ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННЫЕ ЗАЯВКИ
По настоящей заявке испрашивается приоритет на основании предварительной японской патентной заявки № 37475/2010, поданной 23 февраля 2010 года, полное содержание которой вводится в настоящее изобретение с помощью ссылки.
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ НАСТОЯЩЕЕ ИЗОБРЕТЕНИЕ
Изобретение относится к стабильной кристаллической форме 2-этил-3,7-диметил-6-(4-(трифторметокси)фенокси)хинолин-4-илметилкарбоната, обладающей высокой инсектицидной активностью, и способу ее получения. Настоящее изобретение также относится к агрохимической композиции, содержащей данную стабильную кристаллическую форму.
УРОВЕНЬ ТЕХНИКИ
Когда определенное соединение находится в двух или более кристаллических состояниях, данные различные кристаллические состояния называют кристаллическим полиморфизмом. Общеизвестно, что, когда существует кристаллический полиморфизм, может происходить изменение кристаллической формы и, кроме того, кристаллические полиморфы (кристаллические формы) при кристаллическом полиморфизме являются отличными друг от друга по стабильности и физическим свойствам.
Изменение кристаллической формы при кристаллическом полиморфизме представляет собой явление, которое часто обнаруживается, например, при сушке, измельчении и хранении в области химической промышленности. Изменение кристаллической формы иногда приводит к серьезным проблемам, таким как затвердевание, увеличение кристаллов, превращение в порошок и выделение тепла. В частности, в области агрохимикатов, изменение кристаллической формы после формулирования иногда оказывает значительное воздействие на свойства и качества агрохимикатов, например эффективность агрохимикатов и стабильность при хранении составов. Разрабатываются кристаллические формы с высокой стабильностью для того, чтобы избежать данных проблем.
Например, патентный документ 1 описывает кристаллический полиморфизм метазаклора, который является активным ингредиентом для уничтожения сорняков. В частности, композиция в виде водной суспензии, содержащая кристаллический полиморф (1A), полученный кристаллизацией из циклогексана или толуола, вызывает агломерацию, и, впоследствии, композиция становится гетерогенной и не поддающейся распылению. С другой стороны, композиция в виде водной суспензии, содержащая кристаллический полиморф (1B), полученный кристаллизацией в присутствии органического растворителя, который является смешиваемым с водой, полярным и инертным, не вызывает агломерации и может сохранять хорошие свойства.
В патентном документе 2, проблемы, такие как затвердевание и выделение тепла, вызванное твердофазным переходом полиморфного кристалла квизалофоп-P-этила, избегаются получением стабилизированных кристаллов. Далее, в патентных документах 3 и 4, эффективность тифлузамида, который является фунгицидным активным ингредиентом, увеличивается за счет улучшения элюции тифлузамида в воде изменением кристаллической формы тифлузамида.
2-Этил-3,7-диметил-6-(4-(трифторметокси)фенокси)хинолин-4-илметилкарбонат (иногда называемый в настоящем изобретении просто "соединением I") представляет собой соединение, обладающее высокой инсектицидной активностью (патентные документы 5 и 6). Соединение I проявляет высокую инсектицидную активность, в частности, против чешуекрылых, полужесткокрылых, жесткокрылых, клещей, перепончатокрылых, прямокрылых, двукрылых и отряда бахромчатокрылых, и ожидают, что оно будет пригодным в качестве активного ингредиента для сельскохозяйственных и плодовоовощных инсектицидов. До настоящего времени не сообщалось о каких-либо кристаллических формах соединения I, обладающих высокой физико-химической стабильностью.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Патентные документы
Патентный документ 1: Опубликованная японская патентная заявка № 66671/1991
Патентный документ 2: Опубликованная японская патентная заявка № 114707/2001
Патентный документ 3: Опубликованная японская патентная заявка № 227538/1997
Патентный документ 4: Опубликованная японская патентная заявка № 1476/1998
Патентный документ 5: WO 2006/013896
Патентный документ 6: WO 2010/007964
СУЩНОСТЬ НАСТОЯЩЕГО ИЗОБРЕТЕНИЯ
Проблемы, которые будет решать настоящее изобретение
Когда агрохимический состав, содержащий соединение I, получают, используя соединение I, обладающее высокой инсектицидной активностью, в качестве активного ингредиента агрохимиката, необходимо располагать исходной субстанцией соединения I, которое может обеспечить получение составов, обладающих высокой стабильностью при хранении. В частности, когда соединение I формулируют в виде дозированной формы, которая содержит кристаллы активного ингредиента для агрохимикатов, например порошков, гранул и смачивающихся порошков, возможно снижение качества агрохимических составов за счет изменения кристаллической формы в процессе хранения составов.
Соответственно, задача настоящего изобретения состоит в представлении кристаллической формы соединения I, которая не подвергается изменениям кристаллических свойств, вызванных изменением кристаллической формы в условиях хранения, способа получения стабильной кристаллической формы соединения I и агрохимической композиции, обладающей высокой стабильностью при хранении.
Способы решения проблем
Авторы настоящего изобретения обнаружили, что кристаллические соединения I, обладающие стабильными физико-химическими свойствами (называемые иногда в настоящем изобретение просто "кристаллической формой В"), получают растворением выделенной субстанции соединения I в растворителе и осаждением кристаллов соединения I при температуре 40°C или выше из раствора. Авторы настоящего изобретения также обнаружили, что агрохимические составы, содержащие кристаллическую форму В, могут подвергаться очень незначительному ухудшению качества при хранении, т.е. затвердеванию и увеличению кристаллов соединения I и снижению эффективности из-за затвердевания и увеличения кристаллов. Указанные разработки были положены в основу настоящего изобретения.
Настоящее изобретение будет изложено ниже.
(1) Кристаллическая форма 2-этил-3,7-диметил-6-(4-(трифторметокси)фенокси)хинолин-4-илметилкарбоната, которая характеризуется дифракционными пиками по меньшей мере при следующих дифракционных углах (2θ), как определено порошковой рентгеновской дифрактометрией:
(2) Кристаллическая форма 2-этил-3,7-диметил-6-(4-(трифторметокси)-фенокси)хинолин-4-илметилкарбоната по (1), которая дополнительно характеризуется дифракционными пиками при следующих углах дифракции (2θ), как определено порошковой рентгеновской дифрактометрией:
(3) Способ получения кристаллической формы по (1) или (2), где способ включает осаждение кристаллов из алканолового раствора 2-этил-3,7-диметил-6-(4-(трифторметокси)фенокси)хинолин-4-илметилкарбоната при температуре 40°C или выше.
(4) Способ получения кристаллической формы по (1) или (2), где способ включает добавление воды к алканоловому раствору 2-этил-3,7-диметил-6-(4-(трифторметокси)фенокси)хинолин-4-илметилкарбоната и осаждение кристаллов из него при температуре 40°C или выше.
(5) Агрохимическая композиция, содержащая кристаллическую форму по (1) или (2) в качестве активного ингредиента.
(6) Агрохимическая композиция по (5), дополнительно содержащая поверхностно-активное вещество и воду.
(7) Агрохимическая композиция по (5), дополнительно содержащая поверхностно-активное вещество и твердый носитель.
Кристаллическая форма согласно настоящему изобретению обладает высокой физико-химической стабильностью и позволяет избежать таких явлений, как увеличение, затвердение и агломерация соединения I, в условиях хранения в кристаллическом состоянии. Соответственно, настоящее изобретение может выгодно обеспечивать агрохимическими составами соединения I, которые позволяют избежать явления изменения кристаллической формы активного ингредиента и обладают высокой стабильностью при хранении и инсектицидной активностью.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
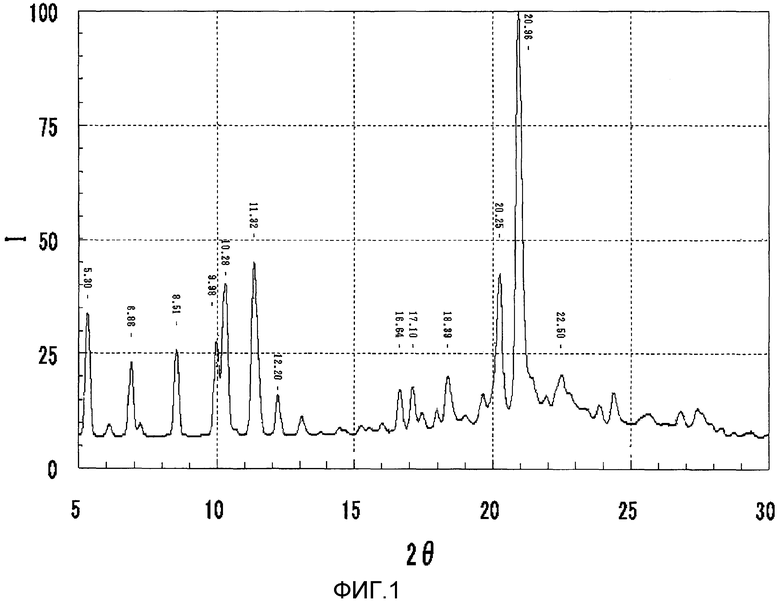
На фиг.1 представлена порошковая рентгеновская дифрактограмма кристаллической формы В соединения I.
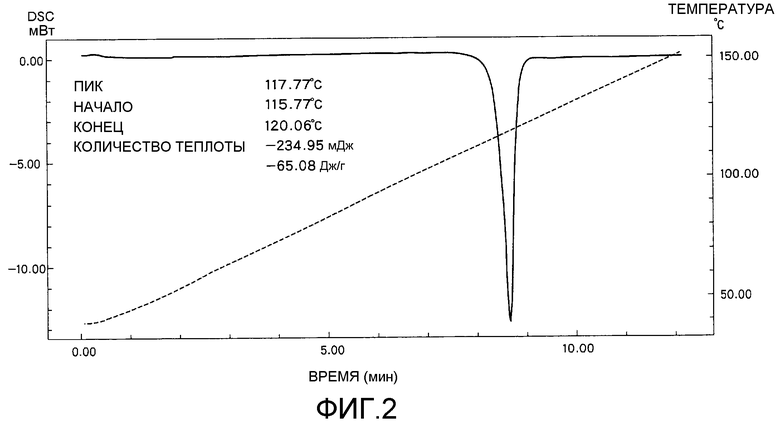
На фиг.2 представлена аналитическая диаграмма дифференциальной сканирующей калориметрии (DSC) кристаллической формы В соединения I.
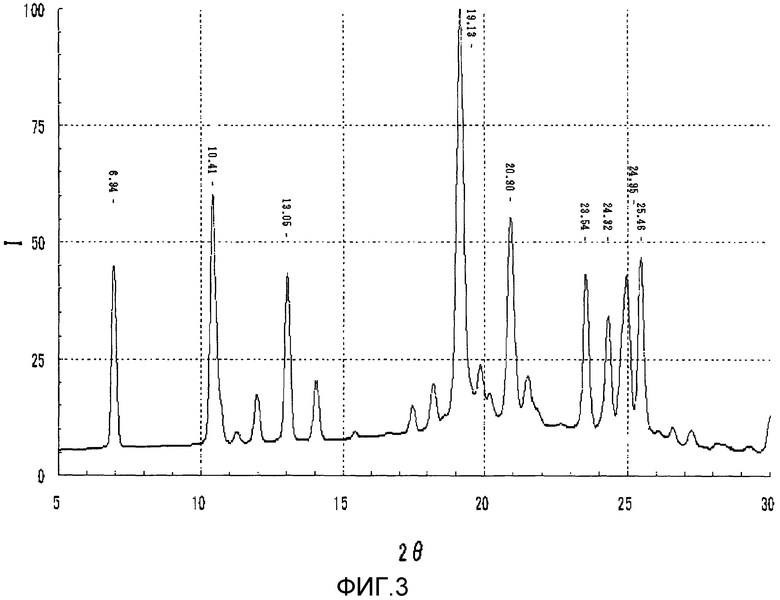
На фиг.3 представлена порошковая рентгеновская дифрактограмма кристаллической формы А соединения I.
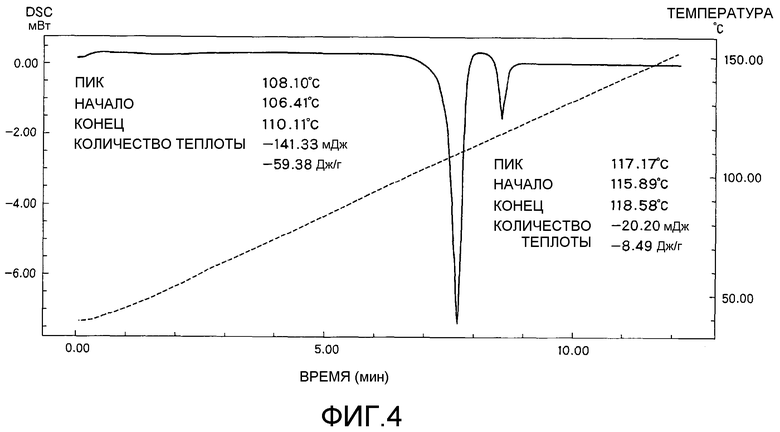
На фиг.4 представлена аналитическая диаграмма дифференциальной сканирующей калориметрии (DSC) кристаллической формы А соединения I.
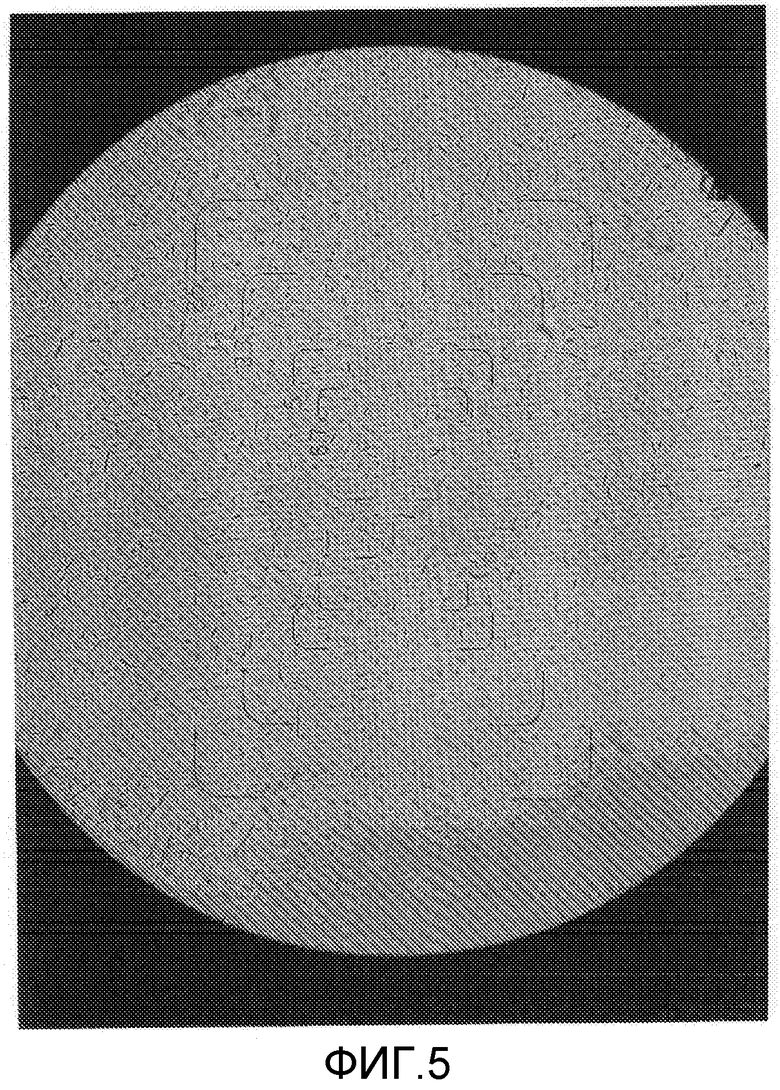
На фиг.5 представлена фотография микроскопического изображения (1500-кратное увеличение) кристаллического образца примера 2 (кристаллическая форма В) перед хранением в тестовом примере 1.
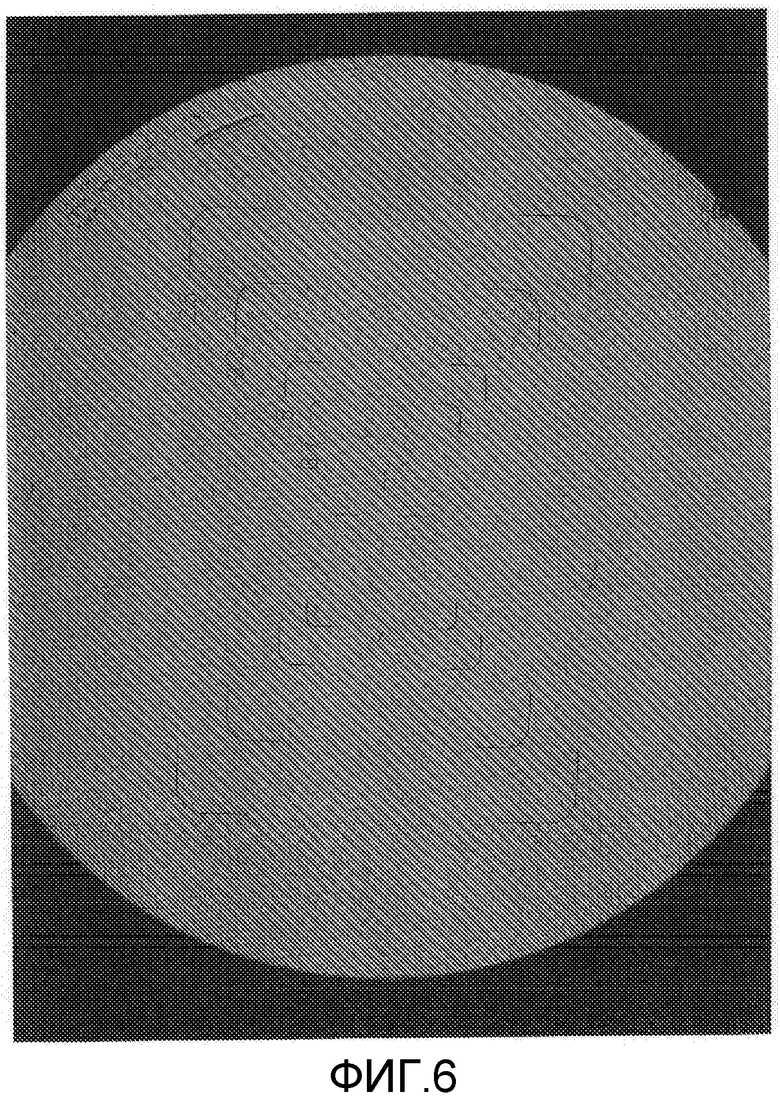
На фиг.6 представлена фотография микроскопического изображения (1500-кратное увеличение) кристаллического образца сравнительного примера 2 (кристаллическая форма А) перед хранением в тестовом примере 1.
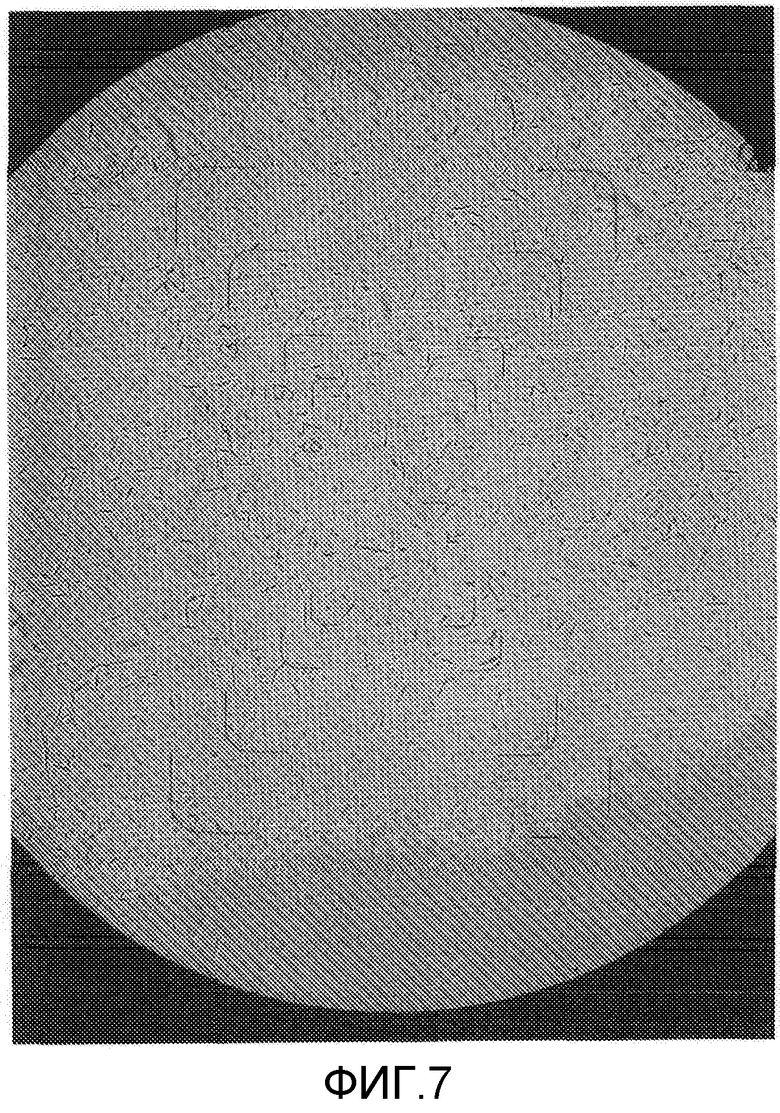
На фиг.7 представлена фотография микроскопического изображения (1500-кратное увеличение) кристаллического образца примера 2 (кристаллическая форма В) после хранения в тестовом примере 1.
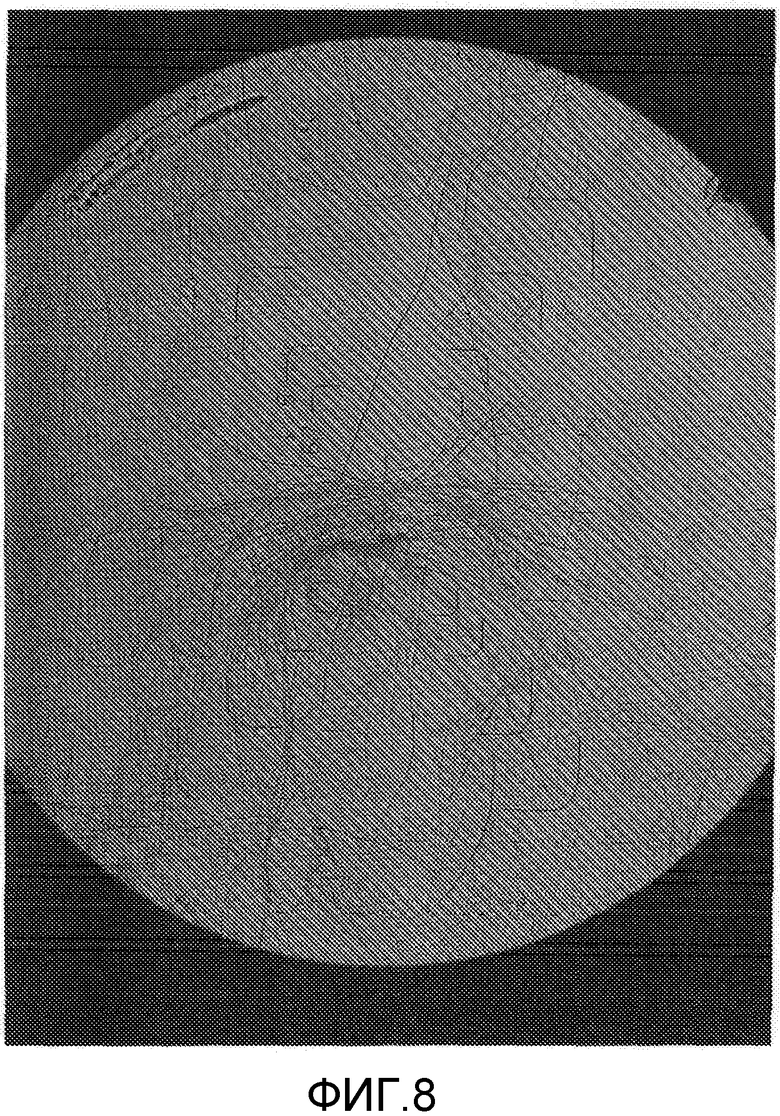
На фиг.8 представлена фотография микроскопического изображения (1500-кратное увеличение) кристаллического образца сравнительного примера 1 (кристаллическая форма A) после хранением в тестовом примере 1.
ПОДРОБНОЕ ОПИСАНИЕ НАСТОЯЩЕГО ИЗОБРЕТЕНИЯ
Кристаллы (кристаллическая форма В соединения I) согласно настоящему изобретению характеризуются спектром дифракционных пиков, показанных на фиг.1, как определено порошковой рентгеновской дифрактометрией. Т.е. кристаллическая форма В соединения I имеет пики при 2θ=5,3°, 6,9°, 8,5°, 10,0°, 10,3°, 11,3°, 20,3° и 21,0° с относительной интенсивностью, не меньшей чем 15%, как определено допущением, что интенсивность пика при 2θ=21,0° равна 100. Кристаллическая форма В соединения I дополнительно имеет пики при 2θ=5,3°, 6,9°, 8,5°, 10,0°, 10,3°, 11,3°, 12,2°, 16,6°, 17,1°, 18,4°, 20,3°, 21,0° и 22,5° с относительной интенсивностью, не меньшей чем 10%, как определено допущением, что интенсивность пика при 2θ=21,0° равна 100. Дифракционный пик, полученный порошковой рентгеновской дифрактометрией, включает интервал погрешности ±0,2°, связанный с измеряющим устройством, окружением при анализе и другими причинами.
Далее, кристаллическая форма В соединения I характеризуется наличием аналитического графика, показанного на фиг.2, как определено дифференциальной сканирующей калориметрией (DSC). Т.е. кристаллическая форма В соединения I характеризуется наличием эндотермического пика в области 117,8°C и не обладает другими эндотермическими пиками.
С другой стороны, WO 2006/013896 описывает соединение I, но не описывает кристалличность выделенного продукта, и таким образом, свойства кристаллов до настоящего времени были не выяснены. Выделенный продукт соединения I, полученный способом, описанным в документе предшествующего уровня техники, исследовали на кристалличность. Как результат, была получена дифрактограмма, показанная на фиг.3, порошковой рентгеновской дифрактометрией. Выделенные кристаллы соединения I, полученные согласно WO 2006/013896 (называемого в настоящем изобретении просто "кристаллическая форма А"), при анализе порошковой рентгеновской дифрактометрией имеют пики при 2θ=6,9°, 10,4°, 19,1°, 23,5°, 24,3°, 25,0° и 25,5° с относительной интенсивностью, не меньшей чем 15%, как определено допущением, что интенсивность пика при 2θ=19,1° равна 100. Далее, кристаллическая форма А соединения I имела пики 2θ=6,9°, 10,4°, 13,1°, 19,1°, 20,9°, 23,5°, 24,3°, 25,0° и 25,5° с относительной интенсивностью, не меньшей чем 10%, как определено допущением, что интенсивность пика при 2θ=19,1° равна 100. Дифракционный пик, полученный порошковой рентгеновской дифрактометрией, включает интервал ошибок приблизительно ±0,2°, связанный с измерительным устройством, окружением при анализе и другими причинами.
Кристаллическая форма А соединения I характеризуется наличием аналитической диаграммы, показанной на фиг.4, как определено дифференциальной сканирующей калориметрией (DSC). Т.е. кристаллическая форма А соединения I имеет эндотермический пик в области 108,1°C и дополнительный в области 117,2°C.
Кристаллическая форма В соединения I согласно настоящему изобретению и кристаллическая форма А, описанная в WO 2006/013896, имеют различные дифрактограммы, как определено порошковой рентгеновской дифрактометрией, и диаграммы эндотермических пиков, как определено дифференциальной сканирующей калориметрией (DSC).
Результаты измерения порошковой рентгеновской дифрактометрией для кристаллической формы В и кристаллической формы А соединения I являются результатами, полученными анализом в следующих условиях измерения.
Данные рентгеновской дифракции кристаллической формы В получали измерением рентгеновским дифрактометром типа рентгенографическая пластина (R-AXIS VII, полученный Rigaku Industrial Corporation), применяя излучение Cu-Kα (50 кВ, 100 мА, λ=1,5418 ангстрем). Конкретно, образец помещали в стеклянный капилляр с внутренним диаметром 0,7 мм и в сумме получали шесть дифракционных изображений в условиях длины рентгеновской камеры 300 мм, осцилляционного 30-градусного расстояния и продолжительности выдержки 45 минут для сбора данных.
Данные рентгеновской дифракции кристаллической формы А получали измерением рентгеновским дифрактометром типа рентгенографическая пластина (R-AXIS VII, полученный Rigaku Industrial Corporation), применяя излучение Cu-Kα (50 кВ, 100 мА, λ=1,5418 ангстрем). Конкретно, образец помещали в стеклянный капилляр с внутренним диаметром 0,7 мм и в сумме получали шесть дифракционных изображений в условиях длины рентгеновской камеры 300 мм, осцилляционного 45-градусного расстояния и продолжительности выдержки 90 минут для сбора данных.
Интегрирование по контуру дифракционных изображений проводили программным обеспечением R-AXIS Display (Rigaku) (диапазон интегрирования: 45-135 градусов). Относительную интенсивность интегрированных интенсивностей рассчитывали допущением, что максимальная интегрированная интенсивность равна 100, и наносили на график относительно угла дифракции 2θ для получения дифрактограммы.
Кристаллы согласно настоящему изобретению (кристаллическая форма В соединения I) можно получить осаждением кристаллов соединения I из раствора соединения I, поддерживая температуру равной 40°C или выше. Конкретно, кристаллическую форму В соединения I согласно настоящему изобретению можно получить добавлением первого растворителя, который является подходящим растворяющим растворителем, к соединению I, нагреванием смеси для получения раствора соединения I, добавлением второго растворителя к нагретому раствору и осаждением кристаллов, поддерживая температуру в сборнике для кристаллизации равной 40°C или выше.
Кристаллическую форму В соединения I можно, в частности, получить следующим образом. А именно, соединение I растворяют в первом растворителе и раствор греют для получения нагретого раствора соединения I. Альтернативно, можно также применять способ, в котором добавляют первый растворитель, смесь греют для растворения для получения нагретого раствора соединения I. Кристаллическую форму В можно получить необязательным смешением нагретого раствора соединения I с первым растворителем, добавлением и смешением второго растворителя, в котором растворимость соединения I является меньшей, чем растворимость в первом растворителе, и осаждением кристаллов. Для того чтобы селективно осадить кристаллическую форму В, предпочтительно осуществлять способ осаждения кристаллов соединения I в условиях нагревания. Температуру нагревания можно установить равной температуре выше комнатной температуры. Более предпочтительно, процесс кристаллизации осуществляют при температуре 40°C или выше. Еще более предпочтительно, процесс кристаллизации регулируют при температуре 45°C или выше. Для того чтобы с большой точностью регулировать температуру на стадии кристаллизации, предпочтительно, при добавлении второго растворителя к раствору соединения I, чтобы содержимое системы для кристаллизации заранее тщательно перемешивалось. Скорость добавления второго растворителя к раствору соединения I, температура растворителя, количество растворителя и скорость перемешивания и подобные условия специально не ограничены при условии, что при них протекает кристаллизация.
Что касается нагревания, упоминаемого в настоящем изобретении, максимальная температура может представлять собой температуру кипения первого растворителя. При получении нагретого раствора соединения I в способе получения, предпочтительно, чтобы соединение I было в растворенном состоянии при кипячении с нагреванием. Способы регулирования температуры в процессе кристаллизации включают способ, в котором второй растворитель добавляют к раствору соединения I, предварительно нагретому до 40°C или выше при перемешивании, и содержимое сборника для кристаллизации держат при температуре 40°C или выше, применяя температуру окружающей среды без нагревания, и способ, в котором второй растворитель добавляют при непрерывном перемешивании внутри резервуара для осаждения, или способ, в котором второй растворитель, предварительно нагретый до 40°C или выше, добавляют к раствору соединения I, предварительно нагретому до 40°C или выше. В процессе кристаллизации, стадия нагревания специально не ограничена при условии, что температура поддерживается равной температуре 40°C или выше.
Раствор соединения I, полученный из соединения I и первого растворителя, можно предварительно нагреть до 40°C или выше. Предпочтительно, чтобы раствор был истинным при нагревании первого растворителя при температуре рефлюкса. Более предпочтительно, чтобы раствор представлял собой высококонцентрированный раствор соединения I при кипячении с нагреванием. Особенно предпочтительно, чтобы раствор представлял собой раствор соединения I, который имеет концентрацию, доведенную близко к растворимости при насыщении при кипячении с нагреванием. Например, нагретый раствор можно получить стадией добавления к соединению I растворителя в 1-5-кратном количестве по объему, предпочтительно в 1-2-кратном количестве по объему в расчете на объем минимального количества, которое полностью растворяет соединение I при температуре кипения первого растворителя, и перемешиванием смеси при нагревании для растворения соединения I. Второй растворитель в 0,1-10-кратном количестве по объему, предпочтительно 0,3-3-кратном количестве по объему в расчете на количество первого растворителя можно добавлять к нагретому раствору соединения I для кристаллизации. В данном случае, в процессе кристаллизации, кристаллическую форму В соединения I можно селективно осадить регулированием температуры внутри сборника для кристаллизации, равной температуре 40°C или выше.
Далее, рост кристаллов можно стимулировать без изменения кристаллической формы B непрерывным перемешиванием после осаждения кристаллической формы В при нагревании суспензии для кристаллизации в течение нескольких часов. Кристаллическая форма В, полученная с помощью стадии перемешивания суспензии для кристаллизации при нагревании, хорошо отфильтровывается и имеет большую объемную плотность и, таким образом, не является объемной, приводя в результате к преимуществу превосходной обрабатываемости.
Кристаллическую форму В соединения I согласно настоящему изобретению можно получить фильтрацией таким образом полученной суспензии кристаллической формы В соединения I для сбора влажных кристаллов и сушкой влажных кристаллов. Физико-химическая стабильность кристаллической формы В соединения I является такой высокой, что условия для стадии сушки после сбора кристаллов конкретно не ограничены и можно допускать любую стадию сушки и условия сушки. Конкретно, можно применять способ горячей сушки, способ вакуумной сушки или способ горячей вакуумной сушки.
Первый растворитель, применяемый в настоящем изобретении, конкретно не ограничен при условии, что растворитель является инертным для соединения I и может растворять соединение I. Растворители, которые могут растворять соединение I, могут представлять собой один из или смешанные растворители, состоящие из двух или более из спиртов, кетонов, простых эфиров, ацеталей, сложных эфиров, нитрилов, N-алкилпирролидона, N,N-диметилформамида, N,N-диметилацетамида, диметилсульфоксида, ароматических углеводородов и галогенированных углеводородов. Среди растворителей, которые могут растворять соединение I, спирты можно упомянуть в качестве предпочтительных растворителей в качестве первого растворителя для получения кристаллической формы В соединения I согласно настоящему изобретению, когда учитываются растворимость соединения I, режим стадии кристаллизации и режим стадии выделения. Применение спиртов, т.е. ациклических насыщенных углеводородных молекул, в которых в качестве заместителя имеется одна или более гидроксильных групп, является более предпочтительным. Спирты включают метанол, этанол, 1-пропанол, изопропиловый спирт, бутанол, этиленгликоль, 1,3-бутандиол, 1,4-бутандиол, глицерин, пропиленгликоль, диглицидиловый эфир этиленгликоля, монометиловый эфир этиленгликоля, моноэтиловый эфир этиленгликоля, 1,3-диметиловый эфир глицерина, диэтиленгликоль, моноэтиловый эфир диэтиленгликоля, монометиловый эфир дипропиленгликоля и 1,5-пентандиол. Особенно предпочтительные растворители включают метанол, этанол, 1-пропанол и изопропиловый спирт. Можно применять один из или смешанный растворитель, состоящий из двух или более из них.
Можно правильно подобрать количество применяемого первого растворителя. Раствор соединения I в первом растворителе предпочтительно представляет собой сильно концентрированный раствор с точки зрения режима стадии осаждения кристаллов и более предпочтительно представляет собой насыщенный раствор соединения I. Далее, количество первого растворителя более предпочтительно представляет собой количество, которое позволяет получить насыщенный раствор соединения I при температуре кипения первого растворителя, и еще более предпочтительно представляет собой количество, которое позволяет полностью растворить соединение I при кипячении с нагреванием. Конкретно, количество применяемого первого растворителя составляет 1-20-кратное количество по весу относительно количества соединения I.
Растворители, пригодные в качестве второго растворителя, который способствует кристаллизации, в настоящем изобретении специально не ограничены при условии, что они являются инертными для соединения I, растворяют соединение I с низкой растворимостью и являются смешиваемыми с первым растворителем в любом соотношении. Соответственно, подходящий второй растворитель следует выбирать на основании соотношения совместимости между первым растворителем и вторым растворителем. Когда применяют спирт, который является предпочтительным в качестве первого растворителя, подходящий второй растворитель представляет собой воду. Вода может представлять собой очищенную воду или содержащую соль воду, которая содержит подходящую неорганическую соль или органическую соль. Соли включают хлорид натрия, хлорид калия, хлорид кальция, хлорид магния, хлорид аммония, карбонат натрия, калия карбонат, сульфат натрия, сульфат магния, фосфат натрия и фосфат калия. Данные соли применяют при условии, что они являются инертными для соединения I и смешение первого растворителя со вторым растворителем не дает соли.
Второй растворитель можно добавлять в количестве, достаточно большом для осаждения кристаллов соединения I из раствора соединения I. Например, второй растворитель можно применять в 0,1-10-кратном количестве по объему, предпочтительно 0,3-3-кратном количестве по объему количества первого растворителя.
Кристаллическую форму А соединения I можно получить и выделить согласно способу получения хинолиновых производных, описанному в документе международной публикации WO2006/013896. Кристаллическую форму А соединения I можно селективно получить растворением кристаллической формы А соединения I, которая выделена реакцией для получения соединения I, в диметилацетамидном растворителе при комнатной температуре, выливанием раствора соединения I в воду, сохраняя температуру равной или меньшей комнатной температуры, например ледяную воду для осаждения кристаллов соединения I, и фильтрованием раствора для выделения кристаллов.
Кристаллическая форма В соединения I в настоящем изобретении обладает большой физико-химической стабильностью и не вызывает неблагоприятные явления, такие как увеличение кристаллов, затвердевание и агломерация в условиях хранения в кристаллическом состоянии. Соответственно, агрохимические композиции, содержащие кристаллическую форму В соединения I в качестве активного ингредиента, свободны от явления изменения кристаллической формы активного ингредиента и, таким образом, обладают большой стабильностью свойств в виде составов и хорошим сохранением инсектицидной активности в состоянии хранения. Форма составов агрохимических композиций, содержащих кристаллическую форму В соединения I в качестве активного ингредиента, конкретно не ограничена. В форме состава, в которой кристаллы активного ингредиента сохраняются в составе, состав проявляет полезный эффект состава посредством физико-химических свойств кристаллической формы В соединения I. Соответственно, данная форма является предпочтительной. Подходящие формы составов включают порошки, смачивающиеся порошки, гранулярные диспергируемые в воде порошки, мелкие гранулы и водные суспензии. Данные формы можно формулировать соответствующими общими способами. Любые добавки, отличные от кристаллической формы В соединения I, добавляемые к агрохимической композиции согласно настоящему изобретению, можно применять без конкретных ограничений при условии, что стабильность соединения I не уменьшается, и твердые носители, поверхностно-активные вещества и другие вспомогательные вещества можно применять в зависимости от формы состава.
Например, кристаллическую форму В соединения I можно смешивать с твердыми носителями и необязательно другими добавками и формулировать в виде порошков, смачивающихся порошков, гранулярных дисперигируемых в воде порошков и мелких гранул общепринятыми способами. Далее, кристаллическую форму В соединения I можно смешивать с поверхностно-активным веществом и растворителем (например, водой) и необязательно другими добавками и формулировать в виде водных суспензий общепринятыми способами.
Твердые носители, пригодные в настоящем изобретении, включают, например, тальк, бентонит, глину, каолин, кизельгур, вермикулит, белую сажу, карбонат кальция, кислую глину, кварцевый песок, кварцит, цеолит, аттапульгит, пемзу, сульфат аммония, сульфат натрия, мочевину, хлорид калия, хлорид натрия, кристаллическую целлюлозу, карбоксиметилцеллюлозу и ксантановую камедь.
Неионные и/или анионные поверхностно-активные вещества являются применимыми в качестве поверхностно-активных веществ. Неионные поверхностно-активные вещества, пригодные в настоящем изобретении, включают простые полиоксиэтиленалкиловые эфиры, простые полиоксиэтиленстирилфениловые эфиры, сложные полиоксиэтиленалкиловые эфиры, полиоксиэтиленфенилэфирные полимеры, простые полиоксиэтиленалкиленарилфениловые эфиры, полиоксиэтиленполиоксипропиленовые блокполимеры, полиоксиэтиленированное касторовое масло, полиоксиэтиленированное гидрогенизированное касторовое масло, глицерилмоностеараты и глицерилдистеараты. Анионные поверхностно-активные вещества включают сульфат простого полиоксиэтиленстирилфенилового эфира, лигнинсульфонаты, алкиларилсульфонаты, алкилнафталинсульфонаты, поликарбоксилаты, сульфаты и фосфаты простого полиоксиэтиленполистирилфенилового эфира. Неионные и анионные поверхностно-активные вещества не ограничены при условии, что активный ингредиент сельскохозяйственного ингредиента не теряет своих свойств. В настоящем изобретении можно применять одно из или комбинацию двух или более поверхностно-активных веществ, выбранных из группы, состоящей из данных неионных и анионных поверхностно-активных веществ.
Другие вспомогательные вещества для препаратов включают добавки, препятствующие осаждению, и вещества, понижающие температуру замерзания. Добавки, препятствующие осаждению, включают, но не ограничиваются перечисленным, бентонит, смектит, ксантановую камедь, кристаллическую целлюлозу и карбоксиметилцеллюлозу. Можно применять одно или комбинацию двух или более из них.
Вещества, понижающие температуру замерзания, включают, но не ограничиваются перечисленным, этиленгликоль, пропиленгликоль, глицерин, диэтиленгликоль и полиэтиленгликоль. Можно применять одно или комбинацию двух или более из них.
Агрохимическую композицию согласно настоящему изобретению можно применять в виде смеси или в комбинации, например, с другими инсектицидами, фунгицидами, майтицидами, гербицидами, агентами, регулирующими рост растений, или удобрениями. Агенты, которые можно смешивать или применять в комбинации, включают агенты, описанные, например, в The Pesticide Manual, 13th edition, опубликованном The British Crop Protection Council, и SHIBUYA INDEX, the 14th edition, 2009, опубликованном SHIBUYA INDEX RESEARCH GROUP.
Более конкретно, примеры инсектицидов включают органические соединения, представляющие собой сложные эфиры фосфорной кислоты, такие как ацефат, диклорфос, EPN, фениторотион, фенамифос, протиофос, профенофос, пираклофос, хлорпирифос-метил и диазинон; карбаматные соединения, такие как метомил, тиодикарб, алдикарб, оксамил, пропоксур, карбарил, фенобукарб, этиофенкарб, фенотиокарб, пиримикарб, карбофуран и бенфуракарб; нереистоксиновые производные, такие как картап и тиоциклам; хлорорганические соединения, такие как дикофол и тетрадифон; пиретроидные соединения, такие как перметрин, тефлутрин, циперметрин, дельтаметрин, цигалотрин, фенвалерат, флувалинат, этофенпрокс и силафлуофен; бензоилкарбаматные соединения, такие как дифлубензурон, тефлубензурон, флуфеноксурон и хлорфлуазурон; и соединения, подобные ювенильным гормонам, такие как метопрен. Другие инсектициды включают такие соединения, как бупрофезин, гекситиазокс, хлордимеформ, пиридабен, амитраз, фенпироксимат, пиримидифен, тебуфенпирад, флаукрипирим, ацеквиноцил, фипронил, этоксазол, имидаклоприд, хлотианидин, пиметрозин, бифеназат, спиродиклофен, хлорфенапир, пирипроксифен, индоксакарб, пиридалил или спиносад, авермектин, милбемицин, металлоорганические соединения и динитросоединения. Далее, сельскохозяйственные и плодовоовощные инсектициды согласно настоящему изобретению можно применять в виде смеси или в комбинации с микробными пестицидами, такими как BT составы, и вирусными агентами, патологическими для насекомых.
Фунгициды, пригодные в настоящем изобретении, включают, например, стробилриновые соединения, такие как азоксистробин, крезоксим-метил и трифлоксистробин; анилинопиримидиновые соединения, такие как мепанипирим, пириметанил и ципродинил; азоловые соединения, такие как триадимефон, битертанол, трифлумизол, этаконазол, пропиконазол, пенконазол, флузилазол, миклобутанил, ципроконазол, тебуконазол, гексаконазол, прохлораз и симеконазол; хиноксалиновые соединения, такие как квинометионат; дитиокарбаматные соединения, такие как манеб, зинеб, манкоцеб, поликарбамат и пропинеб; фенилкарбаматные соединения, такие как диэтофенкарб; хлорорганические соединения, такие как хлорталонил и квинтозен; бензимидазольные соединения, такие как беномил, тиофанат-метил и карбендазол; фениламидные соединения, такие как металаксил, оксадиксил, офураз, беналаксил, фуралаксил и ципрофурам; соединения, являющиеся производными сульфеновой кислоты, такие как дихлофлуанид; соединения меди, такие как гидроксид меди и оксихинолин меди; изоксазольные соединения, такие как гидроксиизоксазол; фосфорорганические соединения, такие как фосэтил-алюминий и толклофос-метил; N-галогентиоалкильные соединения, такие как каптан, каптафол и фолпет; дикарбоксиимидные соединения, такие как процимидон, ипродион и винклозолин; бензанилидные соединения, такие как флутоланил и мепронил; морфолиновые соединения, такие как фенпропиморф и диметоморф; оловоорганические соединения, такие как фентингидроксид и фентинацетат; и цианопирроловые соединения, такие как флудиоксонил и фенпиклонил. Другие фунгициды включают фталид, флуазинам, цимоксанил, трифорин, пирифенокс, фенаримол, фенпропидин, пенцикурон, циазофамид, ипроваликарб и бентиаваликарб-изопропил.
Агрохимические композиции, содержащие кристаллическую форму В соединения I, полученные согласно настоящему изобретению, обладают преимуществом по сравнению с агрохимическими композициями, содержащими кристаллическую форму А, т.к. обладают повышенной стабильностью при хранении, увеличение кристаллов в значительной степени ингибировано, и неблагоприятные явления, такие как пониженная эффективность в результате увеличения кристаллов и затвердевание агрохимической композиции, не наблюдаются.
ПРИМЕРЫ
Настоящее изобретение дополнительно иллюстрируется следующими справочным примером, примерами и сравнительным примером, и эффект настоящего изобретения иллюстрируется тестовыми примерами. Однако следует отметить, что настоящее изобретение не ограничивается данными примерами.
Аналитический прибор и условия измерения
Аналитический прибор и условия измерения для измерений порошковой рентгеновской дифрактометрией справочного примера и примера 1 являются следующими. Данные рентгеновской дифракции кристаллической формы А получали измерением с помощью рентгеновского дифрактометра типа рентгенографической пластины (R-AXIS VII, полученного Rigaku Industrial Corporation), применяя излучение Cu-Kα (50 кВ, 100 мА, λ=1,5418 ангстрем). Конкретно, образец помещали в стеклянный капилляр с внутренним диаметром 0,7 мм и в сумме шесть дифракционных изображений получали в условиях длины рентгеновской камеры 300 мм, осцилляционного 45-градусного расстояния и продолжительности выдержки 90 минут для сбора данных.
Данные рентгеновской дифракции кристаллической формы В получали измерением с помощью рентгеновского дифрактометра типа рентгенографической пластины (R-AXIS VII, полученного Rigaku Industrial Corporation), применяя излучение Cu-Kα (50 кВ, 100 мА, λ=1,5418 ангстрем). Конкретно, образец помещали в стеклянный капилляр с внутренним диаметром 0,7 мм и в сумме шесть дифракционных изображений получали в условиях длины рентгеновской камеры 300 мм, осцилляционного 30-градусного расстояния и продолжительности выдержки 45 минут для сбора данных.
Интегрирование по контуру дифракционных изображений проводили программным обеспечением R-AXIS Display (Rigaku) (диапазон интегрирования: 45-135 градусов). Относительную интенсивность интегрированных интенсивностей рассчитывали допущением, что максимальная интегрированная интенсивность равна 100, и наносили на график относительно угла дифракции 2θ для получения дифрактограммы.
Справочный пример: получение кристаллической формы А соединения I
Диметилформамид (980 мл) и 98 г 2-этил-3,7-диметил-6-(4-(трифторметокси)фенокси)хинолин-4(1H)-она, полученного согласно способу, описанному в WO 2006/013896, помещали в атмосфере азота в стеклянную колбу (объем: 2000 мл), снабженную мешалкой, термометром, противоточным холодильником и хлоркальциевой трубкой, и смесь охлаждали до 15°C. Добавляли порциями к смеси гидрид натрия (55%, 18,2 г), и смесь реагировала при комнатной температуре в течение одного часа. Добавляли порциями к смеси метилхлорформиат (32,1 г), и смесь реагировала при комнатной температуре в течение одного часа. Реакционную смесь выливали в 5 л ледяной воды в 10-литровом пластиковом контейнере и перемешивали при комнатной температуре в течение 2 часов. Выпавший осадок собирали фильтрацией через вакуумный фильтр и промывали н-гексаном и водой. Твердый остаток сушили при пониженном давлении для получения 103,3 г (выход 91,4%) соединения I (2-этил-3,7-диметил-6-(4-(трифторметокси)фенокси)хинолин-4-илметилкарбоната) (кристаллическая форма А). Таким образом, полученный продукт идентифицировали как соединение I на основании следующих спектральных данных.
1H-ЯМР (CDCl3) 1,38 (т, 3H), 2,31 (с, 3H), 2,41 (с, 3H), 3,01 (кв, 2H), 3,88 (с, 3H), 6,97 (д, 2H), 7,14 (с, 1H), 7,20 (д, 2H), 7,94 (с, 1H).
Дифрактограмма, полученная порошковой рентгеновской дифрактометрией соединения I (кристаллическая форма А), показана на фиг.3. Соединение I (кристаллическая форма А) имело пики при 2θ=6,94°, 10,41°, 19,13°, 23,54°, 24,32°, 24,95° и 25,46° с относительной интенсивностью, не меньшей чем 15%, как определено допущением, что интенсивность пика при 2θ=19,13° равна 100, и имело пики при 2θ=6,94°, 10,41°, 13,05°, 19,13°, 20,90°, 23,54°, 24,32°, 24,95° и 25,46° с относительной интенсивностью, не меньшей чем 10%.
Соединение I (кристаллическая форма А) анализировали дифференциальной сканирующей калориметрией (DSC), и диаграмма полученных результатов показана на фиг.4. Соединение I (кристаллическая форма А) имело эндотермические пики в области 108,1°C и 117,2°C.
Пример 1: получение кристаллической формы В соединения I
Кристаллическую форму А соединения I (148,8 г), полученную в справочном примере, и 1200 мл метанола добавляли в 3000-мл четырехгорбую колбу, снабженную мешалкой, охлаждающей трубкой, капельной воронкой и термометром. Перемешиваемый раствор кипятили с обратным холодильником до полного растворения кристаллов. Добавляли по каплям дистиллированную воду (520 мл) к раствору в диапазоне температур внутри колбы 40-65°C для кристаллизации. Внутреннюю температуру перемешиваемого раствора сохраняли равной 40-65°C. Затем суспензию кипятили с обратным холодильником в течение 2 часов. Суспензию охлаждали и кристаллы собирали фильтрацией, промывали 410 мл 50% (об./об.) водно-метанольного раствора и сушили при 60°C при пониженном давлении для получения 140,6 г кристаллической формы В соединения I. Таким образом, полученный продукт идентифицировали как соединение I на основании спектральных данных, показанных ниже.
1H-ЯМР (CDCl3) 1,38 (т, 3H), 2,31 (с, 3H), 2,41 (с, 3H), 3,01 (кв, 2H), 3,88 (с, 3H), 6,97 (д, 2H), 7,14 (с, 1H), 7,20 (д, 2H), 7,94 (с, 1H).
Дифрактограмма, полученная порошковой рентгеновской дифрактометрией соединения I (кристаллическая форма В), показана на фиг.1. При порошковой рентгеновской дифрактометрии соединение I (кристаллическая форма В) имело пики при 2θ=5,30°, 6,86°, 8,51°, 9,98°, 10,28°, 11,32°, 20,25° и 20,96° с относительной интенсивностью, не меньшей чем 15%, как определено допущением, что интенсивность пика при 2θ=20,96° равна 100, и имело пики при 2θ=5,30°, 6,86°, 8,51°, 9,98°, 10,28°, 11,32°, 12,20°, 16,64°, 17,10°, 18,39°, 20,25°, 20,96° и 22,50° с относительной интенсивностью, не меньшей чем 10%.
Соединение I (кристаллическая форма В) анализировали дифференциальной сканирующей калориметрией (DSC), и диаграмма полученных результатов показана на фиг.2. Соединение I (кристаллическая форма В) имело эндотермический пик в области 117,8°C, как определено DSC, и не наблюдали отчетливых пиков, отличных от пика в области 117,8°C.
Пример 2: получение состава, содержащего кристаллическую форму В соединения I (состав в виде водной суспензии)
Кристаллическую форму В соединения I (10,8 частей по весу), полученную в примере 1, 1,0 части по весу Newcalgen FS-3PG (торговое название, поверхностно-активное вещество, полученное Takemoto Oils & Fats Co., Ltd.), 1,0 части по весу Demol N (торговое название, поверхностно-активное вещество, полученное Kao Corp.), 0,1 части по весу ANTIFOAM E-20 (торговое название, противовспенивающее вещество, полученное Kao Corp.) и 30 частей по весу воды смешивали вместе и смесь измельчали вертикальной мельницей мокрого помола (1000 об/мин, 90 мин), полученной IMEX Co., Ltd. Измельченный продукт добавляли к смеси, состоящей из 0,2 части по весу ксантановой камеди, 0,3 части по весу Kunipia F (торговое название, загуститель, полученный Kunimine Industries Co., Ltd.), 7,5 части по весу пропиленгликоля, 0,2 части по весу Proxel GXL(S) (торговое название, фунгицид, полученный Arch Chemicals Japan, Inc.) и 49,1 части по весу воды, для получения состава в виде водной суспензии (жидкотекучий состав), содержащей 10,8% по весу кристаллической формы В соединения I.
Пример 3: получение состава (смачивающийся порошок), содержащего кристаллическую форму В соединения I
Кристаллическую форму В соединения I (10,5 части по весу), полученную в примере 1, 10,0 части по весу Carplex #80 (торговое название, белая сажа, полученная Degussa Japan), 73,5 части по весу лактозы, 3,0 части по весу Newcalgen BX-C (торговое название, поверхностно-активное вещество, полученное Takemoto Oils & Fats Co., Ltd.) и 3,0 части по весу Newcalgen WG-4 (торговое название, поверхностно-активное вещество, полученное Takemoto Oils & Fats Co., Ltd.) смешивали вместе и смесь измельчали SampleMill для получения смачивающегося порошка, содержащего 10,5% по весу кристаллической формы В соединения I.
Сравнительный пример 1: получение состава, содержащего кристаллическую форму А соединения I (состав в виде водной суспензии)
Кристаллическую форму А соединения I (10,8 части по весу), 1,0 части по весу Newcalgen FS-3PG (торговое название, поверхностно-активное вещество, полученное Takemoto Oils & Fats Co., Ltd.), 1,0 части по весу Demol N (торговое название, поверхностно-активное вещество, полученное Kao Corp.), 0,1 части по весу ANTIFOAM E-20 (торговое название, противовспенивающее вещество, полученное Kao Corp.) и 30 частей по весу воды смешивали вместе и смесь измельчали вертикальной мельницей мокрого помола (1000 об/мин, 90 мин), полученной IMEX Co., Ltd. Измельченный продукт добавляли к смеси, состоящей из 0,2 части по весу ксантановой камеди, 0,3 части по весу Kunipia F (торговое название, загуститель, полученный Kunimine Industries Co., Ltd.), 7,5 части по весу пропиленгликоля, 0,2 части по весу Proxel GXL(S) (торговое название, фунгицид, полученный Arch Chemicals Japan, Inc.) и 49,1 части по весу воды, для получения состава в виде водной суспензии (жидкотекучий состав), содержащей 10,8% по весу кристаллической формы А соединения I.
Тестовый пример 1: тест на хранение
Составы в виде водной суспензии примера 2 и сравнительного примера 1 хранили при 54°C в течение 2 недель и наблюдали за изменением качества до и после хранения. Размер кристаллов до и после хранения измеряли измерительным прибором на основе лазерной дифракции для определения распределения частиц по размеру и под оптическим микроскопом.
Измерение измерительным прибором на основе лазерной дифракции для определения распределения частиц по размеру
Применяемый прибор: измерительный прибор на основе лазерной дифракции для определения распределения частиц по размеру SALD-2200, полученный Shimadzu Corp.
Жидкотекучий состав, разбавленный водой после теста на хранение, применяли для измерения размера частиц с помощью вышеуказанного измерительного прибора. Кристаллическая форма А после хранения не могла образовывать суспензию в воде, и таким образом, вместо суспензии, перед применением в тесте, ее подвергали ультразвуковой обработке до исчезновения крупных частиц.
Измерение с помощью оптического микроскопа
Применяемый прибор: цифровой микроскоп VHX-200, полученный Keyence Corp.
Изображения разбавленных водой жидкотекучих составов после теста на хранение получали с помощью вышеуказанного прибора (увеличение: 1500-кратное). Основные оси кристаллов на изображениях измеряли и рассчитывали углы. Кристаллическая форма А после хранения не могла образовывать суспензию в воде, и таким образом, вместо суспензии, перед применением в тесте, ее подвергали ультразвуковой обработке до исчезновения крупных частиц.
Результаты измерения показаны в таблице 1.
Результаты теста на хранение
Y: Результаты измерения с помощью оптического микроскопа
Составы в виде водной суспензии примера 2 (состав, содержащий кристаллическую форму В) и сравнительного примера 1 (состав, содержащий кристаллическую форму А) хранили в течение 2 недель в камере с автоматическим регулированием температуры при 54°C. Как результат, состав примера 2 обладал свойствами, остающимися неизменными, и сохранял жидкотекучесть в виде состава в виде водной суспензии. Диаметр частиц состава примера 2 после теста на хранение оставался практически неотличным от диаметра частиц до теста на хранение, и изображения, полученные с помощью микроскопа (смотрите фиг.5 и 7), показывают, что рост кристаллов ингибировался. С другой стороны, состав сравнительного примера 1 после теста на хранение был в агломерированном состоянии, терял жидкотекучесть в виде состава в виде водной суспензии и, таким образом, плохо растворялся в воде, осложняя применение разбавленного состава в качестве сельскохозяйственных и плодовоовощных инсектицидов. Изображения, полученные с помощью микроскопа, частиц (смотрите фиг.6 и 8) показали увеличение кристаллов, связанное с ростом кристаллов в процессе хранения, создавая проблему стабильности при хранении в виде составов в виде водной суспензии. Диаметр частиц состава сравнительного примера 1 после теста на хранение заметно увеличивался по сравнению с диаметром частиц до теста на хранение. Состав сравнительного примера 1 после теста на хранение был в агломерированном состоянии и не суспендировался в воде. Соответственно, состав обрабатывали ультразвуком перед измерением. Следовательно, считается, что реальный размер частиц состава сравнительного примера 1 после хранения является большим, чем измеренная величина, показанная в таблице 1.
Кристаллическая форма В соединения I согласно настоящему изобретению обладает высокой физико-химической стабильностью и обладает высокой стабильностью при хранении. Агрохимические составы, обладающие высокой стабильностью при хранении, можно предоставить, выбирая кристаллическую форму В соединения I в качестве исходного вещества активного фармацевтического ингредиента.
Тестовый пример 2: тест на подтверждение эффективности
Составы в виде водной суспензии примера 2 и сравнительного примера 1 хранили при 54°C в течение 2 недель. Составы в виде водной суспензии разбавляли водой для получения разбавленных водой растворов, имеющих заранее определенную концентрацию. Разбавленные растворы (2 мл) наносили на листовой диск огурца. После сушки на воздухе, выпускали 25-40 взрослых особей белокрылки табачной. Крышку из гигроскопической ваты помещали на контейнер и контейнер переворачивали вверх дном. Через 2 дня после обработки считали число умерших взрослых особей для расчета LC50 (м.д.). Диаметр частиц измеряли с помощью измерительного прибора на основе лазерной дифракции для определения распределения частиц по размеру и согласно способу, описанному в тестовом примере 1.
Результаты измерения показаны в таблице 2.
Результаты теста на подтверждение эффективности
Что касается состава, содержащего кристаллическую форму В примера 2, размер частиц соединения I в составе после хранения оставался практически неизменным, тогда как для состава, содержащего кристаллическую форму А сравнительного примера 1, наблюдали увеличение диаметра частиц, вызванное ростом кристаллов соединения I при хранении. В испытании на педикулицидную активность обоих образцов составов против белокрылки табачной, для сравнительного примера 1 наблюдали снижение педикулицидной активности. Считают, что снижение эффективности объясняется снижением вероятности контакта, как результат уменьшения удельной площади поверхности за счет увеличения размера частиц кристаллической формы А при хранении. Соответственно, было показано, что в составе в виде водной суспензии изменение кристаллического состояния соединения I, которое представляет собой активный ингредиент для инсектицидной активности, оказывает значительное влияние на инсектицидную активность.
Как ясно из результатов тестовых примеров 1 и 2, при получении агрохимических составов, содержащих соединение I в качестве активного ингредиента агрохимикатов, агрохимические составы, которые, в условиях продолжительного хранения, менее вероятно подвергаются изменениям свойств, могут сохранять заранее определенную инсектицидную активность и обладать высокой стабильностью при продолжительном хранении и их можно получить, применяя кристаллическую форму В соединения I согласно настоящему изобретению в качестве исходного вещества активного ингредиента агрохимикатов.
| название | год | авторы | номер документа |
|---|---|---|---|
| СТАБИЛЬНАЯ КРИСТАЛЛИЧЕСКАЯ ФОРМА ТИПИРАЦИЛА ГИДРОХЛОРИДА И СПОСОБ ЕЕ КРИСТАЛЛИЗАЦИИ | 2014 |
|
RU2640417C2 |
| СТАБИЛЬНАЯ КРИСТАЛЛИЧЕСКАЯ ФОРМА ТИПИРАЦИЛА ГИДРОХЛОРИДА И СПОСОБ ЕЕ КРИСТАЛЛИЗАЦИИ | 2014 |
|
RU2674441C1 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ ПРОИЗВОДНЫХ ДИГИДРОПИРИМИДИНА | 2013 |
|
RU2646599C2 |
| КРИСТАЛЛИЧЕСКАЯ ФОРМА БИСУЛЬФАТНОГО ИНГИБИТОРА JAK-КИНАЗЫ И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2015 |
|
RU2716260C2 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ 3-(2,4-ДИХЛОРБЕНЗИЛ)-2-МЕТИЛ-N-(ПЕНТИЛСУЛЬФОНИЛ)-3H- БЕНЗИМИДАЗОЛ-5-КАРБОКСАМИДА | 1999 |
|
RU2228931C2 |
| КРИСТАЛЛИЧЕСКАЯ ФОРМА ПРОИЗВОДНОГО 1,2-ДИГИДРОПИРИДИНА И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2005 |
|
RU2323930C1 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ БИЛАСТИНА И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 2016 |
|
RU2772222C2 |
| КРИСТАЛЛИЧЕСКОЕ ВЕЩЕСТВО | 2018 |
|
RU2834293C2 |
| СОЛЬ ИНГИБИТОРА SYK И ЕЕ КРИСТАЛЛИЧЕСКАЯ ФОРМА | 2019 |
|
RU2818103C2 |
| КРИСТАЛЛИЧЕСКАЯ ФОРМА БИСУЛЬФАТА ИНГИБИТОРА JAK И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2015 |
|
RU2704795C2 |
Изобретение относится к области органической химии, а именно к кристаллической форме 2-этил-3,7-диметил-6-(4-(трифторметокси)фенокси)хинолин-4-илметилкарбоната. Также изобретение относится к способу получения указанной выше кристаллической формы и агрохимической композиции на основе указанной кристаллической формы. Технический результат: получена стабильная при хранении кристаллическая форма 2-этил-3,7-диметил-6-(4-(трифторметокси)фенокси)хинолин-4-илметилкарбоната, обладающая инсектицидной активностью. 3 н. и 3 з.п. ф-лы, 8 ил., 2 табл., 5 пр.
1. Кристаллическая форма 2-этил-3,7-диметил-6-(4-(трифторметокси)фенокси)хинолин-4-илметилкарбоната, которая имеет дифракционные пики при по меньшей мере следующих углах дифракции (2θ), как определено порошковой рентгеновской дифрактометрией:
2. Кристаллическая форма 2-этил-3,7-диметил-6-(4-(трифторметокси)фенокси)хинолин-4-илметилкарбоната по п.1, которая дополнительно имеет дифракционные пики при следующих углах дифракции (2θ), как определено порошковой рентгеновской дифрактометрией:
3. Способ получения кристаллов по п.1 или 2, включающий добавление воды к алканоловому раствору 2-этил-3,7-диметил-6-(4-(трифторметокси)фенокси)хинолин-4-илметилкарбоната и осаждение кристаллов при температуре 40°C или выше.
4. Агрохимическая композиция, обладающая инсектицидной активностью, содержащая кристаллы по п.1 или 2 в качестве активного ингредиента.
5. Агрохимическая композиция по п.4, дополнительно содержащая поверхностно-активное вещество и воду.
6. Агрохимическая композиция по п.4, дополнительно содержащая поверхностно-активное вещество и твердый носитель.
| Приспособление для суммирования отрезков прямых линий | 1923 |
|
SU2010A1 |
| ДИГАЛОИДПРОПЕНОВЫЕ СОЕДИНЕНИЯ, ИНСЕКТИЦИДНО-АКАРИЦИДНЫЕ СРЕДСТВА, СОДЕРЖАЩИЕ ИХ, И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ ДЛЯ ИХ ПОЛУЧЕНИЯ | 1995 |
|
RU2144526C1 |

