Объектом данного изобретения является фармацевтическая комбинация для применения при гликемическом контроле у пациентов с диабетом 2 типа, при этом указанная комбинация содержит (a) desPro36Эксендин-4(1-39)-Lys6-NH2 (AVE0010, ликсисенатид) и/или его фармацевтически приемлемую соль, (b) базальный инсулин и/или его фармацевтически приемлемую соль и (c) необязательно, метформин и/или его фармацевтически приемлемую соль. Дополнительным объектом данного изобретения является фармацевтическая комбинация для применения с целью предотвращения гипогликемии, при этом указанная комбинация содержит (a) desPro36Эксендин-4(1-39)-Lys6-NH2 и/или его фармацевтически приемлемую соль, (b) базальный инсулин и/или его фармацевтически приемлемую соль и (c) необязательно, метформин и/или его фармацевтически приемлемую соль. Еще одним объектом представленного изобретения является фармацевтическая комбинация для применения с целью индуцирования снижения массы тела у пациентов с диабетом 2 типа и/или для предотвращения увеличения массы тела у пациентов с диабетом 2 типа, при этом указанная комбинация содержит (a) desPro36Эксендин-4(1-39)-Lys6-NH2 и/или его фармацевтически приемлемую соль, (b) базальный инсулин и/или его фармацевтически приемлемую соль и (c) необязательно, метформин и/или его фармацевтически приемлемую соль.
У здорового человека высвобождение инсулина поджелудочной железой строго связано с концентрацией глюкозы крови. Повышенный уровень глюкозы крови, который возникает после приемов пищи, быстро уравновешивается соответствующим увеличением секреции инсулина. В состоянии натощак уровень инсулина в плазме понижается до базального значения, которое является достаточным, чтобы обеспечивать непрерывное снабжение глюкозой чувствительных к инсулину органов и тканей, а по ночам поддерживать выработку глюкозы печенью на низком уровне.
В отличие от диабета 1 типа, при диабете 2 типа в целом нет недостатка инсулина, но во многих случаях, в частности в прогрессирующих случаях, лечение инсулином считается наиболее подходящей терапией, при необходимости, в комбинации с вводимыми перорально противодиабетическими лекарственными средствами.
Повышенный уровень глюкозы в крови в течение нескольких лет без первоначальных симптомов представляет собой значительный риск для здоровья. Посредством крупномасштабного исследования DCCT в США (The Diabetes Control and Complications Trial Research Group (1993) N. Engl. J. Med. 329, 977-986) может быть четко продемонстрировано, что хронически повышенные уровни глюкозы крови являются основной причиной для развития осложнений диабета. Примерами осложнений диабета являются микро- и макрососудистые повреждения, которые, возможно, проявляются ретинопатиями, нефропатиями или нейропатиями и приводят к слепоте, почечной недостаточности и потере конечностей и сопровождаются повышенным риском сердечно-сосудистых заболеваний. Таким образом, можно заключить, что улучшенная терапия диабета в первую очередь должна быть нацелена на сохранение глюкозы крови как можно ближе к физиологическому диапазону.
Особый риск существует для пациентов с избыточной массой тела, страдающих диабетом 2 типа, например, пациентов с индексом массы тела (BMI) ≥30. У данных пациентов риск диабета в сочетании с рисками избыточной массы тела приводит, например, к увеличению сердечно-сосудистых заболеваний по сравнению с пациентами с диабетом 2 типа, имеющими нормальную массу тела. Соответственно, особенно необходимо лечить диабет у данных пациентов, уменьшая при этом избыточную массу тела.
Метформин представляет собой гипогликемическое средство бигуанид, применяемое при лечении инсулинонезависимого сахарного диабета (сахарного диабета 2 типа), не отвечающего на модификацию диеты. Метформин улучшает гликемический контроль за счет улучшения чувствительности к инсулину и уменьшения кишечной абсорбции глюкозы. Метформин, как правило, вводят перорально. Однако контроль за сахарным диабетом 2 типа у страдающих ожирением пациентов с помощью метформина может быть недостаточным. Соответственно, для данных пациентов могут потребоваться дополнительные меры для контроля за сахарным диабетом 2 типа.
Инсулин представляет собой полипептид, имеющий 51 аминокислотный остаток. Инсулин состоит из цепи A, имеющей 21 аминокислотный остаток, и цепи B, имеющей 30 аминокислотных остатков. Цепи соединены 2 дисульфидными мостиками. Для лечения сахарного диабета 1 и 2 типа на протяжении длительного времени применяют препараты инсулина. В последнее время используются производные инсулина и аналоги инсулина.
Соединение desPro36Эксендин-4(1-39)-Lys6-NH2 (AVE0010, ликсисенатид) представляет собой производное Эксендина-4. В WO 01/04156 AVE0010 раскрыт, как SEQ ID NO:93:
SEQ ID NO: 1: AVE0010 (44 AS)
H-G-E-G-T-F-T-S-D-L-S-K-Q-M-E-E-E-A-V-R-L-F-I-E-W-L-K-N-G-G-P-S-S-G-A-P-P-S-K-K-K-K-K-K-NH2
SEQ ID NO: 2: Эксендин-4 (39 AS)
H-G-E-G-T-F-T-S-D-L-S-K-Q-M-E-E-E-A-V-R-L-F-I-E-W-L-K-N-G-G-P-S-S-G-A-P-P-P-S- NH2
Эксендины представляют собой группу пептидов, которые могут понижать концентрацию глюкозы в крови. Аналог эксендина AVE0010 отличается отсечением С-конца последовательности исходного Эксендина-4. AVE0010 содержит шесть С-концевых лизиновых остатков, не присутствующих в Эксендине-4.
В контексте представленного изобретения AVE0010 включает его фармацевтически приемлемые соли. Квалифицированному специалисту в данной области известны фармацевтически приемлемые соли AVE0010. Предпочтительной фармацевтически приемлемой солью AVE0010, используемого в представленном изоретении, является ацетат.
В примере представленного изобретения было продемонстрировано, что AVE0010 (Ликсисенатид) в дополнительной терапии к базальному инсулину и, необязательно, метформину значительно улучшал гликемический контроль и снижал массу:
- HbA1c значительно снижался;
- концентрация глюкозы в плазме после приема пищи с ликсисенатидом значительно улучшалась;
- стимулировалась значительная потеря массы;
- можно наблюдать значительное снижение ежедневной дозы базального инсулина;
- не наблюдалось значительного увеличения заболеваемости гипогликемией.
Первым аспектом представленного изобретения является фармацевтическая комбинация для применения при гликемическом контроле у пациентов с диабетом 2 типа, при этом указанная комбинация содержит
(а) desPro36Эксендин-4(1-39)-Lys6-NH2 и/или его фармацевтически приемлемую соль,
(b) базальный инсулин и/или его фармацевтически приемлемую соль и
(с) необязательно, метформин и/или его фармацевтически приемлемую соль.
Как продемонстрировано Примером, раскрытым в данном описании, комбинация, которая раскрыта в данном описании, может применяться для улучшения гликемического контроля. В представленном изобретении «улучшение гликемического контроля» или «гликемический контроль» относится, в частности, к улучшению концентрации глюкозы в плазме после приема пищи, улучшению концентрации глюкозы в плазме натощак и/или улучшению значения HbA1c.
Метформин представляет собой международное непатентованное название 1,1-диметилбигуанида (CAS-номер657-24-9). В представленном изобретении термин «метформин» включает его любую фармацевтически приемлемую соль.
В представленном изобретении метформин можно вводить перорально. Квалифицированному специалисту известны препараты метформина, подходящие для лечения диабета 2 типа посредством перорального введения. Метформин можно вводить нуждающемуся в этом пациенту в количестве, достаточном, чтобы вызвать терапевтический эффект. Метформин можно вводить в дозе, составляющей по меньшей мере 1,0 г/день или по меньшей мере 1,5 г/день. Для перорального введения метформин может быть приготовлен в виде твердой лекарственной формы, такой как таблетка или пилюля. Метформин может быть приготовлен с подходящими фармацевтически приемлемыми носителями, адъювантами и/или вспомогательными веществами.
В представленном изобретении desPro36Эксендин-4(1-39)-Lys6-NH2 и/или фармацевтически приемлемую соль можно применять в дополнительной терапии для введения базального инсулина и, необязательно, метформина.
В представленном изобретении термины «дополнительно», «дополнительное лечение» и «дополнительная терапия» относятся к лечению сахарного диабета 2 типа метформином, AVE0010 и базальным инсулином. Метформин, AVE0010 и базальный инсулин можно вводить в пределах временного интервала, равного 24 ч. Метформин, AVE0010 и базальный инсулин, каждый, можно вводить с дозировкой один раз в день. Метформин, AVE0010 и базальный инсулин можно вводить с помощью различных путей введения. Метформин можно вводить перорально, а AVE0010 и базальный инсулин можно вводить парентерально.
В представленном изобретении термины «дополнительно», «дополнительное лечение» и «дополнительная терапия» также относятся к лечению сахарного диабета 2 типа AVE0010 и базальным инсулином. AVE0010 и базальный инсулин можно вводить в пределах временного интервала, равного 24 ч. AVE0010 и базальный инсулин, каждый, можно вводить с дозировкой один раз в день. AVE0010 и базальный инсулин можно вводить парентерально.
В представленном изобретении «базальный инсулин» включает его подходящие фармацевтически приемлемые соли. В представленном изобретении может применяться любой базальный инсулин. В частности, базальный инсулин может быть выбран из инсулина Гларгин, Детемир, NPH, Ленте, Ультраленте, Новолин, Хумалог и их смесей. Смесь может содержать два различных базальных инсулина. Например, может быть использована смесь, содержащая Детемир и Гларгин, или смесь, содержащая NPH и Новолин. Предпочтительно, базальным инсулином является инсулин Гларгин (Лантус) или смесь, содержащая инсулин Гларгин.
Инсулин гларгин (Лантус) представляет собой Gly(A21)-Arg(B31)-Arg(B32)-человеческий инсулин. В представленном изобретении инсулин Гларгин включает его фармацевтически приемлемые соли.
Базальный инсулин и/или его фармацевтически приемлемую соль можно вводить парентерально, например, посредством инъекции (например, посредством внутримышечной или посредством подкожной инъекции). Известны подходящие инъекционные устройства, например, так называемые «шприцы-ручки», содержащие картридж, содержащий активный ингредиент, и инъекционную иглу. Базальный инсулин и/или его фармацевтически приемлемую соль можно вводить в подходящем количестве, например, в количестве в диапазоне, составляющем 15-80 единиц на дозу.
В представленном изобретении базальный инсулин и/или его фармацевтически приемлемую соль можно вводить в ежедневной дозе в диапазоне, составляющем 15-80 Ед. Инсулин гларгин и/или его фармацевтически приемлемую соль можно вводить один раз в день, например, посредством одной инъекции в день.
Квалифицированному специалисту известны препараты базального инсулина, содержащие подходящие фармацевтически приемлемые носители, адъюванты и/или вспомогательные вещества.
В представленном изобретении базальный инсулин и/или его фармацевтически приемлемая соль могут быть предоставлены в жидкой композиции. Квалифицированному специалисту известны жидкие композиции базальных инсулинов, подходящие для парентерального введения.
В представленном изобретении базальный инсулин и/или его фармацевтически приемлемую соль можно вводить нуждающемуся в этом пациенту в количестве, достаточном, чтобы вызвать терапевтический эффект.
Пациентом, подлежащим лечению медицинским препаратом представленного изобретения, страдающим от диабета 2 типа, может быть пациент, страдающий от диабета 2 типа, у которого диабет 2 типа не управляется должным образом посредством лечения одним базальным инсулином и, необязательно, метформином, например, дозой, составляющей 15-80 Ед инсулина/день в течение 3 месяцев, и, необязательно, дозой, составляющей по меньшей мере 1,0 г метформина/день или по меньшей мере 1,5 г метформина/день в течение 3 месяцев. В представленном изобретении пациент, диабет 2 типа которого не управляется должным образом, может иметь значение HbA1c в диапазоне, составляющем от 7% до 10%.
Пациентом, подлежащим лечению медицинским препаратом представленного изобретения, страдающим от диабета 2 типа, может быть страдающий ожирением пациент. В представленном изобретении страдающий ожирением пациент может иметь индекс массы тела, равный по меньшей мере 30 кг/м2.
Пациент, подлежащий лечению медицинским препаратом представленного изобретения, страдающий от диабета 2 типа, может иметь нормальную массу тела. В представленном изобретении пациент, имеющий нормальную массу тела, может иметь индекс массы тела в диапазоне, составляющем от 17 кг/м2 до 25 кг/м2 или от 17 кг/м2 до <30 кг/м2.
Пациентом, подлежащим лечению медицинским препаратом представленного изобретения, может быть взрослый человек. Пациент может иметь возраст, составляющий по меньшей мере 18 лет, может иметь возраст в диапазоне, составляющем 18-80 лет, 18-50 лет, или 40-80 лет, или 50-60 лет. Пациент может быть моложе, чем 50 лет.
Пациент, подлежащий лечению медицинским препаратом представленного изобретения, может страдать от сахарного диабета 2 типа в течение по меньшей мере 1 года или по меньшей мере 2 лет. В частности, у пациента, подлежащего лечению, сахарный диабет 2 типа был диагностирован по меньшей мере за 1 год или по меньшей мере за 2 года до начала терапии медицинским препаратом представленного изобретения.
Пациент, подлежащий лечению, может иметь значение HbA1c, равное по меньшей мере приблизительно 8% или по меньшей мере приблизительно 7,5%. Пациент также может иметь значение HbA1c, составляющее от приблизительно 7 до приблизительно 10%. Пример представленного изобретения демонстрирует, что у пациентов с диабетом 2 типа лечение AVE0010 приводит к снижению значения HbA1c.
В еще одном аспекте представленного изобретения комбинация, которая раскрыта в данном описании, может применяться для улучшения значения HbA1c у пациента, страдающего диабетом 2 типа. Улучшение значения HbA1c означает, что значение HbA1c снижается ниже 6,5% или 7%, например, после лечения в течение по меньшей мере одного месяца, по меньшей мере двух месяцев или по меньшей мере трех месяцев.
В еще одном аспекте представленного изобретения комбинация, которая раскрыта в данном описании, может применяться для улучшения толерантности к глюкозе у пациента, страдающего диабетом 2 типа. Улучшение толерантности к глюкозе означает, что концентрация глюкозы в плазме после приема пищи снижается активным агентом представленного изобретения. Снижение, в частности, означает, что концентрация глюкозы в плазме достигает нормогликемических значений или по меньшей мере приближается к данным значениям.
В представленном изобретении нормогликемическими значениями являются концентрации глюкозы в крови, составляющие, в частности, 60-140 мг/дл (соответствующие 3,3-7,8 мМ/л). Данный диапазон относится, в частности, к концентрациям глюкозы в крови в состоянии натощак и в состоянии после приема пищи.
Пациент, подлежащий лечению, 2 часа после приема пищи может иметь концентрацию глюкозы в плазме, равную по меньшей мере 10 ммоль/л, по меньшей мере 12 ммоль/л или по меньшей мере 14 ммоль/л. Данные концентрации глюкозы в плазме превышают нормогликемические концентрации.
Пациент, подлежащий лечению, может иметь колебание уровня глюкозы, равное по меньшей мере 2 ммоль/л, по меньшей мере 3 ммоль/л, по меньшей мере 4 ммоль/л или по меньшей мере 5 ммоль/л. В представленном изобретении колебанием глюкозы является, в частности, разница концентрации глюкозы в плазме через 2 часа после приема пищи и концентрации глюкозы в плазме за 30 минут перед тестовым приемом пищи.
«После приема пищи» представляет собой термин, хорошо известный квалифицированному специалисту в области диабетологии. Термин «после приема пищи» описывает, в частности, фазу после приема пищи и/или воздействие глюкозы в экспериментальных условиях. У здорового человека данная фаза характеризуется увеличением и последующим снижением концентрации глюкозы в крови. Термин «после приема пищи» или «фаза после приема пищи» обычно заканчивается через 2 ч после приема пищи и/или воздействия глюкозы.
Пациент, подлежащий лечению, как раскрыто в данном описании, может иметь концентрацию глюкозы в плазме натощак, равную по меньшей мере 8 ммоль/л, по меньшей мере 8,5 ммоль/л или по меньшей мере 9 ммоль/л. Данные концентрации глюкозы в плазме превышают нормогликемические концентрации.
В еще одном аспекте представленного изобретения комбинация, которая раскрыта в данном описании, может применяться для улучшения (т.е. снижения) глюкозы в плазме натощак у пациента, страдающего диабетом 2 типа. Снижение означает, в частности, что концентрация глюкозы в плазме достигает нормогликемических значений или по меньшей мере приближается к данным значениям.
Дополнительным аспектом представленного изобретения является способ улучшения гликемического контроля у пациентов с диабетом 2 типа, при этом указанный способ включает введение нуждающемуся в этом пациенту desPro36Эксендин-4(1-39)-Lys6-NH2 и/или его фармацевтически приемлемой соли в комбинации с метформином. В частности, можно вводить комбинацию, которая раскрыта в данном описании. В способе представленного изобретения пациентом может быть пациент по определению данного описания.
Комбинация представленного изобретения может применяться при лечении одного или более медицинских показаний, раскрытых в данном описании, например, при лечении пациентов с диабетом 2 типа, или для состояний, связанных с диабетом 2 типа, например, для улучшения гликемического контроля, снижения концентрации глюкозы в плазме натощак, для улучшения амплитуды колебаний уровня глюкозы, снижения концентрации глюкозы в плазме после приема пищи, улучшения толерантности к глюкозе, улучшения значения HbA1c, для предотвращения гипогликемии, для потери массы и/или предотвращения увеличения массы.
В представленном изобретении desPro36Эксендин-4(1-39)-Lys6-NH2 и/или его фармацевтически приемлемую соль можно вводить нуждающемуся в этом пациенту в количестве, достаточном, чтобы вызвать терапевтический эффект.
В представленном изобретении desPro36Эксендин-4(1-39)-Lys6-NH2 и/или его фармацевтически приемлемая соль могут быть разработаны с подходящими фармацевтически приемлемыми носителями, адъювантами и/или вспомогательными веществами.
Соединение desPro36Эксендин-4(1-39)-Lys6-NH2 и/или его фармацевтически приемлемую соль можно вводить парентерально, например, посредством инъекции (такой как внутримышечная или подкожная инъекция). Известны подходящие инъекционные устройства, например, так называемые «шприцы-ручки», содержащие картридж, содержащий активный ингредиент, и инъекционную иглу. Соединение desPro36Эксендин-4(1-39)-Lys6-NH2 и/или его фармацевтически приемлемую соль можно вводить в подходящем количестве, например, в количестве в диапазоне, составляющем 10-15 мкг на дозу или 15-20 мкг на дозу.
В представленном изобретении desPro36Эксендин-4(1-39)-Lys6-NH2 и/или его фармацевтически приемлемую соль можно вводить с ежедневной дозой в диапазоне, составляющем 10-20 мкг, в диапазоне, составляющем 10-15 мкг, или в диапазоне, составляющем 15-20 мкг. desPro36Эксендин-4(1-39)-Lys6-NH2 и/или его фармацевтически приемлемую соль можно вводить посредством одной инъекции в день.
В представленном изобретении desPro36Эксендин-4(1-39)-Lys6-NH2 и/или его фармацевтически приемлемая соль могут быть предоставлены в жидкой композиции. Квалифицированному специалисту известны жидкие композиции AVE0010, подходящие для парентерального введения. Жидкая композиция представленного изобретения может иметь кислый или физиологический pH. Кислый pH предпочтительно находится в диапазоне pH 1-6,8, pH 3,5-6,8 или pH 3,5-5. Физиологический pH предпочтительно находится в диапазоне pH 2,5-8,5, pH 4,0-8,5 или pH 6,0-8,5. pH можно регулировать фармацевтически приемлемой разбавленной кислотой (обычно HCl) или фармацевтически приемлемым разбавленным основанием (обычно NaOH).
Жидкая композиция, содержащая desPro36Эксендин-4(1-39)-Lys6-NH2 и/или его фармацевтически приемлемую соль, может содержать подходящий консервант. Подходящий консервант может быть выбран из фенола, м-крезола, бензилового спирта и сложного эфира п-гидроксибензойной кислоты. Предпочтительным консервантом является м-крезол.
Жидкая композиция, содержащая desPro36Эксендин-4(1-39)-Lys6-NH2 и/или его фармацевтически приемлемую соль, может содержать вещество, регулирующее тоничность. Подходящее вещество, регулирующее тоничность, может быть выбрано из глицерина, лактозы, сорбита, маннита, глюкозы, NaCl, кальций- или магнийсодержащих соединений, таких как CaCl2. Концентрация глицерина, лактозы, сорбита, маннита и глюкозы может находиться в диапазоне, составляющем 100-250 мМ. Концентрация NaCl может составлять до 150 мМ. Предпочтительным веществом, регулирующим тоничность, является глицерин.
Жидкая композиция, содержащая desPro36Эксендин-4(1-39)-Lys6-NH2 и/или его фармацевтически приемлемую соль, может содержать метионин от 0,5 мкг/мл до 20 мкг/мл, предпочтительно от 1 мкг/мл до 5 мкг/мл. Предпочтительно жидкая композиция содержит L-метионин.
Еще одним аспектом представленного изобретения является фармацевтическая комбинация для применения с целью вызвать потерю массы у пациентов с диабетом 2 типа и/или для предотвращения увеличения массы у пациентов с диабетом 2 типа, при этом указанная комбинация содержит
(а) desPro36Эксендин-4(1-39)-Lys6-NH2 и/или его фармацевтически приемлемую соль,
(b) базальный инсулин и/или его фармацевтически приемлемую соль и
(с) необязательно, метформин и/или его фармацевтически приемлемую соль.
Дополнительным аспектом представленного изобретения является способ стимулирования потери массы у пациентов с диабетом 2 типа и/или предотвращения увеличения массы у пациентов с диабетом 2 типа, при этом указанный способ включает введение нуждающемуся в этом пациенту desPro36Эксендин-4(1-39)-Lys6-NH2 и/или его фармацевтически приемлемой соли в комбинации с метформином. В частности, можно вводить комбинацию, которая раскрыта в данном описании. В способе представленного изобретения пациентом может быть пациент по определению данного описания.
Еще одним аспектом представленного изобретения является фармацевтическая комбинация для применения с целью предотвращения гипогликемии у пациентов с сахарным диабетом 2 типа, при этом указанная комбинация содержит
(а) desPro36Эксендин-4(1-39)-Lys6-NH2 и/или его фармацевтически приемлемую соль,
(b) базальный инсулин и/или его фармацевтически приемлемую соль и
(с) необязательно, метформин и/или его фармацевтически приемлемую соль.
В частности, фармацевтическая комбинация применяется для предотвращения симптоматической гипогликемии и/или тяжелой симптоматической гипогликемии у пациента с сахарным диабетом 2 типа.
В представленном изобретении гипогликемия представляет собой состояние, при котором пациент с сахарным диабетом 2 типа имеет концентрацию глюкозы в плазме ниже 60 мг/дл (или ниже 3,3 ммоль/л), ниже 50 мг/дл, ниже 40 мг/дл или ниже 36 мг/дл.
С помощью способа представленного изобретения гипогликемия может быть снижена у ниже 12%, ниже 11%, ниже 10%, ниже 9%, ниже 8%, ниже 7%, ниже 6% или ниже 5% пациентов с диабетом 2 типа, получающих комбинацию ликсисенатида и/или его фармацевтически приемлемой соли, базального инсулина и/или его фармацевтически приемлемой соли и, необязательно, метформина и/или его фармацевтически приемлемой соли, которая раскрыта в данном описании.
В представленном изобретении «симптоматическая гипогликемия» представляет собой состояние, связанное с клиническим симптомом, который обусловлен гипогликемией, в котором концентрация глюкозы в плазме ниже 60 мг/дл (или ниже 3,3 ммоль/л), ниже 50 мг/дл или ниже 40 мг/дл. Клиническими симптомами могут быть, например, потоотделение, учащенное сердцебиение, чувство голода, возбужденное состояние, патологическое состояние тревоги, усталость, раздражительность, головная боль, потеря концентрации, сонливость, психические расстройства, глазные нарушения, транзиторные нарушения ощущения, транзиторные двигательные дефекты, спутанность сознания, судороги и кома. В представленном изобретении могут быть выбраны один или более клинических симптомов симптоматической гипогликемии, которые указаны в данном описании.
Симптоматическая гипогликемия может быть связана с быстрой нормализацией после перорального введения углеводов.
В представленном изобретении «тяжелая симптоматическая гипогликемия» представляет собой состояние с клиническим симптомом, который указан в данном описании, которое обусловлено гипогликемией и в котором концентрация глюкозы в плазме ниже 36 мг/дл (или ниже 2,0 ммоль/л). Тяжелая симптоматическая гипогликемия может быть связана с острым неврологическим дефицитом, обусловленным гипогликемическим событием. При тяжелой симптоматической гипогликемии пациенту может потребоваться помощь другого человека, если, например, пациент не может лечить или помогать себе сам вследствие острого неврологического дефицита. Определение тяжелой симптоматической гипогликемии может включать все эпизоды, в которых неврологический дефицит является достаточно тяжелым, чтобы препятствовать самолечению и которые поэтому, как считается, подвергают пациентов риску ранения себя или других. Острым неврологическим дефицитом может быть по меньшей мере острый неврологический дефицит, выбранный из сонливости, психических расстройств, нарушений зрения, транзиторных нарушений ощущения, транзиторных двигательных дефектов, спутанности сознания, судорог и комы.
Тяжелая симптоматическая гипогликемия может быть связана с быстрой нормализацией после перорального введения углеводов, внутривенного введения глюкозы и/или глюкагона.
Дополнительным аспектом представленного изобретения является способ предотвращения гипогликемии у пациентов с диабетом 2 типа, при этом указанный способ включает введение нуждающемуся в этом пациенту desPro36Эксендин-4(1-39)-Lys6-NH2 и/или его фармацевтически приемлемой соли, в комбинации с базальным инсулином и/или его фармацевтически приемлемой соли и, необязательно, метформином. В частности, можно вводить комбинацию, которая раскрыта в данном описании. В способе представленного изобретения пациентом может быть пациент по определению данного описания.
Еще один аспект представленного изобретения относится к применению комбинации, которая раскрыта в данном описании, для изготовления медицинского препарата для лечения медицинского показания, которое раскрыто в данном описании. Например, комбинация представленного изобретения может быть применена для изготовления медицинского препарата для лечения пациентов с диабетом 2 типа или для лечения состояний, связанных с диабетом 2 типа, например, для улучшения гликемического контроля, снижения концентрации глюкозы в плазме натощак, для улучшения амплитуды колебаний уровня глюкозы, снижения концентрации глюкозы в плазме после приема пищи, улучшения значения HbA1c и/или улучшения толерантности к глюкозе. В другом примере комбинация, которая раскрыта в данном описании, может применяться для изготовления медицинского препарата для стимулирования потери массы у пациентов с диабетом 2 типа и/или для предотвращения увеличения массы у пациентов с диабетом 2 типа. В еще одном примере комбинация, которая раскрыта в данном описании, может применяться для изготовления медицинского препарата для предотвращения гипогликемии у пациентов с диабетом 2 типа. Может быть разработан медицинский препарат, который раскрыт в данном описании. Например, медицинский препарат может содержать парентеральную готовую форму AVE0010 и/или его фармацевтически приемлемую соль, парентеральную готовую форму базального инсулина и/или его фармацевтически приемлемую соль, и необязательную пероральную готовую форму метформина и/или его фармацевтически приемлемую соль.
Изобретение дополнительно проиллюстрировано с помощью следующего примера и фигур.
Обозначения фигур
Фиг.1 - Дизайн исследования.
Фиг.2 - График Каплана-Майера периода до прекращения лечения по любой причине - Рандомизированная популяция.
Фиг.3 - График среднего изменения HbA1c (%) к 24 неделе по сравнению с исходным уровнем при осмотре и в конечной точке – mITT.
Фиг.4 - График среднего изменения среднего значения по 7 точкам самоконтролируемой глюкозы в плазме (SMPG) (ммоль/л) к 24 неделе по сравнению с исходным уровнем при осмотре и в конечной точке - mITT популяция.
Фиг.5 - График среднего изменения глюкозы в плазме натощак (ммоль/л) к 24 неделе по сравнению с исходным уровнем при осмотре и в конечной точке - mITT популяция.
Фиг.6 - График среднего изменения массы тела (кг) к 24 неделе по сравнению с исходным уровнем при осмотре и в конечной точке - mITT популяция.
Фиг.7 - График среднего изменения дозы базального инсулина (Ед) к 24 неделе по сравнению с исходным уровнем при осмотре и в конечной точке - mITT популяция.
Фиг.8 - График среднего изменения HbA1c (%) от исходного уровня при осмотре и в конечной точке - mITT популяция.
Фиг.9 - График среднего изменения глюкозы в плазме через 2 часа после приема пищи (ммоль/л) от исходного уровня при осмотре и в конечной точке - mITT популяция.
Фиг.10 - График среднего изменения среднего значения по 7 точкам самоконтролируемой глюкозы в плазме (SMPG) (ммоль/л) от исходного уровня при осмотре и в конечной точке - mITT популяция.
Фиг.11 - График среднего изменения глюкозы в плазме натощак (ммоль/л) от исходного уровня при осмотре и в конечной точке - mITT популяция.
Фиг.12 - График среднего изменения массы тела (кг) от исходного уровня при осмотре и в конечной точке - mITT популяция.
Фиг.13 - График среднего изменения дозы базального инсулина (Ед) от исходного уровня при осмотре и в конечной точке - mITT популяция.
Пример
Пример относится к рандомизированному, двойному слепому, плацебо-контролируемому, международному исследованию в двух параллельных группах для оценки эффективности и безопасности ликсисенатида по сравнению с плацебо в качестве дополнительного лечения к базальному инсулину в комбинации с метформином или без него у пациентов с диабетом 2 типа.
Пример относится к рандомизированному, двойному слепому, плацебо-контролируемому, международному исследованию в двух параллельных группах для оценки эффективности и безопасности ликсисенатида по сравнению с плацебо в качестве дополнительного лечения к базальному инсулину в комбинации с метформином или без него у пациентов с диабетом 2 типа. Приблизительная минимальная продолжительность исследования на пациента составляла 79 недель (до 3 недель скрининг + 24-недельное основное лечение + варьируемое продление + 3 дня последующее наблюдение). Исследование проводили в 111 центрах в 15 странах. Основная цель исследования состояла в оценке эффективности ликсисенатида при гликемическом контроле по сравнению с плацебо в показателях снижения HbA1c (абсолютное изменение) в течение периода, составляющего 24 недели.
В общей сложности 496 пациентов рандомизировали в одну из двух лечебных групп (329 в группе ликсисенатида и 167 в группе плацебо) и 495 рандомизированных пациентов подвергали воздействию исследуемого продукта (IP). Демографические данные и исходные характеристики были в целом аналогичными по всем лечебным группам. Четырех пациентов (2 на ликсисенатиде и 2 на плацебо) исключили из mITT популяции для анализа эффективности вследствие отсутствия данных эффективности после исходного уровня. Во время всего периода лечения исследования 115 (35,0%) пациентов, лечащихся ликсисенатидом, досрочно прекратили получать IP, при том, что прекратили получать IP 52 (31,1%) пациентов, лечащихся плацебо. Для обеих лечебных групп основной причиной для прекращения лечения была «другие причины» (15,8% для ликсисенатида против 13,2% для плацебо), за которыми следуют «нежелательные явления» (11,2% для ликсисенатида против 7,2% для плацебо).
Анализы эффективности основаны на 24-недельном лечении: рассчитанные методом наименьших квадратов (LS) средние изменения HbA1c к 24 неделе по сравнению с исходным уровнем составляли -0,74% для группы ликсисенатида и -0,38% для группы плацебо (средняя разность LS против плацебо = -0,36%; p-значение = 0,0002). В общей сложности 86 пациентов (28,3%) в группе ликсисенатида достигли HbA1c <7% на 24 неделе по сравнению с 19 пациентами (12,0%) в группе плацебо, и 44 (14,5%) пациента, лечащихся ликсисенатидом, имели HBA1c ≤6,5% по сравнению с 6 (3,8%) пациентами, лечащимися плацебо. Анализ HbA1c пациентов, ответивших на лечение (HbA1c ≤6,5 или <7% на 24 неделе), с использованием метода Кохрана-Мантеля-Хенселя (CMH) показал существенное различие лечения против плацебо для группы ликсисенатида на 24 неделе (p-значение=0,0003 и p-значение <0,0001, соответственно).
Лечение ликсисенатидом также улучшало гликемический контроль после приема пищи, как показано результатами для глюкозы в плазме через 2 часа после приема пищи (PPG) и оценкой колебания уровня глюкозы. Статистически значимое улучшение PPG после тестового приема пищи было продемонстрировано в группе ликсисенатида в сравнении с группой плацебо со средней разностью LS, равной -3,81 ммоль/л (p-значение <0,0001). Кроме того, лечение ликсисенатидом продемонстрировало статистически значимое улучшение среднего значения в профиле по 7 точкам самоконтролируемой глюкозы в плазме (SMPG) (средняя разность LS равна -0,88 ммоль/л; p-значение <0,0001) в сравнении с группой плацебо. Для глюкозы в плазме натощак, между лечебными группами не наблюдалось никакого статистически значимого различия (средняя разность LS против плацебо = 0,08 ммоль/л; значение p =0,7579). Пациенты, принимающие лечение ликсисенатидом, показали статистически значимое снижение массы тела (Средняя разность LS равна -1,28 кг; p-значение <0,0001) в сравнении с группой плацебо без поправки на многократность сравнений. В общей сложности 30 пациентов (12 [7,3%] в группе плацебо и 18 [5,5%] в группе ликсисенатида) получали резервный вариант лечения. Наряду с достижением более хорошего гликемического контроля, пациенты, принимающие лечение ликсисенатидом, также показали статистически значимое снижение ежедневной дозы базального инсулина по сравнению с пациентами, принимающими лечение плацебо. (средняя разность LS равна -3,69 Ед, p-значение = 0,0120).
Анализы безопасности основаны на всем исследовании лечения: Ликсисенатид переносился хорошо. Доли пациентов с возникшими после начала лечения нежелательными явлениями (TEAE) были в целом сопоставимы между двумя лечебными группами (87,5% в группе ликсисенатида против 85,6% в группе плацебо). Два пациента в группе ликсисенатида и два пациента в группе плацебо имели TEAE, приведшие к смерти. Количество пациентов с серьезными TEAE составляло 46 (14,0%) в группе ликсисенатида и 17 (10,2%) в группе плацебо. Сто тридцать восемь (42,1%) пролеченных ликсисенатидом пациентов имели симптоматические гипогликемические события, как определено в протоколе в течение периода применения исследуемого препарата, тогда как 65 (38,9%) пациентов в группе плацебо сообщали о симптоматической гипогликемии. Помимо гипогликемии, наиболее часто сообщаемыми TEAE была тошнота (29,3%) для группы ликсисенатида и назофарингит (12,6%) для группы плацебо. Семь пациентов в группе ликсисенатида (2,1%) и 1 пациент в группе плацебо (0,6%) испытывали тяжелую симптоматическую гипогликемию по определению протокола. В общей сложности 11 пациентов (8 [2,4%] пролеченных ликсисенатидом пациентов и 3 [1,8%] пациента, лечившихся плацебо) сообщали об 11 TEAE, расцененных как аллергическая реакция Комитетом по оценке аллергических реакций (ARAC), и три из данных событий (2 события анафилактической реакции в группе ликсисенатида и 1 ангиоэдема в группе плацебо) были расценены как возможно связанные с IP. Один лечившийся ликсисенатидом пациент сообщал о событии панкреатита, которое было оценено исследователем как рецидивирующий панкреатит и не связанное с IP.
1 ЦЕЛИ
1.1 Основная цель
Основная цель данного исследования состояла в оценке эффективности ликсисенатида при гликемическом контроле по сравнению с плацебо в качестве дополнительного лечения к базальному инсулину у пациентов с диабетом 2 типа, получающих лечение базальным инсулином в показателях абсолютного снижения HbA1c в течение периода, составляющего 24 недели.
1.2 Дополнительная цель(и)
Дополнительные цели данного исследования состояли
в оценке воздействия ликсисенатида на
- массу тела,
- глюкозу в плазме через 2 часа после приема пищи после стандартизированного постпрандиального теста,
- процентную долю пациентов, достигших HbA1c <7%,
- процентную долю пациентов, достигших HbA1c ≤6,5%,
- глюкозу в плазме натощак (FPG),
- изменение профилей 7 точек самоконтролируемой глюкозы в плазме (SMPG),
- изменение доз базального инсулина и общего инсулина;
в оценке безопасности и переносимости ликсисенатида;
в оценке ФК ликсисенатида;
в оценке формирования антител против ликсисенатида.
2 ДИЗАЙН ИССЛЕДОВАНИЯ
Провели двойное слепое, рандомизированное, плацебо-контролируемое, международное исследование в двух параллельных группах с несбалансированным 2:1 отношением рандомизации. Исследование было двойное слепое в отношении активного и плацебо лечения. Объем лекарственного средства исследования (т.е. доза активного лекарственного средства или соответствующего плацебо) не был слепым.
Пациенты были стратифицированы по скрининговым значениям гликозилированного гемоглобина A1c (HbA1c) (<8%, ≥8%) и по использованию метформина при скрининге (Да, Нет). После скринингового периода пациентов централизованно рандомизировали посредством системы интерактивного голосового взаимодействия (IVRS) в отношении 2:1 либо к ликсисенатиду, либо к плацебо.
Приблизительная минимальная продолжительность исследования на пациента составляла 79 недель (до 3 недель скрининг + 24 недели основное двойное слепое лечение + варьируемое продление + 3 дня последующее наблюдение). Пациенты, которые завершили 24-недельный основной двойной слепой период, подвергались варьируемому двойному слепому периоду продления, который заканчивался для всех пациентов приблизительно на запланированную дату осмотра 76 недели (V25) для последнего рандомизированного пациента.
Пациенты, которые досрочно прекратили IP, оставались в исследовании до запланированной даты завершения исследования. За ними проводилось последующее наблюдение согласно процедурам исследования в соответствии с поправкой к протоколу (за исключением 3-дневного последующего наблюдения за безопасностью после лечения, оценки фармакокинетики и постпрандиального теста).
Фиг.1-Дизайн исследования.
3 ОСНОВНОЙ И КЛЮЧЕВОЙ ДОПОЛНИТЕЛЬНЫЕ КОНЕЧНЫЕ ПОКАЗАТЕЛИ
3.1 ОСНОВНОЙ КОНЕЧНЫЙ ПОКАЗАТЕЛЬ
Основным показателем эффективности было абсолютное изменение HbA1c к 24 неделе по сравнению с исходным, которое определяли как HbA1c на 24 неделе - HbA1c в начале исследования.
Если пациент прервал лечение досрочно, или принял резервный вариант лечения в течение основного 24-недельного периода двойного слепого лечения, или не имел значения HbA1c при осмотре на 24 неделе, в качестве значения HbA1c на 24 неделе использовали последнее после исходного измерение HbA1c в течение основного 24-недельного периода двойного слепого применения исследуемого продукта (метод замены пропущенных данных последним значением [LOCF]).
3.2 ДОПОЛНИТЕЛЬНЫЕ КОНЕЧНЫЕ ПОКАЗАТЕЛИ
3.2.1 Конечные показатели эффективности
Для дополнительных показателей эффективности применялась точно такая же методика обработки отсутствующей оценки/раннего прекращения, как для основного показателя.
Непрерывные показатели
- Изменение глюкозы в плазме через два часа после приема пищи (ммоль/л) после стандартизированного приема пищи к 24 неделе по сравнению с исходным уровнем
- Изменение профилей 7 точек SMPG (ммоль/л) (т.е., среднее значение и каждый момент времени по 7 точкам) к 24 неделе по сравнению с исходным уровнем
- Изменение FPG (содержание глюкозы в плазме натощак) (ммоль/л) к 24 неделе по сравнению с исходным уровнем
- Изменение массы тела (кг) к 24 неделе по сравнению с исходным уровнем
- Изменение колебания уровня глюкозы (глюкоза в плазме через 2 часа после приема пищи - глюкоза в плазме за 30 минут до тестового приема пищи перед введением исследуемого лекарственного средства) (ммоль/л) после стандартизированного постпрандиального теста к 24 неделе по сравнению с исходным уровнем
- Изменение ежедневной дозы базального инсулина (Ед) и общей дозы инсулина (Ед) к 24 неделе по сравнению с исходным уровнем.
Категориальные переменные
- Процентная доля пациентов с HbA1c <7% на 24 неделе
- Процентная доля пациентов с HbA1c ≤6,5% на 24 неделе
- Процентная доля пациентов, нуждающихся в резервном варианте лечения в течение основного 24недельного периода двойного слепого лечения
- Процентная доля пациентов с ≥5% потерей массы (кг) к 24 неделе по сравнению с исходным уровнем
3.2.2 КОНЕЧНЫЕ ПОКАЗАТЕЛИ БЕЗОПАСНОСТИ
Анализ безопасности проводили на основании сообщаемых TEAE и другой информации о безопасности, включая симптоматическую гипогликемию и тяжелую симптоматическую гипогликемию, местное раздражающее действие в месте инъекции, аллергические события (по решению ARAC), предполагаемый панкреатит, повышенный кальцитонин, показатели жизнедеятельности, ЭКГ в 12 отведениях и лабораторные испытания.
Главные сердечно-сосудистые явления также собирали и посылали для рассмотрения Комитетом по оценке сердечно-сосудистых явлений (CAC). Рассмотренные и подтвержденные CAC события из данного исследования и 3 других исследований фазы ликсисенатида будут объединяться при необходимости для анализа и суммироваться в отдельном отчете на основании плана статистического анализа для общей сердечно-сосудистой оценки ликсисенатида. KRM/CSR не будет отображать сущность рассмотренных и подтвержденных CV явлений из данного исследования.
4. РАСЧЕТНЫЕ ПРЕДПОЛОЖЕНИЯ РАЗМЕРОВ ВЫБОРКИ
Расчеты размера/мощности выборки выполняли на основании основного показателя, изменения HbA1c к 24 неделе по сравнению с исходным.
Ожидалось, что триста пациентов в группе лечения ликсисенатидом и 150 в группе лечения плацебо обеспечат мощность, равную 96% (или 86%) для выявления различий в изменении HbA1c к 24 неделе между ликсисенатидом и плацебо, равных 0,5% (или 0,4%) по сравнению с исходным уровнем, принимая, что общее стандартное отклонение (SD) составляло 1,3% с двусторонним критерием при 5% уровне значимости.
5 СТАТИСТИЧЕСКИЕ МЕТОДЫ
5.1 АНАЛИЗ ПОПУЛЯЦИЙ
mITT популяция состоит из всех рандомизированных пациентов, получивших по меньшей мере одну дозу двойного слепого IP, и имевших как исходную оценку, так и по меньшей мере одну оценку после исходной любого основного или дополнительного показателей эффективности, независимо от соблюдения протокола и процедур исследования.
Выборка для оценки безопасности определяется, как все рандомизированные пациенты, которые получали по меньшей мере одну дозу двойного слепого IP.
5.2 ОСНОВНОЙ АНАЛИЗ ЭФФЕКТИВНОСТИ
Основной показатель эффективности (изменение HbA1c к 24 неделе по сравнению с исходным уровнем) анализировали с использованием модели ковариационного анализа (ANCOVA) с лечением, рандомизационными стратами уровня HbA1c при скрининге (<8,0, ≥8,0%), рандомизационными стратами использования метформина при скрининге (Да, Нет) и страной в качестве фиксированных эффектов и с использованием значения исходного уровня в качестве ковариаты. Разницу между ликсисенатидом и плацебо и двусторонний 95% доверительный интервал также как p-значение оценивали в рамках программы ANCOVA.
Методику LOCF использовали, принимая в качестве значения HbA1c на 24 неделе последнее доступное после исходного уровня измерение HbA1c в процессе лечения (перед началом приема нового лекарственного препарата в случае резервного варианта лечения).
Основной анализ основного показателя эффективности проводили на основании mITT популяции и измерений, полученных в течение основного 24-недельного периода двойного слепого применения исследуемого продукта для оценки показателей эффективности. Основной 24недельный период двойного слепого применения исследуемого продукта для оценки показателей эффективности за исключением показателей эффективности из постпрандиального теста, SMPG по 7 точкам, дозы базального инсулина и общего инсулина, определяли, как время от инъекции первой дозы двойного слепого IP до 3 дней (за исключением FPG центральной лабораторией, который составлял до 1 дня) после последней дозы двойного слепого IP при или перед осмотром V12/24 недели (или D169, если осмотр V12/24 недели был пропущен), или до введения резервного варианта лечения, в зависимости от того, что было раньше. Основной 24недельный период двойного слепого применения исследуемого продукта для оценки показателей эффективности из постпрандиального теста, включая 2-часовой PPG и колебание уровня глюкозы, SMPG по 7 точкам, дозу базального инсулина и общий инсулин, определяли как время от инъекции первой дозы двойного слепого IP до даты последней дозы двойного слепого IP при или перед осмотром V12/24 недели (или D169, если осмотр V12/24 недели был пропущен), или до введения резервного варианта лечения, в зависимости от того, что было раньше.
5.3 ДОПОЛНИТЕЛЬНЫЙ АНАЛИЗ ЭФФЕКТИВНОСТИ
Если основной показатель был статистически значимым при α=0,05, проводили процедуру тестирования для тестирования следующих дополнительных показателей эффективности с помощью следующего порядка установки приоритета. Тестирования останавливали, как только обнаруживали, что конечный показатель не является статистически значимым при α=0,05.
1. Изменение глюкозы в плазме через два часа после приема пищи (ммоль/л) после стандартизированного тестового приема пищи к 24 неделе по сравнению с исходным уровнем,
2. Изменение среднего значения SMPG по 7 точкам к 24 неделе по сравнению с исходным уровнем,
3. Изменение FPG (ммоль/л) к 24 неделе по сравнению с исходным уровнем,
4. Изменение массы тела (кг) к 24 неделе по сравнению с исходным уровнем,
5. Процентная доля пациентов, нуждающихся в резервном варианте лечения в течение основного 24недельного периода двойного слепого лечения.
Не будет сделана поправка на множественность по другим дополнительным показателям эффективности, которые не упоминаются выше.
Все непрерывные дополнительные показатели эффективности на 24 неделе, которые описаны в разделе 3.2.1, анализировали с использованием аналогичного подхода и модели ANCOVA, как описано выше для основного анализа основной конечной точки эффективности. Были предоставлены оценки среднего различия в лечении между ликсисенатидом и плацебо и двусторонний 95% доверительный интервал.
Следующие категориальные дополнительные показатели эффективности на 24 неделе анализировали с использованием метода Кохрана-Мантеля-Хенселя (СМН) со стратифицированием на рандомизационные страты (скрининг HbA1c [<8,0, ≥8%] и использование метформина при скрининге [Да, Нет]):
- Процентная доля пациентов с HbA1c <7,0% на 24 неделе
- Процентная доля пациентов с HbA1c ≤6,5% на 24 неделе
- Процентная доля пациентов, нуждающихся в резервном варианте лечения в течение основного 24недельного периода двойного слепого лечения
Количество и процентная доля пациентов с потерей массы ≥5% по сравнению с исходным уровнем на 24 неделе отображены по лечебным группам.
Все дополнительные показатели в конце лечения оценивали только посредством описательной статистики (среднее, стандартное отклонение, срединное значение и диапазоны, предусмотренные в CSR).
5.4 АНАЛИЗ БЕЗОПАСНОСТИ
Анализы безопасности были основаны главным образом на периоде применения исследуемого продукта для всего исследования. Период применения исследуемого продукта для всего исследования определяли как время от введения первой дозы двойного слепого IP до 3 дней после последней дозы IP во время всего периода исследования независимо от статуса резервного лечения. 3-дневный интервал выбрали, исходя из периода полувыведения IP (приблизительно 5-кратный период полувыведения).
В дополнение, в CSR будут подытожены анализы безопасности для 24-недельного периода двойного слепого лечения.
Итог результатов безопасности (описательная статистика или Таблицы частоты) представлен по группам лечения.
6 РЕЗУЛЬТАТЫ
6.1 ПАЦИЕНТЫ ИССЛЕДОВАНИЯ
6.1.1 Учет пациентов
Исследование проводили в 111 центрах в 15 странах (Бразилия, Канада, Чили, Египет, Франция, Германия, Индия, Италия, Корея, Мексика, Пуэрто-Рико, Российская Федерация, Турция, Соединенное Королевство и Соединенные Штаты Америки). В общей сложности скринировали 879 пациентов и 496 рандомизировали в одну из двух лечебных групп. Основной причиной сбоя при скрининге было значение HbAlc при скрининговом осмотре за пределами определенного диапазона протокола (205 [23,3%] из 879 подвергшихся скринингу пациентов).
Из 496 рандомизированных пациентов 495 подвергли IP. Одного пациента из группы ликсисенатида не подвергали IP. Четырех пациентов (2 в группе ликсисенатида и 2 в группе плацебо) исключили из mITT популяции для анализа эффективности вследствие отсутствия данных эффективности после исходного уровня. Таблица 1 предоставляет количество пациентов, включенных в каждую популяцию анализа.
Для выборок для оценки эффективности пациенты приведены в таблице согласно их рандомизированному лечению (по рандомизации).
6.1.2 Распределение исследования
Таблица 2 предоставляет сущность распределения пациентов для каждой группы лечения.
Во время всего периода лечения исследования 115 (35,0%) пролеченных ликсисенатидом пациентов досрочно прекратили IP, при этом 52 (31,1%) пролеченных плацебо пациентов прекратили IP. Для обеих лечебных групп основной причиной для прекращения лечения была «другие причины» (52 пациента [15,8%] для ликсисенатида и 22 пациента [13,2%] для плацебо), причем главным образом это были личные причины, такие как отъезд из города, потеря работы, семейные трудности или большая занятость, чтобы придерживаться графиков осмотра, за которыми следуют «нежелательные явления» (37 пациентов [11,2%], включая 2 не связанных с TEAE для ликсисенатида против 12 пациентов [7,2%] для плацебо).
Для 24-недельного основного периода лечения 53 (16,1%) пациента в группе ликсисенатида и 20 (12,0%) в плацебо досрочно прекратили IP, причем основной причиной были нежелательные явления (26 пациентов, 7,9%) для группы ликсисенатида и «другая» (8 пациентов, 4,8%) для группы плацебо. Время до наступления прекращения лечения по любой причине для общего периода лечения изображено на фиг.2. Более высокая частота прекращения наблюдалась для группы ликсисенатида.
(N=167)
(N=329)
Фиг.2 - График Каплана-Майера периода до прекращения лечения по любой причине - Рандомизированная популяция.
6.1.3 Демографические данные и исходные характеристики
Демографические и исходные характеристики пациентов были в целом одинаковыми между двумя лечебными группами для выборки для оценки безопасности (таблица 3). Средний возраст популяции исследования составлял 58,0 лет. Большинство пациентов были представители белой европеоидной расы (77,6%). Группа ликсисенатида имела больше пациентов-женщин в процентном соотношении (55,5% женщин и 44,5% мужчин), чем группа плацебо (50,9% женщин и 49,1% мужчин).
(N=167)
(N=328)
(N=495)
Характеристики заболевания, включая историю заболевания сахарным диабетом, были в целом сопоставимы между двумя лечебными группами (таблица 4). Один пролеченный плацебо пациент (#840608010) имел диабет 1 типа и был исключен из исследования вскоре после постановки диагноза.
Средняя продолжительность лечения базальным инсулином для популяции исследования составляла 3,11 года (таблица 5). Большинство пациентов принимали во время скринига либо аналоги инсулина пролонгированного действия (гларгин 50,1%, детемир 8,7%), либо NPH (40,0%), и продолжали в период лечения исследования за несколькими исключениями (1,6%), которые принимали вместо этого предварительно смешанный инсулин. Восемь пациентов (5 на ликсисенатиде и 3 на плацебо) принимали предварительно смешанный инсулин при скрининге и продолжали при исследовании. Два пациента, лечившихся плацебо, принимали два типа инсулина (один детемир + гларгин и еще один NPH + Новолин смесь 70/30) во время скринига и продолжали при исследовании. Все использование инсулина, включая предварительно смешанный инсулин, отображено в таблице 5.
Триста девяносто два пациента (79,2%) принимали метформин при скрининговом осмотре с аналогичной долей использования в двух лечебных группах (ликсисенатид 79,6% и плацебо 78,4% (таблица 6). Было два противоречия в количестве пациентов между «рандомизационной стратой использования метформина при скрининге» и фактическим «использованием метформина при скрининге» вследствие ошибок в рандомизационных стратах.
(N=167)
(N=328)
(N=495)
Значение креатининового клиренса получено с использованием уравнения Кокрофта-Голта.
(N=167)
(N=328)
(N=495)
Общая
2Отклонение от протокола.
3Предварительно смешанный инсулин включал Новолин 70/30 смесь и Хумалог 75/25 смесь.
4Два пациента (840612006 ликсисенатид и 630625001 плацебо) не принимали свой базальный инсулин в день скрининга.
(N=167)
(N=328)
(N=495)
Показатели эффективности исходного уровня, включая HbA1c, были в целом сопоставимы между двумя лечебными группами для выборки для оценки безопасности (таблица 7).
(N=167)
(N=328)
(N=495)
SMPG = Самоконтролирующаяся глюкоза в крови.
Колебание уровня глюкозы = глюкоза в плазме через 2 часа после приема пищи - глюкоза в плазме за 30 минут до тестового приема пищи перед введением исследуемого лекарственного средства.
6.1.4 Дозировка и продолжительность
Среднее значение продолжительности лечения составило 491,5 дня (70,2 недели) для группы ликсисенатида и 510,4 дня (72,9 недели) для группы плацебо (таблица 8). Из 495 пациентов 270 (82,3%) пациентов в группе ликсисенатида и 146 (87,4%) пациентов в группе плацебо получили по меньшей мере 169 дней (24 недели) лечения; более того, 171 (52,1%) пациент в группе ликсисенатида и 89 (53,3%) пациентов в группе плацебо имели по меньшей мере 547 дней (18 месяцев) лечения. У шести пациентов (3 для ликсисенатида и 3 для плацебо) отсутствовала дата последнего введения; из них три выбыли из последующего наблюдения (2 для ликсисенатида и 1 для плацебо) и, следовательно, следуя конвенции SAP по обработке данных, продолжительность их лечения была установлена с учетом отсутствия.
Для группы ликсисенатида, 286 (87,2%) пациентов принимали намеченную общую суточную дозу, составляющую 20 мкг, как в конце 24-недельного периода двойного слепого лечения, так и в конце всего двойного слепого лечения (Таблицы 9 и 10). Для группы плацебо, 161 (96,4%) пациент и 162 (97,0%) пациента принимали намеченную общую суточную дозу, составляющую 20 мкг как в конце 24-недельного периода двойного слепого лечения, так и в конце всего двойного слепого лечения, соответственно (таблицы 9 и 10).
(N=167)
(N=328)
Запланированным осмотром для окончания титрования по протоколу должен быть 5 осмотр/2 неделя.
Примечание: Процентные доли рассчитаны с использованием в качестве знаменателя количества пациентов для оценки безопасности.
Примечание: Процентные доли рассчитаны с использованием в качестве знаменателя количества пациентов для оценки безопасности.
(N=167)
(N=328)
Примечание: Процентные доли рассчитаны с использованием в качестве знаменателя количества пациентов для оценки безопасности.
6.2 ЭФФЕКТИВНОСТЬ
6.2.1 Основной конечный показатель эффективности
Основной анализ
Таблица 11 приводит результаты основного параметра эффективности, изменение HbA1c к 24 неделе по сравнению с исходным уровнем (LOCF) с использованием анализа ANCOVA.
Первичный анализ по переменным, заранее предусмотренным в плане, показал, что лечение ликсисенатидом в сравнении с группой плацебо приводило к статистически значимому снижению HbA1c к 24 неделе по сравнению с исходным уровнем (средняя разность LS против группы плацебо = -0,36%; значение p=0,0002).
(N=165)
(N=326)
(a)Модель ковариационного анализа (ANCOVA) с группами лечения (ликсисенатид и плацебо), рандомизационными стратами скрининга HbA1c (<8,0, ≥8,0%), рандомизационными стратами использования метформина при скрининге (Да, Нет), страной в качестве фиксированных эффектов и значением исходного уровня HbA1c в качестве ковариаты.
Анализ включал измерения, полученные перед введением резервного лечения и вплоть до 3 дней после последней дозы двойной слепой инъекции исследуемого продукта при или перед 12 осмотром (24 неделя), или на 169 день, если 12 осмотр (24 неделя) невозможен. Включали пациентов с измерениями исходного уровня и 24 недели (LOCF).
Фиг.3 иллюстрирует среднее (±стандартная ошибка) изменение HbA1c по сравнению с исходным уровнем в течение основного 24-недельного периода двойного слепого лечения. Фиг.8 в приложении иллюстрирует среднее (±стандартная ошибка) изменение HbA1c по сравнению с исходным уровнем с течением времени вплоть до 76 недели. Снижение HbA1c относительно сохранялось с течением времени за пределами 24 недель.
Фиг.3 - График среднего изменения HbA1c (%) от исходного уровня при осмотре вплоть до 24 недели и в конечной точке - mITT популяция.
LOCF = Перенос вперед данных последнего наблюдения.
Примечание: График включал измерения, полученные перед введением резервного лечения и до 3 дней после последней дозы двойной слепой инъекции исследуемого продукта при или перед 12 осмотром (24 неделя), или на 169 день, если 12 осмотр (24 неделя) невозможен.
Таблица 13 приводит долю пациентов с лечебным эффектом в виде HbA1c ≤6,5% или <7% на 24 неделе, соответственно. Анализ HbA1c пациентов, у которых достигнут лечебный эффект, с использованием метода CMH, показал существенное различие лечения против плацебо для группы, пролеченной ликсисенатидом (значение p=0,0003 и значение p<0,0001, соответственно) для обеих категорий. На 24 неделе 14,5% пролеченных ликсисенатидом пациентов и 3,8% пролеченных плацебо пациентов достигли значений HbA1c ≤6,5%; 28,3% пациентов в группе ликсисенатида и 12,0% пациентов в группе плацебо достигли значений HbA1c<7%.
(N=165)
(N=326)
Анализ включал измерения, полученные перед введением резервного лечения и до 3 дней после последней дозы двойной слепой инъекции исследуемого продукта при или перед 12 осмотром (24 неделя), или на 169 день, если 12 осмотр (24 неделя) невозможен.
6.2.2 Дополнительные конечные показатели эффективности
Таблицы 14-18 и таблицы 20-21 представляют анализы ANCOVA глюкозы в плазме через 2 часа после приема пищи, среднее значение SMPG по 7 точкам, FPG, массу тела, базальный инсулин и колебание уровня глюкозы, соответственно. Фиг.4-7 иллюстрируют среднее (±стандартная ошибка) изменение по сравнению с исходным уровнем среднего значения SMPG по 7 точкам, FPG, массы тела и базального инсулина с течением времени в течение основного 24-недельного двойного слепого периода лечения. Фиг.9-13 в приложении иллюстрируют среднее (±стандартная ошибка) изменение по сравнению с исходным уровнем глюкозы в плазме через 2 часа после приема пищи, среднего значения SMPG по 7 точкам, FPG, массы тела и базального инсулина с течением времени до 76 недели.
Результаты глюкозы в плазме через 2 часа после приема пищи после тестового приема пищи показали статистически значимое улучшение к 24 неделе по сравнению с исходным уровнем в группе ликсисенатида в сравнении с группой плацебо (средняя разность LS против плацебо = 3,81 ммоль/л; значение p<0,0001). Более того, лечение ликсисенатидом по существу уменьшало колебание уровня глюкозы в плазме после приема пищи к 24 неделе по сравнению с исходным уровнем в сравнении с группой плацебо (средняя разность LS = -3,80 ммоль/л, 95% CI = от -4,57 до -3,03) (таблица 21).
Для среднего значения SMPG по 7 точкам, статистически значимое улучшение к 24 неделе по сравнению с исходным уровнем наблюдалось в группе ликсисенатида в сравнении с группой плацебо (средняя разность LS против плацебо = 0,88 ммоль/л; значение p<0,0001) (таблица 15).
Пациенты в обеих лечебные группах продемонстрировали умеренное снижение FPG к 24 неделе по сравнению с исходным уровнем (LS среднее составило -0,63 для ликсисенатида против -0,55 для плацебо) со статистически незначимым различием, наблюдаемым между группой ликсисенатида и группой плацебо (средняя разность LS против плацебо = 0,08 ммоль/л; значение p=0,7579) (таблица 16).
Согласно стратегии тестирования с поправкой на многократность сравнений логически вытекающее тестирование потери массы тела по сравнению с исходным уровнем на 24 неделе и процентные доли пациентов, нуждающихся в резервном варианте лечения на 24 неделе, было экспериментальным, поскольку предшествующий тест (FPG) не показал статистически значимую разницу между группами.
Средняя LS потеря массы тела к 24 неделе по сравнению с исходным уровнем составила -1,80 кг для пролеченных ликсисенатидом пациентов и -0,52 кг для пролеченных плацебо пациентов, со статистически значимым различием, наблюдаемым между двумя лечебными группами (средняя разность LS против плацебо = -1,28 кг, значение p<0,0001) без поправки на многократность сравнений (таблица 17). Потерю массы, составившую 5% или более к 24 неделе по сравнению с исходным уровнем, имели больше пролеченных ликсисенатидом пациентов (13,2%), чем пролеченных плацебо пациентов (3,1%) (таблица 18).
Процентная доля пациентов, нуждающихся в резервном варианте лечения на 24 неделе, была незначительно ниже в группе ликсисенатида, чем в группе плацебо (18 пациентов [5,5%] в группе ликсисенатида и 12 пациентов [7,3%] в группе плацебо) (таблица 19).
Наряду с достижением более значительного снижения HbA1c, пациенты в группе ликсисенатида показали стабильное снижение ежедневной дозы базального инсулина на протяжении периода лечения (фиг.7) и достигли статистически значимого снижения среднего изменения в конечной точке (24 неделя) в сравнении с группой плацебо (средняя разность LS против плацебо =3,09 Ед; значение p=0,0412) (таблица 20). Результаты анализа изменения «общей дозы инсулина» (не показано) являются идентичными результатам анализа «дозы базального инсулина» вследствие факта, что из анализа было исключено использование инсулина для купирующей терапии.
(N=165)
(N=326)
(a)Модель ковариационного анализа (ANCOVA) с лечебными группами (ликсисенатид и плацебо), рандомизационными стратами скрининга HbA1c (<8,0, ≥8,0%), рандомизационными стратами использования метформина при скрининге (Да, Нет), страной в качестве фиксированных эффектов и значением исходного уровня глюкозы в плазме через 2 часа после приема пищи в качестве ковариаты.
Анализ включал измерения, полученные перед введением резервного лечения и до даты последней дозы двойной слепой инъекции исследуемого продукта при или перед 12 осмотром (24 неделя), или на 169 день, если 12 осмотр (24 неделя) невозможен. Включены пациенты с измерениями как исходного уровня, так и 24 недели (LOCF).
(N=326)
(a)Модель ковариационного анализа (ANCOVA) с лечебными группами (ликсисенатид и плацебо), рандомизационными стратами скрининга HbA1c (<8,0, ≥8,0%), рандомизационными стратами использования метформина при скрининге (Да, Нет), страной в качестве фиксированных эффектов и значением исходного уровня среднего значения SMPG по 7 точкам в качестве ковариаты.
Анализ включал измерения, полученные перед введением резервного лечения и до даты последней дозы двойной слепой инъекции исследуемого продукта при или перед 12 осмотром (24 неделя), или на 169 день, если 12 осмотр (24 неделя) невозможен. Включены пациенты с измерениями как исходного уровня, так и 24 недели (LOCF).
Фиг.4 График среднего изменения среднего значения по 7 точкам самоконтролируемой глюкозы в плазме (SMPG) (ммоль/л) к 24 неделе по сравнению с исходным уровнем при осмотре и в конечной точке - mITT популяция.
LOCF = Перенос вперед данных последнего наблюдения.
Примечание: График включал измерения, полученные перед введением резервного лечения и до даты последней дозы двойной слепой инъекции исследуемого продукта при или перед 12 осмотром (24 неделя), или на 169 день, если 12 осмотр (24 неделя) невозможен.
(N=165)
(N=326)
(a)Модель ковариационного анализа (ANCOVA) с лечебными группами (ликсисенатид и плацебо), рандомизационными стратами скрининга HbA1c (<8,0, ≥8,0%), рандомизационными стратами использования метформина при скрининге (Да, Нет), страной в качестве фиксированных эффектов и исходным уровнем глюкозы в плазме натощак в качестве ковариаты.
Анализ включал измерения, полученные перед введением резервного лечения и до 1 дня после последней дозы двойной слепой инъекции исследуемого продукта при или перед 12 осмотром (24 неделя), или на 169 день, если 12 осмотр (24 неделя) невозможен. Включены пациенты с измерениями как исходного уровня, так и 24 недели (LOCF).
Фиг.5 График среднего изменения глюкозы в плазме натощак (ммоль/л) к 24 неделе по сравнению с исходным уровнем при осмотре и в конечной точке - mITT популяция.
LOCF = Перенос вперед данных последнего наблюдения.
Примечание: График включал измерения, полученные перед введением резервного лечения и до 1 дня после последней дозы двойной слепой инъекции исследуемого продукта при или перед 12 осмотром (24 неделя), или на 169 день, если 12 осмотр (24 неделя) невозможен.
(N=165)
(N=326)
(a)Модель ковариационного анализа (ANCOVA) с лечебными группами (ликсисенатид и плацебо), рандомизационными стратами скрининга HbA1c (<8,0, ≥8,0%), рандомизационными стратами использования метформина при скрининге (Да, Нет), страной в качестве фиксированных эффектов и исходным уровнем массы тела в качестве ковариаты.
Анализ включал измерения, полученные перед введением резервного лечения и до 3 дней после последней дозы двойной слепой инъекции исследуемого продукта при или перед 12 осмотром (24 неделя), или на 169 день, если 12 осмотр (24 неделя) невозможен. Включены пациенты с измерениями как исходного уровня, так и 24 недели (LOCF).
Фиг.6 График среднего изменения массы тела (кг) к 24 неделе по сравнению с исходным уровнем при осмотре и в конечной точке - mITT популяция.
LOCF = Перенос вперед данных последнего наблюдения.
Примечание: График включал измерения, полученные перед введением резервного лечения и до 3 дней после последней дозы двойной слепой инъекции исследуемого продукта при или перед 12 осмотром (24 неделя), или на 169 день, если 12 осмотр (24 неделя) невозможен.
(N=165)
(N=326)
Анализ включал измерения, полученные перед введением резервного лечения и до 3 дней после последней дозы двойной слепой инъекции исследуемого продукта при или перед 12 осмотром (24 неделя), или на 169 день, если 12 осмотр (24 неделя) невозможен. Включены пациенты с измерениями как исходного уровня, так и 24 недели (LOCF).
(N=165)
(N=326)
(N=165)
(N=326)
(a)Модель ковариационного анализа (ANCOVA) с лечебными группами (ликсисенатид и плацебо), рандомизационными стратами скрининга HbA1c (<8,0, ≥8,0%), рандомизационными стратами использования метформина при скрининге (Да, Нет), страной в качестве фиксированных эффектов и исходным уровнем дозы базального инсулина в качестве ковариаты.
Анализ включал измерения, полученные перед введением резервного лечения и до даты последней дозы двойной слепой инъекции исследуемого продукта при или перед 12 осмотром (24 неделя), или на 169 день, если 12 осмотр (24 неделя) невозможен. Включены пациенты с измерениями как исходного уровня, так и 24 недели (LOCF).
Фиг.7 График среднего изменения дозы базального инсулина (Ед) к 24 неделе по сравнению с исходным уровнем при осмотре и в конечной точке - mITT популяция.
LOCF = Перенос вперед данных последнего наблюдения.
Примечание: График включал измерения, полученные перед введением резервного лечения и до даты последней дозы двойной слепой инъекции исследуемого продукта при или перед 12 осмотром (24 неделя), или на 169 день, если 12 осмотр (24 неделя) невозможен.
(N=165)
(N=326)
(a)Модель ковариационного анализа (ANCOVA) с лечебными группами (ликсисенатид и плацебо), рандомизационными стратами скрининга HbA1c (<8,0, ≥8,0%), рандомизационными стратами использования метформина при скрининге (Да, Нет), страной в качестве фиксированных эффектов и значением исходного уровня колебания уровня глюкозы в качестве ковариаты.
Колебание уровня глюкозы = глюкоза в плазме через 2 часа после приема пищи - глюкоза в плазме за 30 минут до тестового приема пищи перед введением исследуемого лекарственного средства.
Анализ включал измерения, полученные перед введением резервного лечения и до даты последней дозы двойной слепой инъекции исследуемого продукта при или перед 12 осмотром (24 неделя), или на 169 день, если 12 осмотр (24 неделя) невозможен. Включены пациенты с измерениями как исходного уровня, так и 24 недели (LOCF).
6.3 БЕЗОПАСНОСТЬ
Обзор нежелательных явлений, наблюдаемых в течение периода применения исследуемого продукта для всего исследования, предоставлен в таблице 22. Доли пациентов с возникшими после начала лечения нежелательными явлениями (TEAE) были в целом сопоставимы между двумя лечебными группами (87,5% для ликсисенатида против 85,6% для плацебо). Четыре пациента (2 в группе ликсисенатида и 2 в плацебо) имели TEAE, приведшие к смерти. Процентная доля пациентов, которые испытывали серьезные TEAE, была более высокая в группе ликсисенатида (14,0%), чем в группе плацебо (10,2%). Процентная доля пациентов с TEAE, приведшими к прекращению лечения, составляла 10,7% в группе ликсисенатида по сравнению с 7,2% в группе плацебо. Таблицы 23, 24 и 25 представляют TEAE, приведшие к смерти, серьезные TEAE и TEAE, приведшими к прекращению лечения, по основным SOC, HLGT, HLT и PT, соответственно. Наиболее обычным TEAE, приводившим к прекращению лечения, была тошнота в группе ликсисенатида (11 пациентов [3,4%]), при этом в группе плацебо ни один пациент не прервал лечение вследствие тошноты.
Таблица 35 в приложении представляет случаи возникновения TEAE, встречающихся по меньшей мере у 1% пациентов в любой лечебной группе в течение периода применения исследуемого продукта для всего исследования. Наиболее часто сообщалось о TEAE гипогликемии как для группы ликсисенатида (138 [42,1%]), так и для группы плацебо (68 [40,7%]). Помимо гипогликемии, наиболее обычным TEAE в группе ликсисенатида была тошнота (96 пациентов [29,3%] для ликсисенатида против 16 пациентов [9,6%] для плацебо), за которыми следуют головная боль (41 пациент [12,5%] для ликсисенатида против 17 [10,2%] для плацебо) и диарея (37 пациентов [11,3%] для ликсисенатида против 10 [6,0%] для плацебо).
(N=167)
(N=328)
Период применения исследуемого продукта для всего исследования = время от первой дозы лекарственного средства в двойном слепом исследовании до 3 дней после введения последней дозы.
n (%) = Количество и процентная доля пациентов по меньшей мере с одним нежелательным явлением.
HLGT: Групповой термин высокого уровня
HLT: Термин высокого уровня
Предпочтительный термин
(N=167)
(N=328)
Период применения исследуемого продукта для всего исследования = время от первой дозы лекарственного средства в двойном слепом исследовании до 3 дней после введения последней дозы.
MedDRA, версия: 13.1.
n (%) = Количество и процентная доля пациентов по меньшей мере с одним TEAE, приведшим к смерти.
Примечание: Таблица отсортирована в соответствии с международно согласованным порядком SOC и алфавитным порядком HLGT, HLT, PT.
HLGT: Групповой термин высокого уровня
HLT: Термин высокого уровня
Предпочтительный термин
(N=328)
Период применения исследуемого продукта для всего исследования = время от первой дозы лекарственного средства в двойном слепом исследовании до 3 дней после введения последней дозы.
MedDRA, версия: 13.1.
n (%) = Количество и процентная доля пациентов по меньшей мере с одним серьезным TEAE.
Примечание: Таблица отсортирована в соответствии с международно согласованным порядком SOC и алфавитным порядком HLGT, HLT, PT.
HLGT: Групповой термин высокого уровня
HLT: Термин высокого уровня
Предпочтительный термин
(N=167)
(N=328)
Период применения исследуемого продукта для всего исследования = время от первой дозы лекарственного средства в двойном слепом исследовании до 3 дней после введения последней дозы.
MedDRA версия: 13.1.
n (%) = Количество и процентная доля пациентов по меньшей мере с одним TEAE, приведшим к необратимому прекращению лечения.
Примечание: Таблица отсортирована в соответствии с международно согласованным порядком SOC и алфавитным порядком HLGT, HLT, PT.
Согласно определению протокола для симптоматической гипогликемии 138 (42,1%) пролеченных ликсисенатидом пациентов и 65 (38,9%) пролеченных плацебо пациентов сообщали по меньшей мере об одном симптоматическом гипогликемическом событии в течение периода применения исследуемого продукта на протяжении всего исследования (таблица 26). Из данных пациентов, имеющих симптоматические гипогликемические события по определению протокола, 4 пролеченных ликсисенатидом пациента сообщали исследователю о терминах AE, не являющихся гипогликемией (гипогликемическое бессознательное состояние, нечувствительность к развитию гипогликемии, пониженная глюкоза в крови и тремор), которые не отображены в сводной таблице TEAE (таблица 35) в качестве гипогликемических PT. Наоборот, 7 пациентов (4 для ликсисенатида и 3 для плацебо), которые сообщали о гипогликемических TEAE, исключены из определяемых протоколом симптоматических гипогликемических событий в таблице 26, вследствие либо неудовлетворения определению протокола по гипогликемии (случай значения глюкозы ≥60 мг/дл) или отсутствия соответствующей информации для анализа (для одного пролеченного плацебо пациента отсутствовала дополняющая форма).
Семь (2,1%) пролеченных ликсисенатидом пациентов сообщали о 8 тяжелых симптоматических гипогликемических событиях по определению протокола, тогда как 1 (0,6%) пролеченный плацебо пациент сообщал об 1 тяжелом симптоматическом гипогликемическом событии в течение того же самого периода (таблица 27).
(N=167)
(N=328)
Период применения исследуемого продукта для всего исследования = время от первой дозы лекарственного средства в двойном слепом исследовании до 3 дней после введения последней дозы.
1Рассчитано как (количество пациентов с событием*100, деленное на суммарную величину воздействия + 3 дня в пересчете на общее количество лет пациентов).
(N=167)
(N=328)
Период применения исследуемого продукта для всего исследования = время от первой дозы лекарственного средства в двойном слепом исследовании до 3 дней после введения последней дозы.
1Рассчитано как (количество пациентов с событиями*100, деленное на суммарную величину воздействия + 3 дня в пересчете на общее количество лет пациентов).
Восемь пациентов (2,4%) из группы ликсисенатида и один пациент (0,6%) из группы плацебо испытывали AE реакции в месте инъекции (таблица 28). AE реакции в месте инъекции идентифицировали посредством поиска термина «место инъекции» либо в PT AE, сообщаемых исследователю, либо в PT из диагноза ARAC после признания аллергической реакции. Интенсивность ни одной из реакций не была серьезной или тяжелой, и данные AE не привели к прекращению IP.
Предпочтительный термин
ARAC = Комитет по оценке аллергических реакций.
В общей сложности для 28 пациентов сообщалось о 33 случаях, когда исследователи предполагали аллергические события и посылали в ARAC для оценки в течение периода применения исследуемого продукта для всего исследования. Из них 11 событий у 11 пациентов (8 [2,4%] у пролеченных ликсисенатидом пациентов, и 3 [1,8%] у пролеченных плацебо пациентов) были расценены ARAC как аллергические реакции, но только 3 события у 3 пациентов (два события анафилактических реакций в группе ликсисенатида и одно событие ангиоэдемы в группе плацебо) были расценены как возможно связанные с IP (таблица 29).
Пациент 840635031 (ликсисенатид): пациент женского пола 52 лет, в истории болезни дислипидемия, астма, аллергический ринит, аллергические реакции на лекарственное средство, пищу, пыльцу и пыль, а также в прошлом аллергическая сыпь и ангиоэдема, сообщала о легких реакциях в месте инъекции при каждой дозе с 8 июня 2009 года (21 день получения IP). Пациент жаловался на местный и общий зуд, и опухание в месте инъекции с эритемой. Также она получила хрипоту, изменение высоты голоса, хрипы и чувство стеснения в груди. Показатели жизнедеятельности во время реакции (17 июня 2009 года, 9:08) были: АД 129/62 мм рт.ст., ЧСС 67 ударов в минуту. Она получила лечение пероральным бенадрилом и поправилась 18 июня 2009 года. 17 июня 2009 года IP было прекращено. Оценка причины была сделана исследователем. Аллергическая реакция была расценена ARAC как анафилактическая реакция, возможно связанная с IP.
Пациент 840635033 (ликсисенатид): пациент женского пола 58 лет, в истории болезни гипертензия, дислипидемия, астма, аллергический ринит и в прошлом аллергические реакции на лекарственное средство и домашнюю пыль, а также шинил, 19 июля 2009 года (25 день получения IP) вслед за применением IP и перорального метоклопрамида развился зуд и аллергическая сыпь высокой интенсивности (вновь начался 19 июля 2009 года и закончился 20 июля 2009 года). Пациент жаловался на общий зуд, прилив крови к лицу, припухлость на губах, глазах и лице, заложенность носа и ощущение тошноты. Показатели жизнедеятельности во время реакции (23 июля 2009 года, 10:10) были: АД 134/66 мм рт.ст., ЧСС 95 ударов в минуту. Она получила лечение пероральным бенадрилом, и произошло быстрое улучшение. 23 июля 2009 года IP было прекращено. Оценка причины была связана исследователем с IP, но также, возможно, с метоклопрамидом. Аллергическая реакция была расценена ARAC как анафилактическая реакция, возможно связанная с IP.
(N=167)
(N=328)
IP = Исследуемый препарат.
Период применения исследуемого продукта для всего исследования = время от первой дозы лекарственного средства в двойном слепом исследовании до 3 дней после введения последней дозы.
По протоколу, любое увеличение амилазы и/или липазы более чем в два раза над верхней границей нормального диапазона (ULN), которое было подтверждено повторным измерением, необходимо отслеживать и документально подтверждать в предварительно утвержденной форме: «форма нежелательных явлений для предполагаемого панкреатита». В течение периода применения исследуемого продукта для всего исследования 6 (1,8%) пролеченных ликсисенатидом пациентов и 1 (0,6%) пролеченный плацебо пациент сообщили о 7 TEAE с помощью предварительно утвержденной формы AE (таблица 30). Среди них 7 пациентов, один лечившийся ликсисенатидом пациент (#840614004) имели AE панкреатита.
Пациент 840614004 (ликсисенатид): пациент мужского пола 60 лет, в истории болезни гиперплазия предстательной железы, вывих поясничного диска (L4), кислотный рефлюкс, гипертензия, депрессия, резекции печени вследствие абсцесса печени и хронического курения, получал инсулин гларгин, метформин и другие сопутствующие лекарственные препараты, включая антигипертензивные, антацидные и противовоспалительные и болеутоляющие лекарственные препараты.
5 апреля 2010 года (272 дня получения IP) пациент был госпитализирован с панкреатитом тяжелой интенсивности, и IP было прекращено. Пациент жаловался на периодическую рецидивирующую диарею, связанную с тошнотой, рвотой, и схваткообразную эпигастральную боль в течение одной недели. Амилаза составляла 166 МЕ/л, а липаза составляла 40 Ед/л. Симптомы пациента улучшились, и он был выписан из больницы 08 апреля 2010 года, но липаза продолжала повышаться. 06 мая 2010 года пациент проходил гастроэнтерологическую оценку, при этом компьютерная томография (CT) показала расширение протоков поджелудочной железы, по-видимому вследствие хронического панкреатита. 24 июня 2010 года, через 11 недель после последней дозы IP, у пациента развилось тяжелое обострение панкреатита, и он был госпитализирован. У пациента появилась острая абдоминальная боль с множеством эпизодов рвоты. уровень амилазы пациента составлял 159 Ед/л, а липаза составляла 1108 Ед/л. Пациент оправился от обострения панкреатита 27 июня 2010 года, и причинная оценка исследователя не была связана с IP.
Пациенты, которые имели по меньшей мере одно значение липазы или амилазы ≥3 ULN в течение периода применения исследуемого продукта, сведены в таблицу 31. Наблюдалось десять пациентов (7 [2,2%] пациентов в группе ликсисенатида и 3 [1,8%] в группе плацебо) с повышенной липазой (≥3 ULN). Один пациент в группе ликсисенатида имел повышенную амилазу (≥3 ULN), и ни одного не было в группе плацебо.
(N=167)
(N=328)
n (%) = Количество и процентная доля пациентов с любыми случаями, сообщаемыми в форме AE для предполагаемого панкреатита наряду с дополняющей формой.
Исходный уровень по критерию PCSA n/N1 (%)
(N=167)
(N=328)
Всего*
Всего*
Период применения исследуемого продукта для всего исследования = время от первой дозы лекарственного средства в двойном слепом исследовании до 3 дней после введения последней дозы.
*Независимо от исходного уровня.
Примечание: Количество (n) отображает подмножество общего количества пациентов, которые по меньшей мере один раз соответствуют интересующему критерию. Знаменателем (/Nl) для каждого параметра в пределах лечебной группы является количество пациентов для группы лечения, которые имели такой параметр, оцененный после исходного уровня посредством состояния PCSA исходного уровня. Только ухудшение самого плохого случая для каждого пациента отображается исходным состоянием.
По протоколу, любое значение кальцитонина ≥20 пг/мл, подтвержденное повторным измерением, следует отслеживать и сообщать в предварительно заданной форме о нежелательных явлениях для «повышенного кальцитонина ≥20 пг/мл». В течение периода применения исследуемого продукта для всего исследования, 5 пациентов (4 [1,2%] для ликсисенатида и 1 [0,6%] для плацебо) сообщали о 5 TEAE увеличения кальцитонина в крови (таблица 32). Из этих 5 пациентов 1 лечившийся ликсисенатидом пациент (#840636032) имел значения кальцитонина ≥50 нг/л, а 4 другие имели значения кальцитонина ≥20 нг/л, но <50 нг/л. В дополнение, один пролеченный плацебо пациент имел TEAE «повышенного кальцитонина в крови», о котором сообщал исследователь (таблица 35), хотя значения были <20нг/л. Данное AE не было задокументировано в предварительно согласованной форме AE по протоколу и, вследствие этого, не включено в таблицу 32. Описание случая со значением кальцитонина ≥50 нг/л предоставлено ниже:
Пациент 840636032 (ликсисенатид): пациент мужского пола 55 лет, в истории болезни депрессия, дислипидемия и гипертензия, принимал инсулин гларгин, метформин и другие сопутствующие лекарственные препараты, включая поливитамины, гемфиброзил, валсартан, бупропион, флуоксетин и метилфенидат. 24 сентября 2009 года (24 неделя), сообщалось об AE «повышенного кальцитонина в крови» со значением, равным 47,5 нг/л, которое было результатом первого измерения кальцитонина для данного пациента. AE не сопровождалось какими-либо симптомами. В январе и феврале 2010 года кальцитонин повышался до 75,9 и 70,7 нг/л, поэтому IP было прекращено. 23 февраля 2010 года пациент посетил врача-эндокринолога и был подвергнут ультразвуковому тиреосканированию, которое выявило «нормальный» результат. При последующем наблюдении (27 мая 2010 года) кальцитонин составлял 50 нг/л. Повторное ультразвуковое тиреосканирование (28 июля 2010 года) снова было «нормальным». Врач-эндокринолог оценил событие, как «случайно выявленное повышение уровня кальцитонина неизвестного значения» и рекомендовал серийный мониторинг кальцитонина через некоторое время и повторное ультразвуковое тиреосканирование в течение года. Исследователь не установил причинно-следственной связи с IP. кальцитонин при последующем исследовании 23 декабря 2010 года составлял 60 пг/мл (локальное лабораторное значение <11 пг/мл).
n (%) = Количество и процентная доля пациентов с любыми случаями, о которых сообщается в форме AE, для повышенного кальцитонина ≥20 пг/мл.
Пациенты по меньшей мере с одним кальцитонином в сыворотке, измеренным в течение периода применения исследуемого продукта всего исследования, представлены в таблице 33 согласно 4 категориям уровня кальцитонина на исходном уровне. Восемь (2,8%) пациентов в группе ликсисенатида и 1 (0,7%) пациент в группе плацебо имели значения кальцитонина ≥20 нг/л (таблица 33), включая тех 5 пациентов (4 принимающих ликсисенатид), которые сообщали о TEAE с предварительно установленной формой AE (Таблица 32). Четыре из 8 пролеченных ликсисенатидом пациентов со значением кальцитонина ≥20 нг/л (таблица 33) не сообщали о TEAE с предварительно установленной формой AE по причине неподтвержденного уровня; 2 имели отдельное значение ≥50 нг/л (все другие измерения <20 нг/л), а 2 имели значение между ≥20 - <50 нг/л. Следует указать, что измерение кальцитонина было внесено в протокол посредством поправки к протоколу после того, как большая часть пациентов была рандомизирована. Вследствие этого, значения исходного уровня отсутствуют для большинства пациентов (209 [88,2%] для ликсисенатида и 101 [88,6%] для плацебо).
Статус исходного уровня
После исходного
(N=167)
(N=328)
Всего*
Период применения исследуемого продукта для всего исследования = время от первой дозы лекарственного средства в двойном слепом исследовании до 3 дней после введения последней дозы.
*Независимо от исходного уровня.
Примечание: числитель представляет количество пациентов, которые были в предварительно оговоренных категориях в каждой категории исходного уровня. Знаменателем для каждого параметра в пределах лечебной группы является количество пациентов для лечебной группы, которые имели данный параметр, оцененный после исходного по статусу исходного уровня.
Пациента считают только в самой плохой категории.
7. ПРИЛОЖЕНИЕ
Из анализа исключены измерения, полученные после введения резервного лечения и/или после прекращения лечения плюс 3 дня.
Для 24 недели (LOCF), в анализ включены измерения, полученные до 3 дней после последней дозы двойной слепой инъекции исследуемого продукта при или перед 12 осмотром (24 неделя), или на 169 день, если 12 осмотр (24 неделя) невозможен.
Фиг.8 - График среднего изменения HbA1c (%) по сравнению с исходным уровнем при осмотре и в конечной точке - mITT популяция.
LOCF = Перенос вперед данных последнего наблюдения.
EOT = Последнее значение в процессе лечения.
Из анализа исключены измерения, полученные после введения резервного лечения и/или после прекращения лечения плюс 3 дня.
Для 24 недели (LOCF), в анализ включены измерения, полученные до 3 дней после последней дозы двойной слепой инъекции исследуемого продукта при или перед 12 осмотром (24 неделя), или на 169 день, если 12 осмотр (24 неделя) невозможен.
Фиг.9 График среднего изменения глюкозы в плазме через 2 часа после приема пищи (ммоль/л) по сравнению с исходным уровнем при осмотре и в конечной точке - mITT популяция.
LOCF = Перенос вперед данных последнего наблюдения.
EOT = Последнее значение в процессе лечения.
Из анализа исключены измерения, полученные после введения резервного лечения и/или после прекращения лечения.
Для 24 недели (LOCF), в анализ включены измерения, полученные до даты последней дозы двойной слепой инъекции исследуемого продукта при или перед 12 осмотром (24 неделя), или на 169 день, если 12 осмотр (24 неделя) невозможен.
Фиг.10 График среднего изменения среднего значения по 7 точкам самоконтролируемой глюкозы в плазме (SMPG) (ммоль/л) по сравнению с исходным уровнем при осмотре и в конечной точке - mITT популяция.
LOCF = Перенос вперед данных последнего наблюдения.
EOT = Последнее значение в процессе лечения.
Из анализа исключены измерения, полученные после введения резервного лечения и/или после прекращения лечения.
Для 24 недели (LOCF), в анализ включены измерения, полученные до даты последней дозы двойной слепой инъекции исследуемого продукта при или перед 12 осмотром (24 неделя), или на 169 день, если 12 осмотр (24 неделя) невозможен.
Фиг.11 График среднего изменения глюкозы в плазме натощак (ммоль/л) по сравнению с исходным уровнем при осмотре и в конечной точке - mITT популяция.
LOCF = Перенос вперед данных последнего наблюдения.
EOT = Последнее значение в процессе лечения.
Из анализа исключены измерения, полученные после введения резервного лечения и/или после прекращения лечения плюс 1 день.
Для 24 недели (LOCF), в анализ включены измерения, полученные до одного дня после последней дозы двойной слепой инъекции исследуемого продукта при или перед 12 осмотром (24 неделя), или на 169 день, если 12 осмотр (24 неделя) невозможен.
Фиг.12 График среднего изменения массы тела (кг) по сравнению с исходным уровнем при осмотре и в конечной точке - mITT популяция.
LOCF = Перенос вперед данных последнего наблюдения.
EOT = Последнее значение в процессе лечения.
Из анализа исключены измерения, полученные после введения резервного лечения и/или после прекращения лечения плюс 3 дня.
Для 24 недели (LOCF), в анализ включены измерения, полученные до 3 дней после последней дозы двойной слепой инъекции исследуемого продукта при или перед 12 осмотром (24 неделя), или на 169 день, если 12 осмотр (24 неделя) невозможен.
Фиг.13 График среднего изменения дозы базального инсулина (Ед) по сравнению с исходным уровнем при осмотре и в конечной точке - mITT популяция.
LOCF = Перенос вперед данных последнего наблюдения.
EOT = Последнее значение в процессе лечения.
Из анализа исключены измерения, полученные после введения резервного лечения и/или после прекращения лечения.
Для 24 недели (LOCF), в анализ включены измерения, полученные до даты последней дозы двойной слепой инъекции исследуемого продукта при или перед 12 осмотром (24 неделя), или на 169 день, если 12 осмотр (24 неделя) невозможен.
HLGT: Групповой термин высокого уровня
HLT: Термин высокого уровня
Предпочтительный термин
(N=167)
(N=328)
Период применения исследуемого продукта для всего исследования = время от первой дозы лекарственного средства в двойном слепом исследовании до 3 дней после введения последней дозы.
MedDRA версия: 13,1.
n (%) = Количество и процентная доля пациентов по меньшей мере с одним TEAE.
Примечание: Таблица отсортирована в соответствии с международным согласованным порядком SOC и алфавитным порядком HLGT, HLT, PT.
Отображены только SOC по меньшей мере с одним PT≥1% по меньшей мере в одной группе.





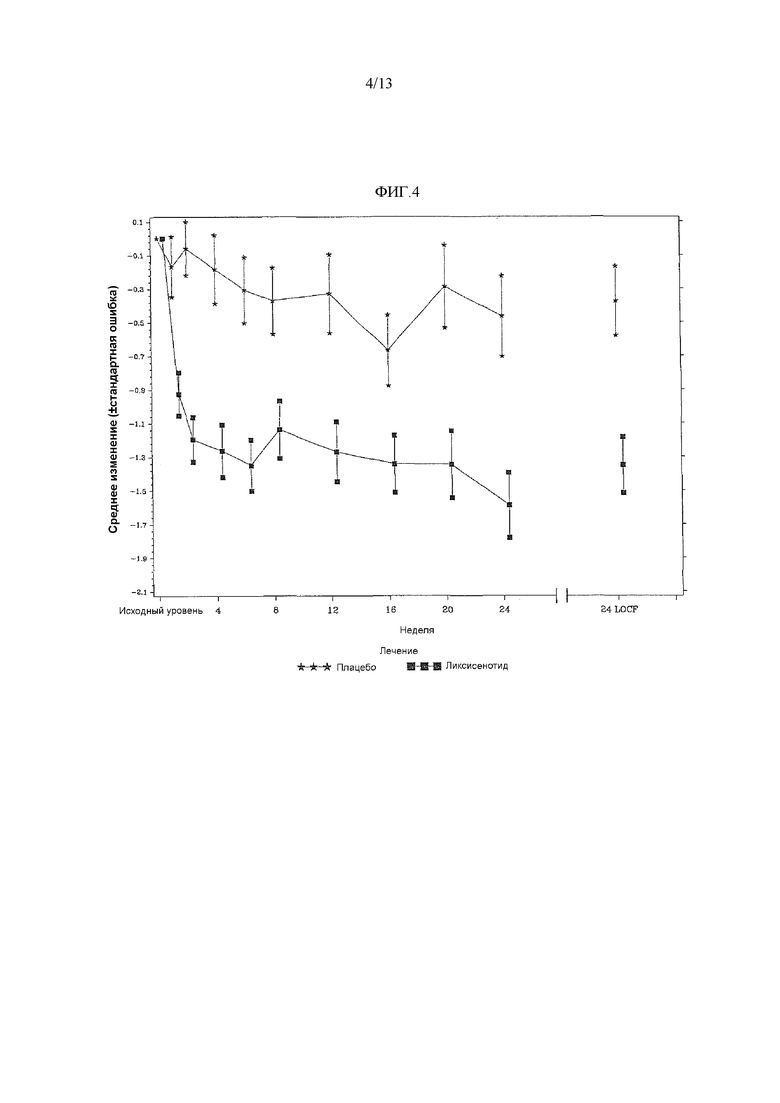
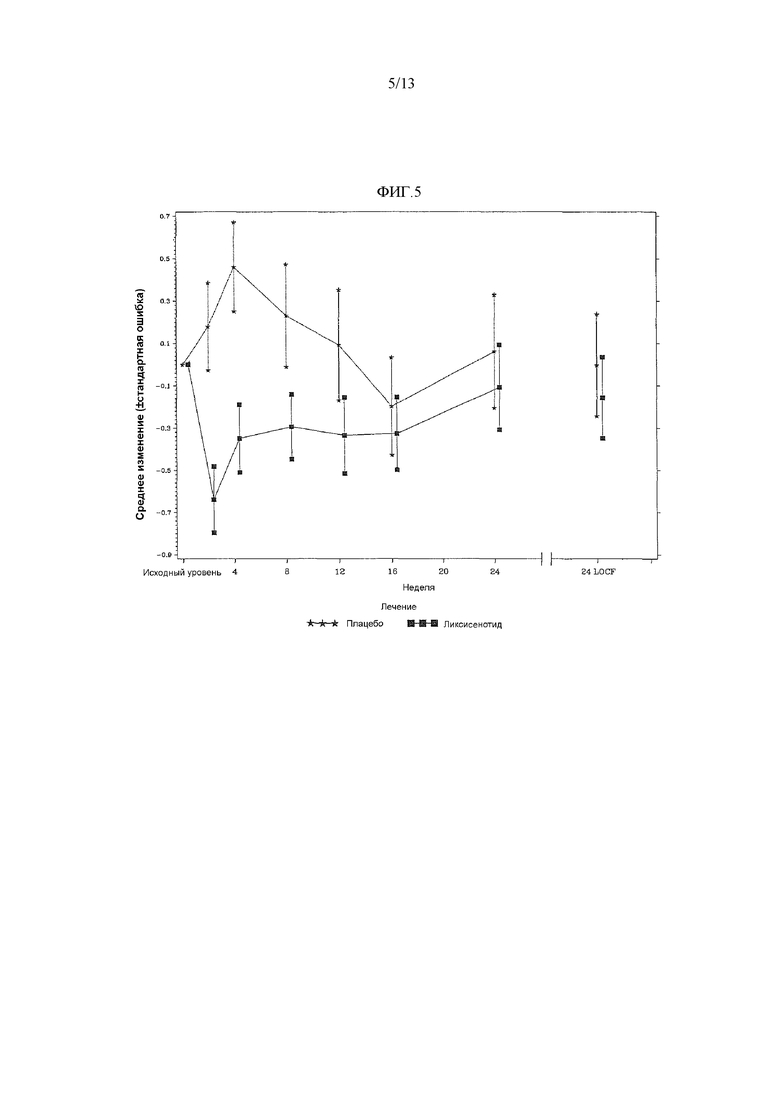
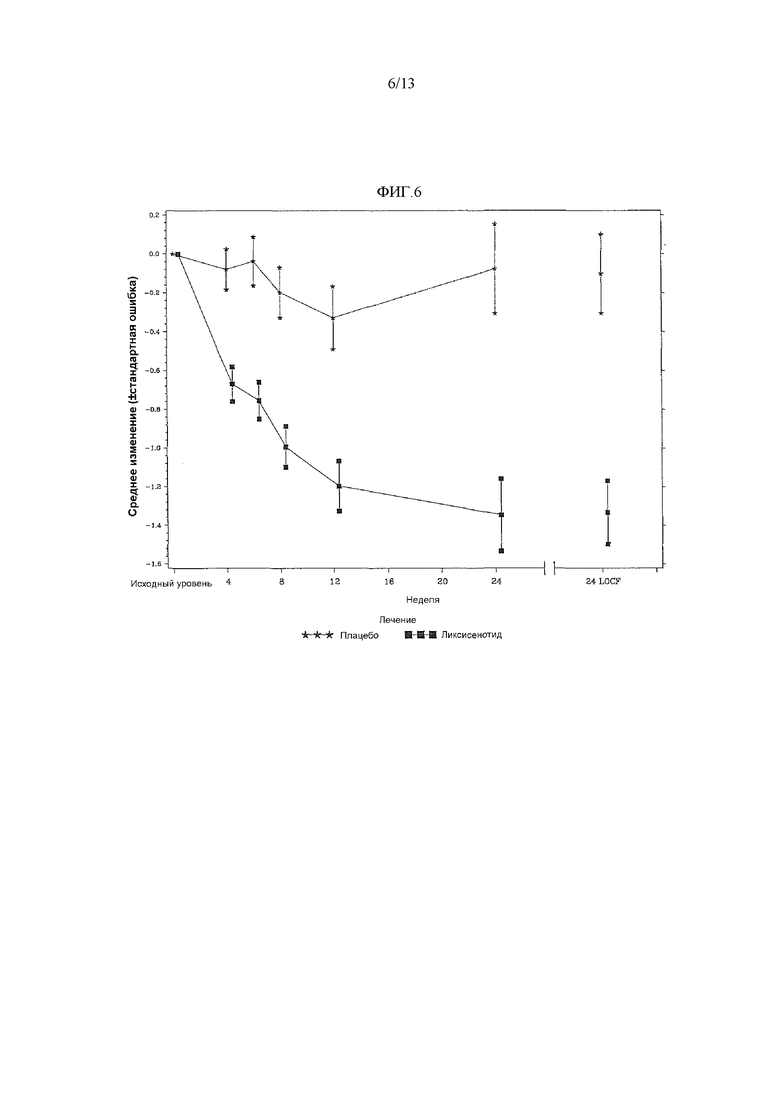







Группа изобретений относится к гликемическому контролю пациентов с диабетом 2 типа с уровнем глюкозы через 2 часа после приёма пищи по меньшей мере 14 ммоль/л, у которых гликемия не контролируется должным образом только базальным инсулином. Предложены: фармацевтическая комбинация для применения по указанному назначению, содержащая ликсисенатид (AVE0010, desPro36Эксендин-4(1-39)-Lys6-NH2 и/или его фармацевтически приемлемую соль), базальный инсулин или его фармацевтически приемлемую соль, и соответствующий способ улучшения гликемического контроля; комбинация и способ могут необязательно включать метформин или его соль. Технический результат состоит в реализации заявленного назначения и в значимом снижении гликозилированного гемоглобина HbA1c. 2 н. и 15 з.п. ф-лы, 35 табл., 13 ил.
1. Фармацевтическая комбинация для применения при гликемическом контроле у пациентов с диабетом 2 типа, где указанная комбинация содержит
(a) desPro36Эксендин-4(1-39)-Lys6-NH2 и/или его фармацевтически приемлемую соль и
(b) базальный инсулин и/или его фармацевтически приемлемую
соль,
при этом пациент, подлежащий лечению, имеет концентрацию глюкозы в плазме через 2 часа после приема пищи, равную по меньшей мере 14 ммоль/л, и при этом диабет 2 типа, подлежащий лечению, не контролируется должным образом только базальным инсулином.
2. Фармацевтическая комбинация по п. 1, где пациент, подлежащий лечению, имеет колебание уровня глюкозы, равное по меньшей мере 2 ммоль/л, по меньшей мере 3 ммоль/л, по меньшей мере 4 ммоль/л или по меньшей мере 5 ммоль/л, где колебанием глюкозы является разница концентрации глюкозы в плазме через 2 часа после приема пищи и концентрации глюкозы в плазме за 30 минут перед тестовым приемом пищи.
3. Фармацевтическая комбинация по п. 1 или 2, где пациент, подлежащий лечению, страдает ожирением.
4. Фармацевтическая комбинация по п. 1 или 2, где пациент, подлежащий лечению, имеет индекс массы тела, равный по меньшей мере 30 кг/м2.
5. Фармацевтическая комбинация по п. 1 или 2, где пациентом, подлежащим лечению, является взрослый человек.
6. Фармацевтическая комбинация по п. 1 или 2, где у пациента, подлежащего лечению, сахарный диабет 2 типа был диагностирован по меньшей мере за 1 год или по меньшей мере за 2 года до начала терапии.
7. Фармацевтическая комбинация по п. 1 или 2, где пациент, подлежащий лечению, имеет значение HbA1c, составляющее от приблизительно 7 до приблизительно 10%.
8. Фармацевтическая комбинация по п. 1 или 2, где пациент, подлежащий лечению, имеет концентрацию глюкозы в плазме натощак, равную по меньшей мере 8 ммоль/л.
9. Фармацевтическая комбинация по п. 1 или 2, где указанная комбинация дополнительно содержит
(с) метформин и/или его фармацевтически приемлемую соль,
и при этом диабет 2 типа, подлежащий лечению, не контролируется должным образом только базальным инсулином и метформином.
10. Фармацевтическая комбинация по п. 1 или 2, где desPro36Эксендин-4(1-39)-Lys6-NH2 и/или его фармацевтически приемлемая соль приготовлены для парентерального введения.
11. Фармацевтическая комбинация по п. 1 или 2, где desPro36Эксендин-4(1-39)-Lys6-NH2 и/или его фармацевтически приемлемая соль приготовлены для введения в ежедневной дозе, выбранной из диапазона, составляющего от 10 мкг до 20 мкг.
12. Фармацевтическая комбинация по п. 1 или 2, где базальный инсулин выбран из инсулина Гларгин, Детемир, NPH, Ленте, Ультраленте, Новолин, Хумалог и их смесей.
13. Фармацевтическая комбинация по п. 1 или 2, где базальный инсулин и/или его фармацевтически приемлемая соль приготовлены для парентерального введения.
14. Фармацевтическая комбинация по п. 1 или 2, где метформин и/или его фармацевтически приемлемая соль приготовлены для перорального введения.
15. Способ улучшения гликемического контроля у пациентов с диабетом 2 типа, включающий введение комбинации по любому из пп. 1-14 нуждающемуся в этом пациенту, где пациент, подлежащий лечению, имеет концентрацию глюкозы в плазме через 2 часа после приема пищи, равную по меньшей мере 14 ммоль/л, и при этом диабет 2 типа, подлежащий лечению, не контролируется должным образом только базальным инсулином.
16. Способ по п. 15, где пациентом является пациент, как определено в любом из пп. 2-9.
17. Способ по п. 15 или 16, где указанная комбинация дополнительно содержит метформин и/или его фармацевтически приемлемую соль,
и при этом диабет 2 типа, подлежащий лечению, не контролируется должным образом только базальным инсулином и метформином.
| DE102008053048 A1 29.04.2010 CHRISTENSEN M | |||
| et al | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Expert Opin.Investig.Drugs | |||
| Способ приготовления лака | 1924 |
|
SU2011A1 |
| et al | |||
| Furthet improvement in postprandial glucose control with addition of exenatide or sitagliptin to combination thherapy with insulin glargine and metformin: a proof-of-concept study | |||
| Diabetes Care | |||
| Приспособление для суммирования отрезков прямых линий | 1923 |
|
SU2010A1 |

