Область техники
Настоящее изобретение относится к новым соединениям, обладающим активирующим действием на α-подтип, δ-подтип и γ-подтип рецепторов, активируемых пролифератором пероксисом (PPARs), способу их получения и применению лекарственных средств, содержащих указанные соединения, для лечения диабета и сердечно-сосудистых заболеваний. Настоящее изобретение также относится к промежуточным соединениям новых соединений и способу их получения.
Уровень техники
С развитием производства и улучшением качества жизни метаболический синдром, характеризующийся ожирением, инсулиннезависимым диабетом (диабетом II типа), нарушением липидного обмена и гипертензией, встречается во всем мире и угрожает здоровью людей в значительной степени вследствие чрезмерного потребления жиров и белков. Метаболический синдром не только связан с генетическими особенностями, возрастом, полом, физиологическими особенностями, пищевым статусом, пищевыми привычками и т.д. индивидуумов, но также вовлечен в нарушение балансов липидного обмена, энергии и углеводного обмена in vivo. Поэтому, терапевтический режим, нацеленный на поддержание балансов энергии, жиров и углеводов in vivo, становится эффективным способом для лечения метаболического синдрома. Ядерные рецепторы (ЯР) находятся в фокусе исследований, поскольку они играют важную роль в поддержании баланса энергии и балансов жиров и углеводов в клетках, даже у всего индивидуума. Ядерные рецепторы могут регулировать системы транскрипции зависимых генов и таким образом проявлять свою физиологическую активность только после активации различными физиологическими лигандами (например, насыщенными и ненасыщенными жирными кислотами, метаболитами и их различными синтетическими соединениями) (Kasuga, J. et al., Bioorg. Med. Chem. 2007, 15, 5177-5190).
В семействе ядерных рецепторов, рецепторы, активируемые пролифератором пероксисом (PPARs), ядерные транскрипционные факторы, активируемые лигандами, привлекают внимание исследователей на протяжении более десяти лет и представляют собой важные регуляторные факторы при метаболическом синдроме (Guan, Y. J. Am. Soc. Nephrol, 2004, 15, 2801-2815). Таким образом, рецепторы PPARs играют важную роль в патогенезе, развитии, лечении и предотвращении заболеваний, например, инсулинорезистентности, нарушения толерантности к глюкозе, диабета II типа, ожирения, гиперлипидемии, гипертензии, сердечно-сосудистых заболеваний, атеросклероза и т.д.
Рецепторы PPARs классифицируют на три подтипа: PPARα, PPARδ и PPARγ, которые регулируют экспрессию гена за счет связывания с конкретными последовательностями ДНК (Berger, J. et al., The Journal of Biological Chemistry, 1999, 274 (10), 6718-6725). Рецептор PPARα экспрессируется преимущественно в печени, сердце, кишечнике, почках и макрофагах и может ускорять обмен жирных кислот, частично снимать воспалительный ответ макрофагов и понижать уровень холестерина липопротеидов низкой плотности после активации; рецептор PPARγ экспрессируется в адипоцитах, хориокарциноме и других тканях и не только может понижать уровень глюкозы крови и увеличивать чувствительность к инсулину, но также играет важную роль в липидном обмене, ингибировании цитокинов, противовоспалительном ответе, регуляции иммунного ответа, регуляции кровяного давления и т.д. после активации (Kasuga, J. et al., Bioorg. Med. Chem. 2007, 15, 5177-5190). По сравнению с другими двумя подтипами, физиологическая функция рецептора PPARδ неизвестна до сих пор. Однако недавние исследования на животных моделях для фармакологических экспериментов показывают, что рецептор PPARδ может ускорять катаболизм жирных кислот и увеличивать разобщение процессов окисления и фосфорилирования в жировых тканях и мышцах и подавлять воспаление, опосредуемое макрофагами. Путем контроля увеличения массы тела, повышения толерантности организма человека, увеличения чувствительности к инсулину и улучшения состояния при атеросклерозе в различных аспектах лиганды рецептора PPARδ могут таким образом стать эффективным лекарственным средством для лечения гиперлипидемии, ожирения, инсулинорезистентности и атеросклероза.
В настоящее время ни один из "трехканальных" агонистов, оказывающих действие на все из рецепторов PPARα, PPARδ и PPARγ, не является коммерчески доступным в качестве терапевтического агента где-либо в мире. "Трехканальный" агонист рецепторов PPARα, PPARδ, и PPARγ, разработанный изобретателями, может быть применен для лечения метаболического синдрома, который в основном характеризуется диабетом. Указанный агонист обладает функцией, аналогичной функции глитазонов или других агентов, увеличивающих чувствительность к инсулину, но может быть применен более широко. Несмотря на то, что агонисты рецептора PPARγ, глитазоны, могут увеличивать чувствительность к инсулину, недавние клинические результаты показывают, что эти агонисты способны повышать риск возникновения сердечно-сосудистых заболеваний. Кроме того, глитазоны имеют дополнительные распространенные побочные эффекты, включающие увеличение массы тела и гепатотоксичность. Таким образом, изобретатели прилагают большие усилия для того, чтобы найти новое лекарственное средство, которое не только может лечить диабет, но также имеет некоторое протекторное действие на сердечно-сосудистую систему.
Исследование и разработка новых лекарственных средств для лечения метаболического синдрома находится в центре внимания многих фармацевтических компаний. Китайские фармацевтические компании также на конкурентой основе концентрируют свои исследования на новых мишенях в разработке лекарственных средств для лечения диабета. Таким образом, большое клиническое значение будет иметь обеспечение новых соединений, обладающих активирующим действием на рецепторы, активируемые пролифератором пероксисом (PPARα, PPARδ и PPARγ).
Краткое описание изобретения
Одной задачей настоящего изобретения является обеспечение соединения формулы I или его фармацевтически приемлемых солей:

где
X представляет собой O, S, NR11 или (CR11Rn11 ,)n, где n представляет собой целое число, выбранное из 1, 2, 3 и 4;
Y представляет собой О, S, NR11 или (CR11R11 ,)m, где m представляет собой целое число, выбранное из 1, 2, 3 и 4;
R1 независимо представляет собой H, алкил или циклоалкил;
G1 независимо представляет собой алкил или циклоалкил;
G2 и G3 каждый независимо выбраны из H, алкила, алкокси, трифторметила, галогена (F, Cl, Br), нитро, NR11R11 ,, алкилтио, амидо, циано, карбоксила и тетразолила;
R11 и R11, каждый независимо выбраны из H и C1-С6 алкила.
В предпочтительном соединении формулы I X представляет собой S, O или NR11, Y представляет собой O, и другие заместители имеют значения, определенные выше. В другом предпочтительном соединении формулы I R1 независимо представляет собой Н или C1-С6 алкил, и другие заместители имеют значения, определенные выше. В другом предпочтительном соединении формулы I G1 выбран из C1-С6 алкила, и другие заместители имеют значения, определенные выше.
В другом предпочтительном соединении формулы I G2 и G3 каждый независимо выбраны из Н, C1-C6 алкила, C1-C6 алкокси, трифторметила, F, Cl, Br, нитро, NR11R11 ,, C1-C6 алкилтио, амидо, циано, карбоксила и тетразолила, и другие заместители имеют значения, определенные выше.
В другом предпочтительном соединении формулы I G1 представляет собой этил; G2, G3 представляют собой F, CF3 или метил, и другие заместители имеют значения, определенные выше.
В другом предпочтительном соединении формулы I R1 представляет собой метил или Н, и другие заместители имеют значения, определенные выше.
Следующие соединения формулы I или их фармацевтически приемлемые соли являются более предпочтительными:



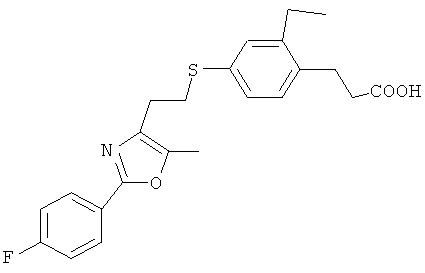



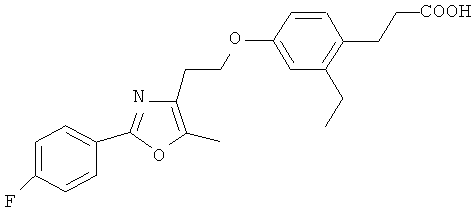

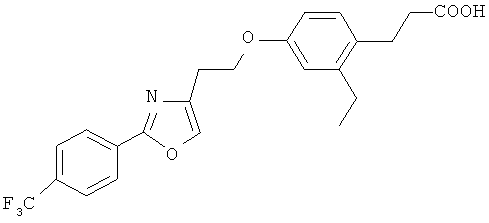
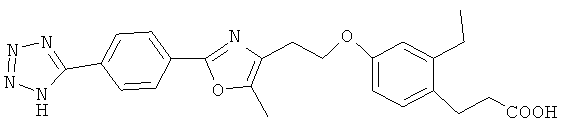




В настоящей заявке термин "алкил" относится к одновалентной линейной или разветвленной насыщенной алифатической углеводородной группе, содержащей атомы углерода в пределах некоторого диапазона. Так, например, "С1-6 и алкил" (или "C1-C6 алкил") относится к любому изомеру гексила и пентила; н-бутилу, изо-бутилу, втор-бутилу и трет-бутилу; н-пропилу и изо-пропилу; этилу; и метилу. В качестве дополнительного примера, "С1-4 алкил" относится к н-бутилу, изо-бутилу, втор-бутилу и трет-бутилу; н-пропилу и изо-пропилу; этилу; и метилу.
Термин "алкенил" относится к одновалентной линейной или разветвленной алифатической углеводородной группе, содержащей одну углерод-углеродную двойную связь и содержащей атомы углерода в пределах некоторого диапазона. Так, например, "С2-С6 алкенил" (или "С2-6 алкенил") относится ко всем изомерам гексенила и пентенила; 1-бутенилу, 2-бутенилу, 3-бутенилу, изо-бутенилу; 1-пропенилу, 2-пропенилу; и этенилу. Что касается настоящего изобретения, алкенил предпочтительно имеет формулу -CH=CH-(CH2)1-3CH3.
Термин "алкинил" относится к одновалентной линейной или разветвленной алифатической углеводородной группе, содержащей одну углерод-углеродную тройную связь и содержащей атомы углерода в пределах некоторого диапазона. Так, например, "С2-С6 алкинил" (или "С2-6 алкинил") относится ко всем изомерам гексинила и пентинила; 1-бутинилу, 2-бутинилу, 3-бутинилу; 1-пропинилу, 2-пропинилу; и этинилу.
Термин "алкилен" относится к двухвалентной линейной или разветвленной алифатической углеводородной группе, содержащей атомы углерода в пределах некоторого диапазона. Так, например, "-C1-6 алкилен-" относится к любому C1-С6 линейному или разветвленному алкилену, и "-C1-4 алкилен-" относится к любому С1-С4 линейному или разветвленному алкилену. Что касается настоящего изобретения, алкилен предпочтительно представляет собой -(CH2)1-6. Более предпочтительно, алкилен включает -(CH2)1-4-, -(CH2)2-4-, -(CH2)1-3-, -(CH2)2-3-, -(CH2)1-2- и -CH2-. Также предпочтительно, алкилен выбран из -CH2-, -CH(CH3)- и -C(CH3)2-.
Термин "циклоалкил" относится к любому моноциклическому алкану, содержащему число атомов углерода в пределах некоторого диапазона. Так, например, "C3-8 циклоалкил" (или "C3-C8 циклоалкил") относится к циклопропилу, циклобутилу, циклопентилу, циклогексилу, циклогептилу и циклооктилу.
Термин "циклоалкенил" относится к любому моноциклическому алкену, содержащему число атомов углерода в пределах некоторого диапазона. Так, например, "C5-8 циклоалкенил" (или "C5-C8 циклоалкенил") относится к циклопентенилу, циклогексенилу, циклогептенилу и циклооктенилу.
Термин "галоген" (или "гало") относится к фтору, хлору, брому и йоду.
Термин "галоалкил" относится к алкилу, как определено выше, в котором один или более атом водорода был замещен атомом галогена (т.е. F, Cl, Br и/или I). Так, например, "C1-6 галогеналкил" (или "C1-C6 галогенированный алкил") относится к C1-С6 линейному или разветвленному алкилу, как определено выше, содержащему один или более атомов галогена в качестве заместителей. Термин "фторалкил" имеет определение, аналогичное приведенному выше, за исключением того, что галогеновый заместитель определен как фтор. Подходящий фторалкил включает группу из (CH2)0-4CF3 (т.е. трифторметила, 2,2,2-трифторэтила, 3,3,3-трифтор-н-пропила и т.д.). Более предпочтительно, фторалкил представляет собой CF3.
Соединение может быть введено в форме его фармацевтически приемлемой соли. Термин "фармацевтически приемлемая соль" относится к соли, которая имеет эффективность ее родоначального соединения, но не проявляет нежелательных биологических свойств или нежелательных свойств в других аспектах (т.е. которая является нетоксичной и безопасной для субъекта). Подходящие соли включают кислотно-аддитивные соли, которые могут быть, например, образованы путем смешивания раствора соединения согласно настоящему изобретению и раствора фармацевтически приемлемых кислот (таких как соляная кислота, серная кислота, уксусная кислота или бензойная кислота). В случае, когда соединение, применяемое согласно настоящему изобретению, содержит кислотный фрагмент (такой как -COOH или фенольная группа), его подходящая фармацевтически приемлемая соль может включать соли щелочных металлов (такие как натриевые соли или калиевые соли), соли щелочноземельных металлов (такие как кальциевые соли или магниевые соли) и соли, образованные из соединения и подходящих органических лигандов (такие как четвертичные аммониевые соли). Кроме того, в присутствии кислотной группы (-COOH) или спиртовой группы может быть применен фармацевтически приемлемый сложный эфир с целью улучшения растворимости или гидролитических свойств соединения.
Другой задачей настоящего изобретения является обеспечение фармацевтической композиции, содержащей соединение формулы (I). При этом, лекарственная форма фармацевтической композиции выбрана из следующих лекарственных форм: таблеток, таблеток, покрытых пленочной оболочкой, таблеток, покрытых сахарной оболочкой, таблеток с кишечнорастворимым покрытием, диспергируемых таблеток, капсул, гранул, растворов для перорального введения и суспензий для перорального введения.
Еще одной задачей настоящего изобретения является обеспечение способа для лечения заболеваний, ассоциированных с α-подтипом, δ-подтипом и γ-подтипом рецепторов, активируемых пролифератором пероксисом (PPARα, PPARδ и PPARγ). При этом, заболевания, ассоциированные с α-подтипом, δ-подтипом и γ-подтипом рецепторов, активируемых пролифератором пероксисом (PPARα, PPARδ и PPARγ), выбраны из гипергликемии, инсулинорезистентности, гиперлипидемии, ожирения и т.д.
В соответствии со способом лечения согласно настоящему изобретению, соединение формулы I, возможно в форме его соли или пролекарства, может быть введено в качестве единственного терапевтического агента или комбинации терапевтических агентов путем любого применимого традиционного способа введения. Несмотря на то, что соединение может быть введено отдельно, обычно его вводят в комбинации с фармацевтическими переносящими средами, которые выбирают в соответствии с выбранным путем введения и стандартной фармацевтической практикой. Соединение согласно настоящему изобретению может быть, например, введено перорально, парентерально (включая подкожную инъекцию, внутривенную инъекцию, внутримышечную инъекцию, внутригрудинную инъекцию или инфузию), с помощью ингаляционного спрея или ректально, в форме единичной дозы лекарственного средства, содержащей эффективное количество соединения и нетоксичные традиционные фармацевтически приемлемые переносящие среды, адъюванты и вспомогательные вещества. Жидкие лекарственные формы, подходящие для перорального введения (такие как суспензии, сиропы, эликсир и т.д.), могут быть приготовлены в соответствии с любым способом, известным в данной области техники, с применением любой традиционной среды, такой как вода, гликоли, масла, спирты и т.д. Твердые составы, подходящие для перорального введения (такие как порошки, пилюли, капсулы и таблетки), могут быть приготовлены в соответствии со способом, известным в данной области техники, с применением твердых вспомогательных веществ, таких как крахмал, сахара, каолин, смазывающие вещества, связующие вещества, дезинтегранты и т.д. Парентеральные композиции могут быть приготовлены в соответствии со способом, известным в данной области техники, традиционно с применением стерилизованной воды в качестве переносящей среды и возможно других компонентов, таких как сорастворители. Растворы для инъекций могут быть приготовлены в соответствии со способом, известным в данной области техники, согласно которому переносящая среда включает солевые растворы, растворы глюкозы или растворы, содержащие смесь солевого раствора и глюкозы. Дополнительное описание способа, подходящего для получения фармацевтической композиции согласно настоящему изобретению и компонентов, подходящих для применения в композиции, можно найти в Remington's Pharmaceutical Sciences, 18th Edition, Edited by A.R.Gennaro, Mack Publishing Co., 1990 и Remington - The Science and Practice of Pharmacy, 21st Edition, Lippincott Williams & Wilkins, 2005.
Другой задачей настоящего изобретения является обеспечение способа для получения соединения формулы (I) со ссылкой на следующую схему.

где
X, Y, R1, G1, G2 и G3 имеют значения, определенные для соединения формулы I, R3 представляет собой уходящую группу, выбранную из ОН, Cl, Br, I, OTs, OMs и т.д.
Предпочтительно, алкил, алкоксил и алкилтио в соответствии с настоящим изобретением содержат 1-6 атомов углерода; и циклоалкил содержит 3-8 атомов углерода.
В предпочтительном варианте реализации настоящего изобретения способ получения включает: нагревание соединения III и соединения IV в ацетонитриле с обратным холодильником в присутствии карбоната калия с целью получения соединения II; омыление соединения II в спиртовом растворе в присутствии щелочи; и подкисление реакционной смеси после завершения реакции с целью получения целевого соединения I.
При этом, соединение IV может быть получено в соответствии с описанием в ссылке на патент WO/2005/054176 с применением следующей схемы:
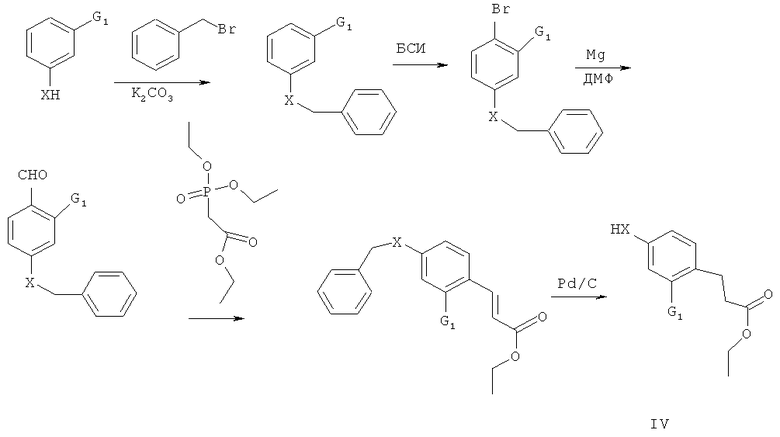
Еще задачей целью настоящего изобретения является обеспечения способа для получения соединения формулы III с применением следующей схемы:
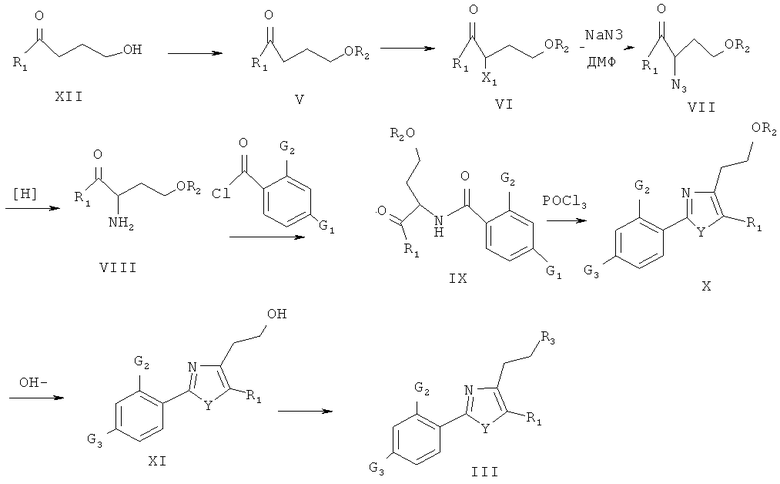
где R1= Η или C1-С6 алкил; X = уходящая группа, выбранная из Cl, Br, I, OTs, OMs и т. д.; R2 = гидроксизащищающая группа; R3 = уходящая группа, выбранная из Cl, Br, 1, OTs, OMs и т. д.
Соединение XII, выступающее в качестве исходного вещества, подвергают защите гидроксильной группы, и затем галоген или гидроксил (может быть превращен в сульфонат) вводят в молекулу соединения в α-положение к ацильному заместителю с целью образования уходящей группы. Далее, полученный продукт подвергают реакции с NaN3 с образованием азида. Азид подвергают гидрогенизации с получением амина и затем подвергают реакции с соответствующим ацилхлоридом с целью получения амида. Амид обрабатывают хлорокисью фосфора с целью получения продукта циклизации. Гидроксизащищающую группу удаляют из продукта циклизации, и незащищенный гидроксил превращали в галоген или сульфат с целью получения соединения III.
Модель скрининга лекарственных средств
Экспериментальную процедуру модели скрининга лекарственных средств проводят следующим образом:
1) Краткое описание модели скрининга, связанного с ядерными рецепторами
Применяя метод гена-репортера, создают модель скрининга для скрининга агониста ядерных рецепторов в живых клетках на основе принципа, заключающегося в том, что ядерный рецептор после активации может активировать транскрипцию регулируемого им гена. Конструируют плазмиду, содержащую ген-репортер, в которой последовательность ДНК, связывающуюся с ядерным рецептором (nuclear receptor element, NRE), вставляют перед геном люциферазы, так что экспрессия гена люциферазы регулируется ядерным рецептором. Плазмиду, содержащую ген-репортер, переносят в клетку одновременно с ядерным рецептором. Ядерный рецептор будет активирован, когда агонист ядерного рецептора будет присутствовать в культуральной среде для клеток. Активируемый рецептор может индуцировать экспрессию гена люциферазы, в то время как количество вырабатываемой люциферазы может быть определено с помощью ее люминесцентного субстрата. Таким образом, интенсивность активации ядерного рецептора соединением может быть определена путем наблюдения интенсивности люминесценции. Для проверки ошибки эксперимента, вызываемой такими факторами, как эффективность трансфекции, количество посеянных клеток, токсичность соединения и т.д., клетки одновременно котрансфицируют плазмидой с геном зеленого флуоресцентного белка (green fluorescent protein, GFP) в качестве внутренней меры. При анализе результатов эксперимента величины интенсивности люминесценции, определяемые для всех лунок, выверяют по величинам для GFP. Результаты эксперимента выражают как относительную кратность активации, при этом за 1 принимают контроль растворителя. Чем больше кратность, тем выше способность к активации.
2) Процедура эксперимента
Подробный протокол для эксперимента по модели скрининга можно найти по следующей ссылке: "Design, synthesis and evaluation of a new class of noncyclic 1,3-dicarbonyl compounds as PPARa selective activators" Bioorg Med Chem Lett. 2004; 14 (13): 3507-11. Подробная процедура описана следующим образом: Необходимые реагенты: соединения, подлежащие проверке (в ДМСО).
(1) День 1: Культивирование и посев клеток
Клетки гепатокарциномы HepG2 (от АТСС, American Type Culture Collection) культивировали в культуральной среде DMEM (модифированной по способу Дульбекко среде Игла) с добавлением 10% инактивированной нагреванием фетальной бычьей сыворотки (FBS, Invitogen, Grand Island, NY, USA) в культуральном флаконе Т-75 (Greiner, Germany), помещенном в инкубатор с 5% CO2 при 37°C и относительной влажности 100%. Когда клетки в культуральном флаконе достигали 80-90% слияния, их обрабатывали 0,25% раствором панкреатического фермента (с ЭДТА) в течение 3 мин и высеивали в 96-луночный культуральный планшет для клеток с плотностью посева, составляющей 2000 клеток/100 мкл/ лунка.
(2) День 2: Трансфекция клеток
На следующий день, когда клетки в 96-луночном культуральном планшете вырастали до 50-80% слияния, выполняли трансфекцию клеток. Система котрансфекции клеток содержала агент для трансфекции FuGene6 (Roche Molecular Biochemicals, Indianapolis, IN, U.S.A.) и 60 нг ДНК (10 нг ретиноидного рецептора X человека (hRXR), 10 нг плазмиды pCMV βGal, 10 нг плазмиды для экспрессии ядерных рецепторов RXR/PPARα, δ, γ, 30 нг плазмиды, содержащей ген-репортер и ген флуоресцентного белка GFP, соответственно).
(3) Обработка лекарственными средствами
Культуральную среду для клеток удаляли немедленно через 24 часа после трансфекции и заменяли 200 мкл свежей среды DMEM, содержащей исследуемое лекарственное средство (с 10% FBS, обработанной активированным углем). Конечные градиенты концентрации исследуемых лекарственных средств составляли 10 мкМ, 5 мкМ, 1 мкМ, 0,1 мкМ, 0,01 мкМ, 0,001 мкМ и 0 мкМ. 0,05 мкМ 2-бромстеариновой кислоты (приобретенной у Sigma, USA) применяли в качестве положительного контроля. Конечная концентрация ДМСО в каждой лунке составляла 0,1%.
(4) Анализ киназной активности
Через 24 ч после обработки лекарственными средствами клетки лизировали с помощью лизирующего раствора (Cell Culture Lysis buffer, Promega) и центрифугировали, и собирали супернатант.Супернатант подвергали взаимодействию с реагентами набора для анализа флуоресценции (Promega), и подсчитывали флуоресценцию с помощью флуориметра (Ascent Fluoroskan FL reader, Thermo Labsystems, Finland) и определяли относительную интенсивность люциферазы. Для анализа активности β-галактозидазы, применяемой в эксперименте в качестве внутренней меры (внутренняя мера для проверки эффективности трансфекции), 50 мкл супернатанта переносили в новый микропланшет, обрабатывали набором Promega и считывали значения с помощью спектрофотометра для прочтения микропланшетов при длине волны 405 нм (Bio-tech Instruments Inc., Winooski, VT, USA) (Sauerberg, P.; Olsen, G.S.; Jeppesen, L.; Mogensen, J. P. et al., J. Med. Chem., 2007, 50, 1495-1503).
3) Анализ:
Средняя эффективная концентрация (EC50) образца представляет собой концентрацию, при которой образец проявляет 50% фармакологический эффект, и является одним из важных параметров для оценки фармакологических эффектов соединения. В настоящем протоколе скрининга данную концентрацию рассчитывали в соответствии с активацией рецептора образцом при 6 разных концентрациях последнего.
4) Результаты скринингового теста
Результаты скринингового теста показывают, что соединения формулы I согласно настоящему изобретению активируют рецепторы α, δ и γ, активируемые пролифератором пероксисом.
1) Активность соединения формулы I in vitro
Активность соединения формулы I in vitro исследовали в соответствии со следующей процедурой:
образец (соединение формулы I) растворяли и разбавляли в различных концентрациях, активность образца по активации рецепторов PPARα, δ, γ исследовали исходя из градиентов концентрации, получали зависимость между концентрацией и активностью и рассчитывали соответствующую величину средней эффективной концентрации (ЕС50).
Результаты:

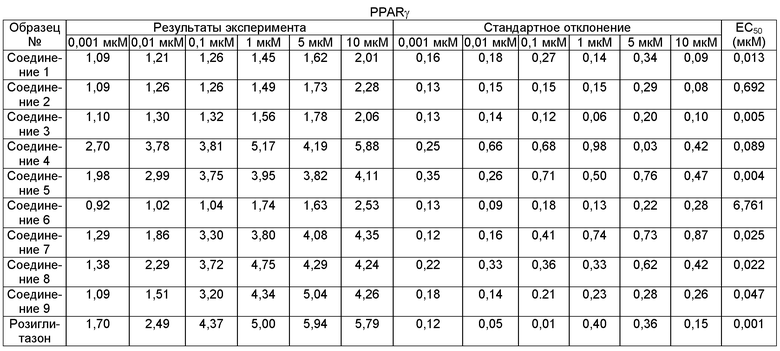
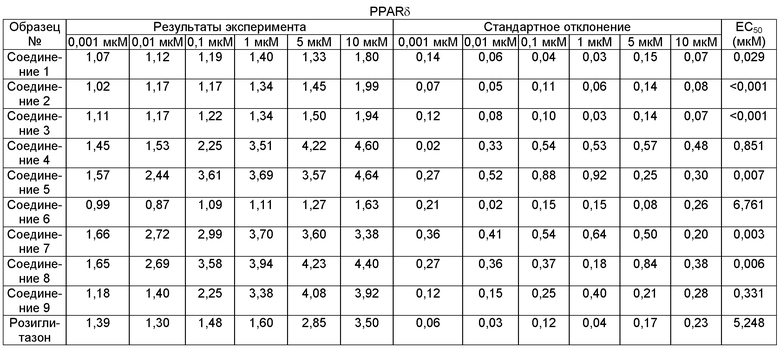
Чем ниже величина ЕС50, тем более высокую активность in vitro проявляет соединение.
Анализ:
Средняя эффективная концентрация (ЕС50) является одним из важных параметров для оценки фармакологических эффектов соединения. В протоколе настоящей модели скрининга наблюдали активирующее действие образца на рецептор при 6 разных концентрациях, и это может дать общее представление для фармакологических характеристик соединения. Профиль концентрация-активность эффектов соединения был подобран путем выполнения итерационного вычисления в соответствии со следующей формулой, и были рассчитаны соответствующие ЕС50:

Как можно увидеть из результатов скрининга, исследуемые соединения 1, 3 и 5 обладают лучшим активирующим действием на рецептор PPARα, рецептор PPARδ и рецептор PPARγ.

Как можно увидеть из приведенной выше таблицы: что касается соединения формулы I, соединения, в которых заместитель G3 представляет собой электрофильную группу, имеют значительно лучшую ЕС50 для рецепторов PPARα, PPARγ и PPARδ, чем соединения, в которых заместитель G3 представляет собой электронодонорную группу, К тому же, EC50 в значительной степени связана с размером заместителя G3. Чем больше заместитель, тем больше ЕС50. Наряду с прочим, наибольшему влиянию подвержена ЕС50 для рецептора PPARα. Кроме того, соединения, в которых заместитель R1 представляет собой метил, имеют значительно более высокую активность in vitro, чем соединения, в которых R1 представляет собой Н.
2) Скрининг части соединений формулы (I) согласно настоящему изобретению на активность in vivo
Соединения, показавшие относительно высокую активность в скрининге in vitro, были исследованы на активность in vivo. Были применены животные модели, такие как крысы линии ZDF, диабетические мыши с генотипом db/db, мыши линии DIO с ожирением, индуцированным диетой, и т.д., с целью определения воздействия лекарственных средств.
В настоящее время изобретатели завершили фармакодинамические исследования на 3 животных моделях диабета, т.е. DB/DB, DIO и ZDF. Что касается ключевых показателей при диабете II типа, включая толерантность к сахару, инсулин плазмы, триглицериды плазмы и т.д., некоторые соединения имеют сходные или лучшие фармакологические эффекты по снижению уровня сахара в крови по сравнению с розиглитазоном. Соединения, разработанные изобретателями, также имеют лучший эффект по снижению уровня холестерина и эффект по снижению массы тела, чем розиглитазон, который является агонистом исключительно рецептора PPARγ. Изобретатели также изучили безопасность взятых для скрининга соединений.
Что касается LD50, основного показателя для оценки острой токсичности лекарственного средства, большинство соединений близки к розиглитазону (3-4 г/кг, перорально). При вскрытии не было найдено видимых явных повреждений в основных внутренних органах, и не было статистического различия в изменении коэффициентов органов.
Согласно данным фармакодинамического исследования в сочетании с проверкой на токсичность, изобретатели предположили, что соединения формулы обладают потенциалом быть разработанными в новые лекарственные средства для лечения диабета II типа.
Примеры
Настоящее изобретение будет объяснено со ссылкой на следующие примеры. Специалисты в данной области техники примут во внимание, что примеры не ограничивают способ получения согласно настоящему изобретению.
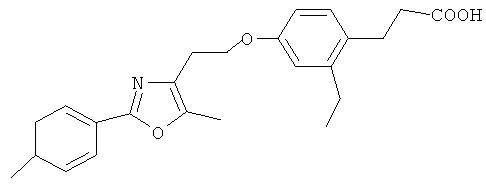
Пример 1: Получение 3-(2-этил-4-{2-[2-(4-фторфенил)-5-метилоксазол-4-ил]этокси} фенил)пропионовой кислоты (соединение 1)
Соединение 1
а) Получение 2-[2-(4-фторфенил)-5-метилоксазол-4-ил]этилметансульфоната
Стадия 1: Получение 4-карбонилпентилбензоата
5-Гидрокси-2-пентанон (0,647 моль) растворили в дихлорметане (400 мл), затем добавили 104 мл (1,294 моль) пиридина. Смесь охладили до температуры ниже 10°C в водно-ледяной бане, и затем медленно добавили по каплям бензоилхлорид (90,15 мл, 0,076 моль), поддерживая температуру реакционной смеси ниже 10°С. После того, как добавление было завершено, смесь нагрели до комнатной температуры и перемешивали в течение 2 дней для протекания реакции. После завершения реакции реакционную смесь промывали водой и экстрагировали дихлорметаном. Органическую фазу сушили с помощью безводного сульфата магния, дихлорметан концентрировали. Продукт перегоняли при пониженном давлении с масляным насосом с получением промежуточного соединения, т.е. 4-карбонилпентилбензоата, с выходом 75%.
1Н ЯМР (400 МГц, CDCl3) δ: 2,04-2,06 (м, 2Н, СН2), 2,15 (с, 3Н, СН3), 2,58-2,61 (м, 2Н, СН2), 4,29-4,33(м, 2Н, ОСН2), 7,41-7,44(м, 2Н, ArH), 7,53-7,55 (м, Н, ArH), 8,00-8,02 (м, 2Н, ArH);
13С ЯМР (75 МГц, CDCl3) δ: 22,9, 30,0, 39,9, 64,1, 128,4, 129,5, 130,2, 133,0, 166,5, 207,6;
МС (+С, ЭС): М=206, найдено 207 (М+1).
Стадия 2: Получение 3-бром-4-карбонилпентилбензоата
Промежуточное соединение, 4-карбонилпентилбензоат (72 ммоль), растворили в 75 мл дихлорметана. Температуру системы поддерживали ниже 5°C, затем медленно добавили по каплям бром (3,7 мл, 72 ммоль). После того, как добавление было завершено, продолжали перемешивание в течение получаса до тех пор, пока окраска реакционной системы не исчезла. Далее продукт промывали водой, сушили с помощью безводного сульфата магния и концентрировали с получением промежуточного соединения, т.е. 3-бром-4-карбонилпентилбензоата, который непосредственно применяли для следующей стадии без дополнительной очистки.
Стадия 3: Получение 3-азидо-4-карбонилпентилбензоата
Промежуточное соединение, 3-бром-4-карбонилпентилбензоат (продукт, полученный согласно предыдущей стадии), растворили в 100 мл ДМФ и затем добавили 9,3 г (143 ммоль) NaN3. Смесь перемешивали в течение ночи при комнатной температуре. После завершения реакции продукт экстрагировали этилацетатом, промывали водой, сушили с помощью безводного сульфата магния и затем концентрировали с получением промежуточного соединения, т.е. 3-азидо-4-карбонилпентилбензоата, который непосредственно применяли для следующей стадии без дополнительной очистки. Стадия 4: Получение 3-(4-фторбензамидо)-4-карбонилпентилбензоата
Промежуточное соединение, 3-азидо-4-карбонилпентилбензоат (продукт, полученный согласно предыдущей стадии), растворили в 200 мл метанола, добавили 2 г катализатора для гидрирования палладия на угле и затем пропускали водород через раствор. После завершения реакции отфильтровывали палладий на угле, и продукт концентрировали с получением промежуточного соединения, т.е. 3-амино-4-карбонилпентилбензоата. Промежуточное соединение, 3-амино-4-карбонилпентилбензоат, растворили в 200 мл этилацетата, охладили до 0°C, добавили карбонат калия (30 г, 216 ммоль) и добавили по каплям и-фторбензоилхлорид (72 ммоль), поддерживая температуру ниже 5°С.После завершения реакции продукт промывали водой и экстрагировали этилацетатом. Органическую фазу сушили с помощью безводного сульфата магния и концентрировали с получением промежуточного соединения, т.е. неочищенного 3-(4-фторбензамидо)-4-карбонилпентилбензоата. Неочищенный продукт отделяли и очищали с получением 16,1 г промежуточного соединения с выходом 65% (общий выход на 3 стадии, т.е. стадию 2, стадию 3 и стадию 4).
1Н ЯМР (400 МГц, CDCl3) δ: 2,32-2,41 (м, 4Н, СН3 и CH2), 2,58-2,65 (м, Н, СН2), 4,30 (т, J=6,0 Гц, 2Н, ОСН2), 4,92 (м, 1Н, NCH), 7,08-7,12 (м, 2Н, ArH), 7,27 (с, уш, Н, NH), 7,41-7,45 (м, 2Н, ArH), 7,53-7,59 (м, Н, ArH), 7,85 (д, J=8,4 Гц, 2Н, ArH), 7,95 (д, J=8,4 Гц, 2Н, ArH);
МС (+С, ЭС): М=343, найдено 344 (М+1).
Стадия 5: Получение 2-[2-(4-фторфенил)-5-метилоксазол-4-ил]этилбензоата
Промежуточное соединение, 3-(4-фторбензамидо)-4-карбонилпентилбензоат (15,4 г, 45 ммоль), полученный согласно предыдущей стадии, растворили в 350 мл толуола и медленно добавили по каплям хлорокись фосфора (8,5 мл, 90 ммоль) при комнатной температуре. После того, как добавление было завершено, смесь нагревали в течение 2,5 часов с обратным холодильником. После завершения реакции реакционный раствор вылили в смесь воды со льдом, экстрагировали толуолом, концентрировали и затем отделяли и очищали с получением промежуточного соединения, т.е. 2-[2-(4-фторфенил)-5-метилоксазол-4-ил]этилбензоата (13,2 г, с выходом 90%).
1Н ЯМР (400 МГц, CDCl3) δ: 2,33 (с, 3Н, СН3), 2,96 (т, J=6,4 Гц, 2Н, CH2), 4,59 (т, J=6,4 Гц, 2H, ОСН2), 7,09-7,13 (м, 2Н, ArH), 7,41-7,44 (м, 2Н, ArH), 7,53-7,55 (м, Н, ArH), 7,95-8,03 (м, 4Н, ArH);
13С ЯМР (75 МГц, CDCl3) δ: 10,2, 25,8, 63,7, 115,7, 115,9, 124,1, 124,2, 127,9, 128,0, 128,4, 129,6, 130,3, 132,5, 132,9, 144,9, 158,8, 162,5, 165,0, 166,5;
МС (+С, ЭС): М=325, найдено 326 (M+1).
Стадия 6: Получение 2-[2-(4-фторфенил)-5-метилоксазол-4-ил]этанола
Промежуточное соединение, 2-[2-(4-фторфенил)-5-метилоксазол-4-ил] этилбензоат (19,5 г, 60 ммоль) растворили в 70 мл этанола и перемешали. Медленно добавили по каплям 10% раствор гидроксида натрия (NaOH: 4,8 г, 120 ммоль), и смесь перемешивали в течение ночи при комнатной температуре. После завершения реакции этанол сконцентрировали, и продукт экстрагировали толуолом, а затем последовательно промыли водой и солевым раствором, сушили с помощью безводного сульфата магния и концентрировали с получением промежуточного соединения, т.е. 2-[2-(4-фторфенил)-5-метилоксазол-4-ил]этанола, который непосредственно применяли для следующей стадии без дополнительной очистки.
Стадия 7: Получение 2-[2-(4-фторфенил)-5-метилокасазол-4-ил] этилметансульфоната
Промежуточное соединение, 2-[2-(4-фторфенил)-5-метилоксазол-4-ил]этанол (продукт, полученный согласно предыдущей стадии), растворили в 200 мл дихлорметана и затем охладили до 0°С. Добавили триэтиламин (100 ммоль) при этой температуре, и медленно добавили по каплям MsCl (60 ммоль). После того, как добавление было завершено, смесь нагрели до комнатной температуры и перемешивали в течение ночи. После завершения реакции продукт промывали водой, экстрагировали дихлорметаном, сушили с помощью безводного сульфата магния, концентрировали, отделяли и очищали с получением соединения 2-[2-(4-фторфенил)-5-метилокасазол-4-ил]этилметансульфоната (15,6 г, с выходом 87% на 2 стадии).
1Н ЯМР (400 МГц, CDCl3) δ: 2,32 (с, 3Н, СН3), 2,91-2,96 (м, 5Н, СН2 и SO2CH3), 4,51 (т, 2Н, ОСН2), 7,09-7,13 (м, 2Н, ArH), 7,92-7,94 (м, Н, ArH);
13СЯМР (75 МГц, CDCl3) δ: 10,1, 26,2, 37,2, 116,0, 123,9, 128,0, 131,1, 145,5, 158,9, 162,5, 162,6, 165,0;
МС (+С, ЭС): М=299, найдено 300 (М+1).
б) Получение этил-3-(2-этил-4-гидроксифенил)пропионата
Соединение может быть получено в соответствии с описанием в ссылке на патент WO/2005/054176.
в) Получение этил-3-(2-этил-4-{2-[2-(4-фторфенил)-5-метилоксазол-4-ил]этокси} фенил)пропионата и 3-(2-этил-4-{2-[2-(4-фторфенил)-5-метилоксазол-4-ил]этокси}фенил)пропионовой кислоты
Исходное фенольное соединение, этил-3-(2-этил-4-гидроксифенил)пропионат (10,4 г, 50 ммоль) и 120 мл ацетонитрила добавили в одногорлую колбу объемом 250 мл и перемешали. Затем добавили исходное сульфонатное соединение, 2-[2-(4-фторфенил)-5-метилоксазол-4-ил]этилметансульфонат (15 г, 50 ммоль) и карбонат калия (К2СО3) (100 ммоль). Смесь нагревали в течение 16 часов с обратным холодильником. После завершения реакции продукт отфильтровали. Осадок на фильтре промыли этилацетатом (3×50 мл) и затем удалили. Фильтраты объединили, и растворитель отогнали при пониженном давлении с получением неочищенного этил-3-(2-этил-4-{2-[2-(4-фторфенил)-5-метилоксазол-4-ил]этокси}фенил)пропионата. Неочищенный этил-3-(2-этил-4-{2-[2-(4-фторфенил)-5-метилоксазол-4-ил]этокси}фенил)пропионат, без очистки, растворили в 120 мл этанола. Добавили 10% NaOH (30 мл) при комнатной температуре, и продолжали перемешивание в течение 3 часов при комнатной температуре. После завершения реакции продукт подкислили добавлением 10% разбавленной соляной кислоты с получением твердого осадка. Осадок отфильтровали посредством фильтрования с отсасыванием с получением неочищенной 3-(2-этил-4-{2-[2-(4-фторфенил)-5-метилоксазол-4-ил]этокси}фенил)пропионовой кислоты. Неочищенный продукт кристаллизовали из смеси этилацетат-петролейный эфир с получением 12,3 г белого твердого вещества с выходом 62% на 2 стадии.
1H ЯМР (400 МГц, CDCl3) δ: 1,22 (т, J=7,6 Гц, 3H, CH3), 2,37 (с, 3H, CH3), 2,59-2.65 (м, 4H, 2CH2), 2,90-2,94 (м, 2H, CH2), 2,97 (т, J=6,4 Гц, 2H, CH2), 4,22 (т, J=6,4 Гц, 2Н, OCH2), 6,69-6,76 (м, 2H, ArH), 7,05-7,15 (м, 3Н, ArH), 7,96-7,98 (м, 2Н, ArH);
13C ЯМР (75 МГц, CDCl3) δ: 10,2, 15,1, 25,7, 26,3, 26,7, 35,4, 66,6, 111,7, 114,9, 115,0, 115,7, 115,9, 124,0, 128,1, 130,0, 132,7, 143,3, 145,1, 157,5, 158,8, 162,5, 165,0, 179,1;
МС (+С, ЭС): М=397, найдено 398 (М+1), 420 (M+Na).
Пример 2:
Следующие соединения были получены путем применения соответствующих исходных веществ в соответствии со способом согласно примеру 1.

Соединение 2, белое твердое вещество, выход: 63%
1H ЯМР (400 МГц, CDCl3) δ: 1,22 (т, J=7,6 Гц, 3H, CH3), 2,59-2,65 (м, 4H, 2CH2), 2,89-2,93 (м, 2H, CH2), 3,09 (т, J=6,4 Гц, 2H, CH2), 4,27 (т, J=6,4 Гц, 2H, OCH2), 6,70-6,73 (м, 2H, ArH), 6,77 (д, J=2,4 Гц, 1H, ArH), 7,07 (д, J=8,4 Гц, 1H, ArH), 7,62 (с, 1H, OCH=), 7,70 (д, J=8,4 Гц, 2H, ArH), 8,13 (д, J=8,4 Гц, 2Н, ArH);
13C ЯМР (75 МГц, CDCl3) δ: 15,1, 25,7, 26,7, 26,9, 35,2, 66,0, 111,7, 115,1, 125,7, 125,8, 126,7, 129,4, 130,2, 130,6, 136,1, 139,4, 143,4, 157,4, 178,0;
МС (+С, ЭС): М=433, найдено 438 (М+1), 456 (M+Na).
Соединение 3, белое твердое вещество, выход: 58%
1H ЯМР (400 МГц, CDCl3) δ: 1,20 (т, J=7,6 Гц, 3H, CH3), 2,40 (с, 3H, CH3), 2,57-2,64 (м, 4H, 2CH2), 2,88-2,92 (м, 2H, CH2), 2,98 (т, J=6,4 Гц, 2H, CH2), 4,22 (т, J=6,4 Гц, 2H, OCH2), 6,67-6,74 (м, 2H, ArH), 6,73 (д, J=2,4 Гц, 1H, ArH), 7,67 (д, J=8,4 Гц, 2H, ARH), 8,08 (д, J=8,4 Гц, 2Н, ArH);
13C ЯМР (75 МГц, CDCl3) δ: 10,3, 15,2, 25,7, 26,3, 26,7, 35,5, 66,5, 111,7, 115,0, 125,6, 125,7, 126,1, 129,7, 130,0, 130,8, 131,2, 133,5, 143,3, 146,1, 157,4, 158,2, 178,7;
МС (+С, ЭС): М=447, найдено 448 (М+1), 470 (M+Na).
Соединение 4, белое твердое вещество, выход: 72%
1H ЯМР (400 МГц, CDCl3) δ: 1.17 (т, J=7,6 Гц, 3H, CH3), 1,27 (с, 9H, C(CH3)3), 2,35 (с, 3H, CH3), 2,57 (м, 4H, 2CH2), 2,86 (м, 2H, CH2), 2,96 (т, J=6,4 Гц, 2H, CH2), 4,17 (т, J=6,4 Гц, 2H, -OCH2), 6,67 (м, 2H, ArH), 7,02 (д, J=4,0 Гц, 1H, ArH), 7,43 (д, J=4,4 Гц, 2Н, ArH), 7,89 (д, J=4,4 Гц, 2Н, ArH);
13C ЯМР (100 МГц, CDCl3) 5: 10,2, 15,1, 25,6, 26,2, 26,8, 31,1 (3С), 34,8, 35,6, 66,5, 111,5, 114,8, 124,6, 125,6 (2С), 125,7 (2С), 129,6, 130,0, 132,3, 143,2, 144,7, 153,1, 157,3, 159,6, 178,5;
МС (+С, ЭС): М=435, найдено 436 (М+1).

Соединение 5, белое твердое вещество, выход: 71%
1H ЯМР (400 МГц, CDCl3) δ:1,19 (т, J=7,6 Гц, 3H, CH3), 2,35 (с, 3H, CH3), 2,37 (с, 3H, CH3), 2,59 (м, 4H, 2CH2), 2,88 (т, J=7,6 Гц, 2H, CH2), 2,96 (т, J=6,4 Гц, 2H, CH2), 4,18 (т, J=6,4 Гц, 2H, CH2), 6,68 (м, 2H, ArH), 7,03 (д, J=8,0 Гц, 1H, ArH), 7,23 (д, J=8,0 Гц, 2Н, ArH), 7,85 (д, J=7,6 Гц, 2Н, ARH);
13C ЯМР (100 МГц, CDCl3) δ: 10,3, 15,2, 21,5, 25,7, 26,2, 26,9, 35,7, 66,6, 111,6, 114,9, 124,7, 126,0 (2С), 129,4 (2С), 129,7, 130,1, 132,3, 140,2, 143,3, 144,8, 157,4, 159,8, 178,3;
МС (+С, ЭС): М=393, найдено 394 (М+1).
Соединение 6, белое твердое вещество, выход: 62%
1H ЯМР (400 МГц, d-ДМСО) δ 1,11 (т, J=7,6 Гц, 3H, CH3), 2,35 (с, 3H, CH3), 2,43-2,63 (м, 4H, 2CH2), 2,74 (т, J=6,4 Гц, 2H, CH2), 2,90 (т, J=6,4 Гц, 2H, CH2), 4,17 (т, J=6,4 Гц, 2H, OCH2), 6,61-6,68 (м, 2H, ArH), 6,70 (д, J=2,4 Гц, 1H, ArH), 7,67 (д, J=8,4 Гц, 2H, ArH), 8,08 (д, J=8,4 Гц, 2H, ArH);
13C ЯМР (100 МГц, d-ДМСО) δ: 9,8, 15,1, 25,0, 25,6, 26,2, 35,0, 65,9, 111,5, 114,4, 126,1, 127,3, 128,7, 129,5, 130,2, 133,2, 142,8, 145,6, 155,6, 156,7, 157,5, 173,8;
МС (+С, ЭС): М=447, найдено 448 (М+1), 470 (M+Na).
Соединение 7, белое твердое вещество, выход: 70%
1H ЯМР (400 МГц, d-ДМСО) δ: 1,20 (т, J=7,6 Гц, 3H, CH3), 2,42 (с, 3H, CH3), 2,57-2,61 (м, 4H, 2CH2), 2,89 (т, J=7,6 Гц, 2H, CH2), 2,99 (т, J=6,4 Гц, 2H, CH2), 4,21 (т, J=6,4 Гц, 2H, OCH2), 6,67-6,73 (м, 2H, ArH), 7,05 (д, J=8,0 Гц, 1H, ARH), 8,13 (д, J=8,0 Гц, 2H, ArH), 8,29 (д, J=7,6 Гц, 2H, ArH);
13С ЯМР (100 МГц, d-ДМСО) δ: 9,8, 15,0, 25,0, 25,5, 26,2, 35,0, 65,7, 111,4, 114,3, 124,1, 126,2, 129,4, 130,2, 132,2, 134,0, 142,7 146,9, 147,5, 156,4, 156,6, 173,9;
МС (+С, ЭС): М=424, найдено 425 (М+1), 447 (M+Na).
Соединение 8, белое твердое вещество, выход: 52%
1H ЯМР (400 МГц, CDCl3) 5: 0,87 (т, J=7,6 Гц, 3H, CH3), 2,40 (с, 3H, CH3), 2,59-2,61 (м, 4H, 2CH2), 2,74-2,98 (м, 4H, 2CH2), 4,20 (т, J=6,4 Гц, 2H, OCH2), 6,66-6,72 (м, 2H, ArH), 7,05 (д, J=2,4 Гц, 1H, ArH), 7,70 (д, J=8,4 Гц, 2H, ArH), 8,06 (д, J=8,4 Гц, 2H, ArH);
13C ЯМР (100 МГц, CDCl3) δ: 10,3, 15,2, 25,6, 26,3, 26,7, 35,4, 66,4, 111,6, 112,9, 114,9, 118,5, 126,2, 129,7, 130,0, 131,3, 132,5, 133,9, 143,3, 146,7, 157,3, 157,6, 178,5;
MC (+C, ЭС): M=404, найдено 405 (М+1), 427 (M+Na).

Соединение 9, белое твердое вещество, выход: 64%
1H ЯМР (400 МГц, CDCl3) δ: 1,12 (т, J=7,2 Гц, 3H, CH3), 2,37 (с, 3H, CH3), 2,43 (т, J=7,2 Гц, 2H, CH2), 2,51-2,56 (м, 4H, 2CH2), 2,72 (т, J=7,2 Гц, 2H, CH2), 2,91 (м, 2H, CH2), 4,17 (т, J=7,2 Гц, 2H, OCH2), 6,68-6,70 (м, 2H, ArH), 7,04 (д, J=7,6 Гц, 1H, ArH), 7,51 (с, уш, 1H, ArH), 7,93-8,04 (м, 4H, ArH), 8,12 (с, уш, 1H, ArH);
13C ЯМР (100 МГц, CDCl3) δ: 9,8, 15,2, 25,0, 25,6, 26,3, 35,1, 65,9, 111,6, 114,5, 125,2, 128,2, 129,2, 129,6, 130,3, 133,0, 135,2, 142,8, 145,7, 156,7, 157,7, 167,2, 173,9;
MC (+C, ЭС): M=422, найдено 423 (M+1), 445 (M+Na).
Соединение 10, белое твердое вещество, выход: 65%
1H ЯМР (400 МГц, CDCl3) δ: 1,21 (т, J=7,6 Гц, 3H, CH3), 2,58-2,64 (м, 4H, 2CH2), 2,89-2,93 (м, 2H, CH2), 3,07 (т, J=6,4 Гц, 2H, CH2), 4,25 (т, J=6,4 Гц, 2H, OCH2), 6,69-6,77 (м, 2H, ArH), 7,06-7,15 (м, 3H, ArH), 7,55 (с, 1H, OCH=), 7,98-8,02 (м, 2H, ArH);
13C ЯМР (75 МГц, CDCl3) δ: 15,2, 25,7, 26,7, 26,8, 35,4, 66,1, 111,7, 115,0, 115,8, 116,1, 123,8, 128,5, 129,7, 130,2, 135,3, 138,9, 143,4, 157,4, 160,8, 162,8, 165,3, 178,5;
MC (+C, ЭС): M=383, найдено 384 (M+1).
Соединение 11, белое твердое вещество, выход: 54%
1Н ЯМР (400 МГц, CDCl3) δ: 1,22 (т, J=7,6 Гц, 3H, CH3), 1,33 (т, J=7,6 Гц, 3H, CH3), 2,59-2,66 (м, 4H, 2CH2), 2,79 (к, J=7,6 Гц, 2H, CH2), 2,90-2,94 (м, 2H, CH2), 3,01 (т, J=6,4 Гц, 2H, CH2), 4,23 (т, J=6,4 Гц, 2H, OCH2), 6,69-6,75 (м, 2H, ArH), 7,07 (д, J=7,2 Гц, 1H ArH), 7,70 (д, J=8,0 Гц, 2H, ArH), 8,11 (д, 7=8,0 Гц, 2H, ArH);
MC (+С, ЭС): M=461, найдено 462 (M+1), 484 (M+Na).
Группа изобретений относится к соединению формулы (I) или его фармацевтически приемлемым солям формулы (I), где X представляет собой О, S; Υ представляет собой О, S; R1 независимо представляет собой Н, алкил; G1 представляет собой этил; G2 и G3 каждый независимо выбраны из Н, алкила, трифторметила, галогена, нитро, амидо, циано и тетразолила. Также изобретение относится к фармацевтической композиции, обладающей активирующим действием на α-подтип, δ-подтип и γ-подтип рецепторов, активируемых пролифератором пероксисом, содержащей эффективное количество соединения формулы (I)или его фармацевтически приемлемых солей. Соединения формулы (I) применяют для лечения или получения лекарственного средства для лечения или предотвращения заболеваний, ассоциированных с α-подтипом, δ-подтипом и γ-подтипом рецепторов, активируемых пролифератором пероксисом. Соединения формулы (I) получают путем взаимодействия соединения формулы (III) и соединения формулы (IV) при нагревании в ацетонитриле с обратным холодильником в присутствии карбоната калия с получением соединения формулы (II), с последующим омылением соединения формулы (II) в спиртовом растворе в присутствии щелочи и подкисления реакционной смеси после завершения реакции с получением соединения формулы (I). При этом Χ, Υ, R1, G1, G2 и G3 имеют значения, определенные выше, R3 представляет собой уходящую группу, выбранную из ОН, Cl, Br, I, OTs, OMs. Технический результат - соединения на основе фенилпропионовой кислоты, обладающие активирующим действием на рецепторы, активируемые пролифератором пероксисом (PPARα, δ, γ). 5 н. и 10 з.п. ф-лы, 2 пр.


1. Соединение формулы (I) или его фармацевтически приемлемые соли:
где
X представляет собой О, S;
Υ представляет собой О, S;
R1 независимо представляет собой Н, алкил;
G1 представляет собой этил;
G2 и G3 каждый независимо выбраны из Н, алкила, трифторметила, галогена, нитро, амидо, циано и тетразолила.
2. Соединение по п. 1, отличающееся тем, что X представляет собой S или О, Υ представляет собой О.
3. Соединение по п. 1, отличающееся тем, что R1 независимо представляет собой Η или С1-С6 алкил.
4. Соединение по п. 3, отличающееся тем, что R1 представляет собой метил или Н.
5. Соединение по п. 1, отличающееся тем, что G2 и G3 каждый независимо выбраны из Н, C1-С6 алкила, трифторметила, F, Cl, Вr, нитро, амидо, циано и тетразолила.
6. Соединение по п. 5, отличающееся тем, что G1 представляет собой этил; G2 и G3 представляют собой F, CF3 или метил.
7. Соединение по п. 1, выбранное из:


или его фармацевтически приемлемые соли.
8. Фармацевтическая композиция, обладающая активирующим действием на α-подтип, δ-подтип и γ-подтип рецепторов, активируемых пролифератором пероксисом, содержащая эффективное количество соединения по любому из пп.1-7 или его фармацевтически приемлемые соли.
9. Фармацевтическая композиция по п.8 с лекарственной формой, выбранной из
таблеток, таблеток, покрытых пленочной оболочкой, таблеток, покрытых сахарной оболочкой, таблеток с кишечнорастворимым покрытием, диспергируемых таблеток, капсул, гранул, растворов для перорального введения и суспензий для перорального введения.
10. Применение соединения по любому из пп.1-7 для получения лекарственного средства для лечения или предотвращения заболеваний, ассоциированных с α-подтипом, δ-подтипом и γ-подтипом рецепторов, активируемых пролифератором пероксисом.
11. Применение по п.10, отличающееся тем, что заболевания, ассоциированные с α-подтипом, δ-подтипом и γ-подтипом рецепторов, активируемых пролифератором пероксисом, выбраны из гипергликемии, инсулинорезистентности, гиперлипидемии и ожирения.
12. Применение соединения по любому из пп. 1-7 для лечения и предотвращения заболеваний, ассоциированных с α-подтипом, δ-подтипом и γ-подтипом рецепторов, активируемых пролифератором пероксисом.
13. Способ получения соединения формулы (I) по п. 1, включающий
где
Χ, Υ, R1, G1, G2 и G3 имеют значения, определенные в п. 1, R3 представляет собой уходящую группу, выбранную из ОН, Cl, Br, I, OTs, OMs.
14. Способ по п. 13, включающий нагревание соединения формулы (III) и соединения формулы (IV) в ацетонитриле с обратным холодильником в присутствии карбоната калия с получением соединения формулы (II); омыление соединения формулы (II) в спиртовом растворе в присутствии щелочи; и подкисление реакционной смеси после завершения реакции с получением целевого соединения формулы (I).
15. Способ по п. 13 или 14, отличающийся тем, что соединение (III) получают в соответствии со следующим способом:

где
R1 представляет собой Η или C1-С6 алкил;
X представляет собой уходящую группу, выбранную из Cl, Br, I, OTs и OMs;
R2 представляет собой гидроксизащищающую группу;
R3 представляет собой уходящую группу, выбранную из Cl, Br, I, OTs, OMs;
Y, R1, G2 и G3 имеют значения, определенные в п. 1.
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Способ модификации полимерных изделий | 1989 |
|
SU1666472A1 |
| НОВЫЕ СОЕДИНЕНИЯ 2-АРИЛТИАЗОЛА В КАЧЕСТВЕ АГОНИСТОВ PPAR α И PPAR γ | 2003 |
|
RU2296754C2 |
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| Приспособление для суммирования отрезков прямых линий | 1923 |
|
SU2010A1 |
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |

