Объектом представленного изобретения является фармацевтическая комбинация для применения для индуцирования потери массы тела у субъектов с диабетом 2 типа или/и для предотвращения набора массы тела у субъектов с диабетом 2 типа, при этом указанная комбинация содержит (a) desPro36Эксендин-4(1-39)-Lys6-NH2 (AVE0010, ликсисенатид) или/и его фармацевтически приемлемую соль, и (b) метформин или/и его фармацевтически приемлемую соль. Другим аспектом является способ индуцирования потери массы тела у субъектов с диабетом 2 типа или/и для предотвращения набора массы тела у субъектов с диабетом 2 типа, включающий введение комбинации представленного изобретения нуждающемуся в этом субъекту.
У здорового человека высвобождение инсулина поджелудочной железой строго связано с концентрацией глюкозы крови. Повышенный уровень глюкозы крови, который возникает после приемов пищи, быстро уравновешивается соответствующим увеличением секреции инсулина. В состоянии натощак уровень инсулина в плазме понижается до базального значения, которое является достаточным, чтобы обеспечивать непрерывное снабжение глюкозой чувствительных к инсулину органов и тканей, а по ночам поддерживать выработку глюкозы печенью на низком уровне.
В отличие от диабета 1 типа, при диабете 2 типа в целом нет недостатка инсулина, но во многих случаях, в частности, в прогрессирующих случаях, лечение инсулином считается наиболее подходящей терапией, при необходимости, в комбинации с вводимыми перорально противодиабетическими лекарственными средствами.
Повышенный уровень глюкозы в крови в течение нескольких лет без первоначальных симптомов представляет собой значительный риск для здоровья. Посредством крупномасштабного исследования DCCT в США (The Diabetes Control and Complication Trial Research Group (1993) N. Engl. J. Med. 329, 977-986) может быть четко продемонстрировано, что хронически повышенные уровни глюкозы крови являются основной причиной для развития осложнений диабета. Примерами осложнений диабета являются микро- и макрососудистые повреждения, которые, возможно, проявляются ретинопатиями, нефропатиями или нейропатиями и приводят к слепоте, почечной недостаточности и потере конечностей и сопровождаются повышенным риском сердечно-сосудистых заболеваний. Таким образом, можно заключить, что улучшенная терапия диабета в первую очередь должна быть нацелена на сохранение глюкозы крови как можно ближе к физиологическому диапазону.
Особый риск существует для субъектов с избыточной массой тела, страдающих диабетом 2 типа, например, субъектов с индексом массы тела (BMI) ≥30. У данных субъектов риск диабета в сочетании с рисками избыточной массы тела, приводит, например, к увеличению сердечно-сосудистых заболеваний по сравнению с субъектами с диабетом 2 типа, имеющими нормальную массу тела. Соответственно, у данных субъектов особенно необходимо лечить диабет, уменьшая при этом избыточную массу тела.
Метформин представляет собой гипогликемическое средство бигуанид, применяемое при лечении инсулинонезависимого сахарного диабета (сахарного диабета 2 типа), не отвечающего на модификацию диеты. Метформин улучшает гликемический контроль за счет улучшения чувствительности к инсулину и уменьшения кишечной абсорбции глюкозы. Метформин, как правило, вводят перорально. Однако, контроль за сахарным диабетом 2 типа у страдающих ожирением субъектов с помощью метформина может быть недостаточным. Соответственно, для данных субъектов, могут потребоваться дополнительные меры для контроля за сахарным диабетом 2 типа.
Соединение desPro36Эксендин-4(1-39)-Lys6-NH2 (AVE0010, ликсисенатид) представляет собой производное Эксендина-4. В WO 01/04156 AVE0010 раскрыт, как SEQ ID NO:93:
SEQ ID NO:1: AVE0010 (44 AS)
H-G-E-G-T-F-T-S-D-L-S-K-Q-M-E-E-E-A-V-R-L-F-I-E-W-L-K-N-G-G-P-S-S-G-A-P-P-S-K-K-K-K-K-K-NH2
SEQ ID NO:2: Эксендин-4 (39 AS)
H-G-E-G-T-F-T-S-D-L-S-K-Q-M-E-E-E-A-V-R-L-F-I-E-W-L-K-N-G-G-P-S-S-G-A-P-P-P-S-NH2
Эксендины представляют собой группу пептидов, которые могут понижать концентрацию глюкозы в крови. Аналог эксендина AVE0010 отличается отсечением С-конца последовательности исходного Эксендина-4. AVE0010 содержит шесть С-концевых лизиновых остатков, не присутствующих в Эксендине-4.
В контексте представленного изобретения, AVE0010 включает его фармацевтически приемлемые соли. Квалифицированному специалисту в данной области известны фармацевтически приемлемые соли AVE0010. Предпочтительной фармацевтически приемлемой солью AVE0010, используемого в представленном изобретении, является ацетат.
В примере 1 представленного изобретения, у субъектов с диабетом 2 типа было продемонстрировано, что AVE0010 (Ликсисенатид) в дополнительной к метформинуу терапии значительно улучшал гликемический контроль и снижал массу:
• HbA1c значительно снижался в обеих группах
- 2-стадийное титрование: средняя разница, рассчитанная методом наименьших квадратов, в HbA1c против плацебо равно -0,41% (p<0,0001)
- 1-стадийное титрование: средняя разница, рассчитанная методом наименьших квадратов, в HbA1c против плацебо равно -0,49% (p<0,0001)
• Значительно больше получавших ликсисенатид субъектов достигали целей по HbA1c (≤ 6,5% & <7,0%)
• С ликсисенатидом значительно улучшалась глюкоза в плазме натощак (FPG)
• Индуцировалась существенная потеря массы
- 2-стадия: средняя разница, рассчитанная методом наименьших квадратов, равна -1,05 кг против плацебо (p=0,0025)
- 1-стадия: средняя разница, рассчитанная методом наименьших квадратов, равна -1,00 кг против плацебо (p=0,0042)
Длительная эффективность наблюдалась на протяжении всего периода лечения.
В примере 2 представленного изобретения, у страдающих ожирением субъектов моложе 50 лет с диабетом 2 типа с неадекватным контролем посредством метформина,
• Ликсисенатид (AVE0010) демонстрировал значительное снижение HbA1c и массы у молодых страдающих ожирением субъектов с диабетом 2 типа в течение периода, равного 24 неделям,
• Ликсисенатид (AVE0010) демонстрировал существенное преимущество перед ситаглиптином в показателях потери массы тела и аналогичную величину снижения HbA1c,
• Ликсисенатид (AVE0010) демонстрировал очень благоприятный профиль безопасности и переносимости перед ситаглиптином, а конкретно отсутствие разницы в частоте встречаемости гипогликемии,
• Эффективность Ликсисенатида (AVE0010) обусловлена его двойной эффективностью в снижении как PPG, так и FPG,
при этом Ликсисенатид вводили в качестве дополнительной терапии к метформину.
Первым аспектом представленного изобретения является фармацевтическая комбинация для применения для индуцирования потери массы тела у субъектов с диабетом 2 типа или/и для предотвращения набора массы тела у субъектов с диабетом 2 типа, при этом указанная комбинация содержит
(a) desPro36Эксендин-4(1-39)-Lys6-NH2 или/и его фармацевтически приемлемую соль, и
(b) метформин или/и его фармацевтически приемлемую соль.
Метформин представляет собой международное непатентованное название 1,1-диметилбигуанида (CAS-номер657-24-9). В представленном изобретении, термин «метформин» включает его любую фармацевтически приемлемую соль.
В представленном изобретении, метформин можно вводить перорально. Квалифицированному специалисту известны препараты метформина, подходящие для лечения диабета 2 типа посредством перорального введения. Метформин можно вводить нуждающемуся в этом субъекту в количестве, достаточном, чтобы вызвать терапевтический эффект. Метформин можно вводить в дозе, составляющей по меньшей мере 1,0 г/день или по меньшей мере 1,5 г/день. Для перорального введения, метформин может быть приготовлен в виде твердой лекарственной формы, такой как таблетка или пилюля. Метформин может быть приготовлен с подходящими фармацевтически приемлемыми носителями, адъювантами и/или вспомогательными веществами.
В представленном изобретении, desPro36Эксендин-4(1-39)-Lys6-NH2 и/или фармацевтически приемлемую соль можно применять в дополнительной терапии к введению метформина.
В представленном изобретении, термины «дополнительно», «дополнительное лечение» и «дополнительная терапия» относятся к лечению сахарного диабета 2 типа метформином и AVE0010. Метформин и AVE0010 можно вводить в пределах временного интервала, равного 24 ч. Метформин и AVE0010 каждый можно вводить с дозировкой один раз в день. Метформин и AVE0010 можно вводить с помощью различных путей введения. Метформин можно вводить перорально, а AVE0010 можно вводить парентерально.
Субъектом, подлежащим лечению лекарственным средством представленного изобретения, страдающим от диабета 2 типа, может быть субъект, страдающий от диабета 2 типа. Пример 1 демонстрирует у данных субъектов, что введение AVE0010 в комбинации с метформином обеспечивает предпочтительную терапию.
Субъектом, подлежащим лечению лекарственным средством представленного изобретения, страдающим от диабета 2 типа, может быть субъект, страдающий от диабета 2 типа, при этом диабет 2 типа имеет неадекватный контроль за счет лечения одним метформином, например, дозой, составляющей по меньшей мере 1,0 г/день метформина или по меньшей мере 1,5 г/день метформина в течение 3 месяцев. В представленном изобретении, субъект, диабет 2 типа которого имеет неадекватный контроль, может иметь значение HbA1c в диапазоне, составляющем 7%-10%.
Субъектом, подлежащим лечению лекарственным средством представленного изобретения, страдающим от диабета 2 типа, может быть страдающий ожирением субъект. В представленном изобретении, страдающий ожирением субъект может иметь индекс массы тела, составляющий по меньшей мере 30 кг/м2.
Субъектом, подлежащим лечению лекарственным средством представленного изобретения, страдающим от диабета 2 типа, может быть страдающий ожирением субъект, как описано в данном документе, который может быть моложе, чем 50 лет, например, с возрастом по меньшей мере 18 лет и моложе, чем 50 лет, при этом субъект имеет неадекватный контроль посредством метформина. Пример 2 демонстрирует у данных субъектов, что введение AVE0010 в комбинации с метформином обеспечивает предпочтительную терапию.
Субъект, подлежащий лечению лекарственным средством представленного изобретения, страдающий от диабета 2 типа, может иметь нормальную массу тела. В представленном изобретении, субъект, имеющий нормальную массу тела, может иметь индекс массы тела в диапазоне, составляющем от 17 кг/м2 до 25 кг/м2 или от 17 кг/м2 до <30 кг/м2.
Субъектом, подлежащим лечению лекарственным средством представленного изобретения, может быть взрослый человек. Субъект может иметь возраст, составляющий по меньшей мере 18 лет, может иметь возраст в диапазоне, составляющем 18-80 лет, 18-50 лет или 40-80 лет или 50-60 лет. Субъект может быть моложе 50 лет.
Субъект, подлежащий лечению лекарственным средством представленного изобретения, предпочтительно, не получает противодиабетическое лечение, например, инсулином или/и родственными соединениями.
Субъект, подлежащий лечению лекарственным средством представленного изобретения, может страдать от сахарного диабета 2 типа в течение по меньшей мере 1 года или по меньшей мере 2 лет. В частности, у субъекта, подлежащего лечению, сахарный диабет 2 типа был диагностирован по меньшей мере за 1 год или по меньшей мере за 2 года до начала терапии лекарственным средством представленного изобретения.
Субъект, подлежащий лечению, может иметь значение HbA1c, равное по меньшей мере приблизительно 8% или по меньшей мере приблизительно 7,5%. Субъект также может иметь значение HbA1c, составляющее от приблизительно 7 до приблизительно 10%. Пример представленного изобретения демонстрирует, что у субъектов с диабетом 2 типа лечение AVE0010 приводит к снижению значения HbA1c.
В еще одном аспекте представленного изобретения, комбинация, которая раскрыта в данном описании, может использоваться для улучшения гликемического контроля. В представленном изобретении, улучшение гликемического контроля, в частности, относится к улучшению концентрации глюкозы в плазме после приема пищи, улучшению концентрации глюкозы в плазме натощак или/и улучшению значения HbA1c.
В еще одном аспекте представленного изобретения, комбинация, которая раскрыта в данном описании, может использоваться для улучшения значения HbA1c у субъекта, страдающего от диабета 2 типа. Улучшение значения HbA1c означает, что значение HbA1c снижается ниже 6,5% или 7%, например, после лечения в течение по меньшей мере одного месяца, по меньшей мере двух месяцев или по меньшей мере трех месяцев.
В еще одном аспекте представленного изобретения, комбинация, которая раскрыта в данном описании, может применяться для улучшения толерантности к глюкозе у субъекта, страдающего диабетом 2 типа. Улучшение толерантности к глюкозе означает, что концентрация глюкозы в плазме после приема пищи снижается активным агентом представленного изобретения. Снижение, в частности, означает, что концентрация глюкозы в плазме достигает нормогликемических значений или по меньшей мере приближается к данным значениям.
В представленном изобретении, нормогликемическими значениями являются концентрации глюкозы в крови, составляющие, в частности, 60-140 мг/дл (соответствующие 3,3-7,8 мМ/л). Данный диапазон относится, в частности, к концентрациям глюкозы в крови в состоянии натощак и в состоянии после приема пищи.
Субъект, подлежащий лечению, 2 часа после приема пищи может иметь концентрацию глюкозы в плазме, равную по меньшей мере 10 ммоль/л, по меньшей мере 12 ммоль/л или по меньшей мере 14 ммоль/л. Данные концентрации глюкозы в плазме превышают нормогликемические концентрации.
Субъект, подлежащий лечению, может иметь колебание уровня глюкозы, равное по меньшей мере 2 ммоль/л, по меньшей мере 3 ммоль/л, по меньшей мере 4 ммоль/л или по меньшей мере 5 ммоль/л. В представленном изобретении, колебанием глюкозы является, в частности, разница концентрации глюкозы в плазме через 2 часа после приема пищи и концентрации глюкозы в плазме за 30 минут перед тестовым приемом пищи.
«После приема пищи» представляет собой термин, хорошо известный квалифицированному специалисту в области диабетологии. Термин «после приема пищи» описывает, в частности, фазу после приема пищи и/или воздействие глюкозы в экспериментальных условиях. У здорового человека данная фаза характеризуется увеличением и последующим снижением концентрации глюкозы в крови. Термин «после приема пищи» или «фаза после приема пищи» обычно заканчивается через 2 ч после приема пищи и/или воздействия глюкозы.
Субъект, подлежащий лечению, как раскрыто в данном описании, может иметь концентрацию глюкозы в плазме натощак, равную по меньшей мере 8 ммоль/л, по меньшей мере 8,5 ммоль/л или по меньшей мере 9 ммоль/л. Данные концентрации глюкозы в плазме превышают нормогликемические концентрации.
В еще одном аспекте представленного изобретения, комбинация, которая раскрыта в данном описании, может применяться для улучшения (т.е. снижения) глюкозы в плазме натощак у субъекта, страдающего диабетом 2 типа. Снижение означает, в частности, что концентрация глюкозы в плазме достигает нормогликемических значений или по меньшей мере приближается к данным значениям.
Комбинация представленного изобретения может применяться при лечении одного или более медицинских показаний, раскрытых в данном описании, например, при лечении субъектов с диабетом 2 типа, или для состояний, связанных с диабетом 2 типа, например для улучшения гликемического контроля, снижения концентрации глюкозы в плазме натощак, для улучшения амплитуды колебаний уровня глюкозы, снижения концентрации глюкозы в плазме после приема пищи, улучшения толерантности к глюкозе, улучшения значения HbA1c, для предотвращения гипогликемии, для потери массы тела у субъектов и/или предотвращения увеличения массы тела у субъектов.
В представленном изобретении, desPro36Эксендин-4(1-39)-Lys6-NH2 и/или его фармацевтически приемлемую соль можно вводить нуждающемуся в этом субъекту, в количестве, достаточном, чтобы вызвать терапевтический эффект.
В представленном изобретении, desPro36Эксендин-4(1-39)-Lys6-NH2 и/или его фармацевтически приемлемая соль может быть разработана с подходящими фармацевтически приемлемыми носителями, адъювантами и/или вспомогательными веществами.
Соединение desPro36Эксендин-4(1-39)-Lys6-NH2 и/или его фармацевтически приемлемую соль можно вводить парентерально, например, посредством инъекции (такой как внутримышечная или подкожная инъекция). Известны подходящие инъекционные устройства, например, так называемые «шприцы-ручки», содержащие картридж, содержащий активный ингредиент, и инъекционную иглу. Соединение desPro36Эксендин-4(1-39)-Lys6-NH2 и/или его фармацевтически приемлемую соль можно вводить в подходящем количестве, например, в количестве в диапазоне, составляющем 10-15 мкг на дозу или 15-20 мкг на дозу.
В представленном изобретении, desPro36Эксендин-4(1-39)-Lys6-NH2 и/или его фармацевтически приемлемую соль можно вводить с ежедневной дозой в диапазоне, составляющем 10-20 мкг, в диапазоне, составляющем 10-15 мкг, или в диапазоне, составляющем 15-20 мкг. desPro36Эксендин-4(1-39)-Lys6-NH2 и/или его фармацевтически приемлемую соль можно вводить посредством одной инъекции в день.
В представленном изобретении, desPro36Эксендин-4(1-39)-Lys6-NH2 и/или его фармацевтически приемлемая соль могут быть предоставлены в жидкой композиции. Квалифицированному специалисту известны жидкие композиции AVE0010, подходящие для парентерального введения. Жидкая композиция представленного изобретения может иметь кислый или физиологический pH. Кислый pH предпочтительно находится в диапазоне pH 1-6,8, pH 3,5-6,8 или pH 3,5-5. Физиологический pH, предпочтительно, находится в диапазоне pH 2,5-8,5, pH 4,0-8,5 или pH 6,0-8,5. pH можно регулировать фармацевтически приемлемой разбавленной кислотой (обычно HCl) или фармацевтически приемлемым разбавленным основанием (обычно NaOH).
Жидкая композиция, содержащая desPro36Эксендин-4(1-39)-Lys6-NH2 и/или его фармацевтически приемлемую соль, может содержать подходящий консервант. Подходящий консервант может быть выбран из фенола, m-крезола, бензилового спирта и сложного эфира p-гидроксибензойной кислоты. Предпочтительным консервантом является m-крезол.
Жидкая композиция, содержащая desPro36Эксендин-4(1-39)-Lys6-NH2 и/или его фармацевтически приемлемую соль, может содержать вещество, регулирующее тоничность. Подходящее вещество, регулирующее тоничность, может быть выбрано из глицерола, лактозы, сорбитола, маннитола, глюкозы, NaCl, кальций или магний содержащих соединений, таких как CaCl2. Концентрация глицерола, лактозы, сорбитола, маннитола и глюкозы может быть в диапазоне, составляющем 100-250 мМ. Концентрация NaCl может быть до 150 мМ. Предпочтительным веществом, регулирующим тоничность, является глицерол.
Жидкая композиция, содержащая desPro36Эксендин-4(1-39)-Lys6-NH2 и/или его фармацевтически приемлемую соль, может содержать метионин от 0,5 мкг/мл до 20 мкг/мл, предпочтительно, от 1 мкг/мл до 5 мкг/мл. Предпочтительно, жидкая композиция содержит L-метионин.
Дополнительным аспектом представленного изобретения является способ стимулирования потери массы тела у субъектов с диабетом 2 типа и/или предотвращения увеличения массы тела у субъектов с диабетом 2 типа, при этом указанный способ включает введение нуждающемуся в этом субъекту desPro36Эксендин-4(1-39)-Lys6-NH2 и/или его фармацевтически приемлемой соли в комбинации с метформином. В частности, можно вводить комбинацию, которая раскрыта в данном описании. В способе представленного изобретения, субъектом может быть субъект по определению данного описания.
Еще один аспект представленного изобретения относится к применению комбинации, которая раскрыта в данном описании, для изготовления лекарственного средства для лечения медицинского показания, которое раскрыто в данном описании. Например, комбинация представленного изобретения может применяться для стимулирования потери массы тела у субъектов с диабетом 2 типа и/или предотвращения увеличения массы тела у субъектов с диабетом 2 типа. Комбинация представленного изобретения также может применяться для изготовления лекарственного средства для лечения субъектов с диабетом 2 типа или для лечения состояний, связанных с диабетом 2 типа, например, для улучшения гликемического контроля, снижения концентрации глюкозы в плазме натощак, для улучшения амплитуды колебаний уровня глюкозы, снижения концентрации глюкозы в плазме после приема пищи, улучшения значения HbA1c и/или улучшения толерантности к глюкозе. Может быть составлено лекарственное средство, которое раскрыто в данном описании. Например, лекарственное средство может содержать парентеральную готовую форму AVE0010 и/или его фармацевтически приемлемую соль, и пероральную готовую форму метформина и/или его фармацевтически приемлемую соль.
Изобретение дополнительно проиллюстрировано с помощью следующего примера и фигур.
Обозначения Фигур
Пример 1
Фиг.1 - Дизайн исследования
Фиг.2: Общая пошаговая процедура тестирования
Фиг.3: - График Каплана-Майера периода до прекращения лечения по любой причине - Рандомизированная популяция
Фиг.4: График среднего изменения HbA1c (%) по сравнению с исходным уровнем при осмотре во время основного 24-недельного периода лечения - mITT популяция. LOCF = перенос вперед данных последнего наблюдения. Примечание: график включал измерения, полученные перед введением резервного препарата и до 3 суток после последней дозы двойной слепой инъекции исследуемого препарата при 12 осмотре или перед ним (24 неделя) или на 169 день, если 12 осмотр (24 неделя) невозможен.
Фиг.5: График среднего изменения глюкозы в плазме натощак (ммоль/л) по сравнению с исходным уровнем при осмотре во время основного 24-недельного периода лечения - mITT популяция. LOCF = перенос вперед данных последнего наблюдения. Примечание: график включал измерения, полученные перед введением резервного препарата и до 1 суток после последней дозы двойной слепой инъекции исследуемого препарата при 12 осмотре или перед ним (24 неделя) или на 169 день, если 12 осмотр (24 неделя) невозможен.
Фиг.6: График среднего изменения массы тела (кг) по сравнению с исходным уровнем при осмотре во время основного 24-недельного периода лечения - mITT популяция. LOCF = перенос вперед данных последнего наблюдения. Примечание: график включал измерения, полученные перед введением резервного препарата и до 3 суток после последней дозы двойной слепой инъекции исследуемого препарата при 12 осмотре или перед ним (24 неделя) или на 169 день, если 12 осмотр (24 неделя) невозможен.
Фиг.7: График среднего изменения HbA1c (%) по сравнению с исходным уровнем при осмотре и в конечной точке - mITT популяция. LOCF = перенос вперед данных последнего наблюдения, EOT = последнее значение в период терапии. Примечание: анализ исключал измерения, полученные после введения резервного препарата и/или после прекращения лечения плюс 3 суток. Для 24 недели (LOCF), анализ включал измерения, полученные за 3 суток после последней дозы двойной слепой инъекции исследуемого препарата при 12 осмотре или перед ним (24 неделя) или на 169 день, если 12 осмотр (24 неделя) невозможен.
Фиг.8: График среднего изменения глюкозы в плазме натощак (ммоль/л) по сравнению с исходным уровнем при осмотре и в конечной точке - mITT популяция. LOCF = перенос вперед данных последнего наблюдения, EOT = последнее значение в период терапии. Примечание: анализ исключал измерения, полученные после введения резервного препарата и/или после прекращения лечения плюс 1 сутки. Для 24 недели (LOCF), анализ включал измерения, полученные за 1 сутки после последней дозы двойной слепой инъекции исследуемого препарата при 12 осмотре или перед ним (24 неделя) или на 169 день, если 12 осмотр (24 неделя) невозможен.
Фиг.9: График среднего изменения массы тела (кг) по сравнению с исходным уровнем при осмотре и в конечной точке - mITT популяция. LOCF = перенос вперед данных последнего наблюдения, EOT = последнее значение в период терапии. Примечание: анализ исключал измерения, полученные после введения резервного препарата и/или после прекращения лечения плюс 3 суток. Для 24 недели (LOCF), анализ включал измерения, полученные за 3 суток после последней дозы двойной слепой инъекции исследуемого препарата при 12 осмотре или перед ним (24 неделя) или на 169 день, если 12 осмотр (24 неделя) невозможен.
Пример 2:
Фиг.10 - Дизайн исследования.
Фиг.11 - График Каплана-Майера периода до прекращения лечения по любой причине - Рандомизированная популяция.
Фиг.12 - График субъектов, ответивших на лечение (субъектов с HbA1c <7% и потерей массы ≥5% исходной массы тела) при осмотрах и в конечной точке - mITT популяция. LOCF = перенос вперед данных последнего наблюдения. Примечание: анализ исключал измерения, полученные после введения резервного препарата и/или после прекращения лечения плюс 3 суток. Для 24 недели (LOCF), субъекты, у которых отсутствуют значения периода терапии после исходного уровня (HbA1c и массы тела), разделенные не больше чем 30 днями, считаются не ответившими на лечение.
Фиг.13 - График среднего изменения HbA1c (%) по сравнению с исходным уровнем при осмотре и в конечной точке - mITT популяция. LOCF = перенос вперед данных последнего наблюдения. Примечание: анализ исключал измерения, полученные после введения резервного препарата и/или после прекращения лечения плюс 3 суток.
Фиг.14 - График субъектов, ответивших на лечение снижением HbA1c (≤6,5% или <7%, соответственно) при выбранных осмотрах и в конечной точке - mITT популяция. LOCF = перенос вперед данных последнего наблюдения. Примечание: анализ исключал измерения, полученные после введения резервного препарата и/или после прекращения лечения плюс 3 суток.
Фиг.15 - График среднего изменения массы тела (кг) по сравнению с исходным уровнем при осмотре и в конечной точке - mITT популяция. LOCF = перенос вперед данных последнего наблюдения. Примечание: анализ исключал измерения, полученные после введения резервного препарата и/или после прекращения лечения плюс 3 суток.
Фиг.16 - График среднего изменения глюкозы в плазме натощак (ммоль/л) по сравнению с исходным уровнем при осмотре и в конечной точке - mITT популяция. LOCF = перенос вперед данных последнего наблюдения. Примечание: анализ исключал измерения, полученные после введения резервного препарата и/или после прекращения лечения плюс 1 сутки.
Пример 1
Рандомизированное, двойное слепое, плацебо-контролируемое, многоцентровое, многонациональное исследование в параллельных группах по оценке эффективности и безопасности ликсисенатида по сравнению с плацебо в качестве дополнительного лечения к метформину у субъектов с диабетом 2 типа.
Сущность
Пример относится к рандомизированному, двойному слепому, плацебо-контролируемому, многоцентровому, многонациональному исследованию в параллельных группах по оценке эффективности и безопасности ликсисенатида по сравнению с плацебо в качестве дополнительного лечения к метформину у субъектов с диабетом 2 типа. Приблизительная минимальная продолжительность исследования на субъекта составляла 79 недель (до 3 недель скрининг + 24-недельное основное лечение + варьируемое продление + 3 дня последующее наблюдение). Исследование проводили в 75 центрах в 15 странах. Основная цель данного исследования состояла в оценке воздействия ликсисенатида в качестве дополнительного лечения к метформину на гликемический контроль, с использованием режима двухстадийного титрования дозы по сравнению с плацебо в показателях снижения HbA1c (абсолютное изменение) в течение периода, равного 24 неделям.
В общей сложности 484 субъекта рандомизировали в одну из четырех лечебных групп (161 в группе ликсисенатида двухстадийного титрования, 161 в группе ликсисенатида одностадийного титрования, 80 в группе двухстадийного титрования плацебо и 82 в группе одностадийного титрования плацебо). Из 484 рандомизированных субъектов, 482 субъектов подвергали лечению исследования. Два субъекта (по 1 в каждой группе плацебо) были рандомизированы по ошибке и не подвергались никакому лечению исследования. Обоих субъектов исключили из анализов как эффективности, так и безопасности. В анализах объединили группы одностадийного и двухстадийного титрования плацебо. Демографические данные и исходные характеристики были в целом аналогичными по всем лечебным группам за исключением того, что получавшие лечение плацебо субъекты немного старше, чем получавшие лечение ликсисенатидом субъекты. Из 482 рандомизированных и получавших лечение субъектов, 5 субъектов (1 субъект в группе ликсисенатида двухстадийного титрования, 3 субъекта в группе ликсисенатида одностадийного титрования и 1 субъект в группе одностадийного титрования плацебо) исключили из mITT популяции для анализов эффективности вследствие отсутствия данных эффективности после исходного уровня. В течение всего периода лечения исследования, 103 (21,3%) субъекта досрочно прекратили лечение исследования. Процентная доля субъектов, которые прекратили лечение исследования, была более высокой в группе ликсисенатида двухстадийного титрования (24,8%), чем в группе ликсисенатида одностадийного титрования (18,6%) и в объединенной группе плацебо (20,4%). Для групп лечения ликсисенатидом, основной причиной прекращения лечения было «нежелательные события» (11,8% для ликсисенатида двухстадийного титрования и 8,7% для ликсисенатида одностадийного титрования, против 6,2% для объединенного плацебо), за которым следовало «другие причины» (9,9% и 7,5%, против 9,9% для объединенного плацебо).
Рассчитанные методом наименьших квадратов (LS) средние изменения HbA1c к 24 неделе по сравнению с исходным уровнем составляли -0,83% для группы ликсисенатида двухстадийного титрования (LS среднее различие против объединенного плацебо = -0,41%; p-значение < 0,0001), -0,92% для группы ликсисенатида одностадийного титрования (LS среднее различие против объединенного плацебо = -0,49%; p-значение < 0,0001), по сравнению с -0,42% для объединенной группы плацебо. Процентная доля достижения субъектами HbA1c ≤6,5 или <7% на 24 неделе была значительно более высокой в обеих группах лечения ликсисенатидом, чем в группе лечения плацебо (для HbA1c ≤6,5%, 20,4% для ликсисенатида двухстадийного титрования и 25,6% для ликсисенатида одностадийного титрования, против 7,6% для объединенного плацебо; для HbA1c <7%, 42,1% для ликсисенатида двухстадийного титрования и 47,4% для ликсисенатида одностадийного титрования, против 24,1% для объединенного плацебо). Обе группы, получавшие лечение ликсисенатидом, продемонстрировали статистически значимое снижение глюкозы в плазме натощак по сравнению с объединенной группой плацебо (для группы ликсисенатида двухстадийного титрования, средняя разница, рассчитанная методом наименьших квадратов = -0,67 ммоль/л, а p-значение = 0,0004; для группы ликсисенатида одностадийного титрования, средняя разница, рассчитанная методом наименьших квадратов = -0,65 ммоль/л, а p-значение = 0,0007). Статистически значимое снижение массы тела к 24 неделе по сравнению с исходным уровнем также было продемонстрировано в обеих группах лечения ликсисенатидом, по сравнению с объединенной группой плацебо (для ликсисенатида двухстадийного титрования, средняя разница, рассчитанная методом наименьших квадратов = -1,05 кг, а p-значение 0,0025; для ликсисенатида одностадийного титрования, средняя разница, рассчитанная методом наименьших квадратов = -1,00 кг, а p-значение 0,0042). Обе группы, получавшие лечение ликсисенатидом, показали немного более низкие процентные доли субъектов, нуждавшихся в резервном варианте лечения во время основного 24-недельного двойного слепого периода лечения (3,1% для двухстадийного титрования и 1,3% для одностадийного титрования), по сравнению с объединенной группой плацебо (4,4%). Не было подтверждения существенного различия между каждой группой ликсисенатида и объединенной группой плацебо вследствие низкой частоты встречаемости субъектов, получивших резервный вариант лечения.
Ликсисенатид имел хорошую переносимость. частота встречаемости возникших после начала лечения нежелательных явлений (TEAE) была сопоставима между лечебными группами (87,6% в группе ликсисенатида двухстадийного титрования, 85,7% в группе ликсисенатида одностадийного титрования, и 86,3% в объединенной группе плацебо). Пять субъектов (1 субъект в группе ликсисенатида двухстадийного титрования, 2 субъекта в группе ликсисенатида одностадийного титрования и 2 субъекта в объединенной группе плацебо) имели в течение периода лечения исследуемым препаратом TEAE, приведшие к смерти. Пятьдесят девять субъектов имели по меньшей мере одно серьезное TEAE, произошедшее в течение периода лечения исследуемым препаратом на протяжении всего исследования с аналогичной частотой встречаемости между группой ликсисенатида двухстадийного титрования (13,0%) и объединенной группой плацебо (13,8%), но с немного более низкой частотой встречаемости в группе ликсисенатида одностадийного титрования (9,9%). Наиболее часто сообщаемым TEAE для получавших лечение ликсисенатидом субъектов была тошнота (62 [38,5%] субъекта для двухстадийного титрования и 47 [29,2%] для одностадийного титрования, против 13 [8,1%] для объединенного плацебо), за которой следовала рвота (29 [18,0%] субъектов для двухстадийного титрования и 21 [13,0%] для одностадийного титрования, против 1 [0,6%] для объединенного плацебо). Двенадцать (7,5%) субъектов в группе ликсисенатида двухстадийного титрования и 6 (3,7%) субъектов в группе ликсисенатида одностадийного титрования имели события симптоматической гипогликемии по определению протокола в течение периода лечения исследуемым препаратом на протяжении всего исследования, по сравнению с 12 (7,5%) получавшими лечение плацебо субъектами, которые имели события симптоматической гипогликемии в течение того же самого периода. Ни одно из явлений симптоматической гипогликемии не было тяжелым по интенсивности. В общей сложности 15 субъектов (6 [3,7%] для ликсисенатида двухстадийного титрования, 3 [1,9%] для ликсисенатида одностадийного титрования и 6 [3,8%] для объединенного плацебо) имели сообщаемые аллергические события, которые были расценены как аллергические реакции комитетом по оценке аллергических реакций (ARAC), но только 2 аллергических события (по 1 в каждой группе ликсисенатида) были расценены как возможно связанные с исследуемым препаратом. В исследовании не наблюдалось ни одного случая острого панкреатита. Не было существенного различия в показателях безопасности и переносимости между 2 режимами титрования для ликсисенатида (одностадийным и двухстадийным).
1 ЦЕЛИ
1.1 Основная цель
Основной целью данного исследования была оценка воздействия ликсисенатида в качестве дополнительного лечения к метформину на гликемический контроль с использованием режима двухстадийного титрования дозы по сравнению с плацебо в показателях снижения HbA1c (абсолютное изменение) в течение периода, равного 24 неделям у субъектов с диабетом 2 типа.
1.2 дополнительная цель (цели)
Дополнительные цели данного исследования состояли:
• В оценке воздействия AVE0010 на:
- Гликемический контроль по сравнению с плацебо в показателях снижения HbA1c, когда его используют в режиме одностадийного титрования дозы,
- Процентную долю достижения субъектами HbA1c <7% или HbA1c ≤6,5%,
- Массу тела,
- Глюкозу в плазме натощак,
• В оценке безопасности и переносимости AVE0010,
• В оценке ФК AVE0010 и формирования антител анти-AVE0010.
2 ДИЗАЙН ИССЛЕДОВАНИЯ
Провели рандомизированное, двойное слепое, плацебо-контролируемое, 4-групповое, многоцентровое, многонациональное исследование в параллельных группах с несбалансированным дизайном: двухстадийное титрование (150 получавших лечение ликсисенатидом и 75 получавших лечение плацебо субъектов) и одностадийное титрование (150 получавших лечение ликсисенатидом и 75 получавших лечение плацебо субъектов). Исследование было двойное слепое относительно активного и плацебо лечения. Объем лекарственного средства исследования (т.е. доза активного лекарственного средства или эквивалентного плацебо) и режимы титрования (т.е. одностадийного и двухстадийного) не были слепыми.
Субъекты были стратифицированы по скрининговым значениям HbA1c (<8%, ≥8%) и индексу массы тела (BMI <30 кг/м2, ≥30 кг/м2). После скринингового периода, субъектов централизованно рандомизировали посредством системы интерактивного голосового взаимодействия (IVRS) в отношении 2:1:2:1 к одной из четырех групп (двухстадийное титрование ликсисенатида, двухстадийное титрование плацебо, одностадийное титрование ликсисенатида и одностадийное титрование плацебо).
Посредством поправки 4 к протоколу (датирована 19 января 2010 года), приблизительная минимальная продолжительность исследования на субъекта составляла 79 недель (до 3 недель скрининга + 24 недели основное двойное слепое лечение + показатель продолжение двойного слепого лечения + 3 дня последующее наблюдение). Субъектов, которые завершили 24-недельный основной двойной слепой период, подвергали изменяемому периоду продолжения двойного слепого лечения, который заканчивался для всех субъектов приблизительно на запланированную дату осмотра 76 недели (V24) для последнего рандомизированного субъекта.
Субъекты, которые досрочно прекратили лечение исследования, оставались в исследовании вплоть до запланированной даты завершения исследования согласно поправке 3 к протоколу (датирована 03 июля 2009 года). Они следовали процедурам исследования, точно определенным в поправках к протоколу (за исключением 3-дневного последующего наблюдения за безопасностью после лечения, оценки фармакокинетики и постпрандиального теста). (Фиг.1)
3 Основной и ключевой дополнительные КОНЕЧНЫЕ показатели
3.1 Основной КОНЕЧНЫЙ показатель
Основным показателем эффективности было абсолютное изменение HbA1c к 24 неделе по сравнению с исходным, которое определяли как: HbA1c на 24 неделе - HbA1c в начале исследования.
Если субъект прервал лечение досрочно или принял резервный вариант лечения в течение основного 24-недельного периода двойного слепого лечения или не имел значения HbA1c при осмотре на 24 неделе, в качестве значения HbA1c на 24 неделе использовали последнее после исходного измерение HbA1c в течение основного 24-недельного периода двойного слепого применения исследуемого препарата (метод замены пропущенных данных последним значением [LOCF]).
3.2 дополнительные КОНЕЧНЫЕ показатели
3.2.1 КОНЕЧНЫЕ Показатели эффективности
Для дополнительных показателей эффективности, применялась точно такая же методика обработки отсутствующей оценки/раннего прекращения, как для основного показателя.
Непрерывные показатели
• Изменение к 24 неделе FPG (ммоль/л) по сравнению с исходным уровнем
• Изменение к 24 неделе массы тела (кг) по сравнению с исходным уровнем
Категориальные переменные
• Процентная доля субъектов с HbA1c <7% на 24 неделе
• Процентная доля субъектов с HbA1c ≤6,5% на 24 неделе
• Процентная доля субъектов, нуждавшихся в резервном варианте лечения во время основного 24-недельного периода двойного слепого лечения
• Процентная доля субъектов с потерей массы (кг) ≥5% к 24 неделе по сравнению с исходным уровнем
3.2.2 Конечные показатели безопасности
Анализ безопасности был основан на сообщениях о TEAE и другой информации о безопасности, включая симптоматическую гипогликемию и тяжелую симптоматическую гипогликемию, местную переносимость в месте инъекции, аллергические события (которые рассматриваются ARAC), подозреваемый панкреатит, повышенный кальцитонин, жизненно важные функции, ЭКГ в 12 отведениях и лабораторные испытания. Главные сердечно-сосудистые события также собирались и рассматривались Комитетом по оценке сердечно-сосудистых явлений (CAC). Рассмотренные и подтвержденные CAC события из данного исследования и 2-3 других исследований фазы ликсисенатида будут объединяться для анализа и суммироваться в отдельном отчете на основании плана статистического анализа для общей сердечно-сосудистой оценки ликсисенатида. KRM/CSR не будет отображать сущность рассмотренных и подтвержденных CV явлений из данного исследования.
4. Расчетные предположения Размеров выборки
Расчеты размера/мощности выборки выполняли на основании основного показателя эффективности, абсолютного изменения HbA1c к 24 неделе по сравнению с исходным уровнем.
150 субъектов для одной группы ликсисенатида и 2x75 субъектов для объединенной группы плацебо обеспечивали мощности, составляющие 91% (или 75%) для события разницы абсолютного изменения HbA1c к 24 неделе, составляющей 0,5% (или 0,4%), по сравнению с исходным уровнем между ликсисенатидом и плацебо, принимая, что общее стандартное отклонение составляет 1,3% с двусторонним критерием при 5% уровне значимости. Рассчеты размера выборки основаны на двухвыборочном критерии стьюдента и сделаны с применением nQuery® Advisor 5,0. Стандартное отклонение оценивали консервативным образом из ранее проведенных исследований сахарного диабета (на основании опубликованных данных аналогично разработанного исследования и внутренних данных, не опубликованных), принимая в расчет более ранний отсев.
5 статистические методы
5.1 Анализ популяций
Модифицированная популяция субъектов, включенных в испытание (mITT), состояла из всех рандомизированных субъектов, получивших по меньшей мере одну дозу двойного слепого исследуемого препарата (IP) и имевших как исходную оценку, так и по меньшей мере одну оценку после исходных показателей эффективности.
Выборка для оценки безопасности определяется, как все рандомизированные субъекты, которые получали по меньшей мере одну дозу двойного слепого IP.
5.2 ОСНОВНОЙ АНАЛИЗ ЭФФЕКТИВНОСТИ
Основной показатель эффективности (изменение HbA1c к 24 неделе по сравнению с исходным уровнем) анализировали с использованием модели ковариационного анализа (ANCOVA) с лечебными группами (группами ликсисенатида двухстадийного титрования и плацебо, группами ликсисенатида одностадийного титрования и плацебо), рандомизированной стратой уровня HbA1c при скрининге (<8,0, ≥8,0%), рандомизированной стратой уровня BMI при скрининге (<30, ≥30 кг/м2) и страной в качестве фиксированных эффектов и с использованием в качестве ковариаты исходного значения HbA1c. Разницу между каждой группой ликсисенатида и объединенной группой плацебо и двусторонний 95% доверительный интервал, а также p-значение оценивали в рамках ANCOVA. В модели ANCOVA, две группы титрования плацебо включили в качестве отдельных лечебных уровней, но их объединили в виде одной группы при проведении сравнений с использованием соответствующего сопоставления (например, для сравнения ликсисенатида двухстадийного титрования с объединенным плацебо [-0,5, -0,5, 0, +1] в порядке одностадийного титрования плацебо, двухстадийного титрования плацебо, ликсисенатида одностадийного титрования и ликсисенатида двухстадийного титрования).
Для того, чтобы обеспечить контроль погрешности I типа, применяли пошаговую методику тестирования. Сперва группу ликсисенатида двухстадийного титрования сравнивали с объединенной группой плацебо (основная цель). Если тест был статистически значимым, тогда группу ликсисенатида одностадийного титрования сравнивали с объединенной группой плацебо (дополнительная цель).
Основной анализ основного показателя эффективности проводили на основании mITT популяции и измерений, полученных в течение основного 24-недельного периода двойного слепого применения исследуемого препарата для показателей эффективности. Основной 24-недельный период двойного слепого лечения исследуемым препаратом для показателей эффективности определяли как время от первой дозы двойного слепого IP до 3 дней (за исключением определения FPG центральной лабораторией, которая занимала до 1 дня) после последней дозы двойной слепой инъекции IP при или перед осмотром V12/24 недели (или D169, если осмотр V12/24 недели был пропущен) или до введения резервного варианта лечения, в зависимости от того, что было раньше. Методику LOCF используют, принимая в качестве значения HbA1c на 24 неделе данное последнее доступное после исходного уровня измерение HbA1c при лечении исследуемым препаратом (перед началом нового препарата в случае резервного варианта лечения).
5.3 ДОПОЛНИТЕЛЬНЫЙ АНАЛИЗ ЭФФЕКТИВНОСТИ
Если основной показатель был статистически значимым при α=0,05 для обоих сравнений, для тестирования следующих дополнительных показателей эффективности процедуру тестирования выполняли с помощью следующего порядка установки приоритета:
1. Изменение глюкозы в плазме натощак (FPG) (ммоль/л) к 24 неделе по сравнению с исходным уровнем,
2. Изменение массы тела (кг) к 24 неделе по сравнению с исходным уровнем,
3. Процентная доля субъектов, нуждавшихся в резервном варианте лечения во время основного 24-недельного периода двойного слепого лечения.
На фиг.2 показана схема, которая предоставляет подробное описание данной общей пошаговой процедуры тестирования.
Все непрерывные дополнительные показатели эффективности на 24 неделе, которые описаны в разделе 3.2.1, анализировали с использованием аналогичного подхода и модели ANCOVA, которая описана в разделе 5.2 для основного анализа основного показателя эффективности. Были предоставлены скорректированные оценки среднего различия в лечении между каждой группой ликсисенатида и объединенной группой плацебо и двусторонние 95% доверительные интервалы.
Следующие категориальные дополнительные показатели эффективности на 24 неделе анализировали с использованием метода Кохрана-Мантеля-Хенселя (CMH) со стратифицированием на рандомизационные страты (значения уровня HbA1c при скрининге [<8,0, ≥8%] и уровня BMI при скрининге (<30 кг/м2, ≥30 кг/м2)):
• Процентная доля субъектов с HbA1c <7,0% на 24 неделе,
• Процентная доля субъектов с HbA1c ≤6,5% на 24 неделе,
• Процентная доля субъектов, нуждавшихся в резервном варианте лечения во время основного 24-недельного периода двойного слепого лечения.
Количество и процентная доля субъектов с потерей массы ≥5% по сравнению с исходным уровнем на 24 неделе отображены по лечебным группам.
Все дополнительные показатели в конце лечения оценивали только посредством описательной статистики (среднее, стандартное отклонение, срединное значение и диапазоны, предусмотренные в CSR).
См. Фиг.2
5.4 Анализ безопасности
Анализ безопасности главным образом был основан на периоде лечения исследуемым препаратом на протяжении всего исследования. Период лечения исследуемым препаратом на протяжении всего исследования определяли как время от первой дозы двойного слепого IP до 3 дней после введения последней дозы IP на протяжении всего периода исследования независимо от статуса резервного лечения. 3-дневный интервал был выбран на основании периода полувыведения IP (приблизительно 5-кратный период полувыведения).
В дополнение, в CSR будут подытожены анализы безопасности для 24-недельного периода лечения.
Итог результатов безопасности (описательная статистика или Таблицы частоты) представлен по группам лечения.
6 результаты
6.1 субъекты Исследования
6.1.1 Учет субъектов
Исследование проводили в 75 центрах в 15 странах (Бразилия, Чили, Колумбия, Эстония, Германия, Италия, Литва, Малазия, Мексика, Филиппины, Польша, Румыния, Словакия, Украина и Соединенные Штаты Америки). В общей сложности скринировали 884 субъекта и 484 рандомизировали в одну из четырех лечебных групп (161 в группе ликсисенатида двухстадийного титрования, 161 в группе ликсисенатида одностадийного титрования, 80 в группе двухстадийного титрования плацебо и 82 в группе одностадийного титрования плацебо). Основной причиной отказа при скрининге было значение HbAlc при скрининговом осмотре за пределами определенных диапазонов протокола (257 [29,1%] из 884 подвергшихся скринингу субъектов).
Из 484 рандомизированных субъектов, 482 субъекта подвергли лечению исследования и включили в анализ. Два субъекта (по 1 в каждой группе плацебо) были рандомизированы по ошибке и не подвергались какому-либо лечению исследования. Обоих субъектов исключили из анализов как эффективности, так и безопасности. Из 482 рандомизированных и получавших лечение субъектов, 5 субъектов (1 субъекта в группе ликсисенатида двухстадийного титрования, 3 субъектов в группе ликсисенатида одностадийного титрования и 1 субъекта в группе одностадийного титрования плацебо) исключили из mITT популяции для анализов эффективности вследствие отсутствия данных эффективности после исходного уровня. Таблица 1 предоставляет количество субъектов, включенных в каждую популяцию, участвующую в анализе.
Популяции, участвующие в анализе - Рандомизированная популяция
Примечание: Субъекты выборки для оценки безопасности и PK приведены в таблице согласно фактически полученному лечению (по лечению).
Для выборки с целью оценки эффективности, субъекты приведены в таблице согласно их рандомизированному лечению (по рандомизации).
6.1.2 Распределение Исследования
Таблица 2 предоставляет сущность распределения субъектов для каждой лечебной группы. В течение всего периода лечения, 103 (21,3%) субъекта досрочно прекратили лечение исследования. Процентная доля субъектов, которые прекратили лечение исследования, была выше в группе ликсисенатида двухстадийного титрования (24,8%), чем в группе ликсисенатида одностадийного титрования (18,6%) и в объединенной группе плацебо (20,4%). Для групп лечения ликсисенатидом, основной причиной прекращения лечения были «нежелательные события» (11,8% для ликсисенатида двухстадийного титрования и 8,7% для ликсисенатида одностадийного титрования, против 6,2% для объединенного плацебо), за которым следовали «другие причины» (9,9% и 7,5%, против 9,9% для объединенного плацебо). Аналогичные результаты наблюдались в течение основного 24-недельного периода лечения, когда в общей сложности 40 (8,3%) субъектов досрочно прекратили лечение исследования, при этом основной причиной также были нежелательные события для групп лечения ликсисенатидом (6,8% для двухстадийного титрования и 5,0% для одностадийного титрования, против 1,9% для объединенного плацебо). На фиг.3 отображено время до наступления прекращения лечения по любой причине в течение всего периода лечения Фиг.2. Аналогичные тренды прекращения наблюдались между двумя группами ликсисенатида в течение первых 6 месяцев, по сравнению с немного более низкой нормой прекращения в объединенной группе плацебо. После 6 месяцев, группа ликсисенатида одностадийного титрования показала тренд, аналогичный объединенной группе плацебо, тогда как двухстадийное титрование ликсисенатида все-таки сохраняло более высокую норму.
Из 4 получавших лечение плацебо двухстадийного титрования субъектов, которые прекратили лечение вследствие AE (Таблица 2), один имел отсутствующую дату последнего введения на «Конец лечения» CRF и прекратил лечение вследствие AE, которые были классифицированы как AE после лечения согласно SAP конвенция по обработке данных, тогда как 3 имели TEAE, ведущие к прекращению лечения (Таблица 16).
Распределение субъектов - Рандомизированная популяция
(N=162)
стадийное титрование (N=161)
ненное
(N=322)
6.1.3 Демографические данные и исходные характеристики
Демографические и исходные характеристики субъектов были в целом одинаковыми между лечебными группами для выборки для оценки безопасности (Таблица 3) за исключением того, что получавшие лечение плацебо субъекты были немного старше, чем получавшие лечение ликсисенатидом субъекты. Средний возраст популяции исследования составлял 57,0 лет. Большинство субъектов были представители европеоидной расы/белые (90,2%).
Характеристики заболевания, включая историю заболевания сахарным диабетом, были в общем сравнимы между лечебными группами за исключением того, что средний возраст при наступлении диабета 2 типа у получавших лечение плацебо субъектов немного выше, чем у получавших лечение ликсисенатидом субъектов (Таблица 4).
HbA1c, FPG и масса тела на исходном уровне были в общем сравнимы между лечебными группами для выборки для оценки безопасности (Таблица 5).
Демографические данные и характеристики субъектов при скрининге или на исходном уровне - Выборка для оценки безопасности
(N=79)
(N=81)
(N=160)
(N=161)
(N=161)
(N=322)
(N=482)
[n (%)]
Характеристики заболевания при скрининге или на исходном уровне - Выборка для оценки безопасности
(N=81)
(N=160)
(N=161)
(N=161)
(N=322)
(N=482)
3400
360,5
Значение креатининового клиренса получено с использованием уравнения Кокрофта-Голта
Исходные показатели эффективности - Выборка для оценки безопасности
(N=81)
(N=160)
(N=161)
(N=322)
(N=482)
6.1.4 Дозировка и продолжительность
Средняя продолжительность лечения была аналогична между лечебными группами (552,8 дней (79,0 недель) для объединенного плацебо, 518,6 дней (74,1 недели) для ликсисенатида двухстадийного титрования и 538,1 дней (76,9 недели) для ликсисенатида одностадийного титрования) (Таблица 6). Из 482 субъектов для оценки безопасности, 439 (93,8% в объединенной группе плацебо, 88,2% в группе ликсисенатида двухстадийного титрования и 91,3% в группе ликсисенатида одностадийного титрования) имели по меньшей мере 169 дней (24 недели) лечения, и 298 (63,8% в объединенной группе плацебо, 59,0% в группе ликсисенатида двухстадийного титрования и 62,7% в группе ликсисенатида одностадийного титрования) имели по меньшей мере 547 дней (18 месяцев) лечения. Два субъекта (по 1 в каждой группе плацебо) имели отсутствующую дату последнего введения на «Конец лечения» CRF и следовательно, следуя SAP конвенции по обработке данных, продолжительность их лечения была устанавлена как отсутствующая.
Для группы ликсисенатида двухстадийного титрования, 141 (87,6%) субъект получали намеченную общую ежедневную дозу, равную 20 мкг, в конце двойного слепого лечения, в конце титрования и в конце 24-недельного периода двойного слепого лечения (Таблица 7, Таблица 8 и Таблица 9). Для группы ликсисенатида одностадийного титрования, 147 (91,3%) субъектов, 150 (93,2%) субъектов и 150 (93,2%) субъектов получали намеченную общую ежедневную дозу, равную 20 мкг, в конце двойного слепого лечения, в конце титрования и в конце 24-недельного периода двойного слепого лечения, соответственно (Таблица 7, Таблица 8 и Таблица 9). Для объединенной группы плацебо, 156 (97,5%) субъектов, 155 (96,9%) субъектов и 156 (97,5%) субъектов получали намеченную общую ежедневную дозу 20 мкг в конце двойного слепого лечения, в конце титрования и в конце 24-недельного периода двойного слепого лечения, соответственно (Таблица 7, Таблица 8 и Таблица 9).
Воздействие исследуемого препарата - Выборка для оценки безопасности
(N=79)
(N=81)
(N=160)
(N=161)
(N=161)
(N=322)
Количество (%) субъектов по конечной общей суточной дозе в конце двойного слепого лечения - Выборка для оценки безопасности
(N=79)
(N=81)
(N=160)
(N=161)
(N=161)
(N=322)
Примечание: Процентные доли рассчитаны с использованием в качестве знаменателя количества субъектов для оценки безопасности.
Количество (%) субъектов по дозе в конце титрования - Выборка для оценки безопасности
(N=79)
(N=81)
(N=160)
(N=161)
(N=161)
(N=322)
Запланированным осмотром для конца титрования по протоколу должен быть 5 осмотр/2 неделя
Примечание: Процентные доли рассчитаны с использованием в качестве знаменателя количества субъектов для оценки безопасности.
Количество (%) субъектов по конечной общей суточной дозе в конце 24-недельного лечения - Выборка для оценки безопасности
(N=79)
(N=81)
(N=160)
(N=161)
(N=161)
(N=322)
Примечание: Процентные доли рассчитаны с использованием в качестве знаменателя количества субъектов для оценки безопасности.
6.2 Эффективность
6.2.1 Основной конечный показатель эффективности
Основной анализ
Таблица 10 приводит результаты основного параметра эффективности, изменение HbA1c к 24 неделе по сравнению с исходным уровнем (LOCF) с использованием анализа ANCOVA.
Предварительно утвержденный основной анализ показал, что обе группы, получавшие лечение ликсисенатидом, продемонстрировали статистически значимое снижение к 24 неделе HbA1c по сравнению с исходным уровнем, по сравнению с объединенной группой плацебо (для группы ликсисенатида двухстадийного титрования, средняя разница, рассчитанная методом наименьших квадратов = -0,41%; p-значение <0,0001; для группы ликсисенатида одностадийного титрования, средняя разница, рассчитанная методом наименьших квадратов = -0,49%; p-значение <0,0001).
Среднее изменение HbA1c (%) к 24 неделе по сравнению с исходным уровнем - mITT популяция
(N=159)
(N=160)
(N=158)
a Модель ковариационного анализа (ANCOVA) с лечебными группами (группы ликсисенатида двухстадийного титрования и плацебо, группы ликсисенатида одностадийного титрования и плацебо), рандомизированными стратами уровня HbA1c при скрининге (<8,0, ≥8,0%), рандомизированными стратами индекса массы тела при скрининге (<30, ≥30 кг/м2) и страной в качестве фиксированных эффектов и исходным значением HbA1c в качестве ковариаты. Сравнение между каждой группой ликсисенатида и объединенной группой плацебо достигалось через соответствующие сопоставления.
Примечание: анализ включал измерения, полученные перед введением резервного лечения и до 3 дней после последней дозы двойной слепой инъекции исследуемого препарата при или перед 12 осмотром (24 Неделя) или на 169 день, если 12 осмотр (24 неделя) невозможен. Включены субъекты с измерениями как исходного уровня, так и на 24 неделе (LOCF).
Фигура 4 иллюстрирует среднее изменение HbA1c (±стандартная ошибка) по сравнению с исходным уровнем с течением времени во время основного 24-недельного периода двойного слепого лечения. Фигура 7 в приложении иллюстрирует среднее изменение HbA1c (±стандартная ошибка) по сравнению с исходным уровнем с течением времени вплоть до 76 недели. Снижение HbA1c относительно сохранялось с течением времени за пределами 24 недель.
Дополнительный анализ
Таблица 11 приводит долю субъектов с лечебным эффектом в виде HbA1c ≤6,5% или <7% на 24 неделе, соответственно. Лечебные эффекты были аналогичны между группами лечения ликсисенатидом. Анализ субъектов, ответивших на лечение снижением HbA1c, с использованием метода CMH показал существенное отличие лечения между каждой группой ликсисенатида против объединенного плацебо (для HbA1c ≤6,5% на 24 неделе, p-значение = 0,0009 для ликсисенатида двухстадийного титрования, и p-значение <0,0001 для ликсисенатида одностадийного титрования; для HbA1c < 7% на 24 неделе, p-значение = 0,0005 для ликсисенатида двухстадийного титрования, и p-значение <0,0001 для ликсисенатида одностадийного титрования).
Количество (%) субъектов со значением HbA1c ≤6,5% или <7%, соответственно, на 24 неделе - mITT популяция
(N=159)
(N=160)
(N=158)
Примечание: анализ включал измерения, полученные перед введением резервного лечения и до 3 дней после последней дозы двойной слепой инъекции исследуемого препарата при или перед 12 осмотром (24 Неделя) или на 169 день, если 12 осмотр (24 неделя) невозможен.
6.2.2 Дополнительные конечные показатели эффективности
Таблица 12 и Таблица 13 приводят ANCOVA анализы FPG и массы тела, соответственно. Фигура 5 и Фигура 6 иллюстрируют среднее изменение (±стандартная ошибка) по сравнению с исходным уровнем FPG и массы тела с течением времени во время основного 24-недельного периода двойного слепого лечения, соответственно. Среднее изменение FPG и массы тела (±стандартная ошибка) по сравнению с исходным уровнем с течением времени вплоть до 76 недели отображены на фиг.8 и фиг.9 в приложении, соответственно.
Для FPG обе группы, получавшие лечение ликсисенатидом, показали статистически значимое снижение к 24 неделе по сравнению с исходным уровнем в сравнении с объединенной группой плацебо (для группы ликсисенатида двухстадийного титрования средняя разница, рассчитанная методом наименьших квадратов = -0,67 ммоль/л, а p-значение = 0,0004; для группы ликсисенатида одностадийного титрования, средняя разница, рассчитанная методом наименьших квадратов = -0,65 ммоль/л, а p-значение = 0,0007).
LS среднее изменение массы тела по сравнению с исходным уровнем на 24 неделе составляло -2,68 кг для группы ликсисенатида двухстадийного титрования, -2,63 кг для группы ликсисенатида одностадийного титрования, и -1,63 кг для объединенной группы плацебо, со статистически значимым различием, наблюдаемым в обеих группах лечения ликсисенатидом по сравнению с объединенной группой плацебо (для ликсисенатида двухстадийного титрования, средняя разница, рассчитанная методом наименьших квадратов = -1,05 кг, а p-значение 0,0025; для ликсисенатида одностадийного титрования, средняя разница, рассчитанная методом наименьших квадратов = -1,00 кг, а p-значение 0,0042).
Среднее изменение глюкозы в плазме натощак (ммоль/л) к 24 неделе по сравнению с исходным уровнем - mITT популяция
(N=159)
(N=160)
(N=158)
aМодель ковариационного анализа (ANCOVA) с лечебными группами (группы ликсисенатида двухстадийного титрования и плацебо, группы ликсисенатида одностадийного титрования и плацебо), рандомизированными стратами уровня HbA1c при скрининге (<8,0, ≥8,0%), рандомизированными стратами индекса массы тела при скрининге (<30, ≥30 кг/м2) и страной в качестве фиксированных эффектов и исходным уровнем глюкозы в плазме натощак в качестве ковариаты. Сравнение между каждой группой ликсисенатида и объединенной группой плацебо достигалось через соответствующие сопоставления.
Примечание: анализ включал измерения, полученные перед введением резервного лечения и до 1 дня после последней дозы двойной слепой инъекции исследуемого препарата при или перед 12 осмотром (24 Неделя) или на 169 день, если 12 осмотр (24 неделя) невозможен. Включены субъекты с измерениями как исходного уровня, так и на 24 неделе (LOCF).
Среднее изменение к 24 неделе массы тела (кг) по сравнению с исходным уровнем - mITT популяция
(N=159)
(N=160)
(N=158)
a Модель ковариационного анализа (ANCOVA) с лечебными группами (группы ликсисенатида двухстадийного титрования и плацебо, группы ликсисенатида одностадийного титрования и плацебо), рандомизированными стратами уровня HbA1c при скрининге (<8,0, ≥8,0%), рандомизированными стратами индекса массы тела при скрининге (<30, ≥30 кг/м2) и страной в качестве фиксированных эффектов и исходной массой тела в качестве ковариаты. Сравнение между каждой группой ликсисенатида и объединенной группой плацебо достигалось через соответствующие сопоставления.
Примечание: анализ включал измерения, полученные перед введением резервного лечения и до 3 дней после последней дозы двойной слепой инъекции исследуемого препарата при или перед 12 осмотром (24 Неделя) или на 169 день, если 12 осмотр (24 неделя) невозможен. Включены субъекты с измерениями как исходного уровня, так и на 24 неделе (LOCF).
25,8% получавших лечение ликсисенатидом двухстадийного титрования субъектов, 19,6% получавших лечение ликсисенатидом одностадийного титрования субъектов и 15,2% получавшие лечение плацебо субъектов имели к 24 неделе потерю массы ≥5% по сравнению с исходным уровнем (Таблица 14). В обеих группах лечения ликсисенатидом масса тела продолжала уменьшаться после 24 недельного основного периода лечения (Фигура 9).
Количество (%) субъектов с потерей массы к 24 неделе >=5% по сравнению с исходным уровнем - mITT популяция
(N=158)
Обе группы, получавшие лечение ликсисенатидом, показали немного более низкие процентные доли субъектов, нуждавшихся в резервном варианте лечения во время основного 24-недельного периода двойного слепого лечения (3,1% для двухстадийного титрования и 1,3% для одностадийного титрования) по сравнению с объединенной группой плацебо (4,4%) (Таблица 15). Не было подтверждения существенного различия между каждой группой ликсисенатида и объединенной группой плацебо вследствие низкой частоты встречаемости субъектов, получивших резервный вариант лечения во время основного 24-недельного периода двойного слепого лечения.
Количество (%) субъектов, нуждавшихся в резервном варианте лечения во время 24-недельного периода лечения - mITT популяция
(N=159)
(N=160)
(N=158)
6.3 Безопасность
В Таблице 16 предоставлен обзор нежелательных явлений, наблюдаемых в течение периода лечения исследуемым препаратом на протяжении всего исследования. Количественное соотношение субъектов, которые испытывали TEAE, было сопоставимо между лечебными группами (87,6% в группе ликсисенатида двухстадийного титрования, 85,7% в группе ликсисенатида одностадийного титрования и 86,3% в объединенной группе плацебо). Пять субъектов (1 субъект в группе ликсисенатида двухстадийного титрования, 2 субъекта в группе ликсисенатида одностадийного титрования и 2 субъекта в объединенной группе плацебо) имели в течение периода лечения исследуемым препаратом TEAE, приведшие к смерти. Пятьдесят девять субъектов имели по меньше мере одно серьезное TEAE, произошедшее в течение периода лечения исследуемым препаратом на протяжении всего исследования с аналогичной частотой встречаемости между группой ликсисенатида двухстадийного титрования (13,0%) и объединенной группой плацебо (13,8%), но с немного более низкой частотой встречаемости в группе ликсисенатида одностадийного титрования (9,9%). Процентная доля субъектов с TEAE, приведшими к прекращению лечения, была немного выше в группах лечения ликсисенатидом (11,8% для двухстадийного титрования; 8,7% для одностадийного титрования), чем в объединенной группе плацебо (5,6%). Между двумя группами ликсисенатида, немного более низкая норма TEAE, приведших к прекращению лечения, наблюдалась при одностадийном титровании, чем при двухстадийном титровании. Таблица 17, Таблица 18 и Таблица 19 приводят TEAE, приведшие к смерти, серьезные TEAE и TEAE, приведшие к прекращению лечения по основным SOC, HLGT, HLT и РT, соответственно. Наиболее обычным TEAE в обеих группах лечения ликсисенатидом, приведшим к прекращению лечения, была тошнота (по 6 [3,7%] субъектов в каждой группе ликсисенатида). Ни один субъект не прекратил лечение вследствие тошноты в объединенной группе плацебо.
Таблица 28 в приложении приводит частоту встречаемости TEAE в течение периода лечения исследуемым препаратом на протяжении всего исследования, произошедших по меньшей мере у 1% субъектов в объединенной группе плацебо или любой отдельной группе ликсисенатида. Тошнота была наиболее часто сообщаемым TEAE в обеих группах лечения ликсисенатидом (62 [38,5%] субъекта для ликсисенатида двухстадийного титрования и 47 [29,2%] субъектов для ликсисенатида одностадийного титрования). Тринадцать получавших лечение плацебо субъектов (8,1%) сообщали о тошноте. Вторым наиболее часто сообщаемым TEAE у получавших лечение ликсисенатидом субъектов была рвота (29 [18,0%] субъектов для ликсисенатида двухстадийного титрования и 21 [13,0%] субъект для ликсисенатида одностадийного титрования), за которым следовала головная боль (23 [14,3%] субъекта для ликсисенатида двухстадийного титрования и 20 [12,4%] субъектов для ликсисенатида одностадийного титрования) и диарея (24 [14,9%] субъекта для ликсисенатида двухстадийного титрования и 16 [9,9%] субъектов для ликсисенатида одностадийного титрования). Соответствующее количество субъектов (%) в объединенной группе плацебо было 1 (0,6%) для рвоты, 20 (12,5%) для головной боли и 21 (13,1%) для диареи.
Обзор профиля нежелательных явлений: возникшие после начала лечения нежелательные события в течение периода применения исследуемого препарата в течение всего исследования - Выборка для оценки безопасности
(N=79)
(N=81)
(N=160)
(N=161)
(N=161)
(N=322)
n (%) = Количество и процентная доля субъектов по меньшей мере с одним нежелательным событием.
Примечание: Период применения исследуемого препарата в течение всего исследования = время от первой дозы двойного слепого лечения исследования до 3 дней после введения последней дозы.
Количество (%) субъектов, испытавших TEAE, приведшее (приведшие) к смерти в течение всего периода лечения по основным SOC, HLGT, HLT и PT - Выборка для оценки безопасности
HLGT: Групповой термин высокого уровня
HLT: Термин высокого уровня
Предпочтительный термин
(N=79)
(N=81)
(N=160)
(N=161)
(N=161)
(N=322)
n (%) = Количество и процентная доля субъектов по меньшей мере с одним TEAE, приведшим к смерти.
Примечание: Период применения исследуемого препарата в течение всего исследования = время от первой дозы двойного слепого лечения исследования до 3 дней после введения последней дозы.
Таблица отсортирована в соответствии с международно согласованным порядком SOC и алфавитным порядком HLGT, HLT, PT.
Количество (%) субъектов, испытавших серьезное (серьезные) TEAE в течение всего периода лечения, представленные по основным SOC, HLGT, HLT и PT - Выборка для оценки безопасности
HLGT: Групповой термин высокого уровня
HLT: Термин высокого уровня
Предпочтительный Термин
(N=79)
(N=81)
(N=160)
(N=161)
(N=161)
(N=322)
n (%) = Количество и процентная доля субъектов по меньшей мере с одним серьезным TEAE.
Примечание: Период применения исследуемого препарата в течение всего исследования = время от первой дозы двойного слепого лечения исследования до 3 дней после введения последней дозы.
Таблица отсортирована в соответствии с международно согласованным порядком SOC и алфавитным порядком HLGT, HLT, PT.
Количество (%) субъектов, перенесших TEAE, приведших к необратимому прекращению лечения в течение всего периода лечения по основным SOC, HLGT, HLT и PT - Выборка для оценки безопасности
HLGT: Групповой термин высокого уровня
HLT: Термин высокого уровня
Предпочтительный Термин
(N=79)
(N=81)
(N=160)
(N=161)
(N=161)
(N=322)
n (%) = Количество и процентная доля субъектов по меньшей мере с одним TEAE, приведшим к необратимому прекращению лечения.
Примечание: Период применения исследуемого препарата в течение всего исследования = время от первой дозы двойного слепого лечения исследования до 3 дней после введения последней дозы.
Таблица отсортирована в соответствии с международно согласованным порядком SOC и алфавитным порядком HLGT, HLT, PT.
Двенадцать (7,5%) субъектов в группе ликсисенатида двухстадийного титрования и 6 (3,7%) субъектов в группе ликсисенатида одностадийного титрования в течение периода лечения исследуемым препаратом на протяжении всего исследования имели события симптоматической гипогликемии по определению протокола по сравнению с 12 (7,5%) получавшими лечение плацебо субъектами, которые имели события симптоматической гипогликемии в течение того же самого периода (Таблица 20). Ни одно из явлений симптоматической гипогликемии не было тяжелым по интенсивности. Один дополнительный субъект (в группе двухстадийного титрования плацебо) сообщал о событии симптоматической гипогликемии на специальной странице AE для «симптоматической гипогликемии», но данное событие не соответствовало указанному в протоколе определению (т.е. ассоциированным значениям глюкозы ≥60 мг/дл).
Девять (5,6%) субъектов в каждой группе ликсисенатида и 3 (1,9%) в объединенной группе плацебо испытывали AE реакции в месте инъекции (Таблица 21). AE реакции в месте инъекции идентифицировали посредством поиска термина «место инъекции» либо в закодированных PT из сообщаемых исследователю терминов, либо в PT из диагноза ARAC в процессе экспертизы аллергических реакций. Интенсивность ни одной из реакций не была серьезной или тяжелой.
В общей сложности от исследователей были сообщения о 30 событиях, как о возможных аллергических событиях в течение периода лечения исследуемым препаратом на протяжении всего исследования, которые посылали в ARAC для экспертизы. Из них, 16 событий у 15 субъектов (6 [3,7%] получавших лечение ликсисенатидом двухстадийного титрования субъектов, 3 [1,9%] получавших лечение ликсисенатидом одностадийного титрования субъектов и 6 [3,8%] получавших лечение плацебо субъектов) были расценены ARAC как аллергические реакции, но только 2 события анафилактической реакции (по 1 в каждой группе ликсисенатида) были расценены как возможно связанные с IP (Таблица 22).
Субъект # 276303004 (одностадийное титрование ликсисенатида), без персонального или семейного аллергологического анамнеза, обнаружил кожную реакцию спустя 30 мин. после первой дозы рандомизированного лечения. О событии сообщалось как об «аллергической экзантеме», и оно было закодировано PT «аллергический дерматит». IP необратимо прекратили. Применили корректирующее лечение антигистаминными средствами и стероидами, и событие было устранено в тот же самый день. Событие было расценено ARAC, как анафилактическая реакция, возможно связанная с IP.
У субъекта # 642307010 (двухстадийное титрование ликсисенатида), без персонального или семейного аллергологического анамнеза, через 5,5 месяцев после начала IP появилась тошнота и головокружение спустя несколько секунд после введения IP, за которым последовала кожная реакция. Событие сопровождалось гипотонией. О событии сообщалось как об «аллергическом дерматите», и оно было закодировано PT «аллергический дерматит». Вслед за событием IP необратимо прекратили. Применили корректирующее лечение антигистаминными средствами и стероидами, и день спустя событие было устранено. Событие было расценено ARAC как анафилактическая реакция, возможно связанная с IP.
По протоколу, любое подтвержденное увеличение амилазы и/или липазы выше двойного верхнего предела нормального диапазона (ULN) необходимо было отслеживать и документально подтверждать в предварительно утвержденной форме: «форма нежелательных явлений для подозрения на панкреатит». В течение периода лечения исследуемым препаратом на протяжении всего исследования, данная форма было заполнена для 4 (2,5%) субъектов в каждой группе ликсисенатида и для 5 (3,1%) субъектов в объединенной группе плацебо (Таблица 23). В исследовании не наблюдалось ни одного случая панкреатита.
В Таблице 24 приведены субъекты, которые имели по меньшей мере одно значение липазы или амилазы ≥3 ULN в течение периода лечения исследуемым препаратом. В общей сложности с повышенной липазой (≥3ULN) наблюдалось 17 субъектов (8 [5,0%] в группе ликсисенатида двухстадийного титрования, 5 [3,1%] в группе ликсисенатида одностадийного титрования и 4 [2,5%] в объединенном плацебо). Повышенную амилазу ≥3ULN имел один (0,6%) субъект в группе ликсисенатида одностадийного титрования и ни одного в группе ликсисенатида двухстадийного титрования и в объединенной группе плацебо.
По протоколу, любое подтвержденное значение кальцитонина, составляющее ≥20 пг/мл, необходимо было отслеживать и документально подтверждать в предварительно заданной форме о нежелательных явлениях для «повышенного кальцитонина ≥ 20 пг/мл». В течение периода лечения исследуемым препаратом на протяжении всего исследования, данная форма была оформлена для 1 (0,6%) субъекта в каждой группе ликсисенатида и для 1 (0,6%) субъекта в объединенной группе плацебо (Таблица 25). Для 2 из данных 3 субъектов (по 1 в каждой группе ликсисенатида), значение кальцитонина составляло ≥20 нг/л, но < 50 нг/л, а pT был «повышенный кальцитонин в крови». Обоим субъектам провели дальнейшее обследование, как рекомендовано протоколом, включая ультразвуковое тиреосканирование и освидетельствование специалиста, и результат был нормальным. Для третьего субъекта (в группе двухстадийного титрования плацебо), значение кальцитонина составляло ≥50 нг/л, а PT был «рак щитовидной железы». У данного субъекта был диагностирован медуллярный рак щитовидной железы, оставшийся с лимфогенными метастазами, который исследователь оценил как не связанный с IP.
Два (1,4%) субъекта в группе ликсисенатида двухстадийного титрования, 2 (1,3%) в группе ликсисенатида одностадийного титрования и 4 (2,5%) в объединенной группе плацебо в течение периода лечения исследуемым препаратом имели по меньшей мере одно значение кальцитонина ≥20 нг/л (Таблица 26). Из данных субъектов, в дополнение к субъектам, описанным в предшествующем абзаце, два других субъекта (1 в группе ликсисенатида одностадийного титрования и 1 в объединенной группе плацебо) имели неблагоприятное событие после прекращения IP, о котором сообщалось в специальной форме для неблагоприятных событий для «повышенного кальцитонина ≥20 пг/мл». Для получавшего лечение ликсисенатидом субъекта, о «высоком уровне кальцитонина» сообщалось спустя 5,5 месяцев после прекращения IP, а с помощью ультразвукового тиреосканирования, выполненного спустя 2,5 месяца, были выявлены узлы щитовидной железы. Для получавшего лечение плацебо субъекта, спустя 7 месяцев после прекращения IP сообщалось о «периодическом повышении кальцитонина». Спустя один месяц было проведено ультразвуковое тиреосканирование и освидетельствование специалиста, и результаты были нормальными. Следует отметить, что измерения кальцитонина осуществляли по поправке к протоколу после того, как большинство субъектов уже были рандомизированы в данном исследовании. Вследствие этого, для большинства субъектов отсутствуют исходные значения кальцитонина.
Сводная информация о симптоматической гипогликемии в течение периода лечения исследуемым препаратом на протяжении всего исследования - Выборка для оценки безопасности
(N=79)
(N=81)
(N=160)
(N=161)
(N=161)
(N=322)
b Рассчитано как (количество субъектов с событиями*100, деленное на суммарную величину воздействия + 3 дня в субъекто-годах).
Симптоматическая гипогликемия = симптоматическая гипогликемия, как определено по протоколу.
Примечание: Период применения исследуемого препарата для всего исследования = время от первой дозы двойного слепого лечения исследования до 3 дней после введения последней дозы.
Количество (%) субъектов, перенесших реакции в месте инъекции в течение периода лечения исследуемым препаратом на протяжении всего исследования - Выборка для оценки безопасности
Предпочтительный Термин n (%)
(N=79)
(N=81)
(N=160)
(N=161)
(N=161)
ненное
(N=322)
Примечание: Период применения исследуемого препарата для всего исследования = время от первой дозы двойного слепого лечения исследования до 3 дней после введения последней дозы.
Количество (%) субъектов с событиями, расцененными как аллергические реакции ARAC в течение периода лечения исследуемым препаратом на протяжении всего исследования - Выборка для оценки безопасности
(по ARAC)
(N=79)
(N=81)
(N=160)
(N=161)
(N=161)
(N=322)
аллергическая реакция
Примечание: Период применения исследуемого препарата для всего исследования = время от первой дозы двойного слепого лечения исследования до 3 дней после введения последней дозы.
Количество (%) субъектов со специальной формой для неблагоприятных событий для подозрения на панкреатит, заполненной в течение периода лечения исследуемым препаратом на протяжении всего исследования - Выборка для оценки безопасности
(N=79)
(N=81)
(N=160)
(N=161)
(N=161)
(N=322)
Примечание: Период применения исследуемого препарата для всего исследования = время от первой дозы двойного слепого лечения исследования до 3 дней после введения последней дозы.
Ферменты поджелудочной железы: Количество (%) субъектов по меньшей мере с одним PCSA после исходного в течение периода применения исследуемого препарата для всего исследования согласно статусу исходного PCSA - Выборка для оценки безопасности
Исходный уровень
По критерию PCSA n/N1 (%)
(N=79)
(N=81)
(N=160)
(N=161)
(N=161)
(N=322)
*Независимо от исходного уровня.
Примечание: Период применения исследуемого препарата для всего исследования = время от первой дозы двойного слепого лечения исследования до 3 дней после введения последней дозы.
Количество (n) отображает подмножество общего количества субъектов, которые по меньшей мере один раз соответствуют интересующему критерию. Знаменателем (/N1) для каждого параметра в пределах лечебной группы является количество субъектов для лечебной группы, которые имели такой параметр, оцененный после исходного уровня по состоянию PCSA исходного уровня. Только ухудшение самого плохого случая для каждого субъекта отображается исходным состоянием.
Количество (%) субъектов с повышенным кальцитонином в течение периода лечения исследуемым препаратом на протяжении всего исследования - Выборка для оценки безопасности
(N=79)
(N=81)
(N=160)
(N=161)
(N=161)
(N=322)
Примечание: Период применения исследуемого препарата для всего исследования = время от первой дозы двойного слепого лечения исследования до 3 дней после введения последней дозы.
Кальцитонин сыворотки - Количество (%) субъектов по предварительно определенным категориям в течение периода применения исследуемого препарата всего исследования согласно категории исходного уровня - Выборка для оценки безопасности
Статус исходного уровня
После исходного
(N=79)
(N=81)
(N=160)
(N=161)
(N=161)
(N=322)
*Независимо от исходного уровня.
Примечание: Период применения исследуемого препарата для всего исследования = время от первой дозы двойного слепого лечения исследования до 3 дней после введения последней дозы.
Числитель представляет количество субъектов, которые были в предварительно оговоренных категориях в каждой категории исходного уровня. Знаменателем для каждого параметра в пределах лечебной группы является количество субъектов для лечебной группы, которые имели данный параметр, оцененный после исходного по статусу исходного уровня.
Субъекта считают только в самой плохой категории.
7 Приложение
Среднее изменение HbA1c (%) по сравнению с исходным уровнем при осмотре - mITT популяция
Временная точка
объединенное (N=159)
Примечание: Из анализа исключены измерения, полученные после введения резервного лечения и/или после прекращения лечения плюс 3 дня.
Для 24 недели (LOCF), в анализ включены измерения, полученные до 3 дней после последней дозы двойной слепой инъекции исследуемого препарата при или перед 12 осмотром (24 неделя), или на 169 день, если 12 осмотр (24 неделя) невозможен.
Количество (%) субъектов, испытавших общее TEAE (общие TEAE) (PT ≥1% в объединенном плацебо или любой отдельной группе ликсисенатида), представленные по основным SOC, HLGT, HLT и PT в течение периода лечения исследуемым препаратом на протяжении всего исследования - Выборка для оценки безопасности
HLGT: Групповой термин высокого уровня
HLT: Термин высокого уровня
Предпочтительный Термин
(N=79)
(N=81)
(N=160)
(N=161)
(N=161)
(N=322)
n (%) = Количество и процентная доля субъектов по меньшей мере с одним TEAE.
Примечание: Период применения исследуемого препарата в течение всего исследования = время от первой дозы двойного слепого лечения исследования до 3 дней после введения последней дозы.
Таблица отсортирована в соответствии с международно согласованным порядком SOC и алфавитным порядком HLGT, HLT, PT.
Представлены только SOC по меньшей мере с одним PT ≥1 в группе объединенного плацебо или в группе ликсисенатида одно- или двухстадийного титрования.
Пример 2.
Рандомизированное, двойное слепое, с двойной маскировкой, с активным контролем, с 2 параллельными группами, многонациональное исследование по оценке эффективности и безопасности ликсисенатида по сравнению с ситаглиптином в качестве дополнительного лечения к метформину у страдающих ожирением субъектов с диабетом 2 типа моложе, чем 50 и с неадекватным контролем посредством метформина.
Пример относится к рандомизированному, двойному слепому, с двойной маскировкой, с активным контролем, с 2 параллельными группами, многонациональному исследованию по оценке эффективности и безопасности ликсисенатида по сравнению с ситаглиптином в качестве дополнительного лечения к метформину у страдающих ожирением субъектов с диабетом 2 типа моложе, чем 50 и с неадекватным контролем посредством метформина. Приблизительная продолжительность исследования на субъекта составляла 27 недель (до 3 недель скрининг + 24 недели двойное слепое лечение + 3 дня последующее наблюдение). Исследование проводили в 92 центрах в 13 странах. Основная цель исследования состояла в оценке эффективности ликсисенатида по комбинированному показателю гликемического контроля (гликированному гемоглобину A1c [HbA1c]) и массы тела по сравнению с ситаглиптином в течение периода, равного 24 неделям.
В общей сложности 319 субъектов рандомизировали в одну из двух лечебных групп (158 в группе ликсисенатида и 161 в группе ситаглиптина). Всех рандомизированных субъектов подвергали лечению исследования и включили в модифицированную популяцию субъектов, включенных в испытание (mITT). Демографические данные и исходные характеристики были в целом аналогичны между лечебными группами, причем в группе ликсисенатида было больше субъектов-женщин. В течение периода лечения исследования, 27 (8,5%) субъектов досрочно прекратили лечение исследования с более высокой процентной долей в группе ликсисенатида (10,1%) по сравнению с группой ситаглиптина (6,8%). У получавших лечение ликсисенатидом субъектов, основной причиной прекращения лечения было «другие причины» (7 субъектов: 4,4% против 5 субъектов: 3,1% для ситаглиптина), за которым следовало «нежелательные события» (4 субъекта: 2,5% против 5 субъектов: 3,1% для ситаглиптина).
Процентная доля субъектов с HbA1c<7% на 24 неделе и потерей массы, составляющей по меньшей мере 5% исходной массы тела, на 24 неделе (основной показатель эффективности) была более высокой для группы ликсисенатида (12,0%), чем для группы ситаглиптина (7,5%). Различие лечения не было статистически значимым (средневзвешенное значение различия эффективности лечения против ситаглиптина = 4,6%; p-значение = 0,1696 на основании основного анализа с использованием метода Кохрана-Мантеля-Хенселя [CMH]), вследствие более низкой, чем ожидалось эффективности лечения для группы ликсисенатида. Вспомогательный анализ с использованием регрессионной логистической модели показал непротиворечивый результат (p-значение = 0,1160). В общей сложности 61 субъект (40,7%) в группе ликсисенатида имел HbA1c <7% на 24 неделе по сравнению с 64 субъектами (40,0%) в группе ситаглиптина, а 36 (24,0%) получавших лечение ликсисенатидом субъектов имели HbA1c ≤6,5% по сравнению с 42 (26,3%) получавшими лечение ситаглиптином субъектами. Больше получавших лечение ликсисенатидом субъектов (28 [18,4%]) имели к 24 неделе потерю массы ≥5% по сравнению с исходным уровнем, чем получавших лечение ситаглиптином субъектов (19 [11,9%]).
Субъекты, получавшие лечение ликсисенатидом, имели значительно большее снижение массы тела, чем субъекты, получавшие лечение ситаглиптином (LS среднее различие равна -1,34 кг; 95% CI [-2,101; -0,575]). Средние изменения HbA1c к 24 неделе по сравнению с исходным уровнем в группах ликсисенатида и ситаглиптина были аналогичны: LS среднее равно -0,66% и -0,72%, соответственно. Лечение ликсисенатидом значительно улучшало гликемический контроль после приема пищи по сравнению с ситаглиптином, как показано результатами для оценки глюкозы в плазме через 2 часа после приема пищи (PPG) (LS среднее различие равна -1,91 ммоль/л; 95% CI [-2,876; -0,941]) и для колебания глюкозы (LS среднее различие равна -2,13 ммоль/л; 95% CI [-2,819; -1,434]). Для изменения на 24 неделе по сравнению с исходным уровнем HbA1c, глюкозы в плазме натощак (FPG), инсулинорезистентности, оцениваемых с помощью HOMA-IR, и функции β-клеток, оцениваемой с помощью HOMA-β, между лечебными группами ликсисенатида и ситаглиптина не наблюдалось существенного различия. Процентные доли субъектов, нуждавшихся в резервном варианте лечения, составляли 9,5% в группе ликсисенатида и 6,8% в группе ситаглиптина, без существенного различия между двумя данными группами.
Ликсисенатид имел хорошую переносимость. Частота встречаемости возникших после начала лечения нежелательных явлений (TEAE) была немного более высокой для получавших лечение ликсисенатидом субъектов против получавших лечение ситаглиптином субъектов (63,9% для ликсисенатида против 60,9% для ситаглиптина). Шесть субъектов (3 [1,9%] в каждой группе) имели серьезное TEAE. Не было сообщений о смертности в данном исследовании. Наиболее часто сообщаемым TEAE для получавших лечение ликсисенатидом субъектов против получавших лечение ситаглиптином субъектов была тошнота (17,7% против 6,8%, соответственно) за которой следовала головная боль (12,7% против 9,3%, соответственно). Один (0,6%) получавший лечение ликсисенатидом субъект имел 2 события симптоматической гипогликемии, которые определены в протоколе в течение периода лечения исследуемым препаратом по сравнению с 3 (1,9%) субъектами с 3 событиями в группе ситаглиптина. Ни одно из данных событий симптоматической гипогликемии не было тяжелым согласно определению протокола. В общей сложности для 3 субъектов (2 [1,3%] в группе ликсисенатида и 1 [0,6] в группе ситаглиптина) сообщалось о 3 событиях, расцененных комитетом по оценке аллергических реакций (ARAC), как аллергические реакции, но только одно событие (анафилактическая реакция) у получавшего лечение ликсисенатидом субъекта было расценено, как возможно связанное с исследуемым препаратом. В исследовании не сообщалось ни об одном случае панкреатита.
1 ЦЕЛИ
1.1 Основная цель
Основной целью данного исследования была оценка эффективности ликсисенатида по комбинированному показателю гликемического контроля (HbA1c) и массы тела по сравнению с ситаглиптином в качестве дополнительного лечения к метформину в течение периода, равного 24 неделям, у страдающих ожирением субъектов с диабетом 2 типа моложе, чем 50, и с неадекватным контролем одним метформином.
1.2 Ключевая дополнительная цель (цели)
Дополнительные цели данного исследования состояли:
• В оценке воздействия ликсисенатида на:
- Абсолютные изменения значений HbA1c и массы тела
- Глюкозу в плазме натощак (FPG)
- Глюкозу в плазме, инсулин, C-пептид, глюкагон и проинсулин во время 2-часового стандартизированного тестового приема пищи
- Инсулинорезистентность, оцениваемую посредством HOMA-IR
- функцию бетта-клеток, оцениваемую посредством HOMA-бетта
• В оценке безопасности и переносимости ликсисенатида
• В оценке формирования антител против ликсисенатида
2 ДИЗАЙН ИССЛЕДОВАНИЯ
Провели двойное слепое, с двойной маскировкой, рандомизированное, с активным контролем, многоцентровое, многонациональное исследование в 2 параллельных группах с запланированными 150 получавшими лечение ликсисенатидом субъектами и 150 получавшими лечение ситаглиптином субъектами. Исследование было двойным слепым относительно лечебной группы. объем лекарственного средства исследования (т.е., доза активного лекарственного средства или эквивалентного плацебо) не был слепым.
Субъектов стратифицировали по скрининговым значениям гликированного гемоглобина A1c (HbA1c) (<8%, ≥8%) и индекса массы тела (BMI) (<35 кг/м2, ≥35 кг/м2). После скринингового периода, субъектов централизованно рандомизировали посредством системы интерактивного голосового взаимодействия (IVRS) в соотношении 1:1 либо к ликсисенатиду, либо к ситаглиптину.
Исследование состояло из 3 периодов: 1) скринингового периода до 3 недель, который включал скрининговую фазу до 2 недель и 1-недельную вводную фазу простого-слепого плацебо; 2) 24-недельный период двойного слепого лечения, с двойной маскировкой, с активным контролем; 3) период из 3 дней после лечебного последующего наблюдения для всех субъектов после необратимого прекращения лечения (за исключением субъектов, которые досрочно прекратили лечение исследования).
В соответствии с поправкой 1 к протоколу (датирована 30 июня 2009 года), субъекты, которые досрочно прекратили лечение исследования оставались в исследовании до заключительной оценки на 24 неделе. Они следовали процедурам исследования, определенным в протоколе (за исключением постпрандиального теста и 3-дневного последующего наблюдения за безопасностью после лечения).
3 Основной и ключевой дополнительные конечные показатели
3.1 Основной конечный показатель
Основным показателем эффективности была процентная доля субъектов с HbA1c <7% на 24 неделе и потерей массы, составляющей по меньшей мере 5% исходной массы тела, на 24 неделе.
Если субъект досрочно прекратил лечение в течение 24-недельного периода двойного слепого лечения или не имел на 24 неделе значения HbA1c или массы тела, для расчета HbA1c или массы тела на 24 неделе использовали, соответственно, последнее после исходного уровня измерение HbA1c или массы тела во время лечения исследуемым препаратом в течение 24-недельного периода (процедура переноса вперед данных последнего наблюдения [LOCF]). Если во время лечения исследуемым препаратом последние после исходного уровня значения HbA1c и массы тела измеряли с интервалом друг от друга более чем 30 дней, в качестве комбинированного конечного показателя на 24 неделе использовали последние после исходного уровня значения (HbA1c и массы тела) во время лечения исследуемым препаратом, которые были разделены интервалом не более чем 30 дней. Субъектов, которые не имели после исходного уровня значений (HbA1c и массы тела) во время лечения исследуемым препаратом, которые во время 24-недельного периода были разделены интервалом не более чем 30 дней, считали субъектами, не ответившими на лечение по комбинированному основному показателю. Для всех субъектов, получивших в течение 24-недельного периода резервное лечение, в качестве HbA1c и массы тела на 24 неделе использовали последние перед резервным лечением измерения HbA1c и массы тела после исходного уровня во время лечения исследуемым препаратом.
3.2 Ключевые дополнительные конечные показатели
3.2.1 Конечные показатели эффективности
Для дополнительных показателей эффективности, применялась точно такая же методика обработки отсутствующей оценки/раннего прекращения, как для основного показателя эффективности. То есть, методику LOCF использовали, принимая в качестве значения 24 недели последнее доступное после исходного уровня измерение во время лечения исследуемым препаратом (перед резервным лечением в случае резервного варианта лечения).
Непрерывные показатели
• Абсолютное изменение HbA1c (%) к 24 неделе по сравнению с исходным уровнем
• Абсолютное изменение массы тела (кг) к 24 неделе по сравнению с исходным уровнем
• Изменение 2 часовой постпрандиальной глюкозы в плазме (ммоль/л) после стандартизированного тестового приема пищи к 24 неделе по сравнению с исходным уровнем
• Изменение FPG (ммоль/л) к 24 неделе по сравнению с исходным уровнем
• Изменение колебания глюкозы (глюкоза в плазме через 2 часа после приема пищи - глюкоза в плазме за 30 минут перед тестовым приемом пищи перед введением лекарственного средства исследования) (ммоль/л) во время стандартизированного постпрандиального теста к 24 неделе по сравнению с исходным уровнем
• Изменение следующих показателей в условиях натощак (за 30 минут перед тестовым приемом пищи перед введением лекарственного средства исследования) и через 2 часа после приема пищи, собранных во время стандартизированного тестового приема пищи: инсулина (пмоль/л), C-пептида (нмоль/л), глюкагона (нг/л), проинсулина (пмоль/л) и соотношения проинсулина-к-инсулину к 24 неделе по сравнению с исходным уровнем
• Изменение инсулинорезистентности, оцениваемой посредством HOMA-IR к 24 неделе по сравнению с исходным уровнем
• Изменение функции бета-кеток, оцениваемой посредством HOMA-бетта к 24 неделе по сравнению с исходным уровнем
Категориальные переменные
• Процентная доля субъектов с HbA1c<7% на 24 неделе
• Процентная доля субъектов с HbA1c≤6,5% на 24 неделе
• Процентная доля субъектов, нуждавшихся в резервном варианте лечения в течение периода двойного слепого лечения
• Процентная доля субъектов с потерей массы ≥5% (кг) к 24 неделе по сравнению с исходным уровнем
3.2.2 Конечные показатели безопасности
Анализ безопасности проводили на основании сообщаемых TEAE и другой информации о безопасности, включая симптоматическую гипогликемию и тяжелую симптоматическую гипогликемию, местное раздражающее действие в месте инъекции, аллергические события (по решению ARAC), предполагаемый панкреатит, повышенный кальцитонин, показатели жизнедеятельности, ЭКГ в 12 отведениях и лабораторные испытания.
Главные сердечно-сосудистые явления также собирались и рассматривались Комитетом по оценке сердечно-сосудистых явлений (CAC). Рассмотренные и подтвержденные CAC события из данного исследования и 3 других исследований фазы ликсисенатида будут объединяться для анализов и суммироваться в отдельном отчете на основании плана статистического анализа для общей сердечно-сосудистой оценки ликсисенатида. Заключение о Ключевых Результатах (KRM)/Отчет о клиническом исследовании (CSR) не будет отображать сущность рассмотренных и подтвержденных сердечно-сосудистых явлений данного исследования.
4. Расчетные предположения размеров выборки
Расчет размера выборки был основан на основном конечном показателе эффективности: процентной доле субъектов с HbA1c <7% на 24 неделе и потере массы, равной по меньшей мере 5% исходной массы тела на 24 неделе. Данный расчет, принимавший процентную долю субъектов, определенных как субъекты, ответившие на лечение по HbA1c (<7%) и массе (потеря по меньшей мере 5%) составлял 25% с ликсисенатидом и 10% с ситаглиптином с 2-сторонним критерием при 5% уровне значимости.
Размер выборки, равный 300 субъектам (по 150 субъектов на группу) считали достаточным для демонстрации превосходства ликсисенатида перед ситаглиптином по процентной доле субъектов, определенных как субъекты, ответившие на лечение по HbA1c (<7%) и массе (потеря по меньшей мере 5%) на 24 неделе с мощностью, равной 90%.
5 Статистические методы
5.1 Анализ популяций
Модифицированная популяция субъектов, включенных в испытание (mITT), состояла из всех рандомизированных субъектов, которые получили по меньшей мере одну дозу двойного слепого исследуемого препарата (IP). Субъектов анализировали в лечебной группе, в которую они были рандомизированы.
Выборку для оценки безопасности определяли, как все рандомизированные субъекты, которые получили по меньшей мере одну дозу двойного слепого IP.
5.2 Основной анализ эффективности
Основной показатель эффективности (процентную долю субъектов с HbA1c <7% на 24 неделе и потерей массы, составляющей по меньшей мере 5% исходной массы тела, на 24 неделе) анализировали с использованием критерия Кохрана-Мантеля-Хенселя (CMH) со стратификацией по рандомизированным стратам уровня HbA1c при скрининге (<8%, ≥8%) и рандомизированным стратам уровня BMI при скрининге (<35 кг/м2, ≥35 кг/м2). P-значение было основано на критерии CMH из PROC FREQ. Точечная оценка различия лечения (ликсисенатид по сравнению с ситаглиптином) в количественном соотношении, а также соответствующий 95% доверительный интервал (CI) предоставляли на основании средневзвешенного значения различия лечения из каждой страты с использованием весовых коэффициентов CMH.
В случае отсутствия события в ячейке для страты, делали поправку на непрерывность (т.е., в каждую ячейку таблицы 2×2 добавляли 0,5) для данной страты только для расчета отклонений. Следует заметить, что корректировку на непрерывность не применяли для оценки средневзвешенного значения различия лечения. Поправку на непрерывность применяли для оценки отклонения для построения 95% CI для общего различия.
Основной анализ основного показателя эффективности выполняли на основании mITT популяции и измерений, полученных в течение периода лечения исследуемым препаратом для показателей эффективности. Период лечения исследуемым препаратом для показателей эффективности за исключением показателей из постпрандиального теста определяли как время от первой дозы двойного слепого IP (ликсисенатида или ситаглиптина) до 3 дней (за исключением FPG центральной лабораторией, которое занимало до 1 дня) после последней дозы двойного слепого IP, или до введения резервного варианта лечения в зависимости от того, что было раньше. Период лечения исследуемым препаратом для показателей эффективности из постпрандиального теста, включая глюкозу в плазме через 2 часа после приема пищи, инсулин, C-пептид, глюкагон, проинсулин, колебание глюкозы, HOMA-индексы, и отношение проинсулина к инсулину, определяли как время от первой дозы двойного слепого IP (ликсисенатида или ситаглиптина) до даты последней дозы двойного слепого IP или до введения резервного варианта лечения, в зависимости от того, что было раньше.
Методику LOCF использовали, принимая в качестве комбинированного конечного показателя на 24 неделе последние доступные после исходного уровня значения во время лечения исследуемым препаратом (HbA1c и массы тела перед введением резервного варианта лечения), которые были отделены друг от друга интервалом не более чем 30 дней. Субъектов, которые не имели подобных значений, рассматривали как субъектов, не ответивших на лечение по комбинированному основному конечному показателю.
5.3 Ключевой дополнительный анализ эффективности
Никакой корректировки на множественность не делали для обеих какого-либо дополнительного показателя эффективности.
Все непрерывные дополнительные показатели эффективности, которые описаны в разделе 3.2.1, анализировали с помощью модели ковариационного анализа (ANCOVA) с лечебными группами (ликсисенатида и ситаглиптина), рандомизированными стратами уровня HbA1c при скрининге (<8,0, ≥8,0%), рандомизированными стратами уровня BMI при скрининге (<35 кг/м2, ≥35 кг/м2) и страной в качестве фиксированных эффектов и используя соответствующее исходное значение в качестве ковариаты. Были предоставлены скорректированные оценки среднего различия лечения между ликсисенатидом и ситаглиптином и двусторонние 95% доверительные интервалы.
Для следующих категориальных дополнительных показателей эффективности, сводные статистические данные были представлены по лечебным группам:
• Процентная доля субъектов с HbA1c<7% на 24 неделе,
• Процентная доля субъектов с HbA1c≤6,5% на 24 неделе,
• Процентная доля субъектов, нуждавшихся в резервном варианте лечения в течение 24-недельного периода лечения,
• Процентная доля субъектов с потерей массы ≥5% (кг) по сравнению с исходным уровнем на 24 неделе.
5.4 Анализ безопасности
Анализы безопасности главным образом были основаны на периоде лечения исследуемым препаратом. Период лечения исследуемым препаратом определяли как время от первой дозы двойного слепого IP (ликсисенатида или ситаглиптина) до 3 дней после введения последней дозы IP независимо от статуса резервного лечения. 3-дневный интервал был выбран на основании периода полувыведения IP (приблизительно 5-кратный период полувыведения).
Итог результатов безопасности (описательная статистика или Таблицы частоты) представлен по группам лечения.
6. Результаты
6.1 Субъекты исследования
6.1.1 Учет субъектов
Исследование проводили в 92 центрах в 13 странах (Австралия, Бразилия, Канада, Чили, Германия, Гватемала, Мексика, Перу, Польша, Румыния, Российская Федерация, Украина и Соединенные Штаты Америки). Скринингу подвергли в общей сложности 620 субъектов, из которых 319 рандомизировали в одну из двух лечебных групп. Наиболее обычной причиной отказа от рандомизации было значение HbA1c при скрининговом осмотре за пределами диапазона, как определено по протоколу (203 [32,7%] из 620 подвергшихся скринингу субъектов).
Всех 319 рандомизированных субъектов подвергли лечению исследования и включили в mITT популяцию. Таблица 1 предоставляет количество субъектов, включенных в каждую выборку для анализа.
Выборки для анализа - Рандомизированная популяция
Для выборки для оценки эффективности, субъекты сведены в таблицу согласно их рандомизированному лечению (по рандомизации).
6.1.2 Распределение Исследования
Таблица 2 представляет сущность распределения субъектов для каждой лечебной группы. Из 319 рандомизированных субъектов, 27 (8,5%) субъектов досрочно прекратили лечение исследования с более высокой процентной долей в группе ликсисенатида (16 субъектов: 10,1%) по сравнению с группой ситаглиптина (11 субъектов: 6,8%). Для группы ликсисенатида, основной причиной прекращения лечения было «другие причины» (7 субъектов: 4,4% против 5 субъектов: 3,1% для ситаглиптина), за которым следовало «нежелательные события» (4 субъекта: 2,5% против 5 субъектов: 3,1% для ситаглиптина). Время до наступления прекращения лечения по любой причине отображено на фиг.11. Более высокая норма прекращения лечения наблюдалась в группе ликсисенатида на всем протяжении исследования.
Распределение субъектов - Рандомизированная популяция
6.1.3 Демографические данные и исходные характеристики
Демографические и исходные характеристики субъектов были в целом одинаковыми между двумя лечебными группами для выборки для оценки безопасности (Таблица 3), однако при этом в группе ликсисенатида было больше субъектов-женщин (103 [65,2%]) по сравнению с группой ситаглиптина (88 [54,7%]). Средний возраст составлял 44,0 года. Популяцию исследования составляли главным образом представители европеоидной расы (81,2%).
Демографические данные и характеристики субъектов при скрининге или на исходном уровне - Выборка для оценки безопасности
(N=161)
В таблице 4 описаны характеристики заболевания, включая историю заболевания сахарным диабетом. В популяции исследования, средняя продолжительность диабета составляла 3,31 года, а средний возраст при наступлении диабета составлял 40 лет, с аналогичными величинами в обеих лечебных группах. Числа немного отличались для следующего: более высокая средняя суточная доза метформина на исходном уровне в группе ликсисенатида (2000 мг против 1700 мг для ситаглиптина), больше женщин с анамнезом гестационного диабета (12,6% против 6,8% для ситаглиптина) и больше субъектов с макроальбуминурией при рандомизации (4,6% против 1,9% для ситаглиптина), но меньше субъектов с диабетической сенсорной или двигательной нейропатией в группе ликсисенатида (12,2% против 17,5% для ситаглиптина) и меньше субъектов с микроальбуминурией при рандомизации (17,8% против 26,4% для ситаглиптина).
Характеристики заболевания при скрининге или на исходном уровне - Выборка для оценки безопасности
(N=158)
(N=161)
Значение клиренса креатинина получено с использованием Формулы Кокрофта-Гаулта.
Соотношение Альбумин/креатинин выражено в мкг/мг креатинина, эквивалентно мг/г креатинина, а коэффициент пересчета в стандартные международные единицы мг/ммоль креатинина составляет 0,1130.
HbA1c, PPG через 2 часа, и FPG были сравнимы между двумя лечебными группами для выборки для оценки безопасности (5). Средний HbA1c на исходном уровне составлял 8,12%. Более низкая средняя масса тела на исходном уровне наблюдалась в группе ликсисенатида (98,51 кг) по сравнению с группой ситаглиптина (100,56 кг), но средний исходный BMI для двух лечебных групп был одинаковым (36,76 кг/м2), как показано в Таблице 3.
Исходные показатели эффективности - Выборка для оценки безопасности
FPG = Глюкоза в плазме натощак.
Колебание глюкозы = глюкоза в плазме через 2 часа после приема пищи - глюкоза в плазме за 30 минут перед тестовым приемом пищи перед введением лекарственного средства исследования.
6.1.4 Дозировка и продолжительность
Средняя продолжительность лечения была немного короче для группы ликсисенатида по сравнению с группой ситаглиптина: 160,2 дней (22,9 недели) воздействия ликсисенатидом для группы ликсисенатида (Таблица 6) и 164,8 дней (23,5 недели) воздействия ситаглиптином для группы ситаглиптина (Таблица 7). Подавляющее большинство субъектов в обеих лечебных группах имели по меньшей мере 85 дней лечения (90,5% и 96,3% в группах ликсисенатида и ситаглиптина, соответственно). Пять субъектов (4 для ликсисенатида и 1 для ситаглиптина) не зарегистрировали дату последнего введения на страницах «Конец лечения» и «Исследуемый препарат введение» CRF, следовательно, следуя SAP конвенции по обработке данных, их продолжительности воздействия были установлены как отсутствующие.
В конце двойного слепого лечения, количественное соотношение субъектов, которые достигли намеченной суточной дозы ликсисенатида (активного лекарственного средства или совпадающего по объему плацебо), равной 20 мкг, было ниже в группе ликсисенатида (94,9%) по сравнению с группой ситаглиптина (100%) (Таблица 8).
Воздействие ликсисенатидом (или плацебо ликсисенатида) - Выборка для оценки безопасности
Воздействие ситаглиптина (или плацебо ситаглиптина) - Выборка для оценки безопасности
(N=161)
Количество (%) субъектов по заключительной дозе ликсисенатида в конце двойного слепого лечения - Выборка для оценки безопасности
(N=158)
(N=161)
Примечание: Процентные доли рассчитаны с использованием в качестве знаменателя количества субъектов для оценки безопасности.
6.2 Эффективность
6.2.1 Основной конечный показатель эффективности
Основной анализ
Таблица 9 приводит результаты основного показателя эффективности, процентной доли субъектов с HbA1c<7% на 24 неделе и потери массы, составляющей по меньшей мере 5% исходной массы тела, на 24 неделе с использованием метода CMH.
Предварительно утвержденный основной анализ, основанный на методе CMH, не показал статистически значимого различия между 2 лечебными группами в процентной доле субъектов с HbA1c <7% на 24 неделе и потерей массы, составляющей по меньшей мере 5% исходной массы тела, на 24 неделе (средневзвешенное значение различия эффективности лечения против ситаглиптина = 4,6%; p-значение = 0,1696). Процентная доля субъектов, которые соответствовали критерию (эффективность лечения), была численно выше в группе ликсисенатида (12,0%), чем группа ситаглиптина (7,5%).
Количество (%) субъектов с HbA1c <7% на 24 неделе и потерей массы тела, составляющей ≥5% исходной массы тела, на 24 неделе - mITT популяция
(N=158)
bНа основании метода CMH со стратификацией по рандомизированным стратам уровня HbA1c при скрининге (<8,0 или ≥8,0%) и рандомизированным стратам уровня BMI при скрининге (<35 или ≥35 кг/м2).
Субъекты, которые не имеют значений после исходного уровня во время лечения исследуемым препаратом (HbA1c и массы тела), которые разделены интервалом не более чем 30 дней, считаются субъектами, не ответившими на лечение.
Фигура 12 иллюстрирует процентную долю субъектов с HbA1c<7% и потерей массы, составляющей по меньшей мере 5% исходной массы тела, при осмотре в течение периода лечения исследуемым препаратом.
Вспомогательный анализ
Вспомогательный анализ с использованием регрессионной логистической модели показал непротиворечивый результат с данными основного анализа для основного показателя эффективности (p-значение = 0,1160) (Таблица 10).
Количество (%) субъектов с HbA1c <7% на 24 неделе и потерей массы, составляющей ≥5% исходной массы тела, на 24 неделе - mITT популяция
Примечание: из анализа исключены измерения, полученные после введения резервного лечения и/или после прекращения лечения плюс 3 дня.
Субъекты, которые не имеют значений после исходного уровня во время лечения исследуемым препаратом (HbA1c и массы тела), которые разделены интервалом не более чем 30 дней, считаются субъектами, не ответившими на лечение.
6.2.2 Другие ключевые конечные показатели эффективности
В данном разделе представлены анализы ANCOVA HbA1c, массы тела, PPG через 2 часа, FPG, колебания глюкозы, HOMA-IR и HOMA-β. Фигура 13, Фигура 15 и Фигура 16 иллюстрируют среднее изменение HbA1c (±стандартная ошибка) по сравнению с исходным уровнем, массы тела и FPG с течением времени во время 24-недельного периода двойного слепого лечения. Фиг.14 иллюстрирует субъектов, ответивших на лечение снижением HbA1c (≤6,5% или <7%, соответственно) при выбранных осмотрах. В таблице 20 представлена процентная доля субъектов, которые перешли на резервную терапию в течение периода двойного слепого лечения.
Для HbA1c, LS средние изменения к 24 неделе по сравнению с исходным уровнем составляли -0,66% для группы ликсисенатида и -0,72% для группы ситаглиптина, без существенного различия между лечебными группами (LS среднее различие против ситаглиптина = 0,06%, 95% CI [-0,179; 0,308]) (Таблица 11). HbA1c достигал плато после 12 недели для обеих лечебных групп (Фигура 13). В общей сложности 61 субъект (40,7%) в группе ликсисенатида имел HbA1c<7% на 24 неделе по сравнению с 64 субъектами (40,0%) в группе ситаглиптина, а 36 (24,0%) получавших лечение ликсисенатидом субъектов имели HbA1c ≤6,5% по сравнению с 42 (26,3%) получавшими лечение ситаглиптином субъектами (Таблица 12).
Результатом лечения ликсисенатидом являлось существенное снижение массы тела (LS среднее различие против ситаглиптина = -1,34 кг, 95% CI [-2,101; -0,575]) (Таблица 13). Масса тела достигала плато после 16 недели для получавших лечение ситаглиптином субъектов, тогда как она непрерывно снижалась для получавших лечение ликсисенатидом субъектов (Фигура 15). Больше получавших лечение ликсисенатидом субъектов (28 [18,4%]) имели потерю массы к 24 неделе ≥5% по сравнению с исходным уровнем, чем получавших лечение ситаглиптином субъектов (19 [11,9%]) (Таблица 14).
Результаты оценки PPG через 2 часа показали существенное улучшение к 24 неделе по сравнению с исходным уровнем в группе ликсисенатида по сравнению с группой ситаглиптина (LS среднее различие против ситаглиптина = -1,91 ммоль/л, 95% CI [-2,876; -0,941]) (Таблица 15).
LS средние изменения FPG к 24 неделе по сравнению с исходным уровнем составляли -0,45 ммоль/л для группы ликсисенатида и -0,69 ммоль/л для группы ситаглиптина, без существенного различия между лечебными группами (LS среднее различие против ситаглиптина = 0,25 ммоль/л, 95% CI [-0,254; 0,744]) (Таблица 16).
Лечение ликсисенатидом значительно уменьшало колебание глюкозы к 24 неделе по сравнению с исходным уровнем в сравнении с группой ситаглиптина (LS среднее различие против ситаглиптина = -2,13 ммоль/л, 95% CI [-2,819; -1,434]) (Таблица 17).
Для инсулинорезистентности, оцениваемой с помощью HOMA-IR, LS среднее изменение к 24 неделе по сравнению с исходным уровнем составляло -0,52 в группе ликсисенатида и -0,57 в группе ситаглиптина, без существенного различия между лечебными группами (LS среднее различие против ситаглиптина = 0,05, 95% CI [-0,823; 0,918]) (Таблица 18).
Для функции β-клеток, оцениваемой с помощью HOMA-β, LS среднее изменение к 24 неделе по сравнению с исходным уровнем составляло 17,66 в группе ликсисенатида и 17,79 в группе ситаглиптина, без существенного различия между лечебными группами (LS среднее различие против ситаглиптина = -0,13, 95% CI [-23,108; 22,842]) (Таблица 19).
Процентные доли субъектов, нуждавшихся в резервном варианте лечения, составляли 9,5% в группе ликсисенатида и 6,8% в группе ситаглиптина, без существенного различия между двумя данными группами. (Таблица 20).
Среднее изменение HbA1c (%) к 24 неделе по сравнению с исходным уровнем - mITT популяция
aМодель ковариационного анализа (ANCOVA) с лечебными группами (ликсисенатида и ситаглиптина), рандомизированными стратами уровня HbA1c при скрининге (<8,0, ≥8,0%), рандомизированными стратами уровня BMI при скрининге (<35, ≥35кг/м2) и страной в качестве фиксированных эффектов и исходным значением HbA1c в качестве ковариаты.
Примечание: из анализа исключены измерения, полученные после введения резервного лечения и/или после прекращения лечения плюс 3 дня.
Включены субъекты с измерениями как исходного уровня, так и на 24 неделе (LOCF).
Количество (%) субъектов со значением HbA1c ≤6,5% или <7%, соответственно, на 24 неделе - mITT популяция
(N=158)
(N=161)
Среднее изменение массы тела (кг) к 24 неделе по сравнению с исходным уровнем - mITT популяция
(N=158)
a Модель ковариационного анализа (ANCOVA) с лечебными группами (ликсисенатида и ситаглиптина), рандомизированными стратами уровня HbA1c при скрининге (<8,0, ≥8,0%), рандомизированными стратами уровня BMI при скрининге (<35, ≥35кг/м2) и страной в качестве фиксированных эффектов и исходной массой тела в качестве ковариаты.
Примечание: из анализа исключены измерения, полученные после введения резервного лечения и/или после прекращения лечения плюс 3 дня.
Включены субъекты с измерениями как исходного уровня, так и на 24 неделе (LOCF).
Количество (%) субъектов с потерей массы тела >=5% к 24 неделе по сравнению с исходным уровнем - mITT популяция
(N=158)
(N=161)
Включены субъекты с измерениями как исходного уровня, так и на 24 неделе (LOCF).
Среднее изменение глюкозы в плазме через 2 часа после приема пищи (ммоль/л) к 24 неделе по сравнению с исходным уровнем - mITT популяция
(N=158)
a Модель ковариационного анализа (ANCOVA) с лечебными группами (ликсисенатида и ситаглиптина), рандомизированными стратами уровня HbA1c при скрининге (<8,0, ≥8,0%), рандомизированными стратами уровня BMI при скрининге (<35, ≥35 кг/м2) и страной в качестве фиксированных эффектов и значением исходного уровня глюкозы в плазме через 2 часа после приема пищи в качестве ковариаты.
Примечание: из анализа исключены измерения, полученные после введения резервного лечения и/или после прекращения лечения.
Включены субъекты с измерениями как исходного уровня, так и на 24 неделе (LOCF).
Среднее изменение глюкозы в плазме натощак (ммоль/л) к 24 неделе по сравнению с исходным уровнем - mITT популяция
aМодель ковариационного анализа (ANCOVA) с лечебными группами (ликсисенатида и ситаглиптина), рандомизированными стратами уровня HbA1c при скрининге (<8,0, ≥8,0%), рандомизированными стратами уровня BMI при скрининге (<35, ≥35 кг/м2) и страной в качестве фиксированных эффектов и значением исходного уровня глюкозы в плазме натощак в качестве ковариаты.
Примечание: из анализа исключены измерения, полученные после введения резервного лечения и/или после прекращения лечения плюс 1 день.
Включены субъекты с измерениями как исходного уровня, так и на 24 неделе (LOCF).
Среднее изменение колебания глюкозы (ммоль/л) к 24 неделе по сравнению с исходным уровнем - mITT популяция
aМодель ковариационного анализа (ANCOVA) с лечебными группами (ликсисенатида и ситаглиптина), рандомизированными стратами уровня HbA1c при скрининге (<8,0, ≥8,0%), рандомизированными стратами уровня BMI при скрининге (<35, ≥35 кг/м2) и страной в качестве фиксированных эффектов и значением исходного уровня колебания глюкозы в качестве ковариаты.
Включены субъекты с измерениями как исходного уровня, так и на 24 неделе (LOCF).
Среднее изменение HOMA-IR к 24 неделе по сравнению с исходным уровнем - mITT популяция
aМодель ковариационного анализа (ANCOVA) с лечебными группами (ликсисенатида и ситаглиптина), рандомизированными стратами уровня HbA1c при скрининге (<8,0, ≥8,0%), рандомизированными стратами уровня BMI при скрининге (<35, ≥35 кг/м2) и страной в качестве фиксированных эффектов и значением исходного уровня HOMA-IR в качестве ковариаты.
Примечание: из анализа исключены измерения, полученные после введения резервного лечения и/или после прекращения лечения.
Включены субъекты с измерениями как исходного уровня, так и на 24 неделе (LOCF).
Среднее изменение HOMA-бетта к 24 неделе по сравнению с исходным уровнем - mITT популяция
a Модель ковариационного анализа (ANCOVA) с лечебными группами (ликсисенатида и ситаглиптина), рандомизированными стратами уровня HbA1c при скрининге (<8,0, ≥8,0%), рандомизированными стратами уровня BMI при скрининге (<35, ≥35 кг/м2) и страной в качестве фиксированных эффектов и значение исходного уровня HOMA-β в качестве ковариаты.
Примечание: из анализа исключены измерения, полученные после введения резервного лечения и/или после прекращения лечения.
Включены субъекты с измерениями как исходного уровня, так и на 24 неделе (LOCF).
Количество (%) субъектов, нуждавшихся в резервном варианте лечения в течение периода двойного слепого лечения - mITT популяция
(N=158)
(N=161)
6.3 Безопасность
В Таблице 21 предоставлен обзор нежелательных явлений, наблюдаемых в течение периода лечения исследуемым препаратом. Количественное соотношение субъектов, которые испытывали TEAE было немного выше для группы ликсисенатида против группы ситаглиптина (63,9% для ликсисенатида против 60,9% для ситаглиптина). Шесть субъектов (3 [1,9%] в каждой группе) имели серьезное TEAE. Не было сообщений о смертности в данном исследовании. Процентная доля субъектов с TEAE, приведшими к прекращению лечения, была аналогичной в каждой группе (2,5% для ликсисенатида и 3,1% для ситаглиптина). Таблица 22 и Таблица 23 приводят серьезные TEAE и TEAE, приведшие к прекращению лечения, по основным SOC, HLGT, HLT и РT, соответственно.
Таблица 34 в приложении приводит частоту встречаемости TEAE, произошедших по меньшей мере у 1% субъектов в любой лечебной группе. Наиболее часто сообщалось о TEAE тошнота в группе ликсисенатида (28 [17,7%] субъектов) против 11 (6,8%) получавших лечение ситаглиптином субъектов. Вторым наиболее часто упоминаемым TEAE в группе ликсисенатида была головная боль (20 [12,7%] субъектов для ликсисенатида против 15 [9,3%] субъектов для ситаглиптина), за которой следовала диарея (14 [8,9%] субъектов для ликсисенатида против 12 [7,5%] субъектов для ситаглиптина).
Обзор профиля неблагоприятных событий: возникшие после начала лечения нежелательные события - Выборка для оценки безопасности
(N=158)
(N=161)
n (%) = количество и процентная доля субъектов по меньшей мере с одним неблагоприятным событием
Количество (%) субъектов, испытавших серьезное TEAE (серьезные TEAE), представленных по основным SOC, HLGT, HLT и РT - Выборка для оценки безопасности
HLGT: Групповой термин высокого уровня
HLT: Термин высокого уровня
Предпочтительный Термин
n (%) = Количество и процентная доля субъектов по меньшей мере с одним серьезным TEAE.
Примечание: Период применения исследуемого препарата = время от первой дозы двойного слепого лечения исследования до 3 дней после введения последней дозы.
Таблица отсортирована в соответствии с международно согласованным порядком SOC и алфавитным порядком HLGT, HLT, PT.
Количество (%) субъектов, испытавших TEAE, приведшее (приведшие) к необратимому прекращению лечения по основным SOC, HLGT, HLT и РT - Выборка для оценки безопасности
HLGT: Групповой термин высокого уровня
HLT: Термин высокого уровня
Предпочтительный Термин
(N=161)
n (%) = Количество и процентная доля субъектов по меньшей мере с одним серьезным TEAE, приведшим к необратимому прекращению лечения.
Примечание: Период применения исследуемого препарата = время от первой дозы двойного слепого лечения исследования до 3 дней после введения последней дозы.
Таблица отсортирована в соответствии с международно согласованным порядком SOC и алфавитным порядком HLGT, HLT, PT.
В течение периода лечения исследуемым препаратом, 1 (0,6%) получавший лечение ликсисенатидом субъект сообщил о 2 событиях симптоматической гипогликемии, которые определены в протоколе. В течение того же самого периода, 3 (1,9%) получавших лечение ситаглиптином субъекта испытали событие симптоматической гипогликемии (Таблица 24). Ни одно из данных событий симптоматической гипогликемии не было тяжелым согласно определению протокола. Три дополнительных субъекта (1 для ликсисенатида и 2 для ситаглиптина) сообщили о гипогликемии (Таблица 34), которая не соответствовала определению протокола (ассоциированные значения глюкозы составляли ≥60 мг/дл).
Сводный обзор симптоматической гипогликемии в течение периода лечения исследуемым препаратом - Выборка для оценки безопасности
Симптоматическая гипогликемия = Симптоматическая гипогликемия, как определено по протоколу.
Примечание: Период лечения исследуемым препаратом = время от первой дозы двойного слепого лечения исследования до 3 дней после введения последней дозы.
Одиннадцать субъектов (9 [5,7%] получавших лечение ликсисенатидом субъектов и 2 [1,2%] получавших лечение ситаглиптином субъекта) испытали AE реакции в месте инъекции (Таблица 25). AE реакции в месте инъекции идентифицировали посредством поиска термина «место инъекции» либо в закодированных PT из сообщаемых исследователю терминов, либо в PT из диагноза ARAC в процессе экспертизы аллергических реакций. Интенсивность ни одной из реакций не была серьезной или тяжелой.
Количество (%) субъектов, испытавших реакции в месте инъекции в течение периода лечения исследуемым препаратом - Выборка для оценки безопасности
Предпочтительный Термин
(N=161)
Примечание: Период лечения исследуемым препаратом = время от первой дозы двойного слепого лечения исследования до 3 дней после введения последней дозы.
В общей сложности в течение периода лечения исследуемым препаратом исследователи сообщали о 5 событиях как о возможных аллергических событиях, которые послали в ARAC для экспертизы. Из них 3 события у 3 субъектов (2 [1,3%] получавших лечение ликсисенатидом субъекта и 1 [0,6%] получавший лечение ситаглиптином субъект) были расценены ARAC как аллергические реакции, но только одно событие (анафилактическая реакция) у одного получавшего лечение ликсисенатидом субъекта (#320001015) было расценено как возможно связанное с IP (Таблица 26).
Субъект # 320001015, возраста 48 лет, 48-летняя женщина в постменопаузе, с гипертонией и ожирением в анамнезе, испытала на второй день лечения, через 10 минут после введения IP, аллергическую реакцию умеренной интенсивности (у нее обнаружился генерализованный зуд, отек глаз, лица, рук и ног, сопровождающиеся стеснением в груди. При обследовании было замечено, что у нее также имеется покраснение, сыпь и отек в месте инъекции. Она приняла дексаметазон и хлорфенамин. Спустя 30 минут, без улучшения, ее поместили в реанимационное отделение для получения в/в дексаметазона, эпинефрина подкожно и кислорода трансназально. Реакция была устранена через 90 минут после данного лечения, а спустя 1 час субъект был выписан домой без осложнений. IP необратимо прекратили в день аллергической реакции. По мнению исследователя, данное AE было связано с ликсисенатидом (или его плацебо), в то же время по мнению заказчика клинического исследования, связь не была исключена. ARAC расценил данное AE как «аллергическую реакцию (анафилактическую реакцию)». При осмотре последующего наблюдения 5 дней спустя, у субъекта больше не было реакции и он чувствовал себя хорошо.
Количество (%) субъектов с событиями, расцененными ARAC как аллергические реакции в течение периода лечения исследуемым препаратом - Выборка для оценки безопасности
Примечание: Период лечения исследуемым препаратом = время от первой дозы двойного слепого лечения исследования до 3 дней после введения последней дозы.
По протоколу, любое увеличение амилазы и/или липазы более чем в два раза выше верхней границы нормального диапазона (ULN), которое было подтверждено повторным измерением, необходимо отслеживать и документально подтверждать в предварительно утвержденной форме: «форма нежелательных явлений для предполагаемого панкреатита». В течение периода применения исследуемого препарата, данная форма была заполнена для 6 (3,8%) получавших лечение ликсисенатидом субъектов и 2 (1,2%) получавших лечение ситаглиптином субъектов (Таблица 27). Среди данных 8 субъектов, PT было повышенная амилаза крови для одного получавшего лечение ситаглиптином субъекта и повышенная липаза для 6 получавших лечение ликсисенатидом субъектов и одного получавшего лечение ситаглиптином субъекта. В процессе исследования не сообщалось ни об одном случае панкреатита.
Количество (%) субъектов со специальной формой для неблагоприятных событий для подозрения на панкреатит, заполненной в течение периода лечения исследуемым препаратом - Выборка для оценки безопасности
(N=158)
(N=161)
Примечание: Период лечения исследуемым препаратом = время от первой дозы двойного слепого лечения исследования до 3 дней после введения последней дозы.
В Таблица 28 приведены субъекты, которые имели по меньшей мере одно значение липазы или амилазы ≥3 ULN в течение периода лечения исследуемым препаратом. В общей сложности наблюдалось 5 субъектов (2 [1,3%] субъекта в группе ликсисенатида и 3 [1,9%] в группе ситаглиптина) с повышенной липазой (≥3 ULN). В течение периода лечения исследуемым препаратом ни один не имел повышенной амилазы (≥3 ULN).
Ферменты поджелудочной железы: Количество (%) субъектов по меньшей мере с одним PCSA после исходного в течение периода лечения исследуемым препаратом согласно статусу исходного уровня - Выборка для оценки безопасности
Исходный уровень
По критерию PCSA n/N1 (%)
ULN = Верхняя граница нормы.
*Независимо от исходного уровня.
Примечание: Период применения исследуемого препарата = время от первой дозы двойного слепого лечения исследования до 3 дней после введения последней дозы.
Количество (n) отображает подмножество общего количества субъектов, которые по меньшей мере один раз соответствуют интересующему критерию. Знаменателем (/N1) для каждого параметра в пределах лечебной группы является количество субъектов для лечебной группы, которые имели такой параметр, оцененный после исходного уровня по состоянию PCSA исходного уровня.
Только ухудшение самого плохого случая для каждого субъекта отображается исходным состоянием.
По протоколу, любое значение кальцитонина ≥20 пг/мл, подтвержденное повторным измерением, необходимо было отслеживать и документально подтверждать в предварительно утвержденной форме нежелательных явлений для «повышенного кальцитонина ≥20 пг/мл». В течение периода лечения исследуемым препаратом, данная форма была заполнена для 1 (0,6%) получавшего лечение ситаглиптином субъекта (Таблица 29). PT был повышенный кальцитонин в крови. Соответствующее значение кальцитонина составляло 22 нг/л.
Количество (%) субъектов со специальной формой для неблагоприятных событий для повышенного кальцитонина ≥20 пг/мл, заполненной в течение периода лечения исследуемым препаратом - Выборка для оценки безопасности
(N=158)
(N=161)
Примечание: Период лечения исследуемым препаратом = время от первой дозы двойного слепого лечения исследования до 3 дней после введения последней дозы.
В Таблице 30 приведены субъекты по меньшей мере с одним измерением кальцитонина в сыворотке в течение периода лечения исследуемым препаратом согласно 4 предварительно определенным категориям исходного уровня кальцитонина. Один (0,7%) субъект с исходным значением кальцитонина «≥20 нг/л - <50 нг/л» в группе ликсисенатида и 1 (0,6%) субъект с исходным значением кальцитонина «>ULN - <20 нг/л» в группе ситаглиптина в течение периода лечения исследуемым препаратом имели по меньшей мере одно значение кальцитонина «≥20 нг/л - <50 нг/л». Из двух субъектов, получавший лечение ситаглиптином субъект сообщал о TEAE с предварительно утвержденной AE формой (Таблица 29), тогда как получавший лечение ликсисенатидом субъект нет вследствие неподтвержденного повышения с последующим значением кальцитонина <20 нг/л.
Кальцитонин в сыворотке - Количество (%) субъектов по предварительно определенным категориям в течение периода лечения исследуемым препаратом согласно категории исходного уровня - Выборка для оценки безопасности
Статус исходного уровня
После исходного уровня
(N=158)
(N=161)
*Независимо от исходного уровня.
Примечание: Период применения исследуемого препарата = время от первой дозы двойного слепого лечения исследования до 3 дней после введения последней дозы.
Числитель представляет количество субъектов, которые были в предварительно оговоренных категориях после исходного уровня в каждой исходной категории.
Знаменателем для каждого параметра в пределах лечебной группы является количество субъектов для лечебной группы, которые имели данный параметр, оцененный после исходного по статусу исходного уровня.
Пациента считают только в самой плохой категории.
7 Приложение
Количество (%) субъектов, ответивших на лечение (субъекты с HbA1c <7% и потерей массы тела, составляющей ≥5% исходной массы тела) при осмотре - mITT популяция
Примечание: из анализа исключены измерения, полученные после введения резервного лечения и/или после прекращения лечения плюс 3 дня.
Для 24 недели (LOCF), субъекты, которые не имеют значений после исходного уровня во время лечения исследуемым препаратом (HbA1c и массы тела), которые разделены интервалом не более чем 30 дней, считаются субъектами, не ответившими на лечение.
Среднее изменение HbA1c (%) при осмотре по сравнению с исходным уровнем - mITT популяция
Временная точка
Примечание: из анализа исключены измерения, полученные после введения резервного лечения и/или после прекращения лечения плюс 3 дня.
Среднее изменение массы тела (кг) при осмотре по сравнению с исходным уровнем - mITT популяция
Временная точка
Примечание: из анализа исключены измерения, полученные после введения резервного лечения и/или после прекращения лечения плюс 3 дня.
Количество (%) субъектов, испытавших обычное TEAE (PT ≥1% в любой лечебной группе), представленное по основным SOC, HLGT, HLT и РT - Выборка для оценки безопасности
HLGT: Групповой термин высокого уровня
HLT: Термин высокого уровня
Предпочтительный Термин
n (%) = Количество и процентная доля субъектов по меньшей мере с одним TEAE.
Примечание: Таблица отсортирована в соответствии с международно согласованным порядком SOC и алфавитным порядком HLGT, HLT, PT.
Представлены только SOC по меньшей мере с одним PT ≥1% по меньшей мере в одной группе.
--->
СПИСОК ПОСЛЕДОВАТЕЛЬНОСТЕЙ
<110> Sanofi-Aventis Deustchland GmbH
<120> Фармацевтическая комбинация для ПРИМЕНЕНИЯ ДЛЯ индуцироваНИЯ потерИ
массы ТЕЛА у субъектов с диабетом 2 типа или/и для предотвращения
набора массы ТЕЛА у субъектов с диабетом 2 типа
<130> 50537P EP
<160> 2
<170> BiSSAP 1.0
<210> 1
<211> 44
<212> Белок
<213> Искусственная последовательность
<220>
<221> ИСТОЧНИК
<222> 1..44
<223> / тип молекулы = «белок»
/ замечание="desPro36Эксендин -4(1-39)-Lys6-NH2"
/ организм = “искусственная последовательность”
<400> 1
His Gly Glu Gly Thr Phe Thr Ser Asp Leu Ser Lys Gln Met Glu Glu
1 5 10 15
Glu Ala Val Arg Leu Phe Ile Glu Trp Leu Lys Asn Gly Gly Pro Ser
20 25 30
Ser Gly Ala Pro Pro Ser Lys Lys Lys Lys Lys Lys
35 40
<210> 2
<211> 39
<212> Белок
<213> Искусственная последовательность
<220>
<221> ИСТОЧНИК
<222> 1..39
<223> / тип молекулы = «белок»
/ замечание="нативная последовательность Эксендин -4"
/ организм = “искусственная последовательность”
<400> 2
His Gly Glu Gly Thr Phe Thr Ser Asp Leu Ser Lys Gln Met Glu Glu
1 5 10 15
Glu Ala Val Arg Leu Phe Ile Glu Trp Leu Lys Asn Gly Gly Pro Ser
20 25 30
Ser Gly Ala Pro Pro Pro Ser
35
<---
| название | год | авторы | номер документа |
|---|---|---|---|
| ФАРМАЦЕВТИЧЕСКАЯ КОМБИНАЦИЯ ДЛЯ ПРИМЕНЕНИЯ ПРИ ГЛИКЕМИЧЕСКОМ КОНТРОЛЕ У ПАЦИЕНТОВ С САХАРНЫМ ДИАБЕТОМ 2 ТИПА | 2012 |
|
RU2650616C2 |
| ПРЕДОТВРАЩЕНИЕ ГИПОГЛИКЕМИИ У ПАЦИЕНТОВ С САХАРНЫМ ДИАБЕТОМ 2 ТИПА | 2012 |
|
RU2572703C2 |
| ЛИКСИСЕНАТИД В КАЧЕСТВЕ ДОПОЛНИТЕЛЬНОЙ ТЕРАПИИ К БАЗАЛЬНОМУ ИНСУЛИНУ ПРИ ДИАБЕТЕ 2 ТИПА | 2012 |
|
RU2606154C2 |
| ПРОФИЛАКТИКА ГИПОГЛИКЕМИИ У ПАЦИЕНТОВ С САХАРНЫМ ДИАБЕТОМ 2 ТИПА | 2012 |
|
RU2583134C2 |
| ПРИМЕНЕНИЕ AVE0010 ДЛЯ ПРОИЗВОДСТВА ЛЕКАРСТВЕННОГО СРЕДСТВА ДЛЯ ЛЕЧЕНИЯ САХАРНОГО ДИАБЕТА 2 ТИПА | 2010 |
|
RU2546520C2 |
| ЛИКСИСЕНАТИД И МЕТФОРМИН ДЛЯ ЛЕЧЕНИЯ ДИАБЕТА ТИПА 2 | 2012 |
|
RU2623023C2 |
| ЛЕЧЕНИЕ САХАРНОГО ДИАБЕТА С ПОМОЩЬЮ СОСТАВОВ ИНСУЛИНОВ ДЛИТЕЛЬНОГО ДЕЙСТВИЯ | 2014 |
|
RU2834367C2 |
| СОСТАВ С ПОСТОЯННЫМ СООТНОШЕНИЕМ ИНСУЛИНА ГЛАРГИНА/ЛИКСИЗЕНАТИДА | 2014 |
|
RU2684398C2 |
| ЛЕКАРСТВЕННЫЙ КОМПЛЕКС ДЛЯ ЛЕЧЕНИЯ ДИАБЕТА 2 ТИПА И ДИАБЕТИЧЕСКОЙ ДИСЛИПИДЕМИИ | 2017 |
|
RU2721406C1 |
| СПОСОБЫ ИСПОЛЬЗОВАНИЯ ДИАЦЕРЕИНА В ДОПОЛНИТЕЛЬНОМ ЛЕЧЕНИИ ДИАБЕТА | 2011 |
|
RU2563988C2 |
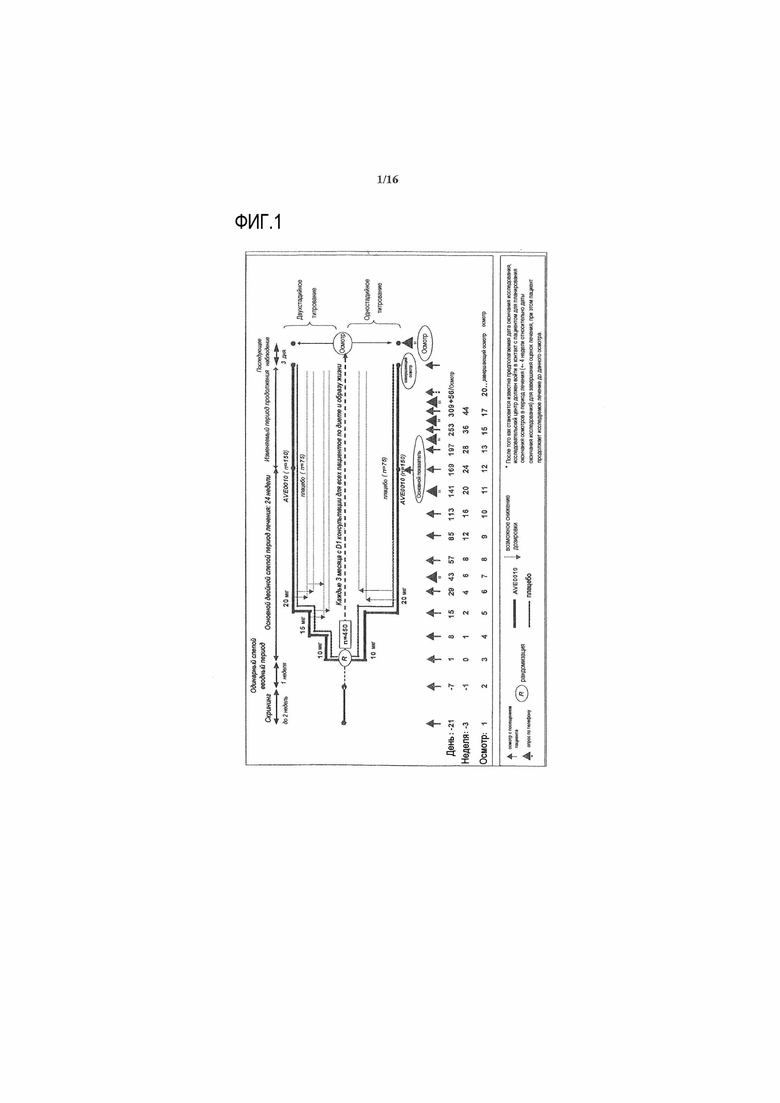
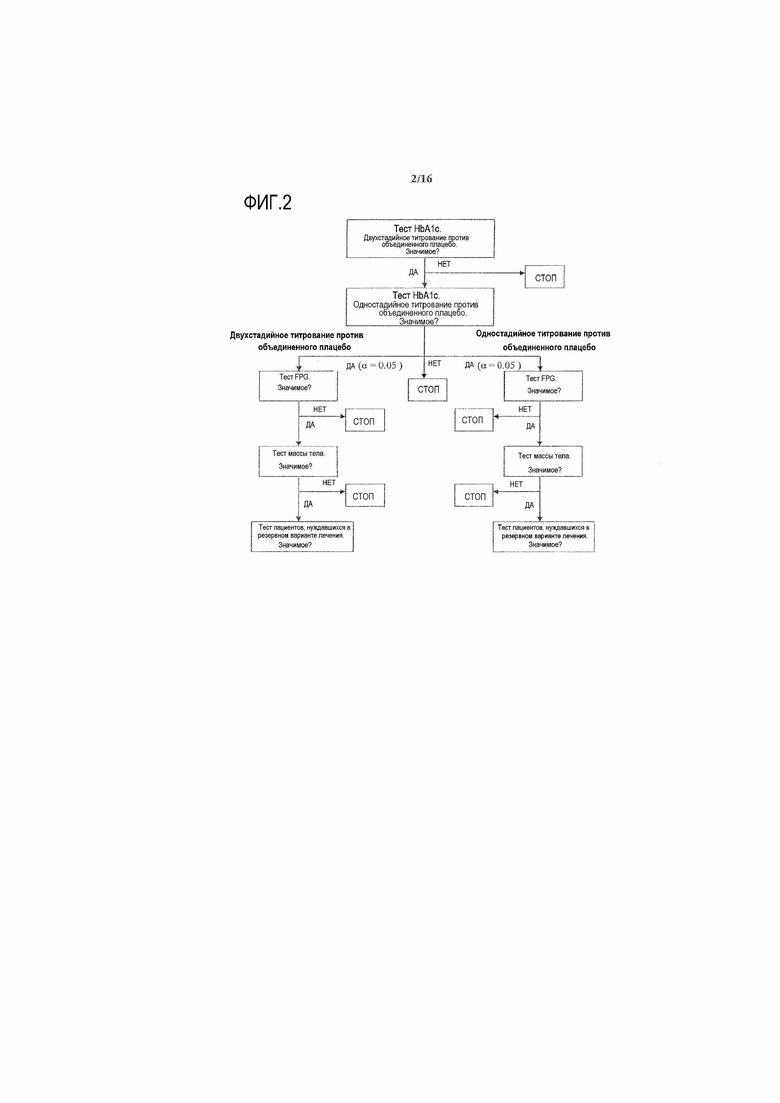

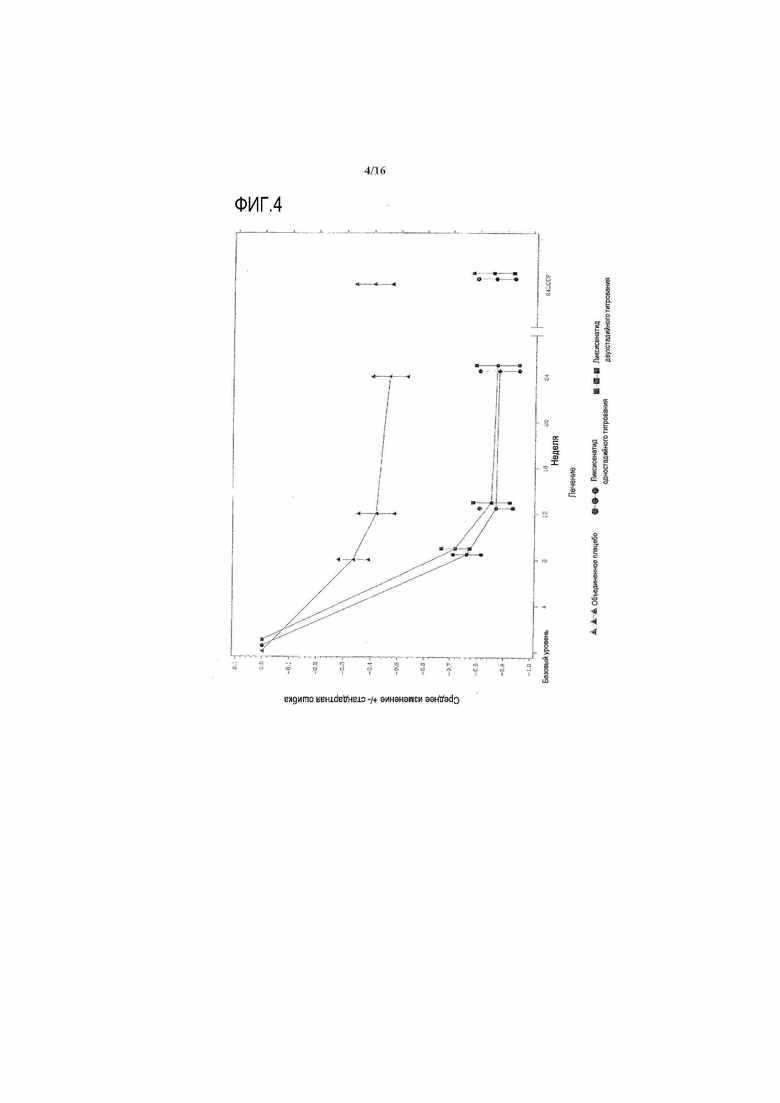
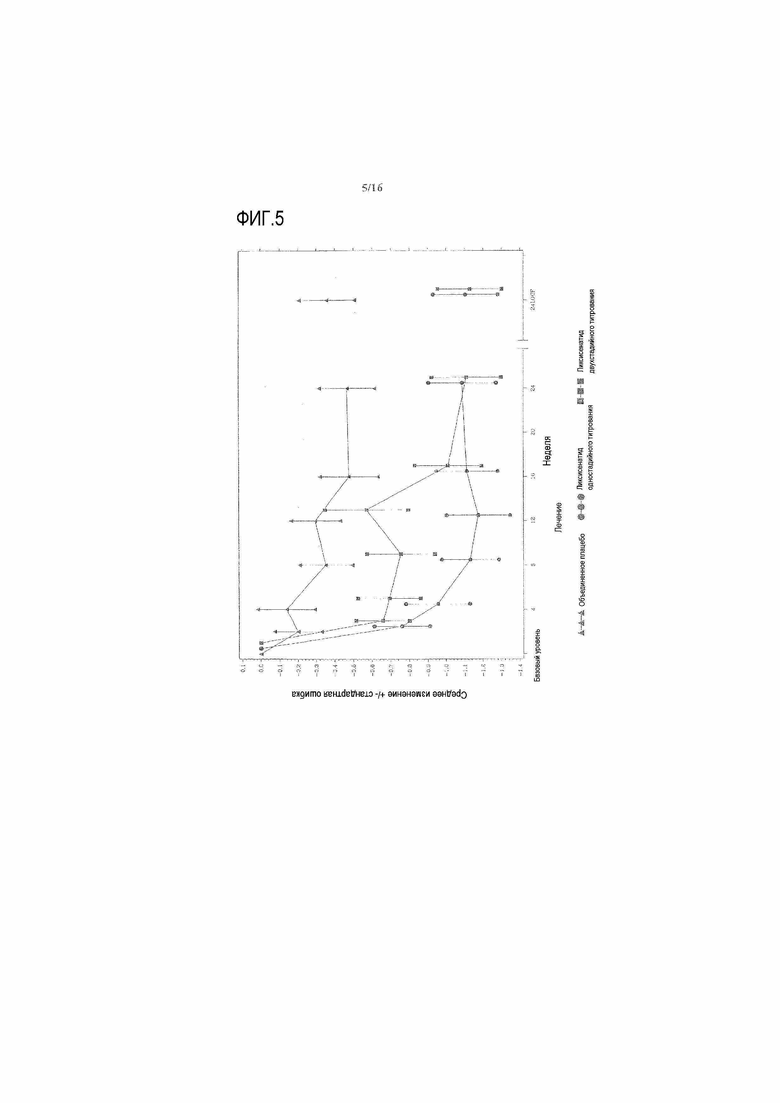
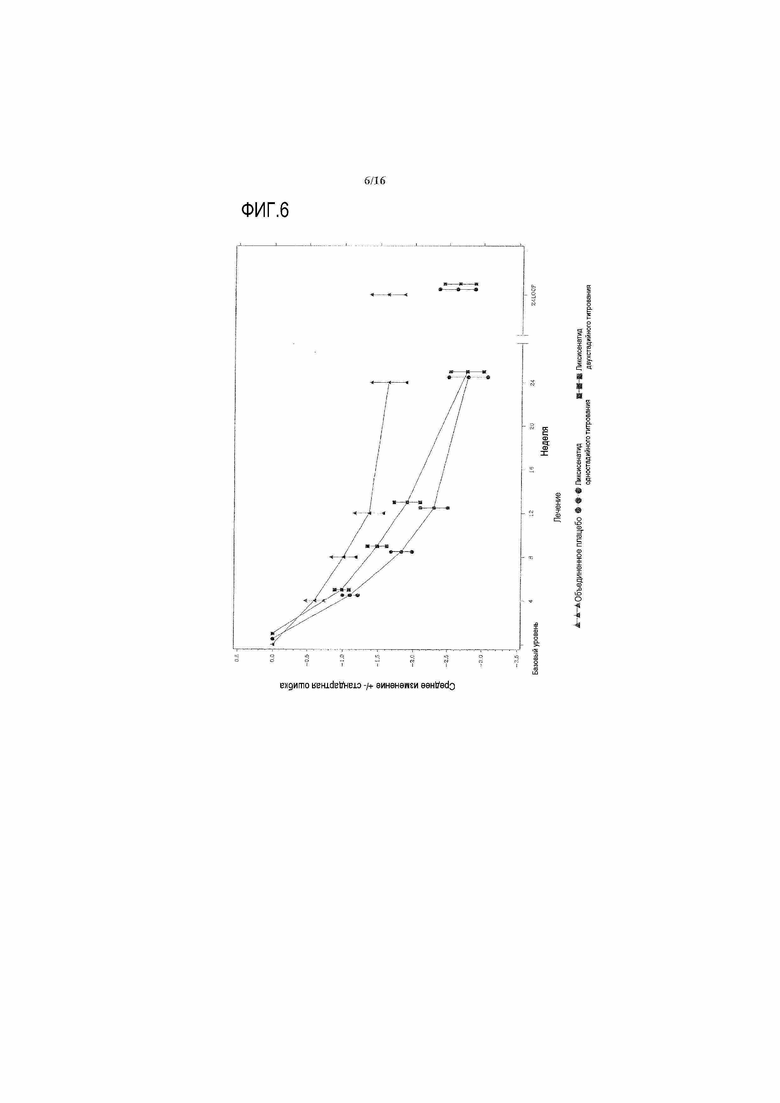
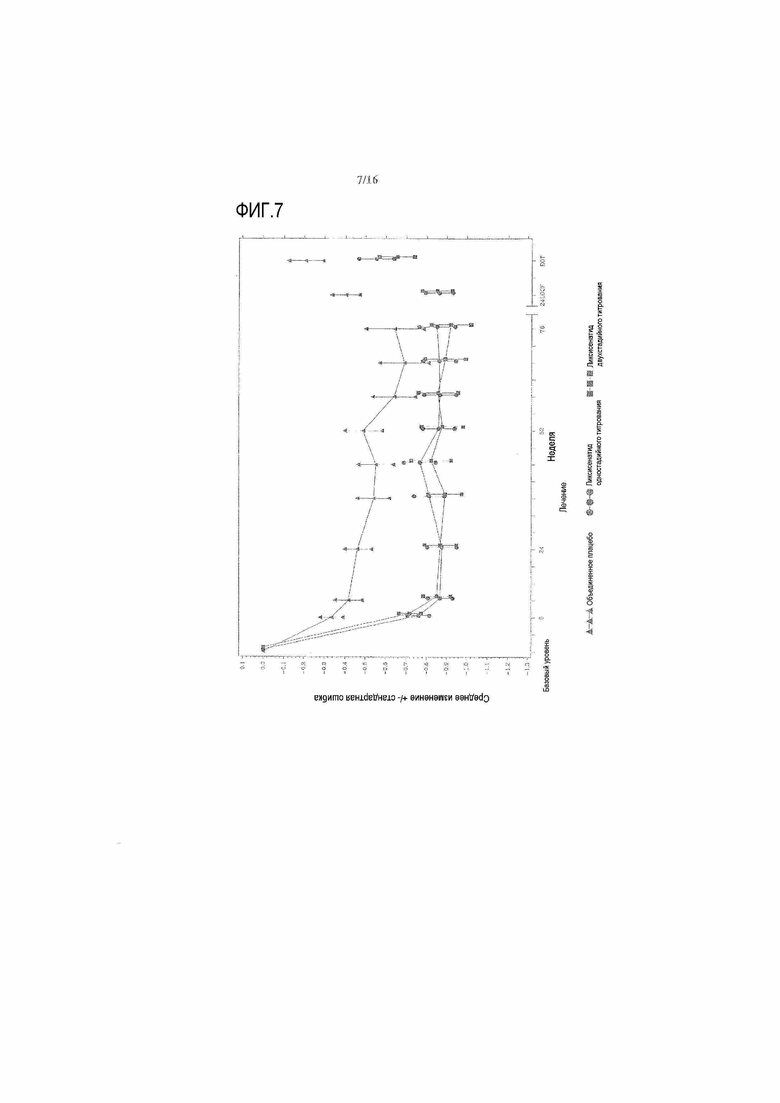

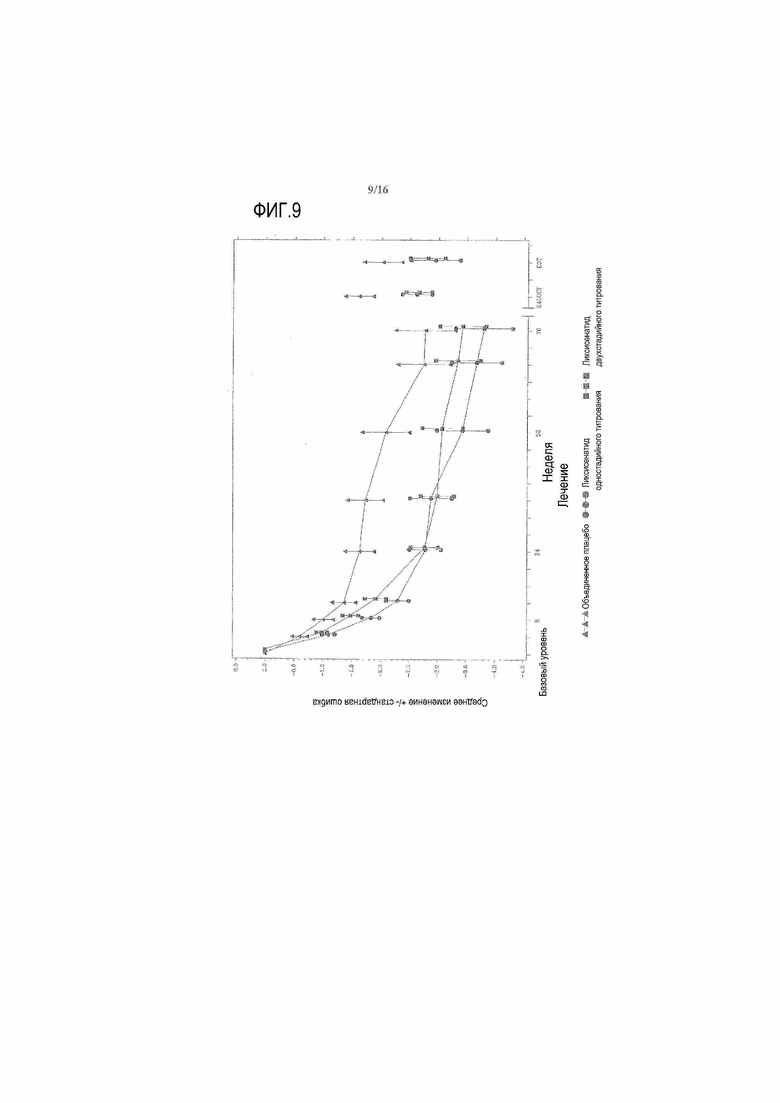
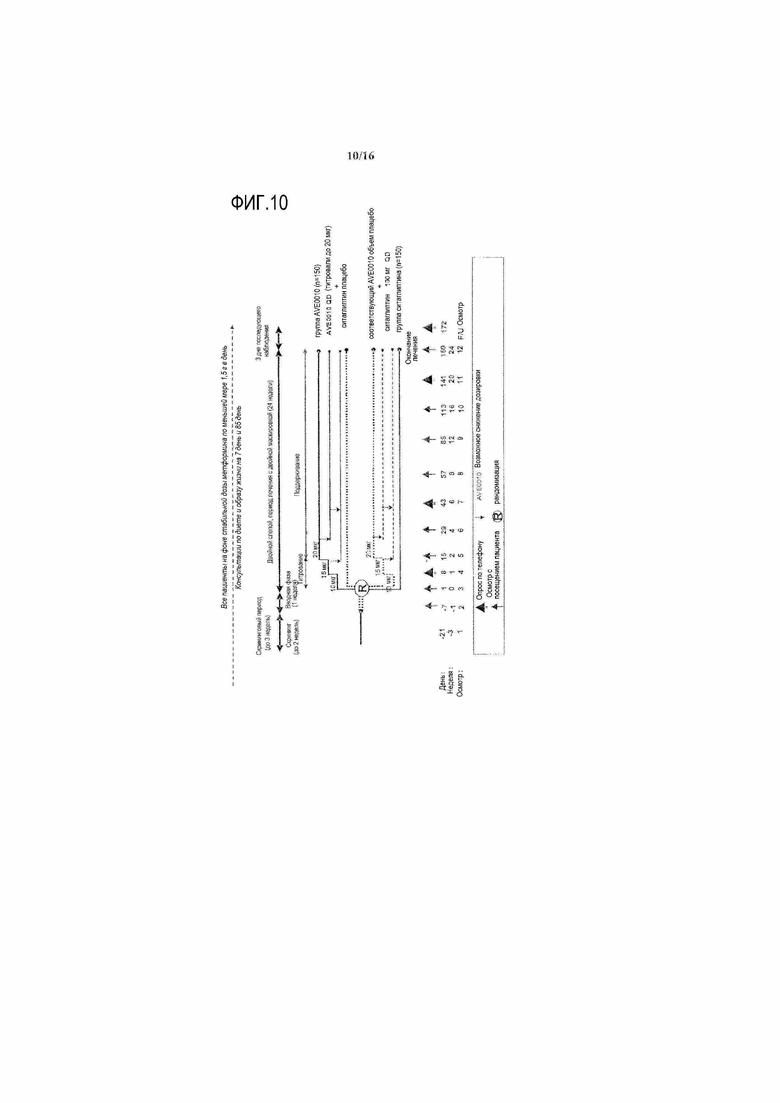
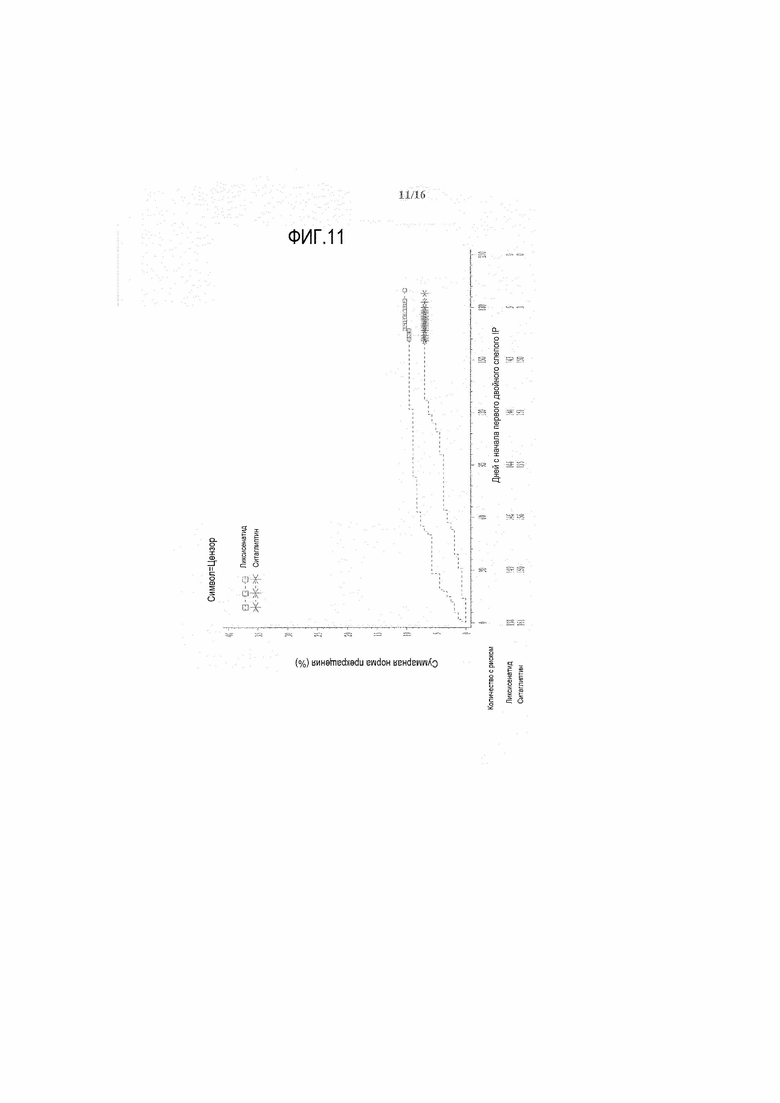
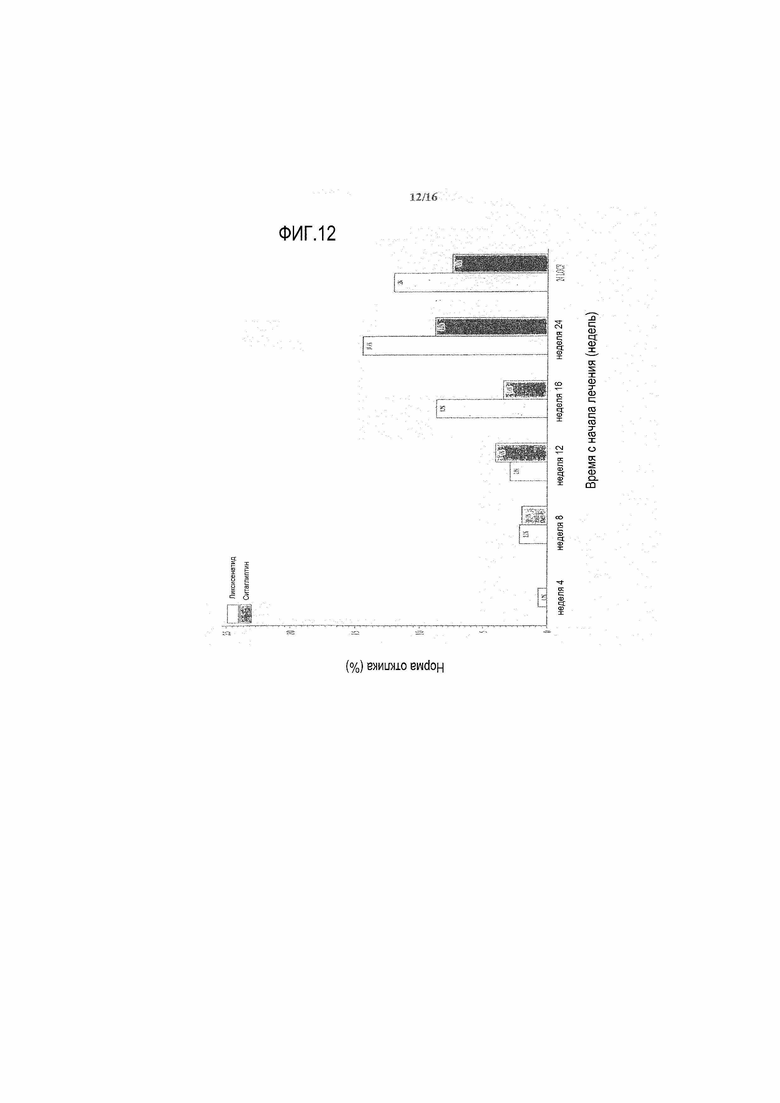

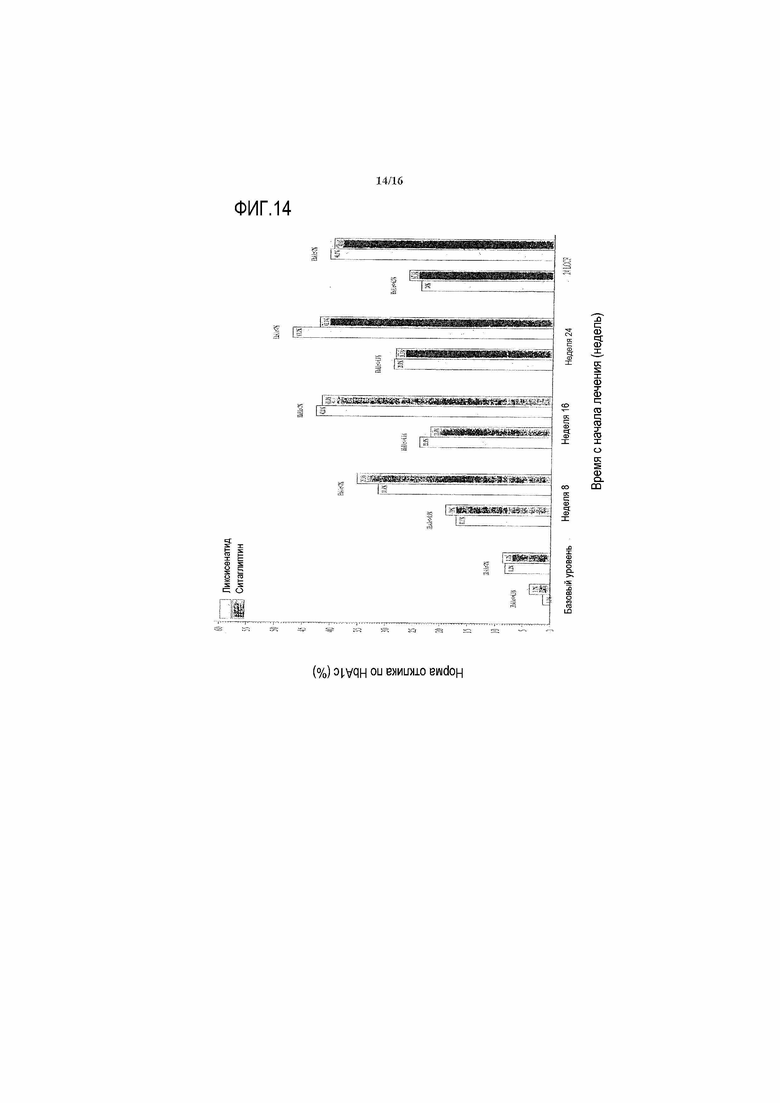
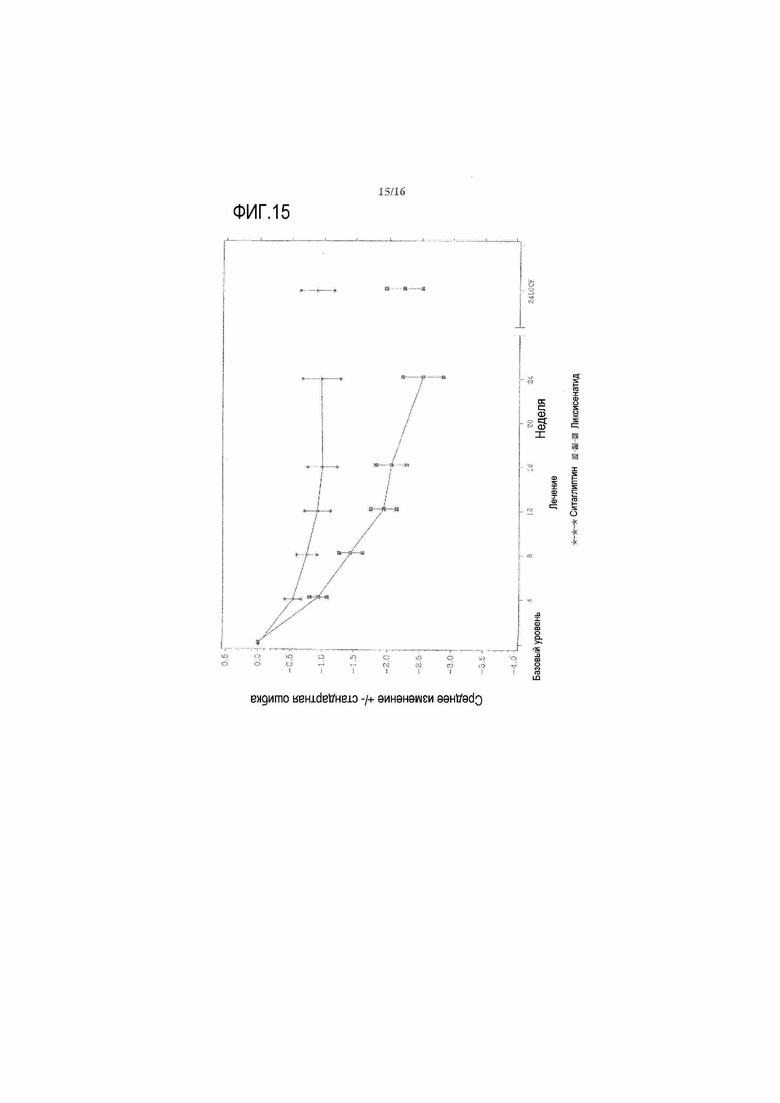
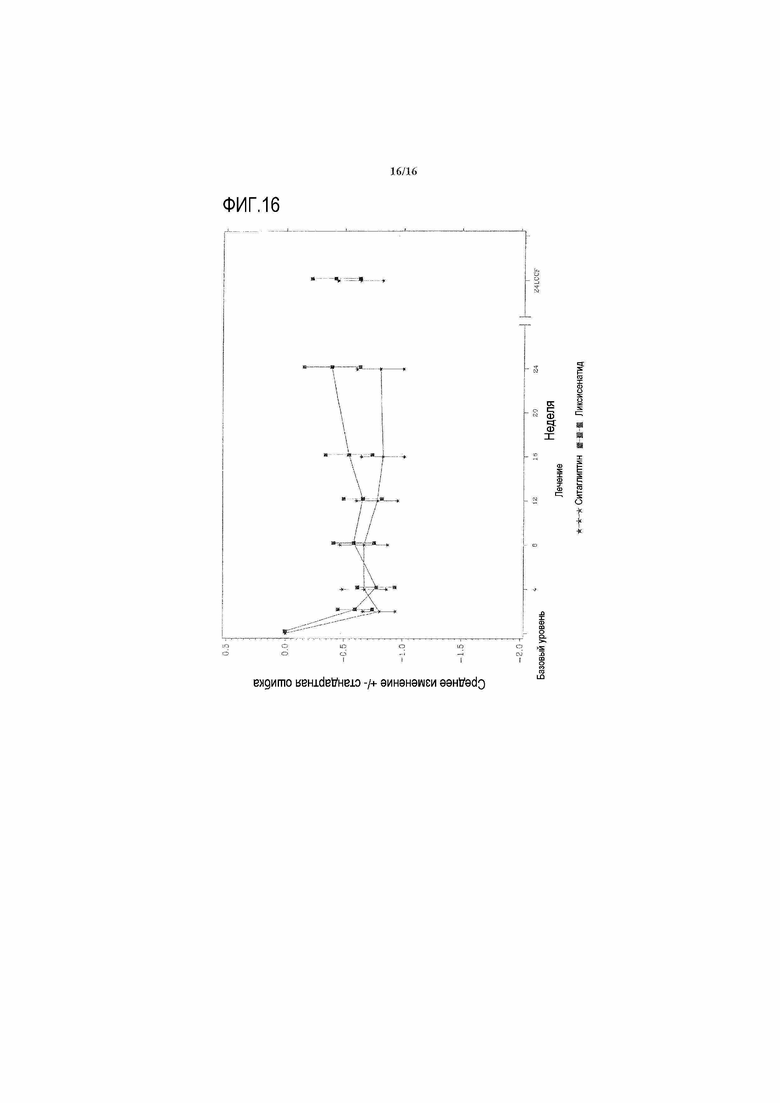
Изобретение относится к области фармацевтики, а именно к способу улучшения толерантности к глюкозе у субъектов с диабетом 2 типа, который неадекватно контролируется за счет лечения одним метформином. Способ включает парентеральное введение desPro36Эксендин-4(1-39)-Lys6-NH2 или/и его фармацевтически приемлемой соли в ежедневной дозе 20 мкг и пероральное введение метформина или/и его фармацевтически приемлемой соли в дозе от 1,0 до 1,5 г/день субъекту моложе 50 лет, который страдает ожирением, имеет концентрацию глюкозы в плазме через 2 часа после приема пищи по меньшей мере 14 ммоль/л и значение HbA1c по меньшей мере 8%, имеет колебание глюкозы по меньшей мере 4 ммоль/л. Изобретение обеспечивает улучшение толерантности к глюкозе, снижение HbA1c, массы тела у определенной группы пациентов с диабетом 2 типа, который неадекватно контролируется за счет лечения одним метформином, имеющих значение HbA1c по меньшей мере 8%, концентрацию глюкозы в плазме через 2 часа после приема пищи по меньшей мере 14 ммоль/л и колебание глюкозы по меньшей мере 4 ммоль/л. 5 з.п. ф-лы, 16 ил., 34 табл., 2 пр.
1. Способ улучшения толерантности к глюкозе у субъектов с диабетом 2 типа, включающий парентеральное введение desPro36Эксендин-4(1-39)-Lys6-NH2 или/и его фармацевтически приемлемой соли в ежедневной дозе 20 мкг и пероральное введение метформина или/и его фармацевтически приемлемой соли в дозе от 1,0 до 1,5 г/день субъекту, при этом субъект, подлежащий лечению, страдает ожирением, моложе 50 лет, имеет концентрацию глюкозы в плазме через 2 часа после приема пищи, составляющую по меньшей мере 14 ммоль/л, и значение HbA1c по меньшей мере 8%, имеет колебание глюкозы, составляющее по меньшей мере 4 ммоль/л, при этом диабет 2 типа, подлежащий лечению, неадекватно контролируется за счет лечения одним метформином.
2. Способ по п. 1, при этом субъект, подлежащий лечению, дополнительно характеризуется тем, что имеет колебание глюкозы, составляющее по меньшей мере 5 ммоль/л, при этом колебанием глюкозы является разница концентрации глюкозы в плазме через 2 часа после приема пищи и концентрации глюкозы в плазме за 30 минут перед тестовым приемом пищи.
3. Способ по п. 1 или 2, при этом субъект, подлежащий лечению, имеет индекс массы тела, составляющий по меньшей мере 30 кг/м2.
4. Способ по п. 1 или 2, при этом субъектом, подлежащим лечению, является взрослый человек.
5. Способ по п. 1 или 2, при этом у субъекта, подлежащего лечению, сахарный диабет 2 типа был диагностирован по меньшей мере за 1 год или по меньшей мере за 2 года перед началом терапии.
6. Способ по п. 1 или 2, при этом субъект, подлежащий лечению, дополнительно характеризуется тем, что имеет концентрацию глюкозы в плазме натощак, равную по меньшей мере 8 ммоль/л.
| RATNER R | |||
| E | |||
| et al | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Приспособление для суммирования отрезков прямых линий | 1923 |
|
SU2010A1 |
| - V | |||
| Прибор с двумя призмами | 1917 |
|
SU27A1 |
| - N | |||
| Разборный с внутренней печью кипятильник | 1922 |
|
SU9A1 |
| - P | |||
| АСИНХРОННЫЙ ДВИГАТЕЛЬ С КОРОТКОЗАМКНУТЫМ РОТОРОМ, ПУСКАЕМЫЙ В ХОД БЕЗ РЕОСТАТА | 1923 |
|
SU1024A1 |
| CHRISTENSEN M | |||
| et al | |||
| Lixisenatide, a novel GLP-1 receptor | |||

