Область техники, к которой относится изобретение
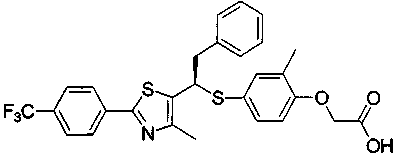
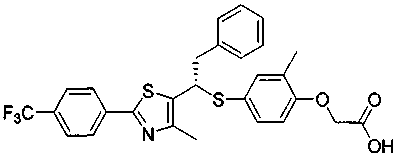
Данное изобретение относится к производному тиазола, представленному формулой 1, как PPARδ лиганду (рецептор δ, активируемый пролифераторами пероксисом), который может быть использован для лечения ожирения, гиперлипидемии, артериосклероза, диабета, деменции, болезни Альцгеймера и болезни Паркинсона, и использован для укрепления мышц или улучшения памяти, и к фармацевтической композиции, косметической композиции, диетическому пищевому продукту, диетическим напиткам, добавке к пищевому продукту и кормам для животных, содержащих указанное соединение.
Формула 1
Уровень техники
Среди ядерных рецепторов, как известно, PPAR (рецептор, активируемый пролифераторами пероксисом) имеет три подтипа, которые представляют собой PPARα, PPARγ и PPARδ (Nature, 1990, 347, p645-650., Proc. Natl. Acad. Sci. USA 1994, 91, p7335-7359). PPARα, PPARγ, и PPARδ обладают тканеспецифическими функциями in vivo и различные области для экспрессии. PPARα, главным образом, экспрессирован в сердце, почке, скелетной мышце, толстом кишечнике у людей (Mol. Pharmacol. 1998, 53, p14-22., Toxicol. Lett. 1999, 110, p119-127., J. Biol. Chem. 1998, 273, p16710-16714), и включен в β-окисление пероксисомы и митохондрии (Biol. Cell. 1993, 77, p67-76., J. Biol. Chem. 1997, 272, p27307-27312). PPARγ экспрессирован в скелетной мышце в малом количестве, но, главным образом, экспрессирован в жировой ткани, чтобы вызывать дифференцировку адипоцитов и сохранять энергию в форме жира, и включен в гомеостатическое регулирование инсулина и глюкозы (Moll. Cell. 1999, 4, p585-594., р597-609., p611-617). PPARδ эволюционно сохраняется в теплокровных, включая людей и позвоночных, включающих грызунов и асцидий. Предшествующие исследования подтверждают, что PPARδ играет важную роль в репродуктивной экспрессии клеток (Genes Dev. 1999, 13, p1561-1574) и имеет физиологические функции по дифференцировке нейронных клеток (J. Chem. Neuroanat. 2000, 19, p225-232) в центральной нервной системе (ЦНС/CNS) и заживлению раны с противовоспалительным эффектом (Genes Dev. 2001, 15, p3263-3277., Proc. Natl. Acad. Sci. USA 2003, 100, p6295-6296). Недавние исследования также подтверждают, что PPARδ включен в дифференцировку адипоцитов и метаболизм липидов (Proc. Natl. Acad. Sci. USA 2002, 99, p303-308., Mol. Cell. Biol. 2000, 20, p5119-5128). Например, PPARδ активирует экспрессию ключевого гена, включенного в β-окисление в катаболизме жирных кислот, и разобщение белков (UCP), гена, включенного в энергетический метаболизм, который вносит эффект в положительную динамику ожирения (Nature, 2000, 406, p415-418., Cell 2003, 113, p159-170). Активация PPARδ повышает уровень HDL (липопротеин высокой плотности), дает положительную динамику по диабету типа 2 без изменений массы (Proc. Natl. Acad. Sci. USA 2001, 98, p5306-5311, 2003, 100, p15924-15929, 2006, 103, p3444-3449) и способствует лечению артериосклероза ингибированием гена, связанного с артериосклерозом (Science, 2003, 302, p453-457). Поэтому PPARδ лиганд может разрабатываться как лекарственное средство для лечения метаболических заболеваний, таких как ожирение, диабет, гиперлипидемия и артериосклероз.
PPARδ регулирует митохондриальный биосинтез. Когда PPARδ искусственно продолжительно экспрессировали в мышцах мыши, митохондриальный биосинтез повышался и значительно увеличивалось мышечное волокно типа I в дополнение к увеличению β-оксидазы жирных кислот. Поэтому продолжительность постоянного беганья и расстояние повышались соответственно до 67% и 92% при сравнении с мышью дикого типа (PloS Biology, 1004, 1:e294). Повышение митохондриального биосинтеза имеет положительное влияние на усиление функции головного мозга. Если митохондрия в клетке головного мозга разрушена окислительным стрессом, то значительно снижается память (Proc. Natl. Acad. Sci. USA 2002, 99, p2356-2361). Деменция, болезнь Альцгеймера и болезнь Паркинсона представляют собой соответствующие дегенеративные заболевания, которые показывают значительное снижение к обучению и памяти. Поэтому митохондриальное пролиферирующее средство, разработанное в данном изобретении, не только дает вклад в улучшение памяти, но также может быть разработано как терапевтическое средство для болезни Альцгеймера и болезни Паркинсона.
Разработанные синтетические PPARδ лиганды до сих пор имеют меньшую селективность по сравнению с другими PPARα и PPARγ лигандами. Ранее созданный селективный лиганд представлял собой L-631033, разработанный фирмой Merk (J. Steroid Biochem. Mol. Biol. 1997, 63, p1-8), который был получен введением функциональной группы, способной фиксировать боковую цепь на основе морфологии ее природной жирной кислоты. Та же самая группа исследователей позже сообщала о более эффективном лиганде L-165041 (J. Med. Chem. 1996, 39, p2629-2654), в котором соединение, известное как лейкотриеновый агонист, функционирует, чтобы активировать PPARδ человека. Данное соединение проявляет высокую селективность к hPPARδ, которая равна 10-кратной селективности PPARα или PPARγ. И ЕС50 данного соединения составляла 530 нМ. Другие лиганды L-796449 и L-783483 имеют улучшенную аффинность (ЕС50=7,9 нМ), но едва имеют селективность к другим hPPAR подтипам.
Glaxo-Smith-Kline опубликовал сведения о GW2433 (Chem Biol. 1997, 4, p909-918), PPARα активатора, который представляет собой лиганд Y-типа, содержащий структуру, аналогичную кристаллической структуре кармана PPARδ лиганда. В отличие от обычных лигандов, известных до сих пор, данный лиганд имеет структуру Y-типа, содержащую бензольное кольцо, которое способствует пространственному присоединению к карману PPARδ лиганда. Однако данный лиганд представляет собой двойственно-активный лиганд, имеющий активность к hPPARα, также предполагая, что селективность к PPARδ понижена. PPARδ селективный лиганд GW501516 ([2-метил-4-[[[4-метил-2-[4-(трифторметил)фенил]1,3-тиазол-5-ил]метил]сульфанил]фенокси]уксусная кислота), недавно разработанный GlaxoSmithKline, проявляет лучший физиологический эффект, чем любые другие лиганды, созданные ранее (Proc. Natl. Acad. Sci. USA 2001, 98, p5306-5311).
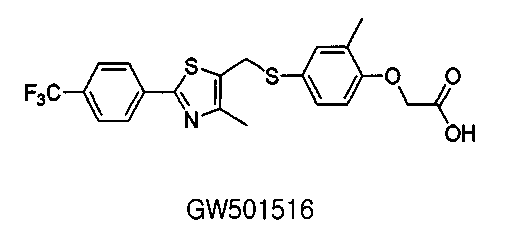
Данное соединение GW501516 обладает отличной аффинностью (1-10 нМ) к PPARδ и отличной селективностью к PPARα или PPARγ также, которая составляет, по меньшей мере, 1000-кратную селективность более ранних лигандов.
Однако PPARδ активность, вызываемая всеми лигандами, созданными до сих пор, является результатом только 30-40% общих лигандносвязывающих карманов.
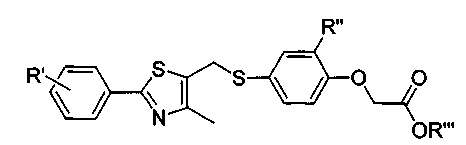
WO 2001-00603, заявленная группой Glaxo, описывает соединение, представленное следующей формулой А, содержащей GW501516 в качестве селективного активатора PPARδ. Однако данное описание включает только часть тест-результатов соединения GW501516 с использованием резус/Rhesus модели.
Формула А
в которой R'означает CF3 или F, R'' означает H, CH3 или Cl, и R''' означает H, CH3 или CH2CH3.
Производное тиазола, представленное формулой В, в качестве PPARδ селективного активатора было описано в заявке WO 2002-62774, поданной группой Glaxo.
Формула В
Заявка WO 2003-072100, поданная фирмой Eli Lilly, описывает фармацевтическую композицию для селективного регулирования PPARδ, содержащую соединение, представленное следующей формулой С:
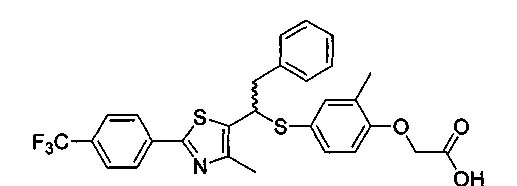
Формула С
Однако данное описание только декларирует, что данная композиция была получена, которая получена как рацемат, но не как оптический изомер, содержащий два типа. И данный документ только описывает М++1 величину по данным масс-спектрометрии полученного рацемата, который подтвержден с помощью 1Н-ЯМР и действует как селективный активатор PPARδ, но отсутствует упоминание о фармакологическом эффекте в качестве селективного активатора PPARδ.
Раскрытие
Техническая проблема
Авторы настоящего изобретения получили оптически-активное соединение, обладающее высокой PPARδ селективностью среди рацемических соединений производного тиазола, описанного в WO 2003-072100. Поэтому предмет данного изобретения состоит в том, чтобы предложить оптически-активное соединение, обладающее высокой PPARδ селективностью, и фармацевтическую композицию, функциональную косметическую композицию и композицию для диетического пищевого продукта и кормов для животных, содержащую оптически-активное соединение производного тиазола.
Техническое решение
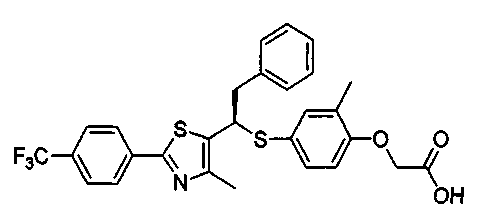
Данное изобретение относится к производному тиазола, представленному формулой 1, как PPARδ лиганду (рецептор δ, активируемый пролифераторами пероксисом), который может быть использован для лечения ожирения, гиперлипидемии, артериосклероза, диабета, деменции, болезни Альцгеймера и болезни Паркинсона, и для укрепления мышц или улучшения памяти и для фармацевтической композиции, косметической композиции, и композиции диетического пищевого продукта и кормов для животных, содержащей указанное соединение.
Формула 1
Заявка WO 2003-072100 описывает производное тиазола, представленное формулой С. Но она только описывает М++1 величину по данным масс-спектрометрии полученного рацемата, который подтвержден с помощью 1Н-ЯМР и действует как селективный активатор PPARδ; она не описывает фармакологический эффект в качестве селективного активатора PPARδ.
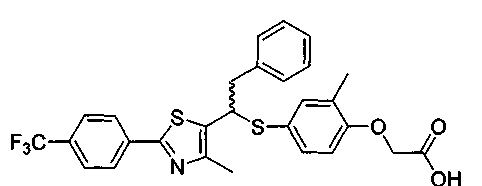
Формула С
Вышеприведенное соединение формулы С содержит хиральный атом углерода и таким образом имеются его стереоизомеры.
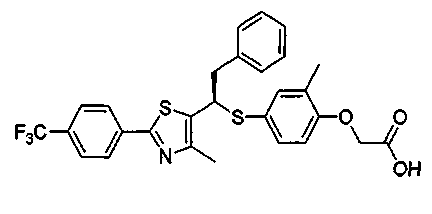
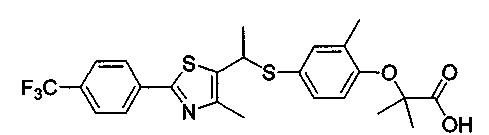
Авторы настоящего изобретения утверждают, что изомер R-формы, представленный формулой 1, оптически активный изомер рацемического соединения формулы С, обладает высокой селективной активностью к PPARδ, но изомер S-формы, представленный формулой 2, показывает значительно пониженную активность к PPARδ.
Формула 2
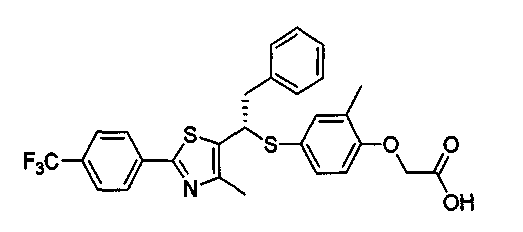
Таким образом, соединение формулы 1 данного изобретения рассматривается как селективное изобретение в отношении заявки WO 2003-072100.
Соединение, представленное формулой 1 данного изобретения, может быть получено следующими реакциями для формулы 1.
Реакции для формулы 1
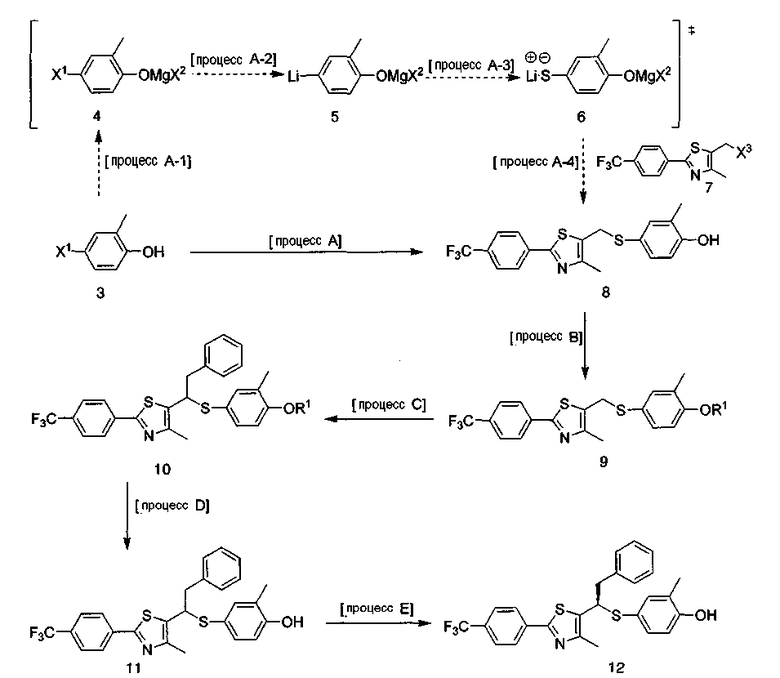
где R1 представляет собой защитную группу фенола, которая может быть С1-С4низшей алкильной, аллильной, алкилсилильной, алкиларилсилильной или тетрагидропиранильной; R2 представляет собой защитную группу карбоновой кислоты, содержащую С1-С4-алкил или аллил; Х1 представляет собой атом брома или атом иода; Х2 и Х3 независимо представляют собой атом хлора, атом брома, атом иода или удаляемую группу, обладающую реакционной способностью с нуклеофильным замещением.
Далее в настоящем документе способ получения соединения изобретения описан подробно. Однако нижеследующие описания не могут ограничивать ни объем, ни сущность данного изобретения.
[Процесс А] Получение соединения, представленного формулой 8
Для получения соединения, представленного формулой 8, 4-галоген-2-метилфенол, соединение, представленное формулой 3, обрабатывали реактивом Гриньяра, чтобы защитить фенольную группу без самостоятельного процесса выделения, и подвергали взаимодействию с металлорганическим реагентом и S постадийно, и наконец подвергали взаимодействию с соединением, представленным формулой 7. Данный процесс имеет 4 подстадии реакций, протекающих подряд.
Данные подстадии реакций описаны подробно в настоящем документе далее.
(Процесс А-1): безводный растворитель, используемый в данном процессе, выбирают из группы, состоящей из таких отдельных растворителей, как диэтиловый эфир, тетрагидрофуран, гексан и гептан, и из смешанных растворителей, содержащих, по меньшей мере, два из данных растворителей. В большей степени предпочитают выбирать диэтиловый эфир, тетрагидрофуран, или смешанный растворитель, содержащий диэтиловый эфир и тетрагидрофуран в качестве безводного растворителя.
Реактив Гриньяра, используемый в данном описании, может быть выбран из группы, состоящей из метил-, этил-, н-пропил-, изо-пропил-, н-бутил-, втор-бутилмагнийхлорида (R2MgCl) и алкилмагнийбромида (R2MgBr). Среди перечисленных соединений изо-пропилмагнийхлорид [(CH3)2CHMgCl] является самым предпочтительным.
Температура реакции зависит от растворителя, но обычно ее устанавливают при -20~40°С, и предпочтительно при 0°С~комнатная температура (25°С). Время реакции зависит от температуры реакции и растворителя, но обычно оно составляет 10-60 минут и предпочтительно 10-30 минут.
(Процесс А-2 и процесс А-3): металлорганический реагент, используемый для замещения галогена литием, может быть выбран из группы, состоящей из н-бутиллития, втор-бутиллития и трет-бутиллития. Среди перечисленных соединений трет-бутиллитий является самым предпочтительным.
Предпочитают S в виде мелких частиц, которую непосредственно добавляют в растворитель.
Температура реакции зависит от растворителя, но обычно ее устанавливают при -78 ~ 25°С. Температура реакции для замещения галогена на металл предпочтительно равна -75°С, и температура для введения S находится в интервале -75 ~ комнатная температура (25°С). Для реакции замещения галогена на металл требуется 10-30 минут и для реакции введения S требуется 30-90 минут.
(Процесс А-4): 5-галогенметил-4-метил-2-[4-(трифторметил)фенил]тиазол, соединение, представленное формулой 7, используемое в данном процессе, получали известным способом (WO 03/106442). Галоген данного соединения, представленного формулой 7, выбирают из группы, состоящей из хлора, брома и иода. И среди перечисленных галогенов хлор является самым предпочтительным.
Температура реакции зависит от растворителя, но обычно ее устанавливают при -78 ~ 25°С, предпочтительнее, при 0 ~ 10°С. Время реакции обычно составляет 10-120 минут и, предпочтительно, 10-60 минут.
[Процесс В] Получение соединения, представленного формулой 9
Для получения соединения, представленного формулой 9, соединение, представленное формулой 8, предпочтительно подвергают взаимодействию с соединением, используемым в качестве защитной группы фенола, в присутствии основания.
Апротонный полярный растворитель, используемый в данном процессе, выбирают из группы, состоящей из N,N-диметилформамида, N,N-диметилацетамида, диметилсульфоксида, ацетонитрила, ацетона, этилацетата, четыреххлористого углерода, хлороформа и дихлорметана. Простой эфир в данном описании может быть выбран из группы, состоящей из тетрагидрофурана, диоксана, диметоксиэтана, диэтиленгликольдиметилового эфира и триэтиленгликольдиметилового эфира. Ароматический углеводород представлен бензолом, толуолом и ксилолом. В качестве растворителя в данном описании предпочитают апротонный полярный растворитель, и особенно N,N-диметилформамид, хлороформ или дихлорметан являются более предпочтительными.
Основание в данном описании представляет собой амин, включающий пиридин, триэтиламин, имидазол и N,N-диметиламинопиридин. Для реакции алкильной или аллильной этерифицированной защитной группы используют такие основания как гидроксид натрия, гидроксид калия, карбонат натрия и карбонат калия. В особенности, имидазол и карбонат калия являются более предпочтительными.
Алкилсилилгалогенид или алкиларилсилилгалогенид используют в качестве силильной защитной группы и 3,4-дигидро-2Н-пиран используют в качестве тетрагидропиранильной защитной группы.
Температура реакции зависит от растворителя, но обычно ее устанавливают при 10 ~ 80°С, предпочтительнее при 0°С ~ комнатная температура (25°С). Время реакции зависит от температуры реакции и растворителя, но обычно требуется от одного часа до одних суток. В большей степени предпочитают заканчивать реакцию в течение 4 часов.
[Процесс С] Получение соединения, представленного формулой 10
Для получения соединения, представленного формулой 10, α-протон простого тиоэфира соединения, представленного формулой 9, обрабатывают сильной щелочью с получением нуклеофила, который подвергают взаимодействию с различными электрофилами.
Безводный растворитель, используемый в данном процессе, выбирают из группы, состоящей из таких отдельных растворителей, как диэтиловый эфир, тетрагидрофуран, гексан и гептан, и из смешанных растворителей, содержащих, по меньшей мере, два из данных растворителей. В большей степени предпочитают выбирать диэтиловый эфир, тетрагидрофуран или смешанный растворитель, содержащий диэтиловый эфир и тетрагидрофуран в качестве безводного растворителя.
Сильную щелочь, используемую для извлечения α-протона, выбирают из группы, состоящей из трет-бутоксида калия (t-BuOK), диизопропиламида лития (LDA), н-бутиллития, втор-бутиллития и трет-бутиллития и среди данных соединений диизопропиламид лития (LDA) является самым предпочтительным.
Электрофил, реагирующий с нуклеофилом тиоэфира, представляет собой бензилгалогенид.
Температура реакции зависит от растворителя, но обычно находится в интервале -78 ~ 25°С. В большей степени предпочитают выполнять реакцию извлечения α-протона в присутствии сильной щелочи при -75°С, при которых добавляют электрофил. Затем температуру медленно поднимают до комнатной температуры (25°С). Временной период реакции отличается в зависимости от каждой стадии реакции. Например, для извлечения α-протона сильной щелочью требуется 10-30 минут, и для реакции с электрофилом требуется 30-90 минут.
[Процесс D] Получение соединения, представленного формулой 11
Соединение, представленное формулой 11, получают элиминированием защитной группы фенола из соединения, представленного формулой 10.
Полярный растворитель, используемый в данном процессе, выбирают из группы, состоящей из N,N-диметилформамида, N,N-диметилацетамида, диметилсульфоксида, ацетонитрила, ацетона, этилацетата, четыреххлористого углерода, хлороформа и дихлорметана. Простой эфир в данном описании может быть выбран из группы, состоящей из тетрагидрофурана, диоксана, диметоксиэтана и диэтиленгликольдиметилового эфира. Спирт может представлять собой метанол или этанол. Ароматический углеводород представлен бензолом, толуолом и ксилолом. В качестве растворителя в данном описании предпочитают полярный растворитель и, особенно, тетрагидрофуран является более предпочтительным.
Для элиминирования защитной группы фенола, особенно для элиминирования метильной, этильной, трет-бутильной, бензильной или аллилэфирной защитной группы, используют триметилсилилиодид, натриевую соль этантиоспирта, лития иодид, алюминия галогенид, бора галогенид или кислоту Льюиса, такую как трифторуксусная кислота, и для элиминирования силильной защитной группы, такой как триметилсилильная, трет-бутилдифенилсилильная, триизопропилсилильная и трет-бутилдиметилсилильная, используют фторид, такой как фторид тетрабутиламмония (Bu4N+F-), галогенводородную кислоту (фтористоводородную кислоту, хлористоводородную кислоту, бромистоводородную кислоту или иодистоводородную кислоту) или фторид калия. Предпочитают применять фторид, чтобы элиминировать силильную защитную группу и в большей степени предпочитают применять фторид тетрабутиламмония.
Температура реакции зависит от способа и растворителя, но обычно находится в интервале 0 ~ 120°С, и, предпочтительно, в интервале 10 ~ 25°С. Временной период реакции зависит от температуры реакции, но обычно требуется от 30 минут до одних суток. В большей степени предпочитают заканчивать реакцию в течение 2 часов.
[Процесс Е] Получение соединения, представленного формулой 12
Данное соединение получают выделением R-изомера, представленного формулой 12, и другого изомера, S-изомера, из рацемического соединения формулы 11. Данное разделение выполняют с помощью ВЭЖХ, используя колонку с нормальной хиральной фазой. Для этого случая данный растворитель представляет собой смешанный растворитель, содержащий неполярные растворители, такие как гексан, гептан и пентан, и полярные растворители, такие как этанол и изопропиловый спирт.
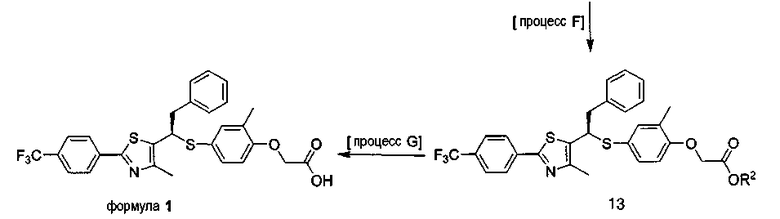
[Процесс F] Получение соединения, представленного формулой 13
Для получения соединения, представленного формулой 13, соединение, представленное формулой 12, подвергают взаимодействию предпочтительно с галогенацетаталкиловым сложным эфиром или с галогенацетаталлиловым сложным эфиром в присутствии основания.
Галогенацетаталкиловый сложный эфир или галогенацетаталлиловый сложный эфир представляет собой обычное соединение, которое может быть легко получено. Галоген в данном случае представлен атомом хлора, атомом брома и атомом иода. Предпочтительно используют бромацетатметиловый сложный эфир, бромацетаталлиловый сложный эфир или бромацетатэтиловый сложный эфир в качестве галогенацетаталкилового сложного эфира или галогенацетаталлилового сложного эфира.
Используемый растворитель в данном процессе может представлять собой отдельный растворитель, выбираемый из группы, состоящей из N,N-диметилформамида, N,N-диметилацетамида, диметилсульфоксида, ацетонитрила, ацетона, этанола и метанола, или смешанный растворитель, полученный смешиванием данных растворителей с 1-10% воды. Самый предпочтительный растворитель является смешанным растворителем, полученным смешиванием ацетона или диметилсульфоксида с 1-5 % воды.
Основание, используемое в данном процессе, не ограничивают, поскольку оно не оказывает вредное влияние на данную реакцию, безотносительно к тому, сильное оно или слабое, и оно представлено гидридом щелочного металла, таким как гидрид натрия и гидрид лития, гидридом щелочноземельного металла, таким как гидрид калия, гидроксидом щелочного металла, таким как гидроксид натрия и гидроксид калия, и карбонатом щелочного металла, таким как карбонат лития, карбонат калия, бикарбонат калия и карбонат цезия. Среди данных соединений, предпочитают карбонат щелочного металла, и карбонат калия является в большей степени предпочтительным.
Температуру реакции не ограничивают, но только вплоть до температуры кипения растворителя. Однако высокая температура не является предпочтительной, чтобы ингибировать побочные реакции. Предпочитаемая температура реакции находится в интервале 0-60°С. Временной период реакции отличается от температуры реакции, но обычно составляет 30 минут-1 сутки и, предпочтительно, 30-90 минут.
[Процесс G] Получение соединения, представленного формулой 1
Соединение, представленное формулой 1, получают из соединения, представленного формулой 13, гидролизом сложного эфира карбоновой кислоты данного соединения в смешанном растворе растворимой неорганической соли и спирте, или соли получают из аллилового сложного эфира в присутствии палладиевого катализатора.
Растворитель, используемый в данном процессе, представляет собой растворитель, который может быть смешан с водой, например спирт, такой как метанол или этанол. Неорганическая соль, используемая в данном процессе, представляет собой водный раствор, полученный смешиванием гидроксида щелочного металла, такого как гидроксид лития, гидроксид натрия и гидроксид калия, с водой в концентрации 0,1-3 Н, с учетом типа щелочной соли карбоновой кислоты. Кислота, используемая для получения соединения, представленного формулой 13 в форме карбоновой кислоты, представляет собой предпочтительно водный раствор уксусной кислоты или 0,1-3 Н водный раствор хлористоводородной кислоты.
Данную реакцию предпочтительно проводят при низкой температуре, чтобы ингибировать побочные реакции, которая обычно находится в интервале 0°С-комнатная температура. Временной период реакции зависит от температуры реакции, но обычно составляет 10 минут-3 часа и, предпочтительнее, 30 минут-1 час.
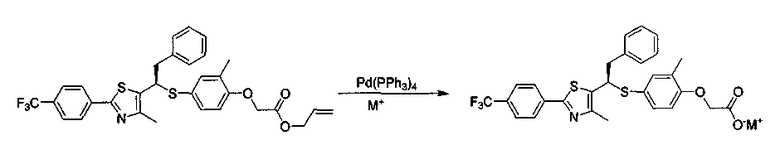
Фармацевтически приемлемую соль соединения, представленного формулой 1, получают замещением соли сложного аллилового эфира из соединения, представленного формулой 13, используя палладийтетракистрифенилфосфиновый катализатор и соль металла согласно реакции формулы 2. Растворитель, используемый в данном процессе, выбирают из группы, состоящей из хлороформа, четыреххлористого углерода, дихлорметана, тетрагидрофурана и этилацетата. Катализатор, используемый в данном процессе, представляет собой палладийтетракистрифенилфосфин. Соль для замещения соли представляет собой 2-этилгексаноат калия или 2-этилгексаноат натрия.
Реакция для формулы 2
Данное полученное соединение, содержащее S, представленное формулой 1, является очень важным продуктом в качестве PPARδ белкового лиганда.
Данное изобретение включает соединение, представленное формулой 1, его сольваты и соли, которые могут быть эффективно применены в качестве активаторной композиции рецептора δ (PPARδ), активируемого пролифераторами пероксисом. Определенно соединение, представленное формулой 1 данного изобретения, и его фармацевтически приемлемые соли могут быть очень эффективными в качестве фармацевтической композиции для лечения и профилактики артериосклероза или гиперлипидемии; для повышения липопротеида высокой плотности (HDL); для лечения и профилактики диабета; для лечения и профилактики ожирения; для укрепления мышцы или усиления выносливости; для улучшения памяти; для лечения и профилактики деменции или болезни Паркинсона; и в качестве композиции для добавок в диетические пищевые продукты, диетические напитки, добавок в пищевые продукты и корма для животных. Соединение, представленное формулой 1, или его фармацевтически приемлемые соли данного изобретения могут быть использованы для функциональной косметической композиции с целью профилактики или положительной динамики ожирения и для функциональной косметической композиции с целью укрепления мышцы и усиления выносливости. Функциональная косметическая композиция для укрепления мышцы и усиления выносливости может быть приготовлена как мазь, лосьон, крем или гель, предназначенная для применения на часть тела перед/после физической нагрузки, и может быть использована в течение длительного периода, чтобы вызывать желаемый эффект. Соединение, представленное формулой 1, или его фармацевтически приемлемые соли данного изобретения могут быть приготовлены как мазь и применены на часть тела для профилактики или лечения диабета или язвы диабетической стопы, так называемой диабетической язвы.
Данное изобретение также относится к активаторной композиции рецептора δ (PPARδ), активируемого пролифераторами пероксисом, содержащей производное тиазола, представленное формулой 1, или его фармацевтически приемлемые соли в качестве активного ингредиента.
Фармацевтически приемлемая соль в данном описании включает все соли, которые способны образовать соль с карбоновой кислотой соединения формулы 1 и ионами щелочного металла или ионами щелочноземельного металла, которые иллюстрированы посредством Li+, Na+, K+, Ca2+ и т.д.
Эффективная доза соединения, представленного формулой 1, или его фармацевтически приемлемых солей данного изобретения может быть определена согласно типу соединения, методу введения, целевому субъекту и целевому заболеванию, но определяется на основе обычного медицинского стандарта. Предпочитаемая доза соединения, представленного формулой 1, находится в интервале 1-100 мг/кг (масса тела)/сутки. Частота введения может быть один или несколько раз в сутки в пределах допускаемой дозировки в сутки. Композицию данного изобретения можно вводить перорально или парентерально и можно использовать в обычных формах фармацевтического препарата. Например, композиция данного изобретения может быть приготовлена в виде таблеток, порошков, сухих сиропов, жевательных таблеток, гранул, капсул, мягких капсул, пилюль, напитков, подъязычных препаратов и т.д. Таблетки данного изобретения могут быть введены субъекту способом или путем, который доставляет эффективную дозу таблетки с биодоступностью, которая равна таковой при пероральном пути. И способ введения или путь может быть определен согласно характеристикам, стадиям целевого заболевания и другим состояниям. Когда композицию данного изобретения формируют в виде таблеток, она может дополнительно включать один или несколько фармацевтически приемлемых эксципиентов. Содержание и характеристики эксципиента могут определяться растворимостью и химическими свойствами выбранной таблетки, путем введения и обычной фармацевтической практикой.
Соединение формулы 1 данного изобретения может быть внесено в добавки для диетических пищевых продуктов или в диетические напитки для профилактики или положительной динамики гиперлипидемии; для повышения липопротеина высокой плотности (HDL); для профилактики и положительной динамики диабета; для профилактики и положительной динамики ожирения; для укрепления мышцы или усиления выносливости; для улучшения памяти; для профилактики и положительной динамики деменции или болезни Паркинсона. В данном случае содержание соединения в этих диетических пищевых продуктах и напитках может быть установлено согласно цели и применению. Кроме ингредиентов, упоминаемых выше, соединение, представленное формулой 1 данного изобретения, может содержать разнообразные питательные вещества, витамины, минералы (электролиты), ароматизаторы/вкусовые добавки, включающие природные ароматизаторы и синтетические ароматизаторы, окрашивающие средства и наполнители (пластовый мармелад, шоколад и т.д.), пектиновую кислоту и ее соли, альгиновую кислоту и ее соли, органическую кислоту, защитные коллоидные загустители, регуляторы рН, стабилизаторы, антисептики, глицерин, спирты, карбонизаторы (науглероживатели), которые используются для добавления к соде, и т.д.
Наилучший способ
Практические и в настоящее время предпочтительные варианты осуществления данного изобретения проиллюстрированы, как показано в следующих примерах.
Однако следует определить, что специалисты в данной области при рассмотрении данного описания могут делать модификации и улучшения в пределах сущности и объема данного изобретения.
Пример 1: получение соединения формулы 8 (процесс А)
500 мг (2,14 ммоль) 4-иод-2-метилфенола растворяли в 20 мл безводного тетрагидрофурана в присутствии азота и в этот период температуру устанавливали при 0°С. К содержимому медленно добавляли 1,1 мл изопропилмагнийхлорида (2 М эфирный раствор, 2,16 ммоль), затем следовала реакция в течение 10 минут. Реакционный раствор охлаждали ниже -78°С, к которому медленно добавляли 2,77 мл трет-бутиллития (1,7 М-гептановый раствор, 4,70 ммоль). После взаимодействия в течение 20 минут к содержимому медленно добавляли 69 мг S (2,14 ммоль), затем следовала дополнительная реакция до тех пор, пока температура реагирующего вещества не достигала 15°С. 40 минут спустя, 624 мг (2,14 ммоль) 5-хлорметил-4-метил-2-[(4-трифторметил)фенил]тиазола, представленного формулой 7, растворяли в 2 мл безводного ТГФ, и этот раствор медленно добавляли в реакционную смесь при той же самой температуре. Спустя более одного часа взаимодействия, реакцию завершали добавлением 20 мл раствора хлорида аммония. Органический слой отделяли и сушили над сульфатом магния. После фильтрования смеси растворитель отгоняли при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле (гексан/этилацетат=3/1, об./об.) с получением 728 мг (выход: 86%) требуемого соединения.
1H ЯМР (300 МГц, CDCl3) δ 7,96 (д, 2H, J=8,1 Гц), 7,65 (д, 2H, J=8,3 Гц), 7,19 (д, 1Н, J=1,5 Гц), 7,01 (дд, 1Н, J=8,2, 2,0 Гц), 6,62 (д, 1Н, J=8,2 Гц), 5,86 (ушир.с, 1Н), 4,07 (с, 2H), 2,19 (с, 3H), 2,12 (с, 3H).
13C ЯМР (75,5 МГц, CDCl3) δ 163,9, 155,5, 151,7, 137,4, 136,9, 133,5, 131,9 (кв, J=32,6 Гц), 131,7, 126,8, 126,3, (кв, J=3,9 Гц), 125,8, 123,8, 115,7, 33,2, 16,2, 14,8.
Пример 2: получение соединения формулы 9 (R 1 =трет-Bu(CH 3 ) 2 Si-, процесс B)
500 мг (1,26 ммоль) соединения формулы 8 и 171 мг (2,52 ммоль) имидазола полностью растворяли в 5 мл диметилформамида. В данный раствор медленно добавляли 209 мг (1,38 ммоль) трет-бутилдиметилсилилхлорида с последующим перемешиванием при комнатной температуре в течение 4 часов. После завершения реакции органический растворитель экстрагировали с использованием раствора хлорида аммония и этилацетата. Влагу органического слоя сушили над сульфатом магния. После фильтрования смеси растворитель отгоняли при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле (гексан/этилацетат=10/1, об./об.) с получением 610 мг (выход: 95%) требуемого соединения.
1H ЯМР (300 МГц, CDCl3): 7,97 (д, 2H, J=8,1 Гц), 7,65 (д, 2H, J=8,2 Гц), 7,19 (д, 1Н, J=1,9 Гц) 7,07 (м, 1Н), 6,69 (д, 1Н, J=8,3 Гц), 4,11 (с, 2H), 2,21 (с, 3H), 2,11 (с, 3H), 1,01 (с, 9H), 0,21 (с, 6H).
13C ЯМР (75,5 МГц, CDCl3): δ 163,2, 154,7, 151,5, 137,1, 136,6, 133,3, 132,4, 131,7, 131,2, 131,0, 130,2, 129,2, 126,6, 126,1, 126,06, 126,01, 125,9, 124,9, 119,4, 32,8, 25,9, 18,5, 16,9, 15,0, -4,0.
Пример 3: получение соединения формулы 10 (R 1 =трет-Bu(CH 3 ) 2 Si- процесс С)
300 мг (0,59 ммоль) соединения (R1=трет-Bu(CH3)2-) формулы 9, приготовленного в примере 2, растворяли в 5 мл безводного тетрагидрофурана в присутствии азота и температуру снижали до -78°С. В реакционную смесь медленно добавляли 619 мкл (2,0 М эфирный раствор, 1,24 ммоль) раствора диизопропиламида лития, затем следовала реакция в течение 10 минут. Затем к реакционному раствору добавляли 77 мкл (0,65 ммоль) бензилбромида с последующим перемешиванием в течение 30 минут при той же самой температуре (-78°С). Реакцию завершали добавлением 5 мл раствора хлорида аммония. Влагу органического слоя сушили над сульфатом магния. После фильтрования смеси растворитель отгоняли при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле (гексан/этилацетат=10/1, об./об.) с получением 265 мг (выход: 75%) требуемого соединения.
1H ЯМР (300 МГц, CDCl3): δ 7,97 (д, 2H, J=8,1 Гц), 7,65 (д, 2H, J=8,2 Гц), 7,03-7,26 (м, 7H) 6,63 (д, 1Н, J=8,3 Гц), 4,51 (дд, 1Н, J=9,8, 5,3 Гц), 3,37 (дд, 1Н, J=9,8, 5,3 Гц), 3,05 (дд, 1Н, J=13,6, 9,9 Гц), 2,10 (с, 3H), 1,83 (с, 3H), 0,98 (с, 9H), 0,18 (с, 6H).
13C ЯМР (75 МГц, CDC13): δ 163,5, 155,1, 151,8, 138,4, 137,7, 136,5, 133,5, 130,3, 129,3, 128,9, 127,2, 126,8, 126,7, 126,2, 126,1, 124,5, 119,4, 49,2, 44,4, 26,1, 18,7, 17,1, 15,1, -3,81, -3,84.
Пример 4: получение соединения формулы 11 (процесс D)
200 мг (0,33 ммоль) соединения (R1=трет-Bu(CH3)2Si-) формулы 10, приготовленного в примере 3, растворяли в 5 мл безводного тетрагидрофурана в присутствии азота. В реакционную смесь при комнатной температуре медленно добавляли 660 мкл (0,66 ммоль) тетрабутиламмонийфторида в виде 1 М тетрагидрофуранового раствора с последующим перемешиванием в течение 1 часа. После завершения реакции органический слой отделяли добавлением этилацетата и воды. Влагу органического слоя сушили над сульфатом магния. После фильтрования смеси растворитель отгоняли при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле (гексан/этилацетат=5/1, об./об.) с получением 146 мг (выход: 91%) требуемого соединения.
1H ЯМР (500 МГц, CDCl3): δ 7,92 (д, 2H, J=8,2 Гц), 7,50 (с, 1Н), 7,59 (д, 2H, J=8,2 Гц), 7,07-7,22 (м, 6H), 6,85(м, 1Н), 6,44 (д, 1Н, J=8,2 Гц), 4,47 (дд, 1Н, J=9,7, 5,4 Гц), 3,39 (дд, 1Н, J=13,8, 5,4 Гц), 3,06 (дд, 1Н, J=13,8, 9,8 Гц), 2,12 (с, 3H), 1,73 (с, 3H).
13C ЯМР (125 МГц, CDCl3): δ 164,1, 155,7, 151,2, 138,0, 137,9, 137,0, 136,4, 133,8, 131,8, 131,5, 129,0, 128,6, 127,2, 127,0, 126,6, 126,14, 126,11, 125,7, 125,0, 122,9, 122,7, 115,2, 60,8, 49,1, 43,7, 21,2, 15,9, 14,3, 14,2.
Пример 5: получение соединения формулы 12 (процесс Е)
90 мг соединения формулы 11, полученного в примере 4, поступало для полуразделения на колонку ВЭЖХ с хиральной фазой (chiralpack AD-H) c получением 45 мг требуемого соединения, представленного формулой 12 в R-форме и 45 мг его соответствующего изомера в S-форме.
Подвижная фаза: гексан/изопропиловый спирт: 90/10.
Скорость потока: 3 мл/мин.
Пример 6: получение соединения формулы 13 (процесс F)
20 мг соединения формулы 12, полученного в примере 5, хорошо смешивали с 10 мл ацетона, содержащего 5% воды и 127 мг (0,9 ммоль, 2,3 эквивалента) карбоната калия при комнатной температуре. В реакционную смесь добавляли 67 мкл (0,6 ммоль, 1,5 эквивалента) бромацетатэтилового сложного эфира с последующим интенсивным перемешиванием в течение 4 часов. После завершения реакции органический растворитель экстрагировали с использованием раствора хлорида аммония и этилацетата, который сушили над сульфатом магния для устранения влаги из органического слоя. После фильтрования смеси растворитель отгоняли при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле (гексан/этилацетат=5/1, об./об.) с получением 22 мг (выход: 95%) требуемого соединения формулы 13.
1H ЯМР (300 МГц, CDCl3): 7,98 (д, 2H, J=8,1 Гц), 7,65 (д, 2H, J=8,3 Гц), 7,06-7,27 (м, 7H), 6,55 (д, 1Н, J=8,4 Гц), 4,59 (с, 2H), 4,53 (дд, 1Н, J=9,7, 5,3 Гц), 4,22 (кв, 2H, J=7,1 Гц), 3,37 (дд, 1Н, J=13,7, 5,3 Гц), 3,17 (м, 1Н) 2,20 (с, 3H), 1,83 (с, 3H), 1,26 (т, 3H, J=7,2 Гц).
13C ЯМР (75 МГц, CDCl3): 169,1, 163,6, 156,9, 151,8, 138,3, 137,4, 137,3, 136,4, 133,4, 129,3, 128,9, 128,6, 127,2, 126,8, 126,3, 126,2, 126,1, 125,1, 111,8, 65,9, 61,7, 49,1, 44,4, 16,5, 15,1, 14,5.
Пример 7: получение соединения формулы 1 (процесс G)
20 мг (0,03 ммоль) соединения формулы 13, полученного в примере 6, хорошо перемешивали с 15 мл этанола, к которому добавляли 15 мкл 3 Н раствора гидроксида натрия. После перемешивания при комнатной температуре в течение 20 минут рН реакционной смеси доводили до 2,0 с помощью 2 Н HCl. Вакуумную перегонку проводили для удаления примерно 80% этанола. Органический растворитель экстрагировали с использованием раствора хлорида аммония и этилацетата. После фильтрования смеси растворитель отгоняли при пониженном давлении и остаток очищали LH-20 колоночной хроматографией с получением 16 мг (выход: 98%) требуемого соединения формулы 1.
1H ЯМР (500 МГц, CDCl3): δ 7,92 (д, 2H, J=8,2 Гц), 7,50 (с, 1Н), 7,59 (д, 2H, J=8,2 Гц), 7,07-7,22 (м, 6H), 6,85(м, 1Н), 6,44 (д, 1Н, J=8,2 Гц), 4,47 (дд, 1Н, J=9,7, 5,4 Гц), 3,39 (дд, 1Н, J=13,8, 5,4 Гц), 3,06 (дд, 1Н, J=13,8, 9,8 Гц), 2,12 (с, 3H), 1,73 (с, 3H).
13C ЯМР (125 МГц, CDCl3): δ 164,1, 155,7, 151,2, 138,0, 137,9, 137,0, 136,4, 133,8, 131,8, 131,5, 129,0, 128,6, 127,2, 127,0, 126,6, 126,14, 126,11, 125,7, 125,0, 122,9, 122,7, 115,2, 60,8, 49,1, 43,7, 21,2, 15,9, 14,3, 14,2.
Экспериментальный пример 1: оценка активности и цитотоксичности
PPARδ активности R-формы соединения, представленного формулой 1 данного изобретения, и S-формы соединения, представленного формулой 2, измеряли в анализе трансфекции. Кроме того, исследовали селективность к PPAR подтипам, PPARα и PPARγ. С помощью ММТ метода исследовали цитотоксичность, in vivo активность и токсичность исследовали в опыте с животными, используя мышей.
Анализ трансфекции
В данном анализе использовали CV-1 клетки. Данные клетки инокулировали в 96-луночном планшете, содержащем DMEM, дополненную 10% FBS, DBS (делипидизированная) и 1% пенициллином/стрептомицином и культивировали в 37°С, 5% СО2 инкубаторе. Опыт проводили согласно стадиям инокуляции, трансфекции, обработке образца и апробации. В частности, CV-1 клетки инокулировали в 96-луночном планшете (5000 клетки/лунка) с последующей трансфекцией 24 часа спустя. Плазмидная ДНК PPAR полной длины, репортерная ДНК, подтверждающая PPAR активность благодаря их люциферазной активности, β-галактозидазная ДНК, обеспечивающая информацию по эффективности трансфекции, и реагент трансфекции были использованы для трансфекции. Соединения формулы 1 и формулы 2 растворяли в диметилсульфоксиде (ДМСО), которыми обрабатывали клетки со средой в различных концентрациях. После культивирования клеток в инкубаторе в течение 24 часов клетки лизировали применением лизисного буфера. Активность люциферазы и активность β-галактозидазы измеряли люминометром и микропланшетным ридером. Полученные значения люциферазы модифицировали до величин β-галактозидазы. Строили график с данными величинами и вычисляли ЕС50.
Как показано в таблице 1, соединение, представленное формулой 1 данного изобретения, является высокоселективным к PPARδ. Производное тиазола формулы 1 данного изобретения показало в 100000 раз более высокую селективность к PPARα и PPARγ. ЕС50 соединения формулы 1 к PPARδ была равна 0,6 нМ и ЕС50 соединения формулы 2 к PPARδ была равна 5,1 нМ. R-изомер, соединение формулы 1 данного изобретения, проявил в 10 раз более сильную активность, чем S-изомер, соединение формулы 2. Выше представленные результаты указывают, что соединение формулы 1 данного изобретения имеет высокую селективность.
МТТ анализ
МТТ анализ выполняли для оценки цитотоксичности соединений данного изобретения. МТТ представляет собой желтое вещество, растворимое в воде, но когда оно введено в живую клетку, оно возвращается в пурпурный нерастворимый кристалл под действием дегидрогеназы в митохондрии. Цитотоксичность может быть подтверждена измерением OD550 после растворения МТТ в диметилсульфоксиде. Опыт проводили, как указано ниже.
CV-1 клетки инокулировали в 96-луночном планшете (5000 клетки/лунка). Клетки культивировали в 37°С, 5% СО2 инкубаторе в течение 24 часов и обрабатывали соединениями формулы 1 и формулы 2 в различных концентрациях. Затем клетки снова культивировали в течение 24 часов, к которым добавляли МТТ реагент. После культивирования в течение 15 минут полученные пурпурные кристаллы растворяли в диметилсульфоксиде. Оптическую плотность измеряли микропланшетным ридером для подтверждения цитотоксичности. В результате этого было подтверждено, что соединение, представленное формулой 1 данного изобретения, и соединение, представленное формулой 2, их оптические изомеры не обладают цитотоксичностью даже в концентрации, кратной величине ЕС50 в 100000 раз.
Тест на токсичность
Тесты на острую токсичность и репродуктивную токсичность выполняли, используя мышей для оценки токсичности соединения данного изобретения. Соединение формулы 1 перорально вводили ICR мышам возрастом 6 недель в дозе 50 мг/кг, 300 мг/кг и 2000 мг/кг с последующим наблюдением токсичности в течение 14 суток. В результате этого не наблюдалось гибели при введении соединения формулы 1 даже при самой высокой концентрации 2000 мг/кг и не наблюдались значительные изменения ни в массе, ни в требующемся корме. Результаты вскрытия трупов не показали никаких аномальных признаков. Результат теста на репродуктивную токсичность с использованием C57BL/6 мышей также согласовывался с вышеприведенным в том, что не наблюдалось проявление токсичности под действием соединения формулы 1. После перорального введения данного соединения беременным самкам мышей исследовали увеличение массы и скорости роста плода. В результате этого не наблюдали значительных изменений из-за введения и никаких изменений в развитии скелета и материалов, связанных с заболеванием, не наблюдали.
Тест на животных
Ингибирующий эффект на ожирение
Тест на животных, использующий мышей, выполняли для подтверждения данного эффекта in vivo соединения данного изобретения. Использовали C57BL/6 (SLC Co.) мышей в возрасте 14 недель. Чтобы вызвать ожирение, давали корма, содержащие 35% жира. При кормлении такими кормами с высоким содержанием жира в течение 35 дней перорально вводили наполнитель, GW501516 (10 мг/кг/сутки) и соединение формулы 1 данного изобретения (10 мг/кг/сутки). В результате этого, только 26,5% группы, обработанной с помощью GW501516, 23,1% группы, обработанной соединением данного изобретения, показали увеличение массы по сравнению с группой, обработанной наполнителем (58,9%), что составило приблизительно ½ увеличение по сравнению с группой, обработанной наполнителем. Поэтому соединением формулы 1 данного изобретения подтверждено, что оно обладает сильным ингибирующим эффектом на ожирение, который был намного лучше эффекта GW501516.
Влияние на положительную динамику диабета
GTT (проба на переносимость глюкозы) выполняли для подтверждения влияния на положительную динамику диабета соединения данного изобретения. Глюкозу (1,5 г/кг) вводили интраабдоминально мышам, предварительно обработанным перорально образцами в течение 78 суток. Глюкозу в крови измеряли каждый час. Глюкоза в крови натощак была ниже в группе, обработанной соединением данного изобретения, чем в группе, обработанной наполнителем или GW501516. Группа, обработанная соединением данного изобретения, показала быстрое снижение глюкозы в крови за 20-40 минут и клиренс глюкозы в 100 минут. Между тем уровень глюкозы в крови не возвращался до нормального в группе, обработанной наполнителем, даже после 120 минут. Группа, обработанная GW501516, показала более низкую глюкозу, чем группа, обработанная наполнителем, но уровень глюкозы в крови не восстанавливался до нормального. Вышепредставленные результаты указывают, что соединение формулы 1 данного изобретения обладало отличным влиянием на положительную динамику диабета.
Влияние на положительную динамику гиперлипидемии
In vivo выполняли тест на животных, использующий C57BL/6 (SLC Co.) мышей в возрасте 6 недель для подтверждения влияния соединения данного изобретения на положительную динамику гиперлипидемии. Животным перорально вводили 10 мг/кг/сутки соединения формулы 1 данного изобретения и GW501516, причем кормили их кормами с высоким содержанием жира. Через 6 недель брали кровь из глазничных вен. Отделяли сыворотку и измеряли уровень HDL крови биохимическим методом. В результате этого, HDL уровень был на 36,3% выше в группе, обработанной GW501516, по сравнению с контролем. HDL уровень составлял на 44,6% выше в группе, обработанной соединением формулы 1 данного изобретения. Вышепредставленные результаты указывают, что соединение формулы 1 данного изобретения повышает HDL крови более эффективно, чем GW501516.
Ингибирующий эффект на артериосклероз
In vivo выполняли тест на животном, использующий АроЕ-/-мышь, который представляет собой животную модель для артериосклероза, для подтверждения ингибирующего эффекта на артериосклероз соединения данного изобретения. Животным перорально вводили 2 мг/кг/сутки соединения формулы 1 данного изобретения при кормлении кормами с высоким содержанием жира, высоким холестерином (20% жир, 1,25% холестерин; AIN-93G диета). Через 28 суток проводили окрашивание тромбоцитов на протяжении всей артерии, используя судан IV, для исследования ингибирующего эффекта на артериосклероз соединения сравнением результатов между экспериментальной и контрольной группами. В результате этого артериосклероз ингибировался на 30% в АроЕ-/-мыши, обработанной соединением формулы 1 данного изобретения по сравнению с контролем.
Влияние на усиление выносливости мышц и на усиление функции мышц
Тест на животном проводили для подтверждения влияния на усиление выносливости мышц и на усиление функции мышц соединения данного изобретения. Большинство мышц зарождаются на стадии развития. Таким образом, соединение формулы 1 (10 мг/кг/сутки) перорально вводили беременным мышам либо в период беременности, либо лактации, или в оба периода - беременности и лактации. Увеличение в массе и скорость роста немного отличались между плодами контрольной группы и обработанной группы. Мышцы наблюдали после удаления кожи. В результате этого мышцы из обработанной группы были красными в отличие от контрольной группы. Проводили окрашивание АТФазы и иммуноокрашивание. В результате этого волокно мышцы типа I увеличивалось в группе, обработанной соединением формулы 1. Влияние изменений мышечного волокна на усиление выносливости мышцы и функцию мышцы исследовали применением тредмил-теста. В результате этого период беганья намного увеличивается в группе, обработанной соединением формулы 1.
Результаты теста на выносливость мышц
Когда соединением данного изобретения обрабатывали взрослых особей, выносливость мышц и функция мышц также усиливалась. В частности, соединение формулы 1 перорально вводили C57BL/6 мышам в возрасте 10 недель в концентрации 10 мг/кг, в процессе чего мыши были усилены для физических нагрузок. Физическую нагрузку осуществляли тредмилом в течение 30 минут раз в сутки в течение 30 суток, конкретно, при скорости 2 м/мин в течение первых 5 минут, при 5 м/мин в течение 5 минут, при 8 м/мин в течение 5 минут и при 20 м/мин в течение последних 5 минут. В конце выносливость мышц и влияние на усиление функции мышц оценивали, применяя тредмил. В результате этого, время (1,5 раза) и дистанция (1,6 раза) физической нагрузки все были увеличены в группе, обработанной соединением данного изобретения по сравнению с контролем.
Улучшение памяти
Тест на животных выполняли для исследования терапевтического эффекта соединения данного изобретения на деменцию и болезнь Паркинсона, с учетом его влияния на улучшение памяти. Для подтверждения влияния соединения данного изобретения в период развития головного мозга данное соединение перорально вводили беременным мышам в концентрации 10 мг/кг в периоды беременности и лактации. Тест Морриса с водным лабиринтом осуществляли для определения любых изменений функций головного мозга в обработанной группе и в контрольной группе. В результате этого, среднее время, затрачиваемое для нахождения платформы, было намного короче в группе, обработанной соединением формулы 1 по сравнению с контрольной группой; конкретно, обработанная группа тратит 6,8 сек для нахождения платформы, и контрольная группа тратит 24,2 сек в среднем, причем предполагается, что соединение формулы 1 имело отличное влияние на улучшение памяти.
Терапевтический эффект соединения данного изобретения на деменцию и болезнь Паркинсона с оценкой его влияния на улучшение памяти исследовали, используя животную модель заболевания головного мозга (C57BL/6 мыши в возрасте 10 недель). Вначале LPS вводили инъекцией в головной мозг мыши, чтобы создать животную модель заболевания головного мозга. Мышей разделяли на четыре группы согласно введению и нагрузке. Нагрузку осуществляли тредмилом при скорости 2 м/мин в течение первых 5 минут, при 5 м/мин в течение 5 минут, при 8 м/мин в течение 5 минут и при 20 м/мин в течение последних 5 минут. В конце выполняли тест Морриса водный лабиринт. Результаты суммированы в таблице 3. В результате этого, терапевтический эффект соединения данного изобретения на деменцию и болезнь Паркинсона через усиление памяти данным соединением и нагрузки был подтвержден в животной модели заболевания головного мозга.
Результаты теста с водным лабиринтом
Промышленная применимость
Производное тиазола - соединение данного изобретения как PPARδ лиганд, который, в отличие от предыдущих изобретений, является селективным, может быть эффективно использовано в качестве фармацевтической композиции для лечения и профилактики артериосклероза или гиперлипидемии; для увеличения липопротеина высокой плотности (HDL); для лечения и профилактики диабета; для лечения и профилактики ожирения; для укрепления мышц или усиления выносливости; для улучшения памяти; для лечения и профилактики деменции или болезни Паркинсона; и в качестве композиции для добавок в диетические пищевые продукты, диетические напитки, пищевые добавки, функциональную косметику и корма для животных.
| название | год | авторы | номер документа |
|---|---|---|---|
| СЕЛЕНОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2005 |
|
RU2371437C2 |
| АРИЛЬНЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ЛИГАНДОВ PPAR И ИХ ПРИМЕНЕНИЕ | 2007 |
|
RU2444514C2 |
| ТИАЗОЛЬНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ЛИГАНДОВ PPAR-ДЕЛЬТА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2006 |
|
RU2392274C2 |
| ПРОИЗВОДНОЕ СЕЛЕНАЗОЛА, ИМЕЮЩЕЕ ЛИГАНД, КОТОРЫЙ АКТИВИРУЕТ РЕЦЕПТОР, АКТИВИРУЕМЫЙ ПРОЛИФЕРАТОРОМ ПЕРОКСИСОМ ( PPAR ), СПОСОБ ЕГО ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ ХИМИЧЕСКИХ СОЕДИНЕНИЙ | 2010 |
|
RU2510394C1 |
| ЭНАНТИОМЕР ЗАМЕЩЕННОЙ ФЕНИЛПРОПИОНОВОЙ КИСЛОТЫ И СПОСОБ ЕГО ПОЛУЧЕНИЯ, ЕГО КОМПОЗИЦИЯ И ПРИМЕНЕНИЕ | 2017 |
|
RU2735524C2 |
| АГЕНТ, АКТИВИРУЮЩИЙ РЕЦЕПТОР, АКТИВИРУЕМЫЙ ПРОЛИФЕРАТОРАМИ ПЕРОКСИСОМ δ | 2007 |
|
RU2435764C2 |
| ДИАРИЛЬНЫЕ СОЕДИНЕНИЯ С МОСТИКОВОЙ СВЯЗЬЮ | 2002 |
|
RU2297409C2 |
| АМИДНОЕ ПРОИЗВОДНОЕ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1998 |
|
RU2185381C2 |
| СОЕДИНЕНИЯ, ОБЛАДАЮЩИЕ АКТИВИРУЮЩИМ ДЕЙСТВИЕМ НА ПОДТИПЫ РЕЦЕПТОРОВ, АКТИВИРУЕМЫХ ПРОЛИФЕРАТОРОМ ПЕРОКСИСОМ (PPARs), И СПОСОБ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ УКАЗАННЫХ СОЕДИНЕНИЙ | 2011 |
|
RU2546117C2 |
| ПРОИЗВОДНЫЕ ДИАРИЛОВОЙ КИСЛОТЫ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 2000 |
|
RU2267484C2 |
Изобретение относится к производному тиазола формулы 1, как активатору рецептора 5, активируемому пролифераторами пероксисом (PPARδ), или его фармацевтически приемлемым солям и к фармацевтической композиции для профилактики и лечения артериосклероза или гиперлипидемии, для повышения уровня липопротеина высокой плотности (HDL), для профилактики и лечения диабета, ожирения, для укрепления мышцы или усиления выносливости, для улучшения памяти или для профилактики и лечения деменции или болезни Альцгеймера или болезни Паркинсона, содержащим такое производное тиазола. 7 н.п. ф-лы, 3 табл., 8 пр.
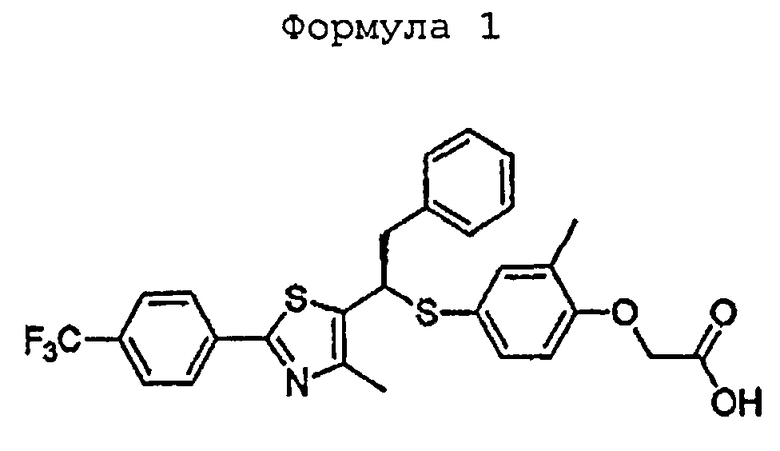
1. Производное тиазола, представленное формулой 1, или его фармацевтически приемлемые соли
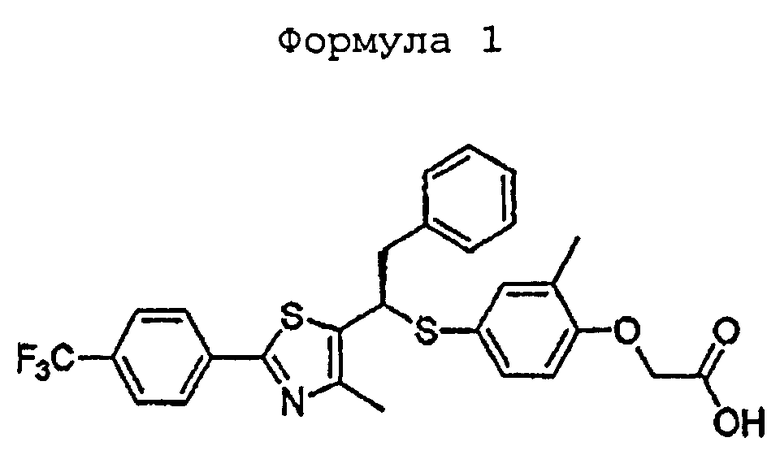
2. Фармацевтическая композиция для профилактики и лечения артериосклероза или гиперлипидемии, содержащая производное тиазола, представленное формулой 1, или его фармацевтически приемлемые соли по п.1 в качестве активного ингредиента и фармацевтически приемлемые эксципиенты.
3. Фармацевтическая композиция для повышения уровня липопротеина высокой плотности (HDL), содержащая производное тиазола, представленное формулой 1, или его фармацевтически приемлемые соли по п.1 в качестве активного ингредиента и фармацевтически приемлемые эксципиенты.
4. Фармацевтическая композиция для профилактики и лечения диабета, содержащая производное тиазола, представленное формулой 1, или его фармацевтически приемлемые соли по п.1 в качестве активного ингредиента и фармацевтически приемлемые эксципиенты.
5. Фармацевтическая композиция для профилактики и лечения ожирения, содержащая производное тиазола, представленное формулой 1, или его фармацевтически приемлемые соли по п.1 в качестве активного ингредиента и фармацевтически приемлемые эксципиенты.
6. Фармацевтическая композиция для укрепления мышцы или усиления выносливости, содержащая производное тиазола, представленное формулой 1, или его фармацевтически приемлемые соли по п.1 в качестве активного ингредиента и фармацевтически приемлемые эксципиенты.
7. Фармацевтическая композиция для улучшения памяти или для профилактики и лечения деменции или болезни Альцгеймера или болезни Паркинсона, содержащая производное тиазола, представленное формулой 1, или его фармацевтически приемлемые соли по п.1 в качестве активного ингредиента и фармацевтически приемлемые эксципиенты.
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| ПРОФИЛАКТИЧЕСКОЕ ИЛИ ТЕРАПЕВТИЧЕСКОЕ СРЕДСТВО И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ГЕТЕРОЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ | 2001 |
|
RU2259361C2 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |

