Область техники, к которой относится изобретение
Это изобретение относится к производным бис-безимидазола, которые являются ингибиторами вируса гепатита C (HCV), их синтезу и их применению, отдельно или в сочетании с другими ингибиторами HCV, для лечения или профилактики HCV.
Предшествующий уровень техники
HCV представляет собой одноцепочечный положительный РНК-вирус, относящийся к семейству вирусов Flaviviridae рода hepacivirus. Геном вируса транслируется в единую открытую рамку считывания, которая кодирует множество структурных и неструктурных белков.
После первоначальной острой инфекции у большинства инфицированных индивидов развивается хронический гепатит, поскольку HCV реплицируется предпочтительно в гепатоцитах, но не является непосредственно цитопатическим. В частности, отсутствие интенсивного T-лимфоцитарного ответа и высокая способность вируса к мутациям, по-видимому, обеспечивают высокий уровень хронической инфекции. Хронический гепатит может прогрессировать в печеночный фиброз, приводящий к циррозу, заболеванию печени конечной стадии, и в HCC (печеночно-клеточную карциному), что делает его основной причиной трансплантаций печени.
Существуют 6 основных генотипов HCV и более 50 подтипов, которые географически распределены по-разному. HCV 1 типа представляет собой преобладающий генотип в Европе и США. Выраженная генетическая гетерогенность HCV имеет высокое диагностическое и клиническое значение, являясь возможным объяснением трудностей в разработке вакцины и отсутствия ответа на лечение.
HCV может передаваться через контакт с зараженной кровью или продуктами крови, например, после переливания крови или внутривенного введения лекарственного средства. Внедрение диагностических тестов, используемых для скрининга крови, вызвало тенденцию к понижению встречаемости посттрансфузионного заражения HCV. Однако учитывая медленное прогрессирование в заболевание печени конечной стадии существующие инфекции будут оставаться серьезной медицинской и экономической проблемой в течение десятилетий.
Современные способы лечения HCV основаны на (пегилированном) интерфероне-альфа (IFN-α) в сочетании с рибавирином. Эта комбинированная терапия приводит к замедленному вирусологическому ответу более чем у 40% пациентов, инфицированных вирусами 1 генотипа и приблизительно у 80% пациентов, инфицированных 2 и 3 генотипами. Помимо ограниченной эффективности в отношении HCV 1 генотипа, эта комбинированная терапия обладает значительными побочными эффектами, включая гриппоподобные симптомы, гематологические нарушения и нейропсихиатрические симптомы. Таким образом, существует необходимость в более эффективных, более удобных и лучше переносимых способах лечения.
Опыт, связанный с лекарственными средствами против ВИЧ, и, в частности, с ингибиторами протеазы ВИЧ, показал, что субоптимальная фармакокинетика и комплексные схемы дозирования быстро приводят к непредусмотренным проблемам, связанным с режимом лечения. Это, в свою очередь, означает, что 24-часовая минимальная концентрация (минимальная концентрация в плазме) соответствующих лекарственных средств в схеме лечения ВИЧ часто снижается ниже порога IC90 или ED90 на большую часть дня. Полагают, что 24-часовой минимальный уровень по меньшей мере IC50 и, более реалистично, IC90 или ED90 является необходимым для замедления возникновения устойчивых к лекарственному средству мутантных форм. Достижение необходимой фармакокинетики и метаболизма лекарственного средства для обеспечения таких минимальных уровней представляет собой важную задачу при разработке лекарственного средства.
Белок NS5A HCV расположен ниже белка NS4B и выше белка NS5B. После посттрансляционного расщепления вирусной сериновой протеазой NS3/4A, NS5A созревает в цинксодержащий фосфопротеин из трех доменов, который существует либо в виде гипофосфорилированной (56-кДа, p56), либо гиперфосфорилированной формах (58-кДа, p58). NS5A HCV вовлечен во множество аспектов жизненного цикла вируса, включая репликацию вируса и сборку инфекционных частиц, а также модулирование окружающей среды его клетки-хозяина. Хотя этому белку не приписывается ферментативная функция, описано, что он взаимодействует с многочисленными вирусными и клеточными факторами.
В ряде патентов и патентных заявок описаны соединения с ингибирующей активностью в отношении HCV, в частности нацеленные на NS5A. В WO2006/133326 описаны производные стилбена, а в WO2008/021927 и WO2008/021928 описаны бифенильные производные, обладающие активностью ингибирования NS5A HCV. В WO2008/048589 описаны производные 4-(фенилэтинил)-1H-пиразола и их противовирусное применение. В WO2008/070447 описан широкий диапазон ингибирующих HCV-соединений, включая бензимидазольную часть. Как в WO2010/017401, так и в WO2010/065681 описаны бис-имидазольные ингибиторы NS5A HCV.
Существует потребность в ингибиторах HCV, которые могут преодолеть недостатки современной терапии HCV, такие как побочные эффекты, ограниченная эффективность, возникновение устойчивости и несоблюдение пациентом режима лечения, а также улучшить ответ в виде постоянной вирусной нагрузки.
Настоящее изобретение относится к группе ингибирующих HCV бис-бензимидазольных производных с пригодными свойствами в отношении одного или нескольких из следующих параметров: противовирусная эффективность, благоприятный профиль развития устойчивости, снижение или отсутствие токсичности или генотоксичности, благоприятная фармакокинетика и фармакодинамика, простота изготовлении и введения и ограниченное взаимодействие лекарственное средство - лекарственное средство с другими лекарственными веществами, в частности с другими средствами против HCV, или его отсутствие.
Соединения по изобретению также могут быть привлекательными вследствие того факта, что они лишены активности против других вирусов, в частности против ВИЧ. ВИЧ-инфицированные пациенты часто страдают от коинфекций, таких как HCV. Лечение таких пациентов ингибитором HCV, который также ингибирует ВИЧ, может привести к появлению устойчивых к ВИЧ штаммов.
Описание изобретения
В одном аспекте настоящее изобретение относится к соединениям, которые могут быть представлены формулой I:
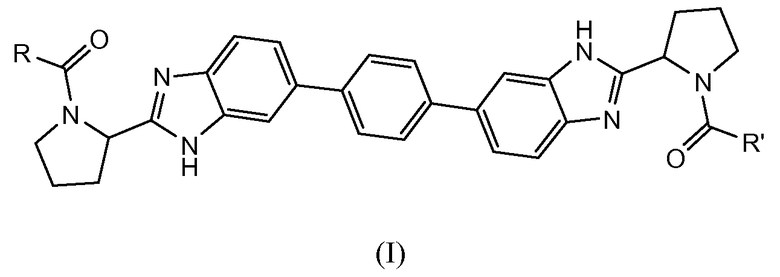
включая любые их возможные стереоизомеры, где
R и R' независимо выбраны из -CR1R2R3, арила, необязательно замещенного 1 или 2 заместителями, выбранными из галогена и метила, и гетероС4-7циклоалкила, где R1 выбран из C1-4алкила, необязательно замещенного метокси, гидроксилом или диметиламино; C3-6циклоалкила; тетрагидропиранила; фенила, необязательно замещенного 1, 2 или 3 заместителями, независимо выбранными из галогена, C1-4алкокси, трифторметокси, или 2 заместителя на соседних атомах кольца образуют 1,3-диоксолановую группу; бензила, необязательно замещенного галогеном или метокси; гетероарила и гетероарилметила;
R2 выбран из водорода, гидроксила, амино, моно- и ди-C1-4алкиламино, (C3-6циклоалкил)(C1-4алкил)амино, C1-4алкилкарбониламино, фениламино, C1-4алкилоксикарбониламино, (C1-4алкилоксикарбонил)(C1-4алкил)амино, C1-4алкиламинокарбониламино, тетрагидро-2-оксо-1(2H)-пиримидинила, пирролидин-1-ила, пиперидин-1-ила, 3,3-дифторпиперидин-1-ила, морфолин-1-ила, 7-азабицикло[2.2.1]гепт-7-ила и имидазол-1-ила; и
R3 представляет собой водород или C1-4алкил, или
CR2R3 вместе образуют карбонил; или
CR1R3 образует циклопропильную группу;
и их фармацевтически приемлемым солям и сольватам.
В следующем аспекте изобретение относится к применению соединений формулы I или их подгрупп, как описано в настоящем документе, для ингибирования HCV. Альтернативно предусмотрено применение указанных соединений для изготовления лекарственного средства для ингибирования HCV.
Варианты осуществления настоящего изобретения относятся к соединениям формулы (I) или любой их подгруппе, как описано в настоящем документе, где применимо одно или несколько определений для R, R', R1, R2 и R3, как указано в настоящем документе.
Подгруппы соединений формулы I представляют собой соединения формулы I или подгруппы соединений формулы I, как определено в настоящем документе, где R и R' независимо представляют собой -CR1R2R3 или арил, где арил представляет собой 5-членный гетероарил; в частности, где R и R' независимо представляют собой -CR1R2R3; более конкретно, где R и R' представляют собой -CR1R2R3 и являются одинаковыми.
Подгруппы соединений формулы I представляют собой соединения формулы I или подгруппы соединений формулы I, как определено в настоящем документе, где R2 представляет собой гидроксил, амино, моно- или ди-C1-4алкиламино, C1-4алкилкарбониламино, C1-4алкилоксикарбониламино; в частности, R2 представляет собой C1-4алкилкарбониламино или C1-4алкилоксикарбониламино.
Подгруппы соединений формулы I представляют собой соединения формулы I или подгруппы соединений формулы I, как определено в настоящем документе, где R1 выбран из C1-4алкила; фенила, необязательно замещенного 1 или 2 заместителями, независимо выбранными из галогена, метила, метокси, или 2 заместителя на соседних атомах кольцах образуют 1,3-диоксолановую группу; и гетероарила. В частности, R1 выбран из разветвленного C3-4алкила; фенила, необязательно замещенного 1 заместителем, выбранным из галогена и метила; и гетероарила. Более конкретно, R1 выбран из разветвленного C3-4алкила; фенила, необязательно замещенного 1 заместителем, выбранным из галогена.
В первом варианте осуществления R и R' независимо выбраны из -CR1R2R3, арила, необязательно замещенного 1 или 2 заместителями, выбранными из галогена и метила, и гетероС4-7циклоалкила, где
R1 выбран из C1-4алкила, необязательно замещенного метокси или диметиламино; фенила, необязательно замещенного 1, 2 или 3 заместителями, независимо выбранными из галогена, C1-4алкокси, трифторметокси, или 2 заместителя на соседних атомах кольцах образуют 1,3-диоксолановую группу; бензила, необязательно замещенного галогеном или метокси; гетероарила и гетероарилметила;
R2 выбран из водорода, гидроксила, амино, моно- и ди-C1-4алкиламино, C1-4алкилкарбониламино, C1-4алкилоксикарбониламино, C1-4алкиламинокарбониламино, пиперидин-1-ила и имидазол-1-ила; и
R3 представляет собой водород, или R1 и R3 вместе образуют оксо или циклопропильную группу; или их фармацевтически приемлемая соль и/или сольват.
Во втором варианте осуществления R и R' независимо выбраны из -CR1R2R3, арила, необязательно замещенного 1 или 2 заместителями, выбранными из галогена и метила, и гетероС4-7циклоалкила, где
R1 выбран из C1-4алкила, необязательно замещенного метокси, гидроксилом или диметиламино; C3-6циклоалкила; фенила, необязательно замещенного 1, 2 или 3 заместителями, независимо выбранными из галогена, C1-4алкокси, трифторметокси, или 2 заместителя на соседних атомах кольцах образуют 1,3-диоксолановую группу; бензила, необязательно замещенного галогеном или метокси; гетероарила и гетероарилметила;
R2 выбран из водорода, гидроксила, амино, моно- и ди-C1-4алкиламино, (C3-6циклоалкил)(C1-4алкил)амино, C1-4алкилкарбониламино, фениламино, C1-4алкилоксикарбониламино, (C1-4алкилоксикарбонил)(C1-4алкид)амино, C1-4алкиламинокарбониламино, тетрагидро-2-оксо-1(2H)-пиримидинила, пирролидин-1-ила, пиперидин-1-ила, 3,3-дифторпиперидин-1-ила, морфолин-1-ила, 7-азабицикло[2.2.1]гепт-7-ила и имидазол-1-ила; и
R3 представляет собой водород или C1-4алкил, или
CR2R3 вместе образуют карбонил; или
CR1R3 образует циклопропильную группу;
и их фармацевтически приемлемые соли и сольваты;
при условии, что (a) когда R и R' являются идентичными и представляют собой -CR1R2R3, где (a-1) R2 представляет собой C1-4алкилоксикарбониламино и R3 представляет собой водород, тогда R1 отличен от незамещенного C1-4алкила, или этила, замещенного гидроксилом или метокси; или где (a-2) R2 представляет собой метилоксикарбониламино и R3 представляет собой водород, тогда R1 отличен от незамещенного фенила; и
(b) когда R и R' отличаются и каждый из них независимо представляет собой -CR1R2R3, где R1 представляет собой фенил или 2-пропил, R2 представляет собой диметиламин и R3 представляет собой водород в одной группе -CR1R2R3, тогда в другой группе -CR1R2R3 R1 не может принимать значение 2-пропила и R2 не может принимать значение метилоксикарбониламино и R3 не может принимать значение водорода.
В третьем варианте осуществления R и R' независимо выбраны из -CR1R2R3, где R1 выбран из фенила, необязательно замещенного 1, 2 или 3 заместителями, независимо выбранным из галогена, C1-4алкокси, трифторметокси, или 2 заместителя на соседних атомах кольцах образуют 1,3-диоксолановую группу;
R2 выбран из гидроксила, моно- или ди-C2-4алкиламино, (C3-6циклоалкил)(C1-4алкил)амино, C1-4алкилкарбониламино, (C1-4алкилоксикарбонил)(C1-4алкил)амино, C1-4алкиламинокарбониламино, тетрагидро-2-оксо-1(2H)-пиримидинила, пирролидин-1-ила, пиперидин-1-ила, 3,3-дифторпиперидин-1-ила, морфолин-1-ила, 7-азабицикло[2.2.1]гепт-7-ила и имидазол-1-ила; и
R3 представляет собой водород или C1-4алкил, или
CR2R3 вместе образуют карбонил; или
CR1R3 образует циклопропильную группу;
и их фармацевтически приемлемые соли и сольваты;
В четвертом варианте осуществления R1 выбран из гетероарила и гетероарилметила;
R2 выбран из водорода, моно- или ди-C1-4алкиламино, (C3-6циклоалкил)(C1-4алкил)амино, C1-4алкилкарбониламино, C1-4алкилоксикарбониламино, (C1-4алкилоксикарбонил)(C1-4алкил)амино, C1-4алкиламинокарбониламино, тетрагидро-2-оксо-1(2H)-пиримидинила, пирролидин-1-ила, пиперидин-1-ила, 3,3-дифторпиперидин-1-ила, морфолин-1-ила, 7-азабицикло[2.2.1]гепт-7-ила и имидазол-1-ила; и
R3 представляет собой водород;
и их фармацевтически приемлемые соли и сольваты.
В пятом варианте осуществления
R1 представляет собой C1-4алкил;
R2 выбран из C1-4алкиламинокарбониламино или тетрагидро-2-оксо-1(2H)-пиримидинила; и
R3 представляет собой водород или C1-4алкил;
и их фармацевтически приемлемые соли и сольваты.
В шестом варианте осуществления
R1 представляет собой C3-6циклоалкил;
R2 представляет собой водород
и R3 представляет собой водород;
и их фармацевтически приемлемые соли и сольваты.
В следующем аспекте изобретение относится к соединению формулы I или его фармацевтически приемлемой соли, гидрату или сольвату для применения для лечения или профилактики (или для изготовления лекарственного средства для лечения или профилактики) инфекции HCV. Типичные генотипы HCV в контексте лечения или профилактики согласно изобретению включают, но не ограничиваются ими, генотип 1b (преобладающий в Европе) и 1a (преобладающий в Северной Америке). Также изобретение относится к способу лечения или профилактики инфекции HCV, в частности генотипа 1a или 1b, включающему введение индивидууму, нуждающемуся в этом, терапевтически эффективного количества соединения, как определено в настоящем документе выше.
Чистые стереоизомерные формы соединений и промежуточных соединений, упоминаемых в настоящем документе, определяют как изомеры, по существу не содержащие других энантиомерных или диастереомерных форм той же основной молекулярной структуры указных соединений или промежуточных соединений. В частности, термин "стереоизомерно чистый" относится к соединениям или промежуточным соединениям, имеющим стереоизомерный избыток по меньшей мере 80% (т.е. минимум 90% одного изомера и максимум 10% других возможных изомеров) вплоть до стереоизомерного избытка 100% (т.е. 100% одного изомера и отсутствие другого изомера), более конкретно, соединения или промежуточные соединения, имеющие стереоизомерный избыток от 90% вплоть до 100%, еще более конкретно имеющие стереоизомерный избыток от 94% вплоть до 100% и наиболее конкретно имеющие стереоизомерный избыток от 97% вплоть до 100%. Термины "энантиомерно чистый" и "диастереомерно чистый" следует понимать аналогично, но рассматривая энантиомерный избыток и диастереомерный избыток, соответственно, данной смеси.
Чистые стереоизомерные формы или стереоизомеры соединений и промежуточных соединений по настоящему изобретению можно получать с использованием известных в данной области способов. Например, энантиомеры можно отделять друг от друга селективной кристаллизацией их диастереомерных солей с оптически активными кислотами или основаниями. Их примерами являются винная кислота, дибензоилвинная кислота, дитолуолвинная кислота и камфорсульфоновая кислота. Альтернативно энантиомеры можно разделять хроматографическими способами с использованием хиральных стационарных фаз. Указанные чистые стереохимически изомерные формы также можно получать из соответствующих чистых стереоизомерных форм соответствующих исходных материалов при условии, что реакция протекает стереоспецифично. Предпочтительно, если является желательным конкретный стереоизомер, указанное соединение синтезируют стереоспецифическими способами получения. В этих способах преимущественно используются энантиомерно чистые исходные материалы.
Диастереомерные рацематы соединений формулы I можно получать по отдельности общепринятыми способами. Соответствующими способами физического разделения, которые можно преимущественно использовать, являются, например, селективная кристаллизация и хроматография, например колоночная хроматография или сверхкритическая жидкостная хроматография.
Соединения формулы имеют несколько центров хиральности. Интерес представляют стереогенные центры кольца пирролидина на 2 атоме углерода. Конфигурация в этом положении может соответствовать L-пролину, т.е.
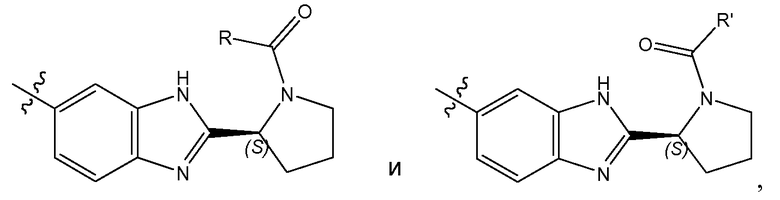
или соответствовать D-пролину, т.е.
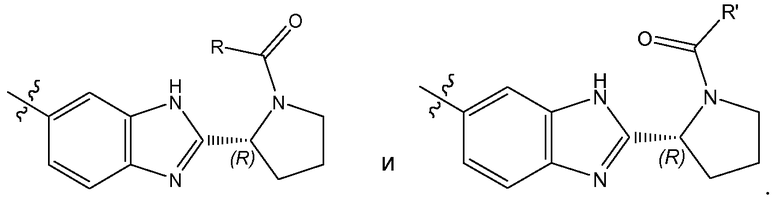
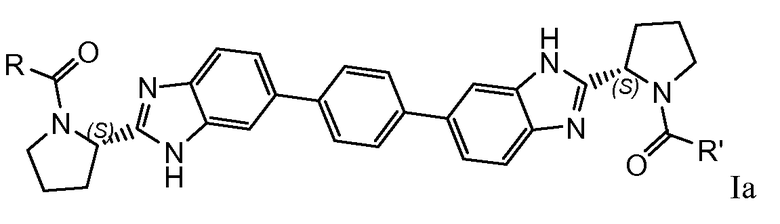
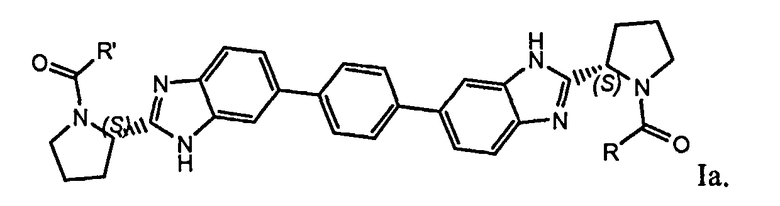
Особый интерес представляют соединения формулы I или их подгруппы, как определено в настоящем документе, которые соответствуют формуле Ia.
Также интерес представляет конфигурация группы -CR4R2R3: когда R1 выбран из C1-4алкила, необязательно замещенного метокси, гидроксилом или диметиламино; C3-6циклоалкила и тетрагидропиранила, тогда предпочтительной является S-конфигурация; когда R1 выбран из фенила, необязательно замещенного 1, 2 или 3 заместителями, независимо выбранными из галогена, C1-4алкокси, трифторметокси, или 2 заместителя на соседних атомах кольцах образуют 1,3-диоксолановую группу; и гетероарила; тогда предпочтительной является R-конфигурация.
Фармацевтически приемлемые аддитивные соли включают терапевтически активные нетоксичные кислотные и основно-аддитивные солевые формы соединений формулы (I) или их подгрупп. Интерес представляют свободные, т.е. несолевые формы соединений формулы I, или любой подгруппы соединений формулы I, указанных в настоящем документе.
Фармацевтически приемлемые кислотно-аддитивные соли можно удобным образом получать путем обработки основной формы такой подходящей кислотой. Подходящие кислоты включают, например, неорганические кислоты, такие как галогенводородные кислоты, например, хлористоводородная или бромистоводородная кислота, серная, азотная, фосфорная кислоты и т.п.; или органические кислоты, например, такие как уксусная, пропионовая, гидроксиуксусная, молочная, пировиноградная, щавелевая (т.е. этандиовая), малоновая, янтарная (т.е. бутандиовая кислота), малеиновая, фумаровая, яблочная (т.е. гидроксибутандиовая кислота), винная, лимонная, метансульфоновая, этансульфоновая, бензолсульфоновая, п-толуолсульфоновая, цикламовая, салициловая, п-аминосалициловая, памовая кислоты и т.п. Наоборот, указанные солевые формы можно превращать обработкой соответствующим основанием в форму свободного основания.
Соединения формулы (I), содержащие кислотный протон, также можно превращать в их основно-аддитивные соли, в частности аддитивные солевые формы металлов или аминов, путем обработки подходящими органическими и неорганическими основаниями. Подходящие солевые формы включают, например, соли аммония, соли щелочных и щелочноземельных металлов, например соли лития, натрия, калия, магния, кальция и т.п., соли с органическими основаниями, например, бензатином, N-метил-D-глюкамином, соли гидрабамина и соли с аминокислотами, например, такими как аргинин, лизин и т.п.
Термин "сольваты" охватывает любые фармацевтически приемлемые сольваты, которые соединения формулы I, а также их соли способны образовывать. Такие сольваты представляют собой, например, гидраты, алкоголяты, например, этаноляты, пропаноляты и т.п.
Некоторые из соединений формулы I также могут существовать в таутомерных формах. Например, таутомерными формами амидных (-C(=O)-NH-) групп являются иминоспирты (-C(OH)=N-). Подразумевается, что таутомерные формы, хотя и не указаны явно в структурных формулах, представленных в настоящем документе, включены в объем настоящего изобретения.
Как используют в настоящем документе, "C1-4алкил" в качестве группы или части группы определяет насыщенные прямые или разветвленные углеводородные группы, имеющие от 1 до 4 атомов углерода, например, такие как метил, этил, 1-пропил, 2-пропил, 1-бутил, 2-бутил, 2-метил-1-пропил, 2-метил-2-пропил. Для целей настоящего изобретения среди C1-4алкилов интерес представляют C3-4алкилы, т.е. прямые или разветвленные углеводородные группы, имеющие 3 или 4 атома углерода, такие как 1-пропил, 2-пропил, 1-бутил, 2-бутил, 2-метил-1-пропил, 2-метил-2-пропил. Особый интерес может представлять разветвленный C3-4алкил, такой как 2-пропил, 2-бутил, 2-метил-1-пропил, 2-метил-2-пропил.
Термин "C3-6циклоалкил" в качестве группы или ее части определяет насыщенные циклические углеводородные группы, имеющие от 3 до 6 атомов углерода, которые вместе образуют циклическую структуру. Примеры C3-6циклоалкила включают циклопропил, циклобутил, циклопентил и циклогексил.
"C1-4алкокси" в качестве группы или части группы означает группу формулы -O-C1-4алкил, где C1-4алкил является таким, как определено выше. Примерами C1-4алкокси являются метокси, этокси, н-пропокси, изопропокси.
Термин "галоген" является общим для фтора, хлора, брома и йода.
Как используют в настоящем документе, термин "(=O)" или "оксо" образует карбонильную группу, когда она присоединена к атому углерода. Следует отметить, что атом может быть замещен только оксогруппой, когда валентность этого атома допускает это.
Как используют в настоящем документе "арил" для цели определения в качестве группы или ее части означает ароматическую кольцевую структуру, необязательно содержащую один или два гетероатома, выбранных из N, O и S, в частности из N и O. Указанная ароматическая кольцевая структура может иметь 5 или 6 атомов в кольце.
Как используют в настоящем документе, приставка "гетеро-" в определении группы означает, что группа содержит по меньшей мере 1 гетероатом, выбранный из N, O и S, в частности N и O. Например, термин "гетероарил" означает ароматическую кольцевую структуру, как определено для термина "арил", содержащую по меньшей мере 1 гетероатом, выбранный из N, O и S, в частности из N и O, например, фуранил, оксазолил, пиридинил. Альтернативно термин "гетероС4-7циклоалкил" означает насыщенные углеводородные группы, содержащие по меньшей мере 1 гетероатом, выбранный из N, O и S, в частности из N и O, например, тетрагидрофуранил, тетрагидропиранил, пиперидинил.
Когда положение группы на молекулярной части не указано (например, заместитель на фениле) или представлено нефиксированной связью, такая группа может быть расположена на любом атоме такой части при условии, что полученная структура является химически стабильной. Когда какая-либо переменная присутствует в молекуле более одного раза, каждое определение является независимым.
Когда термин "соединения формулы I", или "настоящие соединения" или сходные термины используют в настоящем документе, он включает соединения формулы I, включающие возможные его стереоизомерные формы и фармацевтически приемлемые соли и сольваты.
Общие способы синтеза
Схема 1
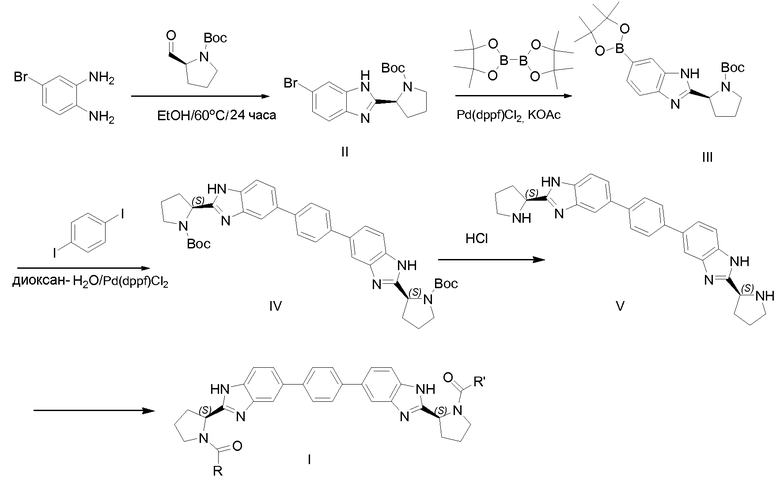
Соединения формулы I, где R и R' являются одинаковыми, можно получать с использованием синтетических реакций, проиллюстрированных на схеме 1 выше. Окислительная циклизация N-(трет-бутоксикарбонил)-L-пролиналя с 4-бромбензол-1,2-диамином приводит к производному бензимидазола II, которое превращается в бороновый сложный эфир III в катализируемых Pd условиях в присутствии бис(пинаколято)дибора. Затем бороновый сложный эфир III превращают в соединение IV путем связывания с 1,4-дийодбензолом с использованием условий Сузуки-Майяра. Альтернативно вместо 1,4-дийодбензола можно использовать 1,4-дибромбензол. Пригодным Pd-катализатором является дихлор-((бис-дифенилфосфино)ферроценил)палладий(II) (Pd(dppf)Cl2). Соединение V получают после удаления трет-бутоксикарбонильной (Boc) защитной группы азота пирролидина в кислотных условиях, например, с использованием HCl в изопропаноле. Затем полученное соединение V можно превращать в соединение формулы I ацилированием соответствующей кислотой формулы R-C(=O)-OH, где R имеет значения R и R', как определено для соединений формулы I или любой их подгруппы.
Указанное ацилирование можно проводить путем реакции исходных материалов в присутствии агента реакции сочетания или путем превращения карбоксильной функциональной группы в активную форму, такую как активный сложный эфир, смешанный ангидрид или хлорид, или бромид карбоновой кислоты. Общие описания таких реакций сочетания и реагентов, используемых в них, могут быть найдены в общих справочниках по химии пептидов, например, M. Bodanszky, "Peptide Chemistry", 2nd rev. ed., Springer-Verlag, Berlin, Germany, (1993).
Примеры реакций сочетания для ацилирования по аминогруппе или образования амидной связи включают азидный способ, способ смешанного ангидрида угольной-карбоновой кислоты (изобутилхлорформиат), карбодиимидный способ (дициклогексилкарбодиимид, диизопропилкарбодиимид или растворимый в воде карбодиимид, такой как N-этил-N'-[3-(диметиламино)пропил]карбодиимид), способ активного сложного эфира (например, п-нитрофенилового, п-хлорфенилового, трихлорфенилового, пентахлорфенилового, пентафторфенилового, N-гидроксиянтарного имидо- сложных эфиров и т.п.), способ с реагентом K Вудводрда, способ 1,1-карбонилдиимидазола (CDI или N,N'-карбонилдиимидазол), способы фосфорных реагентов или окислительно-восстановительные способы. Некоторые из этих способов можно усиливать добавлением пригодных катализаторов, например в способе карбодиимида - добавлением 1-гидроксибензотриазола или 4-DMAP. Следующими агентами реакции сочетания являются гексафторфосфат (бензотриазол-1-илокси)-трис-(диметиламино)фосфония либо самостоятельно, либо в присутствии 1-гидроксибензотриазола или 4-DMAP; или тетрафторборат 2-(1H-бензотриазол-1-ил)-N,N,N',N'-тетраметилурония (TBTU), или гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (HATU). Эти реакции сочетания можно проводить либо в фазе раствора (жидкая фаза), либо в твердой фазе. Для целей настоящего изобретения предпочтительный способ ацилирования проводят с использованием HATU.
Реакции сочетания предпочтительно проводят в инертных растворителях, таких как галогенированные углеводороды, например, дихлорметан, хлороформ, диполярные апротонные растворители, такие как ацетонитрил, диметилформамид, диметилацетамид, DMSO, HMPT или простые эфиры, такие как тетрагидрофуран (THF).
Во многих случаях реакции сочетания проводят в присутствии пригодного основания, такого как третичный амин, например, триэтиламин, диизопропилэтиламин (DIPEA), N-метилморфолин, N-метилпирролидин, 4-DMAP или 1,8-диазабицикло[5.4.0]ундец-7-ен (DBU). Температура реакции может находиться в диапазоне от 0°С до 50°С и время реакции может находиться диапазоне от 15 мин до 24 ч.
Схема 2
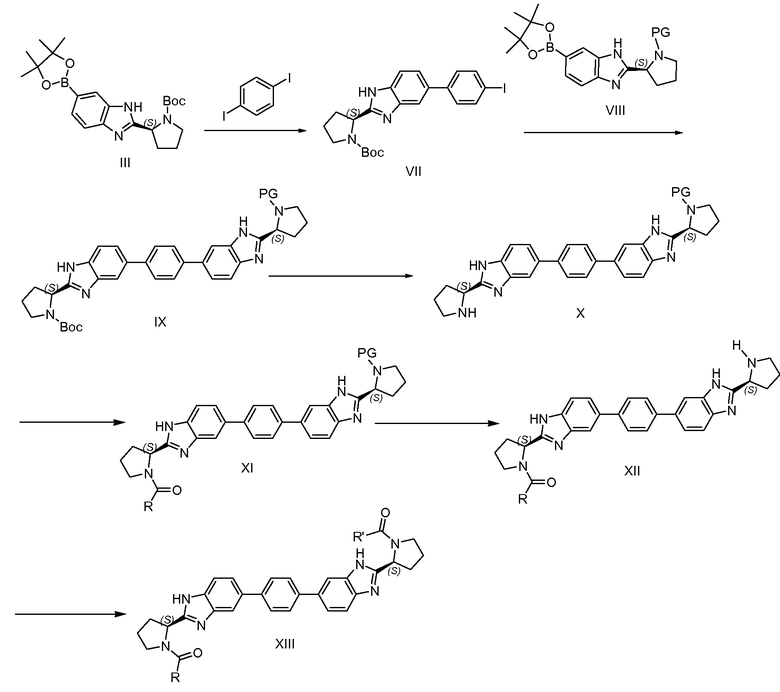
Альтернативно соединения формулы I, где R и R' не являются одинаковыми, т.е. соединения формулы XIII, можно получать с использованием синтетического пути, проиллюстрированного на схеме 2. С использованием стандартных условий Сузуки-Майяра, можно проводить реакцию сочетания боронового сложного эфира III и 1,4-дийодбензола в условиях, сравнимых с условиями, используемыми для превращения III в IV (схема 1), за исключением того, что соотношение 1,4-дийодбензола и боронового сложного эфира III составляет приблизительно 1 к 1, возможно выше, для получения монойодида VII. Альтернативно вместо 1,4-дийодбензола можно использовать 1,4-дибромбензол. Затем VII можно подвергать реакции сочетания с бороновым сложным эфиром VIII. Следует понимать, что аминозащитную группу PG на азоте пирролидина в бороновом сложном эфире VIII следует выбирать так, чтобы ее можно было удалить в условиях, которые не влияют на Boc-группу или R-C(=O)-группу на другом азоте в молекуле. Также следует понимать, что PG также может представлять собой R'-C(=O)-группу конечного синтезируемого соединения формулы I. Реакцию сочетания VII и VIII можно вновь проводить с использованием стандартных условий Сузуки-Майяра, и получать соединение IX. Затем из соединения IX можно селективно удалять защитную группу с получением соединения X с использованием условий, подходящих для удаления Boc-защитной группы. Например, в случае, когда PG представляет собой бензилоксикарбонил или бензил, Boc-защитную группу можно селективно удалять в стандартных условиях удаления защитной группы Boc, т.е. путем обработки кислотой.
Более того, в случае, когда PG представляет собой бензилоксикарбонил или бензил, PG можно селективно удалять восстановительной обработкой, оставляющей R-C(=O)-группу в соединении XI незатронутой. Другие пригодные защитные группы PG и соответствующие условия селективного удаления защитных групп могут быть найдены в Greene, "Protective groups in organic synthesis", Peter G. M. Wuts, Fourth Edition, Chapter 7: "Protection for the Amino group". Затем соединение X можно ацилировать соответствующей кислотой формулы R-C(=O)-OH, где R имеет значения R, как определено для соединений формулы I или любой их подгруппы. Получают соединение XI.
Для соединения XI, в случае, когда PG представляет собой -(C=O)-R', соединение XI совпадает с соединением XIII. В случае, когда PG представляет собой аминозащитную группу, PG можно удалять в условиях, совместимых с -(C=O)-R, например путем гидрогенизации, когда PG представляет собой бензил или бензилоксикарбонил, или в основных условиях, таких как диэтиламин, в случае, когда PG представляет собой флуоренилметилоксикарбонил, с получением соединения XII. Другие способы селективного удаления защитных групп могут быть найдены в справочнике Greene.
В случае, когда -(C=O)-R несовместим с условиями удаления защитной группы PG, можно использовать путь, представленный на схеме 5, где защитная группа PG выбрана так, чтобы она была совместима с группой Boc.
Соединения XII можно превращать в соединение XIII ацилированием, сходным с превращением XIV в XV, V в I и X в XI, и как подробно описано для превращения V в I согласно схеме 1.
Схема 3
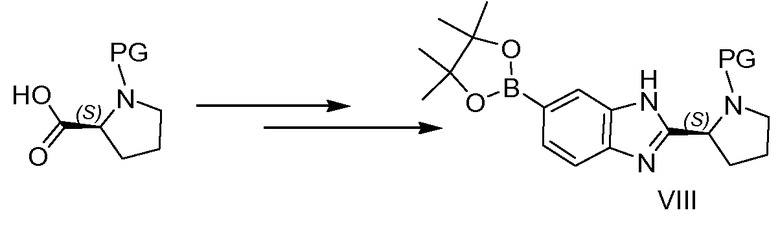
Схема 4
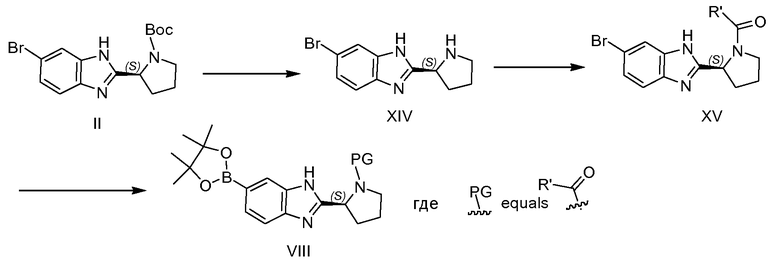
Бороновый сложный эфир VIII можно получать по меньшей мере двумя различными путями, как проиллюстрировано на схемах 3 и 4. В случае, когда PG представляет собой защитную группу, например, такую как бензилоксикарбонил, флуоренилметилоксикарбонил, бензил или другую пригодную защитную группу PG, как описано в справочнике Greene, соединение можно синтезировать способами, используемыми для синтеза промежуточных соединений II и IIa (см. пример 2), и боронового сложного эфира III, начиная с соответствующим образом защищенного производного пирролидина. В случае, когда PG представляет собой -(C=O)-R', соединение можно получать из промежуточного соединения II, как представлено на схеме 4, путем удаления защитной группы азота пролина в кислых условиях, таких как обработка HCl, например, в iPrOH, или трифторуксусной кислотой с получением соединения XIV, с последующей реакцией конденсации в стандартных условиях ацилирования, таких как применение HATU в присутствии основания, такого как DIPEA. Далее полученный бромид XV можно превращать в бороновую кислоту VIII (где PG представляет собой -(C=O)-R'), как например, как превращение II в III.
Схема 5
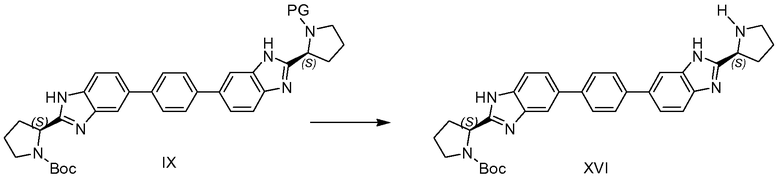
Альтернативно, как показано на схеме 5, защитную группу соединения IX можно удалять в условиях, совместимых с защитной группой Boc, например, путем гидрогенизации, когда PG представляет собой бензил или бензилоксикарбонил, или в основных условиях, таких как диэтиламин, в случае, когда PG представляет собой флуоренилметилоксикарбонил, с получением соединения XVI. Другие способы селективного удаления защитных групп могут быть найдены в справочнике Greene. В этом случае соединение XVI совпадает с соединением X, причем PG представляет собой Boc. В этом случае удаление защитной группы из XI-XII можно проводить в условиях, сходных с превращением II в XIV и IV в V.
Способы синтеза, представленные выше на схемах 1-5, также можно проводить с использованием рацемических производных пролина или производных D-пролина вместо L-пролина. Таким образом, можно получать соединения формулы I с альтернативной стереохимией.
В следующем аспекте настоящее изобретение относится к фармацевтической композиции, содержащей терапевтически эффективное количество соединения формулы I, как описано в настоящем описании, и фармацевтически приемлемый носитель. Терапевтически эффективное количество в данном случае представляет собой количество, достаточное для стабилизации или снижения инфекции HCV у инфицированных индивидуумов, или количество, достаточное для предупреждения инфекции HCV у индивидуумов, обладающих риском инфицирования. В следующем аспекте это изобретение относится к способу получения фармацевтической композиции, как описано в настоящем описании, который включает смешение в однородную смесь фармацевтически приемлемого носителя с терапевтически эффективным количеством соединения формулы I, как описано в настоящем описании.
Таким образом, для целей введения соединения по настоящему изобретению или любую их подгруппу можно изготавливать в виде различных фармацевтических форм. В качестве пригодных композиций можно назвать все композиции, обычно используемые для системного введения лекарственных средств. Для получения фармацевтических композиций по настоящему изобретению, эффективное количество конкретного соединения необязательно в форме аддитивной соли или комплекса с металлом в качестве активного ингредиента смешивают в однородную смесь с фармацевтически приемлемым носителем, который может находиться в различных в формах в зависимости от желательной формы препарата для введения. Желательно, чтобы эти фармацевтические композиции были представлены в единичной дозированной форме, в частности для перорального, ректального, подкожного введения или для парентеральной инъекции. Например, для получения композиций в пероральной дозированной форме можно использовать любую обычную фармацевтическую среду, например такую как вода, гликоли, масла, спирты и т.п. в случае пероральных жидких препаратов, таких как суспензии, сиропы, эликсиры, эмульсии и растворы; или твердые носители, такие как крахмалы, сахара, каолин, лубриканты, связующие вещества, дезинтегрирующие вещества и т.п. в случае порошков, пилюль, капсул и таблеток. Вследствие простоты введения таблетки и капсулы представляют собой наиболее предпочтительные пероральные единичные дозированные формы, в случае которых обычно используют твердые фармацевтические носители. В случае парентеральных композиций носитель, как правило, содержит, стерильную воду, составляющую по меньшей мере большую его часть, хотя в него могут быть включены другие ингредиенты, например, для обеспечения растворимости. Например, можно получать инъецируемые растворы, в которых носитель содержит солевой раствор, раствор глюкозы или смесь солевого раствора и раствора глюкозы. Также можно получать инъецируемые суспензии, в случае которых можно использовать пригодные жидкие носители, суспендирующие вещества и т.п. Также предусмотрены препараты в твердой форме, которые необходимо превращать, непосредственно перед применением, в препараты в жидкой форме. В композициях, пригодных для чрескожного введения, носитель необязательно содержит усиливающее проницаемость вещество и/или пригодное смачивающее вещество, необязательно в сочетании с пригодными добавками любой структуры в небольших пропорциях, которые не оказывают значительных неблагоприятных эффектов на кожу. Соединения по настоящему изобретению также можно вводить посредством оральной ингаляции или инсуффляции в форме раствора, суспензии или сухого порошка с использованием любой из известных в данной области системы для доставки.
Особенно предпочтительным является составление упомянутых выше фармацевтических композиций в единичную дозированную форму для простоты введения и единообразия дозирования. Как используют в настоящем описании, единичная дозированная форма относится к физически дискретным единицам, пригодным в качестве единичных доз, где каждая единица содержит определенное количество активного ингредиента, вычисленное для обеспечения требуемого терапевтического эффекта, совместно с требуемым фармацевтическим носителем. Примерами таких единичных дозированных форм являются таблетки (включая шероховатые или покрытые таблетки), капсулы, пилюли, суппозитории, пакеты с порошком, вафли, инъецируемые растворы или суспензии и т.п., и отдельные их типы.
Соединения формулы I демонстрируют активность против HCV, и их можно использовать для лечения или профилактики инфекции HCV или заболеваний, обусловленных с HCV. Заболевания, обусловленные HCV, включают прогрессирующий фиброз печени, воспаление и некроз, ведущие к циррозу, заболевание печени конечной стадии и печеночно-клеточную карциному. Более того, о некоторых соединениях по настоящему изобретению известно, что они являются активными в отношении мутантных штаммов HCV. Кроме того, соединения по настоящему могут обладать привлекательными свойствами с точки зрения биодоступности, демонстрируют благоприятный фармакокинетический профиль, включая приемлемое время полужизни, AUC (площадь под кривой) и максимальные и минимальные значения, и лишены неблагоприятных эффектов, таких как недостаточное быстрое начало действия и задерживание в тканях.
Противовирусную активность соединений формулы I в отношении HCV in vitro тестировали в клеточной системе с репликоном HCV на основе Lohmann et al. (1999) Science 285:110-113, с дополнительными модификациями, описанными Krieger et al. (2001) Journal of Virology 75: 4614-4624, которые далее проиллюстрированы в разделе "Примеры". Несмотря на то, что эта модель не является полной моделью инфекции HCV, эта модель широко распространена в качестве наиболее надежной и эффективной модели автономной репликации РНК HCV, доступной в настоящее время. Соединения, обладающие активностью против HCV в этой клеточной модели, рассматривают в качестве кандидатов для дальнейшей разработки для лечения инфекции HCV у млекопитающих. Будет понятно, что важно отличать соединения, которые специфично препятствуют функционированию HCV, от соединений, которые оказывают цитотоксические или цитостатические эффекты в модели репликона HCV, и вследствие этого приводят к снижению РНК HCV или концентрации связанного с ней репортерного фермента. В данной области известны способы анализа для оценки клеточной цитотоксичности на основе, например, активности митохондриальных ферментов с использованием флуорогенных окислительно-восстановительных красителей, таких как резазурин. Более того, существуют способы скрининга с подсчетом клеток, для оценки неселективного ингибирования активности связанного репортерного гена, такого как ген люциферазы светляков. Пригодные типы клеток можно адаптировать посредством стабильной трансфекции репортерным геном люциферазы, экспрессия которого зависит от конститутивно активного промотора гена, и такие клетки можно использовать в качестве способа скрининга с подсчетом для устранения неселективных ингибиторов.
Вследствие свойств, направленных против HCV, соединения формулы I или их подгруппы, как описано в настоящем документе, являются пригодными для ингибирования репликации HCV, в частности для лечения теплокровных животных, в частности людей, инфицированных HCV, и для профилактики инфекций HCV у теплокровных животных, в частности у людей. Более того, настоящее изобретение относится к способу лечения теплокровного животного, в частности человека, инфицированного HCV или обладающего риском инфицирования HCV, причем указанный способ включает введение терапевтически или профилактически эффективного количества соединения формулы I, как определено в настоящее документе выше.
Таким образом, соединения формулы I, как описано в настоящем документе, можно использовать в качестве лекарственного средства, в частности в качестве лекарственного средства против HCV. Указанное применение в качестве лекарственного средства или способа лечения включает системное введение инфицированным HCV индивидуумам или восприимчивым к инфекции HCV индивидуумам количества, эффективного для смягчения или предупреждения симптомов и состояний, ассоциированных с инфекцией HCV.
Настоящее изобретение также относится к применению представленных соединений для изготовления лекарственного средства для лечения или профилактики инфекции HCV.
Главным образом, предусматривается, что эффективное противовирусное суточное количество может составлять от приблизительно 0,01 до приблизительно 50 мг/кг, или от приблизительно 0,02 до приблизительно 30 мг/кг массы тела. Может быть пригодным введение требуемой дозы в качестве одной, двух, трех, четырех или более субдоз через соответствующие интервалы на протяжении суток. Указанные субдозы можно изготавливать в виде единичных дозированных форм, например, содержащих от приблизительно 1 до приблизительно 1000 мг, или от приблизительно 1 до приблизительно 500 мг, или от приблизительно 1 до приблизительно 100 мг, или от приблизительно 2 до приблизительно 50 мг активного ингредиента на единичную дозированную форму.
Комбинированная терапия
Также настоящее изобретение относится к комбинации соединения формулы I, его фармацевтически приемлемой соли или сольвата, и другого противовирусного соединения, в частности другого соединения против HCV. Термин "комбинация" относится к продукту, содержащему модулирующее (a) соединение формулы I, как определено в настоящем документе выше, и (b) другой ингибитор против HCV в качестве комбинированного препарата для одновременного, отдельного или последовательного применения при лечении инфекций HCV.
Комбинации по настоящему изобретению можно использовать в качестве лекарственных средств. Таким образом, настоящее изобретение относится к применению соединения формулы (I) или его подгруппы, как определено выше, для изготовления лекарственного средства, пригодного для ингибирования активности HCV у млекопитающего, инфицированного вирусами HCV, где указанное лекарственное средство используют в комбинированной терапии, причем указанная комбинированная терапия, в частности, включает соединение формулы (I) и по меньшей мере одно другое средство против HCV, например IFN-α, пегилированный IFN-α, рибавирин, альбуферон, тарибавирин, нитазоксанид Debio025 или их комбинацию.
Другие средства, которые можно комбинировать с соединениями по настоящему изобретению, включают, например, нуклеозидные и ненуклеозидные ингибиторы полимеразы HCV, ингибиторы протеазы, ингибиторы хеликазы, ингибиторы NS4B и средства, которые функционально ингибируют участок внутренней посадки рибосомы (IRES) и другие средства, которые ингибируют прикрепление HCV к клетке или проникновение вируса внутрь, трансляцию РНК HCV, транскрипцию РНК HCV, репликацию или созревание HCV, сборку или высвобождение вируса. Конкретные соединения в этих классах включают ингибиторы протеазы HCV, такие как телапревир (VX-950), боцепревир (SCH-503034), нарлапревир (SCH-900518), ITMN-191 (R-7227), TMC435350 (TMC435), MK-7009, BI-201335, BI-2061 (цилупревир), BMS-650032, ACH-1625, ACH-1095, GS 9256, VX-985, IDX-375 (ингибитор кофактора протеазы NS4A HCV), VX-500, VX-813, PHX-1766, PHX2054, IDX-136, IDX-316, ABT-450, EP-013420 (и родственные соединения) и VBY-376; нуклеозидные ингибиторы полимеразы HCV, пригодные для изобретения, включают R7128, PSI-7851, PSI 7977, IDX-189, IDX-184, IDX-102, R1479, UNX-08189, PSI-6130, PSI-938 и PSI-879 и различные другие аналоги нуклеозидов и нуклеотидов и ингибиторы HCV, включая ингибиторы, полученные в качестве 2'-C-метил-модифицированных нуклеозидов, 4'-аза-модифицированных нуклеозидов, и 7'-деаза-модифицированных нуклеозидов, например, 4-амино-1-[5-азидо-4-гидрокси-5-гидроксиметил-3-метилтетрагидрофуран-2-ил]пиримидин-2(1H)-он (ссылка 1) и его бис-2-метилпропаноатный сложный эфир (ссылка 2). Ненуклеозидные ингибиторы полимеразы HCV, пригодные для изобретения, включают HCV-796, HCV-371, VCH-759, VCH-916, VCH-222, ANA-598, MK-3281, ABT-333, ABT-072, PF-00868554, BI-207127, GS-9190, A-837093, JKT-109, GL-59728, GL-60667, ABT-072, AZD-2795 и 13-циклогексил-3-метокси-17,23-диметил-7H-10,6-(метаноиминотиоиминоэтанооксиэтаноиминометано)индоло[2,1-a][2]бензазепин-14,24-диона 16,16-диоксид (ссылка 3).
Следующие примеры предоставлены для иллюстрации изобретения и их не следует истолковывать как ограничение его объема.
ПРИМЕРЫ
Пример 1 - синтез соединения V
1.1 Получение промежуточного соединения II
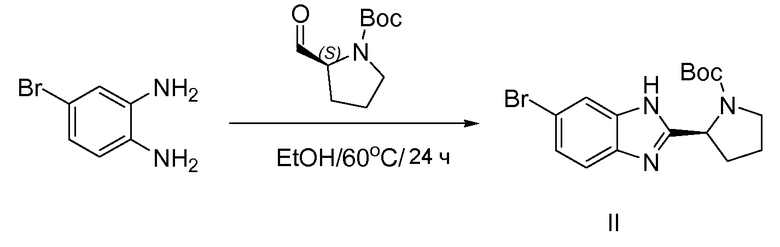
К раствору 4-бромбензол-1,2-диамина (170 г, 0,91 моль) в этаноле (2 л) добавляли (S)-трет-бутил-2-формилпирролидин-1-карбоксилат (258 г, 1,3 моль) при 25°С, смесь нагревали до 60°С в течение 24 часов, TLC показала, что реакция завершилась. Раствор концентрировали и неочищенный продукт очищали колоночной хроматографией (петролейный эфир: этилацетат от 10:1 до 2:1) с получением 215 г II в виде желтого твердого вещества.
1H-ЯМР: CDCl3 400 МГц
δ 7,95-7,4 (м, 3H), 5,35-5,25 (м, 1H), 3,85-3,70 (м, 1H), 3,6-3,45 (м, 1H), 2,6-2,45 (м, 1H), 2,20-1,95 (м, 3H), 1,48-1,38 (м, 5H), 1,2-1,1 (м, 4H).
1.2 Получение промежуточного соединения III
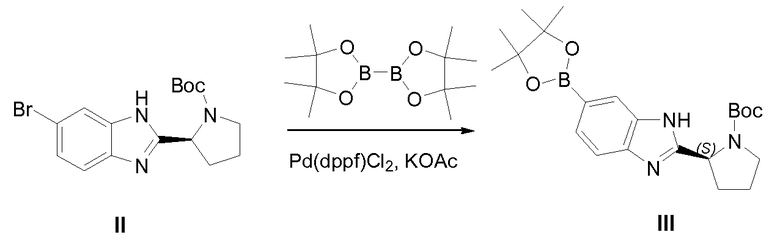
К смеси II (200 г, 546 ммоль), ацетата калия (160,8 г, 1,64 моль) и 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолана) (416 г, 1,64 моль) в DMF (3 л) добавляли Pd(dppf)Cl2 (20 г) в газообразном азоте. Реакционную смесь перемешивали при 85°С в течение 15 часов. Смесь разбавляли этилацетатом, промывали водой и рассолом, сушили над сульфатом магния, твердые вещества удаляли фильтрацией и растворители фильтрата удаляли при пониженном давлении. Осадок очищали колоночной хроматографией на силикагеле (петролейный эфир: этилацетат от 10:1 до 2:1) с получением 125 г III в виде белого твердого вещества (содержит 15% бороновой кислоты).
1.3 Получение промежуточного соединения IV
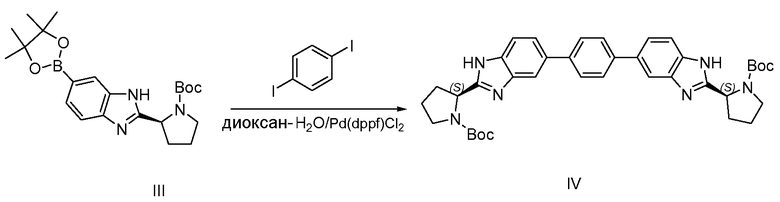
К раствору 1,4-дийодбензола (1,7 г, 5,15 ммоль), III (6 г, 14,4 ммоль) и K2CO3 (2,14 г, 15,5 ммоль) в смеси диоксан-H2O (50 мл, 5:1) добавляли Pd(dppf)Cl2 (300 мг) в атмосфере азота. Смесь нагревали до 85°С в течение 15 часов. Смесь охлаждали до комнатной температуры, концентрировали, добавляли воду и смесь экстрагировали этилацетатом, сушили над сульфатом магния, твердые веществ удаляли фильтрацией и растворители фильтрата удаляли при пониженном давлении. Неочищенный продукт очищали обращенно-фазовой ВЭЖХ с получением 2 г, 96% (IV).
1H-ЯМР: d-метанол 400 МГц
δ 7,84-7,70 (м, 6H), 7,67-7,52 (м, 4H), 5,15-4,99 (м, 2H), 3,70-3,83 (м, 2H), 3,62-3,52 (м, 2H), 2,54-2,35 (м, 2H), 2,19-1,93 (м, 6H), 1,44-1,54 (м, 6H), 1,12-1,25 (м, 12H).
1.4 Получение промежуточного соединения V
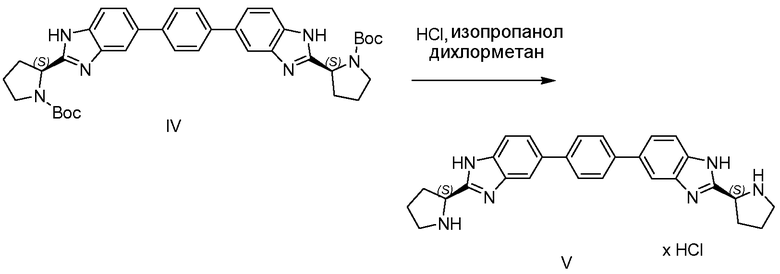
В 20-мл колбу помещали IV (500 мг), дихлорметан (3 мл) и HCl в изопропаноле (3 мл от 5 до 6 M раствора, Acros). Этой смеси позволяли перемешаться в течение 4 часов при комнатной температуре, LCMS подтверждал полное превращение в V. Растворитель удаляли азеотропной перегонкой при пониженном давлении с толуолом и метанолом с получением желтовато-коричневого твердого вещества, которое использовали как есть на следующей стадии.
Пример 2 - Получение IIA, бензилзащищенного промежуточного соединения
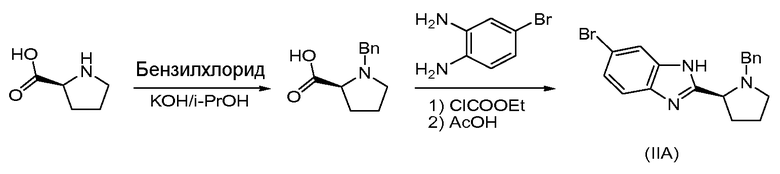
Раствор 10 г L-пролина и KOH (10 г) в 120 мл изопропанола перемешивали при 40°С, затем капельно добавляли бензилхлорид (13,5 мл). Реакционную смесь перемешивали в течение 6 дополнительных часов, затем нейтрализовывали концентрированной HCl до pH 5-6. Реакционную смесь экстрагировали с помощью CH2Cl2 (3×70 мл), объединенные органические слои промывали рассолом, сушили над безводным сульфатом магния, твердые вещества удаляли фильтрацией и растворители фильтрата удаляли при пониженном давлении. Осадок обрабатывали ацетоном с получением N-бензилпролина (15 г).
N-бензилпролин (10,2 г, 50 ммоль) и триэтиламин (50 ммоль) растворяли в THF (250 мл) и охлаждали до 0°С. К этому раствору капельно добавляли этилхлорформиат (ClCOOEt, 50 ммоль) в течение 15 минут и перемешивали в течение дополнительных 30 минут. К этому раствору добавляли 4-бромбензол-1,2-диамин (75 ммоль) в течение 15 минут. Реакционную смесь перемешивали при 0°С в течение 1 часа, затем позволяли ей достигнуть комнатной температуры и перемешивали в течение 16 часов, а затем кипятили с обратным холодильником в течение 3 часов. После завершения этой реакции смесь охлаждали до комнатной температуры и разбавляли этилацетатом (3×70 мл). Органические слои объединяли, сушили (сульфат магния), твердые вещества удаляли фильтрацией и растворители фильтрата выпаривали при пониженном давлении. Осадок очищали колоночной хроматографией (гексан/этилацетат: 7/3) с получением 11 г промежуточного соединения.
11 г промежуточного соединения растворяли в уксусной кислоте (50 мл) при 20°С и перемешивали в течение 15 часов. Растворитель удаляли при пониженном давлении, а затем добавляли NaHCO3 (насыщенный водный, 200 мл). Полученную смесь экстрагировали этилацетатом (3×80 мл). Объединенные органические слои концентрировали и осадок очищали колоночной хроматографией с получением 3 г IIA.
Пример 3 - синтез соединений формулы I
3.1 Получение соединения No. 1
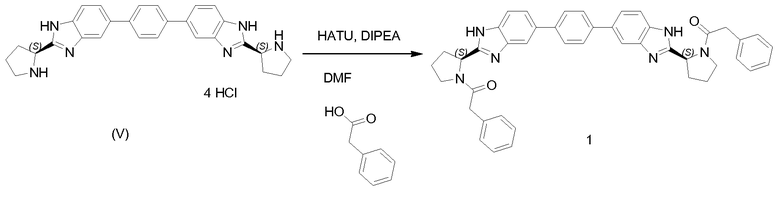
К раствору V (400 мг, 0,77 ммоль) в DMF (10 мл) добавляли DIPEA (0,5 мл, 3 ммоль), HATU (0,73 г, 1,9 ммоль) и фенилуксусную кислоту (2,2 экв, 230 мг, 1,7 ммоль). Смесь перемешивали в течение 2 часов при комнатной температуре, а затем очищали твердофазной экстракцией (Waters PoraPak CX 60cc, промытая 3 объемами метанола перед применением). Наносили неочищенную реакционную смесь, промывали метанолом (4 объема), а затем элюировали 7 M аммиаком в метаноле (раствор от Aldrich, 4 объема). Элюат упаривали при пониженном давлении с получением желтовато-коричневой пены. Для получения чистого твердого вещества добавляли HCl (3 мл, от 5 до 6 M в изопропаноле, Acros), а затем растворители удаляли азеотропной перегонкой с толуолом с получение желтовато-коричневого твердого вещества.
LCMS (M+H) m/z=685 для VI, имеющего формулу C44H40N6O2
Альтернативно очистку и обработку реакционной смеси можно проводить следующим образом: добавить CH2Cl2, промывать насыщенным NaHCO3, высушить органическую фазу с помощью Na2SO4, отфильтровать и концентрировать в вакууме. Затем осадок очищают хроматографией на силикагеле (0-10% MeOH в CH2Cl2) или препаративной ВЭЖХ.
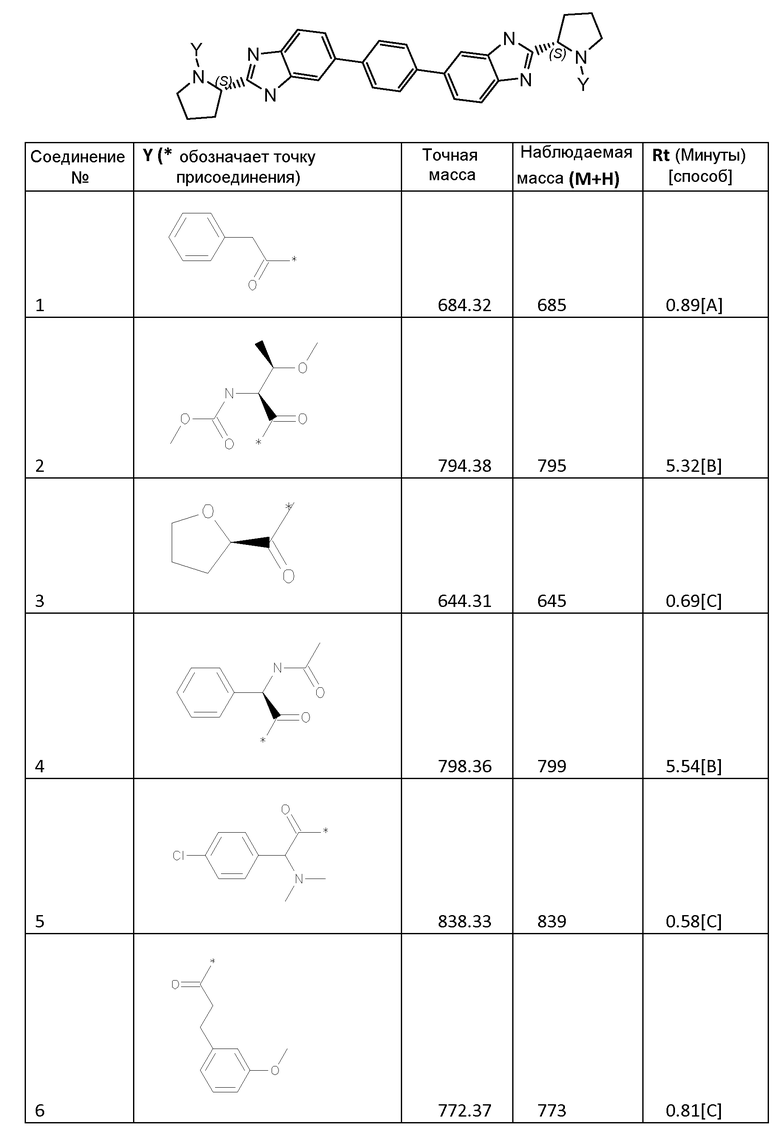
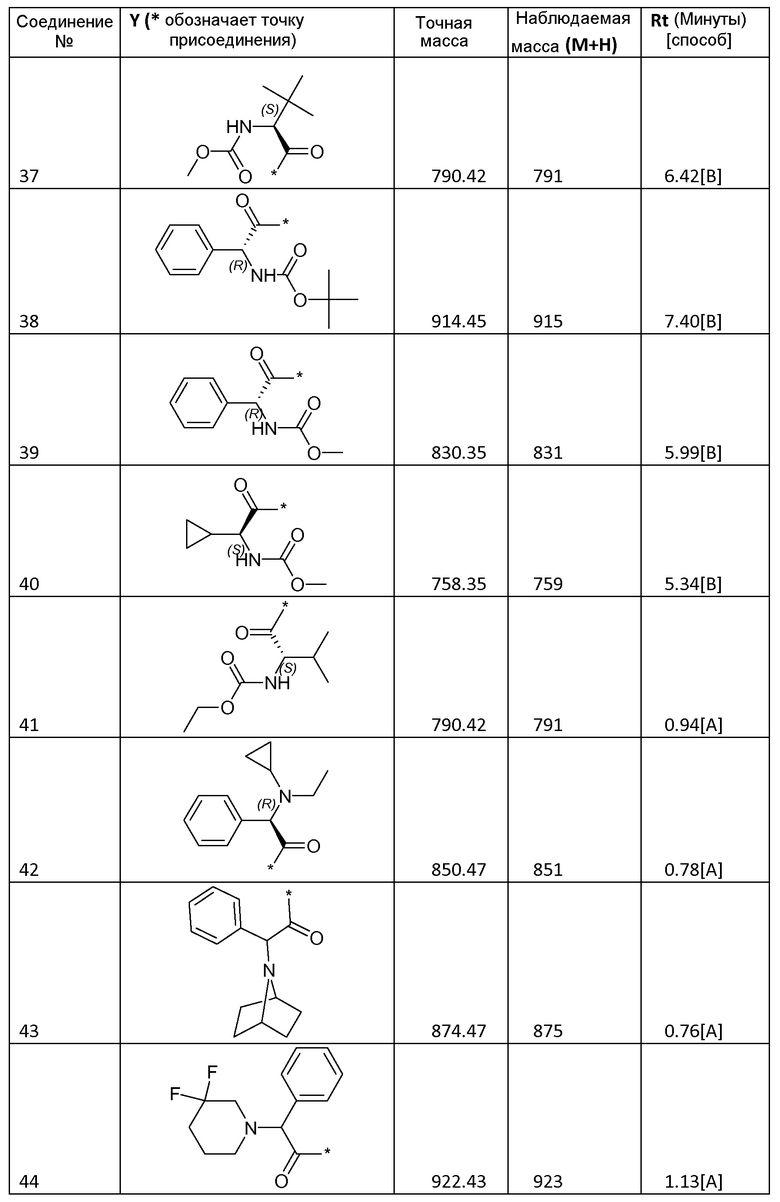
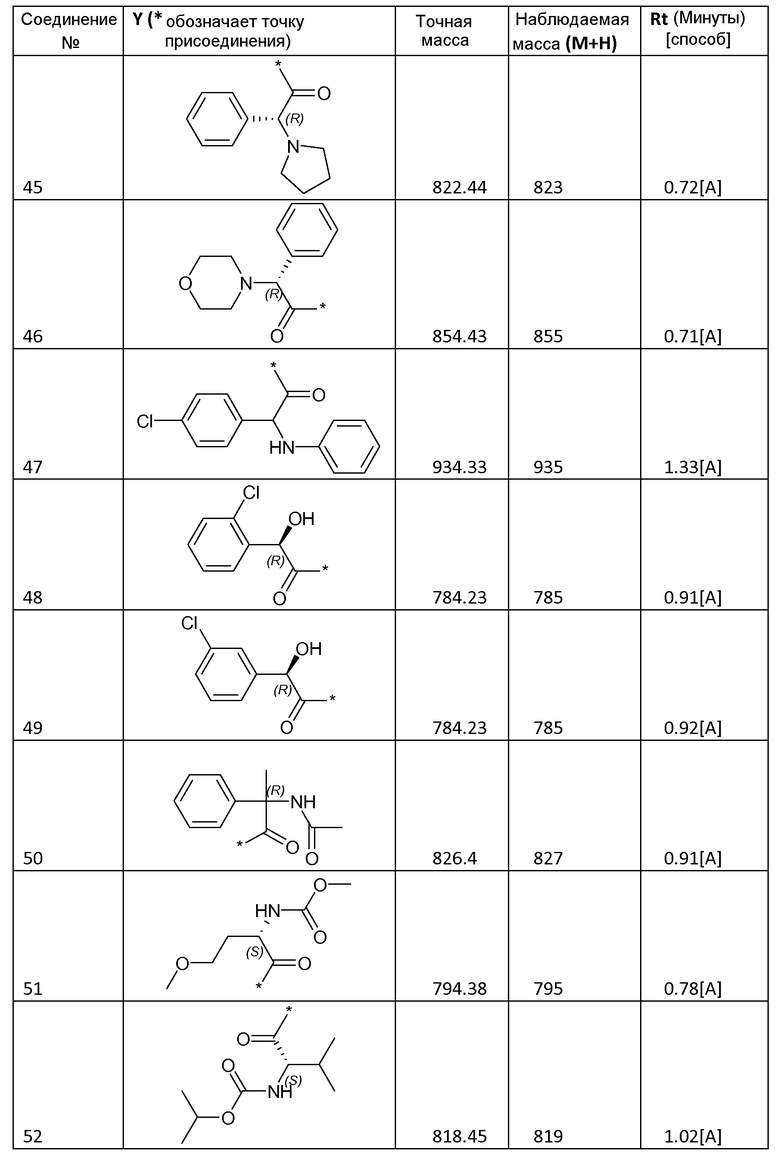
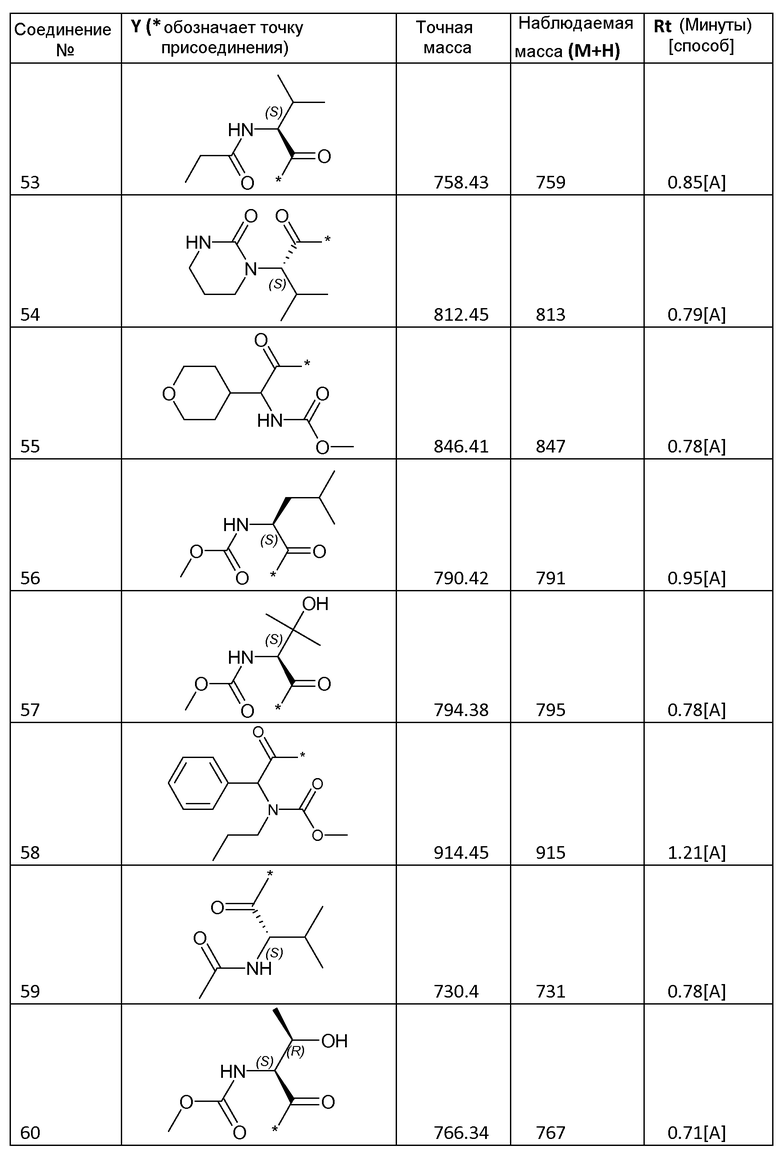
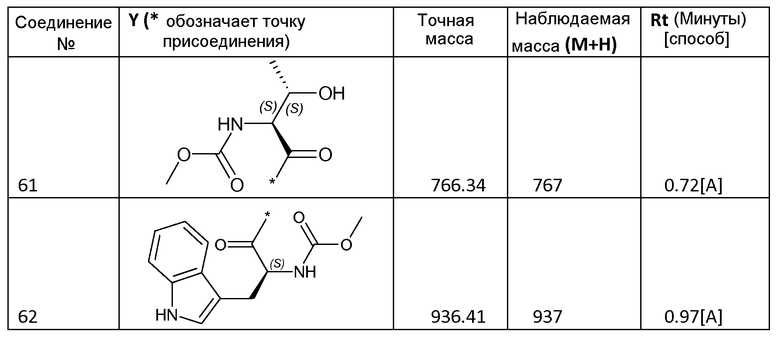
3.2 Получение соединений 2-62
Соединения 2-62, приведенные в таблице 1, синтезировали с использованием способа для соединения 1, описанного в примере 3.1, с использованием соответствующей карбоновой кислоты формулы R-C(=O)-OH.
Все соединения охарактеризовывали с помощью LC/MS. Использовали следующие способы LC/MS:
Способ A: система Waters Acquity UPLC, оборудованная детектором PDA (диапазон 210-400 нм) и Waters SQD с двухрежимным источником ионов ES+/-. Использованная колонка представляла собой Halo C18, 2,7 мк, 2,1×50 мм, и ее поддерживали при 50°С. Градиент от 95% водного раствора муравьиной кислоты (0,1%)/5% ацетонитрила до 100% ацетонитрила пропускали в течение 1,5 минуты, удерживали в течение 0,6 минуты, а затем возвращали к 100% водному раствору муравьиной кислоты (0,1%) в течение 0,5 минуты. Скорость потока составляла 0,6 мл/мин.
Способ B: жидкостная хроматография: Waters Alliance 2695, УФ-детектор: Waters 996 PDA, диапазон: 210-400 нм; детектор масс: Waters ZQ, источник ионов: ES+, ES-. Использованная колонка: SunFire C18 3,5 мк 4,6×100 мм, подвижная фаза A: 10 мМ NH4OOCH+0,1% HCOOH в H2O; подвижная фаза B: CH3OH; температура колонки: 50°С; скорость потока: 1,5 мл/мин. Время градиента (мин) [%A/%B]: от 0 [65/35] до 7[5/95] до 9,6[5/95] до 9,8[65/35] до 12 [65/35].
Способ C: система Waters Acquity UPLC, оборудованная детектором PDA (диапазон 210-400 нм) и Waters SQD с двухрежимным источником ионов ES+/-. Использованная колонка представляла собой XS Strategy 1,7 мк, 2,1×20 мм, и ее поддерживали при 50°С. Градиент от 100% водного раствора муравьиной кислоты (0,1%) до 100% ацетонитрила пропускали в течение 1,5 минуты, затем удерживали в течение 0,6 минуты, а затем возвращали к 100% водному раствору муравьиной кислоты (0,1%) на 0,5 минуты. Скорость потока составляла 0,6 мл/мин.
Способ D: XTerra MS C18 2,5 мк 4,6×50 мм; подвижная фаза A: 10 мМ NH4OOCH+0,1% HCOOH в H2O; подвижная фаза B: CH3OH; температура колонки: 50°С; скорость потока: 1,5 мл/мин; время градиента (мин) [%A/%B]: от 0 [65/35] до 3,8 [5/95] до 5,5[5/95] до 5,6[65/35] до 7 [65/35].
Некоторые соединения охарактризовывали с помощью 1H-ЯМР:
Соединение 2: 1H-ЯМР (400 МГц, ДМСО-d6, основной описанный изомер): 12,14-12,30 (2H, м), 7,70-7,85 (6H, м), 7,44-7,62 (4H, м), 7,24 (2H, д, J=8,0 Гц), 5,15-5,28 (2H, м), 4,26-4,38 (2H, м), 3,81-3,98 (4H, м), 3,56 (6H, с), 3,47-3,54 (2H, м), 3,20 (6H, с), 2,15-2,32 (4H, м), 2,15-2,32 (4H, м), 1,09 (6H, д, J=6,0 Гц).
Соединение 30: 1H-ЯМР (400 МГц, ДМСО-d6, основной описанный изомер): 12,13-12,77 25 (2H, м), 7,73-7,80 (6H, м), 7,47-7,60 (4H, м), 7,32 (2H, д, J=8,6 Гц), 5,17-5,24 (2H, м), 4,05-4,15 (2H, м), 3,80-3,91 (4H, м), 3,55 (6H, с), 2,18-2,31 (4H, м), 1,87-2,13 (6H, м), 0,80-0,92 (12H, м).
Соединение 40: 1H-ЯМР (400 МГц, ДМСО-d6, основной описанный изомер): 12,13-12,25 (2H, м), 7,42-7,87 (12H, м), 5,12-5,26 (2H, м), 3,97-4,07 (2H, м), 3,69-3,89 (4H, м), 3,55 (6H, с), 2,15-2,31 (4H, м), 1,89-2,14 (4H, м), 1,05-1,19 (2H, м) 0,31-0,50 (8H, м).
Соединения формулы I
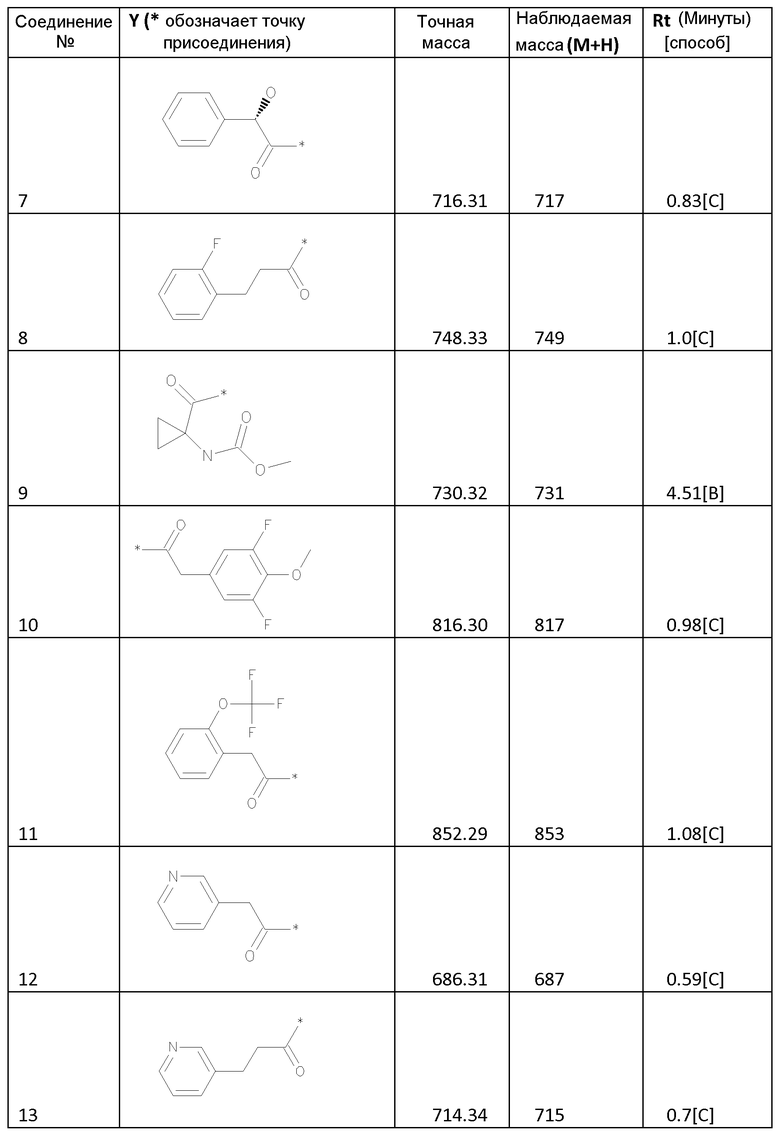
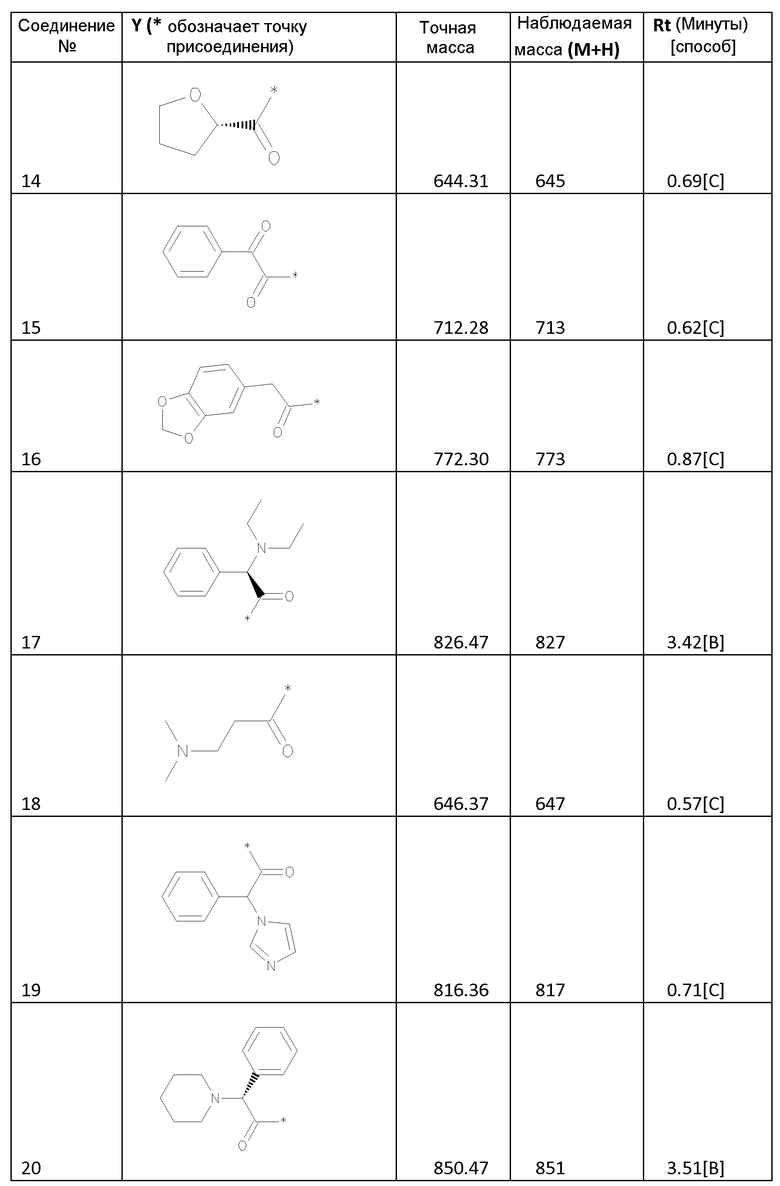
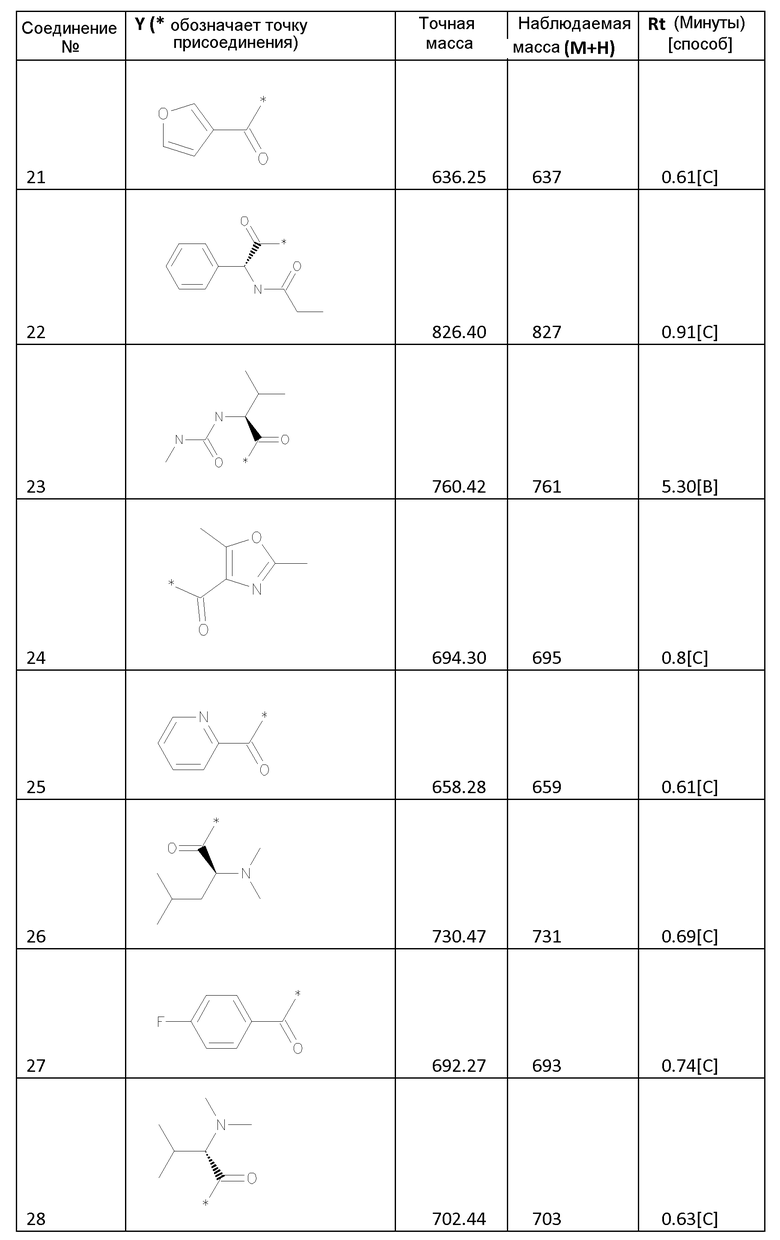
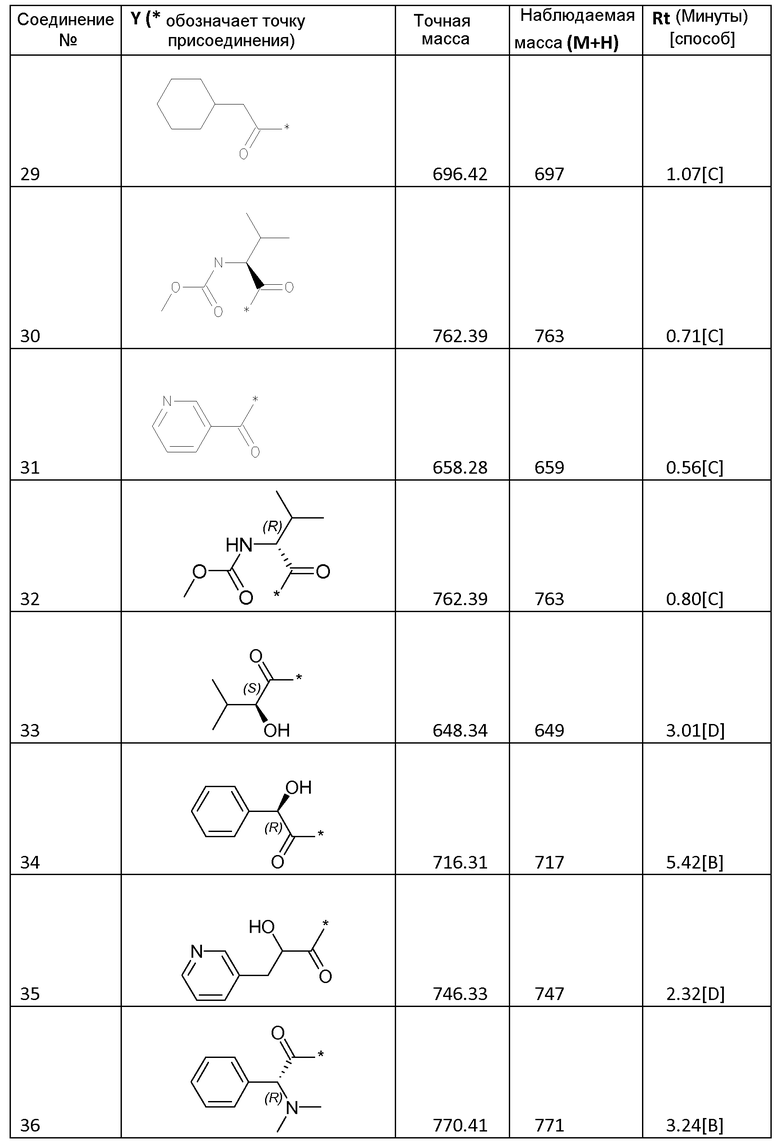
Биологические примеры - активность соединений формулы I против HCV
Анализ репликонов
Соединения формулы (I) исследовали на ингибирующую активность в репликоне HCV. Этот клеточный анализ основан на бицистронной экспрессирующей конструкции, как описано Lohmann et al. (1999) Science vol. 285 pp. 110-113 с модификациями, описанными Krieger et al. (2001) Journal of Virology 75: 4614-4624, в стратегии скрининга множества мишеней. В сущности, способ был следующим.
По существу, метод был следующим.
В анализе использовали стабильно трансфицированную клеточную линию Huh-7 luc/neo (в дальнейшем в настоящем документе обозначаемую как Huh-Luc). Эта клеточная линия содержит РНК, кодирующую бицистронную экспрессирующую конструкцию, содержащую участки NS3-NS5B HCV типа 1b дикого типа, транслируемые с внутреннего сайта связывания рибосомы (IRES) вируса энцефаломиокардита (EMCV), с предшествующим репортерным участком (FfL-люцифераза), и участком селективного маркера (neoR, неомицинфосфотрансфераза). Конструкция фланкировалась 5' и 3'-NTR (нетранслируемые области) из HCV 1b типа. Сохранение культуры клеток с репликоном в присутствии G418 (neoR) зависит от репликации РНК HCV. Для скрининга противовирусных соединений использовали стабильно трансфицированные репликоном клетки, которые экспрессируют РНК HCV, которая реплицируется автономно и на высоком уровне и которая кодирует, в частности, люциферазу.
Клетки с репликоном помещали в 384-луночные планшеты в присутствии тестируемых и контрольных соединений, которые добавляли в различных концентрациях. После инкубации в течение трех суток определяли репликацию HCV посредством анализа активности люциферазы (с использованием стандартных субстратов и реагентов для анализа люциферазы и устройства для визуализации микропланшетов Perkin Elmer ViewLuxTm ultraHTS). Клетки с репликоном в контрольных культурах обладали высокой экспрессией люциферазы в отсутствие какого-либо ингибитора. Мониторинг ингибирующей активности соединения в отношении активности люциферазы проводили на клетках Huh-Luc, получая кривую "доза-эффект" для каждого тестируемого соединения. Затем вычисляли значения EC50, которые соответствуют количеству соединения, необходимому для снижения уровня выявляемой активности люциферазы на 50%, или более конкретно, способности генетически связанного с ней репликона РНК HCV к репликации.
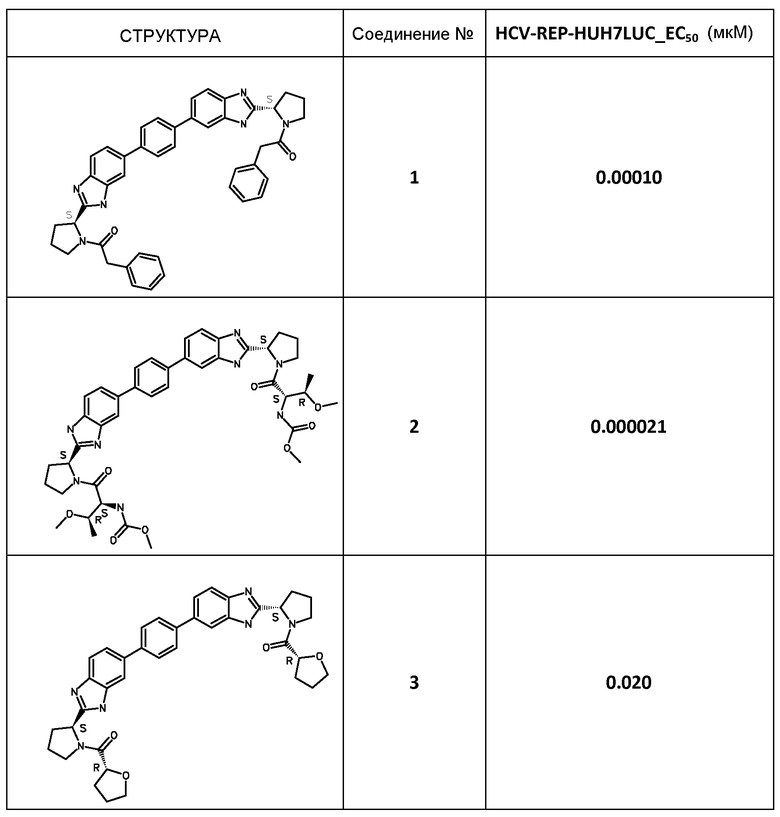
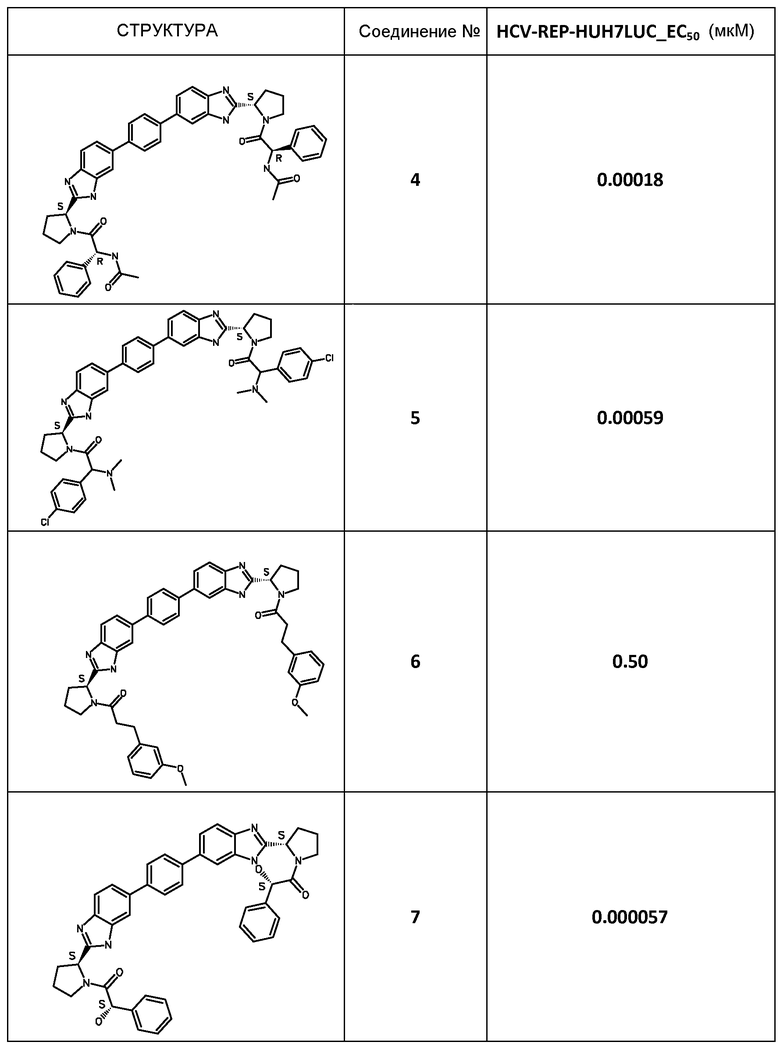
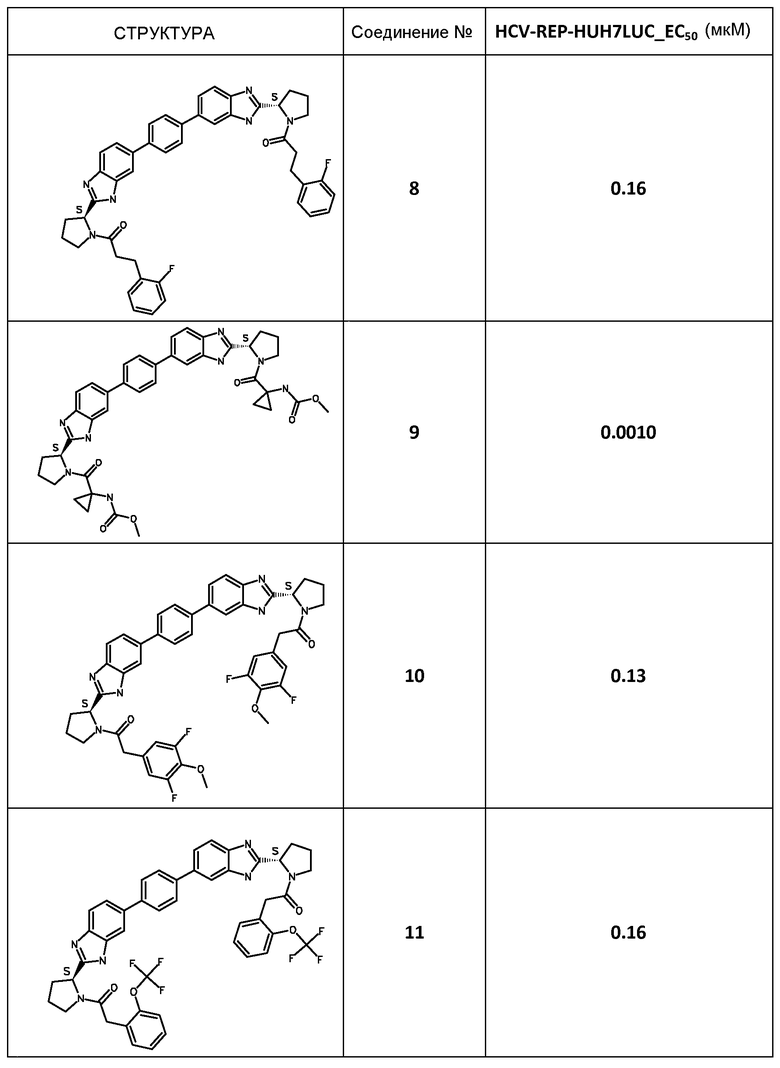
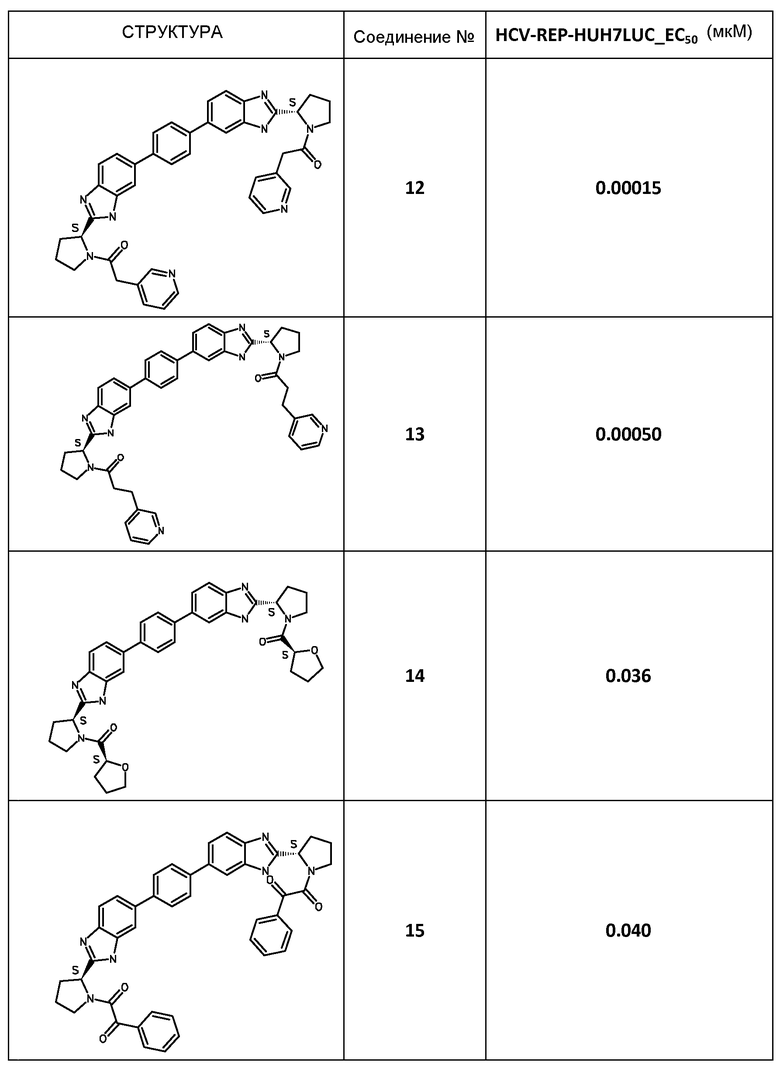
Результаты
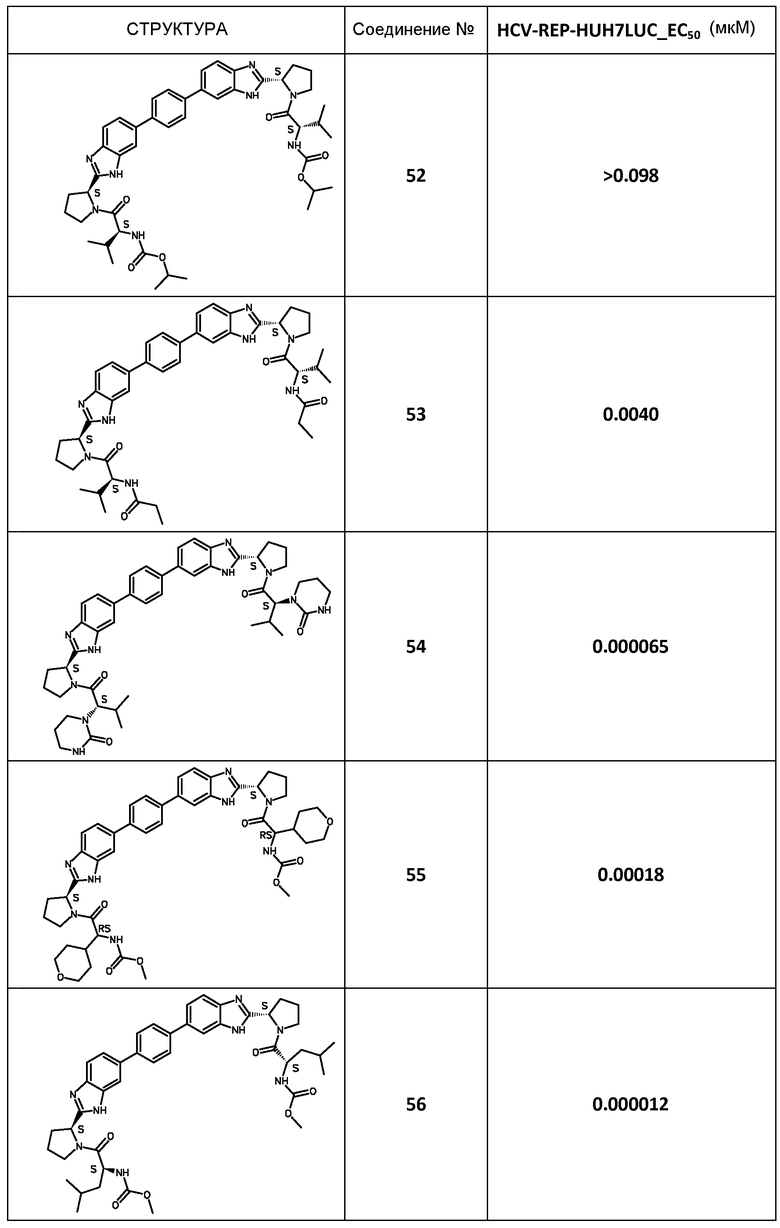
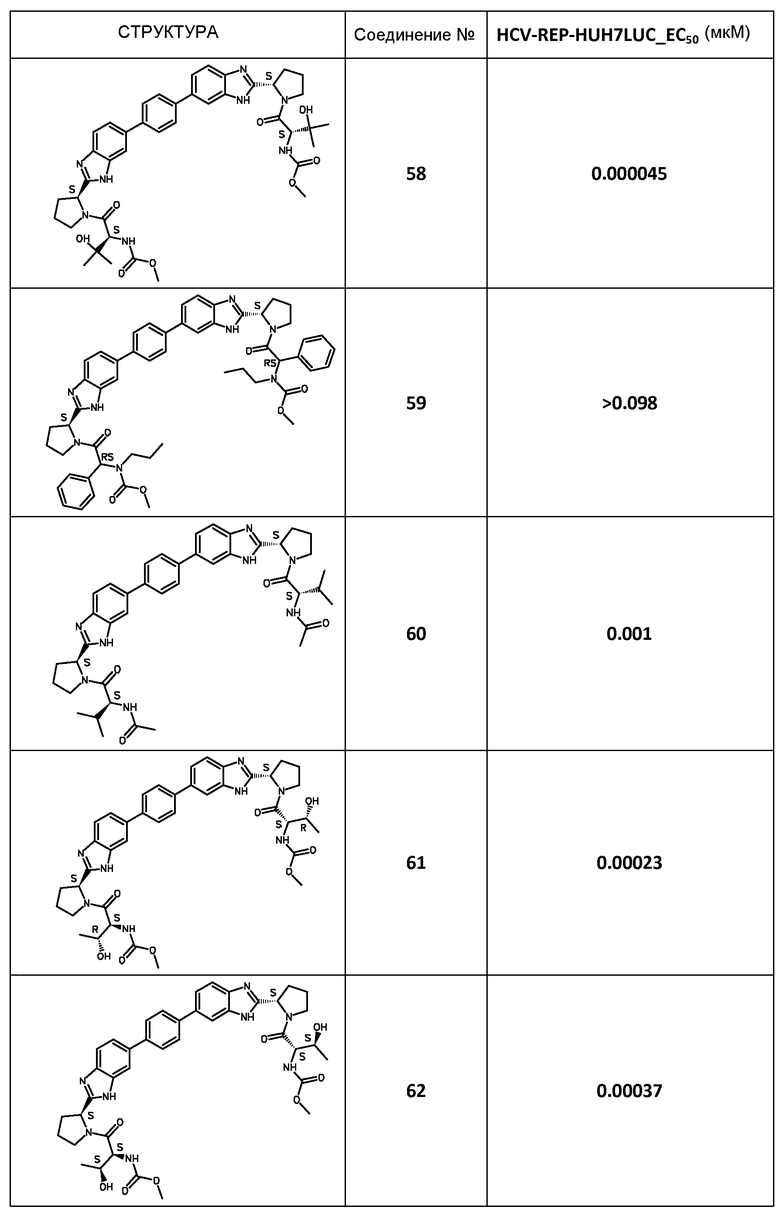
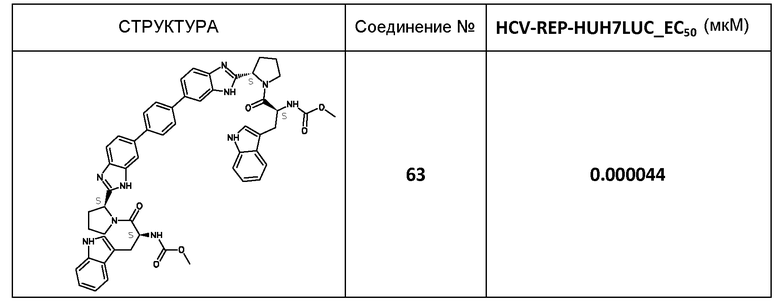
В таблице 2 представлены результаты анализа с репликоном, полученные для соединений примеров, приведенных выше.
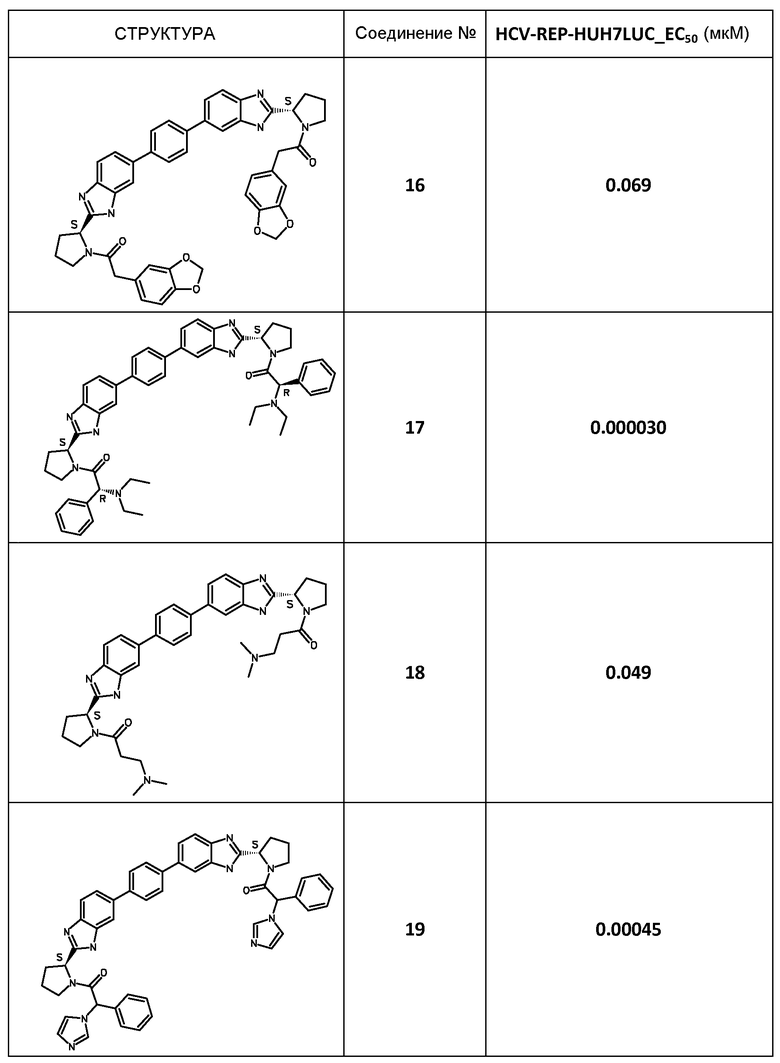
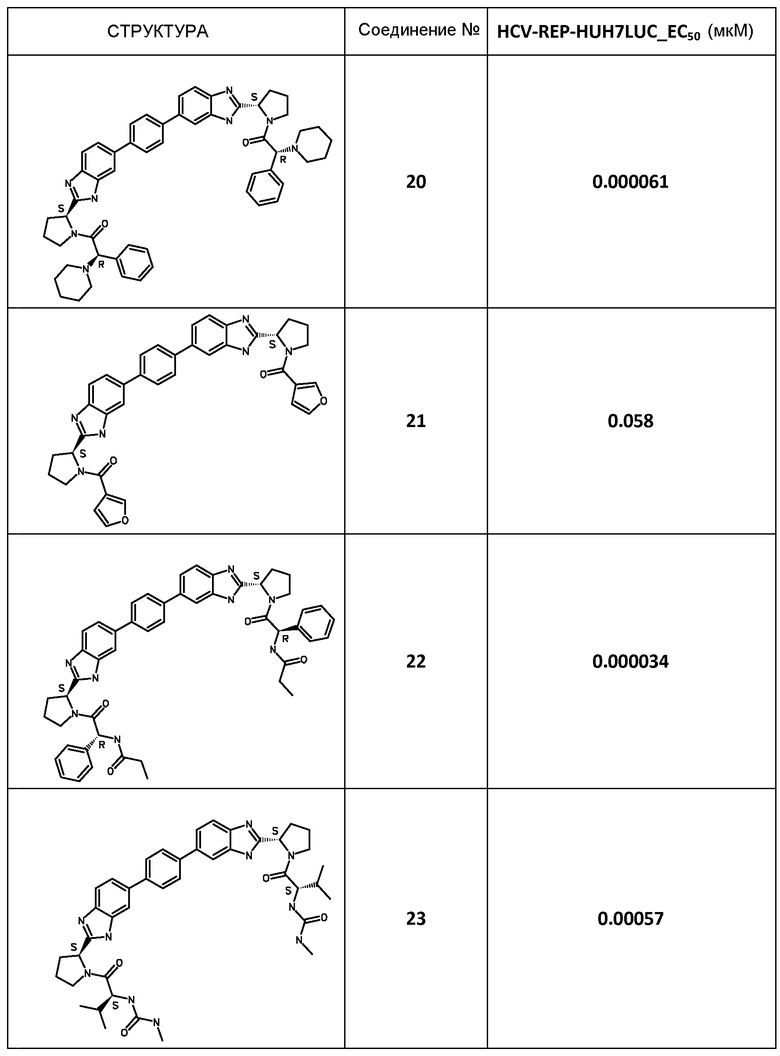
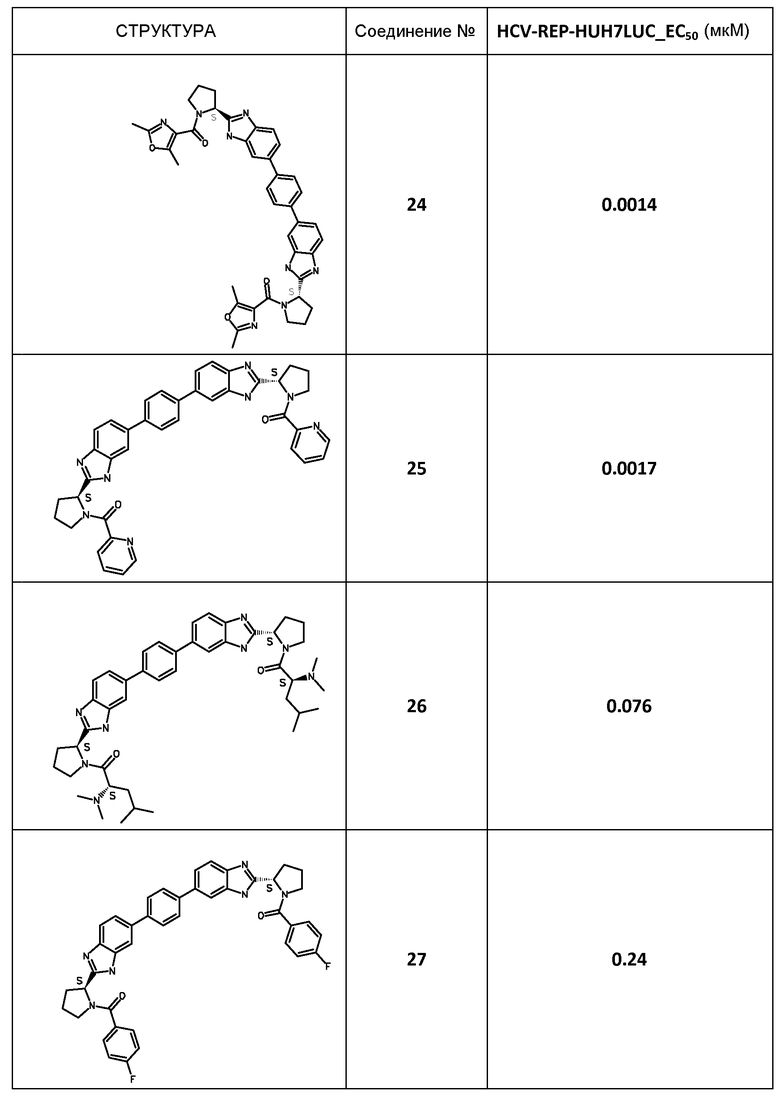
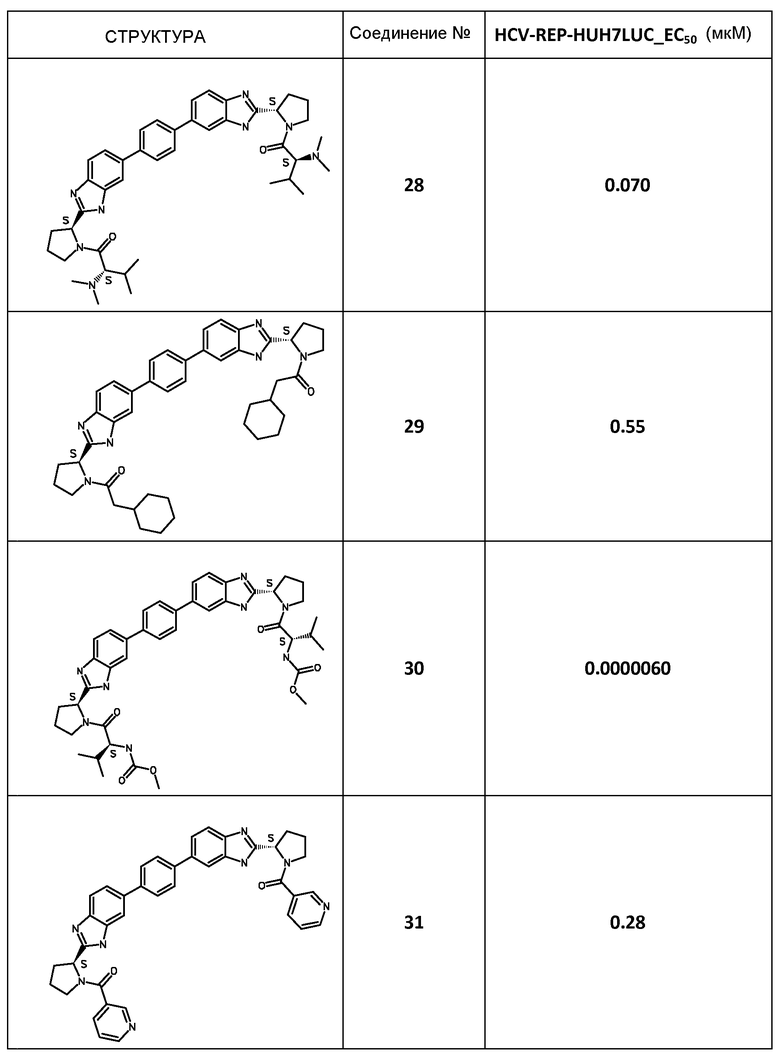
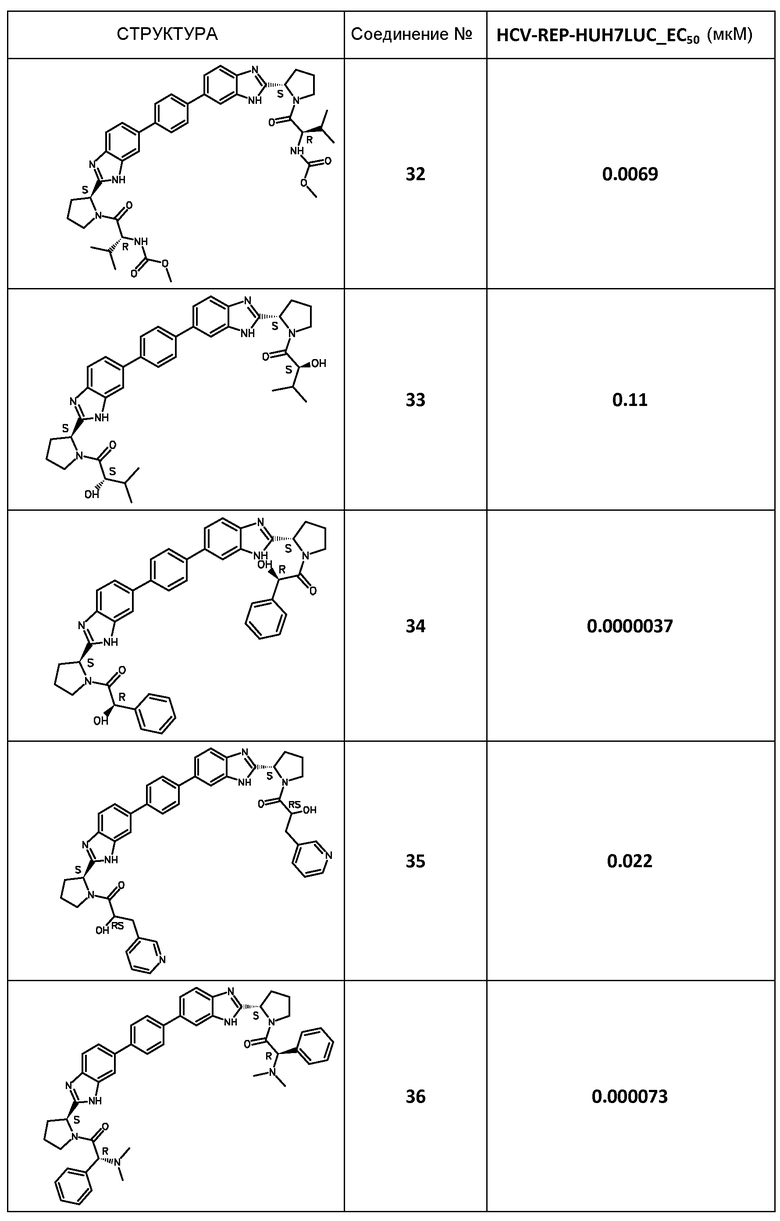
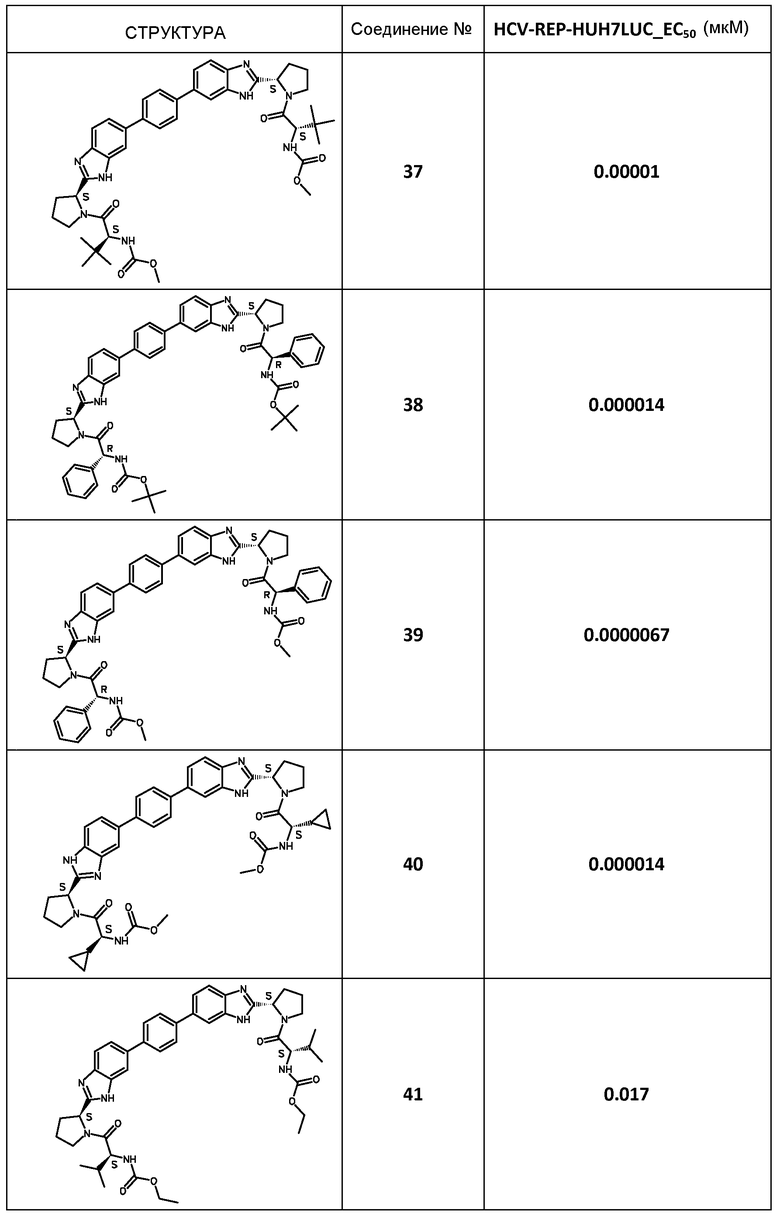
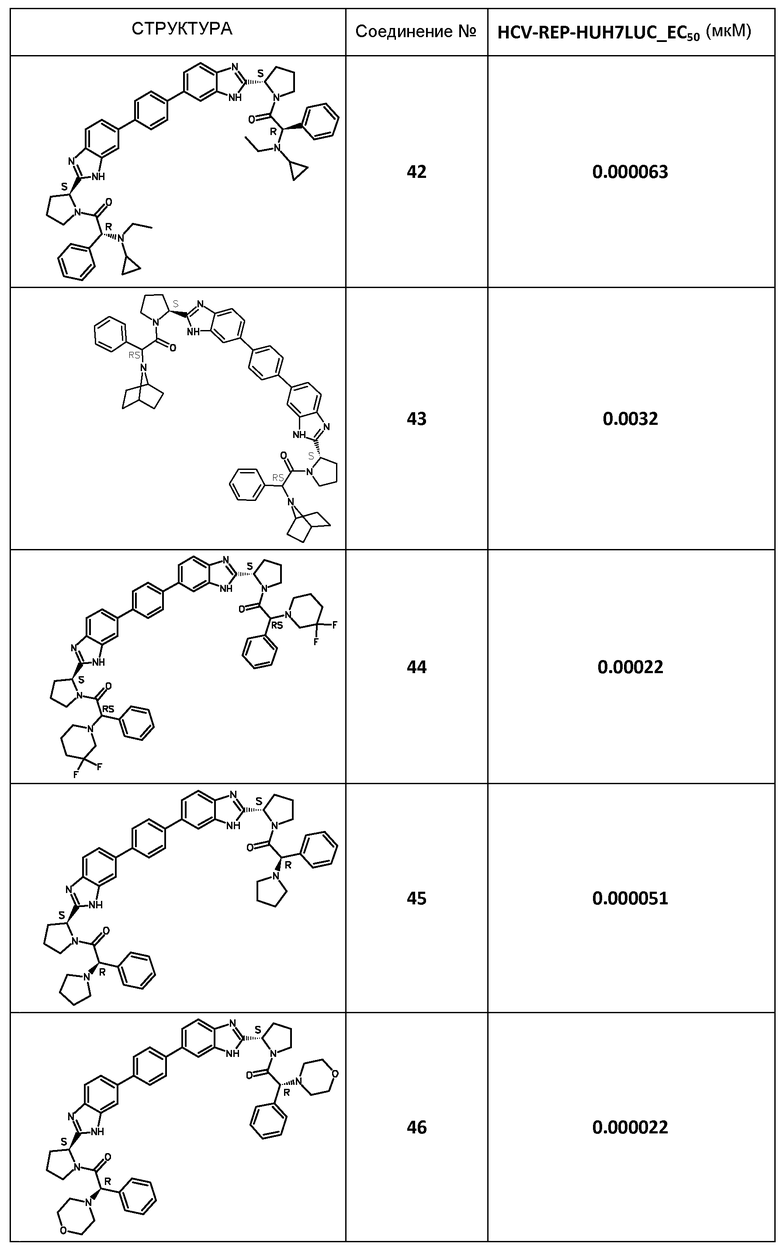
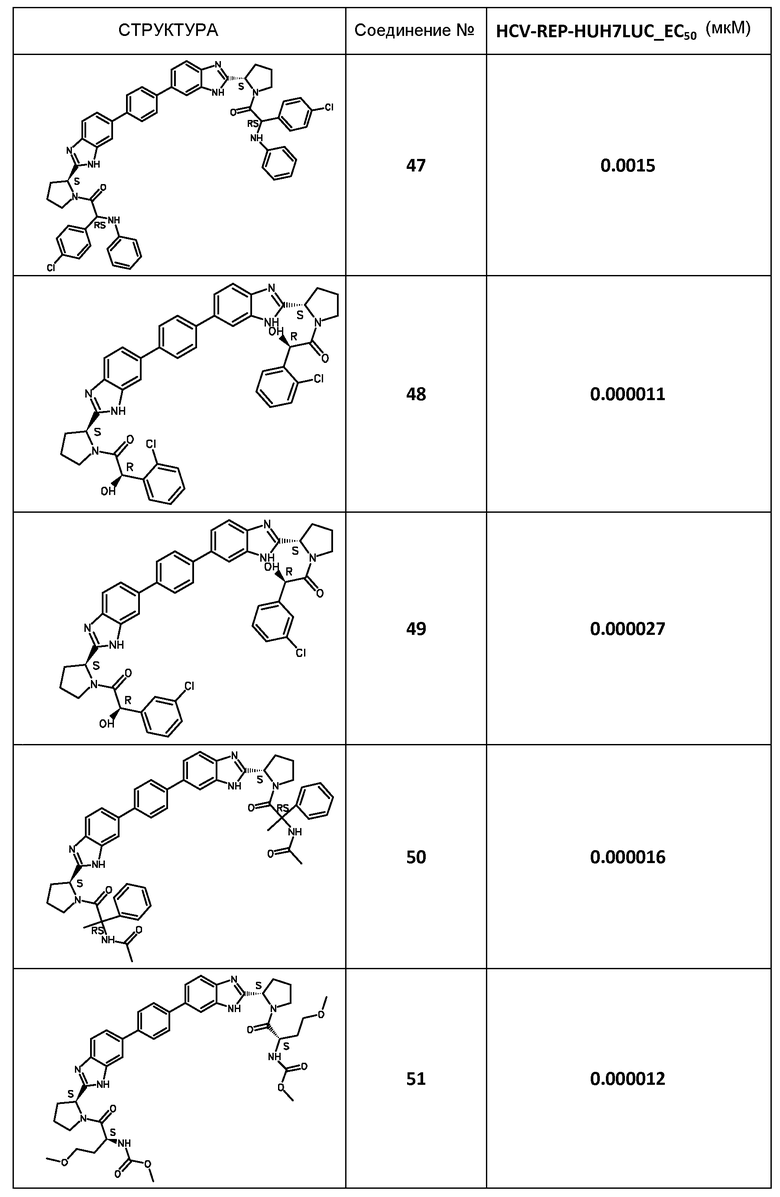
Исследования комбинаций ингибиторов
В предпочтительном варианте осуществления комбинация соединений, описанных в настоящем документе, с другим средством, которое изменяет репликацию вируса HCV, может действовать синергически или антагонистически. Взаимодействия соединений можно анализировать различными механистическими и эмпирическими способами.
Один подход анализа таких комбинаций осуществляют с помощью трехмерных графиков и вычисления синергических объемов, проводимого MacSynergyTM II на основе модели Bliss Independency model (Dr. Mark Pritchard, University of Alabama, Tuscaloosa, AL). По существу, соединения, описанные в настоящем документе, в сочетании с другим средством, которое изменяет репликацию вируса HCV, называют действующими синергично или имеющими синергичный эффект, когда величины, выраженные в нМ2% (объем синергии) составляют от 25 до 50 нМ2% (небольшая, но существенная величина синергии), между 50 и 100 нМ2% (умеренная синергия) или свыше 100 нМ2% (сильная синергия).
В определенных вариантах осуществления соединение 2 комбинируют с соединением, которое ингибирует репликацию вируса гепатита C. Примеры таких соединений включают ингибитор протеаз (TMC435350) или ингибитор полимеразы (ингибитор на основе нуклеозидов: ссылка 1; ненуклеозидный ингибитор: ссылка 3). Эксперимент было организован в "шахматном" порядке, когда одно лекарственное средство титровали горизонтально, а другое титровали вертикально, на клетках Huh-Luc, содержавших стабильно трансфицированный репликон HCV типа 1b. Каждую двухстороннюю комбинацию осуществляли в четырех экземплярах и анализировали с помощью программного обеспечения MacSynergyTM II для получения объемов процентной синергии/антагонизма (выраженных как нМ2%).
В MacSynergyTM II теоретические вычисления аддитивных взаимодействий получают из кривых "доза-эффект" для каждого отдельного соединения. Затем вычисленную аддитивную поверхность вычитают из экспериментальной поверхности с получением поверхности синергии. Только аддитивные взаимодействия могут приводить к горизонтальной плоскости на уровне 0%. Пик выше плоскости 0% указывает на синергию, а снижение ниже плоскости 0% относится к антагонизму. 95% доверительный интервал для экспериментальных поверхностей "доза-эффект" вычисляли для оценки статистической значимости синергии или антагонизма.
Комбинации тестировали в диапазоне концентраций, упомянутых в таблице 3. Объемы, полученные с помощью MacSynergyTM II при комбинировании соединения 2 с TMC435350, эталонным соединением 1 или эталонным соединением 3, являются небольшими, умеренными или высокосинергическими соответственно. Учитывая, что диапазон объемов синергии, полученный при 95% доверительном интервале для независимости в Bliss для комбинаций с TMC435350 и эталонным соединением 1, охватывает диапазоны объемов, определенные как синергические и независимые в Bliss, эти тестированные комбинации считаются обладающими действием от аддитивного до синергического. В случае комбинации с эталонным соединением 3 этот диапазон объемов синергии является синергическим (таблица 4). Во всех случаях не наблюдали значимого антагонизма.
Протестированный диапазон соединений
Объемы синергии/антагонизма при комбинировании, полученные с помощью MacSynergy
TM
II
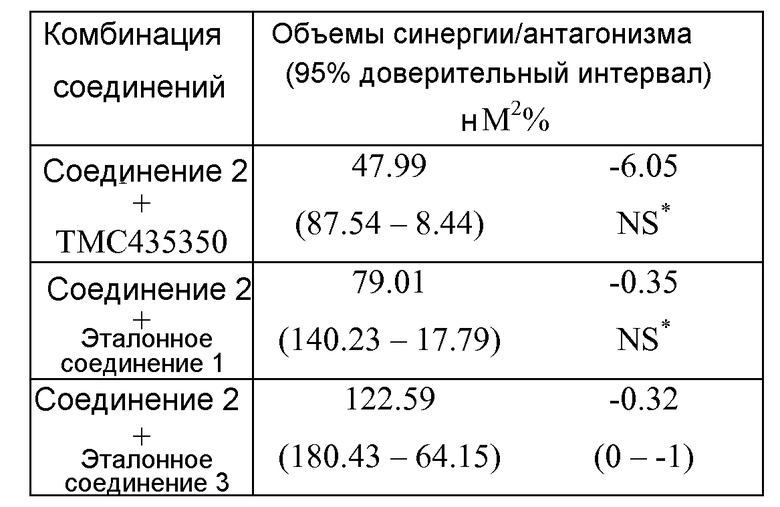
*NS="не значимо", как обозначалось MacSynergyTM II
| название | год | авторы | номер документа |
|---|---|---|---|
| ФЕНИЛЭТИНИЛЬНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ВИРУСА ГЕПАТИТА С | 2010 |
|
RU2538507C2 |
| ПРОИЗВОДНЫЕ ПИПЕРИДИНА/ПИПЕРАЗИНА | 2008 |
|
RU2478628C2 |
| НОВЫЕ ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ АЗАИНДОЛИНА В КАЧЕСТВЕ ИНГИБИТОРОВ NIK | 2018 |
|
RU2780577C2 |
| Имидазопиридиновые соединения в качестве ингибиторов PAD | 2018 |
|
RU2782743C2 |
| ДИАРИЛОВЫЕ ЭФИРЫ | 2010 |
|
RU2528231C2 |
| ГЕТЕРОБИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ HCV | 2012 |
|
RU2621734C1 |
| ПИРИМИДИНОВЫЕ СОЕДИНЕНИЯ ДЛЯ ИСПОЛЬЗОВАНИЯ В КАЧЕСТВЕ МОДУЛЯТОРОВ ДЕЛЬТА-ОПИОИДНЫХ РЕЦЕПТОРОВ | 2010 |
|
RU2568434C2 |
| СОЕДИНЕНИЯ ТИЕНОПИРИДИНОНА | 2019 |
|
RU2827549C2 |
| Необязательно конденсированные гетероциклил-замещенные производные пиримидина, пригодные для лечения воспалительных, метаболических, онкологических и аутоиммунных заболеваний | 2015 |
|
RU2719422C2 |
| аЭКЗО-АЗАСПИРО-ИНГИБИТОРЫ ВЗАИМОДЕЙСТВИЯ МЕНИН-MLL | 2018 |
|
RU2795096C2 |
Изобретение относится к области органической химии, а именно к производным бис-бензимидазола формулы I и к его возможным стереоизомерам, фармацевтически приемлемым солям и сольватам, где R и R' независимо выбраны из -CR1R2R3, фенила, замещенного 1 заместителем, выбранным из галогена; и тетрагидрофуранила, где R1 выбран из С1-4алкила, необязательно замещенного метокси, гидроксилом или диметиламино; С3-6циклоалкила; фенила, необязательно замещенного 1, 2 или 3 заместителями, независимо выбранными из галогена, С1-4алкокси, трифторметокси, или 2 заместителя на соседних атомах кольца образуют 1,3-диоксолановую группу; бензила, замещенного галогеном или метокси; пиридинила; индолила; пиридинилметила и индолилметила; R2 выбран из водорода, гидроксила, ди-С1-4алкиламино, (С3-6циклоалкил) (С1-4алкил)амино, С1-4алкилкарбониламино, фениламино, С1-4алкилоксикарбониламино, (С1-4алкилоксикарбонил)(С1-4алкил)амино, С1-4алкиламинокарбониламино, тетрагидро-2-оксо-1(2Н)-пиримидинила, пирролидин-1-ила, пиперидин-1-ила, 3,3-дифторпиперидин-1-ила, морфолин-1-ила, 7-азабицикло[2.2.1]гепт-7-ила и имидазол-1-ила; и R3 представляет собой водород или С1-4алкил или CR2R3 вместе образуют карбонил; или CR1R3 образует циклопропильную группу. Также изобретение относится к фармацевтической композиции на основе соединения формулы I. Технический результат: получены производные бис-бензимидазола, обладающие ингибирующей активностью в отношении вируса гепатита С. 2 н. и 7 з.п. ф-лы, 4 табл., 3 пр.
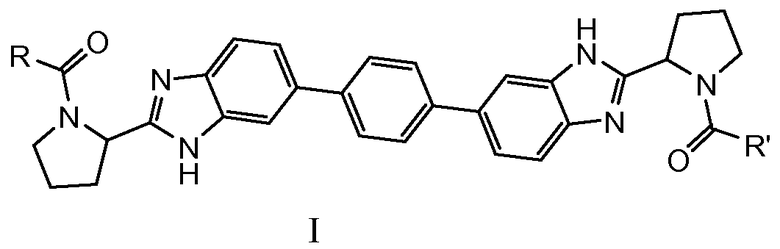
1. Соединение формулы I:
включая любые его возможные стереоизомеры, где
R и R' независимо выбраны из -CR1R2R3, фенила, замещенного 1 заместителем, выбранным из галогена; и тетрагидрофуранила, где
R1 выбран из С1-4алкила, необязательно замещенного метокси, гидроксилом или диметиламино; С3-6циклоалкила; фенила, необязательно замещенного 1, 2 или 3 заместителями, независимо выбранными из галогена, С1-4алкокси, трифторметокси, или 2 заместителя на соседних атомах кольца образуют 1,3-диоксолановую группу; бензила, замещенного галогеном или метокси; пиридинила; индолила; пиридинилметила и индолилметила;
R2 выбран из водорода, гидроксила, ди-С1-4алкиламино, (С3-6циклоалкил) (С1-4алкил)амино, С1-4алкилкарбониламино, фениламино, С1-4алкилоксикарбониламино, (С1-4алкилоксикарбонил)(С1-4алкил)амино, С1-4алкиламинокарбониламино, тетрагидро-2-оксо-1(2Н)-пиримидинила, пирролидин-1-ила, пиперидин-1-ила, 3,3-дифторпиперидин-1-ила, морфолин-1-ила, 7-азабицикло[2.2.1]гепт-7-ила и имидазол-1-ила; и
R3 представляет собой водород или С1-4алкил, или
CR2R3 вместе образуют карбонил; или
CR1R3 образует циклопропильную группу;
и его фармацевтически приемлемые соли и сольваты;
при условии, что (а), когда R и R' идентичны и представляют собой -CR1R2R3, где
(а-1) R2 представляет собой С1-4алкилоксикарбониламино и R3 представляет собой водород, тогда R1 отличен от незамещенного С1-4алкила или этила, замещенного гидроксилом или метокси; или где
(а-2) R2 представляет собой метилоксикарбониламино и R3 представляет собой водород, тогда R1 отличен от незамещенного фенила; и,
(b) когда R и R' отличаются и каждый из них независимо представляет собой -CR1R2R3, где R1 представляет собой фенил или 2-пропил, R2 представляет собой диметиламино и R3 представляет собой водород в одной группе -CR1R2R3, тогда в другой группе -CR1R2R3 R1 не может принимать значение 2-пропила, и R2 не может принимать значение метилоксикарбониламино, и R3 не может принимать значение водорода.
2. Соединение формулы I по п.1, где
R и R' независимо выбраны из -CR1R2R3, где
R1 выбран из фенила, необязательно замещенного 1, 2 или 3 заместителями, независимо выбранными из галогена, С1-4алкокси, трифторметокси, или 2 заместителя на соседних атомах кольцах образуют 1,3-диоксолановую группу;
R2 выбран из гидроксила, ди-С2-4алкиламино, (С3-6циклоалкил)(С1-4алкил)амино, С1-4алкилкарбониламино, (С1-4алкилоксикарбонил)(С1-4алкил)амино, С1-4алкиламинокарбониламино, тетрагидро-2-оксо-1(2Н)-пиримидинила, пирролидин-1-ила, пиперидин-1-ила, 3,3-дифторпиперидин-1-ила, морфолин-1-ила, 7-азабицикло[2.2.1]гепт-7-ила и имидазол-1-ила; и
R3 представляет собой водород или С1-4алкил или CR2R3 вместе образуют карбонил; или CR1R3 образует циклопропильную группу;
и его фармацевтически приемлемые соли и сольваты.
3. Соединение формулы I по п.1, где
R1 выбран из пиридинила, индолила, пиридинилметила и индолилметила;
R2 выбран из водорода, ди-С1-4алкиламино, (С3-6циклоалкил)(C1-4алкил)амино, С1-4алкилкарбониламино, С1-4алкилоксикарбониламино, (С1-4алкилоксикарбонил)(С1-4алкил)амино, С1-4алкиламинокарбониламино, тетрагидро-2-оксо-1(2Н)-пиримидинила, пирролидин-1-ила, пиперидин-1-ила, 3,3-дифторпиперидин-1-ила, морфолин-1-ила, 7-азабицикло[2.2.1]гепт-7-ила и имидазол-1-ила; и
R3 представляет собой водород;
и его фармацевтически приемлемые соли и сольваты.
4. Соединение формулы I по п.1, где
R1 представляет собой С1-4алкил;
R2 выбран из С1-4алкиламинокарбониламино и тетрагидро-2-оксо-1(2Н)-пиримидинила; и
R3 представляет собой водород или С1-4алкил;
и его фармацевтически приемлемые соли и сольваты.
5. Соединение формулы I по п.1, где
R1 представляет собой С3-6циклоалкил;
R2 представляет собой водород
и R3 представляет собой водород;
и его фармацевтически приемлемые соли и сольваты.
6. Соединение по п.1, где R и R' являются одинаковыми.
7. Соединение по п.1, где соединение имеет формулу Ia
8. Фармацевтическая композиция, обладающая ингибирующей активностью в отношении вируса гепатита С, содержащая соединение по любому из пп.1-7 и фармацевтически приемлемый носитель.
9. Соединение по любому из пп.1-7 или фармацевтическая композиция по п.8 для применения для предупреждения или лечения инфекции HCV у млекопитающего.
| Станок для изготовления деревянных ниточных катушек из цилиндрических, снабженных осевым отверстием, заготовок | 1923 |
|
SU2008A1 |
| Способ обработки строительных растворов или бетона с целью сообщения им водонепроницаемости | 1926 |
|
SU4939A1 |
| RU 2006106272 Ф, 27.08.2006 | |||

