Изобретение относится к полипептид-экспрессирующим системам, требующим расщепления продукта-предшественника, а также к протеазам, используемым в этих системах. Кроме того, настоящее изобретение относится к новому полипептиду, способному к восстановлению дихлороиндолфенола и окисленного глутатиона, к ДНК, кодирующей указанный новый полипептид, к векторам, содержащим указанную ДНК, к клеткам-хозяевам, трансформированным указанными векторами, и к фармацевтическим композициям, содержащим указанный полипептид. Настоящее изобретение также относится к моноклональным антителам против указанного полипептида и к способу выделения и очистки этого полипептида с использованием указанных антител.
Предшествующий уровень техники
Потивирусы представляют собой группу вирусов, которые имеют геном, состоящий приблизительно из 10 000 оснований одноцепочечной РНК, и которые инфицируют растения, например растения семейства Пасленовых (Solancede). Геном потивируса отличается тем, что он имеет исключительно длинную открытую рамку считывания (ORF) (Dougherty, W.G. & Hiebert, E. (1980) Virology 101, 466-474, Allison, R. et. al. (1986), Virology 154: 9-20). Для экспрессии отдельных белков, кодируемых внутри ORF, транслированный полипротеин переваривается двумя типами протеаз, которые также кодируются внутри (Dougherty, W. G & Carrington, R. et. al., (1988), Ann. Rev. Phutopath 26: 23-143).
Вирус табачной гравировки (TEV) является членом семейства потивирусов и продуцирует ядерные включения, которые в инфицированных клетках могут быть окрашены трипановым синим. Эти ядерные включения состоят, по-видимому, из двух видов белка, один из которых, как было установлено, является вирусной протеазой, обозначенной "ядерным включением a" или Nla (J. Virol., 61 2540-2548 (1987)).
Протеазы потивирусов, именуемые ядерным включением a, распознают и расщепляют пептидную последовательность, которая включает в себя один из Gln-Cly, Gln-Ser, и Cln-Ala, и которая является, очевидно, гексамерной последовательностью, находящейся у C-конца рассматриваемого Nla внутри полипротеина. Это расщепление происходит между двумя остатками, образующими вышеуказанные димеры.
Были определены полные геномные последовательности TEV и вируса стеблевой пятнистости табака (TYMV), другого члена семейства потивирусов, и в результате гомологических исследований этих последовательностей было установлено, что Nla этих вирусов локализуются в их соответствующих геномах. (Virology 154, 9-20 (1986), Nucleic Acids Res., 14: 5417-5430 (1986)).
Вирус желтой прожилковой мозаики клевера, или CYVV, также является потивирусом. До настоящего времени был секвенирован только лишь ген, расположенный у 3'-конца генома CYVV, вместе с белком оболочки, который этот ген кодирует (Uyeda, 1. et al., (1991), Intervirol 32: 234-245). Однако до сих пор структура Nla-области этого генома остается невыявленной, и, кроме того, само включение Nla не было выделено.
Продуцирование экзогенных белков с помощью экспрессирующих систем может быть осуществлено непосредственно с использованием хорошо известной техники. Однако существует множество полипептидов, которые не могут быть легко экспрессированы в экзогенных системах. Проблема заключается в том, что эти полипептиды не могут быть синтезированы в больших количествах, и простое введение регуляторного элемента выше указанного гена не приводит к желаемому результату. Альтернативно, может оказаться так, что посттранскрипционный процессинг, необходимый для образования зрелой формы белка, либо вовсе не происходит, либо происходит неправильно.
Так, например, трансляция многих эукариотических полипептидов начинается с N-концевого метионина, который затем делетируется с образованием зрелой формы. В прокариотах такого процессинга не происходит, а поэтому необходимо было найти альтернативный способ достижения экспрессии. Один из таких способов предусматривает сшивание нужного экзогенного белка, например, с мальтозу-связывающим белком или глутатион-S-трансферазов, а затем очистку синтезированного гибридного белка с последующим его расщеплением протеазой, такой как фактор Xa, энтерокиназа, или тромбин. Основной недостаток этого трудоемкого способа заключается в том, что он требует две стадии очистки, что приводит к значительной потере конечного продукта.
В патенте США N 5162601 раскрывается использование TEV-протеазы в синтезе полипротеина, содержащего между белками линкерные последовательности, что является желательным для синтеза полипептида, такого как чел. tPA. Однако в этом патенте описывается лишь клонирование мультигена, кодирующего указанный полипротеин, в хозяйскую клетку, но не описываются ни экспрессия, ни очистка протеолитически расщепленного конечного продукта.
Кислород для метаболической энергии, обычно, поставляется окисляющими агентами, присутствующими в окружающей клетку среде. Активированной формой, в виде которой обычно потребляется кислород, являются свободные радикалы, такие как супероксид (O2 -), пероксид (H2O2), или гидроксильный радикал (OH-), при этом все указанные радикалы, после их использования, восстанавливаются с образованием воды (H2O). Газообразный кислород, сам по себе, является очень хорошим окислителем, однако используемый в настоящем описании термин "активированный кислород" относится к кислороду или кислородсодержащим молекулам, обладающим более высоким окисляющим потенциалом, чем атмосферный кислород. Наиболее эффективной формой активированного кислорода является свободный радикал, который представляет собой молекулу или атом, имеющие один или несколько неспаренных электронов.
Свободные радикалы, обычно, являются нестабильными, и без соответствующего контроля они могут денатурировать липиды, белки и нуклеиновые кислоты. А поэтому, хотя активированный кислород необходим для жизнеобеспечения, он может представлять значительную опасность для здоровья человека, в результате чего его количество в организме должно быть строго регулируемым. Благодаря своей высокой реакционной способности активированный кислород, даже в ничтожно малых количествах, может вызывать определенные нарушения в организме. Поэтому высокоактивные формы кислорода способны даже убить живую клетку, если только эта клетка не обладает механизмами защиты от указанного поврежденного действия кислорода.
В окружающей клетку среде локализация, количество и время генерации активированного кислорода должно быть тщательно сбалансировано в отношении способности клетки нейтрализовать его повреждающее действие. Эта способность, обычно, обеспечивается механизмами защиты клетки, использующей в этих целях свои собственные антиоксиданты или антиокислительные ферменты. В контексте настоящего описания термин "антиоксидант" используется как общее название для всех природных соединений, обладающих способностью предупреждать или ингибировать самоокисление, например, липидов. Термин "антиокислительный фермент" означает в основном фермент, который катализирует реакцию элиминирования активированного кислорода, а термин "антиокисляющее действие" означает, соответственно, действие, направленное на элиминирование указанного активированного кислорода.
Избыточное количество активированного кислорода в организме может индуцировать ряд патологических явлений в этом организме, сходных с явлениями, вызванными стрессом, принятием лекарственного средства, курением, хирургической операцией, трансплантацией какого-либо органа, либо явлениями, вызванными ишемией, или даже инфарктом головного мозга или миокарда. Под большими количествами кислорода подразумеваются количества, превышающие тот уровень кислорода, который регулирующие системы организма способны элиминировать, а поэтому указанные избыточные количества кислорода обладают токсическим действием на организм, вызывая серьезные нарушения в жизнедеятельности его клеток. Это токсическое действие, названное иначе окислительным стрессом, является ответственным за возникновение многих патологических состояний организма.
Так, например, считается, что одна из причин возникновения атеросклероза является продуцирование липопротеинов низкой плотности, которые были окислены активированным кислородом (Steinberg, D. (1983), Arterioslerosis 3, 283-301). Считается также, что окислительный стресс имеет непосредственное отношение к механизмам возникновения метаболических нарушений и васкулярных осложнений диабета (Kondo, M. ed., "Approaches from Modern Medicine (4) Free Radicals", Medical View Pub., pp. 138-146).
Активированный кислород также является причиной возникновения других патологических состояний и нарушений, например, таких, как ишемические нарушения (реперфузионные повреждения, ишемическая болезнь сердца, ишемия головного мозга, ишемический энтерит и т.п.), отеки, сосудистая сверхпроницаемость, воспалительные заболевания, повреждения слизистой желудка, острый панкреатит, болезнь Крока, язвенный колит, заболевания печени, болезнь Paraquat, эмфизема легких, химический карцерогенез, метастазы рака, респираторный дистресс-синдром у взрослых, диссеминированное внутрисосудистое свертывание крови (ДВС-синдром), катаракта, ретролетальная фиброплазия, аутоиммунные заболевания, порфиремия, гемолиз, эритробластическая (среднеземноморская) анемия, болезнь Паркинсона, болезнь Альцгеймера, эпилепсия, нарушения, вызванные ультрафиолетовым излучением, нарушения, вызванные радиоактивным излучением (лучевая болезнь), отморожения и ожоги.
Клетки имеют несколько внутренних и внешних механизмов защиты, направленных исключительно на элиминирование активированного кислорода, генерируемого внутри организма.
Известно, что внутриклеточные средства защиты, а именно антиоксиданты и антиокислительные ферменты, примеры которых приведены ниже, утилизируют и элиминируют активированный кислород. Так, например, в пероксисомах клетки присутствует каталаза, которая восстанавливает и удаляет пероксид водорода. Глутатион-пероксидаза, присутствующая в цитоплазме и митохондриях, катализирует восстановление и детоксикацию пероксида водорода и пероксидов липида в присутствии восстановленного глутатиона. Трансферрин, ферритин и лактоферрин, например, ингибируют генерирование активированного кислорода путем стабилизации ионов железа, тогда как церулоплазмин осуществляет аналогичную функцию в отношении ионов меди. Кроме того, в цитоплазме и митохондриях имеется супероксиддисмутаза, катализирующая восстановление супероксидов с образованием перекиси водорода, который затем элиминируется каталазой. Помимо вышеуказанных ферментов, способностью к восстановлению и элиминированию активированного кислорода обладают также витамины C и E, восстановленный глутатион и другие низкомолекулярные соединения.
С другой стороны, такие агенты, как внеклеточная супероксиддисмутаза, внеклеточная глутатионпероксидаза, и восстановленный глутатион присутствуют во внеклеточном пространстве, где они имеют те же самые функции, что и их внутриклеточные аналоги, описанные выше. Однако по сравнению с ситуацией, имеющей место внутри клетки, во внеклеточной среде присутствует гораздо меньшее число различных видов антиоксидантов и антиокислительных ферментов, и, кроме того, лишь немногие из них обладают антиокислительным действием.
Восстановленный глутатион, формула которого представлена ниже, играет главную роль в поддержании восстановительного состояния как вне, так и внутри клетки. Впервые глутатион был обнаружен в дрожжах в 1888 году de-Rey-Pailhade, и свое название глутатион получил после его выделения как соединения в 1921 году Хопкинсом.
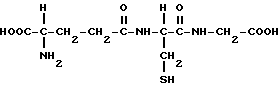
Глутатион состоит из трех аминокислот: глутаминовой кислоты, цистеина и глицина. Тиоловые группы двух молекул глутатиона могут быть окислены с образованием дисульфидной связи в присутствии активированного кислорода, что приводит к восстановлению указанного активированного кислорода.
Глутатион продуцируется главным образом в печени, после чего он попадает в кровоток и циркулирует в организме. В нормальном организме глутатион, почти полностью, присутствует в восстановленной форме. В случае увеличения уровней окисленной формы происходит регенерация восстановленной формы благодаря действию глутатионредуктазы в присутствии никотинамидаденин-динуклеотидфосфата (НАДФ). Таким образом, восстановленный глутатион защищает клеточную мембрану от повреждающего действия активированного кислорода путем восстановления указанного активированного кислорода и свободных радикалов. Благодаря своим антиокислительным свойствам восстановленный глутатион обладает также противорадиационным действием и может быть использован как терапевтическое средство против катаракты. Кроме того, недавно было установлено, что у пациентов, страдающих СПИДом, системные уровни восстановленного глутатиона понижены, что указывает на исключительно важную роль восстановленного глутатиона в организме человека. Однако в аномальных условиях количество активированного кислорода может быть настолько велико, что фактически весь глутатион присутствует в окисленном состоянии, и активированный кислород удаляется гораздо медленней, чем это обычно происходит в нормальных условиях.
Вторым примером соединения, обладающего, благодаря своим окислительным свойствам, различными физиологическими функциями, является тиоредоксин, присутствующий как во внутриклеточном, так и во внеклеточном пространстве. Тиоредоксин человека (известный также, как фактор T-клеточного лейкоза взрослых, ADF) был клонирован как фактор, способный индуцировать рецепторы интерлейкина-2 (1L-2R) в клеточных линиях T-клеточного лейкоза взрослых. Этот фактор представляет собой тиол-зависимую редуктазу, имеющую в своем активном центре два цистеиновых остатка и обладающую способностью восстанавливать активированный кислород и свободные радикалы.
Помимо индуцирования 1L-2-рецепторов, тиоредоксин человека также обладает действием, стимулирующим рост клеток B-клеточного штамма 3B6, инфицированного вирусом Эпштейна - Барра (EBV), защитным действием против фактора некроза опухоли (TNF), происходящего от моноцитарной клеточной линии U937, и защитным действием против повреждения васкулярных эндотелиальных клеток нейтрофилами. Кроме того, благодаря своей восстановительной активности внутри клетки, тиоредоксин человека воздействует на транскрипционные факторы NFkB, JUN и FOS, что способствует стимулированию ДНК-связывающей активности и тем самым повышению транскрипционной активности. В настоящее время разрабатывается способ использования тиоредоксина человека в качестве защитного средства от радиационных поражений, а также в качестве терапевтического средства для лечения реперфузионных повреждений, ревматоидного артрита и воспалительных заболеваний, одним словом, таких повреждений и заболеваний, которые могут быть устранены или уменьшены посредством восстанавливающей активности указанного тиоредоксина человека.
Как указывалось выше, для нормальной физиологической деятельности организма крайне важно, чтобы внутриклеточное и внеклеточное пространство поддерживалось в восстановительных условиях, что может быть достигнуто путем элиминирования активированного кислорода и свободных радикалов. Очевидно, что во внутриклеточном и внеклеточном пространстве присутствует много пока еще неизвестных антиоксидантов и антиокислительных ферментов, выполняющих функцию удаления активированного кислорода и свободных радикалов. Поэтому было бы крайне важно обнаружить эти восстановительные агенты, способные регенерировать, например, восстановленный глутатион. Такие вещества модно было использовать для лечения различных патологических состояний, например заболеваний и нарушений, описанных выше.
Краткое описание изобретения.
Первой целью настоящего изобретения является получение новой протеазы; нуклеотидной последовательности, кодирующей эту протеазу; вектора, содержащего ДНК-последовательность, кодирующую указанную протеазу, и клетки-хозяина, трансформированной указанным вектором.
Второй целью настоящего изобретения является получение ДНК, кодирующей нужный белок, а также ДНК, кодирующей новую протеазу, расположенную выше от указанного белка, причем нуклеотидная последовательность, находящаяся между указанными двумя ДНК-последовательностями, кроме того, кодирует пептид, расщепляемый указанной протеазой, а все указанные последовательности находятся в одной и той же открытой рамке считывания. Кроме того, целью настоящего изобретения является получение белка, кодируемого указанной ДНК, а также вектора, содержащего указанную ДНК, и экспрессирующей системы, включающей в себя указанный вектор, причем указанный вектор обладает способностью к автономной репликации в соответствующей клетке-хозяине, например, в клетке, содержащей нуклеотидную последовательность, необходимую для осуществления автономной репликации.
В альтернативном варианте, первой целью настоящего изобретения является получение нуклеотидной последовательности, кодирующей новый полипептид, обладающий восстанавливающей активностью in vivo. Кроме того, целью настоящего изобретения является также получение такой ДНК-последовательности, которая кодирует пептид, способный восстанавливать дихлороиндофенол и окисленный глутатион.
Другой целью настоящего изобретения является получение рекомбинантного вектора, содержащего вышеуказанную ДНК и обладающего способностью к автономной репликации в клетке-хозяине, а именно в клетке, включающей в себя последовательность оснований, обеспечивающую указанную автономную репликацию.
Еще одной целью настоящего изобретения является продуцирование микроорганизма-хозяина, трансформированного с использованием вышеуказанного рекомбинантного вектора. Кроме того, целью настоящего изобретения является получение вышеупомянутого пептида в качестве продукта экспрессии трансформированной клетки-хозяина, а также получение моноклональных антител против этого пептида.
В данной работе мы идентифицировали и клонировали новую CYVV-протеазу (Nla), и при этом, неожиданно обнаружили, что CYVV-Nla может быть использована как часть гибридного белка, способного экспрессироваться в таком хозяине, как E.coli, что позволяет продуцировать большие количества указанного гибридного белка, который, кроме того, обладает способностью к саморасщеплению с образованием нужного экзогенного продукта. При этом CYVV-Nla-ген может стабильно сохраняться и экспрессироваться в Esherichia coli, а экспрессированный Nla-продукт сохраняет свою активность как специфическую протеазную активность даже в том случае, если указанный белок составляет лишь часть от данного гибридного белка.
Кроме того, мы также обнаружили ДНК-последовательность, кодирующую новый полипептид, обладающий способностью восстанавливать дихлороиндофенол (известный также, как дихлорофенолиндофенол, 2,6-дихлороиндофенол, или 2,6-дихлоро-4-(4-гидроксифенил)имино-2,5-циклогексадиен-1-он), и окисленный глутатион, причем указанный полипептид может быть получен в больших количествах путем использования техники генной инженерии. Этот полипептид может быть с особой эффективностью использован для лечения заболеваний, вызванных или опосредованных окислительным стрессом, или любых других заболеваний, вызванных активированным кислородом, например, таких, как атеросклероз, диабет, и ишемические состояния (включая реперфузионные нарушения, ишемическая болезнь сердца, цереброишемия и ишемический энтерит).
Так, например, в первом аспекте первого варианта своего осуществления, настоящее изобретение относится к полинуклеотидной последовательности, которая, в направлении 5′_→ 3′ и в той же самой открытой рамке считывания, включает в себя:
а) последовательность, кодирующую белок "ядерного включения а" вируса желтой прижилковой мозаики клевера, либо его мутант или вариант, обладающий такой же протеолитической специфичностью, что и белок "ядерного включения а" вируса желтой прижилковой мозаики клевера, b) последовательность, кодирующую пептид, распознаваемый и расщепляемый указанным белком "ядерного включения а" вируса желтой прижилковой мозаики клевера, либо его мутантом или вариантом, с) по крайней мере одну последовательность, кодирующую полипептид.
Настоящее изобретение также относится к последовательности, кодирующей белок "ядерного включения а" вируса желтой прижилковой мозаики клевера, либо его мутант или вариант, обладающий такой же протеолитической специфичностью, что и указанный белок "ядерного включения а" вируса желтой прижилковой мозаики клевера.
Кроме того, настоящее изобретение относится к вектору, в частности к экспрессирующему вектору, содержащему последовательность, определенную выше.
Настоящее изобретение также относится к хозяину, трансформированному вектором, определенным выше; к экспрессирующей системе, включающей в себя указанного хозяина и указанный экспрессирующий вектор, а также к полипептиду, продуцированному указанной экспрессирующей системой.
В первом аспекте альтернативного варианта своего осуществления, настоящее изобретение относится к полинуклеотидной последовательности, кодирующей полипептид, имеющий аминокислотную последовательность из аминокислот 1-526 последовательности ID N 12, либо кодирующей мутант или вариант указанного полипептида при условии, что полипептид, кодированный указанной полинуклеотидной последовательностью, обладает способностью восстанавливать дихлороиндофенол и окисленный глутатион.
Кроме того, настоящее изобретение относится к вектору, в частности к экспрессирующему вектору, содержащему последовательность, определенную выше.
Настоящее изобретение также относится к хозяину, трансформированному вектором, определенным выше, к экспрессирующей системе, включающей в себя указанного хозяина и указанный экспрессирующий вектор, а также к полипептиду, продуцированному указанной экспрессирующей системой.
Кроме того, настоящее изобретение относится к вышеуказанному полипептиду, который может быть использован в терапевтических целях, к использованию указанного полипептида для лечения и профилактики заболеваний или состояний, вызванных или опосредованных окислительным стрессом, или любых других заболеваний, вызванных активированным кислородом, а также к фармацевтической композиции, содержащей указанный полипептид.
Помимо вышеуказанного, настоящее изобретение относится к моноклональному антителу и его эквивалентам, направленным против указанного полипептида, к способу продуцирования этого антитела, а также к способу очистки полипептида с использованием указанного антитела.
Краткое описание фиг. 1 - 7, 8а, 8b, 9 - 13
Настоящее изобретение иллюстрируется фиг. 1 - 7, 8а, 8b, 9 - 13, описание которых приводится ниже.
На фиг. 1 показана рестрикционная карта кДНК Nla-области, выделенной из CYVV-кДНК.
На фиг. 2 проиллюстрировано конструирование плазмиды pKN15', содержащей 5'-область "ядерного включения а" (Nla).
На фиг. 3 проиллюстрировано конструирование плазмиды pKN151L, содержащей часть 1L-11-гена и 5'-область Nla.
На фиг. 4 показаны праймеры, которые были использованы для получения 5' 1L-ДНК-фрагмента и C1N3-ДНК-фрагмента, и в который 3'-конец Nla-гена был сшит с 5'-концом 1L-11-гена.
На фиг. 5 проиллюстрировано лигирование C1N3-ДНК-фрагмента и 5'-1L-ДНК фрагмента с помощью полимеразно-цепной реакции.
На фиг. 6 проиллюстрировано конструирование плазмиды pK SUN 9.
На фиг. 7 показана рестрикционная карта плазмиды PUCKM31-7.
На фиг. 8a и b проиллюстрировано сравнение нуклеотидных последовательностей 3'-концов в pUCKM31-7 и pcD-31.
На фиг. 9 показана диаграмма конструирования pSR α 31-7.
На фиг. 10 показана схема введения последовательности, кодирующей гистидиновый гексамер, в плазмиду pUCKM31-7.
На фиг. 11 показана схема конструирования плазмиды pMAL31-7.
На фиг. 12 проиллюстрирован анализ дихлорфенол-индофенол-восстанавливающей активности.
На фиг. 13 проиллюстрировано определение активности, направленной на восстановление окисленного глутатиона.
Подробное описание изобретения
Нижеприведенное описание настоящего изобретения относится главным образом к первому варианту его осуществления, однако это описание может быть также отнесено и ко второму его варианту, за исключением тех случаев, когда совершенно очевидно, что данное обсуждение не может быть применено к указанному второму варианту.
При этом следует отметить, что предпочтительной формой полинуклеотидной последовательности настоящего изобретения является ДНК, а поэтому дальнейшее описание будет относиться в основном к ДНК. Однако там, где это необходимо, в описании имеются также указания на РНК. РНК не является предпочтительной формой нуклеотидной последовательности для настоящего изобретения, поскольку ее использование практически ограничено. Например, мРНК может быть экспрессирована в социтах Xenopus (шпорцевой лягушки) или в системе лизатов пшеничных проростков, однако ни одна из этих экспрессирующих систем не способна экономически эффективно продуцировать большие количества гибридного белка.
Используемый в настоящем описании термин "пептид" означает любую молекулу, содержащую 2 или более аминокислот, связанных между собой пептидной связью. Этот термин может означать олигопептид, полипептид или белок. Термин "гибридный белок" относится к любому отдельно взятому полипептиду, полученному путем объединения двух или более различных пептидных последовательностей.
Следует также отметить, что предпочтительной последовательностью настоящего изобретения является двухцепочечная последовательность; при этом в настоящем изобретении также рассматривается антисмысловая последовательность, соответствующая последовательности настоящего изобретения. Двухцепочечная (дц) последовательность настоящего изобретения может иметь один или два "липких" конца, и в этом случае, необязательно, чтобы антисмысловая и смысловая нити точно соответствовали друг другу.
При этом имеется в виду, что белок, кодируемый последовательностью (а), определенной выше, расщепляет пептид, кодируемый последовательностью (b), определенной выше, в результате чего высвобождается полипептид(ы), кодируемый последовательностью (с), определенной выше, при условии, что указанная последовательность настоящего изобретения экспрессируется в подходящей экспрессирующей системе.
Расщепление гибридного белка может иметь место в любое время после трансляции последовательностей (а) и (b), определенных выше. В этой связи следует заметить, что полипептид, кодированный последовательностью (c), определенной выше, необязательно должен быть полностью транслирован перед расщеплением. На практике, однако, было обнаружено, что по крайней мере некоторая часть, а иногда и большая часть гибридного белка является полностью транслированной перед его расщеплением.
Если последовательность (c), определенная выше, кодирует более чем один полипептид, то в этом случае необходимо, чтобы, кроме того, кодировались расщепляемые последовательности, расположенные между каждыми из кодированных полипептидов, за исключением лишь тех случаев, когда требуется получить нерасщепленный гибрид из множества полипептидов.
Если последовательность (c), определенная выше, кодирует более чем один полипептид, то эта последовательность может быть восприимчивой к аттенюации в условиях транскрипции. При аттенюации транскрипция мРНК-последовательности настоящего изобретения прекращается до того, как будет прочитана вся мРНК, в результате чего полипептиды, кодируемые ближе к 3'-концу последовательности, будут продуцироваться в меньших количествах, чем полипептиды, кодируемые ближе к 5'-концу. Однако если не кодируется несколько полипептидов и/или, если эти полипептиды не являются очень длинными, то каких-либо проблем, связанных с аттенюацией, не возникает.
В основном предпочтительно, чтобы последовательность (c), определенная выше, кодировала только один полипептид, за исключением тех случаев, когда необходимо получить несколько пептидов для их совместного использования, либо, когда необходимо получить гибрид из нескольких полипептидов. В противном случае, после расщепления потребуется выделять каждый отдельный полипептид, а эта операция может оказаться весьма трудоемкой, и, кроме того, процедуры очистки могут привести к значительным потерям конечного продукта.
Однако если последовательность (c), определенная выше, кодирует более чем один полипептид, и если между каждыми из этих кодируемых полипептидов имеется расщепляемая последовательность, то совсем необязательно, чтобы эти расщепляемые последовательности распознавались CYVV-Nla. Все, что требуется в этом случае, это, чтобы расщепляемая последовательность, находящаяся между последовательностью (а) и 5'-концом последовательности (b), распознавалась протеазой, кодируемой последовательностью (a). Однако если необходимо или допустимо, чтобы гибридный белок подвергался саморасщеплению с образованием нескольких полипептидов, то любая другая из расщепляемых последовательностей может быть распознаваемой протеазой, кодированной последовательностью (a). Причем указанные другие последовательности могут быть выбраны таким образом, чтобы они расщеплялись другими путями. Например, гибридный белок будет расщепляться своей Nla-частью после транскрипции с образованием в результате Nla и полипротеина, после чего Nla может быть удалена, а полипротеин может быть расщеплен, например, фактором Xa или трипсином.
Расщепляемый пептид, кодированный последовательностью (b), определенной выше (именуемой также "расщепляемой последовательностью" и "расщепляемым пептидом") может представлять собой последовательность, которая целиком или частично входит в состав любой из последовательностей, кодируемых последовательностями (a) и (c) ("Nla" или "протеаза" и "полипептид" соответственно). Так, например, N-конец расщепляемого пептида может быть также включен в C-концевую последовательность протеазы, а C-концевая часть расщепляемого пептида может быть включена в N-концевую часть полипептида. В этом случае расщепляемый пептид не может существовать независимо, и от C-конца протеазы и до N-конца полипептида должны быть сконструированы сайты рестрикции для протеазы.
Расщепляемый пептид может быть также включен лишь в часть либо протеазы, либо полипептида. В этом случае N-конец расщепляемого пептида будет включен в C-концевую часть протеазы, а N-часть полипептида будет либо непосредственно связана с расщепляемой последовательностью, либо между этим полипептидом и линкером может присутствовать один или несколько аминокислотных остатков.
Если между расщепляемой последовательностью и протеазой и/или между расщепляемой последовательностью и полипептидом присутствует один или несколько аминокислотных остатков, то число и природа этих остатков должны быть такими, чтобы эти остатки не препятствовали действию протеазы. Избыточное количество аминокислотных остатков на N-конце полипептида в основном нежелательно, поскольку эти остатки должны быть удалены для получения зрелой формы полипептида. Обычно предпочтительно, если, это возможно, и если не имеется каких-либо противопоказаний, сконструировать расщепляемый пептид таким образом, чтобы зрелая форма нужного белка образовывалась после расщепления гибридного белка. Однако вполне возможно получить белок, имеющий, например, Gly, Ser или Ala, присоединенные к N-концу, которые могут быть затем удалены, если это необходимо, с помощью соответствующей аминопептидазы.
В некоторых случаях может оказаться предпочтительным между N-концом полипептида и расщепляемой последовательностью кодировать пролин. Это позволит, например, с помощью аминопептидазы P (3.4.11.9) отщепить остатки, находящиеся перед пролином, но не после него. А затем пролиновый остаток может быть удален с помощью пролин-иминопептидазы с получением зрелого белка. Этот вариант осуществления настоящего изобретения является предпочтительным.
Последовательность (a) кодирует протеазу, способную расщеплять гибридный белок, кодируемый последовательностью настоящего изобретения. В соответствии с настоящим изобретением, такой протеазой является "ядерное включение a" (Nla) вируса желтой прижилковой мозаики клевера либо его мутант или вариант, обладающий такой же протеолитической специфичностью, что и указанная Nla-протеаза вируса желтой мозаики клевера.
Эта протеза, а именно "ядерное включение а" (Nla) вируса желтой прижилковой мозаики клевера (CYVV), кодируется нуклеотидами 10-1311 последовательности ID N 1 (см. ниже, список последовательностей), а первичная последовательность Nla представлена аминокислотами 4-437 последовательности ID N 2 (см. ниже, список последовательностей). Эти последовательности являются новыми, а поэтому так же, как и их мутанты и варианты, они входят в объем настоящего изобретения.
"Ядерное включение а" обладает протеолитической активностью, способствующей специфическому гидролизу пептидной связи между Gln-Ala, Gln-Gly или Gln-Ser в пептидном субстрате. Кроме того, было также обнаружено, что Nla может расщепляться Gln-Val. Таким образом, авторами настоящей заявки было обнаружено, что в противоположность другим протеазам семейства потивирусов, Nla CYVV (далее, если это не указывается особо, обозначение Nla будет означать Nla CYVV) может расщеплять последовательность AsnCys SerPheGlnX, где X представляет собой любой аминокислотный остаток, предпочтительно Gly, Ala, Val или Ser, а более предпочтительно Gly, Ala или Ser.
Следует также отметить, что пептиды, включающие в себя последовательность AsnCys SerPheGlnX могут быть расщеплены Nla-протеазой, особенно, если эта последовательность подвергается стерическому воздействию Nla-протеазы. Таким образом, настоящее изобретения также относится к системе, используемой для получения полипептида, где предшественник полипептида, содержащий последовательность AsnCys SerPheClnX, расщепляется Nla-протеазой. Эта система может также включать в себя любые другие стадии процессинга, осуществляемые, если это необходимо, перед, после или одновременно с расщеплением Nla-протеазой.
Хотя в основном предпочтительно использовать последовательность, кодирующую натуральную Nla (последовательность ID N 2), однако в настоящем изобретении с таким же успехом могут быть использованы мутанты или варианты Nla.
Как указывалось выше, эти мутанты или варианты должны иметь специфичность, аналогичную специфичности натуральной Nla. В соответствии с этим данный мутант или вариант должен иметь последовательность, в основном гомологичную последовательности аминокислот 4-437 в SEQ ID N2, за исключением разве что тех случаев, где, как очевидно каждому специалисту, возможно некоторые изменения, не затрагиваемые распознающую последовательность или не снижающие активность протеазы ниже допустимого уровня.
В целом, можно отметить, что активность данного белка зависит от определенных консервативных областей этой молекулы, тогда как другие области не играют такой важной роли и могут быть фактически или абсолютно лишними. В соответствии с этим, как уже указывалось выше, настоящее изобретение включает в себя любые варианты и мутанты, которые обладают специфичностью, аналогичной специфичности природной Nla-протеазы. Указанные варианты или мутанты могут включать в себя, например, делеции, инсерции, добавления, инверсии, повторы и замещения (например, один гидрофильный остаток на другой гидрофильный остаток, но не такие замещения, как сильно гидрофобный остаток на сильно гидрофильный остаток). Небольшие изменения обычно не оказывают значительного влияния на активность молекулы, если только они не приходятся на главную часть молекулы, и такие изменения могут возникать как побочные продукты генетических манипуляций, например, при конструировании лишних рестрикционных сайтов, если это необходимо.
Вообще говоря, каких-либо веских причин для изменения структуры Nla, обычно, не возникает, за исключением, может быть, отдельных совершенно очевидных случаев. Действительно, большинство мутаций или вариаций в аминокислотной последовательности либо в кодирующей последовательности возникает в результате выделения новых вариантов на природном вирусе дикого типа. И тем не менее намеренные и даже случайные модификации также должны быть включены в настоящее изобретение при условии, что они обладают необходимой специфичностью и достаточной протеолитической активностью.
Используемый в настоящем описании термин "неблагоприятное действие" означает любое воздействие на специфичность или активность протеазы, приводящие к значительному снижению ее эффективности по сравнению с природной Nla (см. выше), обусловленному уменьшением ее активности ниже необходимого уровня.
При этом могут быть осуществлены многие замещения, инсерции и т.п., если только они не оказывают неблагоприятного воздействия на активность протеазы. Как правило, серьезное неблагоприятное воздействие на активность протеазы имеет место лишь в том случае, когда 3-D (третичная) структура Nla подвергается значительной модификации.
Используемый в настоящем описании термин "мутанты" относится к делециям, добавлениям, инсерциям и замещениям аминокислотных остатков в последовательности протезы, активность которой при этом не подвергается неблагоприятному воздействию. Используемый в настоящем описании термин "варианты" относится к природным Nla CYVV, имеющим последовательность, сходную с последовательностью ID N 2, но тем не менее отличающуюся от этой последовательности в пределах, ожидаемых для данной популяции. Понятие "варианты" включает в себя параллельные варианты, и пептиды от тех видов, которые обнаруживают аналогичный тип активности и имеют родственную последовательность
При этом следует отметить, что ни Nla, ни белок, относящийся ко второму варианту осуществления настоящего изобретения, не должны полностью соответствовать последовательностям ID N 2 и 12 соответственно. Единственное, что необходимо, так это то, чтобы каждый из указанных полипептидов обладал нужной активностью, независимо от того, является ли этот полипептид какой-либо частью указанной природной последовательности, либо он является всего лишь мутантом или вариантом этой части последовательности.
Считается, что гены эукариотов, такие как ген интерферона, обычно обнаруживают полиморфизм (см. Nishi T. et al, 1985), J. Biochem 97, 153-159). Этот полиморфизм приводит к тому, что в одних случаях одна или несколько аминокислот в полипептиде являются замещенными, а в других случаях такого замещения не наблюдается, несмотря на замещение в нуклеотидной последовательности.
В соответствии с этим следует отметить, что полинуклеотидная кодирующая последовательность может быть также модифицирована любым способом при условии, что такая модификация не будет оказывать неблагоприятного воздействия на протеазную активность. Например, могут быть осуществлены точечные мутации и другие изменения с добавлением или делецией рестрикционных сайтов, например, для осуществления тех или иных генетических манипуляций/экспрессий или для более эффективного проведения тех или иных модификаций Nla-молекулы.
Термин "мутант" или "вариант", используемые в настоящем описании, относятся также к полинуклеотидной последовательности, которая может быть сконструирована с соответствующими необходимыми изменениями. При этом следует отметить, что хотя мутации или вариации пептидной последовательности всегда отражены в кодирующей нуклеотидной последовательности, однако обратное утверждение необязательно справедливо. Поэтому может оказаться так, что значительные изменения в нуклеотидной последовательности (см. ниже дискуссию о вырожденности генетического кода) не будут оказывать какого-либо влияния на пептидную последовательность. В соответствии с этим указанные мутанты и варианты нуклеотидной последовательности также входят в объем настоящего изобретения.
Так, например, было установлено, что белок, полученный путем замены цистеинового кодона в гене интерлейкина (ID-2) сериновым кодом, способен тем не менее продуцировать 1L-2активность (Wang A. et al., (1984), Science 224: 1431-1433). Поэтому в объем настоящего изобретения входит любая нуклеотидная последовательность при условии, что она кодирует природный или синтетический белок, обладающий соответствующей Nla-активностью.
Ген, кодирующий Nla-протеазу настоящего изобретения, может быть легко получен любым специалистом путем обратного конструирования исходя из последовательности ID N2.
При этом следует отметить, что из-за вырожденности генетического кода любая конкретно сконструированная последовательность необязательно будет хорошо, а то и вообще не будет гибридизироваться с любой данной комплементарной последовательностью, сконструированной исходя из одного и того же пептида. Этот фактор известен каждому специалисту, причем вырожденность любой конкретной последовательности часто оказывается настолько явной, что синтез даже очень короткой комплементарной олигонуклеотидной последовательности в целях ее использования в качестве зонда для натуральной олигонуклеотидной последовательности представляет значительные трудности.
Так, например, вырожденность кода может быть таковой, что большинству часто встречающихся аминокислот может соответствовать 4 или более кодонов. Поэтому отсюда очевидно, что число возможных кодирующих последовательностей для данного пептида будет экспоненциально возрастать с числом остатков. В соответствии с этим следует отметить, что число возможных кодирующих последовательностей для Nla настоящего изобретения может быть выражено шестизначными цифрами или даже цифрами более высокого порядка. Однако может оказаться желательным сбалансировать содержание G+C в соответствии с рассматриваемой экспрессирующей системой, и в этом случае необходимо учитывать другие факторы, такие как частота встречаемости кодона для данной экспрессирующей системы.
Как указывалось выше, гибридизация не может служить надежным показателем гомологии последовательностей, но тем не менее в основном предпочтительными являются те последовательности, гомология которых по отношению к последовательности ID N 1 составляет более 50%, предпочтительно 70%, а более предпочтительно 80%. Однако в любом случае протеаза, определенная выше, должна быть кодирована.
Настоящее изобретение также относится к возможному использованию лидерной последовательности, кодированной "выше" от протеазы. Эта последовательность способствует экстернализации гибридного белка из клетки хозяина и его высвобождению в супернатант культуры. Экстернализованный гибридный белок может затем подвергаться саморасщеплению с образованием полипептида. Для указанных сигнальных последовательностей может быть использована любая подходящая последовательность, в частности такая последовательность, которая была специально получена для данной экспрессирующей системы.
Настоящее изобретения также относится к векторам, содержащим последовательность настоящего изобретения. В соответствии с настоящим изобретением природа вектора не имеет решающего значения. Подходящие векторы экспрессии и конструкции, необходимые для их получения, могут быть выбраны самим специалистом в зависимости от преследуемых целей, например, клонирования или экспрессии.
Подходящими экспрессирующими векторами являются фаговые или плазмидные векторы, оба из которых являются хозяин-специфичными, хотя часто они используются и для других хозяев. Другими походящими векторами являются космиды и ретровирусы или какие-либо иные носители, которые могут быть специфичными или неспецифичными для данной системы. Подходящие регуляторные последовательности, такие как распознающая последовательность, промотор, оператор, индуктор, терминатор и другие последовательности, обычно используемые для регуляции экспрессии, могут быть легко выбраны самим специалистом, причем эти последовательности могут быть ассоциированы с CYVV или с используемым вектором, либо они могут происходить от любого другого источника. Указанные векторы могут быть модифицированы или сконструированы соответствующим образом.
Следует отметить, что последовательность ID 2 представляет собой последовательность, достаточную для кодирования полной Nla-протеазы. Для облегчения легирования в соответствующий вектор или экспрессии либо того и другого к этой последовательности могут быть добавлены терминатор, промотор и какая-либо другая регуляторная последовательность.
Следует отметить, что ДНК-фрагмент, кодирующий Nla настоящего изобретения вместе с любым фрагментом, кодирующим расщепляемую последовательность, а также фрагмент, кодирующий полипептид (ы), могут быть легко вставлены в любой подходящий сектор. В идеальном случае, для облегчения инсерции желательно использовать вектор, имеющий соответствующие рестрикционные сайты, но можно также проводить лигирование по тупым концам, хотя это может привести к неопределенности в отношении открытой рамки считывания и направлении инсерции. В таких случаях, конечно, желательно провести экспериментальную трансформацию хозяев трансфецированным вектором в целях отбора векторов, имеющих нужные фрагменты, инсертированные в правильном направлении и в правильной рамке считывания. Для полной гарантии того, что данные фрагменты будут находиться в правильной рамке считывания, желательно создать соответствующую конструкцию из последовательностей (a), (b) и (c), которая может быть затем непосредственно вставлена в вектор, что позволит уменьшить неопределенность в получении нужного экспрессирующего вектора. Подходящие векторы могут быть выбраны самим специалистом в соответствии с нужной экспрессирующей системой.
Нужный белок может быть продуцирован, например, путем трансформации E. coli, полученной плазмидой с последующим отбором трансформантов с использованием ампициллина или других подходящих средств и добавлением триптофана или другого подходящего индуктора промотора (такого, как индолилакриловая кислота). Степень экспрессии может быть оценена с помощью ДСН-электрофореза в полиакриламидном геле (ДСН-ПААГ) (Laemmli et al., Nature, (1970), 227, pp. 680-685).
При этом следует также отметить, что при использовании другого вектора может быть использован любой другой подходящий селективный маркер или маркеры либо альтернативный метод селекции, и/или, если это необходимо или удобно, может быть использован любой подходящий промотор.
Если гибридный белок необходимо выделить из клеток-хозяев, то после культивирования трансформированные клетки собирают, дезинтегрируют, например, ультразвуковой обработкой и центрифугируют. Разрушение клеток может быть также осуществлено путем ферментного гидролиза, например, с использованием целлюлозы либо путем перемешивания со стеклянными сферами на шейкере, однако ультразвуковой способ является более предпочтительным, поскольку он не требует использования дополнительных ингредиентов. Полученные супернатанты могут быть проанализированы на полипептидную активность, а продукты расщепления могут быть оценены, например, с помощью ДСП-ПААГ-электрофореза.
Для получения продукта экспрессии проводят очистку белка стандартными методами.
ДНК настоящего изобретения может быть получена путем выделения РНК-генома из вируса желтой прижилковой мозаики клевера (Uyeda, 1. et al., (1975) Ann Phytopath. Soc. Japan 41: 92-203). Подходящим источником CYVV является Американская коллекция типовых культур (ATCC, N PV 123). Вирус желтой прижилковой мозаики клевера может быть определен как вирус, вызывающий некроз у Vicia faba.
Геномная РНК может быть получена из CYVV-частиц, выделенных из инфицированных растений, а затем с помощью обратной транскрипции указанной РНК и с использованием известной методики может быть получена двухцепочечная кДНК.
Для определения является ли данный вирус мутантным или вариантным штаммом CYVV, хорошим индикатором может служить гомология последовательностей. Специалистам известны многие типы потивирусов, и все они различаются по своей патогенности. Принадлежность данного вируса к семейству потивирусов может быть установлена на основании серологического родства белков оболочки вируса и гомологии их аминокислотных последовательностей. В соответствии с этим вирусы, аминокислотные последовательности которых имеют 90%-ную гомологию, рассматриваются как вирусы, принадлежащие к одному и тому же штамму, тогда как вирусы, аминокислотные последовательности которых имеют гомологию менее чем 70%, рассматриваются
как члены разных семейств (Shukla, D.D. & Ward, C.W. (1989), Arch, Virol 106: 171-200). Исходя из различных свойств оболочечного белка CYVV, используемого в настоящем изобретении (Uyeda, 1, et al. (1991), 32: 234-Intervirol 245), CYVV рассматривается как независимый член семейства потивирусов. Поэтому любой вирус, обнаруживающий 90%-ную или более гомологию первичной аминокислотной последовательности оболочечного белка, или любой вирус, положительный на ELISA-анализ с использованием антисыворотки против CYVV (например, ATCC N PVA-123: антисыворотка к CYVV), рассматривается как штамм вируса желтой прижилковой мозаики клевера.
Растениями, на которых могут быть культивированы CYVV, являются Phaseolus vulgaris (фасоль обыкновенная), Vicia faba (кормовые бобы) и Pisum sativum (горох посевной), из них предпочтительным является Vicia faba, сорт Wase-soramme.
Предпочтительный способ очистки вирусных частиц предусматривает гомогенизацию листьев инфицированного растения, и их разведение в соответствующем буфере с последующей экстракцией органическим растворителем и многократным дифференциальным центрифугированием с окончательным центрифугированием в градиенте плотности сахарозы.
Одним из способов подтверждения того, что данная вирусная частица принадлежит к виду CYVV, является анализ этой вирусной частицы под электронным микроскопом. Другим способом является инокуляция Vicia faba данной вирусной частицей с последующим наблюдением развивающихся симптомов.
Подходящими методами экстракции геномной РНК из вирусных частиц является метод с использованием тиоцината гуанидиния/фенола, метод с использованием тиоционата гуанидиния/трифтороцезия и метод с использованием фенола/ДСН. Однако предпочтительным методом является центрифугирование в градиенте плотности щелочной сахарозы (Dougherty, W.G. & Hiebert, E. (1980), Virology 101: 466-474).
Для того, чтобы убедиться, действительно ли полученная РНК кодирует протеазу, эту РНК подвергают тестированию путем трансляции в бесклеточной системе трансляции. Аутолиз (саморасщепление) может быть затем обнаружен в том случае, когда РНК кодирует не только одну протеазу в полном трансляционном продукте, путем контролирования любого изменения молекулярной массы продуктов, собранных из лизатов. Такой контроль может быть осуществлен, например, с использованием антитела против белка оболочки.
Если необходимо, то продуцирование трансляционного продукта может быть проконтролировано, например, в течение определенного периода времени с использованием антитела против белка оболочки, причем геномная РНК может быть транслирована путем ее инъецирования в ооциты Xenopus laevis (шпорцевая лягушка) (Gurdon J. B. (1872), Nature 233: 177-182) либо путем использования кроличьих ретикулоцитов или системы лизатов пшеничных проростков (Schleif. P. F. & Wensink P. C. (1981), в "Practical Methods in Molecular Biology; Springer-Verlag, N.Y.).
Одноцепочечная ДНК может быть синтезирована с использованием полученной таким образом PHK в качестве матрицы и с использованием обратной транскриптазы, а двухцепочечная (дц) кДНК может быть синтезирована из одноцепочечной(оц) ДНК в соответствии со стандартными процедурами. Подходящими для этой цели методами являются метод с использованием нуклеазы s1 (Efstratidiatis, A. et. al. (1976), Cell 7: 279-288: Okayama H. & Berg, P. (1982), Mol. Cell. Biol. 2: 161-170 и др.), метод Ланда (Land H. et.al., (1981), Nucleis Acid Res, 9, 2251-2266) и метод O.Joon Yoo (Yoo O.J. et al., (1983), Proc. Natl. Acao. Sci USA 79, 1049-1053). В целях настоящего изобретения, предпочтительно использовать метод Gubler-Hoffman (Gubber, U. & Hoffman, B.J. (1983), Gee 25: 263-269).
Полученная таким образом дцДНК может быть затем введена в клонирующий вектор, такой как плазмида или лямбда-фаг, и продуцированный, в результате, рекомбинантный вектор может быть трансформирован в микроорганизм, такой как Escherichia coli. При этом особенно предпочтительным является штамм DH5 E. coli. После этого трансформанты могут быть отобраны на их резистентность к бактерицидным агентам, таким как тетрациклин или ампициллин, с использованием стандартной техники.
Трансформация может быть осуществлена, например, способом Hanahan (Hanahan, D (1983), J. Mol. Biol. 166: 557-580), заключающимся в том, что рекомбинантный ДНК-вектор вводят в клетку, которую делали предварительно компетентной путем обработки хлоридом кальция, хлоридом магния или хлоридом рубидия.
Селекцию трансформантов, содержащих ДНК, кодирующую Nla, осуществляли методом, описанным ниже.
(1) Скрининг с использованием зондов
Если в качестве источника РНК желательно использовать вирус CYVV дикого типа, то для выделения соответствующей РНК необходимо использовать кДНК-зонд при условии, что предварительно будет выявлена аминокислотная последовательность Nla (часть используемой последовательности может происходить от любой области Nla). Таким образом синтезируют олигонуклеотид, соответствующий определенной аминокислотной последовательности. Вообще говоря, выбранная аминокислотная последовательность должна содержать, по возможности, минимальное количество вырожденностей, в противном случае необходимо продуцировать несколько зондов с использованием, по возможности, различных кодонов. В таких случаях, кроме того, следует принимать во внимание частоту встречаемости используемого кодона. Альтернативно, может быть рассмотрено множество нуклеотидных последовательностей, и варьирующиеся нуклеотиды могут быть заменены инозином. Затем зонды могут быть подвергнуты мечению радиоактивным изотопом, таким как 32P, 35S, или биотином. После этого трансформантные штаммы могут быть идентифицированы путем фиксации денатурированной плазмидной ДНК на нитроцеллюлозных фильтрах с использованием радиоактивно меченных зондов, при этом позитивные клоны могут быть обнаружены с помощью авторадиографии.
(2) Использование PCR-зондов
В этом способе из смысловой цепи и антисмысловой цепи синтезируют олигонуклеотиды, соответствующие части известной аминокислотной последовательности, и осуществляют полимеразную цепную реакцию (Saiki P.K. et al. (1988), Science 239, 487-491). Указанные олигонуклеотиды используют для амплификации ДНК-фрагмента, кодирующего Nla. Подходящей ДНК-матрицей является кДНК, полученная посредством обратной транскрипции вирусной геномной РНК, которая, как известно, кодирует Nla. Полученные таким образом фрагменты ДНК подвергают мечению, например, 32P, 35S или биотином и для отбора нужного клона осуществляют гибридизацию колоний или бляшек.
(3) Скрининг путем экзогенного продуцирования в клетке животного
Этот метод предусматривает культивирование трансформантного штамма для амплификации гена, а затем трансфекцию этого гена в клетку животного (обычно, с использованием плазмиды, обладающей транскрипционной компетентностью и содержащей область транскрипционного промотора, либо с использованием плазмиды, которая может быть интегрирована в соответствующую хромосому), в результате чего осуществляется продуцирование белка, кодированного указанным геном. Трансформантный штамм может быть отобран путем оценки соответствующей активности.
(4) Селекция с использованием антитела против Nla
Из растения, инфицированного вирусом CYVV, продуцируют антитело против ядерных включений (Nla и Nlo) либо антитело против белка, продуцированного с помощью экспрессирующего вектора в соответствующей системе. Нужное Nla или нужный штамм могут быть затем обнаружены с помощью антитела или его антиантитела.
(5) Трансляционная система для селективной гибридизации
Трансформантную кДНК гибридизировали с мРНК, происходящей из клеток, экспрессирующих Nla, и описанной выше и, мРНК, связанную с кДНК, диссоциировали и выделяли. Выделенную мРНК транслировали в белок в трансляционной системе, например, путем инъекции в ооциты Xenopus laevis или в бесклеточные системы, такие как лизат кроличьих ретикулоцитов или лизат пшеничных проростков. Нужный штамм отбирали путем оценки активности Nla или путем детекции Nla с помощью антитела против Nla.
ДНК, кодирующая CYVV-Nla, может быть получена из трансформированных штаммов известными способами (см. Maniatis T. et al,. (1982), в "Molecular cloning A Laboratory Manual" Cold Spring Harbor Laboratory, N.Y.).
Для этого, например, из клеток выделяют фракцию, соответствующую векторной ДНК, и из указанной плазмидной ДНК вырезают область ДНК, кодирующую нужный белок.
Определение полученной таким образом ДНК-последовательности может быть осуществлено, например, методом химической модификации Максама-Гилберта (Maxam A. M & Gilbert, W. (1980), в "Methods in Enzymology" 65: 499-276) либо методом дидезокси-терминации цепи с использованием фага M13 (Messing J. & Vieira, J (1982), Gene 19: 269-276).
В последнее время, для определения ДНК-последовательностей имеется тенденция использовать флуорохром вместо более опасных радиоактивных изотопов. Кроме того, в настоящее время процедуру дидезокси-терминации цепи осуществляют с использованием робототехники с компьютерным управлением. Широко применяются также системы для прочитывания последовательностей после электрофореза, примерами таких систем могут служить "CATALYST 800" (робот для секвенирования) и ДНК-секвенатор 373A (Perkin-Elmer Japan Applied Biosystems). Эти системы позволяют осуществлять детерминацию ДНК-последовательностей с высокой степенью эффективности и надежности.
Векторы настоящего изобретения могут быть организованы таким образом, что они будут экспрессироваться в "стандартных клетках", либо эукариотических, либо прокариотических. Кроме того, путем введения в этот вектор соответствующего промотора им последовательности для фенотипической экспрессии нужный ген может быть экспрессирован в выбранных клетках-хозяевах.
Подходящими прокариотическими хозяевами являются Escherichia coli и Bacillus subtilis. Для фенотипической экспрессии соответствующего гена в вектор может быть введен репликон, происходящий от вида, который является совместимым с хозяином. В случае использования E. coli плазмида может содержать сайт инициации репликации и промоторную последовательность, такую как lac или Uv5. При этом предпочтительными являются векторы, которые на основании своего фенотипического признака могут сообщать трансформированной клетке избирательность.
В большинстве случаев в качестве хозяина используют Escherichia coli, а особенно предпочтительным хозяином является штамм JM 109, происходящий от штамма E. coli K12. Векторы для E.coli обычно выбирают из серии плазмид pBP322 или pUC, однако, если необходимо, могут быть использованы и другие векторы и штаммы. Подходящими промоторами для E.coli являются лактозный промотор (lac), триптофановый промотор (trp триптофан-лактозный промотор (tac), липопротеиновый промотор (lpp), лямбда ( λ )-промотор (PL) (от λ -фага) и промотор Tu-фактора элонгации полипептидной цепи (tufB), однако настоящее изобретение не ограничивается указанными промоторами.
Подходящими штаммами Bacillus subtilis является штамм 207-25. Подходящими векторами является pT B228 (Ohmura K. et al (1984), J. Biochem 95: 87-93) и др. Подходящим промотором является регуляторная последовательность гена α -амилазы Bacillus subtilis. Сигнальная пептидная последовательность (от α -амилазы) может быть использована для внеклеточной секреции.
Если необходимо экспрессировать гибридный белок в эукариотических клетках, то для этой цели могут быть использованы клетки позвоночных, насекомых, дрожжей, растений и т.п. Предпочтительными клетками позвоночных являются COS-клетки, в частности клеток COS-1 (см., например, Gluzman Y. (1981), Cell 23: 175-182), или клеточная линия яичника китайского хомячка (CHO), в которой отсутствует гидрофолат-редуктаза (Urlaub G & Chasin L.A. (1980), Proc. Natl. Acad Sci. USA 77, 4216-4220), хотя настоящее изобретение не ограничивается только этими примерами.
Как указывалось выше, COS-клетки являются подходящими клетками-хозяевами позвоночных, и они могут быть использованы в качестве примера. Экспрессирующие векторы, содержащие репликон SV40, способны в автономной репликации и снабжены транскрипционным промотором, транскрипционным терминатором и одним или несколькими сайтами сплайсинга. Экспрессирующие векторы, содержащие нужную ДНК-последовательность, могут быть использованы для трансформации COS-клеток различными методами, например, такими, как метод с использованием DEAE-декстрана (Luthman H. & Magnusson G (1983), Nucleic Acids Res. 11, 1295-1308), метод копреципитации фосфата кальция-ДНК (Graham F.L. & Vander Eb. , A.J. (1973), и методом электропорации (Neumann E. et. al. (1982) EMBO J. 1, 841-845).
Если в качестве клеток-хозяев используются клетки CHO, то желательно использовать вектор, способный экспрессировать neo-ген, сообщающий G418-резистентность. Подходящими маркерами, несущими этот маркер, являются ppSVneo (Sambrook J. et. al. (1989), "Molecular Cloning-A Laboratory Manual", Cold Spring Harbor Laboramory), u pSV2-neo (Southern P.J. & Berg P. (1982), J. Mol. Appl. Genet, 1, 327-341).
Трансформанты могут быть затем отобраны на их резистентность к G418.
Отобранные трансформанты могут быть затем культивированы стандартными методами, а в случае осуществления второго варианта настоящего изобретения полипептид продуцируют как внутриклеточно, так и внеклеточно. Культуральная среда может быть выбрана в соответствии с используемыми клетками-хозяевами. Например, для COS-клеток, к такой среде, как PRM1-1640 или модифицированной по методу Дульбеко среде Игла (DMEM), по необходимости, может быть добавлена среда, содержащая компоненты крови, например фетальная телячья сыворотка.
Если необходимо использовать клетки насекомого, то для этой цели могут быть использованы клетки, происходящие от Spodoptera frugipedra (Smith G.E. et al. (1983), Mol. Cell. Biol. 3: 2156-2165).
Подходящими дрожжами являются хлебопекарные дрожжи (Saccharomyces cereviside) и делящиеся дрожжи (Schizosaccharomyces pomle).
Подходящими растительными клетками являются клетки от Nicotiana tabacum. (табак) и Oryza sativa (рис посевной).
При этом следует отметить, что все вышеуказанные хозяева являются стандартными клетками-хозяевами, обычно используемыми на практике, однако специалисты могут выбрать и другие клетки, подходящие для использования в качестве хозяев.
Подходящими векторами экспрессии для клеток позвоночных являются такие векторы, которые содержат промотор, расположенный выше экспрессируемого гена, вместе с такими сайтами, как сайт РНК-сплайсинга, сайт полиаденилирования, и сайт терминации транскрипции, и которые, кроме того, если этот необходимо, содержат сайт инициации репликации. Примером подходящего экспрессирующего вектора является pSV2dnfr, который содержит ранний промотор SV40 (Subramani S. et al, (1981), Mol. Cell. Biol: 854-864).
Подходящей экспрессирующей системой для клеток насекомых являются культивируемые клетки Spodoptera frugipedra. Подходящие экспрессирующие векторы содержат, например, промотор полиэдрина бакуловируса, расположенный "выше" экспрессируемого гена, вместе с сайтом полиаденилирования, а также часть AcMNPV-генона (вируса ядерного полиэдроза Acutographa californica), необходимого для гомологичной рекомбинации. Примером такого вектора является pBacPAK8 (Matuura Y. et al. (1987), J. Gen. Virol 68: 1233-1250).
Для эукариотической экспрессии обычно используют дрожжи, например хлебопекарные дрожжи (S. cerevisiae). Подходящие экспрессирующие векторы для дрожжей могут содержать алкогольдегидрогеназный промотор (Bennetzen J.L & Hall B. D. (1982), J. Biol. Chem. 257: 3018-3025), промотор кислотной фосфатазы (Miyahara A. et al. (1983). Proc. Natl. Acad. Sci USA, 80, 1-5) или промотор карбоксипептидазы Y (Ichikawa K. et al. (1993), Biosci. Biotech. Biochem. 57: 1686-1690). В этом случае для осуществления секреции во внеклеточное пространство может быть использована сигнальная пептидная последовательность от карбоксипептидазы Y.
Подходящим экспрессирующим вектором для растений является, например, pB1121, содержащий 35S-промотор (происходящий от раннего промотора вируса мозаики цветной капусты), сайт полиаденилирования гена синтеза нопалина от Agrobacterium tumefaciens; и сайт переноса гена Agrobacterium tumefaciens (Jefferson, P.A. et al. (1987), EMBOJ 6: 3901-3907). Такой вектор может быть введен в растительные клетки, например, путем инфицирования бактерий Agrobacterium tumefaciens и путем электропорации.
Плазмида pKK388-1 (сконструированная Clonetech Co.), содержащая trc-промотор, является подходящей для использования в E. coli. Этот экспрессирующий вектор способен к автономной репликации в штамме, происходящем от штамма K12 Escherichia coli такого, как штамм JM109. этот вектор может быть легко введен в E. coli хорошо известными методами, упомянутыми выше. Полученный таким образом штамм может быть инокулирован в среду, например, такую хорошо известную среду, как LB-среда, и культивирован в этой среде в течение определенного периода времени.
trc-Промотор может быть затем активирован путем добавления, например, изопропил- β -галактопиранозида (IPTG). После последующего культивирования продуцированный белок может быть затем экстрагирован из клеток путем их лизиса, например, с помощью ультразвукового аппарата. Если необходимо продуцировать белок, имеющий Pro на N-конце, то, в этом случае, в качестве хозяина желательно использовать E. coli.
Если необходимо, то в соответствии с примерами, сопровождающими настоящее описание, фракция, содержащая нужный полипептид, может быть выделена из культуры трансформированных клеток и очищена известными методами, выбранными в зависимости от физических и химических свойств полипептида. Подходящими для этой цели методами является обработка преципитирующим агентом, ультрафильтрация, хроматография различного типа (например, хроматография на молекулярных ситах (гель-фильтрация), адсорбционная хроматография, ионообменная хроматография, аффинная хроматография и высокоразрешающая жидкостная хроматография (ВРЖХ), диализ и комбинированные методы.
Для того, чтобы убедиться в том, что экспрессированный Nla-белок имеет нужную протеазную активность, очищенный Nla-белок может быть подвергнут реакции с субстратом, содержащим расщепляемую последовательность для протеазы, например, таким, как продукт экспрессии гена, кодирующего гибридный белок, включающий в себя белок вирусной оболочки (природный субстрат для 1a) вместе "ядерным включением b" (Nlb) (Dougherty, W.G. at al. (1988), EMBO J, 7: 1281-1287). Указанный гибридный белок может быть выделен из вирусной геномной кДНК и вставлен в экспрессирующий вектор. Полученный в результате экспрессирующий вектор затем вводят в соответствующую хозяйскую клетку для продуцирования указанного гибридного белка. Полученный гибридный белок может быть выделен и очищен стандартными способами, например, с помощью преципитации с использованием соответствующего осаждающего агента или с помощью хроматографии.
Выделенный и очищенный таким образом субстратный белок может быть затем подвергнут реакции с указанной протеазой в соответствующем буферном растворе при подходящей температуре с выделением реакционного продукта. Полученный реакционный продукт может быть затем подвергнут электрофорезу или Вестерн - блоттингу с использованием антитела против белка оболочки в целях оценки протеазной активности, обнаруживаемой по различию в мобильности полосы. В этом методе можно также использовать синтетический олигопептид, содержащий соответствующую расщепляемую последовательность.
Протеолитическая активность протеазы настоящего изобретения может быть также определена и без очистки. Так, например, указанная активность может быть определена путем присоединения гена протеазы в последовательности и с сохранением рамки считывания так, чтобы он был расположен "ниже" промотора, например, trc-промотора, и "выше" расщепляемой последовательности для протеазы. Если протеаза является активной в продукте экспрессии, то при вестерн-блоттинге будет наблюдаться полоса с более высокой мобильностью, чем гибридный белок.
Путем выделения этой полосы из геля, в котором наблюдалось расщепление, и путем анализа ее аминокислотной последовательности стандартными методами можно установить, расщепляется ли этот белок в нужной пептидной связи.
Нужный белок может быть продуцирован, например, в виде гибридного белка вместе с таким белком, как глутатион-S-трансфераза, а Nla настоящего изобретения может быть затем использована для расщепления линкерной последовательности in vitro. В одном из альтернативных способов нужный белок непосредственно продуцируют в клетке-хозяине в виде гибридного белка вместе с указанной протеазой, после чего нужный белок образуется посредством саморасщепления.
Если необходимо, то с использованием вышеописанной методики Nla может быть легко продуцировано с высоким выходом и с высокой степенью чистоты, и полученное таким образом Nla ("ядерное включение а") настоящего изобретения может быть использовано как протеаза.
По желанию, с использованием стандартного метода, например триэфирфосфитного метода (Hunkapiller M. et al. (1984), Nature 310: 105-111), или метода химического синтеза нуклеиновых кислот (Grantham P. et al. (1981) Nucleic Acids Res 9: г43-г74) может быть осуществлен химический синтез ДНК настоящего изобретения. Если необходимо, то может быть осуществлена частичная модификация указанных нуклеотидных последовательностей с использованием стандартной методики, такой как сайт-специфический мутагенез, в котором используется праймер, содержащий синтетический олигонуклеотид, кодирующий нужную модификацию (Mark D. F. et al. (1984), Proc. Natl. Acad, Sci. 81: 5662-5666).
Гибридизация, упомянутая выше, может быть установлена, например, с использованием зонда, меченного, например, [ α -32P] oCTP, методом рассеянной затравки (Feinberg, A.P. & Vagelstein в (1983) Anal. Biochem 132: 6-13), или путем ник-трансляции (Maniatis T, et al. (1982) в "Molecular Cloning A Laboratory Manual" Cold Spring Harbor Laboratort, N.Y.). ДНК фиксируют на твердой фазе стандартным способом, например, путем адсорбции на нитроцеллюлозной мембране или на найлоновой мембране, а затем нагревают или подвергают УФ-излучению. После этого твердую фазу погружают в раствор для предварительной гибридизации, содержащей 6 х SSC, 5% раствор Денхардта и 0,1% ДСН, и инкубируют при 55oC в течение 4 часов или более. Затем к раствору для предварительной гибридизации добавляют заранее полученный зонд до достижения конечной удельной активности 1 • 106 имп./мин на мл, после чего смесь инкубируют при 60oC в течение ночи. После этого твердую фазу пять раз, при комнатной температуре и в течение 5 минут, промывают 6 х SSC, с последующим 20-минутным промыванием при 57oC, а затем, для того, чтобы определить, имела место гибридизация ДНК или нет, проводят авторадиографию. При этом следует отметить, что описанный выше пример является чисто иллюстративным, и в целях настоящего изобретения с таким же успехом могут быть использованы другие методы.
Нужный белок может быть получен либо методом внутриклеточного прямого расщепления, либо методом внеклеточного расщепления.
Метод внутриклеточного прямого расщепления
ДНК, кодирующую Nla, присоединяют к ДНК, кодирующей нужный белок, посредством расщепляемой последовательности, предпочтительно Gln-Gly, Gln-Ser или Gln-Ala. Полученную ДНК, которая кодирует гибридный белок, вставляют в вектор, содержащий промотор и терминатор, и указанный вектор используют в соответствующей клетке-хозяине. В результате экспрессированный гибридный белок самостоятельно расщепляется благодаря протеазной активности, образуя тем самым пептид, в котором Gly, Ser или Ala связаны пептидной связью у N-конца нужного белка. Особенно предпочтительным белком для экспрессии является 1L-11.
Если от N-конца полученного белка необходимо отщепить какой-либо остаток, то это может быть сделано, как описано выше, то есть, например, для удаления N-концевого Pro-остатка может быть использована аминопептидаза P.
В некоторых системах экспрессии уже присутствует аминопептидаза P. Однако если указанная аминопептидаза P отсутствует, но при этом необходимо, чтобы N-концевые аминокислотные остатки были отщеплены in situ, то в этом случае, в клетку-хозяин может быть введен вектор экспрессии для аминопептидазы P. Аминопептидаза P является известным белком, и генная последовательность для этого фермента является легко доступной для каждого специалиста.
Если необходимо получить белок, N-конец которого начинается с Pro, то для этого желательно продлить время культивирования, чтобы обеспечить действие клеточной аминопептидазы P.
Как описано выше, N-концевой Pro-остаток может быть удален посредством каталитического действия пролин-иминопептидазы (3.4.11.5), эндогенно продуцированной или продуцированной экзогенным экспрессирующим вектором (этот фермент является известным белком, а генная последовательность для этого фермента легко доступна специалистам). Альтернативно, указанный пролиновый остаток может быть удален после выделения из экспрессирующей системы, например, такой, где гибридный белок секретируется из клетки.
Таким образом, благодаря настоящему изобретению могут быть продуцированы белки, имеющие любой нужный N-концевой остаток.
Если последовательность настоящего изобретения продуцирует полипептид, имеющий N-концевой Ala, то этот аланиновый остаток может быть удален посредством каталитического действия аланин-аминопептидазы (3.4.11.14), которая также является известным ферментом, и генная последовательность которой легко доступна для каждого специалиста. Описанная техника также позволяет продуцировать полипептид, имеющий свободно выбранный N-концевой остаток.
Затем нужный белок может быть выделен и очищен в соответствии со стандартной методикой.
Метод внеклеточного расщепления.
Ядерное включение a (Nla) может быть продуцировано путем введения соответствующей ДНК в вектор, который, в свою очередь, вводят в подходящую экспрессирующую систему. Nla, продуцированное в трансформированных клетках, может быть затем очищено путем ионообменной хроматографии, гель-фильтрации или обращенно-фазовой хроматографии. Альтернативно, Nla может быть продуцировано в виде гибрида с другим полипептидом, таким как глутатион-S-трансфераза или связывающийся с мальтозой белок, причем указанный гибридный белок может быть разделен и выделен с использованием колонки с глутатионом или мальтозой соответственно. После расщепления очищенного продукта энтерокиназой или фактором Xa Nla может быть очищено и использовано в соответствующих целях.
Между тем, получают белок-предшественник, который содержит последовательность распознавания Nla (отщепляемая последовательность). Она может присутствовать в данном участке, либо этот белок может образовывать часть гибридного белка, например, с белком, связывающимся с мальтозой, или с глутатион-S-трансферазой соединенными посредством подходящего линкера (отщепляемая последовательность). Реакция указанного предшественника с Nla в соответствующем буферном растворе способствует расщеплению предшественника in vitro. Если необходимо, то N-концевые аминокислотные остатки могут быть удалены, как описано выше.
Как указывалось выше, полученный белок может быть выделен и очищен известными методами.
Что касается второго варианта осуществления настоящего изобретения, то авторы считают, что природный восстановительный пептид имеет последовательность, показанную в списке последовательностей (см. ниже) как SEQ ID N 12, и состоит из 526 аминокислот с Val-остатком на N-конце.
Авторы считают также, что природная ДНК, кодирующая этот пептид, имеет нуклеотидную последовательность, состоящую из нуклеотидов 70-1647, и показанную в списке последовательностей как SEQ ID N 11.
Пептид настоящего изобретения способен восстанавливать окисленный глутатион и дихлороиндофенол. Приводимое в настоящей заявке описание указанного пептида является довольно точным, но несколько громоздким, а поэтому, далее, пептид настоящего изобретения будет обозначаться как KM31-7-пептид или белок.
KM31-7-белок, или его мутант, или вариант продуцируется в самом организме, а поэтому проблемы, связанные токсичностью и/или антигенностью белков, в данном случае практически отсутствуют.
В соответствии со вторым вариантом осуществления настоящего изобретения предполагается, что полипептид, имеющий последовательность аминокислот от -23 до 526 (последовательность ID 12), является предшественником KM31-7-белка. В связи с этим следует отметить, что настоящее изобретение также охватывает указанный предшественник, его мутанты, и варианты, и полинуклеотидную последовательность, кодирующую любой из указанных пептидов.
Нижеследующее обсуждение относится в основном ко второму варианту осуществления настоящего изобретения. При этом следует отметить, что процедуры, описанные выше для CYVV-Nla, могут быть также использованы для выделения, клонирования и экспресии ДНК, кодирующей пептид настоящего изобретения. В основе всех возможных расхождений лежит тот факт, что CY Nla является белком вируса растений, тогда как пептид настоящего изобретения является белком млекопитающих. Ниже будут описаны основные стадии получения конкретного полипептида, продуцированного из клеток млекопитающего способами генетической рекомбинации.
мРНК, кодирующая KM31-7-пептид, может быть получена, а затем обратно транскрибирована в дцДНК в соответствии с хорошо известной техникой. В качестве источника мРНК могут быть использованы любые подходящие клетки, клеточные линии или ткани млекопитающих, однако предпочтительно использовать клеточную линию KM-102, происходящую от стромальных клеток головного мозга человека (Harigaya K. & Handa H. (1985), Proc, Natl, Acad, Sci, USA, 82, 3447-3480).
Для экстракции мРНК из клеток млекопитающих могут быть использованы любые подходящие методы, например метод с использованием гуанидинтиоцианата - горячего фенола, либо метод с использованием гуанидинтиоцианата и гуанидин-соляной кислоты, однако в основном предпочтительным является метод с использованием гуанидинтиоцианата - хлорида цезия.
Поскольку в большинстве случаев мРНК, присутствующая в цитоплазме эукариотических клеток, имеет, как известно, 3'-концевую (polyA)-последовательность, то очистка мРНК млекопитающих может быть осуществлена путем адсорбции на колонке с олиго(dT)-целлюлозой. Элюированная мРНК может быть затем фракционирована путем центрифугирования в градиенте плотности сахарозы.
Убедиться в том, что мРНК действительно кодирует нужный пептид, можно путем трансляции мРНК в соответствующей системе, например в системе ооцитов Xenopus laevis системе кроличьих ретикулоцитов или в системе пшеничных проростков (см. выше).
Измерение восстановительной активности продукта экспрессии может быть осуществлено в соответствии с нижеприведенным описанием.
I. Определение дихлороиндофенол-восстанавливающей активности
Методика такого определения описана Beinert H, в "Methods in Enzymology" (1962), 5, 546.
50 мкМ-препарат дихлороиндофенола (Sigma) получают с использованием 20 мМ фосфатного буфера и 0,5 М NaCl (pH 7,8), 1 мл этого препарата затем помещают в кювету (SARSTEDT, 10х4х45 мм) и после этого добавляют образец для измерения. 15 микролитров 1 мМ NADPH (НАДФ) (Boeringer-Mannheim) изготавливают в том же буфере и добавляют в кювету при комнатной температуре для инициации реакции. Редуктазную активность затем определяют по последующему снижению абсорбции окисленного дихлороиндофенола при 600 нм или по последующему снижению абсорбции A PH при 340 нм.
II) Определение активности восстановления окисленного глутатиона.
Методика этого анализа описана Nakajima T и др. в "New Basic Experimental Methods in Biochemistry (6) - Assay Methods Using Biological Activity", 3-34.
Препарат 10 мМ окисленного глутатиона (Boeringer-Manheim) изготавливают с использованием 20 мМ фосфатного буфера и 0,5 М NaCl (pH 7,8), 15 мкл этого препарата помещают в кювету (10х4х4 мм) после того, как туда был помещен образец для измерения. 15 мкл-препарат 1 мМ NADPH получают в том же самом буфере, а затем добавляют в кювету при комнатной температуре для инициации реакции. Глутатионредуктазная активность может быть затем определена по последующему уменьшению абсорбции при 340 нм.
Для получения дцДНК из мРНК могут быть использованы различные методы, описанные выше. Такими методами являются S1-нуклеазный метод, метод Ланда и метод O.Joo Yoo (см., выше), однако в настоящей работе авторы предпочли использовать метод Okayama-Bero (Okayama H. & Bero P. (1982), Mol. Cell. Biol. 2, 161-170).
Полученная таким образом дц-кДНК может быть затем введена в вектор клонирования, а полученная в результате рекомбинантная плазмида может быть введена в Escherichia coli, как описано выше.
Трансформантный штамм, содержащий нужную ДНК, кодирующую пептид настоящего изобретения, может быть отобран различными методами, например, такими, которые были описаны выше в связи с отбором трансформантов, содержащих Nla-ДНК, с соответствующими модификациями. Например, если для получения зонда используется полимеразно-цепная реакция (PCR), то соответствующей ДНК-матрицей может быть кДНК, описанная выше, или геномная ДНК.
Во втором варианте осуществления настоящего изобретения для снижения числа штаммов-трансформантов, подвергаемых испытанию, может быть проведен первичный скрининг. Проведение такого первичного скрининга вполне возможно, поскольку пептид настоящего изобретения относится к цитокинам, и его мРНК имеет общий с мРНК цитокинов элемент AUUUA (Shaw. G. & Kamen P. (1986), Cell 46, 659-667). Таким образом, для первичного скрининга может быть использован синтетический олигонуклеотидный зонд, комплементарный элементу AUUUA. Затем может быть проведен скрининг путем анализа продуцирования экзогенного белка в клетках млекопитающих.
ДНК, кодирующая пептид, может быть затем вырезана из вектора и секвенирована в соответствии с методикой, описанной выше для Nla-ДНК. И так же, как описано выше, фрагмент ДНК может быть затем введен в соответствующий вектор и использован для трансформации прокариотического или эукариотического хозяина.
KM31-7-белок может быть использован либо отдельно, либо в комбинации с одним или несколькими лекарственным средствами для предупреждения и лечения заболеваний, вызванных или опосредованных окислительным стрессом, или любых других заболеваний, вызываемых активированным кислородом. Такими заболеваниями или состояниями являются (но не ограничиваются ими) атеросклероз, диабет, ишемические нарушения (такие, как реперфузионные повреждения, ишемическая болезнь сердца, ишемия головного мозга и ишемический энтерит), отеки, сосудистая сверхпроницаемость, воспалительные заболевания, повреждения слизистой желудка, острый панкреатит, болезнь Крона, язвенный колит, заболевания печени, болезнь Paraguat, эмфизема легких, химический канцерогенез, метастазы рака, респираторный дистресс-синдром у взрослых, диссеминированное внутрисосудистое свертывание крови (ДВС-синдром), катаракта, ретролетальная фиброплазия, аутоиммунные заболевания, порфиремия, гемолиз, эритробластическая (средиземноморская) анемия, болезнь Паркинсона, болезнь Альцгеймера, эпилепсия, нарушения, связанные с ультрафиолетовым облучением, нарушения, вызванные радиоактивным облучением (лучевая болезнь), обморожения и ожоги.
В соответствии со вторым вариантом осуществления настоящего изобретения фармацевтические композиции включают в себя фармацевтически активное количество KM31-7-пептида и его фармацевтически приемлемый носитель.
Композиции настоящего изобретения могут быть введены перорально, например, в виде таблеток, капсул, гранул, порошков или сиропов, либо парентерально в виде инъекций, вливаний и суппозиториев. Если необходимо, могут быть использованы и другие способы введения.
Если пептид настоящего изобретения необходимо ввести в виде инъекции или вливания, то для этой цели может быть изготовлен апирогенный препарат указанного пептида, разведенного в фармацевтически приемлемом водном растворе, подходящем для парентерального введения. Этот препарат полипептидного раствора должен иметь соответствующий pH, изотоничность и стабильность, согласно заключению технической экспертизы.
Доза и форма вводимого препарата могут быть легко установлены самим специалистом на основании таких критериев, как состояние пациента, его масса, пол, возраст и режим питания, наличие других инфекций, время введения и другие клинически важные факторы. Однако в основном нормальная пероральная доза для взрослого человека составляет порядка от около 0,01 мг до около 1000 мг в день. Это количество может быть введено в виде разовой дозы или в виде дробных доз в течение 24 часов. Если указанный пептид необходимо ввести парентерально, то он может быть введен подкожно, внутримышечно или внутривенно в дозе, составляющей от около 0,01 мг до около 100 мг.
Для полной характеристики KM31-7-белка очень важно продуцировать антитело к этому белку. Такое антитело может быть использовано для анализа функций белка, его количественного анализа и очистки, а также для оценки распределения KM31-7-белка в тканях.
Для этого, сначала, путем инокуляции лабораторных животных полипептидом, продуцированным E. coli, трансформированной плазмидой pMA 31-7, получали гибридому, продуцирующую антитело против KM31-7, а затем эту гибридому, образованную антитело-продуцирующими клетками и миеломными клетками, скринировали и клонировали. Антитела, продуцируемые указанной гибридомой, обладали способностью к распознаванию полипептида, выделенного из бесклеточного супернатанта COS-1-клеток, трансформированных плазмидой pSP 31-7.
Таким образом, настоящее изобретение относится к антителу, предпочтительно к моноклональному антителу, или к его эквиваленту, способному специфически распознавать KM31-7-белок, или его мутант, или вариант.
Антитело настоящего изобретения направлено против KM31-7-белка, или его мутанта, или варианта. При этом указанное антитело может быть поликлональным или моноклональным, хотя предпочтительным является моноклональное антитело. Причиной такого предпочтения является неопределенность, связанная с использованием поликлональных антител. Поэтому для получения надежных результатов, независимо от того, используется ли это антитело в терапевтических целях или для очистки KM31-белка, предпочтительно использовать моноклональные антитела.
Антитела настоящего изобретения могут быть получены от любого подходящего животного. Однако если данное антитело происходит не от человека, то в этом случае могут возникнуть проблемы, связанные с антигенностью этого антитела. Поэтому, чтобы получить антитела, более близкие к человеческим антителам, желаемые антитела можно сконструировать. Это может быть достигнуто с помощью химической или генетической модификации методами, хорошо известными специалистам.
В настоящем изобретении также рассматриваются антиидиотипические антитела, то есть антитела, сайт узнавания которых распознает сайт узнавания вышеуказанных моноклональных антител. Такие антиидиотипические антитела могут быть получены путем введения исходного антитела подходящему животному. При этом следует заметить, что этот процесс может продолжаться фактически до бесконечности, где каждая генерация будет соответствовать либо исходному, либо антиидиотипу. Однако если эти антитела не будут отобраны на правильное распознавание после каждой генерации, то их специфичность может быть в значительной степени утрачена. Указанные антиидиотипические антитела могут быть использованы, например, для анализа свободного антитела против KM31-7.
Настоящее изобретение также относится к фрагментам антител настоящего изобретения, способным к узнаванию KM31-7-белка, и к молекулам, несущим сайт узнавания указанных антител. Далее, такие фрагменты будут именоваться "эквивалентами" антител настоящего изобретения.
Плазмиду pMA 31-7 использовали для трансформации E.coli, а продукты экспрессии очищали и использовали для иммунизации лабораторных животных. Клетки селезенки от иммунизированных животных использовали для получения гибридомы путем слияния этих клеток с клетками миеломы, в результате чего был получен клон, продуцирующий моноклональные антитела против KM31 с высокой концентрацией и с хорошей стабильностью (этот клон был обозначен MKM150-2 и депонирован в Научно-исследовательском институте ферментации Управления промышленных наук и технологий Японии (Fermentation Research of the Agency of Industrial Science and Technology, Japan) под входящим номером ГЕРМ ВР-5086). В результате культивирования этого клона из супернатанта культуры получали моноклональные антитела против KM31-7.
Полученные моноклональные антитела против KM31-7 иммунохимически реагируют с гибридным белком, полученным путем введения и экспрессии pMA131-7 в E. coli. Это антитело также иммунохимически реагирует с KM31-7-белком, полученным из супернатанта клеток млекопитающих, трансформированных с помощью кДНК, кодирующий KM31-7.
Продуцирование моноклонального антитела осуществляли в соответствии с нижеследующими процедурами, включающими в себя:
(a) очистку биополимера, используемого в качестве антигена,
(b) иммунизацию мышей путем инъекции антигена и получения из селезенки антитело-продуцирующих клеток за соответствующий период времени путем отбора проб и исследования крови,
(c) получение миеломных клеток,
(d) проведение слияния клеток селезенки и клеток миеломы,
(e) скрининг для отбора гибридом, продуцирующих нужные антитела,
(f) получение клона из одной клетки (клонирование),
(g) культивирование гибридомы для крупномасштабного продуцирования моноклонального антитела или разведение мышей, инфицированных гибридомой, в зависимости от обстоятельств,
(h) исследование физиологической активности полученного моноклонального антитела или оценка его свойства как реагента для мечения.
Продуцирование моноклональных антител против KM31-7 осуществляли в соответствии с описанием, представленным ниже. При этом следует отметить, что нет необходимости придерживаться этого описания с абсолютной точностью, и там, где это требуется, можно использовать любую подходящую процедуру. Например, в качестве антитело-продуцирующих клеток, вместо клеток селезенки можно использовать другие клетки, а вместо мышиной миеломы можно использовать миеломы от других млекопитающих. Нижеприведенная процедура представляет собой предпочтительный метод получения моноклональных антител против KM31-7.
(a) Очистка антигена
Гибридный белок, полученный путем экспрессии pMAL31-7 в E.coli с последующей очисткой продукта экспрессии, является эффективным антигеном. Экспрессию E. coli TB-1, трансформированную плазмидой pMAL31-7, индуцировали с помощью изопропил-N-тиогалактопиранозида (IPTG). Затем экспрессированный гибридный белок очищали с помощью аффинной хроматографии, используя колонку с амилозной смолой (New England Biolabs). KM31-7-белок, очищенный из бессывороточного культурального супернатанта COS-1-клеток, трансформированных плазмидой pSR α 31-7, также представляет собой эффективный антиген.
(b) Получение антитело-продуцирующих клеток
Очищенный гибридный белок, полученный в (a), смешивали с полным или неполным адъювантом Фрейнда, или таким адъювантом, как калиевые квасцы, и полученной в результате этого вакциной иммунизировали лабораторных животных. В качестве лабораторных животных предпочтительно использовать мышки BALB/c, поскольку в большинстве случаев используемые мышиные миеломы происходят от мышей BALB/c. Кроме того, эти мыши были хорошо и детально исследованы, и их характеристики подробно описаны. Более того, если антитело-продуцирующие клетки и миелома происходят от мыши BALB/c, то полученная гибридома может быть культивирована в брюшной полости BALB/c-мыши. Таким образом, использование мышей BALB/c имеет значительные преимущества, позволяющие легко получить моноклональные антитела из асцитов без использования громоздких и сложных процедур. Тем не менее следует упомянуть, что настоящее изобретение не ограничивается использованием мышей BALB/c.
Указанный антиген может быть введен в любой подходящей форме, например, путем подкожной инъекции, внутрибрюшинной инъекции, чрезкожной инъекции или внутримышечной инъекции, при этом предпочтительной является подкожная или внутрибрюшинная инъекция.
Иммунизация мышей может быть осуществлена с помощью одной инъекции или несколько инъекций, вводимых через соответствующие промежутки времени. Предпочтительная схема иммунизации предусматривает введение первой инъекции антигена, а затем повторной инъекции, один или несколько раз, через интервалы времени от 1 до 5 недель. Эффективность последующих процедур может быть увеличена, если регулярно анализировать титр антитела к указанному антигену в сыворотке иммунизированных животных, и животные, имеющие высокий титр антитела, могут быть затем использованы для получения антитело-продуцирующих клеток. Антитело-продуцирующие клетки, используемые для последующего слияния клеток, предпочтительно выделять через 3-5 дней после конечной иммунизации животного.
Для анализа титра антител могут быть использованы различные известные методы, например, такие, как иммуноанализ с использованием радиоактивного изотопа (RIA), твердофазный иммуноферментный анализ (ELISA), метод с использованием флуоресцирующих антител, и пассивная гемагглютинация, однако предпочтительным являются анализы RIA и ELISA из-за их высокой чувствительности, скорости, точности и возможности автоматизации процесса.
Процедуру иммуноанализа ELISA осуществляют следующим образом. Антиген адсорбируют на твердую фазу, а затем твердофазную поверхность экспонируют белком, который не является родственным антигену, например, альбумином бычьей сыворотки (BSA) для блокирования любых участков поверхности, на которых не был адсорбирован антиген. Затем твердую фазу промывают и подвергают обработке серийно разведенным образцом "первичного антитела (например, мышиной сывороткой). Любое антитело против KM31-7 в образце связывается с антигеном. После промывания добавляют "второе" связанное с ферментом антимышиное антитело и оставляют для связывания со связанным мышиным антителом. После промывания добавляют ферментный субстрат, а затем может быть подсчитан титр антитела путем определения степени развития окраски, возникающей в результате разложения субстрата.
(c) Получение клеток миеломы
В качестве источника миеломных клеток предпочтительно использовать зрелые клетки мышиных клеточных линий. Подходящими примерами таких клеточных линий являются клеточная линия миеломы P3-X63 Ag8-U1 (P3-U1), происходящая от 8-азагуанин-резистентных мышей BALB/c (Current Topics in Microbiology and Immunology, 81, 1-7 (1978)), P3-NS 1/1-Ag4.1 (NS-1) (European J. Immunology, 6, 511-519 (1976)), SP2/0-Ag14 (SP-2) (Nature, 276, 269-270 (1978)), P3-X63-Ag8.653 (653) (J.Immunology, 123, 1548-1550 (1978)), и P3-X64-Ag8 (X63) (Nature 256, 495-497 (1975)). Эти клеточные линии могут быть субкультивированы в подходящих средах, таких как 8-азагуаниновая среда (RPM1-1640-среда, содержащая 8-азагуанин, 1,5 мМ глутамин, 5•10-5 М 2-меркаптоэтанол, 10 мкг/мл гентамицина и 10% фетальную телячью сыворотку), среда Дульбекко, модифицированная по способу Исков (IMDM), или модифицированная по способу Дульбекко среда Игла (DMEM). При субкультивировании на нормальной среде известной так же, как полная GIT-среда (5,5 мл MEM-раствора аминокислот, не являющихся незаменимыми (NEAA, Gibco), 27,5 мл CTC109 (Cibco), 6 мл раствора пенициллина-стрептомицина (Sigma) и 11 мл 200 мМ-раствора глутамина в 500 мл GIT-среды (Wako Pure Chemical Industry)) за 3-4 дня до слияния клеток, число клеток на день слияния возрастало по крайней мере до 2•107.
(O) Слияние клеток
Антитело-продуцирующими клетками являются клетки плазмы и их лимфоциты-предшественники. Они могут быть получены
из соответствующих органов животного, таких как селезенка, лимфатические узлы, периферическая кровь или любые их комбинации. Однако обычно используются клетки селезенки.
Антитело-продуцирующие клетки брали через 3 - 5 дней после конечной иммунизации у мышей с заранее определенным высоким титром антител. Затем проводили слияние антитело-продуцирующих клеток с миеломными клетками, полученными, как описано выше в (c). Слияние клеток селезенки с клетками миеломы обычно проводили методом с использованием полиэтиленгликоля, благодаря относительно низкому уровню клеточной токсичности этого соединения и удобству его использования. Процедуру слияния клеток проводили следующим образом.
Клетки селезенки и миеломы тщательно промывали средой или фосфатно-буферным раствором (PBS), а затем смешивали так, чтобы отношение клеток селезенки в клетках миеломы составляло приблизительно 5-10:1, после чего их осаждали центрифугированием. Супернатант удаляли, а скопление клеток тщательно расщепляли, а затем, перемешивая, добавляли размешанный раствор полиэтиленгликоля (ПЭГ), молекулярная масса: 1000 - 4000. Через несколько минут клетки подвергали осаждению путем центрифугирования. Затем супернатант снова отбрасывали, осажденные клетки суспендировали в соответствующем количестве полной GIT-среды, содержащей 5-10 нг/мл мышиного 11-6, и полученную клеточную суспензию переносили в лунки планшетов для культивирования. После проверки каждой лунки на положительный рост клеток среду заменяли HAT-средой (полная GIT-среда, содержащая 5-10 нг/мл мышиного 11-6, 10-6 - 10-3 M гипоксантина, 10-8 - 10-7 M аминоптерина и 10-6 - 10-4 M тимидина).
(e) Селекция колоний гибридом
Планшет для культивирования инкубировали в CO2-инкубаторе в течение 10-14 дней при 35-40oC. В течение этого времени, каждый 1-3 дня, добавляли свежую HAT-среду в количестве, эквивалентном половине количества среды.
Клетки миеломы происходили от 8-азагуанин-резистентной клеточной линии, и клетки миеломы, а также клетки гибридомы, состоящие лишь из одних миеломных клеток, не могут выживать в HAT-среде. С другой стороны, гибридомы, содержащие антитело-продуцирующие клетки, то есть гибридомы, состоящие из антитело-продуцирующих клеток и миеломных клеток, способны выживать на этой среде. Гибридомы, содержащие лишь антитело-продуцирующие клетки, являются нежизнеспособными, а поэтому гибридомы, состоящие из клеток миеломы и антитело-продуцирующих клеток, могут быть отобраны путем культивирования на HAT-среде.
В тех лунках, в которых наблюдалось развитие колоний гибридом, HATИ-среду заменяли HT-средой (которая отличалась от HAT-среды тем, что в ней отсутствовал аминоптерин). Затем часть супернатанта удаляли, а титр антитела против KM31-7 оценивали, например, путем ELISA-анализа.
Описанные выше процедуры относятся к 8-азагуанин-резистентной клеточной линии, однако в целях настоящего изобретения могут быть также использованы и другие клеточные линии при условии, что такое использование позволяет осуществлять селекцию гибридом. Само собой разумеется, что в этом случае необходимо также использовать другой состав среды.
(f) Клонирование
Гибридомы, скринированные, как описано в (e), и проанализированные на продуцирование специфических антител против KM31-7, переносили на другой планшет для клонирования. Существует много различных методов клонирования, которые могут быть использованы в целях настоящего изобретения, например метод рассева (в котором гибридомы подвергают анализу путем серийного разведения так, чтобы каждая лунка содержала лишь одну гибридому), метод клонирования в мягком агаре (в котором засеянные колонии выращивают в мягкой агаровой среде), метод посева (в котором одиночную клетку удаляют с помощью микроманипулятора) и метод клонирования с использованием клеточного сортера (в котором отдельные клетки "сортируют" с помощью клеточного сортера). Чаще всего используется анализ методом серийного разведения благодаря его простоте.
При осуществлении анализа путем серийного разведения клонирование повторяют в 2 до 4 раз для тех лунок, в которых продолжает наблюдаться титр антител. Клон, постоянно продуцирующий антитело против KM31-7, отбирают как целевую гибридому.
(g) Получение моноклонального антитела с помощью культивирования гибридомы
Гибридомы, отобранные, как описано в (f), затем культивируют в ординарной среде. Крупномасштабное культивирование осуществляют путем роторного культивирования либо в большом сосуде для культивирования, либо в центрифуге. Моноклональные антитела к KM31-7 могут быть получены посредством гель-фильтрации клеточного супернатанта, с последующими сбором и очисткой IgG-фракции. Кроме того, гибридома может быть также выращена в брюшной полости мыши того же самого штамма (например, вышеупомянутой мыши BALB/c) или, например, "голой" мыши (NU/NU-мыши). Этот метод может быть очень просто осуществлен, если использовать специальный набор для получения моноклонального антитела (например, MAbTrap GlI of Pharmacia)
(h) Идентификация моноклонального антитела
Определение изотипа и подкласса моноклонального антитела, полученного в (g), может быть осуществлено в соответствии с нижеследующим описанием. В качестве примеров техники идентификации, которая может быть использована в целях настоящего изобретения, могут служить метод Ухтерлони, ELISA-метод и RIA-метод. Хотя метод Ухтерлони является очень простым, однако он имеет тот недостаток, что в нем моноклональное антитело должно быть концентрировано в тех случаях, когда его концентрация является слишком низкой.
В случае использования ELISA или RIA культуральный супернатант может быть непосредственно экспонирован твердой фазой, на которой был адсорбирован антиген. Для идентификации подкласса может быть затем использовано "второе" антитело, специфичное для каждого типа подкласса IgG. Альтернативно, может быть использован набор для изотипирования (например, набор Amersham для изотипирования мышиного моноклонального антитела). Количественная оценка белка может быть осуществлена методом Фолина-Лоури (Folin-Lowry), или путем измерения оптической плотности при 280 нм (1,4 (ОП280)=1 мг/мл иммуноглобулина).
В примерах, приведенных ниже, моноклональное антитело, полученное из гибридомы, обозначенной MKM150-2, оценивали как изотил класса IgG и идентифицировали как антитело, принадлежащее к подклассу IgG1.
Моноклональное антитело, полученное в соответствии с настоящим изобретением, обладает высокой специфичностью по отношению к белку KM31-7. Кроме того, поскольку моноклональное антитело непрерывно и в больших количествах продуцируется путем культивирования вышеописанных гибридом, оно может быть использовано для выделения и очистки белка KM31-7, а поэтому такое выделение и очистка белка KM31-7 при помощи указанного моноклонального антитела путем иммунной преципитации, где используется реакция "антиген-антитело", также является частью настоящего изобретения.
Настоящее изобретение проиллюстрировано нижеприведенными примерами, которые, однако, не должны рассматриваться как некое ограничение изобретения. Все растворы являются водными и получены с использованием деионизированной воды (их количественные соотношения даны в процентах (мас./объем), если это не оговорено особо). Если методы не описываются конкретно, то они могут быть найдены в "Molecular Cloning-A Laboratory Manual (второе издание, Sambrook, J.Fritisch, E.F. & Maniatis (1989), Cold Spring Harbor Laboratory Press). Методы получения растворов и других сред, если это не указано в примерах, даны в последней главе данного описания под названием "Среды". Методы, упоминаемые в примерах без описания их осуществления, объясняются в предыдущих примерах.
A) Исходный материал для CYVV
Листья, инфицированные вирусом желтой прижилковой мозаики клевера и сохраняемые в замороженном состоянии (CY-изолят N 30: Uyeda, 1 и др., (1975), Ann. Phytopath. Soc. Japan 41: 192-203) размалывали с 10 объемами буфера для инокуляции.
Размножение CYVV осуществляли с использованием Vicia faba (сорт Wase-Sora mame) (кормовые бобы), выращенных до второго развитого листа. Развитый лист опрыскивали карборундом (400 меш), а затем инокулировали жидкостью, полученной, как описано выше, с использованием стеклянного шпателя. Через 8-10 дней после инокуляции инокулированный лист как бы покрывался пятнообразной мозаикой. Этот лист вынимали и использовали в качестве источника для CYVV.
B) Очистка вируса
Листья, выделенные, как описано в AX, измельчали в специальной машине для резки, содержащей 3 объема буфера для экстрации. После резки листья измельчали в ступке. Полученный препарат медленно размешивали в течение 1 часа при комнатной температуре, используя мотор, снабженный лопастями для размешивания из нержавеющей стали. Жидкий препарат затем отжимали через двойной слой марли.
Все последующие процедуры осуществляли при 4oC.
Неочищенную жидкость смешивали с половинным объемом хлороформа и полученную смесь размешивали в смесителе Уоринга (Waring), после чего препарат центрифугировали в течение 15 минут при 6000 х g. После центрифугирования водный слой собирали и к этой фракции добавляли полиэтиленгликоль (ПЭГ 6000) до конечной концентрации 4% об/об.
Полученную смесь размешивали на льду в течение 1 часа, а затем оставляли на льду еще на один час. Полученную таким образом жидкость центрифугировали 15 минут при 6000 х g, а преципитат выделяли как вирус-содержащую фракцию. Этот преципитат суспендировали в 50 мл 10 мМ фосфатного буфера (pH 7,4), содержащего 0,5 M мочевины, а затем смешивали с равным объемом тетрахлорметана и энергично размешивали в течение 5 минут, после чего препарат центрифугировали в течение 10 минут при 3000 х g. Затем водную фазу подвергали ультрацентрифугированию при 30000 об/мин в течение 90 минут при 4oC, используя2 при этом ротор Hitachi PP-30. Полученный осадок выделяли как вирус-содержащую фракцию.
Этот осадок суспендировали в 10 мМ фосфатном буфере, содержащем 1% Тритона X100 (pH 7,4), и суспензию центрифугировали в течение 1 минуты при 4oC и при 8000 х g. Полученный супернатант наносили слоем на градуированную колонку с градиентом плотности сахарозы 10-40% (40%:10 мл, 30%:10 мл, 20%:10 мл, 10%: 10 мл, и оставляли в течение ночи при 4oC), который был ранее получен с использованием 10 мМ фосфатного буфера (pH 7,4). Затем пробирку покрывали слоем супернатанта и центрифугировали при 23000 об/мин и при 4oC в течение 120 минут в роторе PP 25 Hitachi.
После центрифугирования колоночные пробирки с градиентом плотности сахарозы фракционировали в аппарате для фракционирования (модель UA-2, изготовленная ISCO Co)., снабженном ОП260 нм-детектором. Фракции, имеющие плотность сахарозы от около 20 до 30% и адсорбирующиеся при ОП260нм, собирали как вирус-содержащие фракции.
Выделенную вирус-содержащую фракцию 2-кратно разводили 10 мМ фосфатным буфером (pH 7,4), после чего подвергали центрифугированию при 40000 об/мин и при 4oC в течение 90 минут с использованием ротора PP-65 Hitachi. Полученный осадок ресуспендировали в 10 мМ фосфатном буфере (pH 7,4), в результате чего получали очищенный вирусный раствор.
C) Выделение вирусной РНК
Геномную РНК вируса CYVV, содержащую примерно 10000 оснований, подвергали полиаденилированию у ее 3'-конца. Сочетание длины и полиаденилирования затрудняет экстракцию полноразмерной РНК без каких-либо потерь, если экстракцию проводят стандартным методом с использованием фенола/додецилсульфата натрия или методом с использованием гуанидинтиоцианата. Поэтому вирусную геномную РНК получали методом центрифугирования в градиенте плотности щелочной сахарозы.
500 мкл вирусного раствора (который содержал 2 мг вируса, как было подсчитано исходя из того, что 1 мг/мл вируса имеет ОП260нм= 0,5), добавляли к 500 мкл раствора для денатурации и полученную смесь оставляли на 20 минут при комнатной температуре. После этого смесь наслаивали на пробирку колонки с градиентом плотности 0% - 34% сахарозы (33,4%:1,4 мл, 30,4%:7,6 мл, 27%: 7,0 мл, 23%:6,3 мл, 18,7%:5 мл, 12%:3,2 мл, 0%:2,7 мл, и оставляли в течение ночи при 4oC), который был получен с использованием 1хSSC. В качестве буфера также использовали 1хSSC. Затем пробирки с нанесенной смесью подвергали ультрацентрифугированию в роторе RPS-27 Hitachi при 24000 об/мин и при 15oC в течение 9 часов. После этого дно пробирки прокалывали для фракционирования градиентов плотности сахарозы. Фракция вирусной геномной РНК идентифицировали путем измерения оптической плотности каждой фракции при 260 нм. Седиментацию вирусной геномной РНК осуществляют при концентрации сахарозы от около 20 до 30%. Выход геномной РНК составлял примерно 25 мкг.
D) Синтез вирусной кДНК
кДНК синтезировали с использованием в качестве матрицы геномной РНК, полученной, как описано выше в стадии (C). Синтез кДНК осуществляли с использованием системы cDNA Synthesis System Plus (изготовленной Amersham). Полученную кДНК очищали на колонке с Сефадексом C50 (зарегистрированный товарный знак, Pharmacia). И с использованием dCTP и концевой дезоксинуклеотид-трансферазы (Bethesda Research Laboratories) добавляли polyG-цепь к 5'-концу очищенной кДНК. Плазмидный препарат pBP322 (Bethesda Research Laboratories) получали путем переваривания указанной плазмиды рестриктирующим ферментом Pst1 с последующим добавлением 3' polyG-цепи с обоих концов. Затем к этому препарату добавляли кДНК и полученную смесь инкубировали 5 минут при 65oC, а после этого инкубировали 2 часа при 57oC. Эту смесь постепенно охлаждали для гибридизации polyG-цепи кДНК с polyG-цепью плазмиды.
E) Трансформация
Штамм HB 101 E/coli трансформировали при помощи новой плазмиды, полученной как описано в стадии (D), методом с использованием хлорида кальция. Посевную культуру штамма HB 101 E.coli получали путем встряхивания в течение ночи в жидкой B-среде. 0,5 мл этой посевной культуры использовали для инокуляции 50 мл свежей жидкой B-среды, и полученный инокулят культивировали, встряхивая, при 37oC до тех пор, пока ОП550нм не становилась равной 0,5. Бактериальные клетки выделяли путем центрифугирования в течение 5 минут при 5000 х g и при 4oC, а полученный осадок слегка суспендировали в 25 мл Трис-кальциевого буфера и оставляли на льду на 5 минут. Образовавшуюся суспензию снова центрифугировали при 4000 х g в течение 4 минут, а остаток суспендировали в 5 мл Трис-кальциевого буфера и оставляли на 2 часа на льду для получения компетентных клеток.
К 200 мкл полученных компетентных клеток добавляли 100 мкл новой плазмиды, полученной в стадии (D), и образовавшуюся смесь оставляли на льду в течение 30 минут. По истечении этого времени смесь инкубировали в течение 2 минут при 42oC, после чего добавляли 1 мл жидкой LB-среды и полученную смесь культивировали, встряхивая, при 37oC в течение еще одного часа. Затем эту смесь наносили на твердую LB-среду, содержащую 12,5 мкг/мл гидрохлорида тетрациклина и 1,5% агар. Клетки культивировали в течение ночи при 37oC для получения библиотеки кДНК-клона.
F) Получение и селекция плазмиды pNS 51
Плазмидную библиотеку рекомбинантных кДНК анализировали методом с использованием щелочного ДСН. Более подробно этот метод описан ниже.
2 мл жидкой LB-среды инокулировали колонией бактерий, резистентных к тетрациклину, но восприимчивых к ампициллину и полученных из твердой LB-культуры, как описано в (E), после чего полученную суспензию культивировали, встряхивая при этом в течение ночи при 37oC. Клетки осаждали путем центрифугирования при 10000 х g и суспендировали в 70 мкл буфера для лизиса, содержащего 10 мкл раствора лизоцима (10 мг/мл). После перемешивания в течение 5 секунд для образования суспензии полученную суспензию оставляли на 5 минут при комнатной температуре. После этого к суспензии добавляли 160 мкл щелочного раствора ДСН и полученную смесь сначала смешивали путем переворачивания пробирки несколько раз, а затем составляли в течение 5 минут на льду. К этой смеси добавляли 120 мкл 5 M ацетата калия, а затем снова оставляли в течение 5 минут на льду. По истечении этого времени смесь центрифугировали при 10000 х g и при 4oC в течение 5 минут.
После центрифугирования супернатант переносили в свежую пробирку и добавляли 250 мкл изопропанола, а затем оставляли на льду в течение 30 минут. По истечении этого времени смесь снова центрифугировали в течение 5 минут при 10000 х g и полученный осадок растворяли в 100 мкл TE-буфера (10 мМ Трис, 1 мМ EDTA (pH 8,0). Затем проводили экстракцию фенолом путем добавления разного количества смеси фенола/хлороформа/изоамилового спирта (25:24: 1) к полученной выше смеси, энергично размешивая при этом, после чего эту смесь центрифугировали при 10000 х g и при 4oC в течение 5 минут (далее, вся эта процедура будет именоваться как экстракция фенолом).
100 мкл водного слоя, полученного в результате центрифугирования, смешивали с 10 мкл 3 М ацетата натрия (pH 5,2) и с 250 мкл этанола, после чего смесь оставляли на сухом льду в течение 10 минут, а затем центрифугировали при 10000 х g в течение 5 минут с выделением фракции нуклеиновой кислоты (далее, эта процедура с использованием тех же самых относительных объемов супернатанта, этанола и раствора ацетата натрия будет называться "осаждение этанолом").
Полученный осадок суспендировали в 50 мл раствора РНКазы A (10 мкг/мл в TE-буфере), а затем инкубировали в течение 1 часа при 37oC. После этого 30 мкл раствора полиэтиленгликоля (2,5 М хлорид натрия, 20% полиэтиленгликоль 8000) добавляли к инкубированной суспензии и полученную смесь оставляли на один час на льду. По истечении этого времени смесь центрифугировали при 10000 х g и при 4oC и выделяли ДНК-фракцию в виде осадка, после чего два раза проводили процедуру осаждения этанола и получали очищенную плазмидную ДНК.
Очищенную плазмидную ДНК переваривали рестриктирующим ферментом PSt 1 и подвергали электрофорезу на 1% агарозном геле с использованием TBE-раствора. После проведения электрофореза гель подвергали Саузерн-блот-гибридизации (Southern E.M. (1975), J. Mol. Biol. 89: 503-517). В частности, гель встряхивали в течение 40 минут в растворе для денатурации, а затем переносили на 2 часа, встряхивая при этом в буфер для нейтрализации.
После встряхивания гель переносили на полиуретановую губку, содержащую 20 х SSC для переноса ДНК на части мембраны Hybond-N (зарегистрированный товарный знак Amersham), находящийся на губке. После перенесения ДНК на мембрану эту мембрану встряхивали в течение 10 минут в 1 х SSC, а затем осушали в течение 1 часа при 80oC для фиксации ДНК. После этого мембрану помещали в раствор для предварительной гибридизации (5 мл формамида, 1 мл 50 х раствора Денхардта, 2,5 мл 20 х SSC, 100 мкл тРНК дрожжей (50 мг/мл), 100 мкл 10% ДСН и 1,3 мл бидистиллированной воды) и инкубировали при 50oC в течение 3 часов.
Вирусную геномную РНК, которая была расщеплена с помощью Mg2+, использовали в качестве зонда. В частности, к раствору (16 мкл), полученному как описано в (C) и содержащему 1 мкг вирусной РНК, добавляли 4 мкл 5x буфера для денатурации и полученную смесь инкубировали 3 часа при 37oC. 5 мкл раствора денатурированной РНК, содержащего 100 нг РНК (расчеты проводили на основании того, что 1 мг/мл РНК имеет ОП260=20, осаждали путем осаждения этанолом с последующим нагреванием при 70oC в течение 5 минут и охлаждением на льду.
4 мкл 5 х буфера для мечения 1 мкл 3,3 мМ [ ν - 32P]АТР (0,37 МБк) и 10 мкл бидистиллированной воды добавляли к раствору денатурированной РНК. Затем к смеси добавляли раствор 20 ед. T4-полинуклеотид-кинаты (поставляемой Takara Shuzo) и инкубировали 30 минут при 37oC. Невключенный [ ν - 32P]АТР удаляли путем 5-кратного осаждения этанолом.
Полученный меченный зонд добавляли к раствору для гибридизации (5 мл формамида, 2 мл 50% сульфата декстрана, 200 мкл 50 х раствора Денхардта, 2,5 мл 20 х SSC, 50 мкл тРНК дрожжей (50 мг/мл), 100 мкл 10% ДСН, 1,3 мл бидистиллированной воды) до конечной концентрации 5 х 105 имп. в мин/мл, после чего в этот раствор переносили мембрану Hybond-N, полученную выше, и подвергали гибридизации путем инкубирования в течение ночи при 50oC, с последующим промыванием при встряхивании, 2 х SSC, содержащим 0,1% додецилсульфата натрия (ДСН). Эту процедуру повторяли три раза, после чего мембрану Hybond-N еще раз промывали 0,1 х SSC, содержащим 0,1% ДСН, в течение 1 часа при комнатной температуре и при встряхивании, а затем осушали.
Авторадиография показала, что плазмида, которая была затем обозначена PNS 51, имела вставленный фрагмент, состоящий примерно из 6500 пар оснований (п.о.), который гибридизировался с вирусной геномной РНК.
G) Определение последовательности вставки pNS51
Для того, чтобы построить рестрикционную карту кДНК, клонированную в pNS51, указанную плазмиду переваривали каждым из рестриктирующих ферментов: BamHI, EcoRI, HindIII, KpnI, NcoI, RstI, SaII, SphI, SacI, SmaI, XhoI, XbaI, Полученная в результате карта показана на фиг.1.
Затем каждый из фрагментов, полученный путем переваривания рестриктирующими ферментами SalI и PstI, вставляли в RF-ДНК M13 mp19. Для этого плазмиду pNS 51 переваривали рестриктирующими ферментами PStI и SaII, а расщепленные продукты подвергали электрофорезу на 5% полиакриламидном геле с использованием 1 х TBE. После проведения электрофореза, гель окрашивали в 1 мкг/мл этидийбромида для того, чтобы можно было идентифицировать полосы под действием УФ-излучения. Полоски геля, содержащие каждую ДНК-полосу, вырезали бритвой и подвергали электроэлюции с использованием устройства для электроэлюции (Amicon), снабженного Centricon-30 (Amicon).
Затем, с использованием набора для лигирования ДНК (изготовливаемого Taka a Shuzo), каждый фрагмент вставляли в M13 mp19, который был предварительно рестриктирован либо одним ферментом SaII, либо двумя ферментами PstI и SaII, и который был обработан щелочной фосфатазой.
Полученную рекомбинантную M13mp19-RF-ДНК (где фрагменты были инсертированы с использованием T4-ДНК-лигазы) вводили в штамм JM109 E.coli методом с использованием хлорида рубидия. Одиночную колонию штамма JM109, которая была культивирована на минимальной агаровой среде M9, инокулировали и культивировали в жидкой SOB-среде в течение ночи при встряхивании. 0,6 мл ночной культуры инокулировали в 50 мл свежей жидкой SOB-среды и культивировали при 37oC и при встряхивании до тех пор, пока ОП600нм не достигала 0?5. Затем клетки осаждали путем центрифугирования при 5000 х g и при 4oC в течение 10 минут и осадок слегка суспендировали в 25 мл ТГВ1-буфера, после чего оставляли на льду в течение 20 минут.
После этого суспензию снова центрифугировали при 3000 х g в течение 5 минут, а остаток суспендировали в 2 мл ТГВ2-буфере и оставляли на льду в течение 20 минут для получения компетентных клеток.
После лигирования рекомбинантную RF-ДНК M13 mp19, полученную, как описано выше, использовали в количестве от 1 до 10 мкл для смешивания с 100 мкл компетентных клеток, после чего смесь оставляли на один час на льду. По истечении этого времени смесь инкубировали при 42oC в течение 1,5 минут, а затем к смеси добавляли 500 мкл жидкой SOC-среды, после чего полученную смесь культивировали в течение 1 часа при встряхивании и при 37oC.
К полученной культуре, размешивая, добавляли 2 х УТ-среду, содержащую 0,8% агар, 30 мкл 100 мМ IRTG, 10 мкл 10% 5-бромо-4-хлоро-3-индолил-1-галактозида (X-gal), и 100 мкл бактерии-индикатора (ночная культура штамма JM109, культивированного при встряхивании в 2 х УТ-среде при 37oC), и полученную смесь выливали на твердую 2 • УТ-среду, содержащую 1,2% агар. После отверждения смесь культивировали в течение ночи при 37oC. Рекомбинантные фаги затем обнаруживали как белые бляшки.
Каждую из полученных белых бляшек инокулировали в жидкую 2 х УТ-среду, содержащую бактерию-индикатор в количестве 1/100, и культивировали в течение ночи при 37oC и при встряхивании. После центрифугирования в течение 1 минуты при 10000 х g супернатант замораживали или хранили при 4oC в виде фагового раствора, а полученный осадок обрабатывали с использованием стандартной процедуры для получения плазмиды с выделением RF-дцДНК (обозначаемой далее RF-ДНК), которая представляет собой промежуточный продукт репликации фаза. Присутствие вставленного фрагмента подтверждалось путем переваривания рестриктирующим ферментом.
Таким образом, получали 51SS/M13mp19 (т. е. M13mp 19, содержащий SaII/SaII-фрагмент, который, в свою очередь, является частью 51PS5'/M13mp19-генома, содержащего RstI-SaII-фрагмент от 5'-области CYVV-генома). RF-ДНК очищали из фаговых клонов с использованием щелочного ДСН-раствора и с помощью центрифугирования в градиенте плотности хлорида цезия в соответствии со стандартными процедурами.
5 мкг полученной очищенной ДНК сначала расщепляли ферментом BamHI с получением липкого 5'-конца, а затем расщепляли ферментом KpnI с получением липкого 3'-конца. После этого, в соответствии со схемой, прилагаемой к системе Erase-a-base (Promega), добавляли экзонуклеазу в целях постадийной делеции оснований из липкого 5'-конца каждой кДНК линеаризованной плазмиды. Обработку экзонуклеазой осуществляли в течение 10 минут, а образцы удаляли в различные промежутки времени от 1 до 10 минут. Поскольку вектор имеет липкий 3'-конец, он не подвергается воздействию экзонуклеазы. Затем различные образцы, полученные в интервале времени 1-10 минут, были затуплены по концам путем обработки S1-нуклеазой и лигированы в кольцевую форму с использованием T4-ДНК-лигазы.
Полученную кольцевую рекомбинантную M13mp-19-РГ-ДНК вводили в штамм JM 109 E.coli методом с использованием хлорида рубидия, описанным выше. В соответствии с этим методом рекомбинантные фаги, имеющие кДНК-вставки различной длины, были получены в виде белых бляшек.
RF-ДНК экстрагировали из субклонов рекомбинантного фага, полученного как описано выше, с использованием стандартной техники в целях отбора клонов, имеющих вставки различной длины. Отобранные клоны отдельно инокулировали в жидкую 2 х УТ-среду, содержащую индикаторный штамм E.coli, и культивировали в течение ночи при 37oC. По истечении этого времени 1,5 мл каждой культуры центрифугировали в течение 5 минут при 10000 х g и при 4oC. 1 мл супернатанта переносили в свежую пробирку и в эту пробирку, размешивая, добавляли 250 мкл 20% полиэтиленгликоля (водный раствор 20% полиэтиленгликоля 8000, 2,5 хлорида натрия), после чего смесь оставляли на 30 минут на льду. Затем эту смесь центрифугировали в течение 5 минут при 10000 х g и при 4oC, а полученный осадок суспендировали в 100 мкл TE-буфера. Затем путем экстракции фенолом и преципитации этанолом (как описано выше), из каждого препарата выделяли фаговые геномные одноцепочечные ДНК (сокращенно обозначаемые далее оцДНК).
После этого методом дидезокси-терминации цепи и с использованием в качестве матрицы оцДНК осуществляли секвенирование нуклеотидов каждого из полученных субклонов. В частности, фаговую оцДНК метили [ α -32P]dCTP (220 ТБк/мМ, Amersham) с использованием набора, содержащего 7-деаза-секвеназу, версия 2,0 dCTP (изготавливаемого USB). Полученный реакционный продукт подвергали электрофорезу в 5% полиакриламидном геле, содержащем 8 М мочевины, с использованием 1 х TBE-буфера. Затем гель осушали, а нуклеотидную последовательность определяли с помощью авторадиографии.
Путем анализа нуклеотидной последовательности 5'-Pst1 SalI-фрагмента и M-SaII-SaII-фрагмента PNS51, нам удалось определить в целом 3839 оснований.
В результате исследований, направленных на определение открытой рамки считывания (обозначаемой далее ORF) в последовательности, полученной выше, на (+)-цепи вирусного генома была обнаружена одна рамка считывания для полипептида. После этого была определена последовательность полипептида, кодируемая в ORF. Исследования, проводимые путем сравнения этой полипептидной последовательности с известными последовательностями с использованием генного банка последовательностей (GeneBank), выявили гомологию этого полипептида с TEV-HAT.
Известно, что такие вирусы, как TEV, "оспа" сливы (скрытая мозаика сливы), или полиовирус (вирус животных) созревают под действием протеазы, и что эта протеаза обычно расщепляет пептидную связь, содержащую Gln-Ala, Gln-Ser или Gln-Gly-связь (Willink J. & Van Kammen A. (1988), Arch. Virol 98: 1-26). В результате анализа сайтов расщепления белка оболочки вируса CY - N 30 (Uyeda I. et al. (1991), Intervirology 32: 234-245) также удалось обнаружить три расщепляемые последовательности в выведенной полипептидной последовательности. Исходя из одного из этих сайтов расщепления, было установлено, что полипептид, простирающийся от Gly в 4 положении до Gln в 437 положении, представляет собой "ядерное включение a", происходящее от вируса CYVV.
H) Получение гибридного белка CYVV-Nla/1L-11
ДНК, идентифицированная выше (см. п. (G)) как последовательность: кодирующая CYVV-Nla, тандемно связана, с сохранением рамки считывания, у своего 3'-конца с ДНК, кодирующей интерлейкин 1L (IL-11 человека, посредством линкерной последовательности, кодирующей Gln-Ala.
При этом были проведены следующие исследования:
1) кодирует ли идентифицированная в п. (G) ДНК "ядерное включение а" (Nla), обладающее протеолитической активностью,
2) расщепляет или нет указанная протеаза (т.е. Nla) нужную аминокислотную последовательность,
3) обладает ли Nla протеазной активностью в том случае, если оно является частью гибридного белка вместе с нужным гетероциклическим белком, и является ли эта активность такой же, какой является активность в вирус-инфицированной клетке,
4) сохраняет ли гетероциклический белок, полученный посредством расщепления указанного гибридного белка, свою целостность, структуру и функцию.
Поскольку Nla-белок, сам по себе, продуцируется посредством отщепления от вирусного полипептида-предшественника, то Nla-цистрон не имеет инициирующего кодона. Поэтому в 5'-концу Nla-гена необходимо добавить инициирующий кодон ATG. Кроме того, 3'-конец Nla-гена должен быть также присоединен к 5'-концу гена IL-11 с сохранением рамки считывания. Для того, чтобы решить все поставленные задачи, Nla модифицировали при помощи полимеразно-цепной реакции (PCR) с использованием рестрикционного XhoI-сайта, присутствующего в центре Nla-гена.
Для добавления инициирующего кодона ATG и сайта узнавания для рестриктазы NcoI, необходимого для клонирования в 5'-конец Nla (Nla5'), получали PCR-праймеры. Эти праймеры имели следующие последовательности: 5' GTCCATGGGGAAAAGTAAGAGAACA 3' (обозначаемая далее как NSATG, последовательность ID N 3) и 5' ACTCTGAGACCGTGCTCGAG 3' (обозначаемая далее как NSX1, последовательность ID N 4).
0,8 мкл dNTR-раствор (25 мМ каждого из dATA, dTTP, dCTP и dCTP), 10 х Tag-буфер (изготавливаемый Promega) и 1 мкг каждого из полученных праймеров добавляли к 1 мкг pNS51-плазмидной ДНК и полученную смесь доводили до 100 мкл путем добавления бидистиллированной воды. Затем для осуществления полимеразно-цепной реакции добавляли 5 ед ДНК-полимеразы в 10 х Tag-буфере. PCR проводили по следующей схеме: цикл - 1 мин при 92oC, 1 мин при 37oC и 2 мин при 72oC - повторяли 20 раз после одного цикла - 1 мин, 92oC, 1 мин, 37oC и 30 мин 72oC.
Полученную амплифицированную ДНК подвергали процедурам экстракции фенолом и осаждению этанолом, а затем электрофорезу на 5% полиакриламидном геле. С помощью окрашивания бромидом этидия обнаруживали одну полосу, содержащую ДНК, эту полосу вырезали из геля и подвергали электроэлюции с использованием аппарата для электроэлюции Centreluter (Amicon), снабженного Centricon-30 (Amicon). После проведения элюции элюированный ДНК-фрагмент концентрировали и выделяли путем центрифугирования при 7500 х g (с использованием Centricon, Amicon) и при 4oC в течение 45 минут. Полученный ДНК-концентрат затем очищали путем экстракции фенолом и осаждение этанолом.
Независимо от этого 1 мкг плазмиды pKK388 (Clonetech, в которой SacI-сайт узнавания был заменен XhoI-сайтом узнавания, расщепляли рестриктирующими ферментами NcoI и XhoI, а затем дефосфорилировали кишечной щелочной фосфатазой теленка (обозначаемой далее CIAR и изготавливаемой Takara Shuzo. Полученную ДНК лигировали при помощи набора для легирования Takara Shuzo) с 100 нг плазмиды pKK388-1, которая также была рестриктирована ферментами NcoI и XhoI и дефосфорилирована, как описано выше, в результате чего получали рекомбинантную ДНК, которую затем клонировали в JM 109 E.coli. Таким образом получали плазмиду pKN15', которая имела вставку, находящуюся в нормальной ориентации и расположенную "ниже" от trc-промотора плазмиды pKK388-1 (фиг. 2).
Плазмида pCD20-2 (Kawashima 1. et. al., (1991), FEBS L 283: 199-202) содержала кДНК, кодирующую предшественник IL-11 (pre-IL-11) и имеющую последовательность сигнала секреции. Эту плазмиду переваривали рестриктирующими ферментами BamHI и ApaI и вырезали область, содержащую pre-IL-11 и промотор SV40. Полученный фрагмент лигировали в BamHI- и ApaI-сайты вектора pBLUESCRIPT 11 SK+, а затем снова расщепляли рестриктирующими ферментами XhоI и KpnI, в результате чего получали ген, кодирующий белок, лишенный N-конца зрелого IL-11 (Mat-IL-11).
Полученный Mat-IL-11-фрагмент (лишенный своего N-конца) интегрировали с использованием T4-лигазы в pKN15', которая была предварительно рестриктирована ферментами XnoI и KpnI, а также обработана CIAP. Полученная конструкция была обозначена pKN141 (фиг.3). Для осуществления лигирования специфической последовательности в C-конце Nla с фрагментом Mat-IL-11 посредством Ala и с сохранением рамки считывания были синтезированы четыре вида PCR-праймеров:
5'AGGAAAAGACTTCCTCGACC 3'
(обозначенный NSX2, последовательность ID N 5),
5' AATTGTTCATTCCAAGCACCTGGGCCACCACCTGGC 3'
(обозначенный NSJ001P, последовательность ID N 6),
5' GCCAGGTGGTGGCCCAGGTGCTTGGAATGAACAATT 3'
(обозначенный NSJ002) последовательность ID N 7), и
5' TTGTCAGCACACCTGGGAGCTGTAGAGCTC 3'
(обозначенный ILSAC последовательность ID N 8).
Первую PCR-реакцию осуществляли (с использованием пары праймеров NSX2 и NSJ002N и с использованием ДНК pNS51 в качестве матрицы) для амплификации области вставки, кодирующей C-конец Nla (обозначенной CN13-областью). Затем отдельно осуществляли другую PCR-реакцию (с использованием пары праймеров NSJ001 и 11SAC и с использованием в качестве матрицы ДНК pNS51) для амплификации области вставки, кодирующей N-конец-IL-11-пептида (обозначенной 5' IL-областью). PCR осуществляли по следующей схеме: цикл - 1 мин при 92oC, 1 мин при 37oC и 2 мин при 72oC - повторяли 20 раз, а затем проводили один цикл / 1 мин при 92oC, 1 мин при 37oC и 30 мин при 72oC. Продукты, полученные в результате полимеразно-цепной реакции, экстрагировали фенолом и осаждали этанолом, а затем подвергали электрофорезу на 5% полиакриламидном геле. Полоску геля, содержащую амплифицированную ДНК-полосу, вырезали и ДНК отделяли от геля методом электроэлюции, описанным выше. Результаты показаны на фиг. 4.
Два полученных амплифицированных ДНК-фрагмента представляли собой области праймеров NSJ001P и NSJ002N, т.е. 3'-концевую часть CIN 3-ДНК-фрагмента и 5'-часть 5'IL-ДНК-фрагмента, нуклеотидны, последовательности которых обнаруживали частичную гомологию. Так, например, если снова провести полимеразно-цепную реакцию с использованием праймеров NSX2 и ILSAC, то гомологичные участки будут гибридизироваться с образованием гибридной ДНК, содержащей оба гена, при этом гомологичный участок будет действовать как сшивающая область (фиг. 5). В соответствии с этим, выделенный CIN3-ДНК-фрагмент и 5'II - ДНК-фрагмент смешивали и снова проводили PCP с использованием в качестве праймеров лишь NSX2 и ILSAC. PCR осуществляли по следующей схеме: сначала проводили 10 циклов в режиме: 1 мин при 92oC, 1 мин при 37oC и 2 мин при 72oC, затем 20 циклов в режиме: 1 мин при 92oC, 1 мин при 45oC и 2 мин при 72oC, а после этого проводили один цикл в режиме: 1 мин при 92oC, 1 мин при 55oC и 30 мин при 72oC. После обработки фенолом и осаждения этанолом осуществляли электрофорез на 5% полиакриламидном геле, в результате чего получали CNI3IL-ДНК-фрагмент, в котором CNI3 был присоединен к 5'IL. Этот метод показан на фиг. 5.
CNI3IL-ДНК-фрагмент расщепляли рестриктирующим ферментом XhoI и полученный фрагмент, с использованием T4-ДНК-лигазы, вставляли в pKN15', которая была предварительно расщеплена рестриктирующим ферментом XhoI и дефосфорилирована CIAP. Полученную плазмиду клонировали в штамм JM 109 E. coli. Для отбора клона, в котором CNI3IL-фрагмент был вставлен в правильной ориентации, плазмиды экстрагировали из полученных клонов и с использованием праймеров NSX2 и ILSAC осуществляли PCR. В соответствии с используемой системой обнаруживали лишь одну ДНК-полосу от клона, в котором данный фрагмент был инсертирован в правильной ориентации. В результате была получена плазмида pKSUN9, в которой Nla и Mat-IL-11 соединены посредством расщепляемой последовательности Gln-Ala с сохранением рамки считывания (фиг. 6).
1) Экспрессия Ala-IL-11 в бактериальной клетке
E.coli, несущая плазмиду pK 9 (обозначенная далее штаммом KSUN9), культивировали при встряхивании и при 37oC в течение ночи в 5 мл LB-среде, содержащей 42 мкг/мл ампициллина. Затем к 250 мл свежей LB-среды (содержащей такое же количество ампициллина) добавляли 2,5 мл полученной KSUN 9 - культуры и культивировали при встряхивании и при 37oC до тех пор, пока ОП600нм не достигала значения 1,0. После того, как ОП600нм становилось равным 0,1 мМ, добавляли IPTG до конечной концентрации 0,1 мМ и полученную смесь культивировали при встряхивании и при 28oC в течение 12 часов.
Второе культивирование проводили в полном соответствии с вышеописанной процедурой, за исключением того, что последнее культивирование осуществляли при 28oC в течение 36 часов или более с концентрацией IPTG 1 мМ, в результате чего получали зрелый IL-11 (который имел N-концевой пролин).
J) Вестерн-блоттинг
Вестерн-блоттинг осуществляли для того, чтобы определить является ли pKSUN 9 функциональной в E. coli, а также определить экспрессируется ли сконструированный рекомбинантный ген.
Каждую из 12- и 36-часовых культур, индуцированных с помощью IPTG, подвергали последующей обработке, описанной ниже. Для этого из 2 мл культуры путем 5-минутного центрифугирования при 3000 х g и при 4oC выделяли клетки, полученный в результате центрифугирования осадок суспендировали в 100 мкл 20 мМ натрийборатного буфера (доведенного до pH 9,0 с помощью 0,1 н. NaOH). Полученную суспензию обрабатывали ультразвуком с использованием специального аппарата (Handysonic UR-20P, изготовленного Tomy Seiko Co.) в течение 2 минут при работе аппарата на позиции 8, для дезинтеграции клеток, а затем, в течение 10 минут, осуществляли центрифугирование при 10000 х g и при 4oC. Супернатант, содержащий растворимую белковую фракцию, выделяли. 5 мкл этого супернатанта подвергали электрофорезу в 12% полиакриламидном геле с ДСН в соответствии с методом Лаэммли (Laemmli (1970) Nature 227, 680 - 685).
После электрофореза гель в течение 5 минут встряхивали в буфере для переноса (25 мМ Трис, 192 мМ глицина, 20% метанола) и блотировали на часть PVDF-мембраны (Thans-Blot Transfer Medium - среда для переноса, изготовленная Bio-Rad Laboratories) путем обработки при 15 В в течение 1 часа в специальной камере Trans-Blot-SD-Semi-Dry Transfer Cell (изготовленной Bio-Rad Laboratories). Блотированную PVDF-мембрану промывали в PBS-TW-среде в течение 10 минут. PVDF-мембрану переносили на PBS-TW-среду, содержащую 5% сепарированное молоко (изготовленное Snow Brand Milk Products), и оставляли на 1 час при 37oC для блотирования.
После блотирования PVDF-мембрану снова промывали в PBS-TW-среде, один раз в течение 10 минут и два раза в течение 5 минут, а затем переносили на кроличью сыворотку против IL-11, предварительно 10000-кратно разведенную в PBS-TW, и инкубировали 20 минут при 37oC. По истечении этого времени PVDF-мембрану снова промывали в PBS-TW, один раз в течение 10 минут и 4 раза по 5 минут.
После этой промывки PVDF-мембрану обрабатывали реагентом для усиленного продуцирования хемилюминесценции (ECL) (изготовленного Amersham) и полосы, которые реагируют с антителом против IL-11, обнаруживают путем экспонирования указанной PVDF-мембраны с рентгеновской пленкой в течение периода времени от 30 сек до 5 мин.
В результате проведения Вестерн-блоттинга, как описано выше, сигнальные полосы, соответствующие молекулярным массам около 50 кДа и около 23 кДа, обнаруживали от 12-часовой и 36-часовой культур. 23 кДа-полоса имела подвижность в основном аналогичную подвижности зрелого IL-11, используемого в качестве контроля. Таким образом, очевидно, что 230-кДа-сигнальная полоса представляет собой IL-11, который был отщеплен от Nla/IL-11 гибридного белка Gln-Ala-линкерной последовательности благодаря протеолитической активности Nla. Отсюда можно сделать вывод, что более тяжелая полоса (50 кДа) представляет собой нерасщепленный гибридный белок.
Далее, 23 кДа-белок, полученный от 12-часовой культуры, будет обозначаться 23 кДа-ON, а 23-кДа-белок, полученный от 36-часовой культуры, будет обозначаться 23-кДа-36hr.
K) Очистка белков 23 кДа-ON и 23 кДа-hr
Белки 23 кДа-ON и 23 кДа-36hr были очищены для определения аминокислотной последовательности их N-концов. В соответствии с процедурой, описанной в п. (1), получали 250 мл каждой из 12-часовой и 36-часовой культур. Затем каждую культуру обрабатывали следующим образом. Культуру центрифугировали 15 минут при 5000 х g, при 4oC, а осадок суспендировали в 10 мл 20 мМ боратного буфера (pH 9,0), а затем подвергали дезинтеграции с использованием пресса Френча и аппарата для обработки ультразвуком (того и другого). После центрифугирования в течение 30 минут при 15000 х g и при 4oC растворимую белковую фракцию выделяли в виде супернатанта.
Полученную растворимую белковую фракцию подвергали слабой ионообменной колоночной хроматографии с использованием FPLC (Pharmacia) при следующих условиях:
Колонка: CM Toyopearl Pack 650 М (2,2 х 20 см, изготовлена Toso)
Буферы для элюирования: A = 10 мМ борной кислоты - гидроксид натрия (pH 9,0), 13 мМ хлорида калия, B = 10 мМ борной кислоты - гидроксида натрия (pH 9,0), 13 мМ хлорида калия, 400 мМ хлорида натрия.
Скорость потока: 2,5 мл/мин.
Объем фракции: 5 мл на пробирку
Используемый градиент концентрации (в течение 120 мин) представлял собой линейный градиент "элюент A - элюент B"
Затем каждую элюированную фракцию подвергали твердофазному иммуноферментному анализу (ELISA) для идентификации фракций, содержащих IL-11.
ELISA осуществляли следующим образом. Каждую лунку 96-луночного иммунопланшета (Max oap, изготовленный Nunc.) загружали 100 мкл 50 мМ натрийкарбонатного буфера (pH 9,6), содержащего 1 мкг/мл мышиного моноклонального антитела против IL-11, а затем инкубировали в течение 1 часа при 37oC. По истечении этого времени каждую лунку промывали 4 раза PBS-T-средой (PBS, содержащий 0,1% Твин 20).
Каждую из FPLC-фракций, полученных как описано выше, разбавляли 1:100 PBS-T и загружали в лунки в аликвотах 100 мкл на лунку. Планшеты инкубировали в течение 1 часа при 37oC, после чего лунки снова промывали PBS-T и в каждую лунку добавляли 100 мкл кроличьего IgG против IL-11, разбавленного PBS-T до конечной концентрации 1 мкг/мл.
Затем планшет инкубировали в течение 1 часа при 37oC и промывали PBS-T, после чего в каждую лунку добавляли 100 мкл меченного щелочного фосфатазой козьего антитела против кроличьих IgG (Gibco Bethesda Research Laboratories), которое было 3000-кратно разбавлено в PBS-T. Планшет снова инкубировали в течение 1 часа при 37oC, а затем промывали PBS-T. После этого в каждую лунку добавляли 100 мкл раствора субстрата щелочной фосфатазы. Планшет инкубировали при комнатной температуре еще от 30 минут до 1 часа, после чего окрашивание каждой лунки измеряли как оптическую плотность при 450 нм в целях идентификации нужной (или нужных) фракции (фракций).
Фракции, полученные в результате FPLC-процедуры и идентифицированные в соответствии с вышеописанным анализом ELISA как фракции, содержащие вещество, реагирующее с кроличьим антителом против IL-11 (фракции NN 19-25), объединяли 100-кратно концентрировали с использованием Centprep-10 (Amicon) и подвергали ДСН-электрофорезу в 12% полиакриламидном геле по методу Лаэммли (см. выше). Затем гель подвергали электроблотированию на PVDF-мембране в соответствии с вышеописанной процедурой для Вестерн-блоттинга, за исключением того, что в качестве PVDF-мембраны использовали мембрану Problot (изготовленную Applied Biosystems).
После проведения блотирования мембрану тщательно промывали бидистиллированной водой, окрашивали кумасси бриллиантовым голубым P-250, обрабатывали 50% метанолом для отмывки окраски и полосу, содержащую белок, который вступает в реакцию с антителом против IL-11 в Вестерн-блот-анализе, вырезали.
Аминокислотную последовательность N-конца данного белка анализировали с использованием белкового секвенатора (Applied-Biosyntems). В результате этого анализа было установлено, что N-концевая последовательность полосы от 23 кДа-ON, реагирующей с антителом против IL-11, представляет собой:
Ala-Pro-Gly-Pro-Pro-Pro-Gly- (последовательность ID N 9)
Эта последовательность соответствует последовательности от -1 до +6 аминокислотной последовательности зрелого IL-11-белка. Исходя из этого, можно сделать вывод, что 23 кДа-белок, полученный от 12-часовой культуры и вступающий в реакцию с антителом против IL-11, представляет собой Ala-IL-11. В соответствии с этим очевидно, что этот белок был продуцирован посредством протеолитической активности Nla-белка, расщепляющего Nla/IL-11-гибридный белок в Gln-Ala-сайте в специфической расщепляемой последовательности.
Кроме того, было установлено, что N-концевая последовательность полосы от 23 кДа-36hr, реагирующей с антителом против IL-11, представляет собой:
Pro-Gly-Pro-Pro-Pro-Gly-Pro- (последовательность ID N 10)
Эта последовательность соответствует аминокислотной последовательности от +1 до +7 зрелого IL-11-белка. Исходя из этого, были сделаны следующие выводы:
В результате индуцирования с помощью IPTG, гибридный белок Nla/IL-11 экспрессируется в E.coli.
В результате 12-часового культивирования после индуцирования продуцированный Nla/IL-11-гибридный белок расщепляется в Gln-Ala-пептидной связи со специфической расщепляемой
последовательности благодаря протеазной активности Nla.
После расщепления пептидной связи Nla-белком зрелый, но имеющий дополнительный Ala на своем N-конце IL-11 продуцируется в E.coli.
При культивировании еще 24 часа после экспрессии Ala-IL-11 было установлено, что Ala-IL-11 "созревает" до IL-11, в котором Ala-остаток, присутствующий в N-конце Ala-IL-11, делетируется.
На основании полученных результатов был сделан вывод, что 23 кДа-белок, полученный после 36-часового культивирования, представляет собой зрелый тип IL-11-белка, в котором N-концом является Pro.
В соответствии с этим было установлено, что IL-11 может быть продуцирован как гибридный с Nla белок, и что благодаря активности Nla этот белок должен расщепляться в специфической линкерной последовательности, содержащей Gln-Ala, с образованием Ala-IL-11. Последующее культивирование приводит к образованию зрелого IL-11, в котором аланиновый остаток делетируется благодаря фактору, присутствующему в E.coli, в результате чего N-концом становится Pro.
Для того, чтобы убедиться в том, что продуцированные IL-11 и Ala-IL-11 являются биологически активными, в качестве показателя такой активности определяли активность адипогенез-ингибирующего фактора (AGIF). Тот факт, что IL-11 обладает адипогенез-ингибирующей активностью, был установлен ранее (Kawashima, 1, et al. (1991), FEBSL. 283: 199-202).
Оценка ингибирующего действия на морфологические изменения от ЗТЗ-LI-клеток до адипоцитов
Ниже описывается метод определения AGIF-активности, используемый в настоящем изобретении. В этом методе использовали клеточную линию ЗТЗ-L1 эмбриональных мышиных фибробластов (Green H. & Kehinde (1974), Cell 1: 113-116), взятую из ATCC. Во всех случаях эти клетки культивировали в смешанной влажной атмосфере (10 CO2 - 90% воздух) при 37oC и пересевали в среду A. Индуцирование адипогенной дифференцировки осуществляли в соответствии с процедурой, описанной Rubin и др. (Rubin C.S. et al. (1978), J. Biol. Che,. 253: 7570-7578).
Клетки ЗТЗ-L1 суспендировали в среде A до достижения плотности 1,0 х 104 клю/мл, засевали в 48-луночный планшет для культивирования микроколоний (0,5 мл/лунка, изготавливаемый Coaster), а затем культивировали. После 3-дневного культивирования клетки достигали сплошности. Затем среду заменяли свежей средой A и после культивирования в течение еще 2 дней эту среду заменяли на адипогенез-индуцирующую среду (среду B), одновременно добавляли при этом 0,5 мл испытуемого образца. Эту среду заменяли свежей средой B с добавлением свежего образца каждые два дня.
В различные промежутки времени для различных лунок, начиная с 4 и по 7 день после добавления первого испытуемого образца, истощенную среду B в лунках заменяли адипоцит-поддерживающей средой C.
После культивирования в среде C в течение 2 дней клетки фиксировали 5% формальдегидом и все жирные частицы, которые были аккумулированы в клетках и клеточных ядрах, окрашивали масляным красным O и гематоксилином соответственно. По полученным микрофотографиям подсчитывали число ядер и число клеток, которые содержали аккумулированные окрашенные жировые частицы. Адипогенное отношение (AR) вычисляли по следующему уравнению:
Фиксацию клеток и окрашивание масляным красным O и гематоксилином осуществляли в соответствии с процедурами, описанными Yoshio Mitomo и Shojiro Takayama в "Lectures on Clinical Testing" Vol. 12, "Pathology" (1982), опуб. Ishiyaku Shuppan
M) Метод определения ингибирующей активности липопротеин-липазы
Определение ингибирующей активности липопротеин-липазы осуществляли в соответствии с методом, описанным Beutler и др. (Beutler B.A. et al. (1985) J. Immunol 135: 3972-3977). Адипогенетически дифференцированные клетки ЗТЗ-L1 получали, как описано в п. (1), за исключением того, что при индуцировании дифференцировки клеток в адипоциты испытуемого образца не добавляли. Вместо этого испытуемый образец добавляли вместе со свежей средой C, после чего клетки культивировали в течение 18 часов. По истечении этого времени среду удаляли, а клетки дважды промывали PBS(-) (фосфатно-буферным раствором, изготавливаемым Nissui Seiyaku), после чего в каждую лунку добавляли 3000 мкл среды D и культивировали еще 1 час. Затем от каждого из супернатантов культуры брали 100-микролитровые аликвоты для измерения активности липопротеинлипазы, которую измеряли в тройном дубликате для каждого образца для получения среднего значения.
Активность липопротеин-липазы (LPL) измеряли, как описано Nilsson-Ehle & Schotz (Nilsson-Ehle P. & Schotz M.C. (1976), J. Lipid Res. 17: 536 - 541). Каждую из аликвот супернатанта, полученные как описано выше, смешивали с равным объемом субстратного раствора и оставляли на 120 минут при 37oC для осуществления реакции. Реакцию прекращали путем добавления 1,05 мл 0,1 М калийкарбонатного буфера (доведенного до pH 10,5 с помощью 0,1 М бората калия) и 3,25 мл смеси метанола/хлороформа/гептана (141 : 125 : 100 (объем/объем)) при интенсивном размешивании. Затем полученную смесь центрифугировали в течение 15 минут при 3000 х g и при комнатной температуре. После этого с помощью жидкостного сцинтилляционного счетчика подсчитывали содержание 3H в воднометаноловой фазе.
За одну единицу LPL-активности принимали такую активность, которая генерирует 1 мкМ жирной кислоты в минуту. 13 мМ три[9.10(n)-3H] олеата глицерина в субстратном растворе получали путем разбавления три-[9.10(n)-3H] олеата глицерина (37,0 ГБк/М) (изготавливаемого Amersham) триолеином (изготавливаемого Sigma), с последующей очисткой с помощью колоночной хроматографии на силикагеле.
В нижеприведенных примерах проиллюстрирован вариант осуществления настоящего изобретения, который относится к глутатион-восстанавливающему белку.
Пример 1
Экстрагирование Poly(A)±РНК из клеток KM-102
Клетки KM-102 выращивали в 36 пластиковых чашках для культивирования с диаметром 15 см, в минимальной эссенциальной среде, модифицированной по методу Исков (Boeringer Mannheim) и содержащей 10% фетальную бычью сыворотку. После выращивания клеток до сплошности добавляли форболмиристилацетат (PMA) и инофор кальция A23187 (Sigma) до конечных концентраций 10 нг/мл и 0,2 мкМ соответственно и культивирование осуществляли при температуре 37oC. Через 3, 6 и 14 часов собирали партии из 12 чашек и содержимое каждой чашки отдельно растворяли в растворе гуанидинтиоцианата, а жидкую фазу собирали.
Выделение Pol(A)+РНК осуществляли в основном в соответствии с описанием, приведенным в "Molecular Cloning A Laboratory Manual [Maniatis, T. et al. (1982) стр. 196-198]. Ниже приводится подробное описание этой процедуры.
Каждую выделенную жидкую фазу отдельно обрабатывали следующим образом. Жидкость повторно набирали и выливали из поршня 10 мл шприца, снабженного шприцевой иглой 21G. 3-Миллилитровый раствор 5,7 М CsCl - 0,1 М ЭДТА (pH 7,5) предварительно добавляли в пробирку центрифуги Polyoromar, подобранную в соответствии с размером лопасти ротора центрифуги RPS40-T (Hitachi Koki). Полученные клетки наносили поверх раствора, содержащегося в пробирке, до тех пор, пока пробирка не становилась заполненной.
После 18-часового центрифугирования при 3000 об/мин и при температуре 20oC полученный осадок суспендировали в 400 мкл дистиллированной воды и осаждали путем добавления этанола. Образовавшийся осадок растворяли в 400 мкл дистиллированной воды и полученный раствор, перемешивая, добавляли к равному объему смеси хлороформа/1-бутанола (4:1, об/об), а затем водный слой собирали путем центрифугирования. Осажденный этанол снова центрифугировали один раз и полученный осадок растворяли в 600 мкл дистиллированной воды, в результате чего получали целую РНК. Через 3, 6 и 14 часов из каждого собранного PMA/A23187-стимулированного образца было получено приблизительно 4,5 мг целой РНК.
600 мкг каждой из трех типов целой РНК, полученной способом, описанным выше, объединяли и подвергали хроматографии на колонке с олиго(dT)-целлюлозой для очистки poly(A)+РНК.
Целую РНК растворяли в буфере для адсорбции, а затем нагревали в течение 5 минут при 65oC. Полученный раствор наносили на колонку с олиго(dT)-целлюлозой (Pharmacia, тип 7), загруженную адсорбционным буфером, и элюировали элюирующим раствором, в результате чего выделяли 100 мкг poly(A)+РНК.
Пример 2
Получение кДНК-библиотеки
кДНК-библиотеку получали способом, описанным Okayama Beg.
5 мкг poly(A)+РНК и 24 единицы обратной транскриптазы (Seikagaku Corp.) оставляли для реакции на один час при 42oC в 20 мкл реакционного раствора обратной транскриптазы.
После этого реакцию прекращали путем добавления 2 мкл 0,25 М ЭДТА и 1 мкл 10% ДСН, а затем полученный раствор депротеинизировали с использованием 20 мкл смеси фенола и хлороформа (1:1, об/об). После центрифугирования в целях удаления фракций, содержащих белок, к супернатанту добавляли 20 мкл 4 М ацетата аммония и 80 мкл этанола, а затем полученную смесь охлаждали в течение 15 минут при температуре -70oC. По окончании этой процедуры осадок собирали посредством осаждения центрифугированием, промывали 75%-ным этанолом и осушали при пониженном давлении.
Осушенный осадок растворяли в 15 мкл реакционного раствора концевой трансферазы и нагревали при температуре 37oC в течение 3 минут. По истечении этого времени 18 единиц концевой дезоксинуклеотидилтрансферазы (Pharmacia) добавляли к реакционному раствору и полученную смесь оставляли для реакции на 5 минут. После этого реакцию прекращали путем добавления 1 мкл 0,25 М ЭДТА и 0,5 мкл 10% ДСН и полученный раствор депротеинизировали смесью фенола/хлороформа (как описано выше), а затем центрифугировали для удаления фракций, содержащих белок. Супернатант собирали и энергично перемешивали с 15 мкл и 4 М ацетата аммония и 60 мкл этанола. Полученную таким образом смесь охлаждали при температуре -70oC в течение 15 минут, а осадок собирали путем центрифугирования.
Полученный осадок растворяли в 10 мкл буфера для рестрикции ферментом и к этому раствору добавляли 2,5 единиц рестрикционного фермента HindIII, после чего раствор оставляли на 1 час при температуре 37oC для переваривания.
Затем реакционный раствор депротеинизировали смесью фенола/хлороформа, после чего осаждали этанолом и полученный супернатант охлаждали в течение 15 минут при температуре -70oC. Полученный осадок собирали путем центрифугирования и растворяли в 10 мкл TE-буфера [10 мМ Трис-HCl при pH 7,5 и 1 мМ ЭДТА] . 1 мкл полученного раствора добавляли к 10 мкл реакционного раствора, содержащего 10 мМ Трис-HCl (pH 7,5), 1 мМ ЭДТА, 100 мМ хлорида натрия (NaCl), и 10 нг линкерной ДНК с олиго(dT)-хвостом [pL-1-HindIII-линкер с 3'-олиго(dG)-хвостом, Pharmacia, после чего полученную смесь нагревали при температуре 65oC в течение 5 минут, а затем нагревали при температуре 42oC в течение 30 минут. Эту реакционную смесь охлаждали ледяной водой с последующим добавлением 10 мкл 10х лигазного буфера, 78 мкл дистиллированной воды и 8 единиц T4-ДНК-лигазы. Полученный реакционный раствор выдерживали в течение ночи при температуре 12oC.
На следующий день реакционную смесь объединяли с 10 мкл 1 М KCl, рибонуклеазой H (1 единица), 33 единицами ДНК-полимеразы 1, 4 единицами T4-ДНК-лигазы, 0,5 мкл раствора dNTP (20 мМ dATP, 20 мМ dCTP, 20 мМ dGTP и 20 мМ dTTP) и 0,1 мкл 50 мкг/мл альбумина бычьей сыворотки (BSA). Полученную таким образом смесь нагревали в течение часа при температуре 12oC, а затем в течение часа при температуре 25oC. По истечении этого времени реакционный раствор пять раз разводили дистиллированной водой, а затем непосредственно использовали для трансформирования DH5 E.coli методом [Hana-Hanahan D.D. (1983 J. Mol. Biol 166, 557 - 580], в результате чего получали кДНК-библиотеку клеток КМ-102.
Пример 3
Получение олигонуклеотидного зонда
Исходя из последовательности AUUUA в 3'-нетранслированной области мРНК цитокинов, был химически синтезирован олигонуклеотид 5'-TAAATAAATAAATAA-3', состоящий из 15 оснований (последовательность ID N 13) и обозначенный как ATT-3. Синтез проводили с использованием автоматического ДНК-синтезатора 380B (Perkin-Elmer Japan Applied Biosystems) в соответствии с указаниями, прилагаемыми в сопровождающей инструкции. Эту процедуру осуществляли фосфитамидным способом, описанным Caruther et al. (Matteucci, M.D. и Caruthers, M.H. (1981) J. Am. Chem. Soc. 103, 3185-2191]. После синтеза 15-мера полученный олигонуклеотид отделяли от носителя, а защитные группы удаляли. Затем раствор олигонуклеотида лиофилизовали и получали порошок, который растворяли в дистиллированной воде и хранили при температуре -20oC вплоть до его использования.
Пример 4
Скринирование кДНК-библиотеки
6500 колоний, генерированных из кДНК-библиотеки, полученной в примере 2, фиксировали на нитроцеллюлозном фильтре в соответствии со способом, описанным Grunstein & Hogness (Grunstein, M & Hogness, D.S. (1975) Proc. Acad. Sci США 72, 3691-3695] . ATT-3-зонд, полученный в примере 3, помечали по 5'-концу с помощью 32P и в соответствии со стандартными процедурами (см. "Molecular Cloning-A Laborotory Manual") и меченный зон использовали для гибридизации колоний.
Предварительную гибридизацию осуществляли при температуре 37oC в течение 3 часов, используя следующие компоненты: 6 х SSC, 1 х раствор Денхардта, 0,25% ДСН, 0,05% пирофосфата натрия и 100 мкг/мл денатурированной ДНК спермы лосося. Затем, в течение ночи, при температуре 31oC осуществляли гибридизацию с использованием следующих компонентов: 6 х SSC, 1 х раствора Денхардта, 17 мкг/мл дрожжевой транспортной РНК и 0,05% пирофосфата натрия, содержащего 32P-меченный зонд ATTT-3.
На следующий день нитроцеллюлозный фильтр промывали при температуре в течение 2 часов 6 х SSC-раствором, содержащим 0,05% пирофосфата натрия. После авторадиографии было выявлено 33 положительных клона.
ДНК-плазмиду экстрагировали из положительных клонов в соответствии со стандартными процедурами. Затем произвольно отбирали несколько клонов и их частичные нуклеотидные последовательности кДНК определяли с помощью дидезокси-метода. Эти частичные последовательности оценивали на гомологию с нуклеотидными последовательностями, зарегистрированными в банке данных EMBL или Geneank, с помощью персонального компьютера, в результате чего было установлено, что некоторые частичные последовательности клонов, обнаруженные с помощью ATT-3, имели гомологию с частями AIu-повтора (Schmid, C.W. & Jelinek, W.R. (1982) Science, 216, 1065-1070).
ДНК-фрагмент, содержащий последовательность AIu-повтора, получали из геномной ДНК человека и помечали с помощью 32P с использованием стандартных процедур. Эту меченную ДНК использовали в качестве зонда при гибридизации колоний с использованием 33 клонов, идентифицированных выше, в результате чего было установлено, что 12 клонов имели AIu-повтора. Затем определяли длину кДНК-вставки каждого из оставшихся 21 клонов, и было установлено, что длина кДНК-вставки менялась в пределах от 50 до 3600 оснований.
Картирование рестрикционного фермента осуществляли на кДНК-вставках из оставшегося 21 клона, а частичные нуклеотидные последовательности были определены способом, описанным выше. Эти частичные последовательности оценивали на гомологию (как описано выше) с нуклеотидными последовательностями, зарегистрированными в банке данных EMB или GenBank, с помощью персонального компьютера, в результате чего были отобраны клоны, имеющие новые последовательности.
Пример 5
Нозерн-гибридизация клона N 31
Один из клонов, а именно клон N 31 (обозначенный как pcD-31), имел кДНК-вставку, имеющую примерно 560 пар оснований. В соответствии с процедурой, описанной в примере 1, PST1-Aat1-фрагмент (292 п.о.), полученный из кДНК-вставки pcD-31 и помещенный 32P, служил в качестве зонда в Нозернблот-процедуре, с использованием poly(A)+РНК, полученной из клеток KM-102. Эту гибридизацию использовали для определения длины природной мРНК, соответствующей вставке клона pcD-31.
Процедуру Нозерн-гибридизации осуществляли следующим образом. 5,5 мкг poly(A)+ РНК получали из клеток KM-102, а затем инкубировали в течение часа при температуре 50oC в смеси 1 М гликосаля, 50% диметилсульфоксида (ДМСО) и 0,01 М динайтрийбифосфата (pH 7,0). По истечении этого времени 4 мкл пигмента для электрофореза добавляли к инкубированной смеси, которую затем подвергали электрофорезу на 1% агарозном геле в 1 х TAE-растворе.
После электрофореза, РНК на агарозном геле переносили и оставляли на ночь в найлоновом мембранном фильтре (BioPad, Z-зонд) (с использованием 20 х SSC) методом капиллярного переноса (см. "Molecular Cloning-A Laboratory Manual"). После процедуры переноса фильтр слегка промывали 2 х SSC-раствором, осушали воздухом, а затем дополнительно осушали при температуре 80oC в течение 2 часов для фиксации мРНК.
PstI-Aat1-фрагмент клона pcD-31 помечали с помощью 32P с использованием стандартной системы мечения ДНК (Amersham)
Предварительную гибридизацию осуществляли на фильтре в течение 3 часов при температуре 37oC в растворе, содержащем 5 х SSCP, 2,5 х раствор Денхардта, 50% формамида, 10 мМ динатрийбифосфат (pH 7,0), 0,5% ДСН и 100 мкг/мл денатурированной ДНК спермы лосося.
Затем гибридизацию осуществляли на фильтре в течение ночи при температуре 42oC в растворе, содержащем 32P-меченный зонд, 5 х SSCP, 1 х раствор Денхардта, 50% формамид, 10 мМ динатрийбифосфат (pH 7,0), 0,1% ДСН и 100 мкг/мл денатурированной ДНК спермы лосося. На следующий день фильтр промывали в течение ночи при температуре 37oC раствором, содержащем 50% формамид, 5 х SSC и 0,1% ДСН, а затем промывали в течение 2 часов при той же температуре раствором, содержащем 40% формамида, 5 х SSC и 0,1% ДСН, и наконец промывали при комнатной температуре в течение 15 минут раствором 2 х SSC. В результате авторадиографии было установлено, что кДНК-вставка клона pcD-31 имеет неполную длину, а полная длина соответствующей мРНК составляет 3,9 kb (определено с помощью калибровочной кривой на основании маркера молекулярной массы).
Пример 6
Получение свежей библиотеки для скрининга кДНК клона pcD-31
Свежую кДНК-библиотеку получали с использованием системы синтеза кДНК (cDNA Synthesis Systems Plus) и системы клонирования кДНК ( λ gt10, адаптерный метод, Amersham
5 мкг poly(A)+РНК экстрагировали из клеток KM-102 (в соответствии с процедурой, описанной в примере 1) и 100) единиц обратной транскриптазы подвергали реакции в течение 40 минут при температуре 42oC в 50 мкл раствора для реакции обратной транскриптазы. По истечении этого времени 20 мкл [ α -32P] dCTP, 93,5 мкл буфера 4 единицы рибонуклеазы H и 115 единиц ДНК-полимеразы 1 (все они были включены в набор) добавляли к реакционному раствору, который сначала инкубировали при температуре 12oC в течение часа, а затем при температуре 22oC в течение часа, и наконец нагревали в течение 10 минут при температуре 70oC. После нагревания к реакционному раствору добавляли 10 единиц T4-ДНК-полимеразы (включенной в набор), и полученную смесь оставляли для реакции на 10 минут при температуре 37oC.
Затем реакционную смесь депротеинизировали с использованием смеси фенола/хлороформа. Реакционный раствор подвергали центрифугированию и супернатант собирали и перемешивали тщательно с 250 мкл 4 М ацетата аммония и 1 мл этанола. Полученную смесь охлаждали в течение ночи при температуре -20oC, а образовавшийся осадок собирали посредством центрифугирования. После этого осадок растворяли в 30 мкл стерилизованной воды. 10 мкл полученного раствора удаляли и добавляли к смеси 2 мкл лигазного/киназного буфера. 250 пМ EcoRI-адаптера и 5 единиц T4-ДНК-лигазы (все они были включены в набор), после чего полученную смесь инкубировали в течение ночи при температуре 15oC.
Весь реакционный раствор наносили на колонку для фракционирования по размерам, с использованием набора для удаления EcoRI-адаптера. Затем реакционный раствор собирали в 120 мкл аликвот и каждую аликвоту смешивали с 200 мкл 0,25 х TE-буфера. Фракции (с 10 по 17) объединяли и концентрировали с использованием бутанола до получения полного объема 120 мкл.
Весь объем концентрированного продукта смешивали с 55 мкл стерилизованной воды, 20 мкл лигазного/киназного буфера и 40 единицами T4-полинуклеотид-киназы (все они были включены в раствор), после чего полученную смесь инкубировали в течение 30 минут при температуре 37oC. По истечении этого времени реакционную смесь депротеинизировали 3 раза с использованием смеси фенола/хлороформа, а затем осаждали этанолом и охлаждали в течение ночи при температуре -20oC. Полученный осадок собирали посредством центрифугирования и растворяли в 10 мкл стерилизованной воды, в результате чего получали образец кДНК.
1 мкг EcoRI ( λ gt10), 1 мкл лигазного/киназного буфера и 2,5 единиц T4-ДНК-лигазы (все они были включены в набор) добавляли к 2 мкл полученного выше образца кДНК и эту смесь инкубировали в течение ночи при 15oC. Образец, содержащий 4 мкл образца кДНК, был получен тем же самым образом. После этого каждый образец обрабатывали следующим образом. Весь полученный реакционный раствор сначала добавляли к 10 мкл Экстракта A (включенного в набор, а затем смесь добавляли к 15 Экстракта B (включенного в набор) и полученную таким образом смесь инкубировали при 20oC в течение 20 минут для осуществления реакции in vitro-упаковки.
По истечении этого времени к реакционному раствору добавляли 470 мкл SM-буфера и полученную смесь хранили при температуре 4oC. Штамм M514 E.coli обрабатывали сульфатом магния (10 мМ) и инфицировали с использованием вышеописанного раствора, с последующим получением λ gt-10-кДНК-библиотеки из клеток KM-102.
Пример 7
Скринирование кДНК-библиотеки
2 • 105 бляшек, полученных из объединенных кДНК-библиотек (полученных в примере 6), фиксировали на найлоновых фильтрах (Hyobnd N, Amersham) в соответствии со следующей процедурой.
Инфицированные E.coli, полученные в примере 6, культивировали в десяти 9 см-планшетах, содержащих твердую LB-срезу, таким образом, чтобы на одном планшете количество бляшек составляло от 1 до 2 • 104. После этого бляшки переносили на планшет путем мягкого вдавливания найлонового фильтра в планшет. Иглу шприца 18G использовали для прокалывания фильтра и маркировки геля в 3 местах для сравнения. Затем фильтр оставляли на 5-10 минут при температуре 4oC, после чего очищали и погружали в щелочной раствор (0,1 н. гидроксид натрия и 1,5 М хлорид натрия) на 20 секунд. Фильтр переносили в нейтральный раствор [0,2 М Трис-HCl (pH 7,5), 2 х SSCP] на период времени от 20 секунд до одной минуты и осушали воздухом при комнатной температуре в течение 2 часов. Затем этот фильтр осушали при температуре 80oC в течение 2 часов.
Пример 8
Получение зонда и гибридизация
32P-меченные зонды конструировали из клона pcD-31 (полученного в примере 4) путем мечения pstI-Aat-фрагмента и EcoT221-Aat1-фрагмента (223 п.о.) с использованием стандартной системы для мечения ДНК (Multiprime). Гибридизацию бляшек осуществляли на фильтре, полученном в примере 7, с использованием вышеописанных зондов.
Предварительную гибридизацию осуществляли путем помещения фильтра в баню, содержащую 50% формамида, 5 х SSCP, 2,5 х раствора Денхардта, 0,01 М динатрийбифосфат (pH 7,0), 0,5% ДСН, и 100 мкг/мл денатурированной ДНК спермы лосося. Полученную смесь инкубировали в течение 2 часов при температуре 37oC.
Затем гибридизацию осуществляли путем помещения фильтра в реакционный раствор, содержащий 32P-меченные зонды, полученные, как описано выше, в также, 50% формамида, 5 х SSCP, 1 х раствор Денхардта, 0,01% М динатрийбифосфат (pH 7,0), 0,1% ДСН и 100 мкг/мл денатурированной ДНК спермы лосося, а затем полученную смесь инкубировали в течение ночи при 37oC.
На следующий день фильтр промывали в течение трех часов при комнатной температуре раствором, содержащим 50% формамида, 5 х SSC-раствор, и T, 1%-ный ДСН, а затем промывали в течение пяти минут при комнатной температуре 2 х SSC-раствором. В результате авторадиографии было выявлено 80 положительных клонов, полученных при первичном скрининге.
С использованием клонов, идентифицированных как положительные (в каждому случае), процедуры примеров 7 и 8 повторяли еще три часа (четвертичный скрининг) и в конце концов получали всего 17 положительных клонов. Из каждого из 17 клонов выделяли кДНК, а вставки вырезали ферментом EcoR1. Длину каждой кДНК-вставки исследовали с использованием электрофореза на агарозном геле, после чего был выделен клон N 31-7, имеющий кДНК-вставку, состоящую из 3,9 п.о., и соответствующую полной длине исходной мРНК.
Пример 9
Рестрикционное картирование клонов N 31-7
Клоны N 31-7 переваривали ферментом EcoR1 для выделения и очистки 3,9 kb-фрамгента, содержащего кДНК-вставку. Этот фрагмент вставляли в pUC18 с использованием T4-ДНК-лигазы. α DH5. E.coli трансформировали с помощью новой плазмиды. Трансформированные клетки отбирали по мере их устойчивости к ампициллину, а клон pUCKM31-7, имеющий вставку кДНК, с 3,9 к.о., идентифицировали путем переваривания ДНК ферментом EcoRI и полученную расщепленную ДНК подвергали электрофорезу на агарозном геле.
pUCKM31-7 расщепляли с помощью каждого рестрикционного фермента HindIII, SacI, XbaI, SmaI, BgIII, EcoT22I и AatI или их частей. Полученные фрагменты подвергали электрофорезу на агарозном геле, после чего длину каждого фрагмента измеряли с использованием λ HindIII/фX174HaeIII-маркера, применяемого в качестве индикатора. Полученная рестрикционная карта представлена на фиг. 7.
Пример 10
Определение последовательности клона N 31-7
Полную нуклеотидную последовательность кДНК-вставки плазмиды pUCKM-31-7 определяли дидезокси-методом с использованием фага M13. Кроме того, часть данной последовательности анализировали с помощью ДНК-секвентатора 373A (Perkin Elmer Japan Applied Biosystems). Полученная нуклеотидная последовательность проиллюстрирована в Списке последовательностей под номером 1D N 11.
кДНК-вставка плазмиды pUCKM-31-7 имеет длину 3815 оснований и, кроме того, имеет, очевидно, открытую рамку считывания, состоящую из 549 аминокислот, начиная с метионина. poly(A)-хвост, очевидно, отсутствует. Посредством сравнения последовательности оснований у 3'-конца вставки плазмиды pcD-31 с последовательностью клона pUCKM-31-7 было установлено, что во вставке клона pUCKM-31-7 отсутствует лишь часть poly(A)-хвоста (фиг. 8) и не более того.
Нуклеотидный банк данных EMBL и GenBank и банк данных NBRF и SWISS-PROT позволяет сравнивать последовательности оснований и аминокислот соответственно. При этом было обнаружено, что наиболее близким соответствием является 35,3% гомология данной пептидной последовательности и последовательности редуктазы глутатиона человека. В соответствии с этим был сделан вывод, что ORF кДНК-вставки плазмиды pUCKM31-7, очевидно, кодирует новый полипептид. Этот новый полипептид показан как последовательность в нижеприведенном списке последовательностей под номер ID N 12.
Пример 11
Экспрессирование и очистка нового полипептида
Построение высокоэкспрессирующего вектора и экспрессия его в клетках COS-1.
Плазмиду pUCKM31-7 переваривали ферментом HindIII, а 3003 п.о.-фрагмент, содержащий кДНК-вставку, выделяли и очищали в соответствии со стандартными процедурами. Концы полученного фрагмента затупляли с использованием набора для затупления ДНК (Takara Shuzo).
Между тем, высокоэкспрессирующий вектор pcDL-SR α 296 [Takabe, Y. et al. (1988) Mol. Cell. Biol. 8, 466-472] переваривали ферментами PstI и KPnI, а концы затупляли с использованием набора для затупления ДНК. После этого затупленную вставку легировали в затупленную плазмиду путем реакции с использованием T4-ДНК-лигазы. E.coli трансформировали с помощью полученной ДНК методом с применением хлорида кальция и полученные AmpR-трансформанты отбирали, а ДНК-плазмиду, удерживаемую микроорганизмами, анализировали.
В частности, штамм, в котором направление транскрипции кДНК идентично направлению промотора SR α, отбирали путем переваривания плазмиды ферментами HindIII и BgIII, с последующим электрофорезом на агарозном геле для выделения 800 п. о. -фрагмента и эту отобранную плазмиду обозначали pUR α 31-7 (фиг. 9). Промотор SR α содержит исходный промотор SV40 и последовательность R-U5 длинного концевого повтора (LTP) HTLV-1, а также обладает промоторной активностью, в 10 и 100 раз превышающей активность лишь одного исходного промотора SV 40.
Клетки COS-1 трансфецировали полученной плазмидой pSR α 31-7. Трансфекцию клеток COS-1 осуществляли путем электропорации с использованием устройства для введения гена GTE-1 (Shimadzu).
COS-1-клетки культивировали до состояния полусплошности по всей поверхности дна семи 150 см3-колб, каждая из которых содержала 25 мл DMEM (содержащей 10% фетальной сыворотки теленка). Затем культуры собирали и каждую из них обрабатывали 3 миллилитрами раствора Трипсин-EDTA (10 х раствор, после чего оставляли при комнатной температуре до тех пор, пока клетки не отделялись. Затем добавляли 1 мл инактивированной фетальной телячьей сыворотки и 9 мл свежего раствора трипсина-EDTA и клетки собирали путем центрифугирования. Собранные клетки два раза промывали PBS(-)-буфером и суспендировали в PBS(-)-буфере до достижения плотности 6 • 107 кл./мл.
Кроме того, методом с использованием хлорида цезия получали плазмидную ДНК, которую разводили до концентрации 200 мкг/мл в PBS(-)-буфере.
20 мкл каждого из вышеупомянутых препаратов клеток и плазмиды смешивали и полученную смесь помещали в камеру, содержащую электроды, расположенные друг от друга на расстоянии 2 мм. После этого смесь подвергали воздействию двух импульсов в 600 B через интервал времени 1 сек и продолжительностью 30 мксек.
Камеру с электродами охлаждали при 4oC в течение 5 минут, после чего смесь клеток и ДНК, находящуюся внутри, смешивали с 10 мл DMEM, содержащей 10% фетальной телячьей сыворотки. Затем эту смесь переносили в чашки Петри и культивировали в течение ночи при 37oC в 5% CO2-атмосфере. На следующий день супернатант культуры отбрасывали, а клетки промывали бессывороточной DMEM, суспендировали в 10 мл DMEM и культивировали 3 дня при 37oC. По истечении этого времени клеточный супернатант собирали.
Культуральный супернатант также собирали от негативного контроля. Плазмида, используемая для негативного контроля, а именно плазмида pcDL-SR α 296, не содержала кДНК-вставки, но в остальном она была получена так же, как и испытуемая культура.
1 мл каждого из культуральных супернатантов негативного и контроля и испытуемой культуры отдельно обрабатывали следующим образом. Сначала супернатант обрабатывали трихлороуксусной кислоты (TCA) для осаждения белка и полученный в результате осадок собирали путем центрифугирования. Этот осадок промывали охлажденным льдом ацетоном, осушали воздухом, а затем растворяли в ДСН-ПААГ-буфере для образца, содержащем 2-меркаптоэтанол. После этого проводили ДСН-ПААГ-электрофорез на 12%-ном геле в восстановительных условиях.
Полосы, полученные после электрофореза, окрашивали серебром с использованием реагента "Daiichi" (Daiichi Chemical Detection). Несколько специфических полос (молекулярная масса около 60000) от культурального супернатанта испытуемого образца были окрашены.
Поскольку молекулярная масса полипептида, кодируемого в pSR α 31, составляет около 60000, и поскольку, исходя их аминокислотной последовательности, маловероятно, что посттранскрипционная модификация способствовала добавлению сахаридных боковых цепей, то можно сделать вывод, что эти несколько специфических полос относятся к полипептиду, кодируемому кДНК pSR α 31-7.
Пример 12
Получение высокоэкспрессирующей плазмиды для клеток COS-1
Нижеописанную стадию осуществляли для того, чтобы убедиться в том, что несколько специфических 60 кДа-полос, идентифицированных в примере 11, аналогичны полипептиду, кодированному вставкой SR α 31-7. Было бы также желательно определить N-концевую аминокислотную последовательность этого полипептида. В соответствии с этим был получен клон, в котором были кодированы шесть дополнительных His-остатков для C-конца полипептида, кодируемого pSR α 31-7-вставкой, расположенных непосредственно перед стоп-кодоном. Гистидиновые остатки имеют высокую степень сродства к Ni2+, а поэтому были бы желательно продуцировать полипептид, содержащий гистидиновый гессамер (6 х His), который мог бы быть очищен с использованием аффинной колонки, загруженной Ni2+.
Первый олигонуклеотид, состоящий из 66 оснований, 5'-CTAGCGCTCTGGGGCAAGCAТCCTCCAGGCTGGCTGCCACCACCACCACCACCACT- GATCTAGACT-3' (последовательность ID N 14) и комплементарную цепь (66 оснований) синтезировали и очищали с использованием автоматического ДНК-синтезатора 394 (Perkin-Elmer Japan Applied Biosystems). Оба олигонуклеотидных препарата смешивали и инкубировали 3 минуты при 70oC, а затем для осуществления отжига дополнительно нагревали еще 30 мин при 37oC. После этого концы фосфорилировали с использованием T4-полинуклеотид-киназы.
Полученный двухцепочечный (дц) фрагмент лигировали в pUCKM31-7 с использованием T4-ДНК-лигазы, причем указанную pUCKM31-7 предварительно переваривали ферментом Eco47III. Эта конструкция показана на фиг. 10. С использованием кальцийхлоридного метода и указанной ДНК трансформировали DH5 α E. coli и полученные трансформированные штаммы отбирали и скринировали, в результате чего получали pUCKM31-7His. Путем анализа соответствующей последовательности оснований указанной pUCKM31-7His, в которую был встроен фрагмент, было установлено, что в этой части pUCKM31-7His какие-либо отклонения отсутствуют.
Путем субклонирования вставки pUCKM31-7His в pcDL-SR α 296 была получена высокоэкспрессирующая плазмида для COS-1-клеток.
pUCKM31-7His переваривали ферментами XbaI и HindIII, полученные фрагменты очищали, а концы фрагментов затупляли с использованием 1 ед. фрагмента Кленова и присутствии 2 нМ dATP, 2 мМ dCTP, 2 мМ dGTP, 2 мМ dTTP, 50 мМ Трис-HCl (pH 7,2), 10 мМ MgSO4, 0,1 мМ дитиотреитола и 50 мкг/мл BSA.
Высокоэкспрессирующий вектор pcDL-SR α 296 переваривали ферментами PstI и KpnI и затупляли по концам с использованием набора для затупления ДНК. Затем затупленные фрагменты лигировали в затупленную плазмиду с использованием T4-ДНК-лигазы. Полученную плазмиду использовали для трансформации DH5 α E. coli. Трансформанты отбирали и скринировали. Отбирали штамм, в котором направление транскрипции кДНК идентично направлению SR α - промотора, и плазмиду этого штамма обозначали pSR α 31-7His. COS-1-клетки трансфецировали полученной плазмидой pSR α 31-7His, а бессывороточный супернатант получали в соответствии с процедурой, описанной в примере 11.
Пример 13
Очистка и анализ N-концевой аминокислотной последовательности
600 мл супернатанта, полученного в примере 12, подвергали диализу против 17 объемов буфера для диализа в течение 15 часов при 4oC. Затем этот буфер заменяли еще 17 объемами буфера для диализа, после чего диализ продолжали еще 4 часа при 4oC.
Диализованный препарат подвергали аффинной хроматографии с использованием FPLC (Жидкостная экспресс-хроматография белков - Pharmacia) при следующих условиях:
Колонка: 20 мл смолы ProBondTM (Invitrogen), загруженной в XK16/20 (2.0 x 20 см, Pharmacia)
Элюирующий буфер
A) 20 нМ фосфатного буфера (pH 7,8), содержащего 200 мМ имидазола, 0,5 M NaCl
B) 20 мМ фосфатного буфера (pH 7,8), содержащего 300 мМ имидазола, 0,5 M NaCl
Скорость потока: 1 мл/мин.
Раствор фракции: 5 мл/пробирка
Условия элюирования: после сбора 4 фракций элюирующим буфером (A) собирали 16 фракций элюирующим буфером (B) и эти фракции пронумеровывали от 1 до 20.
От каждой из полученных фракций брали 300 мкл образца и каждый образец отдельно обрабатывали TCA, после чего полученный осадок подвергали ДСН-ПААГ-электрофорезу с использованием 12,5%-ного геля в условиях восстановления, как описано выше. Путем окрашивания серебром обнаруживали три полосы, сосредоточенные вокруг фракции N 10. Существование трех полос указывали на то, что pSR α 31-7His-вставка кодирует полипептид, имеющий 3 последовательности различной длины с различными N-концевыми последовательностями.
Оставшиеся фракции 7-14 концентрировали путем TCA-осаждения и полученный осадок подвергали ДСН-ПААГ-электрофорезу с использованием 10%-ного геля в восстановительных условиях. Затем полосы белка переносили из полиакриламидного геля на поливиниллидиндифторидную (PVDF) пленку (ProBlotTM, Applied Biosystems), используя устройство для переноса геля на мембраны (Marisol, KS-8441), работающего при 9 B, в присутствии буфера для переноса (0,02% ДСН, 20% метанол, 25 мМ Трис-бората (pH 9,5)), на 2,5 часа, при 4oC.
После этого мембрану окрашивали 0,2% нафтоловым темно-синим (Sigma), три полосы, соответствующие предварительно идентифицированным полосам, вырезали из мембраны, после чего с помощью газофазного белкового секвенатора определяли последовательность каждый из них до 6-ой аминокислоты от N-конца. N-конец полосы, имеющей вторую самую большую кажущуюся молекулярную массу (мол. мас. около 60000), имел следующую последовательность:
Val-Val-Phe-Val-Lys-Gln (аминокислоты NN 1-6 последовательности ID N 12).
Эти шесть аминокислот соответствовали первым шести аминокислотам ORF от клона 31-7, а также соответствовали последовательности из шести аминокислот, начиная с 24 аминокислоты (VAl) с N-конца предшественника полипептида, кодируемого кДНК-вставкой pSR α 31-7H и pSR α 31-7. В соответствии с этим делеция аминокислот 1-23 из N-конца этого предшественника должна приводить к высвобождению зрелой формы белка, начинающегося с Val-остатка.
Пример 14
Определение восстанавливающей активности
i) Конструирование вектора экспрессии
Полипептид, очищенный, как описано в предыдущих примерах, мог быть получен лишь в исключительно малых количествах, поскольку он был экспрессирован из COS-1-клеток. Поэтому этот полипептид не может быть использован в других целях, например для анализа активности. В соответствии с этим было бы желательно найти способ экспрессии полипептида, кодируемого кДНК-вставкой pSR α 31-7, в альтернативном хозяине, что позволило бы продуцировать этот полипептид в количествах, достаточных для очистки и анализа. Для достижения этой цели осуществляли следующую процедуру.
pUCKM31-7 переваривали ферментом HindIII, 3003 п.о.-фрагмент, содержащий кДНК-вставку, выделяли и очищали, а концы затупляли с использованием набора для затупления ДНК. Затем фрагмент переваривали ферментом XbaI.
Экспрессирующий вектор pMAL-c (Guan, C. et al. (1987) Gene 67, 21-30) переваривали ферментами XoaI и StuI, а затем вышеуказанный XbaI-модифицированный HindII-фрагмент лигировали в эту рестриктированную плазмиду с использованием T4-ДНК-лигазу. Полученная конструкция показана на фиг. 11. Эту конструкцию затем использовали для трансформации ЕИ-1 E.coli и полученные AmpP-трансформанты отбирали и скринировали. После этого отбирали штамм, в котором направление кДНК-транскрипции было идентично направлению промотора, и полученную таким образом плазмиду обозначали pMAL31-7.
II) Экспрессия и очистка гибридного белка.
Посевную культуру E.coli, содержащую pMAL31-7, получали путем культивирования в течение ночи, встряхивая при этом при 37oC в 3 мл LB-среде, содержащей 50 мкг/мл ампициллина. На следующий день 1 мл посевной культуры добавляли к 100 мл свежей культуральной LB-среды, содержащей 50 мкг/мл ампициллина, и культивировали при встряхивании и при 37oC до тех пор, пока ОП600нм не достигнет значения 0,5. На этой стадии к культуре добавляли IPTG до конечной концентрации 0,1 мМ, после чего культуральный бульон культивировали при встряхивании и при 37oC в течение ночи.
На следующий день из ночной культуры выделяли бактериальные клетки путем центрифугирования при 6500 об/мин и при 4oC в течение 20 минут. Полученный осадок суспендировали в 10 мл колоночного буфера, а клетки в этой суспензии дезинтегрировали путем обработки ультразвуком. Целые клетки и клеточные фрагменты удаляли путем центрифугирования в течение 30 минут при 8800 об/мин и при 0oC, после чего фракцию растворимого белка выделяли в виде супернатанта. 1 мл этой растворимой фракции подвергали хроматографии на колонке с амилозной смолой (New England Biolabs).
Элюирующий буфер для хроматографии получали путем добавления мальтозы к 10 мл колоночного буфера до конечной концентрации 10 мМ.
Образец негативного контроля также хроматографировали. Этот негативный контроль получали в соответствии с той же самой процедурой, за исключением того, что вектор pMAL-c не содержал какой-либо кДНК-вставки. Затем анализировали восстанавливающую активность белка в хроматографических образцах.
iii) Определение восстанавливающей активности
Определение восстанавливающей активности осуществляли в кювете (SARSTEDT, 10 x 4 x 45 мм) с использованием дихлорофенола-индофенола (DCIP) и окисленного глутатиона.
a) Определение восстанавливающей активности с использованием DCIP
90,4 мкг (как было определено с использованием набора для анализа белков (Bio-Rad) от каждого хроматографического образца, полученных в (11) отдельно смешивали с 1 мл 50 мкМ DCIP (Sigma). Затем к каждому из образцов добавляли 15 мкл 1 мМ NADPH (Boehringer-Mannheim) и постоянно контролировали ОП600нм и ОП340нм. Полученное снижение оптической плотности при обеих длинах волн проиллюстрировано на фиг. 12, откуда видно, что лишь pMAL31-7-образец содержит фактор, который восстанавливает DCIP.
b) Определение восстанавливающей активности с использованием окисленного глутатиона
15 мл 10 мМ окисленного глутатиона (Boerihger-Mannheim) добавляли к 90,4 мкг каждого из хроматографических образцов, полученные как описано в (11), и предварительно загруженных в отдельные кюветы. К каждой кювете добавляли 15 мкл 1 мМ NADPH и следили за изменением оптической плотности при 340 нм (ОП340) с течением времени. Результаты представлены на фиг. 13, из которого видно, что только белок от pMAL31-образца обладает способностью восстанавливать окисленный глутатион. По наблюдениям было установлено, что в том случае, если окисленный глутатион отсутствует, то поглощения NADPH не происходит, и исходя из этого был сделан вывод, что белок от pMAL31-7-образца может восстанавливать окисленный глутатион лишь в присутствии NADPH.
Пример 15
Очистка и анализ N-концевой аминокислотной последовательности
Исходя из примера 13 был сделан вывод, что COS-1-клетки, трансфецированные pSR α 31-7H-вставкой, продуцируют полипептид, имеющий три типа N-концов.
В отдельном эксперименте, для получения препарата поликлонального антитела против KM31-7-блока проводили иммунизацию кроликов с использованием гибридного белка, выделенного из E.coli, трансформированных pMAL31-7. Затем с использованием этого поликлонального антитела осуществляли Вестерн-блоттинг, в котором указанные три типа полос были также обнаружены в бессывороточном культуральном супернатанте, полученном от COS-1-клеток, трансфецированных pSR α 31-7. Эти результаты аналогичны результатам, полученным в примере 13.
В соответствии с этим COS-1-клетки были трансфецированы pSRα 31-7 в целях получения большого объема бессывороточного культурального супернатанта для очистки и анализа N-концевой последовательности KM31-7.
COS-1-клетки трансфецировали pSR α 31-7 и культивировали в течение 3 дней в 150 мм-чашках Петри, каждая из которых содержала 30 мл DMEM. По истечении этого времени культуральный супернатант собирали и в каждую чашку добавляли 30 мл свежей среды, после чего культивирование продолжали еще 3 дня. Затем культуральный супернатант снова собирали. В примере 11 были описаны другие варианты трансфекции и культивирования, но независимо от этого было культивировано 199 чашек.
Собранные супернатанты объединяли и после центрифугирования собирали 10 литров бессывороточного культурального супернатанта, который затем диализировали в течение ночи против 10 мМ Трис-HCl (pH 9,0). Полученные диализованные препараты восемь раз подвергали ионообменной хроматографии с использованием FPLC (Pharmacia) при следующих условиях:
Колонка: 20 мл DEAE-сефарозы для экспресс-хроматографии (Pharmacia), загруженной в XK16/20 (φ 2,0 x 29 см, Pharmacia)
Элюирующие буферы:
A) 10 мМ Трис-HCl (pH 9,0)
B) 10 мМ Трис-HCl (pH 9,0) - 0,5M NaCl
Скорость потока: 1 мл/мин.
Раствор фракций: 3 мл/пробирка
Условия элюции: Изменение элюирующего буфера A на элюирующий буфер B в линейном градиенте концентраций в течение 60 минут.
Эти фракции, элюированные при каждой концентрации NaCl от 0,1 M до 0,4 M, собирали и объединяли, а затем в течение ночи диализовали против буфера для диализа, содержащего 0,1 M Трис-HCl, 5 мМ EDTA (pH 7,6) и 1 мМ 2-меркаптоэтанола. Диализованный препарат подвергали аффинной хроматографии с использованием 2', 5', -ADP-Сефарозы 4B (Pharmacia) при следующих условиях:
Колонка: 20 мл 2',5'-ADP-Сефарозы, загруженные в XK16/20 (φ 2,0 x 20 см, Pharmacia).
A) 0,1 M Трис-HCl, 5 мМ EDTA (pH 7,6), 1 мМ 2-меркаптоэтанол
B) 0,1 M Трис-HCl, 5 мМ EDTA (pH 7,6), 1 мМ 2-меркаптоэтанол, 10 мМ NADPH.
Скорость потока: 0,5 мл/мин.
Раствор фракции: 2 мл/пробирка
Условия элюции: Изменение элюирующего буфера A на элюирующий буфер B в линейном градиенте концентрацией в течение 120 минут.
100 мкл-аликвоты от каждой из полученных фракций осаждали с помощью ТСА, а осадки подвергали ДСР-ПААГ-электрофорезу с использованием 12,5% геля в восстановительных условиях.
После электрофореза гель окрашивали серебром для обнаружения полос белка. Исходя из фракции N 11, были получены три полосы.
Все остальные фракции N 11 - N 14 затем концентрировали путем ТСА-преципитации и полученный осадок подвергали ДСН-ПААГ-электрофорезу на 12,5%-ном геле в условиях восстановления. После электрофореза белок переносили с геля на PVDF-мембрану Problot (Applied Biosystems). После переноса белка на мембрану эту мембрану окрашивали 0,2% нафтоловым иссине-черным и три белковые полосы вырезали. Анализ N-концевой последовательности осуществляли с использованием газофазного белкового секвенатора.
Было установлено, что N-конец полосы, по всей видимости, с наименьшей молекулярной массой трех типов имел последовательность Lys-Leu-Leu-Lys-Met. Эти пять аминокислот соответствуют пяти аминокислотам, начиная с 49-ой аминокислоты от N-конца последовательности полипептида, кодируемого кДНК-вставкой pSR α 31-7. В соответствии с этим был сделан вывод, что расщепление пептида в 48-м остатке приводит к образованию одной зрелой формы белка, начинающегося с N-концевого
Пример 16
Получение моноклонального антитела против белка KM31-7
а) Получение антигенного белка
Посевную культуру E. coli, содержащую pMAL31-7, получали путем культивирования петли клеток в 3 мл LB-среды, содержащей 50 мкг/мл амципиллина, при встяхивании и при 37oC в течение ночи. 1 мл полученной посевной культуры инокулировали в 100 мл свежей LB-среды, содержащей 50 мкг/мл ампициллина, и культивировали при встряхивании и при 37oC до тех пор, пока ОП600нм не достигала 0,5. На этой стадии к культуральному бульону добавляли IPTG до конечной концентрации 0,1 мМ, а затем культивировали и встряхивали при встряхивании и при 37oC в течение ночи.
Из полученной ночной культуры выделяли клетки путем центрифугирования в течение 20 минут при 6500 об/мин и при 4oC, а осадок суспендировали в 10 мл колоночного буфера. Клетки в полученной суспензии дезинтегрировали с помощью ультразвукового дезинтегратора, а полученную жидкость центрифугировали 30 минут при 8000 об/мин и при 0oC. Полученный в результате супернатант содержат растворимую белковую фракцию.
Эту растворимую белковую фракцию подвергали хроматографии на колонке с 1 мл амилозной смолы. Элюирование проводили с использованием 10 мл колоночного буфера, содержащего 10 мМ мальтозы. Полученный после хроматографии гибридный белок оставляли на хранение, а затем использовали в качестве антигена.
b) Получение клеток селезенки от иммунизированных мышей
2 мл полного адъюванта Фрейнда добавляли к 2 мл антигена (эквивалентным 200 мкг), очищенного, как описано выше в стадии (а), в виде эмульсии. Эту эмульсию растворяли в 5 мл цилиндре шприца, снабженного стеклянным поршнем, а затем использовали для иммунизации 8-недельных самцов мышей BALB/c путем подкожной инъекции.
В последующих циклах иммунизации осуществляли точно такую же процедуру, что и при первой иммунизации, за исключением того, что в этом случае антиген вводили в неполном адъюванте Фрейнда. Иммунизацию осуществляли четыре раза, при этом каждую инъекцию делали примерно через каждые 2 недели.
Начиная со второй иммунизации, непосредственно черед иммунизацией у мышей брали кровь из венозного сплетения глазного дна и с помощью твердофазного иммуноферментного анализа (ELISA) определяли титр антител к KM31-7 в сыворотке.
Твердофазный анализ ELISA антитела против KM31-7
150-200 микролитров (соответствующих примерно 200 нг гибридного белка) бессывороточного супернатанта, полученного от COS-1-клеток, трансфецированных pSR α/ 31-7, помещали в каждую лунку 96-луночного ELISA-планшета (Costar) и использовали в качестве антигена. Затем планшет оставляли на ночь при 4oC для покрытия поверхностей дна лунок планшета. На следующий день планшет промывали 3 раза 0,1% Твин 20/фосфатно-буферным раствором (0,1% Твин 20/PBS), 0 и в каждую лунку добавляли 100 мкл BSA, доведенного до 10 мкг/мл фосфатно-буферным раствором, после чего планшет оставляли на 1 час при комнатной температуре.
По истечении этого времени планшет промывали еще три раза 0,1% Твин 20/PBS. Затем в каждую лунку добавляли 30-100 мкл первичного антитела в виде серийно разведенного образца (например, мышиной сыворотки, супернатант от культуры гибридомы или моноклональное антитело), после чего планшет оставляли на 1 час при комнатной температуре.
После этого планшет опять три раза промывали 0,1% Твин /PBS и в каждую лунку добавляли 100 мкл "второго" антитела. Второе антитело получали в виде раствора с 3000-кратным разведением комплекса "козий антимышиный IgG-пероксидаза" (Amersham) или в виде раствора с 3000-кратным разведением комплекса "козий антимышиный IgG-щелочная фосфатаза" (B10-RAD). Затем этот планшет оставляли на два часа при комнатной температуре.
По истечении этого времени планшет снова промывали три раза 0,1% Твин 20/PBS, а затем в каждую лунку добавляли 100 мкл либо раствора субстрата пероксидазы (B10-RAD, Peroxidase Substrade Kit ABTC), либо 10% диэтаноламина, содержащего 0,001% пара-нитрофенилфосфатного раствора. Затем планшет оставляли на 15 - 30 минут при комнатной температуре, после чего путем измерения оптической плотности при 415 нм с использованием планшет-ридера (B10-RAD) может быть подсчитан титр антител.
(c) Получение клеток мышиной миеломы
8-Азагуанин-резистентные клетки мышиной миеломы P3-X63-Ag8.653 (653) (ATCC N CPD-1580) культивировали в нормальной среде (полная GIT-среда) до получения по крайней мере 2 • 107 клеток.
(d) Получение гибридомы
1,4 • 108 клеток селезенки иммунизированных мышей, полученных после иммунизации, проведенной по схеме, описанной выше в (b), тщательно промывали DMEM-средой (Nissui Pharmaceutical). Промытые клетки смешивали с 1,5 • 107 клетками мышиной миеломы P3-X63-Ag8.653 (653), полученными, как описано выше в стадии (c), и эту смесь центрифугировали в течение 69 минут при 800 об/мин.
Группу клеток, состоящую из клеток селезенки и клеток P3-X63-Ag8.653 (653), собирали в виде осадка и дезинтегрировали. Предварительно получали 50%-ный раствор полиэтиленгликоля 4000 (полиэтиленгликоль N 4000) и DMEM и этот раствор выливали в дезинтегрированные клетки со скоростью 2 мл/мин в течение 1 мин, размешивания при этом. Затем аналогичным образом (т.е. в течение 1 мин при скорости 2 мл/мин) к клеточному препарату добавляли DMEM. Эту процедуру повторяли еще один раз как для полиэтиленового раствора, так и для DMEM-раствора. И наконец, в течение 3 минут постепенно добавляли 16 мл DMEM. Затем полученный клеточный препарат центрифугировали в течение 6 минут при 800 об./мин. Полученный супернатант отбрасывали, а затем суспендировали в 35 мл полной GIT-среды, содержащей 5-10 нг/мл мышиного 1D-6.
(e) Скриннинг гибридом
100 мкл суспензии, полученной, как описано выше в стадии (b), добавляли в каждую лунку 96-луночного планшета (Sumimoto Bakelite), а затем культивировали в 7,0%-CO2-инкубаторе при 37oC. Через 7 дней после инкубирования к каждой лунке добавляли 50 мкл HAT-среды. После инкубирования еще в течение 4 дней к каждой лунке добавляли еще 50 мкл HAT-среды. Затем планшет инкубировали еще 3 дня. По истечении этого времени из лунок, в которых наблюдался рост колоний гибридных клеток, отбирали образец культурального супернатанта и с использованием твердофазного иммуноферментного анализа (ELISA, описанного в п.(о)) анализировали титр антитела против KM31-7. Отобранную для образцов среду сразу же заменяли HT-средой.
(f) Клонирование
Клонирование клеток, взятых из лунок, тестированных как положительные, проводили три раза с помощью анализа методом предельного разведения. Клоны, которые обнаруживали, как было установлено по наблюдениям, постоянный титр антител, были отобраны для использования в качестве клеточных линий гибридомы продуцирующей моноклональное антитело против KM31-7. В частности, на этой стадии, ELISA осуществляли не только, как описано в (b), но при этом осуществляли контрольный ELISA с использованием бессывороточного культурального супернатанта, полученного от COS-1-клеток, трансфецированных pcDL-pSR α 296, для получения твердой фазы. В соответствии с этим клеточные линии, которые являются реактивными в случае первого ELISA, но не являются реактивными в случае контрольного ELISA, были отобраны для клонирования.
(o) Очистка моноклонального антитела
Культуральный супернатант от клеточной линии гибридомы, продуцирующей моноклональное антитела против KM37-7, собирали, фильтр стерилизовали 0,22 мкл фильтра Millipore, а затем антитела очищали с использованием MabTrap GII (Pharmacia).
(h) Анализ моноклонального антитела
1) Антигенная специфичность моноклонального антитела
Посредством иммунопреципитации, проводимой с использованием бессывороточного культурального супернатанта, полученного от COS-1-клеток, трансфецированных pSR α 31-7, было установлено, что моноклональные антитела обладали специфичностью по отношению к белку KM31-7.
2) Классификация моноклонального антитела
Этот тест осуществляли с использованием набора для изотипирования мышиного моноклонального антитела (Amersham), и в результате этого теста было установлено, что данное антитело принадлежит к подклассу IgG1.
Пример 17
Выделение и очистка белка KM31-7 с использованием реакции "антиген-антитело"
Этот тест осуществляли в соответствии с процедурой, описанной выше в примере 16 (h), (1). Этот тест также повторяли с использованием антитела и бессывороточного супернатанта, полученного от COS-1-клеток, трансфецированных pcDL-pSR α 296.
1,4 мкг моноклонального антитела добавляли к 1,7 мл каждого из бессывороточных супернатантов и оставляли на 1 час при комнатной температуре для осуществления реакции с последующим центрифугированием в микроцентрифужных 2,2 мл-пробирках при 20 об/мин. Контроль осуществляли с использованием бессывороточного супернатанта, полученного от COS-1-клеток, трансфецированных pSR α 31-7, но без добавления моноклонального антитела.
30 мкл протеин-G-сефарозы Fast Flow (Pharmacia), предварительно промытой 0,1% Твин 20/PBS, добавляли к каждой пробирке для абсорбции антитела, а затем центрифугировали в течение 30 минут на скорость 20 об/мин при комнатной температуре.
По истечении этого времени каждую смесь центрифугировали в течение нескольких секунд в микроцентрифуге при 10000 об/мин, после чего супернатант осторожно удаляли так, чтобы это не повлекло за собой какой-либо потери сегмента. Затем осадки отдельно промывали 0,1% Твин 20/PBS, а после этого подвергали микроцентрифугированию и промывали еще 5 раз аналогичным образом.
Полученный седимент суспендировали в буферном растворе ДСН-ПААГ-образца, содержащем 10 мкл 2-меркаптоэтанола. Каждую суспензию нагревали 2 минуты при 90oC, а затем подвергали ДСН-ПААГ-электрофорезу с использованием 12,5% геля в восстановительных условиях. После электрофореза продукт переносили с полиакриламидного геля на нитроцеллюлозную пленку (B10-RAD). Вестерн-блоттинг осуществляли с использованием поликлонального антитела против KM31-7, описанного в примере 1 (а), и в результате этого анализа было установлено, что моноклональное антитело против KM31 специфически осаждает KM31-белок из бессывороточного культурального супернатанта CO-1/pSR α 31-7.
Пример 18
Получение CYVV-Nla/KM31-7-гибридного белка
Для продуцирования белка KM31-7 с помощью CYVV-Nla-протеазы, необходимо, чтобы 3'-конец Nla-гена был лигирован с ДНК белка KM31 в одной и той же рамке считывания. Для этого осуществляли следующую двухстадийную процедуру:
1) Введение 3'-боковой цепи (SmaI-XbaI, 1005 п.о.) KM31-7-кДНК в pKSUN9
Для того, чтобы получить SmaI-XbaI-фрагмент (1006 п.о.), содержащий 3'-конец KM32-7-кДНК, 7 мкг pSR α 31-7 плазмидной, ДНК переваривали рестриктирующими ферментами SmaI и XbaI и полученный фрагмент выделяли и очищали с помощью GENECLEAN 11 (Funakoshi Japan) с использованием 0,8% агарового геля.
Между тем, 5 мкг ДНК плазмиды pKSUN 9 аналогичным образом переваривали ферментами SmaI и XbaI и расщепленный фрагмент дефосфорилировали бычьей щелочной фосфатазой (щелочная фосфатаза E.coli C 75, Takara Shuso, Япония). Полученную дефосфорилированную линеаризованную ДНК лигировали с SmaI-XbaI-KM-31- фрагментом с использованием набора для лигирования (Takara Shuso) и полученную конструкцию использовали для трансформирования штамма JM109 E. coli. Трансформанты отбирали и скринировали, в результате чего получали клон pU1a31- 7X, содержащий SmaI-XbaI-фрагмент.
11) Сшивание Nla-протеазы и KM31-7
Для сшивания C-концевой последовательности N1a с N-концевой последовательностью подтипа KM31-7, имеющего валиновый N-концевой остаток, в одной и той же рамке считывания, конструировали четыре типа PCR-праймеров с использованием ДНК-синтезатора (Perkin-Elmer Japan Applied Biosystems Model 392). Эти праймеры имели следующие последовательности:
5' GGT CAG CAC AAA TTT CCA 3' SEQ 1 N 15 (1)
5' AAA CAC AAC TTG GAA TGA ACA ATT 3' SEQ 1 N 16 (2)
5' TCA TTC CAA GTT GTG TTT GTG AAA 3' SEQ 1 N 17 (3)
5' CAT AGG ATG CTC CAA CAA 3' SEQ 1 N 18 (4)
Первый цикл PCR осуществляли с использованием 1 мкг ДНК плазмиды pK SUN9 в качестве матрицы. К реакционному раствору добавляли последовательно 100 пМ каждого из праймеров (1) и (2) и 1/10 объем 10х - концентрированного реакционного буферного раствора для полимеразы Tag, а затем 5 ед. Tag-полимеразы (Takara Shuzo). PCP-реакцию осуществляли по следующей схеме: сначала реакцию проводили при 72oC в течение 3 минут, а затем 30 циклов проводили 1 мин при 94oC, 2 мин при 55oC и 3 мин при 72oC, с последующей обработкой при 72oC в течение 10 минут. После PCR-реакции амплифицированный ДНК-продукт подвергали электрофорезу на 8%-ном полиакриламидном геле. Полоски геля, содержащего ДНК, идентифицировали путем окрашивания бромидом этидия и измельчали. Затем к каждой из раздробленных полосок добавляли 300 мкл буфера для элюирования (0,5 ацетата аммония, 1 мМ EDTA, pH 8,0) и инкубировали в течение ночи при 37oC. После центрифугирования получали супернатант, содержащий очищенную и амплифицированную ДНК.
Затем PCR проводили еще раз аналогичным способом, за исключением того, что на этот раз в качестве матрицы использовали 1 мгк ДНК плазмиды p CKM31, а в качестве праймеров использовали праймеры (3) и (4) и в результате получали очищенную ДНК, как описано выше.
Амплифицированный фрагмент от первой PCR-реакции содержал последовательность, кодирующую остатки N-концевого KM31-7-белка, начиная с Val-Val-Phe и далее ( на протяжении 31 п.о.) и расположенную "выше" от XhoI-сайта Nla. Амплифицированный фрагмент от второй PCP-реакции содержал последовательность, кодирующую Asn-Cys-Ser-Phe-Gln- от C-конца Nla и далее (на протяжении 32 п.о.) и расположенную "ниже" от SmaI-сайта KM31-7-кДНК.
В соответствии с этим при проведении PCR с использованием обоих ДНК-фрагментов, полученных из двух PCR, вместе с праймерами (1) - (4), получали гибридицированную нить, состоящую из 9 п. о. 3 - концевой Nla и 15 п.о. последовательности, кодирующей нужный конец KM31-7. Таким образом, можно генерировать ДНК-последовательность, содержащую эту часть в качестве связующего звена.
Исходя из этого, был осуществлен второй "раунд" PCR точно таким же способом, что и первый, после чего полученный амплифицированный фрагмент был выделен из геля.
III) Введение Nla/KM31-7-ДНК в PUla31-7X
5 мкг pUla31-7 X-плазмидной ДНК, полученной в части (1), переваривали ферментами XhbI и SmaI и полученную ДНК дефосфорилировали путем обработки бычьей щелочной фосфатазой. PCR-продукт, полученный в части (II), также переваривали ферментами XhoI и SmaI, после чего полученный фрагмент лигировали с переваренной дефосфорилированной pNla31-7SX с использованием набора для лигирования. Полученную конструкцию использовали для трансформации штамма J M109 E.coli.
Затем AmpR-трансформанты отбирали и скринировали.
Скрининг осуществляли путем переваривания ферментом XhoI с последующим электрофорезом. После этого отбирали клоны, имеющие лишь полосу 8,0 п.о. Затем плазмиды отобранных клонов переваривали ферментом HindIII и снова подвергали электрофорезу. Отобранная плазмида имела 330 п.о.-полосу, соответствующую части Nla-кДНК-вставки. Эта плазмида, обозначенная pNla31-7V, содержала Xho1- и Va1-PCR-продукт.
Затем определяли нуклеотидную последовательность клона pNla31-7V, в результате чего было установлено, что последовательности, кодирующие Nla и KM31-7, лигировали с сохранением ORF, а необходимая расщепляемая G1n-Na1-последовательность расположена между Nla и KM31-7-белком.
IV) Продуцирование белка KM31-7
Вестерн-блот-анализ подтвердил, что pNla31-7V функционирует в E.coli и что белок KM31-7 может быть экспрессирован сконструированным рекомбинантным геном.
Посевную культуру E.coli, содержащую плазмиду pNla31-7, культивировали в течение ночи при встряхивании в 3 мл LB-среде, содержащей 50 мкг/мл ампициллина. Затем 1 мл этой посевной культуры добавляли к 100 мл свежей LB-среды, содержащей 50 мкг/мл ампициллина, и культивировали при 38oC и при встряхивании до тех пор, пока оп600нм не достигала значения 1,0. На этой стадии к культуральному бульону добавляли 1PTG до конечной концентрации 1 мМ, после чего культуру инкубировали при 28oC и при встряхивании в течение еще двух ночей.
По истечении этого времени 1 мл культуры переносили в центрифужную пробирку и центрифугировали при 15000 об/мин в течение 5 минут. Затем супернатант удаляли, а осадок смешивали с 300 мкл стерильной воды и 300 мкл буферного раствора для ДСН-ПААГ-образца, содержащего 2-меркаптоэтанол для диспергирования осажденных клеточных включений. Полученную суспензию нагревали 2 минуты при 95oC, а затем 10 мкл этой суспензии подвергали ДСН-ПААГ-электрофорезу на 8%-геле в восстановительных условиях.
После электрофореза белок переносили с геля на нитроцеллюлозную мембрану. Эту процедуру осуществляли путем объединения геля с мембраной с последующим 2,5-часовым инкубированием при 4oC в присутствии буферного раствора для трансляции (25 мМ Трис-HCl, 1,4% глицин и 20% метанол) при 19 B с использованием устройства для переноса геля на мембраны (Marisol Japan).
Затем нитроцеллюлозную мембрану промывали 20 мл PBS-T-среды, после чего в течение 1 часа осуществляли блокирование в 20 мл PBS-T, содержащей 5% сепарированного молока (Snow, Brand Co., Ltd). По истечении этого времени мембрану промывали двумя партиями 20 мл PBS-T, а затем оставляли на 90 мин в 20 мл PBS-T, содержащем 1 мкл 100 х-разведенной в стерильной воде кроличьей Mab-сыворотки ("первого" антитела) против KM31-7, для осуществления реакции. После этого нитроцеллюлозную пленку промывали один раз в течение 15 минут и два раза в течение 5 минут (каждый раз 20 мл PBS-T).
Затем промытую мембрану помещали в баню с 3000х-разведенным и помеченным пероксидазой козьим антителом против кроличьих иммуноглобулинов (IgG) (B10-RAD) в PBS-T (используемым в качестве "второго" антитела) и оставляли на 1 час. По истечении этого времени мембрану промывали 20 мл PBS-T и переносили в баню с реагентом для ECI-детекции (Amersham), и с помощью авторадиографии обнаруживали полосы, реагирующие с антителом против KM31-7.
Осуществляли Вестерн-блоттинг, в результате которого было установлено, что молекулярная масса полосы составляет около 60000. Эта полоса обнаруживала такую же подвижность, что и белок, имеющий вторую из наиболее больших молекулярных масс трех KM31-7-белков, обнаруженных в бессывороточном культуральном супернатанте, полученном путем трансфекции клеток COS-1 клоном pSR α 31-7, используемым в качестве контроля.
Среды
x M фосфатный буфер
х M раствор Na2HPO4 доводили до нужного значения pH с использованием x M раствора NaH2PO4.
Буфер для инокуляции
0,1 М Трис-HCl буфер, pH 7,0 0,05 М ЭДТА, 1% 2-меркаптоэтанол.
Буфер для экстракции
0,1 М Трис-HCl буфер, 0,05 М ЭДТА, 1% 2-меркаптоэтанол, pH 7,0.
Раствор для деградации
200 мМ карбонат аммония, 2% ДСН, 2 мМ ЭДТА, 400 мкг/мл бентонита и 20 мкг/мл протеазы К (pH 9,0).
1 x SSc
0,15 м NaCl, 0,015 М тринатрийцитрат, pH 7,0.
Жидкая LB-среда
10 г Bacto-Трипотона (Difco) 5 г Bacto-дрожжевого экстракта (Difco) и 5 г хлорида натрия доводили до объема 1 л с использованием дистиллированной воды.
Трис-Кальциевый буфер
10 мМ Трис, 50 мМ хлорид кальция доводили до pH 7,4 путем добавления соляной кислоты.
Буфер для лизиса
0,17 г сахарозы, 250 мкл 1М Трис-HCl-буфера (pH 8,0) и 200 мкл 0,5 - M ЭДТА (pH 8,0) доводили до объема 20 мл с использованием бидистиллированной воды.
Щелочно-ДСН-раствор
0,2 М гидроксид натрия, 1% ДСН.
TBE-раствор
100 мМ Трис, 100 мМ борная кислота, 1 мМ ЭДТА.
Раствор для денатурации
1,5 М хлорид натрия, 0,5 М гидроксид натрия.
Буфер для нейтрализации
0,5 М трис, 3 М хлорид натрия (pH 7,4).
50 x Раствор Денхардта
1% поливинилпирролидон, 1% альбумин бычьей сыворотки и 1% Ficoll 400. Этот раствор разводили бидистиллированной водой соответственно и получали нужную концентрацию.
5x Буфер для денатурации
125 мкл 1 М глицина (pH 9,0), 25 мкл 1 М хлорида магния и 850 мкл бидистиллированной воды.
5x Буфер для мечения
25 мкл 1 М Трис-HCl буфера (pH 7,9), 5 мкл 1 М хлорида магния, 2,5 мкл 1 M дитиотреитола и 9,2 мкл бидистиллированной воды.
10 x М9 Солевой раствор
0,145 M динатрийфосфат, 0,172 М дигидрофосфат калия, 0,187 М хлорид аммония и 0,137 М хлорид натрия (pH 7,0).
М9 Минимальный агар
10 мл 10x M9 солевого раствора, 100 мкл 1 М сульфата магния, 1 мл 20% глюкозы, 50 мкл 1% гидрохлоридной соли тиамина 1 мл 0,01 М хлорида кальция и 13 мл бидистиллированной воды, перемешивали, стерилизовался путем фильтрации, а затем выливали в планшеты непосредственно после добавления 50 мл 3% Bacto-агара.
Жидкая SOB-среда
10 г Bacto-Триптона, 2,5 г Bacto-дрожжевого экстракта, 100 мл 5 М хлорида натрия и 125 мкл 1 М хлорида калия перешивали и доводили до объема 500 мл с использованием дистиллированной воды. После обработки в автоклаве к смеси добавляли 5 мл 1 М хлорида магния и 5 мл 1 М сульфата магния.
TFB1-буфер
5 мл 1 М 2-(N-Морфолино)этансульфоновой кислоты (MES) (доведенной до pH 6,2 с использованием 1 н7 HCl), 6,045 г хлорида рубидия, 0,735 г дигидрата хлорида кальция и 4,94 г тетрагидрата хлорида марганца перемешивали, доводили до pH 5,8 с использованием ледяной уксусной кислоты, а затем полученную смесь доводили до объема 500 мл с использованием бидистиллированной воды и стерилизовали путем фильтрации.
TFB2-буфер
1 мл 1 М 2-(N-морфолино)пропансульфоновой кислоты (MOPS), 1,102 г бигидрата хлорида кальция, 0,12 хлорида рубидия и 15 мл глицерина перемешивали и доводили до pH 6,5 с использованием ледяной уксусной кислоты, после чего полученную смесь доводили до объема 100 мл с использованием бидистиллированной воды и стерилизовали путем фильтрации.
Жидкая SOC-среда
5 мл жидкой SOB-среды, 90 мкл 20% глюкозы.
2x УТ-среда
16 г Bacto-Триптона, 5 г Bacto-дрожжевого экстракта и 5 г хлорида натрия перемешивали и доводили до объема 1 л с использованием бидистиллированной воды.
PBS-TW-среда
80 мМ динатрийбифосфат, 20 мМ бифосфат натрия, 100 мМ хлорид натрия и 0,1% Tween 20.
TBS-T-среда
4 г хлорида натрия, 0,1 г бифосфата калия, 1,45 г додекагидрида бифосфата натрия, 0,1 г хлорида калия, 0,1 г азида натрия доводили до объема в 1 л с использованием бидистиллированной воды (pH 7,4).
Раствор щелочно фосфатазного субстрата
0,01% п-нитрофенилфосфат растворяли в 10% водном растворе диэтаноламина и доводили до pH 9,8 с использованием соляной кислоты.
Среда A
D MEM (среда Игла, модифицированная по способу Дульбекко, и содержащая 4,5 г/л глюкозы), 10% инактивированная фетальная бычья сыворотка (изготовленная фирмой Hyclone) и 10 мМ HEPES (pH 7,2).
Среда B
D MEM (содержащая 4,5 г/л глюкозы), 10 мМ HEPES (pH 7,2) 3% инактивированная фетальная бычья сыворотка, 5 мкг/мл бычьего инсулина (изготовленного Sigma) 8 мкг/мл d-биотина (изготовленного Sigma), 4 мкг/мл пантотеновой кислоты (изготовленной Sigma), 1,0 мМ дексаметазон (изготовленный Sigma) и 0,5 мМ изобутилметилксантин (изготовленный Aldrich).
Среда C
G MEM (содержащая 4,5 г/л глюкозы), включающая в себя 5% инактивированную фетальную бычью сыворотку 10 мМ HEPES (pH 7,2), и 100 нг/мл бычьего инсулина.
Среда D
D MEM (содержащая 4,5 г/л глюкозы), 5% инактивированная фетальная бычья сыворотка, 10 мМ HEPES (pH 7,2) 100 нг/мл бычьего инсулина и 10 U/мл гепарина натрия (изготовленного Novo Industry Co.).
Раствор LPL -субстрата
13 мМ глицерин-три 9,10 (н)-3Н олеата (51,8 КВкв./мкМ, изготовленного Amersham), 1,3 мг/мл дисетароил-L- α - фосфатидилхолина (изготовленного Sigma Co.), 20 мг/мл альбумина бычьей сыворотки (изготовленной Sigma Co.), 135 мМ Трис гидрохлорид Трис-HCl, pH 8,1, изготовленный Sigma Co., 16,5% (об/об) глицерина и 16,5% (об/об) инактивированной фетальной бычьей сыворотки.
Раствор тиоцианата гуанидина
4 М тиоцианат гуанидина, 1% Sarkosyl, 20 мМ этилендиаминтетрауксусная кислота (ЭДТА), 25 мМ цитрат натрия (pH 7,0), 100 мМ 2-меркаптоэтанол и 0,1% противопенистый реагент A(Sigma).
Буфер для адсорбции
0,5 m NaCl, 20 мМ Трис-HCl (pH 7,5), 1 мМ ЭДТА и 0,1% ДСН.
Элюирующий раствор
10 мМ Трис-HCl (при 7,5), 1 мМ ЭДТА и 0,05% ДСН.
Реакционный раствор обратной транскриптазы примера 2
50 мл Трис-HCl (pH 8,3), 8 мМ MgCl2, 30 мМ KCl, 0,3 мМ дитиотреитол, 2 мМ dATP, 2 мМ dGTP, 2 мМ dTTP, 10 мкг [ α - 32P] dCTP и 1,4 мкг векторной плазмидной ДНК (pcDV-1 c 3'-олигонуклеотидным (dT)-хвостом, Pharmacia).
Реакционный раствор концевой трансферазы
140 мМ какодилат калия, 30 мМ Трис-HCl (pH 6,8), 1 мМ CoCl2, 0,5 мМ битиотреитол, 0,2 мкг полинуклеотида A и 100 мМ dCTP.
Буфер для рестрикации
50 мМ NaCl, 10 мМ Трис-HCl (pH 7,5), 10 мМ MgCl2 и 1 мМ дитиотреитол.
10-Объемный лигазный буфер
10 мМ ATP, 660 мМ Трис-HCl (pH 7,5), 66 мМ MgCl2 и 100 мМ дитиотреитол.
Пигмент для электрофореза
50% глицерина, 0,01 М динатрийбифосфат (pH 7,0) и 0,4% бромфенолового синего.
1х TAE
0,014 М Трис-ацетат и 0,001 М ЭДТА
1x SSCP
120 мМ NaCl, 15 мМ цитрат натрия, 13 мМ бифосфат калия и 1 мМ ЭДТА.
Реакционный раствор для обратной транскриптазы примера 6
1x Буфер для синтеза первой цепи, 5% пирофосфата натрия, 100 единиц ингибитора рибонуклеазы, 1 мМ dATP, 1 мМ dGTP, 1 мМ dTTP, 0,5 dCTP и 3,75 мг олигонуклеотидного праймера получали с использованием Системы Клонирования кДНК (Amersham).
SM Буфер
100 мМ NaCl, 8 мМ MgSO4 • 7H2O, 50 мМ Трис-HCl (pH 7,5) и 0,01% желатина.
Буфер для анализа
20 мМ фосфатный буфер (PH 7,8) и 0,5 М NaCl.
Колоночный буфер
10 мМ Трис-HCl (pH 7,4), 200 мМ NaCl и 1 мМ ЭДТА.
Буферный раствор для Tag-полимеразной реакции
500 мМ Трис-HCl (pH 8,3), 500 мМ хлорид калия, 15 мМ MgCl2, 100 мМ dATP, 100 мМ dCTP, 100 мМ dGTP, 100 мМ dTTP и - мг/мл желатина.














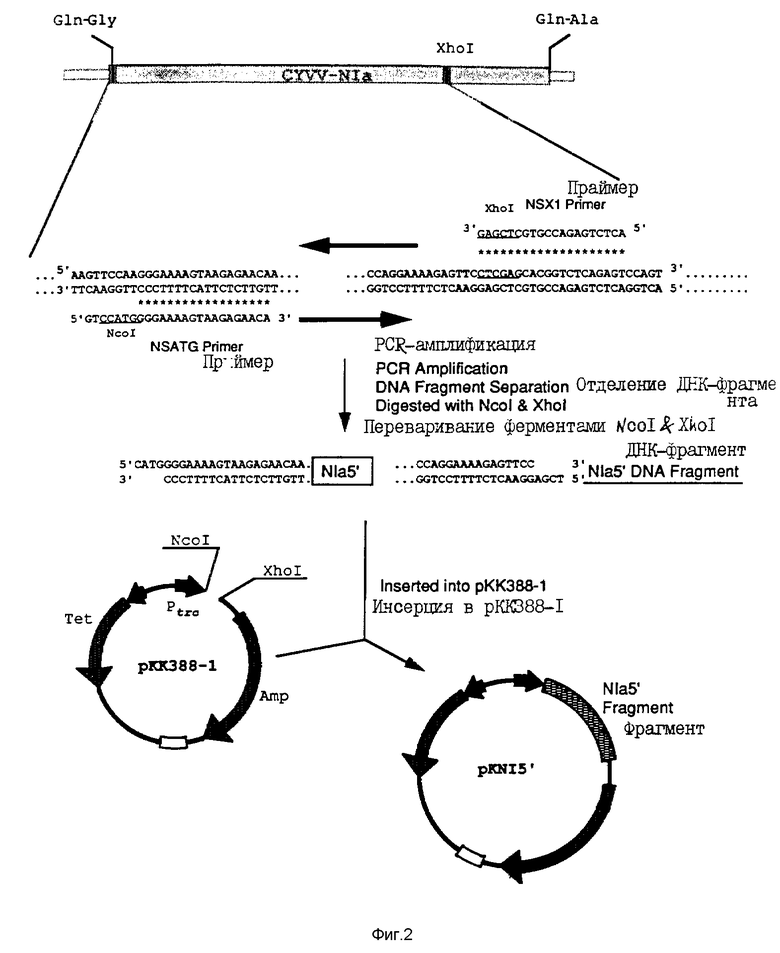
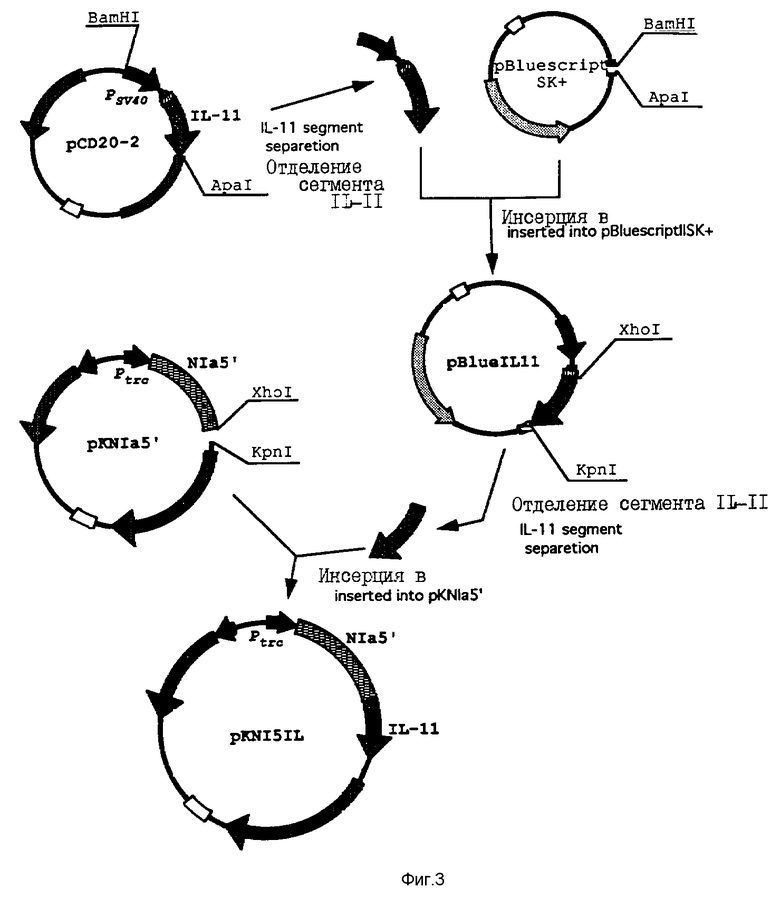
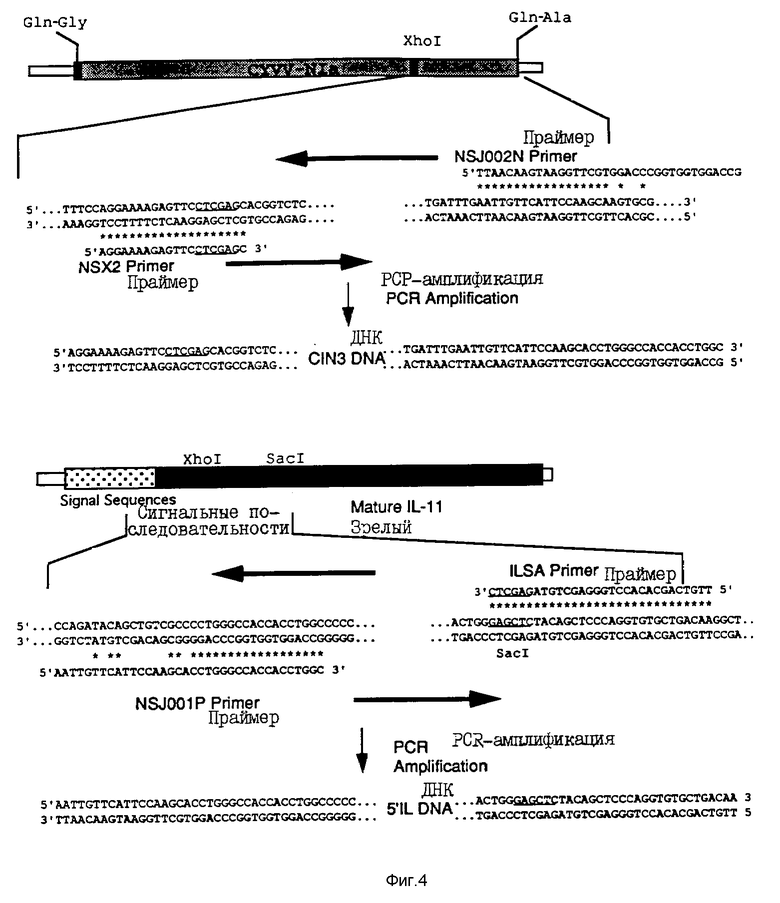
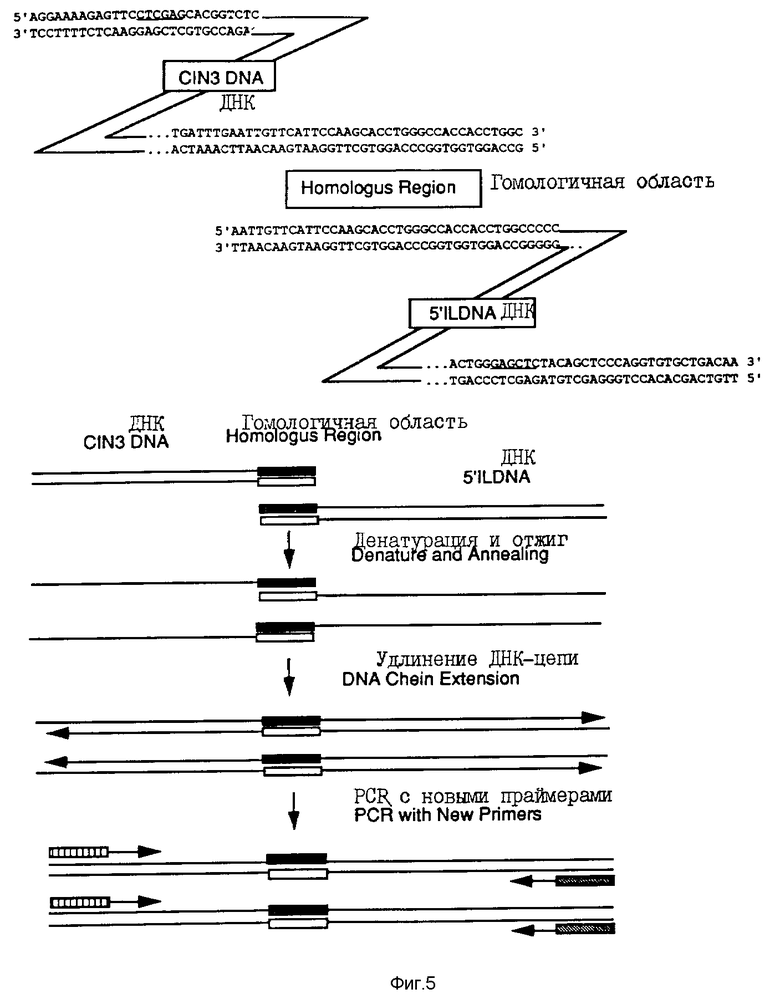
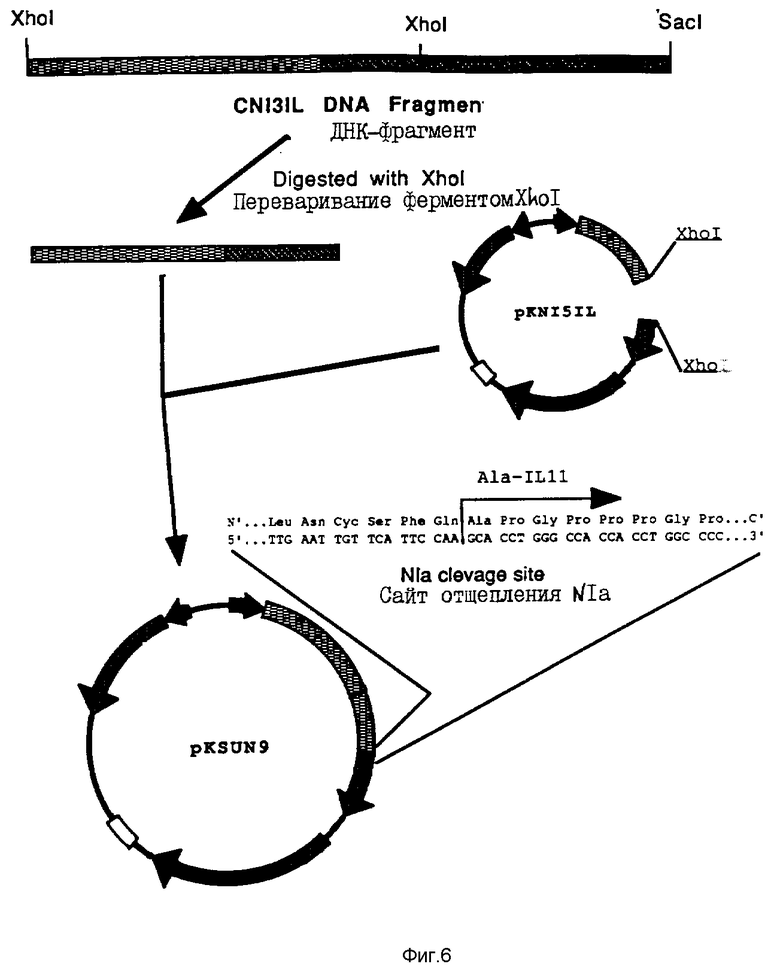
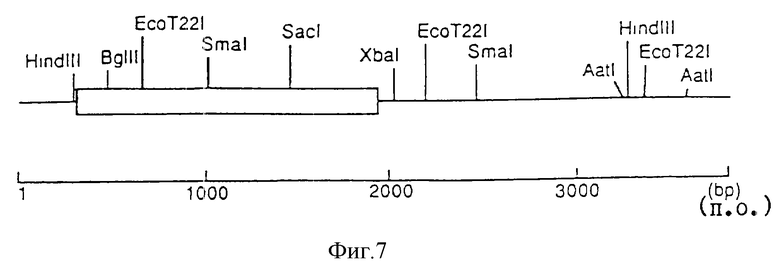
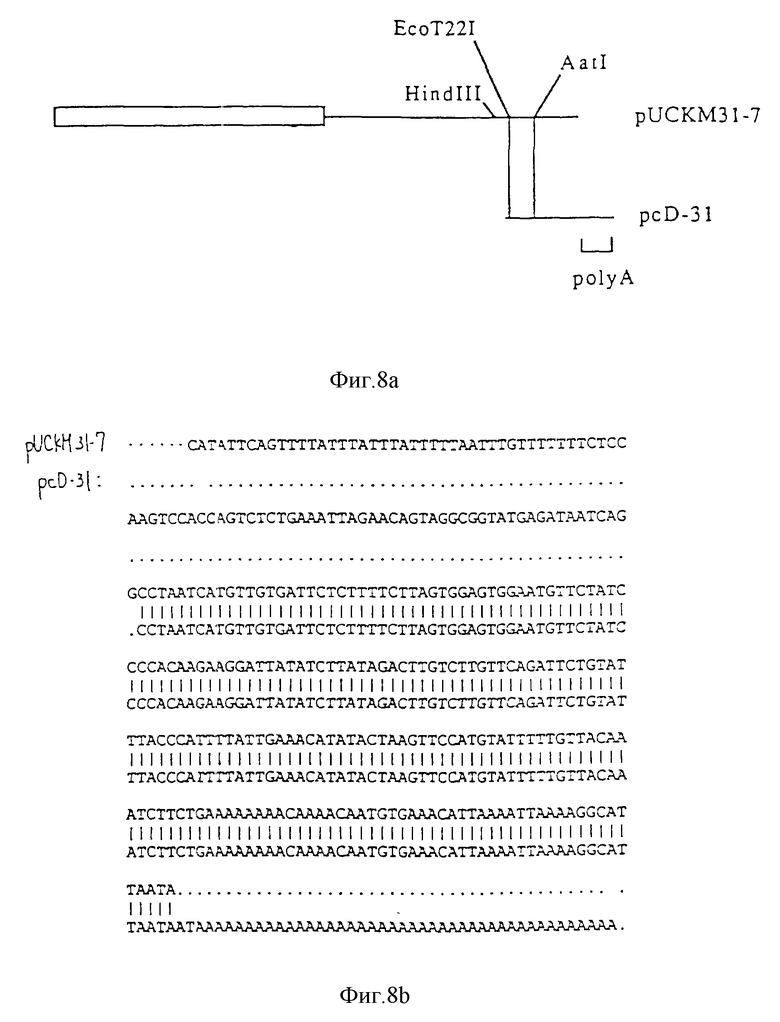
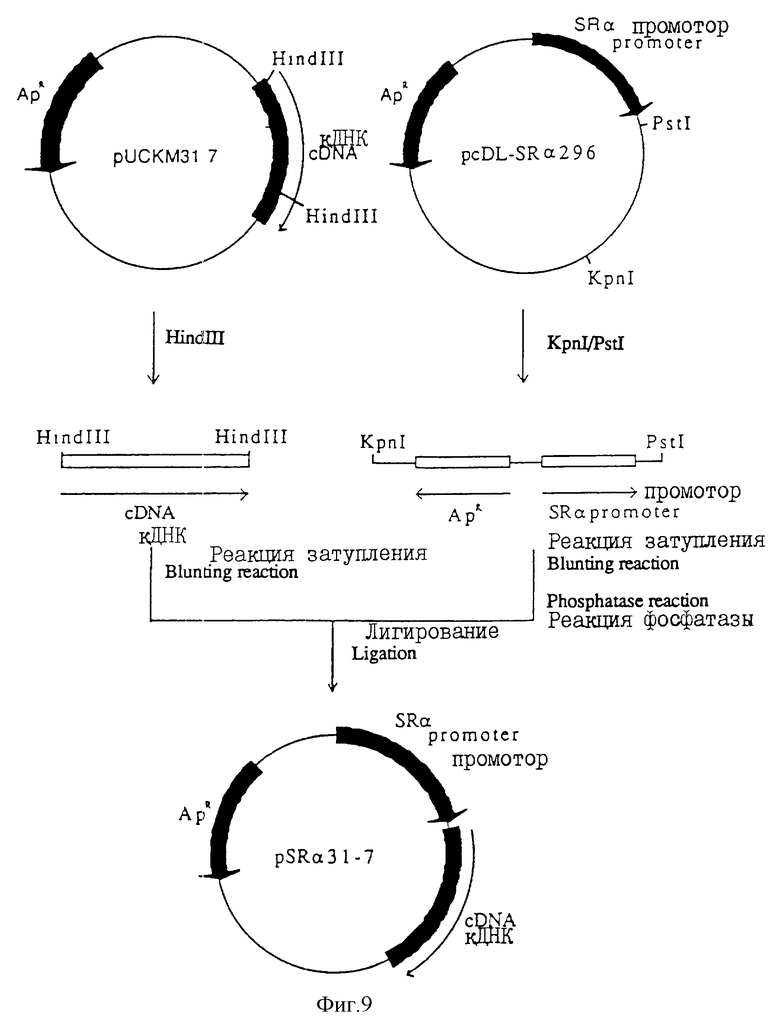
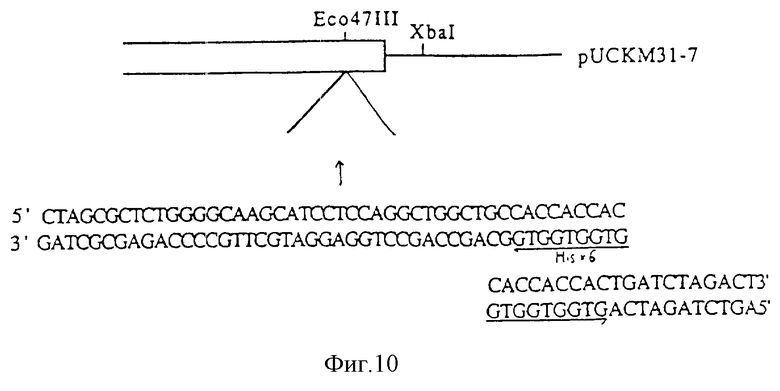
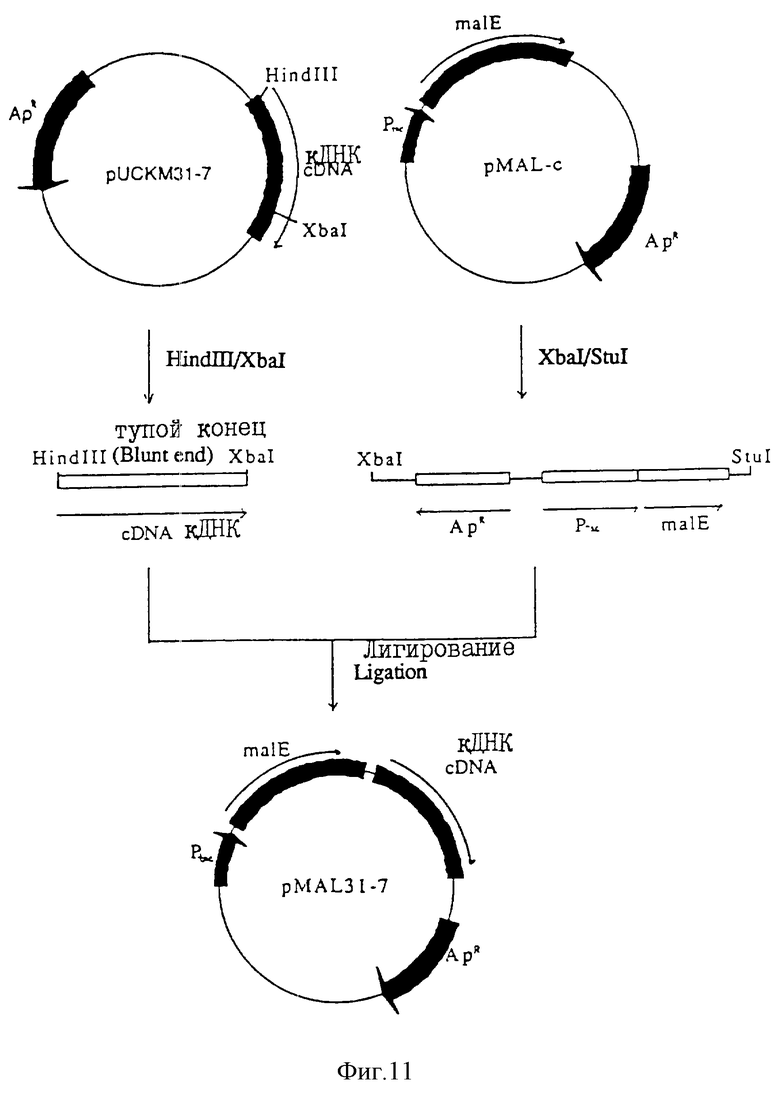
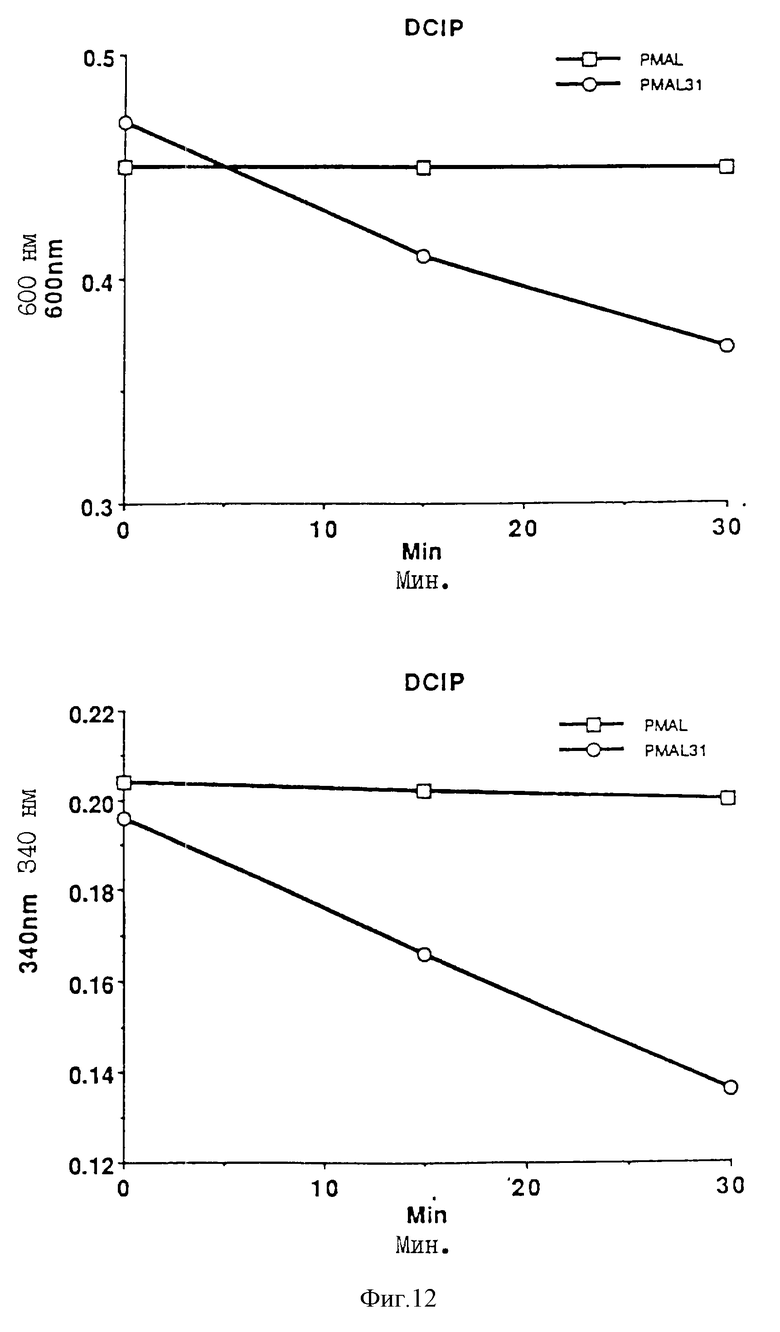
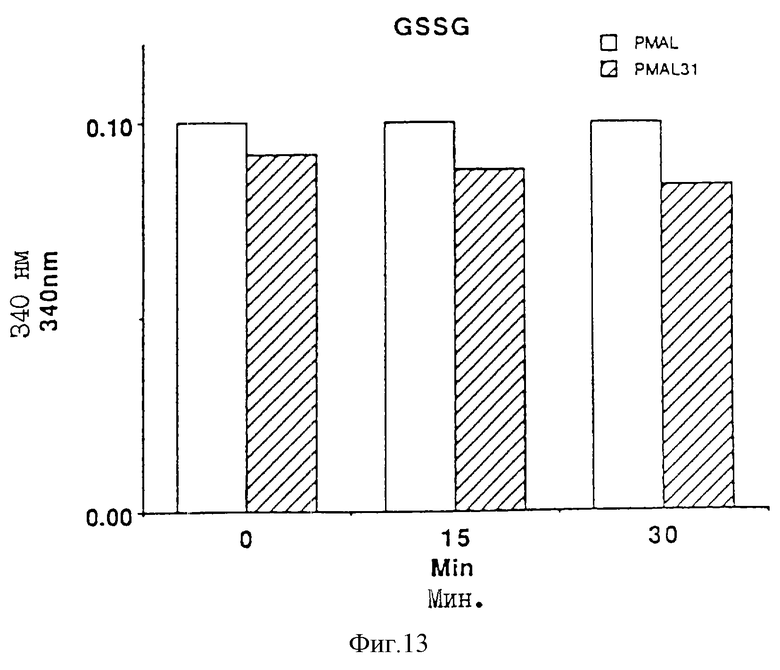
Изобретение относится к полипептидэкспрессирующим системам, требующим расщепления продукта-предшественника, к новому полипептиду, способному к восстановлению дихлориндофенола и окисленного глутатиона, к ДНК, кодирующей этот полипетид, фармкомпозициям, включающим данный полипептид, моноклональным антителам против указанного полипептида. Полинуклеотидная последовательность, кодирующая слитый белок, характеризуется структурной формулой А-В-С. А представляет последовательность нуклеотидов 10-1311 в SEQ ID NO. В представляет собой Asn-Cys-Ser-Phe-Gln. С представляет собой последовательность, кодирующую полипептид КМ31-7. Полипептид КМ31-7 имеет аминокислотную последовательность 1-526 в SEQ ID 12. Фармацевтическая композиция содержит полипетид КМ3 1-7 в сочетании с фармацевтически приемлемым носителем для него. Моноклональное антитело специфически взаимодействует с полипептидом КМ31-7. Оно продуцируется штаммом гибридных культивируемых клеток Mus musculus МКМ 150-2 FERM ВР-5086. Относится к изотипу Ig G1. Изобретение позволяет получить новый полипептид, обладающий восстанавливающей активностью in vivo и может быть использован для лечения заболеваний, вызванных окислительным стрессом. 9 c. и 2 з.п.ф-лы, 14 ил.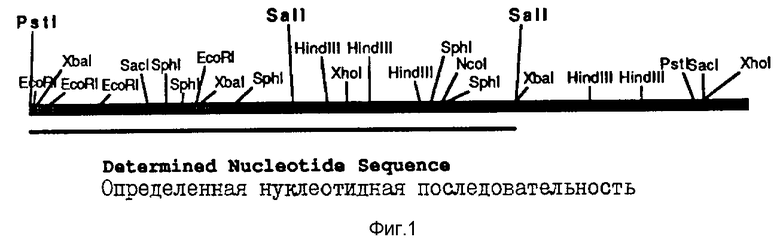
A-B-C,
где A представляет собой последовательность нуклеотидов 10-1311 в SEQ ID NO;
B представляет собой последовательность, кодирующую пептид: Asn-Cys-Ser-Phe-Gln;
C представляет собой, по меньшей мере, одну последовательность, кодирующую целевой полипептид.
Приоритет по пунктам:
13.07.94 по пп.1, 3, 4;
13.09.94 по пп.2, 5 - 9;
13.07.95 по пп.10 и 11.
| US 5162601 A, 10.11.92 | |||
| SU 1396603 A, 07.05.90. |

