Область техники, к которой относится изобретение
Перекрестная ссылка на родственные заявки
Настоящая заявка испрашивает приоритет описания заявки на патент Японии 2013-172746 (полное раскрытие включено в настоящее описание изобретения путем ссылки), поданной 22 августа 2013 г.
Настоящее изобретение относится к хинолин-замещенным соединениям, обладающим ингибирующим действием в отношении рецептора эпидермального фактора роста (EGFR), и фармацевтическим композициям, содержащим эти соединения в качестве активного ингредиента.
Предпосылки создания изобретения
EGFR представляет собой тирозинкиназу рецепторного типа, проявляет свою физиологическую функцию в нормальной ткани, когда связан с эпидермальным фактором роста (EGF), который является лигандом, и способствует росту, ингибированию апоптоза и т.д. в эпителиальных тканях (непатентный документ (NPD) 1).
В дополнение, EGFR является одним из онкогенов, и амплификация гена EGFR и высокая экспрессия или мутация его белка найдены в различных типах раковых заболеваний, как например рак головы и шеи, рак молочной железы, колоректальный рак, рак пищевода, рак поджелудочной железы, рак легких, овариальный рак, рак почек, рак мочевого пузыря, рак кожи и опухоль головного мозга (непатентный документ (NPD) 2). В Японии и западных странах приблизительно от 170 до 375 человек на каждые 100000 населения умирают вследствие рака каждый год и раковое заболевание занимает высокое место в качестве причины смерти (непатентный документ (NPD) 3). Среди типов раковых заболеваний, смертельный исход вследствие рака легких составляет приблизительно 1400000 в год во всем мире, и так как немелкоклеточный рак легких составляет 80% или более раковых заболеваний легких существует потребность в разработке эффективной терапии для этого заболевания (непатентный документ (NPD) 4).
В последние годы были идентифицированы ответственные за эти раковые заболевания гены, и мутация в гене EGFR также является одной из них и приводит к активному мутированному белку EGFR. Активный мутированный белок EGFR представляет собой, например, делецию аминокислот в положениях 746-750 (EGFR (d746-750)), мутацию аминокислоты в положении 858 от лейцина до аргинина (EGFR (L858R)) или т.п. О таких мутациях сообщалось, например, в 20-40% случаев немелкоклеточного рака легких в Японии и в 10-15% случаев немелкоклеточного рака легких в западных странах. Так как немелкоклеточный рак легких, имеющий эти мутации, является высокочувствительным по отношению к гефитинибу (название продукта: Iressa (зарегистрированный торговый знак)) и эрлотинибу (название продукта: Tarceva (зарегистрированный торговый знак)), которые представляют собой химические агенты (ингибиторы EGFR), ингибирующие киназную активность EGFR, эти химические агенты использовали в качестве терапевтических агентов в Японии и западных странах. Однако, раковое заболевание приобретает резистентность по отношению к гефитинибу и эрлотинибу спустя от 6 до 12 месяцев с начала использования и терапевтический эффект становится слабым. Поэтому эта приобретенная резистентность представляет собой серьезную проблему для лечения немелкоклеточного рака легких, имеющего высокочувствительный мутированный EGFR. Обнаружено, что приблизительно 50% приобретенной резистентности обусловлены появлением резистентного мутированного белка EGFR (EGFR (d746-750/T790M) или EGFR (T790M/L858R)), имеющего вторую мутацию в гене EGFR, приводящую к замене аминокислоты в положении 790 треонина на метионин. Важной задачей является нахождение терапевтического агента, который является эффективным против немелкоклеточного рака легких, имеющего этот резистентный к лекарственному средству мутированный EGFR (непатентный документ (NPD) 5).
С другой стороны, сообщалось о анормальности кожи и нарушении пищеварительного тракта как о обычных побочных эффектах ингибиторов EGFR гефитиниба и эрлотиниба, которые клинически используют в качестве терапевтических агентов в настоящее время, и ингибиторов EGFR, как например BIBW2992 и т.д., которые используют при клиническом исследовании. Во всех случаях полагают, что эти побочные эффекты вызваны ингибиторами EGFR, ингибирующими активность не только мутированного EGFR, экспрессируемого в случае немелкоклеточного рака легких, но и также активность EGFR дикого типа (EGFR (WT)), экспрессируемого в коже или пищеварительном тракте (непатентный документ (NPD) 1). С точки зрения уменьшения побочного эффекта считают предпочтительным иметь слабую ингибирующую активность по отношению к EGFR (WT) в нормальных тканях.
Таким образом, существует вероятность возможного подавления роста клеток немелкоклеточного рака легких, имеющих резистентный к лекарственному средству мутированный EGFR, за счет введения химического агента, обладающего более слабой ингибирующей активностью по отношению к EGFR дикого типа по сравнению с ингибирующей активностью по отношению к резистентному к лекарственному средству мутированному EGFR, аминокислота которого в положении 790 мутирована до метионина, при вводимой дозе, где побочный эффект на коже или в пищеварительном тракте сильно не проявляется. Это прогнозировано для способствования лечению ракового заболевания и продолжительной жизни и улучшения коэффициента QOL пациентов. В дополнение, если химический агент обладает слабой ингибирующей активностью по отношению к EGFR дикого типа, однако, является сильным по ингибирующей активности не только по отношению к резистентному к лекарственному средству мутированному EGFR, но и также по отношению к высокочувствительным мутированным EGFRs, как например EGFR (d746-750) и EGFR (L858R) и т.д., которые являются высокочувствительными по отношению к гефитинибу и эрлотинибу; существует вероятность возможного подавления роста клеток немелкоклеточного рака легких, экспрессирующих высокочувствительный мутированный EGFR или резистентный к лекарственному средству мутированный EGFR, при вводимой дозе, где побочный эффект на коже или в пищеварительном тракте сильно не проявляется, или вероятность возможного уменьшения частоты появления резистентного к лекарственному средству EGFR, в качестве приобретенной резистентности, от клеток немелкоклеточного рака легких, экспрессирующих высокочувствительный мутированный EGFR. Это прогнозировано для способствования лечению ракового заболевания и продолжительной жизни и улучшения коэффициента QOL пациентов. Кроме того, так как экспрессии высокочувствительного мутированного EGFR и резистентного к лекарственному средству мутированного EGFR могут быть использованы в современной области терапии в качестве показателей стратификации для возможного отбора пациентов, они значительно способствуют этому с этической точки зрения.
В качестве соединения, имеющего структуру, аналогичную соединению согласно настоящему изобретению, известно производное N-(3-(4-амино-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-5-ил)фенил)бензамид (патентный документ (PTD) 1). Хотя в патентном документе 1 описывается использование амидного соединения для лечения заболеваний, характеризующихся B-RAF-киназой, в документе не раскрываются конкретные тесты и их результаты, подтверждающие ингибирующую киназу активность, и также не подтверждена активность.
Перечень ссылок
Патентные документы
PTD 1: Международная публикация WO 2006/102079, брошюра
Непатентные документы
NPD 1: Nature Rev. Cancer, том 6, сс. 803-811 (2006)
NPD 2: J. Clin. Oncol., том 19, сс. 32-40 (2001)
NPD 3: Ministry of Internal Affairs and Communications Statistics Bureau homehage/statistical data/world statistics “World Statistics 2011”
NPD 4: Lung Cancer, том 69, сс. 1-12 (2010)
NPD 5: Nature Rev. Cancer, том 10, сс. 760-774 (2010)
Краткое изложение сущности изобретения
Техническая проблема
Как описано выше, ингибиторы EGFR, хотя полагают, что являются эффективными в терапии ракового заболевания, в настоящее время являются клинически недостаточно эффективными.
Поэтому, целью настоящего изобретения является получение нового соединения, которое сильно ингибирует EGFR, или его соли. Дальнейшей целью настоящего изобретения является получение нового соединения, которое ингибирует мутированный EGFR, например, EGFR (d746-750), EGFR (L858R), EGFR (d746-750/T790M) и EGFR (T790M/L858R), но не ингибирует EGFR (WT); или его соли.
Решение проблемы
Авторы настоящего изобретения провели всестороннее исследование для достижения вышеуказанной цели. В результате, авторами настоящего изобретения найдено, что группа хинолин-замещенных соединений согласно настоящему изобретению обладает превосходной ингибирующей активностью в отношении EGFR и проявляет ингибирующее рост раковой клетки действие, и эти соединения пригодны в качестве лекарственного средства для лечения ракового заболевания. Авторы настоящего изобретения, таким образом, пришли к настоящему изобретению.
Таким образом, в настоящем изобретении предусмотрены следующие пункты.
1. Соединение, представленное нижеприводимой формулой (I), или его соль:
где группа:
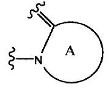
представляет собой
(1) группу, представленную формулой А1:
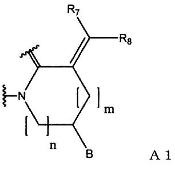
(в формуле А1, В означает группу, представленную формулой:
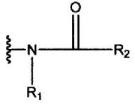
[R1 означает атом водорода или С1-С6-алкильную группу;
и R2 означает группу, представленную формулой:
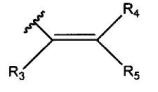
где R3, R4 и R5 являются одинаковыми или различными, и каждый означает атом водорода, атом галогена, С1-С6-алкильную группу, С6-С12-арильную группу, С4-С9-гетероарильную группу, аминометильную группу, которая может быть замещена С1-С6-алкильной групой, или 1-пиперидинометильную группу,
или группу, представленную формулой:

где R6 означает атом водорода или С1-С6-алкильную группу],
R7 и R8 являются одинаковыми или различными, и каждый означает атом водорода или С1-С6-алкильную группу; m означает 0 или 1; n означает 1 или 2);
(2) группу, представленную формулой А2:
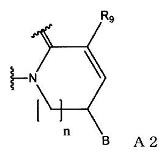
(в формуле А2, В и n имеют значения, как описано в формуле А1; и R9 означает атом водорода или С1-С6-алкильную группу); или
(3) группу, представленную формулой А3:
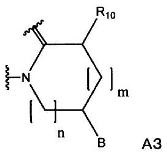
(в формуле А3, В, m и n имеют значения, как определено в формуле А1; и R10 означает С1-С6-алкильную группу).
2. Соединение, или его соль, по п.1, где R2 представляет собой группу, представленную формулой:
где R3, R4 и R5 являются одинаковыми или различными, и каждый означает атом водорода, атом галогена, С1-С6-алкильную группу, аминометильную группу, которая может быть замещена С1-С6-алкильной группой, или 1-пиперидинометильную группу,
или группу, представленную формулой:
где R6 означает атом водорода или С1-С6-алкильную группу.
3. Соединение, или его соль, по п.1 или 2, где R2 представляет собой группу, представленную формулой:
где R3, R4 и R5 являются одинаковыми или различными, и каждый означает атом водорода, атом галогена, аминометильную группу, которая может быть замещена метильной группой, или 1-пиперидинометильную группу.
4. Соединение или его соль по любому одному из пп.1-3, где группа:
представляет собой
(1) группу, представленную формулой А1:
(в формуле А1, В означает группу, представленную формулой:
где R1 означает атом водорода или С1-С6-алкильную группу; и R2 означает группу, представленную формулой:
где R3, R4 и R5 являются одинаковыми или различными, и каждый означает атом водорода или атом галогена,
R7 и R8 являются одинаковыми или различными, и каждый означает атом водорода или С1-С6-алкильную группу; m означает 0 или 1; n означает 1); или
(2) группу, представленную формулой А2:
(в формуле А2, В и n имеют значения, как определено в формуле А1; и R9 означает атом водорода или С1-С6-алкильную группу).
5. Соединение, или его соль, по любому одному из пп.1-4, где группа:
представляет собой
(1) группу, представленную формулой А1:
(в формуле А1, В означает группу, представленную формулой:
где R1 означает атом водорода; и R2 означает группу, представленную формулой:
где R3, R4 и R5, каждый, означают атом водорода,
R7 и R8, каждый, означает атом водорода; m означает 0; и n означает 1); или
(2) группу, представленную формулой А2:
(в формуле А2, В и n имеют значения, как определено в формуле А1; и R9 означает С1-С6-алкильную группу).
6. Соединение, или его соль, по любому одному из пп.1-5, где соединение выбирают из следующей группы соединений, состоящей из:
(S)-N-(4-амино-6-метилен-5-(хинолин-3-ил)-7,8-дигидро-6Н-пиримидо[5,4-b]пирролизин-7-ил)акриламида (Соединение 1);
(S)-N-(4-амино-6-метилен-5-(хинолин-3-ил)-7,8-дигидро-6Н-пиримидо[5,4-b]пирролизин-7-ил)метакриламида (Соединение 2);
(S)-N-(4-амино-6-метилен-5-(хинолин-3-ил)-7,8-дигидро-6Н-пиримидо[5,4-b]пирролизин-7-ил)бут-2-енамида (смесь Е- и Z- изомеров) (Соединение 3);
(S,Е)-N-(4-амино-6-метилен-5-(хинолин-3-ил)-7,8-дигидро-6Н-пиримидо[5,4-b]пирролизин-7-ил)-4-(диметиламино)бут-2-енамида (Соединение 4);
(S,Е)-N-(4-амино-6-метилен-5-(хинолин-3-ил)-7,8-дигидро-6Н-пиримидо[5,4-b]пирролизин-7-ил)-3-хлоракриламида (Соединение 5);
(S,Z)-N-(4-амино-6-метилен-5-(хинолин-3-ил)-7,8-дигидро-6Н-пиримидо[5,4-b]пирролизин-7-ил)-3-хлоракриламида (Соединение 6);
(S,E)-N-(4-амино-6-метилен-5-(хинолин-3-ил)-7,8-дигидро-6Н-пиримидо[5,4-b]пирролизин-7-ил)-4-(пиперидин-1-ил)бут-2-енамида (Соединение 7);
(S)-N-(4-амино-6-метилен-5-(хинолин-3-ил)-7,8-дигидро-6Н-пиримидо[5,4-b]пирролизин-7-ил)пропиоламида (Соединение 8);
(S)-N-(4-амино-6-метилен-5-(хинолин-3-ил)-7,8-дигидро-6Н-пиримидо[5,4-b]пирролизин-7-ил)бут-2-инамида (Соединение 9);
(S,E)-N-(4-амино-6-метилен-5-(хинолин-3-ил)-7,8-дигидро-6Н-пиримидо[5,4-b]пирролизин-7-ил)-4-(диэтиламино)бут-2-енамида (Соединение 10);
(S,E)-N-(4-амино-6-метилен-5-(хинолин-3-ил)-7,8-дигидро-6Н-пиримидо[5,4-b]пирролизин-7-ил)-4-(этил(метил)амино)бут-2-енамида (Соединение 11);
(S,E)-N-(4-амино-6-метилен-5-(хинолин-3-ил)-7,8-дигидро-6Н-пиримидо[5,4-b]пирролизин-7-ил)-4-(изопропил(метил)амино)бут-2-енамида (Соединение 12);
(R)-N-(4-амино-6-метилен-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-7-ил)акриламида (Соединение 13);
(S)-N-(4-амино-6-метил-5-(хинолин-3-ил)-8,9-дигидропиримидо[5,4-b]индолизин-8-ил)акриламида (Соединение 14);
(S)-N-(4-амино-6-метилен-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)акриламида (Соединение 15);
(R)-N-(4-амино-6-метил-5-(хинолин-3-ил)-8,9-дигидропиримидо[5,4-b]индолизин-8-ил)акриламида (Соединение 16);
(R)-N-(4-амино-6-метилен-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)акриламида (Соединение 17);
(S)-N-(4-амино-6-метилен-5-(хинолин-3-ил)-7,8,9,10-тетрагидро-6Н-пиримидо[5’,4’:4,5]пирроло[1,2-a]азепин-8-ил)акриламида (Соединение 18);
(S,E)-N-(4-амино-6-этилиден-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)акриламида (Соединение 19);
(S)-N-(4-амино-6-изопропил-5-(хинолин-3-ил)-8,9-дигидропиримидо[5,4-b]индолизин-8-ил)акриламида (Соединение 20А);
(S)-N-(4-амино-6-(пропан-2-илиден)-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)акриламида (Соединение 20В);
(R)-N-(4-амино-6-метилен-5-(хинолин-3-ил)-7,8-дигидро-6Н-пиримидо[5,4-b]пирролизин-7-ил)-N-метилакриламида (Соединение 21);
(R)-N-(4-амино-6-метилен-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)-N-метилакриламида (Соединение 22);
(R)-N-(4-амино-6-метил-5-(хинолин-3-ил)-8,9-дигидропиримидо[5,4-b]индолизин-8-ил)-N-метилакриламида (Соединение 23);
N-((7S)-4-амино-6-метил-5-(хинолин-3-ил)-7,8-дигидро-6Н-пиримидо[5,4-b]пирролизин-7-ил)акриламида (Соединение 24);
(R)-N-(4-амино-6-метилен-5-(хинолин-3-ил)-7,8-дигидро-6Н-пиримидо[5,4-b]пирролизин-7-ил)акриламида (Соединение 25);
(S)-N-(4-амино-6-метилен-5-(хинолин-3-ил)-7,8-дигидро-6Н-пиримидо[5,4-b]пирролизин-7-ил)-N-метилакриламида (Соединение 26);
(S)-N-(4-амино-5-(хинолин-3-ил)-8,9-дигидропиримидо[5,4-b]индолизин-8-ил)акриламида (Соединение 27);
(R)-N-(4-амино-5-(хинолин-3-ил)-9,10-дигидро-8Н-пиримидо[5’,4’:4,5]пирроло[1,2-a]азепин-8-ил)акриламида (Соединение 28);
N-((6R*,8S)-4-амино-6-метил-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)акриламида (Соединение 29А); и
N-((6S*,8S)-4-амино-6-метил-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)акриламида (Соединение 29В).
7. Соединение, или его соль, по любому одному из пп.1-5, где соединение выбирают из следующей группы соединений, состоящей из:
(S)-N-(4-амино-6-метилен-5-(хинолин-3-ил)-7,8-дигидро-6Н-пиримидо[5,4-b]пирролизин-7-ил)акриламида (Соединение 1);
(S,Е)-N-(4-амино-6-метилен-5-(хинолин-3-ил)-7,8-дигидро-6Н-пиримидо[5,4-b]пирролизин-7-ил)-3-хлоракриламида (Соединение 5);
(S,Z)-N-(4-амино-6-метилен-5-(хинолин-3-ил)-7,8-дигидро-6Н-пиримидо[5,4-b]пирролизин-7-ил)-3-хлоракриламида (Соединение 6);
(S)-N-(4-амино-6-метил-5-(хинолин-3-ил)-8,9-дигидропиримидо[5,4-b]индолизин-8-ил)акриламида (Соединение 14);
(S)-N-(4-амино-6-метилен-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)акриламида (Соединение 15);
(S,E)-N-(4-амино-6-этилиден-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)акриламида (Соединение 19);
(R)-N-(4-амино-6-метилен-5-(хинолин-3-ил)-7,8-дигидро-6Н-пиримидо[5,4-b]пирролизин-7-ил)-N-метилакриламида (Соединение 21);
(R)-N-(4-амино-6-метилен-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)-N-метилакриламида (Соединение 22);
(R)-N-(4-амино-6-метил-5-(хинолин-3-ил)-8,9-дигидропиримидо[5,4-b]индолизин-8-ил)-N-метилакриламида (Соединение 23); и
(R)-N-(4-амино-5-(хинолин-3-ил)-9,10-дигидро-8Н-пиримидо[5’,4’:4,5]пирроло[1,2-a]азепин-8-ил)акриламида (Соединение 28).
8. (S)-N-(4-Амино-6-метилен-5-(хинолин-3-ил)-7,8-дигидро-6Н-пиримидо[5,4-b]пирролизин-7-ил)акриламид (Соединение 1) или его соль.
9. (S)-N-(4-Амино-6-метил-5-(хинолин-3-ил)-8,9-дигидропиримидо[5,4-b]индолизин-8-ил)акриламид (Соединение 14) или его соль.
10. Ингибитор EGFR, содержащий соединение, или его соль, по любому одному из пп.1-9, в качестве активного ингредиента.
11. Фармацевтическая композиция, содержащая соединение, или его соль, по любому одному из пп.1-9.
12. Противоопухолевый агент, содержащий соединение, или его соль, по любому одному из пп.1-9, в качестве активного ингредиента.
13. Способ лечения или предупреждения ракового заболевания, причем этот способ включает стадию введения млекопитающему соединения, или его соли, по любому одному из пп.1-9, в дозе, эффективной для лечения или предупреждения ракового заболевания.
14. Применение соединения или его соли, по любому одному из пп.1-9, для получения противоопухолевого агента.
15. Соединение или его соль, по любому одному из пп.1-9, для применения в целях предупреждения или лечения ракового заболевания.
Полезные эффекты данного изобретения
Согласно настоящему изобретению, получают новое соединение, представленное вышеприведенной формулой (I), или его соль, пригодное в качестве ингибитора EGFR.
Выяснено, что соединение согласно настоящему изобретению или его соль, обладает превосходной, ингибирующей EGFR, активностью и ингибирующим рост штаммов раковых клеток эффектом. В дополнение, соединение согласно настоящему изобретению, или его соль, благоприятно имеет незначительное число побочных эффектов в результате превосходной селективности против EGFR. Следовательно, соединение согласно настоящему изобретению, или его соль, являются пригодными в качестве агента для предупреждения и/или лечения ракового заболевания.
Описание вариантов осуществления
Соединение, представленное формулой (I), согласно настоящему изобретению, представляет собой хинолин-замещенное соединение, имеющее структуру хинолина и структуру ненасыщенного α,β-амида, и представляет собой новое соединение, нигде не описанное в каком-либо из вышеуказанных документов уровня техники и т.д.
В особенности, соединение, конкретно раскрытое в PTD 1, представляет собой N-(3-(4-амино-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-5-ил)фенил)бензамид производное.
Соединение согласно настоящему изобретению отличается от соединения, раскрытого в PTD 1, так как соединение согласно настоящему изобретению имеет структуру хинолина и структуру ненасыщенного α,β-амида.
В описании настоящего изобретения, примеры «атома галогена» включают фтор, хлор, бром и иод.
В описании настоящего изобретения, термин «С1-С6-алкильная группа» относится к алкильной группе с линейной или разветвленной цепью, имеющей 1-6 атомов углерода. Конкретные примеры алкильной группы включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, пентил и гексил.
В описании настоящего изобретения, термин «С6-С12-арильная группа» относится к арильной группе, имеющей 6-12 атомов углерода. Конкретные примеры арильной группы включают фенил, нафтил и бифенил.
В описании настоящего изобретения, термин «С4-С9-гетероарильная группа» относится к моноциклической или бициклической С4-С9-гетероарильной группе, содержащей от 1 до 3 одинаковых или различных гетероатомов, выбираемых из атомов азота, кислорода и серы; и, предпочтительно, представляет собой моноциклическую или бициклическую С4-С9-гетероарильную группу, содержащую 1-3 атома азота. Конкретные примеры гетероарильной группы включают тиенил, фурил, пирролил, триазолил, имидазолил, пиразолил, изотиазолил, изоксазолил, пиридил, пиразинил, пиримидинил, пиридазинил, изобензофурил, индолизинил, изоиндолил, индолил, индазолил, хинолил, изохинолил, фталазинил и нафтиридинил.
В описании настоящего изобретения, термин «аминометильная группа, которая может быть замещена С1-С6-алкильной группой» относится к аминометильной группе, в которой, по меньшей мере, один атом водорода аминогруппы может быть замещен алкильной группой с линейной или разветвленной цепью, имеющей 1-6 атомов углерода. Конкретные примеры включают аминометил, N-метиламинометил, N,N-диметиламинометил, N-этиламинометил, N,N-диэтиламинометил, N-метил-N-этиламинометил, N-метил-N-изопропиламинометил, N-пропиламинометил, N-бутиламинометил, N-пентиламинометил и N-гексиламинометил.
В вышеприведенной формуле (I), фрагмент, представленный формулой:
представляет собой (1) группу, представленную формулой А1:
(в формуле А1, В, m, n, R7 и R8 имеют значения, как определено выше);
(2) группу, представленную формулой А2:
(в формуле А2, В, n и R9 имеют значения, как определено выше);
или
(3) группу, представленную формулой А3:
(в формуле А3, В, m, n, R7 и R8 имеют значения, как определено выше);
и, предпочтительно, представляет собой (1) группу, представленную формулой А1:
(в формуле, В, m, n, R7 и R8 имеют значения, как определено выше); или
(2) группу, представленную формулой А2:
(в формуле, В, n и R9 имеют значения, как определено выше);
m, в формуле (I), предпочтительно, означает 0.
n, в формуле (I), предпочтительно, означает 1.
R1, в формуле (I), предпочтительно, означает атом водорода.
R2, в формуле (I), предпочтительно, означает группу, представленную формулой:
(где R3, R4 и R5 имеют значения, как определено выше).
R3, R4 и R5, в формуле (I), предпочтительно, являются одинаковыми или различными, и каждый означает атом водорода, атом галогена, С1-С6-алкильную группу, аминометильную группу, которая может быть замещена С1-С6-алкильной группой, или 1-пиперидинометильную группу; более предпочтительно, R3, R4 и R5 являются одинаковыми или различными, и каждый означает атом водорода, атом галогена, аминометильную группу, которая может быть замещена метильной группой, или 1-пиперидинометильную группу; далее, предпочтительно, R3, R4 и R5 являются одинаковыми или различными, и каждый означает атом водорода или атом галогена, в особенности, предпочтительно, атом водорода.
R7 и R8, в формуле (I), предпочтительно, являются одинаковыми или различными, и каждый означает атом водорода или С1-С6-алкильную группу (предпочтительно, С1-С3-алкильную группу, более предпочтительно, метильную группу); далее, предпочтительно, по меньшей мере, один из R7 и R8 означает атом водорода; в особенности, предпочтительно, оба R7 и R8 означают атомы водорода.
R9, в формуле (I), предпочтительно, означает С1-С6-алкильную группу, более предпочтительно, С1-С3-алкильную группу, далее, предпочтительно, метильную группу.
Соединение согласно настоящему изобретению или его соль, предпочтительно, обладает сильной, ингибирующей фермент, активностью против EGFR (T790M/L858R); более предпочтительно, концентрация соединения, при которой фермент на 50% может быть ингибирован, составляет 2 нМ или менее. Далее, соединение согласно настоящему изобретению или его соль, предпочтительно, обладает сильной, ингибирующей фермент, активностью против EGFR (d746-750/T790M); ингибирующая на 50% концентрация соединения также, предпочтительно, составляет 2 нМ или менее. Далее, соединение согласно настоящему изобретению или его соль, предпочтительно, обладает сильным, ингибирующим рост клеток, эффектом против опухолевых клеток с EGFR (T790M/L858R); более предпочтительно, ингибирующая на 50% концентрация соединения составляет 200 нМ или менее, далее, предпочтительно, 100 нМ или менее, и, особенно предпочтительно, 40 нМ или менее.
Далее раскрывается способ получения соединения согласно настоящему изобретению.
Соединение (I) согласно настоящему изобретению можно получать, например, с помощью следующего способа получения, способов, описанных в Примерах, и т.п. Однако, способ получения соединения согласно настоящему изобретению не ограничен этими примерами осуществления реакций.
Способ получения 1
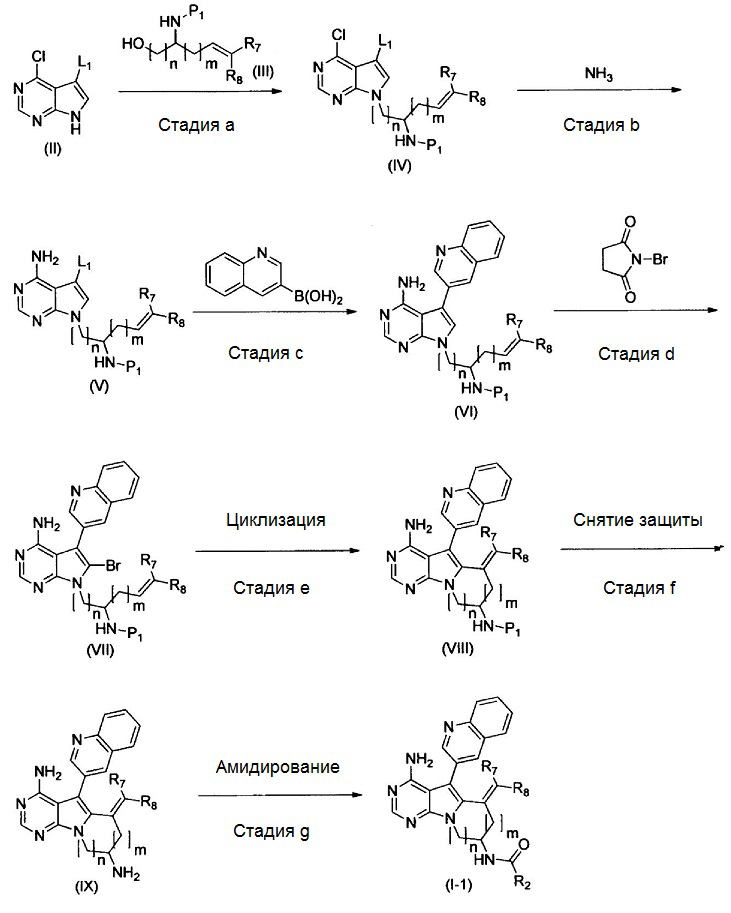
где Р1 представляет собой защитную группу аминогруппы; L1 представляет собой удаляемую группу; и R2, R7, R8, m и n имеют значения, как определено выше.
Стадия а
На этой стадии, соединение, представленное формулой (IV), получают с помощью реакции Mitsunobu, используя соединения, представленные формулами (II) и (III).
Примеры удаляемой группы, представленной L1, в соединении, представленном формулой (II), включают атом брома или атом иода. Соединение, представленное формулой (II), может быть коммерчески доступным продуктом или его можно получать с помощью известного способа. Примеры защитной для аминогруппы группы, представленной P1, в формуле (III), включают трет-бутоксикарбонильную группу и бензоильную группу. Соединение, представленное формулой (III), может быть коммерчески доступным продуктом или его можно получать с помощью известного способа. Соединение, представленное формулой (III), можно использовать в количестве от 1 моль до 10 моль и, предпочтительно, 1-5 моль, в расчете на моль соединения, представленного формулой (II).
Реакцию Mitsunobu можно осуществлять в соответствии с известным способом (например, способ, раскрытый в Synthesis, с.1, 1981) или с помощью подобного способа.
Примеры эфиров азодикарбоновой кислоты включают диэтилазодикарбоксилат и диизопропилазодикарбоксилат. Такой эфир азодикарбоновой кислоты можно использовать в количестве 1-10 моль и, предпочтительно, 1-5 моль, в расчете на моль соединения, представленного формулой (II).
В качестве фосфинового соединения можно использовать трифенилфосфин, трибутилфосфин или т.п. Фосфиновое соединение можно использовать в количестве 1-10 моль и, предпочтительно, 1-5 моль, в расчете на моль соединения, представленного формулой (II).
В качестве растворителя можно использовать тетрагидрофуран, 1,2-диметоксиэтан, 1,4-диоксан, толуол, N,N-диметилформамид, N,N-диметилацетамид, диметилсульфоксид, N-метилпирролидин-2-он и т.п., индивидуально или в виде смеси. Время реакции составляет от 0,1 часа до 100 часов и, предпочтительно, 0,1-24 часа. Температура реакции составляет от 0°С до температуры кипения растворителя и, предпочтительно, от 0°С до 100°С.
Таким образом полученное соединение, представленное формулой (IV), можно подвергать последующей стадии, после или без выделения или очистки посредством известных способов выделения и очистки, таких как концентрирование, концентрирование в вакууме, кристаллизация, экстракция растворителем, переосаждение и хроматография.
Стадия b
На этой стадии, соединение, представленное формулой (IV), вводят во взаимодействие с аммиаком, или его солью, до получения соединения, представленного формулой (V).
Количество аммиака или его соли, используемое на этой стадии, обычно составляет от эквимолярного количества до избыточного молярного количества на моль соединения, представленного формулой (IV).
Можно использовать любой растворитель для реакции, который не оказывает неблагоприятного воздействия на реакцию. Примеры пригодных реакционных растворителей включают воду, метанол, этанол, изопропанол, трет-бутиловый спирт, тетрагидрофуран, 1,2-диметоксиэтан, 1,4-диоксан, N,N-диметилформамид, N-метилпирролидин-2-он, диметилсульфоксид и получаемые из них смешанные растворители.
Температура реакции обычно составляет от 0°С до 200°С, предпочтительно, от комнатной температуры до температуры 150°С. Время реакции обычно составляет от 5 мин до 7 суток и, предпочтительно, от 30 мин до 24 часов.
Таким образом полученное соединение, представленное формулой (V), можно подвергать последующей стадии, после или без выделения или очистки посредством известных способов выделения и очистки, таких как концентрирование, концентрирование в вакууме, кристаллизация, экстракция растворителем, переосаждение и хроматография.
Стадия с
На этой стадии, соединение, представленное формулой (V), подвергают реакции связывания с 3-хинолинбороновой кислотой или эфиром 3-хинолинбороновой кислоты до получения соединения, представленного формулой (VI).
Эту стадию можно осуществлять в соответствии с общеизвестным способом (например, Chemical Reviews, том 95, с. 2457, 1995). Эту стадию можно осуществлять в присутствии катализатора на основе переходного металла и основания, в растворителе, который не оказывает неблагоприятного воздействия на реакцию.
Используемое количество 3-хинолинбороновой кислоты или эфира 3-хинолинбороновой кислоты может составлять 1-10 моль и, предпочтительно, 1-3 моль, в расчете на моль соединения, представленного формулой (V).
Примеры катализаторов на основе переходного металла включают катализаторы на основе палладия (например, ацетат палладия, хлорид палладия, тетракис(трифенилфосфин)палладий, 1,1’-бис(дифенилфосфино)ферроценпалладий(II)дихлорид и трис(дибензилиденацетон)дипалладий(0)), катализаторы на основе никеля (например, хлорид никеля), и т.п. Если необходимо, можно добавлять лиганд (например, трифенилфосфин, три-трет-бутилфосфин или 2-дициклогексилфосфино-2’,4’,6’-триизопропилбифенил) и, в качестве сокатализатора, можно использовать оксид металла (такой как оксид меди или оксид серебра). Используемое количество катализатора на основе переходного металла может варьироваться в зависимости от типа катализатора. Катализатор на основе переходного металла обычно используют в количестве от 0,0001 моль до 1 моль и, предпочтительно, от 0,01 моль до 0,5 моль, в расчете на моль соединения, представленного формулой (V). Количество используемого лиганда обычно составляет от 0,0001 моль до 4 моль и, предпочтительно, от 0,01 моль до 2 моль, в расчете на моль соединения, представленного формулой (V). Количество используемого сокатализатора обычно составляет от 0,0001 моль до 4 моль и, предпочтительно, от 0,01 моль до 2 моль, в расчете на моль соединения, представленного формулой (V).
Примеры пригодных оснований включают органические амины (например, триметиламин, триэтиламин, диизопропилэтиламин, N-метилморфолин, 1,8-диазабицикло[5,4,0]ундец-7-ен, пиридин и N,N-диметиланилин), соли щелочных металлов (например, гидрокарбонат натрия, гидрокарбонат калия, карбонат натрия, карбонат калия, карбонат цезия, фосфат натрия, фосфат калия, гидроксид натрия и гидроксид калия), гидриды металлов (например, гидрид калия и гидрид натрия), алкоксиды щелочных металлов (например, метоксид натрия, этоксид натрия, трет-бутоксид натрия и трет-бутоксид калия), дисилазиды щелочных металлов (например, дисилазид лития, дисилазид натрия и дисилазид калия) и т.п. Среди них, предпочтительными являются соли щелочных металлов, такие как карбонат натрия, карбонат калия, карбонат цезия, фосфат натрия и фосфат калия; алкоксиды щелочных металлов, такие как трет-бутоксид натрия и трет-бутоксид калия; и органические амины, такие как триэтиламин и диизопропилэтиламин. Количество используемого основания обычно составляет 0,1-10 моль и, предпочтительно, 1-5 моль, в расчете на моль соединения, представленного формулой (V).
Можно использовать любой растворитель, который не оказывает неблагоприятного воздействия на реакцию. Примеры пригодного растворителя включают углеводороды (например, бензол, толуол и ксилол), галогенированные углеводороды (например, хлороформ и 1,2-дихлорэтан), нитрилы (например, ацетонитрил), простые эфиры (например, 1,2-диметоксиэтан, тетрагидрофуран и 1,4-диоксан), спирты (например, метанол и этанол), апротонные полярные растворители (например, N,N-диметилформамид, диметилсульфоксид и гексаметилфосфориламид), воду и получаемые из них смешанные растворители. Время реакции составляет от 0,1 часа до 100 часов и, предпочтительно, от 0,5 часа до 24 часов. Температура реакции составляет от 0°С до температуры кипения растворителя и, предпочтительно, от 20°С до 150°С.
Таким образом полученное соединение, представленное формулой (VI), можно подвергать последующей стадии, после или без выделения или очистки посредством известных способов выделения и очистки, таких как концентрирование, концентрирование в вакууме, кристаллизация, экстракция растворителем, переосаждение и хроматография.
Стадия d
На этой стадии, соединение, представленное формулой (VI), подвергают бромированию путем введения во взаимодействие с N-бромсукцинимидом до получения соединения, представленного формулой (VII).
Галогенирование можно осуществлять с помощью способа, раскрытого в заявке на патент под номером WO 2006/102079 или с помощью подобного способа.
Используемое на этой стадии количество N-бромсукцинимида составляет от 0,5 моль до 2,0 моль и, предпочтительно, от 0,9 моль до 1,2 моль, в расчете на моль соединения, представленного формулой (VI).
Можно использовать любой растворитель для реакции, который не оказывает неблагоприятного воздействия на реакцию. Например, предпочтительно, можно использовать тетрагидрофуран, 1,4-диоксан, N,N-диметилформамид, N-метилпирролидин-2-он или получаемые из них смешанные растворители.
Температура реакции обычно составляет от -20°С до 50°С и, предпочтительно, от 0°С до комнатной температуры. Время реакции обычно составляет от 1 мин до 2 суток и, предпочтительно, от 5 мин до 12 часов.
Таким образом полученное соединение, представленное формулой (VI), можно подвергать последующей стадии, после или без выделения или очистки посредством известных способов выделения и очистки, таких как концентрирование, концентрирование в вакууме, кристаллизация, экстракция растворителем, переосаждение и хроматография.
Стадия е
На этой стадии соединение, представленное формулой (VII), подвергают реакции внутримолекулярной циклизации до получения соединения, представленного формулой (VIII).
Эту стадию можно осуществлять в соответствии с общеизвестным способом (например, способ, раскрытый в Chemical Reviews, том 103, с. 2945, 2003).
Примеры катализаторов на основе переходных металлов включают катализаторы на основе двухвалентного палладия (например, ацетат палладия, хлорид палладия, 1,1’-бис(дифенилфосфино)ферроценпалладий(II)дихлорид и т.д.) и катализаторы на основе палладия с нулевой валентностью (например, тетракис(трифенилфосфин)палладий, трис(дибензилиденацетон)дипалладий и т.д.). Если необходимо, можно добавлять лиганд (например, трифенилфосфин, три-трет-бутилфосфин и т.д.). Используемое количество катализатора на основе переходного металла может варьироваться в зависимости от типа катализатора. Катализатор на основе переходного металла обычно используют в количестве от 0,0001 моль до 1 моль и, предпочтительно, от 0,01 моль до 0,5 моль, в расчете на моль соединения, представленного формулой (VII). Используемое количество лиганда обычно составляет от 0,0001 моль до 4 моль и, предпочтительно, от 0,01 моль до 2 моль, в расчете на моль соединения, представленного формулой (VII).
Примеры пригодных оснований включают неорганические основания, такие как гидрокарбонат натрия, карбонат натрия, карбонат калия, карбонат цезия, гидроксид натрия, гидроксид калия, гидроксид цезия, гидрид натрия или гидрид калия. Такое основание можно использовать в количестве от 1 моль до 100 моль и, предпочтительно, от 2 моль до 20 моль, в расчете на моль соединения, представленного формулой (VII).
Можно использовать любой растворитель, который не оказывает неблагоприятного воздействия на реакцию. Примеры пригодных растворителей включают углеводороды (например, бензол, толуол и ксилол), простые эфиры (например, 1,2-диметоксиэтан, тетрагидрофуран и 1,4-диоксан), апротонные полярные растворители (например, N,N-диметилформамид, диметилсульфоксид и гексаметилфосфориламид), воду и получаемые из них смешанные растворители. Время реакции составляет от 0,1 часа до 100 часов и, предпочтительно, от 0,5 часа до 24 часов. Температура реакции составляет от -20°С до температуры кипения растворителя и, предпочтительно, от 0°С до 150°С.
Таким образом полученное соединение, представленное формулой (VIII), можно подвергать последующей стадии, после или без выделения или очистки посредством известных способов выделения и очистки, таких как концентрирование, концентрирование в вакууме, кристаллизация, экстракция растворителем, переосаждение и хроматография.
Стадия f
На этой стадии, снимают защиту с защищенной аминогруппы соединения, представленного формулой (VIII), до получения соединения, представленного формулой (IX).
Снятие защиты можно осуществлять с помощью известного способа, как например способ, описанный в Protective Groups in Organic Synthesis, T.W. Greene, John Wiley & Sons (1981); или с помощью подобного способа.
Если трет-бутоксикарбонильную группу используют в качестве защитной группы, то соляную кислоту, серную кислоту, метансульфоновую кислоту, трифторуксусную кислоту и т.п. используют в качестве реагента для снятия защиты. Используемое количество реагента, предпочтительно, составляет от 1 моль до 100 моль, в расчете на моль соединения (VIII).
Можно использовать любой растворитель, который не оказывает неблагоприятного воздействия на реакцию. Примеры пригодных растворителей включают воду, метанол, этанол, метиленхлорид, хлороформ и т.п., и получаемые из них смешанные растворители. Время реакции составляет от 0,1 часа до 100 часов и, предпочтительно, от 0,5 часа до 24 часов. Температура реакции составляет от 0°С до температуры кипения растворителя и, предпочтительно, от 0°С до 50°С.
Таким образом полученное соединение, представленное формулой (IX), можно подвергать последующей стадии, после или без выделения или очистки посредством известных способов выделения и очистки, таких как концентрирование, концентрирование в вакууме, кристаллизация, экстракция растворителем, переосаждение и хроматография.
Стадия g
На этой стадии, соединение, представленное формулой (IX), амидируют с помощью α,β-ненасыщенной карбоновой кислоты или хлорангидрида или бромангидрида α,β-ненасыщенной карбоновой кислоты до получения соединения, представленного формулой (I-1), согласно настоящему изобретению.
Если карбоновую кислоту используют в качестве реагента амидирования, то карбоновую кислоту можно использовать в количестве от 0,5 моль до 10 моль, предпочтительно, от 1 моль до 3 моль, в расчете на моль соединения, представленного формулой (IX), в присутствии подходящего конденсирующего агента. Карбоновая кислота может представлять собой коммерчески доступный продукт или ее можно получать в соответствии с известным способом.
Можно использовать любой растворитель для реакции, который не оказывает неблагоприятного воздействия на реакцию. Примеры пригодного растворителя включают толуол, бензол, метиленхлорид, хлороформ, тетрагидрофуран, 1,4-диоксан, N,N-диметилформамид, диметилацетамид, N-метилпирролидин-2-он, диметилсульфоксид и получаемые из них смешанные растворители. Температура реакции обычно составляет от -78°С до 200°С и, предпочтительно, от 0°С до 50°С. Время реакции обычно составляет от 5 мин до 3 суток и, предпочтительно, от 5 мин до 10 часов.
Примеры конденсирующих агентов включают дифенилфосфорилазид, N,N’-дициклогексилкарбодиимид, соли бензотриазол-1-илокси-трисдиметиламинофосфония, 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4-метилморфолинийхлорид, 1-этил-3-(3-диметиламинопропил)карбодиимид, комбинацию 1-этил-3-(3-диметиламинопропил)карбодиимида и 1-гидроксибензотриазола, 2-хлор-1,3-диметилимидазолинийхлорид, O-(7-азабензотриазо-1-ил)-N,N,N’,N’-тетраметилгексауронийгексафторфосфат и т.п.
Если необходимо, при осуществлении реакции можно необязательно добавлять основание. Примеры пригодных оснований включают органические основания, такие как триэтиламин, диизопропилэтиламин, пиридин, лутидин, коллидин, 4-(N,N-диметиламино)пиридин, трет-бутират калия, трет-бутират натрия, метоксид натрия, этоксид натрия, гексаметилдисилазид лития, гексаметилдисилазид натрия, гексаметилдисилазид калия и бутиллитий; и неорганические основания, такие как гидрокарбонат натрия, карбонат натрия, карбонат калия, карбонат цезия, гидроксид натрия и гидрид натрия. Такое основание можно добавлять в количестве от 1 моль до 100 моль и, предпочтительно, от 1 моль до 10 моль, в расчете на моль соединения, представленного формулой (IX).
Если хлорангидрид кислоты или бромангидрид кислоты используют в качестве реагента амидирования, то галогенангидрид кислоты используют в количестве от 0,5 моль до 5 моль и, предпочтительно, от 0,9 моль до 1,1 моль, в расчете на моль соединения, представленного формулой (IX). Галогенангидрид кислоты может преставлять собой коммерчески доступный продукт или его можно получать в соответствии с известным способом.
Можно использовать любой растворитель для реакции, который не оказывает неблагоприятного воздействия на реакцию. Примеры таких растворителей включают толуол, бензол, метиленхлорид, хлороформ, тетрагидрофуран, 1,4-диоксан, N,N-диметилформамид, диметилацетамид, N-метилпирролидин-2-он, ацетонитрил, воду и получаемые из них смешанные растворители. Температура реакции обычно составляет от -78°С до 200°С, предпочтительно, от 0°С до 50°С. Время реакции обычно составляет от 5 мин до 3 суток и, предпочтительно, от 5 мин до 10 часов.
Если необходимо, при осуществлении реакции можно добавлять основание. Примеры пригодных оснований включают органические основания, такие как триэтиламин, диизопропилэтиламин, пиридин, лутидин, коллидин, 4-(N,N-диметиламино)пиридин, трет-бутират калия, трет-бутират натрия, метоксид натрия, этоксид натрия, гексаметилдисилазид лития, гексаметилдисилазид натрия, гексаметилдисилазид калия и бутиллитий; и неорганические основания, такие как гидрокарбонат натрия, карбонат натрия, карбонат калия, карбонат цезия, гидроксид натрия и гидрид натрия. Такое основание можно добавлять в количестве от 1 моль до 100 моль и, предпочтительно, от 1 моль до 20 моль, в расчете на моль соединения, представленного формулой (IX).
Таким образом полученное соединение, представленное формулой (I-1), можно выделять и очищать посредством известных способов выделения и очистки, таких как концентрирование, концентрирование в вакууме, кристаллизация, экстракция растворителем, переосаждение и хроматография.
Способ получения 2
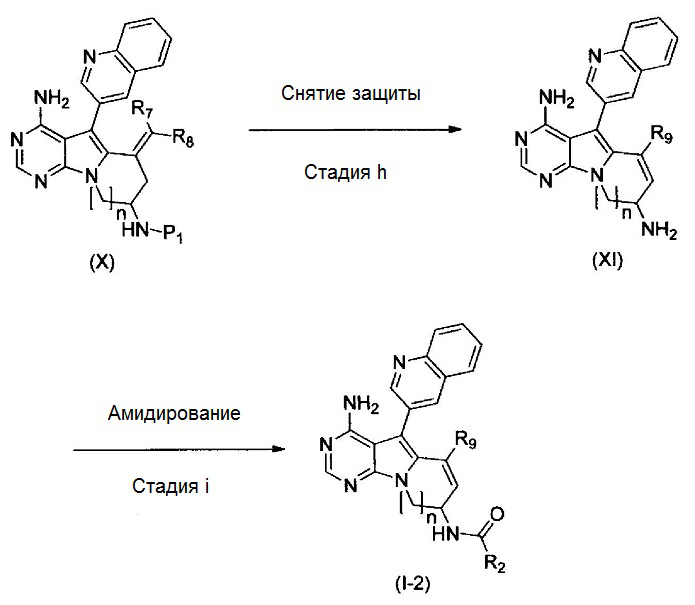
где Р1 представляет собой защитную для аминогруппы группу; и R2, R7, R8, R9 и n имеют значения, как определено выше.
Стадия h
На этой стадии, снимают защиту с защищенной аминогруппы соединения, представленного формулой (Х), до получения соединения, представленного формулой (XI).
Снятие защиты можно осуществлять с помощью известного способа, как например способ, описанный в Protective Groups in Organic Synthesis, T.W. Greene, John Wiley & Sons (1981); или с помощью подобного способа.
Если трет-бутоксикарбонильную группу используют в качестве защитной группы, то соляную кислоту, серную кислоту, метансульфоновую кислоту, трифторуксусную кислоту и т.п. можно использовать в качестве реагента для снятия защиты. Предпочтительно, реагент используют в количестве от 1 моль до 100 моль, в расчете на моль соединения (Х).
Можно использовать любой растворитель, который не оказывает неблагоприятного воздействия на реакцию. Примеры пригодных растворителей включают воду, метанол, этанол, метиленхлорид, хлороформ и их смешанные растворители. Время реакции составляет от 0,1 часа до 100 часов и, предпочтительно, от 0,5 часа до 24 часов. Температура реакции составляет от 0°С до температуры кипения растворителя и, предпочтительно, от 50°С до температуры кипения растворителя.
Таким образом полученное соединение, представленное формулой (XXI), можно подвергать последующей стадии, после или без выделения или очистки посредством известных способов выделения и очистки, таких как концентрирование, концентрирование в вакууме, кристаллизация, экстракция растворителем, переосаждение и хроматография.
Стадия i
Эту стадию можно осуществлять таким же образом, как и стадию g.
Способ получения 3
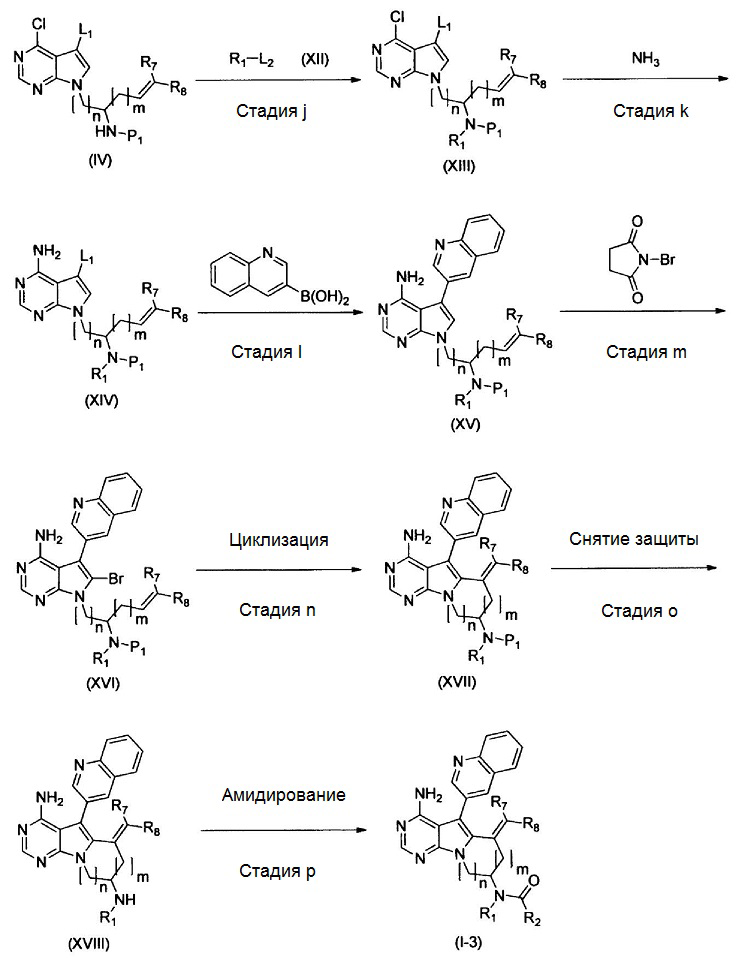
где Р1 представляет собой защитную для аминогруппы группу; L1 и L2 представляют собой удаляемую группу; и R1, R2, R7, R8, m и n имеют значения, как определено выше.
Стадия j
На этой стадии, соединение, представленное формулой (ХIII), получают путем реакции алкилирования, используя соединения, представленные формулами (IV) и (XII), в присутствии основания.
В соединении, представленном формулой (XII), примеры удаляемой группы, представленной L2, включают атом брома, атом иода, эфир метансульфоновой кислоты, эфир п-толуолсульфоновой кислоты и т.п. Соединение, представленное формулой (XII), может представлять собой коммерчески доступный продукт или его можно получать с помощью известного способа. Соединение, представленное формулой (XII), можно использовать в количестве от 1 моль до 10 моль и, предпочтительно, от 1 моль до 5 моль, в расчете на моль соединения, представленного формулой (IV). Примеры пригодных оснований включают органические амины (например, триметиламин, триэтиламин, диизопропилэтиламин, N-метилморфолин, 1,8-диазабицикло[5,4,0]ундец-7-ен, пиридин и N,N-диметиланилин), соли щелочных металлов (например, гидрокарбонат натрия, гидрокарбонат калия, карбонат натрия, карбонат калия, карбонат цезия, фосфат натрия, фосфат калия, гидроксид натрия и гидроксид калия), гидриды металлов (например, гидрид калия и гидрид натрия), алкоксиды щелочных металлов (например, метоксид натрия, этоксид натрия, трет-бутоксид натрия и трет-бутоксид калия), дисилазиды щелочных металлов (например, дисилазид лития, дисилазид натрия и дисилазид калия) и т.п.. Среди них, предпочтительными являются соли щелочных металлов, такие как карбонат натрия, карбонат калия, карбонат цезия, фосфат натрия и фосфат калия; и гидриды металлов, такие как гидрид натрия; алкоксиды щелочных металлов, такие как трет-бутоксид натрия и трет-бутоксид калия. Количество используемого основания обычно составляет от 0,1 моль до 10 моль и, предпочтительно, от 1 моль до 5 моль, в расчете на моль соединения, представленного формулой (V).
В качестве растворителя можно использовать тетрагидрофуран, 1,2-диметоксиэтан, 1,4-диоксан, N,N-диметилформамид, N,N-диметилацетамид, диметилсульфоксид, N-метилпирролидин-2-он и т.п., индивидуально или в виде смеси. Время реакции составляет от 0,1 часа до 100 часов и, предпочтительно, от 0,5 часа до 24 часов. Температура реакции составляет от 0°С до температуры кипения растворителя и, предпочтительно, от 20°С до 150°С.
Таким образом полученное соединение, представленное формулой (XIII), можно подвергать последующей стадии, после или без выделения или очистки посредством известных способов выделения и очистки, таких как концентрирование, концентрирование в вакууме, кристаллизация, экстракция растворителем, переосаждение и хроматография.
Стадия k
Эту стадию можно осуществлять таким же образом, как стадию b.
Стадия l
Эту стадию можно осуществлять таким же образом, как стадию с.
Стадия m
Эту стадию можно осуществлять таким же образом, как стадию d.
Стадия n
Эту стадию можно осуществлять таким же образом, как стадию e.
Стадия о
Эту стадию можно осуществлять таким же образом, как стадию f.
Стадия р
Эту стадию можно осуществлять таким же образом, как стадию g.
Способ получения 4
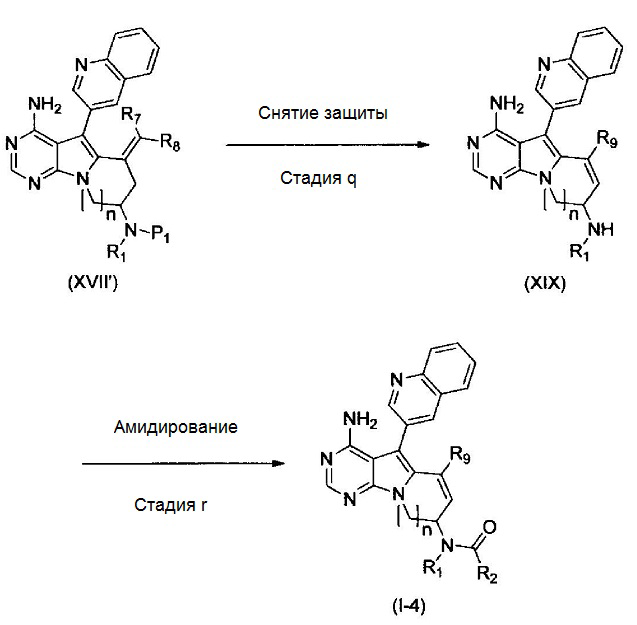
где Р1 представляет собой защитную для аминогруппы группу; и R1, R2, R7, R8, R9 и n имеют значения, как определено выше.
Стадия q
Эту стадию можно осуществлять таким же образом, как стадию h.
Стадия r
Эту стадию можно осуществлять таким же образом, как стадию g.
Способ получения 5
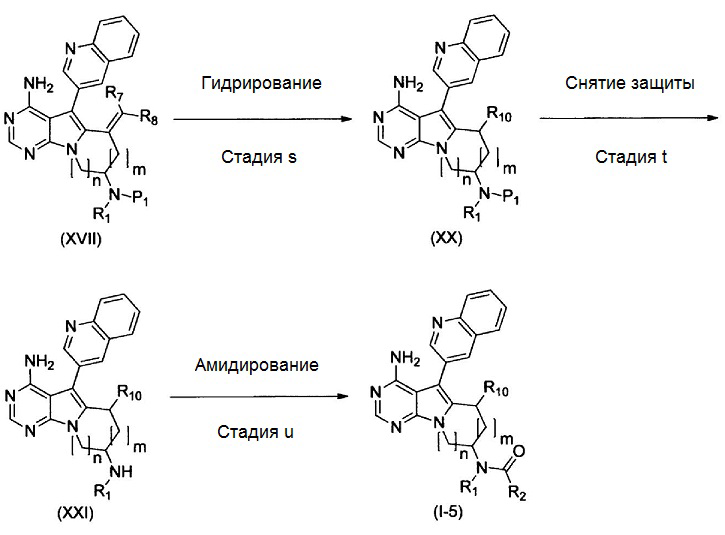
где Р1 представляет собой защитную для аминогруппы группу; и R1, R2, R7, R8, R10, m и n имеют значения, как определено выше.
Стадия s
На этой стадии, соединение, представленное формулой (ХХ), получают путем гидрирования соединения, представленного формулой (XVII), в присутствии катализатора.
Примеры катализатора включают палладий-на-угле, гидроксид палладия-на-угле и никель Ренея. Катализатор можно использовать в количестве от 0,01 моль до избыточного количества, предпочтительно, от 0,1 моль до 10 моль, в расчете на моль соединения, представленного формулой (XVII).
Гидрирование можно осуществлять при давлении от 1 атм. до 100 атм., предпочтительно, от 1 до 10 атм. В качестве растворителя можно использовать метанол, этанол, этилацетат, тетрагидрофуран, 1,2-диметоксиэтан, 1,4-диоксан, N,N-диметилформамид, N,N-диметилацетамид, N-метилпирролидин-2-он, воду и т.п., индивидуально или в виде смеси. Время реакции составляет от 0,1 часа до 100 часов и, предпочтительно, от 0,5 часа до 48 часов. Температура реакции находится в диапазоне от комнатной температуры до температуры кипения растворителя, предпочтительно, от комнатной температуры до 100°С.
Таким образом полученное соединение, представленное формулой (ХХ), можно подвергать последующей стадии, после или без выделения или очистки посредством известных способов выделения и очистки, таких как концентрирование, концентрирование в вакууме, кристаллизация, экстракция растворителем, переосаждение и хроматография.
Стадия t
Эту стадию можно осуществлять таким же образом, как стадию f.
Стадия u
Эту стадию можно осуществлять таким же образом, как стадию g.
Способ получения 6
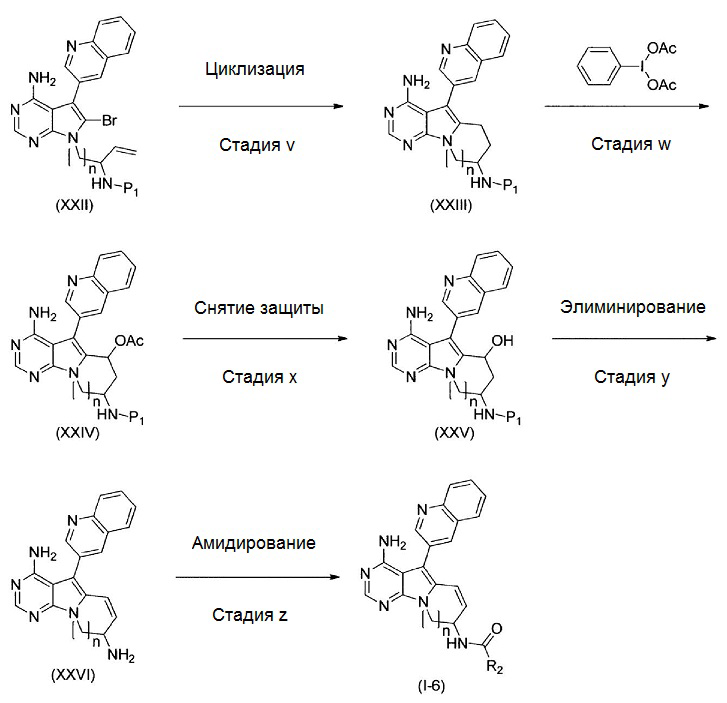
где Р1 представляет собой защитную для аминогруппы группу; и R2 и n имеют значения, как определено выше.
Стадия v
На этой стадии, органический борановый реагент вводят во взаимодействие с соединением, представленным формулой (XXII), до получения промежуточного алкилборанового соединения в системе и затем, в присутствии катализатора на основе переходного металла и основания, получают соединение, представленное формулой (XXIII).
Эту стадию можно осуществлять в соответствии с общеизвестным способом (например, WO 2006/102079).
Примеры органических борановых реагентов включают 9-BBN(9-борабицикло[3,3,1]-нонан), димер 9-BBN(9-борабицикло[3,3,1]-нонана), дисиамилборан(бис(1,2-диметилпропил)боран), тексилборан(1,1,2-триметилпропил)боран) и т.п. Органический борановый реагент представляет собой, предпочтительно, 9-BBN(9-борабицикло[3,3,1]-нонан или димер 9-BBN(9-борабицикло[3,3,1]-нонана) и, особенно предпочтительно, 9-BBN(9-борабицикло[3,3,1]-нонан). Используемое количество органического боранового реагента не особенно ограничено, до тех пор, пока может быть получено помежуточное алкилборановое соединение. Органический борановый реагент можно использовать в количестве от 1 моль до 20 моль, в расчете на моль соединения, представленного формулой (XXII); количество органического боранового реагента составляет, предпочтительно, от 6 моль до 10 моль, с точки зрения облегчения прогрессирования реакции.
В качестве катализатора на основе переходного металла можно использовать, например, катализатор на основе двухвалентного палладия (например, ацетат палладия, хлорид палладия и 1,1’-бис(дифенилфосфино)ферроценпалладий(II)дихлорид). Если необходимо, можно использовать лиганд (например, трифенилфосфин и три-трет-бутилфосфин). Используемое количество катализатора на основе переходного металла может изменяться в зависимости от типа катализатора. Катализатор на основе переходного металла типично используют в количестве от 0,0001 моль до 1 моль и, предпочтительно, от 0,01 моль до 0,5 моль, в расчете на моль соединения, представленного формулой (XXII). Лиганд типично используют в количестве от 0,0001 моль до 4 моль и, предпочтительно, от 0,01 моль до 2 моль, в расчете на моль соединения, представленного формулой (XXII).
Альтернативно, например, может быть использован катализатор на основе палладия с нулевой валентностью. Примеры катализаторов на основе палладия с нулевой валентностью включают тетракис(трифенилфосфин)палладий, трис(дибензилиденацетон)дипалладий, палладий-на-угле и т.п. Предпочтительным является тетракис(трифенилфосфин)палладий или трис(дибензилиденацетон)дипалладий и, особенно предпочтительным, является тетракис(трифенилфосфин)палладий. Используемое количество катализатора на основе палладия с нулевой валентностью не особенно ограничено, до тех пор, пока можно осуществлять реакцию внутримолекулярной циклизации, и может изменяться в зависимости от типа катализатора. Катализатор на основе палладия с нулевой валентностью можно использовать в количестве от 0,0001 моль до 1 моль и, предпочтительно, от 0,01 моль до 0,5 моль, в расчете на моль соединения, представленного формулой (XXII).
Если необходимо, вместе с катализатором на основе палладия с нулевой валентностью можно добавлять лиганд. Примеры таких лигандов включают трифенилфосфин, 1,1’-бис(дифенилфосфино)ферроцен, три-трет-бутилфосфин, трициклогексилфосфин, 2-дициклогексилфосфино-2’,6’-диметоксибифенил, 2-дициклогексилфосфино-2’,4’,6’-триизопропилбифенил, 2-(ди-трет-бутилфосфино)бифенил, 2-дициклогексилфосфино-2’-(N,N-диметиламино)бифенил, 4,5’-бис(дифенилфосфино)-9,9’-диметилксантен и т.п. Когда используют трис(дибензилиденацетон)дипалладий в качестве катализатора на основе палладия с нулевой валентностью, в качестве лиганда можно добавлять трифенилфосфин. Используемое количество лиганда не особенно ограничено, до тех пор, пока можно осуществлять реакцию внутримолекулярной циклизации. Лиганд можно использовать в количестве от 0,0001 моль до 4 моль и, предпочтительно, от 0,01 моль до 2 моль, в расчете на моль соединения, представленного формулой (XXII).
Примеры оснований включают неорганические основания, как например гидрокарбонат натрия, карбонат натрия, карбонат калия, карбонат цезия и гидроксиды щелочных металлов. Предпочтительными являются гидроксиды щелочных металлов. Примеры гидроксидов щелочных металлов включают гидроксид лития, гидроксид натрия, гидроксид калия и гидроксид цезия. Предпочтительно используют гидроксид лития, гидроксид натрия, гидроксид калия или гидроксид цезия. Особенно предпочтительным является гидроксид лития или гидроксид натрия. Используемое количество основания не особенно ограничено, до тех пор, пока протекает реакция. Основание можно использовать в количестве от 1 моль до 100 моль и, предпочтительно, от 2 моль до 20 моль, в расчете на моль соединения, представленного формулой (XXII). Гидроксид щелочного металла можно использовать в форме водного раствора гидроксида щелочного металла.
В качестве комбинации из органического боранового реагента, гидроксида щелочного металла и катализатора на основе палладия с нулевой валентностью предпочтительной является комбинация из предпочтительного органического боранового реагента, предпочтительного гидроксида щелочного металла и предпочтительного катализатора на основе палладия с нулевой валентностью. Особенно предпочтительной является комбинация из особенно предпочтительного органического боранового реагента, особенно предпочтительного гидроксида щелочного металла и особенно предпочтительного катализатора на основе палладия с нулевой валентностью.
Можно использовать любой растворитель, который не оказывает неблагоприятного влияния на реакцию. Примеры такого растворителя включают углеводороды (например, бензол, толуол и ксилол), простые эфиры (например, 1,2-диметоксиэтан, тетрагидрофуран и 1,4-диоксан), апротонные полярные растворители (например, N,N-диметилформамид, диметилсульфоксид и гексаметилфосфориламид), воду и их смеси. Предпочтительно используют 1,2-диметоксиэтан или тетрагидрофуран. Тетрагидрофуран является особенно предпочтительным с точки зрения стабильности органического боранового реагента и генерируемого алкилборанового промежуточного соединения. Используемое количество растворителя не особенно ограничено, до тех пор, пока протекает реакция. Растворитель можно использовать в количестве, являющимся 1-300-кратным и, предпочтительно, 10-96-кратным массе соединения, представленного формулой (XXII).
Время реакции не особенно ограничено, до тех пор, пока может быть получено соединение формулы (XXIII). Время реакции может составлять от 0,1 часа до 100 часов и, предпочтительно, от 0,5 часа до 24 часов.
Температура реакции не особенно ограничена, до тех пор, пока, в конечном счете, может быть получено соединение, представленное формулой (XXIII). Температура реакции может составлять от -20°С до температуры кипения растворителя и, предпочтительно, от 0°С до 150°С. В случае реакции внутримолекулярной циклизации промежуточного алкилборанового соединения, при использовании катализатора на основе палладия с нулевой валентностью и водного раствора гидроксида щелочного металла, низкая температура реакции приводит к возникновению побочных реакций, которые, в свою очередь, приводят к низкому выходу. Поэтому температура составляет предпочтительно 61°С или выше.
Таким образом полученное соединение, представленное формулой (XXIII), можно подвергать последующей стадии, после или без выделения или очистки известными способами выделения и очистки, как например концентрирование, концентрирование в вакууме, кристаллизация, экстракция растворителем, переосаждение и хроматография.
На этой стадии, может быть подтверждено образование алкилборанового промежуточного соединения в системе. Например, в качестве способа подтверждения можно использовать спектры LCMS.
Стадия w
На этой стадии, соединение, представленное формулой (XXIV), получают путем введения во взаимодействие соединения, представленного формулой (XXIII), с иодбензолдиацетатом.
Количество иодбензолдиацетата, используемое на этой стадии, составляет от 1 моль до 10 моль, предпочтительно, от 1 моль до 2 моль, в расчете на моль соединения, представленного формулой (XXIII).
В случае вышеуказанной реакции, если необходимо, можно добавлять тетрабутиламмонийиодид. Тетрабутиламмонийиодид можно добавлять в количестве от 0,01 моль до 10 моль, предпочтительно, от 0,1 моль до 1 моль, в расчете на моль соединения, представленного формулой (XXIII).
Можно использовать любой растворитель для реакции, который не оказывает неблагоприятного воздействия на реакцию. Примеры пригодных растворителей для реакции включают метиленхлорид, хлороформ, 1,2-дихлорэтан, уксусную кислоту и получаемые из них смешанные растворители.
Температура реакции обычно составляет от 0°С до температуры кипения растворителя и, предпочтительно, от 0°С до комнатной температуры. Время реакции составляет от 0,1 часа до 100 часов и, предпочтительно, от 0,1 часа до 24 часов.
Таким образом полученное соединение, представленное формулой (XXIV), можно подвергать последующей стадии, после или без выделения или очистки посредством известных способов выделения и очистки, таких как концентрирование, концентрование в вакууме, кристаллизация, экстракция растворителем, переосаждение и хроматография.
Стадия х
На этой стадии, снимают защиту с защищенной гидроксильной группы соединения, представленного формулой (XXIV), до получения соединения формулы (XXV).
Снятие защиты можно осуществлять с помощью известного способа, такого как способ, описанный в Protective Groups in Organic Synthesis, T.W. Greene, John Wiley & Sons (1981); или с помощью подобного способа.
Когда снимают защиту с ацетильной группы, примеры реагента для снятия защиты включают гидроксид натрия, гидроксид калия и т.п. Используемое количество реагента предпочтительно составляет от 1 моль до 100 моль, в расчете на моль соединения, представленного формулой (XXIV).
Можно использовать любой растворитель, который не оказывает неблагоприятного воздействия на реакцию. Примеры растворителя включают воду, метанол, этанол, тетрагидрофуран и получаемые из них смешанные растворители. Время реакции составляет от 0,1 часа до 100 часов и, предпочтительно, от 0,5 часа до 24 часов. Температура реакции составляет от 0°С до температуры кипения растворителя и, предпочтительно, от 0°С до 50°С.
Таким образом полученное соединение, представленное формулой (XXV), можно подвергать последующей стадии, после или без выделения или очистки посредством известных способов выделения и очистки, таких как концентрирование, концентрирование в вакууме, кристаллизация, экстракция растворителем, переосаждение и хроматография.
Стадия y
На этой стадии, соединение, представленное формулой (XXVI) получают, подвергая соединение, представленное формулой (XXV), реакции элиминирования.
В случае реакции элиминирования используют кислоты, такие как моногидрат п-толуолсульфоновой кислоты, 10-камфорсульфоновая кислота и т.п. Используемое количество кислоты составляет от 0,1 моль до 100 моль, предпочтительно, от 1 моль до 10 моль, в расчете на моль соединения, представленного формулой (XXV).
Можно использовать любой растворитель, который не оказывает неблагоприятного воздействия на реакцию. Примеры растворителя включают толуол, ксилол и получаемые из них смешанные растворители. Время реакции составляет от 0,1 часа до 100 часов и, предпочтительно, от 0,5 часа до 24 часов. Температура реакции находится в диапазоне от комнатной температуры до температуры кипения растворителя.
Таким образом полученное соединение, представленное формулой (XXVI), можно подвергать последующей стадии, после или без выделения или очистки посредством известных способов выделения и очистки, таких как концентрирование, концентрирование в вакууме, кристаллизация, экстракция растворителем, переосаждение и хроматография.
Стадия z
Эту стадию можно осуществлять таким же образом, как стадию g.
В случае вышеуказанных способов получения 1-6, для функциональных групп, имеющих активный протон, таких как аминогруппа, иминогруппа, гидроксильная группа, карбоксильная группа, карбонильная группа и амидная группа, и индол, можно использовать защитные реагенты или защитную группу можно вводить в такую функциональную группу в соответствии с обычным способом; впоследствии, защитную группу можно удалять на соответствующей стадии в случае каждого способа получения.
«Защитная для аминогруппы группа или защитная для иминогруппы группа» не является особо ограниченной, до тех пор, пока эта группа обладает защитной функцией. Примеры таких защитных групп включают аралкильные группы, такие как бензил, п-метоксибензил, 3,4-диметоксибензил, о-нитробензил, п-нитробензил, бензгидрил, тритил и кумил; низшие алканоильные группы, такие как формил, ацетил, пропионил, бутирил, пивалоил, трифторацетил и трихлорацетил; бензоил; арилалканоильные группы, такие как фенилацетил и феноксиацетил; низшие алкоксикарбонильные группы, такие как метоксикарбонил, этоксикарбонил, пропилоксикарбонил и трет-бутоксикарбонил; аралкилоксикарбонильные группы, такие как п-нитробензилоксикарбонил и фенетилоксикарбонил; низшие алкилсилильные группы, такие как триметилсилил и трет-бутилдиметилсилил; тетрагидропиранил; триметилсилилэтоксиметил; низшие алкилсульфонильные группы, такие как метилсульфонил, этилсульфонил и трет-бутилсульфонил; низшие алкилсульфинильные группы, такие как трет-бутилсульфинил; арилсульфонильные группы, такие как бензолсульфонил и толуолсульфонил; и имидогруппы, такие как фталимидо. В особенности, предпочтительными являются трифторацетил, ацетил, трет-бутоксикарбонил, бензилоксикарбонил, триметилсилилэтоксиметил, кумил и т.п.
«Защитная для гидроксильной группы группа» не является особо ограниченной, до тех пор, пока эта группа обладает защитной функцией. Примеры таких защитных групп включают низшие алкильные группы, такие как метил, этил, пропил, изопропил и трет-бутил; низшие алкилсилильные группы, такие как триметилсилил и трет-бутилдиметилсилил; низшие алкоксиметильные группы, такие как метоксиметил и 2-метоксиэтоксиметил; тетрагидропиранил; триметилсилилэтоксиметил; аралкильные группы, такие как бензил, п-метоксибензил, 2,3-диметоксибензил, о-нитробензил, п-нитробензил и тритил; и ацильные группы, такие как формил, ацетил и трифторацетил. В особенности, предпочтительными являются метил, метоксиметил, тетрагидропиранил, триметилсилилэтоксиметил, трет-бутилдиметилсилил и ацетил.
«Защитная для карбоксильной группы группа» не является особо ограниченной, до тех пор, пока эта группа обладает защитной функцией. Примеры таких защитных групп включают низшие алкильные группы, такие как метил, этил, пропил, изопропил и трет-бутил; низшие галогеналкильные группы, такие как 2,2,2-трихлорэтил; низшие алкенильные группы, такие как аллил; триметилсилилэтоксиметил; и аралкильные группы, такие как бензил, п-метоксибензил, п-нитробензил, бензгидрил и тритил. В особенности, предпочтительными являются метил, этил, трет-бутил, аллил, бензил, п-метоксибензил, триметилсилилэтоксиметил и т.п.
«Защитная для карбонильной группы группа» не является особо ограниченной, до тех пор, пока эта группа обладает защитной функцией. Примеры таких защитных групп включают этиленкеталь, триметиленкеталь, диметилкеталь и подобные кетали и ацетали.
Способ удаления такой защитной группы может варьироваться, в зависимости от типа защитной группы, стабильности желательного соединения (I) и т.д. Например, можно использовать следующие способы: сольволиз с использованием кислоты или основания, в соответствии со способом, раскрытым в публикации (Protective Groups in Organic Synthesis, третье изд., T.W. Greene, John Wiley & Sons (1999)), или подобный способ, то есть, способ, включающий введение во взаимодействие с 0,01 моль или большим избытком кислоты, предпочтительно, трифторуксусной кислоты, муравьиной кислоты или соляной кислоты, или с от эквимолярного до большого избыточного молярного количеством основания, предпочтительно, гидроксидом калия или гидроксидом кальция; химическое восстановление с использованием комплекса гидрида металла, и т.д.; или каталитическое восстановление с использованием катализатора палладий-на-угле, катализатора никель Ренея и т.д.
Соединение согласно настоящему изобретению можно выделять и очищать с помощью обычных способов выделения и очистки. Примеры таких способов включают экстракцию растворителем, перекристаллизацию, препаративную высокоэффективную жидкостную хроматографию с обращенными фазами, колоночную хроматографию, препаративную тонкослойную хроматографию и т.п.
Если соединение согласно настоящему изобретению имеет изомеры, такие как оптические изомеры, стереоизомеры, региоизомеры и поворотные изомеры, любые изомеры и их смеси включены в рамки соединения согласно настоящему изобретению. Например, если соединение имеет оптические изомеры, то оптический изомер, выделенный из рацемической смеси, также включен в рамки соединения согласно настоящему изобретению. Каждый из таких изомеров можно получать в виде индивидуального соединения с помощью известного синтеза и способов разделения (например, концентрирование, экстракция растворителем, колоночная хроматография, перекристаллизация и т.д.).
Согласно настоящему изобретению, атом углерода, связанный с заместителем В, в формуле (I), представляет собой асимметрический атом углерода; следовательно, соединение включает изомеры. Как указано выше, за исключением иначе указанного, соединение согласно настоящему изобретению включает все энантиомеры и их смеси. Соединение согласно настоящему изобретению может представлять собой смесь R- и S-энантиомеров. Такая смесь может представлять собой смесь, содержащую 90% или более, 95% или более, или 99% или более R-энантиомера; или смесь, содержащую 90% или более, 95% или более, или 99% или более S-энантиомера.
Способы хирального разделения включают, например: диастереомерный способ, основанный на введении во взаимодействие хирального агента разделения с соединением согласно настоящему изобретению до образования соли и выделении одного из энантиомеров, используя различие в растворимости и т.д., из полученной соли; предпочтительный способ кристаллизации путем добавления одного из энантиомеров к перенасыщенному раствору рацемата, в качестве затравки, для кристаллизации; и колоночную хроматографию, такую как ВЭЖХ, с использованием хиральной колонки. Хиральный агент разделения, который можно использовать при диастереомерном способе, можно, соответственно, выбирать, например, из группы, состоящей из кислотных агентов разделения, таких как винная кислота, яблочная кислота, молочная кислота, миндальная кислота, 10-камфорсульфоновая кислота и их производные; и основных агентов разделения, таких как бруцин, стрихнин, хинин и подобные алкалоидные соединения, аминокислотные производные, цинхонидин и α-метилбензиламин. В дополнение, один из энантиомеров соединения согласно настоящему изобретению можно получать не только путем получения соединения согласно настоящему изобретению в виде смеси из каждого из энантиомеров и затем осуществления вышеуказанных способов хирального разделения, но и также путем получения, посредством хирального разделения с помощью вышеуказанных способов и т.д., и использования одного энантиомера соединения согласно настоящему изобретению в качестве синтетического сырья. Кроме того, способы получения одного из энантиомеров соединения согласно настоящему изобретению или соединения в виде его сырья включают способ предпочтительного получения одного из энантиомеров путем регулирования условий реакции для катализатора, или т.п., на стадии реакции получения асимметрического атома углерода.
Соединение согласно настоящему изобретению, или его соль, может быть в форме кристаллов. Отдельные кристаллы и полиморфные смеси включены в рамки соединения согласно настоящему изобретению или его соли. Такие кристаллы можно получать путем кристаллизации в соответствии со способом кристаллизации, само по себе известным в данной области. Соединение согласно настоящему изобретению, или его соль, может быть в виде сольвата (например, гидрат) или в несольватированной форме. Любые такие формы включены в рамки соединения согласно настоящему изобретению, или его соли. Соединения, меченые изотопом (например, 3Н, 14С, 35S и 125I) также включены в рамки соединения согласно настоящему изобретению или его соли.
Соль соединения согласно настоящему изобретению или его промежуточного продукта относится к обычной соли, используемой в области органической химии. Примеры таких солей включают аддитивные соли с основаниями по карбоксильной группе, когда соединение имеет карбоксильную группу, и аддитивные соли с кислотами по аминогруппе или основной гетероциклической группе, когда соединение имеет аминогруппу или основную гетероциклическую группу.
Примеры аддитивных солей с основаниями включают соли щелочных металлов, как например соли натрия и соли калия; соли щелочноземельных металлов, как например соли кальция и соли магния; соли аммония; и соли органических аминов, как например соли триметиламина, соли триэтиламина, соли дициклогексиламина, соли этаноламина, соли диэтаноламина, соли триэтаноламина, соли прокаина и соли N,N’-дибензилэтилендиамина.
Примеры аддитивных солей с кислотами включают соли неорганических кислот, как например гидрохлориды, сульфаты, нитраты, фосфаты и перхлораты; соли органических кислот, как например ацетаты, формиаты, малеаты, фумараты, тартраты, цитраты, аскорбаты и трифторацетаты; и сульфонаты, как например метансульфонаты, изетионаты, бензолсульфонаты и п-толуолсульфонаты.
Соединение согласно настоящему изобретению, или его соль, обладает превосходной ингибирующей активностью по отношению к EGFR и является пригодным в качестве противоопухолевого агента. Далее, соединение согласно настоящему изобретению или его соль обладает превосходной селективностью по отношению к EGFR и, преимущественно, более незначительным числом побочных эффектов, вызываемых другими киназами. Тип злокачественной опухоли, которую подвергают лечению с помощью соединения согласно настоящему изобретению или его соли не является особо ограниченным. Примеры злокачественных опухолей включают эпителиальные раковые заболевания (например, раковые заболевания системы дыхания, раковые заболевания пищеварительной системы, раковые заболевания репродуктивной системы, раковые заболевания системы секреции и т.п.), саркомы, гемопоэтические опухоли, опухоли центральной нервной системы и опухоли периферической нервной системы. Предпочтительные примеры включают эпителиальные раковые заболевания. Более предпочтительные примеры включают раковые заболевания системы дыхания. Далее, орган, из которого развивается опухоль, не является особо ограниченным. Примеры включают раковые заболевания головы и шеи, рак пищевода, рак желудка, рак толстой кишки, рак прямой кишки, рак печени, рак желчного пузыря, холангиокарциному, рак желчных протоков, рак поджелудочной железы, рак легких, рак молочной железы, рак яичников, цервикальный рак, внутриматочный рак, рак почек, рак мочевого пузыря, рак предстательной железы, тестикулярную опухоль, остеосаркому, саркому мягких тканей, рак крови, множественную миелому, рак кожи, опухоль головного мозга и мезотелиому. Предпочтительно, раковое заболевание - мишень представляет собой раковые заболевания головы и шеи, рак желудка, рак толстой кишки, рак прямой кишки, рак печени, рак поджелудочной железы, рак легких, рак молочной железы, рак яичников, рак почек или рак предстательной железы, в особенности, предпочтительным является рак легких.
Далее, соединение согласно настоящему изобретению, или его соль, обладает превосходной ингибирующей активностью против мутированного EGFR. Примеры такого мутированного EGFR включают толерантный к лекарственному средству мутированный EGFR и гиперчувствительный мутированный EGFR. Следовательно, соединение согласно настоящему изобретению, или его соль, является пригодным в качестве противоопухолевого агента для лечения вышеуказанных злокачественных опухолей, имеющих мутированный EGFR.
Если соединение согласно настоящему изобретению, или его соль, используют в виде фармацевтического препарата, можно добавлять фармацевтический носитель, если необходимо, таким образом получая подходящую дозированную форму согласно целям предупреждения и лечения. Примеры дозированной формы включают пероральные препараты, инъекции, суппозитории, мази, пластыри и т.п. Из них предпочтительными являются пероральные препараты. Такие дозированные формы можно получать посредством способов, обычно известных квалифицированному специалисту в данной области.
В качестве фармацевтического носителя, можно применять различные стандартные органические или неорганические носители, используемые в качестве веществ для получения фармацевтического препарата, как эксципиент, связующее вещество, дезинтегрирующий агент, лубрикант или краситель, в случае твердых препаратов; или как растворитель, солюбилизирующий агент, суспендирующий агент, изотонизирующий агент, буфер или успокаиващее средство, в случае жидких препаратов. Кроме того, также можно использовать, если необходимо, добавки к фармацевтическому препарату, такие как антисептики, антиоксиданты, красители, подсластители и стабилизаторы.
Пероральные твердые препараты получают следующим образом.
После эксципиента, к соединению согласно настоящему изобретению необязательно добавляют связующее вещество, дезинтегрирующий агент, лубрикант, краситель, маскирующий вкус агент или ароматизатор и т.д., полученную в результате смесь доводят до готовой дозированной формы в виде таблеток, таблеток с нанесенным покрытием, гранул, порошков, капсул или т.п., посредством стандартных способов.
Примеры эксципиентов включают лактозу, сахарозу, D-маннит, глюкозу, крахмал, карбонат кальция, каолин, микрокристаллическую целлюлозу и ангидрид кремниевой кислоты. Примеры связующих веществ включают воду, этанол, 1-пропанол, 2-пропанол, сахарный сироп, раствор глюкозы, жидкий α-крахмал, жидкий желатин, D-маннит, карбоксиметилцеллюлозу, гидроксипропилцеллюлозу, гидроксипропилкрахмал, метилцеллюлозу, этилцеллюлозу, шеллак, фосфат кальция, поливинилпирролидон и т.п. Примеры дезинтегрирующих агентов включают сухой крахмал, альгинат натрия, порошкоообразный агар, гидрокарбонат натрия, карбонат кальция, лаурилсульфат натрия, моноглицерид стеариновой кислоты, лактозу и т.п. Примеры лубрикантов включают очищенный тальк, натриевую соль стеариновой кислоты, стеарат магния, буру, полиэтиленгликоль и т.п. Примеры красителей включают оксид титана, оксид железа и т.п. Примеры маскирующих вкус агентов или ароматизаторов включают сахарозу, горькую апельсиновую цедру, лимонную кислоту, винную кислоту и т.п.
Когда получают жидкий препарат для перорального введения, к соединению согласно настоящему изобретению можно добавлять маскирующий вкус агент, буфер, стабилизатор, ароматизатор и т.п.; и полученную смесь можно доводить до готовой дозированной формы в виде перорального жидкого препарата, сиропа, эликсира и т.д., в соответствии со стандартным способом.
В этом случае, можно использовать такой же маскирующий вкус агент или ароматизатор, как таковой, указанный выше. Примером буфера является цитрат натрия и примеры стабилизаторов включают трагакант, гуммиарабик и желатин. Если необходимо, на эти препараты для перорального введения можно наносить покрытие в соответствии со способами, известными в данной области, в виде энтеросолюбильного покрытия или другого покрытия для цели, например, стойкости эффектов. Примеры таких агентов для нанесения покрытия включают гидроксипропилметилцеллюлозу, этилцеллюлозу, гидроксиметилцеллюлозу, гидроксипропилцеллюлозу, полиоксиэтиленгликоль и Tween 80 (зарегистрированная торговая марка).
Когда получают агент для инъекции, к соединению согласно настоящему изобретению можно добавлять регулятор рН, буфер, стабилизатор, изотонизирующий агент, местное анестезирующее средство и т.п.; и смесь можно доводить до готовой дозированной формы в виде подкожной, внутримышечной или внутривенной инъекции, в соответствии со стандартным способом.
Примеры регулятора рН и буфера, используемых согласно данному контексту, включают цитрат натрия, ацетат натрия и фосфат натрия. Примеры стабилизатора включают пиросульфит натрия, EDTA, тиогликолевую кислоту и тиомолочную кислоту. Примеры местного анестезирующего средства включают прокаингидрохлорид и лидокаингидрохлорид. Примеры тонизирующего агента включают хлорид натрия, декстрозу, D-маннит и глицерин.
Когда получают суппозиторий, к соединению согласно настоящему изобретению можно добавлять фармацевтически приемлемые носители, известные в данной области, такие как полиэтиленгликоль, ланолин, масло какао и триглицерид жирной кислоты; и, если необходимо, поверхностно-активные вещества, такие как Tween 80 (зарегистрированная торговая марка), и полученную смесь можно доводить до готовой дозированной формы в виде суппозитория, в соответствии со стандартным способом.
Когда получают мазь, обычно используемую(мый) основу, стабилизатор, смачивающий агент, консервант и т.п. можно смешивать с соединением согласно настоящему изобретению, когда необходимо; и полученную смесь можно перемешивать и доводить до готовой дозированной формы в виде мази, в соответствии со стандартным способом.
Примеры основы включают жидкий парафин, белый вазелин, белый воск, октилдодециловый спирт и парафин.
Примеры консервантов включают метилпараоксибензоат, этилпараоксибензоат и пропилпараоксибензоат.
Когда получают пластырь, к стандартному субстрату можно добавлять вышеуказанную мазь, крем, гель, пасту или т.п., в соответствии со стандартным способом.
В качестве субстрата пригодными являются тканые или нетканые материалы, включающие хлопок, штапельные волокна или химические волокна; и пленки или листовые пенопласты из пластичного поливинилхлорида, полиэтилена, полиуретана и т.д.
Количество соединения согласно настоящему изобретению, включенное в каждую из таких единичных дозированных форм, зависит от состояния пациента, которому вводят соединение, дозированной формы этого соединения и т.д. Вообще, в случае перорального агента, количество соединения составляет от 0,05 мг до 1000 мг на единичную дозированную форму. В случае инъекции, количество соединения составляет от 0,01 мг до 500 мг на единичную дозированную форму; и, в случае суппозитория, количество соединения составляет от 1 мг до 1000 мг на единичную дозированную форму.
Суточная доза лекарственного средства в такой дозированной форме зависит от состояния, массы тела, возраста, анатомического пола и т.д. пациента и не может быть генерализована. Например, суточная доза для взрослого пациента (масса тела: 50 кг) в большинстве случаев может составлять 0,05-5000 мг и, предпочтительно, 0,1-1000 мг; и, предпочтительно, эту дозу вводят в виде одной дозы или в виде от двух до трех разделенных доз, в сутки.
Примеры млекопитающих, которым вводят соединение согласно настоящему изобретению, включают людей, обезьян, мышей, крыс, кроликов, собак, кошек, коров, лошадей, свиней и овец.
Примеры
Настоящее изобретение более конкретно поясняется ниже, со ссылкой на примеры; однако, настоящее изобретение не ограничено этими примерами.
В примерах, за исключением иначе указанного, использовали коммерчески доступные реагенты. Для колоночной хроматографии на силикагеле использовали Purif-Pack® SI, выпускаемый фирмой Moritex Corp. (производимый фирмой Shoko Scientific Co., Ltd.); предварительно заполненную KP-Sil® Silica колонку, выпускаемую фирмой Biotage; предварительно заполненную HP-Si® Silica колонку, выпускаемую фирмой Biotage, или предварительно заполненную HP-Sphere® Silica колонку, выпускаемую фирмой Biotage. Для колоночной хроматографии на основном силикагеле использовали Purif-Pack® NH, выпускаемый фирмой Moritex Corp. (производимый фирмой Shoko Scientific Co., Ltd.); или предварительно заполненную KP-NH® колонку, выпускаемую фирмой Biotage. Для препаративной тонкослойной хроматографии использовали Kieselgel TM 60F254, Art. 5744, выпускаемый фирмой Merk, или пластинку NH2 Silica Gel 60F254, выпускаемую фирмой Wako. ЯМР-спектр снимали, используя спектрометр AL400 (400 МГц; выпускаемый фирмой JEOL) или Mercury 400 (400 МГц; выпускаемый фирмой Agilent Technologies, Inc.). Когда дейтерированный растворитель содержит тетраметилсилан, тетраметилсилан используют в качестве внутреннего стандарта. Другим образом, растворитель для ЯМР использовали в качестве внутреннего стандарта. Все значения δ представляли в м.д. Реакцию под воздействием микроволнового излучения осуществляли, используя инициатор, выпускаемый фирмой Biotage.
Далее, спектр LCMS получали, используя прибор Acquity SQD (квадрупольный), выпускаемый фирмой Waters Corporation, при следующих условиях.
Колонка: Acquity UPLC® BEH C18, размером 2,1×50 мм, 1,7 мкм, выпускаемая фирмой Waters Corporation;
MS-детектирование: ESI с образованием положительных ионов;
УФ-детектирование: 254 и 210 нм;
Объемная скорость потока через колонку: 0,5 мл/мин;
Подвижная фаза: вода/ацетонитрил (0,1% муравьиной кислоты);
Объем введенной пробы: 1 мкл;
Градиент
Очистку с помощью препаративной ВЭЖХ с обращенными фазами осуществляли, используя препаративную систему разделения, доступную от фирмы Gilson.
Колонка: CombiPrep Pro C18, размером 50×30 мм (внутренний диаметр), S-5 мкм (выпускаемая фирмой YMC);
УФ-детектирование: 254 нм;
Объемная скорость потока через колонку: 40 мл/мин;
Подвижная фаза: вода/ацетонитрил (0,1% трифторуксусной кислоты);
Объем введенной пробы: от 0,1 до 1 мл.
Каждый символ представляет собой следующее.
s (с): синглет
d (д): дублет
t (т): триплет
q (кв.): квартет
dd (дд): дублет дублетов
dt (дт): дублет триплетов
ddd (ддд): дублет дублетов дублетов
m (м): мультиплет
brs (уш.с): уширенный синглет
DMSO-d6 (ДМСО-d6): дейтерированный диметилсульфоксид
CDCl3: дейтерированный хлороформ
CD3OD: дейтерированный метанол
THF (ТГФ): тетрагидрофуран
DMF (ДМФА): N,N-диметилформамид
DME: 1,2-диметоксиэтан
DMSO (ДМСО): диметилсульфоксид
HATU: О-(7-азабензотриазол-1-ил)-N,N,N’,N’-тетраметилуронийгексафторфосфат
Пример 1
(S)-N-(4-Амино-6-метилен-5-(хинолин-3-ил)-7,8-дигидро-6Н-пиримидо[5,4-b]пирролизин-1-ил)акриламид (Соединение 1)
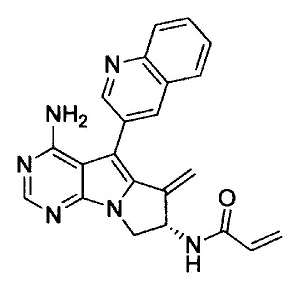
Стадия 1: Синтез (S)-трет-бутил(1-(4-хлор-5-иод-7Н-пирроло[2,3-d]пиримидин-7-ил)бут-3-ен-2-ил)карбамата
Диизопропилазодикарбоксилат (2,44 мл) медленно добавляют к раствору трифенилфосфина (13,1 г) в ТГФ (70 мл), при охлаждении льдом. Реакционную смесь перемешивают, при охлаждении льдом, в течение 1 часа и затем к ней медленно добавляют раствор (S)-трет-бутил(1-гидроксибут-3-ен-2-ил)карбамата (7,0 г), синтезированного в соответствии со способом, описанным в NPD Org. Lett., 2005, том 7, №5, сс.847-849, и 4-хлор-5-иод-7Н-пирроло[2,3-d]пиримидин (6,97 г), в ТГФ (35 мл). Потом реакционную смесь перемешивают при комнатной температуре, в течение 2 часов, растворитель отгоняют при пониженном давлении. Полученный остаток очищают с помощью колоночной хроматографии на силикагеле (элюирующий растворитель: гексан/этилацетат), таким образом получая указанное в заголовке соединение (20,84 г) в виде маслянистого вещества светло-желтого цвета.
ESI-MS m/z: 448, 450 [M+H]+.
Стадия 2: Синтез (S)-трет-бутил(1-(4-амино-5-иод-7Н-пирроло[2,3-d]пиримидин-7-ил)бут-3-ен-2-ил)карбамата
8 н Раствор аммиака в метаноле (89,4 мл) добавляют к (S)-трет-бутил(1-(4-хлор-5-иод-7Н-пирроло[2,3-d]пиримидин-7-ил)бут-3-ен-2-ил)карбамату (20,84 г), полученному на стадии 1, и смесь перемешивают в автоклаве при температуре 120°С, в течение 6 часов. Реакционную смесь охлаждают льдом и растворитель отгоняют при пониженном давлении. Потом полученный остаток разбавляют небольшим количеством метанола, выпавший осадок собирают путем фильтрации, промывают холодным метанолом (11 мл) и затем высушивают при пониженном давлении, таким образом получая указанное в заголовке соединение (8,28 г), в виде твердого вещества молочно-белого цвета.
ESI-MS m/z: 430 [M+H]+.
Стадия 3: Синтез (S)-трет-бутил(1-(4-амино-5-(хинолин-3-ил)-7Н-пирроло[2,3-d]пиримидин-7-ил)бут-3-ен-2-ил)карбамата
Смесь (S)-трет-бутил(1-(4-амино-5-иод-7Н-пирроло[2,3-d]пиримидин-7-ил)бут-3-ен-2-ил)карбамата (8,26 г), полученного на стадии 2, 3-хинолинбороновой кислоты (4,99 г), карбоната цезия (12,54 г), 1,1’-бис(дифенилфосфино)ферроцен-палладий(II)дихлорида (II) (785,6 мг), DME (66 мл) и воды (33 мл) перемешивают в атмосфере азота при температуре 100°С, в течение 2 часов. После охлаждения реакционной смеси, туда добавляют воду и этилацетат для отделения органического слоя. Водный слой затем дважды экстрагируют этилацетатом. Полученный органический слой сушат над безводным сульфатом магния и растворитель отгоняют при пониженном давлении. Полученный остаток очищают с помощью колоночной хроматографии на силикагеле (элюирующий растворитель: гексан/этилацетат, этилацетат/метанол), таким образом получая указанное в заголовке соединение (8,0 г), в виде твердого вещества светло-оранжевого цвета.
ESI-MS m/z: 431 [M+H]+.
Стадия 4: Синтез (S)-трет-бутил(1-(4-амино-6-бром-5-(хинолин-3-ил)-7Н-пирроло[2,3-d]пиримидин-7-ил)бут-3-ен-2-ил)карбамата
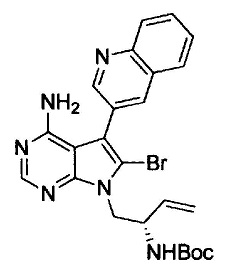
N-Бромсукцинимид (3,63 г) добавляют к раствору (S)-трет-бутил(1-(4-амино-5-(хинолин-3-ил)-7Н-пирроло[2,3-d]пиримидин-7-ил)бут-3-ен-2-ил)карбамата (7,98 г), полученного на стадии 3, в ДМФА (64 мл), при температуре -15°С, и смесь перемешивают при температуре -15°С, в течение 1 часа. К реакционной смеси добавляют 10%-ный водный раствор тиосульфата натрия и этилацетат, и перемешивают при комнатной температуре в течение 10 мин. Органический слой отделяют, а водный слой дважды экстрагируют этилацетатом. Полученный органический слой дважды промывают насыщенным раствором хлорида натрия и сушат над безводным сульфатом магния. Растворитель отгоняют при пониженном давлении. Полученный остаток очищают с помощью колоночной хроматографии на силикагеле (элюирующий растворитель: этилацетат/метанол), таким образом получая указанное в заголовке соединение (6,30 г), в виде твердого вещества светло-коричневого цвета.
ESI-MS m/z: 509, 511 [M+H]+.
Стадия 5: Синтез (S)-трет-бутил(4-амино-6-метилен-5-(хинолин-3-ил)-7,8-дигидро-6Н-пиримидо[5,4-b]пирролизин-7-ил)карбамата
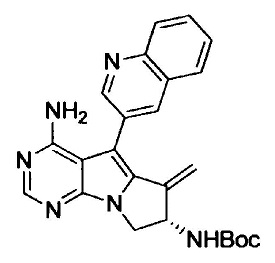
4 н Водный раствор гидроксида натрия (28,8 мл) добавляют к раствору (S)-трет-бутил(1-(4-амино-6-бром-5-(хинолин-3-ил)-7Н-пирроло[2,3-d]пиримидин-7-ил)бут-3-ен-2-ил)карбамата (5,00 г), полученного на стадии 4, в ТГФ (100 мл), и смесь дегазируют при пониженном давлении, затем продувают азотом. Потом добавляют тетракис(трифенилфосфин)палладий (1,13 г), смесь перемешивают в течение ночи при кипячении с обратным холодильником. Затем реакционную смесь охлаждают до комнатной температуры, осуществляют экстракцию, используя этилацетат, и полученный органический слой промывают насыщенным раствором хлорида натрия и сушат над безводным сульфатом натрия. После фильтрации и концентрирования, полученный остаток очищают с помощью хроматографии на силикагеле (элюирующий растворитель: этилацетат/метанол), таким образом получая указанное в заголовке соединение, в виде твердого вещества оранжевого цвета (5,01 г).
ESI-MS m/z: 429 [M+H]+.
Стадия 6: Синтез Соединения 1
4 н Раствор хлороводорода в диоксане (1 мл) и метанол (1 мл) добавляют к раствору (S)-трет-бутил(4-амино-6-метилен-5-(хинолин-3-ил)-7,8-дигидро-6Н-пиримидо[5,4-b]пирролизин-7-ил)карбамата (100 мг), полученного на стадии 5, в хлороформе (1 мл), и смесь перемешивают в течение 5 часов, при комнатной температуре. Реакционную смесь концентрируют при пониженном давлении, таким образом получая (S)-6-метилен-5-(хинолин-3-ил)-7,8-дигидро-6Н-пиримидо[5,4-b]пирролизин-4,7-диамингидрохлорид.
Диизопропилэтиламин (0,187 мл) добавляют к раствору (S)-6-метилен-5-(хинолин-3-ил)-7,8-дигидро-6Н-пиримидо[5,4-b]пирролизин-4,7-диамингидрохлорида в хлороформе (4 мл) и, при охлаждении льдом, добавляют 100 мг/мл раствора акрилоилхлорида в хлороформе (0,19 мл). Смесь перемешивают в течение 40 мин. Реакционную смесь очищают с помощью хроматографии на силикагеле (элюирующий растворитель: этилацетат/метанол); после концентрирования, полученный остаток суспендируют и промывают при использовании смеси этилацетат/гексан. Полученное твердое вещество собирают путем фильтрации и высушивают при пониженном давлении, таким образом получая указанное в заголовке соединение (49 мг), в виде твердого вещества светло-зеленого цвета.
1Н-ЯМР (ДМСО-d6) δ: 3,88-3,93 (1Н, м), 4,57-4,63 (1Н, м), 5,03 (1Н, д, J=2,4 Гц), 5,24 (1Н, д, J=2,4 Гц), 5,55-5,62 (1Н, м), 5,68 (1Н, дд, J=10,0, 2,4 Гц), 6,12-6,38 (4Н, м), 7,65 (1Н, дд, J=7,8, 7,8 Гц), 7,77-7,83 (1Н, м), 8,04-8,11 (2Н, м), 8,15 (1Н, с), 8,41 (1Н, д, J=2,2 Гц), 8,82 (1Н, д, J=7,8 Гц), 8,98 (1Н, д, J=2,2 Гц).
ESI-MS m/z: 383 [M+H]+.
Пример 2
(S)-N-(4-Амино-6-метилен-5-(хинолин-3-ил)-7,8-дигидро-6Н-пиримидо[5,4-b]пирролизин-7-ил)метакриламид (Соединение 2)
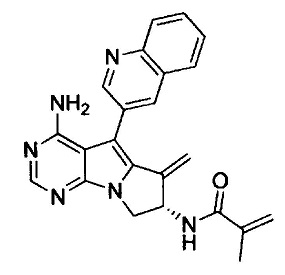
Стадия 1: Синтез (S)-6-метилен-5-(хинолин-3-ил)-7,8-дигидро-6Н-пиримидо[5,4-b]пирролизин-4,7-диамина
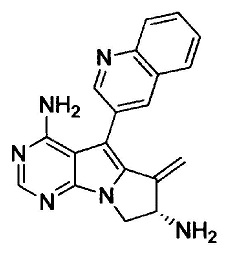
5 н Соляную кислоту (12 мл) добавляют к раствору (S)-трет-бутил(4-амино-6-метилен-5-(хинолин-3-ил)-7,8-дигидро-6Н-пиримидо[5,4-b]пирролизин-7-ил)карбамата (5,01 г), полученного на стадии 5 Примера 1, в этаноле (25 мл), и полученную смесь перемешивают в течение 1 часа, при наружной температуре от 70°С до 80°С. Потом реакционную смесь охлаждают до комнатной температуры, промывают хлороформом и значение рН водного слоя доводят примерно до 10, используя 5 н водный раствор гидроксида натрия, затем экстрагируют хлороформом. Полученный органический слой сушат над безводным сульфатом натрия, потом отфильтровывают и концентрируют. Полученный остаток очищают с помощью хроматографии на основном силикагеле (элюирующий растворитель: хлороформ/метанол), таким образом получая указанное в заголовке соединение (2,10 г), в виде твердого вещества желтого цвета.
ESI-MS m/z: 329 [M+H]+.
Стадия 2: Синтез Соединения 2
Диизопропилэтиламин (0,0318 мл) и метакрилоилхлорид (0,0148 мл) последовательно добавляют к раствору (S)-6-метилен-5-(хинолин-3-ил)-7,8-дигидро-6Н-пиримидо[5,4-b]пирролизин-4,7-диамина (50,0 мг), полученного на стадии 1, в ацетонитриле (2,0 мл) и воде (2,0 мл), при температуре 0°С, и полученную смесь перемешивают в течение 45 мин, при той же температуре. Реакционную смесь выливают в насыщенный водный раствор бикарбоната натрия, потом экстрагируют хлороформом. Затем органический слой сушат над безводным сульфатом натрия, растворитель отгоняют при пониженном давлении. Полученный остаток очищают с помощью колоночной хроматографии на силикагеле (элюирующий растворитель: этилацетат/метанол), таким образом получая указанное в заголовке соединение (38,0 мг), в виде твердого вещества светло-желтого цвета.
1Н-ЯМР (CDCl3) δ: 2,02 (3Н, с), 4,03 (1Н, дд, J=11,6, 4,9 Гц), 4,70 (1Н, дд, J=11,6, 8,2 Гц), 5,20-5,22 (3Н, м), 5,44 (2Н, д, J=1,7 Гц), 5,73-5,75 (1Н, м), 5,82 (1Н, с), 6,89 (1Н, д, J=7,6 Гц), 7,61-7,65 (1Н, м), 7,76-7,80 (1Н, м), 7,87 (1Н, д, J=8,3 Гц), 8,15 (1Н, д, J=8,5 Гц), 8,21-8,25 (2Н, м), 9,06 (1Н, д, J=2,2 Гц).
ESI-MS m/z: 397 [M+H]+.
Пример 3
(S)-N-(4-Амино-6-метилен-5-(хинолин-3-ил)-7,8-дигидро-6Н-пиримидо[5,4-b]пирролизин-7-ил)бут-2-енамид (смесь Е- и Z- изомеров) (Соединение 3)
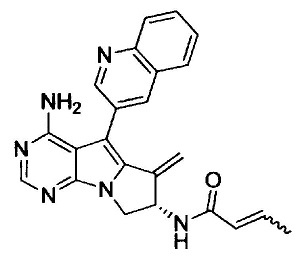
Такой же синтез, как на стадии 2 Примера 2, осуществляют, используя кротоноилхлорид вместо метакрилоилхлорида, используемого на стадии 2 Примера 2, таким образом получая указанную в заголовке смесь (8,8 мг), в виде твердого вещества светло-желтого цвета.
1Н-ЯМР (CDCl3) δ: 1,83-1,90 (3Н, м), 4,01 (1Н, дд, J=11,6, 5,2 Гц), 4,73 (1Н, дд, J=11,6, 8,2 Гц), 5,00 (2Н, с), 5,19 (1Н, д, J=1,5 Гц), 5,44 (1Н, д, J=2,0 Гц), 5,75-5,77 (1Н, м), 5,87-5,91 (1Н, м), 6,27 (1Н, д, J=8,3 Гц), 6,92-7,01 (1Н, м), 7,62-7,66 (1Н, м), 7,78-7,82 (1Н, м), 7,88 (1Н, д, J=7,3 Гц), 8,18 (1Н, д, J=8,0 Гц), 8,28-8,29 (2Н, м), 9,08 (1Н, д, J=2,2 Гц).
ESI-MS m/z: 397 [M+H]+.
Пример 4
(S,Е)-N-(4-Амино-6-метилен-5-(хинолин-3-ил)-7,8-дигидро-6Н-пиримидо[5,4-b]пирролизин-7-ил)-4-(диметиламино)бут-2-енамид (Соединение 4)
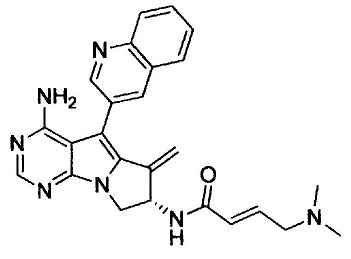
Гидрохлорид (Е)-4-(диметиламино)бут-2-еновой кислоты (60,6 мг), HATU (139 мг), диизопропилэтиламин (0,106 мл) и ДМФА (1,0 мл) последовательно добавляют к суспензии (S)-6-метилен-5-(хинолин-3-ил)-7,8-дигидро-6Н-пиримидо[5,4-b]пирролизин-4,7-диамина (100 мг), полученного на стадии 1 Примера 2, в метиленхлориде (3,0 мл), при комнатной температуре, и смесь перемешивают в течение 1,5 часов, при той же температуре. Реакционную смесь выливают в насыщенный водный раствор бикарбоната натрия, затем экстрагируют этилацетатом. Потом органический слой сушат над безводным сульфатом натрия, растворитель отгоняют при пониженном давлении. Полученный остаток очищают с помощью колоночной хроматографии на силикагеле (элюирующий растворитель: этилацетат/метанол), таким образом получая указанное в заголовке соединение (114 мг), в виде твердого вещества светло-желтого цвета.
1Н-ЯМР (CDCl3) δ: 2,23 (6Н, с), 3,07 (2Н, дд, J=6,0, 1,3 Гц), 4,01 (1Н, дд, J=11,6, 5,0 Гц), 4,70 (1Н, дд, J=11,6, 8,2 Гц), 5,09 (2Н, уш.с), 5,20 (1Н, д, J=2,0 Гц), 5,43 (1Н, д, J=2,0 Гц), 5,73-5,79 (1Н, м), 6,07 (1Н, дт, J=15,4, 1,3 Гц), 6,72 (1Н, уш.с), 6,94 (1Н, дт, J=15,4, 6,0 Гц), 7,61-7,65 (1Н, м), 7,77-7,81 (1Н, м), 7,87 (1Н, д, J=8,0 Гц), 8,16 (1Н, д, J=8,5 Гц), 8,25-8,26 (2Н, м), 9,05 (1Н, д, J=2,0 Гц).
ESI-MS m/z: 440 [M+H]+.
Пример 5
(S,Е)-N-(4-Амино-6-метилен-5-(хинолин-3-ил)-7,8-дигидро-6Н-пиримидо[5,4-b]пирролизин-7-ил)-3-хлоракриламид (Соединение 5)
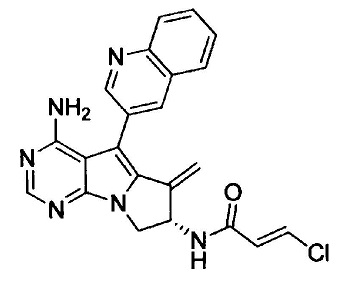
Такой же синтез, как в Примере 4, осуществляют, используя транс-3-хлоракриловую кислоту вместо гидрохлорида (Е)-4-(диметиламино)бут-2-еновой кислоты, используемого в Примере 4, таким образом получая указанное в заголовке соединение (49,2 мг), в виде твердого вещества желтого цвета.
1Н-ЯМР (CDCl3) δ: 4,04 (1Н, дд, J=11,7, 4,6 Гц), 4,69 (1Н, дд, J=11,7, 8,0 Гц), 5,03 (2Н, с), 5,22 (1Н, д, J=1,7 Гц), 5,44 (1Н, д, J=1,7 Гц), 5,72-5,75 (1Н, м), 6,34 (1Н, д, J=13,0 Гц), 6,89 (1Н, уш.с), 7,41 (1Н, д, J=13,0 Гц), 7,63-7,65 (1Н, м), 7,79-7,81 (1Н, м), 7,87 (1Н, д, J=8,0 Гц), 8,16 (1Н, д, J=8,3 Гц), 8,25-8,26 (2Н, м), 9,02 (1Н, д, J=2,0 Гц).
ESI-MS m/z: 417, 419 [M+H]+.
Пример 6
(S,Z)-N-(4-Амино-6-метилен-5-(хинолин-3-ил)-7,8-дигидро-6Н-пиримидо[5,4-b]пирролизин-7-ил)-3-хлоракриламид (Соединение 6)
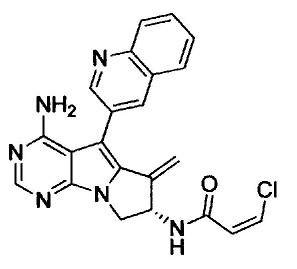
Такой же синтез, как в Примере 4, осуществляют, используя цис-3-хлоракриловую кислоту вместо гидрохлорида (Е)-4-(диметиламино)бут-2-еновой кислоты, используемого в Примере 4, таким образом получая указанное в заголовке соединение (74,7 мг), в виде твердого вещества желтого цвета.
1Н-ЯМР (CDCl3) δ: 4,07 (1Н, дд, J=11,6, 5,4 Гц), 4,82 (1Н, дд, J=11,6, 8,2 Гц), 4,96 (2Н, с), 5,26 (1Н, д, J=2,0 Гц), 5,48 (1Н, д, J=2,0 Гц), 5,75-5,78 (1Н, м), 6,28 (1Н, д, J=8,3 Гц), 6,61 (1Н, д, J=8,3 Гц), 6,81 (1Н, д, J=7,3 Гц), 7,64-7,67 (1Н, м), 7,79-7,83 (1Н, м), 7,90 (1Н, д, J=7,1 Гц), 8,19 (1Н, д, J=8,5 Гц), 8,31 (1Н, д, J=2,0 Гц), 8,33 (1Н, с), 9,12 (1Н, д, J=2,2 Гц).
ESI-MS m/z: 417, 419 [M+H]+.
Пример 7
(S,E)-N-(4-Амино-6-метилен-5-(хинолин-3-ил)-7,8-дигидро-6Н-пиримидо[5,4-b]пирролизин-7-ил)-4-(пиперидин-1-ил)бут-2-енамид (Соединение 7)
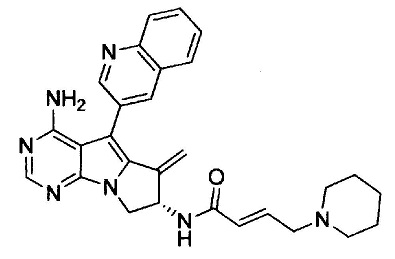
Такой же синтез, как в Примере 4, осуществляют, используя гидрохлорид (Е)-4-(пиперидин-1-ил)бут-2-еновой кислоты вместо гидрохлорида (Е)-4-(диметиламино)бут-2-еновой кислоты, используемого в Примере 4, таким образом получая указанное в заголовке соединение (122 мг), в виде твердого вещества желтого цвета.
1Н-ЯМР (CDCl3) δ: 1,42-1,59 (5Н, м), 2,14 (1Н, с), 2,39 (4Н, уш.с), 3,10 (2Н, дд, J=6,0, 1,4 Гц), 4,01 (1Н, дд, J=11,6, 5,0 Гц), 4,69 (1Н, дд, J=11,5, 8,0 Гц), 5,10 (2Н, уш.с), 5,20 (1Н, д, J=2,1 Гц), 5,43 (1Н, д, J=2,1 Гц), 5,73-5,79 (1Н, м), 6,06 (1Н, дт, J=15,3, 1,4 Гц), 6,76 (1Н, уш.с), 6,96 (1Н, дт, J=15,3, 6,0 Гц), 7,62-7,64 (1Н, м), 7,77-7,80 (1Н, м), 7,87 (1Н, д, J=8,0 Гц), 8,16 (1Н, д, J=8,3 Гц), 8,24-8,25 (2Н, м), 9,04 (1Н, д, J=2,0 Гц).
ESI-MS m/z: 480 [M+H]+.
Пример 8
(S)-N-(4-Амино-6-метилен-5-(хинолин-3-ил)-7,8-дигидро-6Н-пиримидо[5,4-b]пирролизин-7-ил)пропиоламид (Соединение 8)
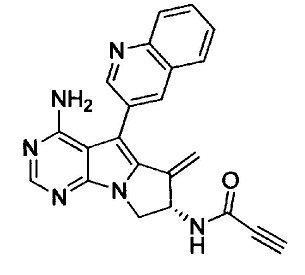
Такой же синтез, как в Примере 4, осуществляют, используя пропиоловую кислоту вместо гидрохлорида (Е)-4-(диметиламино)бут-2-еновой кислоты, используемого в Примере 4, таким образом получая указанное в заголовке соединение (30,0 мг), в виде твердого вещества желтого цвета.
1Н-ЯМР (CDCl3) δ: 2,91 (1Н, с), 4,08 (1Н, дд, J=11,7, 4,9 Гц), 4,76 (1Н, дд, J=11,7, 8,0 Гц), 4,91 (2Н, с), 5,24 (1Н, д, J=1,2 Гц), 5,49 (1Н, д, J=1,7 Гц), 5,68-5,69 (1Н, м), 6,34-6,37 (1Н, м), 7,65-7,67 (1Н, м), 7,81-7,83 (1Н, м), 7,90 (1Н, д, J=8,3 Гц), 8,20 (1Н, д, J=8,5 Гц), 8,30 (1Н, д, J=2,0 Гц), 8,34 (1Н, с), 9,11 (1Н, д, J=2,2 Гц).
ESI-MS m/z: 381 [M+H]+.
Пример 9
(S)-N-(4-Амино-6-метилен-5-(хинолин-3-ил)-7,8-дигидро-6Н-пиримидо[5,4-b]пирролизин-7-ил)бут-2-инамид (Соединение 9)
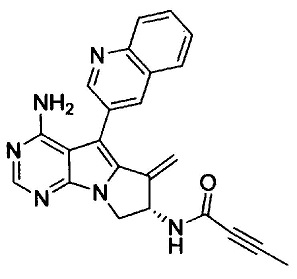
Такой же синтез, как в Примере 4, осуществляют, используя бут-2-иновую кислоту вместо гидрохлорида (Е)-4-(диметиламино)бут-2-еновой кислоты, используемого в Примере 4, таким образом получая указанное в заголовке соединение (98,0 мг), в виде твердого вещества желтого цвета.
1Н-ЯМР (CDCl3) δ: 1,96 (3Н, с), 4,02 (1Н, дд, J=11,6, 5,0 Гц), 4,70 (1Н, дд, J=11,6, 8,3 Гц), 5,05 (2Н, с), 5,24 (1Н, д, J=1,6 Гц), 5,45 (1Н, д, J=1,6 Гц), 5,70-5,72 (1Н, м), 6,95-7,01 (1Н, уш.с), 7,63-7,67 (1Н, м), 7,78-7,83 (1Н, м), 7,88 (1Н, д, J=8,3 Гц), 8,21 (1Н, д, J=8,5 Гц), 8,26-8,28 (2Н, м), 9,02 (1Н, д, J=2,0 Гц).
ESI-MS m/z: 395 [M+H]+.
Пример 10
(S,E)-N-(4-Амино-6-метилен-5-(хинолин-3-ил)-7,8-дигидро-6Н-пиримидо[5,4-b]пирролизин-7-ил)-4-(диэтиламино)бут-2-енамид (Соединение 10)
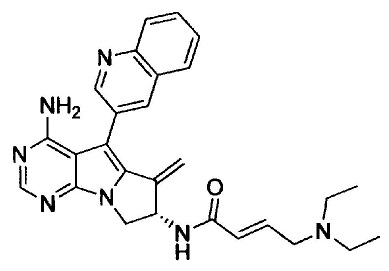
Такой же синтез, как в Примере 4, осуществляют, используя гидрохлорид (Е)-4-(диэтиламино)бут-2-еновой кислоты вместо гидрохлорида (Е)-4-(диметиламино)бут-2-еновой кислоты, используемого в Примере 4, таким образом получая указанное в заголовке соединение (38,2 мг), в виде твердого вещества желтого цвета.
1Н-ЯМР (CDCl3) δ: 1,00 (6Н, т, J=7,2 Гц), 2,51 (4Н, кв., J=7,2 Гц), 3,22 (2Н, дд, J=5,9, 1,5 Гц), 4,00 (1Н, дд, J=11,5, 4,9 Гц), 4,62 (1Н, дд, J=11,5, 8,2 Гц), 5,22-5,24 (3Н, м), 5,40 (1Н, д, J=2,0 Гц), 5,74-5,80 (1Н, м), 6,12 (1Н, дт, J=15,4, 1,5 Гц), 6,98 (1Н, дт, J=15,4, 5,9 Гц), 7,50 (1Н, уш.с), 7,58-7,62 (1Н, м), 7,74-7,78 (1Н, м), 7,84 (1Н, д, J=7,6 Гц), 8,12-8,14 (2Н, м), 8,22 (1Н, д, J=1,7 Гц), 8,99 (1Н, д, J=2,0 Гц).
ESI-MS m/z: 468 [M+H]+.
Пример 11
(S,E)-N-(4-Амино-6-метилен-5-(хинолин-3-ил)-7,8-дигидро-6Н-пиримидо[5,4-b]пирролизин-7-ил)-4-(этил(метил)амино)бут-2-енамид (Соединение 11)
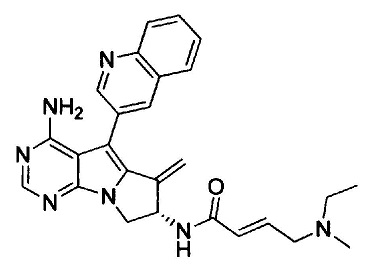
Такой же синтез, как в Примере 4, осуществляют, используя гидрохлорид (Е)-4-(этил(метил)амино)бут-2-еновой кислоты вместо гидрохлорида (Е)-4-(диметиламино)бут-2-еновой кислоты, используемого в Примере 4, таким образом получая указанное в заголовке соединение (13,5 мг), в виде твердого вещества желтого цвета.
1Н-ЯМР (CDCl3) δ: 1,06 (3Н, т, J=7,2 Гц), 2,24 (3Н, с), 2,44 (2Н, кв., J=7,2 Гц), 3,15 (2Н, дд, J=6,0, 1,6 Гц), 4,03 (1Н, дд, J=11,6, 5,1 Гц), 4,73 (1Н, дд, J=11,6, 8,2 Гц), 5,01 (2Н, с), 5,20 (1Н, д, J=1,7 Гц), 5,44 (1Н, д, J=1,7 Гц), 5,75-5,77 (1Н, м), 6,05 (1Н, дт, J=15,4, 1,6 Гц), 6,42 (1Н, д, J=6,8 Гц), 6,96 (1Н, дт, J=15,4, 6,0 Гц), 7,62-7,66 (1Н, м), 7,78-7,82 (1Н, м), 7,88 (1Н, д, J=8,0 Гц), 8,18 (1Н, д, J=8,5 Гц), 8,29 (2Н, с), 9,08 (1Н, д, J=2,2 Гц).
ESI-MS m/z: 454 [M+H]+.
Пример 12
(S,E)-N-(4-Амино-6-метилен-5-(хинолин-3-ил)-7,8-дигидро-6Н-пиримидо[5,4-b]пирролизин-7-ил)-4-(изопропил(метил)амино)бут-2-енамид (Соединение 12)
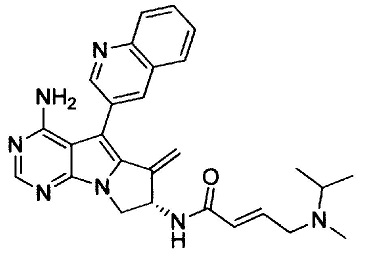
Такой же синтез, как в Примере 4, осуществляют, используя гидрохлорид (Е)-4-(изопропил(метил)амино)бут-2-еновой кислоты вместо гидрохлорида (Е)-4-(диметиламино)бут-2-еновой кислоты, используемого в Примере 4, таким образом получая указанное в заголовке соединение (25,8 мг), в виде твердого вещества желтого цвета.
1Н-ЯМР (CDCl3) δ: 1,00 (6Н, д, J=6,6 Гц), 2,15-2,36 (3Н, м), 2,79-2,88 (1Н, м), 3,18 (2Н, дд, J=5,9, 1,5 Гц), 4,01 (1Н, дд, J=11,5, 5,0 Гц), 4,70 (1Н, дд, J=11,5, 8,0 Гц), 5,11 (2Н, с), 5,21 (1Н, д, J=1,8 Гц), 5,43 (1Н, д, J=1,8 Гц), 5,73-5,79 (1Н, м), 6,10 (1Н, дт, J=15,4, 1,5 Гц), 6,74 (1Н, уш.с), 6,94 (1Н, дт, J=15,4, 5,9 Гц), 7,61-7,65 (1Н, м), 7,77-7,81 (1Н, м), 7,87 (1Н, д, J=8,5 Гц), 8,16 (1Н, д, J=8,8 Гц), 8,23-8,26 (2Н, м), 9,05 (1Н, д, J=2,2 Гц).
ESI-MS m/z: 468 [M+H]+.
Пример 13
(R)-N-(4-Амино-6-метилен-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-7-ил)акриламид (Соединение 13)
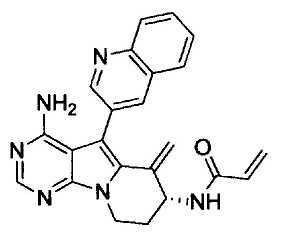
Стадия 1: Синтез (R)-трет-бутил(1-гидрокси-5-(метилтио)пентан-3-ил)карбамата
N-метилморфолин (3,63 мл) и этилхлорформиат (3,01 мл) добавляют к раствору (R)-3-((трет-бутоксикарбонил)амино)-5-(метилтио)пентановой кислоты (7,92 г), в ТГФ (79,2 мл), при температуре -10°С. После перемешивания при температуре -10°С, в течение 15 мин, полученное нерастворимое вещество отфильтровывают. К фильтрату добавляют водный раствор боргидрида натрия (1,55 г) (15 мл), при температуре -10°С, и смесь перемешивают при температуре -10°С, в течение 1 часа. К реакционной смеси добавляют насыщенный водный раствор хлорида аммония и смесь перемешивают при комнатной температуре в течение 30 мин. К реакционной смеси добавляют этилацетат для отделения органического слоя. Органический слой промывают 0,5 н водным раствором гидросульфата калия, водой, 0,5 н водным раствором гидроксида натрия и насыщенным раствором хлорида натрия и сушат над безводным сульфатом натрия. Растворитель затем отгоняют при пониженном давлении. Полученный остаток очищают с помощью колоночной хроматографии на силикагеле (элюирующий растворитель: хлороформ/этилацетат), таким образом получая указанное в заголовке соединение, в виде маслянистого вещества светло-желтого цвета (7,18 г).
Стадия 2: Синтез трет-бутил((3R)-1-гидрокси-5-(метилсульфинил)пентан-3-ил)карбамата
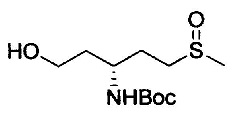
Суспензию периодата натрия (7,0 г), в воде (32 мл), добавляют к раствору (R)-трет-бутил(1-гидрокси-5-(метилтио)пентан-3-ил)карбамата (8,16 г), полученного на стадии 1, в метаноле (98 мл), при температуре 10°С или ниже, и смесь перемешивают при комнатной температуре, в течение 2 часов.
Полученное нерастворимое вещество отфильтровывают и фильтрат подвергают дистилляции при пониженном давлении. Полученный остаток растворяют в насыщенном растворе хлорида натрия, затем 3 раза экстрагируют хлороформом. Органический слой сушат над безводным сульфатом натрия и растворитель отгоняют при пониженном давлении, таким образом получая указанное в заголовке соединение (9,38 г), в виде твердого вещества светло-желтого цвета.
Стадия 3: Синтез (R)-трет-бутил(5-гидроксипент-1-ен-3-ил)карбамата
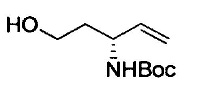
Ацетат натрия (13,45 г) добавляют к раствору трет-бутил((3R)-1-гидрокси-5-(метилсульфинил)пентан-3-ил)карбамата (9,38 г), полученного на стадии 2, в 1,2-дихлорбензоле (140 мл), при комнатной температуре. Смесь перемешивают при внутренней температуре 166°С, в течение 18 часов. После охлаждения реакционной смеси, нерастворимое вещество отфильтровывают и 1,2-дихлорбензол отгоняют при пониженном давлении. Полученный остаток растворяют в этилацетате, промывают водным раствором гипохлорита натрия, водой и насыщенным раствором хлорида натрия, и сушат над безводным сульфатом натрия. Растворитель затем отгоняют при пониженном давлении. Полученный остаток очищают с помощью колоночной хроматографии на силикагеле (элюирующий растворитель: гексан/этилацетат), таким образом получая указанное в заголовке соединение (2,50 г), в виде маслянистого вещества светло-желтого цвета.
Стадия 4: Синтез (R)-трет-бутил(5-(4-хлор-5-иод-7Н-пирроло[2,3-d]пиримидин-7-ил)пент-1-ен-3-ил)карбамата
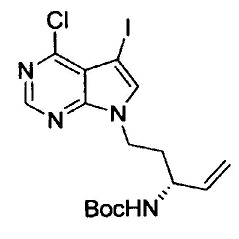
Трифенилфосфин (3,25 г) добавляют и растворяют в растворе (R)-трет-бутил(5-гидроксипент-1-ен-3-ил)карбамата (2,5 г), полученного на стадии 3, и 4-хлор-5-иод-7Н-пирроло[2,3-d]пиримидина (2,31 г), в DME (23 мл), при охлаждении льдом. После этого, постепенно добавляют диизопропилазодикарбоксилат (2,44 мл). Реакционную смесь перемешивают при охлаждении льдом в течение 30 мин и, при комнатной температуре, в течение 1 часа, и растворитель затем отгоняют при пониженном давлении. Полученный остаток растворяют в этилацетате, промывают водой и сушат над безводным сульфатом натрия. Растворитель потом отгоняют при пониженном давлении. Полученный остаток очищают с помощью колоночной хроматографии на силикагеле (элюирующий растворитель: гексан/этилацетат), таким образом получая указанное в заголовке соединение (3,49 г), в виде твердого вещества светло-желтого цвета.
ESI-MS m/z: 463, 465 [M+H]+.
Стадия 5: Синтез (R)-трет-бутил(5-(4-амино-5-иод-7Н-пирроло[2,3-d]пиримидин-7-ил)пент-1-ен-3-ил)карбамата
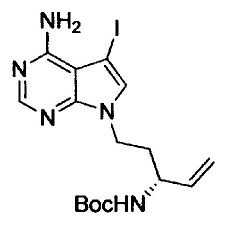
28%-ный Водный раствор аммиака (17,5 мл) добавляют к раствору (R)-трет-бутил(5-(4-хлор-5-иод-7Н-пирроло[2,3-d]пиримидин-7-ил)пент-1-ен-3-ил)карбамата (3,49 г), полученного на стадии 4, в DME (17,5 мл), и смесь перемешивают в автоклаве при внутренней температуре 105°С, в течение 8 часов. После охлаждения реакционной смеси, добавляют воду (70 мл) и смесь перемешивают при комнатной температуре, в течение 4 часов. Полученный осадок собирают путем фильтрации, промывают водой и высушивают, таким образом получая указанное в заголовке соединение (3,20 г), в виде твердого вещества светло-желтого цвета.
ESI-MS m/z: 444 [M+H]+.
Стадия 6: Синтез (R)-трет-бутил(5-(4-амино-5-(хинолин-3-ил)-7Н-пирроло[2,3-d]пиримидин-7-ил)пент-1-ен-3-ил)карбамата
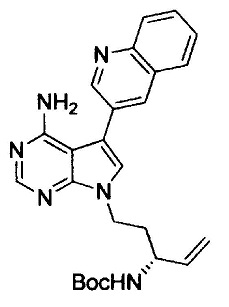
Смесь (R)-трет-бутил(5-(4-амино-5-иод-7Н-пирроло[2,3-d]пиримидин-7-ил)пент-1-ен-3-ил)карбамата (3,2 г), полученного на стадии 5, 3-хинолинбороновой кислоты (1,37 г), карбоната натрия (843 мг), тетракис(трифенилфосфин)палладия (250 мг), DME (32 мл) и воды (32 мл) перемешивают в атмосфере азота, при температуре 100°С, в течение 6 часов. После охлаждения реакционной смеси, добавляют насыщенный водный раствор бикарбоната натрия и этилацетат. Полученную смесь перемешивают при комнатной температуре, в течение 30 мин. После отфильтровывания нерастворимого вещества, органический слой отделяют и сушат над безводным сульфатом натрия. Растворитель затем отгоняют при пониженном давлении. Полученный остаток очищают с помощью колоночной хроматографии на силикагеле (элюирующий растворитель: гексан/этилацетат, этилацетат/метанол), таким образом получая указанное в заголовке соединение (3,21 г), в виде твердого вещества светло-оранжевого цвета.
ESI-MS m/z: 445 [M+H]+.
Стадия 7: Синтез (R)-трет-бутил(5-(4-амино-6-бром-5-(хинолин-3-ил)-7Н-пирроло[2,3-d]пиримидин-7-ил)пент-1-ен-3-ил)карбамата
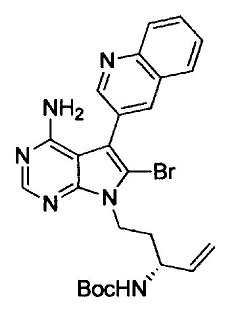
Раствор N-бромсукцинимида (1,35 г), в ТГФ (23 мл), добавляют к раствору (R)-трет-бутил(5-(4-амино-5-(хинолин-3-ил)-7Н-пирроло[2,3-d]пиримидин-7-ил)пент-1-ен-3-ил)карбамата (3,21 г), полученного на стадии 6, в ТГФ (26 мл), при охлаждении льдом, в течение 30 мин. Смесь перемешивают при охлаждении льдом в течение 30 мин. После добавления 5%-ного водного раствора тиосульфата натрия к реакционной смеси, смесь выливают в насыщенный водный раствор бикарбоната натрия, затем экстрагируют этилацетатом. Органический слой промывают насыщенным раствором хлорида натрия и сушат над безводным сульфатом натрия. Растворитель потом отгоняют при пониженном давлении. Полученный остаток очищают с помощью колоночной хроматографии на силикагеле (элюирующий растворитель: гексан/этилацетат, этилацетат/метанол), таким образом получая указанное в заголовке соединение (3,15 г), в виде твердого вещества светло-коричневого цвета.
ESI-MS m/z: 523, 525 [M+H]+.
Стадия 8: Синтез (R)-трет-бутил(4-амино-6-метилен-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-7-ил)карбамата
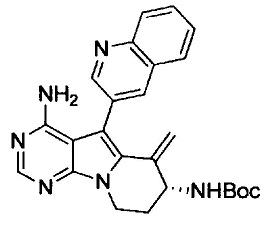
4 н Водный раствор гидроксида натрия (0,454 мл) добавляют к раствору (R)-трет-бутил(5-(4-амино-6-бром-5-(хинолин-3-ил)-7Н-пирроло[2,3-d]пиримидин-7-ил)пент-1-ен-3-ил)карбамата (475 мг), полученного на стадии 7, в ТГФ (5 мл). После этого, смесь дегазируют при пониженном давлении, затем продувают азотом. Потом добавляют тетракис(трифенилфосфин)палладий (41,6 мг), смесь перемешивают в течение ночи при кипячении с обратным холодильником. Затем реакционную смесь охлаждают до комнатной температуры, выливают в воду и экстрагируют этилацетатом. Полученный органический слой промывают насыщенным раствором хлорида натрия и сушат над безводным сульфатом натрия. После фильтрации и концентрирования, полученный остаток очищают с помощью хроматографии на силикагеле (элюирующий растворитель: этилацетат/метанол), таким образом получая указанное в заголовке соединение (293 мг), в виде твердого вещества желтого цвета.
ESI-MS m/z: 443 [M+H]+.
Стадия 9: Синтез (R)-6-метилен-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-4,7-диамина
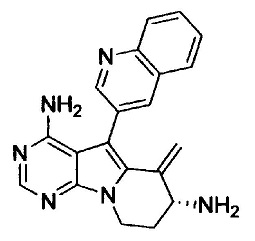
5 н Соляную кислоту (1 мл) добавляют к раствору (R)-трет-бутил(4-амино-6-метилен-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-7-ил)карбамата (290 мг), полученного на стадии 8, в этаноле (4 мл). После этого, смесь перемешивают в течение 6 часов, при температуре 70°С. Потом реакционную смесь охлаждают до комнатной температуры, значение рН реакционной смеси доводят до примерно 10, используя 5 н водный раствор гидроксида натрия, затем экстрагируют хлороформом. Полученный органический слой сушат над безводным сульфатом натрия, потом отфильтровывают и концентрируют. Полученный остаток очищают с помощью хроматографии на основном силикагеле (элюирующий растворитель: хлороформ/метанол), таким образом получая указанное в заголовке соединение (218 мг), в виде твердого вещества светло-коричневого цвета.
ESI-MS m/z: 343 [M+H]+.
Стадия 10: Синтез Соединения 13
Диизопропилэтиламин (0,129 мл) и раствор акрилоилхлорида (56,1 мг), в хлороформе (0,5 мл), добавляют к раствору (R)-6-метилен-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-4,7-диамина (215 мг), полученного на стадии 9, в хлороформе (4 мл), при охлаждении льдом, и полученную смесь перемешивают в течение 30 мин. Потом реакционную смесь концентрируют, остаток очищают с помощью хроматографии на силикагеле (элюирующий растворитель: этилацетат/метанол), таким образом получая указанное в заголовке соединение (117 мг), в виде твердого вещества желтого цвета.
1Н-ЯМР (CDCl3) δ: 2,28-2,41 (2Н, м), 4,32-4,50 (2Н, м), 4,64 (2Н, уш.с), 4,97 (1Н, с), 5,07 (1Н, с), 5,05-5,12 (1Н, м), 5,72 (1Н, дд, J=10,2, 1,2 Гц), 5,75-5,85 (1Н, м), 6,14 (1Н, дд, J=16,8, 10,2 Гц), 6,36 (1Н, дд, J=16,8, 1,2 Гц), 7,61-7,68 (1Н, м), 7,77-7,84 (1Н, м), 7,87 (1Н, д, J=8,4 Гц), 8,18 (1Н, д, J=8,4 Гц), 8,27 (1Н, д, J=2,0 Гц), 8,33 (1Н, с), 8,98 (1Н, д, J=2,0 Гц).
ESI-MS m/z: 397 [M+H]+.
Пример 14
(S)-N-(4-Амино-6-метил-5-(хинолин-3-ил)-8,9-дигидропиримидо[5,4-b]индолизин-8-ил)акриламид (Соединение 14)
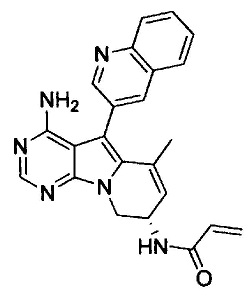
Стадия 1: Синтез (S)-метил-2-((трет-бутоксикарбонил)амино)пент-4-еноата
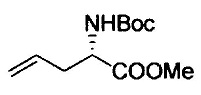
5 н Водный раствор гидроксида натрия (2,1 мл) и ди-трет-бутилдикарбонат (2,128 мл) добавляют к суспензии (S)-2-амино-4-пентеновой кислоты (1,016 г), в метаноле (20 мл), при комнатной температуре, и полученную смесь перемешивают в течение 8 часов, при такой же температуре. Добавляют ди-трет-бутилдикарбонат (0,304 мл), при комнатной температуре, и смесь перемешивают в течение 1 часа, при такой же температуре. К реакционной смеси добавляют 4-гидрокси-1Н-бензотриазол (1,796 г) и 1-(3-диметиламинопропил)-3-этилкарбодиимидгидрохлорид (3,882 г), при комнатной температуре, и смесь перемешивают в течение ночи, при такой же температуре. Реакционную смесь концентрируют при пониженном давлении и затем выливают в насыщенный водный раствор бикарбоната натрия, потом экстрагируют этилацетатом. Полученный органический слой сушат над безводным сульфатом натрия, затем отфильтровывают и концентрируют. Полученный остаток очищают с помощью хроматографии на силикагеле (элюирующий растворитель: хлороформ/метанол), таким образом получая указанное в заголовке соединение (2,0311 г), в виде маслянистого вещества светло-желтого цвета.
Стадия 2: Синтез (S)-трет-бутил(1-гидроксипент-4-ен-2-ил)карбамата
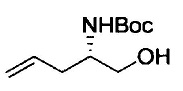
Литийалюминийгидрид (668,2 мг) добавляют к раствору (S)-метил-2-((трет-бутоксикарбонил)амино)пент-4-еноата (1,983 г), полученного на стадии 1, в ТГФ (50 мл), при охлаждении льдом, и смесь перемешивают в течение 1,5 часов, при такой же температуре. К реакционной смеси добавляют декагидрат сульфата натрия (1,1375 г) и ТГФ (10 мл), при комнатной температуре, и смесь перемешивают в течение ночи, при такой же температуре. Нерастворимое вещество отфильтровывают. После промывки с помощью ТГФ и этилацетата, фильтрат концентрируют при пониженном давлении. Полученный остаток очищают с помощью хроматографии на силикагеле (элюирующий растворитель: гексан/этилацетат), таким образом получая указанное в заголовке соединение (1,2601 г), в виде маслянистого вещества светло-желтого цвета.
Стадия 3: Синтез (S)-трет-бутил(1-(4-хлор-5-иод-7Н-пирроло[2,3-d]пиримидин-7-ил)пент-4-ен-2-ил)карбамата
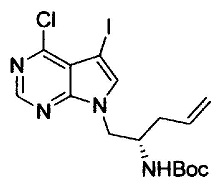
Такой же синтез, как на стадии 4 Примера 13, осуществляют, используя (S)-трет-бутил(1-гидроксипент-4-ен-2-ил)карбамат, полученный на стадии 2, вместо (R)-трет-бутил(5-гидроксипент-1-ен-3-ил)карбамата, используемого на стадии 4 Примера 13, таким образом получая указанное в заголовке соединение (2,7679 г), в виде твердого вещества светло-желтого цвета.
ESI-MS m/z: 463, 465 [M+H]+.
Стадия 4: Синтез (S)-трет-бутил(1-(4-амино-6-бром-5-(хинолин-3-ил)-7Н-пирроло[2,3-d]пиримидин-7-ил)пент-4-ен-2-ил)карбамата
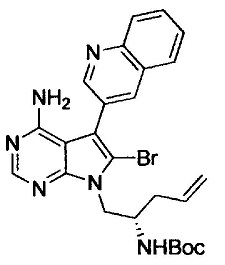
Такой же синтез, как на стадиях 5-7 Примера 13, осуществляют, используя (S)-трет-бутил(1-(4-хлор-5-иод-7Н-пирроло[2,3-d]пиримидин-7-ил)пент-4-ен-2-ил)карбамат, полученный на стадии 3, вместо (R)-трет-бутил(5-(4-хлор-5-иод-7Н-пирроло[2,3-d]пиримидин-7-ил)пент-1-ен-3-ил)карбамата, используемого на стадии 5 Примера 13, таким образом получая указанное в заголовке соединение (2,669 г), в виде твердого вещества светло-коричневого цвета.
ESI-MS m/z: 523, 525 [M+H]+.
Стадия 5: Синтез (S)-трет-бутил(4-амино-6-метил-5-(хинолин-3-ил)-8,9-дигидропиримидо[5,4-b]индолизин-8-ил)карбамата и (S)-трет-бутил(4-амино-6-метилен-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)карбамата
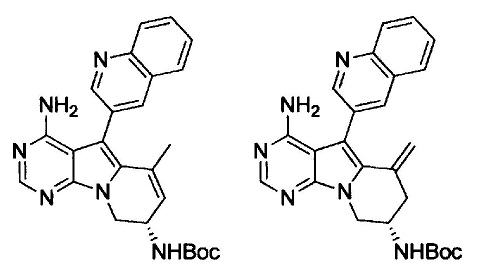
4 н Водный раствор гидроксида натрия (5,1 мл) и тетракис(трифенилфосфин)палладий (235,4 мг) добавляют к раствору (S)-трет-бутил(1-(4-амино-6-бром-5-(хинолин-3-ил)-7Н-пирроло[2,3-d]пиримидин-7-ил)пент-4-ен-2-ил)карбамата (2,669 г), полученного на стадии 4, в ТГФ (50 мл), и смесь дегазируют при пониженном давлении, затем продувают азотом. Реакционную смесь перемешивают в течение ночи при кипячении с обратным холодильником. Потом реакционную смесь охлаждают до комнатной температуры, выливают в насыщенный водный раствор бикарбоната натрия и экстрагируют этилацетатом. Полученный органический слой сушат над безводным сульфатом натрия. После фильтрации и концентрирования, полученный остаток очищают с помощью хроматографии на силикагеле (элюирующий растворитель: хлороформ/метанол), таким образом получая смесь указанных в заголовке соединений (2,427 г), в виде твердого вещества желтого цвета.
ESI-MS m/z: 443 [M+H]+.
Стадия 6: Синтез (S)-6-метил-5-(хинолин-3-ил)-8,9-дигидропиримидо[5,4-b]индолизин-4,8-диамина и (S)-6-метилен-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-4,8-диамина
5 н Соляную кислоту (5,1 мл) добавляют к раствору смеси (2,427 г) (S)-трет-бутил(4-амино-6-метил-5-(хинолин-3-ил)-8,9-дигидропиримидо[5,4-b]индолизин-8-ил)карбамата и (S)-трет-бутил(4-амино-6-метилен-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)карбамата, полученной на стадии 5, в этаноле (20 мл), и смесь перемешивают в течение ночи при температуре 60°С. Потом добавляют дополнительное количество 5 н соляной кислоты (5,1 мл), смесь перемешивают в течение 10 часов при кипячении с обратным холодильником. Затем реакционную смесь охлаждают до комнатной температуры, разбавляют водой и промывают хлороформом. К водному слою добавляют 5 н водный раствор гидроксида натрия (10 мл), потом экстрагируют хлороформом. Полученный органический слой сушат над безводным сульфатом натрия, затем отфильтровывают и концентрируют. Полученный остаток очищают с помощью хроматографии на основном силикагеле (элюирующий растворитель: этилацетат/метанол), таким образом получая смесь указанных в заголовке соединений (1,4098 г), в виде твердого вещества светло-желтого цвета.
ESI-MS m/z: 343 [M+H]+.
Стадия 7: Синтез Соединения 14
Диизопропилэтиламин (0,8452 мл) и раствор акрилоилхлорида (0,35 мл), в ацетонитриле (3,5 мл), последовательно добавляют к раствору смеси (1,407 г) (S)-6-метил-5-(хинолин-3-ил)-8,9-дигидропиримидо[5,4-b]индолизин-4,8-диамина и (S)-6-метилен-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-4,8-диамина, полученной на стадии 6, в ацетонитриле (10 мл) и воде (10 мл), при температуре 0°С, и смесь перемешивают в течение 45 мин, при такой же температуре. Потом реакционную смесь разбавляют водой, выливают в насыщенный водный раствор бикарбоната натрия, затем экстрагируют этилацетатом. Потом органический слой сушат над безводным сульфатом натрия, растворитель отгоняют при пониженном давлении. Полученный остаток очищают с помощью колоночной хроматографии на силикагеле (элюирующий растворитель: этилацетат/метанол), таким образом получая указанное в заголовке соединение (897,1 мг), в виде твердого вещества светло-желтого цвета.
1Н-ЯМР (ДМСО-d6) δ: 1,51 (3Н, с), 4,17 (1Н, дд, J=13,2, 5,1 Гц), 4,25 (1Н, дд, J=13,2, 5,1 Гц), 4,73-4,83 (1Н, м), 5,61 (1Н, дд, J=9,8, 2,7 Гц), 5,65-6,00 (2Н, уш.с), 5,81 (1Н, дд, J=5,1, 1,2 Гц), 6,14 (1Н, дд, J=17,1, 2,4 Гц), 6,24 (1Н, дд, J=17,1, 9,8 Гц), 7,67 (1Н, ддд, J=8,1, 7,1, 1,0 Гц), 7,81 (1Н, ддд, J=8,3, 7,1, 1,5 Гц), 8,05 (1Н, д, J=8,1 Гц), 8,09 (1Н, д, J=8,3 Гц), 8,13 (1Н, с), 8,39 (1Н, уш.с), 8,46 (1Н, д, J=7,3 Гц), 8,94 (1Н, с).
ESI-MS m/z: 397 [M+H]+.
Пример 15
(S)-N-(4-Амино-6-метилен-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)акриламид (Соединение 15)
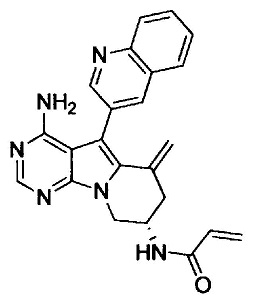
Смешанную фракцию, полученную посредством очистки с помощью колоночной хроматографии на силикагеле на стадии 7 Примера 14, очищают с помощью препаративной ВЭЖХ с обращенными фазами, таким образом получая указанное в заголовке соединение (32,2 мг), в виде твердого вещества светло-желтого цвета.
1Н-ЯМР (ДМСО-d6) δ: 2,56-2,70 (1Н, м), 2,72-2,82 (1Н, м), 4,19 (1Н, дд, J=14,1, 8,3 Гц), 4,25-4,36 (1Н, м), 4,72 (1Н, д, J=13,7 Гц), 5,62 (1Н, дд, J=10,0, 2,2 Гц), 5,80 (1Н, дт, J=12,2, 4,6 Гц), 6,00-6,20 (3Н, м), 6,20-6,31 (2Н, м), 7,65 (1Н, т, J=8,1 Гц), 7,80 (1Н, т, J=8,3 Гц), 8,05 (1Н, д, J=8,5 Гц), 8,08 (1Н, д, J=8,3 Гц), 8,15 (1Н, с), 8,28-8,39 (2Н, м), 8,87 (1Н, д, J=2,2 Гц).
ESI-MS m/z: 397 [M+H]+.
Пример 16
(R)-N-(4-Амино-6-метил-5-(хинолин-3-ил)-8,9-дигидропиримидо[5,4-b]индолизин-8-ил)акриламид (Соединение 16)
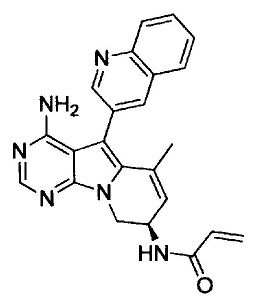
Стадия 1: Синтез (R)-трет-бутил(1-(4-хлор-5-иод-7Н-пирроло[2,3-d]пиримидин-7-ил)пент-4-ен-2-ил)карбамата
Такой же синтез, как на стадиях 1-3 Примера 14, осуществляют, используя (R)-2-амино-4-пентеновую кислоту вместо (S)-2-амино-4-пентеновой кислоты, используемой на стадии 1 Примера 14, таким образом получая указанное в заголовке соединение (1,4217 г), в виде твердого вещества светло-желтого цвета.
ESI-MS m/z: 463, 465 [M+H]+.
Стадия 2: Синтез Соединения 16
Такой же синтез, как на стадиях 4-7 Примера 14, осуществляют, используя (R)-трет-бутил(1-(4-хлор-5-иод-7Н-пирроло[2,3-d]пиримидин-7-ил)пент-4-ен-2-ил)карбамат, полученный на стадии 1, таким образом получая указанное в заголовке соединение (21,0 мг), в виде твердого вещества светло-желтого цвета.
1Н-ЯМР (ДМСО-d6) δ: 1,51 (3Н, с), 4,17 (1Н, дд, J=13,2, 5,1 Гц), 4,25 (1Н, дд, J=13,2, 5,1 Гц), 4,73-4,83 (1Н, м), 5,61 (1Н, дд, J=9,8, 2,7 Гц), 5,65-6,00 (2Н, уш.с), 5,81 (1Н, дд, J=5,1, 1,2 Гц), 6,14 (1Н, дд, J=17,1, 2,4 Гц), 6,24 (1Н, дд, J=17,1, 9,8 Гц), 7,67 (1Н, т, J=7,1 Гц), 7,81 (1Н, т, J=7,6 Гц), 8,05 (1Н, д, J=8,1 Гц), 8,09 (1Н, д, J=8,6 Гц), 8,13 (1Н, с), 8,39 (1Н, уш.с), 8,46 (1Н, д, J=7,1 Гц), 8,94 (1Н, с).
ESI-MS m/z: 397 [M+H]+.
Пример 17
(R)-N-(4-Амино-6-метилен-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)акриламид (Соединение 17)
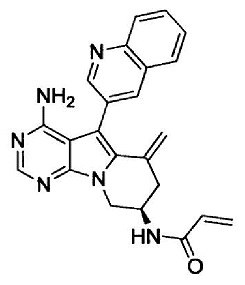
Смешанную фракцию, полученную посредством очистки с помощью колоночной хроматографии на силикагеле в Примере 16, очищают с помощью такой же методики, как в Примере 15, таким образом получая указанное в заголовке соединение (8,4 мг), в виде твердого вещества светло-желтого цвета.
1Н-ЯМР (ДМСО-d6) δ: 2,57-2,70 (1Н, м), 2,71-2,82 (1Н, м), 4,19 (1Н, дд, J=13,2, 8,3 Гц), 4,25-4,36 (1Н, м), 4,72 (1Н, д, J=13,2 Гц), 5,62 (1Н, дд, J=10,0, 2,2 Гц), 5,80 (1Н, дт, J=12,2, 4,6 Гц), 6,00-6,20 (3Н, м), 6,20-6,31 (2Н, м), 7,65 (1Н, т, J=7,6 Гц), 7,79 (1Н, ддд, J=8,3, 7,1, 1,5 Гц), 8,05 (1Н, д, J=8,5 Гц), 8,08 (1Н, д, J=8,5 Гц), 8,15 (1Н, с), 8,33 (1Н, д, J=2,2 Гц), 8,36 (1Н, д, J=7,6 Гц), 8,87 (1Н, д, J=2,2 Гц).
ESI-MS m/z: 397 [M+H]+.
Пример 18
(S)-N-(4-Амино-6-метилен-5-(хинолин-3-ил)-7,8,9,10-тетрагидро-6Н-пиримидо[5’,4’:4,5]пирроло[1,2-a]азепин-8-ил)акриламид (Соединение 18)
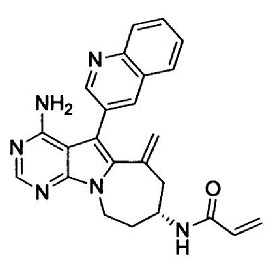
Стадия 1: Синтез (S)-трет-бутил(1-гидроксигекс-5-ен-3-ил)карбамата
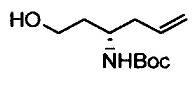
N-Метилморфолин (2,1 г) добавляют к раствору (S)-3-((трет-бутоксикарбонил)амино)гекс-5-еновой кислоты (4,0 г), в ТГФ (40 мл), и постепенно добавляют этилхлорформиат (1,75 мл), при температуре -10°С. После этого, смесь перемешивают в течение 20 минут, при такой же температуре, и полученное нерастворимое вещество отфильтровывают через целит. К полученному фильтрату постепенно добавляют раствор тетрагидробората натрия (904 мг) в воде (8 мл), при температуре -10°С. Потом смесь перемешивают в течение 30 мин, при такой же температуре, добавляют насыщенный водный раствор хлорида аммония. После экстракции этилацетатом, промывают, используя 0,5 н водный раствор гидросульфата калия, насыщенный водный раствор бикарбоната натрия и насыщенный раствор хлорида натрия, в таком порядке. Полученный органический слой сушат над безводным сульфатом натрия и затем отфильтровывают, потом концентрируют. Полученный остаток очищают с помощью хроматографии на силикагеле (элюирующий растворитель: гексан/этилацетат), таким образом получая указанное в заголовке соединение (3,54 г), в виде маслянистого вещества.
Стадия 2: Синтез Соединения 18
Такой же синтез, как на стадиях 4-10 Примера 13, осуществляют, используя (S)-трет-бутил(1-гидроксигекс-5-ен-3-ил)карбамат, полученный на стадии 1, вместо (R)-трет-бутил(5-гидроксипент-1-ен-3-ил)карбамата, используемого на стадии 4 Примера 13, таким образом получая указанное в заголовке соединение (147 мг), в виде бесцветного твердого вещества.
1Н-ЯМР (ДМСО-d6) δ: 1,68-1,82 (1Н, м), 2,17-2,25 (1Н, м), 2,42-2,53 (1Н, м), 2,75-2,83 (1Н, м), 3,86-4,16 (2Н, м), 4,64-4,78 (1Н, м), 4,84 (1Н, д, J=1,7 Гц), 5,26 (1Н, с), 5,60 (1Н, дд, J=10,0, 2,4 Гц), 6,00 (2Н, уш.с), 6,11 (1Н, дд, J=17,1, 2,4 Гц), 6,27 (1Н, дд, J=17,1, 10,0 Гц), 7,60-7,64 (1Н, м), 7,73-7,77 (1Н, м), 7,99-8,04 (2Н, м), 8,17 (1Н, с), 8,22 (1Н, д, J=8,0 Гц), 8,30 (1Н, д, J=2,0 Гц), 8,78 (1Н, д, J=2,0 Гц).
ESI-MS m/z: 411 [M+H]+.
Пример 19
(S,E)-N-(4-Амино-6-этилиден-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)акриламид (Соединение 19)
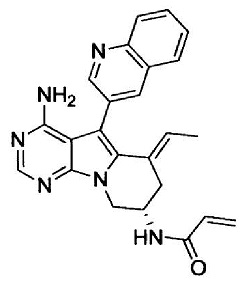
Стадия 1: Синтез (S)-метил-3-((трет-бутоксикарбонил)амино)-4-гидроксибутаноата
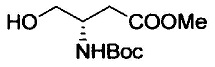
N-Метилморфолин (9,78 мл) и этилхлорформиат (8,09 мл) добавляют к раствору (S)-2-((трет-бутоксикарбонил)амино)-4-метокси-4-оксобутановой кислоты (20,0 г), в ТГФ (200 мл), при охлаждении льдом, и смесь перемешивают в течение 1 часа, при охлаждении льдом. Полученное нерастворимое вещество отфильтровывают. К фильтрату добавляют раствор боргидрида натрия (4,14 г), в воде (41,4 мл), при охлаждении льдом, и смесь перемешивают в течение 30 мин, при охлаждении льдом. К реакционной смеси добавляют 0,5 н водный раствор гидросульфата калия и этилацетат. Органический слой отделяют, промывают насыщенным водным раствором бикарбоната натрия и насыщенным раствором хлорида натрия, и сушат над безводным сульфатом магния. Растворитель затем отгоняют при пониженном давлении, таким образом получая указанное в заголовке соединение (10,82 г).
Стадия 2: Синтез (S)-трет-бутил-4-(2-метокси-2-оксоэтил)-2,2-диметилоксазолидин-3-карбоксилата
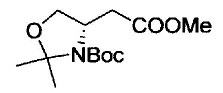
2,2-Диметоксипропан (28,52 мл) и комплекс трифторид бора - диэтиловый эфир (0,294 мл) добавляют к раствору (S)-метил-3-((трет-бутоксикарбонил)амино)-4-гидроксибутаноата (10,82 г), полученного на стадии 1, в ацетоне (108,2 мл), при комнатной температуре, и смесь перемешивают в течение 4 часов, при такой же температуре. Реакционную смесь выливают в насыщенный водный раствор бикарбоната натрия и экстрагируют этилацетатом. Полученный органический слой сушат над безводным сульфатом магния и затем отфильтровывают, потом концентрируют. Полученный остаток очищают с помощью хроматографии, на силикагеле (элюирующий растворитель: гексан/этилацетат), таким образом получая указанное в заголовке соединение (8,72 г).
Стадия 3: Синтез (S)-трет-бутил-4-(2-гидроксиэтил)-2,2-диметилоксазолидин-3-карбоксилата
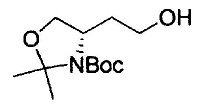
1 М Раствор диизобутилалюминийгидрида в толуоле (65,7 мл) добавляют к раствору (S)-трет-бутил-4-(2-метокси-2-оксоэтил)-2,2-диметилоксазолидин-3-карбоксилата (8,71 г), полученного на стадии 2, в метиленхлориде (87,1 мл), при охлаждении льдом, и смесь перемешивают в течение 2 часов, при охлаждении льдом. К реакционной смеси добавляют 5%-ный водный раствор смешанного калий-натрий-тартрата и этилацетат, и смесь перемешивают в течение ночи при комнатной температуре. Полученный органический слой сушат над безводным сульфатом магния и затем отфильтровывают, потом концентрируют. Полученный остаток очищают с помощью хроматографии, на силикагеле (элюирующий растворитель: гексан/этилацетат), таким образом получая указанное в заголовке соединение (5,94 г).
Стадия 4: Синтез (S)-трет-бутил-2,2-диметил-4-(2-оксоэтил)оксазолидин-3-карбоксилата
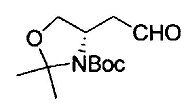
Триэтиламин (16,9 мл) и комплекс триоксид серы - пиридин (12,56 г) добавляют к раствору (S)-трет-бутил-4-(2-гидроксиэтил)-2,2-диметилоксазолидин-3-карбоксилата (5,94 г), полученного на стадии 3, в ДМСО (59,4 мл), при комнатной температуре, и смесь перемешивают в течение 1 часа, при такой же температуре. Реакционную смесь выливают в насыщенный водный раствор бикарбоната натрия и экстрагируют этилацетатом. Полученный органический слой сушат над безводным сульфатом магния и затем отфильтровывают, потом концентрируют. Полученный остаток очищают с помощью хроматографии на силикагеле (элюирующий растворитель: гексан/этилацетат), таким образом получая указанное в заголовке соединение (6,06 г).
Стадия 5: Синтез (S)-трет-бутил-4-(бут-2-ен-1-ил)-2,2-диметилоксазолидин-3-карбоксилата
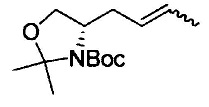
2,69 М Раствор н-бутиллития в гексане (7,0 мл) добавляют к суспензии этилтрифенилфосфонийбромида (5,60 г), в ТГФ (25,2 мл), при охлаждении льдом, и смесь перемешивают в течение 30 мин, при охлаждении льдом. К реакционной смеси добавляют раствор (S)-трет-бутил-2,2-диметил-4-(2-оксоэтил)оксазолидин-3-карбоксилата (3,06 г), полученного на стадии 4, в ТГФ (3,06 мл), при охлаждении льдом, и смесь перемешивают в течение 14 часов, при комнатной температуре. К реакционной смеси добавляют гексан и нерастворимое вещество отфильтровывают, потом промывают смесью ТГФ-гексан = 2/1. Фильтрат концентрируют при пониженном давлении. Полученный остаток очищают с помощью хроматографии на силикагеле (элюирующий растворитель: гексан/этилацетат), таким образом получая указанное в заголовке соединение (1,81 г).
Стадия 6: Синтез (S)-трет-бутил(1-гидроксигекс-4-ен-2-ил)карбамата
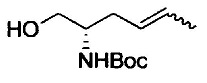
4 н Раствор хлороводорода в диоксане (18,1 мл) добавляют к (S)-трет-бутил-4-(бут-2-ен-1-ил)-2,2-диметилоксазолидин-3-карбоксилату (1,81 г), полученному на стадии 5, при комнатной температуре, и смесь перемешивают в течение 2 часов, при температуре 70°С. Реакционную смесь охлаждают и затем концентрируют при пониженном давлении. Полученный остаток растворяют в ТГФ (18,1 мл) и добавляют туда насыщенный водный раствор бикарбоната натрия (18,1 мл) и ди-трет-бутилдикарбонат (1,53 г), при комнатной температуре. Смесь перемешивают в течение ночи, при такой же температуре. Реакционную смесь выливают в воду и экстрагируют этилацетатом. Полученный органический слой сушат над безводным сульфатом магния и затем отфильтровывают, потом концентрируют. Полученный остаток очищают с помощью хроматографии на силикагеле (элюирующий растворитель: гексан/этилацетат), таким образом получая указанное в заголовке соединение (1,57 г).
Стадия 7: Синтез Соединения 19
Такой же синтез, как на стадиях 4-10 Примера 13, осуществляют, используя (S)-трет-бутил(1-гидроксигекс-4-ен-2-ил)карбамат, полученный на стадии 6, вместо (R)-трет-бутил(5-гидроксипент-1-ен-3-ил)карбамата, используемого на стадии 4 Примера 13, таким образом получая указанное в заголовке соединение (327 мг).
1Н-ЯМР (ДМСО-d6) δ: 1,47 (3Н, д, J=7,1 Гц), 2,64-2,70 (1Н, м), 2,81-2,85 (1Н, м), 4,00-4,09 (1Н, м), 4,32-4,43 (2Н, м), 5,41-5,46 (1Н, м), 5,60-5,63 (1Н, м), 5,82 (2Н, уш.с), 6,11-6,16 (1Н, м), 6,21-6,31 (1Н, м), 8,05 (1Н, д, J=7,3 Гц), 8,08 (1Н, д, J=8,5 Гц), 8,13 (1Н, с), 8,35-8,39 (2Н, м), 8,87-8,92 (1Н, м).
ESI-MS m/z: 411 [M+H]+.
Пример 20
Смесь (S)-N-(4-амино-6-изопропил-5-(хинолин-3-ил)-8,9-дигидропиримидо[5,4-b]индолизин-8-ил)акриламида (Соединение 20А) и (S)-N-(4-амино-6-(пропан-2-илиден)-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)акриламида (Соединение 20В)
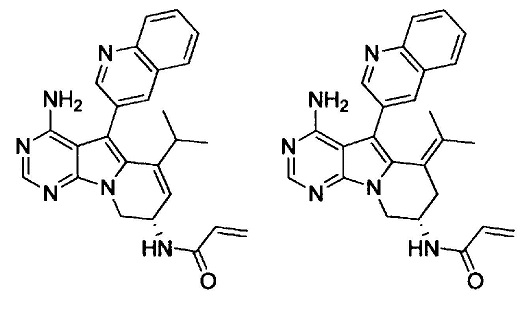
Такой же синтез, как в Примере 19, осуществляют, используя изопропилтрифенилфосфонийиодид вместо этилтрифенилфосфонийбромида, используемого на стадии 5 Примера 19, таким образом получая смесь (18,9 мг) указанных в заголовке соединений.
1Н-ЯМР (ДМСО-d6) δ: 0,85-4,71 (11Н, м), 5,63-5,68 (1Н, м), 5,88 (1Н, уш.с), 6,03 (1Н, уш.с), 6,12-6,19 (1Н, м), 6,24-6,31 (1Н, м), 7,59-7,64 (1Н, м), 7,72-7,78 (1Н, м), 7,96-7,99 (1Н, м), 8,01-8,05 (1Н, м), 8,16 (1Н, д, J=3,4 Гц), 8,24-8,28 (1Н, м), 8,39-8,44 (1Н, м), 8,90 (1Н, д, J=2,0 Гц).
ESI-MS m/z: 425 [M+H]+.
Пример 21
(R)-N-(4-Амино-6-метилен-5-(хинолин-3-ил)-7,8-дигидро-6Н-пиримидо[5,4-b]пирролизин-7-ил)-N-метилакриламид (Соединение 21)
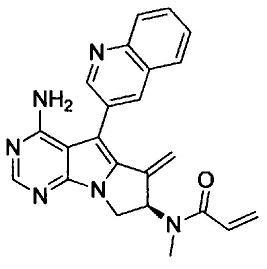
Стадия 1: Синтез (R)-трет-бутил(1-(4-хлор-5-иод-7Н-пирроло[2,3-d]пиримидин-7-ил)бут-3-ен-2-ил)карбамата
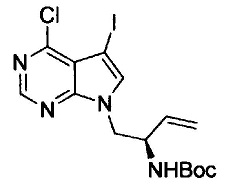
Такой же синтез, как на стадии 1 Примера 1, осуществляют, используя (R)-трет-бутил(1-гидроксибут-3-ен-2-ил)карбамат вместо (S)-трет-бутил(1-гидроксибут-3-ен-2-ил)карбамата, используемого на стадии 1 Примера 1, таким образом получая указанное в заголовке соединение (1,083 г), в виде твердого вещества светло-желтого цвета.
ESI-MS m/z: 448, 450 [M+H]+.
Стадия 2: Синтез (R)-трет-бутил(1-(4-хлор-5-иод-7Н-пирроло[2,3-d]пиримидин-7-ил)бут-3-ен-2-ил)(метил)карбамата
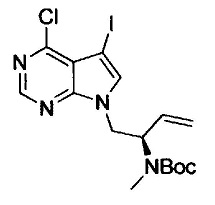
Метилиодид (1,58 мл) и гидрид натрия (224 мг), диспергированный в жидком парафине, добавляют к раствору (R)-трет-бутил(1-(4-хлор-5-иод-7Н-пирроло[2,3-d]пиримидин-7-ил)бут-3-ен-2-ил)карбамата (2,28 г), полученного на стадии 1, в ДМФА (11,4 мл), при комнатной температуре, и смесь перемешивают в течение 1 часа, при такой же температуре. Полученную смесь выливают в воду и экстрагируют этилацетатом. Полученный органический слой промывают водой и насыщенным раствором хлорида натрия и сушат над безводным сульфатом натрия; после этого, растворитель отгоняют при пониженном давлении. Полученный остаток очищают с помощью колоночной хроматографии на силикагеле (элюирующий растворитель: гексан/этилацетат), таким образом получая указанное в заголовке соединение (2,41 г), в виде твердого вещества светло-желтого цвета.
ESI-MS m/z: 462, 464 [M+H]+.
Стадия 3: Синтез Соединения 21
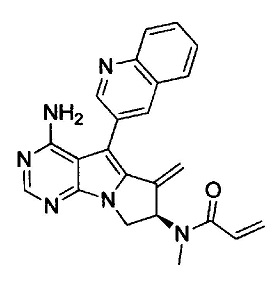
Такой же синтез, как на стадиях 5-10 Примера 13, осуществляют, используя (R)-трет-бутил(1-(4-хлор-5-иод-7Н-пирроло[2,3-d]пиримидин-7-ил)бут-3-ен-2-ил)(метил)карбамат, полученный на стадии 2, вместо (R)-трет-бутил(5-(4-хлор-5-иод-7Н-пирроло[2,3-d]пиримидин-7-ил)пент-1-ен-3-ил)карбамата, используемого на стадии 5 Примера 13, таким образом получая указанное в заголовке соединение (256 мг), в виде твердого вещества светло-желтого цвета.
1Н-ЯМР (ДМСО-d6) δ: 2,75 и 2,94 (общий 3Н, каждый с), 4,06-4,20 (1Н, м), 4,46-4,72 (1Н, м), 4,81-4,89 (1Н, м), 5,26-5,35 (1Н, м), 5,70-5,80 (1Н, м), 5,98-6,37 (4Н, м), 6,77-7,01 (1Н, м), 7,62-7,68 (1Н, м), 7,77-7,83 (1Н, м), 8,03-8,10 (2Н, м), 8,14-8,18 (1Н, м), 8,41-8,45 (1Н, м), 8,96-9,01 (1Н, м).
ESI-MS m/z: 397 [M+H]+.
Пример 22
(R)-N-(4-Амино-6-метилен-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)-N-метилакриламид (Соединение 22)
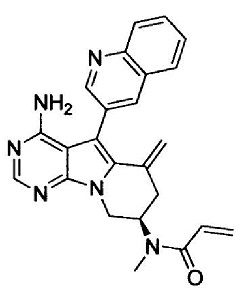
Стадия 1: Синтез (R)-трет-бутил(1-(4-хлор-5-иод-7Н-пирроло[2,3-d]пиримидин-7-ил)пент-4-ен-2-ил)(метил)карбамата
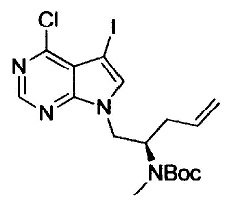
Такой же синтез, как на стадии 2 Примера 21, осуществляют, используя (R)-трет-бутил(1-(4-хлор-5-иод-7Н-пирроло[2,3-d]пиримидин-7-ил)пент-4-ен-2-ил)карбамат, полученный на стадии 1 Примеров 16 и 17, вместо (R)-трет-бутил(1-(4-хлор-5-иод-7Н-пирроло[2,3-d]пиримидин-7-ил)бут-3-ен-2-ил)карбамата, используемого на стадии 2 Примера 21, таким образом получая указанное в заголовке соединение (2,743 г), в виде твердого вещества светло-желтого цвета.
ESI-MS m/z: 477, 479 [M+H]+.
Стадия 2: Синтез (R)-трет-бутил(4-амино-6-метилен-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)(метил)карбамата
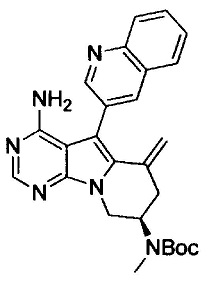
Такой же синтез, как на стадиях 5-8 Примера 13, осуществляют, используя (R)-трет-бутил(1-(4-хлор-5-иод-7Н-пирроло[2,3-d]пиримидин-7-ил)пент-4-ен-2-ил)(метил)карбамат, полученный на стадии 1, вместо (R)-трет-бутил(5-(4-хлор-5-иод-7Н-пирроло[2,3-d]пиримидин-7-ил)пент-1-ен-3-ил)карбамата, используемого на стадии 5 Примера 13, таким образом получая указанное в заголовке соединение (1,366 г), в виде твердого вещества светло-коричневого цвета.
ESI-MS m/z: 457 [M+H]+.
Стадия 3: Синтез (R)-N8-метил-6-метилен-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-4,8-диамина
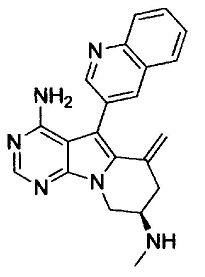
5 н Соляную кислоту (0,5 мл) добавляют к раствору (R)-трет-бутил(4-амино-6-метилен-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)(метил)карбамата (228 мг), полученного на стадии 2, в этаноле (3 мл), и смесь перемешивают в течение 4 суток, при температуре 50°С. После охлаждения, реакционную смесь подщелачивают 5 н водным раствором гидроксида натрия, затем экстрагируют хлороформом. Потом полученный органический слой сушат над безводным сульфатом натрия, растворитель отгоняют при пониженном давлении. Полученный остаток очищают с помощью колоночной хроматографии на основном силикагеле (элюирующий растворитель: хлороформ/метанол), таким образом получая указанное в заголовке соединение (197 мг), в виде твердого вещества светло-коричневого цвета.
ESI-MS m/z: 357 [M+H]+.
Стадия 4: Синтез Соединения 22
Такой же синтез, как на стадии 10 Примера 13, осуществляют, используя (R)-N8-метил-6-метилен-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-4,8-диамин, полученный на стадии 3, вместо ((R)-6-метилен-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-4,7-диамина, используемого на стадии 10 Примера 13, таким образом получая указанное в заголовке соединение (68,5 мг), в виде твердого вещества светло-желтого цвета.
1Н-ЯМР (ДМСО-d6) δ: 2,53-2,68 (1Н, м), 2,88-3,11 (4Н, м), 4,04-4,20 (1Н, м), 4,34-4,96 (4Н, м), 5,58-6,21 (4Н, м), 6,72-7,01 (1Н, м), 7,62-7,68 (1Н, м), 7,78-7,83 (1Н, м), 8,02-8,10 (2Н, м), 8,15 (1Н, уш.с), 8,37-8,44 (1Н, м), 8,86-8,90 (1Н, м).
ESI-MS m/z: 411 [M+H]+.
Пример 23
(R)-N-(4-Амино-6-метил-5-(хинолин-3-ил)-8,9-дигидропиримидо[5,4-b]индолизин-8-ил)-N-метилакриламид (Соединение 23)
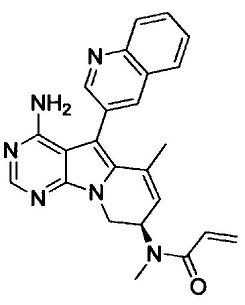
Стадия 1: Синтез (R)-N8,6-диметил-5-(хинолин-3-ил)-8,9-дигидропиримидо[5,4-b]индолизин-4,8-диамина
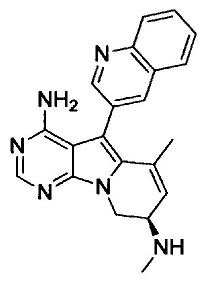
5 н Соляную кислоту (1 мл) добавляют к раствору (R)-трет-бутил(4-амино-6-метилен-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)(метил)карбамата (228 мг), полученного на стадии 2 Примера 22, в этаноле (4 мл), и смесь перемешивают в течение 24 часов при кипячении с обратным холодильником. После охлаждения, реакционную смесь подщелачивают 5 н водным раствором гидроксида натрия, потом экстрагируют хлороформом. Затем полученный органический слой сушат над безводным сульфатом натрия, растворитель отгоняют при пониженном давлении. Полученный остаток очищают с помощью колоночной хроматографии на основном силикагеле (элюирующий растворитель: хлороформ/метанол), таким образом получая указанное в заголовке соединение (190,8 мг), в виде твердого вещества светло-коричневого цвета.
ESI-MS m/z: 357 [M+H]+.
Стадия 2: Синтез Соединения 23
Такой же синтез, как на стадии 10 Примера 13, осуществляют, используя (R)-N8,6-диметил-5-(хинолин-3-ил)-8,9-дигидропиримидо[5,4-b]индолизин-4,8-диамин, полученный на стадии 1, вместо ((R)-6-метилен-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-4,7-диамина, используемого на стадии 10 Примера 13, таким образом получая указанное в заголовке соединение (150,9 мг), в виде твердого вещества желтого цвета.
1Н-ЯМР (ДМСО-d6) δ: 1,52 (3Н, с), 2,74 и 2,88 (общий 3Н, каждый уш.с), 4,14-4,42 (2Н, м), 5,21-5,98 (5Н, м), 6,09-6,22 (1Н, м), 6,71-7,03 (1Н, м), 7,64-7,69 (1Н, м), 7,79-7,85 (1Н, м), 8,02-8,07 (1Н, м), 8,08-8,12 (1Н, м), 8,14 (1Н, с), 8,43-8,49 (1Н, м), 8,89-8,96 (1Н, м).
ESI-MS m/z: 411 [M+H]+.
Пример 24
N-((7S)-4-Амино-6-метил-5-(хинолин-3-ил)-7,8-дигидро-6Н-пиримидо[5,4-b]пирролизин-7-ил)акриламид (Соединение 24)
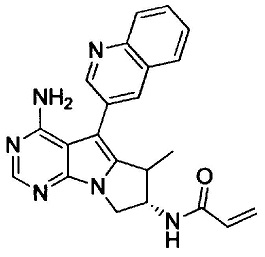
Стадия 1: Синтез трет-бутил((7S)-4-амино-6-метил-5-(хинолин-3-ил)-7,8-дигидро-6Н-пиримидо[5,4-b]пирролизин-7-ил)карбамата
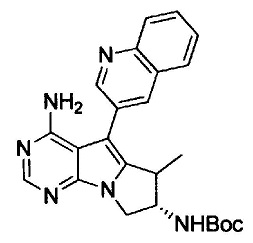
10%-ный Палладий-на-угле (50% масс., 15,0 мг) добавляют к раствору (S)-трет-бутил(4-амино-6-метилен-5-(хинолин-3-ил)-7,8-дигидро-6Н-пиримидо[5,4-b]пирролизин-7-ил)карбамата (15,0 мг), полученного на стадии 5 Примера 1, в смеси этилацетат (2 мл) - этанол (1 мл), и смесь перемешивают в течение 12 часов, при комнатной температуре, в атмосфере водорода. Реакционную смесь отфильтровывают через целит и фильтрат концентрируют при пониженном давлении. Полученный остаток очищают с помощью препаративной тонкослойной хроматографии (элюирующий растворитель: хлороформ/метанол), таким образом получая указанное в заголовке соединение (10,0 мг).
ESI-MS m/z: 357 [M+H]+.
Стадия 2: Синтез Соединения 24
Такой же синтез, как на стадиях 9 и 10 Примера 13, осуществляют, используя трет-бутил((7S)-4-амино-6-метил-5-(хинолин-3-ил)-7,8-дигидро-6Н-пиримидо[5,4-b]пирролизин-7-ил)карбамат, полученный на стадии 1, вместо (R)-трет-бутил(4-амино-6-метилен-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-7-ил)карбамата, используемого на стадии 9 Примера 13, таким образом получая указанное в заголовке соединение (3,10 мг), в виде бесцветного твердого вещества.
1Н-ЯМР (ДМСО-d6) δ: 0,86 (2,7Н, д, J=7,6 Гц), 1,08 (0,3Н, д, J=7,6 Гц), 3,43-4,02 (2Н, м), 4,34-4,62 (1Н, м), 5,08-5,19 (1Н, м), 5,59-5,68 (1Н, м), 5,99-6,17 (3Н, м), 6,18-6,33 (1Н, м), 7,62 (1Н, дд, J=7,6, 7,6 Гц), 7,75 (1Н, дд, J=7,6, 7,6 Гц), 7,99-8,07 (2Н, м), 8,12 (1Н, с), 8,29-8,36 (1Н, м), 8,53-8,76 (1Н, м), 8,98 (1Н, д, J=2,2 Гц).
ESI-MS m/z: 385 [M+H]+.
Пример 25
(R)-N-(4-Амино-6-метилен-5-(хинолин-3-ил)-7,8-дигидро-6Н-пиримидо[5,4-b]пирролизин-7-ил)акриламид (Соединение 25)
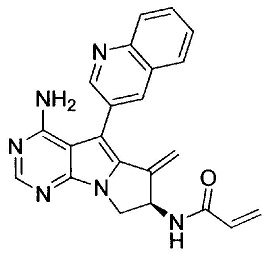
Такой же синтез, как на стадиях 2-6 Примера 1, осуществляют, используя (R)-трет-бутил(1-(4-хлор-5-иод-7Н-пирроло[2,3-d]пиримидин-7-ил)бут-3-ен-2-ил)карбамат, полученный на стадии 1 Примера 21, вместо (S)-трет-бутил(1-(4-хлор-5-иод-7Н-пирроло[2,3-d]пиримидин-7-ил)бут-3-ен-2-ил)карбамата, используемого на стадии 2 Примера 1, таким образом получая указанное в заголовке соединение (44,6 мг), в виде твердого вещества светло-желтого цвета.
1Н-ЯМР (ДМСО-d6) δ: 3,88-3,93 (1Н, м), 4,57-4,63 (1Н, м), 5,03 (1Н, д, J=2,4 Гц), 5,24 (1Н, д, J=2,4 Гц), 5,55-5,62 (1Н, м), 5,68 (1Н, дд, J=10,0, 2,4 Гц), 6,12-6,38 (4Н, м), 7,65 (1Н, дд, J=7,8, 7,8 Гц), 7,77-7,83 (1Н, м), 8,04-8,11 (2Н, м), 8,15 (1Н, с), 8,41 (1Н, д, J=2,2 Гц), 8,82 (1Н, д, J=7,8 Гц), 8,98 (1Н, д, J=2,2 Гц).
ESI-MS m/z: 383 [M+H]+.
Пример 26
(S)-N-(4-Амино-6-метилен-5-(хинолин-3-ил)-7,8-дигидро-6Н-пиримидо[5,4-b]пирролизин-7-ил)-N-метилакриламид (Соединение 26)
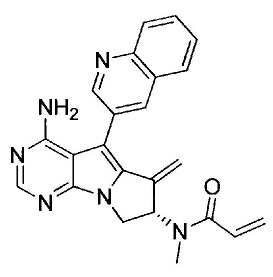
Такой же синтез, как на стадиях 2 и 3 Примера 21, осуществляют, используя (S)-трет-бутил(1-(4-хлор-5-иод-7Н-пирроло[2,3-d]пиримидин-7-ил)бут-3-ен-2-ил)карбамат, полученный на стадии 1 Примера 1, вместо (R)-трет-бутил(1-(4-хлор-5-иод-7Н-пирроло[2,3-d]пиримидин-7-ил)бут-3-ен-2-ил)карбамата, используемого на стадии 2 Примера 21, таким образом получая указанное в заголовке соединение (143 мг), в виде твердого вещества светло-желтого цвета.
1Н-ЯМР (ДМСО-d6) δ: 2,75 и 2,94 (общий 3Н, каждый с), 4,06-4,20 (1Н, м), 4,46-4,72 (1Н, м), 4,81-4,89 (1Н, м), 5,26-5,35 (1Н, м), 5,70-5,80 (1Н, м), 5,98-6,37 (4Н, м), 6,77-7,01 (1Н, м), 7,62-7,68 (1Н, м), 7,77-7,83 (1Н, м), 8,03-8,10 (2Н, м), 8,14-8,18 (1Н, м), 8,41-8,45 (1Н, м), 8,96-9,01 (1Н, м).
ESI-MS m/z: 397 [M+H]+.
Пример 27
(S)-N-(4-Амино-5-(хинолин-3-ил)-8,9-дигидропиримидо[5,4-b]индолизин-8-ил)акриламид (Соединение 27)
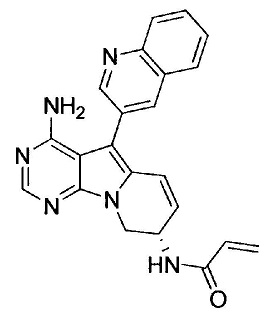
Стадия 1: Синтез (S)-трет-бутил(4-амино-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)карбамата
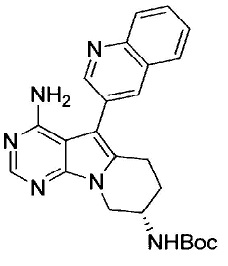
0,5 М Раствор 9-борабицикло[3,3,1]нонана в тетрагидрофуране (141,3 мл) добавляют к раствору (S)-трет-бутил(1-(4-амино-6-бром-5-(хинолин-3-ил)-7Н-пирроло[2,3-d]пиримидин-7-ил)бут-3-ен-2-ил)карбамата (6,0 г), полученного на стадии 4 Примера 1, в тетрагидрофуране (42 мл), в атмосфере азота. Смесь перемешивают при комнатной температуре, в течение 2 часов. К реакционной смеси постепенно добавляют 2 н водный раствор гидроксида натрия (84,8 мл), при комнатной температуре, и дегазируют при пониженном давлении. В атмосфере азота, добавляют (тетракистрифенилфосфин)палладий(0) (1,70 г) и смесь перемешивают при температуре 66°С, в течение 12 часов. Потом реакционную смесь охлаждают, органический слой отделяют и промывают 20%-ным водным раствором хлорида аммония (60 мл). Затем к органическому слою добавляют силикагель SH (6,0 г) и смесь перемешивают при температуре 50°С, в атмосфере азота, в течение 14 часов, и потом отфильтровывают. К фильтрату снова добавляют силикагель SH (выпускаемый фирмой Fuji Silysia Chemical Ltd.) (6,0 г) и смесь перемешивают в атмосфере азота при температуре 50°С, в течение 14 часов, и потом отфильтровывают. Растворитель отгоняют из фильтрата при пониженном давлении. Полученный остаток очищают с помощью колоночной хроматографии на силикагеле (элюирующий растворитель: этилацетат/метанол), таким образом получая указанное в заголовке соединение (4,46 г; выход = 88%), в виде твердого вещества светло-желтого цвета.
ESI-MS m/z: 431 [M+H]+.
Стадия 2: Синтез (8S)-4-амино-8-((трет-бутоксикарбонил)амино)-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-6-илацетата
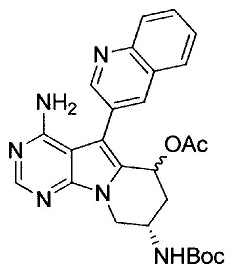
Тетрабутиламмонийиодид (37 мг) и иодбензолдиацетат (241 мг) добавляют к раствору (S)-трет-бутил(4-амино-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)карбамата (215 мг), полученного на стадии 1, в уксусной кислоте (2 мл) и метиленхлориде (2 мл), при охлаждении льдом. Смесь перемешивают в течение 2 часов, при охлаждении льдом, и, в течение 2 часов, при комнатной температуре. После концентрирования при пониженном давлении, реакционную смесь подщелачивают насыщенным водным раствором бикарбоната натрия, затем экстрагируют этилацетатом. Полученный органический слой сушат над безводным сульфатом натрия, потом отфильтровывают. После этого растворитель отгоняют при пониженном давлении. Полученный остаток очищают с помощью хроматографии на силикагеле (элюирующий растворитель: этилацетат/метанол), таким образом получая указанное в заголовке соединение (216 мг), в виде твердого вещества светло-коричневого цвета.
ESI-MS m/z: 489 [M+H]+.
Стадия 3: Синтез трет-бутил((8S)-4-амино-6-гидрокси-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)карбамата
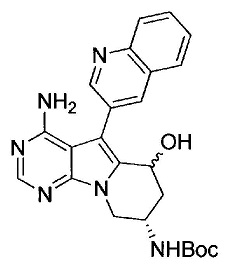
2 н Водный раствор гидроксида натрия (1 мл) добавляют к раствору (8S)-4-амино-8-((трет-бутоксикарбонил)амино)-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-6-илацетата (215 мг), полученного на стадии 2, в метаноле (3 мл), при комнатной температуре, и смесь перемешивают в течение 1 часа, при комнатной температуре. Реакционную смесь выливают в воду и экстрагируют хлороформом. Полученный органический слой сушат над безводным сульфатом натрия, затем отфильтровывают. После этого растворитель отгоняют при пониженном давлении. Полученный остаток очищают с помощью хроматографии на силикагеле (элюирующий растворитель: этилацетат/метанол), таким образом получая указанное в заголовке соединение (166,7 мг), в виде твердого вещества светло-желтого цвета.
ESI-MS m/z: 447 [M+H]+.
Стадия 4: Синтез (S)-5-(хинолин-3-ил)-8,9-дигидропиримидо[5,4-b]индолизин-4,8-диамина
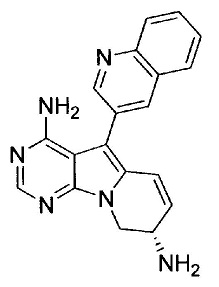
Смесь трет-бутил((8S)-4-амино-6-гидрокси-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)карбамата (44,6 мг), полученного на стадии 3, моногидрата п-толуолсульфоновой кислоты (95 мг) и толуола (3 мл) перемешивают в течение 4 часов, при температуре 100°С. После охлаждения реакционной смеси, растворитель отгоняют при пониженном давлении. Полученный остаток очищают с помощью хроматографии на силикагеле (элюирующий растворитель: хлороформ/метанол), таким образом получая указанное в заголовке соединение (39 мг), в виде маслянистого вещества светло-коричневого цвета.
ESI-MS m/z: 329 [M+H]+.
Стадия 5: Синтез Соединения 27
Такой же синтез, как на стадии 10 Примера 13, осуществляют, используя (S)-5-(хинолин-3-ил)-8,9-дигидропиримидо[5,4-b]индолизин-4,8-диамин, полученный на стадии 4, вместо (R)-6-метилен-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-4,7-диамина, используемого на стадии 10 Примера 13, таким образом получая указанное в заголовке соединение (44,1 мг), в виде твердого вещества желтого цвета.
1Н-ЯМР (ДМСО-d6) δ: 4,19-4,30 (2Н, м), 4,86-4,94 (1Н, м), 5,63 (1Н, дд, J=9,8, 2,7 Гц), 6,08-6,32 (5Н, м), 6,70 (1Н, дд, J=9,8, 1,1 Гц), 7,62-7,69 (1Н, м), 7,75-7,82 (1Н, м), 8,04-8,09 (2Н, м), 8,17 (1Н, с), 8,34 (1Н, д, J=2,0 Гц), 8,55 (1Н, д, J=7,3 Гц), 8,91 (1Н, д, J=2,4 Гц).
ESI-MS m/z: 383 [M+H]+.
Пример 28
(R)-N-(4-Амино-5-(хинолин-3-ил)-9,10-дигидро-8Н-пиримидо[5’,4’:4,5]пирроло[1,2-a]азепин-8-ил)акриламид (Соединение 28)
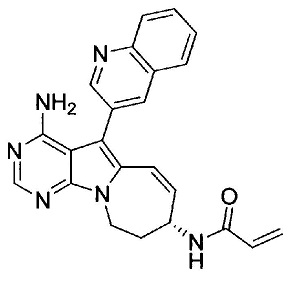
Такой же синтез, как в Примере 27, осуществляют, используя (R)-трет-бутил(5-(4-амино-6-бром-5-(хинолин-3-ил)-7Н-пирроло[2,3-d]пиримидин-7-ил)пент-1-ен-3-ил)карбамат, полученный на стадии 7 Примера 13, вместо (S)-трет-бутил(1-(4-амино-6-бром-5-(хинолин-3-ил)-7Н-пирроло[2,3-d]пиримидин-7-ил)бут-3-ен-2-ил)карбамата, используемого на стадии 1 Примера 27, таким образом получая указанное в заголовке соединение (101,8 мг), в виде твердого вещества желтого цвета.
1Н-ЯМР (ДМСО-d6) δ: 2,01-2,12 (1Н, м), 2,23-2,33 (1Н, м), 4,28-4,38 (1Н, м), 4,55-4,63 (1Н, м), 4,84-4,92 (1Н, м), 5,62 (1Н, дд, J=10,0, 2,4 Гц), 5,68 (1Н, дд, J=12,6, 3,8 Гц), 6,08-6,31 (5Н, м), 7,63-7,69 (1Н, м), 7,78-7,83 (1Н, м), 8,03-8,10 (2Н, м), 8,18 (1Н, с), 8,31 (1Н, д, J=2,0 Гц), 8,51 (1Н, д, J=8,2 Гц), 8,84 (1Н, д, J=2,2 Гц).
ESI-MS m/z: 397 [M+H]+.
Пример 29
N-((6R*,8S)-4-Амино-6-метил-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)акриламид (Соединение 29А) и N-((6S*,8S)-4-амино-6-метил-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)акриламид (Соединение 29В)
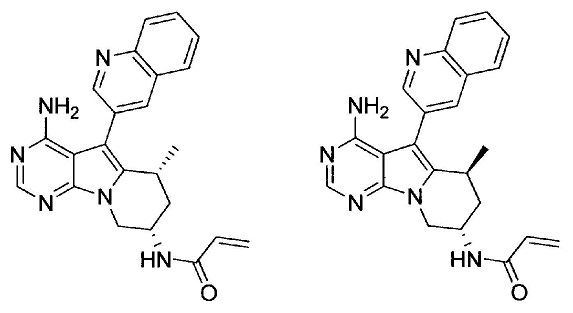
Стадия 1: Синтез (S)-трет-бутил(4-амино-6-метилен-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)карбамата
Смесь (S)-трет-бутил(4-амино-6-метил-5-(хинолин-3-ил)-8,9-дигидропиримидо[5,4-b]индолизин-8-ил)карбамата и (S)-трет-бутил(4-амино-6-метилен-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)карбамата, полученная путем такого же синтеза, как на стадии 5 Примера 14, с использованием (S)-трет-бутил(1-(4-амино-6-бром-5-(хинолин-3-ил)-7Н-пирроло[2,3-d]пиримидин-7-ил)пент-4-ен-2-ил)карбамата (1,263 г), полученного на стадии 4 Примера 14, очищают с помощью хроматографии на силикагеле (элюирующий растворитель: этилацетат/метанол), таким образом получая указанное в заголовке соединение (0,805 г), в виде твердого вещества светло-желтого цвета.
ESI-MS m/z: 443 [M+H]+.
Стадия 2: Синтез трет-бутил((8S)-4-амино-6-метил-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)карбамата
10%-ный Палладий-на-угле (50% масс., 100,6 мг) добавляют к раствору (S)-трет-бутил(4-амино-6-метилен-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)карбамата (100,0 мг), полученного на стадии 1, в метаноле (10 мл), и смесь перемешивают в течение 12 часов, при комнатной температуре, в атмосфере водорода. Реакционную смесь отфильтровывают через стекловолоконный фильтр и фильтрат концентрируют при пониженном давлении. Полученный остаток очищают с помощью колоночной хроматографии на основном силикагеле (элюирующий растворитель: хлороформ/метанол), таким образом получая указанное в заголовке соединение (122,1 мг).
ESI-MS m/z: 445 [M+H]+.
Стадия 3: Синтез (8S)-6-метил-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-4,8-диамина
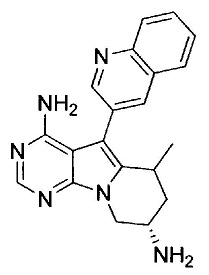
Трифторуксусную кислоту (1 мл) добавляют к раствору трет-бутил((8S)-4-амино-6-метил-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)карбамата (122,1 мг), полученного на стадии 2, в хлороформе (3 мл), и смесь перемешивают в течение 30 мин, при комнатной температуре. Реакционную смесь концентрируют при пониженном давлении и полученный остаток растворяют в метаноле. Затем обессоливают при использовании колонки для основной твердофазной экстракции (VARIAN BondElut), смесь концентрируют при пониженном давлении, таким образом получая указанное в заголовке соединение (58,9 мг), в виде твердого вещества светло-желтого цвета.
ESI-MS m/z: 345 [M+H]+.
Стадия 4: Синтез N-((8S)-4-амино-6-метил-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)акриламида
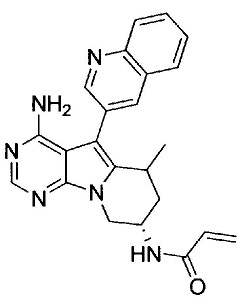
Такой же синтез, как на стадии 7 Примера 14, осуществляют, используя (8S)-6-метил-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-4,8-диамин (58,9 мг), полученный на стадии 3, таким образом получая указанное в заголовке соединение (30,3 мг), в виде твердого вещества светло-желтого цвета.
ESI-MS m/z: 399 [M+H]+.
Стадия 5: Синтез Соединения 29А и Соединения 29В
Смесь диастереоизомеров, содержащую (6R*,8S)-изомер и (6S*,8S)-изомер, полученную на стадии 4, разделяют с помощью препаративной хиральной колоночной хроматографии (колонка: CHIRALCEL OZ-H (20 мм ×250 мм ×5 мкм), выпускаемая фирмой Daicel Chemical Industries, Ltd., элюирующий растворитель: гексан/этанол/триэтиламин = 60/40/0,01), таким образом получая Соединение 29А и Соединение 29В, в виде первой фракции (15,8 мг) и второй фракции (6,1 мг), соответственно, в виде бесцветных твердых веществ.
Соединение 29А
1Н-ЯМР (CDCl3) δ: 0,96 (3Н, д, J=6,6 Гц), 1,68 (1Н, м), 2,29-2,39 (1Н, м), 3,49-3,63 (1Н, м), 3,81 (1Н, дд, J=11,7, 10,2 Гц), 4,56-4,69 (1Н, м), 4,76 (1Н, дд, J=12,2, 5,1 Гц), 4,81 (2Н, уш.с), 5,70 (1Н, дд, J=10,5, 1,2 Гц), 6,17 (1Н, дд, J=16,6, 10,2 Гц), 6,15-6,25 (1Н, м), 6,36 (1Н, дд, J=17,1, 1,2 Гц), 7,64 (1Н, т, J=7,6 Гц), 7,75-7,82 (1Н, м), 7,88 (1Н, д, J=8,0 Гц), 8,18 (1Н, д, J=8,5 Гц), 8,21-8,29 (2Н, м), 9,05 (1Н, д, J=2,0 Гц).
ESI-MS m/z: 399 [M+H]+.
Соединение 29В
1Н-ЯМР (CDCl3) δ: 1,06 (3Н, д, J=6,8 Гц), 1,83-2,08 (1Н, м), 2,18-2,31 (1Н, м), 3,46-3,59 (1Н, м), 4,00 (1Н, дд, J=12,4, 7,6 Гц), 4,59 (1Н, дд, J=12,4, 4,6 Гц), 4,73-4,93 (3Н, м), 5,70 (1Н, д, J=10,5 Гц), 6,18 (1Н, дд, J=16,8, 10,2 Гц), 6,26-6,42 (2Н, м), 7,63 (1Н, т, J=7,6 Гц), 7,78 (1Н, т, J=8,3 Гц), 7,88 (1Н, д, J=8,0 Гц), 8,17 (1Н, д, J=8,0 Гц), 8,20-8,27 (2Н, м), 9,02 (1Н, д, J=1,7 Гц).
ESI-MS m/z: 399 [M+H]+.
Сравнительный пример 1
N-(3-(4-Амино-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-5-ил)фенил)бензамид (Сравнительное Соединение 1)
Соединение синтезируют, в соответствии с методикой, раскрытой в статье Международной публикации под номером WO 2006/102079.
ESI-MS m/z: 384 [M+H]+.
Тест-примеры
Соединения согласно настоящему изобретению оценивали, используя нижеприводимые тест-способы.
Тест-пример 1: Измерение ингибирующего действия против различных EGFR-киназных активностей ( in vitro )
1) Измерение ингибирующей EGFR (T790M/L858R)-киназу активности
Измеряли ингибирующие активности соединений согласно настоящему изобретению против активности EGFR (T790M/L858R)-киназы.
Среди веществ для измерения ингибирующей активности, субстратный пептид и киназный белок получали следующим образом. Пептид с биотинилированным N-концом (биотин-EEPLYWSFPAKKK) синтезировали относительно FL-Peptide 22, реагента серии LabChip® PerkinElmer, Inc. Для киназного белка, у фирмы Carna Biosciences, Inc., приобретали очищенный рекомбинантный человеческий EGFR (T790M/L858R)-белок.
Измерение ингибирующей активности осуществляли следующим образом. Соединения согласно настоящему изобретению индивидуально растворяли в диметилсульфоксиде (ДМСО) и последовательные разведения этих соединений приготовляли, используя ДМСО. Затем, раствор, содержащий субстратный пептид (конечная концентрация: 250 нМ), хлорид магния (конечная концентрация: 10 мМ), хлорид марганца (конечная концентрация: 10 мМ)и АТФ (конечная концентрация: 1 мкМ), в буферном растворе для киназной реакции (Carna Biosciences, Inc.), смешивали с разведениями соединений (конечная концентрация ДМСО после киназной реакции: 2,5%) или ДМСО (конечная концентрация: 2,5%). Туда, далее, добавляли EGFR (T790M/L858R)-белок и смеси инкубировали при температуре 25°С, в течение 120 минут, для осуществления киназной реакции. Затем туда добавляли EDTA до конечной концентрации 24 мМ для завершения, таким образом, реакции. К реакционным смесям добавляли раствор для детектирования фосфорилированного тирозина, содержащий меченое европием (Eu) антитело РТ66, против фосфорилированного тирозина (PerkinElmer, Inc.) и SureLight APC-SA (PerkinElmer, Inc.), и смеси оставляли стоять при комнатной температуре в течение двух часов или более. Для фона, образец, к которому добавляли EDTA до добавления EGFR (T790M/L858R)-белка, инкубировали при температуре 25°С, в течение 120 мин, используя ДМСО вместо раствора соединения в ДМСО. Также к этому образцу добавляли раствор для детектирования и смесь оставляли стоять в течение двух часов или более. Окончательно, в случае всех тест-образцов, измеряли интенсивность флуоресценции во время облучения возбуждающим светом, имеющим длину волны 337 нм, при двух длинах волн 620 нм и 665 нм, с помощью прибора PHERAstar FS (BMG LABTECH) и, в качестве данных, получали соотношение интенсивностей флуоресценции при двух длинах волн.
При анализе измеренных данных, соотношение интенсивностей флуоресценции образца, в случае которого осуществляли киназную реакцию с добавлением ДМСО в конечной концентрации 2,5%, устанавливали, как степень ингибирования фосфорилирования 0%, а соотношение интенсивностей флуоресценции фона устанавливали, как степень ингибирования фосфорилирования 100%. Основываясь на вышеуказанных степенях, степени ингибирования фосфорилирования (%) рассчитывали для образцов, к которым добавляли различные концентрации растворов соединений. В заключение, в случае каждого соединения вычерчивали кривые для полученных степеней ингибирования реакции (%), при соответственных концентрациях, и определяли значение IC50 (нМ), которое представляет собой концентрацию соединения, при которой фосфорилирование посредством EGFR (T790M/L858R) ингибировано на 50%, используя программное обеспечение для Xlfit регрессионного анализа кривой (IDBS).
2) Измерение ингибирующей EGFR (d746-750/T790M)-киназу активности
Измеряли ингибирующие активности соединений согласно настоящему изобретению против активности EGFR (d746-750/T790M)-киназы.
Вещества, способ измерения и метод анализа данных фактически были такими же, как и таковые, указанные в вышеприведенном описании относительно «Измерения ингибирующей EGFR (T790M/L858R)-киназу активности», за исключением того, что очищенный рекомбинантный человеческий EGFR (d746-750/T790M)-белок, приобретенный у фирмы Carna Biosciences, Inc., использовали в качестве киназного белка и измерение осуществляли с конечной концентрацией АТФ 1,5 мкМ. В заключение, значение IC50 (нМ) каждого соединения, что касается EGFR (d746-750/T790M), определяли посредством анализа данных.
3) Измерение ингибирующей EGFR (L858R)-киназу активности
Измеряли ингибирующие активности соединений согласно настоящему изобретению против активности EGFR (L858R)-киназы.
Вещества, способ измерения и метод анализа данных фактически были такими же, как и таковые, указанные в вышеприведенном описании относительно «Измерения ингибирующей EGFR (T790M/L858R)-киназу активности», за исключением того, что очищенный рекомбинантный человеческий EGFR (L858R)-белок, приобретенный у фирмы Carna Biosciences, Inc., использовали в качестве киназного белка и измерение осуществляли с конечной концентрацией АТФ 4 мкМ. В заключение, значение IC50 (нМ) каждого соединения, что касается EGFR (L858R), определяли посредством анализа данных.
4) Измерение ингибирующей EGFR (d746-750)-киназу активности
Измеряли ингибирующие активности соединений согласно настоящему изобретению против активности EGFR (d746-750)-киназы.
Вещества, способ измерения и метод анализа данных фактически были такими же, как и таковые, указанные в вышеприведенном описании относительно «Измерения ингибирующей EGFR (T790M/L858R)-киназу активности», за исключением того, что очищенный рекомбинантный человеческий EGFR (в746-750)-белок, приобретенный у фирмы Carna Biosciences, Inc., использовали в качестве киназного белка, измерение осуществляли с конечной концентрацией АТФ 5 мкМ и инкубацию для киназной реакции осуществляли, в течение 90 мин. В заключение, значение IC50 (нМ) каждого соединения, что касается EGFR (d746-750), определяли посредством анализа данных.
5) Измерение ингибирующей EGFR (WT)-киназу активности
Измеряли ингибирующие активности соединений согласно настоящему изобретению против активности EGFR (WT)-киназы.
Вещества, способ измерения и метод анализа данных фактически были такими же, как и таковые, указанные в вышеприведенном описании относительно «Измерения ингибирующей EGFR (T790M/L858R)-киназу активности», за исключением того, что киназный белок, получаемый путем экспрессии цитоплазматического домена человеческого EGFR (WT) с его N-концом, гибридизированным по FLAG-метке с клетками насекомых Sf9, используя бакуловирусную систему экспрессии, и затем подвергаемый очистке с помощью анти-FLAG-антитело-агарозы (Sigma Aldrich), использовали в качестве киназного белка, конечная концентрация субстратного белка составляла 500 нМ и конечная концентрация АТФ составляла 4,7 мкМ. В заключение, значение IC50 (нМ) каждого соединения, что касается EGFR (WT), определяли посредством анализа данных.
В таблице 1 представлены результаты тестов, используя 30 различных EGFR.
Подтверждено, что соединения согласно настоящему изобретению обладают сильными ингибирующими активностями не только против EGFR (L858R) и EGFR (d746-750), но и также против EGFR (T790M/L858R) и EGFR (d746-750/T790M). Также подтверждено, что их ингибирующие активности по отношению к EGFR (WT) являются более низкими, чем таковые по отношению к вышеуказанным EGFR-белкам.
В противоположность, подтверждено, что соединение сравнительного примера 1, которое представляет собой соединение, имеющее структуру, подобную таковой соединения согласно настоящему изобретению, почти не проявляет ингибирующей активности против этих EGFR-киназ.
L858R)
T790M)
Тест-пример 2: Тест на активность ингибирования роста против клеточных линий, экспрессирующих дикого типа и мутированный EGFR ( in vitro )
Человеческую клеточную линию мелкоклеточного рака легких NCI-H1975, экспрессирующую EGFR (T790M/L858R), (2) человеческую клеточную линию немелкоклеточного рака легких HCC827, экспрессирующую EGFR (d746-750), и (3) человеческую клеточную линию эпителиоидного рака А431, экспрессирующую EGFR (WT), каждую, суспендировали в полной питательной среде, рекомендованной ATCC. Клеточные суспензии высевали в каждую лунку 384-луночного плоского микропланшета или 96-луночного плоского планшета и культивировали в инкубаторе, содержащем 5% газообразного диоксида углерода, при температуре 37°С, в течение одних суток. Каждое соединение согласно настоящему изобретению растворяли в ДМСО и последовательные разведения тест-соединений приготовляли, используя ДМСО (до концентрации, 500-кратной конечной концентрации). Раствор в ДМСО тест-соединения или ДМСО разводили, с помощью полной питательной среды, и полученный раствор добавляли в каждую лунку клеточного культурального планшета, так, чтобы конечная концентрация ДМСО составляла 0,2%. Затем клетки культивировали в инкубаторе, содержащем 5% газообразного диоксида углерода, при температуре 37°С, в течение трех суток. Число клеток измеряли до добавления ДМСО-раствора тест-соединения, а также после добавления и культивирования путем использования CellTiter-Glo Assay® (выпускаемый фирмой Promega) согласно протоколу, рекомендованному фирмой Promega.
Для каждой клетки, степени ингибирования клеточного роста в лунках, в которые добавлены тест-соединения в различных концентрациях, рассчитывали по нижеследующей формуле. Затем по полученным степеням ингибирования (%) при соответствующих концентрациях вычерчивали кривые для каждого тест-соединения и значение GI50 (нМ), которое представляет собой концентрацию тест-соединения, при которой клеточный рост может быть ингибирован на 50%, определяли, используя программное обеспечение для XLfit регрессионного анализа кривой (IDBS).
Степень ингибирования клеточного роста (%)=(С-Т)/(С-С0)×100
Т: интенсивность люминесценции в лунке, культивированной в течение трех суток после добавления раствора тест-соединения
С: интенсивность люминесценции в лунке, культивированной в течение трех суток после добавления ДМСО
С0: интенсивность люминесценции в лунке до добавления раствора тест-соединения
Результаты представлены в таблице 2.
Подтверждено, что соединения согласно настоящему изобретению оказывают сильные, ингибирующие рост, воздействия не только на клетки, экспрессирующие EGFR (d746-750), но и также на клетки, экспрессирующие EGFR (T790M/L858R). Также подтверждено, что их ингибирующие рост воздействия на клетки, экспрессирующие EGFR (WT), являются более низкими, чем таковые в случае вышеуказанных EGFR-белков.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВОЕ ХИНОЛИН-ЗАМЕЩЕННОЕ СОЕДИНЕНИЕ | 2014 |
|
RU2797117C2 |
| ХИНОЛИЛПИРРОЛПИРИМИДИЛЬНОЕ КОНДЕНСИРОВАННОЕ СОЕДИНЕНИЕ ИЛИ ЕГО СОЛЬ | 2013 |
|
RU2581039C1 |
| ПИРИМИДО[5,4-b]ИНДОЛИЗИНОВОЕ ИЛИ ПИРИМИДО[5,4-b]ПИРРОЛИЗИНОВОЕ СОЕДИНЕНИЕ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ | 2017 |
|
RU2721774C1 |
| СЕЛЕКТИВНЫЙ ИНГИБИТОР МУТАНТА EGFR СО ВСТАВКОЙ В ЭКЗОНЕ 20 | 2017 |
|
RU2774629C2 |
| ПРОТИВОВИРУСНЫЕ СРЕДСТВА ДЛЯ ЛЕЧЕНИЯ И ПРОФИЛАКТИКИ ВИЧ ИНФЕКЦИИ | 2021 |
|
RU2780103C1 |
| ПРОТИВОВИРУСНЫЕ СРЕДСТВА ДЛЯ ЛЕЧЕНИЯ И ПРОФИЛАКТИКИ ВИЧ-ИНФЕКЦИИ | 2020 |
|
RU2780101C2 |
| ПРОИЗВОДНЫЕ ПИРИДОПИРРОЛИЗИНА И ПИРИДОИНДОЛИЗИНА | 2003 |
|
RU2342386C2 |
| МОЧЕВИНОЗАМЕЩЕННЫЕ ИМИДАЗОХИНОЛИНЫ, ФАРМАЦЕВТИЧЕСКИЙ ПРЕПАРАТ И СПОСОБ ИНДУКЦИИ БИОСИНТЕЗА ЦИТОКИНОВ НА ИХ ОСНОВЕ | 2000 |
|
RU2265020C2 |
| ПИРИМИДИНОВЫЕ ПРОИЗВОДНЫЕ, ИСПОЛЬЗУЕМЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕИНКИНАЗЫ | 2013 |
|
RU2623221C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ 1,2,3,4-ТЕТРАГИДРОПИРИМИДО{1,2-a}ПИРИМИДИН-6-ОНА, ИХ ПОЛУЧЕНИЕ И ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2010 |
|
RU2561130C2 |
Изобретение относится к новому соединению формулы (I) или его фармацевтически приемлемой соли, которые обладают свойствами ингибитора рецептора эпидермального фактора роста (EGFR). Соединения могут быть использованы для получения противоопухолевого лекарственного средства для лечения или предупреждения ракового заболевания, базирующегося на ингибирующих эффектах по отношению к EGFR. В формуле (I) 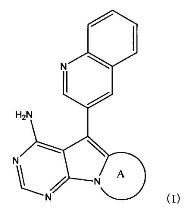 группа
группа 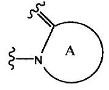 представляет собой (1) группу, представленную формулой А1
представляет собой (1) группу, представленную формулой А1
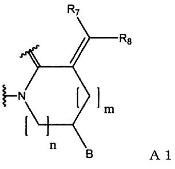
(в формуле А1 В означает группу, представленную формулой
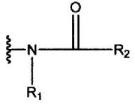
[R1 означает атом водорода или С1-С6-алкильную группу; и R2 означает группу, представленную формулой
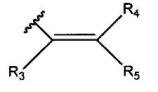 ,
,
где R3, R4 и R5 являются одинаковыми или различными и каждый означает атом водорода, атом галогена, C1-C6-алкильную группу, фенил, нафтил или бифенил, моноциклическую или бициклическую C2-C9-гетероарильную группу, содержащую от 1 до 3 одинаковых или различных гетероатомов, выбираемых из атомов азота, кислорода и серы, аминометильную группу, которая может быть замещена C1-C6-алкильной группой, или 1-пиперидинометильную группу, или группу, представленную формулой  , где R6 означает атом водорода или С1-С6-алкильную группу], R7 и R8 являются одинаковыми или различными и каждый означает атом водорода или С1-С6-алкильную группу; m означает 0 или 1; n означает 1 или 2); (2) группу, представленную формулой А2:
, где R6 означает атом водорода или С1-С6-алкильную группу], R7 и R8 являются одинаковыми или различными и каждый означает атом водорода или С1-С6-алкильную группу; m означает 0 или 1; n означает 1 или 2); (2) группу, представленную формулой А2:
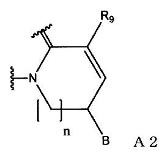
(в формуле А2 В и n имеют значения, как определено в формуле А1; и R9 означает атом водорода или С1-С6-алкильную группу); или (3) группу, представленную формулой А3:
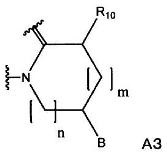
(в формуле А3 В, m и n имеют значения, как определено в формуле А1; и R10 означает С1-С6-алкильную группу). 6 н. и 9 з.п. ф-лы, 3 табл., 31 пр.
1. Соединение, представленное нижеприводимой формулой (I), или его фармацевтически приемлемая соль
где группа
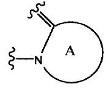
представляет собой
(1) группу, представленную формулой А1:
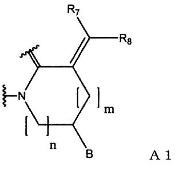
(в формуле А1 В означает группу, представленную формулой:
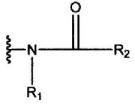
[R1 означает атом водорода или С1-С6-алкильную группу;
и R2 означает группу, представленную формулой:
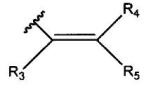
где R3, R4 и R5 являются одинаковыми или различными и каждый означает атом водорода, атом галогена, C1-C6-алкильную группу, фенил, нафтил или бифенил, моноциклическую или бициклическую C2-C9-гетероарильную группу, содержащую от 1 до 3 одинаковых или различных гетероатомов, выбираемых из атомов азота, кислорода и серы, аминометильную группу, которая может быть замещена C1-C6-алкильной группой, или 1-пиперидинометильную группу,
или группу, представленную формулой:

где R6 означает атом водорода или С1-С6-алкильную группу],
R7 и R8 являются одинаковыми или различными и каждый означает атом водорода или С1-С6-алкильную группу; m означает 0 или 1; n означает 1 или 2);
(2) группу, представленную формулой А2:
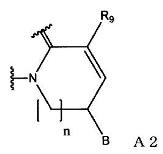
(в формуле А2 В и n имеют значения, как определено в формуле А1; и R9 означает атом водорода или С1-С6-алкильную группу); или
(3) группу, представленную формулой А3:
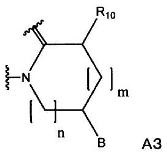
(в формуле А3 В, m и n имеют значения, как определено в формуле А1; и R10 означает С1-С6-алкильную группу).
2. Соединение или его фармацевтически приемлемая соль по п.1, где R2 представляет собой группу, представленную формулой:
где R3, R4 и R5 являются одинаковыми или различными и каждый означает атом водорода, атом галогена, С1-С6-алкильную группу, аминометильную группу, которая может быть замещена С1-С6-алкильной группой, или 1-пиперидинометильную группу,
или группу, представленную формулой:
где R6 означает атом водорода или С1-С6-алкильную группу.
3. Соединение или его фармацевтически приемлемая соль по п.1, где R2 представляет собой группу, представленную формулой:
где R3, R4 и R5 являются одинаковыми или различными и каждый означает атом водорода, атом галогена, аминометильную группу, которая может быть замещена метильной группой, или 1-пиперидинометильную группу.
4. Соединение или его фармацевтически приемлемая соль по п.1, где группа:
представляет собой
(1) группу, представленную формулой А1:
(в формуле А1 В означает группу, представленную формулой:
где R1 означает атом водорода или С1-С6-алкильную группу; и R2 означает группу, представленную формулой:
где R3, R4 и R5 являются одинаковыми или различными и каждый означает атом водорода или атом галогена,
R7 и R8 являются одинаковыми или различными и каждый означает атом водорода или С1-С6-алкильную группу; m означает 0 или 1; n означает 1); или
(2) группу, представленную формулой А2:
(в формуле А2 В и n имеют значения, как определено в формуле А1; и R9 означает атом водорода или С1-С6-алкильную группу).
5. Соединение или его фармацевтически приемлемая соль по п.1, где группа:
представляет собой
(1) группу, представленную формулой А1:
(в формуле А1 В означает группу, представленную формулой:
где R1 означает атом водорода; и R2 означает группу, представленную формулой:
где R3, R4 и R5 каждый означает атом водорода,
R7 и R8 каждый означает атом водорода; m означает 0; и n означает 1); или
(2) группу, представленную формулой А2:
(в формуле А2 В и n имеют значения, как определено в формуле А1; и R9 означает С1-С6-алкильную группу).
6. Соединение или его фармацевтически приемлемая соль по п.1, где соединение выбирают из следующей группы соединений, состоящей из:
(S)-N-(4-амино-6-метилен-5-(хинолин-3-ил)-7,8-дигидро-6Н-пиримидо[5,4-b]пирролизин-7-ил)акриламида;
(S)-N-(4-амино-6-метилен-5-(хинолин-3-ил)-7,8-дигидро-6Н-пиримидо[5,4-b]пирролизин-7-ил)метакриламида;
(S)-N-(4-амино-6-метилен-5-(хинолин-3-ил)-7,8-дигидро-6Н-пиримидо[5,4-b]пирролизин-7-ил)бут-2-енамида (смесь Е- и Z-изомеров);
(S,Е)-N-(4-амино-6-метилен-5-(хинолин-3-ил)-7,8-дигидро-6Н-пиримидо[5,4-b]пирролизин-7-ил)-4-(диметиламино)бут-2-енамида;
(S,Е)-N-(4-амино-6-метилен-5-(хинолин-3-ил)-7,8-дигидро-6Н-пиримидо[5,4-b]пирролизин-7-ил)-3-хлоракриламида;
(S,Z)-N-(4-амино-6-метилен-5-(хинолин-3-ил)-7,8-дигидро-6Н-пиримидо[5,4-b]пирролизин-7-ил)-3-хлоракриламида;
(S,E)-N-(4-амино-6-метилен-5-(хинолин-3-ил)-7,8-дигидро-6Н-пиримидо[5,4-b]пирролизин-7-ил)-4-(пиперидин-1-ил)бут-2-енамида;
(S)-N-(4-амино-6-метилен-5-(хинолин-3-ил)-7,8-дигидро-6Н-пиримидо[5,4-b]пирролизин-7-ил)пропиоламида;
(S)-N-(4-амино-6-метилен-5-(хинолин-3-ил)-7,8-дигидро-6Н-пиримидо[5,4-b]пирролизин-7-ил)бут-2-инамида;
(S,E)-N-(4-амино-6-метилен-5-(хинолин-3-ил)-7,8-дигидро-6Н-пиримидо[5,4-b]пирролизин-7-ил)-4-(диэтиламино)бут-2-енамида;
(S,E)-N-(4-амино-6-метилен-5-(хинолин-3-ил)-7,8-дигидро-6Н-пиримидо[5,4-b]пирролизин-7-ил)-4-(этил(метил)амино)бут-2-енамида;
(S,E)-N-(4-амино-6-метилен-5-(хинолин-3-ил)-7,8-дигидро-6Н-пиримидо[5,4-b]пирролизин-7-ил)-4-(изопропил(метил)амино)бут-2-енамида;
(R)-N-(4-амино-6-метилен-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-7-ил)акриламида;
(S)-N-(4-амино-6-метил-5-(хинолин-3-ил)-8,9-дигидропиримидо[5,4-b]индолизин-8-ил)акриламида;
(S)-N-(4-амино-6-метилен-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)акриламида;
(R)-N-(4-амино-6-метил-5-(хинолин-3-ил)-8,9-дигидропиримидо[5,4-b]индолизин-8-ил)акриламида;
(R)-N-(4-амино-6-метилен-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)акриламида;
(S)-N-(4-амино-6-метилен-5-(хинолин-3-ил)-7,8,9,10-тетрагидро-6Н-пиримидо[5’,4’:4,5]пирроло[1,2-a]азепин-8-ил)акриламида;
(S,E)-N-(4-амино-6-этилиден-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)акриламида;
(S)-N-(4-амино-6-изопропил-5-(хинолин-3-ил)-8,9-дигидропиримидо[5,4-b]индолизин-8-ил)акриламида;
(S)-N-(4-амино-6-(пропан-2-илиден)-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)акриламида;
(R)-N-(4-амино-6-метилен-5-(хинолин-3-ил)-7,8-дигидро-6Н-пиримидо[5,4-b]пирролизин-7-ил)-N-метилакриламида;
(R)-N-(4-амино-6-метилен-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)-N-метилакриламида;
(R)-N-(4-амино-6-метил-5-(хинолин-3-ил)-8,9-дигидропиримидо[5,4-b]индолизин-8-ил)-N-метилакриламида;
N-((7S)-4-амино-6-метил-5-(хинолин-3-ил)-7,8-дигидро-6Н-пиримидо[5,4-b]пирролизин-7-ил)акриламида;
(R)-N-(4-амино-6-метилен-5-(хинолин-3-ил)-7,8-дигидро-6Н-пиримидо[5,4-b]пирролизин-7-ил)акриламида;
(S)-N-(4-амино-6-метилен-5-(хинолин-3-ил)-7,8-дигидро-6Н-пиримидо[5,4-b]пирролизин-7-ил)-N-метилакриламида;
(S)-N-(4-амино-5-(хинолин-3-ил)-8,9-дигидропиримидо[5,4-b]индолизин-8-ил)акриламида;
(R)-N-(4-амино-5-(хинолин-3-ил)-9,10-дигидро-8Н-пиримидо[5’,4’:4,5]пирроло[1,2-a]азепин-8-ил)акриламида;
N-((6R*,8S)-4-амино-6-метил-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)акриламида; и
N-((6S*,8S)-4-амино-6-метил-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)акриламида.
7. Соединение или его фармацевтически приемлемая соль по п.1, где соединение выбирают из следующей группы соединений, состоящей из:
(S)-N-(4-амино-6-метилен-5-(хинолин-3-ил)-7,8-дигидро-6Н-пиримидо[5,4-b]пирролизин-7-ил)акриламида;
(S,Е)-N-(4-амино-6-метилен-5-(хинолин-3-ил)-7,8-дигидро-6Н-пиримидо[5,4-b]пирролизин-7-ил)-3-хлоракриламида;
(S,Z)-N-(4-амино-6-метилен-5-(хинолин-3-ил)-7,8-дигидро-6Н-пиримидо[5,4-b]пирролизин-7-ил)-3-хлоракриламида;
(S)-N-(4-амино-6-метил-5-(хинолин-3-ил)-8,9-дигидропиримидо[5,4-b]индолизин-8-ил)акриламида;
(S)-N-(4-амино-6-метилен-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)акриламида;
(S,E)-N-(4-амино-6-этилиден-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)акриламида;
(R)-N-(4-амино-6-метилен-5-(хинолин-3-ил)-7,8-дигидро-6Н-пиримидо[5,4-b]пирролизин-7-ил)-N-метилакриламида;
(R)-N-(4-амино-6-метилен-5-(хинолин-3-ил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)-N-метилакриламида;
(R)-N-(4-амино-6-метил-5-(хинолин-3-ил)-8,9-дигидропиримидо[5,4-b]индолизин-8-ил)-N-метилакриламида; и
(R)-N-(4-амино-5-(хинолин-3-ил)-9,10-дигидро-8Н-пиримидо[5’,4’:4,5]пирроло[1,2-a]азепин-8-ил)акриламида.
8.(S)-N-(4-Амино-6-метилен-5-(хинолин-3-ил)-7,8-дигидро-6Н-пиримидо[5,4-b]пирролизин-7-ил)акриламид или его фармацевтически приемлемая соль.
9.(S)-N-(4-Амино-6-метил-5-(хинолин-3-ил)-8,9-дигидропиримидо[5,4-b]индолизин-8-ил)акриламид или его фармацевтически приемлемая соль.
10. Cоединение или его фармацевтически приемлемая соль по любому одному из пп.1-9, обладающее свойствами ингибитора EGFR.
11. Фармацевтическая композиция, обладающая ингибирующим действием в отношении рецептора эпидермального фактора роста (EGFR), содержащая эффективное количество соединения или его фармацевтически приемлемой соли по любому одному из пп.1-9.
12. Соединение или его фармацевтически приемлемая соль по любому одному из пп.1-9, обладающее свойствами ингибитора EGFR для получения противоопухолевого лекарственного средства.
13. Способ лечения или предупреждения ракового заболевания, включающий стадию введения млекопитающему соединения или его фармацевтически приемлемой соли по любому одному из пп.1-9, обладающих активностью ингибитора EGFR, в дозе, эффективной для лечения или предупреждения ракового заболевания.
14. Применение соединения или его фармацевтически приемлемой соли по любому одному из пп.1-9, обладающих активностью ингибитора EGFR, для получения противоопухолевого лекарственного средства.
15. Соединение или его фармацевтически приемлемая соль по любому одному из пп.1-9, обладающие активностью ингибитора EGFR, для применения при предупреждении или лечении ракового заболевания.
| WO 2013118817 A1, 15.08.2013 | |||
| WO 2006102079 A1, 28.09.2006 | |||
| WO 2011046964 A2, 21.04.2011 & EA 021715 B1, 31.08.2015 | |||
| WO 2013125709 A1, 29.08.2013 & RU 2581039 C1, 10.04.2016 | |||
| WO 2014129596 A1, 28.08.2014 & US 20160115172 A1, 28.04.2016 & RU 2015140075A1. |

