Настоящее изобретение относится к новым производным подофиллотоксина, замещенного по положению 4 возможно замещенной цепью (поли)аминоалкиламиноалкиламида, или алкил-сульфонамида, или алкил-мочевины, способу их получения и их применению в качестве лекарственного средства, в частности в качестве противораковых средств.
Соединения по настоящему изобретению представляют собой производные подофиллотоксина, природного лигнана, известного в качестве терапевтического средства в лечении рака. Другие синтетические производные, такие как этопозид или тенипозид, составляют часть терапевтического арсенала для лечения, в частности, мелкоклеточного рака легкого. Эти различные соединения действуют путем ингибирования каталитической активности топоизомеразы II.
Таким образом, алкиламинный заместитель в положении 4β 4'-деметилподофиллотоксинового остова представляет собой спермин- или спермидин-алкиламидную единицу или в более общем случае (поли)аминоалкиламиноалкиламидную единицу. Кроме того, этот заместитель представляет собой спермин- или спермидин-алкил-сульфонамидную единицу или в более общем случае (поли)аминоалкиламиноалкилсульфонамидную единицу. Помимо всего прочего, этот заместитель представляет собой спермин- или спермидин-алкилмочевинную единицу или в более общем случае единицу (поли)аминоалкиламиноалкилмочевины.
Производные 4'-деметилэпиподофиллотоксина известны в качестве ингибиторов 2-топоизомеразы. Их цитотоксическая и противоопухолевая активности были обнаружены и показаны, в частности, для этопозида, ТОР 53 (Drugs of the Future 1996, 21, 1136), GL 331 (Medicinal Research Reviews, 1997, 17, 367) и NK 611 (Cancer Chemother. Pharmacol. 1996, 38, 217 and 541). Описаны соединения, имеющие аминные цепи бензиламиннного типа, напрямую присоединенные по положению 4β подофиллотоксина (J. Med. Chem. 1991, 34, 3346). В заявке на патент FR 2810321 описываются производные подофиллотоксина карбаматного или тиокарбаматного типа, полезные в лечении рака. Также описаны соединения с амидом в положении 4β (US 6566393; Acta Pharmaceutica Sinica (Yaoxue Xuebao), 1993, 28, 422; Acta Chem. Scand. 1993, 47, 1190; Anti-Cancer Drug Design 2001, 16, 305). Описаны соединения с мочевиной в положении 4β (Heterocycles 1994, 39, (1), 361; J. Med. Chem. 2002, 45, 2294).
В патенте ЕР 0876374 описывается способ деметилирования подофиллотоксина и несложного получения 4'-деметилэпиподофиллотоксина, представляющего собой синтетическое промежуточное соединение при получении этопозида и тенипозида.
В международной заявке WO 03/082876 описываются 4β-4''-[{2''-бензоил-замещенные}анилино]-производные подофиллотоксина, обладающие противораковой активностью.
Необходимость в обеспечении более эффективными видами терапии побуждает к поиску новых молекул, имеющих различные механизмы действия, направленные при этом на виды опухолей, плохо поддающихся или не поддающихся лечению в настоящее время, а также действующих в обход проблем резистентности. Наличие этих новых продуктов также позволяет разрабатывать протоколы, включающие в себя совместное применение терапий, которые более активны в отношении некоторых опухолей.
Новые соединения по настоящему изобретению обеспечивают путь преодоления этой проблемы.
Соединения, описанные в заявке на патент WO 2005/100363, имеют ацетамидную группировку в положении 4β подофиллотоксиновой единицы, причем указанная группировка связана с аминной или полиаминной цепью. Авторы синтезировали другие производные, имеющие алкиламидную, мочевинную или сульфонамидную группировку и документально подтвердили их цитотоксическую и противораковую активность.
Настоящее изобретение относится к соединениям общей формулы 1:
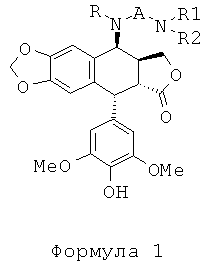
где:
R представляет собой водород или С1-4алкил,
А представляет собой СО(СН2)n, где n равно 2, 3, 4 или 5, либо А представляет собой CONH(СН2)n, где n имеет те же значения, что описаны выше, либо
А представляет собой SO2(CH2)n, где n имеет те же значения, что описаны выше,
R1 представляет собой Н или С1-4алкил,
R2 представляет собой Н или С1-4алкил, либо
R2 также может представлять собой (CH2)m-NR3R4, где R3 представляет собой Н или С1-4алкил, и m равно 2, 3, 4 или 5,
R4 представляет собой Н или С1-4алкил, либо
R4 также может представлять собой (CH2)p-NR5R6, где R5 представляет собой Н или С1-4алкил, и р равно 2, 3, 4 или 5, и
R6 представляет собой Н или С1-4алкил, либо
R6 также может представлять собой (CH2)q-NH2, где q равно 2, 3, 4 или 5,
за исключением соединения, где А представляет собой СО(СН2)2, где оба R1 и R2 представляют собой Н.
Подразумевается, что термин "С1-4алкил", как он определен в настоящем изобретении, относится к насыщенной линейной или разветвленной углеводородной цепи, содержащей 1-4 атома углерода. Пример включает группы метил, этил, пропил, изопропил, бутил, изобутил, втор-бутил и трет-бутил. Таким образом, во всем этом описании под С3- и C4-алкильными группами понимаются как линейные, так и разветвленные группы.
Данное изобретение также относится к их солям, в частности их фармацевтически приемлемым водорастворимым солям, в особенности их солям присоединения неорганических или органических кислот, а также содержащим их фармацевтическим композициям и их применению в качестве лекарственного средства, в частности предназначенного для лечения рака.
Мочевинные, амидные, карбаматные или сульфонамидные производные подофиллотоксина описаны в литературе и патентах (Zhongguo Yaoke Daxue Xuebao 1993, 24, 134; WO 2004/000859; US 2004/0106676; J. Med. Chem. 2004, 47, 2365; Org. Biomol. Chem. 2005, 3, 1074; WO 2004/073375; Bioorg. Med. Chem. 2003, 11, 5135). Их активность указывает на ингибирующее действие в отношении 2-топоизомеразы и на их полезность в качестве соединений, обладающих противоопухолевой активностью. Однако низкая растворимость в воде этих соединений затрудняет их применение. Хотя наличие в данной молекуле основного атома азота дает возможность получения растворимой соли, не всегда очевидно, каким образом можно получить активное соединение, обладающее требуемыми противоопухолевыми свойствами.
В литературе, за исключением заявки на патент WO 2005/100363, нет описания какого-либо соединения, имеющего полиаминную цепь, "пришитую" через спейсер в 4β-положение 4'-деметил-4-дезоксиподофиллотоксина.
Соответственно, в настоящем изобретении дается описание новых полиаминных производных подофиллотоксина.
Соединения по настоящему изобретению имеют структуру эпиподофиллотоксина, замещенного в положении 4β мочевинной единицей, соединенной с полиаминной цепью, такой как, в частности, у путресцина, спермина или спермидина, а также и у других полиаминов. Подобным же образом это положение 4β может быть соединено с амидной группой, затем независимо от того, соединена или не соединена последняя с линейным спейсером, содержащим от 2 до 5 атомов углерода, с полиамином, таким как путресцин, спермин или спермидин либо другие полиамины.
Кроме того, положение 4β может быть замещено сульфамидоэтильной единицей, соединенной в свою очередь с полиаминной цепью, такой как цепь путресцина, спермина или спермидина. Системы транспорта полиаминов уже были использованы для целевой доставки цитотоксических полиаминных аналогов (Anna. Rev. Pharmacol. Toxicol. 1995, 35, 55; Medicine/Sciences 1996, 12, 745), но, похоже, без особого успеха.
Описаны соединения, имеющие полиаминную цепь, "пришитую" к интеркалирующей в ДНК единице акридинового (J. Org. Chem. 2000, 65, 5590; J. Med. Chem. 2002, 45, 5098) или инденизохинолинового (J. Med. Chem. 2003, 46, 5712) типа.
Особенность соединений по настоящему изобретению заключается в том, что они являются ДНК-направленными агентами и успешно индуцируют повреждения в указанной ДНК, как качественно, так и количественно отличающиеся от других известных противораковых соединений, таких как этопозид.
Наличие полиаминной цепи, такой как, например, цепь путресцина, спермина или спермидина, имеет то преимущество, что она распознается системой транспорта природных полиаминов, используемых раковой клеткой для пролиферации (J. Cell. Physiol. 1993, 155, 399; Annu. Rev. Biochem. 1984, 53, 749). Это дает преимущество соединениям по настоящему изобретению в виде предпочтительного пути прохождения к раковой клетке по сравнению с другими клетками. Таким образом, соединения по настоящему изобретению обладают цитотоксическими свойствами in vitro и противоопухолевыми свойствами in vivo.
Кроме того, важным преимуществом этих соединений является наличие функциональных аминогрупп, обеспечивающих хорошую растворимость в воде, что делает эти соединения пригодными с точки зрения технологии изготовления лекарственных средств, введения, способности к распределению и биодоступности в организме. Ввиду этого фармакокинетические параметры являются улучшенными.
Предпочтительные соединения по изобретению выбраны из следующих далее соединений:
амидного ряда:
соединения 1: 3-(2-диметиламиноэтиламино)-N-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро[3',4':6,7]нафто[2,3-d][1,3]диоксол-5-ил]-пропионамида
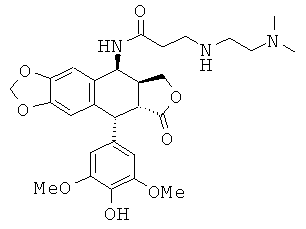
соединения 2: 4-(2-диметиламиноэтиламино)-N-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро[3',4':6,7]нафто[2,3-d][1,3]диоксол-5-ил]-бутирамида
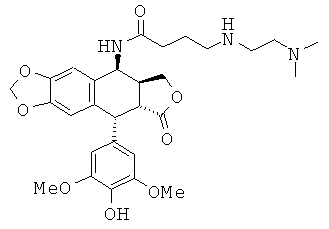
соединения 3: 3-[(2-диметиламиноэтил)-метиламино]-N-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро[3',4':6,7]нафто[2,3-d][1,3]диоксол-5-ил]-пропионамида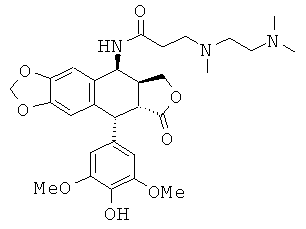
соединения 4: 4-[(2-диметиламиноэтил)-метиламино]-N-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро[3',4':6,7]нафто[2,3-d][1,3]диоксол-5-ил]-бутирамида
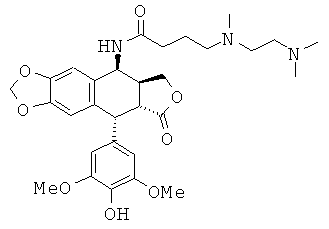
соединения 5: 3-диметиламино-N-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5аД8,8а,9-гексагидрофуро[3',4':6,7]нафто[2,3-d][1,3]диоксол-5-ил]-пропионамида
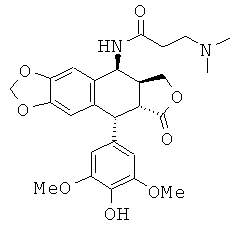
соединения 6: 4-диметиламино-N-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро[3',4':6,7]нафто[2,3-d][1,3]диоксол-5-ил]-бутирамида
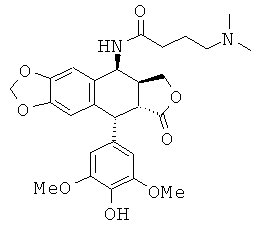
соединения 7: 5-диметиламинопентановой кислоты N-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро[3',4':6,7]нафто[2,3-d][1,3]диоксол-5-ил]-амида

соединения 8: 3-(2-диэтиламиноэтиламино)-N-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро[3',4':6,7]нафто[2,3-d][1,3]диоксол-5-ил]-пропионамида
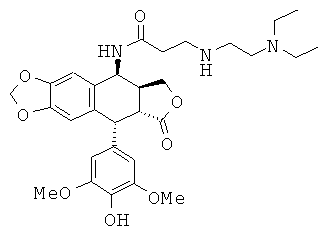
соединения 9: 4-(2-диэтиламиноэтиламино)-N-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро[3',4':6,7]нафто[2,3-d][1,3]диоксол-5-ил]-бутирамида
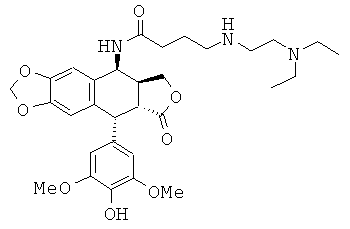
соединения 10: 3-(2-диэтиламинопропиламино)-N-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро[3',4':6,7]нафто[2,3-d][1,3]диоксол-5-ил]-пропионамида
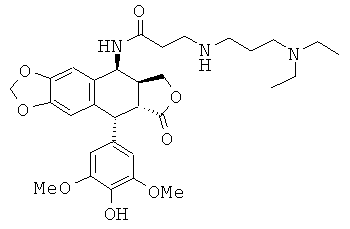
соединения 11: 4-(2-диэтиламинопропиламино)-N-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро[3',4':6,7]нафто[2,3-d][1,3]диоксол-5-ил]-бутирамида
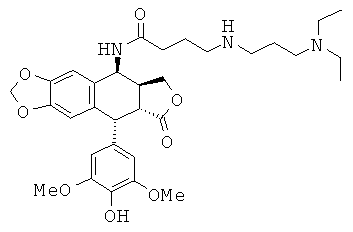
соединения 12: 3-(2-аминоэтиламино)-N-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро[3',4':6,7]нафто[2,3-d][1,3]диоксол-5-ил]-пропионамида
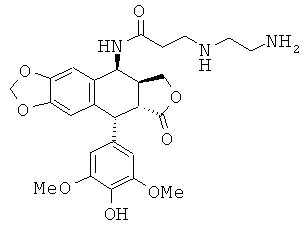
соединения 13: 3-(3-аминопропиламино)-N-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро[3',4':6,7]нафто[2,3-d][1,3]диоксол-5-ил]-пропионамида
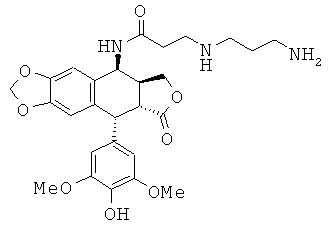
соединения 14: 3-(4-аминобутиламино)-N-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро[3',4':6,7]нафто[2,3-d][1,3]диоксол-5-ил]-пропионамида
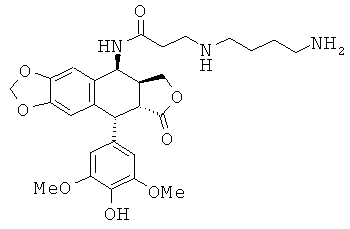
соединения 15: 4-(3-аминопропиламино)-N-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро[3',4':6,7]нафто[2,3-d][1,3]диоксол-5-ил]-бутирамида
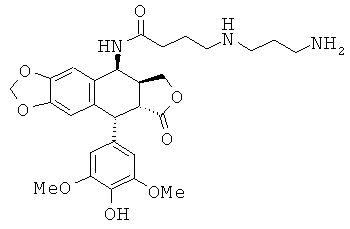
соединения 16: 4-(4-аминобутиламино)-N-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро[3',4':6,7]нафто[2,3-d][1,3]диоксол-5-ил]-бутирамида
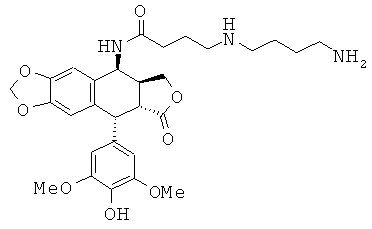
соединения 17: 5-(4-аминобутиламино)пентановой кислоты N-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро[3',4':6,7]нафто[2,3-d][1,3]диоксол-5-ил]-амида
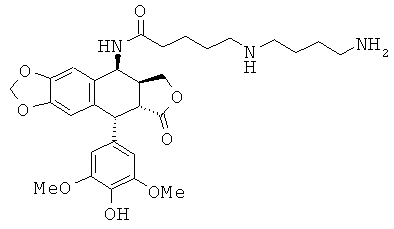
соединения 18: 3-{3-[4-(3-аминопропиламино)-бутиламино]-пропиламино}-N-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро-[3',4':6,7]нафто[2,3-d][1,3]диоксол-5-ил]-пропионамида
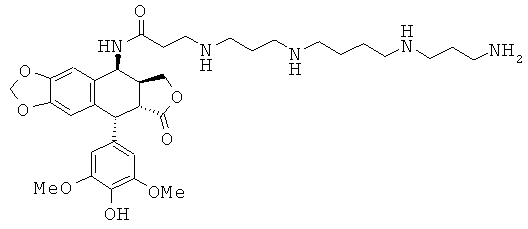
соединения 19: 3-{3-[3-(3-аминопропиламино)-пропиламино]-пропиламино}-N-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро-[3',4':6,7]нафто[2,3-d][1,3]диоксол-5-ил]-пропионамида
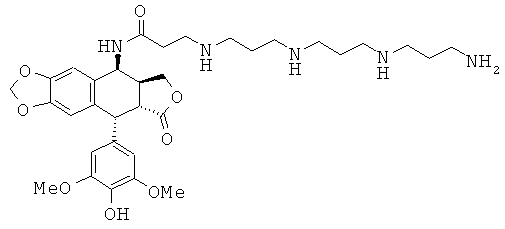
соединения 20: 3-{4-[4-(4-аминобутиламино)-бутиламино]-бутиламино}-N-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро-[3',4':6,7]нафто[2,3-d][1,3]диоксол-5-ил]-пропионамида
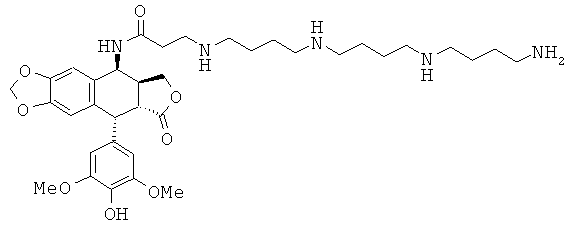
соединения 21: 4-{3-[4-(3-аминопропиламино)-бутиламино]-пропиламино}-N-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро-[3',4':6,7]нафто[2,3-d][1,3]диоксол-5-ил]-бутирамида
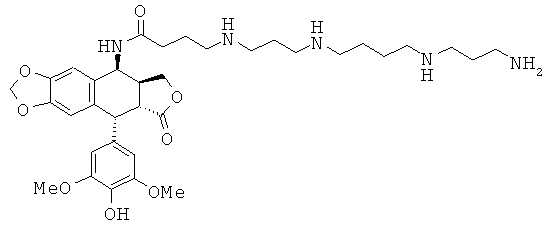
соединения 22: 4-{3-[3-(3-аминопропиламино)-пропиламино]-пропиламино}-N-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро-[3',4':6,7]нафто[2,3-d][1,3]диоксол-5-ил]-бутирамида
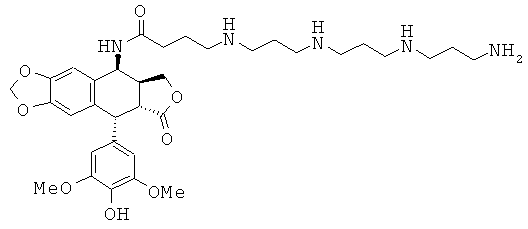
соединения 23: 4-{4-[4-(4-аминобутиламино)-бутиламино]-бутиламино}-N-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро[3',4':6,7]-нафто[2,3-d][1,3]диоксол-5-ил]-бутирамида
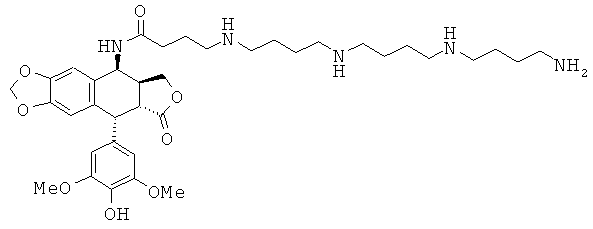
соединения 24: 5-{3-[4-(3-аминопропиламино)-бутиламино]-пропиламино} пентановой кислоты N-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро[3',4':6,7]нафто[2,3-d][1,3]диоксол-5-ил]-амида
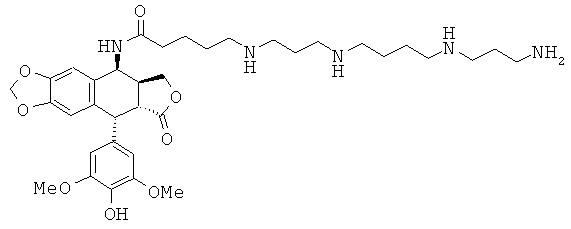
соединения 25: 5-{3-[3-(3-аминопропиламино)-пропиламино]-пропиламино}пентановой кислоты N-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро[3',4':6,7]нафто[2,3-d][1,3]диоксол-5-ил]-амида
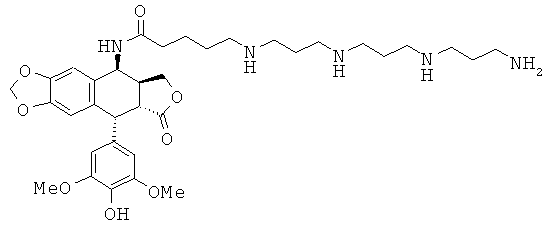
соединения 26: 5-{4-[4-(4-аминобутиламино)-бутиламино]-бутиламино}пентановой кислоты N-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро[3',4':6,7]нафто[2,3-d][1,3]диоксол-5-ил]-амида
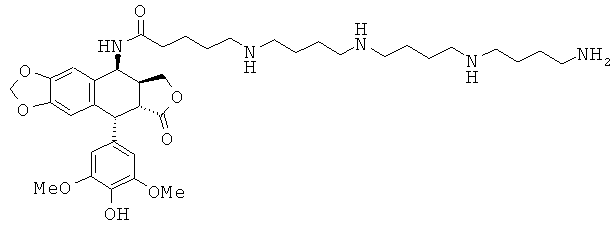
соединения 27: 3-[3-(4-аминобутиламино)-пропиламино]-N-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро[3',4':6,7]нафто[2,3-d][1,3]диоксол-5-ил]-пропионамида
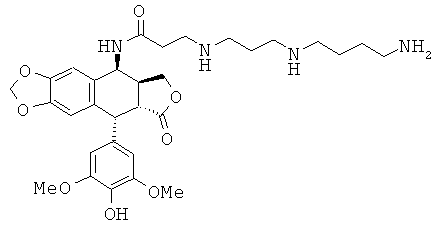
соединения 28: 3-[4-(3-аминопропиламино)-бутиламино]-N-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро[3',4':6,7]нафто[2,3-d][1,3]диоксол-5-ил]-пропионамида
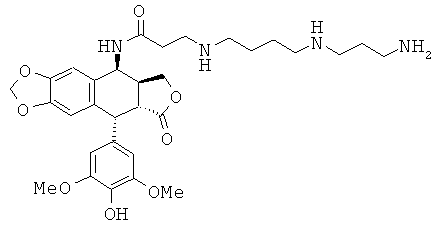
соединения 29: 3-[3-(3-аминопропиламино)-пропиламино]-N-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро[3',4':6,7]нафто[2,3-d][1,3]диоксол-5-ил]-пропионамида
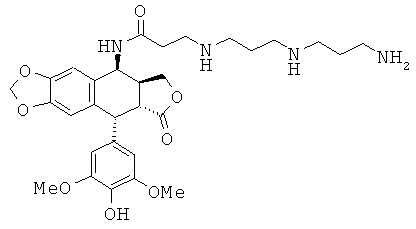
соединения 30: 3-[4-(4-аминобутиламино)-бутиламино]-N-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро[3',4':6,7]нафто[2,3-d][1,3]диоксол-5-ил]-пропионамида
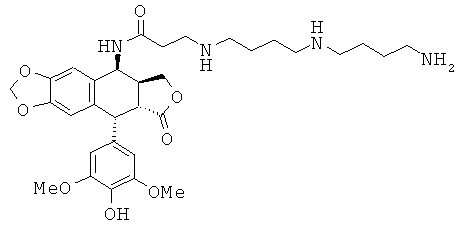
соединения 31: 4-[3-(4-аминобутиламино)-пропиламино]-N-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро[3',4':6,7]нафто[2,3-d][1,3]диоксол-5-ил]-бутирамида
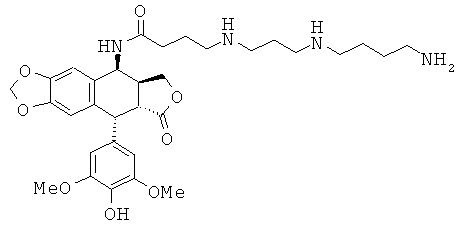
соединения 32: 4-[4-(3-аминопропиламино)-бутиламино]-N-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро[3',4':6,7]нафто[2,3-d][1,3]диоксол-5-ил]-бутирамида
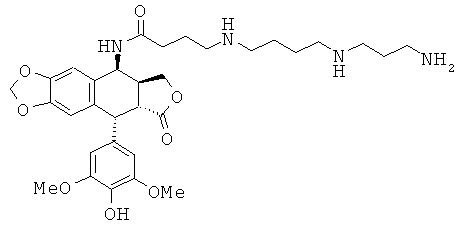
соединения 33: 4-[3-(3-аминопропиламино)-пропиламино]-N-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро[3',4':6,7]нафто[2,3-d][1,3]диоксол-5-ил]-бутирамида
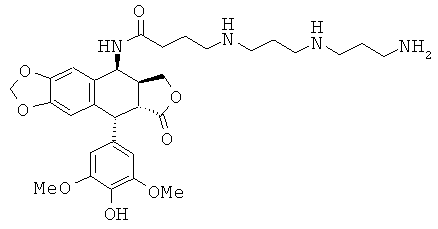
соединения 34: 4-[4-(4-аминобутиламино)-бутиламино]-N-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро[3',4':6,7]нафто[2,3-d][1,3]диоксол-5-ил]-бутирамида
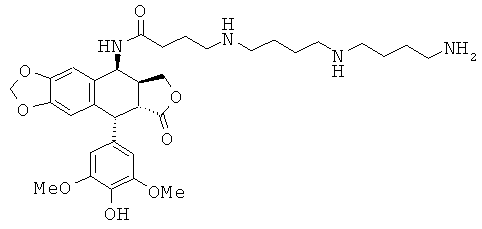
соединения 35: 5-[3-(4-аминобутиламино)-пропиламино]пентановой кислоты N-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро-[3',4':6,7]-нафто[2,3-d][1,3]диоксол-5-ил]-амида
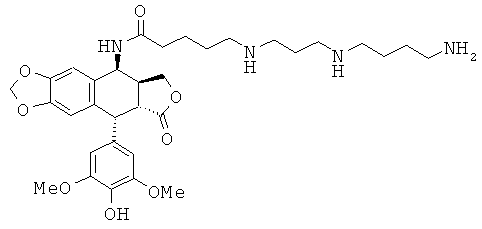
соединения 36: 5-[4-(3-аминопропиламино)-бутиламино]пентановой кислоты N-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро-[3',4':6,7]нафто[2,3-d][1,3]диоксол-5-ил]-амида
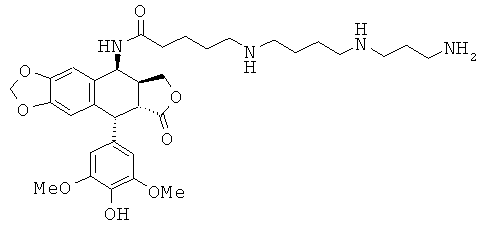
соединения 37: 5-[3-(3-аминопропиламино)-пропиламино]пентановой кислоты N-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро[3',4':6,7]-нафто[2,3-d][1,3]диоксол-5-ил]-амида
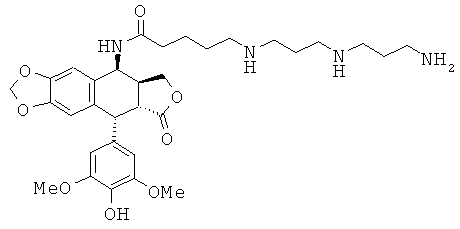
соединения 38: 5-[4-(4-аминобутиламино)-бутиламино]пентановой кислоты N-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро-[3',4':6,7]нафто[2,3-d][1,3]диоксол-5-ил]-амида

соединения 55: 5-[(2-диметиламиноэтил)-метиламино]пентановой кислоты N-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро-[3',4':6,7]нафто[2,3-d][1,3]диоксол-5-ил]-амида
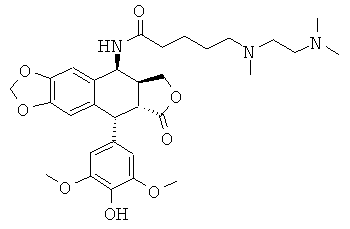
соединения 56: 4-амино-N-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро[3',4':6,7]нафто[2,3-d][1,3]диоксол-5-ил]-бутирамида
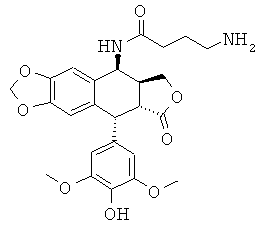 ,
,
соединения 57: 5-аминопентановой кислоты N-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро[3',4':6,7]нафто[2,3-d][1,3]-диоксол-5-ил]-амида
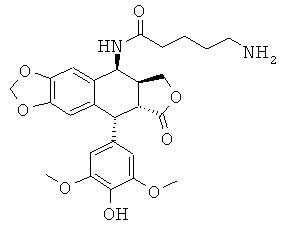
соединения 58: 3-(5-аминопентиламино)-N-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро[3',4':6,7]нафто[2,3-d][1,3]диоксол-5-ил]-пропионамида
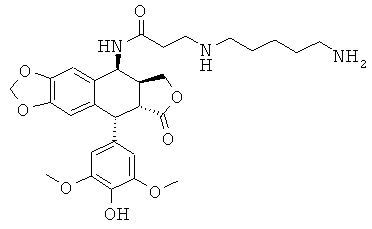
ряда мочевины:
соединения 39: 1-(4-аминобутил)-3-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро[3',4':6,7]нафто[2,3-d][1,3]диоксол-5-ил]-мочевины

соединения 40: 1-[4-(3-аминопропиламино)-бутил]-3-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро[3',4':6,7]нафто[2,3-d][1,3]диоксол-5-ил]-мочевины
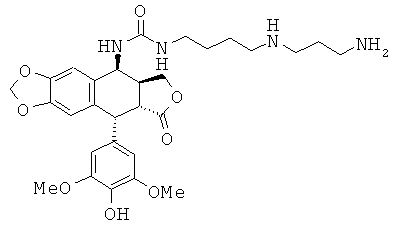
соединения 41: 1-[3-(4-аминобутиламино)-пропил]-3-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро[3',4':6,7]нафто[2,3-d][1,3]диоксол-5-ил]-мочевины
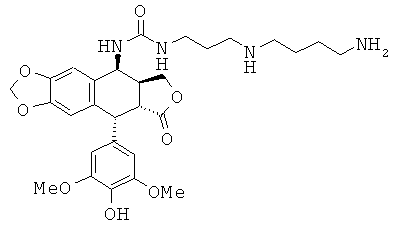
соединения 42: 1-[3-(3-аминопропиламино)-пропил]-3-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро[3',4':6,7]нафто[2,3-d][1,3]диоксол-5-ил]-мочевины
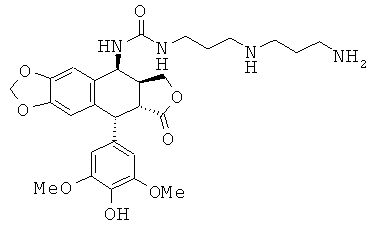
соединения 43: 1-[4-(4-аминобутиламино)-бутил]-3-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро[3',4':6,7]нафто[2,3-d][1,3]диоксол-5-ил]-мочевины
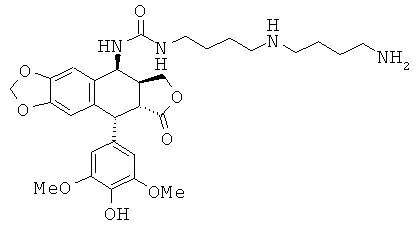
соединения 44: 1-{2-[3-(4-аминобутиламино)-пропиламино]-этил}-3-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро[3',4':6,7]-нафто[2,3-d][1,3]диоксол-5-ил]-мочевины
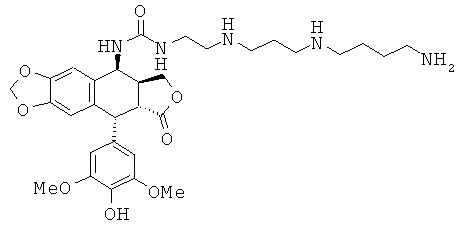
соединения 45: 1-{2-[4-(3-аминопропиламино)-бутиламино]-этил}-3-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро[3',4':6,7]-нафто[2,3-d][1,3]диоксол-5-ил]-мочевины
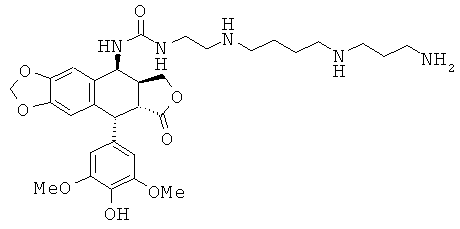
соединения 46: 1-{4-[4-(4-аминобутиламино)-бутиламино]-бутил}-3-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро[3',4':6,7]-нафто[2,3-d][1,3]диоксол-5-ил]-мочевины

соединения 47: 1-{3-[3-(3-аминопропиламино)-пропиламино]-пропил}-3-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро[3',4':6,7]-нафто[2,3-d][1,3]диоксол-5-ил]-мочевины
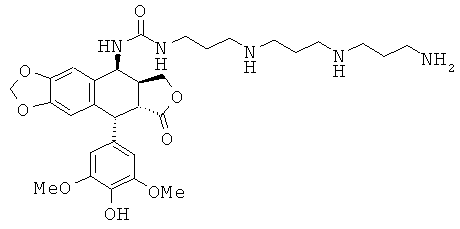
соединения 48: 1-{3-[4-(3-аминопропиламино)-бутиламино]-пропил}-3-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро[3',4':6,7]-нафто[2,3-d][1,3]диоксол-5-ил]-мочевины
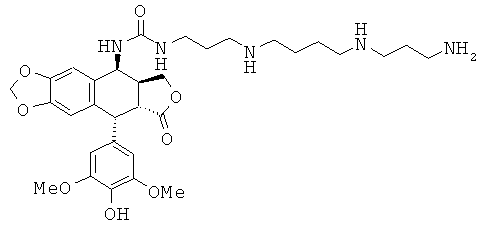
соединения 49: 1-[2-{3-[4-(3-аминопропиламино)-бутиламино]-пропиламино}-этил]-3-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро[3',4':6,7]нафто[2,3-d][1,3]диоксол-5-ил]-мочевины
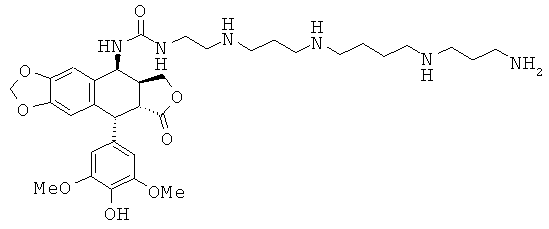
соединения 50: 1-[3-{3-[4-(3-аминопропиламино)-бутиламино]-пропиламино}-пропил]-3-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро[3',4':6,7]нафто[2,3-d][1,3]диоксол-5-ил]-мочевины
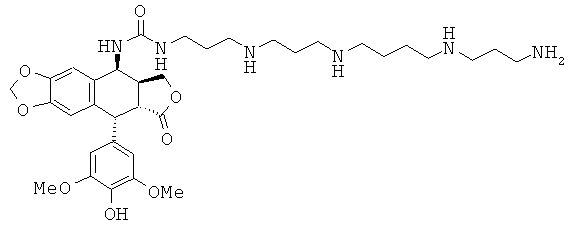
соединения 64: 1-[4-{3-[4-(3-аминопропиламино)-бутиламино]-пропиламино}-бутил]-3-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро[3',4':6,7]нафто[2,3-d][1,3]диоксол-5-ил]-мочевины
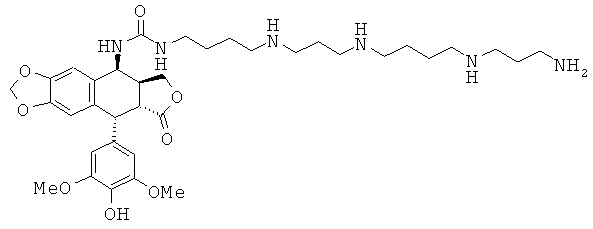
соединения 65: 1-(5-{3-[4-(3-аминопропиламино)-бутиламино]-пропиламино}-пентил)-3-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро[3',4':6,7]нафто[2,3-d][1,3]диоксол-5-ил]-мочевины
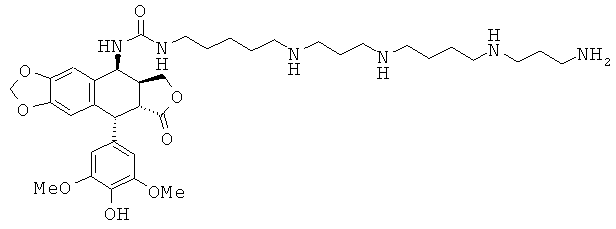
соединения 66: 1-{3-[3-(4-аминобутиламино)-пропиламино]-пропил}-3-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро[3',4':6,7]-нафто[2,3-d][1,3]диоксол-5-ил]-мочевины
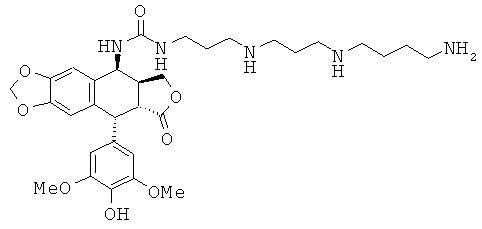
соединения 67: 1-{4-[3-(4-аминобутиламино)-пропиламино]-бутил}-3-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро[3',4':6,7]-нафто[2,3-d][1,3]диоксол-5-ил]-мочевины
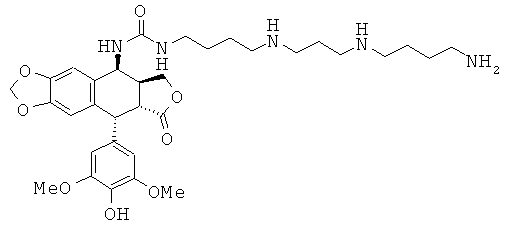
соединения 68: 1-{4-[4-(3-аминопропиламино)-бутиламино]-бутил}-3-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро[3',4':6,7]-нафто[2,3-d][1,3]диоксол-5-ил]-мочевины
сульфамидного ряда:
соединения 61: 2-(4-аминопентиламино)-этансульфоновой кислоты N-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро-[3',4':6,7]нафто[2,3-d][1,3]диоксол-5-ил]-амида
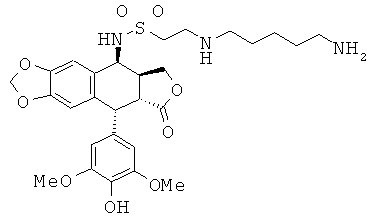
соединения 62: 2-[4-(4-аминобутиламино)-бутиламино]-этансульфоновой кислоты N-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро-[3',4':6,7]нафто[2,3-d][1,3]диоксол-5-ил]-амида
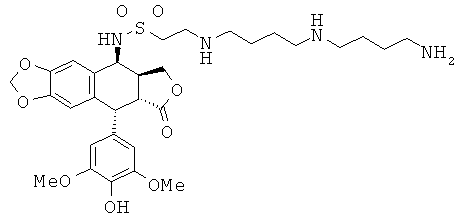
соединения 63: 2-[3-(3-аминопропиламино)-пропиламино]-этансульфоновой кислоты N-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро-[3',4':6,7]нафто[2,3-d][1,3]диоксол-5-ил]-амида
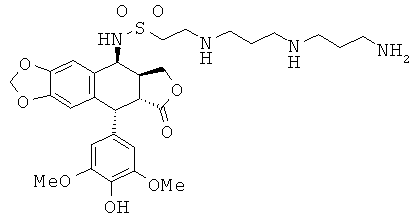
соединения 51: 2-(4-аминобутиламино)-этансульфоновой кислоты N-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро[3',4':6,7]нафто[2,3-d][1,3]диоксол-5-ил]-амида
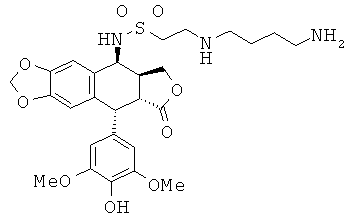
соединения 52: 2-[3-(4-аминобутиламино)-пропиламино]-этансульфоновой кислоты N-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро-[3',4':6,7]нафто[2,3-d][1,3]диоксол-5-ил]-амида
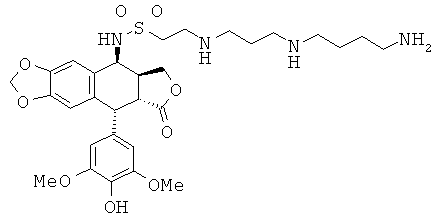
соединения 53: 2-[4-(3-аминопропиламино)-бутиламино]-этансульфоновой кислоты N-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро-[3',4':6,7]нафто[2,3-d][1,3]диоксол-5-ил]-амида
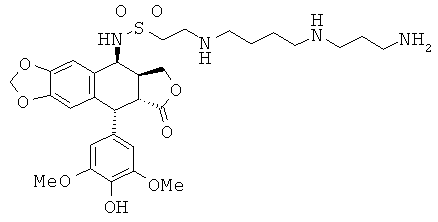
соединения 54: 2-{3-[4-(3-аминопропиламино)-бутиламино]-пропиламино}-этансульфоновой кислоты 3-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро[3',4':6,7]нафто[2,3-d][1,3]диоксол-5-ил]-амида
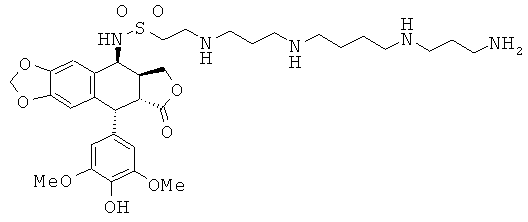
соединения 59: 2-{3-[3-(3-аминопропиламино)-пропиламино]-пропиламино}-этансульфоновой кислоты 3-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро[3',4':6,7]нафто[2,3-d][1,3]диоксол-5-ил]-амида
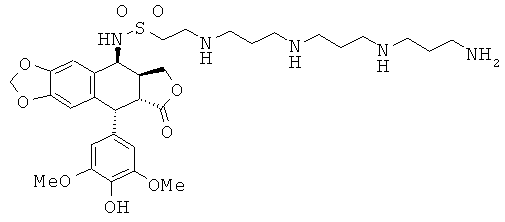
соединения 60: 2-{4-[4-(4-аминобутиламино)-бутиламино]-бутиламино}-этансульфоновой кислоты 3-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро[3',4':6,7]нафто[2,3-d][1,3]диоксол-5-ил]-амида

и их солей присоединения неорганических или органических кислот.
Соединения по изобретению представляют собой, например, соединения общей формулы 1, где:
- R представляет собой атом водорода или С1-4алкил,
- А представляет собой CO(CH2)n, CONH(CH2)n или SO2(CH2)n, где n равно 2, 3, 4 или 5,
- R1 представляет собой атом водорода или С1-4алкил,
- R2 представляет собой атом водорода или С1-4алкил или также может представлять собой (CH2)m-NR3R4, где m равно 2, 3, 4 или 5,
- R3 представляет собой атом водорода или С1-4алкил,
- R4 представляет собой атом водорода или С1-4алкил или также может представлять собой (CH2)p-NR5R6, где р равно 2, 3, 4 или 5,
- R5 представляет собой атом водорода или С1-4алкил, и
- R6 представляет собой атом водорода или С1-4алкил или также может представлять собой (CH2)q-NH2, где q равно 2, 3, 4 или 5,
за исключением соединений, где А представляет собой СО(СН2)2 или А представляет собой SO2(CH2)3, и оба R1 и R2 представляют собой Н.
Предпочтительно, R1 представляет собой Н, R3 представляет собой Н и R5 представляет собой Н. R также предпочтительно может представлять собой атом водорода.
Соединениями по изобретению являются, например, соединения общей формулы 1, где А представляет собой СО(СН2)n или CONH(СН2)n, где n равно 2, 3, 4 или 5, за исключением соединений, где А представляет собой СО(СН2)2 и оба R1 и R2 представляют собой Н.
Соединениями по изобретению являются, например, такие соединения общей формулы 1, которые определены выше, где R представляет собой Н.
Соединениями по изобретению являются, например, такие соединения общей формулы 1, которые определены выше, где R1 и R2 одновременно не являются Н, когда R представляет собой Н, и А представляет собой O(СН2)n, где n равно 2, 3 или 4.
Одно конкретное воплощение изобретения относится к таким соединениям общей формулы 1, которые определены выше, где R2 представляет собой (CH2)m-NR3R4, предпочтительно R4 представляет собой (CH2)p-NR5R6 и, в частности, m равно 3 или 4, и р равно 3 или 4, например соединениям общей формулы 1, где R6 представляет собой Н, С1-4алкил или также (CH2)q-NH2, где q равно 3.
Настоящее изобретение относится, в частности, к соединениям формулы 1, выбранным из группы, состоящей из соединений 1-50, 55-58 и 64-68, описанных выше, и их солям присоединения неорганических или органических кислот.
Более конкретно, соединения по изобретению могут быть выбраны из группы, состоящей из соединений 14-50 и 64-68, которые определены выше, и их солей присоединения неорганических или органических кислот.
Например, соединения по изобретению могут быть выбраны из группы, состоящей из соединений 14, 16-18, 21-24, 27, 28, 31-36, 39-41, 44-50, 54, 64-68, которые определены выше, и их солей присоединения неорганических или органических кислот.
Изомерные соединения по изобретению включены в объем изобретения.
В настоящем изобретении термин "фармацевтически приемлемый", как он использован в данном описании, подразумевает то, что полезно в изготовлении фармацевтической композиции, обычно являющейся безвредной, нетоксичной и не являющейся биологически или в других отношениях нежелательной и являющейся подходящей для фармацевтического применения как в ветеринарии, так и для человека.
Как использовано в данном описании, термин "фармацевтически приемлемые соли" соединения означает соли, которые являются фармацевтически приемлемыми, как определено в данном описании, и которые имеют желаемую фармакологическую активность исходного соединения. В рамках настоящего изобретения этот термин более конкретно означает соли присоединения фармацевтически приемлемых неорганических или органических кислот.
Фармацевтически приемлемые кислоты включают соляную, бромистоводородную, серную, фосфорную, уксусную, трифторуксусную, молочную, пировиноградную, малоновую, янтарную, глутаровую, фумаровую, винную, малеиновую, лимонную, аскорбиновую, щавелевую, метансульфоновую, камфарную и сульфамовую кислоты, но этим не ограничиваются. Соединения по изобретению характеризуются тем, что они являются растворимыми в воде благодаря образованию неорганических или органических солей совместно с основными атомами азота боковой цепи в положении 4.
Еще одной задачей настоящего изобретения является использование соединений формулы 1 для противораковой терапии несолидных ("жидких") опухолей и солидных опухолей, таких как меланомы, колоректальный рак, рак легких, рак предстательной железы, рак мочевого пузыря, рак молочной железы, рак матки, рак пищевода, рак желудка, рак поджелудочной железы, рак печени, рак яичников, лейкозы, в частности лимфомы и миеломы, рак уха-горла-носа (ENT (ear, nose and throat) cancer) и рак головного мозга.
Эти соединения могут быть использованы в комбинации с другими противораковыми средствами лечения, которые могут быть цитотоксическими или цитостатическими, такими как производные платины, таксаны, винкаалкалоиды, 5-FU (5-фторурацил), для усиления их терапевтической эффективности с целью лечения опухолей, резистентных к обычным видам терапии.
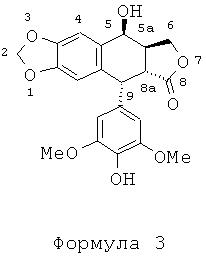
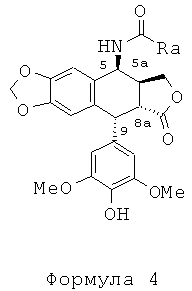
Другой задачей настоящего изобретения является способ получения этих соединений. Этот способ включает в себя применение подофиллотоксина формулы 2 в качестве исходного вещества. В частности, в соответствии со способом, описанным во французском патенте FR 2742439, используют реакцию деметилирования подофиллотоксина под действием пары реагентов - метионина (или диметилсульфида) и метансульфоновой кислоты - в присутствии либо трифторуксусной кислоты, либо ацетона и воды с получением 4'-деметилэпиподо-филлотоксина (4'-DMEP) формулы 3. Это соединение может быть подвергнуто реакции Риттера в присутствии серной кислоты или другой сильной кислоты с органическим нитрилом формулы Ra-CN, где Ra представляет собой -(CH2)n-X или -СН=СН2, где n равно 3, 4 или 5, и Х представляет собой атом галогена, такой как атом хлора, с получением соединения формулы 4. Органическим нитрилом может быть, в частности, хлорацетонитрил или в более общем случае галогеноалкилонитрил формулы NC-(CH2)n-X.
Таким образом может быть образовано промежуточное соединение, амид формулы 4а, где n равно 3-5. Когда вместо галогеноалкилонитрилов во взаимодействие приводят акрилонитрил, получают промежуточный виниламид формулы 4b.
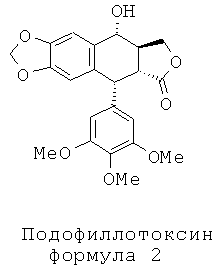
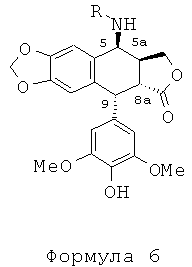
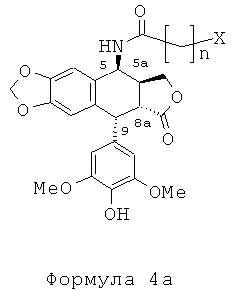
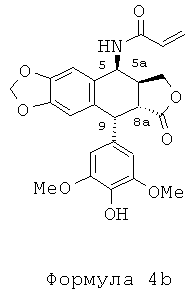
Промежуточный амид формулы 4а, где n равно 1 и Х представляет собой Cl, который является известным промежуточным соединением, обрабатывают тиомочевиной при кипячении с обратным холодильником в уксусной кислоте с получением с превосходным выходом 4β-амино-4-дезокси-4'-деметилподофилло-токсина, соединения формулы 6, где R представляет собой Н, в соответствии со способом, описанным в заявке на патент WO 2007/010007.
Амидные соединения формулы 1, где А представляет собой (СН2)n-Х и R представляет собой Н, получают следующим образом.
Промежуточные соединения формулы 4 (4а или 4b) могут быть подвергнуты алкилированию амином (моноамином, диамином или полиамином), в частности путресцином, спермидином или спермином, в защищенной форме. Полиамины содержат несколько функциональных аминогрупп, поэтому они должны быть защищены защитными группами с оставлением свободного положения первичной аминогруппы для хорошей избирательности взаимодействия. Специалисту в данной области известен выбор защитных групп, таких как бензилоксикарбонильная или трет-бутилоксикарбонильная группы, для защиты тех функциональных аминогрупп, которые не должны вступать во взаимодействие.
Например, описан спермин, защищенный бензилоксикарбонильной (Z) или трет-бутилоксикарбонильной (ВОС) группами. Также описан спермидин, защищенный группами Z или ВОС.
Таким образом, реакция алкилирования будет осуществляться между соединением формулы 4 и амином формулы HNR1R2a в защищенной форме, где:
- R1 такой, как определено выше,
- R2a представляет собой С1-4алкил, защитную группу для амина или (СН2)m-NR3aR4a, где m такой, как определено выше,
- R3a представляет собой С1-4алкил или защитную группу для амина,
- R4a представляет собой С1-4алкил, защитную группу для амина или (СН2)р-NR5aR6a, где р такой, как определено выше,
- R5a представляет собой С1-4алкил или защитную группу для амина,
- R6a представляет собой С1-4алкил, защитную группу для амина или (CH2)q-NR7aR8a, где q такой, как определено выше,
- R7a представляет собой Н или защитную группу для амина, и
- R8a представляет собой защитную группу для амина.
Применение защищенных функциональных аминогрупп подходит для предотвращения синтеза нежелательных побочных продуктов в процессе реакции сочетания, поскольку в этом случае имеется только один реакционно-способный сайт.
Получение различных аминов с защитными группами изложено в следующих публикациях: Protective Groups in Organic Synthesis (Th. W. Greene, 2-е изд., John Wiley and Sons, 1991) или в Synthesis 2002, 15, 2195; Bull. Chem. Soc. Jpn. 1998, 71, 699; Jet. Let. 1998, 39, 439 and 443; Tet. Let. 2001, 42, 2709; OPPI 1994, 26, 599; Synthesis 1994, 37; J. Org. Chem. 1998, 63, 9723; Tet. Let. 1994, 35, 2057 и 2061, J. Med. Chem. 2004, 47, 6055; J. Med. Chem. 2003, 46, 5712; Tet. Let. 1995, 36, 9401; Tet. 2000, 56, 2449.
Защитные группы для амина могут, в частности, представлять собой Z или ВОС. Предпочтительно, когда все защитные группы на защищенном амине будут одинаковыми.
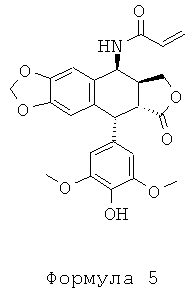
В результате реакции алкилирования между защищенным амином и соединением формулы 4 получают соединение формулы 5, затем, после удаления защит с функциональных аминогрупп, защищенных защитными группами для амина (если такие группы имеются), соединение формулы 7а.
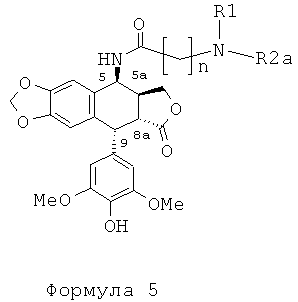
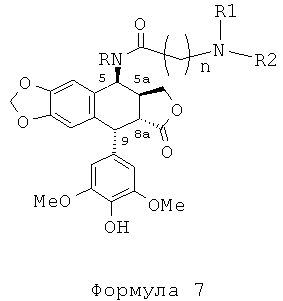
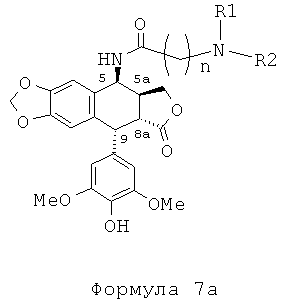
Таким образом, с использованием набора избирательных защит с помощью защитных групп для амина, например ВОС или Z, специалисты в данной области могут получить соединения формулы 7а.
Возможной последней стадией способа по изобретению является удаление защиты с функциональных аминогрупп, защищенных соответствующими группами.
Полученные соединения затем будут выделены из реакционной смеси согласно методам, хорошо известным специалистам в данной области техники.
Соединения по настоящему изобретению содержат хиральные центры природного подофиллотоксина. В соединении формулы 2 (4'-DMEP) атомы водорода в положениях 5, 5а, 8а и 9 характеризуются следующей стехиометрией: Н5α, Н5аα, Н8аβ, 1-19β. В соединении формулы 3 конфигурация асимметрических атомов углерода предпочтительно следующая: 5S, 5aS, 8aR, 9R.
Соединения мочевины формулы 10 получают из 4р-хлорацетамидо-4'-деметилподофиллотоксина формулы 4а (n равно 1, Х представляет собой Cl), в котором 4'-фенол защищен защитной группой для гидроксила Y, такой как бензилоксикарбонильная группа. В результате обработки тиомочевиной получается аминосоединение формулы 8, где R представляет собой Н, в котором группа в положении 4' защищена защитной группой Y, такой как группа Z (бензилоксикарбонил), при этом соединения формулы 8, где R не является Н, могут быть получены в соответствии со способом, изложенным в US 7378419.
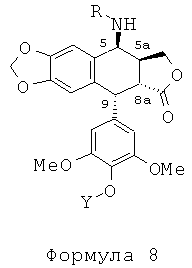
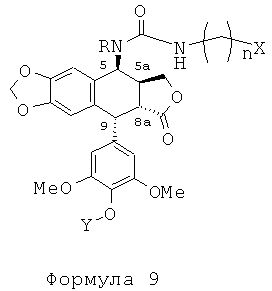
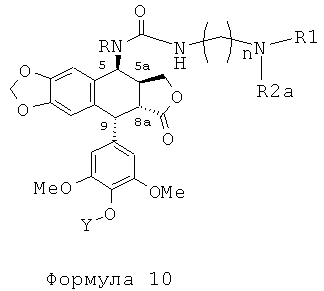
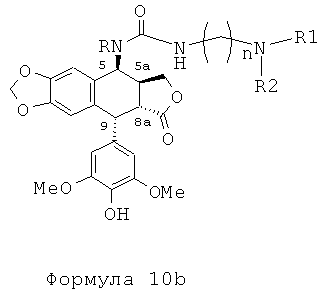
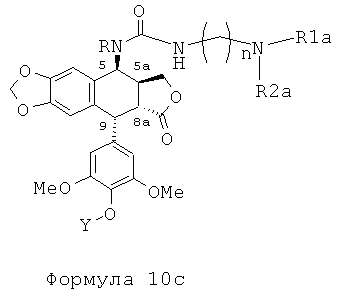
Это соединение формулы 8 (в частности, где R представляет собой Н) далее приводят во взаимодействие с изоцианатами, такими как галогеноалкилизоцианаты формулы O=С=N-(СН2)n-Х, где Х представляет собой галоген и n обозначает цепь, содержащую от 2 до 5 СН2, с получением соединения формулы 9 (в соответствии с методикой, изложенной в Heterocycles 1994, 39, 361). Это промежуточное соединение формулы 9 приводят во взаимодействие с защищенными моно-, ди-, три- или тетрааминами (формулы HNR1R2a), как упомянуто выше в традиционных условиях алкилирования, то есть, в частности, при комнатной температуре в DMF в присутствии триэтиламина и KI с получением соединений формулы 10а, затем, после удаления защит в 4'-положении подофиллотоксинового остова и с защищенных функциональных аминогрупп, с получением соединений формулы 10b.
Полученные соединения затем будут выделены из реакционной смеси согласно методам, хорошо известным специалистам в данной области.
Мочевины можно получить, используя соединение формулы 8 (в частности, где R представляет собой Н) и фосген или трифосген, с получением без выделения активированного карбонилированного промежуточного соединения. Эти промежуточные соединения соответствуют следующей далее формуле:

Это промежуточное соединение далее приводят во взаимодействие непосредственно с защищенным амином, диамином или полиамином формулы H2N-(CH2)n-NR1aR2a, где R1a представляет собой Н, С1-4алкил или защитную группу для амина и где R2a и n такие, как определено выше (где R1a не является Н, когда R2a представляет собой С1-4алкил или (CH2)m-NR3aR4a), с получением соединения формулы 10с, при этом оставшиеся стадии синтеза выполняют, как описано выше (удаление защит с функциональных аминогрупп и фенола). Последняя стадия удаления защит, проводимая либо в кислотной среде в случае группы ВОС или посредством каталитического гидрирования в случае группы Z, приводит к получению свободного полиаминного соединения общей формулы 1, где А представляет собой CONH(CH2)n.
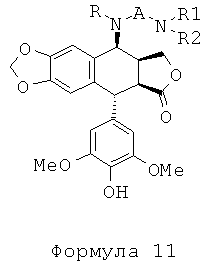
Однако алкилирование полиаминов, проводимое на подофиллотоксин-галогеноалкиламидах, не является однозначной реакцией. Одним из условий проведения процесса этого классически используемого алкилирования является щелочная среда. Важно проводить реакцию в слабощелочной среде, как, например, в присутствии триэтиламина. Основность среды может приводить наряду с данным процессом к получению побочного продукта, получающегося в результате эпимеризации протона в положении 2, приводящей к получению цис-лактонового производного формулы 11, то есть изомера формулы 1. Однако с помощью прецизионной хроматографии возможно выделение желаемого транс-лактонового производного. В следующих далее примерах показан альтернативный способ предупреждения такой возможной эпимеризации. Он состоит в образовании цепи алкановой кислоты на защищенном полиамине (соединение формулы 12, где R1a такой, как определено выше), затем сочетании полученного продукта посредством пептидного сочетания с 4β-амино-4-дезокси-4'-деметилподофиллотоксином формулы 6 (в частности, где R представляет собой Н) в соответствии с приведенной ниже реакционной схемой:
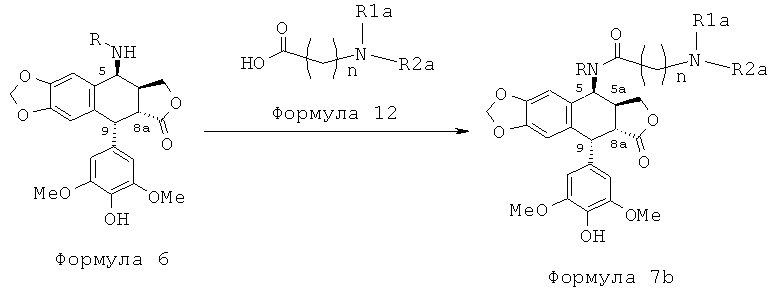
Это пептидное сочетание проводят предпочтительно в присутствии TBTU, предпочтительно с полиамином, защищенным бензилоксикарбонильными группами и содержащим группировку пропионовой, масляной или пентановой кислот. Промежуточные кислоты, содержащие группировку с 2-мя атомами углерода (формула 12, n=2), получают посредством конденсации с метилакрилатом аналогично получению продуктов, описанных в Tet. 2006, 62, 8332. Промежуточные кислоты формулы 12, где n равно 3-5, получают общепринятым алкилированием амина, защищенного галогеноалкиловым сложным эфиром, который далее омыляют до карбоновой кислоты. В дальнейшем получают соединения формулы 7b, чтобы после удаления защит с защищенных функциональных аминогрупп получить соединения формулы 7.
Полученные соединения затем будут выделены из реакционной смеси согласно методам, хорошо известным специалистам в данной области.
Сульфонамидные соединения получают следующим образом.
Соединение формулы 8 (в частности, где R представляет собой Н) приводят во взаимодействие с хлорэтилсульфонилхлоридом с получением винилсульфонамидного промежуточного соединения, в противоположность получению различных защищенных полиаминов удаление защит проводят посредством традиционного гидрирования в присутствии палладия на угле в случае защитной группы Z или в кислотной среде в случае защитной группы ВОС.
В следующих далее неограничивающих примерах проиллюстрированы используемые методики данного способа
1. Получение промежуточных соединений
Промежуточное соединение I 4-амино-4'-деметил-4-дезоксиподофилло-токсин (формула 6, где R=Н)
Это соединение получают, как описано в заявке на патент WO 2007/010007.
Стадия 1: реакция Риттера: получение 4β-хлорацетамидо-4'-деметил-4-дезоксиподофиллотоксина Формулы 4а (где n=1, и Х=Cl)
К суспензии 30 г (0,075 моль) 4'-деметилэпиподофиллотоксина формулы 3 в 47,5 мл (0,75 моль) хлорацетонитрила при комнатной температуре по каплям добавляют 0,5 мл концентрированной серной кислоты. Смесь оставляют перемешиваться при этой температуре в течение 1 часа, в течение этого периода времени наблюдается растворение с последующим повторным образованием осадка. Добавляют 300 мл 2-пропанола. Осадок отфильтровывают, промывают 200 мл 2-пропанола и водой до рН 7. Полученное белое твердое вещество сушат под вакуумом при 40°С с получением 32,9 г хлорацетамидсодержащего соединения формулы 4а (n=1, Х=Cl), то есть с выходом 93%. Т.пл.=240°С.
Стадия 2: получение 4-амино-4'-деметил-4-дезоксиподофиллотоксина (Формула 6, где R=Н)
Суспензию 17 г (0,0358 моль) 4(3-хлорацетамидо-4'-деметил-4-дезоксиподофиллотоксина, полученного выше, в 75 мл ледяной уксусной кислоты нагревают до 80°С с перемешиванием. Добавляют в виде одной порции 4,2 г (0,0537 моль) тиомочевины. Смесь оставляют перемешиваться при этой температуре в течение 1 ч 30 мин, в течение этого периода времени наблюдается растворение с последующим повторным образованием осадка. Реакционную смесь фильтруют в горячем состоянии, промывают 75 мл ледяной уксусной кислоты и диизопропиловым эфиром. Полученное белое твердое вещество сушат под вакуумом при 40°С с получением 14,6 г соединения формулы 6 в форме гидрохлорида, что соответствует молярному выходу 93%. Т.пл.>260°С. 1H-ЯМР (DMSO) δ 8.63 (m, 2H), 8,32 (m, 1H), 7.23 (s, 1H, Н5), 6.60 (s, 1H, H8), 6.18 (s, 2H, H2', Н6'), 6.05 (d, 2H, J=2.1 Гц, OCH2O), 4.73 (d, 1H, J=4.5 Гц, Н4), 4.56 (d, 1H, J=5.2 Гц, H1), 4,34 (m, 2H, H11a и H11b), 3.65 (dd, 1H, J=5.2 Гц, Н2), 3.62 (s, 6H, 2×ОСН3), 3.06 (m, 1Н, Н3).
Промежуточное соединение II: получение 4β-акриламидо-4'-деметил-4-дезоксиподофиллотоксина
К суспензии 3 г (0,0075 моль) 4'-деметилэпиподофиллотоксина формулы 3 в 10 мл акрилонитрила при комнатной температуре добавляют несколько капель концентрированной серной кислоты. Смесь оставляют перемешиваться при этой температуре в течение 3 часов, в течение этого периода времени наблюдается растворение с последующим повторным образованием осадка. Добавляют 50 мл 2-пропанола. Осадок отфильтровывают, промывают 2-пропанолом и водой до рН 7. Полученное белое твердое вещество сушат под вакуумом при 40°С с получением 2,64 г акриламидного соединения. Т.пл.=180°С. TLC SiO2 (30:70 гептан:AcOEt): Rf=0,25. Анал. C24H23NO3, H2O (MW=471,464): рассчитано С% 61,14, Н% 5,63, N% 2,66; обнаружено: С% 60,84, Н% 5,34, N% 2,97.
Промежуточное соединение III: получение 4β-хлорбутирамидо-4'-деметил-4-дезоксиподофиллотоксина
Стадия 1: получение 4β-хлорбутирамидо-4-дезоксиподофиллотоксина
Это соединение получают из подофиллотоксина и 4-хлорбутиронитрила в соответствии с методикой, описанной на стадии 1 получения промежуточного соединения I для взаимодействия хлорацетонитрила с 4'-деметилэпиподофиллотоксином. TLC SiO2 (9:1 CH2Cl2:ацетон): Rf=0,38; выход=71%.
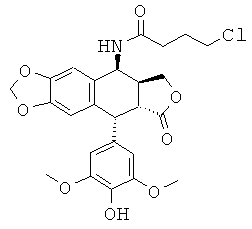
Стадия 2: получение 4β-хлорбутирамидо-4'-деметил-4-дезокси-подофиллотоксина (Формула 4а, где n=3 и Х=Cl)
4,46 г соединения, полученного на стадии 1 выше, суспендируют с перемешиванием в 21,16 мл метансульфоновой кислоты. Затем добавляют 1,93 г D,L-метионина и перемешивание продолжают в течение 2 ч. Реакционную смесь выливают с перемешиванием в воду и образуется осадок. В результате фильтрования и промывания водой до нейтрального значения рН получают после сушки и удаления воды 2,26 г (выход=52%) продукта деметилирования. TLC SiO2 (9:1 CH2Cl2:ацетон): Rf=0,20. Продукт используют непосредственно без очистки на следующих далее стадиях алкилирования.
Промежуточное соединение IV: получение 4β-бромпентанамидо-4'-деметил-4-дезоксиподофиллотоксина
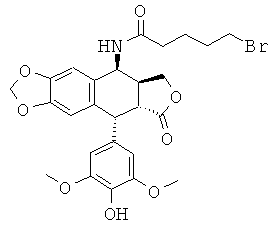
Это соединение получают аналогично получению 4β-хлорацетамидо-4'-деметил-4-дезоксиподофиллотоксина, стадии 1 для промежуточного соединения I, но с использованием соответствующего реагента, то есть 5-бромбутиронитрила. 4β-Бромпентанамидо-4'-деметил-4-дезоксиподофиллотоксин получают с выходом 57%. TLC SiO2 (95:5 CH2Cl2:MeOH): Rf=0,28. Характерные сигналы спектра 1H-ЯМР (DMSO) δ 5.39 (t, 2H, J=6.4 Гц, CH2Br), 2.17 (t, 2H, J=7.2 Гц, CH2CO), 1.79 (m, 2H, CH2), 1.66 (m, 2H, CH2).
2. Получение соединений по изобретению
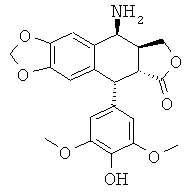
Пример 1: получение 3-диметиламино-N-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро[3',4':6,7]нафто[2,3-d][1,3]диоксол-5-ил]-пропионамида или (4β-диметиламинопропионамидо)-4'-деметил-4-дезоксиподофиллотоксина) (Соединение 5)
500 мг 4β-амино-4'-деметил-4-дезоксиподофиллотоксина формулы 6 и 146 мг 3-диметиламинопропионовой кислоты растворяют с перемешиванием в 50 мл ацетонитрила вместе с 0,21 мл триэтиламина. Добавляют 400 мг TBTU и перемешивание продолжают в течение 6 ч при комнатной температуре. Реакционную смесь выливают в воду (300 мл) и экстрагируют этилацетатом (3×100 мл). Органические фазы сушат над Na2SO4, фильтруют и упаривают. Остаток подвергают флэш-хроматографии на SiO2 (элюирование смесью 78:20:2 CH2Cl2:MeOH:NH4OH). После упаривания остаток еще раз подвергают хроматографии на препаративной HPLC-колонке (X Bridge, OBD, C18, 30×250 мм, 10 мкм), градиент элюирования CH3CN/5 мМ HCI (от 10/90 до 80/20). Фракции экстрагируют этилацетатом (2×100 мл), сушат и упаривают. Остаток переводят в соль, используя смесь HCl в изопропаноле и этилового эфира, отфильтровывают и сушат с получением 246 мг гидрохлорида в виде белого порошка. Выход=37%. TLC SiO2 (90:9:1 CH2Cl2:MeOH:NH4OH): Rf=0,38. ЯМР основания: 1H-ЯМР (DMSO) δ 8.36 (d, 1Н, NH), 8.28 (s, 1H, ОН), 6.76 (s, 1Н, H5), 6.53 (s, 1H, Н8), 6.24 (s, 2H, H2', Н6'), 5.99 (d, 2H, J=8.4 Гц, OCH2O), 5.16 (dd, 1H, H4), 4.50 (d, 1H, J=5 Гц, Н1), 4.25 (t, 1H, H11a), 3,87 (t, 1H, H11b), 3.62 (s, 6H, ОМе), 3.11 (dd, 1H, Н2), 2,93 (m, 1H, Н3), 2.42-2.55 (m, 2H, CH2N), 2.24-2.33 (m, 2H, CH2N), 2.12 (s, 6H, NMe2). Масс-спектр (APCI), m/z=499, M-H+.
Пример 2: получение 4-диметиламино-N-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро-[3',4':6,7]-нафто[2,3-d][1,3]диоксол-5-ил]-бутирамида или (4β-диметиламинобутирамидо)-4'-деметил-4-дезоксиподофиллотоксина) (Соединение 6)
Раствор 570 мг промежуточного соединения III, полученного выше, перемешивают в течение 12 ч в 25 мл ацетонитрила вместе с 0,28 мл (5 экв.) диметиламина. Затем реакционную смесь выливают на лед и добавляют 1 н. раствор HCl до рН 4. Экстракцию проводят, используя метиленхлорид, и затем водную фазу подщелачивают раствором NaHCO3 до рН 8. Эту фазу повторно экстрагируют CH2Cl2, сушат над Na2SO4, фильтруют и упаривают, получая 100 мг оранжевой пены. Гидрохлорид образуют в метилэтилацетоне, добавляя раствор HCl в изопропаноле (3 н.). Затем гидрохлорид отфильтровывают, промывают метилэтилкетоном, затем этиловым эфиром. После сушки полученные кристаллы представляют собой 90 мг беловатого порошка. TLC SiO2 (90:10:1 CH2Cl2:MeOH:NH4OH): Rf=0,47. Т.пл.=169°С. 1Н-ЯМР (DMSO) δ 8.35 (d, 1H, NH), 6.75 (s, 1H, Н5), 6.51 (s, 1H, H8), 6.20 (s, 2H, H2', H6'), 5.96 (d, 2H, J=6.36 Гц, OCH2O), 5.15 (dd, 1H, H4), 4.47 (d, 1H, J=5 Гц, Н1), 4.26 (t, 1H, H11a), 3.68 (t, 1H, H11b), 3.59 (s, 6H, ОМе), 3,34 (m, 2H, CH2N), 3.08 (dd, 1H, Н2), 2,93 (m, 1H, Н3), 2.72 (s, 6H, NMe2), 2.22 (m, 2H, CH2CO), 1,86 (m, 2H, CH2). Анал. C27H33ClN2O8, рассчитано С% 55,43; Н% 6,37; N% 6,06; обнаружено С% 55,74, Н% 6,01, N% 4,68.
Пример 3: получение 3-[(2-диметиламиноэтил)-метиламино]-N-[9-(4-гидрокси-3,5-диметоксисренил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро-[3',4':6,7]-нафто[2,3-d][1,3]диоксол-5-ил]-пропионамида или (4β-[3-[2-(N-метил-N,N-диметиламино-2-этил)]пропионамид)]-4'-деметил-4-дезоксиподофиллотоксина) (Соединение 3)
200 мг промежуточного соединения II растворяют в 20 мл THF и в реакционную смесь по каплям вносят 1,15 мл N,N,N'-триметилэтилендиамина. Смесь перемешивают в течение 12 ч при комнатной температуре и затем упаривают досуха. На этой стадии получают смесь 2 эпимеров по положению 2 (цис-лактон и транс-лактон). Флэш-хроматография (элюирование смесью CH2Cl2:MeOH:NH4OH 90:10:0.5) позволяет получить 70 мг эпимерного производного по положению 2 (цис-лактона). Т.пл.=178°С. 1H-ЯМР (DMSO) δ 8.41 (d, 1H, J=8.96 Гц, CONH), 8.29 (m, 1H, ОН), 6.95 (s, 1H, Н8), 6,89 (s, 1H, Н5), 6.42 (s, 2H, H2', H6'), 6.01 (d, 2H, J=4.04 Гц, OCH2O), 5.08 (dd, 1H, J=6.6 Гц, Н4), 4,37 (s, 1H, H1), 4.28 (t, 1H, J=9.2 Гц, H11a), 4.01 (dd, 1H, J=4 Гц, J'=9.6, H11b), 3.79 (dd, 1H, J=1.6 Гц, J'=10.8, Н2), 3.69 (s, 6H, ОМе), 3,32 (m, 3Н, Н3, COCH2), 2.63-2.27 (m, 6H, CH2N), 2.194 (s, 3Н, NMe), 2.101 (s, 6H, NMe2).
Пример 4: получение 5-диметиламинопентановой кислоты N-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро-[3',4':6,7]-нафто[2,3-d][1,3]-диоксол-5-ил]-амида или (4β-диметиламинопентанамидо-4'-деметил-4-дезокси-подофиллотоксина) (Соединение 7)
700 мг бромированного промежуточного соединения IV, полученного выше, перемешивают в 3,3 мл 2 М раствора диметиламина в THF в течение 4 суток в атмосфере азота. Раствор выливают в лед и добавляют раствор HCl (0,1 н.) до рН 7. Раствор экстрагируют этилацетатом, получая после сушки над Na2SO4, фильтрования и упаривания 341 мг масла, которое затем очищают флэш-хроматографией на SiO2 (97:7:0,7 CH2Cl2:MeOH:NH4OH) с получением 200 мг чистого масла. Гидрохлорид образуют, добавляя к основанию, растворенному в изопропаноле, раствор солянокислого этанола до кислого значения рН. TLC SiO2 (90:10:1 CH2Cl2:MeOH:NH4OH): Rf=0,23. Т.пл. (клейкое вещество)=224°С. 1H-ЯМР (DMSO) δ 8,38 (d, 1H, J=8.56 Гц, NH), 6.77 (s, 1H, Н5), 6.53 (s, 1H, H8), 6.23 (s, 2H, H2', H6'), 5.99 (d, 2H, J=12.4 Гц, OCH2O), 5.18 (dd, 1H, J=8.16 Гц, J'=4.76 Гц, Н4), 4.49 (d, 1H, J=5.12 Гц, H1), 4.29 (t, 1H, J=8 Гц, H11a), 3.73 (t, 1H, J=10.34 Гц, H11b), 3.63 (s, 6H, ОМе), 3.22 (dd, 1H, J=5.16 Гц, J'=14.3 Гц, Н2), 3,3 (t, 2H, J=7.08 Гц, CH2N), 2.93 (m, 1H, Н3), 2.71 (s, 6H, NMe2), 2.20 (t, 2H, J=6.88 Гц, СН2СО), 1.59 (m, 4H, СН2).
Пример 5: получение 5-[(2-диметиламиноэтил)-метиламино]пентановой кислоты N-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро-[3',4':6,7]-нафто[2,3-d][1,3]диоксол-5-ил]-амида или 4β-[5-[2-(N-метил-N-диметиламино-2-этил)]пентанамид)]-4'-деметил-4-дезоксиподофилло-токсина (Соединение 55)
Это соединение получают аналогично получению соединения из Примера 2, но используя бромированное промежуточное соединение IV и N,N,N'-триметилэтилендиамин. Получают 4β[5-[2-(N-метил-N-диметиламино-2-этил)]пентанамид)]-4'-деметил-4-дезокси-подофиллотоксин с примесью его 8а-эпимера. Флэш-хроматография (элюирование смесью CH2Cl2:MeOH:NH4OH, 95:5:0,5, затем 90:10:0,6) позволяет выделить указанное в заголовке соединение. Дигидрохлорид кристаллизуют из изопропанола, добавляя солянокислый этанол. HPLC (C8 Symmetry; элюирование смесью буфера на основе KH2PO4 (3,4 г/л), подведенного до рН 4 добавлением H3PO4, и CH3CN (80/20)). Время удерживания (RT): 4,95 мин. 1H-ЯМР (DMSO) δ 8,38 (d, 1Н, J=8.56 Гц, NH), 6.78 (s, 1H, H5), 6.53 (s, 1H, H8), 6.24 (s, 2H, H2', H6'), 6.00 (d, 2H, J=11.3 Гц, OCH2O), 5.19 (dd, 1H, J=8.15 Гц, J'=4.6 Гц, Н4), 4.50 (d, 1H, J=4.8 Гц, H1), 4.29 (t, 1H, J=8 Гц, H11a), 3.72 (dd, 1H, H11b), 3.63 (s, 6H, ОМе), 3.53 (m, 2H, CH2N), 3.08-3.24 (m, 5H, CH2N, Н2), 2.93 (m, 1H, Н3), 2.84 (s, 6H, NMe2), 2.79 (s, 3H, NMe), 2.20 (t, 2H, J=6.88 Гц, CH2CO), 1.69 (t, 2H, CH2), 1.57 (m, 2H, CH2).
Пример 6: получение 4-амино-N-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро-[3',4':6,7]-нафто[2,3-d][1,3]диоксол-5-ил]-бутирамида или (4β-аминобутанамидо-4'-деметил-4-дезоксиподофиллотоксина) (Соединение 56)
Стадия 1: к раствору 1 г 4β-амино-4'-деметил-4-дезоксиподофиллотоксина формулы 6 в 50 мл ацетонитрила при перемешивании добавляют 510 мг 4-трет-бутоксикарбониламиномасляной кислоты (Bioorg. Med. Chem. Lett. 2005, 15, 1969) вместе с 0,40 мл триэтиламина. Затем добавляют 800 мг TBTU и перемешивание продолжают при комнатной температуре в течение 5 ч. Реакционную смесь выливают в воду и экстрагируют этилацетатом. После выпаривания растворителя остаток очищают флэш-хроматографией на SiO2 (градиент элюирования от чистого CH2Cl2 до смеси 90:9:1 CH2Cl2:MeOH:NH4OH). Препаративная хроматография на Х Bridge C18, OBD, 30×250 мм, 10 мкм, элюент: градиент от 10:90 CH3CN/H2O до 90:10 CH3CN/Н2О, позволила получить после упаривания содержащих чистое соединение фракций 460 мг бесцветного масла. Выход=31%. TLC SiO2 (90:9:1 CH2Cl2:MeOH:NH4OH): Rf=0,20, 1H-ЯМР (DMSO) δ 8.25 (s, 1H, ОН), 8.22 (d, 1H, J=8 Гц, NH амида), 6.79 (m, 1H, NH карбамата), 6.76 (s, 1H, H5), 6.52 (s, 1H, Н8), 6.24 (s, 2H, H2', H6'), 6.00 (d, 2H, J=13.2 Гц, OCH2O), 5.17 (dd, 1H, J=8 Гц, J'=4.4 Гц, Н4), 4.49 (d, 1H, J=4.8 Гц, H1), 4.27 (t, 1H, J=8 Гц, H11a), 3.74 (t, 1H, J=9.6 Гц, H11b), 3.62 (s, 6H, ОМе), 3.15 (dd, 1H, J=14.4 Гц и J'=5.2 Гц, Н2), 2.89-2.96 (m, 3H, Н3 и CH2N), 2.13 (t, 2Н, J=7.2 Гц, СН2СО), 1.62 (m, 2H, CH2), 1.36 (s, 9H, t-Bu).
Стадия 2: промежуточный карбамат, полученный на стадии 1 выше, перемешивают при комнатной температуре в течение 4 ч в 25 мл CH2Cl2 в присутствии 25 мл HCl в изопропаноле (3,3 M). После упаривания получают белый осадок, который затем фильтруют, промывают этиловым эфиром и сушат, получая 275 мг гидрохлорида в виде белого порошка. Выход 67%. Т.пл.=284°С. TLC SiO2 (90:9:1 CH2Cl2:MeOH:NH4OH): Rf=0,18; MS (ESI+) m/z=485 (M-H+). 1H-ЯМР (DMSO) δ 8.44 (d, 1H, J=7.6 Гц, NH амида), 8.27 (m, 1H, ОН), 7.91 (m, 2H, NH2 и HCl), 6.77 (s, 1H, Н5), 6.53 (s, 1H, Н8), 6.24 (s, 2H, H2', H6'), 6.00 (d, 2H, J=11.6 Гц, OCH2O), 5.19 (d, 1H, J=4.4 Гц, Н4), 4.52 (d, 1H, J=5.2 Гц, H1), 4,31 (t, 1H, J=8 Гц, H11a), 3.74 (m, 1H, H11b), 3.63 (s, 6H, ОМе), 3.17 (dd, 1H, J=14 Гц и J'=4.8 Гц, Н2), 2.95 (m, 1H, Н3), 2.81 (t, 2H, J=7.6 Гц, CH2N), 2.27 (t, 2H, J=7.2 Гц, CH2CO), 1.83 (m, 2H, CH2).
Пример 7: получение 5-аминопентановой кислоты N-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро-[3',4':6,7]-нафто[2,3-d][1,3]-диоксол-5-ил]-амида или (4β-аминопентанамидо-4'-деметил-4-дезоксиподофиллотоксина) (Соединение 57)
Это соединение получают аналогично получению соединения из Примера 6 выше, но используя 5-трет-бутоксикарбониламинопентановую кислоту.
Пример 8: таким же образом, как и в Примере 6, но с использованием вместо 4-трет-бутоксикарбониламиномасляной кислоты соответствующих защищенных диамино-, триамино- или тетрааминосодержащих кислот с пропионовой цепью (которые получают с использованием метилакрилата аналогично способу в публикации Tetrahedron 2006, 62, 8335), синтезированы соединения 8, 10, 12, 13, 14, 29, 30, 27, 28, 58, 19, 20 и 18 формулы 1 (где А=CO(CH2)n, n=2).
Пример 9: получение 2-{3-[4-(3-аминопропиламино)-бутиламино]-пропиламино}-этансульфоновой кислоты 3-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро-[3',4':6,7]-нафто[2,3-d][1,3]диоксол-5-ил]-амида или (4β-2-{3-[4-(3-аминопропиламино)-бутиламино1-пропиламино}-этансульфон-амидо-4'-деметил-4-дезоксиподофиллотоксина) (Соединение 54)
Стадия 1: получение 4β-винилсульфониламино-4'-бензилоксикарбонил-4'-деметил-4-дезоксиподофиллотоксина
500 мг 4β-амино-4-дезокси-4'-бензилоксикарбонил-4'-деметилэпиподофиллотоксина формулы 8 растворяют в 20 мл CH2Cl2 вместе с 0,4 мл триэтиламина. По каплям с перемешиванием добавляют 0,1 мл 2-хлорэтан-сульфонилхлорида в 5 мл CH2Cl2 при -15°С. Перемешивание продолжают в течение 15 мин, затем смесь оставляют нагреваться до температуры окружающей среды и перемешивание продолжают в течение 4 ч. Затем реакционную смесь выливают в воду и экстрагируют CH2Cl2. Органические фазы объединяют, сушат над Na2SO4, фильтруют и упаривают. Остаток очищают флэш-хроматографией, элюируют градиентом от чистого гептана до чистого AcOEt. Полученные фракции, содержащие чистое соединение, упаривают, получая 220 мг пены. Выход=55%. TLC SiO2 (90:9:1 CH2Cl2:MeOH:NH4OH): Rf=0,7; 1H-ЯМР (DMSO) δ 8.03 (d, 1H, J=8.56 Гц, NH), 7.40 (m, 5H, Ar), 7.02 (dd, 1H, J=16.36 Гц, J'=9.8 Гц, НС=), 6.91 (s, 1H, Н5), 6.53 (s, 1H, H8), 6,33 (s, 2H, H2', H6'), 6.15 (d, 1H, J=16.4 Гц, НС=), 6.09 (d, 1H, J=9.8 Гц, НС=), 6.01 (d, 2H, J=11.3 Гц, OCH2O), 5.23 (s, 2H, CH2Ph), 4.67 (dd, 1H, J=8.24 Гц, J'=4.4 Гц, Н4), 4.59 (d, 1H, J=5.4 Гц, H1), 4,31 (t, 1H, J=8.04 Гц, H11a), 4.13 (t, 1H, H11b), 3.63 (s, 6H, OMe), 3.28 (dd, 1H, J=5.36 Гц и J'=18.48 Гц, Н2), 2.97 (m, 1Н, Н3).
Стадия 2: добавление N1,N2,N3-трибензилоксикарбонилспермина
220 мг винильного производного, полученного на приведенной выше стадии, растворяют в 10 мл метанола. К реакционной смеси добавляют 220 мг N1,N2,N3-трибензилоксикарбонилспермина и перемешивание продолжают в течение 5 суток при комнатной температуре. После упаривания под вакуумом добавляют воду и смесь экстрагируют этилацетатом. После сушки органической фазы, фильтрования и упаривания проводят очистку флэш-хроматографией (градиент элюирования от чистого гептана до чистого AcOEt и затем до смеси 90:9:1 AcOEt:MeOH:NH4OH). Получают 70 мг тетразамещенного соединения, полученного в результате присоединения, то есть с выходом 16% (транс-лактонового соединения). Также получают другое эпимерное по положению 2 соединение (цис-лактоновое соединение).
Анализ транс-лактонового соединения: TLC SiO2 (90:9:1 CH2Cl2:MeOH:NH4OH): Rf=0,6. Аналитическая HPLC: X Bridge C8, 4,6×250 мм, 5 мкм, элюент: 80:20 CH3CN:H2O-KH2PO4 6,8 г/л, рН=4, скорость потока 1 мл/мин, RRT=3,55 мин. MS (ESI+) m/z=1094.
Стадия 3: гидрогенолиз защитных групп
70 мг полученного выше транс-лактонового производного помещают в атмосфере водорода в смесь 10 мл метанола и 5 мл CH2Cl2. Также добавляют 0,25 мл HCl в изопропаноле вместе с 50 мг 10%-ного палладия на угле. Энергичное перемешивание продолжают в течение 5 ч. Катализатор отфильтровывают, промывают метанолом, остаток упаривают под вакуумом и переносят в этиловый эфир для кристаллизации гидрохлорида, который отфильтровывают и сушат под вакуумом. Получают 30 мг кристаллов в виде гидрохлорида (выход 63%). Т.пл.=191°С. Аналитическая HPLC: X Bridge C8, 4,6×250 мм, 5 мкм, элюент: 15:85 CH3CN:H2O-KH2PO4 6,8 г/л, рН 4, скорость потока 1 мл/мин, RRT=14,08 мин. MS (ESI+) m/z=692 (М-Н+).
Пример 10: соединения 51, 52, 53, 59, 60, 61, 62 и 63 могут быть синтезированы таким же образом, как и в Примере 9, с использованием соответствующих защищенных моноаминов, диаминов, триаминов и тетрааминов посредством конденсации с 4-β-винилсульфониламино-4'-бензилоксикарбонил-4'-деметил-4-дезоксиподофиллотоксином, полученным на стадии 1 Примера 9.
Пример 11: получение 4-{3-[4-(3-аминопропиламино)-бутиламино]-пропиламино}-N-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6.8,8а,9-гексагидрофуро-[3',4':6,7]-нафто[2,3-d][1,3]диоксол-5-ил]-бутирамида или (4-{3-4-{3-[4-(3-аминопропиламино)-бутиламино]-пропиламино}-бутирамидо-4'-деметил-4-дезоксиподофиллотоксина) (Соединение 21)
Это соединение синтезируют в соответствии с одним из 2 следующих способов.
Способ 1: алкилирование хлорированного производного (промежуточного соединения III)
Это соединение получают способом, аналогичным Примеру 2. Используют полученное выше хлорированное промежуточное соединение III и его конденсируют с трибензилоксикарбонил-спермином (описанным в Tet. Let. 1998, 39, 439) с получением 4-β-4-{3-[4-(3-аминопропиламино)-бутиламино]-пропиламино}-бутирамидо-4'-деметил-4-дезоксиподофиллотоксина после гидрогенолиза в соответствии с тем же способом, что и на стадии 3 Примера 9.
Способ 2: пептидное сочетание
Стадия 1: 7,51 г трибензилоксикарбонилспермина (триZ-спермина) (Tet. Let. 1998, 39, 439) с перемешиванием растворяют в 150 мл ацетонитрила. Добавляют 2,1 мл триэтиламина, затем 2,25 г метилбромбутирата и после этого 900 мг карбоната цезия. Реакционную смесь кипятят с обратным холодильником в течение 20 ч при перемешивании. Смесь выливают в воду и экстрагируют этилацетатом (3×200 мл), органические фазы сушат над Na2SO4, фильтруют и упаривают. Остаток подвергают флэш-хроматографии на SiO2 (градиент элюирования от чистого CH2Cl2 до смеси 70% CH2Cl2 и 30% смеси 9:1 MeOH:NH4OH). Выделяют 2,48 г моноалкилированного сложноэфирного производного спермина: метил-4-[3-(бензилоксикарбонил-{4-[бензилоксикарбонил-(3-бензилоксикарбониламино-пропил)-амино]бутил}-амино)-пропиламино]-бутирата формулы 12а (в виде сложного метилового эфира, где В=Н)
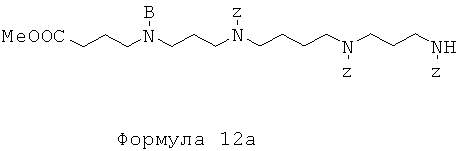
TLC SiO2 (90:9:1 CH2Cl2:MeOH:NH4OH): Rf=0,4. Другие хроматографические фракции содержат диалкилированное производное производного спермина.
Стадия 2: 2,48 г этого моноалкилированного промежуточного соединения со Стадии 1 помещают в 30 мл ацетонитрила вместе с 0,45 мл триэтиламина. По каплям с перемешиванием добавляют 0,55 мл бензилхлорформиата в 5 мл ацетонитрила при комнатной температуре и смесь оставляют перемешиваться в течение 2 ч. Реакционную смесь выливают в воду, экстрагируют этилацетатом, органические фазы сушат над Na2SO4, фильтруют и упаривают. Проводят флэш-хроматографию (градиент элюирования от чистого CH2Cl2 до смеси, состоящей из 90% CH2Cl2 и 10% MeOH:NH4OH (9:1)). Получают 0,95 г тетразамещенного производного спермина: метил-4-{бензилоксикарбонил-[3-(бензилоксикарбонил-{4-[бензилоксикарбонил-(3-бензилоксикарбониламинопропил)-амино]-бутил}-амино)-пропил]-амино}-бутирата, соответствующего формуле 12а, где В=Z, в виде бесцветного масла. Выход 32%. TLC SiO2 (90:10:1 CH2Cl2:MeOH:NH4OH) Rf=0,79. 1H-ЯМР (DMSO) δ 7.32 (m, 21 Н, NH и 4Ph), 5.03 и 5.00 (2s, 8H, CH2 бензила), 3.54 (m, 3Н, ОМе), 3.14 (m, 12Н, CH2N), 2.97 (m, 2Н, CH2N), 2.27 (m, 2H, СН2СО), 1.62-1.67 (m, 6Н, СН2), 1.37 (m, 2H, CH2).
Стадия 3. Полученный выше сложный эфир (0,95 г) кипятят с обратным холодильником с перемешиванием в 60 мл смеси МеОН:вода (50:50) и в присутствии 1,7 мл 1 н. NaOH в течение 1 ч. После охлаждения смесь подкисляют, используя 1 н. HCl, до рН 2 и экстрагируют этилацетатом, получая соответствующую карбоновую кислоту: 4-{бензилоксикарбонил-[3-(бензилоксикарбонил-{4-[бензилоксикарбонил-(3-бензилоксикарбониламинопропил)-амино]-бутил}-амино)-пропил]-амино}-масляную кислоту в виде бесцветного масла с количественным выходом. TLC SiO2 (95:5 CH2Cl2:MeOH): Rf=0,32. 1H-ЯМР (DMSO) δ 7.32 (m, 21Н, NH и 4Ph), 4.99 и 5.04 (2s, 8H, СН2 бензила), 3.14 (m, 12H, CH2N), 2.97 (m, 2H, CH2N), 2.15 (m, 2H, CH2CO), 1.66 (m, 6Н, СН2), 1.37 (m, 4H, СН2). ESI-MS m/z=825 М-Н+.
Стадия 4: 510 мг 4β-амино-4'-деметил-4-дезоксиподофиллотоксина (Промежуточное соединение I), полученного, как упомянуто выше, растворяют в 20 мл ацетонитрила в присутствии 950 мг кислоты, полученной на приведенной выше стадии, и 0,34 мл триэтиламина. Добавляют в виде одной порции 370 мг TBTU и перемешивание продолжают при комнатной температуре в течение 2 ч. Реакционную смесь выливают в воду и экстрагируют этилацетатом, органический раствор промывают b (рассолом), сушат, фильтруют и упаривают. Остаток очищают флэш-хроматографией (элюирование CH2Cl2:MeOH (90:10)) и затем препаративной HPLC (X Bridge, C18, 10 мкм, OBD, 30×250 мм), градиент элюирования CH3CN:H2O от 10:90 до 50:50. Получают 430 мг (выход 30%) защищенного производного спермина и подофиллотоксина. 1H-ЯМР (DMSO) δ 8.23 (d, 1H, J=8.16 Гц, NH), 7.32 (m, 4 Ph), 6.78(s, 1H, H5), 6.52 (s, 1H, Н8), 6.23 (s, 2H, H2', Н6'), 5.98 (d, 2H, J=17.08 Гц, OCH2O), 5.19 (dd, 1H, Н4), 4.99 и 5.03 (2s, 8H, CH2 бензила), 4.49 (d, 1H, J=4.8 Гц, H1), 4.29 (t, 1H, J=7.2 Гц, H11a), 3.73 (m, 1H, H11b), 3.62 (s, 6H, OMe), 3.15 (m, 13H, CH2N и Н2), 2.96 (m, 3H, CH2N и Н3), 2.10 (m, 2H, СН2СО), 1.61-1.68 (m, 6H, CH2), 1.37 (m, 4H, CH2).
Стадия 5: Это очищенное тетрабензилоксикарбонилированное соединение (430 мг) растворяют в смеси метанола (20 мл) и CH2Cl2 (10 мл). Добавляют 5 эквивалентов раствора HCl в изопропаноле. Среду в присутствии 50 мг 10%-ного палладия на угле помещают на 8 ч в атмосферу водорода с энергичным перемешиванием. Катализатор отфильтровывают, промывают метанолом и затем фильтрат упаривают. Остаток подвергают хроматографии на препаративной HPLC-колонке (Xbridge C18, 10 мкм, OBD, 30×250 мм) с элюированием 5 мМ раствором HCl. Фракции, содержащие соединение, подвергают сублимационной сушке с получением 115 мг белого твердого вещества. Т.пл.=229°С. Аналитическая чистота: 98,25% (аналитич. HPLC: Xbridge C8, элюирование (15:85) CH3CN:H2O-KHPO4 6,8 г/л при рН 4).
1H-ЯМР (DMSO) δ 8.47(d, 1H, J=8.28 Гц, NH), 6.78 (s, 1H, Н5), 6.53 (s, 1H, H8), 6.23 (s, 2H, H2', H6'), 6.00 (d, 2H, J=10.2 Гц, OCH2O), 5.19 (dd, 1H, J=7.72 Гц, J'=4.68 Гц, Н4), 4.49 (d, 1H, J=4.96 Гц, H1), 4.29 (t, 1H, J=7.8 Гц, H11a), 3.75 (t, 1H, J=9.8 Гц, H11b), 3.62 (s, 6H, OMe), 3.24 (dd, 2H, J=14.28 Гц, J'=4.8 Гц, H2), 2.89-3.00 (m, 15H, Н3 и CH2N), 2.29 (m, 2H, СН2СО), 1.88-2.08 (m, 6H, CH2), 1.73 (m, 4H, СН2).
Пример 12: аналогично тому, как описано в Примере 11, следуя Способу 2 пептидного сочетания, получают следующие соединения формулы 1 (где А=CO(CH2)n, n=3, 4 или 5): 2, 4, 6, 15, 16, 17, 33, 34, 32, 31, 37, 38, 35, 36, 22, 23, 25, 26, 24, 9, 11, 55 и 56, используя соответствующие защищенные моноамины, диамины, триамины или тетраамины, "пришитые", как указано на Стадии 1 Примера 11, Способ 2, к этил-бромбутирату или этил-бромпропионату. Стадии введения защиты (Стадию 2), омыления (Стадию 3), сочетания (Стадию 4) и удаления защиты (Стадию 5) проводят таким же образом.
Пример 13: получение 1 -{3-[4-(3-аминопропиламино)-бутиламино]-пропил}-3-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро-[3',4':6,7]-нафто[2,3-d][1,3]диоксол-5-ил]-мочевины (Соединение 48)
Стадия 1: получение 4β-хлорацетамидо-4'-деметил-4'-бензилоксикарбонил-4-дезоксиподофиллотоксина
19,6 г 4β-хлорацетамидо-4'-деметил-4-дезоксиподофиллотоксина формулы 4а (X=Cl, n=1), промежуточного соединения I, полученного на стадии 1, растворяют в 400 мл THF, затем добавляют 10 мл пиридина. Далее с перемешиванием при комнатной температуре добавляют 6,5 мл бензилхлорформиата, растворенного в 50 мл THF. Реакционную смесь перемешивают при температуре окружающей среды в течение 6 ч. По окончании реакции раствор выливают в 300 мл 1 н. HCl, затем экстрагируют этилацетатом (2×200 мл). Органические фазы сушат над Na2SO4, фильтруют и упаривают, получая 27,9 г неочищенного промежуточного соединения. TLC (95:5 CH2Cl2:MeOH): Rf=0,29. Это промежуточное соединение используют непосредственно на Стадии 2.
Стадия 2: получение 4β-амино-4'-деметил-4'-бензилоксикарбонил-4-дезоксиподофиллотоксина
27,9 г промежуточного соединения, полученного на Стадии 1 выше, растворяют в 120 мл диметилацетамида, 24 мл уксусной кислоты и 24 мл воды. Все это нагревают до 80°С с перемешиванием. На этой стадии добавляют тиомочевину (4,81 г) и реакционную смесь поддерживают при этой температуре в течение 12 ч. После охлаждения реакционную смесь медленно выливают в насыщенный раствор NaHCO3 (500 мл). Затем экстрагируют этилацетатом (200 мл) и органические фазы промывают насыщенным раствором NaHCO3 и затем насыщенным раствором NaCl. Органические фазы отделяют, сушат над Na2SO4, фильтруют и упаривают. Остаток подвергают флэш-хроматографии (градиент от чистого гептана, CH2Cl2 до 90:10 CH2Cl2:MeOH), получая 13,8 г 4β-амино-4'-деметил-4'-бензилоксикарбонил-4-дезоксиподофиллотоксина формулы 8. Выход за 2 стадии=63%. TLC (95:5 CH2Cl2:MeOH): Rf 0,55.
Стадия 3: получение 1-{3-[4-(3-трет-бутоксикарбониламинопропил)-трет-бутоксикарбониламинобутил]-трет-бутоксикарбониламинопропил}-3β-[9-(4-бензилоксикарбонилокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро-[3',4':6,7]-нафто[2,3-d][1,3]диоксол-5-ил]-мочевины
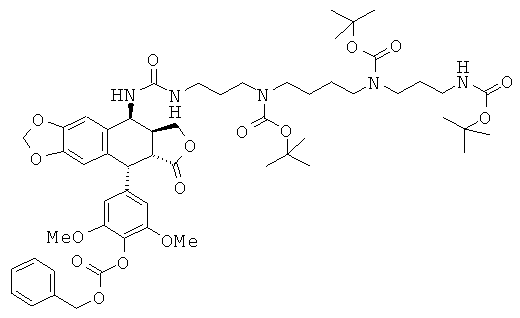
500 мг промежуточного соединения, полученного на Стадии 2 выше, растворяют с перемешиванием в 30 мл CH2Cl2 вместе с 0,13 мл триэтиламина. С перемешиванием при 0°С в атмосфере азота добавляют раствор 100 мг трифосгена в 20 мл CH2Cl2. После достижения комнатной температуры по каплям добавляют раствор 500 мг триВОС-спермина (Tet. 2000, 56, 2449) и 0,13 мл триэтиламина в 20 мл CH2Cl2. Реакционную смесь перемешивают при комнатной температуре в течение 3 ч. Затем реакционную смесь выливают в насыщенный раствор NaHCO3 и после этого экстрагируют, используя CH2Cl2. Органические фазы сушат над Na2SO4, фильтруют и упаривают. Остаток подвергают флэш-хроматографии на SiO2 (с элюированием от чистого CH2Cl2 до смеси 90:10 CH2Cl2:MeOH), получая 370 мг желтого масла. Выход 37%. TLC SiO2 (CH2Cl2:MeOH 90:10): Rf 0,65. 1H-ЯМР (DMSO) δ 7.4 (s, 5H, аром. Н), 6.81 (s, 1Н, H5), 6.53 (s, 1H, Н8), 6,35 (s, 2Н, Н2', Н6'), 5.98 (d, 2H, J=7 Гц, OCH2O), 5.23 (s, 2H, CH2Ar), 5.03 (dd, 1H, H4), 4.60 (d, 1H, J=5.2 Гц, H1), 4,32 (t, 1H, J=7.6 Гц, H11a), 3.81 (t, 1H, J=10 Гц, H11a), 3.63 (s, 6H, OMe), 2.87 (m, 14H, Н2, Н3, CH2N), 1.56 (m, 2H, СН2), 1.37(m, 33H, CH2, СН3).
Стадия 4: получение 1-{3-[4-(3-трет-бутоксикарбониламинопропил)-трет-бутоксикарбониламинобутил]-трет-бутоксикарбониламинопропил}-3-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро-[3',4':6,7]-нафто[2,3-d][1,3]-диоксол-5-ил]-мочевины
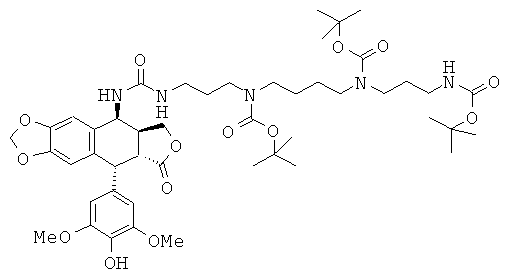
740 мг защищенного (триВОС- и 4'O-бензилоксикарбонильного) промежуточного соединения, полученного на Стадии 3 выше, растворяют с перемешиванием в 20 мл метанола с 100 мг 10%-ного палладия на угле и выдерживают в атмосфере водорода в течение 2 ч с энергичным перемешиванием. Раствор отфильтровывают от катализатора, промывают МеОН и затем упаривают досуха. Остаток подвергают флэш-хроматографии на SiO2 (градиент от CH2Cl2 до 90:10 CH2Cl2:MeOH), затем препаративной HPLC (X Bridge, OBD, C18, 10 мкм, 30×250 мм) с элюированием от 20.80 CH3CN:H2O до 100% CH3CN включительно. После экстракции фракций этилацетатом, сушки над Na2SO4, фильтрования и упаривания получают 630 мг 1-{3-[4-(3-трет-бутоксикарбониламинопропил)-трет-бутоксикарбониламино-бутил]-трет-бутоксикарбониламинопропил}-3-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро-[3',4':6,7]-нафто[2,3-d][1,3]диоксол-5-ил]-мочевины в виде бесцветного масла. Выход 97%. TLC SiO2 (90:10:1 CH2Cl2:MeOH:NH4OH): Rf=0,58.
Стадия 5: получение 1-{3-[4-(3-аминопропиламино)-бутиламино]-пропил}-3-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро-[3',4':6,7]-нафто[2,3-d][1,3]диоксол-5-ил]-мочевины
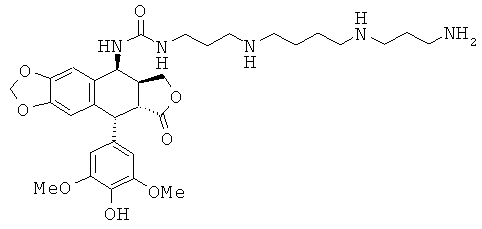
630 мг триВОС-защищенного промежуточного соединения, упомянутого выше, растворяют в 10 мл HCl в изопропаноле (3 М) и затем оставляют перемешиваться в течение 6 ч при комнатной температуре. Среду упаривают досуха и затем остаток переносят в этанол. Выпавший в осадок гидрохлорид отфильтровывают, промывают абсолютным этанолом и этиловым эфиром. Получают 391 мг соли, то есть выход составляет 78%. TLC SiO2 (40:40:20 CH2Cl2:MeOH:NH4OH): Rf=0,55. Т.пл.=166°C. Чистота по HPLC 97% (аналитическая HPLC X Bridge, 15:85 CH3CN:H2O; 6,8 г/л KH2PO4 - pH 4, RT=8,08). ESI-MS, m/z=628 (M-H+). Анал. C32H45N5O8·3HCl·4,4H2O=813,39, рассчитано: С% 52,14, Н% 6,56, N% 9,50; обнаружено: С% 51,89, Н% 5,95, N% 9,58. 1H-ЯМР (DMSO, D2O) δ 6.82 (s, 1H, H5), 6.52 (s, 1H, Н8), 6.24 (s, 2H, H2', H6'), 5.98 (d, 2H, J=10 Гц, ОСН2О), 5.01 (d, 1H, J=4 Гц, Н4), 4.51 (d, 1H, J=4.8 Гц, H1), 4,35 (t, 1H, J=8 Гц, H11a), 3.84-3,94 (m, Н11а, H2O), 3.63 (s, 6Н, ОМе), 3.17 (m, 3Н, Н2, CH2N), 2.95 (m, 11H, Н3, CH2N), 1.94 (m, 2H, СН2), 1.76 (m, 2H, CH2), 1.68 (m, 2H, CH2).
Пример 14: аналогично тому, как описано в Примере 13, соединения 39, 40, 41, 42, 43, 47 и 48 могут быть получены, следуя той же методике, которая описана на Стадии 3, но с использованием соответствующих защищенных диаминов, триаминов и тетрааминов. Стадии удаления защиты проводят, как описано на Стадии 4 в отношении гидрогенолиза группы Z или как описано на Стадии 5 в отношении отщепления групп ВОС.
Пример 15: соединения мочевины 44, 45 и 49 получают в соответствии с Примером 2 путем алкилирования, но используя защищенные триамины или тетраамины вместо диметиламина и производное 1-хлорэтил-3-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро-[3',4':6,7]-нафто[2,3-d][1,3]диоксол-5-ил]-мочевины (описанное в Heterocycles 1994, 39, 361) вместо промежуточного соединения III. Стадии удаления защиты проводят так же, как описано в Примере 13 на Стадии 4 и Стадии 5.
Пример 16: соединения мочевины 46, 47, 64, 65, 66, 67 и 68 получают в соответствии с методикой, описанной на Стадии 3 Примера 13, но используя 3-хлорпропилизоцианат, 4-хлорбутилизоцианат, 5-хлорпентилизоцианат (Bull. Soc. Chim. Fr. 1959, 611) вместо трифосгена, что приводит к получению соответствующих алкилмочевин. Последующие стадии проводят, как указано в Примере 15.
Пример 17: соединение 50: 1-[3-{3-[4-(3-аминопропиламино)-бутиламино]-пропиламино}-пропил]-3-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро-[3',4':6,7]-нафто[2,3-d][1,3]диоксол-5-ил]-мочевину также получают в соответствии с принципом процесса, изложенного в Примере 13, но используя соответствующий реагент.
Стадия 1: получение (3-трет-бутоксикарбониламинопропил)-[4-(трет-бутоксикарбонил-{3-[3-(1,3-диоксо-1,3-дигидроизоиндол-2-ил)пропиламино]-пропил}-амино)-бутил]-карбаминовой кислоты сложного трет-бутилового эфира
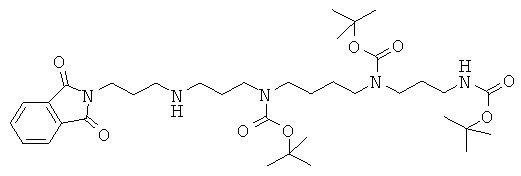
К раствору триВОС-спермина (Tet. 2000, 56, 2449) (6 г; 11,2 ммоль; 1 экв.) в 100 мл ацетонитрила добавляют N-(3-бромпропил)фталимид (3 г; 11,2 ммоль; 1 экв.) и карбонат цезия (7,2 г; 22,4 ммоль; 2 экв.). Среду кипятят с обратным холодильником с перемешиванием в течение 8 ч. После упаривания ее выливают в воду (400 мл) и экстрагируют AcOEt (3×200 мл). Органические фазы промывают насыщенным водным раствором NaCl, разделяют, сушат над Na2SO4 и упаривают. Остаток подвергают флэш-хроматографии на SiO2 и элюируют градиентом от чистого CH2Cl2 до смеси CH2Cl2:MeOH:NH4OH (80:18:2), получая после упаривания 2,31 г бесцветного масла (выход 30%). TLC SiO2 (90:9:1 CH2Cl2:MeOH:NH4OH): Rf=0,5. MS: m/z=690 (M-H+).
Стадия 2: получение [3-(трет-бутоксикарбонил-{4-[трет-бутоксикарбонил-(3-трет-бутоксикарбониламинопропил)-амино]-бутил}-амино)-пропил]-[3-(1,3-диоксо-1,3-дигидро-изоиндол-2-ил)-пропил]-карбаминовой кислоты сложного трет-бутилового эфира
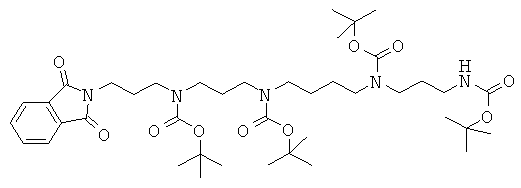
Соединение, полученное на приведенной выше Стадии 1 (2,31 г; 3,3 ммоль; 1 экв.), растворяют в 50 мл THF с перемешиванием. Затем по каплям при комнатной температуре добавляют раствор BOC2O (0,8 г; 3,7 ммоль; 1,1 экв.) в 10 мл THF. Перемешивание продолжают в течение 4 ч, затем среду выливают в воду, экстрагируют AcOEt (3×100 мл), сушат (Na2SO4), фильтруют и упаривают. Остаток подвергают флэш-хроматографии с градиентом от чистого гептана до чистого AcOEt. После упаривания получают 1,49 г (выход 56%).
Стадия 3: получение (3-аминопропил)-[3-(трет-бутоксикарбонил-{4-[трет-бутоксикарбонил-(3-трет-бутоксикарбонил-аминопропил)-амино]-бутил}-амино)-пропил]-карбаминовой кислоты сложного трет-бутилового эфира (аминопропил-тетраВОС-спермина)
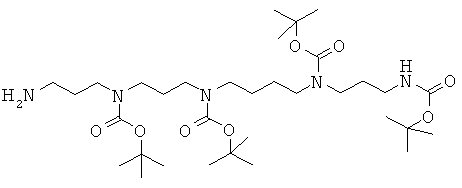
Соединение с приведенной выше Стадии 2 (1,49 г; 1,88 ммоль; 1 экв.) кипятят с обратным холодильником в 50 мл EtOH в присутствии гидразина гидрата (0,5 мл; 16,1 ммоль; 8,5 экв.) в течение 6 ч. Охлажденную среду фильтруют, промывают EtOH и упаривают. Остаток подвергают флэш-хроматографии на SiO2 (градиент элюирования от чистого CH2Cl2 до смеси 80:18:2 CH2Cl2:MeOH:NH4OH включительно). После упаривания фракций, содержащих чистое соединение, получают 1,09 г бесцветного масла (выход 88%). TLC SiO2 (90:9:1 CH2Cl2:MeOH:NH4OH): Rf=0,34. MS: m/z=660 (M-H+).
Стадия 4: сочетание аминопропил-тетраВОС-спермина с 4β-амино-4'-деметил-4'-бензилоксикарбонил-4-дезоксиподофиллотоксином
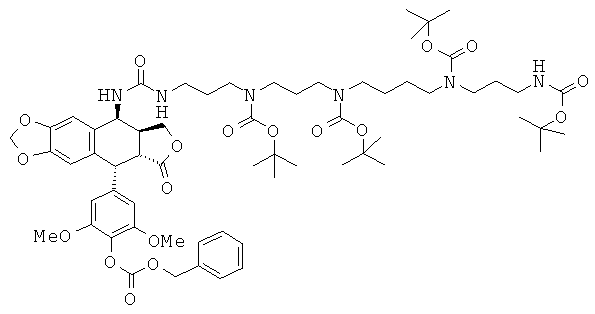
4β-Амино-4'-деметил-4'-бензилоксикарбонил-4-дезоксиподофиллотоксин, полученный на Стадии 2 Примера 13, (0,88 г; 1,6 ммоль; 1 экв.) растворяют с перемешиванием в 100 мл CH3CN вместе с 0,23 мл (1,6 ммоль; 1 экв.) триэтиламина и охлаждают до 0°С. Далее по каплям добавляют раствор трифосгена (0,17 г; 0,58 ммоль; 0,35 экв.). Затем после достижения комнатной температуры по каплям добавляют раствор смеси промежуточного соединения, аминопропил-тетраВОС-спермина, полученного на приведенной выше Стадии 3 (1,09 г; 1,6 ммоль; 1 экв.), и 0,23 мл (1,6 ммоль; 1 экв.) триэтиламина в 30 мл CH2Cl2. После перемешивания в течение 3 ч смесь выливают в раствор NaHCO3 и экстрагируют CH2Cl2 (3×100 мл). Органические фазы отделяют, сушат над Na2SO4, фильтруют и упаривают, получая остаток, который очищают флэш-хроматографией (градиент от чистого CH2Cl2 до 90:10 CH2Cl2:MeOH). После упаривания получают 1,37 г (68%) защищенного производного мочевины в виде белой пены. TLC SiO2 (95:5 CH2Cl2:MeOH): Rf=0,62. Аналитическая HPLC: колонка Xbridge C8, 5 мкм, 4,6×250 мм, элюирование 80:20 CH3CN:H2O, RRT=7,7 мин.
Стадия 5: удаление защиты в положении 4'
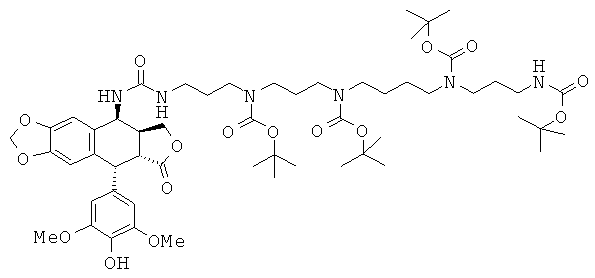
Гидрогенолиз производного, полученного на Стадии 4, (1,37 г), проводят, используя 50 мг 10%-ного палладия на угле в 100 мл МеОН с энергичным перемешиванием в течение 8 ч. Катализатор отфильтровывают и фильтрат упаривают досуха. Остаток сначала очищают флэш-хроматографией на SiO2 с элюированием градиентом от чистого гептана до чистого AcOEt, а затем HPLC (X bridged 8, OBD, 30×250 мм) с градиентом от CH3CN:H2O (50:50) до чистого CH3CN. Получают 0,95 г (выход=78%) соединения, у которого удалена защита в положении 4'. TLC SiO2 (95:5 CH2Cl2:MeOH): Rf=0,33. Аналитическая HPLC: колонка Xbridge C8, 5 мкм, 4,6×250 мм, элюирование 80:20 CH3CN:H2O, RRT=4,7 мин.
Стадия 6: получение 1-[3-{3-[4-(3-аминопропиламино)-бутиламино]-пропиламино}-пропил]-3-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро-[3',4':6,7]-нафто[2,3-d][1,3]диоксол-5-ил]-мочевины посредством удаления защитных групп ВОС

Соединение, полученное на приведенной выше Стадии 5 (0,95 г; 0,72 ммоль), растворяют в 10 мл CH2Cl2 в присутствии HCl в изопропаноле (3 М, 10 мл) с перемешиванием в течение 4 ч. Полученный осадок отфильтровывают и затем промывают Et2O, получая 0,6 г (94%) белого порошка 1-[3-{3-[4-(3-аминопропиламино)-бутиламино]-пропиламино}-пропил]-3-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро-[3',4':6,7]-нафто[2,3-d][1,3]диоксол-5-ил]-мочевины. Т.пл.=213°С. MS: m/z=685 (М-Н+).
3. Фармакологический тест
Пример 18: фармакологические тесты in vitro
Используют тест на цитотоксичность. В нем измеряют ингибирование клеточного роста на линии А549 человека (немелкоклеточный рак легкого).
Опухолевые клетки А549 высевают в 96-луночный планшет в среде RPMI 1640, не содержащей фенолового красного (Seromed), в которую добавляют 5% фетальной телячьей сыворотки (100 мкл/лунка; 1,25×104 клеток/мл). После инкубации в течение 24 ч при 37°С в инкубаторе при 5% CO2 среду заменяют на среду, содержащую тестируемое соединение, затем планшеты инкубируют в течение следующих 48 ч. Выживаемость клеток оценивают, измеряя люминесценцию после удаления осаждением АТФ из среды с использованием раствора для лизиса клеток, раствора люциферазы и раствора люциферина, входящих в состав набора ATP-lite-M™, следуя рекомендациям производителя (Packard, Rungis, France). Каждое экспериментальное условие тестировали по меньшей мере три раза в шести повторах.
Результаты показывают, что соединения по изобретению обладают сильными цитотоксическими свойствами. Ингибирующая концентрация 50 (IC50), представляющая собой концентрацию тестируемого соединения, обеспечивающую ингибирование клеточной пролиферации на 50%, составляет, например, для соединения 21: IC50=1,7×10-9 М или для соединения 48: IC50=1,2×10-8 М.
Пример 19: фармакологические тесты in vivo
Экспериментальная модель опухоли Р388. Используемая модель представляет собой модель мышиного лейкоза Р388 (Tumor Models in Cancer Research. Teicher, B.A. ed., Humana Press Inc., Totowa, NJ. pp.23-40, 2002), которую поддерживают посредством последовательных интраперитонеальных трансплантаций на мышах DBA/2 (мышах DBA/2JIco, Charles River), как изложено ранее (Classic in vivo cancer models: Three examples of mouse models used in experimental therapeutics. Current Protocols in Pharmacology Unit 5.24: 5.24.1-5.24.16, 2001).
Эксперимент проводят в соответствии с изложенным ранее протоколом (Cancer Chemother. Pharmacol. 1998, 41, 437-447). Он включает в себя имплантирование по 106 клеток лейкоза Р388 на одну мышь гибридным мышам C2DF1 (CD2F1/CrlIBR, Charles River, St Aubin-les-Elbeuf, France) внутривенно на нулевые сутки. После этого животных случайным образом распределяют в клетки для лечения и контрольные клетки, тестируемые соединения вводят в виде единичной инъекции интраперитонеальным путем на следующие сутки после трансплантации опухоли, на 1-е сутки. Затем каждые сутки осуществляют мониторинг животных, взвешивают два раза в неделю и регистрируют некоторые клинические реакции. Коэффициент выживаемости (survival rate) представляет собой параметр, используемый для оценки противоопухолевой активности. Увеличение коэффициента выживаемости определяют как отношение Т/Csurvival (%), которое соответствует: (среднее значение коэффициента выживаемости для подвергаемой лечению группы/среднее значение коэффициента выживаемости для контрольной группы)×100. Отношение Т/Csurvival рассчитывают для каждой дозы, и наибольшее полученное значение представляет собой достигнутое максимальное увеличение коэффициента выживаемости (максимальную активность), которое по определению называется оптимальным отношением T/Csurvival.
Результаты показывают, что данные соединения вызывают значительное увеличение коэффициента выживаемости для животных с лейкозом Р388.
Например, соединение 21 из Примера 11 демонстрирует величину оптимального Т/Csurvival, равную 186%, в дозе 0,16 мг/кг, что указывает на то, что лечение этим соединением приводит к увеличению коэффициента выживаемости животных на 86%. Фактически в соответствии с критериями NCI (Национального института рака) величина Т/Csurvival считается существенной, если она превышает по меньшей мере 120% (Semin. Oncol. 1981, 8, 349-361).
Относительное уменьшение массы тела животных наряду с оптимальной активностью соединений лежит гораздо ниже порога токсичности в соответствии с критериями NCI (Ann. Oncol. 1994, 5, 415-422).
СОКРАЩЕНИЯ
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ СИНТЕЗА ПРОТИВОРАКОВЫХ ПРОИЗВОДНЫХ (ПОЛИ)АМИНОАЛКИЛАМИНОАЦЕТАМИДА ЭПИПОДОФИЛЛОТОКСИНА | 2008 |
|
RU2450009C2 |
| ПРОИЗВОДНЫЕ ХРОМОНОВ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ТЕРАПЕВТИЧЕСКИЕ ПРИМЕНЕНИЯ | 2010 |
|
RU2545214C2 |
| АГОНИСТ ГЛЮКОКОРТИКОИДНОГО РЕЦЕПТОРА И ЕГО ИММУНОКОНЪЮГАТЫ | 2017 |
|
RU2745748C2 |
| ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛА В КАЧЕСТВЕ МОДУЛЯТОРОВ ROR-ГАММА | 2017 |
|
RU2760366C2 |
| ЗАМЕЩЕННЫЕ ТРИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И СПОСОБ ПРИМЕНЕНИЯ | 2016 |
|
RU2744766C2 |
| 6,7-ДИГИДРО-5H-ПИРАЗОЛО[1,2-a]ПИРАЗОЛ-1-ОНЫ (ВАРИАНТЫ), ИНГИБИРУЮЩИЕ ВЫСВОБОЖДЕНИЕ ВОСПАЛИТЕЛЬНЫХ ЦИТОКИНОВ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ИНГИБИРОВАНИЯ | 2002 |
|
RU2299885C2 |
| ПРОИЗВОДНЫЕ 2-АМИНО-1,2,3,4-ТЕТРАГИДРОНАФТАЛИНА, ОБЛАДАЮЩИЕ АКТИВНОСТЬЮ В ОТНОШЕНИИ СЕРДЕЧНО-СОСУДИСТОЙ СИСТЕМЫ, И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1995 |
|
RU2149158C1 |
| ЗАМЕЩЕННЫЕ ПУРИНОВЫЕ И 7-ДЕАЗАПУРИНОВЫЕ СОЕДИНЕНИЯ | 2011 |
|
RU2606514C2 |
| МОДУЛЯТОРЫ ТРАНСПОРТЕРОВ АТФ-СВЯЗЫВАЮЩЕЙ КАССЕТЫ | 2005 |
|
RU2528046C2 |
| ЗАМЕЩЕННЫЕ ГЕТЕРОАРИЛОМ ПИРИДИНЫ И СПОСОБЫ ПРИМЕНЕНИЯ | 2017 |
|
RU2756743C2 |
Настоящее изобретение относится к новым производным эпиподосриллотоксина, замещенного по положению 4 возможно замещенной цепью (поли)аминоалкиламиноалкиламида, или алкил-мочевины, или алкил-сульфонамида, формулы 1,
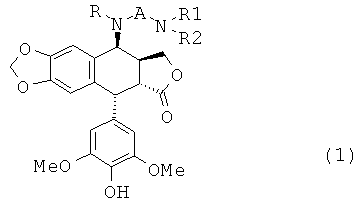
где R представляет собой водород или C1-4алкил,
А представляет собой CO(СН2)n или CONH(CH2)n, где n равно 2, 3, 4 или 5,
R1 представляет собой H или C1-4алкил,
R2 представляет собой (CH2)m-NR3R4, где m равно 2, 3, 4 или 5,
R3 представляет собой H или C1-4алкил,
R4 представляет собой H, C1-4алкил или (CH2)p-NR5R6, где p равно 2, 3, 4 или 5,
R5 представляет собой H или C1-4алкил и
R6 представляет собой H, C1-C4алкил или (CH2)q-NH2, где q равно 2, 3, 4 или 5 или их фармацевтически приемлемым солям, а также к способам их получения и к их применению в качестве противоракового средства. 6 н. и 5 з.п. ф-лы., 19 пр.
Соединение общей формулы 1:
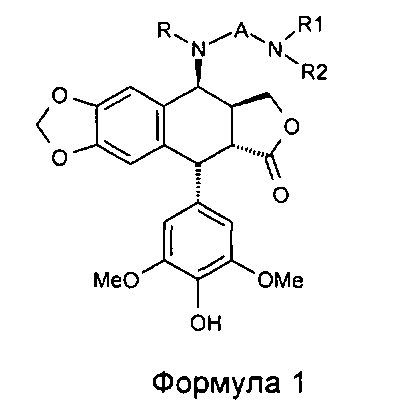
где:
- R представляет собой водород или C1-4алкил,
- А представляет собой CO(СН2)n или CONH(CH2)n, где n равно 2, 3, 4 или 5,
- R1 представляет собой H или C1-4алкил,
- R2 представляет собой (CH2)m-NR3R4, где m равно 2, 3, 4 или 5,
- R3 представляет собой H или C1-4алкил,
- R4 представляет собой H, C1-4алкил или (CH2)p-NR5R6, где p равно 2, 3, 4 или 5,
- R5 представляет собой H или C1-4алкил, и
- R6 представляет собой H, C1-C4алкил или (CH2)q-NH2, где q равно 2, 3, 4 или 5,
или его фармацевтически приемлемая соль.
2. Соединение общей формулы 1 по п.1, в котором R представляет собой H.
3. Соединение общей формулы 1 по п.1, в котором m равно 3 или 4, p равно 3 или 4, и q равно 3.
4. Соединение общей формулы 1 по п.1, где оно выбрано из следующих соединений:
соединения 3: 3-[(2-диметиламиноэтил)-метиламино]-N-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро-[3′,4′:6,7]-нафто[2,3-d][1,3]диоксол-5-ил]-пропионамида,
соединения 21: 4-{3-[4-(3-аминопропиламино)-бутиламино]-пропиламино}-N-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро-[3′,4′:6,7]-нафто[2,3-d][1,3]диоксол-5-ил]-бутирамида,
соединения 48: 1-{3-[4-(3-аминопропиламино)-бутиламино]-пропил}-3-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро-[3′,4′:6,7]-нафто[2,3-d][1,3]диоксол-5-ил]-мочевины,
соединения 50: 1-[3-{3-[4-(3-аминопропиламино)-бутиламино]-пропиламино}-пропил]-3-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро-[3′,4′:6,7]-нафто-[2,3-d][1,3]диоксол-5-ил]-мочевины,
соединения 55: 5-[(2-диметиламиноэтил)-метиламино]пентановой кислоты N-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро-[3′,4′:6,7]-нафто[2,3-d][1,3]диоксол-5-ил]-амида.
5. Соединение формулы 1 по любому из пп.1-4 для применения в качестве лекарственного средства для лечения рака.
6. Соединение формулы 1 по п.5 для применения в качестве лекарственного средства, предназначенного для противораковой терапии несолидных (″жидких″) опухолей и солидных опухолей, таких как меланомы, колоректальный рак, рак легких, рак предстательной железы, рак мочевого пузыря, рак молочной железы, рак матки, рак пищевода, рак желудка, рак поджелудочной железы, рак печени, рак яичников, лейкозы, в частности лимфомы и миеломы, рак уха-горла-носа и рак головного мозга.
7. Применение соединения формулы 1 по любому из пп.1-4 для изготовления лекарственного средства, предназначенного для противораковой терапии несолидных (″жидких″) опухолей и солидных опухолей, таких как меланомы, колоректальный рак, рак легких, рак предстательной железы, рак мочевого пузыря, рак молочной железы, рак матки, рак пищевода, рак желудка, рак поджелудочной железы, рак печени, рак яичников, лейкозы, в частности лимфомы и миеломы, рак уха-горла-носа и рак головного мозга.
8. Способ получения соединения формулы 1 по любому из пп.1-4, где А представляет собой СО(СН2)n и R представляет собой H, где n такой, как определено в п.1, включающий нижеперечисленные последовательные стадии:
(a) проведение реакции Риттера между соединением следующей ниже формулы 3:
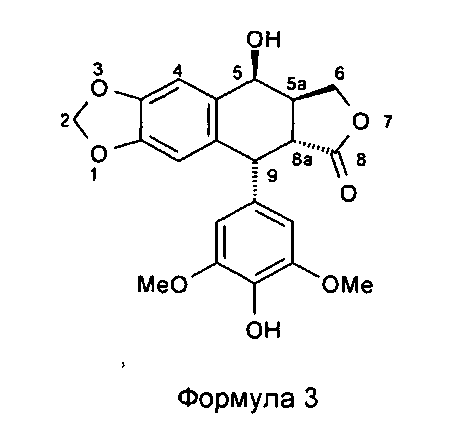
и нитрилом формулы Ra-CN, где Ra представляет собой -(СН2)n-Х или -СН=CH2, n представляет собой 3, 4 или 5 и X представляет собой атом галогена, такой как атом хлора, с получением соединения следующей ниже формулы 4:

(b) проведение реакции алкилирования между амином в защищенной форме формулы HNR1R2a, где:
- R1 такой, как определено в п.1,
- R2a представляет собой (CH2)m-NR3aR4a, где m такой, как определено в п.1,
- R3a представляет собой C1-4алкил или защитную группу для амина,
- R4a представляет собой C1-4алкил, защитную группу для амина или (СН2)p-NR5aR6a, где p такой, как определено в п.1,
- R5a представляет собой C1-4алкил или защитную группу для амина,
- R6a представляет собой C1-4алкил, защитную группу для амина или (CH2)q-NR7aR8a, где q такой, как определено в п.1,
- R7a представляет собой H или защитную группу для амина и
- R8a представляет собой защитную группу для амина,
и соединением формулы 4, полученным на предыдущей стадии, с получением соединения следующей ниже формулы 5:

где R2a такой, как определено выше, и R1 и n такие, как определено в п.1,
(c) возможно удаление защиты с функциональных аминогрупп, защищенных защитными группами для амина, с получением соединения следующей ниже формулы 5а:
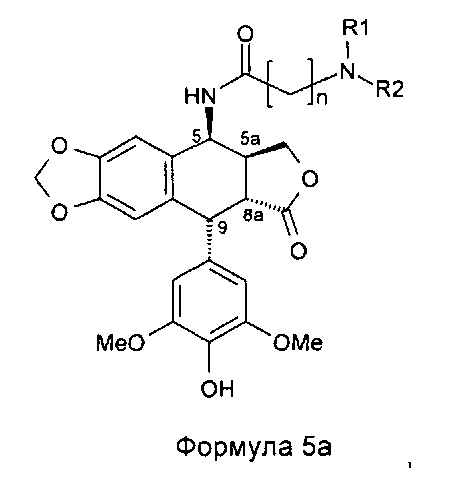
где R1, R2 и n такие, как определено в п.1, и
(d) выделение соединения, полученного на предыдущей стадии, из реакционной смеси.
9. Способ получения соединения формулы 1 по любому из пп.1-4, где А представляет собой CONH(CH2)n, где n такой, как определено в п.1, включающий нижеперечисленные последовательные стадии:
(a) взаимодействие соединения следующей ниже формулы 8:
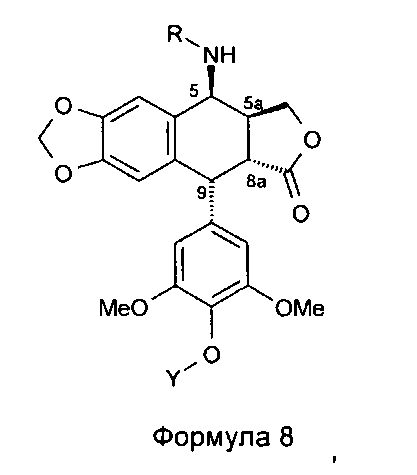
где R такой, как определено в п.1, и Y представляет собой защитную группу для гидроксила, такую как бензилоксикарбонил,
с изоцианатом формулы O=C=N-(CH2)n-X, где n такой, как определено в п.1, и X представляет собой атом галогена, такой как атом хлора, с получением соединения следующей ниже формулы 9:
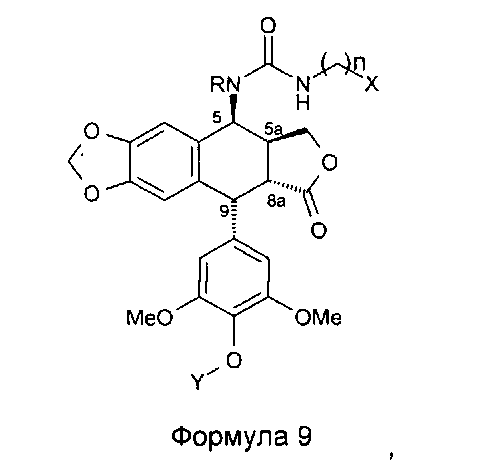
(b) проведение реакции алкилирования между амином в защищенной форме формулы HNR1R2a, где
- R1 такой, как определено в п.1,
- R2a представляет собой C1-4алкил, защитную группу для амина или (СН2)m-NR3aR4a, где m такой, как определено в п.1,
- R3a представляет собой C1-4алкил или защитную группу для амина,
- R4a представляет собой C1-4алкил, защитную группу для амина или (СН2)p-NR5aR6a, где p такой, как определено в п.1,
- R5a представляет собой C1-4алкил или защитную группу для амина,
- R6a представляет собой C1-4алкил, защитную группу для амина или (CH2)q-NR7aR8a, где q такой, как определено в п.1,
- R7a представляет собой H или защитную группу для амина и
- R8a представляет собой защитную группу для амина,
и соединением формулы 9, полученным на предыдущей стадии, с получением соединения следующей ниже формулы 10а:
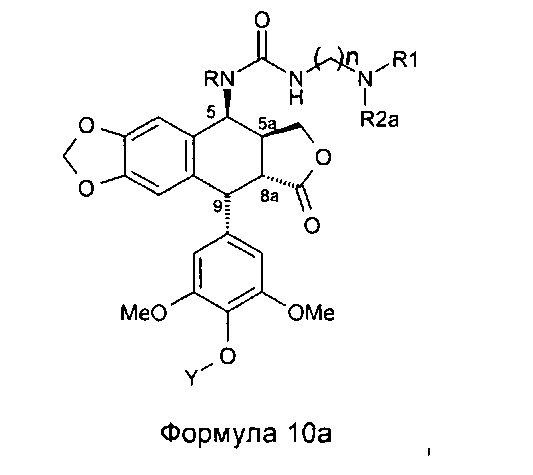
где Y и R2a такие, как определено выше, и R1 и n такие, как определено в п.1,
(c) удаление защиты с функциональной фенольной группы и возможно удаление защиты с функциональных аминогрупп, защищенных защитными группами для амина в соединении формулы 10а, полученном на предыдущей стадии, с получением соединения следующей ниже формулы 10b:
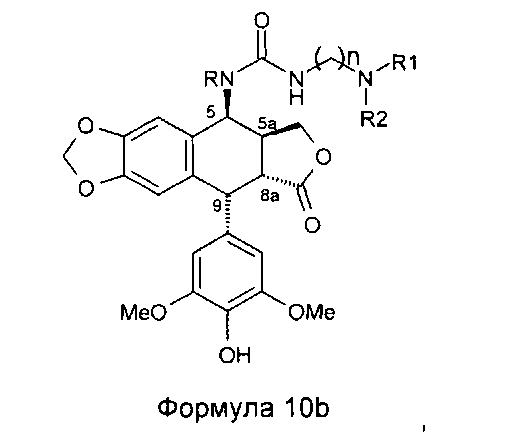
где R, R1, R2 и n такие, как определено в п.1, и
(d) выделение соединения, полученного на предыдущей стадии, из реакционной смеси.
10. Способ получения соединения формулы 1 по любому из пп.1-4, где А представляет собой CONH(CH2)n, где n такой, как определено в п.1, включающий нижеперечисленные последовательные стадии:
(a) взаимодействие соединения формулы 8,
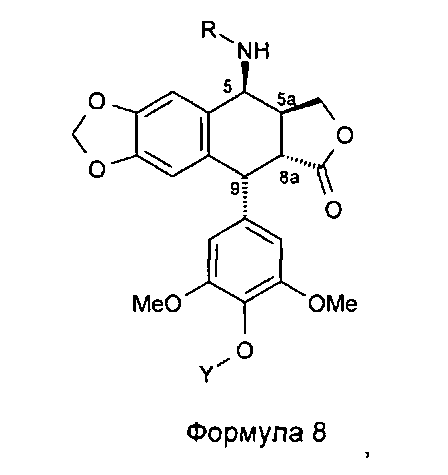
где R такой, как определено в п.1, и Y представляет собой защитную группу для гидроксила, такую как бензилоксикарбонил,
с фосгеном или трифосгеном с получением промежуточного активированного карбонилированного соединения,
(b) проведение реакции алкилирования между амином в защищенной форме формулы H2N-(CH2)n-NR1aR2a, где
- R1a представляет собой C1-4алкил или защитную группу для амина и
- R2a представляет собой C1-4алкил, защитную группу для амина или (СН2)m-NR3aR4a, где m такой, как определено в п.1,
- R3a представляет собой C1-4алкил или защитную группу для амина,
- R4a представляет собой C1-4алкил, защитную группу для амина или (СН2)p-NR5aR6a, где p такой, как определено в п.1,
- R5a представляет собой C1-4алкил или защитную группу для амина,
- R6a представляет собой C1-4алкил, защитную группу для амина или (CH2)q-NR7aR8a, где q такой, как определено в п.1,
- R7a представляет собой H или защитную группу для амина и
- R8a представляет собой защитную группу для амина,
и промежуточным активированным карбонилированным соединением, полученным на предыдущей стадии, с получением соединения следующей ниже формулы 10с:
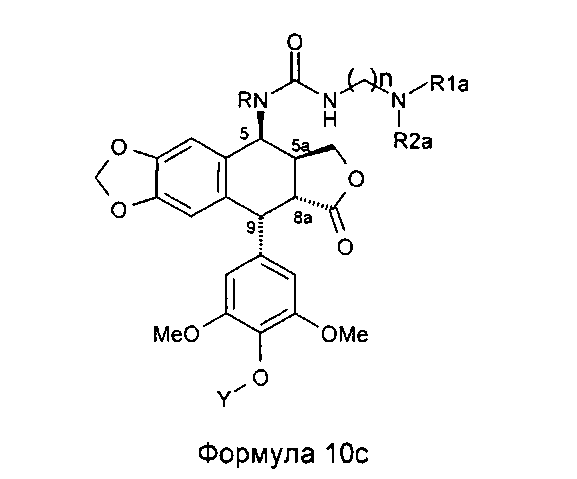
где R и n такие, как определено в п.1, и Y, R1a и R2a такие, как определено выше,
(c) удаление защиты с функциональной фенольной группы и возможно удаление защиты с функциональных аминогрупп, защищенных защитными группами для амина в соединении формулы 10с, полученном на предыдущей стадии, с получением соединения формулы 10b,
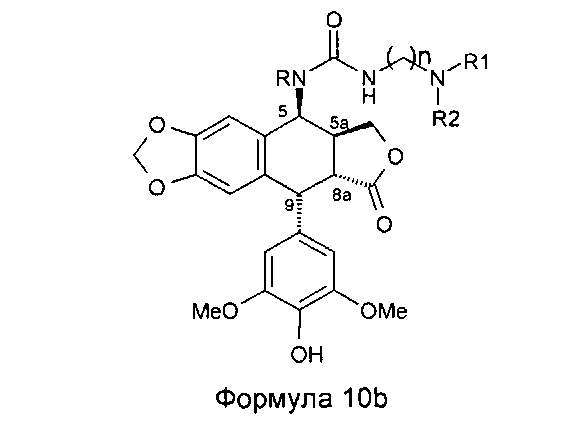
где R, R1, R2 и n такие, как определено в п.1, и
(d) выделение соединения, полученного на предыдущей стадии, из реакционной смеси.
11. Способ получения соединения формулы 1 по любому из п.п.1-4, где А представляет собой СО(CH2)n, где n такой, как определено в п.1, включающий нижеперечисленные последовательные стадии:
(a) проведение пептидного сочетания между соединением следующей ниже формулы 6:
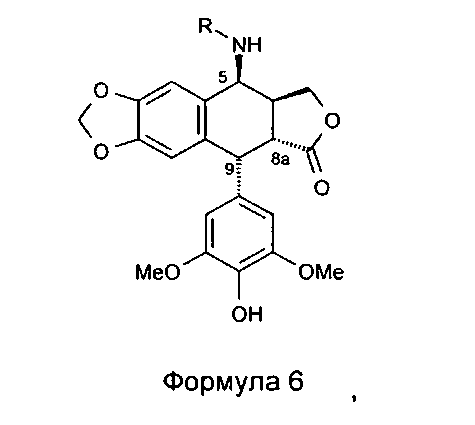
где R такой, как определено в п.1,
и кислотой следующей ниже формулы 12:
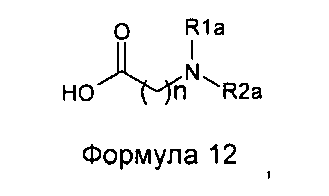
где
- R1a представляет собой C1-4алкил или защитную группу для амина и
- R2a представляет собой C1-4алкил, защитную группу для амина или (CH2)m-NR3aR4a, где m такой, как определено в п.1,
- R3a представляет собой C1-4алкил или защитную группу для амина,
- R4a представляет собой C1-4алкил, защитную группу для амина или (СН2)p-NR5aR6a, где p такой, как определено в п.1,
- R5a представляет собой C1-4алкил или защитную группу для амина,
- R6a представляет собой C1-4алкил, защитную группу для амина или (CH2)q-NR7aR8a, где q такой, как определено в п.1,
- R7a представляет собой Н или защитную группу для амина и
- R8a представляет собой защитную группу для амина,
и n такой, как определено в п.1,
с получением соединения следующей ниже формулы 7b:
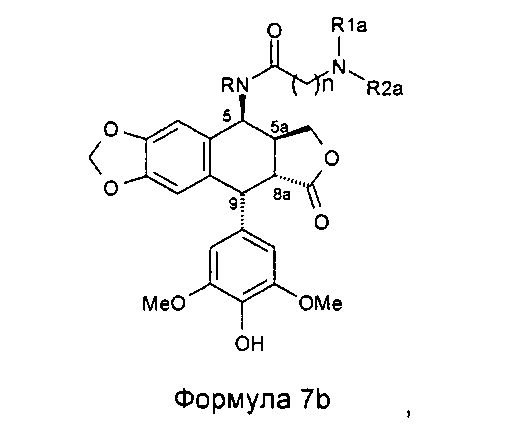
где R1a и R2a такие, как определено выше, и R и n такие, как определено в п.1,
(b) возможно удаление защиты с функциональных аминогрупп, защищенных защитными группами для амина, с получением соединения следующей ниже формулы 7:
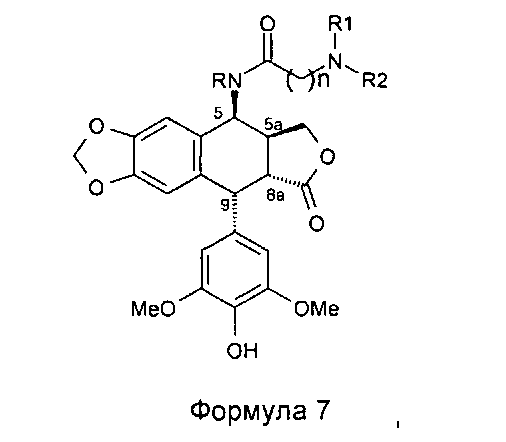
где R, R1, R2 и n такие, как определено в п.1, и
(c) выделение соединения, полученного на предыдущей стадии, из реакционной смеси.
| WO 2005100363 A1 27.10.2007 |

