Целью настоящего изобретения является новый способ получения производных (поли)аминоалкиламиноацетамида эпиподофиллотоксина формулы 1 и их фармацевтически приемлемых солей.
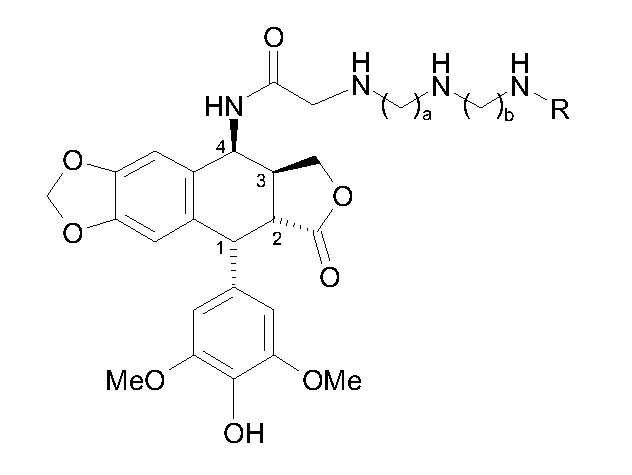
Формула 1
в которой R представляет собой атом водорода или группу -(СН2)с-NH2 при 2≤a,b,c≤5.
Данные соединения состоят из линейного фрагмента типа эпиподофиллотоксина и полиаминного фрагмента, связанного с 4 положением эпиподофиллотоксина через ацетамидное звено.
Наличие полиаминной цепи придает свойства водорастворимости молекуле, в частности ее хлоргидратам, а также фармакологические свойства, особенно интересные в отношении лечения рака.
Эти соединения, описанные в заявке WO 2005/100363, представляют собой противораковые соединения, особенно полезные для лечения солидных опухолей, или не солидных опухолей, таких, как меланомы, рак кишечника, предстательной железы, мочевого пузыря, груди, матки, желудка, поджелудочной железы, печени, яичников, а также для лечения лейкемии, лимфом и миелом, рака области ОЛЛ (ORL) и опухолей мозга.
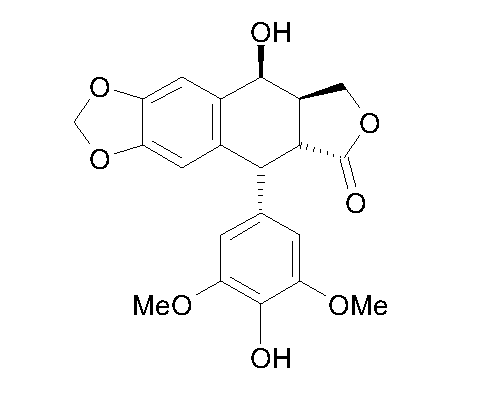
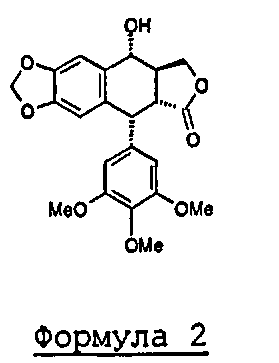
В способе синтеза, описанном в WO 2005/100363, для получения соединений формулы 1 в качестве исходного соединения используют подофиллотоксин формулы 2:
Формула 2
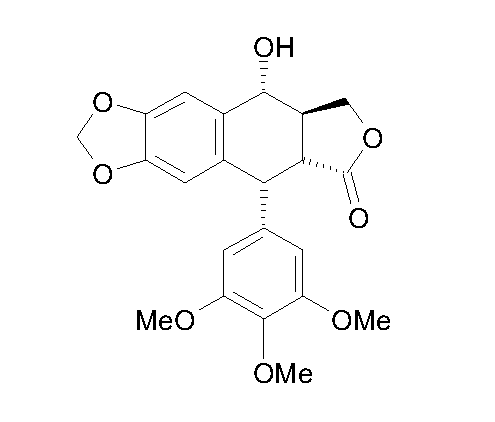
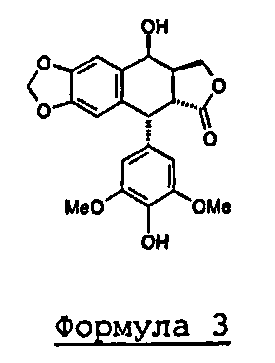
затем 4'-деметилэпиподофиллотоксин формулы 3:
Формула 3
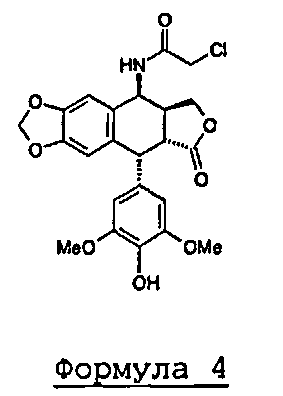
на который действуют хлорацетонитрилом в кислой среде, получая промежуточный продукт синтеза 4-хлорацетамидо-4'-деметилэпиподофиллотоксин формулы 4:
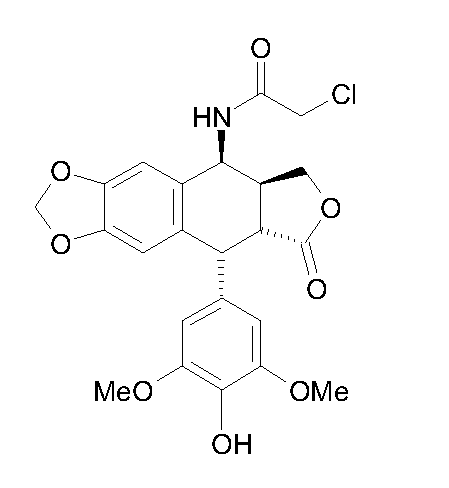
Формула 4
После этого данное соединение конденсируют с реагентом первичного амина формулы 5:
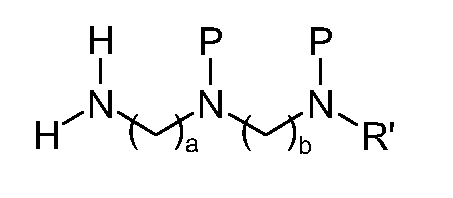
Формула 5
в которой R' представляет собой водород, или цепь -(СН2)с-NHP, и в которой Р представляет собой защитную группу для функциональных аминных групп.
Подходящие защитные группы могут представлять собой радикал: бензильный, бензилоксикарбонильный или третбутоксикарбонильный. Данную конденсацию проводят в смеси растворителей, включающей в себя полярный апротонный растворитель (ацетонитрил, ДМФА) в присутствии основания Льюиса (триэтиламин).
Однако данный способ, помимо того факта, что он включает в себя повышенное количество стадий и, следовательно, довольно невысокий общий выход, представляет два неудобства.
С одной стороны, условия, использованные в патенте WO 2005/100363, благоприятствуют эпимеризации атома углерода во 2 положении производного эпиподофиллотоксина формулы 1, приводя к форме цис-лактона, названной «пикро», формулы 7:
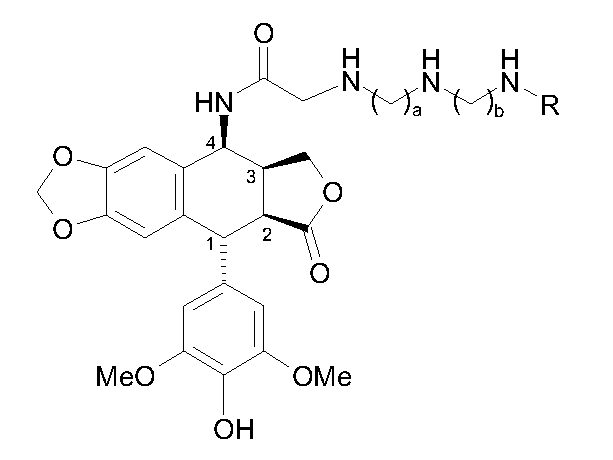
Формула 7
Очистка же целевого продукта транс-лактона сложна и требует трудоемких и дорогостоящих хроматографических операций.
С другой стороны, в описанном выше методе образуются также побочные продукты типа бисалкилирования за счет взаимодействия другой молекулы 4β-хлорацетамидо-4'-деметилэпиподофиллотоксина с уже образовавшимся продуктом формулы 1. В этом случае требуется использование избытка реагента первичного амина формулы 5, чтобы наиболее полное превращение исходных соединений свело к минимуму образование побочных продуктов, для чего необходима сложная стадия выделения избытка амина, делающая данный способ малоэкономичным.
Неожиданным образом, заявитель установил, что при использовании реагента первичного амина, представленного формулой 6, в которой a, b и R имеют те же значения, что и раньше, но в которой функциональные аминные группы не защищены, потенциальные побочные продукты сведены к минимуму и их присутствие не представляет большого препятствия для получения чистого конечного соединения.
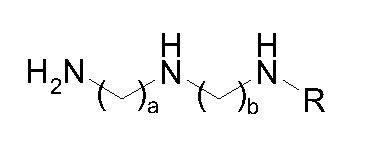
Формула 6
В таком случае, непосредственная конденсация 4-хлорацетамидо-4'-деметилэпиподофиллотоксина формулы 4 с реагентом первичного амина, без дополнительной стадии защиты функциональных аминных групп последнего, происходит в условиях, удовлетворительных в отношении выхода и чистоты полученного продукта. Таким образом, в случае соединения, для которого a=3, b=4, c=3, при использовании методики, такой как описанная в WO 2005/100363, проводят синтез, включающий в себя 11 стадий, с общим выходом приблизительно 15%; при этом синтез, который является целью настоящего изобретения, позволяет получать общий выход 30% лишь за 3 стадии.
В рамках целевого синтеза настоящего изобретения использовали стехиометрические количества реагентов, что минимизирует затраты.
Таким образом, доминирующий продукт реакции представляет собой продукт алкилирования, составляющий соединение с линейной цепью за счет замещения первичного амина преобладающим образом. Подобная избирательность реакционноспособности является неожиданной с учетом побочных продуктов, полученных при использовании способа синтеза, описанного в WO 2005/100363. Использование незащищенного амина приводит, главным образом, к искомому продукту.
В то же время данный способ представляет преимущество в отношении снижения количества проведенных стадий, поскольку стадии защиты реагента первичного амина более не являются необходимыми.
Таким образом, настоящее изобретение относится к способу синтеза соединений формулы 1, включающему в себя стадию конденсации промежуточного соединения 4β-хлорацетамидо-4'-деметилэпиподофиллотоксина формулы 4 с реагентом первичного амина формулы 6, без предварительной защиты функциональных аминных групп.
Предпочтительным образом, соединения формулы 1 получают в виде хлоргидрата.
Данная реакция конденсации протекает непосредственным образом, так что для аминной группы используемого реагента первичного амина формулы 6 не требуется какая бы то ни было защита при помощи подходящей защитной группы.
В предпочтительном варианте осуществления стадию конденсации 4β-хлорацетамидо-4'-деметилэпиподофиллотоксина формулы 4 с реагентом незащищенного первичного амина формулы 6 проводят в полярном апротонном растворителе.
В предпочтительном варианте осуществления используемый на стадии конденсации полярный апротонный растворитель выбирают из числа диметилформамида, диметилацетамида, N-метилпирролидона или еще диметилсульфоксида.
Также в предпочтительном варианте осуществления реакцию конденсации осуществляют в интервале температуры от -20°С до 30°С. При проведении процесса в количестве нескольких десятков грамм наблюдается разогревание, и, следовательно, предпочтителен контроль температуры реакции. В еще более предпочтительном варианте осуществления температуру поддерживают при 0°С.
Изобретение относится также к способу синтеза соединений формулы 1, в которых за стадией конденсации промежуточного соединения 4β-хлорацетамидо-4'-деметилэпиподофиллотоксина формулы 4 с реагентом первичного амина формулы 6 следует стадия выделения соединения 1.
В предпочтительном варианте осуществления стадию выделения соединения формулы 1 проводят путем осаждения в спиртовом растворителе, таком как метанол или этанол, с последующей стадий хроматографирования с обращенной фазой в кислой среде. Соединение очищают в растворе кислоты, такой как соляная кислота. Оно не подвергается опасности эпимеризации на стадии лактона, приводящего к «пикро» производному. Конечная лиофилизация позволяет выделять соль требуемого соединения.
В еще одном предпочтительном варианте осуществления в качестве реагента незащищенного первичного амина формулы 6 используют спермин или спермидин приведенных ниже формул 8 и 9.
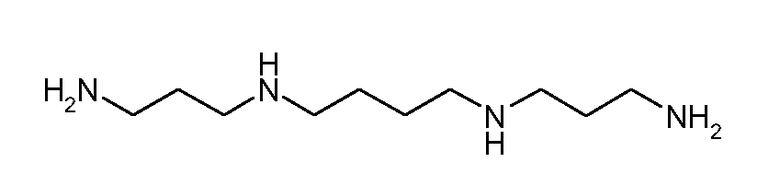
Формула 8
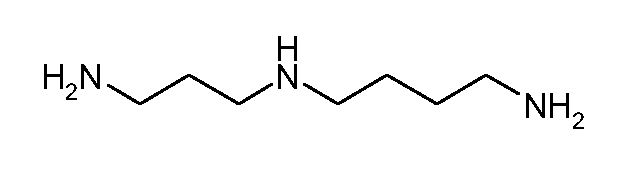
Формула 9
В случае конденсации со спермидином, несимметричным полиамином, получают 2 изомерных соединения формулы 1 в равных количествах (соединения, в которых a=3, b=4, R=H и a=4, b=3, R=H).
В случае конденсации со спермином получают соединение, в котором a=3, b=4, а R=(CH2)3-NH2.
Кроме того, настоящее изобретение относится к способу синтеза соединения, в котором a=3, b=4, R=H, а именно 2-[3-(4-аминобутиламино)пропиламино]-N-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро[3',4':6,7]нафто[2,3-d][1,3]диоксол-5-ил]ацетамида, включающему в себя стадию конденсации 4β-хлорацетамидо-4'-деметилэпиподофиллотоксина формулы 4 со спермидином с последующей стадией выделения данного соединения.
Кроме того, настоящее изобретение относится к способу синтеза соединения, в котором a=4, b=3, R=H, а именно 2-[4-(3-аминопропиламино)бутиламино]-N-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро[3',4':6,7]нафто[2,3-d][1,3]диоксол-5-ил]ацетамида, включающему в себя стадию конденсации 4β-хлорацетамидо-4'-деметилэпиподофиллотоксина формулы 4 со спермидином с последующей стадией выделения данного соединения.
Кроме того, настоящее изобретение относится к способу синтеза соединения, в котором a=3, b=4, а R=(CH2)3-NH2, а именно 2-{3-[4-(3-аминопропиламино)бутиламино]пропиламино}-N-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро[3',4':6,7]нафто[2,3-d][1,3]диоксол-5-ил]ацетамида, включающему в себя стадию конденсации 4β-хлорацетамидо-4'-деметилэпиподофиллотоксина формулы 4 со спермином с последующей стадией выделения данного соединения.
Кроме того, настоящее изобретение относится к использованию реагента первичного амина формулы 6 для получения общей формулы 1 в соответствии с методикой, в которой вводят стадию конденсации реагента первичного амина с 4β-хлорацетамидо-4'-деметилэпиподофиллотоксином формулы 4.
Настоящее изобретение относится также к способу получения соединений формулы 1 исходя из подофиллотоксина формулы 2, который включает в себя следующие стадии и характеризуется тем фактом, что используемый на стадии с) реагент первичного амина представляет собой реагент формулы 6. Конденсация стадии с) происходит непосредственным образом без стадии защиты функциональных аминных групп используемого реагента первичного амина при помощи какой-либо защитной группы.
а) Получение 4'-деметилэпиподофиллотоксина формулы 3 исходя из подофиллотоксина формулы 2.
b) Превращение 4'-деметилэпиподофиллотоксина в 4β-хлорацетамидо-4'-деметилэпиподофиллотоксин формулы 4.
с) Конденсация 4β-хлорацетамидо-4'-деметилэпиподофиллотоксина с реагентом первичного амина формулы 6.
Стадию а) осуществляют предпочтительным образом по методике, описанной в заявке патента WO 97/21713, то есть путем обработки подофиллотоксина парой сильная кислота - алифатический, ароматический или функционализированный сульфид в присутствии органической или минеральной кислоты, или в присутствии воды при наличии, или в отсутствие органического растворителя, смешивающегося с водой.
Стадию b) осуществляют предпочтительным образом в соответствии с методикой, описанной в заявке WO 2005/100363, то есть по реакции 4'-деметилэпиподофиллотоксина, полученного на предшествующей стадии, с хлорацетонитрилом в кислой среде.
Так же, как и последующую стадию выделения, стадию с) осуществляют, как описано ранее, при этом используемый реагент первичного амина не содержит никакой защиты для своих функциональных аминных групп.
В случае необходимости, после стадии выделения из соединения общей формулы 1 можно получить соль действием минеральной или органической кислоты.
Предпочтительный вариант осуществления стадии конденсации с) проводят в полярном апротонном растворителе, выбранном из числа диметилформамида, диметилацетамида, N-метилпирролидона или еще диметилсульфоксида.
В еще одном предпочтительном варианте осуществления реакцию конденсации проводят в интервале температуры от -20°С до 30°С, более предпочтительно при 0°С.
В предпочтительном варианте осуществления стадию выделения соединения формулы 1 проводят путем осаждения в спиртовом растворителе, таком как метанол или этанол, с последующей стадией хроматографирования с обращенной фазой в кислой среде. Соединение очищают в растворе кислоты, такой как хлористоводородная кислота.
Кроме того, в предпочтительном варианте осуществления не содержащий защиты реагент первичного амина формулы 6, используемый на стадии конденсации с), представляет собой спермин или спермидин.
В случае использования на стадии с) спермидина получают 2 изомерных соединения формулы 1, для которых a=3, b=4, R=H и a=4, b=3, R=H в равном соотношении.
В случае конденсации со спермином, получают соединение, для которого a=3, b=4, а R=(CH2)3-NH2.
Изобретение также относится к использованию реагента первичного амина формулы 6 для получения соединений общей формулы 1 исходя из подофиллотоксина в соответствии с описанными ранее стадиями а), затем b), затем с).
Кроме того, настоящее изобретение относится к способу синтеза соединения, для которого a=3, b=4, R=H, или соединения, для которого a=4, b=3, R=H, а именно 2-[3-(4-аминобутиламино)пропиламино]-N-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро[3',4':6,7]нафто[2,3-d][1,3]диоксол-5-ил]ацетамида, или 2-[4-(3-аминопропиламино)бутиламино]-N-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро[3',4':6,7]нафто[2,3-d][1,3]диоксол-5-ил]ацетамида, который включает в себя следующие стадии.
а) Получение 4'-деметилэпиподофиллотоксина формулы 3 исходя из подофиллотоксина формулы 2.
b) Превращение 4'-деметилэпиподофиллотоксина в 4β-хлорацетамидо-4'-деметилэпиподофиллотоксин формулы 4.
с) Конденсация 4β-хлорацетамидо-4'-деметилэпиподофиллотоксина формулы 4 со спермидином.
За этими 3 стадиями следует стадия выделения 2-[3-(4-аминобутиламино)пропиламино]-N-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро[3',4':6,7]нафто[2,3-d][1,3]диоксол-5-ил]ацетамида, или 2-[4-(3-аминопропиламино)бутиламино]-N-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро[3',4':6,7]нафто[2,3-d][1,3]диоксол-5-ил]ацетамида, при этом, в случае необходимости, из данных продуктов получали соль действием минеральной или органической кислоты.
Кроме того, настоящее изобретение относится к способу синтеза соединения, для которого a=3, b=4, а R=(CH2)3-NH2, а именно 2-{3-[4-(3-аминопропиламино)бутиламино]пропиламино}-N-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро[3',4':6,7]нафто[2,3-d][1,3]диоксол-5-ил]ацетамида, который включает в себя следующие стадии.
а) Получение 4'-деметилэпиподофиллотоксина формулы 3 исходя из подофиллотоксина формулы 2.
b) Превращение 4'-деметилэпиподофиллотоксина в 4β-хлорацетамидо-4'-деметилэпиподофиллотоксин формулы 4.
с) Конденсацию 4β-хлорацетамидо-4'-деметилэпиподофиллотоксина формулы 4 со спермином
с последующей стадией выделения 2-{3-[4-(3-аминопропиламино)бутиламино]пропиламино}-N-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро[3',4':6,7]нафто[2,3-d][1,3]диоксол-5-ил]ацетамида и, в случае необходимости, получения из него соли.
Следующие примеры иллюстрируют изобретение, не ограничивая его рамки.
Пример 1:
Синтез соединения формулы 1 при a=3, b=4, c=3, а именно
2-{3-[4-(3-аминопропиламино)бутиламино]пропиламино}-N-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро[3',4':6,7]нафто[2,3-d][1,3]диоксол-5-ил]ацетамида в виде хлоргидрата исходя из 4β-хлорацетамидо-4'-деметилэпиподофиллотоксина и спермина при незащищенных аминных функциональных группах
Схема синтеза следующая:
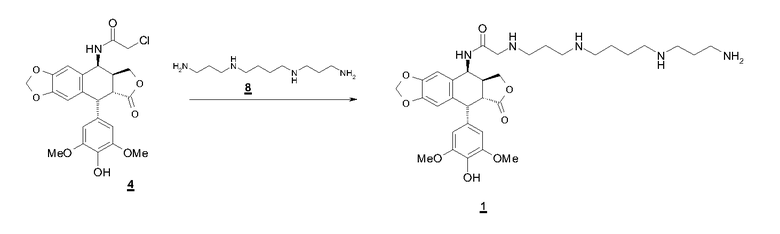
К раствору 1 г (2,1 ммоль) 4β-хлорацетамидо-4'-деметилэпиподофиллотоксина в 5 мл ДМФА добавляли 0,43 г (2,1 ммоль) спермина формулы 8 в 5 мл ДМФА. Перемешивали в течение 5 ч. Затем добавляли 20 мл EtOH, потом добавляли раствор изопропанол/HCl до слабокислого рН. Выпадал хлоргидрат. Кристаллы фильтровали и сушили, получая 1,9 г сырого соединения 1. Проводили очистку препаративной ВЭЖХ (Lichrospher 100 RP 18, элюирование HCl:c=5 мМ). Фракции лиофилизировали, затем экстрагировали диэтиловым эфиром и получали хлоргидрат соединения 1 в аморфной форме с чистотой 97%, выходом 40%.
Т пл 267°С. 1Н ЯМР: (DMSO) δ 9,07(д, 1H, J=8,32 Гц, NHCO), 8,27(с, 1H, ArOH), 6,80(с, 1H, H5), 6,55(с, 1H, H8), 6,23(с, 2H, H2', H6',), 6,01 (д, 2H, J=12 Гц, OCH20), 5,23(дд, 1H, J=5,3 и 8,1 Hz, H4), 4,52(д, 1H, J=5,2 Гц, H1), 4,28(т, 1H, J=8 Гц, H11a), 3,94(дд, 1H, J=8,8 и 10,4 Гц, H11b), 3,8(м, 2H, CH2CO), 3,63(с, 6H, 2xOCH3), 3,22(дд, 1H, j=5 и 14,4 Гц, H2), 3,06(м, 3H, H3 и CH2NH), 2,99(м, 4H, CH2NH), 2,89(м, 6H, CH2NH), 2,08(т, J=7,6 Hz, 2H, насыщ., CH2), 1,99(кв, 2H, J=7,2 Гц, насыщ., CH2), 1,73(м, 4H, насыщ., CH2), MS-ESI (m/z) 642,2 (MH+), анализ: C33H47N5O8, 4HC1, вычислено C% 50,32, H% 6,53, N% 8,89, найдено C% 50,264, H% 6,57, N% 8,66.
Пример 2
Синтез соединения формулы 1 при a=3, b=4, R=H, а именно хлоргидрата 2-[3-(4-аминобутиламино)пропиламино]-N-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро[3',4':6,7]нафто[2,3-d][1,3]диоксол-5-ил]ацетамида, и соединения формулы 1 при a=4, b=3, R=H, а именно 2-[4-(3-аминопропиламино) бутиламино]-N-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро[3',4':6,7]нафто[2,3-d][1,3]диоксол-5-ил]ацетамида в форме хлоргидрата исходя из 4β-хлорацетамидо-4'-деметилэпиподофиллотоксина и спермидина при незащищенных функциональных аминных группах
Схема синтеза следующая:
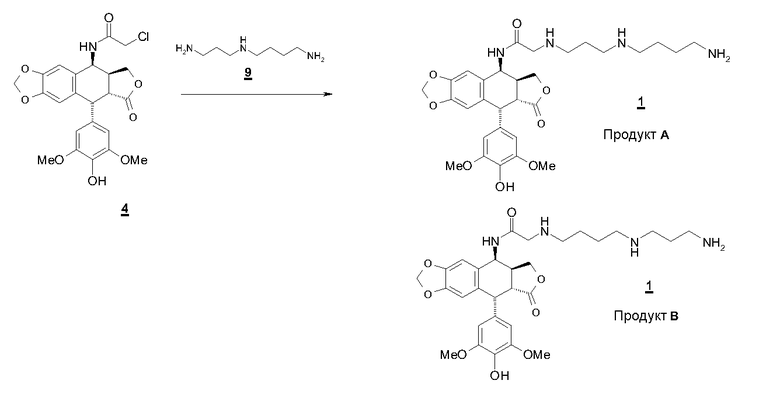
В тех же условиях, что и в случае производного примера 1, но заменяя спермин на спермидин формулы 9, получают соединения формулы 1 (продукт А: a=3, b=4, R=H и продукт В: a=4, b=3, R=H). По аналогии с примером 1 выделяют 2 продукта в равном соотношении с общим выходом 40%.
Данные продукты по всем пунктам идентичны соединениям, полученным соответственно в примерах 31 и 32 в заявке патента WO 2005/100363.
Данные примеры 1 и 2 можно преобразовать для синтеза всех соединений формулы 1 при использовании вместо спермина или спермидина соответствующего реагента незащищенного первичного амина формулы 6.
Пример 3
Синтез соединения формулы 1 при a=3, b=4, с=3, а именно
2-{3-[4-(3-аминопропиламино)бутиламино]пропиламино}-N-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро [3',4':6,7]нафто[2,3-d][1,3]диоксол-5-ил]ацетамида, в форме хлоргидрата, исходя из подофиллотоксина в 3 стадии
1 стадия
Схема синтеза следующая:
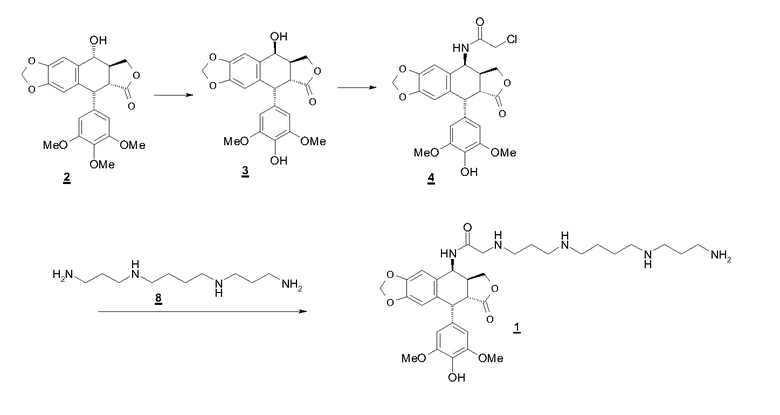
10 г (24 ммоль) подофиллотоксина растворяют в 60 мл трифторуксусной кислоты. Постепенно прибавляют 5,4 мл (72 ммоль) метансульфокислоты. Перемешивание продолжают в течение 9 ч, вновь прибавляют 5,4 мл (72 мл) диметилсульфида и продолжают перемешивание в течение 9 ч. Содержимое выливают на лед (600 мл) и экстрагируют этилацетатом (3×300 мл). Органические фазы промывают водой, затем раствором NaHCO3 до нейтральной реакции. После сушки над сульфатом натрия, фильтрования и упаривания получают 6,3 г 4'-деметилэпиподофиллотоксина, который непосредственно вводят в следующую стадию.
2 стадия
К 47,4 мл хлорацетонитрила прибавляют 30 г 4'-деметилэпиподофиллотоксина, затем при перемешивании добавляют 3 капли концентрированной серной кислоты. Перемешивают 3 ч при температуре окружающей среды. Затем добавляют 300 мл изопропанола при перемешивании. Полученный осадок фильтруют и промывают 200 мл изопропанола. Осадок промывают водой до нейтральной реакции, затем диэтиловым эфиром. После высушивания в вакууме получают 34,2 г (выход 96%) твердого вещества белого цвета (т.пл.=240°С) соответствующего 4β-хлорацетамидо-4'-деметилэпиподофиллотоксина.
3 стадия
Исходя из 4β-хлорацетамидо-4'-деметилэпиподофиллотоксина, полученного на предшествующей стадии, синтез продолжают в соответствии с методикой, описанной в примере 1 для получения продукта формулы 1 (a=3, b=4, с=3).
Этот пример можно преобразовать для всех соединений формулы 1 при использовании соответствующих реагентов незащищенных первичных аминов формулы 6.
Изобретение относится к новому способу получения противораковых препаратов, представляющих собой производные (поли)аминоалкиламиноацетамида эпиподофиллотоксина формулы 1.
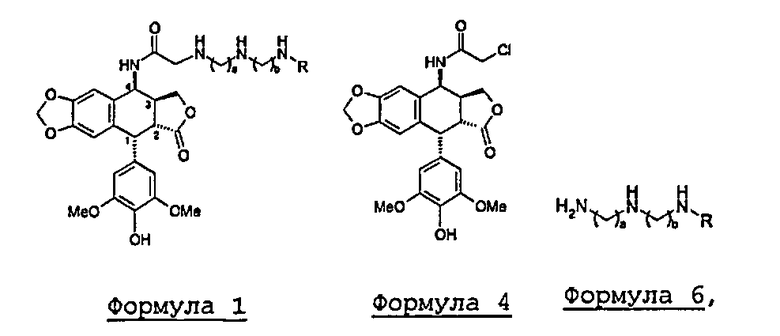
Способ включает стадию конденсации в полярном апротонном растворителе соединений формул 4 и 6 без предварительной защиты функциональных аминных групп с получением соединения формулы 1, где в формулах 1,4 и 6 R представляет собой атом водорода или -(CH2)C-NH2, 2≤а, b, с≤5. Предложен новый эффективный способ получения соединений с противораковой активностью. 3 н. и 12 з.п. ф-лы, 3 пр.
1. Способ получения производных (поли)аминоалкиламиноацетамида эпиподофиллотоксина формулы 1 и их фармацевтически приемлемых солей, в которых R представляет собой атом водорода или группу -(CH2)C-NH2 при 2≤а, b, с≤5
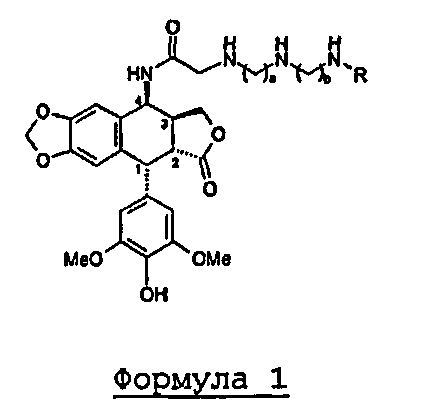
отличающийся тем, что он включает в себя стадию конденсации 4β-хлорацетамидо-4'-деметилэпиподофиллотоксина формулы 4
с реагентом первичного амина формулы 6: а, b, с и R имеют значения, аналогичные указанным выше, и не включает в себя предварительную защиту функциональных аминных групп 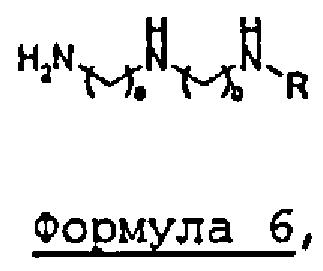
отличающийся тем, что стадию конденсации 4β-хлорацетамидо-4'-деметилэпиподофиллотоксина и реагента первичного амина формулы 6 проводят в полярном апротонном растворителе.
2. Способ получения производных (поли)аминоалкиламиноацетамида эпиподофиллотоксина формулы 1 по п.1, отличающийся тем, что их получают в виде хлоргидрата.
3. Способ по любому из пп.1 и 2, отличающийся тем, что полярный апротонный растворитель выбран из диметилформамида, диметилацетамида, N-метилпирролидона или диметилсульфоксида.
4. Способ по любому из пп.1 и 2, отличающийся тем, что реакция конденсации 4β-хлорацетамидо-4'-деметилэпиподофиллотоксина и реагента первичного амина формулы 6 сопровождается стадией выделения продукта общей формулы 1.
5. Способ по любому из пп.1 и 2, отличающийся тем, что выделение продукта формулы 1 проводят путем осаждения в спиртовом растворителе с последующей хроматографией с обращенной фазой в кислой среде.
6. Способ по любому из пп.1 и 2, отличающийся тем, что реагент первичного амина формулы 6, используемый на стадии конденсации с 4β-хлорацетамидо-4'-деметилэпиподофиллотоксином, представляет собой спермин или спермидин.
7. Способ получения соединения формулы 1, в котором а=3, b=4 и с=3, а именно 2-{3-[4-(3-аминопропиламино)бутиламино]пропиламино}-N-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-
гексагидрофуро[3',4':6,7]нафто[2,3-d][1,3]диоксол-5-ил]ацетамида, по любому из пп.1-6, отличающийся тем, что он включает в себя стадию конденсации 4β-хлорацетамидо-4'-деметилэпиподофиллотоксина со спермином.
8. Способ получения соединения формулы 1, в котором а=3, b=4, R=H, а именно 2-[3-(4-аминобутиламино)пропиламино]-N-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро[3',4':6,7]нафто[2,3-d][1,3]диоксол-5-ил]ацетамида, по любому из пп.1-6, отличающийся тем, что он включает в себя стадию конденсации 4β-хлорацетамидо-4'-деметилэпиподофиллотоксина формулы 4 со спермидином.
9. Способ получения соединения формулы 1, в котором а=4, b=3, R=H, а именно 2-[4-(3-аминопропиламино)бутиламино]-N-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро[3',4':6,7]нафто[2,3-d][1,3]диоксол-5-ил]ацетамида, по любому из пп.1-6, отличающийся тем, что он включает в себя стадию конденсации 4β-хлорацетамидо-4'-деметилэпиподофиллотоксина формулы 4 со спермидином.
10. Способ получения соединения формулы 1 по любому из пп.1 и 2, отличающийся тем, что он включает в себя следующие стадии:
а) получение 4'-деметилэпиподофиллотоксина формулы 3, исходя из подофиллотоксина формулы 2
b) превращение 4'-деметилэпиподофиллотоксина формулы 3, полученного на стадии а), в 4β-хлорацетамидо-4'-деметилэпиподофиллотоксин формулы 4 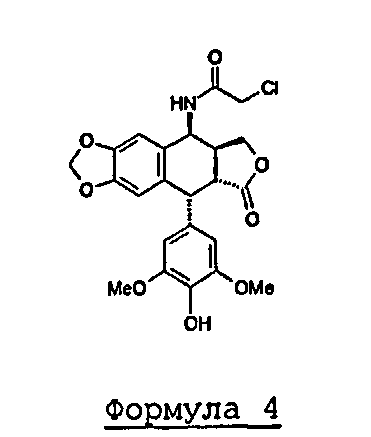
с) конденсация 4β-хлорацетамидо-4'-деметилэпиподофиллотоксина, полученного на стадии b), с реагентом первичного амина формулы 6.
11. Способ получения соединения формулы 1, в котором а=3, b=4 и с=3, а именно 2-{3-[4-(3-аминопропиламино)бутиламино]пропиламино}-N-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро[3',4':6,7]нафто[2,3-d][1,3]диоксол-5-ил]ацетамида, по п.10, отличающийся тем, что он включает в себя следующие стадии:
a) получение 4'-деметилэпиподофиллотоксина, исходя из подофиллотоксина,
b) превращение 4'-деметилэпиподофиллотоксина, полученного на стадии а), в 4β-хлорацетамидо-4'-деметилэпиподофиллотоксин,
c) конденсация 4β-хлорацетамидо-4'-деметилэпиподофиллотоксина, полученного на стадии b), со спермином.
12. Способ получения соединения формулы 1, в котором а=3, b=4 и R=H, а именно 2-[3-(4-аминобутиламино)пропиламино]-N-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро[3',4':6,7]нафто[2,3-d][1,3]диоксол-5-ил]ацетамида, по п.11, отличающийся тем, что он включает в себя следующие стадии:
a) получение 4'-деметилэпиподофиллотоксина, исходя из подофиллотоксина,
b) превращение 4'-деметилэпиподофиллотоксина, полученного на стадии а), в 4β-хлорацетамидо-4'-деметилэпиподофиллотоксин,
c) конденсация 4β-хлорацетамидо-4'-деметилэпиподофиллотоксина, полученного на стадии b), со спермидином.
13. Способ получения соединения формулы 1, в котором а=3, b=4 и R=H, а именно 2-[4-(3-аминопропиламино)бутиламино]-N-[9-(4-гидрокси-3,5-диметоксифенил)-8-оксо-5,5а,6,8,8а,9-гексагидрофуро[3',4':6,7]нафто[2,3-d][1,3]диоксол-5-ил]ацетамида, по п.10, отличающийся тем, что он включает в себя следующие стадии:
a) получение 4'-деметилэпиподофиллотоксина, исходя из подофиллотоксина,
b) превращение 4'-деметилэпиподофиллотоксина, полученного на стадии а), в 4β-хлорацетамидо-4'-деметилэпиподофиллотоксин,
c) конденсация 4β-хлорацетамидо-4'-деметилэпиподофиллотоксина, полученного на стадии b), со спермидином.
14. Применение соединений общей формулы 6 как определена в п.1 для получения соединений общей формулы 1 по пп.1-6.
15. Применение соединений общей формулы 6 для получения соединений общей формулы 1 в соответствии со способами по пп.10-13.
| WO 2005100363 А1, 27.10.2005 | |||
| Способ получения гидрохлорида метиламиноацетопирокатехина | 1976 |
|
SU598871A1 |
| WO 9721713 А, 19.06.1997 | |||
| WO 2007010007 А, 25.01.2007 | |||
| WO 2004073375 А, 02.09.2004. | |||

