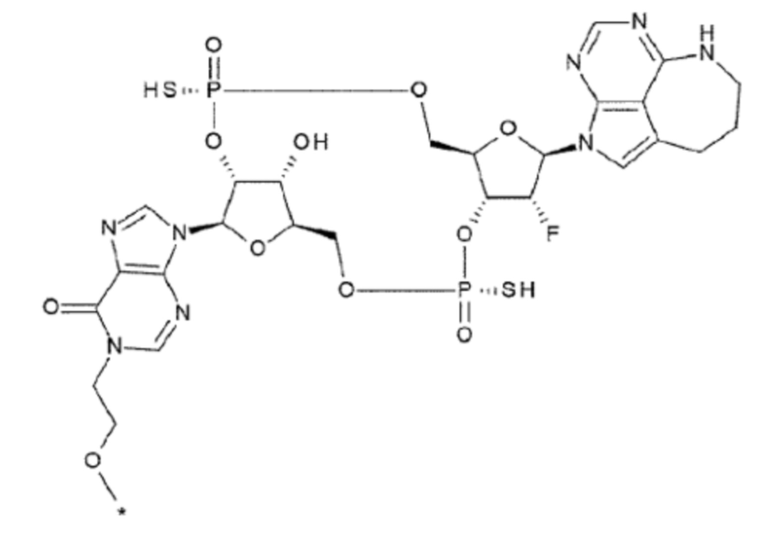 ,
,
где звездочка указывает на связывание с L.
66. Конъюгат антитела с лекарственным средством по п. 65, отличающийся тем, что антитело представляет собой любое из следующих (i)-(iii):
(i) антитело, содержащее легкую цепь, состоящую из последовательности аминокислот, представленной в SEQ ID NO: 28, и тяжелую цепь, состоящую из последовательности аминокислот, представленной в SEQ ID NO: 29, или антитело, содержащее легкую цепь, состоящую из последовательности аминокислот, представленной в SEQ ID NO: 28, и тяжелую цепь, состоящую из последовательности аминокислот, представленной в SEQ ID NO: 30;
(ii) антитело, как определено в (i), причем часть аминокислотных остатков в константной области заменена; и
(iii) белок, полученный в клетках-хозяевах с применением гена (a), модифицированного из гена методами генной инженерии (b) антитела, как определено в (i).
67. Конъюгат антитела с лекарственным средством по п. 66, отличающийся тем, что замена включает замену лейцина аланином в положениях 234 и 235, определенных согласно нумерации EU-индекс.
68. Конъюгат антитела с лекарственным средством по п. 65 или 66, отличающийся тем, что антитело представляет собой антитело, содержащее легкую цепь, состоящую из последовательности аминокислот, представленной в SEQ ID NO: 28, и тяжелую цепь, состоящую из последовательности аминокислот, представленной в SEQ ID NO: 29,
причем часть аминокислотных остатков в константной области антитела заменена.
69. Конъюгат антитела с лекарственным средством по п. 68, отличающийся тем, что антитело представляет собой IgG1.
70. Конъюгат антитела с лекарственным средством по п. 65 или 66, отличающийся тем, что антитело представляет собой антитело, содержащее легкую цепь, состоящую из последовательности аминокислот, представленной в SEQ ID NO: 28, и тяжелую цепь, состоящую из последовательности аминокислот, представленной в SEQ ID NO: 29,
причем константная область антитела содержит замену лейцина аланином в положениях 234 и 235, определенных согласно нумерации EU-индекс.
71. Конъюгат антитела с лекарственным средством по п. 65 или 66, отличающийся тем, что антитело представляет собой антитело, содержащее легкую цепь, состоящую из последовательности аминокислот, представленной в SEQ ID NO: 28, и тяжелую цепь, состоящую из последовательности аминокислот, представленной в SEQ ID NO: 30,
причем часть аминокислотных остатков в константной области антитела заменена.
72. Конъюгат антитела с лекарственным средством по п. 71, отличающийся тем, что антитело представляет собой IgG1.
73. Конъюгат антитела с лекарственным средством по п. 65 или 66, отличающийся тем, что антитело представляет собой антитело, содержащее легкую цепь, состоящую из последовательности аминокислот, представленной в SEQ ID NO: 28, и тяжелую цепь, состоящую из последовательности аминокислот, представленной в SEQ ID NO: 30,
причем константная область антитела содержит замену лейцина аланином в положениях 234 и 235, определенных согласно нумерации EU-индекс.
74. Конъюгат антитела с лекарственным средством по любому из пп. 65-73, отличающийся тем, что одна или две аминокислоты удалены на карбоксильном конце тяжелой цепи антитела.
75. Конъюгат антитела с лекарственным средством по любому из пп. 65-74, отличающийся тем, что указанный конъюгат антитела с лекарственным средством обладает активностью агониста STING.
76. Фармацевтическая композиция для лечения заболеваний, связанных с активностью агониста STING, содержащая эффективное количество конъюгата антитела с лекарственным средством по любому из пп. 65-74 и один или более фармацевтически приемлемых вспомогательных веществ.
77. Конъюгат антитела с лекарственным средством по любому из пп. 65-74, обладающий противоопухолевой активностью.
78. Конъюгат антитела с лекарственным средством по п. 77, отличающийся тем, что опухоль представляет собой рак легких, рак почки, уротелиальный рак, колоректальный рак, рак предстательной железы, мультиформную глиобластому, рак яичников, рак поджелудочной железы, рак молочной железы, меланому, рак печени, рак мочевого пузыря, рак желудка, рак пищевода, рак эндометрия, рак яичка, рак шейки матки, плацентарную хориокарциному, мультиформную глиобластому, опухоль головного мозга, рак головы и шеи, рак щитовидной железы, мезотелиому, стромальную опухоль желудочно-кишечного тракта (GIST), рак желчного пузыря, рак желчного протока, рак надпочечников, плоскоклеточную карциному, лейкоз, злокачественную лимфому, плазмоцитому, миелому или саркому.
79. Конъюгат антитела с лекарственным средством по п. 77 или 78, отличающийся тем, что опухоль представляет собой рак легких, колоректальный рак, рак предстательной железы, рак яичников, рак поджелудочной железы, рак молочной железы, рак мочевого пузыря, рак желудка, рак пищевода, рак эндометрия, рак шейки матки, рак головы и шеи, рак желчного протока, миелому или саркому.
80. Способ лечения рака, связанного с активностью агониста STING, где указанный способ включает введение эффективного количества конъюгата антитела с лекарственным средством по любому из пп. 65-75 и 77-79 или фармацевтической композиции по п. 76.
81. Способ по п. 80, отличающийся тем, что рак представляет собой рак легких, рак почки, уротелиальный рак, колоректальный рак, рак предстательной железы, мультиформную глиобластому, рак яичников, рак поджелудочной железы, рак молочной железы, меланому, рак печени, рак мочевого пузыря, рак желудка, рак пищевода, рак эндометрия, рак яичка, рак шейки матки, плацентарную хориокарциному, мультиформную глиобластому, опухоль головного мозга, рак головы и шеи, рак щитовидной железы, мезотелиому, стромальную опухоль желудочно-кишечного тракта (GIST), рак желчного пузыря, рак желчного протока, рак надпочечников, плоскоклеточную карциному, лейкоз, злокачественную лимфому, плазмоцитому, миелому или саркому.
82. Способ по п. 80 или 81, отличающийся тем, что рак представляет собой рак легких, колоректальный рак, рак предстательной железы, рак яичников, рак поджелудочной железы, рак молочной железы, рак мочевого пузыря, рак желудка, рак пищевода, рак эндометрия, рак шейки матки, рак головы и шеи, рак желчного протока, миелому или саркому.
| WO 2017161349 A1, 21.09.2017 | |||
| WO 2017027645 A1, 16.02.2017 | |||
| ПОДВИЖНОЙ ВОЗВЫШЕННЫЙ КОНТРРЕЛЬС ТУПОЙ КРЕСТОВИНЫ | 1928 |
|
SU29856A1 |
| WO 2018100558 A2,07.06.2018. | |||
