ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к области фармацевтики. В частности, настоящее изобретение относится к новому дейтерированному соединению хиназолинона и содержащим его фармацевтическим композициям.
УРОВЕНЬ ТЕХНИКИ
Фосфоинозитид-3-киназы (PI3K) представляют собой ферменты, специфически катализирующие фосфорилирование гидроксильной группы в 3-м положении фосфатидилинозитола (PI) и его производных и получение фосфатидилинозитол-3,4,5-трифосфата (PI3P), играющего роль вторичного мессенджера. Передача сигнала, опосредованная PI3K, вовлечена в регуляцию нескольких клеточных функций, таких как клеточное деление, дифференциация, апоптоз, метаболизм, ангиогенез, и играет важную роль в активации ряда биологических клеточных функций. Исследования, проведенные в последние годы, показали, что сигнальные пути, состоящие из PI3K и нижеследующей молекулярной протеинкиназы В (PKB или Akt) тесно связаны с образованием и развитием рака, регуляцией пролиферации опухолевых клеток, апоптозом и стимуляцией опухолевого ангиогенеза, и т.д.
Соединения хиназолинона и их производные представляют собой класс ингибиторов фосфоинозитид-3-киназы. Серия производных хиназолина была раскрыта в WO 03035075 и WO 2005113556. Среди них соединение GS-1101, химическое название которого (S)-2-(1-(9Н-пурин-6-ил-амино)пропил)-5-фтор-3-фенилхиназолин-4(3Н)-кетон, представляет собой селективный ингибитор PI3K-киназы, и может применяться в лечении рака и заболеваний, связанных с клеточной пролиферацией, а также других связанных заболеваний. В настоящее время данное соединение находится в III стадии клинических исследований лечения хронической лимфоцитарной лейкемии и неходжкинских лимфом.
Фосфоинозитид-3-киназа (PI3K) представляет собой одну из важных мишеней при разработке новых противоопухолевых лекарственных препаратов. Однако, за исключением рапамицина и гомологов, исследования ингибирования пути трансдукции сигналов PI3K продвигаются достаточно медленно, особые трудности все еще представляет разработка специфических ингибиторов подтипов PI3K (таких как тип I PI3K, включающий р110α, р110β, р110δ, и т.д.).
Таким образом, все еще существует потребность в получении соединений, обладающих ингибирующей активностью в отношении PI3K-киназы или улучшенными фармакодинамическими/фармакокинетическими свойствами.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Объектом настоящего изобретения является получение класса новых соединений, обладающих активностью ингибиторов PI3K-киназ и/или улучшенными фармакодинамическими/фармакокинетическими свойствами, и их применения.
В первом аспекте настоящего изобретения предложено дейтерированное соединение хиназолинона формулы (I), или его кристаллическая форма, фармацевтически приемлемая соль, гидрат или сольват:
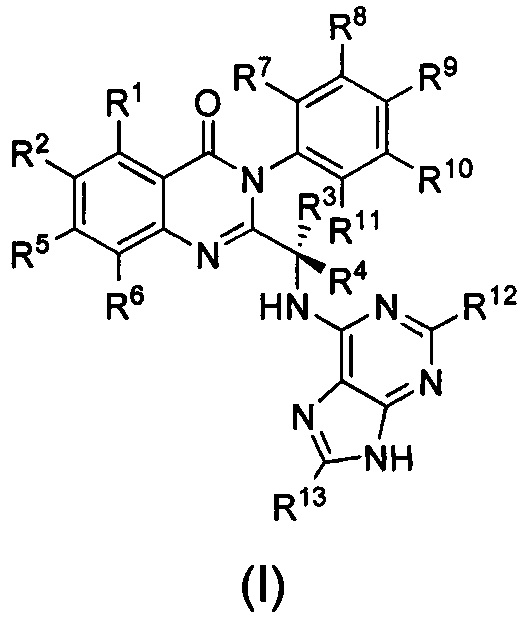
где R1 и R2 независимо представляют собой водород, дейтерий или галоген;
R3 выбран из водорода, дейтерия, СН3, CH2D, CHD2, CD3, СН2СН3, CD2CH3, CH2CD3 и CD2CD3;
каждый из R4, R5, R6, R7, R8, R9, R10, R11, R12 и R13 независимо представляет собой водород или дейтерий;
при условии, что по меньшей мере один из R1, R2, R3, R4, R5, R6, R7, R8, R9, R10, R11, R12 или R13 является дейтерированным или представляет собой дейтерий.
В другом предпочтительном воплощении содержание изотопа дейтерия в положении, замещенном дейтерием, составляет по меньшей мере больше, чем природное содержание изотопа дейтерия (приблизительно 0,015%), предпочтительно больше чем 30%, более предпочтительно больше чем 50%, более предпочтительно больше чем 75%, более предпочтительно больше чем 95%, более предпочтительно больше чем 99%.
В другом предпочтительном воплощении соединение формулы (I) содержит по меньшей мере один атом дейтерия, более предпочтительно три атома дейтерия, более предпочтительно четыре атома дейтерия, более предпочтительно 6 атомов дейтерия.
В другом предпочтительном воплощении энантиомерная чистота соединения формулы (I) составляет больше чем 95%, более предпочтительно больше чем 98%, более предпочтительно больше чем 99%.
В другом предпочтительном воплощении R1 представляет собой фтор и/или R2 представляет собой водород.
В другом предпочтительном воплощении R4 представляет собой водород или дейтерий.
В другом предпочтительном воплощении R5, R6, R7, R8, R9, R10, R11, R12 и R13 независимо представляют собой водород или дейтерий.
В другом предпочтительном воплощении R1 представляет собой галоген, такой как фтор, хлор, бром или иод.
В другом предпочтительном воплощении R1 представляет собой фтор.
В другом предпочтительном воплощении R3 представляет собой СН3, CH2D, СНD2, CD3, СН2СН3, CD2CH3, CH2CD3 или CD2CD3.
В другом предпочтительном воплощении R4 представляет собой дейтерий.
В другом предпочтительном воплощении R12 представляет собой дейтерий и/или R13 представляет собой дейтерий.
В другом предпочтительном воплощении R12 представляет собой дейтерий.
В другом предпочтительном воплощении R13 представляет собой дейтерий.
В другом предпочтительном воплощении соединение представляет собой одно из следующих соединений или его фармацевтически приемлемую соль:
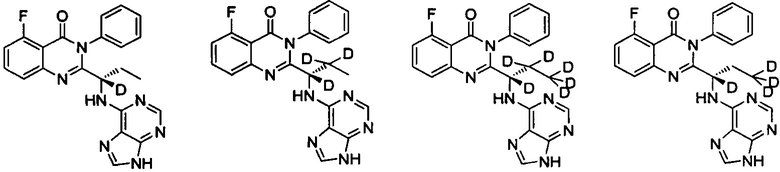
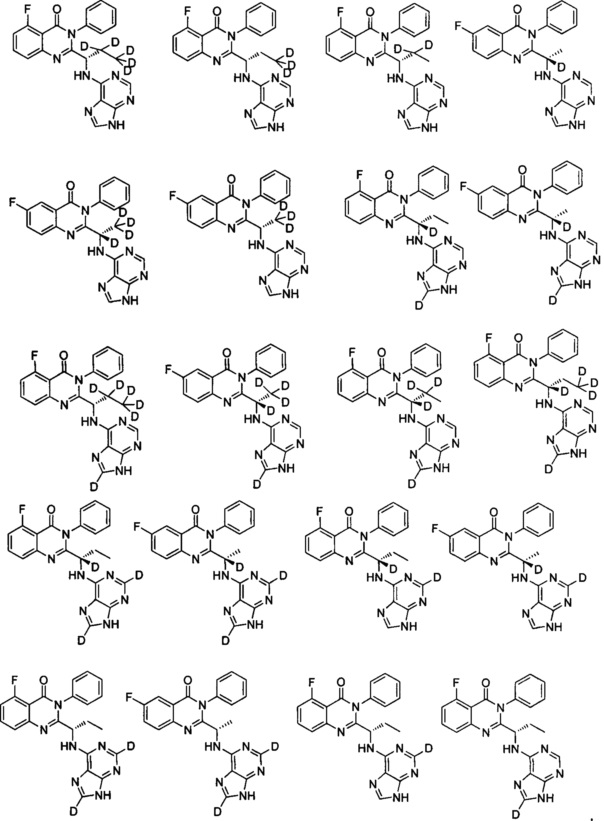
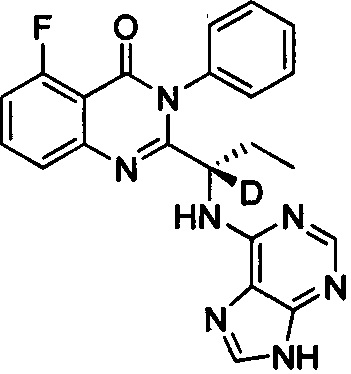
В другом предпочтительном воплощении соединение представляет собой одно из следующих соединений или его фармацевтически приемлемую соль:
(S)-2-(1-(9Н-пурин-6-ил-амино)-(1-d-пропил))-5-фтор-3-фенилхиназолин-4(3Н)-кетон;
(S)-2-(1-(9Н-пурин-6-ил-амино)-(1,2,2-d3-пропил))-5-фтор-3-фенилхиназолин-4(3Н)-кетон;
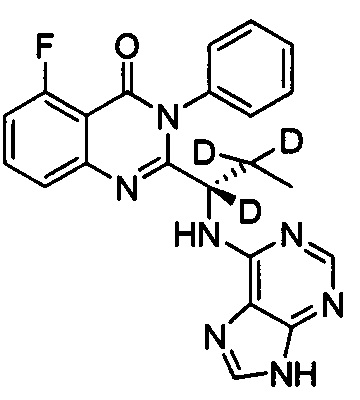
(S)-2-(1-(9Н-пурин-6-ил-амино)-(1,2,2,3,3,3-d6-пропил))-5-фтор-3-фенилхиназолин-4
(3H)-кетон;
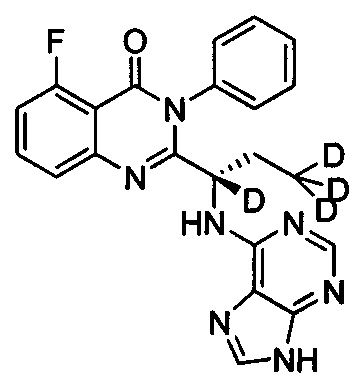
(S)-2-(1-(9Н-пурин-6-ил-амино)-(1,3,3,3-d4-пропил))-5-фтор-3-фенилхинозалин-4(3Н)-кетон;
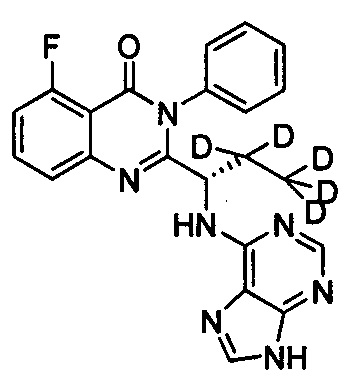
(S)-2-(1-(9Н-пурин-6-ил-амино)-(2,2,3,3,3-d5-пропил))-5-фтор-3-фенилхиназолин-4
(3Н)-кетон;
(S)-2-(1-(9Н-пурин-6-ил-амино)-(3,3,3-d3-пропил))-5-фтор-3-фенилхиназолин-4(3Н)-кетон;
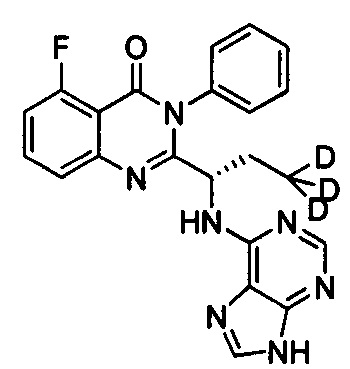
(S)-2-(1-(9Н-пурин-6-ил-амино)-(2,2-d2-пропил))-5-фтор-3-фенилхиназолин-4(3Н)-кетон;
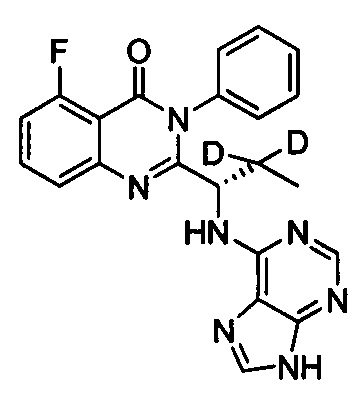
(S)-2-(1-(9Н-пурин-6-ил-амино)-(1-d-этил))-6-фтор-3-фенилхиназолин-4(3Н)-кетон;
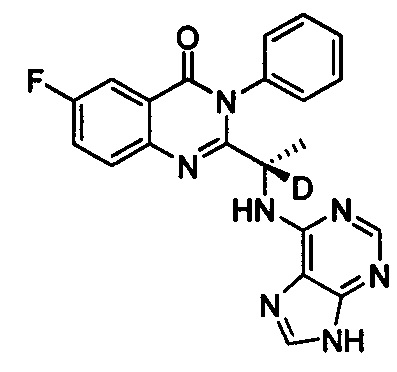
(S)-2-(1-(9Н-пурин-6-ил-амино)-(2,2,2-d3-этил))-6-фтор-3-фенилхиназолин-4(3Н)-кетон;
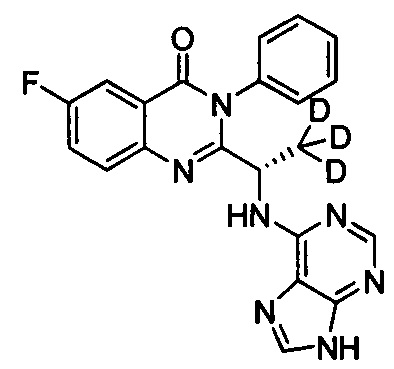
(S)-2-(1-(9Н-пурин-8-d-6-ил-амино)-(1-d-пропил))-5-фтор-3-фенилхиназолин-4(3Н)-кетон;
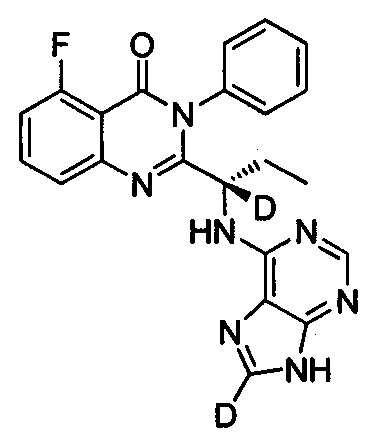
(S)-2-(1-(9Н-пурин-2,8-d2-6-ил-амино)-(1-d-пропил))-5-фтор-3-фенилхиназолин-4(3Н)-кетон;
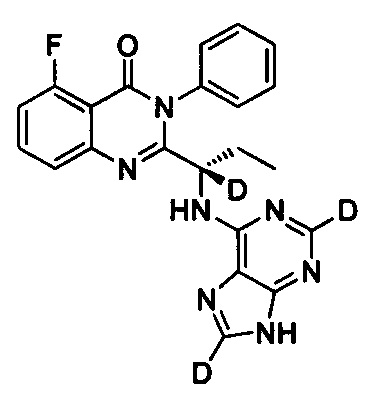
(S)-2-(1-(9Н-пурин-2,8-d2-6-ил-амино)-пропил)-5-фтор-3-фенилхиназолин-4(3Н)-кетон;
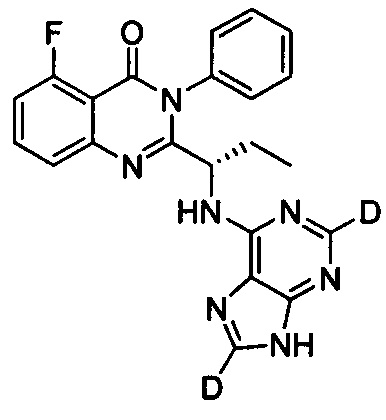
(S)-2-(1-(9Н-пурин-8-d-6-ил-амино)-пропил)-5-фтор-3-фенилхинозалин-4(3Н)-кетон;
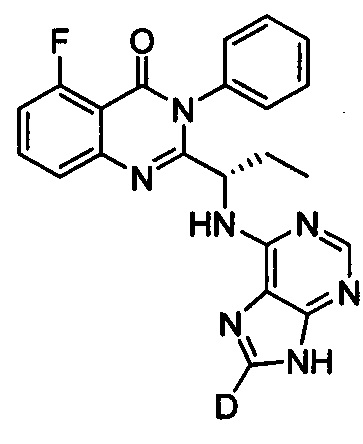 .
.
В другом предпочтительном воплощении соединение представляет собой
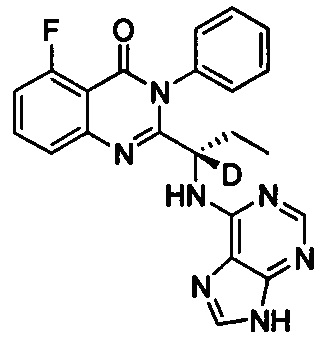 , обладающее следующими характеристиками: MS, рассчитано: 416; MS, найдено: 417 (М+Н)+, 439 (M+Na)+.
, обладающее следующими характеристиками: MS, рассчитано: 416; MS, найдено: 417 (М+Н)+, 439 (M+Na)+.
В другом предпочтительном воплощении соединение представляет собой
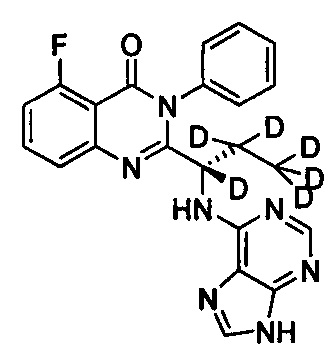 , обладающее следующими характеристиками: MS, рассчитано: 421; MS, найдено: 422 (М+Н)+, 444 (M+Na)+.
, обладающее следующими характеристиками: MS, рассчитано: 421; MS, найдено: 422 (М+Н)+, 444 (M+Na)+.
В другом предпочтительном воплощении соединение представляет собой
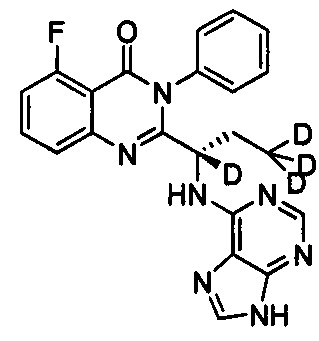 , обладающее следующими характеристиками: MS, рассчитано: 419; MS, найдено: 420 (М+Н)+, 442 (M+Na)+.
, обладающее следующими характеристиками: MS, рассчитано: 419; MS, найдено: 420 (М+Н)+, 442 (M+Na)+.
В другом предпочтительном воплощении соединение представляет собой
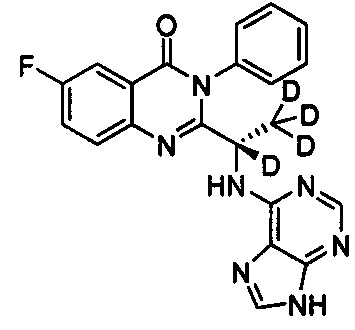 , обладающее следующими характеристиками: MS, рассчитано: 405; MS, найдено: 406 (М+Н)+, 428 (M+Na)+.
, обладающее следующими характеристиками: MS, рассчитано: 405; MS, найдено: 406 (М+Н)+, 428 (M+Na)+.
В другом предпочтительном воплощении соединение представляет собой
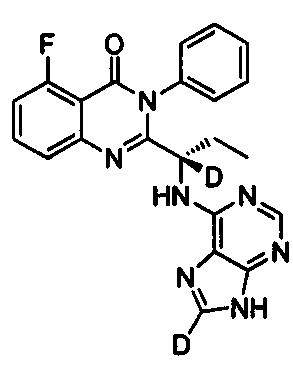 , обладающее следующими характеристиками: MS, рассчитано: 417; MS, найдено: 418 (М+Н)+, 440 (M+Na)+.
, обладающее следующими характеристиками: MS, рассчитано: 417; MS, найдено: 418 (М+Н)+, 440 (M+Na)+.
В другом предпочтительном воплощении соединение представляет собой
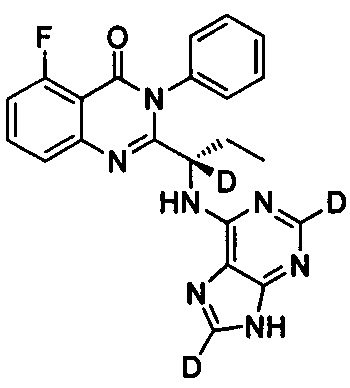 , обладающее следующими характеристиками: MS, рассчитано: 418; MS, найдено: 419 (М+Н)+, 441 (M+Na)+.
, обладающее следующими характеристиками: MS, рассчитано: 418; MS, найдено: 419 (М+Н)+, 441 (M+Na)+.
В другом предпочтительном воплощении соединение представляет собой
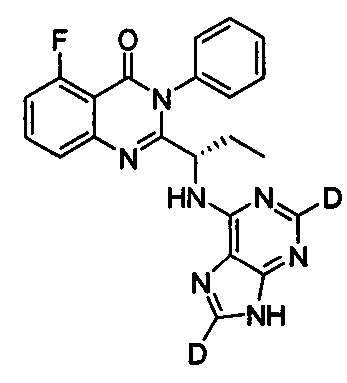 , обладающее следующими характеристиками: MS рассчитано: 417; MS, найдено: 418 (М+Н)+, 440 (M+Na)+.
, обладающее следующими характеристиками: MS рассчитано: 417; MS, найдено: 418 (М+Н)+, 440 (M+Na)+.
В другом предпочтительном воплощении соединение представляет собой
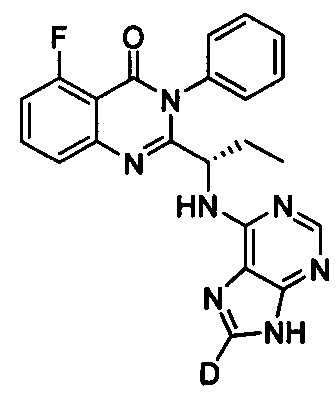 , обладающее следующими характеристиками: MS, рассчитано: 416; MS, найдено: 417 (М+Н)+, 439 (M+Na)+.
, обладающее следующими характеристиками: MS, рассчитано: 416; MS, найдено: 417 (М+Н)+, 439 (M+Na)+.
В другом предпочтительном воплощении недейтерированные соединения не включены в соединение.
В другом предпочтительном воплощении недейтерированное соединение представляет собой (S)-2-(1-(9Н-пурин-6-ил-амино)пропил)-5-фтор-3-фенилхиназолин-4(3Н)-кетон.
В другом предпочтительном воплощении соединение получено способом, описанным в Примерах 1-12.
Во втором аспекте настоящего изобретения предложен способ получения фармацевтической композиции, включающий следующую стадию: смешивания соединений согласно первому аспекту настоящего изобретения или их кристаллических форм, фармацевтически приемлемых солей, гидратов или сольватов с фармацевтически приемлемым носителем с образованием фармацевтической композиции.
В третьем аспекте настоящего изобретения предложена фармацевтическая композиция, содержащая фармацевтически приемлемый носитель и соединение согласно первому аспекту настоящего изобретения, или его кристаллическую форму, фармацевтически приемлемую соль, гидрат или сольват.
В другом предпочтительном воплощении фармацевтическая композиция находится в форме для инъекции, в виде капсулы, таблетки, пилюли, порошка или гранулы.
В другом предпочтительном воплощении фармацевтическая композиция содержит другие терапевтические средства, и другие терапевтические средства представляют собой средства для лечения раковых заболеваний, сердечно-сосудистых заболеваний, воспалений, инфекций, аутоимунных заболеваний, нарушений клеточной пролиферации, вирусных заболеваний, нарушений обмена веществ, или средство для трансплантации органов.
Более предпочтительно, другие терапевтические средства включают (без ограничения): 5-фторурацил, FOLFOX (Фольфокс), Avastin™ (авастин, бевацизумаб), бексаротен, бортезомиб, кальцитриол, канертиниб, капецитабин, гемцитабин, карбоплатин, целекоксиб, цетуксимаб, цисплатин, дасатиниб, дигоксин, энзастаурин, эрлотиниб, этопосид, эверолимус, фулвестрант, гефитиниб, генистейн, иматиниб, иринотекан, лапатиниб, леналидомид, летрозол, лейковорин, матуцумаб, оксалиплатин, Таксол (паклитаксел), доцетаксел, панитумумаб, пегилированный гранулоцит-колониестимулирующий фактор (пегфилграстин), пегилированный альфа-интерферон, пеметрексед, Polyphenon® Е (полифенон Е), сатраплатин, сиролимус, сунитиниб (сутент), сулиндак, таксотер, темозоломид (темодар), Торисел, темсиролимус, типифарниб, трастузумаб, вальпроевую кислоту, винфлунин, Волоциксимаб, Вориностат, сорафениб, Кризотиниб, Леотиниб, лапатиниб, Тофацитиниб, PD-0332991 (Палбоциклиб), амбризентан, доксорубицин, метотрексат, Преднизон, ритуксимаб, CD40 и/или СD154-специфические антитела, слитые белки, ингибиторы NF-kB, нестероидные противовоспалительные лекарственные средства, ингибиторы фактора свертывания FXa (такие как ривароксабан и т.д.), антитела против TNF, антибиотики, такие как калихеамицин, актиномицин, Адриамицин (доксорубицин), и т.д.
В четвертом аспекте настоящего изобретения предложено применение соединения согласно первому аспекту настоящего изобретения или его кристаллической формы, фармацевтически приемлемой соли, гидрата или сольвата для получения фармацевтических композиций, ингибирующих PI3K-киназы.
В другом предпочтительном воплощении фармацевтические композиции по изобретению могут применяться для лечения следующих заболеваний: раковых заболеваний, нарушений клеточной пролиферации, воспалений, инфекций или аутоиммунных заболеваний.
В другом предпочтительном воплощении раковые заболевания включают (без ограничения): рак легких, рак молочной железы, рак предстательной железы, рак пищевода, колоректальный рак, рак толстой кишки, рак крови (или злокачественное гематологическое заболевание), остеосаркому, рак почки, рак желудка, рак печени или колоректальный рак.
В другом предпочтительном воплощении рак крови (или злокачественное гематологическое заболевание) представляет собой лейкемию и лимфому.
В другом предпочтительном воплощении лимфома представляет собой хронический лимфобластный лейкоз, острый лимфобластный лейкоз, острую миелоидную лейкемию, множественную миелому и хроническую миелоидную лейкемию.
В пятом аспекте настоящего изобретения предложен способ ингибирования PI3K-киназ или способ лечения заболеваний (таких как рак, нарушения клеточной пролиферации, воспаление, инфекция, иммунные заболевания), включающий следующие стадии: введения соединения согласно первому аспекту настоящего изобретения или его кристаллической формы, фармацевтически приемлемой соли, гидрата или сольвата, или введения фармацевтической композиции согласно третьему аспекту настоящего изобретения субъекту, нуждающемуся в этом.
Следует иметь в виду, что в настоящем изобретении каждый из технических признаков, конкретно описанных выше и ниже (таких, как приведенные в Примерах) может быть комбинирован с другими, образуя, таким образом, новые или предпочтительные технические решения, которые могут не упоминаться в настоящем описании.
ВОПЛОЩЕНИЯ ДЛЯ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
Изобретателем посредством проведения исследований было неожиданно обнаружено, что дейтерированное соединение хиназолинона или его фармацевтически приемлемые соли очевидно превосходят недейтерированное соединение по фармакокинетическим и/или фармакодинамическим свойствам, и, следовательно, являются более подходящими для применения в качестве соединений, ингибирующих PI3K-киназы, и более подходящими для применения в получении лекарственных средств для лечения раковых заболеваний и заболеваний, связанных с PI3K-киназами. Настоящее изобретение выполнено на этой основе.
ОПРЕДЕЛЕНИЯ
В контексте данного документа термин «галоген» относится к F, Cl, Br и I. Более предпочтительно, галоген выбран из F, Cl и Br.
В контексте данного документа термин «превосходящие фармакокинетические и/или фармакодинамические свойства» относится к более длительному периоду полувыведения лекарственного препарата (t1/2) или к более высокой экспозиции лекарственного препарата (AUC (площадь под кривой зависимости концентрации от времени), или к более высокой максимальной концентрации лекарственного препарата (Cmax), или к пониженному клиренсу лекарственного препарата.
В контексте данного документа термин «дейтерированный» означает, что один или более атом водорода является(ются) замещенным(и) дейтерием. «Дейтерированный» может предполагать монозамещение, дизамещение, мультизамещение или полное замещение. Термины «одно- или мультидейтерированный» и «дейтерированный один или более раз» могут использоваться взаимозаменяемо.
В контексте данного документа термин «недейтерированное соединение» относится к соединению, содержание дейтерированных атомов в котором не больше, чем природное содержание изотопа дейтерия (приблизительно 0,015%).
В другом предпочтительном воплощении содержание изотопа дейтерия в положениях, замещенных дейтерием, составляет больше, чем природное содержание изотопа дейтерия (0,015%), более предпочтительно больше чем 50%, более предпочтительно больше чем 75%, более предпочтительно больше чем 95%, более предпочтительно больше чем 97%, более предпочтительно больше чем 99%, более предпочтительно больше чем 99,5%.
В другом предпочтительном воплощении соединение формулы (I) содержит по меньшей мере один атом дейтерия, более предпочтительно два атома дейтерия, более предпочтительно три атома дейтерия, более предпочтительно шесть атомов дейтерия.
Предпочтительно в соединении формулы (I) N представляет собой 14N и/или О представляет собой 16О.
В другом предпочтительном воплощении содержание изотопа 14N в соединении в положении атома азота составляет ≥95%, предпочтительно ≥99%.
В другом предпочтительном воплощении содержание изотопа 16O в соединении в положении атома кислорода составляет ≥95%, предпочтительно ≥99%.
АКТИВНЫЕ ИНГРЕДИЕНТЫ
В контексте данного документа термин «соединение по настоящему изобретению» относится к соединению формулы (I). Данный термин также включает кристаллические формы, фармацевтически приемлемые соли, гидраты или сольваты соединения формулы (I).
Среди них термин «фармацевтически приемлемая соль» относится к подходящей для лекарственного средства соли, образованной соединением по настоящему изобретению и кислотой или щелочью. Фармацевтически приемлемые соли включают неорганические и органические соли. Предпочтительный тип солей представляет собой соли, образованные соединениями по настоящему изобретению и кислотой. Подходящие солеобразующие кислоты включают, без ограничения: неорганические кислоты, такие как соляная кислота, бромоводородная кислота, фтороводородная кислота, серная кислота, азотная кислота, фосфорная кислота и тому подобные; органические кислоты, такие как муравьиная кислота, уксусная кислота, трифторуксусная кислота, пропионовая кислота, щавелевая кислота, малоновая кислота, янтарная кислота, фумаровая кислота, малеиновая кислота, молочная кислота, яблочная кислота, винная кислота, лимонная кислота, пикриновая кислота, бензойная кислота, метансульфоновая кислота, этансульфоновая кислота, р-толуолсульфоновая кислота, бензолсульфоновая кислота, нафталинсульфоновая кислота и тому подобные; и аминокислоты, такие как пролин, фенилаланин, аспарагиновая кислота, глютаминовая кислота, и тому подобные. Другой предпочтительный тип солей представляет собой соли, образованные соединениями по настоящему изобретению и основаниями, например, соли щелочных металлов (например, соли натрия или калия), соли щелочноземельных металлов (например, соли кальция или магния), соли аммония (например, соли низшего алканоламмония или другие фармацевтически приемлемые аминные соли), например, соль метиламина, соль этиламина, соль пропиламина, соль диметиламина, соли триметиламина, соли диэтиламина, соли триэтиламина, соли трет-бутиламина, соли этилендиамина, соли гидроксиэтиламина, соли бис-гидроксиэтиламина, соли трис-гидроксиэтиламина и аминные соли, образованные морфолином, пиперазином и лизином.
В контексте данного документа термин «сольват» относится к комплексу с определенным стехиометрическим соотношением, образованному в результате координации соединения по настоящему изобретению с молекулами растворителя. «Гидрат» относится к комплексу, образованному в результате координации соединения по настоящему изобретению с водой.
Более того, соединения по настоящему изобретению дополнительно содержат хиральные энантиомеры или рацематы соединений хиназолинона формулы (I).
Более того, соединения по настоящему изобретению дополнительно содержат пролекарства соединений хиназолинона формулы (I). Термин «пролекарство» включает тип соединений, биологически активных или неактивных, претерпевающих превращения в соединение формулы (I) посредством метаболизма или химических реакций в организме человека при введении надлежащим способом, или соль или сольват, образованные соединением формулы (I). Пролекарства включают (без ограничения) сложный эфир карбоновой кислоты, сложный эфир угольной кислоты, фосфат, нитрат, сульфат, сложный сульфоновый эфир, сложные эфиры сульфоксида, аминосоединения, карбаматы, азосоединения, фосфорамиды, глюкозид, простой эфир, ацеталь соединения и т.д.
СПОСОБ ПОЛУЧЕНИЯ
В дальнейшем в этом документе получение соединений формулы (I) будет подробно описано, однако такие конкретные способы не представляют собой какого-либо ограничения настоящего изобретения. Соединения по изобретению могут также быть легко получены путем, при необходимости, комбинирования различных способов синтеза, приведенных в настоящем описании или известных из уровня техники, и такая комбинация может быть легко осуществлена специалистом в области, к которой относится настоящее изобретение.
Способы, используемые в настоящем изобретении для получения недейтерированных соединений хиназолинона и его физиологически совместимых солей являются известными. Получение соответствующих дейтерированных соединений хиназолинона может быть осуществлено путем использования соответствующего дейтерированного исходного соединения посредством такого же пути синтеза. Например, соединение формулы (I) по настоящему изобретению может быть получено в соответствии со способом, описанным в WO 03035075, за исключением того, что вместо недейтерированных реагентов используются дейтерированные.
Как правило, в процессе получения каждая реакция обычно протекает в инертном растворителе при температуре от комнатной до температуры кипения реакционной смеси (такой как 0°C-200°C, предпочтительно 0°C-100°C). Время реакции обычно составляет от 0,1 часа до 60 часов, предпочтительно от 0,5 до 48 часов.
Для синтеза соединений формулы (I) по настоящему изобретению может быть использован следующий общий путь получения.
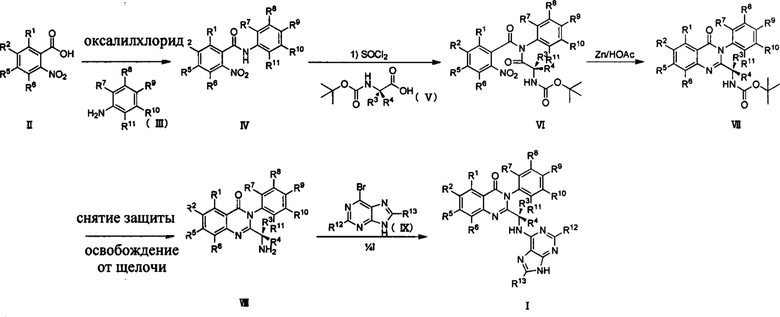
Путь синтеза I
Где R2, R1 выбраны из Н, D, F, Cl, Br, I; R3, R4, R5, R6, R7, R8, R9, R10, R11, R12 и R13 являются такими, как определено выше.
Как показано в Пути синтеза I, соединение II замещенной m-нитробензойной кислоты, ацилированное оксалилхлоридом, было использовано для проведения реакции с замещенным анилином III в щелочных условиях с получением соединения IV. Амидный водород соединения IV замещается хлором в присутствии тионилхлорида, и затем соединение IV реагирует с дейтерированной Вос-защищенной аминокислотой V с получением соединения VI. Ариламин получают из арильной нитроструктуры соединения VI в восстанавливающих условиях, таких как порошок цинка/уксусная кислота, двухлористое олово, порошок восстановленного железа, или в условиях каталитического гидрирования, таких как палладий, платина, никель Ренея и тому подобное. Структуру хиназолинкетона VII получают посредством реакции закрытия кольца. Вос-защиту соединения VII удаляют в условиях трифторуксусной кислоты/метиленхлорида и соляной кислоты/диоксана, после чего оно освобождается от щелочи с получением соединения VIII. Наконец, соединение I по настоящему изобретению получают из соединения VIII при кипячении с 6-бромпурином или 6-бром дейтерированным пурином в щелочных условиях в спиртовом растворителе (таком как этанол, n-бутанол и третичный бутиловый спирт) или тетрагидрофурановом растворителе. Вышеприведенные реакции проводят в инертном растворителе, таком как дихлорметан, дихлорэтан, ацетонитрил, n-гексан, толуол, тетрагидрофуран, N,N-диметилформамид, диметилсульфоксид, уксусная кислота, бутанол, пропиловый спирт и т.д., при температуре 0-200°C.
Дейтерированное соединение V может быть получено следующими путями:
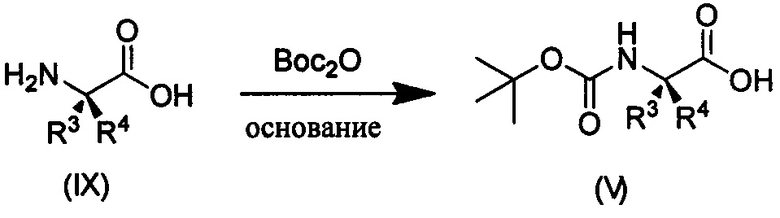
Путь синтеза II
где R3, R4 являются такими, как определено выше.
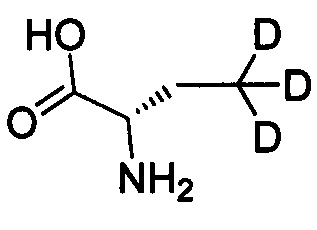
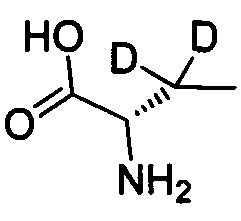
Как показано в Пути синтеза II, в щелочных условиях дейтерированная аминокислота IX реагирует с ди-трет-бутилдикарбонатом с получением N-Boc-защищенной аминокислоты V. Некоторые дейтерированные аминокислоты IX получают путем традиционного способа дейтерирования. Другие дейтерированные аминокислоты IX могут быть приобретены, как, например, (2S)-2-амино-4,4,4-d3-масляная кислота, (2S)-2-амино-3,3-d2-масляная кислота, (2S)-2-амино-2-d-масляная кислота и (2S)-2-амино-2,3,3-d3-масляная кислота.
 ,
,  ,
,  ,
,  .
.
ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ЕЕ ВВЕДЕНИЕ
Соединения по настоящему изобретению обладают выдающейся активностью ингибирования PI3K-киназ. Следовательно, соединение по настоящему изобретению и его кристаллические формы, фармацевтически приемлемые неорганические или органические соли, гидраты или сольваты, и фармацевтические композиции, содержащие соединение по настоящему изобретению в качестве основного активного ингредиента, могут применяться для лечения, предотвращения и облегчения заболеваний, опосредованных PI3K-каназами. На основании уровня техники соединения по настоящему изобретению могут применяться для лечения следующих заболеваний: раковых заболеваний, нарушений клеточной пролиферации, воспалений, инфекций и аутоиммунных заболеваний.
Фармацевтическая композиция по настоящему изобретению содержит соединение по настоящему изобретению или его фармацевтически приемлемые соли в безопасном и эффективном диапазоне доз, и фармацевтически приемлемые эксципиенты или носители. Термин «безопасная и эффективная доза» относится к такому количеству соединения, которое является достаточным для улучшения состояния пациента без каких-либо серьезных побочных эффектов. Как правило, фармацевтическая композиция содержит 1-2000 мг соединений по изобретению на дозу, предпочтительно 10-1000 мг соединений по изобретению на дозу. Предпочтительно, термин «на дозу» означает одну капсулу или таблетку.
«Фармацевтически приемлемый носитель» означает один или более совместимых твердых или жидких наполнителей или гелевых материалов, подходящих для человека, которые должны обладать достаточной чистотой и достаточно низкой токсичностью. Термин «совместимость» в контексте данного документа означает, что компоненты композиции могут быть смешаны с соединениями по настоящему изобретению или друг с другом, при этом не уменьшая существенно эффективность соединений. Некоторые примеры фармацевтически приемлемых носителей включают целлюлозу и ее производные (такие как натрий карбоксиметилцеллюлоза, натрий этилцеллюлоза, ацетат целлюлозы, и т.д.), желатин, тальк, твердые смазывающие вещества (такие как стеариновая кислота, стеарат магния), сульфат кальция, растительные масла (такие как соевое масло, кунжутное масло, арахисовое масло, оливковое масло, и т.д.), полиолы (такие как пропиленгликоль, глицерин, маннит, сорбит, и т.д.), эмульгаторы (такие как Tween®), увлажняющие агенты (такие как додецилсульфат натрия), красящие вещества, вкусовые добавки, стабилизаторы, антиоксиданты, консерванты, апирогенную воду и т.д.
В способе введения соединения или фармацевтических композиций по настоящему изобретению нет каких-либо определенных ограничений, и типичные способы введения включают (без ограничения): пероральный, внутриопухолевый, ректальный, парентеральный (внутривенный, внутримышечный или подкожный), и местное применение.
Твердые лекарственные формы для перорального введения включают капсулы, таблетки, пилюли, порошки и гранулы. В этих твердых лекарственных формах активные соединения смешаны с по меньшей мере одним традиционно применяемым инертным эксципиентом (или носителем), таким как цитрат натрия или CaHPO4, или смешаны с любым из следующих компонентов: (а) наполнители или агент, улучшающий совместимость, например, крахмал, лактоза, сахароза, глюкоза, маннит и кремниевая кислота; (б) связывающие вещества, например, гидроксиметилцеллюлоза, альгинаты, желатин, поливинилпирролидон, сахароза и аравийская камедь; (в) влагоудерживатель, такой как глицерин; (г) разрыхлитель, такой как агар, карбонат кальция, картофельный крахмал или тапиоковый крахмал, альгиновая кислота, некоторые смешанные силикаты и карбонат натрия; (д) замедляющие растворение агенты, такие как парафин; (е) усилители абсорбции, например, четвертичные аммониевые соединения; (ж) увлажняющие агенты, такие как цетиловый спирт и глицерилмоностеарат; (з) адсорбенты, например, каолин; и (и) смазывающие вещества, такие как тальк, стеарат кальция, стеарат магния, твердый полиэтиленгликоль, лаурилсульфат натрия, или их смеси. В капсулах, таблетках и пилюлях лекарственные формы также могут содержать буферные агенты.
Твердые лекарственные формы, такие как таблетки, пилюли с сахарной оболочкой, капсулы, пилюли и гранулы могут быть получены путем использования покрывающих материалов и материалов оболочки, таких как энтеросолюбильное покрытие и любые другие материалы, известные из уровня техники. Они могут содержать агент, придающий непрозрачность. Высвобождение активных соединений или соединений в составе композиций может происходить с задержкой в определенной части желудочно-кишечного тракта. Примеры капсулирующих компонентов включают полимеры и воски. При необходимости активные соединения и один или более указанный эксципиент могут образовывать микрокапсулы.
Жидкие лекарственные формы для перорального введения включают фармацевтически приемлемые эмульсии, растворы, суспензии, сиропы или настойки. В дополнение к активным ингредиентам жидкие лекарственные формы могут содержать любые традиционно применяемые инертные разбавители, известные из уровня техники, такие как вода или другие растворители, солюбилизаторы и эмульгаторы, например, этанол, изопропанол, этилкарбонат, этилацетат, пропиленгликоль, 1,3-бутандиол, диметилфомамид, а также масло, в частности, хлопковое масло, арахисовое масло, масло зародышей кукурузы, оливковое масло, касторовое масло и кунжутное масло, или их комбинацию.
Помимо этих инертных разбавителей композиция также может содержать добавки, такие как увлажняющие агенты, эмульгаторы и суспендирующие агенты, подсластители, вкусовые добавки и отдушки.
В дополнение к активным соединением суспензия также может содержать суспендирующий агент, например, этоксилированный изооктадеканол, полиоксиэтиленсорбит и сложные эфиры сорбитана, микрокристаллическую целлюлозу, метанол алюминий и агар, или их комбинацию.
Композиции для парентеральной инъекции могут содержать физиологически приемлемые стерильные водные или безводные растворы, дисперсии, суспензии или эмульсии, и стерильные порошки, которые могут быть повторно растворены с получением стерильных инъецируемых растворов или дисперсий. Подходящие водные и безводные носители, разбавители, растворители или эксципиенты включают воду, этанол, полиолы и любые их подходящие смеси.
Лекарственные формы для местного применения соединений по изобретению включают мази, порошки, пластыри, аэрозоли и лекарственные формы для ингаляции. Активные ингредиенты смешивают с физиологически приемлемыми носителями и любыми консервантами, буферными веществами или, при необходимости, пропеллентом, в стерильных условиях.
Соединение по настоящему изобретению могут быть введены отдельно или в комбинации с любыми другими фармацевтически приемлемыми соединениями.
При применении фармацевтических композиций безопасное и эффективное количество соединения по настоящему изобретению применяется к млекопитающему (такому, как человек), нуждающегося в этом, где вводимая доза представляет собой фармацевтически эффективную дозу. Для человека массой 60 кг дневная доза обычно составляет 1-2000 мг, предпочтительно 50-1000 мг. Безусловно, конкретная доза должна также зависеть от различных факторов, таких как путь введения, состояние здоровья пациента, которые находятся в рамках компетенции опытного врача.
По сравнению с недейтерированными соединениями, известными из уровня техники, соединения по настоящему изобретению обладают рядом преимуществ. Основные преимущества настоящего изобретения состоят в следующем:
(1) Соединения по настоящему изобретению обладают хорошей ингибирующей активностью в отношении протеинкиназы (такой как PI3K-киназа).
(2) Метаболизм дейтерированных соединений в организме изменяется в связи с дейтерированием, что придает соединению лучшую характеристику фармакокинетических параметров. В этом случае дозу можно варьировать и получать препарат длительного действия для улучшения применимости.
(3) Концентрация лекарственного препарата соединения у животных может быть увеличена путем замещения в соединении дейтерия на водород в связи с эффектом изотопа дейтерия, тем самым улучшая эффективность препарата.
(4) Защита соединения может быть улучшена посредством замены в нем дейтерия водородом, так как в таком случае подавляются некоторые метаболиты.
Настоящее изобретение будет дополнительно проиллюстрировано ниже со ссылкой на конкретные примеры. Следует иметь в виду, что эти примеры приведены только для иллюстрации изобретения, и ни в коем случае не ограничивают его объем. Описанные в следующих примерах методики проведения экспериментов, для которых не указаны конкретные условия, как правило проводятся при традиционных условиях или в соответствии с указаниями производителя. Если не указано иное, части и проценты рассчитаны по массе.
Пример 1: Получение (S)-2-(1-(9Н-пурин-6-ил-амино)-(1-d-пропил))-5-фтор-3-фенилхиназолин-4(3Н)-кетона (соединение 6)
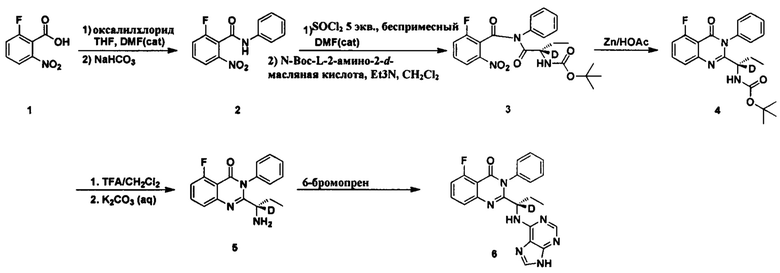
1. Получение 2-фтор-6-нитро-N-фенилбензамида (соединение 2)
Соединение 2-фтор-6-нитробензойная кислота (5,0 г, 0,027 моль) и N,N-диметилформамид (0,5 мл) последовательно добавляли в колбу и растворяли в 20 мл дихлорметана. Затем медленно по каплям добавляли оксалилхлорид (5,1 г, 0,04 моль, 1,5 экв.). После перемешивания в течение 2 часов при комнатной температуре реакционный раствор концентрировали. Образец суспензии растворяли в диоксане (10 мл) и охлаждали до 5°C. Раствор по каплям добавляли к смешанной системе диоксана и воды (1:1, v:v, 30 мл), содержащей анилин (5 мл, 0,027 моль, 1 экв.) и бикарбонат натрия (4,5 г, 0,054 моль, 2 экв.). После добавления реакционный раствор нагревали до комнатной температуры и перемешивали в течение 60 мин. Добавляли воду, и большое количество твердых соединений осаждалось. Твердые соединения фильтровали и промывали осадок водой. После фильтрования с отсасыванием фильтрат сушили в глубоком вакууме при 50°C в течение 24 часов с получением беловатого твердого целевого продукта (6,0 г, 85%). 1H ЯМР (300 МГц, DMSO-d6) δ 10,82 (s, 1Н), 8,12 (d, J=7,7 Гц, 1Н), 7,91-7,77 (m, 2Н), 7,64 (d, J=7,7 Гц, 2Н), 7,38 (t, J=7,9 Гц, 2Н), 7,15 (t, J=7,4 Гц, 1Н); ESI-MS (тандемная масс-спектрометрия с ионизацией электрораспылением) m/z 261 (М+Н)+.
2. Получение трет-бутилового эфира (S)-[1-(2-фтор-6-нитро-бензилформил)-фенил-аминоформил]-(1-d-пропил)аминокислоты (соединение 3)
2-фтор-6-нитро-N-фенилбензамид (7,8 г, 0,03 моль), N,N-диметилформамид (0,5 мл) и тионилхлорид (17,8 г, 0,15 моль, 5 экв.) последовательно добавляли в колбу, нагревали до 40°C, и затем реакционную смесь перемешивали в течение 5 часов. Реакционную жидкость концентрировали с получением вязкой коричневой субстанции. Вязкую коричневую субстанцию растворяли в 20 мл дихлорметана и по каплям добавляли к 50 мл раствора дихлорметана, содержащего (S)-2-t-бутилоксикарбониламид-2-d-бутират (6,7 г, 0,033 моль, 1,1 экв.) и триэтиламин (3,4 г, 0,033 моль, 1,1 экв.). Реакционную смесь перемешивали в течение 3 часов при комнатной температуре и затем фильтровали для удаления твердой фазы. Водный слой промывали чистой водой, насыщенным раствором бикарбоната натрия, чистой водой, 5% лимонной кислотой и насыщенным соленым раствором в указанном порядке. Органический слой сушили над безводным сульфатом магния и концентрировали с получением красной вязкой субстанции. Сырой продукт очищали колоночной хроматографией на силикагеле (10%-25% N-гексан/этилацетат) с получением целевого белого твердого конечного соединения 3 (8,0 г, 60%). ESI-MS m/z 447 (М+Н)+.
3. Получение трет-бутилового эфира (S)-[1-(5-фтор-4-оксо-3-фенил-3,4-дигидрокси-хиназолин-2-ил)-(1-d-пропил)]-карбаминовой кислоты (соединение 4)
Соединение трет-бутиловый эфир (S)-[1-(2-фтор-6-нитро-бензолформил)-фенил-аммоний формил]-(1-d-пропил)-карбаминовой кислоты (4,5 г, 0,01 моль, 1 экв.) и уксусную кислоту (50 мл) последовательно добавляли в колбу. Температуру поддерживали ниже 20°C и тремя частями добавляли цинковый порошок (48,4 г, 740 ммоль, 6 экв.). Реакционную смесь перемешивали в течение 2 часов при комнатной температуре и затем фильтровали с отсасыванием. Осадок промывали уксусной кислотой, фильтрат концентрировали и растворяли в этилацетате, промывали чистой водой, насыщенным раствором бикарбоната натрия и соленым раствором в указанном порядке, сушили над безводным сульфатом магния, концентрировали с получением остатка. Остаток очищали колоночной хроматографией на силикагеле (10%-25% N-гексан/этилацетат) с получением целевого беловатого пористого твердого продукта. 1H ЯМР (400 МГц, DMSO-d6) δ 7,80 (s, 1Н), 7,62-7,44 (m, 5Н), 7,38 (d, J=7,6 Гц, 1Н), 7,30 (m, 1Н), 7,23 (d, J=7,6 Гц, 1Н), 1,76-1,68 (m, 1Н), 1,60-1,46 (m, 1Н), 1,31 (s, 9Н), 0,62 (t, J=7,2 Гц, 3Н). ESI-MS m/z 399 (М+Н)+.
4. Получение (S)-2-(1-амино-1-d-пропил)-5-фтор-3-фенилхиназолин-4(3Н)-кетона (соединение 5)
Трет-бутиловый эфир
(S)-[1-(5-фтор-4-оксо-3-фенил-3,4-дигидрокси-хиназолин-2-ил)-(1-d-пропил)]-карбаминовой кислоты (1,99 г, 5 ммоль) и дихлорметан (6 мл) последовательно добавляли в колбу. Добавляли трифторуксусную кислоту (6 мл) при перемешивании. Перемешивали смесь при комнатной температуре в течение 1 часа и затем концентрировали посредством глубокого вакуумного концентрирования. Остаток растворяли в метиленхлориде и затем добавляли 10% раствор карбоната калия до достижения рН, равного 9, и проводили разделение на слои. Водный слой экстрагировали дихлорметаном. Органические слои объединяли, последовательно промывали водой и соленым раствором, и сушили над безводным сульфатом магния, концентрировали с получением беловатого твердого целевого продукта (1,4 г, 93%). 1H ЯМР (400 МГц, CDCl3) δ 7,74-7,66 (m, 1Н), 7,62-7,50 (m, 4Н), 7,30-7,20 (m, 2Н), 7,12-7,06 (m, 1Н), 1,88-1,72 (m, 1Н), 1,58-1,41 (m, 1Н), 0,78 (t, J=7,2 Гц, 3Н). ESI-MS m/z 299,1 (М+Н)+.
5. Получение (S)-2-(1-(9Н-пурин-6-ил-амино)-1-d-пропил)-5-фтор-3-фенилхиназолин-4(3Н)-кетона (Соединение 6)
(S)-2-(1-амино-1-d-пропил)-5-фтор-3-фенилхиназолин-4(3Н)-кетон (1,2 г, 4 ммоль, 1 экв.), 6-бромпурин (0,88 г, 4,4 ммоль, 1,1 экв), диизопропилэтиламин (1,04 г, 8 ммоль, 2 экв.) и третичный бутиловый спирт последовательно добавляли в колбу. Реакционную смесь перемешивали в течение 30 часов при 80°C. Образец концентрировали с получением твердого сырого продукта. Сырой продукт разделяли и очищали колоночной хроматографией на силикагеле (4% метанол/дихлорметан) с получением продукта в виде желтоватого твердого вещества (1,0 г, 60%). 1H ЯМР (400 МГц, DMSO-d6) δ 12,70 (s, 1Н), 8,12 (s, 1Н), 8,02 (s, 1Н), 7,82-7,74 (m, 1Н), 7,62-7,40 (m, 6Н), 7,26-7,15 (m, 2Н), 2,03-1,75 (m, 2Н), 0,78 (t, J=7,2 Гц, 3Н). ESI-MS m/z 417 (M+H)+.
Пример 2: Получение (S)-2-(1-(9Н-пурин-6-ил-амино)-1,2,2-d3-пропил)-5-фтор-3-фенилхиназолин-4(3Н)-кетона (Соединение 7)
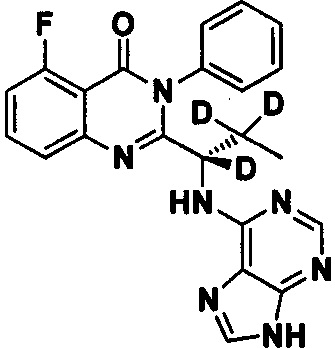
Синтез проводили в соответствии со способом, приведенным в Примере 1. Различие заключалось в следующем: целевой продукт (соединение 7) получали путем замены (S)-2-(t-бутилоксикарбониламид)-2-d-масляной кислоты на (S)-2-(t-бутилоксикарбониламид)-2,3,3-d3-масляную кислоту. ESI-MS m/z 419 (М+Н)+.
Пример 3: Получение (S)-2-(1-(9Н-пурин-6-ил-амино)-1,2,2,3,3,3-d6-пропил)-5-фтор-3-фенилхиназолин-4(3Н)-кетона (Соединение 8)
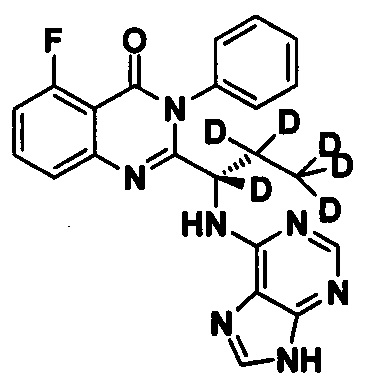
Синтез проводили в соответствии со способом, приведенным в Примере 1. Единственное различие заключалось в следующем: целевой продукт (соединение 8) получали путем замены (S)-2-(t-бутилоксикарбониламид)-2-d-масляной кислоты на (S)-2-(t-бутилоксикарбониламид)-2,3,3,4,4,4-d6-масляную кислоту. ESI-MS m/z 422 (М+Н)+.
Пример 4: Получение (S)-2-(1-(9Н-пурин-6-ил-амино)-1,3,3,3-d4-пропил)-5-фтор-3-фенилхиназолин-4(3Н)-кетона (Соединение 9)
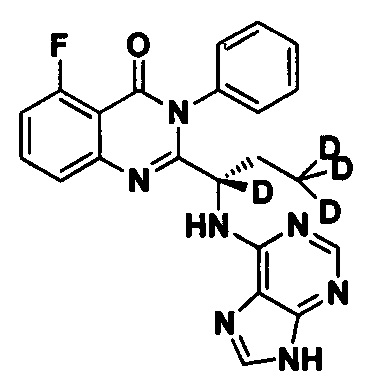
Синтез проводили в соответствии со способом, приведенным в Примере 1. Единственное различие заключалось в следующем: целевой продукт (соединение 9) получали путем замены (S)-2-(t-бутилоксикарбониламид)-2-d-масляной кислоты (S)-2-(t-бутоксикарбониламид)-2,4,4,4-d4-масляной кислотой. ESI-MS m/z 422 (М+Н)+.
Пример 5: Получение (S)-2-(1-(9Н-пурин-6-ил-амино)-2,2,3,3,3-d5-пропил)-5-тор-3-фенилхиназолин-4(3Н)-кетона (Соединение 10)
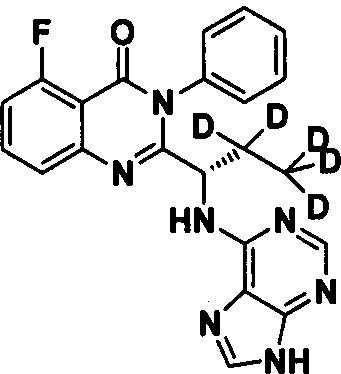
Синтез проводили в соответствии со способом, приведенным в Примере 1. Единственное различие заключалось в следующем: целевой продукт (соединение 10) получали путем замены (S)-2-(t-бутилоксикарбониламид)-2-d-масляной кислоты на (S)-2-(t-бутилоксикарбониламид)-3,3,4,4,4-d5-масляную кислоту. ESI-MS m/z 421 (М+Н)+.
Пример 6: Получение (S)-2-(1-(9Н-пурин-6-ил-амино)-3,3,3-d3-пропил)-5-фтор-3-фенилхиназолин-4(3Н)-кетона (Соединение 11)
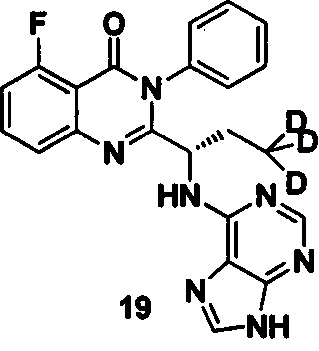
Синтез проводили в соответствии со способом, приведенным в Примере 1. Единственное различие заключалось в следующем: целевой продукт (соединение11) получали путем замены (S)-2-(t-бутилоксикарбониламид)-2-d-масляной кислоты на (S)-2-(t-бутилоксикарбониламид)-4,4,4-d3-масляную кислоту. ESI-MS m/z 419 (М+Н)+.
Пример 7: Получение (S)-2-(1-(9Н-пурин-6-ил-амино)-2,2-d2-пропил)-5-фтор-3-фенилхиназолин-4(3Н)-кетона (Соединение 12)
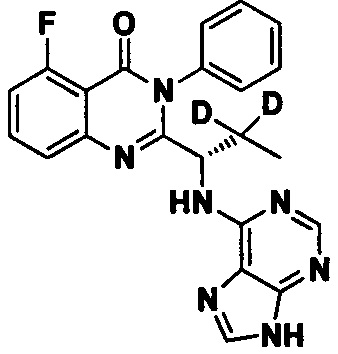
Синтез проводили в соответствии со способом, приведенным в Примере 1. Единственное различие заключалось в следующем: целевой продукт (соединение 12) получали путем замены (S)-2-(t-бутилоксикарбониламид)-2-d-масляной кислоты на (S)-2-(t-бутилоксикарбониламид)-3,3-d2-масляную кислоту. ESI-MS m/z 418 (М+Н)+.
Пример 8: Получение (S)-2-(1-(9Н-пурин-6-ил-амино)-1-d-этил)-6-фтор-3-фенилхиназолин-4(3H)-кетона (Соединение 13)
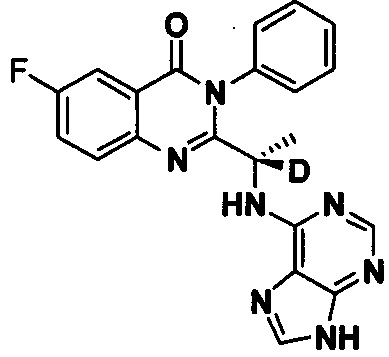
Синтез проводили в соответствии со способом, приведенным в Примере 1. Единственное различие заключалось в следующем: целевой продукт (соединение 13) получали путем замены 2-фтор-6-нитробензойной кислоты на 3-фтор-6-нитробензойную кислоту, и замены (S)-2-(t-бутилоксикарбониламид)-2-d-масляной кислоты на (S)-2-(t-бутилоксикарбониламид)-2-d-пропионовую кислоту. ESI-MS m/z 403 (М+Н)+.
Пример 9: Получение (S)-2-(1-(9Н-пурин-6-ил-амино)-1,2,2,2-d4-этил)-6-фтор-3-фенилхиназолин-4(3Н)-кетона (Соединение 14)
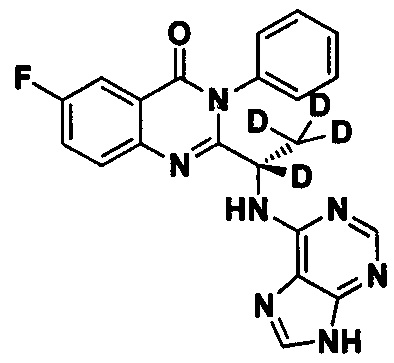
Синтез проводили в соответствии со способом, приведенным в Примере 1. Единственное различие заключалось в следующем: целевой продукт (соединение 14) получали путем замены 2-фтор-6-нитробензойной кислоты на 3-фтор-6-нитробензойную кислоту, и замены (S)-2-(t-бутилоксикарбониламид)-2-d-масляной кислоты на (S)-2-(t-бутилоксикарбониламид)-2,3,3,3-d4-пропионовую кислоту. ESI-MS m/z 406 (М+Н)+.
Пример 10: Получение (S)-2-(1-(9Н-пурин-6-ил-амино)-2,2,2-d3-этил)-6-фтор-3-фенилхиназолин-4(3Н)-кетона (Соединение 15)
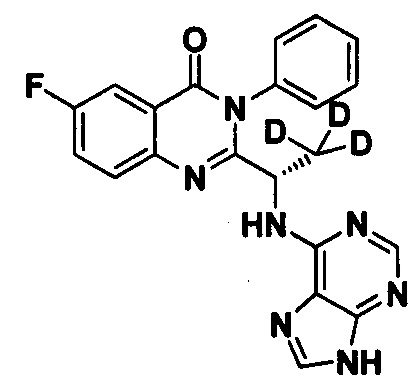
Синтез проводили в соответствии со способом, приведенным в Примере 1. Единственное различие заключалось в следующем: целевой продукт (соединение 15) получали путем замены 2-фтор-6-нитробензойной кислоты на 3-фтор-6-нитробензойную кислоту, и замены (S)-2-(t-бутилоксикарбониламид)-2-d-масляной кислоты на (S)-2-(t-бутилоксикарбониламид)-3,3,3-d3-пропионовую кислоту. ESI-MS m/z 405 (М+Н)+.
Пример 11: Получение (S)-2-(1-(9Н-пурин-8-d-6-ил-амино)-(1-d-пропил)-5-фтор-3-фенилхиназолин-4(3Н)-кетона (Соединение 16)
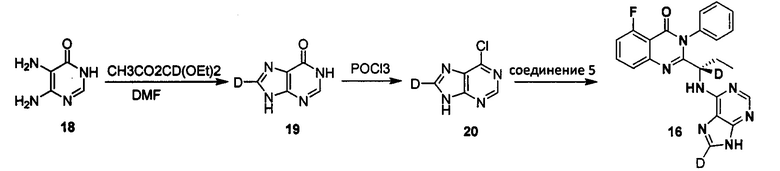
Получение 1,9-дигидро-6Н-пурин-6-кетона-8-d (Соединение 19)
5,6-диаминопириидин-4(3Н)-кетон (3,52 г, 0,028 моль) суспендировали в муравьиной кислоте (25 мл). Смесь перемешивали при нагревании с обратным холодильником в течение 2 часов и затем концентрировали с получением желтого твердого вещества. К смеси добавляли Диэтоксиацетат (метил-d) эфир (9,13 г, 0,056 моль), муравьиную кислоту (2 мл) и N.N-диметилформамид (50 мл). Реакционную смесь нагревали с обратным холодильником в течение 4 часов, и затем концентрировали. Концентраты растворяли в ацетонитриле и нагревали с обратным холодильником в течение 30 минут, затем охлаждали до 0°C, фильтровали и сушили с получением беловатого твердого продукта (35 г, выход: 78.3%). 1Н ЯМР (D2O/NaOD) 8,10 (s, 1Н).
Получение 6-хлор-9Н-пурина-8-d (Соединение 20)
Соединение 1,9-дигидро-6Н-пурин-6-кетон-8-d (0,26 г, 1,9 ммоль), оксихлорид фосфора (7 мл) и N,N-диметиланилин (0,7 мл) последовательно добавляли в колбу. Смесь нагревали с обратным холодильником в течение 25 минут. Летучие органические соединения удаляли посредством глубокого вакуумного концентрирования. Концентраты охлаждали до -15°C и растворяли в аммиаке. После диатомитовой фильтрации полученную смесь дважды экстрагировали этилацетатом и диэтиловым эфиром. Раствор охлаждали до 0°C и разбавляли чистой водой. Для доведения значения рН приблизительно до 2 использовали концентрированную соляную кислоту. Раствор экстрагировали диэтиловым эфиром. Органический слой нейтрализовали аммиаком и затем концентрировали. Концентраты обрабатывали при помощи препаративной хроматографии с получением целевого продукта в виде беловатого твердого вещества (0,22 г). 1H ЯМР (DMSO-d6) 8,75 (s, 1Н).
Получение (S)-2-(1-(9Н-пурин-8-d-6-ил-амино)-(1-d-пропил))-5-фтор-3-фенилхиназолин-4(3Н)-кетона (Соединение 16)
Соединение (S)-2-(1-амино-1-d-пропил)-5-фтор-3-фенилхиназолин-4(3Н)-кетон (0,24 г, 0,8 ммоль), 6-хлор-9Н-пурин-8-d (0,18 г, 0,88 ммоль), диизопропилэтиламин (0,21 г, 1,6 ммоль) и третичный бутанол (2 мл) добавляли в колбу, нагревали до 80°C и перемешивали в течение 30 часов. Реакционный раствор концентрировали с получением сырого продукта. Концентраты очищали колоночной хроматографией на силикагеле (4% метанол/дихлорметан) с получением целевого желтоватого твердого продукта (0,25 г). ESI-MS m/z 418 (М+Н)+, 440 (M+Na)+.
Пример 12: Получение (S)-2-(1-(9Н-пурин-2,8-d2-6-ил-амино)-(1-d-пропил))-5-фтор-3-фенилхиназолин-4(3Н)-кетона (Соединение 17)
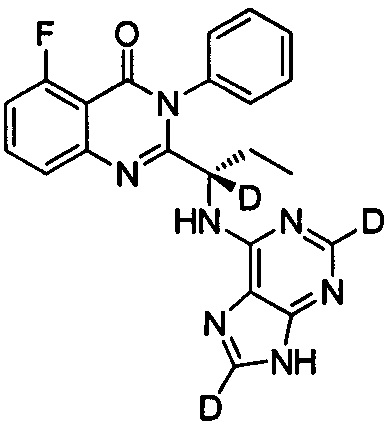
Синтез проводили в соответствии со способом, приведенным в Примере 11. Единственное различие заключалось в следующем: целевой продукт (соединение 17) получали путем замены 6-хлор-9Н-пурина-8-d на 6-хлор-2,8-d2-пурин. ESI-MS m/z 419(M+H)+.
Пример 13: Получение (S)-2-(1-(9Н-пурин-2,8-d2-6-ил-амино)-пропил)-5-фтор-3-фенилхиназолин-4(3Н)-кетона (Соединение 21)
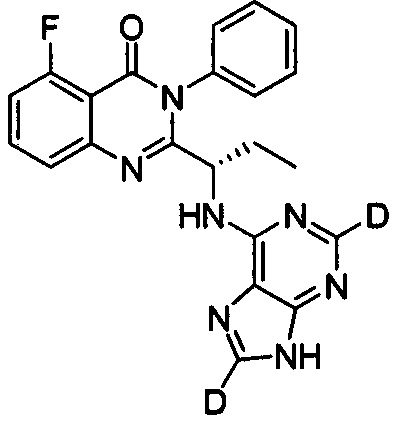
Синтез проводили в соответствии со способом, приведенным в Примере 11. Единственное различие заключалось в следующем: целевой продукт (соединение 21) получали путем замены (S)-2-(1-амино-1-d-пропил)-5-фтор-3-фенилхиназолин-4(3Н)-кетона на (S)-2-(1-аминопропил)-5-фтор-3-фенилхиназолин-4(3Н)-кетон, и замены 6-хлор-9Н-пурина-8-d на 6-хлор-2,8-d2-пурин. ESI-MS m/z 418 (М+Н)+.
Пример 14: Получение (S)-2-(1-(9Н-пурин-8-d-6-ил-амино)-пропил)-5-фтор-3-фенилхиназолин-4(3Н)-кетона (Соединение 22)
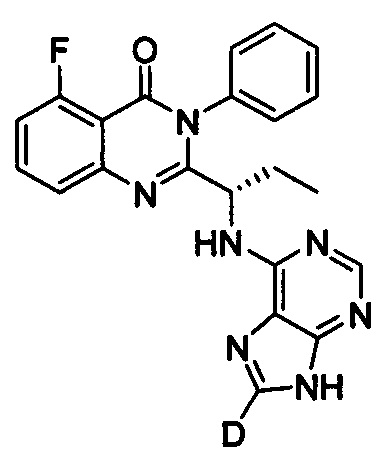
Синтез проводили в соответствии со способом, приведенным в Примере 11. Единственное различие заключалось в следующем: целевой продукт (соединение 22) получали путем замены (S)-2-(1-амино-1-d-пропил)-5-фтор-3-фенилхиназолин-4(3Н)-кетона на (S)-2-(1-аминопропил)-5-фтор-3-фенилхиназолин-4(3Н)-кетон, и замены 6-бромпурина на 6-хлор-9Н-пурин-8-d. ESI-MS m/z 417 (М+Н)+.
Пример 15: Оценка фармакокинетики у крыс
4 самца крыс линии Спрег-Доули (возрастом 7-8 недель, с массой тела приблизительно 210 г) разделяли на две группы, по четыре крысы в каждой. Каждый раз перорально вводили дозу 3 мг/кг (а) контрольного соединения (S)-2-(1-(9Н-пурин-6-ил-амино)пропил)-5-фтор-3-фенилхиназолин-4(3Н)-кетона и (б) исследуемого соединения - соединений, полученных в Примерах 1-14, и сравнивали разницу в фармакокинетике между двумя группами.
Крыс кормили стандартной пищей, по желанию давали воду и прекращали кормление за 16 часов до проведения теста. Лекарственный препарат растворяли в ПЭГ-400 (полиэтиленгликоле 400) и диметилсульфоксиде. Сбор крови из глазничной вены производили через 0,25 часа, 0,5 часа, 1 час, 2 часа, 4 часа, 6 часов, 8 часов, 12 часов, 24 часов и 36 часов после введения.
Крыс быстро анестезировали ингаляцией эфира; в пробирку собирали 300 мл крови из глазничной вены. В пробирке находилось 30 мкл 1% раствора гепарина в физиологическом растворе. Перед использованием пробирки сушили в течение ночи при 60°C. После сбора образца крови в следующий контрольный момент времени крыс анестезировали эфиром и усыпляли.
Немедленно после взятия образца крови пробирки осторожно переворачивали по меньшей мере 5 раз для обеспечения равномерного перемешивания и помещали в емкость со льдом. При 4°C образцы крови центрифугировали при 5000 об./мин в течение 5 минут для разделения плазмы и красных кровяных телец. 100 мкл плазмы при помощи пипетки переносили в чистую пластиковую центрифужную пробирку, на которой указывали название соединения и контрольный момент времени. Плазму перед проведением анализа хранили при -80°C. Концентрацию соединения по изобретению в плазме определяли при помощи ЖХ-МС/МС (жидкостной хроматографии с тандемной масс-спектрометрией). Фармакокинетические параметры оценивали на основании концентрации соединения в плазме каждого животного в различные контрольные моменты времени.
Из результатов анализа можно видеть, что в сравнении с контрольным соединением соединения по настоящему изобретению обладают более длительным периодом полувыведения и более высоким уровнем экспозиции в плазме крови животных, лучшей фармакодинамикой и лучшими терапевтическими эффектами.
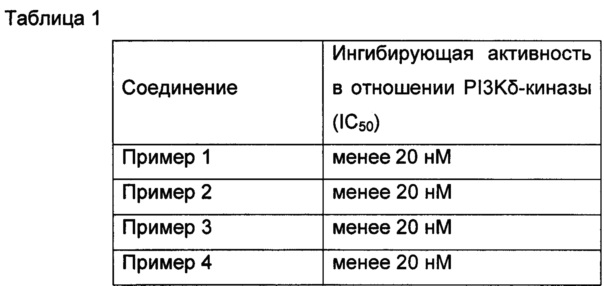
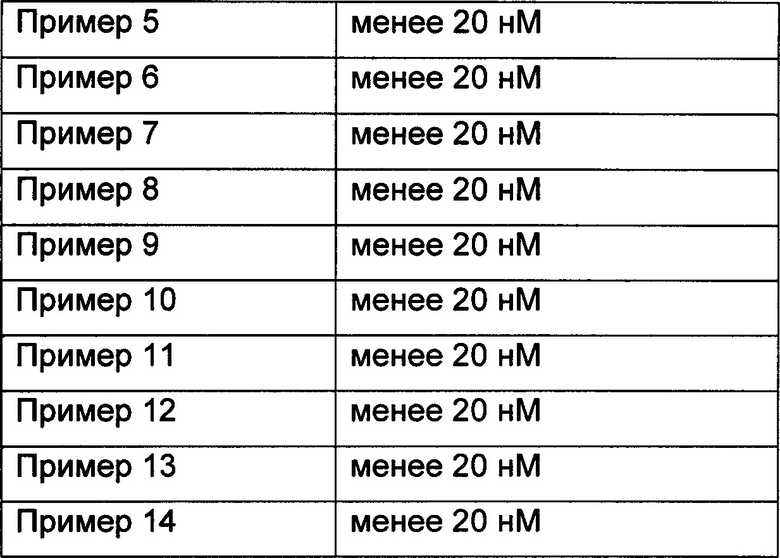
Пример 16: In vitro оценка фармакодинамики соединений по изобретению в отношении PI3K-киназ
Исследование in vitro оценки фармакодинамики было специально разработано в соответствии со ссылкой J. Med. Chem. 2013, 56, 1922-1939.
Результаты приведены в Таблице 1. Как можно видеть, соединения по настоящему изобретению обладают превосходной ингибирующей активностью в отношении PI3K-киназы.
Пример 17 Фармацевтическая композиция
Вышеприведенные вещества смешивали общепринятыми способами, и затем заполняли ими обычные желатиновые капсулы с получением 1000 капсул.
Все литературные источники, упомянутые в настоящей заявке, включены в нее посредством ссылки, как если бы каждый из них был отдельно включен посредством ссылки. Кроме того, следует иметь в виду, что после прочтения вышеуказанных источников специалисты могут привносить в настоящее изобретение различные изменения и модификации. Эти эквиваленты также находятся в объеме, определяемом прилагаемой формулой изобретения.
Настоящее изобретение относится к новому дейтерированному соединению хиназолинона формулы (I) или его фармацевтически приемлемой соли, обладающим свойствами ингибитора PI3К. Соединения могут найти применение для лечения или предотвращения таких заболеваний, как раковые заболевания, нарушения клеточной пролиферации, воспаления, инфекции, аутоиммунные заболевания. В формуле (I)
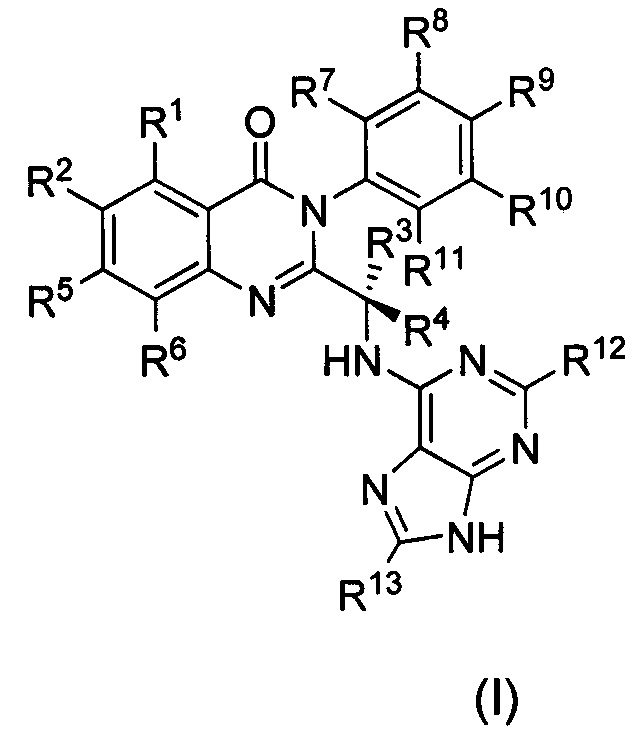
R1 и R2 независимо представляют собой водород или галоген; R3 выбран из водорода, дейтерия, СН3, CH2D, CHD2, CD3, СН2СН3, CD2CH3, CH2CD3 и CD2CD3; каждый из R4, R11, R12 и R13 независимо представляет собой водород или дейтерий; каждый из R5, R6, R7, R8, R9 и R10 независимо представляет собой водород; при условии, что по меньшей мере один из R3, R4, R11, R12 или R13 является дейтерированным или представляет собой дейтерий. Дейтерированные соединения настоящего изобретения обладают повышенной активностью по сравнению с недейтерированными или соединениями дейтерированными в другие положения. 3 н. и 7 з.п. ф-лы, 1 табл., 17 пр.
1. Дейтерированное соединение хиназолинона формулы (I) или его фармацевтически приемлемая соль
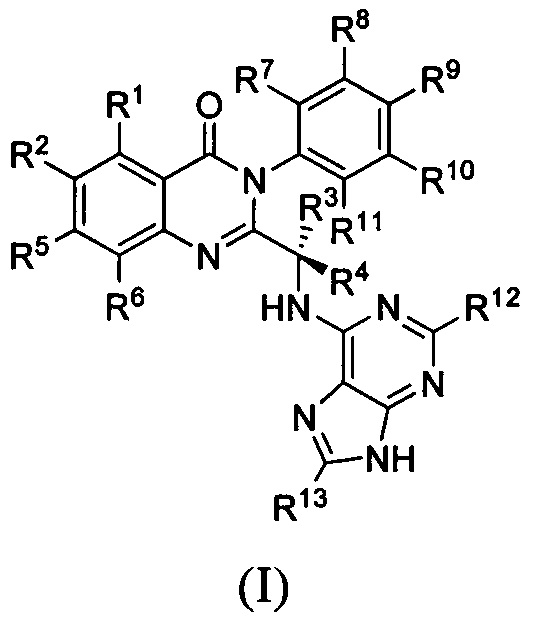
где R1 и R2 независимо представляют собой водород или галоген;
R3 выбран из водорода, дейтерия, СН3, CH2D, CHD2, CD3, СН2СН3, CD2CH3, CH2CD3 и CD2CD3;
каждый из R4, R11, R12 и R13 независимо представляет собой водород или дейтерий;
каждый из R5, R6, R7, R8, R9 и R10 независимо представляет собой водород;
при условии, что по меньшей мере один из R3, R4, R11, R12 или R13 является дейтерированным или представляет собой дейтерий.
2. Соединение по п. 1, где R1 представляет собой фтор и/или R2 представляет собой водород.
3. Соединение по п. 1, где R3 представляет собой СН3, CH2D, CHD2, CD3, СН2СН3, CD2CH3, CH2CD3 или CD2CD3.
4. Соединение по п. 1, где R4 представляет собой дейтерий или водород.
5. Соединение по любому из пп. 1-4, где R12 представляет собой дейтерий и/или R13 представляет собой дейтерий.
6. Соединение по п. 1, где соединение представляет собой одно из следующих соединений или его фармацевтически приемлемую соль
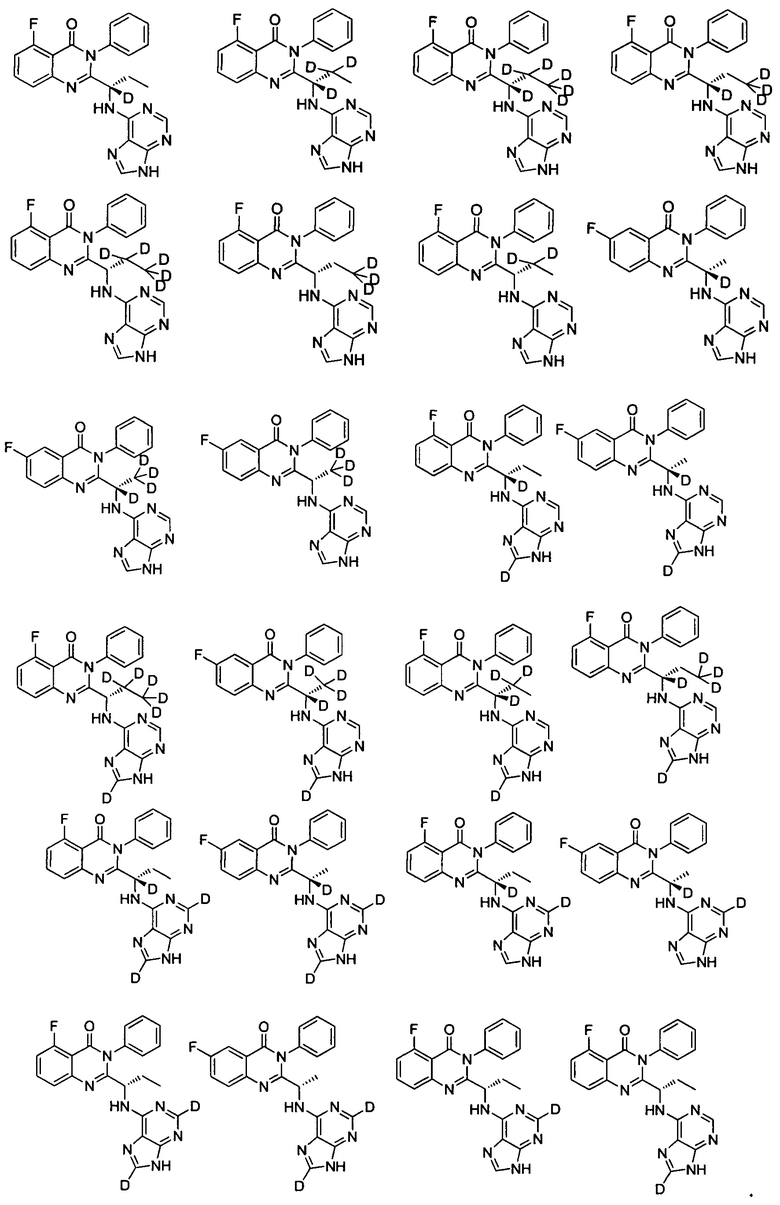
7. Соединение по п. 1, где недейтерированные соединения не включены в соединение.
8. Фармацевтическая композиция, обладающая свойствами ингибитора PI3К, содержащая фармацевтически приемлемый носитель и в эффективном количестве соединение по п. 1 или его фармацевтически приемлемую соль.
9. Применение соединения по п. 1 или его фармацевтически приемлемой соли для получения фармацевтической композиции для ингибирования PI3К.
10. Применение по п. 9, где фармацевтические композиции применяются для лечения или предотвращения любого из следующих заболеваний: раковые заболевания, нарушения клеточной пролиферации, воспаления, инфекции, аутоиммунные заболевания.
| EA 201101507 A, 30.05.2012 & CN 102458410 A, 16.05.2012 | |||
| Подъемник | 1988 |
|
SU1606444A1 |
| CN 101031569 A, 05.09.2007 & WO 2005113554 A2, 01.12.2005 | |||
| ЕA 201270184 A1, 30.08.2012 & CN 102647987 A, 22.08.2012 | |||
| Clement Chung et al., Ibrutinib, Obinutuzumab, Idelalisib, and Beyond: Review of Novel and Evolving Therapies for Chronic Lymphocytic Leukemia, PHARMACOTHERAPY, 2014, страницы 1298-1316 | |||
| Изложница с суживающимся книзу сечением и с вертикально перемещающимся днищем | 1924 |
|
SU2012A1 |
| Способ приготовления лака | 1924 |
|
SU2011A1 |

