Ссылка на родственные заявки
В данной заявке заявлен приоритет предварительной заявки US 61/047464 под названием «ПЕПТИДИЛ-ДИАЦИЛГЛИЦЕРИДЫ», поданной 24 апреля 2008, описание которой включено посредством ссылки в данное описание изобретения.
Предшествующий уровень техники
Биологическая роль диацилглицеринов хорошо описана в литературе. Например, хорошо известно, что диацилглицерины участвуют в транспорте липидов в виде триглицеридов и в ассоциации с растворимыми белками, такими как аполипопротеины, являются переносчиками холестерина. Также известно, что диацилглицерины обладают функцией внутриклеточной передачи сигналов. Внутриклеточный мембраносвязанный фосфатидил-инозитол-4,5-бифосфат расщепляется под действием фермента фосфолипазы C с высвобождением двух внутриклеточных молекул-мессенджеров, инозитолтрифосфата и мембраносвязанного диацилглицерина (конкретно 1-стеароил-2-арахидоноил-глицерина). Диацилглицерин активирует протеинкиназу С, которая активирует транскрипционный фактор NFkB для активирования экспрессии гена различных цитокинов и хемокинов. Пептидил-2,3-диацилглицерид или PDAG вовлечен в иммунную функцию. Более конкретно, PDAG играет роль в стимулировании иммунного ответа. Заявка US 11/459772 (публикация US 2007/0197436) и другие публикации, используемые в данном описании изобретения для освещения предшествующего уровня техники или предоставления дополнительных подробностей, касающихся биологических механизмов, включены посредством ссылки.
Краткое изложение сущности изобретения:
Воплощения изобретения, описанные в данном описании изобретения, включают соединения, которые могут стимулировать иммунный ответ, включающие иммунореактивный пептид из от примерно 5 до примерно 25 аминокислот, ковалентно связанных с липидом. В некоторых воплощениях липид может представлять собой глицерид общей формулы (I):
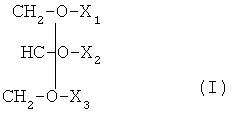
где X1, X2 и Х3 выбраны из водорода, жирной кислоты C2-C25 и пептида, и по меньшей мере один из X1, X2 и Х3 представляет собой пептид. Жирная кислота из воплощений может представлять собой насыщенные, ненасыщенные или полиненасыщенные жирные кислоты, и пептид может быть ковалентно связан с липидом сложноэфирной связью на С-конце пептида.
В некоторых воплощениях иммунореактивный пептид представляет собой аминокислотную последовательность XLYDKGYTSKEQKDCVGIX или XLYDKGYTPKEQKDCVGIX или их инверсии или миметики, где Х отсутствует или представляет собой природную аминокислоту или ее миметик, дериватизированную аминокислоту или неаминокислотную простетическую группу.
Соединения согласно воплощениям изобретения дополнительно могут содержать фармацевтически приемлемый эксципиент и могут быть предложены в форме стандартной дозы, которая соответствует эффективному количеству соединения, и такие воплощения можно считать фармацевтическими композициями.
Другие воплощения изобретения включают способы, которые могут включать введение эффективного количества агента, образованного пептидом, имеющим от примерно 5 до примерно 25 аминокислот, ковалентно связанным с липидом, субъекту, нуждающемуся в этом, и стимулирование иммунного ответа. Пептид согласно воплощениям изобретения может включать пептид или пептидная группировка с аминокислотной последовательностью XLYDKGYTSKEQKDCVGIX или XLYDKGYTPKEQKDCVGIX или ее инверсии или миметики, где Х отсутствует или представляет собой природную аминокислоту или ее миметики, дериватизированную аминокислоту или неаминокислотную простетическую группу.
Агенты согласно воплощениям изобретения могут включать глицерид общей формулы (I):
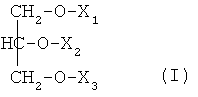
где X1, X2 и Х3 выбраны из водорода, жирной кислоты C2-C25 и пептида, и по меньшей мере один из X1, X2 и Х3 представляет собой пептид. Жирная кислота согласно воплощениям может представлять собой насыщенные, ненасыщенные или полиненасыщенные жирные кислоты. Пептид может быть ковалентно связан с липидом сложноэфирной связью на С-конце пептида. Агент может быть введен для стимулирования врожденного иммунного ответа, и можно стимулировать резидентные иммунные клетки тканей, включая, но без ограничения ими, γδ Т-клетки, моноциты, NK-клетки (естественные клетки-киллеры), нейтрофилы, CD5+ В-клетки и их комбинации. В некоторых воплощениях резидентные иммунные клетки тканей стимулируются при контакте с пептидом.
Агенты согласно воплощениям изобретения могут включать глицерид общей формулы (I), где агент может быть введен для стимулирования врожденного иммунного ответа, и можно стимулировать неиммунные клетки, включая, но без ограничения ими, эпителиальные клетки, эндотелиальные клетки, фибробласты, кератиноциты, гепатоциты и их комбинации. В некоторых воплощениях тканевые клетки стимулируются при контакте с пептидом.
В некоторых воплощениях изобретения стимулированный иммунный ответ может включать стимулирование иммунного ответа в течение по меньшей мере 1-3 суток после введения агента, и в некоторых других воплощениях иммунный ответ можно стимулировать в течение недели или более.
В дополнительных воплощениях изобретения агент может осаждаться в жировой ткани субъекта, и агент может быть введен любым способом, включая энтеральную, парентеральную и местную доставку, инфузию в молочную железу и их комбинации. Парентеральное введение может включать, без ограничения им, внутрисуставное, внутрисиновиальное, интратекальное, внутриартериальное, внутривенное, внутримышечное, подкожное и их комбинации. Энтеральное введение может включать, без ограничения им, внутриротовое, пероральное, ректальное и их комбинации, и может включать местное введение, без ограничения ей, интраназальную, в дыхательные пути, накожную, трансдермальную доставку и их комбинации.
В некоторых воплощениях изобретения агент можно вводить до воздействия образующего заболевание агента, и он может в таких воплощениях по существу предупреждать начало или развитие заболевания. В других воплощениях агент можно вводить до заболевания. Агент также можно доставлять после воздействия образующего заболевание агента, и он может в таких воплощениях по существу подавлять развитие заболевания. В дополнительных воплощениях агент может быть доставлен субъекту после начала заболевания для уменьшения тяжести заболевания или реверсии течения заболевания.
Дополнительные способы согласно воплощениям изобретения включают способы лечения инфекции, способы стимулирования иммунного ответа, способы предупреждения заболевания, способы предупреждения инфекции и их комбинации, такие как способы лечения заболевания и предупреждения вторичной инфекции с использованием агентов и композиций по изобретению, описанных выше.
Воплощения изобретения дополнительно включают антитела к соединениям по изобретению и способы лечения воспаления, такого как, но без ограничения ими, системное воспаление, хроническое воспалительное заболевание и воспаление вследствие сепсиса, несептическая рана, травма, операция или их комбинации, путем введения антител согласно воплощениям изобретения. Антитела согласно воплощениям изобретения можно вводить субъекту любым способом, известным в данной области техники, включая, но без ограничения ими, энтеральную, парентеральную и местную доставку.
Антитела согласно воплощениям изобретения также могут быть связаны с селектируемым маркером, таким как, но без ограничения им, флуоресцентный маркер, такой как белок, или квантовой точкой для создания зонда, и способы использования таких зондов для определения и идентификации пептидов в образце, таком как биологический, клеточный или тканевый образец и клетки или белки, которые взаимодействуют с такими пептидами.
Описание графических материалов
Для более полного понимания сущности и преимуществ настоящего изобретения следует сделать ссылку на следующее подробное описание вместе с прилагаемыми графическими материалами, в которых:
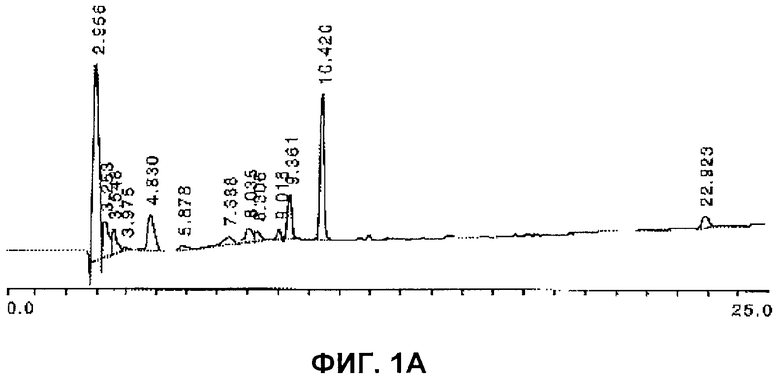
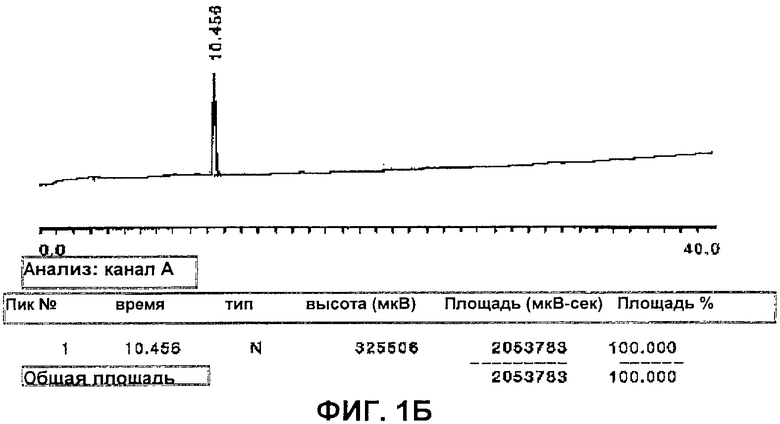
На Фиг.1 представлена типичная ВЭЖХ хроматограмма сывороточной фракции, содержащей PDAG (Фиг.1А) и очищенный 1-пептидил-2,3-диацилглицерид по настоящему изобретению (Фиг.1Б).
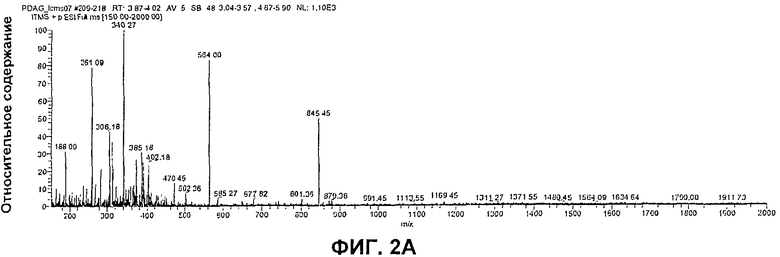
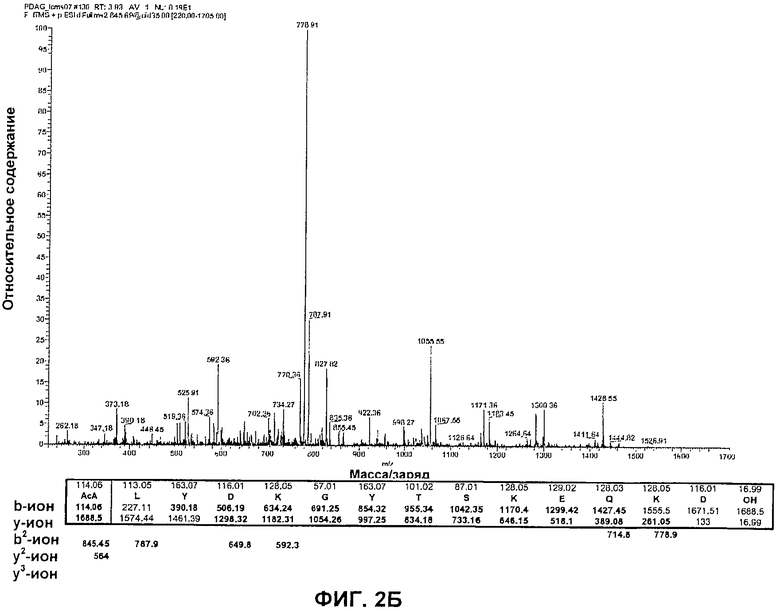
На Фиг.2 представлен масс-спектр с ESI (ионизация посредством электрораспыления) фрагмента из PDAG козьего происхождения (Фиг.2А), и масс-спектр MS/MS многозарядных видов ионов (m/z 845,45), подтверждающий аминокислотную последовательность по настоящему изобретению (Фиг.2 В).
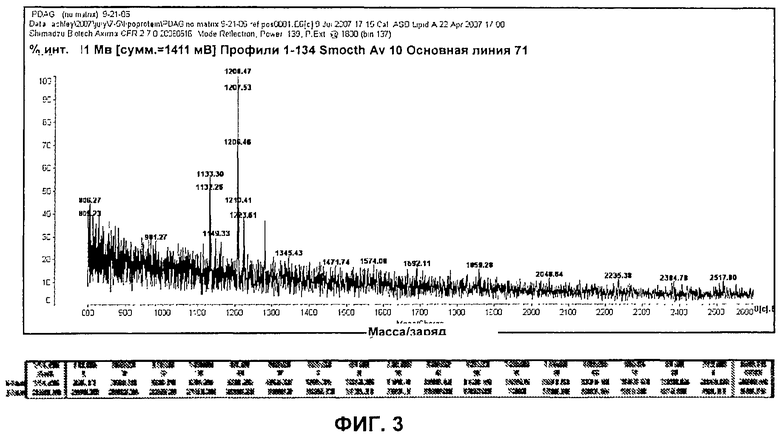
На Фиг.3 представлен PSD (деструкция после источника) MALDI-TOF (времяпролетная масс-спектрометрия с ионизацией методом лазерной десорбции из матрицы) масс-спектр очищенного PDAG в матрице из дигидроксибензойной кислоты.
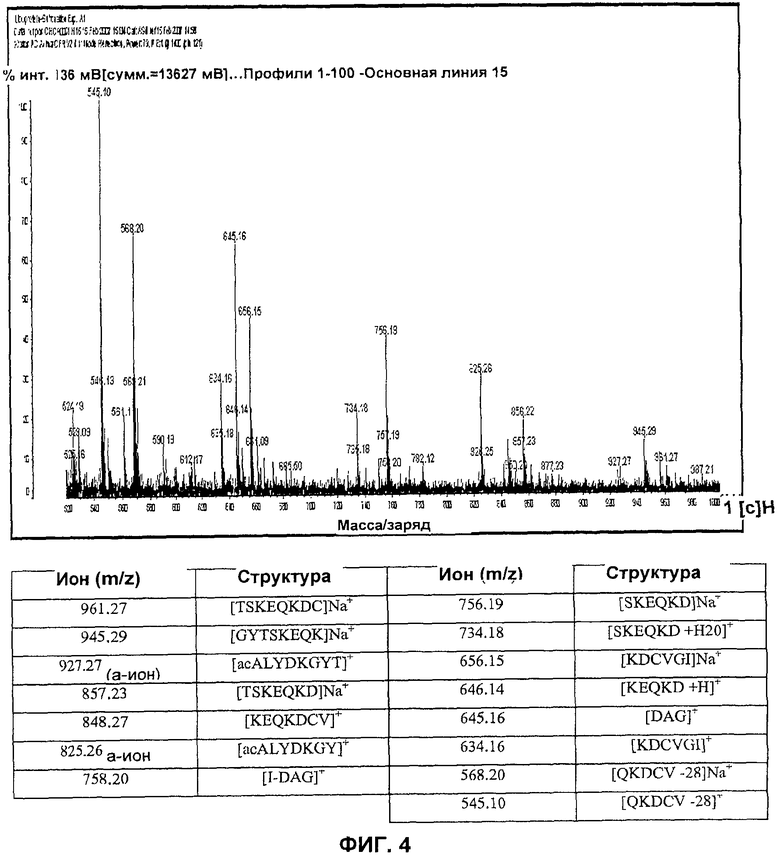
На Фиг.4 представлен PSD MALDI-TOF масс-спектр продуктов гидролиза PDAG после мягкого кислотного гидролиза. В таблице показаны распределения основных ионов для главных пептидных фрагментов, составляющих целую молекулу PDAG в настоящем изобретении.
На Фиг.5 показано стимулирование экспрессии мРНК IL-6 и белка IL-6 в фибробластах, которые инкубировали с PDAG.
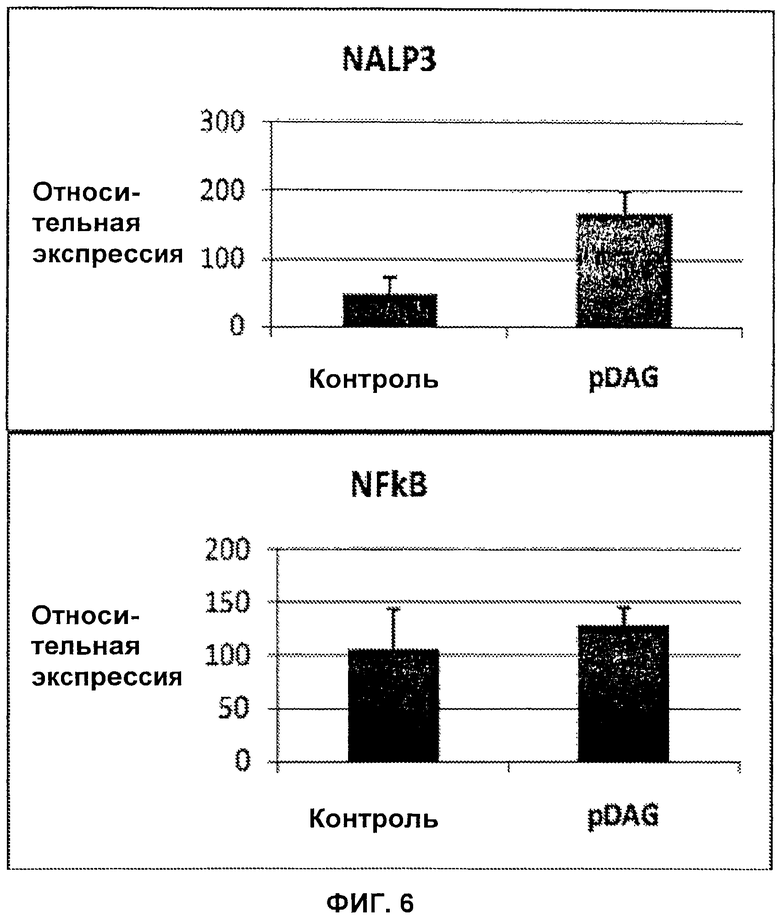
На Фиг.6 показано, что PDAG стимулирует экспрессию мРНК NALP3 (NACHT-, LRR- (богатый лейцином повтор) и PYD (пиридиновый домен)-содержащий белок 3) в фибробластах, но не NFkB.
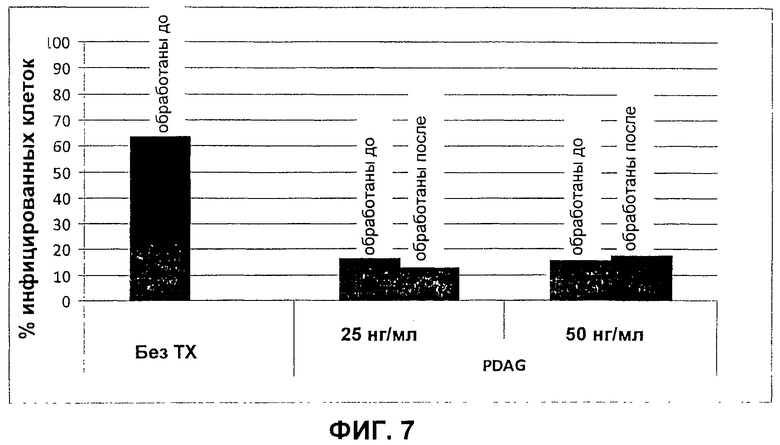
На Фиг.7 показано, что и PDAG, и PDAG-пептид (25 нг/мл и 50 нг/мл) индуцируют выведение Chlamydia pneumoniae из инфицированных моноцитов.
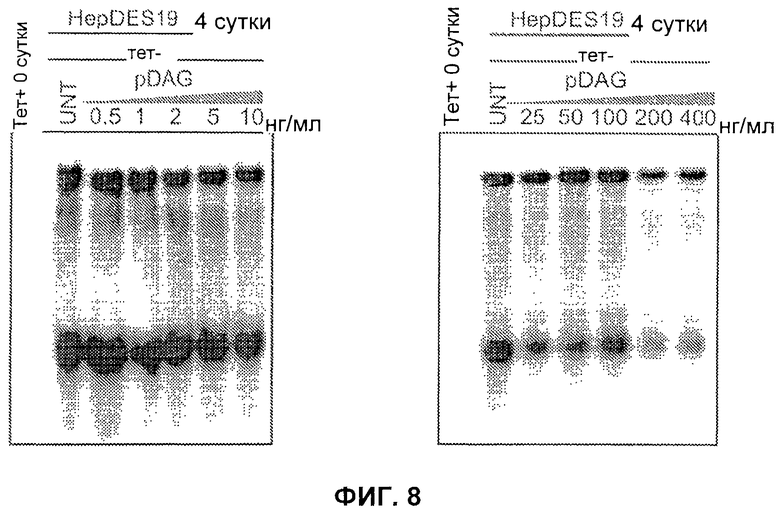
На Фиг.8 показано, что PDAG подавляет репликацию вируса гепатита B в клетках HepDES 19.
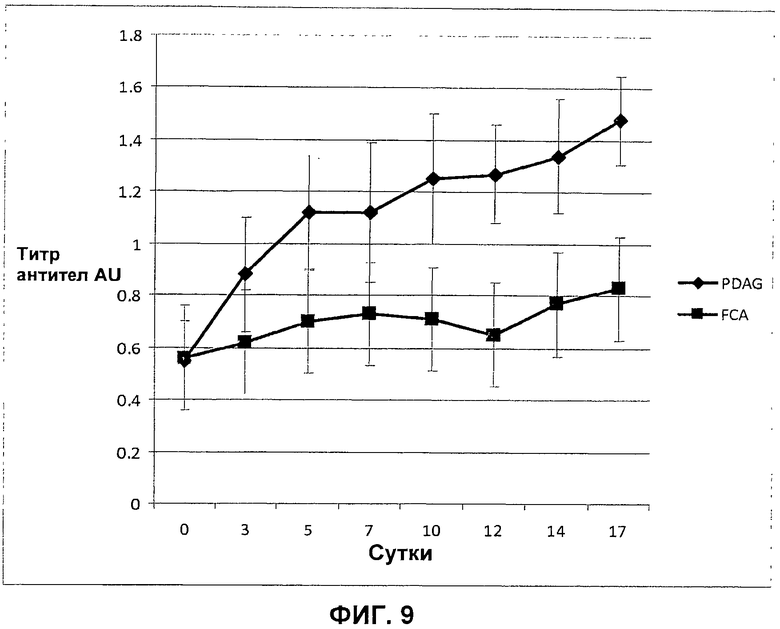
На Фиг.9 показано, что PDAG индуцирует двукратное увеличение продуцирования антиген-специфического IgM у кроликов, привитых убитой нагреванием М.tuberculosis (FCA).
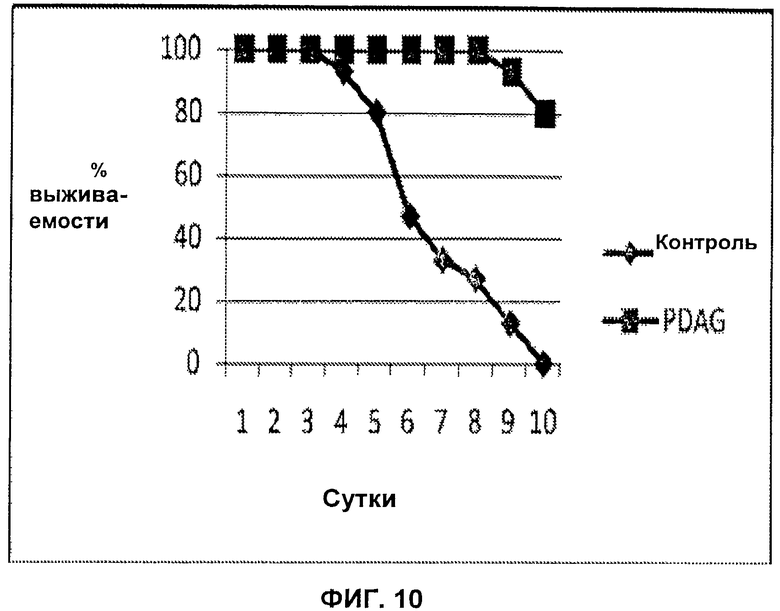
На Фиг.10 показано, что введение PDAG значительно задерживает развитие заболевания у мышей, инфицированных летальной дозой Salmonella typhimurium.
Подробное описание изобретения
Также следует отметить, что при использовании в данном описании изобретения и в прилагаемой формуле изобретения формы единственного числа охватывают множественную ссылку, если контекст ясно не указывает иное. Таким образом, например, ссылка на «клетку» является ссылкой на одну или более чем одну клетку и ее эквиваленты, известные специалисту в данной области техники, и так далее. Если не определено иное, все технические и научные термины, используемые в данном описании изобретения, имеют такие же значения, которые обычно подразумеваются специалистом в данной области техники. Хотя в практическом воплощении или испытании воплощений настоящего изобретения можно использовать любые способы и вещества, аналогичные или эквивалентные описанным в данном описании изобретения, здесь описаны предпочтительные способы, устройства и вещества. Все публикации, указанные в данном описании изобретения, включены посредством ссылки. Ничего в данном описании изобретения не следует истолковывать как признание того, что изобретение не имеет права относить такое описание к более ранней дате на основании предшествующего изобретения.
«Адъювант» относится к любому веществу, которое усиливает иммуностимулирующие свойства антигена или фармакологический эффект лекарственного средства.
При использовании в данном описании изобретения термин «примерно» означает плюс или минус 10% от численного значения, с которыми его используют. Таким образом, примерно 50% означает диапазон 45-55%.
Термины «миметик», «пептидные миметики» и «пептидомиметик» используют взаимозаменяемо в данном описании изобретения, и они в основном относят к пептиду, частичному пептиду или непептидной молекуле, которые имитируют третичную связывающую структуру или активность выбранного нативного пептида или функциональный домен белка (например связывающий мотив или активный сайт). Такие пептидные миметики включают рекомбинантно или химически модифицированные пептиды, а также непептидные агенты, такие как миметики низкомолекулярных лекарственных средств, как дополнительно описано ниже.
При использовании в данном описании изобретения термин «фармацевтически приемлемые соли, сложные эфиры, амиды и пролекарства» относится к тем карбоксилатным солям, солям присоединения аминокислот, сложным эфирам, амидам и пролекарствам соединений по настоящему изобретению, которые являются, в рамках обоснованного медицинского суждения, пригодными для использования в контакте с тканями пациентов без нежелательной токсичности, раздражения, аллергической реакции и тому подобного, в соответствии с разумным соотношением польза /риск, и эффективными для их предусмотренного использования, а также, где это возможно, к цвиттер-ионным формам соединений по изобретению.
При использовании в данном описании изобретения термин «физиологически приемлемый» и его грамматические варианты в отношении к композициям, носителям, разбавителям и реагентам, используют взаимозаменяемо, и он означает, что вещества можно вводить млекопитающему без или с минимальным образованием нежелательных физиологических эффектов, таких как тошнота, головокружение, сыпь или расстройство желудка. В предпочтительном воплощении терапевтическая композиция не является антигенной при введении пациенту-человеку или другому животному для терапевтических целей.
«Обеспечение» при использовании вместе с терапевтическими средствами означает введение терапевтического средства непосредственно в ткань-мишень или на нее, или введение терапевтического средства пациенту, посредством чего терапевтическое средство положительно воздействует на ткань, на которую оно нацелено.
Использование в данном описании изобретения терминов «субъект» или «пациент» относится к животному или млекопитающему, включая, без ограничения ими, человека, собаку, кошку, лошадь, корову, свинью, овцу, козу, цыпленка, обезьяну, кролика, крысу, мышь и так далее.
«Заболевание» для целей настоящего изобретения может представлять собой любой инфекционный агент, такой как, например, вирусные частицы, бактериальные патогены и тому подобное. «Заболевший» при использовании применительно к «заболевшему субъекту» может относиться к любому человеческому или животному субъекту, инфицированному инфекционным агентом. «Заболевший субъект» может проявлять или может не проявлять признаки инфекции, такие как, например, известные симптомы.
Использование в данном описании изобретения термина «образец» включает биологический образец, который можно испытывать способами по настоящему изобретению, и включает, без ограничения ими, биологические жидкости, такие как сыворотка, плазма, цельная кровь, спинномозговая жидкость, лимфатические жидкости, различные внешние секреции (моча, секреции дыхательных путей, желудочно-кишечного или мочеполового тракта, слезы и так далее)и так далее.
При использовании в данном описании изобретения термин «терапевтическое средство» означает агента, используемого для лечения, противодействия, улучшения, предупреждения или оздоровления нежелательного состояния или заболевания пациента. Воплощения настоящего изобретения направлены на стимулирование врожденного иммунного ответа или модулирование воспалительного ответа. В данном описании изобретения способы использования предполагают профилактическое применение, а также куративное применение в терапии имеющегося состояния.
Термины «терапевтически эффективный» или «эффективный» при использовании в данном описании изобретения можно использовать взаимозаменяемо и они относятся к количеству терапевтических композиционных воплощений настоящего изобретения, например к одному или более пептидил-диацилглицеридам или их миметикам. Например, терапевтически эффективное количество композиции, содержащей 1-пептидил-2,3-диацилглицерид или его миметики, представляет собой заранее определенное количество, рассчитанное для достижения желаемого эффекта, то есть для эффективного стимулирования врожденного иммунного ответа у животного, которому вводят эту композицию.
Термин «стандартная доза» при использовании в применении к терапевтической композиции по настоящему изобретению относится к физически дискретным единицам, пригодным в качестве однократной дозировки для субъекта, где каждая единица содержит заранее определенное количество активного вещества, рассчитанное для создания желаемого терапевтического эффекта вместе с нужным разбавителем, то есть эксципиентом, наполнителем или носителем.
Одно воплощение настоящего изобретения может быть направлено на 1-пептидил-2,3-диацилглицерид или PDAG. Другие воплощения изобретения могут включать композиции, содержащие PDAG, композиции, которые содержат участки PDAG, композиции, содержащие аналоги PDAG-пептидов, и композиции, содержащие пептидные миметики PDAG.
В воплощениях изобретения PDAG, участки PDAG, аналоги PDAG и миметики PDAG могут быть предложены субъекту и могут стимулировать терапевтические эффекты, такие как, без ограничения ими, индуцирование иммунного ответа, и в некоторых воплощениях иммунный ответ может представлять собой врожденный иммунный ответ у субъекта, которому они предоставлены. В других воплощениях изобретения PDAG, участки PDAG, аналоги PDAG и миметики PDAG можно вводить субъекту, проходящему лечение заболевания, субъекту, который здоров, или субъекту, который здоров и может подвергаться воздействию заболевания, образующих заболевание агентов или заболевших людей и/или животных. В воплощениях изобретения, когда PDAG, участки PDAG, аналоги PDAG и миметики PDAG, и терапевтические средства, содержащие PDAG, участки PDAG, аналоги PDAG и миметики PDAG, предоставлены субъекту, который здоров, PDAG, участки PDAG, аналоги PDAG и миметики PDAG могут способствовать профилактической активации иммунной системы субъекта, и в некоторых воплощениях профилактической активации врожденного иммунитета.
Не желая быть связанными теорией, PDAG, участки PDAG, аналоги PDAG и миметики PDAG в воплощениях изобретения при предоставлении субъекту могут активировать резидентные иммунные клетки тканей субъекта. В некоторых воплощениях PDAG, участки PDAG, аналоги PDAG и миметики PDAG могут инициировать иммунный ответ, а в других воплощениях иммунный ответ может представлять собой врожденный иммунный ответ.
Воплощения настоящего изобретения также могут включать способы введения PDAG, участков PDAG, аналогов PDAG и миметиков PDAG и терапевтических средств, содержащих PDAG, участки PDAG, аналоги PDAG и миметики PDAG, такие как, без ограничения ими, парентеральное, энтеральное или местное введение.
Другие воплощения настоящего изобретения включают антитела со специфичностью к PDAG и способы использования таких антител в обеднении системных или локальных концентраций PDAG у субъекта. В некоторых воплощениях субъект, обеспеченный антителами, специфическими к PDAG, может проявлять симптомы иммунного заболевание, такого как, без ограничения ими, системное воспаление, хронические воспалительные заболевания, атеросклеротическое заболевание, ревматоидные заболевания, аутоиммунные заболевания и тому подобное.
Другие воплощения изобретения включают флуоресцентно меченые PDAG, аналоги PDAG и флуоресцентно меченые антитела или фрагменты антител со специфичностью к PDAG, и способы создания таких флуоресцентно меченых PDAG, аналогов PDAG и антител. В воплощениях изобретения флуоресцентно меченые PDAG, аналоги PDAG и антитела можно использовать в качестве диагностических средств для оценки аспектов иммунной системы и иммунопатологии как in vitro, так и in vivo, и в некоторых воплощениях субъекты могут включать, без ограничения ими, субъекта, проявляющего симптомы, соответствующие хроническому воспалительному заболеванию, аутоиммунному заболеванию, атеросклеротическому заболеванию, диабету и тому подобные.
PDAG, описанные в различных воплощениях изобретения, могут включать пептидил-диацилглицерид, имеющий по меньшей мере одну пептидную группировку, ковалентно присоединенный к липидной группировке, и имеют общую формулу 1-пептидил-2,3-диацилглицерид. В некоторых воплощениях липидный фрагмент представляет собой 1-стеароил-2-арахидоноил-глицерин.
PDAG, описанные в воплощениях изобретения, могут быть представлены общей формулой (I):
где X1, X2, и Х3 могут представлять собой водород, пептид, пептидный миметик, или аналог пептида, или насыщенную, ненасыщенную или полиненасыщенную жирную кислоту, имеющую от примерно 1 до примерно 20 атомов углерода. В некоторых воплощениях изобретения по меньшей мере один из X1, X2 и Х3 может представлять собой пептид, пептидный миметик или аналог пептида, а в других более чем один из X1, X2, и Х3 могут представлять собой пептид, пептидный миметик, аналог пептида.
Пептидная группировка согласно воплощениям настоящего изобретения может быть составлен из от примерно 5 до примерно 25 аминокислот и может включать одну или более из природных, неприродных и химически модифицированных аминокислот. Такие пептидные фрагменты могут иметь массу от примерно 1000 до примерно 3000 а.е.м. (атомных единиц массы). Пептиды, охваченные настоящим изобретением, или их миметики могут включать любую аминокислотную последовательность, которая дает желаемый эффект активации иммунитета. Пептидные фрагменты можно создавать и/или выделять до присоединения к липидной группировке и можно синтезировать и выделять из природного источника, такого как, например, человеческий, животный, бактериальный источники и тому подобное, или можно синтезировать любым способом, известным в данной области техники. Пептиды, пептидные миметики и аналоги пептидов, которые синтезированы и выделены таким образом, можно очищать или концентрировать способами, известными специалисту в данной области техники, такими как например фильтрация, хроматография и тому подобное. Пептидная группировка может быть ковалентно соединена с липидной группировкой посредством этерификации по карбокси-концевой карбоновой кислоте пептида, и в некоторых воплощениях пептидная группировка может быть соединена с липидной группировкой посредством фосфоэфира на карбокси-концевой карбоновой кислоте пептида.
Последовательность пептидного участка PDAG может варьироваться в разных воплощениях. Например, в одном воплощении пептидная последовательность может представлять собой XLYDKGYTSKEQKDCVGIX или XLYDKGYTPKDCVGIX или их синтетические эквиваленты, пептидные аналоги или пептидомиметики. В таких воплощениях Х может отсутствовать или представлять собой природную аминокислоту или ее миметик, дериватизированные аминокислоты или неаминокислотные простетические группы, и в конкретных воплощениях N-конец большинства аминокислот может представлять собой N-ацетил-аланин (асА). Не желая быть связанными теорией, пептидные последовательности, представленные выше, могут быть представителями еще большего класса пептидов, которые могут вызывать иммунный ответ у млекопитающего и, в частности, у человека при введении в одиночку или конъюгированными с липидной группировкой, например, как PDAG. Таким образом, любой пептид, выделенный практически из любого источника, который выполняет такую функцию, может быть охвачен воплощениями изобретения, включая, например, пептиды, ассоциированные с PDAG, выделенными из источников, таких как, без ограничения ими, животные, млекопитающие, люди, приматы, коровы, лошади, свиньи, птицы, рептилии, насекомые, микроорганизмы, бактерии и так далее.
Пептиды, пептидные миметики, или аналоги пептидов, которые ковалентно конъюгированы с липидной группировкой с получением PDAG, могут считаться и в дальнейшем именоваться как «PDAG-липопептиды».
Например, PDAG можно синтезировать следующим образом. PDAG-пептиды или PDAG-пептидные миметики можно создавать, используя способы твердофазного синтеза, такие как, например, протокол синтеза FMOC (9-флуоренил-метилоксикарбонил) с продолжительными циклами HBTU/HOBt (N,N,N",N"-тетраметил-O-(бензотриазол-1-ил)урония гексафторфосфат/1-гидроксибензотриазол) сочетания и предварительно загруженными смолами Ванга. После синтеза защитные группы боковой цепи пептида можно отщепить, и пептид может быть высвобожден со смолы К-реагентом. Затем пептид можно экстрагировать из буферов для синтеза, используя, например, экстракцию диэтиловым эфиром и лиофилизацию. Для дополнительной очистки полученных неочищенных пептидов можно использовать очистку на обращенной фазе С-18, после чего может следовать MALDI-TOF анализ для подтверждения аминокислотной последовательности.
PDAG-пептид или пептидный миметик может быть ковалентно присоединен к липидному участку PDAG, используя двухстадийный способ, где сначала синтезируют PDAG-пептид, как описано выше, но без стадии снятия защиты. Затем C-концевая карбоновая кислота может быть активирована, например, дициклогексилкарбамидом, и пептид можно инкубировать в присутствие диацилглицерина и каталитического агента, такого как диметиламинопиридин (DMAP). Этерификация может происходить на стадии инкубирования, обеспечивая возможность образования пептидил- диацилглицерида. Затем защитные группы боковой цепи пептида отщепляют при помощи Reagent K, и пептидил-диацилглицеридовый продукт может быть выделен и очищен хроматографически. Затем можно использовать анализ MALDI-TOF или ESI-MSn для подтверждения структуры очищенного продукта.
Пептидный участок PDAG согласно воплощениям настоящего изобретения можно модифицировать, например, путем замещения одной или более природных боковых цепей 20 генетически закодированных аминокислот (или D-аминокислот) на неприродные боковые цепи, примеры неприродных боковых цепей включают, без ограничения ими, алкил, низший алкил, 4-, 5-, 6-, 7-членный алкарил, амид, амид-низший алкил, амид-ди(низший алкил), низший алкокси, гидрокси, карбокси и их низшие сложноэфирные производные; и с 4-, 5-, 6-, 7-членными гетероциклами с получением пептидных миметиков. Например, могут быть получены аналоги пролина, в которых размер кольца остатка пролина меняется от 5-членного до 4, 6 или 7-членного. Циклические группы могут быть насыщенными или ненасыщенными, и когда они ненасыщенные, могут быть ароматическими или неароматическими. Гетероциклические группы могут содержать один или более чем один гетероатом, такой как, например, азот, кислород, и/или сера и тому подобное, и могут образовать группы, включая, без ограничения ими, фуразинил, фурил, имидазолидинил, имидазолил, имидазолинил, изотиазолил, изоксазолил, морфолинил (например морфолино), оксазолил, пиперазинил (например 1-пиперазинил), пиперидил (например 1-пиперидил, пиперидино), пиранил, пиразинил, пиразолидинил, пиразолинил, пиразолил, пиридазинил, пиридил, пиримидинил, пирролидинил (например 1-пирролидинил), пирролинил, пирролил, тиадиазолил, тиазолил, тиенил, тиоморфолинил (например тиоморфолино) и триазолил. Такие гетероциклические группы могут быть замещенными или незамещенными. Замещенные гетероциклические группы могут содержать заместители, такие как, но без ограничения ими, алкил, алкокси, галоген, кислород, или замещенный или незамещенный фенил. Пептидомиметики PDAG-пептидов также могут иметь аминокислотную остатки, которые были химически модифицированы, например, посредством фосфорилирования, сульфонирования, биотинилирования и подобным образом.
Доступно множество способов создания пептидомиметиков с такой же или подобной желательной биологической активностью, как у соответствующего нативного пептида, но с лучшей растворимостью, стабильностью, и/или подверженностью гидролизу или протеолизу. Таким образом, данные характеристики пептидомиметических соединений способствуют их использованию в терапевтических применениях, так как они могут обладать повышенной клеточной проницаемостью, большим сродством и/или авидностью к клеточным рецепторам и пролонгированным биологическим периодом полувыведения. Некоторые пептидомиметические соединения основаны на аминокислотной последовательности пептидов по изобретению. Часто пептидомиметические соединения представляют собой синтетические соединения с трехмерной структурой (то есть «пептидный мотив) на основе трехмерной структуры выбранного пептида. Пептидный мотив обеспечивает пептидомиметические соединения с нужной биологической активностью, то есть усиливающие или стимулирующие иммунный ответ, где связывающая активность миметического соединения существенно не снижена, и является часто такой же, как или больше, чем активность нативного пептида, по которому моделируют миметик.
Стратегии конструирования пептидомиметиков являются общедоступными в данной области техники. Один класс пептидомиметиков содержит каркас, который частично или полностью является непептидным, но имитирует пептидную основу «атом за атомом» и содержит боковые группы, которые подобным образом имитируют функциональные свойства боковых групп нативных аминокислотных остатков. Некоторые типы химических связей, такие как сложноэфирные, тиоэфирные, тиоамидные, ретроамидные, восстановленные карбонильные, диметиленовые и кетометиленовые связи, известны в данной области техники как обычно полезные заместители пептидных связей в конструировании устойчивых к протеазе пептидомиметиков. Другой класс пептидомиметиков представляет собой небольшую непептидную молекулу, которая связывается с другим пептидом или белком, но которая необязательно является структурным миметиком нативного пептида. Еще один класс пептидомиметиков появился из комбинаторной химии и генерации больших химических библиотек. Они обычно представляют собой новые матрицы, которые, не являясь структурно близкими нативному пептиду, обладают необходимыми функциональными группами, расположенными на непептидном каркасе, чтобы служить «топографическими» миметиками исходного пептида.
Не желая быть связанными теорией, PDAG, участки PDAG, аналоги PDAG, или миметики PDAG могут быть активированы в целевых тканях, продуцирующих активированный PDAG («PDAG»). Затем группировка «PDAG» может связываться с молекулами конкретного рецептора в иммунных клетках или на них и/или неиммунных клетках, как указано ранее, и инициировать высвобождение цитокинов, таких как, без ограничения ими, IL-6, IL-8, МСР-1, МIР-1а и β, INF-γ, TNF-α, Гранзим и RANTES (регулируемый активацией фактор, экспрессируемый и секретируемый нормальными Т-клетками) рекрутинговые и активирующие маркофаги, фагоцитарных NK-клеток или нейтрофилов и стимулирование высвобождение других стимулирующих цитокинов и пептидов. Высвобождение цитокинов также может стимулировать CD5+ В-клетки (также известные как резидентные В1-клетки ткани) продуцировать иммуноглобулин (IgM, эффективный опсонизирующий иммуноглобулин) и другие иммуноглобулины, происходящие из В-клеток, такие как IgA или IgG, цитокины и хемокины. Таким образом, воплощения настоящего изобретения включают PDAG-пептиды, участки PDAG-пептидов, аналоги PDAG-пептидов и пептидные миметики PDAG-пептидов, которые при введении субъекту могут присутствовать в целевой ткани в неактивной форме (то есть ковалентно присоединенные к липидной группировке) и активируются под действием липопротеинлипазы со временем, таким образом позволяя поддерживать повышенные концентрации PDAG-пептида и иммуноактивацию в целевой ткани субъекта с течением времени. Замедленное высвобождение активного PDAG-пептида может обеспечить замедленную активацию врожденной иммунной системы, таким образом предоставляя профилактически лечимому субъекту усиленную возможность борьбы с заболеванием при иммунологической провокации с течением времени, или терапевтически лечимому субъекту с помощью композиции PDAG с пролонгированным действием.
В других воплощения настоящего изобретения более чем один PDAG-пептид может быть ковалентно соединен с липидной группировкой. Не желая быть связанными теорией, продолжительность эффективного высвобождения PDAG-пептида или пептидных миметиков может быть непосредственно связана с количеством PDAG-пептидов или пептидных миметиков, соединенных с липидной группировкой. Таким образом, введение PDAG, участков PDAG, аналогов PDAG или миметиков PDAG, имеющих три пептидных фрагмента PDAG, соединенных с одной липидной группировкой, может высвобождать PDAG-пептид в течение более длительного периода времени, чем аналогичным образом введенные PDAG, участки PDAG, аналоги PDAG, или миметики PDAG, имеющие только одну PDAG-группировку, соединенную с липидной группировкой.
В некоторых воплощениях PDAG, участки PDAG, аналоги PDAG и миметики PDAG могут быть доставлены непосредственно субъекту, а в других PDAG, участки PDAG, аналоги PDAG и миметики PDAG могут быть объединены с фармацевтически приемлемым носителем с получением фармацевтической композиции, которую можно доставлять или предлагать субъекту.
В воплощениях изобретения доступно множество путей введения. Конкретный выбранный способ будет зависеть от конкретного выбранного химиотерапевтического лекарственного средства, тяжести состояния, подлежащего лечению, и дозировки, необходимой для терапевтической эффективности. Способы по изобретению в общем случае можно применять на практике, используя любой способ введения, который является приемлемым с медицинской точки зрения, что означает любой способ, который обеспечивает эффективные уровни активных соединений без вызывания клинически неприемлемых побочных эффектов. Такие способы введения включают, без ограничения ими, пероральный, ректальный, местный, назальный, внутрикожный, ингаляционный, внутрибрюшинный или парентеральный пути. Термин «парентеральный» включает подкожный, внутривенный, внутримышечный или инфузию. Внутривенный, подкожный или внутримышечный пути особенно пригодны для целей настоящего изобретения.
Фармацевтические композиции согласно воплощениям изобретения могут включать буферные агенты, такие как, например, уксусная кислота в виде соли, лимонная кислота в виде соли, борная кислота в виде соли, фосфорная кислота в виде соли и тому подобное и, возможно, консерванты, такие как бензалкония хлорид, хлорбутанол, парабены, тимеросал и подобные.
Фармацевтические композиции могут быть удобно представлены в стандартной лекарственной форме и могут быть изготовлены любым способом, хорошо известным в фармацевтической области. Все способы могут включать стадию приведения активного агента в ассоциацию с носителем, который состоит из одного или более вспомогательных ингредиента. В общем случае, композиции могут быть изготовлены посредством равномерного и близкого приведения активного соединения в ассоциацию с жидким наполнителем, тонкоизмельченным твердым наполнителем, или тем и другим, и затем, при необходимости, формования продукта.
Различные другие вещества-носители могут также успешно присутствовать в назальном спрее в подходящих количествах. Раствор можно быть изготовлен умеренно солевым путем растворения небольшого количества хлорида натрия в водной среде. Концентрация соли может находиться в диапазоне примерно 0,1-2,0% и предпочтительно составляет порядка примерно 0,65%. Другие вещества, такие как поверхностно-активные вещества, витамины и производные витаминов, антигистамины, смачивающие агенты, консерванты, увлажнители, эмульгаторы, пахучие вещества и тому подобные также могут присутствовать в обычных концентрациях. Многочисленные описания подходящих веществ можно найти в литературе, также как и описания эффективных концентраций в водной среде. Специалисту в данной области техники не составит труда определить подходящие вещества и концентрации для их известных функций. Доставку спрея в носовую полость можно выполнять при помощи любой обычной методики распыления или устройства.
В воплощениях изобретения также предлагаются композиции, подходящие для парентерального введения, где стерильный водный препарат PDAG, участков PDAG, аналогов PDAG и миметиков PDAG является предпочтительно изотоническим по отношении к крови реципиента. Такой водный препарат может быть приготовлен известными способами с использованием подходящих диспергирующих или смачивающих агентов и суспендирующих агентов. Стерильный инъецируемый препарат также может представлять собой стерильный инъецируемый раствор или суспензию в нетоксичном парентерально-приемлемом разбавителе или растворителе, например таком как раствор в 1,3-бутандиоле. В число приемлемых носителей и растворителей, которые можно использовать, входит вода, раствор Рингера и изотонический раствор хлорида натрия. Кроме того, в качестве растворителя или суспендирующей среды обычно используют стерильные нелетучие масла. Для этой цели можно использовать любое мягкое нелетучее масло, включая синтетические моно- или диглицериды. Кроме того, в изготовлении инъецируемых средств можно использовать жирные кислоты, такие как олеиновая кислота. Композицию наполнителя, подходящую для перорального, подкожного, внутривенного, внутримышечного и тому подобных введений, можно найти в Remington's Pharmaceutical Sciences, Mack Publishing Co., Easton, PA, который включен в данное описание изобретения посредством ссылки во всей его полноте.
Системы доставки согласно воплощениям изобретения можно конструировать так, чтобы они включали системы доставки с постепенным высвобождением, отсроченным высвобождением или замедленным высвобождением. PDAG, участки PDAG, аналоги PDAG и миметики PDAG также можно использовать в сочетании с дополнительными иммуностимулирующими или усиливающими иммунитет агентами. Используя такие системы, можно избежать повторных введений PDAG, участков PDAG, аналогов PDAG и миметиков PDAG, увеличивая удобство для субъекта, и их использованием может быть особенно подходящим для некоторых композиций по настоящему изобретению,
PDAG, участки PDAG, аналоги PDAG, миметики PDAG и фармацевтические композиции, включающие PDAG, участки PDAG, аналоги PDAG и миметики PDAG, можно вводить в эффективным количестве, которое усиливает или стимулирует иммунный ответ, и в некоторых воплощениях в эффективном количестве для стимулирования врожденного иммунного ответа.
В общем случае, в клинических испытаниях можно использовать обычное экспериментирование для определения конкретных диапазонов для оптимального эффекта для каждого агента или фармацевтической композиции и административного протокола. Введение PDAG, участков PDAG, аналогов PDAG, миметиков PDAG и фармацевтических композиций, включающих PDAG, участки PDAG, аналоги PDAG и миметики PDAG, конкретным субъектам можно скорректировать до эффективных и безопасных диапазонов в зависимости от состояния субъекта и восприимчивости к первичным введениям. Однако окончательный протокол введения можно регулировать в соответствии с решением оказывающего помощь врача, учитывая такие факторы, как возраст, состояние и размер субъекта, эффективность PDAG, участков PDAG, аналогов PDAG, миметиков PDAG и фармацевтических композиций, включающих PDAG, участки PDAG, аналоги PDAG и миметики PDAG, продолжительность лечения и тяжесть заболевания, подлежащего лечению.
В воплощениях изобретения схема приема PDAG, участков PDAG, аналогов PDAG, миметиков PDAG и фармацевтических композиций, включающих PDAG, участки PDAG, аналоги PDAG и миметики PDAG, может осуществляться введением с помощью назального спрея или ингалятора. Для композиций назального спрея или ингалятора размер измельченных частиц для эффективного растворения или диспергирования PDAG, участков PDAG, аналогов PDAG, миметиков PDAG и фармацевтических композиций, включающих PDAG, участки PDAG, аналоги PDAG и миметики PDAG, может составлять порядка от примерно 0,1 до примерно 20 микрон, от примерно 0,2 до примерно 10 микрон, и в некоторых воплощениях, от примерно 0,2 до примерно 5 микрон. Включение PDAG, участков PDAG, аналогов PDAG или миметиков PDAG в водный наполнитель можно облегчить сначала диспергированием PDAG, участков PDAG, аналогов PDAG или миметиков PDAG в растворе, таком как, например, 4% концентрация в растворе лактона. После того как PDAG, участки PDAG, аналоги PDAG или миметики PDAG тщательно смешали, диспергировали и/или растворили, они могут присутствовать в концентрации от примерно 0,001% до примерно 2,0%, от примерно 0,01% до примерно 0,35%, и в некоторых воплощениях примерно 0,10%. (Все процентные соотношения в данном описании изобретения приведены по массе, если не указано иное).
В других воплощениях PDAG, участки PDAG, аналоги PDAG, или миметики PDAG можно вводить перорально для достижения общих уровней в крови в диапазоне от примерно 25 мкг до примерно 2000 мкг/сутки, от примерно 25 до 500 мкг/сутки, или в некоторых воплощениях от примерно 50 до примерно 250 мкг/сутки, в двух-четырех отдельных дозах. В некоторых воплощениях можно использовать прерывистую терапию (например, одну неделю из трех недель или три из четырех недель).
В случае, когда ответ субъекта является недостаточным при начальных используемых дозах, можно использовать более высокие дозы (или эффективно более высокие дозы другим, более локализованным путем доставки) в степени, которую позволяет переносимость пациента. Для достижения подходящих системных уровней соединений можно использовать многократные дозы в сутки. Обычно можно использовать максимальную дозу. Максимальной дозой можно считать наивысшую безопасную дозу в соответствии с обоснованным медицинским решением. Специалисту в данной области техники понятно, однако, что субъект может настаивать на более низкой дозе или переносимой дозе по медицинским причинам, физиологическим причинам или практически по любой причине.
В других воплощениях изобретения по меньшей мере один PDAG, участок PDAG, аналог PDAG, миметик PDAG, или PDAG-пептид может быть ковалентно присоединен к антигенному пептиду или просто смешан с антигенным пептидом или вакциной перед введением антигенного пептида или вакцины субъекту. Не желая быть связанными теорией, добавление PDAG или PDAG-пептида может усиливать иммуногенность антигенного пептида или вакцины путем стимулирования врожденного иммунной системы во время введения антигенного пептида или вакцины. Синтетические антигены, имеющие ковалентно присоединенные один или более чем один PDAG, или PDAG-пептид или PDAG- или PDAG-пептидный антиген или вакцинные смеси, можно вводить субъекту для индуцирования длительного адаптивного иммунного ответа у субъекта.
В других воплощениях изобретения антитела можно генерировать к природным PDAG и PDAG-пептидам, и в других воплощениях генерированные таким образом антитела можно вводить субъекту для истощения концентрации PDAG, к которому генерировали антитело. Не желая быть связанными теорией, введение PDAG- и/или PDAG-пептид- истощающих антител может являться полезной терапевтической стратегией для субъектов с неконтролируемым системным воспалением, таким как, например, сепсис, атеросклероз, ревматоидные заболевания, аутоиммунные заболевания, воспалительное заболевание кишечника, диабет 1 типа и тому подобное. В аналогичных воплощениях PDAG- и PDAG-пептид-истощающие антитела можно использовать для лечения несептического повреждения, такого как, например, травма, воспаление вследствие обширных хирургических вмешательств и тому подобное.
Антитела к PDAG и PDAG-пептидам согласно воплощениям изобретения можно генерировать в кроликах, мышах, козах, лошадях или других видах способами, хорошо известными специалисту в данной области техники. Например, моноклональные антитела к PDAG или PDAG-пептидам можно генерировать, используя методики слияния гибридом, и выбранные гибридомы можно поддерживать в клеточной культуре или в биореакторе для непрерывного получения моноклональных антител. В некоторых воплощениях связывающий участок моноклонального антитела, специфичный к PDAG-пептиду, можно селективно получать посредством специфического химического расщепления целого антитела или рекомбинантными способами, известными в данной области техники. В других воплощениях специфичный участок связывания PDAG можно конъюгировать с Fc-участком человеческого антитела с получением гуманизированной химеры для введения PDAG-истощающего антитела субъектам-людям. Химерные антитела хорошо известны в данной области техники и могут быть созданы с использованием синтетических, полусинтетических или рекомбинантных способов. Гуманизированные PDAG-химерные антитела могут быть полезными для использования у субъектов людей, так как не могут вызвать по существу никакой вторичной реакции антител в субъектах-людях.
В других воплощениях изобретения можно изготовить флуоресцентно меченые PDAG, PDAG-пептиды или антитела к PDAG. В таких воплощениях флуоресцентные красители, такие как, но без ограничения ими, фикоэритрин (РЕ), красный флуоресцирующий краситель и флуоресцеинизотиоцианат (FITC), зеленый флуоресцирующий краситель, можно активировать присоединением к N-концу пептидильного участка PDAG, свободного сульфгидрила или амино или карбоксила в PDAG-пептиде или свободной аминогруппы антитела к PDAG.
Способы изготовления таких конъюгатов хорошо известны в данной области техники, например PDAG или PDAG-пептид можно конъюгировать с флуоресцентным красителем через его N-конец путем активирования пептида присоединением тиол-реакционоспособного аналога сукцинимидил-транс-4-(малеимидил-метил)циклогексан-1-карбоксилата (LC-SMCC) с вытянутой цепью и отделения непровзаимодействовавшего LC-SMCC от дериватизированного PDAG-пептида эксклюзионной хроматографией. Пиридилдисульфидное производное R-РЕ или FITC до свободного тиола можно активировать инкубированием R-PE или FITC в течение 10-15 минут в трис-(2-карбоксиэтил)фосфине (ТСЕР). Затем очищенное производное LC-SMCC-PDAG-пептида может быть объединено с активированным R-PE или FITC и смешиваться при 4°С в течение ночи. Взаимодействие можно остановить добавлением N-этилмалеимида (NEM), который закрывает любые оставшиеся тиольные группы. Конъюгат R-PE или FITC-PDAG можно очищать эксклюзионной хроматографией и лиофилизировать с получением конечного продукта.
В других воплощениях изобретения флуоресцентно меченые PDAG или PDAG-пептиды можно использовать для анализа образцов ткани. Например, флуоресцентно меченые PDAG или PDAG-пептиды можно смешивать ex vivo с образцами от субъекта для определения и выполнения количественного анализа иммунных клеток, включающих флуоресцентно меченые PDAG или PDAG-пептиды. В других воплощениях флуоресцентно меченые антитела к PDAG или PDAG-пептидам можно использовать для определения и выполнения количественного анализа уровней PDAG или PDAG-пептида ех vivo в образцах от субъектов, с использованием способов, таких как способы флуоресцентной микроскопии, ELISA (иммуноферментный твердофазный анализ) и тому подобные. Такие способы анализа хорошо известны практикующим специалистам в данной области техники.
Пример 1
В данном примере описано выделение и структурный анализ PDAG по настоящему изобретению. PDAG может быть обычным способом выделен в количествах, достаточных для научных исследований, из сывороточной фракции коагулированной крови диализом против дистиллированной воды через кассету для диализа с отсекаемой молекулярной массой 7-10 кДа (Slide-A-Lyzer, Pierce Biotechnology, Inc.) и путем концентрирования упариванием в вакууме и лиофилизацией диализата.
Фракцию неочищенной сыворотки дополнительно очищают эксклюзионной хроматографией или фильтрацией путем пропускания через смолу для эксклюзионной хроматографии или фильтр для удаления соли и других низкомолекулярных загрязняющих веществ. Окончательную очистку выполняют посредством экстракции органическим растворителем и ВЭЖХ с обращенной фазой. Такой способ дает после очистки достаточное количество вещества для проведения исследований биологической активности и начала химической идентификации биоактивного(ых) компонента(ов). Анализ LC/MS характеризует компонент PDAG как липопептид массой 2-3 кДа. Количественный анализ, основанный на общей распространенности по массам, показывает, что чистота PDAG составляет >98% после ВЭЖХ. ВЭЖХ/ESI-тандемную масс-спектроскопию можно использовать для подтверждения аминокислотной последовательности пептида и идентификации липидного участка PDAG по настоящему изобретению. Характерный профиль ВЭЖХ очищенного PDAG неприматов представлен на Фиг.1. ВЭЖХ хроматографию выполняли на Ultimate 3000 HPLC (Dionex, Sunnyvale, CA). Колонка представляла собой Zorbax C8 1×150 мм (Agilent, Santa Clara, CA). 200 мкл образца в воде впрыскивали в 200 мкл петлю. Градиент составлял 5-65% растворителя А к растворителю В в течение 60 минут, с последующим 5 минутным промыванием 90%-ным растворителем В. Растворитель А представлял собой 5% ацетонитрил + 0,1% TFA, а растворитель В представлял собой 90% ацетонитрил + 0,1% TFA при скорости потока 50 мкл/мин. Детектирование выполняли при 214 нм и 280 нм.
Козью сыворотку (2 л) экстрагировали с получением приблизительно 100 мкг очищенного PDAG. Фиг.1А представляет собой типичную хроматограмму фракции, растворимой в смеси метанол-хлороформ (Technical Grade PDAG). Пик, элюируемый номинально в 10,4 минуты, определяют как родительский PDAG, и пики, элюируемые номинально между 9,4 минутами и 4,8 минутами, представляют собой продукты гидролиза родительского PDAG. Пик, элюируемый в 2,9 минут, был охарактеризован как содержащий в основном олигосахариды (данные не показаны). Очищенный PDAG (пик в 10,4 минуты) собирали препаративной хроматографией с обращенной фазой, и растворитель удаляли в вакууме. Данные результаты представлены на Фиг.1А и 1В.
Пример 2
В данном примере описано определение структуры нативного PDAG. Анализ последовательности посредством расщепления по Эдману очищенного PDAG идентифицировал аминокислотную последовательность (X1)LYDKGYTSKEQKDCVGI(X2) и рассчитанную молекулярную массу 1883,57 а.е.м. (атомных единиц массы) для предполагаемого PDAG-пептида. X1 и Х2 представляли собой неидентифицированные дериватизированные аминокислоты или неаминокислотные простетические группы. Жидкостную хроматографию/тандемную масс-спектрометрию (LC/MSn) использовали для анализа очищенного PDAG, чтобы (а) идентифицировать N-концевые и C-концевые простетические группы и (б) подтвердить аминокислотную последовательность пептида. ESI-MS родительского пика PDAG (10,4 минут) выявила 3 основных ионных фрагмента. Определили, что наиболее распространенный (Фрагмент А) представляет собой триптофан (m/z 205), который совместно элюировался с PDAG в виде аддукта PDAG-триптофан.
Масс-спектр второго наиболее распространенного иона, Фрагмента В (молекулярная масса 1688,8) имел два многозарядных иона. Ион с m/z 564,00 представляет собой [М+3Н]3+ и ион с 845,45 представляет собой [М+2Н]2+. Масс-спектр иона продукта MS/MS с m/z 845,5 имел аминокислотную последовательность acALYDKGYTSKEQKD (m/z 1688,8). Данная последовательность соответствует первым 13 аминокислотам из анализа последовательности посредством расщепления по Эдману, где X1 представляет собой N-ацетилаланин (асА). Масс-спектр для Фрагмента В представлен на Фиг.2А и 2В.
В качестве дополнительного экспериментального подтверждения структуры, определенной для PDAG, проводили ряд экспериментов посредством масс-спектрометрии MALDI-TOF. Так как PDAG выделяли в виде аддукта триптофан: PDAG (предположительно 200:1), PDAG анализировали с помощью MALDI-TOF MS с добавлением и без добавления матрицы. Наблюдали отсутствие молекулярного иона, соответствующего неповрежденной молекуле PDAG, однако в спектре положительных ионов без добавленной матрицы были обнаружены четыре основные ионные фрагмента, относящиеся к целой молекуле PDAG (триптофан служил в качестве матрицы). Фрагмент с большой массой (m/z 1282,71) получался при потере нейтрального NH3 с N-концевого фрагмента иона [acALYDKGYTSKE]+. Фрагмент с низкой массой (m/z 1133,30) соответствует у-иону [DCVGI-(DAG)]+, содержащему C-концевой диацилглицерин («DAG») [DCVGI-(DAG)]. Основной пик (m/z 1208,47) соответствует аддукту с триптофаном фрагмента внутреннего z-иона [KEQKDCVGI]W+. Соответствующий у-ион (+15 а.е.м.) наблюдали при m/z 1223,61. у-Ион с m/z 1207,53 представляет собой C-концевой фрагмент [TSKEQKDCVGI]+
В результате совместной кристаллизации аддукта триптофан: PDAG и дигидроксибензойной кислотой (DHB) и повторного анализа при помощи масс-спектрометрии MALDI-TOF получили расширенный ряд ионных фрагментов, который также давал информацию об общей структуре PDAG (Фиг.3).
PDAG-продукт, происходящий из сыворотки, особенно подвержен гидролизу в кислых условиях, и это может объяснить трудности в получении молекулярного иона в условиях ионизации или ES1, или MALDI. Пики, элюируемые между 4,83 и 9,36 минутами на Фиг.1А, представляют собой продукты гидролиза родительского PDAG с пиком, элюируемым в 10,42 минуты (данные не показаны). Таким образом, очищенный PDAG подвергали мягкому кислотному гидролизу, N-концевому сульфонированию in situ после анализа продуктов гидролиза для получения информации об общей структуре PDAG. Анализ PSD MALDI-TOF N-концевых сульфонированных пептидных фрагментов, которые были образованы после мягкого кислотного гидролиза PDAG, показал многочисленные у-ионы, соответствующие ранее наблюдаемым пептидным фрагментам и объясняющим полную структуру PDAG (Фиг.4). Таким образом, предполагаемая структура PDAG acAL YDKGYTSKEQKDCVGI-DAG соответствует анализу последовательности посредством расщепления по Эдману, ESI-MS/MS анализу последовательности и PSD MALDI-TOF MS анализу природного продукта.
Для данной предполагаемой последовательности пептида в PDAG поиск гомологичных последовательностей осуществляли по базе данных статистически определимых последовательностей, используя NCBI BLAST средство поиска BLASTP (Altshul, 1997). PDAG-пептид имеет идентичную последовательность, гомологичную внутренней последовательности аминокислот в положениях 558-574 белка 1, относящегося к каналам транзиторного рецепторного потенциала (TRPC1). Отмечено отсутствие значительной гомологии с другими известными белками или пептидами. Сравнение аминокислотной последовательности в положениях 557-574 из бычьего, мышиного и человеческого TRPC1 с предполагаемой последовательностью PDAG-пептида выявило идентичные аминокислотные последовательности за исключением того, что 9-серин в бычьем PDAG был замещен пролином.
Пример 3
pDAG-пептид синтезировали по протоколу стандартного твердофазного синтеза с длительным HBTU-соединением на Н-изолейцин-2-хлортритиловой смоле, используя пептидный синтезатор ААРРТЕС 348 Sigma (Advanced Automated Peptide Protein Technologies, Inc., Louisville, KY). Полностью защищенную пептидную последовательность, соединенную со смолой [Ala-Leu-Tyr(But)-Asp(OBut)-Lys(Boc)-Gly-Tyr(But)-Thr(But)-Ser(But)-Lys(Boc)-Glu(OBut)-Gln(Trt)-Lys(Boc)-Asp(OBut)-Cys(Trt)-Val-Gly-lle-СМОЛА], восстанавливали после промывания дихлорметаном. Пептид, соединенный со смолой, ацетилировали по N-концевому аланину пептида добавлением 10%-ного уксусного ангидрида в N,N-диизопропилэтиламине (20%) и N,N-диметилацетамиде (70%). Через два часа при комнатной температуре смолу отфильтровали, промывали последовательно N,N-диметилацетамидом и дихлорметаном и лиофилизировали. Ацетилированный и полностью защищенный пептид отщепляли от смолы, используя 20 мл раствора 1,1,1,3,3,3-гексафторо-2-пропанола в дихлорметане 1:4. Через два часа при комнатной температуре смолу отфильтровали и промывали 2 мл отщепляющего раствора. Фильтрат упаривали в вакууме, используя роторный испаритель. Образец анализировали масс-спектрометрией для подтверждения предполагаемой массы при m/z 3286 (данные не показаны).
1-Стеароил-2-арахидоноил-sn-глицерин (Sigma Aldrich) соединяли с C-концевым изолейциновым карбоксилом полностью защищенного ацетилированного пептида, используя реакцию сочетания дициклогексилкарбодиимид/диметиламинопиридин (DCC/DMAP). 1-Стеароил-2-арахидоноил-sn-глицерин (5 мг) растворяли в 2 мл дихлорметана и смешивали с 2 эквивалентами полностью защищенного ацетилированного пептида, растворенного в 2 мл дихлорметана, и 1 эквивалентом DMAP, также растворенного в 2 мл дихлорметана. Реакционную смесь оставляли взаимодействовать в течение ночи при комнатной температуре. Реакционную смесь сушили в вакууме, и защитные группы удаляли in situ добавлением 8 мл раствора, снимающего защитные группы (2,5% 1,2-этандиола, 94% трифторуксусная кислота, 0,1% триизопропилсилана и 2,5% воды). После двух часового инкубационного периода при комнатной температуре в присутствии снимающего защитные группы раствора реакционную смесь фильтровали, и фильтрат упаривали в вакууме на роторном испарителе.
Очистку неочищенного продукта выполняли препаративной ВЭЖХ с обращенной фазой, используя колонку Jupiter® Proteo (Phenomenex, Inc., Torrance, CA) и градиент бинарной подвижной фазы (Растворитель А: 0,1% TFA в воде; Растворитель В: 0,1% TFA в ацетонитриле), образованный от 5% до 95% Растворителем B в течение 20 минут (4,5% в минуту) при скорости потока 1 мл/минуту, и вытекающий поток непрерывно контролировали при 220 нм. Продукт pDAG элюировали в 22,9 минуты при данных условиях. Неочищенный продукт (63 мг) растворяли в 4 мл ацетонитрила и 2 мл воды и загружали в препаративную колонку. Пик, элюируемый в 22,9 минуты, собирали в 22,5 мл подвижной фазы (95% ацетонитрил/0,1% TFA). Вытекающий поток выпаривали в вакууме, и измеряли конечную массу pDAG-пептида, равную 2,5 мг (выход 4%), с чистотой, определяемой анализом ВЭЖХ, равной 98,9%.
Пример 4
Фибробласты стимулировали синтетическим PDAG в течение 24 ч. РНК экстрагировали из фибробластов, и IL-6 транскрипты измеряли при помощи ПЦР в режиме реального времени, и белок IL-6 измеряли в среде при помощи ELISA. Авторы изобретения обнаружили, что 100 пг/мл фибробластов, стимулированных PDAG, увеличивали уровни мРНК IL-6 по сравнению с контролем (необработанные клетки), Р=0,001. Аналогичным образом измерили приблизительно в 2 раза больше белка IL-6 в среде обработанных PDAG фибробластов (Р=0,0001). Данные результаты представлены на Фиг.5.
Пример 5
мРНК экстрагировали из первичных фибробластов человека, инкубированных в течение с 100 пг/мл синтетического PDAG. Измеряли транскрипты NALP3 и NFKB. Было обнаружено, что транскрипты NALP3 увеличились в 1,75 раз (Р=0,007), в то время как транскрипты NFkB не увеличились (Р=0,4). Данные результаты представлены на Фиг.6.
Пример 6
Моноциты ТНР-1 (5×104 клеток/лунку) культивировали в забуференном солевом растворе Хэнка 1 с 10%-ным FBS в 24-луночном микропланшете. Клетки инфицировали 2,5×104 бактериальных клеток AR39 Chlamydia pneumoniae (CPn) с получением MOI=1, и инфицированные клетки поддерживали в течение 72 часов после инфицирования с ежедневной заменой среды. Клетки обрабатывали или носителем PDAG (0,01% Твин-20 в DMSO) или PDAG (25 нг/мл и 50 нг/мл) за 24 часа до инфицирования или через 24 часа после инфицирования. После 72 часового инкубационного периода клетки собирали и делали проницаемыми с помощью Cytoffx/Cytoperm (BD Pharmingen) и добавляли мышиный α-CPn моноклональный IgG (клон 61С75), и клетки с нарушенной проницаемостью мембран инкубировали в течение 1 часа. После инкубирования клетки промывали и добавляли FITC-меченый α-мышиный IgG и инкубировали в течение 1 часа. Клетки снова собирали и промывали. Количественную оценку инфицированных клеток осуществляли посредством проточной цитометрии. Жизнеспособность клеток определяли по исключению трипанового синего. Данные результаты представлены на Фиг.7.
Пример 7
Тетрациклин-индуцируемую HBV-стабильную клеточную линию, клетки HepDES19 (Guo, 2007), поддерживали в среде DMEM/F-12 среде с пенициллином и стрептомицином (Invitrogen), 10% FBS, 500 мкг/мл G418 (Invitrogen) и 1 мкг/мл тетрациклином (Sigma-Aldrich, St. Louis, МО). Для начала репликации HBV в клетках HepDES19 тетрациклин выводили из среды, и клетки культивировали в течение 4-5 суток до анализа вирусной ДНК. Для обработки клеток pDAG в не содержащую тетрациклин среду добавляли пептид, и доставляли в клеточную культуру ежедневно. Концентрацию растворителя DMSO доводили до 0,01% в каждой обработке.
ДНК ядра HBV экстрагировали из клеток HepDES19, обработанных pDAG, как описано ранее (Guo, 2009). Кратко, клетки из одной 35 мм чашки лизировали при помощи 0,5 мл лизирующего буфера (10 мМ Трис-HCl рН 8.0, 10 мМ EDTA, 1% NP40 и 2% сахарозы) при 37°С в течение 10 минут. Клеточный дебрис и ядра удаляли центрифугированием, и надосадочную жидкость смешивали с 130 мкл 35% полиэтиленгликоля (PEG-8000), содержащего 1,5 М NaCl. Через 1 час инкубирования во льду вирусные нуклеокапсиды осаждали центрифугированием при 10000 об/мин в течение 5 мин при 4°С, с последующим расщеплением в течение 1 час при 37°С в 200 мкл расщепляющего буфера [0,5 мг/мл проназы (Calbiochem), 0,5% SDS, 150 мМ NaCl, 25 мМ Трис-HCl рН 8,0, и 10 мМ EDTA]. Подвергнутую расщеплению смесь дважды экстрагировали фенолом, и ДНК осаждали этанолом и растворяли в буфере ТЕ (10 мМ Трис-HCl, рН 8,0, 1 мМ EDTA). Образец ДНК разделяли электрофорезом в 1,5%-ном агарозном геле. Затем гель подвергали депуринизации в 0,2 н. HCl в течение 10 мин при комнатной температуре, затем денатурировали в растворе, содержащем 0,5 М NaOH и 1,5 М NaCl в течение 1 ч с последующей нейтрализацией в течение 1 ч в буфере, содержащем 1 М Трис-HCl (рН 7,4) и 1,5 М NaCl. Затем ДНК блотировали на мембрану Hybond-XL (GE Healthcare) в 20Х SSC буфере. Мембраны зондировали с помощью α-32P-UTP (800 Ки/ммоль, Perkin Elmer)-меченым HBV минус нитеспецифический полноразмерный рибозонд. Гибридизацию выполняли в 5 мл EKONO буфера для гибридизации (Genotech) с 1 ч предварительной гибридизацией при 65°С и гибридизации в течение ночи при 65°С, с последующим 1 ч промывания с помощью 0,1Х SSC и 0,1% SDS при 65°С. Мембрану подвергали скринингу на Phosphorimager, и сигналы гибридизации выявляли и количественно анализировали при помощи программы QuantityOne (Bio-Rad).
Пример 8
Самкам белого новозеландского кролика инокулировали FCA в присутствии (n=3) и в отсутствии (n=3) нативного PDAG. Образцы крови отбирали за сутки до инокуляции (фон) и снова на 3, 5, 7, 10, 12, 14, и 17 сутки после инокуляции. Сыворотки анализировали в трех параллелях в отношении продуцирования IgM М.tuberculosis при помощи ELISA. Из титров антител для каждых суток после заражения вычитали фон, используя титр -1 суток. В 5 сутки после инокуляции титр IgM у кроликов, инокулированных FCA+PDAG, был значительно выше, чем титры у кроликов, получивших только FCA (Р=0,0004). Данные результаты представлены на Фиг.9.
Пример 9
Мышей Swiss Webster заражали летальной дозой Salmonella typhimurium, 5×103 KOE (колониеобразующих единиц)/мышь. Пятнадцать мышей обработали 5 мкг/мл PDAG за 24 часа до летальной инокуляции. Мышей поддерживали и наблюдали ежедневно в течение 10 суток. Смертность наблюдалась в контрольной группе на 4 сутки, но не в группе, обработанной PDAG, вплоть до 7 суток. На 10 сутки в группе, обработанной PDAG, 12 мышей оставались живыми, в то время как в необработанной группе умерли все. Р<0,0001. Данные результаты представлены на Фиг.10.
Хотя настоящее изобретение были описано со ссылкой на конкретные воплощения, специалисты в данной области техники определят, что на их основании можно создать много вариантов, например в конкретных экспериментальных условиях, описанных в данном описании изобретения, и понятно и следует понимать, что описания в соответствии с изобретением показывают только некоторые предпочтительные воплощения и задачи и преимущества изобретения без отклонения от более широкого объема и сущности изобретения. Следует понимать и принимать во внимание, что данные раскрытиями согласно данному изобретению являются только те, которые иллюстрируют множество дополнительных возможных применений соединения, которые может представить специалист в данной области техники, и, таким образом, не предполагается, что они каким-либо образом ограничивают изобретение. Соответственно, другие цели и преимущества изобретения будут очевидны специалисту в данной области техники из подробного описания вместе с формулой изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| БИОЛОГИЧЕСКИ АКТИВИРУЕМЫЕ ЛЕКАРСТВЕННЫЕ СРЕДСТВА НА ОСНОВЕ ЦИТОКИНОВ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2019 |
|
RU2819307C2 |
| МОДИФИЦИРОВАННЫЕ ПЕПТИДЫ MELK И СОДЕРЖАЩИЕ ИХ ВАКЦИНЫ | 2011 |
|
RU2580035C2 |
| АНТИТЕЛА ПРОТИВ TNF-α И ИХ ПРИМЕНЕНИЯ | 2010 |
|
RU2595379C2 |
| РАСТВОРИМЫЙ МЕДИАТОР | 2012 |
|
RU2660580C2 |
| АНТИТЕЛА ПРОТИВ VEGF И ИХ ПРИМЕНЕНИЯ | 2010 |
|
RU2567639C2 |
| НОВЫЕ ПЕПТИДЫ ДЛЯ ЛЕЧЕНИЯ И ПРОФИЛАКТИКИ ИММУНОПАТОЛОГИЧЕСКИХ ЗАБОЛЕВАНИЙ, ВКЛЮЧАЯ ЛЕЧЕНИЕ И ПРОФИЛАКТИКУ ИНФЕКЦИИ ПОСРЕДСТВОМ МОДУЛИРОВАНИЯ ВРОЖДЕННОГО ИММУНИТЕТА | 2006 |
|
RU2507213C2 |
| СЛИТЫЕ КОНСТРУКЦИИ ЛЕКАРСТВЕННОГО СРЕДСТВА И КОНЪЮГАТЫ | 2005 |
|
RU2428431C2 |
| ПРИМЕНЕНИЕ КАЛЬЦИТОНИНА В КАЧЕСТВЕ КОМБИНИРОВАННОЙ ТЕРАПИИ ДЛЯ ЛЕЧЕНИЯ ВОСПАЛИТЕЛЬНЫХ БОЛЕЗНЕННЫХ СОСТОЯНИЙ | 2006 |
|
RU2394591C2 |
| АНТИГЕННЫЕ TAU-ПЕПТИДЫ И ИХ ПРИМЕНЕНИЯ | 2010 |
|
RU2518291C2 |
| РЕЦЕПТОР ДЛЯ VISTA | 2019 |
|
RU2812846C2 |


Изобретение относится к области биотехнологии, конкретно к иммуностимулирующим соединениям, и может быть использовано в медицине. Иммуностимулирующий пептид с аминокислотной последовательностью XLYDKGYTSKEQKDCVGI, где N-концевой X представляет собой N-ацетилаланин, ковалентно связывают с жирными кислотами, выбранными из С2-С25, с получением PDAG (пептидил-2,3-диацилглицерида). Полученное соединение используют в составе фармацевтической композиции для стимуляции иммунного ответа. Изобретение позволяет эффективно стимулировать иммунный ответ у индивидуума и усиливать иммуногенность антигенного пептида при совместном введении его с PDAG. 6 н. и 23 з.п. ф-лы, 10 ил., 9 пр.
1. Соединение, стимулирующее иммунный ответ, имеющее общую формулу (I):
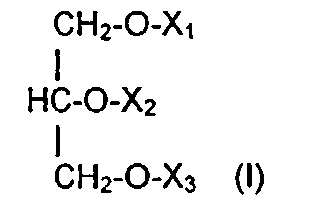
где X1 представляет собой пептид, имеющий аминокислотную последовательность XLYDKGYTSKEQKDCVGI, где N-концевой X представляет собой N-ацетилаланин;
Х2 и Х3 независимо выбраны из С2-С25 жирной кислоты.
2. Фармацевтическая композиция, стимулирующая иммунный ответ, содержащая эффективную дозу соединения по п.1 и фармацевтически приемлемый носитель.
3. Фармацевтическая композиция по п.2, дополнительно содержащая фармацевтически приемлемый буферный агент.
4. Фармацевтическая композиция по п.3, где указанный буферный агент выбран из группы, состоящей из уксусной кислоты в виде соли, лимонной кислоты в виде соли, борной кислоты в виде соли, фосфорной кислоты в виде соли и их комбинаций.
5. Фармацевтическая композиция по п.2, где указанный носитель выбран из группы, пригодной для перорального, подкожного, внутривенного, внутримышечного, внутрибрюшинного, трансбуккального или глазного пути введения, введения ректально, парентерально, системно, вагинально, местно, перорально в виде перорального или назального спрея, и их комбинаций.
6. Фармацевтическая композиция по п.2, которая находится в форме пилюли, таблетки, леденца, покрытой оболочкой таблетки, гранулы, капсулы, твердой или мягкой желатиновой капсулы, водного раствора, спиртового раствора, масляного раствора, сиропа, эмульсии, суспензии, пастилки, суппозитория, раствора для инъекции, мази, настойки, крема, лосьона, порошка, спрея, чрескожных терапевтических систем, назального спрея, аэрозольной смеси, микрокапсулы, имплантата, палочки или пластыря.
7. Фармацевтическая композиция по п.5, где указанная парентеральная композиция содержит смачивающие агенты, суспендирующие агенты, разбавители, растворители или их комбинации.
8. Фармацевтическая композиция по п.7, где указанный разбавитель представляет собой 1,3-бутандиол.
9. Фармацевтическая композиция по п.7, где указанный растворитель выбран из группы, состоящей из воды, раствора Рингера, изотонического раствора хлорида натрия и стерильных нелетучих масел.
10. Фармацевтическая композиция по п.9, где указанное нелетучее масло представляет собой мягкое нелетучее масло.
11. Фармацевтическая композиция по п.6, где указанный раствор для инъекций содержит жирные кислоты.
12. Фармацевтическая композиция по п.2, дополнительно содержащая консервант.
13. Фармацевтическая композиция по п.12, где указанный консервант выбран из группы, содержащей бензалкония хлорид, хлорбутанол, парабены, тимеросал и их комбинации.
14. Фармацевтическая композиция по п.5, где указанный носитель обеспечивает слабый солевой раствор в указанном назальном спрее.
15. Фармацевтическая композиция по п.14, где указанный солевой раствор является примерно 0,1%-2,0%-ным.
16. Фармацевтическая композиция по п.14, где указанный солевой раствор является примерно 0,65%-ным.
17. Фармацевтическая композиция по п.5, где указанная композиция спрея имеет размер измельченных частиц для эффективного диспергирования указанного соединения.
18. Фармацевтическая композиция по п.17, где указанный размер частиц составляет примерно 0,1-20 микрон.
19. Фармацевтическая композиция по п.17, где указанный размер частиц составляет примерно 0,2-10 микрон.
20. Фармацевтическая композиция по п.17, где указанный размер частиц составляет примерно 0,2-5 микрон.
21. Фармацевтическая композиция по п.2, дополнительно содержащая добавки из группы, состоящей из поверхностно-активных веществ, витаминов, производных витаминов, антигистаминов, смачивающих агентов, консервантов, увлажнителей, эмульгаторов, ароматизаторов и их комбинаций.
22. Фармацевтическая композиция для доставки соединения по п.1 субъекту, содержащая:
а) фармацевтическую композицию по п.2; и
б) средство высвобождения, выбранное из группы, состоящей из постепенного высвобождения, отсроченного высвобождения, замедленного высвобождения и их комбинаций.
23. Способ изготовления фармацевтической композиции по п.2, включающий:
а) получение соединения по п.1; и
б) объединение указанного соединения с фармацевтически приемлемым носителем.
24. Способ по п.23, где указанное объединение облегчается диспергированием указанного соединения в 4%-ном растворе лактона.
25. Способ усиления иммуногенности антигенного пептида, включающий введение субъекту антигенного пептида совместно с фармацевтической композицией по п.2.
26. Способ синтезирования соединения по п.1, представляющего собой PDAG (пептидил-2,3-диацилглицерид), включающий:
а) синтез пептида, имеющего аминокислотную последовательность XLYDKGYTSKEQKDCVGI, где N-концевой X представляет собой N-ацетилаланин, твердофазным способом, где указанный синтез не содержит стадии снятия защитной группы;
б) активирование С-концевой карбоновой кислоты указанного пептида;
в) инкубирование с DAG и каталитическим агентом, где в результате указанного инкубирования получают PDAG; и
г) отщепление защитных групп боковой цепи пептида с помощью К-реагента;
где полученный PDAG может быть очищен посредством хроматографии.
27. Способ по п.26, где указанное активирование осуществляют при помощи дициклогексилкарбодиимида.
28. Способ по п.26, где указанный каталитический агент представляет собой диметиламинопиридин.
29. Способ по п.26, где PDAG очищают посредством хроматографии.
| US6812339 A1, 02.11.2004 | |||
| US2003109437 A1, 12.06.2003 | |||
| US2007197436 А1, 23.08.2007 | |||
| СРЕДСТВО, ОБЛАДАЮЩЕЕ ЛИПИДКОРРИГИРУЮЩИМИ И ИММУНОМОДУЛИРУЮЩИМИ СВОЙСТВАМИ | 2003 |
|
RU2259824C2 |

