Область техники, к которой относится изобретение
Изобретение относится к фенилэтинильным производным, которые являются ингибиторами вируса гепатита C (HCV), и к их применению, отдельно или в комбинации с другими ингибиторами HCV, при лечении или профилактике HCV.
Предшествующий уровень техники
HCV представляет собой одноцепочечный (+)РНК вирус, принадлежащий к вирусному семейству Flaviviridae, к роду гепатовирусов. Вирусный геном транслируется в одиночную открытую рамку считывания, кодирующую разнообразные структурные и неструктурные белки.
Как последствие первоначальной острой инфекции, у большинства инфицированных пациентов развивается хронический гепатит из-за того, что происходит репликация HCV преимущественно в гепатоцитах, но без явного цитопатологического проявления. В частности, недостаток интенсивного ответа Т-лимфоцитов и сильная предрасположенность вируса к мутации, по всей видимости, способствуют высокой скорости развития хронического инфекционного процесса. Хронический гепатит может развиваться в фиброз печени, приводящий к циррозу, конечной стадии заболевания печени, и в гепатоклеточную карциному, что делает его основной причиной необходимости трансплантации печени.
Существует шесть основных генотипов HCV и более 50 подтипов, которые географически по-разному распределены. HCV генотипа 1 представляет собой генотип, преобладающий в Европе и США. Экстенсивная генетическая гетерогенность HCV обуславливает существенные осложнения при диагностике и изучении симптомов течения болезни, может служить объяснением трудностей в создании вакцины и недостаточную действенность существующей в настоящее время терапии.
Передача HCV может происходить при контакте с зараженной кровью или препаратами крови, например, вследствие переливания крови или внутривенного применения лекарственного средства. Внедрение диагностических тестов, применяемых при скрининге крови, приводит к тенденции понижения числа случаев постинфузионного HCV. Несмотря на то что обеспечивается медленное развитие конечной стадии заболевания печени, существующие инфекции будут оставаться, представляя собой серьезную медицинскую проблему и экономическое бремя на десятилетия.
Существующая в настоящее время терапия HCV основывается на (пегилированном) интерфероне-альфа (IFN-α) в комбинации с рибавирином. Эта комбинированная терапия дает поддерживающий вирусологический контроль у более чем 40% пациентов, инфицированных HCV генотипа 1, и приблизительно у 80% пациентов, инфицированных HCV генотипов 2 и 3. Кроме ограниченной эффективности против HCV генотипа 1, эта комбинированная терапия имеет существенные побочные эффекты и плохо переносима многими пациентами. Большинство побочных эффектов включают подобные гриппу симптомы, гематологические нарушения и психоневрологические симптомы. Следовательно, существует потребность в более эффективных, удобных и лучше переносимых методах лечения.
Опыт применения лекарственных средств против HIV, в частности с ингибиторами HIV протеазы, указывает на то, что недостаточно оптимальная фармакокинетика и сложный режим дозирования незамедлительно приводят к непреднамеренным ошибкам в соблюдении терапевтических рекомендаций. Это, в свою очередь, означает, что на 24 часу самая низшая точка концентрации (минимальная концентрации в плазме) для соответствующих лекарственных средств при HIV состоянии часто опускается ниже предельной величины IC90 или ED90 для большей части суток. Следует принять во внимание, что на 24 часу самый низший уровень значений по меньшей мере для IC50 и, более реально, для IC90 или ED90, весьма важен для сдерживания проявления «ускользнувших» от лекарственного средства мутантов. Достижение необходимой фармакокинетики и метаболизма лекарственного средства, которые обеспечивают такие самые низшие уровни значений, определяют точные перспективы создания лекарственного средства.
NS5A белок HCV располагается после NS4B белка и перед NS5B белком. На стадии послетрансляционного расщепления вирусной сериновой протеазой NS3/4A белок NS5A доводится до зрелого состояния в виде цинксодержащего трехдоменного фосфорилированного белка, присутствующего либо в форме гипофосфорилированной (56-кДа, p56), либо в форме гиперфосфорилированной (58-кДа, p58) его разновидностей. NS5A белок HCV вовлечен в многообразные аспекты вирусного жизненного цикла, включая вирусную репликацию и сборку инфекционных частиц, а также модуляцию окружения его клетки-хозяина. Несмотря на то, что белку не приписывали какой-либо ферментативной функции, сообщалось, что он взаимодействует с многочисленными вирусными и клеточными факторами.
В ряде патентов и патентных заявок раскрыты соединения с ингибирующей активностью в отношении NS5A HCV. В WO 2006/133326 раскрыты производные стильбена, в то время как в WO 2008/021927 и WO 2008/021928 раскрыты бифенильные производные, обладающие ингибирующей активностью в отношении NS5A HCV. В WO 2008/048589 раскрыты производные 4-(фенилэтинил)-1Н-пиразола и их противовирусное применение. В WO-2010/065668 раскрыты 5-(фенилэтинил)-1Н-имидазолы и их противовирусная активность.
Существует потребность в ингибиторах HCV, которые могут преодолеть недостатки существующей в настоящее время терапии HCV, такие как побочные эффекты, ограниченная эффективность, возникновение устойчивости и отсутствие соблюдения режима терапии, а также обеспечение устойчивого вирусологического ответа.
Настоящее изобретение относится к группе ингибирующих HCV фенилэтинильных производных с полезными свойствами, касающихся одного или более следующих параметров: антивирусная эффективность, благоприятный профиль развития устойчивости, пониженный уровень или отсутствие токсичности и генотоксичности, подходящая фармакокинетика и фармакодинамика, простота изготовления лекарственной формы и введения и ограниченные или отсутствующие межлекарственные взаимодействия с другими лекарственными средствами, а именно с другими противо-HCV средствами.
Соединения изобретения также могут быть перспективны благодаря тому факту, что у них отсутствует активность против других вирусов, а именно против HIV. HIV-инфицированные пациенты часто страдают сопутствующими инфекциями, такими как HCV. Лечение таких пациентов с помощью ингибитора HCV, который также ингибирует и HIV, может привести к появлению устойчивых HIV штаммов.
Описание сущности изобретения
В одном из аспектов настоящее изобретение предоставляет соединения, которые могут быть представлены формулой I-а

включая любые возможные их стереоизомеры, где
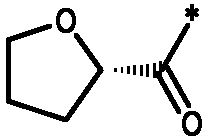
R представляет собой -CR1R2R3, алкиламино, бензиламино, ариламино, арил, необязательно замещенный 1 или 2 заместителями, выбранными из галогена и метила, гетеро(C4-7)циклоалкил;
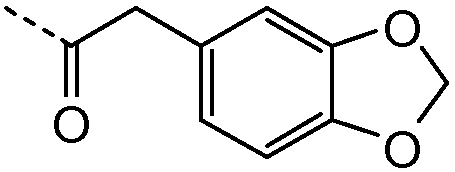
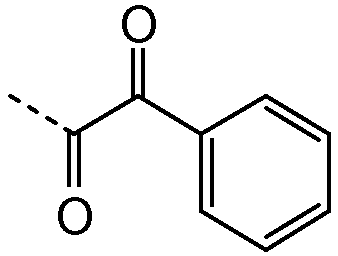
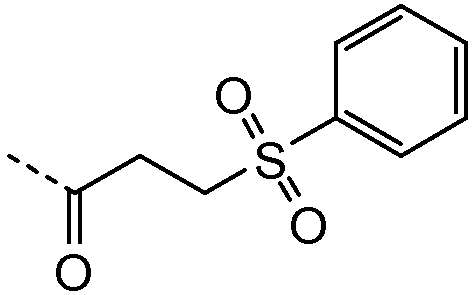
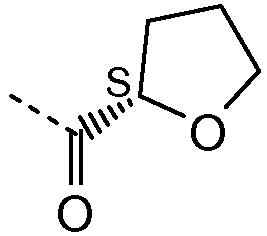
R1 выбирают из C1-4алкила; фенила, необязательно замещенного 1, 2 или 3 заместителями, независимо выбранными из галогена, C1-4алкила, C1-4алкокси, трифторметокси или 2 заместителей у соседних атомов кольца, образующих 1,3-диоксолановую группу; бензила, необязательно замещенного галогеном или метокси; фенилсульфонилметила; гетероарила; гетероарилметила и C3-6циклоалкила;
R2 выбирают из водорода, гидроксила, амино, моно- и ди-C1-4алкиламино, C1-4алкилкарбониламино, C1-4алкилоксикарбониламино, C1-4алкиламинокарбониламино, пиперидин-1-ила или имидазол-1-ила;
R3 представляет собой водород или, альтернативно, R2 и R3 вместе образуют оксогруппу или циклопропил;
или их фармацевтически приемлемые соль и/или сольват.
В дополнительном аспекте изобретение относится к применению соединений формулы I, как указано в данном описании, для ингибирования HCV. Альтернативно, предусматривается применение для производства лекарственного средства соединения формулы I или любой его подгруппы, как указано в данном описании.
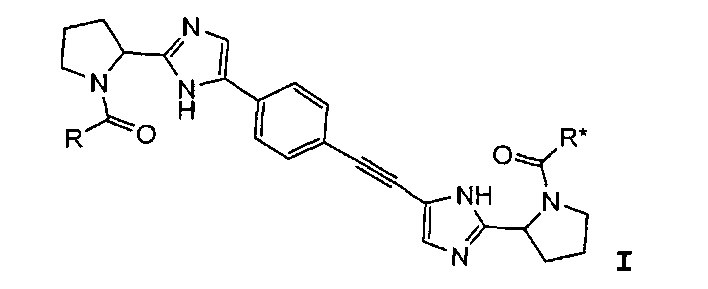
Первый вариант осуществления настоящего изобретения относится к соединению формулы I
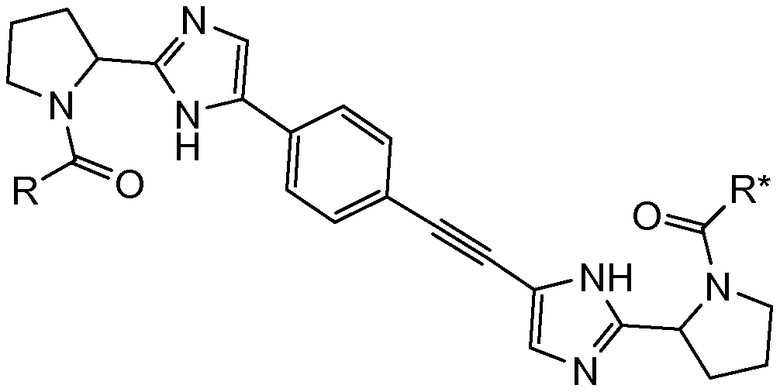 I
I
или его стереоизомеру, где
R и R* представляют собой, каждый независимо, -CR1R2R3, C1-4алкиламино, бензиламино, ариламино, арил, необязательно замещенный 1 или 2 заместителями, выбранными из галогена и метила, гетеро(C3-6)циклоалкил; где
R1 выбирают из C1-4алкила; фенила, необязательно замещенного 1, 2 или 3 заместителями, независимо выбранными из галогена, C1-4алкила, C1-4алкокси, трифторметокси или 2 заместителей у соседних атомов кольца, образующих 1,3-диоксолановую группу; бензила, необязательно замещенного галогеном или метокси; фенилсульфонилметила; гетероарила; гетероарилметила и C3-6циклоалкила;
R2 выбирают из водорода, гидроксила, амино, моно- и ди-C1-4алкиламино, C1-4алкилкарбониламино, C1-4алкилоксикарбониламино, C1-4алкиламинокарбониламино, пиперидин-1-ила или имидазол-1-ила; и
R3 представляет собой водород, или
R2 и R3 вместе образуют оксогруппу; или
R1 и R3 вместе образуют циклопропил;
при условии, что если один из R или R* представляет собой -CH(C6H5)N(CH3)2, тогда другой не может представлять собой -CH(C6H5)NHC(=O)OCH3; и если R и R* являются одинаковыми, тогда R1 отличен от фенила, когда R2 представляет собой гидроксил, ацетиламино, метоксикарбониламино или трет-бутоксикарбониламино, и R3 представляет собой водород; и R1 отличается от C1-4алкила, когда R2 представляет собой C1-4алкилоксикарбониламино, и R3 представляет собой водород;
или его фармацевтически приемлемой соли или его сольвату.
Подгруппы соединений формулы I представляют собой такие соединения формулы I или подгруппы соединений формулы I, как определено в данном описании, где R и R* представляют собой -CR1R2R3 или бензиламино; в частности, где R и R* представляют собой -CR1R2R3.
Подгруппы соединений формулы I представляют собой такие соединения формулы I или подгруппы соединений формулы I, как определено в данном описании, где R2 представляет собой водород, гидроксил, диметиламино, ацетиламино, метоксикарбониламино, метиламинокарбониламино, пиперидин-1-ил или имидазол-1-ил.
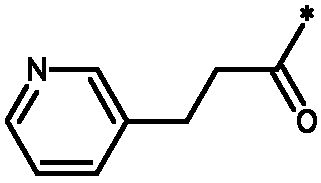
Подгруппы соединений формулы I представляют собой такие соединения формулы I или подгруппы соединений формулы I, как определено в данном описании, где R1 выбирают из C1-4алкила; фенила, необязательно замещенного 1 или 2 заместителями, независимо выбранными из галогена, метила, метокси или 2 заместителей у соседних атомов кольца, образующих 1,3-диоксолановую группу; гетероарила и гетероарилметила. В частности, гетероарил представляет собой пиридинил, а именно пиридин-3-ил.
В дополнительном аспекте изобретение предоставляет соединение формулы I или его фармацевтически приемлемую соль, гидрат или сольват для применения при лечении или профилактике (или в производстве лекарственного средства для лечения или профилактики) HCV инфекции. Репрезентативные генотипы HCV в контексте лечения или профилактики, в соответствии с изобретением, включают генотип 1b (преобладающий в Европе) или 1a (преобладающий в Северной Америке). Изобретение также предоставляет способ лечения или профилактики HCV инфекции, а именно для случаев генотипа 1a или 1b, включающий введение нуждающемуся в этом пациенту терапевтически эффективного количества соединения, как определено в данном описании.
Стереоизомерно чистые формы соединений и промежуточных продуктов, приведенных в данном описании, определяются как изомеры, по существу свободные от других энантиомерных или диастереомерных форм таких же базовых молекулярных структур указанных соединений или промежуточных продуктов. А именно термин "стереоизомерно чистые" имеет отношение к соединениям или к промежуточным продуктам, имеющим стереоизомерный избыток по меньшей мере от 80% (т.e. не менее 90% одного изомера и не более 10% других возможных изомеров) и до стереоизомерного избытка 100% (т.e. 100% одного изомера и полное отсутствие других), более конкретно, имеет отношение к соединениям или к промежуточным продуктам, имеющим стереоизомерный избыток от 90% вплоть до 100%, еще более конкретно, к имеющим стереоизомерный избыток от 94% вплоть до 100%, и наиболее конкретно, к имеющим стереоизомерный избыток от 97% вплоть до 100% или от 98% вплоть до 100%. Термины "энантиомерно чистые" и "диастереоизомерно чистые" следует понимать аналогичным образом, но в данном случае как имеющие отношение к энантиомерному избытку и к диастереоизомерному избытку, соответственно, при анализе смеси.
Чистые стереоизомерные формы соединений и промежуточных продуктов настоящего изобретения могут быть получены посредством применения методик, известных в данной области техники. Например, энантиомеры могут быть отделены один от другого путем селективной кристаллизации их диастереомерных солей с оптически активными кислотами или основаниями. Их примерами являются виноградная кислота, дибензоилвиноградная кислота, дитолилвиноградная кислота и камфорсульфоновая кислота. Альтернативно, энантиомеры могут быть разделены хроматографическими методами с использованием хиральных неподвижных фаз. Указанные химически чистые стереоизомерные формы также могут быть получены из соответствующих химически чистых стереоизомерных форм подходящих исходных веществ, при условии, что реакция протекает стереоспецифично. Предпочтительно, в том случае, если необходим определенный стереоизомер, указанное соединение синтезируют посредством стереоспецифических способов получения. Для таких способов предпочтительно использовать энантиомерно чистые исходные вещества.
Диастереомерные рацематы соединений формулы I могут быть получены в разделенном виде посредством общепринятых способов. Подходящие физические способы разделения, которые успешно могут быть использованы, представляют собой, например, селективную кристаллизацию и хроматографию, например колоночную хроматографию или хроматографию со сверхкритической подвижной фазой.
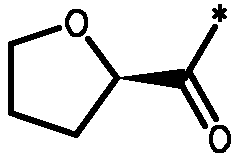
Соединения формулы I имеют несколько центров хиральности. Представляют интерес стереогенные центры пирролидиновых колец, т.e. при 2-углероде. Конфигурация в этом положении может быть такой, которая соответствует L-пролину, т.e.
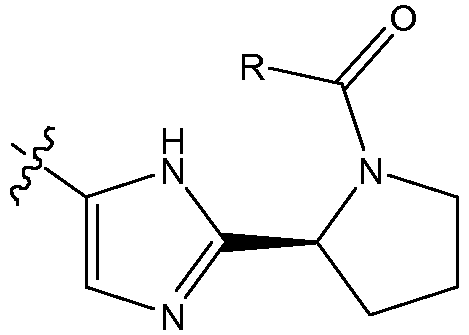 ,
,
или такой, которая соответствует D-пролину, т.e.
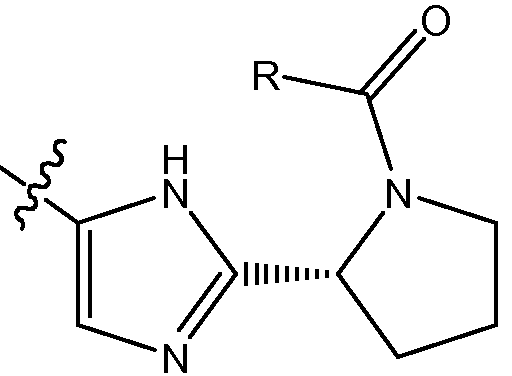 .
.
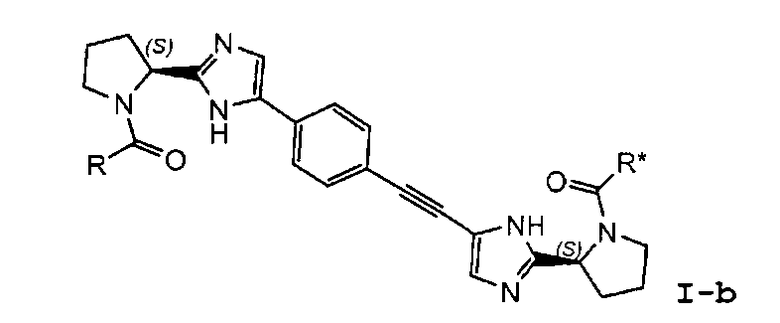
В частности, представляют интерес соединения формулы I или подгруппы соединений формулы I, как определено в данном описании, которые соответствуют формуле I-b:
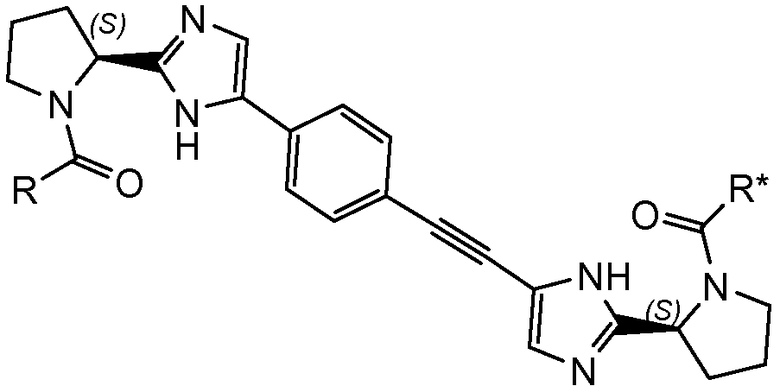 I-b
I-b
Также представляет интерес конфигурация группы -CR1R2R3: если R1 выбирают из C1-4алкила, необязательно замещенного метоксигруппой, гидроксилом или диметиламиногруппой; C3-6циклоалкила и тетрагидропиранила, тогда предпочтительной является S-конфигурация; если R1 выбирают из фенила, необязательно замещенного 1, 2 или 3 заместителями, независимо выбранными из галогена, C1-4алкокси, трифторметокси или 2 заместителей у соседних атомов кольца, образующих 1,3-диоксолановую группу, и гетероарила; тогда предпочтительной является R-конфигурация.
Фармацевтически приемлемые аддитивные соли включают терапевтически активные нетоксичные формы соединений формулы I или их подгрупп в виде аддитивных солей кислот или оснований. Представляют интерес и свободные, т.e. несолевые формы соединений формулы I или любой подгруппы соединений формулы I, указанных в данном описании.
Фармацевтически приемлемые кислотно-аддитивные соли могут быть получены общепринятыми способами, путем обработки основной формы подходящей кислотой. Подходящие кислоты включают, например, неорганические кислоты, такие как галогеноводородные кислоты, например хлористоводородная или бромистоводородная кислота, серная, азотная, фосфорная и подобные кислоты; или органические кислоты, такие как, например, уксусная, пропионовая, гидроксиуксусная, молочная, пировиноградная, щавелевая (т.e. этандиовая), малоновая, янтарная (т.e. бутандиовая кислота), малеиновая, фумаровая, яблочная (т.e. гидроксибутандиовая кислота), виноградная, лимонная, метансульфоновая, этансульфоновая, бензолсульфоновая, п-толуолсульфоновая, цикламовая, салициловая, п-аминосалициловая, памовая и подобные кислоты. В противоположность этому, указанные солевые формы могут быть преобразованы в свободные основные формы обработкой подходящим основанием. Одно соединение формулы I, в зависимости от количества присутствующих в его структуре основных функциональных групп, может образовать соль с одной, двумя или несколькими молекулами кислоты.
Соединения формулы I, содержащие кислотный протон, могут также быть преобразованы в их аддитивные соли оснований, в частности в формы аддитивных солей металла или амина, при обработке подходящими органическими или неорганическими основаниями. Подходящие аддитивные соли оснований включают, например, аммонийные соли, соли щелочного или щелочноземельного металла, например литиевые, натриевые, калиевые, магниевые, кальциевые соли и подобные, соли с органическими основаниями, например соли бензатина, N-метил-D-глюкамина, гидрабамина, и соли с аминокислотами, такими как, например, аргинин, лизин и подобные.
Термин "сольваты" охватывает любые фармацевтически приемлемые сольваты, которые способны образовывать соединения формулы I, а также их соли. Такие сольваты представляют собой, например, гидраты, алкоголяты, например этаноляты, пропаноляты и подобные.
Некоторые соединения формулы I также могут существовать в виде их таутомерных форм. Например, таутомерные формы амидных групп (-C(=O)-NH-) представляют собой иминоспирты (-C(OH)=N-). Таутомерные формы, несмотря на то, что они точно не указаны в структурных формулах, представленных в данном описании, подразумеваются включенными в объем настоящего изобретения.
Как используется в данном описании, "C1-4алкил", как группа или часть группы, означает насыщенные, с прямой или разветвленной цепью углеводородные группы, имеющие от 1 до 4 атомов углерода, такие как, например, метил, этил, 1-пропил, 2-пропил, 1-бутил, 2-бутил, 2-метил-1-пропил, 2-метил-2-пропил. Для целей настоящего изобретения из числа C1-4алкилов представляет интерес C3-4алкил, т.е. углеводородная группа с прямой или с разветвленной цепью, имеющая 3 или 4 атома углерода, такая как 1-пропил, 2-пропил, 1-бутил, 2-бутил, 2-метил-1-пропил, 2-метил-2-пропил. Особый интерес представляет разветвленный C3-4алкил, такой как 2-пропил, 2-бутил, 2-метил-1-пропил, 2-метил-2-пропил.
Термин "C3-6циклоалкил", как группа или ее часть, означает насыщенные циклические углеводородные группы, имеющие от 3 до 6 атомов углерода, которые вместе образуют циклическую структуру. Примеры C3-6циклоалкила включает циклопропил, циклобутил, циклопентил и циклогексил.
"C1-4алкокси", как группа или часть группы, означает группу формулы -O-C1-4алкил, где C1-4алкил является таким, как определено выше. Примеры C1-4алкокси представляют собой метокси, этокси, н-пропокси или изопропокси.
Термин "галоген" является общим для фтора, хлора, брома и йода.
Как использовано в данном описании, термин "(=O)" или "оксо" создает карбонильную функциональную группу при связывании с атомом углерода. Следует отметить, что этот атом только тогда может быть с заместителем в виде оксогруппы, когда валентность этого атома дает такую возможность.
Как использовано в данном описании, с целью определения термина "арил", как группу или ее часть, значения ароматической кольцевой структуры необязательно включают один или два гетероатома, выбранных из N, O и S, в частности, из N и O. Указанная ароматическая кольцевая структура может иметь 5 или 6 атомов в кольце. Примеры представляют собой фенил, пиридинил, оксазолил и подобные.
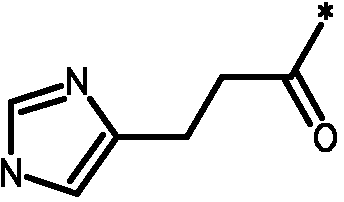
Как использовано в данном описании, приставка "гетеро-" означает, что группа включает или дополнительно имеет по меньшей мере 1 гетероатом, выбранный из N, O и S, в частности из N и O. Например, термин "гетероарил" означает ароматическую кольцевую структуру, как определено для термина "арил", включающую по меньшей мере 1 гетероатом, выбранный из N, O и S, в частности из N и O, например имидазолил, фуранил, пиридинил, оксазолил и подобные. Альтернативно, термин "гетероС4-7циклоалкил" означает насыщенную циклическую углеводородную группу, как определено для "C3-6циклоалкила", дополнительно включающую по меньшей мере 1 гетероатом, выбранный из N, O и S, в частности из N и O, например тетрагидрофуранил, тетрагидропиранил, пирролидинил, пиперидинил, морфолинил, пиперазинил и подобные.
Если положение группы на молекулярном фрагменте точно не указано (например, заместитель в фениле) или представлено плавающей связью, тогда такая группа может располагаться у любого атома такого фрагмента, при условии, что полученная в результате структура является химически стабильной. Если какие-либо переменные присутствуют в молекуле более чем один раз, каждое определенное является независимым.
Всякий раз, когда используется в данном описании термин "соединения формулы I", или "настоящие соединения", или аналогичные термины, подразумевается, что он охватывает соединения формулы I, включая возможные стереоизомерные формы и их фармацевтически приемлемые соли и сольваты.
Общие методы синтеза
Соединения изобретения, где R и R* являются одинаковыми, могут быть получены путем ацилирования фенилэтинильного остова формулы III подходящей кислотой формулы R-C(=O)-OH, где R имеет значение, как определено для соединений формулы I или любой их подгруппы.
 III
III
Указанное ацилирование может быть осуществлено путем взаимодействия исходных веществ в присутствии агента сочетания или путем преобразования карбоксильной функциональной группы в активную форму, такую как активированный сложный эфир, смешанный ангидрид, или хлорангидрид, или бромангидрид карбоновой кислоты. Общее описание таких реакций сочетания и реагентов, используемых при их осуществлении, можно найти в общих руководствах по химии пептидов, например, M.Bodanszky, "Peptide Chemistry", 2nd rev. ed., Springer-Verlag, Berlin, Germany, (1993).
Примеры реакций сочетания для ацилирования аминогруппы или для образования амидной связи включают азидный метод, метод смешанных ангидридов карбонильно-карбоксильный (изобутилхлорформиат), карбодиимидный метод (дициклогексилкарбодиимид, диизопропилкарбодиимид или водорастворимый карбодиимид, такой как N-этил-N'-[3-(диметиламино)пропил]карбодиимид), метод активированных сложных эфиров (например, п-нитрофенил, п-хлорфенил, трихлорфенил, пентахлорфенил, пентафторфенил, N-гидроксисукцинимид и подобные сложные эфиры), метод с К-реагентом Вудворда, 1,1-карбонилдиимидазольный метод (CDI или N,N'-карбонилдиимидазол), метод с фосфорсодержащими реагентами или окислительно-восстановительные методы. Некоторые из этих методов могут быть улучшены посредством добавления подходящих катализаторов, например в карбодиимидном методе путем добавления 1-гидроксибензотриазола или 4-диметиламинопиридина (4-DMAP). Кроме того, агенты сочетания представляют собой гексафторфосфат (бензотриазол-1-илокси)трис(диметиламино)фосфония, либо сам по себе, либо в присутствии 1-гидроксибензотриазола или 4-DMAP; или тетрафторборат 2-(1H-бензотриазол-1-ил)-N,N,N',N'-тетраметилурония (TBTU), или гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (HATU). Такие реакции сочетания можно осуществлять либо в растворе (жидкая фаза), либо в твердой фазе. Для целей настоящего изобретения предпочтительным методом ацилирования является такой, который проводят с использованием HATU.
Реакции сочетания предпочтительно проводить в инертном растворителе, таком как галогенированные углеводороды, например дихлорметан, хлороформ, биполярные апротонные растворители, такие как ацетонитрил, диметилформамид, диметилацетамид, диметилсульфоксид (ДМСО), гексаметилфосфортриамид (HMPT), простые эфиры, такие как тетрагидрофуран (ТГФ).
Во многих случаях реакции сочетания проводят в присутствии подходящего основания, такого как третичный амин, например триэтиламин, диизопропилэтиламин (DIPEA), N-метилморфолин, N-метилпирролидин, 4-DMAP или 1,8-диазабицикло[5.4.0]ундец-7-ен (DBU). Температура реакции может находиться в диапазоне от 0°С до 50°С, и время реакции может составлять диапазон от 15 мин до 24 ч.
Фенилэтинильный остов формулы III может быть получен согласно пути синтеза, проиллюстрированного в примере 1, описанном в данном описании. Промежуточное соединение I-9 представляет собой соединение формулы III, имеющее (S,S) конфигурацию пирролидиновых фрагментов. Альтернативные соединения формулы III, где конфигурация пирролидиновых фрагментов не относится к (S,S), могут быть получены, используя такой же путь синтеза, с использованием N-Boc-пролина другой конфигурации при синтезе промежуточного соединения I-11.
В дополнительном аспекте настоящее изобретение относится к фармацевтической композиции, содержащей терапевтически эффективное количество соединения формулы I, как указано в данном описании, и фармацевтически приемлемый носитель. Терапевтически эффективное количество в этом контексте означает такое количество, которое является достаточным для воздействия в качестве профилактической меры против инфекции HCV, для стабилизации или ослабления инфекции HCV у инфицированных субъектов или субъектов в состоянии риска заражения. В еще дополнительном аспекте изобретение относится к способу получения фармацевтической композиции, как указано в данном описании, включающему тщательное смешивание фармацевтически приемлемого носителя с терапевтически эффективным количеством соединения формулы I, как указано в данном описании.
Следовательно, соединения настоящего изобретения или любая их подгруппа могут быть включены в состав различных фармацевтических форм, предназначенных для целей введения. В качестве подходящих композиций можно привести все композиции, обычно используемые для систематически вводимых лекарственных средств. Для того чтобы получить фармацевтические композиции настоящего изобретения, эффективное количество отдельного соединения, необязательно в виде аддитивной соли или в виде металлокомплекса, в качестве активного ингредиента объединяют при тщательном смешивании с фармацевтически приемлемым носителем, который можно использовать в широко разнообразных формах, в зависимости от требуемой для введения формы. Эти фармацевтические композиции желательны в единичной дозированной форме, в особенности удобной для перорального введения, для ректального введения, для введения через кожу или для парентеральной инъекции. Например, при получении композиций в пероральной дозированной форме может применяться любая, обычно используемая фармацевтическая среда, такая как, например, вода, гликоли, масла, спирты и подобные, в случае пероральных жидких лекарственных средств, таких как суспензии, сиропы, эликсиры, эмульсии и растворы; или твердые носители, такие как крахмалы, сахара, каолин, лубриканты, связующие вещества, дезинтегранты и подобные в случае порошков, пилюль, капсул и таблеток. Из-за простоты введения таблетки и капсулы являются преимущественными пероральными единичными дозированными формами, в случае которых, конечно, используют твердые фармацевтические носители. Для парентеральных композиций, как правило, носитель будет включать стерильную воду, по меньшей мере, по большей части, однако могут быть включены и другие ингредиенты, например, способствующие растворимости. Растворы для инъекций, например, могут быть получены таким образом, чтобы носитель состоял из солевого раствора, раствора глюкозы или смеси солевого и глюкозного растворов. Суспензии для инъекций также могут быть получены таким образом, чтобы в этом случае могли использоваться подходящие жидкие носители, суспендирующие агенты и подобные. Также включены твердые лекарственные формы, предназначенные для преобразования непосредственно перед применением в жидкие лекарственные формы. В композициях, удобных для введения через кожу, носитель необязательно включает усиливающее проникновение вещество и/или подходящее увлажняющее вещество, необязательно комбинированные с подходящими добавками любой природы в незначительных пропорциях, причем эти добавки не привносят ощутимого вредного воздействия на кожу. Соединения настоящего изобретения также могут вводиться посредством пероральной ингаляции или инсуффляции в форме раствора, суспензии или сухого порошка с использованием любых известных в данной области систем доставки.
Особо предпочтительным является составление указанных выше фармацевтических композиций в виде единичной дозированной формы для простоты введения и равномерности дозирования. Единичная дозированная форма, как используется в данном описании, означает физически дискретные единицы, подходящие в качестве одноразовых дозировок, причем каждая единица содержит заранее определенное количество активного ингредиента, рассчитанное таким образом, чтобы вызвать требуемый терапевтический эффект в сочетании с требуемым фармацевтическим носителем. Примерами таких единичных дозированных форм являются таблетки (включая таблетки с насечкой или покрытые оболочкой таблетки), капсулы, пилюли, суппозитории, пакетики с порошком, пластинки, растворы или суспензии для инъекций и подобные и их многочисленные отдельные виды.
Соединения формулы I проявляют активность против HCV и могут применяться для лечения и профилактики инфекции HCV или заболеваний, связанных с HCV. Последние включают прогрессирующий фиброз печени, воспаление и некроз, приводящие к циррозу, последней стадии заболевания печени и гепатоклеточной карциноме. Кроме того, известно, что ряд соединений настоящего изобретения активен против мутантных штаммов HCV. Кроме того, соединения настоящего изобретения могут обладать благоприятными свойствами в отношении биодоступности, демонстрировать подходящий фармакокинетический профиль, включая приемлемый период полувыведения, площадь под кривой (AUC) и пиковые и минимальные значения характеристик, и характеризоваться отсутствием неблагоприятных проявлений, таких как недостаточно быстрое проникновение и задержка в тканях.
Противовирусная активность in vitro в отношении HCV для соединений формулы I может быть протестирована на клеточной модели HCV в системе контроля репликации, основываясь на публикации Lohmann et al. (1999) Science 285:110-113, с дополнительными модификациями, описанными в публикации Krieger et al. (2001) Journal of Virology 75:4614-4624 (включены в данное описание посредством ссылок), что в дальнейшем подтверждено примерами в разделе примеров. Эта модель, несмотря на то что не представляет собой в полной мере всеохватывающую модель инфекции HCV, является широко используемой в качестве наиболее понятной и эффективной модели автономной репликации РНК вируса HCV, доступной в настоящее время. Следует принять во внимание, что важно отличать соединения, которые специфически препятствуют функционированию HCV, от таких соединений, которые вызывают цитотоксическое или цитостатическое воздействие на модели репликации HCV и, как следствие, вызывают снижение концентрации РНК вируса HCV или концентрации связанного репортерного фермента. Анализы, известные в области определения цитотоксичности на клеточном уровне, базируются, например, на активности митохондриальных ферментов с использованием красителей, флуоресцирующих при окислительно-восстановительных процессах, таких как резазурин. Более того, обратный скрининг клеток показывает оценку неселективного ингибирования активности связанного репортерного гена, такого как ген люциферазы светлячка. Подходящие типы клеток можно снабдить посредством стабильной трансфекции геном-репортером люциферазы, у которого экспрессия зависит от конститутивно активного генного промотора, и такие клетки можно использовать в качестве обратного скрининга при проведении отбора для исключения неселективных ингибиторов.
Благодаря их противовирусным свойствам, в частности их противо-HCV свойствам, соединения формулы I, как указано в данном описании, могут быть использованы для ингибирования HCV репликации, в частности, при лечении теплокровных животных, в частности людей, инфицированных HCV, и для профилактики инфекций HCV. Настоящее изобретение, кроме того, относится к способу лечения теплокровного животного, в частности человека, инфицированного HCV или находящегося в состоянии риска инфицирования HCV, включающему введение противо-HCV эффективного количества соединения формулы I, как указано в данном описании.
Соединения формулы I, как указано в данном описании, следовательно, могут применяться в качестве лекарственных средств, в частности в качестве лекарственных средств против HCV. Указанное применение в качестве лекарственного средства или способ лечения включает системное введение инфицированным вирусом HCV субъектам или субъектам, восприимчивым к инфекции HCV, количества лекарственного средства, которое эффективно для борьбы с состояниями, связанными с инфекцией HCV.
Настоящее изобретение также относится к применению настоящих соединений в производстве лекарственного средства для лечения или предотвращения инфекции HCV.
Кроме того, соединения изобретения также могут представлять интерес благодаря тому факту, что они не имеют активности против других вирусов, в частности против HIV. Инфицированные HIV пациенты часто страдают сопутствующими инфекциями, такими как HCV. Лечение таких пациентов с помощью ингибитора HCV, который также ингибирует и HIV, может приводить к появлению устойчивых HIV штаммов.
Как правило, предполагают, что противовирусное эффективное дневное количество должно составлять приблизительно от 0,01 до приблизительно 50 мг/кг, или приблизительно от 0,02 до приблизительно 30 мг/кг массы тела. Может быть уместно вводить необходимую дозу в виде двух, трех, четырех или более субдоз через определенные интервалы времени в течение дня. Указанные субдозы могут быть сформулированы в виде единичных дозированных форм, например, содержащих приблизительно от 1 до приблизительно 1000 мг, или приблизительно от 1 до приблизительно 500 мг, или приблизительно от 1 до приблизительно 100 мг, или приблизительно от 2 до приблизительно 50 мг активного ингредиента на единичную дозированную форму.
Комбинированная терапия
Изобретение также относится к комбинации соединения формулы I, его фармацевтически приемлемой соли или сольвата и другого противовирусного соединения, а именно другого противо-HCV соединения. Термин "комбинация" относится к продукту, содержащему (a) соединение формулы I, как определено выше, и (b) другой противо-HCV ингибитор, в качестве комбинированного лекарственного средства для одновременного, раздельного или последовательного применения при лечении инфекций HCV.
Комбинации настоящего изобретения могут быть использованы в качестве лекарственных средств. Таким образом, настоящее изобретение относится к применению соединения формулы (I) или любой его подгруппы, как определено выше, для производства лекарственного средства, пригодного для ингибирования активности HCV у инфицированных вирусом HCV млекопитающих, где указанное лекарственное средство применяется в комбинированной терапии, причем указанная комбинированная терапия, в частности, включает соединение формулы (I) и по меньшей мере один другой противо-HCV агент, например IFN-α, пегилированный IFN-α, рибавирин, альбуферон, тарибавирин, нитазоксанид, Debio025 или их комбинацию.
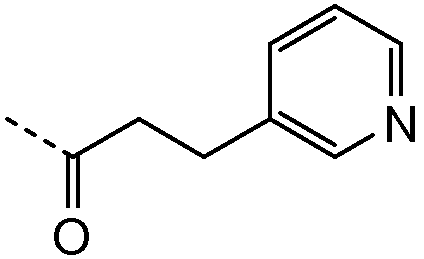
Другие агенты, которые могут быть комбинированы с соединениями настоящего изобретения, включают, например, нуклеозидные и ненуклеозидные ингибиторы полимеразы вируса HCV, ингибиторы протеазы, ингибиторы геликазы, ингибиторы NS4B и агенты, которые функционально ингибируют доступность внутририбосомального участка (IRES), и другие агенты, которые ингибируют клеточное присоединение HCV или вирусное проникновение, трансляцию РНК вируса HCV, транскрипцию РНК вируса HCV, репликацию или созревание HCV, сборку или вирусное высвобождение. Характерные соединения этих классов включают ингибиторы протеазы HCV, такие как телапревир (VX-950), боцепревир (SCH-503034), нарлапревир (SCH-900518), ITMN-191 (R-7227), TMC435350 (TMC435), MK-7009, BI-201335, BI-2061 (цилупревир), BMS-650032, ACH-1625, ACH-1095, GS 9256, VX-985, IDX-375 (HCV NS4A ингибитор кофактора протеазы), VX-500, VX-813, PHX-1766, PHX2054, IDX-136, IDX-316, ABT-450, EP-013420 (и представители того же рода) и VBY-376; нуклеозидные ингибиторы полимеразы HCV, которые могут быть использованы в изобретении, включают R7128, PSI-7851, PSI 7977, IDX-189, IDX-184, IDX-102, R1479, UNX-08189, PSI-6130, PSI-938 и PSI-879 и всевозможные другие нуклеозиды и аналоги нуклеотидов, и ингибиторы HCV, включающие такие производные, как 2'-C-метил-модифицированные нуклеозиды, 4'-аза-модифицированные нуклеозиды и 7'-деаза-модифицированные нуклеозиды, например 4-амино-1-[5-азидо-4-гидрокси-5-гидроксиметил-3-метилтетрагидрофуран-2-ил]пиримидин-2(1H)-он (ссылка 1) и его сложный бис-2-метилпропаноатный эфир (ссылка 2). Ненуклеозидные ингибиторы полимеразы HCV, пригодные для применения в изобретении, включают HCV-796, HCV-371, VCH-759, VCH-916, VCH-222, ANA-598, MK-3281, ABT-333, ABT-072, PF-00868554, BI-207127, GS-9190, A-837093, JKT-109, GL-59728, GL-60667, ABT-072, AZD-2795 и 13-циклогексил-3-метокси-17,23-диметил-7H-10,6-(метаноиминотиоиминоэтанооксиэтаноиминометано)индоло[2,1-a][2]бензазепин-14,24-дион-16,16-диоксид (ссылка 3).
Приведенные ниже примеры предназначены для иллюстрации изобретения, и их не следует истолковывать как ограничивающие его объем.
Примеры
Пример 1 - Синтез фенилэтинильного остова формулы III
Схема 1
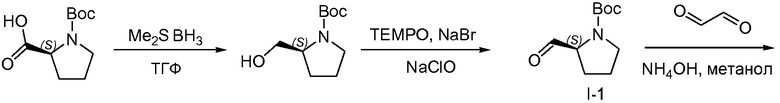
1.1 Получение L-Boc-пролинола
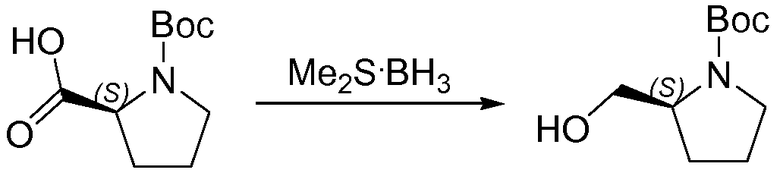
Боргидрид-метилсульфидный комплекс (180 мл, 1,80 моль) добавляли по каплям к раствору N-Boc-L-пролина (300 г, 1,39 моль) в безводном тетрагидрофуране (ТГФ) (3,0 л), который был охлажден до 0°C. Когда прекратилось выделение газа, баню со льдом удаляли и раствор перемешивали при 10°C в течение 18 часов. Тонкослойная хроматография (ТСХ) показала, что исходное вещество осталось и что образовался требуемый продукт. Раствор охлаждали до 0°C и медленно добавляли метанол (2,4 л). Растворители удаляли при пониженном давлении. Остаток снова растворяли в дихлорметане (1 л), промывали NaHCO3 (500 мл, насыщенный водный раствор) и насыщенным раствором соли (500 мл), сушили над MgSO4. Твердые вещества удаляли фильтрованием и растворители из фильтрата удаляли при пониженном давлении, что давало возможность получить белое твердое вещество, 260 г (93%), использованное на следующей стадии без дополнительной очистки.
1.2 Получение L-Boc-пролиналя, I-1
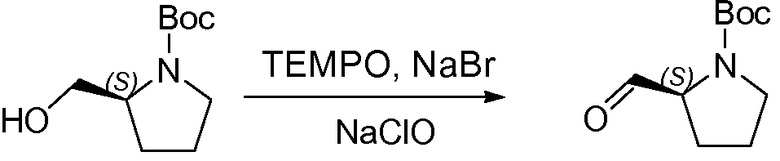
К раствору L-Boc-пролинола (100 г, 500 ммоль) в CH2Cl2 (1,5 л) при 0°C последовательно добавляли при энергичном перемешивании 2,2,6,6-тетраметилпиперидин-1-оксил (TEMPO; 1,56 г, 10 ммоль) и NaBr (5,14 г, 50 ммоль). К полученной в результате смеси по каплям добавляли раствор NaHCO3 (6 г, 110 ммоль, растворенные в 1,5 л 6% раствора NaClO) и 6% NaClO в активном хлоре (1,5 л, 750 ммоль) при 0°C в течение периода времени, равного 1 часу. ТСХ показала, что не осталось исходного вещества и что требуемый продукт образовался. Смесь быстро экстрагировали дихлорметаном (2×1,5 л). Органические слои объединяли, промывали NaHSO4 (10%, 1 л) и KI (4%, 200 мл), затем Na2S2O3 (10%, 1 л) и насыщенным раствором соли (1,5 л), сушили над MgSO4, твердые вещества удаляли фильтрованием и растворители удаляли при пониженном давлении, что давало возможность получить желтое масло, Boc-пролиналь, I-1 (89 г, 92%), использованное на следующей стадии без дополнительной очистки.
1.3 Получение промежуточного соединения I-2
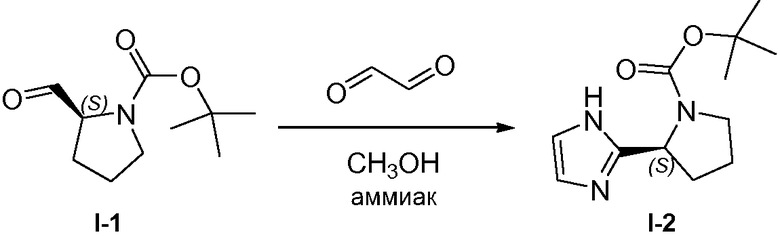
Водный раствор аммиака (25-28%, 200 мл) по каплям добавляли к раствору промежуточного соединения I-1 (89 г, 0,44 mol) и глиоксаля (183 мл, 40% раствор в воде) в 1 л метанола. Реакционную смесь герметично закрывали и проводили реакцию при 10°C. Спустя 16 часов дополнительный глиоксаль (20 мл) и водный раствор аммиака (20 мл) добавляли и реакцию проводили в течение дополнительных 6 часов. Растворители удаляли при пониженном давлении и неочищенное вещество повторно растворяли в этилацетате (1,0 л), промывали водой и насыщенным раствором соли, сушили над MgSO4, твердые вещества удаляли фильтрованием и растворители удаляли при пониженном давлении. Неочищенное вещество очищали колоночной хроматографией (силикагель, дихлорметан, затем до смеси метанол/дихлорметан, 1:70), с получением 73 г (70%) промежуточного соединения I-2 в виде белого твердого вещества.
1H ЯМР: CDCl3 400 МГц δ 6,95 (с, 2H), 4,82-4,94 (м, 1H), 3,60-3,70 (м, 1H), 3,41-3,50 (м, 1H), 2,20-2,39 (м, 1H), 1,91-2,03 (м, 3H), 1,47 (с, 3H), 1,25 (с, 6H).
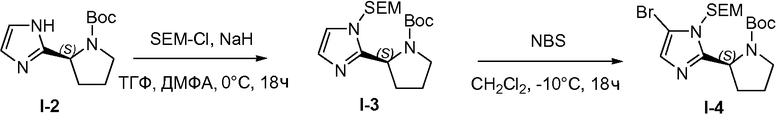
1.4 Получение промежуточного соединения I-3

К раствору промежуточного соединения I-2 (500 г, 2,1 моль) в безводных тетрагидрофуране (ТГФ) (1,8 л) и диметилформамиде (ДМФА; 0,8 л) добавляли гидрид натрия (55 г, 60% дисперсия в масле, 2,3 моль) порциями при перемешивании при 0°C в течение периода времени, равного 4 часам. [2-(триметилсилил)этокси]метилхлорид (SEM-Cl) (245,8 г, 2,3 моль) добавляли по каплям при 0°C. Полученную в результате смесь перемешивали при 25°C в течение 14 часов. Реакцию гасили добавлением воды (100 мл). Затем растворитель удаляли при пониженном давлении и остаток растворяли в этилацетате (2,5 л), промывали водой (2 л), насыщенным раствором соли (2 л) и сушили над MgSO4, твердые вещества удаляли фильтрованием и растворители из фильтрата удаляли при пониженном давлении. Остаток очищали хроматографией на силикагеле (петролейный эфир/этилацетат, 4:1), с получением масла, 530 г (68%) промежуточного соединения I-3.
1.5 Получение промежуточного соединения I-4
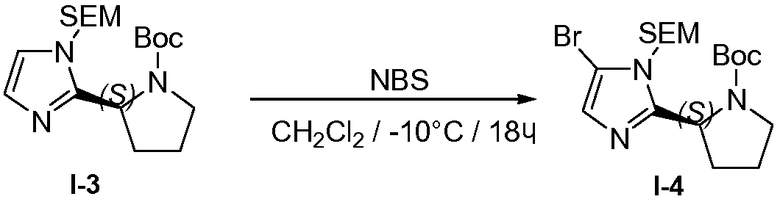
К раствору промежуточного соединения I-3 (530 г, 1,44 моль) в дихлорметане (2 л) добавляли N-бромсукцинимид (NBS; 256 г, 1,44 моль) порциями при перемешивании при -10°C в течение периода времени, равного 4 часам. Смесь перемешивали в течение 14 часов при 20°C. Реакционную смесь промывали водой (2 л), насыщенным раствором соли (2 л) и сушили над MgSO4, твердые вещества удаляли фильтрованием, и растворители из фильтрата удаляли при пониженном давлении. Остаток очищали хроматографией (силикагель, петролейный эфир/этилацетат, 10:1). Фракции с целевым веществом объединяли и растворители удаляли при пониженном давлении, что давало возможность получить желтое масло, 460 г (71%) промежуточного соединения I-4.
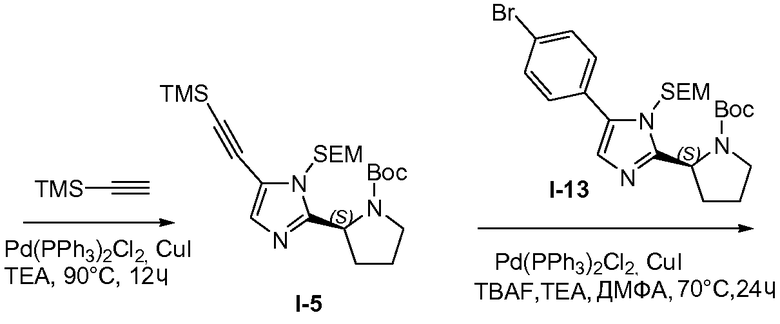
1.6 Получение промежуточного соединения I-5

Смесь промежуточного соединения I-4 (272 г, 609,2 ммоль), триметилсилилацетилена (TMS-ацетилен; 77,8 г, 792 ммоль), Pd(OAc)2 (13,7 г, 60,9 ммоль) и трифенилфосфина (Ph3P; 31,9 г, 121,8 ммоль) в триэтиламине (TEA, 2 л) перемешивали при 80°C в течение 18 часов в атмосфере N2. Затем TMS-ацетилен (41,7 г, 426,3 ммоль) добавляли после охлаждения до 20°C. Полученную в результате смесь затем перемешивали при 80°C в течение 8 часов в атмосфере N2. Затем реакционную смесь фильтровали через диатомовую землю и растворители из фильтрата удаляли при пониженном давлении. Остаток очищали хроматографией на силикагеле (петролейный эфир/этилацетат, от 30:1 до 10:1) дважды, с получением 98 г (34,7%) промежуточного соединения I-5 в виде коричневого твердого вещества.
1H ЯМР: CDCl3 400 МГц δ 6,87 (с, 1H), 5,16-5,65 (м, 1H), 4,61-4,87 (м, 2H), 3,24-3,42 (м, 4H), 1,64-2,21 (м, 4H), 1,02-1,17 (д, 9H), 0,59-0,73 (м, 2H), 0,01 (с, 9H), -0,35 (с, 9H).
1.7 Получение промежуточного соединения I-8
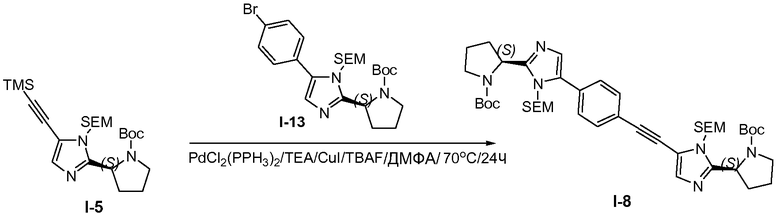
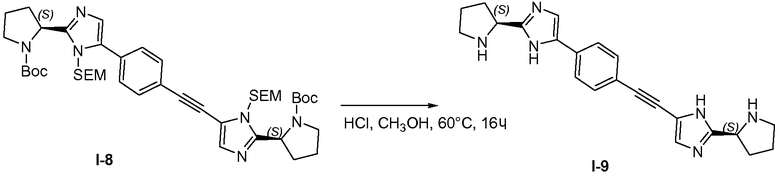
Промежуточное соединение I-5 (55 г, 118,6 ммоль), промежуточное соединение I-13 (68 г, 130,46 ммоль), CuI (2,26 г, 11,86 ммоль) и TEA (36 г, 355,79 ммоль) объединяли в ДМФА (500 мл), затем через реакционную смесь барботировали N2 и добавляли PdCl2(Ph3P)2 (8,32 г, 11,86 ммоль) в атмосфере N2. Смесь нагревали до 70°C и продолжали барботирование N2. Тетра-н-бутиламмонийфторид (TBAF; 1M в ТГФ, 118 мл, 118 ммоль) добавляли с помощью нагнетания шприцом в течение 10 часов. Реакционную смесь разбавляли водой (1 л) и экстрагировали смесью петролейного эфира и этилацетата (1:1 (об.:об.), 2×500 мл). Объединенные органические слои промывали водой и насыщенным раствором соли, сушили над MgSO4, твердые вещества удаляли фильтрованием и растворители из фильтрата удаляли при пониженном давлении. Остаток очищали хроматографией на силикагеле (петролейный эфир/этилацетат 2:1), с получением 35 г неочищенного продукта с чистотой 80%, и еще раз очищали препаративной ВЭЖХ, с получением 19,2 г (19,4%) промежуточного соединения I-8 в виде желтого твердого вещества.
1H ЯМР: CDC13 400 МГц δ 7,70-7,72 (д, J=8,0 Гц, 2H), 7,49-7,51 (д, J=8,0 Гц, 2H), 7,14-7,20 (м, 2H), 5,38-5,90 (м, 2H), 4,87-5,19 (м, 4H), 3,50-3,70 (м, 8H), 1,91-2,36 (м, 8H), 1,21-1,47 (м, 18H), 0,85-0,97 (м, 4H), 0,01 (с, 18H).
1.8 Получение I-9
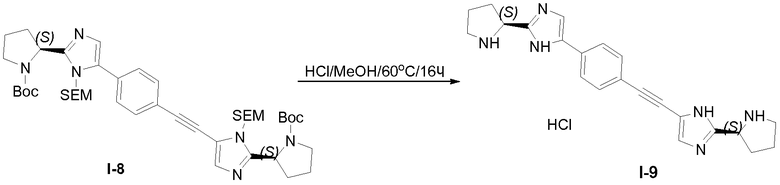
Промежуточное соединение I-9 (53,9 г, 64,6 ммоль) растворяли в метаноле (250 мл) и добавляли смесь HCl/метанол (250 мл, 4M). Смесь перемешивали при 60°C в течение 18 часов. Удаляли 80% растворителя и добавляли ацетонитрил (200 мл). Полученную в результате смесь перемешивали при 60°C в течение 2 часов. Полученное в результате твердое вещество выделяли фильтрованием и сушили в вакууме, с получением 25,2 г (75,2%) твердого вещества в виде гидрохлоридной соли.
1H ЯМР: MeOD 400 МГц δ 7,97 (с, 1H), 7,85-7,87 (д, J=8,2 Гц, 2H), 7,66-7,68 (д, J=8,2 Гц, 2H), 7,62 (с, 1H), 4,90-5,19 (м, 2H), 3,50-3,60 (м, 4H), 2,16-2,76 (м, 8H).
1.9 Получение промежуточного соединения I-11
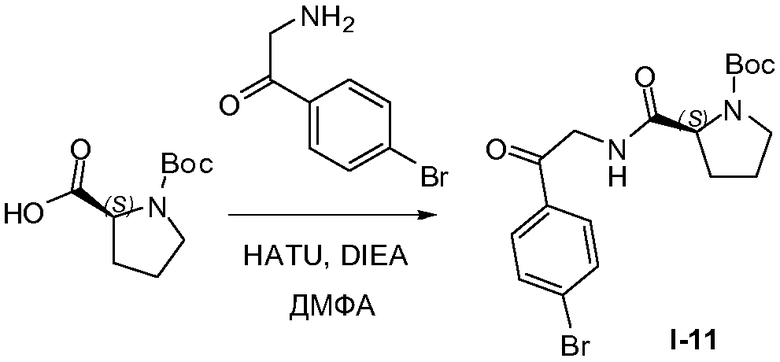
N,N-Диизопропилэтиламин (80,0 г, 0,62 моль) по каплям добавляли в течение 30 минут к смеси 2-амино-1-(4-бромфенил)этанона (50 г, 0,2 моль), гексафторфосфата 2-(1H-7-азабензотриазол-1-ил)-1,1,3,3-тетраметилурония (гидроксид (карбоксиметил)триметиламмония, внутренняя соль) (HATU; 53 г, 0,21 моль), N-Boc-L-пролина (43,0 г, 0,2 моль) в диметилформамиде (ДМФА) (600 мл). Реакционную смесь перемешивали при 5°C в течение 1 часа. Большинство летучих компонентов удаляли в вакууме и полученный в результате остаток распределяли между этилацетатом (600 мл) и водой (300 мл). Органический слой промывали насыщенным водным раствором NaHCO3 (500 мл) и насыщенным раствором соли (500 мл), сушили над MgSO4, твердые вещества удаляли фильтрованием, растворители из фильтрата удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (силикагель, петролейный эфир/этилацетат, от 3:1 до 1:1), с получением бледно-желтого твердого вещества, 60 г (62%) промежуточного соединения I-11.
1H ЯМР: CDC13 400 МГц δ 7,85 (д, J=8,4 Гц, 2H), 7,66 (д, J=8,4 Гц, 2H), 4,67-4,80 (м, 2H), 4,33-4,41 (м, 1H), 3,42-3,53 (м, 2H), 2,19-2,31 (м, 2H), 1,90-2,00 (м, 2H), 1,50 (с, 9H).
1.10 Получение I-12
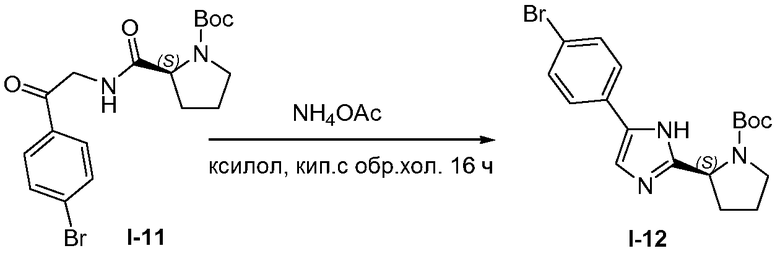
Смесь промежуточного соединения I-11 (60 г, 0,14 моль) и ацетата аммония (89 г, 1,4 моль) в ксилоле (800 мл) нагревали при кипячении с обратным холодильником в течение 16 часов. Реакционную смесь распределяли между этилацетатом (700 мл) и насыщенным раствором NaHCO3 (500 мл). Слои разделяли и водный слой экстрагировали дополнительным количеством этилацетата (2×300 мл). Органические слои объединяли, промывали насыщенным раствором соли (500 мл), сушили над MgSO4, твердые вещества удаляли фильтрованием и растворители из фильтрата упаривали при пониженном давлении. Полученное в результате вещество перекристаллизовывали из смеси этилацетат/петролейный эфир, что давало возможность получить желтое твердое вещество I-12, 25 г (43%).
1H ЯМР (400 МГц, MeOD) δ м.д. 1,23 (с, 6H), 1,46 (с, 3H), 1,84-2,10 (м, 3H), 2,36 (м, J=5,80 Гц, 1H), 3,50 (м, J=10,40, 5,10 Гц, 1H), 3,60-3,73 (м, 1H), 4,94-5,00 (м, 1H), 7,28-7,39 (м, 1H), 7,49 (д, J=8,28 Гц, 2H), 7,61 (д, J=8,03 Гц, 2H).
1.11 Получение промежуточного соединения I-13
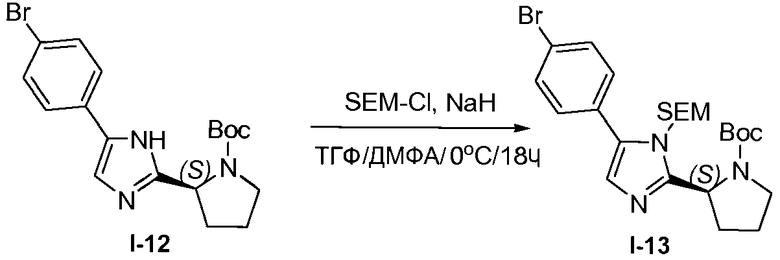
К раствору промежуточного соединения I-12 (212 г, 540,4 ммоль) в безводных тетрагидрофуране (ТГФ) (1,6 л) и диметилформамиде (ДМФА) (0,8 л) добавляли гидрид натрия (14,27 г, 60% дисперсия в масле, 540,4 моль) порциями при перемешивании при 0°C на протяжении периода времени, равного 4 часам. SEM-Cl (90,1 г, 540,4 моль) затем добавляли по каплям при 0°C. Полученную в результате смесь перемешивали при 25°C в течение 14 часов. Реакцию гасили добавлением воды (100 мл). Растворитель удаляли в вакууме и остаток растворяли в этилацетате (2 л), промывали водой, насыщенным раствором соли и сушили над MgSO4, твердые вещества удаляли фильтрованием и растворители из фильтрата удаляли при пониженном давлении. Остаток очищали хроматографией на силикагеле (петролейный эфир/этилацетат 4:1), с получением желтого твердого вещества, 200 г, которое перекристаллизовывали из 1 л петролейного эфира, что давало возможность получить белый порошок, 150 г (53,4%) промежуточного соединения I-13.
1H ЯМР: ДМСО 400 МГц δ 7,76 (с, 1H), 7,69-7,71 (д, J=8,4 Гц, 2H), 7,54-7,56 (д, J=8,4 Гц, 2H), 4,87-5,69 (м, 3H), 3,41-3,57 (м, 4H), 1,81-2,47 (м, 4H), 1,14-1,38 (д, 9H), 0,87-0,98 (м, 2H), 0,01 (с, 9H).
Пример 2 - Синтез соединения формулы I
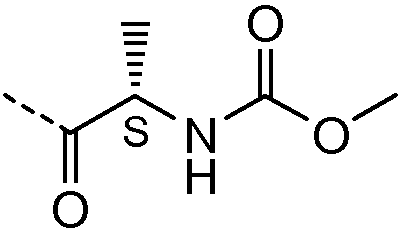
2.1 Получение соединения 2
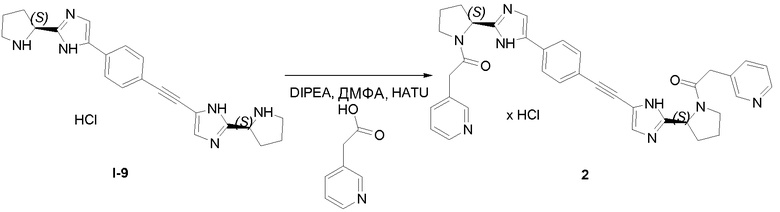
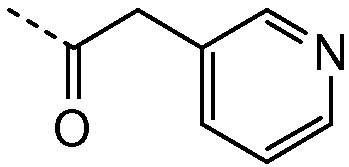
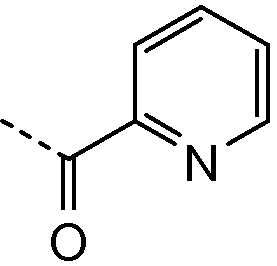
В колбу на 20 мл помещали промежуточное соединение I-9 (259 мг, 0,5 ммоль), 3-пиридилуксусную кислоту (171 мг, 1,25 ммоль), HATU (475 мг, 1,25 ммоль), N,N-диизопропилэтиламин (DIPEA; 0,826 мл, 5 ммоль) в ДМФА (5 мл) и смесь встряхивали в течение 15 часов. Жидкостная хроматография-масс-спектрометрия (ЖХМС) показала полное превращение в целевой продукт. Растворитель удаляли при пониженном давлении и остаток очищали с использованием сильного колоночного катионообменника SCX SPE (5 г сорбента, 20 см3). Колонку промывали метанолом (2 объема), затем на нее наносили соединение в остаточном ДМФА. Примеси элюировали метанолом (4 объема) и затем продукт вымывали из колонки, элюируя 7M раствором аммиака в метаноле (2 объема). Растворитель и избыток аммиака удаляли упариванием в вакууме. Соединение 2 выделяли в виде желтого порошка.
1H ЯМР (400 МГц, Хлороформ-d) δ м.д. 1,13 (д, J=6,44 Гц, 2H), 2,06-2,22 (м, 4H), 2,30-2,52 (м, 2H), 3,50 (с, 4H), 3,53-3,78 (м, 6H), 3,62-3,62 (м, 0H), 5,17-5,28 (м, 2H), 7,18 (с, 1H), 7,21 (с, 1H), 7,28-7,33 (м, 3H), 7,49 (д, J=7,80 Гц, 2H), 7,63 (д, J=7,61 Гц, 2H), 8,50 (д, J=1,95 Гц, 2H), 8,55 (дт, J=3,17, 1,44 Гц, 3H).
Альтернативно, очистка и выделение продукта реакции может быть осуществлена следующим образом. Добавляли CH2Cl2 к остатку и полученный в результате раствор промывали насыщенным раствором NaHCO3. Органическую фазу сушили над безводным Na2SO4, фильтровали и концентрировали в вакууме. Соединение затем очищали колоночной хроматографией на силикагеле (0-10% MeOH в CH2Cl2) или препаративной ВЭЖХ.
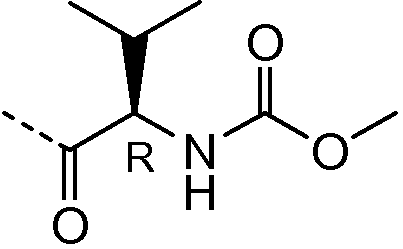
Получение соединения 4
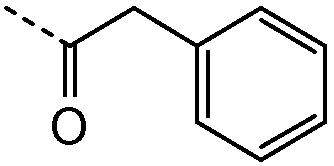
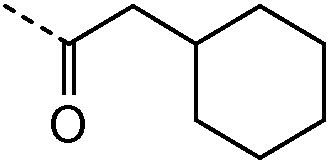
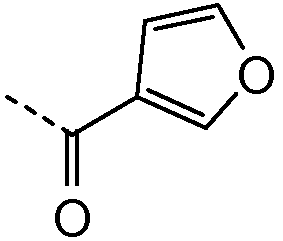
Получение и очистку указанного в заголовке соединения 4 осуществляли, следуя методике, изложенной в примере 2.1 (получение соединения 2), используя 2-циклогексилуксусную кислоту на первой стадии реакции.
1H ЯМР (400 МГц, Хлороформ-d) δ м.д. 0,97 (д, J=13,07 Гц, 8H), 1,09-1,21 (м, 2H), 1,22-1,37 (м, 5H), 1,69 (м, 9H), 1,83-1,93 (м, 2H), 2,01-2,12 (м, 4H), 2,15-2,43 (м, 6H), 3,49-3,61 (м, 4H), 5,11-5,34 (м, 2H), 7,12-7,24 (м, 2H), 7,36-7,56 (м, 2H), 7,65-7,81 (м, 2H).
Получение соединения 21
Получение и очистку указанного в заголовке соединения 21 осуществляли, следуя методике, изложенной в примере 2.1 (получение соединения 2), используя (S)-2-(диметиламино)-3-метилбутановую кислоту на первой стадии реакции.
1H ЯМР (400 МГц, Хлороформ-d) δ м.д. 0,65-0,79 (м, 5H), 1,00 (д, J=6,63 Гц, 12H), 1,99-2,23 (м, 6H), 2,42 (д, J=3,71 Гц, 12H), 2,95-3,03 (м, 2H), 3,04-3,18 (м, 1H), 3,53-3,71 (м, 4H), 5,28-5,45 (м, 2H), 7,15-7,25 (м, 2H), 7,51 (д, J=8,39 Гц, 2H), 7,68-7,80 (м, 2H).
Получение соединения 28
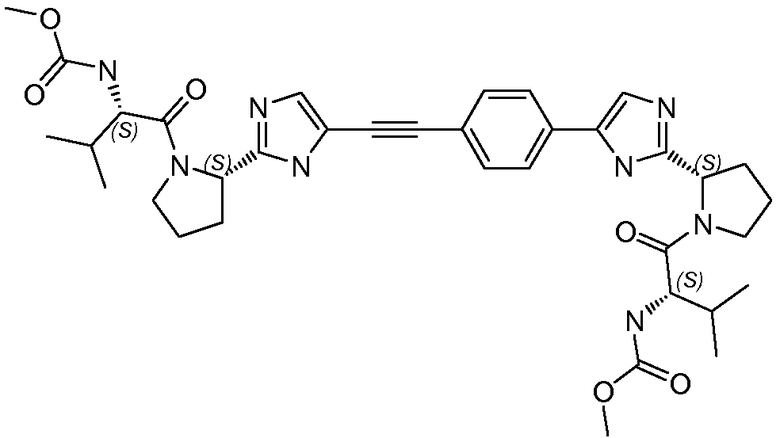
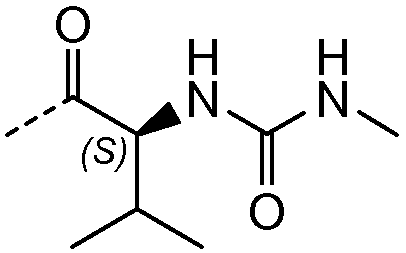
Получение и очистку указанного в заголовке соединения 28 осуществляли, следуя методике, изложенной в примере 2.1 (получение соединения 2), используя (S)-2-(метоксикарбониламино)-3-метилбутановую кислоту на первой стадии реакции.
1H ЯМР (400 МГц, ДМСО-d6) δ м.д. 0,77-0,92 (м, 4H), 1,03 (д, J=6,05 Гц, 12H), 1,19-1,30 (м, 2H), 1,83-2,05 (м, 6H), 2,06-2,22 (м, 2H), 3,53 (с, 6H), 3,56-3,65 (м, 2H), 3,68-3,87 (м, 1H), 4,04 (м, J=11,10 Гц, 1H), 4,98-5,02 (м, 1H), 5,06 (м, J=3,50 Гц, 1H), 7,28 (с, 1H), 7,30 (с, 1H), 7,42 (м, 2H), 7,50-7,59 (м, 2H), 7,71 (м, 2H).
Получение соединения 32
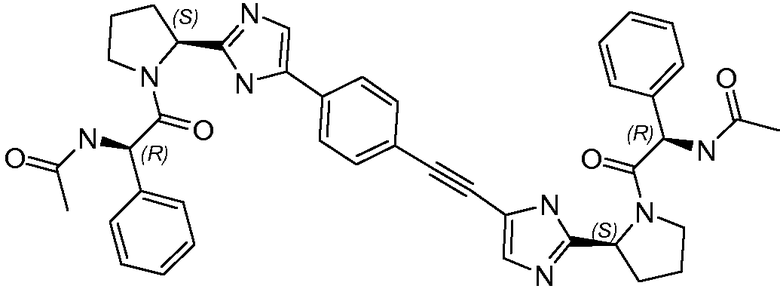
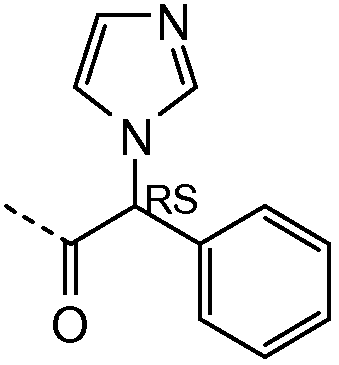
Получение и очистку указанного в заголовке соединения 32 осуществляли, следуя методике, изложенной в примере 2.1 (получение соединения 2), используя (R)-2-ацетамидо-2-фенилуксусную кислоту на первой стадии реакции.
1H ЯМР (400 МГц, Хлороформ-d) δ м.д. 1,41 (ушир.с, 2H), 1,91 (ушир.с, 2H), 1,98-2,10 (м, 6H), 2,86-3,06 (м, 3H), 3,09-3,35 (м, 3H), 3,53-3,88 (м, 4H), 5,16-5,33 (м, 2H), 5,62 (д, J=6,44 Гц, 1H), 5,71-5,83 (м, 1H), 6,58-6,68 (м, 1H), 6,69-6,88 (м, 1H), 7,30 (д, J=2,54 Гц, 2H), 7,37-7,61 (м, 12H), 7,63-7,87 (м, 2H).
Получение соединения 36
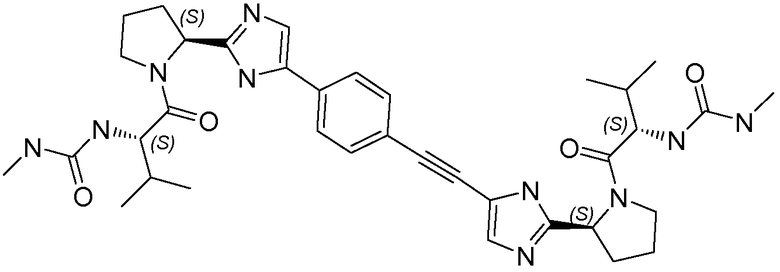
Получение и очистку указанного в заголовке соединения 36 осуществляли, следуя методике, изложенной в примере 2.1 (получение соединения 2), используя (S)-3-метил-2-(3-метилуреидо)бутановую кислоту на первой стадии реакции.
1H ЯМР (400 МГц, ДМСО-d6) δ м.д. 0,71-0,83 (м, 7H), 0,84-0,95 (м, 5H), 1,75-2,04 (м, 7H), 2,13 (ушир.с, 3H), 2,52-2,60 (м, 12H), 3,44 (дд, J=6,63, 3,71 Гц, 2H), 3,67-3,84 (м, 2H), 4,13-4,29 (м, 2H), 4,96-5,12 (м, 2H), 5,90 (м, J=4,70 Гц, 1H), 6,02 (д, J=8,98 Гц, 1H), 7,43 (д, J=6,24 Гц, 2H), 7,72 (д, J=7,61 Гц, 2H).
Получение соединения 37

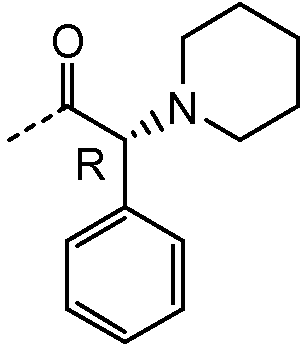
Получение и очистку указанного в заголовке соединения 37 осуществляли, следуя методике, изложенной в примере 2.1 (получение соединения 2), используя (R)-2-фенил-2-(пиперидин-1-ил)уксусную кислоту на первой стадии реакции.
1H ЯМР (400 МГц, ДМСО-d6) δ м.д. 1,04-1,56 (м, 15H), 1,70-2,03 (м, 8H), 2,10 (ушир.с, 2H), 2,20-2,44 (м, 9H), 3,80-4,02 (м, 1H), 4,28 (с, 2H), 4,84-5,05 (м, 1H), 6,87 (д, J=6,24 Гц, 1H), 7,14 (м, J=3,50 Гц, 2H), 7,23-7,48 (м, 10H), 7,56 (ушир.с, 1H), 7,76 (д, J=6,44 Гц, 2H).
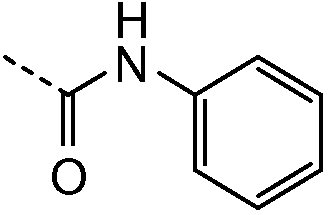
2.1 Получение соединения 3
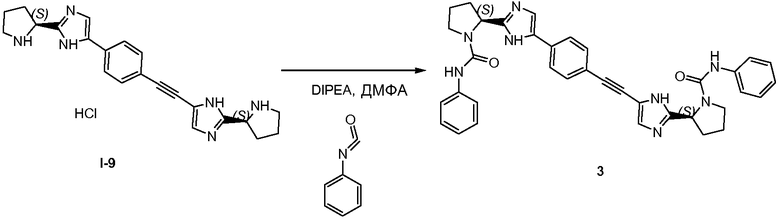
В колбу на 10 мл помещали промежуточное соединение I-9 (200 мг, 0,386 ммоль), N,N-диизопропилэтиламин (0,638 мл, 3,859 ммоль), фенилизоцианат (0,126 мл, 1,158 ммоль). Колбу встряхивали в течение ночи при комнатной температуре. Растворители удаляли при пониженном давлении, остаток растворяли в дихлорметане (1 мл) и очищали колоночной хроматографией на силикагеле, используя градиент от дихлорметана до 10% метанола в дихлорметане. Лучшие фракции объединяли и растворители удаляли при пониженном давлении, что давало возможность получить чистый продукт 3.
Все соединения были охарактеризованы с помощью ЖХ/МС. Использовали следующие способы ЖХ/МС:
Способ A: Прибор Waters Acquity UPLC, снабженный спектрофотометрическим детектором PDA (диапазон 210-400 нм), и Waters SQD с источником ионизации, работающим в двух режимах ES+/-. Используемая колонка представляла собой Halo C18, 2,7 мкм, 2,1×50 мм, и поддерживалась рабочая температура 50°C. Градиент от 95% водной муравьиной кислоты (0,1%)/5% ацетонитрила до 100% ацетонитрила линейно изменялся за период времени, равный 1,5 минутам, поддерживался в течение 0,6 минут, затем снова использовали 100% водную муравьиную кислоту (0,1%) в течение 0,5 минут. Скорость потока составляла 0,6 мл/мин.
Способ B: Жидкостная хроматография: прибор Waters Alliance 2695, УФ-детектор: Waters 996 PDA, диапазон: 210-400 нм; масс-детектор: Waters ZQ, источник ионизации: ES+, ES-. Используемая колонка: SunFire C18 3,5 мкм 4,6×100 мм, подвижная фаза A: 10 мМ NH4OOCH+0,1% HCOOH в H2O; подвижная фаза B: CH3OH; рабочая температура колонки: 50°C; скорость потока: 1,5 мл/мин градиент во времени (мин) [%A/%B] от 0 [65/35] до 7[5/95], до 9,6[5/95], до 9,8[65/35], до 12 [65/35].
Способ C: Приборы Waters Acquity UPLC, снабженный спектрофотометрическим детектором PDA (диапазон 210-400 нм), и Waters SQD с источником ионизации, работающим в двух режимах, ES+/-. Используемая колонка представляла собой XS Strategy 1,7 мкм, 2,1×20 мм, и поддерживалась рабочая температура 50°C. Градиент от 100% водной муравьиной кислоты (0,1%) до 100% ацетонитрила линейно изменялся за период времени, равный 1,5 минутам, поддерживался в течение 0,6 минут, затем снова использовали 100% водную муравьиную кислоту (0,1%) в течение 0,5 минут. Скорость потока составляла 0,6 мл/мин.
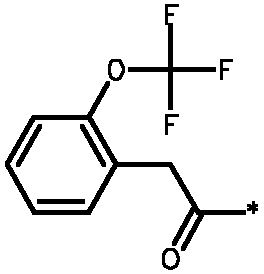
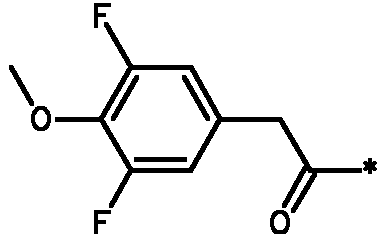
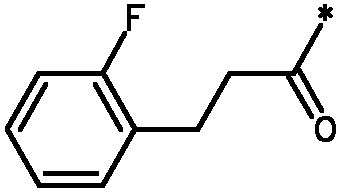
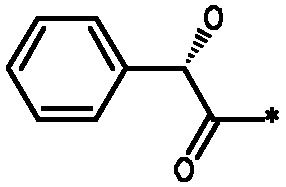
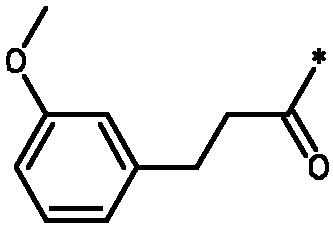
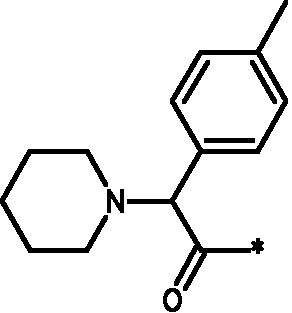
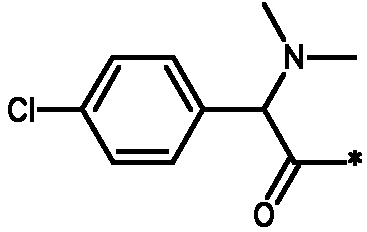
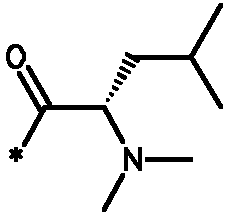
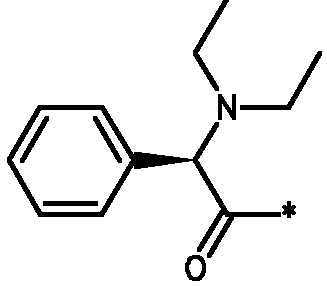
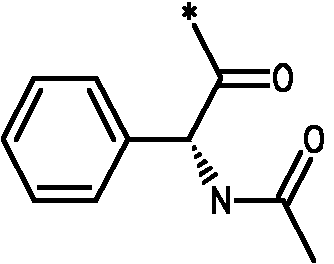
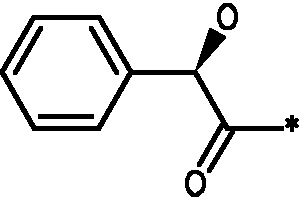
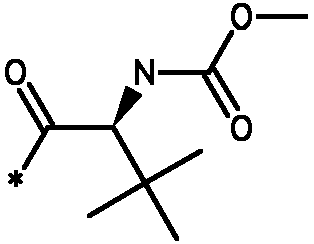
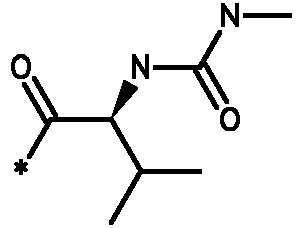
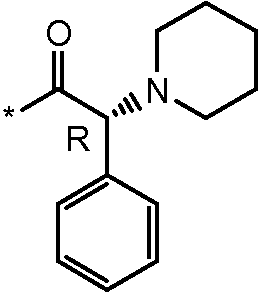
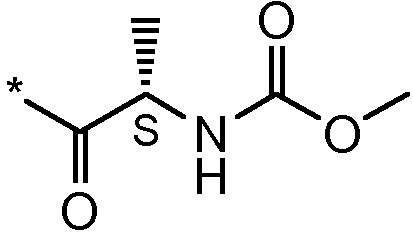
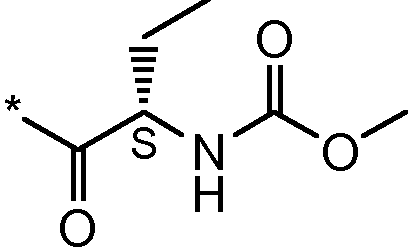
Соединения формулы I
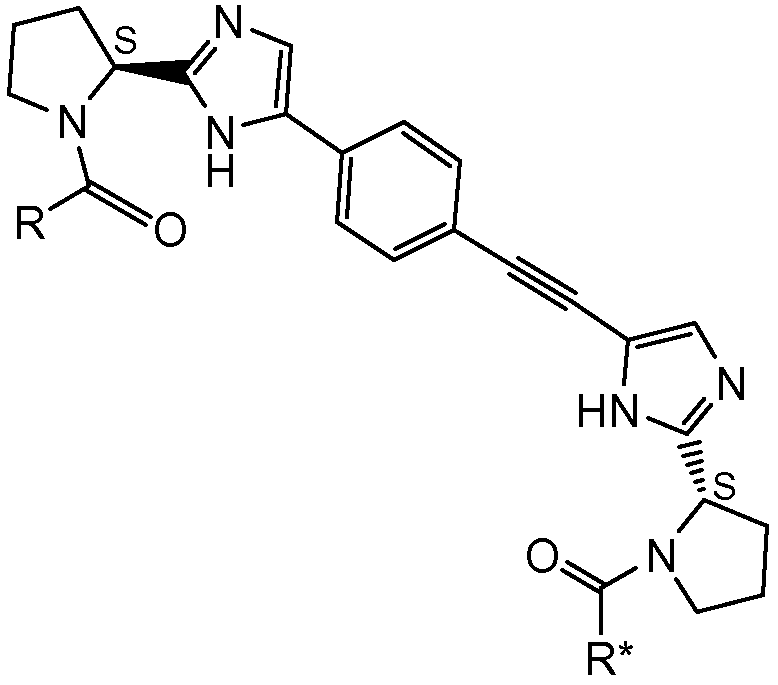
* Обозначение места присоединения радикала
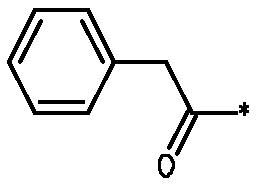
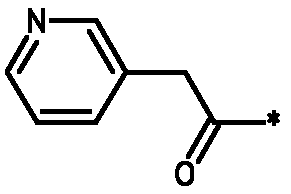
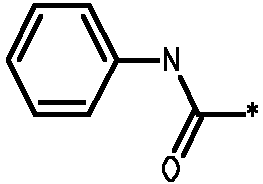
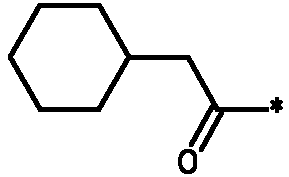
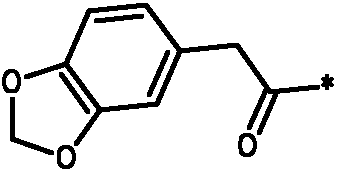
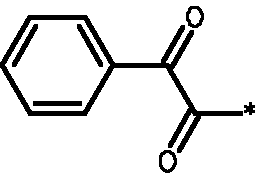
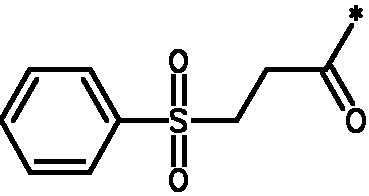
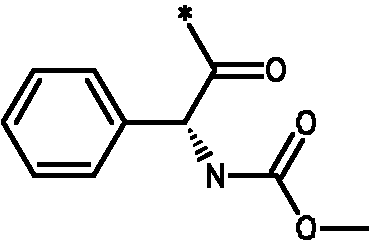

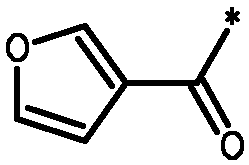
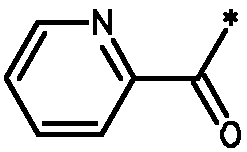

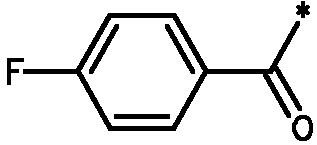

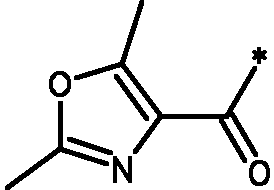
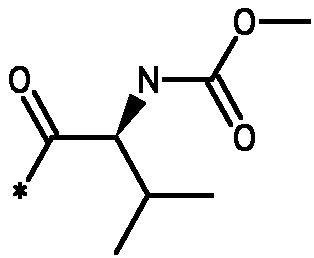
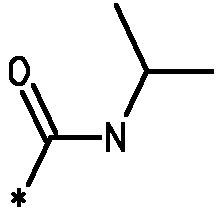
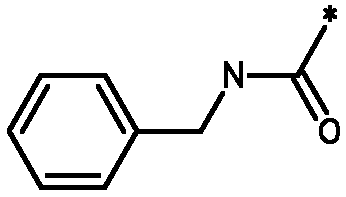


Пример 3 - Биологическая активность соединений формулы I
Анализ репликации
Соединения формулы (I) проверяли на их ингибирующую активность в процессе репликации вируса HCV. Клеточный анализ основывался на экспрессии гена в составе бицистронной конструкции, как описано Lohmann et al. (1999) Science vol. 285 pp. 110-113, с использованием модификаций, описанных Krieger et al. (2001) Journal of Virology 75: 4614-4624, при стратегии многоцелевого скрининга.
По существу, способ заключался в следующем.
При анализах использовали стабильно трансфицированную клеточную линию Huh-7 luc/neo (далее в данном описании обозначаемую как Huh-Luc). Данная клеточная линия содержит кодирующую РНК экспрессируемую бицистронную конструкцию, включающую NS3-NS5B области дикого типа немутантного HCV типа Ib, полученные при трансляции внутририбосомального доступного участка (IRES) из вируса энцефаломиокардита (EMCV), находящиеся перед участком репортерного гена (FfL-люциферазы), и участок гена селектируемого маркера (neoR, неомицинфосфотрансферазы). На ограничивающих конструкцию 5' и 3' концах содержатся NTR (нетранслируемые участки) генома HCV генотипа 1b. Функционирующая культура клеток, содержащих репликон, в присутствии G418 (neoR) зависит от репликации РНК вируса HCV. Стабильно трансфицированные репликоном клетки, в которых экспрессируется HCV РНК, имеющая высокий уровень автономной репликации, кодирующая, помимо всего прочего, люциферазу, использовали для скрининга антивирусных соединений.
Содержащие репликон клетки высевали в 384-луночные планшеты в присутствии тестируемых и контрольных соединений, которые добавляли в различных концентрациях. Далее проводили инкубацию в течение трех дней, HCV репликацию определяли путем измерения активности люциферазы (используя стандартные субстраты и реагенты для анализа люциферазы и микропланшетного визуализатора Perkin Elmer ViewLux™ ultraHTS). Содержащие репликон клетки в контрольных культурах имели высокий уровень экспрессии гена люциферазы в отсутствие какого-либо ингибитора. Ингибирующую активность соединения в отношении активности люциферазы контролировали на клетках Huh-Luc, обеспечивая кривую доза-ответ для каждого тестируемого соединения. Затем рассчитывали величины EC50, которые представляют собой количество соединения, требуемое для снижения уровня определяемой люциферазной активности на 50%, или, более конкретно, представляют способность генетически связанного с HCV репликона РНК к репликации.

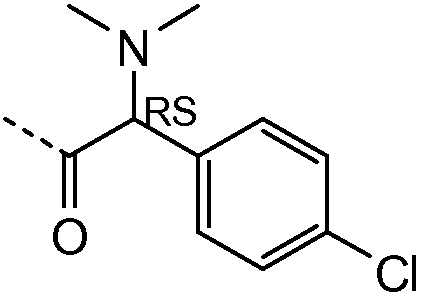
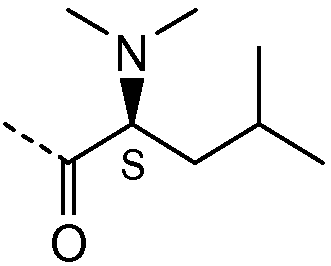
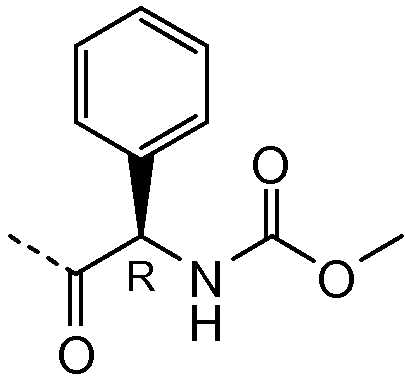
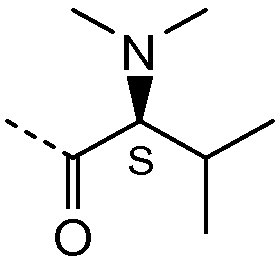


| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ БИС-БЕНЗИМИДАЗОЛА В КАЧЕСТВЕ ИНГИБИТОРОВ ВИРУСА ГЕПАТИТА С | 2010 |
|
RU2540897C2 |
| ДИАРИЛОВЫЕ ЭФИРЫ | 2010 |
|
RU2528231C2 |
| МАКРОЦИКЛИЧЕСКИЕ ИНГИБИТОРЫ ВИРУСА ГЕПАТИТА С | 2006 |
|
RU2437886C2 |
| МАКРОЦИКЛИЧЕСКИЕ ИНГИБИТОРЫ ВИРУСА ГЕПАТИТА С | 2006 |
|
RU2441870C2 |
| ГЕТЕРОБИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ HCV | 2012 |
|
RU2621734C1 |
| МАКРОЦИКЛИЧЕСКИЕ ИНДОЛЬНЫЕ ПРОИЗВОДНЫЕ, ПРИМЕНИМЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ВИРУСА ГЕПАТИТА С | 2009 |
|
RU2518471C2 |
| МАКРОЦИКЛИЧЕСКИЕ ИНГИБИТОРЫ ВИРУСА ГЕПАТИТА С | 2006 |
|
RU2486189C2 |
| СОЕДИНЕНИЯ НА ОСНОВЕ ПИРАЗОЛОПИРИДИНОНА | 2018 |
|
RU2806625C2 |
| 1,5,6-ЗАМЕЩЕННЫЕ 2-ОКСО-3-ЦИАНО-1,6А-ДИАЗАТЕТРАГИДРОФЛУОРАНТЕНЫ | 2006 |
|
RU2389730C2 |
| НОВЫЕ ХИНОЛИНОВЫЕ СОЕДИНЕНИЯ | 2018 |
|
RU2810113C2 |
Изобретение относится к области органической химии, а именно к гетероциклическим соединениям общей формулы I или к их фармацевтически приемлемым солям, или сольватам, или их стереоизомерам, где R и R*, каждый независимо, представляет собой -CR1R2R3, C1-4алкиламино, бензиламино, C6-10ариламино, гетероС4-7циклоалкил, содержащий 1 гетероатом, выбранный из O; где R1 выбирают из C1-4алкила; фенила, необязательно замещенного 1, 2 или 3 заместителями, независимо выбранными из галогена, C1-4алкила, C1-4алкокси, трифторметокси или 2 заместителей у соседних атомов кольца, образующих 1,3-диоксолановую группу; бензила, необязательно замещенного галогеном или метокси; фенилсульфонилметила; C3-5гетероарила, содержащего от 1 до 2 гетероатомов, независимо выбранных из N и O; C3-5гетероарилметила, содержащего от 1 до 2 гетероатомов, выбранных из N и C3-6циклоалкила; R2 выбирают из водорода, гидроксила, ди-C1-4алкиламино, C1-4алкилкарбониламино, C1-4алкилоксикарбониламино, C1-4алкиламинокарбониламино, пиперидин-1-ила или имидазол-1-ила; R3 представляет собой водород или, альтернативно, R2 и R3 вместе образуют оксогруппу; или R1 и R3 вместе образуют циклопропил; при условии, что если один из R или R* представляет собой - CH(C6H5)N(CH3)2, тогда другой не может представлять собой - CH(C6H5)NHC(=O)OCH3; и если R и R* являются одинаковыми, тогда R1 отличен от фенила, когда R2 представляет собой гидроксил, ацетиламино, метоксикарбониламино или трет-бутоксикарбониламино, и R3 представляет собой водород; и R1 отличается от C1-4алкила, когда R2 представляет собой C1-4алкилоксикарбониламино, и R3 представляет собой водород. Также изобретение относится к фармацевтической композиции на основе соединения формулы I и его применению. Технический результат: получены новые соединения, полезные для профилактики или лечения инфекции HCV. 3 н. и 6 з.п. ф-лы, 2 табл., 3 пр.
1. Соединение формулы I
или его стереоизомер, где
R и R*, каждый независимо, представляет собой -CR1R2R3, C1-4алкиламино, бензиламино, C6-10ариламино, гетероС4-7циклоалкил, содержащий 1 гетероатом, выбранный из O; где
R1 выбирают из C1-4алкила; фенила, необязательно замещенного 1, 2 или 3 заместителями, независимо выбранными из галогена, C1-4алкила, C1-4алкокси, трифторметокси или 2 заместителей у соседних атомов кольца, образующих 1,3-диоксолановую группу; бензила, необязательно замещенного галогеном или метокси; фенилсульфонилметила; C3-5гетероарила, содержащего от 1 до 2 гетероатомов, независимо выбранных из N и O; C3-5гетероарилметила, содержащего от 1 до 2 гетероатомов, выбранных из N и C3-6циклоалкила;
R2 выбирают из водорода, гидроксила, ди-C1-4алкиламино, C1-4алкилкарбониламино, C1-4алкилоксикарбониламино, C1-4алкиламинокарбониламино, пиперидин-1-ила или имидазол-1-ила;
R3 представляет собой водород или, альтернативно,
R2 и R3 вместе образуют оксогруппу; или
R1 и R3 вместе образуют циклопропил;
при условии, что если один из R или R* представляет собой - CH(C6H5)N(CH3)2, тогда другой не может представлять собой - CH(C6H5)NHC(=O)OCH3; и если R и R* являются одинаковыми, тогда R1 отличен от фенила, когда R2 представляет собой гидроксил, ацетиламино, метоксикарбониламино или трет-бутоксикарбониламино, и R3 представляет собой водород; и R1 отличается от C1-4алкила, когда R2 представляет собой C1-4алкилоксикарбониламино, и R3 представляет собой водород;
или его фармацевтически приемлемая соль или сольват.
2. Соединение по п.1, где R и R* представляют собой -CR1R2R3 или бензиламино.
3. Соединение по п.2, где R и R* представляют собой -CR1R2R3.
4. Соединение по п.1 или 3, где R2 представляет собой водород, гидроксил, диметиламино, ацетиламино, метоксикарбониламино, метиламинокарбониламино, пиперидин-1-ил или имидазол-1-ил.
5. Соединение по п.1 или 3, где R1 выбирают из C1-4алкила; фенила, необязательно замещенного 1 или 2 заместителями, независимо выбранными из галогена, метила, метокси или 2 заместителей у соседних атомов кольца, образующих 1,3-диоксолановую группу; гетероарила и гетероарилметила.
6. Соединение по п.5, где гетероарил представляет собой пиридинил.
7. Соединение по п.1, где соединение представлено формулой I-b
8. Фармацевтическая композиция для профилактики или лечения инфекции HCV у млекопитающих, содержащая соединение, как определено в любом из пп.1-7, и фармацевтически приемлемый носитель.
9. Применение соединения по любому из пп.1-7 для профилактики или лечения инфекции HCV у млекопитающих.
| Станок для изготовления деревянных ниточных катушек из цилиндрических, снабженных осевым отверстием, заготовок | 1923 |
|
SU2008A1 |
| Станок для изготовления деревянных ниточных катушек из цилиндрических, снабженных осевым отверстием, заготовок | 1923 |
|
SU2008A1 |
| Станок для изготовления деревянных ниточных катушек из цилиндрических, снабженных осевым отверстием, заготовок | 1923 |
|
SU2008A1 |
| Приспособление для смазывания бумаги клеем | 1927 |
|
SU10577A1 |

