Предметом настоящего изобретения является применение десPro36эксендин-4(1-39)-Lys6-NH2 (AVE0010, ликсисенатид) или/и его фармацевтически приемлемой соли для производства лекарственного средства для лечения сахарного диабета 2 типа. Другим предметом изобретения является фармацевтическая композиция, содержащая десPro36эксендин-4(1-39)-Lys6-NH2 или/и его фармацевтически приемлемую соль и необязательно содержащая фармацевтически приемлемые носители, адъюванты или/и вспомогательные вещества. Еще одним аспектом является способ лечения сахарного диабета 2 типа, включающий введение десPro36эксендин-4(1-39)-Lys6-NH2 или/и его фармацевтически приемлемой соли субъекту, нуждающемуся в этом.
У здорового человека высвобождение инсулина поджелудочной железой строго связано с концентрацией глюкозы в крови. Повышенный уровень глюкозы в крови, который появляется после приема пищи, быстро компенсируется соответствующим увеличением секреции инсулина. В состоянии натощак уровень инсулина в плазме снижается до базального значения, которое является достаточным для обеспечения непрерывного поступления глюкозы в инсулин-чувствительные органы и ткани и для сохранения продуцирования глюкозы в печени на низком уровне в ночное время.
В отличие от диабета 1 типа, при диабете 2 типа, как правило, нет недостатка в инсулине, однако во многих случаях, особенно в случаях прогрессирования заболевания, лечение инсулином рассматривается в качестве наиболее подходящей терапии, если оно необходимо в сочетании с пероральным приемом антидиабетических лекарственных средств.
Повышенный уровень глюкозы в крови в течение нескольких лет без начальных симптомов представляет собой значительный риск для здоровья. Результаты крупномасштабного исследования DCCT (Исследовательской группы, занимающейся изучением контроля над диабетом и его осложнениями (The Diabetes Control and Complications Trial Research Group) (1993) N. Engl. J. Med. 329, 977-986) в США продемонстрировали, что хронически повышенный уровень глюкозы в крови является основной причиной развития осложнений диабета. Примерами осложнений диабета являются микро- и макроповреждения сосудов, которые могут проявляться в виде ретинопатии, нефропатии и невропатии и приводить к слепоте, почечной недостаточности и потере конечностей, и которые сопровождаются повышенным риском развития сердечно-сосудистых заболеваний. Таким образом, можно сделать вывод, что усовершенствованная терапия диабета в первую очередь должна быть направлена на поддержание уровня глюкозы в крови как можно ближе к физиологическому диапазону.
Особый риск существует для пациентов с избыточной массой тела, страдающих диабетом 2 типа, например пациентов с индексом массы тела (ИМТ) ≥30. У этих пациентов риск диабета перекрывается с рисками, сопровождающими избыточную массу тела, что приводит, например, к росту числа сердечно-сосудистых заболеваний по сравнению с пациентами с диабетом 2 типа, имеющими нормальную массу тела. Таким образом, крайне необходимо лечить диабет у таких пациентов, при этом снижая их избыточную массу тела.
Соединение десPro36эксендин-4(1-39)-Lys6-NH2 (AVE0010, ликсисенатид) представляет собой производное эксендина-4. AVE0010 приведено в виде SEQ ID NO: 93 в WO 01/04156:
SEQ ID NO: 1: AVE0010 (44 AS)
H-G-E-G-T-F-T-S-D-L-S-K-Q-M-E-E-E-A-V-R-L-F-I-E-W-L-K-N-G-G-P-S-S-G-A-P-P-S-K-K-K-K-K-K-NH2
SEQ ID NO: 2: Эксендин-4 (39 AS)
H-G-E-G-T-F-T-S-D-L-S-K-Q-M-E-E-E-A-V-R-L-F-I-E-W-L-K-N-G-G-P-S-S-G-A-P-P-P-S-NH2
Эксендины представляют собой группу пептидов, способных понижать концентрацию глюкозы в крови. Аналог эксендина AVE0010 имеет укороченную с C-конца последовательность природного эксендина-4. AVE0010 содержит шесть С-концевых остатков лизина, отсутствующих в эксендине-4.
В контексте настоящего изобретения AVE0010 включает его фармацевтически приемлемые соли. Специалистам в данной области известны фармацевтически приемлемые соли AVE0010. Предпочтительной фармацевтически приемлемой солью AVE0010, используемой в настоящем изобретении, является ацетат.
Первым аспектом настоящего изобретения является применение десPro36эксендин-4(1-39)-Lys6-NH2 или/и его фармацевтически приемлемой соли для производства лекарственного средства для лечения сахарного диабета 2 типа.
Субъект, подлежащий лечению лекарственным средством по настоящему изобретению, страдающий от диабета 2 типа, может быть субъектом с ожирением. Согласно настоящему изобретению субъект с ожирением может иметь индекс массы тела по меньшей мере 30 кг/м2.
Субъект, подлежащий лечению лекарственным средством по настоящему изобретению, может быть взрослым субъектом. Возраст субъекта может составлять по меньшей мере 18 лет или возраст субъекта может находиться в диапазоне от 18 до 80 лет или от 40 до 80 лет, или от 50 до 60 лет.
Субъект, подлежащий лечению лекарственным средством по настоящему изобретению, предпочтительно не получает антидиабетическое лечение, например, с использованием инсулина или/и родственных соединений.
Субъект, подлежащий лечению лекарственным средством по настоящему изобретению, может страдать от сахарного диабета 2 типа в течение по меньшей мере 1 года или по меньшей мере 2 лет. В частности, у субъекта, подлежащего лечению, сахарный диабет 2 типа был диагностирован по меньшей мере за 1 год или по меньшей мере за 2 года до начала терапии лекарственным средством по настоящему изобретению.
Субъект, подлежащий лечению, может иметь показатель HbA1c по меньшей мере примерно 8% или по меньшей мере примерно 7,5%. Субъект может также иметь показатель HbA1c от примерно 7 до примерно 10%. Пример по настоящему изобретению демонстрирует, что лечение с использованием AVE0010 приводит к снижению показателя HbA1c у пациентов с диабетом 2 типа (см. таблицы 9, 10).
Активное вещество по настоящему изобретению предпочтительно используют для улучшения толерантности к глюкозе при лечении пациентов, страдающих от диабета 2 типа. Улучшение толерантности к глюкозе означает, что постпрандиальная концентрация глюкозы в плазме снижается при помощи активного вещества по настоящему изобретению. Снижение означает, в частности, что концентрация глюкозы в плазме крови достигает нормогликемических значений или по меньшей мере приближается к этим значениям.
Согласно настоящему изобретению нормогликемические значения представляют собой концентрации глюкозы в крови, составляющие, в частности, 60-140 мг/дл (что соответствует 3,3-7,8 мМ/л). Этот диапазон относится, в частности, к концентрациям глюкозы в крови в условиях натощак и после приема пищи.
Субъект, подлежащий лечению, может иметь концентрацию глюкозы в плазме натощак по меньшей мере 8 ммоль/л, по меньшей мере 8,5 ммоль/л или по меньшей мере 9 ммоль/л. Эти концентрации глюкозы в плазме превышают нормогликемические концентрации. Пример по настоящему изобретению демонстрирует, что лечение с применением AVE0010 приводит к снижению концентрации глюкозы в крови у пациентов с диабетом 2 типа (см. таблицу 15).
Субъект, подлежащий лечению, может иметь 2-часовую постпрандиальную концентрацию глюкозы в плазме, составляющую по меньшей мере 10 ммоль/л, по меньшей мере 12 ммоль/л или по меньшей мере 14 ммоль/л. Эти концентрации глюкозы в плазме превышают нормогликемические концентрации. Пример по настоящему изобретению демонстрирует, что лечение с применением AVE0010 приводит к снижению 2-часовой постпрандиальной концентрации глюкозы в плазме у пациентов с диабетом 2 типа (см. таблицу 11).
Субъект, подлежащий лечению, может иметь экскурсию глюкозы по меньшей мере 2 ммоль/л, по меньшей мере 3 ммоль/л, по меньшей мере 4 ммоль/л или по меньшей мере 5 ммоль/л. Согласно настоящему изобретению экскурсия глюкозы представляет собой, в частности, разницу между 2-часовой постпрандиальной концентрацией глюкозы в плазме и концентрацией глюкозы в плазме за 30 минут до теста с приемом пищи. В контексте настоящего изобретения тест с приемом пищи представляет собой... Пример по настоящему изобретению демонстрирует, что лечение с применением AVE0010 приводит к уменьшению экскурсии глюкозы у пациентов с диабетом 2 типа (см. таблицу 12).
Термин «постпрандиальная» хорошо известен специалисту в области диабетологии. Термин «постпрандиальная» описывает, в частности, фазу после приема пищи или/и воздействия глюкозы в экспериментальных условиях. У здорового человека эта фаза характеризуется увеличением и последующим снижением концентрации глюкозы в крови. Термин «постпрандиальная» или «постпрандиальная фаза», как правило, относится к фазе, которая заканчивается в период до 2 часов после приема пищи или/и воздействия глюкозы.
Вторым аспектом настоящего изобретения является применение десPro36эксендин-4(1-39)-Lys6-NH2 или/и его фармацевтически приемлемой соли для производства лекарственного средства, предназначенного для стимуляции снижения массы тела у пациентов с диабетом 2 типа или/и для предотвращения увеличения массы тела у пациентов с диабетом 2 типа. Пример по настоящему изобретению демонстрирует, что лечение с применением AVE0010 приводит к снижению массы тела у пациентов с диабетом 2 типа (см. таблицы 13 и 14).
Активное вещество, лекарственное средство или/и фармацевтическую композицию по настоящему изобретению можно использовать для лечения по одному или более медицинским показаниям, описанным в данном документе, например, для лечения пациентов с диабетом 2 типа, или с состояниями, связанными с диабетом 2 типа, например, для снижения концентрации глюкозы в плазме натощак, снижения постпрандиальной концентрации глюкозы в плазме, улучшения толерантности к глюкозе, снижения массы тела или/и предотвращения увеличения массы тела.
Согласно настоящему изобретению десPro36эксендин-4(1-39)-Lys6-NH2 можно вводить субъекту, нуждающемуся в этом, в количестве, достаточном для оказания терапевтического эффекта.
Соединение десPro36эксендин-4(1-39)-Lys6-NH2 или/и его фармацевтически приемлемую соль можно вводить парентерально, например, путем инъекции (такой как внутримышечная или подкожная инъекция). Подходящие устройства для инъекций, например, так называемые «ручки», содержащие картридж, в котором заключен активный ингредиент, и инъекционную иглу, известны. Соединение десPro36эксендин-4(1-39)-Lys6-NH2 или/и его фармацевтически приемлемую соль можно вводить в соответствующем количестве, например в количестве в диапазоне от 10 до 15 мкг на дозу или от 15 до 20 мкг на дозу.
По настоящему изобретению десPro36эксендин-4(1-39)-Lys6-NH2 или/и его фармацевтически приемлемую соль можно вводить в суточной дозе в диапазоне от 10 до 20 мкг, в диапазоне от 10 до 15 мкг или в диапазоне от 15 до 20 мкг. ДесPro36эксендин-4(1-39)-Lys6-NH2 или/и его фармацевтически приемлемую соль можно вводить одной инъекцией в сутки.
Еще одним аспектом настоящего изобретения является фармацевтическая композиция, содержащая десPro36эксендин-4(1-39)-Lys6-NH2 или/и его фармацевтически приемлемую соль и необязательно содержащая фармацевтически приемлемые носители, адъюванты или/и вспомогательные вещества.
Фармацевтическую композицию по настоящему изобретению можно готовить для применения в лечении сахарного диабета 2 типа.
Фармацевтическую композицию по настоящему изобретению можно также готовить для применения с целью стимуляции снижения массы тела у пациентов с диабетом 2 типа или/и для применения с целью предотвращения увеличения массы тела у пациентов с диабетом 2 типа.
Фармацевтическую композицию по настоящему изобретению можно также готовить для применения в лечении субъекта, описанного в данном документе.
Согласно настоящему изобретению фармацевтическая композиция или/и лекарственное средство, описанное в данном документе, может представлять собой жидкую композицию, содержащую десPro36эксендин-4(1-39)-Lys6-NH2 или/и его фармацевтически приемлемую соль. Квалифицированному специалисту известны жидкие композиции AVE0010, подходящие для парентерального введения. Жидкая композиция по настоящему изобретению может иметь кислое или физиологическое значение pH. Кислое значение pH предпочтительно находится в диапазоне pH 1-6,8, pH 3,5-6,8 или pH 3,5-5. Физиологическое значение pH предпочтительно находится в диапазоне pH 2,5-8,5, pH 4,0-8,5 или pH 6,0-8,5. Предпочтительным является диапазон pH 4,5-5,0.
Значение pH можно регулировать при помощи фармацевтически приемлемой разбавленной кислоты (как правило, HCl) или фармацевтически приемлемого разбавленного основания (как правило, NaOH).
Жидкая композиция по настоящему изобретению может содержать подходящий консервант. Подходящий консервант можно выбирать из фенола, м-крезола, бензилового спирта и сложного эфира п-гидроксибензойной кислоты. Предпочтительным консервантом является м-крезол.
Жидкая композиция по настоящему изобретению может содержать средство, регулирующее тоничность. Подходящее средство, регулирующее тоничность, можно выбирать из глицерина, лактозы, сорбита, маннита, глюкозы, NaCl, содержащих кальций или магний соединений, таких как CaCl2. Концентрация глицерина, лактозы, сорбита, маннита и глюкозы может быть в диапазоне 100-250 мМ. Концентрация NaCl может составлять до 150 мМ. Предпочтительным средством, регулирующим тоничность, является глицерин.
Жидкая композиция по настоящему изобретению может содержать метионин.
Еще одним аспектом настоящего изобретения является способ лечения сахарного диабета 2 типа, включающий введение десPro36эксендин-4(1-39)-Lys6-NH2 или/и его фармацевтически приемлемой соли субъекту, нуждающемуся в этом.
Следующим аспектом настоящего изобретения является способ стимуляции снижения массы тела у пациентов с диабетом 2 типа или/и предотвращения увеличения массы тела у пациентов с диабетом 2 типа, включающий введение десPro36эксендин-4(1-39)-Lys6-NH2 или/и его фармацевтически приемлемой соли субъекту, нуждающемуся в этом.
В способе по настоящему изобретению субъект может быть субъектом, определенным в данном документе.
В способе по настоящему изобретению можно вводить фармацевтическую композицию или/и лекарственное средство, описанные в данном документе.
Далее изобретение проиллюстрировано следующими примерами и фигурами.
Подписи к фигурам
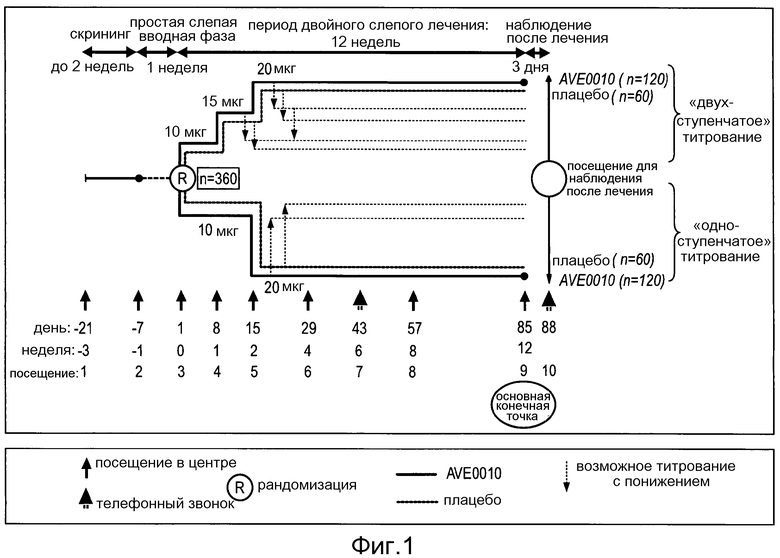
Фигура 1: Дизайн исследования.
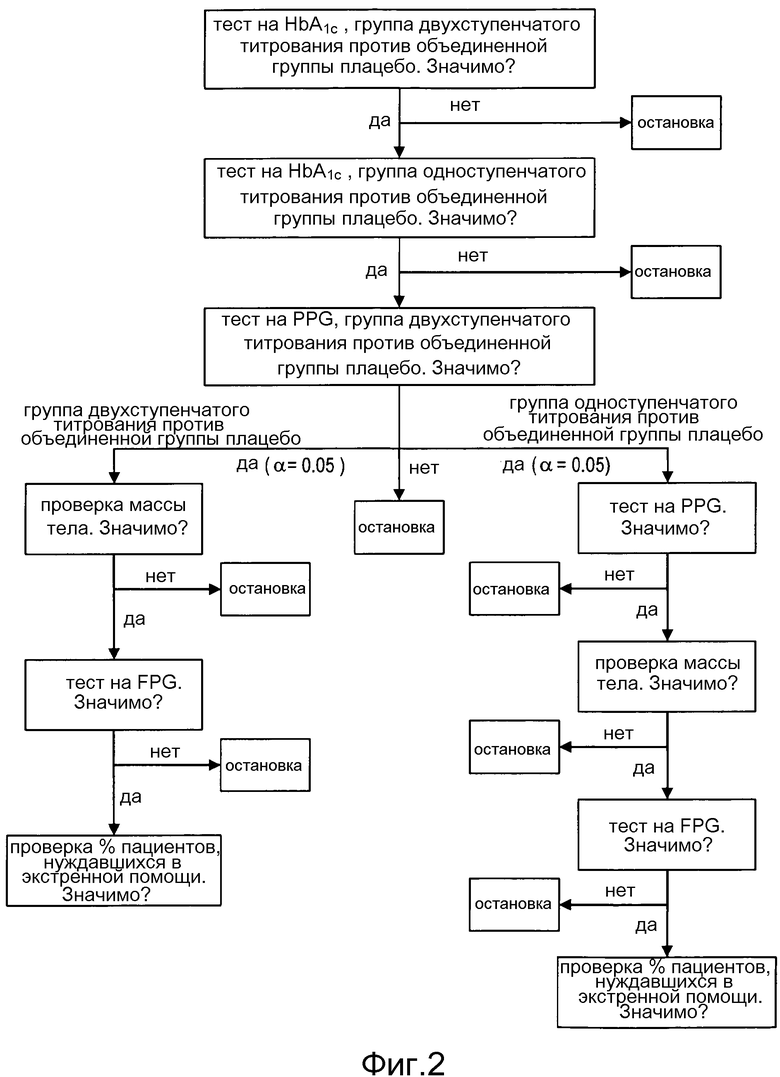
Фигура 2: Общая процедура тестирования с пошаговым понижением.
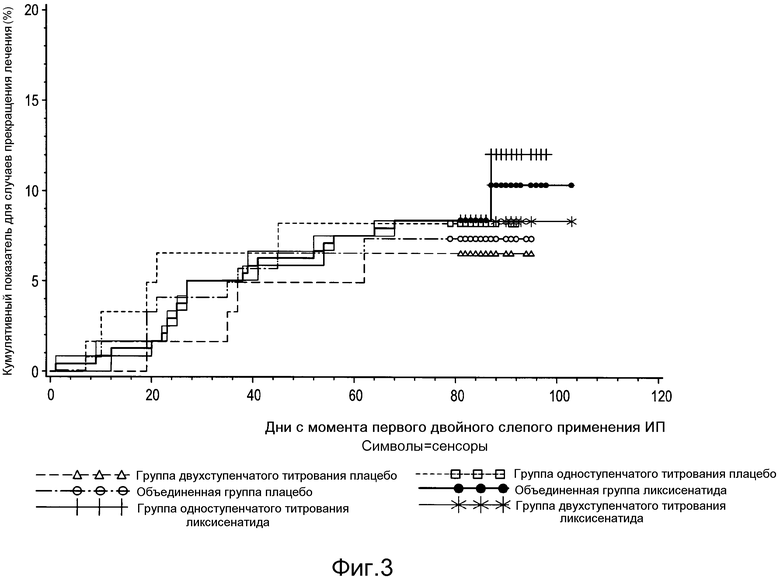
Фигура 3: График Каплана-Мейера, отражающий время до прекращения лечения по любой причине - рандомизированная популяция.
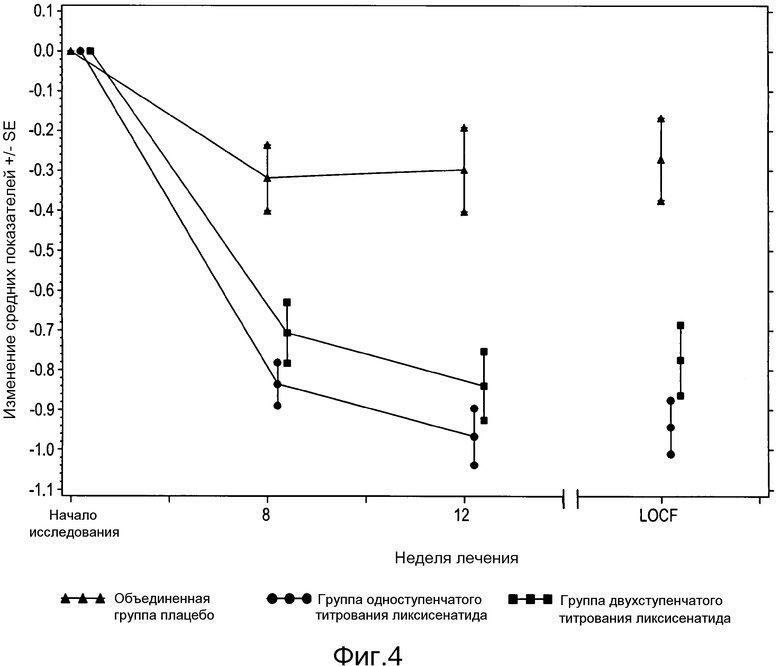
Фигура 4: График изменения среднего показателя HbA1c (%) ± SE от исходного значения по посещениям и по окончании исследования - популяция мITT. Из анализа исключены измерения, полученные после введения резервного препарата или/и после прекращения лечения плюс 3 дня.
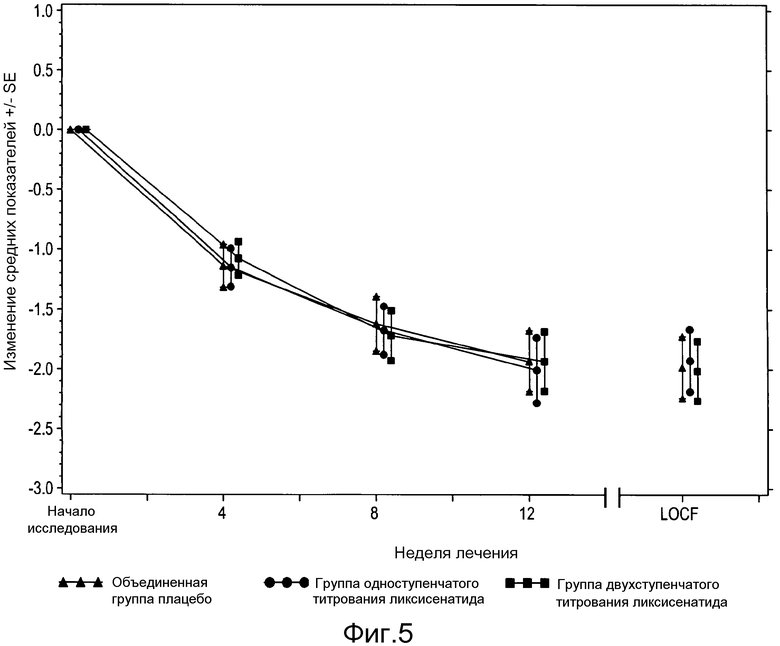
Фигура 5: График изменения среднего показателя массы тела (кг) ± SE от исходного значения по посещениям и по окончании исследования - популяция мITT. Из анализа исключены измерения, полученные после введения резервного препарата или/и после прекращения лечения плюс 3 дня.
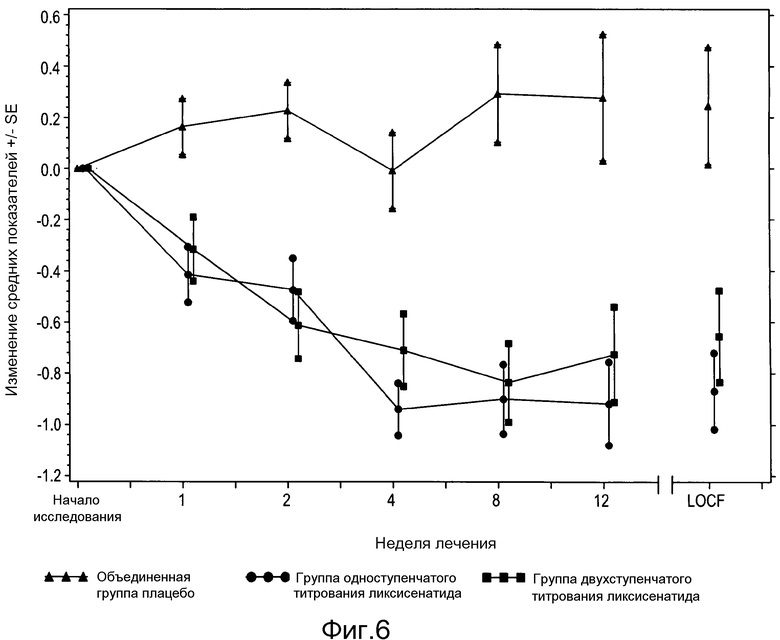
Фигура 6: График изменения среднего показателя уровня глюкозы в плазме натощак (ммоль/л) ± SE от исходного значения по посещениям и по окончании исследования - популяция мITT. Из анализа исключены измерения, полученные после введения резервного препарата или/и после прекращения лечения плюс 1 день.
ПРИМЕР: Рандомизированное, двойное слепое, плацебо-контролируемое, многоцентровое 12-недельное исследование в параллельных группах для оценки эффективности и безопасности ликсисенатида у пациентов с диабетом 2 типа, не получающих антидиабетические препараты.
РЕЗЮМЕ
Пример относится к рандомизированному, двойному слепому, плацебо-контролируемому, многоцентровому 12-недельному исследованию в параллельных группах для оценки эффективности и безопасности ликсисенатида у пациентов с диабетом 2 типа, не получающих антидиабетические препараты, проведенному в 61 центре в 12 странах. Основная цель исследования заключалась в оценке эффекта на гликемический контроль ликсисенатида, используемого в режиме двухступенчатого титрования дозы, в сравнении с плацебо с точки зрения снижения HbA1c (абсолютное изменение) на протяжении 12-недельного периода.
В общей сложности 361 пациент был рандомизирован в одну из четырех групп лечения (61 в группу двухступенчатого титрования плацебо, 61 в группу одноступенчатого титрования плацебо, 120 в группу двухступенчатого титрования ликсисенатида и 119 в группу одноступенчатого титрования ликсисенатида). В анализе группы одноступенчатого и двухступенчатого титрования плацебо были объединены. Два пациента были исключены из популяции мITT для анализов эффективности вследствие отсутствия пост-исходных данных по эффективности. Из 361 рандомизированных пациентов 331 (91,7%) полностью прошли курс 12-недельного двойного слепого лечения. Тридцать пациентов прекратили лечение преждевременно и 9 из этих пациентов прекратили лечение из-за побочных эффектов. Демографические и исходные характеристики, как правило, были сходными у групп лечения.
Среднеквадратичные (LS) изменения среднего от исходного до конечного значения для HbA1c составляли -0,19% для группы плацебо, -0,73% для группы 2-ступенчатого титрования ликсисенатида (LS изменение среднего против плацебо = -0,54%; p-величина = <0,0001), и -0,85% для группы 1-ступенчатого титрования ликсисенатида (LS изменение среднего против плацебо = -0,66%; p-величина = <0,0001). Анализ пациентов с терапевтическим эффектом в отношении HbA1c (HbA1c ≤6,5 или <7% в конце исследования) при помощи метода CMH также показал значительные отличия лечения в сравнении с плацебо для обеих групп пациентов, получавших ликсисенатид.
В отношении 2-часовых постпрандиальных уровней глюкозы в плазме, каждая из групп, получавших ликсисенатид, продемонстрировала значительное улучшение по сравнению с группой плацебо. Межгрупповое различие в массе тела в сравнении с группой плацебо не было статистически значимым для любой из групп пациентов, получавших ликсисенатид, вследствие аналогичного уменьшения в группе плацебо. В обеих группах пациентов, получавших ликсисенатид, наблюдали значимые улучшения по сравнению с группой плацебо в уровнях глюкозы в плазме натощак при использовании анализа ANCOVA без правки на множественность. В общей сложности 3 пациента, получавших ликсисенатид (2 [1,7%] при 2-ступенчатом титровании и 1 [0,8%] при 1-ступенчатом титровании) получили резервную терапию, и 3 пациента [2,5%] в группе плацебо.
Ликсисенатид (AVE0010) хорошо переносился в течение 12 недель лечения. Частота появления TEAE (возникающих во время лечения побочных эффектов) была в целом сопоставима между группами лечения. Сообщалось только об одном серьезном TEAE у пациента, получавшего ликсисенатид (2-ступенчатое титрование), тогда как у 5 пациентов, получавших плацебо, наблюдали серьезные TEAE. В данном исследовании не было случаев смерти. В общей сложности 8 пациентов, получавших ликсисенатид (5 [4,2%] при 2-ступенчатом титровании и 3 [2,5%] при 1-ступенчатом титровании), прекратили лечение, в основном из-за заболевания желудочно-кишечного тракта (GI), при том, что лечение прекратил один пациент, получавший плацебо (0,8%). Не было очевидной разницы с точки зрения GI переносимости у пациентов, получавших ликсисенатид при 1-ступенчатом и 2-ступенчатом титровании. Наиболее часто отмечаемым TEAE была тошнота (24,2% при 2-ступенчатом титровании ликсисенатида, 20,2% при 1-ступенчатом титровании ликсисенатида и 4,1% при использовании плацебо).
В общей сложности наблюдали 6 случаев (3 [2,5%] при 2-ступенчатом титровании ликсисенатида; 1 [0,8%] при 1-ступенчатом титровании ликсисенатида; 2 [1,6%] при использовании плацебо) симптоматической гипогликемии по определению протокола и ни один из них не был серьезным. Ни одного случая повышения уровня липазы или амилазы (≥3 верхних границ нормы (ULN)) не наблюдали ни в одной из групп лечения.
1. ЦЕЛИ ИССЛЕДОВАНИЯ
1.1. ОСНОВНАЯ ЦЕЛЬ
Основная цель данного примера заключалась в оценке эффекта на гликемический контроль ликсисенатида, используемого в режиме двухступенчатого титрования дозы, в сравнении с плацебо с точки зрения снижения HbA1c (абсолютное изменение) на протяжении 12-недельного периода у пациентов с диабетом 2 типа, не получавших антидиабетических препаратов.
1.2. ВТОРИЧНАЯ ЦЕЛЬ(И)
Вторичными целями данного исследования были:
• Оценка эффекта ликсисенатида на:
- на гликемический контроль в сравнении с плацебо с точки зрения снижения HbA1c при использовании в режиме одноступенчатого титрования дозы на протяжении 12-недельного периода,
- массу тела в неделю 12,
- уровень глюкозы в плазме натощак (FPG) в неделю 12,
- 2-часовой постпрандиальный уровень глюкозы в плазме после стандартизированного провокационного теста с приемом пищи в неделю 12 в подгруппе всех пациентов в выбранных центрах (примерно 50% рандомизированных пациентов),
• Оценка безопасности и переносимости ликсисенатида на протяжении 12-недельного периода,
• Оценка ФК ликсисенатида с использованием популяционного ФК подхода,
• Оценка выработки антител против ликсисенатида.
2. ДИЗАЙН ИССЛЕДОВАНИЯ
Это было двойное слепое, рандомизированное, плацебо-контролируемое, в четырех группах, с несбалансированным планом, многонациональное исследование в параллельных группах: двухступенчатое титрование (120 получающих ликсисенатид и 60 получающих плацебо пациентов) и одноступенчатое титрование (120 получающих ликсисенатид и 60 получающих плацебо пациентов). Исследование было двойным слепым в отношении лечения активным веществом и плацебо. Информация об объеме лекарственного средства (то есть, дозе активного лекарственного средства или соответствующего плацебо) и режимах титрования (то есть, одноступенчатое и двухступенчатое) была открытой.
Пациенты были стратифицированы на основании значений при скрининге гликозилированного гемоглобина A1c (HbA1c) (<8%, ≥8%) и индекса массы тела (ИМТ <30 кг/м2, ≥30 кг/м2). После периода скрининга пациенты были рандомизированы централизованно с помощью системы интерактивного голосового ответа (IVRS) в отношении 2:1:2:1 в одну из четырех групп (двухступенчатое титрование ликсисенатида, двухступенчатое титрование плацебо, одноступенчатое титрование ликсисенатида и одноступенчатое титрование плацебо).
Исследование состояло из 3 периодов: 1) период скрининга продолжительностью до 3 недель, который включал фазу скрининга продолжительностью до 2 недель и 1-недельную простую слепую вводную фазу с плацебо; 2) основной 12-недельный период двойного слепого плацебо-контролируемого лечения; 3) 3-дневный свободный от лекарств период наблюдения после лечения.
Дизайн исследования описан на фигуре 1.
Введение выполняли следующим образом...
3. ОСНОВНАЯ И ВТОРИЧНЫЕ КОНЕЧНЫЕ ТОЧКИ ИССЛЕДОВАНИЯ
3.1. ОСНОВНАЯ КОНЕЧНАЯ ТОЧКА
Основная переменная эффективности представляла собой абсолютное изменение HbA1c от начала исследования до недели 12, которое определяли как: значение HbA1c в неделю 12 - значение HbA1c в начале исследования.
Если пациент безвозвратно прекращал лечение преждевременно или получал резервную терапию в течение 12-недельного периода двойного слепого лечения, или не имел показателя HbA1c в неделю 12, последнее пост-исходное измерение HbA1c во время периода применения исследуемого препарата на протяжении 12-недельного периода двойного слепого лечения следовало использовать в качестве значения HbA1c в неделю 12 (метод перенесения последнего наблюдения [LOCF]).
3.2. ВТОРИЧНЫЕ КОНЕЧНЫЕ ТОЧКИ
Для вторичных переменных эффективности применяли тот же метод обращения с отсутствующими показателями/ранним прекращением исследования на протяжении 12-недельного периода двойного слепого лечения, что и для основной переменной эффективности.
Длительные переменные:
• Изменение 2-часовой постпрандиальной концентрации глюкозы в плазме (ммоль/л) после стандартизированного теста с приемом пищи от начала исследования до недели 12,
• Изменение массы тела (кг) от начала исследования до недели 12,
• Изменение концентрации глюкозы в плазме натощак (ммоль/л) от начала исследования до недели 12,
• Изменение экскурсии глюкозы (ммоль/л) (2-часовая постпрандиальная концентрация глюкозы в плазме - концентрация глюкозы в плазме за 30 минут до теста с приемом пищи, перед введением исследуемого лекарственного средства) после стандартизированного теста с приемом пищи от начала исследования до недели 12.
Категориальные переменные:
• Процентная доля пациентов с HbA1c <7% в неделю 12,
• Процентная доля пациентов с HbA1c ≤6,5% в неделю 12,
• Процентная доля пациентов, нуждающихся в резервной терапии на протяжении периода двойного слепого лечения,
• Процентная доля пациентов с ≥5% снижением массы тела (кг) от начала исследования до недели 12.
4. РАССЧЕТНАЯ ГИПОТЕЗА О РАЗМЕРЕ ВЫБОРКИ
Расчет размера выборки/статистической мощности проводили на основании основной переменной эффективности, изменения HbA1c от начала исследования до недели 12.
Для выявления разницы в 0,5% в изменении HbA1c от исходного значения между одной группой ликсисенатида и объединенной группой плацебо в неделю 12, 120 пациентов на группу (то есть, 120 пациентов на группу ликсисенатида и 2·60 пациентов для объединенной группы плацебо) обеспечивали статистическую мощность исследования 90%. Этот расчет предполагал общее стандартное отклонение 1,2% с 2-сторонним критерием на 5% уровне значимости. Расчеты размера выборки были основаны на двухвыборочном критерии Стьюдента и были выполнены при помощи программы nQuery Advisor 5.0.
5. СТАТИСТИЧЕСКИЕ МЕТОДЫ
5.1. ПОПУЛЯЦИИ ДЛЯ АНАЛИЗА
Модифицированная-ITT популяция состояла из всех пациентов, которые были рандомизированы (анализировались «как рандомизированные»), получали по меньшей мере одну дозу исследуемого препарата двойным слепым образом и проходили как исходную оценку, так и по меньшей мере одну пост-исходную оценку любой основной или вторичной переменной эффективности, независимо от соблюдения протокола и процедур исследования.
Популяция для изучения безопасности представляла собой всю подвергающуюся лечению популяцию, определенную как все пациенты, которые были рандомизированы (через центральную систему рандомизации в соответствии с протоколом) и получали по меньшей мере одну дозу исследуемого препарата, независимо от количества введенного лекарственного средства.
5.2. АНАЛИЗ ОСНОВНОЙ ЭФФЕКТИВНОСТИ
Основную переменную эффективности (изменение HbA1c от начала исследования до недели 12) анализировали при помощи модели ковариационного анализа (ANCOVA) с группами лечения (группы двухступенчатого титрования ликсисенатида и плацебо, группы одноступенчатого титрования ликсисенатида и плацебо), рандомизированными стратами по уровню HbA1c при скрининге (<8,0, ≥8,0%), рандомизированными стратами по значениям ИМТ при скрининге (<30, ≥30 кг/м2) и страной в качестве фиксированного эффекта, и использованием исходных значений HbA1c в качестве ковариата. В модели ANCOVA две группы титрования плацебо были включены в качестве отдельных уровней лечения, но они были объединены в одну группу при проведении сравнений с соответствующей противоположностью, например для сравнения группы двухступенчатого титрования ликсисенатида с объединенной группой плацебо [-0,5, -0,5, 0, +1] в порядке групп одноступенчатого титрования плацебо, двухступенчатого титрования плацебо, одноступенчатого титрования ликсисенатида и двухступенчатого титрования ликсисенатида.
Использовали поэтапную процедуру тестирования в целях обеспечения контроля ошибок I типа. Во-первых, группу двухступенчатого титрования ликсисенатида сравнивали с объединенной группой плацебо (основная цель). Если тест был статистически значимым, тогда группу одноступенчатого титрования ликсисенатида сравнивали с объединенной группой плацебо (вторичная цель).
Как упомянуто в разделе 3.1, основная конечная точка представляет собой абсолютное изменение HbA1c от начала исследования до недели 12 при использовании LOCF во время периода применения исследуемого препарата. Период применения исследуемого препарата для переменных эффективности, за исключением полученных из провокационного теста с приемом пищи, представляет собой время от первой дозы исследуемого препарата до 3 дней (за исключением определения уровня глюкозы в плазме натощак (FPG) центральной лабораторией, в этом случае до 1 дня) после последней дозы исследуемого препарата или до введения резервной терапии, в зависимости от того, что раньше. Период применения исследуемого препарата для переменных эффективности, полученных из провокационного теста с приемом пищи, включая постпрандиальный уровень глюкозы в плазме (PPG) и экскурсию глюкозы, представляет собой время от первой дозы до даты применения последней дозы исследуемого препарата или до введения резервной терапии, в зависимости от того, что раньше.
5.3. АНАЛИЗ ВТОРИЧНОЙ ЭФФЕКТИВНОСТИ
После того как основная переменная оказалась статистически значимой при α=0,05 для обоих сравнений, проводили процедуру тестирования для проверки вторичных переменных эффективности, см. фигуру 2.
Все длительные вторичные переменные эффективности в неделю 12 анализировали с использованием аналогичной модели ANCOVA, как описано в разделе 5.2, для сравнения группы двухступенчатого титрования ликсисенатида с объединенной группой плацебо, и группы одноступенчатого титрования ликсисенатида с объединенной группой плацебо.
Следующие категориальные вторичные переменные эффективности в неделю 12 анализировали с использованием метода Кохрана-Мантеля-Хензеля (CMH), со стратификацией на рандомизированные страты (значения HbA1c при скрининге (<8,0, ≥8,0%) и ИМТ при скрининге (<30 кг/м2, ≥30 кг/м2)):
• Процентная доля пациентов с HbA1c <7,0% в неделю 12,
• Процентная доля пациентов с HbA1c ≤6,5% в неделю 12,
• Процентная доля пациентов, нуждающихся в резервной терапии на протяжении 12-недельного периода лечения.
Количество и процентная доля пациентов с ≥5% снижением массы тела от начала исследования до недели 12 представлены по группам лечения.
5.4. АНАЛИЗ НА БЕЗОПАСНОСТЬ
Возникающие во время лечения AE (TEAE) определяют как AE, которые возникли или усугубились (по мнению исследователя), либо стали серьезными в процессе периода применения исследуемого препарата. Период применения исследуемого препарата был определен как время от первой дозы исследуемого препарата (ИП), примененного двойным слепым методом, до 3 дней после последнего введения инъекцией ИП. 3-дневный интервал был выбран на основе периода полувыведения ИП (примерно в 5 раз больше периода полувыведения).
6. РЕЗУЛЬТАТЫ
6.1. ПАЦИЕНТЫ ИССЛЕДОВАНИЯ
6.1.1. Учет пациентов
Из 795 пациентов, прошедших скрининг, 434 (54,6%) пациентов не были рандомизированы для двойного слепого лечения. Главной причиной была величина HbA1c при скрининговом посещении, находящаяся за пределами определенного протоколом диапазона (318 (40,0%) пациентов).
В общей сложности 361 пациент был рандомизирован в одну из четырех групп лечения (61 в группу двухступенчатого титрования плацебо, 61 в группу одноступенчатого титрования плацебо, 120 в группу двухступенчатого титрования ликсисенатида и 119 в группу одноступенчатого титрования ликсисенатида) в 61 центре в 12 странах (Бельгия, Индия, Израиль, Япония, Корея, Мексика, Польша, Румыния, Россия, Тунис, Украина и Соединенные Штаты). Все 361 рандомизированные пациенты подвергались двойному слепому лечению. Два пациента были исключены из мITT популяции для анализов эффективности вследствие отсутствия пост-исходных данных по эффективности. В таблице 1 ниже приведено количество пациентов, включенных в каждую анализируемую популяцию.
Анализируемые популяции - Рандомизированная популяция
Примечание: Пациенты популяций для изучения безопасности и ФК приведены в таблице в соответствии с фактически полученным лечением (какое лечение получали).
6.1.2 Распределение участников клинического исследования
В таблице 2 ниже приведены сводные данные по распределению участников клинического исследования для каждой группы лечения. Из 361 рандомизированных пациентов 30 (8,3%) пациентов преждевременно прекратили исследуемое лечение, главным образом по причинам, классифицируемым как «другие» (то есть, по решению субъекта, 18 пациентов), с последующими побочными эффектами (9 пациентов). Время до начала прекращения лечения приведено на фигуре 3, при этом не наблюдали никакой определенной закономерности.
Распределение участников клинического исследования - Рандомизированная популяция
6.1.3 Демографические и исходные характеристики
В таблице 3 ниже приведены сводные данные по исходным и демографическим характеристикам для каждой группы лечения и в целом. Демографическая и исходная информация, как правило, была схожей у групп лечения для популяции, предназначенной для изучения безопасности. Исследуемая популяция была сбалансирована по половой принадлежности, и средний возраст составлял 54 года. Большинство пациентов были белыми (72,9%).
Демография и характеристики пациентов при скрининге - Популяция для изучения безопасности
[n (%)]
восточная
[n (%)]
10,0
[n (%)]
58,7
В таблице 4 ниже приведена история заболевания диабетом для каждой группы лечения и в целом для всей популяции, предназначенной для изучения безопасности. Истории заболевания диабетом, как правило, были сопоставимыми для групп лечения.
Таблица 4
Характеристики заболевания при скрининге - Популяция для изучения безопасности
максимум
[n (%)]
[n (%)]
324,1
[n (%)]
В таблице 5 ниже приведены описательные сводные данные по переменным эффективности в начале исследования для каждой группы лечения и в целом для популяции, предназначенной для изучения безопасности. Переменные эффективности в начале исследования, как правило, сопоставимы для групп лечения.
Исходные переменные эффективности - Популяция для изучения безопасности
FPG = Глюкоза в плазме натощак.
Экскурсия глюкозы = 2-часовая постпрандиальная концентрация глюкозы в плазме - концентрация глюкозы в плазме за 30 минут до теста с приемом пищи, перед введением исследуемого лекарственного средства.
6.1.4 Дозировка и продолжительность лечения
Продолжительность лечения и дозировки приведены в таблице 6, таблице 7 и таблице 8 ниже. Средняя продолжительность лечения была одинаковой для групп лечения. Из 361 пациента в популяции для изучения безопасности, к 335 (92,8%) применяли лечение в течение 57 дней или более, у 349 (96,7%) была достигнута целевая доза 20 мкг в конце титрования, и 335 (92,8%) получили конечную дозу с целевой дозой 20 мкг по окончании двойного слепого лечения.
Продолжительность приема исследуемого препарата - Популяция для изучения безопасности
[n (%)]
[n (%)]
Количество (%) пациентов по конечной дозе в конце двойного слепого лечения - Популяция для изучения безопасности
Примечание: Проценты рассчитаны с использованием количества пациентов в популяции для изучения безопасности в качестве знаменателя.
Количество (%) пациентов по дозе в конце титрования - Популяция для изучения безопасности
Запланированным посещением для окончания титрования по протоколу должно было стать посещение 5/неделя 2.
Примечание: Проценты рассчитаны с использованием количества пациентов в популяции для изучения безопасности в качестве знаменателя.
6.2. ЭФФЕКТИВНОСТЬ
6.2.1. Основной параметр эффективности
Главный анализ
В таблице 1 приведены результаты по основному параметру эффективности, изменению HbA1c от исходного до конечного значения, полученные при помощи анализа LOCF ANCOVA. На фигуре 4 показано среднее (±SE) изменение HbA1c от исходного значения с течением времени на протяжении 12-недельного двойного слепого лечения.
Исходя из заранее определенного основного анализа, обе группы получавших ликсисенатид пациентов продемонстрировали статистически значимое снижение HbA1c от исходного до конечного значения, по сравнению с группой плацебо (для группы двухступенчатого титрования ликсисенатида LS изменение среднего = -0,54%; p-величина= <,0001; для группы одноступенчатого титрования ликсисенатида LS изменение среднего = -0,66%; p-величина = <,0001). Кроме того, значение HbA1c, судя по всему, достигло плато после недели 8 в группе плацебо, тогда как значение HbA1c непрерывно снижалось в обеих группах получавших ликсисенатид пациентов.
Изменение средних показателей HbA1c (%) от исходного до конечного значения - мITT популяция
(N=121)
Из анализа исключены измерения, полученные после введения резервного препарата или/и после прекращения лечения плюс 3 дня.
Вторичные анализы
В таблице 10 приведено количество в процентах пациентов, ответивших на лечение (HbA1c ≤6,5 или <7% в конце исследования, соответственно). Ответы на лечение были сходными у групп получавших ликсисенатид пациентов, и разница в зависимости от лечения между каждой из групп получавших ликсисенатид и группой получавших плацебо пациентов была статистически значимой.
Количество (%) пациентов, имеющих показатель HbA1c ≤6,5% или <7% в конце исследования -мITT популяция
(N=121)
(N=120)
(N=118)
6.2.2. Вторичные параметры эффективности
В таблице 11, таблице 12, таблице 13 и таблице 15 приведены данные анализов ANCOVA для 2-часовой постпрандиальной концентрации глюкозы в плазме, экскурсии глюкозы, массы тела и FPG, соответственно.
В таблице 14 и таблице 16 представлено количество в процентах пациентов со снижением массы тела ≥5% от исходного до конечного значения, и процентная доля пациентов, которым потребовалась резервная терапия, соответственно. На фигуре 5 и фигуре 6 представлено среднее (±SE) изменение от исходного значения массы тела и FPG с течением времени на протяжении 12-недельного периода двойного слепого лечения.
Обе группы получавших ликсисенатид пациентов продемонстрировали статистически значимое улучшение по сравнению с группой плацебо в отношении 2-часовой постпрандиальной концентрации глюкозы в плазме, что подтвердилось аналогичным анализом ANCOVA экскурсии глюкозы.
Изменение средних показателей 2-часовой постпрандиальной концентрации глюкозы в плазме (ммоль/л) от исходного до конечного значения в избранных центрах - мITT популяция
Изменение средних показателей экскурсии глюкозы (ммоль/л) от исходного до конечного значения в избранных центрах - мITT популяция
Сравнение между каждой группой ликсисенатида и объединенной группой плацебо достигалось за счет соответствующих противопоставлений.
Из анализа исключены измерения, полученные после введения резервного препарата или/и после прекращения лечения.
Экскурсия глюкозы = 2-часовая постпрандиальная концентрация глюкозы в плазме - концентрация глюкозы в плазме за 30 минут до теста с приемом пищи, перед введением исследуемого лекарственного средства.
Никакой разницы в изменении массы тела не наблюдалось между группами ликсисенатида и плацебо (таблица 13), также сопоставим процент пациентов среди групп лечения, у которых масса тела снизилась на 5% или более (таблица 14).
Изменение средних показателей массы тела (кг) от исходного до конечного значения - мITT популяция
Сравнение между каждой группой ликсисенатида и объединенной группой плацебо достигалось за счет соответствующих противопоставлений.
Из анализа исключены измерения, полученные после введения резервного препарата или/и после прекращения лечения плюс 3 дня.
Количество (%) пациентов с ≥5% снижением массы тела от исходного до конечного значения - мITT популяция
(N=121)
(N=120)
(N=118)
Согласно стратегии тестирования правки на множественность (пошаговое понижение), было выполнено логически выведенное тестирование на FPG в поисковой манере, поскольку предшествующий тест (масса тела) не показал статистически значимых различий между группами. Для обеих групп получавших ликсисенатид пациентов было продемонстрировано значимое улучшение по сравнению с группой плацебо показателей FPG с помощью анализа ANCOVA без правки на множественность.
Изменение средних показателей концентрации глюкозы в плазме натощак (ммоль/л) от исходного до конечного значения - мITT популяция
Сравнение между каждой группой ликсисенатида и объединенной группой плацебо достигалось за счет соответствующих противопоставлений.
Из анализа исключены измерения, полученные после введения резервного препарата или/и после прекращения лечения плюс 1 день.
Не было никаких доказательств наличия разницы между группами пациентов, получавших ликсисенатид и плацебо, в процентной доле пациентов, которым потребовалась резервная терапия, вследствие крайне редких случаев экстренного спасения пациентов на протяжении периода двойного слепого лечения.
Количество (%) пациентов, нуждающихся в резервной терапии на протяжении периода двойного слепого лечения - мITT популяция
6.3 БЕЗОПАСНОСТЬ
В таблице 17 ниже приведены обобщенные данные пациентов, у которых проявились побочные эффекты в процессе двойного слепого лечения, а в таблице 18 и таблице 19 приведены серьезные TEAE, и TEAE, приводящие к прекращению лечения, соответственно. Процентные доли пациентов, у которых проявились TEAE, были в целом сопоставимы для группы получавших плацебо и групп, получавших ликсисенатид пациентов. Частота возникновения серьезных TEAE была низкой, 5 случаев (4,1%) в группе плацебо, 1 (0,8%) в группе двухступенчатого титрования ликсисенатида и 0 в группе одноступенчатого титрования ликсисенатида. В данном исследовании не было случаев смерти. Больше пациентов в получавших ликсисенатид группах (5 [4,2%] при двухступенчатом титровании; 3 [2,5%] при одноступенчатом титровании) прекратили лечение, чем в группе плацебо (1 [0,8%]), в основном из-за заболевания желудочно-кишечного тракта.
В таблице 25 представлены случаи TEAE в процессе двойного слепого лечения, имевшие место у по меньшей мере 1% пациентов в любой из групп лечения. Тошнота была наиболее часто отмечаемым TEAE в группе получавших ликсисенатид: 29 пациентов (24,2%) при двухступенчатом титровании и 24 пациента (20,2%) при одноступенчатом титровании. Пять получавших плацебо пациентов (4,1%) сообщали о тошноте. Вторым наиболее часто отмечаемым TEAE у получавших ликсисенатид пациентов была головная боль (10 пациентов (8,3%) при двухступенчатом титровании и 9 пациентов (7,6%) при одноступенчатом титровании), сопровождаемая рвотой (9 пациентов [7,5%] при двухступенчатом титровании и 8 пациентов [6,7%] при одноступенчатом титровании). Соответствующее количество пациентов (%) в группе плацебо было 14 (11,5%) с головной болью и ни одного с рвотой.
Обзор профиля побочных эффектов: возникающие во время лечения побочные эффекты - Популяция для изучения безопасности
n (%)=количество и процентная доля пациентов, имеющих по меньшей мере один побочный эффект.
Количество (%) пациентов, испытавших серьезные TEAE(s), представленные по основным SOC, HLGT, HLT и PT в период применения исследуемого препарата - Популяция для изучения безопасности
HLGT: групповые термины высокого уровня
HLT: термины высокого уровня
предпочтительный термин n (%)
Период применения исследуемого препарата = время от первой дозы препарата в двойном слепом исследовании до 3 дней после введения последней дозы.
MedDRA версия: 12.1
n (%)=количество и процентная доля пациентов с по меньшей мере одним серьезным TEAE.
Примечание: Таблица сортирована в соответствии с международным согласованным регламентом SOC и алфавитным порядком HLGT, HLT, PT.
Количество (%) пациентов, испытавших TEAE(s), приводящие к безвозвратному прекращению лечения, представленные по основным SOC, HLGT, HLT и PT в период применения исследуемого препарата - Популяция для изучения безопасности
HLGT: групповые термины высокого уровня
HLT: термины высокого уровня
предпочтительный термин n (%)
Период применения исследуемого препарата=время от первой дозы препарата в двойном слепом исследовании до 3 дней после введения последней дозы.
MedDRA версия: 12.1
n (%)=количество и процентная доля пациентов с по меньшей мере одним TEAE, приводящим к безвозвратному прекращению лечения.
Примечание: Таблица сортирована в соответствии с международным согласованным регламентом SOC и алфавитным порядком HLGT, HLT, PT.
Как показано в таблице 20 ниже, наблюдали в общей сложности 6 случаев симптоматической гипогликемии по определению протокола (3 [2,5%] в группе двухступенчатого титрования ликсисенатида, 1 [0,8%] в группе одноступенчатого титрования ликсисенатида и 2 [1,6%] в группе плацебо), и ни один из них не был серьезным.
Сводные данные по симптоматической гипогликемии - Популяция для изучения безопасности
1: Проценты рассчитаны с использованием количества пациентов в популяции для изучения безопасности в качестве знаменателя.
2: Количество пациентов с эпизодами на 100 пациенто-лет приема препаратов=100*(количество пациентов с эпизодами/прием препаратов в пациенто-годах).
В общей сложности 11 пациентов, все из получавших ликсисенатид пациентов (4 [3,3%] в группе двухступенчатого титрования и 7 [5,9%] в группе одноступенчатого титрования), сообщали о реакциях в месте инъекции. Ни одна из реакций не была серьезной или тяжелой.
Количество (%) пациентов, испытавших реакции в местах инъекций в процессе периода применения исследуемого препарата - Популяция для изучения безопасности
Исследователи сообщали в общей сложности о 3 случаях аллергических реакций в группе одноступенчатого титрования ликсисенатида в процессе периода двойного слепого лечения, и 2 из них были подтверждены комитетом по оценке аллергических реакций (ARAC).
Количество (%) пациентов с аллергической реакцией, признанной и подтвержденной ARAC - Популяция для изучения безопасности
Для одного пациента в группе плацебо сообщалось о побочном эффекте «повышенный уровень липазы», таблица 23, который имел место в день исследования 1 и, предположительно, до первой инъекции препарата в двойном слепом лечении в соответствии с протоколом исследования. Ни одного случая повышенного уровня липазы или амилазы (≥3 ULN) не наблюдали ни в одной из групп лечения (таблица 24) на протяжении периода двойного слепого лечения.
Количество (%) пациентов с подозрением на панкреатит - Популяция для изучения безопасности
Панкреатические ферменты: количество пациентов с аномалиями (PCSA) в период применения исследуемого препарата в соответствии с исходным статусом - Популяция для изучения безопасности
Период применения исследуемого препарата = время от первой дозы препарата в двойном слепом исследовании до 3 дней после введения последней дозы.
*независимо от исходного значения.
Примечание: Количество (n) представляет собой часть пациентов от общего числа, которые соответствовали рассматриваемому критерию по меньшей мере один раз в процессе лечения.
Знаменатель (/N1) для каждого параметра в группе лечения представляет собой количество пациентов в группе лечения, у которых данный параметр был оценен пост-исходно по исходному статусу PCSA.
Для PCSA, включая состояние, основанное только на изменении от исходного значения, знаменатель ограничен пациентами, имеющими исходное и пост-исходные показатели.
Количество (%) пациентов, испытавших обычные TEAE(s) (PT≥1% в объединенной группе плацебо или любой отдельной группе ликсисенатида), представленное по основным SOC и HLGT, HLT и PT
HLGT: групповые термины высокого уровня
HLT: термины высокого уровня
предпочтительный термин n (%)
Период применения исследуемого препарата = время от первой дозы препарата в двойном слепом исследовании до 3 дней после введения последней дозы.
MedDRA версия: 12.1
n (%)=количество и процентная доля пациентов с по меньшей мере одним TEAE.
Примечание: Таблица сортирована в соответствии с международным согласованным регламентом SOC и алфавитным порядком HLGT, HLT, PT.
Представлены только SOC с по меньшей мере одним PT≥1% в объединенной группе плацебо или любой группе одноступенчатого или двухступенчатого титрования ликсисенатида
Группа изобретений относится к применению десPro36эксендин-4(1-39)-Lys6-NH2 или/и его фармацевтически приемлемой соли для производства лекарственного средства для лечения сахарного диабета 2 типа и/или стимуляции снижения массы тела у пациентов с диабетом 2 типа, где указанный пациент с диабетом 2 типа, подлежащий лечению, имеет 2-часовую постпрандиальную концентрацию глюкозы в плазме по меньшей мере 14 ммоль/л и не получает антидиабетического лечения. Группа изобретений эффективна в лечении сахарного диабета 2 типа и/или стимуляции снижения массы тела у пациентов с диабетом 2 типа. 3 н. и 12 з.п. ф-лы, 6 ил., 25 табл., 1 прим.
1. Применение десPro36эксендин-4(1-39)-Lys6-NH2 или/и его фармацевтически приемлемой соли для производства лекарственного средства для снижения постпрандиальной концентрации глюкозы в плазме у пациентов с диабетом 2 типа, где указанный пациент с диабетом 2 типа, подлежащий лечению, имеет 2-часовую постпрандиальную концентрацию глюкозы в плазме по меньшей мере 14 ммоль/л и не получает антидиабетического лечения.
2. Применение по п. 1, где субъект, подлежащий лечению, страдает ожирением.
3. Применение по п. 1 или 2, где субъект, подлежащий лечению, имеет индекс массы тела по меньшей мере 30 кг/м2.
4. Применение по п. 1, где субъект, подлежащий лечению, является взрослым субъектом.
5. Применение по п. 1, где у субъекта, подлежащего лечению, сахарный диабет 2 типа был диагностирован по меньшей мере за 1 год или по меньшей мере за 2 года до начала терапии.
6. Применение по п. 1, где субъект, подлежащий лечению, имеет показатель HbA1c от примерно 7 до примерно 10%.
7. Применение по п. 1, где субъект, подлежащий лечению, имеет концентрацию глюкозы в плазме натощак по меньшей мере 8 ммоль/л.
8. Применение по п. 1, где субъект, подлежащий лечению, имеет показатель экскурсии глюкозы по меньшей мере 2 ммоль/л, по меньшей мере 3 ммоль/л, по меньшей мере 4 ммоль/л или по меньшей мере 5 ммоль/л, где экскурсия глюкозы представляет собой разницу между 2-часовой постпрандиальной концентрацией глюкозы в плазме и концентрацией глюкозы в плазме за 30 минут до теста с приемом пищи.
9. Применение по п. 1, где десPro36эксендин-4(1-39)-Lys6-NH2 или/и его фармацевтически приемлемую соль вводят парентерально.
10. Применение по п. 1, где лекарственное средство готовят для введения десPro36эксендин-4(1-39)-Lys6-NH2 или/и его фармацевтически приемлемой соли в суточной дозе, выбранной в диапазоне от 10 мкг до 20 мкг.
11. Применение по п. 1, где лекарственное средство готовят для введения один раз в сутки.
12. Фармацевтическая композиция, содержащая десPro36эксендин-4(1-39)-Lys6-NH2 или/и его фармацевтически приемлемую соль и содержащая фармацевтически приемлемые носитель, адъювант или/и вспомогательное вещество, для применения в снижении постпрандиальной концентрации глюкозы в плазме у пациентов с диабетом 2 типа, где указанный пациент с диабетом 2 типа, подлежащий лечению, имеет 2-часовую постпрандиальную концентрацию глюкозы в плазме по меньшей мере 14 ммоль/л и не получает антидиабетического лечения.
13. Композиция по п. 12 для применения в лечении субъекта, определенного в любом из пп. 2-8.
14. Способ снижения постпрандиальной концентрации глюкозы в плазме у пациентов с диабетом 2 типа, где пациент с диабетом 2 типа, подлежащий лечению, имеет 2-часовую постпрандиальную концентрацию глюкозы в плазме по меньшей мере 14 ммоль/л и не получает антидиабетического лечения, включающий введение десPro36эксендин-4(1-39)-Lys6-NH2 или/и его фармацевтически приемлемой соли субъекту, нуждающемуся в этом.
15. Способ по п. 14, в котором субъект представляет собой субъекта, определенного в любом из пп. 2-8.

