[Область техники, к которой относится изобретение]
[0001]
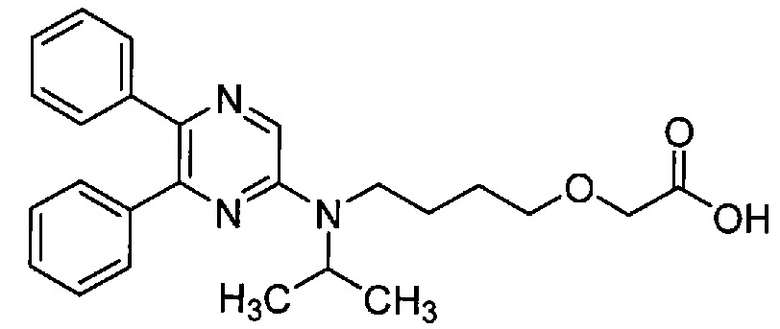
Настоящее изобретение относится к новому кристаллическому веществу 2-{4-[N-(5,6-дифенилпиразин-2-ил)-N-изопропиламино]бутилокси}уксусной кислоте (далее называемому как «Соединение B»).
[Хим. 1]
[Уровень техники]
[0002]
Фармацевтический продукт должен сохранять свое качество в течение длительного периода времени даже при различных условиях распределения, хранения и т.д. Следовательно, соединение, которое служит в качестве активного ингредиента, должно иметь высокую физико-химическую стабильность. Вследствие этого в качестве активного ингредиента фармацевтического продукта обычно используют кристаллическое вещество, которое, как можно ожидать, обладает высокой стабильностью.
В ходе скрининга кристаллического вещества активного ингредиента фармацевтического продукта не только трудно найти оптимальные условия для получения кристаллического вещества, но также, даже если кристаллическое вещество получено, существование полиморфизма часто является проблематичным. Проблема вызвана тем, что существует разница в физико-химической стабильности в зависимости от формы кристаллического вещества.
Кроме того, если кристаллическая форма в качестве активного ингредиента фармацевтического продукта выбрана неправильно, происходит снижение чистоты, превращение кристаллической формы или подобное в зависимости от условий окружающей среды во время хранения, и, таким образом, становится трудно поддерживать постоянное качество соединения, и, следовательно, в зависимости от кристаллической формы может быть вызвано неожиданное явление, такое как снижение эффективности лекарственного средства или неблагоприятное воздействие. В связи с этим, когда кристаллическое вещество соединения, служащего в качестве активного ингредиента фармацевтического продукта, получено успешно, необходимо проводить строгую оценку и проверку физико-химической стабильности полиморфизма.
[0003]
Однако невозможно предсказать существование или отсутствие полиморфизма или стабильной кристаллической формы из структуры соединения, и, кроме того, существует соединение, которое в некоторых случаях не может быть кристаллизовано, и необходимо по-разному изучать условия формирования кристаллического вещества для каждого соединения.
[0004]
С другой стороны, известно, что Соединение B обладает превосходным агонистическим действием в отношении рецептора PGI2 и проявляет различные лечебные эффекты, такие как ингибирующее действие на агрегацию тромбоцитов, сосудорасширяющее действие, расширение гладкой мускулатуры бронхов, ингибирующее действие на отложение липидов и ингибирующее действие на активацию лейкоцитов (см., например, PTL 1-PTL 6). Однако текущая ситуация такова, что неизвестно, может ли кристаллическое вещество быть образовано, тем более существует ли полиморфизм, и важной задачей является получение оптимального кристаллического вещества для его разработки в качестве фармацевтического продукта.
[Список цитирования]
[Патентная литература]
[0005]
[PTL 1] WO 2002/088084
[PTL 2] WO 2009/157396
[PTL 3] WO 2009/107736
[PTL 4] WO 2009/154246
[PTL 5] WO 2009/157397
[PTL 6] WO 2009/157398
[PTL 7] US 2014/0221397
[PTL 8] US 2011/0178103
[PTL 9] US 2011/0015211
[PTL 10] US 2011/0118254
[PTL 11] US 2011/0105518
[Непатентная литература]
[0006]
[NPL 1] Hepatology, 2007, Vol. 45, No. 1, pp. 159-169
[NPL 2] PubMed: Nihon Yakurigaku Zasshi, 2001, Feb, 117(2), pp. 123-130, Abstract
[NPL 3] International Angiology, 29, Suppl. 1 to No. 2, pp. 49-54, 2010
[NPL 4] Japanese Journal of Clinical Immunology, Vol. 16, No. 5, pp. 409-414, 1993
[NPL 5] Japanese Journal of Thrombosis and Hemostasis, Vol. 1, No. 2, pp. 94-105, 1990, Abstract
[NPL 6] The Journal of Rheumatology, Vol. 36, No. 10, pp. 2244-2249, 2009
[NPL 7] The Japanese Journal of Pharmacology, Vol. 43, No. 1, pp. 81-90, 1987
[NPL 8] British Heart Journal, Vol. 53, No. 2, pp. 173-179, 1985
[NPL 9] The Lancet, 1, 4880, pt 1, pp. 569-572, 1981
[NPL 10] European Journal of Pharmacology, 449, pp. 167-176, 2002
[NPL 11] The Journal of Clinical Investigation, 117, pp. 464-72, 2007
[NPL 12] American Journal of Physiology Lung Cellular and Molecular Physiology, 296: L648-L656 2009
[Сущность изобретения]
[Техническая задача]
[0007]
Задачей настоящего изобретения является предоставление кристаллического вещества Соединения B, обладающего превосходной физико-химической стабильностью, а также предоставление фармацевтической композиции, содержащей кристаллическое вещество в качестве активного ингредиента.
[Решение задачи]
[0008]
Способ получения Соединения B раскрыт в Примере 42 PTL 1. Однако в Примере 42 PTL 1 не указано, какая форма Соединения B была получена.
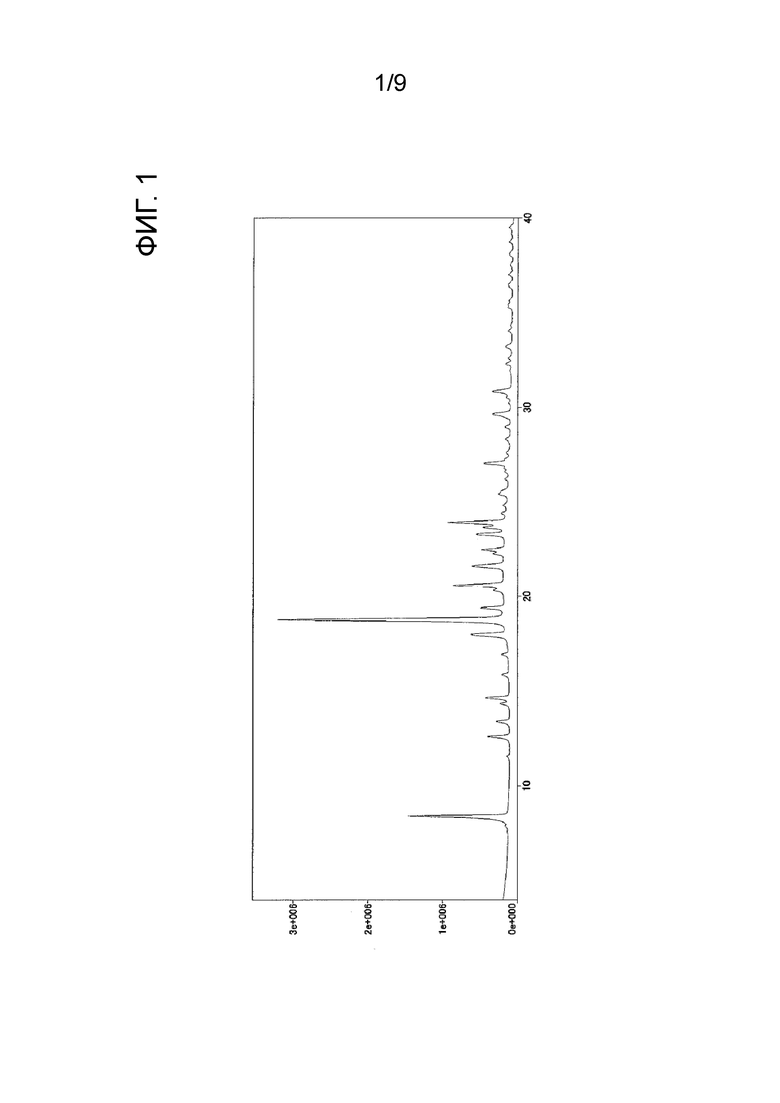
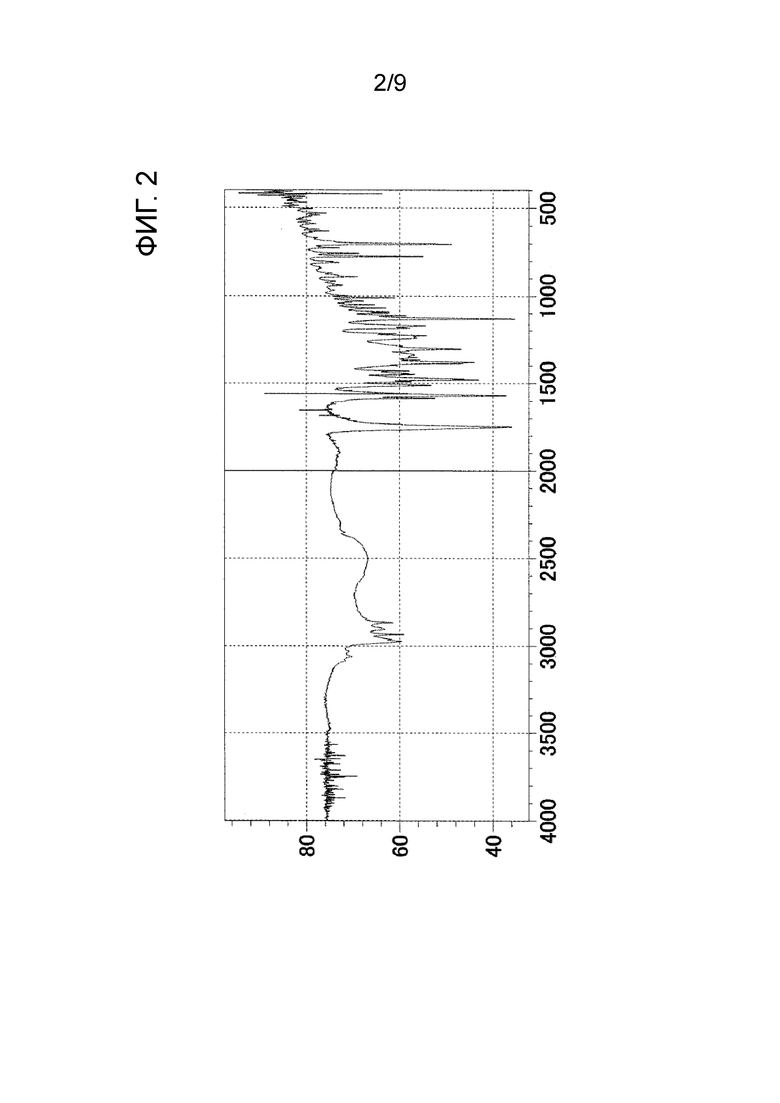
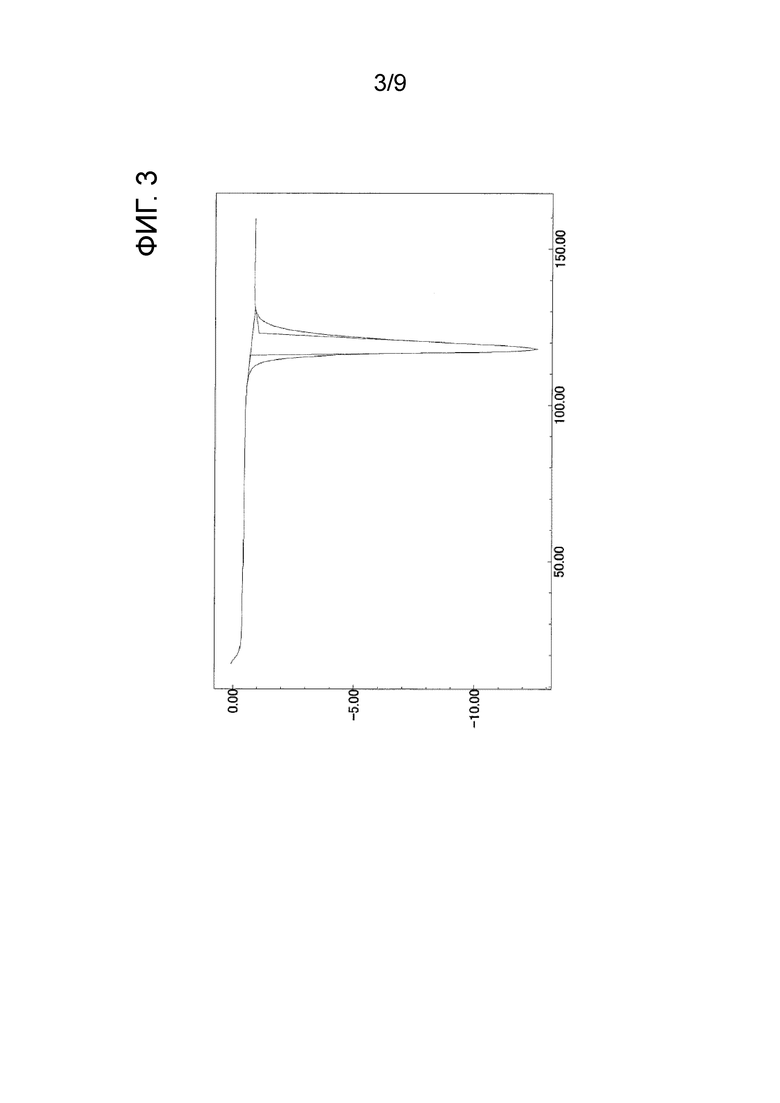
Следовательно, когда автор настоящего изобретения предпринял попытку получить Соединение B в соответствии с той же методикой, что и способ, раскрытый в Примере 42 PTL 1, было обнаружено, что форма представляет собой кристаллическое вещество (далее называемое как «кристаллическое вещество формы-III») (см. нижеприведенный Ссылочный пример 1). Результаты измерения порошковой рентгеновской дифракции, ИК-измерения и ДСК-измерения кристаллического вещества формы-III показаны на ФИГ. 1, ФИГ. 2 и ФИГ. 3 соответственно.
Однако, как показано в нижеприведенном Примере исследования 1, было обнаружено, что кристаллическое вещество формы-III является термодинамически нестабильным, и, следовательно, автор настоящего изобретения провел интенсивные исследования для достижения вышеуказанной цели, и в результате было обнаружено, что существует кристаллическое вещество формы-I и кристаллическое вещество формы-II, каждое из которых является термодинамически более стабильным, и, таким образом, настоящее изобретение было завершено.
[0009]
Настоящее изобретение может включать, например, следующее (1)-(7).
(1) Кристаллическое вещество формы-I Соединения B (далее называемое «кристаллическое вещество формы-I настоящего изобретения»), которое показывает дифракционные пики при углах дифракции (2θ) 6,4, 8,1, 9,5, 10,9, 13,2, 15,7, 17,0, 19,5, 20,3, 21,0 и 22,8 в спектре порошковой рентгеновской дифракции, полученном с использованием Cu-Kα-излучения (λ=1,54 Å).
(2) Кристаллическое вещество формы-I настоящего изобретения, которое показывает пики поглощения при волновых числах 2874 см-1, 1736 см-1, 1558 см-1, 1375 см-1, 1126 см-1 и 696 см-1 в инфракрасном спектре поглощения.
(3) Кристаллическое вещество формы-I настоящего изобретения, которое имеет эндотермический пик при 127°С в дифференциальной сканирующей калориметрии.
(4) Кристаллическое вещество формы-II Соединения B (далее называемое «кристаллическое вещество формы-II настоящего изобретения»), которое показывает дифракционные пики при углах дифракции (2θ) 9,6, 11,4, 11,7, 16,3, 17,5, 18,5, 18,7, 19,9, 20,1, 21,0 и 24,6 в спектре порошковой рентгеновской дифракции, полученном с использованием Cu-Kα-излучения (λ=1,54 Å).
(5) Кристаллическое вещество формы-II настоящего изобретения, которое показывает пики поглощения при волновых числах 2867 см-1, 1749 см-1, 1568 см-1, 1382 см-1, 1131 см-1 и 701 см-1 в инфракрасном спектре поглощения.
(6) Кристаллическое вещество формы-II настоящего изобретения, которое имеет эндотермический пик при 147°С в дифференциальной сканирующей калориметрии.
(7) Фармацевтическая композиция, содержащая кристаллическое вещество по любому одному из (1)-(6) в качестве активного ингредиента (далее называемая «фармацевтическая композиция настоящего изобретения»).
[0010]
При определении угла дифракции (2θ) для дифракционного пика в Примерах и формуле изобретения настоящего изобретения следует понимать, что полученное значение находится в пределах диапазона значения ±0,2, предпочтительно в пределах диапазона значения ±0,1.
Кроме того, при определении пика поглощения в инфракрасном спектре поглощения (далее называемом «ИК-спектр») в Примерах и формуле изобретения настоящего изобретения следует понимать, что полученное значение находится в пределах диапазона значения ±2 см-1, предпочтительно в пределах диапазона значения ±1 см-1.
Кроме того, при определении эндотермического пика с помощью дифференциальной сканирующей калориметрии (далее называемой «ДСК») в Примерах и формуле изобретения настоящего изобретения следует понимать, что полученное значение находится в пределах диапазона значения ±3°C, предпочтительно в пределах диапазона значения ±2°C.
[Краткое описание чертежей]
[0011]
[ФИГ. 1] На ФИГ. 1 показан график спектра порошковой рентгеновской дифракции кристаллического вещества формы-III. Вертикальная ось представляет интенсивность пика (cps), и горизонтальная ось представляет угол дифракции (2θ [°]).
[ФИГ. 2] На ФИГ. 2 показан график ИК-спектра кристаллического вещества формы-III. Вертикальная ось представляет коэффициент пропускания (%), и горизонтальная ось представляет волновое число (см-1).
[ФИГ. 3] На ФИГ. 3 показан график ДСК-измерения, когда температура кристаллического вещества формы-III была увеличена на 10°С в минуту. Вертикальная ось на чертеже представляет экзотермическое количество (мВт) (в случае отрицательного значения это значение представляет эндотермическое количество), и горизонтальная ось представляет температуру (°C).
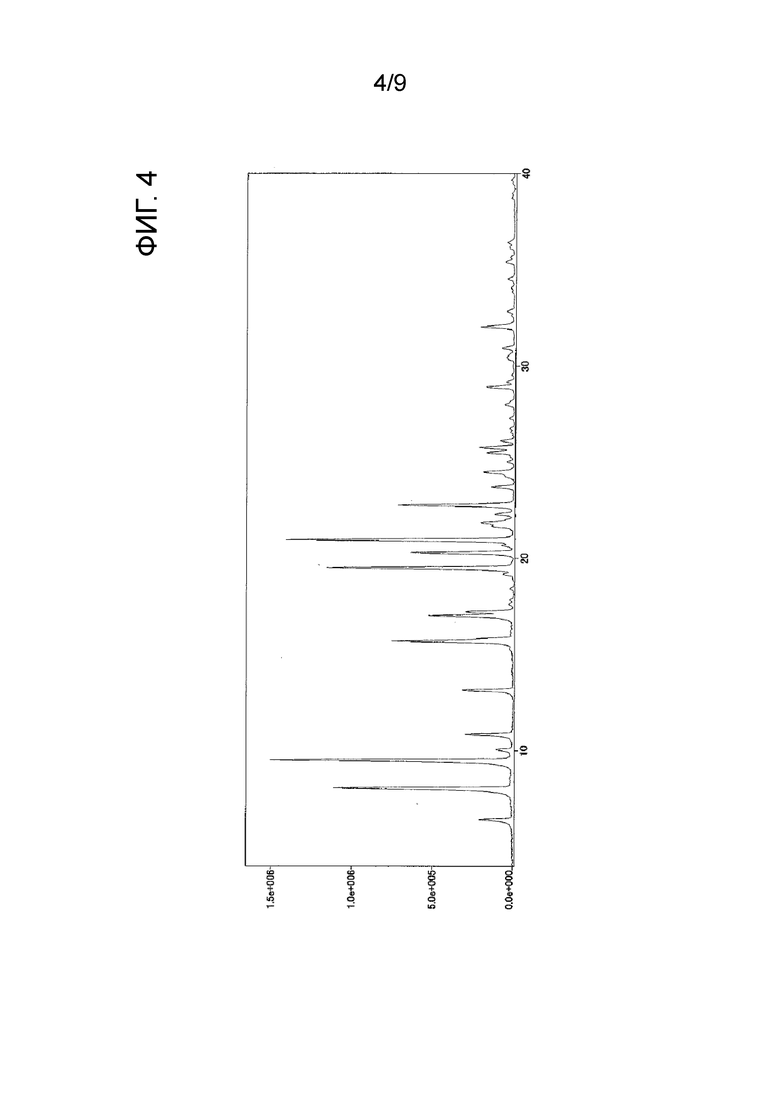
[ФИГ. 4] На ФИГ. 4 показан график спектра порошковой рентгеновской дифракции кристаллического вещества формы-I настоящего изобретения. Вертикальная ось представляет интенсивность пика (cps), и горизонтальная ось представляет угол дифракции (2θ [°]).
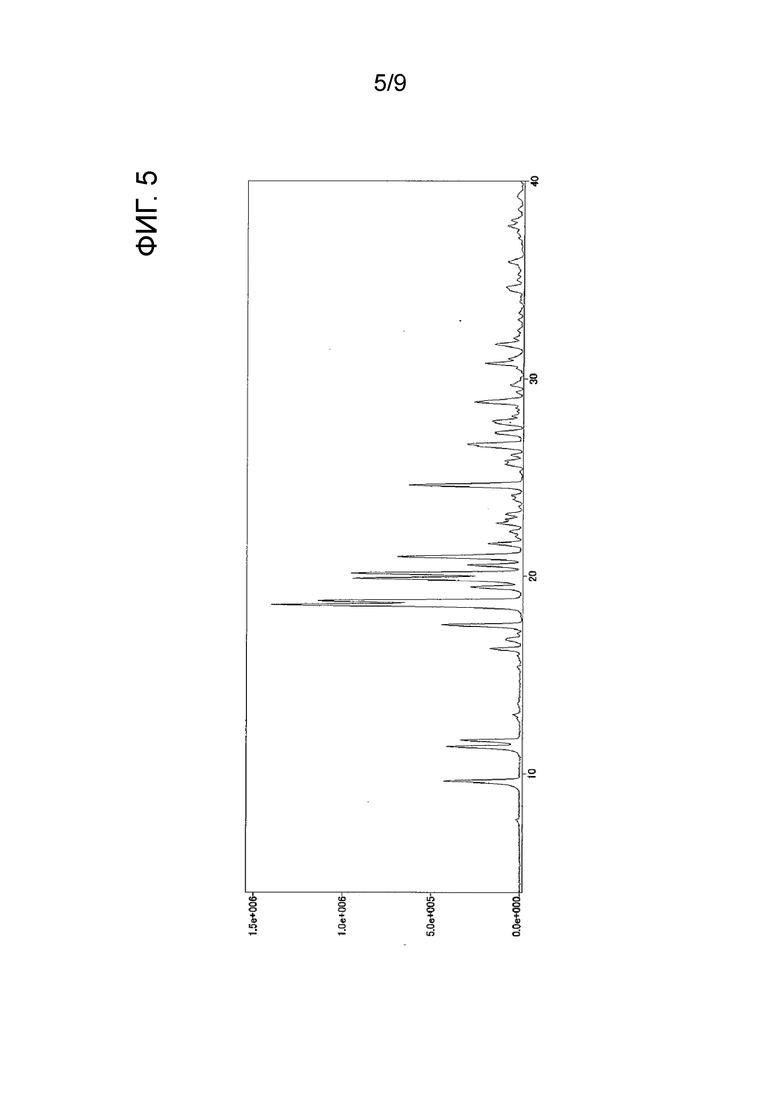
[ФИГ. 5] На ФИГ. 5 показан график спектра порошковой рентгеновской дифракции кристаллического вещества формы-II настоящего изобретения. Вертикальная ось представляет интенсивность пика (cps), и горизонтальная ось представляет угол дифракции (2θ [°]).
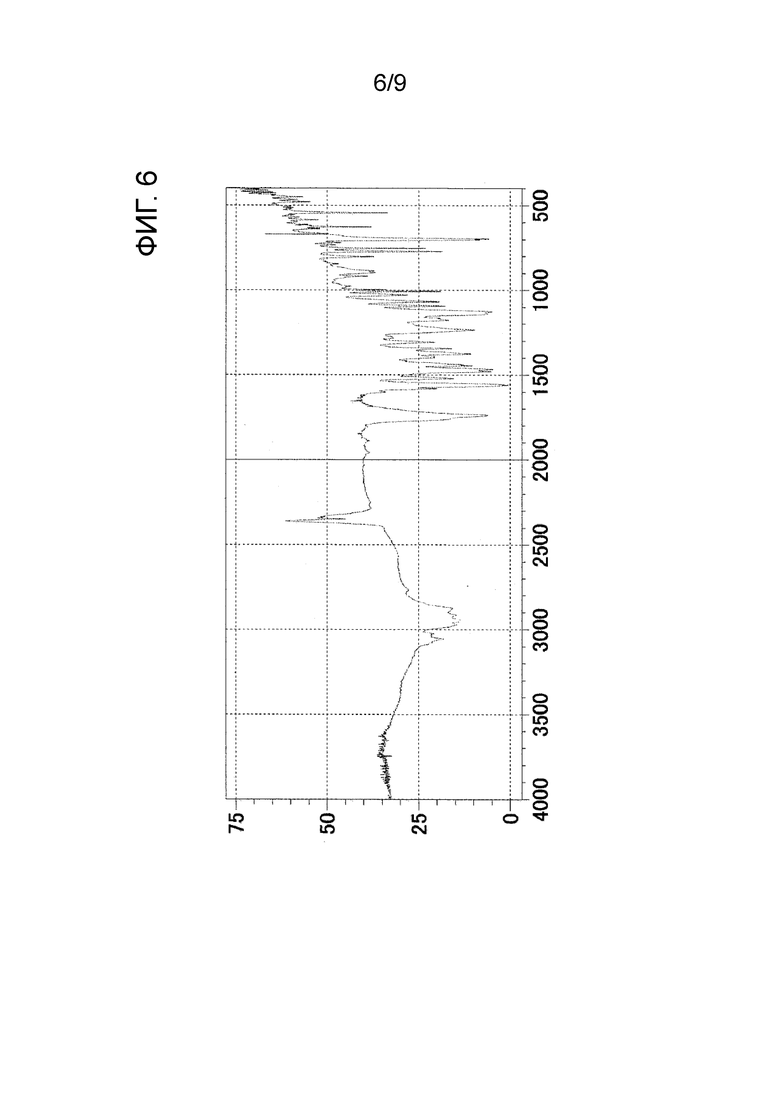
[ФИГ. 6] На ФИГ. 6 показан график ИК-спектра кристаллического вещества формы-I настоящего изобретения. Вертикальная ось представляет коэффициент пропускания (%), и горизонтальная ось представляет волновое число (см-1).
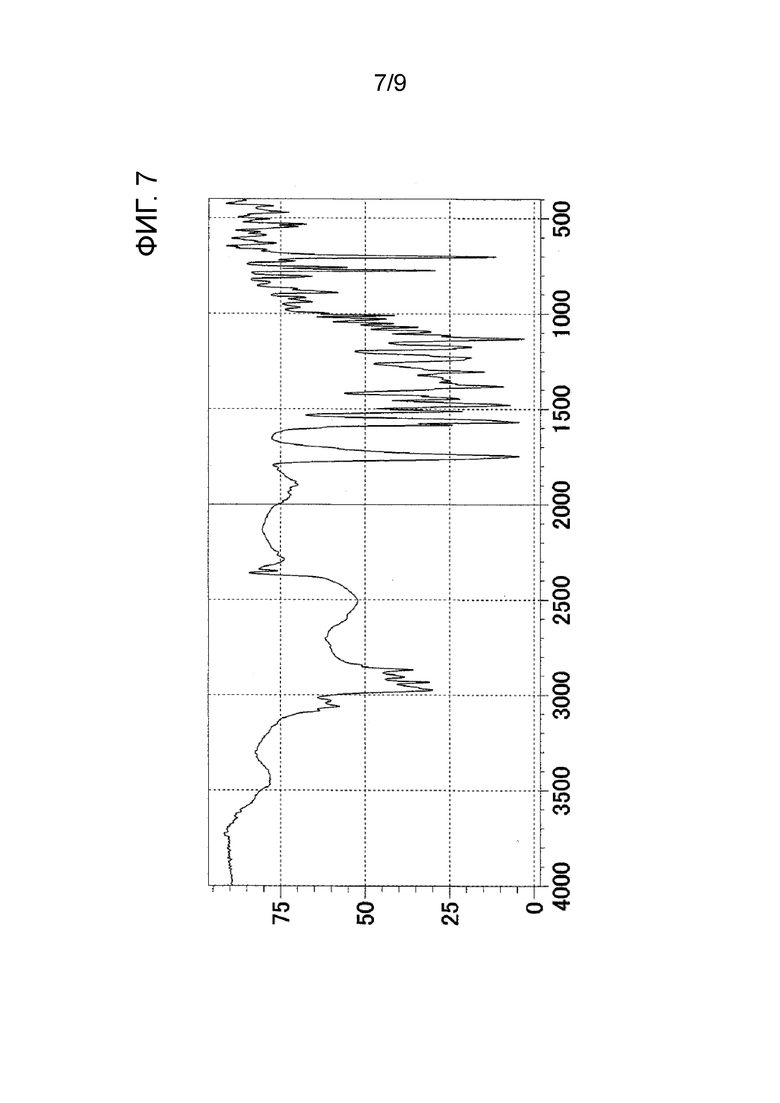
[ФИГ. 7] На ФИГ. 7 показан график ИК-спектра кристаллического вещества формы-II настоящего изобретения. Вертикальная ось представляет коэффициент пропускания (%), и горизонтальная ось представляет волновое число (см-1).
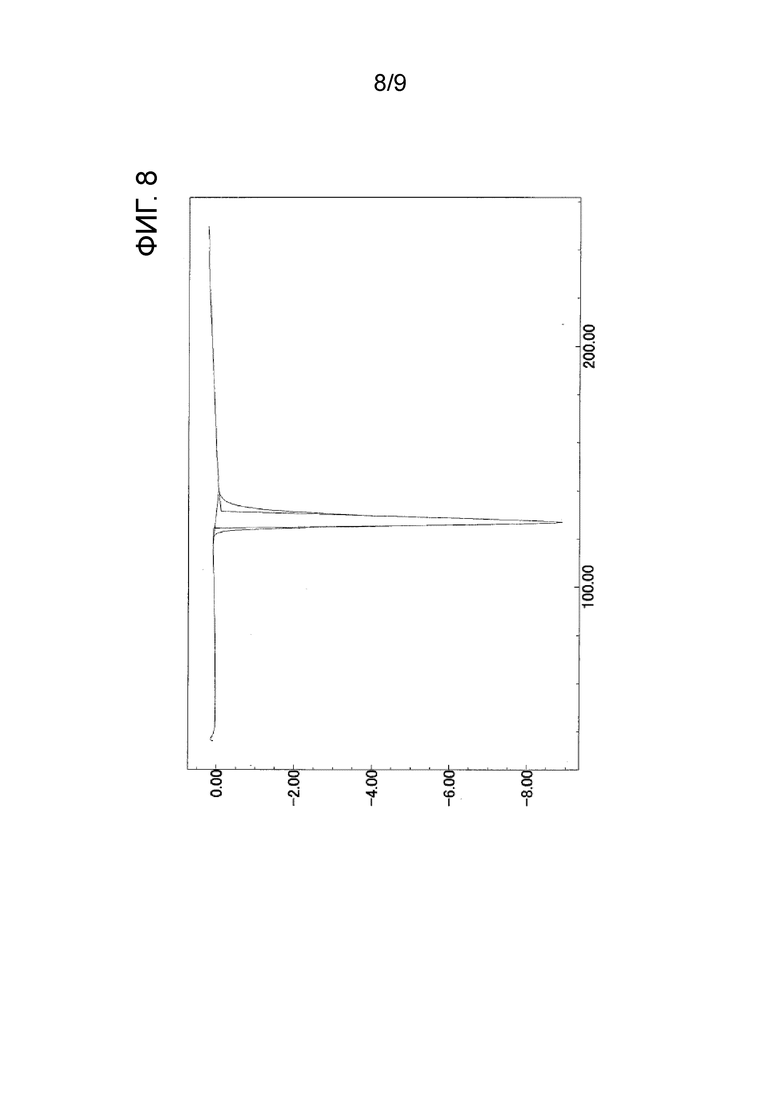
[ФИГ. 8] На ФИГ. 8 показан график ДСК-измерения, когда температура кристаллического вещества формы-I настоящего изобретения была увеличена на 10°С в минуту. Вертикальная ось представляет экзотермическое количество (мВт) в секунду (в случае отрицательного значения это значение представляет эндотермическое количество), и горизонтальная ось представляет температуру (°C).
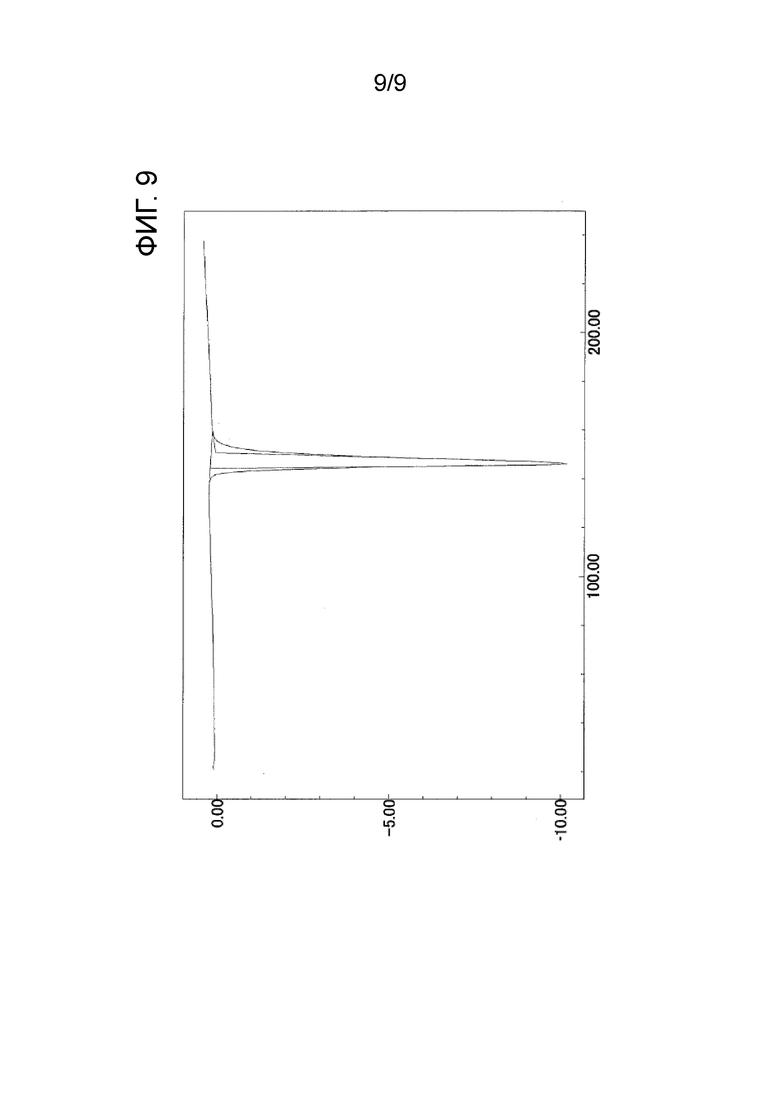
[ФИГ. 9] На ФИГ. 9 показан график ДСК-измерения, когда температура кристаллического вещества формы-II настоящего изобретения была увеличена на 10°С в минуту. Вертикальная ось на чертеже представляет экзотермическое количество (мВт) (в случае отрицательного значения это значение представляет эндотермическое количество), и горизонтальная ось представляет температуру (°C).
[Описание вариантов осуществления]
[0012]
А. Кристаллическое вещество формы-I настоящего изобретения
Кристаллическое вещество формы-I настоящего изобретения характеризуется дифракционными пиками при углах дифракции (2θ) 6,4, 8,1, 9,5, 10,9, 13,2, 15,7, 17,0, 19,5, 20,3, 21,0 и 22,8 в спектре порошковой рентгеновской дифракции, полученном с использованием Cu-Kα-излучения (λ=1,54 Å). Кроме того, предпочтительно характеризуется дифракционными пиками при 15,8, 17,2, 21,9, 23,7, 24,5, 25,5, 25,8, 28,9 и 32,0 в дополнение к вышеупомянутым дифракционным пикам.
Кроме того, кристаллическое вещество формы-I настоящего изобретения характеризуется пиками поглощения при волновых числах 2874 см-1, 1736 см-1, 1558 см-1, 1375 см-1, 1126 см-1 и 696 см-1 в ИК-спектре (метод KBr).
Кроме того, кристаллическое вещество формы-I настоящего изобретения характеризуется наличием эндотермического пика при 127°С в дифференциальной сканирующей калориметрии.
Кристаллическое вещество формы-I настоящего изобретения можно получить, например, способом, описанным в нижеприведенном Примере 1.
[0013]
B. Кристаллическое вещество формы-II настоящего изобретения
Кристаллическое вещество формы-II настоящего изобретения характеризуется дифракционными пиками при углах дифракции (2θ) 9,6, 11,4, 11,7, 16,3, 17,5, 18,5, 18,7, 19,9, 20,1, 21,0 и 24,6 в спектре порошковой рентгеновской дифракции, полученном с использованием Cu-Kα-излучения (λ=1,54 Å). Кроме того, предпочтительно характеризуется дифракционными пиками при 19,4, 20,6, 21,1, 21,7, 22,7, 26,6, 26,7, 28,8 и 30,8 в дополнение к вышеупомянутым дифракционным пикам.
Кроме того, кристаллическое вещество формы-II настоящего изобретения характеризуется пиками поглощения при волновых числах 2867 см-1, 1749 см-1, 1568 см-1, 1382 см-1, 1131 см-1 и 701 см-1 в ИК-спектре (метод KBr).
Кроме того, кристаллическое вещество формы-II настоящего изобретения характеризуется наличием эндотермического пика при 147°С в дифференциальной сканирующей калориметрии.
Кристаллическое вещество формы-II настоящего изобретения может быть получено, например, с помощью способа, описанного в нижеприведенном Примере 2.
[0014]
C. Медицинское применение Фармацевтическая композиция настоящего изобретения
Соединение В в соответствии с настоящим изобретением обладает превосходным агонистическим действием в отношении рецептора PGI2 и проявляет различные лечебные эффекты, такие как ингибирующий эффект агрегации тромбоцитов, сосудорасширяющий эффект, эффект расширения гладкой мускулатуры бронхов, ингибирующий эффект отложения липидов и ингибирующий эффект активации лейкоцитов (см., например, PTL 1).
[0015]
Следовательно, кристаллическое вещество формы-I настоящего изобретения, кристаллическое вещество формы-II настоящего изобретения (далее именуемое совместно «кристаллическое вещество настоящего изобретения») или фармацевтическая композиция настоящего изобретения является пригодной в качестве профилактического средства или терапевтического средства для транзиторной ишемической атаки (ТИА), диабетической невропатии (см., например, NPL 1), диабетической гангрены (см., например, NPL 1), нарушения периферического кровообращения [например, хронической артериальной окклюзии (см. например, NPL 2), перемежающейся хромоты (см., например, NPL 3), периферической эмболии, синдрома вибрации или болезни Рейно] (см., например, NPL 4 и NPL 5), заболевания соединительной ткани [например, системной красной волчанки, склеродермии (см., например, PTL 7 и NPL 6), смешанного заболевания соединительной ткани или васкулитового синдрома], реокклюзии/рестеноза после чрескожной транслюминальной коронарной ангиопластики (ЧТКА), атеросклероза, тромбоза (например, острого церебрального тромбоза или эмболии легких (см., например, NPL 5 и NPL 7), гипертензии, легочной гипертензии, ишемической болезни [например, инфаркта головного мозга или инфаркта миокарда (см., например, NPL 8)], стенокардии (например, стабильной стенокардии или нестабильной стенокардии) (см., например, NPL 9), гломерулонефрита (см., например, NPL 10), диабетической нефропатии (см., например, NPL 1), хронической почечной недостаточности (см., например, PTL 8), аллергии, бронхиальной астмы (см., например, NPL 11), язвы, пролежневой язвы (пролежней), рестеноза после коронарного вмешательства, такого как атерэктомия или имплантация стента, тромбоцитопении путем диализа, заболевания, при котором включен фиброгенез в органе или ткани [например, почечного заболевания {например, тубулоинтерстициального нефрита (см., например, PTL 9)}, респираторного заболевания {например, интерстициальной пневмонии (например, легочного фиброза) (см., например, PTL 9), хронической обструктивной болезни легких (см., например, NPL 12)}, заболевания пищеварительной системы (например, гепатоцирроза, вирусного гепатита, хронического панкреатита или рака желудка), сердечно-сосудистого заболевания (например, фиброза миокарда), заболевания костей или суставов (например, фиброза костного мозга или ревматоидного артрита), кожного заболевания (например, послеоперационного рубца, ожогового рубца, келоидного или гипертрофического рубца), акушерского заболевания (например, миомы матки), заболевания мочевыводящих путей (например, гипертрофии простаты), других заболеваний (например, болезни Альцгеймера, склерозирующего перитонита, диабета I типа и послеоперационной адгезии органов)], эректильной дисфункции (например, диабетической эректильной дисфункции, психогенной эректильной дисфункции, психотической эректильной дисфункции, эректильной дисфункции вследствие хронической почечной недостаточности, эректильной дисфункции после тазовой операции по поводу резекции простаты или сосудистой эректильной дисфункции, связанной со старением или атеросклерозом), воспалительного заболевания кишечника (например, язвенного колита, болезни Крона, кишечного туберкулеза, ишемического колита или кишечной язвы, связанной с болезнью Бехчета) (см., например, PTL 10), гастрита, язвенной болезни желудка, ишемической болезни глаза (например, окклюзии артерии сетчатки, окклюзии вен сетчатки или ишемической оптической нейропатии), внезапной потери слуха, аваскулярного некроза кости, повреждения кишечника, вызванного введением нестероидного противовоспалительного средства (НВПС) (например, диклофенака, мелоксикама, оксапрозина, набуметона, индометацина, ибупрофена, кетопрофена, напроксена или целекоксиба) (особых ограничений нет, если речь идет, например, о повреждении, встречающемся в двенадцатиперстной кишке, тонкой кишке или толстой кишке, однако, например, повреждении слизистой оболочки, такой как эрозия или язва, встречающаяся в двенадцатиперстной кишке, тонкой кишке или толстой кишке), или симптомов (например, паралича, тупости в сенсорном восприятии, боли, онемения или уменьшения способности ходьбы), связанных со стенозом позвоночного канала (например, стенозом шейного позвоночного канала, стенозом грудного позвоночного канала, стенозом поясничного позвоночного канала, сосуществующим шейным и поясничным спинальным стенозом или крестцовым спинальным стенозом) (см. PTL 11).
Кроме того, кристаллическое вещество настоящего изобретения или фармацевтическая композиция настоящего изобретения также является пригодной в качестве ускоряющего агента для генной терапии или ангиогенной терапии, такой как аутологичная трансплантация костного мозга, или ускоряющего агента для ангиогенеза при восстановлении периферической артерии или ангиогенной терапии.
[0016]
Когда кристаллическое вещество настоящего изобретения вводят в виде лекарственного средства, кристаллическое вещество вводят в том виде, в каком он есть, или содержится в фармацевтически приемлемом нетоксичном инертном носителе в количестве в диапазоне, например, от 0,1% до 99,5%, предпочтительно в пределах диапазона от 0,5% до 90%.
Примеры носителя включают твердые, полутвердые или жидкие разбавители, наполнители и другие вспомогательные агенты для фармацевтического состава. Среди них можно использовать один тип, или два, или более типов.
[0017]
Фармацевтическая композиция настоящего изобретения может быть в любой форме препаратов для перорального введения, такой как порошок, капсула, таблетка, таблетка с сахарным покрытием, гранула, порошковый препарат, суспензия, жидкость, сироп, эликсир и пастилка, и парентеральных препаратов, таких как инъекция и суппозиторий в твердой или жидкой единице дозирования. Она может быть в форме препарата с замедленным высвобождением. Среди них, в частности, предпочтительными являются препараты для перорального введения, такие как таблетки.
Порошок может быть получен путем измельчения кристаллического вещества настоящего изобретения до соответствующей степени измельчения.
Порошковый препарат может быть получен путем измельчения кристаллического вещества настоящего изобретения до соответствующей степени измельчения и затем смешивания измельченного кристаллического вещества с аналогично измельченным фармацевтическим носителем, например, пищевым углеводом, таким как крахмал или маннит. В него могут быть произвольно добавлены ароматизатор, консервант, диспергирующее средство, краситель, отдушка или подобное.
Капсула может быть получена путем первоначального заполнения порошка или порошкового препарата, превращенного в порошкообразную форму, как описано выше, или гранулированного материала, как будет описано в разделе, в таблетку, например, в оболочку капсулы, такой как желатиновая капсула. Кроме того, капсула может быть получена путем смешивания скользящего вещества или псевдоожижающего агента, такого как коллоидный диоксид кремния, тальк, стеарат магния, стеарат кальция или твердый полиэтиленгликоль с порошком или порошковым препаратом, превращенным в порошкообразную форму, и после этого выполняют операцию заполнения. Можно улучшить эффективность лекарственного средства, когда капсула принимается, если к нему добавляют дезинтегрирующее средство или солюбилизирующее средство, такое как карбоксиметилцеллюлоза, карбоксиметилцеллюлоза кальция, низкозамещенная гидроксипропилцеллюлоза, кроскармеллоза натрия, карбоксиметилкрахмал натрия, карбонат кальция или карбонат натрия.
Кроме того, также можно сформировать мягкую капсулу путем суспендирования и диспергирования мелкого порошка кристаллического вещества настоящего изобретения в растительном масле, полиэтиленгликоле, глицерине или поверхностно-активном веществе и оборачивания полученного материала желатиновым листом.
Таблетка может быть получена путем добавления эксципиента к порошкообразному кристаллическому веществу настоящего изобретения для приготовления порошковой смеси, гранулирования или шлакования порошковой смеси, а затем добавления к ней дезинтегрирующего средства или скользящего средства с последующим таблетированием.
Порошковая смесь может быть приготовлена путем смешивания подходящего порошкообразного кристаллического вещества настоящего изобретения с разбавителем или основой. При необходимости можно добавить связующее вещество (например, карбоксиметилцеллюлозу натрия, метилцеллюлозу, гидроксипропилметилцеллюлозу, желатин, поливинилпирролидон или поливиниловый спирт), замедляющий растворение агент (например, парафин), реабсорбирующий агент (например, четвертичную соль), адсорбент (например, бентонит или каолин) или подобное.
Гранула может быть получена путем первоначально смачивания порошковой смеси связующим веществом, например, сиропом, крахмальной пастой, аравийской камедью, раствором целлюлозы или раствором полимерного вещества, перемешивания и смешивания влажной смеси, и затем высушивания и дробления смеси. Вместо гранулирования порошка таким способом также можно образовать гранулу, сначала подвергая порошок обработке в таблеточной машине, а затем измельчая шлак, полученный в неполной форме. Добавляя стеариновую кислоту, стеаратную соль, тальк, минеральное масло или подобное в качестве скользящего вещества к полученной таким образом грануле, можно предотвратить прилипание гранул друг к другу.
Кроме того, таблетка также может быть изготовлена путем смешивания кристаллического вещества настоящего изобретения с жидким инертным носителем и последующего непосредственного таблетирования полученной смеси без проведения стадии гранулирования или шлакования, как описано выше.
Полученная таким образом таблетка может быть подвергнута пленочному или сахарному покрытию. Можно также использовать прозрачное или полупрозрачное защитное пленочное покрытие, выполненное из уплотняющего пленочного покрытия шеллака, пленочное покрытие, выполненное из сахара или полимерного материала или полировочное пленочное покрытие, выполненное из воска.
Другой препарат для перорального введения, например, жидкость, сироп, пастилка или эликсир также может быть составлен в единичную дозированную форму, так что его предварительно определенное количество содержит предварительно определенное количество кристаллического вещества настоящего изобретения.
Сироп может быть получен путем растворения кристаллического вещества настоящего изобретения в подходящем водном ароматизированном растворе. Эликсир может быть получен с использованием нетоксичного спиртового носителя.
Суспензия может быть получена путем диспергирования кристаллического вещества настоящего изобретения в нетоксичном носителе. В случае необходимости к нему может быть добавлен солюбилизатор или эмульгатор (например, этоксилированный изостеариловый спирт, сложный эфир полиоксиэтиленсорбита), консервант, придающий вкус агент (например, масло мяты перечной или сахарин) или подобное.
В случае необходимости единичный дозированный состав для перорального введения может быть микроинкапсулирован. Также возможно продлить продолжительность действия или достичь длительного высвобождения путем нанесения покрытия на состав или включения состава в полимер, воск или подобное.
Препарат для парентерального введения может быть в жидкой стандартной лекарственной форме для подкожной, внутримышечной или внутривенной инъекции, например, в форме раствора или суспензии. Препарат для парентерального введения может быть получен путем суспендирования или растворения предварительно определенного количества кристаллического вещества настоящего изобретения в нетоксичном жидком носителе, отвечающем цели инъекции, например, в водной или масляной среде и затем стерилизации суспензии или раствора. Также к нему можно добавить стабилизирующий агент, консервант, эмульгатор или подобное.
Суппозиторий может быть получен путем растворения или суспендирования кристаллического вещества настоящего изобретения в твердом веществе, которое имеет низкую температуру плавления и является растворимым или нерастворимым в воде, например, полиэтиленгликоле, масле какао, полусинтетическом масле или жире [например, Witepsol (зарегистрированный товарный знак)], более сложном эфире (например, сложном эфире миристилпальмитата) или их смеси.
[0018]
Доза варьируется в зависимости от состояния пациента, такого как масса тела или возраст, пути введения, характера и степени тяжести заболевания или подобного, однако доза в виде количества кристаллического вещества настоящего изобретения в день для взрослого соответственно находится в пределах диапазона от 0,001 мг до 100 мг, предпочтительно в пределах диапазона от 0,01 мг до 10 мг.
В некоторых случаях может быть достаточной доза, не превышающая вышеуказанный диапазон или, с другой стороны, может потребоваться доза, не превышающая вышеуказанный диапазон. Кроме того, препарат можно вводить от одного до нескольких раз в день или можно вводить с интервалом от одного до нескольких дней.
[Примеры]
[0019]
Далее настоящее изобретение будет описано более подробно со ссылкой на Примеры и Примеры исследований, однако настоящее изобретение ни в коей мере не ограничивается ими.
[0020]
Спектр порошковой рентгеновской дифракции измеряли с использованием SmartLab (изготовлено Rigaku Corporation) (оптическая система: фокусный метод, напряжение: 45 кВ, ток: 200 мА, длина волны: Cu-Kα, щель Соллера: 5,0, диапазон сканирования: от 4 до 40, скорость сканирования: 47,3/мин, вращение образца: 60/мин).
ИК-спектр измеряли с использованием IR Affinity-1 (изготовлено Shimadzu Corporation) (режим измерения: %пропускания, совокупное количество: 16 раз, разрешение: 2,0, диапазон волновых чисел: от 400 до 4000 см-1).
ДСК измеряли с использованием DSC-50 (изготовлено Shimadzu Corporation) (ячейка: оксид алюминия (открытая), газ: азот (20,0 мл/мин), скорость нагревания: 10,0°С/мин, температура выдерживания: 250°С, время удерживания: 0 мин).
[0021]
Ссылочный пример 1: Получение кристаллического вещества формы-III
После растворения трет-бутил-2-{4-[N-(5,6-дифенилпиразин-2-ил)-N-изопропиламино]бутилокси}ацетата (см., например, PTL 1) (13,15 г) в метаноле (179,7 мл) в него добавляли 1 N водный раствор гидроксида натрия (41,47 мл). После того как полученную смесь нагревали с обратным холодильником в течение 1 часа, растворитель отгоняли при пониженном давлении и к остатку добавляли воду для растворения остатка. После промывания диэтиловым эфиром полученный водный слой нейтрализовали 1 н. хлористоводородной кислотой (44 мл) и проводили экстракцию этилацетатом. Полученный этилацетатный слой высушивали над безводным сульфатом магния, и растворитель отгоняли при пониженном давлении, и затем к остатку добавляли диизопропиловый эфир для осуществления кристаллизации. Полученное кристаллическое вещество отфильтровывали и промывали соответствующим количеством диизопропилового эфира. Кристаллическое вещество высушивали при 40°С при пониженном давлении, в результате чего получали кристаллическое вещество формы-III (9,88 г).
Результаты измерения порошковой рентгеновской дифракции, ИК-измерения и ДСК-измерения кристаллического вещества формы-III показаны на ФИГ. 1, ФИГ. 2 и ФИГ. 3 соответственно.
углы дифракции (2θ): 8,4, 12,6, 13,4, 14,3, 14,6, 15,9, 16,9, 18,0, 18,8, 19,4, 20,3, 20,6, 21,6, 21,7, 22,3, 22,5, 23,3, 23,7, 23,9, 27,0, 29,6 и 30,8
ИК-пик поглощения: 2867 см-1, 1747 см-1, 1558 см-1, 1380 см-1, 1131 см-1 и 701 см-1
Эндотермический пик ДСК: 118°C
[0022]
Ссылочный пример 2: Получение Соединения B
К суспензии 2-{4-[N-(5,6-дифенилпиразин-2-ил)-N-изопропиламино]бутилокси}-N-(метилсульфонил)ацетамида (см., например, PTL 1) (300 г) в изопропиловом спирте (1425 мл) добавляли водный раствор гидроксида натрия (раствор, полученный растворением гидроксида натрия (120,8 г) в воде (570 мл)). После перемешивания при 100°С в течение 11 часов полученную смесь охлаждали до 10°С или ниже. После того как в нее по каплям добавляли концентрированную хлористоводородную кислоту, перемешивание проводили при 10°С или ниже в течение 1 часа, полученный осадок отфильтровывали и промывали соответствующим количеством 50%-ного водного раствора изопропилового спирта, воды и ацетонитрила. Осадок высушивали при 65°С при пониженном давлении, в результате чего получали целевое соединение (208,3 г).
[0023]
Пример 1: Получение кристаллического вещества формы-I настоящего изобретения.
Соединение B (63 г), полученное в Ссылочном примере 2, растворяли в ацетонитриле (315 мл) при 90°C и перемешивание проводили при той же температуре в течение 30 минут. Раствор отфильтровывали, и промывание осуществляли с помощью 5 мл ацетонитрила, и перемешивание с нагреванием проводили снова. В качестве стимуляции к нему добавляли небольшое количество Соединения B, полученного в Ссылочном примере 2, с последующим постепенным охлаждением, и перемешивание проводили при 10°C или ниже в течение 1 часа, и затем кристаллическое вещество отфильтровывали и промывали соответствующим количеством ацетонитрила. Кристаллическое вещество высушивали при 65°C при пониженном давлении, в результате чего получали кристаллическое вещество формы-I настоящего изобретения (59,5 г).
Результаты измерения порошковой рентгеновской дифракции, ИК-измерения и ДСК-измерения в отношении кристаллического вещества формы-I настоящего изобретения показаны на ФИГ. 4, ФИГ. 6 и ФИГ. 8 соответственно.
углы дифракции (2θ): 6,4, 8,1, 9,5, 10,9, 13,2, 15,7, 15,8, 17,0, 17,2, 19,5, 20,3, 21,0, 21,9, 22,8, 23,7, 24,5, 25,5, 25,8, 28,9 и 32,0
ИК-пик поглощения: 2874 см-1, 1736 см-1, 1558 см-1, 1375 см-1, 1126 см-1 и 696 см-1
Эндотермический пик ДСК: 127°C
[0024]
Пример 2: Получение кристаллического вещества формы-II настоящего изобретения
Соединение B (0,5 г), полученное в Ссылочном примере 2, растворяли в изопропиловом спирте (2,5 мл) и 8% водном растворе гидроксида натрия (1,5 мл) при 80°C и перемешивание проводили при той же температуре в течение 30 минут. Раствор постепенно охлаждали до комнатной температуры, и рН раствора доводили до 5-6 с помощью 4 N водного раствора хлористоводородной кислоты при комнатной температуре, и затем перемешивание проводили при 10°С или ниже в течение 1 часа. После этого кристаллическое вещество отфильтровывали и промывали соответствующим количеством воды. Кристаллическое вещество высушивали при 65°C при пониженном давлении, в результате чего получали кристаллическое вещество формы-II настоящего изобретения (0,45 г).
Результаты измерения порошковой рентгеновской дифракции, ИК-измерения и ДСК-измерения кристаллического вещества формы-II настоящего изобретения показаны на ФИГ. 5, ФИГ. 7 и ФИГ. 9 соответственно.
углы дифракции (2θ): 9,6, 11,4, 11,7, 16,3, 17,5, 18,5, 18,7, 19,4, 19,9, 20,1, 20,6, 21,0, 21,1, 21,7, 22,7, 24,6, 26,6, 26,7, 28,8 и 30,8
ИК-пик поглощения: 2867 см-1, 1749 см-1, 1568 см-1, 1382 см-1, 1131 см-1 и 701 см-1
Эндотермический пик ДСК: 147°C
[0025]
Пример исследования 1: Исследование стабильности
Различные кристаллические формы Соединения B помещали в стеклянные бутылки соответственно, и стеклянные бутылки герметично закрывали и хранили при 90°C. Образцы отбирали через 1 день, 5 дней и 14 дней и растворяли в метаноле в концентрации 1 мг/мл для определения родственных веществ с помощью ВЭЖХ. В отношении кристаллических веществ через 14 дней проверяли кристаллическую форму. Результаты показаны в Таблице 1.
[0026]
[Таблица 1]
Из вышеприведенных результатов было выявлено, что в любой из кристаллических форм химическая стабильность является очень высокой, однако форма-I и форма-III постепенно превращаются в форму-II, которая является термодинамически стабильной.
[0027]
Пример исследования 2: Исследование суспендирования растворителя формы-I кристаллического вещества настоящего изобретения в различных растворителях
Кристаллическое вещество формы-I настоящего изобретения смешивали с различными растворителями и перемешивание проводили при комнатной температуре в течение 30 минут. Образовавшиеся кристаллические вещества получали фильтрованием и определяли кристаллические формы. Результаты приведены в Таблице 2.
[0028]
[Таблица 2]
Как описано выше, кристаллическое вещество формы-I настоящего изобретения было частично превращено в кристаллическое вещество формы-II настоящего изобретения при суспендировании во всех растворителях. Из данных результатов было обнаружено, что кристаллическое вещество формы-II настоящего изобретения является термодинамически стабильным при суспендировании в различных растворителях при комнатной температуре.
| название | год | авторы | номер документа |
|---|---|---|---|
| КРИСТАЛЛЫ | 2010 |
|
RU2556206C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ 2-{ 4-[N-(5,6-ДИФЕНИЛПИРАЗИН-2-ИЛ)-N-ИЗОПРОПИЛАМИНО]БУТИЛОКСИ} -N-(МЕТИЛСУЛЬФОНИЛ)АЦЕТАМИД | 2016 |
|
RU2735547C2 |
| НОВАЯ КРИСТАЛЛИЧЕСКАЯ ФОРМА МЕТАНСУЛЬФОНАТ 5-ХЛОР-N-({ (5S)-2-ОКСО-3-[4-(5,6-ДИГИДРО-4H-[1,2,4]ТРИАЗИН-1-ИЛ)ФЕНИЛ]-1,3-ОКСАЗОЛИДИН-5-ИЛ} МЕТИЛ)ТИОФЕН-2-КАРБОКСАМИДА И СОДЕРЖАЩАЯ ЕГО ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2014 |
|
RU2663617C1 |
| ПРОИЗВОДНЫЕ ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ И ЛЕКАРСТВЕННЫЕ СРЕДСТВА | 2002 |
|
RU2283835C2 |
| ПРОИЗВОДНОЕ АЗОЛОБЕНЗОЛА И ЕГО КРИСТАЛЛИЧЕСКАЯ ФОРМА | 2015 |
|
RU2701515C2 |
| Двухстадийный синтез селексипага с применением природосберегающих "зеленых" технологий | 2023 |
|
RU2838750C2 |
| КРИСТАЛЛИЧЕСКАЯ ФОРМА НАТРИЕВОЙ СОЛИ 4-ТРЕТ-БУТИЛ-N-{ 4-ХЛОР-2-(1-ОКСИ-ПИРИДИН-4-КАРБОНИЛ)-ФЕНИЛ} -БЕНЗОЛСУЛЬФОНАМИД | 2012 |
|
RU2607515C2 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ { [1-ЦИАНО-5-(4-ХЛОРОФЕНОКСИ)-4-ГИДРОКСИИЗОХИНОЛИН-3-КАРБОНИЛ]-АМИНО} -УКСУСНОЙ КИСЛОТЫ | 2014 |
|
RU2666144C2 |
| СОЛЬ И ПОЛИМОРФ ФЕНИЛ-ПИРИМИДОНОВОГО СОЕДИНЕНИЯ, ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ПРИМЕНЕНИЕ | 2018 |
|
RU2761213C2 |
| КРИСТАЛЛИЧЕСКАЯ ФОРМА БИСУЛЬФАТНОГО ИНГИБИТОРА JAK-КИНАЗЫ И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2015 |
|
RU2716260C2 |
Изобретение относится к способу получения кристаллической формы-II 2-{4-[N-(5,6-дифенилпиразин-2-ил)-N-изопропиламино]бутилокси}уксусной кислоты (Соединение В). Способ получения включает стадии растворения 2-{4-[N-(5,6-дифенилпиразин-2-ил)-N-изопропиламино]бутилокси}уксусной кислоты в растворе, содержащем смесь изопропилового спирта и водного раствора гидроксида натрия, для получения смешанного раствора; доведения рН полученного смешанного раствора до 5-6, охлаждения полученного смешанного раствора до ниже 10°С до образования кристаллов формы-II 2-{4-[N-(5,6-дифенилпиразин-2-ил)-N-изопропиламино]бутилокси}уксусной кислоты; где кристаллическая форма-II 2-{4-[N-(5,6-дифенилпиразин-2-ил)-N-изопропиламино]бутилокси}уксусной кислоты показывает дифракционные пики при углах дифракции (2θ) 9,6°, 11,4°, 11,7°, 16,3°, 17,5°, 18,5°, 18,7°, 19,9°, 20,1°, 21,0° и 24,6° в спектре порошковой рентгеновской дифракции, полученном с использованием Cu-Kα-излучения (λ=1,54  ), показывает пики поглощения при волновых числах 2867 см-1, 1749 см-1, 1568 см-1, 1382 см-1, 1131 см-1 и 701 см-1 в инфракрасном спектре поглощения, и имеет эндотермический пик при 147°С в дифференциальной сканирующей калориметрии. Также изобретение относится к способу получения кристаллической формы-II 2-{4-[N-(5,6-дифенилпиразин-2-ил)-N-изопропиламино]бутилокси}уксусной кислоты (Соединение В), включающему стадию нагревания кристаллической формы-III 2-{4-[N-(5,6-дифенилпиразин-2-ил)-N-изопропиламино]бутилокси}уксусной кислоты для превращения кристаллической формы-III в кристаллическую форму-II 2-{4-[N-(5,6-дифенилпиразин-2-ил)-N-изопропиламино]бутилокси}уксусной кислоты при температуре 90°С; где кристаллическая форма-III 2-{4-[N-(5,6-дифенилпиразин-2-ил)-N-изопропиламино]бутилокси}уксусной кислоты показывает дифракционные пики при углах дифракции (2θ) 8,4°, 12,6°, 13,4°, 14,3°, 14,6°, 15,9°, 16,9°, 18,0°, 18,8°, и 19,4° в спектре порошковой рентгеновской дифракции, полученном с использованием Cu-Kα-излучения (λ=1,54 ). Технический результат – получение кристаллического вещества соединения B, обладающего физико-химической стабильностью. 4 н. и 9 з.п. ф-лы, 9 ил., 2 табл., 4 пр.
), показывает пики поглощения при волновых числах 2867 см-1, 1749 см-1, 1568 см-1, 1382 см-1, 1131 см-1 и 701 см-1 в инфракрасном спектре поглощения, и имеет эндотермический пик при 147°С в дифференциальной сканирующей калориметрии. Также изобретение относится к способу получения кристаллической формы-II 2-{4-[N-(5,6-дифенилпиразин-2-ил)-N-изопропиламино]бутилокси}уксусной кислоты (Соединение В), включающему стадию нагревания кристаллической формы-III 2-{4-[N-(5,6-дифенилпиразин-2-ил)-N-изопропиламино]бутилокси}уксусной кислоты для превращения кристаллической формы-III в кристаллическую форму-II 2-{4-[N-(5,6-дифенилпиразин-2-ил)-N-изопропиламино]бутилокси}уксусной кислоты при температуре 90°С; где кристаллическая форма-III 2-{4-[N-(5,6-дифенилпиразин-2-ил)-N-изопропиламино]бутилокси}уксусной кислоты показывает дифракционные пики при углах дифракции (2θ) 8,4°, 12,6°, 13,4°, 14,3°, 14,6°, 15,9°, 16,9°, 18,0°, 18,8°, и 19,4° в спектре порошковой рентгеновской дифракции, полученном с использованием Cu-Kα-излучения (λ=1,54 ). Технический результат – получение кристаллического вещества соединения B, обладающего физико-химической стабильностью. 4 н. и 9 з.п. ф-лы, 9 ил., 2 табл., 4 пр.
1. Способ получения кристаллической формы-II 2-{4-[N-(5,6-дифенилпиразин-2-ил)-N-изопропиламино]бутилокси}уксусной кислоты (Соединение В), включающий стадии:
растворения 2-{4-[N-(5,6-дифенилпиразин-2-ил)-N-изопропиламино]бутилокси}уксусной кислоты в растворе, содержащем смесь изопропилового спирта и водного раствора гидроксида натрия, для получения смешанного раствора;
доведения рН полученного смешанного раствора до 5-6,
охлаждения полученного смешанного раствора до ниже 10°С до образования кристаллов формы-II 2-{4-[N-(5,6-дифенилпиразин-2-ил)-N-изопропиламино]бутилокси}уксусной кислоты;
где кристаллическая форма-II 2-{4-[N-(5,6-дифенилпиразин-2-ил)-N-изопропиламино]бутилокси}уксусной кислоты показывает дифракционные пики при углах дифракции (2θ) 9,6°, 11,4°, 11,7°, 16,3°, 17,5°, 18,5°, 18,7°, 19,9°, 20,1°, 21,0° и 24,6° в спектре порошковой рентгеновской дифракции, полученном с использованием Cu-Kα-излучения (λ=1,54  ).
).
2. Способ по п.1, где кристаллическая форма-II 2-{4-[N-(5,6-дифенилпиразин-2-ил)-N-изопропиламино]бутилокси}уксусной кислоты показывает пики поглощения при волновых числах 2867 см-1, 1749 см-1, 1568 см-1, 1382 см-1, 1131 см-1 и 701 см-1 в инфракрасном спектре поглощения.
3. Способ по п.1, где кристаллическая форма-II 2-{4-[N-(5,6-дифенилпиразин-2-ил)-N-изопропиламино]бутилокси}уксусной кислоты имеет эндотермический пик при 147°С в дифференциальной сканирующей калориметрии.
4. Способ по п.2, где кристаллическая форма-II 2-{4-[N-(5,6-дифенилпиразин-2-ил)-N-изопропиламино]бутилокси}уксусной кислоты имеет эндотермический пик при 147°С в дифференциальной сканирующей калориметрии.
5. Способ по любому из пп.1-4, где соединение В, которое использовали на вышеуказанной стадии растворения, получено способом, включающим стадии:
добавления водного раствора гидроксида натрия к суспензии 2-{4-[N-(5,6-дифенилпиразин-2-ил)-N-изопропиламино]бутилокси}-N-(метилсульфонил)ацетамида в изопропиловом спирте,
перемешивания полученной смеси при 100°С и
охлаждения до ниже 10°С с последующим добавлением концентрированной соляной кислоты и дальнейшим перемешиванием при температуре не более 10°С.
6. Способ получения кристаллической формы-II 2-{4-[N-(5,6-дифенилпиразин-2-ил)-N-изопропиламино]бутилокси}уксусной кислоты (Соединение В), включающий стадии:
растворения 2-{4-[N-(5,6-дифенилпиразин-2-ил)-N-изопропиламино]бутилокси}уксусной кислоты в растворе, содержащем смесь изопропилового спирта и водного раствора гидроксида натрия, для получения смешанного раствора;
доведения рН полученного смешанного раствора до 5-6,
охлаждения полученного смешанного раствора до ниже 10°С до образования кристаллов формы-II 2-{4-[N-(5,6-дифенилпиразин-2-ил)-N-изопропиламино]бутилокси}уксусной кислоты;
где кристаллическая форма-II 2-{4-[N-(5,6-дифенилпиразин-2-ил)-N-изопропиламино]бутилокси}уксусной кислоты показывает дифракционные пики при углах дифракции (2θ) 9,6°, 11,4°, 11,7°, 16,3°, 17,5°, 18,5°, 18,7°, 19,9°, 20,1°, 21,0° и 24,6° в спектре порошковой рентгеновской дифракции, полученном с использованием Cu-Kα-излучения (λ=1,54 ), и
где кристаллическая форма-II 2-{4-[N-(5,6-дифенилпиразин-2-ил)-N-изопропиламино]бутилокси}уксусной кислоты показывает пики поглощения при волновых числах 2867 см-1, 1749 см-1, 1568 см-1, 1382 см-1, 1131 см-1 и 701 см-1 в инфракрасном спектре поглощения.
7. Способ по п. 6, где соединение B, которое использовали на вышеуказанной стадии растворения, получено способом, включающим стадии:
добавления водного раствора гидроксида натрия к суспензии 2-{4-[N-(5,6-дифенилпиразин-2-ил)-N-изопропиламино]бутилокси}-N-(метилсульфонил)ацетамида в изопропиловом спирте,
перемешивания полученной смеси при 100°С и
охлаждения до ниже 10°С с последующим добавлением концентрированной соляной кислоты и дальнейшим перемешиванием при температуре не более 10°С.
8. Способ получения кристаллической формы-II 2-{4-[N-(5,6-дифенилпиразин-2-ил)-N-изопропиламино]бутилокси}уксусной кислоты (Соединение В), включающий стадии:
растворения 2-{4-[N-(5,6-дифенилпиразин-2-ил)-N-изопропиламино]бутилокси}уксусной кислоты в растворе, содержащем смесь изопропилового спирта и водного раствора гидроксида натрия, для получения смешанного раствора;
доведения рН полученного смешанного раствора до 5-6,
охлаждения полученного смешанного раствора до ниже 10°С до образования кристаллов формы-II 2-{4-[N-(5,6-дифенилпиразин-2-ил)-N-изопропиламино]бутилокси}уксусной кислоты,
где кристаллическая форма-II 2-{4-[N-(5,6-дифенилпиразин-2-ил)-N-изопропиламино]бутилокси}уксусной кислоты показывает дифракционные пики при углах дифракции (2θ) 9,6°, 11,4°, 11,7°, 16,3°, 17,5°, 18,5°, 18,7°, 19,9°, 20,1°, 21,0° и 24,6° в спектре порошковой рентгеновской дифракции, полученном с использованием Cu-Kα-излучения (λ=1,54 ); и
где кристаллическая форма-II 2-{4-[N-(5,6-дифенилпиразин-2-ил)-N-изопропиламино]бутилокси}уксусной кислоты имеет эндотермический пик при 147°С в дифференциальной сканирующей калориметрии.
9. Способ по п. 8, где соединение B, которое использовали на вышеуказанной стадии растворения, получено способом, включающим стадии:
добавления водного раствора гидроксида натрия к суспензии 2-{4-[N-(5,6-дифенилпиразин-2-ил)-N-изопропиламино]бутилокси}-N-(метилсульфонил)ацетамида в изопропиловом спирте,
перемешивания полученной смеси при 100°С и
охлаждения до ниже 10°С с последующим добавлением концентрированной соляной кислоты и дальнейшим перемешиванием при температуре не более 10°С.
10. Способ получения кристаллической формы-II 2-{4-[N-(5,6-дифенилпиразин-2-ил)-N-изопропиламино]бутилокси}уксусной кислоты (Соединение В), включающий стадию:
нагревания кристаллической формы-III 2-{4-[N-(5,6-дифенилпиразин-2-ил)-N-изопропиламино]бутилокси}уксусной кислоты для превращения кристаллической формы-III в кристаллическую форму-II 2-{4-[N-(5,6-дифенилпиразин-2-ил)-N-изопропиламино]бутилокси}уксусной кислоты при температуре 90°С;
где кристаллическая форма-III 2-{4-[N-(5,6-дифенилпиразин-2-ил)-N-изопропиламино]бутилокси}уксусной кислоты показывает дифракционные пики при углах дифракции (2θ) 8,4°, 12,6°, 13,4°, 14,3°, 14,6°, 15,9°, 16,9°, 18,0°, 18,8°, и 19,4° в спектре порошковой рентгеновской дифракции, полученном с использованием Cu-Kα-излучения (λ=1,54 ); и
где кристаллическая форма-II 2-{4-[N-(5,6-дифенилпиразин-2-ил)-N-изопропиламино]бутилокси}уксусной кислоты показывает дифракционные пики при углах дифракции (2θ) 9,6°, 11,4°, 11,7°, 16,3°, 17,5°, 18,5°, 18,7°, 19,9°, 20,1°, 21,0° и 24,6° в спектре порошковой рентгеновской дифракции, полученном с использованием Cu-Kα-излучения (λ=1,54 ).
11. Способ по п.10, где кристаллическая форма-II 2-{4-[N-(5,6-дифенилпиразин-2-ил)-N-изопропиламино]бутилокси}уксусной кислоты показывает пики поглощения при волновых числах 2867 см-1,
1749 см-1, 1568 см-1, 1382 см-1, 1131 см-1 и 701 см-1 в инфракрасном спектре поглощения.
12. Способ по п.10, где кристаллическая форма-II 2-{4-[N-(5,6-дифенилпиразин-2-ил)-N-изопропиламино]бутилокси}уксусной кислоты имеет эндотермический пик при 147°С в дифференциальной сканирующей калориметрии.
13. Способ по п.11, где кристаллическая форма-II 2-{4-[N-(5,6-дифенилпиразин-2-ил)-N-изопропиламино]бутилокси}уксусной кислоты имеет эндотермический пик при 147°С в дифференциальной сканирующей калориметрии.
| ПРОИЗВОДНЫЕ ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ И ЛЕКАРСТВЕННЫЕ СРЕДСТВА | 2002 |
|
RU2283835C2 |
| Топчак-трактор для канатной вспашки | 1923 |
|
SU2002A1 |
| MINO R.CAIRA, Crystalline polymorphism of organic compounds, TOPICS IN CURRENT CHEMISTRY, Springer Verlag Berlin Heidelberg, 1998, V.198, p.163-208 (doi: 10.1007/3-540-69178-2_5) | |||
| L.Morissette et al.: "High-through put crystallization: polymorphs, salts, co-crystals and solvates of | |||

