Изобретение относится к применению химических соединений в области медицины и может быть использовано как средство при изготовлении фармакологических препаратов для предупреждения и лечения нейродегенеративных заболеваний.
Неконтролируемая агрегация определенных типов белков является ключевым звеном развития нейродегенеративных процессов, лежащих в основе патогенеза многих нейродегенеративных заболеваний. Накопление в нервной системе различных промежуточных (олигомеров, протофибрилл) и конечных (фибриллярных и аморфных отложений) продуктов, многие из которых обладают цитотоксическими свойствами, приводит к функциональным нарушениям и, в конечном итоге, к гибели нейронов. Такой тип патологии, получивший название протеинопатия, объединил в одну группу неврологические расстройства с существенно различающимися клиническими проявлениями патологии [Шелковникова Т.А., Куликова А.А., Цветков Ф.О., Петере О., Бачурин C.O., Бухман В.Л., Нинкина Н.Н. (2012). Протеинопатии - формы нейродегенеративных заболеваний, в основе которых лежит патологическая агрегация белков. Молекулярная биология. Т. 46, №3. С:402-415; Шелковникова Т.А., Устюгов А.А., Смирнов А.П., Скворцова В.И., Бухман В.Л., Бачурин C.O., Нинкина Н.Н. Мутации в гене FUS, ассоциированные с наследственными формами бокового амиотрофического склероза, влияют на клеточную локализацию кодируемого белка и его способность к агрегации. ДАН, 2011, Т.438, №3, С.422-426]. В эту группу заболеваний входят такие широко распространенные и в настоящее время неизлечимые нейродегенеративные расстройства, как болезнь Альцгеймера (БА), болезнь Паркинсона (БП), боковой амиотрофический склероз (БАС), фронтотемпоральная деменция (ФТД). К протеинопатиям относятся и наследственные заболевания, например хорея Гентингтона, а также широкая группа прионовых болезней.
Принято считать, что вероятная причина развития болезни Альцгеймера, являющейся наиболее распространенным нейродегенеративным заболеванием, связана с формирование патологических агрегатов тау-белков, образующих нейрофибриллярные клубки, и амилоидных бляшек [Spillantini M.G. and Goedert М. Tau protein pathology in neurodegenerative diseases Trends Neurosci. (1998) 21, 428-433].
До сих пор лечение протеинопатий является преимущественно симптоматическим, поскольку не разработаны эффективные средства, непосредственно воздействующие на патогенетический процесс, лежащий в основе заболевания.
В настоящее время процесс неконтролируемой агрегации белков рассматривается как одна из важнейших мишеней для создания нового поколения терапевтических средств, которые позволят модифицировать процессы, лежащие в основе развития нейродегенерации, и тем самым замедлить течение болезни или даже остановить патологический процесс [Jacobsen J.S., Reinhart P., Pangalos M.N. NeuroRX. 2005. vol. 2(4). p: 612-626; Walker L.C., Ibegbu C.C., Todd C.W., Robinsona H.L., Juckere M., LeVine H 3rd, Gandy S. Biochemical Pharmacology. 2005. vol.69. p:1001-1008; Christensen D.D. CNS Spectrums. 2007. vol.12. p:113-123].
Известно, что производное гамма-карболина - отечественный антигистаминный препарат димебон, проявляет способность блокировать неконтролируемую агрегацию некоторых белков в нервной системе, которую связывают с развитием целого ряда нейродегенеративных заболеваний. Так, было показано, что димебон избирательно ингибирует агрегацию белка гамма-синуклеина, участвующего в патогенезе БАС [С.О. Бачурин, А.А. Устюгов, О. Петере, Т.А. Шелковникова, В.Л. Бухман, Н.Н. Нинкина. Блокада нейродегенеративных процессов, вызванных протеинопатией, как новый механизм действия нейропротекторных и когнитивно-стимулирующих препаратов. ДАН, 2009, 428(2), 262-265; А.А. Ustyugov, Т.A. Shelkovnikova, V.S. Kokhan, I.V. Khritankova, О. Peters, V.L. Buchman, S.O. Bachurin, and N.N. Ninkina (2012) Dimebon Reduces the Levels of Aggregated Amyloidogenic Protein Forms in Detergent-Insoluble Fractions In Vivo. Bulletin of Experimental Biology and Medicine, Vol.152, No.6, p.731-733; Bachurin S.O., Shelkovnikova T.A., Ustyugov A.A., Peters O., Khritankova I., Afanasieva M.A., Tarasova T.V., Alentov I.I., Buchman V.L., Ninkina N. (2012) Dimebon slows progression of proteinopathy in γ-synuclein transgenic mice. Neurotoxicity Research 22(1), 33-42]. Вместе с тем димебон не влиял на патологически процессы, связанные с системой альфа-синуклеинов [Shelkovnikova Т.А., Ustyugov А.А., Millership S., Peters О., Anichtchik О., Spillantini M.G., Buchman V.L., Bachurin S.O., Ninkina N.N. Dimebon does not ameliorate pathological changes caused by expression of truncated (1-120) human alpha-synuclein in dopaminergic neurons of transgenic mice. Neurodegenerative Diseases. 2011. T.8, №6. P.430-437]. Установлено, что димебон способен улучшать состояние экспериментальных животных, у которых генетически вызвано усиленное образование тау-агрегатов - характерных патологических образований при БА [Peters О.М., Connor-Robson N., Sokolov V.B., Aksinenko A.Y., Kukharsky M.S., Bachurin S.O., Ninkina N.N., Buchman V.L. Chronic administration of dimebon ameliorates pathology in TauP301S transgenic mice. J. Alzheimers Dis. 2013; 33(4): 1041-9].
Недавно было показано, что димебон способен также уменьшать образование патологических агрегатов белка TDP-43, связанного с развитием БАС и ФТД. Причем это действие усиливается при совместном введении димебона и лекарственного препарата - метиленовый синий, относящегося к группе производных фенотиазина [Makiko Yamashita, Takashi Nonaka, Fuyuki Kametani, Tetsuaki Arai, Haruhiko Akiyama, Vladimir L. Buchman, Natalia Ninkina, Sergey O. Bachurin, Shahin Zibaee, Michel Goedert, Masato Hasegawa. Methylene blue and dimebon inhibit aggregation of TDP-43 in cellular models. FEBS Letters, 2009. 583 (2009) 2419-2424]. Bo многих работах последних лет показано, что метиленовый синий эффективно блокирует развитие патологий, связанных с агрегацией тау-белка, что имеет значение в плане терапии БА [Congdon Е.Е., Wu J.W., Myeku N., Figueroa Y.H., Herman M., Marinec P.S., Gestwicki J.E., Dickey C.A., Yu W.H., Duff K.E. Methylthioninium chloride (methylene blue) induces autophagy and attenuates tauopathy in vitro and in vivo. Autophagy. 2012 Apr;8(4):609-22; Oz M., Lorke D.E., Hasan M., Petroianu G.A. Cellular and molecular actions of Methylene Blue in the nervous system. Med. Res. Rev. 2011 Jan; 31(1):93-117].
Предлагаемое изобретение решает задачу расширения арсенала средств, которые могут быть использованы в качестве новых эффективных протекторов, защищающих нервные клетки от образования патологических белковых агрегатов, связанных с развитием нейродегенеративных процессов в центральной и перифирической нервной системе.
Поставленная цель достигается применением следующих фенотиазинсодержащих 1,2,3,4-тетрагидропиридо[4,3-b]индолов общей формулы 1 и их хлоргидратов в качестве средства для снижения неконтролируемой агрегации белков в нервной системе.
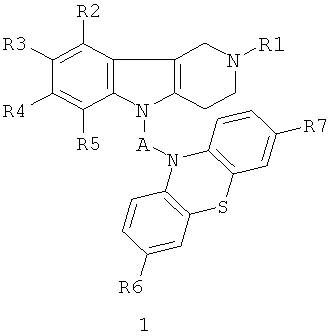
в которой R1=H, (C1-C6)алкил;
R2, R3, R4, R5=Н, F, Cl, Br, (C1-C6)алкил, (C1-C6)алкокси,
R6, R7=H, F, Cl, Br, (C1-C6)алкил, (C1-C6)алкокси, NH2, NHAlkyl, NAlkyl2;
A=CH2CH2, CH=CHCH2, CH2CH2CH2, CH2CH[(C1-C6)алкил]C(O), CH2CH2C(O)NHCH2CH2); CH2CH[(C1-C6)алкил]C(O)N[(C1-C6)алкил]CH2CH2), CH2CH(OH)CH2, CH2CH(OH)CH2NHCH2CH2, CH2CH(OH)CH2N[(C1-C6)алкил]CH2CH2, CH2CHFCH2NHCH2CH2, CH2CHFCH2N[(C1-C6)алкил]CH2CH2.
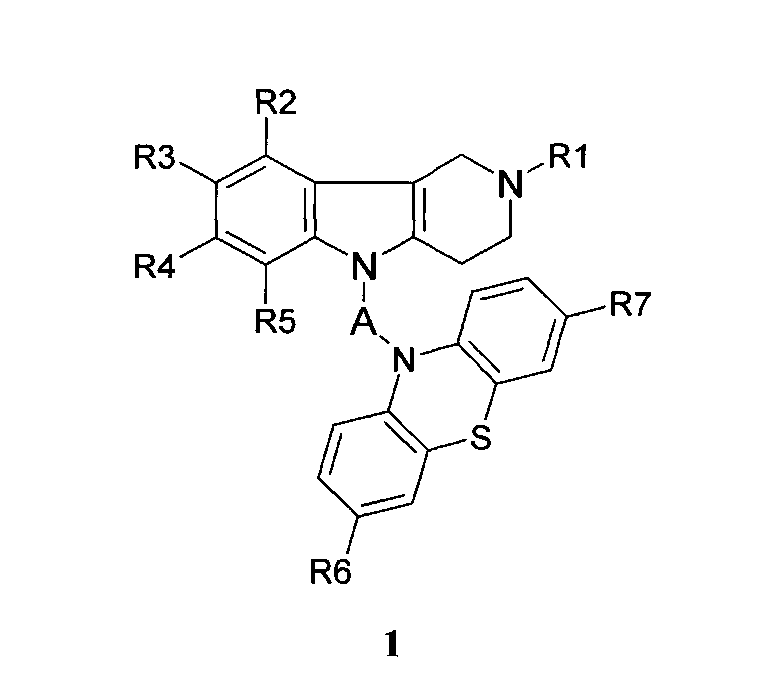
Производные фенотиазинсодержащих 1,2,3,4-тетрагидропиридо[4,3-b]индолов могут представлять собой соединения общей формулы 1.1
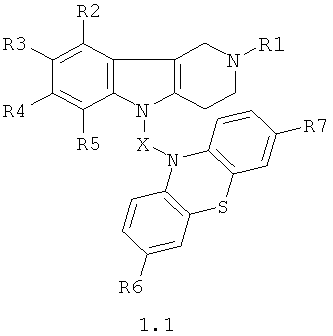
в которой R1, R2, R3, R4, R5, R6, R7 имеют значения, определенные выше для формулы 1;
X=CH2CH2, CH=CHCH2, CH2CH2CH2, CH2CH[(C1-C6)алкил]C(O), CH2CH2C(O)NHCH2CH2); CH2CH[(C1-C6)алкил]C(O)N[(C1-C6)алкил]CH2CH2).
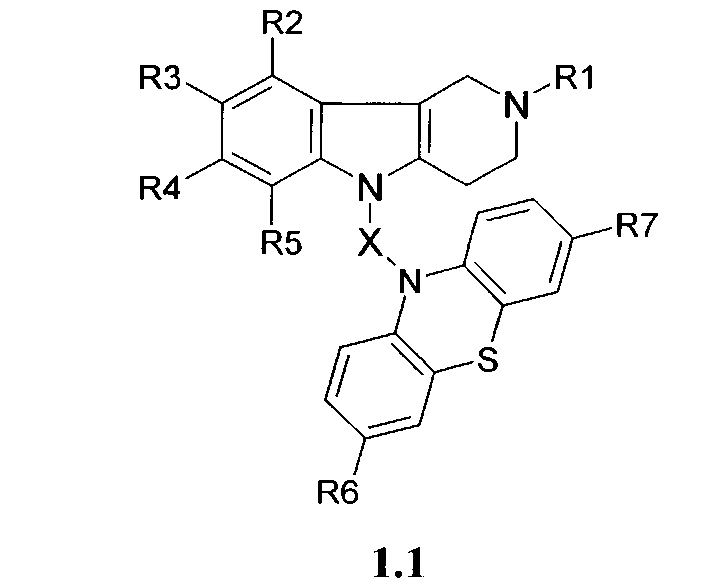
Производные фенотиазинсодержащих 1,2,3,4-тетрагидропиридо [4,3-b] индолов могут представлять собой также соединения общей формулы 1.2
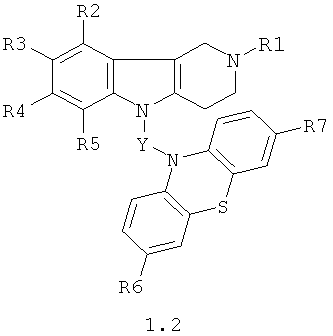
в которой R1, R2, R3, R4, R5, R6, R7 имеют значения, определенные выше для формулы 1;
Y=CH2CH(OH)CH2, CH2CH(OH)CH2NHCH2CH2,
CH2CH(OH)CH2N[(C1-C6)алкил]CH2CH2.
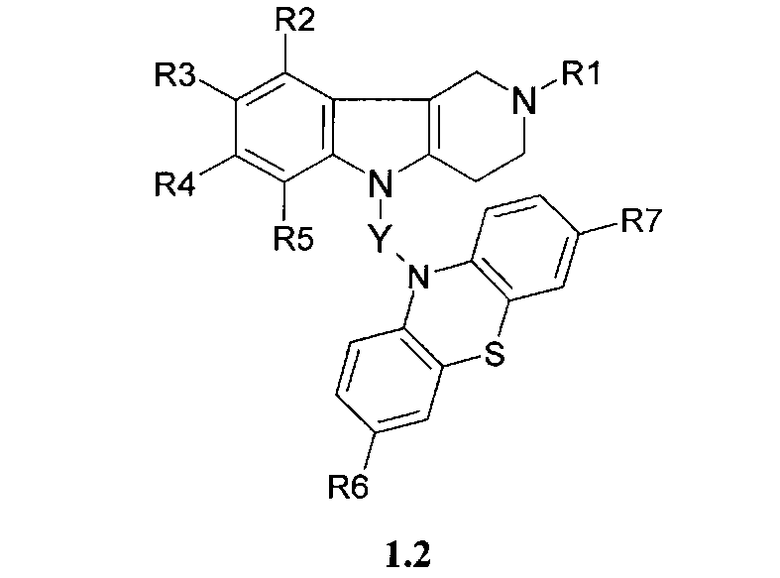
Производные фенотиазинсодержащих 1,2,3,4-тетрагидропиридо [4,3-b] индолов могут представлять собой также соединения общей формулы 1.3
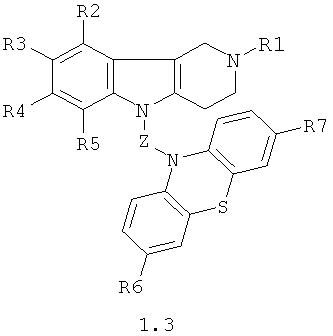
в которой R1, R2, R3, R4, R5, R6, R7 имеют значения, определенные выше для формулы 1;
Z=CH2CHFCH2NHCH2CH2 или
CH2CHFCH2N[(C1-C6)алкил]CH2CH2,
Еще одним аспектом изобретения являются способы получения соединений формулы 1.1, формулы 1.2, формулы 1.3, а также их хлоргидратов.
Способ получения соединений формулы 1.1 заключается в том, что эквимольную смесь гамма-карболинов общей формулы 2
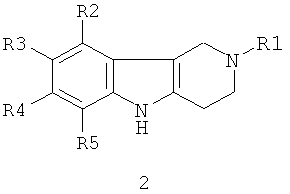
в которой R1, R2, R3, R4, R5, имеют значения, определенные выше для формулы 1;
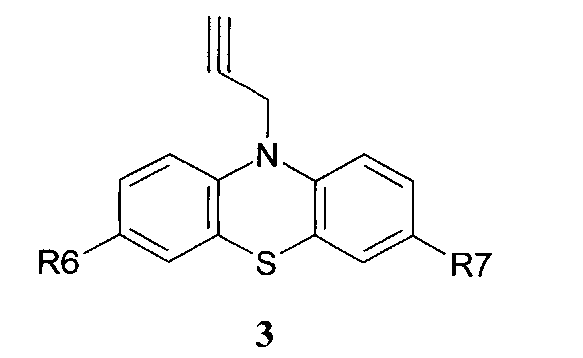
и ацетиленсодержащих фенотиазинов общей формулы 3,
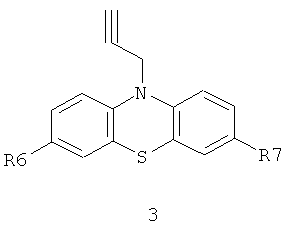
или винилсодержащих фенотиазинов общей формулы 4,
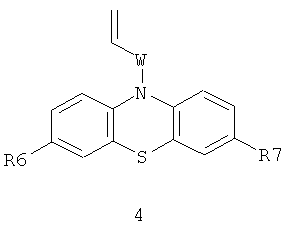
в которых R6, R7 имеют значения, определенные выше для формулы 1;
W=(CH2)n, где n=0 или 1; C(O); CH[(C1-C6)алкил]C(O); C(O)NHCH2CH2); C(O)N[(C1-C6)алкил]CH2CH2; CH[(C1-C6)алкил]C(O)N[(C1-C6)алкил]CH2CH2),
нагревают в диметилсульфоксиде или диметилформамиде в присутствии катализатора, выбранного из ряда: метилат натрия, трет-бутилат калия, гидроокись калия или фторид цезия при температуре 120-160°C в течение 6-24 часов.
Способ получения соединений 1.2 заключается в том, что эквимольную смесь гамма-карболинов общей формулы 2
в которой R1, R2, R3, R4, R5, имеют значения, определенные выше для формулы 1;
и N-эпоксидсодержащих фенотиазинов общей формулы 5
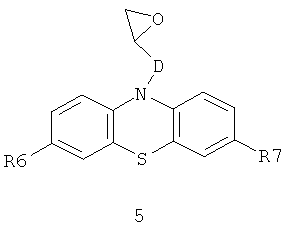
в которых R6, R7 имеют значения, определенные выше для формулы 1;
D=CH2; CH2NHCH2CH2; CH2N[(C1-C6)алкил]CH2CH2
нагревают в диметилсульфоксиде или диметилформамиде в присутствии катализатора, выбранного из ряда: метилат натрия, трет-бутилат калия, гидроокись калия или фторид цезия при температуре 100-140°C в течение 2-12 часов.
Способ получения соединений 1.3 заключается в том, что гидроксилсодержащие конъюгаты гамма-карбонилов и фенотиазинов общей формулы 6
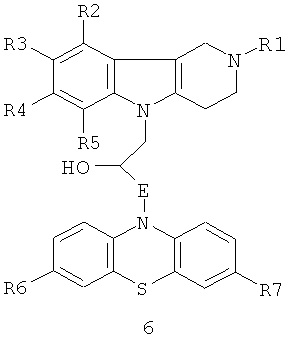
в которой R1, R2, R3, R4, R5, R6, R7 имеют значения, определенные выше для формулы 1;
E=CH2; CH2NHCH2CH2; CH2N[(C1-C6)алкил]CH2CH2.
фторируют эквимольным количеством диэтиламиносульфотрифторида (Et2NSF3) в диметилсульфоксиде или диметилформамиде при температурах (-78)-0°C в течение 1-3 часов.
Способ получения хлоргидратов любого из соединений формул 1.1, 1.2, 1.3 заключается в растворении соответствующих соединений формул 1.1, 1.2, 1.3 в 10%-ном растворе HCl в изопропаноле.
Производные фенотиазинсодержащих 1,2,3,4-тетрагидропиридо [4,3-b] индолов общей формулы 1, обладающие заявленными свойствами, получали путем конъюгации индольной и фенотиазиновой составляющих, соответствующих заявляемому составу и объединенных через заявляемый молекулярный спейсер. Синтетические способы для получения соединений формулы 1 представляют четыре общие группы, иллюстрированные примерами.
Приведенные примеры подтверждают, но не ограничивают предлагаемое изобретение.
Синтез соединений формулы 1.1 (общая методика)
Процесс проводят нагреванием эквимольной смеси гамма-карболинов 2 и ацетиленсодержащих фенотиазинов 3 (схема 1) или винилсодержащих фенотиазинов 4 (схема 2) в диметилсульфоксиде (ДМСО) или диметилформамиде (ДМФА) в присутствии катализатора, выбранного из ряда: метилат натрия (MeONa), трет-бутилат калия (трет.-BuOK), гидроокись калия (KOH) и фторид цезия (CsF) при температуре 120-160°C в течение 6-24 часов.
Схема 1.
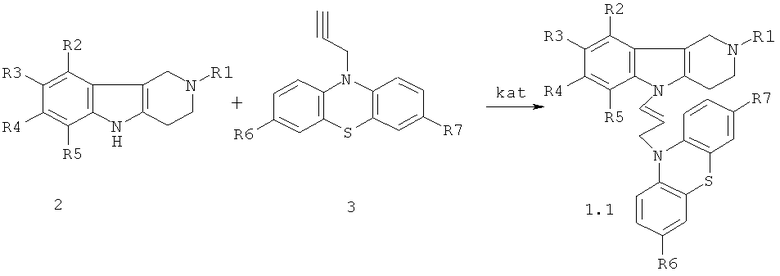
R1=H, (C1-C6)алкил;
R2, R3, R4, R5 = Н, F, Cl, Br, (C1-C6)алкил, (C1-C6)алкокси,
R6, R7=. Н, F, Cl, Br, (C1-C6)алкил, (C1-C6)алкокси, NH2, NHAlkyl, NAlkyl2;
kat=MeONa, трет.-BuOK), KOH, CsF
Схема 2.
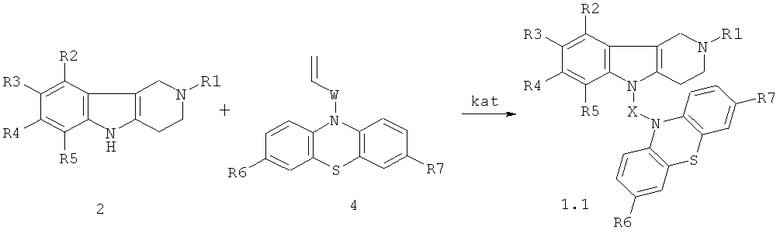
R1=H, (C1-C6)алкил;
R2, R3, R4, R5=H, F, Cl, Br, (C1-C6)алкил, (C1-C6)алкокси,
R6, R7=. H, F, Cl, Br, (С1-С6)алкил, (С1-С6)алкокси, NH2, NHAlkyl, NAlkyl2;
W= нет, CH2; CH[(C1-C6)алкил]C(O); C(O)NHCH2CH2); C(O)N[(C1-C6)алкил]CH2CH2; CH[(C1-C6)алкил]C(O)N[(C1-C6)алкил]CH2CH2),
X=CH2CH2, CH2CH2CH2, CH2CH[(C1-C6)алкил]C(O), CH2CH2C(O)NHCH2CH2); CH2CH[(C1-C6)алкил]C(O)N[(C1-C6)алкил]CH2CH2).
kat= MeONa, трет.-BuOK), KOH, CsF
Пример 1: Способ получения 10-[3-(2,8-Диметил-1,2,3,4-тетрагидропиридо[4,3-b]индол-5-ил)-аллил]N,N,N,N-тетраметил-10H-фенотиазин-3,7-диамина (I).
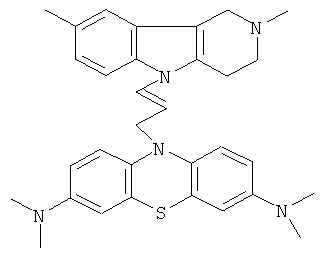
В качестве исходных веществ брали 1 ммоль 2,8-диметил-2,3,4,5-тетрагидро-1H-пиридо [4,3-b] индола и 1 ммоль N,N,N,N-тетралетил-10-проп-2-инил-10H-фенотиазин-3,7-диамина. В качестве катализатора брали 200 мг CsF в 1.5 мл ДМСО. Нагревали при перемешивании при 140-160°C в течение 8 ч. ДМСО удаляли в вакууме 3 мм рт.ст., из остатка продукт экстрагировали хлористым метиленом. Хлористый метилен упаривали и остаток хроматографировали на силикагеле (60 меш), элюент метанол/хлороформ =1/5. Полученный продукт представляет собой желтое твердое вещество, выход: 82%. Спектр ЯМР 1H (CDCl3) δ, м.д.: 2.48 и 2.53 оба с (3H, Me); 2.65 т (2H, CH2, J=6.4 Гц); 2.70-2.86 м (4H, CH2); 2.94 (с, 12H, Me); 3.64 с (2H, CH2); 6.3 (д, 1H, CH=, J=12.2 Гц), 6.5 (м, 1H, CH=), 6.70-7.00 (м, 3H), 7.00-7.25 (м, 2H) 7.30 (д, 2H, J=8.8 Гц), 7.54 (д, 2H, J=2.2 Гц).
Пример 2: Способ получения 10-[3-(2,8-Диметил-1,2,3,4-тетрагидропиридо[4,3-b]индол-5-ил)-аллил]-N,N,N,N-тетраметил-10H-фенотиазин-3,7-диамина (I).
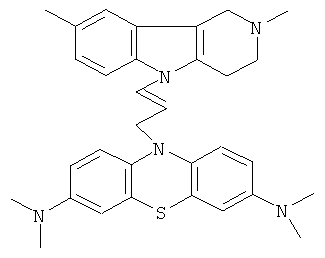
В качестве исходных веществ брали 1 ммоль 2,8-диметил-2,3,4,5-тетрагидро-1H-пиридо[4,3-b]индола и 1 ммоль замещенного N,N,N,N-тетраметил-10-проп-2-инил-10H-фенотиазин-3,7-диамина, в качестве катализатора брали 300 мг MeONa в 1.5 мл ДМФА. Нагревали при перемешивании при 130-140°C в течение 8 ч. Затем ДМФА удаляли в вакууме 3 мм рт. ст., из остатка продукт экстрагировали хлористым метиленом. Хлористый метилен упаривали и остаток хроматографировали на силикагеле (60 меш), элюент метанол/хлороформ =1/5. Полученный продукт представляет собой желтое твердое вещество, выход: 78%. Спектр ЯМР 1H (CDCl3) δ, м.д.: 2.48 и 2.53 оба с (3H, Me); 2.65 т (2H, CH2, J=6.4 Гц); 2.70-2.86 м (4H, CH2); 2.94 (с, 12H, Me); 3.64 с (2H, CH2); 6.3 (д, 1Н, CH=, J=12.2 Гц), 6.5 (м, 1H, CH=), 6.70-7.00 (м, 3H), 7.00-7.25 (м, 2H) 7.30 (д, 2H, J=8.8 Гц), 7.54 (д, 2H, J=2.2 Гц).
Пример 3: Способ получения 10-[3-(2,8-Диметил-1,2,3,4-тетрагидропиридо[4,3-b]индол-5-ил)-аллил]-N,N,N,N-тетраметил-10H-фенотиазин-3,7-диамина (I).
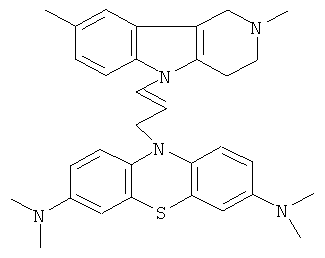
В качестве исходных веществ брали 1 ммоль 2,8-диметил-2,3,4,5-тетрагидро-1H-пиридо[4,3-b]индола, 1 ммоль N,N,N,N-тетраметил-10-проп-2-инил-10H-фенотиазин-3,7-диамина, в качестве катализатора брали 300 мг трет.-BuOK в 1.5 мл ДМСО. Нагревали при перемешивании при 120-130°C в течение 8 ч. ДМСО удалили в вакууме 3 мм рт.ст., из остатка продукт экстрагировали хлористым метиленом. Хлористый метилен упаривали и остаток хроматографировали на силикагеле (60 меш), элюент метанол/хлороформ =1/5. Полученный продукт представляет собой желтое твердое вещество, выход: 80%. Спектр ЯМР 1H (CDCl3) δ, м.д.: 2.48 и 2.53 оба с (3H, Me); 2.65 т (2H, CH2, J=6.4 Гц); 2.70-2.86 м (4H, CH2); 2.94 (с, 12H, Me); 3.64 с (2H, CH2); 6.3 (д, 1H, CH=, J=12.2 Гц), 6.5 (м, 1H, CH=), 6.70-7.00 (м, 3H), 7.00-7.25 (м, 2H) 7.30 (д, 2H, J=8.8 Гц), 7.54 (д, 2H, J=2.2 Гц).
Пример 4: Способ получения 10-[3-(2,8-Диметил-1,2,3,4-тетрагидропиридо[4,3-b]индол-5-ил)-аллил]-N,N,N,N-тетраметил-10H-фенотиазин-3,7-диамина (I).
В качестве исходных веществ брали 1 Ммоль 2,8-диметил-2,3,4,5-тетрагидро-1H-пиридо[4,3-b]индола, 1 ммоля N,N,N,N-траметил-10-проп-2-инил-10H-фенотиазин-3,7-диамина, в качестве катализатора брали 100 мг KOH в 1.5 мл ДМФА нагревали при перемешивании при 120-140°C в течение 8 ч. ДМФА удалили в вакууме 3 мм рт.ст., из остатка продукт экстрагировали хлористым метиленом. Хлористый метилен упаривали и остаток хроматографировали на силикагеле (60 меш), элюент метанол/хлороформ =1/5. Желтое твердое вещество, выход: 79%. Спектр ЯМР 1H (CDCl3) δ, м.д.: 2.48 и 2.53 оба с (3H, Me); 2.65 т (2H, CH2, J=6.4 Гц); 2.70-2.86 м (4H, CH2); 2.94 (с, 12H, Me); 3.64 с (2H, CH2); 6.3 (д, 1H, CH=, J=12.2 Гц), 6.5 (м, 1H, CH=), 6.70-7.00 (м, 3H), 7.00-7.25 (м, 2H) 7.30 (д, 2H, J=8.8 Гц), 7.54 (д, 2H, J=2.2 Гц).
Пример 5: Способ получения 10-[3-(2,8-Диметил-1,2,3,4-тетрагидропиридо[4,3-b]индол-5-ил)-этил]N,N,N,N-тетраметил-10H-Фенотиазин-3,7-диамина (II).
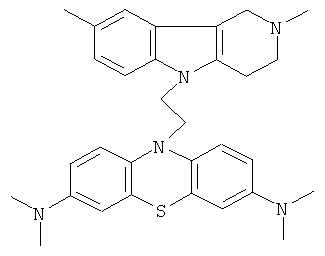
В качестве исходных веществ брали 1 ммоль 2,8-диметил-2,3,4,5-тетрагидро-1H-пиридо[4,3-b]индола, 1 ммоль 10-аллил-N,N,N,N-тетраметил-10H-фенотиазин-3,7,-диамина, в качестве катализатора брали 200 мг CsF в 1.5 мл ДМСО. Нагревали при перемешивании при 140-150°C в течение 8 ч. ДМСО удалили в вакууме 3 мм рт.ст., из остатка продукт экстрагировали хлористым метиленом. Хлористый метилен упаривали и остаток хроматографировали на силикагеле (60 меш), элюент метанол/хлороформ =1/5. Полученный продукт представляет собой желтое твердое вещество, выход: 86%. Спектр ЯМР 1H (CDCl3) δ, м.д.: 2.50 и 2.54 оба с (3H, Me); 2.63 т (2H, CH2, J=6.4 Гц); 2.60-2.86 м (6H, CH2); 2.96 (с, 12H, Me); 3.64 с (2H, CH2); 3.68 (т, 2H, J=6.5 Гц), 6.70-7.00 (м, 3H), 7.00-7.25 (м, 2H), 7.31 (д, 2H, J=8.8 Гц), 7.55 (д, 2H, J=2.2 Гц).
Пример 6: Способ получения 10-[3-(2,8-Диметил-1,2,3,4-тетрагидропиридо[4,3-b]индол-5-ил)этил]-N,N,N,N-тетраметил-10H-фенотиазин-3,7-диамина (II).
В качестве исходных веществ брали 1 ммоль 2,8-диметил-2,3,4,5-тетрагидро-1H-пиридо[4,3-b]индола, 1 ммоль 10-аллил-N,N,N,N-тетраметил-10H-фенотиазин-3,7,-диамина, в качестве катализатора брали 300 мг MeONa в 1.5 мл ДМФА. Нагревали при перемешивании при 150-160°C в течение 8 ч. ДМФА удалили в вакууме 3 мм рт.ст., из остатка продукт экстрагировали хлористым метиленом. Хлористый метилен упаривали и остаток хроматографировали на силикагеле (60 меш), элюент метанол/хлороформ =1/5. Полученный продукт представляет собой желтое твердое вещество, выход: 82%. Спектр ЯМР 1Н (CDCl3) δ, м.д.: 2.50 и 2.54 оба с (3H, Me); 2.63 т (2H, CH2, J=6.4 Гц); 2.60-2.86 м (6H, CH2); 2.96 (с, 12H, Me); 3.64 с (2H, CH2); 3.68 (т, 2H, J=6.5 Гц), 6.70-7.00 (м, 3H), 7.00-7.25 (м, 2H), 7.31 (д, 2H, J=8.8 Гц), 7.55 (д, 2H, J=2.2 Гц).
Пример 7: Способ получения 10-[3-(2,8-Диметил-1,2,3,4-тетрагидропиридо[4,3-b]индол-5-ил)-этил]-N,N,N,N-тетраметил-10H-фенотиазин-3,7-диамина (II).
В качестве исходных веществ брали 1 ммоль 2,8-диметил-2,3,4,5-тетрагидро-1H-пиридо[4,3-b]индола, 1 ммоль 10-аллил-N,N,N,N-тетраметил-10H-фенотиазин-3,7,-диамина, 300 мг трет.-BuOK в 1.5 мл ДМСО. Нагревали при перемешивании при 140-150°C в течение 18 ч. ДМСО удалили в вакууме 3 мм рт.ст., из остатка продукт экстрагировали хлористым метиленом. Хлористый метилен упаривали и остаток хроматографировали на силикагеле (60 меш), элюент метанол/хлороформ = 1/5. Полученный продукт представляет собой желтое твердое вещество, выход: 85%. Спектр ЯМР 1Н (CDCl3) δ, м.д.: 2.50 и 2.54 оба с (3H, Me); 2.63 т (2H, CH2, J=6.4 Гц); 2.60-2.86 м (6H, CH2); 2.96 (с, 12H, Me); 3.64 с (2H, CH2); 3.68 (т, 2H, J=6.5 Гц), 6.70-7.00 (м, 3H), 7.00-7.25 (м, 2H), 7.31 (д, 2H, J=8.8 Гц), 7.55 (д, 2Н, J=2.2 Гц).
Пример 8: Способ получения 10-[3-(2,8-Диметил-1,2,3,4-тетрагидропиридо[4,3-b]индол-5-ил)-этил]-N,N,N,N-тетраметил-10H-фенотиазин-3,7-диамина (II).
В качестве исходных веществ брали 1 ммоль 2,8-диметил-2,3,4,5-тетрагидро-1H-пиридо[4,3-b]индола, 1 ммоль 10-аллил-N,N,N,N-тетраметил-10H-фенотиазин-3,7,-диамина, в качестве катализатора брали 100 мг KOH в 1.5 мл ДМФА. Нагревали при перемешивании при 150-160°C в течение 12 ч. ДМФА удалили в вакууме 3 мм рт.ст., из остатка продукт экстрагировали хлористым метиленом. Хлористый метилен упаривали и остаток хроматографировали на силикагеле (60 меш), элюент метанол/хлороформ =1/5. Полученный продукт представляет собой желтое твердое вещество, выход: 78%. Спектр ЯМР 1H (CDCl3) δ, м.д.: 2.50 и 2.54 оба с (3H, Me); 2.63 т (2H, CH2, J=6.4 Гц); 2.60-2.86 м (6H, СН2); 2.96 (с, 12Н, Me); 3.64 с (2H, CH2); 3.68 (т, 2H, J=6.5 Гц), 6.70-7.00 (м, 3H), 7.00-7.25 (м, 2H), 7.31 (д, 2H, J=8.8 Гц), 7.55 (д, 2H, J=2.2 Гц).
Пример 9: Способ получения 4-(2-Метил-1,2,3,4-тетрагидро-пиридо[43-b]индол-5-ил)-1-(фенотиазин-10-ил)пропан-1-она (III).
В качестве исходных веществ брали 1 ммоль 2-метил-2,3,4,5-тетрагидро-1H-пиридо[4,3-b]индола, 1 ммоль 1-(3,7-бис-диметиламино-фенотиазин-10-ил)пропенона, в качестве катализатора брали 200 мг CsF в 1.5 мл ДМСО. Нагревали при перемешивании при 120-140°C в течение 20 ч. ДМСО удалили в вакууме 3 мм рт.ст., из остатка продукт экстрагировали хлористым метиленом. Хлористый метилен упаривали и остаток хроматографировали на силикагеле (60 меш), элюент метанол/хлороформ =1/5. Полученный продукт представляет собой желтое твердое вещество, выход: 83%. Спектр ЯМР 1H (CDCl3) δ, м.д.: 2.43 (с, 3H, MeN); 2.68 (с, 6H, CH2); 3.49 (с, 2H, CH2); 4.17 (уш. с, 2H, CH2); 6.67-6.95 (м, 8H, CHAr); 7.00-7.16 (м, 4H, CHAr).
Пример 10: Способ получения 4-(2-Метил-1,2,3,4-тетрагидро-пиридо[4,3-b]индол-5-ил)-1-(фенотиазин-10-ил)пропан-1-она (III).
В качестве исходных веществ брали 1 ммоль 2-метил-2,3,4,5-тетрагидро-1H-пиридо[4,3-b]индола, 1 ммоль 1-(3,7-бис-диметиламино-фенотиазин-10-ил)пропенона, в качестве катализатора брали 300 мг MeONa в 1.5 мл ДМСО. Нагревали при перемешивании при 120-140°C в течение 11 ч. ДМСО удалили в вакууме 3 мм рт.ст., из остатка продукт экстрагировали хлористым метиленом. Хлористый метилен упаривали и остаток хроматографировали на силикагеле (60 меш), элюент метанол/хлороформ =1/5. Желтое твердое вещество, выход: 80%. Спектр ЯМР 1H (CDCl3) δ, м.д.: 2.43 (с, 3H, MeN); 2.68 (с, 6H, CH2); 3.49 (с, 2H, CH2); 4.17 (уш. с, 2H, CH2); 6.67-6.95 (м, 8H, CHAr); 7.00-7.16 (м, 4H, CHAr).
Пример 11: Способ получения 4-(2-Метил-1,2,3,4-тетрагидро-пиридо[4,3-b]индол-5-ил)-1-(фенотиазин-10-ил)пропан-1-она (III).
В качестве исходных веществ брали 1 ммоль 2-метил-2,3,4,5-тетрагидро-1H-пиридо[4,3-b]индола, 1 ммоль 1-(3,7-бис-диметиламино-фенотиазин-10-ил)пропенона, в качестве катализатора брали 300 мг трет.-BuOK в 1.5 мл ДМФА. Нагревали при перемешивании при 120-130°C в течение 6 ч. ДМФА удалили в вакууме 3 мм рт.ст., из остатка продукт экстрагировали хлористым метиленом. Хлористый метилен упаривали и остаток хроматографировали на силикагеле (60 меш), элюент метанол/хлороформ =1/5. Желтое твердое вещество, выход: 81%. Спектр ЯМР 1H (CDCl3) δ, м.д.: 2.43 (с, 3H, MeN); 2.68 (с, 6H, CH2); 3.49 (с, 2H, CH2); 4.17 (уш. с, 2H, CH2); 6.67-6.95 (м, 8Н, CHAr); 7.00-7.16 (м, 4H, CHAr).
Пример 12: Способ получения 4-(2-Метил-1.2.3,4-тетрагидро-пиридо[4,3-b]индол-5-ил)-1-(фенотиазин-10-ил)пропан-1-она (III).
В качестве исходных веществ брали 1 ммоль 2-метил-2,3,4,5-тетрагидро-1H-пиридо[4,3-b]индола, 1 ммоль 1-(3,7-бис-диметиламино-фенотиазин-10-ил)пропенона, в качестве катализатора брали 100 мг KOH в 1.5 мл ДМСО нагревали при перемешивании при 130-140°C в течение 10 ч. ДМСО удалили в вакууме 3 мм рт.ст., из остатка продукт экстрагировали хлористым метиленом. Хлористый метилен упаривали и остаток хроматографировали на силикагеле (60 меш), элюент метанол/хлороформ =1/5. Полученный продукт представляет собой желтое твердое вещество, выход: 79%. Спектр ЯМР 1H (CDCl3) δ, м.д.: 2.43 (с, 3H, MeN); 2.68 (с, 6H, CH2); 3.49 (с, 2H, CH2); 4.17 (уш. с, 2H, CH); 6.67-6.95 (м, 8H, CHAr); 7.00-7.16 (м, 4H, CHAr).
Пример 13: Способ получения 4-(2-Этил-1,2,3,4-тетрагидро-пиридо[4,3-b]индол-5-ил)-1-(фенотиазин-10-ил)пропан-1-она (IV).
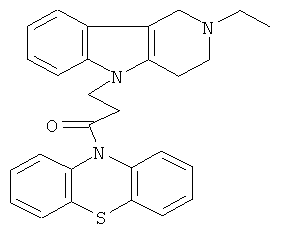
В качестве исходных веществ брали 1 ммоль 2-этил-2,3,4,5-тетрагидро-1H-пиридо[4,3-b]индола, 1 ммоль 1-(3,7-бис-диметиламино-фенотиазин-10-ил)пропенона, в качестве катализатора брали 200 мг CsF в 1.5 мл ДМСО. Нагревали при перемешивании при 120-140°C в течение 8 ч. ДМСО удалили в вакууме 3 мм рт.ст., из остатка продукт экстрагировали хлористым метиленом. Хлористый метилен упаривали и остаток хроматографировали на силикагеле (60 меш), элюент метанол/хлороформ =1/5. Полученный продукт представляет собой желтое твердое вещество, выход: 88%. Спектр ЯМР 1H (CDCl3) δ, м.д.: 1.12 (т, 3H, J=6.7, Me); 2.49 (к, 2Н, J=6.7, MeCH2); 2.65 (м, 6H, CH2); 3.46 (с, 2H, CH2); 4.09 (уш. с, 2H, CH2); 6.62-6.82 (м, 8H, CHAr); 6.90-7.05 (м, 4H, CHAr).
Пример 14: Способ получения 4-(2-Этил-1,2,3,4-тетрагидро-пиридо[4,3-b]индол-5-ил)-1-(фенотиазин-10-ил)пропан-1-она (IV).
В качестве исходных веществ брали 1 Ммоль 2-этил-2,3,4,5-тетрагидро-1H-пиридо[4,3-b]индола, 1 ммоля 1-(3,7-бис-диметиламино-фенотиазин-10-ил)пропенона, в качестве катализатора брали 300 мг MeONa в 1.5 мл ДМФА. Нагревали при перемешивании при 140-150°C в течение 6 ч. ДМФА удалили в вакууме 3 мм рт.ст., из остатка продукт экстрагировали хлористым метиленом. Хлористый метилен упаривали и остаток хроматографировали на силикагеле (60 меш), элюент метанол/хлороформ =1/5. Полученный продукт представляет собой желтое твердое вещество, выход: 83%. Спектр ЯМР 1H (CDCl3) δ, м.д.: 1.12 (т, 3H, J=6.7, Me); 2.49 (к, 2H, J=6.7, MeCH2); 2.65 (м, 6H, CH2); 3.46 (с, 2H, CH2); 4.09 (уш. с, 2H, CH2); 6.62-6.82 (м, 8H, CHAr); 6.90-7.05 (м, 4H, CHAr).
Пример 15: Способ получения 4-(2-Этил-1,2,3,4-тетрагидро-пиридо[4,3-b]индол-5-ил)-1-(фенотиазин-10-ил)пропан-1-она (IV).
В качестве исходных веществ брали 1 ммоль 2-этил-2,3,4,5-тетрагидро-1H-пиридо[4,3-b]индола, 1 ммоль 1-(3,7-бис-диметиламино-фенотиазин-10-ил)пропенона, в качестве катализатора брали 300 мг трет.-BuOK в 1.5 мл ДМСО. Нагревали при перемешивании при 120-130°C в течение 8 ч. ДМСО удалили в вакууме 3 мм рт.ст., из остатка продукт экстрагировали хлористым метиленом. Хлористый метилен упаривали и остаток хроматографировали на силикагеле (60 меш), элюент метанол/хлороформ =1/5. Полученный продукт представляет собой желтое твердое вещество, выход: 81%. Спектр ЯМР 1H (CDCl3) δ, м.д.: 1.12 (т, 3H, J=6.7, Me); 2.49 (к, 2H, J=6.7, MeCH2); 2.65 (м, 6H, CH2); 3.46 (с, 2H, CH2); 4.09 (уш. с, 2H, CH2); 6.62-6.82 (м, 8H, CHAr); 6.90-7.05 (м, 4H, CHAr).
Пример 16: Способ получения 4-(2-Этил-1,2,3,4-тетрагидро-пиридо[4.3-b]индол-5-ил)-1-(фенотиазин-10-ил)пропан-1-она (IV).
В качестве исходных веществ брали 1 ммоль 2-этил-2,3,4,5-тетрагидро-1H-пиридо[4,3-b]индола, 1 ммоль 1-(3,7-бис-диметиламино-фенотиазин-10-ил)пропенона, в качестве катализатора брали 100 мг KOH в 1.5 мл ДМФА. Нагревали при перемешивании при 130-140°C в течение 8 ч. ДМФА удалили в вакууме 3 мм рт.ст., из остатка продукт экстрагировали хлористым метиленом. Хлористый метилен упаривали и остаток хроматографировали на силикагеле (60 меш), элюент метанол/хлороформ =1/5. Полученный продукт представляет собой желтое твердое вещество, выход: 77%. Спектр ЯМР 1H (CDCl3) δ, м.д.: 1.12 (т, 3H, J=6.7, Me); 2.49 (к, 2H, J=6.7, MeCH2); 2.65 (м, 6H, CH2); 3.46 (с, 2H, CH2); 4.09 (уш. с, 2H, CH2); 6.62-6.82 (м, 8H, CHAr); 6.90-7.05 (м, 4H, CHAr).
Пример 17: Способ получения 4-(2,8-Диметил-1,2,3,4-тетрагидро-пиридо[4,3-b]индол-5-ил)-1-(фенотиазин-10-ил)пропан-1-она (V).
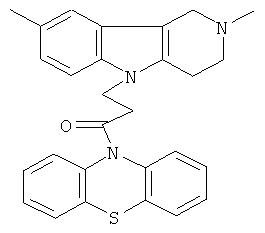
В качестве исходных веществ брали 1 ммоль 2,8-диметил-2,3,4,5-тетрагидро-1H-пиридо[4,3-b]индола, 1 ммоль 1-(3,7-бис-диметиламино-фенотиазин-10-ил)пропенона, в качестве катализатора брали 200 мг CsF в 1.5 мл ДМСО. Нагревали при перемешивании при 120-130°C в течение 12 ч. ДМСО удалили в вакууме 3 мм рт.ст., из остатка продукт экстрагировали хлористым метиленом. Хлористый метилен упаривали и остаток хроматографировали на силикагеле (60 меш), элюент метанол/хлороформ =1/5. Полученный продукт представляет собой желтое твердое вещество, выход: 85%. Спектр ЯМР 1H (CDCl3) δ, м.д.: 2.43 (с, 3H, MeC); 2.54 (с, 3H, MeN); 2.79 (м, 6H, CH2); 3.64 (с, 2H, CH2); 4.34 (уш. с, 2H, CH2); 6.92 (д, 1H, J=8.3, CHAr); 7.02 (д, 1H, J=8.3, CHAr); 7.14-7.28 (м, 6Н, CHAr); 7.36-7.47 (м, 3H, CHAr).
Пример 18: Способ получения 4-(2,8-Диметил-1,2,3,4-тетрагидро-пиридо[4,3-b]индол-5-ил)-1-(фенотиазин-10-ил)пропан-1-она (V).
В качестве исходных веществ брали 1 ммоль 2,8-диметил-2,3,4,5-тетрагидро-1H-пиридо[4,3-b]индола, 1 ммоль 1-(3,7-бис-диметиламино-фенотиазин-10-ил)пропенона, в качестве катализатора брали 300 мг MeONa в 1.5 мл ДМСО. Нагревали при перемешивании при 120-130°C в течение 8 ч. ДМСО удалили в вакууме 3 мм рт.ст., из остатка продукт экстрагировали хлористым метиленом. Хлористый метилен упаривали и остаток хроматографировали на силикагеле (60 меш), элюент метанол/хлороформ =1/5. Полученный продукт представляет собой желтое твердое вещество, выход: 83%. Спектр ЯМР 1H (CDCl3) δ, м.д.: 2.43 (с, 3H, MeC); 2.54 (с, 3H, MeN); 2.79 (м, 6H, CH2); 3.64 (с, 2H, CH2); 4.34 (уш. с, 2H, CH2); 6.92 (д, 1H, J=8.3, CHAr); 7.02 (д, 1H, J=8.3, CHAr); 7.14-7.28 (м, 6H, CHAr); 7.36-7.47 (м, 3H, CHAr).
Пример 19: Способ получения 4-(2,8-Диметил-1,2,3,4-тетрагидро-пиридо[4,3-b]индол-5-ил)-1-(фенотиазин-10-ил)пропан-1-она (V).
В качестве исходных веществ брали 1 ммоль 2,8-диметил-2,3,4,5-тетрагидро-1H-пиридо[4,3-b]индола, 1 ммоль 1-(3,7-бис-диметиламино-фенотиазин-10-ил)пропенона, в качестве катализатора брали 300 мг трет.-BuOK в 1.5 мл ДМФА. Нагревали при перемешивании при 120-140°C в течение 8 ч. ДМСО (или ДМФА) удалили в вакууме 3 мм рт.ст., из остатка продукт экстрагировали хлористым метиленом. Хлористый метилен упаривали и остаток хроматографировали на силикагеле (60 меш), элюент метанол/хлороформ =1/5. Полученный продукт представляет собой желтое твердое вещество, выход: 84%. Спектр ЯМР 1H (CDCl3) δ, м.д.: 2.43 (с, 3H, MeC); 2.54 (с, 3H, MeN); 2.79 (м, 6H, CH2); 3.64 (с, 2H, CH2); 4.34 (уш. с, 2H, CH2); 6.92 (д, 1H, J=8.3, CHAr); 7.02 (д, 1H, J=8.3, CHAr); 7.14-7.28 (м, 6H, CHAr); 7.36-7.47 (м, 3H, CHAr).
Пример 20: Способ получения 4-(2,8-Диметил-1,2,3,4-тетрагидро-пиридо[4,3-b]индол-5-ил)-1-(фенотиазин-10-ил)пропан-1-она (V).
В качестве исходных веществ брали 1 ммоль 2,8-диметил-2,3,4,5-тетрагидро-1H-пиридо[4,3-b]индола, 1 ммоль 1-(3,7-бис-диметиламино-фенотиазин-10-ил)пропенона, в качестве катализатора брали 100 мг KOH в 1.5 мл ДМФА. Нагревали при перемешивании при 140-150°C в течение 8 ч. ДМФА удалили в вакууме 3 мм рт.ст., из остатка продукт экстрагировали хлористым метиленом. Хлористый метилен упаривали и остаток хроматографировали на силикагеле (60 меш), элюент метанол/хлороформ =1/5. Полученный продукт представляет собой желтое твердое вещество, выход: 80%. Спектр ЯМР 1H (CDCl3) δ, м.д.: 2.43 (с, 3H, MeC); 2.54 (с, 3H, MeN); 2.79 (м, 6H, CH2); 3.64 (с, 2H, CH2); 4.34 (уш. с, 2H, CH2); 6.92 (д, 1H, J=8.3, CHAr); 7.02 (д, 1H, J=8.3, CHAr); 7.14-7.28 (м, 6H, CHAr); 7.36-7.47 (м, 3H, CHAr).
Пример 21: Способ получения 4-(8-Метил-2-этил-1,2,3,4-тетрагидропиридо[4,3-b]индол-5-ил)-1-(фенотиазин-10-ил)пропан-1-она (VI).
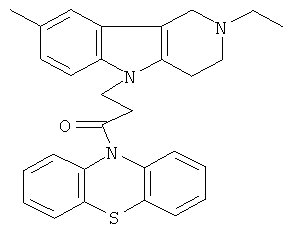
В качестве исходных веществ брали 1 ммоль 8-метил-2-этил-2,3,4,5-тетрагидро-1H-пиридо[4,3-b]индола, 1 ммоль 1-(3,7-бис-диметиламино-фенотиазин-10-ил)пропенона, в качестве катализатора брали 200 мг CsF в 1.5 мл ДМСО. Нагревали при перемешивании при 120-130°C в течение 8 ч. ДМСО удалили в вакууме 3 мм рт.ст., из остатка продукт экстрагировали хлористым метиленом. Хлористый метилен упаривали и остаток хроматографировали на силикагеле (60 меш), элюент метанол/хлороформ =1/5. Полученный продукт представляет собой желтое твердое вещество, выход: 84%. Спектр ЯМР 1H (CDCl3) δ, м.д.: 1.21 (т, 3H, J=7.1, MeCH2); 2.42 (с, 3H, MeC); 2.65 (к, 2H, J=6.7, MeCH2); 2.80 (м, 6H, CH2); 3.65 (с, 2H, CH2); 4.32 (уш. с, 2H, CH2); 6.92 (д, 1H, J=8.4, CH2); 7.00 (д, 1H, J=8.4, CHAr); 7.08-7.28 (м, 7H, CHAr); 7.33-7.44 (м, 2H, CHAr).
Пример 22: Способ получения 4-(8-Метил-2-этил-1,2,3,4-тетрагидропиридо[4,3-b]индол-5-ил)-1-(фенотиазин-10-ил)пропан-1-она
В качестве исходных веществ брали 1 ммоль 8-метил-2-этил-2,3,4,5-тетрагидро-1H-пиридо[4,3-b]индола, 1 ммоль 1-(3,7-бис-диметиламино-фенотиазин-10-ил)пропенона, в качестве катализатора брали 300 мг MeONa в 1.5 мл ДМСО. Нагревали при перемешивании при 130-140°C в течение 8 ч. ДМСО (или ДМФА) удалили в вакууме 3 мм рт.ст., из остатка продукт экстрагировали хлористым метиленом. Хлористый метилен упаривали и остаток хроматографировали на силикагеле (60 меш), элюент метанол/хлороформ =1/5. Полученный продукт представляет собой желтое твердое вещество, выход: 81%. Спектр ЯМР 1H (CDCl3) δ, м.д.: 1.21 (т, 3H, J=7.1, МеСН2); 2.42 (с, 3H, MeC); 2.65 (к, 2H, J=6.7, MeCH2); 2.80 (м, 6H, CH2); 3.65 (с, 2H, CH2); 4.32 (уш. с, 2H, CH2); 6.92 (д, 1H, J=8.4, CHAr); 7.00 (д, 1H, J=8.4, CHAr); 7.08-7.28 (м, 7H, CHAr); 7.33-7.44 (м, 2H, CHAr).
Пример 23: Способ получения 4-(8-Метил-2-этил-1,2,3,4-тетрагидропиридо[4,3-b]индол-5-ил)-1-(фенотиазин-10-ил)пропан-1-она (IV).
В качестве исходных веществ брали 1 ммоль 8-метил-2-этил-2,3,4,5-тетрагидро-1H-пиридо[4,3-b]индола, 1 ммоль 1-(3,7-бис-диметиламино-фенотиазин-10-ил)пропенона, в качестве катализатора брали 300 мг трет.-BuOK в 1.5 мл ДМФА. Нагревали при перемешивании при 120-130°C в течение 8 ч. ДМФА удалили в вакууме 3 мм рт.ст., из остатка продукт экстрагировали хлористым метиленом. Хлористый метилен упаривали и остаток хроматографировали на силикагеле (60 меш), элюент метанол/хлороформ =1/5. Полученный продукт представляет собой желтое твердое вещество, выход: 86%. Спектр ЯМР 1H (CDCl3) δ, м.д.: 1.21 (т, 3H, J=7.1, MeCH2); 2.42 (с, 3H, MeC); 2.65 (к, 2H, J=6.7, MeCH2); 2.80 (м, 6H, CH2); 3.65 (с, 2H, CH2); 4.32 (уш. с, 2H, CH2); 6.92 (д, 1H, J=8.4, CHAr); 7.00 (д, 1Н, J=8.4, CHAr); 7.08-7.28 (м, 7Н, CHAr); 7.33-7.44 (м, 2H, CHAr).
Пример 24: Способ получения 4-(8-Метил-2-этил-1,2,3,4-тетрагидропиридо[4,3-b]индол-5-ил)-1-(фенотиазин-10-ил)пропан-1-она (VI).
В качестве исходных веществ брали 1 ммоль 8-метил-2-этил-2,3,4,5-тетрагидро-1H-пиридо[4,3-b]индола, 1 ммоль 1-(3,7-бис-диметиламино-фенотиазин-10-ил)пропенона, в качестве катализатора брали 100 мг KOH в 1.5 мл ДМФА. Нагревали при перемешивании при 130-140°C в течение 8 ч. ДМФА удалили в вакууме 3 мм рт.ст., из остатка продукт экстрагировали хлористым метиленом. Хлористый метилен упаривали и остаток хроматографировали на силикагеле (60 меш), элюент метанол/хлороформ =1/5. Полученный продукт представляет собой желтое твердое вещество, выход: 82%. Спектр ЯМР 1H (CDCl3) δ, м.д.: 1.21 (т, 3H, J=7.1, MeCH2); 2.42 (с, 3H, MeC); 2.65 (к, 2H, J=6.7, MeCH2); 2.80 (м, 6H, CH2); 3.65 (с, 2H, CH2); 4.32 (уш. с, 2H, CH2); 6.92 (д, 1H, J=8.4, CH2); 7.00 (д, 1H, J=8.4, CHAr); 7.08-7.28 (м, 7Н, CHAr); 7.33-7.44 (м, 2H, CHAr).
Пример 25: Способ получения 4-(2-Метил-8-фтор-1,2,3-4-тетрагидропиридо[4,3-b]индол-5-ил)-1-(фенотиазин-10-ил)пропан-1-она (VII).
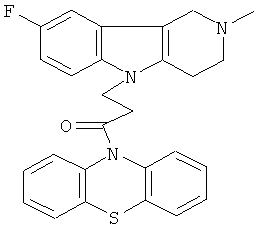
В качестве исходных веществ брали 1 ммоль 2-метил-8-фтор-2,3,4,5-тетрагидро-1H-пиридо[4,3-b]индола, 1 ммоль 1-(3,7-бис-диметиламино-фенотиазин-10-ил)пропенона, в качестве катализатора брали 200 мг CsF в 1.5 мл ДМСО. Нагревали при перемешивании при 130-140°C в течение 8 ч. ДМСО удалили в вакууме 3 мм рт.ст., из остатка продукт экстрагировали хлористым метиленом. Хлористый метилен упаривали и остаток хроматографировали на силикагеле (60 меш), элюент метанол/хлороформ = 1/5. Полученный продукт представляет собой желтое твердое вещество, выход: 78%. Спектр ЯМР 1H (CDCl3) δ, м.д.: 2.44 (с, 3H, MeN); 2.65 (м, 4H, CH2); 2.72 (м, 2Н, CH2); 3.49 (с, 2H, CH2); 4.25 (уш. с, 2H, CH2); 6.79 (дт, 1H, J=9.0, 2.2, CHAr); 6.94-7.11 (м, 7Н, CHAr); 7.23-7.30 (м, 2H, CHAr); 7.54 (д, 1H, J=2.2, CHAr).
Пример 26: Способ получения 4-(2-Метил-8-Фтор-1,2,3,4-тетрагидропиридо[4,3-b]индол-5-ил)-1-(фенотиазин-10-ил)пропан-1-она (VII).
В качестве исходных веществ брали 1 ммоль 2-метил-8-фтор-2,3,4,5-тетрагидро-1H-пиридо[4,3-b]индола, 1 ммоль 1-(3,7-бис-диметиламино-фенотиазин-10-ил)пропенона, в качестве катализатора брали 300 мг MeONa в 1.5 мл ДМФА. Нагревали при перемешивании при 130-140°C в течение 8 ч. ДМФА удалили в вакууме 3 мм рт.ст., из остатка продукт экстрагировали хлористым метиленом. Хлористый метилен упаривали и остаток хроматографировали на силикагеле (60 меш), элюент метанол/хлороформ =1/5. Полученный продукт представляет собой желтое твердое вещество, выход: 81%. Спектр ЯМР 1H (CDCl3) δ, м.д.: 2.44 (с, 3H, MeN); 2.65 (м, 4H, CH2); 2.72 (м, 2H, CH2); 3.49 (с, 2H, CH2); 4.25 (уш. с, 2H, CH2); 6.79 (дт, 1H, J=9.0, 2.2, CHAr); 6.94-7.11 (м, 7Н, CHAr); 7.23-7.30 (м, 2Н, CHAr); 7.54 (д, 1H, J=2.2, CHAr).
Пример 27: Способ получения 4-(2-Метил-8-фтор-1,2,3,4-тетрагидропиридо[4,3-b]индол-5-ил)-1-(фенотиазин-10-ил)пропан-1-она (VII).
В качестве исходных веществ брали 1 ммоль 2-метил-8-фтор-2,3,4,5-тетрагидро-1H-пиридо[4,3-b]индола, 1 ммоля 1-(3,7-бис-диметиламино-фенотиазин-10-ил)пропенона, в качестве катализатора брали 300 мг трет.-BuOK 1.5 мл ДМСО. Нагревали при перемешивании при 130-140°C в течение 8 ч. ДМСО удалили в вакууме 3 мм рт.ст., из остатка продукт экстрагировали хлористым метиленом. Хлористый метилен упаривали и остаток хроматографировали на силикагеле (60 меш), элюент метанол/хлороформ =1/5. Полученный продукт представляет собой желтое твердое вещество, выход: 82%. Спектр ЯМР 1H (CDCl3) δ, м.д.: 2.44 (с, 3H, MeN); 2.65 (м, 4H, CH2); 2.72 (м, 2H, CH2); 3.49 (с, 2H, CH2); 4.25 (уш. с, 2H, CH2); 6.79 (дт, 1H, J=9.0, 2.2, CHAr); 6.94-7.11 (м, 7H, CHAr); 7.23-7.30 (м, 2H, CHAr); 7.54 (д, 1H, J=2.2, CHAr).
Пример 28: Способ получения 4-(2-Метил-8-фтор-1,2,3,4-тетрагидропиридо[4,3-b]индол-5-ил)-1-(фенотиазин-10-ил)пропан-1-она (VII).
В качестве исходных веществ брали 1 ммоль 2-метил-8-фтор-2,3,4,5-тетрагидро-1H-пиридо[4,3-b]индола, 1 ммоля 1-(3,7-бис-диметиламино-фенотиазин-10-ил)пропенона, в качестве катализатора брали 100 мг KOH в 1.5 мл ДМФА. Нагревали при перемешивании при 120-130°C в течение 8 ч. ДМФА удалили в вакууме 3 мм рт.ст., из остатка продукт экстрагировали хлористым метиленом. Хлористый метилен упаривали и остаток хроматографировали на силикагеле (60 меш), элюент метанол/хлороформ =1/5. Полученный продукт представляет собой желтое твердое вещество, выход: 75%. Спектр ЯМР 1H (CDCl3) δ, м.д.: 2.44 (с, 3H, MeN); 2.65 (м, 4H, CH2); 2.72 (м, 2H, CH2); 3.49 (с, 2H, CH2); 4.25 (уш. с, 2H, CH2); 6.79 (дт, 1H, J=9.0, 2.2, CHAr); 6.94-7.11 (м, 7H, CHAr), 7.23-7.30 (м, 2H, CHAr); 7.54 (д, 1H, J=2.2, CHAr).
Пример 29: Способ получения 1-(3,7-Бис-диметиламино-фенотиазин-10-ил)-3-(2,8-диметил-1,2,3,4-тетрагидропиридо[4,3-b]индол-5-ил)пропан-1-она (VIII).
В качестве исходных веществ брали 1 ммоль 2,8-диметил-2,3,4,5-тетрагидро-1H-пиридо[4,3-b]индола, 1 ммоль 1-(3,7-бис-диметиламино-фенотиазин-10-ил)пропенона, в качестве катализатора брали 200 мг CsF в 1.5 мл ДМСО. Нагревали при перемешивании при 140-150°C в течение 6 ч. ДМСО удалили в вакууме 3 мм рт.ст., из остатка продукт экстрагировали хлористым метиленом. Хлористый метилен упаривали и остаток хроматографировали на силикагеле (60 меш), элюент метанол/хлороформ =1/5. Полученный продукт представляет собой желтое твердое вещество, выход: 83%. Спектр ЯМР 1H (CDCl3) δ, м.д.: 2.43 (с, 3H, MeC); 2.54 (с, 3H, MeN); 2.79 (м, 6H, CH2); 2.96 (с, 12H, Me); 3.64 (с, 2H, CH2); 4.34 (уш. с, 2H, CH2); 6.92 (д, 1H, J=8.3, CHAr); 7.02 (д, 1H, J=8.3, CHAr); 7.14-7.28 (м, 4Н, CHAr); 7.36-7.47 (м, 3H, CHAr).
Пример 30: Способ получения 1-(3,7-Бис-диметиламино-фенотиазин-10-ил)-3-(2,8-диметил-1,2,3,4-тетрагидропиридо[4,3-b]индол-5-ил)пропан-1-она (VIII).
В качестве исходных веществ брали 1 ммоль 2,8-диметил-2,3,4,5-тетрагидро-1H-пиридо[4,3-b]индола, 1 ммоль 1-(3,7-бис-диметиламино-фенотиазин-10-ил)пропенона, в качестве катализатора брали 300 мг MeONa в 1.5 мл ДМСО. Нагревали при перемешивании при 130-140°C в течение 12 ч. ДМСО удалили в вакууме 3 мм рт.ст., из остатка продукт экстрагировали хлористым метиленом. Хлористый метилен упаривали и остаток хроматографировали на силикагеле (60 меш), элюент метанол/хлороформ =1/5. Полученный продукт представляет собой желтое твердое вещество, выход: 81%. Спектр ЯМР 1H (CDCl3) δ, м.д.: 2.43 (с, 3H, MeC); 2.54 (с, 3H, MeN); 2.79 (м, 6H, CH2); 2.96 (с, 12H, Me); 3.64 (с, 2H, CH2); 4.34 (уш. с, 2H, CH2); 6.92 (д, 1H, J=8.3, CHAr); 7.02 (д, 1H, J=8.3, CHAr); 7.14-7.28 (м, 4H, CHAr); 7.36-7.47 (м, 3H, CHAr).
Пример 31: Способ получения 1-(3,7-Бис-диметиламино-фенотиазин-10-ил)-3-(2,8-диметил-1,2,3,4-тетрагидропиридо[4,3-b]индол-5-ил)пропан-1-она (VIII).
В качестве исходных веществ брали 1 ммоль 2,8-диметил-2,3,4,5-тетрагидро-1H-пиридо[4,3-b]индола, 1 ммоль 1-(3,7-бис-диметиламино-фенотиазин-10-ил)пропенона, в качестве катализатора брали 300 мг трет.-BuOK в 1.5 мл ДМСО. Нагревали при перемешивании при 120-130°C в течение 10 ч. ДМСО удалили в вакууме 3 мм рт.ст., из остатка продукт экстрагировали хлористым метиленом. Хлористый метилен упаривали и остаток хроматографировали на силикагеле (60 меш), элюент метанол/хлороформ =1/5. Полученный продукт представляет собой желтое твердое вещество, выход: 83%. Спектр ЯМР 1H (CDCl3) δ, м.д.: 2.43 (с, 3H, MeC); 2.54 (с, 3H, MeN); 2.79 (м, 6H, CH2); 2.96 (с, 12H, Me); 3.64 (с, 2H, CH2); 4.34 (уш. с, 2H, CH2); 6.92 (д, 1H, J=8.3, CHAr); 7.02 (д, 1H, J=8.3, CHAr); 7.14-7.28 (м, 4H, CHAr); 7.36-7.47 (м, 3H, CHAr).
Пример 32: Способ получения 1-(3,7-Бис-диметиламино-фенотиазин-10-ил)-3-(2,8-диметил-1,2,3,4-тетрагидропиридо[4,3-b]индол-5-ил)пропан-1-она (VIII).
В качестве исходных веществ брали 1 ммоль 2,8-диметил-2,3,4,5-тетрагидро-1H-пиридо[4,3-b]индола, 1 ммоль 1-(3,7-бис-диметиламино-фенотиазин-10-ил)пропенона, в качестве катализатора брали 100 мг KOH в 1.5 мл ДМФА. Нагревали при перемешивании при 130-140°C в течение 12 ч. ДМФА удалили в вакууме 3 мм рт.ст., из остатка продукт экстрагировали хлористым метиленом. Хлористый метилен упаривали и остаток хроматографировали на силикагеле (60 меш), элюент метанол/хлороформ =1/5. Полученный продукт представляет собой желтое твердое вещество, выход: 82%. Спектр ЯМР 1H (CDCl3) δ, м.д.: 2.43 (с, 3H, MeC); 2.54 (с, 3H, MeN); 2.79 (м, 6H, CH2); 2.96 (с, 12H, Me); 3.64 (с, 2H, CH2); 4.34 (уш. с, 2H, CH2); 6.92 (д, 1H, J=8.3, CHAr); 7.02 (д, 1H, J=8.3, CHAr); 7.14-7.28 (м, 4H, CHAr); 7.36-7.47 (м, 3H, CHAr).
Синтез соединений общей формулы 1.2 (общая методика)
Процесс проводят нагреванием эквимольной смеси гамма-карболинов общей формулы 2 и эпоксидсодержащих фенотиазинов общей формулы 5 (схема 3) в диметилсульфоксиде или диметилформамиде в присутствии катализатора, выбранного из ряда: метилат натрия (MeONa), третбутилат калия (трет.-BuOK), гидроокись калия (KOH) и фторид цезия (CsF) при температуре 100-140°C в течение 2-12 часов.
Схема 3.
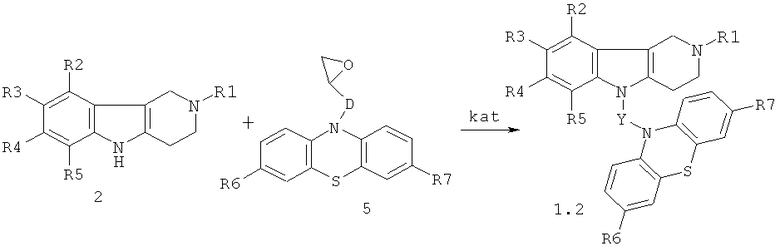
R1=H, (C1-C6)алкил;
R2, R, R4, R5=H, F, O, Br, (C1-C6)алкил, (C1-C6)алкокси,
R6, R7=. H, F, Cl, Br, (C1-C6)алкил, (C1-C6)алкокси, NH2, NHAlkyl, NAlkyl2;
D=CH2; CH2NHCH2CH2; CH2N[(C1-C6)алкил]CH2CH2
Y=CH2CH(OH)CH2, CH2CH(OH)CH2NHCH2CH2, CH2CH(OH)CH2N[(C1-C6)алкил]CH2CH2,
kat=MeONa, трет.-BuOK), KOH, CsF.
Пример 33: Способ получения 1-(3,7-Бис-диметиламинофенотиазин-10-ил)-3-(2,8-диметил-1,2,3,4-тетрагидропиридо[4,3-b]индол-5-ил)-пропан-2-она (IX).
В качестве исходных веществ брали 1 ммоль 2,8-диметил-2,3,4,5-тетрагидро-1H-пиридо[4,3-b]индола, 1 ммоль N,N,N,N-тетраметил-10-оксиранилметил-10Н-фенотиазин-3,7,-диамина, в качестве катализатора брали 200 мг CsF, мг в 1.5 мл ДМСО. Нагревали при перемешивании при 100-120°С в течение 6 ч. ДМСО удалили в вакууме 3 мм рт.ст., из остатка продукт экстрагировали хлористым метиленом. Хлористый метилен упаривали и остаток хроматографировали на силикагеле (60 меш), элюент метанол/хлороформ =1/5. Полученный продукт представляет собой желтое твердое вещество, выход: 88%. Спектр ЯМР 1H (CDCl3) δ, м.д.: 2.50 и 2.52 оба с (3H, Me); 2.64 т (2H, CH2, J=6.4 Гц); 2.70-2.85 м (4H, CH2); 2.95 (с, 12H, Me); 3.16 (дд, 1H, J=13.3, 6.2 Гц), 3.30 (дд, 1H, J=13.3, 6.2 Гц), 3.42 (м, 1H), 4.30-4.43 (м, 3H), 6.70-7.00 (м, 2H), 7.01-7.26 (м, 3H), 7.30 (д, 2H, J=8.8 Гц), 7.56 (д, 2H, J=2.2 Гц).
Пример 34: Способ получения 1-(3,7-Бис-диметиламинофенотиазин-10-ил)-3-(2,8-диметил-1,2,3,4-тетрагидропиридо[4,3-b]индол-5-ил)-пропан-2-она (IX).
В качестве исходных веществ брали 1 ммоль 2,8-диметил-2,3,4,5-тетрагидро-1H-пиридо[4,3-b]индола, 1 ммоль N,N,N,N-тетраметил-10-оксиранилметил-10Н-фенотиазин-3,7,-диамина, в качестве катализатора брали 200 мг MeONa в 1.5 мл ДМФА. Нагревали при перемешивании при 110-120°C в течение 8 ч. ДМФА удалили в вакууме 3 мм рт.ст., из остатка продукт экстрагировали хлористым метиленом. Хлористый метилен упаривали и остаток хроматографировали на силикагеле (60 меш), элюент метанол/хлороформ =1/5. Полученный продукт представляет собой желтое твердое вещество, выход: 83%. Спектр ЯМР 1H (CDCl3) δ, м.д.: 2.50 и 2.52 оба с (3H, Me); 2.64 т (2H, CH2, J=6.4 Гц); 2.70-2.85 м (4H, CH2); 2.95 (с, 12H, Me); 3.16 (дд, 1H, J=13.3, 6.2 Гц), 3.30 (дд, 1H, J=13.3, 6.2 Гц), 3.42 (м, 1H), 4.30-4.43 (м, 3H), 6.70-7.00 (м, 2H), 7.01-7.26 (м, 3H), 7.30 (д, 2H, J=8.8 Гц), 7.56 (д, 2H, J=2.2 Гц).
Пример 35: Способ получения 1-(3,7-Бис-диметиламинофенотиазин-10-ил)-3-(2,8-диметил-1,2,3,4-тетрагидропиридо[4,3-b]индол-5-ил)-пропан-2-она (IX).
В качестве исходных веществ брали 1 ммоль 2,8-диметил-2,3,4,5-тетрагидро-1H-пиридо[4,3-b]индола, 1 ммоль N,N,N,N-тетраметил-10-оксиранилметил-10Н-фенотиазин-3,7,-диамина, в качестве катализатора брали 200 мг трет.-BuOK 1.5 мл ДМСО. Нагревали при перемешивании при 100-120°C в течение 12 ч. ДМСО удалили в вакууме 3 мм рт.ст., из остатка продукт экстрагировали хлористым метиленом. Хлористый метилен упаривали и остаток хроматографировали на силикагеле (60 меш), элюент метанол/хлороформ =1/5. Полученный продукт представляет собой желтое твердое вещество, выход: 80%. Спектр ЯМР 1H (CDCl3) δ, м.д.: 2.50 и 2.52 оба с (3H, Me); 2.64 т (2H, CH2, J=6.4 Гц); 2.70-2.85 м (4H, CH2); 2.95 (с, 12H, Me); 3.16 (дд, 1H, J=13.3, 6.2 Гц), 3.30 (дд, 1H, J=13.3, 6.2 Гц), 3.42 (м, 1H), 4.30-4.43 (м, 3H), 6.70-7.00 (м, 2H), 7.01-7.26 (м, 3H), 7.30 (д, 2H, J=8.8 Гц), 7.56 (д, 2H, J=2.2 Гц).
Пример 36: Способ получения 1-(3,7-Бис-диметиламинофенотиазин-10-ил)-3-(2,8-диметил-1,2,3,4-тетрагидропиридо[4,3-b]индол-5-ил)-пропан-2-она (IX).
В качестве исходных веществ брали 1 ммоль 2,8-диметил-2,3,4,5-тетрагидро-1H-пиридо[4,3-b]индола, 1 ммоль N,N,N,N-тетраметил-10-оксиранилметил-10H-фенотиазин-3,7,-диамина, в качестве катализатора брали в качестве катализатора брали 200 мг КОН в 1.5 мл ДМФА. Нагревали при перемешивании при 100-110°C в течение 2 ч. ДМФА удалили в вакууме 3 мм рт.ст., из остатка продукт экстрагировали хлористым метиленом. Хлористый метилен упаривали и остаток хроматографировали на силикагеле (60 меш), элюент метанол/хлороформ =1/5. Полученный продукт представляет собой желтое твердое вещество, выход: 76%. Спектр ЯМР 1H (CDCl3) δ, м.д.: 2.50 и 2.52 оба с (3H, Me); 2.64 т (2H, CH2, J=6.4 Гц); 2.70-2.85 м (4H, CH2); 2.95 (с, 12H, Me); 3.16 (дд, 1H, J=13.3, 6.2 Гц), 3.30 (дд, 1H, J=13.3, 6.2 Гц), 3.42 (м, 1H), 4.30-4.43 (м, 3H), 6.70-7.00 (м, 2H), 7.01-7.26 (м, 3H), 7.30 (д, 2H, J=8.8 Гц), 7.56 (д, 2H, J=2.2 Гц)
Синтез соединений формулы 1.3 (общая методика)
Процесс проводят смешением эквимольных количеств гидроксилсодержащих конъюгатов гамма-карболинов и фенотиазинов общей формулы 6 и диэтиламиносульфотрифторида (Et2NSF3) (схема 4) в диметилсульфоксиде или диметилформамиде при температурах (-78)-0°C в течение 1-3 часов:
Схема 4.
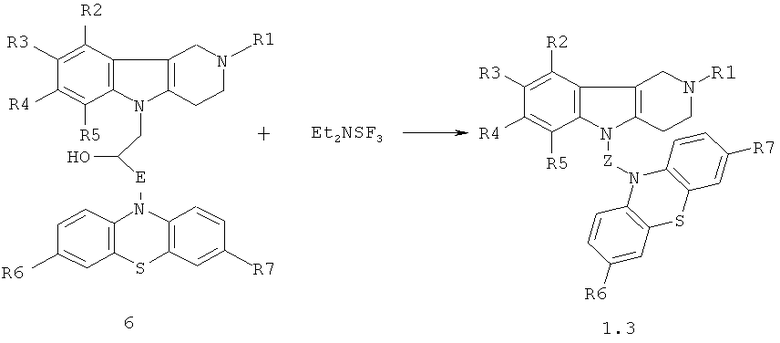
R1=H, (C1-C6)алкил;
R2, R3, R4, R5=H, F, Cl, Br, (C1-C6)алкил, (C1-C6)алкокси,
R6, R7=. H, F, Cl, Br, (C1-C6)алкил, (C1-C6)алкокси, NH2, NHAlkyl, NAlkyl2;
E=CH2; CH2NHCH2CH2; CH2N[(C1-C6)алкил]СН2СН2.
Z=CH2CHFCH2NHCH2CH2, CH2CHFCH2N[(C1-C6)алкил]СН2СН2.
Пример 37: Способ получения 10-[3-(2,8-Диметил-1,2,3,4-тетрагидропиридо[4,3-b]индол-5-ил)-2-фторпропил]-N,N,N,N-тетраметил-10H-фенотиазин-3,7-диамина (X).
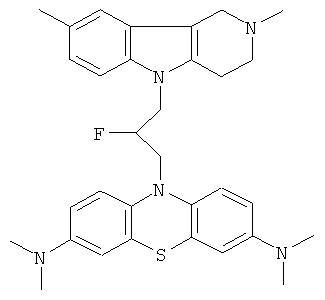
В качестве исходного вещества брали 1 ммоль 1-(3,7-Бис-диметиламинофенотиазин-10-ил)-3-(2,8-диметил-1,2,3,4-тетрагидропиридо[4,3-b]индол-5-ил)-пропан-2-она в 3.0 мл ДМСО и 1 ммоль диэтиламиносульфотрифторида. Реагенты смешивали при температуре (-78)°C. Реакционную массу перемешивали 3 часа, повышая температуру до 0°C, выливали в 10 мл воды, экстрагировали хлористым метиленом. Хлористый метилен упаривали и остаток хроматографировали на силикагеле (60 меш), элюент метанол/хлороформ =1/5. Полученный продукт представляет собой желтое твердое вещество, выход: 63%. Спектр ЯМР 1H (CDCl3) δ, м.д.: 2.50 и 2.53 оба с (3H, Me); 2.65 т (2H, CH2, J=6.4 Гц); 2.70-2.85 м (4H, CH2); 2.94 (с, 12H, Me); 3.16 (дд, 1H, J=13.3, 6.2 Гц), 3.30 (дд, 1H, J=13.3, 6.2 Гц), 3.42 (м, 1H), 3.90 (т, 1H, J=6.3 Гц), 4.40 (м, 1H), 5.10 (дм, 1H, JHF=48 Гц), 6.88 д (1H, CHAr, J=8.8 Гц); 6.90 (дд, 2H, J=8.8, 2.2 Гц), 7.01-7.20 (м, 2H), 7.31 (д, 2H, J=8.8 Гц), 7.54 (д, 2H, J=2.2 Гц).
Синтез хлоргидратов соединений формулы 1.1, 1.2, 1.3 (общая методика).
Процесс проводят растворением соединений формулы 1.1, 1.2, 1.3 в 10%-ном растворе HCl в изопропаноле при температуре 50°C.
Пример 38: Способ получения Хлоргидрата 4-(2,8-диметил-1,2,3,4-тетрагидропиридо[4,3-b]индол-5-ил)-1-(фенотиазин-10-ил)пропан-1-она (XI).
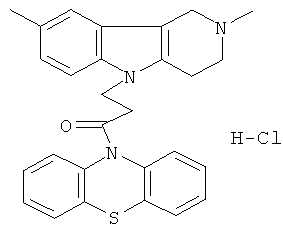
В качестве исходного вещества берут 1 ммоль 4-(2,8-диметил-1,2,3,4-тетрагидропиридо[4,3-b]индол-5-ил)-1-фенотиазин-10-ил-пропан-1-она, растворяют в 2 мл 10%-ного раствора HCl в изопропаноле при температуре 50°C. Охлаждают до 0°C, выпавший осадок отфильтровывают. Полученный продукт представляет собой белое твердое вещество, выход: 95%. Спектр ЯМР 1H (CDCl3) δ, м.д.: 2.59 (с, 3H, МеС); 2.65 (с, 3H, MeN); 2.73-2.97 (м, 4H, CH2); 3.03-3.16 (м, 2H, CH2); 3.44-3.65 (м, 2H, CH2); 4.33 (уш. с, 2H, CH2); 6.96-7.15 (м, 2H, CHAr); 7.19-7.32 (м, 5H, CHAr); 7.38-7.49 (м, 4H, CHAr); 10.90 (с, 1H).
Еще одним аспектом изобретения является фармакологическое средство для снижения неконтролируемой агрегации белков в нервной системе при лечении нейродегенеративных заболеваний, содержащее активное начало и фармацевтически приемлемый носитель, новизна которого заключается в том, что в качестве активного начала используется эффективное количество соединения формулы 1.
Понятие «фармакологическое средство» подразумевает использование любой лекарственной формы, содержащей соединение формулы 1, которая могла бы найти профилактическое или лечебное применение в медицине в качестве средства для снижения неконтролируемой агрегации белков в нервной системе
Понятие «эффективное количество», используемое в данной заявке, подразумевает использование того количества соединений формулы 1, которое в сочетании с его показателями активности и токсичности, а также на основании знаний специалиста должно быть эффективным в данной лекарственной форме.
Для получения фармакологического средства одно или несколько соединений формулы 1 смешиваются как активный ингредиент с фармацевтически приемлемым носителем, известным в медицине, согласно принятым в фармацевтике способам. В зависимости от лекарственной формы препарата носитель может иметь различные виды.
Техническим эффектом настоящего изобретения является создание новых соединений, в которых объединены гамма-карболиновое ядро и фенотиазиновое ядро для получения средства для снижения неконтролируемой агрегации белков в нервной системе, что позволяет увеличить эффективность лечения нейродегенеративных заболеваний.
В Таблице 1 представлены результаты 5 независимых экспериментов, в которых было изучено влияние заявляемых веществ на содержание крупных Т5 включений после трансфекции клеточной культуры SH-SY5Y.
В Таблице 2 представлено влияние хронического введения заявляемых соединений на число амилоидных включений в переднем роге грудного отдела спинного мозга у гамма-синуклеин трансгенных мышей на симптоматической стадии протеинопатии.
Примечание: * - уровень значимости (p-level), в сравнении с группой соединения II,
На Фиг.1 представлены репрезентативные фотографии гистопатологических отложений на поперечных срезах через шейный отдел спинного мозга мышей линии Thy1mgSN; где А - Амилоидные отложения, окрашенные Конго красным, Б - Нейроны, содержащие скопления гиперфосфорилированного белка tau. Гистопатологические отложения указаны стрелкой.
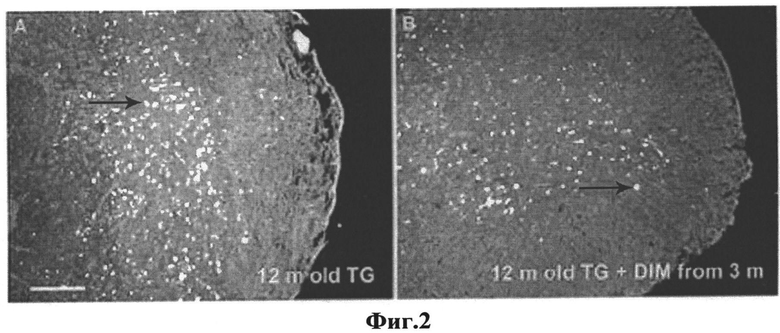
На Фиг.2 представлены изображения гистологических срезов спинного мозга 12-месячных контрольных Thy1mγSN мышей (А) и Thy1mγSN мышей, получавших соединение I с 3-х месячного возраста (В). Шкала =100 мкм.
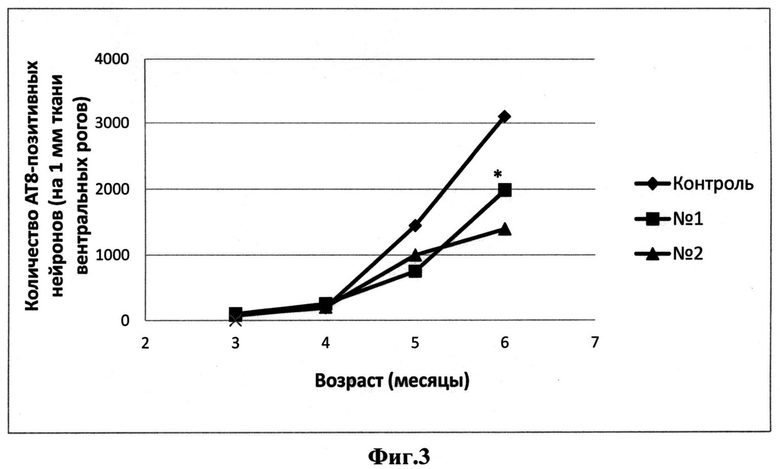
На Фиг.3 представлены нейроны с включениями гиперфосфорилированного белка tau у контрольной и опытных групп мышей линии tauP301S. * - р<0,05, критерий Манна-Уитни.
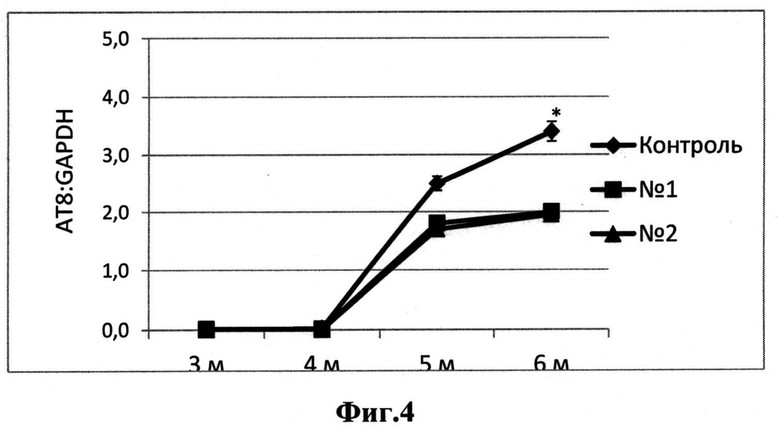
На Фиг.4 представлен количественный анализ гиперфосфорилированного белка tau в спинном мозге мышей линии tauP301S, принимавших соединение I (квадраты), соединение II (треуголиники) и контрольной группы (ромб) разного возраста (3-6 месяцев). Нормализация по GAPDH. * - р<0,05, критерий Манна-Уитни.
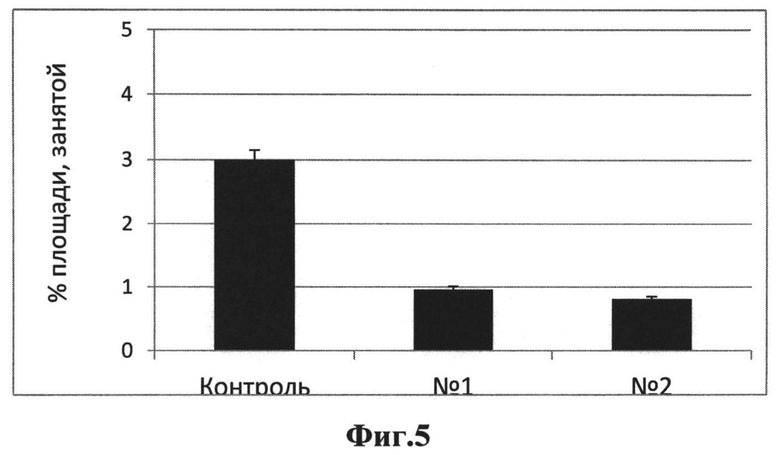
На Фиг.5 представлен эффект исследуемых соединений на амилоидную нагрузку в коре головного мозга 12-месячных мышей линии 5xFAD, получавших препараты с возраста 3 месяцев, по сравнению с контрольной группой, не получавшей препарата (кора головного мозга).
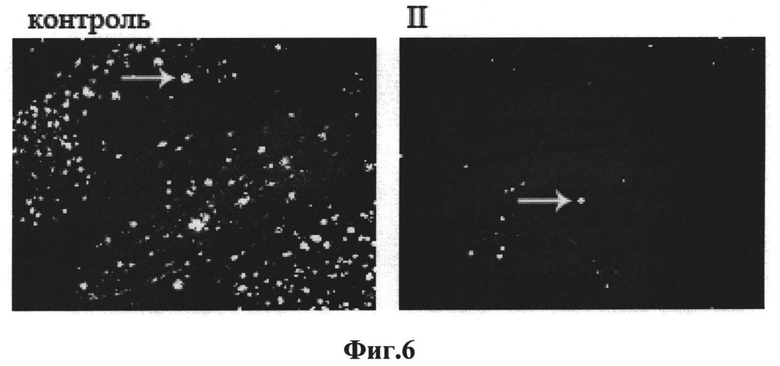
На Фиг.6 представлены включения амилоидного типа в коре головного мозга мышей линии 5xFAD в возрасте 12 месяцев, получавших (II) и не получавших (контроль) соединение II.
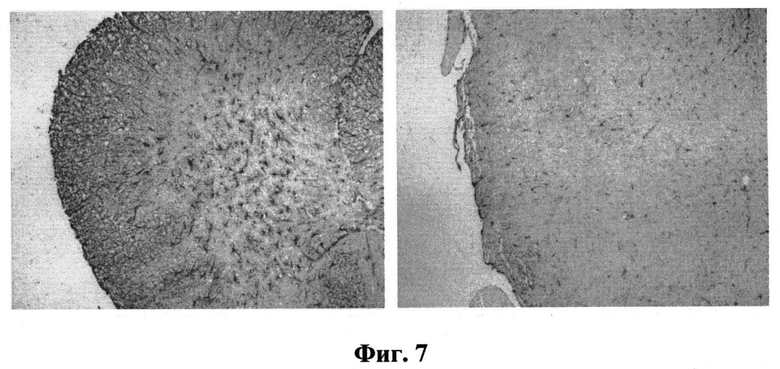
На Фиг.7 представлено влияние соединения I на выраженность астроглиоза (маркер GFAP) в линии Thy1mgSN (препарат с возраста 3 месяцев до 9 месяцев).
На Фиг.8 представлено влияние вещества II на выраженность астроглиоза (маркер GFAP) в спинном мозге мышей линии P301S в контрольной группе (А) и после получения получения соединения II с возраста 1 месяцев до 5 месяцев (Б) по данным иммуногистохимии.
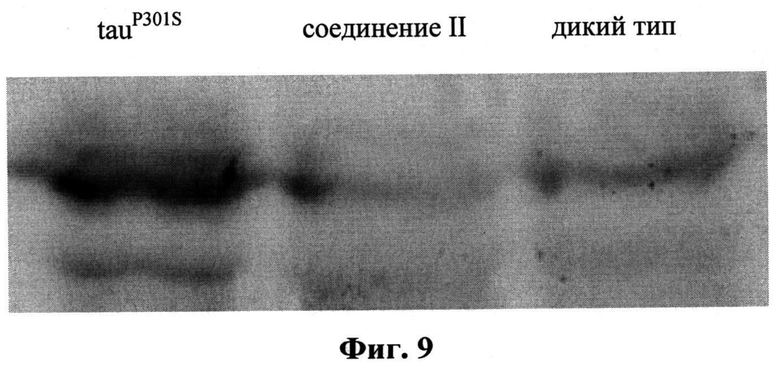
На Фиг.9 представлены данные о влиянии препарата II на выраженность астроглиоза (маркер GFAP) в спинном мозге мышей линии P301S, принимавших препарат II с возраста 1 месяцев до 5 месяцев (Б), по сравнению с контрольными нетрансгенными мышами (А) по данным иммуноблоттинга.
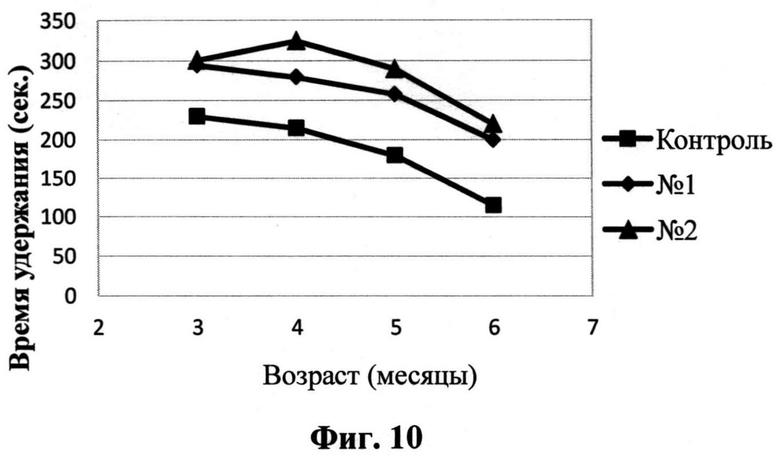
На Фиг.10 приведены результаты теста оценки баланса и координации на вращающемся стержне животных в разные возрастные периоды. Квадрат - контрольная группа трансгенных животных; Ромб - опытная группа трансгенных животных, получавших соединение I; Треугольник - опытная группа трансгенных животных, получавших соединение II. На графике представлено среднее время удержания на стержне в группе на каждый срок измерения.
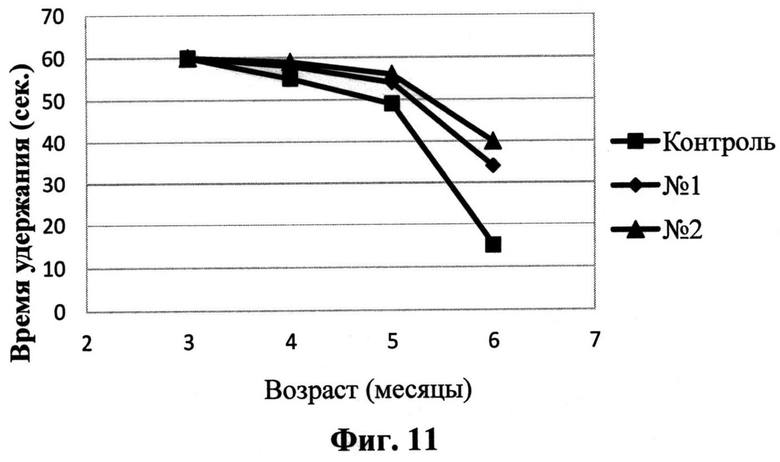
На Фиг.11 представлены результаты тестирования на «перевернутой сетке» животных линии tauP301S, получавших соединения I (ромб), II (треугольник) и контрольных животных (квадраты) в разные возрастные периоды.
Влияние исследуемых веществ на формирование TDP43-реактивных включений в недифференцированной клеточной культуре SH-SY5Y
Было исследовано действие соединений I, полученных в примерах 1-4, II, полученных в примерах 5-8, III, полученных в примерах 9-12, IV, полученных в примерах 13-16, V, полученных в примерах 17-20, XI полученного в примере 38, на количество, морфологию и кинетику формирования депозитов, образуемых Т5 патогенной формой белка TDP43 в трансфецированных SH-SY5Y клетках. Клетки засевали на обработанные полилизином стекла диаметром 10 мм, помещенные в 24-луночные культуральные планшеты. Плазмидная ДНК, кодирующая Т5 форму, была введена в недифференцированные клетки SH-SY5Y методом транзиторной трансфекции. Через 3 часа после трансфекции к культивируемым клеткам добавляли питательную среду, содержащую исследуемые вещества до конечной концентрации 10 мкг/мл; в контрольные образцы добавляли среду без препаратов. Через 16 часов клеточные культуры фиксировали и подсчитывали число флюоресцентных включений, размер которых был не менее пяти микрометров. Считали включения на всей площади стекла, для каждого препарата брали среднее значение трех стекол. В каждом эксперименте за 100% принимали количество включений в контрольных культурах, которые инкубировались после трансфекции без добавления препаратов. Число включений в образцах, культивируемых с добавлением исследуемых соединений, выражали в процентном отношении к контрольным.
В таблице 1 представлены результаты 5 независимых экспериментов, в которых было изучено влияние исследуемых веществ на содержание крупных Т5 включений после трансфекции клеточной культуры SH-SY5Y. Полученные данные свидетельствуют, что соединения I и II способны ингибировать агрегацию мутантной формы Т5 белка TDP43, при этом число клеток, содержащих агрегаты, уменьшается практически в два раза. Исследование соединений III-V и XI показало, что они в меньшей степени обладают этой способностью. Из представленных данных видно, что заявляемые соединения препятсвуют неконтролируемой агрегаци белков в культуре клеток SH-SY5Y, причем наиболее эффективны соединения I, II.
Исследование количества амилоидных отложений у модельных животных.
Было неоднократно продемонстрировано, что число амилоидных отложений в нервной ткани пациентов с нейродегенеративными заболеваниями коррелирует со степенью прогрессии неврологической симптоматики (Braak et al., 2006). В частности, такая зависимость была обнаружена для сенильных бляшек при болезни Альцгеймера и телец Леви при болезни Паркинсона.
Исследования на мышах линии Thy1mgSN
Окрашивание амилоидных агрегатов в спинном мозге у животных линий Thy1mgSN проводилось амилоид-специфическим красителем конго красным, а линии P301S - антителами против тау-белка.
Было установлено, что при прогрессии неврологической симптоматики (возрастные точки 3, 6 и 9 месяцев для линии Thy1mgSN; 2, 4 и 6 месяцев для линии P301S) происходит постепенное увеличение числа агрегатов - накопление амилоидных отложений в нервной системе (спинной мозг). При этом максимальное число патологических белковых структур наблюдается на последних временных точках и далее (на терминальных стадиях) достоверно не увеличивалось. Репрезентативные фотографии гистопатологических отложений в нервной ткани животных представлены на Фиг.1.
Гистохимическое исследование срезов спинного мозга двенадцатимесячных животных линии Thy1mgSN, которые получали исследуемые соединения в течение девяти месяцев, показало значительное уменьшение числа окрашиваемых Конго красным амилоидных отложений (группа, получающая соединение I, - 19,02±1,72; группа, получающая соединение II, - 22,60±1,10) по сравнению с животными такого же возраста, которые не получали препарата (38,06±4,35) (Таблица 2, Фиг.2).
Таким образом полученные данные сведетельствуют, что соединения I, II препятствуют накоплению патологических агрегатов (амилоидных отложений) в нервной системе модельных животных линии Thy1mgSN.
Исследования на мышах линии tauP301S
Для оценки количества нейронов, содержащих гиперфосфорилированные формы tau-белка, был использован иммуногистохимический анализ поперечных срезов торакального отдела спинного мозга мышей линии tauP301S, с использованием антител anti-tau АТ8. Анализ образцов нервной ткани с помощью иммуноблоттинга с данными антителами показал, что в процессе развития нерйодегенерации у животных линии tauP301S наблюдается прогрессивное увеличение количества нейронов, содержащих гиперфосфорилированный тау-белок. При исследовании действия соединений I и II на количество таких нейронов был выявлен тренд к уменьшению количества АТ8-позитивных нейронов в группах животных, принимавших исследуемые препараты, по сравнению с группой контроля. В группах 6-месячных животных эти различия были статистически значимыми (Фиг.3).
Исследование уровня фосфорилирования тау-белка
Анализ уровня фосфорилирования тау-белка проводился методом количественного Вестерн блоттинга. Было показано, что общее количество гиперфосфорилированного тау-белка в спинном мозге мышей линии tauP301S, принимавших исследуемые соединения, снижается. На временной точки 6-месяцев эти различия были статистически достоверными (Фиг.4).
Исследования на мышах линии 5xFAD
В связи с тем, что отложения бета-амилоида (сенильные бляшки) являются внеклеточными, в отличие от внутриклеточных нейрофибриллярных белков в случае линии с таупатией P301S, и очень многочисленны у животных линии 5xFAD на терминальных стадиях заболевания, для оценки эффекта исследуемых соединений была выполнена оценка общей площади (в процентом отношении), занятой отложениями. Определение амилоидной нагрузки было выполнено в коре головного мозга - структуре головного мозга, характеризующейся наиболее выраженной амилоидной патологией. Было обнаружено существенное и статистически достоверное снижение площади, занятой амилоидными отложениями в коре головного мозга (Фиг.5). На 32% и 27% для соединения I и II соответственно.
Репрезентативные микрофотографии соответствующих отделов головного мозга животных, получавших соединение II, и контрольной группы приведены на Фиг.6 (амилоидные отложения визуализированы при помощи флуоресцентного красителя Конго красный).
Данные, полученные в описанной серии экспериментов, показывают, что заявляемые соединения могут уменьшать количество и/или действовать на стабильность агрегатов, тем самым замедляя прогрессирование патологии, вызванной агрегацией соответствующего белка.
Исследование состояния специфических нейроинфламматорных реакций у модельных животных
Было исследовано действие изучаемых соединений I и II в отношении воспалительной реакции в нервной системе на продвинутой стадии протеинопатии (6 месяцев в линии Thy1mgSN и 5 месяцев в линии P301S). Согласно данным, полученным при исследовании спинного мозга мышей, получавших соединения I, II в течение 6 месяцев, происходит существенное ослабление реакции астроглиоза, что выражается в общем снижении GFAP-иммунореактивности и числа активированных астроцитов (Фиг.7).
Данный эффект был также подтвержден в линии P301S, у животных происходило снижение числа активированных астроцитов после 4 месяцев получения препарата. На Фиг.8 приведены репрезентативные фотографии спинного мозга животных, не получавших препарат и получавших соединение II с возраста 1 месяца до 5 месяцев.
Обнаруженный при иммуногистохимическом исследовании эффект был также подтвержден с помощью иммуноблоттинга: содержание GFAP (две изоформы, 55 и 45 кДа) было существенно снижено у получавших исследуемые соединения мышей по сравнению с трансгенными мышами (tauP301S) того же возраста и было сходным с таковым у мышей дикого типа (Фиг.9).
Таким образом, данные, полученные при исследовании специфических нейроинфламматорных реакций в двух линиях трансгенных животных, показывают, что заявляемые соединения обладают нейропротекторными свойствами у животных моделирующих протеинопатии.
Поведенческое тестирование животных
Тест оценки баланса и координации на вращающемся стержне
С помощью инструментальных методов исследования удается выявить нарушения локомоторной функции у модельных мышей на ранней пре-симптоматической стадии заболевания, начиная уже с двухмесячного возраста. Так, при тестировании 2-месячных мышей линии P301S на вращающемся стержне в режиме ускорения вращения мы обнаружили, что трансгенные животные не были способны удержаться на стержне в течение всего интервала тестирования, который составлял 300 секунд, и падали до завершения теста. В то же время контрольные животные дикого типа в этом возрасте свободно удерживались на вращающемся стержне в течение 300 секунд.
В дальнейшем были получены данные о действии соединений I и II на показатели моторной функции в линии P301S (Фиг.10). Тестирование проводилось ежемесячно, между третьим и шестым месяцем жизни. Было показано, что данные вещества улучшают способность опытных животных удерживаться на вращающемся стержне.
Тест «перевернутая сетка»
Для инструментальной оценки локомоторной дисфункции трансгенных животных был также использован тест «перевернутая сетка». В норме животные дикого типа свободно могут удерживаться на перевернутой сетке в течение 60 сек, сохраняя эту способность даже в возрасте 2-х лет. Однако при дисфункции двигательного аппарата время пребывания на сетке может быть значительно снижено вплоть до полной невозможности даже начать тест. У исследованных P301S мышей разного возраста (от 3 до 6 мес) было зарегистрировано прогрессивное ухудшение показателей способности удерживаться на сетке, при этом в возрасте 6 мес гомозиготные трансгенные животные были фактически не способны удерживаться на перевернутой сетке, тогда как в контрольной группе более 80% животных дикого типа в возрасте 18 мес были способны удерживаться на перевернутой сетке в течение всего 60-секундного теста.
На Фиг.11 представлены результаты тестирования на «перевернутой сетке» животных линии tauP301S, получавших исследуемые соединения, в сравнении с контролем. Из приведенных данных видно, что у мышей данной линии способность удерживаться на сетке ухудшается с возрастом. В группе животных, получавших опытные препараты, также наблюдается прогрессия в моторной дисфункции, однако темп прогрессирования значимо ниже.
Таким образом, приведенные данные указывают на способность заявляемых соединений ингибировать прогрессию нейродегенеративного процесса в нервной системе модельных животных, вызванного неконтролируемой агрегацией патогенных белков.

| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ 1,2,3,4-ТЕТРАГИДРОПИРИДО[4,3-b]ИНДОЛСОДЕРЖАЩИХ ФЕНОТИАЗИНОВ В КАЧЕСТВЕ ИНГИБИТОРОВ ХОЛИНЭСТЕРАЗ И БЛОКАТОРОВ СЕРОТОНИНОВЫХ РЕЦЕПТОРОВ 5-HT, СПОСОБЫ ПОЛУЧЕНИЯ ИХ ХЛОРГИДРАТОВ И ФАРМАКОЛОГИЧЕСКОЕ СРЕДСТВО НА ИХ ОСНОВЕ | 2013 |
|
RU2530881C1 |
| ХЛОРГИДРАТЫ ФТОРСОДЕРЖАЩИХ ЗАМЕЩЕННЫХ 5-[2-(ПИРИД-3-ИЛ)-ЭТИЛ]-2,3,4,5-ТЕТРАГИДРО-1Н-ПИРИДО[4,3-b]ИНДОЛОВ, В КАЧЕСТВЕ СРЕДСТВ СНИЖЕНИЯ НЕКОНТРОЛИРУЕМОЙ АГРЕГАЦИИ БЕЛКОВ В НЕРВНОЙ СИСТЕМЕ, ФАРМАКОЛОГИЧЕСКОЕ СРЕДСТВО НА ИХ ОСНОВЕ И СПОСОБ ЕГО ПРИМЕНЕНИЯ | 2011 |
|
RU2490268C2 |
| ПРОИЗВОДНЫЕ АЗОЛА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ С АКТИВНОСТЬЮ В ОТНОШЕНИИ СЕРОТОНИНОВОГО (5-HT)РЕЦЕПТОРА | 1995 |
|
RU2146256C1 |
| АДАМАНТАНСОДЕРЖАЩИЕ ИНДОЛЫ И ИХ ГИДРОХЛОРИДЫ, ОБЛАДАЮЩИЕ СВОЙСТВОМ СТАБИЛИЗАЦИИ МИКРОТРУБОЧЕК, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ФАРМАКОЛОГИЧЕСКОЕ СРЕДСТВО НА ИХ ОСНОВЕ И СПОСОБ ЛЕЧЕНИЯ И ПРЕДУПРЕЖДЕНИЯ ЗАБОЛЕВАНИЙ, СВЯЗАННЫХ С НАРУШЕНИЯМИ СИСТЕМЫ МИКРОТРУБОЧЕК | 2015 |
|
RU2608631C1 |
| ПРОИЗВОДНЫЕ ГИДРИРОВАННЫХ ПИРИДО(4,3-B)ИНДОЛОВ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЛЕЧЕНИЯ | 1995 |
|
RU2140417C1 |
| ИЗОКСАЗОЛИНЫ И ИЗОКСАЗОЛЫ, СПОСОБ ПОДАВЛЕНИЯ АГРЕГАЦИИ ТРОМБОЦИТОВ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ПОДАВЛЯЮЩАЯ АГРЕГАЦИЮ ТРОМБОЦИТОВ | 1994 |
|
RU2149871C1 |
| ПРОИЗВОДНЫЕ ТРИАЗОЛО[1,4]ДИАЗЕПИНА И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 1989 |
|
RU2117670C1 |
| ЗАМЕЩЕННЫЕ ПИРАЗОЛЫ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ, СПОСОБ ЛЕЧЕНИЯ, ПРОМЕЖУТОЧНЫЙ ПРОДУКТ | 1993 |
|
RU2130453C1 |
| ЗАМЕЩЕННЫЕ ПИРИМИДИНТИОАЛКИЛЬНЫЕ ИЛИ АЛКИЛЭФИРНЫЕ СОЕДИНЕНИЯ И СПОСОБ ИНГИБИРОВАНИЯ ОБРАТНОЙ ТРАНСКРИПТАЗЫ ВИРУСОВ | 1996 |
|
RU2167155C2 |
| ПРОИЗВОДНЫЕ ПИРИМИДИН-4(3H)-ОНА В КАЧЕСТВЕ АНТАГОНИСТОВ TRPV4 | 2021 |
|
RU2840769C1 |


Изобретение относится к производным фенотиазинсодержащих 1,2,3,4-тетрагидропиридо[4,3-b] индолов общей формулы 1 и их хлоргидратов в качестве средства для снижения неконтролируемой агрегации белков в нервной системе, способам их получения, фармакологическому средству на их основе и способу снижения неконтролируемой агрегации белков в нервной системе. В общей формуле 1: R1=H, (C1-C6) алкил; R2, R3, R4, R5=H, F, O, Br, (C1-C6)алкил, (C1-C6)алкокси, R6, R7=. H, F, Cl, Br, (C1-C6)алкил, (C1-C6)алкокси, NH2, NHAlkyl, NAlkyl2, A=CH2CH2, CH=CHCH2, CH2CH2CH2, CH2CH[(C1-C6)алкил]C(O), CH2CH2C(O)NHCH2CH2); CH2CH[((C1-C6)алкил]C(O)N[(C1-C6))алкил]CH2CH2), CH2CH(OH)CH2, CH2CH(OH)CH2NHCH2CH2, CH2CH(OH)CH2N[(C1-C6)алкил]CH2CH2, CH2CHFCH2NHCH2CH2, CH2CHFCH2N[(C1-C6)алкил]CH2CH2. 6 н. и 3 з.п. ф-лы, 2 табл., 11 ил.
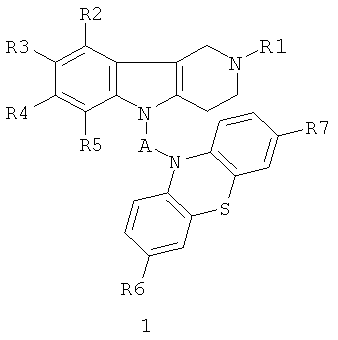
1. Производные фенотиазинсодержащих 1,2,3,4-тетрагидропиридо[4,3-b]индолов общей формулы 1 и их хлоргидраты в качестве средства для снижения неконтролируемой агрегации белков в нервной системе
в которой R1=H, (C1-C6) алкил;
R2, R3, R4, R5=H, F, Cl, Br, (C1-C6) алкил, (C1-C6) алкокси,
R6, R7=H, F, Cl, Br, (C1-C6) алкил, (C1-C6) алкокси, NH2,
NHAlkyl, NAlkyl2;
A=CH2CH2, CH=CHCH2, CH2CH2CH2, CH2CH[(C1-C6)алкил]C(O), CH2CH2C(O)NHCH2CH2); CH2CH[(C1-C6) алкил]C(O)N[(C1-C6) алкил]CH2CH2), CH2CH=(OH)CH2, CH2CH(OH)CH2NHCH2CH2,
CH2CH(OH)CH2N[(C1-C6) алкил]CH2CH2, CH2CHFCH2NHCH2CH2, CH2CHFCH2N[(C1-C6) алкил]CH2CH2.
2. Производные фенотиазинсодержащих 1,2,3,4-тетрагидропиридо[4,3-b]индолов по п.1, представляющие собой соединения общей формулы 1.1
в которой R1, R2, R3, R4, R5, R6, R7 имеют значения, определенные выше для формулы 1;
X=CH2CH2, CH=CHCH2, CH2CH2CH2, CH2CH[(C1-C6) алкил]C(O), CH2CH2C(O)NHCH2CH2); CH2CH[(C1-C6) алкил]C(O)N[(C1-C6)])алкил]CH2CH2).
3. Производные фенотиазинсодержащих 1,2,3,4-тетрагидропиридо[4,3-b]индолов по п.1, представляющие собой соединения общей формулы 1.2
в которой R1, R2, R3, R4, R5, R6, R7 имеют значения, определенные выше для формулы 1;
Y=CH2CH(OH)CH2, CH2CH(OH)CH2NHCH2CH2,
CH2CH(OH)CH2N[(C1-C6) алкил]CH2CH2.
4. Производные фенотиазинсодержащих 1,2,3,4-тетрагидропиридо[4,3-b]индолов по п.1, представляющие собой соединения общей формулы 1.3
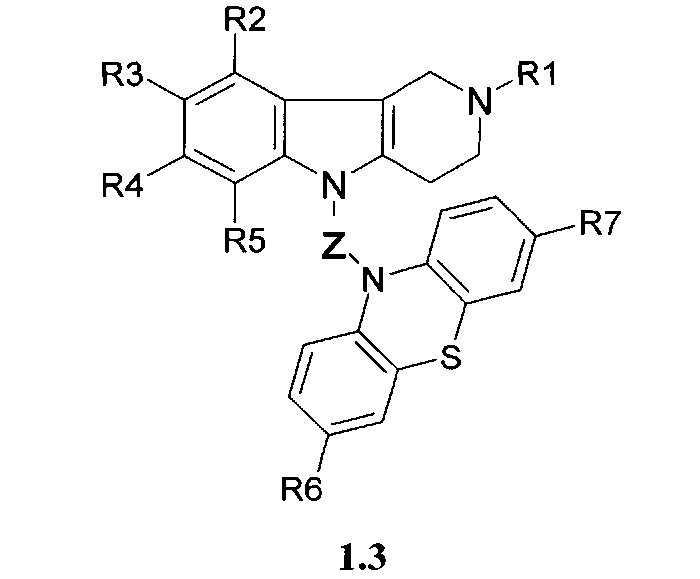
в которой R1, R2, R3, R4, R5, R6, R7 имеют значения, определенные выше для формулы 1;
Z=CH2CHFCH2NHCH2CH2CH2 или CH2CHFCH2N [(C1-C6) алкил]CH2CH2,
5. Способ получения соединений формулы 1.1, заключающийся в том, что эквимольную смесь гамма-карболинов общей формулы 2
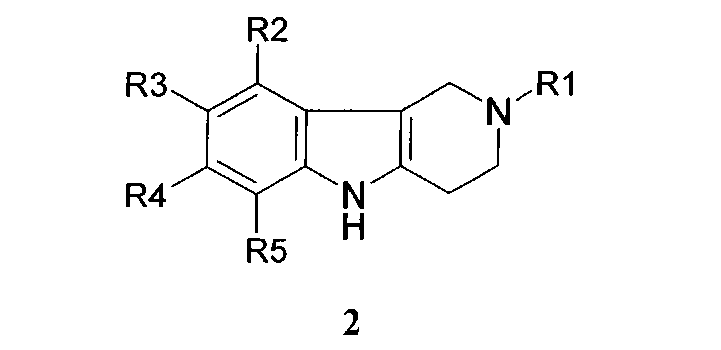
в которой R1, R2, R3, R4, R5 имеют значения, определенные выше для формулы 1;
и ацетиленсодержащих фенотиазинов общей формулы 3
или винилсодержащих фенотиазинов общей формулы 4
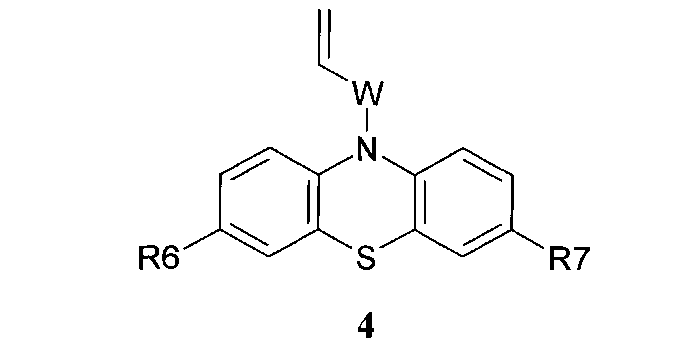
в которых R6, R7 имеют значения, определенные выше для формулы 1;
W=(CH2)n, где n=0 или 1; C(O); CH[(C1-C6) алкил]C(O);
C(O)NHCH2CH2); C(O)N[(C1-C6)алкил]CH2CH2; CH[(C1-C6)
алкил]C(O)N(C1-C6)алкил]CH2CH2),
нагревают в диметилсульфоксиде или диметилформамиде в присутствии катализатора, выбранного из ряда: метилат натрия, трет-бутилат калия, гидроокись калия или фторид цезия при температуре 120-160°C в течение 6-24 часов.
6. Способ получения соединений формулы 1.2, заключающийся в том, что эквимольную смесь гамма-карболинов общей формулы 2
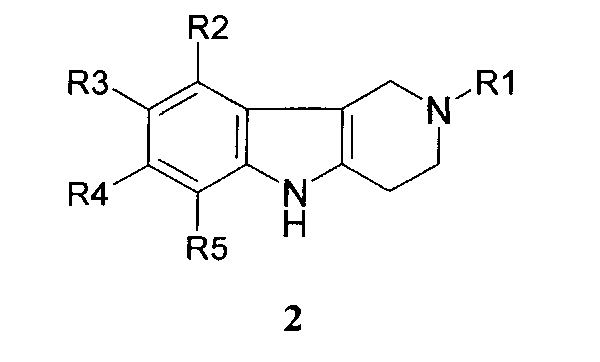
в которой R1, R2, R3, R4, R5 имеют значения, определенные выше для формулы 1;
и N-эпоксидсодержащих фенотиазинов общей формулы 5
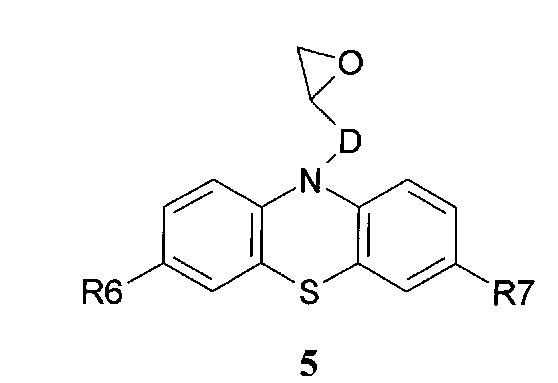
в которых R6, R7 имеют значения, определенные выше для формулы 1;
D=CH2; CH2NHCH2CH2; CH2N[(C1-C6)алкил]CH2CH2 нагревают в диметилсульфоксиде или диметилформамиде в присутствии катализатора, выбранного из ряда: метилат натрия, трет-бутилат калия, гидроокись калия или фторид цезия при температуре 100-140°C в течение 2-12 часов.
7. Способ получения соединений 1.3, заключающийся в том, что гидроксилсодержащие конъюгаты гамма-карболинов и фенотиазинов общей формулы 6
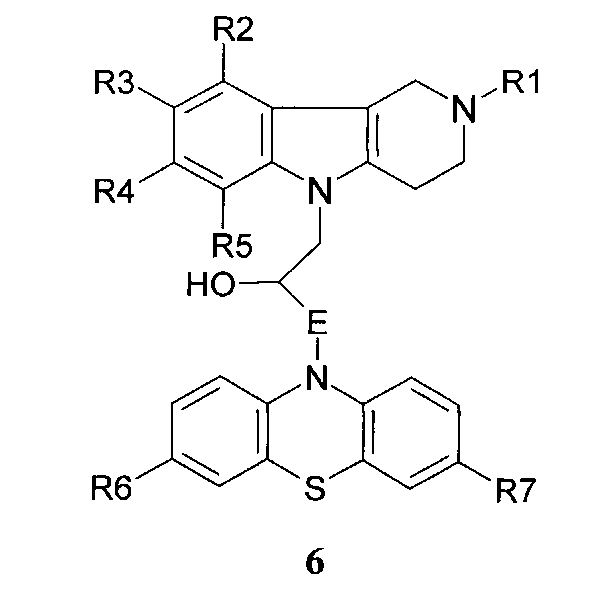
в которой R1, R2, R3, R4, R5, R6, R7 имеют значения, определенные выше для формулы 1;
E=CH2; CH2NHCH2CH2; CH2N[(C1-C6)алкил]CH2CH2
фторируют эквимольным количеством диэтиламиносульфотрифторида в диметилсульфоксиде или диметилформамиде при температурах (-78)-0°C в течение 1-3 часов.
8. Фармакологическое средство для снижения неконтролируемой агрегации белков в нервной системе, содержащее активное начало и фармацевтически приемлемый носитель, отличающееся тем, что в качестве активного начала содержит эффективное количество соединения формулы 1.
9. Способ снижения неконтролируемой агрегации белков в нервной системе, заключающийся во введении пациенту фармакологического средства, содержащего эффективное количество соединения формулы 1 в дозе 0,01-1.5 мг/кг массы тела по крайней мере один раз в день в течение периода, необходимого для достижения терапевтического эффекта.
| Смоляр Н.Н., Волчков А.С., Ютилов Ю.М "Синтез 3-алкил- и 3,9-диалкил-1,2,3,4-тетрагидро-γ-карболинов", Химико-фармацевтический журнал, том 35, ч.9, стр | |||
| Способ изготовления звездочек для французской бороны-катка | 1922 |
|
SU46A1 |
| Bacu, Elena; Belei, Dalila; Couture, Axel; Grandclaudon, Pierre "Synthesis of new N-acylphenothiazinic derivatives with potential activity in chemotherapy", Revue Roumaine de | |||

