Изобретение относится к генной инженерии, в частности к способам получения пептидов, которые способны специфично распознавать определенные типы клеток и могут быть использованы в диагностике, терапии и фармацевтических приложениях.
В настоящее время одной из главных проблем является повышение уровня сердечно-сосудистых, раковых и иммунных заболеваний. В большинстве случаев первоисточниками таких нарушений является быстротечная неконтролируемая пролиферация тканево-клеточных структур, что затрудняет и осложняет их диагностику и терапию. Поэтому, первоочередным условием является своевременное выявление патологического состояния, разработка и доставка лекарственных препаратов, блокирующих клеточную активность в очагах заболеваний.
Известны терапевтические ингибиторы для сосудистых гладкомышечных клеток, которые используют для подавления клеточной активности и/или уничтожения клетки путем целевой доставки терапевтических агентов в очаги активной пролиферации (см. пат .США №6515009 от 04.02.2003 г., МКП А61К 31/40). Для ингибирования роста сосудистых гладкомышечных клеток, согласно данному патенту, используются терапевтические конъюгаты, которые содержат лекарственный препарат, ассоциированный с пептидом или белком, специфическим образом связывающийся с клеточной мембраной сосудистых гладкомышечных клеток (например, моноклональные и поликлональные антитела, факторы роста, полипептидные гормоны, цитокины и им подобные) или с элементом внутри- или внеклеточного матрикса (например, макромолекулы, распознающие рецепторы интегринов и фибронектинов, а также эпитопы коллагена и внеклеточных гликопротеинов) этих же клеток. Один из таких пептидов, например, содержит последовательность аминокислот, специфически узнающую хондроитинсульфат протеогликан, с предположительным молекулярным весом около 250 килодальтон, экспрессируемым на мембранах сосудистых гладкомышечных клеток. Такая же последовательность аминокислот находится в регионах, обуславливающих комплементарность моноклонального антитела NR-AN-01 и/или его функциональных эквивалентов.
Недостатками данного изобретения является использование пептидов, которые распознают белки с выраженной экспрессией в ряде других функционально важных клеток и тканей, это ведет к тому, что данный терапевтический конъюгат может связываться и с другими типами клеток не гладкомышечного происхождения, как, например, клетки нейроглии, костной ткани и другие, что приводит к нежелательной аккумуляции лекарственного препарата в данных клетках и побочному цитотоксическому эффекту (нежелательной клеточной гибели). Кроме того, при использовании антител в качестве ведущих составляющих конъюгатов, их размер, ввиду плотного внеклеточного матрикса, может стать препятствием при проникновении конъюгата в очаг пролиферации, причем кросс-реактивность антител к другим молекулярным структурам снижает их специфичность.
Известен также способ целенаправленной доставки пептидов к сосудистым гладкомышечным клеткам in vitro и in vivo, с целью доставки лекарственных препаратов к рестенозной бляшке посредством пептидов, встроенных в фаговые частицы, которые связываются с пролиферирующими сосудистыми гладкомышечными клетками из библиотеки синтетических 15-ти членных пептидов, сконструированной случайным образом (см. статью Ingrid N. Michon et al. "Targeting of peptides to restenotic vascular smooth muscle cells using phage display in vitro and in vivo" в журнале "Biochimica et Biophysica Acta", вып. №1591, 2002 г., стр.87-97). Отобранные пептиды посредством многократного повторения одноэтапного скрининга, в дальнейшем проходят подтверждение специфичности, исключительно, методом иммуноферментного анализа и визуализируются с помощью иммунофлюоресценции, при этом обнаруживают два пептида с консервативной аминокислотной последовательностью, такие как 5G6 (N'-CNIWGVVLSWIGVFPEC-C') и 5Е5 (N'-CESLWGGLMWTIGLSDC-C').
Недостатками данного способа являются использование одной исходной библиотеки на основе синтетических случайно ориентированных пептидов, не имеющих аналогов в природе, что определяет их первично низкую специфичность и селективность, а также высокую иммуногенность. Кроме того, многократное повторение одноэтапного скрининга приводит к ослаблению специфических взаимодействий и наращиванию неспецифических.
В основу заявленного изобретения поставлена задача создать способ получения пептидов, специфично распознающих определенные типы клеток с высокой степенью селективности, которые предназначены для терапевтических целей.
Поставленная задача достигается путем осуществления нового способа получения пептидов, специфично распознающих определенные типы клеток, при котором предусматривают конструирование фаговой библиотеки случайных пептидов на основе кодирующих их олигонуклеотидных фрагментов, ее скрининг для получения пептидов, связывающихся с определенным типом клеток, и подтверждают специфичность отобранных пептидов, при этом, согласно изобретению,
- олигонуклеотидные фрагменты, кодирующие случайные пептиды, получают реакцией обратной транскрипции с использованием случайных праймеров и суммарных РНК клеток определенного типа и клеток других типов, которые могут быть вовлечены в патологический процесс или способны оказывать влияние на его диагностику и терапию,
- указанные олигонуклеотидные фрагменты встраивают в бактериофаговые векторы в правильной ориентации и используют для создания фаговых библиотек случайных пептидов для клеток всех типов, использованных для выделения суммарных РНК,
- скрининг полученных фаговых библиотек случайных пептидов проводят в два этапа, на первом из которых отбирают пептиды, способные связываться с клетками определенного типа, а на втором - из отобранных на первом этапе пептидов отбирают пептиды, не связывающиеся с клетками других использованных типов,
- подтверждают специфичность пептидов в составе бактериофаговых частиц к клеткам определенного типа, используя комбинации различных тестов,
- определяют первичные последовательности олигонуклеотидных фрагментов, кодирующих пептиды в составе отобранных фаговых частиц, и транслируют их в аминокислотные последовательности с дальнейшим проведением анализа полученных последовательностей,
- конструируют матрицы из аминокислотных последовательностей пептидов, на основе их физико-химических характеристик, включающие линкер и сигнал обнаружения,
- считывают последовательности новых пептидов с матриц и синтезируют такие пептиды химическим путем, с последующим подтверждением специфичности полученных пептидов в отношении клеток определенного типа.
При осуществлении данного способа получают пептиды, специфично распознающие клетки определенных типов, которые могут использоваться для доставки лекарственных препаратов в места непосредственного их действия, с целью профилактики и/или терапии нарушений, связанных с функционированием клеток определенных типов, или для непосредственного использования полученных пептидов как лекарственной формы в терапевтических приложениях. Кроме того, использование таких пептидов, или их фрагментов, позволит диагностировать нарушения функционирования клеток определенных типов и прогнозировать их лечение.
Заявленный способ позволяет получать высокоспецифичные и высокоселективные пептиды к клеткам определенного типа.
Способ получения пептидов поясняется чертежами, где:
на фиг.1 показано синтез кДНК и конструирование библиотек;
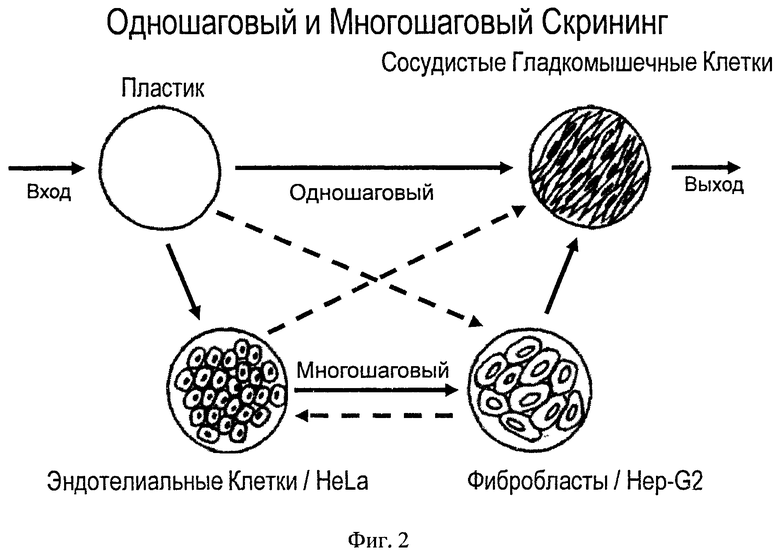
на фиг.2 изображена схема одно- и многошагового скрининга;
на фиг.3 изображена схема пептидного конструкта (матрицы);
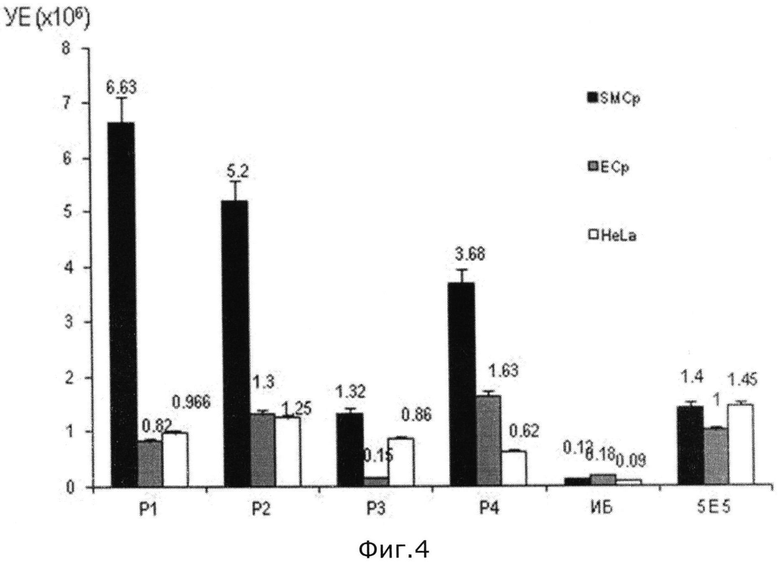
на фиг.4 - график зависимости специфичности отобранных в скрининге пептидов (Р1-Р4), в отношении сосудистых гладкомышечных клеток человека (обозначены как SMCp);
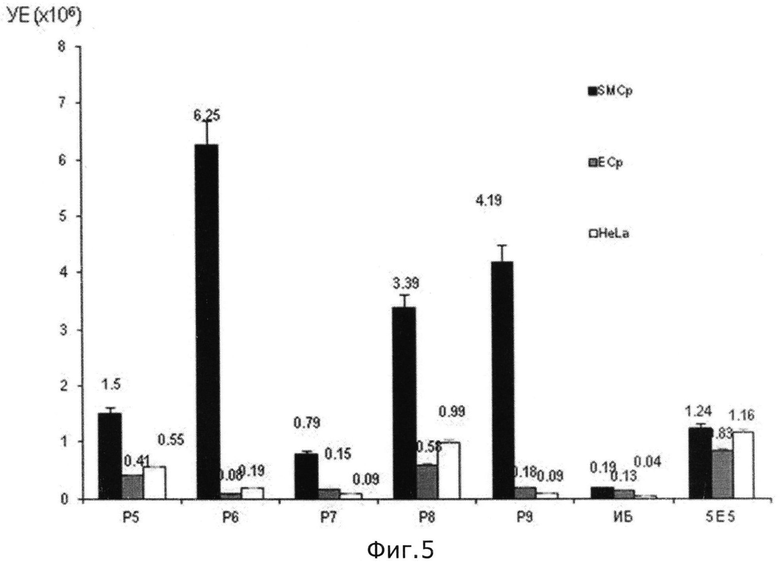
на фиг.5 - график зависимости специфичности новых пептидов (Р5-Р9), в отношении сосудистых гладкомышечных клеток человека (обозначены как SMCp).
Способ получения пептидов осуществляют таким образом.
Вначале проводят отбор и разделение клеточных популяций на типы: клетки, которые должны быть специфично распознаны пептидами, и клетки, которые могут быть вовлечены в патологический процесс или способны оказывать влияние на его течение, диагностику и терапию, распознавание которых при этом является нежелательным. Затем, конструируют серии библиотек случайных пептидов с использованием суммарной клеточной РНК в векторе вирусного типа на основе ДНК бактериофагов.
Сначала изолируют суммарную РНК общеизвестными методами с адаптационными модификациями, с последующей, в случае необходимости, очисткой ее и выделением фракции матричной РНК. Качество такой РНК анализируют и в дальнейшем, используют РНК в реакциях обратной транскрипции, направляемых праймерами, как случайного происхождения, так и комплементарными к поли А участку матричных РНК.
На этом этапе осуществляют первичное разделение суммарной клеточной РНК посредством контролируемого синтеза фрагментов кДНК ограниченных в размере и гетерогенности, что достигается использованием различных концентраций ионов в реакционной смеси, комбинацией праймеров, количеством исходного материала и режимов температур.
Вторичное разделение клеточной РНК осуществляют на этапе фракционирования и очистки полученных молекул кДНК, для чего используют гель-фильтрацию таким образом, что после элюции пул кДНК молекул состоит из 2-х частей: фракции с количеством нуклеотидных звеньев менее 500 и фракции, состоящей из молекул размером более 500 пар оснований.
Такое разделение позволяет, в первом случае, создание библиотек, направленных на определение точных мест взаимодействия молекулы пептида с молекулами на поверхности клеток распознаваемого типа, а во втором случае, идентифицировать фрагмент нуклеиновой кислоты, ответственный за синтез данного пептида или белкового фрагмента.
Далее на стадии встраивания фрагментов кДНК в бактериофаговый вектор в правильной ориентации, полученные в результате деления и синтеза молекулы кДНК легируют с олигонуклеотидными линкерами, синтетически синтезированными и содержащими места узнавания для эндонуклеаз рестрикции, используемых для обработки и подготовки вектора для клонирования. Для этого концы кДНК вначале обрабатывают с помощью ДНК полимеразы и только потом такие фрагменты легируют с линкерами и инкубируют с эндонуклеазами рестрикции. Используя данные линкеры, становится возможным получить фрагменты кДНК, которые содержат на разных концах места узнавания для используемых эндонуклеаз рестрикции, в таком расположении, что могут быть беспрепятственно клонированы в вектор в определенной ориентации (смысловое клонирование).
Одновременно с этим, бактериофаговый вектор обрабатывают эндонуклеазами рестрикции, с целью получения фрагментов, пригодных для легирования с синтезированными фрагментами кДНК. Полученные, в результате легирования фрагментов кДНК и молекул вектора, рекомбинантные молекулы упаковывают in vitro и инициируют экспрессию данного фрагмента посредством одного цикла репликации. Затем, определяют количество рекомбинантов в библиотеке и абсолютное число частиц.
После этого проводят скрининг (отсев) таких библиотек, используя комбинации клеток определенного типа и клеток других типов, которые могут быть вовлечены в патологический процесс или способны оказывать влияние на его течение, диагностику и терапию. При этом скрининг осуществляют многократным повторением раундов инкубации с клетками, отмывок фаговых частиц, которые не связались с клетками, растворами различной ионной силы и растворами различной способности нарушать белок - белковые взаимодействия, с последующей элюцией и амплификацией искомых фаговых частиц. Причем, в случае прямого скрининга определенно рассчитанное количество фаговых частиц в буфере сначала инкубируют с материалом, который используют для культивирования клеток, а потом остаточную фракцию (супернатант), содержащую свободные фаги, переносят на клетки определенного типа. В случае, многошагового скрининга, а именно, использования комбинаций нескольких типов клеток, которые могут быть вовлечены в патологический процесс или способны оказывать влияние на его течение, диагностику и терапию, супернатант после инкубации с материалом направляют на эти клетки, после которых супернатант направляют либо на клеток распознаваемого типа, либо на следующий тип клеток, которые могут быть вовлечены в патологический процесс или способны оказывать влияние на его течение, диагностику и терапию, а потом супернатант инкубируют с клетками распознаваемого типа. Аналогично проводят скрининг, если используются клетки определенного типа и клетки других типов, которые могут быть вовлечены в патологический процесс или способны оказывать влияние на его течение, диагностику и терапию, в различных структурно-функциональных состояниях.
Таким образом, результатом многошагового скрининга является группа пептидов в составе фаговых частиц, обладающая выраженной специфичностью в отношении клеток определенного типа.
Далее из вышеуказанных групп выделяют индивидуальные фаговые частицы и их тестируют.
В одном случае, тестируемый фаг нарабатывают (наращивают) в необходимом количестве, очищают и испытывают как первое антитело в прямом иммуноферментном анализе (ИФА), где антигеном выступают культивируемые клетки определенных типов в различных функциональных состояниях.
При исследовании большого количества фаговых частиц, проводят микроселекцию. Для этого исходную группу рассеивают до получения индивидуальных бляшек, которые затем элюируют буфером для инкубации в ИФА и используют непосредственно, как источник первого антитела в ИФА, без предварительного наращивания и очистки.
В другом случае, индивидуальный клон аналогично первому случаю амплифицируют, очищают и инкубируют одновременно с клетками распознаваемого типа и клетками, которые могут быть вовлечены в патологический процесс или способны оказывать влияние на его течение, диагностику и терапию. После чего отмывают клетки, элюируют связанные частицы и определяют их титр, затем сравнивают его с титром, полученным для клеток другого типа.
В третьем случае используют полимеразную цепную реакцию (ПЦР), в анализе специфичности пептидов, в составе фаговых частиц, при этом, несколько фагов очищают и смешивают в одинаковых количествах, потом инкубируют с клетками распознаваемого типа и клетками, которые могут быть вовлечены в патологический процесс или способны оказывать влияние на его течение, диагностику и терапию, элюируют, рассеивают до отдельных бляшек, которые отбирают парами, выделяют ДНК и используют ее в качестве матрицы для ПЦР. Так определяют количественное распределение и степень специфичности пептидов, в составе фаговых частиц.
Также определяют стабильность пептидов и способность интернализироваться в составе фаговых частиц, которые проверяют, инкубируя фаг с клетками определенного типа при различных режимах температуры и времени, с последующим элюированием жизнеспособных фагов.
Для получения индивидуальных пептидов, распознающих клетки определенного типа вне фаговых частиц, ДНК бактериофага выделяют, и посредством полимеразной цепной реакции амплифицируют олигонуклеотидные фрагменты, кодирующие пептиды в составе фаговой частицы, специфично распознающие клетки определенного типа. Первичные нуклеотидные последовательности этих фрагментов определяют методом дидезокси-секвенирования по Сэнгеру, используя автоматический секвенатор. Полученные нуклеотидные последовательности транслируют в рамке считывания для получения аминокислотных последовательностей пептидов, находящихся в составе фаговых частиц. Далее проводят компьютерный анализ полученных аминокислотных последовательностей, используя общедоступные пакеты программ для анализа биологических макромолекул, при этом определяют физико-химические и структурные характеристики пептидов. На основе данных компьютерного анализа конструируют матрицы из аминокислотных последовательностей пептидов, специфично распознающих клетки определенных типов. Для чего на первом этапе аминокислотные последовательности группируют в короткие (до 5 аминокислотных звеньев) матрицы, которые на втором этапе объединяют в длинные матрицы (не менее 15 аминокислотных звеньев), комбинируя минимум по три и более коротких матриц. На завершающем этапе к полученным матрицам из аминокислотных последовательностей добавляют последовательности линкера и сигнала обнаружения. Для получения пептидов в чистом виде, с конструированных матриц считывают последовательности новых пептидов и синтезируют такие пептиды химическим путем. Далее пептиды очищают до готовности, с последующим подтверждением специфичности полученных пептидов к клеткам определенных типов.
Примеры конкретного выполнения заявленного способа
Пример 1. Выбор клеточных популяций
Для получения пептидов, высокоспецифичных к, например, сосудистым гладкомышечным клеткам (СГМК), используют их в качестве клеток распознаваемого типа, а клетки, взаимодействующие с ними, как в составе стенки кровеносного сосуда (эндотелиальные), так и переносимые током крови (фибробласты) используют в качестве клеток, которые могут быть вовлечены в патологический процесс или способны оказывать влияние на его течение, диагностику и терапию. Клетки карциномы цервикального канала - HeLa и клетки карциномы печени - Hep-G2 также используют как клетки, которые могут быть вовлечены в патологический процесс или способны оказывать влияние на его течение, диагностику и терапию.
Культуру СГМК инициируют из магистральных кровеносных сосудов пациентов с патологиями сердечно-сосудистой системы. Целый ряд клеточных линий доступен в виде коммерческих препаратов. Например, параллельно культурам клеток, полученным при обработке артерий и вен пациентов, использовали гладкомышечные клетки коронарной артерии (Cell Systems GmbH, Lot# 17151, Catalog # CC-2583). Препараты исходной культуры СГМК являются первичными в своем происхождении и имеют ограниченное количество дупликаций, что лимитирует количество последующих пересевов. Поэтому, во всех случаях инициируют культуры, для последующего исследования используя образцы клеток, имеющих не более чем 6 делений. Для культивирования используют пластиковые флаконы, произведенные исключительно для работы с культурами тканей, общей площадью поверхности для роста от 25 до 182 квадратных сантиметров (например, флаконы типов - Т25, Т75, Т182).
Сосудистые гладкомышечные клетки культивируют, используя питательную среду, которая состоит на 1/3 из каждого следующего компонента - Waymouth (GIBCO BRL Cat. #31220-023), F12 (GIBCO BRL Cat. #31220-023), SMCBM (Promocell Cat. # C-22260). Такую среду дополняют предварительно инактивированной инкубацией 30 минут при температуре 56°C на водяной бане, сывороткой плода коровы (GIBCO BRL Cat. # СС-4102) до конечной концентрации - 15%, а также раствором пенициллина/стрептомицина (GIBCO BRL Cat. #15140-114) до конечной концентрации -1,5%. Среду, содержащую в своем составе сыворотку плода коровы, называют обогащенной. Сосудистые гладкомышечные клетки, блокированные на стадии роста, культивируют, используя обедненную питательную среду, т.е. без добавления сыворотки плода коровы, но с добавлением раствора пенициллина/стрептомицина в вышеуказанной концентрации. Для блокирования сосудистых гладкомышечных клеток на стадии роста, их сначала культивируют, пока размер монослоя не достигнет 90% от всей площади роста, используя обогащенную питательную среду, и лишь, затем проводят замену обогащенной питательной среды на обедненную и продолжают инкубацию в течение 72 часов, проводя замену старой питательной среды на новую, каждые 24 часа.
Эндотелиальные клетки культивируют, используя питательную среду - EGM-2 (Clonetics Company, Cat. # CC-3202), дополненную прилагающимся к ней пакетом комплементации - EGM-2 MV (Clonetics Company, Cat # CC-4147), который содержит факторы роста, гормоны и инактивированную сыворотку плода коровы, конечная концентрация которой в среде после комплементации составляет 10%. Эндотелиальные клетки инициируют из препарата (Promocell Cat. # СС-2519). Блокируют эндотелиальные клетки на стадии роста аналогично тому, как в случае сосудистых гладкомышечных клеток.
Фибробласты культивируют, используя питательную среду, основой которой является стандартная среда DMEM (GIBCO BRL), дополненная инактивированной сывороткой плода коровы до конечной концентрации 10%, и раствором пенициллина/стрептомицина до конечной концентрации 1,5%. Инициируют культуру клеток фибробластов из препарата пациентов с патологиями сердечно-сосудистой системы и блокируют так же, как это описано для сосудистых гладкомышечных клеток.
Клетки HeLa 3T3 и клетки Hep-G2 представляют собой иммортализованные линии, поэтому практически любой доступный образец клеток используют для инициирования культуры, однако, стараются использовать клетки, прошедшие не более 100 делений. Для культивирования этих клеток используют питательную среду на основе какой-либо из MEM - композиций (GIBCO BRL), дополненную сывороткой плода коровы в конечной концентрации 15-20% (что определяется желаемыми скоростями роста) и раствором пенициллина/стрептомицина в конечной концентрации 1,5%. Для получения достоверных результатов скрининга данные типы клеток так же подвергают культивированию с использованием обедненной питательной среды, имитируя блокирование роста, сходное с таковым наблюдаемым для первичных типов клеток (СГМК, эндотелиальные и фибробласты).
Пример 2. Выделение РНК и реакции обратной транскрипции (см. фиг.1)
На основе первичного отбора клеточных популяций выделяют РНК, как из клеток определенного типа, так и из клеток, которые могут быть вовлечены в патологический процесс или способны оказывать влияние на его течение, диагностику и терапию.
Клетки всех типов наращивают в течение 5-7 суток, используя флаконы типа Т75 и 18 мл питательной среды, замену которой производят каждые 24 часа. Сосудистые гладкомышечные клетки культивируют в 40 флаконах, из расчета 3,5*105 клеток на флакон. Эндотелиальные клетки культивируют в 20 флаконах, из расчета 3*105 клеток на флакон. Клетки HeLa и Hep-G2 культивируют в 20 флаконах, из расчета 1,6*105 клеток на флакон. Клетки интенсивно промывают 2 раза, используя 12 мл буфера HBSS (сбалансированный солевой раствор по Хэнксу) каждый раз, далее обрабатывают в течение 3 минут, используя 6 мл раствора, содержащего 0,05% трипсина и 0,53 мМ ЭДТА (этилендиаминтетраацетата), и нейтрализуют добавлением 9 мл буфера HBSS. Собирают клетки центрифугированием в течение 10 минут на 3000*g и промывают 3 раза, используя 20 мл буфера HBSS. Число клеток определяют с помощью камеры для счета клеток, для этого смешивают 50 мкл суспензии клеток с равным объемом 15%-ного раствора трипанового голубого, являющегося витальным красителем. Число живых клеток фиксируют и рассчитывают общее число клеток в суспензии.
В случае использования методов грубой экстракции гуанидинизотиоцианатом, как с последующим осаждением солями лития, так и без осаждения, клетки концентрируют центрифугированием, супернатант удаляют, а осадок ресуспендируют интенсивно в буфере для лизиса (5,25М гуанидинизотиоцианата; 50 мМ Трис-Cl, pH 6,4; 20 мМ ЭДТА; 1% Тритон Х-100; 0,1М 2-меркаптоэтанола) из расчета 1 мл на 107 клеток. К данному раствору добавляют 0,1 объема 2М р-ра ацетата натрия, 1 объем р-ра водного фенола, 0,3 объема смеси хлороформ - изоамиловый спирт (в соотношении 24:1, по объему) и интенсивно перемешивают в течение 30-60 секунд. Инкубируют полученную смесь 15 минут при температуре 0°C, после чего центрифугируют в течение 30 минут на 8000*g при температуре 4°C, с целью разделения водной и органической фаз. Водную фазу после осаждения переносят в новую пробирку и добавляют к ней 0,6 объема изопропилового спирта, предварительно охлажденного до температуры -20°C, перемешивают и помещают на период не менее 60 минут в охладитель с температурой -70°C. После инкубации раствор центрифугируют 30 минут на 12000*g при температуре 4°C, супернатант тщательно удаляют, а осадок промывают несколько раз центрифугированием по 5 минут в присутствии 1 мл 70%-ного раствора этилового спирта. Полученный осадок растворяют в 0,4 мл воды, обработанной диэтилпирокарбонатом (ДЭПК).
В случае применения методов мягкой экстракции с использованием протеиназы К, клетки концентрируют центрифугированием, супернатант удаляют, а осадок ресуспендируют интенсивно в предварительно охлажденном буфере для лизиса (0,15М хлорида натрия; 10 мМ Трис-Cl, pH 8,5; 0,05% NP-40; 0,01% 3-карбоксиауриновой кислоты; 0,1М 2-меркаптоэтанола) из расчета 1 мл на 5*107 клеток. Затем, смесь центрифугируют 2 минуты на 12000*g и температуре 4°C, супернатант переносят в новую пробирку и добавляют равный объем буфера для протеиназы К (0,2М Трис-Ацетат; 0,3М натрия ацетат; 0,05М ЭДТА, pH 7,5; 2% (по весу) натрия додецилсульфата), содержащего 400 мг/мл протеиназы К, и инкубируют 60 минут при температуре 37°C. Далее добавляют 1 объем смеси фенол - хлороформ - изоамиловый спирт (в соотношении 25:24:1) и интенсивно перемешивают 5 минут при комнатной температуре. После чего смесь центрифугируют 2 минуты на 12000*g при комнатной температуре, полученную водную фазу переносят в новую пробирку и повторяют экстракцию смесью фенол - хлороформ - изоамиловый спирт (в соотношении 25:24:1) несколько раз. К водной фазе, полученной в результате экстракции, добавляют 2,5 объема абсолютного этилового спирта, интенсивно перемешивают и осаждают РНК, помещая образец на 30 минут в охладитель с температурой -70°C. Далее раствор, содержащий РНК, центрифугируют 10 минут на 12000*g при температуре 4°C, и полученный осадок промывают 3 раза центрифугированием 5 минут на 12000*g, в присутствии 700 мкл 70%-ного раствора этилового спирта. Осадок осушают 2,5 минуты под вакуумом и растворяют в 0,4 мл воды, обработанной ДЭПК.
Полученный в результате первичной экстракции раствор суммарной клеточной РНК подвергают обработке дезоксирибонуклеазой первого типа, с целью удаления фрагментов ДНК, способных попасть в раствор РНК в процессе ее экстракции и способных затруднять дальнейшее фракционирование такой РНК и использование ее в реакциях обратной транскрипции. Для этого к 0,4 мл раствора, содержащего 4 мг/мл РНК, добавляют 12 мкл (480 единиц) ингибитора рибонуклеаз (Invitrogen, Cat #10777-019), 50 мкл 10-ти кратного стандартного буфера для дезоксирибонуклеазы и 50 мкл (500 единиц) дезоксирибонуклеазы первого типа (GIBCO BRL). Затем, полученную смесь инкубируют 2 часа при температуре 37°C, и реакцию останавливают одновременным добавлением 0,1 объема 3М р-ра ацетата натрия (pH 5,2), 1,1 объема раствора водного фенола, 0,3 объема смеси хлороформ - изоамиловый спирт (в соотношении 24:1). Раствор интенсивно перемешивают в течение 1 минуты, инкубируют 15 минут при температуре 0°C и центрифугируют 10 минут на 12000*g, при температуре 4°C. Водную фазу переносят в новую пробирку и осаждают РНК с помощью абсолютного этилового спирта, как описано выше.
Для выделения фракции матричной РНК (мРНК), 1-2 мг суммарной РНК растворяют в 5 мл буфера для нанесения (прилагается к колонке) и наносят на колонку, содержащую олиго - (дТ) - целлюлозу (Sigma, Cat #03131), предварительно промытую 3 раза 1 мл буфера для нейтрализации (прилагается к колонке). После экстракции колонку промывают 5 раз, используя 1 мл буфера для нейтрализации, и связанную мРНК элюируют 1,5 мл буфера для элюции. В дальнейшем, мРНК, содержащуюся в полученном растворе, осаждают добавлением 150 мкл р-ра 3М ацетата натрия (pH 5,2) и 3,75 мл абсолютного этилового спирта.
Пример 3. Синтез кДНК и фрагментация суммарной РНК
С целью получения молекул кДНК для клонирования, суммарную РНК и/или мРНК используют в реакциях обратной транскрипции, направляемых РНК - зависимой - ДНК-полимеразой.
Для этого смешивают 0,001-5,000 мг суммарной РНК и 1 мкл праймера (500 мкг олиго - (дТ) или 0,050-0,250 мкг случайного связывающегося праймера, 5'-TTNNNNNN-3'), к которым добавляют 1 мкл 10 мМ р-ра дезоксирибонуклеотидов (10 мМ каждого в нейтральном растворе), и доводят объем смеси до 12 мкл, используя дважды дистиллированную воду, обработанную диэтилпирокарбонатом. Полученную смесь инкубируют 5 минут при температуре 65°C, быстро охлаждают и добавляют 4 мкл 5-ти кратного буфера для синтеза первичной цепи кДНК (250 мМ Трис-Cl, pH 8,3; 375 мМ калия хлорида; 15 мМ хлорида магния), 2 мкл 0,1М р-ра дитиотриэтола (ДТТ), 1 мкл (40 единиц) ингибитора рибонуклеаз. Содержимое пробирки интенсивно перемешивают и инкубируют 2 минуты при температуре 42°C, после чего, добавляют 1 мкл (200 единиц) Superscript II РНК - зависимой - ДНК-полимеразы (GIBCO BRL, Cat. #18064-014), интенсивно перемешивают и инкубируют 60 минут при температуре 42°C. Реакцию обратной транскрипции останавливают посредством инактивации фермента, для чего смесь инкубируют 15 минут при температуре 70°C. С целью удаления фрагментов РНК, комплементарных новосинтезированным молекулам кДНК и затрудняющих последующую амплификацию кДНК с помощью ПЦР, к раствору добавляют 1 мкл (2 единицы) рибонуклеазы Н и инкубируют 20 минут при температуре 37°C.
На этом этапе осуществляют первичное разделение суммарной РНК посредством контролируемого синтеза фрагментов кДНК, ограниченных в размере и гетерогенности, что достигается использованием различных концентраций ионов в реакционной смеси, комбинацией праймеров, количеством исходного материала и режимов температур.
Синтез 2-й цепи кДНК осуществляют с помощью полимеразной цепной реакции (ПЦР), для чего используют не более 10% конечного раствора после обработки рибонуклеазой Н. Собирают смесь последовательным добавлением 5 мкл 10-ти кратного буфера для ПЦР (200 мМ Трис-Cl, pH 8,4; 500 мМ хлорида калия), 1,5 мкл 50 мМ р-ра хлорида магния, 1 мкл 10 мМ р-ра дезоксирибонуклеотидов, 0,5 мкл (2,5 единиц) Taq - ДНК-полимеразы, 2 мкл р-ра кДНК (конечный продукт реакции синтеза первичной цепи кДНК), 38,1 мкл воды обработанной ДЭПК. Раствор интенсивно перемешивают и инкубируют в течение 2 минут при температуре 94°C. Затем проводят 15-40 стандартных циклов амплификации ПЦР.
С целью получения фрагментов, пригодных для клонирования, концы молекул кДНК обрабатывают и легируют с синтетическими олигонуклеотидными линкерами, которые содержат в своем составе место узнавания для эндонуклеазы рестрикции - Есо R1 и частично место узнавания для эндонуклеазы рестрикции - Hind III. Это позволяет получить после обработки таких линкеров соответствующими эндонуклеазами рестрикции, молекулы кДНК, способные эффективно легироваться с 5′ - конца только с Eco R1, обработанной ДНК, и с 3′ - конца только с Hind III, обработанной ДНК. Для этого, к 20 мкл (не более 10 мкг) раствора молекул кДНК добавляют 3 мкл 10-ти кратного буфера для Т4 ДНК-полимеразы (200 мМ Трис-Cl, pH 8,4; 500 мМ хлорида калия), 1,5 мкл 100 мМ р-ра ДТТ, 3 мкл 1 мМ р-ра дезоксирибонуклеотидов и 1,0 мкл (1,5 единиц) Т4 - ДНК-полимеразы, доводят конечный объем раствора до 30,0 мкл, используя дважды дистиллированную воду, обработанную ДЭПК. Раствор медленно перемешивают и инкубируют в течение 20 минут при температуре 11°C. Реакцию останавливают добавлением равного объема смеси фенол - хлороформ - изоамиловый спирт (в соотношении 25:24:1) и затем ДНК, которая содержится в водной фазе, осаждают добавлением растворов ацетата натрия и абсолютного этилового спирта, как описано выше. Полученный осадок, который содержит молекулы кДНК, растворяют в 10 мкл дважды дистиллированной воды, обработанной ДЭПК. Олигонуклеотидные линкеры фосфорилируют посредством инкубации с Т4 - полинуклеотидкиназой, непосредственно перед использованием в реакции легирования. Для этого собирают смесь последовательным добавлением к 10 мкл р-ра молекул кДНК (с предыдущего этапа), 2 мкл 10-ти кратного буфера для легирования (10х=200 мМ Трис-Cl, pH 7,6; 50 мМ хлорида калия), 2 мкл 1 мМ р-ра АТФ, 2 мкл (100 пикомоль) р-ра олигонуклеотидных линкеров (5′-GCTTGAATTCAAGC-3′/3′-CGAACTTAAGTTCG-5′), 2 мкл 100 мМ р-ра ДТТ, 2,0 мкл (5 единиц) Т4 - полинуклеотидкиназы. Раствор перемешивают и инкубируют в течение 5 минут при температуре 37°C. Затем добавляют 6-8 единиц Т4 - ДНК-лигазы и инкубируют в течение 6-20 часов при температуре 16°C.
Далее молекулы кДНК расщепляют с помощью 2-х эндонуклеаз рестрикции - Hind III и EcoR1. Для этого к раствору кДНК добавляют 10 мкл 10-ти кратного буфера для эндонуклеазы рестрикции Hind III и 10 мкл (100 единиц) эндонуклеазы рестрикции Hind III (Invitrogen), объем такой смеси доводят до 100 мкл с помощью дважды дистиллированной воды. Полученную смесь инкубируют в течение 2-х часов при температуре 37°C. Затем добавляют 10 мкл 10-ти кратного буфера для эндонуклеазы рестрикции Eco R1 и 10 мкл (100 единиц) эндонуклеазы рестрикции Eco R1 (Invitrogen) и снова инкубируют в течение 4 часов при температуре 37°C. Реакцию останавливают добавлением равного объема смеси фенол - хлороформ - изоамиловый спирт (в соотношении 25:24:1) и затем ДНК, которая содержится в водной фазе, осаждают добавлением растворов ацетата натрия и абсолютного этилового спирта, как описано выше.
Вторичное разделение суммарной РНК осуществляют на этапе фракционирования и очистки полученных молекул кДНК, для чего используют гель-фильтрацию. Хроматографическую колонку, содержащую частицы сефарозы определенного размера, промывают 5 раз, используя 1 мл буфера для нейтрализации каждый раз. Наносят разделяемый материал, популяцию кДНК, в объеме 1 мл и собирают фракции по 25 мкл.
Пример 4. Клонирование и создание множества библиотек
Создание библиотек проводят по одной модели, как для клеток определенного типа, так и для клеток, которые могут быть вовлечены в патологический процесс или способны оказывать влияние на его течение, диагностику и терапию.
Для этого используют, например, ДНК бактериофага Т7 (Novagen, T7 Select 10-3 вектор), обработанную с использованием 2-х эндонуклеаз рестрикции - Eco R1 и Hind III, т.е. того же типа, которые использовались для обработки концов кДНК. Раствор молекул ДНК вектора и раствор молекул кДНК смешивают в отношении 1:3, в пересчете на моль вещества, добавляют эквивалентное количество Т4 - ДНК-лигазы (GIBCO BRL) и инкубируют не более 28 часов при температуре 16°C. Реакцию останавливают добавлением равного объема смеси фенол - хлороформ - изоамиловый спирт (в соотношении 25:24:1), и затем ДНК, которая содержится в водной фазе, осаждают добавлением растворов ацетата натрия и абсолютного этилового спирта, как описано выше.
Полученные рекомбинантные молекулы кДНК, в количестве не более 1 мкг, смешивают с 250 мкл р-ра упаковочных белков бактериофага и инкубируют в течение 5 минут при температуре 0°C. Затем добавляют питательную среду и продолжают инкубацию еще 60 минут при температуре 37°C.
Пример 5. Амплификация бактериофагов
Бактериофаг Т7 культивируют, используя штамм E.coli BLT5403 в качестве хозяина. При этом амплификацию проводят несколькими путями.
В одном случае, микроамплификация, единичную бляшку или элюат после скрининга, или какое-либо другое малое количество бактериофагов, растворяют в 1 мл питательной среды, приготовленной по методу Луриа - Бертани (ЛБ). Затем добавляют 1 мл культуры штамма-хозяина с оптической плотностью 0,4-0,6 при длине волны 600 нм (ОП600=0,4-0,6), которую инициируют в 50 мл питательной среды ЛБ из 0,5 мл суспензии бактерий, предварительно культивированных в течение 16 часов, и наращивают в течение 2-3 часов. Смесь фагов и бактерий общим объемом 2 мл инкубируют при температуре 37°C и 200 оборотах в минуту на шейкере в течение 3-4 часов, наблюдая полный лизис, выражающийся просветлением содержимого пробирки.
В другом случае, макроамплификацию проводят, используя колбы посредством инокуляции необходимого числа фагов в 135 мл культуры штамма-хозяина (ОП600=0,4-0,6) и последующим ростом в течение 4-5 часов до момента полного лизиса.
К лизированным культурам добавляют 0,1 объема 5М р-ра хлорида натрия, интенсивно перемешивают 30 секунд и центрифугируют в течение 3 минут на 7000*g и при температуре 4°C. Супернатант переносят в новую пробирку и добавляют 1/6 объема раствора, содержащего 20% полиэтиленгликоля и 2,5М хлорида натрия, интенсивно перемешивают и инкубируют не менее 3 часов при температуре 0°C. Далее раствор центрифугируют 10 минут на 8000*g, а осадок, содержащий фаги, растворяют в 1 мл буфера TBS (Трис - Боратный Буфер) и снова осаждают фаговые частицы добавлением раствора полиэтиленгликоля - хлористого натрия с последующей инкубацией и центрифугированием в течение 10 минут на 12000*g и при температуре 4°C. Полученный осадок растворяют в 0,2 мл буфера TBS.
Пример 6. Скрининг (см. фиг.2)
Скрининг библиотек осуществляют, как многократным повторением одношаговой процедуры, так и с использованием многошаговой комплексной процедуры, включающей селекцию на комбинациях различных типов клеток, которые могут быть вовлечены в патологический процесс или способны оказывать влияние на его течение, диагностику и терапию.
Для этого инициируют культуру сосудистых гладкомышечных клеток в чашках Петри диаметром 60 мм из расчета 64000 клеток на чашку и растят в течение 48 часов. Одновременно, в аналогичных чашках инициируют культуры эндотелиальных клеток, фибробластов, HeLa, Hep-G2, соответственно в количествах - 48000, 24000, 16000, 16000 клеток на чашку, и растят в течение 48 часов.
Клетки промывают интенсивно 5 раз по 5 минут, используя каждый раз 3 мл буфера HBSS.
При использовании одношаговой процедуры, 1010 фаговых частиц из любой библиотеки, растворяют в 2 мл буфера HBSS и инкубируют 20 минут с преинкубационным материалом (пластик). После этого, верхнюю фазу, содержащую не связавшиеся с пластиком фаги, переносят на предварительно промытые сосудистые гладкомышечные клетки и инкубируют 40 минут при температуре 4°C. После инкубации, с целью удаления несвязанного материала, клетки интенсивно отмывают 5 раз по 5 минут, используя каждый раз 3 мл буфера HBSS, с последующей элюцией специфично связывающихся фагов с помощью 1 мл буфера для элюции, который содержит в своем составе не более чем 1,0% додецилсульфата натрия, или с помощью 1 мл свежеприготовленной дневной культуры штамма-хозяина. Титр фагов в элюате определяют, и элюат амплифицируют для получения как минимум 1010 фагов, которые инкубируют с новой порцией преинкубационного материала и новой порцией сосудистых гладкомышечных клеток, повторяя селекционный цикл 3-5 раз.
При использовании многошаговой процедуры, 1010 фаговых частиц из любой библиотеки, растворяют в 2 мл буфера HBSS и инкубируют 20 минут с преинкубационным материалом (чашкой Петри, в которой вместо клеток содержалась лишь среда). После этого, верхнюю фазу, содержащую не связавшиеся с пластиком фаги, переносят на предварительно промытые эндотелиальные клетки и инкубируют в течение 40 минут при температуре 4°C, с последующим переносом верхней фазы на новую порцию эндотелиальных клеток и аналогичной инкубацией, что повторяют 2-3 раза и лишь после этого, верхнюю фазу направляют на фибробласты и проводят сходную селекцию 2-3 раза. Или верхнюю фазу после инкубации на эндотелиальных клетках, без дополнительной селекции на фибробластах, переносят непосредственно на сосудистые гладкомышечные клетки и инкубируют в течение 40 минут при температуре 4°C. Отмывку и элюцию проводят, как описано выше.
В другом случае, верхнюю фазу после инкубации с преинкубационным материалом (чашкой Петри, в которой вместо клеток содержалась лишь среда) переносят на предварительно промытые клетки HeLa и инкубируют с ними в течение 40 минут при температуре 4°C, а далее верхнюю фазу переносят на клетки Hep-G2 и инкубируют с ними в течение 40 минут при температуре 4°C, после чего верхнюю фазу переносят на новую порцию клеток HeLa и клеток Hep-G2, повторяя комбинированную селекцию 2-3 раза. Конечную верхнюю фазу переносят на сосудистые гладкомышечные клетки и инкубируют в течение 40 минут при температуре 4°C.
В следующем варианте скрининга, материал, после селекции на иммортализованных типах клеток, переносят на эндотелиальные клетки и/или фибробласты и лишь затем на сосудистые гладкомышечные клетки.
При выполнении скрининга, в ходе проведения экспериментальных работ, получили пептиды - P1 N′-Thr-Asn-His-Tyr-(Ile)2-Ser-Pro-Tyr-Asn-Ile-C′, P2 N′-Ala-Trp-Lys-Leu-Pro-Ile-His-Gln-Ser-Arg-Val-Tyr-C′, P3 N′-Asn-Thr-Tyr-Gln-His-Ser-Trp-Pro-Ala-Ser-Arg-Pro-C′, P4 N′-Thr-His-Thr-Leu-Asn-(Thr)2-Lys-Met-Arg-Val-Leu-C′, которые распознают клетки определенного типа, например, сосудистые гладкомышечные клетки человека.
Пример 7. Выполнение комбинированного тестирования пептидов в составе бактериофаговых частиц
А. Иммуноферментный анализ
Специфичность индивидуальных пептидов в составе фаговых частиц оценивают, используя их вместо первого антитела в прямом иммуноферментном анализе, где в качестве антигена выступают клетки определенных типов и клетки других типов, которые могут быть вовлечены в патологический процесс или способны оказывать влияние на его течение, диагностику и терапию.
Для этого клетки всех используемых типов культивируют в 96-луночных планшетах, инициируя культуру из расчета 10000 клеток на лунку. Клетки растят в течение 48 часов, при этом проводят замену питательной среды каждые 24 часа. Затем, планшеты промывают интенсивно 5 раз по 5 минут, используя каждый раз 200 мкл буфера HBSS. Далее инкубируют 106-107 фаговых частиц с клетками в течение 1 часа при температуре 4°C, с интенсивной последующей отмывкой буфером HBSS и инкубацией со вторым антителом, в течение 1 часа при комнатной температуре. В качестве второго антитела используют моноклональное антитело, конъюгированное с пероксидазой хрена и распознающее мотив из 11 аминокислот (N′-Met-Ala-Ser-Met-Thr-(Gly)2-(Gln)2-Met-Gly-C′) (Novagen, Cat. No.69522), расположенный на N-конце белка №10, входящего в состав капсида бактериофага Т7. Затем, планшеты промывают вновь буфером HBSS и производят обнаружение второго антитела, используя субстрат для пероксидазы, расщепление которого останавливают добавлением 50 мкл 1М р-ра серной кислоты.
Б. Анализ на связывание
Связывание индивидуальных пептидов в составе фаговых частиц с клетками различных типов исследуют, используя одношаговую селекцию, при которой анализируемый пептид тестируют параллельно на связывание с клетками распознаваемого типа и с клетками других типов, которые могут быть вовлечены в патологический процесс или способны оказывать влияние на его течение, диагностику и терапию.
Для этого инициируют культуру клеток СГМК в чашках Петри диаметром 60 мм в расчете 64000 клеток на чашку и культивируют в течение 48 часов. Одновременно, в аналогичных чашках инициируют культуры эндотелиальных клеток, фибробластов, HeLa, Hep-G2, соответственно в количествах - 48000, 24000, 16000, 16000 клеток на чашку, и растят в течение 48 часов. Используют 109 фаговых частиц, несущих уникальный пептид, которые растворяют в 2 мл буфера HBSS. Такой раствор инкубируют сначала в течение 20 минут с преинкубационным материалом (чашкой Петри, в которой вместо клеток содержалась лишь среда) и потом переносят верхнюю фракцию на испытуемые клетки, предварительно промытые 5 раз буфером HBSS, длительностью по 5 минут каждый раз. При этом процедуру параллельно совершают, как для сосудистых гладкомышечных клеток (распознаваемый тип клеток), так и для эндотелиальных клеток, фибробластов, клеток HeLa и Hep-G2 (клетки, которые могут быть вовлечены в патологический процесс или способны оказывать влияние на его течение, диагностику и терапию), используя для клеток всех типов 2 режима температур: 4°C и 37°C. После инкубации, не связавшиеся частицы интенсивно отмывают, используя буфер HBSS, после чего элюируют, используя 1 мл буфера для элюции. Определяют титр фагов в элюате и сравнивают полученные данные для всех типов клеток. При этом, чем больше отношение титра фагов, полученных после элюции с СГМК, к титру фагов, полученных с других типов клеток, тем более специфичен пептид в составе фаговой частицы к СГМК, и соответственно, менее специфичен к другим типам клеток.
В. Использование полимеразной цепной реакции (ПЦР)
Специфичность индивидуальных пептидов в составе фаговых частиц тестируют, используя ПЦР для идентификации клонов, а также для анализа их количественного распределения при взаимодействии с клетками распознаваемого типа и клетками других типов, которые могут быть вовлечены в патологический процесс или способны оказывать влияние на его течение, диагностику и терапию.
Для этого инициируют культуру клеток СГМК в чашках Петри диаметром 60 мм в расчете 64000 клеток на чашку и культивируют в течение 48 часов. Одновременно, в аналогичных чашках инициируют культуры эндотелиальных клеток, фибробластов, HeLa, Hep-G2, соответственно в количествах - 48000, 24000, 16000, 16000 клеток на чашку, и культивируют в течение 48 часов. Фаговые частицы смешивают в эквивалентных количествах, обычно 107 частиц каждого типа, из расчета на конечный объем раствора 2 мл. Полученный раствор фаговых частиц инкубируют сначала в течение 20 минут с преинкубационным материалом (чашкой Петри, в которой вместо клеток содержалась лишь среда) и потом переносят верхнюю фракцию на испытуемые клетки, предварительно отмытые 5 раз буфером HBSS, длительностью по 5 минут каждый. При этом процедуру одновременно совершают, как для сосудистых гладкомышечных клеток (распознаваемый тип клеток), так и для эндотелиальных клеток, фибробластов, клеток HeLa и Hep-G2 (клетки, которые могут быть вовлечены в патологический процесс или способны оказывать влияние на его течение, диагностику и терапию), используя для клеток всех типов 2 режима температур: 4°C и 37°C. После инкубации, не связавшиеся частицы интенсивно отмывают, используя буфер HBSS, после чего связанные фаги элюируют, используя 1 мл буфера для элюции. Затем определяют титр фагов в элюате и рассеивают равные аликвоты элюата до получения единичных бляшек. Отбирают с каждой чашки 100 бляшек, распределяют их группами по две и выделяют ДНК посредством лизиса фаговых частиц. Для этого инкубируют фаговые частицы в 10 мМ растворе ЭДТА в течение 10 минут при температуре 65°C, с последующим охлаждением и центрифугированием в течение 3 минут на 14000*g. Полученный раствор, содержащий ДНК бактериофага, используют в качестве матрицы для ПЦР по стандартной для данного вектора схеме, направляемой праймерами к участкам вектора (Т7 ″Forward″ праймер: 5′-AACCCCTCAAGACCCGTTTA-3′; T7 ″Reverse″ праймер: 5′-GGAGCTGTCGTATTCCAGTC-3′), или специально разработанными праймерами. После этого, определяют абсолютную частоту встречаемости каждого из пептидов в составе фаговых частиц в зависимости от типа клеток.
Г. Тест на стабильность пептида и способность интернализироваться в составе фаговой частицы
Стабильность индивидуального пептида в составе фаговой частицы, т.е. способность максимально долго сохранять свою функциональную эффективность в клетке, которую он специфически распознает и в самом организме хозяина, напрямую пересекается со способностью эффективно проникать в клетку, сохранять активность и специфически бомбардировать внутриклеточные мишени.
Для этого инициируют культуру клеток СГМК в чашках Петри диаметром 60 мм в расчете 64000 клеток на чашку и культивируют в течение 48 часов. Одновременно, в аналогичных чашках инициируют культуры эндотелиальных клеток, фибробластов, HeLa, Hep-G2, соответственно в количествах - 48000, 24000, 16000, 16000 клеток на чашку, и культивируют в течение 48 часов. 1010 фаговых частиц, несущих пептиды, распознающие клетки определенного типа в своем составе и представляющие фракцию, полученную после элюции фагов в результате 3-х раундов селекции многошаговым скринингом, инкубируют с сосудистыми гладкомышечными клетками при 2-х режимах температур: 4°C и 37°C и в течение различных периодов времени. Далее, клетки отмывают и лизируют в течение 10 минут, используя 2 мл буфера для лизиса (1 мл 2%-го р-ра дезоксихолиевой к-ты; 10 мМ Трис; 2 мМ ЭДТА, pH 8,0). После нейтрализации, раствор, содержащий фаги, рассеивают до одиночных бляшек, которые затем отбирают с целью определения первичной последовательности олигонуклеотидных фрагментов, кодирующих пептиды в составе отобранных фаговых частиц, а также для проведения дополнительного тестирования.
На основании экспериментальных данных при проведении комбинации тестов (А-Г), указанных выше, составлен график (см. фиг.4) зависимости специфичности отобранных в скрининге пептидов (Р1-Р4), в отношении сосудистых гладкомышечных клеток человека (обозначены как SMCp). Специфичность связывания пептидов выражена в условных единицах - УЕ. Для сравнения в этом графике используют сосудистые эндотелиальные клетки человека (ЕСр) и клетки опухоли (HeLa). При этом специфичность отобранных пептидов оценивается по отношению к исходной библиотеке (ИБ), сконструированной в заявляемом способе, и одним из пептидов (5Е5) прототипа полученных в работе Ingrid N. Michon et al. ″Targeting of peptides to restenotic vascular smooth muscle cells using phage display in vitro and in vivo″ в журнале ″Bio-chimica et Biophysica Acta″ вып. №1591, 2002 г., стр.87-97.
На основании графика на фиг.4 видно, что специфичность отобранных пептидов (Р1-Р4) по заявляемому способу намного превышает специфичность пептида из прототипа (5Е5), а также показывает высокую степень селективности в отношении сосудистых гладкомышечных клеток человека по сравнению с сосудистыми эндотелиальными клетками человека и/или клетками опухоли HeLa. При этом сконструированная библиотека по заявляемому способу не выявляет специфичности ни к одному типу клеток, тем самым подтверждая высокую степень эффективности скрининга, которая оценена в 500 и более раз.
Пример 8. Определение первичной последовательности олигонуклеотидных фрагментов и кодируемых ими аминокислотных последовательностей пептидов, с последующим компьютерным анализом
Олигонуклеотидные фрагменты ДНК участка генома вектора, представляющие вставки кДНК и кодирующие специфичные пептиды, и ограниченные праймерами вектора, амплифицируют, используя ПЦР, и определяют их первичную последовательность, методом дидезокси-секвенирования по Сэнгеру используя автоматический секвенатор, например, ABI Prism 310 (Applied Biosystems Inc.), руководствуясь рекомендованные производителем протоколами проведения реакций.
Используя сигналы распознавания в векторе, первичную последовательность олигонуклеотидных фрагментов вставок транслируют в рамке считывания в аминокислотные последовательности пептидов, специфично связывающихся с активно пролиферирующими и арестованными в росте формами гладкомышечных клеток, и полученные в результате аминокислотные последовательности перераспределяют согласно их длине и происхождению. В первую группу помещают пептиды с короткой аминокислотной цепью (до 30 звеньев), которые распознают исключительно линейные эпитопы (участки) рецепторных белков, расположенных на поверхности гладкомышечных клеток. Во вторую группу при этом отбираются пептиды с длинной аминокислотной цепью и фрагментами белков, которые связываются с гладкомышечными клетками, распознавая сложные вторичные и третичные структуры на их поверхности. Аминокислотные последовательности в каждой из двух групп далее последовательно анализируют согласно следующему алгоритму: 1) ищут гомологию с известными аминокислотными последовательностями, зарегистрированными в международных молекулярно-биологических базах данных, используя алгоритмы совмещения, предоставляемые через сервера BLAST (www.ncbi.nlm.nih.gov/BLAST/) и ExPaSy (www.expasy.org/tools/#align); 2) определяют первичную структуру и физико-химические характеристики пептидов, используя при этом алгоритмы NHH, предоставляемые через сервер ExPaSy (http://www.expasy.org/tools/) в комбинации с автономными пакетами, находящимися в свободном доступе. При этом определяют такие параметры, как частота встречаемости различных классов аминокислот (структурообразующих, гидрофобных, заряженных позитивно, заряженных негативно, полярных без заряда) в каждой из позиций полипептида, начиная с N-конца молекулы. Дополнительно определяют профиль гидрофобности, а также средний заряд и среднюю гидрофобность в пересчете на звено цепи для всей молекулы пептида; 3) определяют элементы вторичной и третичной структуры пептидов, сравнивая последовательности из базы данных по белковым молекулам (http://www.wwpdb.org/) с последовательностями специфических пептидов, например, используя пакет программ WebLab ViewerPro v4.0 (Molecular Simulations Inc., San Diego, CA, U.S.A.).
Возможные участки связывания специфических пептидов с молекулами белков на поверхностях гладкомышечных клеток выявляют, посредством сравнения совмещений фрагментов пептидов в многомерном пространстве, используя данные полученных для первой и второй групп аминокислотных последовательностей одновременно.
Пример 9. Проектирование матриц из аминокислотных последовательностей, создание пептидного конструкта и считывание новых пептидов (см. фиг.3).
Конструирование матриц осуществляется за счет унификации полипептидных цепей, предполагающее использование наименьшего количества аминокислотных звеньев для получения пептидов с минимальной комплексностью (для уменьшения времени и затрат на синтез пептидов) и максимальной специфичностью связывания клетками избранного типа (для уменьшения количества пептидов при использовании в диагностических и терапевтических приложениях).
Конструируют матрицы из аминокислотных последовательностей в два этапа. На первом этапе создают набор коротких матриц (не более 5 аминокислот) посредством перекрытия коротких участков, встречающихся с наибольшей частотой в анализируемых аминокислотных последовательностях, используя, например, пакет программ Clastal W: (http://www.ebi.ac.uk/Tools/clustalw2/index.html).
На втором этапе короткие матрицы комбинируют в группы по 3 и более с участками перекрывания на C- и N-концах пептидов не более 2-х аминокислот. Полученные таким образом матрицы, как например, вариант, приведенный в таблице, представляют собой синтетически выведенные детерминированные аминокислотные последовательности с множеством вариантов замещения в различных позициях. Полноразмерный пептидный конструкт формируют, добавляя к C-концу матрицы, полученной на втором этапе, последовательности линкера и сигнала обнаружения.
Полученные пептидные конструкты используют для считывания, т.е. создания новых аминокислотных последовательностей специфичных пептидов. Считывание производят с помощью доступных программ компьютерного моделирования, предназначенных для работы с аминокислотными последовательностями пептидов и белков, как например, CAVER (Caver Soft s.r.o., Czech Republic), ExPaSy и т.д. Для этого в буфер программы помещают, во-первых, последовательности всех экспериментально отобранных пептидов, используемых для конструирования матриц, во-вторых, последовательность полноразмерного пептидного конструкта и далее выполняют компьютерное моделирование введенных данных, а именно, программное обеспечение создает последовательности новых пептидов и представляет собой получение вариантов аминокислотных последовательностей путем случайного подбора аминокислот по всем позициям в матрице. При этом, как минимум, один из считанных пептидов должен содержать оригинальную последовательность пептида, отобранного на этапе селекции, а остальные последовательности пептидов должны представлять собой максимально различающиеся комбинации аминокислотных звеньев. Кроме того, считывание новых специфичных пептидов производят блоками, включающими как минимум 2 триплета (6 пептидов).
Пример 10. Химический синтез новых пептидов
Синтез пептидов, специфически распознающих определенный тип клеток, осуществляют на твердофазном носителе с участием аппарата для автоматизации химического синтеза пептидов, например РЕ Biosystems Pioneer® (Foster City, CA, U.S.A.), используя термостатичную колонку, наполненную 2-хлоротритил хлоридным носителем (замещение 0,27 ммоль/г носителя глицином), поддерживая постоянную температуру 50°C. Присоединение первой аминокислоты с C-конца пептида осуществляют согласно технической документации аппарата для автоматизации химического синтеза. Присоединение каждой последующей аминокислоты производят, используя четырехкратный избыток смеси флуорил-9-илметоксикарбонил аминокислоты, ТБТУ (О-(Бензотриазол-1-ил)-N,N,N′,N′-тетраметилурониум тетрафлуороборат) и диизопропилэтиламин (в соотношении 1:1:1,7, по объему) в растворе, предотвращающем агрегацию (диметилформамид / N-метилпирролидон (3:1, по объему), 1% Тритон Х-100 и 1М этиленкарбонат). Цикл присоединения активированных аминокислот повторяют дважды на тех участках пептидной молекулы, для которых синтез является сложным из-за комплексной вторичной структуры цепи, что предварительно выявляют с помощью пакета программ, например, Peptide Companion (CoshiSoft, AZ, U.S.A.).
Процесс синтеза периодически останавливают и отбирают пробу для оценки качества реакции с помощью масс-спектрометрии с ионизирующим распылением, например (ESI)-MS, (Perkin Elmer/Sciex API I, PE Biosystems, Foster City, CA, U.S.A.). Полученные пептиды отщепляют от носителя и деактивируют, используя смесь из трифторуксусной кислоты, воды, тиоанизола, фенола, этандитиола и 3-изопропилсилана (в соотношении 82,5:5:5:2,5:2,5:2,5, по объему). Неочищенные пептиды восстанавливают в течение 60 минут, при температуре 60°C в 10-кратном избытке раствора трис-(2-карбоксиэтил)-фосфин гидрохлорида, приготовленном в буферном растворе, содержащем 6М гунидинхлорида и 0,1М цитрата натрия (pH 3,0).
Затем пептиды немедленно опресняют посредством жидкостной хроматографии на C18 колонке, например, Waters Delta-Pak® C18 (15 µм, 300 Å, 25 мм ×100 мм) в растворителях, насыщенных молекулярным азотом, а сопутствующее очистке окисление пептидных молекул контролируют.
Концентрацию пептидов после очистки предварительно оценивают по поглощению молекул триптофана и тирозина на длине волны 280 нм, а точные значения вычисляют с помощью теста на окисление протеинов бихинольными соединениями.
После считывания с матриц и осуществления химического синтеза последовательностей новых пептидов, получили, например, новые пептиды, специфически распознающие сосудистые гладкомышечные клетки человека - Р5 N′-Asn-Ser-Trp-Pro-Leu-Ser-Arg-Gln-Arg-Leu-Leu-Gln-Leu-His-Pro-Ser-Leu-Leu-C′, P6 N′-His-Ala-Tyr-Lys-Ala-Pro-His-Ser-Pro-Ala-Ile-Pro-Leu-His-Pro-Arg-Pro-Gly-C′, P7 N′-(Arg)2-Tyr-Pro-Leu-Pro-Arg-Pro-Arg-(Leu)2-(Pro)2-Arg-Pro-Arg-(Pro)2-C′, P8 N′-His-Ser-Trp-Lys-Leu-Pro-His-Pro-Arg-(Leu)2-Ser-Pro-His-Pro-(Ser)2-Pro-Gly-C′, P9 N′-Asn-Ser-Tyr-His-Ile-Ser-His-Ser-Arg-Ala-Val-Tyr-Pro-His-Arg-(Leu)2-C′.
Пример 11. Подтверждение специфичности новых пептидов
Подтверждение специфичного распознавания гладкомышечных клеток новыми синтезированными пептидами проводят, например, методом иммуноферментного анализа, описанным в примере 7, используя вместо первого антитела не фаговую частицу, а полученный пептид.
При этом 40 мкг пептида инкубируют с клетками в течение 40 минут при температуре 37°C, после чего клетки фиксируют, помещая их в 100%-ный раствор метилового спирта на 10 минут при температуре -20°C. Затем клетки промывают буфером HBSS, содержащим 0,1% сапонина и 1,0% сухого молока (по массе), и инкубируют в течение 10 минут с раствором второго антитела.
Специфичность распознавания гладкомышечных клеток новыми пептидами выражают в относительных единицах и сравнивают ее со специфичностью любых фаговых частиц, выбранных случайным образом из множества библиотек, описанных в примере 4, до проведения селекции.
На основании экспериментальных данных при проведении иммуноферментного анализа составлен график (см. фиг.5) зависимости специфичности новых пептидов (Р5-Р9), в отношении сосудистых гладкомышечных клеток человека (обозначены как SMCp). Специфичность связывания пептидов выражена в условных единицах - УЕ. Для сравнения в этом графике используют сосудистые эндотелиальные клетки человека (ЕСр) и клетки опухоли (HeLa). При этом специфичность новых пептидов оценивается по отношению к исходной библиотеке (ИБ), сконструированной в заявляемом способе, и одним из пептидов (5Е5) прототипа, полученных в работе Ingrid N. Michon et al. ″Targeting of peptides to restenotic vascular smooth muscle cells using phage display in vitro and in vivo″ в журнале ″Biochimica et Biophysica Acta″ вып. №1591, 2002 г, стр.87-97.
На основании графика на фиг.5 видно, что специфичность новых пептидов (Р5-Р9) значительно превышает специфичность пептида из прототипа (5Е5), а также показывает высокую степень селективности в отношении сосудистых гладкомышечных клеток человека по сравнению с сосудистыми эндотелиальными клетками человека и клетками опухоли HeLa. При этом сконструированная библиотека по заявляемому способу не выявляет специфичности ни к одному типу клеток, тем самым подтверждая высокую степень эффективности считывания с матриц последовательностей новых пептидов.

Изобретение относится к области биохимии, в частности к способу получения пептидов, специфично распознающих клетки определенных типов. Указанный способ предполагает конструирование множества библиотек случайных пептидов на основе олигонуклеотидных фрагментов, кодирующих их, путем фрагментации суммарной РНК клеток определенного типа и клеток других типов, которые могут быть вовлечены в патологический процесс и оказывать влияние на его диагностику и терапию. При этом клонируют указанные фрагменты в правильной ориентации в векторе на основе бактериофага. Затем выполняют скрининг библиотек с использованием комбинаций клеток определенного типа и клеток других типов с получением групп пептидов, специфично распознающих клетки выбранного типа в составе фаговых частиц. Из полученных пептидов выделяют и тестируют индивидуальные пептиды в составе фаговых частиц для подтверждения их специфичности. Затем для получения пептидов в чистом виде создают матрицы на основе аминокислотных последовательностей пептидов в составе фаговых частиц, с которых затем считывают последовательности новых пептидов и синтезируют их химическим путем, с последующим подтверждением специфичного распознавания клеток определенного типа. Изобретение позволяет получать высокоспецифичные и высокоселективные пептиды, распознающие клетки определенного типа. 5 ил., 1 табл., 11 пр.
Способ получения пептидов, специфично распознающих клетки определенных типов и предназначенных для терапевтических целей, при котором предусматривают конструирование фаговой библиотеки случайных пептидов на основе кодирующих их олигонуклеотидных фрагментов, ее скрининг для получения пептидов, связывающихся с клетками определенного типа, и подтверждают специфичность отобранных пептидов, отличающийся тем, что:
- олигонуклеотидные фрагменты, кодирующие случайные пептиды, получают реакцией обратной транскрипции с использованием случайных праймеров и суммарных РНК клеток определенного типа и клеток других типов, которые могут быть вовлечены в патологический процесс или способны оказывать влияние на его диагностику и терапию,
- указанные олигонуклеотидные фрагменты встраивают в бактериофаговые векторы в правильной ориентации и используют для создания фаговых библиотек случайных пептидов для всех типов клеток, использованных для выделения суммарных РНК,
- скрининг полученных фаговых библиотек случайных пептидов проводят в два этапа, на первом из которых отбирают пептиды, способные связываться с клетками определенного типа, а на втором - из отобранных на первом этапе пептидов отбирают пептиды, не связывающиеся с клетками других использованных типов,
- подтверждают специфичность пептидов в составе фаговых частиц к клеткам определенного типа, используя комбинации иммуноферментного анализа, полимеразной цепной реакции, анализа на связывание и теста на стабильность пептида и его способность интернализироваться в составе фаговой частицы,
- определяют первичные последовательности олигонуклеотидных фрагментов, кодирующих пептиды в составе отобранных фаговых частиц, и транслируют их в аминокислотные последовательности с дальнейшим проведением анализа полученных последовательностей,
- конструируют матрицы из аминокислотных последовательностей пептидов, селективно выбранных и распознающих клетки определенного типа, к которым присоединяют с помощью линкера сигнал обнаружения, при этом сигнал обнаружения используют как для одного вида, так и для различных видов новых пептидов,
- считывают последовательности новых пептидов с матриц, синтезируют новые пептиды химическим путем с последующим подтверждением специфичности полученных пептидов к клеткам определенного типа.
| MICHON I.N | |||
| et al., Targeting of Peptides to Restenotic Vascular Smooth Muscle Cells Using Phage Display In Vitro and In Vivo, Biochim Biophys | |||
| Acta, 2002, vol | |||
| Раскладочно-вытяжной оческовый стан | 1925 |
|
SU1591A1 |
| Торфодобывающая машина с вращающимся измельчающим орудием | 1922 |
|
SU87A1 |

