Область техники, к которой относится изобретение
Изобретение относится к способу получения тиоэфира пептида.
Уровень техники
Известно, что для синтеза белков применяют разнообразные способы, такие как биосинтез, химический синтез и бесклеточный синтез. В биосинтетическом способе белок получают, используя внутреннее содержимое клетки, такой как клетка Escherichia coli, и применяя введение и экспрессию ДНК, кодирующей белок, который намереваются синтезировать в клетке. В химическом синтезе целевой белок синтезируют путем последовательного присоединения аминокислот с помощью приемов органической химии. В бесклеточном синтезе белок синтезируют в бесклеточной системе, применяя фермент и т.п., присутствующий в различных клетках, таких как клетка Escherichia coli. Эти способы соответственно применяют по отдельности или в комбинации в зависимости от предполагаемого использования, размера белка и свойств, которые хотят ему придать.
В настоящее время для того, чтобы синтезировать гомогенный белок, имеющий определенные модификации, такие как полисахаридные цепи или липид и т.п., в средней части своей аминокислотной последовательности, аминокислоты заранее модифицируют полисахаридными цепями или липидом и т.п. и затем химическим способом синтезируют пептидную цепь, включающую модифицированные аминокислоты.
В качестве способа химического синтеза пептидной цепи, главным образом, применяют твердофазный синтез. Однако пептидная цепь, полученная с помощью твердофазного синтеза, обычно представляет собой короткую цепь и составлена самое большее из примерно 50-ти остатков.
Таким образом, получают по отдельности короткие пептидные цепи и затем их лигируют с целью синтезировать длинную пептидную цепь, имеющую модификации. В литературе описаны различные методики лигирования пептидных цепей, и одна из широко применяемых методик представляет собой способ нативного химического лигирования (NCL-способ). Способ NCL также может быть применен для незащищенных пептидных цепей, и известно, что его применяют для образования нативной амидной связи (пептидной связи) в месте лигирования (например, Патентная литература 1). Способ NCL представляет собой хемоселективную реакцию между первым пептидом, несущим α-карбокситиоэфирную группировку на своем С-конце, и вторым пептидом, несущим остаток цистеина на своем N-конце, и тиоловая группа (SH-группа, также обозначаемая как сульфгидрильная группа) цистеина боковой цепи селективно реагирует с углеродом карбонила тиоэфирной группы, и посредством связывания через тиоэфир в реакции обмена тиолов образуется исходный интермедиат. Этот интермедиат претерпевает спонтанную внутримолекулярную перегруппировку с получением нативной амидной связи в месте лигирования, в то же время в нем происходит регенерация тиола цистеина боковой цепи.
В этом способе две пептидные цепи могут быть лигированы через пептидную связь только путем смешивания незащищенных пептидов в буферном растворе. В способе NCL, даже если реагируют соединения, имеющие много функциональных групп, такие как пептиды, то С-конец одного из пептидов может быть селективно лигирован к N-концу другого пептида. С этой точки зрения важно установить, каким образом следует применять способ NCL, для того, чтобы химически синтезировать белок.
Однако при применении способа NCL возникает проблема получения тиоэфира пептида, имеющего α-карбокситиоэфирную группировку на своем С-конце, необходимого для применения в качестве исходного материала. В литературе известны различные способы получения тиоэфира пептида, и эти способы, основанные на твердофазном синтезе, в целом, могут быть подразделены на два типа.
Первый способ представляет собой способ создания тиоэфира пептида на смоле. В этом способе тиоэфира пептида может быть получен вместе с отщеплением пептидной цепи от смолы после конструирования пептида (например, твердофазный синтез Boc, твердофазный синтез Fmoc). Второй способ представляет собой способ конструирования пептидной цепи на твердой фазе через линкер, эквивалентный тиоэфиру (надежно связанный линкер, способ Фуджии, способ Доусон, способ с меркаптопропанолом, способ Каваками, способ Данишевского, способ Годжо, способ Аимото и т.п.). В этом способе тиоэфир получают путем активирования пептидной цепи С-конца, сконструированной с помощью подходящей обработки с линкером, с последующим тиолизом пептидной цепи (Непатентная литература 1).
Помимо этих способов также сообщалось о способе, в котором защищенный пептид, такой, что его боковая цепь защищена путем твердофазного синтеза и только карбоксильная группа на С-конце свободна, синтезируют с последующей тиоэтерификацией в подходящих условиях конденсации (например, Патентная литература 2). Любой из этих способов представляет собой общепринятый способ и его применяют для различных белковых синтезов. Однако размер тиоэфира пептида, который возможно получить с помощью синтеза, ограничен, потому что эти способы ограничены недостатками твердофазного синтеза. Кроме того, в способе с применением линкера необходимо отдельно химически синтезировать ненативное производное аминокислоты или специфическое производное. Таким образом, такие процедуры не всегда могут быть названы простыми.
Способ с применением интеина позволяет преодолеть проблему ограничений при тиоэтерификации в твердофазном синтезе (Непатентная литература 2). В этом способе полипептидный фрагмент, биосинтез которого проводят в клетке, может быть получен в виде тиоэфира. В способе с применением интеина пептидную цепь тиоэтерифицируют, применяя функцию белкового сплайсинга, который имеет место в определенной белковой последовательности, и полипептидную цепь получают в виде тиоэфира. Преимущество этого способа заключается в том, что может быть получен тиоэфир пептида с длинной цепью. Синтез большого модифицированного белка, который как считалось до настоящего времени было трудно выполнить, стал возможен в результате комбинирования этого способа со способом химического синтеза (Непатентная литература 3). Способ экспрессии полипептидной цепи и ее получения был всесторонне исследован и стал общепринятым в качестве основной методики в биологии.
Однако если применяют способ с применением интеина, то необходимо иметь целевую пептидную последовательность, и эксперессируемый сложный белок с интеином должен свернуться так, чтобы иметь присущую ему трехмерную структуру, потому что он не только представляет собой экспрессированный полипептид, но также должен пройти сплайсинг белка. Таким образом, в зависимости от последовательности полипептида, которую нужно экспрессировать, не всегда удается найти оптимальные условия для получения тиоэфира пептида и избежать сопутствующих осложнений в работе.
При этом способ расщепления пептидной цепи в том положении, где находится остаток цистеина, путем взаимодействия соединения с SH-группой остатка цистеина в пептиде (Непатентная литература 4 и 5) и способ расщепления пептида, связанного с твердой фазой с помощью линкера (Непатентная литература 6 и 7), известны как способы расщепления пептида. Кроме того, известен способ расщепления пептидной связи в С-концевой части метионинового остатка с применением бромистого циана (CNBr). Однако эти способы не представляют собой способы получения фрагмента пептида в виде тиоэфира.
Литература, имеющая отношение к этой области техники
Патентная литература
Патентная литература 1: Международная публикация заявки WO 96/34878.
Патентная литература 2: Международная публикация заявки WO 2007/114454.
Непатентная литература
Непатентная литература 1: Ingenito et al., J. Am. Chem. Soc., 121: 11369-11374, 1999.
Непатентная литература 2: Schwartz et al., CHEM COMMUN., 2087-2090, 2003.
Непатентная литература 3; Muir, Annu. Rev. Biochem., 72: 249-289, 2003.
Непатентная литература 4: Stark GR, Methods of Enzymology, 47: 129-132, 1977.
Непатентная литература 5: Nakagawa et al., J. Am. Chem. Soc., 116: 5513-5514, 1994.
Непатентная литература 6: Sola et al., J. Chem. Soc. Chem. Commun., 1786-1788, 1993.
Непатентная литература 7: Pascal et al., Tetrahedron Letters, Vol.35, No. 34: 6291-6294, 1994.
Раскрытие изобретения
Проблемы, которые будут решены с помощью изобретения
При тиоэтерификации пептидов, описанной в приведенном выше уровне техники, пептид, который может быть тиоэтерифицирован, ограничен пептидной цепью, синтезируемой на твердой фазе, и пептидной цепью, к которой будет применен сплайсинг белка. Это связано с тем, что любой из этих способов требует не природного производного аминокислоты, линкера и определенной трехмерной структуры и т.п.
Таким образом, целью настоящего изобретения является обеспечение нового способа химического превращения полипептидной цепи в тиоэфир пептида.
Способы решения проблем
Авторы настоящего изобретения считают, что необходим способ селективной активации С-конца пептида, направленный на природный аминокислотный остаток в последовательности пептида. В таком способе пептидная цепь в любом пептиде, полученном с помощью любого способа, такого как биосинтез, может быть селективно активирована и тиоэтерифицирована.
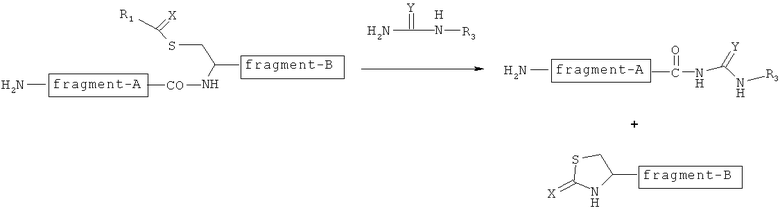
Таким образом, авторы настоящего изобретения сосредоточили свои усилия на остатке цистеина, который представляет собой особую серосодержащую аминокислоту среди природных аминокислот. И авторы настоящего изобретения обнаружили, что как показано на следующей фигуре, группу -C(=X)-R1 вводят в тиоловую группу остатка цистеина, и соединение, имеющее замещаемую группу, представленную формулой -NH-C(=Y)NHR3, реагирует при этом в органическом растворителе с присоединением группы -NH-C(=Y)NHR3 к карбоксильной группе пептидной связи на N-концевой стороне остатка цистеина, посредством чего пептидную связь расщепляют и фрагмент пептида на С-концевой стороне вырезают. Кроме того, авторы настоящего изобретения обнаружили, что получаемая пептидная цепь может быть превращена в тиоэфир пептида в обменной реакции с тиолом, в которой соединение тиола реагирует с пептидной цепью, к которой в буферном растворе была присоединена группа -NH-C(=Y)NHR3, что позволяет, таким образом, связать тиоловую группу соединения тиола с углеродом карбонила, к которому группа -NH-C(=Y)NHR3 была присоединена, и удалить группу -NH-C(=Y)NHR3.
В особенности в качестве одного из примеров к сказанному выше в тиоловую группу остатка цистеина первой вводят тионоформиатную группу. И пептидная цепь, в которой к ее С-концу была добавлена N-ацетилгуанидидо-группа, была получена в реакции N-ацетилгуанидина с этой тионоформиатной группой в органическом растворителе, чтобы привести к расщеплению пептидной цепи с N-концевой стороны остатка цистеина. Кроме того, эта пептидная цепь с присоединенной N-ацетилгуанидидо-группой вступает в реакцию с тиолом R4-SH в буферном растворе для превращения в тиоэфир пептида.
Авторы настоящего изобретения также обнаружили, что в способе NCL могут быть применены полученная выше пептидная цепь с присоединенной N-ацетилгуанидидо-группой и тиоэфир пептида.
[Химическая формула 1]

Следовательно, настоящее изобретение специфически обеспечивает следующие пункты [1]-[14].
[1] Способ получения тиоэфира пептида, включающий следующие стадии (а)-(с):
(а) стадия получения первого интермедиата путем взаимодействия соединения А, представленного следующей формулой (I), с тиоловой группой остатка цистеина в пептидной цепи, имеющей остаток цистеина, для отщепления R2:
[Химическая формула 2]
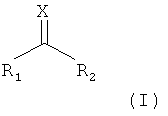
в которой X представляет собой атом серы или атом кислорода, R1 и R2 представляют собой замещаемые группы;
(b) стадия взаимодействия соединения В, представленного следующей формулой (II), с первым интермедиатом в органическом растворителе для добавления группы -NH-C(=Y)NHR3 к карбоксильной группе, образующей пептидную связь с аминокислотой, прилегающей к N-концевой стороне остатка цистеина, и расщепления пептидной связи, с получением, таким образом, с N-концевой стороны фрагмента пептида, расположенного ближе к N-концевой стороне, чем расщепленная пептидная связь, в качестве второго интермедиата:
[Химическая формула 3]

в которой Y представляет собой атом кислорода, атом серы или NH-группу, и R3 представляет собой атом водорода, ацильную группу или алкоксикарбонильную группу; и
(c) стадия тиоэтерификации С-конца второго интермедиата в реакции второго интермедиата с тиолом для обмена группы -NH-C(=Y)NHR3 на тиоловую группу на С-конце.
[2] Способ по вышеприведенному пункту [1], в котором Х представляет собой атом серы.
[3] Способ по вышеприведенным пунктам [1] или [2], в котором R1 представляет собой -O-C6-арильную группу.
[4] Способ по любому из вышеприведенных пунктов [1]-[3], в котором R2 представляет собой атом галогена или замещенную или незамещенную -S-C6-10-арильную группу.
[5] Способ по любому из вышеприведенных пунктов [1]-[4], в котором Y представляет собой NH-группу.
[6] Способ по любому из вышеприведенных пунктов [1]-[5], в котором R3 представляет собой ацетильную группу.
[7] Способ по любому из вышеприведенных пунктов [1]-[6], в котором тиол представляет собой тиол, представленный следующей формулой (III) в стадии (с):
R4-SH (Формула III)
в которой R4 представляет собой любой заместитель из группы, которую выбирают из замещенной или незамещенной бензильной группы, замещенной или незамещенной арильной группы и замещенной или незамещенной алкильной группы.
[8] Способ по любому из вышеприведенных пунктов [1]-[7], в котором пептидная цепь представляет собой рекомбинантный белок.
[9] Способ по любому из вышеприведенных пунктов [1]-[8], в котором пептидная цепь представляет собой рекомбинантный белок, включающий метку для очистки.
[10] Способ получения полипептида, включающий стадию связывания тиоэфира пептида, полученного с помощью способа по любому из вышеприведенных пунктов [1]-[9], с пептидной цепью, имеющей цистеин на N-конце, способом дотирования.
[11] Способ получения второго интермедиата, применяемого в способе получения тиоэфира пептида, по любому из следующих пунктов [1]-[9], приведенных выше, включающий:
(a) стадию получения первого интермедиата путем взаимодействия соединения А, представленного следующей формулой (I), с тиоловой группой остатка цистеина в пептидной цепи, имеющей остаток цистеина, для отщепления R2:
[Химическая формула 4]
в которой Х представляет собой атом серы или атом кислорода, R1 и R2 представляют собой замещаемые группы; или
(b) стадию реагирования соединения В, представленного следующей формулой (II), с первым интермедиатом в органическом растворителе для добавления группы -NH-C(=Y)NHR3 к карбоксильной группе, образующей пептидную связь между остатком цистеина и аминокислотой, прилегающей к N-концевой стороне остатка цистеина, и расщепления пептидной связи, с получением, таким образом, с N-концевой стороны фрагмента пептида, расположенного ближе к N-концевой стороне, чем расщепленная пептидная связь, в качестве второго интермедиата:
[Химическая формула 5]
в которой Y представляет собой атом кислорода, атом серы или NH-группу, и R3 представляет собой атом водорода, ацильную группу или алкоксикарбонильную группу.
[12] Пептидная цепь, имеющая группу -NH-C(=Y)NHR3 на С-конце, в которой Y представляет собой атом кислорода или NH-группу, и R3 представляет собой атом водорода, ацильную группу или алкоксикарбонильную группу.
[13] Способ получения полипептида, включающий стадию связывания пептидной цепи, имеющей группу -NH-C(=Y)NHR3 на С-конце по вышеприведенному пункту [12], с пептидной цепью, имеющей цистеин на N-конце, способом лигирования.
[14] Способ удаления метки для очистки, добавленной к С-концевой части рекомбинантного белка, включающий следующие стадии (а)-(с):
(a) стадия получения первого интермедиат путем взаимодействия соединение А, представленного следующей формулой (I), с тиоловой группой остатка цистеина в рекомбинантном белке, содержащем метку для очистки в С-концевой части для отщепления R2:
[Химическая формула 6]
в которой Х представляет собой атом серы или атом кислорода, R1 и R2 представляют собой замещаемые группы;
(b) стадия взаимодействия соединения В, представленного следующей формулой (II), с первым интермедиатом в органическом растворителе для добавления группы -NH-C(=Y)NHR3 к карбоксильной группе, образующей пептидную связь между остатком цистеина и аминокислотой, прилегающей к N-концевой стороне остатка цистеина, и расщепления пептидной связи, с получением, таким образом, с N-концевой стороны фрагмента пептида, расположенного ближе к N-концевой стороне, чем расщепленная пептидная связь, в качестве второго интермедиата:
[Химическая формула 7]
в которой Y представляет собой атом кислорода, атом серы или NH-группу, и R3 представляет собой атом водорода, ацильную группу или алкоксикарбонильную группу; и
(с) стадия тиоэтерификации С-конца второго интермедиата в реакции второго интермедиата с тиолом для обмена группы -NH-C(=Y)NHR3 на тиоловую группу на С-конце.
Эффекты изобретения
В соответствии с настоящим изобретением обеспечивают новый способ химического превращения полипептидной цепи в тиоэфир пептида.
В способе настоящего изобретения может быть тиоэтерифицирована пептидная цепь, не имеющая ненативное производное аминокислоты, линкера и определенной трехмерной структуры и т.п., необходимых для традиционных способов тиоэтерификации. Следовательно, даже длинноцепочечный полипептидный фрагмент, полученный с помощью биосинтеза и т.п., может быть легко тиоэтерифицирован.
Кроме того, путем комбинирования способа настоящего изобретения с традиционным способом пептидного синтеза имеющий частичные пептидные модификации длинноцепочечный полипептид, который до настоящего времени было трудно синтезировать, может быть легко и просто получен путем изготовления фрагмента той части, которая не имеет модификаций, биосинтетическим способом, с помощью которого длинная цепь может быть сравнительно легко синтезирована, и изготовления способом твердофазного синтеза фрагмента той части, которая имеет модификацию, и их дотирования.
Точнее говоря, более длинный пептид с полисахаридной цепью может быть получен легко и просто с помощью химического синтеза отдельного фрагмента, содержащего аминокислоты, к которым добавлена природная связывающаяся форма полисахаридной цепи, если модификацию проводят с цепью полисахарида, получения другой части с помощью биосинтеза и тиоэтерификации с помощью способа настоящего изобретения, и их лигирования.
Способ последовательного добавления полисахаридной цепи и тому подобного к пептидной цепи через линкер также общеизвестен, и, таким образом, также можно последовательно добавлять цепь полисахарида к биосинтезированному длинноцепочечному пептиду. Однако этот способ присоединения полисахаридной цепи через линкер позволяет связывать цепь полисахарида и тому подобное с помощью определенной аминокислоты и ее структуры. Следовательно, например, если в пептиде присутствуют множество участков, способных связывать цепь полисахарида, то цепь полисахарида может быть добавлена более легко и просто сайт-специфическим способом по сравнению с традиционными способами путем вырезания фрагмента пептида, содержащего единственный желаемый участок связывания из длинноцепочечного пептида после получения длинноцепочечного пептида путем биосинтеза, добавления к нему полисахаридной цепи, тиоэтерификации фрагмента пептида с добавленной полисахаридной цепью с помощью способа тиоэтерификации настоящего изобретения и снова лигирования с ним оставшейся части.
Кроме того, при биосинтезе, даже если полноразмерный белок нормально экспрессируется, то его пептидный фрагмент может быть ошибочно распознан, деградирован или неправильно экспрессирован в клетке. Также возможно, что после синтеза полноразмерного белка вырезают только отдельный его фрагмент, который будет модифицирован, с ним проводят необходимую обработку, такую как модификация, модифицированный фрагмент пептида тиоэтерифицируют с помощью способа настоящего изобретения, и тиоэтерифицированный фрагмент снова лигируют с оставшейся частью и получают желаемый модифицированный белок.
Как описано выше, способ тиоэтерификации пептидов настоящего изобретения обычно применяют для синтеза белков.
Осуществление предпочтительных воплощений
Ниже будут описаны соответствующие воплощения настоящего изобретения.
Настоящее изобретение обеспечивает новый способ получения тиоэфира пептида, включающий следующие стадии (а)-(с):
(а) стадия получения первого интермедиата путем взаимодействия соединения А, представленного следующей формулой (I), с тиоловой группой остатка цистеина в пептидной цепи, имеющей остаток цистеина, для отщепления R2:
[Химическая формула 8]
в которой Х представляет собой атом серы или атом кислорода, R1 и R2 представляют собой замещаемые группы;
(b) стадия взаимодействия соединения В, представленного следующей формулой (II), с первым интермедиатом в органическом растворителе для добавления группы -NH-C(=Y)NHR3 к карбоксильной группе, образующей пептидную связь между остатком цистеина и аминокислотой, прилегающей к N-концевой стороне остатка цистеина, и расщепления пептидной связи, с получением, таким образом, фрагмента пептида с N-концевой стороны, ближе к N-концевой стороне, чем расщепленная пептидная связь, в качестве второго интермедиата:
[Химическая формула 9]
в которой Y представляет собой атом кислорода, атом серы или NH-группу, и R3 представляет собой атом водорода, ацильную группу или алкоксикарбонильную группу; и
(с) стадия тиоэтерификации С-конца второго интермедиата в реакции второго интермедиата с тиолом для обмена группы -NH-C(=Y)NHR3 на тиоловую группу на С-конце.
В настоящем изобретении термин «пептид» особенно не ограничен пептидом, длиной в две или более аминокислоты, связанными через амидную связь(амидные связи), и включает общеизвестные пептиды, новые пептиды и модифицированные пептиды. Пептиды, обычно обозначаемые как белок, включены в термин «пептиды» в настоящем изобретении. Также в настоящем изобретении термин «полипептид» включен в термин «пептиды». Пептидная цепь, применяемая в способе настоящего изобретения, может представлять собой нативный белок или пептидную цепь, полученную с помощью таких способов, как биосинтез, химический синтез или бесклеточный синтез.
В настоящем изобретении «модифицированный пептид» включает природные варианты пептидов, пептиды, претерпевшие посттрансляционную модификацию, или искусственно модифицированные соединения. Такая модификация включает, например, алкилирование, ацилирование (например, ацетилирование), амидирование (например, амидирование С-конца пептида), карбоксилирование, образование сложного эфира, образование дисульфидной связи, гликозилирование, присоединение липидов, фосфорилирование, гидроксилирование, присоединение меченого компонента и т.п. к одному или нескольким аминокислотным остаткам в пептиде.
В настоящем изобретении термин «аминокислота» применяют в наиболее широком его значении, и он включает не только природные аминокислоты, такие как серии (Ser), аспарагин (Asn), валин (Val), лейцин (Leu), изолейцин (Ile), аланин (Ala), тирозин (Tyr), глицин (Gly), лизин (Lys), аргинин (Arg), гистидин (His), аспарагиновая кислота (Asp), глутаминовая кислота (Glu), глутамин (Gln), треонин (Thr), цистеин (Cys), метионин (Met), фенилаланин (Phe), триптофан (Trp) и пролин (Pro), но также и неприродные аминокислоты, такие как варианты и производные аминокислот. Специалистам в этой области техники ясно, что аминокислоты в настоящем изобретении включают, например, L-аминокислоты; D-аминокислоты, химически модифицированные аминокислоты, такие как варианты и производные аминокислот; такие аминокислоты, как норлейцин, β-аланин и орнитин, которые не представляют собой материал для построения белков in vivo; и химически синтезированные соединения, обладающие свойствами аминокислот, известные специалистам в этой области техники, и т.п., в соответствии с этим широким определением.
В настоящем изобретении пептидная цепь, которую предполагается тиоэтерифицировать, особым образом не ограничена, при условии, что пептидная цепь содержит остаток цистеина. Например, происхождение, способ синтеза, размер и т.п. пептидной цепи особым образом не ограничены. Пептидная цепь также может иметь модификацию и защитную группу.
Число остатков цистеина, содержащихся в пептидной цепи, примененной в настоящем изобретении, особым образом не ограничено, и пептидную цепь расщепляют, нацеливаясь на остатки цистеина. Следовательно, необходимо сконструировать основной углеродный скелет конечного синтезированного белка в зависимости от участков, имеющих остаток цистеина, и специалисты в этой области техники могут легко создать такой основной скелет. Остатки цистеина, отличные от желаемых остатков цистеина, могут быть защищены защитными группами заранее, для того чтобы тиоэтерификация проходила только по желаемым остаткам цистеина, а остальные остатки цистеина не будут вступать в реакцию в пептидной цепи, содержащей множество остатков цистеина. Примеры такой защитной группы включают Acm-группу.
Пептидная цепь, примененная в настоящем изобретении, может иметь жирорастворимую защитную группу на N-концевой стороне. Предпочтительные защитные группы могут включать, но не ограничиваться, ацильные группы, такие как ацетильная (Ас) группа, карбонилсодержащие группы, такие как mpem-бутилоксикарбонильная (Boc) группа, 9-флуоренилметоксикарбонильная (Fmoc) группа и аллилоксикарбонильная (Alloc) группа, и аллилльную группу и бензильную группу.
Пептидная цепь, применяемая в способе настоящего изобретения, может представлять собой природный белок или пептидную цепь, полученную с помощью таких способов как биосинтез, химический синтез или бесклеточный синтез, и, предпочтительно, представляет собой рекомбинантный белок, экспрессируемый в бактериальной клетке или в клетке. Рекомбинантный белок может быть белком, имеющим такую же пептидную последовательность как в природном белке, или белком с пептидной последовательностью, имеющей модификацию, такую как метку для мутации или очистки, при условии, что белок искусственно экспрессируют в бактериальной клетке или в клетке.
Рекомбинантный белок, примененный в настоящем изобретении, может быть получен способом, известным специалистам в этой области техники. Например, рекомбинантный белок может быть экспрессирован путем введения целевого гена в рекомбинантный вектор. Рекомбинантный вектор, примененный в настоящем изобретении, может быть вектором, способным к трансформированию клетки-хозяина, и применяют плазмиду для Escherichia coli, плазмиду для Bacillus subtilis, плазмиду для дрожжей и векторы, представляющие собой вирусы животных, такие как ретровирус, вирус осповакцины и бакуловирус. Эти векторы предпочтительно имеют регуляторную последовательность, такую как промотор, способную к экспрессии соответствующим образом белка в клетке-хозяине. Кроме того, клетка-хозяин может представлять собой клетку, способную к экспрессии чуждого гена в рекомбинантном векторе, и обычно применяют клетки Escherichia coli. Bacillus subtilis, дрожжей, клетки насекомых и животные клетки.
Обычно применяемый способ, в общем, может быть применен как способ трансфецирования клетки-хозяина рекомбинантным вектором. Например, в случае Escherichia coli может быть применен способ с применением хлорида кальция и способ электропорации, а в случае дрожжей - способ с применением хлорида лития и способ электропорации. Трансформация животной клетки может быть выполнена с помощью физического способа, такого как способ электропорации, химического способа, такого как способ с применением липосом и способ с применением фосфата кальция, или с помощью вирусного вектора, такого как ретровирус. Условия культивирования клетки-хозяина, которую трансформируют, могут быть выбраны с учетом потребностей в питании и физиологических свойств клетки-хозяина.
Предпочтительно, чтобы пептид, примененный в настоящем изобретении, был очищен. Пептид может быть очищен с помощью традиционных способов очистки. Например, в случае рекомбинантного белка, если культивируют бактериальную клетку или клетку, экспрессирующую рекомбинантный белок, примененный в настоящем изобретении, то бактериальные клетки или клетки последовательно собирают с помощью известных способов, затем суспендируют в подходящем буферном растворе, разрушают под действием ультразвука, лизосом и/или замораживания и оттаивания и затем центрифугированием или фильтрацией получают раствор неочищенного экстракта пептида. Буферный раствор может содержать денатурирующее белок средство, такое как мочевина и гуанидин гидрохлорид, и поверхностно-активное соединение, такое как Triton Х-100 ТМ. Пептид, содержащийся в растворе экстракта или культуральном супернатанте и полученный как описано выше, может быть очищен с помощью известных способов очистки. Например, пептид может быть выделен и очищен с помощью соответственным образом подобранных и скомбинированных способов аффинной хроматографии, ионно-обменной хроматографии, фильтрации, ультрафильтрации, гель-фильтрации, электрофореза, высаливания, диализа и т.п.
Метка для очистки может быть введена в вектор экспрессии с целью облегчить очистку рекомбинантного белка. Примеры метки для очистки включают, например, His-метку, GST-метку, Мус-метку, FLAG-метку и мальтоза-связывающий белок (МВР). В настоящем изобретении N-концевую сторону Cys, размещенного внутри пептидной цепи, тиоэтерифицируют, следовательно, метку для очистки добавляют к С-концевой стороне пептида, и пептид после очистки тиоэтерифицируют, в результате чего С-концевую сторону Cys в пептидной цепи, которая включает метку, вырезают, и тиоэфир пептида может быть эффективно получен. Поместив Cys в желаемом положении на пептидной цепи, также возможно применить способ настоящего изобретения для удаления метки на С-концевой стороне.
Следовательно, способ удаления метки для очистки, добавленной на С-конце рекомбинантного белка, способом получения тиоэфира пептида настоящего изобретения также включен в настоящее изобретение.
В настоящем изобретении термин «тиоэфир пептида» (в дальнейшем в этой заявке иногда также просто называемый «тиоэфиром») означает пептид, имеющий карбокситиоэфирную группировку (-C=O-SR) на С-конце. Тиоэфир пептида, примененный в настоящем изобретении, особым образом не ограничен, поскольку тиоэфир может вызывать обменную реакцию с другими тиоловыми группами. R-группа включает, например, группы, приведенные ниже в качестве примера для R4.
В способе настоящего изобретения первой (а) проводят стадию реагирования соединения А с тиоловой группой остатка цистеина в пептидной цепи, имеющей остаток цистеина, для получения первого интермедиата.
В настоящем изобретении соединение А представлено следующей формулой (I).
[Химическая формула 10]
В этой формуле Х представляет собой атом серы или атом кислорода и предпочтительно атом серы.
R1 и R2 особым образом не ограничены при условии, что они имеют сниженную нуклеофильность по сравнению с атомом или группой атомов, которые будут замещены, и они будут удалены в условиях реакции следующей стадии (а) как замещаемые группы, и предпочтительно, чтобы R1 и R2 представляли собой замещаемые группы, отличающиеся друг от друга. Примеры R1 и R2 специфически включают атомы галогенов, замещенные или незамещенные -O-алкильные группы, замещенные или незамещенные -O-алкенильные группы, замещенные или незамещенные -O-алкинильные группы, замещенные или незамещенные -O-арильные группы, замещенные или незамещенные -O-гетероарильные группы, замещенные или незамещенные -S-алкильные группы, замещенные или незамещенные -S-алкенильные группы, замещенные или незамещенные -S-алкинильные группы, замещенные или незамещенные -S-арильные группы, или замещенные или незамещенные -S-гетероарильные группы. Более предпочтительно примеры R1 и R2 включают комбинацию R1, которая представляет собой замещаемую группу, которую выбирают из группы, состоящей из замещенных или незамещенных -0-С6-10 арильных групп и замещенных или незамещенных -S-C1-8 алкильных групп и R2, которая представляет собой замещаемую группу, которую выбирают из группы, состоящей из атомов галогена, замещенных или незамещенных -S-C1-8 алкильных групп и замещенных или незамещенных -S-С6-10 арильных групп.
В настоящем изобретении термин «алкильная группа» представляет собой моновалентную группу, получаемую из алифатического углеводорода путем удаления любого одного атома водорода, и которая имеет подмножество гидрокарбилов или углеводорода, содержащего водород и атомы углерода. Алкильная группа включает структуру с неразветвленной цепью или с разветвленной цепью. Алкильная группа настоящего изобретения предпочтительно включает алкильные группы, имеющие 1-8 атомов углерода. Термин «C1-8-алкильная группа» обозначает алкильную группу, имеющую 1-8 атомов углерода, и его специфические примеры включают метальные, этильные, пропильные, бутильные, пентильные, гексильные, гептильные и октильные группы.
В настоящем изобретении термин «алкенильная группа» представляет собой моновалентную группу, имеющую, по меньшей мере, одну двойную связь. Геометрические формы двойных связей могут принимать противоположную (Entgegen (Е)), одностороннюю (Zusammen (Z)), цис- или транс-конфигурации в зависимости от конфигурации двойных связей и заместителей. Алкенильная группа включает форму с неразветвленной цепью или с разветвленной цепью. Алкенильная группа настоящего изобретения предпочтительно включает алкенильные группы, имеющие 2-8 атомов углерода. Термин «C2-8 -алкенильная группа» обозначает алкенильную группу, имеющую 2-8 атомов углерода, и ее специфические примеры включают винильную, аллилльную, пропенильную, бутенильную, пентенильную, гексенильную, гептенильную и октенильную группы.
В настоящем изобретении термин «алкинильная группа» представляет собой моновалентную группу, имеющую, по меньшей мере, одну тройную связь. Алкинильная группа включает алкинильные группы с неразветвленной цепью или с разветвленной цепью. Алкинильная группа настоящего изобретения предпочтительно включает алкинильные группы, имеющие 2-8 атомов углерода. Термин «С2-8-алкинильная группа» обозначает алкинильную группу, имеющую 2-8 атомов углерода, и ее специфические примеры включают этинильные, 1-пропинильные, 2-пропинильные, бутинильные, пентинильные, гексинильные, гептинильные и октинильные группы.
В настоящем изобретении термин «арильная группа» означает группу ароматического углеводородного кольца. Арильная группа настоящего изобретения предпочтительно включает арильные группы, имеющие 6-10 атомов углерода. Термин «С6-10-арильная группа» обозначает арильную группу, имеющую 6-10 атомов углерода, и ее специфические примеры включают фенильные, 1-нафтильные и 2-нафтильные группы.
В настоящем изобретении «гетероарильная группа» означает моновалентную или бивалентную группу, получаемую из гетероарильного кольца путем удаления одного или двух атомов водорода в любом положении(в любых положениях). В настоящем изобретении термин «гетероарильное кольцо» означает ароматическое кольцо, имеющее один или несколько гетероатомов в атомах, составляющих кольцо, и предпочтительно 5-9-членные кольца. Кольцо может представлять собой моноциклическую или бициклическую гетероарильную группу, полученную с помощью слияния с бензольным кольцом или моноциклическим гетероарильным кольцом. Ее специфические примеры включают фуранильные, тиофенильные, пирролильные, бензофуранильные, бензотиофенильные, индолильные, пиридильные и хинолильные группы.
Типы, число и положения заместителей, которые имеют вышеупомянутые замещаемые группы, особым образом не ограничены, и примеры заместителей включают алкильные, алкенильные, алкокси-, арильные, формильные, карбонильные, карбоксильные, алкилкарбоксильные, алкоксикарбонильные, галогены, сульфонильные или нитрогруппы.
Точнее, соединение А настоящего изобретения включает следующее.
[Химическая формула 11]

[Химическая формула 12]
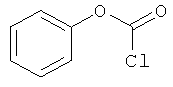
[Химическая формула 13]
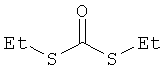
[Химическая формула 14]

Также вышеописанное соединение [Химическая формула 15]
или [Химическая формула 16]
может реагировать с МРАА ((4-карбоксиметил)тиофенол) для получения следующего тионоформиатного реагента, который также быть применен:
[Химическая формула 17]

и
[Химическая формула 18]

Первый интермедиат, в котором группа -C(=X)-R1 связывается с SH-группой в остатке цистеина, как показано на следующей фигуре, может быть получен путем реакции соединения А настоящего изобретения с остатком цистеина в пептиде.
[Химическая формула 19]
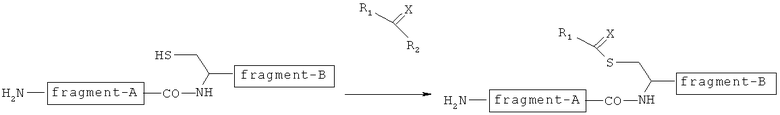
В настоящем изобретении стадию (а) предпочтительно проводят в кислых условиях, в особенности при рН 3-5. Реакцию предпочтительно проводят в смешанном растворителе, состоящем из буферного раствора и ацетонитрила при 0-50ºС, предпочтительно при 15-25ºС, в течение примерно 0,1-3 часов, предпочтительно от 10 минут до одного часа, но не ограничиваясь этим.
Затем в настоящем изобретении проводят стадию (b), в которой получают в качестве второго интермедиата фрагмент пептида с N-концевой стороны, который расположен ближе к N-концевой стороне, чем расщепленная пептидная связь, путем реакции соединения В с первым интермедиатом в органическом растворителе для добавления группы -NH-C(=Y)NHR3 к карбоксильной группе, формирующей пептидную связь с аминокислотой, прилегающей к N-концевой стороне остатка цистеина, и расщепления пептидной связи.
В настоящем изобретении соединение В представлено следующей формулой (II).
[Химическая формула 20]
В этой формуле Y представляет собой атом кислорода, NH-группу или атом серы, и R3 представляет собой атом водорода, ацильную группу или алкоксикарбонильную группу.
В настоящем изобретении термин «ацильная группа» означает группу атомов, получаемую с помощью удаления ОН-группы из карбоксильной группы карбоновой кислоты. Ацильная группа настоящего изобретения предпочтительно включает ацильную группу, имеющую 1-4 атома углерода, и ее специфические примеры включают ацетильные, пропионильные и бутироильные группы.
В настоящем изобретении термин «алкокси-группа» означает окси-группу, присоединенную к «алкильной группе». Алкокси-группа настоящего изобретения может быть группой с неразветвленной цепью или с разветвленной цепью. Алкокси-группа настоящего изобретения предпочтительно включает алкокси-группы с неразветвленной цепью, имеющие 1-14 атомов углерода, и алкокси-группы с разветвленной цепью, имеющие 3-14 атомов углерода. В особенности могут быть включены, например, метокси-, этокси-, н-пропилокси, изопропокси-, н-бутокси-, 2-метил-2-пропилокси-, н-пентилокси- и н-гексилокси-группы.
Также термин «С2-n-алкоксикарбонильная группа» означает карбонильную группу, имеющую С1-(n-1)-алкокси-группу. Алкоксикарбонильная группа настоящего изобретения предпочтительно включает алкоксикарбонильную группу, имеющую 2-15 атомов углерода. В особенности могут быть включены, например, метоксикарбонильные, этоксикарбонильные, н-пропилоксикарбонильные, изопропоксикарбонильные, н-бутоксикарбонильные, 2-метил-2-пропилоксикарбонильные, н-пентилоксикарбонильные и н-гексилоксикарбонильные группы.
Ацильная группа предпочтительно включает ацетильную группу. Также алкоксикарбонильная группа предпочтительно включает отреот-бутоксикарбонильную (Boc) группу.
В особенности соединение В настоящего изобретения включает следующее.
[Химическая формула 21]

[Химическая формула 22] NH

[Химическая формула 23]

[Химическая формула 24]

В настоящем изобретении стадию (b) предпочтительно проводят в присутствии органического растворителя. Предпочтительно, чтобы органический растворитель обладал высокой растворяющей способностью и низкой нуклеофильностью. Такой органический растворитель может включать, например, DMSO, DMF и диоксан. Реакцию предпочтительно проводят при 0-50ºС, предпочтительно при 15-25ºС, в течение примерно 1-24 часов, предпочтительно в течение 5-10 часов, но не ограничиваясь этим.
Пептидную цепь расщепляют на N-концевой стороне остатка цистеина, как показано на следующей фигуре, путем добавления группы -NH-C(=Y)NHR3 к карбоксильной группе, формирующей пептидную связь между остатком цистеина и аминокислотой, расположенной рядом с N-концевой стороной остатка цистеина.
[Химическая формула 25]
Если пептид имеет аминогруппу в своей боковой цепи, то жирорастворимая защитная группа может быть введена в аминогруппу в боковой цепи перед проведением стадии (b) настоящего изобретения. Жирорастворимая защитная группа может включать, но не ограничиваться, такие защитные группы, как карбонилсодержащие группы, такие как 9-флуоренилметоксикарбонильная (Fmoc) группа, mpem-бутилоксикарбонильная (Boc) группа и аллилоксикарбонильная (Alloc) группа, как ацильные группу, такие как ацетильная (Ас) группа, и аллильная группа и бензильная группа.
С целью введения жирорастворимой защитной группы, например, Fmoc-группа может быть введена путем добавления 9-флуоренилметил-N-сукцинимидилкарбоната и гидрокарбоната натрия и проведения реакции с ними. Реакцию предпочтительно проводят при 0-50ºС, предпочтительно при комнатной температуре в течение примерно 1-5 часов, но не ограничиваясь этим.
Фрагмент пептида на N-концевой стороне, который находится ближе к N-концевой стороне, чем расщепленный участок расщепленной пептидной цепи, может быть получен в качестве второго интермедиата по следующей формуле (1) в стадии (b).
[Химическая формула 26]

Способ получения тиоэфира пептида настоящего изобретения дополнительно включает стадию (с) тиоэтерификации С-конца второго интермедиата в реакции тиола со вторым интермедиатом для обмена группа -NH-C(=Y)NHR3 на С-конце на тиоловую группу.
Второй интермедиат, применяемый в стадии (с), может быть выделен или его не нужно выделять после стадии (b).
В предпочтительных воплощениях в стадии (с) применяют тиол, представленный следующей формулой (III):
R4-SH (Формула III).
R4 особым образом не ограничено при условии, что он не ингибирует реакцию тиольного обмена и становится замещаемой группой в реакции замещения на углероде карбонила. Предпочтительно, R4 представляет собой любую группу, которую выбирают из замещенных или незамещенных бензильных групп, замещенных или незамещенных арильных групп и замещенных или незамещенных алкильных групп. Более предпочтительно, R4 представляет собой любую группу, которую выбирают из замещенных или незамещенных бензильных групп, замещенных или незамещенных С6-10-арильных групп и замещенных или незамещенных C1-8-алкильных групп. В особенности R4 может быть выбран из замещаемых групп бензильного типа, таких как бензилмеркаптан, из замещаемых групп арильного типа, таких как тиофенол и 4-(карбоксиметил)тиофенол, из замещаемых групп алкильного типа, таких как группа 2-меркаптоэтансульфоновой кислоты и амид 3-меркаптопропионата и т.п. Тип, число и положения заместителей, которые эти замещаемые группы имеют, особым образом не ограничены.
Второй интермедиат полностью превращают в тиоэфир согласно следующей фигуре путем проведения стадии (с).
[Химическая формула 27]

Тиоэфир пептида, полученный так, как описано выше, может быть лигирован с пептидом (или модифицированным пептидом), который содержит остаток аминокислоты, имеющей -SH-группу на N-конце среди пептидов или модифицированных пептидов с помощью способа лигирования. Следовательно, настоящее изобретение также обеспечивает способ получения полипептида, включающий стадию связывания тиоэфира пептида, полученного с помощью способа настоящего изобретения, с пептидной цепью, имеющей цистеин на N-конце способом лигирования.
Также возможно применять второй интермедиат, полученный в стадии (b), вместо описанного выше тиоэфира пептида для способа лигирования.
В настоящем изобретении термин «способ лигирования» включает не только нативный химический способ лигирования (способ NCL), описанный в Патентной литературе 1, но также случаи применения нативного химического способа лигирования к пептидам, содержащим неприродные аминокислоты и производные аминокислот (например, производное треонина А, защищенный метионин, аминокислоты с присоединенной полисахаридной цепью и т.п.). Пептид, имеющий природную амидную связь (пептидную связь) в месте лигирования, может быть получен способом лигирования.
Лигирование с помощью способа лигирования может быть проведено в любом случае между пептидом и пептидом, между пептидом и модифицированным пептидом и между модифицированным пептидом и модифицированным пептидом.
Термины настоящей заявки применяют для описания определенных аспектов, и они не предназначены для ограничения настоящего изобретения.
Термин «включающий» (также «содержащий» и «включающий»), примененные в настоящей заявке, означают, что описанные аспекты (участники, стадии, элементы и цифры и т.п.) присутствуют, исключая случаи, очевидно отличные, как это ясно из контекста, и не исключая то, что аспекты (участники, стадии, элементы и цифры и т.п.), отличные от указанных, присутствуют.
Если не определено иначе, все термины (включая технические термины и научные термины), примененные в настоящей заявке, имеют те же значения, как их широко понимают специалисты в той области техники, к которой настоящее изобретение относится. Термины, примененные в настоящей заявке, должны быть истолкованы как имеющие значения, когерентные значениям в этом описании и в родственной области техники пока не появится другое определение и не должны быть истолкованы в идеализованных или слишком формальных значениях.
Аспекты настоящего изобретения иногда были описаны с отсылками к схемам. При описании с помощью схемы воплощение иногда выражено в чрезмерно расширенной манере с целью его более ясного описания.
Такие термины, как первый и второй, применяют для обозначения различных элементов, но понятно, что эти элементы не ограничены этими терминами. Эти термины применяют только для того, чтобы отличить один элемент от другого элемента, и без отступления от объема настоящего изобретения возможно, чтобы первый элемент был описан как второй элемент, также как второй элемент был описан как первый элемент.
Настоящее изобретение будет описано более подробно с отсылкой к следующим примерам. Однако настоящее изобретение может быть осуществлено с помощью различных аспектов и не должно быть истолковано как ограниченное описанными в этой заявке примерами.
Примеры
Пример 1. Введение тионоформиатной группы (Синтез МРАА фенилтионоформиата) [Химическая формула 28]

МРАА ((4-карбоксиметил)тиофенол) (98 мг, 0,583 ммоль) и фенилхлортионоформиат (103 мкл, 0,76 ммоль) растворяют в дихлорметане (400 мкл) и смесь перемешивают при комнатной температуре в течение одного часа. Через один час реакционный раствор разводят 2,0 мл хлороформа, добавляют 1,0 мл водного раствора насыщенного бикарбоната натрия и смесь экстрагируют и промывают хлороформом. Слой хлороформа промывают насыщенным солевым раствором, высушивают на сульфате магния и затем концентрируют при пониженном давлении и получают желтый прозрачный остаток в виде сиропа. Затем полученное применяют в качестве тионоформиатного реагента (МРАА фенилтионоформиат) (MW: 305,3, MS: нет доступных данных).
(Введение тионоформиатной группы с помощью МРАА фенилтионоформиатного реагента)
Пептид (Ac-Val Try Ala Xaa Cys Gly-OH) (SEQ ID NO: 1), Xaa=Lys (SEQ ID NO: 2), Ser (SEQ ID NO: 3), Asp (SEQ ID NO: 4), Ala (SEQ ID NO: 5), Val (SEQ ID NO: 6), неочищенный (смесь Lys, Ser, Asp, Ala и Val), 6 мг) растворяют в буферном растворе при рН 5,5 (1,0 мл 0,2 М Na2HPO4 и 6 М Gn-HCl) и затем к этому добавляют общее количество МРАА фенилтионоформиата (15 мкл), растворенного в ацетонитриле (230 мкл). Через один час реакционный раствор промывают Et2O. Очистки проводят с помощью HPLC и получают целевое соединение. Реакция проходит количественно благодаря применению HPLC.
(Xaa=Lys, ESIMS по расчету [М+Н]+ 818,3, найдено [М+Н]+ 818,4)
(Xaa=Ser, ESIMS по расчету [М+Н]+ 777,3, найдено [М+Н]+ 777,3)
(Xaa=Asp, ESIMS по расчету [М+Н]+ 805,3, найдено [М+Н]+ 805,3)
(Хаа=Ala, ESIMS по расчету [M+H]+ 761,3, найдено [М+Н]+ 761,3)
(Xaa=Val, ESIMS по расчету [М+Н]+ 789,3, найдено [M+H]+ ---)
Пептид (Ac-Val Try Ala Xaa Cys Gly-OH) (SEQ ID NO: 1), Xaa-Ser (SEQ ID NO: 3), Phe (SEQ ID NO: 8), Leu (SEQ ID NO: 7), неочищенный (смесь Ser, Phe и Leu), 10 мг) растворяют в буферном растворе при рН 5,0 (2,0 мл 0,2 М Na2HPO4 и 6 М Gn-HCl) и затем к этому добавляют МРАА фенилтионоформиат (5 мкл), растворенный в ацетонитриле (700 мкл). Через 1,5 часа реакционный раствор промывают Et2O. Очистку проводят с помощью HPLC и получают целевое соединение. Реакция проходит количественно благодаря применению HPLC.
(Xaa=Ser, ESIMS по расчету [М+Н]+ 777,3, найдено [М+Н]+ 777,3)
(Xaa=Leu, ESIMS по расчету [М+Н]+ 803,4, найдено [M+H]+ 803,3)
(Xaa=Phe, ESIMS по расчету [М+Н]+ 837,4, найдено [М+Н]+ 837,3)
Тионоформиатную группу вводят в -SH-группу цистеина независимо от того, аминокислота какого типа прилегает к N-концевой стороне цистеина.
Пример 2. Реакция N-ацетилгуанидинилирования
[Химическая формула 29]
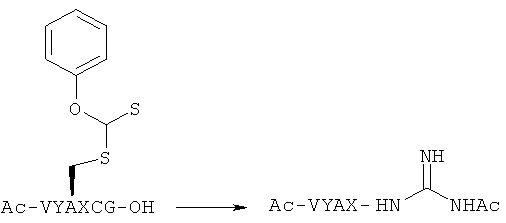
(Проводят с Хаа=Ala, Leu, Phe, Ser и Lys)
(Случай, когда Хаа=Ala)
Ac-Val Tyr Ala Ala Cys(C(S)OPh)Gly-OH (SEQ ID NO: 9) (0,2 мг, 0,28 мкмоль) растворяют в растворе 250 мМ N-ацетилгуанидина/DMSO (260 мкл). Через два часа соединение осаждают и промывают Et2O. Целевое соединение очищают с помощью HPLC и получают целевой N-ацетилгуанидидо-(Ас-Val Tyr Ala Ala-NHC(NH)NHAc (SEQ ID No: 10) (выход: 80%, рассчитан по площади пика HPLC).
(ESIMS по расчету [М+Н]+ 548,3, найдено [М+Н]+ 548,4)
(Случай, когда Xaa=Leu или Phe)
Смесь Ac-Val Tyr Ala Leu Cys(C(S)OPh)Gly-OH (SEQ ID NO: 11) (0,1 мг, 0,12 мкмоль) и Ac-Val Tyr Ala Phe Cys(C(S)OPh)Gly-OH (SEQ ID NO: 12) (0,1 мг, 0,12 мкмоль) растворяют в растворе 250 мМ N-ацетилгуанидина/DMSO (100 мкл). Через 4,5 часа соединения осаждают и промывают Et2O. Целевые соединения очищают с помощью HPLC и получают целевые N-ацетилгуанидидо-(Ас-Val Tyr Ala Leu-NHC(NH)NHAc (SEQ ID No: 13) и Ac-Val Tyr Ala Phe-NHC(NH)NHAc (SEQ ID No: 14) (выход: 80%, рассчитан по площади пика HPLC).
(Xaa=Leu, ESIMS по расчету [М+Н]+ 590,3, найдено [М+Н]+ 590,3)
(Xaa=Phe, ESIMS по расчету [M+H]+ 624,3, найдено [М+Н]+ 624,3)
(Случай, когда Xaa=Ser)
Ac-Val Tyr Ala Ser Cys(C(S)OPh)Gly-OH (SEQ ID NO: 15) (0,2 мг, 0,26 мкмоль) растворяют в растворе 250 мМ N-ацетилгуанидина/DMSO (100 мкл). Через 3,5 часа соединение осаждают и промывают Et2O. Целевое соединение очищают с помощью HPLC и получают целевой N-ацетилгуанидидо-(Ас-Val Tyr Ala Ser-NHC(NH)NHAc (SEQ ID No: 16) (выход: 70%, по площади пика HPLC).
(Случай, когда Xaa=Lys)
Пептид (Ac-Val Tyr Ala Lys Cys(C(S)OPh)Gly-OH (SEQ ID NO: 17) (0,1 мг) растворяют в растворе DMSO (30 мкл), содержащем Вос2О (0,3 мг) и триэтаноламин (0,14 мкл). Через 1,5 часа реакционный раствор осаждают и промывают Et2O. Полученный осадок растворяют в растворе 250 мМ N-ацетилгуанидина/DMSO (100 мкл). Через 2,5 часа целевое соединение очищают с помощью HPLC и получают целевой N-ацетилгуанидидо-(Ас-Val Tyr Ala Lys(Boc)-NHC(NH)NHAc (SEQ ID No: 18) (выход: 70%, рассчитан по площади пика HPLC).
Было установлено, что этот остаток цистеина, к которому присоединен тионоформиат, обладает реактивностью с гуанидином к пептидной связи на N-концевой стороне независимо от типа аминокислоты, прилегающей к N-концевой стороне цистеина.
Пример 3. Тиоэтерификация 24 аа пептида
[Химическая формула 30]

Подходящее количество (оценочно, примерно 1 мг) неочищенного (грубого) пептида (H2N-Leu Ile Cys(Acm)Asp Ser Arg Val Leu Glu Arg Tyr Leu Leu Glu Ala Lys Glu Ala Glu Asn Ile Thr Thr Gly Cys Gly-OH (SEQ ID NO: 19) растворяют в буферном растворе при рН 5,0 (300 мкл 0,2 М Na2HPO4 и 6 М Gn-HCl) и затем к этому добавляют общее количество МРАА фенилтионоформиата (1 мкл), растворенного в ацетонитриле (100 мкл). Через 50 минут реакционный раствор промывают Et2O. Очистку проводят с помощью HPLC и получают целевое соединение (H2N-Leu Ile Cys(Acm)Asp Ser Arg Val Leu Glu Arg Tyr Leu Leu Glu Ala Lys Glu Ala Glu Asn Ile Thr Thr Gly Cys(C(S)OPh)Gly-OH (SEQ ID NO: 20)). Cys в положении 3, предварительно защищенный Acm, не подвергается действию тионоформиатного реагента.
(ESIMS по расчету [M+2H]2+ 1553,8, [М+3Н]3+ 1035,8, найдено [M+2H]2+ 1552,9, [М+3Н]3+ 1035,7)
Пептид (H2N-Leu Ile Cys(Acm)Asp Ser Arg Val Leu Glu Arg Tyr Leu Leu Glu Ala Lys Glu Ala Glu Asn Ile Thr Thr Gly Cys(C(S)OPh)Gly-OH (SEQ ID NO: 20), прим. 0,3 мг) растворяют в DMSO (20 мкл), содержащем Boc2O (0,4 мг) и триэтаноламин (0,03 мкл). Через 1,5 часа реакционный раствор осаждают и промывают Et2O. Полученный осадок растворяют в растворе 250 мМ N-ацетилгуанидина/DMSO (50 мкл). Через 2,5 часа целевое соединение очищают с помощью HPLC и получают целевой N-ацетилгуанидидо-(BocHN-Leu Ile Cys(Acm)Asp Ser Arg Val Leu Glu Arg Tyr Leu Leu Glu Ala Lys(Boc)Glu Ala Glu Asn Ile Thr Thr Gly-NHC(NH)NHAc (SEQ ID NO: 21)).
(ESIMS по расчету [М+2H]2+ 1546,8, [М+3Н]3+ 1031,5, найдено [М+2H]2+ 1547,0, [М+3Н]3+ 1031,4)
24 аа пептид (BocHN-Leu Ile Cys(Acm)Asp Ser Arg Val Leu Glu Arg Tyr Leu Leu Glu Ala Lys(Boc)Glu Ala Glu Asn Ile Thr Thr Gly-NHC(NH)NHAc (SEQ ID NO: 21), прим. 0,1 мг>) растворяют в буферном растворе при рН 7,05 (0,2 М фосфорная кислота, 6 М гуанидин, 50 мкл), содержащем MES-Na (2-сульфанилэтансульфонат натрия) (1 мг, 2% объем/объем). Через 3,5 часа целевое соединение очищают с помощью HPLC и получают тиоэфир (BocHN-Leu Ile Cys(Acm)Asp Ser Arg Val Leu Glu Arg Tyr Leu Leu Glu Ala Lys(Boc)Glu Ala Glu Asn Ile Thr Thr Gly-SCH2CH2SO3 (SEQ ID NO: 22)) (выход не известен, примерно 70% по HPLC).
(ESIMS по расчету [М+2H]2+ 1567,3, найдено [М+2H]2+ 1566,8)
Пример 4. Введение тионоформиатной группы с помощью хлортионоформиатного реагента
Пептид (Ac-Val Tyr Ala Ala Cys Gly-OH (SEQ ID NO: 5), 6 мг) растворяют в буферном растворе при рН 5,0 (961 мкл 0,2 М Na2HPO4 и 6 М Gn-HCl) и к этому добавляют фенилхлортионоформиат (6,5 мкл), растворенный в ацетонитриле (320 мкл). Через один час реакционный раствор промывают Et2O. Очистку проводят с помощью HPLC и получают целевой пептид с присоединенным тионоформиатом (SEQ ID NO: 9) (6,4 мг, 88%).
(Xaa=Ala, ESIMS по расчету [М+Н]+ 761,3, найдено [М+Н]+ 761,3)
Пептид (Ac-Val Tyr Ala Leu Cys Gly-OH (SEQ ID NO: 7), 3,4 мг) растворяют в буферном растворе при рН 5,0 (510 мкл 0,2 М Na2HPO4 и 6 М Gn-HCl) и к этому добавляют фенилхлортионоформиат (3,5 мкл), растворенный в ацетонитриле (170 мкл). Через один час реакционный раствор промывают Et2O. Очистку проводят с помощью HPLC и получают целевой пептид с присоединенным тионоформиатом (SEQ ID N0: 11) (3,8 мг, 92%).
(Xaa=Leu, ESIMS по расчету [M+H]+ 803,4, найдено [М+Н]+ 803,3)
Пептид (Ac-Val Tyr Ala Phe Cys Gly-OH (SEQ ID NO: 8), 5,1 мг) растворяют в буферном растворе при рН 5,0 (729 мкл 0,2 М Na2HPO4 и 6 М Gn-HCl) и к этому добавляют фенилхлортионоформиат (5,0 мкл), растворенный в ацетонитриле (243 мкл). Через один час реакционный раствор промывают Et2O. Очистку проводят с помощью HPLC и получают целевой пептид с присоединенным тионоформиатом (SEQ ID NO: 12) (5,1 мг, 84%).
(Xaa=Phe, ESIMS по расчету [M+H]+ 837,4, найдено [М+Н]+ 837,3)
Пептид (Ac-Val Tyr Ala Ser Cys Gly-OH (SEQ ID NO: 3), 4,9 мг) растворяют в буферном растворе при рН 5,0 (766 мкл 0,2 М Na2HPO4 и 6 М Gn-HCl) и к этому добавляют фенилхлортионоформиат (5,2 мкл), растворенный в ацетонитриле (265 мкл). Через один час реакционный раствор промывают Et2O. Очистку проводят с помощью HPLC и получают целевой пептид с присоединенным тионоформиатом (SEQ ID NO: 15) (5,5 мг, 92%).
(Xaa=Ser, ESIMS по расчету [М+Н]+ 777,3, найдено [М+Н]+ 777,3)
Пептид (Ac-Val Tyr Ala Lys Cys Gly-OH (SEQ ID NO: 2), 5,5 мг) растворяют в буферном растворе при рН 5,0 (810 мкл 0,2 М Na2HPO4 и 6 М Gn-HCl) и к этому добавляют фенилхлортионоформиат (5,5 мкл), растворенный в ацетонитриле (270 мкл). Через один час реакционный раствор промывают Et2O. Очистку проводят с помощью HPLC и получают целевой пептид с присоединенным тионоформиатом (SEQ ID NO: 17) (6,1 мг, 94%).
(Xaa=Lys, ESIMS по расчету [М+Н]+ 818,3, найдено [М+Н]+ 818,4)
С помощью хлортионоформиатного реагента получают такую же пептидную цепь с присоединенным тионоформиатом как та, что была получена в примере 1.
Следовательно, было обнаружено, что тиоэфир пептида также может быть получен из пептидной цепи, в которую тионоформиатную группу вводят с помощью хлортионоформиатного реагента путем присоединения N-ацетилгуанидидо-группы и затем тиоэтерификации таким же способом как в пептидной цепи, в которую вводят тионоформиатную группу в приведенных выше примерах 1 и 3.
Промышленная применимость
Согласно настоящему изобретению обеспечен новый способ химического превращения полипептидной цепи в тиоэфир пептида.
В способе настоящего изобретения тиоэтерификацию можно проводить в пептидной цепи, которая не имеет неприродного производного аминокислоты, линкера или определенной трехмерной структуры и т.п., необходимых для традиционного способа тиоэтерификации, и возможно легко провести тиоэтерификацию даже в длинноцепочечном полипептидном фрагменте, полученном с помощью биосинтеза и т.п. Следовательно, способ тиоэтерификации настоящего изобретения может быть в целом применен для синтеза белков.












Изобретение относится к новому способу химического превращения пептидной цепи в тиоэфир пептида. Группу -C(=X)-R1 вводят в тиоловую группу остатка цистеина, и затем полученный пептид в органическом растворителе реагирует с соединением, имеющим замещаемую группу, представленную формулой: -NH-C(=Y)NHR3, и группа -NH-C(=Y)NHR3 связывается в реакции присоединения с карбоксильной группой пептидной связи на N-концевой стороне остатка цистеина, посредством чего пептидную связь расщепляют и фрагмент пептида на С-концевой стороне вырезают. Когда полученная пептидная цепь, имеющая группу -NH-C(=Y)NHR3, реагирует с тиолом в буферном растворе, то происходит реакция обмена тиолов, а именно тиоловая группа соединения тиола связывается с углеродом карбонила, к которому была присоединена группа -NH-C(=Y)NHR3, посредством чего группу -NH-C(=Y)NHR3 удаляют. Таким образом, достигают превращения в тиоэфир пептида. 6 н. и 8 з.п. ф-лы, 4 пр.
1. Способ получения тиоэфира пептида, включающий следующие стадии (a)-(c):
(a) стадия получения первого интермедиата путем взаимодействия соединения A, представленного следующей формулой (I), с тиоловой группой остатка цистеина в пептидной цепи, имеющей остаток цистеина, для отщепления R2:

в которой Х представляет собой атом серы или атом кислорода, и R1 и R2 представляют собой замещаемые группы;
(b) стадия взаимодействия соединения В, представленного следующей формулой (II), с указанным первым интермедиатом в органическом растворителе для добавления группы -NH-C(=Y)NHR3 к карбоксильной группе, образующей пептидную связь между остатком цистеина и аминокислотой, прилегающей к N-концевой стороне указанного остатка цистеина, и расщепления указанной пептидной связи, с получением, таким образом, фрагмента пептида с N-концевой стороны, который расположен ближе к N-концевой стороне, чем расщепленная пептидная связь, в качестве второго интермедиата:
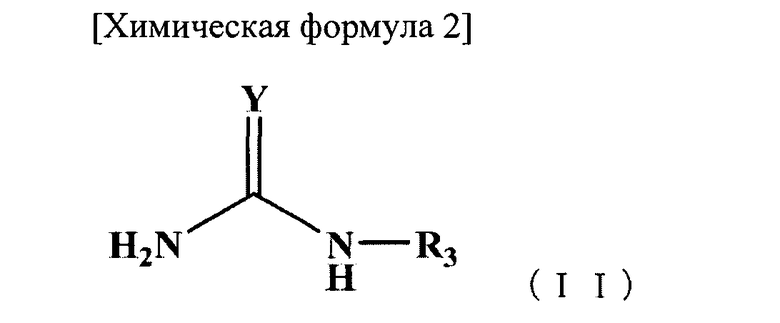
в которой Y представляет собой атом кислорода, атом серы или NH-группу, и R3 представляет собой атом водорода, ацильную группу или алкоксикарбонильную группу; и
(c) стадия тиоэтерификации C-конца второго интермедиата в реакции тиола со вторым интермедиатом для обмена группы -NH-C(=Y)NHR3 на C-конце на тиоловую группу.
2. Способ по п.1, в котором X представляет собой атом серы.
3. Способ по п.1, в котором R1 представляет собой -O-C6 арильную группу.
4. Способ по п.1, в котором R2 представляет собой атом галогена или замещенную или незамещенную -S-C6-10 арильную группу.
5. Способ по п.1, в котором Y представляет собой NH-группу.
6. Способ по п.1, в котором R3 представляет собой ацетильную группу.
7. Способ по п.1, в котором тиол в стадии (c) представляет собой тиол, представленный следующей формулой (III):
R4-SH (Формула III)
в которой R4 представляет собой любую группу, которую выбирают из замещенной или незамещенной бензильной группы, замещенной или незамещенной арильной группы и замещенной или незамещенной алкильной группы.
8. Способ по п.1, в котором пептидная цепь представляет собой рекомбинантный белок.
9. Способ по любому из пп.1-8, в котором пептидная цепь представляет собой рекомбинантный белок, включающий метку для очистки.
10. Способ получения полипептида, включающий:
(a) стадию получения первого интермедиата путем взаимодействия соединения A, представленного следующей формулой (I), с тиоловой группой остатка цистеина в пептидной цепи, имеющей остаток цистеина, для отщепления R2:

в которой X представляет собой атом серы или атом кислорода, и R1 и R2 представляют собой замещаемые группы;
(b) стадию взаимодействия соединения В, представленного следующей формулой (II), с указанным первым интермедиатом в органическом растворителе для добавления группы -NH-C(=Y)NHR3 к карбоксильной группе, образующей пептидную связь между остатком цистеина и аминокислотой, прилегающей к N-концевой стороне указанного остатка цистеина, и расщепления указанной пептидной связи, с получением, таким образом, фрагмента пептида с N-концевой стороны, который расположен ближе к N-концевой стороне, чем расщепленная пептидная связь, в качестве второго интермедиата:

в которой Y представляет собой атом кислорода, атом серы или NH-группу, и R3 представляет собой атом водорода, ацильную группу или алкоксикарбонильную группу; и
(c) стадию тиоэтерификации C-конца второго интермедиата в реакции тиола со вторым интермедиатом для обмена группы -NH-C(=Y)NHR3 на C-конце на тиоловую группу;
(в) стадию связывания полученного тиоэфира пептида с пептидной цепью, имеющей цистеин на N-конце, способом лигирования.
11. Способ получения второго интермедиата, применяемого в способе получения тиоэфира пептида по любому из следующих пп.1-9, включающий следующую стадию (a) или (b):
(a) стадия получения первого интермедиата путем взаимодействия соединения A, представленного следующей формулой (I), с тиоловой группой остатка цистеина в пептидной цепи, имеющей остаток цистеина для отщепления R2:

в которой X представляет собой атом серы или атом кислорода, и R1 и R2 представляют собой замещаемые группы; или
(b) стадия взаимодействия соединения B, представленного следующей формулой (II), с указанным первым интермедиатом в органическом растворителе для добавления группы -NH-C(=Y)NHR3 к карбоксильной группе, образующей пептидную связь между остатком цистеина и аминокислотой, прилегающей к N-концевой стороне указанного остатка цистеина, и расщепление указанной пептидной связи, с получением, таким образом, фрагмента пептида с N-концевой стороны, который расположен ближе к N-концевой стороне, чем расщепленная пептидная связь, в качестве второго интермедиата:


в котором Y представляет собой атом кислорода, атом серы или NH-группу, и R3 представляет собой атом водорода, ацильную группу или алкоксикарбонильную группу.
12. Пептидная цепь, имеющая группу -NH-C(=Y)NHR3, в которой Y представляет собой атом кислорода или NH-группу, и R3 представляет собой атом водорода, ацильную группу или алкоксикарбонильную группу, на ее C-конце.
13. Способ получения полипептида, включающий стадию связывания пептидной цепи, имеющей группу -NH-C(=Y)NHR3 на своем C-конце, по п.12 с пептидной цепью, имеющей цистеин на своем N-конце способом лигирования.
14. Способ удаления метки для очистки, присоединенной к C-концевой части рекомбинантного белка, включающий следующие стадии (a)-(c):
(a) стадия получения первого интермедиата путем взаимодействия соединения A, представленного следующей формулой (I), с тиоловой группой остатка цистеина в рекомбинантном белке, имеющим остаток цистеина и включающем метку для очистки на своем C-конце, для отщепления R2:

в которой X представляет собой атом серы или атом кислорода, и R1 и R2 представляют собой замещаемые группы;
(b) стадия взаимодействия соединения B, представленного следующей формулой (II), с указанным первым интермедиатом в органическом растворителе для добавления группы -NH-C(=Y)NHR3 к карбоксильной группе, образующей пептидную связь между остатком цистеина и аминокислотой, прилегающей к N-концевой стороне указанного остатка цистеина, и расщепление указанной пептидной связи, с получением, таким образом, фрагмента пептида с N-концевой стороны, который расположен ближе к N-концевой стороне, чем расщепленная пептидная связь, в качестве второго интермедиата:


в которой Y представляет собой атом кислорода, атом серы или NH-группу, и R3 представляет собой атом водорода, ацильную группу или алкоксикарбонильную группу; и
(c) стадия тиоэтерификации C-конца второго интермедиата в реакции тиола со вторым интермедиатом для обмена группы -NH-C(=Y)NHR3 на C-конце на тиоловую группу.
| WO 2007043615 A1, 19.04.2007 | |||
| WO 2002098902 A2, 12.12.2002 | |||
| WO 1996034878 A1, 07.11.1996 | |||
| WO 2007114454 A1, 11.10.2007 | |||
| СПОСОБ ПОЛУЧЕНИЯ ПЕПТИДА | 2008 |
|
RU2478105C2 |
| BRASK J | |||
| ET AL.: 'Fmoc solid-phase synthesis of peptide thioesters by masking as trithioortho esters' ORG | |||
| Способ и приспособление для нагревания хлебопекарных камер | 1923 |
|
SU2003A1 |
| Кипятильник для воды | 1921 |
|
SU5A1 |
| Устройство для электрической сигнализации | 1918 |
|
SU16A1 |
| JOHNSON EC | |||
| ET AL.: 'Insights into the | |||

