Объектом настоящего изобретения является фармацевтическая композиция, содержащая (a) ликсизенатид или/и его фармацевтически приемлемую соль, и (b) инсулин гларгин или/и его фармацевтически приемлемую соль, причем соединение (b) и соединение (a) присутствуют в соотношении от приблизительно 1,6 до приблизительно 2,4 ед. соединения (b) на мкг соединения (a).
У здорового человека высвобождение инсулина поджелудочной железой строго связано с концентрацией глюкозы в крови. Повышенный уровень глюкозы в крови, который появляется после приема пищи, быстро нейтрализуется соответствующим повышением секреции инсулина. В состоянии натощак уровень инсулина в плазме крови падает до исходного значения, что является достаточным для обеспечения постоянной доставки глюкозы к инсулин-чувствительным органам и тканям и для поддержания низкого уровня выработки глюкозы в печени ночью.
В отличие от диабета 1 типа, обычно при диабете 2 типа недостаток инсулина отсутствует, но во многих случаях, особенно в случаях прогрессирования, лечение при помощи инсулина считают наиболее подходящей терапией, в случае необходимости, в комбинации с перорально вводимыми противодиабетическими лекарственными средствами.
Повышенный уровень глюкозы в крови на протяжении нескольких лет без первичных симптомов является признаком значительного риска для здоровья. Это было явно продемонстрировано в масштабном исследовании DCCT в США (The Diabetes Control and Complications Trial Research Group (1993) N. Engl. J. Med. 329, 977-986), в котором хронически повышенные уровни глюкозы в крови являются главной причиной развития диабетических осложнений. Примерами диабетических осложнений являются микро- и макрососудистые повреждения, которые могут проявляться в виде ретинопатий, нефропатий или невропатий и приводить к слепоте, почечной недостаточности и потере конечностей, и при этом они сопровождаются повышенным риском сердечно-сосудистых заболеваний. Таким образом, можно сделать вывод, что целью улучшенной терапии диабета, главным образом, должно быть поддержание уровня глюкозы, настолько близкого к физиологическому диапазону, насколько это возможно.
Существует особый риск для пациентов с избыточным весом, страдающих диабетом 2 типа, например, пациентов с индексом массы тела (BMI) ≥30. У таких пациентов риски диабета перекрываются с рисками избыточного веса, что приводит, например, к повышению частоты возникновения сердечно-сосудистых заболеваний по сравнению с пациентами с диабетом 2 типа, имеющими нормальный вес. Таким образом, особенно необходимо лечение диабета у таких пациентов одновременно с уменьшением избыточного веса.
Метформин представляет собой гипогликемическое средство группы бигуанидов, применяемое для лечения инсулиннезависимого сахарного диабета (сахарного диабета 2 типа), не поддающегося коррекции модификацией рациона. Метформин улучшает гликемический контроль путем повышения чувствительности к инсулину и снижения всасывания глюкозы в кишечнике. Метформин обычно вводят перорально. Однако контроль сахарного диабета 2 типа у страдающих ожирением пациентов посредством метформина может быть недостаточным. Таким образом, для таких пациентов могут требоваться дополнительные меры контроля сахарного диабета 2 типа.
Соединение desPro36эксендин-4(1-39)-Lys6-NH2 (AVE0010, ликсизенатид) является производным эксендина-4. Ликсизенатид раскрыт как SEQ ID NO:93 в WO 01/04156.
SEQ ID NO: 1: ликсизенатид (44 аминокислоты)
H-G-E-G-T-F-T-S-D-L-S-K-Q-M-E-E-E-A-V-R-L-F-I-E-W-L-K-N-G-
G-P-S-S-G-A-P-P-S-K-K-K-K-K-K-NH2.
SEQ ID NO: 2: эксендин-4 (39 аминокислот)
H-G-E-G-T-F-T-S-D-L-S-K-Q-M-E-E-E-A-V-R-L-F-I-E-W-L-K-N-G-
g-p-s-s-g-a-p-p-p-s-nh2.
Эксендины представляют собой группу пептидов, которые могут понижать концентрацию глюкозы в крови. Аналог эксендина ликсизенатид характеризуется C-концевым усечением нативной последовательности эксендина-4. Ликсизенатид содержит шесть C-концевых остатков лизина, которые отсутствуют в эксендине-4.
В контексте настоящего изобретения ликсизенатид предусматривает его фармацевтически приемлемые соли. Специалисту в данной области техники известны фармацевтически приемлемые соли ликсизенатида. Предпочтительной фармацевтически приемлемой солью ликсизенатида, используемой согласно настоящему изобретению, является ацетат.
Инсулин гларгин представляет собой человеческий инсулин 31B-32B-Di-Arg, аналог человеческого инсулина с дополнительной заменой аспарагина в положении A21 на глицин. Инсулин гларгин также называют человеческий инсулин Gly(A21)-Arg(B31)-Arg(B32). В настоящем изобретении инсулин гларгин предусматривает его фармацевтически приемлемые соли.
Инсулин гларгин раскрыт в US 5656722.
Lantus® является инсулиновым продуктом, содержащим инсулин гларгин, обеспечивающим доставку инсулина в течение 24 часов с поддержанием исходного уровня после подкожной инъекции однократной дозы.
Для дозы, составляющей 100 ед. инсулина гларгина, требуется 1 мл Lantus® U100, каждый мл Lantus® U100 содержит 100 ед. инсулина гларгина. 100 ед. инсулина гларгина соответствуют 3,6378 мг инсулина гларгина.
В WO 2011/147980 раскрыта получаемая на месте использования смесь, содержащая постоянную концентрацию инсулина гларгина и изменяемую концентрацию ликсизенатида. В данном документе также раскрыт иллюстративный смешиваемый на месте использования препарат, содержащий 100 ед./мл инсулина гларгина и 66,67 мкг/мл (или 800/300*25 мкг/мл) ликсизенатида, 60,6 мкг/мл (или 800/330*25 мкг/мл) ликсизенатида, 55,56 мкг/мл (или 800/360*25 мкг/мл) ликсизенатида, 51,28 мкг/мл ликсизенатида (или 800/390*25 мкг/мл ликсизенатида), 47,62 мкг/мл (или 800/420*25) ликсизенатида, 44,44 мкг/мл (или 800/450*25 мкг/мл) ликсизенатида, 41,67 мкг/мл (или 800/480*25 мкг/мл) ликсизенатида или 39,22 мкг/мл (или 800/510*25 мкг/мл) ликсизенатида.
В примере 1 настоящего изобретения эффективность состава, содержащего 100 ед./мл инсулина гларгина и 50 мкг/мл ликсизенатида, была протестирована в сравнении с составом, содержащим 100 ед./мл инсулина гларгина, с участием пациентов с диабетом 2 типа.
Было продемонстрировано, что в группе комбинации (которую лечили при помощи состава с постоянным соотношением инсулина гларгина/ликсизенатида) итоговая суточная доза в конце периода лечения уменьшалась по сравнению с группой, получавшей состав для достижения концентрации глюкозы в плазме крови натощак при самостоятельном контроле от ≥80 до <100 мг/дл. В группе комбинации 0% получали дозу >60 ед./30 мкг, и 42,2% получали дозу от >40 ед./20 мкг до ≤60 ед./30 мкг. В контрольной группе 28,4% пациентов получали дозу от >40 ед./20 мкг до ≤60 ед./30 мкг, и 16,7% пациентов получали дозу >60 ед./30 мкг. В группе комбинации 14,3% пациентов получали дозу <20 ед./10 мкг, при этом только 9,9% пациентов в контрольной группе получали такую дозу (таблица 6).
Кроме того, лечение при помощи комбинации с постоянным соотношением инсулина гларгина/ликсизенатида значимо улучшало постпрандиальный гликемический контроль в сравнении с инсулином гларгином, как показано при помощи результатов для оценки уровня PPG через 2 часа и отклонения уровня глюкозы через 2 часа. В дополнение к этому, у пациентов, которых лечили при помощи комбинации с постоянным соотношением инсулина гларгина/ликсизенатида, наблюдалось статистически значимо большее снижение средних значений в профиле уровня глюкозы в плазме крови при самостоятельном контроле (SMPG) по 7 моментам времени по сравнению с пациентами, которых лечили при помощи инсулина гларгина.
Было обнаружено статистически значимое различие по изменению массы тела от начала исследования до недели 24 между 2 группами лечения: масса тела снижалась в группе комбинации с постоянным соотношением инсулина гларгина/ликсизенатида и повышалась в группе инсулина гларгина.
В группе комбинации у более высокой процентной доли пациентов целевое значение HbA1c достигало ≤6,5% (71,9% по сравнению с 64,6%) или <7% (84,4% по сравнению с 78,3%) по сравнению с группой инсулина гларгина (таблица 8).
Таким образом, комбинация с постоянным соотношением инсулина гларгина/ликсизенатида приводит в результате к улучшению гликемического контроля и массы тела при уменьшенной дозе инсулина гларгина по сравнению с инсулином гларгином отдельно. Это указывает на преимущество комбинации с постоянным соотношением инсулина гларгина/ликсизенатида по сравнению с инсулином гларгином.
В примере 2 описано рандомизированное многоцентровое открытое исследование с 3 группами лечения, с использованием активного контрольного препарата, с параллельными группами, продолжительностью 30 недель, в котором сравнивали эффективность и безопасность комбинации с постоянным соотношением инсулина гларгина/ликсизенатида согласно настоящему изобретению с инсулином гларгином отдельно и с ликсизенатидом отдельно в дополнение к метформину у пациентов с сахарным диабетом 2 типа (T2DM).
Одним аспектом настоящего изобретения является фармацевтическая композиция, содержащая
ликсизенатид (desPro36эксендин-4(1-39)-Lys6-NH2) или/и его фармацевтически приемлемую соль, и
инсулин гларгин или/и его фармацевтически приемлемую соль,
где соединение (b) и соединение (a) присутствуют в соотношении от приблизительно 1,6 до приблизительно 2,4 ед. соединения (b) на мкг соединения (a).
Соединение (b) и соединение (a) также могут присутствовать в соотношении от приблизительно 1,8 до приблизительно 2,2 ед. соединения (b) на мкг соединения (a). Соединение (b) и соединение (a) также могут присутствовать в соотношении от приблизительно 1,9 до приблизительно 2,1 ед. соединения (b) на мкг соединения (a). Соединение (b) и соединение (a) также могут присутствовать в соотношении приблизительно 2 ед. соединения (b) на мкг соединения (a).
Соотношение концентрации соединения (b) к соединению (a) в фармацевтической композиции согласно настоящему изобретению является постоянным соотношением.
Согласно настоящему изобретению соединение (a) и соединение (b) предоставлены в одной композиции в предварительно определенном постоянном соотношении. Также объем настоящего изобретения охватывает две отдельные композиции, причем первая композиция содержит соединение (a), а вторая композиция содержит соединение (b), для введения пациенту, нуждающемуся в этом, как определено в данном документе, в постоянном соотношении, которое определено в данном документе.
В композиции согласно настоящему изобретению концентрация соединения (a) может находиться в диапазоне 40-60 мкг/мл. Соотношение концентрации соединения (b) к соединению (a) может находиться в диапазоне от 1,6 до 2,4 ед./мкг, от 1,8 до 2,2 ед./мкг, от 1,9 до 2,1 ед./мкг или может составлять приблизительно 2 ед./мкг.
В композиции согласно настоящему изобретению концентрация соединения (b) может находиться в диапазоне 64-144 ед./мл, 72-132 ед./мл, 76-126 ед./мл или 80-120 ед./мл.
В композиции согласно настоящему изобретению концентрация соединения (a) может находиться в диапазоне 40-60 мкг/мл, а концентрация соединения (b) может находиться в диапазоне 64-144 ед./мл, 72-132 ед./мл, 76-126 ед./мл или 80-120 ед./мл.
В композиции согласно настоящему изобретению концентрация соединения (a) может находиться в диапазоне 45-55 мкг/мл. Соотношение концентрации соединения (b) к соединению (a) может находиться в диапазоне от 1,6 до 2,4 ед./мкг, от 1,8 до 2,2 ед./мкг, от 1,9 до 2,1 ед./мкг или может составлять приблизительно 2 ед./мкг.
В композиции согласно настоящему изобретению концентрация соединения (b) может находиться в диапазоне 72-132 ед./мл, 81-121 ед./мл, 85,5-115,5 ед./мл или 90-110 ед./мл.
В композиции согласно настоящему изобретению концентрация соединения (a) может находиться в диапазоне 45-55 мкг/мл, а концентрация соединения (b) может находиться в диапазоне 72-132 ед./мл, 81-121 ед./мл, 85,5-115,5 ед./мл или 90-110 ед./мл.
В фармацевтической композиции концентрация соединения (a) также может составлять приблизительно 50 мкг/мл. Соотношение концентрации соединения (b) к соединению (a) может находиться в диапазоне от 1,6 до 2,4 ед./мкг, от 1,8 до 2,2 ед./мкг, от 1,9 до 2,1 ед./мкг или может составлять приблизительно 2 ед./мкг. Концентрация соединения (b) может находиться в диапазоне 80-120 ед./мл, 90-110 ед./мл, 95-105 ед./мл или может составлять приблизительно 100 ед./мл.
В частности, в композиции согласно настоящему изобретению концентрация соединения (a) составляет приблизительно 50 мкг/мл, а концентрация соединения (b) составляет приблизительно 100 ед./мл.
Фармацевтическая композиция предпочтительно не является смешиваемой на месте использования композицией или составом. Смешиваемую на месте использования композицию или состав получают “на месте использования”, например, незадолго до введения. В таком контексте, смешиваемая на месте использования композиция или состав может представлять собой композицию или состав, полученный по меньшей мере из двух отдельных композиций, при этом каждая содержит по меньшей мере одно из ликсизенатида и инсулина гларгина. В частности, смешиваемый на месте использования состав или композиция представляет собой композицию, полученную из двух отдельных композиций, при этом первая композиция содержит ликсизенатид и инсулин гларгин, а вторая композиция содержит инсулин гларгин. Более конкретно, смешиваемая на месте использования композиция или состав может содержать постоянный объем первой композиции и изменяемый объем второй композиции.
Если фармацевтическая композиция содержит соединение (a) при концентрации в диапазоне от 40 до 60 мкг/мл, то концентрация соединения (a) предпочтительно не равна концентрации, выбранной из 55,56 мкг/мл, 51,28 мкг/мл, 47,62 мкг/мл, 44,44 мкг/мл и 41,67 мкг/мл. При концентрации в диапазоне от 40 до 60 мкг/мл концентрация соединения (a) предпочтительно не равна концентрации, выбранной из 800/360*25 мкг/мл, 800/390*25 мкг/мл, 800/420*25 мкг/мл, 800/450*25 мкг/мл и 800/480*25 мкг/мл.
Если фармацевтическая композиция содержит соединение (a) при концентрации в диапазоне от 45 до 55 мкг/мл, то концентрация соединения (a) предпочтительно не равна концентрации, выбранной из 51,28 мкг/мл и 47,62 мкг/мл. При концентрации в диапазоне от 45 до 55 мкг/мл концентрация соединения (a) предпочтительно не равна концентрации, выбранной из 800/390*25 мкг/мл и 800/420*25 мкг/мл.
Композицию согласно настоящему изобретению можно применять для лечения пациентов с сахарным диабетом 1 или/и 2 типа или/и для лечения состояний, ассоциированных с диабетом, относящимся к сахарному диабету 1 или/и 2 типа.
В частности, композицию согласно настоящему изобретению можно применять для лечения пациентов с сахарным диабетом 2 типа или/и для лечения состояний, ассоциированных с диабетом, относящимся к сахарному диабету 2 типа. Такие состояния включают уменьшение толерантности к глюкозе, повышенную концентрацию глюкозы в плазме крови после приема пищи, повышение концентрации глюкозы в плазме крови натощак или/и повышенное значение HbA1c по сравнению, например, с людьми, не страдающими диабетом 2 типа.
Композицию согласно настоящему изобретению можно применять для гликемического контроля у пациентов c диабетом 2 типа. Как продемонстрировано в примере 1 настоящего изобретения, композицию, которая описана в данном документе, можно применять для улучшения гликемического контроля. Согласно настоящему изобретению “улучшение гликемического контроля” или “гликемический контроль”, в частности, относится к улучшению толерантности к глюкозе, улучшению концентрации глюкозы в плазме крови после приема пищи, улучшению концентрации глюкозы в плазме крови натощак или/и улучшению значения HbA1c.
В частности, улучшение толерантности к глюкозе предусматривает улучшение концентрации глюкозы в плазме крови после приема пищи, улучшение отклонения уровня глюкозы в плазме крови после приема пищи или/и улучшение концентрации глюкозы в плазме крови натощак. Более конкретно, улучшение толерантности к глюкозе предусматривает улучшение концентрации глюкозы в плазме крови после приема пищи.
В частности, улучшение концентрации глюкозы в плазме крови после приема пищи представляет собой уменьшение концентрации глюкозы в плазме крови после приема пищи. Уменьшение означает, в частности, что концентрация глюкозы в плазме крови достигает нормогликемических значений или по меньшей мере приближается к таким значениям.
В частности, улучшение отклонения уровня глюкозы в плазме крови после приема пищи представляет собой уменьшение отклонения уровня глюкозы в плазме крови после приема пищи. Уменьшение означает, в частности, что отклонение уровня глюкозы в плазме крови достигает нормогликемических значений или по меньшей мере приближается к таким значениям.
В частности, улучшение концентрации глюкозы в плазме крови натощак представляет собой уменьшение концентрации глюкозы в плазме крови натощак. Уменьшение означает, в частности, что концентрация глюкозы в плазме крови достигает нормогликемических значений или по меньшей мере приближается к таким значениям.
В частности, улучшение значения HbA1c представляет собой уменьшение значения HbA1c. Уменьшение значения HbA1c, в частности, означает, что значение HbAlc уменьшается до уровня ниже 6,5% или 7%, например, после лечения на протяжении по меньшей мере одного месяца, по меньшей мере двух месяцев, по меньшей мере трех месяцев, по меньшей мере четырех месяцев, по меньшей мере пяти месяцев, по меньшей мере шести месяцев или по меньшей мере одного года.
Фармацевтическую композицию согласно настоящему изобретению можно вводить в качестве дополнения к лечению при помощи метформина или/и его фармацевтически приемлемой соли. Метформин является международным непатентованным названием 1,1-диметилбигуанида (номер согласно CAS 657-24-9). Согласно настоящему изобретению термин "метформин" охватывает его любую фармацевтически приемлемую соль.
Согласно настоящему изобретению метформин можно вводить перорально. Специалисту в данной области техники известны составы на основе метформина, пригодные для лечения диабета 2 типа посредством перорального введения. Метформин можно вводить пациенту, нуждающемуся в этом, в количестве, достаточном для того, чтобы вызвать терапевтический эффект. Метформин можно вводить при дозе по меньшей мере 1,0 г/сутки или по меньшей мере 1,5 г/сутки. Для перорального введения метформин может быть составлен в твердой лекарственной форме, такой как таблетка или пилюля. Метформин может быть составлен с подходящими фармацевтически приемлемыми носителями, вспомогательными средствами или/и вспомогательными веществами.
Согласно настоящему изобретению термины "дополнение", "дополнительное лечение", "дополнительная терапия" и "в дополнение к" относятся к лечению сахарного диабета 2 типа при помощи метформина и композиции согласно настоящему изобретению, как описано в данном документе. Композицию согласно настоящему изобретению и метформин можно вводить посредством разных путей введения. Метформин можно вводить перорально, а композицию согласно настоящему изобретению можно вводить парентерально.
Пациент, подлежащий лечению посредством композиции согласно настоящему изобретению, может представлять собой пациента, страдающего диабетом 2 типа.
Пациент, страдающий диабетом 2 типа, подлежащий лечению посредством композиции согласно настоящему изобретению, может представлять собой пациента, страдающего диабетом 2 типа, причем диабет 2 типа надлежащим образом не контролируется посредством лечения метформином отдельно, например, посредством лечения метформином на протяжении по меньшей мере 2 или по меньшей мере 3 месяцев, например, при помощи дозы метформина по меньшей мере 1,0 г/сутки или по меньшей мере 1,5 г/сутки. В частности, диабет 2 типа надлежащим образом не контролируется посредством лечения метформином отдельно в начале лечения при помощи композиции согласно настоящему изобретению.
Пациент, страдающий диабетом 2 типа, подлежащий лечению посредством композиции согласно настоящему изобретению, может представлять собой пациента, страдающего диабетом 2 типа, причем диабет 2 типа надлежащим образом не контролируется посредством лечения инсулином гларгином отдельно, например, посредством лечения инсулином гларгином на протяжении по меньшей мере 2 или по меньшей мере 3 месяцев. В частности, диабет 2 типа надлежащим образом не контролируется посредством лечения инсулином гларгином отдельно в начале лечения при помощи композиции согласно настоящему изобретению.
Пациент, страдающий диабетом 2 типа, подлежащий лечению посредством композиции согласно настоящему изобретению, может представлять собой пациента, страдающего диабетом 2 типа, причем диабет 2 типа надлежащим образом не контролируется посредством лечения ликсизенатидом отдельно, например, посредством лечения ликсизенатидом на протяжении по меньшей мере 2 или по меньшей мере 3 месяцев. В частности, диабет 2 типа надлежащим образом не контролируется посредством лечения ликсизенатидом отдельно в начале лечения при помощи композиции согласно настоящему изобретению.
Пациент, страдающий диабетом 2 типа, подлежащий лечению посредством композиции согласно настоящему изобретению, может представлять собой пациента, страдающего диабетом 2 типа, причем диабет 2 типа надлежащим образом не контролируется посредством лечения метформином и инсулином гларгином отдельно или метформином и ликсизенатидом отдельно, например, посредством лечения на протяжении по меньшей мере 2 или по меньшей мере 3 месяцев. В частности, диабет 2 типа надлежащим образом не контролируется посредством лечения метформином и инсулином гларгином отдельно или метформином и ликсизенатидом отдельно в начале лечения при помощи композиции согласно настоящему изобретению.
Согласно настоящему изобретению у пациента надлежащим образом не контролируется диабет 2 типа, если значения для по меньшей мере одного физиологического параметра, описывающего концентрацию глюкозы в крови (т.е. значение HbA1c, концентрация глюкозы в плазме крови после приема пищи, отклонение уровня глюкозы в плазме крови после приема пищи или/и концентрация глюкозы в плазме крови натощак) превышают нормогликемические, как описано в данном документе. В частности, у пациента, у которого диабет 2 типа надлежащим образом не контролируется, может быть
значение HbA1c в диапазоне от 7% до 10% или даже больше,
отклонение уровня глюкозы после приема пищи, в частности, отклонение уровня глюкозы через 2 часа после приема пищи, по меньшей мере 2 ммоль/л,
концентрация глюкозы в плазме крови после приема пищи, в частности, концентрация глюкозы через 2 часа после приема пищи, по меньшей мере 10 ммоль/л, или/и
концентрация глюкозы в плазме крови натощак по меньшей мере 7,0 ммоль/л или 8,0 ммоль/л.
Пациент, страдающий диабетом 2 типа, подлежащий лечению посредством композиции согласно настоящему изобретению, может представлять собой пациента, страдающего ожирением. Согласно настоящему изобретению пациент, страдающий ожирением, может иметь индекс массы тела по меньшей мере 30 кг/м2, по меньшей мере 31 кг/м2, по меньшей мере 32 кг/м2 или по меньшей мере 33 кг/м2.
Пациент, страдающий диабетом 2 типа, подлежащий лечению посредством композиции согласно настоящему изобретению, может иметь нормальную массу тела. Согласно настоящему изобретению пациент, имеющий нормальную массу тела, может иметь индекс массы тела в диапазоне от 17 кг/м2 до 25 кг/м2, от 17 кг/м2 до <30 кг/м2, или составляющий <30 кг/м2.
Пациент, подлежащий лечению посредством композиции согласно настоящему изобретению, может быть взрослым пациентом. Возраст пациента может быть по меньшей мере 18 лет, или возраст может находиться в диапазоне от 18 до 80 лет, от 18 до 50 лет, или от 40 до 80 лет, или от 50 до 60 лет. Возраст пациента может быть по меньшей мере 50 лет. Пациент может быть младше 50 лет.
Пациент, подлежащий лечению посредством композиции согласно настоящему изобретению, может представлять собой пациента, который не получает противодиабетическое лечение, например, инсулином или/и родственными соединениями, метформином или агонистами GLP-1, такими как ликсизенатид. В частности, пациент, подлежащий лечению, не получает агонист рецептора GLP-1 или/и инсулин.
Пациент, подлежащий лечению посредством композиции согласно настоящему изобретению, может страдать от сахарного диабета 2 типа на протяжении по меньшей мере 1 года или по меньшей мере 2 лет. В частности, у пациента с диабетом 2 типа, сахарный диабет 2 типа был диагностирован по меньшей мере за 1 год или по меньшей мере за 2 года до начала терапии посредством композиции согласно настоящему изобретению.
Значение HbA1c у пациента c диабетом 2 типа может составлять по меньшей мере приблизительно 9%, по меньшей мере 8%, по меньшей мере приблизительно 7,5% или по меньшей мере 7,0% в начале лечения посредством композиции. Значение HbA1c у пациента также может составлять от приблизительно 7% до приблизительно 10% в начале лечения посредством композиции. В примере 1 настоящего изобретения продемонстрировано, что лечение ликсизенатидом приводит в результате к уменьшению значения HbA1c у пациентов c диабетом 2 типа.
Согласно еще одному аспекту настоящего изобретения композицию, которая описана в данном документе, можно применять для улучшения значения HbA1c у пациента, страдающего диабетом 2 типа, как описано в данном документе.
Согласно еще одному аспекту настоящего изобретения композицию, которая описана в данном документе, можно применять для улучшения толерантности к глюкозе у пациента, страдающего диабетом 2 типа, как описано в данном документе. В примере 1 настоящего изобретения продемонстрировано улучшенное отклонение уровня глюкозы через 2 часа.
Согласно еще одному аспекту настоящего изобретения композицию, которая описана в данном документе, можно применять для улучшения концентрации глюкозы в плазме крови после приема пищи у пациента, страдающего диабетом 2 типа, как описано в данном документе. В примере 1 настоящего изобретения продемонстрирована улучшенная концентрация глюкозы через 2 часа после приема пищи.
Согласно еще одному аспекту настоящего изобретения композицию, которая описана в данном документе, можно применять для улучшения отклонения уровня глюкозы в плазме крови после приема пищи, в частности, отклонения уровня глюкозы через 2 часа после приема пищи, у пациента, страдающего диабетом 2 типа, как описано в данном документе.
Согласно еще одному аспекту настоящего изобретения композицию, которая описана в данном документе, можно применять для улучшения концентрации глюкозы в плазме крови натощак у пациента, страдающего диабетом 2 типа, как описано в данном документе.
Согласно еще одному аспекту настоящего изобретения композицию, которая описана в данном документе, можно применять для улучшения профиля средних значений SMPG по 7 моментам времени. В примере 1 согласно настоящему изобретению продемонстрирован улучшенный профиль средних значений SMPG по 7 моментам времени при введении пациентам с диабетом 2 типа композиции согласно настоящему изобретению. Как используется в данном документе, “уровень глюкозы в плазме крови при самостоятельном контроле (SMPG)” представляет собой, в частности, “уровень глюкозы в плазме крови при самостоятельном контроле по 7 моментам времени”. “Уровень глюкозы в плазме крови при самостоятельном контроле по 7 моментам времени”, в частности, относится к измерению уровня глюкозы в плазме крови семь раз в сутки и расчету, исходя из этого, средней концентрации глюкозы в плазме крови. Значение “уровня глюкозы в плазме крови при самостоятельном контроле по 7 моментам времени” представляет собой, в частности, среднюю концентрацию глюкозы в плазме крови, в том числе в состоянии натощак и после приема пищи. В частности, измерения концентрации глюкозы в плазме крови осуществляют перед завтраком, после завтрака (например, через 2 часа после завтрака), перед вторым завтраком, после второго завтрака (например, через 2 часа после второго завтрака), перед обедом, после обеда (например, через 2 часа после обеда) и перед сном (см. также фигуру 3). Лечение посредством комбинации согласно настоящему изобретению, как описано в данном документе, может улучшить уровень глюкозы в плазме крови при самостоятельном контроле.
Согласно еще одному аспекту настоящего изобретения композицию, которая описана в данном документе, можно применять для улучшения массы тела у пациента, страдающего диабетом 2 типа, как описано в данном документе. В примере 1 согласно настоящему изобретению продемонстрировано улучшение массы тела при введении композиции согласно настоящему изобретению.
Согласно настоящему изобретению нормогликемические значения представляют собой значения концентрации глюкозы в крови, в частности, 60-140 мг/дл (соответствующие 3,3-7,8 мМ/л). Этот диапазон относится, в частности, к значениям концентрации глюкозы в крови в состоянии натощак или/и в состоянии после приема пищи.
У пациента c диабетом 2 типа концентрация глюкозы в плазме крови через 2 часа после приема пищи может составлять по меньшей мере 10 ммоль/л, по меньшей мере 12 ммоль/л, по меньшей мере 13 ммоль/л, по меньшей мере 14 ммоль/л, по меньшей мере 15 ммоль/л, по меньшей мере 16 ммоль/л или по меньшей мере 17 ммоль/л в начале лечения при помощи композиции согласно настоящему изобретению. Такие значения концентрации глюкозы в плазме крови превышают нормогликемические значения концентрации.
У пациента c диабетом 2 типа отклонение уровня глюкозы (в частности, отклонение уровня глюкозы через 2 часа после приема пищи) может составлять по меньшей мере 2 ммоль/л, по меньшей мере 3 ммоль/л, по меньшей мере 4 ммоль/л, по меньшей мере 5 ммоль/л, по меньшей мере 5,5 ммоля/л, по меньшей мере 6 ммоль/л, по меньшей мере 6,5 ммоля/л или по меньшей мере 7 ммоль/л в начале лечения при помощи композиции согласно настоящему изобретению. Согласно настоящему изобретению отклонение уровня глюкозы представляет собой, в частности, разность между концентрацией глюкозы в плазме крови через 2 часа после приема пищи и концентрацией глюкозы в плазме крови за 30 минут до теста толерантности к пище.
“После приема пищи” является термином, хорошо известным специалисту в области диабетологии. Термин “после приема пищи” описывает, в частности, фазу после еды или/и приема глюкозы в экспериментальных условиях. У здорового человека эта фаза характеризуется повышением и последующим понижением концентрации глюкозы в крови. Термин “после приема пищи” или “фаза после приема пищи” обычно охватывает 2 часа после еды или/и приема глюкозы.
У пациента c диабетом 2 типа, как раскрыто в данном документе, концентрация глюкозы в плазме крови натощак может составлять по меньшей мере 7 ммоль/л, по меньшей мере 8 ммоль/л, по меньшей мере 9 ммоль/л, по меньшей мере 10 ммоль/л или по меньшей мере 11 ммоль/л в начале лечения при помощи композиции согласно настоящему изобретению. Такие значения концентрации глюкозы в плазме крови превышают нормогликемические значения концентрации в начале лечения при помощи композиции согласно настоящему изобретению.
У пациента c диабетом 2 типа, как раскрыто в данном документе, концентрация глюкозы в плазме крови при самостоятельном контроле может составлять по меньшей мере 8 ммоль/л, по меньшей мере 9 ммоль/л, по меньшей мере 10 ммоль/л или по меньшей мере 11 ммоль/л в начале лечения при помощи композиции согласно настоящему изобретению.
Согласно настоящему изобретению композицию, которая описана в данном документе, можно вводить пациенту, нуждающемуся в этом, в количестве, достаточном для того, чтобы индуцировать терапевтический эффект.
Согласно настоящему изобретению композиция, которая описана в данном документе, может содержать по меньшей мере одно из подходящих фармацевтически приемлемых носителей, вспомогательных средств или/и вспомогательных веществ.
Композицию, которая описана в данном документе, можно вводить парентерально, например, посредством инъекции (к примеру, внутримышечной инъекцией или подкожной инъекции). Известны подходящие устройства для инъекций, например, так называемые "шприц-ручки", которые содержат картридж, содержащий активный ингредиент, и инъекционную иглу.
Фармацевтическую композицию согласно настоящему изобретению можно предоставлять в емкости, например, ампуле, флаконе или “шприц-ручке”, как описано в данном документе, предназначенной для использования пациентом. Например, фармацевтическую композицию, представляющую собой жидкий состав, можно предоставлять во флаконе. Из такого флакона пациент может набирать необходимую дозу в шприц (в частности, в одноразовый шприц).
Композицию, которая описана в данном документе, можно вводить в подходящем количестве.
Дозу композиции согласно настоящему изобретению можно определять по одному из активных средств композиции, введение которой предусматривается, т.е. по количеству инсулина гларгина или по количеству ликсизенатида. Полагают, что в таком случае второе активное средство композиции вводят в количестве, определяемом постоянным соотношением доз в композиции.
Дозу композиции согласно настоящему изобретению можно определять по количеству ликсизенатида, введение которого предусматривается.
Согласно настоящему изобретению композицию, которая описана в данном документе, можно вводить в количестве в диапазоне от 10 до 15 мкг ликсизенатида на дозу или от 15 до 20 мкг ликсизенатида на дозу.
Согласно настоящему изобретению композицию, которая описана в данном документе, можно вводить при суточной дозе в диапазоне от 10 до 20 мкг ликсизенатида, в диапазоне от 10 до 15 мкг ликсизенатида или в диапазоне от 15 до 20 мкг ликсизенатида.
Композицию, которая описана в данном документе, можно вводить посредством одной инъекции в сутки.
Фармацевтическую композицию согласно настоящему изобретению можно вводить при дозе ликсизенатида от 0,05 до 0,5 мкг/кг массы тела.
Дозу композиции согласно настоящему изобретению также можно определять по количеству необходимого инсулина гларгина. Например, доза инсулина гларгина, инъекция которой предусматривается, может составлять 40 ед. или менее или находиться в диапазоне от 10 до 40 ед. инсулина гларгина или от 20 ед. до 40 ед. инсулина гларгина. Доза инсулина гларгина, инъекция которой предусматривается, также может составлять 60 ед. или менее или находиться в диапазоне от 10 ед. до 60 ед. инсулина гларгина или от 30 ед. до 60 ед. инсулина гларгина. Суточная доза инсулина гларгина, инъекция которой предусматривается, может составлять 40 ед. или менее или находиться в диапазоне от 10 до 40 ед. инсулина гларгина или от 20 ед. до 40 ед. инсулина гларгина. Суточная доза инсулина гларгина, инъекция которой предусматривается, также может составлять 60 ед. или менее или находиться в диапазоне от 10 ед. до 60 ед. инсулина гларгина или от 30 ед. до 60 ед. инсулина гларгина.
Композицию согласно настоящему изобретению можно вводить при дозе инсулина гларгина от 0,25 до 1,5 ед./кг массы тела.
Согласно настоящему изобретению, композиция, которая описана в данном документе, может представлять собой жидкую композицию. Специалисту в данной области техники известны жидкие композиции ликсизенатида, подходящие для парентерального введения. Специалисту в данной области техники известны жидкие композиции инсулина гларгина, подходящие для парентерального введения. Жидкая композиция согласно настоящему изобретению может иметь кислое или физиологическое значение pH. Кислое значение pH предпочтительно находится в диапазоне значений pH 1-6,8, pH 3,5-6,8 или pH 3,5-5. Физиологическое значение pH предпочтительно находится в диапазоне значений pH 2,5-8,5, pH 4,0-8,5 или pH 6,0-8,5. Значение pH можно регулировать с использованием фармацевтически приемлемой разбавленной кислоты (обычно HCl) или фармацевтически приемлемого разбавленного основания (обычно NaOH).
Жидкая композиция согласно настоящему изобретению может содержать подходящий консервант. Подходящий консервант можно выбрать из фенола, м-крезола, бензилового спирта и эфира п-гидроксибензойной кислоты. Предпочтительным консервантом является м-крезол.
Жидкая композиция согласно настоящему изобретению может содержать средство, регулирующее тоничность. Подходящее средство, регулирующее тоничность, можно выбрать из глицерина, лактозы, сорбита, маннита, глюкозы, NaCl, соединений, содержащих кальций или магний, таких как CaCl2. Концентрация глицерина, лактозы, сорбита, маннита и глюкозы может находиться в диапазоне 100-250 мМ. Концентрация NaCl может доходить до 150 мМ. Предпочтительным средством, регулирующим тоничность, является глицерин.
Жидкая композиция согласно настоящему изобретению может содержать от 0,5 мкг/мл до 20 мкг/мл, предпочтительно от 1 мкг/мл до 5 мкг/мл метионина. Предпочтительно жидкая композиция содержит L-метионин.
Согласно еще одному аспекту настоящее изобретение относится к способу лечения медицинского симптома, заболевания или состояния, как описано в данном документе. Например, способ может предусматривать введение композиции, которая описана в данном документе. Способ может представлять собой способ лечения пациентов с диабетом 2 типа или/и лечения состояний, ассоциированных с диабетом 2 типа, как описано в данном документе. Пациент может представлять собой пациента в соответствии с определением, приведенным в данном документе.
Согласно дополнительному аспекту настоящее изобретение относится к способу улучшения гликемического контроля у пациентов c диабетом 2 типа, причем указанный способ предусматривает введение композиции согласно настоящему изобретению пациенту, нуждающемуся в этом. Применительно к способу согласно настоящему изобретению пациент может представлять собой пациента в соответствии с определением, приведенным в данном документе.
Согласно еще одному аспекту настоящее изобретение относится к применению композиции, которая описана в данном документе, для получения композиции для лечения медицинского симптома, заболевания или состояния, как описано в данном документе. Например, композицию согласно настоящему изобретению можно применять для получения композиции для лечения пациентов c диабетом 2 типа или/и для лечения состояний, ассоциированных с диабетом 2 типа. В частности, композицию согласно настоящему изобретению можно применять для получения композиции для улучшения гликемического контроля, улучшения толерантности к глюкозе, улучшения концентрации глюкозы в плазме крови после приема пищи, улучшения отклонения уровня глюкозы в плазме крови после приема пищи, улучшения концентрации глюкозы в плазме крови натощак или/и улучшения значения HbA1c. Пациент может представлять собой пациента в соответствии с определением, приведенным в данном документе.
Настоящее изобретение дополнительно проиллюстрировано при помощи следующих примеров и фигур.
Условные обозначения к фигурам
Фигура 1 - кривая кумулятивной частоты по Каплану-Мейеру для времени до прекращения лечения по любой причине - рандомизированная популяция. INS/LIXI = комбинация с постоянным соотношением инсулина гларгина/ликсизенатида, INS = инсулин гларгин.
Фигура 2 - график среднего значения HbA1c (%) в зависимости от визита - популяция mITT. LOCF = перенос данных последнего наблюдения вперед. Примечание: график охватывал результаты измерения, полученные до введения резервной терапии и до 14 суток после последней инъекции исследуемого лекарственного препарата. INS/LIXI = комбинация с постоянным соотношением инсулина гларгина/ликсизенатида, INS = инсулин гларгин.
Фигура 3 - график профилей среднего уровня глюкозы в плазме крови при самостоятельном контроле по 7 моментам времени (SMPG) (ммоль/л) в начале исследования и на 24-й неделе (LOCF) - популяция mITT. LOCF = перенос данных последнего наблюдения вперед. Анализ охватывал результаты измерения, полученные до введения резервной терапии и до даты последней инъекции исследуемого лекарственного препарата. INS/LIXI = комбинация с постоянным соотношением инсулина гларгина/ликсизенатида, INS = инсулин гларгин.
Фигура 4 - график среднего значения изменения массы тела (кг) от начала исследования в зависимости от визита - популяция mITT. LOCF = перенос данных последнего наблюдения вперед. Анализ охватывал результаты измерения, полученные до введения резервной терапии и до 3 суток после последней инъекции исследуемого лекарственного препарата. INS/LIXI = комбинация с постоянным соотношением инсулина гларгина/ликсизенатида, INS = инсулин гларгин.
Фигура 5 - график среднего значения средней суточной дозы инсулина гларгина (ед.) в зависимости от визита - популяция mITT. LOCF = перенос данных последнего наблюдения вперед. Анализ охватывал результаты измерения, полученные до введения резервной терапии и до даты последней инъекции исследуемого лекарственного препарата. INS/LIXI = комбинация с постоянным соотношением инсулина гларгина/ликсизенатида, INS = инсулин гларгин.
Пример 1
Рандомизированное открытое многоцентровое исследование продолжительностью 24 недели с 2 параллельными группами, в котором сравнивали эффективность и безопасность комбинации с постоянным соотношением инсулина гларгина/ликсизенатида по сравнению с инсулином гларгином в дополнение к метформину у пациентов с диабетом 2 типа.
АББРЕВИАТУРЫ
AE: нежелательное явление
ANCOVA: ковариационный анализ
BMI: индекс массы тела
CI: доверительный интервал
CMH: критерий Кохрана-Мантеля-Хензеля
ECG: электрокардиограмма
FPG: уровень глюкозы в плазме крови натощак
GFR: скорость клубочковой фильтрации
GLP-1: глюкагоноподобный пептид-1
HLGT: групповой термин высокого уровня
HLT: термин высокого уровня
IMP: исследуемый лекарственный препарат
LOCF: перенос данных последнего наблюдения вперед
LS: метод наименьших квадратов
MDRD: модификация диеты при заболевании почек
mITT: модифицированная группа, сформированная в зависимости от назначенного лечения
PG: уровень глюкозы в плазме крови
PPG: уровень глюкозы в плазме крови после приема пищи
PT: предпочтительный термин
SAE: серьезное нежелательное явление
SMPG: уровень глюкозы в плазме крови при самостоятельном контроле
SOC: системно-органный класс
TEAE: нежелательное явление, возникшее в ходе лечения.
2. КРАТКОЕ ИЗЛОЖЕНИЕ СОДЕРЖАНИЯ
Название исследования: рандомизированное открытое многоцентровое исследование продолжительностью 24 недели с 2 параллельными группами, в котором сравнивали эффективность и безопасность комбинации с постоянным соотношением инсулина гларгина/ликсизенатида по сравнению с инсулином гларгином в дополнение к метформину у пациентов с диабетом 2 типа.
Исследовательский центр(центры): многоцентровое (67 центров).
Публикации (ссылка): не применимо.
Фаза разработки: фаза 2.
Цели
Первичная цель: продемонстрировать не меньшую эффективность комбинации с постоянным соотношением инсулина гларгина/ликсизенатида по сравнению с инсулином гларгином в отношении гликемического контроля на протяжении 24 недель, что оценивали по уменьшению значения HbA1c у пациентов c диабетом 2 типа, который надлежащим образом не контролируется при помощи метформина.
Вторичные цели
• Продемонстрировать преимущество комбинации с постоянным соотношением инсулина гларгина/ликсизенатида по сравнению с инсулином гларгином в отношении гликемического контроля в связи с питанием на протяжении 24 недель, что оценивали при помощи уровня глюкозы в плазме крови через 2 часа после приема пищи (PPG) и отклонения уровня глюкозы в ходе стандартизированного теста толерантности к пище.
• Оценить эффективность комбинации с постоянным соотношением инсулина гларгина/ликсизенатида при помощи следующего:
процента пациентов, у которых значение HbA1c достигает <7% или ≤6,5% на 24-й неделе;
профиля уровня глюкозы в плазме при самостоятельном контроле по 7 моментам времени (в каждый момент времени и среднее суточное значение) на 24-й неделе;
массы тела на 24-й неделе;
дозы инсулина гларгина на 24-й неделе;
уровня глюкозы в плазме крови натощак (FPG) на 24-й неделе;
процента пациентов, нуждающихся в резервной терапии, в течение 24-недельного периода открытого лечения;
уровня PPG через 30 минут и 1 час, и отклонения уровня глюкозы в плазме в ходе стандартизированного теста толерантности к пище на 24-й неделе;
процента пациентов, у которых значение HbA1c достигает <7% на 24-й неделе без задокументированной симптоматической гипогликемии на протяжении 24-недельного периода открытого лечения;
процента пациентов, у которых значение HbA1c достигает <7% без увеличения массы тела на 24-й неделе.
• Оценить безопасность и переносимость комбинации с постоянным соотношением инсулина гларгина/ликсизенатида.
• Оценить концентрацию ликсизенатида в плазме крови (в группе комбинации с постоянным соотношением инсулина гларгина/ликсизенатида) после инъекции в день 1 и на 24-й неделе.
• Оценить выработку антител к ликсизенатиду (для комбинации с постоянным соотношением инсулина гларгина/ликсизенатида) и к инсулину (для обеих групп лечения).
Методика: открытое исследование продолжительностью 24 недели с рандомизацией в соотношении 1:1 с использованием активного контрольного препарата с 2 параллельными группами, в котором сравнивали:
• комбинацию с постоянным соотношением инсулина гларгина/ликсизенатида (2 ед. инсулина гларгина на 1 мкг ликсизенатида);
• инсулин гларгина отдельно.
Пациентов стратифицировали исходя из значений HbA1c при скрининге (<8, ≥8%) и индекса массы тела (BMI) (<30, ≥30 кг/м2). Исследование предусматривает 3 периода: период скрининга в пределах 2 недель; период рандомизированного лечения в течение 24 недель; период последующего наблюдения в отношении безопасности в течение 3 дней.
Диагностика и критерии включения: пациенты с сахарным диабетом 2 типа, диагностированным на протяжении по меньшей мере 1 года, которых лечили при помощи стабильной дозы метформина по меньшей мере 1,5 г/сутки в течение по меньшей мере 3 месяцев до скринингового визита, и со значением HbA1c ≥7% и ≤10% при скрининге.
Исследуемые средства для лечения
Исследуемые лекарственные препараты (IMP): комбинация с постоянным соотношением инсулина гларгина/ликсизенатида и инсулин гларгин.
Состав
• Изучаемое лекарственное средство: комбинация с постоянным соотношением инсулина гларгина/ликсизенатида (100 ед./мл инсулина гларгина/50 мкг/мл ликсизенатида [соотношение 2 ед./1 мкг]) поставлялась в виде стерильного водного раствора в 3 мл картриджах для применения в многоразовой шприц-ручке с адаптируемой дозой (TactiPen®).
• Контрольное лекарственное средство: инсулин гларгин поставлялся в виде стерильного водного раствора в одноразовом устройстве для самостоятельного введения Lantus® SoloSTAR® (3 мл 100 ед./мл). Путь введения: подкожная инъекция.
Режим дозирования: в обеих группах изначальная суточная доза инсулина гларгина для введения на протяжении первой недели лечения составляла 10 ед. Впоследствии дозу адаптировали для достижения целевого уровня SMPG натощак в диапазоне от 80 до 100 мг/дл (от 4,4 до 5,6 ммоля/л). Дозу подбирали каждую неделю до достижения целевого уровня SMPG натощак у пациента. С этого момента и до конца исследования дозу корректировали по необходимости для поддержания уровня SMPG натощак от 80 до 100 мг/дл (от 4,4 до 5,6 ммоля/л), включительно. Дозы можно уменьшать или модифицировать в любое время по причине гипогликемии.
В группе комбинации с постоянным соотношением инсулина гларгина/ликсизенатида дозу ликсизенатида автоматически повышали или понижали после повышения или понижения дозы инсулина гларгина в соответствии с постоянным соотношением 2 ед./1 мкг, применяемым в комбинированной терапии, и при этом максимальная допустимая доза инсулина гларгина составляла 60 ед. (что соответствует дозе ликсизенатида 30 мкг). Если доза 60 ед./30 мкг не была достаточной для поддержания значения FPG/HbA1c ниже предварительно определенных пороговых значений, следует поддерживать дозу на уровне 60 ед. и применять резервную терапию.
Номер партии(партий): не применимо к KRM.
Лекарственный препарат(препараты), не являющийся исследуемым (фоновая терапия): метформин
Состав: метформин ≥1,5 г/сутки.
Путь введения: пероральный
Режим дозирования: метформин следует поддерживать при стабильной дозе на протяжении всего исследования до тех пор, пока существует определенная проблема безопасности, связанная с таким лечением.
Номер партии(партий): не применимо к KRM.
Продолжительность лечения: 24 недели
Продолжительность наблюдения: максимальная продолжительность приблизительно 27 недель.
Критерии оценки
Эффективность
Первичная конечная точка:
• изменение значения HbA1c от начала исследования до 24-й недели
Вторичные конечные точки:
• изменение PPG через 2 часа в ходе теста толерантности к пище от начала исследования до 24-й недели;
• изменение отклонения уровня глюкозы в плазме крови через 2 часа в ходе теста толерантности к пище от начала исследования до 24-й недели;
• процент пациентов, у которых значение HbA1c достигает ≤6,5% или <7% на 24-й неделе;
• изменение профилей уровня SMPG по 7 моментам времени от начала исследования до 24-й недели (в каждый момент времени и среднее суточное значение);
• изменение массы тела от начала исследования до 24-й недели;
• средняя суточная доза инсулина гларгина на 24-й неделе;
• изменение FPG от начала исследования до 24-й недели;
• процент пациентов, нуждающихся в резервной терапии, в течение 24-недельного периода открытого лечения;
• изменение уровня PPG через 30 минут и 1 час, и отклонение уровня глюкозы в плазме крови в ходе теста толерантности к пище от начала исследования до 24-й недели;
• процент пациентов, у которых значение HbA1c достигает <7% на 24-й неделе без задокументированной симптоматической гипогликемии на протяжении 24-недельного периода открытого лечения;
• процент пациентов, у которых значение HbA1c достигает <7% без увеличения массы тела на 24-й неделе.
Безопасность: нежелательные явления, серьезные нежелательные явления, симптоматическая гипогликемия, основные показатели состояния организма, электрокардиограмма (ECG), значения результатов анализов, проведенных для оценки безопасности.
Оценки антител: не применимо к KRM.
Фармакокинетика: не применимо к KRM.
Статистические методы
Эффективность. Популяция для оценки первичной эффективности представляла собой модифицированную популяцию, сформированную в зависимости от назначенного лечения (mITT), которая охватывала всех рандомизированных пациентов, которые получали по меньшей мере одну дозу исследуемого лекарственного средства и для которых определяли как оценку в начале исследования, так и по меньшей мере одну последующую оценку любых первичных или вторичных переменных эффективности, независимо от соблюдения протокола и процедур исследования.
Первичную конечную точку (изменение значения HbA1c от начала исследования до 24-й недели) анализировали при помощи модели на основе ковариационного анализа (ANCOVA) с использованием лечения (при помощи комбинации с постоянным соотношением инсулина гларгина/ликсизенатида, инсулина гларгином отдельно), рандомизационных страт исходя из значения HbA1c при скрининге (<8%, ≥8%), рандомизационных страт исходя из значения BMI при скрининге (<30 кг/м2, ≥30 кг/м2) и страны в качестве фиксированных эффектов и с использованием значения HbA1c в начале исследования в качестве ковариаты.
Не меньшую эффективность комбинации с постоянным соотношением инсулина гларгина/ликсизенатида по сравнению с инсулином гларгином отдельно тестировали при помощи 1-стороннего статистического теста с альфа-уровнем 0,025 и пределом не меньшей эффективности 0,4% HbA1c. Не меньшая эффективность будет продемонстрирована, если верхняя граница двухстороннего 95% доверительного интервала (CI) для различия между комбинацией с постоянным соотношением инсулина гларгина/ликсизенатида и инсулином гларгином отдельно в отношении популяции mITT составляет ≤0,4%.
Если не меньшая эффективность установлена, то затем можно провести соответствующую проверку преимущества комбинации с постоянным соотношением инсулина гларгина/ликсизенатида по сравнению с инсулином гларгином отдельно, определенного при помощи статистического анализа, для первичной конечной точки.
Все последующие вторичные конечные точки оценки эффективности анализировали при помощи сходной модели ANCOVA с использованием лечения, рандомизационных страт исходя из значения HbA1c при скрининге (<8%, ≥8%), рандомизационных страт исходя из значения BMI при скрининге (<30 кг/м2, ≥30 кг/м2) и страны в качестве фиксированных эффектов и с использованием значения в начале исследования для соответствующего параметра в качестве ковариаты. Дозу инсулина гларгина не включали в модель на основе ANCOVA в качестве ковариаты, поскольку пациенты, участвующие в исследовании, не подвергались воздействию инсулина.
Все вторичные конечные точки оценки эффективности, соответствующие категории, анализировали с помощью способа Кохрана-Мантеля-Хензеля (CMH) с применением стратификации на рандомизационные страты по значениям HbA1c (<8%, ≥8%) и значениям BMI (<30 кг/м2, ≥30 кг/м2) при скрининге.
Безопасность: анализ безопасности проводили в отношении популяции для оценки безопасности, к которой относили, согласно определению, всех рандомизированных пациентов, которые получали по меньшей мере одну дозу IMP (не принимая во внимание количество вводимых средств для лечения). Оценка AE, лабораторных показателей, основных показателей состояния организма и ECG была описательной.
Краткое описание
Характеристики популяции: всего 323 пациента рандомизировали в одну из двух групп лечения (161 в группу комбинации с постоянным соотношением инсулина гларгина/ликсизенатида и 162 в группу инсулина гларгина). Всех рандомизированных пациентов подвергали воздействию исследуемого средства для лечения и включали в популяцию mITT. Демографические данные и исходные характеристики в группах лечения, в целом, были сходными. Медианный возраст составлял 58 лет. Исследуемую популяцию преимущественно составляли европеоиды (98,5%).
Результаты в отношении эффективности
Средние значения изменения HbA1c, рассчитанные при помощи метода наименьших квадратов (LS), от начала исследования до 24-й недели, составляли -1,82% для группы комбинации с постоянным соотношением инсулина гларгина/ликсизенатида и -1,64% для группы инсулина гларгина (различие по среднему значению, рассчитанному при помощи LS, по сравнению с группой гларгина = -0,17%; 95% CI = от -0,312% до -0,037%). Исходя из ранее описанного первичного анализа была продемонстрирована не меньшая эффективность комбинации с постоянным соотношением инсулина гларгина/ликсизенатида по сравнению с инсулином гларгином в отношении изменения значений HbA1c от начала исследования до 24-й недели, поскольку верхняя граница двухстороннего 95% CI для различия по среднему значению, рассчитанному при помощи LS, была меньше предварительно определенного предельного значения для не меньшей эффективности, составляющего 0,4%.
Преимущество комбинации с постоянным соотношением инсулина гларгина/ликсизенатида по сравнению с инсулином гларгином, определенное при помощи статистического анализа, также было продемонстрировано для этих первичных конечных точек (различие по среднему значению, рассчитанному при помощи LS, по сравнению с группой гларгина = -0,17%; p-значение = 0,0130),
Лечение при помощи комбинации с постоянным соотношением инсулина гларгина/ликсизенатида значимо улучшало постпрандиальный гликемический контроль по сравнению с инсулином гларгином, как показано при помощи результатов для оценки уровня PPG через 2 часа (различие по среднему значению, рассчитанному при помощи LS, составляло -3,17 ммоля/л; p-значение <0,0001) и для отклонения уровня глюкозы через 2 часа (различие по среднему значению, рассчитанному при помощи LS, составляло -3,24 ммоля/л; p-значение <0,0001). В дополнение к этому, у пациентов, которых лечили при помощи комбинации с постоянным соотношением инсулина гларгина/ликсизенатида, наблюдалось статистически значимо большее снижение средних значений в профиле SMPG по 7 моментам времени по сравнению с пациентами, которых лечили при помощи инсулина гларгина (различие по среднему значению, рассчитанному при помощи LS, составляло -0,30 ммоля/л; p-значение = 0,0154).
Статистически значимое различие по изменению массы тела от начала исследования до 24-й недели наблюдалось в 2 группах лечения: масса тела понижалась в группе комбинации с постоянным соотношением инсулина гларгина/ликсизенатида и повышалась в группе инсулина гларгина (среднее значение изменения массы тела, рассчитанное при помощи LS, от начала исследования до 24-й недели, составляло -0,97 кг и +0,48 кг, соответственно; различие по среднему значению, рассчитанному при помощи LS, для комбинации с постоянным соотношением инсулина гларгина/ликсизенатида по сравнению с инсулином гларгином составляло -1,44 кг; 95% CI: -2,110 кг, -0,773 кг; p<0,0001).
Для средней суточной дозы инсулина гларгина на 24-й неделе различие между группами лечения при помощи комбинации с постоянным соотношением инсулина гларгина/ликсизенатида и лечения инсулином гларгином характеризовалось пограничной значимостью (различие по среднему значению, рассчитанному при помощи LS, составляло -3,24 ед.; 95% CI: [от -6,592 ед. до 0,114 ед.]; p =0,0583). Наблюдали сходное уменьшение среднего значения изменения уровня FPG от начала исследования до 24-й недели (среднее значение, рассчитанное при помощи LS, составляло: -3,35 ммоля/л в группе комбинации; -3,51 ммоля/л в группе инсулина гларгина). Только 1 пациент (в группе инсулина гларгина) нуждался в резервной терапии.
Результаты в отношении безопасности
Комбинация с постоянным соотношением инсулина гларгина/ликсизенатида была в целом хорошо переносимой. В группе комбинации с постоянным соотношением инсулина гларгина/ликсизенатида немного большее количество пациентов (86 [53,4%]) сообщали о нежелательных явлениях, возникших в ходе лечения (TEAE), чем в группе инсулина гларгина (82 [50,6%]). Наиболее часто сообщаемым TEAE в группе комбинации была тошнота (12 [7,5%] по сравнению с 0 в группе инсулина гларгина).
У пятнадцати пациентов (9 [5,6%] для группы комбинации и 6 [3,7%] для группы инсулина гларгина) наблюдались серьезные нежелательные явления (SAE), возникшие в ходе лечения, которые распространялись на различные системно-органные классы (SOC) без значительного увеличения для любого конкретного SOC. У шести (3,7%) пациентов, которых лечили при помощи комбинации, и ни у одного из получавших инсулин гларгин, наблюдались TEAE, приводящие к досрочному прекращению лечения: у 2 из этих пациентов TEAE, которые приводили к досрочному прекращению лечения, возникали в результате нарушений со стороны SOC желудочно-кишечного тракта (тошнота и/или рвота).
В ходе данного исследования не сообщалось о случаях смерти.
Всего 2 пациента (по 1 [0,6%] в каждой группе) сообщали о 6 случаях, квалифицированных как аллергические реакции Комитетом по оценке аллергических реакций (ARAC). Ни одни из них не был квалифицирован как возможно связанный с IMP. Всего у 5 (3,1%) пациентов в группе комбинации и 1 (0,6%) в группе инсулина гларгина наблюдались реакции в месте инъекции, при этом ни одну из них не квалифицировали как серьезную, или тяжелую, или приводящие к досрочному прекращению лечения.
В ходе исследования не сообщали о TEAE, связанных с поджелудочной железой, или повышенном уровне кальцитонина ≥20 пг/мл.
У сорока (24,8%) пациентов, которых лечили при помощи комбинации, наблюдали 81 случай симптоматической гипогликемии (включая задокументированную, тяжелую и вероятную симптоматическую гипогликемию) по сравнению с 40 (24,7%) пациентами с 84 случаями в группе инсулина гларгина. Количество случаев на пациента в год для симптоматической гипогликемии составляло 1,11 в обеих группах лечения. Не сообщали о тяжелой симптоматической гипогликемии.
Предварительные заключения
У этих пациентов с T2DM, который не контролируется с использованием метформина, была продемонстрирована не меньшая эффективность комбинации с постоянным соотношением инсулина гларгина/ликсизенатида по сравнению с инсулином гларгином в отношении изменения значений HbA1c от начала исследования до 24-й недели, поскольку верхняя граница двухстороннего 95% CI для различия по среднему значению, рассчитанному при помощи LS, была меньше определенного предельного значения для не меньшей эффективности, составляющего 0,4%. Преимущество комбинации с постоянным соотношением инсулина гларгина/ликсизенатида по сравнению с инсулином гларгином, определенное при помощи статистического анализа, также было продемонстрировано для этой первичной конечной точки.
По сравнению с инсулином гларгином, лечение при помощи комбинации с постоянным соотношением инсулина гларгина/ликсизенатида приводило к статистически значимому улучшению постпрандиального гликемического контроля (что продемонстрировано при помощи результатов для уровня PPG через 2 часа и отклонения уровня глюкозы после приема стандартной жидкой пищи на завтрак) и среднего значения в профиле SMPG по 7 моментам времени. Кроме того, данная комбинация оказывала статистически значимо лучший эффект в отношении массы тела по сравнению с инсулином гларгином.
В целом, комбинация с постоянным соотношением инсулина гларгина/ликсизенатида была хорошо переносимой. Профиль безопасности в группе комбинации в целом согласовывался с известным профилем безопасности для класса агонистов рецептора GLP-1 без существенных различий по сравнению с группой инсулина гларгина. Тошнота была наиболее часто сообщаемым нежелательным явлением в группе комбинации. Частота возникновения симптоматической гипогликемии (включая задокументированную, тяжелую и вероятную симптоматическую гипогликемию) была сходной в обеих группах лечения.
В заключение, комбинация с постоянным соотношением инсулина гларгина/ликсизенатида в дополнение к метформину для пациентов с недостаточным контролем при помощи такого лечения значимо улучшала значение HbA1c и уменьшала уровень PPG и массу тела по сравнению с инсулином гларгином. Профиль безопасности согласовывался с известными эффектами агонистов рецептора GLP-1, при этом основным AE являлась тошнота.
3. РЕЗУЛЬТАТЫ
ПАЦИЕНТЫ, УЧАСТВУЮЩИЕ В ИССЛЕДОВАНИИ
Учет пациентов
Из 520 пациентов, подвергнутых скринингу, 323 (62,1%) пациента были рандомизированы в одну из двух групп лечения (161 в группу комбинации, 162 в группу инсулина гларгина) в 67 центрах, распределенных по 13 странам (Чили, Чешская Республика, Германия, Дания, Франция, Венгрия, Литва, Мексика, Польша, Румыния, Словакия, Швеция и Соединенные Штаты Америки). Основной причиной, по которой не проходили скрининг, было значение HbA1c при скрининговом визите, выходящее за пределы установленного протоколом диапазона (133 [25,6%] из 520 пациентов, подвергнутых скринингу). Всех 323 рандомизированных пациентов подвергали открытому лечению и включали в популяции mITT для анализов эффективности (таблица 1).
Анализируемые популяции
Модифицированная группа, сформированная в зависимости от назначенного лечения (mITT)
Примечание: данные пациенты из популяции для оценки безопасности объединены в таблицу в соответствии с фактически полученным лечением (как получавшие лечение)
Для других популяций данные пациентов объединены в таблицу в соответствии с их лечением, назначенным при рандомизации.
Не было ни одного пациента, рандомизированного в группу и получавшего другое исследуемое лечение.
Распределение в ходе исследования
В таблице 2 представлено краткое описание распределения пациентов для каждой группы лечения.
В течение 24-недельного периода исследуемого лечения 11 (6,8%) пациентов, которых лечили при помощи комбинации, досрочно прекращали прием IMP по сравнению с 3 (1,9%) пациентами, которых лечили при помощи инсулина гларгина. Для пациентов, которых лечили при помощи комбинации, наиболее распространенными причинами досрочного прекращения лечения было “нежелательное явление” (6 пациентов [3,7%] по сравнению с 0 пациентов в группе инсулина гларгина), за которыми следовали “другие причины” (4 пациента [2,5%] по сравнению с 2 пациентами [1,2%] в группе инсулина гларгина).
Досрочное прекращение из-за отсутствия эффекта терапии в связи с любой причиной отображено на фигуре 1.
Распределение пациентов - рандомизированная популяция
(N=161)
161 (100%)
162 (100%)
Примечание: процентные значения рассчитаны с использованием количества рандомизированных пациентов в качестве знаменателя.
Демографические и исходные характеристики
В таблице 3 представлено краткое описание демографических данных и характеристик пациентов при скрининге или в начале исследования. Демографические данные и характеристики пациентов в целом были сходными в двух группах лечения для рандомизированной популяции. Медианный возраст составлял 58 лет. Исследуемую популяцию преимущественно составляли европеоиды (98,5%).
Демографические данные и характеристики пациентов при скрининге или в начале исследования - рандомизированная популяция
ликсизенатида
(N=161)
(N=162)
(N=323)
ликсизенатида
(N=161)
(N=323)
История болезни диабетом и характеристики заболевания в группах лечения, в целом, были сравнимыми, как показано в таблице 4. Длительность применения и средняя суточная доза метформина были сходными в двух группах лечения; при этом в начале исследования средняя доза составляла 2084,75 мг для рандомизированной популяции. Переменные эффективности в начале исследования были сходными в двух группах лечения, и при этом они показаны в разделе 3.2 ЭФФЕКТИВНОСТЬ.
Характеристики заболевания при скрининге или в начале исследования - рандомизированная популяция
(N=161)
(N=323)
3000,0
3000,0
(N=161)
(N=323)
(N=161)
(N=323)
[n (%)]
3.1.4 Доза и продолжительность
Воздействие лечения и конечная доза инсулина кратко изложены в таблицах 5 и 6. Медианная продолжительность воздействия лечения составляла 169,0 дней в каждой группе лечения.
Воздействие исследуемого препарата - популяция для оценки безопасности
(N=161)
(N=162)
В группе комбинации конечная суточная доза в конце периода лечения составляла >20 ед./10 мкг и ≤40 ед./20 мкг для 70 (43,5%) пациентов и >40 ед./20 мкг и ≤60 ед./30 мкг для 68 (42,2%) пациентов. Для большего количества пациентов (23[14,3%]) в группе комбинации, чем в группе инсулина гларгина (16 [9,9%]), конечная суточная доза в этой категории составляла ≤20 ед. Для большего количества пациентов в группе инсулина гларгина (27 [16,7%]) конечная суточная доза составляла >60 ед. по сравнению с группой комбинации (0 пациентов, что соответствовало протоколу).
Количество (%) пациентов в зависимости от конечной дозы инсулина в конце открытого лечения - популяция для оценки безопасности
3.2 ЭФФЕКТИВНОСТЬ
3.2.1 Первичные конечные точки оценки эффективности
Основной анализ
В таблице 7 кратко описаны результаты для первичных конечных точек эффективности, изменение значения HbA1c от начала исследования до 24-й недели, определенное при помощи анализа ANCOVA с пропущенными данными, условно рассчитанными при помощи подхода c переносом данных последнего наблюдения вперед (LOCF). Средние значения изменения HbA1c, рассчитанные при помощи метода наименьших квадратов (LS), от начала исследования до 24-й недели, составляли -1,82% для группы комбинации и -1,64% для группы инсулина гларгина (различие по среднему значению, рассчитанному при помощи LS, по сравнению с инсулином гларгином = -0,17%; 95% CI: от -0,312% до -0,037%). Исходя из ранее описанного первичного анализа была продемонстрирована не меньшая эффективность для группы комбинации по сравнению с группой инсулина гларгина, поскольку верхняя граница двухстороннего 95% CI для различия по среднему значению, рассчитанному при помощи LS, была меньше предварительно определенного предельного значения для не меньшей эффективности, составляющего 0,4%. Также было продемонстрировано преимущество комбинации по сравнению с инсулином гларгином, определенное при помощи статистического анализа, (различие по среднему значению, рассчитанному при помощи LS, по сравнению с инсулином гларгином = -0,17%, p-значение = 0,0130).
На фигуре 2 показано среднее значение HbA1c (±SE) в зависимости от времени на протяжении 24-недельного периода лечения. В обеих группах лечения наибольшее снижение среднего значения HbA1c наблюдали на 24-й неделе.
Среднее значение изменение HbA1c (%) от начала исследования до 24-й недели - популяция mITT
(N=161)
(N=162)
(N=161)
a модель на основе ковариационного анализа (ANCOVA) с использованием групп лечения (комбинация с постоянной дозой инсулина гларгина/ликсизенатида, инсулин гларгин), рандомизационных страт исходя из значения HbA1c при скрининге (<8,0%, >8,0%), рандомизационных страт исходя из значения BMI при скрининге (<30, >30 кг/м2) и страны в качестве фиксированных эффектов и с использованием значения HbA1c в начале исследования в качестве ковариаты.
Анализ охватывал результаты измерения, полученные до введения резервной терапии и до 14 суток после последней инъекции исследуемого лекарственного препарата (см. фиг. 2).
Охвачены измерения у пациентов, как в начале исследования, так и на 24-й неделе (LOCF).
В таблице 8 представлена доля пациентов, у которых наблюдается ответ на лечение, со значением HbA1c ≤6,5% или <7% на 24-й неделе, соответственно. Несмотря на то, что различия между группами не были статистически значимыми, как продемонстрировано при помощи 95% CI для различия по доле пациентов, у большего процента пациентов в группе комбинации целевое значение HbA1c достигало ≤6,5% (71,9% по сравнению с 64,6%) или <7% (84,4% по сравнению с 78,3 %) по сравнению с группой инсулина гларгина.
Количество (%) пациентов со значением HbA1c ≤6,5% или <7%, соответственно, на 24-й неделе - популяция mITT
(N=161)
3.2.2 Другие ключевые конечные точки оценки эффективности
В таблицах 9-14 изложено краткое описание результатов анализов при помощи ANCOVA отклонения уровня PPG, PG через 2 часа, профиля средних значений SMPG по 7 моментам времени, массы тела, средней суточной дозы инсулина и уровня FPG, соответственно. На фигурах 3-5 показан профиль средних значений SMPG по 7 моментам времени, масса тела и средняя суточная доза инсулина в зависимости от времени на протяжении периода лечения.
Лечение при помощи комбинации значимо улучшало постпрандиальный гликемический контроль в сравнении с инсулином гларгином, как продемонстрировано при помощи результатов для отклонения уровня PPG и PG через 2 часа. Средние значения изменения уровня PPG через 2 часа (таблица 9) от начала исследования до 24-й недели, рассчитанные при помощи LS, составляли -7,49 ммоля/л для группы комбинации и -4,33 ммоля/л для группы инсулина гларгина (различие по среднему значению, рассчитанному при помощи LS, по сравнению с инсулином гларгином = -3,17 ммоля/л; p-значение <0,0001). Средние значения изменения отклонения уровня PG через 2 часа (таблица 10) от начала исследования до 24-й недели, рассчитанные при помощи LS, составляли -3,91 ммоля/л для группы комбинации и -0,67 ммоля/л для группы инсулина гларгина (различие по среднему значению, рассчитанному при помощи LS, по сравнению с инсулином гларгином = -3,24 ммоля/л; p-значение <0,0001).
Среднее значение изменения уровня глюкозы в плазме крови через 2 часа после приема пищи (ммоль/л) от начала исследования до 24-й недели - популяция mITT
(N=161)
(N=162)
a Анализ модели на основе ковариационного анализа (ANCOVA) с использованием групп лечения (комбинация с постоянной дозой инсулина гларгина/ликсизенатида, инсулин гларгин), рандомизационных страт исходя из значения HbA1c при скрининге (<8,0%, ≥8,0%), рандомизационных страт исходя из значения BMI при скрининге (<30, ≥30 кг/м2) и страны в качестве фиксированных эффектов и значения глюкозы в плазме крови через 2 часа после приема пищи в начале исследования в качестве ковариаты.
Анализ охватывал результаты измерения, полученные до введения резервной терапии и до даты последней инъекции исследуемого лекарственного препарата.
Охвачены результаты измерения у пациентов, как в начале исследования, так и на 24-й неделе (LOCF).
Среднее значение изменения отклонения уровня глюкозы в плазме крови через 2 часа (ммоль/л) от начала исследования до 24-й недели - популяция mITT
a Анализ модели на основе ковариационного анализа (ANCOVA) с использованием групп лечения (комбинация с постоянной дозой инсулина гларгина/ликсизенатида, инсулин гларгин), рандомизационных страт исходя из значения HbA1c при скрининге (<8,0%, ≥8,0%), рандомизационных страт исходя из значения BMI при скрининге (<30, ≥30 кг/м2), и страны в качестве фиксированных эффектов и значения отклонения уровня глюкозы в плазме крови через 2 часа в качестве ковариаты.
Анализ охватывал результаты измерения, полученные до введения резервной терапии и до даты последней инъекции исследуемого лекарственного препарата.
Охвачены результаты измерения у пациентов, как в начале исследования, так и на 24-й неделе (LOCF).
Что касается среднего уровня SMPG по 7 моментам времени (таблица 11), у пациентов, которых лечили при помощи комбинации, наблюдалось статистически значимо большее снижение по сравнению с пациентами, которых лечили инсулином гларгином (различие по среднему значению, рассчитанному при помощи LS, -0,30 ммоль/л; p-значение = 0,0154). На фигуре 3 показан уровень SMPG по 7 моментам времени для каждого момента времени в начале исследования и на 24-й неделе (LOCF).
Среднее значение изменения средних значений в профилях уровня глюкозы в плазме крови при самостоятельном контроле (SMPG) по 7 моментам времени (ммоль/л) от начала исследования до 24-й недели - популяция mITT
a модель на основе ковариационного анализа (ANCOVA) с использованием групп лечения (комбинация с постоянной дозой инсулина гларгина/ликсизенатида, инсулин гларгин), рандомизационных страт исходя из значения HbA1c при скрининге (<8,0%, ≥8,0%), рандомизационных страт исходя из значения BMI при скрининге (<30, ≥30 кг/м2) и страны в качестве фиксированных эффектов и с использованием среднего значения SMPG по 7 моментам времени в начале исследования в качестве ковариаты.
Анализ охватывал результаты измерения, полученные до введения резервной терапии и до даты последней инъекции исследуемого лекарственного препарата (см. фиг. 3).
Охвачены результаты измерения у пациентов, как в начале исследования, так и на 24-й неделе (LOCF).
Среднее значение массы тела, рассчитанное при помощи LS, снижалось от начала исследования до 24-й недели на 0,97 кг для пациентов, которых лечили при помощи комбинации, и повышалось на 0,48 кг для пациентов, которых лечили при помощи инсулина гларгина (различие по среднему значению, рассчитанному при помощи LS, по сравнению c инсулином гларгином = -1,44 кг), причем между группами лечения наблюдали статистически значимое различие (p-значение <0,0001) (таблица 12).
Среднее значение изменения массы тела (кг) от начала исследования до 24-й недели - популяция mITT
(N=161)
a модель на основе ковариационного анализа (ANCOVA) с использованием групп лечения (комбинация с постоянной дозой инсулина гларгина/ликсизенатида, инсулин гларгин), рандомизационных страт исходя из значения HbA1c при скрининге (<8,0%, ≥8,0%), рандомизационных страт исходя из значения BMI при скрининге (<30, ≥30 кг/м2) и страны в качестве фиксированных эффектов и с использованием массы тела в начале исследования в качестве ковариаты.
Анализ охватывал результаты измерения, полученные до введения резервной терапии и до 3 суток после последней инъекции исследуемого лекарственного препарата (см. фиг. 4).
Охвачены результаты измерения у пациентов, как в начале исследования, так и на 24-й неделе (LOCF).
Среднее значение, рассчитанное при помощи LS, средней суточной дозы инсулина гларгина на 24-й неделе составляло 36,08 ед. для группы комбинации и 39,32 ед. для группы инсулина гларгина, и при этом различие между группами лечения характеризовалось пограничной значимостью (различие по среднему значению, рассчитанному при помощи LS, по сравнению с инсулином гларгином = -3,24 ед.; p-значение = 0,0583) (таблица 13).
Средняя суточная доза инсулина гларгина (ед.) на 24-й неделе - популяция mITT
(N=161)
a модель на основе дисперсионного анализа (ANCOVA) с использованием групп лечения (комбинация с постоянной дозой инсулина гларгина/ликсизенатида, инсулин гларгин), рандомизационных страт исходя из значения HbA1c при скрининге (<8,0%, ≥8,0%), рандомизационных страт исходя из значения BMI при скрининге (<30, ≥30 кг/м2) и страны в качестве фиксированных эффектов.
Анализ охватывал результаты измерения, полученные до введения резервной терапии и до даты последней инъекции исследуемого лекарственного препарата (см. фиг. 5).
Наблюдали сходное уменьшение среднего значения изменения уровня FPG от начала исследования до 24-й недели (среднее значение, рассчитанное при помощи LS: -3,35 ммоля/л в группе комбинации; -3,51 ммоля/л в группе инсулина гларгина).
Только один пациент в группе инсулина гларгина нуждался в резервной терапии в течение 24-недельного периода лечения.
Среднее значение изменения уровня глюкозы в плазме крови натощак (ммоль/л) от начала исследования до 24-й недели - популяция mITT
a модель на основе ковариационного анализа (ANCOVA) с использованием групп лечения (комбинация с постоянной дозой инсулина гларгина/ликсизенатида, инсулин гларгин), рандомизационных страт исходя из значения HbA1c при скрининге (<8,0%, ≥8,0%), рандомизационных страт исходя из значения BMI при скрининге (<30, ≥30 кг/м2) и страны в качестве фиксированных эффектов и с использованием уровня глюкозы в плазме крови натощак в начале исследования в качестве ковариаты. Анализ охватывал результаты измерения, полученные до введения резервной терапии и до 1 суток после последней инъекции исследуемого лекарственного препарата.
Охвачены результаты измерения у пациентов, как в начале исследования, так и на 24-й неделе (LOCF).
3.3 БЕЗОПАСНОСТЬ
Случаи симптоматической гипогликемии документировали в специальной форме для случаев гипогликемии, а не на странице CRF для AE, и, таким образом, они не были включены в краткое описание TEAE. Они обобщены отдельно от TEAE (см. раздел 3.3.5).
3.3.1 Нежелательные явления, возникшие в ходе лечения
В таблице 15 представлены обобщенные данные для пациентов, у которых наблюдались случаи нежелательных явлений в течение 24-недельного периода открытого лечения. Немного больше пациентов сообщали о TEAE в группе комбинации (86 [53,4%]), чем в группе инсулина гларгина (82 [50,6%]), что, главным образом, может объясняться различием по случаям нарушений со стороны SOC желудочно-кишечного тракта (25 [15,5%] в группе комбинации по сравнению с 15 [9,3%] в группе инсулина гларгина). В таблице 16 показано, что наиболее часто сообщаемым TEAE в группе комбинации была тошнота (12 [7,5%] по сравнению с 0 в группе инсулина гларгина), а в группе инсулина гларгина - головная боль (12 [7,4%] по сравнению с 8 [5,0%] в группе комбинации).
Обзор профиля нежелательных явлений: нежелательные явления, возникшие в ходе лечения - популяция для оценки безопасности
(N=162)
n (%) = количество и процент пациентов по меньшей мере с одним TEAE.
Количество (%) пациентов с TEAE, которые возникали при PT >=1% в любой группе лечения по главному SOC, HLGT, HLT и PT - популяция для оценки безопасности
HLGT: групповой термин высокого уровня
HLT: термин высокого уровня
Предпочтительный термин, n(%)
гларгин
(N=162)
HLGT: групповой термин высокого уровня
HLT: термин высокого уровня
Предпочтительный термин, n(%)
(N=162)
HLGT: групповой термин высокого уровня
HLT: термин высокого уровня
Предпочтительный термин, n(%)
(N=162)
HLGT: групповой термин высокого уровня
HLT: термин высокого уровня
Предпочтительный термин, n(%)
(N=162)
HLT: термин высокого уровня
Предпочтительный термин, n(%)
(N=161)
(N=162)
n (%) = количество и процент пациентов по меньшей мере с одним TEAE.
Примечание: в таблице SOC приведены в порядке, принятом на международном уровне, а HLGT, HLT и PT - в алфавитном порядке.
Приведены только SOC по меньшей мере с одним PT ≥1% по меньшей мере в одной группе.
3.3.2 Случаи смерти, серьезные нежелательные явления, возникшие в ходе лечения
В ходе этого исследования не сообщалось о случаях смерти. Количество пациентов с SAE, возникшими в ходе лечения, составляло 9 (5,6%) в группе комбинации и 6 (3,7%) в группе инсулина гларгина, которые распределялись среди множества SOC без значительного увеличения в любом конкретном SOC (таблица 17).
Количество (%) пациентов с SAE, возникшими в ходе лечения, представленными по главному SOC, HLGT, HLT и PT - популяция для оценки безопасности
HLT: термин высокого уровня
Предпочтительный термин, [n(%)]
(N=162)
HLGT: групповой термин высокого уровня
HLT: термин высокого уровня
Предпочтительный термин [n(%)]
(N=161)
(N=162)
сердечных сокращений NEC
HLGT: групповой термин высокого уровня
HLT: термин высокого уровня
Предпочтительный термин, [n(%)]
(N=162)
путях (кроме почек)
n (%) = количество и процент пациентов по меньшей мере с одним SAE, возникшим в ходе лечения
Примечание: в таблице SOC приведены в порядке, принятом на международном уровне, а HLGT, HLT и PT - в алфавитном порядке.
Нежелательные явления, которые привели к прекращению участия
Шесть пациентов (3,7%) в группе комбинации досрочно завершили лечение в связи с TEAE по сравнению с отсутствием таковых в группе инсулина гларгина (таблица 18). У 2 из этих пациентов TEAE, которые привели к досрочному завершению лечения, представляли собой нарушения со стороны SOC желудочно-кишечного тракта (тошнота и/или рвота). Один пациент с тошнотой и рвотой и 1 пациент с тошнотой и головной болью досрочно завершили прием IMP в день 66 и 53, и при этом их последняя суточная доза инсулина составляла 52 ед. (26 мкг ликсизенатида) и 18 ед. (9 мкг ликсизенатида), соответственно.
Пациент с гиперчувствительностью досрочно завершил прием IMP в день приема первой дозы. Это явление не было положительно квалифицировано согласно ARAC как аллергическая реакция. Было подтверждено, что спутанность сознания и головокружение у каждого из пациентов не были связаны с симптоматической гипогликемией.
Таблица 18. Количество (%) пациентов с TEAE, которые привели к окончательному прекращению лечения на протяжении периода исследуемого лечения, указанное по главному SOC, HLGT, HLT и PT - популяция для оценки безопасности
HLGT: групповой термин высокого уровня
HLT: термин высокого уровня
Предпочтительный термин, [n(%)]
(N=162)
HLT: термин высокого уровня
Предпочтительный термин, [n(%)]
с постоянным соотношением инсулина гларгина/ликсизенатида (N=161)
гларгин
(N=162)
n (%) = количество и процент пациентов по меньшей мере с одним TEAE, которое привело к окончательному прекращению лечения. Примечание: в таблице SOC приведены в порядке, принятом на международном уровне, а HLGT, HLT и PT - в алфавитном порядке. Гиперчувствительность: не квалифицирована как аллергический случай согласно ARAC.
3.3.4 Другие значимые нежелательные явления
В общей сложности у 6 пациентов (5 пациентов в группе комбинации и 1 пациент в группе инсулина гларгина) наблюдались реакции в месте инъекции (таблица 19). Ни одну из этих реакций не считали серьезной, или тяжелой, или такой, которая привела к досрочному прекращению лечения.
Количество (%) пациентов, у которых наблюдались реакции в месте инъекции в течение периода TEAE - популяция для оценки безопасности
Предпочтительный термин
с постоянным соотношением инсулина гларгина/ликсизенатида
(N=161)
сообщенных исследователями
диагностики согласно ARAC
В общей сложности 2 пациента (1 [0,6%] в каждой группе) сообщали о 6 случаях, положительно квалифицированных как аллергические реакции согласно ARAC с одинаковым диагнозом аллергический ринит. Ни один из них не был квалифицирован как положительно связанный с IMP (таблица 20).
Таблица 20. Количество (%) пациентов со случаями, квалифицированными согласно ABAC как аллергическая реакция, в течение периода TEAE - популяция для оценки безопасности
MedDRA термин (PT)
для диагноза согласно ARAC
ARAC
ликсизенатида
(N=161)
1 (0,6%)
1 (0,6%)
IMP
1 (0,6%)
1 (0,6%)
Согласно протоколу, любое повышение уровня амилазы и/или липазы вдвое выше верхнего предела нормального диапазона (ULN) или кальцитонина ≥20 пг/мл, которое было подтверждено при повторном измерении, необходимо было отслеживать и документировать в специальной форме для AE. На протяжении периода лечения у 3 пациентов (2 [1,2%] в группе комбинации и 1 [0,6%] в группе инсулина гларгина) наблюдалось TEAE, заключающееся в повышенном уровне липазы (>2 ULN), и у 1 пациента (в группе инсулина гларгина) наблюдалось TEAE, заключающееся в повышенном уровне амилазы (> 2 ULN), при этом о них сообщали в специальной форме для AE. Не было пациентов, для которых сообщалось о TEAE, заключающемся в повышенном уровне кальцитонина (≥20 пг/мл).
Также складывали количество пациентов, у которых наблюдалось по меньшей мере 1 значение липазы или амилазы ≥3 ULN или по меньшей мере 1 значение кальцитонина ≥20 пг/мл в течение периода применения исследуемого препарата. У одного пациента в группе инсулина гларгина наблюдалось по меньшей мере 1 значение амилазы ≥3 ULN, и у 5 пациентов (4 в группе комбинации и 1 в группе инсулина гларгина) наблюдалось по меньшей мере 1 значение липазы ≥3 ULN. У одного пациента в группе инсулина гларгина наблюдалось 1 значение кальцитонина ≥20 пг/мл (но <50 пг/мл), при этом повторно протестированные значения находились в пределах нормального диапазона.
У одного пациента в группе комбинации и одного пациента в группе инсулина гларгина наблюдалось, соответственно, два случая (госпитализация по причине нестабильной стенокардии и чрезкожное коронарное вмешательство [PCI]) и один случай (PCI), квалифицированный Комитетом по оценке сердечно-сосудистых осложнений (CAC) как значительные сердечно-сосудистые осложнения.
3.3.5 Другие наблюдения, связанные безопасностью - симптоматическая гипогликемия
Случаи симптоматической гипогликемии (включая задокументированную, вероятную и тяжелую симптоматическую гипогликемию) были зарегистрированы у 40 (24,8%) пациентов, которых лечили при помощи комбинации, по сравнению с 40 (24,7%) пациентами, которых лечили при помощи инсулина гларгина. Количество случаев симптоматической гипогликемии на пациента в год составляло 1,11 в обеих группах лечения. Не было зарегистрировано ни одного случая тяжелой симптоматической гипогликемии ни в одной из групп (таблица 21).
Частота задокументированной симптоматической гипогликемии при уровне глюкозы в плазме ≤70 мг/дл (3,9 ммоля/л) была сходной в обеих группах лечения (35 [21,7%] по сравнению с 37 [22,8%] в группах комбинации и инсулина гларгина, соответственно). Для задокументированной симптоматической гипогликемии при уровне глюкозы в плазме <60 мг/дл (3,3 ммоля/л) частота была выше в группе комбинации по сравнению с группой инсулина гларгина [20 (12,4%) по сравнению с 9 (5,6%)].
Обобщение по симптоматической гипогликемии, учтенной при оценке в специальном протоколе eCRF и соответствующей определению в протоколе, в течение периода TEAE - популяция для оценки безопасности
(N=161)
Симптоматическая гипогликемия
Количество пациентов с задокументированными случаями, n (%)
40 (24,8%)
40 (24,7%)
(N=161)
Симптоматическая гипогликемия = симптоматическая гипогликемия, задокументированная при оценке в специальном протоколе eCRF и соответствующая определению тяжелой, или задокументированной, или вероятной симптоматической гипогликемии в протоколе.
Пример 2
Рандомизированное многоцентровое открытое исследование с 3 группами лечения, с использованием активного контрольного препарата, с параллельными группами, продолжительностью 30 недель, в котором сравнивали эффективность и безопасность комбинации с постоянным соотношением инсулина гларгина/ликсизенатида с инсулином гларгином отдельно и с ликсизенатидом отдельно в дополнение к метформину у пациентов с сахарным диабетом 2 типа (T2DM).
Код соединения: HOE901/AVE0010
НАЗВАНИЕ И ЗАГЛАВИЕ ИССЛЕДОВАНИЯ
МЕДИЦИНСКОЕ СОСТОЯНИЕ
ЦЕЛИ ИССЛЕДОВАНИЯ
эффективность
Объем испытаний
 Диагностика Профилактика
Диагностика Профилактика Терапия
Терапия Эффективность Безопасность
Эффективность Безопасность Фармакодинамика Фармакокинетика
Фармакодинамика Фармакокинетика Биоэквивалентность Реакция на дозу Фармакогенетика Фармакогеномика
Биоэквивалентность Реакция на дозу Фармакогенетика Фармакогеномика Фармакоэкономика
Фармакоэкономика
ПЛАН ИССЛЕДОВАНИЯ
Метка в схеме исследования
Предоставить информацию, специфичную для группы - в частности, подробности о введении IMP и средства, отличного от IMP (например, продукт, доза, частота, продолжительность, условие приема(приемов))
Выбрать “экспериментальная” при введении исследуемого соединения
ликсизенатида
Комбинация с постоянным соотношением инсулина гларгина/ликсизенатида содержала 100 ед./мл инсулина гларгина и 50 мкг/мл ликсизенатида. Состав с инсулином гларгином (Lantus) содержал 100 ед./мл инсулина гларгина. Состав ликсизенатида (Lyxumia) содержал 50 мкг/мл ликсизенатида (для введения дозы 10 мкг ликсизенатида) или 100 мкг/мл ликсизенатида (для введения дозы 20 мкг ликсизенатида). Метформин вводили при дозе по меньшей мере 1,0 г/сутки или по меньшей мере 1,5 г/сутки.
ИССЛЕДУЕМАЯ ПОПУЛЯЦИЯ
Здоровые добровольцы Пациенты NA (без ограничения) NA (без ограничения)
NA (без ограничения)
• подписанное письменное информированное согласие.
• значение HbA1c при скрининговом визите:
<7,5% и >10% для пациентов, которых ранее лечили при помощи метформина отдельно;
<7,0% и >9 % для пациентов, которых ранее лечили при помощи метформина и второго перорального противодиабетического средства для лечения;
• беременные и кормящие женщины и женщины репродуктивного возраста, которые не применяют эффективный способ контрацепции;
• применение других пероральных или вводимых путем инъекции средств, понижающих уровень глюкозы, отличных от заявленных в критериях включения в период за 3 месяца до скрининга;
• лечение при помощи инсулина более 3 месяцев тому назад (за исключением краткосрочного лечения в связи со случайной болезнью, включая гестационный диабет на усмотрение врача, проводящего исследование);
• досрочное прекращение предыдущего лечения при помощи агониста рецептора GLP-1 (GLP-1 RA) в связи с проблемой безопасности/переносимости или отсутствием эффективности в анамнезе;
• пациент, который ранее принимал участие в любом клиническом испытании с использованием ликсизенатида или комбинации с постоянным соотношением инсулина гларгина/ликсизенатида или ранее принимал ликсизенатид;
• любое противопоказание для применения метформина в соответствии с местным вариантом инструкции по применению;
• применения средств для похудения в пределах 3 месяцев перед скрининговым визитом;
• в пределах последних 6 месяцев перед скрининговым визитом: инсульт, инфаркт миокарда, нестабильная стенокардия или сердечная недостаточность, требующая госпитализации в анамнезе; запланированные процедуры коронарной, каротидной или периферической артериальной реваскуляризации, которые должны быть проведены в течение периода исследования;
• в анамнезе панкреатит (не считая панкреатита, связанного с камнями в желчном пузыре, и уже осуществленной холецистэктомии), хронический панкреатит, панкреатит в течение предыдущего лечения при помощи средства для терапии на основе инкретина, панкреатэктомии, оперативного вмешательства в желудок/область желудка;
• личный или непосредственный семейный анамнез медуллярного рака щитовидной железы (MTC) или наследственные состояния, которые обуславливают предрасположенность к MTC (например, синдромы множественных эндокринных неоплазий);
• неконтролируемая или ненадлежащим образом контролируемая гипертензия (артериальное систолическое давление выше 180 мм. рт. ст. или диастолическое артериальное давление выше 95 мм. рт. ст.) при скрининговом визите;
• при скрининговом визите индекс массы тела (BMI) менее или равный 20 или выше 35 кг/м2;
при скрининговом визите уровень амилазы и/или липазы, больше чем в 3 раза превышающий верхний предел нормального (ULN) лабораторного диапазона;
• при скрининговом визите ALT или AST более 3 ULN;
• при скрининговом визите уровень кальцитонина выше или равный 20 пг/мл (5,9 пмоля/л).
• Критерии исключения для рандомизации в конце скринингового периода:
• значение HbA1c <7% или >10% при визите 4 (неделя 1);
• уровень глюкозы в плазме крови натощак при визите 4 (неделя -1) >250 мг/дл (13,9 ммоля/л);
• максимальная переносимая доза метформина <1500 мг/сутки;
• уровень амилазы и/или липазы, измеренный за одну неделю до рандомизации, превышал ULN в >3 раза; Женщины детородного возраста, не применяющие контрацептивы  Женщины детородного возраста, применяющие контрацептивы Беременные женщины Женщины, кормящие грудью
Женщины детородного возраста, применяющие контрацептивы Беременные женщины Женщины, кормящие грудью Чрезвычайное обстоятельство
Чрезвычайное обстоятельство
задействованных (подписанная форма информированного согласия)
плацебо
только соединение Sanofi
например, таблетка, капсула, раствор...
например, пероральный, внутривенный, внутримышечный, подкожный...
гларгин/
ликсизенатид
AVE0010
например, изменение от начала исследования параметра, время до определенного явления, количество пациентов с предварительно указанным явлением, конкретные результаты измерения
ввести моменты времени, в которые оценивают критерий, или длительность оценки
проблему
безопасности? Да Да Да
Да Да Да
Да Да Да Да Да Да Да Да
Да Да Да
Да Да
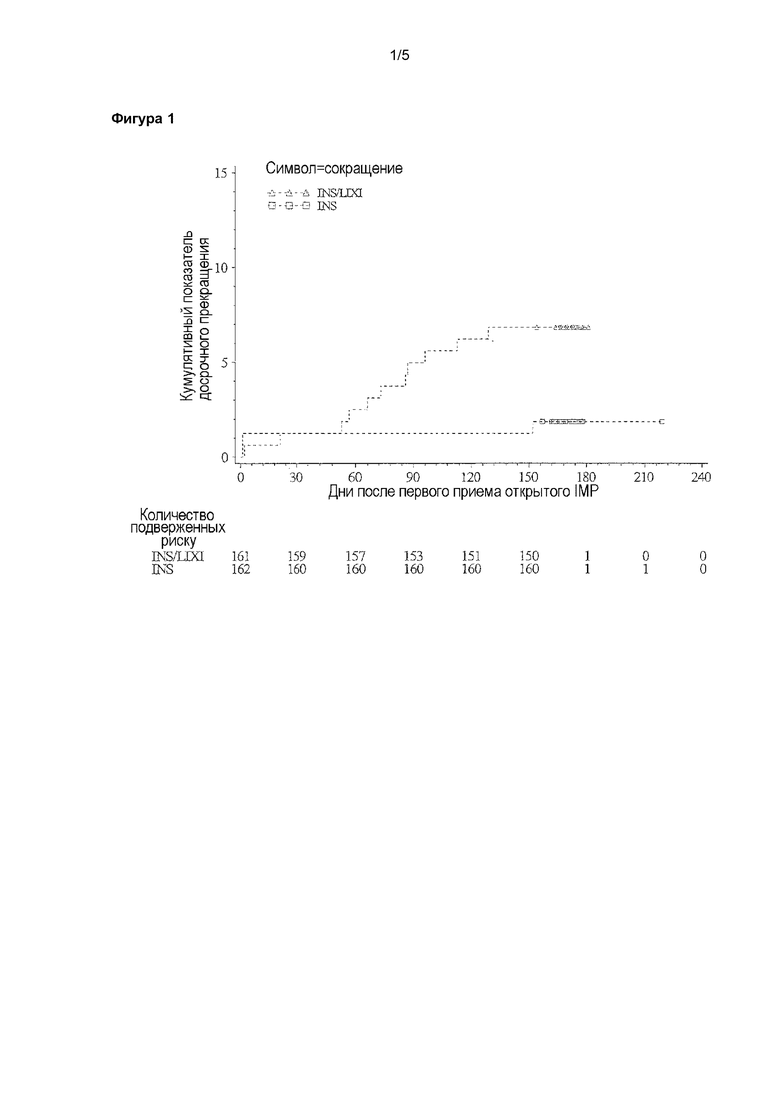
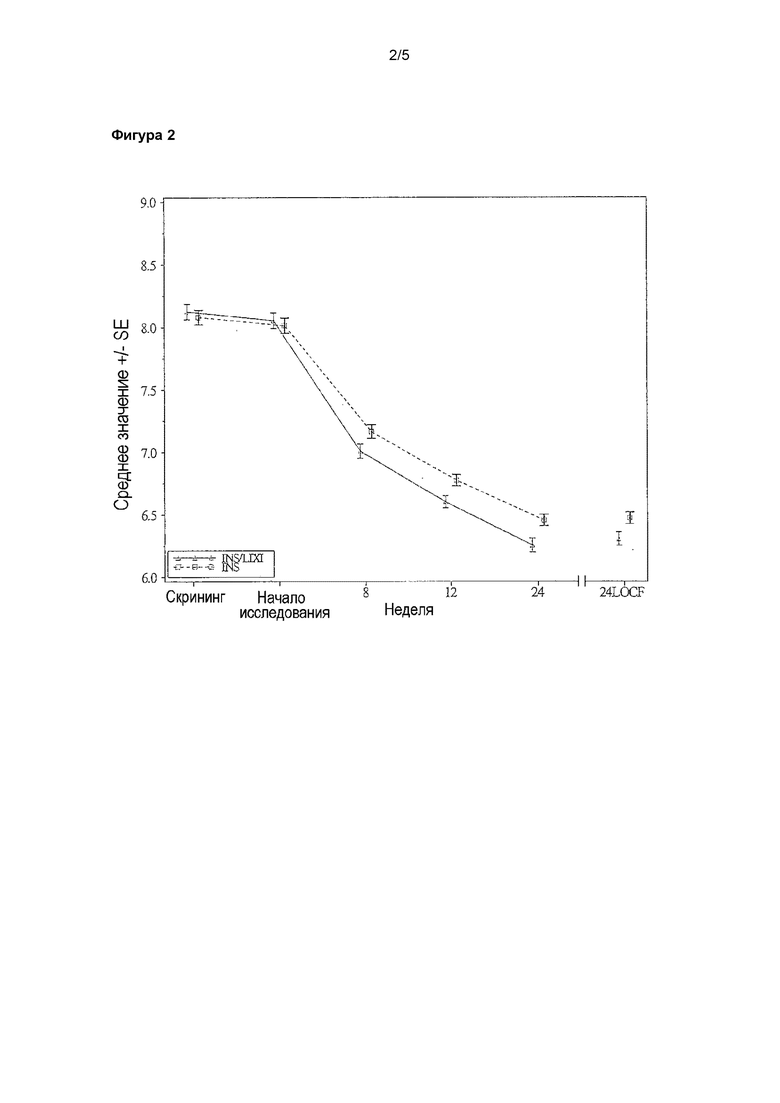
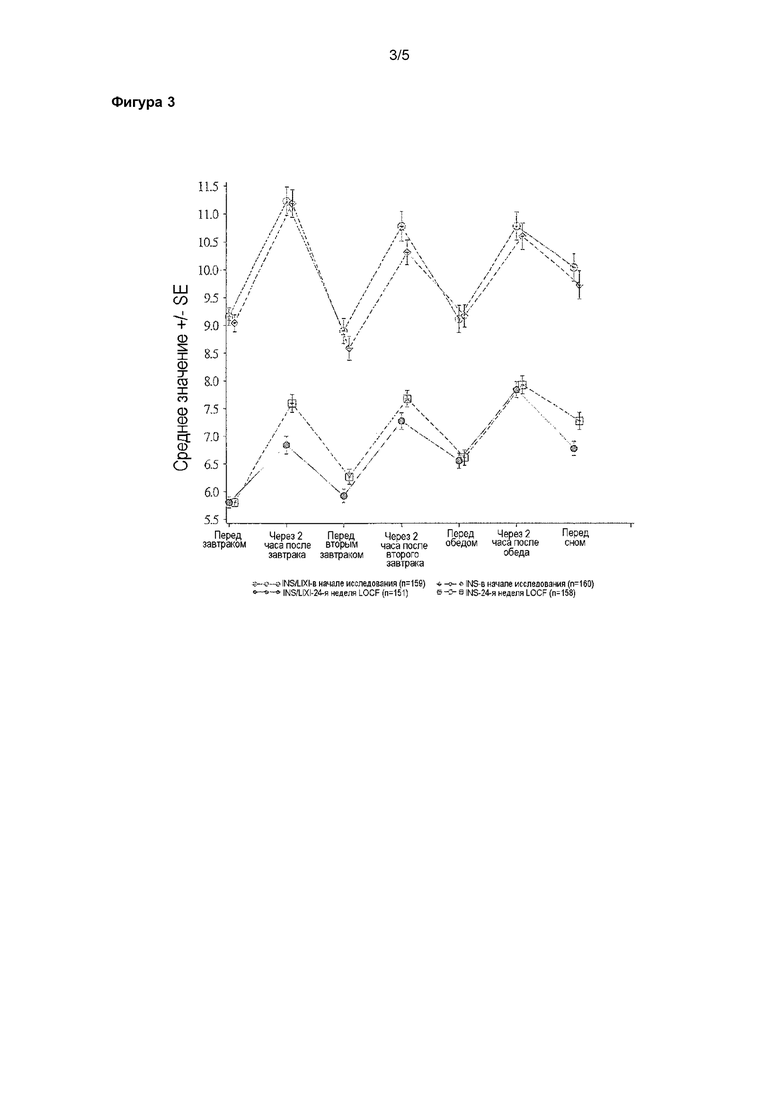
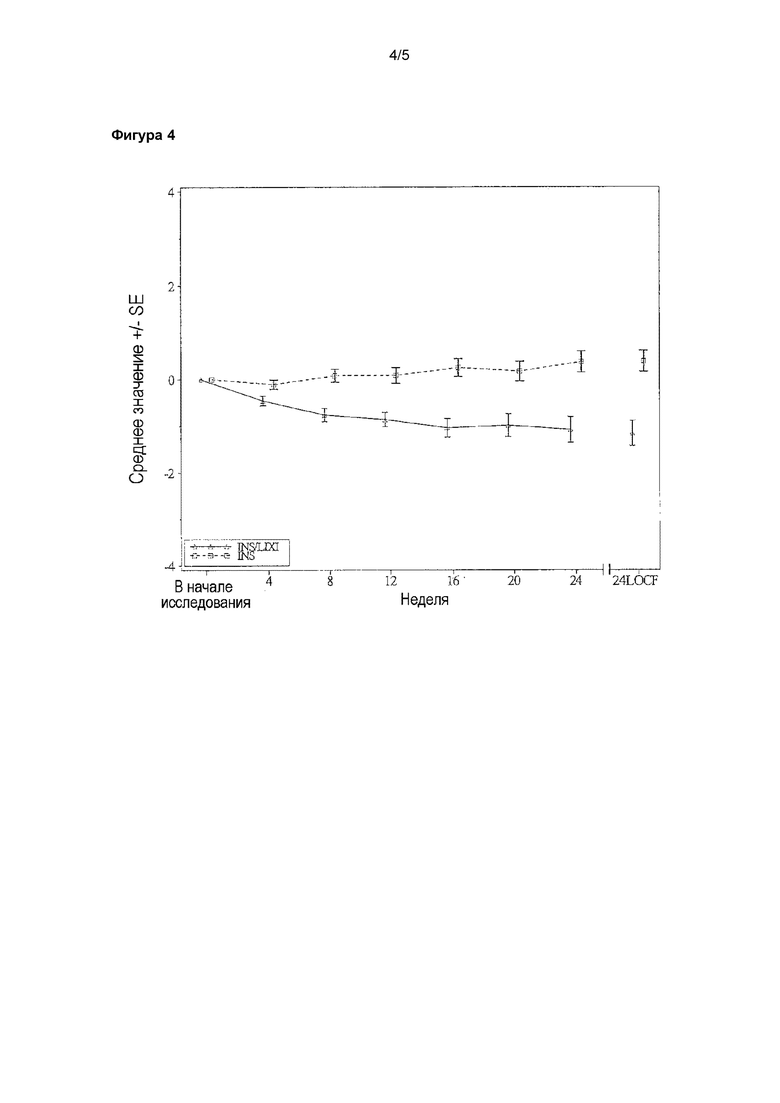
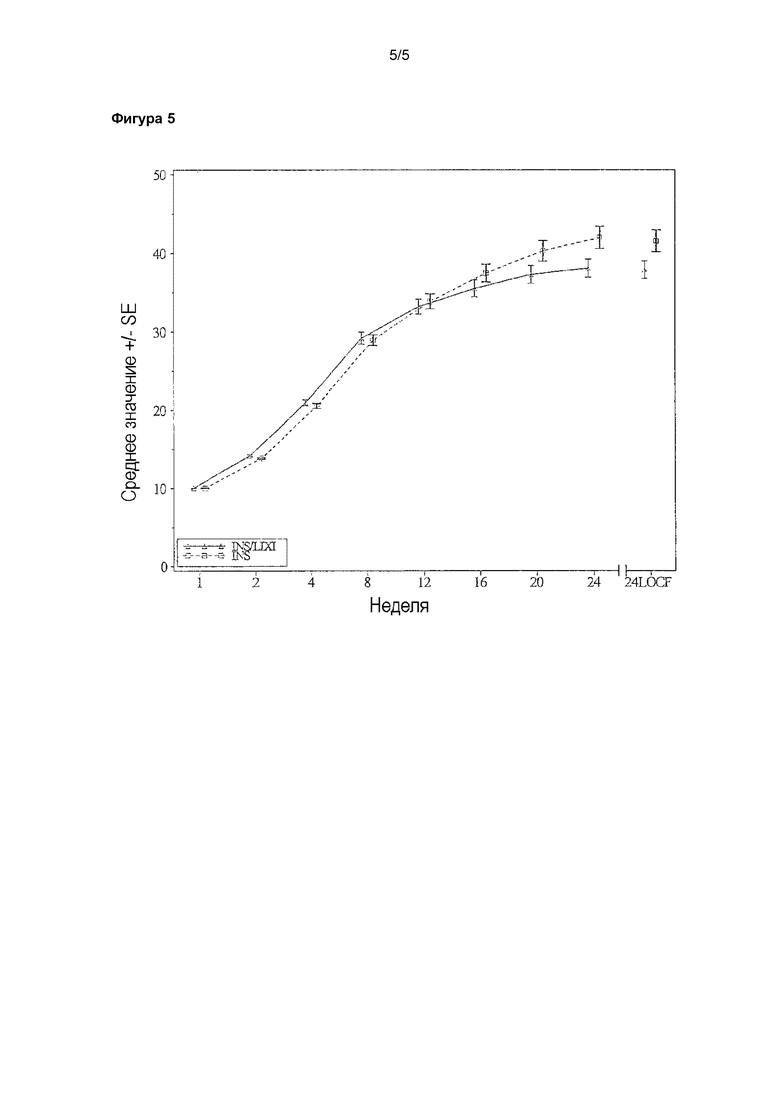
Изобретение относится к фармацевтической композиции, а именно к фармацевтической комбинации для лечения сахарного диабета 2 типа. Представлены: фармацевтическая комбинация для лечения сахарного диабета 2 типа, содержащая фармацевтическую композицию, содержащую ликсизенатид или/и его фармацевтически приемлемую соль, инсулин гларгин или/и его фармацевтически приемлемую соль, взятых в определенных концентрациях; и метформин и/или его фармацевтически приемлемую соль; применение фармацевтической комбинации для лечения сахарного диабета 2 типа и способ лечения сахарного диабета 2 типа, предусматривающий введение субъекту, нуждающемуся в этом, фармацевтической композиции, при этом фармацевтическая композиция вводится в качестве дополнения к лечению метформином или/и его фармацевтически приемлемой солью. Предлагаемая фармацевтическая комбинация для лечения сахарного диабета 2 типа является эффективной комбинацией в качестве дополнения к лечению метформином или/и его фармацевтически приемлемой соли. 3 н. и 13 з.п. ф-лы, 5 ил., 21 табл., 2 пр.
1. Фармацевтическая комбинация для лечения сахарного диабета 2 типа, содержащая
(i) фармацевтическую композицию, содержащую (a) ликсизенатид или/и его фармацевтически приемлемую соль, и (b) инсулин гларгин или/и его фармацевтически приемлемую соль, где соединение (b) присутствует в концентрации 100 ед./мл, и соединение (a) присутствует в концентрации 50 мкг/мл, и
(ii) метформин и/или его фармацевтически приемлемую соль.
2. Фармацевтическая комбинация по п. 1, где субъект, подлежащий лечению, страдает ожирением.
3. Фармацевтическая комбинация по п. 1, где сахарный диабет 2 типа надлежащим образом не контролируется при помощи метформина отдельно.
4. Фармацевтическая комбинация по п. 1, где у субъекта, подлежащего лечению, значение HbA1c находится в диапазоне по меньшей мере 7%, по меньшей мере 8% или по меньшей мере 9% в начале лечения при помощи композиции.
5. Фармацевтическая комбинация по п. 1, где у субъекта, подлежащего лечению, концентрация глюкозы в плазме крови при самостоятельном контроле составляет по меньшей мере 8 ммоль/л, по меньшей мере 9 ммоль/л, по меньшей мере 10 ммоль/л или по меньшей мере 11 ммоль/л в начале лечения при помощи композиции.
6. Фармацевтическая комбинация по п. 1, где у субъекта, подлежащего лечению, уровень глюкозы в плазме крови через 2 часа после приема пищи составляет по меньшей мере 12 ммоль/л, по меньшей мере 13 ммоль/л, по меньшей мере 14 ммоль/л, по меньшей мере 15 ммоль/л, по меньшей мере 16 ммоль/л или по меньшей мере 17 ммоль/л в начале лечения при помощи композиции.
7. Фармацевтическая комбинация по п. 1, где у субъекта, подлежащего лечению, отклонение уровня глюкозы в плазме крови через 2 часа после приема пищи составляет по меньшей мере 5 ммоль/л, по меньшей мере 5,5 ммоль/л, по меньшей мере 6 ммоль/л, по меньшей мере 6,5 ммоль/л или по меньшей мере 7 ммоль/л в начале лечения при помощи композиции.
8. Фармацевтическая комбинация по п. 1, где субъект, подлежащий лечению, не получает агонист рецептора GLP-1 или/и инсулин.
9. Фармацевтическая комбинация по п. 1, где композиция пригодна для парентерального введения.
10. Фармацевтическая комбинация по п. 1, где возраст субъекта, подлежащего лечению, составляет по меньшей мере 50 лет.
11. Фармацевтическая комбинация по п. 1, где индекс массы тела у субъекта, подлежащего лечению, составляет по меньшей мере 30 кг/м2, по меньшей мере 31 кг/м2, по меньшей мере 32 кг/м2 или по меньшей мере 33 кг/м2.
12. Фармацевтическая комбинация по п. 1, где указанная композиция предоставляется в емкости.
13. Применение фармацевтической комбинации, содержащей
(i) фармацевтическую композицию, содержащую (a) ликсизенатид или/и его фармацевтически приемлемую соль, и (b) инсулин гларгин или/и его фармацевтически приемлемую соль, где соединение (b) присутствует в концентрации 100 ед./мл, и соединение (a) присутствует в концентрации 50 мкг/мл, и
(ii) метформин и/или его фармацевтически приемлемую соль для лечения сахарного диабета 2 типа.
14. Применение по п. 13, где субъект, подлежащий лечению, определен в любом из пп. 2-8 и 10, 11.
15. Способ лечения сахарного диабета 2 типа, предусматривающий введение субъекту, нуждающемуся в этом, фармацевтической композиции, содержащей
(a) ликсизенатид или/и его фармацевтически приемлемую соль и
(b) инсулин гларгин или/и его фармацевтически приемлемую соль,
где соединение (b) присутствует в концентрации 100 ед./мл, и соединение (a) присутствует в концентрации 50 мкг/мл, при этом фармацевтическая композиция вводится в качестве дополнения к лечению метформином или/и его фармацевтически приемлемой солью.
16. Способ по п. 15, где субъект, подлежащий лечению, определен в любом из пп. 2-8 и 10, 11.
| WO 2011147980 A1, 01.12.2011 | |||
| СЦЕНА ДЛЯ ТЕАТРАЛИЗОВАННОГО ДЕЙСТВИЯ | 2007 |
|
RU2329848C1 |
| BERLIE H | |||
| et al | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Diabetes Metab Syndr Obes, 2012; N5, с.165-74 | |||
| WO 2011144673 A2, 24.11.2011. | |||

