Область техники, к которой относится изобретение
Данное изобретение относится к способу получения сложных эфиров α-гидроксикарбоновых кислот из амида α-гидроксикарбоновой кислоты и алифатических спиртов.
Уровень техники изобретения
Способ, при котором спирт взаимодействует с нитрилом в присутствии кислотного катализатора, давно известен как способ получения сложных эфиров α-гидроксикарбоновых кислот. Так, например, способ, при котором лактонитрил растворяют в спирте и воде, в которые добавляют серную кислоту, чтобы гидролизовать и этерифицировать его, и при котором в полученную реакционную смесь вводят газообразный спирт, раскрывается как способ получения сложных эфиров молочной кислоты (смотрите патентные документы 1 и 2). Кроме того, способ, при котором ацетонциангидрин взаимодействует со спиртом в присутствии кислотного катализатора, известен как способ получения сложных эфиров α-гидроксиизомасляной кислоты (смотрите патентные документы 3 и 4).
Более того, уже предлагались различные способы в качестве способа получения сложных эфиров α-гидроксикарбоновых кислот из амидов α-гидроксикарбоновой кислоты и алифатических спиртов. В качестве, например, жидкофазных реакций, известны способы, при которых амид α-гидроксиизомасляной кислоты или лактамид взаимодействует со спиртами в присутствии алкоголята металла, нерастворимого твердого кислотного катализатора, растворимого металлокомплекса, содержащего титан и/или олово, и амида α-гидроксикарбоновой кислоты в качестве общих ингредиентов, или металлической соли трифторметансульфокислоты в качестве катализатора (смотрите патентные документы с 5 по 11).
Патентный документ 1: Японская патентная публикация № 8061/1955
Патентный документ 2: Японская патентная публикация № 2333/1965
Патентный документ 3: Патент США № 2041820
Патентный документ 4: Японская выложенная заявка на патент № 230241/1992
Патентный документ 5: Японская выложенная заявка на патент № 3015/1977
Патентный документ 6: Японский патент № 3222639
Патентный документ 7: Японская выложенная заявка на патент № 258154/1995
Патентный документ 8: Японская выложенная заявка на патент № 279120/1999
Патентный документ 9: Японская выложенная заявка на патент № 26370/2000
Патентный документ 10: Японская выложенная заявка на патент № 292824/1999
Патентный документ 11: Японская выложенная заявка на патент № 169432/2000
Сущность изобретения
Способы, раскрытые в патентных документах с 1 по 4, требуют больших производственных затрат, поскольку требуется большое количество кислотных катализаторов, требуется наличие реакционного оборудования с использованием коррозионостойких материалов, чтобы сделать возможным применение вышеуказанных кислотных катализаторов, необходимы большие количества энергии и финансовых затрат для того, чтобы переработать и повторно использовать непрореагировавшие исходные вещества и аммиак, а также возникает необходимость избавляться от ненужных соединений, таких как сульфат аммония и т.п., образующихся в больших количествах, и их надо дополнительно усовершенствовать, прежде чем осуществлять в промышленных масштабах.
К тому же реакции, раскрытые в патентных документах с 5 по 11, являются равновесными реакциями, и аммиак, полученный в реакциях, следует удалять из систем для того, чтобы увеличить степени превращения исходных веществ. Были сделаны различные предложения по вышеуказанному вопросу, но каждый из применяемых способов требует больших количеств энергии и финансовых затрат для переработки аммиака и спиртов, и они не обязательно являются способами, подходящими для промышленного производства с точки зрения экономики.
Данное изобретение создавалось под влиянием вышеописанных обстоятельств, и задачей данного изобретения является обеспечение способа получения сложных эфиров α-гидроксикарбоновых кислот из амида α-гидроксикарбоновой кислоты и алифатических спиртов, отличающегося тем, что этот способ имеет сниженные производственные затраты и увеличенные степень превращения и селективность, и является выгодным с промышленной точки зрения.
Интенсивные исследования, проведенные авторами изобретения для того, чтобы решить задачу, описанную выше, привели к обнаружению того, что высокая степень превращения по сравнению с жидкофазной реакцией достигается при подвергании амида α-гидроксикарбоновой кислоты и алифатических спиртов газофазной реакции в присутствии катализатора на основе диоксида циркония, и было обнаружено, что это дает возможность успешно получать сложные эфиры α-гидроксикарбоновых кислот. В дальнейшем авторы обнаружили, что применение циркониевого катализатора, содержащего особый элемент, дает возможность в значительной степени увеличить срок службы катализатора. Данное изобретение было создано на основе вышеописанных сведений.
Т.е. данное изобретение относится к следующим пунктам с (1) по (7).
(1) Способ получения сложного эфира α-гидроксикарбоновой кислоты, отличающийся тем, что амид α-гидроксикарбоновой кислоты и алифатический спирт подвергают газофазной реакции в присутствии катализатора на основе диоксида циркония.
(2) Способ получения сложного эфира α-гидроксикарбоновой кислоты по вышеописанному пункту (1), в котором катализатор на основе диоксида циркония содержит по меньшей мере один элемент, выбираемый из 2-4 групп, 7 группы и из 9-13 групп Периодической таблицы элементов, лантаноидов, сурьмы (Sb) и висмута (Bi).
(3) Способ получения сложного эфира α-гидроксикарбоновой кислоты по вышеописанному пункту (1), в котором катализатор на основе диоксида циркония содержит по меньшей мере один элемент, выбираемый из бора (B), алюминия (Al), марганца (Mn), кобальта (Co), никеля (Ni), иттрия (Y), ланатана (La) и иттербия (Yb).
(4) Способ получения сложного эфира α-гидроксикарбоновой кислоты по любому из вышеописанных пунктов с (1) по (3), в котором амид α-гидроксикарбоновой кислоты представлен следующей формулой (I):
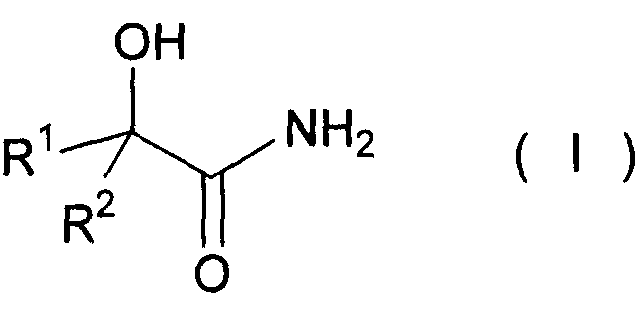
(где R1 и R2, каждый независимо представляет собой атом водорода, алкильную группу, содержащую от 1 до 20 атомов углерода, алкенильную группу, содержащую от 2 до 20 атомов углерода, или циклоалкильную группу, содержащую от 3 до 20 атомов углерода, образующих кольцо), и алифатический спирт представлен формулой R3OH (где R3 представляет собой алкильную группу, содержащую от 1 до 20 атомов углерода, алкенильную группу, содержащую от 2 до 20 атомов углерода, или циклоалкильную группу, содержащую от 3 до 20 атомов углерода, образующих кольцо).
(5) Способ получения сложного эфира α-гидроксикарбоновой кислоты по любому из вышеописанных пунктов с (1) по (3), в котором амид α-гидроксикарбоновой кислоты является лактамидом, или амидом α-гидроксиизомасляной кислоты.
(6) Способ получения сложного эфира α-гидроксикарбоновой кислоты по любому из вышеописанных пунктов с (1) по (5), в котором алифатический спирт является метанолом или этанолом.
(7) Способ получения сложного эфира α-гидроксикарбоновой кислоты по любому из вышеописанных пунктов с (1) по (6), в котором температура реакции составляет от 150 до 270°С, и давление реакции составляет от 1 до 300 кПа.
В соответствии со способом получения, относящимся к данному изобретению, отходы, такие как сульфат аммония, утилизация которых требует больших затрат, не образуются. Также, поскольку в данном изобретении достигнута высокая степень превращения по сравнению с жидкофазной реакцией, нет необходимости удалять из системы вместе с растворителем образующийся аммиак, и энергетические затраты на выпаривание растворителя во время реакции могут быть исключены. Более того, по утверждению авторов изобретения, сложные эфиры α-гидроксикарбоновых кислот можно получать с высокой селективностью.
ЛУЧШИЙ ВАРИАНТ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
Способ получения сложного эфира α-гидроксикарбоновой кислоты
Данное изобретение относится к способу получения сложного эфира α-гидроксикарбоновой кислоты, отличающемуся тем, что амид α-гидроксикарбоновой кислоты и алифатический спирт подвергают газофазной реакции в присутствии катализатора на основе диоксида циркония.
Катализатор на основе диоксида циркония
Катализатор на основе диоксида циркония применяется в качестве катализатора в данном изобретении. С точки зрения увеличения срока службы катализатора, предпочтительно применяют катализатор на основе диоксида циркония, содержащий по меньшей мере один элемент (далее в данном документе именуемый как дополнительный элемент), выбираемый из 2-4 групп, 7 группы и из 9-13 групп Периодической таблицы элементов, лантаноидов, сурьмы (Sb) и висмута (Bi). Вышеуказанный дополнительный элемент более предпочтительно, с точки зрения увеличения срока службы катализатора, является одним из элементов 3 группы, 7 группы, 9 группы, 10 группы и 13 группы Периодической таблицы элементов, еще более предпочтительно бором (B), алюминием (Al), марганцем (Mn), кобальтом (Co), никелем (Ni), иттрием (Y), ланатаном (La) и иттербием (Yb). Помимо указанных выше дополнительных элементов, могут содержаться другие элементы, и их содержание с точки зрения степени превращения и селективности составляет предпочтительно 30% по массе или меньше, более предпочтительно 15% по массе или меньше и еще более предпочтительно 5% по массе или меньше, исходя из массы дополнительного элемента.
Когда катализатор на основе диоксида циркония содержит описанный выше дополнительный элемент, содержание дополнительного элемента с точки зрения увеличения срока службы катализатора составляет от 0,1 до 30% по количеству моль, более предпочтительно от 0,5 до 10% по количеству моль, еще более предпочтительно от 1 до 8% по количеству моль и особенно предпочтительно от 1 до 7% по количеству моль, исходя из суммарного количества моль циркония и дополнительного элемента.
Для катализатора на основе диоксида циркония можно использовать коммерчески доступные ингредиенты, или можно использовать заранее полученные ингредиенты. Также для катализатора на основе диоксида циркония с тем же успехом можно использовать диоксид циркония или гидрат диоксида циркония, полученный путем надлежащей промывки катализатора из гидроксида циркония водой, его сушки и последующего его прокаливания. Более того, диоксид циркония, содержащий гидроксид циркония, можно также использовать в качестве катализатора на основе диоксида циркония.
Коммерчески доступные ингредиенты катализатора на основе диоксида циркония, или катализатора на основе гидроксида циркония, который является его предшественником, включают в себя, например, серию “XZO1501”, серию “XZO632” и серию “XZO882”, изготавливаемые MEL Chemicals Inc., и “NN zirconium hydroxide”, “R zirconium hydroxide”, “RS zirconium hydroxide” и “RSC-HP” (все торговые названия), изготавливаемые закрытой акционерной компанией Daiichi Kigennso Kagaku Kogyo Co.
С точек зрения степени превращения и селективности, а также с точки зрения получения устойчивых результатов реакции предпочтительно используют катализатор на основе диоксида циркония, размер частиц которого контролируют таким образом, чтобы он предпочтительно составлял от 3,5 до 40 меш, более предпочтительно от 5 до 30 меш.
Способ приготовления катализатора на основе диоксида циркония
Способ приготовления катализатора на основе диоксида циркония не должен быть особым образом ограничен, и можно применять способы по выбору. Исходное вещество можно выбрать из, например, элементарного циркония и оксида, гидроксида, хлорида, неорганических и органических солей циркония. Более того, в качестве исходного вещества можно применять все вещества, которые могут стать диоксидом циркония или гидратом диоксида циркония при их химической обработке, прокаливании и т.п. Если быть точнее, вышеупомянутое исходное вещество включает в себя, например, диоксид циркония, ацетилацетонат циркония, хлорид циркония, оксихлорид циркония, оксинитрат циркония, изопропилат циркония, сульфат циркония и т.п.
Способ осаждения также используется в качестве способа получения. Если быть точнее, это способ, при котором вышеописанные исходные вещества, такие как оксихлорид циркония, оксинитрат циркония и т.п. подвергают реакции со щелочами, такими как аммиак, амин, гидроксид натрия, карбонат натрия, бикарбонат натрия, карбонат аммония, бикарбонат аммония и т.п., чтобы получить таким образом белый осадок гидроксида циркония, и при котором полученный таким образом гидроксид циркония промывают достаточным количеством воды, затем сушат и прокаливают с получением гидрата диоксида циркония или диоксида циркония.
Способ добавления дополнительных элементов, описанных выше, в катализатор на основе диоксида циркония не должен быть особым образом ограничен, и можно применять общеизвестные способы. Удобным для применения является, например, способ пропитки, при котором диоксид циркония, гидрат диоксида циркония или гидроксид циркония, которые являются коммерчески доступными или приготовленными, пропитывают солью, содержащей дополнительный элемент, которая переведена в состояние раствора. Кроме того, также удобным для применения является способ, при котором диоксид циркония, гидрат диоксида циркония или гидроксид циркония, которые являются коммерчески доступными или приготовленными, растирают с оксидом металла, гидроксидом металла или солью, содержащей дополнительный элемент. При этом с тем же успехом можно применять способ совместного осаждения, способ совместного гелеобразования, способ ионообмена и т.п.
Катализатор на основе диоксида циркония, содержащий вышеописанный дополнительный элемент, можно получить путем прокаливания смесей, полученных вышеописанными способами.
Способ пропитки, описанный выше, можно осуществить, более конкретно, путем погружения диоксида циркония, гидрата диоксида циркония или гидроксида циркония в раствор, предварительно полученный путем растворения источника дополнительного элемента в растворителе. Вышеуказанным растворителем предпочтительно является вода. При этом любые соединения дополнительного элемента можно применять в качестве источника дополнительного элемента при условии, что они являются растворимыми в применяемом растворителе, и можно применять, например, хлориды, нитраты, сульфаты, соли органических кислот и т.п. Однако, когда хлориды и сульфаты применяют в качестве источника обеспечения элементом, ионы галогена и сульфата, каждые из которых остаются в катализаторе, необходимо удалять путем достаточной промывки для того, чтобы препятствовать ухудшению селективности ионами галогена и сульфата, которые увеличивают кислотность катализатора, и по этой причине применение нитратов и солей органических кислот в качестве источника элемента является особенно предпочтительным. После пропитки растворитель удаляют, и твердое вещество прокаливают или сушат, в результате чего можно получить целевой катализатор на основе диоксида циркония.
Также можно осуществить описанный выше способ совместного осаждения таким образом, чтобы источник элемента присутствовал в системе реакции при проведении операции получения белого осадка гидроксида циркония в описанном выше способе осаждения. Источником элемента являются предпочтительно соли, которые растворимы в воде, и применяют, например, хлориды, нитраты, сульфаты, соли органических кислот и т.п. Нитраты и соли органических кислот являются предпочтительными по той же причине, как и в случае описанного выше способа пропитки.
«Прокаливание», описанное выше, обычно во всех случаях проводят в атмосфере воздуха, но его можно проводить в атмосфере инертного газа. Температура прокаливания обычно во всех случаях составляет от 300 до 700°С, предпочтительно от 400 до 500°С. Обычно время прокаливания составляет предпочтительно от 1 до 6 часов.
Процесс формования катализатора на основе диоксида циркония можно осуществить с помощью способа, известного обычному специалисту в данной области, и, например, можно применять экструзионное формование, формование прессованием и т.п. При формовании можно применять вспомогательные средства для формования, или компоненты катализатора могут наноситься на носитель.
Амид α-гидроксикарбоновой кислоты
Амид α-гидроксикарбоновой кислоты, применяемый в данном изобретении, не должен быть особым образом ограничен при условии, что он является амидным соединением карбоновой кислоты, содержащим гидроксильную группу в α-положении, и с точки зрения его практической ценности в качестве исходного вещества для лекарственных средств и сельскохозяйственных химикатов, он предпочтительно является соединением, представленным в виде следующей формулы (I):
(где R1 и R2, каждый независимо представляет собой атом водорода, алкильную группу, содержащую от 1 до 20 атомов углерода, алкенильную группу, содержащую от 2 до 20 атомов углерода, или циклоалкильную группу, содержащую от 3 до 20 атомов углерода, образующих кольцо).
Алкильная группа, содержащая от 1 до 20 атомов углерода, отображенная независимо при помощи R1 и R2 соответственно, может являться линейной или разветвленной и включает в себя, например, метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, различные пентилы («различные» включает в себя линейную и все разветвленные структуры; то же самое относится к нижеописанному в данном документе), различные гексилы, различные октилы, различные децилы, различные додецилы, различные тетрадецилы, различные гексадецилы и т.п. С точек зрения простоты получения исходных веществ и их практической ценности в качестве исходных веществ для лекарственных средств и сельскохозяйственных химикатов, вышеуказанная алкильная группа предпочтительно является алкильной группой, содержащей от 1 до 10 атомов углерода, более предпочтительно алкильной группой, содержащей от 1 до 5 атомов углерода, а еще более предпочтительно метилом.
Алкенильная группа, содержащая от 2 до 20 атомов углерода, отображенная независимо с помощью R1 и R2 соответственно, может являться линейной или разветвленной и включает в себя, например, винил, различные пропенилы, различные бутенилы, различные гексенилы, различные октенилы, различные деценилы, различные додеценилы, различные тетрадеценилы, различные гексадеценилы, различные октадеценилы и т.п. Вышеуказанная алкенильная группа предпочтительно является алкенильной группой, содержащей от 2 до 10 атомов углерода, более предпочтительно алкенильной группой, содержащей от 2 до 5 атомов углерода.
Циклоалкильная группа, содержащая от 3 до 20 атомов углерода, образующих кольцо, отображенная независимо с помощью R1 и R2 соответственно, включает в себя, например, циклопентил, циклооктил, циклогептил, циклодецил и т.п. Вышеуказанная циклоалкильная группа предпочтительно является циклоалкильной группой, содержащей от 3 до 10 атомов углерода, образующих кольцо, более предпочтительно циклоалкильной группой, содержащей от 3 до 8 атомов углерода, образующих кольцо.
Среди них оба из R1 и R2, с точек зрения простоты получения исходных веществ и их практической ценности в качестве исходных веществ для лекарственных средств и сельскохозяйственных химикатов, предпочтительно являются атомом водорода или алкильной группой, содержащей от 1 до 20 атомов углерода, более предпочтительно атомом водорода или алкильной группой, содержащей от 1 до 10 атомов углерода, еще более предпочтительно атомом водорода или алкильной группой, содержащей от 1 до 5 атомов углерода, и особенно предпочтительно атомом водорода или метилом. Т.е. амид α-гидроксикарбоновой кислоты особенно предпочтительно является лактамидом или амидом α-гидроксиизомасляной кислоты, исходя из тех же соображений.
Алифатический спирт
Алифатический спирт, применяемый в данном изобретении, не должен быть особым образом ограничен, и с точки зрения его практической ценности в качестве исходного вещества для лекарственных средств и сельскохозяйственных химикатов, он предпочтительно является соединением, представленным формулой R3OH (где R3 представляет собой алкильную группу, содержащую от 1 до 20 атомов углерода, алкенильную группу, содержащую от 2 до 20 атомов углерода, или циклоалкильную группу, содержащую от 3 до 20 атомов углерода, образующих кольцо).
Аналогичные группы, как и в случае R1 и R2, могут быть указаны в качестве примеров алкильной группы, содержащей от 1 до 20 атомов углерода, алкенильной группы, содержащей от 2 до 20 атомов углерода, и циклоалкильной группы, содержащей от 3 до 20 атомов углерода, образующих кольцо, каждая из которых отображается с помощью R3. Среди них, R3 с точки зрения практической ценности алифатического спирта в качестве исходного вещества для лекарственных средств и сельскохозяйственных химикатов предпочтительно является алкильной группой, содержащей от 1 до 20 атомов углерода, более предпочтительно алкильной группой, содержащей от 1 до 5 атомов углерода, и еще более предпочтительно метилом или этилом.
Конкретные примеры алифатического спирта, применяемого в данном изобретении, включают в себя метанол, этанол, н-пропиловый спирт, изопропиловый спирт, изобутиловый спирт, н-бутиловый спирт и т.д., и его специально выбирают в соответствии с типом целевого продукта. Метанол или этанол предпочитителен с точки зрения его практической ценности в качестве исходного вещества для лекарственных средств и сельскохозяйственных химикатов.
Используемое количество алифатического спирта составляет предпочтительно от 1 до 50 мольных эквивалентов, более предпочтительно от 2 до 30 мольных эквивалентов и еще более предпочтительно от 5 до 30 мольных эквивалентов в расчете на количество моль амида α-гидроксикарбоновой кислоты.
Газофазная реакция
Данное изобретение - это газофазная реакция, при которой твердый катализатор применяют в неподвижном слое или псевдоожиженном слое. В данном изобретении получена более высокая степень превращения при сравнении со степенью превращения в случае жидкофазной реакции, и было установлено, что это объясняется тем, что в данном изобретении, которое является газофазной реакцией, равновесие между исходными веществами и продуктом в значительной степени смещается в сторону продукта.
В данном изобретении «газофазная реакция» обозначает реакцию, преимущественно проводимую в газовой фазе, доля жидкой фазы в которой составляет 10% по массе или меньше, предпочтительно 5% по массе или меньше, более предпочтительно 2% по массе или меньше и еще более предпочтительно практически 0% по массе, исходя из суммарного количества исходных веществ. Амид α-гидроксикарбоновой кислоты и алифатический спирт, которые являются исходными веществами, можно превратить в пар до подачи в реактор или можно превратить в пар в реакторе. При этом амид α-гидроксикарбоновой кислоты и алифатический спирт можно подавать в реактор каждый по отдельности или можно подавать в реактор после смешивания.
Если реакция проводится в атмосфере инертного газа, такого как газообразный азот, то понижается парциальное давление исходных веществ за счет присутствия инертного газа, и они легко испаряются, и потому это является предпочтительным.
Когда амид α-гидроксикарбоновой кислоты и алифатический спирт, которые являются исходными веществами, испаряются в реакторе, то вышеуказанные исходные вещества можно подавать в реактор совместно с растворителем. Вышеупомянутый растворитель включает в себя, например, эфирный основный растворитель, такой как тетрагидрофуран и т.п.; амидный основный растворитель, такой как N-метилпирролидон и т.п.; и сложноэфирный основный растворитель, такой как метиллактат и т.п. В данном изобретении реакция протекает эффективно без применения вышеупомянутых растворителей.
Температура реакции устанавливается таким образом, чтобы амид α-гидроксикарбоновой кислоты и алифатический спирт, которые являются исходными веществами, испарялись в реакционной системе. Устанавливаемая температура изменяется в соответствии с различными условиями, такими как типы исходных веществ, применяемое мольное соотношение амида α-гидроксикарбоновой кислоты и алифатического спирта, присутствие или отсутствие инертного газа, присутствие или отсутствие растворителя, давление реакции и т.п. Обычно она предпочтительно составляет от 150 до 270°С, более предпочтительно от 170 до 250°С, еще более предпочтительно от 180 до 240°С и особенно предпочтительно от 180 до 210°С.
Когда амид α-гидроксикарбоновой кислоты является, например, амидом α-гидроксиизомасляной кислоты, то удовлетворительная скорость реакции достигается при установке температуры реакции равной 150°С или выше, но для того, чтобы достаточно испарить амид α-гидроксиизомасляной кислоты при атмосферном давлении, эффективным является проведение реакции при 180°С или выше. Кроме того, установка температуры реакции равной 240°С или ниже делает возможным уменьшение разложения амида α-гидроксиизомасляной кислоты на ацетон и некоторые количества таких побочных продуктов, как сложные эфиры α-алкоксиизомасляной кислоты и олефиновые производные, которые являются продуктами дегидрирования.
Обычно реакция проводится при атмосферном давлении или пониженном давлении. При этом реакцию можно проводить под давлением, если это является условием, при котором амид α-гидроксикарбоновой кислоты и алифатический спирт, являющиеся исходными веществами, испаряются.
Амид α-гидроксикарбоновой кислоты имеет высокую температуру кипения, например, амид α-гидроксиизомасляной кислоты имеет температуру кипения, равную 260°С, и по этой причине проще испарять исходные вещества при атмосферном давлении или пониженном давлении. Соответственно, давление реакции предпочтительно составляет от 1 до 300 кПа, более предпочтительно от 10 до 150 кПа, еще более предпочтительно от 30 до 120 кПа и особенно предпочтительно от 30 кПа до атмосферного давления.
Скорость подачи исходных веществ предпочтительно составляет, с точки зрения сохранения высокой степени превращения в течение длительного времени, от 0,01 до 5 ч-1, более предпочтительно от 0,02 до 2 ч-1, еще более предпочтительно от 0,03 до 1 ч-1 и особенно предпочтительно от 0,05 до 0,5 ч-1, исходя из массы амида α-гидроксикарбоновой кислоты на единицу массы катализатора, т.е. объемная скорость (WHSV), исходя из массы амида α-гидроксикарбоновой кислоты.
Аналогичным образом из соотношения используемых количеств амида α-гидроксикарбоновой кислоты и алифатического спирта можно посчитать объемную скорость (WHSV), исходя из массы алифатического спирта, и она составляет предпочтительно приблизительно от 0,01 до 100 ч-1, более предпочтительно от 0,04 до 40 ч-1 и еще более предпочтительно от 0,10 до 2 ч-1.
Способ отделения сложного эфира α-гидроксикарбоновой кислоты от продуктов, полученных при применении способа, описанного выше, не должен особым образом ограничиваться. Например, аммиак удаляется вместе с алифатическим спиртом путем проведения стандартной ректификации, чтобы получить сложный эфир α-гидроксикарбоновой кислоты, а также непрореагировавшие алифатический спирт и амид α-гидроксикарбоновой кислоты из нижней части ректификационной колонны, и эфир α-гидроксикарбоновой кислоты можно отделить путем проведения стандартной ректификации.
ПРИМЕРЫ
Данное изобретение должно быть конкретно изложено ниже со ссылкой на примеры, но данное изобретение никаким образом не должно ограничиваться примерами, показанными ниже.
Результаты реакции в соответствующих примерах были определены с помощью газового хроматографического анализа согласно следующим условиям.
Аналитические условия газовой хроматографии
Оборудование: 6850A (изготовленный Agilent Technologies Inc.)
Применяемая колонка: DB-WAX (изготовленная Agilent Technologies Inc.)
Аналитические условия: температура входного канала хроматографа 200°С, температура детектора 250°С.
Температура колонки: после выдерживания на уровне 50°С в течение 3 минут, температуру поднимали до 250°С со скоростью 15°С/минуту.
Детектор: детектор по теплопроводности (TCD)
Пример 1
Приготовление катализатора
Гидроксид циркония «XZO 1501/03», изготовленный MEL Chemicals Inc., отформовывали прессованием, и отформованное вещество измельчали и контролировали, чтобы размер частиц составлял от 10 до 20 меш. Чтобы приготовить катализатор на основе диоксида циркония, данное вещество сушили при 150°С в течение 3 часов, затем прокаливали при 400°С в течение 3 часов, и снова контролировали, чтобы размер частиц составлял от 10 до 20 меш.
Предварительная обработка
В реакционную трубку из нержавеющей стали марки SUS316, с внутренним диаметром 18 ммφ загружали 14 г приготовленного вышеописанным способом катализатора из диоксида циркония. Реакционную пробирку нагревали в электрической печи, обеспечивая в то же время, чтобы азот протекал при величине расхода 40 мл/минуту, и загруженный катализатор на основе диоксида циркония нагревали при 250°С в течение 3 часов и подвергали предварительной обработке.
Реакция
После доведения температуры каталитического слоя до 220°С подачу азота останавливали и обеспечивали, чтобы через реакционную трубку протекали исходные вещества в виде жидкости, приготовленной путем смешивания амида α-гидроксиизомасляной кислоты и метанола в соотношении 30 частей по массе к 70 частям по массе, при величине расхода потока, равной 4,67 г/час (WHSV, исходя из количества амида α-гидроксиизомасляной кислоты, =0,1 ч-1). Реакция практически 100% исходных веществ по массе проходила в реакционной трубке в газовой фазе.
Когда реакция достигала состояния равновесия по истечении приблизительно 24 часов, образцы продуктов брали с помощью ловушки, охлаждаемой льдом, и анализировали с помощью газовой хроматографии, обнаруживая, что степень превращения амида α-гидроксиизомасляной кислоты составляла 95%, и что селективность по метил α-гидроксиизобутирату составляла 88%. Реакцию продолжали, чтобы обнаружить, что степень превращения амида α-гидроксиизомасляной кислоты может сохраняться на уровне 90% или больше в течение 992 часов от начала реакции. Степень превращения и селективность по истечении приблизительно 24 часов, а также время, в течение которого степень превращения может сохраняться на уровне 90% или больше при продолжении реакции (далее в данном документе называемые просто результатами реакции) показаны в таблице 1.
Примеры 2-5
Приготовление катализатора
Катализаторы, указанные в таблице 1, каждый из которых изготовлен MEL Chemicals Inc., формовали прессованием, и отформованные вещества измельчали и контролировали, чтобы размер частиц составлял от 10 до 20 меш. «XZO 1501/10», «XZO 1501/07» и «XZO 882/03» (примеры 2, 3 и 5), которые представляют собой прокаленные катализаторы из диоксида циркония, сушили при 150°С в течение 12 часов или дольше, и непрокаленный катализатор «XZO 632/03» (пример 4) сушили, как и в случае примера 1, при 150°С в течение 3 часов, и снова контролировали, чтобы размер частиц составлял от 10 до 20 меш.
Катализаторы, описанные выше, были предоставлены MEL Chemicals Inc. в качестве образцов; «XZO 1501/10» является диоксидом циркония; «XZO 1501/07» является диоксидом циркония, полученным путем прокаливания «XZO 1501/03»; «XZO 882/03» является диоксидом циркония, полученным путем прокаливания гидроксида циркония; «XZO 632/03» является диоксидом циркония, полученным путем прокаливания гидроксида циркония, который отличается от серии «XZO 1501» и серии «XZO 632»; и при этом вышеуказанные катализаторы отличаются соответственно удельной площадью поверхности по методу BET, диаметром частиц, следами примесей, включающих в себя преимущественно металлы, и т.п.
Предварительная обработка и реакция
Предварительная обработка и реакция были проведены аналогичным образом, как в примере 1, за исключением того, что были использованы различные катализаторы на основе диоксида циркония, приготовленные вышеописанными способами. Результаты реакции показаны в таблице 1.
Пример 6
Приготовление катализатора
Гидроксид циркония «NN zirconium hydroxide», изготовленный закрытой акционерной компанией Daiichi Kigennso Kagaku Kogyo Co., формовали прессованием, и отформованное вещество измельчали и контролировали, чтобы размер частиц составлял от 10 до 20 меш. Данное вещество сушили, как и в случае примера 1, при 150°С в течение 3 часов, затем прокаливали при 400°С в течение 3 часов, и снова контролировали, чтобы размер частиц составлял от 10 до 20 меш.
Предварительная обработка и реакция
Предварительную обработку и реакцию проводили аналогичным образом, как в примере 1, за исключением того, что был использован катализатор на основе диоксида циркония, приготовленный вышеописанным способом. Результаты реакции показаны в таблице 1.
Пример 7
Приготовление катализатора
Гидроксид циркония «RSC-HP», изготовленный закрытой акционерной компанией Daiichi Kigennso Kagaku Kogyo Co., измельчали, контролировали, чтобы размер частиц составлял от 10 до 20 меш, и сушили при 150°С в течение 3 часов.
Предварительная обработка и реакция
Предварительную обработку и реакцию проводили аналогичным образом, как в примере 1, за исключением того, что был использован катализатор на основе диоксида циркония, приготовленный вышеописанным способом. Результаты реакции показаны в таблице 1.
Справочный пример 1
Предварительную обработку и реакцию проводили при аналогичных условиях, как в примере 3, за исключением того, что температуру (температуру реакции) слоя катализатора изменяли на равную 190°С.
Степень превращения амида α-гидроксиизомасляной кислоты по истечении приблизительно 24 часов составляла 37%, и селективность по метил α-гидроксиизобутирату составляла 94%. Поскольку в условиях вышеупомянутой реакции не было достигнуто достаточного давления паров исходных веществ, часть газообразных исходных веществ превратилась в жидкость в реакционной системе, и предполагалось, что это привело к снижению степени превращения. Результаты реакции по истечении приблизительно 24 часов показаны в таблице 2.
Пример 8
Предварительную обработку и реакцию провели при аналогичных условиях, как в случае справочного примера 1, за исключением того, что вместе с исходными веществами вводили азот при расходе 54 мл/минуту, для того, чтобы исходные вещества в достаточном объеме превратились в пар. Результаты реакции по истечении приблизительно 24 часов показаны в таблице 2. Из результатов, полученных в справочном примере 1 и примере 8, можно обнаружить, что для того, чтобы получить высокую степень превращения, важно довести долю присутствия жидкой фазы практически до 0% по массе.
Примеры 9-12
Предварительную обработку и реакцию проводили при аналогичных условиях, как в примере 3, за исключением того, что температуру (температуру реакции) слоя катализатора изменяли на равную 200°С, 230°С, 240°С и 250°С соответственно. Результаты реакции по истечении приблизительно 24 часов показаны в таблице 2.
Пример 13
Предварительную обработку и реакцию проводили при аналогичных условиях, как в примере 1, за исключением того, что используемое количество катализатора на основе диоксида циркония изменяли на равное 7 г (WHSV, исходя из количества амида α-гидроксиизомасляной кислоты, =0,2 ч-1). Результаты реакции показаны в таблице 3.
Сравнительный пример 1
Приготовление катализатора
56,3 г нитрата лантана (La(NO3)3·6H2O) растворяли в 400 г очищенной воды, после чего раствор нагревали до 50°С. Раствор, приготовленный путем растворения 40,6 г гидрофосфата натрия (Na2HPO4) в 400 г очищенной воды, добавляли к вышеуказанному раствору, и при этом образовывался белый осадок. Вышеуказанный белый осадок отделяли фильтрацией и промывали 600 г очищенной воды, и затем после сушки при 120°С в течение 3 часов, его прокаливали при 400°С в течение 6 часов в атмосфере воздуха, в результате чего получали катализатор на основе фосфата лантана (LaPO4). Этот катализатор измельчали и контролировали, чтобы размер частиц составлял от 10 до 20 меш.
Предварительная обработка и реакция
7 г катализатора, приготовленного вышеописанным способом, использовали для проведения предварительной обработки и реакции при аналогичных условиях, как в примере 13. Результаты реакции показаны в таблице 3.
Сравнительный пример 2
Предварительную обработку и реакцию проводили при аналогичных условиях, как в сравнительном примере 1, за исключением того, что температуру (температуру реакции) каталитического слоя изменяли на равную 250°С. Результаты реакции показаны в таблице 3.
Сравнительный пример 3
Приготовление катализатора
4 г NaH2PO4 растворяли в 20 г воды, при этом добавляли 6 г силикагеля «CARiACTQ-50» (изготовленного Fuji Silysia Chemical Ltd.) и перемешивали при 50°С в течение 30 минут. Затем удаляли воду путем выпаривания, а осадок сушили при 120°С в течение 3 часов, после чего прокаливали при 500°С в течение 6 часов в атмосфере воздуха, чтобы получить катализатор, в котором NaH2PO4 был нанесен на носитель из диоксида кремния.
Предварительная обработка и реакция
Предварительную обработку и реакцию проводили при аналогичных условиях, как в примере 13, за исключением того, что использовали 7 г катализатора, приготовленного вышеописанным способом. Результаты реакции показаны в таблице 3.
Сравнительный пример 4
Предварительную обработку и реакцию проводили при аналогичных условиях, как в сравнительном примере 3, за исключением того, что температуру (температуру реакции) каталитического слоя изменяли на равную 250°С. Результаты реакции показаны в таблице 3.
Сравнительный пример 5
Приготовление катализатора
MgSO4 (изготовленный Wako Pure Chemical Industries Ltd.) отформовывали прессованием, после чего отформованное вещество прокаливали при 500°С в течение 6 часов. Это вещество измельчали и контролировали, чтобы размер частиц составлял от 10 до 20 меш.
Предварительная обработка и реакция
Предварительную обработку и реакцию проводили при аналогичных условиях, как в примере 13, за исключением того, что использовали 7 г катализатора, приготовленного вышеописанным способом. Результаты реакции показаны в таблице 3.
Сравнительный пример 6
Предварительную обработку и реакцию проводили аналогичным образом, как в примере 1, за исключением того, что использовали 14 г катализатора из диоксида титана на носителе из диоксида кремния «HTG-30705» (изготовленного Fuji Silysia Chemical Ltd.) вместо 14 г катализатора из диоксида циркония. Результаты реакции показаны в таблице 3.
Сравнительный пример 7
Предварительную обработку и реакцию проводили аналогичным образом как в примере 1, за исключением того, что использовали 14 г катализатора из диоксида титана «CS-300S-24» (изготовленного Sakai Chemical Industry Co., Ltd.) вместо 14 г катализатора из диоксида циркония. Результаты реакции показаны в таблице 3.
Сравнительный пример 8
Предварительную обработку и реакцию проводили при аналогичных условиях, как в примере 13, за исключением того, что использовали 7 г цеолитового катализатора «ZSM-5 K+ type» (изготовленного N.E. Chemcat Corporation) вместо 7 г катализатора из диоксида циркония, и температуру (температура реакции) каталитического слоя изменяли на равную 250°С. Результаты реакции показаны в таблице 3.
Сравнительный пример 9
Предварительную обработку и реакцию проводили при аналогичных условиях, как в примере 13, за исключением того, что использовали 7 г катализатора из γ-оксида алюминия «KHS-24» (изготовленного Sumitomo Chemical Co., Ltd.) вместо 7 г катализатора из диоксида циркония. Результаты реакции показаны в таблице 3.
Сравнительный пример 10
Предварительную обработку и реакцию проводили при аналогичных условиях, как в примере 13, за исключением того, что использовали 7 г катализатора из оксида олова (катализатора, полученного путем того, что «MM-002», изготовленный Mitsui Mining & Smelting Co., Ltd., подвергали формованию прессованием и контролировали, чтобы размер частиц составлял от 10 до 20 меш) вместо 7 г катализатора из диоксида циркония. Результаты реакции показаны в таблице 3.
Сравнительный пример 11
Предварительную обработку и реакцию проводили при аналогичных условиях, как в примере 13, за исключением того, что использовали 7 г катализатора из оксида магния (катализатора, полученного путем того, что «UCM150», изготовленный Ube Material Industries, Ltd., подвергали формованию прессованием и контролировали, чтобы размер частиц составлял от 10 до 20 меш) вместо 7 г катализатора из диоксида циркония. Результаты реакции показаны в таблице 3.
Сравнительный пример 12
Предварительную обработку и реакцию проводили при аналогичных условиях, как в примере 13, за исключением того, что использовали 7 г катализатора из оксида магния на оксиде кремния «Mizukalife P-1G» (изготовленного Mizusawa Industrial Chemicals, Ltd.) вместо 7 г катализатора из диоксида циркония. Результаты реакции показаны в таблице 3.
Сравнительный пример 13
Предварительную обработку и реакцию проводили при аналогичных условиях, как в примере 13, за исключением того, что использовали 7 г катализатора из триоксида сурьмы (катализатора, полученного путем того, что триоксид сурьмы, изготовленный Soekawa Chemicals Co., Ltd., подвергали формованию прессованием и контролировали, чтобы размер частиц составлял от 10 до 20 меш) вместо 7 г катализатора из диоксида циркония. Результаты реакции показаны в таблице 3.
Сравнительный пример 14
Приготовление катализатора
Цеолитовый катализатор «MCM41» (изготовленный N.E. Chemcat Corporation) перетирали со связующим «Ben-gel 11» (изготовленным Nihon Yuki Nendo Co., Ltd.) в массовом соотношении 9:1. Глинистое вещество, полученное добавлением воды к вышеуказанному растертому веществу, сушили при 150°С в течение 3 часов, после чего прокаливали при 400°С в течение 3 часов, и прокаленное вещество измельчали и контролировали, чтобы размер частиц составлял от 10 до 20 меш.
Предварительная обработка и реакция
Предварительную обработку и реакцию проводили при аналогичных условиях, как в примере 13, за исключением того, что использовали 7 г катализатора, приготовленного вышеописанным способом. Результаты реакции показаны в таблице 3.
Сравнительный пример 15
Предварительную обработку и реакцию проводили при аналогичных условиях, как в примере 13, за исключением того, что использовали 7 г катализатора из гидроксиапатита (катализатора, полученного путем того, что гидроксиапатит, изготовленный Wako Pure Chemical Industries, Ltd., подвергали формованию прессованием и проконтролировали, чтобы размер частиц составлял от 10 до 20 меш) вместо 7 г катализатора из диоксида циркония. Результаты реакции показаны в таблице 3.
Сравнительный пример 16
Предварительную обработку и реакцию проводили при аналогичных условиях, как в примере 13, за исключением того, что использовали 7 г катализатора из оксида свинца (катализатора, полученного путем того, что оксид свинца, изготовленный Wako Pure Chemical Industries, Ltd., подвергали формованию прессованием и контролировали, чтобы размер частиц составлял от 10 до 20 меш) вместо 7 г катализатора из диоксида циркония. Результаты реакции показаны в таблице 3.
Сравнительный пример 17
Приготовление катализатора
Цеолитовый катализатор «HSZ 310NAD» (изготовленный Tosoh Corporation) перетирали со связующим «Ben-gel 11» (изготовленным Nihon Yuki Nendo Co., Ltd.) в массовом соотношении 9:1. Глинистое вещество, полученное добавлением воды к вышеуказанному растертому веществу, сушили при 150°С в течение 3 часов, после чего прокаливали при 400°С в течение 3 часов, и прокаленное вещество измельчали и контролировали, чтобы размер частиц составлял от 10 до 20 меш.
Предварительная обработка и реакция
Предварительную обработку и реакцию проводили при аналогичных условиях, как в примере 13, за исключением того, что использовали 7 г катализатора, приготовленного вышеописанным способом. Результаты реакции показаны в таблице 3.
Сравнительный пример 18
Приготовление катализатора
6,43 г NH4VO3 добавляли в 100 г воды, при этом к раствору постепенно добавляли 10,40 г дигидрат щавелевой кислоты, чтобы растворить NH4VO3. В полученный раствор добавляли 20 г силикагеля «CARiACTQ-50» (изготовленного Fuji Silysia Chemical Ltd.) и перемешивали при 50°С в течение 2 часов. Затем удаляли воду путем выпаривания, и осадок сушили при 150°С в течение 3 часов, после чего прокаливали при 400°С в течение 24 часов в атмосфере воздуха, чтобы получить катализатор.
Предварительная обработка и реакция
Предварительную обработку и реакцию проводили при аналогичных условиях, как в примере 13, за исключением того, что использовали 7 г катализатора, приготовленного вышеописанным способом. Результаты реакции показаны в таблице 3.
Сравнительный пример 19
Приготовление катализатора
Гидроксид висмута (изготовленный Soekawa Chemical Co., Ltd.) отформовывали прессованием и прокаливали при 400°С в течение 3 часов в атмосфере воздуха, после чего катализатор измельчали и контролировали, чтобы размер частиц составлял от 10 до 20 меш, чтобы получить катализатор из оксида висмута.
Предварительная обработка и реакция
Предварительную обработку и реакцию проводили при аналогичных условиях, как в примере 13, за исключением того, что использовали 7 г катализатора, приготовленного вышеописанным способом. Результаты реакции показаны в таблице 3.
Сравнительный пример 20
Приготовление катализатора
Гидроксид кальция (изготовленный Wako Pure Chemical Industries, Ltd.) отформовывали прессованием и прокаливали при 500°С в течение 3 часов в атмосфере воздуха, после чего катализатор измельчали и контролировали, чтобы размер частиц составлял от 10 до 20 меш, чтобы получить катализатор из оксида кальция.
Предварительная обработка и реакция
Предварительную обработку и реакцию проводили при аналогичных условиях, как в примере 13, за исключением того, что использовали 7 г катализатора, приготовленного вышеописанным способом. Результаты реакции показаны в таблице 3.
Сравнительный пример 21
Приготовление катализатора
Цеолитовый катализатор «HSZ 610NAA» (изготовленный Tosoh Corporation) перетирали со связующим «Ben-gel 11» (изготовленным Nihon Yuki Nendo Co., Ltd.) в массовом соотношении 9:1. Глинистое вещество, полученное добавлением воды к вышеуказанному растертому веществу, сушили при 150°С в течение 3 часов, после чего прокаливали при 400°С в течение 3 часов, и прокаленное вещество измельчали и контролировали, чтобы размер частиц составлял от 10 до 20 меш.
Предварительная обработка и реакция
Предварительную обработку и реакцию проводили при аналогичных условиях, как в примере 13, за исключением того, что использовали 7 г катализатора, приготовленного вышеописанным способом. Результаты реакции показаны в таблице 3.
Сравнительный пример 22
Приготовление катализатора
Цеолитовый катализатор «Mizukasieves 13X-15P Na+ type» (изготовленный Mizusawa Industrial Chemicals, Ltd.) перетирали со связующим «Ben-gel 11» (изготовленным Nihon Yuki Nendo Co., Ltd.) в массовом соотношении 9:1. Глинистое вещество, полученное добавлением воды к вышеуказанному растертому веществу, сушили при 150°С в течение 3 часов, после чего прокаливали при 400°С в течение 3 часов, и прокаленное вещество измельчали и контролировали, чтобы размер частиц составлял от 10 до 20 меш.
Предварительная обработка и реакция
Предварительную обработку и реакцию проводили при аналогичных условиях, как в примере 13, за исключением того, что использовали 7 г катализатора, приготовленного вышеописанным способом. Результаты реакции показаны в таблице 3.
Сравнительный пример 23
Приготовление катализатора
Гидроксид цинка (изготовленный Soekawa Chemical Co., Ltd.) отформовывали прессованием и прокаливали при 500°С в течение 3 часов в атмосфере воздуха, после чего катализатор измельчали и контролировали, чтобы размер частиц составлял от 10 до 20 меш, чтобы получить катализатор из оксида цинка.
Предварительная обработка и реакция
Предварительную обработку и реакцию проводили при аналогичных условиях, как в примере 13, за исключением того, что использовали 7 г катализатора, приготовленного вышеописанным способом. Результаты реакции показаны в таблице 3.
Сравнительный пример 24
Приготовление катализатора
Водный раствор, приготовленный путем растворения 0,33 моль моногидрата сульфата марганца в 215 мл воды при перемешивании при 75°С и последующего смешивания этого раствора с 0,958 моль концентрированной серной кислоты, быстро прибавляли к водному раствору, приготовленному путем растворения 0,398 моль перманганата калия в 220 мл воды. Раствор непрерывно перемешивали при 70°С в течение 2 часов, и затем перемешивали при 90°С в течение 4 часов до созревания, после чего в него добавляли смешанный раствор, приготовленный путем суспендирования 0,007 моль оксида висмута(III) в 440 мл воды.
После перемешивания при комнатной температуре в течение 30 минут, полученный осадок отфильтровывали и промывали четыре раза 2000 мл воды, чтобы получить плотный отжатый осадок на фильтре. Полученный таким образом плотный осадок сушили при 110°С в течение ночи, измельчали и контролировали, чтобы размер частиц составлял от 10 до 20 меш, чтобы получить катализатор из диоксида марганца.
Предварительная обработка и реакция
7 г катализатора, приготовленного вышеописанным способом, использовали для проведения предварительной обработки и реакции при аналогичных условиях, как в примере 13. Результаты реакции показаны в таблице 3.
Сравнительный пример 25: Жидкофазная реакция (способ, при котором применяли катализатор на основе титана и при котором аммиак не удаляли из системы)
Приготовление катализатора
65,3 г амида α-гидроксиизомасляной кислоты растворяли в 1000 г изопропанола и добавляли раствор, приготовленный путем растворения 30 г тетраизопропилата титана в 1000 г изопропанола. Изопропанол удаляли из вышеупомянутого раствора с помощью ротационного выпаривания, при этом раствор концентрировали до 840 г и оставляли его стоять при комнатной температуре в течение 24 часов. По истечении 24 часов осевший белый осадок отделяли фильтрацией, промывали гептаном и затем высушивали в вакууме для получения комплекса, в котором амид α-гидроксиизомасляной кислоты был координирован титаном (далее в данном документе называется Ti(HBD)4).
Реакция
В автоклав из нержавеющей стали марки SUS316 емкостью 20 мл загружали 5,4 г амида α-гидроксиизомасляной кислоты, 12,6 г метанола и 0,1 г Ti(HBD)4 в качестве катализатора, и вещества взаимодействовали при 200°С в течение 3 часов при одновременном перемешивании содержимого с помощью магнитного перемешивающего устройства.
Содержимое реактора охлаждали и анализировали с помощью газовой хроматографии, обнаруживая, что концентрация амида α-гидроксиизомасляной кислоты составляла 21,8% по массе; концентрация N-метиламида α-гидроксиизомасляной кислоты составляла 0,3% по массе; концентрация метил α-гидроксиизобутирата составляла 8,8% по массе; концентрация метанола составляла 64,8% по массе; концентрация аммиака составляла 1,2% по массе; степень превращения амида α-гидроксиизомасляной кислоты в вышеописанной реакции составляла 27%; селективность по метил α-гидроксиизобутирату составляла 94%; и селективность по N-метиламиду α-гидроксиизомасляной кислоты составляла 3%.
Справочный пример 2: Жидкофазная реакция (способ, при котором применяли катализатор на основе титана и при котором аммиак удаляли из системы)
В автоклав из нержавеющей стали марки SUS316 емкостью 300 мл, снабженный обратным холодильником с рубашкой и перемешивающим устройством, загружали 30,0 г амида α-гидроксиизомасляной кислоты, 100 г метанола и 6,00 г Ti(HBD)4, приготовленного в сравнительном примере 25. В автоклаве поддерживали температуру 190°С и давление 3,0 МПа при одновременном перемешивании, чтобы проводить реакцию в течение одного часа при одновременной подаче газообразного азота при величине расхода 87 л/час и метанола при величине расхода 158 г/час. В данном случае была обеспечена циркуляция масла с температурой 185°С через рубашку обратного холодильника, чтобы нагревать его, и метанол удаляли из системы вместе с аммиаком при величине расхода 158 г/час, чтобы не вызывать внутреннее орошение.
По завершении реакции реакционную жидкость охлаждали и анализировали с помощью газовой хроматографии, обнаруживая, что степень превращения амида α-гидроксиизомасляной кислоты составляла 93%; селективность по метил α-гидроксиизобутирату составляла 97%; и селективность по N-метиламиду α-гидроксиизомасляной кислоты составляла 3%.
Таким образом, было установлено, что в способе, описанном в сравнительном примере 25, можно увеличить степень превращения путем проведения реакции, при одновременном удалении побочно образующегося аммиака из системы.
Пример 14
Предварительную обработку и реакцию проводили при аналогичных условиях, как в примере 1, за исключением того, что используемое количество катализатора устанавливали равным 7 г и что величину расхода потока исходных веществ в виде жидкости изменяли до 7,00 г/час (WHSV, исходя из количества амида α-гидроксиизомасляной кислоты, =0,3 ч-1). Результаты реакции показаны в таблице 4.
Пример 15
Приготовление катализатора: способ пропитки
Гидроксид циркония «XZO 1501/03» (изготовленный MEL Chemicals Inc.) отформовывали прессованием, и отформованное вещество измельчали и контролировали, чтобы размер частиц составлял от 10 до 20 меш. 14,6 г данного гидроксида циркония (78% по массе в пересчете на ZrO2) добавляли в водный раствор, заранее приготовленный путем растворения 0,377 г ZrO(NO3)2·2H2O (источник дополнительного элемента) в 20 г воды, и смесь перемешивали при 50°С в течение 30 минут. Затем воду удаляли путем выпаривания, и осадок сушили при 150°С в течение 3 часов, затем прокаливали при 400°С в течение 3 часов и повторно контролировали, чтобы размер частиц составлял от 10 до 20 меш, чтобы приготовить катализатор на основе диоксида циркония, содержащий элемент цирконий (Zr).
Предварительная обработка и реакция
Для проведения предварительной обработки и реакции при аналогичных условиях, как в примере 14, применяли 7 г катализатора на основе диоксида циркония, приготовленного вышеописанным способом. Результаты реакции показаны в таблице 4.
Пример 16
Приготовление катализатора: способ пропитки
Катализатор на основе диоксида циркония (мольное соотношение в катализаторе Zr:Y=98,5:1,5) был приготовлен аналогичным способом, как в примере 15, за исключением того, что источник дополнительного элемента изменяли на 0,487 г Y(CH3COO)3·4H2O, чтобы добавить иттрий (Y) в катализатор.
Предварительная обработка и реакция
Для проведения предварительной обработки и реакции при аналогичных условиях, как в примере 14, применяли 7 г катализатора на основе диоксида циркония, приготовленного вышеописанным способом. Результаты реакции показаны в таблице 4.
Пример 17
Приготовление катализатора: способ пропитки
Катализатор на основе диоксида циркония (мольное соотношение в катализаторе Zr:La=98,5:1,5) был приготовлен аналогичным способом, как в примере 15, за исключением того, что источник дополнительного элемента изменяли на 0,611 г La(NO3)3·6H2O, чтобы добавить лантан (La) в катализатор.
Предварительная обработка и реакция
Для проведения предварительной обработки и реакции при аналогичных условиях, как в примере 14, применяли 7 г катализатора на основе диоксида циркония, приготовленного вышеописанным способом. Результаты реакции показаны в таблице 4.
Пример 18
Приготовление катализатора: способ пропитки
Катализатор на основе диоксида циркония (мольное соотношение в катализаторе Zr:Co=98,5:1,5) был приготовлен аналогичным способом, как в примере 15, за исключением того, что источник дополнительного элемента изменяли на 0,410 г Co(NO3)2·6H2O, чтобы добавить кобальт (Co) в катализатор.
Предварительная обработка и реакция
Для проведения предварительной обработки и реакции при аналогичных условиях, как в примере 14, применяли 7 г катализатора на основе диоксида циркония, приготовленного вышеописанным способом. Результаты реакции показаны в таблице 4.
Пример 19
Приготовление катализатора: способ пропитки
Катализатор на основе диоксида циркония (мольное соотношение в катализаторе Zr:Mn=98,5:1,5) был приготовлен аналогичным способом, как в примере 15, за исключением того, что источник дополнительного элемента изменяли на 0,346 г Mn(CH3COO)2·4H2O, чтобы добавить марганец (Mn) в катализатор.
Предварительная обработка и реакция
Для проведения предварительной обработки и реакции при аналогичных условиях, как в примере 14, применяли 7 г катализатора на основе диоксида циркония, приготовленного вышеописанным способом. Результаты реакции показаны в таблице 4.
Пример 20
Приготовление катализатора: способ пропитки
Катализатор на основе диоксида циркония (мольное соотношение в катализаторе Zr:Ni=98,5:1,5) был приготовлен аналогичным способом, как в примере 15, за исключением того, что источник дополнительного элемента изменяли на 0,351 г Ni(CH3COO)2·4H2O, чтобы добавить никель (Ni) в катализатор.
Предварительная обработка и реакция
Для проведения предварительной обработки и реакции при аналогичных условиях, как в примере 14, применяли 7 г катализатора на основе диоксида циркония, приготовленного вышеописанным способом. Результаты реакции показаны в таблице 4.
Пример 21
Приготовление катализатора: способ пропитки
Катализатор на основе диоксида циркония (мольное соотношение в катализаторе Zr:Yb=98,5:1,5) был приготовлен аналогичным способом, как в примере 15, за исключением того, что источник дополнительного элемента изменяли на 0,608 г Yb(NO3)3·4H2O, чтобы добавить иттербий (Yb) в катализатор.
Предварительная обработка и реакция
Для проведения предварительной обработки и реакции при аналогичных условиях, как в примере 14, применяли 7 г катализатора на основе диоксида циркония, приготовленного вышеописанным способом. Результаты реакции показаны в таблице 4.
Пример 22
Приготовление катализатора: способ пропитки
Катализатор на основе диоксида циркония (мольное соотношение в катализаторе Zr:Al=98,5:1,5) был приготовлен аналогичным способом, как в примере 15, за исключением того, что источник дополнительного элемента изменяли на 0,529 г Al(NO3)3·9H2O, чтобы добавить алюминий (Al) в катализатор.
Предварительная обработка и реакция
Для проведения предварительной обработки и реакции при аналогичных условиях, как в примере 14, применяли 7 г катализатора на основе диоксида циркония, приготовленного вышеописанным способом. Результаты реакции показаны в таблице 4.
Пример 23
Приготовление катализатора: способ пропитки
Катализатор на основе диоксида циркония (мольное соотношение в катализаторе Zr:B=98,5:1,5) был приготовлен аналогичным способом, как в примере 15, за исключением того, что источник дополнительного элемента изменяли на 0,087 г H3BO3, чтобы добавить бор (B) в катализатор.
Предварительная обработка и реакция
Для проведения предварительной обработки и реакции при аналогичных условиях, как в примере 14, применяли 7 г катализатора на основе диоксида циркония, приготовленного вышеописанным способом. Результаты реакции показаны в таблице 4.
Пример 24
Приготовление катализатора: способ пропитки
Катализатор на основе диоксида циркония (мольное соотношение в катализаторе Zr:Cu=98,5:1,5) был приготовлен аналогичным способом, как в примере 15, за исключением того, что источник дополнительного элемента изменяли на 0,281 г Cu(CH3COO)2·H2O, чтобы добавить медь (Cu) в катализатор.
Предварительная обработка и реакция
Для проведения предварительной обработки и реакции при аналогичных условиях, как в примере 14, применяли 7 г катализатора на основе диоксида циркония, приготовленного вышеописанным способом. Результаты реакции показаны в таблице 4.
Пример 25
Приготовление катализатора: способ пропитки
Катализатор на основе диоксида циркония (мольное соотношение в катализаторе Zr:Ce=98,5:1,5) был приготовлен аналогичным способом, как в примере 15, за исключением того, что источник дополнительного элемента изменяли на 0,612 г Ce(NO3)3·6H2O, чтобы добавить церий (Ce) в катализатор.
Предварительная обработка и реакция
Для проведения предварительной обработки и реакции при аналогичных условиях, как в примере 14, применяли 7 г катализатора на основе диоксида циркония, приготовленного вышеописанным способом. Результаты реакции показаны в таблице 4.
Пример 26
Приготовление катализатора: способ пропитки
Гидроксид циркония «XZO 1501/03», изготовленный MEL Chemicals Inc., отформовывали прессованием, и отформованное вещество измельчали и контролировали, чтобы размер частиц составлял от 10 до 20 меш. 14,6 г данного гидроксида циркония (78% по массе в пересчете на ZrO2) добавляли в водный раствор, заранее приготовленный путем растворения 0,367 г Bi(OH)3 (источник дополнительного элемента) в 20 г воды с помощью 1 мл 1 н. азотной кислоты, после чего смесь перемешивали при 50°С в течение 30 минут. Затем воду удаляли путем выпаривания, и осадок сушили при 150°С в течение 3 часов, затем прокаливали при 400°С в течение 3 часов и повторно контролировали, чтобы размер частиц составлял от 10 до 20 меш, чтобы приготовить катализатор на основе диоксида циркония, содержащий элемент висмут (Bi).
Предварительная обработка и реакция
Для проведения предварительной обработки и реакции при аналогичных условиях, как в примере 14, применяли 7 г катализатора (мольное соотношение в катализаторе Zr:Bi=98,5:1,5) на основе диоксида циркония, приготовленного вышеописанным способом. Результаты реакции показаны в таблице 4.
Пример 27
Приготовление катализатора: способ пропитки
Гидроксид циркония «XZO 1501/03», изготовленный MEL Chemicals Inc., отформовывали прессованием, и отформованное вещество измельчали и контролировали, чтобы размер частиц составлял от 10 до 20 меш. 14,6 г данного гидроксида циркония (78% по массе в пересчете на ZrO2) добавляли в водный раствор, заранее приготовленный путем растворения 0,401 г Ti(OCH(CH3)2)4 (источник дополнительного элемента) в 20 мл изопропанола, и смесь перемешивали при 50°С в течение 30 минут. Затем в смесь добавляли 1 мл воды, и изопропанол удалили путем выпаривания. Осадок сушили при 150°С в течение 3 часов, затем прокаливали при 400°С в течение 3 часов и повторно контролировали, чтобы размер частиц составлял от 10 до 20 меш, чтобы приготовить катализатор на основе диоксида циркония, содержащий элемент титан (Ti).
Предварительная обработка и реакция
Для проведения предварительной обработки и реакции при аналогичных условиях, как в примере 14, применяли 7 г катализатора (мольное соотношение в катализаторе Zr:Ti=98,5:1,5) на основе диоксида циркония, приготовленного вышеописанным способом. Результаты реакции показаны в таблице 4.
Пример 28
Приготовление катализатора: способ пропитки
Катализатор на основе диоксида циркония (мольное соотношение в катализаторе Zr:Mg=98,5:1,5) был приготовлен аналогичным способом, как в примере 15, за исключением того, что источник дополнительного элемента изменяли на 0,362 г Mg(NO3)2·6H2O, чтобы добавить магний (Mg) в катализатор.
Предварительная обработка и реакция
Для проведения предварительной обработки и реакции при аналогичных условиях, как в примере 14, применяли 7 г катализатора на основе диоксида циркония, приготовленного вышеописанным способом. Результаты реакции показаны в таблице 4.
Пример 29
Приготовление катализатора: способ пропитки
Катализатор на основе диоксида циркония (мольное соотношение в катализаторе Zr:Zn=98,5:1,5) был приготовлен аналогичным способом, как в примере 15, за исключением того, что источник дополнительного элемента изменяли на 0,419 г Zn(NO3)2·6H2O, чтобы добавить цинк (Zn) в катализатор.
Предварительная обработка и реакция
Для проведения предварительной обработки и реакции при аналогичных условиях, как в примере 14, применяли 7 г катализатора на основе диоксида циркония, приготовленного вышеописанным способом. Результаты реакции показаны в таблице 4.
Пример 30
Приготовление катализатора: способ пропитки
Катализатор на основе диоксида циркония (мольное соотношение в катализаторе Zr:Ca=98,5:1,5) был приготовлен аналогичным способом, как в примере 15, за исключением того, что источник дополнительного элемента изменяли на 0,333 г Ca(NO3)2·4H2O, чтобы добавить кальций (Ca) в катализатор.
Предварительная обработка и реакция
Для проведения предварительной обработки и реакции при аналогичных условиях, как в примере 14, применяли 7 г катализатора на основе диоксида циркония, приготовленного вышеописанным способом. Результаты реакции показаны в таблице 4.
Пример 31
Приготовление катализатора: способ пропитки
Катализатор на основе диоксида циркония (мольное соотношение в катализаторе Zr:Fe=98,5:1,5) был приготовлен аналогичным способом, как в примере 15, за исключением того, что источник дополнительного элемента изменяли на 0,570 г Fe(NO3)3·9H2O, чтобы добавить железо (Fe) в катализатор.
Предварительная обработка и реакция
Для проведения предварительной обработки и реакции при аналогичных условиях, как в примере 14, применяли 7 г катализатора на основе диоксида циркония, приготовленного вышеописанным способом. Результаты реакции показаны в таблице 4.
Пример 32
Приготовление катализатора: способ пропитки
Катализатор на основе диоксида циркония (мольное соотношение в катализаторе Zr:Pb=98,5:1,5) был приготовлен аналогичным способом, как в примере 15, за исключением того, что источник дополнительного элемента изменяли на 0,467 г Pb(NO3)2, чтобы добавить свинец (Pb) в катализатор.
Предварительная обработка и реакция
Для проведения предварительной обработки и реакции при аналогичных условиях, как в примере 14, применяли 7 г катализатора на основе диоксида циркония, приготовленного вышеописанным способом. Результаты реакции показаны в таблице 4.
Пример 33
Приготовление катализатора: способ пропитки
Гидроксид циркония «XZO 1501/03», изготовленный MEL Chemicals Inc., отформовывали прессованием, и отформованное вещество измельчали и контролировали, чтобы размер частиц составлял от 10 до 20 меш. 14,6 г данного гидроксида циркония (78% по массе в пересчете на ZrO2) добавляли в водный раствор, заранее приготовленный путем растворения 0,334 г Sn(CH3COO)2·H2O (источник дополнительного элемента) в 20 г воды с помощью 1 мл 1 н. азотной кислоты, и смесь перемешивали при 50°С в течение 30 минут. Затем воду удаляли путем выпаривания, и осадок сушили при 150°С в течение 3 часов, затем прокаливали при 400°С в течение 3 часов и повторно контролировали, чтобы размер частиц составлял от 10 до 20 меш, чтобы приготовить катализатор на основе диоксида циркония, содержащий элемент олово (Sn).
Предварительная обработка и реакция
Для проведения предварительной обработки и реакции при аналогичных условиях, как в примере 14, применяли 7 г катализатора (мольное соотношение в катализаторе Zr:Sn=98,5:1,5) на основе диоксида циркония, приготовленного вышеописанным способом. Результаты реакции показаны в таблице 4.
Пример 34
Приготовление катализатора: способ пропитки
Катализатор на основе диоксида циркония (мольное соотношение в катализаторе Zr:Te=98,5:1,5) был приготовлен аналогичным способом, как в примере 15, за исключением того, что источник дополнительного элемента изменяли на 0,324 г H6TeO6, чтобы добавить теллур (Te) в катализатор.
Предварительная обработка и реакция
Для проведения предварительной обработки и реакции при аналогичных условиях, как в примере 14, применяли 7 г катализатора на основе диоксида циркония, приготовленного вышеописанным способом. Результаты реакции показаны в таблице 4.
Пример 35
Приготовление катализатора: способ пропитки
Катализатор на основе диоксида циркония (мольное соотношение в катализаторе Zr:Cs=98,5:1,5) был приготовлен аналогичным способом, как в примере 15, за исключением того, что источник дополнительного элемента изменяли на 0,275 г CsNO3, чтобы добавить цезий (Cs) в катализатор.
Предварительная обработка и реакция
Для проведения предварительной обработки и реакции при аналогичных условиях, как в примере 14, применяли 7 г катализатора на основе диоксида циркония, приготовленного вышеописанным способом. Результаты реакции показаны в таблице 4.
Пример 36
Приготовление катализатора: способ пропитки
Гидроксид циркония «XZO 1501/03», изготовленный MEL Chemicals Inc., отформовывали прессованием, и отформованное вещество измельчали и контролировали, чтобы размер частиц составлял от 10 до 20 меш. 14,6 г данного гидроксида циркония (78% по массе в пересчете на ZrO2) добавляли в водный раствор, заранее приготовленный путем растворения 0,165 г NH4VO3 (источник дополнительного элемента) в 20 г воды с помощью 0,267 г дигидрата щавелевой кислоты, и смесь перемешивали при 50°С в течение 30 минут. Затем воду удаляли путем выпаривания, и осадок сушили при 150°С в течение 3 часов, затем прокаливали при 400°С в течение 3 часов и повторно контролировали, чтобы размер частиц составлял от 10 до 20 меш, чтобы приготовить катализатор на основе диоксида циркония, содержащий элемент ванадий (V).
Предварительная обработка и реакция
Для проведения предварительной обработки и реакции при аналогичных условиях, как в примере 14, применяли 14 г катализатора (мольное соотношение в катализаторе Zr:V=98,5:1,5) на основе диоксида циркония, приготовленного вышеописанным способом. Результаты реакции показаны в таблице 4.
Пример 37
Приготовление катализатора: способ пропитки
Катализатор на основе диоксида циркония (мольное соотношение в катализаторе Zr:Cr=98,5:1,5) был приготовлен аналогичным способом, как в примере 15, за исключением того, что источник дополнительного элемента изменяли на 0,564 г Cr(NO3)3·9H2O, чтобы добавить хром (Cr) в катализатор.
Предварительная обработка и реакция
Для проведения предварительной обработки и реакции при аналогичных условиях, как в примере 14, применяли 7 г катализатора на основе диоксида циркония, приготовленного вышеописанным способом. Результаты реакции показаны в таблице 4.
Пример 38
Приготовление катализатора
100 г дигидрата оксинитрата циркония, изготовленного Wako Pure Chemical Industries, Ltd., растворяли в 800 мл воды и добавляли 1 мл 1 н. азотной кислоты. В полученный раствор прибавляли 25% водный раствор аммиака до тех пор, пока pH вышеупомянутого раствора не достигал значения 8, чтобы образовался белый осадок. Осадок оставляли стоять в течение ночи, при этом он оседал на дно, и его подвергали декантации, затем отфильтровывали и промывали 600 мл воды. После сушки при 150°С в течение 3 часов, его прокаливали при 400°С в течение 3 часов, измельчали и контролировали, чтобы размер частиц составлял от 10 до 20 меш, чтобы приготовить катализатор из диоксида циркония.
Предварительная обработка и реакция
Для проведения предварительной обработки и реакции при аналогичных условиях, как в примере 14, применяли 7 г катализатора на основе диоксида циркония, приготовленного вышеописанным способом осаждения. Результаты реакции показаны в таблице 4.
Пример 39
Приготовление катализатора: способ совместного осаждения
100 г дигидрата оксинитрата циркония и 8,53 г гексагидрата нитрата лантана, изготовленных Wako Pure Chemical Industries, Ltd., растворяли в 800 мл воды и добавляли 1 мл 1 н. азотной кислоты. В полученный раствор прибавляли 25% водный раствор аммиака до тех пор, пока pH вышеупомянутого раствора не достигал значения 8, чтобы образовался белый осадок. Осадок оставляли стоять в течение ночи, при этом он оседал на дно, и его подвергали декантации, затем отфильтровывали и промывали 600 мл воды. После сушки при 150°С в течение 3 часов, его прокаливали при 400°С в течение 3 часов, измельчали и контролировали, чтобы размер частиц составлял от 10 до 20 меш, чтобы приготовить катализатор на основе диоксида циркония, содержащий элемент лантан (La).
Предварительная обработка и реакция
Для проведения предварительной обработки и реакции при аналогичных условиях, как в примере 14, применяли 7 г катализатора (мольное соотношение в катализаторе Zr:La=98:2) на основе диоксида циркония, приготовленного вышеописанным способом. Результаты реакции показаны в таблице 4.
Пример 40
Катализатор приготавливали по методике, описанной в примере 16, при изменении температуры прокаливания до 500°С, и предварительную обработку и реакцию проводили при аналогичных условиях, за исключением того, что количество используемого катализатора изменяли на равное 4,2 г (WHSV, исходя из количества амида α-гидроксиизомаясляной кислоты, =0,5 ч-1), и того, что температуру реакции изменяли на равную 210°С.
Когда реакция достигала состояния равновесия по истечении приблизительно 24 часов, образцы продуктов брали в ловушку, охлаждаемую льдом, и анализировали с помощью газовой хроматографии, обнаруживая, что степень превращения амида α-гидроксиизомасляной кислоты составляла 95% и что селективность по метил α-гидроксиизобутирату составляла 92%. Реакцию продолжали, и обнаруживали, что степень превращения амида α-гидроксиизомасляной кислоты может сохраняться на уровне 90% или более в течение 216 часов от начала реакции.
Пример 41
Предварительную обработку и реакцию проводили при аналогичных условиях, как в примере 40, за исключением того, что долю содержания (мольная доля) иттрия (Y) регулировали так, чтобы она составляла Zr:Y=94:6. Полученный таким образом катализатор анализировали с помощью рентгеновского флуоресцентного аналитического оборудования «SEA2010» (изготовленного SEICO Electronics Industrial Co., Ltd.) и обнаруживали, что полученное соотношение интенсивностей (Y/Zr) составляло 0,074.
Когда реакция достигала состояния равновесия по истечении приблизительно 24 часов, образцы продуктов брали в ловушку, охлаждаемую льдом, и анализировали с помощью газовой хроматографии, обнаруживая, что степень превращения амида α-гидроксиизомасляной кислоты составляла 94% и что селективность по метил α-гидроксиизобутирату составляла 92%. Реакцию продолжали и обнаруживали, что степень превращения амида α-гидроксиизомасляной кислоты может сохраняться на уровне 90% или более в течение 360 часов от начала реакции.
Пример 42
Приготовление катализатора
К 30,6 г гидроксида циркония «XZO 1501/03», изготовленного MEL Chemicals Inc., прибавляли 500 г воды, чтобы приготовить суспензию, и нагревали ее при температуре от 50 до 60°С при одновременном перемешивании. Водный раствор, заранее приготовленный путем растворения 3,9 г Y(CH3COO)3·4H2O в 100 г воды и нагретый при 50°С, добавляли в суспензию и перемешивали при температуре от 50 до 60°С в течение 1 часа. Вышеуказанное количество добавки соответствовало значению Zr:Y=94:6, которое показывало долю содержания (мольную долю) иттрия (Y) в катализаторе. Раствор охлаждали до комнатной температуры и регулировали pH до значения 8,0 с помощью 25% водного раствора аммиака, и затем перемешивали в течение 30 минут и оставляли стоять в течение приблизительно 16 часов, чтобы осадить гидроксид циркония. Надосадочную жидкость удаляли, и прибавляли к осадку 300 г воды. Смесь снова перемешивали в состоянии суспензии в течение 30 минут и оставляли стоять в течение приблизительно 3 часов, чтобы осадить гидроксид циркония, и после удаления надосадочной жидкости осадок промывали. Осадок гидроксида циркония, полученный путем проведения еще одной промывки водой и последующего удаления надосадочной жидкости, сушили путем постепенного нагревания до 150°С в течение 72 часов в сушильном аппарате. Полученный осадок отформовывали прессованием, и отформованное вещество измельчали и контролировали, чтобы размер частиц составлял от 10 до 20 меш. Измельченное вещество сушили при 150°С в течение 3 часов, затем прокаливали при 400°С в течение 3 часов и повторно контролировали, чтобы размер частиц составлял от 10 до 20 меш, чтобы приготовить катализатор на основе диоксида циркония. Полученный таким образом катализатор анализировали с помощью рентгеновского флуоресцентного аналитического оборудования «SEA2010» (изготовленного SEICO Electronics Industrial Co., Ltd.) и обнаруживали, что полученное соотношение интенсивностей (Y/Zr) составляло 0,069 и что оно являлось почти таким же значением, как значение, полученное для катализатора, приготовленного в примере 40.
Предварительная обработка и реакция
Для проведения предварительной обработки и реакции при аналогичных условиях, как в примере 40, применяли 4,2 г катализатора на основе диоксида циркония (мольное соотношение в катализаторе Zr:Y=94:6).
Когда реакция достигала состояния равновесия по истечении приблизительно 24 часов, образцы продуктов брали в ловушку, охлаждаемую льдом, и анализировали с помощью газовой хроматографии, обнаруживая, что степень превращения амида α-гидроксиизомасляной кислоты составляла 94% и что селективность по метил α-гидроксиизобутирату составляла 92%. Реакцию продолжали и обнаруживали, что степень превращения амида α-гидроксиизомасляной кислоты может сохраняться на уровне 90% или более в течение 793 часов от начала реакции.
Пример 43
Предварительную обработку и реакцию проводили при аналогичных условиях, как в примере 42, за исключением того, что долю содержания (мольную долю) иттрия (Y) регулировали так, чтобы она составляла Zr:Y=96:4.
Когда реакция достигала состояния равновесия по истечении приблизительно 24 часов, образцы продуктов брали в ловушку, охлаждаемую льдом, и анализировали с помощью газовой хроматографии, обнаруживая, что степень превращения амида α-гидроксиизомасляной кислоты составляла 94% и что селективность по метил α-гидроксиизобутирату составляла 91%. Реакцию продолжали и обнаруживали, что степень превращения амида α-гидроксиизомасляной кислоты может сохраняться на уровне 90% или более в течение 699 часов от начала реакции.
Пример 44
Предварительную обработку и реакцию проводили при аналогичных условиях, как в примере 42, за исключением того, что долю содержания (мольную долю) иттрия (Y) регулировали так, чтобы она составляла Zr:Y=91:9.
Когда реакция достигала состояния равновесия по истечении приблизительно 24 часов, образцы продуктов брали в ловушку, охлаждаемую льдом, и анализировали с помощью газовой хроматографии, обнаруживая, что степень превращения амида α-гидроксиизомасляной кислоты составляла 94% и что селективность по метил α-гидроксиизобутирату составляла 93%. Реакцию продолжали и обнаруживали, что степень превращения амида α-гидроксиизомасляной кислоты может сохраняться на уровне 90% или более в течение 194 часов от начала реакции.
Пример 45
Приготовление катализатора
Катализатор готовили при аналогичных условиях, как в примере 42, за исключением того, что используемые количества исходных веществ и воды увеличивали в 13 раз соответственно.
Предварительная обработка
В реакционную трубку, изготовленную из нержавеющей стали марки SUS316, с внутренним диаметром 28 ммφ, загружали 200 г приготовленного вышеуказанным способом катализатора на основе диоксида циркония. Реакционную трубку нагревали с помощью теплообменного масла, обеспечивая в то же время, чтобы азот протекал при величине расхода 100 мл/минуту, и загруженный катализатор на основе диоксида циркония нагревали при 80°С. После остановки подачи азота, обеспечивали прохождение метанола через реакционную трубку при величине расхода 190 г/час, и после того, как метанол выходил из выходного отверстия реакционной трубки, катализатор нагревали до 210°С.
Реакция
После того, как останавливали подачу метанола, обеспечивали, чтобы через реакционную трубку протекали исходные вещества в виде жидкости, приготовленной путем смешивания амида α-гидроксиизомасляной кислоты и метанола в соотношении 30 частей по массе к 70 частям по массе, при величине расхода потока, равной 200 г/час (WHSV, исходя из количества амида α-гидроксиизомасляной кислоты, =0,3 ч-1), одновременно контролируя, чтобы температура каталитического слоя составляла 210°С.
Когда реакция достигала состояния равновесия по истечении приблизительно 29 часов, продукты охлаждали водой, брали образцы и анализировали с помощью газовой хроматографии, обнаруживая, что степень превращения амида α-гидроксиизомасляной кислоты составляла 94% и что селективность по метил α-гидроксиизобутирату составляла 91%. Реакцию продолжали и обнаруживали, что по истечении 143 часов степень превращения амида α-гидроксиизомасляной кислоты составляла 94% и что селективность по метил α-гидроксиизобутирату составляла 94%. Затем реакционную жидкость, охлажденную водой, растворяли в воде и количественно определяли содержание иона аммония с помощью оборудования для капиллярного электрофореза «G1600A» (изготовленного Agilent Technologies Inc.), что селективность по аммиаку, исходя из количества амида α-гидроксиизомасляной кислоты, составляла 94%.
Очистка ректификацией
Реакционный газ по истечении 143 часов от начала реакции подавали в среднюю часть ректификационной колонны, полученной путем заполнения стеклянной трубки, имеющей внутренний диаметр равный 28 мм, 500 мм насыпной насадки из 6 мм седел Мак-Магона, и непрерывно ректифицировали. Условия были отрегулированы таким образом, что температура в верхней части колонны составляла от 60 до 62°С, и температура жидкости в нижней части колонны составляла от 75 до 85°С, в то время как флегмовое число поддерживалось приблизительно равным 3, и величины расходов выходных потоков составляли для дистиллята 99 г/час, и для кубовой жидкости 100 г/ч.
Состав кубовой жидкости (жидкости из нижней части колонны) был следующий: метанол/метил α-гидроксиизобутират/амид α-гидроксиизомасляной кислоты =30/65/4 (массовое соотношение). Массовое соотношение удерживаемого количества жидкости в нижней части колонны и количества получаемой кубовой жидкости в час составляло 1. Количество аммиака в кубовой жидкости составляло 111 м.д., и количество метил α-гидроксиизобутирата, содержащейся в перегнанном метаноле составляло 150 м.д.
Метил α-гидроксиизобутират отделяли от вышеописанной кубовой жидкости и очищали с помощью проведения стандартной ректификации.
Пример 46
Предварительную обработку и реакцию проводили при аналогичных условиях, как в примере 3, за исключением того, что исходные вещества изменяли так, чтобы подавать смешанный раствор, содержащий 10 частей по массе лактамида и 90 частей по массе метанола, при величине расхода 8,8 г/час, а также того, что их проводили при температуре (температуре реакции), равной 230°С в каталитическом слое.
Результаты реакции по истечении приблизительно 24 часов представляли собой то, что степень превращения лактамида составляла 90% и что селективность по метиллактату составляла 85%.
Возможность промышленного применения
Сложные эфиры α-гидроксикарбоновых кислот, полученные путем применения способа получения, описанного в данном изобретении, являются важными соединениями, применяемыми в различных промышленных областях применения, и в случае, например, сложных эфиров молочной кислоты, их применяют в качестве высококипящих растворителей и, в дополнение к этому, применяют в качестве исходных веществ для пищевых добавок, ароматизаторов, лекарственных средств и сельскохозяйственных химикатов, а также биоразлагаемых полимеров. Конкретно сложные эфиры α-гидроксиизомасляной кислоты применяют в качестве исходных соединений для растворителей и лекарственных средств с сельскохозяйственными химикатами и, в дополнение к этому, их также применяют для синтеза сложных эфиров метакриловой кислоты, особенно метилметакрилата, а также применяются в качестве исходных веществ для синтеза α-аминокислот путем аминолиза.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ АЛКИЛ(МЕТ)АКРИЛАТОВ | 2006 |
|
RU2409552C2 |
| СПОСОБ ПОЛУЧЕНИЯ АМИДА КАРБОНОВОЙ КИСЛОТЫ ИЗ КАРБОНИЛЬНОГО СОЕДИНЕНИЯ И ЦИАНИСТОВОДОРОДНОЙ КИСЛОТЫ | 2009 |
|
RU2552619C9 |
| КАТАЛИЗАТОР ДЛЯ ПРЕВРАЩЕНИЯ НИТРИЛОВ КАРБОНОВЫХ КИСЛОТ | 2009 |
|
RU2496575C2 |
| СПОСОБ ПОЛУЧЕНИЯ АЛЬФА-ГИДРОКСИКАРБОНОВЫХ КИСЛОТ | 2007 |
|
RU2454399C2 |
| СПОСОБ ПОЛУЧЕНИЯ СЛОЖНЫХ ЭФИРОВ АЛЬФА-ГИДРОКСИКАРБОНОВЫХ КИСЛОТ В ГАЗОВОЙ ФАЗЕ | 2015 |
|
RU2687751C2 |
| СПОСОБ ПОЛУЧЕНИЯ АМИДА АЛЬФА-ГИДРОКСИИЗОМАСЛЯНОЙ КИСЛОТЫ И РЕАКТОР | 2014 |
|
RU2664112C9 |
| СПОСОБ ФЕРМЕНТАТИВНОГО ПОЛУЧЕНИЯ 2-ГИДРОКСИ-2-МЕТИЛКАРБОНОВЫХ КИСЛОТ | 2007 |
|
RU2459871C2 |
| КАТАЛИЗАТОР ВОССТАНОВЛЕНИЯ МОНООКСИДА УГЛЕРОДА, СПОСОБ ПРИГОТОВЛЕНИЯ КАТАЛИЗАТОРА И СПОСОБ ПОЛУЧЕНИЯ УГЛЕВОДОРОДА | 2007 |
|
RU2436832C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПОЛИАМИДОВ ИЗ СОЕДИНЕНИЙ АМИНОКАРБОНОВОЙ КИСЛОТЫ | 1999 |
|
RU2215754C2 |
| СПОСОБ ПОЛУЧЕНИЯ СВОБОДНЫХ КИСЛОТ ИЗ ИХ СОЛЕЙ | 2010 |
|
RU2533413C2 |
Изобретение относится к способу повышения степени превращения и селективности при получении сложного эфира α-гидроксикарбоновой кислоты из амида α-гидроксикарбоновой кислоты и алифатического спирта, в котором амид α-гидроксикарбоновой кислоты и алифатический спирт подвергают газофазной реакции в присутствии катализатора на основе диоксида циркония при температуре реакции от 150 до 270°C. Способ является эффективным с точки зрения промышленного производства способом получения, так как производственные затраты снижены, в то время как степень превращения и селективность увеличены, срок службы катализатора значительно увеличен. 6 з.п. ф-лы, 4 табл., 46 пр.
1. Способ повышения степени превращения и селективности при получении сложного эфира α-гидроксикарбоновой кислоты, в котором амид α-гидроксикарбоновой кислоты и алифатический спирт подвергают газофазной реакции в присутствии катализатора на основе диоксида циркония при температуре реакции от 150 до 270°C.
2. Способ повышения степени превращения и селективности при получении сложного эфира α-гидроксикарбоновой кислоты по п.1, в котором катализатор на основе диоксида циркония содержит по меньшей мере один элемент, выбираемый из 2-4 групп, 7 группы и 9-13 групп Периодической таблицы элементов, лантаноидов, сурьмы (Sb) и висмута (Bi).
3. Способ повышения степени превращения и селективности при получении сложного эфира α-гидроксикарбоновой кислоты по п.1, в котором катализатор на основе диоксида циркония содержит по меньшей мере один элемент, выбираемый из бора (В), алюминия (Al), марганца (Mn), кобальта (Со), никеля (Ni), иттрия (Y), лантана (La) и иттербия (Yb).
4. Способ повышения степени превращения и селективности при получении сложного эфира α-гидроксикарбоновой кислоты по любому из пп.1-3, в котором амид α-гидроксикарбоновой кислоты представлен следующей формулой (I):
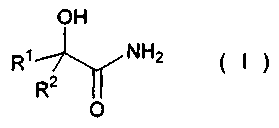
(где R1 и R2 каждый независимо представляет собой атом водорода, алкильную группу, содержащую от 1 до 20 атомов углерода, алкенильную группу, содержащую от 2 до 20 атомов углерода, или циклоалкильную группу, содержащую от 3 до 20 атомов углерода, образующих кольцо), и алифатический спирт представлен формулой R3OH (где R3 представляет собой алкильную группу, содержащую от 1 до 20 атомов углерода, алкенильную группу, содержащую от 2 до 20 атомов углерода, или циклоалкильную группу, содержащую от 3 до 20 атомов углерода, образующих кольцо).
5. Способ повышения степени превращения и селективности при получении сложного эфира α-гидроксикарбоновой кислоты по любому из пп.1-3, в котором амид α-гидроксикарбоновой кислоты является лактамидом или амидом α-гидроксиизомасляной кислоты.
6. Способ повышения степени превращения и селективности при получении сложного эфира α-гидроксикарбоновой кислоты по любому из пп.1-3, в котором алифатический спирт является метанолом или этанолом.
7. Способ повышения степени превращения и селективности при получении сложного эфира α-гидроксикарбоновой кислоты по любому из пп.1-3, в котором температура реакции составляет от 150 до 270°C и давление реакции составляет от 1 до 300 кПа.
| JP6345692 A, 20.12.1994 | |||
| ЕР 0342475 А1, 22.11.1989 | |||
| JP 7258154 A, 09.10.1995 | |||
| US 4464539 А, 07.08.1984 | |||
| WO 2008009503 A1, 24.01.2008 | |||
| JP 8268964 A, 15.10.1996 | |||
| JP 5855444 A, 01.04.1983 | |||
| СПОСОБ ПОЛУЧЕНИЯ Л10ЛОЧНОЙ КИСЛОТЫ | 0 |
|
SU180180A1 |

