Область техники
Данное изобретение относится к способам улучшения доставки лекарственных средств. В частности, изобретение относится к полипептидам, которые при помощи гидролизуемых ковалентных связей связываются с терапевтическим препаратом, таким как подофиллотоксин-производные (напр., этопозид или его производные, такие как этопозид 4'-диметилглицин), либо с доксорубицином или его производными. Эти полипептидные конъюгаты могут быть использованы в качестве векторов для транспортировки терапевтических препаратов через гематоэнцефалитический барьер (ГЭБ) или для доставки в четко определенные виды клеток, такие как яичники, печень, легкие или почки. Эти конъюгаты могут способствовать улучшению физико-химических (например, увеличению растворимости) и фармацевтических свойств (например, расширению спектра целевых органов и тканей, что позволяет применять субтерапевтические дозы, или понижению токсичности, что позволяет применять сверхтерапевтические дозы) по сравнению с неконъюгированными терапевтическими препаратами. Изобретение также относится к фармацевтическим композициям, которые включают соединения по настоящему изобретению, и их применению в различных способах лечения.
Уровень техники
Многие терапевтические препараты (в т.ч. химиотерапевтические) против таких заболеваний оказывают нежелательные побочные эффекты либо, по причине in vivo стабильности, транспорта или других фармакокинетических свойств, представляют затруднения при введении в достаточно высоких концентрациях в целевую ткань, или при длительной продолжительности лечения, без которой невозможно достичь максимального терапевтического эффекта в целевой ткани. Следовательно, возникает необходимость в поиске новых способов и композиций, которые позволили бы увеличить концентрацию терапевтических и диагностических агентов в целевых органах и тканях, таких как головной мозг, яичники, печень или легкие.
Сущность изобретения
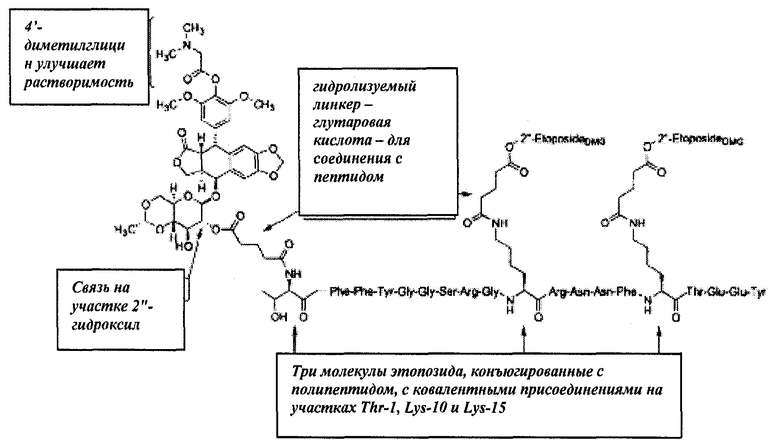
Нами разработаны пептидные терапевтические конъюгаты и их фармацевтически приемлемые соли, где этопозид с помощью ковалентной связи присоединен к полипептиду Angiopep-2 (SEQ ID NO:97) на участке 2"-гидроксил посредством гидролизуемого линкера - глутаровой кислоты (напр., Соединение (1), Схема 1). Также был получен подобный пептидный терапевтический конъюгат, в котором вместо этопозида использовался этопозид 4'-диметилглицин, причем в этом случае удалось добиться улучшенных свойств (напр., растворимости).
Схема 1
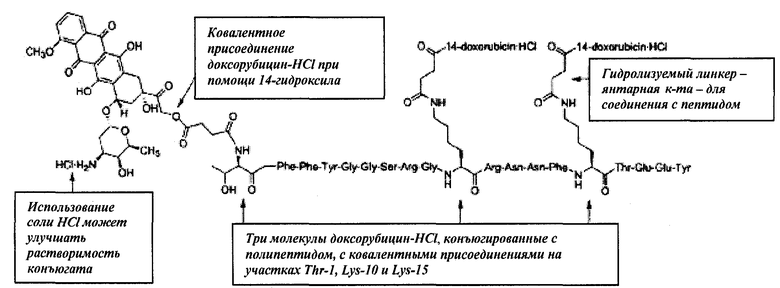
Доксорубицин также был присоединен ковалентной связью на участке 14-гидроксил к полипептиду Angiopep-2 с помощью линкера - янтарной кислоты (напр., треххлористоводородная соль Соединения (2), Схема 2). Ковалентное присоединение хлористоводородной соли доксорубицина также позволяет улучшить свойства (напр., растворимость).
Схема 2
Эти конъюгаты демонстрируют улучшенные свойства по сравнению с соответствующими неконъюгированными терапевтическими препаратами, а именно: улучшенные физико-химические (например, увеличенная растворимость) и фармацевтические (например, расширенный спектр целевых органов и тканей, что позволяет применять субтерапевтические дозы, или пониженная токсичность, что позволяет применять сверхтерапевтические дозы) свойства. Растворимость этопозидаDMG и конъюгатов доксорубицин гидрохлорида также может быть полезна при корректировке режимов дозирования. Поэтому настоящее изобретение охватывает такие соединения, а также родственные им соединения. Кроме того, предоставляются способы получения и применения этих соединений.
Соответственно, в одном аспекте данное изобретение предоставляет соединение или его фармацевтически приемлемую соль, включающие последовательность аминокислот, в значительной мере идентичную последовательности аминокислот, выбранной из группы, которая состоит из SEQ ID NOS: 1-105 и 107-116, или их функциональных производных, где последовательность аминокислот включает ковалентную связь от аминокислоты из последовательности аминокислот до подофиллотоксин-производного. В некоторых вариантах осуществления подофиллотоксин-производное представляет собой соединение, имеющее структуру согласно Формуле (I):
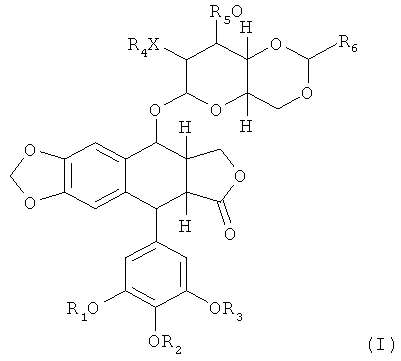 ,
,
или его стереоизомер или фармацевтически приемлемая соль, где каждый R1, R2, и R3 независимо выбраны из: Н, произвольно замещенный C1-6 алкил, C(O)R8, P(O)(OR9)(OR10), S(O)2(OR9) или гидролизуемый линкер Y, который содержит ковалентную связь с аминокислотой полипептида;
Х является О или NR7;
каждый R4, R5 и R7 независимо выбраны из Н, произвольно замещенного C1-6 алкила, C(O)R8 или гидролизуемого линкера Y, который содержит ковалентную связь с аминокислотой полипептида;
R6 - это Н, произвольно замещенный C1-6 алкил, произвольно замещенный арил, произвольно замещенный гетероарил;
R8 выбран из: произвольно замещенный C1-6 алкил или произвольно замещенный арил;
каждый R8 и R10 независимо выбраны из: Н, произвольно замещенный C1-6 алкил или произвольно замещенный арил; и
n равно 1, 2, 3, 4, 5, 6, 7 или 8;
где один из R1, R2, R3, R4, R5 и R7 является Y и не более чем один из R1, R2, R3, R4, R5 и R7 является Y.
В некоторых вариантах осуществления Y является -С(O)(СН2)nC(O)- и n равно 2, 3 или 4. В определенных вариантах осуществления n равно 3.
В некоторых вариантах осуществления фармацевтически приемлемая соль соединения представляет собой моно-, ди- или три- соль добавления кислоты (напр., моно-, ди- или тригидрохлоридную соль).
В некоторых вариантах осуществления каждое соединение согласно Формуле (I) независимо выбрано из:
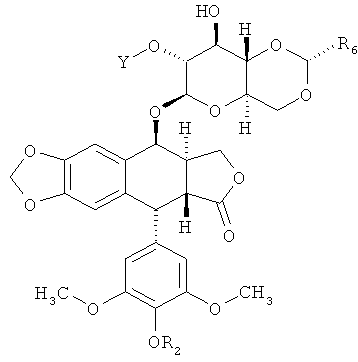 и
и 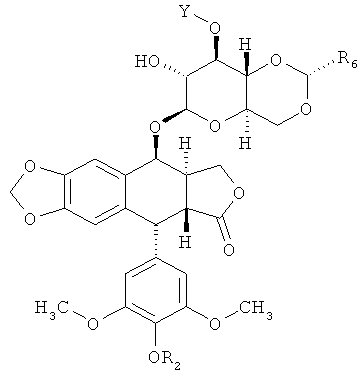 ,
,
где каждый R2 независимо является Н, Р(O)(ОН)2 или C(O)CH2N(CH3)2; каждый R6 независимо является СН3 или 2-тиофеном; каждый Y выбран из -С(O)(СН2)nC(O)-; -[C(O){OCH2CH2}nOC(O)]-; -S(O)2(CH2)nS(O)2-; -[S(O)2{OCH2CH2}nOS(O)2]-[{P(O)(OR9)}(CH2)n{P(O)(OR9)}]-; и -[{P(O)(OR9)}(OCH2CH2)nO{P(O)(OR9)}]-; каждый n независимо равен 1, 2, 3, 4, 5 или 6; и где каждый Y ковалентно связан с аминокислотой. В некоторых вариантах осуществления каждый Y является -С(O)(СН2)nC(O)- или -[С(O){ОСН2СН2}nOC(O)]- и n равен 2, 3 или 4. В некоторых вариантах осуществления каждый R2 является С(O)CH2N(СН3)2. В некоторых вариантах осуществления каждое соединение согласно Формуле (I) представляет собой:
 или
или 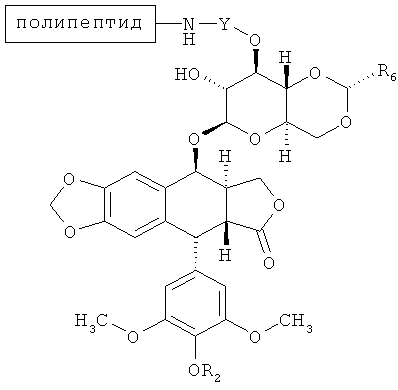
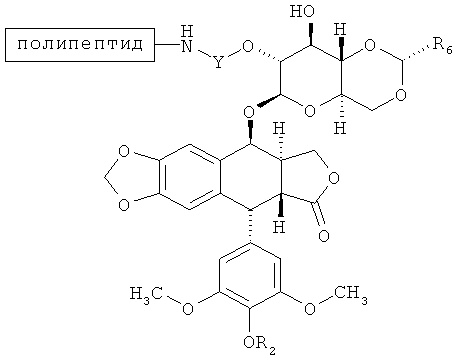
В некоторых вариантах осуществления соединение согласно настоящему изобретению имеет структуру:
где каждая группа согласно (-(Формуле(I)) представляет произвольную ковалентную связь между указанной аминокислотой и соединением по Формуле (I) и где существует по меньшей мере одна ковалентная связь между аминокислотой полипептида и указанным соединением согласно Формуле (I). В некоторых вариантах осуществления два соединения согласно Формуле (I) присоединены к последовательности аминокислот. В других вариантах осуществления треонин в позиции 1 и лизины на позициях полипептида 10 и 15, каждый включает ковалентную связь с соединением, имеющим структуру согласно Формуле (I).
В некоторых вариантах осуществления R2 является Н или -C(O)CH2N(CH3)2 (напр., С-связанный N,N-диметилглицин). В других вариантах осуществления каждый R2 является Н. Еще в некоторых вариантах осуществления каждый R2 является -С(O)CH2N(СН3)2.
В некоторых вариантах осуществления произвольно замещенный C1-6 алкил независимо выбран из: метил, этил, n-пропил, изопропил, n-бутил, изобутил, втор-бутил, втор-пентил, изо-пентил, трет-бутил, n-пентил, неопентил, n-гексил или втор-гексил. В некоторых вариантах осуществления C1-6 алкил замещен по меньшей мере одной произвольно замещенной аминогруппой (напр., NH2 или N(СН3)2) на любом атоме углерода.
В некоторых вариантах осуществления произвольно замещенный С3-10 циклоалкил независимо выбран из циклопропила, циклобутила, циклопентила, циклогексила и циклогептила.
В некоторых вариантах осуществления произвольно замещенная арильная группа независимо выбрана из фенила, нафтила, тетрагидронафтила, инданила или инденила.
В некоторых вариантах осуществления произвольно замещенная гетероциклильная группа независимо выбрана из: азациклопропанил, азациклобутанил, 1,3-диазатидинил, пирролидинил, пиперидинил, пиперазинил, тиранил, тиэтанил, тетрагидротиофенил, дитиоланил, тетрагидротиопиранил, оксиранил, оксетанил, тетрагидрофуранил, тетрагидропиранил, диоксанил, оксатиоланил, морфолинил, тиоморфолинил, тиоксанил и хинуклидинил.
В некоторых вариантах осуществления произвольно замещенная гетероциклильная группа выбрана из: пирролил, пиразолил, имадазолил, пиридинил, пиримидинил, пиразинил, пиридазинил, триазинил, тетразинил, прииролизинил, индолил, хинолинил, изохинолинил, бензимидазолил, индазолил, хинолизинил, циннолинил, хиназолинил, фталазинил, нафтиридинил, хиноксалинил, тиофенил, тиэпинил, фуранил, бензофуранил, тиазолил, изотиазолил, тиадиазолил, оксазолил, изоксазолил и оксадиазолил.
В некоторых вариантах осуществления замещенный алкил, циклоалкил, арил, гетероциклил или гетероарил замещен 1, 2, 3, 4, 5 или 6-ю заместителями, выбранными из: С1-6 алкил; галоген; азидо(-N3), нитро (-NO2), пиано (-CN), ацилокси, ацил (-C(O)R), (-OC(O)R), алкокси (-OR), амидо (-NRC(O)R' или -C(O)NRR'), амино (-NRR'), арил, карбоновая кислота (-СО2Н), сложный эфир карбоновой кислоты (-CO2R), карбамоил (-OC(O)NRR' или -NRC(O)OR'), циклоалкил, гетероциклил, гидрокси (-ОН), изоциано (-NC), фосфат (-P(O)(OR)(OR')), сульфонат (-SO2OR) или сульфонил (-SO2R), где каждый R или R' независимо выбраны из Н, С1-6 алкила, циклоалкила, гетероциклила, арила или гетероарила, согласно данным выше определениям. В некоторых вариантах осуществления эти заместители в дальнейшем не замещаются. В других вариантах осуществления заместители, в свою очередь, и сами могут в дальнейшем замещаться 1, 2, 3, 4, 5 или 6-ю группами заместителей.
В некоторых вариантах осуществления R4 является Y. В других вариантах осуществления R5 является Y.
В других вариантах осуществления последовательность аминокислот ковалентно связана с дополнительными производными подофиллотоксина посредством второй, третьей, четвертой или пятой аминокислоты указанной последовательности аминокислот. В некоторых вариантах осуществления производное подофиллотоксина представляет собой соединение по Формуле (I).
В определенных вариантах осуществления соединение по Формуле I имеет структуру:
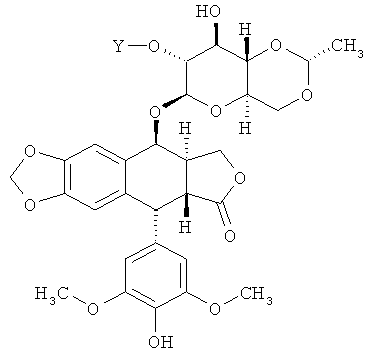 ,
,
где Y - это -C(O)(CH2)nC(O)- или -[С(O){ОСН2СН2}nOC(O)]- и n равно 2, 3 или 4. В других вариантах осуществления соединение по Формуле (I) имеет структуру:
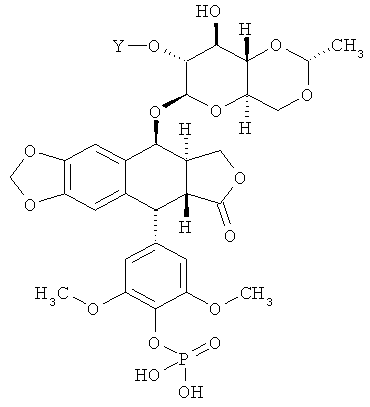 ,
,
где Y - это -С(O)(СН2)nC(O)- или -[С(O){ОСН2СН2}nOC(O)]- и n равно 2, 3 или 4. Еще в некоторых вариантах осуществления соединение по Формуле (I) имеет структуру:
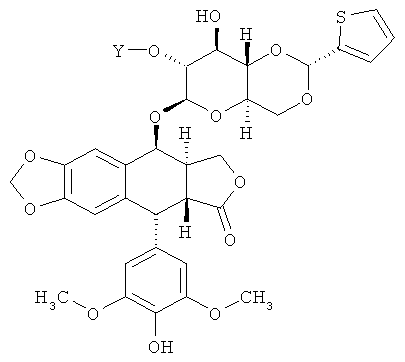 ,
,
где Y - это -С(O)(СН2)nC(O)- или -[С(O){ОСН2СН2}nOC(O)]- и n равно 2, 3 или 4.
В других вариантах осуществления каждое соединение по Формуле (I) независимо выбрано из:
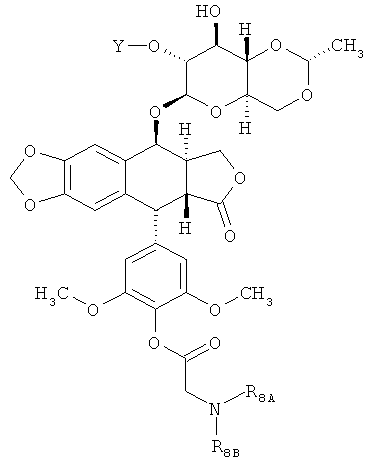 или
или 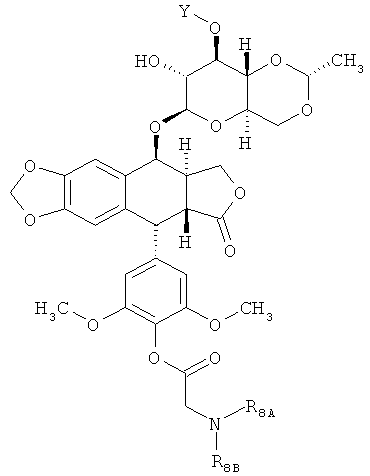 ,
,
где каждые R8A и R8B независимо являются Н или произвольно замещенным C1-6 алкилом, или R8A и R8B объединяются, образуя произвольно замещенное 3-7-членное кольцо. В некоторых вариантах осуществления каждый R8A и R8B представляет собой произвольно замещенный C1-6 алкил. В других вариантах осуществления каждое соединение по Формуле (I) имеет структуру:
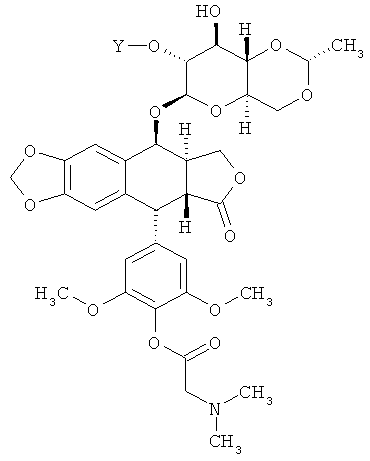
В дальнейших вариантах осуществления соединение имеет такую структуру:
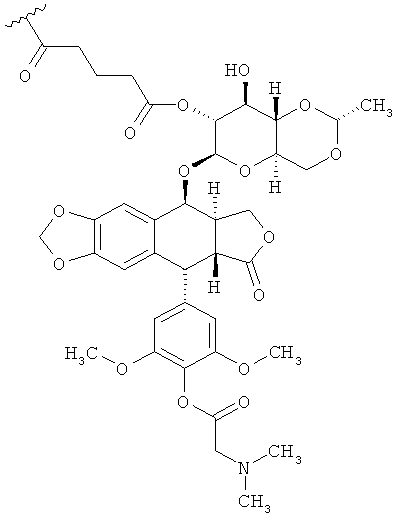
В отдельных вариантах осуществления соединение имеет структуру:
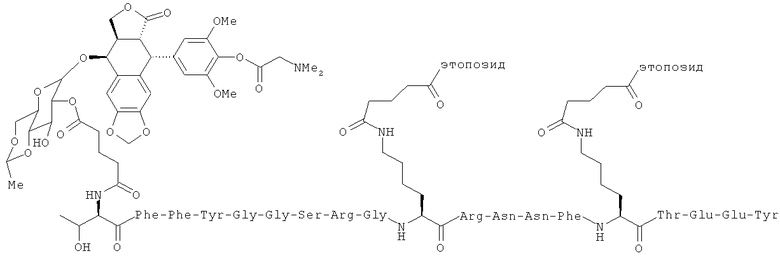
(I), или его фармацевтически приемлемая соль (напр., треххлористоводородная соль), где в Соединении (I) этопозид относится к этопозид 4'-диметилглицину.
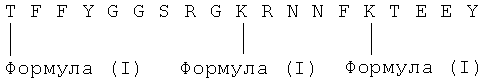
В определенных вариантах осуществления каждая аминокислота, ковалентно связанная с гидролизуемым линкером Y, присоединяется посредством амино-, гуанидино-, гидроксил-, фенол- или тиольной функциональной группы указанной аминокислоты. В некоторых вариантах осуществления аминокислота, ковалентно связанная с гидролизуемым линкером Y, является лизином, тирозином, серином, треонином или аргинином.
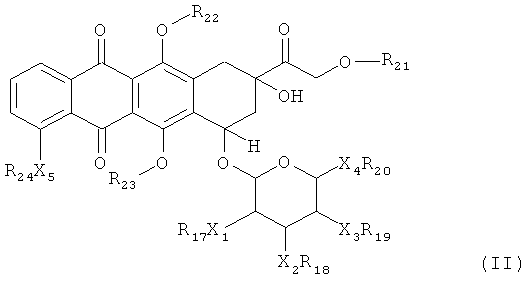
Во втором аспекте, изобретение предоставляет соединение или его фармацевтически приемлемую соль, которое включает последовательность аминокислот, в значительной степени идентичную последовательности аминокислот, выбранной из группы, которая состоит из SEQ ID NOS: 1-105 и 107-116 или их функциональных производных, где указанная последовательность аминокислот включает ковалентную связь от аминокислоты с указанной последовательностью аминокислот до доксорубицин-производного и где указанное доксорубицин-производное представляет собой соединение, имеющее структуру согласно Формуле (II):
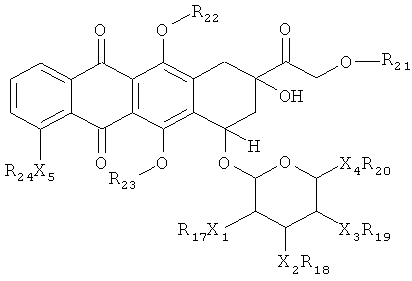
(II), либо его стереоизомер или фармацевтически приемлемая соль, где каждый X1, X2, Х3, Х4 и Х5 независимо выбран из ковалентной связи, О или NR25;
каждый R17, R18, R19, R20, R20, R21, R22, R23, R24 и R25 независимо выбран из: Н, произвольно замещенный C1-6 алкил, произвольно замещенный С2-6 алкенил, произвольно замещенный C2-6 алкинил, произвольно замещенный циклоалкил, произвольно замещенный гетероциклил или он является гидролизуемым линкером Y; и
где один и только один из R17, R18, R19, R20, R20, R21, R22, R23, R24 и R25 является Y.
В некоторых вариантах осуществления фармацевтически приемлемая соль соединения представляет собой моно-, ди- или три-соль добавления кислоты (напр., моно-, ди- или треххлористоводородная соль).
В определенных вариантах осуществления соединение по Формуле (II) имеет структуру:
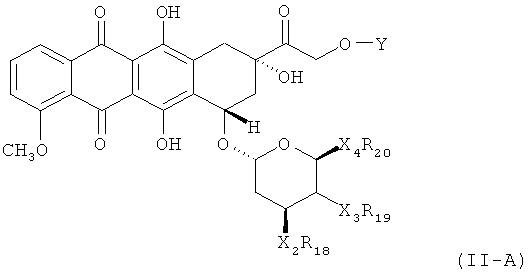 ,
,
где X2R18 - это Н или NH2; X3R19 - это Н или ОН; X4R20 - это Н или произвольно замещенный C1-3 алкил; и Y является гидролизуемым линкером согласно данному выше определению. В дальнейших вариантах осуществления соединение по Формуле (II) имеет структуру:
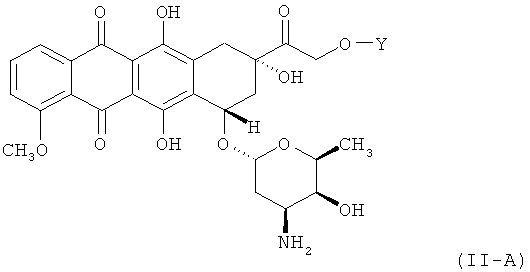 ,
,
или его фармацевтически приемлемая соль.
В дальнейших вариантах осуществления соединение по Формуле (II) имеет структуру:
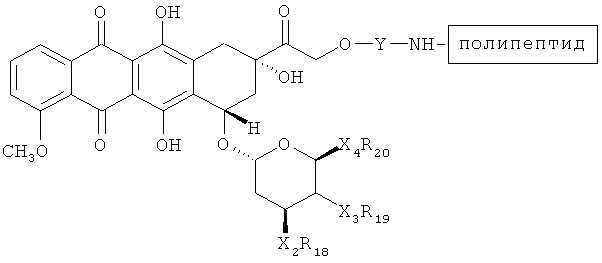
или
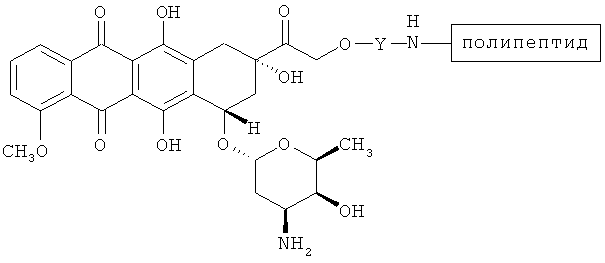 .
.
В определенных вариантах осуществления соединение имеет структуру:
 ,
,
где каждая (-(Формула(II)) представляет произвольную ковалентную связь между указанной аминокислотой и соединением по Формуле (II) и где существует по меньшей мере одна ковалентная связь между аминокислотой полипептида и вышеназванным соединением по Формуле (II). В некоторых вариантах осуществления треонин на позиции 1 и лизины на позициях 10 и 15 полипептида, каждый содержит ковалентную связь с соединением, имеющим структуру согласно Формуле (II).
В некоторых вариантах осуществления Y является -С(O)(СН2)nC(O)- и n равно 2, 3 или 4. В определенных вариантах осуществления n равно 2. В других вариантах осуществления последовательность аминокислот ковалентно связана с соединением, имеющим структуру согласно Формуле (II), с помощью второй, третьей, четвертой или пятой аминокислот последовательности аминокислот. В другом варианте осуществления каждая аминокислота, ковалентно связанная с указанным гидролизуемым линкером Y, присоединена посредством амино-, гуанидино-, гидроксил-, фенол- или тиоловой функциональной группы аминокислоты. В определенных вариантах осуществления аминокислота является лизином или треонином,
В некоторых вариантах осуществления соединение по Формуле (II) имеет структуру:
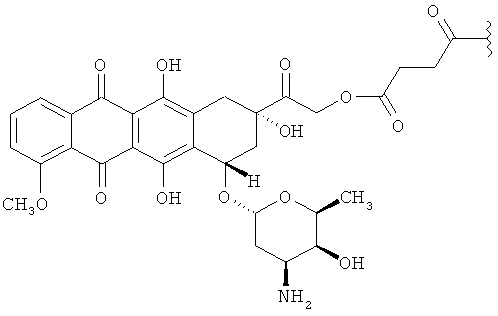 ,
,
или его фармацевтически приемлемая соль.
В других вариантах осуществления соединение имеет структуру:
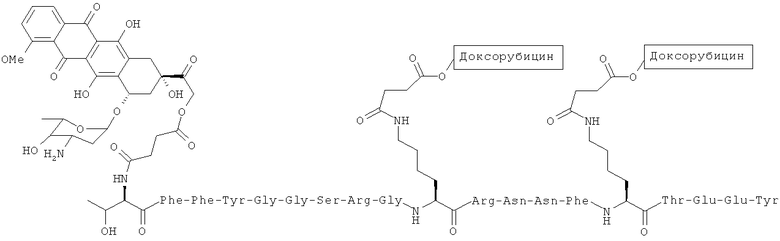
(2), или его фармацевтически приемлемая соль (напр., треххлористоводородная соль).
В другом аспекте, изобретение предоставляет следующее соединение:
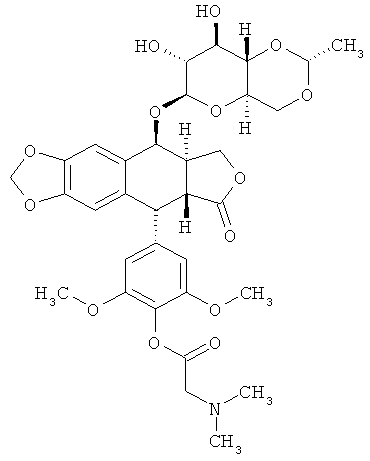
(«этопозид 4'-диметилглицин» или «этопозидDMG») либо любой его стереоизомер или любая фармацевтически приемлемая соль или растворитель этого соединения.
В любом из вышеупомянутых аспектов последовательность аминокислот может быть в значительной степени идентична любой из последовательностей, перечисленных в Табл. 1 либо их фрагментам или их фармацевтически приемлемым солям. В определенных вариантах осуществления последовательность аминокислот имеет последовательность Angiopep-1 (SEQ ID NO: 67), Angiopep-2 (SEQ ID NO: 97), Angiopep-3 (SEQ ID NO: 107), Angiopep-4a (SEQ ID NO: 108), Angiopep-4b (SEQ ID NO: 109), Angiopep-5 (SEQ ID NO: 110), Angiopep-6 (SEQ ID NO: 111) или Angiopep-7 (SEQ ID NO: 112). Последовательность аминокислот или соединений изобретения может быть эффективно транспортирована в конкретный вид клеток (напр., в любой один, два, три, четыре или пять таких видов клеток: печень, яичники, легкие, почки, селезенка и мышцы) либо может эффективно пересекать ГЭБ млекопитающих (напр., Angiopep-1, -2, -3, -4а, -4b, -5 и -6). В некоторых вариантах осуществления клетки являются клетками яичника. В других вариантах осуществления конъюгат способен внедряться в конкретный вид клетки (напр., в любой один, два, три, четыре или пять таких видов клеток: печень, яичники, легкие, почки, селезенка и мышцы), но не пересекая при этом ГЭБ эффективно (напр., конъюгат, включающий Angiopep-7). В некоторых вариантах осуществления клетки являются клетками яичников. Длина полипептида может быть любой, например, по меньшей мере 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 25, 35, 50, 75, 100, 200 или 500 аминокислот. В определенных вариантах осуществления длина полипептида составляет от 10 до 50 аминокислот. Конъюгат может быть практически чистым. Полипептид может быть получен с помощью рекомбинантной генетической технологии или химического синтеза. Конъюгат может быть сформулирован с фармацевтически приемлемым носителем.




Полипептиды с номерами Nos 5, 67, 76 и 91 включают последовательности SEQ ID NOs: 5, 67, 76 и 91 соответственно и амидируются на С-конце.
Полипептиды с номерами Nos 107,109 и 110 включают последовательности SEQ ID NOs: 97, 109 и 110 соответственно и ацетилируются на N-конце.
В любом из вышеперечисленных аспектов полипептид может включать последовательность аминокислот, которая имеет Формулу:
Х1-Х2-Х3-Х4-Х5-Х6-Х7-Х8-Х9-Х10-Х11-Х12-Х13-Х14-Х15-Х16-Х17-Х18-Х19,
где каждый из Х1-Х19 (напр., Х1-Х6, Х8, Х9, Х11-Х14 и Х16-Х19) независимо является любой аминокислотой (напр., встречающейся в природе аминокислотой, такой как Ala, Arg, Asn, Asp, Cys, Gln, Glu, Gly, His, Ile, Leu, Lys, Met, Phe, Pro, Ser, Thr, Trp, Tyr и Val) или отсутствует, и по меньшей мере один (напр., 2 или 3) из X1, Х10 и X15 является аргинином. В некоторых вариантах осуществления Х7 является Ser или Cys; либо X10 и X15 каждый независимо являются Arg или Lys. В некоторых вариантах осуществления остатки с X1 по X19 включительно являются в значительной степени идентичными любой последовательности аминокислот из любой SEQ ID NOS: 1-105 и 107-116 (напр., Angiopep-1, Angiopep-2, Angiopep-3, Angiopep-4a, Angiopep-4b, Angiopep-5, Angiopep-6 и Angiopep-7). В некоторых вариантах осуществления по меньшей мере одна (напр., 2, 3, 4 или 5) из аминокислот Х1-Х19 является Arg. В некоторых вариантах осуществления полипептид имеет один или более дополнительных цистеиновых остатков на N-конце полипептида, С-конце полипептида или на них обоих,
В определенных вариантах осуществления любого из вышеупомянутых аспектов полипептид является модифицированным (как описано в данной публикации). Полипептид может быть амидированным, ацетилированным либо и тем и другим. Такие модификации полипептида могут иметь место как на амино-, так и на карбокси-конце полипептида. Конъюгаты изобретения могут также включать пептидомиметики (напр., как описано в данной публикации) любого из полипептидов, описанных в данной публикации. Полипептид может быть в мультимерной форме, например, димерной форме (напр., образованной посредством дисульфидных связей с помощью цистеиновых остатков).
В определенных вариантах осуществления полипептид имеет последовательность аминокислот, описанную в данной публикации, с по меньшей мере одним аминокислотным замещением (напр., 2, 3, 4, 5, 6, 7, 8, 9, 10, 11 или 12 замещений). Полипептид может содержать, например, от 1 до 12, от 1 до 10, от 1 до 5 или от 1 до 3 аминокислотных замещений, например, от 1 до 10 (напр., до 9, 8, 7, 6, 5, 4, 3, 2) аминокислотных замещений. Аминокислотное(ые) замещение(я) могут быть консервативными или неконсервативными. Например, полипептид может содержать аргинин на одной, двух или трех позициях, соответствующих позициям 1, 10 и 15 последовательности аминокислот любого из SEQ ID NO: 1, Angiopep-1, Angiopep-2, Angiopep-3, Angiopep-4a, Angiopep-4b, Angiopep-5, Angiopep-6 и Angiopep-7.
В любом из вышеупомянутых аспектов конъюгат может специфически исключать полипептид, который включает или состоит из любой из SEQ ID NOS: 1-105 и 107-116 (напр., Angiopep-1, Angiopep-2, Angiopep-3, Angiopep-4a, Angiopep-4b, Angiopep-5, Angiopep-6 и Angiopep-7). В некоторых вариантах осуществления полипептиды и конъюгаты по настоящему изобретению исключают полипептиды с последовательностями SEQ ID NOs: 102, 103, 104 и 105.
В некоторых вариантах осуществления последовательность аминокислот по меньшей мере на 35%, 40%, 50%, 60%, 65%, 70%, 75%, 80%, 85%, 90% или 95% идентична последовательности аминокислот, выбранной из группы, которая состоит из SEQ ID NOS: 1-105 и 107-116 или их функциональных производных, В определенных вариантах осуществления последовательность аминокислот по меньшей мере на 35%, 40%, 50%, 60%, 65%, 70%, 75%, 80%, 85%, 90% или 95% идентична последовательности аминокислот, выбранной из группы, которая состоит из Angiopep-2 (SEQ ID NO: 97), Angiopep-4b, Angiopep-5, Angiopep-6 и Angiopep-7 (SEQ ID NOS: 109-112). Еще в некоторых вариантах осуществления последовательность аминокислот по меньшей мере на 35%, 40%, 50%, 60%, 65%, 70%, 75%, 80%, 85%, 90% или 95% идентична последовательности аминокислот Angiopep-2 (SEQ ID NO: 97).
В некоторых вариантах осуществления последовательность аминокислот содержит последовательность аминокислот, выбранную из группы, которая состоит из SEQ ID NOS: 1-105 и 107-116 или их функциональных производных. В определенных вариантах осуществления последовательность аминокислот соответствует Angiopep-2 (SEQ ID NO: 97), Angiopep-4b, Angiopep-5, Angiopep-6 или Angiopep-7 (SEQ ID NOS: 109-112).
В других вариантах осуществления последовательность аминокислот состоит из последовательности аминокислот, выбранной из группы, которая включает SEQ ID NOS: 1-105 и 107-116 или их функциональные производные. В определенных вариантах осуществления последовательность аминокислот соответствует Angiopep-2 (SEQ ID NO: 97), Angiopep-4b, Angiopep-5, Angiopep-6 или Angiopep-7 (SEQ ID NOS: 109-112).
В некоторых вариантах осуществления соединения по настоящему изобретению могут изменять аккумуляцию биологически активного агента (напр., подофиллотоксин-производных, таких как соединения по Формуле (I), или доксорубицин-производные, такие как соединения по Формуле (II)) в целевых типах клеток и тканей, по сравнению с соответствующим неконъюгированным биологически активным агентом. Еще в некоторых вариантах осуществления соединение по настоящему изобретению ускоряет аккумуляцию биологически активного агента в целевых типах клеток или тканей. В определенных вариантах осуществления концентрация биологически активного агента возрастает на 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 100%, 150%, 200%, 250%, 300%, 350%, 400%, 450%, 500%, 550%, 600%, 650%, 700%, 750%, 800%, 850%, 900%, 950%, 1 000%, 2 000%, 3 000%, 4 000%, 5 000%, 6 000%, 7 000%, 8 000%, 9 000%, 10 000%, 12 500%, 15 000%, 17 500% или 20 000% по сравнению с этим показателем, наблюдаемым при применении неконъюгированного биологически активного агента. В некоторых вариантах осуществления целевыми типами клеток или тканей являются головной мозг, яичники, печень, легкие, почки, селезенка или мышцы. В некоторых вариантах осуществления целевыми типами клеток являются головной мозг или яичники. В определенных вариантах осуществления биологически активный агент выбран из этопозида, этопозид фосфата, этопозидаDMG, тенипозида, доксорубицина или эпирубицина. В других вариантах осуществления соединение по настоящему изобретению содержит последовательность аминокислот Angiopep-2 (SEQ ID NO: 97), Angiopep-4b, Angiopep-5, Angiopep-6 или Angiopep-7 (SEQ ID NOS: 109-112) или их функциональные производные.
В третьем аспекте изобретение предоставляет фармацевтическую композицию, куда входят любое соединение по настоящему изобретению, описанному в данной публикации (напр., соединение, содержащее последовательность аминокислот, в значительной степени идентичную последовательности аминокислот, выбранной из группы, которая включает SEQ ID NOS: 1-105 и 107-116, или его функциональные производные, или фармацевтически приемлемые соли, где последовательность аминокислот содержит ковалентную связь между аминокислотой из последовательности аминокислот и соединением Формул (I) или (II) (напр., Соединение (1) или (2)) и фармацевтически приемлемый носитель. В четвертом аспекте изобретение предоставляет способ лечения или профилактического лечения рака, где способ включает введение пациенту терапевтически эффективного количества любого соединения по настоящему изобретению, описываемого в данной публикации (напр., соединения, содержащего последовательность аминокислот, в значительной степени идентичную последовательности аминокислот, выбранной из группы, которая включает SEQ ID NOS: 1-105 и 107-116 или их функциональные производные, где последовательность аминокислот содержит ковалентную связь между аминокислотой последовательности аминокислот и соединением Формул (I) или (II)).
В некоторых вариантах осуществления соединение представляет собой Соединение (1) или (2). В некоторых вариантах осуществления подофиллотоксин-производное выбрано из:
и ,
где
Y является Н; каждый R2 независимо является Н или Р(O)(ОН)2 или -C(O)R8; каждый R6 независимо является СН3 или 2-тиофеном; каждый Y является -С(O)(СН2)nC(O)-; каждый R8 независимо является произвольно замещенным C1-6 алкилом; и каждый n независимо равен 2, 3 или 4. В некоторых вариантах осуществления n равен 3. В некоторых вариантах осуществления каждый R2 является -C(O)R8. В некоторых вариантах осуществления R8 представляет собой С1-6 алкил, содержащий по меньшей мере одну произвольно замещенную аминогруппу (напр., NH2 или N(СН3)2). В определенных вариантах осуществления -C(O)R8 - это С-связанная аминокислота. В некоторых вариантах осуществления подофиллотоксин-производное - это этопозид, этопозид фосфат, этопозид 4'-диметилглицин (этопозидDMG) или тенипозид. Еще в некоторых вариантах осуществления соединение представляет собой доксорубицин или любое доксорубицин-производное (напр., соединение по Формуле (II)), описываемое в данной публикации.
В некоторых вариантах осуществления способ также включает введение второго агента. В других вариантах осуществления агент является терапевтическим препаратом. В определенных вариантах осуществления второй терапевтический препарат также ковалентно связан с соединением по настоящему изобретению. Еще в некоторых вариантах осуществления второй терапевтический препарат не связан ковалентно с соединением по настоящему изобретению. В некоторых вариантах осуществления терапевтический препарат представляет собой лекарственное вещество, готовый лекарственный препарат; агент, излучающий радиацию, клеточный токсин, их биологически активный фрагмент или их смесь, предназначенные для лечения болезни. В других вариантах осуществления введение лекарственного препарата производится параллельно с другим терапевтическим режимом. В некоторых вариантах осуществления терапевтический режим представляет собой радиационную терапию, химиотерапию, трансплантацию стволовых клеток, трансплантацию костного мозга, хирургическое вмешательство или лечение гипертермии. В некоторых вариантах осуществления второй терапевтический препарат - это полипептид, который включает или состоит из последовательности Angiopep-2 (SEQ ID NO: 97), предпочтительно где Angiopep-2 конъюгирован с противораковым агентом (напр., паклитакселом), напр., ANG1005, который имеет структуру:
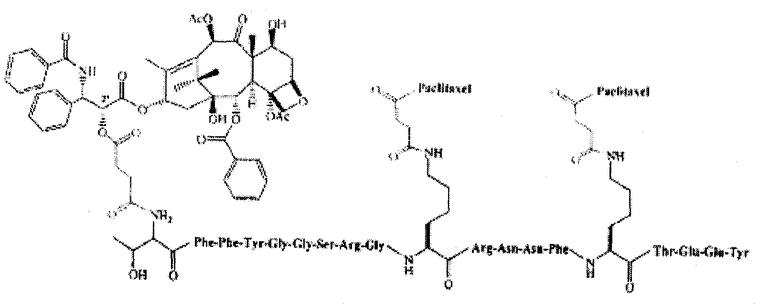
ANG1005:
TxlAn2 (3:1 конъюгат)
Еще одним примером второго терапевтического препарата может служить вещество, описанное в Патенте США №7,557,182, который включен в данную публикацию посредством ссылки.
В некоторых вариантах осуществления рак является раком головного мозга. В других вариантах осуществления рак головного мозга - это глиобластома, глиома, невринома слухового нерва, аденома, астроцитома, папиллома хороидального сплетения, лимфома ЦНС, эпендимома, ганглиоцитома, ганглиоглиома, медуллобластома (mdl), анапластическая (злокачественная) менингиома или нейрофиброматоз. Еще в других вариантах осуществления рак - это острый лимфолейкоз, острый миелобластный лейкоз, адренокортикальный рак, внутривенный и внутрипузырный рак мочевого пузыря, костная саркома, рак молочной железы, карциноид синдром (тонкой кишки), рак эндометрия, саркома Юинга, гинекологические саркомы, рак головы и шеи (плоскоклеточный рак), рак печени, болезнь Ходжкина, рак островковых клеток, лейкемия, рак легких, злокачественная лимфома, множественная миелома, нейробластома, неходжкинская лимфома, рак поджелудочной железы, рак предстательной железы, остеогенная саркома, рак яичников, ретинобластома, рабдомиосаркома, рак желудка, рак яичек, рак щитовидной железы, переходно-клеточный рак мочевого пузыря, саркома мягких тканей или опухоль Вильмса.
Согласно любому терапевтическому способу, описываемому в данной публикации, соединение по настоящему изобретению (Соединение (1) или (2)) либо их фармацевтически приемлемые соли могут вводиться пациенту в виде субтерапевтических или сверхтерапевтических доз, по сравнению с неконъюгированным терапевтическим препаратом (напр., этопозидом, этопозид фосфатом, этопозид 4-диметилглицином или доксорубицином).
В другом аспекте, изобретение предоставляет способ изготовления любого из соединений по настоящему изобретению, описываемых в данной публикации, где способ включает шаг по ковалентному связыванию подофиллотоксин-производного с любой последовательностью аминокислот, описываемой в данной публикации, или их функциональными производными, с использованием дифункциональной гидролизуемой связывающей группы. В некоторых вариантах осуществления последовательность аминокислот выбрана из SEQ ID NOS: 1-105 и 107-116 или их функциональных производных. В других вариантах осуществления последовательность аминокислот содержит последовательность аминокислот Angiopep-2 (SEQ ID NO: 97), Angiopep-4b, Angiopep-5, Angiopep-6 или Angiopep-7 (SEQ ID NOS: 109-112).
В некоторых вариантах осуществления способ изготовления любого из соединений по настоящему изобретению включает такие шаги:
(a) комбинирование упомянутого соединения по Формуле (I) с вышеуказанной дифункциональной гидролизуемой связывающей группой с целью образования ковалентного аддукта; и
(b) комбинирование аддукта по пункту (а) с упомянутой последовательностью аминокислот; и где аддукт по пункту (а) произвольно может быть очищен перед использованием в пункте (b).
В некоторых вариантах осуществления 1.0-10.0 эквиваленты дифункциональной гидролизуемой связывающей группы используются по отношению к соединению по Формуле (I). Например, могут быть использованы эквиваленты 1.1; 1.2; 1.3; 1.4; 1.5; 1.6; 1.7; 1.8; 1.9; 2.0. 2.1; 2.2.; 2.3; 2.4; 2.5; 2.6; 2.7; 2.8; 2.9 или 3.0. В других вариантах осуществления используются эквиваленты дифункциональной гидролизуемой связывающей группы 3.5; 4,0; 4.5; 5.0; 5.5; 6.0; 6.5; 7.0. 7.5; 8.0; 8.5; 9.0; 9.5 или 10.0. В определенных вариантах осуществления способ включает применение пептидного агента реакции сочетания. В некоторых вариантах осуществления пептидный агент реакции сочетания представляет собой N,N,N',N'-Тетраметил-O-(бензотриазол-1-ил)уроний тетрафтороборат (TBTU). В некоторых вариантах осуществления дифункциональная гидролизуемая связывающая группа выбрана из бикарбоновой кислоты, бикарбоната, карбонового ангидрида, диизоцианата или дифосфоновой кислоты. В определенных вариантах осуществления дифункциональная гидролизуемая связывающая группа выбрана из янтарной кислоты, глутаровой кислоты, глутарового ангидрида или масляной кислоты.
В некоторых вариантах осуществления подофиллотоксин-производное выбрано из:
и
где Y является Н; каждый R2 независимо является Н или Р(O)(ОН)2, либо -C(O)R8; каждый R6 независимо является СН3 или 2-тиофеном; каждый Y является -С(O)(СН2)nC(O)-; каждый R8 независимо является произвольно замещенным С1-6 алкилом; и каждый n независимо равен 2, 3 или 4. В некоторых вариантах осуществления n равен 3. В некоторых вариантах осуществления каждый R2 является -C(O)R8. В некоторых вариантах осуществления R8 представляет собой С1-6 алкил, содержащий по меньшей мере одну произвольно замещенную аминогруппу (напр., NH2 или N(СН3)2). В определенных вариантах осуществления -C(O)R8 - это С-связанная аминокислота. В некоторых вариантах осуществления подофиллотоксин-производное - это этопозид, этопозид фосфат, этопозидDMG или тенипозид.
В любых способах или композициях, описываемых в данной публикации, фармацевтически приемлемая соль соединения может быть моно-, ди-, три- или тетра-соль добавления кислоты (напр., треххлористоводородная соль). В любых вариантах осуществления, описываемых в данной публикации, любые соединения по Формуле (I) или (II) (напр., этопозид, этопозидDMG или доксорубицин), которые ковалентно связаны с полипептидом, являются сайтом протонирования. Например, в Соединении (1), протонируются (присоединяется протон) 1, 2 или 3 части молекулы этопозидаDMG, либо в Соединении (2), протонируются 1, 2 или 3 части молекулы доксорубицина с образованием соли добавления кислоты (напр., моно-, ди- или треххлористоводородной соли).
Термин «C1-6 алкил» или «алкил» в данном документе относится к произвольно замещенной C1-6 насыщенной группе углеводородов. Алкильная группа может быть линейной или разветвленной. Примеры алкильных радикалов включают, помимо прочего, метил, этил, n-пропил, изопропил, n-бутил, изо-бутил, втор-бутил, втор-пентил, изо-пентил, трет-бутил, n-пентил, неопентил, n-гексил, втор-гексил, n-гептил, n-октил, n-децил, n-ундецил, додецил и т.п., которые могут нести один или более заместителей. Например, замещенные алкильные группы могут иметь 1, 2, 3, 4, 5 или 6 заместителей.
Термин «арил» в данном документе относится к произвольно замещенной моно- или полициклической, ароматической, углеродной (без заместителей в цикле) части молекулы, имеющей 5-14 атомов углерода. В определенных вариантах осуществления настоящего изобретения «арил» относится к замещенной или незамещенной моноциклической или бициклической группе. Примеры арильных групп включают, помимо прочего, фенил, нафтил, тетрагидронафтил, инданил, инденил и т.п., которые могут нести один или более заместителей. В число арилов также включаются гетероарилы.
Термин «С-связанная аминокислота» в данном документе относится к аминокислоте, которая ковалентно связана с другим соединением (напр., с любым из подофиллотоксин-производных, описываемых в данной публикации) посредством С-конца аминокислоты.
Термин «С3-10 циклоалкил» или «циклоалкил» в данном документе относится к произвольно замещенной насыщенной 3…10-членной моноциклической или бициклической системе углеводородных колец. Примеры циклоалкилов включают циклопропил, циклобутил, циклопентил, циклогексил и циклогептил. Замещенный циклоалкил может иметь, например, 1, 2, 3, 4, 5, 6 или 7 заместителей.
Термин «гетероарил» в данном документе относится к замещенной или незамещенной моно- или полициклической, ароматической части молекулы, которая имеет 5-14 кольцевых атомов, из которых один, два, три или четыре кольцевых атома могут быть выбраны из S, О и N, а остальные кольцевые атомы являются атомами углерода. Примеры гетероарильных групп включают, помимо прочего, пирролил, пиразолил, имадазолил, пиридинил, пиримидинил, пиразинил, пиридазинил, триазинил, тетразинил, прииролизинил, индолил, хинолинил, изохинолинил, бензимидазолил, индазолил, хинолизинил, циннолинил, хиназолинил, фталазинил, нафтиридинил, хиноксалинил, тиофенил, тиепинил, фуранил, бензофуранил, тиазолил, изотиазолил, тиадиазолил, оксазолил, изоксазолил, оксадиазолил и т.п., которые могут нести один или более заместителей. Термин «гетероциклический» или «гетероциклил» в данном документе относится к произвольно замещенной неароматической, частично ненасыщенной или полностью насыщенной, 3…10-членной кольцевой системе, которая содержит одиночные кольца из 3…8 атомов и би- и трициклические кольцевые системы, которые могут содержать ароматические 5- или 6-членные арильные или гетероарильные группы, слитые с неароматическим кольцом. Эти гетероциклические кольца содержат такие, которые имеют от 1 до 3 гетероатомов, независимо выбранных из кислорода, серы и азота, в которых гетероатомы азота и серы произвольно могут быть окислены, а гетероатом азота произвольно может быть кватернизован или замещен. В определенных вариантах осуществления термин гетероциклический относится к неароматическому 5-, 6- или 7-членному моноциклическому кольцу, где по меньшей мере один атом кольца - это гетероатом, выбранный из О, S и N (где гетероатомы азота и серы могут быть произвольно окислены), а остальные атомы кольца являются атомами углерода, и радикал присоединяется к остальной молекуле посредством любого из кольцевых атомов. Примеры гетероциклов включают, помимо прочего, азациклопропанил, азациклобутанил, 1,3-диазатидинил, пирролидинил, пиперидинил, пиперазинил, тиранил, тиэтанил, тетрагидротиофенил, дитиоланил, тетрагидротиопиранил, оксиранил, оксетанил, тетрагидрофуранил, тетрагидропиранил, диоксанил, оксатиоланил, морфолинил, тиоморфолинил, тиоксанил, хинуклидинил и т.п., которые могут нести один или более заместителей. Термин «фармацевтически приемлемая соль» в данном документе охватывает те соли добавления кислоты, которые, в рамках тщательной медицинской оценки, являются пригодными для применения в контакте с тканями человека и животных, не вызывая существенных токсичности, раздражения, аллергических реакций и т.п. и сопоставимы с приемлемым соотношением польза/риск. Фармацевтически приемлемые соли хорошо известны профильным специалистам. Например, фармацевтически приемлемые соли описаны у авторов: Berge et al., J.Pharmaceutical Sciences 66:1-19, 1977 и в работе Pharmaceutical Salts: Properties, Selection и Use, (Eds. P.H.Stahl and C.G.Wermuth), Wiley-VCH, 2008. Соли могут быть получены in situ во время финальной изоляции и очистки соединений, описываемых в данной публикации, или приготовлены отдельно путем реакции соединения, имеющего одну или основную группы (напр., 1, 2, 3, 4, 5, 6, 7, 8, 9 или 10), с желаемыми эквивалентами подходящей органической или неорганической кислоты. Представителями солей добавления кислоты являются такие соли, как ацетат, адипинат, альгинат, аскорбат, аспартат, бензолсульфонат, бензоат, бисульфат, борат, бутират, камфорат, камфорсульфонат, цитрат, циклопентанпропионат, диглюконат, додецилсульфат, этансульфонат, фумарат, глюкогептонат, глицерофосфат, гемисульфат, гептонат, гексаноат, гидробромид, гидрохлорид, гидройодид, 2-гидрокси-этансульфонат, лактобионат, лактат, лаурат, лаурилсульфат, малат, малеат, малонат, метансульфонат, 2-нафталенсульфонат, никотинат, нитрат, олеат, оксалат, пальмитат, памоат, пектинат, персульфат, 3-фенилпропионат, фосфат, пикрат, пивалат, пропионат, стеарат, сукцинат, сульфат, тартрат, тиоцианат, толуолсульфонат, трифторацетат, трифторметилсульфонат, ундеканоат, валерат и т.п. Среди солей щелочных металлов (1-я группа Периодической системы, щелочи) и щелочноземельных металлов (2-я группа, щелочные) можно назвать соли натрия, лития, калия, кальция, магния и т.п., а также катионы нетоксичного аммония, четвертичного аммония и аминов, в том числе, помимо прочего, аммония, тетраметиламмония, тетраэтиламмония, метиламина, диметиламина, триметиламина, триэтиламина, этиламина и т.п. Желательно, чтобы «фармацевтически приемлемая соль добавления кислоты» была моно-, би-, три- или тетра-соль добавления кислоты любого из соединений, описываемых в данной публикации (напр., моно-, би-, три- или тетрахлористоводородная соль любого из соединений, описываемых в данной публикации).
Термин «фосфат» в данном документе относится к пятивалентной фосфористой группе, имеющей формулу -OP(=O)(OR')(OR"), где каждый R' и R" независимо выбраны из водорода, C1-6 алкила, С2-6 алкенил, С2-6 алкинила, С3-10 циклоалкила, гетероциклила, арила или гетероарила.
Если группа охарактеризована как «произвольно замещенная», то произвольные заместители могут быть выбраны независимо из групп, которые включают, помимо прочего: С1-6 алкил; галоген; азидо(-N3), нитро (-NO2), циано (-CN), ацилокси, ацил (-C(O)R), (-OC(O)R), алкокси (-OR), амидо (-NRC(O)R' или -C(O)NRR'), амино (-NRR'), арил, карбоновую кислоту (-CO2H), карбоновый сложный эфир (-CO2R), карбамоил (-OC(O)NRR' или -NRC(O)OR'), циклоалкил, гетероциклил, гидрокси (-ОН), изоциано (-NC), фосфат (-P(O)(OR)(OR')), сульфонат (-SO2OR) или сульфонил (-SO2R), где каждый R или R' независимо выбраны из Н, C1-6 алкила, циклоалкила, гетероциклила, арила или гетероарила. замещенная группа может иметь, например, 1, 2, 3, 4, 5, 6, 7, 8 или 9 заместителей. В некоторых вариантах осуществления группа заместителей может, в свою очередь, в дальнейшем замещаться, при этом атом водорода замещается заместительной группой из числа описываемых в данной публикации.
Под «вектором» понимается соединение или молекула, такая как полипептид, которое можно транспортировать в определенный вид клеток (напр., печень, яичники, легкие, почки, селезенку или мышцы) или через ГЭБ-барьер. Вектор может быть присоединен (в том числе ковалентно) или конъюгирован к агенту и таким образом может перенести этот агент в определенный тип клеток или преодолеть ГЭБ. В определенных вариантах осуществления вектор может связываться с рецепторами, присутствующими в клетках рака или эндотелиальных клетках головного мозга и таким образом попадать в клетки рака или преодолевать ГЭБ путем трансцитоза. Вектор может представлять собой молекулу, для которой может быть достигнут высокий уровень трансэндотелиального транспорта, без вреда для клетки или целостности ГЭБ. Вектор может быть полипептидом или пептидомиметиком и может быть натуральным (встречающимся в природе) или полученным путем химического синтеза или рекомбинантной генетической технологии.
Под «конъюгатом» понимается вектор, связанный с агентом. Конъюгация может быть химической по своему характеру, например, посредством линкера, или генетической, например, с помощью рекомбинантной генетической технологии, такой как в белке слияния с, например, репортерной молекулой (напр., зеленый флуоресцентный белок, (β-галактозидаза, гистидиновый маркер His-tag и т.д.).
Под вектором, который «эффективно транспортируется через ГЭБ» понимается вектор, способный пересекать ГЭБ по меньшей мере так же эффективно, как AngioPep-6 (напр., более, чем 38,5%, характерные для AngioPep-1 (250 нМ) в in situ анализе мозговой перфузии, описываемом в данной публикации). Соответственно, вектор или конъюгат, которые «не транспортируются эффективно через ГЭБ», транспортируются в головной мозг с более низким уровнем эффективности (напр., транспортируются менее эффективно, чем AngioPep-6).
Под вектором или конъюгатом, которые « эффективно транспортируются в определенный вид клеток» понимается вектор или конъюгат, способные аккумулироваться (напр., благодаря увеличению транспорта в клетку либо снижению вытекания из клетки или их комбинации) в этом виде клеток по меньшей мере на 10% (напр., 25%, 50%, 100%, 200%, 500%, 1 000%, 5 000% или 10 000%) больше, чем контрольное вещество, либо, в случае с конъюгатом, сравнительно с неконъюгированным агентом.
Под «в значительной степени чистым» или «изолированным» подразумевается соединение (полипептид или конъюгат), которое было отделено от других химических компонентов. Как правило, соединение является в значительной степени чистым, если оно по меньшей мере на 30% по массе является свободным от других компонентов. В определенных вариантах осуществления препарат является по меньшей мере на 50%, 60%, 75%, 85%, 90%, 95%, 96%, 97%, 98% или 99% по массе свободным от других компонентов. Очищенный полипептид может быть получен, например, путем экспрессии рекомбинантного полинуклеотида, кодирующего этот полипептид, либо в результате химического синтеза полипептида. Чистота может быть измерена с использованием любого приемлемого способа, например, колоночной хроматографии, электрофореза в полиакриламидном геле или ВЭЖХ-анализа.
Под «аналогом» понимается полипептид, образованный из оригинальной последовательности или из части оригинальной последовательности и который может содержать одну или более модификаций; например, одна или более модификаций в последовательности аминокислот (напр., добавление, деление, вставка или замещение аминокислоты), одна или более модификаций в остове или боковой цепи одной или более аминокислот или добавление группы или другой молекулы к одной или более аминокислотам (боковые цепи или остов). Аналог может иметь одну или более аминокислотных вставок, на одном или обоих концах полипептида или внутри последовательности аминокислот полипептида. Последовательность аналога может иметь сходство и(или) идентичность (напр., может быть в значительной степени идентичной) оригинальной последовательности или части оригинальной последовательности. Аналоги могут содержать структурную модификацию, напр., из числа описываемых в данной публикации. Степень подобия между двумя последовательностями базируется на процентном количестве тождественностей (идентичных аминокислот) и консервативных замещений. Аналог может иметь последовательность, по меньшей мере на 35%, 50%, 60%, 70%, 80%, 90% или 95% (напр., 96%, 97%, 98%, 99% и 100%) сходную с оригинальной последовательностью, с сочетанием одной или более модификаций в остове или боковой цепи аминокислоты или с добавлением группы или другой молекулы. Примеры аминокислот, которые следует считать подобными (консервативная аминокислота) другим, хорошо известны специалистам и включают, например, те, что перечислены в Табл. 3.
Под фразой «в значительной степени идентичный» понимается полипептид или нуклеиновая кислота, по меньшей мере на 35%, 40%, 50%, 55%, 60%, 65%, 70%, 75%, 85%, 90%, 95% или даже на 99% идентичная по сравнению с последовательностью аминокислоты или нуклеиновой кислоты. Для полипептидов длина сравниваемых последовательностей составляет по меньшей мере 4 (напр., по меньшей мере 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 25, 50 или 100) аминокислот. Для нуклеиновых кислот, длина сравниваемых последовательностей обычно составляет по меньшей мере 60 нуклеотидов, предпочтительно по меньшей мере 90 нуклеотидов и более предпочтительно по меньшей мере 120 нуклеотидов либо полноразмерную длину. Следует понимать, что могут быть обнаружены пробелы между аминокислотами аналогов, являющихся идентичными или подобными аминокислотам оригинального полипептида. Пробелы могут не содержать аминокислот, либо иметь одну или более аминокислот, не являющихся идентичными или подобными оригинальному полипептиду. Биологически активные аналоги векторов (полипептидов) по настоящему изобретению также считаются охваченными данной заявкой. Процент идентичности можно определить, например, с помощью n алгоритма GAP, BESTFIT или FASTA в программе Wisconsin Genetics Software Package Release 7,0, масса пробелов задана по умолчанию.
Под «функциональным производным» в настоящем изобретении понимается «химическое производное», «фрагмент» или «вариант» биологически активной последовательности или части вектора, агента или конъюгата либо их соли. Функциональное производное вектора может быть способным присоединяться или конъюгироваться с агентом и проникать в определенный вид клеток, транспортируя таким образом агент в клетку.
Под «химическим производным» понимается вектор, агент или конъюгат по настоящему изобретению, который содержит дополнительные химические части молекул, не являющихся частью вектора, агента или конъюгата вектор-агент, включая ковалентные модификации. Химическое производное может быть получено с помощью прямого химического синтеза, способами, известными специалистам. Такие модификации могут быть воплощены в белковом или пептидном векторе, агенте или конъюгате вектор-агент в результате реакции целевых остатков аминокислоты с органическим агентом для получения производных, способных вступать в реакцию с выбранными боковыми цепями или концевыми остатками. Химические производные вектора могут быть способны пересекать ГЭБ-барьер или проникать или аккумулироваться в определенном виде клеток (напр., описываемых в данной публикации, таких как яичники). В предпочтительном варианте осуществления, достигается высокий уровень трансэндотелиального транспорта через ГЭБ, без вреда нарушения целостности ГЭБ.
Под словом «фрагмент» понимается полипептид, образованный от части оригинальной или родительской последовательности или от аналога упомянутой родительской последовательности. Фрагменты охватывают полипептиды, укороченные (усеченные) на одну или более аминокислот, где местом усечения может быть амино-конец (N-конец), карбокси-конец (С-конец) или внутренняя часть белка. Фрагмент может содержать ту же самую последовательность, что и соответствующая часть оригинальной последовательности. Функциональные фрагменты вектора (полипептида), описываемого в данной публикации, охватываются данным изобретением. Фрагменты могут состоять по меньшей мере из 5 (напр., по меньшей мере из 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 25, 28, 30, 35, 40, 45, 50, 60, 75, 100 или 150) аминокислот. Фрагменты по настоящему изобретению могут содержать, например, полипептид длиной от 7, 8, 9 или 10 до 18 аминокислот. Фрагменты могут содержать любую из модификаций, описываемых в данной публикации (напр., ацетилирование, амидирование, аминокислотные замещения).
«Не встречающаяся в природе аминокислота» - это аминокислота, которая не находится и не производится в природных условиях у млекопитающих. Под словом «агент» понимается любое соединение, например, антитело или терапевтический препарат, маркер, метка или соединение для визуализации. «Терапевтический препарат» - это препарат, обладающий биологической активностью. В некоторых случаях, терапевтический препарат используется для лечения симптомов болезни, физического или ментального состояния, повреждения или инфекции и включает противораковые агенты, антибиотики, противоангиогенные агенты и молекулы, активные на уровне центральной нервной системы.
«Низкомолекулярное лекарственное средство» - это лекарственное средство с молекулярной массой 1000 г/моль или менее (напр., меньше чем 800, 600, 500, 400 или 200 г/моль).
Под «субъектом» понимается человек или представитель животного мира (млекопитающее).
Под «лечением» болезни, нарушения или состояния у субъекта понимается уменьшение по меньшей мере одного симптома болезни, нарушения или состояния с помощью введения субъекту терапевтического препарата. «Профилактическое лечение» болезни, нарушения или состояния у субъекта - это снижение частоты возникновения (напр., предотвращение) болезни, нарушения или состояния с помощью введения субъекту терапевтического препарата. Под «раком» понимается любая пролиферация клеток, уникальной особенностью которой является потеря нормального контроля, что может привести к неконтролируемому росту, недостатку дифференциации или способности заражать ткани и давать метастазы. Рак может развиться в любой ткани или любом органе. Рак включает, помимо прочего, рак головного мозга, яичников, печени, легких, почек или селезенки. Дополнительные виды рака описываются в данной публикации.
Слово «предоставлять», употребляемое в контексте вектора или конъюгата по настоящему изобретению, обозначает обеспечивать контакт вектора или конъюгата с целевой клеткой или тканью, in vivo или in vitro. Вектор или конъюгат могут предоставляться путем введения субъекту этого вектора или конъюгата.
«Введение» и «вводить» обозначает способ доставки, включая, помимо прочего, внутриартериально, интраназально, внутрибрюшинно, внутривенно, внутримышечно, подкожно, трансдермально или per os. Суточная дозировка может быть разделена на одну, две или более доз (приемов) в приемлемой форме для введения один, два или более раз за определенный период времени.
«Терапевтически эффективное» или «эффективное количество» - это количество терапевтического препарата, достаточное для улучшения, снижения, предотвращения, отсрочки, подавления или прекращения любого симптома болезни или состояния, подвергающегося лечению. Терапевтически эффективное количество агента не должно излечивать болезнь или устранять состояние, но оно обеспечивает лечение болезни или состояния таким образом, что начало болезни или состояния оказывается отсроченным, заблокированным или предотвращенным, либо симптомы болезни или состояния улучшаются, либо длительность болезни или состояния изменяется, либо, например, становится менее тяжелой, либо выздоровление субъекта наступает быстрее. «Субтерапевтическая доза» - это доза меньшая, чем минимальное эффективное количество терапевтического препарата, утвержденное для клинического применения у пациентов. «Сверхтерапевтическая доза» - это доза большая, чем максимальное эффективное количество терапевтического препарата, утвержденное для клинического применения у пациентов. Количество субтерапевтической или сверхтерапевтической дозы может варьироваться в зависимости от демографических характеристик пациентов (напр., взрослые, дети или пожилые пациенты) либо при применении параллельно с дополнительными терапевтическими препаратами (напр., при введении одновременно с другими терапевтическими препаратами или режимами лечения, например, такими как химиотерапия при раке).
Под «состоянием» понимается любая ситуация, вызывающая боль, дискомфорт, болезнь, заболевание или неработоспособность (умственную или физическую) у субъекта, включая неврологические заболевания, травмы, инфекции или хроническую или острую боль. Неврологические заболевания включают опухоли мозга, метастазы в головной мозг, шизофрению, эпилепсию, болезнь Альцгеймера, болезнь Паркинсона, болезнь Хантингтона и инсульт.
«Фармацевтическая композиция» - это терапевтически эффективное количество агента в комбинации с фармацевтически приемлемыми разбавителями, консервантами, солюбилизаторами, эмульгаторами или адъювантными веществами, например, любыми из описываемых в данной публикации.
«Терапевтическая доза» - это дозировка агента, такого как лекарственное средство (без вектора), подходящее для клинического применения, с учетом его токсичности или эффективности. С помощью конъюгации агента к вектору по настоящему изобретению становится возможным введение агента в дозе либо более низкой, либо более высокой, чем терапевтическая доза.
Если слова «диапазон» или «группа веществ» упоминаются по отношению к определенным характеристикам (напр., температура, концентрация, время и т.п.), то изобретение относится и прямо включает каждый отдельный член этого диапазона и комбинации субдиапазонов или подгрупп в его пределах. Так, например, если указана длина от 9 до 18 аминокислот, то следует понимать, что сюда включаются все без исключения отдельные значения длины, напр., длина 18, 17, 15, 10, 9 и любые значения между указанными.
Поэтому, если не оговорено иное, подразумевается, что каждый упоминаемый в данной публикации диапазон охватывает все без исключения значения в его пределах. Например, при экспрессии длины от 5 до 19 аминокислот это следует понимать как от 5 до 19 включительно. То же самое касается и других параметров, таких как последовательности, длина, концентрация, элементы и т.п.
Последовательности, области, части, описываемые в данной публикации, включают каждую отдельную последовательность, область и часть, а также все без исключения субпоследовательности, субобласти и субчасти, независимо от того, упоминаются ли такие субпоследовательности, субобласти и субчасти как безусловно включающие конкретные возможности, либо как исключающие конкретные возможности, либо как их сочетание. Например, исключающее определение области может звучать так; «при условии, что указанный полипептид не короче, чем 4, 5, 6, 7, 8 или 9 аминокислот. Еще один пример исключающего ограничения может звучать так: последовательность включает SEQ ID NO: X, за исключением (кроме) полипептида SEQ ID NO: Y и т.д. Или еще пример исключающего ограничения: при условии, что указанный полипептид не является (не включает или не состоит из) SEQ ID NO: Z.
Другие особенности и преимущества изобретения будут очевидны из нижеследующих Подробного описания, графических материалов и пунктов формулы изобретения.
Краткое описание чертежей
Фиг.1 отображает подавление роста подкожной U87 (s.c. U87) ксенографтной опухоли с помощью конъюгата доксорубицин-An2(3:1) («Доксорубицин-An2(3:1)»).
Фиг.2А отображает усвоение головным мозгом конъюгата 3:1 Этопозид: Angiopep-2 («Этоп-An2(3:1)»), измеренное in situ в ходе перфузии мозга.
Фиг.2В отображает усвоение головным мозгом конъюгата 3:1 этопозид 4'-диметилглицин:Angiopep-2 («ЭтопDMG-An2(3:1)»), измеренное in situ в ходе перфузии мозга.
Фиг.2С отображает усвоение головным мозгом конъюгата Доксорубицин-An2(3:1), измеренное in situ в ходе перфузии мозга.
Фиг.3 отображает in situ перфузию Этоп-An2(3:1).
Фиг.4А отображает усвоение в паренхиме неконъюгированного этопозида по сравнению с Этоп-An2(3:1).
Фиг.4В отображает перераспределение в головном мозге Этоп-An2(3:1) с последующей деплецией мозговых капилляров.
Фиг.5 отображает in situ мозговую перфузию Этоп-An2(3:1) по сравнению с неконъюгированным этопозидом у CD-I против P-gp нокаутных мышей.
Фиг.6 отображает ингибирование усвоения мозгом Этоп-An2(3:1) с помощью Angiopep-2.
Фиг.7 отображает in situ перфузию в мозге ЭтопDMG-An2 по сравнению с неконъюгированным ЭтопDMG у CD-I против P-gp нокаутных мышей.
Фиг.8 отображает данные по кинетике в плазме Этоп-An2(3:1).
Фиг.9 отображает распределение в головном мозге ЭтопDMG-An2 с последующим внутривенным болюсным введением мышам.
Фиг.10 отображает распределение в головном мозге Этоп-An2(3:1).
Фиг.11 отображает распределение в головном мозге Этоп-An2(3:1) по сравнению с неконъюгированным этопозидом через 30 мин после ВВ болюсного введения.
Фиг.12 отображает распределение в тканях Этоп-An2(3:1) по сравнению с неконъюгированным этопозидом через 30 мин после ВВ болюсного введения. Фиг.13А и Фиг.13В каждая показывает in vivo воздействие (DoxSu)3-An2 на мышей, получавших внутричерепную инъекцию U87 клеток глиобластомы. Фиг.13А отображает результаты, полученные в первом испытании (Испытание 1). Фиг.13В отображает результаты, полученные во втором испытании (Испытание 2). По его результатам, введение Соединения (2) позволяет увеличить среднее время выживаемости в статистически значимых пределах.
Подробное описание изобретения
Изобретение предоставляет соединения или их любые фармацевтически приемлемые соли, которые содержат последовательность аминокислот, в значительной степени идентичную последовательности аминокислот, выбранной из последовательностей аминокислот, описываемых в данной публикации (напр., SEQ ID NOS: 1-105 и 107-116) или их функциональных производных, где упомянутая последовательность аминокислот включает ковалентную связь между аминокислотой последовательности аминокислот и противораковым агентом (напр., подофиллотокси-производные, доксорубицин или доксорубицин-производные). Примеры подофиллотокси-производных включают, например, соединение, имеющее структуру согласно Формуле (I):
либо его стереоизомер или фармацевтически приемлемую соль, где каждый R1, R2 и R3 независимо выбран из Н, произвольно замещенного C1-6 алкила, C(O)R8 (напр., С(O)CH2N(СН3)2), P(O)(OR9)(OR10), S(O)2(OR9)) или гидролизуемого линкера Y, который содержит ковалентную связь с аминокислотой полипептида;
Х является О или NR7;
каждый R4, R5 и R7 независимо выбран из Н, произвольно замещенного С1-6 алкила, C(O)R8 или гидролизуемого линкера Y, который содержит ковалентную связь с аминокислотой полипептида;
R6 - это Н, произвольно замещенный С1-6 алкил, произвольно замещенный арил, произвольно замещенный гетероарил,
R8 выбран из произвольно замещенного C1-6 алкила (напр., CH2N(СН3)2) или произвольно замещенного арила;
каждый R9 and R10 независимо выбран из Н, произвольно замещенного C1-6 алкила или произвольно замещенного арила; и
n равно 2, 3 или 4; и
где один из R1, R2, R3, R4, R5 и R7 является Y. В некоторых вариантах осуществления не более чем один из R1, R2, R3, R4, R5 и R7 является Y. В некоторых вариантах осуществления Y - это -С(O)(СН2)nC(O)-. В некоторых вариантах осуществления каждый R2 является Н или C(O)CH2N(CH3)2. В определенных вариантах осуществления полипептид может быть по меньшей мере на 35%, 40%, 50%, 60%, 70%, 80%, 90%, 95%, 99% или даже на 100% идентичен полипептиду, описываемому в данной публикации. Полипептид может иметь одно или более (напр., 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14 или 15) замещений по сравнению с одной из последовательностей, описываемых в данной публикации. В определенных вариантах осуществления последовательность аминокислот ковалентно связана с дополнительным подофиллотоксин-производным (напр., соединением по Формуле (I)) с помощью второй, третьей, четвертой, пятой или даже шестой аминокислоты упомянутой последовательности аминокислот и на любой позиции последовательности аминокислот.
Примеры соединений по настоящему изобретению включают, помимо прочего, такие, которые имеют последовательность полипептида, соответствующую (SEQ ID NO: 97). В некоторых вариантах осуществления соединения имеют структуру:
где каждая (-(Формула(I)) представляет собой ковалентную связь между указанной аминокислотой и соединением по Формуле (I). В определенных вариантах осуществления соединения по Формуле (I) имеют структуру:
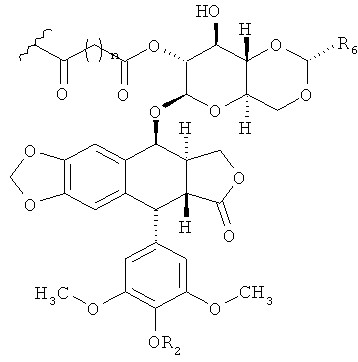 ,
,
где n равно 1, 2 или 3, R6 - это СН3 или 2-тиенил, и R2 - это Н, -ОР(O)(ОН)2 или -С(O)CH2N(СН3)2 или их любые фармацевтически приемлемые соли. В некоторых вариантах осуществления n равно 3, R6 - это СН3 и R2 - это Н. В других вариантах осуществления n равно 3, R6 - это СН3 и R2R6 - это -C(O)CH2N(CH3)2.
Другие варианты осуществления описываются более подробно ниже.
Подофиллотоксин-производные
Подофиллотоксин-производные включают соединения, такие как описываются Формулой (I), т.е. этопозид, тенипозид и их производные или фармацевтически приемлемые соли. Подофиллотоксин-производные - это типичные терапевтические препараты. Они могут ковалентно связываться с аминокислотой в любом полипептиде, описываемом в данной публикации (напр., Angiopep-2). Эти соединения могут обладать, например, противоопухолевой активностью, подавлять активность топоизомеразы II или обладать противовирусной активностью.
Этопозид и Этопозид-производные
Этопозид (также известный как Топозар, Вепезид или VP 16) - это подофиллотоксин-производное, имеющее структуру;
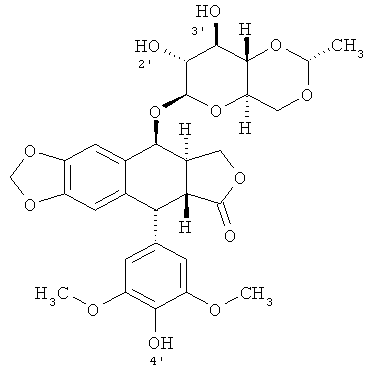 .
.
Химическая структура этопозида может быть изменена для получения его производных. Примером производного от этопозида является этопозид фосфат (ЕТОРОРНО®), где фенольный компонент -ОН заменен на -ОР(O)(ОН)2 или любую его фармацевтически приемлемую соль (напр., -OP(O)(ONa)2). Этопозид фосфат отличается улучшенной растворимостью в воде по сравнению с этопозидом. Другие этопозид-производные включают такие, где фенольный компонент -ОН заменен на ацилокси-группу (напр., -OC(O)R8, как описано в данной публикации), например, следующее соединение:
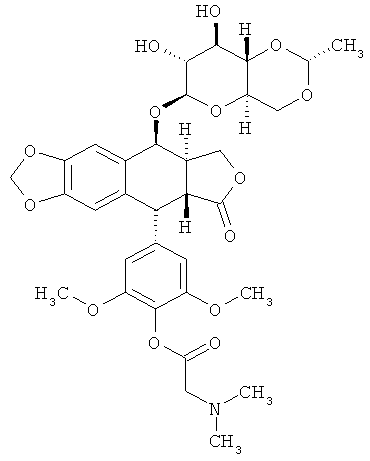
(«этопозид 4'-диметилглицин» или «этопозидDMG»).
Эти ацилированные производные этопозида также демонстрируют улучшенную растворимость в воде по сравнению с этопозидом при ковалентном присоединении к любому полипептиду, описываемому в данной публикации. Этопозид, этопозид фосфат, этопозидDMG или их производные могут быть ковалентно связаны с аминокислотой в полипептиде путем присоединения гидролизуемого ковалентного линкера Y, например, к 2" гидроксилу или 3" гидроксилу молекулы. Типичные линкеры могут быть образованы, например, из дикарбоновых кислот, таких как янтарная, глутаровая и масляная кислоты, или любого их ангидрида. Кроме того, ковалентный линкер может быть присоединен к этопозиду или его производным, в районе фенол -ОН группы. Этопозид-производные обычно описываются следующей Формулой:
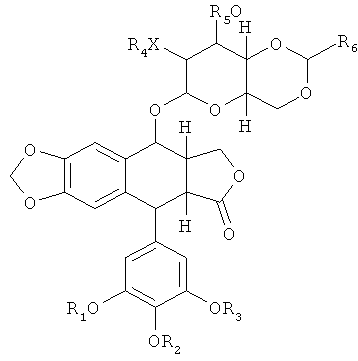
(I-A) или его стереоизомеры, где:
каждый R1, R2 и R3 независимо выбраны из Н, произвольно замещенного C1-6 алкила, C(O)R8, P(O)(OR9)(OR10) или S(O)2(OR9);
Х является О или NR7;
каждый R4, R5 и R7 независимо выбраны из Н, произвольно замещенного C1-6 алкила или C(O)R8;
R6 - это Н, произвольно замещенный C1-6 алкил, произвольно замещенный арил, произвольно замещенный гетероарил,
R8 выбран из произвольно замещенного C1-6 алкила или произвольно замещенного арила; и
каждый R9 и R10 независимо выбраны из Н, произвольно замещенного С1-6 алкила или произвольно замещенного арила.
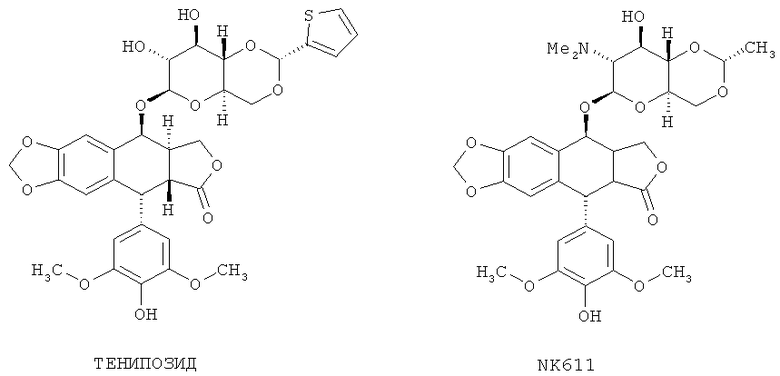
Если соединение по настоящему изобретению включает этопозид-производное в соответствии с Формулой (I), то один из R1-R6 содержит гидролизуемый линкер Y, как описано в данной публикации. В некоторых вариантах осуществления Y - это -С(O)(СН2)nC(O)- и n равно 2, 3 или 4. В типичных, но не ограничивающих данное изобретение вариантах осуществления, где R2 является C(O)R8, R8 может быть C1-6 алкилом, содержащим амино-заместители и имеющим произвольные дополнительные заместители. В некоторых вариантах осуществления C(O)R8 представляет собой С-связанную α-аминокислоту. С-связанная α-аминокислота может быть природной или искусственной аминокислотой. Другие типичные подофиллотоксин-производные по Формуле (I), которые могут быть ковалентно присоединены к любому полипептиду, описываемому в данной публикации, включают тенипозид и NK611 (Схема 3).
Схема 3
Дополнительные подофиллотоксин-производные
Еще некоторые подофиллотоксин-производные, подходящие для использования в данном изобретении, описаны в патентах США №№4,567,253; 4,609,644; 4,900,814; 4,958,010; 5,489,698; 5,536,847; 5,571,914; 6,051,721; 6,107,284; 6,475,486; 6,610,299; 6,878,746; 6,894,075; 7.087,641; 7,176,236; 7,241,595; 7,342,114 и 7,378,419; а также в опубликованных патентах США №№20030064482, 20030162722, 20040044058, 20060148728 и 20070249651, каждый из которых включен в данный документ посредством ссылки.
Например, этопозид-производные, описанные в Патенте США №7,176,236, могут быть ковалентно присоединены к аминокислоте в любом полипептиде, описываемом в данной публикации (напр., Angiopep-2). Соответственно, в одном из вариантов осуществления, соединения по настоящему изобретению включают структуру согласно Формуле (I):
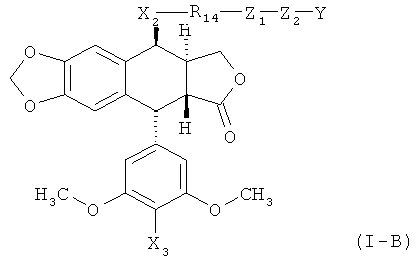 ,
,
где R2 и Y - такие же, как описано для Формулы (I);
Х2 - это -O-, -S-, -NH-, -СО-, -CH=N- или -CH2NH-;
Х3 - это OR2 или N(R2)2;
Z1 - это ковалентная связь, -NHCO-, -CONH-, -ОСО- или -СОО-;
Z2 - это ковалентная -(CH2)oR15, или -(СН2)о инкорпорирован в Z2 как 5-8-членное кольцо;
R14 - это ковалентная связь или произвольно замещенный алкил, алкенил или фенил; и
R15 - это замещенный алкил, замещенный алкенил или замещенный арил, где замещенная группа содержит по меньшей мере одну амино-группу.
В некоторых вариантах осуществления Х3 является -ОН, -OC(O)CH2NH2, -ОС(O)CH2NHCH3 или -OC(O)CH2N(CH3)2. В других вариантах осуществления Х является -NH-. В некоторых вариантах осуществления -R14-Z1-Z2- - это -(р-C6H4-R16)-, где R16 является -NO2, -F, -CONHCH2CH2C6H5 или -CONHCH2CH2(р-C6H4OH). В любом соединении по Формуле (I) или (I-A), группа OR2 может представлять собой -OC(O)R8.
В некоторых вариантах осуществления соединение по Формуле (I) или его фармацевтически приемлемая соль может улучшить физико-химические свойства (напр., растворимость). Например, если необходимо добиться повышенной растворимости, то соединением по Формуле (I) предпочтительно является ЭтопозидDMG.
Доксорубицин-производные
В некоторых вариантах осуществления в качестве противоракового агента выступают: доксорубицин (гидроксидаунорубицин или Adriamycin®), доксорубицин-производное, такое как эпирубицин (Ellence® или Pharmorubicin®), либо их фармацевтически приемлемая соль. Структуры этих типичных соединений показаны на Схеме 4. Доксорубицин и доксорубицин-производные могут быть ковалентно присоединены к аминокислоте в любом полипептиде, описываемом в данной публикации, с помощью гидролизуемого ковалентного линкера Y, согласно приведенному в данном документе определению, который ковалентно связывается, например, с 14-гидроксильной группой.
Схема 4
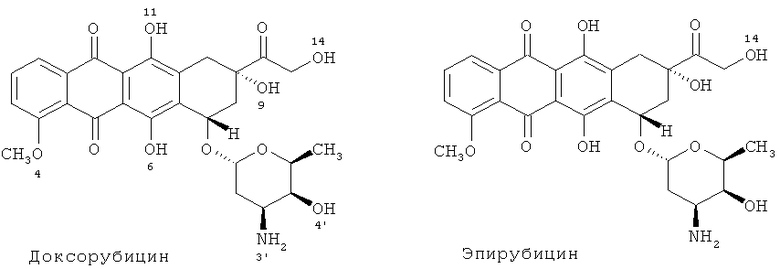
Доксорубицин-производные в общем могут быть описаны следующей Формулой (II):
 ,
,
где каждый X1, Х2, Х3, Х4 и Х5 независимо выбраны из ковалентной связи, О или NR25;
каждый R17, R18, R19, R20, R20, R22, R22, R23, R24 и R25 независимо выбраны из: Н, произвольно замещенный C1-6 алкил, произвольно замещенный C2-6 алкенил, произвольно замещенный С2-6 алкинил, произвольно замещенный циклоалкил, произвольно замещенный гетероциклил, либо является гидролизуемым линкером Y, согласно данному в данной публикации определению.
Если соединение по Формуле (II) присоединено к любому полипептиду, описываемому в данной публикации, то один из R17, R18, R19, R20, R20, R21, R22, R23, R24 и R25 является Y.
В определенных вариантах осуществления R21 является Y. Соединения по Формуле (II) включают соединения, имеющие структуру согласно Формуле (II-A):
где Y - это гидролизуемый линкер, согласно данному в данной публикации определению; X2R18 является Н или NH2; X3R19 является Н или ОН; и X4R20 - это Н или произвольно замещенный C1-3 алкил. В некоторых вариантах осуществления гидролизуемый линкер Y является -С(O)(СН2)nC(O)- и n равно 2, 3 или 4. В определенных вариантах осуществления соединение по Формуле (II) представляет собой:
Другие доксорубицин-производные можно найти в Патентах США №№4,098,884, 4,301,277, 4,314,054. 4,464,529, 4,585,859, 4,672,057, 4,684,629, 4,826,964, 5,200,513, 5,304,687, 5,594,158, 5,625,043 и 5,874,412, каждый их которых включен в данную публикацию посредством ссылки.
В некоторых вариантах осуществления соединение по Формуле (II) или его фармацевтически приемлемая соль, может улучшить физико-химические свойства (напр., растворимость). Например, если необходимо добиться повышенной растворимости, то соединением по Формуле (II) предпочтительно является хлористоводородная соль доксорубицина.
Не только Angiopep-2, но и подофиллотоксин-производные, такие как этопозид, этопозид фосфат, этопозидDMG, тенипозид, NK611 и другие соединения Формул (I) и (I-A), а также доксорубицин, эпирубицин и другие доксорубицин-производные (напр., соединения по Формуле (II)) тоже могут быть конъюгированы с любым полипептидом, описываемым в данной публикации (напр., Angiopep-4b, Angiopep-5, Angiopep-6 или Angiopep-7). Гидролизуемые линкеры, такие как линкеры, включающие группы сложных эфиров, могут быть использованы для ковалентного связывания противоракового агента (напр., подофиллотоксин-производные или доксорубицин-производные) с полипептидом (напр., Пример 1, описываемый в данной публикации). Этопозид, этопозид фосфат, этопозидDMG.Другие его подофиллотоксин-производные, доксорубицин, эпирубицин и другие доксорубицин-производные имеют множество стратегически выгодных позиций (напр., 2" и 3" гидроксилы этопозида, этопозид фосфата и этопозидаDMG, а также 14 гидроксил доксорубицина и эпирубицина). Например, дифункциональная группа (напр., реагент, образованный из янтарной кислоты, глутаровой кислоты, глутарового ангидрида или масляной кислоты или любого ее ангидрида) может быть присоединена к этопозиду на участке 2" гидроксил либо к доксорубицину на участке 14 гидроксил. Эти типичные промежуточные соединения могут затем быть активированы с помощью пептид-связующего реагента, такого как TBTU (O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилуроний тетрафтороборат), и обработаны полипептидом. Среди других пептид-связующих агентов можно назвать карбодиимиды (напр., дициклогексилкарбодиимид (DCC), диизопропилкарбодиимид (DIC) и 1-Этил-3-(3-диметиламинопропил) карбодиимид гидрохлорид) (EDC-HC1)), триазолы (напр., 1-гидрокси-бензотриазол (HOBt) и 1-гидрокси-7-аза-бензотриазол (HOAt)), родственные бензотриазольные пептид-связующие агенты, такие как O-Бензотриазол -N,N,N',N'-тетраметил-уроний-гексафтор-фосфат (HBTU), 2-(6-Хлор-1Н-бензотриазол-1-ил)-1,1,3,3-тетраметиламиний гексафторфосфат (HCTU), 2-(1Н-9-Азобензотриазол-1-ил)-1,1,3,3-тетраметиламиний гексафторфосфат (HATU), Бензотриазол-1-ил-окси-трис-(диметиламино)-фосфоний гексафторфосфат (ВОР реагент) и бензотриазол-1-ил-окситрипирролидинфосфоний гексафторфосфат (РуВОР) и 3-(Диэтоксифосфорилокси)-1,2,3-бензотриазин-4(3Н)-один (DEPBT). Конъюгат впоследствии может быть очищен. Каждое промежуточное соединение или продукт этой процедуры синтеза очищают и производят валидацию с помощью различных способов, таких как ВЭЖХ, тонкослойная жидкостная хроматография, ЯМР (13С или 1Н обмен), точка плавления, масс-спектрометрия. Финальный конъюгат анализируют с помощью масс-спектрометрии и электрофореза в полиакриламидном геле в присутствии додецилсульфата натрия. Такой подход позволяет определить число молекул (напр., этопозида, этопозида фосфата, этопозидаDMG, доксорубицина или эпирубицина), конъюгированных с каждым вектором.
Гидролизуемые линкеры
Если соединение по Формуле (I) ковалентно присоединяется посредством гидролизуемого линкера Y к аминокислоте в полипептиде, то линкер может быть расположен на участках R1, R2, R3, R4, R5 или R7. Аналогично, если соединение по Формуле (II) ковалентно присоединяется посредством гидролизуемого линкера Y к аминокислоте в полипептиде, то линкер может быть расположен на любом из R17, R18, R19, R20, R20, R21, R22, R23, R24 и R25. Типичные, но не ограничивающие изобретение гидролизуемые линкеры могут быть изготовлены из дикарбоновых кислот, бикарбонатов, карбоновых ангидридов, диизоцианатов или дифосфоновых кислот. Соединение, содержащее соединение по Формуле (I) или (II), которое ковалентно присоединено к аминокислоте в любой из последовательностей аминокислот, описываемых в данной публикации, может также быть описано следующей формулой:
D-G-X-G'-A (III),
где каждый G и G' - это группа, независимо выбранная из -С(O)-, -С(O)O-, -ОС(O)-, -S(O)2O-, -OS(O)2-, -S(O)2NH-, -NHS(O)2- и -OP(O)(OR11)O-;
G ковалентно связан с D, где D - это подофиллотоксин-производное (напр., соединение по Формуле (I)), либо доксорубицин или доксорубицин-производное (напр., соединение по Формуле (II);
G' ковалентно связан с А, где А - это аминокислота из последовательности аминокислот, описываемой в данной публикации (напр., последовательности аминокислот, описанные в Табл.1, либо их функциональные производные); и Х - это -(произвольно замещенный арил)-, -(CR12R13)n-, -O{(CR12R13)2O}n-, -{(CR12R13)2O(CR12R13)2}n- или -(CR12R13)oY(CR12R13)p-, где каждый n, о и р независимо являются целыми числами от 1 до 10;
R11 - это Н или низший С1-6 алкил;
R12 и R13 каждый независимо выбраны из Н, ОН или низшего C1-6алкила; и
Y - это О, NH, N(низший С1-5 алкил) или -произвольно замещенный арил.
Каждый n, о и р могут быть независимо 1, 2, 3, 4, 5, 6, 7, 8, 9 или 10.
В некоторых вариантах осуществления молекулярная часть G-X-G' по Формуле (III) выбрана из -С(O)СН2С(O)-, -С(O)(СН2)2С(O)-, -С(O)(СН2)3С(O)-, -С(O)(СН2)4С(O)-, -С(O)(СН2)5С(O)-, -С(O)(СН2)6С(O)-, -С(O)(ОСН2СН2)ОС(O)-, С(O)(ОСН2СН2)2OC(O)-, -С(O)(ОСН2СН2)3ОС(O)- и -С(O)(ОСН2СН2)4OC(O)-.
Полипептиды
Типичные последовательности аминокислот, пригодные для соединения по настоящему изобретению, включают, помимо прочего, последовательности аминокислот, перечисленные в Табл.1.
Помимо последовательностей аминокислот, описанных в Табл.1, настоящее изобретение также предоставляет фрагменты этих последовательностей аминокислот (напр., функциональные фрагменты). В определенных вариантах осуществления фрагменты способны проникать или аккумулироваться в определенном виде клеток (напр., яичники, печень, легкие, почки, селезенка или мышцы) либо способны пересекать ГЭБ. Полипептид может быть укорочен на 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12 или более аминокислот, как на N-конце, так и на С-конце полипептида либо на них обоих. Другие фрагменты включают последовательности, где удалены внутренние части полипептида.
Дополнительные полипептиды по настоящему изобретению могут быть идентифицированы с помощью одного из анализов или способов, описанных в Патентной заявке США №2006/0189515, включенной в данной публикации посредством ссылки, либо любых других способов, известных профессионалам. Например, интересующий вектор может быть получен в ходе общепринятой процедуры синтеза и конъюгирован, например, с соединением по Формуле (I) или (II) и введен лабораторному животному. Биологически активный вектор может быть идентифицирован, например, на основании его эффективности в отношении увеличения выживаемости животных, которым была сделана инъекция опухолевых клеток и которые получали лечение конъюгатом, по сравнению с контрольной группой, которая не получала лечение конъюгатом (напр., получала неконъюгированный агент).
В другом примере, биологически активный полипептид по настоящему изобретению может быть идентифицирован на основании его расположения в паренхиме, которое оценивают в ходе in situ церебрального перфузионного анализа. Для идентификации таких векторов может применяться in vitro ГЭБ-анализ, такой как модель, разработанная CELLIAL™ Technologies.
Кроме того, для определения аккумуляции (накопления) в других тканях могут быть использованы оценочный анализ, а также типичные способы исследования, описываемые в данной публикации. Меченые полипептиды по настоящему изобретению могут быть введены животным, с целью дальнейшего измерения их аккумуляции в различных органах. Например, полипептид, конъюгированный с обнаруживаемой меткой (напр., метка для околоинфракрасной флуоресцентной спектроскопии, такая как Су5,5), позволяет получить «живую» визуализацию in vivo. Такой полипептид может быть введен животному, и исследователь может наблюдать его присутствие в органе, что позволяет определить скорость накопления и количество аккумулированного полипептида в органе-мишени. В других вариантах осуществления полипептид изобретения может быть помечен радиоактивным изотопом (напр., 125I). Полипептид затем вводят животному. Через некоторое время животное умерщвляют, а его органы извлекают. Затем измеряют количество радиоизотопа в каждом органе, используя любые способы, известные в данной научной области. Сравнивая количество меченого исследуемого полипептида в определенном органе за вычетом количества меченого контрольного вещества, устанавливают способность исследуемого полипептида накапливаться, скорость накопления и количество аккумулированного полипептида в конкретной ткани. Среди подходящих веществ негативного контроля можно назвать любые полипептиды, о которых известно, что они обычно не транспортируются в определенный вид клеток.
Например, аминогруппы из Angiopep-1 (SEQ ID NO: 67) и Angiopep-2 (SEQ ID NO:97) могут быть использованы в качестве сайтов при конъюгации агентов. Для исследования роли аминогрупп в конъюгации и влияния на общую транспортную способность этих векторов, были разработаны другие векторы на основе последовательностей Angiopep-1 и Angiopep-2. Эти векторы переменные реакционные аминогруппы и переменный общий заряд. Такие полипептиды перечислены в Табл.2.
Модифицированные полипептиды
Изобретение может также включать полипептиды с модификацией последовательности аминокислот, описанной в данной публикации (напр., полипептид, имеющий последовательность, как описано в любой из SEQ ID NOS: 1-105 и 107-116, такой как AngioPep-3, -4a, -4b, -5, -6 или -7). В этом случае полипептид содержит аминокислоту, которая ковалентно связана с соединением по Формуле (I) или (II). В определенных вариантах осуществления модификация не вредит в значительной степени желаемой биологической активности. В некоторых вариантах осуществления модификация может вызывать снижение биологической активности (напр., по меньшей мере на 5%, 10%, 20%, 25%, 35%, 50%, 60%, 70%, 75%, 80%, 90% или 95%). В других вариантах осуществления модификация не влияет на биологическую активность либо может увеличивать биологическую активность оригинального полипептида (напр., по меньшей мере на 5%, 10%, 25%, 50%, 100%, 200%, 500% или 1000%). Модифицированный полипептид может обладать или может оптимизировать одну или более характеристик полипептида по настоящему изобретению, которые в некоторых случаях могут быть необходимы или желательны. Такие характеристики включают in vivo стабильность, биодоступность, токсичность, иммунологическую активность или иммунологическую идентичность.
Полипептиды по настоящему изобретению могут содержать аминокислоты или последовательности, модифицированные либо в результате естественных процессов, таких как посттрансляционный процессинг, либо в ходе химических техник модификации, хорошо известных специалистам. Модификации могут быть на любом участке полипептида, в том числе в остове полипептида, боковых цепях аминокислот, а также на амино- или карбокси-конце. Тот же тип модификации может присутствовать, в той же или отличной от нее степени, в нескольких сайтах данного полипептида. Полипептид может содержать более одного типа модификаций. Полипептиды могут быть разветвленными в результате убихитинилирования; они могут быть циклическими, с или без разветвления. Циклические, разветвленные и циклические разветвленные полипептиды могут образовываться в результате естественных посттрансляционных процессов, либо в ходе искусственного синтеза. Другие модификации включают пегилирование, ацетилирование, ацилирование, добавление ацетомидометил (Acm) группы, АДФ-рибозилирование, алкилирование, амидирование, биотинилирование, карбамоилирование, карбоксиэтилирование, этерификация, ковалентное присоединение к флавину, ковалентное присоединение к гем-части молекулы, ковалентное присоединение нуклеотидов или нуклеотид-производных, ковалентное присоединение лекарств, ковалентное присоединение маркера (напр., флуоресцентного или радиоактивного), ковалентное присоединение липида или липид-производного, ковалентное присоединение фосфатидилинозитов, перекрестное связывание, циклизация, формирование дисульфидной связи, деметилирование, формирование ковалентных сшивок, формирование цистина, формирование пироглутамата, формилирование, гамма-карбоксилирование, гликозилирование, формирование GPI-якоря (гликозилфосфатидилинозитол), гидроксилирование, йодирование, метилирование, ацилирование остатком миристиновой кислоты, окисление, протеолитический процессинг, фосфорилирование, пренилирование, рацемизация, селенонирование, сульфатация, трансфер-РНК-опосредованное добавление аминокислот к белкам, такое как аргинилирование и убихитинилирование.
Модифицированный полипептид по настоящему изобретению может в дальнейшем содержать вставку, делецию или замещение аминокислоты, либо консервативной, либо неконсервативной (напр., D-аминокислоты, дезаминокислоты) в последовательности полипептида (напр., там, где такие изменения не изменяют существенно биологическую активность полипептида).
Например, в некоторых вариантах осуществления последовательность аминокислот (напр., SEQ ID NOS 1-105 или 107-116) модифицирована путем вставки одного или более дополнительных цистеиновых остатков на N-конце пептида, С-конце пептида или на них обоих. Добавление одного или более цистеиновых остатков к амино- или карбокси-концу любой из последовательностей аминокислот, описываемых в данной публикации, может способствовать конъюгации этих полипептидов с нуклеиновыми кислотами (напр., молекулами малых интерферирующих РНК, или миРНК) либо с липидными векторами с помощью, например, дисульфидного связывания. Так, Angiopep-1 (SEQ ID NO: 67), Angiopep-2 (SEQ ID NO: 97) или Angiopep-7 (SEQ ID NO: 112) могут быть модифицированы так, чтобы они включали одинарный цистеиновый остаток на амино-конце (SEQ ID NOS: 71, 113 и 115 соответственно) или одинарный цистеиновый остаток на карбокси-конце (SEQ ID NOS: 72, 114 и 116 соответственно).
Замещения могут быть консервативными (т.е., где один остаток замещен другим остатком того же общего типа или группы) или неконсервативными (т.е., где остаток замещен аминокислотой другого типа). Кроме того, не встречающаяся в природе аминокислота может быть замещена природной аминокислотой (напр., замещение искусственной консервативной аминокислоты или замещение искусственной неконсервативной аминокислоты).
Синтетические полипептиды могут содержать замещения аминокислот, не кодируемых в природе ДНК-кислотой (напр., не встречающихся в природе или неприродных аминокислот). Примеры не встречающихся в природе аминокислот включают D-аминокислоты; аминокислоты, имеющие ацетиламинометил группу, присоединенную к атому серы в цистеине, пегилированные аминокислоты, омега-аминокислоты с формулой NH2(CH2)nCOOH, где n равно 2-6; нейтральные неполярные аминокислоты, такие как саркозин, т-бутил аланин, т-бутил глицин, N-метил изолейцин и норлейцин, Фенилглицин может замещать Trp, Туг или Phe; цитруллин и метионин сульфоксид являются нейтральными неполярными; цистеиновая кислота является кислой, а орнитин является основным. Пролин может быть замещен на гидроксипролин и сохранять конформационногенные свойства.
С помощью замещающего мутагенеза могут быть разработаны аналоги, сохраняющие биологическую активность оригинального полипептида. Примеры замещений, характеризуемых как «консервативные», приведены в Табл.3. Если такие замещения приводят к возникновению нежелательных изменений, тогда применяют другие виды замещений, именуемые в Табл.3 как «типичные замещения» или как в дальнейшем описывается в данной публикации в отношении классов аминокислот, и продукты отсеивают.
Заместительные модификации в отношении функций или иммунологической идентичности осуществляют, выбирая замещения, которые значительно отличаются по своей способности сохранять: (а) структуру остова полипептида в районе замещения, например, в виде складчатой или спиральной конформации; (b) заряд или гидрофобность молекулы в сайте-мишени или (с) общий объем боковой цепи. Остатки, встречающиеся в естественных условиях, разделяют на группы в зависимости от общих свойств боковых цепей:
(1) гидрофобные: норлейцин, метионин (Met), аланин (Ala), валин (Val), лейцин (Leu), изолейцин (Ile), гистидин (His), триптофан (Trp), тирозин (Tyr), фенилаланин (Phe);
(2) нейтральные гидрофильные: цистеин (Cys), серин (Ser), Треонин (Thr);
(3) кислые/отрицательно заряженные: аспарагиновая кислота (Asp), глутаминовая кислота(Glu);
(4) основные: Аспарагин (Asn), глутамин (Gin), гистидин (His), лизин (Lys), Аргинин (Arg);
(5) остатки, которые влияют на ориентацию цепи: глицин (Gly), пролин (Pro);
(6) ароматические: триптофан (Trp), тирозин (Tyr), фенилаланин (Phe), гистидин (His);
(7) полярные: Ser, Thr, Asn, Gln;
(8) основные, положительно заряженные: Arg, Lys, His; и
(9) заряженные: Asp, Glu, Arg, Lys, His.
Другие консервативные аминокислотные замещения перечислены в Табл.3.
остаток
Дополнительные аналоги полипептидов
Соединения по настоящему изобретению могут содержать полипептидные аналоги апротинина, хорошо известные профильным специалистам, где аналоги включают аминокислоту, ковалентно связанную с подофиллотоксин-производным (напр., соединение по Формуле (I)) либо с доксорубицином или доксорубицин-производным (напр., соединение по Формуле (II)). Например, в Патенте США №5,807,980 описываются ингибиторы, производные от ингибитора трипсина поджелудочной железы быка, а также способ их получения и терапевтического применения, включая полипептид SEQ ID NO: 102. Эти полипептиды использовались для лечения состояния, характеризуемого ненормальным образованием или количеством тканевого фактора и(или) фактора VIIIa, такого как ненормальный тромбоз. В Патенте США №5,780,265 раскрываются ингибиторы серин-протеазы, способные подавлять калликреин плазмы, в т.ч. SEQ ID NO: 103. В Патенте США №5,118,668 описаны варианты ингибитора трипсина поджелудочной железы быка, в т.ч. SEQ ID NO: 105. Последовательность аминокислот апротинина (SEQ ID NO: 98), а также последовательности аминокислот Angiopep-1 (SEQ ID NO: 67) и SEQ ID NO: 104, как и некоторые последовательности биологически активных аналогов, можно найти в Международной заявке №WO 2004/060403.
Типичные последовательности нуклеотидов, кодирующие аналог апротинина, показаны в SEQ ID NO: 106 (atgagaccag atttctgcct cgagccgccg tacactgggc cctgcaaagc tcgtatcatc cgttacttct acaatgcaaa ggcaggcctg tgtcagacct tcgtatacgg cggctgcaga gctaagcgta acaacttcaa atccgcggaa gactgcatgc gtacttgcgg tggtgcttag; № по Базе данных Genbank accession №X04666). Эта последовательность кодирует лизин на позиции 16 вместо валина, как в SEQ ID NO: 98. Мутация в нуклеотидной последовательности SEQ ID NO: 106 может осуществляться способами, хорошо известными профессионалам, с целью изменить воспроизведение полипептида SEQ ID NO: 98, имеющего валин на позиции 16. Дополнительные мутации фрагментов могут быть реализованы с помощью любой техники, известной в данной научной области.
Другие примеры аналогов апротинина можно найти с помощью программы для обнаружения сходства последовательностей белков путем их локального выравнивания «BLAST» (Genebank: www.ncbi.nlm.nih.gov/BLAST/), используя синтетическую последовательность апротинина (или ее часть), раскрываемую в Международной заявке №РСТ/СА2004/000011. Типичные аналоги апротинина представлены под входящими номерами Accession №№САА37967 (GL58005) и 1405218C (GL3604747).
Получение полипептид-производных и пептидомиметиков
Помимо полипептидов, содержащих только природные аминокислоты, настоящее изобретение охватывает также пептидомиметики или аналоги полипептидов. Аналоги полипептидов широко используются в фармацевтической промышленности как неполипептидные лекарственного средства, обладающие свойствами, аналогичными свойствам шаблонного (матричного) полипептида. Неполипептидные соединения называются «миметики полипептидов», или пептидомиметики (Fauchere et al., Infect. Immun. 54:283-287,1986; Evans et al., J. Med. Chem. 30:1229-1239, 1987). Миметики полипептидов, являющиеся родственными по структуре к терапевтически полезным полипептидам, могут быть использованы для получения эквивалентного или усиленного терапевтического или профилактического эффекта. Вкратце, пептидомиметики по структуре напоминают модель полипептида (напр., полипептида, обладающего биологической и фармакологической активностью), например, природные рецептор-связывающие полипептиды, но имеют одну или более пептидных связей, произвольно замещенных такими связями, как -CH2NH-, -CH2S-, -CH2- СН2-, -СН=СН- (цис и транс), -CH2SO-, -СН(ОН)СН2-, -СОСН2- и т.д. Такое замещение осуществляется способами, хорошо известными в науке (Spatola, Peptide Backbone Modifications, Vega Data, 1(3):267, 1983); Spatola et al. (Life Sci. 38:1243-1249, 1986); Hudson et al. {Int. J. Pept. Res. 14:177-185, 1979); and Weinstein. В., 1983, Chemistry and Biochemistry, of Aminoacids, Peptides and Proteins, Weinstein eds, Marcel Dekker, New York). Такие полипептидные миметики могут отличаться существенными преимуществами перед встречающимися в естественных условиях полипептидами, а именно более экономически выгодным производством, большей химической стабильностью, улучшенными фармакологическими свойствами (напр., временем полужизни, всасыванием, активностью, эффективностью), уменьшенной антигенностью и др.
Несмотря на то, что полипептиды по настоящему изобретению могут эффективно проникать в определенные виды клеток (а именно, описываемых в данной публикации), их эффективность может быть снижена из-за присутствия протеаз. Протеазы сыворотки требуют особого субстрата. Субстрат должен иметь связи как L-аминокислот, так и пептидов для расщепления. Более того, экзопептидазы, представляющие собой основной компонент протеазной активности в сыворотке, обычно задействуют первую пептидную связь полипептида и требуют свободного N-конца (Powell et al., Pharm. Res. 10:1268-1273,1993). В свете вышесказанного, наиболее выгодным представляется использование модифицированных версий полипептидов. Модифицированные полипептиды сохраняют структурные характеристики оригинальных L-аминокислот полипептидов, которые дают биологическую активность по отношению к ИФР-1, но преимущественно не так легко поддаются расщеплению протеазой и(или) экзопептидазами.
Систематическое замещение одной или более аминокислот консенсусной последовательности D-аминокислотой того же типа (напр., D-лизин вместо L-лизина) может использоваться для создания более стабильных полипептидов. Так, полипептид-производным, или пептидомиметиком по настоящему изобретению, может быть полипептид со всеми L-, всеми D- или с сочетанием D- и L-аминокислот. Присутствие D-аминокислоты на N-конце или С-конце повышает in vivo стабильность полипептида, так как пептидазы не могут использовать D-аминокислоту как субстрат (Powell et al., Pharm. Res. 10:1268-1273, 1993). Обратные D-полипептиды - это полипептиды, содержащие D-аминокислоты, расположенные в обратной последовательности по сравнению с полипептидами, содержащими L-аминокислоты. Так, С-концевой остаток L-аминокислотного полипептида становится N-концевым для D-аминокислотного полипептида и так далее. Обратные D-полипептиды сохраняют такую же самую третичную конформацию, а значит, имеют ту же активность, что и L-аминокислотные полипептиды, но первые являются более стабильными в отношении ферментативного расщепления in vitro и in vivo, и таким образом становятся терапевтически более эффективными, чем оригинальные полипептиды (Brady and Dodson, Nature 368:692-693, 1994; Jameson et al., Nature 368:744-746, 1994). He только обратные D-полипептиды, но и фиксированные полипептиды, содержащие консенсусную последовательность, или вариации, в значительной степени идентичные консенсусной последовательности, могут быть созданы с помощью общеизвестных способов (Rizo and Gierasch, Ann. Rev. Biochem. 61:387-418, 1992). Например, фиксированные полипептиды можно получить путем добавления цистеиновых остатков, способных образовывать дисульфидные мостики и таким образом давать в результате циклический полипептид. Циклические полипептиды не имеют свободных N- или С-концов.
Соответственно, они не поддаются протеолизу экзопептидазами, несмотря на то, что разумеется, они восприимчивы к эндопептидазам, которые не расщепляются на пептидном конце. Аминокислотные последовательности полипептидов с N-концевыми или С-концевыми D-аминокислотами и циклических полипептидов обычно идентичны последовательностям полипептидов, с которыми они соотносятся, за исключением наличия N-концевых или С-концевых D-аминокислотных остатков или их кольцевых структур, соответственно.
Циклические производные, содержащие внутримолекулярную дисульфидную связь, могут быть получены в ходе обычного твердофазового синтеза, при инкорпорировании подходящего S-защищенного цистеинового или гомоцистеинового остатков на выбранные для циклизации позиции, а именно амино- и карбокси-концы (Sah et al., /.Pharm. Pharmacol. 48:197, 1996). После завершения сборки цепи может быть проведена циклизация - либо (1) путем избирательного удаления S-защищенной группы с последующим окислением с подложкой соответствующих двух свободных SH-функций, с образованием S-S связей, с последующим обычным удалением продукта из подложки и приемлемой процедурой очистки; либо (2) путем удаления полипептида из подложки вместе с полным снятием защиты с боковой цепи, с последующим окислением свободных SH-функций в высокоразбавленном водном растворе.
Циклическое производное, содержащее внутримолекулярную амидную связь, может быть получено в ходе обычного твердофазового синтеза, при инкорпорировании подходящих аминокислотных производных с защищенными амино- и карбоксильной боковыми цепями, на позиции, выбранной для циклизации. Циклические производные, содержащие внутримолекулярные -S-алкильные связи, могут быть получены с помощью обычных твердофазовых химических процессов, при инкорпорировании аминокислотного остатка с подходящей амино-защищенной боковой цепью и подходящим S-защищенным цистеиновым или гомоцистеиновым остатком на позиции, выбранной для циклизации.
Другим эффективным подходом для достижения резистентности к пептидазам, действующим на N-концевых или С-концевых остатках полипептида, является добавление химических групп к концам полипептида, так, чтобы модифицированный полипептид перестал быть субстратом для пептидазы. Одной из разновидностей такой модификации является гликозилирование полипептидов на одном из или на обоих его концах. Доказано, что определенные химические модификации, в частности, N-концевое гликозилирование, способствуют увеличению стабильности полипептидов в сыворотке человека (Powell et al., Pharm. Res. 10:1268-1273, 1993). Другие химические модификации, повышающие стабильность в сыворотке, включают, помимо прочего, добавление N-концевой алкильной группы, состоящей из низшего алкила (от 1 до 20 атомов углерода), такой как ацетил-группа, и(или) добавление С-концевой амидной или замещенной амидной группы. В частности, настоящее изобретение включает модифицированные полипептиды, которые состоят из полипептидов, несущих N-концевую ацетил-группу и(или) С-концевую амидную группу.
Настоящее изобретение включает также другие виды полипептид-производных, содержащих дополнительные химические части молекул, не являющихся частями полипептида в естественных условиях, при условии, что производное сохраняет желаемую функциональную активность полипептида. Примеры таких производных включают (1) N-ацильные производные амино-конца или другой свободной аминогруппы, где ацильная группа может быть алканоил-группой (напр., ацетил, гексаноил, октаноил), ароил-группой (например, бензоил) или блокирующей группой, такой как F-MOC (флуоренилметил-O-СО-), (2) сложные эфиры карбокси-конца или другой свободной карбокси- или гидроксил-группы; (3) амид карбокси-конца или другой свободной карбоксильной группы, полученный в результате реакции с аммиаком или с соответствующим амином; (4) фосфорилированные производные; (5) производные, конъюгированные с антителом или другим биологическим лигандом, а также другие виды производных.
Удлиненные полипептидные последовательности, образованные в результате добавления дополнительных аминокислотных остатков к полипептидам по настоящему изобретению, также охватываются данным изобретением. Такие удлиненные последовательности полипептидов, надо полагать, сохраняют такую же биологическую активность (напр., проникновение в определенный вид клеток), как и полипептиды, описанные выше. Хотя полипептиды, имеющие значительное количество дополнительных аминокислот, не исключаются, следует признать, что некоторые большие полипептиды могут принимать конфигурацию, которая маскирует эффективную последовательность, и таким образом предотвращает связывание с мишенью (напр., представитель семейства рецепторов липопротеина низкой плотности (LRP), такой как LRP или LRP2). Эти производные могут действовать как конкурентные антагонисты. Так, несмотря на то, что настоящее изобретение охватывает полипептиды или производные полипептидов, описываемых в данной публикации, имеющие удлиняющий сегмент, желательно, чтобы этот удлиняющий сегмент не разрушал активность полипептида или производного в отношении клеток-мишеней.
Другие производные, которые включены в настоящее изобретение, представляют собой двойные полипептиды, которые состоят из двух одинаковых или двух разных полипептидов по настоящему изобретению, ковалентно связанных друг с другом либо напрямую, либо с помощью спейсера, такого как короткий участок аланиновых остатков или такого как предполагаемый сайт протеолиза (напр., с помощью катепсина, см. Патент США №5,126,249 и Европейский патент №495049). Мультимеры полипептидов по настоящему изобретению состоят из полимера молекул, образованного из тех же или других полипептидов или их производных.
Настоящее изобретение также посвящено полипептид-производным, которые являются химерными белками или белками слияния и содержат описываемые в данной публикации полипептиды или их фрагменты, связанные на амино- или карбокси-конце или на них обоих, с последовательностью аминокислот другого белка. Такие химерные белки и белки слияния могут быть получены путем рекомбинантной экспрессии нуклеиновой кислоты, кодирующей белок. Например, химерный белок и белок слияния могут содержать по меньшей мере 6 аминокислот полипептида по настоящему изобретению и, желательно, иметь функциональную активность, эквивалентную или превосходящую активность полипептида изобретения.
Полипептид-производные по настоящему изобретению могут быть получены путем изменения последовательности аминокислот с помощью замещения, добавления или делеции аминокислотных остатков, с целью образования функционально эквивалентных молекул либо, при необходимости, функционально превосходящих или уступающих по функциональности молекул. Производные по настоящему изобретению включают, помимо прочего, те, которые в качестве первичной последовательности аминокислот содержат полноразмерную или часть последовательности аминокислот полипептидов, описываемых в данной публикации (напр., любой из SEQ ID NOS:1-105 и 107-112), включая измененные последовательности, содержащие замещения функционально эквивалентных аминокислотных остатков. Например, один или более аминокислотных остатков в последовательности может быть замещен другой аминокислотой такой же полярности, которая выступает как функциональный эквивалент, что приводит к молчащему сдвигу (изменению). Замещения для аминокислоты в последовательности могут быть выбраны из других представителей класса, к которому принадлежит эта аминокислота. Например, положительно заряженные (основные) аминокислоты включают аргинин, лизин и гистидин. Неполярные (гидрофобные) аминокислоты включают лейцин, изолейцин, аланин, фенилаланин, валин, пролин, триптофан и метионин. Незаряженные полярные аминокислоты включают серии, треонин, цистеин, тирозин, аспарагин и глутамин. Отрицательно заряженные (кислые) аминокислоты включают глутаминовую и аспарагиновую кислоты. Аминокислотный глицин может входить в состав либо семейства неполярных аминокислот, либо семейства незаряженных (нейтральных) полярных аминокислот. Замещения, произведенные в семействе аминокислот, как правило, следует понимать как консервативные замещения.
Тесты для идентификации пептидомиметиков
Как говорилось выше, непептидиловые соединения, разработанные для репликации геометрии остова молекулы и отображения фармакофоров (пептидомиметики) полипептидов, идентифицированных с помощью способов настоящего изобретения, часто обладают увеличенной метаболической стабильностью, более высокой активностью, большей продолжительностью действия и лучшей биодоступностью. Соединения-пептидомиметики по настоящему изобретению получают с использованием любого из многочисленных подходов и способов комбинаторной библиотеки, хорошо известных специалистам. В их число входят: биологические библиотеки; библиотеки пространственно доступной параллельной твердой фазы или жидкой фазы; синтетические библиотечные способы, требующие деконволюции; библиотечный способ 'одна гранула - одно соединение'; а также синтетические библиотечные способы с использованием отбора по аффинной хроматографии. Подход биологических библиотек ограничен библиотеками полипептидов, тогда как 4 остальных подхода применимы и к полипептидам, и к непептидным олигомерам и низкомолекулярным библиотекам соединений (Lam, Anticancer Drug Des. 12: 145, 1997). Примеры способов синтеза молекулярных библиотек можно найти в литературе, например, в работах: DeWitt et al. (Proc. Natl. Acad. Sci. USA 90:6909, 1993); Erb et al. (Proc. Natl. Acad. Sci. USA 91:11422, 1994); Zuckermann et al, J.Med.Chem. 37:2678, 1994); Cho et al. (Science 261:1303, 1993); Carell et al. (Angew. Chem, Int. Ed. Engl. 33:2059, 1994 и там же 2061); и Gallop et al. (Med. Chem. 37:1233,1994). Библиотеки соединений могут быть представлены в растворе (напр., Houghten, Biotechniques 13:412-421, 1992) или в гранулах (Lam, Nature 354:82-84, 1991), чипах (Fodor, Nature 364:555-556, 1993), в бактериях или спорах (U.S.Patent No.5,223,409), плазмидах (Cull et al., Proc. Natl. Acad. Sci. USA 89:1865-1869,1992), либо на фагах (Scott and Smith, Science 249:386-390,1990) или люциферазной и ферментной меткой, обнаруживаемой путем определения конверсии подходящего субстрата или продукта.
Когда полипептид по настоящему изобретению идентифицирован, его можно изолировать и очистить любыми стандартными способами, включая, помимо прочего, дифференциальную растворимость (напр., преципитацию), центрифугирование, хроматографию (напр., аффинную, ионообменную, эксклюзионную хроматографию размеров и т.п.), или другими стандартными способами, применяемыми для очистки полипептидов, пептидомиметиков или белков. Функциональные свойства идентифицированного целевого полипептида могут быть оценены с использованием любого функционального анализа, известного в данной научной области. Предпочтительно использовать оценочный анализ функции рецептора, регулирующего последующие звенья сигнальных каскадов, внутриклеточной сигнальной системы (напр., пролиферация клеток).
Например, соединения пептидомиметики по настоящему изобретению могут быть получены в ходе следующего трехфазного процесса: (1) сканирование полипептидов настоящего изобретения с целью идентификации областей вторичной структуры, необходимых для нацеливания на определенные виды клеток, описываемые в данной публикации; (2) использование конформационно фиксированных дипептидных суррогатов, чтобы улучшить геометрию остова и получить органические платформы, соответствующие этим суррогатам; и (3) применение самых лучших органических платформ для отображения органических фармакофоров в библиотеках кандидатов, разработанных для имитации целевой активности природного полипептида. При более детальном рассмотрении вышеназванные 3 фазы выглядят следующим образом. В фазе 1 основные предполагаемые полипептиды сканируют, а их структуру сокращают, чтобы определить требования для их активности. Далее синтезируют серии полипептидов, аналогичных оригиналу. В фазе 2 отборные полипептидные аналоги исследуют с использованием конформационно фиксированных дипептидных суррогатов. Аминокислоты индолизидин-2-один, индолизидин-9-один и хинолизидин один (I2aa, I9aa и Qaa соответственно) используют в качестве платформ для исследования геометрии остова отборных предполагаемых полипептидов. Эти и родственные им платформы (их обзор см. у Halab et al., Biopolymers 55:101-122, 2000; и Hanessian et al. Tetrahedron 53:12789-12854, 1997) могут вводиться в определенные области полипептида, чтобы сориентировать фармакофоры в разных направлениях. Биологическая оценка этих аналогов позволяет идентифицировать отборные полипептиды с улучшенными свойствами, которые воспроизводят геометрические требования к активности. В фазе 3 платформы из наиболее активных основных полипептидов используют для отображения органических суррогатов фармакофоров, которые отвечают за активность природного полипептида. Фармакофоры и остовы комбинируют в формате параллельного синтеза. Деривация полипептидов и вышеперечисленные фазы могут быть реализованы с помощью других средств и способов, известных в данной научной области.
Структурно-функциональные отношения, определенные по полипептидам, полипептид-производным, пептидомиметикам или другим малым молекулам по настоящему изобретению, могут быть использованы для усовершенствования и изготовления аналогичных молекулярных структур с подобными или улучшенными свойствами. Соответственно, соединения по настоящему изобретению также включают молекулы, которые имеют общность структуры, полярности, зарядных характеристик и свойств боковой цепи с полипептидами, описываемыми в данной публикации.
В конечном итоге, приняв за основу все вышесказанное, профильный специалист легко сможет разработать методику скринингового исследования полипептидов и пептидомиметиков для идентификации соединений, подходящих для нацеливания агента на определенный вид клеток (напр., тех, что описываются в данной публикации). Способы анализа по настоящему изобретению могут быть разработаны для низкопроизводительного, высокопроизводительного или ультравысокопроизводительного форматов скрининга. Способы анализа по настоящему изобретению включают такие, которые поддаются автоматизации.
Полипептидные конъюгаты, ковалентно связанные с дополнительными агентами
Описываемые в данной публикации соединения или их функциональные производные не только содержат последовательность аминокислот, ковалентно связанную посредством аминокислоты с подофиллотоксин-производным (напр., соединением, имеющим структуру согласно Формуле (I)), либо с доксорубицином или доксорубицин-производным (напр., соединением по Формуле (II)), - они также могут иметь ковалентную связь с другим агентом (а именно, с другим терапевтическим препаратом, диагностическим агентом или с меткой). В определенных вариантах осуществления последовательность аминокислот также помечена или связана с поддающейся обнаружению меткой, такой как радиоактивное визуализирующее средство, в целях диагностики болезни или состояния. Примеры таких агентов включают конъюгат «радиоактивный агент-антитело-вектор», где антитело связывается со специфическим антигеном болезни или состояния (напр., при диагностике или терапии). Другие связующие молекулы также являются предметом данного изобретения. В иных случаях, соединение по настоящему изобретению или его функциональное производное связано с другим терапевтическим препаратом для лечения болезни или состояния либо оно может быть связано или помечено их смесями. Лечение болезни или состояния может проводиться в виде введения конъюгата «вектор-агент» индивидууму в условиях, допускающих транспорт агента через ГЭБ или в определенный вид клеток. Каждый полипептид может содержать по меньшей мере 1, 2, 3, 4, 5, 6 или 7 дополнительных агентов. В других вариантах осуществления каждый агент имеет по меньшей мере 1, 2, 3, 4, 5, 6 7, 10, 15, 20 или более полипептидов, прикрепленных к нему. Конъюгаты по настоящему изобретению могут способствовать аккумулированию (напр., благодаря увеличенному поглощению или сниженному выведению) агента в определенном виде клеток или тканей, таких как головной мозг, яичники, печень, легкие, почки, селезенка или мышцы субъекта.
Агент (напр., подофиллотоксин-производное, такое как соединение по Формуле (I) либо доксорубицин или доксорубицин-производное (напр., соединение по Формуле (II)), другой терапевтический препарат, диагностический агент или метка), ковалентно связанный с аминокислотой в любой из последовательностей аминокислот, описываемых в данной публикации (напр., перечисленных в Табл. 1 или их функциональных производных), может быть впоследствии, после транспортировки в определенный вид клеток или через ГЭБ, освобожден от вектора. Освобождают агент, например, с помощью ферментного расщепления или другого способа разрывания химической связи между вектором и агентом. После этого освобожденный агент может функционировать согласно своей расчетной мощности, в отсутствии вектора.
Также для связывания полипептидов и агентов РНК-интерференции изобретения могут использоваться и другие способы и кросс-линкеры (перекрестносшивающие агенты). Например, кодирующая нетранскрибируемая нить малой интерферирующей РНК, содержащая 5' или 3' тиол, может быть связана дисульфидной связью с цистеиновым остатком, расположенным либо на амино-, либо на карбокси-конце полипептида. Исследователи Murtovska et al, (FEBSLetters 558:63-68,2004) и Turner et al. (Blood Cells, Molecules и Diseases 38:1-7, 2007) описывают типичные способы химического связывания, используемые для конъюгации полипептидов с молекулами РНК. Эти работы включены в данный документ посредством ссылки.
Терапевтические препараты
Терапевтическим препаратом может быть любой биологически активный агент. Например, терапевтическим может быть лекарственное средство, медицинский препарат, агент, излучающий радиацию, клеточный токсин (например, химиотерапевтический агент), биологически активный фрагмент или смесь любых из них, предназначенные для лечения болезни (напр., для уничтожения раковых клеток); также это может быть агент для лечения болезни или состояния у индивидуума.
Подофиллотоксин-производные (напр., соединения по Формуле (I)), а также доксорубицин и доксорубицин-производные (напр., соединения по Формуле (II)) являются типичными подходящими классами терапевтических препаратов. Терапевтический препарат может быть синтетическим продуктом, либо вести свое происхождение от грибов, бактерии или другого микроорганизма (напр., микоплазмы или вируса), животного (напр., от рептилии) или растения. Терапевтический препарат и(или) его биологически активный фрагмент может быть ферментативно активным агентом и(или) его фрагментом, либо его действие может заключаться в ингибировании или блокировке важных и(или) незаменимых клеточных путей, либо в конкурировании с важными и(или) незаменимыми природными компонентами клетки. Среди других терапевтических препаратов можно назвать антитела и фрагменты антител.
Любой известный противораковый агент может быть частью конъюгата по настоящему изобретению. Подофиллотоксин-производные (напр., соединения по Формуле (I)), а также доксорубицин и доксорубицин-производные (напр., соединения по Формуле (II)) могут быть противораковыми агентами. Дополнительные противораковые агенты также могут быть конъюгированы с соединением по настоящему изобретению, как описывается в данной публикации. Рак головного мозга можно лечить конъюгатом, содержащим вектор, который эффективно пересекает ГЭБ (напр., AngioPep-2, AngioPep-3, AngioPep-4a, AngioPep-4b, AngioPep-5 или AngioPep-6). Рак яичников, печени, легких, почек или селезенки можно лечить противораковым агентом, конъюгированным с вектором, который может эффективно транспортироваться в желаемый вид клеток (напр., AngioPep-7).
Сферы действия конъюгата
Соединения или их фармацевтически приемлемые соли, которые содержат последовательность аминокислот, где последовательность аминокислот ковалентно связана посредством аминокислоты с подофиллотоксин-производным (напр., соединением, имеющим структуру согласно Формуле (I)), либо с доксорубицином или доксорубицин-производным (соединением по Формуле (II)), могут приобретать желаемые свойства, такие как измененные фармакокинетика или распределение в тканях (напр., увеличенная доставка к конкретным тканям или видам клеток, таким как яичники, печень, мозг, легкие, селезенка или почки), по сравнению с неконъюгированным биологически активным агентом. Соответственно, соединения по настоящему изобретению могут быть использованы в качестве векторов. Полипептиды, такие как AngioPep-3, AngioPep-4a, AngioPep-4b, AngioPep-5 и AngioPep-6, эффективно переносят агенты через ГЭБ. Как и AngioPep-2, эти полипептиды также могут нацеливать агенты в другие виды клеток или тканей (напр., яичники, печень, легкие, почки, селезенку или мышцы). Полипептид AngioPep-7 не может эффективно транспортироваться через ГЭБ, но он хорошо переносится в определенные ткани (а именно, в яичники, печень, легкие, почки, селезенку или мышцы). Такое свойство может быть полезным там, где транспорт через ГЭБ не нужен. Например, в результате применения соединения по настоящему изобретению может увеличиться концентрация терапевтического препарата в целевой ткани на любую величину в диапазоне 10%-20000%, по сравнению с этим показателем для неконъюгированного биологически активного агента (например, этопозида, этопозида фосфата, этопозидаDMG, тенипозида, доксорубицина или эпирубицина).
Благодаря тому, что соединения по настоящему изобретению способны транспортировать агенты к отдельным тканям, применение конъюгированных агентов позволяет достичь низкой токсичности (напр., уменьшения побочных эффектов), высокой эффективности (т.к. агент концентрируется в ткани-мишени, что обусловлено увеличенным поглощением или сниженным оттоком из ткани, либо тем, что агент после конъюгирования становится более стабильным) или и того и другого. Такие свойства описаны в данном документе (ниже), а также в Международном патенте №WO 2007/009229, который включен в данную публикацию посредством ссылки.
В некоторых случаях конъюгация агента с вектором позволяет первому избежать воздействия Р-гликопротеина (P-gp) - эффлюксного насоса, который может вытеснять определенные агенты из клетки. Если снизить способность P-gp вытеснять агенты из клетки, сила агента в клетке может быть увеличена. Таким образом, эти конъюгаты активно подавляют пролиферацию раковых клеток. Более того, результаты исследования in vivo роста опухоли показывают, что векторы изобретения могут поражать в качестве мишени рецептор липопротеина низкой плотности (LRP). Кроме того, конъюгация может изменять фармакокинетику или биораспределение неконъюгированных агентов. Взятые вместе, конъюгаты могут быть использованы для борьбы с первичными опухолями, в т.ч. опухолями яичников, молочной железы, легких и рака кожи, а также с метастазами, исходящими от первичных опухолей.
Способы лечения
Настоящее изобретение также относится к способам лечения с использованием описываемых соединений или фармацевтических композиций на их основе. Соединения по настоящему изобретению (напр., соединения, содержащие последовательность аминокислот, которая ковалентно связана посредством аминокислоты с подофиллотоксин-производным, таким как соединение по Формуле (I), либо с доксорубицином или доксорубицин-производным (напр., соединением по Формуле (II)), которые эффективно транспортируются через ГЭБ (напр., AngioPep-2, AngioPep-3, AngioPep-4a, AngioPep-4b, AngioPep-5 и AngioPep-6), могут быть использованы для лечения любого заболевания головного мозга или ЦНС. Типичные неврологические заболевания включают, помимо прочего, рак мозга, в т.ч. опухоль головного мозга, опухоль спинного мозга (напр., хордома) и метастазы в головном мозге.
Опухоли головного мозга могут быть первичными метастатическими опухолями головного мозга. Опухоли, изначально возникшие в головном мозге, являются первичными опухолями головного мозга. Опухоли головного мозга, вызванные распространением рака из какой-либо части тела (напр., рака легких, молочной железы, меланомы, прямой кишки, почек и других видов рака), являются метастатическими опухолями головного мозга. Типичными категориями опухолей, в зависимости от их расположения в головном мозге, являются опухоли ствола мозга, опухоли мозжечково-мостового угла (например, опухоли слухового нерва), опухоли полушарий головного мозга, опухоли лобной доли, опухоли теменной доли, опухоли шишковидной области, опухоли затылочной доли, опухоли височных долей, подкорковые опухоли, опухоли мозговой оболочки, опухоли срединных структур головного мозга (например, краниофарингиома, глиома зрительного нерва и опухоли таламуса и селлярной области), опухоли задней черепной ямки (например, опухоли четвертого желудочка, мозжечковые опухоли).
Типичными опухолями головного мозга являются неврома слухового нерва (неврилеммома, шваннома, невринома), аденома, астроцитома (например, ювенильная пилоцитарная астроцитома, субэпендимальная гигантоклеточная астроцитома, гемистоцитарная астроцитома, анапластическая астроцитома, злокачественная астроцитома, мультиформная глиобластома и глиосаркома); глиома ствола мозга, которая может быть астроцитомой, анапластической астроцитомой, мультиформной глиобластомой или смешанной опухолью; папиллома хороидального сплетения, лимфома ЦНС, эпендимома (напр., анапластическая эпендимома), ганглиоцитома, ганглиоглиома, глиома, мультиформная глиобластома, медуллобластома (mdl), анапластическая (злокачественная) менингиома, смешанная глиома, нейрофиброматоз (болезнь фон Реклингхаузена), олигодендроглиома и глиома зрительного нерва (напр., пилоцитарная астроцитома).
Конъюгаты также можно эффективно транспортировать в печень, яичники, легкие, почки, селезенку или мышцы. Поэтому они, в соединении с необходимым терапевтическим препаратом, могут быть использованы для лечения болезни, связанной с этими тканями (напр., рака). Поскольку AngioPep-7 не транспортируется эффективно в головной мозг, но зато эффективно транспортируется в ткани и клетки, такие как печень, легкие, почки, селезенка и мышцы. Следовательно, соединения по настоящему изобретению, содержащие AngioPep-7, особенно хорошо могут подойти для векторного лечения болезней, ассоциируемых с этими тканями, если нацеливание агента на головной мозг не является необходимым. К типичным заболеваниям печени относятся гепатоцеллюлярная карцинома (гепатома) и рак печени. К типичным заболеваниям легких относятся различные виды рака легких, в т.ч. мелкоклеточная карцинома (напр., овсяноклеточный рак), смешанная мелкоклеточная/ крупноклеточная карцинома, комбинированная мелкоклеточная карцинома и метастатические опухоли. Метастатические опухоли могут вести свое происхождение от рака любой ткани, в т.ч. от рака молочной железы (напр., метастатическая карцинома молочной железы), рака толстой кишки, рак предстательной железы (например, метастатическая карцинома простаты), саркома, рак мочевого пузыря, нейробластома и опухоль Вильмса (нефробластома). К заболеваниям селезенки относятся такие раковые заболевания: лимфома, неходжкинская лимфома и Т-клеточная лимфома.
Кроме того, среди примеров раковых заболеваний, поддающихся лечению с помощью конъюгата или композиции изобретения, можно назвать такие: рак молочной железы, рак головы и шеи, в т.ч. различные лимфомы, такие как лимфома из клеток зоны мантии, аденома, плоскоклеточная карцинома, карцинома гортани, рак сетчатки, рак пищевода, множественная миелома, рак яичников (напр., герминогенные опухоли яичников и карцинома яичников), рак матки, меланома, колоректальный рак, рак мочевого пузыря, рак простаты, рак легких (в т.ч. мелкоклеточная карцинома легких и немелкоклеточная карцинома легких), рак поджелудочной железы, рак шейки матки; рак головы и шеи, рак кожи, карцинома носоглотки, липосаркома, эпителиальная карцинома, почечно-клеточная карцинома, аденокарцинома желчного пузыря, аденокарцинома околоушной железы, саркома эндометрия, мультирезистентный рак; а также пролиферативные заболевания и состояния, такие как неоваскуляризация, связанная с ангиогенезом опухоли, дегенерация желтого пятна (напр., сухая/влажная возрастная дегенерация желтого пятна), неоваскуляризация роговицы, диабетическая ретинопатия, неоваскулярная глаукома, миопическая дегенерация и другие пролиферативные заболевания и состояния, такие как рестенозная и поликистозная болезнь почек.
Как уже говорилось, к разновидностям рака мозга, поддающимся лечению с помощью соединений или композиций изобретения, которые эффективно транспортируются через ГЭБ, относятся астроцитома, пилоцитарная астроцитома, дизэмбриопластическая нейроэпителиальная опухоль, олигодендроглиома, эпендимома, мультиформная глиобластома, смешанные глиомы, олигоастроцитома, медуллобластома, ретинобластома, нейробластома, герминома и тератома. К другим типичным разновидностям рака, поддающимся лечению с помощью соединений или композиций изобретения, относятся фунгоидный микоз (известный также как синдром Альберта-Базена, или фунгоидная гранулема), болезнь Ходжкина (лимфома Ходжкина), острый миелобластный лейкоз, острый лимфобластный лейкоз, хронический миелолейкоз, саркома Капоши, связанная с синдромом приобретенного иммунодефицита (СПИДом); не-Ходжкиновская лимфома, связанная со СПИДом; гестационная трофобластическая опухоль, саркома Юинга, рабдомиосаркома, рефракторный распространенный рак молочной железы, рак яичка (напр., злокачественные опухоли яичка, рефракторные неоплазмы яичка и карцинома - опухоль половых клеток яичка), рефракторные распространенные злокачественные новообразования, диффузная В-крупноклеточная лимфома, остеосаркома, лимфома Беркитта, острый лимфоцитарный лейкоз у взрослых, лейкемия Беркитта, новообразования средостения, лимфобластная лимфома, анапластическая крупноклеточная лимфома, новообразования плазматических клеток. Соединение или композицию по настоящему изобретению вводят субъекту любыми известными в науке способами, напр., перорально, внутриартериально, интраназально, внутрибрюшинно, внутривенно, внутримышечно, подкожно, трансдермально или per os. Агент может быть, например, противоангиогенным соединением.
Комбинированная терапия
Соединения по настоящему изобретению могут вводиться параллельно с другими терапевтическими препаратами или другими терапевтическими режимами. В некоторых вариантах осуществления дополнительный терапевтический препарат или препараты также могут иметь ковалентную связь с полипептидами или их производными, описываемыми в данной публикации (напр., полипептидами из Табл.1 и их производными). В других вариантах осуществления дополнительный терапевтический препарат или препараты не имеют ковалентной связи с полипептидами, описываемыми в данной публикации. Типичные терапевтические режимы и терапевтические препараты, которые могут использоваться при комбинированной терапии с соединениями по настоящему изобретению, включают, помимо прочего, лучевую терапию, химиотерапию, высокодозовую химиотерапию, трансплантацию стволовых клеток (напр., аутологичных стволовых клеток), пересадку костного мозга, хирургическое вмешательство, операции по удалению опухоли, лечение гипертермии; цисплатин, иринотекан, иринотекан гидрохлорид, карбоплатин, хлорамбуцил (Leukeran®), тозитумомаб (Веххаг®), ритуксимаб (Rituxan® и MabThera®), блеомицин, винкристин, винбластин, циклофосфамид, прокарбазин, митоксантрон, преднизон, преднизолон, гемцитабин (Gemzar®), паклитаксел (Taxol®), ифосфамид, метотрексат, доксорубицин (Adriamycin®), дексаметазон, циклоспорин, Rad-001 (Цертикан), цитарабин (Ara-С), даунорубицин, флударабин, идарубицин, вориностат (SAHA), ниацинамид, AZD2171, митотан, гемтузумаб озогамицин (Mylotarg®), митоксантрон, клофарабин, аспарагиназа, меркаптопурин, гранулоцитарный колониестимулирующий фактор (G-CSF или GCSF), виндезин, тиогуанин, VM26, VP 16, дакарбазин, дактиномицин, темозоломид, тиотепа, эпирубицин гидрохлорид, кармустин, филграстим, доцетаксел, гефитиниб или их фармацевтически приемлемые соли, либо любые их комбинации.
Вторым терапевтическим препаратом, используемым согласно способам, описываемым в данной публикации, также может быть полипептид, который включает или состоит из последовательности Angiopep-2 (SEQ ID NO: 97), предпочтительно где Angiopep-2 конъюгирован с противораковым агентом (напр., паклитакселом). Примером терапевтического препарата, который может быть использован в комбинации с любыми соединениями, описываемыми в данной публикации, является ANG1005, с такой структурой:
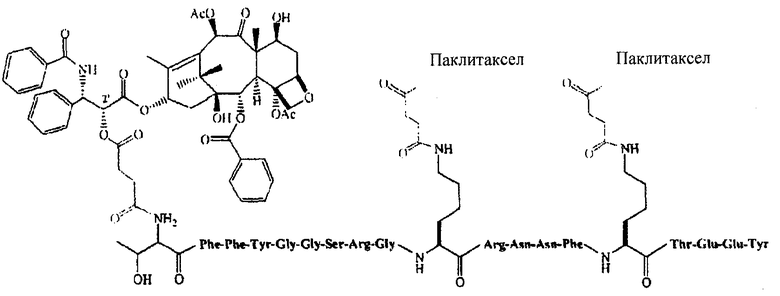
ANG1005:
TxlAn2 (3:1 конъюгат)
Еще несколько типичных веществ, которые могут быть вторым терапевтическим препаратом, описаны в Патенте США №7,557,182, включенном в данной публикации посредством ссылки.
Фармацевтические композиции
Фармацевтические композиции по настоящему изобретению включают соединение по настоящему изобретению в комбинации с фармацевтически приемлемым носителем. Такие композиции представляют собой жидкости либо лиофилизованные или другим способом высушенные лекарственные составы, которые включают растворители различного буферного содержимого (напр., Трис-HCl, ацетат, фосфат), рН и ионной силы; добавки, такие как альбумин или желатин для предотвращения впитывания поверхностями; детергенты (напр., Твин 20, Твин 80, Плюроник F68, соли желчных кислот). Солюбилизирующие агенты (напр., глицерин, полиэтилен глицерин), антиоксиданты (напр., аскорбиновая кислота, метабисульфит натрия), консерванты (напр., тимеросал, бензиловый спирт, парабены), объемообразующие вещества или модификаторы, регулирующее тоничность (напр., лактоза, маннитол), ковалентное присоединение полимеров, таких как полиэтиленгликоль с белком, комплексообразование с ионами металлов или инкорпорирование материала в или на дисперсные препараты полимерных соединений, таких как полимолочная кислота, полигликолевая кислота, гидрогели и т.д., либо на липосомы, микроэмульсии, мицеллы, моноламеллярные или многослойные везикулы, «тени» эритроцита или сферопласты. Такие композиции будут воздействовать на физическое состояние, растворимость, стабильность, скорость in vivo высвобождения и скорость in vivo выведения (клиренса). Композиции с контролируемым или с замедленным высвобождением включают лекарственные составы в липофильных депо (напр., жирные кислоты, воски, масла). Также настоящее изобретение охватывает дисперсные композиции, покрытые полимерами (напр., полоксамерами или полоксаминами). В других вариантах осуществления композиций по настоящему изобретению инкорпорируются защитные покрытия для дисперсных форм, ингибиторы протеазы или усилители проникновения, предназначенные для различных способов введения, в т.ч. парентерального, легочного, назального, перорального, вагинального, ректального. В одном из вариантов осуществления фармацевтическую композицию вводят парентерально, внутрь раковой опухоли, трансмукозально, трансдермально, внутримышечно, внутривенно, интрадермально, подкожно, внутрибрюшинно, внутрижелудочково, интракраниально и внутриопухолево.
Фармацевтически приемлемые носители в дальнейшем включают 0,01-0,1 М или 0,05 М фосфатного буфера либо 0,8% раствор натрия хлорида. Кроме того, такими фармацевтически приемлемыми носителями могут быть водные или неводные растворы, суспензии и эмульсии. Примеры неводных растворителей включают пропиленгликоль, полиэтиленгликоль; растительные масла, такие как оливковое масло, и инъецируемые органические сложные эфиры, такие как этилолеат. Среди водных носителей можно назвать воду, спиртовые/водные растворы, эмульсии или суспензии, в т.ч. раствор натрия хлорида и буферизованную среду.
Парентеральные среды включают раствор натрия хлорида, декстрозу Рингера, декстрозы и натрия хлорид, лактированный раствор Рингера или нелетучие масла. К внутривенным средам относятся жидкие и питательные восполнители (компенсаторы), восполнители электролитов, напр., на основе декстрозы Рингера, и т.п. Также могут присутствовать консерванты и другие добавки, напр., такие как противомикробные средства, антиоксиданты, сопоставляющие агенты, инертные газы и т.п.
Другие лекарственные составы включают полиоксиэтиленовые сложные эфиры жирных кислот (напр., 12-гидроксистеариновой кислоты), такие как Solutol® HS15. Так, в некоторых вариантах осуществления фармацевтическая композиция может содержать: а) конъюгат, описываемый в данной публикации; b) Solutol® HS15 и с) водный раствор или буфер (напр., раствор Рингера/Hepes с рН от 5 до 7). Концентрация Solutol® HS15 в лекарственном составе может составлять по меньшей мере 5%, 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50% или 60% (напр., 30%), или находиться в любом диапазоне между любыми двумя из вышеприведенных значений. Концентрация конъюгата может определяться на основании дозы, необходимой для эффективного лечения субъекта, либо на основании количества сложного эфира, требуемого для растворения вводимого конъюгата. Применение Solutol в лекарственном составе для введения Таксол-конъюгата описано, напр., в Международной заявке №WO 2007/009229, которая включена в данный документ посредством ссылки.
Парентеральные композиции
Фармацевтическая композиция может вводиться парентерально путем инъекции, инфузии или имплантации (подкожно, внутривенно, внутримышечно, внутрибрюшинно или т.п.) в лекарственных формах, лекарственных составах или с помощью приемлемых средств доставки или имплантатов, содержащих общепринятые, нетоксичные фармацевтически приемлемые носители и вспомогательные вещества. Способы разработки лекарственных составов и изготовления таких композиций хорошо известны специалистам в области фармацевтических технологий.
Композиции для парентерального применения могут предоставляться в дозированной лекарственной форме (напр., в ампулах с разовой дозой) или во флаконах, содержащих несколько доз, возможно, с добавлением приемлемого консерванта (см. ниже). Композиция может поставляться в форме раствора, суспензии, эмульсии, средства для инфузии или средства доставки для имплантации; либо она может быть представлена в виде сухого порошка, который перед введением восстанавливают водой или любым другим приемлемым носителем. Не считая активный (действующий) агент(ы), композиция может содержать подходящие парентерально приемлемые носители и(или) эксципиенты. Активный агент(ы) может быть инкорпорирован в микросферы, микрокапсулы, наночастицы, липосомы и т.п. для обеспечения контролируемого высвобождения. Более того, композиция может содержать суспендирующие, солюбилизирующие, стабилизирующие, рН-корректирующие агенты, агенты корректировки тоничности и(или) диспергирующие агенты.
Как говорилось выше, фармацевтические композиции, в соответствии с настоящим изобретением, могут быть представлены в форме, предназначенной для стерильных инъекций. Для приготовления таких композиций подходящий активный агент(ы) растворяют или суспендируют в парентерально приемлемом жидком носителе. Среди приемлемых носителей и растворителей можно назвать воду; воду, приведенную к нужному рН с помощью подходящего количества соляной кислоты, гидроксида натрия или подходящего буфера, 1,3-бутандиола, раствора Рингера, раствора декстрозы и изотонического раствора хлорида натрия. Водные лекарственные составы могут также содержать один или более консервантов (напр., метил, этил или N-пропил р-гидроксибензоат). В случаях, если одно из соединений является трудно- или слаборастворимым в воде, может быть добавлен агент для повышения растворимости или солюбилизации, либо растворитель может содержать 10-60% массовых долей пропиленгликоля и т.п.
Введение пациенту парентеральной композиции или лекарственного состава, включающего соединение по настоящему изобретению, может осуществляться через определенный временной интервал, например, через 10, 15, 20, 25, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, 100, 105, 110, 115 или 120 мин либо, например, через 0,5; 1,0; 1,5; 2,0; 2,5; 3,0; 3,5; 4,0; 4,5 или 5,0 ч.
Парентеральные композиции с контролируемым высвобождением
Парентеральные композиции с контролируемым высвобождением могут быть представлены в форме водных суспензий, микросфер, микрокапсул, магнитных микросфер, масляных растворов, масляных суспензий или эмульсий. Также композиция может быть инкорпорирована в биосовместимые носители, липосомы, наночастицы, имплантаты или устройства для инфузий.
К материалам, которые могут быть использованы для изготовления микросфер и(или) микрокапсул, относятся, напр., биологически разлагаемые/биоразрушаемые полимеры, такие как полигалактин поли-(изобутил цианакрилат), поли(2-гидроксиэтил-L-глутамин), поли(молочная кислота), полигликолевая кислота и их смеси. Из биосовместимых носителей, которые могут быть использованы при разработке парентерального состава с контролируемым высвобождением, можно назвать углеводы (напр., декстраны), белки (напр., альбумин), липопротеины или антитела. Материалами для использования в имплантатах могут быть небиоразлагаемые материалы (напр., полидиметил силоксан) или биоразлагаемые (напр., поли (капролактон), поли(молочная кислота), поли(гликолевая кислота) или поли(ортоэфиры)) либо их комбинации.
Твердые лекарственные формы для перорального применения
Лекарственные составы для перорального применения включают таблетки, содержащие активный(ые) ингредиент(ы), смешанные с нетоксичными фармацевтически приемлемыми эксципиентами. Такие составы хорошо известны профильным специалистам (см., напр.. Патенты США №№: 5,817,307, 5,824,300, 5,830,456, 5,846,526, 5,882,640, 5,910,304, 6,036,949, 6,036,949, 6,372,218, включенные в данную публикацию посредством ссылки). Такими эксципиентами (вспомогательными веществами) могут быть, например, инертные разбавители или наполнители (напр., сахароза, сорбит, сахар, маннит, микрокристаллическая целлюлоза, крахмалы, в т.ч. картофельный крахмал; карбонат кальция, хлорид натрия, лактоза, фосфат кальция, сульфат кальция или фосфат натрия); вещества для гранулирования вещества и для улучшения распадаемости таблеток (напр., производные целлюлозы, в т.ч. микрокристаллическая целлюлоза; крахмалы, в т.ч. картофельный крахмал; кроскармеллоза натрия, альгинаты или альгиновая кислота); связующие агенты (напр., сахароза, глюкоза, сорбитол, акация, альгиновая кислота, альгинат натрия, желатин, крахмал, прежелатинизированный крахмал, микрокристаллическая целлюлоза, алюмосиликат магния, карбоксиметилцеллюлоза натрия, метилцеллюлоза, гидроксипропилметилцеллюлоза, этилцеллюлоза, поливинилпирролидон или полиэтиленгликоль); а также смазывающие агенты, глиданты и антиадгезивные вещества (например, стеарат магния, стеарат цинка, стеариновая кислота, кремний, гидрогенизированные растительные масла или тальк). Среди других фармацевтически приемлемых вспомогательных веществ можно перечислить красители, ароматизаторы, пластификаторы, увлажнители, буферные агенты и т.п.
Таблетки могут быть без оболочки либо они могут быть покрыты оболочкой с помощью известных способов произвольно с целью отсрочить распад и всасывание в желудочно-кишечном тракте и таким образом обеспечить пролонгированное действие в течение более длительного периода. Оболочка может быть устроена так, чтобы агент высвобождался именно определенным образом (напр., чтобы получить лекарственный состав с контролируемым высвобождением). Оболочка также может быть устроена так, чтобы агент(ы) не высвобождались, пока они не пройдут через желудок (кишечнорастворимая оболочка). В качестве оболочки могут использоваться сахарная глазурь, пленочная оболочка (напр., на основе гидроксипропилметилцеллюлозы, метилцеллюлозы, метилгидроксиэтилцеллюлозы, гидроксипропилцеллюлозы, карбоксиметилцеллюлозы, акрилатных сополимеров, полиэтиленгликолей и(или) поливинилпирролидона), либо кишечнорастворимые оболочки (напр., на основе сополимера метакриловой кислоты, ацетатфталата целлюлозы, фталата гидроксипропилметилцеллюлозы, ацетата сукцината гидроксипропилметилцеллюлозы, фталата поливинилацетата, шеллака и(или) этилцеллюлозы). Кроме того, могут быть использованы материалы для задержки (отложенности) во времени, такие как, например, глицерил моностеарат или глицерил дистеарат.
Композиции в виде твердых таблеток могут включать оболочку, разработанную для защиты композиции от нежелательных химических изменений (напр., преждевременного химического распада (деградации) до того, как высвободится активное вещество). Оболочка может быть нанесена на твердую лекарственную форму способами наподобие тех, что описаны в Encyclopedia of Pharmaceutical Technology (см. ссылку выше по тексту).
Композиции изобретения могут присутствовать в таблетке в смешанном виде или быть разделены. В одном из примеров, первый агент может содержаться внутри таблетки, а второй - снаружи, чтобы значительная часть второго агента успела высвободиться до того, как высвободится первый агент.
Лекарственные составы для перорального применения также могут быть представлены в виде таблеток для жевания, либо в виде твердых желатиновых капсул, где активное вещество смешано с инертным твердым разбавителем (напр., картофельным крахмалом, лактозой, микрокристаллической целлюлозой, карбонатом кальция, фосфатом кальция или каолином), либо в виде мягких желатиновых капсул, где активное вещество смешано с водной или масляной средой, например, с арахисовым маслом, жидким парафином или оливковым маслом. Кроме того, возможно изготовление порошков и гранул из ингредиентов, перечисленных для таблеток и капсул, используя общепринятые способы, с помощью, напр., миксера, аппарата для гранулирования и сушки таблеточных смесей в «кипящем слое» или оборудования для распылительной сушки.
Режимы дозирования
Дозировка любого соединения, конъюгата или композиции, описанных в данной публикации, либо идентифицированного по способам, описанным в данной публикации, зависит от нескольких факторов, в т.ч.: способа введения; вида заболевания (напр., рак), подлежащего лечению; тяжести болезни; от цели введения - лечение рака или его предотвращение, а также от возраста, веса и здоровья субъекта, подвергающегося лечению.
В отношении способов лечения, настоящее изобретение не накладывает ограничений на способы введения вектора, конъюгата или композиции субъекту, а также на дозировку и частоту приема; изобретение охватывает все способы введения. Конъюгат или композиция могут вводиться субъекту в однократной или многократной дозах. Например, описываемое в данной публикации соединение или соединение, идентифицированное с помощью способа отбора, предоставляемого данным изобретением, допускает введение конъюгата один раз в неделю на протяжении, напр., 2, 3, 4, 5, 6, 7, 8, 10, 15, 20 или более недель. Соединение изобретения также можно вводить, например, ежедневно в течение 1, 2, 3, 4, 5, 6 или 7 дней или в течение 1, 2, 3, 4, 5, 6, 7, 8, 10, 15, 20 или более недель.
Периоды времени, когда вводится соединение изобретения, могут предшествовать или следовать за периодами, когда соединение не вводится. Например, после введения соединения в соответствии с приведенным в данной публикации описанием, соединение изобретения не вводится пациенту на протяжении 1, 2, 3, 4, 5, 6 или 7 дней или 1, 2, 3, 4, 5, 6, 7, 8, 10, 15, 20 или более недель. В некоторых вариантах осуществления в течение этого периода лечения пациент может получать и другие терапевтические препараты. Такие циклы химиотерапии, которые состоят из периода, когда соединение по настоящему изобретению вводится пациенту, и следующего за ним периода, когда соединение изобретения не вводится этому пациенту, могут повторяться по мере необходимости, согласно медицинским показаниям (например, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19 или 20 раз).
Следует понимать, что в случае каждого конкретного субъекта с течением времени подбирается особый режим дозирования (схема приема), который зависит от индивидуальной потребности и профессионального суждения специалиста, назначающего или контролирующего введение вектора, конъюгата или композиции. Например, дозировка конъюгата может быть увеличена, если более низкие дозы не позволяют достичь достаточной активности при лечении болезней или состояний описываемых в данной публикации (напр., рака). И наоборот, дозировку соединения снижают, если болезнь (напр., рак) облегчена или ликвидирована.
Несмотря на то, что окончательное подходящее количество и режим дозирования определяет лечащий врач, как пример, терапевтически эффективное количество соединения, вектора, конъюгата или композиции, описываемых в данной публикации, может находиться в диапазоне от 0,0035 мкг до 20 мкг/кг массы тела/сут или от 0,010 мкг до 140 мкг/кг массы тела/неделя. Желательно нахождение терапевтически эффективного количества в диапазоне от 0,025 мкг до 10 мкг/кг, например, по меньшей мере 0,025; 0,035; 0,05; 0,075; 0,1; 0,25; 0,5; 1,0; 1,5; 2,0; 2,5; 3,0; 3,5; 4,0; 5,0; 6,0; 7,0; 8,0 или 9,0 мкг/кг массы тела, с приемом ежедневно, через день или 2 раза в неделю. Кроме того, терапевтически эффективное количество может быть в пределах от 0,05 мкг до 20 мкг/кг, например, по меньшей мере 0,05; 0,7; 0,15; 0,2; 1,0; 2,0; 3,0; 4.0; 5,0; 6,0; 7,0; 8,0; 10,0; 12,0; 14,0; 16,0 или 18,0 мкг/кг массы тела, с приемом еженедельно, через неделю или 1 раз в месяц. Более того, терапевтически эффективное количество соединения может быть, например, в пределах от 0,100 мг/м2 до 2000 мг/м2, с введением через день, 1 раз в неделю или через неделю. В наиболее желательном варианте осуществления терапевтически эффективное количество находится в диапазоне от 1 мг/м2 до 1000 мг/м2, например, по меньшей мере 100, 150, 400 или 800 мг/м2 соединения, с введением ежедневно, через день, 2 раза в неделю, 1 раз в неделю или через неделю.
Так, соединения по настоящему изобретению (напр.. Соединение (1)) могут быть использованы для введения подофиллотоксин-производных (напр., соединения по Формуле (I), такого как этопозид, этопозид фосфат, этопозидDMG и тенипозид) или для введения доксорубицина или доксорубицин-производных (напр.. Соединение (2) или соединение изобретения, которое содержит соединение по Формуле (II))) пациенту с помощью любого из способов введения, количества дозы и схем приема, описываемых в данной публикации. Поскольку соединения по настоящему изобретению могут содержать ковалентную связь, например, с 1, 2, 3, 4 или 5 молекулами подофиллотоксин-производного, такого как соединение по Формуле (I) (напр., этопозид, этопозид фосфат, этопозидDMG и тенипозид) или по Формуле (II) (напр., доксорубицин или эпирубицин), эта стехиометрия может быть использована для подсчета и коррекции количества вводимой дозы («эквивалентная доза»). Так, конъюгат Этопозид; Angiopep-2 (3:1) («Этоп-An2(3:1)») имеет молекулярную массу 4354 г/моль, причем содержание этопозида составляет 40% молекулярной массы. Аналогично, конъюгат доксорубицин: Angiopep-2 (3:1) имеет молекулярную массу 4178 г/моль, причем содержание доксорубицина составляет 40% молекулярной массы. Учитывая эти данные, можно рассчитать количество соединения по настоящему изобретению, входящее в состав композиции для введения (напр., парентерального или перорального), чтобы назначить пациенту 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 15, 20, 25, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, 100, 105, 110, 115, 120, 125, 130, 135, 140, 145, 150, 155, 160, 165, 170, 175, 180, 185, 190, 195, 200, 225, 250, 275, 300, 325, 350, 375, 400, 425, 450, 475 или 500 мг/м2/сут подофиллотоксин-производного (напр., этопозида, этопозида фосфата, этопозидаDMG или тенипозида). Такая доза может вводиться пациенту ежедневно в течение 2, 3, 4, 5, 6 или 7 дней. После периода приема можно давать 1, 2, 3, 4, 5, 6 или 7 дней или 2, 3, 4, 5, 6, 7 или 8 недель «отдыха» от приема соединения по настоящему изобретению. Такие циклы периода введения/периода отдыха могут повторяться, напр., 1, 2, 3, 4, 5, 6, 7, 8, 9 или 10 раз.
Соединения по настоящему изобретению (напр.. Соединения (1) и (2) или их фармацевтически приемлемые соли) могут отличаться улучшенными физико-химическими и фармацевтическими качествами по сравнению с соответствующими неконъюгированными терапевтическими препаратами (напр., этопозидом, этопозид 4-диметилглицином или доксорубицином). Например, улучшенное прицеливание на определенные виды клеток, тканей или органов, описываемых в данной публикации (напр., мозг, яичники, печень, легкие, почки, селезенка или мышцы) дает возможность применять субтерапевтические дозы соединения (напр., Соединений (1) или (2)) для введения пациенту. Соединения по настоящему изобретению могут также обладать сниженной токсичностью по сравнению с соответствующими неконъюгированными терапевтическими препаратами, что позволяет вводить пациенту сверхтерапевтические дозы соединения. Например, неконъюгированный доксорубицин обычно вводят согласно схеме приема, находящейся в пределах 60-75 мг/м2, при введении отдельно в виде однократной внутривенной инъекции, или в диапазоне 40-50 мг/м2 при введении в комбинации с другим химиотерапевтическим препаратом. Типичная дозировка неконъюгированного этопозида или этопозида фосфата может варьироваться от 1-5 мг/м2/сут, 1-50 мг/м2/сут, 35-50 мг/м2/сут или 50-100 мг/м2 /сут. Улучшенное прицеливание на определенные виды клеток, тканей или органов, которым отличаются соединения по настоящему изобретению (напр., Соединения (1) или (2)), позволяет применять меньшие дозы терапевтических препаратов, по сравнению с теми, которые используются при введении соответствующих неконъюгированных терапевтических препаратов («субтерапевтическая доза», т.е. эффективная доза, которая может составлять, например, в 25, 50, 75, 100, 125, 150, 175, 200, 225, 250, 275, 300, 325, 350, 375, 400, 425, 450, 475, 500, 525, 550, 575, 600, 625, 650, 675, 700, 725, 750, 775, 800, 825, 850, 875, 900, 925, 950, 975, 1000, 2000, 3000, 4000 или 5000 раз меньше, чем минимальная эффективная доза соответствующего неконъюгированного терапевтического препарата). Сниженная токсичность, обусловленная соединением по настоящему изобретению (Соединениями (1) или (2)), делает возможным безопасное введение пациенту более высоких доз, по сравнению с дозами соответствующих неконъюгированных терапевтических препаратов («сверхтерапевтические дозы», т.е. эффективная доза, которая, например, в 25, 50, 75, 100, 125, 150, 175, 200, 225, 250, 275, 300, 325, 350, 375, 400, 425, 450, 475, 500, 525, 550, 575, 600, 625, 650, 675, 700, 725, 750, 775, 800, 825, 850, 875, 900, 925, 950, 975, 1000, 2000, 3000, 4000 или 5000 раз превышает максимальную допустимую дозу неконъюгированного терапевтического препарата). Аналогично, улучшенные физико-химические качества также могут повлиять на объем дозы, вводимой пациенту. Например, улучшенная растворимость дает возможность вводить субтерапевтические дозы. Такие улучшенные физико-химические фармацевтические характеристики могут способствовать безопасному введению соединения по настоящему изобретению (Соединений (1) или (2)), например, педиатрическим и гериатрическим популяциям пациентов.
Нижеследующие примеры призваны только проиллюстрировать настоящее изобретение и ни в коей мере его не ограничивают.
ПРИМЕРЫ
Пример 1: Синтез конъюгата 3:1 Этопозид: Angiopep-2
Синтез соединения по настоящему изобретению может быть выполнен путем сочетания подофиллотоксин-производного (т.е., соединения по Формуле (I), такого как этопозид, этопозид фосфат, этопозидDMG или тенипозид), дифункциональной гидролизуемой связующей группы (такой как дикарбоновая кислота или диизоцианат) и любой из последовательностей аминокислот, описываемых в данной публикации, или их функциональных производных. Вариации эквивалентов промежуточного соединения, образующего производное подофиллотоксина (напр., 2"-глутарил этопозид), по сравнению с полипептидом, позволяют синтезировать полипептидные конъюгаты с различным стехиометрическим составом (например, «Этоп-An2(1:1)» или «Eto-An2(l:l)», где одна молекула этопозида связана с полипептидом Angiopep-2).
Схема 5 отображает синтез соединения, включающего полипептид Angiopep-2, ковалентно связанный с тремя молекулами этопозида («Этоп-An2(3: 1)» или («Это-An2(3:1)»).
Схема 5
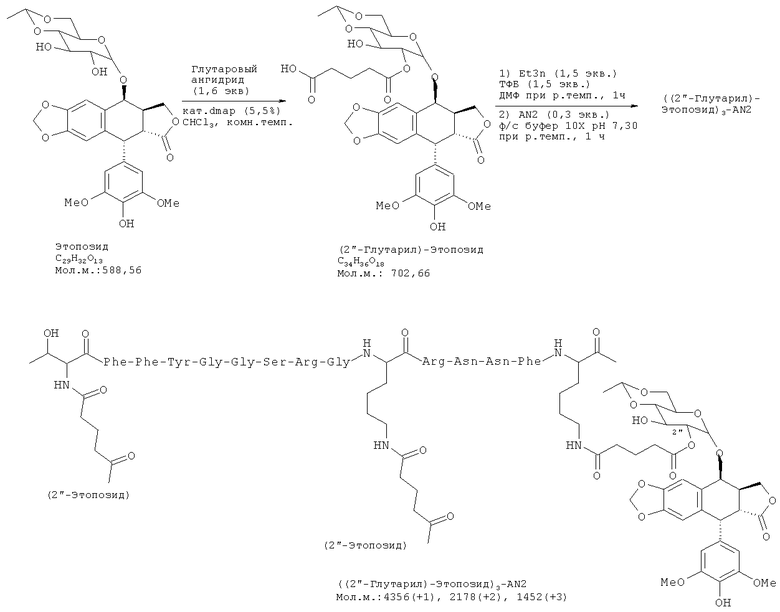
(2"-Глутарил)-Этопозид. К раствору этопозида (10 г, 17 ммоль) в CHCl3 (120 мл) добавляли глутаровый ангидрид (3,13 г, 27,4 ммоль) и DMAP (диметиламинопиридиновый реагент) (115 мг, 0,942 ммоль). Через 72 ч выдерживания при комн. темп. смесь выпаривали и очищали с помощью обращенно-фазовой хроматографии в колонке полистирол/DVB (полидивинилбензол) (от 15 до 35% ацетонитрила (ACN) в H2O, без трифторуксусной кислоты (TFA)). ВЭЖХ сырого продукта показала смесь региоизомеров 2"-Глутарил Этопозид (2"-глу-Этоп) и 3"-Глутарил Этопозид (3"-глу-Этоп) в соотношении 2:1. После выпаривания и лиофилизации получали (2"-Глутарил)-Этопозид в виде белого порошка (4,1 г, 34%). Региоизомерический (3"-глутарил)-этопозид также был изолирован в виде белого твердого вещества (2,4 г, 20%). Чистоту 2"-глу-Этоп и 3"-глу-Этоп определяли с помощью обращенно-фазовой ВЭЖХ. Исследование проводили в колонке MetaChem Taxsil-3 при градиентном элюировании (1 мл/мин; от 10% до 65% (0,05% TFA в H2O):(0,05% TFA в ACN) в течение 15 мин), время удерживания 2"-глу-Этоп составило 9,00 мин, а время удерживания 3"-глу-Этоп - 9,42 мин. Чистота 2"-глу-Этоп (время удерживания = 9.00 мин) была определена на уровне >98% (99,4%), а чистота 3"-глу-Этоп - также >98% (99,6%).
((2"-Глутарил)-Этоп)3-(Angipep2) (Этоп-An2(3:1)). К раствору (2"-Глутарил)-Этопозида (2,83 г, 4,025 ммоль) в безводном DMF (диметилформамиде) (400 мл) добавляли триэтиламин (Et3N; 0,84 мл, 6,037 ммоль) и N,N,N',N'-Тетраметил-О-(бензотриазол-1-ил)уроний тетрафтороборат (TBTU; 1,32 г, 4,628 ммоль). Реакционную смесь перемешивали при комн. темп.в течение 1 ч (рН=9,3). Приготовляли раствор AN2 (Angiopep-2, 75% в долях, 3,68 г, 1,207 ммоль) в фосфатно-солевом буфере (PBS) 10X (рН 7,3; 200 мл) путем корректировки рН с помощью 10N NaOH (до рН 7,3). После охлаждения на ледяной бане, предварительно активированную кислоту добавляли (4×100 мл) к Angiopep-2, и рН смеси доводили до 7,2, добавляя 10N NaOH. Через 1 ч при комн. темп., реакционную смесь фильтровали, чтобы вывести фосфатные соли, и выпаривали, получая 35 мл сырой смеси. С помощью ВЭЖХ сырого продукта была обнаружена смесь конъюгатов (3:1) и (2:1) в соотношении 3:1. Остаток очищали с помощью обращенно-фазовой хроматографии, используя колонку полистирол/DVB (от 15 до 37,5% ACN в Н2О). После очистки к комбинированным фракциям добавляли 0,1% уксусной кислоты, чтобы увеличить растворимость. Выпаривание и лиофилизация смеси позволили получить продукт Этоп-An2(3:1) в виде белого твердого вещества (596 мг, 12%). С помощью колонки MetaChem Taxsil-3 и градиентного элюирования (1 мл/мин; 10% до 65% (0,05% TFA в H2O):(0,05% TFA в ACN) в течение 15 мин), удалось обнаружить, что изолированный продукт имеет время удерживания 9,36 мин и является чистым на >95% (97,3%). Отношение массы к заряду (ESI-TOF (ионизация электрораспылением - время пролета)): 2178 (+2), 1452 (+3).
Пример 2: Синтез конъюгата 3:1 ЭтопозидDMG: Angiopep-2
Процедура, описанная в Примере 1, может быть использована и для получения соединений, включающих пептиды, конъюгированные с другими подофиллотоксин-производными. Например, ЭтопозидDMG можно использовать для создания конъюгата, изображенного на Схеме 7 («ЭтопDMG-An2 (3:1)»), в ходе процедуры синтеза, показанной на Схеме 6.
Схема 6
((2"-Глутарил)-ЭтопозидDMG)3-AN2
Схема 7
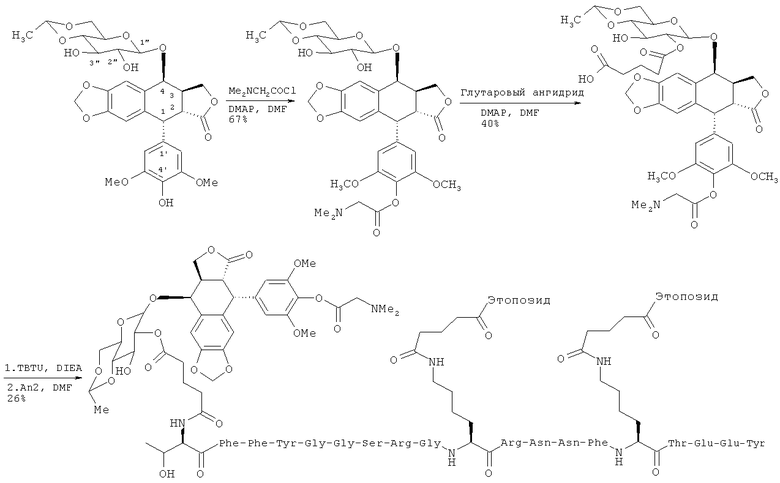
Этопозид 4'-Диметилглицин: Смесь Этопозида (235 мг, 0,4 ммоль) и DMAP (73 мг, 0,6 ммоль) в DMF (4 мл) перемешивали при комн. темп. в течение 20 мин и затем добавляли N,N-диметилацетил хлорид (96 мг, 0,52 ммоль) в одном сосуде, помешивая. Через 30 мин реакция была завершена, согласно ВЭЖХ. Далее добавляли муравьиную кислоту (1М в DMF, 0,5 мл) и растворитель концентрировали до 1 мл. Полученный раствор загружали в АКТА RPC колонку (для обращенно-фазовой хроматографии) для очистки (градиент от 10% до 30% MeCN в Н2О с 0,1% муравьиной кислотой). После лиофилизации получали этопозид 4'-Диметилглицин («ЭтопозидDMG» или «ЭтопDMG»; 180 мг, 67%) в виде бесцветного порошка. 1Н ЯМР (CD3OD) δ 7,01 (1Н, s), 6,56 (1Н, s), 6,39 (2H, s), 5,98 (2H, d, J=2,9 Гц), 5,05 (1Н, d, J=3,4 Гц), 4,77 (1Н, q, J=4,9 Гц), 4,68 (1Н, d, J=5,4 Гц), 4,66 (1Н, d, J=7,8 Гц), 4,46 (2H, s), 4.45 (1Н, dd, J=10,3, 8,8 Гц), 4,31 (1Н, t, J=8,0 Гц), 4,17 (1Н, dd, J=10,3, 4,9 Гц), 3,68 (6Н, s), 3,56 (1Н, q, J=10 Гц), 3,54 (1Н, t, J=9,3 Гц), 3,52 (1Н, dd, J=14,2, 5,6 Гц), 3,32 (1Н, m), 3,26 (1Н, dd, J=9,1, 4,1 Гц), 3,24 (1Н, dd, J=9,2, 5,4 Гц), 3,02 (6Н, s), 2,96 (1Н, m), 1,33 (3H, d, J=4,9 Гц). 13С ЯМР (ДМСО) δ 175,26, 168,68, 151,35, 148,49, 147,01, 139,39, 132,74, 129,6, 127,28, 110,65, 110,45, 108,02, 102,19, 102,02, 99,25, 80,78, 75,06, 73,39, 72,41, 68,43, 68,01, 66,44, 59,73, 56,63, 56,47, 45,03, 43,86, 37,89, 20,99; МСВР (масс-спектрометрия высокого разрешения) (MicroTOF), вычисл. для C33H39NO14 673,2371, найденное значение 274,2534 (М+1).
Этопозид 4'-Диметилглицин 2"-Глутаровая кислота: Смесь этопозид 4'-диметилглицина (655 мг, 0,97 ммоль) и DMAP (18 мг, 0,15 ммоль) в хлороформе (11 мл) охлаждали до 0°С. Затем последовательно добавляли DMF (3 мл) и N,N-диизопропилэтиламин (DIEA; 0,25 мл, 1,46 ммоль), в за ними - глутаровый ангидрид (222 мг, 1,94 ммоль). Реакционную смесь перемешивали при комн. темп., контролируя ее с помощью ВЭЖХ. Через 2 дня растворитель концентрировали до 3 мл. Полученный раствор загружали в колонку АКТА RPC для очистки (градиентное элюирование, от 10% до 30% MeCN в H2O) и после лиофилизации получали Этопозид 4'-Диметилглицин 2"-Глутаровую кислоту (305 мг, 40%) в виде белого порошка. 1Н ЯМР (CD3OD) δ 7,0 (1Н, s), 6,53 (1Н, s), 6,39 (2H, s), 5,99 (2H, d, J=4,6 Гц), 4,97 (1Н, q, J=7,9 Гц), 4,78 (1Н, q, J=4,75 Гц), 4,74 (1Н, d, J=7,9 Гц), 4,68 (1Н, d, J=5,6 Гц), 4,45 (2H, s). 4,41 (1Н, dd, J=9,6, 8,8 Гц), 4,29 (1Н, t, J=8,2 Гц), 4,15 (1Н, dd, J=10,0, 4,5 Гц), 3,78 (1Н, t, J=9.4 Гц), 3,69 (6Н, s), 3,61 (1Н, t, J=10.2 Гц), 3,42 (1Н, td, J=9,6, 5,2 Гц), 3,33 (1Н, dd, J=8,7, 8,2 Гц), 3,3 (1Н, dd, J=13,4, 5,3 Гц), 3,02 (6Н, s), 2,93 (1Н, m), 2,26 (1Н, m), 2,16 (2H, m), 2,02 (1Н, m), 1,64 (2H, m), 1,32 (3Н, d, J=4,9 Гц). 13С ЯМР (ДМСО) δ 175,96, 175,33, 172,46, 163,74, 151,14, 148,96, 147,43, 139,53, 131,90, 129,83, 126,42, 110,20, 109,18, 107,40, 101,91, 100,65, 99,63, 80,39, 74,55, 73,95, 71,55, 71,29, 68,43, 67,82, 66,46, 56,43, 55,35, 43,90, 43,15, 40,82, 38,0, 32,86, 32,59, 19,93,19,39. MCBP (MicroTOF) вычисл. для C38H45NO17 787,2687, найд. зн. 788,2432 (M+1).
Конъюгат (Этопозид-4'-Диметилглицин-2''-Глутаровый)3-Angiopep-2 («Этоп-4'-DMG1y-2»-Глу)3-An2" или «ЭтопDMG-An2(3:1)»):
DIEA (0,17 мл, 0,98 ммоль) добавляли по каплям к смеси Этопозид 4'-Диметилглицин 2"-Глутаровой кислоты (330 мг, 0,42 ммоль) и TBTU (145 мг, 0,46 ммоль) в DMF (24 мл). Смесь перемешивали при комн. темп. в течение 50 мин. Затем добавляли раствор Angpep-2 (422 мг, 0,14 ммоль) в ДМСО (1,5 мл) и DMF (9 мл), а после - DIEA (0,084 мл, 0,48 ммоль). Смесь перемешивали при комн. темп. в течение 20 мин. Аликвотную пробу (10 мл) брали для анализа с помощью сверхэффективной жидкостной хроматографии, которая показала, что реакция была завершена. После перемешивания еще в течение 10 мин реакционный раствор концентрировали до 3 мл и очищали, используя колонку АКТА RPC (градиентное элюирование, от 10% до 25% MeCN в Н2О с 0,05% муравьиной кислоты). После лиофилизации получали (Этоп-4'-DMGly-2"-Глу)3-An2 (172 мг, 26%) в виде бесцветного порошка. МС (MicroTOF), масса/заряд, 2305.9327 (2+), 1537.6443 (3+), 1153.7463 (4+), 922.7970 (5+).
Пример 3: Синтез Конъюгата 3:1 Доксорубицин: Angiopep-2 ("(DoxSu)3-An2")
Конъюгат 3:1 Доксорубицин: Angiopep-2 получают в ходе процесса синтеза, изображенного на Схеме 8 и подробно описанного ниже.
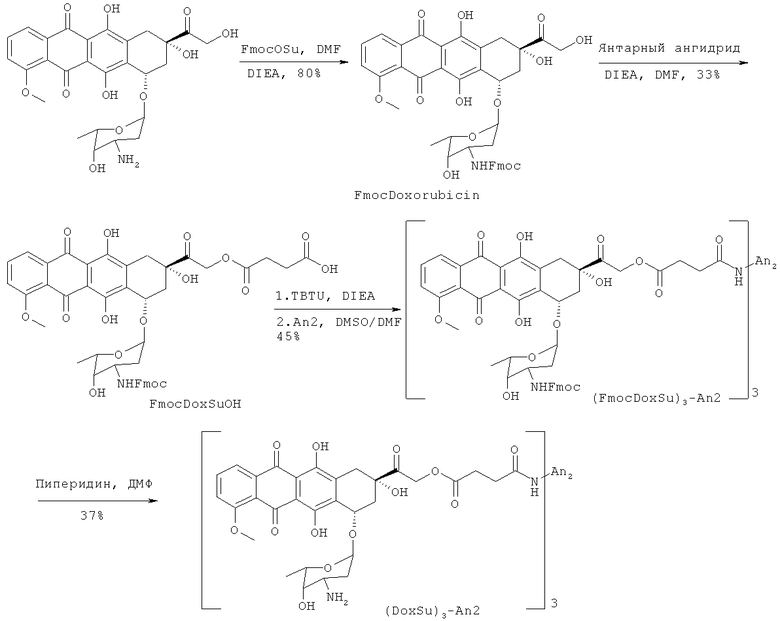
FmocДоксорубицин: DIEA (1,5 мл, 8,63 ммоль) добавляли по каплям к раствору доксорубицина (2,0 г, 3,45 ммоль) и 9-флуоренилметил N-сукцинимидилкарбоната (FmocOSu; 2,32 г, 6,9 ммоль) в DMF (35 мл), помешивая. Смесь перемешивали при комн. темп. в течение 3 ч и концентрировали. Полученный остаток растирали в порошок с 0,1% TFA в Н2О (3×20 мл), промывали с помощью Et2O (8×20 мл). Полученное красное твердое вещество собирали и сушили над вакуумом, получая FmocДоксорубицин в виде красного порошка (2,1 г, выход 80%). Чистота, согласно сверхэффективной жидкостной хроматографии (СЭЖХ), составила 98%. МС (ESI, MicroTOF), 788,2411 (M+Na).
FmocDoxSuOH: DIEA (0,17 мл, 1,0 ммоль) добавляли по каплям к раствору FmocДоксорубицина (0,28 г, 0,366 ммоль) и янтарного ангидрида (0,11 г, 1,1 ммоль) в ДМФ (20 мл), помешивая. Смесь перемешивали при комн. темп. и контролировали с помощью СЭЖХ. Через два дня растворитель удаляли, а полученный остаток очищали с помощью колонки Biotage (силикагель, от 2% до 9% МеОН в DCM), с получением FmocDoxSuOH в виде красного порошка (100 мг, выход 33%). Чистота согласно СЭЖХ: 95%. МС (ESI, MicroTOF), 888,2577 (М+Na).
(FmocDoxSu)3-An2: DIEA (0,25 мл, 1,44 ммоль) добавляли по каплям к раствору FmocDoxSuOH (599 мг, 0,692 ммоль) и TBTU (231 мг, 0,72 ммоль) в DMF (21 мл), постоянно помешивая. Смесь перемешивали при комн. темп. в течение 50 мин, а затем добавляли раствор Angpep-2 (671 мг, 0,229 ммоль) в ДМСО (2 мл) и DMF (12 мл). Смесь перемешивали при комн. темп. в течение 20 мин, после чего ВЭЖХ показала завершение реакции. После перемешивания еще на протяжении 10 мин, растворитель удаляли, а остаток очищали в колонке Biotage С 18 (от 40% до 80% MeCN в воде и 0,05% TFA), с получением (FmocDoxSu)3An-2 в виде красного порошка (500 мг, выход 45%). Чистота по СЭЖХ - 95%. МС (ESI, MicroTOF), м/з 2423,4239 (2+), 1615,6190 (3+).
(DoxSu)3-An2: Пиперидин (20% в DMF, 1,5 мл) добавляли к раствору (FmocDoxSu)3An-2 (260 мг, 0,053 ммоль) в ДМСО (1 мл) и DMF (12 мл). Раствор становился синим. После 10-минутного перемешивания раствор охлаждали до 0°С и обрабатывали муравьиной кислотой (0,5 М в DMF, 6 мл), получая прозрачный красный раствор. Растворитель удаляли с помощью вакуумного насоса, а полученный остаток растирали в порошок с Et2O (3×10 мл) и AcOEt (3×10 мл). Полученное красное твердое вещество очищали на АКТА RPC 30 колонке (от 10% до 40% MeCN в воде и 0,15% муравьиной кислоты), с получением (DoxSu)3An-2 в виде красного порошка (82 мг, выход 37%). Чистота по СЭЖХ: 95%. МС (ESI, MicroTOF), м/з 2089,9674 (2+), 1393,2419 (3+), 1045,4395 (4+).
Пример 3: Воздействие Этопозида, конъюгатов Этопозид-Angiopep, доксорубицина и конъюгатов Доксорубицин-Angiopep на пролиферацию клеток
Для анализа пролиферации клеток in vitro в 24-луночные микропланшеты для культур тканей высевали от 2,5 до 5×104 клеток U87 или SK-HEP-1, с финальным объемом 1 мл среды с 10% сыворотки и инкубировали в течение 24 ч при 37°С и 5% CO2. Затем среду заменяли на бессывороточную среду и инкубировали на протяжении ночи. На следующее утро агент заново растворяли в диметилсульфоксиде (ДМСО), и среду заменяли на полную среду, содержащую агент в различных концентрациях в трех экземплярах. Финальная концентрация ДМСО составила 0,1%, В качестве контрольной использовали лунку микропланшета с клетками без добавления агента. Клетки инкубировали в течение от 48 до 72 ч при 37°С и 5% CO2. После инкубирования среду меняли и заменяли на 1 мл полной среды, содержащей [3H]-тимидин (1 пКи на 1 процедуру анализа). Планшет инкубировали при 37°С и с 5% СО2 в течение 4 ч. Среду удаляли, и клетки промывали с помощью PBS при 37°С. Затем клетки фиксировали смесью этанол:уксусная кислота (3:1), промывали водой и осаждали 3 раза при помощи 10% ледяной ТСА (трихлоруксусной кислоты). Наконец, в лунки добавляли 500 мкл РСА (перхлорной кислоты) и нагревали микропланшеты в течение 30 мин при 65°С и 30 мин при 75°С. Далее содержимое каждой лунки перемещали в сцинтилляционный флакон с 10 мл сцинтилляционного коктейля и измеряли активность в СРМ (число импульсов в минуту) с помощью жидкостного сцинтилляционного счетчика Tri-Carb производства компании Packard. Результаты анализа пролиферации клеток для неконъюгированного этопозида, а также конъюгатов Этоп-An2(1:1) и Этоп-An2(3:1) показаны в Табл.4. Результаты этого же анализа для ЭтопDMG-An2(3:1), неконъюгированного этопозидаDMG, конъюгата доксорубицин/Angiopep-2(3;1) («Доксорубицин-An2 (3:1)») и неконъюгированного доксорубицина отображены в Табл.5.
Ингибирование клеточной пролиферации проверяли не только в ходе in vitro исследований, но и на ксенотрансплантатных моделях опухоли - результаты этих экспериментов показаны на ФИГ.1. Клетки глиобластомы U87 (2,5×106) подкожно имплантировали безтимусным мышам в правый бок. Лечение начинали на 15-й день после имплантации (что соответствует Дню 0 на графике, приведенном на ФИГ.1), когда объем опухоли достигал около 150-200 мм3. Еженедельно мыши получали ВВ болюсную инъекцию доксорубицина (6 мг/кг) и конъюгата Доксорубицин-An2 (20 и 40 мг/кг), курс лечения продолжался три недели. Конъюгат Доксорубицин-An2 разбавляли в подкисленной D5W (5%-ная декстроза в воде) при концентрации 5 мг/мл.
средство
Пример 3: Исследования перфузии мозга in situ
Для исследования мозговой перфузии in situ были использованы процедуры, описанные в Патенте США №20060189515, который включен в данную публикацию посредством ссылки. Описание этих процедур будет дано ниже.
Пример 3а: Этоп-An2(3:1)
Поглощение головным мозгом соединения по настоящему изобретению (напр., Этоп-An2(3:1), ЭтопDMG-An2(3:1) и Доксорубицин-An2 (3:1)) по сравнению с соответствующими неконъюгированными лекарственными средствами измеряли с помощью методик in situ мозговой перфузии, описываемых в данном документе и в работе Dagenais et al., J.Cereb. Blood Flow Metab. 20(2): 381-386 (2000). Всасывание [125I]-полипептидов в области просвета (люминальной области) капилляров мозга мышей измеряли с использованием способа in situ перфузии мозга, адаптированной в нашей лаборатории для исследования поглощения агента в мозге мышей.
Полипептиды йодировали согласно стандартным процедурам, с использованием йодогранул производства Sigma. Вкратце, полипептиды разбавляли в 0,1 М фосфатного буфера, рН 6,5 (ФБ). Для каждого белка использовали по две йодогранулы. Гранулы дважды промывали в 3 мл ФБ на фильтре Ватмана и ресуспендировали в 60 мкл ФБ. 125I (1 мКи) производства Amersham-Pharmacia biotech добавляли к суспензии гранул на протяжении 5 мин при комн. темп. Каждое йодирование начиналось с добавления полипептида (100 мкг). После инкубирования в течение 10 мин при комн. темп. свободный йод удаляли с помощью ВЭЖХ.
Вкратце, мыши, получившие в качестве анестезии кетамин/ксилазин (140/8 мг/кг интраперитонеально) в правую общую сонную артерию, были подвергнуты воздействию; сосуды животных перевязывали на уровне бифуркации (разветвления) общей сонной артерии, рострально к затылочной артерии. Затем в общую сонную артерию вводили катетер рострально с использованием полиэтиленовой системы для внутривенных инфузий, заполненной гепарином (25 ед./мл), и приготавливали препарат для исследования на игле 26 калибра. Шприц, содержащий жидкость для перфузии ([125I]-полипептиды или [14C]-инулин в буфере из раствора Кребса/бикарбоната, с рН 7,4, насыщенном газами 95% 02 и 5% CO2), помещали в инфузионный насос (Harvard pump PHD 2000; Harvard Apparatus) и подсоединяли к катетеру. Перед началом перфузии контралатеральный вклад кровотока устраняли путем отсоединения желудочков сердца. Далее проводили перфузию головного мозга в течение определенного времени при скорости тока жидкости 1,15 мл/мин. Через 14,5 мин после начала перфузии проводили перфузию мозга еще в течение 60 с буферным раствором Кребса, чтобы вымыть избыток [125I]-белков. Затем мышей декапитировали (обезглавливали), чтобы остановить перфузию, и правое полушарие изолировали и помещали на лед, чтобы затем подвергнуть капиллярному истощению. Затем забирали аликвотные количества гомогенатов, надосадочной жидкости, осадков после центрифугирования и перфузируемой жидкости с целью измерения в них содержания [125I]-конъюгатов с помощью реакции преципитации с трихлоруксусной кислотой (ТСА) и оценки кажущегося объема дистрибуции.
Результаты этих экспериментов показаны на ФИГ.2A-D и 3. На ФИГ.2А видно, что по данным, полученным при in situ перфузии мозга, объем распределения в организме (Vd) конъюгата Этоп-An2(3:1) выше, чем неконъюгированного этопозида (напр., наблюдаемый скат кривой для Этоп-An2(3:1) превосходит тот, что наблюдался для неконъюгированного этопозида). Подобная тенденция была характерна и для конъюгата ЭтопDMG-An2(3:1) (ФИГ.2В), по сравнению с неконъюгированным ЭтопозидомDMG, а также для конъюгата доксорубицин-An2(3:1), по сравнению с неконъюгированным доксорубицином (ФИГ.2С). Как видно на ФИГ.2С, коэффициент Kin для соотношения доксорубицин-An2(3:1):неконъюгированный доксорубицин составляет 15. Эта процедура также позволяет провести различие, где соединения, оставшиеся в полостях сосудов головного мозга, а где соединения, которым удалось пересечь аблюминальную эндотелиальную мембрану, чтобы проникнуть в паренхиму головного мозга. ФИГ. 3 отображает in situ перфузию Этоп-An2(3:1). На каждой группе столбцов левая полоса характеризует неконъюгированный этопозид, средняя - ЭтопDMG-An2(3:1) (Серия 1), и правая отображает ЭтопDMG-An2(3:1) (Серия 2). Перераспределение в головном мозге Этоп-An2(3:1), следующее за истощением капилляров мозга, показано на ФИГ. 4А и 4В. Более того, в отличие от этопозида, поглощение головным мозгом конъюгата Этоп-An2(3:1) одинаковое у мышей дикого типа и P-gp нокаутных мышей, что указывает на то, что Этоп-An2(3:1) не является субстратом для P-gp (ФИГ.5). На этой фигуре левая полоса каждой группы отображает результаты, полученные у CD-1 мышей, а правая полоса - результаты, полученные у P-gp нокаутных мышей. Поглощение головным мозгом конъюгата Этоп-An2(3:1) может быть ингибировано с помощью совместного введения неконъюгированного полипептида. ФИГ. 6 показывает, что совместная перфузия из [125I]-Этоп-An2(3:1) и двухкратного избытка неконъюгированного Angiopep-2 вызывает сокращение объема распределения (Vd) в паренхиме на 27%.
Пример 3b: ЭтопDMG-An2 (3:1) и Доксорубицин-An2(3:1)
Поглощение головным мозгом конъюгата ЭтопDMG-An2 (3:1) по сравнению с неконъюгированным Этопомо измеряли с помощью способов, описанных для Примера 3а. Результаты отображены в Табл. 6 и на ФИГ. 7. Кроме того, Табл. 6 включает соответствующие данные для доксорубицин-An2(3:1) и доксорубицина. В отличие от неконъюгированного ЭтопDMG поглощение головным мозгом конъюгата доксорубицин-An2(3:1) одинаковое у мышей дикого типа и P-gp нокаутных мышей, что указывает на то, что доксорубицин-An2(3:1) не является субстратом для P-gp.
Пример 4: Кинетика в плазме конъюгата 3:1 этопозид-Angiopep-2
На ФИГ.8 показана кинетика в плазме Этоп-An2(3:1) после болюсного введения. Меченный радиоактивным изотопом (125I)Этоп-An2 (20 мг/кг) вводили в виде болюсной (струйной) инъекции внутривенно (ВВ) или интраперитонеально (ИП) CD-1 мышам с массой около 25-30 г. Раствор для инъекций состоял из 12,5% диметилсульфоксида (ДМСО), 12,5% безводного этанола, 25% полиэтиленгликоля 400 (PEG400) и 50% буфера NaCl/Глицин. Через некоторые периоды времени (0,5; 1; 2 и 6 ч) у животных брали кровь на анализ с помощью прокола (пункции) сердца, после чего их умерщвляли. После центрифугирования крови измеряли радиоактивность плазмы на счетчике гамма-излучения (Wizard 1470 Automatic Gamma Counter). За показатель радиоактивности был принят % введенной дозы на 1 грамм плазмы. Результаты отображали в графическом виде, используя программу GraphPad prism. Для каждого режима инъекций рассчитывали площадь под фармакокинетической кривой (AUC). Интраперитонеальную биодоступность конъюгата Этоп-An2 оценивали, разделив AUC после ИП инъекции на AUC после ВВ инъекции. Полученный показатель биодоступности после ИП введения, согласно расчетам, составил 46%. Фармакокинетические параметры Этоп-An2(3: 1) после ВВ болюсного введения у мышей приведены в Табл.7.
Время полужизни неконъюгированного этопозида, согласно литературным источникам, составляет T1/2α=0,13 ч (Reddy et al., Journal of drug targeting, 13(10): 543-553 (2005)).
мг/кг)
(мкг/мл)
(ч)
элиминации (ч-1)
Пример 5: Распределение в тканях конъюгата 3:1 этопозидDMG-Angiopep-2 и конъюгата 3:1 этопозид-Angiopep-2
Влияние конъюгирования агента с вектором на распределение агента или фармакокинетика полипептида, конъюгированного с агентом, оценивались таким образом: животным вводили меченый полипептид или конъюгат и измеряли распределение полипептида или конъюгата в органах (напр., с помощью 3H или 125I меченных конъюгатов) мышей. Такие же эксперименты могут быть проведены с соединениями, включающими любой из описываемых в данной публикации полипептидов (напр., полипептидов, перечисленных в Табл.1, таких как AngioPep-3, AngioPep-4a, AngioPep-4b, AngioPep-5, AngioPep-6 и AngioPep-7 или их аналоги). В данном эксперименте неконъюгированный противораковый агент и конъюгаты вводили мышам в виде внутривенной болюсной инъекции. Образцы ткани отбирали в разное время (0,25, 0,5, 1 и 4 ч) и гомогенизировали. Чтобы подсчитать количество 3Н-меченого конъюгата, гомогенаты тканей переваривали с помощью солюбилизатора тканей, и добавляли в образцы 10 мл жидкого сцинтиллятора. Количество 125I меченного конъюгата в различных тканях измеряли после преципитации (осаждения) трихлоруксусной кислотой (ТСА). Затем подсчитывали радиоактивность в тканях. Площадь под фармакокинетической кривой (AUC 0-4) оценивалась в программе Prism; затем строили графики для разных тканей.
ФИГ.9 отображает распределение в головном мозге ЭтопDMG-An2 после ВВ болюсного введения мышам. На этой фигуре левая полоса каждой группы отображает результаты, полученные при исследовании неконъюгированного этопозида, а правая полоса - результаты исследования Этоп-An2(3:1). На ФИГ.10 изображено распределение в головном мозге конъюгата Этоп-An2(3:1) после ВВ или ИП болюсного введения мышам. На ФИГ.11 показано сравнение распределения в мозге Этоп-An2(3:1) и неконъюгированного этопозида через 30 мин после ВВ болюсного введения мышам. ФИГ.12 отображает распределение в тканях Этоп-An2(3:1) по сравнению с неконъюгированным этопозидом. На этой фигуре левая полоса каждой группы отображает результаты, полученные при исследовании неконъюгированного этопозида, а правая полоса - результаты исследования Этоп-An2(3:1). Применение Этоп-An2(3:1) приводит к увеличению концентрации в исследуемых тканях.
Пример 6: Противоопухолевое действие конъюгата Доксорубицин-An2(3:1) на модели опухоли мозга человека у мышей
Всех животных, использованных в этих исследованиях, содержали и ухаживали за ними в соответствии с требованиями Руководства Канадского Совета по уходу за животными (Guidelines of the Canadian Council on Animal Care, CCAC). Протоколы животных были утверждены Институциональным комитетом по содержанию и использованию животных (Institutional Animal Care and Use Committee) Монреальского университета, Квебек.
Модель интрацеребральной опухоли мозга человека создавали путем стереотаксического внедрения 5×105 клеток U87 в мозг безтимусным мышам. Для создания модели опухоли использовали самок бестимусных «голых» мышей (без волосяного покрова) (Crl:Nu/Nu-nuBR; 20-25 г, возраст 4-6 недель; приобретенных в Charles River Canada, Сен-Констан, Квебек Канада). Животных содержали в стерильных (непатогенных) условиях. За один час до хирургического вмешательства мыши получали подкожную инъекцию бупренорфина (0,1 мг кг-1). Перед вживлением опухолевых клеток мышам давали анестезию в виде ИП инъекции кетамина/ксилазина (120/10 мг кг-1) помещали животных в стереотаксический аппарат (Kopf; Туюнга, Калифорния). Затем высверливали трепанационное отверстие не доходя 1,5 мм до и на 2,5 мм в бок от брегмы. Суспензию клеток в 5 мкл бессывороточной культуры клеток вводили путем инъекции на протяжении 5 мин с помощью шприца Гамильтона, вводя иглу на глубину 3,5 мм.
Медикаментозное лечение начинали через 3 дня после внедрения опухоли (Табл.8). Терапевтическое соединение (напр., доксорубицин или конъюгат доксорубицин-An2(3:1)) вводили внутривенно путем болюсной инъекции в хвостовую вену (один раз в неделю). Растворы с лекарственным средством готовили в 5%-ной декстрозе в воде (D5W). Растворы для инъекций приготовляли свежими, непосредственно перед каждой процедурой введения. Клинические признаки прогрессирования болезни и массу тела контролировали каждый день. Когда мыши достигали смертельной конечной точки (снижение массы тела на 20%), их умерщвляли путем асфиксии углекислым газом.
Медиана
соединение
Каждый из ФИГ.13А и 13В показывает эффективность конъюгата (DoxSu)3-An2, введенного отдельно или в комбинации с конъюгатом паклитаксел-Angiopep2. На ФИГ.13А показаны результаты, полученные в первом испытании, а на ФИГ.13В - результаты второй серии испытаний. Статистический анализ данных, полученных в ходе Испытания 2 (Фиг.13 В), показал, что наблюдаемое улучшение на 27% является статистически значимым (р<0,007).
Другие примеры осуществления
Содержимое каждой публикации, патента и патентной заявки, упомянутых в данной заявке, включено в данную публикацию посредством ссылки. Несмотря на то что изобретение было подробно описано и проиллюстрировано сопроводительными графическими материалами, следует понимать, что оно не ограничивается вариантами осуществления, приведенными в данной публикации, и что изобретение может быть реализовано с различными изменениями и модификациями, которые не выходят за пределы сущности и объема изобретения.
Несмотря на то что настоящее изобретение было описано применительно к его конкретным вариантам осуществления, следует понимать, что оно может осуществляться с дальнейшими модификациями, и данная патентная заявка будет охватывать любые вариации, применение или адаптации изобретения, выполненные в соответствии с его общими принципами, и охватывать эти отклонения от настоящего раскрытия, которые подпадают под общеизвестную или общепринятую практику в научной области, к которой настоящее изобретение принадлежит, и которые могут быть применены к существенным характеристикам, изложенным выше, а также в контексте пунктов патентной формулы, прилагаемых ниже.
| название | год | авторы | номер документа |
|---|---|---|---|
| УСИЛЕНИЕ ДЕЙСТВИЯ ПРОТИВОРАКОВЫХ АГЕНТОВ | 2006 |
|
RU2422143C2 |
| ПОЛИПЕПТИДЫ АПРОТИНИНА ДЛЯ ТРАНСПОРТА СОЕДИНЕНИЯ ЧЕРЕЗ ГЕМАТОЭНЦЕФАЛИЧЕСКИЙ БАРЬЕР | 2010 |
|
RU2611193C2 |
| КОМПОЗИЦИЯ, НА ОСНОВЕ ГИДРОФОБНЫХ АГЕНТОВ И СПОСОБ ЕЕ ПОЛУЧЕНИЯ(ВАРИАНТЫ) | 2009 |
|
RU2518240C2 |
| ПОЛИПЕПТИД, СПОСОБНЫЙ ПРЕОДОЛЕВАТЬ ГЕМАТОЭНЦЕФАЛИЧЕСКИЙ БАРЬЕР, И ЕГО КОНЪЮГАТ | 2005 |
|
RU2408605C2 |
| НОВЫЕ ПЕПТИДЫ | 1995 |
|
RU2162855C2 |
| КОНЪЮГАТЫ "ПРОИЗВОДНОЕ КАЛИХЕАМИЦИНА-НОСИТЕЛЬ" | 2011 |
|
RU2602878C2 |
| КОНЪЮГАТЫ "ПРОИЗВОДНОЕ КАЛИХЕАМИЦИНА-НОСИТЕЛЬ" | 2003 |
|
RU2422157C2 |
| КОНЪЮГАТЫ "ПРОИЗВОДНОЕ КАЛИХЕАМИЦИНА-НОСИТЕЛЬ" | 2016 |
|
RU2678818C2 |
| КОНЪЮГАТЫ АНТИ-РТК7 АНТИТЕЛО-ЛЕКАРСТВЕННОЕ СРЕДСТВО | 2015 |
|
RU2708075C2 |
| АНТИТЕЛА ПРОТИВ 5Т4 И ИХ ПРИМЕНЕНИЕ | 2007 |
|
RU2487889C2 |






























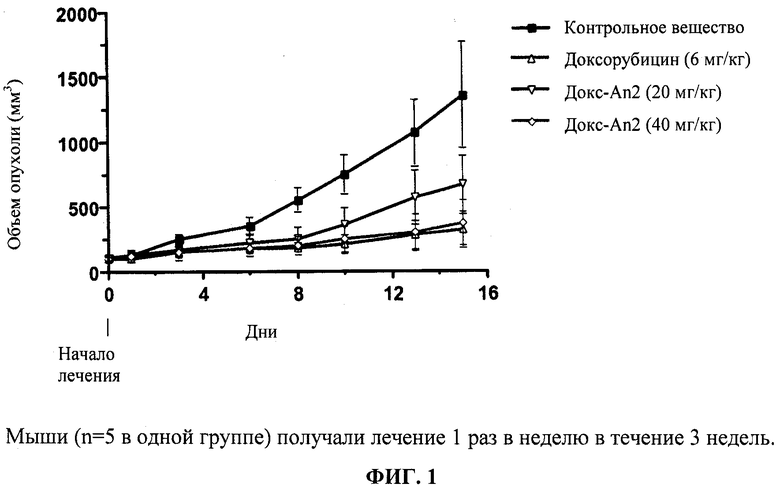
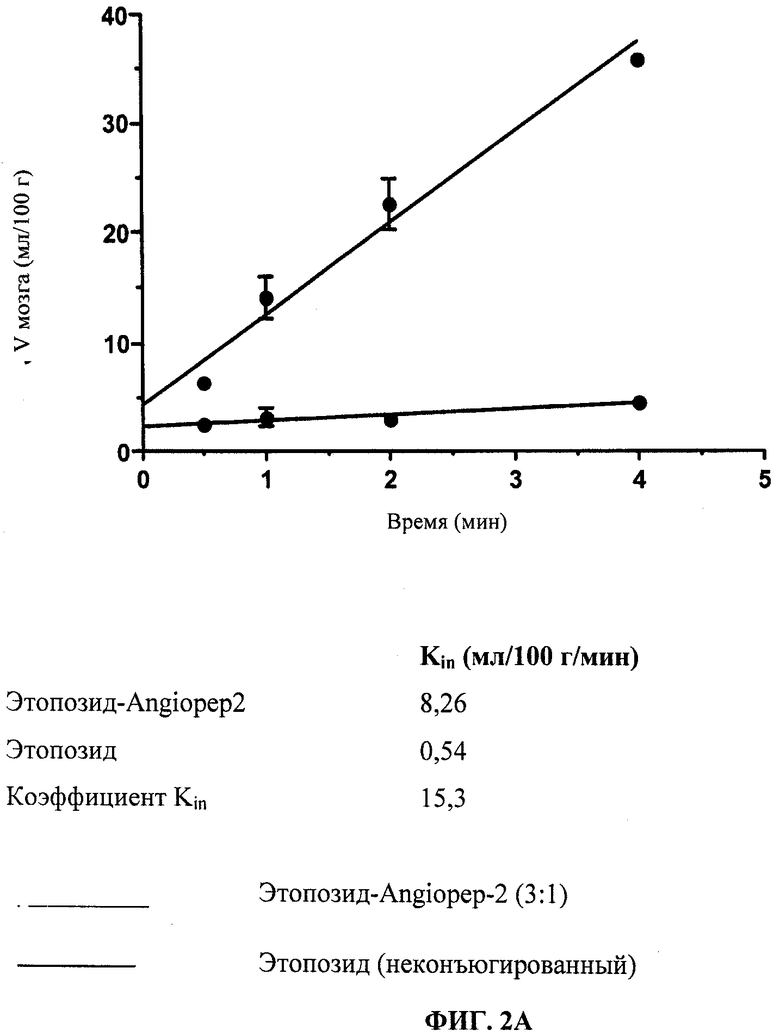
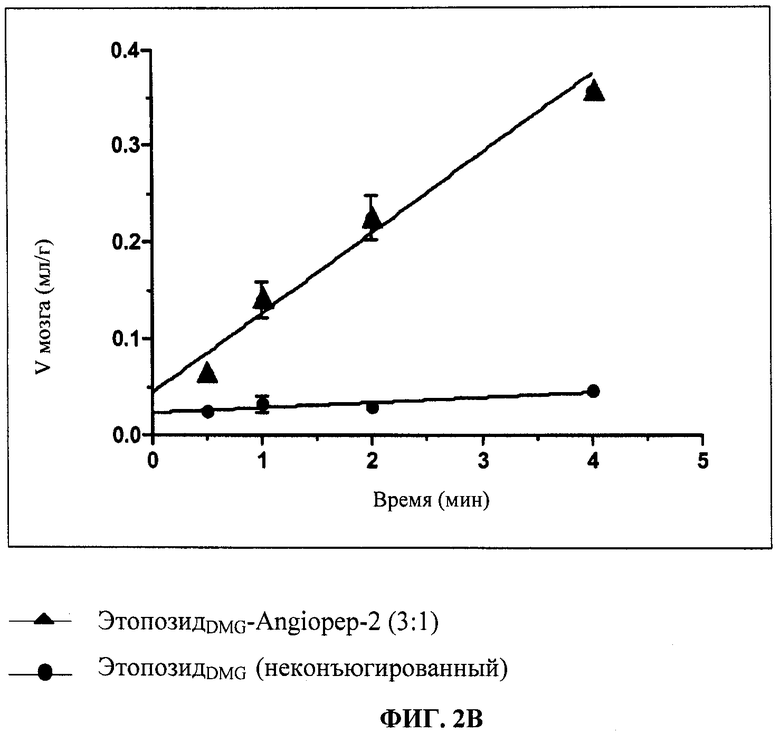
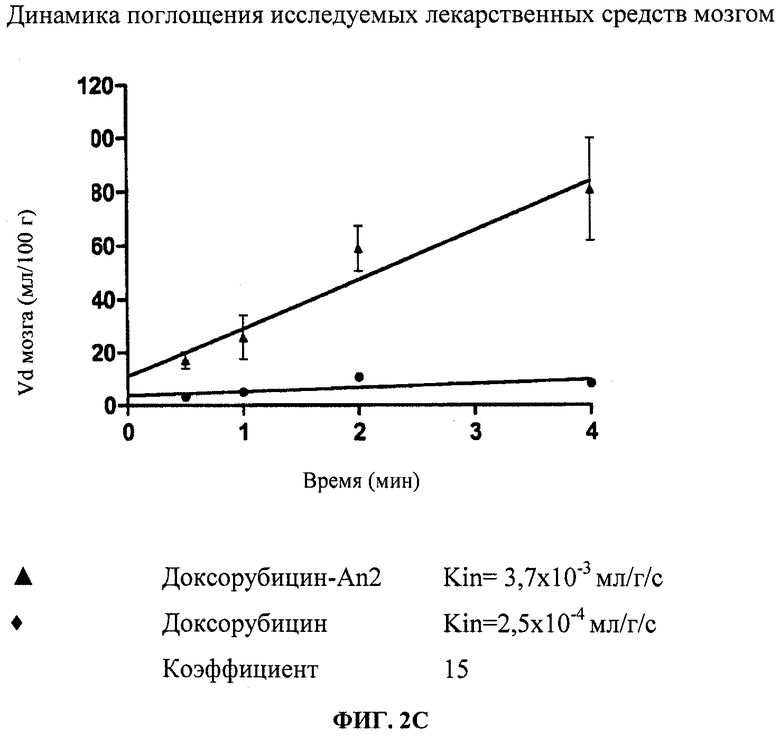
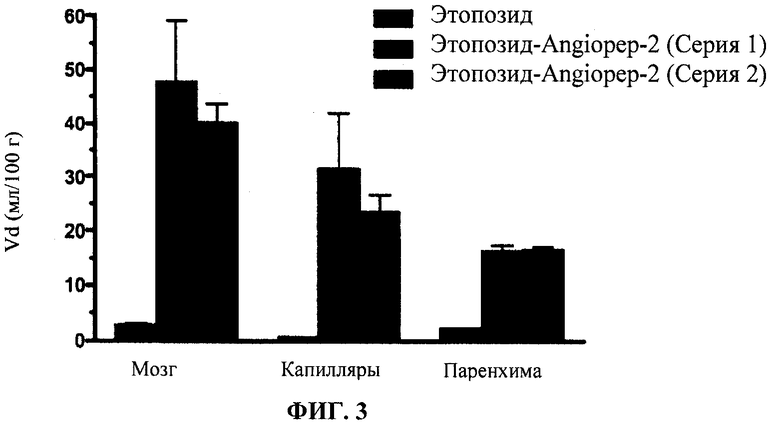
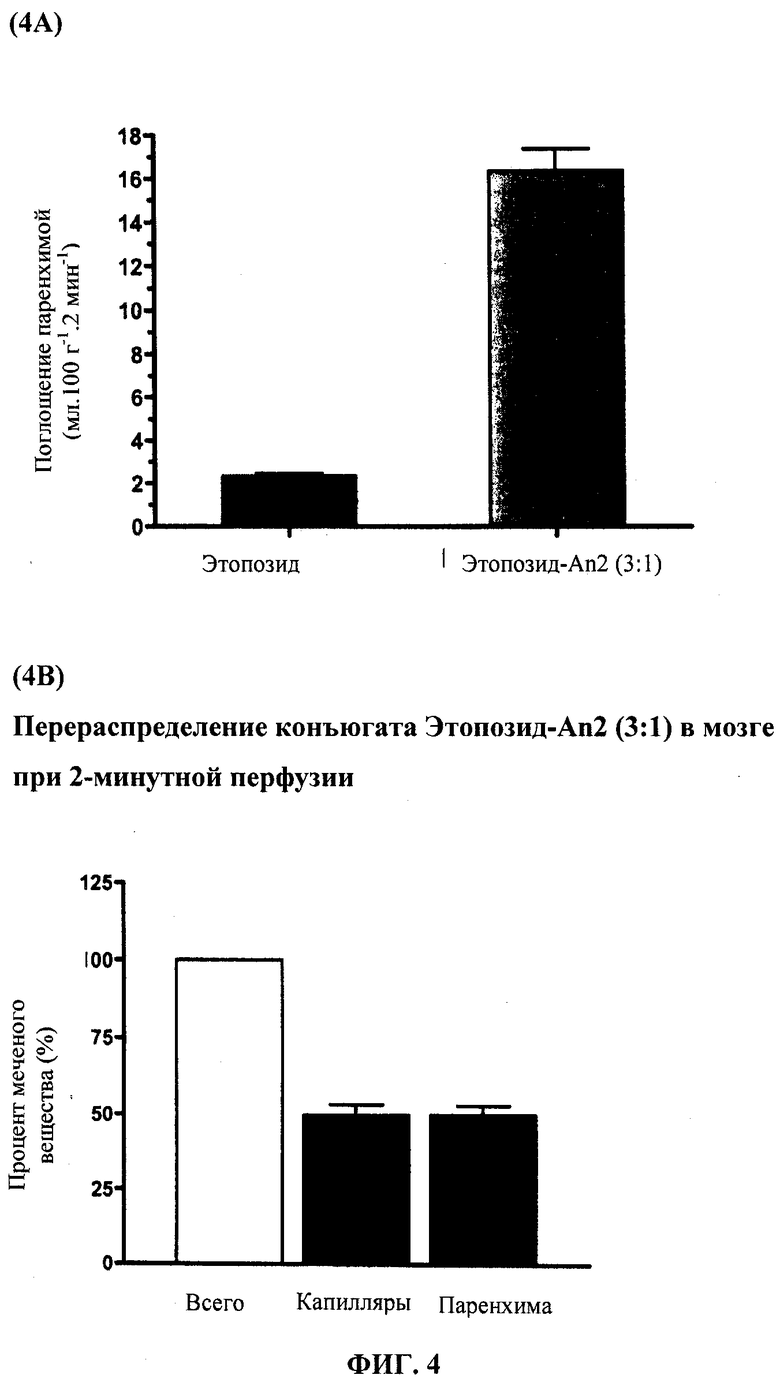
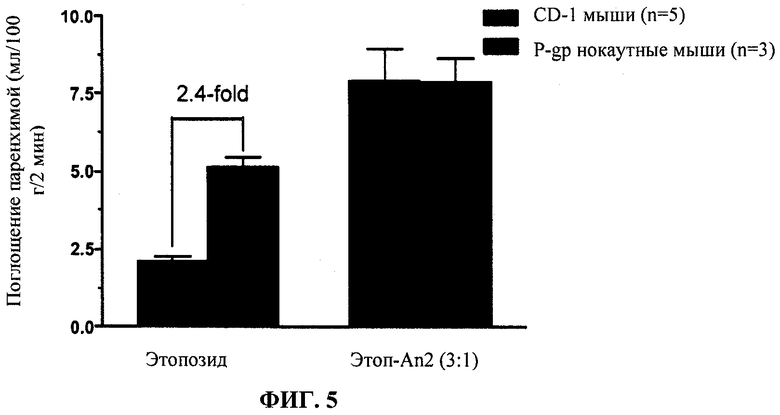
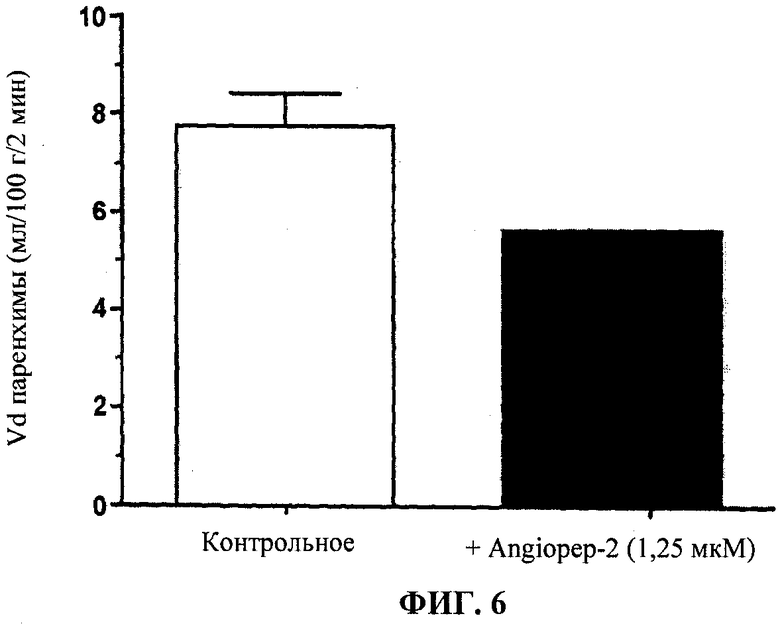
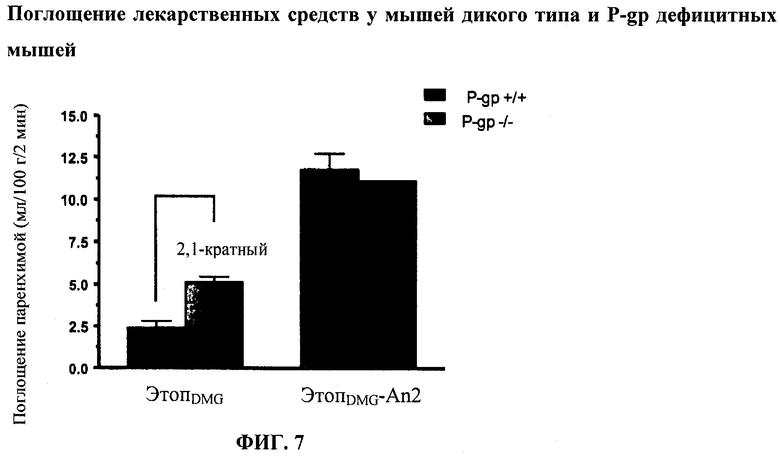
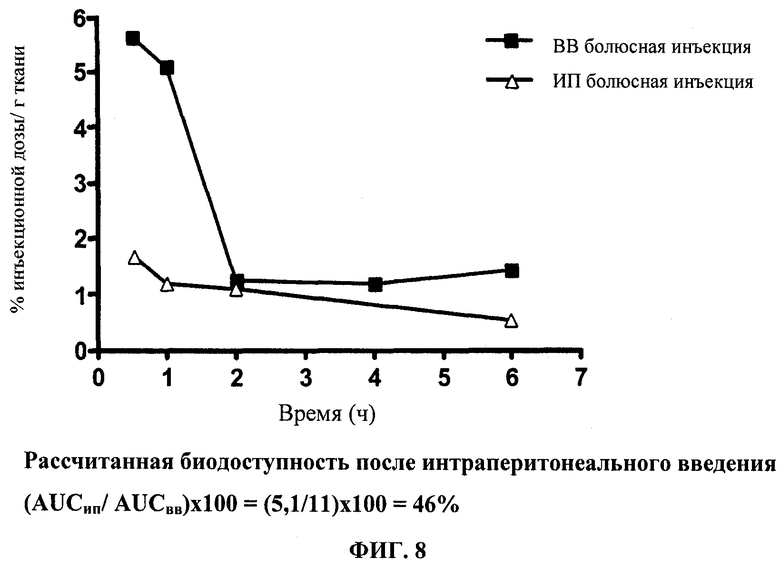
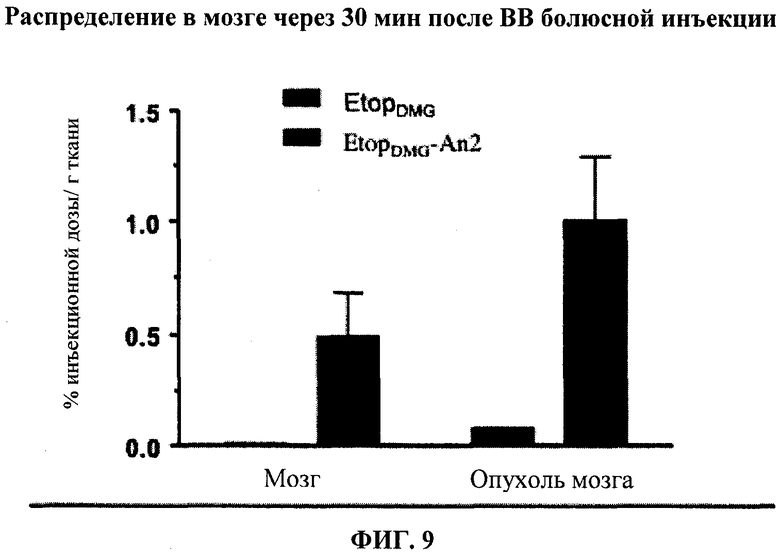
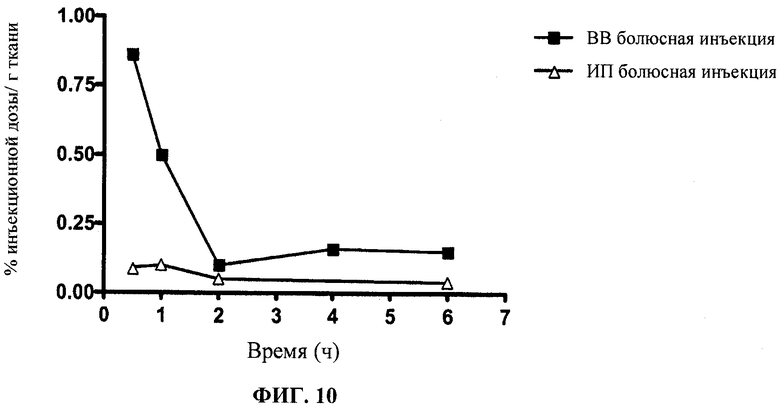
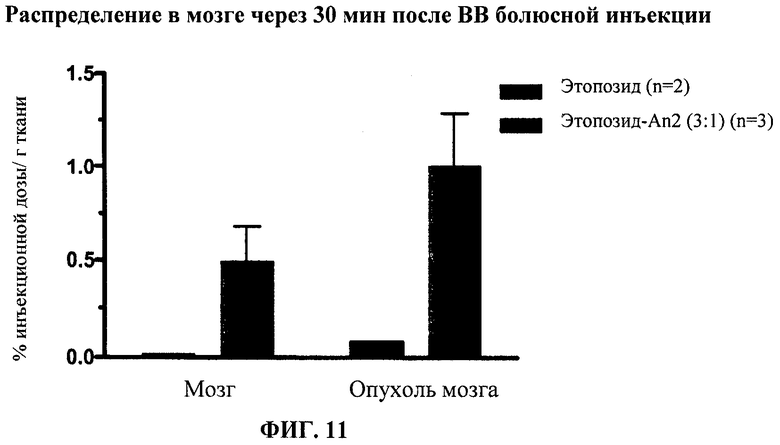
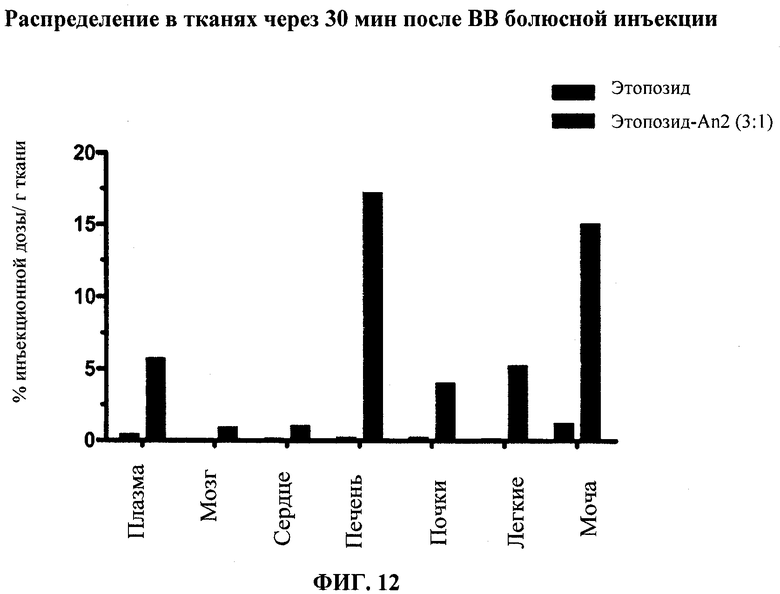
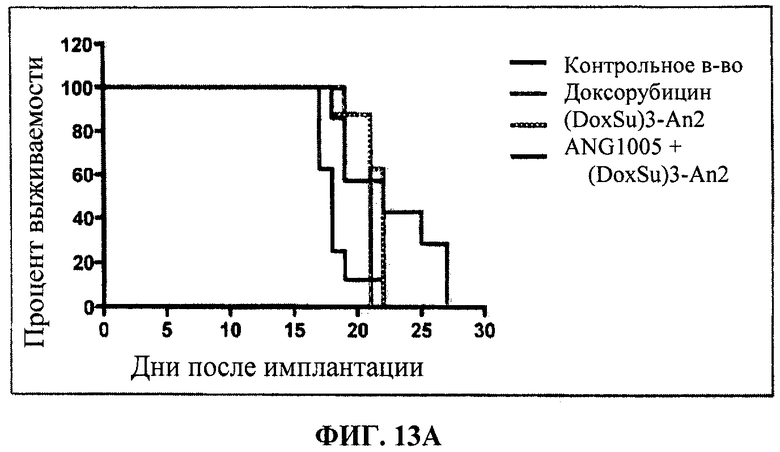
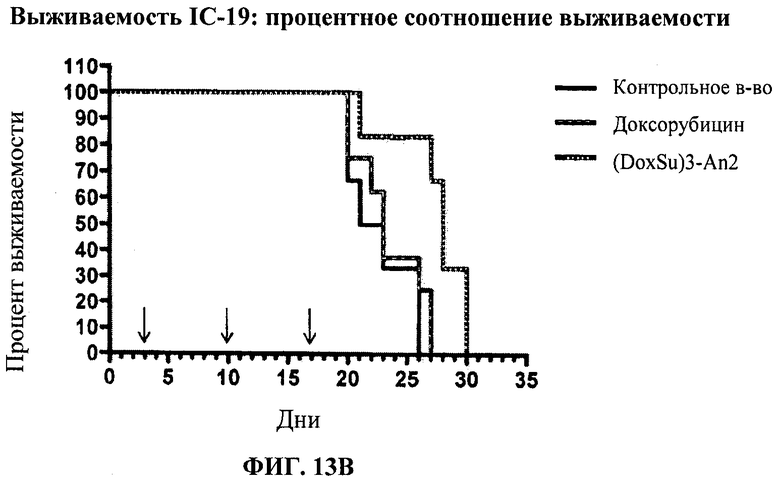
Изобретение относится к полипептидам, имеющим гидролизуемые ковалентные связи с терапевтическими агентами, для доставки лекарственных средств. Эти полипептидные конъюгаты могут быть использованы в качестве векторов для транспортировки терапевтических препаратов через гематоэнцефалитический барьер (ГЭБ) или для доставки в четко определенные виды клеток, такие как яичники, печень, легкие или почки. Изобретение также относится к фармацевтическим композициям, которые включают соединения по изобретению, и к их применению в способах лечения рака. 3 н. и 3 з.п. ф-лы, 16 ил., 8 табл., 6 пр.
1. Соединение, имеющее структуру
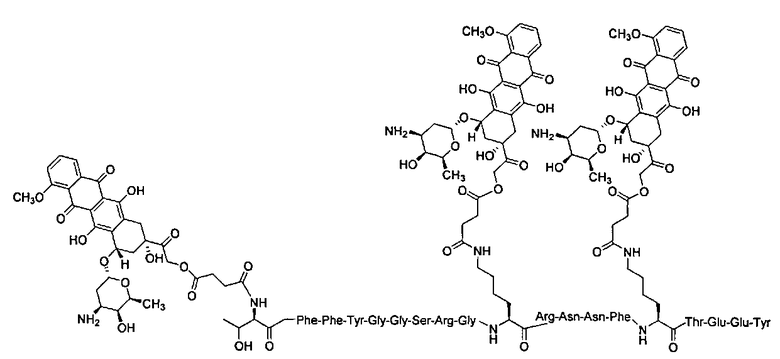
или его фармацевтически приемлемая соль.
2. Соединение по п.1, где соединение содержит гидрохлорид-соль 1,2 или 3 из ковалентно присоединенных частей молекулы доксорубицина.
3. Соединение по п.1, где соединение представляет собой треххлористоводородную соль.
4. Фармацевтическая композиция для лечения или профилактики рака, содержащая терапевтически эффективное количество соединения по любому из пп.1-3 и фармацевтически приемлемый носитель.
5. Способ лечения рака, где способ включает введение пациенту терапевтически эффективного количества соединения по любому из пп.1-3.
6. Способ по п.5, где рак представляет собой острый лимфоцитарный лейкоз, острый миелобластный лейкоз, адренокортикальный рак, внутривенный и внутрипузырный рак мочевого пузыря, костную саркому, рак молочной железы, карциноидный синдром (тонкого кишечника), рак эндометрия, саркому Юинга, гинекологические саркомы, рак головы и шеи (плоскоклеточный рак), рак печени, болезнь Ходжкина, рак островковых клеток, лейкоз, рак легких, злокачественные лимфомы, множественную миелому, нейробластому, неходжкинскую лимфому, рак поджелудочной железы, рак предстательной железы (простаты), остеогенную саркому, рак яичников, ретинобластома, рабдомиосаркома, рак желудка, рак яичек, рак щитовидной железы, карциному «переходных» клеток мочевого пузыря, саркому мягких тканей или опухоль Вильмса.
| УСИЛЕНИЕ ДЕЙСТВИЯ ПРОТИВОРАКОВЫХ АГЕНТОВ | 2006 |
|
RU2422143C2 |
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| GABIUS, H.J | |||
| ET AL.: 'Targeting ofNeoglycoprotein-Drug Conjugates to Cultured Human Embryonal Carcinoma Cells.' J | |||
| CANCER RES | |||
| CLIN | |||
| Кузнечная нефтяная печь с форсункой | 1917 |
|
SU1987A1 |
| Способ обработки грубых шерстей на различных аппаратах для мериносовой шерсти | 1920 |
|
SU113A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| ЦЕНТРОБЕЖНЫЙ ВЕНТИЛЯТОР | 1922 |
|
SU1005A1 |

