Ссылка на родственную заявку
Настоящая заявка заявляет приоритет временной заявки на патент США №61/030407, поданной 21 февраля 2008 г.
Уровень техники
Процесс, который связан с ростом, развитием и метастазированием опухоли, контролируется сигнальными путями, которые активируются в раковых клетках. Путь ERK играет ключевую роль в регуляции роста клеток млекопитающих путем ретрансляции экстраклеточных сигналов от связанных с лигандом тирозинкиназных рецепторов на поверхности клеток, таких как семейство erbB, PDGF, FGF и VEGF тирозинкиназный рецептор. Активация пути ERK происходит через каскад событий фосфориляции, который начинается с активации Ras. Активация Ras приводит к рекрутменту и активации Raf, серин-треонин киназы. Активированная Raf затем фосфорилирует и активирует MEK1/2, которая затем фосфорилирует и активирует ERK1/2. При активации ERK1/2 фосфорилирует несколько нижележащих мишеней, вовлеченных во множество клеточных событий, включая изменения в цитоскелете и транскрипционную активацию. Путь ERK/MAPK является одним из самых важных для клеточной пролиферации, и, как полагают, путь ERK/MAPK часто активируется во многих опухолях. Гены Ras, которые расположены выше ERK1/2, подвергаются мутации при некоторых видах рака, включая колоректальный рак, меланому, опухоли молочной желез и поджелудочной железы. Высокая активность Ras сопровождается повышенной активностью ERK во многих опухолях, встречающихся у человека. Более того, мутации BRAF, серин-треонин киназы семейства Raf, связаны с повышенной киназной активностью. Мутации в BRAF уже обнаружили при меланомах (60%), раке щитовидной железы (выше 40%) и колоректальном раке. Эти наблюдения показывают, что сигнальный путь ERK1/2 является привлекательным путем для противораковой терапии для широкого спектра опухолей, возникающих у человека.
Принимая во внимание все вышесказанное, ценным вкладом в данную область техники были бы «маленькие молекулы» (то есть, соединения), которые ингибирую активность ERK (то есть, активность ERK1 и ERK2), маленькие молекулы которых были бы полезны для лечения широкого спектра раковых заболеваний, таких как, например, меланома, рак поджелудочной железы, рак щитовидной железы, колоректальный рак, рак легких, рак молочной железы и рак яичников. Настоящее изобретение обеспечивает такой вклад.
Краткое описание изобретения
В настоящем изобретении раскрываются соединения, которые ингибируют активность ERK1 и/или активность ERK2.
Соединения согласно настоящему изобретению также ингибируют фосфорилирование ERK1 и ERK2.
Таким образом, настоящее изобретение обеспечивает соединения, которые являются ингибиторами ERK (то есть, ингибиторами ERK1 и/или ингибиторами ERK2), указанные соединения представляют собой соединения формулы 1.0:
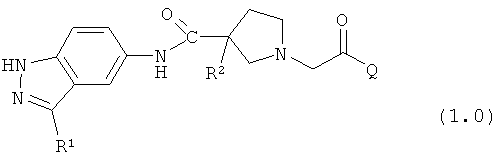
или их фармацевтически приемлемые соли и сольваты, где: Q представляет собой тетрагидропиридинил (например, 1,2,3,6-тетрагидропиридинил) или замещенный тетрагидропиридинил (например, замещенный 1,2,3,6-тетрагидропиридинил); и R1 и R2 определены ниже.
Настоящее изобретение раскрывает соединения формулы 1.0.
Настоящее изобретение раскрывает соединения формулы 1.0 в чистой или выделенной форме.
Настоящее изобретение раскрывает фармацевтически приемлемые соли соединений формулы 1.0.
Настоящее изобретение раскрывает сольваты соединений формулы 1.0.
Настоящее изобретение раскрывает соединения формулы 1.0, в которых от одного до всех атомов водорода (Н) представляют собой дейтерий.
Настоящее изобретение раскрывает соединения формулы 1.0, где, по меньшей мере, один атом водорода представляет собой дейтерий.
Настоящее изобретение раскрывает соединения формулы 1.0, где от 1 до 5 атомов водорода представляют собой дейтерий.
Настоящее изобретение раскрывает соединения формулы 1.0, где один атом водорода представляет собой дейтерий.
Настоящее изобретение раскрывает соединения А1-А16 и А18-А48.
Настоящее изобретение раскрывает соединения А1-А16 и А18-А30.
Настоящее изобретение раскрывает соединения А1-А16 и А18-А26.
Настоящее изобретение раскрывает соединения А31-А48.
Настоящее изобретение раскрывает фармацевтическую композицию, содержащую эффективное количество, по меньшей мере, одного соединения формулы 1.0 и фармацевтически приемлемый носитель.
Настоящее изобретение раскрывает фармацевтическую композицию, содержащую эффективное количество, по меньшей мере, одного соединения формулы 1.0 и эффективное количество, по меньшей мере, одного фармацевтически активного ингредиента (такого как, например, химиотерапевтический агент) и фармацевтически приемлемый носитель.
Настоящее изобретение также раскрывает способ ингибирования ERK (то есть, ингибирования активности ERK) у пациента, которому необходимо такое лечение, содержащий введение указанному пациенту эффективного количества, по меньшей мере, одного соединения формулы 1.0.
Настоящее изобретение также раскрывает способ ингибирования ERK1 (то есть, ингибирования активности ERK1) у пациента, которому необходимо такое лечение, содержащий введение указанному пациенту эффективного количества, по меньшей мере, одного соединения формулы 1.0.
Настоящее изобретение также раскрывает способ ингибирования ERK2 (то есть, ингибирования активности ERK2) у пациента, которому необходимо такое лечение, содержащий введение указанному пациенту эффективного количества, по меньшей мере, одного соединения формулы 1.0.
Настоящее изобретение также раскрывает способ ингибирования ERK1 и ERK2 (то есть, ингибирования активности ERK1 и активности ERK2) у пациента, которому необходимо такое лечение, содержащий введение указанному пациенту эффективного количества, по меньшей мере, одного соединения формулы 1.0.
Настоящее изобретение также раскрывает способ лечения рака у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества, по меньшей мере, одного соединения формулы 1.0.
Настоящее изобретение также раскрывает способ лечения рака у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества фармацевтической композиции, содержащей эффективное количество, по меньшей мере, одного соединения формулы 1.0.
Настоящее изобретение раскрывает способ лечения рака у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества, по меньшей мере, одного соединения формулы 1.0, в комбинации с эффективным количеством, по меньшей мере, одного химиотерапевтического агента.
Настоящее изобретение также раскрывает способ лечения рака у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества фармацевтической композиции, содержащей эффективное количество, по меньшей мере, одного соединения формулы 1.0, в комбинации с эффективным количеством, по меньшей мере, одного химиотерапевтического агента.
Настоящее изобретение также раскрывает способ лечения рака у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества, по меньшей мере, одного соединения формулы 1.0, в комбинации с, по меньшей мере, одним ингибитором сигнальной трансдукции.
Настоящее изобретение также раскрывает способ лечения рака у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества фармацевтической композиции, содержащей эффективное количество, по меньшей мере, одного соединения формулы 1.0, в комбинации с, по меньшей мере, одним ингибитором сигнальной трансдукции.
В способах согласно настоящему изобретению соединения согласно настоящему изобретению могут вводиться одновременно или последовательно (то есть, в последовательном порядке) с химиотерапевтическими агентами или ингибиторами сигнальной трансдукции.
Способы лечения рака, описанные в настоящей заявке, могут, при желании, включать введение эффективного количества облучения (то есть, способы лечения рака, описанные в настоящей заявке, при желании включают введение радиотерапии).
Подробное описание изобретения
Как описывается в настоящем документе, если иного не указано, лекарственное средство или соединение применяется в определенный период цикла лечения. Например, один раз в день означает один раз в день, причем каждый день цикла лечения. Два раза в день означает два раз в день, причем каждый день цикла лечения. Один раз в неделю означает один раз в неделю в течение цикла лечения. Один раз каждые три недели означает один раз за каждые три недели в течение цикла лечения.
Если иного не указано, то следующие аббревиатуры имеют следующие значения:
Как применяется в настоящем документе, если другого не указано, следующие термины имеют следующие значения:
Термин "противораковый агент" означает лекарственное средство (медикамент или фармацевтически активный ингредиент) для лечения рака;
Термин "противоопухолевый агент" означает лекарственное средство (медикамент или фармацевтически активный ингредиент) для лечения рака (то есть, химиотерапевтический агент);
Фраза "по меньшей мере, один" в качестве ссылки на число соединений согласно настоящему изобретению означает, например, 1-6, как правило, 1-4, более часто 1, 2 или 3 и обычно один или два, и более часто один; таким образом, в одном примере "по меньшей мере, один" означает один, в другом примере "по меньшей мере, один" означает два, и в еще одном примере "по меньшей мере, один" означает три;
Термин "химиотерапевтический агент" означает лекарственное средство (медикамент или фармацевтически активный ингредиент), предназначенное для лечения рака (то есть, противоопухолевый агент);
Термин "соединение" при ссылке на противоопухолевые агенты, охватывает агенты, которые представляют собой антитела;
Термин "одновременно" означает (1) одновременно по времени (например, в один и тот же момент); или (2) в разные моменты времени в ходе общего курса лечения;
Термин "последовательно" означает, что одно следует за другим;
Термин "различные", как применяется во фразе "различные противоопухолевые агенты", означает, что агенты не представляют собой одно и то же соединение или структуру; предпочтительно, "различные", как применяется во фразе "различные противоопухолевые агенты", означает, что эти агенты не относятся к одному и тому же классу противоопухолевых агентов; например, один противоопухолевый агент представляет собой таксан, и другой противоопухолевый агент представляет собой координационное соединение платины;
Термин "эффективное количество" или "терапевтически эффективное количество" предназначается для обозначения количества соединения или композиции согласно настоящему изобретению, или количества облучения, эффективного для лечения или ингибирования заболеваний или состояний, описанных в настоящем документе, и, таким образом, для получения желательного терапевтического, мелиоративного, ингибирующего или предупреждающего эффекта; таким образом, например, в способах лечения рака, описанных в настоящей заявке "эффективное количество" (или "терапевтически эффективное количество") означает, например, количество соединения (или лекарственного средства) или облучения, которое приводит к: (а) уменьшению, облегчению или исчезновению одного или более симптомов, вызванных раком, (b) уменьшению размера опухоли, (с) исчезновению опухоли и/или (d) стабилизации опухоли в течение длительного периода (задерживание роста); например, при лечении рака легких (например, немелкоклеточного рака легких), терапевтически эффективное количество означает количество, которое уменьшает или приводит к исчезновению кашля, одышки и/или боли; также, например, эффективное количество или терапевтически эффективное количество ингибитора ERK (то есть, соединения согласно настоящему изобретению) представляет собой такое количество, которое приводит к уменьшению активности и фосфорилирования ERK (ERK1 и/или ERK2); уменьшение активности ERK можно обнаружить с помощью анализа фармакодинамических маркеров, таких как фосфорилированная RSK1,2 и фосфорилированная ERK1,2, с помощью методик, хорошо известных в данной области техники;
Аббревиатура "Ех", приведенная в таблицах, означает "Пример";
Фраза "один или более" имеет тоже значение, что и фраза "по меньшей мере, один";
Термин "пациент" обозначает животное, такое как млекопитающее (например, человека и предпочтительно человек);
Термин "пролекарство" обозначает соединения, которые быстро превращаются in vivo, например, путем гидролиза в крови, в родоначальное соединение, то есть соединение формулы 1.0 или его соль и/или сольват; они подробно раскрываются в Т. Higuchi and V. Stella, Pro-drugs as Novel Delivery Systems, Vol.14 of the A.C.S. Symposium Series, и в Edward В. Roche, ed., Bioreversible Carriers in Drug Design, American Pharmaceutical Association and Pergamon Press, 1987, эти документа включены в настоящий документ посредством ссылки; объем настоящего изобретения охватывает пролекарства соединений согласно настоящему изобретению;
Термин "последовательно" означает (1) введение одного соединения по способу ((а) соединения согласно изобретению или (b) химиотерапевтического агента, ингибитора сигнальной трансдукции и/или радиотерапии), с последующим введением другого компонента или компонентов; после введения одного компонента последующий компонент может вводиться последовательно, сразу после первого компонента, или последующий компонент может вводиться через эффективный период времени после введения первого компонента; эффективный период времени означает количество времени, необходимое для реализации максимальной пользы от введения первого компонента; и
Термин "сольват" обозначает физическую ассоциацию соединения согласно настоящему изобретению с одной или более молекулами растворителя; эта физическая ассоциация содержит различные степени ионного и ковалентного связывания, включая водородное связывание; в определенных случаях сольват будет способен к выделению, например, когда одна или более молекул растворителя включаются в кристаллическую решетку кристаллического твердого вещества; "сольват" охватывает как фазу раствора, так и выделяемые сольваты; неограничивающие примеры подходящих сольватов включают этанолаты, метанолаты и тому подобное; "гидрат" представляет собой сольват, в котором молекулой растворителя является молекула воды (H2O).
Как применяется в настоящем документе, если иного не указано, следующие термины имеют следующие значения, и, если иного не указано, определения каждого термина (то есть, в случае составляющей или заместителя) применяются, когда этот термин используется индивидуально или как компонент другого термина (например, термин арил имеет одно и то же значение в случае арила и в случае арильной части арилалкила, алки-ларила, арилалкинила и тому подобного):
Термин "ацил" означает Н-С(О)-, алкил-С(О)-, алкенил-С(О)-, Алкинил-С(O)-, циклоалкил-С(О)-, циклоалкенил-С(О)- или циклоалкинил-С(О)-группу, в которой различные группы представляют собой группы, определение которым дано ниже, (и, как раскрывается далее, алкильные, алкенильные, алкинильные, циклоалкильные, циклоалкенильные и циклоалкинильные составляющие могут иметь заместители); связь с материнской составляющей осуществляется через карбонил; предпочтительные ацилы содержат низший алкил; неограничивающие примеры подходящих ацильных групп включают формил, ацетил, пропаноил, 2-метилпропаноил, бутаноил и циклогексаноил;
Термин "алкенил" означает алифатическую углеводородную группу (цепь), содержащую, по меньшей мере, одну двойную связь углерод-углерод, причем цепь может быть прямой или разветвленной, и указанная группа содержит от около 2 до около 15 атомов углерода; предпочтительные алкенильные группы содержат от около 2 до около 12 атомов углерода в цепи; и более предпочтительно от около 2 до около 6 атомов углерода в цепи; термин "разветвленный" означает, что к линейной алкенильной цепи присоединены одна или более низших алкильных групп, таких как метил, этил или пропил, или алкенильных групп; термин "низший алкенил" означает алкенильную группу, содержащую от около 2 до около 6 атомов углерода в цепи, и цепь может быть прямой или разветвленной; термин "замещенный алкенил" означает, что алкенильная группа замещена одним или более независимо выбранными заместителями, и каждый заместитель независимо выбирается из группы, состоящей из: гало, алкила, арила, циклоалкила, цианогруппы, алкоксигруппы и -S(алкила); неограничивающие примеры подходящих алкенильных групп включают этенил, пропенил, н-бутенил, 3-метилбут-2-енил, н-пентенил, октенил и деценил;
Термин "алкокси" означает алкил-O- группу (то есть, связь с материнской составляющей осуществляется посредством простоэфирного кислорода), в которой алкильная группа является незамещенной или замещенной, как раскрывается далее; неограничивающие примеры подходящих алкоксигрупп включают метокси, этокси, н-пропокси, изопропокси, н-бутокси и гептокси;
Термин "алкоксикарбонил" означает алкил-O-СО- группу (то есть, связь с материнской составляющей осуществляется посредством карбонила), в которой алкильная группа является незамещенной или замещенной, как раскрывается ранее; неограничивающие примеры алкоксикарбонильных групп включают метоксикарбонил и этоксикарбонил;
Термин "алкил" (включая алкильные части в других составляющих, как например трифторалкил и алкилокси) означает алифатическую углеводородную группу (цепь), которая может быть прямой или разветвленной, где указанная группа содержит от около 1 до около 20 атомов углерода в цепи; предпочтительные алкильные группы содержат от около 1 до около 12 атомов углерода в цепи; более предпочтительные алкильные группы содержат от около 1 до около 6 атомов углерода в цепи; термин "разветвленный" означает, что к линейной алкильной цепи присоединены одна или более низших алкильных групп, таких как метил, этил или пропил; термин "низший алкил" означает группу, содержащую от около 2 до около 6 атомов углерода в цепи, и указанная цепь может быть прямой или разветвленной; термин "замещенный алкил" означает, что алкильная группа замещена одним или более независимо выбранными заместителями, и каждый заместитель независимо выбирается из группы, состоящей из: гало, арила, циклоалкила, циано, гидрокси, алкокси, алкилтио, амино, -NH(алкил), -NH(циклоалкил), -N(алкил)2, карбокси, -С(O)O-алкил и -S(алкил); неограничивающие примеры подходящих алкильных групп включают метил, этил, н-пропил, изопропил, н-бутил, т-бутил, н-пентил, гептил, нонил, децил, фторметил, трифторметил и циклопропилметил;
Термин "алкиларил" (или алкарил) означает алкил-арильную группу (то есть, связь с материнской составляющей осуществляется посредством арильной группы), где алкильная группа является незамещенной или замещенной, как раскрывается выше, и арильная группа является незамещенной или замещенной, как раскрывается далее; предпочтительные алкиларилы содержат низшую алкильную группу; неограничивающие примеры подходящих алкиларильных групп включают о-толил, п-толил и ксилил;
Термин "алкилгетероарил" означает алкил-гетероарильную группу (то есть, связь с материнской составляющей осуществляется посредством гетероарильной группы), где алкил является незамещенным или замещенным, как раскрывается выше, и гетероарильная группа является незамещенной или замещенной, как раскрывается далее;
Термин "алкилсульфинил" означает алкил-S(O)- группу (то есть, связь с материнской составляющей осуществляется через сульфинил), где алкильная группа является незамещенной или замещенной, как раскрывается ранее; предпочтительными группами являются группы, в которых алкильной группой является низший алкил;
Термин "алкилсульфонил" означает алкил-S(O2)- группу (то есть, связь с материнской составляющей осуществляется через сульфонил), где алкильная группа является незамещенной или замещенной, как раскрывается ранее; предпочтительными группами являются группы, в которых алкильной группой является низший алкил;
Термин "алкилтио" означает алкил-S-группу (то есть, связь с материнской составляющей осуществляется через серу), где алкильная группа является незамещенной или замещенной, как описано ранее; неограничивающие примеры подходящих алкилтио групп включают метилтио, этилтио, изо-пропилтио и гептилтио;
Термин "алкинил" означает алифатическую углеводородную группу (цепь), содержащую, по меньшей мере, одну тройную связь углерод-углерод, где цепь может быть прямой или разветвленной, и где указанная группа содержит от около 2 до около 15 атомов углерода в цепи; предпочтительные алкинильные группы содержат от около 2 до около 12 атомов углерода в цепи; и более предпочтительно от около 2 до около 4 атомов углерода в цепи; термин "разветвленный" означает, что к линейной алкинильной цепи присоединены одна или более низших алкильных групп, таких как метил, этил или пропил; термин "низший алкинил" означает алкинильную группу, содержащую от около 2 до около 6 атомов углерода в цепи, и цепь может быть прямой или разветвленной; неограничивающие примеры подходящих алкинильных групп включают этинил, пропинил, 2-бутинил, 3-метилбутинил, н-пентинил и децинил; термин "замещенный алкинил" означает, что алкинильная группа замещена одним или более независимо выбранными заместителями, и каждый заместитель независимо выбирается из группы, состоящей из алкила, арила и циклоалкила;
Термин "амино" означает группу -NH2;
Термин "аралкенил" (или арилалкенил) означает арил-алкенильную группу (то есть, связь с материнской составляющей осуществляется посредством алкенильной группы), где арильная группа является незамещенной или замещенной, как раскрывается далее, и алкенильная группа является незамещенной или замещенной, как раскрывается выше; предпочтительные аралкенилы содержат низшую алкенильную группу; неограничивающие примеры подходящих аралкенильных групп включают 2-фенэтенил и 2-нафтилэтенил;
Термин "аралкил" (или арилалкил) означает арил-алкильную группу (то есть, связь с материнской составляющей осуществляется посредством алкильной группы), где арил является незамещенным или замещенным, как раскрывается далее, и алкил является незамещенным или замещенным, как раскрывается выше; предпочтительные аралкилы содержат низшую алкильную группу; неограничивающие примеры подходящих аралкильных групп включают бензил, 2-фенэтил и нафталинилметил;
Термин "аралкилокси" (или арилалкилокси) означает аралкил-O-группу (то есть, связь с материнской составляющей осуществляется посредством простоэфирного кислорода), где аралкильная группа является незамещенной или замещенной, как описано ранее; неограничивающие примеры подходящих аралкилокси групп включают бензилокси и 1- или 2-нафталинметокси;
Термин "аралкоксикарбонил" означает аралкил-О-С(О)-группу (то есть, связь с материнской составляющей осуществляется через карбонил), где аралкильная группа является незамещенной или замещенной, как раскрывается ранее; неограничивающим примером подходящей аралкоксикарбонильной группы является бензилоксикарбонил;
Термин "аралкилтио" означает аралкил-S-группу (то есть, связь с материнской составляющей осуществляется через серу), где аралкильная группа является незамещенной или замещенной, как описано ранее; неограничивающий пример подходящей аралкилтио группы представляет собой бензилтио;
Термин "ароил" означает арил-С(О)-группу (то есть, связь с материнской составляющей осуществляется через карбонил), где арильная группа является незамещенной или замещенной, как раскрывается далее; неограничивающие примеры подходящих групп включают бензоил и 1- и 2-нафтоил;
Термин "арил" (иногда указывают как "ар") означает ароматическую моноциклическую или мультициклическую кольцевую систему, содержащую от около 6 до около 14 атомов углерода, предпочтительно от около 6 до около 10 атомов углерода; арильная группа необязательно может быть замещена одним или более независимо выбранными "заместителями в кольцевой системе" (определено ниже). Неограничивающие примеры подходящих арильных групп включают фенил и нафтил;
Термин "арилалкинил" означает арил-алкинильную группу (то есть, связь с материнской составляющей осуществляется посредством алкинильной группы), где арильная группа является незамещенной или замещенной, как раскрывается выше, и алкинильная группа является незамещенной или замещенной, как раскрывается выше;
Термин "ариламиногетероарил" означает арил-амино-гетероарильную группу (то есть, связь с материнской составляющей осуществляется через гетероарильную группу), где арильная группа является незамещенной или замещенной, как раскрывается выше, и аминогруппа определена выше (то есть, здесь -NH-), и гетероарильная группа является незамещенной или замещенной, как раскрывается далее;
Термин "арилгетероарил" означает арил-гетероарильную группу (то есть, связь с материнской составляющей осуществляется посредством гетероарильной группы), где арильная группа является незамещенной или замещенной, как раскрывается выше, и гетероарильная группа является незамещенной или замещенной, как раскрывается далее;
Термин "арилокси" означает арил-O-группу (то есть, связь с материнской составляющей осуществляется посредством простоэфирного кислорода), где арильная группа является незамещенной или замещенной, как раскрывается выше; неограничивающие примеры подходящих арилокси групп включают фенокси и нафтокси;
Термин "арилоксикарбонил" означает арил-О-С(О)-группу (то есть, связь с материнской составляющей осуществляется через карбонил), где арильная группа является незамещенной или замещенной, как раскрывается ранее; неограничивающие примеры подходящих арилоксикарбонильных групп включают феноксикарбонил и нафтоксикарбонил;
Термин "арилсульфинил" означает арил-S(O)-группу (то есть, связь с материнской составляющей осуществляется через сульфинил), где арильная группа является незамещенной или замещенной, как раскрывается ранее;
Термин "арилсульфонил" означает арил-S(O2)-группу (то есть, связь с материнской составляющей осуществляется через сульфонил), где арильная группа является незамещенной или замещенной, как раскрывается ранее;
Термин "арилтио" означает арил-S-группу (то есть, связь с материнской составляющей осуществляется через серу), где арильная группа является незамещенной или замещенной, как раскрывается ранее; неограничивающие примеры подходящих арилтиогрупп включают фенилтио и нафтилтио;
Термин "циклоалкенил" означает неароматическую моноциклическую или мультициклическую кольцевую систему, содержащую от около 3 до около 10 атомов углерода, предпочтительно от около 5 до около 10 атомов углерода, которая содержит, по меньшей мере, одну двойную связь углерод-углерод; предпочтительные циклоалкенильные кольца содержат от около 5 до около 7 атомов в кольце; циклоалкенил необязательно может быть замещен одним или более независимо выбранными "заместителями в кольцевой системе" (определено ниже). Неограничивающие примеры подходящих моноциклических циклоалкенилов включают циклопентенил, циклогексенил, циклогептенил и тому подобное; неограничивающим примером подходящего мульти циклического циклоалкенила является норборниленил;
Термин "циклоалкил" означает неароматическую моноциклическую или мульти циклическую кольцевую систему, содержащую от около 3 до около 7 атомов углерода, предпочтительно от около 3 до около 6 атомов углерода; циклоалкил необязательно может быть замещен одним или более независимо выбранными "заместителями в кольцевой системе" (определено ниже). Неограничивающие примеры подходящих моноциклических цикло-алкилов включают циклопропил, циклопентил, циклогексил, циклогептил и тому подобное; неограничивающие примеры подходящих мультициклических циклоалкилов включают 1-декалин, норборнил, адамантил и тому подобное;
Термин "циклоалкилалкил" означает циклоалкил-алкильную группу (то есть, связь с материнской составляющей осуществляется через алкильную группу), где циклоалкильная составляющая является незамещенной или замещенной, как раскрывается выше, и алкильная составляющая является незамещенной или замещенной, как раскрывается выше;
Термин "гало" означает фторо, хлоро, бромо или йодо группы; предпочтительные галозаместители означают фторо, хлоро или бромо, и более предпочтительные означают фторо и хлоро;
Термин "галоген" означает фтор, хлор, бром или йод, предпочтительные галогены представляют собой фтор, хлор и бром;
Термин "галоалкил" означает алкил, как раскрывается выше, где один или более атомов водорода алкила замещены галогруппой, как раскрывается выше;
Термин "гетероаралкенил" означает гетероарил-алкенильную группу (то есть, связь с материнской составляющей осуществляется посредством алкенильной группы), где гетероарильная группа является незамещенной или замещенной, как раскрывается далее, и алкенильная группа является незамещенной или замещенной, как раскрывается выше;
Термин "гетероаралкил" (или гетероарилалкил) означает гетероарил-алкильную группу (то есть, связь с материнской составляющей осуществляется посредством алкильной группы), в которой гетероарил является незамещенным или замещенным, как раскрывается далее, и алкильная группа является незамещенной или замещенной, как раскрывается выше; предпочтительные гетероаралкилы содержит алкильную группу, которая представляет собой низшую алкильную группу; неограничивающие примеры подходящих аралкильных групп включают пиридилметил, 2-(фуран-3-ил)этил и хинолин-3-илметил;
Термин "гетероаралкилтио" означает гетероаралкил-S-группу, где гетероарильная группа является незамещенной или замещенной, как раскрывается выше;
Термин "гетероарил" означает ароматическую моноциклическую или мультициклическую кольцевую систему, содержащую от около 5 до около 14 атомов в кольце, предпочтительно от около 5 до около 10 атомов в кольце, в которой один или более кольцевые атомы являются отличными от углерода элементами, такими как азот, кислород или сера, присутствующими сами по себе или в комбинации; предпочтительные гетероарилы содержат от около 5 до около 6 атомов в кольце; "гетероарил" необязательно может быть замещен одним или более «заместителем в кольцевой системе» (определено ниже); приставка аза, окса или тиа перед корневым именем гетероарила означает, что, по меньшей мере, атом азота, кислорода или серы, соответственно, присутствует в качестве кольцевого атома; атом азота гетероарила может необязательно быть окислен до соответствующего N-оксида; неограничивающие примеры подходящих гетероарилов включают пиридил, пиразинил, фуранил, тиенил, пиримидинил, изоксазолил, изотиазолил, оксазолил, тиазолил, пиразолил, фуразанил, пирролил, пиразолил, триазолил, 1,2,4-тиадиазолил, пиразинил, пиридазинил, хиноксалинил, фталазинил, имидазо[1,2-а]пиридинил, имидазо[2,1-b]тиазолил, бензофуразанил, индолил, азаиндолил, бензимидазолил, бензотиенил, хинолинил, имидазолил, тиенопиридил, хиназолинил, тиенопиримидил, пирролопиридил, имидазопиридил, изохинолинил, бензоазаиндолил, 1,2,4-триазинил, бензотиазолил, фуропиридин
 ,
,
и тому подобное;
Термин "гетероарилалкинил" (или гетероаралкинил) означает гетероарил-алкинильную группу (то есть, связь с материнской составляющей осуществляется посредством алкинильной группы), где гетероарильная группа является незамещенной или замещенной, как раскрывается выше, и акинильная группа является незамещенной или замещенной, как раскрывается выше;
Термин "гетероариларил" (или гетероарарил) означает гетероарил-арильную группу (то есть, связь с материнской составляющей осуществляется посредством арильной группы), где гетероарильная группа является незамещенной или замещенной, как раскрывается выше, и арильная группа является незамещенной или замещенной, как раскрывается выше;
Термин "гетероарилгетероариларил" означает гетероарил-гетероарильную группу (то есть, связь с материнской составляющей осуществляется через последнюю гетероарильную группу), где каждая гетероарильная группа является незамещенной или замещенной, как раскрывается выше;
Термин "гетероарилсульфинил" означает гетероарил-SO-группу, где гетероарильная группа является незамещенной или замещенной, как раскрывается выше;
Термин "гетероарилсульфонил" означает гетероарил-SO2-группу, где гетероарильная группа является незамещенной или замещенной, как раскрывается выше;
Термин "гетероарилтио" означает гетероарил-S-группу, где гетероарильная группа является незамещенной или замещенной, как раскрывается выше;
Термин "гетероцикленил" (или гетероциклоалкенил) означает неароматическую моноциклическую или мультициклическую кольцевую систему, содержащую от около 3 до около 10 кольцевых атомов, предпочтительно от около 5 до около 10 кольцевых атомов, в которой один или более атомов в кольцевой системе являются отличными от углерода элементами (например, один или более гетероатомов независимо выбираются из группы, состоящей из атома азота, кислорода и серы), и которая содержит, по меньшей мере, одну двойную связь углерод-углерод или двойную связь углерод-азот; причем в кольцевой системе нет соседних атомов кислорода и/или серы; Предпочтительные гетероцикленильные кольца содержат от около 5 до около 6 кольцевых атомов; приставка аза, окса или тиа перед корневым именем гетероцикленила означает, что, по меньшей мере, один атом азота, кислорода или серы присутствует в качестве кольцевого атома; гетероцикленил необязательно может быть замещен одним или более «заместителем в кольцевой системе» (определено ниже); атом азота или серы в гетероциклениле может быть необязательно окислен до соответствующего N-оксида, S-оксида или S,S-диоксида; неограничивающие примеры подходящих моноциклических азагетероцикленильных групп включают 1,2,3,4-тетрагидропиридин, 1,2-дигидропиридил, 1,4-дигидропиридил, 1,2,3,6-тетрагидропиридин, 1,4,5,6-тетрагидропиримидин, 2-пирролинил, 3-пирролинил, 2-имидазолинил, 2-пиразолинил и тому подобное; Неограничивающие примеры подходящих оксагетероцикленильных групп включают 3,4-дигидро-2Н-пиран, дигидрофуранил, фтордигидрофуранил и тому подобное; Неограничивающий пример подходящей мультициклической окса-гетероцикленильной группы представляет собой 7-оксабицикло[2.2.1]гептенил; неограничивающие примеры подходящих моноциклических тиагетероцикленильных колец включают дигидротиофенил, дигидротиопиранил и тому подобное;
Термин "гетероциклоалкилалкил" (или гетероциклилалкил) означает гетероциклоалкил-алкильную группу (то есть, связь с материнской составляющей осуществляется посредством алкильной группы), где гетероциклоалкильная группа (то есть, гетероциклильная группа) является незамещенной или замещенной, как указано ниже, и алкильная группа является незамещенной или замещенной, как раскрывается выше;
Термин "гетероциклил" (или гетероциклоалкил) означает неароматическую насыщенную моноциклическую или мультициклическую кольцевую систему, содержащую от около 3 до около 10 кольцевых атомов, предпочтительно от около 5 до около 10 кольцевых атомов, в которой один или более атомов в кольцевой системе являются отличными от углерода элементами, такими как азот, кислород или сера, присутствующими сами по себе или в комбинации, причем в кольцевой системе нет соседних атомов кислорода и/или серы; предпочтительные гетероциклилы содержат от около 5 до около 6 кольцевых атомов; приставка аза, окса или тиа перед корневым именем гетероциклила означает, что, по меньшей мере, один атом азота, кислорода или серы присутствует в качестве кольцевого атома; гетероциклил необязательно может быть замещен одним или более «заместителем в кольцевой системе» (определено ниже); атом азота или серы в гетероциклиле может быть необязательно окислен до соответствующего N-оксида, S-оксида или S,S-диоксида; неограничивающие примеры подходящих моноциклических гетероцикл ильных колец включают пиперидил, пирролидинил, пиперазинил, морфолинил, тиоморфолинил, тиазолидинил, 1,3-диоксоланил, 1,4-диоксанил, тетрагидрофуранил, тетра-гидротиофенил, тетрагидротиопиранил и тому подобное;
Термин "гидроксиалкил" означает НО-алкильную группу, где алкильная группа является замещенной или незамещенной, как раскрывается выше; предпочтительные гидроксиалкилы содержат низший алкил; Неограничивающие примеры подходящих гидроксиалкильных групп включают гидроксиметил и 2-гидроксиэтил; и
Термин "заместитель в кольцевой системе" обозначает заместитель, который присоединен к ароматической или неароматической кольцевой системе, и который, например, замещает доступный водород в кольцевой системе; каждый заместитель в кольцевой системе независимо выбирается из группы, состоящей из: алкила, арила, гетероарила, аралкила, алкиларила, аралкенила, гетероаралкила, алкилгетероарила, гетероаралкенила, гидрокси, гидроксиалкила, алкокси, арилокси, аралкокси, ацила, ароила, гало, нитро, циано, карбокси, алкоксикарбонила, арилоксикарбонила, аралкоксикарбонила, алкилсульфонила, арилсульфонила, гетероарилсульфонила, алкилсульфинила, арилсульфинила, гетероарилсульфинила, алкилтио, арилтио, гетероарилтио, аралкилтио, гетероаралкилтио, циклоалкила, циклоалкенила, гетероциклила, гетероцикленила, R60R65N-, R60R65N-алкил-, R60R65NC(O)- и R60R65NSO2-, где R60 и R65 каждый независимо выбирается из группы, состоящей из: водорода, алкила, арила и аралкила; Термин "заместитель в кольцевой системе" также означает циклическое кольцо, содержащее от 3 до 7 атомов в кольце, где 1-2 кольцевых атомов могут представлять собой гетероатомы, присоединенные к арильному, гетероарильному, гетероциклильному или гетероцикленильному кольцу посредством одновременного замещения двух кольцевых атомов водорода на указанном арильном, гетероарильном, гетероциклильном или гетероцикленильном кольце; неограничивающие примеры включают:
 ,
,  и тому подобное.
и тому подобное.
Линии, которые уходят в кольцо, показываю, что указанная связь может соединяться с любым из подходящих кольцевых атомов углерода.
Любой углерод или гетероатом с ненасыщенными валентностями, приведенный в тексте, схемах, примерах, структурных формулах и любой таблице, раскрытых в настоящем документе, как предполагается, имеет атом водорода или атомы водорода, необходимые для насыщения валентностей. И любой один или более из этих атомов водорода могут представлять собой дейтерий.
Одно или более соединений согласно изобретению могут также существовать в виде сольвата или могут, при желании, быть превращены в сольват. Получение сольватов общеизвестно. Таким образом, например, получение сольватов противогрибкового флуконазола в этилацетате, а также из воды описывается в М. Caira et al, J. Pharmaceutical Sci., 93(3), 601-611 (2004). В Е.С. van Tonder et al, AAPS PharmSciTech., 5(1), article 12 (2004); и A.L. Bingham et al, Chem. Commun., 603-604 (2001) раскрываются подобные методики получения сольватов, гемисольватов, гидратов и тому подобного. Типичный неограничивающий способ получения представляет собой растворение соединения по изобретению в желательных количествах желательного растворителя (органического или водного, или их смесей), при температуре выше температуры окружающей среды, и охлаждение раствора со скоростью, достаточной для образования кристаллов, которые потом выделяют известными способами. Аналитические методики, такие как, например, ИК спектроскопия, позволяют определить присутствие растворителя (или воды) в виде сольвата (или гидрата) в кристаллах.
Термин "фармацевтическая композиция", как предполагается, включает как нерасфасованную композицию, так и отдельные дозированные единицы, содержащие более чем один (например, два) фармацевтически активный агент, такой как, например, соединение согласно настоящему изобретению, и дополнительный агент, выбранный из списка дополнительных агентов, описанных здесь, наряду с любыми фармацевтически неактивными эксципиентами. Нерасфасованная композиция и каждая отдельная дозированная единица могут содержать фиксированные количества вышеупомянутого "более чем одного фармацевтически активного агента". Нерасфасованная композиция представляет собой материал, который еще не был сформирован в отдельные дозированные единицы. Примерами дозированных единиц являются пероральные лекарственные формы, такие как таблетки, капсулы, пилюли и тому подобное. Подобным образом, описанные в настоящей заявке способы лечения пациента введением фармацевтической композиции согласно настоящему изобретению, как также подразумевается, включают введение вышеуказанной нерасфасованной композиции и отдельных лекарственных форм.
Пролекарства соединений согласно настоящему изобретению также раскрываются в настоящей заявке. Термин "пролекарство", применяемый в настоящем изобретении, обозначает соединение, которое является предшественником лекарственного средства, и которое, при введении субъекту, подвергается химическому превращению путем метаболических или химических процессов с получением соединения формулы 1.0 или его соли и/или сольвата. Пролекарства раскрываются в Т. Higuchi and V. Stella, Pro-drugs as Novel Delivery Systems (1987) 14 of the A.C.S. Symposium Series, и в документе Bioreversible Carriers in Drug Design, (1987) Edward B. Roche, ed., American Pharmaceutical Association and Pergamon Press, которые включены в настоящий документ в полном объеме посредством ссылки.
Например, в случае если соединение формулы 1.0, или фармацевтически приемлемая соль, гидрат или сольват соединения, содержит функциональную группу карбоновой кислоты, пролекарство может включать сложный эфир, образованный путем замещения атома водорода кислотной группы на такую группу, как например, (С1-С8)алкил, (C2-С12)алканоилоксиметил, 1-(алканоилокси)этил, содержащий от 4 до 9 атомов углерода, 1-метил-1-(алканоилокси)этил, содержащий от 5 до 10 атомов углерода, алкоксикарбонилоксиметил, содержащий от 3 до 6 атомов углерода, 1-(алкоксикарбонилокси)этил, содержащий от 4 до 7 атомов углерода, 1-метил-1-(алкоксикарбонилокси)этил, содержащий от 5 до 8 атомов углерода, N-(алкоксикарбонил)аминометил, содержащий от 3 до 9 атомов углерода, 1-(N-(алкоксикарбонил)амино)этил, содержащий от 4 до 10 атомов углерода, 3-фталидил, 4-кротонолактонил, гамма-бутиролактон-4-ил, ди-N,N-(С1-С2)алкиламино(C2-C3)алкил (такой как β-диметиламиноэтил), карбамоил-(С1-С2)алкил, N,N-ди(С1-С2)алкилкарбамоил-(С1-С2)алкил и пиперидино-, пирролидино- или морфолино(C2-C3)алкил и тому подобное.
Подобным образом, в случае если соединение формулы 1.0 включает функциональную группу спирта, пролекарство может быть получено путем замещения атома водорода спиртовой группы на такую группу, как например, (С1-С6)алканоилоксиметил, 1-((С1-С6)алканоилокси)этил, 1-метил-1-((С1-С6)алканоилокси)этил, (С1-С6)алкоксикарбонилоксиметил, N-(C1-С6)алкоксикарбониламинометил, сукциноил, (C1-C6)алканоил, α-амино(С1-С4)алканил, арилацил и α-аминоацил или α-аминоацил-α-аминоацил, где каждая α-аминоацильная группа выбирается независимо из природных L-аминокислот, Р(O)(ОН)2, -Р(O)(O(C1-C6)алкил)2 или гликозила (радикал, полученный удалением гидроксильной группы гемиацетальной формы карбогидрата), и тому подобное.
Если соединение формулы 1.0 включает функциональную группу амина, пролекарство может быть получено замещением атома водорода аминогруппы на такую группу, как например, R70-карбонил, R70O-карбонил, NR70R75-карбонил, где R70 и R75 каждый независимо представляет собой (С1-С10)алкил, (С3-С7)циклоалкил, бензил, или R70-карбонил представляет собой природный α-аминоацил или природный α-аминоацил, -C(OH)C(O)OY80, где Y80 представляет собой Н, (С1-С6)алкил или бензил, -C(OY82)Y84, где Y82 представляет собой (C1-C4) алкил, и Y84 представляет собой (C1-C6)алкил, карбокси(С1-С6)алкил, амино(С1-С4)алкил или моно-N- или ди-N,N-(C1-C6)алкиламиноалкил, -C(Y86)Y88, где Y86 представляет собой Н или метил, и Y88 представляет собой моно-n- или ди-N,N-(C1-C6)алкиламино морфолино, пиперидин-1-ил или пирролидин-1-ил и тому подобное.
Настоящее изобретение также включает соединения по изобретению в выделенной и очищенной форме.
Полиморфные формы соединений формулы 1.0 и солей, сольватов и пролекарств соединений формулы 1.0, как предполагается, включены в настоящее изобретение.
Определенные соединения согласно настоящему изобретению могут существовать в виде различных изомерных форм (например, энантиомеры, диастереоизомеры, атропоизомеры). Изобретение включает все такие изомеры, как в чистой форме, так и в виде смеси, включая рацемические смеси. Енольные формы также охватываются настоящим изобретением.
Все стереоизомеры (например, геометрические изомеры, оптические изомеры и тому подобное) соединений согласно настоящему изобретению (включая стереоизомеры солей, сольватов и пролекарств соединений, так же как и солей и сольватов пролекарств), такие как существующие благодаря асимметрическим углеродам различных заместителей, включая энантиомерные формы (которые могут существовать даже в отсутствии асимметрических углеродов), ротамерные формы, атропоизомеры и диастереомерные формы, входят в объем настоящего изобретения. Отдельные стереоизомеры соединений по изобретению могут, например, быть по существу свободны от других изомеров, или могут быть смешанными, например, в виде рацематов, или смешанными со всеми другими или другими выбранными стереоизомерами. Хиральные центры согласно настоящему изобретению могут иметь S или R конфигурацию, определяемую согласно рекомендациям IUPAC 1974. Термины "соль", "сольват" "пролекарство" и тому подобное, как подразумевается, применяются эквивалентно по отношению к солям, сольватам и пролекарствам энантиомеров, стереоизомеров, ротамеров, таутомеров, рацематов или пролекарств соединений по изобретению.
Диастереомерные смеси могут быть разделены на их отдельные диастереомеры, исходя из их физико-химических различий, применяя способы, хорошо известные специалистам в данной области техники, такие как, например, хроматография и/или фракционная кристаллизация. Энантиомеры могут быть разделены путем превращения энантиомерной смеси в диастереомерную смесь, применяя реакцию с соответствующим оптически активным соединением (например, хиральное вспомогательное вещество, такое как хиральный спирт или хлорид кислоты Мошера), путем разделения диастереомеров и превращения (например, путем гидролиза) отдельных диастереомеров в соответствующие чистые энантиомеры. Также некоторые из соединений формулы (I) могут представлять собой атропоизомеры (например, замещенные биарилы) и, в этом случае, также рассматриваются как часть настоящего изобретения. Энантиомеры также можно разделить, применяя хиральные ВЭЖХ колонки.
Соединения формулы 1.0 формируют соли, которые также входят в объем настоящего изобретения. Упоминание соединений формулы 1.0 здесь, как понимают, включает упоминание их солей, если иного не указано. Термин "соль(и)", как применяется здесь, обозначает кислотные соли, образованные с неорганическими и/или органическими кислотами, также как и основные соли, образованные с неорганическими и/или органическими основаниями. Кроме того, когда соединение формулы 1.0 содержит как основную составляющую, такую как, но без ограничения к этому, пиридин или имидазол, так и кислотную составляющую, такую как, но без ограничения к этому, карбоновые кислоты, могут образовываться цвиттерионы ("внутренние соли"), которые входят в объем термина "соль(и)", как применяется в настоящем документе. Фармацевтически приемлемые соли (то есть, нетоксичные, физиологически приемлемые соли) являются предпочтительными. Соли соединений формулы 1.0 можно получить, например, путем реакции соединения формулы 1.0 с количеством кислоты или основания, таким как эквивалентное количество, в среде, такой как среда, в которой соль осаждается, или в водной среде с последующей лиофилизацией. Кислоты (и основания), которые, как правило, рассматриваются как подходящие для образования фармацевтически приемлемых солей из основных (или кислотных) фармацевтических соединений, раскрываются, например, в S. Berge et al, Journal of Pharmaceutical Sciences (1977) 66(1) 1-19; P. Gould, International J. of Pharmaceutics (1986) 33 201-217; Anderson et al, The Practice of Medicinal Chemistry (1996), Academic Press, New York; в The Orange Book (Food & Drug Administration, Washington, D.C. on their website); и P. Heinrich Stahl, Camille G. Wermuth (Eds.), Hubook of Pharmaceutical Salts: Properties, Selection, and Use, (2002) Int′l. Union of Pure and Applied Chemistry, pp.330-331. Эти документы включены в настоящую заявку в полном объеме посредством ссылки.
Примеры солей, полученных добавлением кислоты, включают ацетаты, адипаты, альгинаты, аскорбаты, аспартаты, бензоаты, бензолсульфонаты, бисульфаты, бораты, бутираты, цитраты, камфораты, камфорсульфонаты, циклопентанпропионаты, диглюконаты, додецилсульфаты, этансульфонаты, фумараты, глюкогептаноаты, глицерофосфаты, гемисульфаты, гептаноаты, гексаноаты, гидрохлориды, гидробромиды, гидроиодиды, 2-гидроксиэтансульфонаты, лактаты, малеаты, метансульфонаты, метилсульфаты, 2-нафталинсульфонаты, никотинаты, нитраты, оксалаты, памоаты, пектинаты, персульфаты, 3-фенилпропионаты, фосфаты, пикраты, пивалаты, пропионаты, салицилаты, сукцинаты, сульфаты, сульфонаты (такие, как упомянутые в настоящем документе), тартараты, тиоцианаты, толуолсульфонаты (также известные, как тозилаты), ундеканоаты и тому подобное.
Примерами основных солей являются соли аммония, соли щелочных металлов, такие как соли натрия, лития и калия, соли щелочноземельных металлов, такие как соли кальция и магния, соли алюминия, соли цинка, соли с органическими основаниями (например, органические амины), такие как бензатины, диэтиламины, дициклогексиламины, гидрабамины (образованные с N,N-бис(дегидроабиэтил)этилендиамином), N-метил-D-глюкамины, N-метил-О-глюкамиды, т-бутил амины, пиперазины, фенил-циклогексиламин, холин, трометамин и соли с аминокислотами, такими как аргинин, лизин и тому подобные. Основные азотсодержащие группы могут быть кватернизированы с агентами, такими как галиды низших алкилов (например, метил, этил, пропил и бутил хлориды, бромиды и иодиды), диалкил сульфаты (например, диметил, диэтил, дибутил и диамил сульфаты), длинноцепочечные галиды (например, децил, лаурил, миристил и стеарил хлориды, бромиды и иодиды), галиды аралкилов (например, бензил и фенэтил бромиды) и другие.
Подразумевается, что все такие кислотные и основные соли являются фармацевтически приемлемыми солями, входящими в объем настоящего изобретения, и все кислотные и основные соли рассматриваются как эквивалентные свободным формам соответствующих соединений в целях изобретения.
Соединения формулы 1.0 и их соли, сольваты и пролекарства могут существовать в их таутомерной форме (например, в виде амида или простого иминоэфира). Все такие таутомерные формы рассматриваются здесь как часть настоящего изобретения.
В кольцевых системах согласно настоящему изобретению, включающих гетероатом, на атомах углерода нет никаких гидроксильных групп, соседних с группами N, О или S, и на атомах углерода нет никаких N или S групп, соседних с другим гетероатомом. Таким образом, например, в кольце:
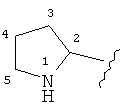
не существует -ОН группа, связанная непосредственно с углеродом, обозначенным как 2 и 5.
Соединения формулы 1.0 могут существовать в различных таутомерных формах, и все эти формы входят в объем настоящего изобретения. Также, например, все кето-енольные и имин-енаминные формы соединений входят в объем настоящего изобретения.
Таутомерные формы, такие как, например, составляющие:
 и
и 
в определенных вариантах выполнения настоящего изобретения рассматриваются как эквивалентные.
Термин "замещенный" означает, что один или более атомов водорода при указанном атоме замещены группой, выбранной из указанных групп, при условии, что при существующих условиях нормальная валентность обозначенного атома не превышается, и что замещение приводит к получению стабильного соединения. Комбинации заместителей и/или переменных допустимы, только если такие комбинации приводят к стабильным соединениям. Термин "стабильное соединение" или "стабильная структура" означает соединение, которое является достаточно прочным, для того чтобы выдержать выделение из реакционной смеси до применяемой степени чистоты и превращение в эффективный фармацевтический агент.
Термин "необязательно замещенный" означает необязательное замещение специфическими группами, радикалами или составляющими.
Термин "очищенный", "в очищенной форме" или "в выделенной и очищенной форме", применяемый в отношении соединения, относится к физическому состоянию указанного соединения после выделения из процесса синтеза или природного источника или их комбинации. Таким образом, термин "очищенный", "в очищенной форме" или "в выделенной и очищенной форме", применяемый по отношению к соединению, относится к физическому состоянию указанного соединения после процесса очистки или процессов, описанных здесь или хорошо известных специалистам в данной области техники, при степени чистоты, достаточной для определения с помощью стандартных аналитических методик, описанных здесь и хорошо известных специалистам в данной области техники.
Когда функциональная группа в соединении обозначается как "защищенная", это означает, что группа находится в модифицированной форме, предназначенной для предотвращения нежелательных побочных реакций по защищенному положению, когда соединение вступает в реакцию. Подходящие защитные группы известны специалистам в данной области техники, и раскрываются в таких документах, как например, Т.W. Greene et al, Protective Groups in organic Synthesis (1991), Wiley, New York.
Когда любая переменная (например, арил, гетероцикл, R3 и тому подобное) более одного раза встречается в любой составляющей или в любом соединение формулы 1.0, то в каждом случае определение этой переменной не зависит от ее определения в каждом другом случае.
Как применяется в настоящем документе, термин "композиция", как предполагается, охватывает продукт, содержащий определенные ингредиенты в определенных количествах, так же как и любой продукт, который получают непосредственно или косвенно, из комбинации определенных ингредиентов в определенных количествах.
Настоящее изобретение также охватывает изотопически меченые соединения согласно настоящему изобретению, которые идентичны указанным здесь соединениям, но один или более атомов заменены атомом, имеющим атомную массу или массовое число, отличные от обычно встречающихся в природе атомной массы или массового числа. Примеры изотопов, которые могут присутствовать в соединение по изобретению, включают изотопы водорода, углерода, азота, кислорода, фосфора, фтора и хлора, такие как 2H, 3H, 13С, 14С, 15N, 18О, 17О, 31P, 32Р, 35S, 18F и 36Cl, соответственно.
Определенные изотопически меченые соединения формулы 1.0 (например, меченые с применением 3H и 14С) подходят для анализа распределения в ткани соединения и/или субстрата. Изотопы тритий (то есть, 3H) и углерод-14 (то есть, 14С) особенно предпочтительны благодаря легкости их получения и определения. Определенные изотопически меченые соединения формулы (I) могут применяться для получения изображения в медицинских целях. Например, такие соединения, меченые позитрон-испускающими изотопами, такими как 11С или 18F, могут быть полезны для применения в Позитронной эмиссионной томографии (ПЭТ), и соединения, меченные изотопами, испускающими гамма-лучи, такими как 123I, могут быть полезны для применения в Однофотонной эмиссионной компьютерной томографии (ОФЭКТ). Кроме того, замещение более тяжелыми изотопами, такими как дейтерий (то есть, 2H) может давать определенные терапевтические преимущества, вытекающие из большей метаболической стабильности (например, увеличенный период полураспада in vivo или уменьшенные требуемые дозировки) и, следовательно, может быть предпочтительно при некоторых условиях. Изотопически меченые соединения формулы (I), в частности, содержащие изотопы с более долгими периодами полураспада (Т1/2 > одного дня), могут, в общем, быть получены по следующим методикам, аналогичным раскрытым в схемах и/или в примерах, приведенных ниже, путем замещения изотопически немеченого реагента соответствующим изотопически меченым реагентом.
Настоящее изобретение раскрывает соединения формулы 1.0:
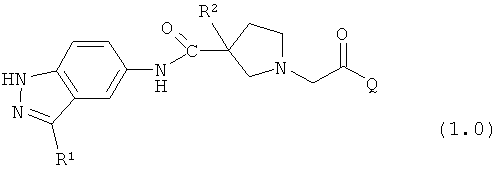
или их фармацевтически приемлемые соли или сольваты, где R1, R2 и Q выбираются независимо и, где:
Q представляет собой: 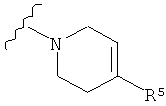 ;
;
R1 выбирается из группы, состоящей из: гетероарила и замещенного гетероарила, где указанный замещенный гетероарил имеет от одного до трех (предпочтительно один) заместителей, независимо выбранных из группы, состоящей из: -ОН, алкокси и -О-алкилен-O-алкила;
R2 выбирается из группы, состоящей из: -O-алкила и -S-алкила; и
R5 выбирается из группы, состоящей из:
(a) триазолилфенила-,
(b) триазолилфенила-, где указанный фенил необязательно имеет от одного до трех заместителей, независимо выбранных из группы, состоящей из: гало (например, Br, Cl, F, и в одном примере F) и алкокси (например, C1-C6алкокси и в одном примере С1-С2алкокси, и в другом примере -ОСН3),
(c) замещенного триазолилфенила-, где указанный фенил необязательно имеет от одного до трех заместителей, независимо выбранных из группы, состоящей из: гало (например, Br, Cl, F, и в одном примере F) и алкокси (например, C1-C6алкокси и в одном примере С1-С2алкокси, и в другом примере -ОСН3), и указанная триазолильная группа имеет один или два заместителя, независимо выбранные из группы, состоящей из: алкила, гидроксизамещенного алкила, -алкилен-O-алкила и амино (то есть, -NH2),
(d) триазолилтиенила-,
(e) триазолилтиенила-, где указанный тиенил необязательно имеет от одного до двух заместителей, независимо выбранных из группы, состоящей из: гало (например, Br, Cl, F, и в одном примере F) и алкокси (например, С1-С6алкокси и в одном примере С1-С2алкокси, и в другом примере -ОСН3),
(f) замещенного триазолилтиенила-, где указанный тиенил необязательно имеет от одного до трех заместителей, независимо выбранных из группы, состоящей из: гало (например, Br, Cl, F, и в одном примере F) и алкокси (например, C1-C6алкокси и в одном примере C1-C2алкокси, и в другом примере -ОСН3), и указанная триазолильная группа имеет один или два заместителя, независимо выбранные из группы, состоящей из: алкила, гидроксизамещенного алкила, -алкилен-O-алкила и амино (то есть, -NH2),
(g) триазолилпиридила-,
(h) триазолилпиридила-, где указанный пиридил необязательно имеет от одного до трех заместителей, независимо выбранных из группы, состоящей из: гало (например, Br, Cl, F, и в одном примере F), алкила и алкокси (например, C1-C6алкокси, и в одном примере, C1-C2алкокси, и в другом примере -OCH3). при условии, что атомы углерода, соседние с атомом азота в указанном пиридиле, не имеют заместитель гало, и
(i) замещенного триазолилпиридила-, где: (1) указанный пиридил необязательно имеет от одного до трех заместителей, независимо выбранных из группы, состоящей из: гало (например, Br, Cl, F, и в одном примере F), алкила и алкокси (например, C1-C6алкокси и в одном примере C1-C2алкокси, и в другом примере -OCH3), при условии, что атомы углерода, соседние с атомом азота в указанном пиридиле, не имеют заместитель гало и, (2) указанная триазолильная группа имеет один или два заместителя, независимо выбранные из группы, состоящей из: алкила, гидроксизамещенного алкила, -алкилен-O-алкила и амино (то есть, -NH2),
(j) триазолилтиазолила-,
(k) триазолилтиазолила-, где указанный тиазолил необязательно имеет один заместитель, независимо выбранный из группы, состоящей из: гало (например, Br, Cl, F, и в одном примере F), алкила и алкокси (например, C1-C6алкокси и в одном примере C1-C2алкокси, и в другом примере -OCH3), амино (то есть, NH2), алкиламино и диалкиламино, где каждый алкил выбирается независимо, и
(l) замещенного триазолилтиазолила-, где (1) указанный тиазолил необязательно имеет один заместитель, независимо выбранный из группы, состоящей из: гало (например, Br, Cl, F, и в одном примере F), алкила и алкокси (например, C1-C6алкокси и в одном примере С1-С2алкокси, и в другом примере -OCH3), амино (то есть, NH2), алкиламино и диалкиламино, где каждый алкил выбирается независимо, и (2) указанная триазолильная группа имеет один или два заместителя, независимо выбранные из группы, состоящей из: алкила, гидроксизамещенного алкила, -алкилен-O-алкила и амино (то есть, -NH2),
(m) пиридазинилтиенила-,
(n) пиридазинилтиенила-, где указанный тиенил необязательно имеет от одного до двух заместителей, независимо выбранных из группы, состоящей из: гало (например, Br, Cl, F, и в одном примере F) и алкокси (например, C1-C6алкокси и в одном примере С1-С2алкокси, и в другом примере -OCH3), и
(о) замещенного пиридазинилтиенила-, где (1) тиенил необязательно имеет от одного до двух заместителей, независимо выбранных из группы, состоящей из: гало (например, Br, Cl, F, и в одном примере F) и алкокси (например, C1-C6алкокси и в одном примере С1-С2алкокси, и в другом примере -OCH3), и (2) указанная пиридазинильная группа имеет от одного до трех заместителей, независимо выбранных из группы, состоящей из: =O, алкила, амино (то есть, -NH2), алкиламино, диалкиламино, где каждый алкил выбирается независимо, и гало (например, Br, Cl, F, и в одном примере F), при условии, что атомы углерода, соседние с атомом азота в указанном пиридазиниле, не имеют заместитель гало, и
при условии, что когда указанная -алкилен-O-алкильная группа связана с атомом азота указанного триазолила в (с), (f), (i) и (l) заместителе группы R5, составляющая алкилен указанной -алкилен-O-алкильной группы не представляет собой -CH2- (то есть, составляющая алкилен имеет 2 или более атомов углерода в длину).
Настоящее изобретение раскрывает соединения формулы 1.0:
или их фармацевтически приемлемые соли или сольваты, где:
Q представляет собой:
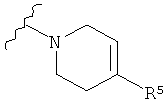 ;
;
R1 выбирается из группы, состоящей из: гетероарила и замещенного гетероарила, где указанный замещенный гетероарил имеет от одного до трех (предпочтительно один) заместителей, независимо выбранных из группы, состоящей из: -ОН, алкокси и -О-алкилен-O-алкила;
R2 выбирается из группы, состоящей из: -O-алкила и -S-алкила; и
R5 выбирается из группы, состоящей из:
(a) триазолилфенила-,
(b) триазолилфенила-, где указанный фенил необязательно имеет от одного до трех заместителей, независимо выбранных из группы, состоящей из: гало (например, Br, Cl, F, и в одном примере F) и алкокси (например, C1-C6алкокси и в одном примере С1-С2алкокси, и в другом примере -OCH3),
(c) замещенного триазолилфенила-, где указанный фенил необязательно имеет от одного до трех заместителей, независимо выбранных из группы, состоящей из: гало (например, Br, Cl, F, и в одном примере F) и алкокси (например, C1-С6алкокси и в одном примере С1-С2алкокси, и в другом примере -OCH3), и указанная триазолильная группа имеет один или два заместителя, независимо выбранные из группы, состоящей из: алкила, гидроксизамещенного алкила, -алкилен-O-алкила и амино (то есть, -NH2),
(d) триазолилтиенила-,
(e) триазолилтиенила-, где указанный тиенил необязательно имеет от одного до двух заместителей, независимо выбранных из группы, состоящей из: гало (например, Br, Cl, F, и в одном примере F) и алкокси (например, C1-C6алкокси и в одном примере С1-C2алкокси, и в другом примере -OCH3),
(f) замещенного триазолилтиенила-, где указанный тиенил необязательно имеет от одного до трех заместителей, независимо выбранных из группы, состоящей из: гало (например, Br, Cl, F, и в одном примере F) и алкокси (например, C1-C6алкокси и в одном примере С1-С2алкокси, и в другом примере -OCH3), и указанная триазолильная группа имеет один или два заместителя, независимо выбранные из группы, состоящей из: алкила, гидроксизамещенного алкила, -алкилен-O-алкила и амино (то есть, -NH2), и
при условии, что когда указанная -алкилен-O-алкильная группа связана с атомом азота указанного триазолила в (с), (f), (i) и (l) заместителе группы R5, составляющая алкилен указанной -алкилен-O-алкильной группы не представляет собой -CH2- (то есть, составляющая алкилен имеет 2 или более атомов углерода в длину).
Специалистам в данной области техники очевидно, что термин "алкилен", используемый в названии заместителей -О-алкилен-O-алкил и -алкилен-O-алкил, означает двухвалентную насыщенную углеводородную группу. Таким образом, пример составляющей алкилен представляет собой -CH2-СН2-, и пример -О-алкилен-O-алкильной составляющей представляет собой -O-(СН2)2-O-CH3, и пример -алкилен-O-алкильной составляющей представляет собой -(СН2)2-O-CH3.
Специалистам в данной области техники очевидно, что термин алкилен также включает составляющую -CH2-.
Примеры R1 гетероарильной группы включают, но без ограничения к этому, пиридил, пирролил, пиразолил, имидазолил, фуранил, тиенил, тиазолил, пиридил N-O и пиримидинил.
Примеры R1 замещенной гетероарильной группы включают, но без ограничения к этому, замещенный пиридил, замещенный пирролил, замещенный пиразолил, замещенный имидазолил, замещенный фуранил, замещенный тиенил, замещенный тиазолил, замещенный пиридил N-O и замещенный пиримидинил.
В одном варианте выполнения настоящего изобретения R1 представляет собой пиридил.
В другом варианте выполнения настоящего изобретения R1 представляет собой замещенный пиридил.
В другом варианте выполнения настоящего изобретения R1 представляет собой пиридил, замещенный одним заместителем.
Заместители на замещенных R1 группах (например, замещенный пиридил) независимо выбираются из группы, состоящей из: -ОН, алкокси и -O-алкилен-O-алкила. Примеры алкоксигруппы включают, например, C1-С6алкоксигруппу (такую как, например, -O-CH3, -O-С2Н5 и -O-СН(CH3)2). Примеры -О-алкилен-O-алкильной группы включают, например, -O-(С1-С4)алкилен-O-(C1-C6)алкил, -O-(С1-С2)алкилен-O-(С1-С3алкил) и -O-(СН2)2-O-CH3).
Примеры R1 включают, например,
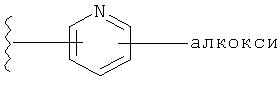 и
и 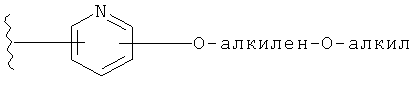 .
.
В одном варианте выполнения настоящего изобретения R1 представляет собой пиридил, замещенный алкоксигруппой.
В другом варианте выполнения настоящего изобретения R1 замещен -ОСН(CH3)2 группой.
В другом варианте выполнения настоящего изобретения R1 замещен -ОС2Н5 группой.
В другом варианте выполнения настоящего изобретения R1 представляет собой:
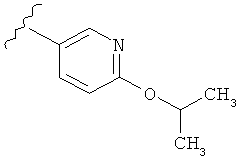 .
.
В другом варианте выполнения настоящего изобретения R1 представляет собой:
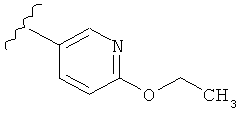 .
.
В другом варианте выполнения настоящего изобретения R1 замещен -O-алкилен-O-алкилом.
В другом варианте выполнения настоящего изобретения R1 замещен -OCH2CH2OCH3.
В другом варианте выполнения настоящего изобретения R1 представляет собой:
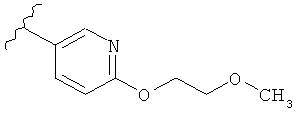 .
.
Примеры -O-алкильной группы R2 включают, например, -O-(С1-С6)алкил, -O-(С1-С2)алкил и -OCH3.
Примеры -S-алкильной группы R2 включают, например, -S-(С1-С6)алкил, -S-(С1-С2)алкил и -SCH3.
В одном варианте выполнения настоящего изобретения R2 представляет собой -O-(С1-С2)алкильную группу.
В другом варианте выполнения настоящего изобретения R2 представляет собой -OCH3.
В другом варианте выполнения настоящего изобретения R2 представляет собой -S-(С1-С2)алкильную группу.
В другом варианте выполнения настоящего изобретения R2 представляет собой -SCH3.
В одном варианте выполнения настоящего изобретения R5 представляет собой триазолилфенильную составляющую, где триазолильная составляющая связана с фенильной составляющей кольцевым атомом углерода триазолильной составляющей.
В одном варианте выполнения настоящего изобретения R5 представляет собой триазолилфенил- составляющую, такую как, например,
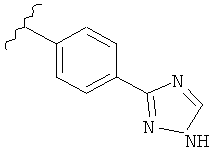 .
.
В другом варианте выполнения настоящего изобретения R5 представляет собой триазолилтиенил- составляющую, такую как, например,
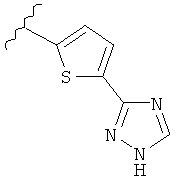 .
.
В другом варианте выполнения настоящего изобретения R5 представляет собой триазолилтиенил- составляющую, такую как, например,
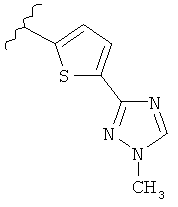 .
.
В другом варианте выполнения настоящего изобретения замещенная триазолильная составляющая указанной R5 группы замещена на кольцевом атоме азота.
Когда триазолильная составляющая R5 замещена алкилом, примеры алкильных групп включают, например, -C1-C6алкил, -С1-С4алкил, -С1-С2алкил и -CH3. И в одном варианте выполнения изобретения присутствует алкильное замещение на триазолильной составляющей R5, и указанный алкил представляет собой -CH3.
Когда триазолильная составляющая R5 замещена -алкилен-O-алкильными группами, примеры -алкилен-O-алкильных групп включают, например, -C1-С4алкилен-O-C1-С6алкил, -С1-С2алкилен-O-С1-С2алкил, -C1-С4алкилен-O-CH3 и -CH2CH2OCH3. И в одном варианте выполнения изобретения присутствует -алкилен-O-алкильное замещение на триазолильной составляющей R5, и указанный -алкилен-O-алкил представляет собой -CH2CH2OCH3. Когда атом азота триазолильной составляющей R5 замещен -алкилен-O-алкильной группой, примеры -алкилен-O-алкильной группы включают, например, -С2-С4алкилен-O-С1-С6алкил, -С2алкилен-O-C1-C2алкил, -С2-С4алкилен-O-CH3 и -CH2CH2OCH3. И в одном варианте выполнения изобретения присутствует -алкилен-O-алкильное замещение на атоме азота триазолильной составляющей R5, и указанный -алкилен-O-алкил представляет собой -CH2CH2OCH3.
Когда триазолильная составляющая R5 замещена гидроксизамещенными алкильными группами, примеры гидроксизамещенных алкильных групп включают, например, гидроксизамещенный -С1-С4алкил, гидроксизамещенный -С1-С2алкил и гидроксизамещенный -CH3. Примеры также включают, например, -СН2СОН(CH3)2 и -CH2CH2OH.
Когда фенильная составляющая R5 замещена атомами галогенов, примеры атомов галогенов включают, например, хлор (Cl), фтор (F) и бром (Br). В одном варианте выполнения настоящего изобретения галоген на фениле представляет собой F. В другом варианте выполнения настоящего изобретения фенил замещен одним атомом F.
В одном варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилфенил-, где указанный триазолил замещен, и указанный фенил является незамещенным.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилфенил-, где указанный триазолил замещен на атоме азота, и указанный фенил является незамещенным.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилфенил-, где указанный триазолил замещен на атоме углерода, и указанный фенил является незамещенным.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилфенил-, где указанный триазолил замещен на атоме азота и на атоме углерода, и указанный фенил является незамещенным.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилфенил-, где указанный триазолил замещен, и указанный фенил замещен.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилфенил-, где указанный триазолил замещен на атоме азота, и указанный фенил замещен.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилфенил-, где указанный триазолил замещен на атоме углерода, и указанный фенил замещен.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилфенил-, где указанный триазолил замещен на атоме азота и на атоме углерода, и указанный фенил замещен.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилфенил-, где указанный триазолил является незамещенным, и указанный фенил замещен.
В другом варианте выполнения настоящего изобретения R5 представляет собой незамещенный триазолилфенил-.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилфенил-, где триазолил замещается группой -СН2СОН(CH3)2 на атоме азота.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилфенил-, где триазолил замещается группой -CH2CH2OH на атоме азота.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилфенил-, где триазолил замещается алкильной группой на атоме азота.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилфенил-, где триазолил замещен алкильной группой на атоме азота и замещен алкильной группой на атоме углерода, где каждая алкильная группа выбирается независимо.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилфени-, где триазолил замещен -CH3 группой на атоме азота.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилфенил-, где триазолил замещен -CH3 группой на атоме азота и -CH3 группой на атоме углерода.
В другом варианте выполнения настоящего изобретения R5 составляющая представляет собой замещенный триазолилфенил-, где триазолил замещен -NH2 группой на атоме углерода.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилфенил-, где триазолил замещен -алкилен-O-алкильной группой на атоме азота.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилфенил-, где триазолил замещен -CH2CH2OCH3 группой на атоме азота.
В другом варианте выполнения настоящего изобретения R5 составляющая представляет собой замещенный триазолилфенил-, где указанная фенильная составляющая замещена галогеном, и указанная триазолильная составляющая замещена, как указанно в любом из приведенных выше вариантов выполнения изобретения.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилфенил-, где указанная фенильная составляющая замещена одним галогеном, и указанная триазолильная составляющая замещена, как указанно в любом из приведенных выше вариантов выполнения изобретения.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилфенил-, где указанная фенильная составляющая замещена атомом фтора, и указанная триазолильная составляющая замещена, как указанно в любом из приведенных выше примеров.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилфенил-, где указанная фенильная составляющая замещена одним атомом фтора, и указанная триазолильная составляющая замещена, как указанно в любом из приведенных выше вариантов выполнения изобретения.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилфенил- группу, где указанная фенильная составляющая замещена одним атомом F, и указанная триазолильная составляющая замещена гидроксил замещенной алкильной группой на атоме азота.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилфенил-, где указанная фенильная составляющая замещена одним атомом F, и указанная триазолильная составляющая замещена группой CH2CH2OH на атоме азота.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилфенил-, где указанная фенильная составляющая замещена одним атомом F, и указанная триазолильная составляющая замещена -алкилен-O-алкильной группой на атоме азота.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилфенил-, где указанная фенильная составляющая замещена одним атомом F, и указанная триазолильная составляющая замещена -CH2CH2OCH3 группой на атоме азота.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилфенил-, где указанная фенильная составляющая замещена галогеном, и указанная триазолильная составляющая является незамещенной.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилфенил-, где указанная фенильная составляющая замещена одним галогеном, и указанная триазолильная составляющая является незамещенной.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилфенил-, где указанная фенильная составляющая замещена F, и указанная триазолильная составляющая является незамещенной.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилфенил-, где указанная фенильная составляющая замещена одним F, и указанная триазолильная составляющая является незамещенной.
В другом варианте выполнения настоящего изобретения R5 составляющая представляет собой замещенный триазолилфенил-, где указанная фенильная составляющая замещена алкоксигруппой, и указанная триазолильная составляющая замещена, как указано в любом из приведенных выше вариантов выполнения изобретения.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилфенил-, где указанная фенильная составляющая замещена одной алкоксигруппой, и указанная триазолильная составляющая замещена, как указано в любом из приведенных выше вариантов выполнения изобретения.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилфенил-, где указанная фенильная составляющая замещена -OCH3 группой, и указанная триазолильная составляющая замещена, как указано в любом из приведенных выше вариантов выполнения изобретения.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилфенил-, где указанная фенильная составляющая замещена одной -OCH3 группой, и указанная триазолильная составляющая замещена, как указано в любом из приведенных выше вариантов выполнения изобретения.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенную триазолилфенил- группу, где указанная фенильная составляющая замещена одной -OCH3 группой, и указанная триазолильная составляющая замещена гидроксил замещенной алкильной группой на атоме азота.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилфенил-, где указанная фенильная составляющая замещена одной -OCH3 группой, и указанная триазолильная составляющая замещена -CH2CH2OH группой на атоме азота.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилфенил-, где указанная фенильная составляющая замещена одной -OCH3 группой, и указанная триазолильная составляющая замещена -алкилен-O-алкильной группой на атоме азота.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилфенил-, где указанная фенильная составляющая замещена одной -OCH3 группой, и указанная триазолильная составляющая замещена -CH2CH2OCH3 группой на атоме азота.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилфенил-, где указанная фенильная составляющая замещена алкоксигруппой, и указанная триазолильная составляющая является незамещенной.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилфенил-, где указанная фенильная составляющая замещена одной алкоксигруппой, и указанная триазолильная составляющая является незамещенной.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилфенил-, где указанная фенильная составляющая замещена группой -OCH3, и указанная триазолильная составляющая является незамещенной.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилфенил-, где указанная фенильная составляющая замещена одной группой -OCH3, и указанная триазолильная составляющая является незамещенной.
В другом варианте выполнения настоящего изобретения R5 представляет собой:
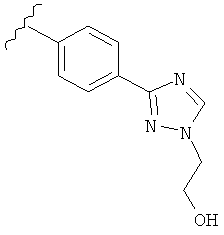 .
.
В другом варианте выполнения настоящего изобретения R5 представляет собой:
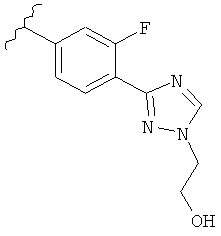 .
.
В другом варианте выполнения настоящего изобретения R5 представляет собой:
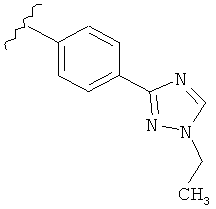 .
.
В другом варианте выполнения настоящего изобретения R5 представляет собой:
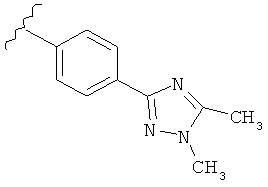 .
.
В другом варианте выполнения настоящего изобретения R5 представляет собой:
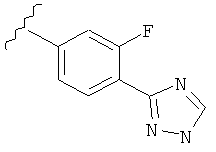 .
.
В другом варианте выполнения настоящего изобретения R5 представляет собой:
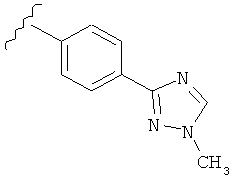
В другом варианте выполнения настоящего изобретения R5 представляет собой:
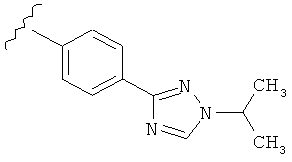
В другом варианте выполнения настоящего изобретения R5 представляет собой:
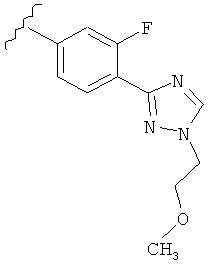 .
.
В другом варианте выполнения настоящего изобретения R5 представляет собой:
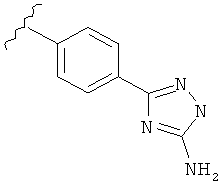 .
.
В другом варианте выполнения настоящего изобретения R5 представляет собой:
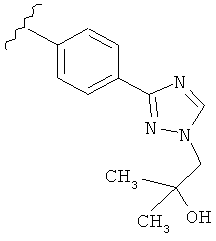 .
.
В другом варианте выполнения настоящего изобретения R5 представляет собой:
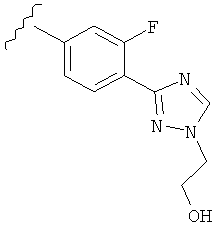 .
.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилтиенил-, где указанный триазолил замещен, и указанный тиенил является незамещенным.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилтиенил-, где указанный триазолил замещен на атоме азота, и указанный тиенил является незамещенным.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилтиенил-, где указанный триазолил замещен на атоме углерода, и указанный тиенил является незамещенным.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилтиенил-, где указанный триазолил замещен на атоме азота и на атоме углерода, и указанный тиенил является незамещенным.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилтиенил-, где указанный триазолил замещен, и указанный тиенил замещен.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилтиенил-, где указанный триазолил замещен на атоме азота, и указанный тиенил замещен.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилтиенил-, где указанный триазолил замещен на атоме углерода, и указанный тиенил замещен.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилтиенил-, где указанный триазолил замещен на атоме азота и на атоме углерода, и указанный тиенил замещен.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилтиенил-, где указанный триазолил является незамещенным, и указанный тиенил замещен.
В другом варианте выполнения настоящего изобретения R5 представляет собой незамещенный триазолилтиенил-.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилтиенил-, где триазолил замещен на атоме азота группой -СН2СОН(CH3)2.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилтиенил-, где триазолил замещен на атоме азота группой -CH2CH2OH.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилтиенил-, где триазолил замещен на атоме азота алкильной группой.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилтиенил-, где триазолил замещен на атоме азота алкильной группой и замещенным на атоме углерода алкильной группой, где каждая алкильная группа выбирается независимо.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилтиенил-, где триазолил замещен на атоме азота группой -CH3.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилтиенил-, где триазолил замещен на атоме азота группой -CH3, и на атоме углерода группой -CH3.
В другом варианте выполнения настоящего изобретения R5 составляющая представляет собой замещенный триазолилтиенил-, где триазолил замещен на атоме углерода группой -NH2.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилтиенил-, где триазолил замещен на атоме азота -алкилен-O-алкильной группой.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилтиенил-, где триазолил замещен на атоме азота группой -CH2CH2OCH3.
В другом варианте выполнения настоящего изобретения R5 составляющая представляет собой замещенный триазолилтиенил-, где указанная тиенильная составляющая замещена галогеном, и указанная триазолильная составляющая замещена, как описано в любом из приведенных выше вариантов выполнения изобретения.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилтиенил-, где указанная тиенильная составляющая замещена одним галогеном, и указанная триазолильная составляющая замещена, как описано в любом из приведенных выше вариантов выполнения изобретения.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилтиенил-, где указанная тиенильная составляющая замещена F, и указанная триазолильная составляющая замещена, как описано в любом из приведенных выше вариантов выполнения изобретения.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилтиенил-, где указанная тиенильная составляющая замещена одним F, и указанная триазолильная составляющая замещена, как описано в любом из приведенных выше вариантов выполнения изобретения.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенную триазолилтиенил- группу, где указанная тиенильная составляющая замещена одним F, и указанная триазолильная составляющая замещена на атоме азота гидроксил замещенной алкильной группой.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилтиенил-, где указанная тиенильная составляющая замещена одним F, и указанная триазолильная составляющая замещена на атоме азота группой -CH2CH2OH.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилтиенил-, где указанная тиенильная составляющая замещена одним F, и указанная триазолильная составляющая замещена на атоме азота -алкилен-O-алкильной группой.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилтиенил-, где указанная тиенильная составляющая замещена одним F, и указанная триазолильная составляющая замещена на атоме азота группой -CH2CH2OCH3.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилтиенил-, где указанная тиенильная составляющая замещена галогеном, и указанная триазолильная составляющая является незамещенной.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилтиенил-, где указанная тиенильная составляющая замещена одним галогеном, и указанная триазолильная составляющая является незамещенной.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилтиенил-, где указанная тиенильная составляющая замещена F, и указанная триазолильная составляющая является незамещенной.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилтиенил-, где указанная тиенильная составляющая замещена одним F, и указанная триазолильная составляющая является незамещенной.
В другом варианте выполнения настоящего изобретения R5 составляющая представляет собой замещенный триазолилтиенил-, где указанная тиенильная составляющая замещена алкоксигруппой, и указанная триазолильная составляющая замещена, как описано в любом из приведенных выше вариантов выполнения изобретения.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилтиенил-, где указанная тиенильная составляющая замещена одной алкоксигруппой, и указанная триазолильная составляющая замещена, как описано в любом из приведенных выше вариантов выполнения изобретения.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилтиенил-, где указанная тиенильная составляющая замещена группой -OCH3, и указанная триазолильная составляющая замещена, как описано в любом из приведенных выше вариантов выполнения изобретения.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилтиенил-, где указанная тиенильная составляющая замещена одной группой -OCH3, и указанная триазолильная составляющая замещена, как описано в любом из приведенных выше вариантов выполнения изобретения.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилтиенил-, где указанная тиенильная составляющая замещена одной группой -OCH3, и указанная триазолильная составляющая замещена на атоме азота гидроксил замещенной алкильной группой.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилтиенил-, где указанная тиенильная составляющая замещена одной группой -OCH3, и указанная триазолильная составляющая замещена на атоме азота группой -CH2CH2OH.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилтиенил-, где указанная тиенильная составляющая замещена одной группой -OCH3, и указанная триазолильная составляющая замещена на атоме азота -алкилен-O-алкильной группой.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилтиенил-, где указанная тиенильная составляющая замещена одной группой -OCH3, и указанная триазолильная составляющая замещена на атоме азота группой -CH2CH2OCH3.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилтиенил-, где указанная тиенильная составляющая замещена алкоксигруппой, и указанная триазолильная составляющая является незамещенной.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилтиенил-, где указанная тиенильная составляющая замещена одной алкоксигруппой, и указанная триазолильная составляющая является незамещенной.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилтиенил-, где указанная тиенильная составляющая замещена группой -OCH3, и указанная триазолильная составляющая является незамещенной.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилтиенил-, где указанная тиенильная составляющая замещена одной группой -OCH3, и указанная триазолильная составляющая является незамещенной.
В другом варианте выполнения настоящего изобретения R5 представляет собой триазолилпиридил-.
В другом варианте выполнения настоящего изобретения R5 представляет собой
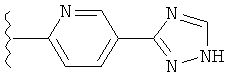 .
.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилпиридил-, где указанный триазолил замещен, и указанный пиридил является незамещенным.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилпиридил-, где указанный триазолил замещен на атоме азота, и указанный пиридил является незамещенным.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилпиридил-, где указанный триазолил замещен на атоме углерода, и указанный пиридил является незамещенным.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилпиридил-, где указанный триазолил замещен на атоме азота и на атоме углерода, и указанный пиридил является незамещенным.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилпиридил-, где указанный триазолил замещен, и указанный пиридил замещен.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилпиридил-, где указанный триазолил замещен на атоме азота, и указанный пиридил замещен.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилпиридил-, где указанный триазолил замещен на атоме углерода, и указанный пиридил замещен.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилпиридил-, где указанный триазолил замещен на атоме азота и на атоме углерода, и указанный пиридил замещен.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилпиридил-, где указанный триазолил является незамещенным, и указанный пиридил замещен.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилпиридил-, где триазолил замещен на атоме азота алкильной группой.
Когда пиридильная составляющая группы R5 замещена алкильной группой, примеры алкильных групп включают, например, -C1-С6алкил, -C1-С4алкил, -C1-С2алкил и -CH3.
Когда пиридильная составляющая группы R5 замещена атомами галогенов, примеры атомов галогенов включают, например, Cl, F и Br, при условии, что атомы углерода, соседние с атомом азота в указанном пиридиле, незамещены галогеном. В одном варианте выполнения настоящего изобретения галоген на пиридиле представляет собой F. В другом варианте выполнения настоящего изобретения пиридил замещен одним F.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилпиридил-, где триазолил замещен на атоме азота группой -СН2СОН(CH3)2, и пиридил является незамещенным.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилпиридил-, где триазолил замещен на атоме азота группой -CH2CH2OH, и пиридил является незамещенным.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилпиридил-, где триазолил замещен на атоме азота алкильной группой, и пиридил является незамещенным.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилпиридил-, где триазолил замещен на атоме азота алкильной группой и замещенным на атоме углерода алкильной группой, где каждая алкильная группа выбирается независимо, и пиридил является незамещенным.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилпиридил-, где триазолил замещен на атоме азота группой -CH3, и пиридил является незамещенным.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилпиридил-, где триазолил замещен на атоме азота группой -CH3 и на атоме углерода группой -CH3, и пиридил является незамещенным.
В другом варианте выполнения настоящего изобретения R5 составляющая представляет собой замещенный триазолилпиридил-, где триазолил замещен на атоме углерода группой -NH2, и пиридил является незамещенным.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилпиридил-, где триазолил замещен на атоме азота -алкилен-O-алкильной группой, и пиридил является незамещенным, при условии, что алкиленовая составляющая указанной -алкилен-O-алкильной группы не представляет собой -СН2- (то есть, алкиленовая составляющая имеет два или более атомов углерода в длину).
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилпиридил-, где триазолил замещен на атоме азота группой -CH2CH2OCH3, и пиридил является незамещенным.
В другом варианте выполнения настоящего изобретения R5 составляющая представляет собой замещенный триазолилпиридил-, где указанная пиридильная составляющая замещена галогеном, при условии, что атомы углерода, соседние с атомом азота в указанном пиридиле, незамещены галогеном, и указанная триазолильная составляющая замещена, как описано в любом из приведенных выше вариантов выполнения изобретения, описывающих замещенные триазолильные группы.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилпиридил-, где указанная пиридильная составляющая замещена одним галогеном, при условии, что атомы углерода, соседние с атомом азота в указанном пиридиле, незамещены галогеном, и указанная триазолильная составляющая замещена, как описано в любом из приведенных выше вариантов выполнения изобретения, описывающих замещенные триазолильные группы.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилпиридил-, где указанная пиридильная составляющая замещена F, при условии, что атомы углерода, соседние с атомом азота в указанном пиридиле, незамещены указанным атомом фтора, и указанная триазолильная составляющая замещена, как описано в любом из приведенных выше вариантов выполнения изобретения, описывающих замещенные триазолильные группы.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилпиридил-, где указанная пиридильная составляющая замещена одним F, при условии, что атомы углерода, соседние с атомом азота в указанном пиридиле, незамещены указанным атомом фтора, и указанная триазолильная составляющая замещена, как описано в любом из приведенных выше вариантов выполнения изобретения, описывающих замещенные триазолильные группы.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенную триазолилпиридил-группу, где указанная пиридильная составляющая замещена одним F, при условии, что атомы углерода, соседние с атомом азота в указанном пиридиле, незамещены указанным F, и указанная триазолильная составляющая замещена на атоме азота гидроксил замещенной алкильной группой.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилпиридил-, где указанная пиридильная составляющая замещена одним F, при условии, что атомы углерода, соседние с атомом азота в указанном пиридиле, незамещены указанным F, и указанная триазолильная составляющая замещена на атоме азота группой -CH2CH2OH.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилпиридил-, при условии, что атомы углерода, соседние с атомом азота в указанном пиридиле, незамещены указанным F, и указанная триазолильная составляющая замещена на атоме азота -алкилен-O-алкильной группой, и алкиленовая составляющая указанный -алкилен-O-алкильной группы не представляет собой -CH2- (то есть, алкиленовая составляющая имеет два или более атомов углерода в длину).
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилпиридил-, где указанная пиридильная составляющая замещена одним F, при условии, что атомы углерода, соседние с атомом азота в указанном пиридиле, незамещены указанным F, и указанная триазолильная составляющая замещена на атоме азота группой -CH2CH2OCH3.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилпиридил-, где указанная пиридильная составляющая замещена галогеном, при условии, что атомы углерода, соседние с атомом азота в указанном пиридиле, незамещены галогеном, и указанная триазолильная составляющая является незамещенной.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилпиридил-, где указанная пиридильная составляющая замещена одним галогеном, при условии, что атомы углерода, соседние с атомом азота в указанном пиридиле, незамещены указанным галогеном, и указанная триазолильная составляющая является незамещенной.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилпиридил-, где указанная пиридильная составляющая замещена F, при условии, что атомы углерода, соседние с атомом азота в указанном пиридиле, незамещены указанным F, и указанная триазолильная составляющая является незамещенной.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилпиридил-, где указанная пиридильная составляющая замещена одним F, при условии, что атомы углерода, соседние с атомом азота в указанном пиридиле, незамещены указанным F, и указанная триазолильная составляющая является незамещенной.
В другом варианте выполнения настоящего изобретения R5 составляющая представляет собой замещенный триазолилпиридил-, где указанная пиридильная составляющая замещена алкоксигруппой, и указанная триазолильная составляющая замещена, как описано в любом из приведенных выше вариантов выполнения изобретения, описывающих триазолильные группы.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилпиридил-, где указанная пиридильная составляющая замещена одной алкоксигруппой, и указанная триазолильная составляющая замещена, как описано в любом из приведенных выше вариантов выполнения изобретения, описывающих триазолильные группы.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилпиридил-, где указанная пиридильная составляющая замещена группой -OCH3, и указанная триазолильная составляющая замещена, как описано в любом из приведенных выше вариантов выполнения изобретения, описывающих триазолильные группы.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилпиридил-, где указанная пиридильная составляющая замещена одной группой -OCH3, и указанная триазолильная составляющая замещена, как описано в любом из приведенных выше вариантов выполнения изобретения, описывающих триазолильные группы.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенную триазолилпиридил- группу, где указанная пиридильная составляющая замещена одной группой -OCH3, и указанная триазолильная составляющая замещена на атоме азота гидроксил замещенной алкильной группой.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилпиридил-, где указанная пиридильная составляющая замещена одной группой -OCH3, и указанная триазолильная составляющая замещена на атоме азота группой -CH2CH2OH.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилпиридил-, где указанная пиридильная составляющая замещена одной группой -OCH3, и указанная триазолильная составляющая замещена на атоме азота -алкилен-O-алкильной группой, и алкиленовая составляющая указанной -алкилен-O-алкильной группы не представляет собой -СН2- (то есть, алкиленовая составляющая имеет два или более атомов углерода в длину).
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилпиридил-, где указанная пиридильная составляющая замещена одной группой -OCH3, и указанная триазолильная составляющая замещена на атоме азота группой -CH2CH2OCH3.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилпиридил-, где указанная пиридильная составляющая замещена алкоксигруппой, и указанная триазолильная составляющая является незамещенной.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилпиридил-, где указанная пиридильная составляющая замещена одной алкоксигруппой, и указанная триазолильная составляющая является незамещенной.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилпиридил-, где указанная пиридильная составляющая замещена -OCH3 группой, и указанная триазолильная составляющая является незамещенной.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилпиридил-, где указанная пиридильная составляющая замещена одной группой -OCH3, и указанная триазолильная составляющая является незамещенной.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилпиридил-, где указанный триазолил замещен 1 или 2 группами, независимо выбранными из группы, состоящей из: (а) гидроксил замещенной алкильной группы (например, -СН2СОН(CH3)2, и -CH2CH2OH), (b) алкила (например, -C1-C6алкил, -С1-С4алкил, -С1-С2алкил и -CH3), (с) -NH2, и (d) -алкилен-O-алкила (например, -CH2CH2OCH3), при условии, что алкиленовая составляющая указанной -алкилен-O-алкильной группы не представляет собой -СН2- (то есть, алкиленовая составляющая имеет два или более атомов углерода в длину), когда указанная -алкилен-O-алкильная группа связана с атомом азота указанного триазолила; и указанный пиридил замещен 1-3 группами, независимо выбранными из группы, состоящей из: (а) алкила (например, -C1-C6алкил, -С1-С4алкил, -С1-С2алкил и -CH3), (b) галогена (например, Cl, F и Br), и при условии, что атомы углерода, соседние с атомом азота в указанном пиридиле, незамещены галогеном, и (с) алкокси (например, -OCH3).
В другом варианте выполнения настоящего изобретения R5 представляет собой
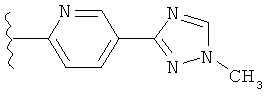 .
.
В другом варианте выполнения настоящего изобретения R5 представляет собой триазолилтиазолил-.
В другом варианте выполнения настоящего изобретения R5 представляет собой:
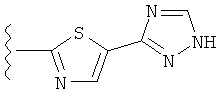 .
.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилтиазолил-, где указанный триазолил замещен, и указанный тиазолил является незамещенным.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилтиазолил-, где указанный триазолил замещен на атоме азота, и указанный тиазолил является незамещенным.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилтиазолил-, где указанный триазолил замещен на атоме углерода, и указанный тиазолил является незамещенным.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилтиазолил-, где указанный триазолил замещен на атоме азота и на атоме углерода, и указанный тиазолил является незамещенным.
Когда тиазолильная составляющая группы R5 замещена алкильной группой, примеры алкильных групп включают, например, -C1-C6алкил, -C1-С4алкил, -С1-С2алкил и -CH3.
Когда тиазолильная составляющая группы R5 замещена атомами галогенов, примеры атомов галогенов включают, например, Cl, F и Br. В одном варианте выполнения настоящего изобретения галоген на тиазолиле представляет собой F. В другом варианте выполнения настоящего изобретения тиазолил замещен одним F.
Когда тиазолильная составляющая группы R5 замещена алкиламино группой, примеры алкиламино группы включают, например, C1-C6алкил-NH-, C1-C2алкил-NH-, CH3-NH- и CH3CH2-NH-.
Когда тиазолильная составляющая группы R5 замещена диалкиламино группой, примеры диалкиламино группы включают, например, (С1-С6алкил)2-N-, где каждый алкил выбирается независимо, (C1-C2алкил)2-N-, где каждый алкил выбирается независимо, (CH3)2N-, (CH3CH2)2-N- и (CH3)(CH3CH2)N-.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилтиазолил-, где указанный триазолил замещен, и указанный тиазолил замещен.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилтиазолил-, где указанный триазолил замещен на атоме азота, и указанный тиазолил замещен.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилтиазолил-, где указанный триазолил замещен на атоме углерода, и указанный тиазолил замещен.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилтиазолил-, где указанный триазолил замещен на атоме азота и на атоме углерода, и указанный тиазолил замещен.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилтиазолил-, где указанный триазолил является незамещенным, и указанный тиазолил замещен.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилтиазолил-, где триазолил замещен на атоме азота алкилом, и указанный тиазолил является незамещенным.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилтиазолил -, где триазолил замещен на атоме азота группой -СН2СОН(CH3)2, и указанный тиазолил является незамещенным.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилтиазолил-, где триазолил замещен на атоме азота группой -CH2CH2OH, и указанный тиазолил является незамещенным.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилтиазолил-, где триазолил замещен на атоме азота алкильной группой и замещенным на атоме углерода алкильной группой, где каждая алкильная группа выбирается независимо, и указанный тиазолил является незамещенным.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилтиазолил-, где триазолил замещен на атоме азота -CH3 группой, и указанный тиазолил является незамещенным.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилтиазолил-, где триазолил замещен на атоме азота группой -CH3 и на атоме углерода группой -CH3, и указанный тиазолил является незамещенным.
В другом варианте выполнения настоящего изобретения R5 составляющая представляет собой замещенный триазолилтиазолил-, где триазолил замещен на атоме углерода группой -NH2, и указанный тиазолил является незамещенным.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилтиазолил-, где триазолил замещен на атоме азота -алкилен-O-алкильной группой, при условии, что алкиленовая составляющая указанной -алкилен-O-алкильной группы не представляет собой -СН2- (то есть алкиленовая составляющая имеет два или более атомов углерода в длину), и указанный тиазолил является незамещенным.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилтиазолил-, где триазолил замещен на атоме азота группой -CH2CH2OCH3, и указанный тиазолил является незамещенным.
В другом варианте выполнения настоящего изобретения R5 составляющая представляет собой замещенный триазолилтиазолил-, где указанная тиазолильная составляющая замещена галогеном, и указанная триазолильная составляющая замещена, как описано в любом из приведенных выше вариантов выполнения изобретения, описывающих замещенные триазолильные группы.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилтиазолил-, где указанная тиазолильная составляющая замещена F, и указанная триазолильная составляющая замещена, как описано в любом из приведенных выше вариантов выполнения изобретения, описывающих замещенные триазолильные группы.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенную триазолилтиазолил- группу, где указанная тиазолильная составляющая замещена одним F, и указанная триазолильная составляющая замещена на атоме азота гидроксил замещенной алкильной группой.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилтиазолил-, где указанная тиазолильная составляющая замещена одним F, и указанная триазолильная составляющая замещена на атоме азота группой -CH2CH2OH.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилтиазолил-, где указанная тиазолильная составляющая замещена одним F, и указанная триазолильная составляющая замещена на атоме азота -алкилен-O-алкильной группой.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилтиазолил-, где указанная тиазолильная составляющая замещена одним F, и указанная триазолильная составляющая замещена на атоме азота группой -CH2CH2OCH3.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилтиазолил-, где указанная тиазолильная составляющая замещена галогеном, и указанная триазолильная составляющая является незамещенной.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилтиазолил-, где указанная тиазолильная составляющая замещена F, и указанная триазолильная составляющая является незамещенной.
В другом варианте выполнения настоящего изобретения R5 составляющая представляет собой замещенный триазолилтиазолил-, где указанная тиазолильная составляющая замещена алкоксигруппой, и указанная триазолильная составляющая замещена, как описано в любом из приведенных выше вариантов выполнения изобретения, описывающих замещенные триазолильные группы.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилтиазолил-, где указанная тиазолильная составляющая замещена группой -OCH3, и указанная триазолильная составляющая замещена, как описано в любом из приведенных выше вариантов выполнения изобретения, описывающих замещенные триазолильные группы.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенную триазолилтиазолил-группу, где указанная тиазолильная составляющая замещена одной группой -OCH3, и указанная триазолильная составляющая замещена на атоме азота гидроксил замещенной алкильной группой.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилтиазолил-, где указанная тиазолильная составляющая замещена одной группой -OCH3, и указанная триазолильная составляющая замещена на атоме азота группой -CH3CH2OH.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилтиазолил-, где указанная тиазолильная составляющая замещена одной группой -OCH3, и указанная триазолильная составляющая замещена на атоме азота -алкилен-O-алкильной группой, и алкиленовая составляющая указанной -алкилен-O-алкильной группы не представляет собой -СН2- (то есть алкиленовая составляющая имеет два или более атомов углерода в длину).
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилтиазолил-, где указанная тиазолильная составляющая замещена группой -OCH3, и указанная триазолильная составляющая замещена на атоме азота группой -CH2CH2OCH3.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилтиазолил-, где указанная тиазолильная составляющая замещена алкоксигруппой, и указанная триазолильная составляющая является незамещенной.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилтиазолил-, где указанная тиазолильная составляющая замещена группой -OCH3, и указанная триазолильная составляющая является незамещенной.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилтиазолил-, где указанная тиазолильная составляющая замещена алкиламино группой, и указанная триазолильная составляющая замещена, как описано в любом из приведенных выше вариантов выполнения изобретения, раскрывающих триазолильные группы.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилтиазолил-, где указанная тиазолильная составляющая замещена диалкиламино группой, и указанная триазолильная составляющая замещена, как описано в любом из приведенных выше вариантов выполнения изобретения, раскрывающих триазолильные группы.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилтиазолил-, где указанная тиазолильная составляющая замещена алкиламино группой, и указанная триазолильная составляющая является незамещенной.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилтиазолил-, где указанная тиазолильная составляющая замещена диалкиламино группой, и указанная триазолильная составляющая является незамещенной.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный триазолилтиазолил-, где указанный триазолил замещен одной или двумя группами, независимо выбранными из группы, состоящей из: (а) гидроксил замещенной алкильной группы (например, -СН2СОН(CH3)2 и -CH2CH2OH), (b) алкила (например, -C1-C6алкил, -C1-С4алкил, -С1-С2алкил и -CH3), -NH2, и (с) -алкилен-O-алкила (например, -CH2CH2OCH3), при условии, что алкиленовая составляющая указанной -алкилен-O-алкильной группы не представляет собой -СН2- (то есть, алкиленовая составляющая имеет два или более атомов углерода в длину), когда указанная -алкилен-O-алкильная группа связана с атомом азота указанного триазолила; и указанный тиазолил замещен одной группой, выбранной из группы, состоящей из: (а) алкила (например, -С1-С6алкил, или -С1-С4алкил или -С1-С2алкил, или -CH3), (b) галогена (например, Cl, F, или Br), (с) алкиламино (например, С1-С6алкил-NH- или С1-C2алкил-NH-, или CH3-NH-, или CH3CH2-NH-), и (d) диалкиламино (например, (С1-С6алкил)2-N-, где каждый алкил выбирается независимо, или (С1-С2алкил)2-N-, где каждый алкил выбирается независимо, или (CH3)2N-, или (CH3CH2)2-N-, или (CH3)(CH3CH2)N-).
В другом варианте выполнения настоящего изобретения R5 представляет собой
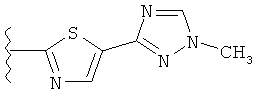 .
.
В другом варианте выполнения настоящего изобретения R5 представляет собой пиридазинилтиенил-.
В другом варианте выполнения настоящего изобретения R5 представляет собой
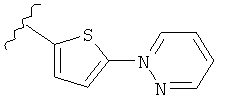 .
.
Когда пиридазинильная составляющая группы R5 замещена алкилом, примеры алкильных групп включают, например, -C1-C6алкил, -С1-С4алкил, -C1-C2алкил и -CH3.
Когда пиридазинильная составляющая группы R5 замещена атомами галогенов, примеры атомов галогенов включают, например, Cl, F и Br. В одном варианте выполнения настоящего изобретения галоген на пиридазиниле представляет собой F. В другом варианте выполнения настоящего изобретения пиридазинил замещен одним атомом F. Когда пиридазинильная составляющая замещена галогенами, атомы углерода, соседние с атомами азота, незамещены галогенами.
Когда пиридазинильная составляющая группы R5 замещена алкиламино группой, примеры алкиламино группы включают, например, C1-C6алкил-NH-, С1-C2алкил-NH-, CH3-NH- и CH3CH2-NH-.
Когда пиридазинильная составляющая группы R5 замещена диалкиламино группой, примеры диалкиламино группы включают, например, (C1-С6алкил)2-N-, где каждый алкил выбирается независимо, (С1-С2алкил)2-N-, где каждый алкил выбирается независимо, (CH3)2N-, (CH3CH2)2-N- и (CH3)(CH3CH2)N-.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный пиридазинилтиенил-, где указанный пиридазинил замещен, и указанный тиенил является незамещенным.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный пиридазинилтиенил-, где указанный пиридазинил замещен группой =O, и указанный тиенил является незамещенным.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный пиридазинилтиенил-, где указанный пиридазинил замещен алкильной группой, и указанный тиенил является незамещенным.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный пиридазинилтиенил-, где указанный пиридазинил замещен метильной группой, и указанный тиенил является незамещенным.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный пиридазинилтиенил-, где указанный пиридазинил замещен группой =O и алкильной группой, и указанный тиенил является незамещенным.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный пиридазинилтиенил-, где указанный пиридазинил замещен группой =O и метильной группой, и указанный тиенил является незамещенным.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный пиридазинилтиенил-, где указанный пиридазинил замещен аминогруппой, и указанный тиенил является незамещенным.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный пиридазинилтиенил-, где указанный пиридазинил замещен алкиламино группой, и указанный тиенил является незамещенным.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный пиридазинилтиенил-, где указанный пиридазинил замещен диалкиламино группой, и указанный тиенил является незамещенным.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный пиридазинилтиенил-, где указанный пиридазинил замещен, и указанный тиенил замещен.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный пиридазинилтиенил-, где указанный пиридазинил является незамещенным, и указанный тиенил замещен.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный пиридазинилтиенил-, где указанный пиридазинил является незамещенным, и указанный тиенил замещен одним или двумя независимо выбранными галогенами.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный пиридазинилтиенил-, где указанный пиридазинил является незамещенным, и указанный тиенил замещен одним или двумя галогенами, независимо выбранными из группы, состоящей из: Br, Cl и F.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный пиридазинилтиенил-, где указанный пиридазинил является незамещенным, и указанный тиенил замещен одной или двумя независимо выбранными алкоксигруппами
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный пиридазинилтиенил-, где указанный пиридазинил является незамещенным, и указанный тиенил замещен одной или двумя группами -OCH3.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный пиридазинилтиенил-, где указанный пиридазинил замещен, как описано в любом из приведенных выше вариантов выполнения изобретения, раскрывающих замещенные пиридазинильные группы, и указанный тиенил замещен.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный пиридазинилтиенил-, где указанный пиридазинил замещен, как описано в любом из приведенных выше вариантов выполнения изобретения, раскрывающих замещенные пиридазинильные группы, и указанный тиенил замещен одним-двумя независимо выбранными галогенами
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный пиридазинилтиенил-, где указанный пиридазинил замещен, как описано в любом из приведенных выше вариантов выполнения изобретения, раскрывающих замещенные пиридазинильные группы, и указанный тиенил замещен одним или двумя галогенами, независимо выбранными из группы, состоящей из: Br, Cl и F.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный пиридазинилтиенил-, где указанный пиридазинил замещен, как описано в любом из приведенных выше вариантов выполнения изобретения, раскрывающих замещенные пиридазинильные группы, и указанный тиенил замещен одной-двумя независимо выбранными алкокси-группами.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный пиридазинилтиенил-, где указанный пиридазинил замещен, как описано в любом из приведенных выше вариантов выполнения изобретения, раскрывающих замещенные пиридазинильные группы, и указанный тиенил замещен одной или двумя -OCH3 группами.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенный пиридазинилтиенил-, где указанный пиридазинил замещен одной или двумя группами, независимо выбранными из группы, состоящей из алкила (например, метил) и =O, и указанный тиенил замещен одной или двумя группами, независимо выбранными из группы, состоящей из: алкокси (например, -OCH3), галогена (например, Br, Cl и F).
В другом варианте выполнения настоящего изобретения R5 представляет собой:
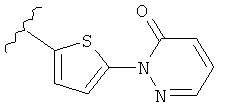 .
.
В другом варианте выполнения настоящего изобретения R5 представляет собой:
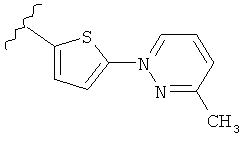 .
.
В другом варианте выполнения настоящего изобретения R5 представляет собой:
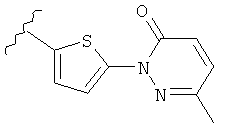 .
.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенную триазолилфенил-группу, где (а) указанная триазолильная составляющая необязательно замещена на атоме азота заместителем, выбранным из группы, состоящей из: -СН2СОН(CH3)2 и -CH2CH2OH, (b) указанная триазолильная составляющая необязательно замещена на атоме азота алкильной группой, (с) указанная триазолильная составляющая необязательно замещена на атоме азота алкильной группой, и на атоме углерода алкильной группой, (d) указанная триазолильная составляющая необязательно замещена на атоме азота группой -CH3, (е) указанная триазолильная составляющая необязательно замещена на атоме азота одной группой -CH3, и на атоме углерода одной группой -CH3, (f) указанная триазолильная составляющая необязательно замещена на атоме углерода группой -NH2, или (g) указанная триазолильная составляющая необязательно замещена на атоме азота группой -CH2CH2OCH3; и, где указанная фенильная составляющая необязательно замещена галогеном.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенную триазолилфенил-группу, где (а) указанная триазолильная составляющая необязательно замещена на атоме азота заместителем, выбранным из группы, состоящей из: -СН2СОН(CH3)2 и -CH2CH2OH, (b) указанная триазолильная составляющая необязательно замещена на атоме азота алкильной группой, (с) указанная триазолильная составляющая необязательно замещена на атоме азота алкильной группой, и на атоме углерода алкильной группой, (d) указанная триазолильная составляющая необязательно замещена на атоме азота группой -CH3, (е) указанная триазолильная составляющая необязательно замещена на атоме азота одной группой -CH3 и на атоме углерода одной группой -CH3, (f) указанная триазолильная составляющая необязательно замещена на атоме углерода группой -NH2, или (g) указанная триазолильная составляющая необязательно замещена на атоме азота группой -CH2CH2OCH3; и, где указанная фенильная составляющая замещена галогеном.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенную триазолилфенил-группу, где (а) указанная триазолильная составляющая замещена на атоме азота заместителем, выбранным из группы, состоящей из: -СН2СОН(CH3)2 и -CH2CH2OH, (b) указанная триазолильная составляющая замещена на атоме азота алкильной группой, (с) указанная триазолильная составляющая замещена на атоме азота алкильной группой и на атоме углерода алкильной группой, (d) указанная триазолильная составляющая замещена на атоме азота группой -CH3, (е) указанная триазолильная составляющая замещена на атоме азота одной группой -CH3 и на атоме углерода одной группой -CH3, (f) указанная триазолильная составляющая замещена на атоме углерода группой -NH2, или (g) указанная триазолильная составляющая замещена на атоме азота группой -CH2CH2OCH3; и, где указанная фенильная составляющая необязательно замещена галогеном.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенную триазолилфенил-группу, где (а) указанная триазолильная составляющая замещена на атоме азота заместителем, выбранным из группы, состоящей из: -СН2СОН(CH3)2 и -CH2CH2OH, (b) указанная триазолильная составляющая замещена на атоме азота алкильной группой, (с) указанная триазолильная составляющая замещена на атоме азота алкильной группой и на атоме углерода алкильной группой, (d) указанная триазолильная составляющая замещена на атоме азота группой -CH3, (е) указанная триазолильная составляющая замещена на атоме азота одной группой -CH3, и на атоме углерода одной группой -CH3, (f) указанная триазолильная составляющая замещена на атоме углерода группой -NH2, или (g) указанная триазолильная составляющая замещена на атоме азота группой -CH2CH2OCH3; и, где указанная фенильная составляющая замещена галогеном.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенную триазолилфенил-группу, где (а) указанная триазолильная составляющая замещена на атоме азота заместителем, выбранным из группы, состоящей из: -СН2СОН(CH3)2 и -CH2CH2OH, (b) указанная триазолильная составляющая замещена на атоме азота алкильной группой, (с) указанная триазолильная составляющая замещена на атоме азота алкильной группой и на атоме углерода алкильной группой, (d) указанная триазолильная составляющая замещена на атоме азота группой -CH3, (е) указанная триазолильная составляющая замещена на атоме азота одной группой -CH3 и на атоме углерода одной группой -CH3, (f) указанная триазолильная составляющая замещена на атоме углерода группой -NH2, или (g) указанная триазолильная составляющая замещена на атоме азота группой -CH2CH2OCH3; и, где указанная фенильная составляющая является незамещенной.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенную триазолилфенил-группу, где (а) указанная триазолильная составляющая необязательно замещена на атоме азота заместителем, выбранным из группы, состоящей из: -СН2СОН(CH3)2 и -CH2CH2OH, (b) указанная триазолильная составляющая необязательно замещена на атоме азота алкильной группой, (с) указанная триазолильная составляющая необязательно замещена на атоме азота алкильной группой и на атоме углерода алкильной группой, (d) указанная триазолильная составляющая необязательно замещена на атоме азота группой -CH3, (е) указанная триазолильная составляющая необязательно замещена на атоме азота одной группой -CH3 и на атоме углерода одной группой -CH3, (f) указанная триазолильная составляющая необязательно замещена на атоме углерода группой -NH2, или (g) указанная триазолильная составляющая необязательно замещена на атоме азота группой -CH3CH3OCH3; и, где указанная фенильная составляющая необязательно замещена алкоксигруппой.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенную триазолилфенил-группу, где (а) указанная триазолильная составляющая необязательно замещена на атоме азота заместителем, выбранным из группы, состоящей из: -СН2СОН(CH3)2 и -CH2CH2OH, (b) указанная триазолильная составляющая необязательно замещена на атоме азота алкильной группой, (с) указанная триазолильная составляющая необязательно замещена на атоме азота алкильной группой и на атоме углерода алкильной группой, (d) указанная триазолильная составляющая необязательно замещена на атоме азота группой -CH3, (е) указанная триазолильная составляющая необязательно замещена на атоме азота одной группой -CH3 и на атоме углерода одной группой -CH3, (f) указанная триазолильная составляющая необязательно замещена на атоме углерода группой -NH2, или (g) указанная триазолильная составляющая необязательно замещена на атоме азота группой -CH2CH2OCH3; и, где указанная фенильная составляющая замещена алкоксигруппой.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенную триазолилфенил-группу, где (а) указанная триазолильная составляющая замещена на атоме азота заместителем, выбранным из группы, состоящей из: -СН2СОН(CH3)2 и -CH2CH2OH, (b) указанная триазолильная составляющая замещена на атоме азота алкильной группой, (с) указанная триазолильная составляющая замещена на атоме азота алкильной группой и на атоме углерода алкильной группой, (d) указанная триазолильная составляющая замещена на атоме азота группой -CH3, (е) указанная триазолильная составляющая замещена на атоме азота одной группой -CH3 и на атоме углерода одной группой -CH3, (f) указанная триазолильная составляющая замещена на атоме углерода группой -NH2, или (g) указанная триазолильная составляющая замещена на атоме азота группой -CH2CH2OCH3; и, где указанная фенильная составляющая необязательно замещена алкоксигруппой.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенную триазолилфенил-группу, где (а) указанная триазолильная составляющая замещена на атоме азота заместителем, выбранным из группы, состоящей из: -СН2СОН(CH3)2 и -CH2CH2OH, (b) указанная триазолильная составляющая замещена на атоме азота алкильной группой, (с) указанная триазолильная составляющая замещена на атоме азота алкильной группой и на атоме углерода алкильной группой, (d) указанная триазолильная составляющая замещена на атоме азота группой -CH3, (е) указанная триазолильная составляющая замещена на атоме азота одной группой -CH3 и на атоме углерода одной группой -CH3, (f) указанная триазолильная составляющая замещена на атоме углерода группой -NH2, или (g) указанная триазолильная составляющая замещена на атоме азота -CH2CH2OCH3 группой; и, где указанная фенильная составляющая замещена ал кокси группой.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенную триазолилтиенил-группу, где (а) указанная триазолильная составляющая необязательно замещена на атоме азота заместителем, выбранным из группы, состоящей из: -СН2СОН(CH3)2 и -CH2CH2OH, (b) указанная триазолильная составляющая необязательно замещена на атоме азота алкильной группой, (с) указанная триазолильная составляющая необязательно замещена на атоме азота алкильной группой и на атоме углерода алкильной группой, (d) указанная триазолильная составляющая необязательно замещена на атоме азота группой -CH3, (е) указанная триазолильная составляющая необязательно замещена на атоме азота одной группой -CH3 и на атоме углерода одной группой -CH3, (f) указанная триазолильная составляющая необязательно замещена на атоме углерода группой -NH2, или (g) указанная триазолильная составляющая необязательно замещена на атоме азота -CH2CH2OCH3 группой; и, где указанная тиенильная составляющая необязательно замещена галогеном.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенную триазолилтиенил-группу, где (а) указанная триазолильная составляющая необязательно замещена на атоме азота заместителем, выбранным из группы, состоящей из: -СН2СОН(CH3)2 и -CH2CH2OH, (b) указанная триазолильная составляющая необязательно замещена на атоме азота алкильной группой, (с) указанная триазолильная составляющая необязательно замещена на атоме азота алкильной группой и на атоме углерода алкильной группой, (d) указанная триазолильная составляющая необязательно замещена на атоме азота группой -CH3, (е) указанная триазолильная составляющая необязательно замещена на атоме азота одной -CH3 группой и на атоме углерода одной -CH3 группой, (f) указанная триазолильная составляющая необязательно замещена на атоме углерода группой -NH2, или (g) указанная триазолильная составляющая необязательно замещена на атоме азота группой -CH2CH2OCH3; и, где указанная тиенильная составляющая замещена галогеном.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенную триазолилтиенил-группу, где (а) указанная триазолильная составляющая замещена на атоме азота заместителем, выбранным из группы, состоящей из: -СН2СОН(CH3)2 и -CH2CH2OH, (b) указанная триазолильная составляющая замещена на атоме азота алкильной группой, (с) указанная триазолильная составляющая замещена на атоме азота алкильной группой и на атоме углерода алкильной группой, (d) указанная триазолильная составляющая замещена на атоме азота группой -CH3, (е) указанная триазолильная составляющая замещена на атоме азота одной группой -CH3 и на атоме углерода одной группой -CH3, (f) указанная триазолильная составляющая замещена на атоме углерода группой -NH2, или (g) указанная триазолильная составляющая замещена на атоме азота -CH2CH2OCH3 группой; и, где указанная тиенильная составляющая необязательно замещена галогеном.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенную триазолилтиенил-группу, где (а) указанная триазолильная составляющая замещена на атоме азота заместителем, выбранным из группы, состоящей из: -СН2СОН(CH3)2 и -CH2CH2OH, (b) указанная триазолильная составляющая замещена на атоме азота алкильной группой, (с) указанная триазолильная составляющая замещена на атоме азота алкильной группой и на атоме углерода алкильной группой, (d) указанная триазолильная составляющая замещена на атоме азота группой -CH3, (е) указанная триазолильная составляющая замещена на атоме азота одной группой -CH3 и на атоме углерода одной группой -CH3, (f) указанная триазолильная составляющая замещена на атоме углерода группой -NH2, или (g) указанная триазолильная составляющая замещена на атоме азота -CH2CH2OCH3 группой; и, где указанная тиенильная составляющая замещена галогеном.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенную триазолилтиенил-группу, где (а) указанная триазолильная составляющая замещена на атоме азота заместителем, выбранным из группы, состоящей из: -СН2СОН(CH3)2 и -CH2CH2OH, (b) указанная триазолильная составляющая замещена на атоме азота алкильной группой, (с) указанная триазолильная составляющая замещена на атоме азота алкильной группой и на атоме углерода алкильной группой, (d) указанная триазолильная составляющая замещена на атоме азота группой -CH3, (е) указанная триазолильная составляющая замещена на атоме азота одной группой -CH3, и на атоме углерода одной группой -CH3, (f) указанная триазолильная составляющая замещена на атоме углерода группой -NH2, или (g) указанная триазолильная составляющая замещена на атоме азота -CH2CH2OCH3 группой; и, где указанная тиенильная составляющая является незамещенной.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенную триазолилтиенил-группу, где (а) указанная триазолильная составляющая необязательно замещена на атоме азота заместителем, выбранным из группы, состоящей из: -СН2СОН(CH3)2 и -CH2CH2OH, (b) указанная триазолильная составляющая необязательно замещена на атоме азота алкильной группой, (с) указанная триазолильная составляющая необязательно замещена на атоме азота алкильной группой и на атоме углерода алкильной группой, (d) указанная триазолильная составляющая необязательно замещена на атоме азота группой -CH3, (е) указанная триазолильная составляющая необязательно замещена на атоме азота одной группой -CH3 и на атоме углерода одной группой -CH3, (f) указанная триазолильная составляющая необязательно замещена на атоме углерода группой -NH2, или (g) указанная триазолильная составляющая необязательно замещена на атоме азота -CH2CH2OCH3 группой; и, где указанная тиенильная составляющая необязательно замещена алкоксигруппой.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенную триазолилтиенил-группу, где (а) указанная триазолильная составляющая необязательно замещена на атоме азота заместителем, выбранным из группы, состоящей из: -СН2СОН(CH3)2 и -CH2CH2OH, (b) указанная триазолильная составляющая необязательно замещена на атоме азота алкильной группой, (с) указанная триазолильная составляющая необязательно замещена на атоме азота алкильной группой и на атоме углерода алкильной группой, (d) указанная триазолильная составляющая необязательно замещена на атоме азота группой -CH3, (е) указанная триазолильная составляющая необязательно замещена на атоме азота одной группой -CH3 и на атоме углерода одной группой -CH3, (f) указанная триазолильная составляющая необязательно замещена на атоме углерода группой -NH2, или (g) указанная триазолильная составляющая необязательно замещена на атоме азота -CH2CH2OCH3 группой; и, где указанная тиенильная составляющая замещена алкоксигруппой.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенную триазолилтиенил-группу, где (а) указанная триазолильная составляющая замещена на атоме азота заместителем, выбранным из группы, состоящей из: -СН2СОН(CH3)2 и -CH2CH2OH, (b) указанная триазолильная составляющая замещена на атоме азота алкильной группой, (с) указанная триазолильная составляющая замещена на атоме азота алкильной группой и на атоме углерода алкильной группой, (d) указанная триазолильная составляющая замещена на атоме азота группой -CH3, (е) указанная триазолильная составляющая замещена на атоме азота одной группой -CH3 и на атоме углерода одной группой -CH3, (f) указанная триазолильная составляющая замещена на атоме углерода группой -NH2, или (g) указанная триазолильная составляющая замещена на атоме азота -CH2CH2OCH3 группой; и, где указанная тиенильная составляющая необязательно замещена алкоксигруппой.
В другом варианте выполнения настоящего изобретения R5 представляет собой замещенную триазолилтиенил-группу, где (а) указанная триазолильная составляющая замещена на атоме азота заместителем, выбранным из группы, состоящей из: -СН2СОН(CH3)2 и -CH2CH2OH, (b) указанная триазолильная составляющая замещена на атоме азота алкильной группой, (с) указанная триазолильная составляющая замещена на атоме азота алкильной группой и на атоме углерода алкильной группой, (d) указанная триазолильная составляющая замещена на атоме азота группой -CH3, (е) указанная триазолильная составляющая замещена на атоме азота одной группой -CH3 и на атоме углерода одной группой -CH3, (f) указанная триазолильная составляющая замещена на атоме углерода -NH2 группой, или (g) указанная триазолильная составляющая замещена на атоме азота -CH2CH2OCH3 группой; и, где указанная тиенильная составляющая замещена алкоксигруппой.
Другие варианты выполнения настоящего изобретения описаны ниже. Варианты выполнения настоящего изобретения были пронумерованы, для того, чтобы на них было проще ссылаться.
Вариант выполнения изобретения №1 представляет собой соединения формулы 1.0, где R2 представляет собой -O-(С1-С2)алкильную группу, и R1 замещен пиридилом.
Вариант выполнения изобретения №2 представляет собой соединения формулы 1.0, где R2 представляет собой -O-(С1-С2)алкильную группу, и R1 представляет собой пиридил, замещенный одним заместителем.
Вариант выполнения изобретения №3 представляет собой соединения формулы 1.0, где R2 представляет собой -O-(С1-С2)алкильную группу, и R1 выбирается из группы, состоящей из:
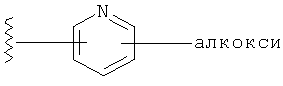 и
и 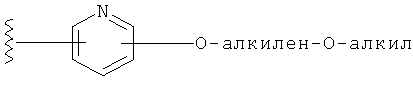 .
.
Вариант выполнения изобретения №4 представляет собой соединения формулы 1.0, где R2 представляет собой -O-(C1-C2) алкильную группу, и R1 представляет собой:
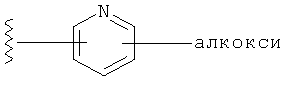 .
.
Вариант выполнения изобретения №5 представляет собой соединения формулы 1.0, где R2 представляет собой -O-(С1-С2)алкильную группу, и R1 представляет собой:
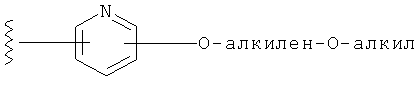
Вариант выполнения изобретения №6 представляет собой соединения формулы 1.0, где R2 представляет собой -O-(С1-С2)алкильную группу, и R1 представляет собой:
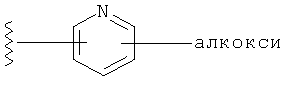 , где указанная алкоксигруппа представляет собой -ОСН(CH3)2.
, где указанная алкоксигруппа представляет собой -ОСН(CH3)2.
Вариант выполнения изобретения №7 представляет собой соединения формулы 1.0, где R2 представляет собой -O-(C1-C2) алкильную группу, и R1 представляет собой:
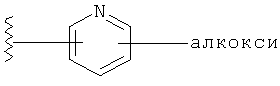 , где указанная алкоксигруппа представляет собой -ОС2Н5.
, где указанная алкоксигруппа представляет собой -ОС2Н5.
Вариант выполнения изобретения №8 представляет собой соединения формулы 1.0, где R2 представляет собой -O-(C1-C2) алкильную группу, и R1 представляет собой:
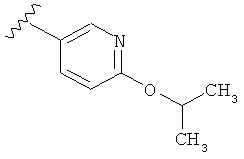 .
.
Вариант выполнения изобретения №9 представляет собой соединения формулы 1.0, где R2 представляет собой -O-(C1-C2) алкильную группу, и R1 представляет собой:
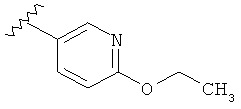 .
.
Вариант выполнения изобретения №10 представляет собой соединения формулы 1.0, где z равно 1, R2 представляет собой -O-(C1-C2) алкильную группу, и R1 представляет собой:
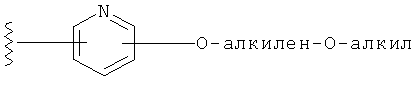 , где указанная -О-алкилен-O-алкильная группа представляет собой -OCH2CH2OCH3.
, где указанная -О-алкилен-O-алкильная группа представляет собой -OCH2CH2OCH3.
Вариант выполнения изобретения №11 представляет собой соединения формулы 1.0, где R2 представляет собой -O-(С1-С2)алкильную группу, и R1 представляет собой:
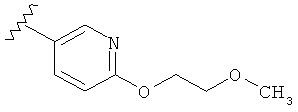 .
.
Вариант выполнения изобретения №12 представляет собой соединения формулы 1.0, где R2 представляет собой -OCH3, и R1 замещен пиридил.
Вариант выполнения изобретения №13 представляет собой соединения формулы 1.0, где R2 представляет собой -OCH3, и R1 представляет собой пиридил, замещенный одним заместителем.
Вариант выполнения изобретения №14 представляет собой соединения формулы 1.0, где R2 представляет собой -OCH3, и R1 выбирается из группы, состоящей из:
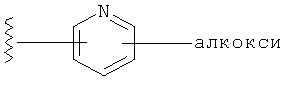 и
и 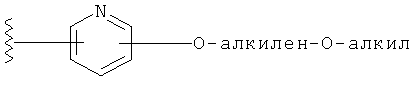 .
.
Вариант выполнения изобретения №15 представляет собой соединения формулы 1.0, где R2 представляет собой -OCH3, и R1 представляет собой:
.
Вариант выполнения изобретения №16 представляет собой соединения формулы 1.0, где R2 представляет собой -OCH3, и R1 представляет собой;
.
Вариант выполнения изобретения №17 представляет собой соединения формулы 1.0, где R2 представляет собой -OCH3, и R1 представляет собой:
, где указанная алкоксигруппа представляет собой -ОСН(CH3)2.
Вариант выполнения изобретения №18 представляет собой соединения формулы 1.0, где R2 представляет собой -OCH3, и R1 представляет собой:
, где указанная алкоксигруппа представляет собой -ОС2Н5.
Вариант выполнения изобретения №19 представляет собой соединения формулы 1.0, где R2 представляет собой -OCH3, и R1 представляет собой:
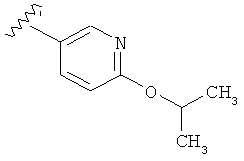 .
.
Вариант выполнения изобретения №20 представляет собой соединения формулы 1.0, где R2 представляет собой -OCH3, и R1 представляет собой:
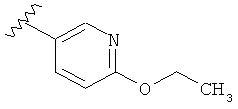 .
.
Вариант выполнения изобретения №21 представляет собой соединения формулы 1.0, где R2 представляет собой -OCH3, и R1 представляет собой:
, где указанная -О-алкилен-O-алкильная группа представляет собой -OCH2CH2OCH3.
Вариант выполнения изобретения №22 представляет собой соединения формулы 1.0, где R2 представляет собой -OCH3, и R1 представляет собой:
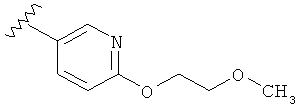 .
.
Вариант выполнения изобретения №23 представляет собой соединения формулы 1.0, где R2 представляет собой -S-(С1-С2)алкильную группу, и R1 замещен пиридилом.
Вариант выполнения изобретения №24 представляет собой соединения формулы 1.0, где R2 представляет собой -S-(С1-С2)алкильную группу, и R1 представляет собой пиридил, замещенный одним заместителем.
Вариант выполнения изобретения №25 представляет собой соединения формулы 1.0, где R2 представляет собой -S-(С1-С2)алкильную группу, и R1 выбирается из группы, состоящей из:
и .
Вариант выполнения изобретения №26 представляет собой соединения формулы 1.0, где R2 представляет собой -S-(С1-С2)алкильную группу, и R1 представляет собой:
.
Вариант выполнения изобретения №27 представляет собой соединения формулы 1.0, где R2 представляет собой -S-(С1-С2)алкильную группу, и R1 представляет собой:
.
Вариант выполнения изобретения №28 представляет собой соединения формулы 1.0, где R2 представляет собой -S-(С1-С2)алкильную группу, и R1 представляет собой:
, где указанная алкоксигруппа представляет собой -ОСН(CH3)2.
Вариант выполнения изобретения №29 представляет собой соединения формулы 1.0, где R2 представляет собой -S-(C1-C2) алкильную группу, и R1 представляет собой:
, где указанная алкоксигруппа представляет собой -ОС2Н5.
Вариант выполнения изобретения №30 представляет собой соединения формулы 1.0, где R2 представляет собой -S-(C1-C2) алкильную группу, и R1 представляет собой:
.
Вариант выполнения изобретения №31 представляет собой соединения формулы 1.0, где R2 представляет собой -S-(С1-С2)алкильную группу, и R1 представляет собой:
.
Вариант выполнения изобретения №32 представляет собой соединения формулы 1.0, где R2 представляет собой -S-(С1-С2)алкильную группу, и R1 представляет собой:
, где указанная -О-алкилен-O-алкильная группа представляет собой -OCH2CH2OCH3.
Вариант выполнения изобретения №33 представляет собой соединения формулы 1.0, где R2 представляет собой -S-(C1-C2) алкильную группу, и R1 представляет собой:
.
Вариант выполнения изобретения №34 представляет собой соединения формулы 1.0, где R2 представляет собой -SCH3, и R1 замещен пиридилом.
Вариант выполнения изобретения №35 представляет собой соединения формулы 1.0, где R2 представляет собой -SCH3, и R1 представляет собой пиридил, замещенный одним заместителем.
Вариант выполнения изобретения №36 представляет собой соединения формулы 1.0, где R2 представляет собой -SCH3, и R1 выбирается из группы, состоящей из:
и .
Вариант выполнения изобретения №37 представляет собой соединения формулы 1.0, где R2 представляет собой -SCH3, и R1 представляет собой:
.
Вариант выполнения изобретения №38 представляет собой соединения формулы 1.0, где R2 представляет собой -SCH3, и R1 представляет собой:
.
Вариант выполнения изобретения №39 представляет собой соединения формулы 1.0, где R2 представляет собой -SCH3, и R1 представляет собой:
, где указанная алкоксигруппа представляет собой -ОСН(CH3)2.
Вариант выполнения изобретения №40 представляет собой соединения формулы 1.0, где R2 представляет собой -SCH3, и R1 представляет собой:
, где указанная алкоксигруппа представляет собой -ОС2Н5.
Вариант выполнения изобретения №41 представляет собой соединения формулы 1.0, где R2 представляет собой -SCH3, и R1 представляет собой:
.
Вариант выполнения изобретения №42 представляет собой соединения формулы 1.0, где R2 представляет собой -SCH3, и R1 представляет собой:
.
Вариант выполнения изобретения №43 представляет собой соединения формулы 1.0, где R2 представляет собой -SCH3, и R1 представляет собой:
, где указанная -О-алкилен-O-алкильная группа представляет собой -OCH2CH2OCH3.
Вариант выполнения изобретения №44 представляет собой соединения формулы 1.0, где R2 представляет собой -SCH3, и R1 представляет собой:
.
Вариант выполнения изобретения №45 представляет собой соединения формулы 1.0, имеющие формулу 1.1:
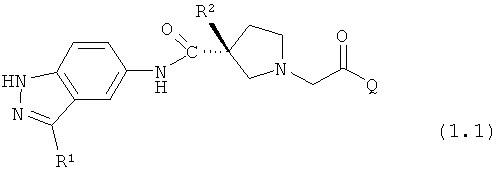
Вариант выполнения изобретения №46 представляет собой любой из вариантов выполнения изобретения №1-44, где соединение формулы 1.0 представляет собой соединение формулы 1.1.
Другие варианты выполнения настоящего изобретения представляют собой соединения, где R5 заместитель описан ниже. Фраза "как описано в любом из вариантов выполнения изобретения №1-46" означает, что вариант выполнения изобретения раскрывается как подходящий для каждого из вариантов выполнения изобретения №1-46. Например, другой вариант выполнения настоящего изобретения представляет собой соединения, описанные в варианте выполнения изобретения №1, где R5 описывается в любом из приведенных ниже абзацев. В другом примере, другой вариант выполнения настоящего изобретения представляет собой соединения, описанные в варианте выполнения изобретения №2, где R5 описывается в любом из приведенных ниже абзацев, и так далее.
Таким образом, другие варианты выполнения настоящего изобретения представляют собой соединения формулы 1.0, как описано в любом из вариантов выполнения изобретения №1-46, где R5 представляет собой замещенный триазолилфенил-, где триазолильная составляющая замещена одной или двумя алкильными группами, выбранными из группы, состоящей из: -C1-C6алкила, -С1-С4алкила, -С1-С2алкила и -CH3.
Другие варианты выполнения настоящего изобретения представляют собой соединения формулы 1.0, как описано в любом из вариантов выполнения изобретения №1-46, где R5 представляет собой замещенный триазолилфенил-, где триазолильная составляющая замещена одной или двумя алкильными группами, выбранными из группы, состоящей из: -С1-С4алкила, -С1-С2алкила и -CH3. Другие варианты выполнения настоящего изобретения представляют собой соединения формулы 1.0, как описано в любом из вариантов выполнения изобретения №1-46, где R5 представляет собой замещенный триазолилфенил-, где триазолильная составляющая замещена одной или двумя -CH3 группами.
Другие варианты выполнения настоящего изобретения представляют собой соединения формулы 1.0, как описано в любом из вариантов выполнения изобретения №1-46, где R5 представляет собой замещенный триазолилфенил-, где триазолильная составляющая замещена на атоме азота алкильной группой, выбранной из группы, состоящей из: -С1-С4алкила, -С1-С2алкила и -CH3. Другие варианты выполнения настоящего изобретения представляют собой соединения формулы 1.0, как описано в любом из вариантов выполнения изобретения №1-46, где R5 представляет собой замещенный триазолилфенил-, где триазолильная составляющая замещена на атоме азота группой -CH3.
Другие варианты выполнения настоящего изобретения представляют собой соединения формулы 1.0, как описано в любом из вариантов выполнения изобретения №1-46, где R5 представляет собой замещенный триазолилфенил-, где триазолильная составляющая замещена на атоме азота -алкилен-O-алкильной группой, выбранной из группы, состоящей из: -C2-С4алкилен-O-С1-С6алкила, -С2алкилен-O-С1-С2алкила, -С2-С4алкилен-O-CH3 и -CH2CH2OCH3.
Другие варианты выполнения настоящего изобретения представляют собой соединения формулы 1.0, как описано в любом из вариантов выполнения изобретения №1-46, где R5 представляет собой замещенный триазолилфенил-, где триазолильная составляющая замещена на атоме азота -алкилен-O-алкильной группой, выбранной из группы, состоящей из: -С2алкилен-O-С1-С2алкила и -CH2CH2OCH3. Другие варианты выполнения настоящего изобретения представляют собой соединения формулы 1.0, как описано в любом из вариантов выполнения изобретения №1-46, где R5 представляет собой замещенный триазолилфенил-, где триазолильная составляющая замещена на атоме азота группой -CH2CH2OCH3.
Другие варианты выполнения настоящего изобретения представляют собой соединения формулы 1.0, как описано в любом из вариантов выполнения изобретения №1-46, где R5 представляет собой замещенный триазолилфенил-, где триазолильная составляющая замещена на атоме азота гидроксизамещенной алкильной группой, выбранной из группы, состоящей из: гидроксизамещенного -С1-C4алкила, гидроксизамещенного -C1-С2алкила и гидроксизамещенного -CH3.
Другие варианты выполнения настоящего изобретения представляют собой соединения формулы 1.0, как описано в любом из вариантов выполнения изобретения №1-46, где R5 представляет собой замещенный триазолилфенил-, где триазолильная составляющая замещена на атоме азота гидроксизамещенной алкильной группой выбранной из группы, состоящей из: СН2СОН(CH3)2 и -CH2CH2OH.
Другие варианты выполнения настоящего изобретения представляют собой соединения формулы 1.0, как описано в любом из вариантов выполнения изобретения №1-46, где необязательные галогеновые заместители для фенильной составляющей R5 независимо выбираются из группы, состоящей из: Cl, F и BR.
Другие варианты выполнения настоящего изобретения представляют собой соединения формулы 1.0, как описано в любом из вариантов выполнения изобретения №1-46, где необязательный галогеновый заместитель для фенильной составляющей R5 представляет собой F.
Другие варианты выполнения настоящего изобретения представляют собой соединения формулы 1.0, как описано в любом из вариантов выполнения изобретения №1-46, где необязательный галогеновый заместитель для фенильной составляющей R5 представляет собой один F.
Другие варианты выполнения настоящего изобретения представляют собой соединения формулы 1.0, как описано в любом из вариантов выполнения изобретения №1-46, где R5 представляет собой замещенную триазолилфенил-группу, где (а) указанная триазолильная составляющая необязательно замещена на атоме азота заместителем, выбранным из группы, состоящей из: -СН2СОН(CH3)2 и -CH2CH2OH, (b) указанная триазолильная составляющая необязательно замещена на атоме азота алкильной группой, (с) указанная триазолильная составляющая необязательно замещена на атоме азота алкильной группой и на атоме углерода алкильной группой, (d) указанная триазолильная составляющая необязательно замещена на атоме азота группой -CH3, (е) указанная триазолильная составляющая необязательно замещена на атоме азота одной -CH3 группой и на атоме углерода одной -CH3 группой, (f) указанная триазолильная составляющая необязательно замещена на атоме углерода -NH2 группой, или (g) указанная триазолильная составляющая необязательно замещена на атоме азота -CH2CH2OCH3 группой; и, где указанная фенильная составляющая необязательно замещена галогеном (например, одним галогеном, таким как, например, F).
Другие варианты выполнения настоящего изобретения представляют собой соединения формулы 1.0, как описано в любом из вариантов выполнения изобретения №1-46, где R5 представляет собой замещенную триазолилфенил- группу, где (а) указанная триазолильная составляющая необязательно замещена на атоме азота заместителем, выбранным из группы, состоящей из: -СН2СОН(CH3)2 и -CH2CH2OH, (b) указанная триазолильная составляющая необязательно замещена на атоме азота алкильной группой, (с) указанная триазолильная составляющая необязательно замещена на атоме азота алкильной группой и на атоме углерода алкильной группой, (d) указанная триазолильная составляющая необязательно замещена на атоме азота -CH3 группой, (е) указанная триазолильная составляющая необязательно замещена на атоме азота одной -CH3 группой и на атоме углерода одной -CH3 группой, (f) указанная триазолильная составляющая необязательно замещена на атоме углерода -NH2 группой, или (g) указанная триазолильная составляющая необязательно замещена на атоме азота -CH2CH2OCH3 группой; и, где указанная фенильная составляющая замещена галогеном (например, одним галогеном, таким как, например, F).
Другие варианты выполнения настоящего изобретения представляют собой соединения формулы 1.0, как описано в любом из вариантов выполнения изобретения №1-46, где R5 представляет собой замещенную триазолилфенил- группу, где (а) указанная триазолильная составляющая замещена на атоме азота заместителем, выбранным из группы, состоящей из: -СН2СОН(CH3)2 и -CH2CH2OH, (b) указанная триазолильная составляющая замещена на атоме азота алкильной группой, (с) указанная триазолильная составляющая замещена на атоме азота алкильной группой и на атоме углерода алкильной группой, (d) указанная триазолильная составляющая замещена на атоме азота -CH3 группой, (е) указанная триазолильная составляющая замещена на атоме азота одной -CH3 группой и на атоме углерода одной -CH3 группой, (f) указанная триазолильная составляющая замещена на атоме углерода -NH2 группой, или (g) указанная триазолильная составляющая замещена на атоме азота -CH2CH2OCH3 группой; и, где указанная фенильная составляющая необязательно замещена галогеном (например, одним галогеном, таким как, например, F).
Другие варианты выполнения настоящего изобретения представляют собой соединения формулы 1.0, как описано в любом из вариантов выполнения изобретения №1-46, где R5 представляет собой замещенную триазолилфенил-группу, где (а) указанная триазолильная составляющая замещена на атоме азота заместителем, выбранным из группы, состоящей из: -СН2СОН(CH3)2 и -CH2CH2OH, (b) указанная триазолильная составляющая замещена на атоме азота алкильной группой, (с) указанная триазолильная составляющая замещена на атоме азота алкильной группой и на атоме углерода алкильной группой, (d) указанная триазолильная составляющая замещена на атоме азота -CH3 группой, (е) указанная триазолильная составляющая замещена на атоме азота одной -CH3 группой и на атоме углерода одной -CH3 группой, (f) указанная триазолильная составляющая замещена на атоме углерода -NH2 группой, или (g) указанная триазолильная составляющая замещена на атоме азота -CH2CH2OCH3 группой; и, где указанная фенильная составляющая замещена галогеном (например, одним галогеном, таким как, например, F).
Другие варианты выполнения настоящего изобретения представляют собой соединения формулы 1.0, как описано в любом из вариантов выполнения изобретения №1-46, где необязательные заместители для фенильной составляющей R5 независимо выбираются из группы, состоящей из: алкокси.
Другие варианты выполнения настоящего изобретения представляют собой соединения формулы 1.0, как описано в любом из вариантов выполнения изобретения №1-46, где необязательный заместитель для фенильной составляющей R5 представляет собой группу -OCH3.
Другие варианты выполнения настоящего изобретения представляют собой соединения формулы 1.0, как описано в любом из вариантов выполнения изобретения №1-46, где необязательный заместитель для фенильной составляющей R5 представляет собой одну группу -OCH3.
Другие варианты выполнения настоящего изобретения представляют собой соединения формулы 1.0, как описано в любом из вариантов выполнения изобретения №1-46, где R5 представляет собой замещенную триазолилфенил- группу, где (а) указанная триазолильная составляющая необязательно замещена на атоме азота заместителем, выбранным из группы, состоящей из: -СН2СОН(CH3)2 и -CH2CH2OH, (b) указанная триазолильная составляющая необязательно замещена на атоме азота алкильной группой, (с) указанная триазолильная составляющая необязательно замещена на атоме азота алкильной группой и на атоме углерода алкильной группой, (d) указанная триазолильная составляющая необязательно замещена на атоме азота -CH3 группой, (е) указанная триазолильная составляющая необязательно замещена на атоме азота одной -CH3 группой и на атоме углерода одной -CH3 группой, (f) указанная триазолильная составляющая необязательно замещена на атоме углерода -NH2 группой, или (g) указанная триазолильная составляющая необязательно замещена на атоме азота -CH3CH2OCH3 группой; и, где указанная фенильная составляющая необязательно замещена алкоксигруппой (например, одной группой -OCH3).
Другие варианты выполнения настоящего изобретения представляют собой соединения формулы 1.0, как описано в любом из вариантов выполнения изобретения №1-46, где R5 представляет собой замещенную триазолилфенил- группу, где (а) указанная триазолильная составляющая необязательно замещена на атоме азота заместителем, выбранным из группы, состоящей из: -СН2СОН(CH3)2 и -CH2CH2OH, (b) указанная триазолильная составляющая необязательно замещена на атоме азота алкильной группой, (с) указанная триазолильная составляющая необязательно замещена на атоме азота алкильной группой и на атоме углерода алкильной группой, (d) указанная триазолильная составляющая необязательно замещена на атоме азота -CH3 группой, (е) указанная триазолильная составляющая необязательно замещена на атоме азота одной -CH3 группой и на атоме углерода одной -CH3 группой, (f) указанная триазолильная составляющая необязательно замещена на атоме углерода -NH2 группой, или (g) указанная триазолильная составляющая необязательно замещена на атоме азота -CH3CH3OCH3 группой; и, где указанная фенильная составляющая замещена алкоксигруппой (например, одной группой -OCH3).
Другие варианты выполнения настоящего изобретения представляют собой соединения формулы 1.0, как описано в любом из вариантов выполнения изобретения №1-46, где R5 представляет собой замещенную триазолилфенил- группу, где (а) указанная триазолильная составляющая замещена на атоме азота заместителем, выбранным из группы, состоящей из: -СН2СОН(CH3)2 и -CH3CH2OH, (b) указанная триазолильная составляющая замещена на атоме азота алкильной группой, (с) указанная триазолильная составляющая замещена на атоме азота алкильной группой и на атоме углерода алкильной группой, (d) указанная триазолильная составляющая замещена на атоме азота -CH3 группой, (е) указанная триазолильная составляющая замещена на атоме азота одной -CH3 группой и на атоме углерода одной -CH3 группой, (f) указанная триазолильная составляющая замещена на атоме углерода -NH2 группой, или (g) указанная триазолильная составляющая замещена на атоме азота -CH2CH2OCH3 группой; и, где указанная фенильная составляющая необязательно замещена алкоксигруппой (например, одной группой -OCH3).
Другие варианты выполнения настоящего изобретения представляют собой соединения формулы 1.0, как описано в любом из вариантов выполнения изобретения №1-46, где R5 представляет собой замещенную триазолилфенил- группу, где (а) указанная триазолильная составляющая замещена на атоме азота заместителем, выбранным из группы, состоящей из: -СН2СОН(CH3)2 и -CH2CH2OH, (b) указанная триазолильная составляющая замещена на атоме азота алкильной группой, (с) указанная триазолильная составляющая замещена на атоме азота алкильной группой и на атоме углерода алкильной группой, (d) указанная триазолильная составляющая замещена на атоме азота -CH3 группой, (е) указанная триазолильная составляющая замещена на атоме азота одной -CH3 группой и на атоме углерода одной -CH3 группой, (f) указанная триазолильная составляющая замещена на атоме углерода -NH2 группой, или (g) указанная триазолильная составляющая замещена на атоме азота -CH2CH2OCH3 группой; и, где указанная фенильная составляющая замещена алкокси-группой (например, одной группой -OCH3).
Другие варианты выполнения настоящего изобретения представляют собой соединения формулы 1.0, как описано в любом из вариантов выполнения изобретения №1-46, где R5 представляет собой замещенную триазолилфенил- группу, где (а) указанная триазолильная составляющая замещена на атоме азота заместителем, выбранным из группы, состоящей из: -СН2СОН(CH3)2 и -CH2CH2OH, (b) указанная триазолильная составляющая замещена на атоме азота алкильной группой, (с) указанная триазолильная составляющая замещена на атоме азота алкильной группой и на атоме углерода алкильной группой, (d) указанная триазолильная составляющая замещена на атоме азота -CH3 группой, (е) указанная триазолильная составляющая замещена на атоме азота одной -CH3 группой и на атоме углерода одной -CH3 группой, (f) указанная триазолильная составляющая замещена на атоме углерода -NH2 группой, или (g) указанная триазолильная составляющая замещена на атоме азота -CH2CH2OCH3 группой; и, где указанная фенильная составляющая является незамещенной.
Другие варианты выполнения настоящего изобретения представляют собой соединения формулы 1.0, как описано в любом из вариантов выполнения изобретения №1-46, где указанный замещенный R5 составляющая представляет собой замещенный триазолилфенил, где указанная триазолильная составляющая замещена: (а) одним заместителем, выбранным из группы, состоящей из: -СН2СОН(CH3)2 и -CH2CH3OH, (b) одной алкильной группой, (с) двумя алкильными группами, (d) одной -CH3 группой, (е) двумя -CH3 группами, (f) одной -NH2 группой или (g) одной -CH2CH2OCH3 группой.
Другие варианты выполнения настоящего изобретения представляют собой соединения формулы 1.0, как описано в любом из вариантов выполнения изобретения №1-46, где R5 представляет собой замещенный триазолилтиенил-, где триазолильная составляющая замещена одной или двумя алкильными группами, выбранными из группы, состоящей из: -C1-C6алкила, -С1-С4алкиал, -С1-С2алкила и -CH3.
Другие варианты выполнения настоящего изобретения представляют собой соединения формулы 1.0, как описано в любом из вариантов выполнения изобретения №1-46, где R5 представляет собой замещенный триазолилтиенил-, где триазолильная составляющая замещена одной или двумя алкильными группами, выбранными из группы, состоящей из: -C1-С4алкила, -С1-С2алкила и -CH3. Другие варианты выполнения настоящего изобретения представляют собой соединения формулы 1.0, как описано в любом из вариантов выполнения изобретения №1-46, где R5 представляет собой замещенный триазолилтиенил-, где триазолильная составляющая замещена одной или двумя -CH3 группами.
Другие варианты выполнения настоящего изобретения представляют собой соединения формулы 1.0, как описано в любом из вариантов выполнения изобретения №1-46, где R5 представляет собой замещенный триазолилтиенил-, где триазолильная составляющая замещена на атоме азота алкильной группой, выбранной из группы, состоящей из: -С1-С4алкил, -C1-С2алкил и -CH3. Другие варианты выполнения настоящего изобретения представляют собой соединения формулы 1.0, как описано в любом из вариантов выполнения изобретения №1-46, где R5 представляет собой замещенный триазолилтиенил-, где триазолильная составляющая замещена на атоме азота группой -CH3.
Другие варианты выполнения настоящего изобретения представляют собой соединения формулы 1.0, как описано в любом из вариантов выполнения изобретения №1-46, где R5 представляет собой замещенный триазолилтиенил-, где триазолильная составляющая замещена на атоме азота -алкилен-O-алкильной группой, выбранной из группы, состоящей из: -C2-С4алкилен-O-С1-С6алкила, -С2алкилен-O-С1-С2алкила, -С2-С4алкилен-O-CH3 и -CH2CH2OCH3.
Другие варианты выполнения настоящего изобретения представляют собой соединения формулы 1.0, как описано в любом из вариантов выполнения изобретения №1-46, где R5 представляет собой замещенный триазолилтиенил-, где триазолильная составляющая замещена на атоме азота -алкилен-O-алкильной группой, выбранной из группы, состоящей из: -С2алкилен-O-С1-С2алкила и -CH2CH2OCH3. Другие варианты выполнения настоящего изобретения представляют собой соединения формулы 1.0, как описано в любом из вариантов выполнения изобретения №1-46, где R5 представляет собой замещенный триазолилтиенил-, где триазолильная составляющая замещена на атоме азота группой -CH2CH2OCH3.
Другие варианты выполнения настоящего изобретения представляют собой соединения формулы 1.0, как описано в любом из вариантов выполнения изобретения №1-46, где R5 представляет собой замещенный триазолилтиенил-, где триазолильная составляющая замещена на атоме азота гидроксизамещенной алкильной группой, выбранной из группы, состоящей из: гидроксизамещенного -С1-С4алкила, гидроксизамещенного -C1-С2алкила и гидроксизамещенной группы -CH3.
Другие варианты выполнения настоящего изобретения представляют собой соединения формулы 1.0, как описано в любом из вариантов выполнения изобретения №1-46, где R5 представляет собой замещенный триазолилтиенил-, где триазолильная составляющая замещена на атоме азота гидроксизамещенной алкильной группой, выбранной из группы, состоящей из: СН2СОН(CH3)2 и -CH2CH2OH.
Другие варианты выполнения настоящего изобретения представляют собой соединения формулы 1.0, как описано в любом из вариантов выполнения изобретения №1-46, где необязательные галогеновые заместители для тиенильной составляющей R5 независимо выбираются из группы, состоящей из: Cl, F и Br.
Другие варианты выполнения настоящего изобретения представляют собой соединения формулы 1.0, как описано в любом из вариантов выполнения изобретения №1-46, где необязательные галогеновые заместители для тиенильной составляющей R5 представляют собой F.
Другие варианты выполнения настоящего изобретения представляют собой соединения формулы 1.0, как описано в любом из вариантов выполнения изобретения №1-46, где необязательный галогеновый заместитель для тиенильной составляющей R5 представляет собой один F.
Другие варианты выполнения настоящего изобретения представляют собой соединения формулы 1.0, как описано в любом из вариантов выполнения изобретения №1-46, где R5 представляет собой замещенную триазолилтиенил группу, где (а) указанная триазолильная составляющая необязательно замещена на атоме азота заместителем, выбранным из группы, состоящей из: -СН2СОН(CH3)2 и -CH2CH2OH, (b) указанная триазолильная составляющая необязательно замещена на атоме азота алкильной группой, (с) указанная триазолильная составляющая необязательно замещена на атоме азота алкильной группой и на атоме углерода алкильной группой, (d) указанная триазолильная составляющая необязательно замещена на атоме азота -CH3 группой, (е) указанная триазолильная составляющая необязательно замещена на атоме азота одной -CH3 группой и на атоме углерода одной -CH3 группой, (f) указанная триазолильная составляющая необязательно замещена на атоме углерода -NH2 группой, или (g) указанная триазолильная составляющая необязательно замещена на атоме азота -CH2CH2OCH3 группой; и, где указанная тиенильная составляющая необязательно замещена галогеном (например, одним галогеном, таким как, например, F).
Другие варианты выполнения настоящего изобретения представляют собой соединения формулы 1.0, как описано в любом из вариантов выполнения изобретения №1-46, где R5 представляет собой замещенную триазолилтиенил группу, где (а) указанная триазолильная составляющая необязательно замещена на атоме азота заместителем, выбранным из группы, состоящей из: -СН2СОН(CH3)2, -CH2CH2OH, (b) указанная триазолильная составляющая необязательно замещена на атоме азота алкильной группой, (с) указанная триазолильная составляющая необязательно замещена на атоме азота алкильной группой и на атоме углерода алкильной группой, (d) указанная триазолильная составляющая необязательно замещена на атоме азота -CH3 группой, (е) указанная триазолильная составляющая необязательно замещена на атоме азота одной -CH3 группой и на атоме углерода одной -CH3 группой, (f) указанная триазолильная составляющая необязательно замещена на атоме углерода -NH2 группой, или (g) указанная триазолильная составляющая необязательно замещена на атоме азота -CH2CH2OCH3 группой; и, где указанная тиенильная составляющая замещена галогеном (например, одним галогеном, таким как, например, F).
Другие варианты выполнения настоящего изобретения представляют собой соединения формулы 1.0, как описано в любом из вариантов выполнения изобретения №1-46, где R5 представляет собой замещенную триазолилтиенил группу, где (а) указанная триазолильная составляющая замещена на атоме азота заместителем, выбранным из группы, состоящей из: -СН2СОН(CH3)2 и -CH2CH2OH, (b) указанная триазолильная составляющая замещена на атоме азота алкильной группой, (с) указанная триазолильная составляющая замещена на атоме азота алкильной группой и на атоме углерода алкильной группой, (d) указанная триазолильная составляющая замещена на атоме азота -CH3 группой, (е) указанная триазолильная составляющая замещена на атоме азота одной -CH3 группой и на атоме углерода одной -CH3 группой, (f) указанная триазолильная составляющая замещена на атоме углерода -NH2 группой, или (g) указанная триазолильная составляющая замещена на атоме азота -CH2CH2OCH3 группой; и, где указанная тиенильная составляющая необязательно замещена галогеном (например, одним галогеном, таким как, например, F).
Другие варианты выполнения настоящего изобретения представляют собой соединения формулы 1.0, как описано в любом из вариантов выполнения изобретения №1-46, где R5 представляет собой замещенную триазолилтиенил группу, где (а) указанная триазолильная составляющая замещена на атоме азота заместителем, выбранным из группы, состоящей из: -СН2СОН(CH3)2 и -CH2CH2OH, (b) указанная триазолильная составляющая замещена на атоме азота алкильной группой, (с) указанная триазолильная составляющая замещена на атоме азота алкильной группой и на атоме углерода алкильной группой, (d) указанная триазолильная составляющая замещена на атоме азота -CH3 группой, (е) указанная триазолильная составляющая замещена на атоме азота одной -CH3 группой и на атоме углерода одной -CH3 группой, (f) указанная триазолильная составляющая замещена на атоме углерода -NH2 группой, или (g) указанная триазолильная составляющая замещена на атоме азота -CH3CH2OCH3 группой; и, где указанная тиенильная составляющая замещена галогеном (например, одним галогеном, таким как, например, F).
Другие варианты выполнения настоящего изобретения представляют собой соединения формулы 1.0, как описано в любом из вариантов выполнения изобретения №1-46, где R5 представляет собой замещенную триазолилтиенил группу, где (а) указанная триазолильная составляющая замещена на атоме азота заместителем, выбранным из группы, состоящей из: -СН2СОН(CH3)2 и -CH2CH2OH, (b) указанная триазолильная составляющая замещена на атоме азота алкильной группой, (с) указанная триазолильная составляющая замещена на атоме азота алкильной группой и на атоме углерода алкильной группой, (d) указанная триазолильная составляющая замещена на атоме азота -CH3 группой, (е) указанная триазолильная составляющая замещена на атоме азота одной -CH3 группой и на атоме углерода одной -CH3 группой, (f) указанная триазолильная составляющая замещена на атоме углерода -NH2 группой, или (g) указанная триазолильная составляющая замещена на атоме азота -CH2CH2OCH3 группой; и, где указанная тиенильная составляющая является незамещенной.
Другие варианты выполнения настоящего изобретения представляют собой соединения формулы 1.0, как описано в любом из вариантов выполнения изобретения №1-46, где указанный замещенный R5 составляющая представляет собой замещенный триазолилтиенил, где указанная триазолильная составляющая замещена: (а) одним заместителем, выбранным из группы, состоящей из: -СН2СОН(CH3)2 и -CH2CH2OH, (b) одной алкильной группой, (с) двумя алкильными группами, (d) одной -CH3 группой, (е) двумя -CH3 группами, (f) одной -NH2 группой, или (g) одной -CH2CH2OCH3 группой.
Другие варианты выполнения настоящего изобретения представляют собой соединения формулы 1.0, как описано в любом из вариантов выполнения изобретения №1-46, где необязательные заместители для тиенильной составляющей R5 независимо выбираются из группы, состоящей из: алкокси.
Другие варианты выполнения настоящего изобретения представляют собой соединения формулы 1.0, как описано в любом из вариантов выполнения изобретения №1-46, где необязательные заместители для тиенильной составляющей R5 представляют собой группу -OCH3.
Другие варианты выполнения настоящего изобретения представляют собой соединения формулы 1.0, как описано в любом из вариантов выполнения изобретения №1-46, где необязательный заместитель для тиенильной составляющей R5 представляет собой одну группу -OCH3.
Другие варианты выполнения настоящего изобретения представляют собой соединения формулы 1.0, как описано в любом из вариантов выполнения изобретения №1-46, где R5 представляет собой замещенную триазолилтиенил группу, где (а) указанная триазолильная составляющая необязательно замещена на атоме азота заместителем, выбранным из группы, состоящей из: -СН2СОН(CH3)2 и -CH2CH2OH, (b) указанная триазолильная составляющая необязательно замещена на атоме азота алкильной группой, (с) указанная триазолильная составляющая необязательно замещена на атоме азота алкильной группой и на атоме углерода алкильной группой, (d) указанная триазолильная составляющая необязательно замещена на атоме азота -CH3 группой, (е) указанная триазолильная составляющая необязательно замещена на атоме азота одной -CH3 группой и на атоме углерода одной -CH3 группой, (f) указанная триазолильная составляющая необязательно замещена на атоме углерода -NH2 группой, или (g) указанная триазолильная составляющая необязательно замещена на атоме азота -CH2CH2OCH3 группой; и, где указанная тиенильная составляющая необязательно замещена алкоксигруппой (например, одной группой -OCH3).
Другие варианты выполнения настоящего изобретения представляют собой соединения формулы 1.0, как описано в любом из вариантов выполнения изобретения №1-46, где R5 представляет собой замещенную триазолилтиенил группу, где (а) указанная триазолильная составляющая необязательно замещена на атоме азота заместителем, выбранным из группы, состоящей из: -СН2СОН(CH3)2 и -CH2CH2OH, (b) указанная триазолильная составляющая необязательно замещена на атоме азота алкильной группой, (с) указанная триазолильная составляющая необязательно замещена на атоме азота алкильной группой и на атоме углерода алкильной группой, (d) указанная триазолильная составляющая необязательно замещена на атоме азота -CH3 группой, (е) указанная триазолильная составляющая необязательно замещена на атоме азота одной -CH3 группой и на атоме углерода одной -CH3 группой, (f) указанная триазолильная составляющая необязательно замещена на атоме углерода -NH2 группой, или (g) указанная триазолильная составляющая необязательно замещена на атоме азота -CH2CH2OCH3 группой; и, где указанная тиенильная составляющая замещена алкоксигруппой (например, одной группой -OCH3).
Другие варианты выполнения настоящего изобретения представляют собой соединения формулы 1.0, как описано в любом из вариантов выполнения изобретения №1-46, где R5 представляет собой замещенную триазолилтиенил группу, где (а) указанная триазолильная составляющая замещена на атоме азота заместителем, выбранным из группы, состоящей из: -СН2СОН(CH3)2 и -CH2CH2OH, (b) указанная триазолильная составляющая замещена на атоме азота алкильной группой, (с) указанная триазолильная составляющая замещена на атоме азота алкильной группой и на атоме углерода алкильной группой, (d) указанная триазолильная составляющая замещена на атоме азота -CH3 группой, (е) указанная триазолильная составляющая замещена на атоме азота одной -CH3 группой и на атоме углерода одной -CH3 группой, (f) указанная триазолильная составляющая замещена на атоме углерода -NH2 группой, или (g) указанная триазолильная составляющая замещена на атоме азота -CH2CH2OCH3 группой; и, где указанная тиенильная составляющая необязательно замещена алкоксигруппой (например, одной группой -OCH3).
Другие варианты выполнения настоящего изобретения представляют собой соединения формулы 1.0, как описано в любом из вариантов выполнения изобретения №1-46, где R5 представляет собой замещенную триазолилтиенил группу, где (а) указанная триазолильная составляющая замещена на атоме азота заместителем, выбранным из группы, состоящей из: -СН2СОН(CH3)2 и -CH2CH2OH, (b) указанная триазолильная составляющая замещена на атоме азота алкильной группой, (с) указанная триазолильная составляющая замещена на атоме азота алкильной группой и на атоме углерода алкильной группой, (d) указанная триазолильная составляющая замещена на атоме азота -CH3 группой, (е) указанная триазолильная составляющая замещена на атоме азота одной -CH3 группой и на атоме углерода одной -CH3 группой, (f) указанная триазолильная составляющая замещена на атоме углерода -NH2 группой, или (g) указанная триазолильная составляющая замещена на атоме азота -CH2CH2OCH3 группой; и, где указанная тиенильная составляющая замещена алкокси-группой (например, одной группой -OCH3).
Другие варианты выполнения настоящего изобретения представляют собой соединения формулы 1.0, как описано в любом из вариантов выполнения изобретения №1-46, где R5 представляет собой замещенный триазолилпиридил-, где триазолильная составляющая замещена одной или двумя алкильными группами, выбранными из группы, состоящей из: -C1-С6алкила, -С1-С4алкила, -С1-С2алкила и -CH3.
Другие варианты выполнения настоящего изобретения представляют собой соединения формулы 1.0, как описано в любом из вариантов выполнения изобретения №1-6, где R5 представляет собой замещенный триазолилпиридил-, где триазолильная составляющая замещена одной или двумя алкильными группами, выбранными из группы, состоящей из: -C1-С4алкила, -C1-C2алкиал и -CH3. Другие варианты выполнения настоящего изобретения представляют собой соединения формулы 1.0, как описано в любом из вариантов выполнения изобретения №1-46, где R5 представляет собой замещенный триазолилпиридил-, где триазолильная составляющая замещена одной или двумя -CH3 группами.
Другие варианты выполнения настоящего изобретения представляют собой соединения формулы 1.0, как описано в любом из вариантов выполнения изобретения №1-46, где R5 представляет собой замещенный триазолилпиридил-, где триазолильная составляющая замещена на атоме азота алкильной группой, выбранной из группы, состоящей из: -C1-С4алкила, -С1-С2алкила и -CH3. Другие варианты выполнения настоящего изобретения представляют собой соединения формулы 1.0, как описано в любом из вариантов выполнения изобретения №1-46, где R5 представляет собой замещенный триазолилпиридил-, где триазолильная составляющая замещена на атоме азота группой -CH3.
Другие варианты выполнения настоящего изобретения представляют собой соединения формулы 1.0, как описано в любом из вариантов выполнения изобретения №1-46, где R5 представляет собой замещенный триазолилпиридил-, где триазолильная составляющая замещена на атоме азота -алкилен-O-алкильной группой, выбранной из группы, состоящей из: -С2-С4алкилен-O-С1-С6алкила, -С2алкилен-O-С1-С2алкиал, -С2-С4алкилен-O-CH3 и -CH2CH2OCH3.
Другие варианты выполнения настоящего изобретения представляют собой соединения формулы 1.0, как описано в любом из вариантов выполнения изобретения №1-46, где R5 представляет собой замещенный триазолилпиридил-, где триазолильная составляющая замещена на атоме азота -алкилен-O-алкильной группой, выбранной из группы, состоящей из: -С2алкилен-O-С1-С2алкила и -CH2CH2OCH3. Другие варианты выполнения настоящего изобретения представляют собой соединения формулы 1.0, как описано в любом из вариантов выполнения изобретения №1-46, где R5 представляет собой замещенный триазолилпиридил-, где триазолильная составляющая замещена на атоме азота группой -CH2CH2OCH3.
Другие варианты выполнения настоящего изобретения представляют собой соединения формулы 1.0, как описано в любом из вариантов выполнения изобретения №1-46, где R5 представляет собой замещенный триазолилпиридил-, где триазолильная составляющая замещена на атоме азота гидроксизамещенной алкильной группой выбранной из группы, состоящей из: гидроксизамещенного -С1-С4алкила, гидроксизамещенного -С1-С2алкила и гидроксизамещенной группы -CH3.
Другие варианты выполнения настоящего изобретения представляют собой соединения формулы 1.0, как описано в любом из вариантов выполнения изобретения №1-46, где R5 представляет собой замещенный триазолилпиридил-, где триазолильная составляющая замещена на атоме азота гидроксизамещенной алкильной группой выбранной из группы, состоящей из: СН2СОН(CH3)2 и -CH2CH2OH.
Другие варианты выполнения настоящего изобретения представляют собой соединения формулы 1.0, как описано в любом из вариантов выполнения изобретения №1-46, где R5 представляет собой замещенную триазолилпиридил-группу, где (а) указанная триазолильная составляющая необязательно замещена на атоме азота заместителем, выбранным из группы, состоящей из: -СН2СОН(CH3)2 и -CH2CH2OH, (b) указанная триазолильная составляющая необязательно замещена на атоме азота алкильной группой, (с) указанная триазолильная составляющая необязательно замещена на атоме азота алкильной группой и на атоме углерода алкильной группой, (d) указанная триазолильная составляющая необязательно замещена на атоме азота -CH3 группой, (е) указанная триазолильная составляющая необязательно замещена на атоме азота одной -CH3 группой и на атоме углерода одной -CH3 группой, (f) указанная триазолильная составляющая необязательно замещена на атоме углерода -NH2 группой, или (g) указанная триазолильная составляющая необязательно замещена на атоме азота -CH2CH2OCH3 группой; и, где указанная пиридильная составляющая является незамещенной.
Другие варианты выполнения настоящего изобретения представляют собой соединения формулы 1.0, как описано в любом из вариантов выполнения изобретения №1-46, где R5 представляет собой замещенную триазолилпиридил-группу, где (а) указанная триазолильная составляющая замещена на атоме азота заместителем, выбранным из группы, состоящей из: -СН2СОН(CH3)2 и -CH2CH2OH, (b) указанная триазолильная составляющая замещена на атоме азота алкильной группой, (с) указанная триазолильная составляющая замещена на атоме азота алкильной группой, и на атоме углерода алкильной группой, (d) указанная триазолильная составляющая замещена на атоме азота -CH3 группой, (е) указанная триазолильная составляющая замещена на атоме азота одной -CH3 группой и на атоме углерода одной -CH3 группой, (f) указанная триазолильная составляющая замещена на атоме углерода -NH2 группой, или (g) указанная триазолильная составляющая замещена на атоме азота -CH2CH2OCH3 группой; и, где указанная пиридильная составляющая является незамещенной.
Другие варианты выполнения настоящего изобретения представляют собой соединения формулы 1.0, как описано в любом из вариантов выполнения изобретения №1-46, где R5 представляет собой замещенный триазолилтиазолил-, где триазолильная составляющая замещена одной или двумя алкильными группами, выбранными из группы, состоящей из: -C1-С6алкила, -С1-С4алкиал, -C1-C2алкил и -CH3.
Другие варианты выполнения настоящего изобретения представляют собой соединения формулы 1.0, как описано в любом из вариантов выполнения изобретения №1-46, где R5 представляет собой замещенный триазолилтиазолил-, где триазолильная составляющая замещена одной или двумя алкильными группами, выбранными из группы, состоящей из: -С1-С4алкила, -C1-C2алкила и -CH3. Другие варианты выполнения настоящего изобретения представляют собой соединения формулы 1.0, как описано в любом из вариантов выполнения изобретения №1-46, где R5 представляет собой замещенный триазолилтиазолил-, где триазолильная составляющая замещена одной или двумя -CH3 группами.
Другие варианты выполнения настоящего изобретения представляют собой соединения формулы 1.0, как описано в любом из вариантов выполнения изобретения №1-46, где R5 представляет собой замещенный триазолилтиазолил-, где триазолильная составляющая замещена на атоме азота алкильной группой, выбранной из группы, состоящей из: -С1-С4алкила, -С1-С2алкила и -CH3. Другие варианты выполнения настоящего изобретения представляют собой соединения формулы 1.0, как описано в любом из вариантов выполнения изобретения №1-46, где R5 представляет собой замещенный триазолилтиазолил-, где триазолильная составляющая замещена на атоме азота группой -CH3.
Другие варианты выполнения настоящего изобретения представляют собой соединения формулы 1.0, как описано в любом из вариантов выполнения изобретения №1-46, где R5 представляет собой замещенный триазолилтиазолил-, где триазолильная составляющая замещена на атоме азота -алкилен-O-алкильной группой, выбранной из группы, состоящей из: -С2-С4алкилен-O-С1-С6алкила, -С2алкилен-O-С1-С2алкила, -С2-С4алкилен-O-CH3 и -CH2CH2OCH3.
Другие варианты выполнения настоящего изобретения представляют собой соединения формулы 1.0, как описано в любом из вариантов выполнения изобретения №1-46, где R5 представляет собой замещенный триазолилтиазолил-, где триазолильная составляющая замещена на атоме азота -алкилен-O-алкильной группой, выбранной из группы, состоящей из: -С2алкилен-O-С1-С2алкила и -CH2CH2OCH3. Другие варианты выполнения настоящего изобретения представляют собой соединения формулы 1.0, как описано в любом из вариантов выполнения изобретения №1-46, где R5 представляет собой замещенный триазолилтиазолил-, где триазолильная составляющая замещена на атоме азота группой -CH2CH2OCH3.
Другие варианты выполнения настоящего изобретения представляют собой соединения формулы 1.0, как описано в любом из вариантов выполнения изобретения №1-46, где R5 представляет собой замещенный триазолилтиазолил-, где триазолильная составляющая замещена на атоме азота гидроксизамещенной алкильной группой, выбранной из группы, состоящей из: гидроксизамещенного -С1-С4алкила, гидроксизамещенного -С1-С2алкила и гидроксизамещенной группы -CH3.
Другие варианты выполнения настоящего изобретения представляют собой соединения формулы 1.0, как описано в любом из вариантов выполнения изобретения №1-46, где R5 представляет собой замещенный триазолилтиазолил-, где триазолильная составляющая замещена на атоме азота гидроксизамещенной алкильной группой, выбранной из группы, состоящей из: СН2СОН(CH3)2 и -CH2CH2OH.
Другие варианты выполнения настоящего изобретения представляют собой соединения формулы 1.0, как описано в любом из вариантов выполнения изобретения №1-46, где R5 представляет собой замещенную триазолилтиазолил-группу, где (а) указанная триазолильная составляющая необязательно замещена на атоме азота заместителем, выбранным из группы, состоящей из: -СН2СОН(CH3)2 и -CH2CH2OH, (b) указанная триазолильная составляющая необязательно замещена на атоме азота алкильной группой, (с) указанная триазолильная составляющая необязательно замещена на атоме азота алкильной группой и на атоме углерода алкильной группой, (d) указанная триазолильная составляющая необязательно замещена на атоме азота -CH3 группой, (е) указанная триазолильная составляющая необязательно замещена на атоме азота одной -CH3 группой и на атоме углерода одной -CH3 группой, (f) указанная триазолильная составляющая необязательно замещена на атоме углерода -NH2 группой, или (g) указанная триазолильная составляющая необязательно замещена на атоме азота -CH3CH2OCH3 группой; и, где указанная тиазолильная составляющая является незамещенной.
Другие варианты выполнения настоящего изобретения представляют собой соединения формулы 1.0, как описано в любом из вариантов выполнения изобретения №1-46, где R5 представляет собой замещенную триазолилтиазолил-группу, где (а) указанная триазолильная составляющая замещена на атоме азота заместителем, выбранным из группы, состоящей из: -СН2СОН(CH3)2 и -CH2CH2OH, (b) указанная триазолильная составляющая замещена на атоме азота алкильной группой, (с) указанная триазолильная составляющая замещена на атоме азота алкильной группой и на атоме углерода алкильной группой, (d) указанная триазолильная составляющая замещена на атоме азота -CH3 группой, (е) указанная триазолильная составляющая замещена на атоме азота одной -CH3 группой и на атоме углерода одной -CH3 группой, (f) указанная триазолильная составляющая замещена на атоме углерода -NH2 группой, или (g) указанная триазолильная составляющая замещена на атоме азота -CH2CH2OCH3 группой; и, где указанная тиазолильная составляющая является незамещенной.
Другие варианты выполнения настоящего изобретения представляют собой соединения формулы 1.0, как описано в любом из вариантов выполнения изобретения №1-46, где R5 представляет собой замещенный пиридизинилтиенил-, где пиридазинильная составляющая замещена одной или двумя группами, независимо выбранными из группы, состоящей из алкила (например, метил) и =O, и указанная тиенильная составляющая является незамещенной.
Другие варианты выполнения настоящего изобретения представляют собой соединения формулы 1.0, как описано в любом из вариантов выполнения изобретения №1-46, где R5 представляет собой замещенный пиридазинилтиенил-, где пиридазинильная составляющая замещена =O составляющей, или указанная пиридазинильная группа замещена алкилом (например, метилом), или указанный пиридазинил замещен =O составляющей и алкилом (например, метилом), и указанная тиенильная составляющая является незамещенной.
Другие варианты выполнения настоящего изобретения представляют собой соединения формулы 1.0, как описано в любом из вариантов выполнения изобретения №1-46, где R5 выбирается из группы, состоящей из:
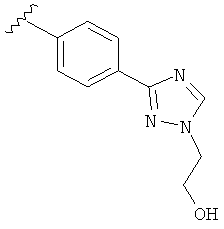 ,
, 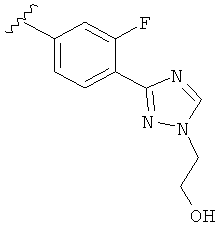 ,
, 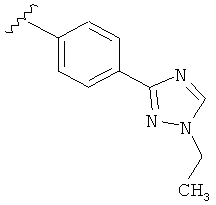 ,
, 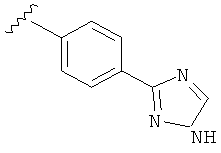 ,
,
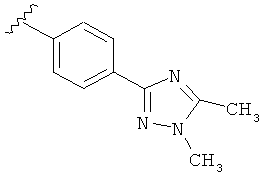 ,
, 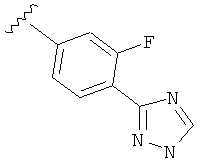 ,
, 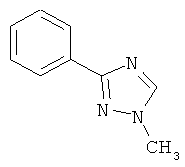 ,
,
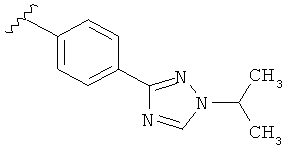 ,
, 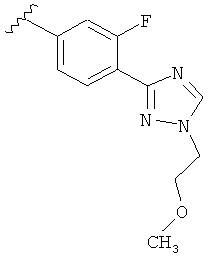 ,
, 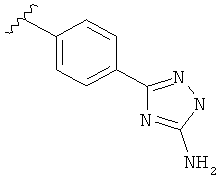 ,
,
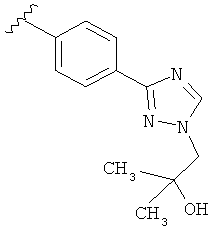 ,
, 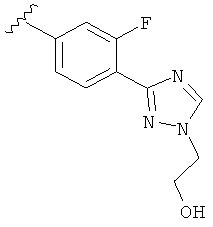 ,
, 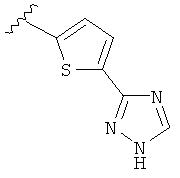 ,
, 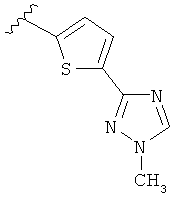 ,
,
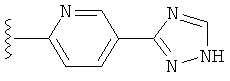 ,
, 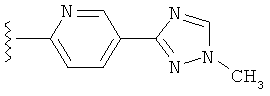 ,
, 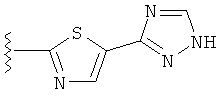 ,
,
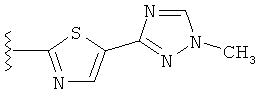 ,
, 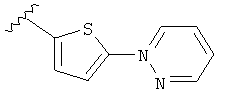 ,
, 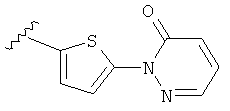 ,
,
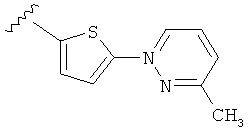 , и
, и 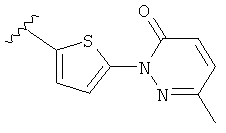 .
.
Другие варианты выполнения настоящего изобретения представляют собой соединения формулы 1.0, как описано в любом из вариантов выполнения изобретения №1-46, где R5 выбирается из группы, состоящей из:
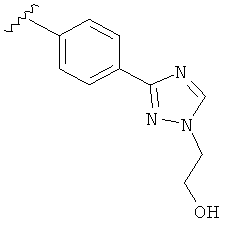 ,
, 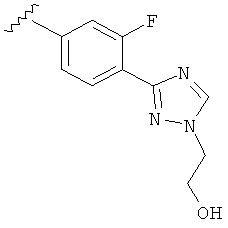 ,
, 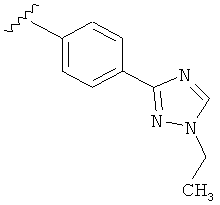 ,
, 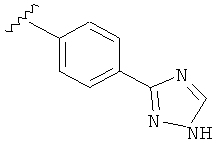 ,
,
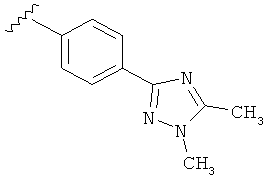 ,
, 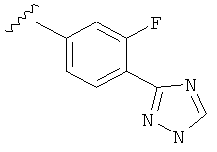 , ,
, ,
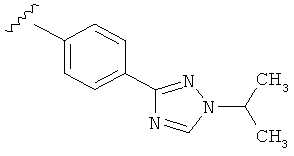 ,
, 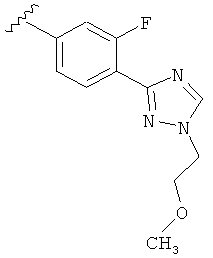 ,
, 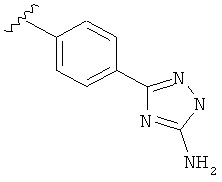 ,
,
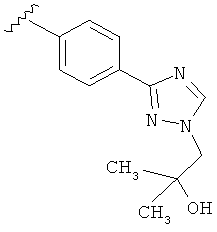 ,
, 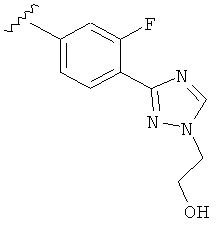 ,
, 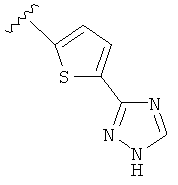 ,
, 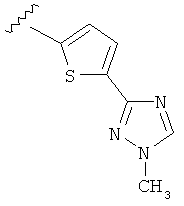 ,
,
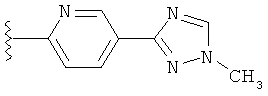 ,
, 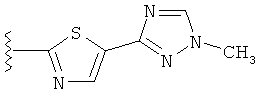 ,
, 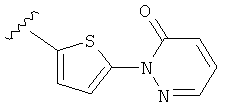 и
и
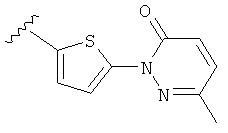 .
.
Другие варианты выполнения настоящего изобретения представляют собой соединения формулы 1.0, как описано в любом из вариантов выполнения изобретения №1-46, где R5 выбирается из группы, состоящей из:
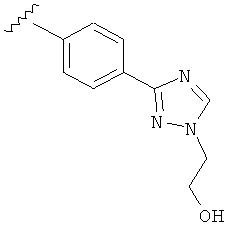 ,
, 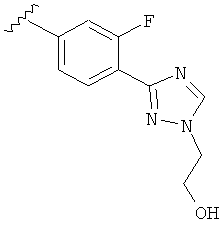 ,
, 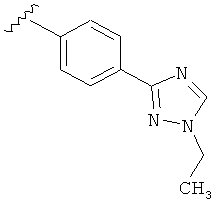 ,
,
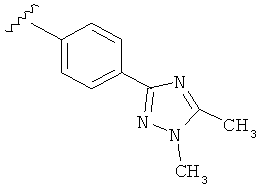 ,
, 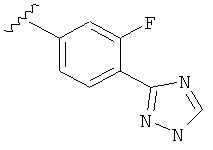 , ,
, ,
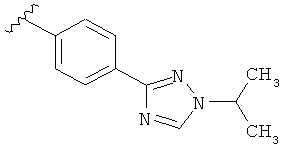 ,
, 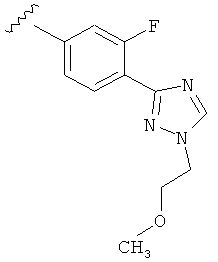 ,
, 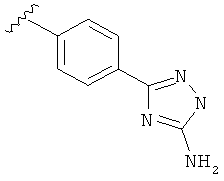 ,
,
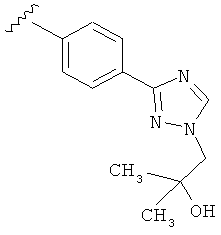 ,
, 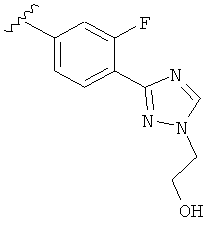 ,
, 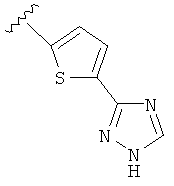 ,
, 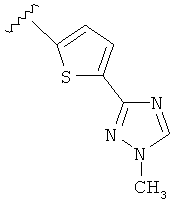 .
.
Другие варианты выполнения настоящего изобретения представляют собой соединения формулы 1.0, как описано в любом из вариантов выполнения изобретения №1-46, где R5 выбирается из группы, состоящей из:
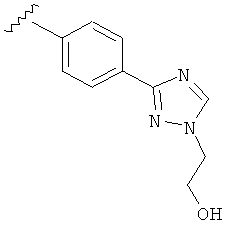 ,
, 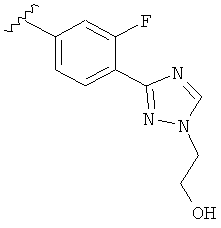 ,
, 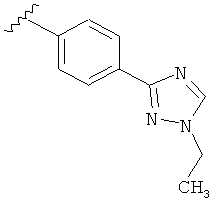 ,
,
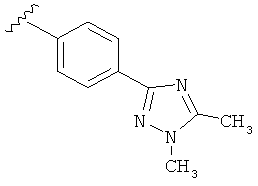 ,
, 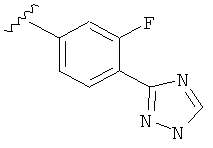 , ,
, ,
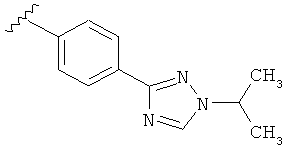 ,
, 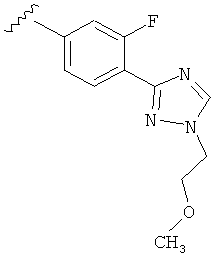 ,
, 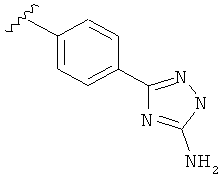 ,
,
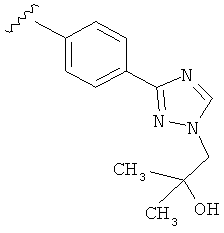 ,
, 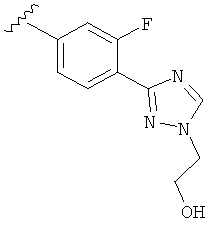 , и
, и 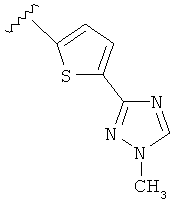 .
.
Другие варианты выполнения настоящего изобретения представляют собой соединения формулы 1.1:
, где:
R1 и R2 определены в любом из вариантов выполнения изобретения №1-44,
Q представляет собой:
 ; и
; и
R5 выбирается из группы, состоящей из:
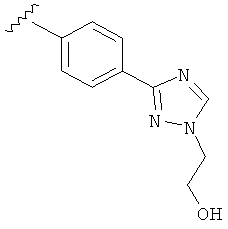 ,
, 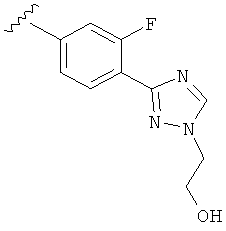 ,
, 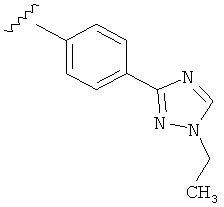 ,
, 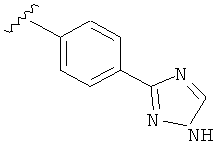 ,
,
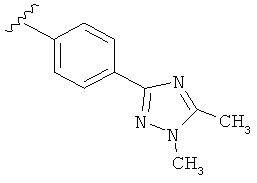 ,
, 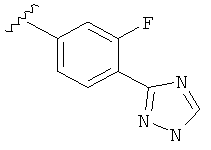 , ,
, ,
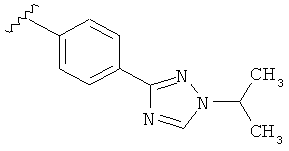 ,
, 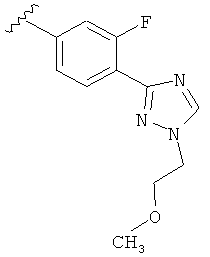 ,
, 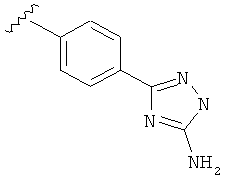 ,
,
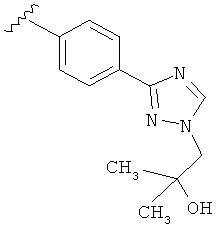 ,
, 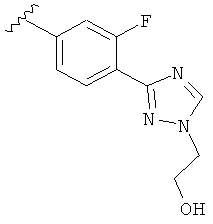 ,
, 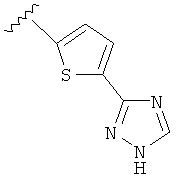 ,
, 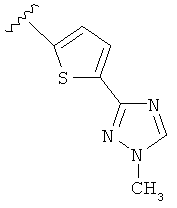 ,
,
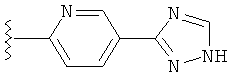 ,
, 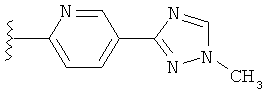 ,
, 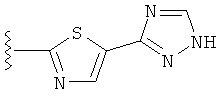 ,
,
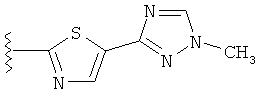 ,
, 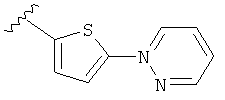 ,
, 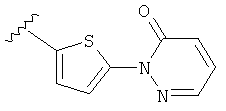 ,
,
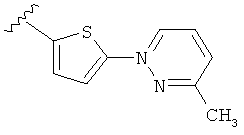 , и
, и 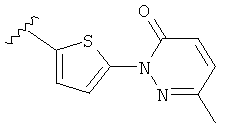 .
.
Другие варианты выполнения настоящего изобретения представляют собой соединения формулы 1.1:
, где:
R2 представляет собой -O-(С1-С2)алкильную группу, и R1 представляет собой:
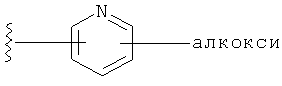 ,
,
Q представляет собой:
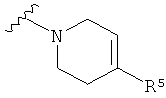 ; и
; и
R5 выбирается из группы, состоящей из:
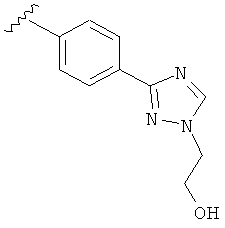 ,
, 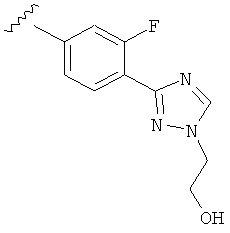 ,
, 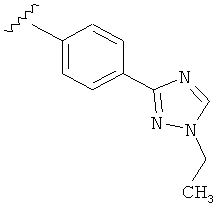 ,
, 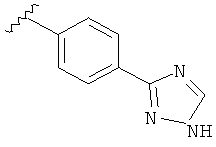 ,
,
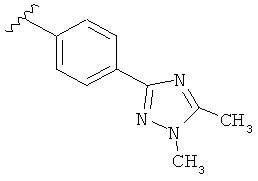 ,
, 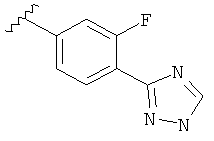 , ,
, ,
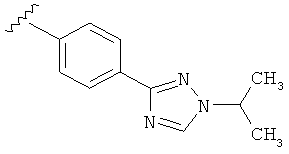 ,
, 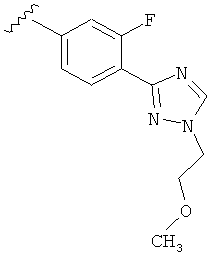 ,
, 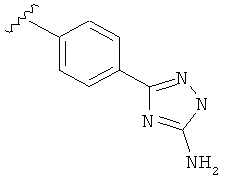 ,
,
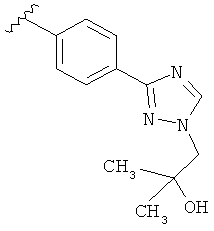 ,
, 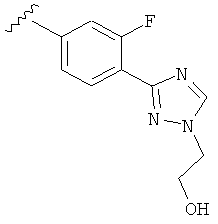 ,
,  ,
, 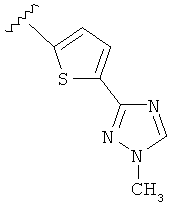 ,
,
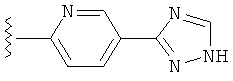 ,
, 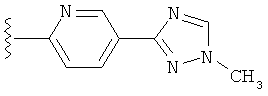 ,
, 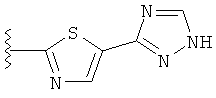 ,
,
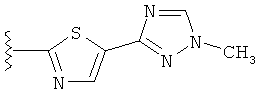 ,
, 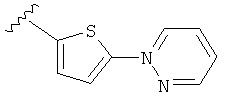 ,
, 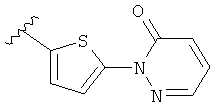 ,
,
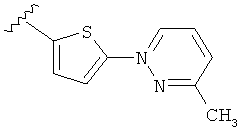 , и
, и 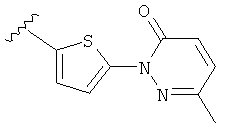 .
.
Другие варианты выполнения настоящего изобретения представляют собой соединения формулы 1.1:
, где:
R2 представляет собой -S-(С1-С2)алкильную группу, и R1 представляет собой:
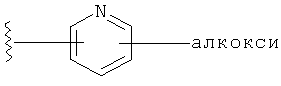 ,
,
Q представляет собой:
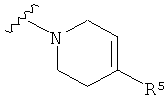 ; и
; и
R5 выбирается из группы, состоящей из:
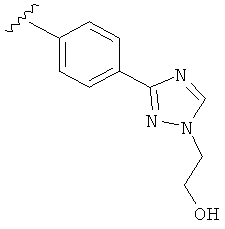 ,
, 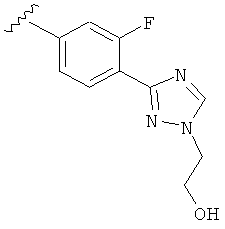 ,
, 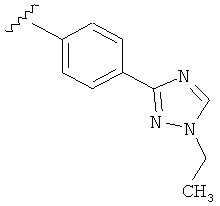 ,
, 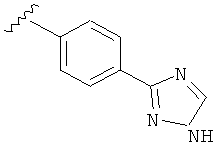 ,
,
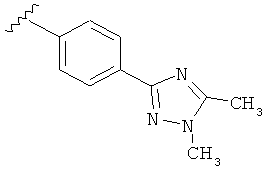 ,
, 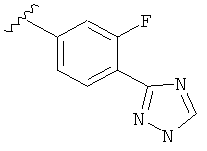 , ,
, ,
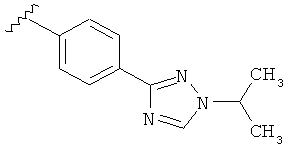 ,
, 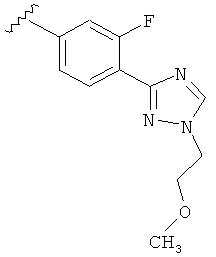 ,
, 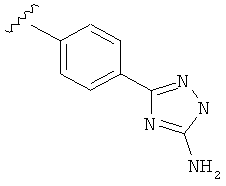 ,
,
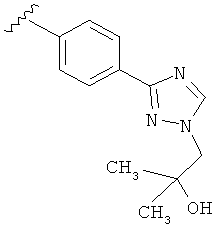 ,
, 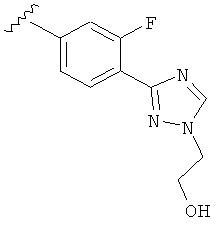 ,
, 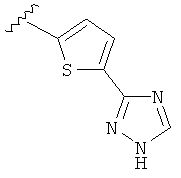 ,
, 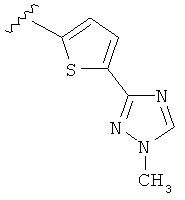 ,
,
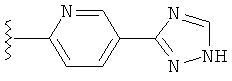 ,
, 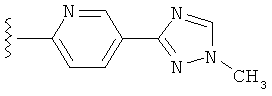 ,
, 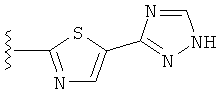 ,
,
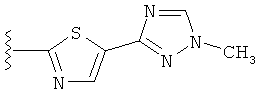 ,
, 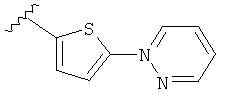 ,
, 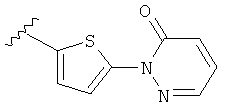 ,
,
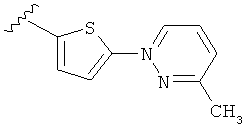 и
и 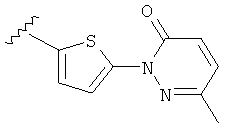 .
.
Другие варианты выполнения настоящего изобретения представляют собой любой из приведенных выше вариантов выполнения изобретения (например, любой из вариантов выполнения изобретения №1-46, или любой другой вариант выполнения изобретения следующий за вариантом выполнения изобретения №46), где один или более атомов водорода представляют собой дейтерий.
Типичные представители соединений согласно настоящему изобретению включают, но без ограничения к этому:
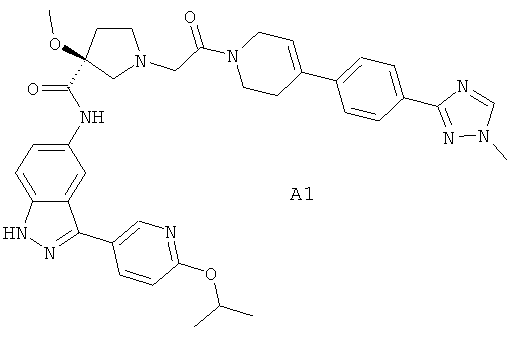 ,
,
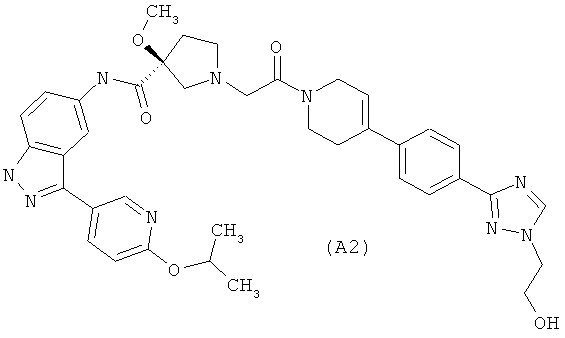 ,
,
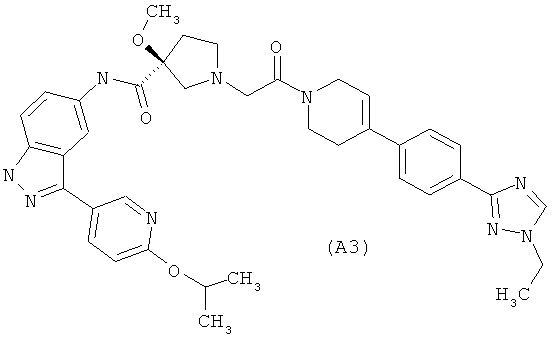 ,
,
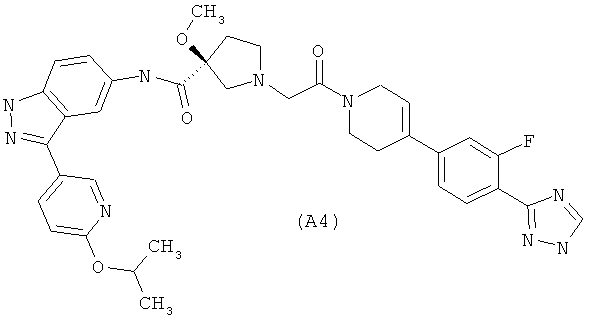 ,
,
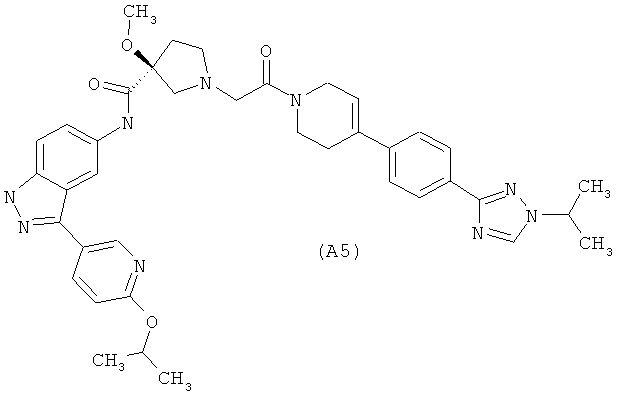 ,
,
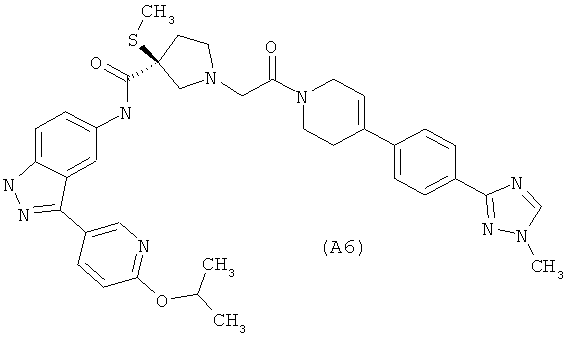 ,
,
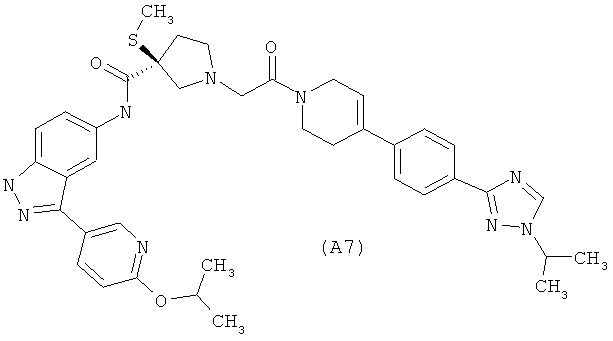 ,
,
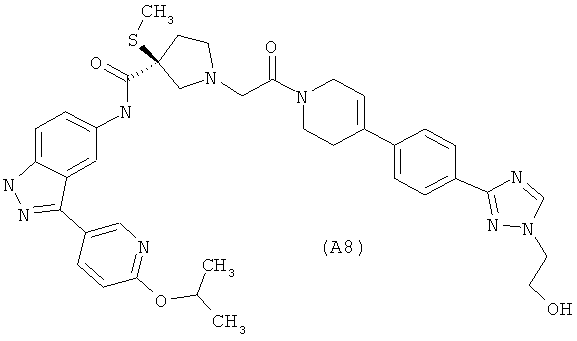 ,
,
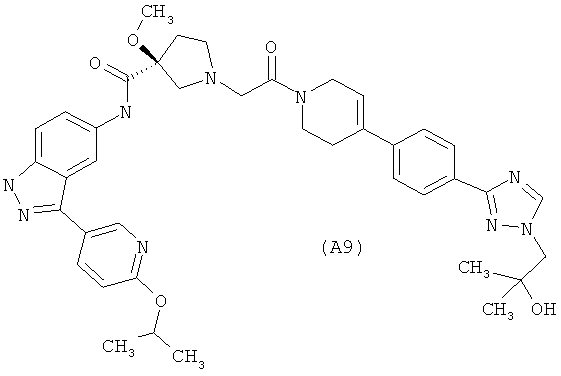 ,
,
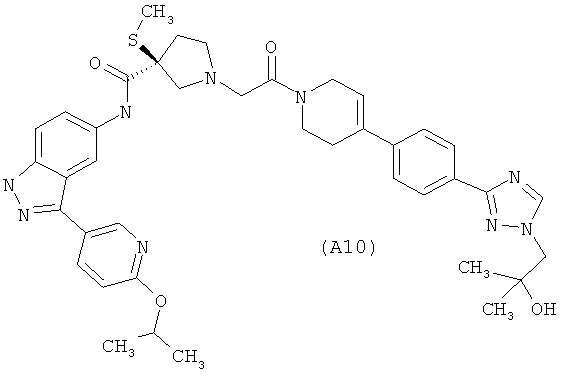 ,
,
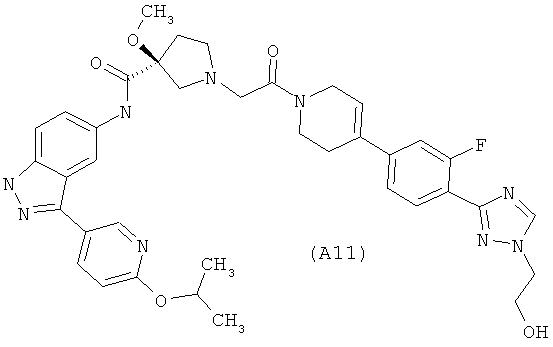 ,
,
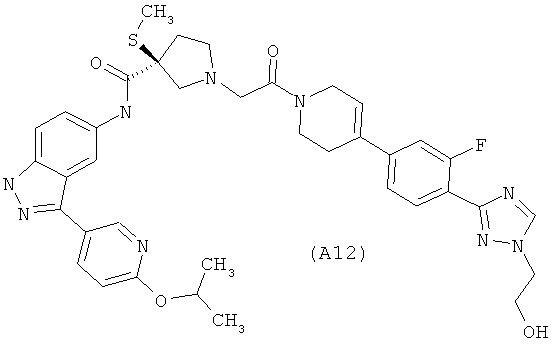 ,
,
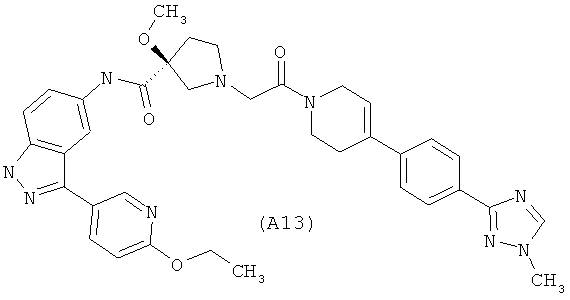 ,
,
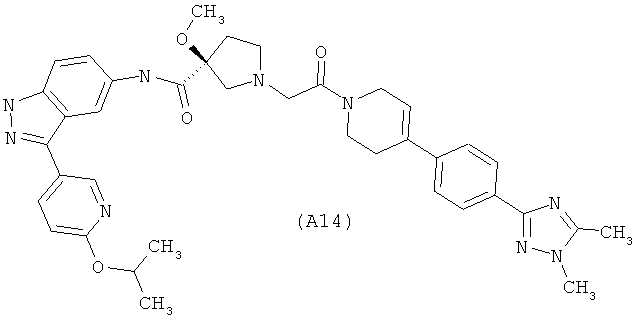 ,
,
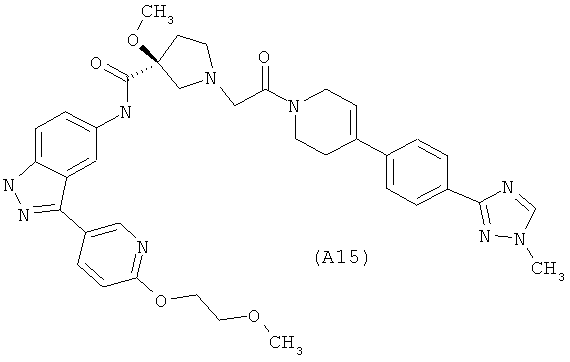 ,
,
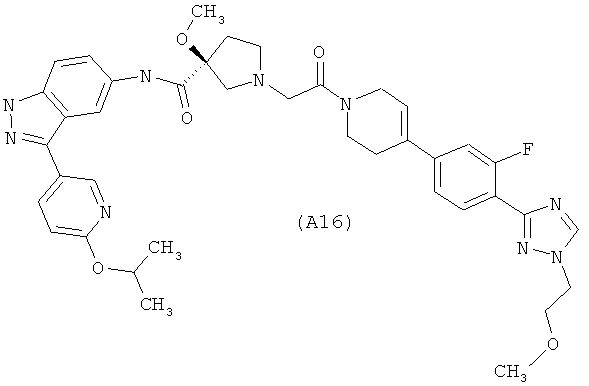 ,
,
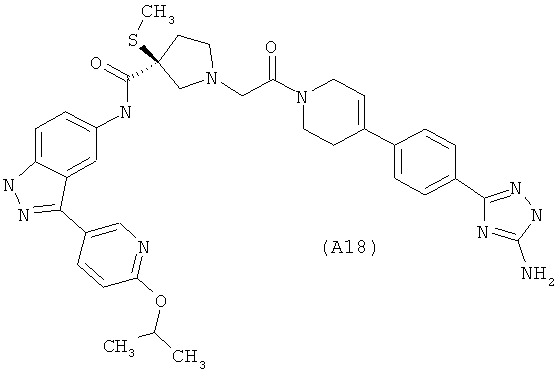 ,
,
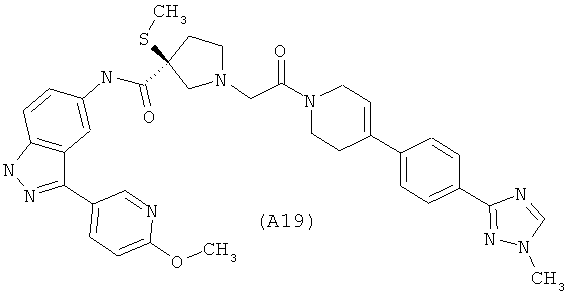 ,
,
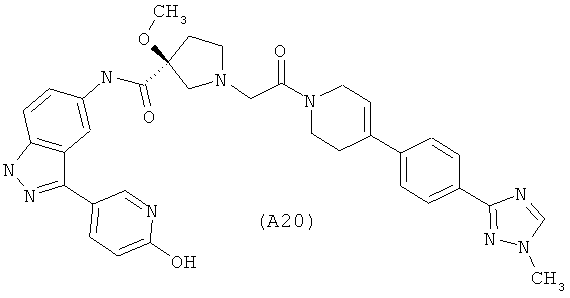 ,
,
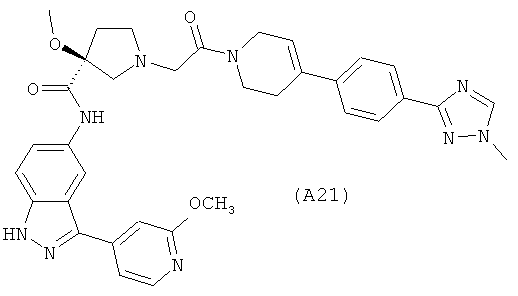 ,
,
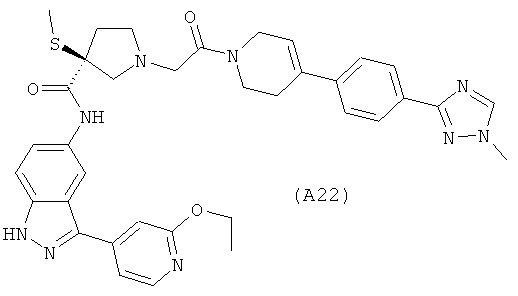 ,
,
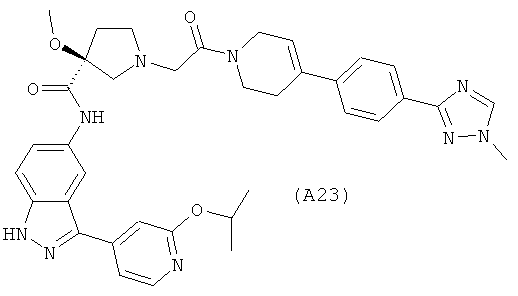 ,
,
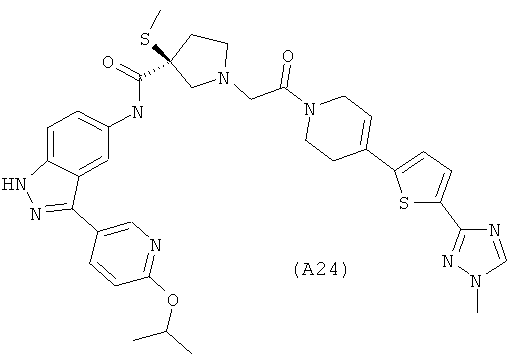 ,
,
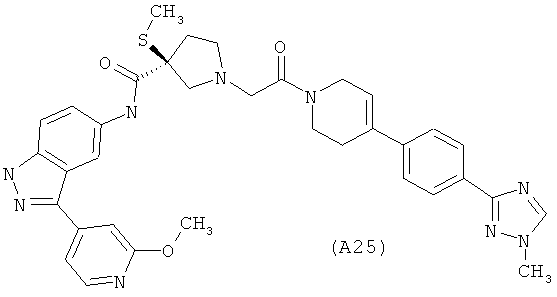 ,
,
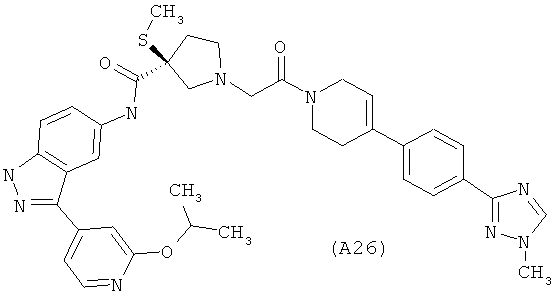 ,
,
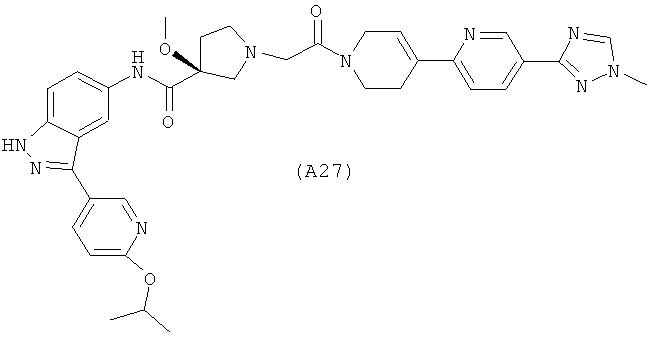 ,
,
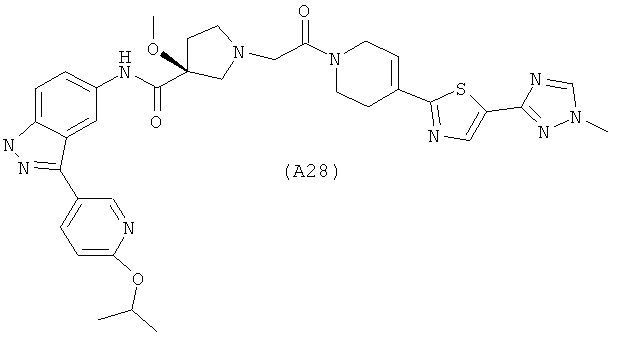 ,
,
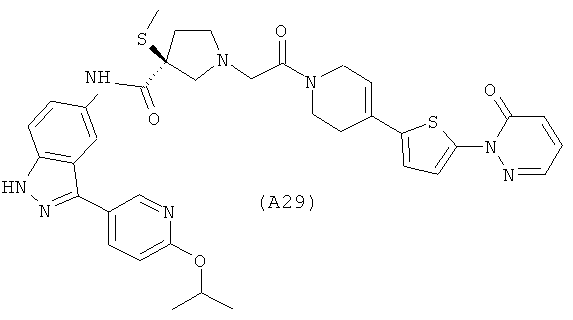 ,
,
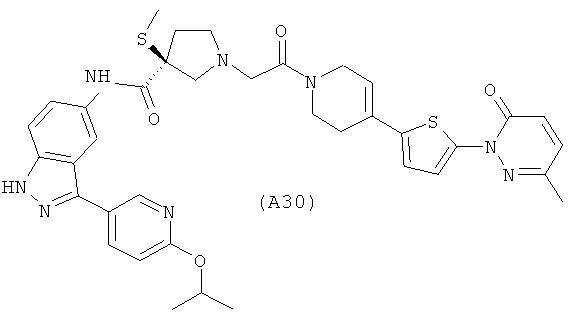 ,
,
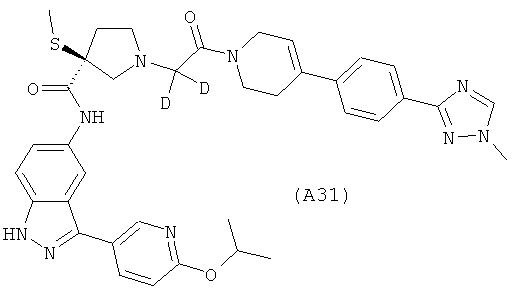 ,
,
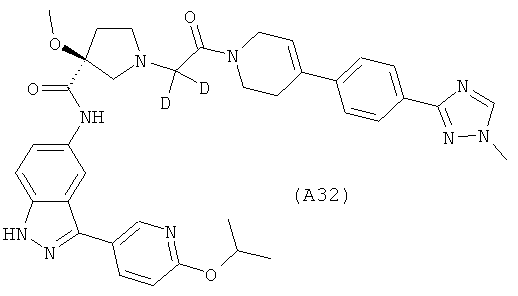 ,
,
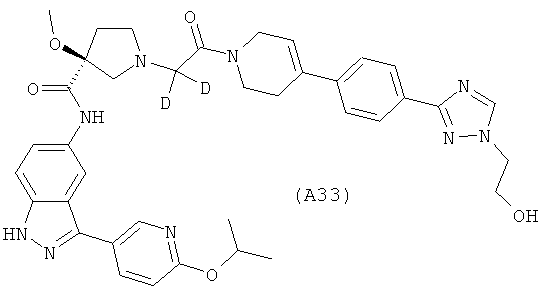 ,
,
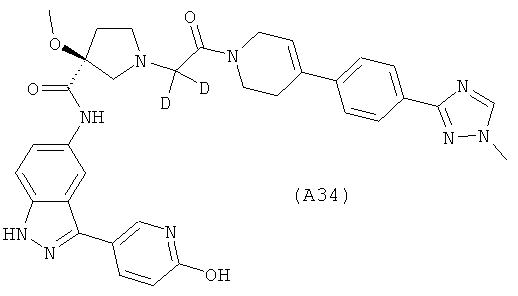 ,
,
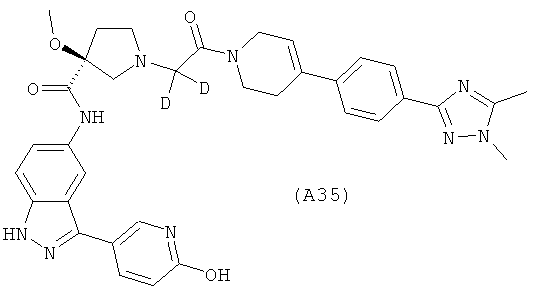 ,
,
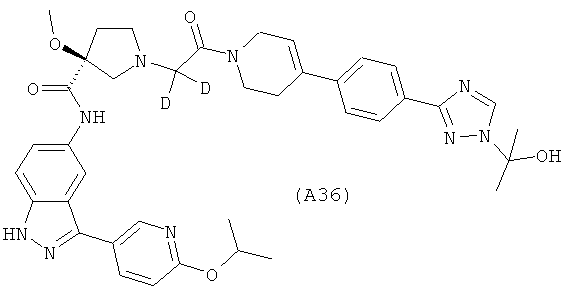 ,
,
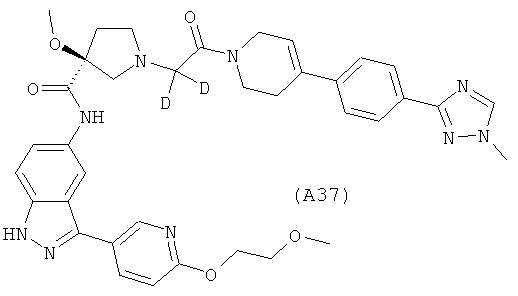 ,
,
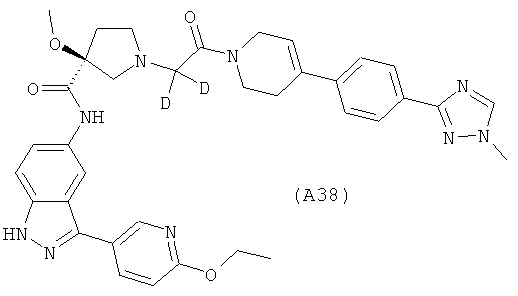 ,
,
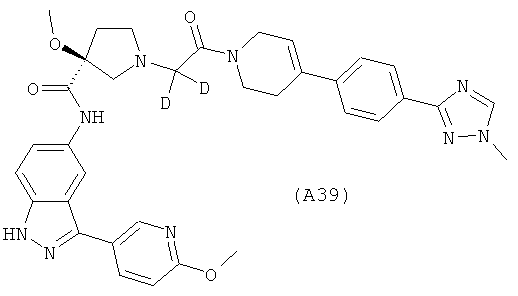 ,
,
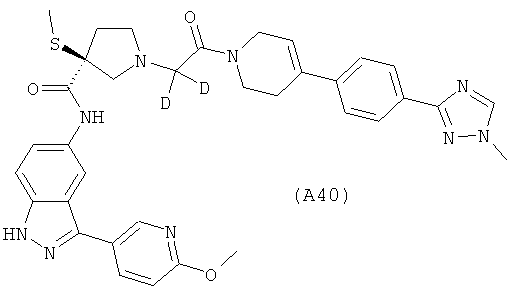 ,
,
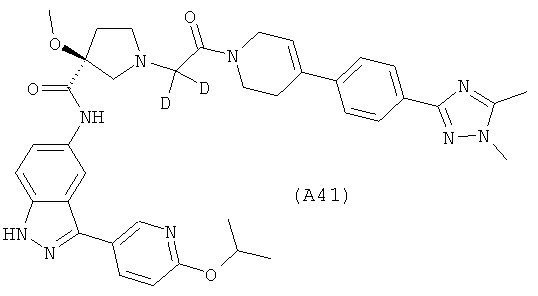 ,
,
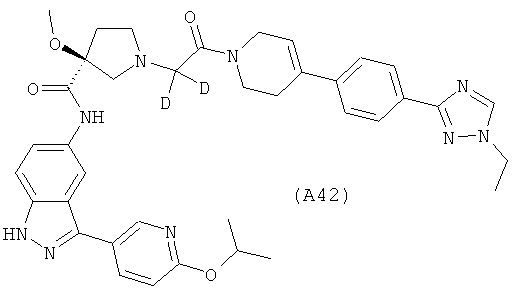 ,
,
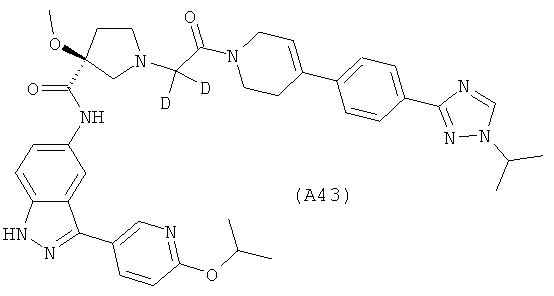 ,
,
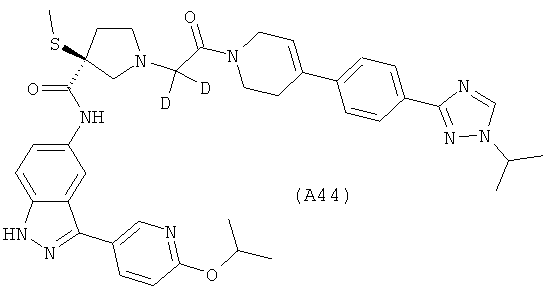 ,
,
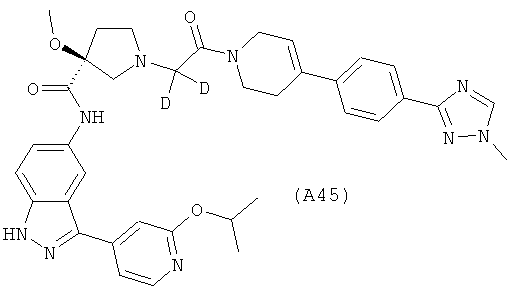 ,
,
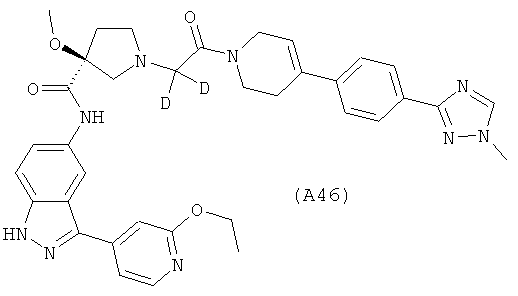 и
и
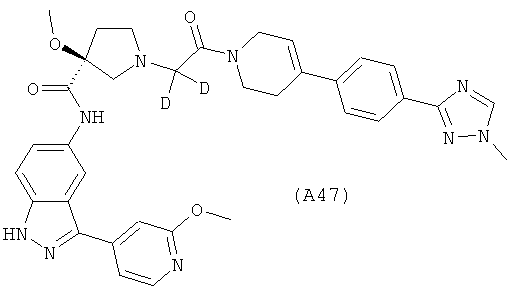 .
.
Типичные представители соединений согласно настоящему изобретению включают, но без ограничения к этому:
,
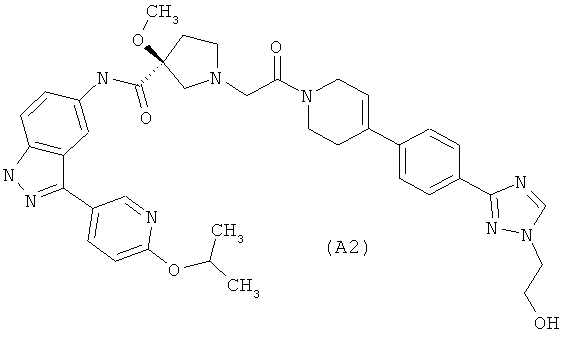 ,
,
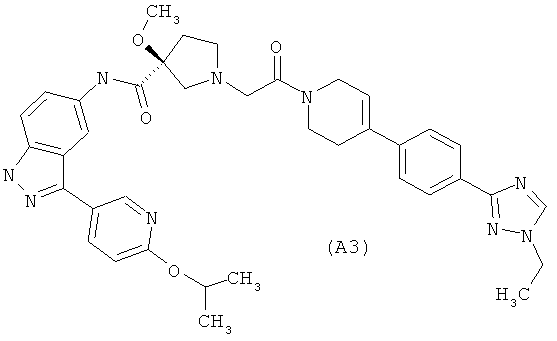 ,
,
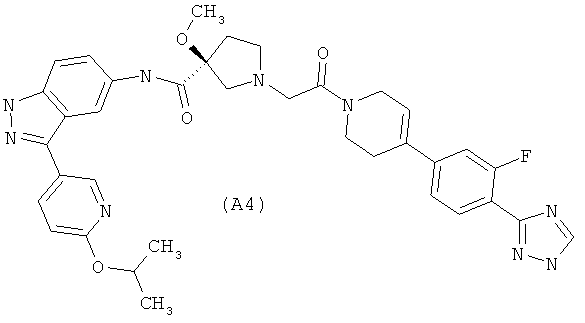 ,
,
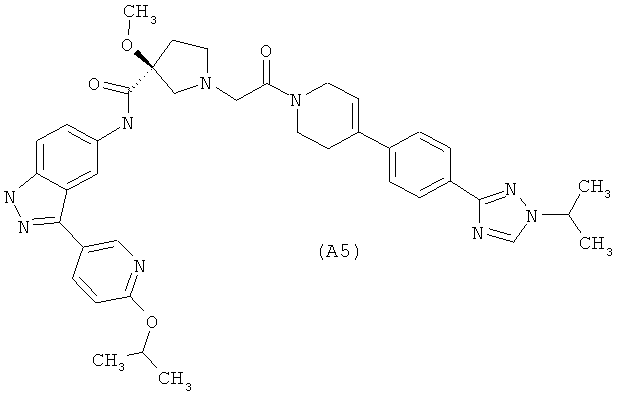 ,
,
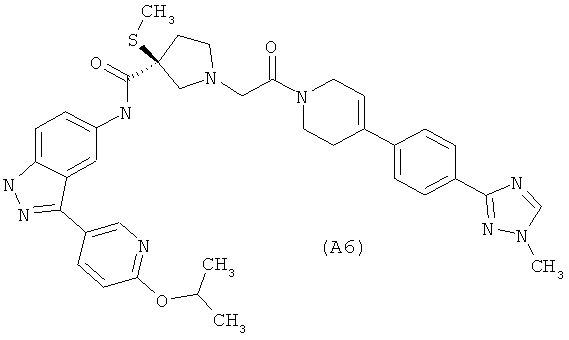 ,
,
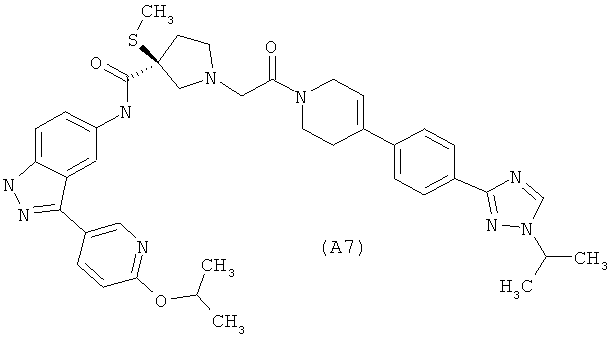 ,
,
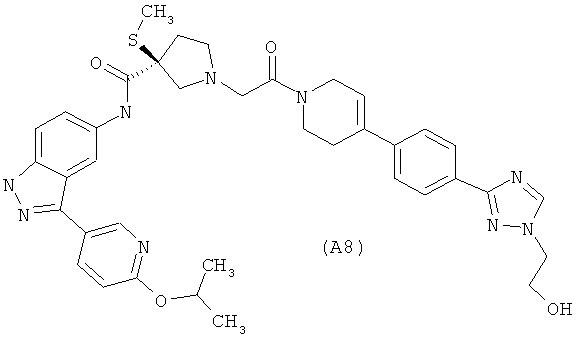 ,
,
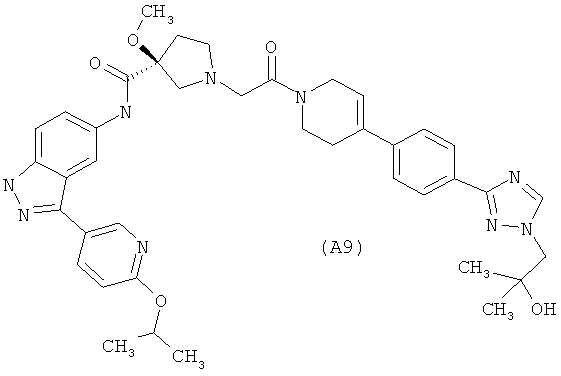 ,
,
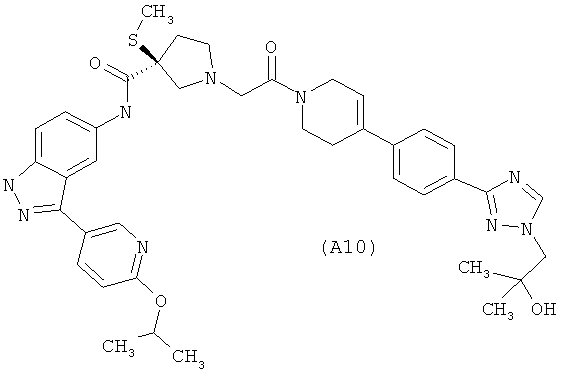 ,
,
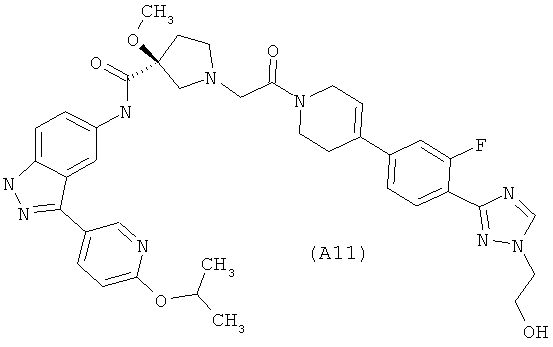 ,
,
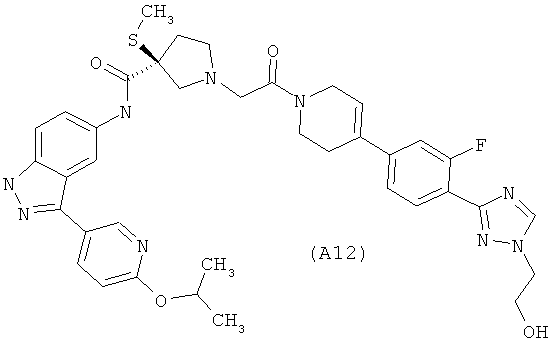 ,
,
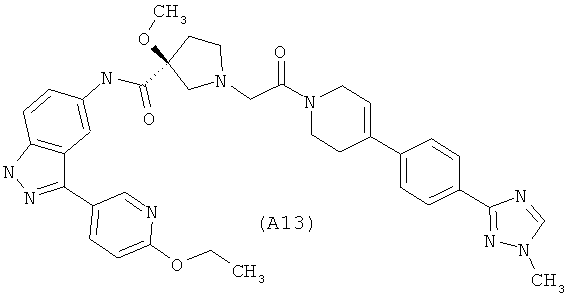 ,
,
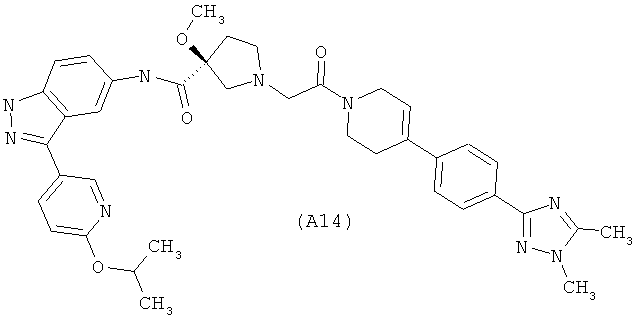 ,
,
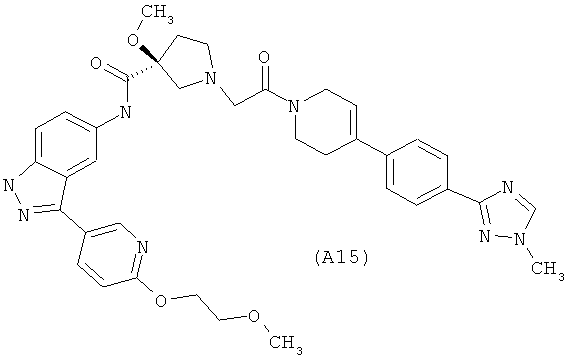 ,
,
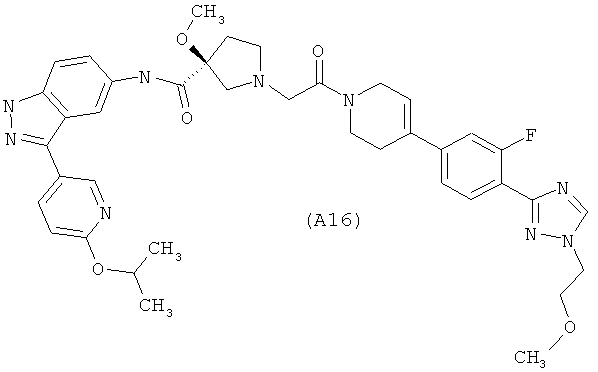 ,
,
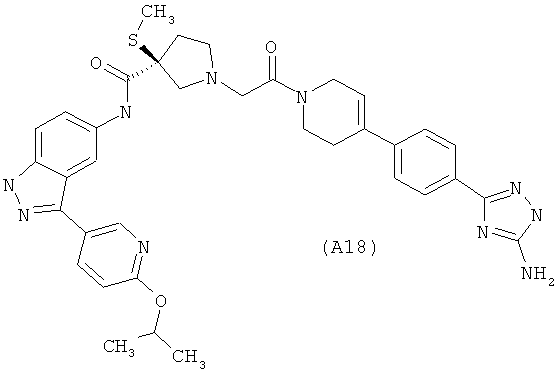 ,
,
,
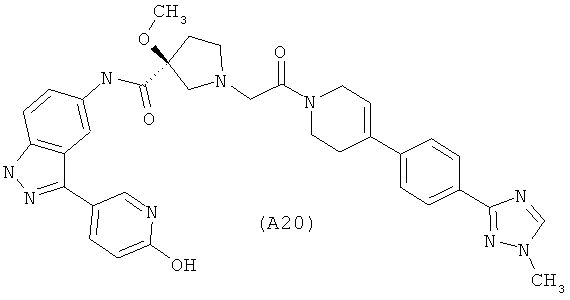 ,
,
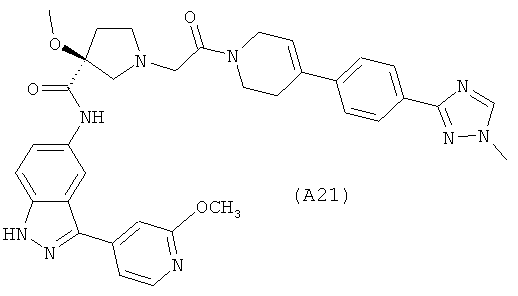 ,
,
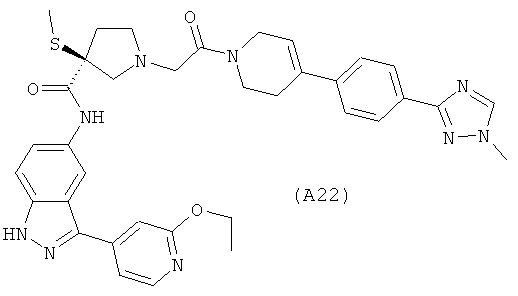 ,
,
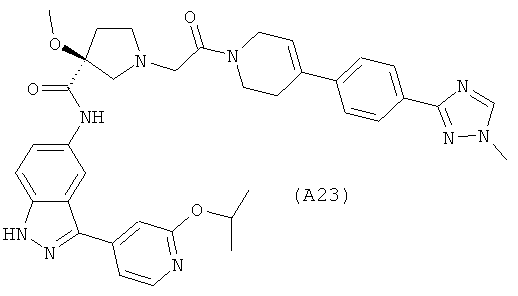 ,
,
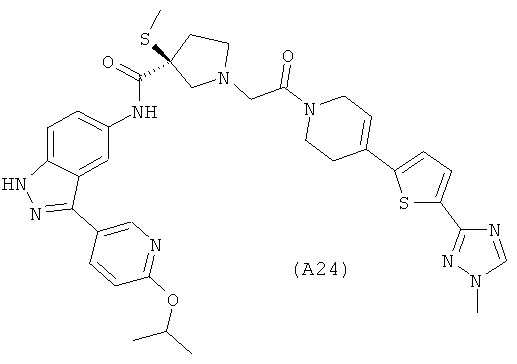 ,
,
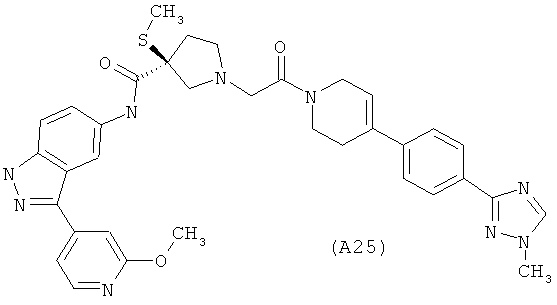 ,
,
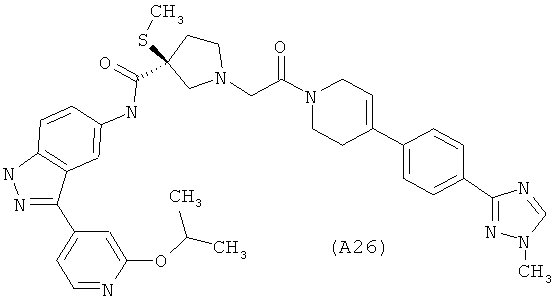 ,
,
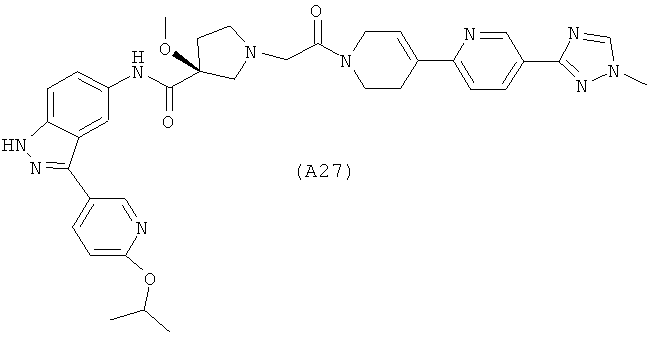 ,
,
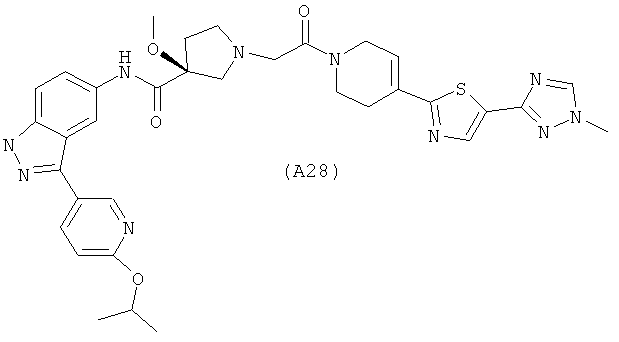 ,
,
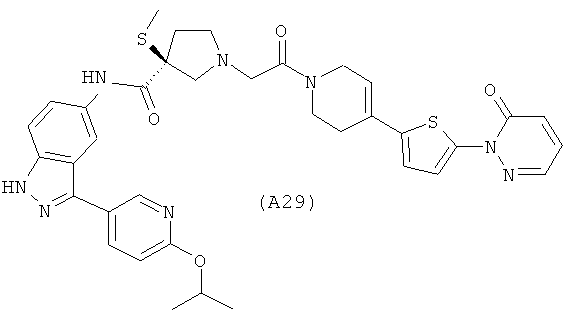 и
и
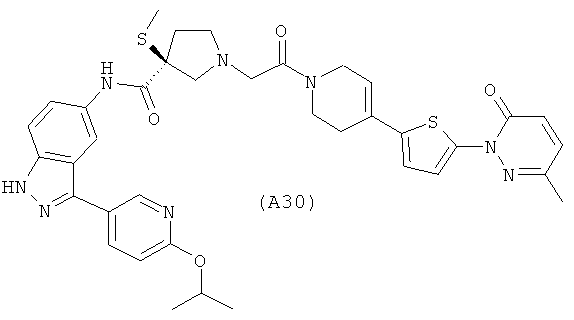 .
.
Типичные представители соединений согласно настоящему изобретению включают, но без ограничения к этому:
,
,
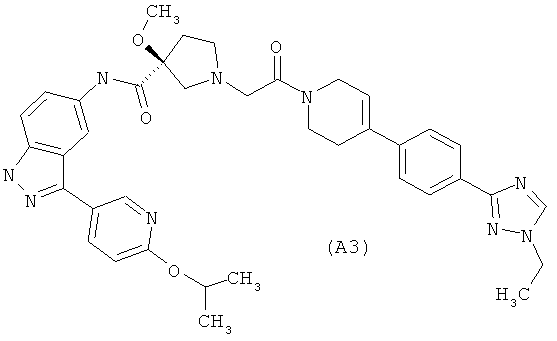 ,
,
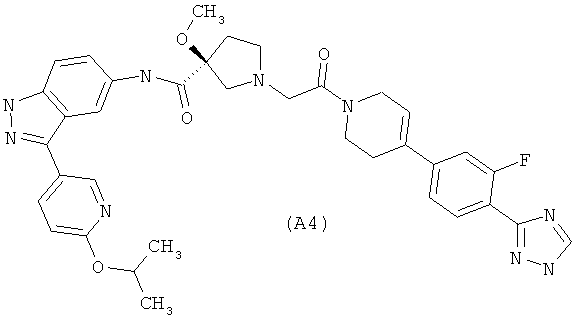 ,
,
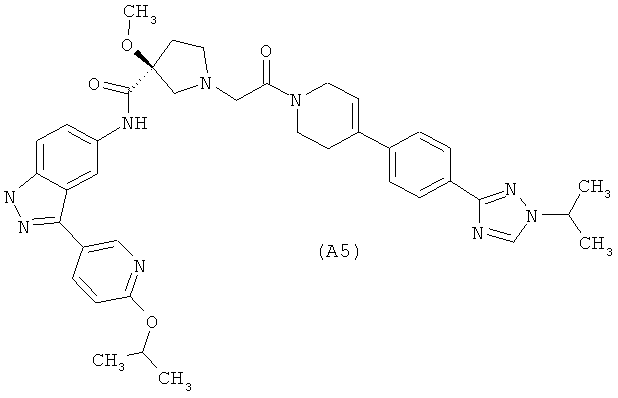 ,
,
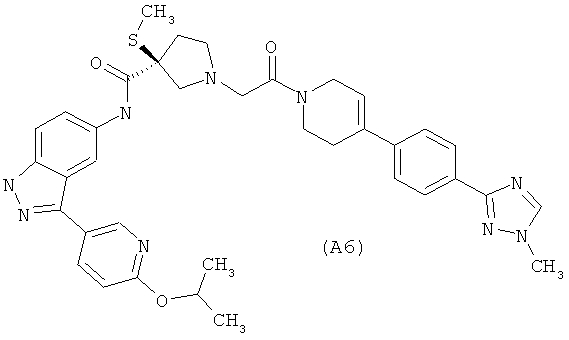 ,
,
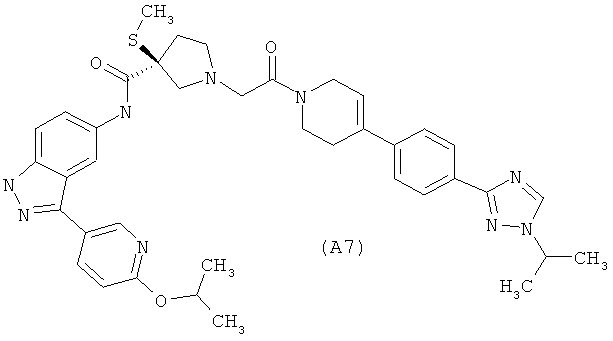 ,
,
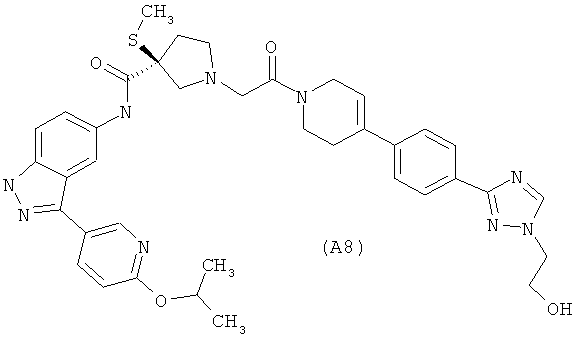 ,
,
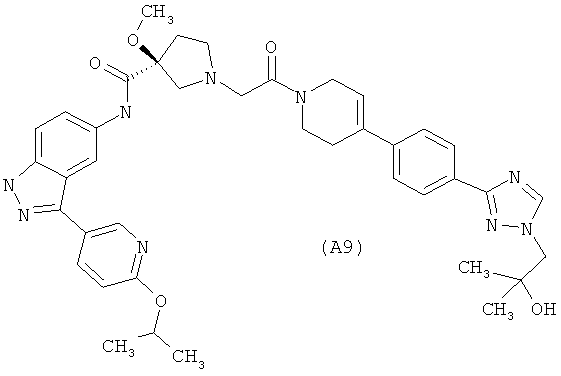 ,
,
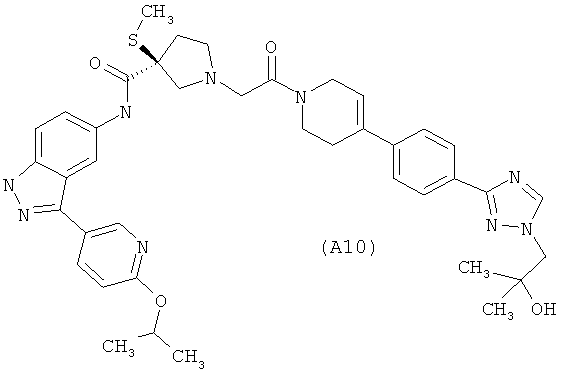 ,
,
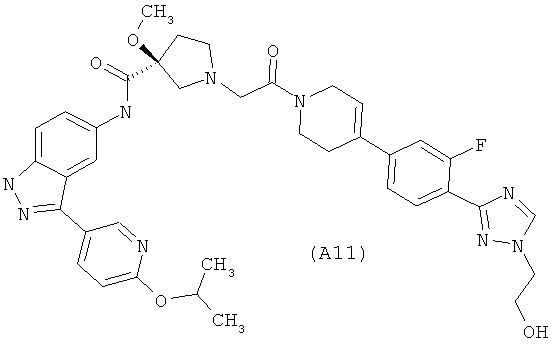 ,
,
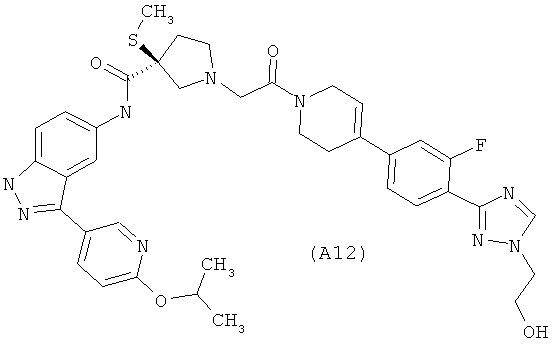 ,
,
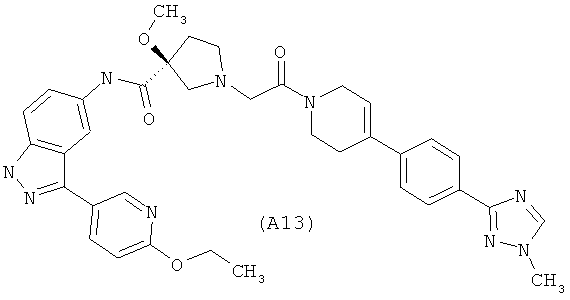 ,
,
,
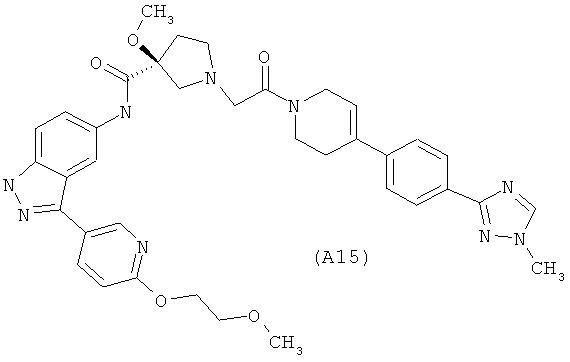 ,
,
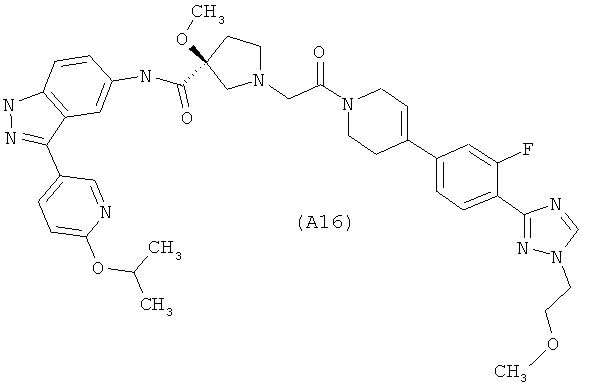 ,
,
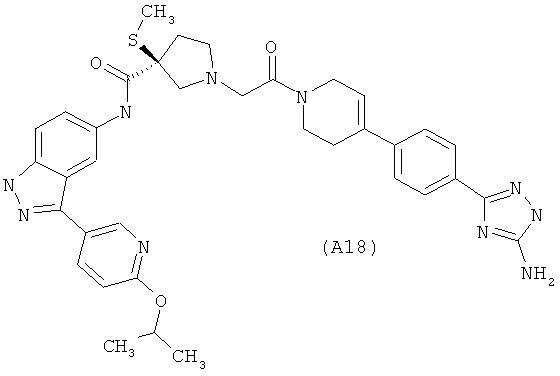 ,
,
,
,
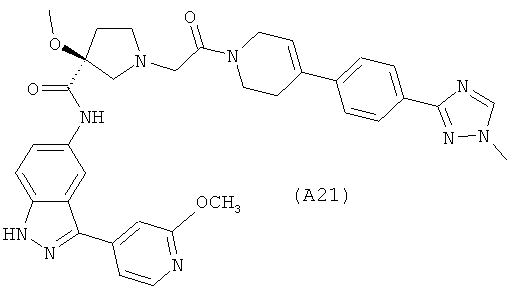 ,
,
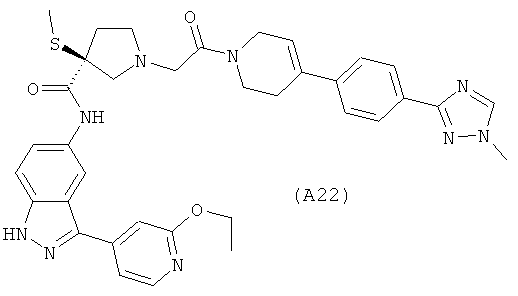 ,
,
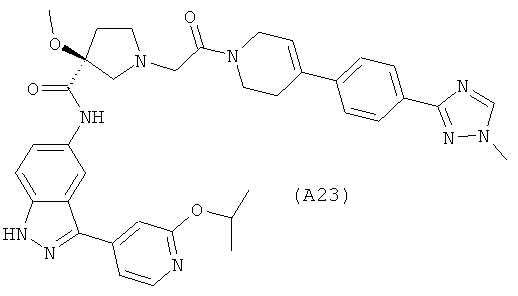 ,
,
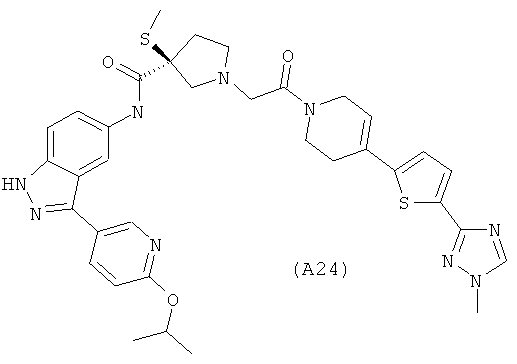 ,
,
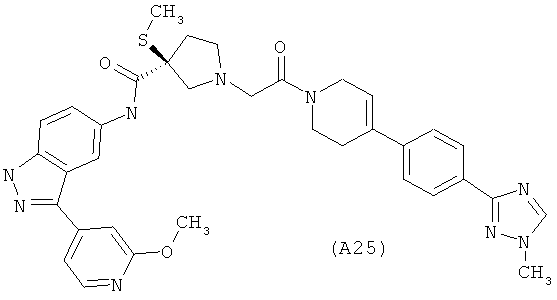 , и
, и
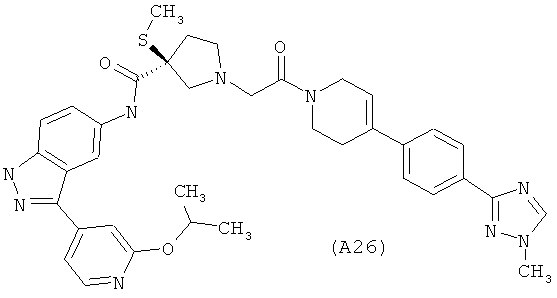 .
.
Типичные представители соединений согласно настоящему изобретению, где водород был замещен дейтерием, включают, но без ограничения к этому:
,
,
,
,
,
,
,
,
,
,
,
,
,
,
,
,
и
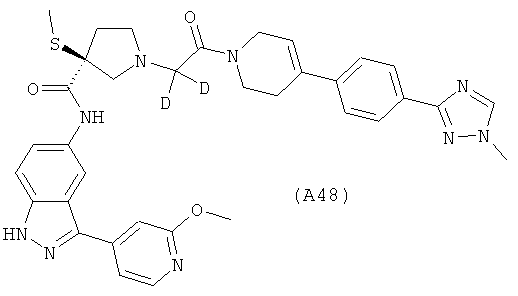 .
.
Другой вариант выполнения настоящего изобретения представляет собой соединение А1.
Другой вариант выполнения настоящего изобретения представляет собой соединение А2.
Другой вариант выполнения настоящего изобретения представляет собой соединение A3.
Другой вариант выполнения настоящего изобретения представляет собой соединение А4.
Другой вариант выполнения настоящего изобретения представляет собой соединение А5.
Другой вариант выполнения настоящего изобретения представляет собой соединение А6.
Другой вариант выполнения настоящего изобретения представляет собой соединение А7.
Другой вариант выполнения настоящего изобретения представляет собой соединение А8.
Другой вариант выполнения настоящего изобретения представляет собой соединение А9.
Другой вариант выполнения настоящего изобретения представляет собой соединение А10.
Другой вариант выполнения настоящего изобретения представляет собой соединение А11.
Другой вариант выполнения настоящего изобретения представляет собой соединение А12.
Другой вариант выполнения настоящего изобретения представляет собой соединение А13.
Другой вариант выполнения настоящего изобретения представляет собой соединение А14.
Другой вариант выполнения настоящего изобретения представляет собой соединение А15.
Другой вариант выполнения настоящего изобретения представляет собой соединение А16.
Другой вариант выполнения настоящего изобретения представляет собой соединение А18.
Другой вариант выполнения настоящего изобретения представляет собой соединение А19.
Другой вариант выполнения настоящего изобретения представляет собой соединение А20.
Другой вариант выполнения настоящего изобретения представляет собой соединение А21.
Другой вариант выполнения настоящего изобретения представляет собой соединение А22.
Другой вариант выполнения настоящего изобретения представляет собой соединение А23.
Другой вариант выполнения настоящего изобретения представляет собой соединение А24.
Другой вариант выполнения настоящего изобретения представляет собой соединение А25.
Другой вариант выполнения настоящего изобретения представляет собой соединение А26.
Другой вариант выполнения настоящего изобретения представляет собой соединение А27.
Другой вариант выполнения настоящего изобретения представляет собой соединение А28.
Другой вариант выполнения настоящего изобретения представляет собой соединение А29.
Другой вариант выполнения настоящего изобретения представляет собой соединение А30.
Другой вариант выполнения настоящего изобретения представляет собой соединение А31.
Другой вариант выполнения настоящего изобретения представляет собой соединение А32.
Другой вариант выполнения настоящего изобретения представляет собой соединение А33.
Другой вариант выполнения настоящего изобретения представляет собой соединение А34.
Другой вариант выполнения настоящего изобретения представляет собой соединение А35.
Другой вариант выполнения настоящего изобретения представляет собой соединение А36.
Другой вариант выполнения настоящего изобретения представляет собой соединение А37.
Другой вариант выполнения настоящего изобретения представляет собой соединение А38.
Другой вариант выполнения настоящего изобретения представляет собой соединение А39.
Другой вариант выполнения настоящего изобретения представляет собой соединение А40.
Другой вариант выполнения настоящего изобретения представляет собой соединение А41.
Другой вариант выполнения настоящего изобретения представляет собой соединение А42.
Другой вариант выполнения настоящего изобретения представляет собой соединение А43.
Другой вариант выполнения настоящего изобретения представляет собой соединение А44.
Другой вариант выполнения настоящего изобретения представляет собой соединение А45.
Другой вариант выполнения настоящего изобретения представляет собой соединение А46.
Другой вариант выполнения настоящего изобретения представляет собой соединение А47.
Другой вариант выполнения настоящего изобретения представляет собой соединение А48.
Другой вариант выполнения настоящего изобретения представляет собой фармацевтически приемлемую соль соединения А1.
Другой вариант выполнения настоящего изобретения представляет собой фармацевтически приемлемую соль соединения А2.
Другой вариант выполнения настоящего изобретения представляет собой фармацевтически приемлемую соль соединения A3.
Другой вариант выполнения настоящего изобретения представляет собой фармацевтически приемлемую соль соединения А4.
Другой вариант выполнения настоящего изобретения представляет собой фармацевтически приемлемую соль соединения А5.
Другой вариант выполнения настоящего изобретения представляет собой фармацевтически приемлемую соль соединения А6.
Другой вариант выполнения настоящего изобретения представляет собой фармацевтически приемлемую соль соединения А7.
Другой вариант выполнения настоящего изобретения представляет собой фармацевтически приемлемую соль соединения А8.
Другой вариант выполнения настоящего изобретения представляет собой фармацевтически приемлемую соль соединения А9.
Другой вариант выполнения настоящего изобретения представляет собой фармацевтически приемлемую соль соединения А10.
Другой вариант выполнения настоящего изобретения представляет собой фармацевтически приемлемую соль соединения А11.
Другой вариант выполнения настоящего изобретения представляет собой фармацевтически приемлемую соль соединения А12.
Другой вариант выполнения настоящего изобретения представляет собой фармацевтически приемлемую соль соединения А13.
Другой вариант выполнения настоящего изобретения представляет собой фармацевтически приемлемую соль соединения А14.
Другой вариант выполнения настоящего изобретения представляет собой фармацевтически приемлемую соль соединения А15.
Другой вариант выполнения настоящего изобретения представляет собой фармацевтически приемлемую соль соединения А16.
Другой вариант выполнения настоящего изобретения представляет собой фармацевтически приемлемую соль соединения А18.
Другой вариант выполнения настоящего изобретения представляет собой фармацевтически приемлемую соль соединения А19.
Другой вариант выполнения настоящего изобретения представляет собой фармацевтически приемлемую соль соединения А20.
Другой вариант выполнения настоящего изобретения представляет собой фармацевтически приемлемую соль соединения А21.
Другой вариант выполнения настоящего изобретения представляет собой фармацевтически приемлемую соль соединения А22.
Другой вариант выполнения настоящего изобретения представляет собой фармацевтически приемлемую соль соединения А23.
Другой вариант выполнения настоящего изобретения представляет собой фармацевтически приемлемую соль соединения А24.
Другой вариант выполнения настоящего изобретения представляет собой фармацевтически приемлемую соль соединения А25.
Другой вариант выполнения настоящего изобретения представляет собой фармацевтически приемлемую соль соединения А26.
Другой вариант выполнения настоящего изобретения представляет собой фармацевтически приемлемую соль соединения А27.
Другой вариант выполнения настоящего изобретения представляет собой фармацевтически приемлемую соль соединения А28.
Другой вариант выполнения настоящего изобретения представляет собой фармацевтически приемлемую соль соединения А29.
Другой вариант выполнения настоящего изобретения представляет собой фармацевтически приемлемую соль соединения А30.
Другой вариант выполнения настоящего изобретения представляет собой фармацевтически приемлемую соль соединения А31.
Другой вариант выполнения настоящего изобретения представляет собой фармацевтически приемлемую соль соединения А32.
Другой вариант выполнения настоящего изобретения представляет собой фармацевтически приемлемую соль соединения А33.
Другой вариант выполнения настоящего изобретения представляет собой фармацевтически приемлемую соль соединения А34.
Другой вариант выполнения настоящего изобретения представляет собой фармацевтически приемлемую соль соединения А35.
Другой вариант выполнения настоящего изобретения представляет собой фармацевтически приемлемую соль соединения А36.
Другой вариант выполнения настоящего изобретения представляет собой фармацевтически приемлемую соль соединения А37.
Другой вариант выполнения настоящего изобретения представляет собой фармацевтически приемлемую соль соединения А38.
Другой вариант выполнения настоящего изобретения представляет собой фармацевтически приемлемую соль соединения А39.
Другой вариант выполнения настоящего изобретения представляет собой фармацевтически приемлемую соль соединения А40.
Другой вариант выполнения настоящего изобретения представляет собой фармацевтически приемлемую соль соединения А41.
Другой вариант выполнения настоящего изобретения представляет собой фармацевтически приемлемую соль соединения А42.
Другой вариант выполнения настоящего изобретения представляет собой фармацевтически приемлемую соль соединения А43.
Другой вариант выполнения настоящего изобретения представляет собой фармацевтически приемлемую соль соединения А44.
Другой вариант выполнения настоящего изобретения представляет собой фармацевтически приемлемую соль соединения А45.
Другой вариант выполнения настоящего изобретения представляет собой фармацевтически приемлемую соль соединения А46.
Другой вариант выполнения настоящего изобретения представляет собой фармацевтически приемлемую соль соединения А47.
Другой вариант выполнения настоящего изобретения представляет собой фармацевтически приемлемую соль соединения А48.
Другой вариант выполнения настоящего изобретения представляет собой сольват соединения А1.
Другой вариант выполнения настоящего изобретения представляет собой сольват соединения А2.
Другой вариант выполнения настоящего изобретения представляет собой сольват соединения A3.
Другой вариант выполнения настоящего изобретения представляет собой сольват соединения А4.
Другой вариант выполнения настоящего изобретения представляет собой сольват соединения А5.
Другой вариант выполнения настоящего изобретения представляет собой сольват соединения А6.
Другой вариант выполнения настоящего изобретения представляет собой сольват соединения А7.
Другой вариант выполнения настоящего изобретения представляет собой сольват соединения А8.
Другой вариант выполнения настоящего изобретения представляет собой сольват соединения А9.
Другой вариант выполнения настоящего изобретения представляет собой сольват соединения А10.
Другой вариант выполнения настоящего изобретения представляет собой сольват соединения А11.
Другой вариант выполнения настоящего изобретения представляет собой сольват соединения А12.
Другой вариант выполнения настоящего изобретения представляет собой сольват соединения А13.
Другой вариант выполнения настоящего изобретения представляет собой сольват соединения А14.
Другой вариант выполнения настоящего изобретения представляет собой сольват соединения А15.
Другой вариант выполнения настоящего изобретения представляет собой сольват соединения А16.
Другой вариант выполнения настоящего изобретения представляет собой сольват соединения А18.
Другой вариант выполнения настоящего изобретения представляет собой сольват соединения А19.
Другой вариант выполнения настоящего изобретения представляет собой сольват соединения А20.
Другой вариант выполнения настоящего изобретения представляет собой сольват соединения А21.
Другой вариант выполнения настоящего изобретения представляет собой сольват соединения А22.
Другой вариант выполнения настоящего изобретения представляет собой сольват соединения А23.
Другой вариант выполнения настоящего изобретения представляет собой сольват соединения А24.
Другой вариант выполнения настоящего изобретения представляет собой сольват соединения А25.
Другой вариант выполнения настоящего изобретения представляет собой сольват соединения А26.
Другой вариант выполнения настоящего изобретения представляет собой сольват соединения А27.
Другой вариант выполнения настоящего изобретения представляет собой сольват соединения А28.
Другой вариант выполнения настоящего изобретения представляет собой сольват соединения А29.
Другой вариант выполнения настоящего изобретения представляет собой сольват соединения А30.
Другой вариант выполнения настоящего изобретения представляет собой сольват соединения А31.
Другой вариант выполнения настоящего изобретения представляет собой сольват соединения А32.
Другой вариант выполнения настоящего изобретения представляет собой сольват соединения А33.
Другой вариант выполнения настоящего изобретения представляет собой сольват соединения А34.
Другой вариант выполнения настоящего изобретения представляет собой сольват соединения А35.
Другой вариант выполнения настоящего изобретения представляет собой сольват соединения А36.
Другой вариант выполнения настоящего изобретения представляет собой сольват соединения А37.
Другой вариант выполнения настоящего изобретения представляет собой сольват соединения А38.
Другой вариант выполнения настоящего изобретения представляет собой сольват соединения А39.
Другой вариант выполнения настоящего изобретения представляет собой сольват соединения А40.
Другой вариант выполнения настоящего изобретения представляет собой сольват соединения А41.
Другой вариант выполнения настоящего изобретения представляет собой сольват соединения А42.
Другой вариант выполнения настоящего изобретения представляет собой сольват соединения А43.
Другой вариант выполнения настоящего изобретения представляет собой сольват соединения А44.
Другой вариант выполнения настоящего изобретения представляет собой сольват соединения А45.
Другой вариант выполнения настоящего изобретения представляет собой сольват соединения А46.
Другой вариант выполнения настоящего изобретения представляет собой сольват соединения А47.
Другой вариант выполнения настоящего изобретения представляет собой сольват соединения А48.
Другие варианты выполнения настоящего изобретения представляют собой любой вариант выполнения изобретения по формуле 1.0, где соединение находится в чистой или выделенной форме.
Другие варианты выполнения настоящего изобретения представляют собой любой вариант выполнения изобретения по формуле 1.0, где соединение находится в чистой форме.
Другие варианты выполнения настоящего изобретения представляют собой любой вариант выполнения изобретения по формуле 1.0, где соединение находится в выделенной форме.
Другие варианты выполнения настоящего изобретения представляют собой любое из соединений А1-А16 и А18-А48 в чистой и выделенной форме.
Другие варианты выполнения настоящего изобретения представляют собой любое соединение из А1-А16 и А18-А30 в чистой и выделенной форме.
Другие варианты выполнения настоящего изобретения представляют собой любое соединение из А1-А16 и А18-А26 в чистой и выделенной форме.
Другие варианты выполнения настоящего изобретения представляют собой любое соединение из А31-А48 в чистой и выделенной форме.
Другие варианты выполнения настоящего изобретения представляют собой любое соединение из А1-А16 и А18-А48 в чистой форме.
Другие варианты выполнения настоящего изобретения представляют собой любое соединение из А1-А16 и А18-А30 в чистой форме.
Другие варианты выполнения настоящего изобретения представляют собой любое соединение из А1-А16 и А18-А26 в чистой форме.
Другие варианты выполнения настоящего изобретения представляют собой любое соединение из А31-А48 в чистой форме.
Другие варианты выполнения настоящего изобретения представляют собой любое соединение из А1-А16 и А18-А48 в выделенной форме.
Другие варианты выполнения настоящего изобретения представляют собой любое соединение из А1-А16 и А18-А30 в выделенной форме.
Другие варианты выполнения настоящего изобретения представляют собой любое соединение из А1-А16 и А18-А26 в выделенной форме.
Другие варианты выполнения настоящего изобретения представляют собой любое соединение из А31-А48 в выделенной форме.
Другой вариант выполнения настоящего изобретения представляет собой фармацевтическую композицию, содержащую эффективное количество, по меньшей мере, одного соединения (например, 1, 2 или 3, или 1 или 2, или 1 и, как правило, 1) формулы 1.0 (предпочтительно формулы 1.1) и фармацевтически приемлемый носитель.
Другой вариант выполнения настоящего изобретения представляет собой фармацевтическую композицию, содержащую эффективное количество соединения формулы 1.0 (предпочтительно формулы 1.1) и фармацевтически приемлемый носитель.
Другой вариант выполнения настоящего изобретения представляет собой фармацевтическую композицию, содержащую эффективное количество, по меньшей мере, одного соединения (например, 1, 2 или 3, или 1 или 2, или 1 и, как правило, 1), выбранного из группы, состоящей из: А1-А16 и А18-А48, и фармацевтически приемлемый носитель.
Другой вариант выполнения настоящего изобретения представляет собой фармацевтическую композицию, содержащую эффективное количество, по меньшей мере, одного соединения (например, 1, 2 или 3, или 1 или 2, или 1 и, как правило, 1), выбранного из группы, состоящей из: А1-А16 и А18-А30, и фармацевтически приемлемый носитель.
Другой вариант выполнения настоящего изобретения представляет собой фармацевтическую композицию, содержащую эффективное количество, по меньшей мере, одного соединения (например, 1, 2 или 3, или 1 или 2, или 1 и, как правило, 1), выбранного из группы, состоящей из: А1-А16 и А18-А26, и фармацевтически приемлемый носитель.
Другой вариант выполнения настоящего изобретения представляет собой фармацевтическую композицию, содержащую эффективное количество, по меньшей мере, одного соединения (например, 1, 2 или 3, или 1 или 2, или 1 и, как правило, 1), выбранного из группы, состоящей из: А31-А48, и фармацевтически приемлемый носитель.
Другой вариант выполнения настоящего изобретения представляет собой фармацевтическую композицию, содержащую эффективное количество соединения, выбранного из группы, состоящей из: А1-А16 и А18-А48, и фармацевтически приемлемый носитель.
Другой вариант выполнения настоящего изобретения представляет собой фармацевтическую композицию, содержащую эффективное количество соединения, выбранного из группы, состоящей из: А1-А16 и А18-А30, и фармацевтически приемлемый носитель.
Другой вариант выполнения настоящего изобретения представляет собой фармацевтическую композицию, содержащую эффективное количество соединения, выбранного из группы, состоящей из: А1-А16 и А18-А26, и фармацевтически приемлемый носитель.
Другой вариант выполнения настоящего изобретения представляет собой фармацевтическую композицию, содержащую эффективное количество соединения, выбранного из группы, состоящей из: А31-А48, и фармацевтически приемлемый носитель.
Другой вариант выполнения настоящего изобретения представляет собой фармацевтическую композицию, содержащую эффективное количество, по меньшей мере, одного соединения (например, 1, 2 или 3, или 1 или 2, или 1 и, как правило, 1) формулы 1.0 (предпочтительно формулы 1.1), по меньшей мере, один (например, 1, 2 или 3, или 1 или 2, или 1 и, как правило, 1) другой фармацевтически активный ингредиент, и фармацевтически приемлемый носитель.
Другой вариант выполнения настоящего изобретения представляет собой фармацевтическую композицию, содержащую эффективное количество соединения формулы 1.0 (предпочтительно формулы 1.1), другой фармацевтически активный ингредиент и фармацевтически приемлемый носитель.
Другой вариант выполнения настоящего изобретения представляет собой фармацевтическую композицию, содержащую эффективное количество, по меньшей мере, одного соединения (например, 1, 2 или 3, или 1 или 2, или 1 и, как правило, 1), выбранного из группы, состоящей из: А1-А16 и А18-А48, по меньшей мере, один (например, 1, 2 или 3, или 1 или 2, или 1 и, как правило, 1) другой фармацевтически активный ингредиент и фармацевтически приемлемый носитель.
Другой вариант выполнения настоящего изобретения представляет собой фармацевтическую композицию, содержащую эффективное количество, по меньшей мере, одного соединения (например, 1, 2 или 3, или 1 или 2, или 1 и, как правило, 1), выбранного из группы, состоящей из: А1-А16 и А18-А30, по меньшей мере, один (например, 1, 2 или 3, или 1 или 2, или 1 и, как правило, 1) другой фармацевтически активный ингредиент и фармацевтически приемлемый носитель.
Другой вариант выполнения настоящего изобретения представляет собой фармацевтическую композицию, содержащую эффективное количество, по меньшей мере, одного соединения (например, 1, 2 или 3, или 1 или 2, или 1 и, как правило, 1), выбранного из группы, состоящей из: А1-А16 и А18-А26, по меньшей мере, один (например, 1, 2 или 3, или 1 или 2, или 1 и, как правило, 1) другой фармацевтически активный ингредиент и фармацевтически приемлемый носитель.
Другой вариант выполнения настоящего изобретения представляет собой фармацевтическую композицию, содержащую эффективное количество, по меньшей мере, одного соединения (например, 1, 2 или 3, или 1 или 2, или 1 и, как правило, 1), выбранного из группы, состоящей из: А31-А48, по меньшей мере, один (например, 1, 2 или 3, или 1 или 2, или 1 и, как правило, 1) другой фармацевтически активный ингредиент, и фармацевтически приемлемый носитель.
Другой вариант выполнения настоящего изобретения представляет собой фармацевтическую композицию, содержащую эффективное количество соединения, выбранного из группы, состоящей из: А1-А16 и А18-А48, другой фармацевтически активный ингредиент и фармацевтически приемлемый носитель.
Другой вариант выполнения настоящего изобретения представляет собой фармацевтическую композицию, содержащую эффективное количество соединения, выбранного из группы, состоящей из: А1-А16 и А18-А30, другой фармацевтически активный ингредиент и фармацевтически приемлемый носитель.
Другой вариант выполнения настоящего изобретения представляет собой фармацевтическую композицию, содержащую эффективное количество соединения, выбранного из группы, состоящей из: А1-А16 и А18-А26, другой фармацевтически активный ингредиент и фармацевтически приемлемый носитель.
Другой вариант выполнения настоящего изобретения представляет собой фармацевтическую композицию, содержащую эффективное количество соединения, выбранного из группы, состоящей из: А31-А48, другой фармацевтически активный ингредиент и фармацевтически приемлемый носитель.
Другой вариант выполнения настоящего изобретения представляет собой фармацевтическую композицию, содержащую эффективное количество, по меньшей мере, одного соединения (например, 1, 2 или 3, или 1 или 2, или 1 и, как правило, 1) формулы 1.0 (предпочтительно формулы 1.1), по меньшей мере, один (например, 1, 2 или 3, или 1 или 2, или 1 и, как правило, 1) химиотерапевтический агент и фармацевтически приемлемый носитель.
Другой вариант выполнения настоящего изобретения представляет собой фармацевтическую композицию, содержащую эффективное количество соединения формулы 1.0 (предпочтительно формулы 1.1), химиотерапевтический агент и фармацевтически приемлемый носитель.
Другой вариант выполнения настоящего изобретения представляет собой фармацевтическую композицию, содержащую эффективное количество, по меньшей мере, одного соединения (например, 1, 2 или 3, или 1 или 2, или 1 и, как правило, 1), выбранного из группы, состоящей из: А1-А16 и А18-А48, по меньшей мере, один (например, 1, 2 или 3, или 1 или 2, или 1 и, как правило, 1) химиотерапевтический агент и фармацевтически приемлемый носитель.
Другой вариант выполнения настоящего изобретения представляет собой фармацевтическую композицию, содержащую эффективное количество, по меньшей мере, одного соединения (например, 1, 2 или 3, или 1 или 2, или 1 и, как правило, 1), выбранного из группы, состоящей из: А1-А16 и А18-А30, по меньшей мере, один (например, 1, 2 или 3, или 1 или 2, или 1 и, как правило, 1) химиотерапевтический агент и фармацевтически приемлемый носитель.
Другой вариант выполнения настоящего изобретения представляет собой фармацевтическую композицию, содержащую эффективное количество, по меньшей мере, одного соединения (например, 1, 2 или 3, или 1 или 2, или 1 и, как правило, 1), выбранного из группы, состоящей из: А1-А16 и А18-А26, по меньшей мере, один (например, 1, 2 или 3, или 1 или 2, или 1 и, как правило, 1) химиотерапевтический агент и фармацевтически приемлемый носитель.
Другой вариант выполнения настоящего изобретения представляет собой фармацевтическую композицию, содержащую эффективное количество, по меньшей мере, одного соединения (например, 1, 2 или 3, или 1 или 2, или 1 и, как правило, 1), выбранного из группы, состоящей из: А31-А48, по меньшей мере, один (например, 1, 2 или 3, или 1 или 2, или 1 и, как правило, 1) химиотерапевтический агент, и фармацевтически приемлемый носитель.
Другой вариант выполнения настоящего изобретения представляет собой фармацевтическую композицию, содержащую эффективное количество соединения, выбранного из группы, состоящей из: А1-А16 и А18-А48, химиотерапевтический агент и фармацевтически приемлемый носитель.
Другой вариант выполнения настоящего изобретения представляет собой фармацевтическую композицию, содержащую эффективное количество соединения, выбранного из группы, состоящей из: А1-А16 и А18-А30, химиотерапевтический агент и фармацевтически приемлемый носитель.
Другой вариант выполнения настоящего изобретения представляет собой фармацевтическую композицию, содержащую эффективное количество соединения, выбранного из группы, состоящей из: А1-А16 и А18-А26, химиотерапевтический агент и фармацевтически приемлемый носитель.
Другой вариант выполнения настоящего изобретения представляет собой фармацевтическую композицию, содержащую эффективное количество соединения, выбранного из группы, состоящей из: А31-А48, химиотерапевтический агент и фармацевтически приемлемый носитель.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения рака у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества, по меньшей мере, одного (1, 2 или 3, или 1 или 2, или 1 и, как правило, 1) соединения формулы 1.0 (предпочтительно формулы 1.6).
Другой вариант выполнения настоящего изобретения представляет собой способ лечения рака у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества одного соединения формулы 1.0 (предпочтительно формулы 1.6).
Другой вариант выполнения настоящего изобретения представляет собой способ лечения рака у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества, по меньшей мере, одного (1, 2 или 3, или 1 или 2, или 1 и, как правило, 1) соединения, выбранного из группы А1-А16 и А18-А48.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения рака у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества, по меньшей мере, одного (1, 2 или 3, или 1 или 2, или 1 и, как правило, 1) соединения, выбранного из группы А1-А16 и А18-А30.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения рака у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества, по меньшей мере, одного (1, 2 или 3, или 1 или 2, или 1 и, как правило, 1) соединения, выбранного из группы А1-А16 и А18-А26.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения рака у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества, по меньшей мере, одного (1, 2 или 3, или 1 или 2, или 1 и, как правило, 1) соединения, выбранного из группы А31-А48.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения рака у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества одного соединения, выбранного из группы А1-А16 и А18-А48.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения рака у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества одного соединения, выбранного из группы А1-А16 и А18-А30.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения рака у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества одного соединения, выбранного из группы А1-А16 и А18-А26.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения рака у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества одного соединения, выбранного из группы А31-А48.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения рака у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества, по меньшей мере, одного (1, 2 или 3, или 1, или 2 или 1 и, как правило, 1) соединения формулы 1.0, и эффективного количества, по меньшей мере, одного (1, 2 или 3, или 1 или 2, или 1 и, как правило, 1) химиотерапевтического агента.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения рака у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества одного соединения формулы 1.0, и эффективного количества, по меньшей мере, одного (1, 2 или 3, или 1 или 2, или 1 и, как правило, 1) химиотерапевтического агента.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения рака у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества, по меньшей мере, одного (1, 2 или 3, или 1 или 2, или 1 и, как правило, 1) соединения, выбранного из группы А1-А16 и А18-А48, и эффективного количества, по меньшей мере, одного (1, 2 или 3, или 1 или 2, или 1 и, как правило, 1) химиотерапевтического агента.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения рака у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества, по меньшей мере, одного (1, 2 или 3, или 1 или 2, или 1 и, как правило, 1) соединения, выбранного из группы А1-А16 и А18-А30, и эффективного количества, по меньшей мере, одного (1, 2 или 3, или 1 или 2, или 1 и, как правило, 1) химиотерапевтического агента.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения рака у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества, по меньшей мере, одного (1, 2 или 3, или 1 или 2, или 1 и, как правило, 1) соединения, выбранного из группы А1-А16 и А18-А26, и эффективного количества, по меньшей мере, одного (1, 2 или 3, или 1 или 2, или 1 и, как правило, 1) химиотерапевтического агента.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения рака у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества, по меньшей мере, одного (1, 2 или 3, или 1 или 2, или 1 и, как правило, 1) соединения, выбранного из группы А31-А48, и эффективного количества, по меньшей мере, одного (1, 2 или 3, или 1 или 2, или 1 и, как правило, 1) химиотерапевтического агента.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения рака у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества одного соединения, выбранного из группы А1-А16 и А18-А48, и эффективного количества, по меньшей мере, одного (1, 2 или 3, или 1 или 2, или 1 и, как правило, 1) химиотерапевтического агента.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения рака у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества одного соединения, выбранного из группы А1-А16 и А18-А30, и эффективного количества, по меньшей мере, одного (1, 2 или 3, или 1 или 2, или 1 и, как правило, 1) химиотерапевтического агента.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения рака у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества одного соединения, выбранного из группы А1-А16 и А18-А26, и эффективного количества, по меньшей мере, одного (1, 2 или 3, или 1 или 2, или 1 и, как правило, 1) химиотерапевтического агента.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения рака у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества одного соединения, выбранного из группы А31-А48, и эффективного количества, по меньшей мере, одного (1, 2 или 3, или 1 или 2, или 1 и, как правило, 1) химиотерапевтического агента.
Соединения согласно настоящему изобретению ингибируют активность ERK1 и ERK2. Таким образом, настоящее изобретение, кроме того, раскрывает способ ингибирования ERK у млекопитающих, особенно людей, путем введения эффективного количества (например, терапевтически эффективного количества) одного или более (например, одного) соединений согласно настоящему изобретению. Введение соединений согласно настоящему изобретению пациентам с целью ингибирования ERK1 и/или ERK2 полезно для лечения рака.
В любом из способов лечения рака, описанных в настоящем изобретении, если иного не указано, способы могут необязательно включать введение одного или более (например, 1, 2 или 3, или 1 или 2, или 1) химиотерапевтического агента. Химиотерапевтические агенты могут вводиться одновременно или последовательно с соединениями согласно настоящему изобретению.
Способы лечения рака, описанные в настоящем изобретении, включают способы, в которых применяется комбинация лекарственных средств (то есть соединений или фармацевтически активный ингредиентов, или фармацевтических композиций) (то есть способы лечения рака согласно настоящему изобретению включают комбинаторные терапии). Специалистам в данной области техники очевидно, что лекарственные средства в общем вводятся индивидуально в виде фармацевтической композиции. Применение фармацевтической композиции, содержащей более чем одно лекарственное средство, входит в объем настоящего изобретения.
В любом из способов лечения рака, описанных в настоящем изобретении, если иного не указано, способы необязательно могут включать эффективное количество радиотерапии. В случае радиотерапии предпочтительным является γ-излучение
Примеры раковых заболеваний, которые можно лечить с помощью способов согласно настоящему изобретению, включают, но без ограничения к этому: (А) рак легких (например, аденокарцинома легких и немелкоклеточный рак легких), (В) рак поджелудочной железы (например, карцинома поджелудочной железы, такая как, например, карцинома экзокринной части поджелудочной железы), (С) рак ободочной кишки (например, колоректальная карцинома, такой как, например, аденокарцинома ободочной кишки и аденома ободочной кишки), (D) миелоидную лейкемию (например, острая миелогенная лейкемия (AML), CML и CMML), (Е) рак щитовидной железы, (F) миелодиспластический синдром (MDS), (G) карциному мочевого пузыря, (Н) эпидермальную карциному, (I) меланому, (J) рак молочной железы, (K) рак предстательной железы, (L) рак головы и шеи (например, плоскоклеточный рак головы и шеи), (М) рак яичников, (N) рак головного мозга (например, глиома, такая как мультиформная глиобластома), (О) рак мезенхимального происхождения (например, фибросаркома и рабдомиосаркома), (Р) саркомы, (Q) тетракарциномы, (R) нейробластомы, (S) рак почек, (Т) гепатомы, (U) неходжкинскую лимфому, (V) множественную миелому и (W) анапластическую карциному щитовидной железы.
Таким образом, другой вариант выполнения настоящего изобретения представляет собой способ лечения рака легких, рака поджелудочной железы, рака ободочной кишки (например, колоректальный рак), миелоидной лейкемии (например, AML, CML и CMML), рака щитовидной железы, миелодиспластического синдрома (MDS), карциномы мочевого пузыря, эпидермальной карциномы, меланомы, рака молочной железы, рака предстательной железы, рака головы и шеи (например, плоскоклеточного рака головы и шеи), рака яичников, рака головного мозга (например, глиом, таких как мультиформная глиобластома), рака мезенхимального происхождения (например, фибросаркома и рабдомиосаркома), саркомы, тетракарциномы, нейробластомы, карциномы почки, гепатом, неходжкинской лимфомы, множественной миеломы или анапластической карциномы щитовидной железы, у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединения формулы 1.0.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения рака легких, рака поджелудочной железы, рака ободочной кишки (например, колоректальный рак), миелоидной лейкемии (например, AML, CML и CMML), рака щитовидной железы, миелодиспластического синдрома (MDS), карциномы мочевого пузыря, эпидермальной карциномы, меланомы, рака молочной железы, рака предстательной железы, рака головы и шеи (например, плоскоклеточного рака головы и шеи), рака яичников, рака головного мозга (например, глиом, таких как мультиформная глиобластома), рака мезенхимального происхождения (например, фибросаркома и рабдомиосаркома), саркомы, тетракарциномы, нейробластомы, карциномы почки, гепатом, неходжкинской лимфомы, множественной миеломы или анапластической карциномы щитовидной железы, у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединения формулы 1.0, в комбинации с эффективным количеством, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2, или 1) химиотерапевтического агента.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения рака легких, рака поджелудочной железы, рака ободочной кишки (например, колоректальный рак), миелоидной лейкемии (например, AML, CML и CMML), рака щитовидной железы, миелодиспластического синдрома (MDS), карциномы мочевого пузыря, эпидермальной карциномы, меланомы, рака молочной железы, рака предстательной железы, рака головы и шеи (например, плоскоклеточного рака головы и шеи), рака яичников, рака головного мозга (например, глиом, таких как мультиформная глиобластома), рака мезенхимального происхождения (например, фибросаркома и рабдомиосаркома), саркомы, тетракарциномы, нейробластомы, карциномы почки, гепатом, неходжкинской лимфомы, множественной миеломы или анапластической карциномы щитовидной железы, у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества фармацевтической композиции, содержащей эффективное количество, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединения формулы 1.0.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения рака легких, рака поджелудочной железы, рака ободочной кишки (например, колоректальный рак), миелоидной лейкемии (например, AML, CML и CMML), рака щитовидной железы, миелодиспластического синдрома (MDS), карциномы мочевого пузыря, эпидермальной карциномы, меланомы, рака молочной железы, рака предстательной железы, рака головы и шеи (например, плоскоклеточного рака головы и шеи), рака яичников, рака головного мозга (например, глиом, таких как мультиформная глиобластома), рака мезенхимального происхождения (например, фибросаркома и рабдомиосаркома), саркомы, тетракарциномы, нейробластомы, карциномы почки, гепатом, неходжкинской лимфомы, множественной миеломы или анапластической карциномы щитовидной железы, у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества фармацевтической композиции, содержащей эффективное количество, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединения формулы 1.0, в комбинации с эффективным количеством, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2, или 1) химиотерапевтического агента.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения рака у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединения формулы 1.0, когда указанный рак выбирается из группы, состоящей из: меланомы, рака поджелудочной железы, рака щитовидной железы, колоректального рака, рака легких, рака молочной железы и рака яичников.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения рака у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединения формулы 1.0, в комбинации с эффективным количеством, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2, или 1) химиотерапевтического агента, когда указанный рак выбирается из группы, состоящей из: меланомы, рака поджелудочной железы, рака щитовидной железы, колоректального рака, рака легких, рака молочной железы и рака яичников.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения рака у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества фармацевтической композиции, содержащей эффективное количество, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединения формулы 1.0, когда указанный рак выбирается из группы, состоящей из: меланомы, рака поджелудочной железы, рака щитовидной железы, колоректального рака, рака легких, рака молочной железы и рака яичников.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения рака у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества фармацевтической композиции, содержащей эффективное количество, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединения формулы 1.0, в комбинации с эффективным количеством, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2, или 1) химиотерапевтического агента, когда указанный рак выбирается из группы, состоящей из: меланомы, рака поджелудочной железы, рака щитовидной железы, колоректального рака, рака легких, рака молочной железы и рака яичников.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения меланомы у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединения формулы 1.0.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения меланомы у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединения формулы 1.0, в комбинации с эффективным количеством, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2, или 1) химиотерапевтического агента.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения меланомы у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества фармацевтической композиции, содержащей эффективное количество, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединения формулы 1.0.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения меланомы у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества фармацевтической композиции, содержащей эффективное количество, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединения формулы 1.0, в комбинации с эффективным количеством, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2, или 1) химиотерапевтического агента.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения рака поджелудочной железы у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединения формулы 1.0.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения рака поджелудочной железы у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединения формулы 1.0, в комбинации с эффективным количеством, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2, или 1) химиотерапевтического агента.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения рака поджелудочной железы у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества фармацевтической композиции, содержащей эффективное количество, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединения формулы 1.0.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения рака поджелудочной железы у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества фармацевтической композиции, содержащей эффективное количество, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединения формулы 1.0, в комбинации с эффективным количеством, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2, или 1) химиотерапевтического агента.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения рака щитовидной железы у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединения формулы 1.0.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения рака щитовидной железы у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединение формулы 1.0, в комбинации с эффективным количеством, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2, или 1) химиотерапевтического агента.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения рака щитовидной железы у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества фармацевтической композиции, содержащей эффективное количество, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединения формулы 1.0.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения рака щитовидной железы у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества фармацевтической композиции, содержащей эффективное количество, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединения формулы 1.0, в комбинации с эффективным количеством, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2, или 1) химиотерапевтического агента.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения колоректального рака у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединения формулы 1.0.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения колоректального рака у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединения формулы 1.0, в комбинации с эффективным количеством, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2, или 1) химиотерапевтического агента.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения колоректального рака у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества фармацевтической композиции, содержащей эффективное количество, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединения формулы 1.0.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения колоректального рака у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества фармацевтической композиции, содержащей эффективное количество, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединения формулы 1.0, в комбинации с эффективным количеством, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2, или 1) химиотерапевтического агента.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения рака легких у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединения формулы 1.0.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения рака легких у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединения формулы 1.0, в комбинации с эффективным количеством, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2, или 1) химиотерапевтического агента.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения рака легких у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества фармацевтической композиции, содержащей эффективное количество, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединения формулы 1.0.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения рака легких у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества фармацевтической композиции, содержащей эффективное количество, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединения формулы 1.0, в комбинации с эффективным количеством, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2, или 1) химиотерапевтического агента.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения рака молочной железы у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединения формулы 1.0.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения рака молочной железы у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединения формулы 1.0, в комбинации с эффективным количеством, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2, или 1) химиотерапевтического агента.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения рака молочной железы у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества фармацевтической композиции, содержащей эффективное количество, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединения формулы 1.0.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения рака молочной железы у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества фармацевтической композиции, содержащей эффективное количество, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединения формулы 1.0, в комбинации с эффективным количеством, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2, или 1) химиотерапевтического агента.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения рака яичников у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединения формулы 1.0.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения рака яичников у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединения формулы 1.0, в комбинации с эффективным количеством, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2, или 1) химиотерапевтического агента.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения рака яичников у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества фармацевтической композиции, содержащей эффективное количество, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединения формулы 1.0.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения рака яичников у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества фармацевтической композиции, содержащей эффективное количество, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединения формулы 1.0, в комбинации с эффективным количеством, по меньшей мере; одного (например, 1, 2 или 3, 1 или 2, или 1) химиотерапевтического агента.
Другие варианты выполнения настоящего изобретения представляют собой способы лечения рака молочной железы (то есть, постклимактерического и предклимактерического рака молочной железы, например, гормонально-зависимого рака молочной железы) у пациента, которому необходимо такое лечение, причем указанное лечение включает введение эффективного количества, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединения формулы 1.0, в комбинации с гормональными терапиями (то есть антигормональными средствами).
Другие варианты выполнения настоящего изобретения представляют собой способы лечения рака молочной железы (то есть, постклимактерического и предклимактерического рака молочной железы, например, гормонально-зависимого рака молочной железы) у пациента, которому необходимо такое лечение, причем указанное лечение включает введение эффективного количества фармацевтической композиции, содержащей эффективное количество, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединения формулы 1.0, в комбинации с гормональными терапиями (то есть антигормональными средствами).
Другие варианты выполнения настоящего изобретения представляют собой способы лечения рака молочной железы (то есть, постклимактерического и предклимактерического рака молочной железы, например, гормонально-зависимого рака молочной железы) у пациента, которому необходимо такое лечение, причем указанное лечение включает введение эффективного количества, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединения формулы 1.0, в комбинации с гормональными терапиями (то есть, антигормональными средствами) и в комбинации с эффективным количеством, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2, или 1) химиотерапевтического агента.
Другие варианты выполнения настоящего изобретения представляют собой способы лечения рака молочной железы (то есть, постклимактерического и предклимактерического рака молочной железы, например, гормонально-зависимого рака молочной железы) у пациента, которому необходимо такое лечение, причем указанное лечение включает введение эффективного количества фармацевтической композиции, содержащей эффективное количество, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединения формулы 1.0, в комбинации с гормональными терапиями (то есть, антигормональными средствами) и в комбинации с эффективным количеством, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2, или 1) химиотерапевтического агента.
Способы лечения рака молочной железы, описанные в настоящей заявке, включают лечение гормонально-зависимого метастатического и запущенного рака молочной железы, адъювантную терапию для гормонально-зависимого первичного и раннего рака молочной железы, in situ лечение дуктальной карциномы и in situ лечение воспалительного рака молочной железы.
Способы лечения гормонально-зависимого рака молочной железы также могут применяться для предотвращения рака молочной железы у пациентов, имеющих высокий риск развития рака молочной железы.
Таким образом, другие варианты выполнения настоящего изобретения представляют собой способы предотвращения рака молочной железы (то есть, постклимактерического и предклимактерического рака молочной железы, например, гормонально-зависимого рака молочной железы) у пациента, которому необходимо такое лечение, причем указанное лечение включает введение эффективного количества, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединения формулы 1.0 в комбинации с гормональными терапиями (то есть, антигормональными средствами).
Другие варианты выполнения настоящего изобретения представляют собой способы предотвращения рака молочной железы (то есть, постклимактерического и предклимактерического рака молочной железы, например, гормонально-зависимого рака молочной железы) у пациента, которому необходимо такое лечение, причем указанное лечение включает введение эффективного количества фармацевтической композиции, содержащей эффективное количество, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединения формулы 1.0, в комбинации с гормональными терапиями (то есть, антигормональными средствами).
Другие варианты выполнения настоящего изобретения представляют собой способы предотвращения рака молочной железы (то есть, постклимактерического и предклимактерического рака молочной железы, например, гормонально-зависимого рака молочной железы) у пациента, которому необходимо такое лечение, причем указанное лечение включает введение эффективного количества, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединения формулы 1.0, в комбинации с гормональными терапиями (то есть, антигормональными средствами) и в комбинации с эффективным количеством, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2, или 1) химиотерапевтического агента.
Другие варианты выполнения настоящего изобретения представляют собой способы предотвращения рака молочной железы (то есть, постклимактерического и предклимактерического рака молочной железы, например, гормонально-зависимого рака молочной железы) у пациента, которому необходимо такое лечение, причем указанное лечение включает введение эффективного количества фармацевтической композиции, содержащей эффективное количество, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединения формулы 1.0, в комбинации с гормональными терапиями (то есть, антигормональными средствами), и в комбинации с эффективным количеством, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2, или 1) химиотерапевтического агента.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения рака головного мозга (например, глиома, такая как мультиформная глиобластома) у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединения формулы 1.0.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения рака головного мозга (например, глиома, такая как мультиформная глиобластома) у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединения формулы 1.0, в комбинации с эффективным количеством, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2, или 1) химиотерапевтического агента.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения рака головного мозга (например, глиома, такая как мультиформная глиобластома) у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества фармацевтической композиции, содержащей эффективное количество, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединения формулы 1.0.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения рака головного мозга (например, глиома, такая как мультиформная глиобластома) у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества фармацевтической композиции, содержащей эффективное количество, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединения формулы 1.0, в комбинации с эффективным количеством, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2, или 1) химиотерапевтического агента.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения рака головного мозга (например, глиома, такая как мультиформная глиобластома) у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединения формулы 1.0, в комбинации с эффективным количеством химиотерапевтического агента, где указанный химиотерапевтический агент представляет собой темозоломид.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения рака головного мозга (например, глиома, такая как мультиформная глиобластома) у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества фармацевтической композиции, содержащей эффективное количество, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединения формулы 1.0, в комбинации с эффективным количеством химиотерапевтического агента, где указанный химиотерапевтический агент, представляет собой темозоломид.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения рака предстательной железы у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединения формулы 1.0.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения рака предстательной железы у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединения формулы 1.0, в комбинации с эффективным количеством, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2, или 1) химиотерапевтического агента.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения рака предстательной железы у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества фармацевтической композиции, содержащей эффективное количество, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединения формулы 1.0.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения рака предстательной железы у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества фармацевтической композиции, содержащей эффективное количество, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединения формулы 1.0, в комбинации с эффективным количеством, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2, или 1) химиотерапевтического агента.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения миелодиспластического синдрома у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединения формулы 1.0.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения миелодиспластического синдрома у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединения формулы 1.0, в комбинации с эффективным количеством, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2, или 1) химиотерапевтического агента.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения миелодиспластического синдрома у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества фармацевтической композиции, содержащей эффективное количество, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединения формулы 1.0.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения миелодиспластического синдрома у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества фармацевтической композиции, содержащей эффективное количество, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединения формулы 1.0, в комбинации с эффективным количеством, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2, или 1) химиотерапевтического агента.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения миелоидной лейкемии у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединения формулы 1.0.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения миелоидной лейкемии у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединения формулы 1.0, в комбинации с эффективным количеством, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2, или 1) химиотерапевтического агента.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения миелоидной лейкемии у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества фармацевтической композиции, содержащей эффективное количество, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединения формулы 1.0.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения миелоидной лейкемии у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества фармацевтической композиции, содержащей эффективное количество, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединения формулы 1.0, в комбинации с эффективным количеством, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2, или 1) химиотерапевтического агента.
Другой вариант выполнения настоящего изобретения представляет собой лечения острой миелогенной лейкемии (AML) у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединения формулы 1.0.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения острой миелогенной лейкемии (AML) у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединения формулы 1.0, в комбинации с эффективным количеством, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2, или 1) химиотерапевтического агента.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения острой миелогенной лейкемии (AML) у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества фармацевтической композиции, содержащей эффективное количество, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединения формулы 1.0.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения острой миелогенной лейкемии (AML) у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества фармацевтической композиции, содержащей эффективное количество, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединения формулы 1.0, в комбинации с эффективным количеством, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2, или 1) химиотерапевтического агента.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения хронической миеломоноцитической лейкемии (CMML) у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединения формулы 1.0.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения хронической миеломоноцитической лейкемии (CMML) у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединения формулы 1.0, в комбинации с эффективным количеством, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2, или 1) химиотерапевтического агента.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения хронической миеломоноцитической лейкемии (CMML) у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества фармацевтической композиции, содержащей эффективное количество, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединения формулы 1.0.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения хронической миеломоноцитической лейкемии (CMML) у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества фармацевтической композиции, содержащей эффективное количество, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединения формулы 1.0, в комбинации с эффективным количеством, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2, или 1) химиотерапевтического агента.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения хронической миелогенной лейкемии (хронической миелоидной лейкемии, CML) у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединения формулы 1.0.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения хронической миелогенной лейкемии (хронической миелоидной лейкемии, CML) у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединения формулы 1.0, в комбинации с эффективным количеством, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2, или 1) химиотерапевтического агента.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения хронической миелогенной лейкемии (хронической миелоидной лейкемии, CML) у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества фармацевтической композиции, содержащей эффективное количество, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединения формулы 1.0.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения хронической миелогенной лейкемии (хронической миелоидной лейкемии, CML) у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества фармацевтической композиции, содержащей эффективное количество, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединения формулы 1.0, в комбинации с эффективным количеством, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2, или 1) химиотерапевтического агента.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения миелоидной лейкемии у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединения формулы 1.0.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения миелоидной лейкемии у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединения формулы 1.0, в комбинации с эффективным количеством, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2, или 1) химиотерапевтического агента.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения миелоидной лейкемии у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества фармацевтической композиции, содержащей эффективное количество, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединения формулы 1.0.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения миелоидной лейкемии у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества фармацевтической композиции, содержащей эффективное количество, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединения формулы 1.0, в комбинации с эффективным количеством, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2, или 1) химиотерапевтического агента.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения рака мочевого пузыря у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединения формулы 1.0.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения рака мочевого пузыря у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединения формулы 1.0, в комбинации с эффективным количеством, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2, или 1) химиотерапевтического агента.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения рака мочевого пузыря у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества фармацевтической композиции, содержащей эффективное количество, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединения формулы 1.0.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения рака мочевого пузыря у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества фармацевтической композиции, содержащей эффективное количество, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединения формулы 1.0, в комбинации с эффективным количеством, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2, или 1) химиотерапевтического агента.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения неходжкинской лимфомы у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединения формулы 1.0.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения неходжкинской лимфомы у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединения формулы 1.0, в комбинации с эффективным количеством, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2, или 1) химиотерапевтического агента.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения неходжкинской лимфомы у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества фармацевтической композиции, содержащей эффективное количество, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединения формулы 1.0.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения неходжкинской лимфомы у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества фармацевтической композиции, содержащей эффективное количество, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединения формулы 1.0, в комбинации с эффективным количеством, по меньшей мере, одного (например, 1, 2 или 3,1 или 2, или 1) химиотерапевтического агента.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения множественной миеломы у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединения формулы 1.0.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения множественной миеломы у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединения формулы 1.0, в комбинации с эффективным количеством, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2, или 1) химиотерапевтического агента.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения множественной миеломы у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества фармацевтической композиции, содержащей эффективное количество, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединения формулы 1.0.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения множественной миеломы у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества фармацевтической композиции, содержащей эффективное количество, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2 и, как правило, 1) соединения формулы 1.0, в комбинации с эффективным количеством, по меньшей мере, одного (например, 1, 2 или 3, 1 или 2, или 1) химиотерапевтического агента.
Химиотерапевтические агенты (противоопухолевые агенты) включают, но без ограничения к этому: агенты, воздействующие на микротрубочки, алкилирующие агенты, антиметаболиты, природные продукты и их производные, гормоны и стероиды (включая синтетические аналоги) и синтетические продукты.
Примерами алкилирующих агентов (включая азотистые иприты, производные этиленимина, алкилсульфонаты, нитрозомочевины и триазены) являются: Урацилмустард, Хлорметин, Циклофосфамид (Цитоксан®), Ифосфамид, Мелфалан, Хлорамбуцил, Пипоброман, Триэтиленмеламин, Триэтилентиофосфорамин, Бусульфан, Кармустин, Ломустин, Стрептозоцин, Дакарбазин и Темозоломид.
Примерами антиметаболитов (включая антагонисты фолиевой кислоты, аналоги пиримидина, пуриновые аналоги и ингибиторы аденозиндезаминазы) являются: Метотрексат, 5-Фторурацил, Флоксуридин, Цитарабин, 6-Меркаптопурин, 6-Тиогуанин, Флударабинфосфат, Пентостатин и Гемцитабин.
Примерами природных продуктов и их производных (включая алкалоиды винка, противоопухолевые антибиотики, ферменты, лимфокины и эпиподофиллотоксины) являются: Винбластин, Винкристин, Виндезин, Блеомицин, Дактиномицин, Даунорубицин, Доксорубицин, Эпирубицин, Идарубицин, Паклитаксел (Паклитаксел представляет собой агент, воздействующий на микротрубочки, и является коммерчески доступным под маркой Таксол®), производные Паклитаксела (например, Таксотер), Митрамицин, Деоксикоформицин, Митомицин-С, L-Аспарагиназа, Интерфероны (особенно IFN-a), Этопозид и Тенипозид.
Примерами гормонов и стероидов (включая синтетические аналоги) являются: 17α-Этинилэстрадиол, Диэтилстилбестрол, Тестостерон, Преднизон, Флуоксиместерон, Дромостанолон пропионат, Тестолактон, Мегестрола ацетат, Тамоксифен, Метилпреднизолон, Метилтестостерон, Преднизолон, Триамцинолон, Хлортрианизен, Гидроксипрогестерон, Аминоглютетимид, Эстрамустин, ацетат Медроксипрогестерона, Леупролид, Флутамид, Торемифен и Золадекс.
Примерами синтетических веществ (включая неорганические комплексы, такие как координационные комплексы платины) являются: Цисплатин, Карбоплатин, Гидроксимочевина, Амсакрин, Прокарбазин, Митотан, Митоксантрон, Левамизол и Гексаметилмеламин.
Примерами других химиотерапевтических агентов являются: Навелбен, СРТ-11, Анастразол, Летразол, Капецитабин, Релоксафин и Дролоксафин.
Агенты, действующие на микротрубочки (например, Паклитаксел, производная Паклитаксела или Паклитаксел - подобное соединение), как применяется в настоящем документе, представляют собой соединение, которое вмешивается в процесс митоза клетки, то есть, оказывает антимитотическое действие, воздействуя на образование и/или работу микротрубочек. Такими агентами могут быть, например, агенты, стабилизирующие микротрубочки или агенты, которые нарушают образование микротрубочек.
Агенты, воздействующие на микротрубочки, полезные в способах согласно настоящему изобретению, хорошо известны специалистам в данной области техники и включают, но без ограничения к этому: Аллоколхицин (NSC 406042), Галихондрин В (NSC 609395), Колхицин (NSC 757), производные Колхицина (например, NSC 33410), Доластатин 10 (NSC 376128), Майтансин (NSC 153858), Ризоксин (NSC 332598), Паклитаксел (Таксол®, NSC 125973), производные Паклитаксела (например, Таксотер, NSC 608832), Тиоколхицин (NSC 361792), Тритилцистеин (NSC 83265), сульфат Винбластина (NSC 49842), сульфат Винкристина (NSC 67574), Эпотилон А, Эпотилон, Дискодермолид (смотрите Service, (1996) Science, 274:2009), Эстрамустин, Нокодазол, МАР4 и тому подобное. Примеры таких агентов раскрываются, например, в Bulinski (1997) J. Cell Sci. 110:3055-3064, Panda (1997) Proc. Natl. Acad. Sci. USA 94:10560-10564, Muhlradt (1997) Cancer Res. 57:3344-3346, Nicolaou (1997) Nature 387:268-272, Vasquez (1997) Mol. Biol. Cell. 8:973-985 и Panda (1996) J. Biol. Chem. 271:29807-29812.
Химиотерапевтические агенты с Паклитаксел - подобной активностью включают, но без ограничения к этому, Паклитаксел и производные Паклитаксела (Паклитаксел - подобные соединения) и их аналоги. Паклитаксел и его производные (например, Таксол и Таксотер) доступны для приобретения коммерческим образом. Кроме того, способы получения Паклитаксела и производных и аналогов Паклитаксела хорошо известны специалистам в данной области техники (смотрите, например, патенты США: 5,569,729; 5,565,478; 5,530,020; 5,527,924; 5,508,447; 5,489,589; 5,488,116; 5,484,809; 5,478,854; 5,478,736; 5,475,120; 5,468,769; 5,461,169; 5,440,057; 5,422,364; 5,411,984; 5,405,972; и 5,296,506).
Более конкретно, термин "Паклитаксел", как применяется в настоящем документе, относится к лекарственному средству, коммерчески доступному под названием Таксол® (NSC номер: 125973). Таксол® ингибирует репликацию эукариотических клеток, усиливая полимеризации тубулиновых составляющих в стабилизированные связки микротрубочек, которые неспособны преобразовываться в соответствующие структуры, необходимые для митоза. Среди многих доступных химиотерапевтических лекарственных средств Паклитаксел вызвал повышенный интерес благодаря своей эффективности, обнаруженной при клинических испытаниях, в отношении опухолей, нечувствительных к лекарственным средствам, включая опухоли яичников и молочной железы (Hawkins (1992) Oncology, 6: 17-23, Horwitz (1992) Trends Pharmacol. Sci. 13: 134-146, Rowinsky (1990) J. Natl. Canc. Inst. 82: 1247-1259).
Дополнительные агенты, воздействующие на микротрубочки, могут быть исследованы, посредством одного или более анализов, известных в области техники, например, полуавтоматического анализа, в котором измеряют тубулин-полимеризующую активность аналогов Паклитаксела, в комбинации с клеточным анализом, применяемым для измерения потенциала этих соединений при блокировании митоза клеток (смотрите Lopes (1997) Cancer Chemother. Pharmacol. 41:37-47).
Как правило, активность тестируемых соединений определяют при контакте клетки с этим соединением, с последующим определением прерывается ли клеточный цикл или нет, в частности, через ингибирование митотического события. Такое ингибирование может заключаться в прерывание митотического аппарата, например, прерыванием нормального образования веретена. Клетки, в которых прерывается митоз, могут отличаться измененной морфологией (например, сжатие микротрубочек, увеличенное число хромосом и т.д.).
Соединения, которые, возможно, способны к полимеризации тубулина, скринировали in vitro. Например, соединения скринировали в отношении культивированных клеток WR21 (полученных из клеточной линии мышей 69-2 wap-ras), на ингибирование пролиферации и/или изменение клеточной морфологии, на сжатие микротрубочек. Затем можно провести in vivo исследование соединений, показавших положительные результаты, с помощью бестимусных мышей, несущих опухолевые клетки WR21. Подробные протоколы этих способов скрининга раскрываются в Porter (1995) Lab. Anim. Sci., 45(2):145-150.
Другие способы скрининга соединений на желательную активность известны специалистам в данной области техники. Как правило, такие анализы включают анализы, позволяющие протестировать ингибирование сборки и/или разборки микротрубочек. Анализы на сборку микротрубочек раскрываются, например, в Gaskin et al. (1974) J. Molec. Biol., 89: 737-758. Патент США №5,569,720 также раскрывает in vitro и in vivo анализы для соединений с Паклитаксел - подобной активностью.
Таким образом, в способах согласно настоящему изобретению, где применяется, по меньшей мере, один химиотерапевтический агент, примеры указанных химиотерапевтических агентов включают агенты, выбранные из группы, состоящей из: действующих на микротрубочки агентов, алкилирующих агентов, антиметаболитов, природных продуктов и их производных, гормонов и стероидов (включая синтетические аналоги) и синтетические продуктов.
В способах согласно настоящему изобретению, в которых применяется, по меньшей мере, один химиотерапевтический агент, примеры указанных химиотерапевтических агентов также включают: (1) таксаны, (2) координационные соединения платины, (3) ингибиторы эпидермального фактора роста (EGF), которые представляют собой антитела, (4) "маленькие молекулы" - ингибиторы EGF (5) ингибиторы васкулярных эндотелиальных факторов роста (VEGF), которые представляют собой антитела, (6) "маленькие молекулы" - ингибиторы киназы VEGF, (7) антагонисты эстрогенового рецептора или селективные модуляторы эстрогенового рецептора (SERMs), (8) противоопухолевые нуклеозидные производны, (9) эпотилоны, (10) ингибиторы топоизомеразы, (11) алкалоиды винка, (12) антитела, которые являются ингибиторами αVβ3 интегринов, (13) антагонисты фолата, (14) ингибиторы рибонуклеотидной редуктазы, (15) антрациклины, (16) биопрепараты; (17) ингибиторы ангиогенеза и/или супрессоры фактора некроза опухоли альфа (TNF-alpha), такие как талидомид (или родственный имид), (18) ингибиторы Bcr/abl киназы, (19) "маленькие молекулы" - ингибиторы MEK1 и/или MEK2, (20) "маленькие молекулы" - ингибиторы IGF-1 и IGF-2, (21) "маленькие молекулы" - ингибиторы RAF и BRAF киназ, (22) "маленькие молекулы" - ингибиторы киназ, зависимых от клеточного цикла, таких как CDK1, CDK2, CDK4 и CDK6, (23) алкилирующие агенты и (24) ингибиторы фарнезилтрансферазы (также известные как ингибиторы FPT или FTI (то есть, ингибиторы фарнезилтрансферазы)).
В способах согласно настоящему изобретению, в которых применяется, по меньшей мере, один химиотерапевтический агент, примеры таких химиотерапевтических агентов включают:
(1) таксаны, такие как Паклитаксел (Таксол®) и/или Доцетаксел (Таксотер®);
(2) координационные соединения платины, такие как, например, Карбоплатин, Цисплатин и Оксалиплатин (например, Элоксатин);
(3) ингибиторы EGF, которые представляют собой антитела, такие как: антитела к гену HER2 (такие как, например, Трастузумаб (Герцептин), Genentech, Inc.), Цетуксимаб (Эрбитукс, IMC-C225, ImClone Systems), EMD 72000 (Merck KGaA), анти-EFGR моноклональные антитела АВХ (Abgenix), TheraCIM-h-R3 (Center of Molecular Immunology), моноклональные антитела 425 (Merck KGaA), моноклональные антитела ICR-62 (ICR, Sutton, England); Херзим (Elan Pharmaceutical Technologies and Ribozyme Pharmaceuticals), PKI 166 (Novartis), EKB 569 (Wyeth-Ayerst), GW 572016 (GlaxoSmithKline), Cl 1033 (Pfizer Global Research and Development), конъюгат Тразтузмаба и майтансиноиди (Genentech, Inc.), Митумомаб (Imclone Systems and Merck KGaA) и Melvax II (Imclone Systems and Merck KgaA);
(4) "маленькие молекулы" - ингибиторы EGF, такие как, Тарцева (ТМ) (OSI-774, OSI Pharmaceuticals, Inc.) и Иресса (ZD 1839, Astra Zeneca);
(5) ингибиторы VEGF, которые представляют собой антитела, такие как: Бевацизумаб (Genentech, Inc.) и IMC-1С11 (ImClone Systems), DC 101 (KDR VEGF Рецептор 2 фирмы ImClone Systems);
(6) "маленькие молекулы" - ингибиторы киназы VEGF, такие как: SU 5416 (фирмы Sugen, Inc), SU 6688 (от Sugen, Inc.), Bay 43-9006 (VEGF и bRAF ингибитор фирмы Вауег Pharmaceuticals и Onyx Pharmaceuticals);
(7) антагонисты эстрогенового рецептора или селективные модуляторы эстрогенового рецептора (SERMs), такие как Тамоксифен, Идоксифен, Ра-локсифен, транс-2,3-Дигидроралоксифен, Левормелоксифен, Дролокси-фен, MDL 103,323 и Аколбифен (Schering Corp.);
(8) противоопухолевые нуклеозидные производные, такие как 5-Фторурацил, Гемцитабин, Капецитабин, Цитарабин (Ara-С), Флударабин (F-Ara-A), Децитабин и Хлордеоксиаденозин (Cda, 2-Cda);
(9) эпотилоны, такие как BMS-247550 (Brisl-Myers Squibb) и EPO906 (Novartis Pharmaceuticals);
(10) ингибиторы топоизомеразы, такие как Топотекан (Glaxo SmithKline) и Камптосар (Pharmacia);
(11) алкалоиды винка, такие как, Навельбин (Anvar and Fabre, France), Винкристин и Винбластин;
(12) антитела, которые являются ингибиторами αVβ3 интегринов, такие как, LM-609 (смотрите Clinical Cancer Research, Vol.6, page 3056-3061, August 2000, который включен в настоящий документ в полном объеме посредством ссылки);
(13) антагонисты фолата, такие как Метотрексат (МТХ) и Преметрексед (Алимта);
(14) ингибиторы рибонуклеотидной редуктазы, такие как Гидроксимочевина (HU);
(15) антрациклины, такие как Даунорубицин, Доксорубицин (Адриамицин) и Идарубицин;
(16) биопрепараты, такие как интерферон (например, Интрон-А и Роферон), пегилированный интерферон (например, Пегинтрон и Пегасис) и Ритуксимаб (Ритуксан, антитело, применяемое для лечения неходжкинской лимфомы);
(17) Талидомид (или родственный имид);
(18) ингибиторы Bcr/abl киназы, такие как, например, Гливек (STI-571), AMN-17, ONO 12380, SU 11248 (Сунитиниб) и BMS-354825
(19) MEK1 и/или MEK2 ингибиторы, такие как PD0325901 и Arry-142886 (AZD6244);
(20) "маленькие молекулы" - ингибиторы IGF-1 и IGF-2, такие как, например, NVP-AEW541;
(21) "маленькие молекулы" - ингибиторы RAF и BRAF киназ, такие как, например, BAY 43-9006 (Сорафениб);
(22) "маленькие молекулы" - ингибиторы киназ, зависимых от клеточного цикла, таких как CDK1, CDK2, CDK4 и CDK6, такие как, например, CYC202, BMS387032 и Флавопиридол;
(23) алкилирующие агенты, такие как, например, Темозоломид торговой марки Темодар;
(24) ингибиторы фарнезилтрансферазы, такие как, например:
(a) Лонифарниб торговой марки Sarasar® (то есть, 4-[2-[4-(3,10-дибром-S-хлор-6,11-дигидро-5Н-бензо[5,6]циклогепта[1,2-b]пиридин-11-ил)-1-пиперидинил)-2-оксоэтил]-1-пиперидинкарбоксамид, смотрите, например, патент США US 5,874,442, опубликованный 23 Февраля 1999, и патент США US 6,632,455, опубликованный 14 Октября, 2003, которые включены в настоящий документ посредством ссылки в полном объеме),
(b) Типифарниб торговой марки Zarnestra® (то есть, (R)-6-амино[(4-хлорфенил)(1-метил-1Н-имидазол-5-ил)метил]-4-(3-хлорфенил)-1-метил-2(1Н)-хинолинон, смотрите например, международную заявку WO 97/16443, опубликованную 9 мая, 1997, и патент США US 5,968,952, выданный 19 октября, 1999, которые включены в настоящий документ в полном объеме посредством ссылки), и
(c) Bristol-Myers Squibb 214662:
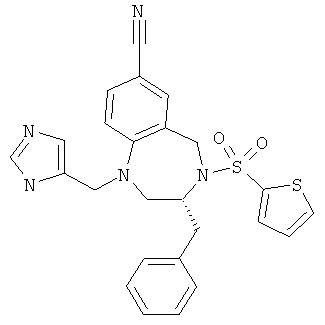
(смотрите международную заявку на патент WO97/30992, опубликованную 28 августа, 1997, патент США US 6,011,029, выданный 4 января 2000, и U.S. 6,455,523, которые включены в настоящий документ посредством ссылки).
Ингибиторы киназы Bcr/abl, ингибиторы рецептора EGF и антитела к гену HER-2 (ингибиторы рецептора EGF, которые представляют собой антитела), которые раскрываются выше, также известны в качестве ингибиторов сигнальной трансдукции. Поэтому, химиотерапевтические агенты, как применяется в настоящей заявке, включают ингибиторы сигнальной трансдукции.
Типичные ингибиторы сигнальной трансдукции, которые представляют собой химиотерапевтические агенты, включают, но без ограничения к этому: (i) ингибиторы Bcr/abl киназы, такие как, например, STI 571 (Гливек), (ii) Ингибитор рецептора эпидермальнога фактора роста (EGF), такие как, например, ингибиторы киназ (Иресса, OSI-774) и антитела (Имклон: С225 [Goldstein et al. (1995), Clin Cancer Res. 1:1311-1318] и Abgenix: ABX-EGF) и (iii) ингибиторы рецептора HER-2/neu, такие как, например, Герцептин® (Трастузумаб).
Способы безопасного и эффективного введения большинства этих химиотерапевтических агентов известны специалистам в данной области техники. Кроме того, эти способы введения раскрываются в литературе. Например, способы введения многих химиотерапевтических агентов раскрываются в справочнике "Physicians′ Desk Reference" (PDR), например, 1996 edition (Medical Economics Company, Montvale, NJ 07645-1742, USA), the Physician′s Desk Reference, 56th Edition, 2002 (опубликованный Medical Economics company, Inc. Montvale, NJ 07645-1742), и the Physician′s Desk Reference, 57th Edition, 2003 (опубликованный Thompson PDR, Montvale, NJ 07645-1742); раскрытие которых включены в настоящий документ в полном объеме посредством ссылки.
Например, соединение формулы 1.0 (например, фармацевтическая композиция, содержащая соединение формулы 1.0); может вводиться пероральным путем (например, в виде капсулы), химиотерапевтические агенты могут вводиться внутривенно, как правило, в виде внутривенного раствора. Применение фармацевтической композиции, содержащей более чем одно лекарственное средство, входит в объем настоящего изобретения.
Соединение формулы 1.0 и химиотерапевтические агенты вводятся в терапевтически эффективных дозах для получения клинически приемлемых результатов, таких как, например, уменьшение или исчезновение симптомов или опухоли. Таким образом, соединение формулы 1.0 и химиотерапевтические агенты могут вводиться одновременно или последовательно по протоколу лечения. Введение химиотерапевтических агентов может осуществляться по протоколами лечения, уже известными в данной области техники.
В общем, когда более чем один химиотерапевтический агент применяется в способах согласно настоящему изобретению, химиотерапевтические агенты могут вводиться в один и тот же день, либо одновременно, либо последовательно, в их стандартной лекарственной форме. Например, химиотерапевтические агенты обычно вводятся внутривенно, предпочтительно посредством внутривенного капельного вливания, применяя внутривенные растворы, хорошо известные в области техники (например, изотонический физиологический раствор (0.9% NaCl) или раствор декстрозы (например, 5% декстроза)).
В случае если применяются два или более химиотерапевтических агента, химиотерапевтические агенты, как правило, вводятся в один и тот же день; однако, специалистам в данной области техники будет очевидно, что химиотерапевтические агенты могут вводиться в разные дни и в разные недели. Квалифицированный клинический врач может вводить химиотерапевтические агенты согласно схеме приема, рекомендованной производителем данного агента, и может регулировать схему в зависимости от нужд пациента, например, принимая во внимание реакцию пациента на лечение. Например, когда Гемцитабин применяется в комбинации с координационным соединением платины, таким как, например, Цисплатин, для лечения рака легких, как Гемцитабин, так и Цисплатин вводятся в один и тот же день, а именно в первый день цикла лечения, и затем Гемцитабин вводится сам по себе в день 8 и снова вводиться сам по себе в день 15.
Соединения согласно настоящему изобретению и химиотерапевтические агенты могут вводиться по протоколу лечения, продолжительность которого составляет от одной до семи недель, и который, как правило, повторяется 6-12 раз. Как правило, протокол лечения рассчитан на период времени от одной до четырех недель. Так же могут применяться протоколы лечения, рассчитанные на период времени от одной до трех недель. Могут применяться режимы лечения, продолжительностью от одной до двух недель. Во время такого протокола лечения или цикла, соединения согласно настоящему изобретению могут вводиться ежедневно, в то время как химиотерапевтические агенты могут вводиться один или более раз в неделю. Как правило, соединение согласно настоящему изобретению может вводиться ежедневно (то есть, один раз в день), и в одном варианте выполнения изобретения соединение согласно настоящему изобретению может вводиться два раза в день, и химиотерапевтический агент вводится один раз в неделю или один раз каждые три недели. Например, таксаны (например, Паклитаксел (например, Таксол®) или Доцетаксел (например, Таксотер®)) могут вводиться один раз в неделю или один раз каждые три недели.
Однако специалистам в области техники очевидно, что протоколы лечения могут изменяться в зависимости от нужд пациента. Таким образом, комбинация соединений (лекарственных средств), применяемых в способах согласно настоящему изобретению, может вводиться в соответствии с вариантами протоколов, раскрытых выше. Например, в течение цикла лечения соединения согласно настоящему изобретению могут вводиться скорее с перерывами, чем непрерывно. Таким образом, например, в течение цикла лечения соединения согласно настоящему изобретению могут вводиться ежедневно в течение недели и затем не вводиться в течение недели, с повторением этого введения во время цикла лечения. Альтернативно, соединения согласно настоящему изобретению могут вводиться ежедневно в течение двух недель и затем не вводиться в течение недели, с повторением этого введения в течение цикла лечения. Таким образом, соединения по изобретению могут вводиться ежедневно в течение одной или более недель и затем не вводиться в течение одной или более недель в течение цикла лечения, с повторением этой модели введения в течение цикла лечения. Такое прерывистое лечение может скорее основываться на числе дней, чем на полной недели. Например, ежедневное ведение дозы в течение 1-6 дней, и затем 1-6 дней без введения дозы, с повторением этой модели введения в течение протокола лечения. Число дней (или недель), в течение которых соединения согласно настоящему изобретению не вводятся, не должно равняться числу дней (или недель), когда соединения согласно настоящему изобретению вводятся. Обычно, если применяется прерывистый протокол дозирования, число дней или недель, в течение которых соединения согласно настоящему изобретению водятся, по меньшей мере, равно или больше, чем число дней или недель, когда соединения согласно настоящему изобретению не вводятся.
Химиотерапевтический агент может вводиться посредством болюсов или непрерывного вливания. Химиотерапевтический агент может вводиться от одного раза в день ежедневно до одного раза каждую неделю, или одного раза каждые две недели, или одного раза каждые три недели, или одного раза каждые четыре недели, за цикл лечения. В случае ежедневного введения в течение цикла лечения, это ежедневное введение дозы может, прерываться на ряд недель цикла лечения. Например, введение дозы в течение недели (или ряда дней), и затем неделя (или ряд дней) без введения дозы, с повторением этой модели введения в течение цикла лечения.
Соединения согласно настоящему изобретению могут вводиться пероральным путем, предпочтительно в виде твердой лекарственной формы, и в одном варианте выполнения изобретения в виде капсулы, и в то же время общая терапевтически эффективная ежедневная доза может вводиться посредством одной-четырех или одной-двух отельных доз в день, как правило, терапевтически эффективная доза вводиться один или два раза в день, и в одном варианте выполнения изобретения терапевтически эффективная доза вводиться два раза в день. Соединения согласно настоящему изобретению могут вводиться в количестве от около 50 до около 400 мг один раз в день, и могут вводиться в количестве от около 50 до около 300 мг один раз в день. Соединения согласно настоящему изобретению, как правило, вводятся в количестве от около 50 до около 350 мг два раза в день, обычно от 50 мг до около 200 мг два раза в день, и в одном варианте выполнения изобретения от около 75 мг до около 125 мг, при введении два раза в день, и в другом варианте выполнения изобретения около 100 мг, при введении два раза в день.
В случае реакции пациента на лечение или стабилизации его состояния терапевтического цикла, этот терапевтический цикл можно повторить согласно оценке лечащего врача. При завершении терапевтических циклов пациент может продолжать принимать соединения согласно настоящему изобретению в тех же дозах, которые вводились по протоколу лечения, или если доза составляла менее чем 200 мг два раза в день, то доза может быть увеличена до 200 мг два раза в день. Эта поддерживающая доза может вводиться до тех пор, пока не произойдет прогресс в состоянии пациента или пациент не сможет более переносить эту дозу (в этом случае доза может быть уменьшена, и пациент продолжит принимать уменьшенную дозу).
Химиотерапевтические агенты, применяемые с соединениями согласно настоящему изобретению, вводятся в их обычно предписываемых дозах в течение цикла лечения (то есть, Химиотерапевтические агенты вводятся в соответствии со стандартной практикой введения этих лекарственных средств). Например: (а) от около 30 до около 300 мг/м2 для таксанов; (b) от около 30 до около 100 мг/м2 для Цисплатина; (с) AUC от около 2 до около 8 для Карбоплатина; (d) от около 2 до около 4 мг/м2 для ингибиторов EGF, которые представляют собой антитела; (е) от около 50 до около 500 мг/м2 для "маленьких молекул" - ингибиторов EGF; (f) от около 1 до около 10 мг/м2 для ингибиторов киназы VEGF, которые представляют собой антитела; (g) от около 50 до около 2400 мг/м2 для "маленьких молекул" - ингибиторов VEGF; (h) от около 1 до около 20 мг для SERMs; (i) от около 500 до около 1250 мг/м2 для противоопухолевых нуклеозидов 5-Фторурацила, Гемцитабина и Капецитабина; (j) для противоопухолевого нуклеозида Цитарабина (Ara-С): 100-200 мг/м2/день в течение 7-10 дней, каждые 3-4 недели, и высокие дозы при рефракторной лейкемии и лимфоме, то есть, 1-3 мг/м2 в течение одного часа, каждые 12 часов за 4-8 доз каждые 3-4 недели; (k) для противоопухолевого нуклеозида Флударабина (F-ara-A): 10-25 мг/м2/день каждые 3-4 недели; (l) для противоопухолевого нуклеозида Дацитабина: 30-75 мг/м2 в течение трех дней каждые 6 недель, максимум 8 циклов; (m) для противоопухолевого нуклеозида Хлордеоксиаденозина (CdA, 2-CdA): 0.05-0.1 мг/кг/день, в виде непрерывного вливания продолжительностью до 7 дней, каждые 3-4 недели; (n) от около 1 до около 100 мг/м2 для эпотилонов; (о) от около 1 до около 350 мг/м2 для ингибиторов топоизомеразы; (р) от около 1 до около 50 мг/м2 для алкалоидов винка; (q) для антагониста фолата Метотрексата (МТХ) 20-60 мг/м2 пероральным путем, внутривенно или внутримышечно, каждые 3-4 недели, режим введения промежуточной дозы составляет 80-250 мг/м2 внутривенно за 60 минут каждые 3-4 недели, режим высокой дозы составляет 250-1000 мг/м2 внутривенно, введение с Лейковорином каждые 3-4 недели; (r) для антагониста фолата Пеметрекседа (Алимта): 300-600 мг/м2 (10 минут внутривенного вливания в день 1) каждые 3 недели; (s) для ингибитора рибонуклеотидной редуктазы Гидроксимочевины (HU): 20-50 мг/кг/день (как необходимо для снижения числа клеток крови); (t) координационное соединение платины Оксалиплатин (Элоксатин) вводится в количестве 50-100 мг/м2 каждые 3-4 недели (предпочтительно применяется для солидных опухолей, таких как немелкоклеточный рак легких, колоректальный рак и рак яичников); (u) для антрациклина Даунорубицина: 10-50 мг/м2/день, внутривенно, в течение 3-5 дней, каждые 3-4 недели; (v) для антрациклина Доксорубицина (Адриамицин): 50-100 мг/м2, внутривенное, непрерывное вливание за 1-4 дня, каждые 3-4 недели, или 10-40 мг/м2 внутривенно, еженедельно; (w) для антрациклина Идарубицин: 10-30 мг/м2, ежедневно, в течение 1-3 дней, в виде медленного внутривенного вливания за 10-20 минут каждые 3-4 недели; (х) для биологического интерферона (Интрон-А, Роферон): 5-20 миллионов иммунизирующих единиц, три раза в неделю; (у) для биологического пегилированного интерферона (Пегинтрон, Пегасис): 3-4 мг/кг/день, постоянно, подкожно (до рецидива или потери активности); (z) для биологического Ритуксимаба (Ритуксана) (антитело, применяемое для неходжкинской лимфомы): 200-400 мг/м2 внутривенно, еженедельно, за 4-8 недель, в течение 6 месяцев; (аа) для алкилирующего агента темозоломида: 75 мг/м2 - 250 мг/м2, например, 150 мг/м2, или, например, 200 мг/м2, как например 200 мг/м2 в течение 5 дней; и (bb) для ингибитора MEK1 и/или MEK2 PD0325901: 15 мг - 30 мг, например, 15 мг ежедневно в течение 21 дня, каждые 4 недели.
Гливек может применяться пероральным путем в количестве от около 200 до около 800 мг/день.
Талидомид (и родственные имиды) могут применяться пероральным путем в количествах от около 200 до около 800 мг/день, и может вводиться непрерывно или применяться до возникновения рецидива или токсичности. Смотрите, например, Mitsiades et al., "Apoptotic signaling induced by immunomodulatory thalidomide analoqs in human multiple myeloma cells; therapeutic implications", Blood, 99(12):4525-30, June 15, 2002, раскрытие которого включено в настоящий документ посредством ссылки.
Ингибитор FPT Sarasar® (Лонафарниб) может вводиться пероральным путем (например, в виде капсулы), в количествах от около 50 до около 200 мг, два раза в день, или в количествах от около 75 до около 125 мг, два раза в день, или в количествах от около 100 до около 200 мг, два раза в день, или в количестве около 100 мг, два раза в день
Паклитаксел (например, Таксол®, например, может вводиться один раз в неделю, в количестве от около 50 до около 100 мг/м2, и в другом примере от около 60 до около 80 мг/м2. В другом примере Паклитаксел (например, Таксол® может вводиться один раз каждые три недели, в количестве от около 150 до около 250 мг/м2, и в другом примере около от 175 до около 225 мг/м2.
В другом примере, Доцетаксел (например, Таксотер®) может вводиться один раз в неделю, в количестве от около 10 до около 45 мг/м2. В другом примере, Доцетаксел (например, Таксотер®) может вводиться один раз каждые три недели, в количестве от около 50 до около 100 мг/м2.
В другом примере Цисплатин может вводиться один раз в неделю, в количестве от около 20 до около 40 мг/м2. В другом примере, Цисплатин может вводиться один раз каждые три недели, в количестве от около 60 до около 100 мг/м2.
В другом примере Карбоплатин может вводиться один раз в неделю, в количестве, необходимом для обеспечения AUC от около 2 до около 3. В другом примере Карбоплатин может вводиться один раз каждые три недели, в количестве, необходимом для обеспечения AUC от около 5 до около 8.
Другие варианты выполнения настоящего изобретения представляют собой любой из способов лечения рака, где соединения формулы 1.0 и химиотерапевтические агенты вводятся в виде фармацевтической композиции, содержащей эффективное количество соединения формулы 1.0, эффективное количество химиотерапевтических агентов, и фармацевтически приемлемый носитель.
Другие варианты выполнения настоящего изобретения представляют собой способ лечения рака, где химиотерапевтический агент выбирается из группы, состоящей из: Паклитаксела, Доцетаксела, Карбоплатина, Цисплатина, Гемцитабина, Тамоксифена, Герцептина, Цетуксимаба, Тарцева, Иресса, Бевацизумаба, Навельбина, IMC-1С11, SU5416 и SU6688.
Другие варианты выполнения настоящего изобретения представляют собой способ лечения рака, где применяется химиотерапевтический агент, и химиотерапевтический агент выбирается из группы, состоящей из: Паклитаксела, Доцетаксела, Карбоплатина, Цисплатина, Навельбина, Гемцитабина и Герцептина.
Другие варианты выполнения настоящего изобретения представляют собой способ лечения рака, где применяется химиотерапевтический агент, и химиотерапевтический агент выбирается из группы, состоящей из: Цикло-фасфамида, 5-Фторурацила, Темозоломида, Винкристина, Цисплатина, Карбоплатина и Гемцитабина.
Другие варианты выполнения настоящего изобретения представляют собой способ лечения рака, где применяется химиотерапевтический агент, и химиотерапевтический агент выбирается из группы, состоящей из: Гемцитабина, Цисплатина и Карбоплатина.
Настоящее изобретение также раскрывает способ лечения рака у пациента, которому необходимо такое лечение, причем указанное лечение включает введение указанному пациенту терапевтически эффективного количества, по меньшей мере, одного (например, 1, 2 или 3, или 1 или 2, или 1 и, как правило, 1) соединения формулы 1.0, и терапевтически эффективного количества, по меньшей мере, одного (например, 1, 2 или 3, или 1 или 2, или 2, или 1) химиотерапевтического агента, выбранного из группы, состоящей из: (1) таксанов, (2) координационных соединений платины, (3) ингибиторов эпидермального фактора роста (EGF), которые представляют собой антитела, (4) "маленьких молекул" - ингибиторов EGF, (5) ингибиторов васкулярных эндотелиальных факторов роста (VEGF), которые представляют собой антитела, (6) "маленьких молекул" - ингибиторов киназы VEGF, (7) антагонистов эстрогенового рецептора или селективных модуляторов эстрогенового рецептора (SERMs), (8) противоопухолевых нуклеозидных производных, (9) эпотилонов, (10) ингибиторов топоизомеразы, (11) алкалоидов винка, (12) антител, которые являются ингибиторами αVβ3 интегринов, (13) антагонистов фолата, (14) ингибиторов рибонуклеотидной редуктазы, (15) антрациклинов, (16) биопрепаратов; (17) ингибиторов ангиогенеза и/или супрессоров фактора некроза опухоли альфа (TNF-alpha), таких как Талидомид (или родственный имид), (18) ингибиторов Bcr/abl киназы, (19) "маленьких молекул" - ингибиторов МЕК1 и/или МЕК2, (20) "маленьких молекул" - ингибиторов IGF-1 и IGF-2, (21) "маленьких молекул" - ингибиторов RAF и BRAF киназ, (22) "маленьких молекул" - ингибиторов киназ, зависимых от клеточного цикла, таких как CDK1, CDK2, CDK4 и CDK6, (23) алкилирующих агентов и (24) ингибиторов фарнезилтрансферазы (также известных как ингибиторы FPT или FTI (то есть, ингибиторы фарнезилтрансферазы)).
Настоящее изобретение также раскрывает способ лечения рака у пациента, которому необходимо такое лечение, причем указанное лечение включает введение указанному пациенту терапевтически эффективного количества, по меньшей мере, одного (например, 1, 2 или 3, или 1 или 2, или 1 и, как правило, 1) соединения формулы 1.0, и терапевтически эффективных количеств, по меньшей мере, двух (например, 2 или 3, или 2, и обычно 2) различных противоопухолевых агентов, выбранных из группы, состоящей из: (1) таксанов, (2) координационных соединений платины, (3) ингибиторов эпидермального фактора роста (EGF), которые представляют собой антитела, (4) "маленьких молекул" - ингибиторов EGF, (5) ингибиторов васкулярных эндотелиальных факторов роста (VEGF), которые представляют собой антитела, (6) "маленьких молекул" - ингибиторов киназы VEGF, (7) антагонистов эстрогенового рецептора или селективных модуляторов эстрогенового рецептора (SERMs), (8) противоопухолевых нуклеозидных производных, (9) эпотилонов, (10) ингибиторов топоизомеразы, (11) алкалоидов винка, (12) антител, которые являются ингибиторами αVβ3 интегринов, (13) антагонистов фолата, (14) ингибиторов рибонуклеотидной редуктазы, (15) антрациклинов, (16) биопрепаратов; (17) ингибиторов ангио-генеза и/или супрессоров фактора некроза опухоли альфа (TNF-alpha), таких как талидомид (или родственный имид), (18) ингибиторов Bcr/abl киназы, (19) "маленьких молекул" - ингибиторов МЕК1 и/или МЕК2, (20) "маленьких молекул" - ингибиторов IGF-1 и IGF-2, (21) "маленьких молекул" - ингибиторов RAF и BRAF киназ, (22) "маленьких молекул" - ингибиторов киназ, зависимых от клеточного цикла, таких как CDK1, CDK2, CDK4 и CDK6, (23) алкилирующих агентов и (24) ингибиторов фарнезилтрансферазы (также известных как ингибиторы FPT или FTI (то есть, ингибиторы фарнезилтрансферазы)).
Настоящее изобретение также раскрывает способ лечения рака у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту терапевтически эффективных количеств, по меньшей мере, одного (например, 1, 2 или 3, или 1 или 2, или 1 и, как правило, 1) соединения формулы 1.0, и противоопухолевого агента, выбранного из группы, состоящей из: (1) ингибиторов EGF, которые представляют собой антитела, (2) "маленьких молекул" - ингибиторов EGF, (3) ингибиторов VEGF, которые представляют собой антитела и (4) "маленьких молекул" - ингибиторов VEGF. Радиотерапия также может применяться в сочетании с вышеуказанной комбинаторной терапией, то есть, вышеуказанный способ, применяющий комбинацию соединений по изобретению и противоопухолевого агента, также может содержать введение терапевтически эффективного количества облучения.
Настоящее изобретение также раскрывает способ лечения лейкемии (например, острой миелоидной лейкемии (AML) и хронической миелоидной лейкемии (CML)) у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту терапевтически эффективных количеств, по меньшей мере, одного (например, 1, 2 или 3, или 1 или 2, или 1 и, как правило, 1) соединения формулы 1.0, и: (1) Гливека или интерферона для лечения CML; (2) Гливека или пегилированного интерферона для лечения CML; (3) Гливека для лечения CML; (4) противоопухолевой нуклеозидной производной (например, Ara-С) для лечения AML; или (5) противоопухолевой нуклеозидной производной (например, Ara-С) в комбинации с антрациклином для лечения AML.
Настоящее изобретение также раскрывает способ лечения неходжкинской лимфомы у пациента, которому необходимо такое лечение, причем указанный способ включает введение терапевтически эффективного количества, по меньшей мере, одного (например, 1, 2 или 3, или 1 или 2, или 1 и, как правило, 1) соединения формулы 1.0 и: (1) биопрепарата (например, Ритуксан); (2) биопрепарата (например, Ритуксан) и противоопухолевой нуклеозидной производной (например, Флударабин); или (3) Генасенса (антисмысловой, по отношению к BCL-2).
Настоящее изобретение также раскрывает способ лечения множественной миеломы у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту терапевтически эффективных количеств, по меньшей мере, одного (например, 1, 2 или 3, или 1 или 2, или 1 и, как правило, 1) соединения формулы 1.0 и: (1) протеосомного ингибитора (например, PS-341 от Millenium); или (2) Талидомида (или родственного имида).
Настоящее изобретение также раскрывает способ лечения рака у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту терапевтически эффективных количеств: (а), по меньшей мере, одного (например, 1, 2 или 3, или 1 или 2, или 1 и, как правило, 1) соединения формулы 1.0, и (b), по меньшей мере, одного (например, 1, 2 или 3, или 1 или 2, или 2, или 1) противоопухолевого агента, выбранного из группы, состоящей из: (1) таксанов, (2) координационных соединений платины, (3) ингибиторов EGF, которые представляют собой антитела, (4) "маленьких молекул" - ингибиторов EGF, (5) VEGF, которые представляют собой антитела, (6) "маленьких молекул" - ингибиторов киназы VEGF, (7) антагонистов эстрогенового рецептора или селективных модуляторов эстрогенового рецептора, (8) противоопухолевых нуклеозидных производных, (9) эпотилонов, (10) ингибиторов топоизомеразы, (11) алкалоидов винка, (12) антител, которые являются ингибиторами αVβ3 интегринов.
Настоящее изобретение также раскрывает способы лечения немелкоклеточного рака легких у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту терапевтически эффективных количеств: (а), по меньшей мере, одного (например, 1, 2 или 3, или 1 или 2, или 1 и, как правило, 1) соединения формулы 1.0, и (b), по меньшей мере, одного (например, 1, 2 или 3, или 1 или 2, или 2, или 1) противоопухолевого агента, выбранного из группы, состоящей из: (1) таксанов, (2) координационных соединений платины, (3) ингибиторов EGF, которые представляют собой антитела, (4) "маленьких молекул" - ингибиторов EGF, (5) VEGF, которые представляют собой антитела, (6) "маленьких молекул" - ингибиторов киназы VEGF, (7) антагонистов эстрогенового рецептора или селективных модуляторов эстрогенового рецептора, (8) противоопухолевых нуклеозидных производных, (9) эпотилонов, (10) ингибиторов топоизомеразы, (11) алкалоидов винка, (12) антител, которые являются ингибиторами αVβ3 интегринов.
Настоящее изобретение также раскрывает способ лечения немелкоклеточного рака легких у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту терапевтически эффективных количеств: (а), по меньшей мере, одного (например, 1, 2 или 3, или 1 или 2, или 1 и, как правило, 1) соединения формулы 1.0, и (b), по меньшей мере, одного (например, 1, 2 или 3, или 1 или 2, или 2, или 1) противоопухолевого агента, выбранного из группы, состоящей из: (1) таксанов, (2) координационных соединений платины, (3) противоопухолевых нуклеозидных производных, (4) ингибиторов топоизомеразы и (5) алкалоидов винка.
Настоящее изобретение также раскрывает способы лечения немелкоклеточного рака легких у пациента, которому необходимо такое лечение, причем указанный способ включает введение терапевтически эффективного количества: (а), по меньшей мере, одного (например, 1, 2 или 3, или 1 или 2, или 1 и, как правило, 1) соединения формулы 1.0, (b) карбоплатина и (с) паклитаксела.
Настоящее изобретение также раскрывает способ лечения немелкоклеточного рака легких у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту терапевтически эффективного количества: (а), по меньшей мере, одного (например, 1, 2 или 3, или 1 или 2, или 1 и, как правило, 1) соединения формулы 1.0, (b) Цисплатина и (с) гемцитабина.
Настоящее изобретение также раскрывает способ лечения немелкоклеточного рака легких у пациента, которому необходимо такое лечение, причем указанный способ включает введение терапевтически эффективного количества: (а), по меньшей мере, одного (например, 1, 2 или 3, или 1 или 2, или 1 и, как правило, 1) соединения формулы 1.0, (b) карболатина и (с) гемцитабина.
Настоящее изобретение также раскрывает способ лечения немелкоклеточного рака легких у пациента, которому необходимо такое лечение, причем указанный способ включает введение терапевтически эффективного количества: (а), по меньшей мере, одного (например, 1, 2 или 3, или 1 или 2, или 1 и, как правило, 1) соединения формулы 1.0, (b) карбоплатина и (с) доцетаксела.
Настоящее изобретение также раскрывает способ лечения рака у пациента, которому необходимо такое лечение, причем указанный способ включает введение терапевтически эффективного количества: (а), по меньшей мере, одного (например, 1, 2 или 3, или 1 или 2, или 1 и, как правило, 1) соединения формулы 1.0, и (b) противоопухолевого агента, выбранного из группы, состоящей из: (1) ингибиторов EGF, которые представляют собой антитела (2) "маленьких молекул" - ингибиторов EGF, (3) ингибиторов VEGF, которые представляют собой антитела, (4) "маленьких молекул" - ингибиторов киназы VEGF.
Настоящее изобретение также раскрывает способ лечения плоскоклеточного рака головы и шеи у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту терапевтически эффективных количеств: (а), по меньшей мере, одного (например, 1, 2 или 3, или 1 или 2, или 1 и, как правило, 1) соединения формулы 1.0, и (b), по меньшей мере, одного (например, 1, 2 или 3, или 1 или 2, или 2, или 1) антинеопластического агента, выбранного из группы, состоящей из: (1) таксанов и (2) координационных соединений платины.
Настоящее изобретение также раскрывает способ лечения плоскоклеточного рака головы и шеи у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту терапевтически эффективных количеств: (а), по меньшей мере, одного (например, 1, 2 или 3, или 1 или 2, или 1 и, как правило, 1) соединения формулы 1.0, и (b), по меньшей мере, одного (например, 1, 2 или 3, или 1 или 2, или 2, или 1) антинеопластического агента, выбранного из группы, состоящей из: (1) таксанов, (2) координационных соединений платины и (3) противоопухолевых нуклеозидных производных (например, 5-Фторурацил).
Настоящее изобретение также раскрывает способ лечения CML у пациента, которому необходимо такое лечение, причем указанный способ включает введение терапевтически эффективного количества: (а), по меньшей мере, одного (например, 1, 2 или 3, или 1 или 2, или 1 и, как правило, 1) соединения формулы 1.0, (b) Гливека и (с) интерферона (например, Интрон-А).
Настоящее изобретение также раскрывает способ лечения CML у пациента, которому необходимо такое лечение, причем указанный способ включает введение терапевтически эффективного количества: (а), по меньшей мере, одного (например, 1, 2 или 3, или 1 или 2, или 1 и, как правило, 1) соединения формулы 1.0, (b) Гливека и (с) пегилированного интерферона (например, Пегинтрон и Пегасис).
Настоящее изобретение также раскрывает способ лечения CML у пациента, которому необходимо такое лечение, причем указанный способ включает введение терапевтически эффективного количества: (а), по меньшей мере, одного (например, 1, 2 или 3, или 1 или 2, или 1 и, как правило, 1) соединения формулы 1.0 и (b) Гливека.
Настоящее изобретение также раскрывает способ лечения CMML у пациента, которому необходимо такое лечение, причем указанный способ включает введение терапевтически эффективного количества, по меньшей мере, одного (например, 1, 2 или 3, или 1 или 2, или 1 и, как правило, 1) соединения формулы 1.0.
Настоящее изобретение также раскрывает способ лечения AML у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту терапевтически эффективных количеств: (а), по меньшей мере, одного (например, 1, 2 или 3, или 1 или 2, или 1 и, как правило, 1) соединения формулы 1.0 и (b) противоопухолевой нуклеозидной производной (например, Цитарабин (то есть, Ara-С)).
Настоящее изобретение также раскрывает способ лечения AML у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту терапевтически эффективных количеств: (а), по меньшей мере, одного (например, 1, 2 или 3, или 1 или 2, или 1 и, как правило, 1) соединения формулы 1.0, (b) противоопухолевой нуклеозидной производной (например, Цитарабин (то есть, Ara-С)) и (с) антрациклина.
Настоящее изобретение также раскрывает способ лечения неходжкинской лимфомы у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту терапевтически эффективных количеств: (а), по меньшей мере, одного (например, 1, 2 или 3, или 1 или 2, или 1 и, как правило, 1) соединения формулы 1.0 и (b) Ритуксимаба (Ритуксан).
Настоящее изобретение также раскрывает способ лечения неходжкинской лимфомы у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту терапевтически эффективных количеств: (а), по меньшей мере, одного (например, 1, 2 или 3, или 1 или 2, или 1 и, как правило, 1) соединения формулы 1.0, (b) Ритуксимаба (Ритуксан) и (с) противоопухолевой нуклеозидной производной (например, Флударабин (то есть, F-ara-А).
Настоящее изобретение также раскрывает способ лечения неходжкинской лимфомы у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту терапевтически эффективных количеств: (а), по меньшей мере, одного (например, 1, 2 или 3, или 1 или 2, или 1 и, как правило, 1) соединения формулы 1.0 и (b) Генасенса (антисмысловой, по отношению к BCL-2).
Настоящее изобретение также раскрывает способ лечения множественной миеломы у пациента, которому необходимо такое лечение, причем указанный способ включает введение терапевтически эффективного количества: (а), по меньшей мере, одного (например, 1, 2 или 3, или 1 или 2, или 1 и, как правило, 1) соединения формулы 1.0 и (b) протеосомного ингибитора (например, PS-341 (Millenium)).
Настоящее изобретение также раскрывает способ лечения множественной миеломы у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту терапевтически эффективных количеств: (а), по меньшей мере, одного (например, 1, 2 или 3, или 1 или 2, или 1 и, как правило, 1) соединения формулы 1.0 и (b) Талидомида или родственного имида.
Настоящее изобретение также раскрывает способ лечения множественной миеломы у пациента, которому необходимо такое лечение, причем указанный способ включает введение терапевтически эффективного количества: (а), по меньшей мере, одного (например, 1, 2 или 3, или 1 или 2, или 1 и, как правило, 1) соединения формулы 1.0 и (b) Талидомида.
Настоящее изобретение направлено на способы лечения рака, описанные в настоящей заявке, особенно раскрытые выше, где в дополнение к введению соединения формулы 1.0 и противоопухолевых агентов также вводится радиотерапия в период до, вовремя или после цикла лечения.
Настоящее изобретение также раскрывает способ лечения рака (например, рак легких, рак предстательной железы и миелоидная лейкемия) у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту: (1) эффективного количества, по меньшей мере, одного (например, 1, 2 или 3, или 1 или 2, или 1 и, как правило, 1) соединения формулы 1.0, в комбинации с (2), по меньшей мере, одним (например, 1, 2 или 3, или 1 или 2, или 2, или 1) противоопухолевым агентом, агентом, действующим на микротрубочки и/или радиотерапией.
Настоящее изобретение также раскрывает способ лечения рака у пациента, которому необходимо такое лечение, причем указанный способ включает введение указанному пациенту эффективного количества, по меньшей мере, одного (например, 1, 2 или 3, или 1 или 2, или 1 и, как правило, 1) соединения формулы 1.0 в комбинации с эффективным количеством, по меньшей мере, одного (например, 1, 2 или 3, или 1 или 2, или 1 и, как правило, 1) ингибитора сигнальной трансдукции.
Таким образом, в одном примере (например, лечение немелкоклеточного рака легких): (1) соединение формулы 1.0 вводится в количестве от около 50 мг до около 200 мг два раза в день, и в другом примере вводится от около 75 мг до около 125 мг два раза в день, и в еще одном примере вводится около 100 мг два раза в день, (2) Паклитаксел (например, Таксол®) вводится один раз в неделю, в количестве от около 50 до около 100 мг/м2, и в другом примере от около 60 до около 80 мг/м2, и (3) Карбоплатин вводится один раз в неделю, в количестве, необходимом для обеспечения AUC от около 2 до около 3.
В другом примере (например, лечение немелкоклеточного рака легких): (1) соединение формулы 1.0 вводится в количестве от около 50 мг до около 200 мг два раза в день, и в другом примере вводится от около 75 мг до около 125 мг два раза в день, и в еще одном примере вводится около 100 мг два раза в день, (2) Паклитаксел (например, Таксол® вводится один раз в неделю, в количестве от около 50 до около 100 мг/м2, и в другом примере от около 60 до около 80 мг/м2, и (3) Цисплатин вводится один раз в неделю, в количестве от около 20 до около 40 мг/м2.
В другом примере (например, лечение немелкоклеточного рака легких): (1) соединение формулы 1.0 вводится в количестве от около 50 мг до около 200 мг два раза в день, и в другом примере соединение формулы 1.0 вводится в количестве от около 75 мг до около 125 мг два раза в день, и в еще одном примере соединение формулы 1.0 вводится в количестве около 100 мг два раза в день, (2) Доцетаксел (например, Таксотер®) вводится один раз в неделю, в количестве от около 10 до около 45 мг/м2, и (3) Карбоплатин вводится один раз в неделю, в количестве, необходимом для обеспечения AUC от около 2 до около 3.
В другом примере (например, лечение немелкоклеточного рака легких): (1) соединение формулы 1.0 вводится в количестве от около 50 мг до около 200 мг два раза в день, и в другом примере соединение формулы 1.0 вводится в количестве от около 75 мг до около 125 мг два раза в день, и в еще одном примере соединение формулы 1.0 вводится в количестве около 100 мг два раза в день, (2) Доцетаксел (например, Таксотер®) вводится один раз в неделю, в количестве от около 10 до около 45 мг/м2, и (3) Цисплатин вводится один раз в неделю, в количестве от около 20 до около 40 мг/м2.
В другом примере (например, лечение немелкоклеточного рака легких): (1) соединение формулы 1.0 вводится в количестве от около 50 мг до около 200 мг два раза в день, и в другом примере соединение формулы 1.0 вводится в количестве от около 75 мг до около 125 мг два раза в день, и в еще одном примере соединение формулы 1.0 вводится в количестве 100 мг два раза в день, (2) Паклитаксел (например, Таксол®) вводится один раз каждые три недели, в количестве от около 150 до около 250 мг/м2, и в другом примере от около 175 до около 225 мг/м2, и в еще одном примере -175 мг/м2, и (3) Карбоплатин вводится один раз каждые три недели, в количестве, необходимом для обеспечения AUC от около 5 до около 8, и в другом примере - AUC 6.
В другом примере лечения немелкоклеточного рака легких: (1) соединение формулы 1.0 вводится в количестве 100 мг два раза в день, (2) Паклитаксел (например, Таксол®) вводится один раз каждые три недели, в количестве 175 мг/м2 и (3) Карбоплатин вводится один раз каждые три недели, в количестве, необходимом для обеспечения AUC 6.
В другом примере (например, лечение немелкоклеточного рака легких): (1) соединение формулы 1.0 вводится в количестве от около 50 мг до около 200 мг два раза в день, и в другом примере соединение формулы 1.0 вводится в количестве от около 75 мг до около 125 мг два раза в день, и в еще одном примере соединение формулы 1.0 вводится в количестве около 100 мг два раза в день, (2) Паклитаксел (например, Таксол®) вводится один раз каждые три недели, в количестве от около 150 до около 250 мг/м2, и в другом примере - от около 175 до около 225 мг/м2, и (3) Цисплатин вводится один раз каждые три недели, в количестве от около 60 до около 100 мг/м2.
В другом примере (например, лечение немелкоклеточного рака легких): (1) соединение формулы 1.0 вводится в количестве от около 50 мг до около 200 мг два раза в день, и в другом примере соединение формулы 1.0 вводится в количестве от около 75 мг до около 125 мг два раза в день, и в еще одном примере соединение формулы 1.0 вводится в количестве около 100 мг два раза в день, (2) Доцетаксел (например, Таксотер®) вводится один раз каждые три недели, в количестве от около 50 до около 100 мг/м2, и (3) Карбоплатин вводится один раз каждые три недели, в количестве, необходимом для обеспечения AUC от около 5 до около 8.
В другом примере (например, лечение немелкоклеточного рака легких): (1) соединение формулы 1.0 вводится в количестве от около 50 мг до около 200 мг два раза в день, в другом примере соединение формулы 1.0 вводится в количестве от около 75 мг до около 125 мг два раза в день, и в еще одном примере соединение формулы 1.0 вводится в количестве около 100 мг два раза в день, (2) Доцетаксел (например, Таксотер®) вводится один раз каждые три недели, в количестве от около 50 до около 100 мг/м2, и (3) Цисплатин вводится один раз каждые три недели, в количестве от около 60 до около 100 мг/м2.
В другом примере, для лечения немелкоклеточного рака легких, применяя соединения формулы 1.0, Доцетаксел и Карбоплатин: (1) соединение формулы 1.0 вводится в количестве от около 50 мг до около 200 мг два раза в день, и в другом примере соединение формулы 1.0 вводится в количестве от около 75 мг до около 125 мг два раза в день, и в еще одном примере соединение формулы 1.0 вводится в количестве около 100 мг два раза в день, (2) Доцетаксел (например, Таксотер®) вводится один раз каждые три недели, в количестве около 75 мг/м2, и (3) Карбоплатин вводится один раз каждые три недели, в количестве, необходимом для обеспечения AUC около 6.
В другом примере лечения немелкоклеточного рака легких, описанного выше, Доцетаксел (например, Таксотер®) и Цисплатин, Доцетаксел (например, Таксотер®) и Карбоплатин, Паклитаксел (например, Таксол® и Карбоплатин, или Паклитаксел (например, Таксол®) и Цисплатин вводятся в один и тот же день.
В другом примере (например, CML): (1) соединение формулы 1.0 вводится в количестве от около 100 мг до около 200 мг два раза в день, (2) Гливек вводится в количестве от около 400 до около 800 мг/день пероральным путем, и (3) интерферон (Интрон-А) вводится в количестве от около 5 до около 20 миллионов иммунизирующих единиц, три раза в неделю.
В другом примере (например, CML): (1) соединение формулы 1.0 вводится в количестве от около 100 мг до около 200 мг два раза в день, (2) Гливек вводится в количестве от около 400 до около 800 мг/день, пероральным путем, и (3) пегилированный интерферон (Пегинтрон или Пегасис) вводится в количестве от около 3 до около 6 микрограмм/кг/день.
В другом примере (например, неходжкинская лимфома): (1) соединение формулы 1.0 вводится в количестве от около 50 мг до около 200 мг два раза в день, и в другом примере соединение формулы 1.0 вводится в количестве от около 75 мг до около 125 мг два раза в день, и в еще одном примере соединение формулы 1.0 вводится в количестве 100 мг два раза в день, и (2) Генасенс (антисмысловой, по отношению к BCL-2) вводится путем непрерывного внутривенного вливания при дозе от около 2 до около 5 мг/кг/день (например, 3 мг/кг/день), в течение 5-7 дней, каждые 3-4 недели.
В другом примере (например, множественная миелома): (1) соединение формулы 1.0 вводится в количестве от около 50 мг до около 200 мг два раза в день, и в другом примере соединение формулы 1.0 вводится в количестве от около 75 мг до около 125 мг два раза в день, и в еще одном примере соединение формулы 1.0 вводится в количестве 100 мг два раза в день, и (2) протеосомный ингибитор (например, PS-341 - Millenium) вводится в количестве от около 1.5 мг/м2, дважды в неделю, в течение двух недель, следующих друг за другом, при перерыве в одну неделю.
В другом примере (например, множественная миелома): (1) соединение формулы 1.0 вводится в количестве от около 50 мг до около 200 мг два раза в день, и в другом примере соединение формулы 1.0 вводится в количестве около 75 мг до около 125 мг два раза в день, и в еще одном примере соединение формулы 1.0 вводится в количестве около 100 мг два раза в день, и (2) Талидомид (или родственный имид) вводится пероральным путем, в количестве от около 200 до около 800 мг/день, причем доза является неизменной до проявления рецидива или токсичности.
В одном варианте способов лечения рака по изобретению, химиотерапевтические агенты выбираются из группы, состоящей из: Паклитаксела, Доцетаксела, Карбоплатина, Цисплатина, Гемцитабина, Тамоксифена, Герцептина, Цетуксимаба, Тарцева, Иресса, Бевацизумаба, навельбина, IMC-1C11, SU5416 и SU6688.
В другом варианте выполнения способов лечения рака по изобретению, химиотерапевтические агенты выбираются из группы, состоящей из: Паклитаксела, Доцетаксела, Карбоплатина, Цисплатина, Навельбина, Гемцитабина и Герцептина.
Таким образом, один вариант выполнения изобретения представляет собой способ лечения рака, содержащий введение пациенту, которому необходимо такое лечение, терапевтически эффективного количества соединения формулы 1.0, таксана и координационного соединения платины.
Другой вариант выполнения изобретения представляет собой способ лечения рака, содержащий введение пациенту, которому необходимо такое лечение, терапевтически эффективного количества соединения формулы 1.0, таксана и координационного соединения платины, где указанное соединение формулы 1.0 вводится каждый день, указанный таксан вводится один раз в неделю на цикл, и указанное координационное соединение платины вводится один раз в неделю на цикл. В другом варианте выполнения изобретения лечение протекает в течение одной-четырех недель на цикл.
Другой вариант выполнения изобретения представляет собой способ лечения рака, содержащий введение пациенту, которому необходимо такое лечение, терапевтически эффективного количества соединения формулы 1.0, таксана и координационного соединения платины, где указанное соединение формулы 1.0 вводится каждый день, указанный таксан вводится один раз каждые три недели на цикл, и указанное координационное соединение платины вводится один раз каждые три недели на цикл. В другом варианте выполнения изобретения лечение протекает в течение одной-трех недель на цикл.
Другой вариант выполнения изобретения представляет собой способ лечения рака, содержащий введение пациенту, которому необходимо такое лечение, терапевтически эффективного количества соединения формулы 1.0, Паклитаксела и Карбоплатина. В другом варианте выполнения изобретения, указанное соединение формулы 1.0 вводится каждый день, указанный Паклитаксел вводится один раз в неделю на цикл, и указанный Карбоплатин вводится один раз в неделю на цикл. В другом варианте выполнения изобретения лечение протекает в течение одной-четырех недель на цикл.
Другой вариант выполнения изобретения представляет собой способ лечения рака, содержащий введение пациенту, которому необходимо такое лечение, терапевтически эффективного количества соединения формулы 1.0, Паклитаксела и Карбоплатина. В другом варианте выполнения изобретения, указанное соединение формулы 1.0 вводится каждый день, указанный Паклитаксел вводится один раз каждые три недели на цикл, и указанный Карбоплатин вводится один раз каждые три недели на цикл. В другом варианте выполнения изобретения лечение протекает в течение одной-трех недель на цикл.
Другой вариант выполнения изобретения представляет собой способ лечения немелкоклеточного рака легких у пациента, которому необходимо такое лечение, содержащий ежедневное введение терапевтически эффективного количества соединения формулы 1.0, введение терапевтически эффективного количества Карбоплатина один раз в неделю на цикл и введение терапевтически эффективного количества Паклитаксела один раз в неделю на цикл, где лечение проводится в течение одной-четырех недель на цикл. В другом варианте выполнения изобретения указанное соединение формулы 1.0 вводится два раза в день. В другом варианте выполнения изобретения указанный Карбоплатин и указанный Паклитаксел вводятся в один и тот же день, и в другом варианте выполнения изобретения указанный Карбоплатин и указанный Паклитаксел вводятся последовательно, и в другом варианте выполнения изобретения указанный Карбоплатин вводится вслед за указанным Паклитакселом.
Другой вариант выполнения изобретения представляет собой способ лечения немелкоклеточного рака легких у пациента, которому необходимо такое лечение, содержащий ежедневное введение терапевтически эффективного количества соединения формулы 1.0, введение терапевтически эффективного количества Карбоплатина один раз каждые три недели на цикл, и введение терапевтически эффективного количества Паклитаксела один раз каждые три недели на цикл, где лечение проводится в течение одной-трех недель. В другом варианте выполнения изобретения соединение формулы 1.0 вводится два раза в день. В другом варианте выполнения изобретения указанный Карбоплатин и указанный Паклитаксел вводятся в один и тот же день, и в другом варианте выполнения изобретения указанный Карбоплатин и указанный Паклитаксел вводятся последовательно, и в другом варианте выполнения изобретения указанный Карбоплатин вводится вслед за указанным Паклитакселом.
Другой вариант выполнения изобретение направлен способ лечения немелкоклеточного рака легких у пациента, которому необходимо такое лечение, содержащий введение от около 50 до около 200 мг соединения формулы 1.0 два раза в день, введение Карбоплатина один раз в неделю на цикл, в количестве, необходимом для обеспечения AUC от около 2 до около 8 (и в другом варианте выполнения изобретения - от около 2 до около 3), и введение один раз в неделю на цикл Паклитаксела, в количестве от около 60 до около 300 мг/м2 (и в другом варианте выполнения изобретения - около 50-100 мг/м2, и в еще одном варианте выполнения изобретения - от около 60 до около 80 мг/м2), причем лечение проводится в течение одной-четырех недель на цикл. В другом варианте выполнения изобретения указанное соединение формулы 1.0 вводится в количестве от около 75 до около 125 мг два раза в день, и в другом варианте выполнения изобретения около 100 мг два раза в день. В другом варианте выполнения изобретения указанный Карбоплатин и указанный Паклитаксел вводятся в один и тот же день, и в другом варианте выполнения изобретения указанный Карбоплатин и указанный Паклитаксел вводятся последовательно, и в другом варианте выполнения изобретения указанный Карбоплатин вводится вслед за указанным Паклитакселом.
В другом варианте выполнения изобретение направлено на способ лечения немелкоклеточного рака легких у пациента, которому необходимо такое лечение, содержащий введение от около 50 до около 200 мг соединения формулы 1.0 два раза в день, введение Карбоплатина один раз каждые три недели на цикл, в количестве, необходимом для обеспечения AUC от около 2 до около 8 (в другом варианте выполнения изобретения - от около 5 до около 8, и в другом варианте выполнения изобретения - 6), и введение один раз каждые три недели на цикл Паклитаксела, в количестве от около 150 до около 250 мг/м2 (и в другом варианте выполнения изобретения - от около 175 до около 225 мг/м2, и в другом варианте выполнения изобретения - 175 мг/м2), причем лечение проводится в течение одной-трех недель. В другом варианте выполнения изобретения указанное соединение формулы 1.0 вводится в количестве от около 75 до около 125 мг два раза в день, и в другом варианте выполнения изобретения около 100 мг два раза в день. В другом варианте выполнения изобретения указанный Карбоплатин и указанный Паклитаксел вводятся в один и тот же день, и в другом варианте выполнения изобретения указанный Карбоплатин и указанный Паклитаксел вводятся последовательно, и в другом варианте выполнения изобретения указанный Карбоплатин вводится вслед за указанным Паклитакселом.
Другие варианты выполнения изобретения представляют собой способы лечения рака как описано в вышеприведенных вариантах выполнения изобретения (то есть, варианты выполнения изобретения, направленные на лечение рака и лечение немелкоклеточного рака легких с помощью таксана и координационного соединения платины), за исключением того, что вместо Паклитаксела и Карбоплатина, таксаны и координационные соединения платины, применяемые вместе в способах, представляют собой: (1) Доцетаксел (Таксотер®) и цисплатин; (2) Паклитаксел и цисплатин; и (3) Доцетаксел и Карбоплатин. В другом варианте выполнения способов согласно настоящему изобретению Цисплатин применяется в количестве от около 30 до около 100 мг/м2. В другом варианте выполнения способов по изобретению Доцетаксел применяется в количествах от около 30 до около 100 мг/м2.
В другом варианте выполнения изобретение направлено на способ лечения рака, содержащий введение пациенту, которому необходимо такое лечение, терапевтически эффективного количества соединения формулы 1.0, таксана и ингибитора EGF, который представляет собой антитело. В другом варианте выполнения изобретения применяемый таксан представляет собой Паклитаксел, и ингибитор EGF представляет собой HER2 антитело (в одном варианте выполнения изобретения Герцептин) или Цетуксимаб, и в другом варианте выполнения изобретения применяется Герцептин. Продолжительность лечения и количества и введение указанного соединения формулы 1.0 и таксана являются такими, как описано в вариантах выполнения изобретения, раскрытых выше. Ингибитор EGF, который представляет собой антитело, вводится один раз в неделю на цикл, и в другом варианте выполнения изобретения вводится в тот же день, что и таксан, и в другом варианте выполнения изобретения вводится последовательно с таксаном. Например, Герцептин вводится в ударной дозе от около 3 до около 5 мг/м2 (в другом варианте выполнения изобретения около 4 мг/м2), и затем вводится в поддерживающей дозе около 2 мг/м2 один раз в неделю на цикл, в течение оставшейся части цикла лечения (обычно цикл составляет 1-4 недели). В одном варианте выполнения изобретения раком, подлежащим лечению, является рак молочной железы.
В другом варианте выполнения изобретение направлено на способ лечения рака, содержащий введение пациенту, которому необходимо такое лечение, терапевтически эффективных количеств: (1) соединения формулы 1.0, (2) таксана и (3) противоопухолевого агента, выбранного из группы, состоящей из: (а) "маленькой молекулы" - ингибитора EGF, (b) ингибитора VEGF, который представляет собой антитело и (с) "маленькой молекулы" - ингибитора киназы VEGF. В другом варианте выполнения изобретения, применяется таксан Паклитаксел или Доцетаксел. В другом варианте выполнения изобретения противоопухолевый агент выбирается из группы, состоящей из: тарцева, Иресса, Бевацизумаб, SU5416, SU6688 и BAY 43-9006. Продолжительность лечения и количества и введение указанного соединения формулы 1.0 и таксана являются такими, как описано в вариантах выполнения изобретения выше. Ингибитор киназы VEGF, который представляет собой антитело, обычно дается один раз в неделю на цикл. "Маленькие молекулы" - ингибиторы EGF и VEGF обычно вводятся ежедневно на цикл. В другом варианте выполнения изобретения, ингибитор VEGF, который представляет собой антитело, вводится в один и тот же день, что и таксан, и в другом варианте выполнения изобретения вводится одновременно с таксаном. В другом варианте выполнения изобретения, "маленькая молекула" - ингибитор EGF или "маленькая молекула" - ингибитор VEGF, вводится в один и тот же день, что и таксан, или вводится одновременно с таксаном. Ингибитор киназы VEGF или EGF, как правило, вводится в количестве от около 10 до около 500 мг/м2.
В другом варианте выполнения изобретение направлено на способ лечения рака, содержащий введение пациенту, которому необходимо такое лечение, терапевтически эффективного количества соединения формулы 1.0, противоопухолевой нуклеозидной производной и координационного соединения платины.
Другой вариант выполнения изобретения представляет собой способ лечения рака, содержащий введение пациенту, которому необходимо такое лечение, терапевтически эффективного количества соединения формулы 1.0, противоопухолевой нуклеозидной производной и координационного соединения платины, где указанное соединение формулы 1.0 вводится каждый день, указанная противоопухолевая нуклеозидная производная вводится один раз в неделю на цикл, и указанное координационное соединение платины вводится один раз в неделю на цикл. Хотя лечение может проводиться в течение одной-четырех недель на цикл, в одном варианте выполнения изобретения лечение проводится в течение одной-семи недель на цикл.
Другой вариант выполнения изобретения представляет собой способ лечения рака, содержащий введение пациенту, которому необходимо такое лечение, терапевтически эффективного количества соединения формулы 1.0, противоопухолевой нуклеозидной производной и координационного соединения платины, где указанное соединение формулы 1.0 вводится каждый день, указанная противоопухолевая нуклеозидная производная вводится один раз в неделю на цикл, и указанное координационное соединение платины вводится один раз каждые три недели на цикл. Хотя лечение может проводиться в течение одной-четырех недель на цикл, в одном варианте выполнения изобретения проводится в течение одной-семи недель на цикл.
Другой вариант выполнения изобретения представляет собой способ лечения рака, содержащий введение пациенту, которому необходимо такое лечение, терапевтически эффективного количества соединения формулы 1.0, гемцитабина и цисплатина. В другом варианте выполнения изобретения, указанное соединение формулы 1.0 вводится каждый день, указанный гемцитабин вводится один раз в неделю на цикл, и указанный цисплатин вводится один раз в неделю на цикл. В одном варианте выполнения изобретения лечение протекает в течение одной-семи недель на цикл.
Другой вариант выполнения изобретения представляет собой способ лечения рака, содержащий введение пациенту, которому необходимо такое лечение, терапевтически эффективного количества соединения формулы 1.0, гемцитабина и цисплатина. В другом варианте выполнения изобретения, указанное соединение формулы 1.0 вводится каждый день, указанный гемцитабин вводится один раз в неделю на цикл, и указанный Цисплатин вводится один раз каждые три недели на цикл. В другом варианте выполнения изобретения лечение проводится в течение одной-семи недель на цикл.
Другой вариант выполнения изобретения представляет собой способ лечения рака, содержащий введение пациенту, которому необходимо такое лечение, терапевтически эффективного количества соединения формулы 1.0, гемцитабина и карбоплатина. В другом варианте выполнения изобретения указанное соединение формулы 1.0 вводится каждый день, указанный гемцитабин вводится один раз в неделю на цикл, и указанный Карбоплатин вводится один раз в неделю на цикл. В другом варианте выполнения изобретения лечение протекает в течение одной-семи недель на цикл.
Другой вариант выполнения изобретения представляет собой способ лечения рака, содержащий введение пациенту, которому необходимо такое лечение, терапевтически эффективного количества соединения формулы 1.0, гемцитабина и карбоплатин. В другом варианте выполнения изобретения указанное соединение формулы 1.0 вводится каждый день, указанный гемцитабин вводится один раз в неделю на цикл, и указанный Карбоплатин вводится один раз каждые три недели на цикл. В другом варианте выполнения изобретения лечение проводится в течение одной-семи недель на цикл.
В вышеуказанных вариантах выполнения изобретения, применяющих гемцитабин, соединение формулы 1.0 и координационное соединение платины вводятся, как раскрывается выше для вариантов выполнения изобретения, применяющих таксаны. Гемцитабин вводится в количестве от около 500 до около 1250 мг/м2. В одном варианте выполнения изобретения гемцитабин вводится в тот же день, что и координационное соединение платины, и в другом варианте выполнения изобретения последовательно с координационным соединением платины, и в другом варианте выполнения изобретения гемцитабин вводится после координационного соединения платины.
Другой вариант выполнения настоящего изобретения представляет собой способ лечения рака у пациента, которому необходимо такое лечение, содержащий введение указанному пациенту соединения формулы 1.0 и противоопухолевого агента, выбранного из: (1) ингибиторов EGF, которые представляют собой антитела, (2) "маленьких молекул" - ингибиторов EGF (3) ингибиторов VEGF, которые представляют собой антитела, и (4) "маленьких молекул" - ингибиторов киназы VEGF, все из которых описаны выше. Лечение проводится в течение одной-семи недель на цикл, и, как правило, в течение одной-четырех недель на цикл. Соединение формулы 1.0 вводится тем же способом, как раскрывается выше для других вариантов выполнения изобретения. Противоопухолевые агенты, в виде маленьких молекул, как правило, вводятся ежедневно, и противоопухолевые агенты в виде антител, как правило, вводятся один раз в неделю на цикл. В одном варианте выполнения изобретения противоопухолевые агенты выбираются из группы, состоящей из: Герцептина, Цетуксимаба, Тарцева, Иресса, Бевацизумаба, IMC-1С11, SU5416, SU6688 и BAY 43-9006.
В других вариантах выполнения настоящего изобретения, где применяется координационное соединение платины, так же как и, по меньшей мере, один другой противоопухолевый агент, и эти лекарственные средства вводятся последовательно, координационное соединение платины, как правило, вводится после других противоопухолевых агентов, которые уже были введены.
Другие варианты выполнения настоящего изобретения включают введение пациенту терапевтически эффективного количества облучения помимо введения соединения формулы 1.0 и противоопухолевых агентов в вариантах выполнения изобретения, раскрытых выше. Облучение вводится согласно методиками и режимами, хорошо известными специалистам в данной области техники.
Другой вариант выполнения этого изобретения представляет собой фармацевтическую композицию, содержащую, по меньшей мере, два различных химиотерапевтических агента и фармацевтически приемлемый носитель для внутривенного введения. Предпочтительно фармацевтически приемлемый носитель представляет собой изотонический солевой раствор (0.9% NaCl) или раствор декстрозы (например, 5% декстрозы).
Другой вариант выполнения этого изобретения представляет собой фармацевтическую композицию, содержащую соединение формулы 1.0 и, по меньшей мере, два различных противоопухолевых агента, и фармацевтически приемлемый носитель для внутривенного введения. Предпочтительно фармацевтически приемлемый носитель представляет собой изотонический солевой раствор (0.9% NaCl) или раствор декстрозы (например, 5% декстрозы).
Другой вариант выполнения этого изобретения представляет собой фармацевтическую композицию, содержащую соединение формулы 1.0 и, по меньшей мере, один противоопухолевый агент и фармацевтически приемлемый носитель для внутривенного введения. Предпочтительно фармацевтически приемлемый носитель представляет собой изотонический солевой раствор (0.9% NaCl) или раствор декстрозы (например, 5% декстрозы).
Другие варианты выполнения настоящего изобретения представляют собой применение комбинации, по меньшей мере, одного (например, одного) соединения формулы 1.0 и лекарственных средств для лечения рака молочной железы, то есть, настоящее изобретение направлено на комбинаторную терапию для лечения рака молочной железы. Специалистам в данной области техники будет очевидно, что соединения формулы 1.0 и лекарственные средства, как правило, вводятся в виде отдельных фармацевтических композиций. Применение фармацевтической композиции, содержащей более чем одно лекарственное средство, входит в объем настоящего изобретения.
Таким образом, другой вариант выполнения настоящего изобретения представляет собой способ лечения (или предотвращения) рака молочной железы (то есть, постклимактерического и предклимактерического рака молочной железы, например, гормонально-зависимого рака молочной железы) у пациента, которому необходимо такое лечение, содержащий введение указанному пациенту терапевтически эффективного количества, по меньшей мере, одного (например, одного) соединения формулы 1.0 и терапевтически эффективного количества, по меньшей мере, одного антигормонального агента, выбранного из группы, состоящей из: (а) ингибиторов ароматазы (b) антиэстрогенов, и (с) LHRH аналогов; и, причем, указанное лечение необязательно включает введение, по меньшей мере, одного химиотерапевтического агента.
Соединение формулы 1.0 предпочтительно вводится пероральным путем, и в одном варианте выполнения изобретения вводится в форме капсулы.
Примерами ингибиторов ароматазы являются, но без ограничения к этому: Анастрозол (например, Аримидекс), Летрозол (например, Фемара), Эксеместан (Аромазин), Фадрозол и Форместан (например, Лентарон).
Примерами антиэстрогенов являются, но без ограничения к этому: Тамоксифен (например, Нолвадекс), Фульвестрант (например, Фаслодекс), Ралоксифен (например, Эвиста) и Аколбифен.
Примерами аналогов LHRH являются, но без ограничения к этому: Гозерелин (например, Золадекс) и Леупролид (например, Леупролида Ацетат, такой как Люпрон или Люпрон Депот).
Примерами химиотерапевтических агентов являются, но без ограничения к этому: Трастузумаб (например, Герцептин), Гефитиниб (например, Иресса), Эрлотиниб (например, Эрлотиниб HCl, такой как Тарцева), Бевацизумаб (например, Авастин), Цетуксимаб (например, Эрбитукс) и Бортезомиб (например, Велкаде).
Предпочтительно, когда применяется более чем один антигормональный агент, причем каждый агент выбирается из различных категорий агентов. Например, одним агентом является ингибитор ароматазы (например, Анастрозол, Летрозол или Эксеместан), а другим агентом является антиэстроген (например, Тамоксифен или Фулвестрант).
Другой вариант выполнения изобретения представляет собой способ лечения или предотвращения рака молочной железы у пациента, которому необходимо такое лечение, где указанное лечение включает введение терапевтически эффективного количества, по меньшей мере, одного (например, одного) соединения формулы 1.0 и, по меньшей мере, одного антигормонального агента, выбранного из группы, состоящей из: (а) ингибиторов ароматазы, (b) антиэстрогенов и (с) LHRH аналогов; и введение эффективного количества, по меньшей мере, одного химиотерапевтического агента.
Другой вариант выполнения изобретения представляет собой способ лечения или предотвращения рака молочной железы у пациента, которому необходимо такое лечение, где указанное лечение включает введение терапевтически эффективного количества, по меньшей мере, одного (например, одного) соединения формулы 1.0 и, по меньшей мере, одного антигормонального агента, выбранного из группы, состоящей из: (а) ингибиторов ароматазы, (b) антиэстрогенов и (с) LHRH аналогов.
Другой вариант выполнения изобретения представляет собой способ лечения или предотвращения рака молочной железы у пациента, которому необходимо такое лечение, где указанное лечение включает введение терапевтически эффективного количества, по меньшей мере, одного (например, одного) соединения формулы 1.0 и, по меньшей мере, одного антигормонального агента, выбранного из группы, состоящей из: (а) ингибиторов ароматазы и (b) антиэстрогенов.
Другой вариант выполнения изобретения представляет собой способ лечения или предотвращения рака молочной железы у пациента, которому необходимо такое лечение, где указанное лечение включает введение терапевтически эффективного количества, по меньшей мере, одного (например, одного) соединения формулы 1.0, по меньшей мере, одного антигормонального агента, выбранного из группы, состоящей из: (а) ингибиторов ароматазы и (b) антиэстрогенов; и, по меньшей мере, одного химиотерапевтического агента.
Другой вариант выполнения изобретения представляет собой способ лечения или предотвращения рака молочной железы у пациента, которому необходимо такое лечение, где указанное лечение включает введение терапевтически эффективного количества, по меньшей мере, одного (например, одного) соединения формулы 1.0 и, по меньшей мере, одного ингибитора ароматазы.
Другой вариант выполнения изобретения представляет собой способ лечения или предотвращения рака молочной железы у пациента, которому необходимо такое лечение, где указанное лечение включает введение терапевтически эффективного количества, по меньшей мере, одного (например, одного) соединения формулы 1.0, по меньшей мере, одного ингибитора ароматазы и, по меньшей мере, одного химиотерапевтического агента.
Другой вариант выполнения изобретения представляет собой способ лечения или предотвращения рака молочной железы у пациента, которому необходимо такое лечение, где указанное лечение включает введение терапевтически эффективного количества: (1), по меньшей мере, одного (например, одного) соединения формулы 1.0; и (2), по меньшей мере, одного антигормонального агента, выбранного из группы, состоящей из: (а) ингибиторов ароматазы, которые выбираются из группы, состоящей из Анастрозола, Летрозола, Эксеместана, Фадрозола и Форместана, (b) антиэстрогенов, которые выбираются из группы, состоящей из: Тамоксифена, Фулвестранта, Ралоксифена и Аколбифена и (с) LHRH аналогов, которые выбираются из группы, состоящей из: Гозерелина и Леупролида; и введение эффективного количества, по меньшей мере, одного химиотерапевтического агента, выбранного из группы, состоящей из: Трастузумаба, Гефитиниба, Эрлотиниба, Бевацизумаба, Цетуксимаба и Бортезомиба.
Другой вариант выполнения изобретения представляет собой способ лечения или предотвращения рака молочной железы у пациента, которому необходимо такое лечение, где указанное лечение включает введение терапевтически эффективного количества: (1), по меньшей мере, одного (например, одного) соединения формулы 1.0; и (2), по меньшей мере, одного антигормонального агента, выбранного из группы, состоящей из: (а) ингибиторов ароматазы, которые выбираются из группы, состоящей из Анастрозола, Летрозола, Эксеместана, Фадрозола и Форместана, (b) антиэстрогенов, которые выбираются из группы, состоящей из: Тамоксифена, Фулвестранта, Ралоксифена и Аколбифена и (с) LHRH аналогов, которые выбираются из группы, состоящей из: Гозерелина и Леупролида.
Другой вариант выполнения изобретения представляет собой способ лечения или предотвращения рака молочной железы у пациента, которому необходимо такое лечение, где указанное лечение включает введение терапевтически эффективного количества: (1), по меньшей мере, одного (например, одного) соединения формулы 1.0; и (2), по меньшей мере, одного антигормонального агента, выбранного из группы, состоящей из: (а) ингибиторов ароматазы, которые выбираются из группы, состоящей из Анастрозола, Летрозола, Эксеместана, Фадрозола и Форместана и (b) антиэстрогенов, которые выбираются из группы, состоящей из: Тамоксифена, Фулвестранта, Ралоксифена и Аколбифена.
Другой вариант выполнения изобретения представляет собой способ лечения или предотвращения рака молочной железы у пациента, которому необходимо такое лечение, где указанное лечение включает введение терапевтически эффективного количества: (1), по меньшей мере, одного (например, одного) соединения формулы 1.0; и (2), по меньшей мере, одного антигормонального агента, выбранного из группы, состоящей из: (а) ингибиторов ароматазы, которые выбираются из группы, состоящей из Анастрозола, Летрозола, Эксеместана, Фадрозола и Форместана, (b) антиэстрогенов, которые выбираются из группы, состоящей из: Тамоксифена, Фулвестранта, Ралоксифена и Аколбифена; и введение эффективного количества, по меньшей мере, одного химиотерапевтического агента, выбранного из группы, состоящей из: Трастузумаба, Гефитиниба, Эрлотиниба, Бевацизумаба, Цетуксимаба и Бортезомиба.
Другой вариант выполнения изобретения представляет собой способ лечения или предотвращения рака молочной железы у пациента, которому необходимо такое лечение, где указанное лечение включает введение терапевтически эффективного количества: (1), по меньшей мере, одного (например, одного) соединения формулы 1.0; и (2), по меньшей мере, одного ингибитора ароматазы, выбранного из группы, состоящей из Анастрозола, Летрозола, Эксеместана, Фадрозола и Форместана.
Другой вариант выполнения изобретения представляет собой способ лечения или предотвращения рака молочной железы у пациента, которому необходимо такое лечение, где указанное лечение включает введение терапевтически эффективного количества: (1), по меньшей мере, одного (например, одного) соединения формулы 1.0; (2), по меньшей мере, одного ингибитора ароматазы, который выбирается из группы, состоящей из Анастрозола, Летрозола, Эксеместана, Фадрозола и Форместан; и (3) введение эффективного количества, по меньшей мере, одного химиотерапевтического агента, выбранного из группы, состоящей из: Трастузумаба, Гефитиниба, Эрлотиниба, Бевацизумаба, Цетуксимаба и Бортезомиба.
Другой вариант выполнения изобретения представляет собой способ лечения или предотвращения рака молочной железы у пациента, которому необходимо такое лечение, где указанное лечение включает введение терапевтически эффективного количества: (1), по меньшей мере, одного (например, одного) соединения формулы 1.0; (2), по меньшей мере, одного ингибитора ароматазы и (3), по меньшей мере, одного LHRH аналога.
Другой вариант выполнения изобретения представляет собой способ лечения или предотвращения рака молочной железы у пациента, которому необходимо такое лечение, где указанное лечение включает введение терапевтически эффективного количества:(1), по меньшей мере, одного (например, одного) соединения формулы 1.0; (2), по меньшей мере, одного антиэстрогена; и (3), по меньшей мере, одного LHRH аналога.
Другой вариант выполнения изобретения представляет собой способ лечения или предотвращения рака молочной железы у пациента, которому необходимо такое лечение, где указанное лечение включает введение терапевтически эффективного количества: (1), по меньшей мере, одного (например, одного) соединения формулы 1.0; (2), по меньшей мере, одного ингибитора ароматазы, который выбирается из группы, состоящей из Анастрозола, Летрозола, Эксеместана, Фадрозола и Форместана; и (3), по меньшей мере, одного LHRH аналога, который выбирается из группы, состоящей из: Гозерелина и Леупролида.
Другой вариант выполнения изобретения представляет собой способ лечения или предотвращения рака молочной железы у пациента, которому необходимо такое лечение, где указанное лечение включает введение терапевтически эффективного количества: (1), по меньшей мере, одного (например, одного) соединения формулы 1.0; (2), по меньшей мере, одного антиэстрогена, который выбирается из группы, состоящей из: Тамоксифена, Фулвестранта, Ралоксифена и Аколбифена; и (3), по меньшей мере, одного LHRH аналога, который выбирается из группы, состоящей из: Гозерелина и Леупролида.
Другой вариант выполнения изобретения представляет собой способ лечения или предотвращения рака молочной железы у пациента, которому необходимо такое лечение, где указанное лечение включает введение терапевтически эффективного количества, по меньшей мере, одного (например, одного) соединения формулы 1.0 и Анастрозола.
Другой вариант выполнения изобретения представляет собой способ лечения или предотвращения рака молочной железы у пациента, которому необходимо такое лечение, где указанное лечение включает введение терапевтически эффективного количества, по меньшей мере, одного (например, одного) соединения формулы 1.0 и Летразола.
Другой вариант выполнения изобретения представляет собой способ лечения или предотвращения рака молочной железы у пациента, которому необходимо такое лечение, где указанное лечение включает введение терапевтически эффективного количества, по меньшей мере, одного (например, одного) соединения формулы 1.0 и Эксеместана.
Другой вариант выполнения изобретения представляет собой способ лечения или предотвращения рака молочной железы у пациента, которому необходимо такое лечение, где указанное лечение включает введение терапевтически эффективного количества, по меньшей мере, одного (например, одного) соединения формулы 1.0 и Фадрозола.
Другой вариант выполнения изобретения представляет собой способ лечения или предотвращения рака молочной железы у пациента, которому необходимо такое лечение, где указанное лечение включает введение терапевтически эффективного количества, по меньшей мере, одного (например, одного) соединения формулы 1.0 и Форместана.
Другой вариант выполнения изобретения представляет собой способ лечения или предотвращения рака молочной железы у пациента, которому необходимо такое лечение, где указанное лечение включает введение терапевтически эффективного количества, по меньшей мере, одного (например, одного) соединения формулы 1.0 и Тамоксифена.
Другой вариант выполнения изобретения представляет собой способ лечения или предотвращения рака молочной железы у пациента, которому необходимо такое лечение, где указанное лечение включает введение терапевтически эффективного количества, по меньшей мере, одного (например, одного) соединения формулы 1.0 Фулвестранта.
Другой вариант выполнения изобретения представляет собой способ лечения или предотвращения рака молочной железы у пациента, которому необходимо такое лечение, где указанное лечение включает введение терапевтически эффективного количества, по меньшей мере, одного (например, одного) соединения формулы 1.0 и Ралоксифена.
Другой вариант выполнения изобретения представляет собой способ лечения или предотвращения рака молочной железы у пациента, которому необходимо такое лечение, где указанное лечение включает введение терапевтически эффективного количества, по меньшей мере, одного (например, одного) соединения формулы 1.0 и Аколбифена.
Другой вариант выполнения изобретения представляет собой способ лечения или предотвращения рака молочной железы у пациента, которому необходимо такое лечение, где указанное лечение включает введение терапевтически эффективного количества, по меньшей мере, одного (например, одного) соединения формулы 1.0 и Гозерелина.
Другой вариант выполнения изобретения представляет собой способ лечения или предотвращения рака молочной железы у пациента, которому необходимо такое лечение, где указанное лечение включает введение терапевтически эффективного количества, по меньшей мере, одного (например, одного) соединения формулы 1.0 и Леупролида.
Другой вариант выполнения изобретения представляет собой способ лечения или предотвращения рака молочной железы у пациента, которому необходимо такое лечение, где указанное лечение включает введение терапевтически эффективного количества, по меньшей мере, одного (например, одного) соединения формулы 1.0, Анастрозола и антиэстрогена, выбранного из группы, состоящей из: Тамоксифена, Фулвестранта, Ралоксифена и Аколбифена.
Другой вариант выполнения изобретения представляет собой способ лечения или предотвращения рака молочной железы у пациента, которому необходимо такое лечение, где указанное лечение включает введение терапевтически эффективного количества, по меньшей мере, одного (например, одного) соединения формулы 1.0, Летрозола и антиэстрогена, выбранного из группы, состоящей из: Тамоксифена, Фулвестранта, Ралоксифена и Аколбифена.
Другой вариант выполнения изобретения представляет собой способ лечения или предотвращения рака молочной железы у пациента, которому необходимо такое лечение, где указанное лечение включает введение терапевтически эффективного количества, по меньшей мере, одного (например, одного) соединения формулы 1.0, Эксеместана и антиэстрогена, выбранного из группы, состоящей из: Тамоксифена, Фулвестранта, Ралоксифена и Аколбифена.
Другой вариант выполнения изобретения представляет собой способ лечения или предотвращения рака молочной железы у пациента, которому необходимо такое лечение, где указанное лечение включает введение терапевтически эффективного количества, по меньшей мере, одного (например, одного) соединения формулы 1.0, Фадрозола и антиэстрогена, выбранного из группы, состоящей из: Тамоксифена, Фулвестранта, Ралоксифена и Аколбифена.
Другой вариант выполнения изобретения представляет собой способ лечения или предотвращения рака молочной железы у пациента, которому необходимо такое лечение, где указанное лечение включает введение терапевтически эффективного количества, по меньшей мере, одного (например, одного) соединения формулы 1.0, Форместана и антиэстрогена, выбранного из группы, состоящей из: Тамоксифена, Фулвестранта, Ралоксифена и Аколбифена.
Другой вариант выполнения изобретения представляет собой способ лечения или предотвращения рака молочной железы у пациента, которому необходимо такое лечение, где указанное лечение включает введение терапевтически эффективного количества, по меньшей мере, одного (например, одного) соединения формулы 1.0, Анастрозола и Тамоксифена.
Другой вариант выполнения изобретения представляет собой способ лечения или предотвращения рака молочной железы у пациента, которому необходимо такое лечение, где указанное лечение включает введение терапевтически эффективного количества, по меньшей мере, одного (например, одного) соединения формулы 1.0, Летрозола и Тамоксифена.
Другой вариант выполнения изобретения представляет собой способ лечения или предотвращения рака молочной железы у пациента, которому необходимо такое лечение, где указанное лечение включает введение терапевтически эффективного количества, по меньшей мере, одного (например, одного) соединения формулы 1.0, Эксеместана и Тамоксифена.
Другой вариант выполнения изобретения представляет собой способ лечения или предотвращения рака молочной железы у пациента, которому необходимо такое лечение, где указанное лечение включает введение терапевтически эффективного количества, по меньшей мере, одного (например, одного) соединения формулы 1.0, Фадрозола и Тамоксифена.
Другой вариант выполнения изобретения представляет собой способ лечения или предотвращения рака молочной железы у пациента, которому необходимо такое лечение, где указанное лечение включает введение терапевтически эффективного количества, по меньшей мере, одного (например, одного) соединения формулы 1.0, Форместана и Тамоксифена.
Другой вариант выполнения изобретения представляет собой способ лечения или предотвращения рака молочной железы у пациента, которому необходимо такое лечение, где указанное лечение включает введение терапевтически эффективного количества, по меньшей мере, одного (например, одного) соединения формулы 1.0, Анастрозола и Фулвестранта.
Другой вариант выполнения изобретения представляет собой способ лечения или предотвращения рака молочной железы у пациента, которому необходимо такое лечение, где указанное лечение включает введение терапевтически эффективного количества, по меньшей мере, одного (например, одного) соединения формулы 1.0, Летрозола и Фулвестранта.
Другой вариант выполнения изобретения представляет собой способ лечения или предотвращения рака молочной железы у пациента, которому необходимо такое лечение, где указанное лечение включает введение терапевтически эффективного количества, по меньшей мере, одного (например, одного) соединения формулы 1.0, Эксеместана и Фулвестранта.
Другой вариант выполнения изобретения представляет собой способ лечения или предотвращения рака молочной железы у пациента, которому необходимо такое лечение, где указанное лечение включает введение терапевтически эффективного количества, по меньшей мере, одного (например, одного) соединения формулы 1.0, Фадрозола и Фулвестранта.
Другой вариант выполнения изобретения представляет собой способ лечения или предотвращения рака молочной железы у пациента, которому необходимо такое лечение, где указанное лечение включает введение терапевтически эффективного количества, по меньшей мере, одного (например, одного) соединения формулы 1.0, Форместана и Фулвестранта.
Другой вариант выполнения изобретения представляет собой способ лечения или предотвращения рака молочной железы у пациента, которому необходимо такое лечение, где указанное лечение включает введение терапевтически эффективного количества, по меньшей мере, одного (например, одного) соединения формулы 1.0, Анастрозола и химиотерапевтического агента, выбранного из группы, состоящей из: Трастузумаба, Гефитиниба, Эрлотиниба, Бевацизумаба, Цетуксимаба и Бортезомиба.
Другой вариант выполнения изобретения представляет собой способ лечения или предотвращения рака молочной железы у пациента, которому необходимо такое лечение, где указанное лечение включает введение терапевтически эффективного количества, по меньшей мере, одного (например, одного) соединения формулы 1.0, Летрозола и химиотерапевтического агента, выбранного из группы, состоящей из: Трастузумаба, Гефитиниба, Эрлотиниба, Бевацизумаба, Цетуксимаба и Бортезомиба.
Другой вариант выполнения изобретения представляет собой способ лечения или предотвращения рака молочной железы у пациента, которому необходимо такое лечение, где указанное лечение включает введение терапевтически эффективного количества, по меньшей мере, одного (например, одного) соединения формулы 1.0, Эксеместана и химиотерапевтического агента, выбранного из группы, состоящей из: Трастузумаба, Гефитиниба, Эрлотиниба, Бевацизумаба, Цетуксимаба и Бортезомиба.
Другой вариант выполнения изобретения представляет собой способ лечения или предотвращения рака молочной железы у пациента, которому необходимо такое лечение, где указанное лечение включает введение терапевтически эффективного количества, по меньшей мере, одного (например, одного) соединения формулы 1.0, Фадрозола и химиотерапевтического агента, выбранного из группы, состоящей из: Трастузумаба, Гефитиниба, Эрлотиниба, Бевацизумаба, Цетуксимаба и Бортезомиба.
Другой вариант выполнения изобретения представляет собой способ лечения или предотвращения рака молочной железы у пациента, которому необходимо такое лечение, где указанное лечение включает введение терапевтически эффективного количества, по меньшей мере, одного (например, одного) соединения формулы 1.0, Форместана и химиотерапевтического агента, выбранного из группы, состоящей из: Трастузумаба, Гефитиниба, Эрлотиниба, Бевацизумаба, Цетуксимаба и Бортезомиба.
Другой вариант выполнения изобретения представляет собой способ лечения или предотвращения рака молочной железы у пациента, которому необходимо такое лечение, где указанное лечение включает введение терапевтически эффективного количества, по меньшей мере, одного (например, одного) соединения формулы 1.0, Тамоксифена и химиотерапевтического агента, выбранного из группы, состоящей из: Трастузумаба, Гефитиниба, Эрлотиниба, Бевацизумаба, Цетуксимаба и Бортезомиба.
Другой вариант выполнения изобретения представляет собой способ лечения или предотвращения рака молочной железы у пациента, которому необходимо такое лечение, где указанное лечение включает введение терапевтически эффективного количества, по меньшей мере, одного (например, одного) соединения формулы 1.0, Фулвестранта и химиотерапевтического агента, выбранного из группы, состоящей из: Трастузумаба, Гефитиниба, Эрлотиниба, Бевацизумаба, Цетуксимаба и Бортезомиба.
Другой вариант выполнения изобретения представляет собой способ лечения или предотвращения рака молочной железы у пациента, которому необходимо такое лечение, где указанное лечение включает введение терапевтически эффективного количества, по меньшей мере, одного (например, одного) соединения формулы 1.0, Ралоксифена и химиотерапевтического агента, выбранного из группы, состоящей из: Трастузумаба, Гефитиниба, Эрлотиниба, Бевацизумаба, Цетуксимаба и Бортезомиба.
Другой вариант выполнения изобретения представляет собой способ лечения или предотвращения рака молочной железы у пациента, которому необходимо такое лечение, где указанное лечение включает введение терапевтически эффективного количества, по меньшей мере, одного (например, одного) соединения формулы 1.0, Аколбифена и химиотерапевтического агента, выбранного из группы, состоящей из: Трастузумаба, Гефитиниба, Эрлотиниба, Бевацизумаба, Цетуксимаба и Бортезомиба.
Другой вариант выполнения изобретения представляет собой способ лечения или предотвращения рака молочной железы у пациента, которому необходимо такое лечение, где указанное лечение включает введение терапевтически эффективного количества, по меньшей мере, одного (например, одного) соединения формулы 1.0, Гозерелина и химиотерапевтического агента, выбранного из группы, состоящей из: Трастузумаба, Гефитиниба, Эрлотиниба, Бевацизумаба, Цетуксимаба и Бортезомиба.
Другой вариант выполнения изобретения представляет собой способ лечения или предотвращения рака молочной железы у пациента, которому необходимо такое лечение, где указанное лечение включает введение терапевтически эффективного количества, по меньшей мере, одного (например, одного) соединения формулы 1.0, Леупролина и химиотерапевтического агента, выбранного из группы, состоящей из: Трастузумаба, Гефитиниба, Эрлотиниба, Бевацизумаба, Цетуксимаба и Бортезомиба.
Другой вариант выполнения изобретения представляет собой способ лечения или предотвращения рака молочной железы у пациента, которому необходимо такое лечение, где указанное лечение включает введение терапевтически эффективного количества, по меньшей мере, одного (например, одного) соединения формулы 1.0, Анастрозола, антиэстрогена, выбранного из группы, состоящей из: Тамоксифена, Фулвестранта, Ралоксифена и Аколбифена, и химиотерапевтического агента, выбранного из группы, состоящей из: Трастузумаба, Гефитиниба, Эрлотиниба, Бевацизумаба, Цетуксимаба и Бортезомиба.
Другой вариант выполнения изобретения представляет собой способ лечения или предотвращения рака молочной железы у пациента, которому необходимо такое лечение, где указанное лечение включает введение терапевтически эффективного количества, по меньшей мере, одного (например, одного) соединения формулы 1.0, Летрозола, антиэстрогена, выбранного из группы, состоящей из: Тамоксифена, Фулвестранта, Ралоксифена и Аколбифена, и химиотерапевтического агента, выбранного из группы, состоящей из: Трастузумаба, Гефитиниба, Эрлотиниба, Бевацизумаба, Цетуксимаба и Бортезомиба.
Другой вариант выполнения изобретения представляет собой способ лечения или предотвращения рака молочной железы у пациента, которому необходимо такое лечение, где указанное лечение включает введение терапевтически эффективного количества, по меньшей мере, одного (например, одного) соединения формулы 1.0, Эксеместана, антиэстрогена, выбранного из группы, состоящей из: Тамоксифена, Фулвестранта, Ралоксифена и Аколбифена, и химиотерапевтического агента, выбранного из группы, состоящей из: Трастузумаба, Гефитиниба, Эрлотиниба, Бевацизумаба, Цетуксимаба и Бортезомиба.
Другой вариант выполнения изобретения представляет собой способ лечения или предотвращения рака молочной железы у пациента, которому необходимо такое лечение, где указанное лечение включает введение терапевтически эффективного количества, по меньшей мере, одного (например, одного) соединения формулы 1.0, Фадрозола, антиэстрогена, выбранного из группы, состоящей из: Тамоксифена, Фулвестранта, Ралоксифена и Аколбифена, и химиотерапевтического агента, выбранного из группы, состоящей из: Трастузумаба, Гефитиниба, Эрлотиниба, Бевацизумаба, Цетуксимаба и Бортезомиба.
Другой вариант выполнения изобретения представляет собой способ лечения или предотвращения рака молочной железы у пациента, которому необходимо такое лечение, где указанное лечение включает введение терапевтически эффективного количества, по меньшей мере, одного (например, одного) соединения формулы 1.0, Форместана, антиэстрогена, выбранного из группы, состоящей из: Тамоксифена, Фулвестранта, Ралоксифена и Аколбифена, и химиотерапевтического агента, выбранного из группы, состоящей из: Трастузумаба, Гефитиниба, Эрлотиниба, Бевацизумаба, Цетуксимаба и Бортезомиба.
Другой вариант выполнения изобретения представляет собой способ лечения или предотвращения рака молочной железы у пациента, которому необходимо такое лечение, где указанное лечение включает введение терапевтически эффективного количества, по меньшей мере, одного (например, одного) соединения формулы 1.0, Анастрозола, Тамоксифена и химиотерапевтического агента, выбранного из группы, состоящей из: Трастузумаба, Гефитиниба, Эрлотиниба, Бевацизумаба, Цетуксимаба и Бортезомиба.
Другой вариант выполнения изобретения представляет собой способ лечения или предотвращения рака молочной железы у пациента, которому необходимо такое лечение, где указанное лечение включает введение терапевтически эффективного количества, по меньшей мере, одного (например, одного) соединения формулы 1.0, Летрозола, Тамоксифена и химиотерапевтического агента, выбранного из группы, состоящей из: Трастузумаба, Гефитиниба, Эрлотиниба, Бевацизумаба, Цетуксимаба и Бортезомиба.
Другой вариант выполнения изобретения представляет собой способ лечения или предотвращения рака молочной железы у пациента, которому необходимо такое лечение, где указанное лечение включает введение терапевтически эффективного количества, по меньшей мере, одного (например, одного) соединения формулы 1.0, Эксеместана, Тамоксифена и химиотерапевтического агента, выбранного из группы, состоящей из: Трастузумаба, Гефитиниба, Эрлотиниба, Бевацизумаба, Цетуксимаба и Бортезомиба.
Другой вариант выполнения изобретения представляет собой способ лечения или предотвращения рака молочной железы у пациента, которому необходимо такое лечение, где указанное лечение включает введение терапевтически эффективного количества, по меньшей мере, одного (например, одного) соединения формулы 1.0, Фадрозола, Тамоксифена и химиотерапевтического агента, выбранного из группы, состоящей из: Трастузумаба, Гефитиниба, Эрлотиниба, Бевацизумаба, Цетуксимаба и Бортезомиба.
Другой вариант выполнения изобретения представляет собой способ лечения или предотвращения рака молочной железы у пациента, которому необходимо такое лечение, где указанное лечение включает введение терапевтически эффективного количества, по меньшей мере, одного (например, одного) соединения формулы 1.0, Форместана, Тамоксифена и химиотерапевтического агента, выбранного из группы, состоящей из: Трастузумаба, Гефитиниба, Эрлотиниба, Бевацизумаба, Цетуксимаба и Бортезомиба.
Другой вариант выполнения изобретения представляет собой способ лечения или предотвращения рака молочной железы у пациента, которому необходимо такое лечение, где указанное лечение включает введение терапевтически эффективного количества, по меньшей мере, одного (например, одного) соединения формулы 1.0, Анастрозола, Фулвестранта и химиотерапевтического агента, выбранного из группы, состоящей из: Трастузумаба, Гефитиниба, Эрлотиниба, Бевацизумаба, Цетуксимаба и Бортезомиба.
Другой вариант выполнения изобретения представляет собой способ лечения или предотвращения рака молочной железы у пациента, которому необходимо такое лечение, где указанное лечение включает введение терапевтически эффективного количества, по меньшей мере, одного (например, одного) соединения формулы 1.0, Летрозола, Фулвестранта и химиотерапевтического агента, выбранного из группы, состоящей из: Трастузумаба, Гефитиниба, Эрлотиниба, Бевацизумаба, Цетуксимаба и Бортезомиба.
Другой вариант выполнения изобретения представляет собой способ лечения или предотвращения рака молочной железы у пациента, которому необходимо такое лечение, где указанное лечение включает введение терапевтически эффективного количества, по меньшей мере, одного (например, одного) соединения формулы 1.0, Эксеместана, Фулвестранта и химиотерапевтического агента, выбранного из группы, состоящей из: Трастузумаба, Гефитиниба, Эрлотиниба, Бевацизумаба, Цетуксимаба и Бортезомиба.
Другой вариант выполнения изобретения представляет собой способ лечения или предотвращения рака молочной железы у пациента, которому необходимо такое лечение, где указанное лечение включает введение терапевтически эффективного количества, по меньшей мере, одного (например, одного) соединения формулы 1.0, Фадрозола, Фулвестранта и химиотерапевтического агента, выбранного из группы, состоящей из: Трастузумаба, Гефитиниба, Эрлотиниба, Бевацизумаба, Цетуксимаба и Бортезомиба.
Другой вариант выполнения изобретения представляет собой способ лечения или предотвращения рака молочной железы у пациента, которому необходимо такое лечение, где указанное лечение включает введение терапевтически эффективного количества, по меньшей мере, одного (например, одного) соединения формулы 1.0, Форместана, Фулвестранта и химиотерапевтического агента, выбранного из группы, состоящей из: Трастузумаба, Гефитиниба, Эрлотиниба, Бевацизумаба, Цетуксимаба и Бортезомиба.
Другой вариант выполнения изобретения представляет собой способ лечения или предотвращения рака молочной железы у пациента, которому необходимо такое лечение, где указанное лечение включает введение терапевтически эффективного количества, по меньшей мере, одного (например, одного) соединения формулы 1.0, Гозерелина и Тамоксифена.
Другой вариант выполнения изобретения представляет собой способ лечения или предотвращения рака молочной железы у пациента, которому необходимо такое лечение, где указанное лечение включает введение терапевтически эффективного количества, по меньшей мере, одного (например, одного) соединения формулы 1.0, Гозерелина и Фулвестранта.
Другой вариант выполнения изобретения представляет собой способ лечения или предотвращения рака молочной железы у пациента, которому необходимо такое лечение, где указанное лечение включает введение терапевтически эффективного количества, по меньшей мере, одного (например, одного) соединения формулы 1.0, Гозерелина и Ралоксифена.
Другой вариант выполнения изобретения представляет собой способ лечения или предотвращения рака молочной железы у пациента, которому необходимо такое лечение, где указанное лечение включает введение терапевтически эффективного количества, по меньшей мере, одного (например, одного) соединения формулы 1.0, Гозерелина и Аколбифена.
Другой вариант выполнения изобретения представляет собой способ лечения или предотвращения рака молочной железы у пациента, которому необходимо такое лечение, где указанное лечение включает введение терапевтически эффективного количества, по меньшей мере, одного (например, одного) соединения формулы 1.0, Леупролида и Тамоксифена.
Другой вариант выполнения изобретения представляет собой способ лечения или предотвращения рака молочной железы у пациента, которому необходимо такое лечение, где указанное лечение включает введение терапевтически эффективного количества, по меньшей мере, одного (например, одного) соединения формулы 1.0, Леупролида и Фулвестранта.
Другой вариант выполнения изобретения представляет собой способ лечения или предотвращения рака молочной железы у пациента, которому необходимо такое лечение, где указанное лечение включает введение терапевтически эффективного количества, по меньшей мере, одного (например, одного) соединения формулы 1.0, Леупролида и Ралоксифена.
Другой вариант выполнения изобретения представляет собой способ лечения или предотвращения рака молочной железы у пациента, которому необходимо такое лечение, где указанное лечение включает введение терапевтически эффективного количества, по меньшей мере, одного (например, одного) соединения формулы 1.0, Леупролида и Аколбифена.
Другой вариант выполнения изобретения представляет собой способ лечения или предотвращения рака молочной железы у пациента, которому необходимо такое лечение, где указанное лечение включает введение терапевтически эффективного количества, по меньшей мере, одного (например, одного) соединения формулы 1.0, Гозерелина и Анастрозола.
Другой вариант выполнения изобретения представляет собой способ лечения или предотвращения рака молочной железы у пациента, которому необходимо такое лечение, где указанное лечение включает введение терапевтически эффективного количества, по меньшей мере, одного (например, одного) соединения формулы 1.0, Гозерелина и Летрозола.
Другой вариант выполнения изобретения представляет собой способ лечения или предотвращения рака молочной железы у пациента, которому необходимо такое лечение, где указанное лечение включает введение терапевтически эффективного количества, по меньшей мере, одного (например, одного) соединения формулы 1.0, Гозерелина и Эксеместана.
Другой вариант выполнения изобретения представляет собой способ лечения или предотвращения рака молочной железы у пациента, которому необходимо такое лечение, где указанное лечение включает введение терапевтически эффективного количества, по меньшей мере, одного (например, одного) соединения формулы 1.0, Гозерелина и Фадрозола.
Другой вариант выполнения изобретения представляет собой способ лечения или предотвращения рака молочной железы у пациента, которому необходимо такое лечение, где указанное лечение включает введение терапевтически эффективного количества, по меньшей мере, одного (например, одного) соединения формулы 1.0, Гозерелина и Форместана.
Другой вариант выполнения изобретения представляет собой способ лечения или предотвращения рака молочной железы у пациента, которому необходимо такое лечение, где указанное лечение включает введение терапевтически эффективного количества, по меньшей мере, одного (например, одного) соединения формулы 1.0, Леупролида и Анастрозола.
Другой вариант выполнения изобретения представляет собой способ лечения или предотвращения рака молочной железы у пациента, которому необходимо такое лечение, где указанное лечение включает введение терапевтически эффективного количества, по меньшей мере, одного (например, одного) соединения формулы 1.0, Леупролида и Летрозола.
Другой вариант выполнения изобретения представляет собой способ лечения или предотвращения рака молочной железы у пациента, которому необходимо такое лечение, где указанное лечение включает введение терапевтически эффективного количества, по меньшей мере, одного (например, одного) соединения формулы 1.0, Леупролида и Эксеместана.
Другой вариант выполнения изобретения представляет собой способ лечения или предотвращения рака молочной железы у пациента, которому необходимо такое лечение, где указанное лечение включает введение терапевтически эффективного количества, по меньшей мере, одного (например, одного) соединения формулы 1.0, Леупролида и Фадрозола.
Другой вариант выполнения изобретения представляет собой способ лечения или предотвращения рака молочной железы у пациента, которому необходимо такое лечение, где указанное лечение включает введение терапевтически эффективного количества, по меньшей мере, одного (например, одного) соединения формулы 1.0, Леупролида и Форместана.
Другой вариант выполнения изобретения представляет собой лечение или предотвращение рака молочной железы у пациента, которому необходимо такое лечение, причем указанное лечение включает введение терапевтически эффективного количества, по меньшей мере, одного (например, одного) соединения формулы 1.0 и Анастрозола.
Другой вариант выполнения изобретения представляет собой лечение или предотвращение рака молочной железы у пациента, которому необходимо такое лечение, причем указанное лечение включает введение терапевтически эффективного количества, по меньшей мере, одного (например, одного) соединения формулы 1.0 и Летрозола.
Другой вариант выполнения изобретения представляет собой лечение или предотвращение рака молочной железы у пациента, которому необходимо такое лечение, причем указанное лечение включает введение терапевтически эффективного количества, по меньшей мере, одного (например, одного) соединения формулы 1.0 и Эксеместана.
Другой вариант выполнения изобретения представляет собой лечение или предотвращение рака молочной железы у пациента, которому необходимо такое лечение, причем указанное лечение включает введение терапевтически эффективного количества, по меньшей мере, одного (например, одного) соединения формулы 1.0 и Тамоксифена.
Другой вариант выполнения изобретения представляет собой лечение или предотвращение рака молочной железы у пациента, которому необходимо такое лечение, причем указанное лечение включает введение терапевтически эффективного количества, по меньшей мере, одного (например, одного) соединения формулы 1.0 и Фулвестранта.
Другой вариант выполнения изобретения представляет собой лечение или предотвращение рака молочной железы у пациента, которому необходимо такое лечение, причем указанное лечение включает введение терапевтически эффективного количества, по меньшей мере, одного (например, одного) соединения формулы 1.0, Анастрозола и Фулвестранта.
Другой вариант выполнения изобретения представляет собой лечение или предотвращение рака молочной железы у пациента, которому необходимо такое лечение, причем указанное лечение включает введение терапевтически эффективного количества, по меньшей мере, одного соединения формулы 1.0 (например, одно), Летрозола и Фулвестранта
Другой вариант выполнения изобретения представляет собой лечение или предотвращение рака молочной железы у пациента, которому необходимо такое лечение, причем указанное лечение включает введение терапевтически эффективного количества, по меньшей мере, одного (например, одного) соединения формулы 1.0, Эксеместана и Фулвестранта.
Другой вариант выполнения изобретения представляет собой лечение или предотвращение рака молочной железы у пациента, которому необходимо такое лечение, причем указанное лечение включает введение терапевтически эффективного количества, по меньшей мере, одного (например, одного) соединения формулы 1.0, Анастрозола и Тамоксифена.
Другой вариант выполнения изобретения представляет собой лечение или предотвращение рака молочной железы у пациента, которому необходимо такое лечение, причем указанное лечение включает введение терапевтически эффективного количества, по меньшей мере, одного (например, одного) соединения формулы 1.0, Летрозола и Тамоксифена.
Другой вариант выполнения изобретения представляет собой лечение или предотвращение рака молочной железы у пациента, которому необходимо такое лечение, причем указанное лечение включает введение терапевтически эффективного количества, по меньшей мере, одного (например, одного) соединения формулы 1.0, Эксеместана и Тамоксифена.
Другие варианты выполнения настоящего изобретения представляют собой любой из вышеперечисленных вариантов выполнения для лечения рака молочной железы, где химиотерапевтический агент, представляет собой Трастузумаб.
Другие варианты настоящего изобретения представляют собой любые из вышеперечисленных вариантов выполнения для лечения или предотвращения рака молочной железы, где способ направлен на лечение рака молочной железы.
Соединение формулы 1.0, антигормональные средства и химиотерапевтические агенты можно вводить одновременно или последовательно.
Антигормональные средства и химиотерапевтические агенты вводятся в соответствии с протоколами, дозами и лекарственными формами, которые хорошо известны специалистам в данной области техники (например, Physician′s Desk Reference или опубликованная литература). Например, для Тамоксифена, Фулвестранта, Ралоксифена, Анастрозола, Летрозола, Эксеместана, Леупролида и Гозерелина, смотрите Physician′s Desk Reference, 57th Edition, 2003, published by Thomas PDR at Montvale, N.J. 07645-1742, который включен в настоящий документ в полном объеме посредством ссылки.
В общем, в вариантах выполнения изобретения, направленных на способы лечения рака молочной железы: (1) соединение формулы 1.0 может вводиться ежедневно (например, один раз в день, и в одном варианте выполнения изобретения соединение формулы 1.0 может вводиться два раза в день), (2) ингибиторы ароматазы могут вводиться по известным протоколам для применяемых ингибиторов ароматазы (например, один раз в день), (3) антиэстрогены могут вводиться по известным протоколам для применяемого антиэстрагена (например, от одного раза в день до одного раза в месяц), (4) LHRH аналог может вводиться по известному протоколу для применяемого аналога LHRH (например, от одного раза в месяц до одного раза каждые три месяца) и (5) химиотерапевтический агент может вводиться по известным протоколам для применяемого химиотерапевтического агента (например, от одного раза в день до одного раза в неделю).
Радиотерапия, в случае введения при вышеуказанных лечениях рака молочной железы, как правило, вводится по известным протоколам до введения соединения формулы 1.0, антигормональных средств и необязательно химиотерапевтических агентов.
Лечение по способами лечения рака молочной железы является непрерывным (то есть, по непрерывной схеме дозирования). Лечение проводится до достижения полного ответа, или пока опытный клинический врач не определит, что пациент не получает пользу от лечения (например, когда заболевание прогрессирует).
Непрерывный протокол лечения для рака молочной железы можно заменить на прерывистую схему лечения, если согласно оценке опытного клинического врача, пациент получит пользу от прерывистой схемы лечения с одним или более вводимыми лекарственными средствами. Например, соединение формулы 1.0 может вводиться по прерывистой схеме лечения, тогда как оставшиеся лекарственные средства, также применяемые при лечении, вводятся как описано здесь. Примером прерывистого протокола лечения, с введением соединения формулы 1.0, является повторяющийся цикл из трех недель с введением соединения формулы 1.0, с последующей одной неделею без введения соединения формулы 1.0.
После того как достигается полный ответ на лечение рака молочной железы, поддерживающая терапия может проводиться с помощью соединения формулы 1.0, при дозировке, описанной в способе согласно настоящему изобретению. Поддерживающая терапия так же может включать введение антигормональных средств, применяя дозировку, описанную в способах согласно настоящему изобретению. Поддерживающая терапия может быть проведена только с помощью антигормональных средств. Например, после того как достигается полный ответ, ингибитор ароматазы (например, Анастрозол, Летрозол или Эксеместан) может вводиться в течение пяти лет. Альтернативно, например, антиэстроген, например, Тамоксифен, может применяться после достижения полного ответа в течение периода, продолжительность которого составляет до пяти лет, с последующим применением ингибитора ароматазы (например, Анастрозол, Летрозол или Эксеместан) в течение периода времени, продолжительность которого составляет до пяти лет.
В вариантах выполнения изобретения, направленных на лечение рака молочной железы, как раскрывается выше, соединение формулы 1.0 вводится непрерывно, при общей дневной дозе от около 100 мг до около 600 мг. Обычно это общее количество вводится в виде отдельных доз, и в одном варианте выполнения изобретения это количество вводится два раза в день. В одном варианте выполнения изобретения соединение формулы 1.0 вводится два раза в день, в количестве от около 50 мг до около 300 мг на дозу. В другом варианте выполнения изобретения соединение формулы 1.0 вводится два раза в день, в количестве от около 100 мг до около 200 мг на дозу. Примеры включают соединение формулы 1.0, которое вводится два раза в день, в количестве 100 мг на дозу. Примеры также включают соединение формулы 1.0, которое вводится два раза в день, в количестве 200 мг на дозу.
Анастрозол вводится пероральным путем, один раз в день, в количестве от около 0.5 до около 10 мг на дозу, и в одном варианте выполнения изобретения, в количестве около 1.0 мг на дозу.
Летрозол вводится пероральным путем, один раз в день, в количестве от около 1.0 до около 10 мг на дозу, и в одном варианте выполнения изобретения, в количестве около 2.5 мг на дозу.
Эксеместан вводится пероральным путем, один раз в день, в количестве от около 10 до около 50 мг на дозу, и в одном варианте выполнения изобретения, в количестве около 25 мг на дозу.
Фадрозол вводится пероральным путем, два раза в день, в количестве от около 0.5 до около 10 мг на дозу, и в одном варианте выполнения изобретения, в количестве около 2.0 мг на дозу.
Форместан вводится один раз каждые две недели, внутримышечно, в количестве от около 100 до около 500 мг на дозу, и в одном варианте выполнения изобретения, в количестве около 250 мг на дозу.
Тамоксифен вводится пероральным путем, один раз в день, в количестве от около 10 до около 100 мг на дозу, и в одном варианте выполнения изобретения, в количестве около 20 мг на дозу.
Фулвестрант вводится внутримышечно, один раз в месяц, в количестве от около 100 до около 1000 мг на дозу, и в одном варианте выполнения изобретения, в количестве от около 250 мг на дозу.
Ралоксифен вводится пероральным путем, один раз в день, в количестве от около 10 до около 120 мг на дозу, и в одном варианте выполнения изобретения, в количестве около 60 мг на дозу.
Аколбифен вводится пероральным путем, один раз в день, в количестве от около 5 до около 20 мг на дозу, и в одном варианте выполнения изобретения, в количестве около 20 мг на дозу.
Гозерелин вводится подкожно, один раз в месяц или один раз каждые три месяца, в количестве от около 2 до около 20 мг на дозу, и в одном варианте выполнения изобретения, в количестве около 3.6 мг на дозу при введении один раз в месяц, и в другом варианте выполнения изобретения, в количестве около 10.8 мг на дозу, при введении один раз каждые три месяца.
Леупролид вводится подкожно, один раз в месяц или один раз каждые три месяца, в количестве от около 2 до около 20 мг на дозу, и в одном варианте выполнения изобретения, в количестве от около 3.75 мг на дозу, при введении один раз в месяц, и в другом варианте выполнения изобретения, в количестве около 11.25 мг на дозу, при введении один раз каждые три месяца.
Трастузумаб вводится внутривенным путем, один раз в неделю, в количестве от около 2 до около 20 мг/кг на дозу, и в одном варианте выполнения изобретения, в количестве около 2 мг/кг на дозу. Трастузумаб, как правило, сначала вводится в ударной дозе, которая, как правило, составляет две еженедельные дозы. Таким образом, например, вводится ударная доза, в количестве 4 мг/кг, и затем вводится 2 мг/кг на дозу на неделю.
Гефитиниб вводится пероральным путем, один раз в день, в количестве от около 100 до около 1000 мг на дозу, и в одном варианте выполнения изобретения, в количестве около 250 мг на дозу.
Эрлотиниб вводится пероральным путем, один раз в день, в количестве от около 100 до около 500 мг на дозу, и в одном варианте выполнения изобретения, в количестве около 150 мг на дозу.
Бевацизумаб вводится внутривенным путем, один раз каждые две недели, в количестве от около 2.5 до около 15 мг на килограмм массы тела на дозу, и в одном варианте выполнения изобретения, в количестве около 10 мг на килограмм на дозу.
Цетуксимаб вводится внутривенным путем, один раз в неделю, в количестве от около 200 до около 500 мг на метр квадратный на дозу, и в одном варианте выполнения изобретения, в количестве около 250 мг на метр квадратный на дозу.
Бортезомиб вводится внутривенным путем, дважды в неделю, в течение двух недель и затем 10 дней перерыв (цикл лечения составляет 21 день), максимум 8 циклов лечения, в количестве от около 1.0 до около 2.5 мг на метр квадратный на дозу, и в одном варианте выполнения изобретения, в количестве около 1.3 мг на метр квадратный на дозу.
Таким образом, в одном варианте выполнения настоящего изобретения рак молочной железы лечится (или предотвращается) у пациента, которому необходимо такое лечение, когда указанное лечение включает введение указанному пациенту: (1) соединения формулы 1.0 пероральным путем, в количестве от около 50 мг до около 300 мг на дозу, причем каждая доза вводится два раза в день, и (2) Анастрозола, пероральным путем, в количестве от около 0.5 до около 10 мг на дозу, причем каждая доза вводится один раз в день.
В другом варианте выполнения настоящего изобретения рак молочной железы лечится (или предотвращается) у пациента, которому необходимо такое лечение, когда указанное лечение включает введение указанному пациенту: (1) соединения формулы 1.0, пероральным путем, в количестве от около 100 до около 200 мг на дозу, причем каждая доза вводится два раза в день, и (2) Анастрозола, в количестве около 1.0 мг на дозу, причем каждая доза вводится один раз в день.
В другом варианте выполнения настоящего изобретения рак молочной железы лечится (или предотвращается) у пациента, которому необходимо такое лечение, когда указанное лечение включает введение указанному пациенту: (1) соединения формулы 1.0, пероральным путем, в количестве от около 50 мг до около 300 мг на дозу, причем каждая доза вводится два раза в день, и (2) Летрозола, пероральным путем, в количестве от около 1.0 до около 10 мг на дозу, причем каждая доза вводится один раз в день.
В другом варианте выполнения настоящего изобретения рак молочной железы лечится (или предотвращается) у пациента, которому необходимо такое лечение, когда указанное лечение включает введение указанному пациенту: (1) соединения формулы 1.0, пероральным путем, в количестве около 100-200 мг на дозу, причем каждая доза вводится два раза в день, и (2) Летрозола, пероральным путем, в количестве около 2.5 мг на дозу, причем каждая доза вводится один раз в день.
В другом варианте выполнения настоящего изобретения рак молочной железы лечится (или предотвращается) у пациента, которому необходимо такое лечение, когда указанное лечение включает введение указанному пациенту: (1) соединения формулы 1.0, пероральным путем, в количестве от около 50 мг до около 300 мг на дозу, причем каждая доза вводится два раза в день, и (2) Эксеместана, пероральным путем, в количестве от около 10 до около 50 мг на дозу, причем каждая доза вводится один раз в день.
В другом варианте выполнения настоящего изобретения рак молочной железы лечится (или предотвращается) у пациента, которому необходимо такое лечение, когда указанное лечение включает введение указанному пациенту: (1) соединения формулы 1.0, пероральным путем, в количестве около 100-200 мг на дозу, причем каждая доза вводится два раза в день, и (2) Эксеместана, в количестве около 25 мг на дозу, причем каждая доза вводится один раз в день.
В другом варианте выполнения настоящего [Изобретения рак молочной железы лечится (или предотвращается) у пациента, которому необходимо такое лечение, когда указанное лечение включает введение указанному пациенту: (1) соединения формулы 1.0, пероральным путем, в количестве от около 50 мг до около 300 мг на дозу, причем каждая доза вводится два раза в день, и (2) Фулвестраната, внутримышечно, в количестве от около 100 до около 1000 мг на дозу, причем каждая доза вводится один раз в месяц.
В другом варианте выполнения настоящего изобретения рак молочной железы лечится (или предотвращается) у пациента, которому необходимо такое лечение, когда указанное лечение включает введение указанному пациенту: (1) соединения формулы 1.0, пероральным путем, в количестве около 100-200 мг на дозу, причем каждая доза вводится два раза в день, и (2) Фулвестранта, внутримышечно, в количестве около 250 мг на дозу, причем каждая доза вводится один раз в месяц.
В другом варианте выполнения настоящего изобретения рак молочной железы лечится (или предотвращается) у пациента, которому необходимо такое лечение, когда указанное лечение включает введение указанному пациенту: (1) соединения формулы 1.0, пероральным путем, в количестве от около 50 мг до около 300 мг на дозу, причем каждая доза вводится два раза в день, и (2) Тамоксифена, пероральным путем, в количестве от около 10 до около 100 мг на дозу, причем каждая доза вводится один раз в день.
В другом варианте выполнения настоящего изобретения рак молочной железы лечится (или предотвращается) у пациента, которому необходимо такое лечение, когда указанное лечение включает введение указанному пациенту: (1) соединения формулы 1.0, пероральным путем, в количестве около 100-200 мг на дозу, причем каждая доза вводится два раза в день, и (2) Тамоксифена, пероральным путем, в количестве около 20 мг на дозу, причем каждая доза вводится один раз в день.
В других вариантах выполнения настоящего изобретения рак молочной железы лечится у пациента, которому необходимо такое лечение, когда указанное лечение включает введение соединения формулы 1.0, одного из ингибиторов ароматазы (например, Анастрозол, Летрозол или Эксеместан, и в одном вариант выполнения изобретения Анастрозол), и одного из антиэстрогенов (например, Фулвестрант или Тамоксифен), где соединение формулы 1.0, ингибитор ароматазы и антиэстроген вводятся в дозах, раскрытых выше.
Таким образом, например, в другом варианте выполнения настоящего изобретения рак молочной железы лечится (или предотвращается) у пациента, которому необходимо такое лечение, когда указанное лечение включает введение указанному пациенту: (1) соединения формулы 1.0, пероральным путем, в количестве от около 50 мг до около 300 мг на дозу, причем каждая доза вводится два раза в день, (2) Анастрозола, пероральным путем, в количестве от около 0.5 до около 10 мг на дозу, причем каждая доза вводится один раз в день, и (3) Фулвестранта, внутримышечно, в количестве от около 100 до около 1000 мг на дозу, причем каждая доза вводится один раз в месяц.
В другом варианте выполнения настоящего изобретения рак молочной железы лечится (или предотвращается) у пациента, которому необходимо такое лечение, когда указанное лечение включает введение указанному пациенту: (1) соединения формулы 1.0, пероральным путем, в количестве около 100-200 мг на дозу, причем каждая доза вводится два раза в день, (2) Анастрозола, пероральным путем, в количестве около 1.0 мг на дозу, причем каждая доза вводится один раз в день, и (3) Фулвестранта, внутримышечно, в количестве около 250 мг на дозу, причем каждая доза вводится один раз в месяц.
В другом варианте выполнения настоящего изобретения рак молочной железы лечится (или предотвращается) у пациента, которому необходимо такое лечение, когда указанное лечение включает введение указанному пациенту: (1) соединения формулы 1.0 пероральным путем, в количестве от около 50 мг до около 300 мг на дозу, причем каждая доза вводится два раза в день, (2) Летрозола, пероральным путем, в количестве от около 1.0 до около 10 мг на дозу, причем каждая доза вводится один раз в день, и (3) Фулвестранта, в количестве от около 100 до около 1000 мг на дозу, причем каждая доза вводится один раз в месяц.
В другом варианте выполнения настоящего изобретения рак молочной железы лечится (или предотвращается) у пациента, которому необходимо такое лечение, когда указанное лечение включает введение указанному пациенту: (1) соединения формулы 1.0, пероральным путем, в количестве около 100-200 мг на дозу, причем каждая доза вводится два раза в день, (2) Летрозола, пероральным путем, в количестве около 2.5 мг на дозу, причем каждая доза вводится один раз в день, и (3) Фулвестранта, внутримышечно, в количестве около 250 мг на дозу, причем каждая доза вводится один раз в месяц.
В другом варианте выполнения настоящего изобретения рак молочной железы лечится (или предотвращается) у пациента, которому необходимо такое лечение, когда указанное лечение включает введение указанному пациенту: (1) соединения формулы 1.0, пероральным путем, в количестве от около 50 мг до около 300 мг на дозу, причем каждая доза вводится два раза в день, (2) Эксеместана, пероральным путем, в количестве от около 10 до около 50 мг на дозу, причем каждая доза вводится один раз в день, и (3) Фулвестранта, внутримышечно, в количестве от около 100 до около 1000 мг на дозу, причем каждая доза вводится один раз в месяц.
В другом варианте выполнения настоящего изобретения рак молочной железы лечится (или предотвращается) у пациента, которому необходимо такое лечение, когда указанное лечение включает введение указанному пациенту: (1) соединения формулы 1.0, пероральным путем, в количестве около 100-200 мг на дозу, причем каждая доза вводится два раза в день, (2) Эксеместана, пероральным путем, в количестве около 25 мг на дозу, причем каждая доза вводится один раз в день, и (3) Фулвестранта, внутримышечно, в количестве около 250 мг на дозу, причем каждая доза вводится один раз в месяц.
В другом варианте выполнения настоящего изобретения рак молочной железы лечится (или предотвращается) у пациента, которому необходимо такое лечение, когда указанное лечение включает введение указанному пациенту: (1) соединения формулы 1.0 в количестве от около 50 мг до около 300 мг на дозу, пероральным путем, причем каждая доза вводится два раза в день, (2) Анастрозола в количестве от около 0.5 до около 10 мг на дозу, пероральным путем, причем каждая доза вводится один раз в день, и (3) Тамоксифена в количестве от около 10 до около 100 мг на дозу, пероральным путем, причем каждая доза вводится один раз в день.
В другом варианте выполнения настоящего изобретения рак молочной железы лечится (или предотвращается) у пациента, которому необходимо такое лечение, когда указанное лечение включает введение указанному пациенту: (1) соединения формулы 1.0 в количестве около 100-200 мг на дозу, пероральным путем, причем каждая доза вводится два раза в день, (2) Анастрозола, в количестве около 1.0 мг на дозу, пероральным путем, причем каждая доза вводится один раз в день, и (3) Тамоксифена в количестве около 20 мг на дозу, пероральным путем, причем каждая доза вводится один раз в день.
В другом варианте выполнения настоящего изобретения рак молочной железы лечится (или предотвращается) у пациента, которому необходимо такое лечение, когда указанное лечение включает введение указанному пациенту: (1) соединения формулы 1.0 в количестве от около 50 мг до около 300 мг на дозу, пероральным путем, причем каждая доза вводится два раза в день, (2) Летрозола в количестве от около 1.0 до около 10 мг на дозу, пероральным путем, причем каждая доза вводится один раз в день, и (3) Тамоксифена в количестве от около 10 до около 100 мг на дозу, пероральным путем, причем каждая доза вводится один раз в день.
В другом варианте выполнения настоящего изобретения рак молочной железы лечится (или предотвращается) у пациента, которому необходимо такое лечение, когда указанное лечение включает введение указанному пациенту: (1) соединения формулы 1.0, в количестве около 100-200 мг на дозу, пероральным путем, причем каждая доза вводится два раза в день, (2) Летрозола в количестве около 2.5 мг на дозу, пероральным путем, причем каждая доза вводится один раз в день, и (3) Тамоксифена в количестве около 20 мг на дозу, пероральным путем, причем каждая доза вводится один раз в день.
В другом варианте выполнения настоящего изобретения рак молочной железы лечится (или предотвращается) у пациента, которому необходимо такое лечение, когда указанное лечение включает введение указанному пациенту: (1) соединения формулы 1.0, в количестве от около 50 мг до около 300 мг на дозу, пероральным путем, причем каждая доза вводится два раза в день, (2) Эксеместана, в количестве от около 10 до около 50 мг на дозу, пероральным путем, причем каждая доза вводится один раз в день, и (3) Тамоксифена, в количестве от около 10 до около 100 мг на дозу, пероральным путем, причем каждая доза вводится один раз в день.
В другом варианте выполнения настоящего изобретения рак молочной железы лечится (или предотвращается) у пациента, которому необходимо такое лечение, когда указанное лечение включает введение указанному пациенту: (1) соединения формулы 1.0, в количестве около 100-200 мг на дозу, пероральным путем, причем каждая доза вводится два раза в день, (2) Эксеместана, в количестве около 25 мг на дозу, пероральным путем, причем каждая доза вводится один раз в день, и (3) Тамоксифена, в количестве от около 20 мг на дозу, пероральным путем, причем каждая доза вводится один раз в день.
Специалистам в данной области техники очевидно, что когда применяются другие комбинации антигормональных агентов, отдельный антигормональный агент применяется в количествах, указанных выше для этого отдельного антигормонального агента.
Другие варианты лечения рака молочной железы представляют собой способы лечения рака молочной железы, раскрытые выше, где соединения формулы 1.0 вводится два раза в день, в количестве около 100 мг на дозу.
Другие варианты лечения рака молочной железы представляют собой способы лечения рака молочной железы, раскрытые выше, где соединения формулы 1.0 вводится два раза в день, в количестве около 200 мг на дозу.
Другие варианты лечения рака молочной железы представляют собой способы лечения рака молочной железы, раскрытые выше, где химиотерапевтический агент вводится в дополнение к соединению формулы 1.0 и антигормональному агенту (или антигормональным агентам). В этих вариантах выполнения изобретения интервалы доз соединения формулы 1.0 и антигормональных агентов являются такими, как раскрывается выше для комбинаторных терапий, или такими, как раскрывается выше для отдельного соединения формулы I и антигормональных агентов, и дозы химиотерапевтических агентов являются такими, как раскрывается выше для отдельного химиотерапевтического агента. Дозы химиотерапевтических агентов хорошо известны из уровня техники.
Другие варианты выполнения настоящего изобретения представляют собой фармацевтические композиции, содержащие соединение формулы 1.0 и, по меньшей мере, один антигормональный агент и фармацевтически приемлемый носитель.
Другие варианты выполнения настоящего изобретения представляют собой фармацевтические композиции, содержащие соединение формулы 1.0, по меньшей мере, один антигормональный агент, по меньшей мере, один химиотерапевтический агент и фармацевтически приемлемый носитель.
Другие варианты настоящего изобретения представляют собой фармацевтические композиции, содержащие соединение формулы 1.0, по меньшей мере, один химиотерапевтический агент и фармацевтически приемлемый носитель.
Специалистам в данной области техники будет очевидно, что соединения (лекарственные средства), применяемые в способах по изобретению, доступны квалифицированному клиническому врачу в фармацевтических композициях (лекарственных формах) от производителей и применяются в этих композициях. Так, приведение соединения или класса соединений в вышеописанных способах может быть заменено приведением фармацевтической композиции, содержащей конкретное соединение или класс соединений. Например, вариант выполнения изобретения, направленный на способ лечения рака, содержащий введение пациенту, которому необходимо такое лечение, терапевтически эффективного количества соединения формулы 1.0, таксана и координационного соединения платины, охватывает способ лечения рака, включающий введение пациенту, которому необходимо такое лечение, терапевтически эффективного количества фармацевтической композиции, содержащей соединение формулы 1.0, фармацевтической композиции, содержащей таксан, и фармацевтической композиции, содержащей координационное соединение платины.
Специалистам в данной области техники очевидно, что точные дозы и протоколы введения, применяемые в способах согласно настоящему изобретению, могут варьироваться в зависимости от оценки квалифицированного клинического врача. Точная применяемая доза может варьироваться в зависимости от нужд пациента и серьезности состояния, подлежащего лечению. Определение соответствующей дозы для конкретного случая находится в компетенции специалиста в данной области техники. Определение того, каким образом следует варьировать дозы и протоколы лечения, может быть сделано после того как квалифицированный клинический врач рассмотрит такие факторы как возраст, состояние и вес пациента, а также серьезность рака подлежащего лечению, и ответная реакция пациента на лечение.
Количество и частота введения соединения формулы 1.0 и химиотерапевтических агентов будут регулироваться согласно оценке квалифицированного клинического врача (лечащего врача), принимающего во внимание такие факторы, как возраст, состояние и вес пациента, так же как и серьезность рака, подлежащего лечению.
Химиотерапевтический агент может вводиться по терапевтическими протоколам, хорошо известным специалистам в данной области техники. Специалистам в данной области техники очевидно, что введение химиотерапевтического агента может изменяться в зависимости от вида рака, подлежащего лечению, и эффектов, оказываемых химиотерапевтическим агентом на заболевание. Также, на основе знаний квалифицированного клинического врача, терапевтические протоколы (например, размеры дозировки и число введений) могут варьироваться в зависимости от наблюдаемых эффектов, оказываемых на пациента при введении терапевтических агентов, и в зависимости от наблюдаемого ответа со стороны рака на введение терапевтических агентов.
Первоначальное введение может осуществляться по установленным в области техники протоколам, а затем дозы, способы введения и количества введений могут изменяться квалифицированным клиническим врачом в зависимости от наблюдаемых эффектов.
Конкретный выбор химиотерапевтического агента будет зависеть от диагноза лечащего врача и его оценки состояния пациента, и соответствующего протокола лечения.
Определение порядка введения и числа повторений введения химиотерапевтического агента при реализации протокола лечения находится в компетенции лечащего врача, и происходит после оценки рака, подлежащего лечению, и состояния пациента.
Таким образом, лечащий врач, основываясь на своем опыте и знаниях, может изменять каждый протокол введения химиотерапевтического агента в зависимости от нужд отдельного пациента, по мере того, как протекает лечение. Все такие изменения находятся в объеме настоящего изобретения.
Конкретный выбор антигормональных агентов, необязательных химиотерапевтических агентов и необязательного облучения зависит от диагноза лечащего врача и его оценки состояния пациента, и соответствующего протокола лечения.
Определение порядка введения и числа повторений введения антигормональных агентов, необязательных химиотерапевтических агентов и необязательного облучения по протоколу лечения, находится в компетенции лечащего врача при соответствующей оценки рака молочной железы, подлежащего лечению, и состояния пациента.
Таким образом, лечащий врач, основываясь на своем опыте и знаниях, может изменять каждый протокол введения антигормональных агентов, необязательных химиотерапевтических агентов и необязательного облучения, в зависимости от нужд отдельного пациента, по мере того, как протекает лечение. Все такие изменения находятся в объеме настоящего изобретения.
Лечащий врач, при оценке эффективности лечения при вводимой дозе, будет рассматривать общее состояние здоровья пациента, так же как и более конкретные показатели, такие как облегчение симптомов, вызванных раком (например, боль, кашель (при раке легких) и одышка (при раке легких)), ингибирование роста опухоли, реальное сжатие опухоли или ингибирование метастаза. Размер опухоли можно измерить стандартными способами, такими как радиологические изучения, например, CAT или MRI сканирование, и последовательные измерения могут применяться для оценки был ли рост опухоли замедлен или даже предотвращен или нет. Облегчение симптомов, связанных с заболеванием, таких как боль, и улучшение общего состояния также могут применяться для оценки эффективности лечения.
Соединения согласно настоящему изобретению можно получить способами, раскрытыми в US 2007/0191604, опубликованной 16 августа, 2007, U.S. №11/810282, поданной 5 июня, 2007, также как и способами, описанными ниже. US 2007/0191604 и U.S. №11/810282 включены в настоящий документ в полном объеме посредством ссылки.
Применяются следующие условия ЖХМС: (1) С-18 обращенофазная колонка, 5 мкм, 4.6×50 мм, (2) MS:PE Sciex API-150EX и (3) ВЭЖХ: Shimadzu LC-10 ADvp, 1 мл/мин, линейный градиент 10% ацетонитрила в воде - 95% ацетонитрила в воде, оба содержат 0.05% TFA.
Пример 1
Синтез 3-Метокси-1-(2-{4-[4-(1-метил-1Н-[1,2,4]триазол-3-ил)-фенил]-3,6-дигидро-2Н-пиридин-1-ил}-2-оксо-этил)-пирролидин-3-карбоновой кислоты [3-(6-изопропокси-пиридин-3-ил)-1Н-индазол-5-ил]-амида
Синтез 2-хлор-1-{4-[4-(1-метил-1Н-[1,2,4]триазол-3-ил)-фенил]-3,6-дигидро-2Н-пиридин-1-ил}-этанона
Стадия 1: Получение 4-бром-бензимидовой кислоты сложного этилового эфира
4-Бром-бензонитрил (5 г) суспендировали в абсолютном EtOH (100 мл) и охладили до 0-5°С. Барботировали газ HCl, сначала интенсивно, а затем медленно в течение 5 часов. Полученный раствор перемешивали всю ночь. Большую часть растворителя удалили, и осадок отфильтровали, дважды промыли EtOH и высушили с получением соединения 2ВН (4.1 г) в виде твердого вещества белого цвета.
Стадия 2: Получение Соединения 3ВН
4-Бром-бензимидовой кислоты сложный этиловый эфир (2.12 г, 8 ммоль) растворили в пиридине (20 мл). При перемешивании добавили метилгидразин (640 мкл, 12 ммоль), полученную смесь перемешивали всю ночь. Реакционную смесь концентрировали при пониженном давлении, добавили диэтиловый эфир, отфильтровали, трижды промыли диэтиловым эфиром и высушили с получением соединения 3ВН (2.2 г).
Стадия 3: Получение 3-(4-бром-фенил)-1-метил-1Н-[1,2,4]триазола
Смесь соединения 3ВН (2.2 г) в муравьиной кислоте (30 мл) кипятили с обратным холодильником и концентрировали. Осадок обработали насыщенным NaHCO, и трижды экстрагировали с помощью EtOAc. Объединенную органическую фазу высушили над MgSO4. После концентрирования получили соединение 4 ВН в виде бесцветных кристаллов (1.39 г). (Необходимо отметить, что было обнаружено, что реакция должна проводиться в течение не более двух часов. При крупномасштабном синтезе необходимо использовать 10% NaOH вместо NaHCO3).
Стадия 4: Получение 4-[4-(1-метил-1H-[1,2,4]триазол-3-ил)-фенил]-3,6-дигидро-2Н-пиридин-1-карбоновой кислоты трет-бутилового сложного эфира
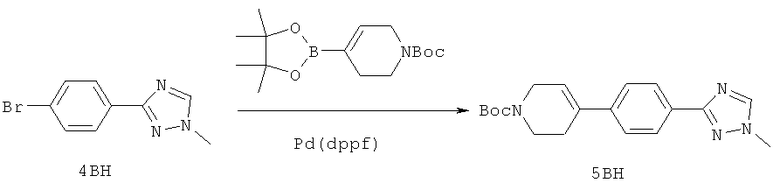
В большую колбу высокого давления загрузили соединение 4 ВН (13.3 г, 55.9 ммоль), 4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-3,6-дигидро-2H-пиридин-1-карбоновой кислоты трет-бутиловый сложный эфир (19 г, 61.5 ммоль), [1,1′-бис(дифенилфосфино)-ферроцен]дихлорпалладия(II) комплекс с дихлорметаном (1:1) (2.3 г, 2.8 ммоль), K2CO3 (23.2 г, 168 ммоль) и DME/вода (5:1, 120 мл). Смесь быстро дегазировали аргоном (Ar) в течение около 0.5 минут, накрыли и перемешивали при 80°С всю ночь. После охлаждения реакционную смесь разбавили EtOAc и соляным раствором. Органический слой выделили и высушили (MgSO4). После концентрирования осадок очистили на силикагеле. Проведение элюции с помощью МеОН/EtOAc (0-10%) дало желаемый продукт 5ВН (13.9 г, 73%).
Стадия 5: Получение 4-[4-(1-метил-1Н-[1,2,4]триазол-3-ил)-фенил]-1,2,3,6-тетрагидро-пиридин гидрохлорида
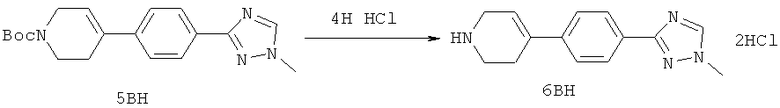
Вое группу можно удалить путем обработки соединения 5ВН раствором 4Н HCl в диоксане при комнатной температуре в течение двух часов. Удаление растворителя в вакууме позволило получить соединение 6ВН.
Стадия 6: Получение 2-хлоро-1-{4-[4-(1-метил-1H-[1,2,4]триазол-3-ил)-фенил]-3,6-дигидро-2Н-пиридин-1-ил}-этанона
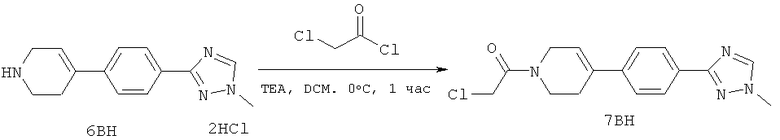
К охлажденному раствору (0°С) 4-[4-(1-метил-1Н-[1,2,4]триазол-3-ил)-фенил]-1,2,3,6-тетрагидро-пиридина 6 ВН (13.7 г, 44 ммоль) в дихлорметане (450 мл) по каплям добавили TEA (37 мл, 264 ммоль). После перемешивания при 0°С в течение 10 минут к реакционной смеси добавили хлор-ацетил хлорид (10.5 мл, 132 ммоль). Полученную смесь перемешивали при 0°С в течение одного часа и погасили водой (165 мл). Реакционную смесь разбавили дихлорметаном (600 мл). Органический слой отделили и промыли соляным раствором, высушили над MgSO4. Реакционную смесь концентрировали до около 50 мл, добавили диэтиловый эфир, и твердое вещество отфильтровали с получением желаемого продукта 7ВН (8.74 г).
Синтез 3-Метокси-пирролидин-3-карбоновой кислоты сложного метилового эфира
Стадия 1: Получение метил α,α-диметоксипропионата
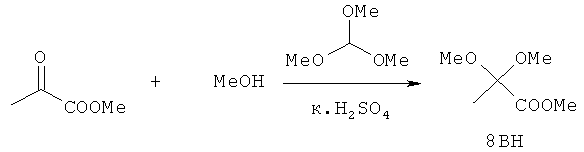
Следовали методике Ernest Wenkert, et al. (JACS, 1983, 105, 2021-2029). Раствор метилпирувата (44 г), триметилортоформиата (62 мл), концентрированной H2SO4 (0.2 мл) в МеОН (120 мл) кипятили с обратным холодильником в течение 4 часов. В течение следующего часа дистиллировали растворитель (около 80 мл). Реакционную смесь охладили до 10°С, перелили в раствор КОН (1.2 г КОН в 600 мл воды) и экстрагировали диэтиловым эфиром (3×). Объединенные экстракты диэтилового эфира промыли соляным раствором и высушили (MgSO4). После концентрирования осадок дистиллировали в вакууме с получением ацеталя (8ВН) (40 г, 62%, 40-43С/1 торр).
Стадия 2: Получение 2-метоксиакрилата
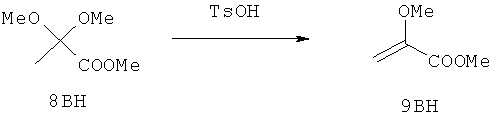
Следовали методике Ernest Wenkert, et al. (JACS, 1983, 105, 2021-2029). Одногорловую колбу загрузили α,α-диметоксипропионатом (8 ВН) (150 г) и толуолсульфоновой кислоты моногидратом (3г), и присоединили дистиллятор для молекулярной перегонки. Смесь нагрели до 140°С (температура водяной бани), и метанол улетучился первым. Продукт (76 г) (9ВН) дистиллировали после того, как температура масляной бани превысила 190°С.
Стадия 3: Получение 1-бензил-3-метокси-пирролидин-3-карбоновой кислоты сложного метилового эфира
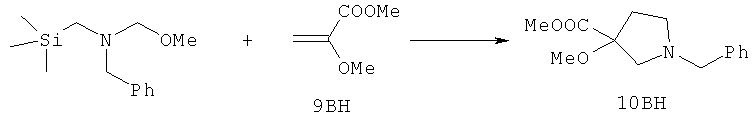
К перемешанному раствору метил 2-метоксиакрилата (20.8 г, 179 ммоль) и N-(метоксиметил)-N-(триметилсилилметил)бензиламина (55 мл, 215 ммоль) в дихлорметане (160 мл) при 0°С добавили раствор трифторуксусной кислоты (2 мл) в дихлорметане (10 мл). Полученный раствор нагрели до комнатной температуры и перемешивали всю ночь. После концентрирования неочищенный продукт очистили с помощью колоночной хроматографии на силикагеле, элюировали раствором этилацетат/гексаны/Et3N (1000:3000:4-1000:1000:3) с получением соединения, название которого указано в заглавии, (10ВН), (17.7 мг, 40%). (Необходимо отметить, что добавление Et3N важно для обеспечения глубокого разделения.)
Стадия 4: Получение соли 3-метокси-пирролидин-3-карбоновой кислоты сложного метилового эфира и винной кислоты
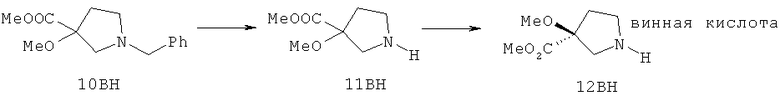
1-Бензил-3-метокси-пирролидин-3-карбоновой кислоты сложный метиловый эфир (10ВН) (2.49 г) гидрогенизировали в этаноле, применяя 10% Pd/C при 55 пси водорода в течение 24 часов Фильтрация на Pd/C с последующим выпариванием этанола дала 1.6 г неочищенного дебензилированного продукта (11ВН). Неочищенный продукт растворили в 95 мл метанола, и добавили 1.35 г L-винной кислоты. Через 24 часа кристаллы отфильтровали и перекристаллизовали из метанола с получением 13.4 г продукта (12ВН), название которого указано в заглавии.
Стадия 5: Получение 3-Метокси-пирролидин-1,3-дикарбоновой кислоты 1-трет-бутилового сложного эфира
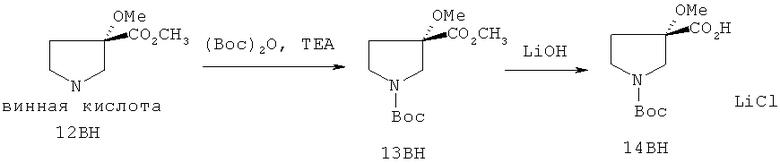
К охлажденному раствору (0°С) соединения 12ВН (28 г, 90.52 ммоль) в сухом CH2Cl2 (250 мл) добавили триэтиламин (31.5 мл, 226.32 ммоль, 2.5 эквив.), а затем (Вос)2O (25.7 г, 117.68 ммоль, 1.3 эквив.). Полученную смесь перемешивали при температуре от 0°С до комнатной температуры всю ночь, а затем разбавили CH2Cl2, полученную смесь промыли насыщенным водным раствором NaHCO3 и соляным раствором, высушили (MgSO4) и концентрировали. Хроматография на силикагеле (гексаны/этилацетат, 4:1) позволила получить соединение 13ВН (23.5 мг, 90.52 ммоль, 100%) в виде бесцветного масла.
К перемешанному раствору соединения 13ВН (23.5 мг, 90.52 ммоль) в THF/MeOH (175 мл/175 мл) добавили 135 мл LiOH (1M в H2O, 135 ммоль, 1.5 эквив.). Реакционную смесь перемешивали при комнатной температуре всю ночь, затем добавили 135 мл 1Н HCl. Полученную смесь перемешивали в течение еще 15 минут и концентрировали, азеотропировали с диоксаном (150 мл × 3) с получением соединения 14ВН (42.32 г) в виде твердого вещества белого цвета, которое применяли на следующей стадии без последующего очищения.
Альтернативно соединение 11ВН может быть получено следующим образом:
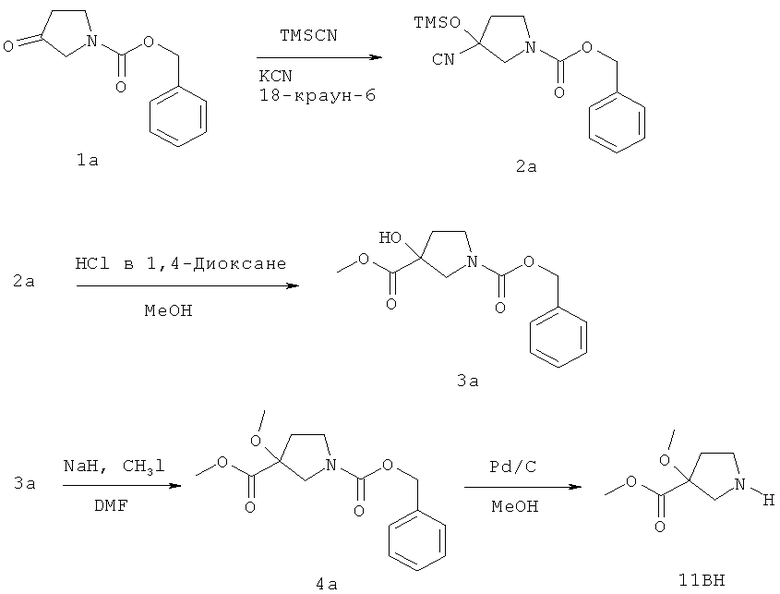
К ледяному раствору 3-оксо-пирролидин-1-карбоновой кислоты сложного бензилового эфира 1а (250 г, 1.14 моль) в 3.5 л безводного дихлорметана добавили KCN (7.5 г, 0.12 моль), затем добавили 18-краун-6 (30 г, 0.11 моль), несмотря на неполное растворение, медленно добавили TMSCN (183 мл, 1.37 моль) в течение 20 минут. Реакционную смесь перемешивали при температуре окружающей среды всю ночь. Полунасыщенный раствор NaHCO3 (2 л) добавили при 15°С, перемешивали в течение 10 минут, и затем органический слой отделили, высушили над сульфатом магния, отфильтровали и выпарили с получением 3-Циано-3-триметилсиланилокси-пирролидин-1-карбоновой кислоты сложного бензилового эфира 2а, 422 г (>100%).
К 3-Циано-3-триметилсиланилокси-пирролидин-1-карбоновой кислоты сложному бензиловому эфиру 2а (422 г) в 4 л безводного МеОН добавили 2.2 л раствора 4Н HCl в диоксане. Реакционную смесь кипятили с обратным холодильником в течение 13 часов и перемешивали при температуре окружающей среды всю ночь. Удалили растворитель, затем суспендировали в 5 л CH2Cl2, промыли 4+3 литрами вода, установили рН 6-7 с помощью водного NaHCO3, высушили над сульфатом магния, отфильтровали и выпарили с получением 3-гидрокси-пирролидин-1,3-дикарбоновой кислоты 1-бензилового сложного эфира 3-метилового сложного эфира 3а, 278 г (2 Стадии, 87%).
К суспензии NaH (52 г, 1.3 моль) в 2.2 л безводного DMF при 8°С добавили раствор 3-гидрокси-пирролидин-1,3-дикарбоновой кислоты 1-бензилового сложного эфира 3-метилового сложного эфира 3а (278 г, 1.0 моль) в 700 DMF, температуру реакции поддерживали ниже 11°С. После завершения добавления (около 20 минут), удалили ледяную баню, перемешали при 16-18°С в течение одного часа, и затем при температуре окружающей среды в течение одного часа. Охладили до 15°С, медленно добавили Mel (81 мл, 1.3 моль). Реакционную смесь перемешивали при температуре окружающей среды всю ночь. Затем реакционную смесь перелили в холодную воду (4 л) и затем экстрагировали Et2O (6 л) и EtOAc (2 л), промыли органический слой водой (5 л), соляным раствором (700 мл), высушили над сульфатом магния, отфильтровали и выпарили с получением неочищенного 3-метокси-пирролидин-1,3-дикарбоновой кислоты 1-бензилового сложного эфира 3-метилового сложного эфира 4а 289 г (99% выход неочищенного продукта, который содержит минеральное масло) (перемешивание/разделение с промывкой пентаном дало соединение 4а, 268.2 г (92%).
К 3-метокси-пирролидин-1,3-дикарбоновой кислоты 1-бензилового сложного эфира 3-метиловому сложному эфиру 4а в МеОН (2200 мл) добавили 14 г 10% Pd/C (около 50% в воде). Реакционную смесь гидрогенизировали, применяя Н2 при давлении около 55 пси (открытый клапан) всю ночь. Реакционную смесь отфильтровали, высушили с получением 3-метокси-пирролидин-3-карбоновой кислоты сложного метилового эфира 11ВН 126 г. (общий выход для четырех стадий составил 70%, без колоночной очистки).
Синтез 3-метокси-пирролидин-3-карбоновой кислоты [3-(6-изопропокси-пиридин-3-ил)-1Н-индазол-5-ил]-амида
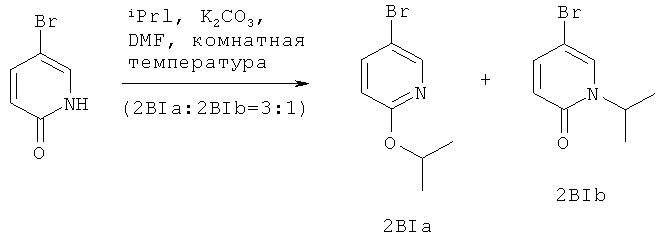
Стадия 1:
5-Бром-1Н-пиридон 1BI (100 г, 0.58 моль), карбонат натрия (238 г, 1.73 моль) и 2-иодпропан (86 мл, 0.86 моль) перемешивали в DMF (1 л) при комнатной температуре в течение одного дня. Смесь разбавили этилацетатом и водой, слои разделили. Отделенный органический слой промыли водой (Х2), высушили (MgSO4) и отфильтровали. Растворители удалили в вакууме, и очистка на колонке [5% этилацетата в гексанах] дала сначала менее полярный 5-изопропоксипиридин 2BIa (73 г, 59%) в виде бесцветной жидкости. Дальнейшая элюция [50% этилацетата в гексанах] дала более полярный 5-бром-1-изопропилпиридон 2BIb в виде твердого вещества белого цвета (22 г, 18%).
Стадия 2:
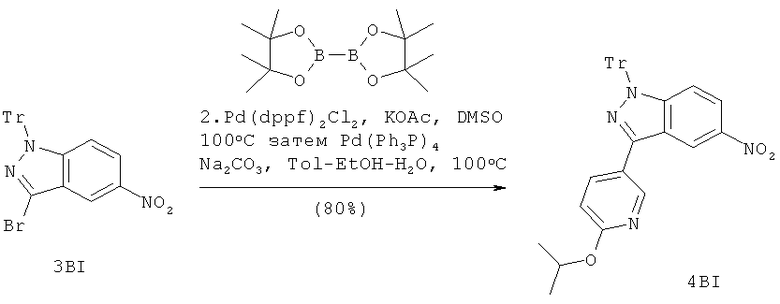
5-Изопропоксипиридин 2BIa (10 г, 0.046 моль), бис(пинаколато)дибор (14.1 г, 0.056 моль), ацетат калия (13.6 г, 0.14 моль) и PdCl2(dppf)2.CH2Cl2 (3.78 г, 0.0046 моль) отвесили в 2-горловой колбе, объемом 1 л, оборудованной водным холодильником. Добавили DMSO (100 мл), и смесь продували азотом в течение 15 минут. Смесь нагревали при 100°С в атмосфере азота, в течение двух часов. После охлаждения до комнатной температуры добавили воду (100 мл), толуол (100 мл), этанол (100 мл), карбонат калия (32 г, 0.23 моль) и броминдазол 3BI (22.4 г, 0.046 моль). Смесь продували азотом в течение 10 минут при комнатной температуре, и добавили Pd(Ph3P)4 (5.35 г, 0.0046 моль). Полученную смесь нагревали при 100°С в течение двух часов и охладили до комнатной температуры. Добавили воду и этилацетат. Твердые вещества отфильтровали через целит. Слои разделили, и отделенный органический слой промыли водой (Х2). Объединенные водные слои подвергли обратимой экстракции этилацетатом (Х1). Объединенные органические слои высушили (MgSO4), отфильтровали, и удалили растворители в вакууме. Очистка на колонке [Гексаны-этилацетат = 9:1 (об/об)] дала изопропоксииндазол 4BI (20 г, 80%) в виде твердого вещества желтого цвета.
Стадия 3:

Изопропоксииндазол 4BI (20 г, 0.037 моль) и Pd/C (10%, 50%, 7.8 г, 0.0037 моль) перемешивали в толуоле (100 мл) и 2-пропаноле (200 мл) в атмосфере Н2 (резиновая камера) при комнатной температуре в течение одного дня. Твердый катализатор отфильтровали через целит, и растворители удалили в вакууме с получением аминоиндазола 5BI (колич.) в виде твердого вещества грязно-белого цвета.
Стадия 4:
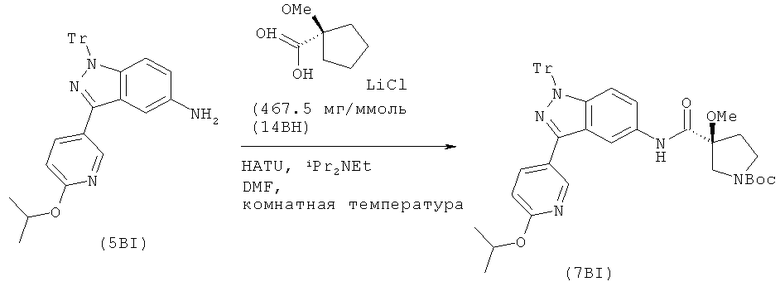
Аминоиндазол 5BI (39 г, 0.076 моль) и пирролидинкарбоновую кислоту 14ВН (32 г, 0.069 моль) растворили в DMF (300 мл) при комнатной температуре. Добавили HATU (29 г, 0.076 моль) и затем iPr2NEt (14.5 мл, 0.083 моль). Смесь перемешивали при комнатной температуре всю ночь и разбавили этилацетатом и водой. Слои разделили. Отделенный органический слой промыли водой (Х2), высушили (MgSO4) и отфильтровали. Концентрировали в вакууме и затем очистили на колонке [гексаны-этилацетат = 4:1 (об/об)] с получением неочищенного соединения 7BI в виде пены грязно-белого цвета.
Стадия 5:
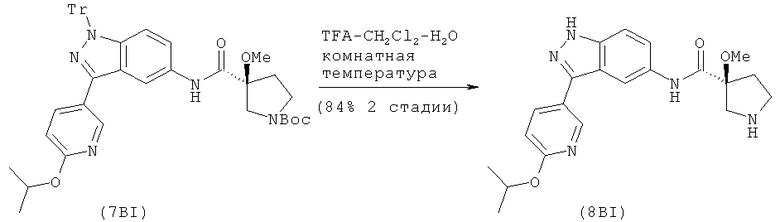
Неочищенное вещество 7BI перемешивали в смеси дихлорметана (300 мл), трифторуксусной кислоты (100 мл) и воды (50 мл) при комнатной температуре всю ночь. Смесь охладили при 0°С и аккуратно погасили насыщенным водным раствором бикарбоната натрия. Растворители удалили в вакууме. Разбавили водой и этилацетатом. Слои разделили, и отделенный водный слой экстрагировали этилацетатом (Х3). Объединенные органические слои высушили (MgSO4), отфильтровали, и растворители удалили в вакууме. Очистка на колонке [5-10% МеОН (7Н аммония) в дихлорметане] дала пирролидин 8BI (23 г, 84%) в виде твердого вещества грязно-белого цвета.
Альтернативно:
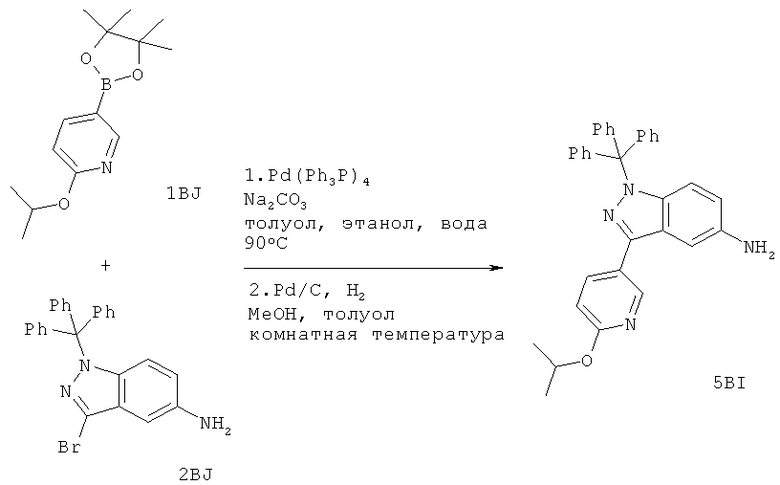
Боронат 1BJ (1.0 г, 3.80 ммоль), броминдазоле 2BJ (1.84 г, 3.80 ммоль), бикарбонат натрия (1.21 г, 11.4 ммоль) загрузили в запаиваемую пробирку. Добавили толуол (30 мл), этанол (30 мл) и воду (15 мл). Суспензию продували азотом в течение 15 минут, и добавили Pd(Ph3P)4 (439 мг, 0.38 ммоль) в виде одной порции. Смесь нагревали в запаянной пробирке при 90°С всю ночь. После охлаждения до комнатной температуры добавили воду и этилацетат, и слои разделили. Отделенный органический слой промыли водой, высушили (MgSO4) и отфильтровали. Растворители удалили в вакууме. Осадок растворили в толуоле (100 мл). Добавили метанол (100 мл) и затем Pd/C (809 мг, 0.38 ммоль, 50%). Смесь перемешивали в атмосфере H2 (резиновая камера) всю ночь. Катализатор отфильтровали, растворители удалили в вакууме. Очистка на колонке [гексаны-этилацетат = 2:1 (об/об)] дала анилин 5BI (1.2 г, 61%, 2 Стадии) в виде твердого вещества грязно-белого цвета.
Синтез 3-Метокси-1-(2-{4-[4-(1-метил-1Н-[1,2,4]триазол-3-ил)-фенил]-3,6-дигидро-2Н-пиридин-1-ил)-2-оксо-этил)-пирролидин-3-карбоновой кислоты [3-(6-изопропокси-пиридин-3-ил)-1Н-индазол-5-ил]-амида
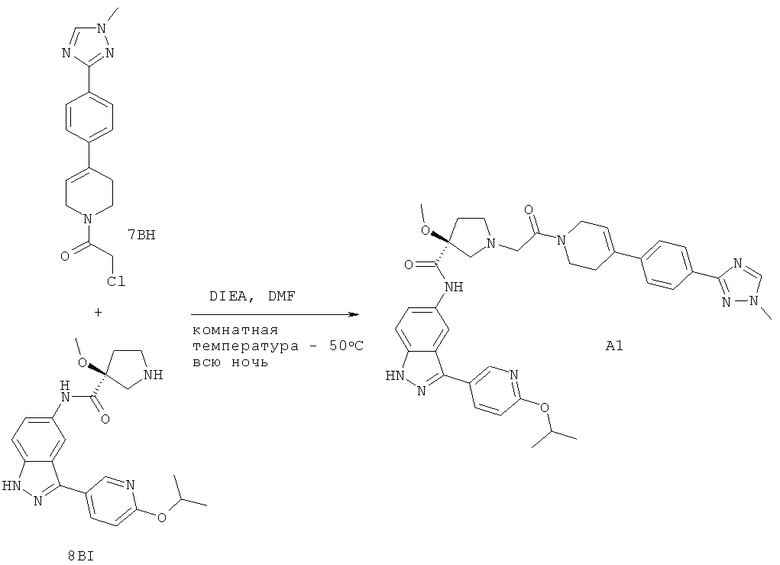
Смесь соединения 7ВН (7.88 г, 24.93 ммоль), соединения 8BI (9.86 г, 24.93 ммоль) и DIEA (26.1 мл, 149.62 ммоль) в DMF (200 мл) перемешивали при комнатной температуре в течение 5 часов. Реакция прошла на около 89% в соответствии с ЖХМС. Реакционную смесь затем нагревали при 50°С всю ночь (16 часов). ЖХМС показала завершение реакции. DMF удалили при пониженном давлении. Неочищенный продукт растворили в 700 мл DCM и один раз промыли 35 мл воды. Водный слой экстрагировали 20%MeOH/DCM (2×120 мл). Объединенные органические экстракты гомогенизировали с помощью МеОН и высушили над MgSO4. Растворитель удалили, и неочищенный продукт очистили с помощью колоночной хроматографии, применяя 20%MeOH/EtoAc с получением целевого продукта А1 в виде твердого вещества желтого цвета (70%). (ЖХМС М+1=674, время удерживания = 2.91 мин.) 1Н ЯМР (400 МГц, CD3OD): δ 8.67 (с, 1Н), 8.42 (с, 1Н), 8.35 (с, 1Н), 8.15-8.19 (м, 1Н), 7.94 (дд, 2Н, J=8.4 & 10 Гц), 7.64 (м, 1Н), 7.49 (м, 3Н), 6.84 (м, 1Н), 6.20 (д, 1Н, J=11.2 Гц), 5.3 (м, 1Н), 4.28 (с, 1Н), 4.23 (м, 1Н), 3.97 (с, 3Н), 3.8 (м, 2Н), 3.56 (д, 1Н, 2.4 Гц), 3.52 (д, 1Н, 6.4 Гц), 3.31 (с, 3Н), 3.17 (т, 1Н, J=10 Гц), 3.07 (т, 1Н, J=12 Гц), 2.81 (м, 1Н), 2.81 (м, 1Н), 2.65 (с, 1Н), 2.58 (с, 1Н), 2.43 (м, 1Н), 2.17 (м, 1Н), 1.32 (д, 6Н, J=6.4 Гц)
Пример 2
Синтез 3-метокси-1-(2-{4-[4-(1-метил-1Н-[1,2,4]триазол-3-ил)-фенил]-3,6-дигидро-2Н-пиридин-1-ил}-2-оксо-этил)-пирролидин-3-карбоновой кислоты [3-(6-гидрокси-пиридин-3-ил)-1Н-индазол-5-ил]-амида
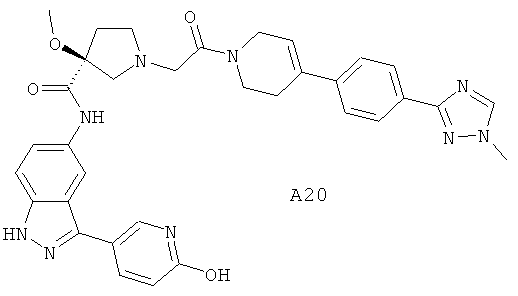
Вышеуказанное соединение (А20) выделили после распадения ди-HCl соли 3-метокси-1-(2-{4-[4-(1-метил-1Н-[1,2,4]триазол-3-ил)-фенил]-3,6-дигидро-2Н-пиридин-1-ил}-2-оксо-этил)-пирролидин-3-карбоновой кислоты [3-(6-изопропокси-пиридин-3-ил)-1Н-индазол-5-ил]-амида.
ЖХМС М+1 634, время удерживания = 2.28 мин. 1H-ЯМР (400 МГц, DMSO-d6): δ 13.09 (уш, 1Н), 11.84 (уш, 1Н), 10.02 (с, 1Н), 8.51 (с, 1Н), 8.37 (с, 1Н), 8.02 (дд, 1Н, J=9.5 Гц & 2.5 Гц), 7.95 (м, 2Н), 7.82 (д, 1Н, J=1.9 Гц), 7.71 (м, 1Н), 7.51 (м, 3Н), 6.52 (д, 1Н, J=9.5 Гц), 6.27 (м, 1Н), 4.08-4.34 (м, 2Н), 3.92 (с, 3Н), 3.70 (м, 2Н), 3.49 (м, 2Н), 3.24 (с, 3Н), 3.16 (д, 1Н, J=5.2 Гц), 2.84-3.13 (м, 3Н), 2.55-2.76 (м, 2Н), 2.36 (м, 1Н), 2.05 (м, 1Н).
Пример 3
Синтез 1-(2-{4-[4-(1,5-Диметил-1Н-[1,2,4]триазол-3-ил)-фенил]-3,6-дигидро-2Н-пиридин-1-ил}-2-оксо-этил)-3-метокси-пирролидин-3-карбоновой кислоты [3-(6-изопропокси-пиридин-3-ил)-1Н-индазол-5-ил]-амида
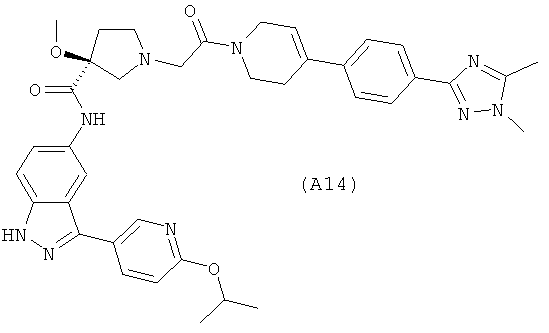
Синтез (S)-1-(2-(4-(4-(1,5-диметил-1Н-1,2,4-триазол-3-ил)фенил)-5,6-дигидропиридин-1(2Н)-ил)-2-оксоэтил)-N-(3-(6-изопропоксипиридин-3-ил)-1H-индазол-5-ил)-3-метоксипирролидин-3-карбоксамида
Стадия 1: Получение 3-(4-бром-фенил)-1,5-диметил-1Н-[1,2,4]триазола
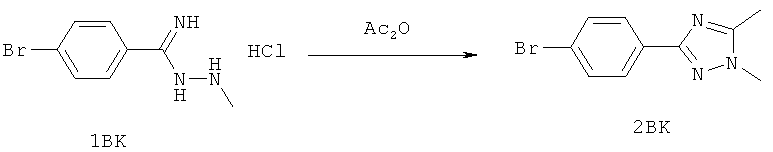
Смесь гидрохлорида 4-бром-N′-метилбензимидгидразида 1 (1.7 г) (полученную согласно методике синтеза Sch-1499895) в уксусном ангидриде (10 мл) нагревали при 100°С в течение 0.5 часов. После охлаждения и концентрирования при пониженном давлении осадок растворили в EtOAc, дважды промыли насыщенным NaHCO3, соляным раствором и высушили (MgSO4). После выпаривания растворителя осадок очистили на силикагеле. Элюция с EtOAc дала 3-(4-бромфенил)-1,5-диметил-1Н-[1,2,4]-триазол 2BK (1.04 г).
Стадия 2: Получение трет-бутил 4-(4-(1,5-диметил-1Н-1,2,4-триазол-3-ил)фенил)-5,6-дигидропиридин-1(2Н)-карбоксилата
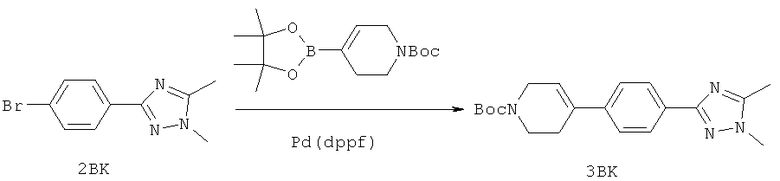
В пробирку высокого давления загрузили соединение 2BK (252 мг, 1 ммоль), 4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-3,6-дигидро-2H-пиридин-1-карбоновой кислоты трет-бутиловый сложный эфир (403 мг, 1.3 ммоль), [1,1′-бис(дифенилфосфино)-ферроцен]дихлорпалладия (II) комплекс с дихлорметаном (1:1) (41 мг, 0.05 ммоль), K2CO3 (410 мг, 3 ммоль) и DME/вода (5:1, 6 мл). Смесь быстро дегазировали аргоном в течение около полуминуты, накрыли и перемешивали при 100°С всю ночь. После охлаждения реакционную смесь разбавили EtOAc и соляным раствором. Органический слой выделили и высушили (MgSO4). После концентрирования осадок очистили на силикагеле. Элюция с помощью МеОН/EtOAc (0-10%) позволила получить целевой продукт 3BK (332 мг).
Стадия 3: Получение 4-(4-(1,5-диметил-1Н-1,2,4-триазол-3-ил)фенил)-1,2,3,6-тетрагидропиридин дигидрохлорида
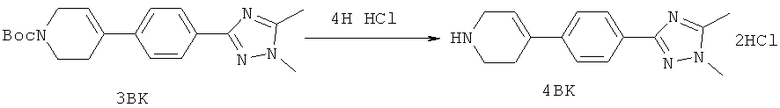
Boc группу можно удалить посредством обработки соединения 3BK раствором 4Н HCl в диоксане при комнатной температуре в течение двух часов. Удаление растворителя в вакууме позволило получить соединение 4BK.
Стадия 4: Получение (S)-трет-бутил 2-(3-(3-(6-изопропоксипиридин-3-ил)-1H-индазол-5-илкарбамоил)-3-метоксипирролидин-1-ил)ацетата
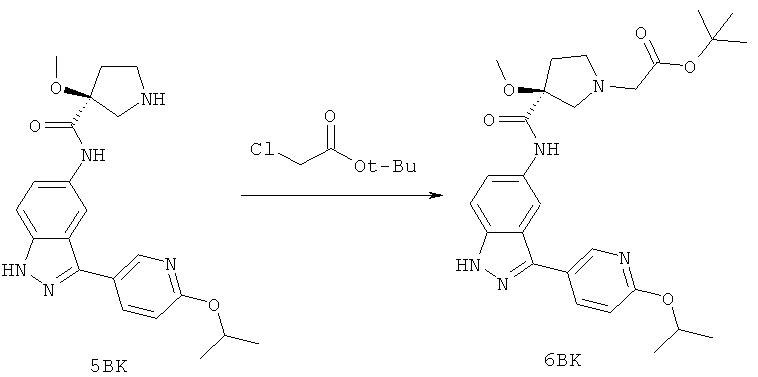
В раствор соединения 5BK (3.5 г, 6.6 ммоль) (полученного согласно методике в синтезе Sch-1499895) в ацетонитриле (26 мл) добавили DIEA (5.7 мл, 32.9 ммоль). Раствор охладили до 0°С, и по каплям добавили 0.47 мл (3.29 ммоль) трет-бутилхлорацетата. После перемешивания в течение 4 часов при 0°С снова добавили 0.47 мл (3.29 ммоль) трет-бутилхлорацетата. Раствор перемешивали еще в течение одного часа при 0°С и затем нагрели до комнатной температуры. После перемешивания в течение ночи при комнатной температуре, растворили в EtOAc (200 мл) и промыли NaHCO3 (1×50 мл), водой (1×50 мл) и соляным раствором (1×50 мл). Органические экстракты высушили над MgSO4, и растворитель удалили. Неочищенный продукт очистили с помощью колоночной хроматографии, применяя 20% МеОН/EtOAC с получением целевого продукта 6ВК (2.4 г).
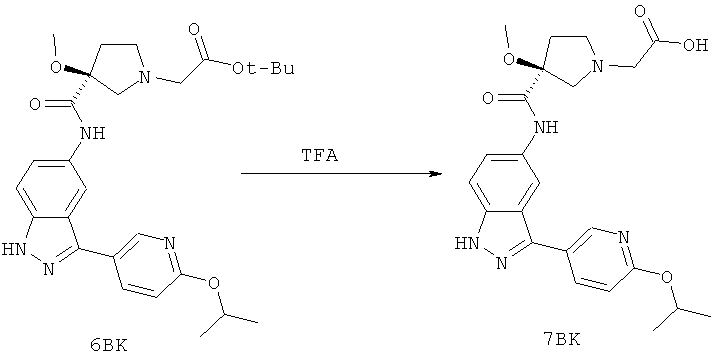
Стадия 5: Получение (S)-2-(3-(3-(6-изопропоксипиридин-3-ил)-1Н-индазол-5-илкарбамоил)-3-метоксипирролидин-1-ил)уксусной кислоты
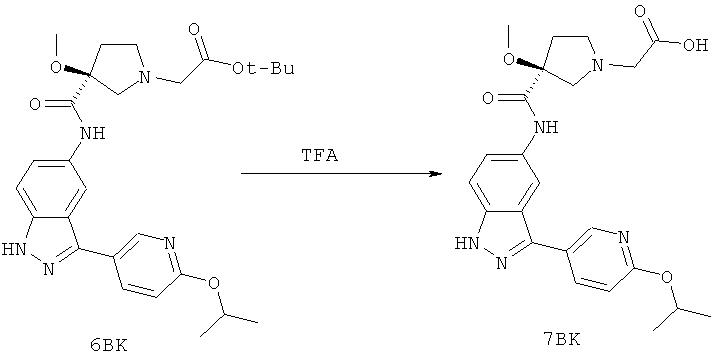
Соединения 6BK (2.4 г) обработали с помощью 40 мл TFA в течение 45 минут при комнатной температуре. TFA удалили при пониженном давлении, и твердое вещество промыли диэтиловым эфиром с получением соединения 7BK в виде соли TFA (4.4 г, 95%).
Стадия 6: Получение (S)-1-(2-(4-(4-(1,5-диметил-1Н-1,2,4-триазол-3-ил)фенил)-5,6-дигидропиридин-1(2Н)-ил)-2-оксоэтил)-N-(3-(6-изопропоксипиридин-3-ил)-1Н-индазол-5-ил)-3-метоксипирролидин-3-карбоксамида
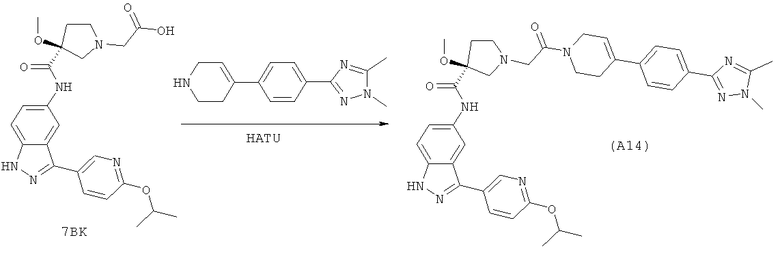
К смеси соединения 7BK (0.12 ммоль), HATU (46 мг, 0.12 ммоль) в DMF (2 мл) добавили соединение 4BK (39 мг, 0.12 ммоль) и DIEA (0.063 мл). Смесь перемешивали в течение двадцати минут и непосредственно очистили с помощью ВЭЖХ с получением соединения А14. Масс-спектр: ЖХМС М+1=690, время удерживания = 3.25 минут;
1H ЯМР А14 HCl соли (400 МГц, DMSO-d6): δ 10.5 (уш, 1Н), 10.23 (д, J=17.6 Гц, 1Н), 8.7 (м, 1Н), 8.45 (д, J=1.2 Гц, 1Н), 8.18 (м, 1Н), 7.94 (м, 2Н), 7.75 (дд, J=8.8, 2.0 Гц, 1Н), 7.56 (м, 3Н), 6.93 (м, 1Н), 6.3 (м, 1Н), 5.3 (м, 1Н), 4.6-4.53 (м, 2Н), 4.2-4.0 (м, 3Н), 3.82 (с, 3Н), 3.7-3.5 (м, 5Н), 3.3 (с, 3Н), 2.67-2.54 (м, 3Н), 2.44 (с, 3Н), 2.4(м, 1Н), 1.33 (д, J=6.4 Гц, 6Н).
Пример 4
Синтез 1-[2-(4-{3-Фтор-4-[1-(2-метокси-этил)-1Н-[1,2,4]триазол-3-ил]-фенил}-3,6-дигидро-2Н-пиридин-1-ил)-2-оксо-этил]-3-метокси-пирролидин-3-карбоновой кислоты [3-(6-изопропокси-пиридин-3-ил)-1Н-индазол-5-ил]-амида
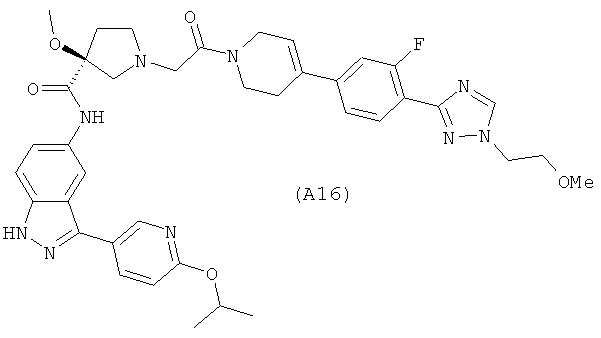
Синтез (S)-1-[2-(4-{3-фтор-4-[1-(2-метокси-этил)-1H-[1,2,4]триазол-3-ил]-фенил}-3,6-дигидро-2Н-пиридин-1-ил)-2-оксо-этил]-3-метокси-пирролидин-3-карбоновой кислоты [3-(6-изопропокси-пиридин-3-ил)-1Н-индазол-5-ил]-амида
Стадия 1: Получение 4-бром-2-фтор-бензамида
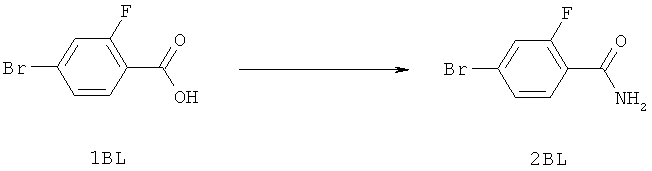
При 0°С по частям добавили 1,1′-карбонилдиимидазол (8.8 г, 54.3 ммоль) к перемешанной смеси 4-бром-2-фторбензойной кислоты (6 г, 27.3 ммоль) в дихлорметане (100 мл). Через двадцать минут получили прозрачный раствор. Добавили гидроксид аммония (28%, 30 мл), и смесь перемешивали всю ночь. Водный слой отделили и дважды экстрагировали дихлорметаном. Объединенные органические экстракты промыли водой, дважды 1 Н HCl, водой, соляным раствором и высушили (MgSO4). Растворитель удалили под вакуумом, и твердое вещество промыли гексаном с получением 4-бром-2-фторбензамида 2BL (5.56 г).
Стадия 2: Получение соединения 3BL
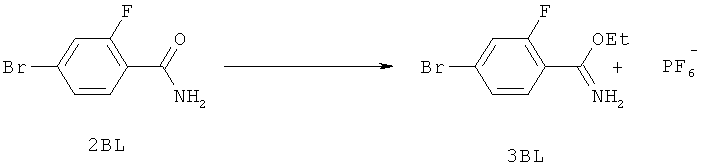
Смесь соединения 2BL (4.66 г, 21.48 ммоль), Et3OPF6 (6.4 г, 25.77 ммоль) в дихлорэтане (86 мл) кипятили с обратным холодильником в течение одного часа. Растворитель удалили при пониженном давлении. Неочищенное вещество охладили до 0°С, растерли в порошок с диэтиловым эфиром и отфильтровали с получением целевого продукта 3BL (7.5 г).
Стадия 3: Получение соединения 4BL
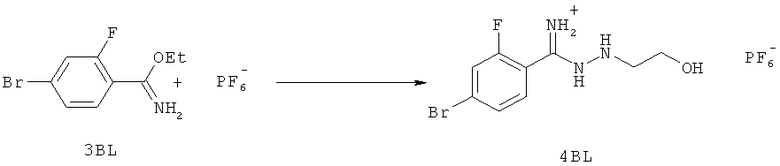
Соединение 3BL (1.96 г) растворили в пиридине (10 мл). Добавили гидроксиэтилгидразин (0.51 мл) при перемешивании, и полученную смесь перемешивали всю ночь. Реакционную смесь концентрировали при пониженном давлении с получением неочищенного продукта 4BL в виде смолы желтого цвета, которую непосредственно применяли на следующей стадии синтеза без последующего очищения.
Стадия 4: Получение соединения 5BL
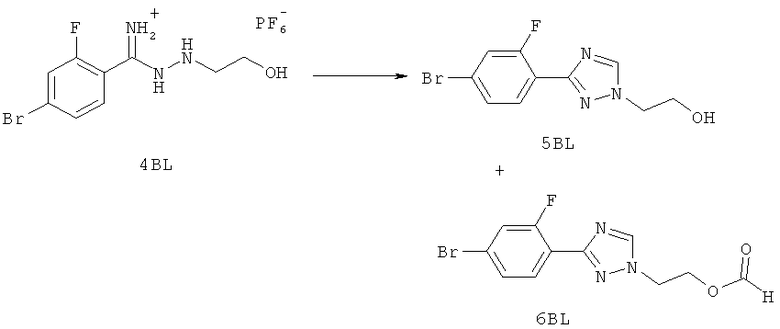
Смесь неочищенного соединения 4BL с предыдущей стадии в муравьиной кислоте (30 мл) кипятили с обратным холодильником всю ночь и концентрировали при пониженном давлении. Осадок обработали насыщенным NaHCO3 и трижды экстрагировали EtOAc. Объединенные органические слои высушили над MgSO4. После концентрирования осадок соединения очистили на силикагеле. Элюция с применением EtOAc позволила получить соединение 6BL (1.1 г), затее соединение 5BL (183 мг).
Соединение 6BL может быть легко превращено в соединение 5BL посредством водного основного гидролиза.
Стадия 5: Получение соединения 7BL
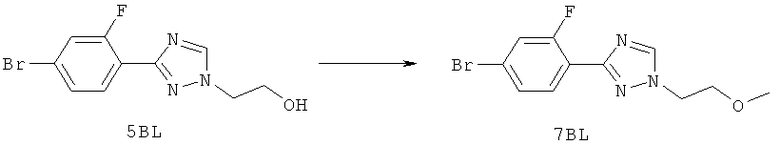
Раствор соединения 5BL (429 мг, 1.5 ммоль) в DMF (3 мл) добавили при перемешивании в колбу, содержащую NaH (60%, 66 мг, 1.65 ммоль). После перемешивания в течение 30 минут медленно добавили Mel (0.103 мл, 1.65 ммоль). Через 30 минут реакционную смесь разбавили EtOAc, трижды промыли водой, соляным раствором и высушили (MgSO4). После концентрирования осадок очистили на силикагеле. Элюция с применением 10% МеОН/EtOAc позволила получить соединение 7BL (88 мг).
Стадия 6: Получение соединения А16
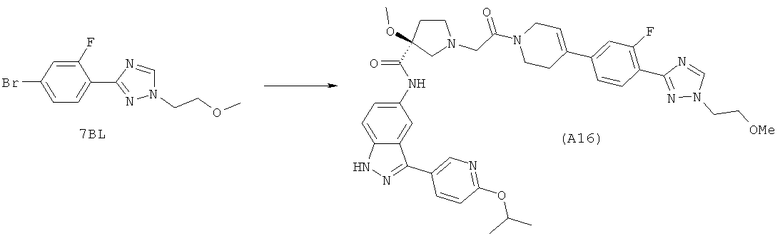
Соединение А16 получили из соединения 7BL, следуя методике, подобной методике для синтеза (S)-1-(2-(4-(4-(1,5-диметил-1Н-1,2,4-триазол-3-ил)фенил)-5,6-дигидропиридин-1(2Н)-ил)-2-оксоэтил)-N-(3-(6-изопропоксипиридин-3-ил)-1Н-индазол-5-ил)-3-метоксипирролидин-3-карбоксамида (А14, Пример 3).
Масс-спектр: ЖХМС М+1=738, время удерживания = 4 минут.
1H ЯМР А16 HCl соль (400 МГц, DMSO-d6): δ 10.45 (уш, 1Н), 10.23 (д, J=16.8 Гц, 1Н), 8.7 (м, 1Н), 8.6 (м, 1Н), 8.46 (м, 1Н), 8.18 (м, 1Н), 8.0 (м, 1Н), 7.74 (д, J=8 Гц, 1Н), 7.57 (д, J=8 Гц, 1Н), 7.42 (м, 1Н), 6.93 (м, 1Н), 6.43 (м, 1Н), 5.3 (м, 1Н), 4.6-4.5 (м, 2Н), 4.41 (м, 2Н), 4.21-4.01 (м, 4Н), 3.8-3.6 (уш, 7Н), 3.33 (м, 3Н), 3.25 (м, 2Н), 2.67-2.54 (м, 3Н), 2.4 (м, 1Н), 1.33 (д, J=6.4 Гц, 6Н).
Пример 5
Синтез 1-(2-{4-[4-(1-Этил-1Н-[1,2,4]триазол-3-ил)-фенил]-3,6-дигидро-2Н-пиридин-1-ил}-2-оксо-этил)-3-метокси-пирролидин-3-карбоновой кислоты [3-(6-изопропокси-пиридин-3-ил)-1Н-индазол-5-ил]-амида
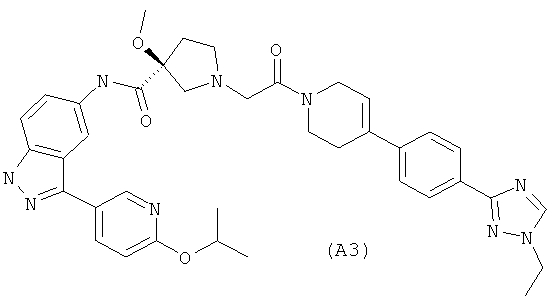
Синтез 4-[4-(1-этил-1Н-[1,2,4]триазол-3-ил)-фенил]-1,2,3,6-тетрагидропиридин гидрохлорида
Стадия 1: Получение 4-бром-бензимидовой кислоты сложного этилового эфира
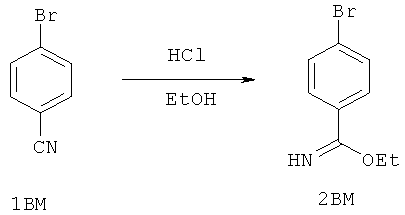
4-Бром-бензонитрил (5 г) суспендировали в абсолютном EtOH (100 мл) и охладили до 0-5°С. Барботировали газ HCl, сначала интенсивно в течение нескольких минут, а затем медленно в течение 5 часов. Полученный раствор перемешивали всю ночь. Большую часть растворителя удалили, и осадок отфильтровали, промыли дважды EtOH и высушили с получением соединения 2ВМ (4.1 г) в виде твердого вещества белого цвета.
Стадия 2: Получение соединения 3ВМ
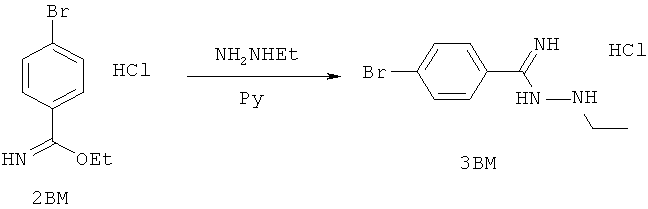
4-бром-бензимидовой кислоты сложный этиловый эфир (1 г) растворили в пиридине (20 мл). Добавили этилгидразин (550 мг) при перемешивании, и полученную смесь перемешивали всю ночь. Реакционную смесь концентрировали при пониженном давлении, добавили диэтиловый эфир, отфильтровали, трижды промыли диэтиловым эфиром и высушили с получением соединения 3ВМ (1 г).
Стадия 3: Получение 3-(4-бром-фенил)-1-этил-1Н-[1,2,4]триазола
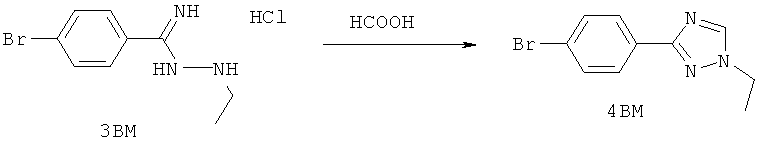
Смесь соединения 3ВМ (1 г) в муравьиной кислоте (10 мл) кипятили с обратным холодильником всю ночь и концентрировали. Осадок обработали насыщенным NaHCO3, и трижды экстрагировали EtOAc. Объединенные органические слои высушили над MgSO4. После концентрирования получили соединение 4 ВМ в виде бесцветных кристаллов (0.9 г).
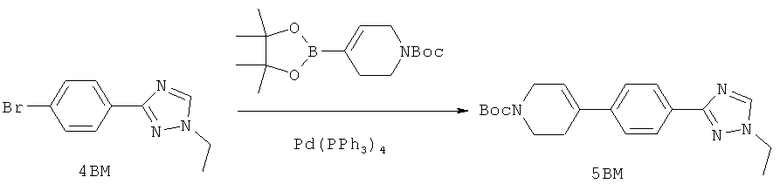
Стадия 4: Получение 4-[4-(1-этил-1Н-[1,2,4]триазол-3-ил)-фенил]-3,6-дигидро-2Н-пиридин-1-карбоновой кислоты сложного трет-бутилового эфира
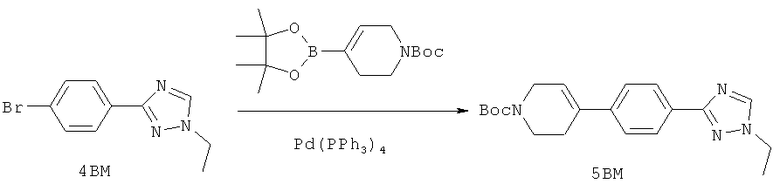
В большую колбу высокого давления загрузили соединение 4ВМ (400 мг), 4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-3,6-дигидро-2H-пиридин-1-карбоновой кислоты сложный трет-бутиловый эфир (540 мг), Pd(PPh3)4 (180 мг), Na2CO3 2N (3 мл) и Диоксан/EtOH/вода (7:3:2, 10 мл). Смесь быстро дегазировали аргоном в течение около полуминуты, накрыли и поместили в микроволновый реактор при 120°С на 20 минут. После охлаждения реакционную смесь разбавили EtOAc и соляным раствором. Органический слой отделили и высушили (MgSO4). После концентрирования осадок очистили на силикагеле. Элюция с МеОН/EtOAc (0-10%) позволила получить целевой продукт 5ВМ (310 мг).
Стадия 5: Получение 4-[4-(1-этил-1Н-[1,2,4]триазол-3-ил)-фенил]-1,2,3,6-тетрагидро-пиридин гидрохлорида
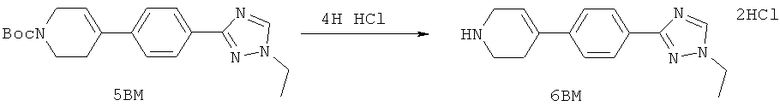
Boc группу можно удалить посредством обработки соединения 5ВМ раствором 4Н HCl в диоксане при комнатной температуре в течение двух часов. Удаление растворителя при пониженном давлении позволило получить соединение 6ВМ.
Синтез 1-(2-{4-[4-(1-Этил-1Н-[1,2,4]триазол-3-ил)-фенил]-3,6-дигидро-2Н-пиридин-1-ил}-2-оксо-этил)-3-метокси-пирролидин-3-карбоновой кислоты [3-(6-изопропокси-пиридин-3-ил)-1Н-индазол-5-ил]-амида
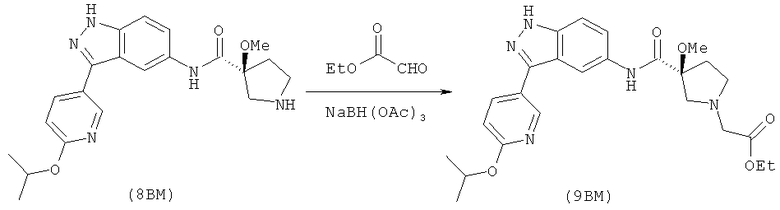
Нечищеное соединение 8ВМ (5.9 ммоль) перемешивали в смеси дихлорметан/МеОН (1:1, 20 мл), оксо-уксусной кислоты сложного этилового эфира (10 мл, 50%) и NaBH(OAc)3 (10 мл) при комнатной температуре всю ночь. Смесь погасили насыщенным водным раствором бикарбоната натрия. Растворители удалили в вакууме. Разбавили водой и этилацетатом. Слои разделили, и отделенный водный слой экстрагировали этилацетатом (Х3). Объединенные органические слои высушили (MgSO4), отфильтровали, и растворители удалили в вакууме. Очистка на колонке позволила получить соединение 8ВМ в виде масла желтого цвета.
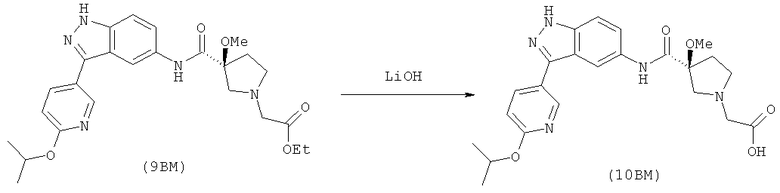
Неочищенное соединение 9ВМ (2.35 г) перемешивали в растворе LiOH (1 м, 10 мл) и THF (10 мл) при комнатной температуре всю ночь. В смеси установили рН 3. Растворители удалили в вакууме. Продукт применяли на следующей стадии без очистки.
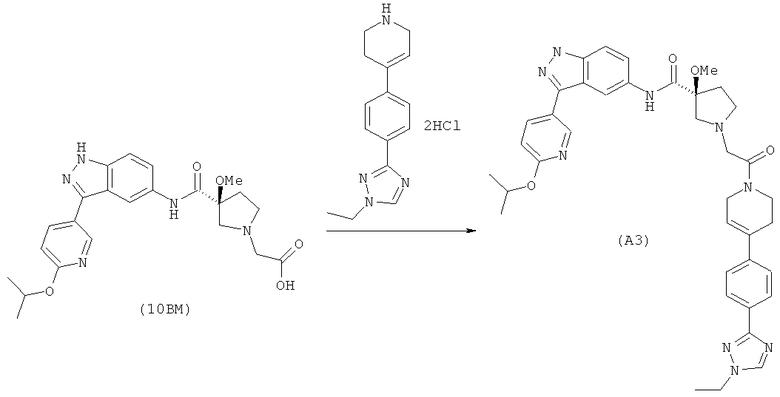
Неочищенный продукт 10ВМ (49 мг), 4-[4-(1-Этил-1Н-[1,2,4]триазол-3-ил)-фенил]-1,2,3,6-тетрагидро-пиридин (25.4 мг), HATU (45 мг) и триэтиламин (0.1 мл) перемешивали в DMF (1 мл, при комнатной температуре, всю ночь). Смесь очистили с помощью ВЭЖХ, с получением соединения A3 в виде масла желтого цвета. Масс-спектр: ЖХМС М+1=690, время удерживания = 3.47 минут.
Пример 6
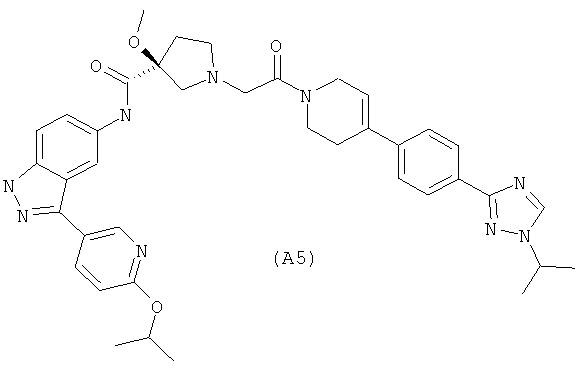
Синтез 4-[4-(1-изопропил-1Н-[1,2,4]триазол-3-ил)-фенил]-1,2,3,6-тетрагидро-пиридин гидрохлорида
Стадия 1: Получение 4-бром-бензимидовой кислоты сложного этилового эфира
4-Бром-бензонитрил (5 г) суспендировали в абсолютном EtOH (100 мл) и охладили до 0-5°С. Барботировали газ HCl, сначала медленно в течение нескольких минут, а затем интенсивно в течение 5 часов. Полученный раствор перемешивали всю ночь. Большую часть растворителя удалили, и осадок отфильтровали, дважды промыли EtOH и высушили с получением соединения 2BN (4.1 г) в виде твердого вещества белого цвета. (Необходимо отметить, что при проведении крупномасштабного синтеза может понадобиться больше времени для завершения реакции. Лучше контролировать исчезновение исходных реагентов для обнаружения конечной точки реакции.)
Стадия 2: Получение соединения 3BN
4-Бром-бензимидовой кислоты сложный этиловый эфир (1 г) растворили в пиридине (20 мл). Добавили изопропилгидразин (550 мг) при перемешивании, и полученную смесь перемешивали всю ночь. Реакционную смесь концентрировали при пониженном давлении, добавили диэтиловый эфир, отфильтровали, трижды промыли диэтиловым эфиром и высушили с получением соединения 3BN (0.9 г).
Стадия 3: Получение 3-(4-бром-фенил)-1-изопропил-1Н-[1,2,4]триазола
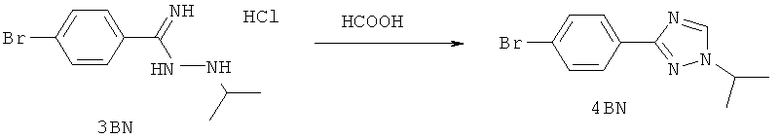
Смесь соединения 3BN (1 г) в муравьиной кислоте (10 мл) кипятили с обратным холодильником всю ночь и концентрировали. Осадок обработали насыщенным NaHCO3 и трижды экстрагировали EtOAc. Объединенные органические слои высушили над MgSO4. После концентрирования соединение 4BN получили в виде бесцветных кристаллов (0.9 г).
Стадия 4: Получение 4-[4-(1-изопропил-1H-[1,2,4]триазол-3-ил)-фенил]-3,6-дигидро-2Н-пиридин-1-карбоновой кислоты сложного трет-бутилового эфира
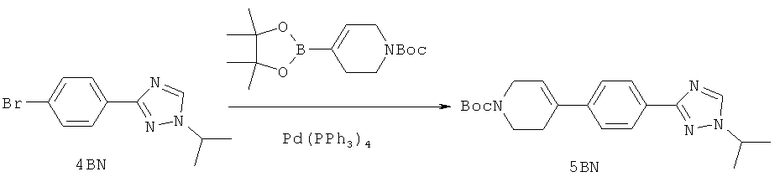
В колбу высокого давления загрузили соединение 4BN (500 мг), 4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-3,6-дигидро-2H-пиридин-1-карбоновой кислоты сложный трет-бутиловый эфир (583 мг), Pd(PPh3)4 (112 мг), Na2CO3 2Н (3 мл) и Диоксан (10 мл). Смесь быстро дегазировали аргоном в течение около полуминуты, накрыли и перемешивали при 100°С всю ночь. После охлаждения реакционную смесь разбавили EtOAc и соляным раствором. Органический слой отделили и высушили (MgSO4). После концентрирования осадок очистили на силикагеле. Элюция с применением МеОН/EtOAc (0-10%) позволила получить целевой продукт 5BN (410 мг).
Стадия 5: Получение 4-[4-(1-изопропил-1Н-[1,2,4]триазол-3-ил)-фенил]-1,2,3,6-тетрагидро-пиридин гидрохлорида
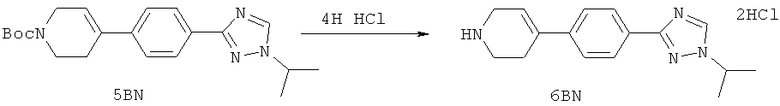
Вое группу можно удалить посредством обработки соединения 5BN раствором 4Н HCl в диоксане, при комнатной температуре, в течение двух часов. Удаление растворителя при пониженном давлении позволило получить соединение 6BN.
Синтез 1-(2-{4-[4-(1-Изопропил-1Н-[1,2,4]триазол-3-ил)-фенил]-3,6-дигидро-2Н-пиридин-1-ил}-2-оксо-этил)-3-метокси-пирролидин-3-карбоновой кислоты [3-(6-изопропокси-пиридин-3-ил)-1Н-индазол-5-ил]-амида
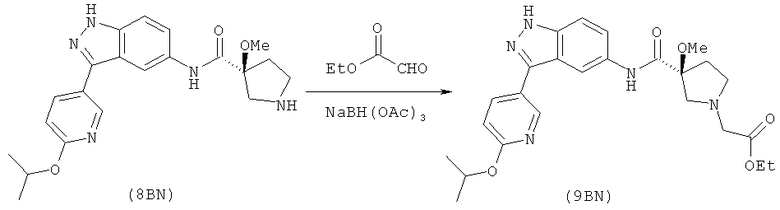
Неочищенное соединение 8BN (5.9 ммоль) перемешивали в смеси дихлорметан/МеОН (1:1, 20 мл), оксо-уксусной кислоты сложного этилового эфира (10 мл, 50%) и NaBH(OAc)3 (10 мл) при комнатной температуре всю ночь. Смесь погасили насыщенным водным раствором бикарбоната натрия. Растворители удалили в вакууме. Разбавили водой и этилацетатом. Слои разделили, и отделенный водный слой экстрагировали этилацетатом (Х3). Объединенный органический слой высушили (MgSO4), отфильтровали, и растворители удалили в вакууме. Очистка на колонке позволила получить соединение 9BN в виде масла желтого цвета, с выходом 65%.
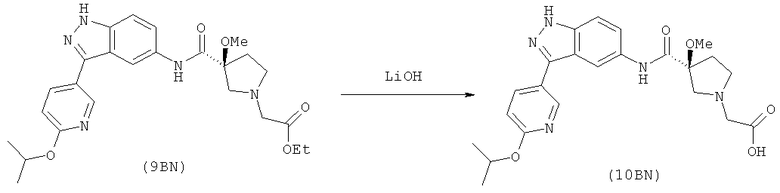
Неочищенное соединение 9BN (2.35 г) перемешивали в растворе LiOH (1 м, 10 мл) и THF (10 мл) при комнатной температуре всю ночь. В смеси установили рН 3. Растворители удалили в вакууме. Продукт применяли на следующей стадии без очистки, выход количественный.
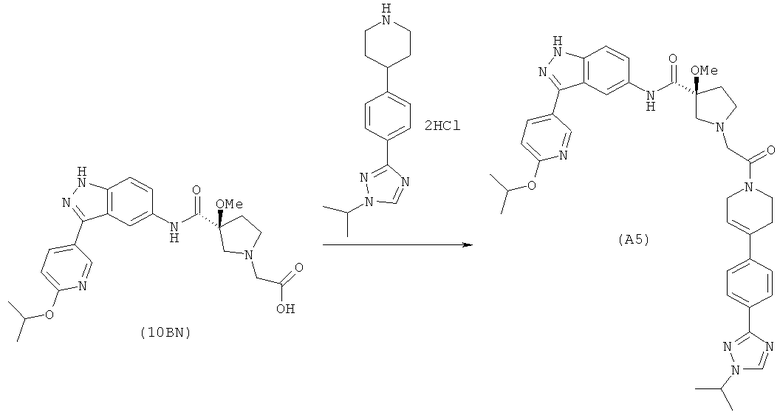
Неочищенное вещество 10BN (60 мг), 4-[4-(1-изопропил-1Н-[1,2,4]триазол-3-ил)-фенил]-1,2,3,6-тетрагидро-пиридин (35 мг), HATU (52 мг) и триэтиламин (0.1 мл) перемешивали в DMF (1 мл) при комнатной температуре всю ночь. Смесь очистили с помощью ВЭЖХ с получением соединения А5 в виде масла желтого цвета. Масс-спектр: ЖХМС М+1=704, время удерживания = 3.56 минут.
Пример 7
Синтез 1-(2-{4-[4-{1-Изопропил-1Н-[1,2,4]триазол-3-ил)-фенил]-3,6-дигидро-2Н-пиридин-1-ил}-2-оксо-этил)-3-метокси-пирролидин-3-карбоновой кислоты [3-(6-изопропокси-пиридин-3-ил)-1Н-индазол-5-ил]-амида
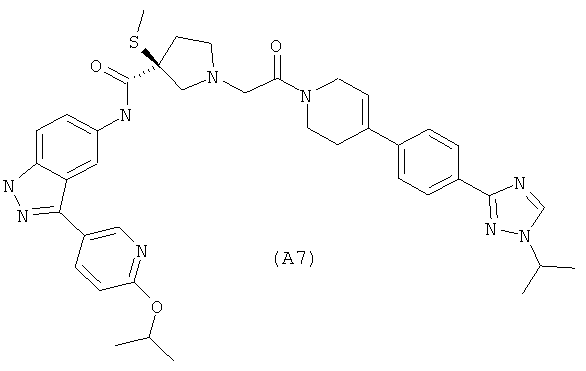
Синтез 4-[4-(1-изопропил-1Н-[1,2,4]триазол-3-ил)-фенил]-1,2,3,6-тетрагидропиридин гидрохлорида
Стадия 1: Получение 4-бром-бензимидовой кислоты сложного этилового эфира
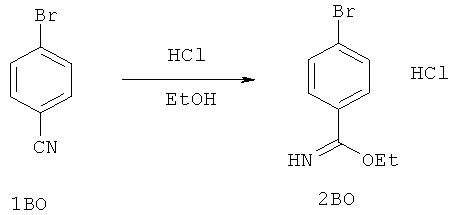
4-Бром-бензонитрил (5 г) суспендировали в абсолютном EtOH (100 мл) и охладили до 0-5°С. Барботировали газ HCl, сначала интенсивно в течение нескольких минут, а затем медленно в течение пяти часов. Полученный раствор перемешивали всю ночь. Большую часть растворителя удалили, и осадок отфильтровали, дважды промыли, применяя EtOH, и высушили с получением соединения 2ВО (4.1 г) в виде твердого вещества белого цвета.
Стадия 2: Получение соединения 3ВО
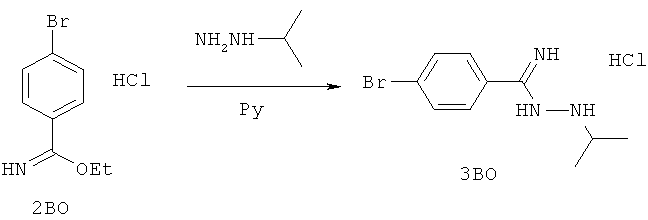
4-Бром-бензимидовой кислоты сложный этиловый эфир (1 г) растворили в пиридине (20 мл). Добавили изопропилгидразин (550 мг) при перемешивании, и полученную смесь перемешивали всю ночь. Реакционную смесь концентрировали при пониженном давлении, добавили диэтиловый эфир, отфильтровали, трижды промыли диэтиловым эфиром и высушили с поучением соединения 3ВО (0.9 г).
Стадия 3: Получение 3-(4-бром-фенил)-1-изопропил-1Н-[1,2,4]триазола:
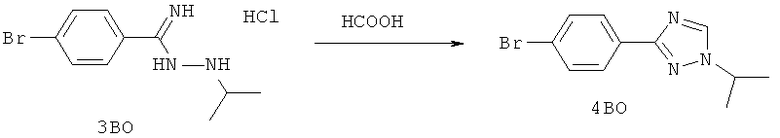
Смесь соединения 3ВО (1 г) в муравьиной кислоте (10 мл) кипятили с обратным холодильником и охладили. Осадок обработали насыщенным NaHCO3 и трижды экстрагировали, применяя EtOAc. Объединенные органические слои высушили над MgSO4. После концентрирования получили соединение 4ВО в виде бесцветных кристаллов (0.9 г). (Необходимо отметить, что было обнаружено, что реакция должна проводиться в течение не более двух часов. При крупномасштабном синтезе необходимо использовать 10% NaOH вместо NaHCO3).
Стадия 4: Получение 4-[4-(1-изопропил-1H-[1,2,4]триазол-3-ил)-фенил]-3,6-дигидро-2Н-пиридин-1-карбоновой кислоты сложного трет-бутилового эфира
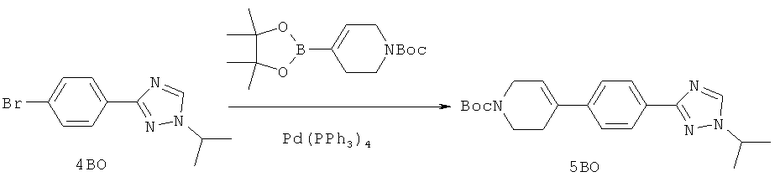
В большую колбу высокого давления загрузили соединение 4ВО (500 мг), 4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-3,6-дигидро-2H-пиридин-1-карбоновой кислоты сложный трет-бутиловый эфир (583 мг), Pd(PPh3)4 (112 мг), Na2CO3 2N (3 мл) и Диоксан (10 мл). Смесь быстро дегазировали аргоном в течение около полуминуты, накрыли и перемешивали при 100С всю ночь. После охлаждения реакционную смесь разбавили EtOAc и соляным раствором. Органический слой отделили и высушили (MgSO4). После концентрирования осадок очистили на силикагеле. Элюция с применением МеОН/EtOAc (0-10%) позволила получить целевой продукт 5ВО (410 мг).
Стадия 5: Получение 4-[4-(1-изопропил-1H-[1,2,4]триазол-3-ил)-фенил]-1,2,3,6-тетрагидропиридин гидрохлорида
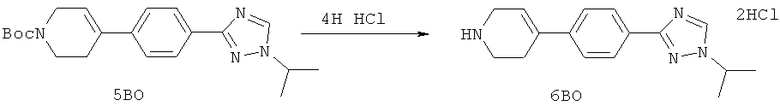
Вое группу можно удалить посредством обработки соединения 5ВО раствором 4Н HCl в диоксане при комнатной температуре в течение двух часов. Удаление растворителя при пониженном давлении позволило получить соединение 6ВО.
Получение и хиральное разделение 3-Метилсульфанил-пирролидин-3-карбоновой кислоты сложного метилового эфира 4ВР
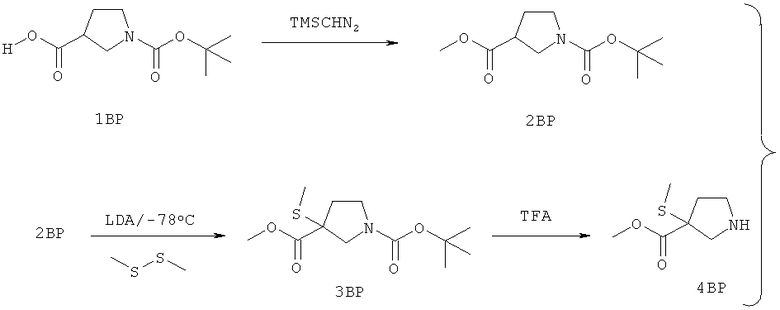
Пирролидин-1,3-дикарбоновой кислоты 1-трет-бутиловый сложный эфир 1ВР (4.3 г, 20 ммоль) растворили в 28 мл толуола и 3.5 мл метанола. 2Н раствор триметилсилилдиазометана в гексанах (13 мл, 26 ммоль) добавили по каплям при 0 С, и реакционную смесь перемешивали в течение 10 мин при температуре окружающей среды. Смесь выпарили с получением 4.3 г масла.
К маслу 2ВР (0.5 г, 2.1 ммоль), растворенному в тетрагидрофуране (15 мл), по каплям добавили 1.2 мл 2Н раствора диизопропиламида лития в гексанах, и реакционную смесь перемешивали в течение одного часа при -78 С. Медленно добавили диметилдисульфид (0.48 мл, 5.4 ммоль), и реакционной смеси позволили постепенно нагреться до температуры окружающей среды. Реакционную смесь перемешивали в течение 18 часов. Добавили насыщенный раствор хлорида аммония (25 мл), и реакционную смесь перемешивали в течение 5 минут. Реакционную смесь трижды экстрагировали этилацетатом (3×25 мл), высушили над сульфатом магния, отфильтровали и выпарили с получением после колоночной хроматографии 0.386 г продукта 3ВР, название которого указано в заглавии.
3-Метилсульфанил-пирролидин-1,3-дикарбоновой кислоты 1-трет-бутилового сложного эфира 3-метиловый сложный эфир (2.15 г, 8.8 ммоль) растворили в 20 мл 50% трифторуксусной кислоты/дихлорметана и перемешивали в течение 2 часов. Реакционную смесь выпаривали с получением 3.35 г 3-метилсульфанил-пирролидин-3-карбоновой кислоты сложного метилового эфира 4 ВР в виде вязкого твердого вещества.
Альтернативно, продукт 4ВР может быть получен следующим образом:
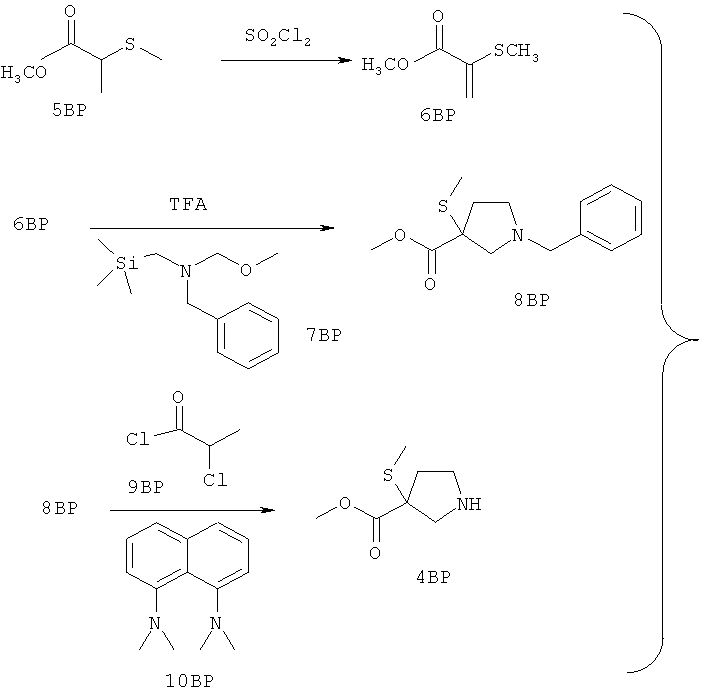
К 2-Метилсульфанил-пропионовой кислоты сложному метиловому эфиру 5ВР (25 г, 0.1 моль), растворенному в хлороформе, медленно добавили сульфурилхлорид (15.1 мл, 0.1 моль) при 0°С. Реакционную смесь перемешивали при 0°С в течение 30 минут, и затем смесь кипятили с обратным холодильником при 65°С в течение 30 минут. Реакционную смесь затем концентрировали до сухости с получением 23.75 г 2-метилсульфанил-акриловой кислоты сложного метилового эфира 6ВР в виде жидкости.
К перемешиваемому раствору 2-метоксиакрилата 2-метилсульфанил-акриловой кислоты сложного метилового эфира 6ВР (136 г, 1.03 моль) и бензил-метоксиметил-триметилсилиланилметил-амина 7ВР (290 г, 1.22 моль) в дихлорметане (2.7 л) при 0°С добавили раствор трифторуксусной кислоты (26 мл, 0.3 моль). Полученный раствор нагрели до комнатной температуры и перемешивали всю ночь. Неочищенный продукт очистили с помощью колоночной хроматографии на силикагеле, элюируя раствором этилацетата в гексане (1:4) с получением 1-бензил-3-метилсульфанил-пирролидин-3-карбоновой кислоты сложного метилового эфира 8ВР (131 г, 47%).
К 1-бензил-3-метилсульфанил-пирролидин-3-карбоновой кислоты сложному метиловому эфиру 8ВР (131 г, 493.7 ммоль), растворенному в дихлорэтане (2.6 л), при 0°С добавили N,N,N′,N′-тетраметил-нафталин-1,8-диамин 9ВР (31.8 г, 0.144 ммоль) и затем добавили 2-хлор-пропионилхлорид 10ВР (64 мл, 593.1 ммоль). Реакционную смесь перемешивали всю ночь при температуре окружающей среды и затем концентрировали до сухости. Осадок растворили в 2.8 л метанола и кипятили с обратным холодильником при 65°С в течение 3.5 часов. Реакционную смесь затем концентрировали до сухости, и осадок очистили с помощью колоночной хроматографии на силикагеле, элюируя раствором метанола в дихлорметане (1:9), с получением 3-метилсульфанил-пирролидин-3-карбоновой кислоты сложного метилового эфира 4ВР (75 г, 86%).
Хиральное разделение 3-метилсульфанил-пирролидин-3-карбоновой кислоты сложного метилового эфира 4ВР
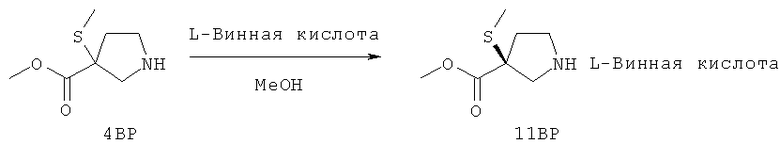
3-Метилсульфанил-пирролидин-3-карбоновой кислоты сложный этиловый эфир 4ВР (42.9 г, 244.8 ммоль) и L-винную кислоту (36.7 г, 244.8 ммоль) поместили в круглодонную колбу, объемом 1 л, и растворили метанолом (250 мл). Колбу затем присоединили к роторному испарителю при 75°С. Колбу мягко вращали при этой температуре в течение около 20 минут для обеспечения полного растворения. После образования прозрачного раствора, добавили около 10 мг кристаллов 3-метилсульфанил-пирролидин-3-карбоновой кислоты сложного метилового эфира 11ВР (для затравки и образования кристаллов) и позволили мягко оседать для образования кристаллов. Через три дня отфильтровали 19.4 г кристаллов, которые затем промыли холодным метанолом (20-30 мл) с получением 18.2 г кристаллического 3-метилсульфанил-пирролидин-3-карбоновой кислоты сложного метилового эфира 11ВР.
Хиральную чистоту кристаллов 3-метилсульфанил-пирролидин-3-карбоновой кислоты сложного метилового эфира 11ВР определяли путем получения производных с 4-нитробензилхлорформиатом и исследования их с помощью аналитической ВЭЖХ (Хиральная колонка AD) в условиях применения системы растворителей 20% изопропанол/гексан со скоростью потока 1 мл/мин. Определенная чистота составила >99.9% при времени удерживания 16.58 минут.
Получение 3-Метилсульфанил-пирролидин-1,3-дикарбоновой кислоты 1-трет-бутилового сложного эфира
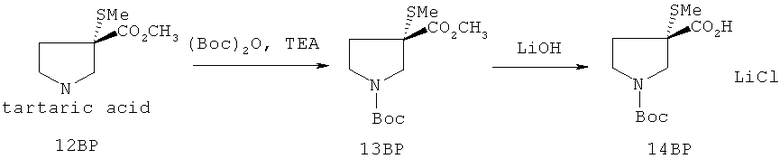
К охлажденному (0°С) раствору 12ВР (28 г, 90.52 ммоль) в сухом CH2Cl2 (250 мл) добавили триэтиламин (31.5 мл, 226.32 ммоль, 2.5 эквив.) и затем добавили (Вос)2O (25.7 г, 117.68 ммоль, 1.3 эквив). Полученную смесь перемешивали при температуре от 0°С до комнатной температуры всю ночь, и затем разбавили, применяя CH2Cl2, промыли насыщенным водным раствором NaHCO3 и соляным раствором, высушили (MgSO4) и концентрировали. Хроматография на силикагеле (гексаны/этилацетат, 4:1) позволила получить соединение 13ВР (23.5 мг, 90.52 ммоль, 100%) в виде бесцветного масла.
К перемешенному раствору вещества 13ВР (23.5 мг, 90.52 ммоль) в THF/MeOH (175 мл/175 мл) добавили 135 мл LiOH (1M в H2O, 135 ммоль, 1.5 эквив.). Реакционную смесь перемешивали при комнатной температуре всю ночь, добавили 135 мл 1Н HCl. Полученную смесь перемешивали в течение еще 15 мин и концентрировали, с получением соединения 14ВР.
Стадия 1:
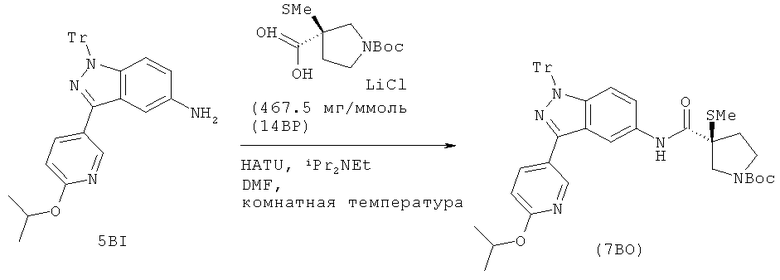
Аминоиндазол 5BI (39 г, 0.076 моль) и пирролидинкарбоновую кислоту 14 ВР (32 г, 0.069 моль) растворили в DMF (300 мл) при комнатной температуре. Добавили HATU (29 г, 0.076 моль) и затем добавили iPr2NEt (14.5 мл, 0.083 моль). Смесь перемешивали при комнатной температуре всю ночь и разбавили этилацетатом и водой. Слои разделили. Отделенный органический слой промыли водой (Х2), высушили (MgSO4) и отфильтровали. Концентрировали в вакууме с последующим очищением на колонке [гексаны-этилацетат = 4:1 (об/об)] с получением неочищенного соединения 7 ВО в виде пены грязно-белого цвета.
Стадия 2:
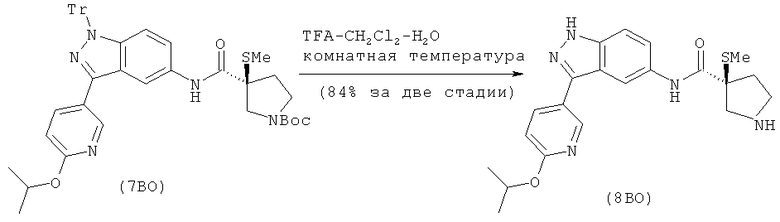
Неочищенное вещество 7 ВО перемешивали в смеси дихлорметана (300 мл), трифторуксусной кислоты (100 мл) и воды (50 мл) при комнатной температуре всю ночь. Смесь охладили при 0°С и аккуратно погасили насыщенным водным раствором бикарбоната натрия. Растворители удалили в вакууме. Разбавили водой и этилацетатом. Слои разделили, и отделенный водный слой экстрагировали этилацетатом (Х3). Объединенные органические слои высушили (MgSO4), отфильтровали, и растворители удалили в вакууме. Очистка на колонке [5-10% МеОН (7Н аммиак) в дихлорметане] позволила получить пирролидин 8ВО (23 г, 84%) в виде твердого вещества грязно-белого цвета.
Синтез 1-(2-{4-[4-(1-изопропил-1Н-[1,2,4]триазол-3-ил)-фенил]-3,6-дигидро-2Н-пиридин-1-ил}-2-оксо-этил)-3-метилсульФанил-пирролидин-3-карбоновой кислоты [3-(6-изопропокси-пиридин-3-ил)-1Н-индазол-5-ил]-амида
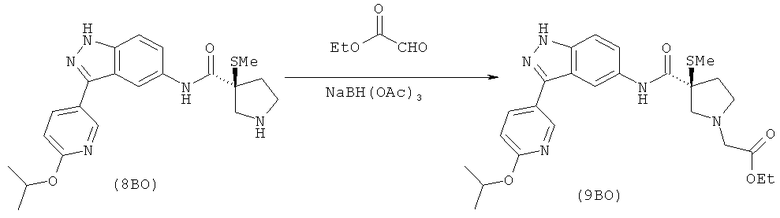
Неочищенное вещество 8ВО (5.9 ммоль) перемешивали в смеси дихлорметан/МеОН (1:1, 20 мл), оксо-уксусной кислоты сложного этилового эфира (10 мл, 50%) и NaBH(OAc)3 (10 мл) при комнатной температуре всю ночь. Смесь погасили насыщенным водным раствором бикарбоната натрия. Растворители удалили в вакууме. Разбавили водой и этилацетатом. Слои разделили, и отделенный водный слой экстрагировали этилацетатом (Х3). Объединенные органические слои высушили (MgSO4), отфильтровали, и растворители удалили в вакууме. Очистка на колонке позволила получить вещество 9ВО в виде масла желтого цвета.
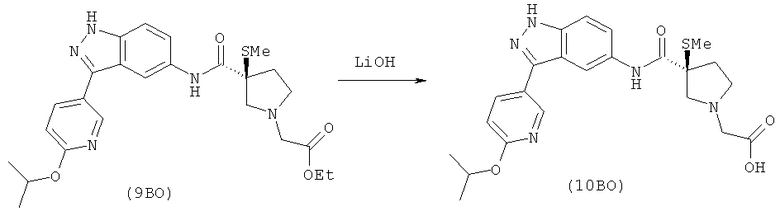
Неочищенное вещество 9ВО (2.35 г) перемешивали в растворе LiOH (1 м, 10 мл) и THF (10 мл) при комнатной температуре всю ночь. В смеси установили рН 3. Растворители удалили в вакууме. Продукт применяли на следующей стадии без очистки.
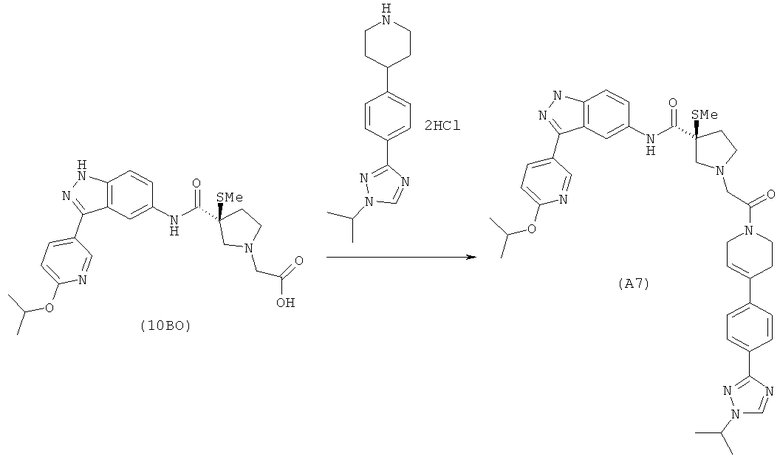
Неочищенное соединение 10ВО (60 мг), 4-[4-(1-изопропил-1Н-[1,2,4]триазол-3-ил)-фенил]-1,2,3,6-тетрагидропиридин (35 мг), HATU (52 мг) и триэтиламин (0.1 мл) перемешивали в DMF (1 мл) при комнатной температуре всю ночь. Смесь очистили с помощью ВЭЖХ с получением соединения ПВО в виде масла желтого цвета. Масс-спектр: ЖХМС М+1=720, время удерживания = 3.68 минут.
Пример 8
Получение 1-[2-(4-{4-[1-(2-гидрокси-2-метил-пропил)-1Н-[1,2,4]триазол-3-ил]-фенил}-3,6-дигидро-2Н-пиридин-1-ил)-2-оксо-этил]-3-метокси-пирролидин-3-карбоновой кислоты [3-(6-изопропокси-пиридин-3-ил)-1Н-индазол-5-ил]амида
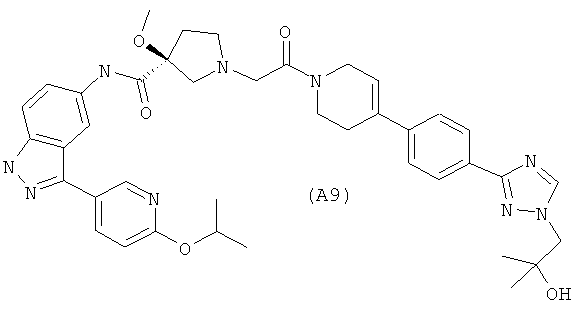
Синтез 2-хлор-1-{4-[4-(1-этил-1Н-[1,2,4]триазол-3-ил)-фенил]-3,6-дигидро-2Н-пиридин-1-ил}-этанона
Стадия 1: Получение 4-бром-бензимидовой кислоты сложного этилового эфира
4-Бром-бензонитрил (5 г) суспендировали в абсолютном EtOH (100 мл) и охладили до 0-5°С. Барботировали газ HCl, сначала интенсивно в течение нескольких минут, а затем медленно в течение 5 часов. Полученный раствор перемешивали всю ночь. Большую часть растворителя удалили, и осадок отфильтровали, промыли EtOH дважды и высушили с получением соединения 2ВО (4.1 г) в виде твердого вещества белого цвета.
Стадия 2: Получение соединения 3BQ:
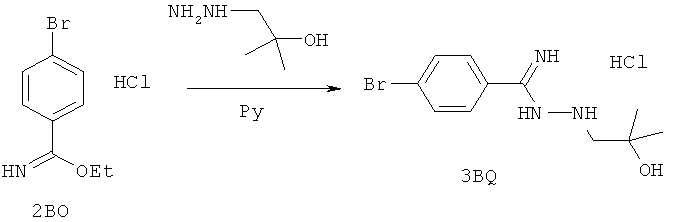
4-Бром-бензимидовой кислоты сложный этиловый эфир (1.68 г) растворили в пиридине (10 мл). 1-Гидразино-2-метил-пропан-2-ол (1 г) добавили при перемешивании, и полученную смесь перемешивали всю ночь. Реакционную смесь концентрировали при пониженном давлении, и добавили диэтиловый эфир, отфильтровали, трижды промыли диэтиловым эфиром и высушили с получением соединения 3BQ (1 г).
Стадия 3: Получение 1-[3-(4-бром-фенил)-[1,2,4]триазол-1-ил]-2-метил-пропан-2-ола:
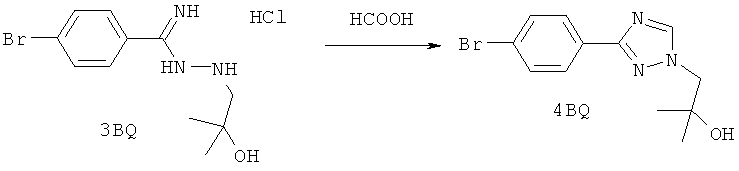
Смесь соединения 3BQ (1 г) в муравьиной кислоте (10 мл) кипятили с обратным холодильником всю ночь и концентрировали. Осадок обработали насыщенным NaHCO3, и экстрагировали EtOAc трижды. Объединенные органические слои высушили над MgSO4. После концентрирования соединение 4BQ получили в виде бесцветных кристаллов (0.9 г).
Стадия 4: Получение 4-{4-[1-(2-гидрокси-2-метил-пропил)-1Н-[1,2,4]триазол-3-ил]-фенил}-3,6-дигидро-2Н-пиридин-1-карбоновой кислоты трет-бутилового сложного эфира
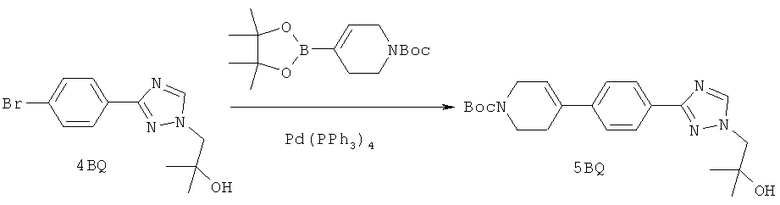
В большую колбу высокого давления загрузили 4BQ (400 мг), 4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-3,6-дигидро-2Н-пиридин-1-карбоновой кислоты трет-бутиловый сложный эфир (540 мг), Pd(PPh3)4 (180 мг), 2Н Na2CO3 (3 мл) и диоксан/EtOH/вода (7:3:2, 10 мл). Смесь быстро дегазировали аргоном в течение около полуминуты, накрыли и поместили в микроволновый реактор при 120С в течение 20 минут. После охлаждения, реакционную смесь разбавили EtOAc и соляным раствором. Органический слой отделили и высушили (MgSO4). После концентрирования, осадок очистили на силикагеле. Элюция с применением МеОН/EtOAc (0-10%) позволила получить целевой продукт 5BQ (310 мг).
Стадия 5: Получение 2-метил-1-{3-[4-(1,2,3,6-тетрагидро-пиридин-4-ил)-фенил]-[1,2,4]триазол-1-ил}-пропан-2-ола
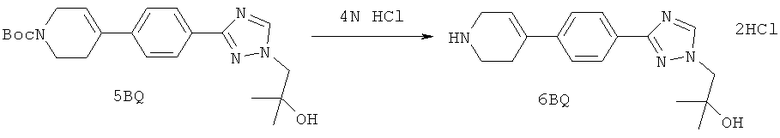
Вое группу можно удалить путем обработки соединения 5BQ 4Н раствором HCl в диоксане при комнатной температуре в течение двух часов. Удаление растворителя под вакуумом позволило получить соединение 6BQ.
Синтез 1-[2-(4-{4-[1-(2-гидрокси-2-метил-пропил)-1Н-[1,2,4]триазол-3-ил]-фенил}-3,6-дигидро-2Н-пиридин-1-ил)-2-оксо-этил]-3-метокси-пирролидин-3-карбоновой кислоты [3-(6-изопропокси-пиридин-3-ил)-1Н-индазол-5-ил]-амида
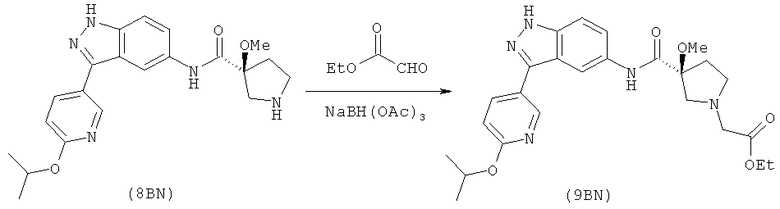
Неочищенное соединение 8BN (5.9 ммоль) перемешивали в смеси дихлорметан/МеОН (1:1, 20 мл), оксо-уксусной кислоты сложного этилового эфира (10 мл, 50%) и NaBH(OAc)3 (10 мл), при комнатной температуре, всю ночь. Смесь погасили насыщенным водным раствором бикарбоната натрия. Растворители удалили в вакууме. Разбавили водой и этилацетатом. Слои разделили, и отделенный водный слой экстрагировали этилацетатом (Х3). Объединенные органические слои высушили (MgSO4), отфильтровали и растворители выпарили под вакуумом. Очистка на колонке позволила получить соединение 9ВМ в виде масла желтого цвета.
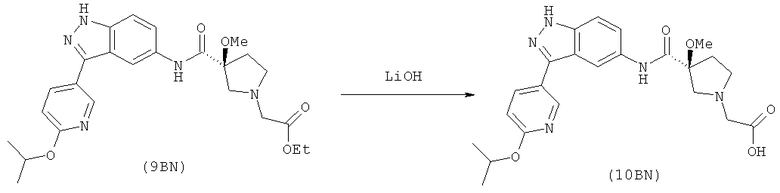
Неочищенное соединение 9ВМ (2.35 г) перемешивали в растворе LiOH (1 м, 10 мл) и THF (10 мл) при комнатной температуре всю ночь. Для смеси установили рН 3. Растворители удалили в вакууме. Продукт применяли на следующей стадии без очистки.
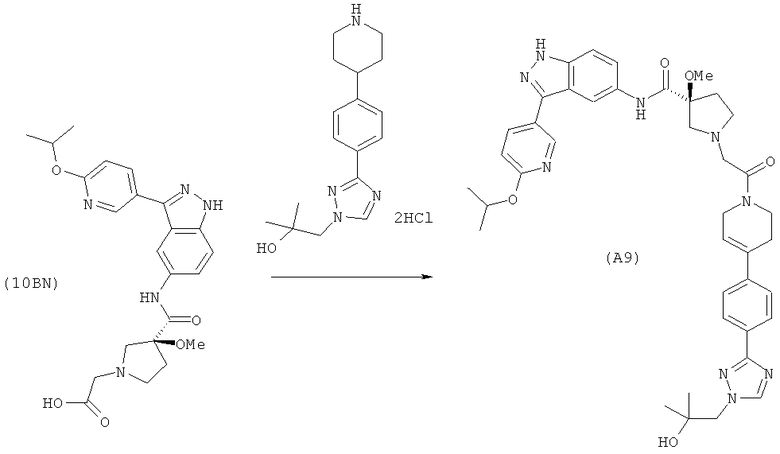
Неочищенное соединение 10BN (60 мг), 2-метил-1-{3-[4-(1,2,3,6-тетрагидропиридин-4-ил)-фенил]-[1,2,4]триазол-1-ил}-пропан-2-ол (38 мг), HATU (50 мг) и триэтиламин (0.1 мл) перемешивали в DMF (1 мл) при комнатной температуре всю ночь. Смесь очистили с помощью ВЭЖХ, что позволило получить соединение А9 в виде масла желтого цвета, выход 50%. Масс-спектр: ЖХМС М+1=734, время удерживания = 3.32 минут.
Пример 9
Получение 3-метокси-1-(2-{4-[4-(1-метил-1Н-[1,2,4]триазол-3-ил)-фенил]-3,6-дигидро-2Н-пиридин-1-ил}-2-оксо-этил)-пирролидин-3-карбоновой кислоты {3-[6-(2-метокси-этокси)-пиридин-3-ил]-1Н-индазол-5-ил}-амида
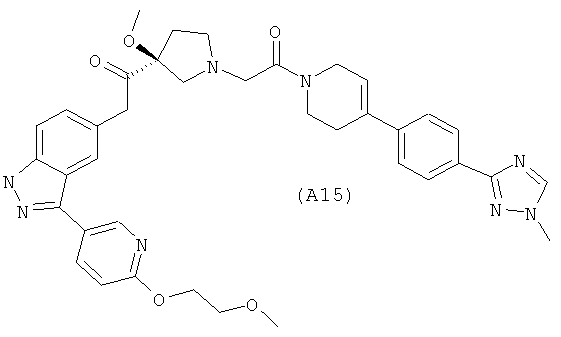
Синтез 3-метокси-пирролидин-3-карбоновой кислоты {3-[6-(2-метокси-этокси)-пиридин-3-ил]-1Н-индазол-5-ил}-амида
Стадия 1:
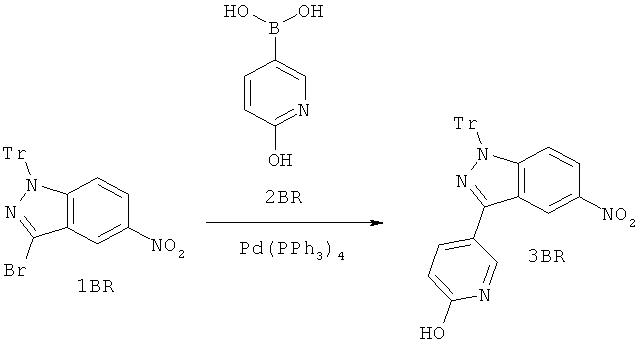
В колбу высокого давления загрузили соединение 1BR (1.75 г), соединение 2BR (0.5 г), Pd(PPh3)4 (210 мг), Na2CO3 2N (10 мл) и диоксан (10 мл). Смесь быстро дегазировали аргоном в течение около полуминуты, накрыли и перемешивали при 100°С всю ночь. После охлаждения, реакционную смесь разбавили EtOAc и соляным раствором. Органический слой отделили, и высушили (MgSO4). После концентрирования осадок очистили на силикагеле. Элюция позволила получить целевой продукт 3BR (0.8 г).
Стадия 2:
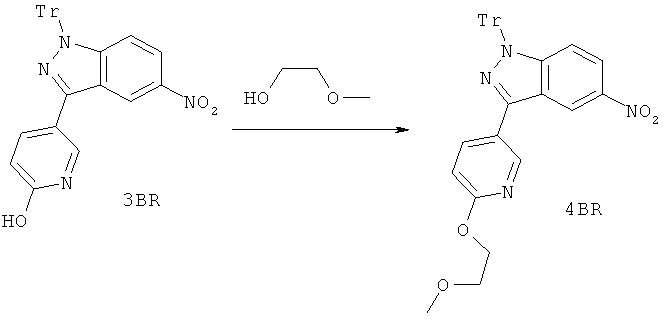
Смесь соединения 3BR (1.8 г), 2-метокси-этанола (1 г), DEAD (0.8 г) и PPh3 (1.2 г) в THF (10 мл) перемешивали всю ночь при комнатной температуре и концентрировали. Осадок очистили на силикагеле, что позволило получить целевой продукт 4BR (0.8 г).
Стадия 3:
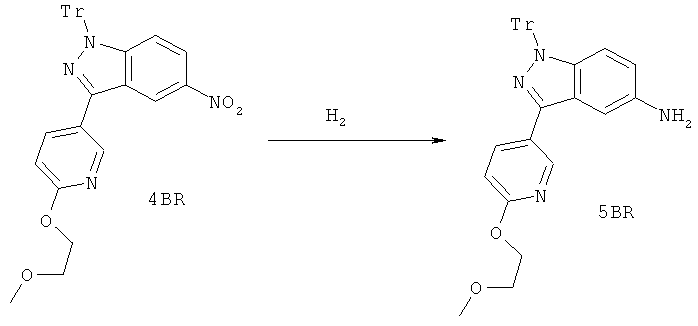
Соединение 4BR (0.5 г) в МеОН (20 мл) восстановили в проточном реакторе H-cube с Pd/C (10%) колонкой.
Стадия 4:
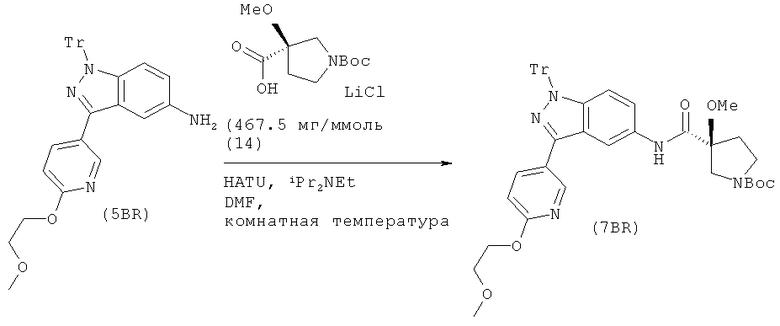
Аминоиндазол 5BR (80 мг) и пирролидинкарбоновую кислоту 14ВН (38 мг) растворили в DMF (1 мл) при комнатной температуре. Добавили HATU (69 мг) и затем iPr2NEt (0.1 мл). Смесь перемешивали при комнатной температуре всю ночь и разбавили этилацетатом и водой. Слои разделили. Отделенный органический слой промыли водой (Х2), высушили (MgSO4) и отфильтровали. Концентрация под вакуумом позволила получить неочищенное соединение 7BR в виде пены грязно-белого цвета.
Стадия 5:
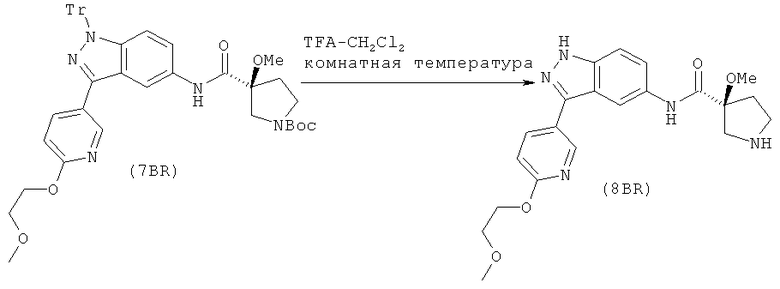
Неочищенное соединение 7BR перемешивали в смеси дихлорметана (1 мл), трифторуксусной кислоты (1 мл), при комнатной температуре, в течение 1 часа. Смесь охладили при 0°С и погасили аккуратно насыщенным водным раствором бикарбоната натрия. Растворители удалили в вакууме. Разбавили водой и этилацетатом. Слои разделили, и отделенный водный слой экстрагировали этилацетатом (Х3). Объединенные органические слои высушили (MgSO4), отфильтровали, и растворители удалили в вакууме, что позволило получить пирролидин 8BR в виде твердого вещества грязно-белого цвета.
Синтез 3-метокси-1-(2-N-[4-(1-метил-1Н-[1,2,4]триазол-3-ил)-фенил]-3,6-дигидро-2Н-пиридин-1-ил}-2-оксо-этил)-пирролидин-3-карбоновой кислоты {3-[6-(2-метокси-этокси)-пиридин-3-ил]-1Н-индазол-5-ил}-амида
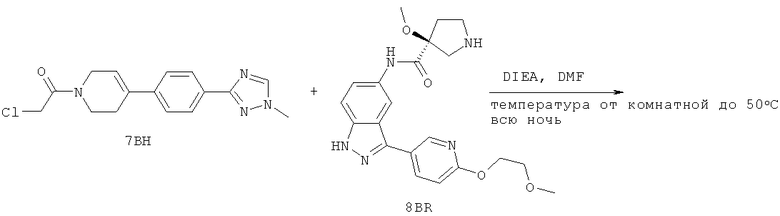
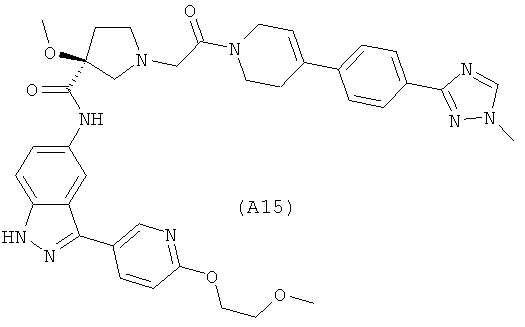
Смесь соединения 7ВН (52 мг), соединения 8BR (62 мг) и DIEA (0.3 мл) в DMF (1.5 мл) перемешивали при комнатной температуре всю ночь. ЖХМС показала завершение реакции. DMF удалили при пониженном давлении. Неочищенное соединение очистили посредством ВЭЖХ. (ЖХМС М+1=692, время удерживания = 3.00 минут) 1Н ЯМР (400 МГц, CDCl3): δ 11.92 (с, 1Н), 11.11 (с, 1Н), 9.14 (с, 1Н), 8.58 (с, 1Н), 8.03 (с, 1Н), 7.99 (д, 1Н, J=8.4 Гц), 7.90 (т, 2Н, J=6.8), 7.39 (с, 2Н), 7.20 (с, 3Н), 6.76 (д, 1Н, J=8.0 Гц), 5.85 (д, 1Н, J=20 Гц), 4.4 (с, 4Н), 4.05 (кв, 4Н, J=7.2 Гц), 3.8 (с, 3Н), 3.50-3.56 (м, 6Н), 2.34-2.62 (м, 5Н), 2.0 (с, 2Н), 1.64-1.49 (м, 3Н).
Пример 10
Получение 1-[2-(4-{4-[1-(2-гидрокси-этил)-1Н-[1,2,4]триазол-3-ил]-фенил}-3,6-дигидро-2Н-пиридин-1-ил)-2-оксо-этил]-3-метокси-пирролидин-3-карбоновой кислоты [3-(6-изопропокси-пиридин-3-ил)-1Н-индазол-5-ил]-амида
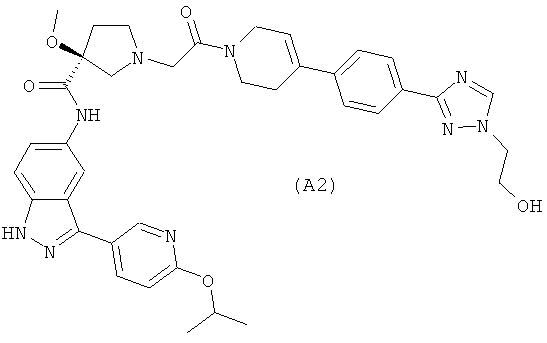
Получение {3-[3-(6-изопропокси-пиридин-3-ил)-1Н-индазол-5-илкарбамоил]-3-метокси-пирролидин-1-ил}-уксусной кислоты
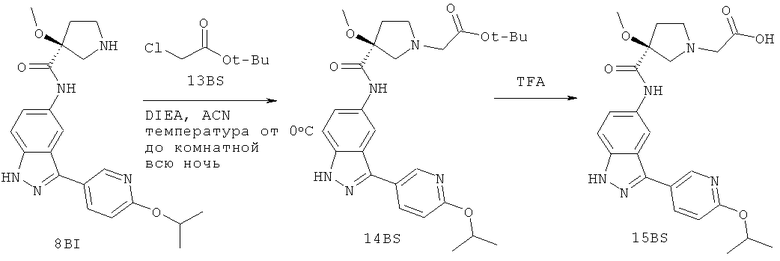
В раствор соединения 8BI (3.5 г, 6.6 ммоль) в ацетонитриле (26 мл) добавили DIEA (5.7 мл, 32.9 ммоль). Раствор охладили до 0°С, и по каплям добавили 0.47 мл (3.29 ммоль) соединения 13BS. После перемешивания в течение 4 часов при 0°С снова добавили 0.47 мл (3.29 ммоль) соединения 13BS. Перемешивали в течение одного часа при 0°С и затем нагрели до комнатной температуры. После перемешивания в течение ночи при комнатной температуре, растворили в EtOAc (200 мл) и промыли NaHCO3 (1×50 мл), водой (1×50 мл) и соляным раствором (1×50 мл). Органический экстракты высушили над MgSO4, и растворитель удалили. Неочищенное соединение очистили с помощью колоночной хроматографии, применяя 20%МеОН/EtOAc с получением целевого продукта 14BS (2.4 г).
Соединение 14BS (2.4 г) обработали, применяя 40 мл TFA в течение 45 минут при комнатной температуре. TFA удалили при пониженном давлении, и твердое вещество промыли эфиром с получением целевого соединения 15BS в виде соли TFA (4.4 г, 95%). Соединение 15BS превратили в соль HCl путем добавления 4Н HCl в воде.
Синтез 2-{3-[[4[(1,2,3,6-тетрагидро-пиридин-4-ил)-фенил]-[1,2,4]триазол-1-ил}-этанол гидрохлорида
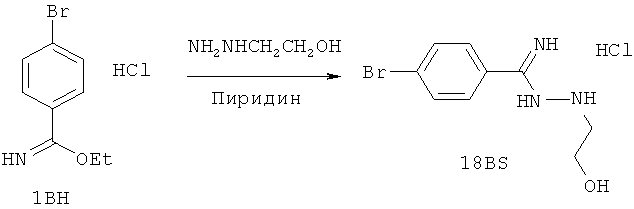
Получение 4-бром-N′-(2-гидроксиэтил)бензимидогидразид гидрохлорида
Сложный этиловый эфир 4-бром-бензимидовой кислоты 1ВН (5 г) растворили в пиридине (100 мл). Гидроксиэтил гидразин (1.92 мл) добавили при перемешивании, и полученную смесь перемешивали всю ночь. Осадок собрали посредством фильтрации, и маточную жидкость концентрировали до сухости, и добавили диэтиловый эфир. Твердое вещество собрали путем фильтрации. Объединенное твердое вещество дважды промыли диэтиловым эфиром и высушили, с получением соединения 18BS (5.1 г).
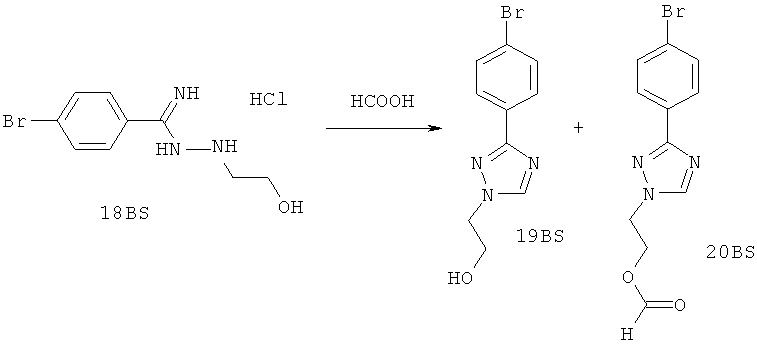
Получение 2-(3-(4-бромфенил)-1Н-1,2,4-триазол-1-ил)этанола
Смесь соединения 18BS (3г) в муравьиной кислоте (50 мл) перемешивали при комнатной температуре в течение 30 минут, при 100°С в течение 1.5 часов и концентрировали. Осадок обработали насыщенным NaHCO3 и экстрагировали EtOAc трижды. Объединенные органические экстракты высушили над MgSO4. После концентрирования неочищенное соединение очистили на силикагеле. Элюция с применением EtOAc позволила получить соединение 20BS (2 г) и соединение 19BS (262 мг).
Соединение 20BS может быть легко превращено в соединение 19BS посредством водного гидролиза
Получение 4-[4-(1-(2-гидрокси-этил)-1H-[1,2,4]триазол-3-ил)-фенил]-3,6-дигидро-2Н-пиридин-1-карбоновой кислоты трет-бутилового сложного эфира
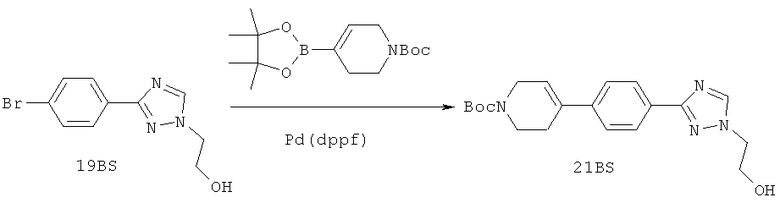
В большую колбу высокого давления загрузили 19BS (1.5 г, 5.6 ммоль), 4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-3,6-дигидро-2Н-пиридин-1-карбоновой кислоты трет-бутиловый сложный эфир (1.9 г, 6.2 ммоль), [1,1′-бис(дифенилфосфино)-ферроцена]дихлорпалладия (II) комплекс с дихлорметаном(1:1) (0.22 г, 0.28 ммоль), K2CO3 (2.32 г, 16.8 ммоль) и DME/вода (5:1, 12 мл). Смесь быстро дегазировали аргоном в течение около полуминуты, накрыли и перемешивали при 80°С всю ночь. После охлаждения реакционную смесь разбавили EtOAc и промыли водой (1Х) и соляным раствором (1x). Органический слой отделили и высушили над MgSO4. После концентрирования осадок очистили на силикагеле. Элюция с применением МеОН/EtOAc (0-10%) позволила получить целевой продукт 21 BS (1.4 г, 73%).
Получение 2-{3-[[4[(1,2,3,6-тетрагидро-пиридин-4-ил)-фенил]-[1,2,4]триазол-1-ил}-этанол гидрохлорида
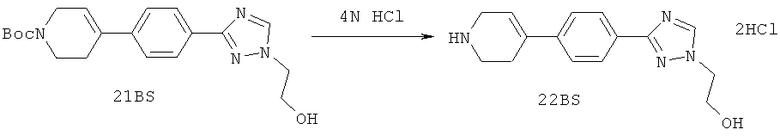
Boc группу можно удалить путем обработки соединения 21BS 4H раствором HCl в диоксане, при комнатной температуре, в течение двух часов. Удаление растворителя под вакуумом позволило получить соединение 22BS.
Получение 1-[2-(4-{4-[1-(2-гидрокси-этил)-1Н-[1,2,4]триазол-3-ил]-фенил}-3,6-дигидро-2Н-пиридин-1-ил)-2-оксо-этил]-3-метокси-пирролидин-3-карбоновой кислоты [3-(6-изопропокси-пиридин-3-ил)-1Н-индазол-5-ил]-амида
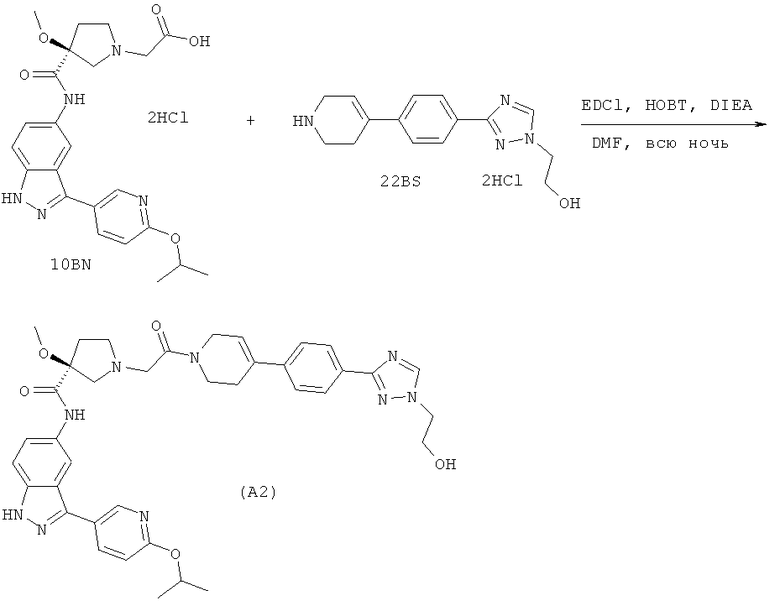
Смесь соединения 10BN (0.49 г, 1 ммоль), 1-(3-диметил-аминопропил)-3-этилкарбоксиимд гидрохлорида (0.38 г, 2 ммоль) и 1-гидроксибензотриазола (0.14 г, 1 ммоль) растворили в DMF (3 мл). После перемешивания в течение 20 минут при комнатной температуре добавили соединение 16BS (0.34 г, 1 ммоль) и DIEA (0.7 мл, 4 ммоль). После перемешивания в течение ночи при комнатной температуре разбавили, применяя DCM (45 мл), и промыли NaHCO3 (1×7 мл), водой (3×7 мл) и соляным раствором (1×10 мл). Органический слой высушили над MgSO4. После концентрирования, осадок очистили на силикагеле. Элюция с применением 2% NH3 в 20%МеОН/EtOAc позволила получить целевой продукт А2 (0.3 г). Это соединение превратили в соль HCl путем добавления 4Н HCl в 1,4-диоксане. (ЖХМС: М+1=706, время удерживания = 3.13 минут), 1H ЯМР (400 МГц, DMSO-d6): δ=10.5-10,8 (м, 1Н), 10.25 (д, 1Н, J=19.2 Гц), 8.60-8.75 (м, 2Н), 8.43 (м, 1Н), 8.18 (м, 1Н), 7.98 (м, 2Н), 7.78 (д, 2Н, J=20 Гц), 7.5-7.6 (м, 4Н), 6.9 (м, 2Н), 6.3 (м, 1Н), 5.3 (м, 1Н), 4.5-4.6 (м, 2Н), 4.15-4.3 (м, 4Н), 4.0-4.15 (м, 2Н), 3.75 (т, 4Н, 5.2 Гц), 3.5-3.6 (м, 2Н), 3.4-3.3 (м, 1Н), 3.29 (с, 2Н), 2.5-2.7 (м, 3Н), 2.3-2.4 (м, 1Н), 1.3 (д, 6Н, J=6.4 Гц).
Пример 11
Синтез 3-метокси-1-(2-{4-[4-(1-метил-1Н-[1,2,4]триазол-3-ил)-фенил]-3,6-дигидро-2Н-пиридин-1-ил}-2-оксо-этил)-пирролидин-3-карбоновой кислоты [3-(6-этокси-пиридин-3-ил)-1Н-индазол-5-ил]-амида (Пример 1)
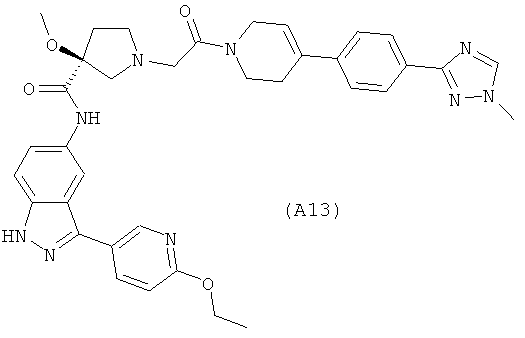
Стадия 1:
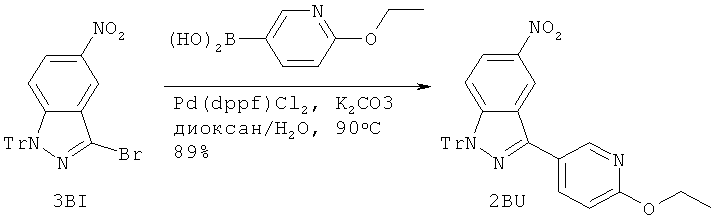
Смесь 6-этоксипиридин-3-бороновой кислоты (2.5 г, 14.97 ммоль), бро-миндазола 3BI (7.25 г, 14.97 ммоль), карбоната калия (6.2 г, 44.91 ммоль), PdCl2(dppf)2.CH2Cl2 (1.22 г, 1.497 ммоль), 1,4-диоксана (40 мл) и воды (10 мл) продували азотом в течение 15 мин при комнатной температуре и затем нагревали при 90°С в течение 18 часов, и охладили до комнатной температуры. Добавили воду (100 мл) и этилацетат (300 мл). Твердые вещества отфильтровали через целит. Слои разделили, и отделенный органический слой промыли водой (100 мл). Объединенные органические слои высушили (Na2SO4), отфильтровали и растворители удалили под вакуумом. Очистка на колонке [Гексаны-этилацетат = 9:1 (об/об)] позволила получить соединение 2BU (7 г, 89%).
Стадия 2:
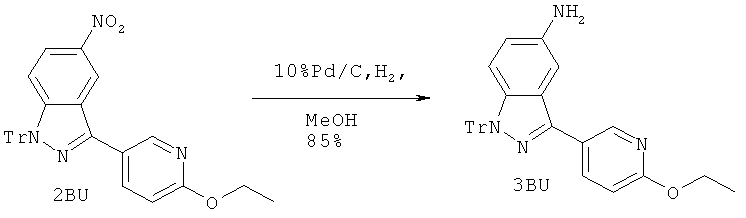
Соединение 2BU (2 г, 3.8 ммоль) и Pd/C (10%, 50 мас.%, 0.7 г) перемешивали в толуоле (30 мл) и МеОН (15 мл), в атмосфере H2 (резиновая камера), при комнатной температуре, в течение 18 часов. Твердый катализатор отфильтровали через целит, и растворители удалили в вакууме. Очистка на колонке с применением 4%MeOH/CH2Cl2 позволила получить соединение 3BU (1.6 г, 85%).
Стадия 3:
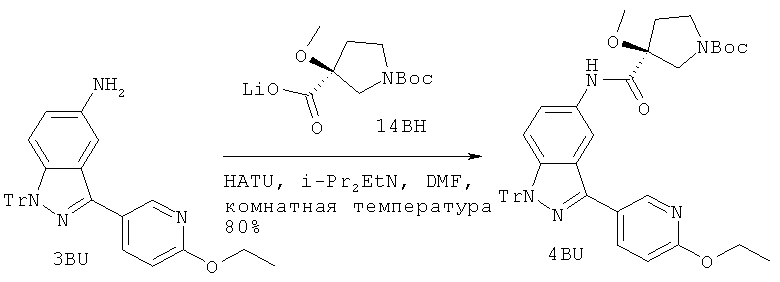
Соединение 3BU (1.46 г, 3.22 ммоль) и пирролидинкарбоновую кислоту 14ВН (0.56 г, 3.22 ммоль) растворили в DMF (300 мл) при комнатной температуре. Добавили HATU (1.68 г, 4.83 ммоль) и затем iPr2NEt (0.78 мл, 4.83 ммоль). Смесь перемешивали при комнатной температуре всю ночь и разбавили этилацетатом (200 мл) и водой (100 мл). Слои разделили. Отделенный органический слой промыли водой (100 мл), высушили (Na2SO4) и отфильтровали. Концентрирование в вакууме с последующей колоночной очисткой [гексаны - этилацетат = 9:1 (об/об)] позволили получить продукт 4BU.
Стадия 4:
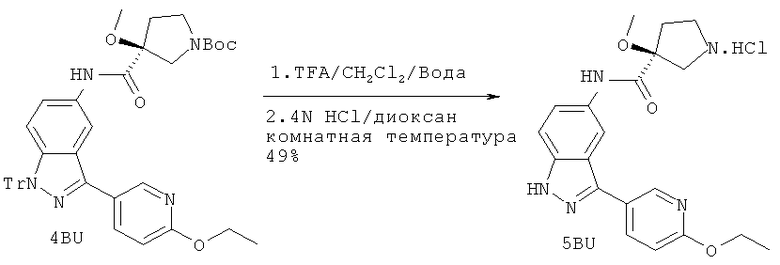
Соединение 4BU (1.6 г) перемешивали в смеси дихлорметана (30 мл), трифторуксусной кислоты (4 мл) и нескольких капелл воды, при комнатной температуре, всю ночь. Смесь охладили при 0°С и аккуратно погасили 7% МеОН (NH3)/CH2Cl2. Растворители удалили в вакууме. Разбавили водой (100 мл) и этилацетатом (200 мл). Слои разделили. Органический слой высушили (Na2SO4), отфильтровали, и растворители удалили в вакууме. Очистка на колонке [7% МеОН (7Н аммиак) в дихлорметане] позволила получить соединение 5BU (0.44 г, 49%), которое превратили в моносоль HCl, посредством обработки 4Н HCl/диоксан и выпаривания раствора до сухости.
Стадия 5:
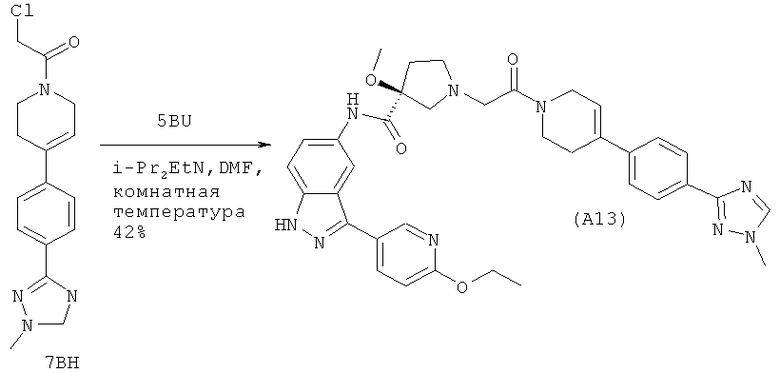
Смесь соединения 7ВН (46 мг, 0.144 ммоль), соединения 5BU (60 мг, 0.144 ммоль), DMF (2 мл) и N,N-диизопропилэтиламина (0.076 мл, 0.432 ммоль) перемешивали при комнатной температуре в течение 18 часов. Разбавили EtOAc (100 мл) и промыли водой (2×100 мл). Органический слой высушили над Na2SO4, отфильтровали и концентрировали. Осадок очистили на силикагеле, элюируя с применением 4% МеОН (NH3)/CH2Cl2, с получением целевого продукта 6BU (40 мг, 42%).
ЖХМС М+1=662 при времени удерживания 2.57 мин
Пример 12
Синтез 3-метилсульфанил-1-(2-{4-[4-(1-метил-1Н-[1,2,4]триазол-3-ил)-фенил]-3,6-дигидро-2Н-пиридин-1-ил}-2-оксо-этил)-пирролидин-3-карбоновой кислоты [3-(6-метокси-пиридин-3-ил)-1Н-индазол-5-ил]-амида
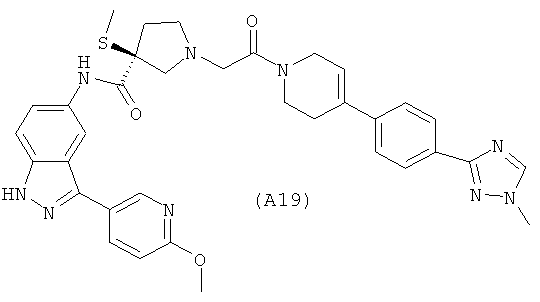
Стадия 1:
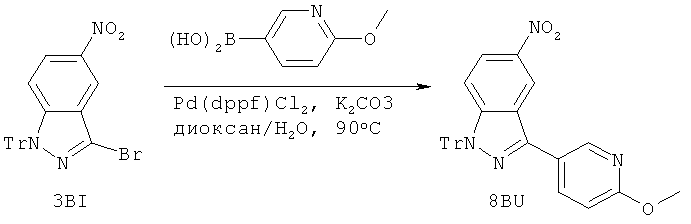
Соединение 8BU получили из соединения 3В1, применяя, по существу, туже самую методику, как описано для получения соединения 2BU из соединения 3В1 (Пример 11, Стадия 1), применяя 6-метоксипиридин-3-бороновую кислоту вместо 6-этоксипиридин-3-бороновой кислоты.
Стадия 2:
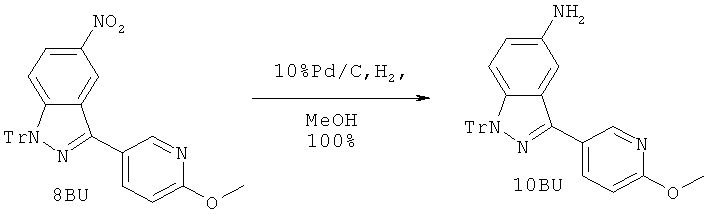
Соединение 8BU (3 г, 3.8 ммоль) и Pd/C (10%, 50 мас.%, 1.2 г) перемешивали в толуоле (30 мл) и МеОН (15 мл) в атмосфере H2 (резиновая камера), при комнатной температуре, в течение 18 часов. Твердые кристаллы отфильтровали через целит, и растворители удалили в вакууме, с получением соединения 10BU (2.8 г, 100%).
Стадия 3:
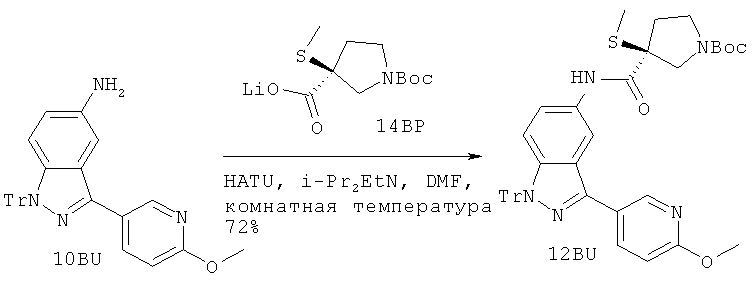
Соединение 10BU (0.6 г, 1.24 ммоль) и пирролидинкарбоновую кислоту 14ВР (0.33 г, 1.24 ммоль) растворили в DMF (5 мл) при комнатной температуре. Добавили HATU (0.71 г, 1.86 ммоль), и затем добавили iPr2NEt (00.66 мл, 3.72 ммоль). Смесь перемешивали при комнатной температуре всю ночь и разбавили этилацетатом (200 мл) и водой (100 мл). Слои разделили. Отделенный органический слой промыли водой (100 мл), высушили (Na2SO4) и отфильтровали. Концентрирование в вакууме с последующей очисткой на колонке [гексаны-этилацетат = 9:1 (об/об)] позволили получить продукт 12BU (0.65 г, 72%).
Стадия 4:
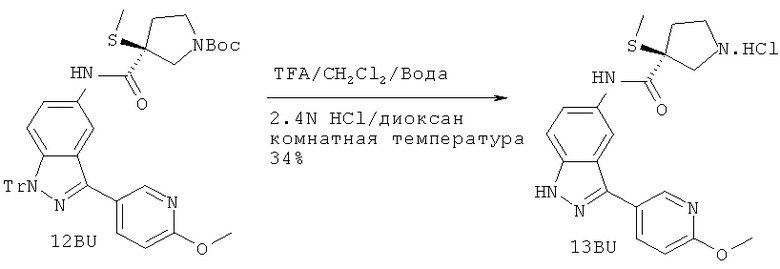
Соединение 12BU (0.65 г) перемешивали в смеси дихлорметана (30 мл), трифторуксусной кислоты (4 мл) и нескольких капель воды при комнатной температуре всю ночь. Смесь охладили при 0°С и аккуратно погасили, применяя 7% МеОН (NH3)/CH2Cl2. Растворители удалили в вакууме. Разбавили водой (100 мл) и этилацетатом (200 мл). Слои разделили. Органический слой высушили (Na2SO4), отфильтровали, и растворители удалили в вакууме. Очистка на колонке [15% МеОН (7Н аммиак) в дихлорметане] позволила получить соединение 13BU (0.22 г, 34%).
Стадия 5:
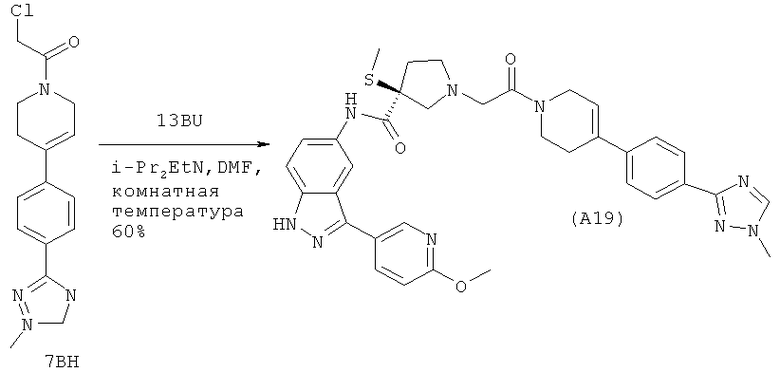
Смесь соединения 7ВН (40 мг, 0.126 ммоль), соединения 13BU (50 мг, 0.126 ммоль), DMF (2 мл) и N,N-диизопропилэтиламина (0.045 мл, 0.25 ммоль) перемешивали при комнатной температуре в течение 18 часов. Разбавили EtOAc (100 мл) и промыли водой (2×100 мл). Органический слой высушили над Na2SO4, отфильтровали и концентрировали. Осадок очистили на силикагеле, элюируя 3% МеОН (NH3)/CH2Cl2, с получением целевого продукта А19 (50 мг, 60%).
ЖХМС М+1=664 при времени удерживания 2.88 мин
Пример 13
Синтез 3-метокси-1-(2-{4-[4-(1-метил-1Н-[1,2,4]триазол-3-ил)-фенил]-3,6-дигидро-2Н-пиридин-1-ил}-2-оксо-этил)-пирролидин-3-карбоновой кислоты [3-(2-метокси-пиридин-4-ил)-1Н-индазол-5-ил]-амида
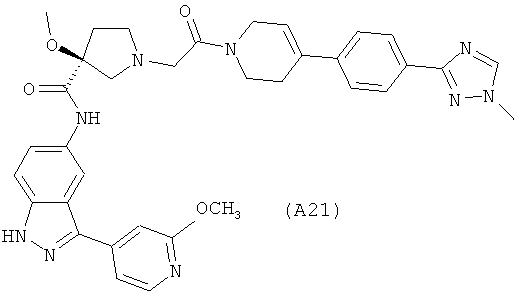
Стадия 1:
Синтез 3-(2-фтор-пиридин-4-ил)-5-нитро-1-тритил-1Н-индазола
3-Бром-5-нитро-1-тритил-1Н-индазол 3BI (15.64 г, 32.3 ммоль), 2-фтор-4-пиридин-бороновую кислоту 2BV (5.0 г, 35.5 ммоль), K3PO4 (17.1 г, 80.7 ммоль) и Pd(dppf)Cl2 (2.64 г, 3.23 ммоль) смешали в диоксане/Н2О (240 мл/60 мл) при комнатной температуре и нагревали при 80°С всю ночь. Реакционную смесь охладили до комнатной температуре и концентрировали до небольшого объема. Остаток распределили между этилацетатом (200 мл) и соляным раствором (150 мл). Органический слой промыли соляным раствором, высушили (MgSO4) и отфильтровали. Полученный фильтрат концентрировали, и осадок очистили на силикагелевой колонке, последовательно элюируя гексанами, 5% этилацетата в гексанах, 10% этилацетата в гексанах, с получением твердого вещества желтого цвета 3BV (3.33 г, 64%).
Стадия 2:
Получение 3-(2-метокси-пиридин-4-ил)-5-нитро-1-тритил-1Н-индазола

В сосуде высокого давления, объемом 250 мл, 3-(2-фтор-пиридин-4-ил)-5-нитро-1-тритил-1Н-индазол 3BV (5.005 г, 10.0 ммоль) растворили в 0.1Н KOCH3/МеОН/Толуол (Acros, 150 мл, 15.0 ммоль) в атмосфере сухого газа N2. Сосуд высокого давления запаяли и нагревали при перемешивании, при 70°С, в течение 24 часов. Сосуд высокого давлением охладили до 0°С на ледяной бане, прежде чем открыть. Содержимое сосуда высокого давления перенесли в круглодонную колбу, объемом 500 мл, и выпарили до сухости. Полученное твердое вещество растворили в CH2Cl2 и промыли насыщенным раствором NaCl и высушили над MgSO4. Растворитель выпарили до сухости и высушили под высоким вакуумом с получением соединения 4BV в виде твердого вещества коричневого цвета (5.12 г, 100%).
Стадия 3:
Получение 3-(2-метокси-пиридин-4-ил)-1-тритил-1Н-индазол-5-иламина
К перемешанной суспензии 3-(2-метокси-пиридин-4-ил)-5-нитро-1-тритил-1H-индазола 4BV (5.125 г, 10.0 ммоль) в МеОН/толуоле (50 мл/50 мл) добавили 10% Pd/C (типа Degussa, 0.5 г) при комнатной температуре в атмосфере сухого газа N2. Смесь дегазировали и перемешивали в резиновой камере, наполненной газом Н2, всю ночь. Катализатор отфильтровали через микроволокнистый фильтр и промыли МеОН и CH2Cl2. Фильтрат выпарили до сухости, и получили твердое вещество коричневого цвета 5BV (5.0 г).
Стадия 4
Получение 3-метокси-3-[3-(2-метокси-пиридин-4-ил)-1-тритил-1Н-индазол-5-илкарбамоил]-пирролидин-1-карбоновой кислоты трет-бутилового сложного эфира
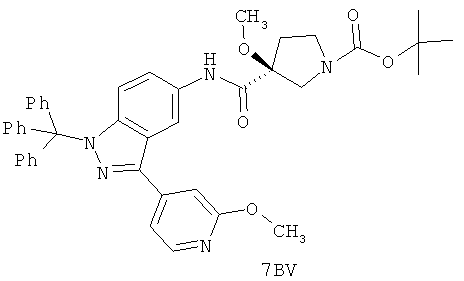
К перемешиваемой суспензии 3-метокси-пирролидин-1,3-дикарбоновой кислоты 1-трет-бутилового сложного эфира 6BV (4.16 ммоль, неочищенный) в DMF/DCM (25 мл/25 мл) добавили 3-(2-метокси-пиридин-4-ил)-1-тритил-1Н-индазол-5-иламин 5BV (2.008 г, 4.16 ммоль) и Et3N (2.9 мл, 21 ммоль) при комнатной температуре в атмосфере сухого газа N2, а затем добавили HATU (3.16 г, 8.32 ммоль). Смесь перемешивали при комнатной температуре в атмосфере сухого газа N2 всю ночь. Смесь распределил между 1:1 EtOAc и насыщенным раствором NaHCO3. Органическую фазу отделили, промыли соляным раствором, высушили над MgSO4 и выпарили до сухости. Неочищенное соединение в виде твердого вещества очистили на RediSep 120 г картридже, элюируя 10-25% EtOAc/Гексаны, с получением твердого вещества коричневого цвета 7BV (2.95 г, 100%).
Стадия 5
Получение 3-метокси-пирролидин-3-карбоновой кислоты [3-(2-метокси-пиридин-4-ил)-1Н-индазол-5-ил]-амида
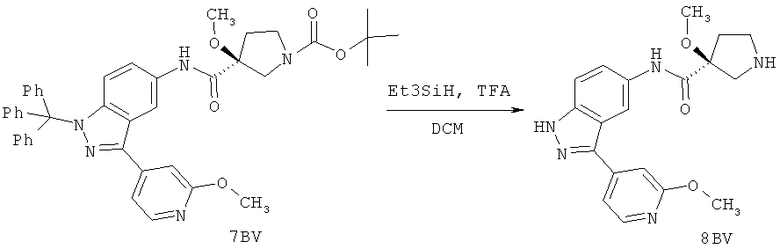
К перемешанному раствору 3-метокси-3-[3-(2-метокси-пиридин-4-ил)-1-тритил-1Н-индазол-5-илкарбамоил]-пирролидин-1-карбоновой кислоты трет-бутилового сложного эфира 7BV (2.95 г, 4.16 ммоль) в DCM (50 мл) при комнатной температуре добавили триэтилсилан (1.00 мл, 6.24 ммоль), а затем TFA (10 мл). Смесь перемешивали при комнатной температуре в атмосфере сухого газа N2 в течение 4-5 часов. Смесь выпарили до сухости и совместно выпарили с сухим толуолом (2×75 мл). Полученное неочищенное твердое вещество очистили на RediSep 120 г картридже, элюируя 2.5%-6% 2М NH3-МеОН/CH2Cl2, с получением твердого вещества грязно-белого цвета 8BV (1.23 г, 80%).
Стадия 6
Синтез 3-метокси-1-(2-{4-[4-(1-метил-1Н-[1,2,4]триазол-3-ил)-фенил]-3,6-дигидро-2Н-пиридин-1-ил}-2-оксо-этил)-пирролидин-3-карбоновой кислоты [3-(2-метокси-пиридин-4-ил)-1Н-индазол-5-ил]-амида
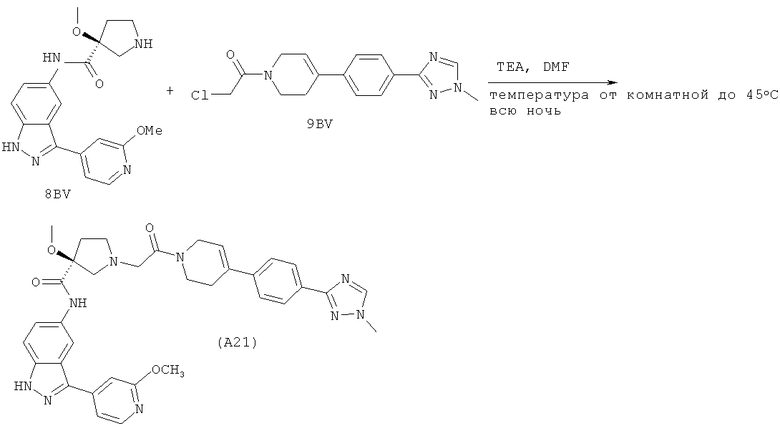
Смесь соединения 8BV (40 мг, 0.11 ммоль), соединения 9BV (41.6 мг, 0.13 ммоль) и триэтиламина (0.1 мл) в DMF (3 мл) нагревали при 45°С, при перемешивании, всю ночь. Реакционную смесь затем концентрировали в вакууме, и полученное неочищенное вещество очистили на силикагелевой колонке, элюируя 2% и 4% 2Н NH3/МеОН в CH2Cl2, с выделением твердого вещества желтого цвета А21 (39.6 мг, 55%). (ЖХМС М+1=648, время удерживания = 2.34 минут) 1H-ЯМР (400 МГц, CDCl3): δ 10.54 (уш, 1Н), 9.51 & 9.44 (с, с, 1Н), 8.46 (дд, 1Н, J=8.2 Гц & 1.4 Гц), 8.27 (д, 1Н, J=5.3 Гц), 8.07 (с, 1Н), 8.05 (м, 2Н), 7.76 (м, 1Н), 7.54 (м, 1Н), 7.44 (м, 3Н), 7.37 (уш, 1Н), 6.20 & 6.11 (т, т, 1Н, J=2.5 Гц), 4.27 (м, 2Н), 4.00 & 3.98 (с, с, 6Н), 3.96 & 3.86 (м, м, 1Н), 3.74 (м, 1Н), 3.47-3.60 (м, 2Н), 3.453 & 3.446 (с, с, 3Н), 3.32 (д, 1Н, 9.9 Гц), 3.02 (м, 1Н), 2.97 (дд, 1Н, J=10.1 Гц & 1.0 Гц), 2.91 (м, 1Н), 2.64 (м, 2Н), 2.47 (м, 1Н), 2.19 (м, 1Н).
Пример 14
Синтез 3-метилсульфанил-1-(2-{4-[4-(1-метил-1Н-[1,2,4]триазол-3-ил)-фенил]-3,6-дигидро-2Н-пиридин-1-ил}-2-оксо-этил)-пирролидин-3-карбоновой кислоты [3-(2-этокси-пиридин-4-ил)-1Н-индазол-5-ил]-амида
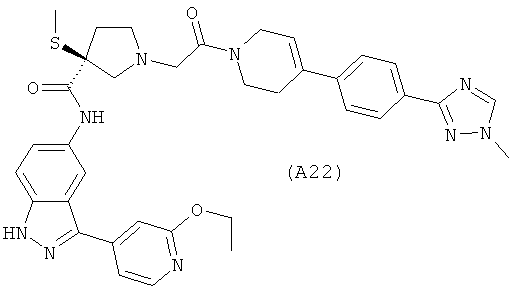
Стадия 1:
Получение 3-(2-Этокси-пиридин-4-ил)-5-нитро-1-тритил-1Н-индазола
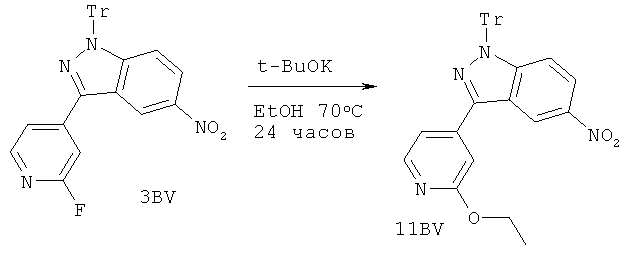
В сосуде высокого давления, объемом 150 мл, к перемешанному раствору 3-(2-фтор-пиридин-4-ил)-5-нитро-1-тритил-1Н-индазола 3BV (2.0 г, 4.0 ммоль) в безводном EtOH (40 мл) добавили твердый трет-бутоксид калия (12 г, 10.0 ммоль) в атмосфере сухого газа N2. Сосуд высокого давления плотно запаяли и нагревали при 80°С в течение 24 часов. Сосуд высокого давления охладили до 0°С на ледяной бане, прежде чем открыть. Содержимое сосуда высокого давления перенесли в круглодонную колбу, объемом 250 мл, и концентрировали до небольшого объема. Полученную смесь распределили между EtOAc и H2O. Органическую фазу отделили, промыли насыщенным раствором NaCl и высушили над MgSO4. Растворитель выпарили до сухости, с получением неочищенного твердого вещества 11BV (1.5 г, 71%).
Стадия 2:
Получение 3-(2-этокси-пиридин-4-ил)-1-тритил-1Н-индазол-5-иламина
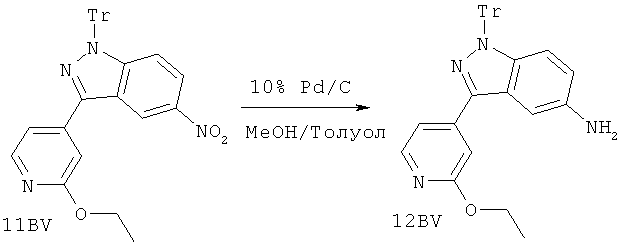
Соединение 12BV получили, применяя, по существу, туже самую методику как в примере 13, Стадия 3, за исключением применения 11BV вместо 4BV.
Стадия 3:
Получение 3-[3-(2-этокси-пиридин-4-ил)-1-тритил-1Н-индазол-5-илкарбамоил]-3-метилсульфанил-пирролидин-1-карбоновой кислоты трет-бутилового сложного эфира
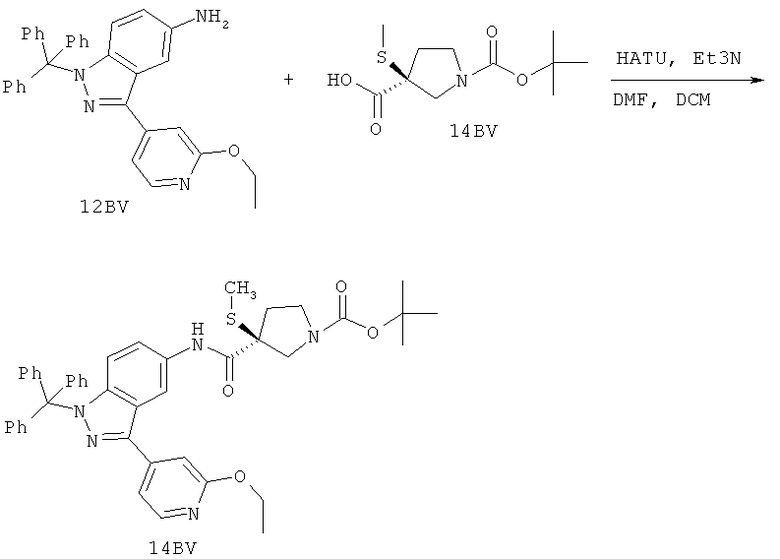
Соединение 14BV, с выходом 88%, получили, применяя, по существу, туже самую методику как в примере 13, Стадия 4, за исключением применения 12BV и 14ВР вместо 5BV и 6BV.
Стадия 4:
Получение 3-метилсульфанил-пирролидин-3-карбоновой кислоты [3-(2-этокси-пиридин-4-ил)-1Н-индазол-5-ил]-амида
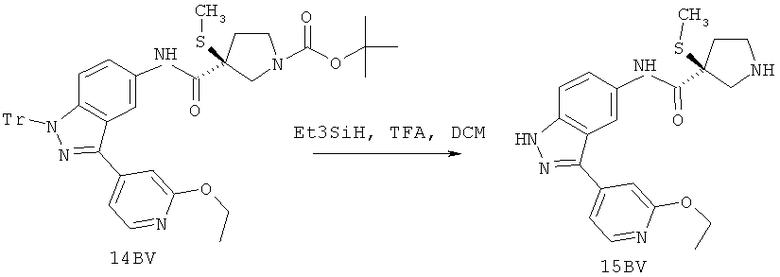
Соединение 15BV, с выходом 80%, получили, применяя, по существу, туже самую методику как в примере 13, Стадия 5, за исключением применения 14BV вместо 7BV.
Стадия 5:
Получение 3-метилсульфанил-1-(2-{4-[4-(1-метил-1Н-[1,2,4]триазол-3-ил)-фенил]-3,6-дигидро-2Н-пиридин-1-ил}-2-оксо-этил)-пирролидин-3-карбоновой кислоты [3-(2-этокси-пиридин-4-ил)-1Н-индазол-5-ил]-амида
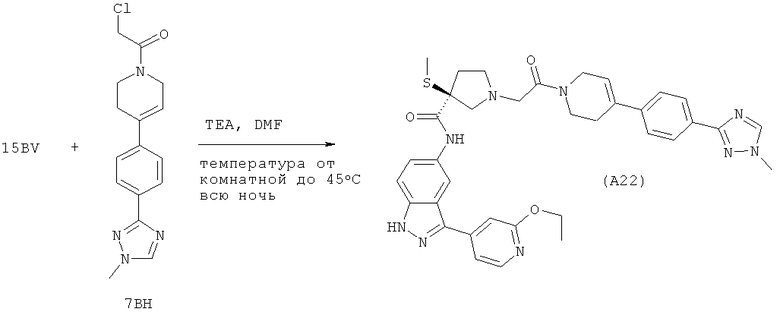
Смесь соединения 15BV (40 мг, 0.10 ммоль), соединения 7 ВН (41.6 мг, 0.13 ммоль) и триэтиламина (0.1 мл) в DMF (3 мл) нагревали при 45°С, при перемешивании, всю ночь. Реакционную смесь затем концентрировали в вакууме, и полученное неочищенное вещество очистили на силикагелевой колонке, элюируя 2% и 4% 2Н NH3/МеОН в CH2Cl2 с выделением твердого вещества желтого цвета А22 (36.0 мг, 53%). (ЖХМС М+1=678, время удерживания = 2.41 минут). 1H-ЯМР (400 МГц), CDCl3): δ 10.55 (уш, 1Н), 10.01 & 9.84 (с, с, 1Н), 8.50 & 8.44 (д, д, 1Н, J=1 Гц), 8.25 (д, 1Н, J=5.2 Гц), 8.08 (с, 1Н), 8.04 (д, 1Н, J=8.2 Гц), 8.00 (д, 1Н, J=8.2 Гц), 7.75 (м, 1Н), 7.53 (м, 1Н), 7.43 (д, 1Н, J=8.2 Гц), 7.39 (м, 2Н), 7.34 (д, 1Н, J=5.2 Гц), 6.18 &6.11 (т, т, 1Н, J=2.5 Гц), 4.42 (кв, 2Н, J=7.0 Гц), 4.35 & 4.30 (м, м, 1Н), 4.22 (м, 1Н), 3.99 (с, 3Н), 3.92 (т, 1Н, J=5.6 Гц), 3.72 (м, 3Н), 3.42 (м, 1Н), 3.12 (м, 1Н), 2.84 (кв, 1Н, J=7.9 Гц), 2.76 (д, 1Н, J=9.9 Гц), 2.70 (м, 1Н), 2.63 (м, 2Н), 2.15 (с, 3Н), 2.11 (м, 1Н), 1.44(т, 3Н, J=7.0 Гц).
Пример 15
Синтез 3-метокси-1-(2-{4-[4-(1-метил-1Н-[1,2,4]триазол-3-ил)-фенил]-3,6-дигидро-2Н-пиридин-1-ил)-2-оксо-этил)-пирролидин-3-карбоновой кислоты [3-(2-изопропокси-пиридин-4-ил)-1Н-индазол-5-ил]-амида
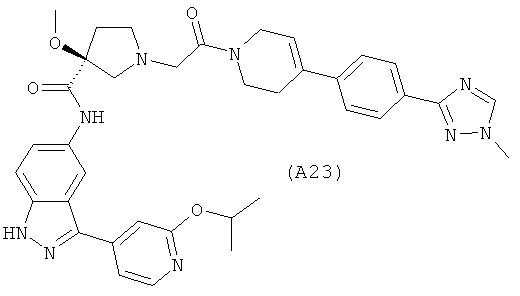
Стадия 1:
Получение 4-бром-2-изопропокси-пиридина
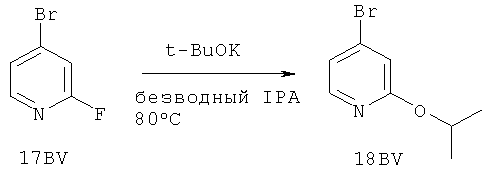
К перемешанному раствору 4-бром-2-фтор-пиридина 17BV (4.12 г, 23.41 ммоль) в 50 мл безводном IPA, в сосуде высокого давления, объемом 150 мл, добавили 2.627 г (23.41 ммоль) твердого трет-бутоксида калия в атмосфере сухого газа N2. Сосуд высокого давления плотно запаяли и нагревали при 80°С в течение 3 часов. Сосуд высокого давления охладили до 0°С на ледяной бане, прежде чем открыть. Содержимое сосуда высокого давления перенесли в круглодонную колбу, объемом 250 мл, и концентрировали до небольшого объема. Полученную смесь распределили между EtOAc и H2O. Органическую фазу отделили, промыли насыщенным раствором NaCl и высушили над MgSO4. Растворитель выпарили, и полученное прозрачное масло очистили на RediSep 80 г картридже, элюируя 20:1 Гексаны/EtOAc, с получением прозрачного масла 18BV (4.2 г, 83%).
Стадия 2
Получение 3-(2-изопропокси-пиридин-4-ил)-5-нитро-1-тритил-1Н-индазола
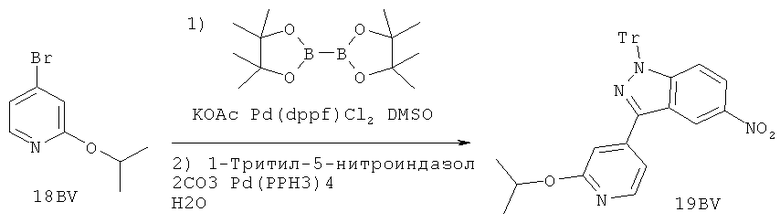
К перемешанному раствору 4.20 г (19.4 ммоль) 4-бром-2-изопропокси-пиридина 18BV в 150 мл безводного DMSO, 7.39 г (29.1 ммоль) (пинаколато)дибора, добавили 5.704 г (58.2 ммоль) ацетата калия и 1.584 г (1.94 ммоль) Pd(dppf)Cl2, при комнатной температуре, в атмосфере сухого газа N2. Смесь дегазировали пару раз сухим газом N2. Смесь темно-оранжевого цвета нагревали при 100°С в течение 2 часов. Смесь темного цвета охладили до комнатной температуры и добавили 75 мл H2O, а затем добавили 9.396 г (19.4 ммоль) 1-тритил-5-нитроиндазола, 13.401 г (96.96 ммоль) карбоната калия и 2.246 г (1.94 ммоль) Тетракис(Трифенилфосфина)палладия, при комнатной температуре, в атмосфере сухого газа N2. Смесь дегазировали пару раз сухим газом N2. Смесь темного цвета нагревали при 100°С всю ночь. Смесь охладили до комнатной температуры и разбавили смесью H2O/EtOAc 1:1. Разбавленную смесь отфильтровали через целит, и целит промыли EtOAc. Содержимое перенесли в делительную воронку и хорошо взболтали. Органическую фазу отделили и пару раз промыли насыщенным раствором NaCl, высушили над MgSO4 и выпарили до сухости. Смолу, окрашенную в темный цвет, очистили на RediSep 330 г картридже, элюируя Гексанами, 5% EtOAc/Гексан и 10% EtOAc/Гексан, что позволило получить 3.24 г (31%) твердого вещества 19BV бледно-желтого цвета.
Стадия 3
Получение 3-(2-изопропокси-пиридин-4-ил)-1-тритил-1Н-индазол-5-иламина
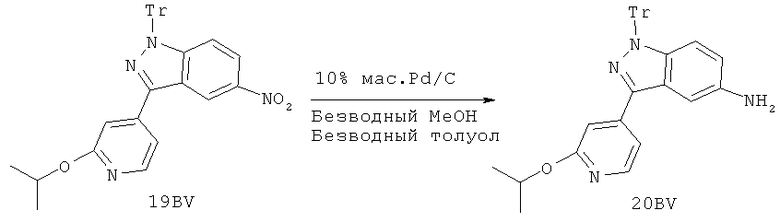
Соединение 20BV, с выходом неочищенного вещества 100%, получили, применяя, по существу, туже самую методику как в примере 13, Стадия 3, за исключением применения 19BV вместо 4BV.
Стадия 4:
Получение 3-[3-(2-изопропокси-пиридин-4-ил)-1-тритил-1Н-индазол-5-илкарбамоил]-3-метокси-пирролидин-1-карбоновой кислоты трет-бутилового сложного эфира
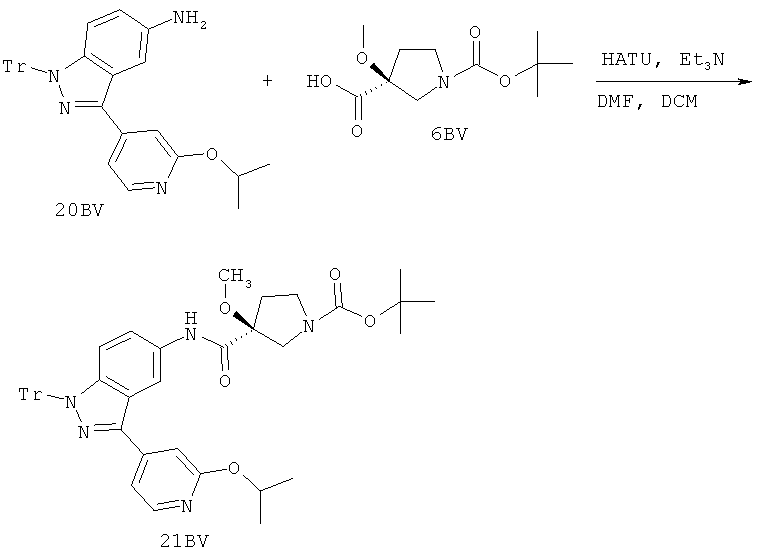
Соединение 21BV получили, применяя, по существу, туже самую методику как в примере 13, Стадия 4, за исключением применения 20BV вместо 5BV.
Стадия 5:
Получение 3-метокси-пирролидин-3-карбоновой кислоты [3-(2-изопропокси-пиридин-4-ил)-1Н-индазол-5-ил]-амида
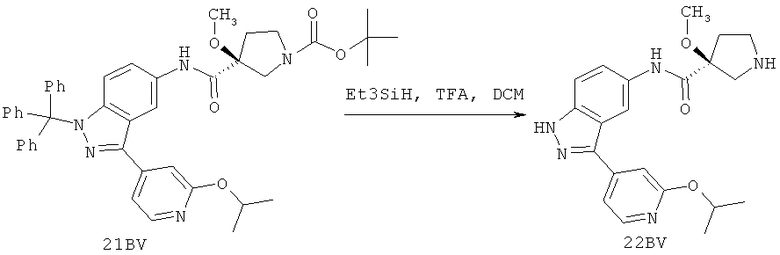
Соединение 22BV получили, применяя, по существу, туже самую методику как в примере 13, Стадия 5, за исключением применения 21BV вместо 7BV.
Стадия 6:
Получение 3-метокси-1-(2-{4-[4-(1-метил-1Н-[1,2,4]триазол-3-ил)-фенил]-3,6-дигидро-2Н-пиридин-1-ил}-2-оксо-этил)-пирролидин-3-карбоновой кислоты [3-(2-изопропокси-пиридин-4-ил)-1Н-индазол-5-ил]-амида
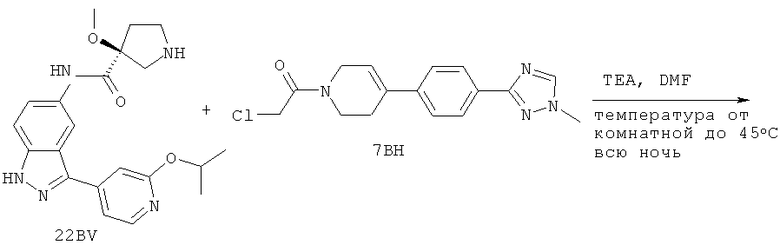
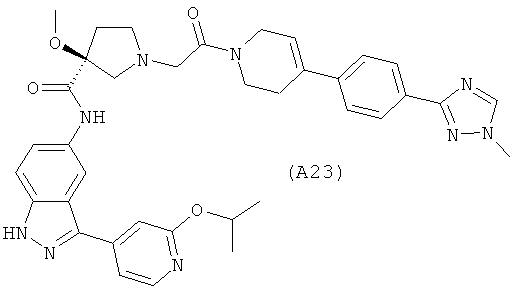
Смесь соединения 22BV (40 мг, 0.10 ммоль), соединения 7 ВН (41.6 мг, 0.13 ммоль) и триэтиламина (0.1 мл) в DMF (3 мл) нагревали при 45°С, при перемешивании, всю ночь. Реакционную смесь затем концентрировали в вакууме, и полученное неочищенное вещество очистили на силикагелевой колонке, элюируя 2% и 4% 2Н NH3/МеОН в CH3Cl2, с выделением твердого вещества желтого цвета 23BV (35.0 мг, 52%). ЖХМС М+1=676, время удерживания = 2.45 минут 1H-ЯМР (400 МГц, CDCl3): δ 10.42 (уш, 1Н), 9.41 & 9.34 (с, с, 1Н), 8.43 (дд, 1Н, J=10.7 Гц & 1.2 Гц), 8.25 (д, 1Н, J=5.2 Гц), 8.07 (с, 1Н), 8.05 (м, 2Н), 7.74 (м, 1Н), 7.49 (м, 1Н), 7.44 (м, 3Н), 7.29 (уш, 1Н), 6.20 & 6.11 (т, т, 1Н, J=2.5 Гц), 5.37 (м, 1Н), 4.27 (м, 2Н), 3.98 (с, 3Н), 3.80-3.98 (м, 1Н), 3.74 (м, 1Н), 3.53 (д, 1Н, J=6.6 Гц), 3.49 (уш, 1Н), 3.47 & 3.46 (с, с, 3Н), 3.28 (д, 1Н, J=10 Гц), 3.02 (м, 2Н), 2.88 (м, 1Н), 2.64 (м, 2Н), 2.47 (м, 1Н), 2.19 (м, 1Н), 1.40 (д, 6Н, J=6.1 Гц).
Пример 16
Синтез 3-метилсульфанил-1-(2-{4-[5-(1-метил-1Н-[1,2,4]триазол-3-ил)-тиофен-2-ил]-3,6-дигидро-2Н-пиридин-1-ил}-2-оксо-этил)-пирролидин-3-карбоновой кислоты [3-(6-изопропокси-пиридин-3-ил)-1Н-индазол-5-ил]-амида
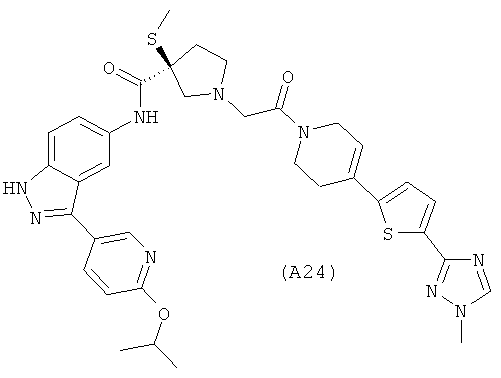
Синтез 2-хлор-1-{4-[5-(1-метил-1Н-[1,2,4]триазол-3-ил)-тиофен-2-ил]-3,6-дигидро-2Н-пиридин-1-ил)-этанона
Стадия 1:
Получение 5-бром-тиофен-2-карбоновой кислоты амида
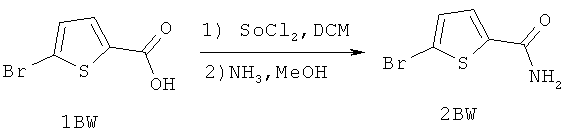
5-Бром-тиофен-2-карбоновую кислоту (1BW) (6 г) суспендировали в дихлорметане (30 мл) и тионилхлориде (30 мл). Полученный раствор кипятили с обратным холодильником всю ночь при 80°С. Растворитель удалили при пониженном давлении, и осадок растворили в дихлорметане, добавили по каплям к охлажденному раствору аммиака в метаноле (100 мл), реакцию контролировали с помощью ТСХ и ЖХМС. Растворитель удалили при пониженном давлении, с получением 5-бром-тиофен-2-карбоновой кислоты амида (2BW).
Стадия 2:
Получение 5-бром-тиофен-2-карбоксимидовой кислоты сложного этилового эфира
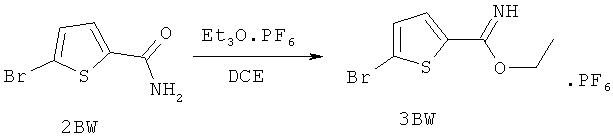
5-Бромтиофен-2-карбоновой кислоты амид (2BW) (7 г, 34.1 ммоль) растворили в дихлорэтане (170 мл). Добавили триэтилоксония гексафторфосфат (10.16 г, 40.97 ммоль), и полученную смесь кипятили с обратным холодильником в течение одного часа при 90°С. Реакционную смесь концентрировали при пониженном давлении и применяли непосредственно на следующей стадии.
Стадия 3:
Получение 5-бром-тиофен-2-карбоксимид-N′-метил-гидразид
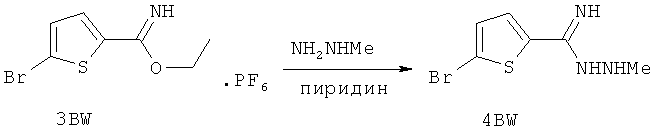
5-Бром-тиофен-2-карбоксимидовой кислоты сложный этиловый эфир (3BW) (13 г, 34.3 ммоль) растворили в пиридине (100 мл). Метилгидразин (2.7, 51.45 ммоль) добавили при перемешивании, и полученную смесь перемешивали всю ночь. Реакционную смесь концентрировали при пониженном давлении, и добавили диэтиловый эфир, отфильтровали, трижды промыли диэтиловым эфиром и высушили с получением соединения, название которого указано в заглавии (4BW) (13 г).
Стадия 4:
Получение 3-(5-бром-тиофен-2-ил)-1-метил-1H-[1,2,4]триазола
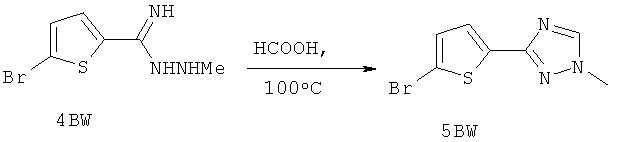
Смесь соединения 5-бром-тиофен-2-карбоксимид-N′-метил-гидразида (4BW) (13 г) в муравьиной кислоте (100 мл) кипятили с обратным холодильником всю ночь и концентрировали. Осадок обработали насыщенным NaHCO3, и трижды экстрагировали EtOAc. Объединенные органические слои высушили над MgSO4. После концентрирования, соединение 5BW очистили на колонке, применяя 80% EtoAc/гексан, с получением твердого вещества, окрашенного в светло-желтый цвет.
Стадия 5:
Получение 4-[5-(1-метил-1Н-(1,2,4]триазол-3-ил)-тиофен-2-ил]-3,6-дигидро-2H-пиридин-1-карбоновой кислоты трет-бутилового сложного эфира
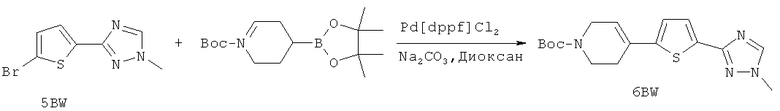
В большую колбу высокого давления загрузили 3-(5-бром-тиофен-2-ил)-1-метил-1Н-[1,2,4]триазол (3.1 г, 12.8 ммоль) (5BW), 4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-3,6-дигидро-2H-пиридин-1-карбоновой кислоты трет-бутиловый сложный эфир (3.6 г, 11.65 ммоль), [1,1′-бис(дифенилфосфино)-ферроцена]дихлорпалладия (II) комплекс с дихлорметаном (1:1) (0.475 г, 0.89 ммоль), Na2CO3 (8.5 мл) и диоксан (40 мл). Смесь быстро дегазировали аргоном в течение около полуминуты, накрыли и перемешивали при 80°С всю ночь. После охлаждения, реакционную смесь разбавили EtOAc и соляным раствором. Органический слой отделили и высушили MgSO4. После концентрирования осадок очистили на силикагеле. Элюция с применением EtOAc (100%) позволила получить целевой продукт (6BW) (3.5 г).
Стадия 6:
Получение 4-[5-(1-метил-1Н-[1,2,4]триазол-3-ил)-тиофен-2-ил]-1,2,3,6-тетрагидропиридина гидрохлорида
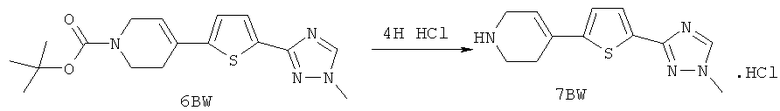
ВОС группу на 6BW можно удалить путем обработки соединения 4-[5-(1-метил-1Н-[1,2,4]триазол-3-ил)-тиофен-2-ил]-3,6-дигидро-2Н-пиридин-1-карбоновой кислоты трет-бутилового сложного эфира с 4Н HCl в диоксане, при комнатной температуре, в течение двух часов. Удаление растворителя под вакуумом с последующей промывкой диэтиловым эфиром позволили получить целевое соединение (7BW).
Стадия 7:
Получение 2-хлор-1-{4-[5-(1-метил-1H-[1,2,4]триазол-3-ил)-тиофен-2-ил]-3,6-дигидро-2Н-пиридин-1-ил}-этанона
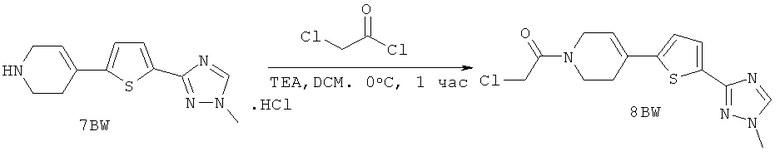
К охлажденному (0°С) раствору 4-[5-(1-метил-1Н-[1,2,4]триазол-3-ил)-тиофен-2-ил]-1,2,3,6-тетрагидропиридина (7BW) (1.5 г, 4.72 ммоль) в дихлорметане (50 мл) по каплям добавили TEA (4.5 мл, 28.32 ммоль). После перемешивания при 0°С в течение 10 минут к реакционной смеси добавили хлорацетилхлорид (1.12 мл, 14.2 ммоль). Полученную смесь перемешивали при 0°С в течение одного часа и погасили водой (15.6 мл). Реакционную смесь разбавили дихлорметаном (200 мл). Органический слой отделили и промыли соляным раствором, высушили над MgSO4. Реакционную смесь концентрировали до около 50 мл, добавили диэтиловый эфир, и твердое вещество отфильтровали с получением целевого продукта (8BW).
Стадия 8:
Синтез 3-метокси-1-(2-{4-[4-(1-метил-1Н-[1,2,4]триазол-3-ил)-фенил]-3,6-дигидро-2Н-пиридин-1-ил}-2-оксо-этил)-пирролидин-3-карбоновой кислоты [3-(6-изопропокси-пиридин-3-ил)-1Н-индазол-5-ил]-амида
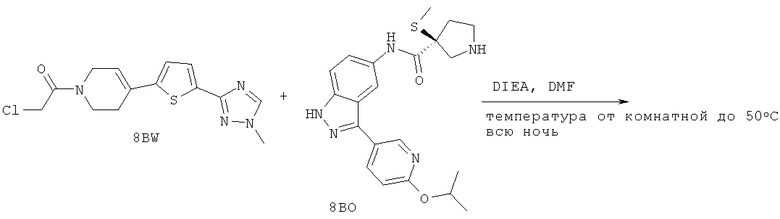
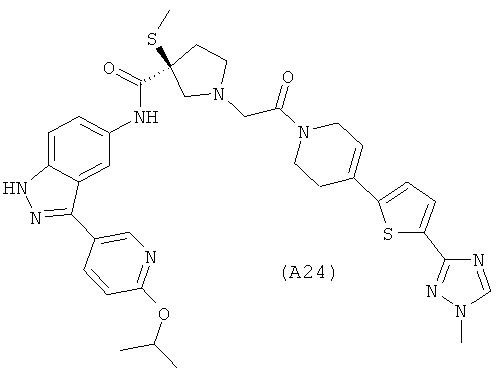
Смесь 2-хлор-1-{4-[5-(1-метил-1Н-[1,2,4]триазол-3-ил)-тиофен-2-ил]-3,6-дигидро-2Н-пиридин-1-ил}-этанона (8BW) (0.522 г, 0.863 ммоль), 3-метилсульфанил-пирролидин-3-карбоновой кислоты [3-(6-изопропокси-пиридин-3-ил)-1Н-индазол-5-ил]-амида (8ВО) (0.463 г, 0.95 ммоль),и DIEA (0.9 мл, 5.2 ммоль) в DMF (10 мл) перемешивали при комнатной температуре всю ночь. DMF удалили при пониженном давлении. Неочищенную реакционную смесь осадили в 30 мл ледяной воды, отфильтровали, высушили и очистили с помощью колоночной хроматографии, применяя 10% МеОН/EtOAc, с получением целевого продукта А24 в виде твердого вещества желтого цвета (70%). (ЖХМС М+1=698, время удерживания = 3.6 минут) 1Н ЯМР (400 МГц, DMSO): δ 8.67 (с, 1Н), 8.64 (с, 1Н), 8.36 (с, 1Н), 8.15 (с, 1Н), 7.64 (д, 1Н, J=8.8 Гц), 7.52 (с, 1Н), 7.49 (д, 1Н J=5.2 Гц), 7.40(д, 1Н J=8.8 Гц), 7.02 (м, 1Н), 6.18 (д, 1Н, J=18.8 Гц), 5.08 (м, 1Н), 4.41 (д, 2Н, J=7.6 Гц), 4.34 (д, 1Н, J=6.4 Гц)), 4.12(с, 1Н), 3.96 (с, 2Н), 3.84 (с, 3Н), 3.76 (с, 2Н), 3.52(с, 2Н), 2.76 (с, 1Н), 2.58 (д, 2Н J=20.4 Гц), 2.13 (с, 3Н), 1.94 (т, 2Н J=2.8 Гц), 1.39 (д, 6Н, J=4.8 Гц).
Примеры 17-25
Соединения А4, А6, А8, А10-А12, А18, А25 и А26 получили, следуя методикам, указанным в Таблице 1 ниже.
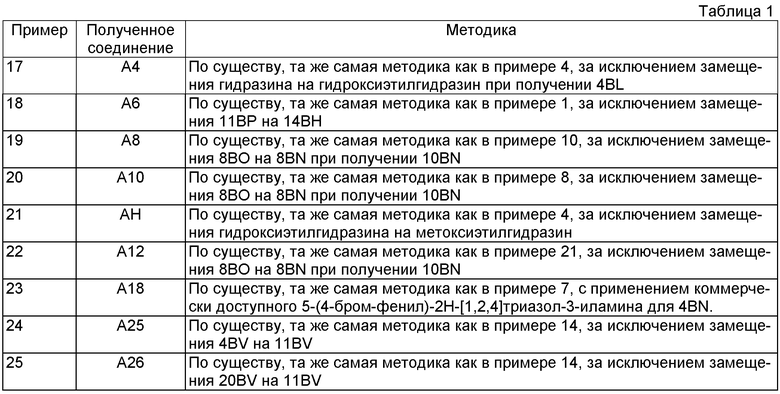
ЖХМС данные для соединений, приведенных в Таблице 1, приведены в Таблице 2 ниже.
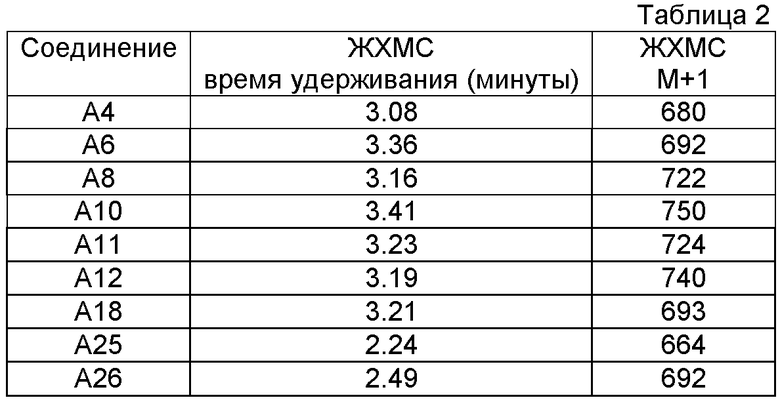
ПРИМЕР 26
Получение (S)-N-(3-(6-изопропоксипиридин-3-ил)-1Н-индазол-5-ил)-3-метокси-1-(2-(4-(5-(1-метил-1Н-1,2,4-триазол-3-ил)пиридин-2-ил)-5,6-дигидропиридин-1(2Н)-ил)-2-оксоэтил)пирролидин-3-карбоксамида:
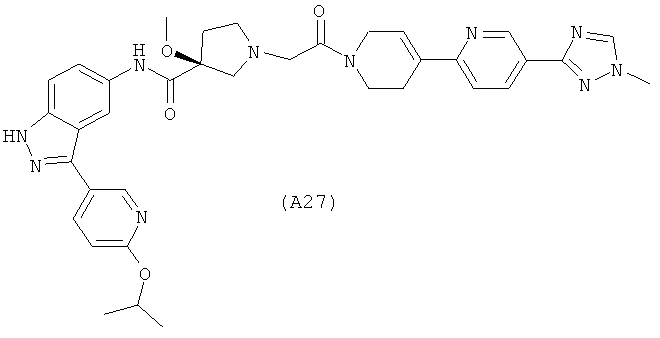
Стадия 1: Метил 6-хлорникотинимидат (1ВХ)
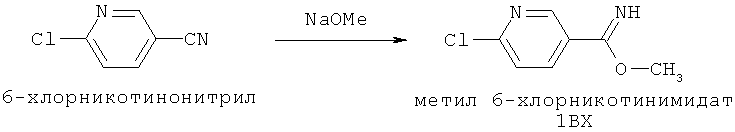
Метоксид натрия (725 мг, 13.42 ммоль) добавили к раствору 2-хлорпиридин-5-карбонитрила (1.8 г, 13.04 ммоль) в МеОН: диоксане (40 мл, 1:1) при 0°С, затем перемешивали в течение 30 минут при 0°С и 1 час при комнатной температуре. Реакционную смесь разбавили EtOAc (200 мл) и H2O (100 мл), органический слой отделили и высушили над Na2SO4, отфильтровали, и растворитель выпарили, с получением соединения 1ВХ в виде твердого вещества белого цвета (2.6 г, 100%). МС (МН 171).
Стадия 2: 6-хлор-N′-метилникотинимидогидразид (2 ВХ)
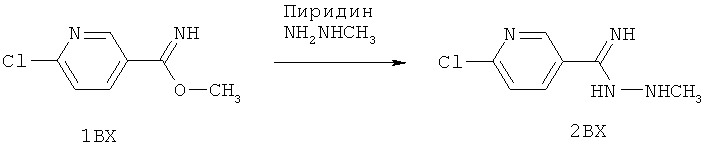
Метилгидразин (750 мг, 16.30 ммоль) добавили в раствор метил 6-хлорникотинимидата (1ВХ) (2.6 г, 15.29 ммоль) в пиридине (10 мл) при комнатной температуре, затем перемешивали в течение одного часа. Растворитель выпарили, и осадок в виде твердого вещества обработали холодным диэтиловым эфиром (2×10 мл), с получением продукта 2ВХ в виде порошка желтого цвета (2.4 г, 85%). МС (МН, 185)
Стадия 3: 2-хлор-5-(1-метил-1Н-1,2,4-триазол-3-ил)пиридина (3ВХ)
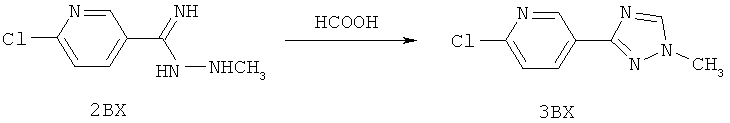
Раствор 6-хлор-N′-метилникотинимидогидразида (2ВХ) (2.4 г, 13 ммоль) в муравьиной кислоте (99%, 10 мл) перемешивали при комнатной температуре в течение одного часа. Реакционную смесь охладили, и растворитель выпарили. Осадок экстрагировали, применяя EtOAc (100 мл) и водный NaHCO3 (50 мл), органический слой отделили, высушили над Na2SO4, отфильтровали, и растворитель выпарили. Осадок хроматографировали на силикагеле, элюируя 10% MeOH:CH2Cl2, с получением продукта в виде твердого вещества (1.8 г, 72%). МС (МН, 195).
Стадия 4: Трет-бутил-4-(5-(1-метил-1Н-1,2,4-триазол-3-ил)пиридин-2-ил)-5,6-дигидропиридин-1(2Н)-карбоксилат (4ВХ)
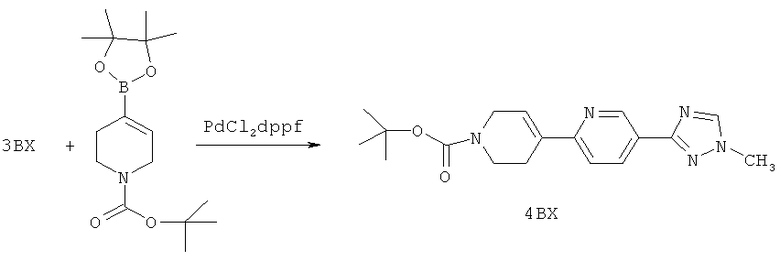
Смесь 2-хлор-5-(1-метил-1Н-1,2,4-триазол-3-ил)пиридина (3ВХ) (400 мг, 2.0 бммоль); N-трет-бутоксикарбонил-1,2,3,6-тетрагидропиридин-4-бороновой кислоты, сложного пинаконового эфира (1.4 г, 4.53 ммоль); карбоната цезия (2.3 г, 7.07 ммоль) и PdCl2dppf (100 мг) в диоксане/H2O (об/об 10:1, 20 мл)перемешивали при 100°С в течение 2 часов. Реакционную смесь охладили, разбавили, применяя CH2Cl2 (300 мл) и H2O (100 мл), органический слой отделили, высушили над Na2SO4, отфильтровали, и растворитель выпарили с получением осадка, который хроматографировали на силикагеле, элюируя EtOAc, с получением продукта 4ВХ, название которого указано в заглавии, в виде твердого вещества (400 мг, 57%). МС (МН, 342).
Стадия 5: 5-(1-метил-1Н-1,2,4-триазол-3-ил)-2-(1,2,3,6-тетрагидропиридин-4-ил)пиридин дигидрохлорида (5ВХ)
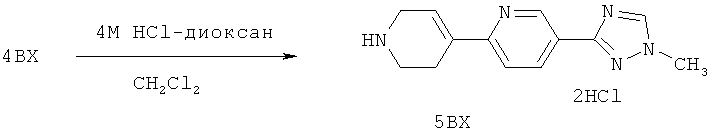
4М HCl в диоксане (20 мл) добавили в раствор трет-бутил-4-(5-(1-метил-1Н-1,2,4-триазол-3-ил)пиридин-2-ил)-5,6-дигидропиридин-1(2Н)-карбоксилата (4ВХ) (4 г, 11.67 ммоль) в CH2Cl2 (50 мл) при комнатной температуре. Смесь перемешивали в течение трех часов, затем растворитель выпарили, с получением продукта, название которого указано в заглавии, в виде твердого вещества белого цвета (3.8 г).
Стадия 6: 2-хлор-1-(4-(5-(1-метил-1Н-1,2,4-триазол-3-ил)пиридин-2-ил)-5,6-дигидропиридин-1(2Н)-ил)этанон (6ВХ)
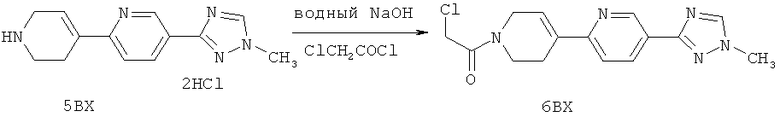
1Н NaOH (50 мл, 50 ммоль) и хлорацетил хлорид (3 мл, 37.7 ммоль) в CH2Cl2 (50 мл) по каплям добавили в раствор 5-(1-метил-1Н-1,2,4-триазол-3-ил)-2-(1,2,3,6-тетрагидропиридин-4-ил)пиридин дигидрохлорида (5ВХ) (1 г, 3.60 ммоль) в CH2Cl2 (50 мл) при 0°С, поддерживая рН>12. Смесь перемешивали в течение двух часов при 0°С, затем реакционную смесь разбавили CH2Cl2 (200 мл) и Н2О (100 мл). Органический слой отделили, промыли H2O (50 мл), высушили (Na2SO4), отфильтровали, и растворитель выпарили с получением продукта, название которого указано в заглавии, в виде твердого вещества белого цвета (1.1 г, 100%). МС (МН, 318).
Стадия 7
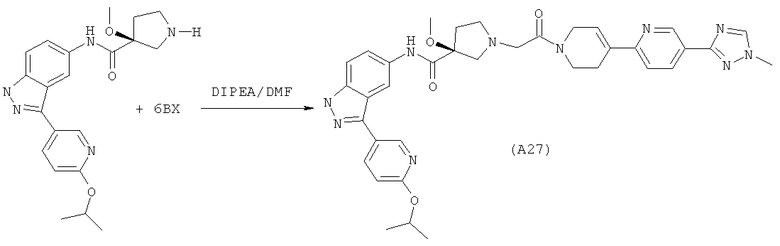
Раствор 2-хлор-1-(4-(5-(1-метил-1Н-1,2,4-триазол-3-ил)пиридин-2-ил)-5,6-дигидропиридин-1(2Н)-ил)этанона (6ВХ) (0.85 г, 2.68 ммоль) в CH2Cl2 (10 мл) добавили в раствор индазола (1 г, 2.53 ммоль) в DMF (10 мл) при комнатной температуре, затем перемешивали при 50°С в течение трех часов. Реакционную смесь разбавили EtOAc (300 мл) и H2O (100 мл), затем органический слой отделили, высушили над Na2SO4, отфильтровали, и выпарили растворитель. Осадок хроматографировали на силикагеле, элюируя 10% об/об MeOH/CH2Cl2/NH4OH, с получением продукта А27 в виде твердого вещества белого цвета (1.3 г, 76%). МС (МН, 677).
ЖХМС, время элюции = 2.61 минут
ПРИМЕР 27
Получение (S)-N-(3-(6-изопропоксипиридин-3-ил)-1Н-индазол-5-ил)-3-метокси-1-(2-(4-(5-(1-метил-1Н-1,2,4-триазол-3-ил)тиазол-2-ил)-5,6-дигидропиридин-1(2Н)-ил)-2-оксоэтил)пирролидин-3-карбоксамида:
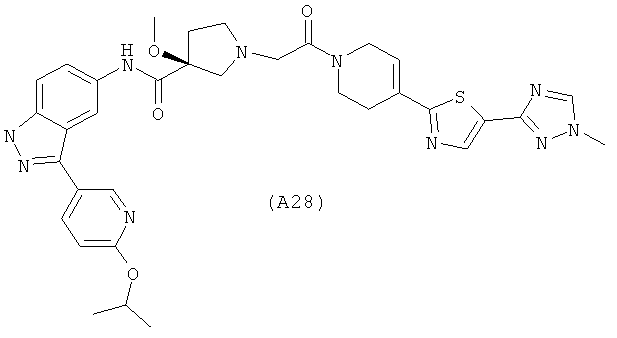
Стадия 1
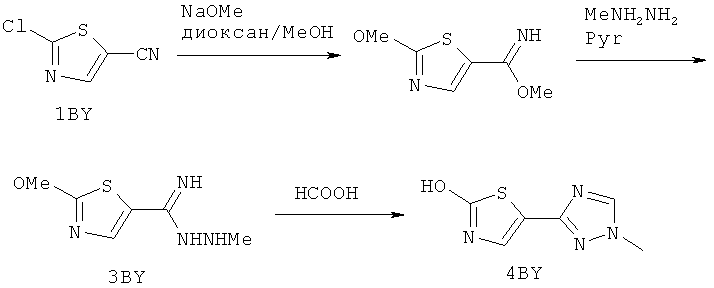
К перемешиваемой смеси 2-хлортиазол-карбонитрила (1 г, 6.92 ммоль) в МеОН добавили NaOMe (745 мг, 13.8 ммоль, 2 эквив.), и реакционную смесь перемешивали при 0°С в течение 15 мин и нагревали до комнатной температуры в течение еще 15 мин, погасили водой, экстрагировали, применяя EtOAc. Объединенный органический слой промыли соляным раствором, высушили над Na2SO4 и концентрировали с получением неочищенного вещества 2BY (1.2 г) в виде масла желтого цвета. К перемешанному раствору неочищенного вещества 2BY в пиридине (1 мл) добавили метилгидразин (363 мкл, 6.9 ммоль, 1 эквив.), полученную смесь перемешивали при 0°С в течение 15 мин и погасили, применяя НСООН (5 мл). Полученную смесь затем перенесли в запаиваемую пробирку и перемешивали при 110°С всю ночь, охладили до комнатной температуры и добавили воду. Полученную смесь экстрагировали, применяя CH2Cl2. Объединенный органический слой промыли соляным раствором, высушили и концентрировали, с получением твердого вещества желтого цвета, которое отфильтровали и промыли, применяя CH2Cl2. Полученное твердое вещество желтого цвета (375 мг) представляет собой целевой продукт 4BY. Фильтрат концентрировали и при желании далее очищали на колонке, с получением большего количества продукта.
Стадия 2
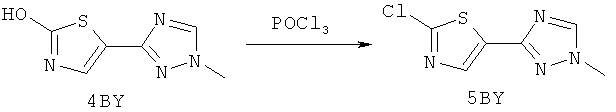
Смесь соединения 4BY (130 мг) в POCl3 перемешивали в атмосфере N2 при 120°С в течение двух дней. Первоначальная гетерогенная смесь превратилась в прозрачный раствор коричневого цвета, который концентрировали и очистили на силикагеле (CH2Cl2/MeOH, 50/1), с получением соединения 5BY (143 мг) в виде твердого вещества желтого цвета.
Стадия 3
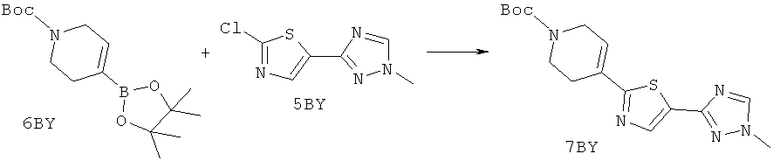
Смесь соединения 5BY (118 мг, 0.59 ммоль), соединения 6BY (274 мг, 0.89 ммоль, 1.5 эквив.), 2 М Na2CO3 (590 мкл, 1.18 ммоль, 2 эквив.) и Pd(PPh3)4 (34 мг, 0.05 эквив.) в бензоле/МеОН (5 мл, 4/1) дегазировали и перемешивали в атмосфере N2 при 80°С всю ночь. Реакционную смесь затем концентрировали и очистили на силикагеле (CH2Cl2/MeOH, 30/1), с получением соединения 7BY (150 мг) в виде твердого вещества желтого цвета.
Стадия 4
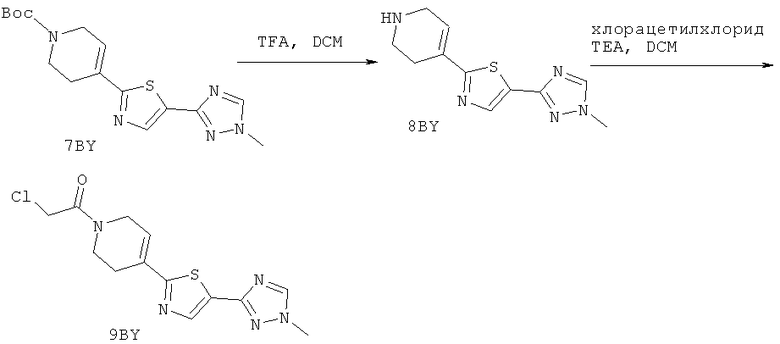
Смесь соединения 7BY (150 мг, 0.43 ммоль) и TFA (1.5 мл) перемешивали при комнатной температуре в течение одного часа и концентрировали. Хроматография на силикагеле (CH2Cl2/MeOH, 15/1) позволила получить соединение 8BY (98 мг). К перемешиваемой смеси соединения 8BY (98 мг, 0.4 ммоль) и трйэтиламина (335 мкл, 2.4 ммоль, 6 эквив.) в CH2Cl2/MeOH (6 мл, 2/1) при 0°С добавили хлорацетилхлорид (126 мкл, 1.6 ммоль, 4 эквив.). Реакционную смесь перемешивали при 0°С в течение одного часа и концентрировали. Хроматография на силикагеле (CH2Cl2/MeOH, 25/1) позволила получить соединение 9BY (103 мг) в виде твердого вещества белого цвета.
Стадия 5
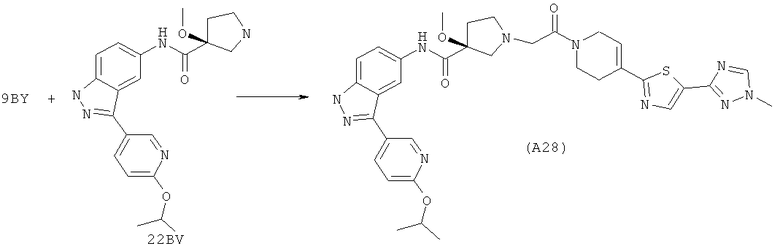
Соединение 9BY заменили соединением 7ВН в Примере 15 на Стадии 6, для того чтобы получить соединение А28. ЖХМС МН=683.4, Время удерживания = 2.77 минут
ПРИМЕР 28
Получение (S)-N-(3-(6-изопропоксипиридин-3-ил)-1Н-индазол-5-ил)-3-(метилтио)-1-(2-оксо-2-(4-(5-(6-оксопиридазин-1(6Н)-ил)-5,6-дигидропиридин-1(2Н)-ил)этил)пирролидин-3-карбоксамида) (А29)
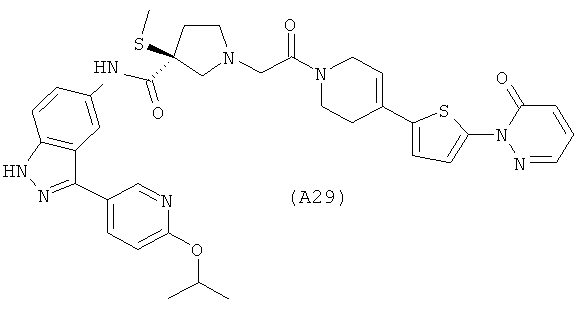
Стадия 1: Получение 2-(5-бромтиофен-2-ил)пиридазин-3(2H)-она
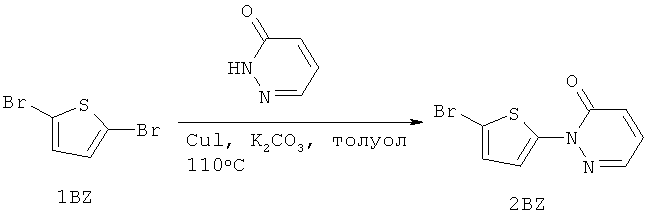
Смесь 2,5-дибромтиофена (1.5 г, 6.2 ммоль), пиридазин-3(2Н)-она (0.4 г, 4.1 ммоль), иодида меди (I) (0.24 г, 1.2 ммоль), карбоната калия (1.7 г, 12.4 ммоль), транс-N,N′-диметилциклогексан-1,2-диамина (0.2 мл, 1.2 ммоль) и толуола (15 мл) дегазировали в течение 15 минут и затем нагревали в запаянной пробирке при 110°С в течение 18 часов. Охладили до комнатной температуры, отфильтровали через целит и промыли, применяя EtOAc. Фильтрат промыли водой (100 мл × 2). Органический слой высушили над Na2SO4, отфильтровали и концентрировали с получением целевого продукта 2BZ (0.9 г, 90%). Осадок очистили на силикагеле, элюируя 80% EtOAc/гексан, с получением целевого продукта 2BZ (0.7 г, 67%).
Стадия 2: Получение трет-бутил-4-(5-(6-оксопиридазин-1(6Н)-ил)тиофен-2-ил)-5,6-дигидропиридин-1(2Н)-карбоксилата
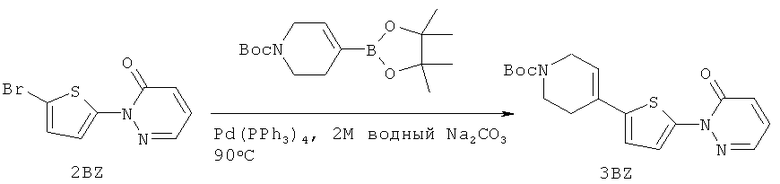
Смесь соединения 2BZ (0.7 г, 2.7 ммоль), 4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-3,6-дигидро-2Н-пиридин-1-карбоновой кислоты трет-бутилового сложного эфира (0.84 г, 2.7 ммоль), 2М водного раствора карбоната натрия (6.8 мл, 13.6 ммоль), Pd(PPh3)4 (0.31 г, 0.27 ммоль) и 1/1 /толуол/этанол (20 мл) дегазировали в течение 15 минут. Затем нагревали при 90°С всю ночь. Охладили до комнатной температуры и разбавили, применяя EtOAc (200 мл). Органический слой промыли водой (100 мл), высушили над Na2SO4, отфильтровали и концентрировали. Осадок очистили на силикагеле, элюируя 60% EtOAc/гексан с получением целевого продукта 3BZ (0.63 г, 65%).
Стадия 4: Получение 2-(5-(1,2,3,6-тетрагидропиридин-4-ил)тиофен-2-ил)пиридазин-3(2Н)-она
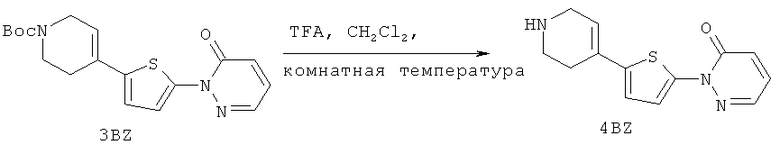
Смесь соединения 3BZ (0.63 г, 1.75 ммоль), CH2Cl2 (20 мл) и TFA (23 мл) перемешивали при комнатной температуре в течение 18 часов. Концентрировали и очистили на силикагеле, элюируя 5% МеОН (NH3)/ CH3Cl2 с получением целевого продукта 4BZ (0.4 г, 89%).
Стадия 5: Получение 2-(5-(1-(2-хлорацетил)-1,2,3,6-тетрагидропиридин-4-ил)тиофен-2-ил)пиридазин-3(2Н)-она
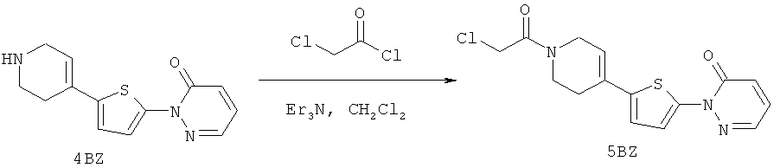
К смеси соединения 4BZ (0.62 г, 2.39 ммоль), CH2Cl2 (10 мл), МеОН (3 мл) и триэтиламина (0.67 мл, 4.78 ммоль) при -78°С добавили хлорацетилхлорид (0.19 мл, 2.39 ммоль). Реакционную смесь перемешивали при -78°С в течение 10 минут, затем нагрели до 0°С и перемешивали в течение одного часа. Разбавили, применяя CH3Cl2 (100 мл), и промыли насыщенным водным раствором NaHCO3 (100 мл). Органический слой высушили над Na2SO4, отфильтровали и концентрировали. Осадок растворили в CH2Cl2 и добавили диэтиловый эфир. Полученное твердое вещество отфильтровали и промыли диэтиловым эфиром, и высушили с получением целевого продукта 5BZ (0.56 г, 70%).
Стадия 6
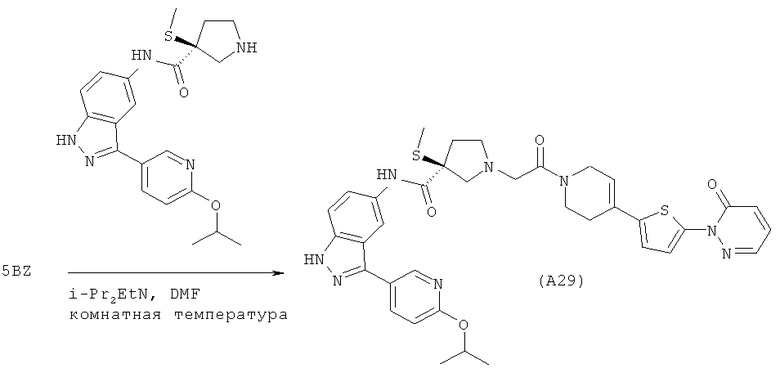
Смесь соединения 5BZ (0.04 г, 0.12 ммоль), соединения 6BZ (0.05 г, 0.12 ммоль), DMF (2 мл) и N,N-диизопропилэтиламина (0.042 мл, 0.24 ммоль) перемешивали при комнатной температуре в течение 18 часов. Разбавили, применяя EtOAc (100 мл), и промыли водой (2×100 мл). Органический слой высушили над Na2SO4, отфильтровали и концентрировали. Осадок очистили на силикагеле, элюируя 3% МеОН (NH3)/CH2Cl2 с получением целевого продукта А29 (0.083 г, 97%).
ЖХМС МН=711, Время удерживания = 3.26 минут.
ПРИМЕР 29
Получение (S)-N-(3-(6-изопропоксипиридин-3-ил)-1Н-индазол-5-ил)-1-(2-(4-(5-(3-метил-6-оксопиридазин-1(6H)-ил)тиофен-2-ил)-5,6-дигидропиридин-1(2Н)-ил)-2-оксоэтил)-3-(метилтио)пирролидин-3-карбоксамида (А30)
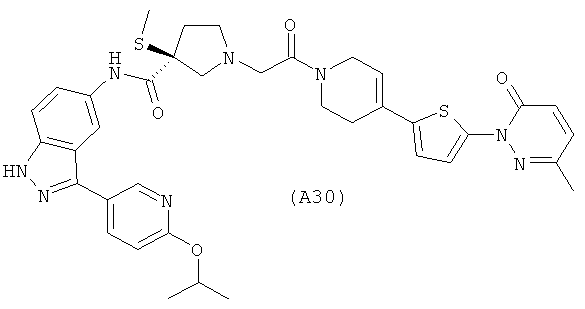
Соединение А30 получили, применяя методику, подобную методике, приведенной в Примере 28, за исключением применения 6-метилпиридазин-3(2H)-она вместо пиридазин-3(2Н)-она на Стадии 1.
ЖХМС МН=725, Время удерживания = 3.21 минут.
ПРИМЕР 30
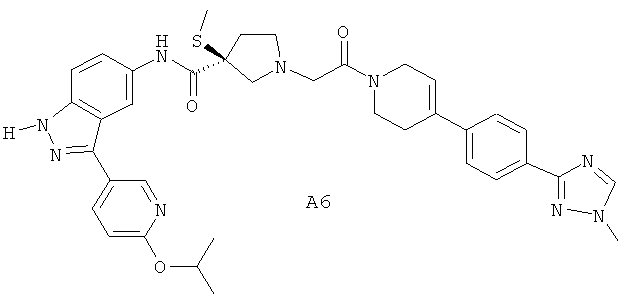
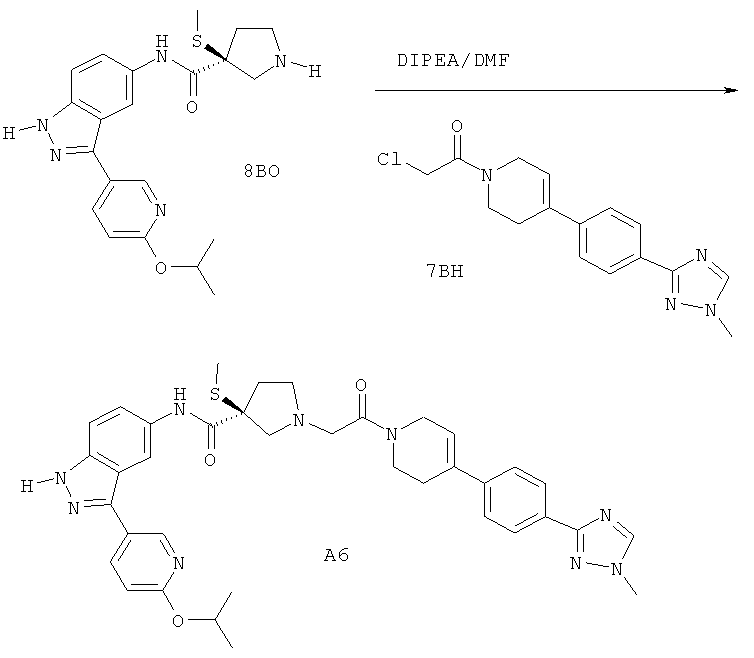
Соединение 8ВО (4.25 г, 10.33 ммоль) растворили в N,N-диметилформамиде (40 мл) при комнатной температуре. Затем добавили диизопропилэтиламин (5.1 мл, 30.99 ммоль), а затем соединение 7ВН (3.27 г, 10.33 ммоль). Реакционную смесь перемешивали в течение 1 часа при температуре окружающей среды. В реакционную смесь добавили соляной раствор и затем экстрагировали этилацетатом (3×100 мл). Экстракты этилацетата высушили над сульфатом магния, отфильтровали и выпарили с получением неочищенного продукта, название которого указано в заглавии. Неочищенный продукт хроматографировали с получением 4.95 г (69%) продукта, название которого указано в заглавии (А6) (10% 2Н NH3 в MeOH:CH2Cl2).
ПРИМЕР 31
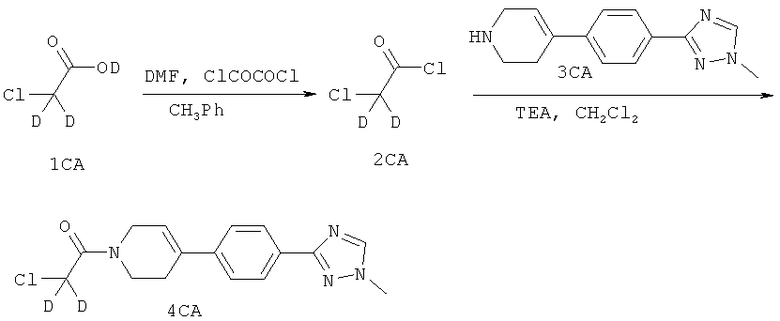
К раствору соединения 1СА (140 мг, 1.4 ммоль) в CH3Ph (3 мл) добавили каплю DMF, затем добавили оксалилхлорид (0.14 мл, 1.6 ммоль) при комнатной температуре. Через 30 минут полученный прозрачный раствор добавили в раствор соединения 3СА (240 мг, 1 ммоль) и триэтиламина (0.5 мл) в CH2Cl2. После перемешивания при комнатной температуре в течение 30 минут, реакцию погасили насыщенным NaHCO3, экстрагировали, применяя CH2Cl2, и концентрировали, с получением неочищенного продукта 4СА (312 мг) в виде твердого вещества белого цвета, которое применяли непосредственно в следующих реакциях. В 1СА, 2СА, и 4СА "D" обозначает дейтерий.
Соединения А31 и А32, получали способом, подобным способу получения соединений А1 и А6, заменяя 4СА на 7ВН. Соединения А45 и А48 получали способом, подобным способу получения соединений А23 и А25, заменяя 4СА на 7ВН. В А31, А32, А45 и А48 "D" обозначает дейтерий.
ЖХМС 3.95 мин М+1=694.4
ЖХМС 4.24 мин М+1=678.4
ЖХМС 3.94 мин М+1=674
ЖХМС 3.61 мин М+1=664
Пример 32
Если следовать методике, подобной методике, приведенной в Примере 31, Соединения А33-А44, А46 и А47, приведенные ниже, будут получены. В А33-А44, А46 и А47 "D" обозначает дейтерий.
,
,
,
,
,
,
,
,
,
,
,
,
и
.
и
В А33-А44, А46 и А47 "D"обозначает дейтерий.
Анализы
Двойные анализы ERK2:
Активность соединений в отношении неактивной ERK2 протестировали, применяя 1МАР парный анализ MEK1/ERK2, следующим образом: Соединения разбавили до 25-кратной конечной тестируемой концентрации в 100% DMSO. 14 мкл киназного буфера (10 мМ Трис. HCl рН 7.2, 10 мМ MgCl2, 0.01% Tween-20, 1 мМ DTT), содержащего 0.4 нг нефосфорилированного мышиного белка ERK2, добавили в каждую лунку черного 384-луночного аналитического планшета. 1 мкл соединения при 25 кратной концентрации добавили в каждую лунку и инкубировали при комнатной температуре в течение 30 минут, чтобы дать возможность соединению связаться с неактивным ферментом. При первичной инкубации концентрация DMSO составляла 6.7%. Активность ERK2, как определили, была нечувствительна к концентрациям DMSO вплоть до 20%. ERK2 затем активировали, и ее киназную активность измеряли путем добавления 10 мкл киназного буфера со следующими компонентами (конечная концентрация за реакцию): 2 нг активного (фосфорилированного) человеческого белка MEK1 и 4 мкМ (всего) IMAP ERK2 субстратных белков (3.9 мкМ немеченного IPTTPITTTYFFFK-CONH2 и 100 нМ IPTTPITTTYFFFK(5-карбоксифлуоресцеин)-CONH2) и 30 мкМ АТР. Концентрация DMSO при активации ERK составила 4%. Через один час реакции были остановлены добавлением 60 мкл IMAP детекционных шариков в связывающем буфере (Молекулярные приборы). Связыванию дали возможность уравновеситься в течение 30 минут перед считыванием планшета на планшетном ридере LJL Analyst Fluorescence Polarizationr. Ингибирование соединением вычисляли по отношению к DMSO и полностью ингибированным стандартам. Активные соединения можно повторно исследовать в независимом анализе.
Анализ активной ERK2:
Активность активированной ERK2 также определили в формате анализа IMAP, применяя методику, описанную выше. 1 мкл Соединения при 25-кратной концентрации добавили к 14 мкл киназного буфера, содержащего 0.25 нг полностью фосфорилированного активного мышиного белка ERK2. Через 30 минут после начала инкубации реакции были инициированы посредством добавления 10 мкл киназного буфера, содержащего 1 мкМ IMAP субстратного белка ERK2 (0.9 мкМ немеченного IPTTPITTTYFFFK-CONH2 и 100 нМ IPTTPITTYFFFK(5-карбоксифлуоресцеин)-CONH2) и 30 мкМ АТР. Реакции протекали в течение 30 минут, прежде чем их завершили добавлением 60 мкл IMAP детекционных шариков в связывающем буфере. Планшеты считывали, как указано выше, после уравновешивания связывания в течение 30 минут. Активные соединения повторно исследовали в независимом анализе.
Анализ на твердом агаре:
Для онкогенных клеточных линий характерен субстрат-независимый рост. Опухолевые клетки человека можно суспендировать в среде для роста, содержащей 0.3% агарозы и указанную концентрацию ингибитора фарнезилтрансферазы. Раствор накладывают на среду для роста, отвержденную 0.6% агарозы, содержащую ту же концентрацию ингибитора ERK1 и ERK2, что и верхний слой. После того, как верхний слой стал твердым, планшеты инкубируют при 37°С, в течение 10-16 дней, в атмосфере 5% CO2, для обеспечения роста колоний. После инкубации колонии были окрашены путем покрытия агара раствором МТТ (3-[4,5-диметил-тиазол-2-ил]-2,5-дифенилтетразолия бромид, Тиазолил синий) (1 мг/мл в фосфатно-солевом буферном растворе). Колонии можно посчитать, и IC50 можно определить.
AUC (площадь под кривой зависимости концентрации от времени в течение первых 6 часов (АЦС6ч) определили, применяя протокол Кассетного прогрессивного быстрого скрининга на крысах (CARRS)
Дозирование животных и получение образцов
Крыс-самцов линии Спраг Доули (Charles River, Co.) предварительно канюлировали (в бедренную артерию) для того, чтобы достичь точных моментов взятия образцов крови и уменьшить нагрузку на животных, вызываемую серийным взятием крови. Двум крысам, которым в течение ночи не давали пищу, пероральным путем ввели одно соединение, при дозе 10 мг/кг, в объеме дозы 5 мл/кг. У каждого животного в момент 0.5, 1, 2, 3, 4 и 6 часов после введения дозы серийно собрали кровь в гепарин-содержащие пробирки и концентрировали, чтобы собрать плазму. Плазму, около 100 мкл, собрали в отдельные моменты времени. Образцы плазмы хранили при -20°С до проведения анализа.
Получение образца плазмы и построение калибровочной кривой
Набор из 12 образцов плазмы крыс собрали для каждой NCE (новая химическая единица) (то есть, 6 временных точек и n=2 крысы). Эти 12 образцов плазмы объединили для двух крыс в каждый момент времени, с получением шести объединенных образцов (один образец для одного момента времени) для каждой NCE. Объединенные образцы проанализировали в формате кассет с шестью отделениями (всего 36 образцов), чтобы получить данные по шести соединениям. Аликвоты, объемом 50 мкл, отобранные из 36 образцов плазмы, поместили в отдельные лунки на 96-луночном планшете. Дополнительное соединение (часто структурный аналог тестируемых соединений) взяли в качестве внутреннего стандарта. Для каждого анализируемого соединения приготовили мини-калибровочную кривую (три точки плюс ноль). Свободную от лекарственного препарата плазму крысы измеряли, при объеме аликвот 1 мл, причем в каждую аликвоту вводили известные концентрации соединений, с получением стандартов с желаемыми концентрациями. Концентрации стандартов выбирали так, чтобы они соответствовали ожидаемым концентрациям объединенных образцов, на основе данных, полученных при ранее проведенных исследованиях для других соединений. Для настоящей работы стандарты содержали концентрации 25, 250 и 2500 нг МСЕ/мл плазмы. Стандарты плазмы наряду с образцами подвергли преципитации в двух экземплярах. Осаждение белка произошло после добавления в каждую лунку с образцом 150 мкл ацетонитрила, содержащего внутренний стандарт при концентрации 1 нг/мл, применяя систему Tomtec Quadra 96. Осажденные образцы и стандарты встряхивали и центрифугировали на 96-луночном планшете. Около 50-100 мкл супернатанта удалили и поместили в свежий 96-луночный планшет, применяя систему Tomtec Quadra 96. 5-10 мкл Супернатанта применяли для анализа посредством ВЭЖХ-МС/МС. По мини-калибровочной кривой пробежались дважды, сразу перед и сразу после образцов. Таким образом, для каждого соединения проанализировали все 14 образцов для изучения плюс стандарты. Кроме того, растворители в качестве стандарта впрыскивали перед и после каждого набора из 14 образцов и после самого высокого стандарта калибровки для каждого соединения, таким образом, всего 103 впрыска было сделано в каждую систему ВЭЖХ для каждого набора из шести соединений. Множественные впрыски растворителя в качестве стандарта могли бы быть сделаны из одной лунки. Двенадцать лунок с растворителем в качестве стандарта были обозначены в каждом 96-луночном планшете. Таким образом, одна серия (кассета) из шести NCEs была получена и проанализирована, применяя 96-луночный планшет.
ВЭЖХ-МС/МС анализ
Все соединения были проанализированы способами селективного мониторинга реакций (SRM) с ЖХ/МС/МС инструментами. Как только разработка способа была завершена, быстро провели анализ, применяя стандартную модель последовательного впрыска для анализа CARRS.
Соединения А1-А16, А18, А20, А21, А23, А25, А26 и А27-А30 показали IC50 для AERK2 в интервале 1.2-50 нМ.
Соединения А1-А3, А6, А8-А11, А13-А16, А20-А24, А26 и А27-А30 показали AUC в интервале 36-50,999 нМ. час в анализе CARRS.
Для приготовления фармацевтических композиций из соединений, описанных в настоящем изобретении, инертные фармацевтически приемлемые носители могут быть либо в твердом, либо в жидком виде. Препараты в твердом виде включают порошки, таблетки, диспергируемые гранулы, капсулы, крахмальные капсулы и суппозитории. Порошки и таблетки могут содержать от около 5 до около 95 процентов активного ингредиента. Подходящие твердые носители известны в данной области техники, например, такие как карбонат магния, стеарат магния, тальк, сахар или лактоза. Таблетки, порошки, крахмальные капсулы и капсулы могут применяться в качестве твердых лекарственных форм, подходящих для перорального введения. Примеры фармацевтически приемлемых носителей и способов получения различных композиций раскрываются в A.Gennaro (ed.), Remington: The Science and Practice of Pharmacy, 20th Edition, (2000), Lippincott Williams & Wilkins, Baltimore, MD.
Препараты в жидком виде включают растворы, суспензии и эмульсии. В качестве примера можно указать водные или водно-пропиленгликолевые растворы для парентеральной инъекции или добавление подсластителей и рентгеноконтрастных веществ для пероральных растворов, суспензий и эмульсий. Препараты в жидком виде могут также включать растворы для интраназального введения.
Аэрозольные препараты, которые подходят для ингаляции, могут включать растворы и твердые вещества в форме порошка, которые можно объединить с фармацевтически приемлемым носителем, таким как инертный сжатый газ, например азот.
Настоящее изобретение также включает препараты в твердом виде, которые, как предполагается, должны превращаться, непосредственно перед применением, в препараты в жидком виде либо для перорального, либо для парентерального введения. Такие жидкие формы включают растворы, суспензии и эмульсии.
Соединения по изобретению также могут доставляться трансдермально. Трансдермальные композиции могут иметь форму кремов, лосьонов, аэрозолей и/или эмульсий, и могут быть включены в трансдермальный пластырь матричного или резервуарного типа, что является обычным в данной области техники для этих целей.
Предпочтительно, соединение вводится пероральным путем.
Предпочтительно, фармацевтический препарат находится в виде дозированной лекарственной формы. В такой форме препараты делятся на стандартные дозы подходящего размера, содержащие соответствующие количества активного компонента, например, количество, эффективное для достижения желаемой цели.
Количество активного соединения в стандартной дозе препарата может варьироваться или устанавливаться от около 0.01 мг до около 1000 мг, предпочтительно от около 0.01 мг до около 750 мг, более предпочтительно от около 0.01 мг до около 500 мг, и наиболее предпочтительно от около 0.01 мг до около 250 мг в зависимости от конкретного применения.
Точная применяемая доза может варьироваться в зависимости от нужд пациента и серьезности состояния, подлежащего лечению. Определение соответствующей схемы приема для конкретного случая находится в компетенции специалиста в данной области техники. Для удобства, общая дневная доза может быть поделена для введения частями в течение дня, по мере возникновения такой необходимости.
Количество и частота введения соединений по изобретению и/или их фармацевтически приемлемых солей будет регулироваться в соответствии с оценкой лечащего врача, принимающего во внимание такие факторы как возраст, состояние и вес пациента, а также серьезность симптомов, подлежащих лечению. Типичная рекомендованная ежедневная схема приема для перорального введения может находиться в интервале от около 0.04 мг/день до около 4000 мг/день, за две-четыре дозы в день.
Несмотря на то, что настоящее изобретение раскрывается выше с указанием конкретных вариантов выполнения изобретения, изложенных выше, многие альтернативы, модификации и вариации настоящего изобретения будут очевидны специалистам в данной области техники. Все такие альтернативы, модификации и вариации, как подразумевается, охватываются сущностью и объемом настоящего изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ГЕМИАСТЕРЛИНА И ИХ ПРИМЕНЕНИЕ ПРИ ЛЕЧЕНИИ РАКА | 2003 |
|
RU2342399C2 |
| НОВЫЕ СОЕДИНЕНИЯ, КОТОРЫЕ ЯВЛЯЮТСЯ ИНГИБИТОРАМИ ERK | 2013 |
|
RU2660429C2 |
| Комбинированная терапия с использованием антагониста хемокинового рецептора 2 (CCR2) и ингибитора PD-1/PD-L1 | 2018 |
|
RU2796863C2 |
| ФАРМАЦЕВТИЧЕСКИЕ СОЕДИНЕНИЯ | 2005 |
|
RU2422448C2 |
| ПИРРОЛО[2,3-b]ПИРИДИНОВЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕИНКИНАЗ | 2006 |
|
RU2565071C2 |
| НИЗКОМОЛЕКУЛЯРНЫЕ ИНГИБИТОРЫ N-КОНЦЕВОЙ АКТИВАЦИИ РЕЦЕПТОРА АНДРОГЕНОВ | 2009 |
|
RU2519948C2 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛА, ИСПОЛЬЗУЕМЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕИНКИНАЗЫ | 2001 |
|
RU2340611C2 |
| НОВЫЙ ИНГИБИТОР КИНАЗЫ PAN-RAF И ЕГО ПРИМЕНЕНИЕ | 2019 |
|
RU2792626C1 |
| КОНЪЮГАТЫ ЭТОПОЗИДА И ДОКСОРУБИЦИНА ДЛЯ ДОСТАВКИ ЛЕКАРСТВЕННЫХ СРЕДСТВ | 2009 |
|
RU2531591C2 |
| ЛЕЧЕНИЕ ЗЛОКАЧЕСТВЕННЫХ И НЕЗЛОКАЧЕСТВЕННЫХ ЗАБОЛЕВАНИЙ С ПОМОЩЬЮ АНТАГОНИСТОВ RAS | 2012 |
|
RU2632097C2 |
Изобретение относится к соединениям формулы 1.0:
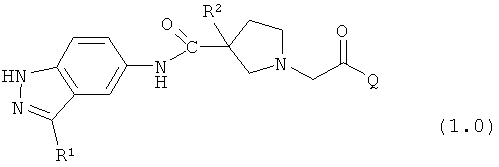
где Q представляет собой тетрагидропиридинильное кольцо замещенное. R5, R1 выбирают из группы, состоящей из: (1) пиридила, замещенного заместителем, выбираемым из группы, состоящей из: -O-СН3, -O-C2H5, -O-СН(СН3)2, и -О-(СН2)2-O-СН3, R2 выбирают из группы, состоящей из: -ОСН3 и -SCH3; и R5 выбирают из группы, состоящей из: (a) замещенного триазолилфенила-, где триазолил замещен одной или двумя алкильными группами, выбранными из группы, состоящей из: -С1-С4алкила, (b) замещенного триазолилфенила-, где триазолил замещен на атоме азота -С1-С4алкилом, (c) замещенного триазолилфенила-, где триазолил замещен на атоме азота -С2алкилен-O-С1-С2алкилом, (d) замещенного триазолилфенила-, где триазолил замещен на атоме азота -С2-С4алкилен-O-СН3, и (e) замещенного триазолилфенила-, где триазолил замещен на атоме азота гидрокси-замещенным -С1-С4алкилом, и где фенил необязательно замещен от 1 до 3 заместителями, независимо выбранными из группы, состоящей из галогена; и их фармацевтически приемлемым солям и сольватам, которые заявлены в качестве ингибиторов ERK. 4 н. и 11 з.п. ф-лы, 2 табл., 32 пр.
1. Соединение формулы 1.0:
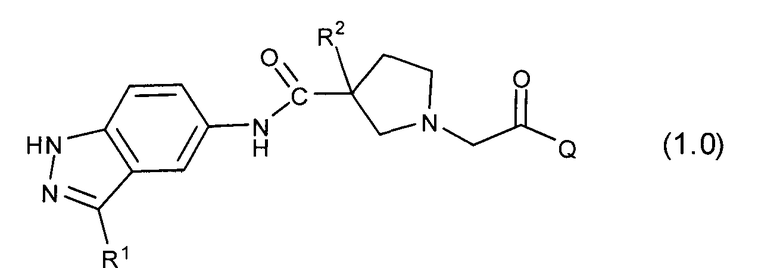
или его фармацевтически приемлемая соль, где R1, R2 и Q независимо выбирают, и где:
Q представляет собой:
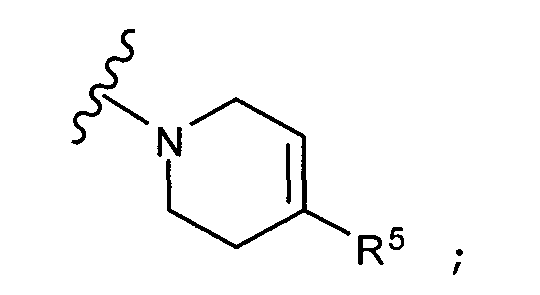
R1 выбирают из группы, состоящей из:
(1) пиридила, замещенного заместителем, выбираемым из группы, состоящей из: -O-СН3, -O-C2H5, -O-СН(СН3)2, и -О-(СН2)2-O-СН3;
(2)
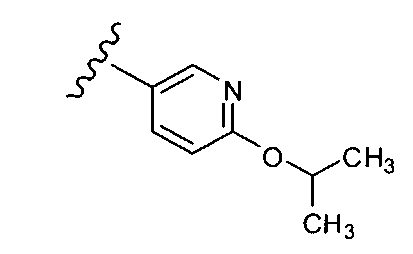 ;
;
(3)
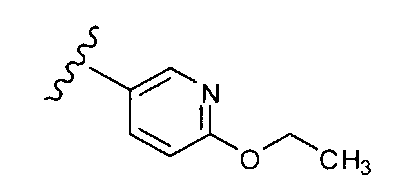 ; и
; и
(4)
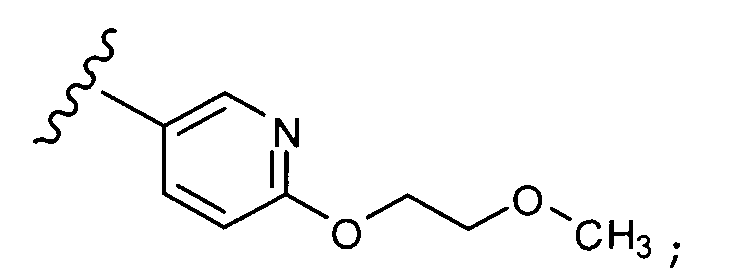
R2 выбирают из группы, состоящей из: -ОСН3 и -SCH3; и
R5 выбирают из группы, состоящей из:
(a) замещенного триазолилфенила-, где триазолил замещен одной или двумя алкильными группами, выбранными из группы, состоящей из: -С1-С4алкила,
(b) замещенного триазолилфенила-, где триазолил замещен на атоме азота -С1-С4алкилом,
(c) замещенного триазолилфенила-, где триазолил замещен на атоме азота -С2алкилен-O-С1-С2алкилом,
(d) замещенного триазолилфенила-, где триазолил замещен на атоме азота -С2-С4алкилен-O-СН3, и
(e) замещенного триазолилфенила-, где триазолил замещен на атоме азота гидрокси-замещенным -С1-С4алкилом, и
где фенил необязательно замещен от 1 до 3 заместителями, независимо выбранными из группы, состоящей из галогена.
2. Соединение по п.1, имеющее формулу:
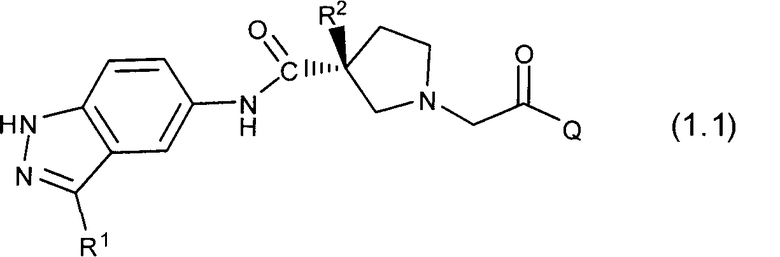
3. Соединение по п.1 или 2, где необязательные галоген-заместители для фенильного фрагмента R5 представляют собой F.
4. Соединение по п.1, где:
(a) указанный триазолил замещен на атоме азота заместителем, выбранным из группы, состоящей из: -СН2СОН(СН3)2 и -СН2СН2ОН,
(b) указанный триазолил замещен на атоме азота -СН3 группой,
(c) указанный триазолил замещен на атоме азота одной -СН3 группой, и на атоме углерода одной -СН3 группой, или
(d) указанный триазолил замещен на атоме азота -CH2CH2OCH3 группой;
и где фенил необязательно замещен галогеном.
5. Соединения по п.1, где:
R5 выбирают из группы, состоящей из:
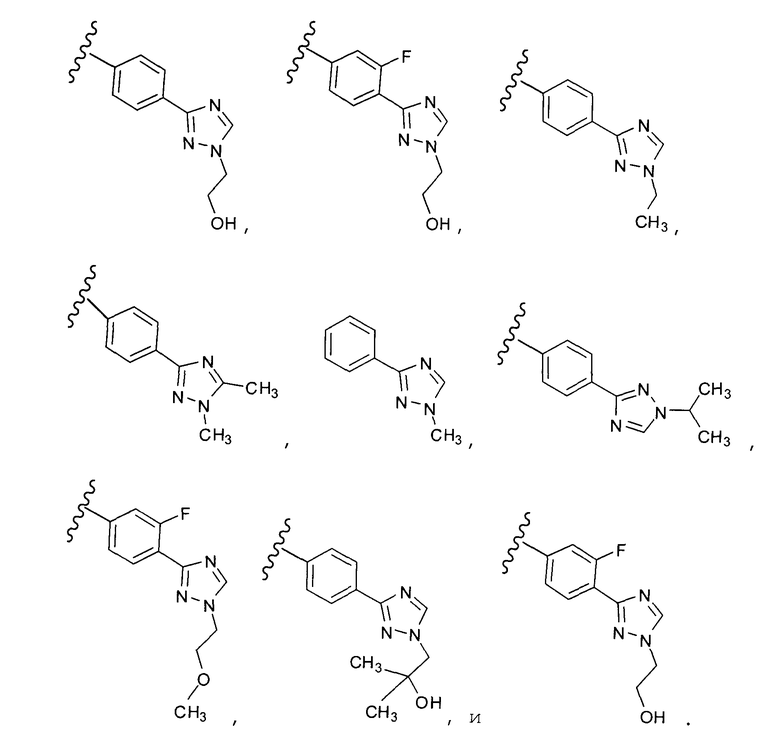
6. Соединение по п.1, где R5 выбирают из группы, состоящей из:
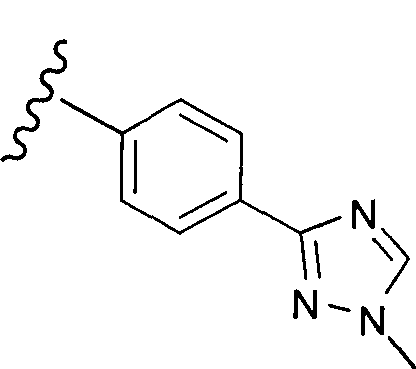 .
.
7. Соединение, выбранное из группы, состоящей из:
 ,
,
 ,
,
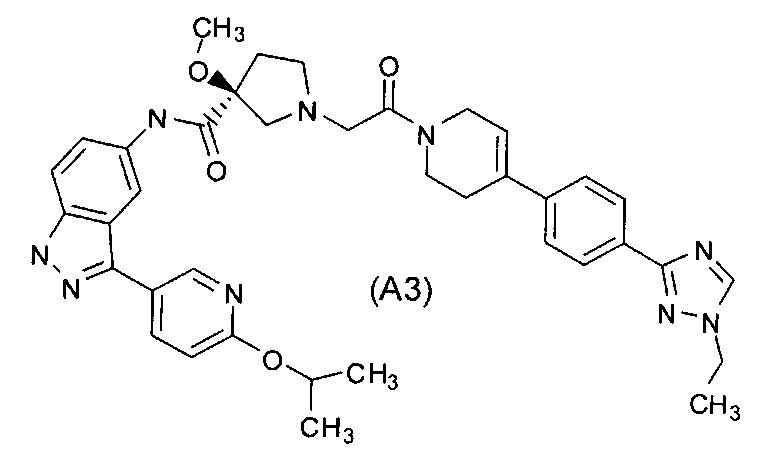 ,
,
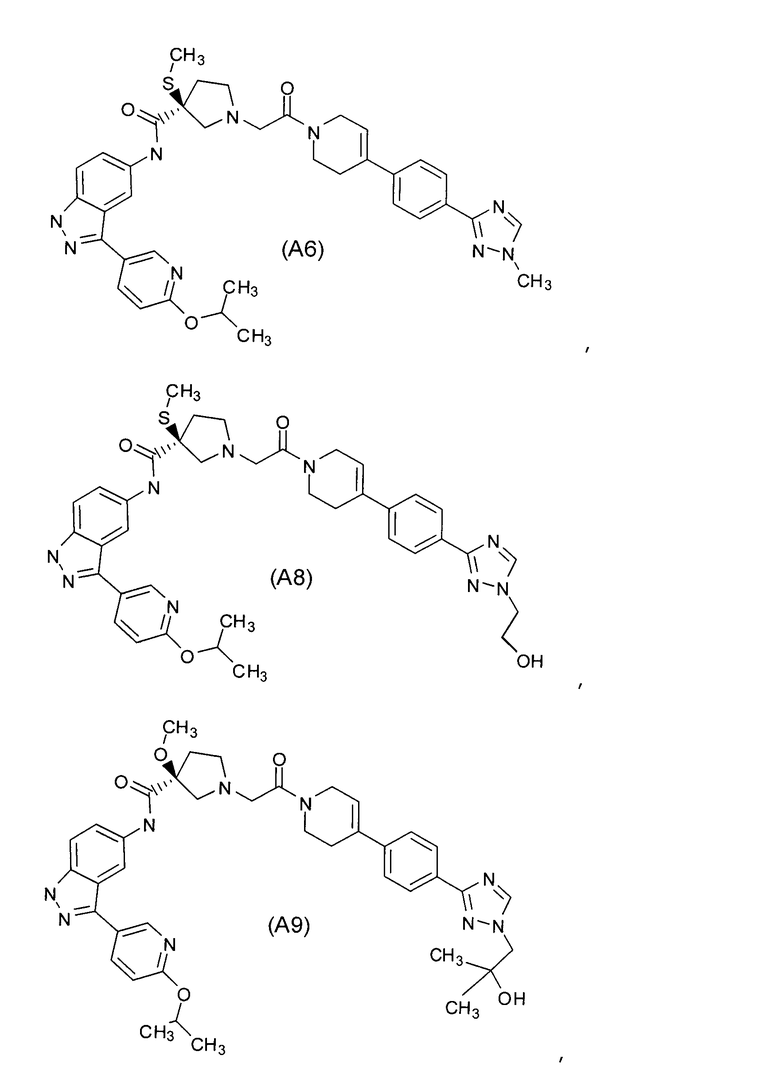
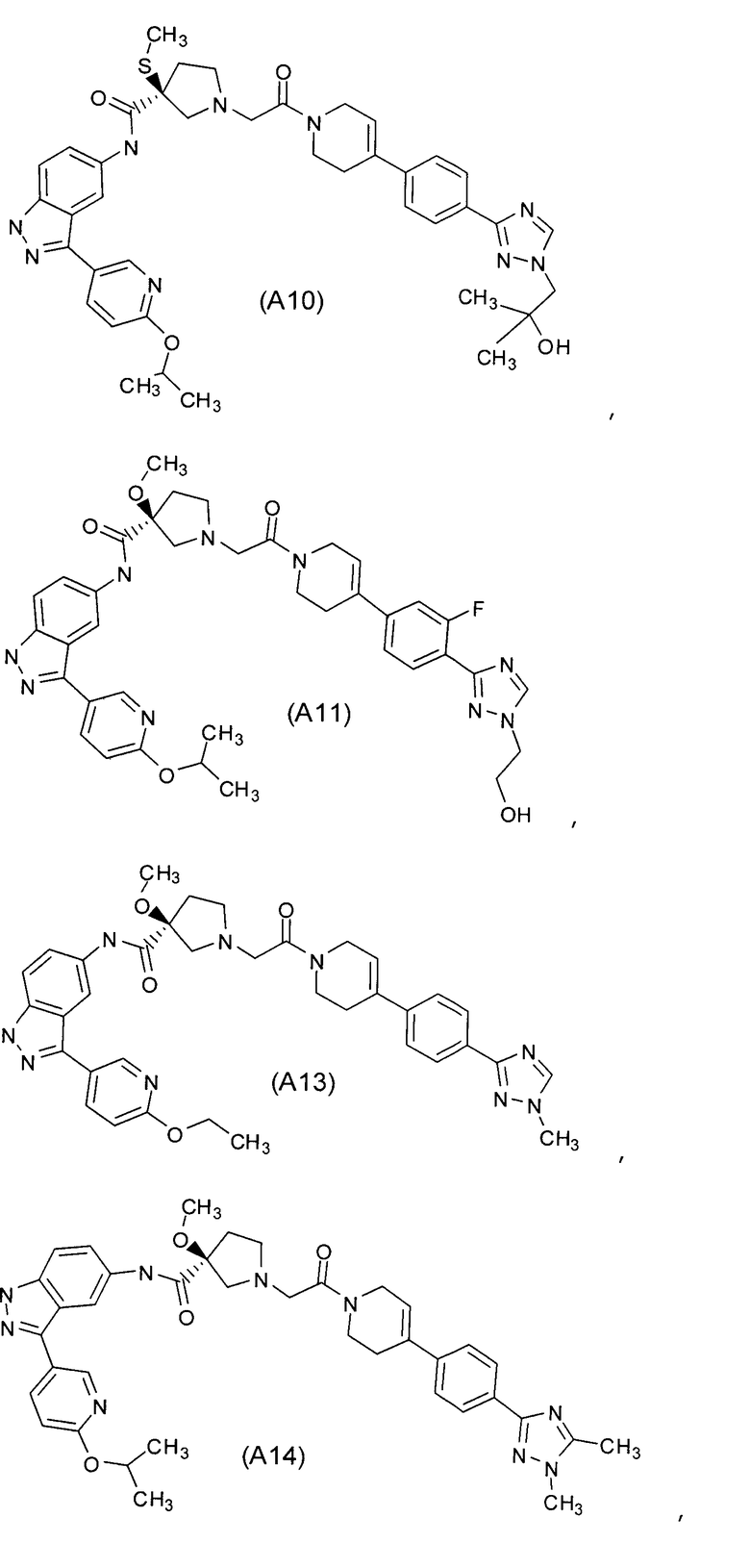
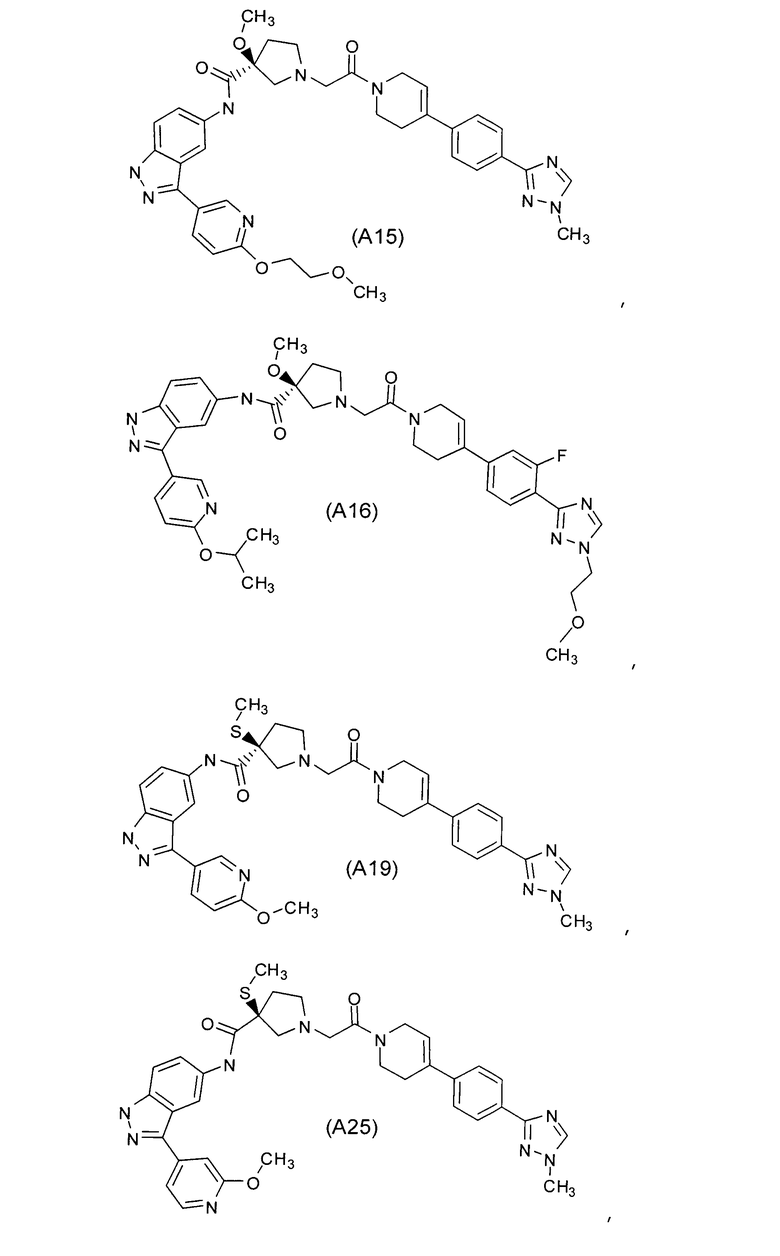
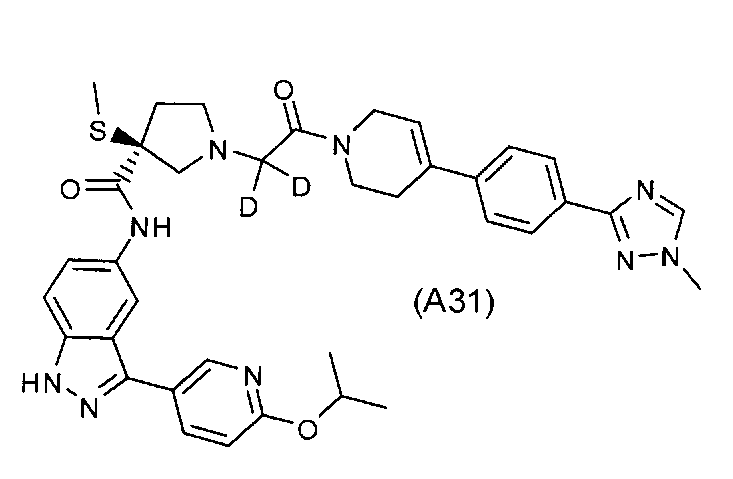 , и
, и
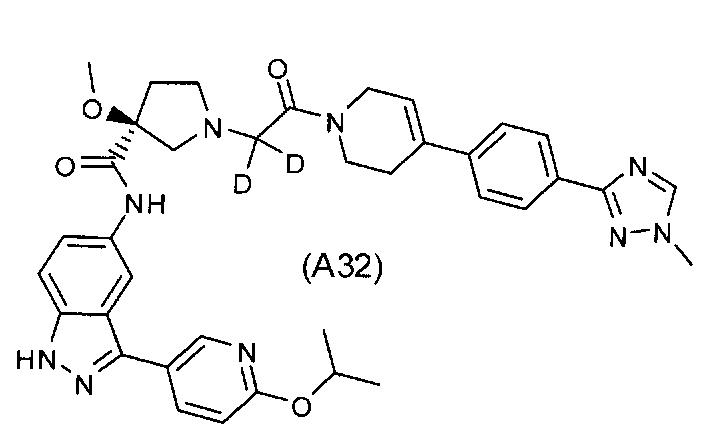 ,
,
и его фармацевтически приемлемые соли.
8. Соединение по п.1, представляющее собой
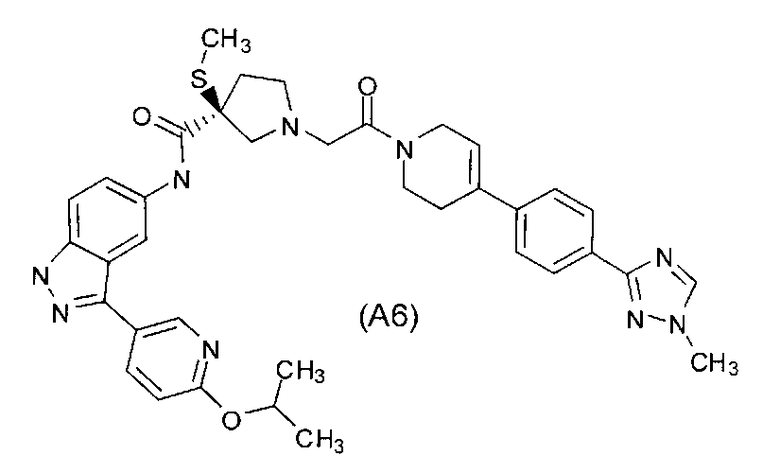
или его фармацевтически приемлемая соль.
9. Соединение по п.1, представляющее собой
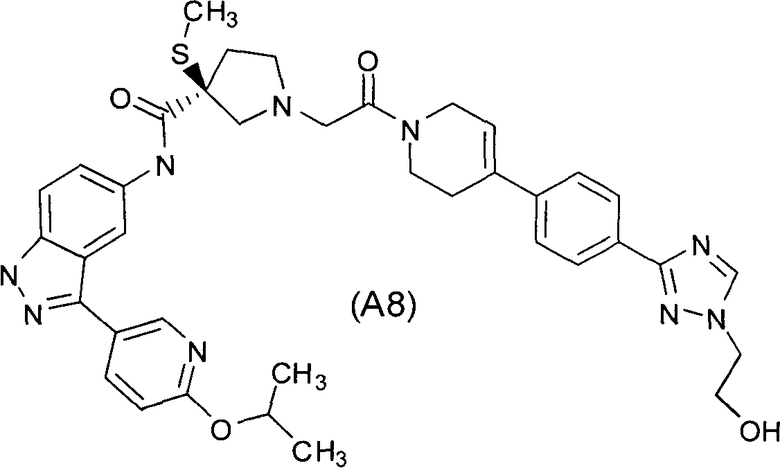
или его фармацевтически приемлемая соль.
10. Соединение по п.1, представляющее собой
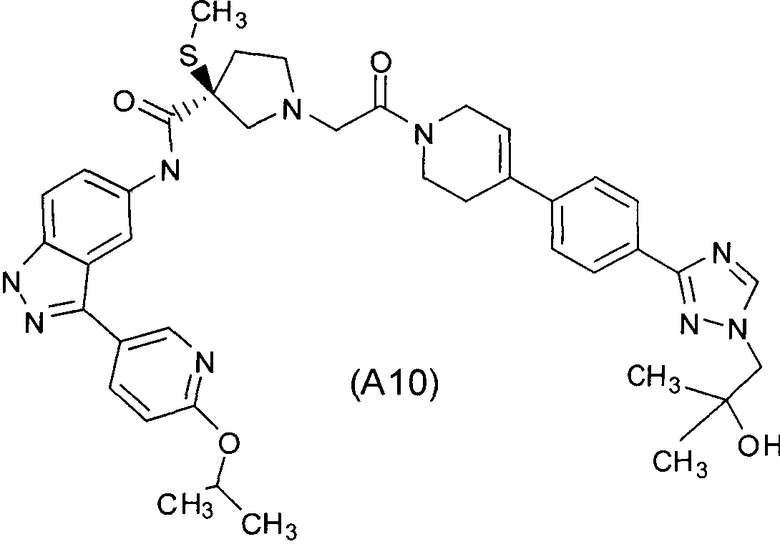
или его фармацевтически приемлемая соль.
11. Соединение по п.1, представляющее собой
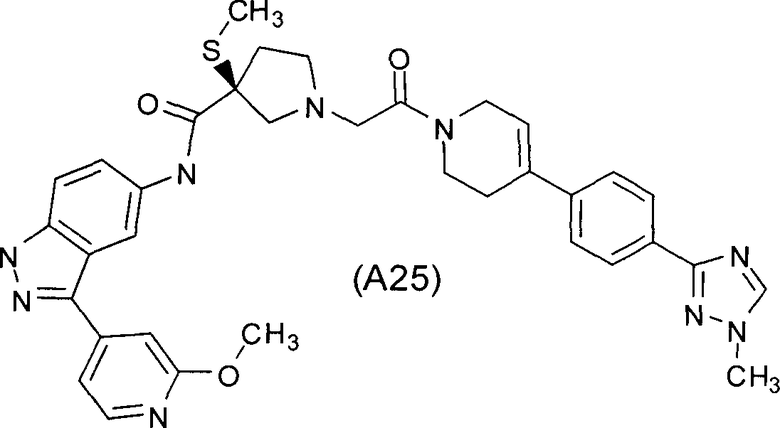
или его фармацевтически приемлемая соль.
12. Соединение по п.1, представляющее собой
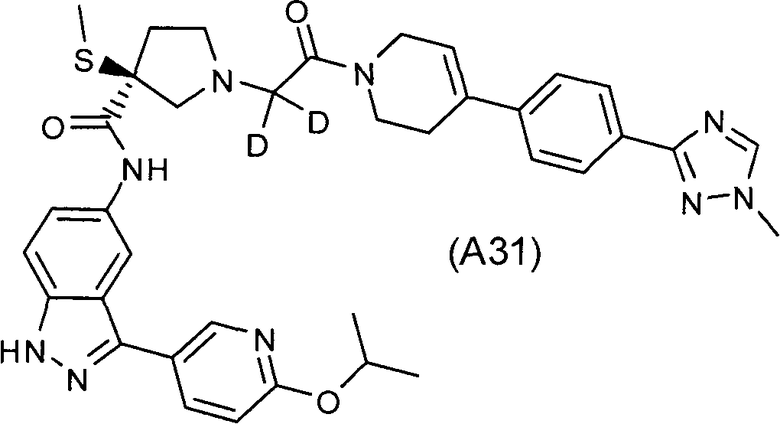
или его фармацевтически приемлемая соль.
13. Соединение по п.1, представляющее собой
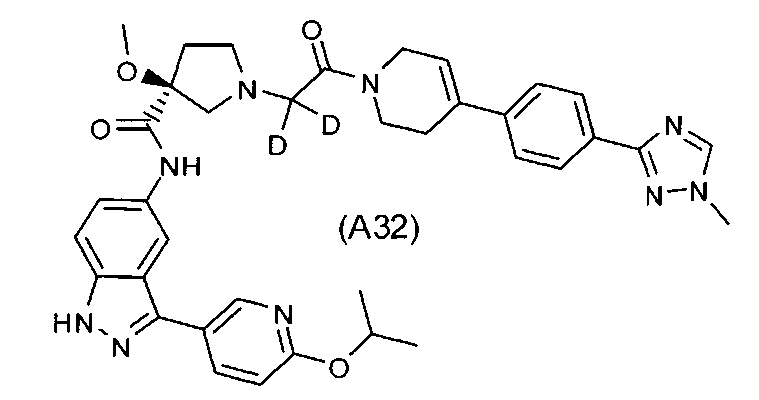
или его фармацевтически приемлемая соль.
14. Фармацевтическая композия, обладающая противораковой активностью, содержащая по меньшей мере одно соединение по любому из пп.1-13 и фармацевтически приемлемый носитель.
15. Применение соединения по п.1 для получения лекарственного средства для лечения рака.
| WO 2007070398, A1, 21.06.2007 | |||
| US 20020103229, A1, 01.08.2002 | |||
| RU 2003127116, A, 27.03.2005 | |||
| Станок для изготовления деревянных ниточных катушек из цилиндрических, снабженных осевым отверстием, заготовок | 1923 |
|
SU2008A1 |

