Согласно настоящему изобретению предложены новые производные изатин-5-сульфонамида и их применение в качестве агентов молекулярной визуализации при визуализации и количественном анализе каспазной активности и каспаз-зависимого апоптоза и в терапевтическом использовании.
Апоптоз или программируемая клеточная смерть (ПКС) представляет собой наиболее распространенный путь клеточной смерти и протекает посредством тонко регулируемого экономичного механизма. В нормальном состоянии апоптоз играет ключевую роль в контролировании клеточного роста, регулировании числа клеток, обеспечении морфогенеза и удалении вредных или аномальных клеток. Дисрегуляция этого процесса вовлечена в ряд болезненных состояний, включая состояния, ассоциированные с ингибированием апоптоза, такие как рак и аутоиммунные расстройства, и состояния, ассоциированные со сверхактивным апоптозом, включая нейродегенеративные заболевания, гематологические заболевания, СПИД, ишемию и отторжение аллотрансплантата. Поэтому визуализация и количественный анализ апоптоза полезны в диагностике такой патофизиологии, связанной с апоптозом.
Виды терапевтического лечения этих заболеваний направлены на восстановление сбалансированного апоптоза посредством либо стимулирования, либо ингибирования этого процесса. Неинвазивная визуализация апоптоза в клетках и ткани имеет, таким образом, огромное значение для ранней оценки реакции на терапевтическое воздействие и может предоставить новое понимание разрушительных патологических процессов. Особый интерес представляет ранний мониторинг эффективности терапии рака для обеспечения того, что злокачественная опухоль будет находиться под контролем до того, как состояние перейдет в терминальную стадию.
Среди зондов, доступных для визуализации клеточной смерти, особого внимания заслуживает меченный радиоактивным изотопом аннексии V [1, 2]. Однако аннексии V может обнаруживать явления только на внешней поверхности клетки, а не внутри клетки, и связывается только с отрицательно заряженными фосфолипидами, что делает его неспособным для различения между апоптозом и некрозом. Что касается взаимодействующих с мембраной зондов, разработан ряд производных ди-дансил-цистеина и нафтил-этил-фтораланина для визуализации апоптоза [3, 4, 5]. Однако такие взаимодействующие с мембраной зонды также страдают низкой специфичностью к апоптозным клеткам и неспособны к различению между апоптозом и некрозом без необходимости в проведении отдельного теста [6]. В последнее время растет интерес к разработке специфических соединений, которые связываются с семейством ферментов, называемых каспазами.
Каспазы представляют собой семейство цистеиновых аспартат-специфичных протеаз, которые играют центральную роль в регуляции апоптоза. Внутренние и внешние сигнальные сети активируют каспазы-«инициаторы» 8 (внешняя) или 9 (внутренняя), которые в свою очередь расщепляют неактивные прокаспазы 3, 6 и 7 до активных каспаз-«убийц» 3, 6 и 7. Каспазы-«убийцы» в итоге вызывают клеточную смерть посредством расщепления клеточных белков, которое происходит правее аспартатных остатков высокоселективным образом. Расщепляемые белки включают ферменты репарации ДНК (например поли-АДФ-рибоза-полимераза (ПАРП)), ключевые сигнальные белки (например, Akt, Ras), ядерные скелетные белки (например, актин, α-фодрин, ламины) и регуляторы клеточного цикла (например, p27Kip1).
Использование пептидных необратимых ингибиторов каспаз, таких как [131I]IZ-VAD-fmk, в качестве агентов молекулярной визуализации оказалось неудовлетворительным, так как они являются лишь умеренно селективными и обладают плохой клеточной проницаемостью, так что накопление в клетках является недостаточным для визуализации in vivo.
Совсем недавно была исследована группа химических веществ, известных как изатины, в качестве потенциальных ингибиторов каспазы. Полагают, что механизм действия изатинов включает образование внутриклеточного комплекса фермент-ингибитор с каспазой 3 и 7 посредством ковалентного связывания с ферментным активным центром активированной каспазы. Дикарбонильная функциональная группа изатинов является существенной для механизма действия; она связывается с остатком цистеина активного центра, образуя тиополукеталь посредством электрофильного карбонильного атома углерода С-3 изатина и нуклеофильного функционального тиолат-иона цистеина [7].
В результате высокопроизводительного скрининга Lee et al. идентифицировали непептидный изатин, N-(1-метил)-5-нитроизатин, в качестве ингибитора каспазы-3. Структурная оптимизация привела к разработке (S)-1-бензил-5-(2-феноксиметил-пирролидин-1-сульфонил)изатин-сульфонамида (2,5 нМ) [8] (обозначенного здесь как соединение 13). Были разработаны другие изатин-сульфонамиды в качестве ингибиторов каспазы 3 и 7 [9-12]. Kopka [13] и Mach [14] независимо разработали 18F-меченный (S)-1-(4-(2-фторэтокси)-бензил)-5-[1-(2-фенилоксиметилпирролидинил)сульфонил]изатин в качестве потенциальной изотопной метки для позитронно-эмиссионной томографии (ПЭТ), и Kopka [13] исследовал биологические свойства меченного радиоактивным йодом аналога [125I]-((S)-1-(4-иодбензил)-5-(2-феноксиметил-пирролидин-1-сульфонил)изатина) (обозначенного здесь как соединение 14). В WO 99/06367 и WO 01/22966 описан ряд производных изатина и их применение для ингибирования каспаз. В WO 2006/074799, US 2005/0250798 и GB 1240648 описан ряд производных изатин-5-сульфонамида и их применение в качестве агентов визуализации апоптоза. Однако эти известные соединения страдают рядом недостатков, включая низкую молекулярную стабильность, относительно низкое сродство к каспазе-3 и высокую липофильность. Низкая молекулярная стабильность приводит к быстрому метаболизму, что ведет к плохо контрастируемым изображениям с низким отношением сигнала к шуму. Низкое сродство к каспазе-3 также может приводить к плохо контрастируемым изображениям. Высокая липофильность приводит к плохой системной элиминации и может повышать общее неспецифическое связывание с макромолекулами.
Таким образом, задача настоящего изобретения заключается в том, чтобы предложить новые производные изатина с повышенной молекулярной стабильностью, повышенным сродством к ферментам каспазам и пониженной липофильностью.
Согласно первому аспекту настоящего изобретения предложены новые производные изатин-5-сульфонамида формулы А:
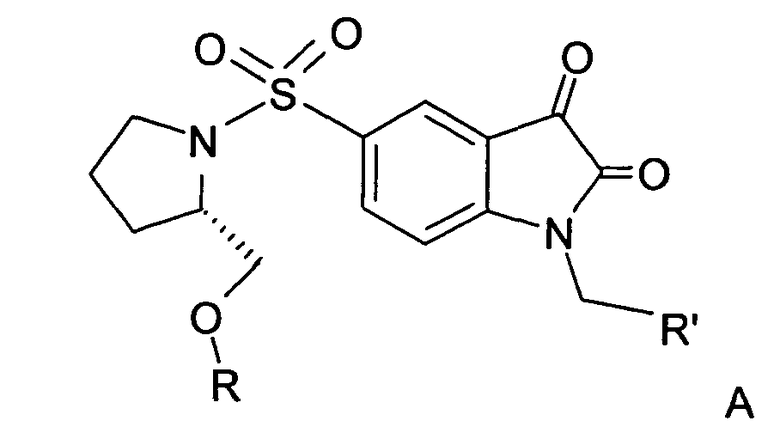
или их соль, гидрат или пролекарство, где:
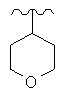
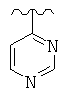
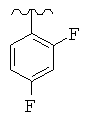
R представляет собой фенил, 3-фторфенил, 2,4-дифторфенил, 3,5-дифторфенил, возможно замещенный тетрагидропиран, возможно замещенный диазин и возможно замещенный триазолилметил; R' представляет собой возможно замещенный фенил или возможно замещенный триазол; где
когда R представляет собой фенил, R' представляет собой возможно замещенный триазол.
В предпочтительном воплощении R включает возможно замещенный триазолилметил и R' включает возможно замещенный фенил. В другом предпочтительном воплощении R включает возможно замещенный фенил и R' включает возможно замещенный триазол.
Предпочтительно, возможно замещенный фенил, возможно замещенный тетрагидропиран и возможно замещенный диазин возможно замещены одной или более чем одной электроноакцепторной группой. Предпочтительно, указанная электроноакцепторная группа выбрана из группы, включающей галоген, нитрогруппу и карбоксильную группу или другую карбонилсодержащую функциональную группу, такую как альдегид или кетон. Наиболее предпочтительно, указанная электроноакцепторная группа представляет собой галоген. Предпочтительно, указанный галоген представляет собой фтор.
Предпочтительно, возможно замещенный фенил представляет собой 2,4-дифторфенил.
В одном воплощении возможно замещенный триазол(триазолилметил) возможно замещен замещенным алкилом. Предпочтительно, указанный замещенный алкил представляет собой галоген-замещенный алкил. Более предпочтительно, указанный галоген-замещенный алкил представляет собой С1-4фторалкил. Предпочтительно, указанный С1-4фторалкил представляет собой фторметил, 2-фторэтил, 3-фторпропил или 4-фторбутил. Наиболее предпочтительно, указанный С1-4фторалкил представляет собой 2-фторэтил.
В альтернативном воплощении возможно замещенный триазол(триазолилметил) возможно замещен алкилом. Предпочтительно, указанный алкил представляет собой метил.
Специалисту в данной области техники будет понятно, что настоящее изобретение также включает все стереоизомеры соединений по настоящему изобретению, включая их энантиомеры и диастереоизомеры. Соединения по настоящему изобретению могут существовать в форме по существу чистых растворов конкретных энантиомеров или в виде рацемических смесей. Предпочтительно, соединения по настоящему изобретению существуют в виде по существу чистого раствора S-энантиомера или раствора, состоящего по существу из S-энантиомера. Предпочтительно, в рацемической смеси, содержащей как S-энантиомер, так и R-энантиомер, указанный S-энантиомер содержит по меньшей мере 50% соединений по настоящему изобретению в указанной рацемической смеси. Более предпочтительно, указанный S-энантиомер содержит по меньшей мере 60%, по меньшей мере 70%, по меньшей мере 80%, по меньшей мере 90%, по меньшей мере 95% или по меньшей мере 99% соединений по настоящему изобретению в указанной рацемической смеси.
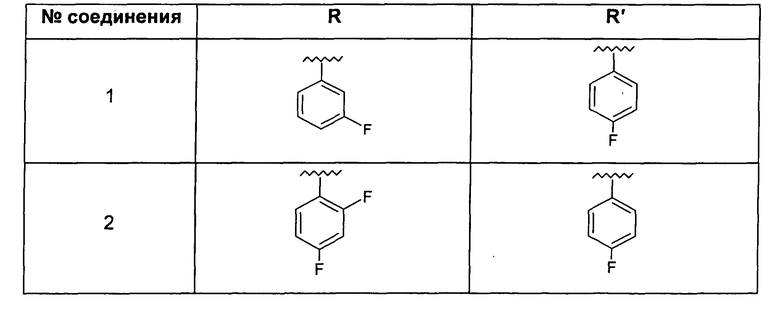
Особо предпочтительные соединения по настоящему изобретению включают соединения формулы А, где R и R' определены следующим образом:
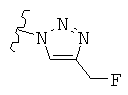
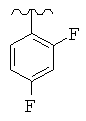
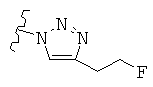
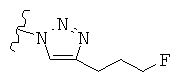
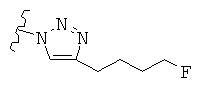
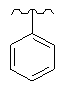
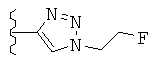
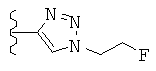
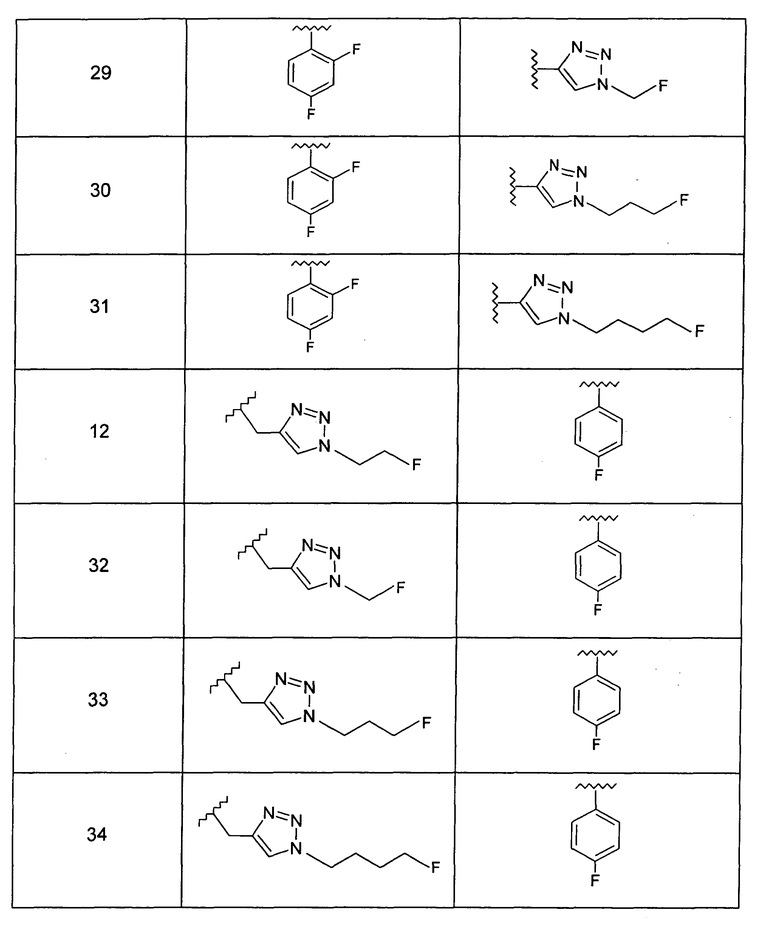
Предпочтительно, термин «соли» включает соли, полученные из органических и неорганических кислот, таких как уксусная, пропионовая, молочная, лимонная, винная, янтарная, фумаровая, малеиновая, малоновая, миндальная, яблочная, фталевая, соляная, бромистоводородная, фосфорная, азотная, серная, метансульфоновая, нафталинсульфоновая, бензолсульфоновая, толуолсульфоновая, камфорсульфоновая, и подобных известных приемлемых кислот, когда соединение по этому изобретению содержит основную группировку. Соли также могут быть образованы из органических и неорганических оснований, предпочтительно соли щелочных металлов, например натрия, лития или калия, когда соединение по этому изобретению содержит карбоксилатную или фенольную группировку или подобную группировку, способную образовать соли присоединения оснований.
Используемый здесь термин «гидрат» относится к формам соединений по настоящему изобретению, которые химически соединены с водой.
Используемый здесь термин «пролекарство» относится к соединению, которое превращается in vivo посредством метаболического пути в соединение по настоящему изобретению.
Используемая здесь фраза «возможно замещенный фенил» относится к фенильной группе, которая возможно может содержать один или более чем один заместитель, находящийся в одном или более чем одном из положений 2, 3, 4, 5 и 6 в кольце.
Используемый здесь термин «тетрагидропиран» относится к органическому соединению, состоящему из насыщенного шестичленного кольца, содержащего пять атомов углерода и один атом кислорода.
Используемый здесь термин «триазол» относится к соединениям с молекулярной формулой C2HN3, имеющим пятичленное кольцо с двумя атомами углерода и тремя атомами азота. Эти соединения включают изомеры 1,4- и 1,5-дизамещенных 1,2,3-триазолов.
Используемый здесь термин «галоген» относится к брому, хлору, фтору и иоду.
Используемый здесь термин «алкил» относится к алифатической углеводородной цепи и включает прямые или разветвленные цепи, имеющие 1, 2, 3, 4, 5 или 6 атомов углерода, но не ограничен ими, если не оговорено особо. Алкилы включают метил, этил, н-пропил, изопропил, м-бутил, втор-бутил, изобутил, трет-бутил и им подобные.
Используемая здесь фраза «замещенный алкил» относится к алкилу, дополнительно содержащему один или более чем один заместитель, выбранный из группы, включающей галоген, гидроксил, тиол, амино, альтернативный гетероатом, ароматическую или гетероароматическую группу и спейсерные группы, такие как полиэфир, включая полиэтиленгликоль, сукцинидил, -NH-(CH2)n-NH- и полиамиды.
Используемый здесь термин «диазин» относится к группе органических соединений, имеющих молекулярную формулу C4H4N2, каждое из которых содержит бензольное кольцо, в котором два атома углерода замещены азотом. Эти соединения включают изомеры пиразина (1,4-диазин), пиримидина (1,3-диазин) и пиридазина (1,2-диазин).
Соединения по настоящему изобретению являются специфичными к активированным каспазам и поэтому обладают высоким сродством. Они являются сильнодействующими и селективными ингибиторами каспаз 3 и 7. В частности, соединения по настоящему изобретению обладают высоким сродством к каспазе-3 и каспазе-7. Они экспрессируются в активной форме только во время апоптоза, и активность каспазы-3 и/или каспазы-7, поэтому, является достоверным индикатором каспаз-зависимого апоптоза.
Соединения по настоящему изобретению обладают пониженной липофильностью по сравнению со многими известными производными изатин-5-сульфонамида, что делает возможной хорошую системную элиминацию и уменьшает общее неспецифическое связывание с макромолекулами. Однако каспазы являются внутриклеточными протеазами, поэтому соединения по настоящему изобретению являются еще и достаточно липофильными, чтобы пересекать клеточные мембраны посредством необлегченной диффузии, для того чтобы они могли свободно проходить в клетку и из клетки. В связи с этим, соединения будут обладать быстрым двусторонним переносом и удерживанием только в клетках с активированной каспазой-3 и/или каспазой-7. Оптимальная липофильность (выраженная в виде Log P) варьирует в зависимости от типа клетки, в которую соединения должны проходить, помимо ряда других факторов. В одном воплощении липофильность находится предпочтительно в пределах от 1,0 до 2,0.
Соединения по настоящему изобретению обладают повышенной метаболической стабильностью, что снижает скорость, с которой соединения подвергаются метаболизму и, в некоторых случаях, экскреции. Повышенная метаболическая стабильность делает возможным накопление соединения по настоящему изобретению в клетках, подвергающихся апоптозу, при связывании с активированными каспазами. Это обеспечивает хорошо контрастируемые изображения с высоким отношением сигнала к шуму. Это является благоприятным для агента молекулярной визуализации, поскольку делает возможной четкую визуализацию и количественный анализ каспазной активности в клетках и тканях.
Предпочтительно, соединения по настоящему изобретению имеют низкое накопление в необработанных опухолях, ткани сердца и головного мозга. Это способствует мониторингу изменений при связывании этих соединений с активированными каспазами в тканях млекопитающих, которые ассоциированы с изменениями в каспазной активности.
Согласно второму аспекту настоящего изобретения предложены соединения по первому аспекту настоящего изобретения, где указанные соединения включают метку визуализирующей группировкой.
Указанное мечение может включать визуализирующую группировку в пределах функциональной группы или присоединение визуализирующей группировки в виде дополнительной группы.
Указанная визуализирующая группировка может включать любую группировку, способную давать детектируемый сигнал. Такие группировки включают флуоресцентные метки и радиоактивные метки.
Флуоресцентные метки содержат ковалентно присоединенный флуорофор. Предпочтительные флуорофоры включают флуоресцеинизотиоцианат, производные родамина, кумариновые и цианиновые красители, Alexa Fluors и DyLight Fluors.
Предпочтительно, соединения метят радиоактивным изотопом. Использование радиоактивного изотопа в качестве визуализирующей группировки обладает преимуществами, так как радиоактивный изотоп значительно не увеличивает молекулярную массу соединения по настоящему изобретению и может быть использован для клинической неинвазивной визуализации.
Предпочтительно, радиоактивный изотоп представляет собой позитронный излучатель. Радиоактивный изотоп может быть выбран из группы, включающей 3H, 14С, 18F, 11С, 120I, 123I, 124I, 125I, 131I, 94mTc, 66Ga, 68Ga, 64Cu, 61Cu, 67Cu, 75Br, 76Br, 94mTc, 99mTc, 201TI, 111In, 86Y и 89Zr.
Если визуализирующая группировка должна быть визуализирована с использованием позитронно-эмиссионной томографии (ПЭТ), радиоактивный изотоп предпочтительно выбран из группы, включающей 18F, 11С, 120I, 124I, 94mTc, 66Ga, 68Ga, 64Cu, 67Cu, 86Y, 75Br и 76Br.
Если визуализирующая группировка должна быть визуализирована с использованием однофотонной эмиссионной компьютерной томографии (ОФЭКТ), радиоактивный изотоп предпочтительно выбран из группы, включающей 123I, 99mTc и 111In.
Предпочтительно, визуализирующая группировка представляет собой 18F или 11С.
Предпочтительно, в любом из соединений от 1 до 12 и от 29 до 34 один или более чем один атом фтора в R и/или R' представляет собой 18F.
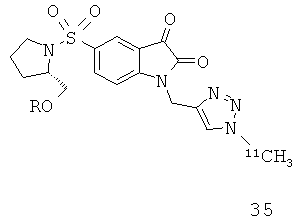
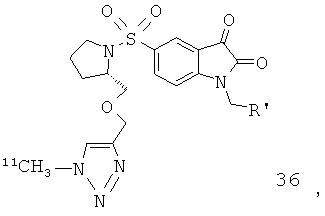
Добавление 1,2,3-триазольной группы в R или R', как, например, в соединениях от 6 до 12 и от 29 до 36, имеет дополнительное преимущество, поскольку в такое соединение может быть легко введена метка 18F или 11С.
Особо предпочтительное воплощение соединения по изобретению имеет формулу:
Другие предпочтительные соединения по настоящему изобретению имеют общие формулы:
и
где R и R' являются такими, как определено выше.
Согласно третьему аспекту настоящего изобретения предложена композиция, содержащая соединение по первому аспекту изобретения, возможно, в комбинации с одним или более чем одним фармацевтически приемлемым носителем, разбавителем или эксципиентом.
Согласно четвертому аспекту настоящего изобретения предложена композиция, содержащая соединение по второму аспекту изобретения, возможно, в комбинации с одним или более чем одним фармацевтически приемлемым носителем, разбавителем или эксципиентом.
Эти композиции могут также содержать один или более чем один дополнительный активный агент. Указанные дополнительные активные агенты могут включать агент, который усиливает сигнал, продуцируемый соединениями по настоящему изобретению. Предпочтительно, указанный активный агент включает соединения, которые уменьшают или модулируют уровни клеточного глутатиона, включая диэтилмалеат (ДЭМ), L-бутионин-(S,R)-сульфоксим (БСО) и их производные.
Композиции по третьему или четвертому аспекту настоящего изобретения могут быть введены любым подходящим способом.
Композиции можно привести в контакт с клетками посредством экспонирования, инкубирования, соприкосновения, ассоциирования или обеспечения соединению доступа в клетки.
Композиции могут быть введены субъекту посредством перорального (включая ингаляцию), парентерального, мукозального (например, трансбуккального, сублингвального, назального), ректального или трансдермального введения, и композиции подобраны соответственно.
Для перорального введения композиции могут быть приготовлены в виде жидкостей или твердых веществ, например растворов, сиропов, суспензий или эмульсий, таблеток, капсул и лепешек.
Жидкий препарат будет обычно состоять из суспензии или раствора соединения или его физиологически приемлемой соли, гидрата или пролекарства в подходящем водном или неводном жидком носителе(ях), например воде, этаноле, глицерине, полиэтиленгликоле или масле. Препарат может также содержать суспендирующий агент, консервант, корригент или краситель.
Композиция в форме таблетки может быть приготовлена с использованием любого(ых) подходящего(их) фармацевтического(их) носителя(ей), обычно используемого(ых) для приготовления твердых препаратов. Примеры таких носителей включают стеарат магния, крахмал, лактозу, сахарозу и микрокристаллическую целлюлозу.
Композиция в форме капсулы может быть приготовлена с использованием стандартных методов инкапсулирования. Например, порошки, гранулы или пеллеты, содержащие активный ингредиент, могут быть приготовлены с использованием стандартных носителей и затем помещены в твердую желатиновую капсулу. Альтернативно, дисперсия или суспензия могут быть приготовлены с использованием любого(ых) подходящего(их) фармацевтического(их) носителя(ей), например жидких камедей, целлюлоз, силикатов или масел, и эту дисперсию или суспензию затем помещают в мягкую желатиновую капсулу.
Композиции для перорального введения могут быть приспособлены для защиты активного ингредиента от разложения, когда он проходит через пищеварительный тракт, например, с помощью наружного покрытия препарата на таблетке или капсуле.
Композиции для назального или перорального введения могут быть легко приготовлены в виде аэрозолей, капель, гелей и порошков. Аэрозольные препараты типично содержат раствор или тонкую суспензию активного вещества в физиологически приемлемом водном или неводном растворителе и обычно представлены в количестве, предназначенном для однократного или многократного дозирования, в стерильной форме в герметичном контейнере, который может иметь форму картриджа или сменного баллончика для применения в распылительном устройстве. Альтернативно, герметичный контейнер может представлять собой единичное распределительное устройство, такое как однодозовый назальный ингалятор или аэрозольный распылитель, снабженный дозатором, которое предназначено на выброс, как только содержимое контейнера будет израсходовано. Когда лекарственная форма содержит аэрозольный распылитель, он будет содержать фармацевтически приемлемый пропеллент. Аэрозольные лекарственные формы могут также иметь форму помпового распылителя.
Композиции, подходящие для трансбуккального или сублингвального введения, включают таблетки, лепешки и пастилки, в которых активный ингредиент включен в состав препарата с носителем, таким как сахар и аравийская камедь, трагакант или желатин и глицерин.
Композиции для ректального или вагинального введения удобны в форме суппозиториев (содержащих традиционную основу для суппозиториев, такую как масло какао), пессариев, вагинальных таблеток, пенок или клизм.
Композиции, подходящие для трансдермального введения, включают мази, гели, пластыри и инъекции, включая порошковые инъекции.
Удобно, чтобы композиция находилась в стандартной лекарственной форме, такой как таблетка, капсула или ампула.
Предпочтительно, соединения или композиции по настоящему изобретению вводят субъекту посредством парентерального введения. В частности, композиции могут быть введены внутривенно, внутрибрюшинно, интратекально, внутрилимфатически или внутримышечно.
Типичные парентеральные композиции состоят из парентерально приемлемого раствора или суспензии соединения или физиологически приемлемой соли в стерильном водном или неводном носителе, который имеет подходящие рН, изотоничность и стабильность. Специалисты в данной области техники могут приготовить подходящие растворы с использованием, например, изотонических носителей, таких как раствор хлорида натрия для инъекций, раствор Рингера для инъекций, раствор Рингера с лактатом для инъекций. В случае необходимости могут быть включены консерванты, стабилизаторы, буферные агенты, антиоксиданты и/или другие вспомогательные вещества.
Согласно пятому аспекту настоящего изобретения предложены соединения по первому или второму аспекту настоящего изобретения или фармацевтические композиции по третьему или четвертому аспекту настоящего изобретения для применения в качестве агентов молекулярной визуализации.
Предпочтительно, указанные агенты молекулярной визуализации предназначены для визуализации и количественного анализа каспазной активности в клетках и тканях. Предпочтительно, указанные агенты молекулярной визуализации предназначены для визуализации и количественного анализа каспазной активности в клетках и тканях млекопитающих, включая клетки и ткани человека. Указанный количественный анализ может включать анализ величины радиоактивности в клетках и тканях или скорости поглощения, диссоциации или распределения.
При образовании комплекса с активными каспазами поглощение и накопление меченого соединения в клетках и тканях можно затем визуализировать для индикации уровня каспазной активности в этих клетках и тканях. Поэтому, согласно шестому аспекту настоящего изобретения, предложен способ молекулярной визуализации каспазной активности, включающий стадии:
а) приведения указанных клеток или тканей в контакт с соединением по второму аспекту настоящего изобретения или композицией по четвертому аспекту настоящего изобретения и
б) детектирования указанной каспазной активности.
Предпочтительно, стадия детектирования указанной каспазной активности включает стадии помещения субъекта в область детекции устройства обнаружения и детектирования указанных соединений у субъекта с помощью указанного устройства обнаружения.
Этот способ может быть осуществлен in vitro. Приведение клеток или тканей в контакт с соединением или композицией по настоящему изобретению может включать экспонирование, инкубирование, соприкосновение, ассоциирование или обеспечение соединению доступа в клетки и ткани.
Когда соединение или композиция по настоящему изобретению включает радиоизотопную метку, указанную каспазную активность можно детектировать in vitro с использованием любого соответствующего устройства радиоизотопной диагностики. Указанное устройство может включать счетчик бета-излучения, такой как Packard Topcount, или счетчик гамма-излучения, такой как Packard Cobra II™ (Perkin Elmer, UK), или сканер для радио-TLC.
Когда соединение или композиция по настоящему изобретению включает флуоресцентную метку, указанную каспазную активность можно детектировать in vitro с использованием любого соответствующего считывающего прибора для измерения флуоресценции. Такие приборы включают флуоресцентный микроскоп, флуорометр или флуоресцентный планшетный ридер, такой как Perkin Elmer Victor.
Альтернативно, этот способ может быть осуществлен in vivo. Соединения или композиции могут быть введены субъекту любым способом, рассмотренным в третьем и четвертом аспектах настоящего изобретения. Предпочтительно, соединения вводят парентерально.
Когда соединение или композиция по настоящему изобретению включает радиоизотопную метку, указанную каспазную активность можно детектировать с использованием устройства радиоизотопной диагностики. Указанное устройство радиоизотопной диагностики может включать сканер позитронно-эмиссионной томографии (ПЭТ) или сканер однофотонной эмиссионной компьютерной томографии (ОФЭКТ). Предпочтительно, указанное устройство радиоизотопной диагностики представляет собой сканер позитронно-эмиссионной томографии (ПЭТ). Указанный ПЭТ-сканер может детектировать пары гамма-лучей, испускаемых опосредованно позитрон-испускающими радиоизотопами, такими как 18F, для получения трехмерного изображения концентрации радиоизотопов в тканях. Таким образом, ПЭТ может быть использована для получения трехмерного изображения локализации меченых радиоактивными изотопами соединений и композиций по настоящему изобретению в клетках млекопитающих.
Когда соединение или композиция по настоящему изобретению включает флуоресцентную метку, указанную каспазную активность можно детектировать с использованием любого соответствующего считывающего прибора для измерения флуоресценции. Указанный считывающий прибор для измерения флуоресценции может включать флуоресцентный эндоскоп.
Предпочтительно, каспазная активность представляет собой активность каспазы-3.
Согласно настоящему изобретению также предложено применение соединений по второму аспекту настоящего изобретения для производства фармацевтической или диагностической композиции для визуализации и количественного анализа каспазной активности.
В альтернативном воплощении шестого аспекта соединения по первому аспекту настоящего изобретения или фармацевтические композиции по третьему аспекту настоящего изобретения могут быть введены согласно стадии (а). Указанные соединения затем могут быть помечены флуоресцентно или радиоактивными изотопами после указанного введения.
Как рассмотрено выше, ферменты каспазы вызывают апоптоз, и как таковая каспазная активность может быть использована в качестве индикатора апоптоза. Поэтому, согласно седьмому аспекту настоящего изобретения, предложено применение соединений по второму аспекту настоящего изобретения или фармацевтических композиций по четвертому аспекту настоящего изобретения для визуализации in vivo каспаз-зависимого апоптоза в клетках или ткани млекопитающих.
Этот способ может включать:
а) введение субъекту соединения по второму аспекту настоящего изобретения или композиции по четвертому аспекту настоящего изобретения;
б) помещение субъекта в область детекции устройства обнаружения; и
в) детектирование указанных соединений у субъекта с помощью указанного устройства обнаружения.
Предпочтительно указанное соединение или композицию вводят субъекту посредством инъекции.
Визуализирующую группировку можно детектировать либо наружно, неинвазивным способом, либо изнутри путем использования детекторов, разработанных для применения in vivo, таких как внутрисосудистые радиационные или оптические детекторы, или радиационных детекторов, разработанных для интраоперационного применения. Предпочтительно, визуализирующую группировку детектируют неинвазивным способом.
В одном воплощении указанные соединения метят флуоресцентно и детектируют путем измерения флуоресценции, излучаемой флуоресцентно мечеными соединениями, с помощью считывающего прибора для измерения флуоресценции, как определено выше в шестом аспекте.
Во втором воплощении, являющемся предпочтительным, указанные соединения метят радиоактивными изотопами и детектируют путем измерения излучения, испускаемого мечеными радиоактивными изотопами соединениями, с помощью устройства радиоизотопной диагностики, как определено выше в шестом аспекте.
Предпочтительно, указанная каспазная активность представляет собой активность каспазы-3.
В альтернативном воплощении седьмого аспекта соединения по первому аспекту настоящего изобретения или фармацевтические композиции по третьему аспекту настоящего изобретения могут быть введены согласно стадии (а). Указанные соединения затем могут быть помечены in vivo флуоресцентно или радиоактивными изотопами после указанного введения.
Как рассмотрено выше, виды терапевтического лечения ряда заболеваний направлены на восстановление нормального сбалансированного апоптоза посредством стимулирования или ингибирования этого процесса. Поэтому, согласно восьмому аспекту настоящего изобретения предложено применение соединений по второму аспекту настоящего изобретения или фармацевтических композиций по четвертому аспекту настоящего изобретения для оценки терапевтического воздействия исследуемого вещества на каспазную активность в клетках или тканях млекопитающих. Этот способ может включать:
а) приведение клеток или тканей млекопитающих в контакт с соединением по второму аспекту настоящего изобретения или композицией по четвертому аспекту настоящего изобретения;
б) помещение указанных клеток или тканей млекопитающих в область детекции устройства обнаружения;
в) детектирование соединений с помощью указанного устройства обнаружения;
г) повторение стадий (а), (б) и (в).
В одном воплощении указанные соединения метят флуоресцентно и детектируют посредством измерения флуоресценции, излучаемой флуоресцентно мечеными соединениями, с помощью считывающего прибора для измерения флуоресценции, как определено выше в шестом аспекте.
Во втором воплощении, являющемся предпочтительным, указанные соединения метят радиоактивными изотопами и детектируют путем измерения излучения, испускаемого мечеными радиоактивными изотопами соединениями, с помощью устройства радиоизотопной диагностики, как определено выше в шестом аспекте.
В альтернативном воплощении восьмого аспекта соединения по первому аспекту настоящего изобретения или фармацевтические композиции по третьему аспекту настоящего изобретения могут быть введены согласно стадии (а). Указанные соединения затем могут быть помечены флуоресцентно или радиоактивными изотопами после указанного введения.
Предпочтительно, этот способ может быть осуществлен in vivo и может включать:
а) введение субъекту соединения по второму аспекту настоящего изобретения или композиции по четвертому аспекту настоящего изобретения;
б) помещение субъекта в область детекции устройства радиоизотопной диагностики;
в) измерение излучения, испускаемого мечеными радиоактивными изотопами соединениями, у субъекта с помощью указанного устройства радиоизотопной диагностики;
г) повторение стадий (а), (б) и (в).
Как для in vitro, так и для in vivo способов по восьмому аспекту, стадию (г) предпочтительно осуществлять в избранные промежутки времени, когда указанное повторение является эффективным для отслеживания изменений в каспазной активности в течение времени. Промежутки времени для стадии (г) должны быть подходящими для субъекта и исследуемого соединения, о которых идет речь. Для людей подходящие промежутки времени включают от 12 до 24 часов, 48 часов, 1 недели, 2 недель, 3 недель, 4 недель, 5 недель или 6 недель, но не ограничены ими.
Каспазную активность, измеряемую при каждом повторении, можно сравнить для оценки количественных изменений величины и локализации каспазной активности, и следовательно, количественных или полуколичественных изменений в каспаз-зависимом апоптозе, в течение времени. Изменения величины и локализации каспазной активности могут служить признаком терапевтического воздействия исследуемого соединения на каспаз-зависимый апоптоз либо посредством стимулирования, либо ингибирования каспазной активности. Этот способ может быть использован для оценки реакции субъекта на терапевтическое лечение с использованием признанных лекарственных средств и оценки эффективности новых лекарственных средств.
Соединение или композицию по настоящему изобретению можно вводить до, после или одновременно с исследуемым веществом. Предпочтительно, стадии от (а) до (в) проводят на субъекте перед введением исследуемого вещества для осуществления первого измерения каспазной активности. Затем вводят исследуемое вещество, и стадии от (а) до (в) повторяют через промежуток времени, как рассмотрено выше, для осуществления второго измерения каспазной активности. Стадии от (а) до (в) можно повторять в последующие моменты до или после последующих введений исследуемого вещества для осуществления последующих измерений каспазной активности.
Предпочтительно, указанное соединение или композицию вводят субъекту посредством инъекции.
Исследуемое вещество может включать лекарственное средство или агент, используемый для лечения заболеваний, включая рак, аутоиммунное заболевание, гематологическое заболевание, ВИЧ-инфекцию (вирус иммунодефицита человека), СПИД (синдром приобретенного иммунодефицита), ишемию, сердечно-сосудистое заболевание, неврологические заболевания и отторжение трансплантата, но не ограничиваясь ими. В частности, исследуемое соединение может включать химиотерапевтические, радиотерапевтические или иммунотерапевтические агенты для лечения рака.
Как указано выше, стадии от (а) до (в) можно повторять два или более раз. В одном предпочтительном воплощении соединение, вводимое субъекту при первом проведении стадии (а), представляет собой соединение по настоящему изобретению, которое включает метку радиоактивным изотопом 11С. Предпочтительно, указанное соединение выбрано из группы, включающей соединения 35 и 36. Соответственно, соединение, вводимое субъекту при втором проведении стадии (а), представляет собой агент визуализации, меченый радиоактивным изотопом. Указанный агент визуализации, меченый радиоактивным изотопом, может представлять собой, например, фтордезоксиглюкозу (ФДГ), 3'-дезокси-3'-18F-фтортимидин (FLT) или соединение по настоящему изобретению. В одном предпочтительном воплощении указанный агент визуализации включает метку 18F. Это воплощение является особенно предпочтительным. Углерод-11 (11С) обладает относительно коротким периодом полураспада, составляющим 20 минут. Таким образом, при использовании соединения, меченного изотопом 11С, имеются либо незначительные, либо полностью отсутствуют фоновые шумы во время второго и последующих сканирований. Это приводит к более высокому отношению сигнала к шуму и более точной визуализации и количественному анализу каспазной активности и каспаз-зависимого апоптоза.
Аналогично, если способы по шестому и седьмому аспектам настоящего изобретения должны быть повторены в пределах относительно короткого промежутка времени, применение 11С-меченого соединения с последующим применением альтернативного агента визуализации, меченого радиоактивным изотопом, такого как 18F-меченое соединение, также будет уменьшать фоновые шумы, присутствующие во время второго сканирования, и увеличивать отношение сигнала к шуму.
Согласно девятому аспекту настоящего изобретения предложены соединения по первому аспекту настоящего изобретения или композиции по третьему аспекту настоящего изобретения для применения в ингибировании каспазной активности. Этот способ может включать введение субъекту, нуждающемуся в таком лечении, эффективного количества соединений по первому аспекту настоящего изобретения или композиций по третьему аспекту настоящего изобретения.
Согласно этому аспекту также предложено применение соединений по первому аспекту настоящего изобретения или композиций по третьему аспекту настоящего изобретения в производстве лекарственного средства для ингибирования каспазной активности.
Поскольку ферменты каспазы вызывают апоптоз, согласно девятому аспекту настоящего изобретения также предложены соединения по первому аспекту настоящего изобретения или композиции по третьему аспекту настоящего изобретения для применения в ингибировании апоптоза.
Ингибирование каспазной активности может быть эффективным для лечения любого заболевания или состояния, вызванного избыточным или неприемлемым апоптозом или связанного с ним. Примеры таких заболеваний или состояний включают нейродегенеративные заболевания, включая болезнь Альцгеймера, гематологические заболевания, гепатоцеллюлярную дегенерацию, остеоартрит, СПИД, ишемию и отторжение аллотрансплантата.
Соединения по первому аспекту настоящего изобретения или композиции по третьему аспекту настоящего изобретения будут обычно вводить субъекту в режиме суточного дозирования. Например, для режима суточного дозирования может потребоваться от примерно 0,001 до примерно 100 мг/кг, предпочтительно от примерно 0,001 до примерно 10 мг/кг массы тела субъекта. Суточная доза для более крупного млекопитающего составляет предпочтительно от примерно 1 мг до примерно 1000 мг, предпочтительно от 1 мг до 500 мг, при этом соединение вводят от 1 до 4 раз в сутки. Альтернативно, принимая во внимание стабильность соединений по настоящему изобретению, дозу можно вводить 1, 2 или 3 раза в неделю.
Понятно, что дозировка, режим дозирования и способ введения соединений и композиций по настоящему изобретению будут варьировать в зависимости от заболевания или состояния и индивидуума, которого лечат, и будет зависеть от заключения лечащего врача.
Согласно десятому аспекту настоящего изобретения предложены соединения по второму аспекту настоящего изобретения или композиции по четвертому аспекту настоящего изобретения для применения в диагностике патофизиологии. В частности, соединения по второму аспекту настоящего изобретения или композиции по четвертому аспекту настоящего изобретения можно применять для диагностирования заболеваний и расстройств, ассоциированных с избыточным или неприемлемым апоптозом, включая хроническую сердечную недостаточность, острый инфаркт миокарда, инсульт, нейродегенеративные расстройства, аутоиммунное заболевание, очаговое гематологическое заболевание, очаговый СПИД, ишемию (включая сердечную ишемию) и отторжение трансплантата, но не ограничиваясь ими.
Этот способ может включать:
а) приведение клеток или тканей млекопитающих в контакт с соединением по второму аспекту настоящего изобретения или композицией по четвертому аспекту настоящего изобретения;
б) помещение указанных клеток или тканей млекопитающих в область детекции устройства обнаружения;
в) детектирование указанных соединений с помощью указанного устройства обнаружения для осуществления количественных измерений каспазной активности в указанных клетках или тканях млекопитающих, где каспазная активность является индикатором уровня апоптоза.
В одном воплощении указанные соединения метят флуоресцентно и детектируют путем измерения флуоресценции, излучаемой флуоресцентно мечеными соединениями, с помощью считывающего прибора для измерения флуоресценции, как определено выше в шестом аспекте.
Во втором воплощении, являющемся предпочтительным, указанные соединения метят радиоактивными изотопами и детектируют путем измерения излучения, испускаемого мечеными радиоактивными изотопами соединениями, с помощью устройства радиоизотопной диагностики, как определено выше в шестом аспекте.
Предпочтительно, указанные измерения затем сравнивают со стандартными значениями, чтобы обеспечить диагностику патофизиологии.
В альтернативном воплощении десятого аспекта соединения по первому аспекту настоящего изобретения или фармацевтические композиции по третьему аспекту настоящего изобретения могут быть введены согласно стадии (а). Указанные соединения затем могут быть мечены флуоресцентно или радиоактивными изотопами после указанного введения.
Согласно одиннадцатому аспекту настоящего изобретения предложены способы синтеза соединений по первому аспекту настоящего изобретения. Соединения по первому аспекту настоящего изобретения могут быть получены любым подходящим способом. Схемы, описанные в примерах 1, 3 и 10 и проиллюстрированные на Фиг.1, 2(i), 2(ii), 12, 19, 20 и 21, иллюстрируют способы, с помощью которых могут быть получены соединения по первому аспекту настоящего изобретения.
На Фиг.1 проиллюстрированы способы, с помощью которых могут быть получены соединения 1-5, 10-12 и промежуточные соединения 19a-h, 20a-h, 21-24. Соединения с 29 по 34 получают аналогичным образом. Дополнительные подробности предложены в примере 1.
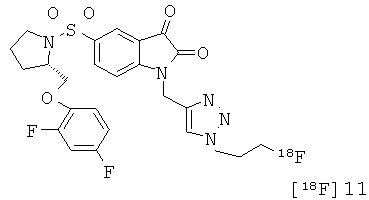
Те соединения по настоящему изобретению, которые содержат триазольные группы, могут также быть получены способом, называемым «клик-мечение», как проиллюстрировано на Фиг.2(i), Фиг.12 и Фиг.19 и описано в примерах 1 и 3. Например, такой способ может быть использован для получения соединений 11, 35 и 36. Для получения соединений по первому аспекту настоящего изобретения конечно будут использованы немеченые соединения вместо меченых радиоактивными изотопами соединений, раскрытых в этих примерах. Например, в схеме, проиллюстрированной на Фиг.2(i) и описанной в примере 3, 2-фторэтилазид (соединение 27) используют вместо 2-[18F]фторэтилазида (соединение [18F]27).
Циклоприсоединение с получением замещенных триазолов также может быть эффективно в обращенном процессе (в способе, называемом «обращенное клик-мечение»), посредством взаимодействия изатина, функционализированного в положении N-1, с терминальным азидом и простетической группой, содержащей терминальный алкин. Например, такой способ может быть использован для получения соединений 6, 7, 8 и 9. Подходящий пример этой методики, которая также включает способы синтеза промежуточного 18F-меченого терминального алкина(ов), показан на Фиг.2(ii) и описан в примере 1, и у Marik, J и Sutcliffe, J.L. (26), Sirion, U et al. (27) и Li, Z. et al. (28), которые включены здесь посредством ссылки.
Согласно двенадцатому аспекту настоящего изобретения предложены способы синтеза соединений по второму аспекту настоящего изобретения. Соединения по второму аспекту настоящего изобретения могут быть получены любым подходящим способом. Схемы, описанные в примерах 1 и 3 и проиллюстрированные на Фиг.1, 2(i), 2(ii) и 12, иллюстрируют способы, с помощью которых могут быть получены соединения по второму аспекту настоящего изобретения, где используют меченые промежуточные соединения.
Схемы, описанные в примере 1 и проиллюстрированные на Фиг.1, могут быть использованы для получения соединений по второму аспекту настоящего изобретения, где одна или более чем одна молекула-предшественник или промежуточное соединение, например соединения 19a-h, 20a-h и/или 21-25, содержат меченую группу R или R'. Например, такой способ может быть использован для получения меченых форм соединений 1-5, 10-12 и с 29 по 34.
Схемы, проиллюстрированные на Фиг.2(i), Фиг.12, Фиг.19 и описанные в примере 1 и примере 3, иллюстрируют способы «клик-мечения», с помощью которых могут быть получены конкретные соединения по второму аспекту настоящего изобретения, включая соединения [18F]11, [11C]35 и [11С]36.
Способ «обращенного клик-мечения», проиллюстрированный на Фиг.2(ii) и описанный в примере 1, также может быть использован для получения некоторых соединений по второму аспекту настоящего изобретения, таких как меченые формы соединений 6, 7, 8 и 9. В этом случае используют одно или более чем одно меченое соединение-предшественник. Например, в схеме, проиллюстрированной на Фиг.2(ii) и описанной в примере 1, 3-[18F]фторпроп-1-ин (соединение [18F]28) используют вместо 3-фторпроп-1-ин (соединение 28).
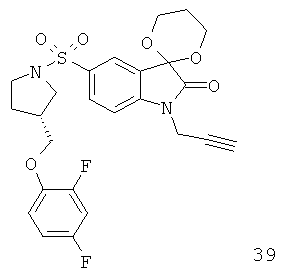
Однако способ «клик-мечения», описанный в примере 3 и проиллюстрированный на Фиг.19, страдает тем недостатком, что образуется стабильная примесь, которая снижает удельную активность продукта, производного изатина. Как описано в примере 10 и проиллюстрировано на Фиг.20 и 21, этот недостаток можно преодолеть посредством синтеза производного изатина, например соединения [18F]11, с использованием защищенного предшественника, такого как показано в общих формулах 38 и 43, где n равен 0, 1, 2, 3, 4, 5 или 6 (то есть n равен 0-6) и где х представляет собой уходящую группу, например мезилат, тозилат, нозилат или другой сульфонатный эфир или галогенид.
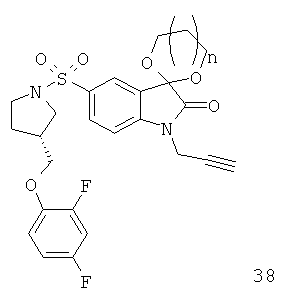
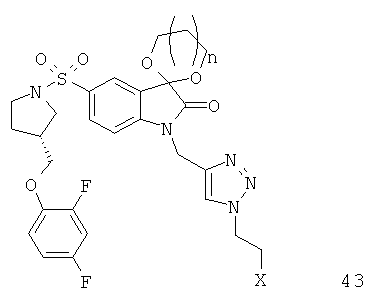
Особо предпочтительным защищенным алкиновым предшественником является (S)-1-{[1'-[1-(2-пропинил)]-(1'2'-дигидро-2'-оксоспиро(1,3-диоксан-2,3'-[3Н]индол)-5'-сульфонил}-2-(2,4-дифторфеноксиметил)-пирролидин (соединение 39). Применение такого защищенного алкинового предшественника предотвращает нежелательные побочные реакции по положению С-3.
Помимо этих способов, соединения по второму аспекту настоящего изобретения могут быть мечены радиоактивными изотопами посредством, например, галогендеметаллирования соответствующего оловосодержащего предшественника, хелатирования с требуемым металлом, предпочтительно с подходящим лигандом, присоединенным к изатину, замещения [18F]-фтором в присутствии подходящей арильной или алкильной уходящей группы.
Описание графических материалов
Фиг.1 является схематическим представлением синтеза целевых соединений. Реагенты и условия являются следующими: (а) фенол/фторзамещенный фенол/4-тетрагидропиран, NaH, ДМФА (диметилформамид), 80°С, 17 часов; (b) 4(3Н)-пиримидон, PPh3, DIAD (диизопропилазодикарбоксилат), ДХМ (дихлорметан), комнатная температура, 48 часов; (с) пропаргилбромид, КОН, ДМФА, комнатная температура, 18 часов; (d) ТФУ (трифторуксусная кислота), ДХМ, 0°С, 1 час; (е) 5-хлорсульфонилизатин, ТЭА (триэтиламин), ТГФ (тетрагидрофуран)УДХМ, комнатная температура, 19 часов; (f) 4-фторбензилбромид, К2СО3, ДМФА, комнатная температура, 2 часа; (g) пропаргилбромид, К2СО3, ДМФА, комнатная температура, 2 часа; (h) 2-фторэтилазид, CuSO4, L-аскорбиновая кислота, ДМФА, комнатная температура, 2 часа.
Фиг.2(i) является схематическим представлением синтеза соединения [18F]11 путем взаимодействия (S)-1-(2-пропинил)-5-(2-(2,4-дифторфенокси-метил)-пирролидин-1-сульфонил)изатина (соединение 25) с [18F]фторэтилазидом (соединение 27). Соединение 26 представляет собой 2-(толуол-4-сульфонил)этилазид. Реагенты являются следующими: (a) [18F]KF, криптофикс[2,2,2] (Kryptofix[2,2,2]), ацетонитрил; (b) CuSO4, аскорбат натрия, фосфатный буфер рН 6,0, соединение 25.
Фиг.2(ii) является схематическим представлением синтеза соединения 6 ((S)-1-[4-(2-фторэтил)-1Н-[1,2,3]-триазол-1-ил]метил-5-(2-(2,4-дифторфенокси-метил)-пирролидин-1-сульфонил)изатина) путем взаимодействия (S)-1-(азидометил)-5-(2-(2,4-дифторфеноксиметил)-пирролидин-1-сульфонил)изатина (соединение 37) с 3-фторпроп-1-ином (соединение 28). Реагенты являются следующими: (а) CuSO4, L-аскорбиновая кислота, ДМФА.
Фиг.3 представляет собой выходную кривую препаративной ВЭЖХ (высокоэффективной жидкостной хроматографии) реакционной смеси, содержащей соединение [18F]11 (слева) и разделение аналитических пиков (справа). Вверху: канал радиоактивности; внизу: канал УФ при 254 нм.
На Фиг.4 показано биораспределение соединения [18F]11 у мышей-носителей опухоли RIF-1 через 2, 10, 30 и 60 минут. Данные представляют собой среднее значение ± SEM (стандартная ошибка среднего); n составляет 3-6 мышей на момент времени.
На Фиг.5 показан метаболизм in vivo соединения [18F]11, оцененный в плазме посредством радио-ВЭЖХ. Вверху слева: соединение [18F]11 стандарт; вверху справа: через 2 минуты в плазме; внизу слева: через 15 минут в плазме; внизу справа: через 60 минут в плазме.
На Фиг.6 показан метаболизм in vivo соединения [18F]11, оцененный в печени и моче посредством радио-ВЭЖХ. Верхний ряд: экстракты печени; нижний ряд: экстракты мочи; слева направо: через 2, 15 и 60 минут, соответственно.
На Фиг.7 показано биораспределение [125I]-меченого (S)-1-(4-иодбензил)-5-(2-феноксиметил-пирролидин-1-сульфонил)изатина (обозначенного здесь как соединение [125I]14) через 2, 10, 30 и 60 минут. Данные представляют собой среднее значение ± SEM; n составляет 3 мыши на момент времени.
На Фиг.8 показан метаболизм in vitro известных соединений (S)-1-(4-иодбензил)-5-(2-феноксиметил-пирролидин-1-сульфонил)изатина (соединение 14), (S)-1-(4-фторбензил)-5-(2-феноксиметил-пирролидин-1-сульфонил)изатина (соединение 15) и 1-(4-фторбензил)-5-(пирролидин-1-сульфонил)изатина (соединение 16), оцененный в 89-фракциях печени мыши посредством ВЭЖХ. Верхний ряд, слева направо: соединения 14, 15 и 16 в нулевой момент времени; нижний ряд, слева направо: соединения 14,15 и 16 после инкубации в течение 60 минут.
На Фиг.9 показано клеточное накопление соединения [18F]11 в течение 60 минут в (А) клетках RIF-1, обработанных 10 мкг/мл CDDP (цисплатин) или 10 мкг/мл СНХ (хлоргексидин) в течение 24 часов; (В) клетках PEO1/4, обработанных 50 мкг/мл CDDP (цисплатин); (С) клетках LNM35, обработанных 100 мкМ CDDP или 100 мкМ VP-16 (этопозид), отдельно или в комбинации. Анализ каспазы-3 (С, правая панель) проводят, как описано в экспериментальном разделе. * Критерий Стьюдента, р<0,005.
На Фиг.10 показаны саггитальные изображения распределения соединения [18F]11 по всему организму у мышей, которым вводили CDDP. Изображения суммировали за период от 30 до 60 минут после внутривенной инъекции приблизительно 100 мкКи соединения [18F]11.
На Фиг.11 показаны изображения распределения соединения [18F]11 по всему организму у мыши, несущей ксенотрансплантат 38С18, которой вводили 100 мг/кг циклофосфамида. Изображения слева направо представляют собой аксиальное, фронтальное и саггитальное, полученные через 24 часа после внутривенной инъекции приблизительно 100 мкКи соединения [18F]11.
Фиг.12 является схематическим представлением синтеза соединений 35 (верхний ряд) и 36 (нижний ряд), соответственно.
На Фиг.13 показан профиль накопления соединения [18F]11 в клетках RIF-1, обработанных носителем или CDDP. Данные выражены в виде усредненного числа скорректированных с учетом затухания импульсов в минуту на миллиграмм общего клеточного белка. Данные представляют собой среднее значение ± SEM, выполнены в трехкратной повторности.
На Фиг.14 показано накопление соединения [18F]11 в опухоли RIF-1, обработанной носителем (50% ДМСО (диметилсульфоксид)) или CDDP (однократная доза 10 мг/кг). Уровни создаваемой [18F] радиоактивности через 60 минут после инъекции радиоактивной метки анализировали и выражали в виде отношения к уровню радиоактивности в крови. Данные представляют собой среднее значение ± SEM, и n составляет 8 мышей на группу.
На Фиг.15 показано биораспределение соединения [18F]11 в опухолях, обработанных циклофосфамидом (ЦФА, 100 мг/кг) или носителем, где плотности импульсов усреднены для каждой изучаемой области в каждый из 19 моментов времени для получения кривой зависимости радиоактивности от времени (ТАС) для изучаемых областей.
Фиг.16 представляет собой гистограмму нормализованного значения накопления (NUV) через 60 минут после инъекции (NUV60) и площадь под кривой NUV (ППК), подсчитанную в виде интеграла NUV от 0 до 60 минут в опухолях, обработанных циклофосфамидом (ЦФА, 100 мг/кг) или носителем (Носит).
Фиг.17 представляет собой репрезентативные трехмерные реконструкции с помощью OSEM (оптическое управление сопряжением) ПЭТ-изображений соединения [18F]11 у двух мышей, несущих ксенотрансплантат 38С18, которым вводили носитель или циклофосфамид. Кружками указана опухоль.
На Фиг.18 показана ингибиторная активность соединения 11 (обозначенного как «изатин-7») и соединения 14 (обозначенного как «изатин-4») в отношении когнатной мишени каспазы-3 поли(АДФ-рибоза)-полимеразы (ПАРП).
Фиг.19 является вторым схематическим представлением синтеза соединения [18F]11 путем взаимодействия (S)-1-(2-пропинил)-5-(2-(2,4-дифторфеноксиметил)-пирролидин-1-сульфонил)изатина (соединение 25) с [18F]фторэтилазидом (соединение 27).
Фиг.20 является общей реакционной схемой синтеза защищенного алкинового предшественника (S)-1-{[1'-[1-(2-пропинил)]-(1',2'-дигидро-2'-оксоспиро(1,3-диоксан-2,3'-[3Н]индол)-5'-сульфонил}-2-(2,4-дифторфенокси-метил)-пирролидина (соединение 39) и защищенного триазола (S)-1-{[1'-[1-(2-фторэтил)-1Н-[1,2,3]-триазол-4-ил]-(1',2'-дигидро-2'-оксоспиро(1,3-диоксан-2,3'-[3Н]индол)-5'-сульфонил}-2-(2,4-дифторфеноксиметил)-пирролидина (соединение 40).
Фиг.21 является схематическим представлением типичной радиохимической реакции получения соединения [18F]11 из защищенного алкинового предшественника (S)-1-{[1'-[1-(2-пропинил)]-(1',2'-дигидро-2'-оксоспиро(1,3-диоксан-2,3'-[3Н]индол)-5'-сульфонил}-2-(2,4-дифторфенокси-метил)-пирролидина (соединение 39).
На Фиг.22 показаны результаты анализа ВЭЖХ меченой смеси перед снятием защиты. Красный: канал радиоактивности; синий: канал УФ при 254 нм. Остаточный [18F]-фторэтилазид: 1,3 минуты, меченый ацеталь изатина: 6,8 минуты.
На Фиг.23 показаны результаты перпаративной ВЭЖХ соединения [18F]11. Вверху: канал радиоактивности. Пик на 11,45 минуты соответствует соединению [18F]11. Внизу: канал УФ при 254 нм. Сигнал на 11,51 минуты соответствует стабильной примеси.
На Фиг.24 показаны результаты аналитической ВЭЖХ приготовленного соединения [18F]11. Вверху: канал УФ при 254 нм, внизу: сигнал радиоактивности, показывающий соединение [18F]11 при 3,3 минуты.
Фиг.25 является схематическим представлением синтеза слабого изатинового ингибитора каспазы-3 N-[1-(2-фторэтил)-1Н-[1,2,3]-триазол-4-ил]изатина (соединение 42) из N-(2-пропинил)изатина (соединение 41).
На Фиг.26 показано связывание соединений [18F]11 и [18F]42 в раковых клетках, подвергнутых индуцированному обработкой апоптозу. (Левая панель): химическая структура и связывание соединения [18F]11 в клетках лимфомы 38С13, обработанных 4-гидропероксициклофосфамидом 4-НС (4-НС; 1 мкг/мл; 24 часа) для индукции апоптоза. Для всех обработанных и контрольных образцов данные радиоактивности выражали в виде числа скорректированных с учетом затухания импульсов на миллиграмм общего клеточного белка. (Правая панель): действие 4-НС на связывание зонда с низкой специфичностью, соединения [18F]42, в клетках лимфомы 38С13. Данные представляют собой среднее значение ± SEM.
ПРИМЕРЫ
Пример 1 - Синтез соединений
Библиотеку целевых соединений создавали с использованием (S)-1-(4-фторбензил)-5-(2-феноксиметил-пирролидин-1-сульфонил)изатина в качестве соединения-прототипа. Модификации проводили по левой боковой эфирной группировке и по положению N-1. Группы фтора включали в левую боковую фенилэфирную группу и исследовали устойчивость гетероциклов и алкинов в этом положении. Также исследовали устойчивость к 1,2,3-триазольным группам в положении N-1. Целевые соединения синтезировали, как показано на Фиг.1, путем конденсации функционализированных пирролидинов с 5-хлорсульфонилизатином и последующим алкилированием атома азота изатина с использованием карбоната калия/ДМФА. Все необходимые исходные вещества имеются в продаже, или их получают, как описано у Lee, D. et al. [8], Chu, W. et al. [9] и Kopka, K. et al. [13].
Взаимодействие имеющихся в продаже фенолов, а также 4-гидрокситетрагидропирана, с тозилатом 17 давало пирролидины 19a-f с хорошим выходом. Проводили сочетание 4(3Н)-пиримидона с (S)-1-(трет-бутоксикарбонил)-2-пирролидинметанолом (соединение 18) с использованием модификации метода, изложенного Wipf et al. [15], с получением пирролидина 19д с выходом 69%. O-алкилирование соединения 18 пропаргилбромидом давало соответствующий эфир 19h с выходом 67%. Снятие защиты с ВОС-защищенных пирролидинов 19a-h с помощью трифторуксусной кислоты с последующей конъюгацией с 5-хлорсульфонилизатином давало сульфонамиды 20a-h с умеренными или хорошими выходами. Последующая обработка соединений 20b-h 4-фторбензилбромидом в щелочных условиях давала новые соединения с 1 по 5 и промежуточные соединения 22 и 23, тогда как обработка соединений 20а и 20d пропаргилбромидом давала промежуточные алкины 24 и 25, соответственно. Новые триазолы 10-12 получали путем катализируемого медью циклоприсоединения 2-фторэтилазида с соответствующими алкиновыми предшественниками 23, 24 и 25. Довольно неожиданно, что остов молекулы изатина разрушался при нагревании при 90°С в присутствии сульфата меди и аскорбиновой кислоты, приводя к низким выходам новых триазолов 10-12. Выход отчасти изменяли посредством увеличения концентрации сульфата меди от 5% до 50% относительно алкинового предшественника, проводя взаимодействие при температуре окружающей среды и уменьшая время взаимодействия до 1 часа, что давало триазолы 10-12 с выходами 48-57%.
Химия: Реагенты и растворители закупали у Sigma-Aldrich (Gillingham, United Kingdom) и использовали без дополнительной очистки. Гидроксид калия и карбонат калия хранили в вакуумном эксикаторе над пентаоксидом фосфора. Все взаимодействия проводили в атмосфере аргона, если не оговорено особо. Петролейный эфир относится к фракции, которую перегоняют при температуре между 40°С и 60°С. Автоматизированную флэш-хроматографию проводили на хроматографе CombiFlash Companion (Companion Presearch Ltd.) при использовании 4 г или 12 г картриджей RediSep с диоксидом кремния с нормальной фазой (скорость потока 12 мл/мин или 26 мл/мин). Неавтоматизированную флэш-хроматографию проводили при использовании нейтрального диоксида кремния Davisil (60 Å, 60-200 микрон, Fisher Scientific, Loughborough, UK), смеси растворителей составлены по объему. Спектры 1Н ЯМР получали на ЯМР-спектрометре Bruker Avance 600 МГц, и спектры соотнесены с остаточным сигналом растворителя. Константы взаимодействия (J) даны в Герцах (Гц). Данные ГХ-МС (газовая хроматография/масс-спектрометрия) получали при ионизации электронов с использованием системы Agilent 6890N. Масс-спектры получали в режиме положительной ионизации с электрораспылением на масс-спектрометре Waters Micromass LCT Premier. Точки плавления определяли в капиллярных трубках на приборе для определения точки плавления Stuart Scientific SMP1, и они являются нескорректированными. Смеси растворителей для тонкослойной хроматографии (ТСХ) составлены по объему, и образцы разделяли на пластинах с нейтральным диоксидом кремния, нанесенным на алюминиевую подложку (толщина 0,2 мм) (Fluka, Seeize, Germany). Анализ чистоты соединений 1-5, 10-16, 20b-h и 22-25 производили посредством аналитической ВЭЖХ; соединения 19b-h анализировали посредством ГХ-МС. Чистота всех соединений составляла более 95%. [18F]Фтор получали с помощью циклотрона (GE РЕТгасе) при использовании ядерной реакции 18О(p,n)18F при облучении протонами с энергией 16,4 МэВ мишени, обогащенной [18O]H2O.
Методы ВЭЖХ:
а) Анализ чистоты нерадиоактивных соединений проводили на системе Agilent серии 1100 с программным обеспечением Laura 3 (Lablogic, Sheffield, UK) с помощью присоединенной ВЭЖХ колонки Phenomenex Luna 50×4,6 мм (3 мкм) и 0,1 М формиата аммония и смеси метанол/ацетонитрил (1,8:1 об./об.) в качестве подвижной фазы, градиента (50% органической фазы за 1 мин; 50→90% органической фазы за 14 мин; 90% органической фазы за 4 мин; 90→50% органической фазы за 4 мин), скорости потока 1 мл/мин и длины волны 254 нм.
б) Препаративную радио-ВЭЖХ проводили на хроматографе Beckman System Gold, снабженном диодным детектором Bioscan Flowcount FC-3400 PIN (Lablogic) и линейным УФ-200 детектором (длина волны 254 нм). ВЭЖХ колонка Phenomenex Onyx C18 100×10 мм и вода и смесь метанол/ацетонитрил (1,8:1 об./об.) в качестве подвижной фазы, градиент 45→90% органической фазы за 20 мин и скорость потока 3 мл/мин.
в) Аналитическую радио-ВЭЖХ проводили, как указано выше, но при использовании детектора гамма-излучения Bioscan Flowcount FC3200 с иодидом натрия в качестве сцинтиллятора и фотоэлектронным умножителем (ФЭУ) (Lablogic) и колонки Phenomenex Luna 50×4,6 мм (3 мкм) с водой и смесью метанол/ацетонитрил (1,8:1 об./об.) в качестве подвижной фазы, градиентом 60→90% органической фазы за 20 мин, скоростью потока 1 мл/мин.
Получение известных соединений
Следующие соединения синтезировали согласно известным в литературе методикам, и спектральные данные 1Н ЯМР соответствовали опубликованным значениям: (S)-1-бензил-5-(2-феноксиметил-пирролидин-1-сульфонил)изатина (соединение 13), (S)-1-(4-иодбензил)-5-(2-феноксиметил-пирролидин-1-сульфонил)изатина (соединение 14) и (S)-1-(4-фторбензил)-5-(2-феноксиметил-пирролидин-1-сульфонил)изатина (соединение 15).
1-(4-Фторбензил)-5-(пирролидин-1-сульфонил)изатин (соединение 16). К охлажденному на льду, перемешиваемому раствору 5-пирролидин-1-сульфонилизатина [8] (0,14 г; 0,5 ммоль) в безводном ДМФА (8 мл) добавляли гидрид натрия (40 мг; 1 ммоль). Через 30 минут добавляли 4-фторбензилбромид (0,38 г; 2 ммоль) и смесь оставляли нагреваться до комнатной температуры. Через 19 часов оранжевый раствор выливали на 10% водн. NH4Cl (25 мл) и экстрагировали ДХМ (3×15 мл). После концентрирования под вакуумом остаток переносили в диэтиловый эфир (10 мл) и промывали водой (3×10 мл) и сушили над Na2SO4. Хроматография (диэтиловый эфир/гексаны) дала указанное в заголовке соединение в виде оранжевой смолы (83 мг; 43%). HRMS (масс-спектрометрия высокого разрешения) (ESI)=389,0988 (М+Н)+. Рассчитано для C19H18FN2O4S 389,0971. 1H ЯМР (600 МГц, CDCl3) δ 8.05 (d, J=1,5 Гц, 1Н), 7.99 (dd, J=8,4 Гц, J=1,5 Гц, 1Н), 7.35-7.32 (m, 2H), 7.09-7.06 (m, 2H), 6.91 (d, J=8,4 Гц, 1H), 4.94 (s, 2H), 3.25-3.23 (4Н, m), 1.84-1.79 (4Н, m). TCX (УФ254) Rf=0,63 (этилацетат/гексаны 4:1). ВЭЖХ tR=6,83 мин.
Получение новых соединений и их промежуточных соединений
(S)-трет-Бутил-2-(4-фторфеноксиметил)пирролидин-1-карбоксилат (соединение 19b). К перемешиваемому раствору 4-фторфенола (0,27 г; 2,4 ммоль) в безводном ДМФА (10 мл) добавляли гидрид натрия (60% масс./масс. в минеральном масле) (0,11 г; 2,8 ммоль). Затем через 30 минут добавляли ((S)-1-(трет-бутоксикарбонил)пирролидин-2-ил)толуол-4-сульфонат [8] (соединение 15) (0,71 г; 2,0 ммоль) в безводном ДМФА (5 мл) и смесь нагревали до 80°С в течение 17 часов. Реакционную смесь оставляли охлаждаться до комнатной температуры и вливали на 1 M NaOH (25 мл) и экстрагировали ДХМ (3×15 мл). Объединенные органические фракции концентрировали под вакуумом и добавляли диэтиловый эфир (20 мл), затем промывали 1 М NaOH (1×20 мл), водой (1×20 мл), затем рассолом (1×20 мл) и сушили над Na2SO4. Хроматография (гексаны/этилацетат) дала продукт в виде бесцветного масла (0,36 г; 61%). HRMS(ESI)=296,1654 (М+Н)+. Рассчитано для C16H23FNO3 296,1656. 1H ЯМР (600 МГц, CDCl3) δ 6.97-6.93 (m, 2H), 6.88-6.84 (m, 2H), 4.18-4.03 (m, 2H), 3.94-3.73 (m, 1H), 3.46-3.32 (m, 2H), 2.07-1.81 (m, 4Н), 1.47 (s, 9H). TCX (УФ254) Rf=0,51 (гексаны/этилацетат 2:1).
(S)-трет-Бутил-2-(3-фторфеноксиметил)пирролидин-1-карбоксилат (соединение 19с). Соединение 19с получали согласно методике получения соединения 19b, за исключением использования 3-фторфенола. Хроматография (гексаны/этилацетат) дала указанное в заголовке соединение в виде бесцветного масла (0,32 г; 54%). HRMS(ESI)=296,1657 (M+H)+. Рассчитано для C16H23HNO3 296,1656. 1Н ЯМР (600 МГц, CDCl3) δ 7.20-7.15 (m, 1H), 6.73-6.69 (m, 1H), 6.65-6.59 (m, 2H), 4.18-4.03 (m, 2H), 3.97-3.74 (m, 1H), 3.48-3.29 (m, 2H), 2.05-1.79 (m, 4Н), 1.48 (s, 9H). TCX (УФ254) Rf=0,40 (гексаны/этилацетат 3:1).
(S)-трет-Бутил-2-(2,4-дифторфеноксиметил)пирролидин-1-карбоксилат (соединение 19d). Соединение 19d получали согласно методике получения соединения 19b, за исключением использования 2,4-дифторфенола. Хроматография (гексаны/этилацетат) дала указанное в заголовке соединение в виде бесцветного масла (1,92 г; 58%). HRMS(ESI)=314,1560 (M+H)+. Рассчитано для C16H22F2NO3 314,1562. 1H ЯМР (600 МГц, CDCl3) δ 7.08-6.76 (m, 3H), 4.21-3.87 (m, 3H), 3.47-3.32 (m, 2H), 2.16-1.85 (m, 4H), 1.48 (s, 9H). ТСХ (УФ254) Rf=0,51 (гексаны/этилацетат2:1).
(S)-трет-Бутил-2-(3,5-дифторфеноксиметил)пирролидин-1-карбоксилат (соединение 19е). Соединение 19е получали согласно методике получения соединения 19b, за исключением использования 3,5-дифторфенола. Хроматография (гексаны/этилацетат) дала указанное в заголовке соединение в виде бесцветного масла (0,31 г; 53%). HRMS(ESI)=314,1563 (M+H)+. Рассчитано для C16H22F2NO3 314,1562. 1H ЯМР (600 МГц, CDCl3) δ 6.44-6.29 (m, 3H), 4.11-3.98 (m, 2H), 3.91-3.72 (m, 1H), 3.42-3.24 (m, 2H), 1.99-1.79 (m, 4H), 1.42 (s, 9H). ТСХ (УФ254) Rf=0,56 (гексаны/этилацетат 2:1).
(S)-трет-Бутил-2-(4-тетрагидропиранилоксиметил)пирролидин-1-карбоксилат (соединение 19f). Соединение 19f получали согласно методике получения соединения 19b, за исключением использования 4-гидрокситетрагидропирана. Хроматография (этилацетат) дала указанное в заголовке соединение в виде бесцветного масла (0,14 г; 25%). HRMS(ESI)=286,2012 (М+Н)+. Рассчитано для C15H28NO4 286,2013. 1H ЯМР (600 МГц, CDCl3) δ 3.97-3.83 (m, 2H), 3.64-3.23 (m, 9H), 1.97-1.72 (m, 5H), 1.60-1.52 (m, 2H), 1.46 (s, 9H). ТСХ (b) Rf=0,62 (этилацетат).
(S)-трет-Бутил-2-(пиримидин-4-илоксиметил)пирролидин-1-карбоксилат (соединение 19g). К перемешиваемому раствору N-трет-бутоксикарбонил-L-пролинола 18 (0,81 г; 4 ммоль) в безводном ДХМ (10 мл) добавляли трифенилфосфин (5,24 г; 20 ммоль) с последующим добавлением 4(3Н)-пиримидона (0,77 г; 8 ммоль). Раствор охлаждали на ледяной бане и по каплям добавляли DIAD (3,24 г; 16 ммоль) в течение 10 минут. Через 48 часов ГХ-МС показала полное превращение соединения 18, и реакционную смесь вливали на воду (30 мл), органическую фракцию собирали, а водную фазу промывали дополнительным количеством ДХМ (2×20 мл). Объединенные органические фракции промывали 1 M NaOH (2×15 мл), затем рассолом (1×15 мл) и сушили над Na2SO4. Удаление большей части растворителя дало оранжевую смолу, добавление смеси гексаны/диэтиловый эфир (1:1) привело к образованию осадка оксида трифенилфосфина, который удаляли посредством фильтрации. Хроматография (этилацетат) дала требуемый продукт в виде бесцветного масла (0,77 г; 69%). HRMS(ESI)=280,1655 (M+H)+. Рассчитано для C14H22N3O3 280,1656. 1Н ЯМР (600 МГц, CDCl3) δ 8.71 (s, 1H), 8.39 (d, J=6 Гц, 1H), 6,69 (d, J=6 Гц, 1H), 4.45-3.91 (m, 3H), 3.40-3.33 (m, 2H), 2.00-1.82 (m, 4H), 1.42 (s, 9H). TCX (УФ254) Rf=0,49 (этилацетат).
(S)-трет-Бутил-2-(2-пропинилоксиметил)пирролидин-1-карбоксилат (соединение 19h). К перемешиваемому раствору соединения 18 (0,40 г; 2 ммоль) в безводном ДМФА (10 мл) добавляли гидроксид калия (0,56 г; 10 ммоль) с последующим добавлением по каплям пропаргилбромида (80 масс.% в толуоле) (0,48 г; 4 ммоль) в течение 5 минут. Через 18 часов реакционную смесь вливали на воду (30 мл) и промывали ДХМ (3×15 мл). Объединенные органические фракции концентрировали под вакуумом и оставшуюся жидкость переносили в диэтиловый эфир (15 мл) и промывали водой (2×10 мл), затем рассолом (1×10 мл) и сушили над Na2SO4. Хроматография (гексаны/этилацетат) дала требуемый продукт в виде бесцветного масла (0,32 г; 67%). HRMS(ESI)=240,1597 (М+Н)+. Рассчитано для C13H22NO3 240,1594. 1H ЯМР (600 МГц, CDCl3) δ 4.13 (s, 2H), 3.96-3.88 (m, 1H), 3.64 (dd, J=9 Гц, J=3,6 Гц, 1H), 3.49-3.22 (m, 3H), 2.40 (s, 1H), 1.94-1.78 (m, 4H), 1.46 (s, 9H). TCX (b) Rf=0,67 (гексаны/этилацетат 1:1).
(S)-5-(2-(4-Фторфеноксиметил)-пирролидин-1-сульфонил)изатин (соединение 20b). К перемешиваемому раствору соединения 19b (0,15 г; 0,5 ммоль) в безводном ДХМ (4 мл), охлажденном на ледяной бане, добавляли ТФУ (0,6 мл; 10 ммоль). Через 1 час большую часть растворителя удаляли под вакуумом и оставшийся остаток переносили в безводный ДХМ (8 мл) и охлаждали на ледяной бане. Затем добавляли безводный триэтиламин (1,5 мл) с последующим добавлением 5-хлорсульфонилизатина [8] (0,16 г; 0,65 ммоль) в безводном ТГФ (4 мл) и этот раствор затем перемешивали. Через 19 часов большую часть растворителя удаляли под вакуумом и повторно растворяли в ДХМ (10 мл), промывали водой (2×10 мл), затем рассолом (1×10 мл) и сушили над Na2SO4. Хроматография (гексаны/этилацетат) дала требуемый продукт в виде оранжевого твердого вещества (104 мг; 51%). Т.пл.: 205-207°С. HRMS(ESI)=405,0941 (М+Н)+. Рассчитано для C19H18FN2O5S 405,0920. 1H ЯМР (600 МГц, CDCl3) δ 8.10 (s, 1H), 8.08 (dd, J=8,4 Гц, J=1,8 Гц, 1Н), 8.00 (уширенный, 1H), 7.00 (d, J=7,8 Гц, 1H), 6.99-6.95 (m, 2H), 6.83-6.81 (m, 2H), 4.17 (dd, J=9,6 Гц, J=3,6 Гц, 1H), 3.98-3.95 (m, 1H), 3.91 (dd, J=9 Гц, J=7,8 Гц, 1Н), 3.54-3.50 (m, 1H), 3.24-3.19 (m, 1H), 2.10-1.99 (m, 2H), 1.87-1.77 (m, 2H). ТСХ (УФ254) Rf=0,27 (этилацетат/гексаны 2:1). ВЭЖХ tR=7,83 мин.
(S)-5-(2-(3-Фторфеноксиметил)-пирролидин-1-сульфонил)изатин (соединение 20с). Соединение 20с получали согласно методике получения соединения 20b, за исключением того, что использовали соединение 19с с получением оранжевого твердого вещества (93 мг; 46%). Т. пл.: 201-203°С. HRMS(ESI)=405,0933 (М+Н)+. Рассчитано для C19H18FN2O5S 405,0920. 1H ЯМР (600 МГц, CDCl3) δ 8.14 (уширенный, 1H), 8.09-8.06 (m, 2H), 7.21 (q, J=7,2 Гц, 1H), 7.03 (d, J=7,8 Гц, 1H), 6.67-6.62 (m, 2H), 6.55 (dt, J=10,8 Гц, J=2,4 Гц, 1H), 4.17 (dd, J=9 Гц, J=3 Гц, 1H), 4.01-3.97 (m, 1H), 3.94 (dd, J=9 Гц, J=7,2 Гц, 1H), 3.54-3.50 (m, 1H), 3.27-3.25 (m, 1H), 2.08-1.96 (m, 2H), 1.88-1.77 (m, 2H). ТСХ (УФ254) Rf=0,36 (этилацетат/гексаны 2:1). ВЭЖХ tR=8,27 мин.
(S)-5-(2-(2,4-Дифторфеноксиметил)-пирролидин-1-сульфонил)изатин (соединение 20d). Соединение 20d получали согласно методике получения соединения 20b, за исключением того, что использовали соединение 19d с получением оранжевого твердого вещества (0,86 г; 34%). Т. пл.: 185-187°С. HRMS(ESI)=423,0834 (М+Н)+. Рассчитано для C19H17F2N2O5S 423,0826. 1H ЯМР (600 МГц, CDCl3) δ 8.08-8.06 (m, 2H), 7.97 (уширенный, 1H), 7.03 (d, J=9 Гц, 1H), 6.97-6.92 (m, 1H), 6.87-6.77 (m, 2H), 4.21 (dd, J=8,4 Гц, J=2,4 Гц, 1H), 4.03-3.97 (m, 2H), 3.56-3.52 (m, 1H), 3.23-3.17 (m, 1H), 2.11-2.01 (m, 2H), 1.88-1.75 (m, 2H). ТСХ (УФ254) Rf=0,46 (этилацетат/гексаны 2:1). ВЭЖХ tR=8,12 мин.
(S)-5-(2-(3,5-Дифторфеноксиметил)-пирролидин-1-сульфонил)изатин (соединение 20е). Соединение 20е получали согласно методике получения соединения 20b, за исключением того, что использовали соединение 19е с получением оранжевого твердого вещества (112 мг; 53%). Т. пл.: 196-198°С. HRMS(ESI)=423,0834 (М+Н)+. Рассчитано для C19H17F2N2O5S 423,0826. 1H ЯМР (600 МГц, CDCl3) δ 8.10-8.06 (m, 2H), 8.04 (уширенный, 1H), 7.05 (d, J=8,4 Гц), 6.45-6.38 (m, 3Н), 4.18 (dd, J=8,4 Гц, J=2,4 Гц, 1H), 3.99-3.92 (m, 2H), 3.56-3.49 (m, 1H), 3.24-3.17 (m, 1H), 2.04-1.92 (m, 2H), 1.86-1.77 (m, 2H). ТСХ (УФ254) Rf=0,36 (этилацетат/гексаны 2:1). ВЭЖХ tR=9,18 мин.
(S)-5-(2-(Тетрагидро-2Н-пиран-4-илоксиметил)-пирролидин-1-сульфонил)изатин (соединение 20f). Соединение 20f получали согласно методике получения соединения 20b, за исключением того, что использовали соединение 19f с получением оранжевой смолы (63 мг; 32%). HRMS(ESI)=395,1282 (М+Н)+. Рассчитано для C18H23N2O6S 395,1277. 1H ЯМР (600 МГц, CDCl3) δ 8.17 (уширенный, 1Н), 8.10-8.08 (m, 2H), 7.07 (d, J=8,4 Гц, 1H), 3.94-3.90 (m, 2H), 3.77-3.73 (m, 1Н), 3.70 (dd, J=9,6 Гц, J=3 Гц, 1Н), 3.56-3.51 (септет, J=4,2 Гц, 1Н), 3.48-3.44 (m, 3Н), 3.13-3.11 (m, 1Н), 2.05-1.87 (m, 4H), 1.72-1.64 (m, 2H), 1.60-1.52 (m, 2H). ТСХ (УФ254) Rf=0,26 (этилацетат/гексаны 4:1). ВЭЖХ tR=2,65 мин.
(S)-5-(2-(Пиримидин-4-илоксиметил)-пирролидин-1-сульфонил)-изатин (соединение 20g). Соединение 20g получали согласно методике получения соединения 20b, за исключением использования соединения 19g. Хроматография (этилацетат) дала оранжевую смолу (51 мг; 27%). HRMS(ESI)=389,025 (М+Н)+. Рассчитано для C17H17N4O5S 389,020. 1H ЯМР (600 МГц, CDCl3) δ 8.79 (s, 1Н), 8.45 (d, J=5,4 Гц, 1Н), 8.12 (d, J=1,8 Гц, 1Н), 8.09 (dd, J=8,4 Гц, J=1,8 Гц, 1Н), 7.95 (уширенный, 1Н), 7.03 (d, J=8,4 Гц, 1Н), 6.72 (d, J=5,4 Гц, 1Н), 4.57 (dd, J=10,8 Гц, J=4,8 Гц, 1Н), 4.39 (dd, J=10,8 Гц, J=7,2 Гц, 1Н), 4.08-4.04 (m, 1Н), 3.53-3.46 (m, 1Н), 3.27-3.22 (m, 1Н), 2.01-1.93 (m, 2H), 1.84-1.74 (m, 2H). ТСХ (УФ254) Rf=0,49 (этила цетат/метанол 9:1). ВЭЖХ tR=1,90 мин.
(S)-5-(2-(2-Пропинилоксиметил)-пирролидин-1-сульфонил)изатин (соединение 20h). Соединение 20h получали согласно методике получения соединения 20b, за исключением того, что использовали соединение 19h с получением оранжевой смолы (92 мг; 53%). HRMS(ESI)=349,0867 (M+H)+. Рассчитано для C16H17N2O5S 349,067. 1H ЯМР (600 МГц, CDCl3) δ 8.10-8.07 (m, 2H), 7.84 (уширенный, 1Н), 7.04 (d, J=8,4 Гц, 1Н), 4.16 (s, 2H), 3.83-3.79 (m, 1Н), 3.72 (dd, J=9,6 Гц, J=3,6 Гц, 1Н), 3.54 (dd, J=9,6 Гц, J=3,6 Гц, 1Н), 3.45-3.43 (m, 1Н), 3.19-3.15 (m, 1Н), 2.46 (t, J=2,4 Гц, 1Н), 1.96-1.89 (m, 2H), 1.74-1.67 (m, 2H). ТСХ (УФ254) Rf=0,28 (этилацетат/гексаны 2:1). ВЭЖХ tR=2,93 мин.
(S)-1-(4-Фторбензил)-5-(2-(4-фторфеноксиметил)-пирролидин-1-сульфонил)изатин (соединение 22). К перемешиваемому раствору соединения 20b (40 мг; 0,1 ммоль) в безводном ДМФА (3 мл) добавляли карбонат калия (21 мг; 0,15 ммоль) с последующим добавлением 4-фторбензилбромида (76 мг; 0,4 ммоль). Через 2 часа ТСХ показала полное превращение соединения 20b, и раствор вливали на 10% водн. NH4Cl (10 мл) и экстрагировали ДХМ (3×10 мл). Объединенные органические фракции концентрировали под вакуумом и переносили в диэтиловый эфир (10 мл), промывали водой (2×10 мл), затем рассолом (1×10 мл) и сушили над Na2SO4. Хроматография (гексаны/этилацетат) дала указанное в заголовке соединение в виде оранжевой смолы (34 мг; 66%). HRMS(ESI)=513,1306 (М+Н)+. Рассчитано для C26H23F2N2O5S 513,1296. 1H ЯМР (600 МГц, CDCl3) δ 8.04 (d, J=1,8 Гц, 1Н), 7.97 (dd, J=8,4 Гц, J=1,8 Гц, 1Н), 7.33-7.30 (m, 2H), 7.09-7.05 (m, 2H), 6.96-6.92 (m, 2H), 6.86 (d, J=8,4 Гц, 1Н), 6.81-6.77 (m, 2H), 4.92 (d, J=15,6, 1Н), 4.91 (d, J=15,6, 1Н), 4.14 (dd, J=9,6 Гц, J=3,6 Гц, 1Н), 3.95-3.92 (m, 1Н), 3.88 (dd, J=9,6 Гц, J=7,2 Гц, 1Н), 3.51-3.47 (m, 1Н), 3.20-3.15 (m, 1Н), 2.06-1.93 (m, 2H), 1.83-1.73 (m, 2H). TCX (УФ254) Rf=0,61 (этилацетат/гексаны 2:1). ВЭЖХ tR=12,25 мин.
(S)-1-(4-Фторбензил)-5-(2-(3-фторфеноксиметил)-пирролидин-1-сульфонил)изатин (соединение 1). Соединение 1 получали согласно методике получения соединения 22, за исключением того, что использовали соединение 20с с получением оранжевой смолы (31 мг; 61%). HRMS (ESI)=513,1298 (М+Н)+. Рассчитано для C26H23F2N2O5S 513,1296. 1H ЯМР (600 МГц, CDCl3) δ 8.02 (d, J=1,8 Гц, 1Н), 7.98 (dd,J=8,4 Гц, J=1,8 Гц, 1Н), 7.33-7.29 (m, 2H), 7.18 (m, 1Н), 7.09-7.04 (m, 2H), 6.86 (d, J=8,4 Гц, 1Н), 6.66-6.62 (m, 2H), 6.53 (dt, J=10,8, J=2,4 Гц, 1Н), 4.88 (s, 2H), 4.15 (dd, J=9,6 Гц, J=3,6 Гц, 1Н), 3.98-3.88 (m, 2H), 3.51-3.47 (m, 1Н), 3.23-3.18 (m, 1Н), 2.08-1.97 (m, 2H), 1.84-1.72 (m, 2H). TCX (УФ254) Rf=0,64 (этилацетат/гексаны 2:1). ВЭЖХ tR=12,57 мин.
(S)-1-(4-Фторбензил)-5-(2-(2,4-дифторфеноксиметил)-пирролидин-1-сульфонил)изатин (соединение 2). Соединение 2 получали согласно методике получения соединения 22, за исключением того, что использовали соединение 20d с получением оранжевой смолы (32 мг; 60%). HRMS (ESI)=531,1204 (М+Н)+. Рассчитано для C26H22F3N2O5S 531,1202. 1H ЯМР (600 МГц, CDCl3) δ 8.03 (d, J=1,8 Гц, 1Н), 7.98 (dd, J=8,4 Гц, J=1,8 Гц, 1Н), 7.33-7.30 (m, 2H), 7.09-7.06 (m, 2H), 6.95-6.91 (m, 1Н), 6.88 (d, J=8,4 Гц, 1Н), 6.81-6.76 (m, 2H), 4.93 (d, J=16,2 Гц, 1Н), 4.92 (d, J=16,2 Гц, 1Н), 4.18 (dd, J=9 Гц, J=3 Гц, 1Н), 4.00-3.95 (m, 2H), 3.51-3.49 (m, 1Н), 3.21-3.17 (m, 1Н), 2.09-1.98 (m, 2H), 1.85-1.74 (m, 2H). TCX (УФ254) Rf=0,67 (этилацетат/гексаны 2:1). ВЭЖХ tR=12,50 мин.
(S)-1-(4-Фторбензил)-5-(2-(3,5-дифторфеноксиметил)-пирролидин-1-сульфонил)изатин (соединение 3). Соединение 3 получали согласно методике получения соединения 22, за исключением того, что использовали соединение 20е с получением оранжевой смолы (31 мг; 58%). HRMS (ESI)=531,1213 (М+Н)+. Рассчитано для C26H22F3N2O5S 531,1202. 1H ЯМР (600 МГц, CDCl3) δ 8.04 (d, J=1,8 Гц, 1Н), 7.99 (dd, J=8,4 Гц, J=1,8 Гц, 1Н), 7.34-7.29 (m, 2H), 7.09-7.06 (m, 2H), 6.90 (d, J=8,4 Гц, 1Н), 6.44-6.38 (m, 3H), 4.93 (s, 2H), 4.18-4.15 (m, 1Н), 3.94-3.89 (m, 2H), 3.51-3.48 (m, 1Н), 3.19-3.15 (m, 1Н), 2.03-1.93 (m, 2H), 1.83-1.73 (m, 2H). ТСХ (УФ254) Rf=0,64 (этилацетат/гексаны 2:1). ВЭЖХ tR=13,00 мин.
(S)-1-(4-Фторбензил)-5-(2-(тетрагидро-2М-пиран-4-илоксиметил)-пирролидин-1-сульфонил)изатин (соединение 4). Соединение 4 получали согласно методике получения соединения 22, за исключением того, что использовали соединение 20f с получением оранжевой смолы (26 мг; 52%). HRMS(ESI)=503,1646 (М+Н)+. Рассчитано для C25H28FN2O6S 503,1652. 1H ЯМР (600 МГц, CDCl3) δ 8.06 (d, J=1,8 Гц, 1Н), 8.01 (dd, J=8,4 Гц, J=1,8 Гц, 1Н), 7.34-7.31 (m, 2H), 7.10-7.06 (m, 2H), 6.91 (d, J=8,4 Гц, 1Н), 4.87 (s, 2H), 3.93-3.88 (m, 2H), 3.73-3.70 (m, 1Н), 3.67 (dd, J=9 Гц, J=3 Гц, 1Н), 3.55-3.49 (m, 1Н), 3.47-3.41 (m, 4H), 3.10-3.05 (m, 1Н), 1.97-1.85 (m, 4H), 1.70-1.63 (m, 2H), 1.59-1.54 (m, 2H). ТСХ (УФ254) Rf=0,55 (этилацетат/гексаны 4:1). ВЭЖХ tR=8,58 мин.
(S)-1-(4-Фторбензил)-5-(2-(пиримидин-4-илоксиметил)-пирролидин-1-сульфонил)изатин (соединение 5). Соединение 5 получали согласно методике получения соединения 22, за исключением того, что использовали соединение 20g с получением оранжевой смолы (12 мг; 24%). HRMS (ESI)=497,1287 (М+Н)+. Рассчитано для C24H22FN4O5S 497,1295. 1H ЯМР (600 МГц, CDCl3) δ 8.77 (s, 1Н), 8.43 (d, J=5,4 Гц, 1Н), 8.08 (d, J=1,8 Гц, 1Н), 8.01 (dd, J=8,4 Гц, J=1,8 Гц, 1Н), 7.34-7.32 (m, 2H), 7.09-7.06 (m, 2H), 6.89 (d, J=8,4 Гц, 1Н), 6.70 (d, J=5,4 Гц, 1Н), 4.90 (s, 2H), 4.55 (dd, J=10,8 Гц, J=4,2 Гц, 1Н), 4.37 (dd, J=10,8 Гц, J=7,8 Гц, 1Н), 4.05-4.01 (m, 1Н), 3.50-3.46 (m, 1Н), 3.22-3.19 (m, 1Н), 1.98-1.87 (m, 2H), 1.81-1.72 (m, 2H). ТСХ (УФ254) Rf=0,32 (этилацетат 2:1). ВЭЖХ tR=7,45 мин.
(S)-1-(4-Фторбензил)-5-(2-(2-пропинилоксиметил)-пирролидин-1-сульфонил)изатин (соединение 23). Соединение 23 получали согласно методике получения соединения 22, за исключением того, что использовали соединение 20h с получением оранжевой смолы. Перекристаллизация из смеси этилацетат/гексаны дала требуемый продукт в виде оранжевых игольчатых кристаллов (93 мг; 58%). HRMS (ESI)=457,1236 (М+Н)+. Рассчитано для C23H22FN2O5S 457,1233. 1H ЯМР (600 МГц, CDCl3) δ 8.07 (d, J=1,8 Гц, 1Н), 8.01 (dd, J=8,4 Гц, J=1,8 Гц, 1Н), 7.43-7.31 (m, 2H), 7.09-7.04 (m, 2H), 6.90 (d, J=8,4 Гц, 1Н), 4.91 (s, 2H), 4.13 (d, J=2,4 Гц, 2H), 3.81-3.77 (m, 1Н), 3.69 (dd, J=9,6 Гц, J=4,2 Гц, 1Н), 3.51 (dd, J=9 Гц, J=7,2 Гц, 1Н), 3.43-3.40 (m, 1Н), 3.16-3.12 (m, 1Н), 2.43 (t, J=2,4 Гц, 1Н), 1.94-1.87 (m, 2H), 1.75-1.66 (m, 2H). TCX (УФ254) Rf=0,62 (этилацетат/гексаны 2:1). ВЭЖХ tR=8,80 мин.
(S)-1-(2-Пропинил)-5-(2-феноксиметил-пирролидин-1-сульфонил)-изатин (соединение 24). К раствору (5)-5-(2-феноксиметил-пирролидин-1-сульфонил)изатина 20а (0,39 г; 1 ммоль) в безводном ДМФА (10 мл) добавляли карбонат калия (0,21 г; 1,5 ммоль) с последующим добавлением пропаргилбромида (80 масс.% в толуоле) (0,14 г; 1,2 ммоль). Через 2 часа TCX показала полное превращение соединения 20а, и раствор вливали на 10% водн. NH4Cl (20 мл) и промывали ДХМ (3×10 мл). Объединенные органические фракции затем концентрировали под вакуумом и остаток переносили в диэтиловый эфир (10 мл) и промывали водой (2×10 мл), затем рассолом (1×10 мл) и сушили над Na2SO4. Хроматография (гексаны/этилацетат) дала продукт в виде оранжевой смолы. Перекристаллизация из смеси этилацетат/гексаны дала продукт в виде оранжевого твердого вещества (0,28 г; 66%). Т. пл.: 115-117°С. HRMS (ESI)=425,1185 (М+Н)+. Рассчитано для C22H21N2O5S 425,1171. 1H ЯМР (600 МГц, CDCl3) δ 8.12 (dd, J=8,4 Гц, J=1,8 Гц, 1Н), 8.05 (d, J=1,8 Гц, 1Н), 7.25-7.21 (m, 2H), 7.16 (d, J=8,4 Гц, 1Н), 6.96-6.93 (m, 1Н), 6.81 (d, J=9 Гц, 2H), 4.56 (dd, J=18 Гц, J=2,4 Гц, 1Н), 4.53 (dd, J=18 Гц, J=2,4 Гц, 1Н), 4.19 (dd, J=9,6 Гц, J=3 Гц, 1Н), 4.07-4.01 (m, 1Н), 3.97 (dd, J=9,6 Гц, J=7,2 Гц, 1Н), 3.55-3.51 (m, 1Н), 3.33-3.28 (m, 1Н), 2.36 (t, J=2,4 Гц, 1Н), 2.09-1.99 (m, 2H), 1.89-1.75 (m, 2H). TCX (УФ254) Rf=0,54 (этилацетат/гексаны 2:1). ВЭЖХ tR=9,00 мин.
(S)-1-(2-Пропинил)-5-(2-(2,4-дифторфеноксиметил)-пирролидин-1-сульфонил)изатин (соединение 25). Соединение 25 получали согласно методике получения соединения 24, за исключением того, что использовали соединение 20d с получением оранжевого твердого вещества (0,48 г; 70%). Т. пл.: 101-103°С. HRMS (ESI)=461,0976 (М+Н)+. Рассчитано для C22H19F2N2O5S 461,0983. 1H ЯМР (600 МГц, CDCl3) δ 8.13 (dd, J=8,4 Гц, J=1,8 Гц, 1Н), 8.07 (d, J=1,8 Гц, 1Н), 7.24 (d, J=8,4 Гц, 1Н), 6.96-6.92 (m, 1Н), 6.85-6.78 (m, 2H), 4.59 (dd, J=18 Гц, J=2,4 Гц, 1Н), 4.57 (dd, J=18 Гц, J=2,4 Гц, 1Н), 4.24-4.20 (m, 1Н), 4.03-3.98 (m, 2H), 3.55-3.52 (m, 1Н), 3.26-3.21 (m, 1Н), 2.37 (t, J=2,4 Гц, 1Н), 2.12-2.01 (m, 2H), 1.88-1.76 (m, 2H). ТСХ (УФ254) Rf=0,63 (этилацетат/гексаны 2:1). ВЭЖХ tR=9,60 мин.
(S)-1-((1-(2-Фторэтил)-1Н-[1,2,3]-триазол-4-ил)метил)-5-(2-феноксиметил-пирролидин-1-сульфонил)изатин (соединение 10). К перемешиваемому раствору соединения 24 (138 мг; 0,3 ммоль) в безводном ДМФА (3 мл) добавляли сульфат меди (38 мг; 0,15 ммоль) в воде (0,2 мл) с последующим добавлением аскорбиновой кислоты (53 мг; 0,3 ммоль) в воде (0,2 мл) и затем 2-фторэтилазида (33 мг; 0,36 ммоль) в безводном ДМФА (1,5 мл) и смесь оставляли для перемешивания в атмосфере аргона. Через 2 часа ТСХ показала завершение реакции, и смесь вливали на 10% водн. NH4Cl (12 мл) и экстрагировали ДХМ (3×10 мл) и сушили над Na2SO4. Хроматография (гексаны/этилацетат) дала оранжевое твердое вещество (26 мг; 51%). Т. пл.: 165-167°С. HRMS (ESI)=514,1557 (M+H)+. Рассчитано для C24H25FN5O5S 514,1560. 1H ЯМР (600 МГц, CDCl3) δ 8.07 (dd, J=8,4 Гц, J=1,8 Гц, 1Н), 8.01 (d, J=1,8 Гц, 1Н), 7.76 (s, 1Н), 7.47 (d, J=8,4 Гц, 1Н), 7.23-7.19 (m, 2H), 6.92 (t, J=7,8 Гц, 1Н), 6.82 (d, J=7,8 Гц, 2H), 5.03 (d, J=15,6 Гц, 1Н), 5.02 (d, J=15,6 Гц, 1Н), 4.79 (dt, J=46,2 Гц, J=4,8 Гц, 2H), 4.66 (dt, J=27 Гц, J=4,8 Гц, 2H), 4.17 (dd, J=9,6 Гц, J=3,6 Гц, 1Н), 4.00-3.97 (m, 1Н), 3.93 (dd, J=9 Гц, J=7,2 Гц, 1Н), 3.53-3.49 (m, 1Н), 3.27-3.24 (m, 1Н), 2.08-1.97 (m, 2H), 1.86-1.75 (m, 2H). ТСХ (УФ254) Rf=0,56 (этилацетат). ВЭЖХ tR=7,93 мин.
(S)-1-((1-(2-Фторэтил)-1Н-[1,2,3]-триазол-4-ил)метил)-5-(2-(2,4-дифторфеноксиметил)-пирролидин-1-сульфонил)изатин (соединение 11). Соединение 11 получали согласно методике получения соединения 10, за исключением того, что использовали соединение 25 с получением оранжевой смолы. Перекристаллизация из смеси этилацетат/гексаны дала оранжевое твердое вещество (94 мг; 57%). Т. пл.: 130-131°С. HRMS (ESI)=550,1381 (М+Н)+. Рассчитано для C24H23F3N5O5S 550,1372. 1H ЯМР (600 МГц, CDCl3) δ 8.08 (dd, J=8,4 Гц, J=1,8 Гц, 1Н), 8.02 (d, J=1,8 Гц, 1Н), 7.77 (s, 1Н), 7.52 (d, J=8,4 Гц, 1Н), 6.95-6.91 (m, 1H), 6.82-6.75 (m, 2H), 5.05 (s, 2H), 4.79 (dt, J=46,2 Гц, J=4,8 Гц, 2Н), 4.67 (dt, J=26,4 Гц, J=4,8 Гц, 2H), 4.20 (dd, J=9,6 Гц, J=3 Гц, 1H), 4.01-3.94 (m, 2H), 3.53-3.50 (m, 1H), 3.22-3.17 (m, 1H), 2.10-1.98 (m, 2H), 1.85-1.74 (m, 2H). TCX (УФ254) Rf=0,47 (этилацетат). ВЭЖХ tR=8,45 мин.
(S)-1-((1-(Фторметил)-1H-[1,2,3]-триазол-4-ил)метил)-5-(2-(2,4-дифторфеноксиметил)-пирролидин-1-сульфонил)изатин (соединение 29) получали согласно методике получения соединения 11, за исключением того, что фторметилазид использовали вместо 2-фторэтилазида.
(S)-1-((1-(3-Фторпропил)-1H-[1,2,3]-триазол-4-ил)метил)-5-(2-(2,4-дифторфеноксиметил)-пирролидин-1-сульфонил)изатин (соединение 30) получали согласно методике получения соединения 11, за исключением того, что 3-фторпропилазид использовали вместо 2-фторэтилазида.
(S)-1-((1-(4-Фторбутил)-1Н-[1,2,3]-триазол-4-ил)метил)-5-(2-(2,4-дифторфеноксиметил)-пирролидин-1-сульфонил)изатин (соединение 31) получали согласно методике получения соединения 11, за исключением того, что 4-фторбутилазид использовали вместо 2-фторэтилазида.
(S)-1-(4-Фторбензил)-5-(2-(1-((2-фторэтил)-1Н-[1,2,3]-триазол-4-ил)-метоксиметил)-пирролидин-1-сульфонил)изатин (соединение 12). Соединение 12 получали согласно методике получения соединения 10, за исключением того, что использовали соединение 23 с получением оранжевой смолы (26 мг; 48%). HRMS (ESI)=546,1632 (М+Н)+. Рассчитано для C25H26F2N5O5S 546,1623. 1Н ЯМР (600 МГц, CDCl3) δ 8.03 (d, J=1,8 Гц, 1Н), 8.00 (dd, J=8,4 Гц, J=1,8 Гц, 1H), 7.67 (s, 1H), 7.35-7.33 (m, 2H), 7.08-7.05 (m, 2H), 6.90 (d, J=8,4 Гц, 1H), 4.95 (s, 2H), 4.81 (dt, J=46,8 Гц, J=4,8 Гц, 2H), 4.67 (dt, J=26,4 Гц, J=4,8 Гц, 2H), 4.62 (d, J=12 Гц, 1H), 4.60 (d, J=12 Гц, 1H), 3.81-3.78 (m, 1H), 3.65 (dd, J=9,6 Гц, J=4,2 Гц, 1H), 3.51 (dd, J=9,6 Гц, J=7,2 Гц, 1H), 3.40-3.37 (m, 1H), 3.19-3.15 (m, 1H), 1.92-1.87 (m, 2H), 1.72-1.66 (m, 2H). TCX (УФ254) Rf=0,35 (этилацетат). ВЭЖХ tR=6,58 мин.
(S)-1-(4-Фторбензил)-5-(2-(1-((фторметил)-1H-[1,2,3]-триазол-4-ил)-метоксиметил)-пирролидин-1-сульфонил)изатин (соединение 32) получали согласно методике получения соединения 12, за исключением того, что фторметилазид использовали вместо 2-фторэтилазида.
(S)-1-(4-Фторбензил)-5-(2-((1-(3-фторпропил)-1Н-[1,2,3]-триазол-4-ил)-метоксиметил)-пирролидин-1-сульфонил)изатин (соединение 33) получали согласно методике получения соединения 12, за исключением того, что 3-фторпропилазид использовали вместо 2-фторэтилазида.
(S)-1-(4-Фторбензил)-5-(2-((1-(4-фторбутил)-1Н-[1,2,3]-триазол-4-ил)-метоксиметил)-пирролидин-1-сульфонил)изатин (соединение 34) получали согласно методике получения соединения 12, за исключением того, что 4-фторбутилазид использовали вместо 2-фторэтилазида.
(S)-1-[4-(Фторметил)-1Н-[1,2,3]-триазол-1-ил]метил-5-(2-(2,4-дифторфеноксиметил)-пирролидин-1-сульфонил)изатин) (соединение 6) получали, как проиллюстрировано на Фиг.2(ii), путем взаимодействия (S)-1-(азидометил)-5-(2-(2,4-дифторфеноксиметил)-пирролидин-1-сульфонил)изатина (соединение 37) с 3-фторпроп-1-ином (соединение 28). Реагенты были следующие: (a) CuSO4, L-аскорбиновая кислота, ДМФА. Для получения соединения [18F]6 использовали 3-[18F]фторпроп-1-ин вместо 3-фторпроп-1-ина.
(S)-1-[4-(2-Фторэтил)-1Н-[1,2,3]-триазол-1-ил]метил-5-(2-(2,4-дифторфеноксиметил)-пирролидин-1-сульфонил)изатин) (соединение 7) получали согласно методике получения соединения 6, за исключением того, что 4-фторбут-1-ин использовали вместо 3-фторпроп-1-ина. Для получения соединения [18F]7 использовали 4-[18F]фторбут-1-ин вместо 3-фторпроп-1-ина.
(S)-1-[4-(3-Фторпропил)-1Н-[1,2,3]-триазол-1-ил]метил-5-(2-(2,4-дифторфеноксиметил)-пирролидин-1-сульфонил)изатин) (соединение 8) получали согласно методике получения соединения 6, за исключением того, что 5-фторпент-1-ин использовали вместо 3-фторпроп-1-ина. Для получения соединения [18F]8 использовали 6-[18F]фторпент-1-ин вместо 3-фторпроп-1-ина.
(S)-1-[4-(4-Фторбутил)-1Н-[1,2,3]-триазол-1-ил]метил-5-(2-(2,4-дифторфеноксиметил)-пирролидин-1-сульфонил)изатин) (соединение 9) получали согласно методике получения соединения 6, за исключением того, что 6-фторгекс-1-ин использовали вместо 3-фторпроп-1-ина. Для получения соединения [18F]9 использовали 6-[18F]фторгекс-1-ин вместо 3-фторпроп-1-ина.
Соединение 35 включает группу соединений, где R является таким, как определено выше. Соединение 36 включает группу соединений, где R' является таким, как определено выше. Эти соединения могут быть получены с использованием способа, схематично представленного на Фиг.12 (верхний ряд для соединения 35, нижний ряд для соединения 36). Эта методика включает взаимодействие [11С]метилиодида с источником азидной группы, либо in situ, либо путем отдельного взаимодействия, с последующим циклоприсоединением соответствующего алкина. Этот способ является модификацией хорошо известной методики клик-химии (Click-chemistry), недавно опубликованной в Schirrmacher, R. et al. (25).
Пример 2 - Измерение липофильности целевых соединений и сродства к каспазам
Сродство целевых соединений к каспазам. Сродство новых фторированных изатинов 1-5 и 10-12 и известных изатиновых производных и промежуточных соединений 22 и 23 к различным активированным каспазам 1, 3, 6, 7 и 8 измеряли посредством флуориметрического анализа in vitro ингибирования каспазы, аналогичного анализу, описанному Kopka и соавторами [13]. Ингибирование рекомбинантных человеческих каспаз оценивали посредством измерения накопления флуорогенного продукта, 7-амино-4-метилкумарина (7-АМС).
Рекомбинантные человеческие каспазы-1, -3, -6, -7 и -8 и их пептид-специфичные субстраты приобретали у Biomol International, UK. Ингибирование рекомбинантных каспаз нерадиоактивными изатинами оценивали с использованием флуориметрического анализа, с помощью которого измеряют накопление флуорогенного продукта, 7-амино-4-метилкумарина (7-АМС). Все анализы проводили в 96-луночных планшетах в объеме 200 мкл на лунку. Анализы проводили при 37°С в соответствующем реакционном буфере, как описано ниже, для каждой каспазы. Для каспазы 1 буфер содержал 0,1% CHAPS (3-[(3-холамидопропил)диметиламмоний]-1-пропансульфонат), 100 мМ NaCl, 5 мМ 2-меркаптоэтанола, 100 мМ HEPES (4-(2-гидроксиэтил)-1-пиперазин-этансульфоновая кислота) (рН 7,4), 2 мМ ЭДТА (этилендиаминтетрауксусная кислота), 10% сахарозы и 10 мкМ пептидного субстрата Ac-YVAD-AMC. Для каспазы 3: 20 мМ HEPES (рН 7,4), 10% сахарозы, 100 мМ NaCl, 0,1% CHAPS, 2 мМ ЭДТА и 10 мкМ Ac-DEVD-AMC. Для каспазы 6: 20 мМ HEPES (рН 7,4), 10% сахарозы, 100 мМ NaCl, 0,1% CHAPS, 2 мМ ЭДТА и 10 мкМ Ac-VEID-AMC. Для каспазы 7: 20 мМ HEPES (рН 7,4), 10% сахарозы, 5 мМ 2-меркаптоэтанола, 100 мМ NaCl, 0,1% CHAPS, 2 мМ ЭДТА, 10 мкМ Ас-DEVD-AMC. Для каспазы 8: 20 мМ HEPES (рН 7,4), 10% сахарозы, 100 мМ NaCl, 0,1% CHAPS, 2 мМ ЭДТА и 10 мкМ Ac-IETD-AMC. Буферы содержали нерадиоактивные изатины в ДМСО в конечной концентрации 500, 50, 5 мкМ; 500, 50, 5 нМ; 500, 50, 5 пМ; конечная концентрация ДМСО во всех лунках составляла 5% от общего объема. Рекомбинантные каспазы использовали в количестве 0,5 единиц на анализ (приблизительно 500 пмоль субстрата, превращаемого за час). Все реагенты, за исключением пептидного субстрата, предварительно инкубировали в течение 10 минут. Затем добавляли пептидный субстрат (конечная концентрация 10 мкМ) и планшет инкубировали в течение дополнительных 30 минут; 30 минут были выбраны после начального исследования линейности, когда реакцию оценивали через 10, 30, 60 или 90 минут. Был выбран момент времени 30 минут. Соответствующие контрольные лунки содержали все компоненты реакции без фермента. Количество продуцируемого 7-АМС измеряли на флуоресцентном микропланшетном ридере (Victor2; Perkin-Elmer Life sciences) при длинах волн возбуждения и излучения 355 нм и 460 нм, соответственно. Концентрацию изатина, при которой каспазная активность ингибируется на 50% (ЕС50), подсчитывали с помощью нелинейного регрессионного анализа при использовании программы GraphPad Prism (версия 4.0 для Windows, GraphPad Software, San Diego California USA). Все изатины анализировали в двух повторностях; анализы повторяли один раз. Известные соединения 13-16 были включены в качестве эталонных соединений. Результаты суммированы в таблице 1 ниже.
Данные представляют собой среднее значение для двух опытов, каждый из которых был дублирован, в девяти концентрациях, находящихся в пределах от 5 пМ до 500 мкМ; концентрацию лекарственного средства, требуемую для ингибирования каждого фермента на 50% (ЕС50), получали из нелинейного регрессионного анализа профилей ингибирования.
Результаты. В этом анализе сродство известного изатина 14 составило 59,9 и 25,3 нМ для каспаз 3 и 7, соответственно. Толерантность к замещающим атомам фтора в фениловом эфире была высокой, и для нового изатина 1 сродство к каспазе 3 возрастало в два-три раза по сравнению с известным нефторированным фениловым эфиром 14. Дифторированные изатины 2 и 3 были еще более эффективными, со сродством 12,4 и 10,4 нМ к каспазе 3, соответственно. Аналогичное сродство было обнаружено для тетрагидропирана 4. Пятикратное увеличение эффективности соединения 4 по сравнению с известным бензильным производным 15 является неожиданным, учитывая то, что это кольцо является полностью насыщенным. Замещение фенилового эфира пиримидинильной группой, как в пиримидильном производном 5, приводило к дополнительному увеличению эффективности, со сродством 5,5 и 2,3 нМ к каспазе 3 и 7, соответственно. Введение 2-фторэтил-1,2,3-триазола по обеим боковым частям молекулы приводило к резкому увеличению эффективности, с измеренными значениями сродства к каспазе 3 16,7 и 12,6 нМ для триазолов 10 и 12, соответственно. Эффективность фторэтилтриазола 12 была сходной с эффективностью соединений 1-3, что является неожиданным, учитывая значительно меньший размер и более высокую полярность этой группы. Lee [8] обозначил домен связывания вблизи атома азота изатина как гидрофильный карман, и большинство групп с тех пор включает бензильные группировки в этом положении. Именно поэтому было совершенно неожиданно, что триазол 10 является в 4 раза более эффективным, чем известное бензильное производное 15. Среди исследуемых соединений триазол 11 оказался самым эффективным из всех, со значениями сродства 0,5 и 2,5 нМ к каспазе 3 и 7, соответственно. Все исследуемые соединения были плохими субстратами для каспазы 1, 6 и 8 (ЕС50>5000 нМ).
Вычисление Log P. Липофильность вычисляли с помощью программного обеспечения ACD/Chemsketch labs software.
Результаты. Результаты представлены в таблицах 1 и 1а ниже.
Таблицы 1 и 1а. Профиль ингибирования каспазы и липофильность (Р) выбранных соединений
b Величины подсчитаны при использовании программного обеспечения ACD Labs;
с Измеренное значение Log P для [18F]11 равно 1,61.
Пример 3 - Синтез соединения [18F]11
Предпочтительно, соединение [18F]11 получают посредством «клик-мечения» (S)-1-(2-пропинил)-5-(2-(2,4-дифторфеноксиметил)-пирролидин-1 -сульфонил)изатина (соединение 25) [18F]фторэтилазидом (соединение 27) [15, 16]. В триазол-(S)-1-((1-(2-фторэтил)-1Н-[1,2,3]-триазол-4-ил)метил)-5-(2-(2,4-дифторфеноксиметил)-пирролидин-1-сульфонил)изатин (соединение 11) включали метку посредством катализируемого медью циклоприсоединения 2-[18F]фторэтилазида (соединение 27) с алкиновым предшественником (S)-1-(2-пропинил)-5-(2-(2,4-дифторфеноксиметил)-пирролидин-1-сульфонил)изатином (соединение 25), как показано на Фиг.2(i) и на Фиг.19. Такой же способ может быть использован для получения немеченого соединения 11, где фторэтилазид используют вместо 2-[18F]фторэтилазида.
Забуференный раствор (натрий-фосфатный буфер; рН 6,0; 250 мМ) аскорбата натрия (50 мкл; 8,7 мг; 43,2 мкмоль) добавляли в атмосфере азота в пробирку Уитона (1 мл), содержащую водный раствор сульфата меди(II) (50 мкл; 1,7 мг пентагидрата; 7,0 мкмоль). Через 1 минуту добавляли раствор алкина 25 (3,0 мг; 6,5 мкмоль) в диметилформамиде (25 мкл) с последующим добавлением перегнанного [18F]FEA ([18F]фторэтилазид) (185-740 МБк) в ацетонитриле (100 мкл). Смесь оставляли при комнатной температуре в течение 30 минут и подвергали препаративной радио-ВЭЖХ после добавления воды (15 мкл). Выделенный с помощью ВЭЖХ раствор продукта разбавляли водой (5 мл) и загружали на картридж SepPak C18-light (Waters), который был предварительно обработан этанолом (5 мл) и водой (10 мл). Картридж затем промывали водой (5 мл) и 18F-9 элюировали этанолом небольшими фракциями (0,1 мл). Содержащую продукт фракцию разбавляли PBS (забуференный фосфатом солевой раствор), получая содержание этанола 10-15% (об./об.).
Результаты. Первоначальные попытки конъюгировать соединение 27 с алкиновым предшественником 25 привели к образованию сложной смеси радиоактивных продуктов. Дальнейшее изучение воздействия каталитической системы, температуры и рН на радиохимический выход показало низкую термостабильность соединения [18F]9, при этом были получены наиболее высокие выходы при комнатной температуре. Добавление натрий-фосфатного буфера (рН 6,0; 250 мМ) к этой реакционной смеси значительно увеличивало радиохимический выход, тогда как лишь незначительные отличия наблюдали для двух изучаемых каталитических систем (медный порошок или сульфат/аскорбат меди). Обнаружили, что оптимальными условиями являются время взаимодействия 30 минут при комнатной температуре в присутствии незначительного избытка сульфата меди относительно алкинового предшественника 25. Это позволило выделить триазол [18F]11 с радиохимическим выходом 65±6% (n равен 26, скорректированный с учетом затухания по соединению 27) с радиохимической чистотой >99% после очистки посредством ВЭЖХ. Типичная хроматограмма ВЭЖХ для смеси продуктов показана на Фиг.3. Когда процесс проводили вручную при низкой радиоактивности, была достигнута лишь незначительная удельная активность (1,2 ГБк/мкмоль) в конце синтеза. Идентичность соединения [18F]11 подтверждали посредством совместного элюирования с нерадиоактивным эталонным соединением. Очищенное соединение [18F]11 получали посредством твердофазной экстракции с эффективностью 91±6% (n равен 26, скорректированный с учетом затухания). Радиохимический синтез, включая получение соединения [18F]11, требовал в общей сложности три часа.
Пример 4 - Липофильность соединения [18F]11
Определение Log P. Липофильность оценивали посредством измерения коэффициента распределения октанол/вода при использовании метода Barthel et al [18]. Вкратце, соединение [18F]11 (приблизительно 180 мкКи в 25 мкл этанола) разводили до конечного объема 100 мкл при использовании воды с получением базового раствора. Аликвоты базового раствора (10 мкл) добавляли в воду (490 мкл) и октан-1-ол (Aldrich, безводный) (500 мкл). Затем эти растворы интенсивно встряхивали в течение 10 минут, затем центрифугировали (13201 g; 20°C; 30 мин). После центрифугирования порции (200 мкл) водных и октанольных слоев осторожно отбирали и помещали для анализа в автоматический счетчик гамма-излучения Cobra II Auto-Gamma (Packard Instruments, Meriden, CT, USA) и сравнивали. Коэффициент распределения октанол/вода получали путем деления значения радиоактивности, содержащейся в октаноле, на значение радиоактивности, содержащейся в воде. Установили, что Log P составлял 1,61, как указано в примечании к таблице 1 выше.
Пример 5 - Распределение в тканях и метаболическая стабильность соединений [18F]11 и [125I]14
Стабильность нового соединения [18F]11 и известного соединения [125I]14 в плазме и печени измеряли в динамике по времени после введения мышам. Соединение [18F]11 вводили внутривенно мышам-носителям опухоли, и его распределение в тканях измеряли в выбранных тканях через 2, 15 и 60 минут после инъекции. Соединение [125I]14 вводили внутривенно мышам, не являющимся носителями опухоли, и его распределение в тканях измеряли в выбранные моменты времени от 2 до 60 минут после инъекции. Кроме того, метаболическую стабильность in vivo радиоактивной метки определяли в образцах плазмы и печени, и для соединения [18F]11 - в образцах мочи.
Изучения метаболизма in vitro под влиянием S9-фракции печени мыши. Исследования метаболизма под влиянием S9-фракции печени проводили, как описано в Aboagye et al. [19]. Печень мышей быстро иссекали и хранили при 4°С на протяжении всего эксперимента. Ткани взвешивали и гомогенизировали в эквивалентном объеме 50 мМ Tris-150 мМ KCl-HCl буфера (рН 7,4) при использовании гомогенизатора Ultra-Thurrax (IKA, Staufen, Germany). Для получения S9-фракции гомогенат центрифугировали (10000 g) в течение 30 минут для удаления ядер, митохондрий и клеточного детрита. Концентрацию белка S9-фракции определяли с помощью имеющегося в продаже набора для анализа содержания белков с использованием бицинхониновой кислоты (ВСА) (Perbio Science, Cheshire, UK). S9-фракции хранили при -80°С вплоть до 6 недель. Выбранные немеченые изатины 14, 15, 16 (10 мМ; 10 мкл) инкубировали с S9-фракцией (33,26 мг/мл; 20 мкл) и 0,5 мМ никотинамидадениндинуклеотидфосфата (восстановленная форма) на воздухе в течение 60 минут при 37°С в 0,1 мМ Tris-HCl буфера (рН 7,4) в общем объеме, равном 1 мл. Контрольные инкубированные образцы не содержали изатина. Реакции останавливали посредством добавления охлажденного на льду ацетонитрила (2 мл); образцы затем немедленно помещали на сухой лед перед экстракцией и анализом ВЭЖХ (ниже).
Изучение биораспределения и метаболизма in vivo. Клетки индуцированной радиационным излучением фибросаркомы (RIF-1) (20) поддерживали в среде RPMI 1640 (Invitrogen Ltd, Paisley, UK) с добавлением 10% фетальной телячьей сыворотки (BioWhittaker Europe Ltd, Verviers, Belgium), 2 мМ L-глутамина, 100 Ед./мл пенициллина, 100 мкг/мл стрептомицина и 0,25 мкг/мл фунгизона (Gibco, UK) при 37°С во влажной камере с 5% CO2. Всю работу с животными проводили дипломированные исследователи согласно United Kingdom's «Guidance on the Operation of Animals (Scientific Procedures) Act 1986» (HMSO, London, United Kingdom, 1990) и в полном соответствии с государственным регулированием и руководящими принципами по благосостоянию животных при экспериментальной неоплазии [21]. Опухоли прививали мышам посредством подкожной инъекции 5×105 клеток в спину самцам мышей С3Н/hej (Harian, Bicester, Oxfordshire, UK). Рост опухоли контролировали каждые двое суток при использовании электронного штангенциркуля, и объем опухоли подсчитывали при использовании уравнения (π/6)×Д×Ш×Г (Д представляет собой длину, Ш представляет собой ширину и Г представляет собой глубину). Животных отбирали для изучения биораспределения соединения [18F]11, когда опухоли достигали приблизительно 100-150 мм3; биораспределение соединения [125I]14 проводили на мышах, не являющихся носителями опухоли.
Мышам внутривенно вводили через латеральную вену 0,08-0,13 мл радиоактивного вещества (~0,37 МБк соединения [125I]14 или 3,7 МБк соединения [18F]11), растворенного в физиологическом растворе с фосфатным буфером. В выбранные моменты времени после инъекции (2-60 мин) мышей умерщвляли посредством обескровливания при помощи пункции сердца под общей анестезией (ингаляция изофлюрана). Аликвоты гепаринизированной крови быстро центрифугировали (2000 g в течение 5 минут) для получения плазмы. Радиоактивность, содержащуюся в тканях, определяли посредством гамма-радиометрии на автоматическом счетчике гамма-излучения Cobra II Auto-Gamma (Packard Instruments, Meriden, CT, USA) и выражали в виде процента вводимой дозы на грамм ткани (% ID/г). Минимум три мыши использовали для каждого момента времени. Все животные ранее не подвергались экспериментам.
Изучение метаболизма in vivo также проводили на мышах С3Н/hej. Мышам, не являющимся носителями опухоли, внутривенно вводили 0,37 МБк соединения [125I]14 или 7,4 МБк соединения [18F]11, и плазму получали, как указано выше. Образцы плазмы, печени и мочи мгновенно замораживали в жидком азоте и хранили в предварительно взвешенных пробирках для сцинтилляционных измерений на сухом льду до анализа.
Анализ ВЭЖХ. Непосредственно перед экстракцией образцы размораживали и помещали на лед. Для экстракции в плазму (0,2 мл) добавляли охлажденный на льду ацетонитрил (1,5 мл); инкубированные образцы S9 (3 мл), содержащие ацетонитрил, также анализировали. Каждую смесь центрифугировали (15493 g; 4°С, 3 мин) и полученный супернатант упаривали досуха под вакуумом при 40°С при использовании роторного испарителя. Образцы печени гомогенизировали с охлажденным на льду ацетонитрилом (1,5 мл) при использовании гомогенизатора IKA Ultra-Turrax T-25 перед центрифугированием. Остатки затем ресуспендировали в подвижной фазе для ВЭЖХ (1,2 мл) и фильтровали через шприцевой фильтр с гидрофильной мембраной Minisart (0,2 мкм) (Sartorius, Goettingen, Germany). Образцы мочи разбавляли подвижной фазой для ВЭЖХ, фильтровали, как указано выше. Затем образцы (1 мл) анализировали посредством радио-ВЭЖХ на системе ВЭЖХ серии Agilent 1100 (Agilent Technologies, Stockport, UK), оснащенной гамма-детектором γ-RAM модель 3 и программным обеспечением Laura (IN/US Systems inc., Florida, USA).
Обращенная фаза Cia колонки µBondapak фирмы Waters (300×7,8 мм) составляет неподвижную фазу. Для анализа соединения [125I]14 образцы плазмы и печени обрабатывали, как указано выше, и анализировали при использовании подвижной фазы, содержащей смесь вода (0,1% ТФУ)/пропан-1-ол (0,1% ТФУ) (35:65), при разделении в изократическом режиме со скоростью потока 2 мл/мин. Для анализа немеченых соединений 14, 15 и 16 после метаболизма in vitro образцы анализировали при использовании подвижной фазы, содержащей смесь вода (0,1% ТФУ)/пропан-1-ол (0,1% ТФУ), с градиентом 2→80% органической фазы за 11 минут, 80→5% органической фазы за 3 минуты, затем 5% органической фазы в течение 6 минут, пропускаемой со скоростью потока 3 мл/мин. Для анализа соединения [18F]9 образцы плазмы, печени и мочи анализировали при использовании подвижной фазы, содержащей смесь 0,1 М формиатаммония/1,8:1 метанол: ацетонитрил, с градиентом 50% органической фазы в течение 1 минуты, 50→90% органической фазы за 14 минут, 90% органической фазы в течение 3 минут, 90→50% органической фазы за 2 минуты, 50% органической фазы в течение 4 минут, пропускаемой со скоростью потока 3 мл/мин. Кроме того, для оценки доли излучаемой соединением [18F]9 радиоактивности, связанной с остатками плазмы и печени при центрифугировании после экстракции, по отношению к радиоактивности, остающейся в вводимой пробе для ВЭЖХ, регистрировали общий объем вводимой пробы для ВЭЖХ и отбирали аликвоту (0,1 мл) для подсчета. Радиоактивность в аликвоте, равной 0,1 мл, и остатке при центрифугировании затем анализировали посредством гамма-радиометрии (Packard Instruments).
Результаты. На Фиг.4 показано, что соединение [18F]11 быстро распределялось в тканях и также быстро элиминировалось. Высокую локализацию излучаемой соединением [18F]11 радиоактивности наблюдали в почках, моче и печени, что говорит о важности как почечного, так и печеночного путей элиминации. Однако даже в этих тканях наблюдали быструю элиминацию радиоактивности. Существенно, с точки зрения визуализации, что накопление соединения [18F]11 в нелеченных опухолях, сердце и головном мозге было низким; это должно способствовать измерению повышенного связывания, ассоциированного с активацией каспазы в этих тканях. Накопление в костной ткани было низким, что говорит об отсутствии дефторирования радиоактивной метки и, следовательно, стабильности фторэтилазидной группировки. Анализ посредством радио-ВЭЖХ показал, что соединение [18F]11 было относительно более стабильным к метаболическому разложению, чем соединение [125I]14 (Фиг.5), образуя пик единственного полярного метаболита в плазме и печени. Исходное соединение все еще находилось в плазме через 60 минут и представляло доминирующий пик. Напротив, в образцах печени метаболит представлял доминирующий пик во все исследуемые моменты времени. Радиоактивность в образцах мочи обусловлена главным образом метаболитом. Доли исходной радиоактивной метки в плазме и печени в выбранные моменты времени вместе с видимой эффективностью экстракции суммированы в таблице 2. После быстрого снижения скорость метаболизма in vivo имела вид плато между 15 и 60 мин. Однако эффективность экстракции из плазмы и печени уменьшалась с течением времени, вероятно, вследствие неспецифического связывания. Имело место также увеличение уровня радиоактивности в зависимости от времени в остаточных осадках после экстракции плазмы, что говорит о повышенном связывании с белками плазмы.
На Фиг.7 показано, что соединение [125I]14 быстро распределялось в главные органы и также быстро элиминировалось. Соединение [125I]14 полностью элиминировалось из плазмы в течение 10 минут и представляло <15% (n. равный 3) радиоактивности, обнаруживаемой в печени в этот момент времени. В течение 30 минут после инъекции исходное соединение [125I]14 полностью метаболизировалось в печени, при этом единственный полярный метаболит составлял большую часть детектируемой радиоактивности. В соответствии с этими результатами авторы наблюдали быстрый клиренс во всех изучаемых тканях, и через 10 минут большая часть введенной радиоактивности экскретировалась в мочу.
Как показано на Фиг.8, метаболические профили известных немеченых изатинов 14 и 15 были сходными, при этом через 60 минут инкубирования наблюдали почти полное разложение.
Пример 6 - Накопление соединения [18F]11 в четырех клеточных линиях рака
Клеточные линии и модель опухоли. Клеточные линии карциномы яичника человека PEO1/4 создавали и характеризовали, как описано в Wolf C.R. et al. [22] и Langdon S.P. et al. [23]. Вкратце, клетки PEO1/4 получали из злокачественного асцита пациента с рецидивирующей муцинозной аденокарциномой яичника до (PEO1) и после (PEO4) наступления клинической резистентности к химиотерапии цисплатином у этого пациента. Клеточную линию индуцированной радиационным излучением фибросаркомы мыши (RIF-1) первоначально характеризовали с помощью Twentyman P.R. et al. [20]. Опухолевые клетки LNM35 представляют собой высоколимфогенную метастатическую сублинию клеточной линии немелкоклеточной карциномы легкого человека NCI-H460, как описано в Kozaki К. et al. [24]. Все клеточные линии поддерживали обычным способом в среде RPMI 1640 (Invitrogen Ltd, Paisley, UK) с добавлением 10% фетальной телячьей сыворотки (BioWhittaker Europe Ltd, Verviers, Belgium), 2 мМ L-глутамина, 100 Ед./мл пенициллина, 100 мкг/мл стрептомицина и 0,25 мкг/мл фунгизона (Gibco, UK) при 37°С во влажной камере с 5% CO2.
Модели опухолей in vivo создавали посредством подкожной инъекции PEO1/4 (10×106 клеток) при об./об. матригеля (matrigel), BD Biosciences, Oxford, UK), RIF-1 (5×105 клеток) и LNM35 (1×106 клеток) в спину самкам nu/nu-BALB/c или самцам С3Н/hej мышей в возрасте 6-8 недель (Harlan, Bicester, Oxfordshire, UK). Рост опухоли контролировали каждые двое суток при использовании электронного штангенциркуля, и объем опухоли подсчитывали при использовании уравнения (π/6)×Д×Ш×Г (Д представляет собой длину, Ш представляет собой ширину и Г представляет собой глубину). Всю работу с животными проводили дипломированные исследователи согласно United Kingdom's «Guidance on the Operation of Animals (Scientific Procedures) Act 1986» (HMSO, London, United Kingdom, 1990) и в полном соответствии с государственным регулированием и руководящими принципами по благосостоянию животных при экспериментальной неоплазии.
Анализы in vitro накопления соединения [18F]11 и активации каспазы 3. Клетки высевали в трех повторностях на 12-луночные планшеты за 2 или 3 суток до экспериментов и обрабатывали противоопухолевым лекарственным средством на основе платины цисплатином, 5-ФУ (5-фторурацилом), ингибитором синтеза белка циклогексимидом (СНХ) или ингибитором топоизомеразы этопозидом (VP-16) (Sigma, UK) или соответствующим носителем в указанной концентрации и в указанное время. В день эксперимента добавляли приблизительно 10 мкКи/лунка соединения [18F]11 и оставляли для накопления в клетках в течение 60 минут при 37°С. Клетки собирали, промывали и ресуспендировали в 400 мкл PBS. 260 мкл каждого образца переносили в сцинтилляционные пробирки и сразу определяли радиоактивность фтора-18 при использовании счетчика гамма-излучения Packard Cobra II (Perkin Elmer, UK). Колориметрический анализ активации каспазы 3 проводили при использовании 40 мкл каждого образца согласно инструкциям производителя (набор для анализа Caspase-Glo 3/7, Promega, UK). Вкратце, клетки переносили в белый непрозрачный 96-луночный планшет, инкубировали в течение периода от 30 минут до 3 часов с 50 мкл реагента Caspase-Glo, и ферментативную активность каспазы 3 измеряли при использовании люминометра Multiskan (Thermo Electron, UK). Анализ содержания белков с помощью ВСА (Pierce, UK) проводили для всех образцов, данные нормализовывали и выражали в виде ССРМА или RLU (относительные световые единицы)/мг белка.
Биораспределение соединения [18F]11 in vivo и ПЭТ-визуализация. Для исследований биораспределения мышам-опухоленосителям вводили 10 мг/кг цисплатина или носитель в течение 24 часов, анестезировали смесью изофлюран/О2/N2O и внутривенно вводили в латеральную хвостовую вену приблизительно 100 мкКи соединения [18F]11 в течение указанного времени. Различные ткани иссекали, взвешивали и помещали в сцинтилляционные пробирки для немедленного подсчета радиоактивности фтора-18.
Мышей-опухоленосителей, которым вводили цисплатин (10 мг/кг в течение 24 часов) или носитель, сканировали на предназначенном для мелких животных ПЭТ-сканере (quad-HIDAC (четырехслойная матрица лавинных камер высокой плотности (quadratic array of high-density avalanche chambers)); Oxford Positron Systems, Weston-on-the-Green, United Kingdom). Анестезированных животных помещали на термостатируемой подложке в положении лежа на животе в пределах сканера. Подложку регулировали для обеспечения ректальной температуры у мышей приблизительно 37°С. Болюсную инъекцию соединения [18F]11 (приблизительно 100 мкКи) проводили внутривенно (i/v) через канюлю в хвостовой вене и начинали сканирование. Динамические эмиссионные сканограммы получали в формате списка в течение 60 минут. Полученные данные затем разбивали на 0,5-мм элементы дискретизации синограммы и 19 временных интервалов (размер векселей 0,5×0,5×0,5 мм; 4×15,4×60 и 11×300 секунд) для реконструкции изображения, которое создавали посредством отфильтрованной обратной проекции при использовании двумерного фильтра Хэмминга (граница пропускания 0,6). Полученные наборы данных изображений переносили на рабочую станцию SUN (Ultra 10; SUN Microsystems, Santa Clara, CA) и визуализировали при использовании программного обеспечения Analyze (версия 6.0; Biomedical Imaging Resource, Mayo Clinic, Rochester, MN). Накопленные изображения скомпонованных динамических данных за период от 0 до 1 минуты после инъекции и от 30 до 60 минут после инъекции использовали для визуализации накопления радиоактивной метки и для определения изучаемых областей (ROI). Плотности импульсов усредняли для каждой ROI в каждый из 19 моментов времени для получения кривой зависимости радиоактивности от времени (ТАС) для ROI. ТАС для опухоли нормализовали по ТАС для сердца в каждый из моментов времени для получения нормализованного значения накопления (NUV). Данные по соединению [18F]9 для мышцы использовали в качестве внутренней входной функции для нормализации данных для опухоли. NUV через 60 минут после инъекции (NUV60), площадь под кривой NUV (ППК), подсчитанную в виде интеграла NUV, от 0 до 60 минут и фракционное удерживание метки (FRT), радиоактивность через 60 минут относительно радиоактивности через 2,5 минуты использовали для сравнения. FRT является полезной переменной, поскольку показывает долю радиоактивной метки, доставленной в опухоль, которая удерживается. Таким образом, эта переменная нормализует накопление соединения [18F]9 в опухоли к доставке.
Результаты. Как показано на Фиг.9, все обработанные лекарственным средством клетки демонстрируют значительно возросшее накопление соединения [18F]11 по сравнению с контролем (обработанные носителем), за исключением клеток рака яичника, резистентных к цисплатину, (PEO4), как и ожидалось. Кроме того, повышенное накопление соединения [18F]9 в обработанных лекарственным средством клетках LNM35 коррелирует с количеством активной каспазы-3 в клетках, и также показан антагонизм между обработками CDDP и VP-16.
В таблице 3 показано значительно возросшее накопление соединения [18F]11 в опухоли у мышей, несущих ксенотрансплантаты PEO1, которым вводили CDDP, тогда как клетки опухолей PEO4, резистентные к CDDP, не демонстрируют различий в накоплении между контролем и обработанными клетками. Накопление соединения [18F]11 в других тканях значительно не отличается между контрольным и обработанным вариантами, за исключением селезенки и мышечной ткани у мышей, несущих клетки опухолей PEO1.
ПЭТ-изображения трех мышей с ксенотрансплантатами PEO1, PEO4 и RIF-1, которым вводили CDDP (Фиг.10), показывали значительное накопление соединения [18F]11 в опухоли.
Все приведенные ниже документы включены здесь посредством ссылки.
Пример 7 - Накопление соединения [18F]11 в апоптозных клетках
Клетки RIF-1 обрабатывали носителем (0,1% ДМСО) или цис-диаминдихлорплатиной(II) (цисплатин) (CDDP) (100 мкМ) в течение 48 часов. Клетки затем инкубировали с соединением [18F]11 в течение 1 часа, промывали и анализировали на радиоактивность. Как продемонстрировано на Фиг.13, клеточный анализ показал приблизительно 1,5-кратное увеличение накопления соединения [18F]11 в апоптозных клетках. Данные выражены в виде усредненного числа скорректированных с учетом затухания импульсов в минуту на миллиграмм общего клеточного белка. Данные представляют собой среднее значение ± SEM, выполнены в трехкратной повторности.
Опухоль RIF-1 обрабатывали носителем (50% ДМСО) или CDDP (однократная доза 10 мг/кг). Уровни создаваемой [18F] радиоактивности через 60 минут после инъекции радиоактивной метки анализировали и выражали в виде отношения к уровню радиоактивности в крови. Как продемонстрировано на Фиг.14, исследования накопления in vivo у мышей, несущих опухоль RIF-1, также показали приблизительно 1,5-кратное увеличение накопления соединения [18F]11 для мышей, которым вводили лекарственное средство, по сравнению с мышами, которым вводили носитель. Данные представляют собой среднее значение ± SEM, и n составляет 8 мышей на группу.
Пример 8 - Функциональные свойства соединения [18F]11 в качестве индикатора визуализации апоптоза
Экспериментальную модель in vivo апоптоза опухоли создавали посредством подкожной инъекции клеток лимфомы мыши 38С13 (5000 клеток) в спину самцам С3Н/hej мышей в возрасте 6-8 недель (Harlan, Bicester, Oxfordshire, UK). Когда ксенотрансплантаты достигали размера приблизительно 100 мм3, мышам вводили циклофосфамид (ЦФА, 100 мг/кг) или носитель в течение 24 часов и затем сканировали на предназначенном для мелких животных ПЭТ-сканере (модуль ПЭТ Siemens Inveon). Анестезированных животных помещали на термостатируемой подложке в положении лежа на животе в пределах сканера. Болюсную инъекцию соединения [18F]11 (приблизительно 100 мкКи) проводили внутривенно (i/v) через канюлю в хвостовой вене и начинали сканирование.
Динамические эмиссионные сканограммы получали в формате списка в течение 60 минут. Полученные данные затем разбивали на 0,5-мм элементы дискретизации синограммы и 19 временных интервалов для реконструкции изображения, которое создавали посредством отфильтрованной обратной проекции. Полученные наборы данных изображений визуализировали при использовании программного обеспечения Siemens Inveon Research Workplace. Накопленные изображения динамических данных использовали для визуализации накопления радиоактивной метки и для определения изучаемых областей (ROI). Плотности импульсов усредняли для каждой ROI в каждый из 19 моментов времени для получения кривой зависимости радиоактивности от времени (ТАС) для ROI (см. Фиг.15).
ТАС для опухоли нормализовали по ТАС для всего организма в каждый из моментов времени для получения нормализованного значения накопления (NUV). NUV через 60 минут после инъекции (NUV60) и площадь под кривой NUV (ППК), подсчитанную в виде интеграла NUV от 0 до 60 минут, использовали для сравнения (см. Фиг.16).
После сканирования различные ткани иссекали, взвешивали и помещали в пробирки для немедленного подсчета радиоактивности фтора-18 для оценки биораспределения соединения [18F]11 (см. таблицу 4 ниже).
Репрезентативные трехмерные реконструкции с помощью OSEM ПЭТ-изображений соединения [18F]11 у двух мышей, несущих ксенотрансплантат 38С18, которым вводили носитель или циклофосфамид, показаны на Фиг.17, где кружками указана опухоль.
Результаты биораспределения, показанные в таблице 4, демонстрируют значительное 2-кратное увеличение накопления соединения [18F]11 в опухолях мышей, которым вводили ЦФА, по сравнению с носителем, которое было подтверждено с помощью полуколичественных данных, генерированных из параметров ПЭТ-изображения, ТАС для опухоли, NUV60 и ППК (см. Фиг.15 и 16). Следовательно, биораспределение и данные ПЭТ-сканирования, показанные на Фиг.15-17 и в таблице 4, демонстрируют эффективность соединения [18F]11 для обнаружения апоптоза опухоли in vivo и впервые описывают пригодность специфического агента ПЭТ-визуализации каспазы-3 для опухолей.
Соотношение % ID в опухоли и в крови
Пример 9 - Активность соединения 11 в клетке
Активность соединения 11 в клетке оценивали с использованием ферментного анализа и сравнивали с активностью соединения 14.
Клетки индуцированной радиационным излучением фибросаркомы мыши (RIF-1) обрабатывали в течение 15 минут ингибиторами каспаз z-VAD-fmk (100 мкМ), соединением 11 и соединением 14 перед индукцией апоптоза цисплатином (100 мкМ) в течение 48 часов. Δ-ПАРП полоса в иммуноблоте соответствует большому фрагменту эндогенного ПАРП массой 89 кДа, образующемуся в результате расщепления каспазой-3. α-Тубулин использовали в качестве контроля нагрузки.
Как показано на Фиг.18, предварительное инкубирование клеток RIF-1 с соединением 11, но не с соединением 14, нарушало индукцию ПАРП, когнатной мишени каспазы-3 посредством индуцированного цисплатином апоптоза, демонстрируя клеточную инактивацию каспазы-3 соединением 11.
Для дальнейшего установления связывания соединения 11 в условиях индуцированного лекарственным средством апоптоза авторы оценивали накопление меченого радиоактивным изотопом соединения [18F]11 в раковых клетках LNM35, RIF-1 и PEO1/4, как описано выше в примере 6 и показано на Фиг.9. Все обработанные лекарственным средством клетки демонстрировали значительно возросшее накопление соединения [18F]11 по сравнению с контролем, за исключением клеток рака яичника, резистентных к цисплатину, (PEO4), как и ожидалось. Кроме того, повышенное накопление соединения [18F]11 в обработанных лекарственным средством клетках LNM35 коррелировало с количеством активной каспазы-3 в клетках, определенным при использовании анализа Caspase-Glo.
Пример 10 - Синтез соединения [18F]11 с использованием защищенного предшественника
Изатиновый радиолиганд [18F]11 может быть синтезирован с использованием двухстадийного способа, как описано на Фиг.2i, Фиг.19 и в примере 3. Недостатком этого радиохимического синтеза является присутствие стабильной примеси, которая снижает удельную активность полученного продукта (1-4 Ки/мкмоль). Более фундаментально стабильная примесь присутствует в значительном количестве (5-10 мкг/мл), величина слишком большая для клинического продвижения. Попытки охарактеризовать примесь, для того чтобы провести испытания на токсичность, оказались безрезультатными, за исключением подтверждения того, что стабильная примесь, не будучи радиоактивным соединением фтор-19-триазола, являлась соединением на основе изатина.
Анализ посредством ВЭЖХ подтвердил, что примесь является результатом побочной реакции во время радиохимического синтеза и материально не присутствует в алкиновом предшественнике. Изучение архитектуры молекулы изатина показало только два участка с высокой химической активностью: терминальная алкиновая функциональная группа, используемая в клик-циклоприсоединении, описанном выше, и карбонильный атом углерода в положении С-3, необходимый для связывания с активным сайтом каспазы-3. Изучали стратегию использования защитных групп для карбонильного атома углерода в положении С-3, в частности защиту карбонильного атома углерода в положении С-3 в виде диоксолана ацеталя, как показано в общих формулах 38 и 43 ниже, где n равен 0, 1, 2, 3, 4, 5 или 6, и где x представляет собой уходящую группу, например мезилат, тозилат, нозилат или другой сульфонатный эфир или галогенид:
Защищенный алкин, (S)-1-{[1'-[1-(2-пропинил)]-(1',2'-дигидро-2'-оксоспиро(1,3-диоксан-2,3'-[3Н]индол)-5'-сульфонил}-2-(2,4-дифторфенокси-метил)-пирролидин (соединение 39) и защищенный триазол (соединение 40) синтезировали, как кратко изложено на Фиг.20.
Основные подробности эксперимента описаны выше в начале раздела «Примеры» и в Smith et al. [29]. Все взаимодействия проводили в атмосфере аргона.
(S)-1-{[1'-[1-(2-Пропинил)]-(1',2'-дигидро-2'-оксоспиро(1,3-диоксан-2,3'-[3Н]индол)-5'-сульфонил}-2-(2,4-дифторфеноксиметил)-пирролидин (соединение 39) получали согласно следующему способу. К раствору соединения 25 (92 мг; 0,2 ммоль) в безводном толуоле (6 мл) добавляли 1,3-пропандиол (0,3 мл) и 4-толуолсульфоновую кислоту (10 мг; 0,005 ммоль). Затем этот раствор кипятили с обратным холодильником в течение 24 часов, при этом образовавшуюся воду удаляли посредством азеотропной перегонки. Реакционную смесь затем оставляли для охлаждения до температуры окружающей среды и большую часть растворителя удаляли при пониженном давлении. Образец затем повторно растворяли в ДХМ (10 мл) и промывали насыщенным Na2CO3 (1×10 мл), водой (1×10 мл) и рассолом (1×10 мл), затем сушили над Na2SO4. Колоночная хроматография (этилацетат/гексаны 2:1) дала требуемый продукт в виде первой фракции (бесцветное масло; 46 мг; 44%). HRMS (ESI)=519,1393 (М+Н)+. Рассчитано для C25H25F2N2O6S 519,1401. 1H ЯМР (400 МГц, CDCl3) δ 7.90 (d, J=1,8 Гц, 1Н), 7.87 (dd, J=8,2 Гц, 1,8 Гц, 1Н), 7.09 (d, J=8,2 Гц, 1Н), 7.04-6.98 (m, 1Н), 6.88-6.78 (m, 1Н), 4.93 (t, J=12,1 Гц, 2Н), 4.45 (d, J=2,4 Гц, 2Н), 4.32-4.27 (m, 1Н), 3.99-3.93 (m, 4H), 3.56-3.49 (m, 1Н), 3.14-3.08 (m, 1Н), 2.43-2.37 (m, 1Н), 2.30 (t, J=2,4 Гц, 1Н), 2.09-1.93 (m, 2Н), 1.78-1.64 (m, 3H).
(S)-1-{[1'-[1-(2-Фторэтил)-1Н-[1,2,3]-триазол-4-ил]-(1',2'-дигидро-2'-оксоспиро(1,3-диоксан-2,3'-[3Н]индол)-5'-сульфонил}-2-(2,4-дифторфенокси-метил)-пирролидин (соединение 40) получали согласно следующему способу. К раствору (S)-1-((1-(2-фторэтил)-1Н-[1,2,3]-триазол-4-ил)метил)-5-(2-(2,4-дифтор-феноксиметил)-пирролидин-1-сульфонил)изатина (соединение 11) (12 мг; 0,02 ммоль) в безводном толуоле (4 мл) добавляли 1,3-пропандиол (0,15 мл) и 4-толуолсульфоновую кислоту (1 мг; 0,005 ммоль). Затем этот раствор кипятили с обратным холодильником в течение 24 часов, при этом образовавшуюся воду удаляли посредством азеотропной перегонки. Реакционную смесь затем оставляли для охлаждения до температуры окружающей среды и большую часть растворителя удаляли при пониженном давлении. Образец затем повторно растворяли в ДХМ (10 мл) и промывали насыщенным Na2CO3 (1×10 мл), водой (1×10 мл) и рассолом (1×10 мл), затем сушили над Na2SO4. Хроматография (этилацетат/гексаны 4:1) дала требуемый продукт в виде первой фракции (бесцветное масло; 7 мг; 58%). HRMS (ESI)=519,1393 (M+H)+. Рассчитано для C25H25F2N2O6S 519,1401. 1H ЯМР (400 МГц, CDCl3) δ 7.84 (s, 1H), 7.81 (d, J=1,6 Гц, 1H), 7.67 (s, 1H), 7.43 (dd, J=8,4 Гц, 1,6 Гц, 1Н), 7.04-6.98 (m, 1H), 6.88-6.79 (m, 1H), 4.96 (s, 1H), 4.92 (t, J=2,8 Гц, 2Н), 4.79 (dt, J=46,6 Гц, 4,8 Гц, 2Н), 4.64 (dt, J=27 Гц, 4,8 Гц, 2Н), 4.29 (dd, J=8,4 Гц, 2,6 Гц, 1H), 4.00-3.89 (m, 4H), 3.53-3.48 (m, 1H), 3.10-3.07 (m, 1H), 2.43-2.37 (m, 1H), 2.05-1.92 (m, 2Н), 1.75-1.65 (m, 3H).
Соединение [18F]11 можно затем синтезировать с использованием одного или более чем одного из этих защищенных предшественников. Типичная радиохимическая реакция с защищенным алкиновым предшественником 39 проиллюстрирована на Фиг.21. Использование такого защищенного алкинового предшественника предотвращает нежелательные побочные реакции по положению С-3.
К водному раствору гексагидрата сульфата меди(II) (0,51 мг; 2,06 мкмоль; 25 мкл) в атмосфере азота добавляли аскорбат натрия (2,53 мг; 12,79 мкмоль) в натрий-фосфатном буфере (рН 6,0; 250 мМ; 25 мкл), ацеталь алкинового предшественника (1,0 мг; 1,93 мкмоль) (соединение 39) в ДМФА (25 мкл) и [18F]-фторэтилазид (1,27 мКи) (соединение 27) в ацетонитриле (100 мкл). Реакционную смесь нагревали до 80°С в течение 30 минут (см. Фиг.22). После добавления соляной кислоты (6 н.; 100 мкл) перемешиваемую смесь нагревали при использовании микроволнового резонатора (2 с; 50 Вт; установленная температура 80°С). После добавления подвижной фазы ВЭЖХ (50 мкл; вода, содержащая 50% МеОН с 35% MeCN) эту смесь очищали посредством препаративной ВЭЖХ (см. Фиг.23). Соединение [18F]11 выделяли с скорректированным с учетом затухания радиохимическим выходом 24% (по эталонному уровню исходного [18F]-фторэтилазида) и готовили с использованием C18-SepPak SPS в смеси PBS/10% EtOH. Уровень стабильной примеси, элюируемой совместно с соединением [18F]11, составлял 0,84 мкг/мл. Удельная радиоактивность составляла 0,05 Ки/мкмоль (2 ГБк/мкмоль) (см. Фиг.24).
Аналогичные защищенные предшественники, конечно, могут быть использованы для синтеза других соединений по настоящему изобретению.
Пример 11 - Сравнение накопления соединения [18F]11 и соединения [18F]42 в апоптозных клетках
Дополнительный аспект данной разработки включает анализ слабого изатинового ингибитора каспазы-3 N-[1-(2-фторэтил)-1Н-[1,2,3]-триазол-4-ил]изатина (соединение 42). Это соединение синтезировали с использованием N-(2-пропинил)изатина (соединение 41), как показано на Фиг.25. Соединение [18F]42 имеет изатиновый структурный каркас, что делает возможным связывание соединения с активированной каспазой-3 посредством традиционного для изатина способа связывания, а также меченую радиоактивными изотопами функциональную триазольную группу. Однако у соединения [18F]42 отсутствует пирролидин-сульфонамидная функциональная группа, которая обеспечивает высокое сродство и селективность к каспазе-3. Соединение [18F]42 исследовали в анализах клеточного накопления для непосредственного сравнения с соединением [18F]11. Задача этого исследования заключалась в изучении профилей накопления соединений с целью определения того, является ли накопление результатом повышенной клеточной проницаемости, характерной особенностью апоптозных клеток, или результатом связывания с каспазой-3. В отличие от соединения [18F]11 не было повышения связывания соединения [18F]42 после обработки лекарственным средством (см. Фиг.26), что подтверждало точку зрения, что связывание соединения [18F]11 происходит вследствие активации каспазы-3.
N-(2-Пропинил)изатин (соединение 41) синтезировали, как описано в Smith et al. [29] (алкины 12 и 13). 1H ЯМР (600 МГц, CDCl3) δ 7.65-7.61 (m, 2H), 7.19-7.12 (m, 2H), 4.61 (s, 2H), 2.30 (s, 1H).
N-[1-(2-Фторэтил)-1H-[1,2,3]-триазол-4-ил]изатин (соединение 42) синтезировали, как описано в Smith et al [29] (триазолы 14 и 15). 1H ЯМР (600 МГц, CDCl3) δ 7.76 (s, 1H), 7.63-7.57 (m, 2H), 7.34 (d, J=8,4 Гц, 1H), 7.13 (t, J=3,6 Гц, 1H), 5.03 (s, 2H), 4.81 (dt, J=46,8 Гц, 4,8 Гц, 2H), 4.68 (dt, J=27 Гц, 4,8 Гц, 2H).
Примечание: из-за ошибки в нумерации соединения 15 и 21 фактически представляют собой одно и то же соединение.
СПИСОК ЛИТЕРАТУРЫ
1. Lahorte et al. (2004) European Journal of Nuclear Medicine and Molecular Imaging. 31, 887-919.
2. Boersma et al. (2005) Journal of Nuclear Medicine. 46, 2035-2050.
3. Aloya, R. et al. (2006) Apoptosis. 11, 2089-2101.
4. Damianovich, M. et al. (2006J European Journal of Nuclear Medicine and Molecular Imaging. 33, 281-291.
5. WO 2005/067388.
6. Neuss, M. et al. (2001). Cardiovascular Drugs and Therapy. 15, 507-523.
7. Lee, D. et al. (2000) Journal of Biological Chemistry. 275, 16007-16014.
8. Lee, D. et al. (2001) Journal of Medicinal Chemistry. 44, 2015-2026.
9. Chu, W. et al. (2005) Journal of Medicinal Chemistry. 48, 7637-7647.
10. Chu, W. et al. (2007) Journal of Medicinal Chemistry. 50, 3751-3755.
11. WO 2005/053752.
12. Faust, A. et al. (2007) Quarterly Journal of Nuclear Medicine and Molecular Imaging. 51, 67-71.
13. Kopka, К. et al. (2006) Journal of Medicinal Chemistry. 49, 6704-6715.
14. Zhou, D. et al. (2006) Bioorganic and Medicinal Chemistry Letters. 16, 5041-5046.
15. WO 2006/067376.
16. Glaser et al. (2007) Bioconjugate Chemistry. 18, 989-993.
17. Wipf, P. et al. (2001) Tetrahedron. 57, 283-296.
18. Barthel, H. et al. (2004) British Journal of Cancer. 90, 2232-2242.
19. Aboagye, E.O. et al. (1997) Biochemical Pharmacology. 54, 1217-1224.
20. Twentyman, P.R. et al. (1980) J Natl. Cancer Inst. 64, 595-604.
21. Workman, P., et al. (1998) British Journal of Cancer. 77, 1-10.
22. Wolf, C.R. et al. (1987) Int. J. Cancer. 39, 695-702.
23. Langdon, S.P. et al. (1988) Cancer Research. 48, 6166-6172.
24. Kozaki, К et al. (2000) Cancer Research. 60, 2535-40.
25. Schirrmacher, R. et al. (2008) Tet. Lett. 49, 4824-4827.
26. Marik, J. and Sutcliffe, J. L. (2006) Click for PET: Rapid preparation of [18F]fluoropeptides using CuI catalyzed 1,3-dipolar cycloaddition. Tet. Lett. 47, 6681-6684.
27. Sirion, U. et al. (2007) An efficient F-18 labeling method for PET study: Huisgen 1,3-dipolar cycloaddition of bioactive substances and F-18-labeled compounds. Tet. Lett., 48, 3953-3957.
28. Li, Z, et al. (2007) Click Chemistry for 18F-Labeling of RGD Peptides and microPET Imaging of Tumor Integrin αvβ3 expression. Bioconjugate Chem., 18, 1987-1994.
29. Smith G. et al. (2008) Design, Synthesis and Biological Characterization of a Caspase 3/7 Selective Isatin Labeled with 2-[18F]fluoroethylazide. Journal of Medicinal Chemistry. 51, 8057-8067.
| название | год | авторы | номер документа |
|---|---|---|---|
| МЕЧЕННЫЕ РАДИОАКТИВНЫМ ИЗОТОПОМ КОНЪЮГАТЫ RGD-СОДЕРЖАЩИХ ПЕПТИДОВ И СПОСОБЫ ИХ ПОЛУЧЕНИЯ С ПОМОЩЬЮ CLICK-ХИМИИ | 2005 |
|
RU2419627C2 |
| НОВЫЕ ПРЕДШЕСТВЕННИКИ ПРОИЗВОДНЫХ ГЛУТАМАТА | 2012 |
|
RU2600981C2 |
| СОЕДИНЕНИЯ, ОБЛАДАЮЩИЕ АКТИВНОСТЬЮ АНТАГОНИСТОВ CRTH2 | 2009 |
|
RU2503672C2 |
| ПРОИЗВОДНЫЕ ХИНОЛИН-2-ОНА В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ C-KIT | 2016 |
|
RU2754858C2 |
| 1-СУЛЬФОНИЛ-1,3-ДИГИДРОИНДОЛ-2-ОНЫ, ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ (ВАРИАНТЫ), СПОСОБ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 2003 |
|
RU2259999C2 |
| ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛА, СОДЕРЖАЩИЕ ИХ КОМПОЗИЦИИ, ИХ ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ | 2004 |
|
RU2346938C2 |
| НАФТИЛУКСУСНЫЕ КИСЛОТЫ | 2009 |
|
RU2539185C2 |
| 1,3-ДИОКСО -2,3-ДИГИДРО-1H-ПИРРОЛО[3,4-C]ХИНОЛИНЫ (ВАРИАНТЫ), ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ (ВАРИАНТЫ), СПОСОБ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ) И СПОСОБЫ ЛЕЧЕНИЯ (ВАРИАНТЫ) | 2003 |
|
RU2257385C2 |
| КОНДЕНСИРОВАННЫЕ ГЕТЕРОАРИЛЫ И ИХ ПРИМЕНЕНИЕ | 2011 |
|
RU2552114C2 |
| ОКТАГИДРО КОНДЕНСИРОВАННЫЕ АЗАДЕКАЛИНОВЫЕ МОДУЛЯТОРЫ ГЛЮКОКОРТИКОИДНОГО РЕЦЕПТОРА | 2014 |
|
RU2674983C1 |
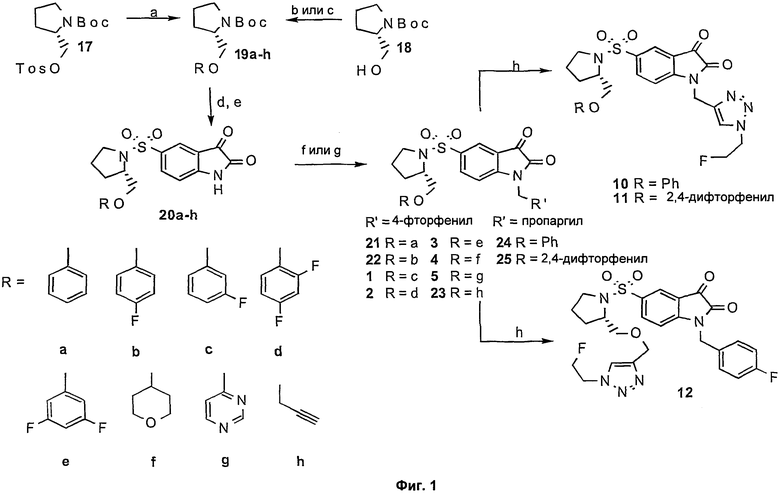
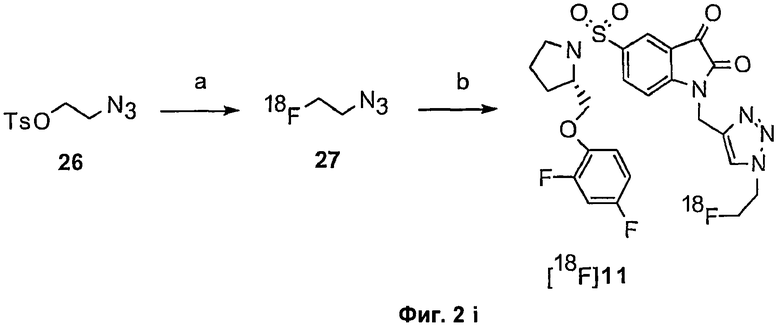
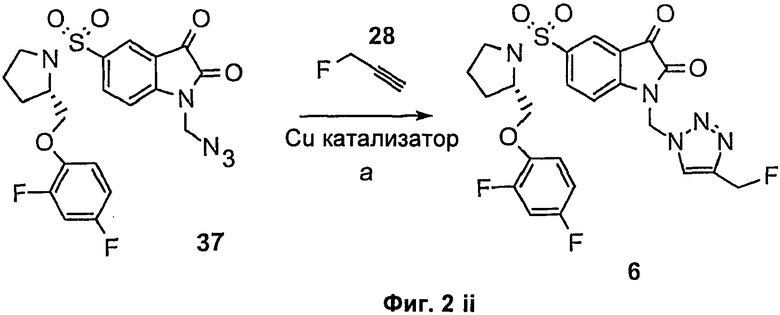
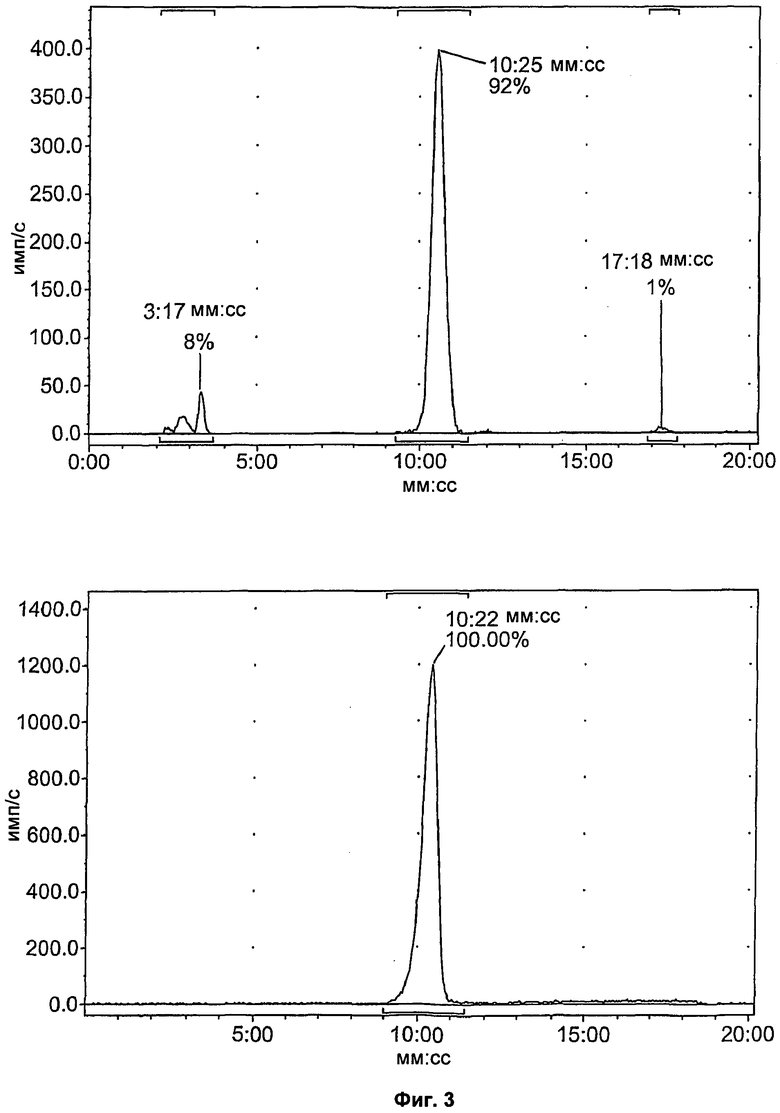
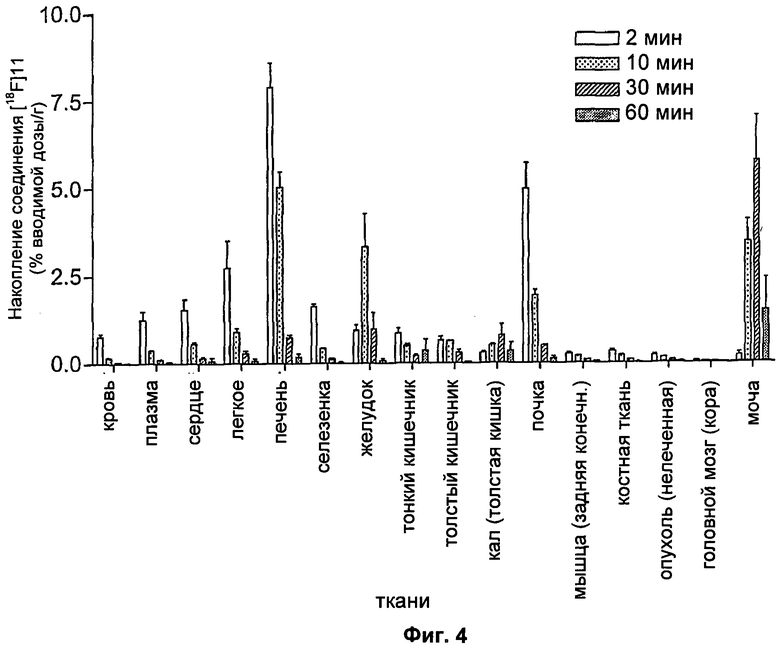
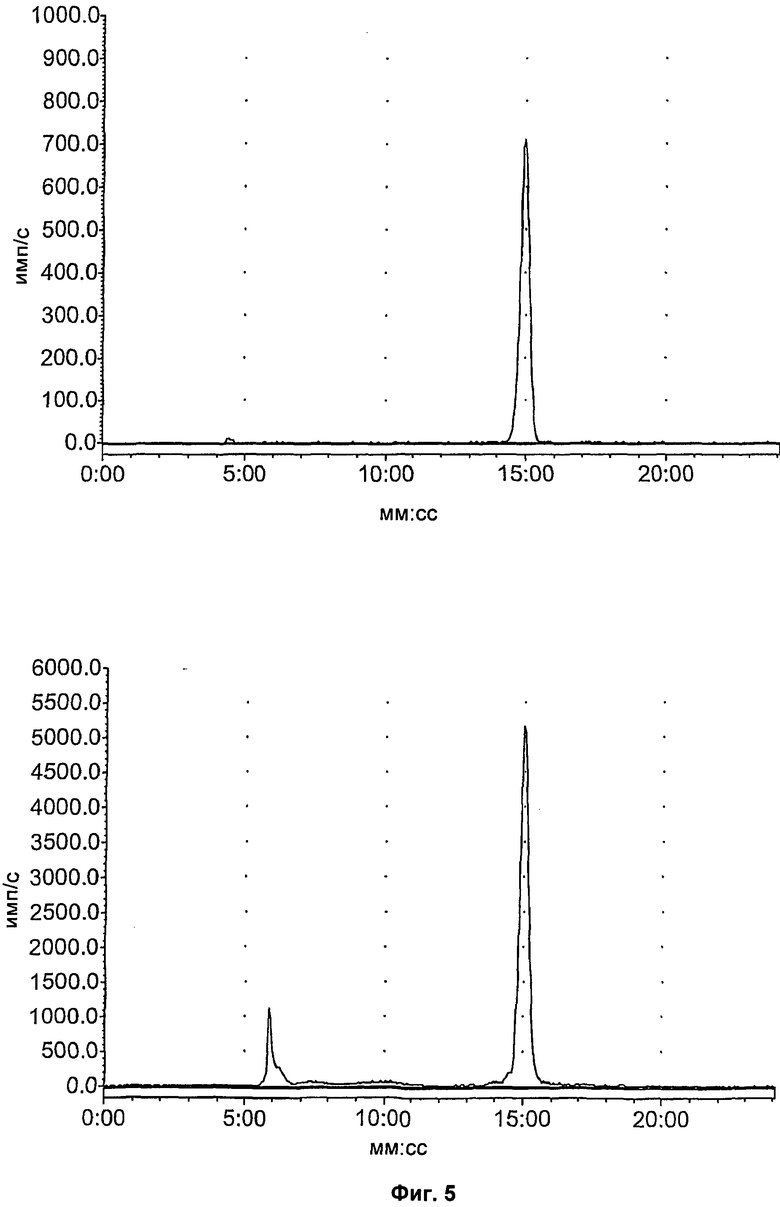
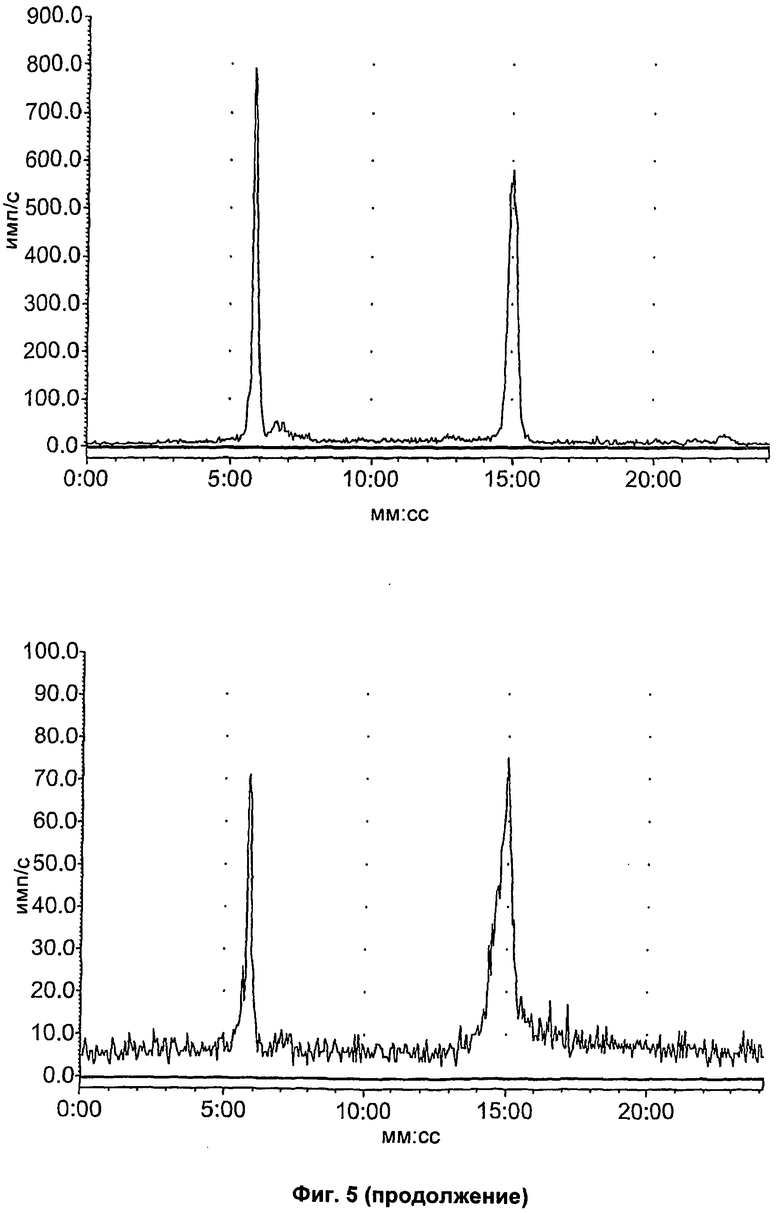
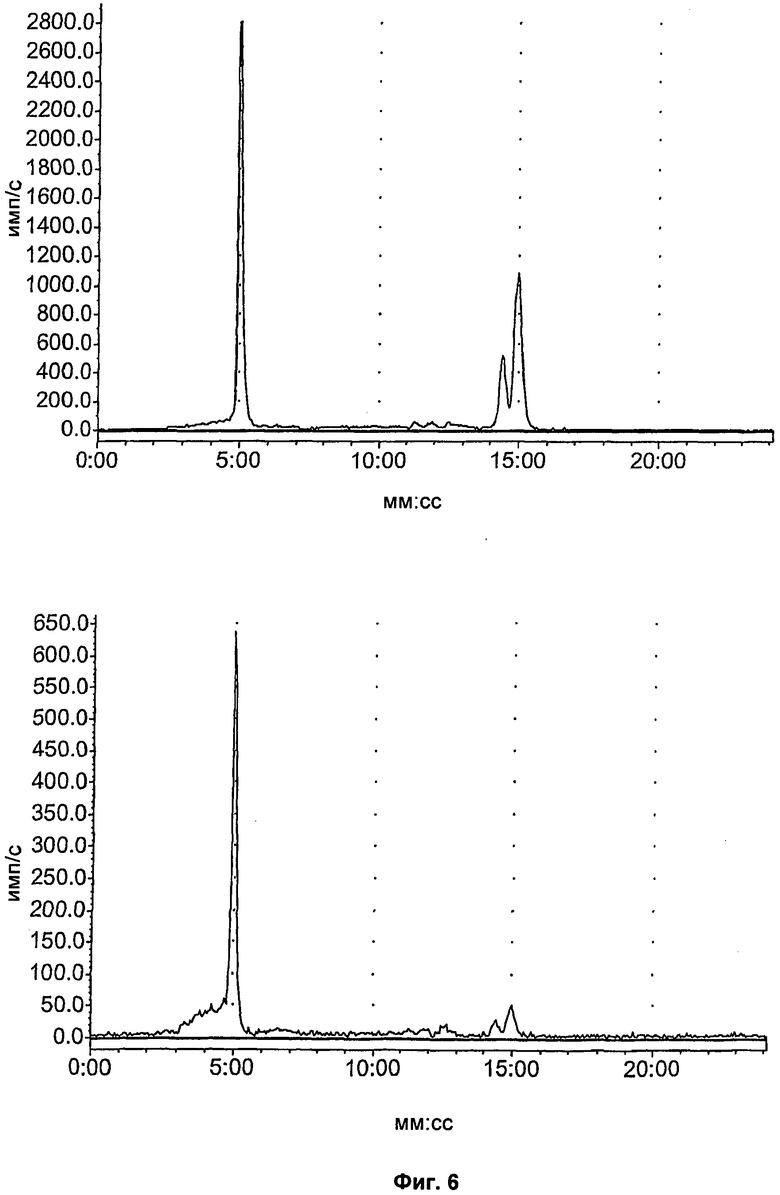
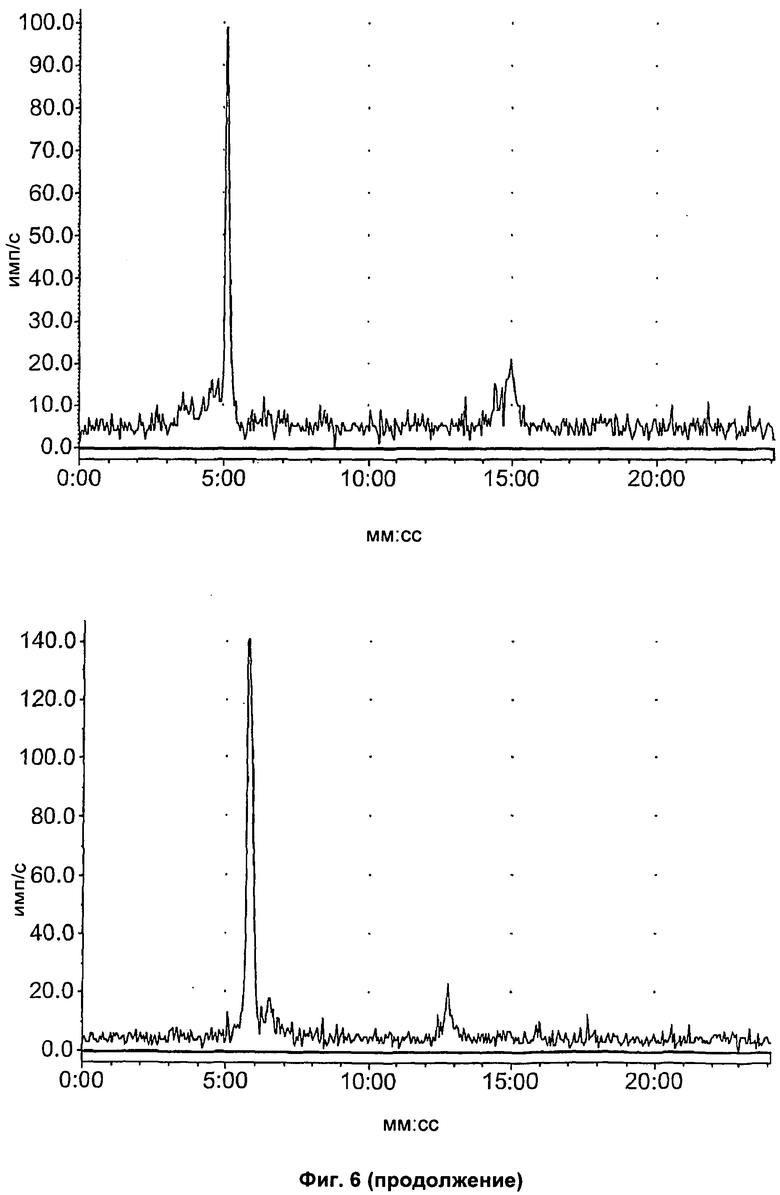
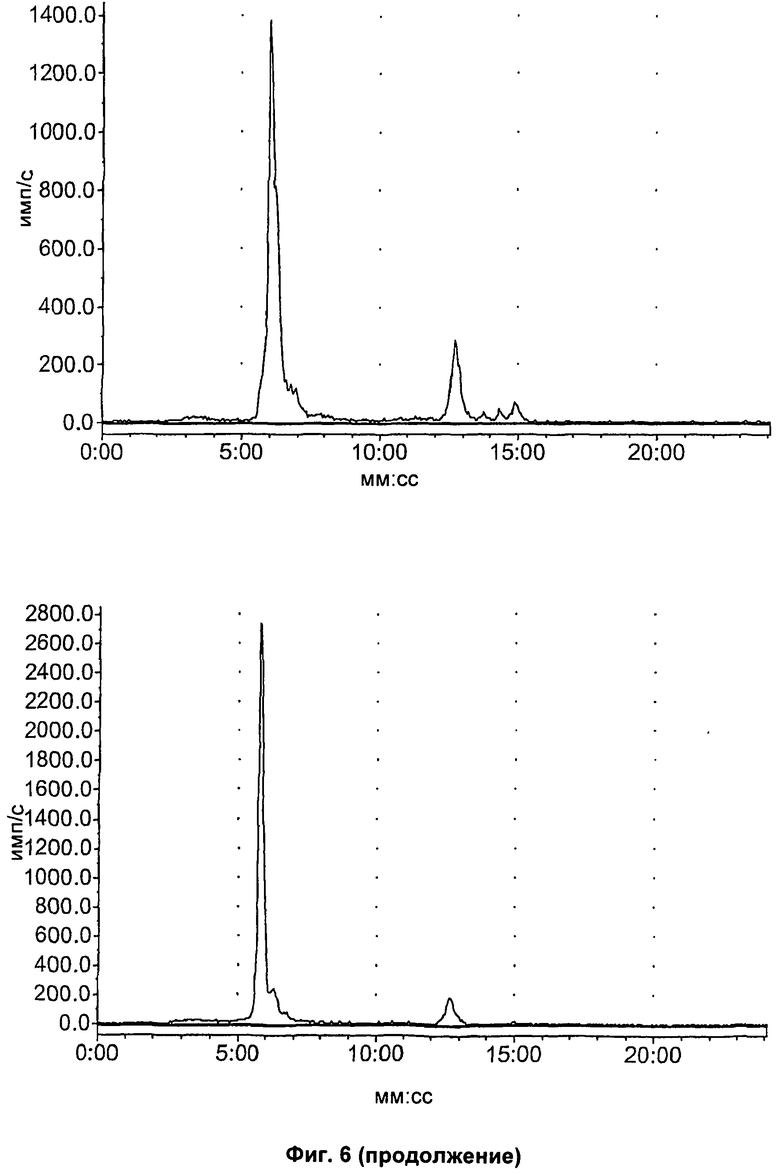
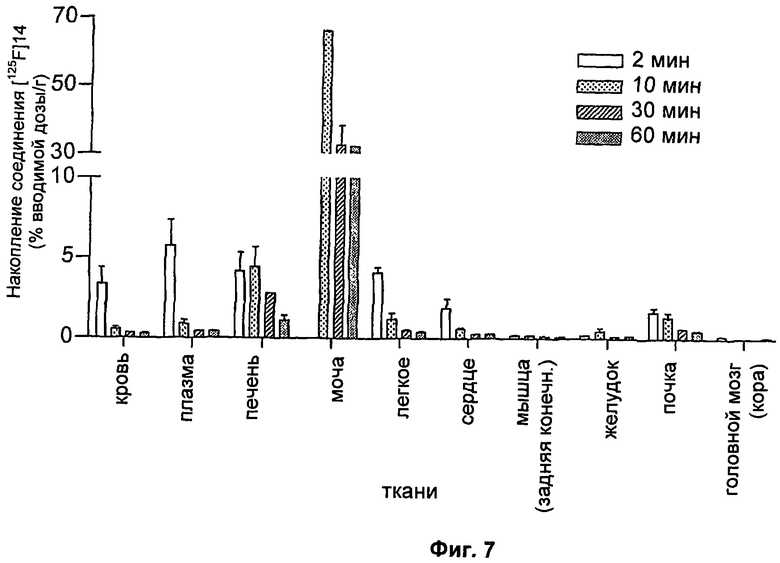
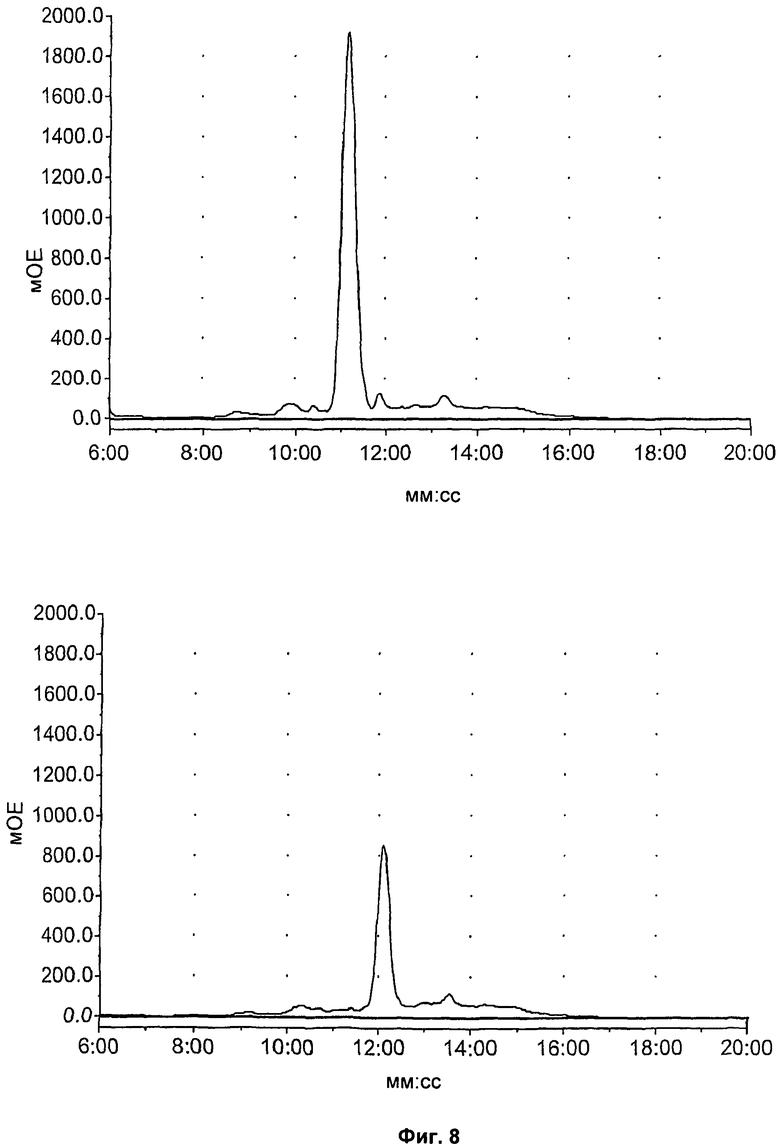
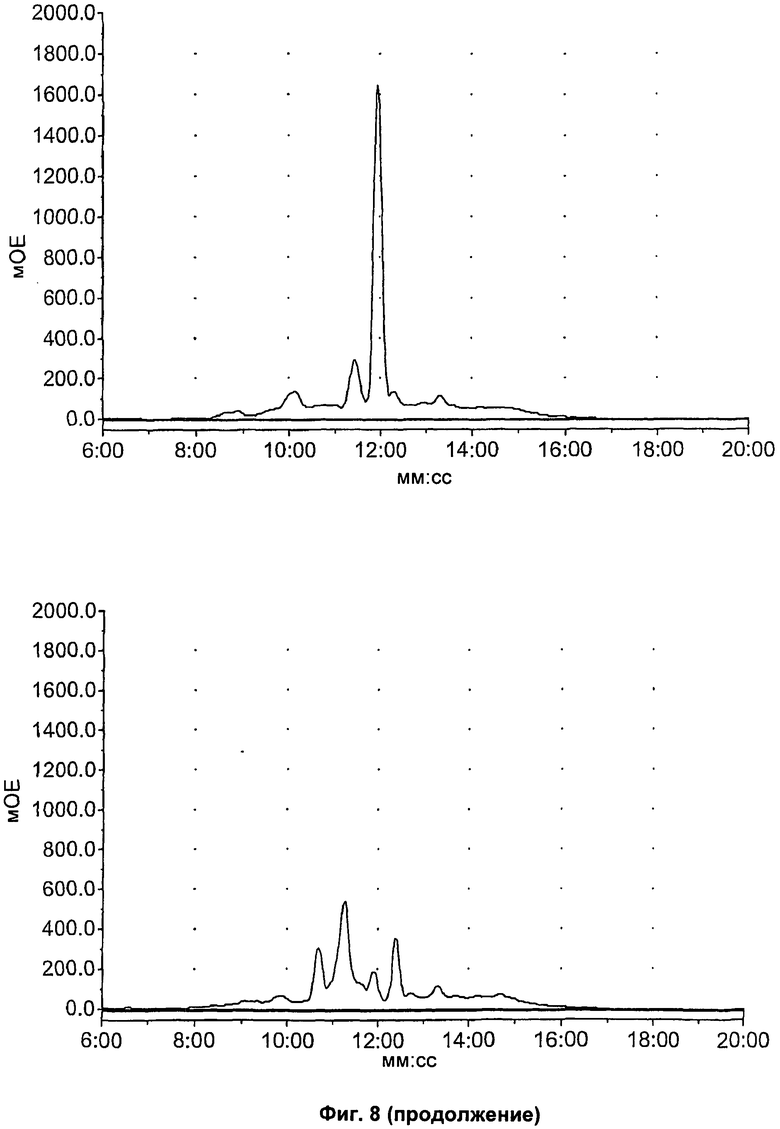

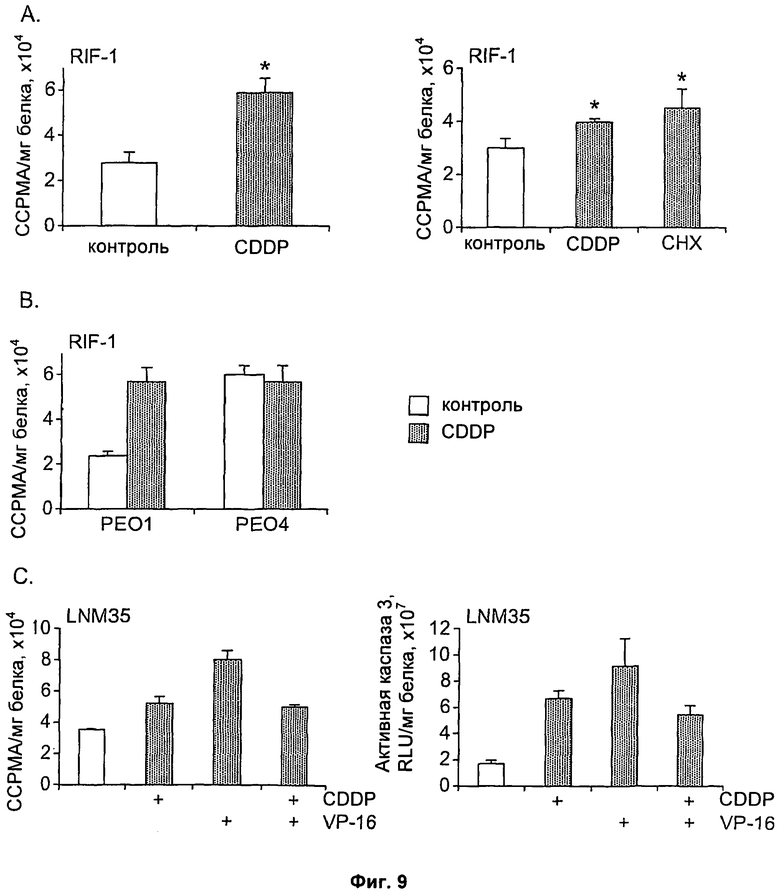
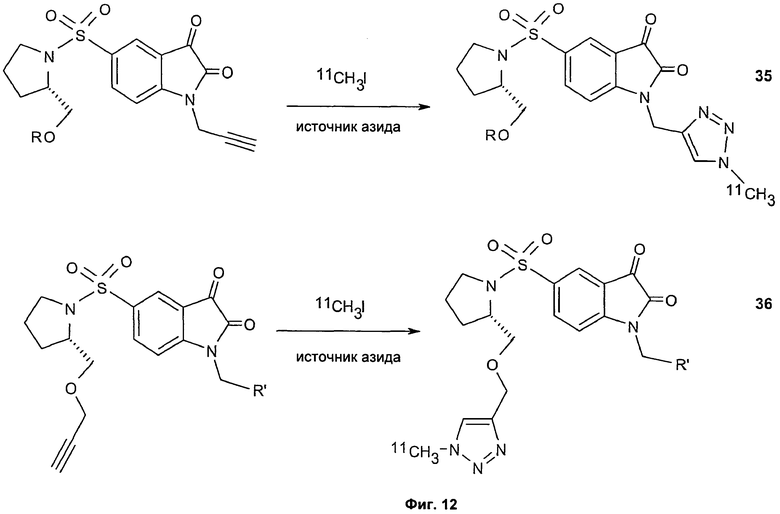
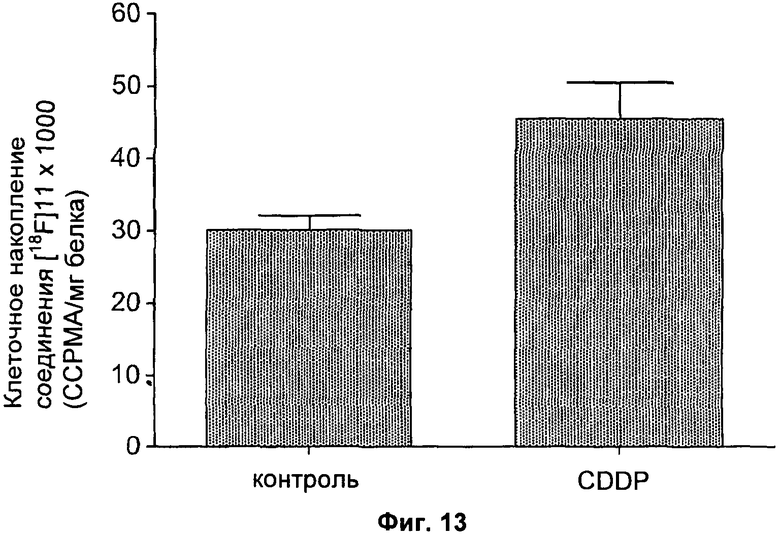
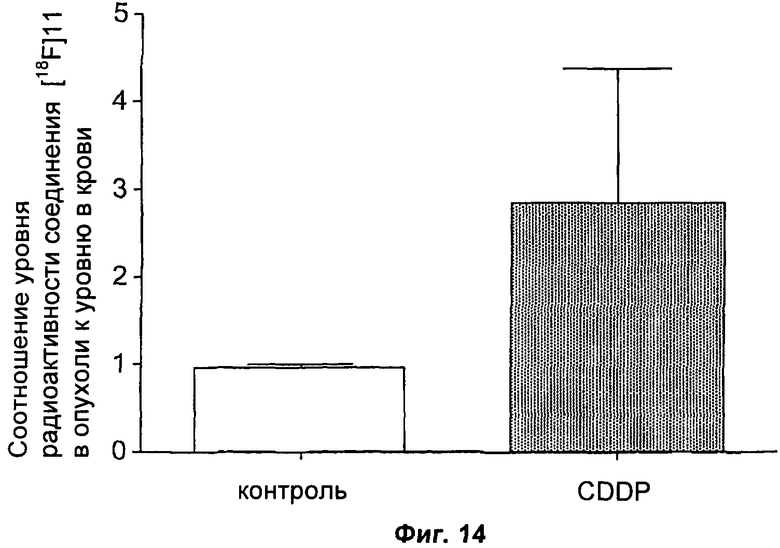
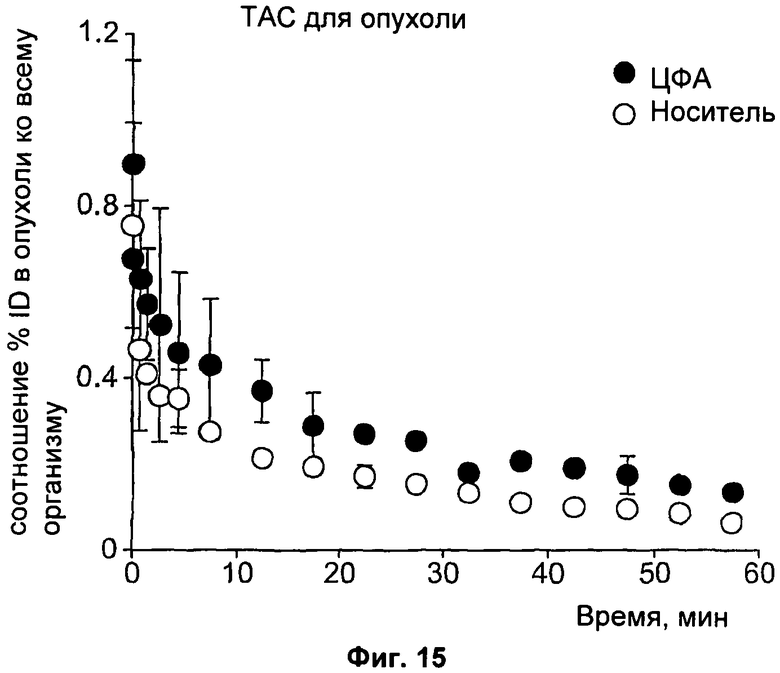
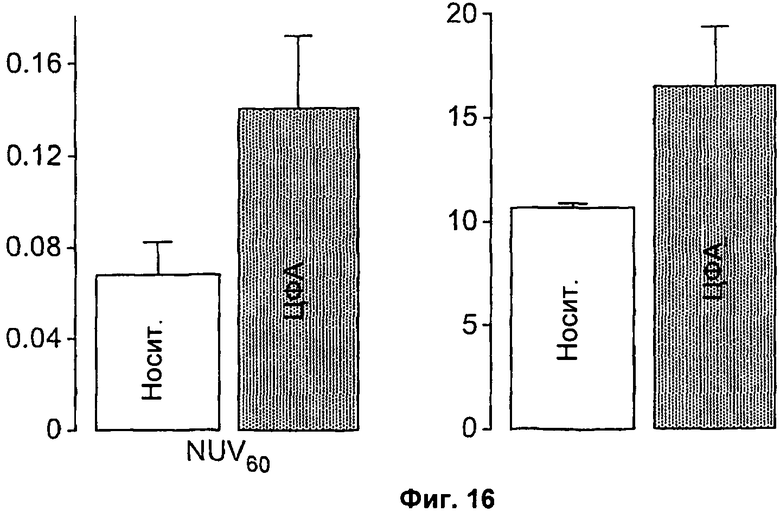
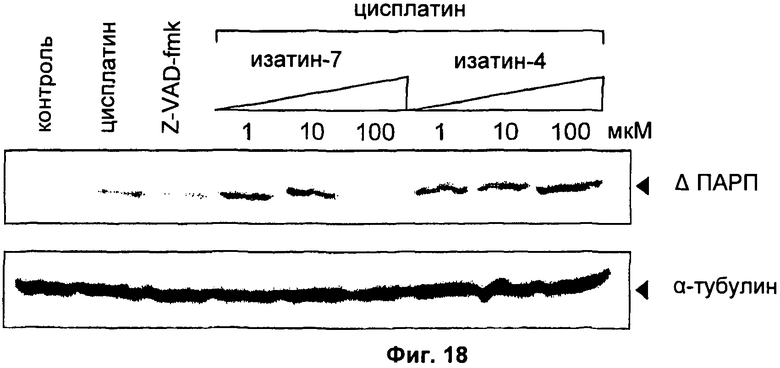
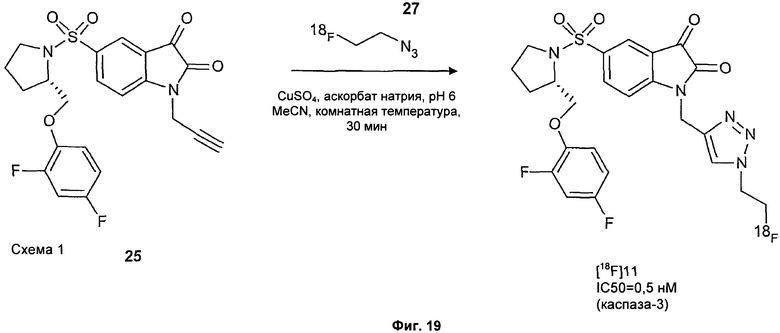
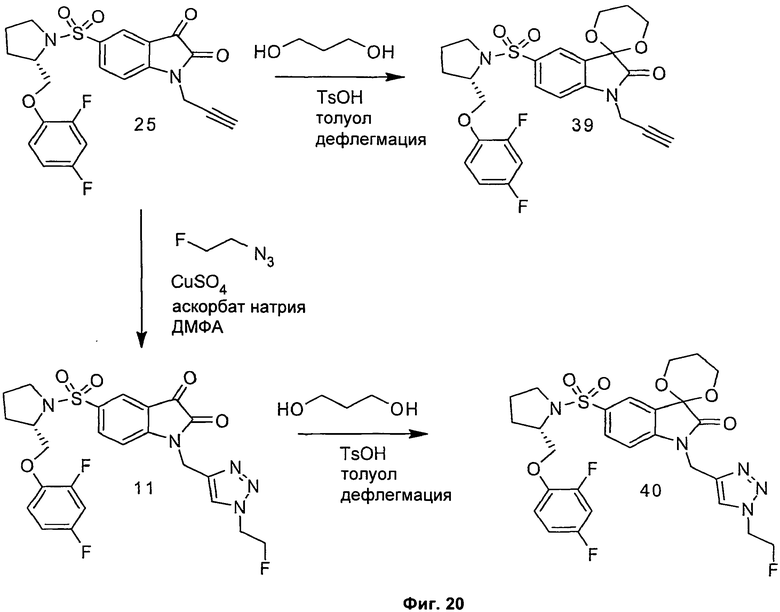
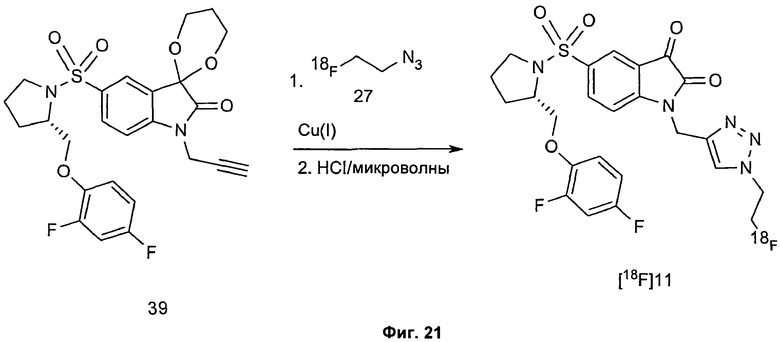
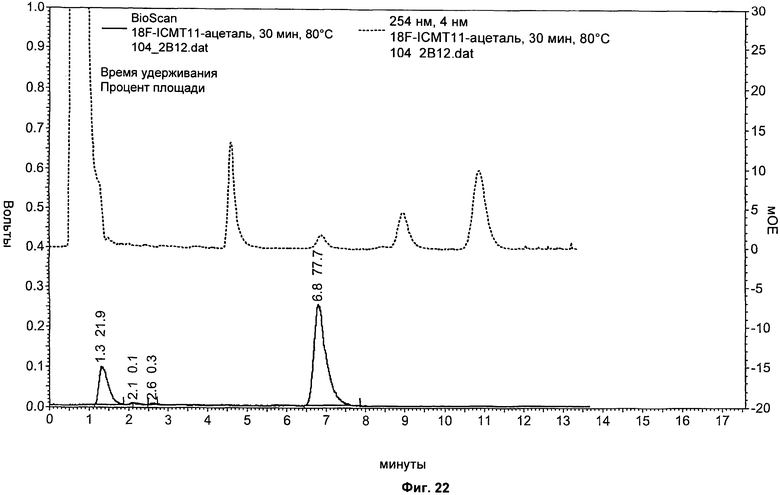
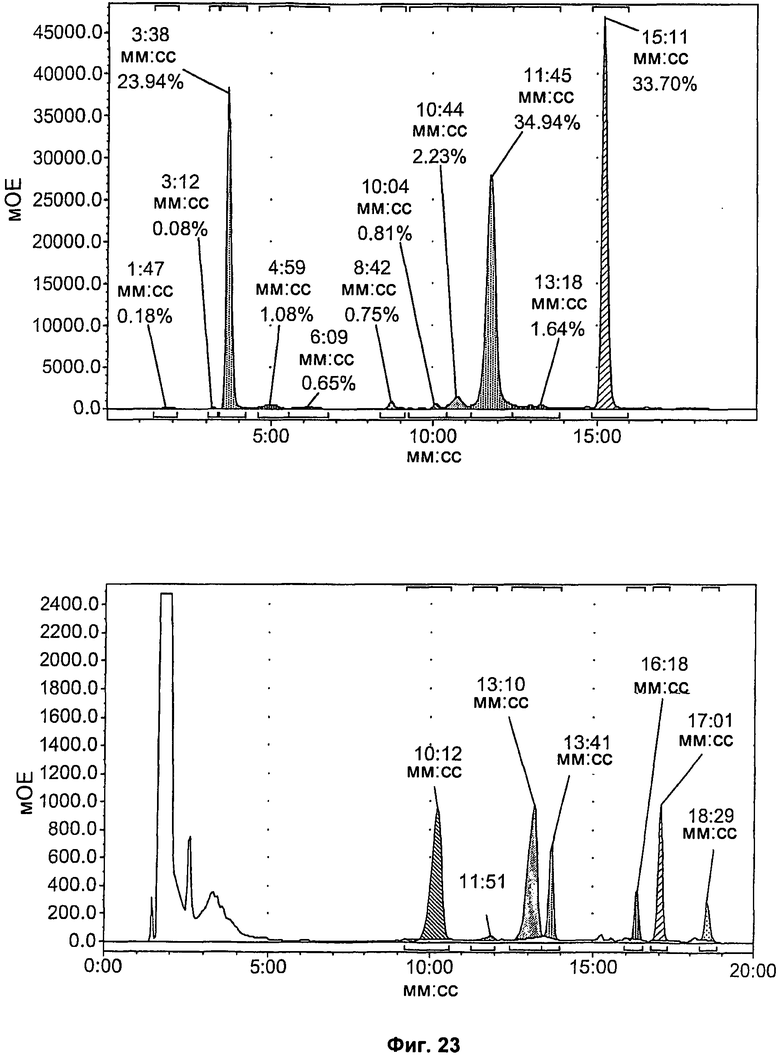
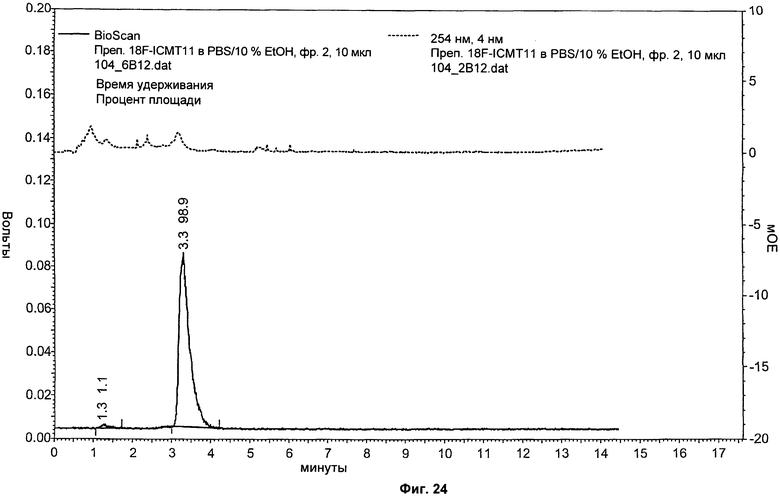
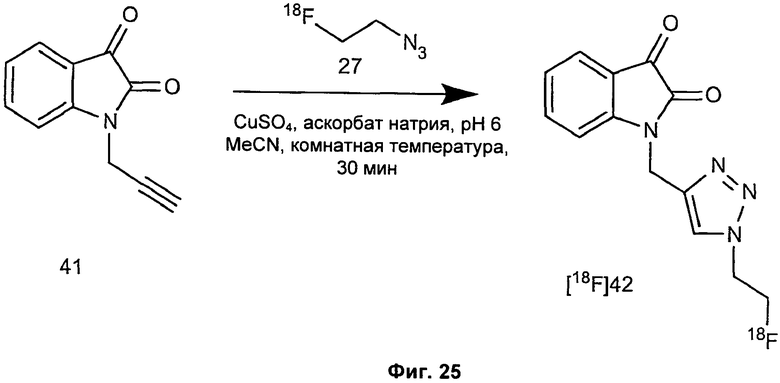
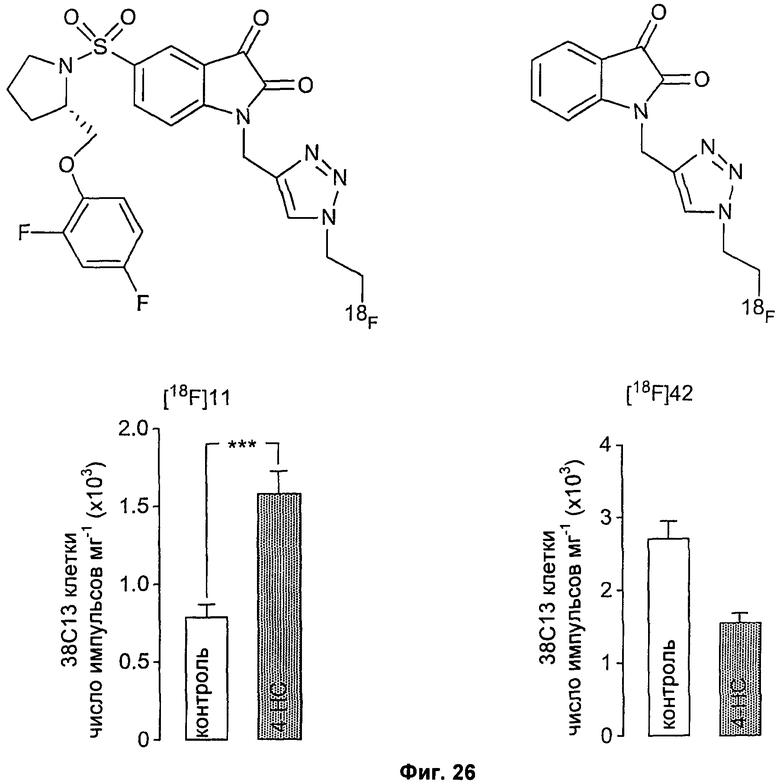
Описываются новые производные изатин-5-сульфонамида общей формулы
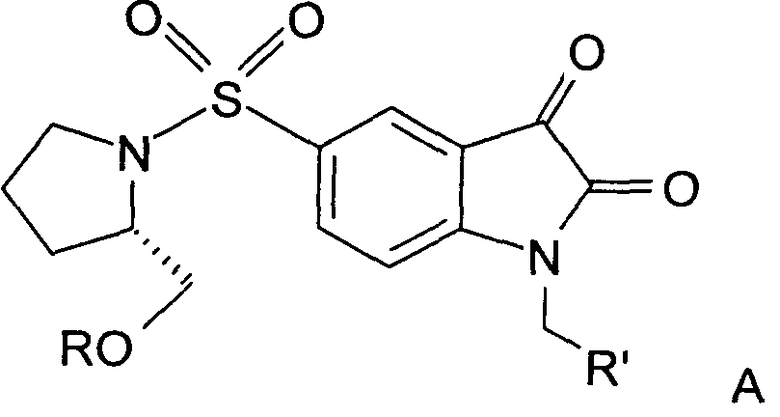
или их физиологически приемлемые соли, где R представляет собой фенил, 3-фторфенил, 2,4-дифторфенил, 3,5-дифторфенил, тетрагидропиранил, диазин или триазолилметил, возможно замещенный одним C1-6алкилом, который дополнительно может быть замещен одним галогеном; R' представляет собой фенил, возможно замещенный одним или двумя галогенами, или триазолил, возможно замещенный одним C1-6алкилом, который дополнительно может быть замещен одним галогеном; причем когда R означает фенил, R' представляет собой возможно замещенный триазолил, фармацевтические композиции, содержащие указанные производные, их применение в качестве агентов молекулярной визуализации, их применение для диагностики или лечения заболеваний или расстройств, связанных с дисрегуляцией апоптоза, способы синтеза указанных производных, способы молекулярной визуализации каспазной активности и апоптоза и способы оценки терапевтического воздействия исследуемого соединения на каспазную активность. 12 н. и 15 з.п. ф-лы, 26 ил., 4 табл., 11 пр.
1. Соединение формулы А:
или его физиологически приемлемая соль, где:
R представляет собой фенил, 3-фторфенил, 2,4-дифторфенил, 3,5-дифторфенил, тетрагидропиран, диазин; или триазолилметил, возможно замещенный одним C1-6алкилом, который также возможно замещен одним галогеном;
R' представляет собой фенил, возможно замещенный одним или двумя галогенами, или триазол, возможно замещенный одним C1-6алкилом,который также возможно замещен одним галогеном;
где, когда R представляет собой фенил, R' представляет собой возможно замещенный триазол.
2. Соединение по п.1, где заместитель галоген представляет собой фтор.
3. Соединение по п.1, где возможно замещенный триазол замещен C1-4фторалкилом, который предпочтительно представляет собой 2-фторэтил.
4. Соединение по п.1, где R выбран из группы, включающей 3-фторфенил, 2,4-дифторфенил и 3,5-дифторфенил, и R' представляет собой 2-фторэтил-замещенный триазол.
5. Соединение по п.1, где R и R' являются такими, как определено ниже:
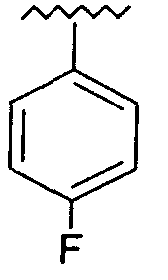
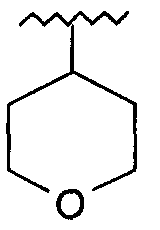
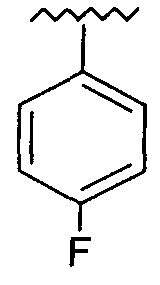
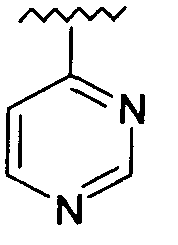
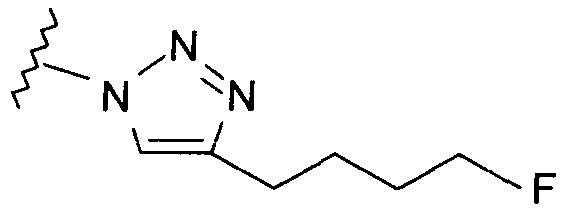
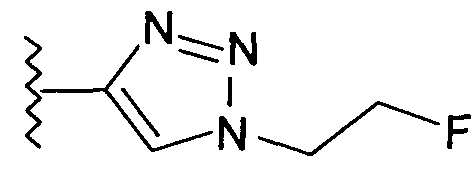
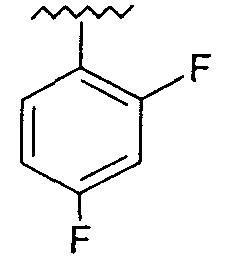
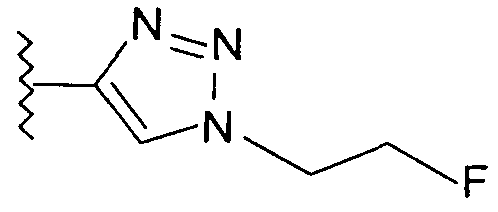
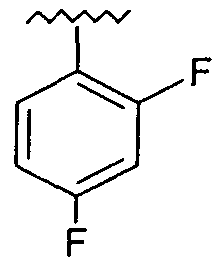
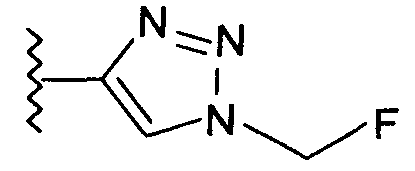
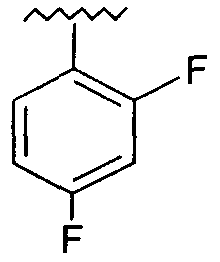
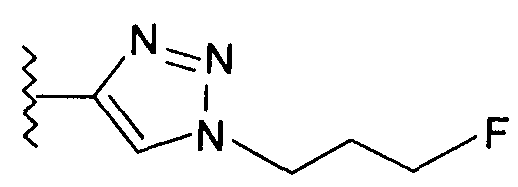
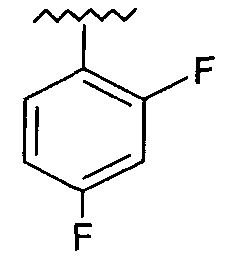
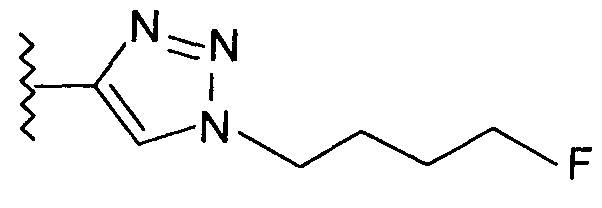
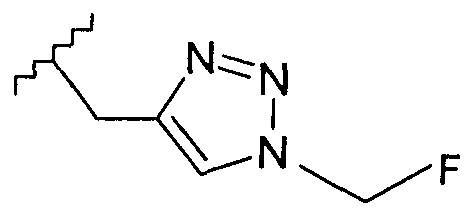
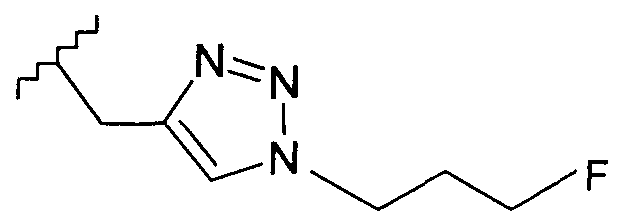
6. Соединение по любому из пп.1-5, дополнительно содержащее визуализирующую группировку, предпочтительно представляющую собой радиоактивную метку, которая предпочтительно представляет собой 18F.
7. Соединение по п.6, представляющее собой соединение формулы [18F]11, формулы 35 или формулы 36:
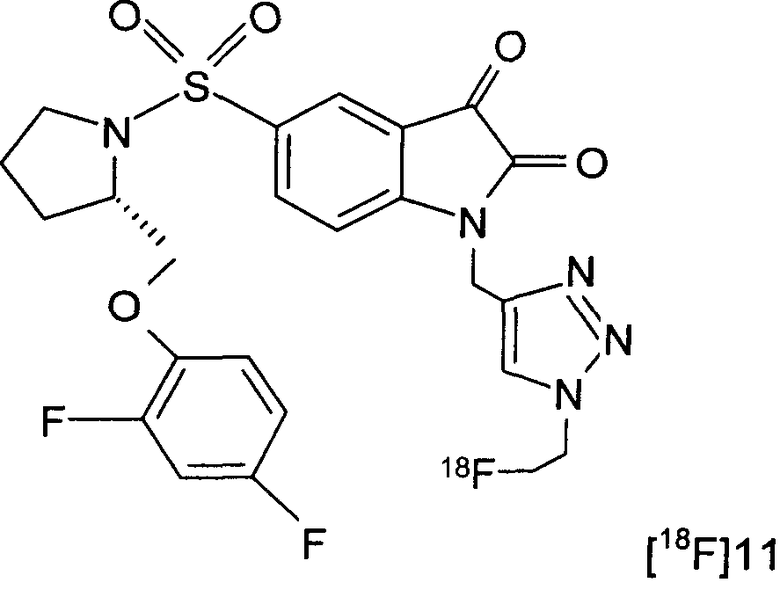 ,
,
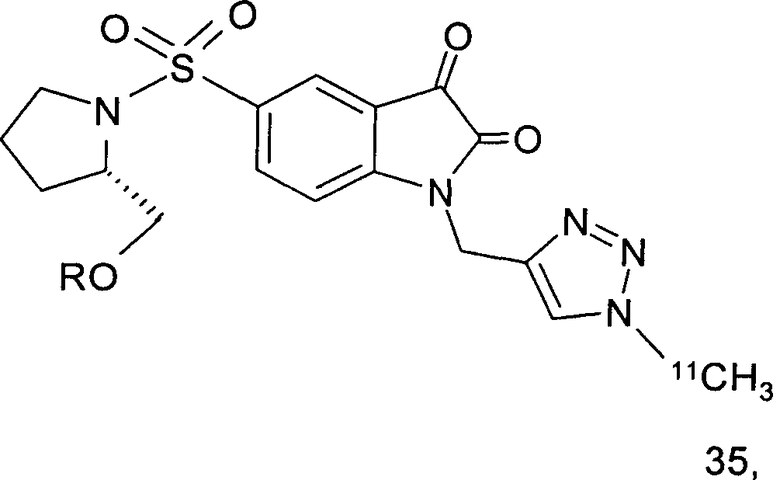
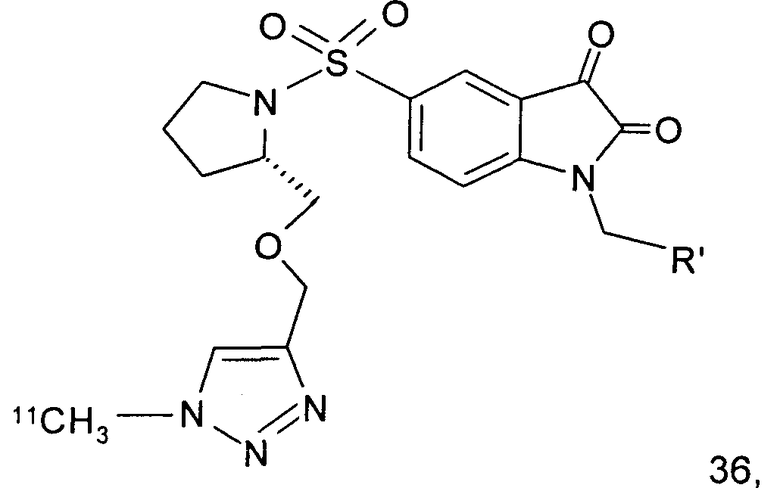
где R и R' являются такими, как определено в любом из пп.1-5.
8. Фармацевтическая композиция, ингибирующая каспазную активность, содержащая соединение по любому из пп.1-5 и возможно один или более чем один фармацевтически приемлемый эксципиент, носитель или разбавитель, где указанное соединение предпочтительно состоит по существу из S-энантиомера.
9. Фармацевтическая композиция для применения в качестве агента молекулярной визуализации, содержащая соединение по п.6 и возможно один или более чем один фармацевтически приемлемый эксципиент, носитель или разбавитель, где указанное соединение предпочтительно состоит по существу из S-энантиомера.
10. Соединение по п.6 для применения в качестве агента молекулярной визуализации, предпочтительно предназначенного для визуализации и количественного анализа каспазной активности.
11. Фармацевтическая композиция по п.9 для применения в качестве агента молекулярной визуализации, предназначенного для визуализации и количественного анализа каспазной активности.
12. Способ молекулярной визуализации активности каспазы-3 и/или каспазы-7 в клетках или тканях, включающий стадии:
а) приведения указанных клеток или тканей в контакт с соединением по п.6 или с фармацевтической композицией по п.9;
б) помещения субъекта в область детекции устройства обнаружения; и
в) детектирования указанных соединений у субъекта с помощью указанного устройства обнаружения,
где указанная каспазная активность предпочтительно представляет собой активность каспазы-3.
13. Способ визуализации апоптоза in vivo у субъекта, включающий стадии:
а) введения субъекту соединения по п.6 или фармацевтической композиции по п.9;
б) помещения субъекта в область детекции устройства обнаружения; и
в) детектирования указанных соединений у субъекта с помощью указанного устройства обнаружения,
где указанные соединения детектируют у субъекта предпочтительно неинвазивным способом.
14. Способ оценки терапевтического воздействия исследуемого вещества на активность каспазы-3 и/или каспазы-7 в клетках или тканях млекопитающих, включающий стадии:
а) приведения клеток или тканей млекопитающих в контакт с соединением по п.6 или с фармацевтической композицией по п.9;
б) помещения указанных клеток или тканей млекопитающих в область детекции устройства обнаружения;
в) детектирования указанных соединений у субъекта с помощью указанного устройства обнаружения; и
г) повторения стадий (а), (б) и (в),
где указанное исследуемое вещество предпочтительно выбрано из группы, включающей лекарственные средства, которые лечат сердечнососудистое заболевание, неврологические заболевания и ВИЧ-инфекцию, и где указанная каспазная активность предпочтительно представляет собой активность каспазы-3.
15. Способ по п.14, где указанные соединения являются меченными радиоактивными изотопами, и указанное устройство обнаружения представляет собой устройство радиоизотопной диагностики, где указанные меченные радиоактивными изотопами соединения детектируют посредством измерения излучения, испускаемого указанными меченными радиоактивными изотопами соединениями, с использованием устройства радиоизотопной диагностики.
16. Способ по п.14 или 15, где соединение или фармацевтическую композицию приводят в контакт с указанными клетками или тканями млекопитающих до, после или одновременно с указанным исследуемым веществом.
17. Способ по п.14 или 15, где стадии от (а) до (в) повторяют в выбранные промежутки времени, где указанное повторение является эффективным для отслеживания изменений в каспазной активности в течение времени, где первое проведение стадий от (а) до (в) предпочтительно осуществляют на субъекте до введения исследуемого вещества для выполнения первого измерения каспазной активности, затем предпочтительно вводят исследуемое вещество, и второе проведение стадий от (а) до (в) предпочтительно осуществляют на указанном субъекте после введения исследуемого вещества в выбранный промежуток времени для выполнения второго измерения каспазной активности.
18. Способ по п.17, где соединение, вводимое при первом проведении стадии (а), представляет собой соединение по п.8.
19. Соединение по любому из пп.1-5 для применения в медицине, предпочтительно для применения в ингибировании каспазной активности, более предпочтительно для применения в лечении заболеваний или состояний, выбранных из группы, включающей нейродегенеративные заболевания, гематологические заболевания, гепатоцеллюлярную дегенерацию, остеоартрит, СПИД, ишемию и отторжение аллотрансплантата.
20. Композиция по п.8 для применения в медицине, предпочтительно для применения в лечении заболеваний или состояний, выбранных из группы, включающей нейродегенеративные заболевания, гематологические заболевания, гепатоцеллюлярную дегенерацию, остеоартрит, СПИД, ишемию и отторжение аллотрансплантата.
21. Способ диагностики заболевания или расстройства, вызванного избыточным или неприемлемым апоптозом или ассоциированного с ним, включающий стадии:
а) приведения клеток или тканей млекопитающих в контакт с соединением по п.6 или с композицией по п.9;
б) помещения указанных клеток или тканей млекопитающих в область детекции устройства обнаружения; и
в) детектирования указанных соединений с помощью указанного устройства обнаружения для выполнения количественных измерений каспазной активности в указанных клетках или тканях млекопитающих, где активность каспазы-3 и/или каспазы-7 является индикатором уровня апоптоза.
22. Способ по п.21, где указанные заболевания или расстройства выбраны из группы, включающей хроническую сердечную недостаточность, острый инфаркт миокарда, инсульт, нейродегенеративные расстройства, аутоиммунное заболевание, очаговое гематологическое заболевание, очаговый СПИД, ишемию, включая сердечную ишемию, и отторжение трансплантата.
23. Способ синтеза соединения формулы:
включающий взаимодействие соединения формулы:
с 4-фторбензилбромидом с использованием карбоната калия/ДМФА(диметилформамид),
где R представляет собой 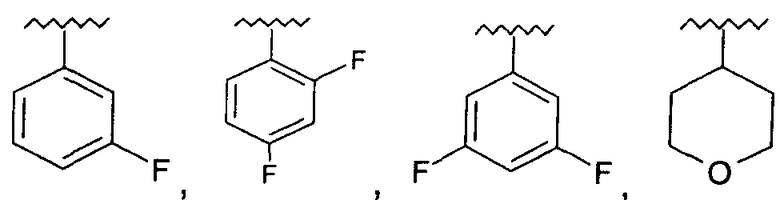 или
или 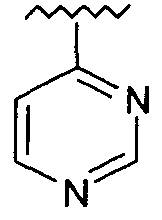
24. Способ синтеза соединения формулы:
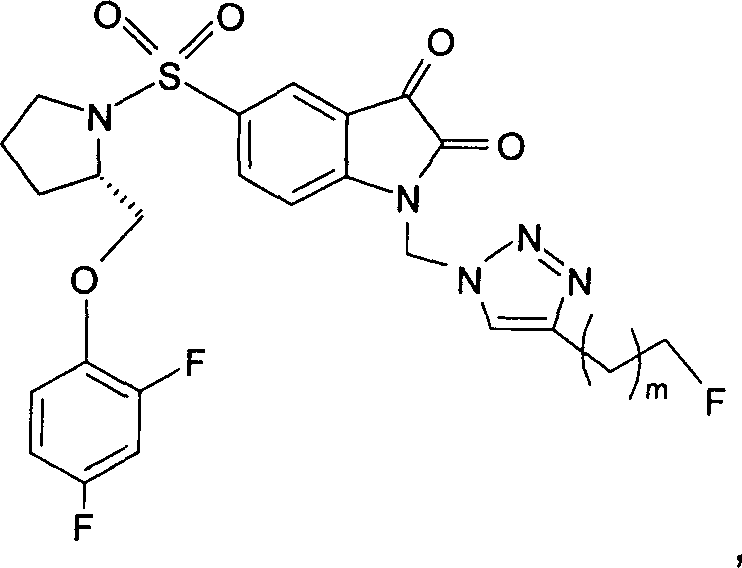
включающий взаимодействие (S)-1-(азидометил)-5-(2-(2,4-дифторфеноксиметил)-пирролидин-1-сульфонил)изатина с соединением формулы: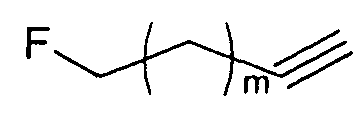 ,
,
где m означает целое число от 0 до 3.
25. Способ синтеза соединения формулы:
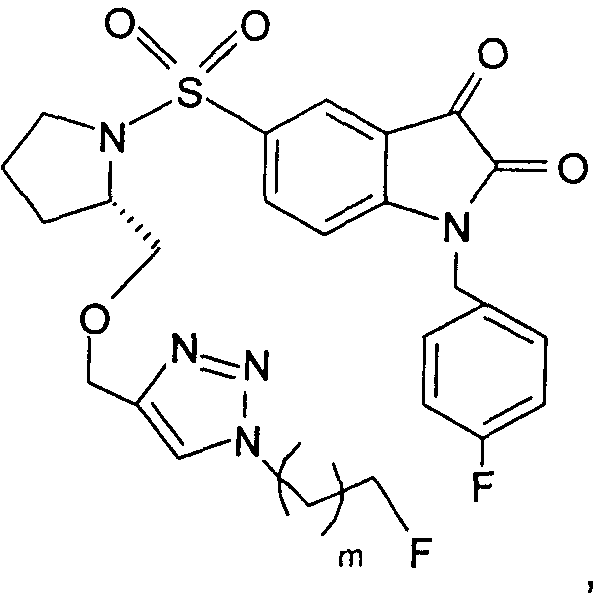
включающий катализируемое медью взаимодействие (S)-1-(4-фторбензил)-5-(2-(2-пропинилоксиметил)-пирролидин-1-сульфонил)изатина с соединением формулы:
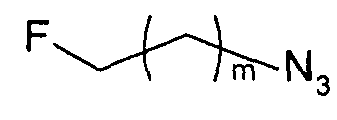 ,
,
где m означает целое число от 0 до 3.
26. Способ синтеза соединения формулы:
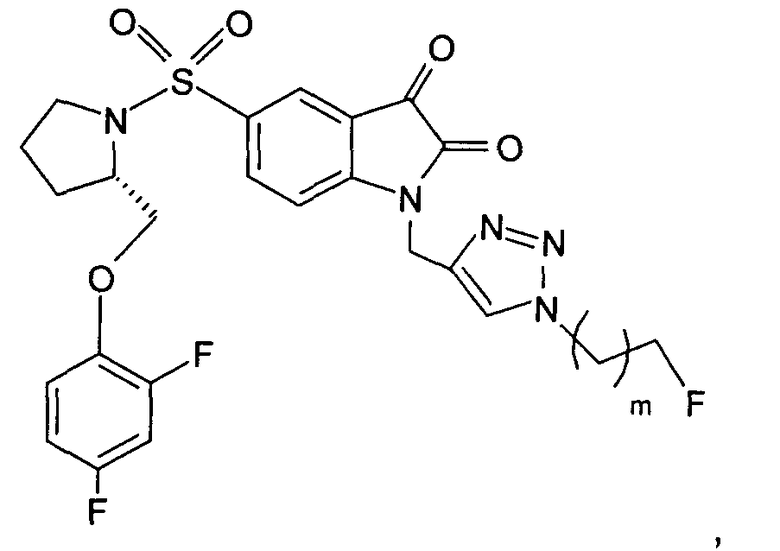
включающий катализируемое медью взаимодействие (S)-1-(2-пропинил)-5-(2-(2,4-дифторфенокси-метил)-пирролидин-1-сульфонил)изатина с соединением формулы:
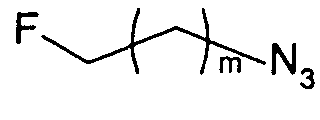 ,
,
где m означает целое число от 0 до 3.
27. Способ синтеза соединения формулы:
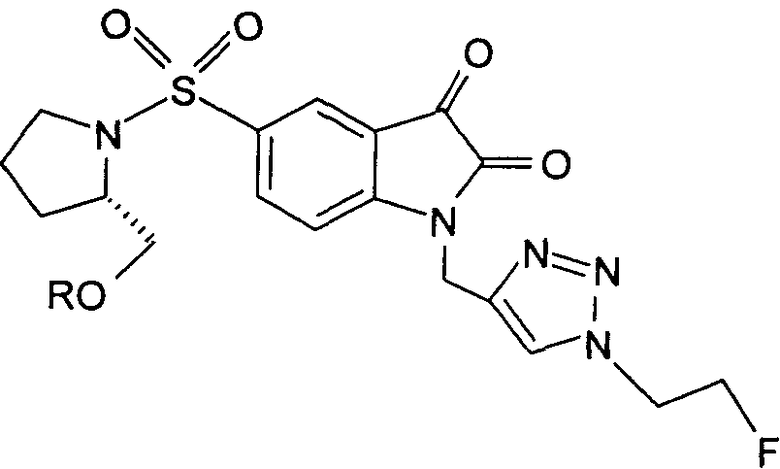 ,
,
включающий катализируемое медью взаимодействие соединения формулы:
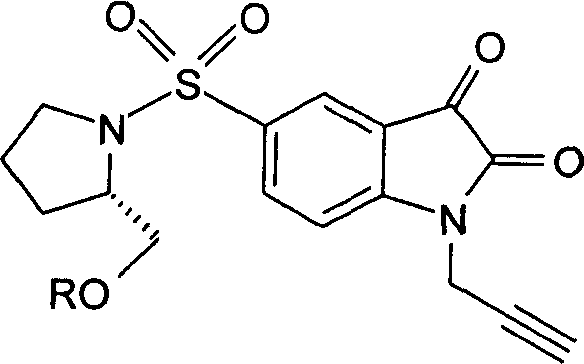
с 2-фторэтилазидом,
где R представляет собой 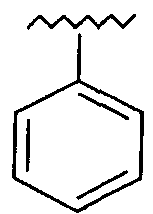 или
или 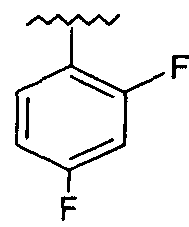 .
.
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |

