Настоящее изобретение относится к способу отделения ацетонитрила от воды, где поток 81, который содержит, по меньшей мере, 95 мас.% ацетонитрила и воды с массовым отношением ацетонитрил:вода более 1, смешивают с потоком Р, который содержит, по меньшей мере, 95 мас.% С3, причем С3 представляет собой либо пропилен, либо смесь пропилена с пропаном, где массовое отношение пропилен:пропан составляет, по меньшей мере, 7:3. Смешанный поток затем подвергают таким условиям температуры и давления, что получают две жидких фазы. Первая жидкая смесь, L1 состоит, по существу, из С3, ацетонитрила и воды. Предпочтительно содержание воды в L1 составляет, самое большее, 12 мас.%, более предпочтительно оно находится в интервале от 1 до 5 мас.%. Вторая жидкая фаза, L2 состоит, по существу, из воды и ацетонитрила с массовым отношением ацетонитрил:вода более 1. Предпочтительно содержание С3 в L2 составляет, самое большее, 5 мас.%. По изобретению, эти две жидкие фазы L1 и L2 подходящим образом отделяют.
По предпочтительному варианту выполнения изобретения настоящее изобретение относится к способу, в котором поток S1 получают из процесса окисления или стадии обработки далее по ходу этого потока, в частности процесса эпоксидирования или стадии обработки далее по ходу этого потока, и еще более предпочтительно из эпоксидирования пропилена пероксидом водорода в присутствии ацетонитрила в качестве растворителя, или стадии обработки далее по ходу этого потока. Поскольку обсуждается этот вариант выполнения изобретения, настоящее изобретение характеризуется несколькими возможностями обеспечения высокоинтегрированного процесса, что касается рециркуляции потоков, получаемых в ходе всего процесса. Таким образом, настоящее изобретение, в особенности, относится к высокоинтегрированному способу производства оксида пропилена.
Международная заявка WO 01/57009 А1 относится к способу каталитического эпоксидирования олефинов. Из реакции эпоксидирования получают поток отработанного газа, который содержит оксид олефина, непрореагировавший олефин и кислород. Этот поток отработанного газа приводят в контакт в поглотительном устройстве с тем же растворителем, что используют в стадии реакции.
Патент США 6350888 В1 относится к способу получения эпоксида, в котором получают смесь продуктов реакции, содержащую эпоксид, растворитель и воду, и необязательно, также не прореагировавшие реагенты. Эту смесь вводят в контакт с экстракционным растворителем так, чтобы получать две отдельные жидкие фазы, где экстракт содержит, по меньшей мере, часть экстракционного растворителя и, по меньшей мере, 10% эпоксида, полученного в реакции эпоксидирования.
Европейская заявка ЕР 1580190 А1 раскрывает способ окисления углерод-углеродной двойной связи соединения А, причем этот способ включает, в качестве первой стадии, окисление углерод-углеродной двойной связи соединения А с использованием пероксида в качестве окислителя и в присутствии титаносиликатного катализатора, чтобы получать окисленную реакционную смесь, в качестве второй стадии - выделение соединения А из указанной реакционной смеси, и в качестве третьей стадии - возвращение отделенного соединения А в первую стадию. В качестве особенно предпочтительных соединений А, раскрыты соединения, которые имеют, по меньшей мере, две или более функциональные группы, причем, по меньшей мере, одна функциональная группа является углерод-углеродной двойной связью. Особенно предпочтительными соединениями А являются простой диаллиловый эфир или аллиловый спирт. Отделение растворителя только кратко упомянуто в заявке ЕР 1580190 А1. Относительно этого отделения растворителя, пример 1 и сравнительный пример 4 заявки ЕР 1580190 А1 описывают отделение через фракционную перегонку, где непрореагировавший простой диаллиловый эфир и метанол возвращают обратно из верхней части дистилляционной колонны. Рециркуляция растворителя не раскрыта в заявке ЕР 1580190 А1.
Европейская заявка ЕР 1602651 А1 описывает способ производства оксида пропилена, где реакционная смесь, полученная в результате эпоксидирования, содержащая оксид пропилена, состоит из двух различных фаз, а именно, водного слоя и масляного слоя. Эти фазы затем разделяют на водный слой и масляный слой. Масляный слой, содержащий органический растворитель, используют в реакции эпоксидирования. Путем разделения реакционной смеси на эти слои, оксид пропилена, который содержался в реакционной смеси, был отделен из водного слоя в органический слой. Следовательно, заявка ЕР 1602651 А1 строго ограничена процессами, где органический слой, отделенный от водного слоя, прежде всего, содержит органический растворитель и оксид пропилена. Хотя нитрилы вообще раскрыты в качестве возможных растворителей, ацетонитрил указывается в качестве растворителя, который приводит к однородному реакционному раствору, который был не разделяем. В этом контексте следует отметить, что в сравнительном примере 3 заявки ЕР 1602651 А1 ошибочно указано, что полученный реакционный раствор был однородным и разделяемым, что, конечно, нужно читать как "однородный и не разделяемый".
Европейская заявка ЕР 2014654 А1 описывает способ получения оксида пропилена, где отходящий газовый поток, который получают из реактора, в котором проводят реакцию эпоксидирования, и этот отходящий газовый поток содержит рециркулируемые соединения, такие как пропилен или оксид пропилена, и его вводят в контакт с растворителем, содержащим нитрил. Путем поглощения в этом растворителе повторно используемые соединения могут быть регенерированы. Далее, европейская заявка ЕР 2014654 А1 раскрывает, что, если смешанный растворитель, такой как ацетонитрил и вода, и воду используют в качестве растворителя в реакции эпоксидирования, часть нитрила, используемая в процессе поглощения, может быть регенерирована из смеси нитрил/вода. Явные способы регенерирования указанной части нитрильного растворителя, раскрытые в заявке ЕР 2014654 А1, представляют собой использование молекулярного сита или цеолита в качестве адсорбирующего агента, либо перегонку. На фигуре 2 описан способ, в котором регенерация растворителя включает перегонку, где в верхней части колонны получают ацетонитрил и воду, где эту газовую смесь затем подвергают стадии колебания давления, из которой получают воду с содержанием ацетонитрила менее 1 мас.% и ацетонитрил с содержанием воды менее 1 мас.%.
В общем, задача настоящего изобретения состоит в том, чтобы обеспечить новый способ отделения ацетонитрила от воды.
Другая задача настоящего изобретения состоит в том, чтобы обеспечить эффективную стадию обработки в получении оксида пропилена, где ацетонитрил применяют в качестве растворителя, а пероксид водорода используют в качестве окислителя для эпоксидирования пропилена.
Следующая задача настоящего изобретения состоит в том, чтобы обеспечить высоко экономичный способ получения оксида пропилена, в котором ацетонитрил применяют в качестве растворителя, а пероксид водорода используют в качестве окислителя для эпоксидирования пропилена.
Настоящее изобретение относится к способу отделения ацетонитрила от воды, который включает
(i) обеспечение потока S1, содержащего, по меньшей мере, 95 мас.%, исходя из общего веса S1, ацетонитрила и воды, где массовое отношение ацетонитрил:вода составляет более 1;
(ii) добавление к S1 потока Р, содержащего, по меньшей мере, 95 мас.% С3, исходя из общего веса потока Р, чтобы получить смешанный поток S2, причем С3 представляет собой пропилен, необязательно, смешанный с пропаном с минимальным массовым отношением пропилен:пропан 7:3;
(iii) подвергание S2 действию температуры, самое большее, 92ºС и давления, по меньшей мере, 10 бар, получая первую жидкую фазу L1, состоящую, по существу, из С3, ацетонитрила и воды, и вторую жидкую фазу L2, состоящую, по существу, из воды и ацетонитрила, где массовое отношение ацетонитрил:вода в L2 составляет менее 1;
(iv) отделения L1 от L2.
В общем, допустимо смешать газообразный поток S1 и газообразный поток Р, или жидкий поток S1 и газообразный поток Р, или газообразный поток S1 и жидкий поток Р, или жидкий поток S1 и жидкий поток Р. Предпочтительно что в (ii), жидкий поток Р добавляют к жидкому потоку S1.
Согласно настоящему изобретению, по меньшей мере, 95 мас.% потока S1 состоит из ацетонитрила и воды, где массовое отношение ацетонитрил:вода составляет более 1. Предпочтительно чтобы, по меньшей мере 96 мас.%, более предпочтительно по меньшей мере 97 мас.%, более предпочтительно по меньшей мере 98 мас.%, и еще более предпочтительно по меньшей мере 99 мас.% потока S1 состоит из ацетонитрила и воды, где массовое отношение ацетонитрил:вода составляет более 1.
Поскольку обсуждается весовое отношение ацетонитрил:вода, предпочтительные отношения составляют по меньшей мере 1,5:1, еще более предпочтительно по меньшей мере 2:1. В частности, S1 содержит от 60 до 85 мас.%, предпочтительно от 65 до 80 мас.%, более предпочтительно от 70 до 80 мас.% ацетонитрила и от 10 до 35 мас.%, предпочтительно от 15 до 30 мас.%, более предпочтительно от 15 до 25 мас.% воды, в каждом случае исходя из общего веса S1. Следовательно, согласно особенно предпочтительному варианту выполнения настоящего изобретения, по меньшей мере, 99 мас.% потока S1 состоит из ацетонитрила и воды, и 81 содержит от 70 до 80 мас.% ацетонитрила и от 15 до 25 мас.% воды.
Согласно одному конкретному варианту выполнения настоящего изобретения, поток S1 содержит, по меньшей мере, один гликоль, предпочтительно, по меньшей мере, один пропиленгликоль, такой как монопропиленгликоль, указываемый просто как пропиленгликоль, и/или дипропиленгликоль, и/или трипропиленгликоль. В частности, S1 содержит, по меньшей мере, один гликоль, предпочтительно, по меньшей мере, один пропиленгликоль, если S1 получают из процесса эпоксидирования, в котором пропилен вступает в реакцию с пероксидом водорода в присутствии ацетонитрила в качестве растворителя. Предпочтительно S1 содержит, по меньшей мере, один гликоль, предпочтительно, по меньшей мере, один пропиленгликоль, который, предпочтительно выбран из группы, состоящей из пропиленгликоля, дипропиленгликоля, трипропиленгликоля, и смеси из двух или трех таковых, в количестве 1 мас.% или меньше.
В соответствии со стадией (ii), поток Р, содержащий С3, предпочтительно жидкий поток Р, добавляют к S1. Обычно, по меньшей мере, 95 мас.% потока Р состоит из пропилена или смеси пропилена с пропаном. Если Р содержит такую смесь пропилена и пропана, массовое отношение пропилен:пропан будет, по меньшей мере, 7:3. Следовательно, потоки пропилена могут быть использованы в качестве Р или С3, которые имеют варьирующие содержания пропана. Например, коммерчески доступный пропилен может быть использован в качестве Р или С3, который может быть либо пропиленом для полимерной промышленности, либо пропиленом химической промышленности. Как правило, пропилен, проклассифицированный для полимерной промышленности, будет иметь содержание пропилена от 99 до 99,8 мас.% и содержание пропана от 0,2 до 1 мас.%. Пропилен, проклассифицированный для химической промышленности, обычно будет иметь содержание пропилена от 92 до 98 мас.% и содержание пропана от 2 до 8 мас.%. По предпочтительному варианту выполнения настоящего изобретения используют поток Р, который, по меньшей мере, на 95 мас.% состоит из С3, где С3 представляет собой смесь пропилена и пропана, и содержание С3 относительно пропилена находится в интервале от 92 до 98 мас.%, предпочтительно в интервале от 94 до 97 мас.%, а содержание С3 относительно пропана находится в интервале от 2 до 8 мас.%, предпочтительно в интервале от 3 до 6 мас.%.
Обычно Р добавляют к S1 в (ii) в таком количестве, чтобы массовое отношение С3:ацетонитрил в S2 находилось в интервале от 0,2:1 до 5:1. По предпочтительным вариантам выполнения настоящего изобретения, Р добавляют к S1 в (ii) в таком количестве, чтобы массовое отношение С3:ацетонитрил в S2 находилось в интервале от 0,5:1 до 2:1, более предпочтительно в интервале от 1,0:1 до 1,5:1.
По настоящему изобретению, поток S2 подвергают действию (iii) таких условий температуры и давления, чтобы получить две жидкие фазы L1 и L2.
Было обнаружено, что благоприятно для разделения на эти фазы L1 и L2 подвергать поток S2 такой низкой температуре, какой возможно, с условием, что температура остается пригодной; например, температура не должна быть настолько низкой, чтобы формировалась твердая фаза, такая как лед. Как правило, S2 будут приводить к температуре, самое большее, 92ºС.По настоящему изобретению, предпочтительно приводить S2 к температуре в интервале от 5 до 90ºС, предпочтительно от 10 до 80ºС, более предпочтительно от 15 до 70ºС, еще более предпочтительно от 20 до 60ºС, и наиболее предпочтительно от 25 до 45ºС. Соответственно, S2 обычно подвергают действию давления, по меньшей мере, 10 бар, для того чтобы S2 присутствовал по существу или полностью в своей жидкой форме. Термин "по существу в своей жидкой форме", как его используют в контексте настоящего изобретения, относится к варианту выполнения изобретения, по которому, по меньшей мере, 95 мас.%, более предпочтительно, по меньшей мере, 99 мас.%, и еще более предпочтительно, по меньшей мере, 99,9 мас.% S2 присутствует в жидкой форме после подвергания указанным выше температурам и давлениям. По настоящему изобретению, предпочтительно подвергать S2 действию давления, по меньшей мере, 15 бар, более предпочтительно давлению в интервале от 15 до 50 бар, более предпочтительно от 15 до 40 бар, еще более предпочтительно от 15 до 30 бар, и наиболее предпочтительно от 15 до 25 бар.
Доведение S2 до указанной выше температуры может быть выполнено любым подходящим способом. По настоящему изобретению, предпочтительным является использование одной или нескольких подходящих теплопередающих сред, например воды, в соответствующем устройстве, например кожухотрубном теплообменнике.
Подвергание S2 действию указанного выше давления может быть выполнено любым подходящим способом. Согласно настоящему изобретению, предпочтительно использование соответствующего насоса, например, центробежного насоса или насоса с радиальным расположением цилиндров.
По настоящему изобретению, температуры и давления, которые описаны выше, позволяют существовать двум отдельным жидким фазам L1 и L2. В соответствии со стадией (iv), две отдельные жидкие фазы L1 и L2 подходящим образом отделены друг от друга. Обычно для этого отделения двух жидких фаз может быть применен любой возможный метод. Возможные устройства, используемые для отделения L1 от L2, представляют собой, например, гравитационные отстойники, отстойники с коалесцирующими средствами, такие как сливы с затвором, сепаратор с наклонными пластинами, коагуляторы, такие как, например, маты, слои, слои из пористых или волокнистых твердых частиц, или мембраны, оборудование для постадийного смешения и отстаивания, гидроциклоны, центрифуги, подходящие колонны с подачей энергии или без нее. Обычно возможен порционный режим или непрерывный режим. Предпочтительно используют гравитационный отстойник, такой как вертикальный или горизонтальный гравитационный отстойник. Еще более предпочтительно используют горизонтальный гравитационный отстойник. Было обнаружено, что вследствие значительной разности плотности и низким вязкостям, достижимым для жидких фаз L1 и L2 в соответствии со способом по изобретению, может быть использован гравитационный отстойник, один из самых простых приборов.
По одному варианту выполнения настоящего изобретения, может быть добавлен, по меньшей мере, один агент, улучшающий разделение жидких фаз, такой как, по меньшей мере, один подходящий агент антиэмульгирования, деэмульгирования или разрушения эмульсии. Обычно можно добавлять указанный агент, улучшающий разделение жидких фаз, к S1 в (i) или к S2 в (ii) или к S1 в (i) и к S2 в (ii). Количество добавляемого агента, улучшающего разделение жидких фаз, составляет, предпочтительно самое большее, 1 мас.%, исходя из общего веса S1 и/или S2. Как правило, количество будет менее 1 мас.%, такое как, например, ниже 0,5 мас.% или ниже 0,1 мас.%. Подходящие агенты будут известны специалистам. Ссылка делается, например, на публикацию K.J.Lissant, Making and Breaking Emulsions, Res. Lab., Petrolite Corp., St. Louis, Missouri, USA, в: K.J.Lissant (ed.), Emulsion Technology (1974), chapter 2, pp.111-124, Dekker, New York; и S.E.Taylor, Chem. Ind. (1992), pp.770-773.
По другому варианту выполнения настоящего изобретения, отделение L1 от L2 проводят без добавления такого дополнительного агента, улучшающего разделение жидких фаз.
Из процесса по настоящему изобретению получают жидкую фазу L1, которая состоит, по существу, из С3, ацетонитрила и воды. Термин "состоит, по существу, из С3, ацетонитрила и воды", как он используется в этом контексте настоящего изобретения, относится к жидкой фазе L1, где, по меньшей мере, 90 мас.% L1 состоит из С3, ацетонитрила и воды.
По предпочтительному варианту выполнения изобретения, по меньшей мере, 95 мас.%, более предпочтительно, по меньшей мере, 98 мас.%, и еще более предпочтительно, по меньшей мере, 99 мас.% L1 состоит из С3, ацетонитрила и воды, где содержание воды в L1 составляет менее 10 мас.%, предпочтительно самое большее, 9 мас.%, более предпочтительно самое большее, 5 мас.%, исходя из общего веса L1. Более предпочтительно содержание воды в L1 находится в интервале от 1 до 9 мас.%, более предпочтительно в интервале от 1 до 5 мас.%, исходя из общего веса L1.
Обычно жидкая фаза L1 может быть использована в любом подходящем процессе. По предпочтительному варианту выполнения настоящего изобретения, жидкая фаза L1 может быть использована в качестве потока, который пропускают в реакцию окисления, где ацетонитрил используют в качестве растворителя и где окисляют пропилен. Еще более предпочтительно жидкая фаза L1 может быть использована в качестве потока, который подводят в реакцию эпоксидирования, где ацетонитрил используют в качестве растворителя и где пропилен окисляют пероксидом водорода, чтобы получать оксид пропилена.
По предпочтительному варианту выполнения настоящего изобретения, жидкую фазу L1 перед использованием в подходящем процессе подвергают, по меньшей мере, одной дополнительной стадии разделения. Предпочтительно эта, по меньшей мере, одна дополнительная стадия разделения служит для отделения С3, предпочтительно части С3 от ацетонитрила.
Возможный способ представляет собой, например, испарение жидкой фазы L1 декомпрессией при подходящем давлении. Предпочтительно температуру жидкой фазы сохраняют, по существу, постоянной в ходе декомпрессии. В результате этой декомпрессии получают С3 в газообразной форме. После этого возможно рециркулировать, по меньшей мере, часть этого газообразного потока С3, чтобы после подходящего сжатия получать жидкий поток, в качестве, по меньшей мере, части потока Р в стадии (ii) способа по изобретению.
Предпочтительный способ указанного отделения С3 от ацетонитрила, по меньшей мере, в одной дополнительной стадии отделения содержит подвергание жидкой фазы L1 стадии перегонки. Предпочтительно перегонку проводят подходящим образом так, чтобы получать поток TL1, который содержит, по меньшей мере, 90 мас.%, предпочтительно, по меньшей мере, 95 мас.% С3, исходя из общего веса TL1.
Также предпочтительно для указанного отделения, что получают 95 мас.% потока BL1, предпочтительно, по меньшей мере, 98 мас.%, который состоит из С3, ацетонитрила и воды, где содержание С3 в BL1 находится в интервале от 7 до 18 мас.%, предпочтительно от 10 до 15 мас.%, в каждом случае исходя из общего веса BL1.
Обычно эта перегонка L1 может быть проведена по любому подходящему способу. Например, могут быть использованы одна, две или несколько перегонных колонн при условии, что получают указанные выше потоки TL1 и BL1. Предпочтительно в указанной стадии перегонки используют одну перегонную колонну. Более предпочтительно указанную перегонку проводят в точке росы в верхней части указанной перегонной колонны, по меньшей мере, 40ºС, предпочтительно в интервале от 40 до 80ºС, более предпочтительно в интервале от 40 до 70ºС. Как правило, число теоретических тарелок находится в интервале от 10 до 20. Типичные флегмовые числа находятся в интервале от 0,01 до 0,2, такие как, например, от 0,05 до 0,15.
Хотя обычно можно использовать жидкую фазу L1 как таковую, как описано выше, в предпочтительном варианте выполнения настоящего изобретения L1 подвергается указанной выше стадии разделения, предпочтительно указанной выше стадии перегонки. Обнаружили, что объединение отделения по изобретению L1 от L2 и отделения ниже по ходу потока TL1 от BL1 позволяет создавать высокоинтегрированный способ по настоящему изобретению. С одной стороны, поток TL1 особенно пригоден для рециркуляции в стадию (ii) способа по изобретению в качестве по меньшей мере части Р. Если в дополнение к, по меньшей мере, части TL1, дополнительный С3 добавляют к S1, этот дополнительный источник С3 может быть выбран приемлемым образом. Например, дополнительный С3 может быть добавлен в виде свежего пропилена, например в виде пропилена категории для химической промышленности, содержащего около 95 мас.% пропилена и около 5 мас.% пропана; конечно, возможны все другие подходящие источники дополнительного С3, такие как, например, поток С3, полученный от поставщика со связанной технологической площадки (Verbund site) или тому подобное. Кроме того, обнаружили, что чем больше С3 рециркулируют в стадию (ii) через TL1, тем более эффективно фазовое отделение согласно стадиям (ii)-(iv) способа по изобретению работает в отношении полноты разделения S1, насколько возможно. Следовательно, предпочтительно что, по меньшей мере, часть TL1, предпочтительно весь TL1 рециркулируют в стадию (ii). Кроме того, в особенности, поскольку дело идет о более предпочтительном варианте выполнения настоящего изобретения, по которому S1 получают ниже по ходу потока в реакции окисления, где пропилен предпочтительно реагирует с пероксидом водорода в присутствии ацетонитрила в качестве растворителя, поток BL1 имеет идеальный состав, позволяя провести прямую рециркуляцию, без любой другой промежуточной обработки, в реакцию эпоксидирования.
Далее, из способа по настоящему изобретению получают жидкую фазу L2, которая состоит, по существу, из воды и ацетонитрила, где массовое отношение ацетонитрил: вода в L2 составляет менее 1. Термин "состоит, по существу, из ацетонитрила и воды", как он используется в контексте настоящего изобретения, относится к жидкой фазе L2, где, по меньшей мере, 90 мас.% L2 состоит из ацетонитрила и воды.
По предпочтительному варианту выполнения изобретения, по меньшей мере, 95 мас.%, более предпочтительно, по меньшей мере, 97 мас.%, и еще более предпочтительно, по меньшей мере, 98 мас.% L2 состоит из С3, ацетонитрила и воды, где содержание С3 в L2 составляет, самое большее, 5 мас.%, предпочтительно самое большее, 3 мас.%, и более предпочтительно самое большее, 2 мас.%, исходя из общего веса L2. Поскольку дело идет об ацетонитриле, относительное содержание его в L2 составляет, предпочтительно менее 45 мас.%, более предпочтительно находится в интервале от 10 до 40 мас.%, еще более предпочтительно от 10 до 35 мас.%, исходя из общего веса L2.
Обычно жидкая фаза L2 может быть использована в любом подходящем процессе. Например, допустимо, что жидкую фазу L2 используют в качестве потока, который подводят в реакцию окисления или стадию обработки ниже по ходу потока указанной реакции окисления, где ацетонитрил используют в качестве растворителя и где окисляют пропилен, такую как реакция эпоксидирования, где ацетонитрил используют в качестве растворителя и где пропилен окисляют пероксидом водорода, чтобы получать оксид пропилена.
По предпочтительному варианту выполнения настоящего изобретения, жидкую фазу L2 до того, как ее используют в подходящем процессе, подвергают, по меньшей мере, одной дополнительной стадии разделения. Предпочтительный способ указанного разделения включает подвергание жидкой фазы L2 стадии перегонки. Предпочтительно перегонку проводят подходящим образом так, чтобы получать поток TL2, который содержит от 75 до 95 мас.%, предпочтительно от 80 до 85 мас.% ацетонитрила, исходя из общего веса TL2.
Обычно перегонка L2 может быть проведена в одной, двух или нескольких перегонных колоннах. Если эту перегонку проводят в одной перегонной колонне, точка росы в верхней части указанной перегонной колонны составляет обычно, по меньшей мере, 40ºС, предпочтительно находится в интервале от 40 до 80ºС, более предпочтительно в интервале от 40 до 65ºС. Как правило, число теоретических тарелок находится в интервале от 10 до 25. Типичные флегмовые числа находятся в интервале от 0,5 до 3. Таким способом получают поток TL2 в качестве верхнего потока из перегонной колонны. Соответствующий нижний поток, BL2, предпочтительно по существу, не содержит ацетонитрила. В этом контексте, термин "по существу, не содержит ацетонитрила" относится к варианту выполнения изобретения, по которому содержание ацетонитрила в BL2 составляет, самое большее, 500 млн. долей по весу, предпочтительно самое большее, 300 млн. долей по весу, более предпочтительно самое большее, 100 млн. долей по весу, исходя из общего веса BL2.
Неожиданно обнаружили, что возможно подвергать жидкую фазу L2 специально разработанной стадии перегонки, которая позволяет проводить высокоинтегрированный по теплу процесс перегонки. Таким образом, обнаружили, что разделение L2, предпочтительно проводят с использованием двух процессов перегонки под давлением, где в первой перегонной колонне проводят перегонку при давлении верха, которое выше, чем давление верха во второй перегонной колонне, в сочетании с указанной первой перегонной колонной, где холодильник, используемый, чтобы конденсировать верхний поток из первой перегонной колонны, используют одновременно в качестве испарителя второй перегонной колонны.
По этому предпочтительному варианту выполнения изобретения, жидкий поток L2, предпочтительно вводят в указанную первую перегонную колонну, из которой получают первый нижний поток и первый верхний поток.
Предпочтительно указанную первую перегонную колонну эксплуатируют в условиях, которые позволяют получение верхнего парового потока VTL2, который содержит от 50 до 70 мас.%, предпочтительно от 55 до 65 мас.% ацетонитрила, исходя из общего веса VTL2. Как правило, указанную первую перегонную колонну эксплуатируют при давлениях в верхней части колонны в интервале от 10 до 20 бар, предпочтительно от 10 до 15 бар. Обычно первая перегонная колонна имеет от 10 до 25, предпочтительно от 15 до 20 теоретических тарелок. Обычно флегмовое число указанной первой перегонной колонны находится в интервале от 0,25 до 2, предпочтительно от 0,25 до 1. Соответствующий нижний поток, получаемый из первой перегонной колонны, предпочтительно по существу, не содержит ацетонитрила. В этом контексте, термин "по существу, не содержит ацетонитрила" относится к варианту выполнения изобретения, по которому содержание ацетонитрила в нижнем потоке первой перегонной колонны составляет, самое большее, 500 млн. долей по весу, предпочтительно самое большее, 300 млн. долей по весу, более предпочтительно самое большее, 100 млн. долей по весу, исходя из общего веса нижнего потока первой перегонной колонны. В последующем, указанный нижний поток, получаемый из указанной первой перегонной колонны, необязательно, смешивают с нижним потоком, получаемым из второй перегонной колонны, как описано здесь ниже, на него ссылаются как на поток BL2.
В условиях процесса перегонки при двух давлениях, по меньшей мере, часть его, предпочтительно весь VTL2 конденсируют подходящим образом, и этот сконденсированный поток вводят во вторую перегонную колонну, из которой получают второй нижний поток и второй верхний поток. Предпочтительно указанную вторую перегонную колонну эксплуатируют при условиях, которые позволяют получение верхнего потока TL2, который содержит от 75 до 95 мас.%, предпочтительно от 80 до 85 мас.% ацетонитрила, исходя из общего веса TL2. Как правило, указанную вторую перегонную колонну эксплуатируют при давлениях в верхней части колонны в интервале от 1 до 5 бар, предпочтительно от 1 до 2 бар. Обычно вторая перегонная колонна имеет от 8 до 20, предпочтительно от 10 до 15 теоретических тарелок. Обычно флегмовое число указанной второй перегонной колонны находится в интервале от 0,5 до 5, предпочтительно от 1 до 3. Соответствующий нижний поток, получаемый из второй перегонной колонны, предпочтительно по существу, не содержит ацетонитрила. В этом контексте, термин "по существу, не содержит ацетонитрила" относится к варианту выполнения изобретения, по которому содержание ацетонитрила в нижнем потоке второй перегонной колонны составляет, самое большее, 500 млн. долей по весу, предпочтительно самое большее, 300 млн. долей по весу, более предпочтительно самое большее, 100 млн. долей по весу, исходя из общего веса нижнего потока второй перегонной колонны.
Предпочтительно TL2, получаемый из соответствующей перегонной колонны, по меньшей мере частично, предпочтительно полностью рециркулируют в процесс по изобретению. Более предпочтительно TL2 объединяют либо с S1, либо с S2, либо с Р.
Если поток S1 содержит, по меньшей мере, один пропиленгликоль, как описано выше, поток BL2, получаемый из указанной перегонки, предпочтительно содержит, по меньшей мере, один пропиленгликоль в количестве от 1 до 5 мас.%, более предпочтительно в количестве от 2 до 5 мас.%, исходя из общего веса BL2, тогда как поток TL2, по существу, не содержит, по меньшей мере, одного пропиленгликоля. В этом контексте настоящего изобретения, термин «TL2, по существу, не содержит, по меньшей мере, одного пропиленгликоля" относится к варианту выполнения изобретения, по которому содержание TL2 относительно, по меньшей мере, одного пропиленгликоля составляет, самое большее, 500 млн. долей по весу. TL2, по существу, не содержит, по меньшей мере, одного пропиленгликоля, предпочтительно содержит, самое большее, 200 млн. долей по весу.
Если BL2 не содержит или, по существу, не содержит пропиленгликоля, предпочтительно подводить BL2 непосредственно на соответствующий завод обработки сточных вод, такой как, например, биологический завод обработки сточных вод. Обнаружили, что не требуется никакой специфической обработки для сточных вод, произведенных способом по изобретению, что придает этому способу еще большую эффективность по стоимости и безопасность для окружающей среды.
Если BL2 содержит, по меньшей мере, один пропиленгликоль в значительных количествах, таких как в количестве от 1 до 5 мас.%, более предпочтительно в количестве от 2 до 5 мас.%, исходя из общего веса BL2, может быть предпочтительным вариант выполнения настоящего изобретения, в котором BL2 подводится в подходящую стадию отделения пропиленгликоля, где, по меньшей мере, один пропиленгликоль подходящим образом отделяют от воды и/или где два или несколько различных пропиленгликолей отделяют друг от друга. Этот способ выделения, по меньшей мере, одного пропиленгликоля из BL2 может быть проведен, например, испарением смеси, по меньшей мере, в две, предпочтительно три стадии испарения и/или перегонки, предпочтительно три стадии испарения, при уменьшенных рабочих давлениях, предпочтительно в интервалах от 1,5 до 5,5 бар при температуре от 111 до 155ºС, затем при давлении от 1,3 до 5,0 бар при температуре от 107 до 152ºС, и затем при от 0,7 до 4,0 бар при температуре от 90 до 144ºС, тем самым получая смесь BL2' и смесь BL2”; и отделяя смесь BL2' в, по меньшей мере, одну дальнейшую стадию перегонки, получая смесь BL2-I, содержащую, по меньшей мере, 70 мас.% воды, и смесь BL2-II, содержащую менее 30 мас.% воды. Особенно предпочтительно далее разделять смесь BL2” на смесь BL2-Ia, содержащую, по меньшей мере, 90 мас.% воды, и смесь BL2-Ib, содержащую менее 95 мас.% воды, посредством обратного осмоса. По предпочтительному варианту выполнения изобретения, по меньшей мере, один пропиленгликоль выделяют из смеси BL2-II, предпочтительно смешанной со смесью BL2-Ib, в, по меньшей мере, одну дополнительную стадию перегонки. По еще одному предпочтительному варианту выполнения изобретения, смеси BL2” и BL2-I объединяют и далее разделяют на смесь BL2-Ia, содержащую, по меньшей мере, 90 мас.% воды, и смесь BL2-Ib, содержащую менее 95 мас.% воды, посредством обратного осмоса.
Следовательно, настоящее изобретение также относится к способу, который описан выше, где
(аа) L2 вводят в первую перегонную колонну, из которой получают верхний паровой поток VTL2, содержащий от 50 до 70 мас.% ацетонитрила, исходя из общего веса верхнего потока VTL2, причем перегонку, предпочтительно проводят при давлении верха от 10 до 20 бар; и
(bb) по меньшей мере, частично конденсируют VTL2, получаемый в (аа), и вводят сконденсированный поток во вторую перегонную колонну, из которой получают TL2 в качестве верхнего потока, причем перегонку, предпочтительно проводят при давлении верха от 1 до 5 бар,
где холодильник, используемый, чтобы сконденсировать VTL2, одновременно используют в качестве испарителя второй перегонной колонны.
Неожиданно обнаружили, что эта концепция, где большинство воды отделяют под давлением в первой колонне и где сравнительно малый поток перегоняют при более низких давлениях, в общем, эта концепция ведет к меньшим потребностям в энергии для проблемы разделения.
По предпочтительному варианту выполнения настоящего изобретения, поток S1, подвергаемый способу по настоящему изобретению, обеспечивают процессом окисления или стадией его обработки ниже по ходу потока. Более предпочтительно поток S1 получают из процесса эпоксидирования или стадии обработки его ниже по ходу потока. Более предпочтительно поток S1 получают из эпоксидирования пропилена или стадии его обработки. Более предпочтительно поток S1 получают из эпоксидирования пропилена, где пероксид водорода используют в качестве окислителя, или стадии его обработки. Еще более предпочтительно поток S1 получают из эпоксидирования пропилена в присутствии ацетонитрила в качестве растворителя, где пероксид водорода используют в качестве окислителя, или из стадии его обработки ниже по ходу потока. Наиболее предпочтительно поток S1 получают из стадии обработки эпоксидирования пропилена в присутствии ацетонитрила в качестве растворителя, где пероксид водорода используют в качестве окислителя. Следовательно, по этим предпочтительным вариантам выполнения изобретения, способ отделения ацетонитрила от воды, который описан выше, может быть рассмотрен в качестве стадии специфической обработки указанного процесса окисления, в частности, указанного процесса эпоксидирования. Следовательно, настоящее изобретение также относится к процессу окисления, предпочтительно процессу эпоксидирования, более предпочтительно процессу производства оксида пропилена, более предпочтительно процессу производства оксида пропилена в присутствии ацетонитрила в качестве растворителя, более предпочтительно процессу производства оксида пропилена, где пропилен вступает в реакцию с пероксидом водорода в присутствии ацетонитрила в качестве растворителя, еще более предпочтительно высокоинтегрированному процессу производства оксида пропилена, где пропилен вступает в реакцию с пероксидом водорода в присутствии ацетонитрила в качестве растворителя, причем указанный процесс содержит описанный выше способ отделения ацетонитрила от воды или любой предпочтительный вариант выполнения этого способа отделения ацетонитрила от воды.
По предпочтительному варианту выполнения изобретения, в указанном процессе эпоксидирования пропилен вступает в реакцию с пероксидом водорода в устройстве для реакции в присутствии ацетонитрила в качестве растворителя, из которого получают поток S0, покидающий устройство для реакции, причем S0 содержит ацетонитрил, воду, оксид пропилена, и необязательно, не прореагировавший пропилен, кислород, и далее необязательно, пропан. В частности, поток S0 содержит пропан, если пропилен, используемый в качестве исходного материала для реакции эпоксидирования, содержится в смеси, которая, в дополнении к пропилену, также содержит пропан. Если такую смесь пропилена и пропана используют в качестве потока, подвергаемого эпоксидированию, массовое отношение пропилен:пропан будет, по меньшей мере, 7:3. Например, может быть использован коммерчески доступный пропилен, в качестве которого может выступать либо пропилен полимерной марки, либо пропилен химической марки. Как правило, пропилен полимерной марки будет иметь содержание пропилена от 99 до 99,8 мас.%, а содержание пропана от 0,2 до 1 мас.%. Пропилен химической марки будет обычно иметь содержание пропилена от 92 до 98 мас.% и содержание пропана от 2 до 8 мас.%. По предпочтительному варианту выполнения настоящего изобретения, эпоксидированию подвергают смесь пропилена и пропана, которая имеет содержание пропилена в интервале от 92 до 98 мас.%, предпочтительно в интервале от 94 до 97 мас.%, и содержание пропана в интервале от 2 до 8 мас.%, предпочтительно в интервале от 3 до 6 мас.%.
Следовательно, настоящее изобретение также относится к способу, который описан выше, где по (i) S1 обеспечивают процессом, содержащим
(а) реакцию пропилена, необязательно, смешанного с пропаном, с пероксидом водорода в устройстве для реакции в присутствии ацетонитрила в качестве растворителя, получая поток S0, покидающий устройство для реакции, причем S0 содержит ацетонитрил, воду, оксид пропилена, и необязательно, не прореагировавший пропилен, кислород, и далее необязательно, пропан.
По предпочтительному варианту выполнения настоящего изобретения, реакцию в (а) проводят в присутствии, по меньшей мере, одного подходящего катализатора, предпочтительно в присутствии, по меньшей мере, одного подходящего гетерогенного катализатора. Еще более предпочтительно, по меньшей мере, один подходящий катализатор содержит, по меньшей мере, один цеолит. Специфическое указание может быть сделано на цеолиты, содержащие титан, германий, теллур, ванадий, хром, ниобий, цирконий, имеющие пентасильную структуру цеолита, в частности, типов, которые могут быть охарактеризованы методом рентгеновской кристаллографии как структуры ABW, AGO, AEI, AEL, AEN, AET, AFG, AFI, AFN, AFO, AFR, AFS, AFT, AFX, AFY, AHT, ANA, APC, APD, AST, ASV, ATN, ATO, ATS, АТТ, ATV, AWO, AWW, ВСТ, ВЕА, ВЕС, BIK, BOG, BPH, BRE, CAN, CAS, CDO, CFI, CGF, CGS, СНА, CHI, CLO, CON, CZP, DAC, DDR, DFO, DFT, DOH, DON, EAB, EDI, EMT, EPI, ERI, ESV, ETR, EUO, FAU, FER, FRA, GIS, GIU, GME, GON, GOO, HEU, IFR, ISV, ITE, ITH, ITW, IWR, IWW, JBW, KFI, LAU, LEV, LIO, LOS, LOV, LTA, LTL, LTN, MAR, MAZ, MEI, MEL, МЕР, MER, MMFI, MFS, MON, MOR, MSO, MTF, MTN, MTT, MTW, RWR, RWY, SAO, SAS, SAT, SAV, SBE, SBS, SBT, SFE, SFF, SFG, SFH, SFN SFO, SGT, SOD, SSY, STF, STI, STT, TER, THO, TON, TSC, UEI, UFI, UOZ, USI, UTL, VET, VFI, VNI, VSV, WEI, WEN, YUG и ZON, и смешанные структуры из двух или нескольких из указанных выше структур. Для целей настоящего изобретения, предпочтение отдают использованию титановых цеолитов, имеющих структуру MFI, структуру MEL, смешанную структуру MFI/MEL или структуру MWW. Специфическое предпочтение отдают использованию цеолитных катализаторов структуры Ti-MWW. Катализаторы, особенно предпочтительно титановые цеолитные катализаторы, и еще более предпочтительно катализаторы, имеющие структуру Ti-MWW, могут быть использованы в виде порошка, в виде гранул, в виде микросфер, в виде формованных тел, имеющих, например, форму таблеток, цилиндров, колес, звезд, сфер и так далее, или в виде экструдатов, таких как экструдаты, имеющие, например, длину от 1 до 10, более предпочтительно от 1 до 7, и еще более предпочтительно от 1 до 5 мм, и диаметр от 0,1 до 5, более предпочтительно от 0,2 до 4 и, особенно предпочтительно от 0,5 до 2 мм. Получение таких предпочтительных катализаторов описано, например, в заявке на патент США 2007043226 А1, в частности в примерах 3 и 5 из заявки на патент США 2007043226 А1.
Обычно реакция в (а) может быть проведена любым подходящим способом. Таким образом, например, она может быть проведена в реакторе периодического действия или в, по меньшей мере, одном полу-непрерывно эксплуатируемом реакторе или в, по меньшей мере, одном непрерывно эксплуатируемом реакторе. В особенности, предпочтителен непрерывный режим работы. В этом особенно предпочтительном варианте выполнения изобретения, реакцию, предпочтительно проводят при температуре от -10 до 120ºС, более предпочтительно от 30 до 90ºС, и особенно предпочтительно от 30 до 65ºС. Как правило, температуру, при которой проводят реакцию, не сохраняют постоянной, но непрерывно или ступенчато регулируют, чтобы позволять постоянную конверсию пероксида водорода, которую определяют в потоке S0, покидающем реактор, в котором проводят эпоксидирование. Если эпоксидирование проводят в, по меньшей мере, одном непрерывно эксплуатируемом реакторе, таком как трубчатый реактор или реактор из связки труб, который содержит, по меньшей мере, одну охлаждающую рубашку, окружающую, по меньшей мере, одну трубу, термин "температура реакции", как его используют здесь, относится к температуре охлаждающей среды при входе в охлаждающую рубашку. Обычно, вследствие дезактивации катализатора, температуру реакции непрерывно или ступенчато повышают.
Как правило, температуру реакции непрерывно или ступенчато повышают со скоростью, самое большее 1ºС/день. Предпочтительная конверсия пероксида водорода составляет, по меньшей мере, 80%, более предпочтительно, по меньшей мере, 85%. Принцип предпочтительного определения конверсии пероксида водорода описан в патенте США 7351587 В2, в частности в примерах 1, 2 и 3 из патента США 7351587 В2. Давления в, по меньшей мере, одном реакторе лежат обычно в интервале от 3 до 100 бар, предпочтительно от 15 до 45 бар. В особенно предпочтительных вариантах выполнения способа по настоящему изобретению, реакцию проводят при температурах и давлениях, при которых реакционная смесь является жидкой, и газовой фазы не присутствует в, по меньшей мере, одном реакторе. Мольное отношение пропилен: пероксид водорода в отношении исходных материалов, пропускаемых в, по меньшей мере, один реактор, в котором проводят эпоксидирование, обычно находится в интервале от 0,9:1 до 3,0:1, причем интервал от 0,98:1 до 1,6:1 предпочтителен, а интервал от 1,0 до 1,5 особенно предпочтителен. Количество ацетонитрила, пропускаемого в, по меньшей мере, один реактор, регулируют так, чтобы концентрация пероксида водорода во всем потоке, пропускаемом в, по меньшей мере, один реактор, в котором проводят эпоксидирование, лежала обычно в интервале от 2 до 20 мас.%, предпочтительно от 5 до 12 мас.%, исходя из общего веса всего потока.
По особенно предпочтительным вариантам выполнения настоящего изобретения, полный поток, пропускаемый в, по меньшей мере, один реактор эпоксидирования, то есть загрузка реактора содержит от 50 до 80, предпочтительно от 60 до 70 мас.% ацетонитрила, от 7 до 14, предпочтительно от 8 до 11 мас.% пропилена, от 5 до 12, предпочтительно от 6 до 10 мас.% пероксида водорода, и от 10 до 25, предпочтительно от 12 до 20 мас.% воды.
В аналогичном предпочтительном варианте выполнения способа по настоящему изобретению, реакция пропилена может быть проведена в две или несколько стадий. Двухстадийная реакция происходит, например, следующим образом:
(A) пропилен, необязательно смешанный с пропаном, вступает в реакцию с пероксидом водорода в присутствии ацетонитрила в качестве растворителя, чтобы дать смесь, содержащую оксид пропилена, кислород и не прореагировавшую пероксид водорода, и необязательно пропан;
(B) не прореагировавший пероксид водорода отделяют от смеси, получающейся в (А);
(C) пероксид водорода, который был отделен в (В), вступает в реакцию с пропиленом, необязательно смешанным с пропаном.
Поскольку дело идет о предпочтительных условиях реакции эпоксидирования стадии (А), ссылку делают на предпочтительную реакцию эпоксидирования, которая обсуждается выше. Пероксид водорода может быть выделен по (В) любыми подходящими методами. Пероксид водорода, предпочтительно выделяют перегонкой, используя одну или несколько перегонных колонн, предпочтительно одну перегонную колонну. Эту перегонную колонну предпочтительно эксплуатируют в условиях, позволяющих получение верхнего потока, который содержит пероксид водорода в количестве, самое большее, 100 млн. долей по весу, исходя из общего веса верхнего потока, предпочтительно не содержащего, по существу, пероксида водорода. Дополнительно эту перегонную колонну предпочтительно эксплуатируют в условиях, позволяющих получение верхнего потока, который содержит, по меньшей мере, 80%, предпочтительно, по меньшей мере, 90%, и наиболее предпочтительно, по меньшей мере, 95% оксида пропилена, содержащегося в подаваемом. Как правило, эта перегонная колонна имеет от 15 до 45, предпочтительно от 20 до 40 теоретических тарелок, и ее эксплуатируют при давлении в верхней части колонны в интервале от 0,5 до 1,2 бар, предпочтительно от 0,7 до 1,1 бар. Флегмовое число этой перегонной колонны лежит обычно в интервале от 0,05 до 0,5, предпочтительно от 0,1 до 0,2. Нижний поток, получаемый из перегонной колонны в (В), содержащий, по существу, весь не прореагировавший пероксид водорода из (А) и далее содержащий ацетонитрил, воду и, необязательно, по меньшей мере, один пропиленгликоль, пропускают в стадию (С). Поскольку дело идет о стадии (С), предпочтительно использовать адиабатический реактор, предпочтительно адиабатический роторный реактор. Условия эпоксидирования в (С) являются предпочтительными, позволяя конверсию пероксида водорода на выходе из (С), по меньшей мере, 99%, предпочтительно, по меньшей мере, 99,5%, относительно пероксида водорода, подаваемого в (А). В (С) предпочтительно использование того же катализатора, как и в (А). Поскольку дело идет о пропилене, который вводят в реактор, используемый в (С), ссылка сделана на пропилен, уже обсужденный выше в контексте (а). Таким образом, например, может быть использован пропилен химической марки или пропилен полимерной марки, причем пропилен химической марки предпочтителен. Если в настоящем изобретении используют стадии (А) и (С), реакторы, предпочтительно эксплуатируют так, чтобы полная конверсия пропилена, принимая во внимание конверсию в (А) и конверсию в (С), была, по меньшей мере, 80%, более предпочтительно, по меньшей мере, 85%.
Как описано выше, предпочтительный вариант выполнения настоящего изобретения включает выделение ацетонитрила из С3 в, по меньшей мере, одной дальнейшей стадии разделения, подвергая жидкую фазу L1 стадии перегонки, из которой получают поток TL1, который содержит, по меньшей мере, 90 мас.%, предпочтительно, по меньшей мере, 95 мас.% С3, исходя из общего веса TL1, и из которого получают поток BL1, 95 мас.%, предпочтительно, по меньшей мере, 98 мас.% которого состоит из С3, ацетонитрила и воды, где содержание С3 в BL1 находится в интервале от 7 до 18 мас.%, предпочтительно от 10 до 15 мас.%, в каждом случае, исходя из общего веса BL1. Неожиданно обнаружили, что BL1, таким образом получаемый, может быть рециркулирован в процесс эпоксидирования, наиболее предпочтительно без промежуточной обработки BL1, вследствие предпочтительного состава полного потока, пропускаемого в, по меньшей мере, один реактор эпоксидирования, и предпочтительного состава BL1. Следовательно, по предпочтительному варианту выполнения изобретения, по меньшей мере, часть BL1, как описано выше, рециркулируют в (а). Следовательно, предпочтительный способ по изобретению отделения ацетонитрила от воды позволяет объединенный процесс эпоксидирования, включающий экономические преимущества повторного использования растворителя эпоксидирования ацетонитрила, отделенного от воды в стадиях обработки и, далее, подходящее применение пропилена, используемого для указанного отделения растворителя ацетонитрила от воды.
Обычно можно себе представить, что, помимо BL1, необходим дополнительный свежий ацетонитрил. Это может быть вследствие потерь ацетонитрила во всем процессе. В качестве дополнительного ацетонитрила, чистый или, по существу, чистый ацетонитрил может быть добавлен, например, к потоку BL1 и/или в другой подходящей точке или точках выше по ходу входа реактора. Также можно себе представить добавление сырого ацетонитрила, такого как азеотроп ацетонитрила и воды, вероятно, также содержащий примеси, где такой сырой ацетонитрил обычно доступен, например, при процессе получения акрилонитрила. В этом последнем случае было бы предпочтительно добавлять ацетонитрил, а именно сырой ацетонитрил, выше по ходу разделения фаз.
По предпочтительному варианту выполнения настоящего изобретения, поток S0, получаемый из (а), в частности, S0, состоящий из верхнего потока, получаемого из перегонной колонны, используемой в (В), объединенный с выходом из реактора, получаемым из (С), пропускают в подходящую стадию обработки ниже по ходу потока. Предпочтительно в этой стадии обработки ниже по ходу потока, пропилен, кислород и пропан выделяют из SO, чтобы получать поток, состоящий, по существу, из ацетонитрила, воды и оксида пропилена. Эту стадию обработки указывают в качестве стадии (b) в контексте настоящего изобретения.
Разделение по стадии (b) может быть проведено любым подходящим способом. Наиболее предпочтительно разделение проводят перегонкой. Разделение по стадии (b), предпочтительно проводят в, по меньшей мере, одной перегонной колонне, более предпочтительно в одной перегонной колонне. Предпочтительно эта колонна имеет от 10 до 30, более предпочтительно от 15 до 25 теоретических тарелок. Перегонную колонну предпочтительно эксплуатируют при давлении верха от 0,5 до 1,2 бар, более предпочтительно от 0,7 до 1,1 бар.
Чтобы облегчить указанную задачу разделения, обнаружили, что благоприятно добавлять либо ацетонитрил, либо смесь ацетонитрила с водой в верхнюю часть колонны. Полагают, что это дополнительное орошение служит в качестве агента для отгонки легких фракций, который, среди других, предотвращает выделение оксида пропилена через верхнюю часть перегонной колонны. По предпочтительному варианту выполнения настоящего изобретения используют часть нижнего потока перегонной колонны, используемого в стадии (с). Также можно себе представить, что поток TL2, либо его часть, используют в качестве агента для отгонки легких фракций. Как правило, количество TL2 не будет достаточным, и будет добавлен другой поток. Как правило, массовое отношение количества ацетонитрила, подаваемого в качестве дополнительного орошения в верхнюю часть перегонной колонны, к весу вводимого потока S0 и подлежащего отделению в перегонной колонне, находится в интервале от 1:1 до 4:1, предпочтительно от 1,5:1 до 3:1. Температура дополнительного орошения лежит обычно в интервале от 2 до 20ºС, предпочтительно в интервале от 5 до 15ºС.
По настоящему изобретению, по меньшей мере, 95% по объему, предпочтительно, по меньшей мере, 90% по объему, и еще более предпочтительно, по меньшей мере, 70% по объему верхнего потока дистилляционной колонны по (b) состоит из пропилена, кислорода, и необязательно, пропана. В зависимости от содержания кислорода в нем, этот верхний поток может быть пропущен в дальнейшую подходящую стадию обработки, где содержание кислорода подходящим образом уменьшают, чтобы позволять, например, чтобы обедненный кислородом поток был рециркулирован в одну или несколько стадий по настоящему изобретению, такой как исходный материал для стадии (а) способа по изобретению, подобно стадии (А) или стадии (В), или в качестве части потока Р. Если содержание кислорода в указанном верхнем потоке снижают, предпочтительно снижать содержание кислорода реакцией с водородом в присутствии подходящего катализатора. Такие катализаторы представляют собой, например, катализаторы, содержащие олово и, по меньшей мере, один благородный металл, как описано в международной заявке WO 2007/000396 А1, в частности в примере 1 из международной заявки WO 2007/000396 А1. Также можно себе представить использование катализаторов, содержащих медь в элементарной и/или оксидной форме на носителе, где медь присутствует на носителе в количестве от 30 до 80 мас.%, относительно всего катализатора, и рассчитанном как CuO. Такие катализаторы могут быть получены, например, по примеру в заявке ЕПВ ЕР 0427062 А2, катализатор 2, страница 4, строки с 41 по 50 (соответствующей патенту США 5194675). Однако чтобы снижать содержание кислорода, также возможны другие подходящие способы. Необязательно, указанный верхний поток перед тем, как быть подвергнутым гидрированию, может быть сжат и частично сконденсирован, где получают жидкий поток, который состоит, по существу, из С3 и ацетонитрила и который содержит незначительные количества воды. Не конденсируемая часть состоит, по существу, из С3 и кислорода и содержит незначительное количество воды, где, по сравнению с основным потоком, содержание кислорода увеличено, хотя оно все еще находится в таком интервале, чтобы смесь была не воспламеняемой. Этот обогащенный кислородом поток затем подвергают гидрированию.
Далее по настоящему изобретению, по меньшей мере, 98 мас.%, предпочтительно, по меньшей мере, 98,5 мас.%, и еще более предпочтительно, по меньшей мере, 99 мас.% нижнего потока из дистилляционной колонны (b) состоит из оксида пропилена, ацетонитрила, воды, и необязательно, по меньшей мере, одного пропиленгликоля. Перед подачей этого нижнего потока в качестве потока S1 в способ по настоящему изобретению особенно предпочтительно выделять оксид пропилена из указанного нижнего потока по стадии (с) способа по изобретению, чтобы получать поток, который, по существу, не содержит оксида пропилена и который может быть частично использован в качестве агента для отгонки легких фракций для стадии (b).
Разделение по стадии (с) может быть проведено любым подходящим способом. Наиболее предпочтительно разделение проводят перегонкой. Разделение по стадии (с), предпочтительно проводят в, по меньшей мере, одной перегонной колонне, более предпочтительно в одной перегонной колонне. Предпочтительно эта колонна имеет от 50 до 80, более предпочтительно от 60 до 70 теоретических тарелок. Эту перегонную колонну предпочтительно эксплуатируют при давлении верха от 0,2 до 2 бар, более предпочтительно от 0,4 до 1 бар. Предпочтительно, по меньшей мере, один подходящий полярный растворитель, такой как, предпочтительно воду добавляют в верхней части колонны в качестве экстрактивного агента. По предпочтительному варианту выполнения изобретения, разделение по стадии (с) может быть проведено введением указанного нижнего потока в колонну экстрактивной дистилляции;
дополнительным введением полярного экстрагирующего растворителя в указанную колонну экстрактивной дистилляции;
- отгонкой оксида пропилена в верхнем погоне из указанной колонны экстрактивной дистилляции в качестве верхнего потока, причем верхний поток содержит только незначительные количества ацетонитрила, такие как 500 млн. долей или меньше;
- сжатием верхнего потока полученного в предыдущей стадии верхнего погона посредством, по меньшей мере, одного компрессора, чтобы получать сжатый пар,
- конденсацией сжатого пара, получаемого в предыдущей стадии, и возвращения, по меньшей мере, части тепла конденсации в, по меньшей мере, один кипятильник, используемый в колонне экстрактивной дистилляции.
Из этой перегонной колонны по (с) получают верхний поток, который содержит, по меньшей мере, 90 мас.%, предпочтительно, по меньшей мере, 95 мас.% оксида пропилена. В зависимости от требований по качеству оксида пропилена можно себе представить использование этой фракции оксида пропилена без дальнейшей очистки. Однако также возможно далее очищать указанную фракцию оксида пропилена, например, в, по меньшей мере, одной дальнейшей стадии перегонки. Из такой дальнейшей стадии перегонки может быть получен поток оксида пропилена, где, по меньшей мере, 99,5 мас.%, более предпочтительно, по меньшей мере, 99,9 мас.% указанного потока состоит из оксида пропилена.
Далее из этой перегонной колонны по (с) получают нижний поток, который обычно содержит, самое большее, 500 млн. долей по весу, предпочтительно самое большее 100 млн. долей по весу, и более предпочтительно самое большее 60 млн. долей по весу оксида пропилена, относительно веса нижнего потока. В частности, по меньшей мере, 98 мас.%, более предпочтительно, по меньшей мере, 98,5 мас.%, и более предпочтительно, по меньшей мере, 99 мас.% нижнего потока состоит из ацетонитрила, воды, и необязательно, по меньшей мере, одного пропиленгликоля.
В зависимости от специфических условий в ходе стадий по настоящему изобретению выше по ходу потока, то есть стадий (а), необязательно, (b) и/или (с), этот нижний поток, получаемый из перегонной колонны по (с), также может содержать некоторые количества гидроперекисей, таких как некоторые количества пероксида водорода и/или некоторые количества органических гидроперекисей, например, 1-гидропероксипропанол-2 и/или 2-гидропероксипропанол-1. Как правило, нижний поток, получаемый из перегонной колонны по (с), может содержать до 2 мас.%, предпочтительно до 1 мас.% этих гидроперекисей в сумме, относительно веса нижнего потока. Чтобы снижать содержание гидроперекисей и, тем самым, чтобы избегать формирования гидроперекисей, которые, как полагают, имеют вредное влияние, когда дело касается формирования нежелательных побочных продуктов и аспектов безопасности в отношении разложения гидроперекисей, предпочтительно подвергать нижний поток, получаемый из перегонной колонны по (с), по меньшей мере, одной дальнейшей стадии обработки. Указанное формирование особенно происходит, если реализуют высокоинтегрированный процесс по изобретению. Хотя каждый подходящий способ, который можно себе представить, по меньшей мере, частично удаляет эти гидроперекиси, особенно предпочтительно каталитическое снижение их содержания, предпочтительно каталитическое гидрирование гидроперекисей. В качестве подходящего катализатора может быть указан катализатор, который описан в заявке на патент США 20040068128 А1, в частности в параграфах с [0053] по [0076]. Предпочтительные катализаторы выбирают из группы, состоящей из гетерогенных катализаторов, содержащих Ru, Ni, Pd, Pt, либо индивидуально, либо в виде смеси из двух или нескольких из них, в качестве активного металла на подходящем материале носителя. Особенно подходящий катализатор, а именно катализатор на носителе, содержащий 5 мас.% Pd на активированном углероде, описан в примере Е2 заявки на патент США 20040068128 А1. Давление в ходе гидрирования лежит обычно в интервале от 1 до 100 бар (абс.), предпочтительно от 1 до 10 бар (абс.), и температура в ходе гидрирования лежит обычно в интервале от 0 до 180ºС, предпочтительно от 25 до 120ºС, более предпочтительно от 65 до 85ºС. Парциальное давление водорода в ходе гидрирования лежит, предпочтительно в интервале от более 1 до 20 бар, более предпочтительно от 2 до 15 бар, и еще более предпочтительно от 3 до 13 бар. Если гидрирование проводят в неподвижном слое, который предпочтителен, время пребывания жидкости, пропускаемой через реактор гидрирования, лежит обычно в интервале от 1 секунды (с) до 1 часа (ч), предпочтительно от 10 с до 20 минут (мин), в особенности, от 30 с до 5 мин.
В зависимости от условий реакции, используемых для восстановления, предпочтительно гидрирования нижнего потока, получаемого из перегонной колонны по (с), может быть необходимым отделять получаемый поток от катализатора, предпочтительно катализатора гидрирования и/или не прореагировавшего восстановителя, предпочтительно водорода и/или побочных продуктов гидрирования, предпочтительно СО и/или метана.
В частности, поток, получаемый от восстановления, предпочтительно гидрирования, содержит, по меньшей мере, 95 мас.% ацетонитрила и воды, исходя из общего веса нижнего потока, где массовое отношение ацетонитрил:вода составляет более 1. Обычно можно себе представить использование нижнего потока, получаемого из перегонной колонны по (с), в качестве потока S1 по настоящему изобретению.
В зависимости от специфических условий в ходе стадий выше по ходу потока по настоящему изобретению, то есть стадий (а), необязательно, (b) и/или (с) и/или восстановления, предпочтительно стадии гидрирования, поток, получаемый от восстановления, предпочтительно гидрирования, может содержать некоторые количества уксусного альдегида, и необязательно, другие низкокипящие соединения, такие как, например, пропионовый альдегид и ацетон. Как правило, этот поток может содержать до 2000 млн. долей по весу, предпочтительно до 1000 млн. долей по весу, более предпочтительно до 300 млн. долей по весу уксусного альдегида и других низкокипящих соединений в сумме, исходя из общего веса этого потока. Чтобы уменьшать содержание уксусного альдегида, и необязательно, содержание в отношении других низкокипящих соединений и, тем самым, чтобы избегать формирования этих соединений, которое особенно происходит, если реализуют высокоинтегрированный процесс по изобретению, предпочтительно подвергать этот поток, по меньшей мере одной стадии дальнейшей обработки. Хотя возможен любой подходящий способ для частичного удаления уксусного альдегида, особенно предпочтительно отделять уксусный альдегид от потока перегонкой. Отделение по этой стадии предпочтительно проводят в, по меньшей мере, одной перегонной колонне, более предпочтительно в одной перегонной колонне. Предпочтительно эта колонна имеет от 15 до 40, более предпочтительно от 15 до 30 теоретических тарелок. Эту перегонную колонну предпочтительно эксплуатируют при давлении верха от 0,7 до 2 бар, более предпочтительно от 1,1 до 1,5 бар.
Из этой перегонной колонны получают нижний поток, который обычно содержит, самое большее, 200 млн. долей по весу, предпочтительно самое большее, 100 млн. долей по весу, и более предпочтительно самое большее, 50 млн. долей по весу уксусного альдегида и других низкокипящих соединений, которые описаны выше, в сумме, относительно веса нижнего потока. В частности, по меньшей мере, 99 мас.%, более предпочтительно, по меньшей мере, 99,5 мас.%, и более предпочтительно, по меньшей мере, 99,7 мас.% нижнего потока состоит из ацетонитрила, воды, и необязательно, по меньшей мере, одного пропиленгликоля. В частности, нижний поток содержит, по меньшей мере, 95 мас.% ацетонитрила и воды, исходя из общего веса нижнего потока, где массовое отношение ацетонитрил: вода составляет более 1. Предпочтительно этот нижний поток используют в качестве потока S1 в способе по настоящему изобретению. По возможному варианту выполнения настоящего изобретения, не требуется никакой стадии перегонки.
Следовательно, настоящее изобретение также относится к способу, который описан выше, далее содержащему
(y) подвергание потока, полученного из (с), стадии гидрирования, предпочтительно стадии каталитического гидрирования, причем катализатор предпочтительно представляет собой гетерогенные катализаторы, содержащие Ru, Ni, Pd, Pt, либо индивидуально, либо в виде смеси из двух или нескольких из них, в качестве активного металла на подходящем материале носителя, в частности Pd на активированном углероде; причем указанное гидрирование предпочтительно проводят при давлении в ходе гидрирования в интервале от 1 до 100 бар (абс.), предпочтительно от 1 до 10 бар (абс.), и температуре в ходе гидрирования в интервале от 0 до 180ºС, предпочтительно от 25 до 120ºС, более предпочтительно от 65 до 85ºС;
(z) подвергание потока, получаемого из (y), стадии перегонки, предпочтительно проводимой в дистилляционной колонне, эксплуатируемой при давлении верха от 0,7 до 2 бар, более предпочтительно от 1,1 до 1,5 бар, чтобы получать поток S1, и подвергание S1 стадии (d), которая обсуждается ниже.
В общем, настоящее изобретение относится к способу, который описан выше, где по (i) S1 обеспечивают процессом, содержащим
(а) реакцию пропилена с пероксидом водорода в устройстве для реакции в присутствии ацетонитрила в качестве растворителя, получая поток S0, покидающий устройство для реакции, причем SO содержит ацетонитрил, воду, оксид пропилена, и необязательно, не прореагировавший пропилен, кислород, и далее необязательно, пропан;
(b необязательно, выделение пропилена, кислорода и пропана из S0, чтобы получать поток, состоящий, по существу, из ацетонитрила, воды и оксида пропилена;
(с) выделение оксида пропилена из S0 или потока, получаемого из (b), тем самым получая поток S1.
Как описано выше, способ по изобретению отделения ацетонитрила от воды предпочтительно объединяют в качестве стадии обработки в процессе получения оксида пропилена. Особенно, в качестве части этого процесса эпоксидирования, способ по изобретению позволяет проводить высокоинтегрированный процесс. Следовательно, настоящее изобретение также относится к высокоинтегрированному способу получения оксида пропилена, причем этот способ содержит
(a) реакцию пропилена, необязательно смешанного с пропаном, с пероксидом водорода в устройстве для реакции в присутствии ацетонитрила в качестве растворителя, получая поток S0, покидающий указанное устройство для реакции, причем S0 содержит ацетонитрил, воду, оксид пропилена, и необязательно, не прореагировавший пропилен, кислород, и необязательно, пропан;
(b) необязательно, отделение пропилена, необязательно вместе с пропаном и кислородом от S0, чтобы получать поток, по меньшей мере, 99 мас.% которого состоит из ацетонитрила, воды и оксида пропилена;
(c) отделение оксида пропилена от S0 или потока, получаемого из (b), таким образом получая поток S1, содержащий, по меньшей мере, 95 мас.% ацетонитрила и воды, исходя из общего веса S1, где массовое отношение ацетонитрил: вода составляет более 1;
(d) добавление потока Р, содержащего, по меньшей мере, 95 мас.% С3, исходя из общего веса потока Р, к S1, чтобы получать смешанный поток S2, причем С3 представляет собой пропилен, необязательно смешанный с пропаном, с минимальным массовым отношением пропилен: пропан 7:3;
(e) подвергание S2 температуре, самое большее, 92ºС и давлению, по меньшей мере, 10 бар, получая первую жидкую фазу L1, состоящую, по существу, из С3, ацетонитрила и воды, и вторую жидкую фазу L2, состоящую, по существу, из воды и ацетонитрила, где массовое отношение ацетонитрил: вода в L2 составляет менее 1;
(f) отделение L1 от L2;
(g) подвергание L1 стадии перегонки, из которой получают поток TL1, который содержит, по меньшей мере, 90 мас.% С3, исходя из общего веса TL1; и из которого получают дальнейший поток BL1, по меньшей мере, 95 мас.% BL1 состоит из С3, ацетонитрила и воды, где содержание С3 в BL1 находится в интервале от 7 до 18 мас.%, исходя из общего веса BL1;
(h) подвергание L2 стадии перегонки, из которой получают поток TL2, который содержит от 75 до 95 мас.%, предпочтительно от 80 до 85 мас.% ацетонитрила, исходя из общего веса TL2;
где, по меньшей мере, часть потока BL1 рециркулируют в (а); и/или
где, по меньшей мере, часть TL1 рециркулируют в (а); и/или
где, по меньшей мере, часть TL2 рециркулируют в (d).
Особенно предпочтительно рециркулировать BL1 в (а), а TL1 в (d), и TL2 в (d).
Описанные выше стадии процесса, включая стадии (y) и (z), могут быть проведены либо непрерывно, полунепрерывно, либо периодически. Предпочтительно все стадии проводят непрерывно.
Что касается предпочтительных составов указанных потоков, предпочтительных условий реакции, предпочтительных условий разделения, предпочтительных условий перегонки или подобного, и далее предпочтительных вариантов выполнения изобретения, что касается, например, предпочтительных и/или возможных дополнительных промежуточных и/или рабочих стадий, делается ссылка на соответствующее раскрытие выше.
Настоящее изобретение далее иллюстрировано на примерах.
Примеры
Пример 1: Эпоксидирование пропилена
Эпоксидирование проводили в непрерывном трубчатом реакторе, имеющем охлаждающую рубашку и каталитический слой. Этот реактор имел длину 1440 мм и внутренний диаметр 7 мм и был изготовлен из нержавеющей стали (спецификация 1.4571). Трубчатый реактор был окружен охлаждающей рубашкой, по которой циркулировал охлаждающий агент (смесь вода/гликоль). Этот реактор эксплуатировали в режиме восходящего потока. Подходящие термоэлементы были расположены непосредственно выше и ниже каталитического слоя и отцентрованы по оси в трубе, чтобы измерять температуры на входе и выходе каталитического слоя. Реактор эксплуатировали при постоянном давлении 20 бар с использованием клапана для регулирования давления на выходе из реактора. Температуру контролировали циркуляцией охлаждающей среды (жидкость) в оболочке реактора параллельно относительно продукта. Температуру на входе охлаждающей среды измеряли термоэлементом и использовали, чтобы регулировать термостат. Эту температуру называют "температурой реакции", и ее поддерживали постоянной в 63ºС на протяжении всего эксперимента. Скорость потока охлаждающей среды регулировали так, чтобы температура на выходе охлаждающей среды была не более чем на 1ºС выше температуры на входе охлаждающей среды.
Реактор заполняли снизу слоем стеклянной дроби (80 мм), затем катализатором, и все остающееся пространство заполняли стеклянной дробью. В качестве катализатора использовали 15 г катализатора, который производили путем смешивания 100 частей порошка Ti-MWW, содержащего 1,5 мас.% Ti, с 40 частями пирогенного кремнезема и воды, смешивая и экструдируя, чтобы сформировать цилиндрические экструдаты диаметром 1,5 мм и длиной между 3 и 5 мм, и прокаливали эти экструдаты при 450ºС перед использованием. Эти экструдаты имели концентрацию Ti 1,1 мас.%. Длина слоя катализатора была около 950 мм.
Эти реагенты помещали в подходящие резервуары и непрерывно отмеряли в реактор, используя стандартные дозировочные насосы. Резервуар 1 содержал ацетонитрил химической марки с чистотой 99,98% (содержащий 0,01% воды, 150 млн. долей по весу пропионитрила и 1 млн. долю по весу акрилонитрила в качестве примесей). Резервуар 2 содержал сырой стандартной марки промытый раствор пероксида водорода с концентрацией 40 мас.%, к которому добавляли 175 млн. долей по весу (NH4)H2PO4. Резервуар 3 содержал сжатый сжиженный пропилен полимерной марки. Дозировочный насос для ацетонитрила начинал сперва при скорости подачи 60,9 г/ч, и температуру реакции устанавливали в 63ºС, а регулятор давления был установлен на 20 бар. Когда реактор заполняли жидкостью и систему уравновешивали, включали дозировочный насос для пропилена со скоростью подачи 8,5 г/ч. Через несколько минут включали насос для пероксида водорода со скоростью подачи 11,5 г/ч (эту точку времени отмечали как t=0). Эти три потока смешивали при комнатной температуре перед тем, как их пропускали в реактор. Температуры, измеренные внутри реактора сразу перед и сразу после каталитического слоя, отличались менее 1ºС от температуры реакции, тем самым демонстрируя, что слой из стеклянной дроби достаточно нагревал реагенты до температуре реакции прежде, чем они входили в контакт с катализатором.
Поток продукта, покидающий реактор, декомпрессировали до окружающего давления в сосуде, где газ и жидкие фазы разделяли. Количество газа определяли волюмометрически, и его состав анализировали методом газовой хроматографии. Жидкую фазу собирали на протяжении всего цикла (около 300 ч) и анализировали. Полную концентрацию перекисей определяли иодометрически. Концентрацию H2O2 определяли колориметрически с использованием метода с сульфатом титанила. Разность между двумя значениями представляет собой обычно хорошую меру концентрации гидропероксипропанолов (1-гидроперокси-2-пропанол и 2-гидроперокси-1-пропанол); это было подтверждено определением количества пропиленгликоля методом газовой хроматографии перед и после восстановления смеси избытком трифенилфосфина. Все другие органические компоненты определяли газовой хроматографией с использованием детектора ионизации в пламени. За исключением ацетонитрила, для которого дается только % площади, все другие компоненты определяли количественно, используя 1,4-диоксан в качестве внутреннего стандарта. Состав жидкости на выходе из реактора дан в таблице 1 ниже.
ГХ
Жидкий поток, получаемый в предыдущей стадии, подвергали перегонке в роторном испарителе с температурой ванны и давлением 200 мбар, пока концентрация оксида пропилена в остатке не становилась ниже 1 мас.%.
Полученный остаток анализировали, как указано выше, причем разность всех не перекисных органических компонентов определяли количественно ЖХ с 1,4-диоксаном в качестве внутреннего стандарта. Воду определяли, используя метод Карла - Фишера. Состав остаточного потока дан в таблице 2 ниже.
Этот поток использовали в качестве потока подачи S1 для процесса по примеру 2 ниже.
Пример 2: Обработка потока, полученного по примеру 1
50 г смеси, которая описана в таблице 2, вводили в вакуумированный, отпущенный автоклав (объем: 315 мл), оборудованный двумя противоположными окнами из сапфира. После уравновешивания, 40 г пропилена (полимерная марка, содержащая 0,1 мас.% пропана) вводили в автоклав. Температуру поддерживали постоянной в 40ºС. Измеренное давление было 14,3 бар.
Содержимое автоклава полностью перемешивали магнитной мешалкой. После того, как мешалку останавливали, смесь немедленно разделялась на две жидких фазы. Нижнюю водную фазу удаляли через выпускное отверстие на дне автоклава и взвешивали. Водная фаза весила 6,12 г, что соответствует 6,8% полного содержания автоклава (90 г). Как водную, так и органическую фазу анализировали, как описано выше, и состав обеих фаз дан в таблице 3 ниже. Как можно видеть из результатов в таблице 3, отношение ацетонитрила к воде (12,1:1) намного ниже, чем массовое отношение в азеотропе ацетонитрил/вода (5,7:1), доступное простой перегонкой.
Пример 3: Отделение ацетонитрила от воды
Во втором эксперименте, снова, количество 50 г потока, полученного по примеру 1, вводили в вакуумированный и отпущенный автоклав, как описано в примере 2. Затем 50 г пропилена (химическая марка, содержащая 0,1 мас.% пропана) вводили в автоклав. Температуру поддерживали постоянной в 40ºС, измеренное давление было 14,8 бар.
Содержимое автоклава полностью перемешивали магнитной мешалкой. После того, как мешалку останавливали, смесь немедленно разделялась на две жидких фазы. Нижнюю водную фазу удаляли через выпускное отверстие на дне автоклава и взвешивали. Водная фаза весила 6,8 г, что соответствует 6,8% полного содержания автоклава (100 г). Как водную, так и органическую фазу анализировали, как описано выше, и состав обеих фаз дан в таблице 4 ниже. Как можно видеть из результатов в таблице 4, отношение ацетонитрила к воде (13,9:1) намного ниже, чем массовое отношение в азеотропе ацетонитрил/вода (5,7:1), доступное простой перегонкой.
Результаты в примерах 2 и 3 показывают, что при добавлении смеси пропилена, в частности, пропилен/пропан, к смеси воды и ацетонитрила, массовое отношение ацетонитрил:вода может быть существенно понижено. Влияние повышения отношения ацетонитрил:вода повышается, когда используют больше пропилена.
Пример 4: Отделение ацетонитрила от воды 4.1 Получение потока S2
Так как в дальнейшем описан полностью непрерывно работающий процесс по настоящему изобретению, составы, раскрытые подробно, должны быть поняты в качестве средних величин, определенных по всему непрерывному процессу, который был проведен за 507 часов.
Из непрерывного процесса получения оксида пропилена, проводимого, в основном, по примеру 1 с использованием тех же исходных материалов, катализатора, и также содержащего стадию обработки ниже по ходу потока в виде отгонки оксида пропилена, причем, однако, указанную проводимую перегонку проводили в перегонной колонне, получали поток со скоростью течения 102 кг/ч и имеющий, в отношении главных компонентов, состав, который дан в таблице 5 ниже.
Этот поток использовали в качестве потока подачи S1 для процесса разделения по изобретению.
Перед пропусканием потока S1 в устройство для разделения, его объединяли с 3 другими потоками:
- Первый поток 
- Второй поток 
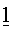
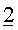
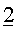
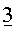
Вместе с указанными потоками 1, 2 и 3, поток S1 формировал поток S2, имеющий скорость потока 169,6 кг/ч и имеющий, относительно главных компонентов, состав, который дан в таблице 6 ниже.
Этот поток подавали в устройство разделения, горизонтальный гравитационный отстойник с емкостью 2 л.
4.2 Разделение по изобретению
В горизонтальном гравитационном отстойнике фазового разделения достигали при температуре в интервале от 30 до 40ºС и при давлении 18 бар. Получаемый верхний слой подавали в качестве потока со скоростью потока 148 кг/час в дистилляционную колонну 
Нижний слой имел состав, относительно главных компонентов, который дан в таблице 8 ниже:
4.3 Обработка верхнего слоя
Поток, получаемый в качестве верхнего слоя из устройства разделения фаз, пропускали, как указано, в дистилляционную колонну 
Дистилляционная колонна 
Верхний поток, получаемый из колонны 
Нижний поток, получаемый из колонны
рециркулировали в устройство разделения в качестве потока 
4.4 Обработка нижнего слоя
Поток, получаемый в качестве нижнего слоя из устройства разделения фаз, пропускали, как указано, в дистилляционную колонну 
Дистилляционная колонна 
Нижний поток, получаемый из дистилляционной колонны
Верхний поток, получаемый из колонны
то есть, по существу, состав азеотропа ацетонитрил/вода. Этот верхний поток из колонны
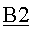
Дистилляционная колонна В2 была сконфигурирована в виде насадочной колонны (DN80*9500) (DN 80=номинальный диаметр 80 по стандарту EN 10255; длина=9500 мм), изготовленной из нержавеющей стали качества 1.4541 с объединенной основой кипятильника. Колонну 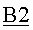
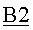
Нижний поток, имеющий скорость потока 3,6 кг/ч, имел содержание воды 99,9 мас.%.
Верхний поток, получаемый из колонны 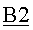
Этот верхний поток колонны 
Сущность изобретения
Главное преимущество способа по изобретению отделения ацетонитрила от воды особенно очевидно в случае, когда отделенный ацетонитрил рециркулируют в качестве исходного материала высокоинтегрированного способа эпоксидирования пропилена, в частности, водным пероксидом водорода. В таком объединенном процессе, способ по изобретению позволяет большое сбережение энергии, в то время как в то же самое время может быть достигнуто отношение ацетонитрил: вода, которое выше, чем отношение ацетонитрил:вода, достижимое простой перегонкой при давлении окружающей среды. Теперь, не желая быть связанными любой теорией, полагают, что, чтобы отделять и рециркулировать ацетонитрил из реакционной смеси, получаемой из такой реакции, как эпоксидирование, или из подходящей смеси, получаемой из одной или нескольких стадий обработки перегонкой при давлении окружающей среды, весь ацетонитрил плюс количество воды, содержащееся в азеотропе, должно было бы испаряться. Однако если желательно более низкое количество воды в ацетонитриле, тогда было бы необходимо устройство перегонки с двумя давлениями, но из этого следовало бы, что все количество ацетонитрила нужно было бы испарять дважды, что обязательно ведет к большой затрате энергии. Таким образом, при использовании способа по настоящему изобретению, необходима только часть энергии по сравнению с указанным выше способом, вследствие нашего открытия, что разделение жидкость/жидкость требует очень небольшой энергии. Далее, возможно разработать желательное устройство разделения в форме сравнительно простой установки. Единственный значительный вклад в потребление энергии необходим, чтобы рекуперировать небольшое количество ацетонитрила, присутствующего в водной фазе после разделения фаз. Настоящее изобретение ясно позволяет большое преимущество как с точки зрения потребления энергии, так и с точки зрения требуемых инвестиций.
Пример 4 далее ясно показывает высокоинтегрированный вариант способа разделения по настоящему изобретению, где ниже по ходу потока процесса разделения слои, получаемые из устройства для разделения, рециркулируют в процесс, либо в процесс разделения, либо выше по ходу потока в процесс эпоксидирования, в максимальной степени, тем самым обеспечивая желательную схему процессов, как с экономической, так и с экологической точки зрения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПЕРЕГОНКА С ЧАСТИЧНЫМ ПОТОКОМ | 2015 |
|
RU2705993C2 |
| ПЕРЕГОНКА С ЧАСТИЧНЫМ ПОТОКОМ | 2014 |
|
RU2665473C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОПИЛЕНОКСИДА | 2017 |
|
RU2734823C2 |
| СПОСОБ РЕГЕНЕРАЦИИ КАТАЛИЗАТОРА НА ОСНОВЕ ТИТАН-СОДЕРЖАЩЕГО ЦЕОЛИТА ДЛЯ ЭПОКСИДИРОВАНИЯ ПРОПИЛЕНА | 2016 |
|
RU2702349C2 |
| ИЗВЛЕЧЕНИЕ ПРОПЕНА ПОСРЕДСТВОМ ОЧИСТКИ В СКРУББЕРЕ СО СМЕСЬЮ РАСТВОРИТЕЛЬ/ВОДА | 2018 |
|
RU2766954C2 |
| СПОСОБ ЭПОКСИДИРОВАНИЯ С - С ОЛЕФИНОВ | 1996 |
|
RU2168504C2 |
| СПОСОБ ОЧИСТКИ ПРОПИЛЕНОКСИДА | 2017 |
|
RU2741991C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОПИЛЕНОКСИДА | 2014 |
|
RU2673676C2 |
| СПОСОБ ОЧИСТКИ ПРОПИЛЕНОКСИДА | 2017 |
|
RU2740188C1 |
| Способ получения пропиленоксида | 2014 |
|
RU2678844C2 |
Изобретение относится к способу отделения ацетонитрила от воды и может найти применение в процессах эпоксидирования пропилена пероксидом водорода. Предлагаемый способ отделения ацетонитрила от воды содержит стадии (i)-(iv). На стадии (i) предоставляют поток S1, содержащий, по меньшей мере, 95 мас.%, исходя из общего веса S1, ацетонитрила и воды, где массовое отношение ацетонитрил:вода составляет более 1. На стадии (ii) добавляют поток Р, содержащий, по меньшей мере, 95 мас.% С3, исходя из общего веса потока Р, к S1, получая смешанный поток S2, причем С3 представляет собой пропилен или пропилен, смешанный с пропаном с минимальным массовым отношением пропилен:пропан 7:3. На стадии (iii) подвергают поток S2 действию температуры, самое большее, 92°С и давления, по меньшей мере, 10 бар, получая первую жидкую фазу L1, состоящую, по существу, из С3, ацетонитрила и воды, и вторую жидкую фазу L2, состоящую, по существу, из воды и ацетонитрила, где массовое отношение ацетонитрил:вода в L2 составляет менее 1. На стадии (iv) отделяют жидкую фазу L1 от жидкой фазы L2. Способ позволяет эффективно отделять ацетонитрил от воды и рециркулировать отделенный ацетонитрил в процесс эпоксидирования пропилена. Изобретение относится также к высокоинтегрированному способу получения оксида пропилена. 2 н. и 26 з.п. ф-лы, 11 табл., 4 пр.
1. Способ отделения ацетонитрила от воды, содержащий
(i) предоставление потока S1, содержащего, по меньшей мере, 95 мас.%, исходя из общего веса S1, ацетонитрила и воды, где массовое отношение ацетонитрил:вода составляет более 1;
(ii) добавление потока Р, содержащего, по меньшей мере, 95 мас.% С3, исходя из общего веса потока Р, к S1, получая смешанный поток S2, причем С3 представляет собой пропилен или пропилен, смешанный с пропаном с минимальным массовым отношением пропилен:пропан 7:3;
(iii) подвергание S2 действию температуры, самое большее, 92°С и давления, по меньшей мере, 10 бар, получая первую жидкую фазу L1, состоящую, по существу, из С3, ацетонитрила и воды, и вторую жидкую фазу L2, состоящую, по существу, из воды и ацетонитрила, где массовое отношение ацетонитрил:вода в L2 составляет менее 1;
(iv) отделение L1 от L2.
2. Способ по п.1, где в (ii) жидкий поток Р добавляют к жидкому потоку S1.
3. Способ по п.1 или 2, где S1 содержит от 60 до 85 мас.% ацетонитрила и от 10 до 35 мас.% воды, в каждом случае исходя из общего веса S1.
4. Способ по п.1 или 2, где в (ii) Р добавляют к S1 в таком количестве, что массовое отношение С3:ацетонитрил в S2 находится в интервале от 0,2:1 до 5:1.
5. Способ по п.1 или 2, где в (iii) S2 подвергают воздействию температуры от 5 до 90°С и давления в интервале от 15 до 50 бар.
6. Способ по п.1 или 2, где, по меньшей мере, один агент, улучшающий разделение жидких фаз, добавляют к S1 в (i) и/или к S2 в (ii).
7. Способ по п.1 или 2, где в (iv) L1 отделяют от L2 в гравитационном отстойнике.
8. Способ по п.1 или 2, где, по меньшей мере, 95 мас.% L1 состоит из С3, ацетонитрила и воды, причем содержание воды в L1 составляет менее 10 мас.%, исходя из общего веса L1.
9. Способ по п.1 или 2, где, по меньшей мере, 95 мас.% L2 состоит из С3, ацетонитрила и воды, причем содержание С3 в L2 составляет, самое большее, 5 мас.%, исходя из общего веса L2, и содержание ацетонитрила в L2 составляет менее 45 мас.%, исходя из общего веса L2.
10. Способ по 1, дополнительно содержащий подвергание L1 стадии перегонки, из которой получают поток TL1, который содержит, по меньшей мере, 90 мас.% С3, исходя из общего веса TL1.
11. Способ по п.10, где из указанной стадии перегонки получают дополнительный поток BL1, причем, по меньшей мере, 95 мас.% BL1 состоит из С3, ацетонитрила и воды, где содержание С3 в BL1 находится в интервале от 7 до 18 мас.%, в каждом случае исходя из общего веса BL1.
12. Способ по п.10 или 11, где в указанной стадии перегонки используют одну перегонную колонну и где указанную перегонку проводят в точке росы в верхней части указанной перегонной колонны, составляющей, по меньшей мере, 40°С.
13. Способ по п.10 или 11, где, по меньшей мере, часть TL1 рециркулируют в (ii).
14. Способ по п.1, дополнительно содержащий подвергание L2 стадии перегонки, из которой получают поток TL2, который содержит от 75 до 95 мас.% ацетонитрила, исходя из общего веса TL2.
15. Способ по п.14, где, по меньшей мере, часть TL2 рециркулируют в (ii).
16. Способ по п.14, где указанная стадия перегонки представляет собой процесс перегонки при двух давлениях, где в первой перегонной колонне проводят перегонку при давлении верха, которое выше, чем давление верха во второй перегонной колонне, и где холодильник, используемый для конденсации верхнего потока первой перегонной колонны, используют одновременно в качестве испарителя второй перегонной колонны.
17. Способ по п.16, где
(aa) L2 вводят в первую перегонную колонну, из которой получают верхний паровой поток VTL2, содержащий от 50 до 70 мас.% ацетонитрила, исходя из общего веса верхнего потока VTL2; и
(bb) по меньшей мере, частично конденсируют VTL2, получаемый в (aa), и вводят сконденсированный поток во вторую перегонную колонну, из которой получают TL2 в качестве верхнего потока,
где холодильник, используемый для конденсации VTL2, одновременно используют в качестве испарителя второй перегонной колонны.
18. Способ по п.1 или 2, где по (i) получают S1 процессом, содержащим
(a) реакцию пропилена или пропилена, смешанного с пропаном, с пероксидом водорода в устройстве для реакции в присутствии ацетонитрила в качестве растворителя, получая поток S0, покидающий устройство для реакции, причем S0 содержит ацетонитрил, воду, оксид пропилена и непрореагировавший пропилен, кислород и пропан, если пропилен, смешанный с пропаном, реагирует с пероксидом водорода;
(b) выделение пропилена, кислорода и пропана из S0 с получением потока, состоящего, по существу, из ацетонитрила, воды и оксида пропилена;
(c) отделение оксида пропилена от S0 или потока, получаемого из (b), тем самым получая поток S1.
19. Способ по п.18, где в (а) пропилен вступает в реакцию с пероксидом водорода в присутствии гетерогенного катализатора.
20. Способ по п.18, где в (b) используют перегонную колонну и в верхнюю часть указанной перегонной колонны добавляют ацетонитрил или ацетонитрил, смешанный с водой, в качестве агента для отгонки легких фракций.
21. Способ по п.18, где, по меньшей мере, часть потока BL1 по п.11 рециркулируют в (а).
22. Способ по п.18, где перед (ii) S1 подвергают стадии гидрирования, причем полученный гидрированный поток подвергают стадии перегонки.
23. Способ по п.1, где S1 дополнительно содержит, по меньшей мере, один гликоль.
24. Способ по п.23, где, по меньшей мере, один гликоль выбран из группы, состоящей из пропиленгликоля, дипропиленгликоля, трипропиленгликоля и смеси из двух или трех из них.
25. Способ по п.23 или 24, где из первой перегонной колонны по п.16 или 17 получают нижний поток, содержащий, по меньшей мере, часть, по меньшей мере, одного гликоля, где указанный нижний поток подвергают стадии разделения гликолей или указанный нижний поток объединяют с нижним потоком, получаемым из второй перегонной колонны по п.16 или 17, перед подверганием стадии разделения гликолей.
26. Высокоинтегрированный способ получения оксида пропилена, причем этот способ содержит
(a) реакцию пропилена или пропилена, смешанного с пропаном, с пероксидом водорода в устройстве для реакции в присутствии ацетонитрила в качестве растворителя, получая поток S0, покидающий указанное устройство для реакции, причем S0 содержит ацетонитрил, воду, оксид пропилена и непрореагировавший пропилен, кислород и пропан, если пропилен, смешанный с пропаном, реагирует с пероксидом водорода;
(b) отделение пропилена или пропилена вместе с пропаном и кислорода из S0, получая поток, по меньшей мере, на 99 мас.% состоящий из ацетонитрила, воды и оксида пропилена;
(c) выделение оксида пропилена из S0 или потока, получаемого из (b), тем самым получая поток S1, содержащий, по меньшей мере, 95 мас.% ацетонитрила и воды, исходя из общего веса S1, где массовое отношение ацетонитрил:вода составляет более 1;
(d) добавление потока Р, содержащего, по меньшей мере, 95 мас.% С3, исходя из общего веса потока Р, к S1, получая смешанный поток S2, причем С3 представляет собой пропилен или пропилен, смешанный с пропаном, с минимальным массовым отношением пропилен:пропан 7:3;
(e) подвергание S2 действию температуры, самое большее, 92°С и давления, по меньшей мере, 10 бар, получая первую жидкую фазу L1, состоящую, по существу, из С3, ацетонитрила и воды, и вторую жидкую фазу L2, состоящую, по существу, из воды и ацетонитрила, где массовое отношение ацетонитрил:вода в L2 составляет менее 1;
(f) отделение L1 от L2;
(g) подвергание L1 стадии перегонки, из которой получают поток TL1, который содержит, по меньшей мере, 90 мас.% С3, исходя из общего веса TL1; и из которой получают другой поток BL1, причем, по меньшей мере, 95 мас.% BL1 состоит из С3, ацетонитрила и воды, где содержание С3 в BL1 находится в интервале от 7 до 18 мас.%, исходя из общего веса BL1;
(h) подвергание L2 стадии перегонки, из которой получают поток TL2, который содержит от 75 до 95 мас.% ацетонитрила, исходя из общего веса TL2;
где, по меньшей мере, часть потока BL1 рециркулируют в (а); и/или
где, по меньшей мере, часть TL1 рециркулируют в (d); и/или
где, по меньшей мере, часть TL2 рециркулируют в (d).
27. Способ по п.26, где BL1 рециркулируют в (а), TL1 рециркулируют в (d) и TL2 рециркулируют в (d).
28. Способ по п.26 или 27, дополнительно содержащий
(y) подвергание потока, получаемого из (с), стадии гидрирования;
(z) подвергание потока, получаемого из (y), стадии перегонки для получения потока S1 и подвергание S1 стадии (d).
| ШТЫРЕВОЙ ИЗОЛЯТОР | 1991 |
|
RU2014654C1 |

