Предметом настоящего изобретения является фармацевтическая комбинация для применения при гликемическом контроле у пациентов с сахарным диабетом 2 типа, причем указанная комбинация содержит (a) desPro36Exendin-4(1-39)-Lys6-NH2 (AVE0010, ликсисенатид) и/или его фармацевтически приемлемую соль, и (b) глитазон и/или его фармацевтически приемлемую соль.
У здорового индивида высвобождение инсулина поджелудочной железой строго связано с концентрацией глюкозы в крови. Повышенный уровень глюкозы в крови, возникающий после приема пищи, быстро уравновешивается соответствующим увеличением секреции инсулина. В состоянии натощак уровень инсулина в плазме падает до базальной величины, которая достаточна для обеспечения непрерывной подачи глюкозы в чувствительные к инсулину органы и ткани и для поддержания печеночной продукции глюкозы на низком уровне в ночное время.
В отличие от сахарного диабета 1 типа, при сахарном диабете 2 типа в целом нет недостатка инсулина, но во многих случаях, в частности, в прогрессирующих случаях, лечение инсулином рассматривается как наиболее подходящее лечение, если требуется, в комбинации с перорально вводимыми противодиабетическими лекарственными средствами.
Повышенный уровень глюкозы в крови в течение нескольких лет без первоначальных симптомов представляет значительный риск для здоровья. Крупномасштабное исследование DCCT (Исследовательской группы, проводящей испытания по контролю сахарного диабета и его осложнений) в США (The Diabetes Control and Complications Trial Research Group (1993) N. Engl. J. Med. 329, 977-986) ясно показало, что хронически повышенные уровни глюкозы крови являются основной причиной развития осложнений сахарного диабета. Примерами осложнений сахарного диабета являются микро- и макрососудистые поражения, которые могут проявляться ретинопатиями, нефропатиями или нейропатиями, и привести к слепоте, почечной недостаточности и потере конечностей, и сопровождаются повышенным риском сердечно-сосудистых заболеваний. Таким образом, можно сделать вывод, что усовершенствованное лечение сахарного диабета в первую очередь должно быть нацелено на удерживание глюкозы крови как можно ближе к физиологическому диапазону.
Особый риск имеется для пациентов с избыточной массой тела, страдающих сахарным диабетом 2 типа, например, пациентов с индексом массы тела (BMI) ≥30. У этих пациентов факторы риска, связанные с сахарным диабетом, перекрываются факторами риска, связанными с избыточной массой тела, приводящими, например, к увеличению сердечно-сосудистой заболеваемости, по сравнению с пациентами, страдающими сахарным диабетом 2 типа, имеющими нормальную массу тела. Таким образом, особенно необходимо лечить сахарный диабет у этих пациентов при одновременном снижении избыточной массы тела.
Глитазоны (также называемые тиазолидиндионами), такие как пиоглитазон, представляют собой антигипергликемические средства, которые снижают устойчивость к инсулину путем сенсибилизации мышц, печени и жировой ткани (Dormandy et al., Lancet 2005, 366:1270-89, Yki-Jarvinen, N Engl J Med 2004, 351: 1106-18).
Метформин представляет собой гипогликемическое средство из группы бигуанидов, применяемое при лечение инсулиннезависимого сахарного диабета (сахарного диабета 2 типа), не реагирующего на модификацию режима питания. Метформин улучшает гликемический контроль повышением чувствительности к инсулину и уменьшением кишечного всасывания глюкозы. Метформин обычно вводится перорально. Однако контроль сахарного диабета 2 типа у страдающих ожирением пациентов с помощью метформина может быть недостаточным. Таким образом, у этих пациентов могут потребоваться дополнительные меры для контроля сахарного диабета 2 типа.
Соединение desPro36Exendin-4(1-39)-Lys6-NH2 (AVE0010, ликсисенатид) представляет собой производное эксендина-4. Ликсисенатид описан в виде SEQ ID NO:93 в документе WO 01/04156:
SEQ ID NO: 1: Ликсисенатид (44 AS)
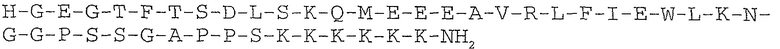
SEQ ID NO: 2: Эксендин-4 (39 AS)
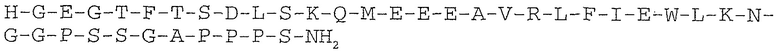
Эксендины представляют собой группу пептидов, которые могут снижать концентрацию глюкозы в крови. Аналог эксендина ликсисенатид характеризуется C-концевым усечением последовательности нативного эксендина-4. Ликсисенатид содержит шесть C-концевых остатков лизина, не присутствующих в эксендине-4.
В контексте настоящего изобретения, ликсисенатид включает фармацевтически приемлемые соли. Специалисту в данной области техники известны фармацевтически приемлемые соли ликсисенатида. Предпочтительной фармацевтически приемлемой солью ликсисенатида, используемой в настоящем изобретении, является ацетат.
В примере настоящего изобретения было продемонстрировано у пациентов с сахарным диабетом 2 типа, что ликсисенатид при дополнительной терапии к глитазону значительно улучшал гликемический контроль:
- Ликсисенатид в комбинации с пиоглитазоном («группа ликсисенатида») значительно уменьшали содержание глюкозы в плазме натощак по сравнению с группой пиоглитазона («группа плацебо») от исходного уровня до 24 недели.
- В группе ликсисенатида величины HbA1c были значительно снижены по сравнению с группой плацебо от исходного уровня до 24 недели.
- В группе ликсисенатида процентная доля пациентов, достигших величин HbA1c ≤6,5% или ≤7% в неделю 24, была значимо выше, чем в группе плацебо.
- Концентрация инсулина в плазме натощак была ниже в группе ликсисенатида по сравнению с группой плацебо.
Один аспект настоящего изобретения относится к фармацевтической комбинации, причем указанная комбинация содержит
(a) desPro36Exendin-4(1-39)-Lys6-NH2 и/или его фармацевтически приемлемую соль, и
(b) глитазон и/или его фармацевтически приемлемую соль.
Комбинация по настоящему изобретению может применяться для лечения пациентов с сахарным диабетом 2 типа и/или для лечения состояний, связанных с сахарным диабетом 2 типа. Такие состояния включают снижение устойчивости к глюкозе, увеличенную концентрацию глюкозы в плазме после приема пищи, увеличенную концентрацию глюкозы в плазме натощак, бόльшую величину HbA1c и/или увеличенную концентрацию инсулина в плазме натощак.
Предпочтительный аспект настоящего изобретения представляет собой фармацевтическую комбинацию для применения в гликемическом контроле у пациентов с сахарным диабетом 2 типа, причем указанная комбинация содержит:
(a) desPro36Exendin-4(1-39)-Lys6-NH2 и/или его фармацевтически приемлемую соль, и
(b) глитазон и/или его фармацевтически приемлемую соль.
Как демонстрируется примером настоящего изобретения, описанная здесь комбинация может применяться для улучшения гликемического контроля. В настоящем изобретении термин «улучшение гликемического контроля» или «гликемический контроль», в частности, относится к повышению устойчивости к глюкозе, коррекции концентрации глюкозы в плазме натощак, коррекции величины HbA1c и/или коррекции концентрации инсулина в плазме натощак.
В частности, повышение устойчивости к глюкозе включает коррекцию концентрации глюкозы в плазме после приема пищи и/или коррекцию концентрации инсулина в плазме натощак. Конкретнее, повышение устойчивости к глюкозе включает коррекцию концентрации глюкозы в плазме после приема пищи.
В частности, коррекция концентрации глюкозы в плазме после приема пищи представляет собой снижение концентрации глюкозы в плазме после приема пищи. Снижение, в частности, значит, что концентрация глюкозы в плазме достигает нормогликемических величин или по меньшей мере приближается к этим величинам.
В частности, коррекция концентрации глюкозы в плазме натощак представляет собой снижение концентрации глюкозы в плазме натощак. Снижение, в частности, значит, что концентрация глюкозы в плазме достигает нормогликемических величин или по меньшей мере приближается к этим величинам.
В частности, коррекция величины HbA1c представляет собой уменьшение величины HbA1c. уменьшение величины HbA1c, в частности, значит, что величина HbA1c снижается ниже 6,5% или 7%, например, после лечения в течение по меньшей мере одного месяца, по меньшей мере двух месяцев, по меньшей мере трех месяцев, по меньшей мере четырех месяцев, по меньшей мере пяти месяцев, по меньшей мере шести месяцев или по меньшей мере одного года.
В частности, коррекция концентрации глюкозы в плазме натощак представляет собой снижение концентрации глюкозы в плазме натощак. Концентрация инсулина в плазме взаимосвязана с концентрацией глюкозы в плазме. В условиях описанного здесь лечения, в состоянии натощак инсулин плазмы может достичь величин по меньшей мере для обеспечения непрерывной подачи глюкозы в чувствительные к инсулину органы и ткани и/или для поддержания на низком уровне печеночной продукции глюкозы в ночное время. В состояниях натощак концентрация инсулина может по меньшей мере достичь или приблизиться к величинам, связанным с нормогликемией, или концентрации глюкозы в плазме, приближающейся к нормогликемии.
В контексте настоящего изобретения, используемый здесь термин «глитазон» включает его фармацевтически приемлемые соли. Глитазон может быть выбран из пиоглитазона, троглитазона, росиглитазона и их фармацевтически приемлемых солей.
В настоящем изобретении, глитазон, в частности, пиоглитазон, может вводиться перорально. Специалисту в данной области техники известны препаративные формы глитазона, в частности, пиоглитазона, пригодные для лечения сахарного диабета 2 типа, пероральным введением. Пиоглитазон можно вводить нуждающемуся в нем пациенту в количестве, достаточном для получения терапевтического эффекта. Глитазон, в частности, пиоглитазон, можно вводить в дозе по меньшей мере 10 мг/день, по меньшей мере 20 мг/день, по меньшей мере 30 мг/день или по меньшей мере 40 мг/день. Максимальная суточная доза глитазона, в частности, пиоглитазона, может составлять 50 мг/день или 60 мг/день. Предпочтительный диапазон дозировки составляет от 10 мг/день до 50 мг/день или от 30 мг/день до 40 мг/день. Более предпочтительная доза составляет примерно 30 мг/день. Для перорального введения глитазон, в частности, пиоглитазон, может быть включен в состав твердой лекарственной формы, такой как таблетка или пилюля. Глитазон, в частности, пиоглитазон, может быть включен в состав с пригодными фармацевтически приемлемыми носителями, адъювантами и/или вспомогательными веществами.
Фармацевтическая комбинация по настоящему изобретению может дополнительно содержать метформин и/или его фармацевтически приемлемую соль. Метформин представляет собой международное непатентованное наименование 1,1-диметилбигуанида (Номер по CAS 657-24-9). В настоящем изобретении термин «метформин» включает его любую фармацевтически приемлемую соль.
В настоящем изобретении метформин можно вводить перорально. Специалисту в данной области техники известны препаративные формы метформина, пригодные для лечения сахарного диабета 2 типа пероральным введением. Метформин можно вводить нуждающемуся в нем пациенту в количестве, достаточном для получения терапевтического эффекта. Метформин можно вводить в дозе по меньшей мере 1,0 г/день или по меньшей мере 1,5 г/день. Для перорального введения метформин может быть включен в состав твердой лекарственной формы, такой как таблетка или пилюля. Метформин можно включать в состав с пригодными фармацевтически приемлемыми носителями, адъювантами и/или вспомогательными веществами.
Если метформин присутствует в комбинации по настоящему изобретению, то метформин и глитазон, в частности, пиоглитазон, могут быть предоставлены в одной препаративной форме, например, в твердой лекарственной форме, такой как таблетка или пилюля. Метформин и глитазон, в частности, пиоглитазон, можно включать в состав с пригодными фармацевтически приемлемыми носителями, адъювантами и/или вспомогательными веществами.
В настоящем изобретении desPro36Exendin-4(1-39)-Lys6-NH2 и/или фармацевтически приемлемую соль можно вводить при терапии, дополнительной к введению глитазона, в частности, пиоглитазона.
В настоящем изобретении термины «дополнительное», «дополнительное лечение» «дополнительная терапия» и «в добавление к» относятся к лечению сахарного диабета 2 типа глитазоном, в частности, пиоглитазоном и ликсисенатидом. Также может быть включено лечение метформином, как описано в настоящей заявке. Глитазон, в частности, пиоглитазон и ликсисенатид, можно вводить в пределах интервала времени 24 ч. Каждый из глитазона, в частности, пиоглитазона и ликсисенатида, можно вводить в дозировке один раз в день. Глитазон, в частности, пиоглитазон и ликсисенатид, можно вводить различными путями введения. Глитазон, в частности, пиоглитазон, можно вводить перорально, а ликсисенатид можно вводить парентерально.
Пациент, подлежащий лечению лекарственным средством по настоящему изобретению, может представлять собой пациента, страдающего сахарным диабетом 2 типа. Пример демонстрирует у этих пациентов, что введение ликсисенатида в комбинации с глитазоном, в частности, пиоглитазоном, обеспечивает имеющую преимущества терапию.
Подлежащий лечению лекарственным средством по настоящему изобретению пациент, страдающий сахарным диабетом 2 типа, может представлять собой пациента, страдающего сахарным диабетом 2 типа, причем сахарный диабет 2 типа неадекватно контролируется лечением одним глитазоном, в частности, пиоглитазоном, например, дозой, выбранной из диапазона от 10 мг/день до 50 мг/день, в частности, примерно 30 мг/день, в течение по меньшей мере 2 или по меньшей мере 3 месяцев. В настоящем изобретении пациент, у которого сахарный диабет 2 тип контролируется неадекватно, может иметь величину HbA1c в диапазоне от 7% до 10%.
Подлежащий лечению лекарственным средством по настоящему изобретению пациент, страдающий сахарным диабетом 2 типа, может представлять собой страдающего ожирением пациента. В настоящем изобретении страдающий ожирением пациент может иметь индекс массы тела по меньшей мере 30 кг/м2.
Подлежащий лечению лекарственным средством по настоящему изобретению пациент, страдающий сахарным диабетом 2 типа, может иметь нормальную массу тела. В настоящем изобретении пациент, имеющий нормальную массу тела, может иметь индекс массы тела в диапазоне от 17 кг/м2 до 25 кг/м2, от 17 кг/м2 до <30 кг/м2 или <30 кг/м2.
Пациент, подлежащий лечению лекарственным средством по настоящему изобретению, может представлять собой взрослого пациента. Возраст пациента может составлять по меньшей мере 18 лет или находиться в диапазоне от 18 до 80 лет, от 18 до 50 лет или от 40 до 80 лет или от 50 до 60 лет. Пациенту может быть меньше 50 лет.
Пациент, подлежащий лечению лекарственным средством по настоящему изобретению, предпочтительно не получает противодиабетическое лечение, например, инсулином и/или родственными соединениями.
Пациент, подлежащий лечению лекарственным средством по настоящему изобретению, может страдать сахарным диабетом 2 типа в течение по меньшей мере 1 года или по меньшей мере 2 лет. В частности, у пациента с сахарным диабетом 2 типа сахарный диабет 2 типа был диагностирован по меньшей мере 1 год или по меньшей мере 2 года до начала терапии лекарственным средством по настоящему изобретению.
У пациента с сахарным диабетом 2 типа величина HbA1c может составлять по меньшей мере примерно 8% или по меньшей мере примерно 7,5%. Величина HbA1c у пациента может также составлять от примерно 7% до примерно 10%. Пример настоящего изобретения демонстрирует, что лечение ликсисенатидом приводит к уменьшению величины HbA1c у пациентов с сахарным диабетом 2 типа.
В еще одном аспекте настоящего изобретения описанную здесь комбинацию можно применять для коррекции величины HbA1c у пациента, страдающего сахарным диабетом 2 типа, как описано здесь.
В еще одном аспекте настоящего изобретения описанную здесь комбинацию можно применять для коррекции устойчивости к глюкозе у пациента, страдающего сахарным диабетом 2 типа, как описано здесь.
В еще одном аспекте настоящего изобретения описанную здесь комбинацию можно применять для коррекции концентрации глюкозы в плазме после приема пищи у пациента, страдающего сахарным диабетом 2 типа, как описано здесь.
В еще одном аспекте настоящего изобретения описанную здесь комбинацию можно применять для коррекции концентрации глюкозы в плазме натощак у пациента, страдающего сахарным диабетом 2 типа, как описано здесь.
В еще одном аспекте настоящего изобретения описанную здесь комбинацию можно применять для коррекции концентрации инсулина в плазме натощак у пациента, страдающего сахарным диабетом 2 типа, как описано здесь.
В настоящем изобретении нормогликемические величины представляют собой концентрации глюкозы крови, в частности, от 60 до 140 мг/дл (соответствующие величинам от 3,3 до 7,8 мМ/л). Этот диапазон относится, в частности, к концентрациям глюкозы в крови в условиях натощак и/или в условиях после приема пищи.
У пациента с сахарным диабетом 2 типа концентрация глюкозы в плазме через 2 часа после приема пищи может составлять по меньшей мере 10 ммоль/л, по меньшей мере 12 ммоль/л или по меньшей мере 14 ммоль/л. Эти концентрации глюкозы в плазме превышают нормогликемические концентрации.
У пациента с сахарным диабетом 2 типа колебание уровня глюкозы может составлять по меньшей мере 2 ммоль/л, по меньшей мере 3 ммоль/л, по меньшей мере 4 ммоль/л или по меньшей мере 5 ммоль/л. В настоящем изобретении колебание уровня глюкозы представляет собой, в частности, разность между концентрацией глюкозы в плазме через 2 часа после приема пищи и концентрацией глюкозы в плазме за 30 минут перед пробным приемом пищи.
«После приема пищи» представляет собой термин, который хорошо известен специалисту в данной области диабетологии. Термин «после приема пищи» описывает, в частности, фазу после приема пищи и/или воздействия глюкозы в экспериментальных условиях. У здорового индивида эта фаза характеризуется увеличением и последующим уменьшением концентрации глюкозы в плазме. Термин «после приема пищи» или «фаза после приема пищи» обычно заканчивается после периода до 2 ч после приема пищи и/или воздействия глюкозы.
Как описано здесь, у пациента с сахарным диабетом 2 типа концентрация глюкозы в плазме натощак может составлять по меньшей мере 8 ммоль/л, по меньшей мере 8,5 ммоль/л или по меньшей мере 9 ммоль/л. Эти концентрации глюкозы в плазме превышают нормогликемические концентрации.
В настоящем изобретении, desPro36Exendin-4(1-39)-Lys6-NH2 и/или его фармацевтически приемлемую соль можно вводить нуждающемуся в нем пациенту в количестве, достаточном для получения терапевтического эффекта. В настоящем изобретении desPro36Exendin-4(1-39)-Lys6-NH и/или его фармацевтически приемлемую соль можно включить в состав с пригодными фармацевтически приемлемыми носителями, адъювантами и/или вспомогательными веществами.
Соединение desPro36Exendin-4(1-39)-Lys6-NH2 и/или его фармацевтически приемлемую соль можно вводить парентерально, например, инъекцией (такой как внутримышечная или подкожная инъекция). Известны пригодные инъекционные устройства, например, так называемые «ручки», содержащие картридж, содержащий активный ингредиент и инъекционную иглу. Соединение desPro36Exendin-4(1-39)-Lys6-NH2 и/или его фармацевтически приемлемую соль можно вводить в подходящем количестве, например, в количестве в диапазоне от 10 до 15 мкг на дозу или от 15 до 20 мкг на дозу.
В настоящем изобретении desPro36Exendin-4(1-39)-Lys6-NH2 и/или его фармацевтически приемлемую соль можно вводить в суточной дозе в диапазоне от 10 до 20 мкг, в диапазоне от 10 до 15 мкг или в диапазоне от 15 до 20 мкг. DesPro36Exendin-4(1-39)-Lys6-NH2 и/или его фармацевтически приемлемую соль можно вводить одной инъекцией в день.
В настоящем изобретении desPro36Exendin-4(1-39)-Lys6-NH2 и/или его фармацевтически приемлемая соль могут быть представлены в виде жидкой композиции. Специалисту в данной области техники известны жидкие композиции ликсисенатида, подходящие для парентерального введения. Жидкая композиция по настоящему изобретению может иметь кислотный или физиологический показатель pH. Кислотный показатель pH предпочтительно находится в диапазоне pH 1-6,8, pH 3,5-6,8 или pH 3,5-5. Физиологический показатель pH предпочтительно находится в диапазоне pH 2,5-8,5, pH 4,0-8,5 или pH 6,0-8,5. Показатель pH можно регулировать фармацевтически приемлемой разбавленной кислотой (обычно, HCl) или фармацевтически приемлемого разбавленного основания (обычно, NaOH).
Жидкая композиция, содержащая desPro36Exendin-4(1-39)-Lys6-NH2 и/или его фармацевтически приемлемую соль, может содержать подходящий консервант. Подходящий консервант может быть вызван из фенола, м-крезола, бензилового спирта и сложного эфира п-гидроксибензойной кислоты. Предпочтительным консервантом является м-крезол. Жидкая композиция, содержащая desPro36Exendin-4(1-39)-Lys6-NH2 и/или его фармацевтически приемлемую соль, может содержать вещество, регулирующее тоничность. Подходящее вещество, регулирующее тоничность, можно выбрать из глицерина, лактозы, сорбита, маннита, глюкозы, NaCl, соединений, содержащих кальций или магний, таких как CaCl2. Концентрация глицерина, лактозы, сорбита, маннита, глюкозы может находиться в диапазоне от 100 до 250 мМ. Концентрация NaCl может составлять вплоть до 150 мМ. Предпочтительным веществом, регулирующим тоничность, является глицерин.
Жидкая композиция, содержащая desPro36Exendin-4(1-39)-Lys6-NH2 и/или его фармацевтически приемлемую соль, может содержать метионин от 0,5 мкг/мл до 20 мкг/мл, предпочтительно от 1 мкг/мл до 5 мкг/мл. Предпочтительно, жидкая композиция содержит L-метионин.
Еще один аспект настоящего изобретения относится к способу лечения медицинского показания, как описано здесь. Например, способ может включать введение комбинации, как описано здесь. Способ может представлять собой способ лечения пациентов с сахарным диабетом 2 типа и/или лечения состояний, связанных с сахарным диабетом 2 типа, как описано здесь. Пациент может представлять собой пациента, как определено здесь.
Дополнительный аспект настоящего изобретения представляет собой способ улучшения гликемического контроля у пациентов с сахарным диабетом 2 типа, причем указанный способ включает введение desPro36Exendin-4(1-39)-Lys6-NH2 и/или его фармацевтически приемлемой соли в комбинации с глитазоном, в частности, пиоглитазоном, нуждающемуся в нем пациенту. В частности, может быть введена комбинация, описанная в настоящем документе. В способе по настоящему изобретению пациент может представлять собой пациента, определенного в настоящем описании.
Еще один аспект настоящего изобретения относится к применению комбинации, как описано здесь, для получения лекарственного средства для лечения медицинского показания, как описано здесь. Например, комбинацию по настоящему изобретению можно применять для получения лекарственного средства для лечения пациентов с сахарным диабетом 2 типа и/или для лечения состояний, связанных с сахарным диабетом 2 типа. В частности, комбинацию по настоящему изобретению можно применять для получения лекарственного средства для улучшения гликемического контроля, коррекции устойчивости к глюкозе, коррекции концентрации глюкозы в плазме после приема пищи, коррекции концентрации глюкозы в плазме натощак, коррекции величины HbA1C и/или коррекции концентрации инсулина в плазме натощак. Пациент может представлять собой пациента, как определено в настоящем описании.
Изобретение дополнительно иллюстрируется следующим примером и чертежами.
НАДПИСИ НА ЧЕРТЕЖАХ
Фиг.1 - Структура исследования.
Фиг.2 - График Каплана-Мейера времени до прекращения лечения по любой причине - Рандомизированная популяция.
Фиг.3 - График среднего изменения HbA1c (%) от исходного уровня по посещениям до недели 24 - mITT-популяция (модифицированная популяция пациентов, включенных в исследование). LOCF = метод переноса вперед данных последнего проведенного наблюдения. Примечание: График включает измерения перед введением средств, используемых при усугублении симптомов заболевания, и до 3 дней после последней инъецированной дозы исследуемого двойным слепым методом продукта во время или перед посещением 12 (неделя 24), или в день 169, если недоступны данные посещения 12 (неделя 24).
Фиг.4 - График среднего изменения глюкозы плазмы натощак (ммоль/л) от исходного уровня до недели 24 - mITT-популяция. LOCF = метод переноса вперед данных последнего проведенного наблюдения. Примечание: График включает измерения перед введением средств, используемых при усугублении симптомов заболевания, и до 1 дня после последней инъецированной дозы исследуемого двойным слепым методом продукта во время или перед посещением 12 (неделя 24), или в день 169, если недоступны данные посещения 12 (неделя 24).
Фиг.5 - График среднего изменения массы тела (кг) от исходного уровня по посещению до недели 24 - mITT-популяция. LOCF = метод переноса вперед данных последнего проведенного наблюдения. Примечание: График включает измерения перед введением средств, используемых при усугублении симптомов заболевания, и до 3 дней после последней инъецированной дозы исследуемого двойным слепым методом продукта во время или перед посещением 12 (неделя 24), или в день 169, если недоступны данные посещения 12 (неделя 24).
Фиг.6 - График среднего изменения HbA1c (%) от исходного уровня по посещению - mITT-популяция. LOCF = метод переноса вперед данных последнего проведенного наблюдения. EOT = Последняя величина при получении исследуемого лечения. Примечание: Анализ исключает измерения, полученные после введения средств, используемых при усугублении симптомов заболевания, и/или после прекращения лечения, плюс 3 дня. Для недели 24 (LOCF), анализ включает измерения, полученные до 3 дней после последней инъецированной дозы исследуемого двойным слепым методом продукта во время или перед посещением 12 (неделя 24), или в день 169, если недоступны данные посещения 12 (неделя 24).
Фиг.7 - График среднего изменения глюкозы в плазме натощак (ммоль/л) от исходного уровня по посещению - mITT-популяция. LOCF = метод переноса вперед данных последнего проведенного наблюдения. EOT = Последняя величина при получении исследуемого лечения. Примечание: Анализ исключает измерения, полученные после применения средств, используемых при усугублении симптомов заболевания, и/или после прекращения лечения, плюс 1 день. Для недели 24 (LOCF), анализ включает измерения, полученные до 1 дня после последней инъецированной дозы исследуемого двойным слепым методом продукта во время или перед посещением 12 (неделя 24), или в день 169, если недоступны данные посещения 12 (неделя 24).
Фиг.8 - График среднего изменения массы тела (кг) от исходного уровня по посещению - mITT-популяция. LOCF - метод переноса вперед данных последнего проведенного наблюдения, EOT = Последняя величина при получении исследуемого лечения. Примечание: Анализ исключает измерения, полученные после применения средств, используемых при усугублении симптомов заболевания, и/или после прекращения лечения, плюс 3 дня. Для недели 24 (LOCF), анализ включает измерения, полученные до 3 дней после последней инъецированной дозы исследуемого двойным слепым методом продукта во время или перед посещением 12 (неделя 24), или в день 169, если недоступны данные посещения 12 (неделя 24).
ПРИМЕР
РЕЗЮМЕ
Пример относится к рандомизированному, двойному слепому, працебо-контролируемому, 2-групповому, несбалансированно структурированному, параллельно-групповому, многоцентровому, многонациональному исследованию, оценивающему эффективность и безопасность ликсисенатида в дополнение к пиоглитазону у пациентов с сахарным диабетом 2 типа, неадекватно контролируемым пиоглитазоном. Приблизительная минимальная длительность двойного слепого исследования на одного пациента составила 79 недель (скрининг до 2 недель + 1-недельный вводный период + 24 недели основного периода лечения, исследуемого двойным слепым методом + варьирующееся продолжение + 3-дневное наблюдение).
Исследование проводили в 150 центрах в 13 странах. Первичной целью исследования была оценка эффективности ликсисенатида в отношении гликемического контроля, по сравнению с плацебо, в качестве дополнительного пиоглитазону терапевтического средства с точки зрения снижения уровня HbA1c (абсолютного изменения) в течение периода 24 недель.
В общей сложности 484 пациента методом рандомизации включали в одну из двух групп лечения (323 в группу ликсисенатида и 161 в группу плацебо). Все рандомизированные пациенты получали исследуемое лечение. Демографические и исходные характеристики были в целом одинаковыми в группах лечения. Пять пациентов (3, получавших ликсисенатид, и 2, получавших плацебо) были исключены из модифицированной популяция пациентов, включенных в исследование (mITT), для анализов эффективности вследствие отсутствия данных эффективности после оценки исходных показателей. В течение всего периода исследуемого лечения 136 (28,1%) пациентов преждевременно прекратили исследуемое лечение (26,0% в группе ликсисенатида и 32,3% в группе плацебо). Для группы ликсисенатида основной причиной прекращения лечения была «другие причины» (10,5%, в сравнении с 12,4% для плацебо), за которой следовала причина «побочные явления» (9,0%, в сравнении с 8,7% для плацебо).
Анализы эффективности базируются на 24-недельном основном периоде лечения, исследуемом двойным слепым методом. Изменения предела среднего (LS) HbA1c от исходного уровня до недели 24 составили -0,90% в группе ликсисенатида и -0,34% в группе плацебо (разность LS среднего в сравнении с плацебо = -0,56%; p-величина <0,0001). Процентные доли пациентов, достигающих уровня HbA1c ≤6,5% или <7% в неделю 24, были значимо выше в группе ликсисенатида, чем в группе плацебо (для HbA1c ≤6,5%, 28,9% в группе ликсисенатида, в сравнении с 10,1% в группе плацебо; для HbA1c <7%, 52,3% в группе ликсисенатида, в сравнении с 26,4% в группе плацебо). Анализ HbA1c у пациентов, реагирующих на лечение (HbA1c ≤6,5% или <7% в неделю 24), с использованием метода Кохрана-Мантеля-Хензеля (CMH) также показал значимое различие результатов лечения ликсисенатидом и плацебо в неделю 24 (p-величина <0,0001).
Для уровня глюкозы в плазме натощак (FPG) значимое снижение от исходного уровня до недели 24 наблюдали в группе ликсисенатида, по сравнению с группой плацебо (разность LS среднего в сравнении с плацебо = -0,84 ммоль/л; p-величина <0,0001). Для массы тела уменьшение LS среднего составила 0,21 кг от исходного уровня в неделю 24 в группе ликсисенатида, по сравнению с увеличением LS среднего на 0,21 кг в группе плацебо, и различие между 2 группами не было статистически значимым (разность LS среднего в сравнении с плацебо = -0,41 кг). В отношении стратегии тестирования для поправки на множественность, дедуктивное тестирование для последующих переменных величин эффективности было поисковым, поскольку анализ массы тела не смог показать статистически значимое различие. Не было релевантного различия, наблюдаемого в функции β-клеток, по данным оценки ΗΟΜΑ-β (модели оценки гомеостаза β-клеток) между ликсисенатидом и плацебо при различии LS среднего -0,25 (95% CI (ДИ, доверительный интервал)): [-6,579 до 6,070]). Процентная доля пациентов, требующих экстренной терапии в неделю 24, была значимо ниже в группе ликсисенатида (12 пациентов [3,8%]), по сравнению с группой плацебо (18 [11,3%]). Для уровня инсулина в плазме натощак (FPI), снижение LS среднего было бόльшим в группе ликсисенатида, чем в группе плацебо, при разности LS среднего -9,36 пмоль/л (95% ДИ: [от -16,586 до -2,124]).
Анализы безопасности базируются на периоде лечения всего исследования. Ликсисенатид был хорошо переносим. Доли пациентов, которые испытали возникновение связанных с лечением побочных эффектов (TEAE) составили 87,9% в группе ликсисенатида и 83,2% в группе плацебо. Случаев смерти пациентов в группе ликсисенатида не было, тогда как в группе плацебо умерли 2 пациента. У одного пациента развился связанный с лечением острый инфаркт миокарда, приведший к смерти, а другой пациент умер вследствие побочного эффекта (AE) после лечения (конечной стадии астения) ввиду дыхательной недостаточности с мультиорганной недостаточностью. Процентная доля пациентов, у которых были тяжелые TEAE, была ниже в группе ликсисенатида (7,4%), чем в группе плацебо (9,3%). Наиболее часто отмечаемыми TEAE в группе ликсисенатида были тошнота (26% в сравнении с 13,7% для плацебо), за которой следует назофарингит (16,4% в сравнении с 14,9% для плацебо) и головная боль (13,3% в сравнении с 11,8% для плацебо). В течение периода проведения лечения всего исследования, у 23 (7,1%) пациентов в группе ликсисенатида была клинически выраженная гипогликемия по протокольному определению, по сравнению с 7 (4,3%) в группе плацебо. Ни одно из клинически выраженных явлений гипогликемии не было тяжелым по интенсивности. 22 (6,8%) пациента в группе ликсисенатида и 8 (5,0%) в группе плацебо испытали побочные явления в виде реакции в участке инъекции. Общее число 12 пациентов (9 [2,8%] в группе ликсисенатида и 3 [1,9%] в группе плацебо) отметили 19 явлений, которые расценивались как аллергические реакции Комитетом по оценке аллергических реакций (ARAC). Из них, 5 явлений у 3 пациентов в группе ликсисенатида (1 пациент с аллергическим дерматитом, 1 с сыпью и 1 с ангиоотеком, анафилактической реакцией и аллергическим конъюнктивитом) расценивались как возможно связанные с IP (исследуемым продуктом). Ни одно явление в группе плацебо не расценивалось как возможно связанное с IP. В исследовании не был отмечено случаев панкреатита или рака щитовидной железы.
1. ЦЕЛИ
1.1. ПЕРВИЧНАЯ ЦЕЛЬ
Первичной целью настоящего исследования была оценка эффективности ликсисенатида в отношении гликемического контроля в качестве дополнительного терапевтического средства к пиоглитазону, по сравнению с плацебо, у пациентов с сахарным диабетом 2 типа, получавших лечение пиоглитазоном, с точки зрения абсолютного снижения HbA1c в течение периода 24 недели.
1.2 КЛЮЧЕВЫЕ ВТОРИЧНЫЕ ЦЕЛИ
Вторичными целями настоящего исследования были:
• Для оценки воздействий ликсисенатида на
- Процентную долю пациентов, достигших HbA1c <7%,
- Процентную долю пациентов, достигших HbA1c ≤6,5%,
- Уровень глюкозы в плазме натощак (FPG),
- Массу тела,
- Функцию β-клеток, по данным оценки ΗΟΜΑ-β,
- Уровень инсулина в плазме натощак (FPI).
• Для оценки безопасности и переносимости ликсисенатида.
2. СТРУКТУРА ИСПЫТАНИЯ
Это было несбалансированное (2:1), рандомизированное, двойное слепое, плацебо-контролируемое, 2-групповое, параллельное, многоцентровое, многонациональное исследование с варьирующимся периодом продолжения, сравнивающее лечение ликсисенатидом и плацебо у пациентов с сахарным диабетом 2 типа (300 пациентов в группе ликсисенатида и 150 пациентов в группе плацебо). Исследование было двойным слепым в отношении лечения активным средством и плацебо. Объем исследуемого лекарственного средства (т.е., доза активного лекарственного средства или соответствующей дозы плацебо) не был скрыт. Пациентов стратифицировали по величинам скрининга HbA1c (<8%, ≥8%) и применению метформина (Да, Нет) при скрининге.
Приблизительная минимальная продолжительность двойного слепого исследования на одного пациента составляла 79 недель (скрининг до 2 недель + 1-недельный вводный период + 24 недели основного периода лечения, исследуемого двойным слепым методом + варьирующееся продолжение + 3-дневное наблюдение). Пациентов, которые завершили 24-недельный основной двойной слепой период лечения, подвергали лечению в течение вариабельного двойного слепого периода продолжения, который заканчивался для всех пациентов приблизительно в запланированную дату посещения недели 76 (V25) для последнего рандомизированного пациента.
Структура испытания иллюстрируется фиг.1.
3. ПЕРВИЧНЫЕ И КЛЮЧЕВЫЕ ВТОРИЧНЫЕ ОЖИДАЕМЫЕ РЕЗУЛЬТАТЫ
3.1 ПЕРВИЧНЫЕ ОЖИДАЕМЫЕ РЕЗУЛЬТАТЫ
Первичным параметром эффективности было абсолютное изменение уровня HbA1c от исходного уровня до недели 24, который определяли как: величина HbA1c в неделю 24 - величина HbA1c в исходном состоянии.
Если пациент постоянно прекращал лечение или получал экстренную терапию в течение 24-недельного основного периода лечения, исследуемого двойным слепым методом, или у него не было данных о величине HbA1c в неделю 24, то последнее измерение после исходного уровня HbA1c в течение основного 24-недельного периода лечения, исследуемого двойным слепым методом, использовали в качестве величины HbA1c в неделю 24 (процедура перенесения вперед данных последнего проведенного наблюдения [LOCF]).
3.2. КЛЮЧЕВЫЕ ВТОРИЧНЫЕ ОЖИДАЕМЫЕ РЕЗУЛЬТАТЫ
3.2.1. Ключевые вторичные ожидаемые результаты эффективности
Для получения вторичных параметров эффективности применяли такую же процедуру для обращения с недостающими оценками/ранним прекращением лечения, как для первичного параметра эффективности.
Непрерывные параметры:
• Изменение FPG (ммоль/л) от исходного уровня до недели 24,
• Изменение массы тела (кг) от исходного уровня до недели 24,
• Изменение функции β-клеток, по данным оценки ΗΟΜΑ-β от исходного уровня до недели 24,
• Изменение FPI (пмоль/л) от исходного уровня до недели 24.
Категориальные параметры:
• Процентная доля пациентов с HbA1c <7% в неделю 24,
• Процентная доля пациентов с HbA1c ≤6,5% в неделю 24,
• Процентная доля пациентов, требующих экстренную терапию в течение основного 24-недельного основного периода лечения, исследуемого двойным слепым методом,
• Процентная доля пациентов с потерей массы тела ≥5% (кг) от исходного уровня до недели 24.
3.2.2. Конечные результаты безопасности
Анализ безопасности основывался на отмеченных TEAE и другой информации о безопасности, включая клинически выраженную гипогликемию и тяжелую клинически выраженную гипогликемию, местную переносимость в участке инъекции, аллергические явления (по оценкам с использованием ARAC), подозреваемый панкреатит, повышенный уровень кальцитонина, жизненно важные признаки, ЭКГ в 12 отведениях и лабораторные тесты.
Также собирали данные о значимых сердечно-сосудистых явлениях, и заключение слепым методом выносил Комитет экспертных заключений по сердечно-сосудистым явлениям (CAC). Рассмотренные и подтвержденные CAC явления в настоящем исследовании и других исследованиях ликсисенатида 3 фазы будут объединены для анализа и резюмированы в отдельном отчете на основании плана статистического анализа для общей оценки ликсисенатида в отношении воздействия на сердечно-сосудистую систему. KRM/CSR (Дистанционный мониторинг/Центр научного анализа) не представит резюме по рассмотренным и подтвержденным CV (сердечно-сосудистым) явлениям по настоящему исследованию.
4. ДОПУЩЕНИЯ ПРИ РАСЧЕТЕ ОБЪЕМА ВЫБОРКИ
Расчеты объема выборки/мощности исследования выполняли на основании первичного показателя, абсолютного изменения HbA1c от исходного уровня до недели 24.
Триста (300) пациентов в группе лечения ликсисенатидом и 150 в группе лечения плацебо обеспечили статистическую мощность 96% (или 86%) для выявления различий 0,5% (или 0,4%) абсолютного изменения HbA1c от исходного уровня до недели 24 между группой ликсисенатида и группой плацебо. Этот расчет допускал общее стандартное отклонение 1,3% при 2-стороннем критерии на уровни значимости различий 5%. Расчеты объема выборки были основаны на 2-выборочном t-критерии и производили с использованием программного обеспечения nQuery Advisor® 5.0. Стандартное отклонение оценивали консервативным образом по ранее проведенным исследованиям лечения сахарного диабета (на основании опубликованных данных аналогичным образом структурированного исследования и на внутренних данных, не опубликованных), принимая во внимание случаи раннего выбывания пациентов из исследования.
5. СТАТИСТИЧЕСКИЕ МЕТОДЫ
5.1. АНАЛИЗИРУЕМАЯ ПОПУЛЯЦИЯ
Модифицированная популяция пациентов, включенных в исследование (mITT), состояла из всех рандомизированных пациентов, которые получали по меньшей мере 1 дозу исследуемого продукта (IP) двойным слепым методом, и у которых была и исходная оценка, и по меньшей мере 1 оценка параметров эффективности после определения исходного уровня.
Популяцию безопасности определяли как всех рандомизированных пациентов, которые приняли по меньшей мере одну дозу IP двойным слепым методом.
5.2. ПЕРВИЧНЫЙ АНАЛИЗ ЭФФЕКТИВНОСТИ
Первичный показатель эффективности (изменение HbA1c от исходного уровня до недели 24) анализировали, используя модель ковариационного анализа (ANCOVA) с группами лечения (ликсисенатид и плацебо), рандомизационные страты, различающиеся уровнем HbA1c при скрининге (<8,0, ≥8,0%), рандомизационные страты, различающиеся применением метформина при скрининге (Да, Нет), и страну в качестве фиксированных воздействий и исходную величину HbA1c в качестве ковариата. Обеспечивали и средние величины, и скорректированные средние величины для ликсисенатида и плацебо, а также 95% доверительные интервалы (CI), выведенные для различий скорректированных средних величин между группами ликсисенатида и плацебо. Различие между группами ликсисенатида и плацебо и двусторонний 95% доверительный интервал, а также p-величину оценивали в пределах сети ANCOVA.
Первичный анализ первичного показателя эффективности осуществляли на основании mITT-популяции и измерений, полученных в течение основного 24-недельного периода лечения, проводимого двойным слепым методом, в отношении показателей эффективности. Основной 24-недельный период лечения, исследуемый двойным слепым методом в отношении показателей эффективности, представлял собой время от введения первой дозы IΡ двойным слепым методом до 3 дней (за исключением, FPG и FPI, которые учитывали до 1 дня) после последней дозы инъекции IP двойным слепым методом в или перед посещением V12/в неделю 24 (или D169, если отсутствуют данные по посещению V12 в неделю 24) или вплоть до проведения экстренной терапии, в зависимости от того, что наступало раньше всего. Процедуру LOCF использовали учетом этого последнего доступного измерения HbA1c в ходе лечения после исходного уровня (перед назначением экстренной терапии) в виде величины HbA1c в неделю 24.
5.3. КЛЮЧЕВОЙ АНАЛИЗ ВТОРИЧНОЙ ЭФФЕКТИВНОСТИ
Процедуру тестирования со ступенчатым понижением применяли для обеспечения контроля над ошибкой 1 типа. Как только первичный параметр был статистически значимым при α=0,05, процедуру тестирования выполняли для испытания следующих параметров вторичной эффективности в следующем приоритетном порядке. Тесты прекращали как только обнаруживали, что ожидаемый результат не был статистически значимым при α=0,05.
• Изменение FPG (ммоль/л) от исходного уровня до недели 24,
• Изменение массы тела (кг) от исходного уровня до недели 24,
• Изменение функции β-клеток, по данным оценки ΗΟΜΑ-β, от исходного уровня до недели 24,
• Процентная доля пациентов, потребовавшая экстренной терапии в течение 24-недельного периода лечения,
• Изменение FPI (ммоль/л) от исходного уровня до недели 24.
Все непрерывные вторичные показатели эффективности в неделю 24, как описано в разделе 3.2.1, анализировали с использованием аналогичного подхода и модели ANCOVA, как описано в разделе 5.2 для первичного анализа первичных ожидаемых результатов эффективности. Были предоставлены скорректированные оценки средних величин показателей эффективности лечения между ликсисенатидом и плацебо и двухсторонние 95% доверительные интервалы.
Следующие категориальные вторичные показатели эффективности в неделю 24 анализировали, используя метод Кохрана-Мантеля-Хензеля (CMH), со стратификацией на рандомизационные страты (HbA1c при скрининге [<8,0, ≥8,0%] и применения метформина при скрининге (Да, Нет):
• Процентная доля пациентов с HbA1c <7,0% в неделю 24,
• Процентная доля пациентов с HbA1c ≤6,5% в неделю 24,
• Процентная доля пациентов, потребовавших экстренного лечения в течение основного 24-недельного периода исследования лечения двойным слепым методом.
Число и процентная доля пациентов с потерей массы ≥5% от исходного уровня в неделю 24 были представлены группами лечения.
Результаты для всех ожидаемых результатов эффективности в течение вариабельного периода продолжения и в конце лечения подлежали оценке только методами описательной статистики.
5.4. АНАЛИЗ БЕЗОПАСНОСТИ
Анализы безопасности были в первую очередь основаны на периоде проведения лечения всего исследования. Период проведения лечения всего исследования определяли как время от введения первой дозы IP, исследуемого двойным слепым методом, до 3 дней после последней дозы введения IP в течение всего периода исследования, независимо от статуса экстренного лечения. 3-дневный интервал был выбран на основании периода полувыведения IP (приблизительно в 5 раз больше периода полувыведения).
Кроме того, анализы безопасности для 24-недельного периода лечения, исследуемого двойным слепым методом, будут резюмированы в CSR.
Резюме результатов определения безопасности (описательная статистика или таблицы частоты) представлено по группам лечения.
6. РЕЗУЛЬТАТЫ
6.1. ИССЛЕДУЕМЫЕ ПАЦИЕНТЫ
6.1.1. Возможность учета пациентов
Исследования проводили в 150 центрах в 13 странах (Австрия, Канада, Франция, Германия, Греция, Гватемала, Индия, Мексика, Перу, Пуэрто-Рико, Румыния, Турция и США). Провели скрининг в общей сложности 906 пациентов, и 484 были рандомизированы в 1 из 2 групп лечения. Самой частой причиной нерандомизации была величина HbA1c вне диапазона при посещении в ходе скрининга, как определено в соответствии с протоколом (283 [31,2%] из 906 прошедших скрининг пациентов).
Все 484 рандомизированных пациента были подвергнуты исследуемому лечению, и 5 пациентов (3 в группе ликсисенатида и 2 в группе плацебо) были исключены из mITT-популяции для анализов эффективности вследствие отсутствия данных эффективности после исходного определения. В таблице 1 представлено число пациентов, включенных в каждую анализируемую популяцию.
Вследствие несоблюдения исследователями клинического протокола и нарушения принципов должной клинической практики, спонсор прекратил лечение одного пациента в группе ликсисенатида. Пациента подвергали лечению в течение 113 дней и включали в анализы безопасности и эффективности.
Анализируемая популяция - Рандомизированная популяция
Модифицированная популяция пациентов, включенных в исследование (mITT)
Для популяции исследования эффективности пациенты внесены в таблицу в соответствии с их определенным рандомизацией лечением (в соответствии с рандомизацией).
6.1.2. Распределение объектов исследования
В таблице 2 представлено резюме распределения пациентов по каждой группе лечения. В течение общего периода лечения 136 (28,1%) пациентов преждевременно прекратили исследуемое лечение (26,0% для ликсисенатида и 32,3% для плацебо). В группе ликсисенатида главной причиной прекращения лечения были «другие причины» (10,5%, в сравнении с 12,4% для плацебо), за которой следовали «побочные явления» (9,0%, в сравнении с 8,7% для плацебо).
Аналогичные результаты наблюдали для 24-недельного периода лечения, где в общей сложности 59 (12,2%) пациентов преждевременно прекратили исследуемое лечение (10,8% для ликсисенатида и 14,9% для плацебо), причем основными причинами в группе ликсисенатида являются «другие причины» (4,0% в сравнении с 5,0% для плацебо) и «побочные явления» (4,0% в сравнении с 5,6% для плацебо). Категория «других причин» была подтверждена исследователями как не связанная с AE и включала без ограничения личные причины, противоречие со схемой лечения, перемещение, неудобство инъекции, закрытие участка инъекции и т.д. Время до начала прекращения лечения по любой причине для всего периода лечения изображено на фиг.2. Более низкую частоту прекращения наблюдали в группе ликсисенатида в течение всего периода лечения, по сравнению с группой плацебо. Увеличение частоты прекращения лечения от примерно 25 до 100% в группе ликсисенатида в конце исследования было связано с наблюдавшимся дольше всех пациентом, прекратившим лечение в 874 день.
Одного пациента в группе ликсисенатида, который прекратил лечение вследствие «увеличенного содержания гликозилированного гемоглобина» в таблице 20, учитывали таблице 2 как не имевшего эффекта, тогда как 2 пациентов в группе плацебо, которые прекратили лечение по причине AE, не учитывали в таблице 20, потому что их AE, ведущие к прекращению лечения, происходили в течение периода до или после лечения.
Распределение пациентов - Рандомизированная популяция
6.1.3 Демографические показатели и исходные характеристики
Демографические показатели и исходные характеристики пациентов были в целом одинаковыми в группах лечения у популяции исследования безопасности (таблица 3). Медианный возраст составил 56 лет, и мужчины составили 52,5% пациентов. Исследуемую популяцию составляли преимущественно европеоиды (83,7%), и у 67,6% популяции исследования безопасности BMI был ≥30 кг/м2.
Характеристики заболевания, включая диабетический анамнез, были в целом сравнимыми в двух группах лечения (таблица 4). Медианная продолжительность сахарного диабета составила 7,22 года, и медианный возраст в начале сахарного диабета составил 48 лет. Пациенты получали пиоглитазон в течение медианного периода 0,83 года, и медианная суточная доза составила 30 мг. При скрининге 81% пациентов применяли метформин при медианной длительности 3,37 лет и медианной суточной дозе 2000 мг.
Величины HbA1c, FPG и ΗΟΜΑ-β в исходном состоянии были в целом сравнимы в группах лечения для популяции исследования безопасности (таблица 5). Более высокую массу тела в исходном состоянии наблюдали в группе плацебо (96,74 кг) по сравнению с группой ликсисенатида (92,93 кг). И средний, и медианный FPI выше в группе плацебо (66,07 пмоль/л и 53,78 пмоль/л, соответственно) по сравнению с группой ликсисенатида (63,32 пмоль/л и 46,14 пмоль/л, соответственно). Средний уровень HbA1c в исходном состоянии составил 8,07%.
Демографические показатели и характеристики пациентов при скрининге или в исходном состоянии - Популяция исследования безопасности
Характеристики заболевания при скрининге или в исходном состоянии - Популяция исследования безопасности
Величина клиренса креатинина получена с использованием уравнения Кокрофта и Голта.
Исходные показатели эффективности - Популяция исследования безопасности
6.1.4. Дозировка и длительность
Средний период проведения лечения составил 560,2 дня (80 недель) в группе ликсисенатида по сравнению с 518,6 днями (74 недели) в группе плацебо (таблица 6). Из 323 пациентов, получавших лечение ликсисенатидом, 286 (88,5%) подвергали IΡ в течение 24 недель (169 дней) или дольше, и 199 (61,6%) подвергали IP в течение 18 месяцев (547 дней) или дольше. Пять пациентов не зарегистрировали дату последнего введения на страницу CRF (Универсального бланка отчетности) «Конец лечения», и, следовательно, длительность лечения у них была отмечена как «пропущенные данные», в соответствии с правилами SAP (Плана статистического анализа) обработки данных.
В конце двойного слепого исследования лечения, 92,3% пациентов достигли целевой суточной дозы 20 мкг в группе ликсисенатида, ниже, чем в группе плацебо (97,5%) (Таблица 7). Аналогичный результат наблюдали в конце 24-недельного периода двойного слепого исследования лечения (92,6% для ликсисенатида, в сравнении с 98,8% для плацебо) (таблица 8). Доза в конце титрования представлена в таблице 28.
Воздействие исследуемого продукта - Популяция исследования безопасности
Число (%) пациентов по конечной общей суточной дозе в конце лечения, исследуемого двойным слепым методом - Популяция исследования безопасности
Примечание: Процентные доли рассчитаны с использованием числа пациентов в популяции исследования безопасности в качестве деноминатора.
Число (%) пациентов по конечной общей суточной дозе в конце лечения, исследуемого двойным слепым методом - Популяция исследования безопасности
Примечание: Процентные доли рассчитаны с использованием числа пациентов в популяции исследования безопасности в качестве деноминатора.
6.2. ЭФФЕКТИВНОСТЬ
6.2.1. Первичный ожидаемый результат эффективности
Основной анализ
В таблице 9 суммированы результаты определения первичного параметра эффективности, изменения содержания HbA1c от исходного уровня до недели 24 (LOCF) с использованием анализа ANCOVA.
Предварительно заданный первичный анализ показал, что лечение ликсисенатидом привело к статистически значимому снижению содержания HbA1c от исходного уровня до недели 24, по сравнению с группой плацебо (разность LS среднего в сравнении с группой плацебо p=-0,56%; p-величина <0,0001).
Среднее изменение HbA1c (%) от исходного уровня до недели 24 - mITT-популяция
Примечание: Анализ включает измерения перед введением лекарственного средства, используемого при усугублении симптомов, и до 3 дней после последней дозы в виде инъекции исследуемого продукта в ходе двойного слепого исследования во время или перед 12 посещением (неделя 24), или 169 днем, если данные 12 посещения (недели 24) были недоступны. Включены пациенты с измерениями и в исходном состоянии, и в неделю 24 (LOCF).
Фиг.3 иллюстрирует среднее (±SE) изменение HbA1c от исходного уровня в течение основного 24-недельного периода лечения двойным слепым методом. На фиг.6 в приложении представлено изменение средней величины HbA1c (±SE) от исходного уровня до недели 76. Снижение HbA1c поддерживалось в течение периода времени более 24 недель.
Вторичный анализ
В таблице 10 суммируется доля пациентов с реакцией на лечение HbA1c ≤6,5% или <7% в неделю 24, соответственно. Анализ пациентов с реакцией HbA1c на лечение с использованием метода CMH показал статистически значимое различие между группой ликсисенатида и группой плацебо (p-величина <0,0001).
Число (%) пациентов с величиной HbA1c ≤6,5% или <7%, соответственно, в неделю 24 - mITT-популяция
Примечание: Анализ включает измерения перед введением лекарственного средства, используемого при усугублении симптомов, и до 3 дней после последней дозы в виде инъекции исследуемого продукта в ходе двойного слепого исследования во время или перед 12 посещением (неделя 24), или 169 днем, если данные 12 посещения (недели 24) были недоступны.
6.2.2. Ключевые вторичные ожидаемые результаты эффективности
Анализы ANCOVA FPG, масса тела, HOMΑ-β и FPI представлены в данном разделе. На фиг.4 и фиг.5 иллюстрируется изменение средних величин (±SE) от исходного уровня FPG и массы тела со временем в течение основного 24-недельного периода лечения, исследуемого двойным слепым методом. Изменения средней величины (±SE) FPG и массы тела от исходного уровня с течением времени до недели 76 изображены соответственно на фиг.7 и фиг.8 в приложении. Процентная доля пациентов, которые получали экстренную терапию в течение основного 24-недельного периода лечения, исследуемого двойным слепым методом, представлена в таблице 15.
В отношении FPG, значимое уменьшение от исходного уровня до недели 24 наблюдалось в группе ликсисенатида по сравнению с группой плацебо (разность LS среднего в сравнении с плацебо = -0,84 ммоль/л; p-величина <0,0001) (таблица 11).
В отношении массы тела уменьшение LS среднего составило 0,21 кг от исходного уровня в неделю 24 в группе ликсисенатида по сравнению с увеличением LS среднего 0,21 кг в группе плацебо, но различие между двумя группами не было значимым (разность LS среднего в сравнении с плацебо = -0,41 кг) (Таблица 12). Примерно у 9,2% пациентов в группе ликсисенатида и у 5,1% в группе плацебо потеря массы составила ≥5% от исходного уровня до недели 24 (Таблица 13).
В отношении стратегии тестирования для поправки на множественность, дедуктивное тестирование для последующих переменных показателей эффективности было поисковым, поскольку анализ массы тела не смог показать статистически значимое различие.
В отношении функции β-клеток, оцененной ΗΟΜΑ-β, не наблюдали релевантного различия между ликсисенатидом и плацебо при различии LS среднего -0,25 (95% CI: [от -6,579 до 6,070]) (Таблица 14).
Процентная доля пациентов, требующих экстренной терапии в неделю 24, была существенно ниже в группе ликсисенатида (12 пациентов [3,8%]) по сравнению с группой плацебо (18 пациентов [11,3%]) (Таблица 15).
В отношении FPI, снижение LS среднего было больше в группе ликсисенатида, чем в группе плацебо, при различии LS среднего -9,36 пмоль/л (95% CI: [от -16,586 до -2,124]) (Таблица 16).
Среднее изменение уровня глюкозы в плазме натощак (ммоль/л) от исходного уровня до недели 24 - mITT-популяция
аМодель ковариационного анализа (ANCOVA) с группами лечения (ликсисенатид и плацебо), рандомизационными стратами по уровню HbA1c (<8,0, ≥8,0%) при скрининге, по применению метформина при скрининге (Да, Нет) и стране в качестве фиксированных воздействий и исходного уровня глюкозы в плазме натощак в качестве ковариата.
Примечание: Анализ включает измерения перед введением лекарственного средства, используемого при усугублении симптомов, и до 1 дня после последней дозы в виде инъекции исследуемого продукта в ходе двойного слепого исследования во время или перед 12 посещением (неделя 24), или 169 днем, если данные 12 посещения (недели 24) были недоступны. Включены пациенты с измерениями и в исходном состоянии, и в неделю 24 (LOCF).
Среднее изменение массы тела (кг) от исходного уровня до недели 24 - mITT-популяция
аМодель ковариационного анализа (ANCOVA) с группами лечения (ликсисенатид и плацебо), рандомизационными стратами по уровню HbA1c (<8,0, ≥8,0%) при скрининге, по применению метформина при скрининге (Да, Нет) и стране в качестве фиксированных воздействий и исходной массы тела в качестве ковариата.
Примечание: Анализ включает измерения перед введением лекарственного средства, используемого при усугублении симптомов, и до 3 дня после последней дозы в виде инъекции исследуемого продукта в ходе двойного слепого исследования во время или перед 12 посещением (неделя 24), или 169 днем, если данные 12 посещения (недели 24) были недоступны. Включены пациенты с измерениями и в исходном состоянии, и в неделю 24 (LOCF).
Число (%) пациентов с потерей массы тела ≥5% от исходного уровня до недели 24 - mITT-популяция
Среднее изменение HOMA-бета от исходного уровня до недели 24 - mITT-популяция
аМодель ковариационного анализа (ANCOVA) с группами лечения (ликсисенатид и плацебо), рандомизационными стратами по уровню HbA1c (<8,0, ≥8,0%) при скрининге, по применению метформина при скрининге (Да, Нет) и стране в качестве фиксированных воздействий и исходной величине HOMA-β в качестве ковариата.
Примечание: Анализ включает измерения перед введением лекарственного средства, используемого при усугублении симптомов, и до 1 дня после последней дозы в виде инъекции исследуемого продукта в ходе двойного слепого исследования во время или перед 12 посещением (неделя 24), или 169 днем, если данные 12 посещения (недели 24) были недоступны. Включены пациенты с измерениями и в исходном состоянии, и в неделю 24 (LOCF).
Число (%) пациентов, требующих экстренной терапии, в течение основного 24-недельного периода лечения, исследуемого двойным слепым методом - mITT-популяция
Среднее изменение уровня инсулина в плазме натощак (пмоль/л) от исходного уровня до недели 24 - mITT-популяция
аМодель ковариационного анализа (ANCOVA) с группами лечения (ликсисенатид и плацебо), рандомизационными стратами по уровню HbA1c (<8,0, ≥8,0%) при скрининге, рандомизационными стратами по применению метформина при скрининге (Да, Нет) и стране в качестве фиксированных воздействий и исходной величине уровня инсулина в плазме натощак в качестве ковариата.
Анализ включает измерения перед введением лекарственного средства, используемого при усугублении симптомов, и до 1 дня после последней дозы в виде инъекции исследуемого продукта в ходе двойного слепого исследования во время или перед 12 посещением (неделя 24), или 169 днем, если данные 12 посещения (недели 24) были недоступны. Включены пациенты с измерениями и в исходном состоянии, и в неделю 24 (LOCF).
6.3. БЕЗОПАСНОСТЬ
Обзор побочных явлений, наблюдавшихся в течение периода проведения лечения для всего исследования представлен в таблице 17. Доля пациентов, которые испытали возникновение при лечении побочных явлений (TEAE), составила 87,9% в группе ликсисенатида и 83,2% в группе плацебо. В группе ликсисенатида не было случаев смерти пациентов, тогда как в группе плацебо умерли 2 пациента. У одного был возникший при лечении острый инфаркт миокарда, приведший к смерти, а другой умер вследствие AE после лечения (конечной стадии астении) после дыхательной недостаточности с мультиорганной недостаточностью. Процентная доля пациентов, у которых были тяжелые TEAE, была ниже в группе ликсисенатида (7,4%), чем в группе плацебо (9,3%). Бόльшая процентная доля пациентов из группы ликсисенатида (9,3%) испытала TEAE, приведшие к прекращению лечения, чем в группе плацебо (7,5%). В таблице 18, таблице 19 и таблице 20 суммированы TEAE, приведшие к смерти, тяжелым TEAE и TEAE, приведшим к прекращению лечения первичным SOC, HLGT, HLT и PT, соответственно. В группе ликсисенатида самым частым TEAE, ведущим к прекращению лечения, была тошнота (6 пациентов [1,9%]), тогда как в группе плацебо ни один пациент не прекратил лечение вследствие тошноты.
В таблице 30 в приложении представлены величины частоты встречаемости TEAE в течение периода проведения лечения во всем исследовании, возникающих по меньшей мере у 1% пациентов в любой группе лечения. Тошнота представляла собой наиболее часто отмечаемое TEAE в группе ликсисенатида (84 пациента [26,0%] в сравнении с 22 [13,7%] для плацебо). Вторым по частоте отмечаемым TEAE в группе ликсисенатида был назофарингит (53 [16.4%] в сравнении с 24 [14,9%] для плацебо), за которым следовала головная боль (43 [13,3%] в сравнении с 19 [11,8%] для плацебо), инфекция верхних дыхательных путей (41 [12,7%] в сравнении с 18 [11,2%] для плацебо), диарея (35 [10,8%] в сравнении с 23 [14,3%] для плацебо) и головокружение (33 [10,2%] в сравнении с 13 [8,1%] для плацебо).
Обзор профиля побочных явлений: возникшие в связи с лечением побочные явления в течение периода проведения лечения всего исследования - Популяция исследования безопасности
n (%) = число и процентная доля пациентов по меньшей мере с одним побочным явлением.
Примечание: период проведения лечения всего исследования = время от введения первой дозы медикаментозной терапии исследования двойным слепым методом до 3 дней после введения последней дозы
Число (%) пациентов, испытавших TEAE, приведшие к смерти, первичными SOC, HLGT, HLT и PT в течение периода проведения лечения всего исследования - Популяция исследования безопасности
Программа MedDRA версия 14.0.
n(%)= число и процентная доля пациентов по меньшей мере с одним TEAE, ведущим к смерти.
Примечание: период проведения лечения всего исследования = время от введения первой дозы медикаментозной терапии исследования двойным слепым методом до 3 дней после введения последней дозы.
Таблица с сортировкой по международно согласованному SOC порядку и алфавитному порядку HLGT, HLT, PT.
Число (%) пациентов, испытавших тяжелые TEAE, проявившиеся первичными SOC, HLGT, HLT и PT в течение периода проведения лечения всего исследования - Популяция исследования безопасности
Программа MedDRA версия 14.0.
n(%)= число и процентная доля пациентов по меньшей мере с одним тяжелым TEAE.
Примечание: период проведения лечения всего исследования = время от введения первой дозы медикаментозной терапии исследования двойным слепым методом до 3 дней после введения последней дозы
Таблица с сортировкой по международно согласованному SOC порядку и алфавитному порядку HLGT, HLT, PT.
Число (%) пациентов, испытавших TEAE, приведшие к постоянному прекращению лечения по первичным SOC, HLGT, HLT и PT в течение периода проводимого лечения всего исследования - Популяция исследования безопасности
Программа MedDRA версия 14.0.
n(%)= число и процентная доля пациентов по меньшей мере с одним тяжелым TEAE, приводящие к постоянному прекращению лечения.
Примечание: период проведения лечения всего исследования = время от введения первой дозы медикаментозной терапии исследования двойным слепым методом до 3 дней после введения последней дозы.
Таблица с сортировкой по международно согласованному SOC порядку и алфавитному порядку HLGT, HLT, PT.
В течение периода проведения лечения всего исследования в общей сложности 36 пациентов (27 в группе ликсисенатида и 9 в группе плацебо) отметили TEAE в предварительно уточненной форме регистрации AE (побочных явлений) для «клинически выраженной гипогликемии» Среди них, у 23 (7,1%) пациентов в группе ликсисенатида имелась клинически выраженная гипогликемия по определению в протоколе, по сравнению с 7 (4,3%) в группе плацебо (Таблица 21). Ни одно из явлений клинически выраженной гипогликемии не было тяжелым по интенсивности. Явления у остальных 6 пациентов (4 в группе ликсисенатида и 2 в группе плацебо) не соответствовали уточненному в протоколе определению клинически выраженной гипогликемии вследствие ассоциированных величин глюкозы, составляющих ≥60 мг/дл, или отсутствия зарегистрированных симптомов.
У 22 (6,8%) пациентов в группе ликсисенатида и 8 (5,0%) в группе плацебо наблюдались AE в виде реакции в участке инъекции (Таблица 22). AE в виде реакции в участке инъекции идентифицировали поиском термина «участок инъекции» или в PT, кодированных из сообщенных исследователем условий, или в PT из диагноза ARAC после экспертного заключения об аллергической реакции. Ни одно из этих явлений реакции в участке инъекции не было серьезным или тяжелым по интенсивности и не привело к прекращению введения IP. Исследователи сообщили в общей сложности о 56 возможных аллергических явлениях у 39 пациентов, и эти данные были направлены в ARAC для экспертного заключения во время периода лечения всего исследования. Из них, по 19 явлениям у 12 пациентов (9 [2,8%] пациентов в группе ликсисенатида и 3 [1,9%] в группе плацебо) ARAC было сделано заключение как об аллергических реакциях, которые включали 5 явлений у 3 пациентов в группе ликсисенатида (1 с аллергическим дерматитом, 1 с сыпью и 1 с ангиоотеком, анафилактической реакцией и аллергическим конъюнктивитом), которые по заключению экспертной комиссии возможно связаны с IP (Таблица 23).
• Пациент 124713001 (группа ликсисенатида): с сыпью и множественными аллергиями, а также зудом в анамнезе в 258 день (07 ноября 2009 г) после начала IP испытал нетяжелое TEAE в виде ВОЛДЫРЯ ОТ ИГЛЫ (закодированного в PT как «сыпь в участке инъекции») легкой интенсивности. Корригирующего лечение не назначали, и побочное явление разрешилось через 7 дней. Это явление не считали связанным с IP. Со дня 264 до дня 368 после начала приема IP пациенту пришлось периодически прекращать введение IP на один день. Со дня 369 до дня 386 пациент не принимал IP. Со дня 387 до дня 393 пациент снова проводил инъекцию 20 мкг IP каждый день. Затем, введение IP снова прекращали на 2 дня и инъецировали в последний раз в день 396. Введение IP полностью прекращали вследствие нетяжелого TEAE легкой интенсивности ВНОВЬ ПОЯВЛЯЮЩИЕСЯ ВОЛДЫРИ ПОСЛЕ ВОЗОБНОВЛЕНИЯ ИССЛЕДУЕМОЙ МЕДИКАМЕНТОЗНОЙ ТЕРАПИИ (закодированного в PT «сыпь») в день 396 после начала введения IP. Это явление рассматривали как связанное с IP. Это явление разрешилось через 11 после введения IP без какого-либо корригирующего лечения. Информацию об обоих явлениях послали в ARAC для заключения экспертной комиссии, но только по второму явлению ARAC вынесла заключение как о сыпи в результате аллергической реакции (крапивнице), возможно, связанной с IP.
• Пациент 642701006 (группа ликсисенатида): Этот пациент без аллергии в анамнезе испытал в день 163 (01 октября 2009 г) после начала введения IP нетяжелое TEAE в виде АЛЛЕРГИИ (закодированной в PT как «гиперчувствительность») легкой интенсивности. Через 30 минут после инъекции IP пациент предъявлял жалобы на общий зуд и красноту глаз, которые самопроизвольно исчезли без лечения. Это явление разрешилось в тот же день. В день 169 после начала IP было отмечено нетяжелое TEAE в виде АЛЛЕРГИЧЕСКОГО ДЕРМАТИТА (закодированное в PT как «дерматит аллергический») умеренной интенсивности. Через 25 минут после введения IP пациент предъявил жалобы на общий зуд, отек глаз и языка и отек в участке инъекции. Это явление разрешилось в тот же день. Пероральное введение лоратадина начали в день 169 после начала IP и проводили в качестве корригирующего лечения в течение 7 дней. В день 170 после начала введения IP было отмечено другое нетяжелое TEAE в виде АЛЛЕРГИЧЕСКОГО ДЕРМАТИТА (закодированное в PT как «дерматит аллергический») умеренной интенсивности, и оно разрешилось в тот же день. Непосредственно после введения IP пациент предъявил жалобы на отек в участке инъекции, общий зуд, отек глаз и языка, тошноту. Все 3 явления расценивали как связанные с IP, и введение IP полностью прекращали вследствие третьего явления после дня 170. Эти 3 явления в соответствии с заключением ARAC расценивали как аллергические реакции (аллергический конъюнктивит, ангиоотек и анафилактическая реакция, соответственно), возможно связанные с IP.
• Пациент 840864001 (группа ликсисенатида): с аллергическим ринитом, пыльцевыми аллергиями, аллергиями, вызванными пылью, отеками (ангиоотеком), лекарственными аллергиями, сыпью и дерматитом в анамнезе испытал нетяжелое TEAE в виде ДВУСТОРОННЕГО ДЕРМАТИТА ПРЕДПЛЕЧИЙ И ЖИВОТА (закодированное в PT как «дерматит») умеренной интенсивности на третий день после начала введения IP. Клиндамицин вводили в качестве корригирующего лечения в течение трех дней. Введение IP временно прекращали в дни с 5 по 8 день после начала введения IP. Проба с повторным введением IP в дни с 9 по 13 вызвала усугубление проявлений на брюшной стенке. Явление считали связанным с IP, и введение IP полностью прекратили после 13 дня. Явление разрешилось через 12 дней (21 декабря 2009 г) после полного прекращения введения IP. Это TEAE по заключению ARAC представляло собой аллергическую реакцию (аллергический дерматит), возможно связанную с IP.
По заключению ARAC, в группе плацебо не было явлений, возможно связанных с IP.
В соответствии с протоколом, любое увеличение уровня амилазы и/или липазы более чем вдвое выше верхнего предела нормального диапазона (ULN), которое было подтверждено повторным измерением, должно было подвергаться мониторингу и документации на бланке AE заранее установленного образца в виде «подозреваемого панкреатита». В течение периода проведения лечения всего исследования 2 (0,6%) пациента в группе ликсисенатида и 2 (1,2%) в группе плацебо отметили TEAE на бланке AE заранее установленного образца (Таблица 24). Случаи панкреатита не были диагностированы или зарегистрированы.
Пациенты, у которых по меньшей мере одна величина липазы или амилазы составила ≥3 ULN (верхней границы нормы) в течение периода получения лечения, суммированы в (Таблице 25). В общей сложности у 7 пациентов отмечено повышение уровня липазы (≥3 ULN): 5 [1,6%] в группе ликсисенатида, 2 [1,3%] в группе плацебо. Ни у одного не было повышения амилазы ≥3 ULN.
В соответствии с протоколом, любую величину кальцитонина ≥20 пг/мл, подтвержденную повторным измерением, необходимо было подвергать мониторингу и документации на бланке AE заранее установленного образца в виде «повышенного уровня кальцитонина ≥20 пг/мл». В течение периода проведения лечения всего исследования у 9 (2,8%) пациентов в группе ликсисенатида и 4 (2,5%) в группе плацебо зарегистрировано увеличение содержания кальцитонина в крови на бланке AE заранее установленного образца (Таблица 26). Среди них у 8 из 9 пациентов в группе ликсисенатида величины кальцитонина были ≥20, но <50 нг/л, и у 1 пациента величина кальцитонина была ≥50 нг/л, тогда как в группе плацебо у 3 из 4 пациентов величины кальцитонина были ≥20, но <50 нг/л, и у 1 пациента величины кальцитонина были ≥50 нг/л. У одного дополнительного пациента в группе ликсисенатида зарегистрировано AE после лечения на бланке заранее установленного образца в виде «повышенного кальцитонина ≥20 пг/мл» при величинах кальцитонина ≥20, но <50 нг/л. У двоих пациентов в группе ликсисенатида и 1 пациента в группе плацебо зарегистрированы AE, которые закодированы в PT как «новообразование щитовидной железы».
• Пациент 642706001 (группа ликсисенатида): не курящий, без заболеваний щитовидной железы и без почечной недостаточности в анамнезе отметил нетяжелое побочное явление в виде УЗЛА ЛЕВОЙ ДОЛИ ЩИТОВИДНОЙ ЖЕЛЕЗЫ легкой интенсивности через 24 дня после последнего введения IP. Медикаментозную терапию по поводу поражения щитовидной железы не назначали. Это явление расценивали как не связанное с IP. В посещение 15, через 255 дней после начала введения IP, впервые измерили содержание кальцитонина, и оно составило 16,4 нг/л. В последний день введения IP уровень кальцитонина составил 22,2 нг/л, а при повторном тестировании через 1 неделю - 18,9 нг/л.
• Пациент 840738004 (группа ликсисенатида): ранее куривший пациент в возрасте 35 лет, без заболеваний щитовидной железы и без почечной недостаточности в анамнезе отметил нетяжелое TEAE в виде УЗЛА ЛЕВОЙ ДОЛИ ЩИТОВИДНОЙ ЖЕЛЕЗЫ размером 6 мм легкой интенсивности через 39 дней после первого приема IP. Медикаментозную терапию по поводу поражения щитовидной железы не назначали. Ультразвуковое исследование щитовидной железы через 177 дней после первого приема IP подтвердило наличие узла размером 7×3 мм в левой доле. Первая величина уровня кальцитонина в начале приема IP составила 19,7 нг/л. Через 262 дня после первого приема IP на определенных страницах для регистрации повышенного уровня кальцитонина было зарегистрировано нетяжелое TEAE в виде ПОВЫШЕННОГО УРОВНЯ КАЛЬЦИТОНИНА легкой интенсивности вследствие измеренной величины кальцитонина 20,1 нг/л. Это явление в виде ПОВЫШЕННОГО УРОВНЯ КАЛЬЦИТОНИНА разрешилось без лечения через 376 дней после первого приема IP. За 9 дней до этого, уровень кальцитонина составил 16,3 нг/л. В последний день приема IP уровень кальцитонина составил 19,4 нг/л. Оба явления рассматривали как не связанные с IP.
Пациенты с по меньшей мере одним измерением кальцитонина в сыворотке во время периода проведения лечения всего исследования суммированы в таблице 27 в соответствии с 4 заранее определенными категориями уровня кальцитонина в исходном состоянии. В общей сложности у 17 пациентов величины кальцитонина составили ≥20 нг/л в течение периода проведения лечения всего исследования: 11 (3,7%) пациентов в группе ликсисенатида, 6 (4,2%) пациентов в группе плацебо. Среди них, 13 пациентов (9 из группы ликсисенатида и 4 из группы плацебо) зарегистрировали TEAE в заранее утвержденном бланке регистрации AE, как описано выше. У двух пациентов в каждой группе лечения величина уровня кальцитонина, по меньшей мере у одного, составляла ≥20 нг/л, но они не сообщали о TEAE в заранее утвержденном бланке регистрации AE в течение периода проведения лечения всего исследования. У одного пациента, у которого имелись множественные величины ≥20, но <50 нг/л в группе плацебо, это было связано с измерениями, проведенными перед поправкой 4 протокола, которая требовала повторного тестирования. У других 3 пациентов это было вызвано неподтвержденным повышением уровня кальцитонина: у 1 пациента в каждой группе одна величина составила ≥20, но <50 нг/л, и у 1 в группе ликсисенатида ≥50 нг/л, но при других предшествующих и последующих измерениях эти величины составили <20 нг/л. Поскольку измерения уровня кальцитонина осуществляли при поправке в протоколе, после того как большинство пациентов были уже рандомизированы в настоящем исследовании. Поэтому для большинства пациентов исходные величины уровня кальцитонина были недоступны.
У одного пациента в группе плацебо и 2 пациентов в группе ликсисенатида величина уровня кальцитонина составила >50 нг/л (Таблица 27).
• Пациент 840782004 (группа ликсисенатида): некурящий, без заболеваний щитовидной железы в анамнезе и без почечной недостаточности в день первого введения IP (03 августа 2009 г) уровень кальцитонина составил 37,8 нг/л, и днем позже было отмечено нетяжелое TEAE в виде ПОВЫШЕНИЯ УРОВНЯ КАЛЬЦИТОНИНА легкой интенсивности. Введение IP продолжили. Корригирующее лечение не проводили. Ультразвуковое исследование щитовидной железы не выполняли. Это явление рассматривали как связанное с IP. Дальнейшие величины уровня кальцитонина во время исследования составили 64,2, 19,3, 50, 36,5, и в 260 день (19 апреля 2010) после начала введения IP она составила 29,6 нг/л. Через 43 дня (24 августа 2010 г) после полного прекращения введения IP (вследствие отсутствия эффективности) уровень кальцитонина составил 48,1 нг/л.
• У пациента 040702004 (группа ликсисенатида): в одно посещение в течение исследования уровень кальцитонина был 104 нг/л. При повторном тестировании через 14 дней уровень кальцитонина составил 3 нг/л. Поскольку при всех более ранних и более поздних посещениях в течение исследования величины содержания кальцитонина составляли от <0,6 нг/л до 3 нг/л, то не было зарегистрировано ни одно TEAE, относящееся к кальцитонину, и дополнительное исследование щитовидной железы не проводили.
Резюме клинически выраженной гипогликемии в течение периода проведения лечения всего исследования - Популяция исследования безопасности
Клинически выраженная гипогликемия = Клинически выраженная гипогликемия, как определено в соответствии с протоколом.
Примечание: период получения лечения всего исследования = время от первой дозы медикаментозной терапии двойного слепого исследования до 3 дней после введения последней дозы.
Число (%) пациентов, имевших реакции в участке инъекции, в течение периода получения лечения всего исследования - Популяция исследования безопасности
Предпочтительное условие
Примечание: период получения лечения всего исследования = время от первой дозы медикаментозной терапии двойного слепого исследования до 3 дней после введения последней дозы.
Число (%) пациентов с явлениями, оцененными ARAC как аллергическая реакция во время периода проведения лечения всего исследования - Популяция исследования безопасности
Примечание: период получения лечения всего исследования = время от первой дозы медикаментозной терапии двойного слепого исследования до 3 дней после введения последней дозы.
Число (%) пациентов со специфической формой побочного явления с подозрением на панкреатит, выявленных в течение периода проведения лечения всего исследования - Популяция исследования безопасности
Примечание: период получения лечения всего исследования = время от первой дозы медикаментозной терапии двойного слепого исследования до 3 дней после введения последней дозы.
Панкреатические ферменты: Число (%) пациентов с патологией (PCSA) в течение периода проведения лечения всего исследования в соответствии с исходным статусом PCSA - Популяция исследования безопасности
Исходный уровень по критериям PCSA n/N1 (%)
*Независимо от исходного уровня.
Примечание: период получения лечения всего исследования = время от первой дозы медикаментозной терапии двойного слепого исследования до 3 дней после введения последней дозы.
Число (n) представляет подгруппу общего числа пациентов, которые по меньшей мере однократно соответствуют рассматриваемым критериям.
Числитель (/N1) для каждого параметра в пределах группы лечения представляет собой число пациентов для группы лечения, у которых этот параметр оценивался после исходного уровня по исходному статусу PCSA. Исходным статусом представлено только усугубление каждого случая для каждого пациента.
Число (%) пациентов с повышенным уровнем кальцитонина во время периода проведения лечения всего исследования - Популяция исследования безопасности
Примечание: период получения лечения всего исследования = время от первой дозы медикаментозной терапии двойного слепого исследования до 3 дней после введения последней дозы.
Уровень кальцитонина в сыворотке: Число (%) пациентов по предварительно заданным категориям в течение периода проведения лечения всего исследования в соответствии с исходной категорией - Популяция исследования безопасности
Исходный статус
После исходного статуса
*Независимо от исходного уровня
Примечание: период получения лечения всего исследования = время от первой дозы медикаментозной терапии двойного слепого исследования до 3 дней после введения последней дозы.
Числитель представляет число пациентов, которые входили в предварительно заданные категории при исследовании после исходного состояния в каждой исходной категории. Знаменатель для каждого параметра в пределах группы лечения представляет собой число пациентов для группы лечения, у которого этот параметр оценивали после исходного уровня по исходному статусу.
Пациента считают только в наихудшей категории.
7. ПРИЛОЖЕНИЕ
Число (%) пациентов по общей суточной дозе в конце титрования - Популяция исследования безопасности
Планируемое посещение на конец периода титрования дозы в соответствии с протоколом должно быть посещением 5/недели 2.
Примечание: Процентные доли рассчитаны с использованием числа пациентов в группе исследования безопасности в качестве деноминатора.
Среднее изменение HbA1c (%) от исходного уровня по посещениям - mITT
Точка времени
LOCF = метод переноса вперед данных последнего проведенного наблюдения.
Примечание: Анализ исключает измерения, полученные после применения экстренной медикаментозной терапии и/или после прекращения лечения плюс 3 дня.
Для недели 24 (LOCF), анализ включает измерения, полученные до 3 дней после введения последней дозы в виде инъекции исследуемого продукта в ходе двойного слепого исследования во время или перед 12 посещением (неделя 24), или 169 днем, если данные 12 посещения (недели 24) недоступны.
Мин - минимальное, Макс - максимальное
Число (%) пациентов, испытавших обычные TEAE (PT ≥ 1% в любой группе лечения), представленное первичным SOC, HLGT, HLT и PT, в течение периода проведения лечения всего исследования - Популяция исследования безопасности
Программа MedDRA версия 14.0.
n(%)= число и процентная доля пациентов по меньшей мере с одним TEAE.
Примечание: период проведения лечения всего исследования = время от введения первой дозы медикаментозной терапии исследования двойным слепым методом до 3 дней после введения последней дозы
Таблица с сортировкой по международно согласованному SOC порядку и алфавитному порядку HLGT, HLT, PT.
Представлены только SOC с по меньшей мере одним PT ≥1% в по меньшей мере одной группе.


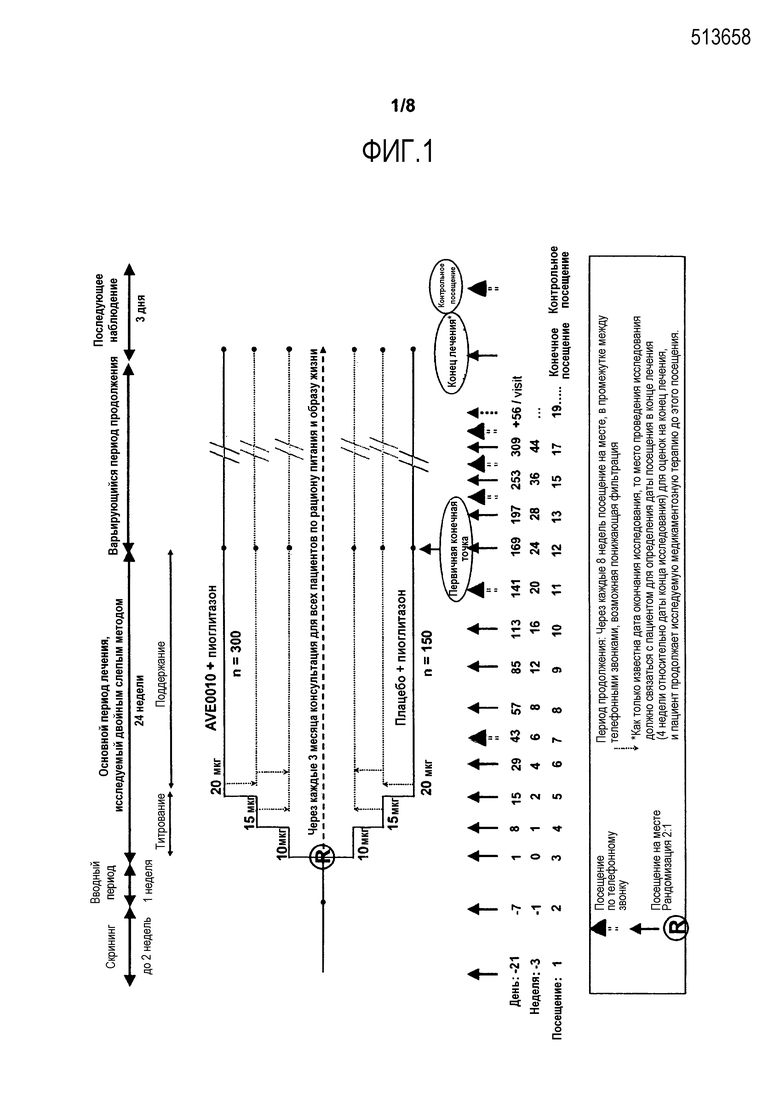
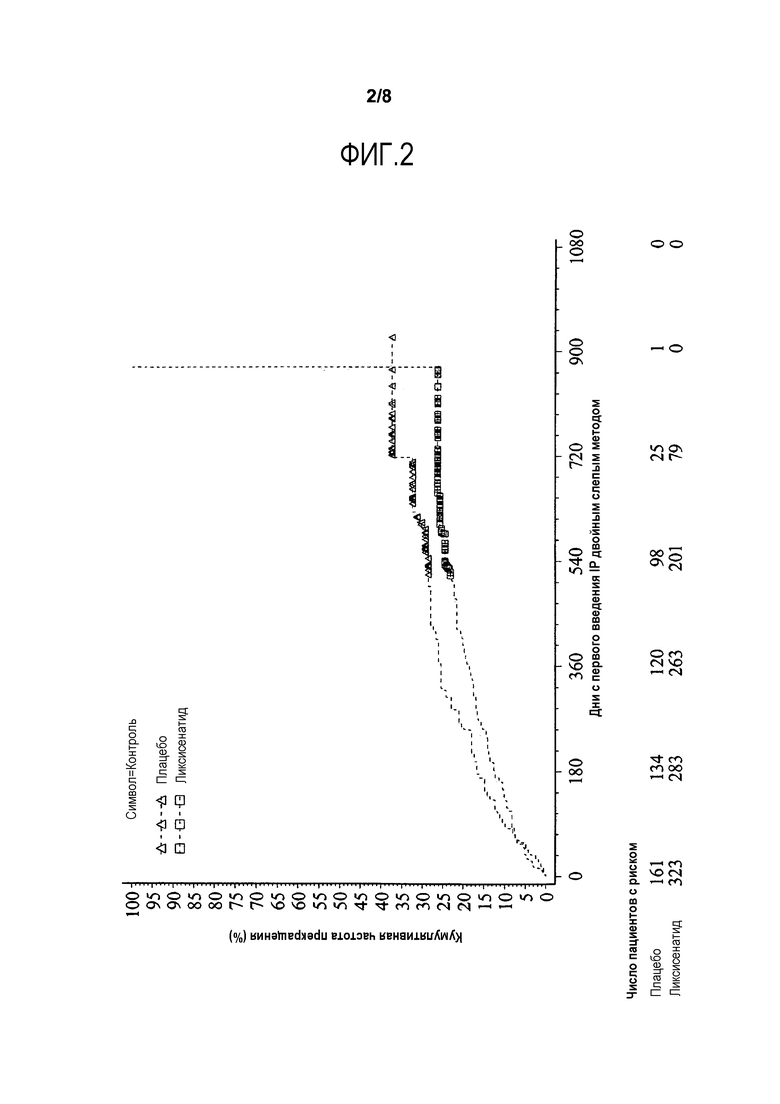
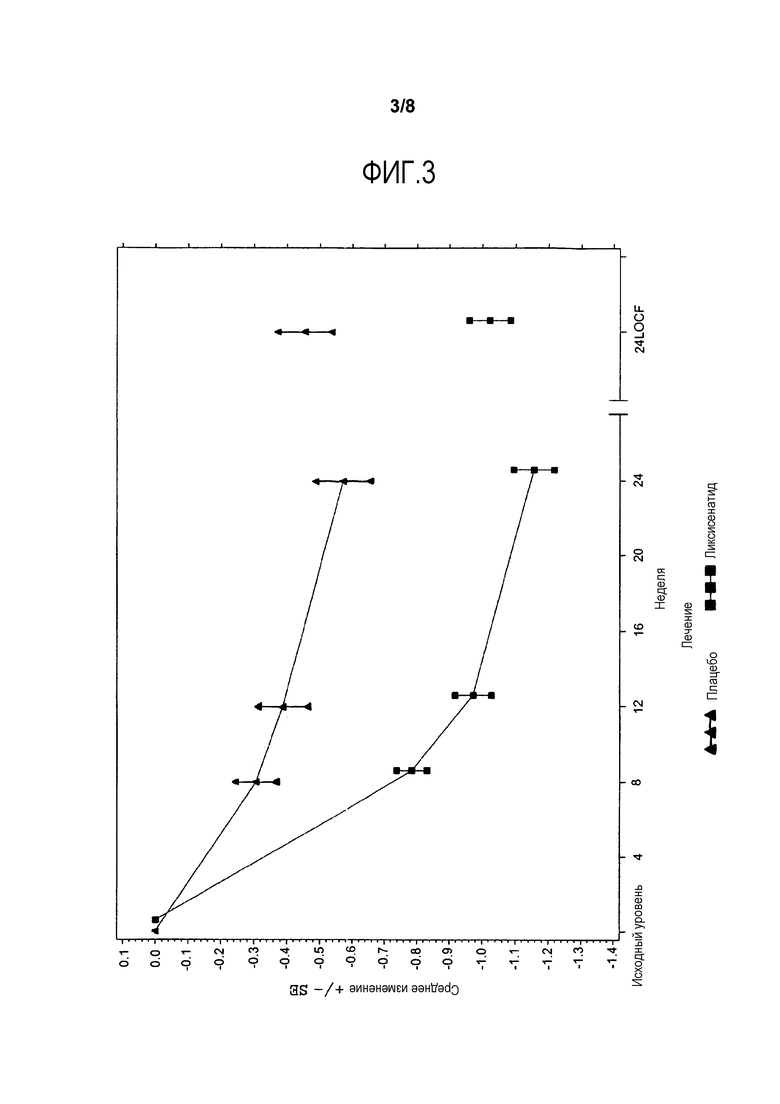
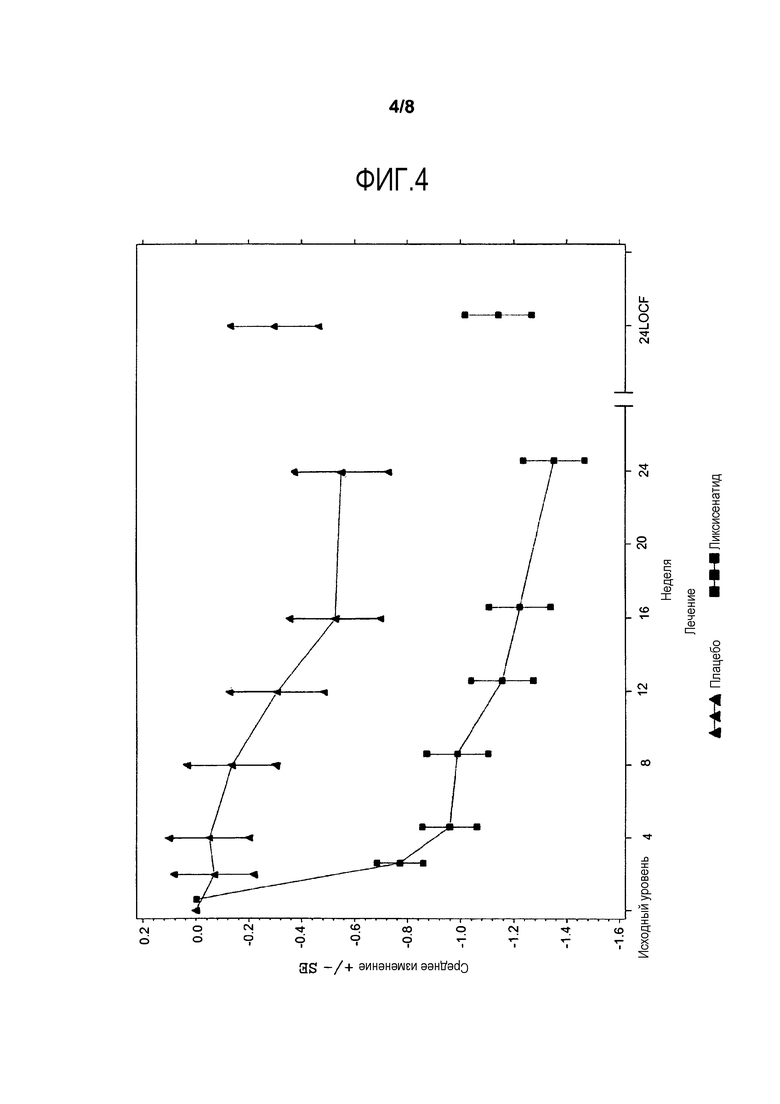
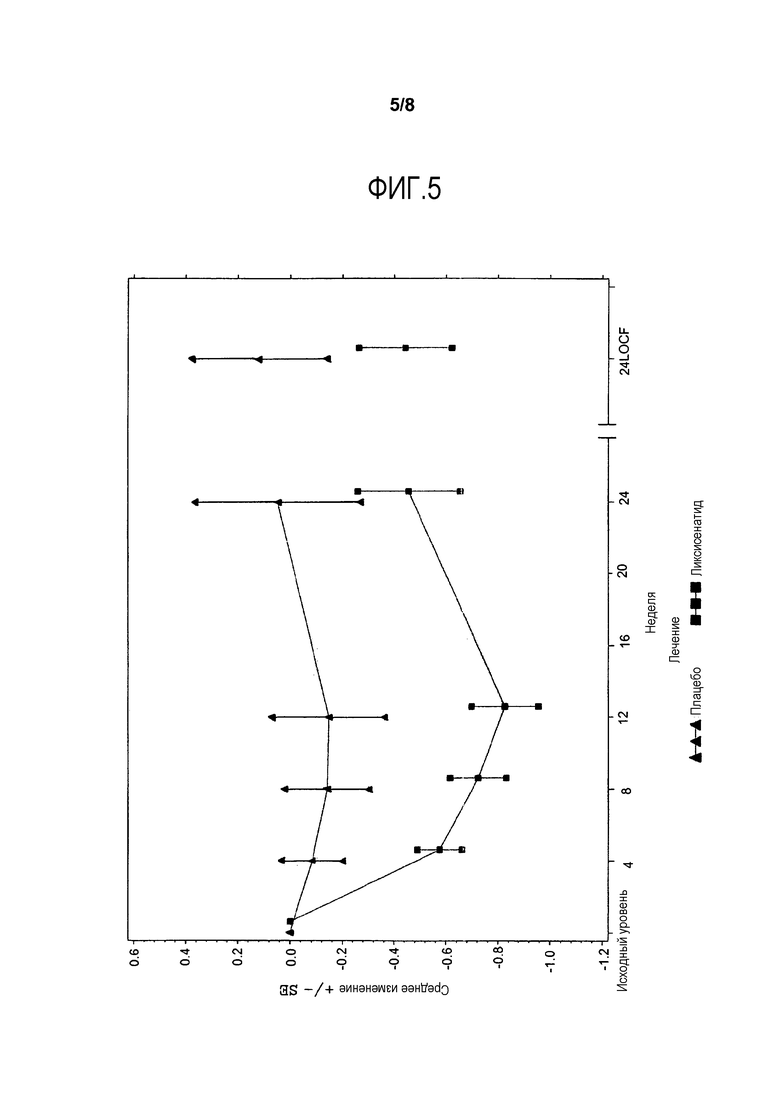
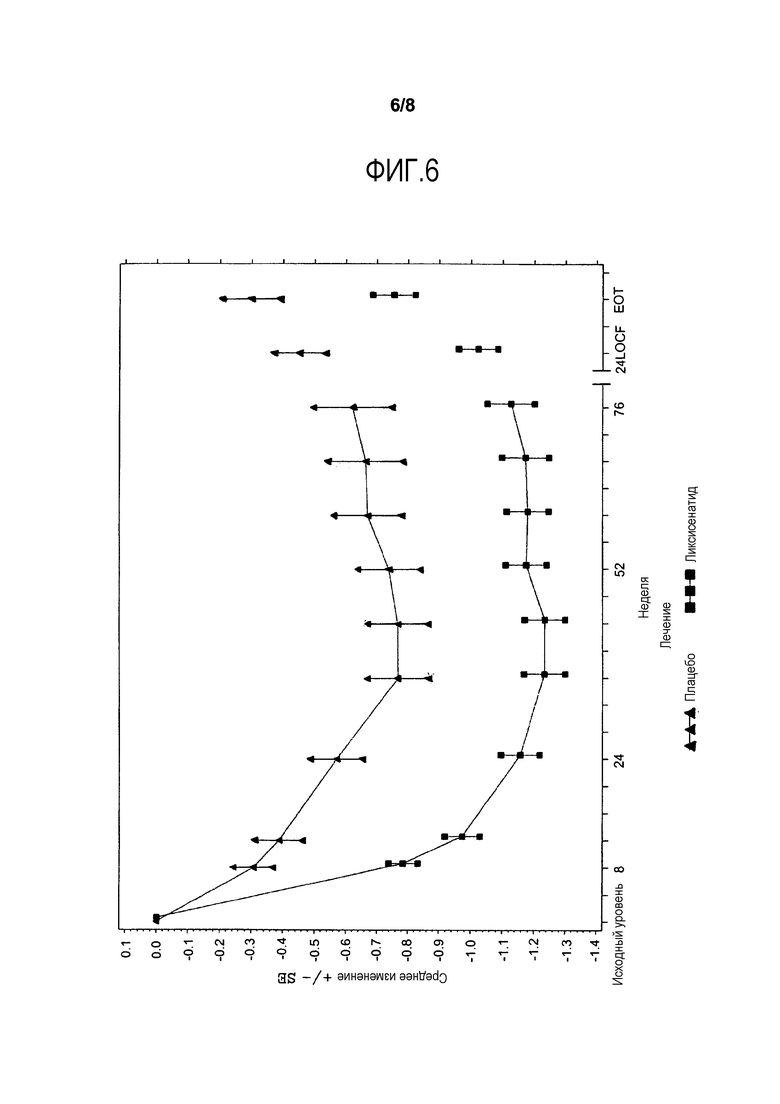
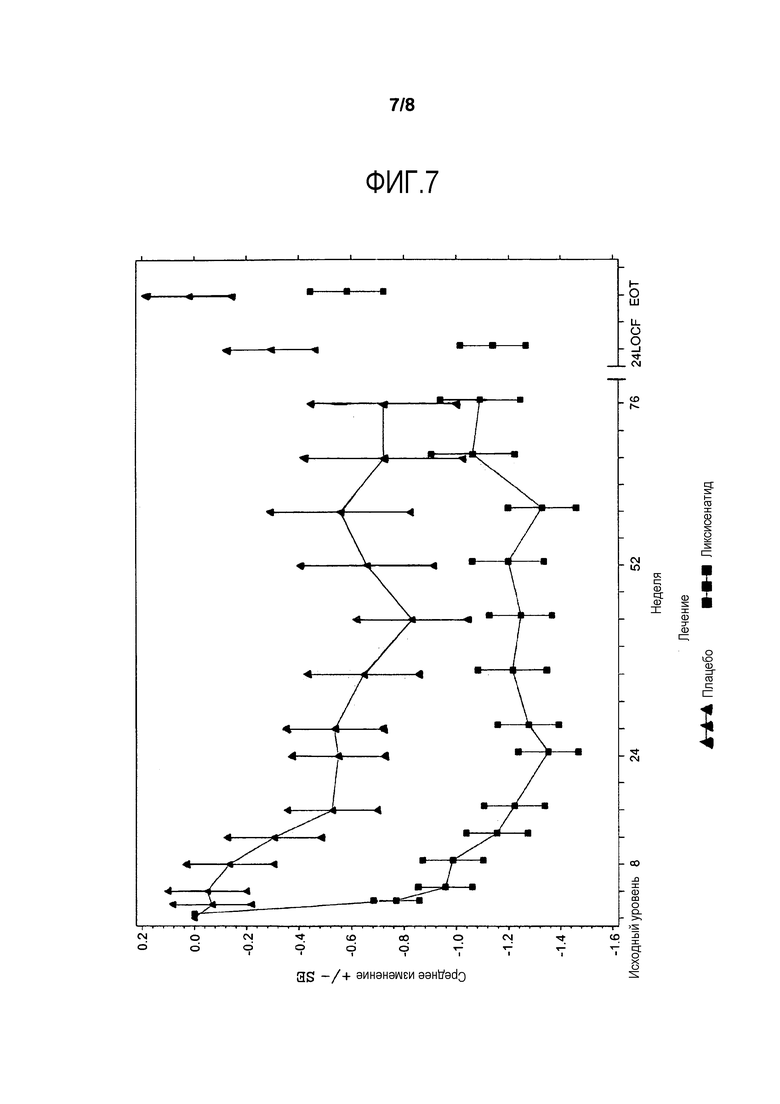
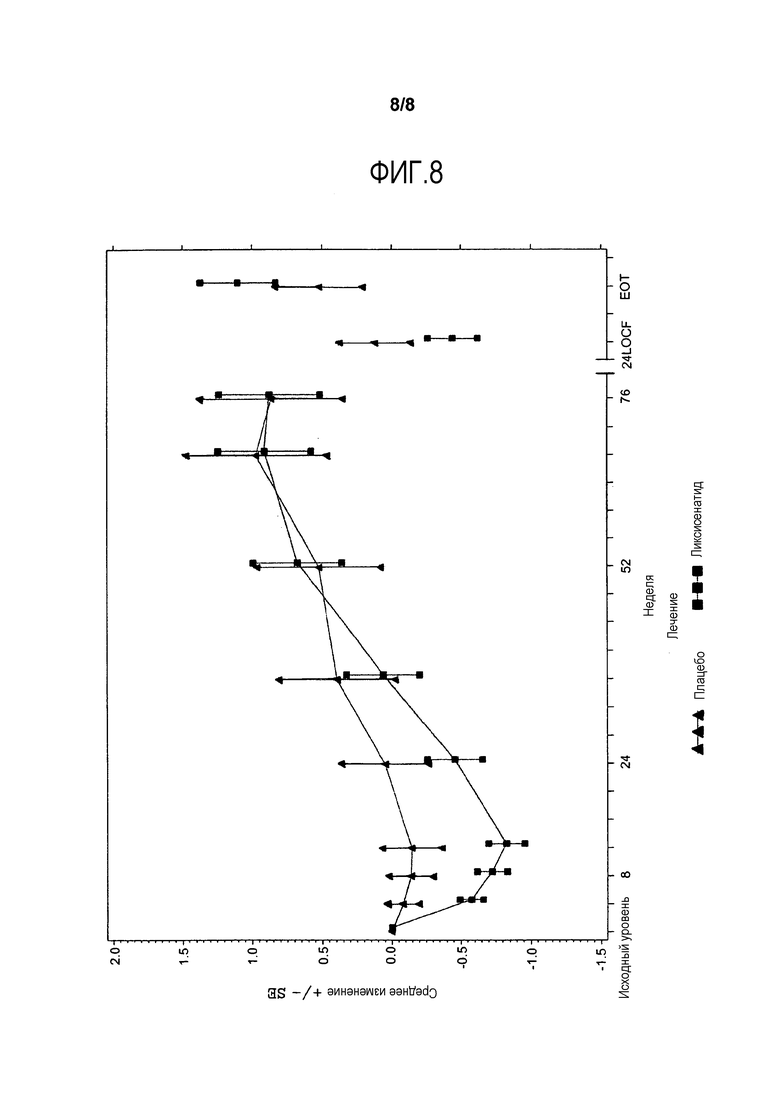
Изобретение относится к медицине и касается способа улучшения гликемического контроля у пациентов с сахарным диабетом 2 типа, у которых диабет 2 типа неадекватно контролируется лечением одним глитазоном, включающего введение нуждающемуся в этом пациенту комбинации, содержащей (a) desPro36Exendin-4(1-39)-Lys6-NH2 и/или его фармацевтически приемлемую соль в суточной дозе 10-20 мкг и (b) глитазон и/или его фармацевтически приемлемую соль в суточной дозе 10-20 мг. Изобретение обеспечивает значимое улучшение гликемического контроля. 14 з.п. ф-лы, 1 пр., 8 ил., 30 табл.
1. Способ улучшения гликемического контроля у пациентов с сахарным диабетом 2 типа, у которых диабет 2 типа неадекватно контролируется лечением одним глитазоном, включающий введение нуждающемуся в этом пациенту комбинации, содержащей
(a) desPro36Exendin-4(1-39)-Lys6-NH2 и/или его фармацевтически приемлемую соль в суточной дозе 10-20 мкг и
(b) глитазон и/или его фармацевтически приемлемую соль в суточной дозе 10-20 мг.
2. Способ по п.1, где дополнительно вводят
(c) метформин и/или его фармацевтически приемлемую соль.
3. Способ по п.2, где метформин и/или его фармацевтически приемлемая соль получены для перорального введения.
4. Способ по п.1 или 2, где desPro36Exendin-4(1-39)-Lys6-NH2 и/или его фармацевтически приемлемая соль получены для парентерального введения.
5. Способ по п.1 или 2, где desPro36Exendin-4(1-39)-Lys6-NH2 и/или его фармацевтически приемлемая соль получены для введения в суточной дозе, выбранной из диапазона от 10 до 20 мкг.
6. Способ по п.1 или 2, где глитазон и/или его фармацевтически приемлемая соль получены для перорального введения.
7. Способ по п.1 или 2, где глитазон представляет собой пиоглитазон.
8. Способ по п.1 или 2, где пациент с сахарным диабетом 2 типа страдает ожирением.
9. Способ по п.8, где у пациента с сахарным диабетом 2 типа индекс массы тела составляет по меньшей мере 30 кг/м2.
10. Способ по п.9, где пациент с сахарным диабетом 2 типа представляет собой взрослого пациента.
11. Способ по п.1, где у пациента с сахарным диабетом 2 типа сахарный диабет 2 типа был диагностирован по меньшей мере за 1 год или по меньшей мере за 2 года до начала терапии.
12. Способ по п.11, где у пациента с сахарным диабетом 2 типа величина HbA1c составляет от примерно 7 до примерно 10%.
13. Способ по п.12, где у пациента с сахарным диабетом 2 типа концентрация глюкозы в плазме натощак составляет по меньшей мере 8 ммоль/л.
14. Способ по п.13, где у пациента с сахарным диабетом 2 типа концентрация глюкозы в плазме через 2 часа после приема пищи составляет по меньшей мере 10 ммоль/л, по меньшей мере 12 ммоль/л или по меньшей мере 14 ммоль/л.
15. Способ по п.14, где у пациента с сахарным диабетом 2 типа колебание уровня глюкозы составляет по меньшей мере 2 ммоль/л, по меньшей мере 3 ммоль/л, по меньшей мере 4 ммоль/л или по меньшей мере 5 ммоль/л, причем колебание уровня глюкозы представляет собой разность между концентрацией глюкозы в плазме через 2 часа после приема пищи и концентрацией глюкозы в плазме за 30 минут перед пробным приемом пищи.
| PANIKAR V., et al., Beneficial effects of triple drug combination of pioglitazone with glibenclamide and metformin in type 2 diabetes mellitus patients on insulin therapy.J Assoc Physicians India | |||
| Способ и приспособление для нагревания хлебопекарных камер | 1923 |
|
SU2003A1 |
| CHRISTENSEN M., et al., Lixisenatide for type 2 diabetes mellitus.Expert Opin Investig Drugs | |||
| Способ приготовления лака | 1924 |
|
SU2011A1 |
| Печь-кухня, могущая работать, как самостоятельно, так и в комбинации с разного рода нагревательными приборами | 1921 |
|
SU10A1 |
| Способ приготовления лака | 1924 |
|
SU2011A1 |
| Приспособление для суммирования отрезков прямых линий | 1923 |
|
SU2010A1 |

