Предметом настоящего изобретения является способ лечения сахарного диабета 2 типа AVE0010 (ликсисенатидом) в качестве дополнительной терапии к введению метформина.
Метформин представляет собой гипогликемический агент бигуанид, применяемый при лечении сахарного диабета 2 типа, не отвечающего на модификацию диеты. Метформин улучшает контроль уровня глюкозы в крови посредством улучшения чувствительности к инсулину. Метформин обычно вводят перорально. Однако, контроль сахарного диабета 2 типа у пациентов, страдающих ожирением, посредством метформина может быть недостаточным. Соответственно, у данных пациентов могут потребоваться дополнительные меры для контроля сахарного диабета 2 типа.
Гипогликемия является критическим ограничивающим фактором как при кратковременном, так и при длительном менеджменте гликемией при диабете. Несмотря на постоянные улучшения в менеджменте гликемией при диабете, основанные на популяции данные показывают, что гипогликемия продолжает являться главной проблемой для людей с диабетом как 1 типа, так и 2 типа (American diabetes association, workgroup on hypoglycemia: Defining and Reporting Hypoglycemia in Diabetes. Diabetes Care 28(5), 2005, 1245-1249).
Первым аспектом настоящего изобретения является способ лечения сахарного диабета 2 типа, включающий введение
(a) desPro36Эксендин-4(1-39)-LyS6-NH2 и/или его фармацевтически приемлемой соли, и
(b) метформина и/или его фармацевтически приемлемой соли,
нуждающемуся в этом пациенту.
В частности, способ представляет собой способ предотвращения гипогликемии у пациента с сахарным диабетом 2 типа. Более конкретно, способ представляет собой способ предотвращения симптоматической гипогликемии или тяжелой симптоматической гипогликемии у пациента с сахарным диабетом 2 типа.
Более конкретно, способ представленного изобретения является способом предотвращения гипогликемии у пациента с сахарным диабетом 2 типа, имеющего повышенный риск гипогликемии, в частности у пациента с диабетом 2 типа, имеющего по меньшей мере одно пережитое гипогликемическое событие. Гипогликемическим событием может быть симптоматическое гипогликемическое событие или тяжелое симптоматическое гипогликемическое событие.
В представленном изобретении, гипогликемия представляет собой состояние, при котором пациент с сахарным диабетом 2 типа имеет концентрацию глюкозы в плазме ниже 60 мг/дл (или ниже 3,3 ммоль/л), ниже 50 мг/дл, ниже 40 мг/дл или ниже 36 мг/дл.
С помощью способа представленного изобретения, гипогликемия может быть снижена ниже 12%, ниже 11%, ниже 10%, ниже 9%, ниже 8%, ниже 7%, ниже 6% или ниже 5% у пациентов с сахарным диабетом 2 типа, принимающих комбинацию ликсисенатида и метформина, как описано в данном документе.
В представленном изобретении, "симптоматическая гипогликемия" или "симптоматическое гипогликемическое событие" представляет собой состояние, связанное с клиническим симптомом, который обусловлен гипогликемией, при этом концентрация глюкозы в плазме находится ниже 60 мг/дл (или ниже 3,3 ммоль/л), ниже 50 мг/дл или ниже 40 мг/дл. Клиническими симптомами могут быть, например, потливость, учащенное сердцебиение, чувство голода, беспокойство, тревога, утомляемость, раздражительность, головная боль, потеря концентрации, сонливость, психические расстройства, нарушения зрения, транзиторные сенсорные нарушения, транзиторные двигательные нарушения, спутанность сознания, судороги и кома. В способе представленного изобретения могут быть выбраны один или более клинических симптомов симптоматической гипогликемии, которые указаны в данном документе. Симптоматическая гипогликемия может быть связана с быстрой нормализацией после перорального введения углеводов.
В представленном изобретении, "тяжелая симптоматическая гипогликемия" или "тяжелое симптоматическое гипогликемическое событие" представляет собой состояние с клиническим симптомом, который указан в данном документе, который обусловлен гипогликемией, при этом концентрация глюкозы в плазме находится ниже 36 мг/дл (или ниже 2,0 ммоль/л). Тяжелая симптоматическая гипогликемия может быть связана с острым неврологическим дефицитом, обусловленным гипогликемическим событием. При тяжелой симптоматической гипогликемии, пациенту может потребоваться помощь другого человека, если, например, пациент не может лечиться или помочь себе вследствие острого неврологического дефицита. Определение тяжелой симптоматической гипогликемии может включать все эпизоды, в которых неврологический дефицит является достаточно тяжелым, чтобы не опустить самолечение и в которых соответственно есть основания полагать, что пациенты имеют риск повреждения себя или других. Острым неврологическим дефицитом может быть по меньшей мере острый неврологический дефицит, выбранный из сонливости, психических расстройств, нарушений зрения, транзиторных сенсорных нарушений, транзиторных двигательных нарушений, спутанности сознания, судорог и комы.
Тяжелая симптоматическая гипогликемия может быть связана с быстрой нормализацией после перорального введения углеводов, внутривенного введения глюкозы и/или глюкагона.
Нормогликемия может относиться к концентрации в плазме крови глюкозы, составляющей от 60 мг/дл до 140 мг/дл (что соответствует от 3,3 ммоль/л до 7,8 ммоль/л).
Неожиданно в клиническом испытании было обнаружено, что в процессе лечения пациентов с сахарным диабетом 2 типа ликсисенатидом, в комбинации с метформином, только 5% пациентов имели симптоматические гипогликемические события, тогда как в сравнительном испытании, 14,6% пациентов с сахарным диабетом 2 типа, подвергавшихся лечению комбинацией эксенатида и метформина, сообщали о симптоматической гипогликемии в течение такого же периода. Данные результаты показывают, что комбинация ликсисенатида и метформина может быть использована для предотвращения гипогликемии.
Комбинация ликсисенатида и метформина, как описано в данном документе, также может быть использована для уменьшения и/или предотвращения побочных эффектов противодиабетического лечения пациентов с сахарным диабетом 2 типа.
В представленном изобретении, побочные эффекты комбинации ликсисенатида и метформина исследовали в клиническом испытании лечения пациентов с сахарным диабетом 2 типа ликсисенатидом, в комбинации с метформином (Пример 2). В данном испытании, побочные эффекты описаны посредством возникших после начала применения препарата нежелательных явлений (TEAE).
Побочный эффект может представлять собой состояние моторики желудочно-кишечного тракта и дефекации, например, диарею, неинфекционную диарею, атоническое и гипокинетическое нарушение желудочно-кишечного тракта, не классифицированное в других рубриках, запор, гастроэзофагеальную рефлюксную болезнь. Побочный эффект может также представлять собой желудочно-кишечный признак и симптом, например, диспептический признак и симптом, диспепсию, флатуленцию, метеоризм, увеличение, вздутие живота, желудочно-кишечную и абдоминальную боль (например, исключая боль в полости рта и горле), абдоминальную боль, боль в верхних отделах живота, абдоминальный дискомфорт, симптом тошноты и/или рвоты, тошноту или рвоту. В частности, побочный эффект представляет собой тошноту или рвоту. Более конкретно, побочный эффект представляет собой тошноту.
Неожиданно было обнаружено, что в клиническом испытании побочные эффекты, например, тошнота уменьшались (см., например, Таблицу 29 примера 2), по сравнению со сравнительным испытанием лечения пациентов с сахарным диабетом 2 типа комбинацией эксенатида и метформина.
Побочным эффектом также может быть панкреатит. В течение периода применения исследуемого препарата, 5 (1,6%) подвергавшихся лечению ликсисенатидом пациентов и 9 (2,8%) подвергавшихся лечению эксенатидом пациентов сообщали о случаях изменений панкреатических ферментов, или липазы или амилазы, при "подозрении на панкреатит" (Таблицы 23 и 24 примера 2). Однако случаев острого панкреатита не наблюдалось.
Побочным эффектом также может быть повышенная концентрация кальцитонина в крови. В клиническом испытании, восемь пациентов (4 [1,3%] в каждой группе) сообщали о значении кальцитонина > 20 нг/л (Таблица 25). О значении > 50 нг/л не сообщалось. Пять (1,8%) пациентов в группе ликсисенатида и 8 (3,0%) пациентов в группе эксенатида имели значение кальцитонина > 20 нг/л в течение периода применения исследуемого препарата (Таблица 26).
Данные результаты показывают, что комбинация ликсисенатида и метформина может быть использована для уменьшения и/или предотвращения побочных эффектов противодиабетического лечения пациентов с сахарным диабетом 2 типа. В частности, данные результаты показывают, что комбинация ликсисенатида и метформина может быть использована для уменьшения и/или предотвращения тошноты, панкреатита и/или повышенной концентрации кальцитонина в крови.
Соединения (a) и (b) могут вводиться нуждающемуся в этом пациенту, в количестве, достаточном для того, чтобы индуцировать терапевтический эффект.
Соединение desPro36Эксендин-4(1-39)-Lys6-NH2 (AVE0010, ликсисенатид) является производным эксендина-4. AVE0010 раскрыто, как SEQ ID NO:93 в WO 01/04156:
SEQ ID NO: 1 AVE0010 (44 AS)
H-G-E-G-T-F-T-S-D-L-S-K-Q-M-E-E-E-A-V-R-L-F-l-E-W-L-K-N-G-G-P-S-S-G-A-P-P-S-K-K-K-K-K-K-NH2
SEQ ID NO: 2 Эксендин-4 (39 AS)
H-G-E-G-T-F-T-S-D-L-S-K-Q-M-E-E-E-A-V-R-L-F-l-E-W-L-K-N-G-G-P-S-S-G-A-P-P-P-S-NH2
Эксендины представляют собой группу пептидов, которые могут понижать концентрацию глюкозы в крови. Аналог эксендина AVE0010 отличается отсечением С-конца последовательности исходного Эксендина-4. AVE0010 содержит шесть С-концевых лизиновых остатков, не присутствующих в Эксендине-4.
В контексте представленного изобретения, AVE0010 включает в себя его фармацевтически приемлемые соли. Квалифицированному специалисту в данной области известны фармацевтически приемлемые соли AVE0010. Предпочтительной фармацевтически приемлемой солью AVE0010, используемого в представленном изобретении, является ацетат.
AVE0010 (desPro36Эксендин-4(1-39)-Lys6-NH2) и/или его фармацевтически приемлемая соль могут вводиться посредством подкожной инъекции. Известны подходящие инъекционные устройства, например, так называемые "ручки", содержащие картридж, содержащий активный ингредиент, и инъекционную иглу. AVE0010 и/или его фармацевтически приемлемая соль могут вводиться в подходящем количестве, например, в количестве в диапазоне, составляющем от 10 до 15 мкг на дозу или от 15 до 20 мкг на дозу один раз в день (нарастание дозы от 10 до 15 и до 20 мкг/день. 20 мкг является эффективной поддерживающей дозой).
В представленном изобретении, AVE0010 и/или его фармацевтически приемлемая соль могут вводиться в ежедневной дозе в диапазоне, составляющем от 10 до 15 мкг или в диапазоне, составляющем от 15 до 20 мкг один раз в день (нарастание дозы от 10 до 15 и до 20 мкг/день. 20 мкг является эффективной поддерживающей дозой). AVE0010 и/или его фармацевтически приемлемая соль могут вводиться в виде одной инъекции в день.
В представленном изобретении, может быть использована жидкая композиция, содержащая desPro36Эксендин-4(1-39)-Lysg-NH2 и/или его фармацевтически приемлемую соль. Квалифицированному специалисту известны жидкие композиции AVE0010, подходящие для парентерального введения. Жидкая композиция представленного изобретения может иметь кислый или физиологический pH. Кислый pH предпочтительно находится в диапазоне pH 1-6,8, pH 3,5-6,8 или pH 3,5-5. Физиологический pH предпочтительно находится в диапазоне pH 2,5-8,5, pH 4,0-8,5 или pH 6,0-8,5. pH может быть отрегулирован с помощью фармацевтически приемлемой разбавленной кислоты (обычно HCL) или фармацевтически приемлемого разбавленного основания (обычно NaOH). Предпочтительный pH находится в диапазоне pH 3,5-5,0.
Жидкая композиция может содержать буфер, такой как фосфатный, цитратный, ацетатный. Предпочтительно, она может содержать ацетатный буфер, в количествах до 5 мкг/мл, до 4 мкг/мл или до 2 мкг/мл.
Жидкая композиция представленного изобретения может включать в себя подходящий консервант. Подходящий консервант может быть выбран из фенола, м-крезола, бензилового спирта и сложного эфира п-гидроксибензойной кислоты. Предпочтительным консервантом является м-крезол.
Жидкая композиция представленного изобретения может включать в себя вещество, регулирующее тоничность. Подходящее вещество, регулирующее тоничность, может быть выбрано из глицерина, лактозы, сорбитола, маннитола, глюкозы, NaCl, кальций- или магнийсодержащих соединений, таких как CaCl. Концентрация глицерина, лактозы, сорбитола, маннитола и глюкозы может находиться в диапазоне, составляющем 100-250 мМ. Концентрация NaCl может составлять до 150 мМ. Предпочтительным веществом, регулирующим тоничность, является глицерин.
В дополнение, жидкая композиция может содержать от 0,5 мкг/мл до 20 мкг/мл L-метионина, предпочтительно от 1 мкг/мл до 5 мкг/мл Предпочтительно, она содержит L-метионин.
Метформин представляет собой международное непатентованное название 1,1-диметилбигуанида (номер CAS 657-24-9). В представленном изобретении, термин "метформин" содержит любую его фармацевтически приемлемую соль.
В представленном изобретении, метформин можно вводить перорально. Квалифицированному специалисту известны готовые формы метформина, подходящие для лечения сахарного диабета 2 типа посредством перорального введения. Метформин можно вводить в дозе, составляющей по меньшей мере 1,0 г/день или по меньшей мере 1,5 г/день. Для перорального введения, метформин может быть приготовлен в виде твердой лекарственной формы, такой как таблетка или пилюля.
В представленном изобретении, desPro36Эксендин-4(1-39)-Lys6-NH2 и/или фармацевтически приемлемую соль вводят в дополнительной терапии к введению метформина.
В представленном изобретении, термины «дополнительная», «дополнительное лечение» и «дополнительная терапия» связаны с лечением сахарного диабета 2 типа метформином и AVE0010. Метформин и AVE0010 могут вводиться в пределах временного интервала, составляющего 24 ч. И метформин, и AVE0010 могут вводиться однократной дозой в день. Метформин и AVE0010 могут вводиться посредством различных способов введения. Метформин можно вводить перорально, а AVE0010 можно вводить подкожно.
Субъект, подвергающийся лечению с помощью способа представленного изобретения, страдающий сахарным диабетом 2 типа, может быть тучным субъектом. В представленном изобретении, тучный субъект может иметь индекс массы тела, равный по меньшей мере 30.
Субъект, подвергающийся лечению с помощью способа представленного изобретения, может иметь значение HbA1c в диапазоне, составляющем 7-10%.
Субъект, подвергающийся лечению с помощью способа представленного изобретения, может иметь значение HbA1c, составляющее по меньшей мере 8%, в частности, субъект, подвергающийся лечению с помощью способа представленного изобретения, может иметь значение HbA1c в диапазоне, составляющем от 8% до 10%.
Субъект, подвергающийся лечению с помощью способа представленного изобретения, может иметь значение HbA1c, составляющее ниже 8%, в частности, субъект, подвергающийся лечению с помощью способа представленного изобретения, может иметь значение HbA1c в диапазоне, составляющем 7-8%.
Субъектом, подвергающемуся лечению с помощью способа представленного изобретения, может быть взрослый субъект. Субъект может иметь возраст в диапазоне, составляющем от 18 до 50 лет.
Способ представленного изобретения предпочтительно является способ лечения субъекта, страдающего сахарным диабетом 2 типа, при этом сахарный диабет 2 типа не регулируется должным образом посредством лечения одним метформином, например, дозой, составляющей по меньшей мере 1,0 г/день метформина или по меньшей мере 1,5 г/день метформина в течение 3 месяцев. В представленном изобретении, субъект, страдающий сахарным диабетом 2 типа, который не регулируется должным образом, может иметь значение HbA1c в диапазоне, составляющем 7%-10%.
Еще одним аспектом представленного изобретения является фармацевтическая комбинация, содержащая
(a) desPro36Эксендин-4(1-39)-Lys6-NH2 и/или его фармацевтически приемлемую соль, и
(b) метформин и/или его фармацевтически приемлемую соль.
Предпочтительно, комбинация представленного изобретения предназначена для использования при лечении сахарного диабета 2 типа.
Предпочтительно, комбинация представленного изобретения предназначена для использования для предотвращения гипогликемии, как описано в данном документе, у пациентов с сахарным диабетом 2 типа.
Более предпочтительно комбинация представленного изобретения предназначена для использования для предотвращения гипогликемии у пациента с сахарным диабетом 2 типа, имеющего повышенный риск гипогликемии, в частности у пациента с диабетом 2 типа, имеющего пережитое по меньшей мере одно гипогликемическое событие. Гипогликемическое событие может быть симптоматическим гипогликемическим событием или тяжелым симптоматическим гипогликемическим событием.
Предпочтительно, комбинация представленного изобретения предназначена для использования для предотвращения побочных эффектов противодиабетического лечения, как описано в данном документе, у пациентов с сахарным диабетом 2 типа. В частности, побочным эффектом является тошнота, панкреатит и/или повышенная концентрация кальцитонина в крови.
Комбинация представленного изобретения может вводиться, как описано в данном документе в контексте способа представленного изобретения. Соединения (a) и (b) комбинации представленного изобретения могут быть получены в виде готовой формы, как описано в данном документе в контексте способа представленного изобретения.
Еще одним аспектом представленного изобретения является применение комбинации, содержащей
(a) desPro36Эксендин-4(1-39)-Lys6-NH2 и/или его фармацевтически приемлемую соль, и
(b) метформин и/или его фармацевтически приемлемую соль,
для изготовления лекарства для лечения сахарного диабета 2 типа.
Лекарство содержит desPro36Эксендин-4(1-39)-Lys6-NH2 и метформин в раздельных готовых формах, как описано в данном документе.
Комбинация представленного изобретения может быть использована для получения лекарства для предотвращения гипогликемии, как описано в данном документе, у пациентов с сахарным диабетом 2 типа.
Комбинация представленного изобретения может быть использована для получения лекарства для предотвращения побочных эффектов противодиабетического лечения пациентов с сахарным диабетом 2 типа, как описано в данном документе. В частности, побочным эффектом является тошнота, панкреатит и/или повышенная концентрация кальцитонина в крови.
Изобретение дополнительно проиллюстрировано посредством последующих Примеров и Чертежей.
ОПИСАНИЕ ЧЕРТЕЖЕЙ
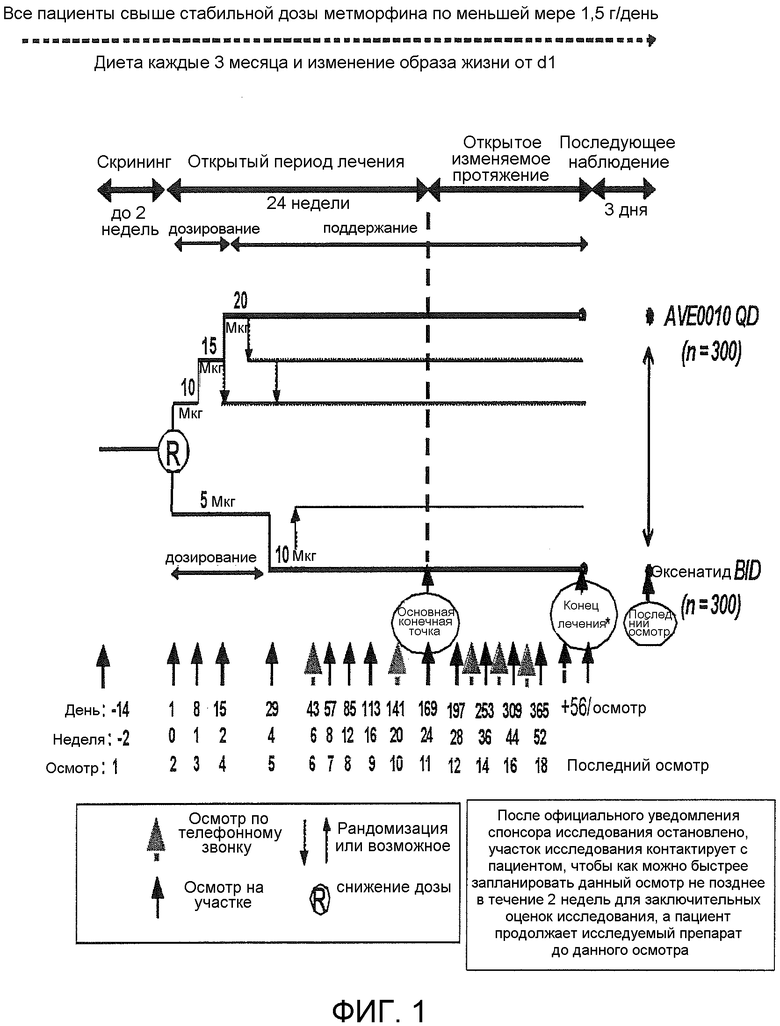
Фиг.1: Дизайн клинического исследования примера 2.
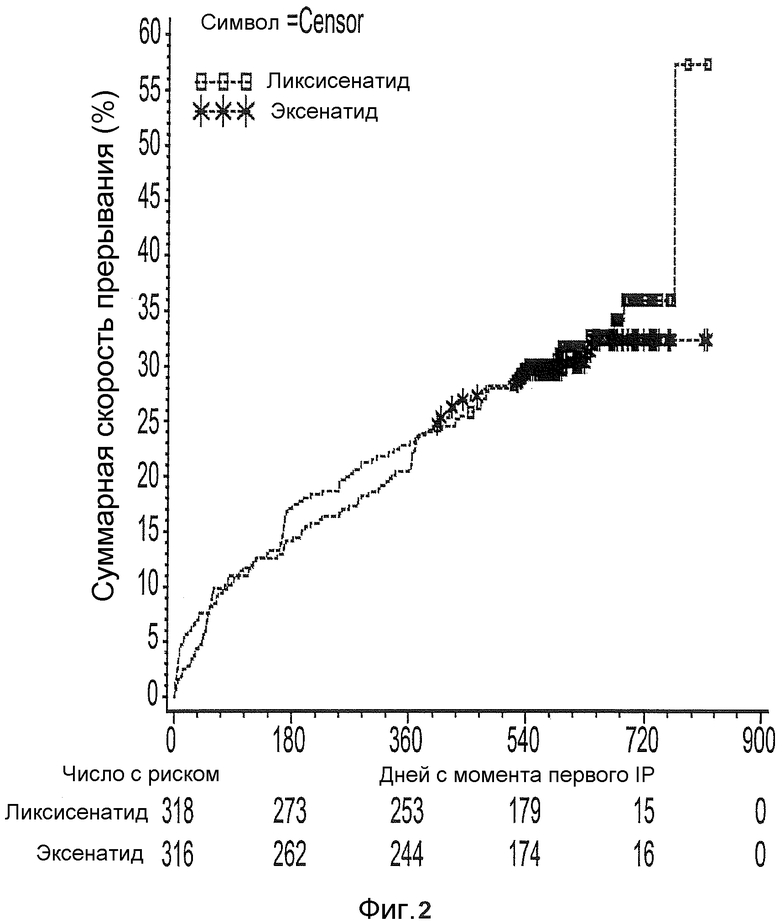
Фиг.2: график Каплана-Майера времени к прекращению лечения вследствие какой-либо причины - рандомизированная популяция.
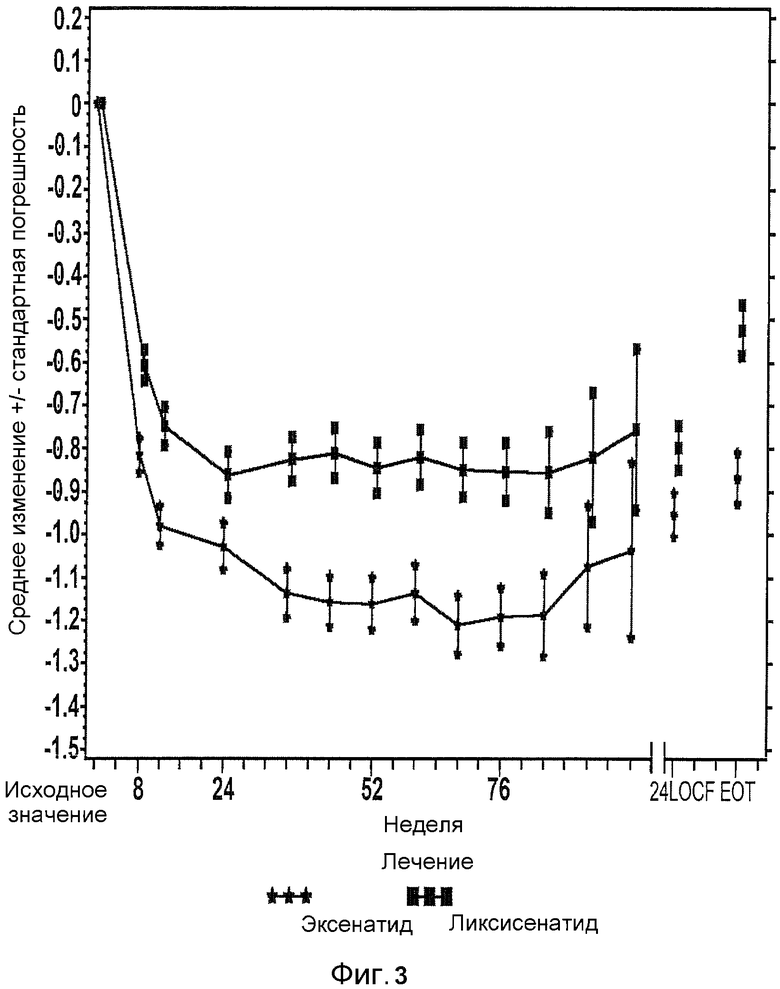
Фиг.3: График среднего изменения HbA1c (%) по сравнению с исходным при осмотре и в конечной точке mITT популяции. EOT = последнее значение в период терапии (LOCF). LOCF = перенос вперед данных последнего наблюдения. Примечание: анализ исключал измерения, полученные после введения резервного препарата и/или после прекращения лечения плюс 3 дня. В течение 24 недели (LOCF), анализ включал измерения, полученные за 3 суток после последней дозы инъекции исследуемого препарата при 11 осмотре или перед ним (24 неделя), или на 169 день, если 11 осмотр (24 неделя) невозможен.
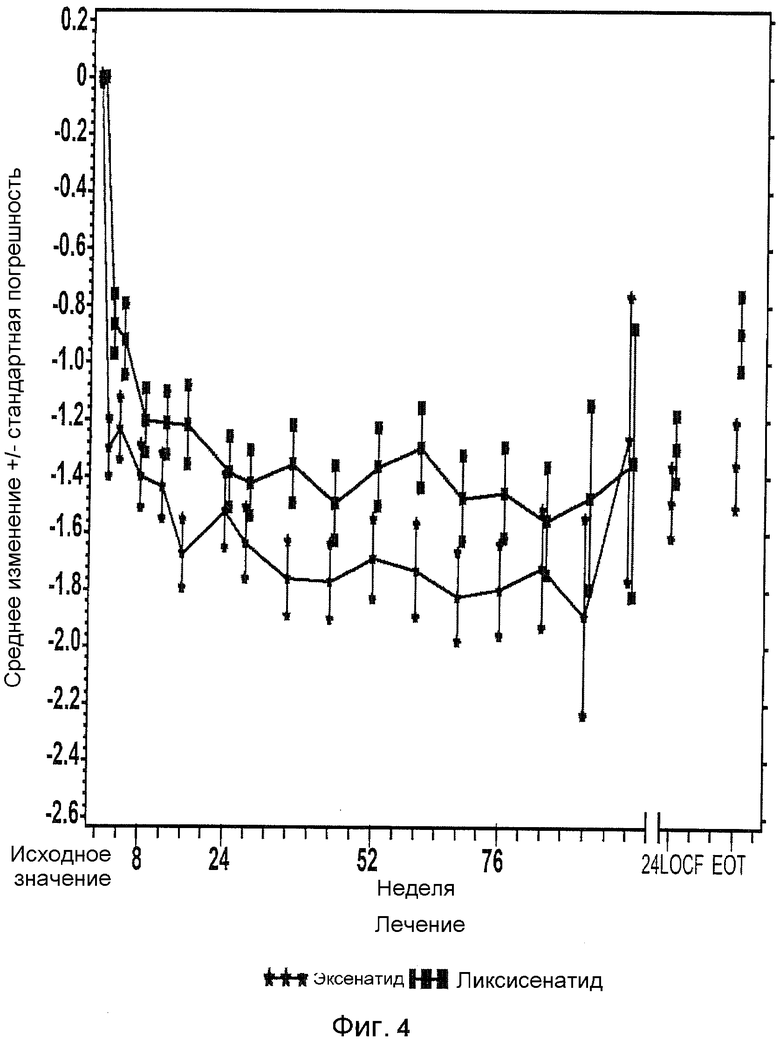
Фиг.4: График среднего изменения глюкозы в плазме натощак (ммоль/л) по сравнению с исходной при осмотре и в конечной точке-mITT популяции. EOT = последнее значение в период терапии (LOCF). LOCF = перенос вперед данных последнего наблюдения. Примечание: анализ исключал измерения, полученные после введения резервного препарата и/или после прекращения лечения плюс 3 дня. В течение 24 недели (LOCF), анализ включал измерения, полученные до 1 дня после последней дозы инъекции исследуемого препарата при 11 осмотре или перед ним (24 неделя), или на 169 день, если 11 осмотр (24 неделя) невозможен.
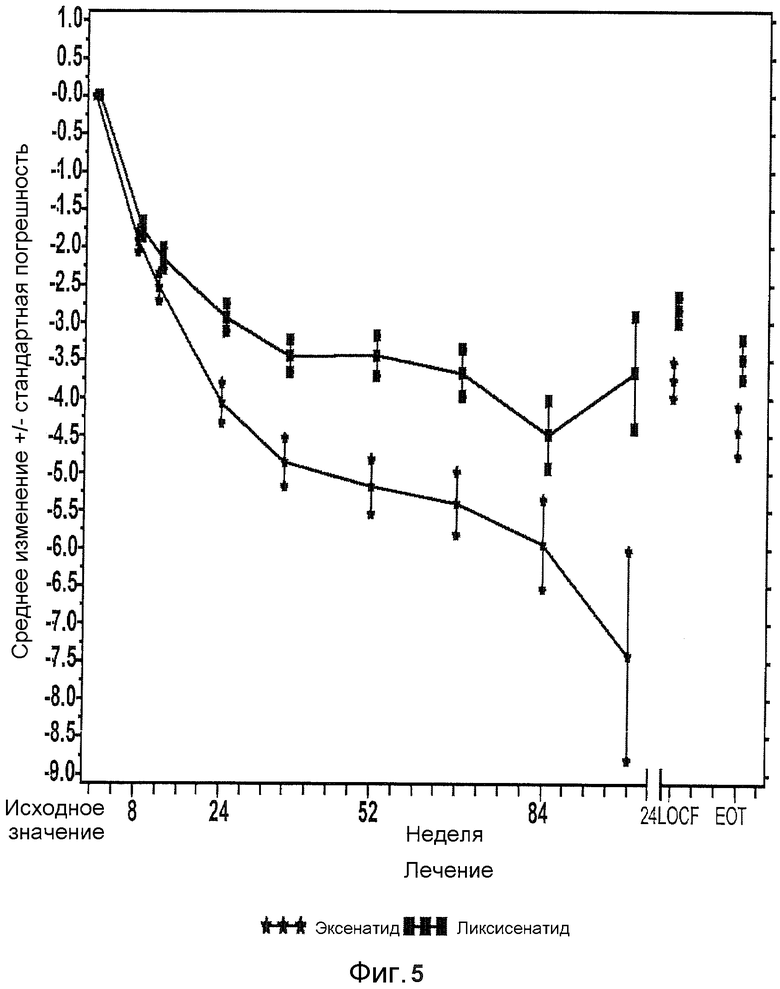
Фиг.5: График среднего изменения массы тела (кг) по сравнению с исходной при осмотре и в конечной точке-mITT популяции. EOT = последнее значение в период терапии (LOCF). LOCF = перенос вперед данных последнего наблюдения. Примечание: анализ исключал измерения, полученные после введения резервного препарата и/или после прекращения лечения плюс 3 дня. В течение 24 недели (LOCF), анализ включал измерения, полученные за 3 суток после инъекции последней дозы исследуемого препарата при 11 осмотре или перед ним (24 неделя), или на 169 день, если 11 осмотр (24 неделя) невозможен.
Пример 1
24-недельное исследование, сравнивающее ликсисенатид (AVE0010) с ситаглиптином в качестве дополнения к метформину для страдающих ожирением пациентов с сахарным диабетом 2 типа моложе, чем 50
Предметом примера является рандомизированное, двойное слепое, с двойной маскировкой, с 2 параллельными группами, многоцентровое, 24-недельное исследование, сравнивающее эффективность и безопасность ликсисенатида (AVE0010) с ситаглиптином (CAS Количество 486460-32-6) в качестве дополнения к метформину у страдающих ожирением пациентов с сахарным диабетом 2 типа моложе, чем 50 лет и не регулируемым должным образом метформином. Ситаглиптин представляет собой противодиабетическое средство, действующее в качестве ингибитора дипептидилпептидазы 4 (DPP4), приводя к повышению уровня глюкагон-подобного пептида 1, снижая тем самым уровень глюкозы в крови у больных сахарным диабетом.
Основные цели исследования
Основная цель данного исследования состоит в оценке эффективности ликсисенатида в комбинированной конечной точке контроля уровня глюкозы в крови (HbA 1c ) и массы тела по сравнению с ситаглиптином в качестве дополнения к лечению метформином в течение периода, составляющего 24 недели, у страдающих ожирением пациентов с сахарным диабетом 2 типа моложе, чем 50.
Дополнительными Целями Исследование является оценка влияния ликсисенатида на:
- абсолютные изменения в HbA1c и массы тела;
- глюкозу в плазме натощак;
- Глюкозу в плазме, инсулин, C пептид, глюкагон и проинсулин в течение 2-часового стандартизированного пробного приема пищи;
- Резистентность к инсулину, оцененную посредством HOMA-IR;
- Функцию бета-клеток, оцененную посредством HOMA-beta;
- Оценку безопасности и переносимости ликсисенатида;
- Оценку фармакокинетики ликсисенатида, используя метод фармакокинетики популяции, чтобы оценить выработку против антител ликсисенатида.
Конкретные уязвимые популяции:
Женщины, способные к деторождению, использующие контрацепцию.
Критерии включения в исследование
Пациенты (мужчины и женщины) с сахарным диабетом 2 типа, по определению ВОЗ (21), диагностированного в течение по крайней мере 1 года во время скринингового осмотра, с недостаточным регулированием метформином при стабильной дозе, составляющей по меньшей мере 1,5 г/день, в течение по меньшей мере 3 месяцев перед скрининговым осмотром. Пациенты с ожирением (ИМТ ≥30 кг/м2) и в возрасте от 18 лет до менее чем 50 лет.
Критерии исключения из исследования
- HbA1c <7,0% или HbA1c >10% во время скрининга
- Сахарный диабет 1 типа
- Беременность или лактация
- Женщины, способные к деторождению, с неэффективным способом контрацепции
- Глюкоза плазмы натощак при скрининге >250 мг/дл (>13,9 ммоль/л)
- Изменение массы тела, составляющее более чем 5 кг, в течение 3 месяцев, предшествующих скрининговому осмотру
- Панкреатит неясной этиологии, хронический панкреатит, панкреатэктомия, хирургические вмешательства на желудке, воспалительные заболевания кишечника в анамнезе
- Метаболический ацидоз, включая диабетический кетоацидоз в течение 1 года перед скринингом в анамнезе
- Гемоглобинопатия или гемолитическая анемия или переливание продуктов крови или плазмы в течение 3 месяцев перед периодом скрининга
- В течение последних 6 месяцев перед скринингом: в анамнезе инфаркт миокарда, инсульт или сердечная недостаточность, требующая госпитализации.
- Известное в анамнезе злоупотребление лекарственными препаратами или алкоголем в течение 6 месяцев перед периодом скрининга.
- Какая-либо клинически значимая патология, выявленная посредством клинического исследования, лабораторных тестов, ЭКГ либо показателей жизненно важных функций в период скрининга, которая при оценке исследователя или любого младшего исследователя могла бы помешать успешному завершению исследования или ограничивает оценку эффективности, такая как основные системные заболевания, наличие клинически значимой диабетической ретинопатии или наличие макулярного отека, который может потребовать лечение лазером в течение периода исследования.
- Неконтролируемая или неадекватно контролируемая гипертензия в период скрининга с систолическим или диастолическим кровяным давлением в покое > 180 мм рт. ст. или > 110 мм рт. ст., соответственно
- Лабораторные находки в период скрининга:
- Амилаза и/или липаза >3 раз выше верхней границы диапазона лабораторной нормы
- Общий билирубин: >1,5 раза выше верхней границы диапазона лабораторной нормы (за исключением случаев синдрома Жильбера)
- Гемоглобин <11 г/дл и/или нейтрофилы <1500/мм3 и/или тромбоциты <100000/мм3
- Положительный тест на поверхностный антиген гепатита В и/или антитела гепатита С
- Положительный сывороточный тест на беременность у женщин репродуктивного возраста
- Применение других пероральных или инъекционных противодиабетических или гипогликемических веществ, не являющихся метформином (например, сульфонилмочевины, ингибитора альфа-гликозидазы, тиазолидиндиона, эксенатида, ингибиторов DPP-IV, инсулина и т.д.) в течение 3 месяцев перед периодом скрининга
- Нестабильная диета или нестабильное лечение ожирения в течение 3 месяцев перед периодом скрининга
- Применение системных глюкокортикоидов (исключая местное применение или ингаляционные формы) в течение одной недели или более в течение 3 месяцев перед периодом скрининга
- Применение каких-либо исследуемых препаратов в течение 3 месяцев перед скринингом
- Клинически отягощенный анамнез заболевания желудочно-кишечного тракта, связанного с продолжительной тошнотой и рвотой, включая, но без ограничения, парез желудка и гастроэзофагеальную рефлюксную болезнь, требующую медицинского лечения, в течение 6 месяцев перед периодом скрининга
- Какое-либо предшествующее лечение ликсисенатидом (например, участие в предыдущем исследовании ликсисенатида)
- Аллергическая реакция на любой GLP 1-агонист в прошлом (например, эксенатид, лираглутид) или на метакрезол
- В анамнезе тяжелая реакция гиперчувствительности на ситаглиптин.
- Умеренное или тяжелое поражение почек (креатининовый клиренс ниже 50 мл/мин)
Продолжительность периода исследования для субъекта
Максимальная продолжительность составляет 27 недель ± 7 дней (3-недельный скрининг + 24-недельное двойное слепое, с двойной маскировкой, активно-контролируемое лечение + 3-дневное последующее врачебное наблюдение)
Количество подразделений: 2
Пример 2
Рандомизированное, открытое, с активным контролем, с 2 параллельными группами, многоцентровое 24-недельное исследование с последующей расширенной оценкой эффективности и безопасности AVE0010 по сравнению с эксенатидомом в дополнение к метформину у пациентов с сахарным диабетом 2 типа, не регулируемым должным образом метформином
СУЩНОСТЬ
Проводили рандомизированное, открытое, с активным контролем, с двумя параллельными группами, многоцентровое, многонациональное исследование оценки эффективности и безопасности ликсисенатида по сравнению с эксенатидомом в качестве дополнительного лечения к метформину у пациентов с сахарным диабетом 2 типа. Приблизительная минимальная продолжительность исследования для пациента составила 78 недель (до 2 недель скрининга +24-недельное основное лечение + варьирующая продолжительность +3-дневное последующее врачебное наблюдение). Исследование проводили в 122 центрах в 18 странах. Основной целью исследования было оценить эффективность ликсисенатида при контроле уровня глюкозы в крови по сравнению с эксенатидомом в показателях уменьшения HbA1c (абсолютное изменение) в течение периода, составляющего 24 недели.
Все 634 пациента были рандомизированы в одну из двух групп лечения (318 в группе ликсисенатида и 316 в группе эксенатида). Всех рандомизированных пациентов подвергли исследуемому виду лечения. Демографические и исходные характеристики в целом были одинаковыми между группами лечения. Восемнадцать пациентов (7 пациентов в группе ликсисенатида и 11 пациентов в группе эксенатида) были исключены из популяции mITT для анализов эффективности вследствие отсутствия данных об эффективности после исходного уровня. В процессе всего периода исследуемого вида лечения, 198 (31,2%) пациентов досрочно прервали исследуемый вид лечения. Процентные доли пациентов, которые прервали лечение, были одинаковыми между группами лечения (32,1% для группы ликсисенатида и 30,4% для группы эксенатида). Основной причиной для прекращения лечения были "нежелательные явления" (14,2% в каждой группе) с последующими "другими причинами" (9,1% для ликсисенатида и 9,8% для эксенатида), "отсутствием эффективности" (6,0% для ликсисенатида и 1,9% для эксенатида) и "плохим соблюдением протокола" (2,2% для ликсисенатида и 4,1% для эксенатида).
Средние изменения наименьших квадратов (LS) HbA1c по сравнению с исходным к 24 неделе составили -0,79% для группы ликсисенатида и -0,96% для группы эксенатида (среднее различие LS vs. эксенатид =0,17%). Была продемонстрирована не меньшая эффективность ликсисенатида по сравнению с эксенатидомом, так как верхняя граница двустороннего 95% CI среднего различия LS была меньше, чем предварительно заданный предел не меньшей эффективности, составляющий 0,4%. Превосходство ликсисенатида над эксенатидом не было продемонстрировано.
Ликсисенатид оказался хорошо переносим. Итоговая частота возникших после начала применения препарата нежелательных явлений (TEAE) была сравнима между двумя группами лечения. Шесть пациентов (3 пациента в каждой группе лечения) имели SAE в период терапии, приведшие к смерти. Сорок восемь тяжелых TEAE произошли во время лечебного периода всего исследования с одинаковой частотой встречаемости в каждой группе лечения (8,2% для ликсисенатида и 7,0% для эксенатида). Наиболее часто сообщаемыми TEAE была тошнота (28,6% для пациентов, подвергавшихся лечению ликсисенатидом, 37,7% пациентов, подвергавшихся лечению эксенатидом). Шестнадцать (5,0%) пациентов, подвергавшихся лечению ликсисенатидом, имели симптоматические гипогликемические события, как определено в протоколе, в течение периода терапии, тогда как 46 (14,6%) пациентов, подвергавшихся лечению эксенатидом, сообщали о симптоматической гипогликемии в течение такого же периода. Ни одно из событий симптоматической гипогликемии не было тяжелым. Всего 9 пациентов (6 [1,9%] пациентов, подвергавшихся лечению ликсисенатидом, и 3 [0,9%] пациента, подвергавшихся лечению эксенатидом) сообщили о случаях признанной Комитетом по оценке Аллергических Реакций (ARAC) как аллергической реакции, но ни одна из них не была признана как возможно связанная с исследуемым препаратом.
1. ЦЕЛИ
1.1 ОСНОВНАЯ ЦЕЛЬ
Основной целью данного исследования было оценить эффективность ликсисенатида при контроле уровня глюкозы крови по сравнению с эксенатидомом в качестве дополнительного лечения к метформину в показателях уменьшения HbA1c в течение периода 24 недель у пациентов с сахарным диабетом 2 типа.
1.2 ДОПОЛНИТЕЛЬНАЯ ЦЕЛЬ (ЦЕЛИ)
• Оценить эффективность ликсисенатида, по сравнению с эксенатидомом, для:
- Процентной доле пациентов, достигших HbA1c <7% или HbA1c ≤6,5%,
- FPG - содержанию глюкозы в плазме крови натощак,
- Массы тела,
• Оценить безопасность и переносимость ликсисенатида,
• Оценить влияние переносимости препарата на качество жизни (Оценка пациентом расстройств верхних отделов желудочно-кишечного тракта - качество жизни, PAGI-QOL).
2. ДИЗАЙН ИССЛЕДОВАНИЯ
Это было рандомизированное, открытое, с активным контролем, с двумя параллельными группами, многоцентровое, многонациональное исследование, запланированное у 300 подвергавшихся лечению ликсисенатидом и 300 подвергавшихся лечению эксенатидом пациентов.
Пациенты были разделены посредством значений скрининга HbA1c (<8,0%, ≥8,0%) и Индекса массы тела (BMI <30, ≥30 кг/м2).
Согласно протоколу Поправки 4 (датированному 18 января 2010 года), приблизительная минимальная продолжительность исследования для пациента составляла 78 недель (до 2 недель скрининга +24 недель основного открытого лечения + варьируемая продолжительность +3 дня последующего врачебного наблюдения). Пациенты, которые завершили 24-недельный основной открытый период, прошли переменный открытый расширенный период, который закончился для всех пациентов приблизительно в запланированную дату осмотра (V24) 76 недели для последнего рандомизированного пациента.
Пациентов, которые досрочно прервали исследуемый вид лечения, продолжили исследование до запланированной даты завершения исследования. Им было выполнено последующее врачебное наблюдение согласно процедурам исследования, как указано в поправке протокола (за исключением 3-дневного последующего врачебного наблюдения за безопасностью после лечения и опросника PAGI-QOL).
3. ОСНОВНАЯ И КЛЮЧЕВАЯ ДОПОЛНИТЕЛЬНЫЕ КОНЕЧНЫЕ ТОЧКИ
3.1 ОСНОВНАЯ КОНЕЧНАЯ ТОЧКА
Основным показателем эффективности было абсолютное изменение HbA1c по сравнению с исходным к 24 неделе, которое определяли как: HbA1c на 24 неделе - HbA1c в начале исследования.
Если пациент прервал лечение досрочно или принял резервный вариант лечения в течение основного 24-недельного открытого периода лечения или не имел значения HbA1c при осмотре на 24 неделе, в качестве значения HbA1c на 24 неделе использовали последнее после исходного уровня в период терапии измерение HbA1c в течение основного 24-недельного периода терапии (метод замены пропущенных данных последним значением [LOCF]).
3.2 ДОПОЛНИТЕЛЬНЫЕ КОНЕЧНЫЕ ТОЧКИ
3.2.1 Конечные точки эффективности
Применялась точно такая же методика обработки отсутствующей оценки/раннего прекращения, как для основной конечной точки.
Дополнительными показателями эффективности были:
• Процентная доля пациентов с HbA1c <7% на 24 неделе,
• Процентная доля пациентов с HbA1c ≤6,5% на 24 неделе,
• Изменение глюкозы плазмы натощак (ммоль/л) (центральной лабораторией) по сравнению с исходным уровнем, к 24 неделе,
• Изменение массы тела (кг) по сравнению с исходной к 24 неделе,
• Процентная доля пациентов, нуждающихся в резервном варианте лечения, в течение основного 24-недельного периода лечения.
• Процентная доля пациентов с потерей массы (кг) по сравнению с исходной на 24 неделе.
Все дополнительные конечные точки в конце лечения подлежали оценке только посредством описательной статистики (представлено в CSR - отчете о клиническом исследовании)
3.2.2 Конечные точки безопасности
Анализ безопасности был основан на сообщениях о возникших после начала лечения нежелательных явлениях и другой информации о безопасности, включая симптоматическую гипогликемию и тяжелую симптоматическую гипогликемию, местную переносимость в месте инъекции, аллергические явления (которые рассматриваются ARAC), подозреваемый панкреатит, повышенный кальцитонин, жизненно важные функции, ЭКГ в 12 отведениях и лабораторные тесты.
Основные сердечно-сосудистые явления также были собраны и рассмотрены Сердечно-сосудистой экспертной комиссией (CAC). Рассмотренные и подтвержденные CAC явления данного исследования и другие исследования ликсисенатида 2-3 фазы будут объединены для анализа и сведены в отдельный отчет на основании плана статистического анализа для общей сердечно-сосудистой оценки ликсисенатида. KRM/CSR не предоставит сущность рассмотренных и подтвержденных CV явлений из данного исследования.
3.2.3 Связанные со здоровьем показатели качества жизни (опросник PAGI-QOL)
Применялась точно такая же методика обработки отсутствующей оценки/раннего прекращения, как для основной конечной точки. Влияние переносимости препарата на связанное со здоровьем качество жизни оценивали с помощью опросника PAGI-QOL, который состоял из 30 вопросов и охватывал пять параметров, включая повседневную деятельность, одежду, диету и пищевые привычки, отношения, психическое здоровье и расстройство. Общая оценка рассчитывалась, беря среднее значение оценок пяти параметров (оценок подшкал) и колебалась от 0 до 5, при этом более низкие оценки показывали более хорошее качество жизни. Анализируется изменение общей оценки PAGI-QOL по сравнению с исходной к 24 неделе.
4. ДОПУЩЕНИЯ РАСЧЕТА РАЗМЕРОВ ВЫБОРКИ
Расчеты размера/мощности выборки выполняли на основании основного показателя, изменения HbA1c по сравнению с исходным к 24 неделе.
Размер выборки, составляющий 600 (300 пациентов в каждой группе) обеспечивал, что верхняя граница достоверности двухстороннего 95% доверительного интервала для скорректированного среднего различия между ликсисенатидом и эксенатидом не будет превышать 0,4% HbA1c, при этом 96% мощность предполагает, что стандартное отклонение составляло 1,3, а истинное различие по HbA1c между ликсисенатидом и эксенатидом составляло ноль. Стандартное отклонение оценивали консервативным образом из ранее проведенных исследований сахарного диабета (на основании опубликованных данных аналогично разработанного исследования и внутренних данных, не опубликованных), принимая в расчет более ранний отсев.
5. СТАТИСТИЧЕСКИЕ МЕТОДЫ
5.1 АНАЛИЗ ПОПУЛЯЦИЙ
Модифицированная популяция больных, включенных в испытание (mITT), состояла из всех рандомизированных пациентов, получивших по меньшей мере одну дозу открытого исследуемого препарата (IP), и имевших как базовую оценку, так и по меньшей мере одну оценку после базовой показателей эффективности.
Выборку для проверки безопасности определяли, как все рандомизированные пациенты, принимавшие по меньшей мере одну дозу исследуемого препарата.
5.2 ОСНОВНОЙ АНАЛИЗ ЭФФЕКТИВНОСТИ
Основную конечную точку (изменение HbA1c по сравнению с исходным к 24 неделе) анализировали с использованием ковариационного анализа (ANCOVA) модели с лечением, рандомизационных страт, различающихся уровнем HbA1c при скрининге (<8,0, ≥8,0%), рандомизационных страт, различающиеся уровнем BMI при скрининге (<30, ≥30 кг/м2), и страны в качестве фиксированных эффектов и с использованием базового значения в качестве ковариаты.
Различия между ликсисенатидом и эксенатидом и двусторонние 95% доверительные интервалы оценивали в рамках ANCOVA. Для оценки не меньшей эффективности, верхнюю границу двустороннего 95% CI для различия скорректированного среднего изменения HbAlc по сравнению с исходным к 24 неделе между ликсисенатидом и эксенатидом сравнивали с предварительно заданным пределом не меньшей эффективности 0,4% HbA1c. Не меньшая эффективность демонстрировалась, если верхняя граница двустороннего 95% CI различия между ликсисенатидом и эксенатидом в выборке mITT составляла ≤0,4%. Если была установлена не меньшая эффективность, необходимо было провести соответствующую проверку статистического превосходства для основной конечной точки.
Основной анализ основного показателя эффективности проводили на основании популяции mITT и измерений, полученных в течение основного 24-недельного периода терапии для показателей эффективности. Основной 24-недельный период терапии определяли как время от первой дозы IP до 3 дней (за исключением FPG центральной лабораторией, который составлял до 1 день) после последней дозы инъекции IP при или перед осмотром V11/24 неделя (или D169, если осмотр V11/24 недели отсутствовал), или до введения резервного варианта лечения, в зависимости от того, что было раньше. В случае прекращения IP перед 24 неделей, HbA1c оценивали на момент прекращения. Использовали методику LOCF, принимая в качестве значения HbA1c на 24 неделе данное последнее доступное измерение HbA1c после базового в период терапии (перед началом нового препарата в случае резервного варианта лечения).
5.3 ДОПОЛНИТЕЛЬНЫЙ АНАЛИЗ ЭФФЕКТИВНОСТИ
Никаких формальных статистических тестов не проводили для каких-либо дополнительных конечных точек эффективности.
Все непрерывные дополнительные показатели эффективности на 24 неделе, которые описаны в разделе 3.2.1, анализировали с использованием аналогичного подхода и модели ANCOVA, как описано выше для основного анализа основной конечной точки эффективности. Были предоставлены скорректированные оценки среднего различия в лечении между ликсисенатидом и эксенатидом и двусторонние 95% доверительные интервалы.
Анализировали следующие категориальные дополнительные показатели эффективности на 24 неделе:
• Процентная доля пациентов с HbA1c <7,0% на 24 неделе;
• Процентная доля пациентов с HbA1c ≤6,5% на 24 неделе;
• Процентная доля пациентов нуждающихся в резервном варианте лечения в течение основного 24-недельного периода лечения.
Количество и процентная доля пациентов с потерей массы на 24 неделе ≥5% по сравнению с исходным представлены группами лечения.
Все дополнительные конечные точки в конце лечения оценивали только посредством описательной статистики (среднее, стандартное отклонение, срединное значение и диапазоны, предусмотренные в CSR).
5.4 АНАЛИЗ БЕЗОПАСНОСТИ
Анализы безопасности были основаны главным образом на периоде терапии всего исследования. Период терапии всего исследования определяли как время от первой дозы инъекции открытого IP до 3 дня после последней дозы введения открытого IP в течение всего периода исследование независимо от состояния резервного лечения. 3-дневный интервал выбрали, исходя из периода полувыведения IP (приблизительно 5-кратный период полувыведения).
В дополнение, анализы безопасности для 24-недельного периода лечения будут подытожены в CSR.
Итог результатов безопасности (описательная статистика или Таблицы частоты) представлен группами лечения.
5.5 АНАЛИЗ СВЯЗАННОГО СО ЗДОРОВЬЕМ КАЧЕСТВА ЖИЗНИ
Для общей оценки PAGI-QOL не проводили никаких формальных статистических тестов.
Общую оценку PAGI-QOL на 24 неделе анализировали с использованием аналогичного подхода и модели ANCOVA, как описано выше для основного анализа основной конечной точки эффективности.
6 РЕЗУЛЬТАТЫ
6.1 ИССЛЕДОВАНИЕ ПАЦИЕНТОВ
6.1.1 Контролируемость пациентов
Исследование проводили в 122 центрах в 18 странах (Аргентина, Австралия, Бразилия, Колумбия, Дания, Финляндия, Германия, Греция, Венгрия, Италия, Нидерланды, Норвегия, Польша, Пуэрто-Рико, Российская Федерация, Испания, Швеция и США). Всего скринировали 1243 пациента и 639 рандомизировали в одну из двух групп лечения. Было обнаружено, что один участок в Германии (#276905), где рандомизировали 5 пациентов (из 8 скринированных пациентов) имеет значительное несоответствие протоколу. До блокировки базы данных, было решено исключить данных пациентов из всех анализов эффективности и безопасности и впоследствии ее передали в FDA США. Данные безопасности с данного участка будут отдельно отображены в CSR. Основной причиной неудачи скрининга было значение HbAlc при скрининговом осмотре вне определенных диапазонов протокола (426 [34,5%] из 1235 подвергшиеся скринингу пациентов, исключая немецкий участок, упомянутый выше).
Шестьсот тридцать четыре рандомизированных пациента были включены в анализ (318 в группе ликсисенатида и 316 в группе эксенатида), и всех пациентов подвергли исследуемому виду лечения.
Восемнадцать пациентов (7 пациентов в группе ликсисенатида и 11 пациентов в группе эксенатида) были исключены из mITT популяции для анализов эффективности вследствие отсутствия данных об эффективности после исходного уровня. Таблица 1 предоставляет количество пациентов, включенных в каждую популяцию, участвующую в анализе.
Для популяции для проверки эффективности, пациентов свели в таблицу согласно их рандомизированному лечению (как рандомизировали).
6.1.2 Распределение Исследования
Таблица 2 предоставляет сущность распределения участников клинического исследования для каждой группы лечения. В течение периода общего лечения, 198 (31,2%) пациентов досрочно прервали исследуемый вид лечения. Процентная доля пациентов, которые прервали лечение, были одинаковыми между группами лечения (32,1% для ликсисенатида и 30,4% для эксенатида). Основной причиной для прекращения лечения были "нежелательные явления" (14,2% в каждой группе) за которыми следуют "другие причины" (9,1% для ликсисенатида и 9,8% для эксенатида), "отсутствие эффективности" (6,0% для ликсисенатида и 1,9% для эксенатида) и "слабое соответствие протоколу" (2,2% для ликсисенатида и 4,1% для эксенатида). Время до начала прекращения лечения по какой-либо причине в течение общего периода лечения изображено на Фиг.2, при этом не наблюдалось различие между 2 группами лечения. Аналогичные результаты наблюдались в течение 24-недельного периода лечения, когда в общей сложности 86 (13,6%) пациентов досрочно прервали исследуемый вид лечения, причем основной причиной также являлись нежелательные явления (9,1% для ликсисенатида и 9,8% для эксенатида).
Демографические и исходные характеристики
Демографические и исходные характеристики пациентов были в общем одинаковыми между двумя группами лечения для популяции для проверки безопасности (Таблица 3). Средний возраст популяции исследования составлял 57,5 лет. Большинство пациентов были с Кавказа (92,7%). Процентная доля пациентов-мужчин (59,2%) в группе эксенатида была выше, чем процентная доля в группе ликсисенатида (47,5%).
(N=634)
[n (%)]
Характеристики заболевания, включая историю заболевания сахарным диабетом, были в общем сравнимы между двумя группами лечения (Таблица 4). Средняя продолжительность лечения метформином была немного дольше в группе эксенатида (4,21 лет) чем в группе ликсисенатида (3,79 лет).
(N=318)
(N=316)
(N=634)
Значение Клиренса креатинина получают с использованием Формулы Кокрофта-Гаулта.
HbA1c и FPG в начале исследования были сравнимы между двумя группами лечения для популяции для проверки безопасности (Таблица 5). Более высокая средняя масса тела в начале исследования наблюдалось в группе эксенатида (96,09 кг) по сравнению с ликсисенатидной группой (94,01 кг).
(N=318)
(N=316)
(N=634)
Оценка расстройств верхнего отдела желудочно-кишечного тракта Пациентов - общая оценка Качества жизни (PAGI-QOL) в начале исследования было одинаковыми между двумя группами лечения (Таблица 6).
(N=318)
(N=316)
(N=634)
6.1.4 Дозировка и продолжительность
Средняя продолжительность лечения было одинаковыми между двумя группами лечения (494,8 дня (70,6 недель) для группы ликсисенатида и 483,0 дня (69 недель) для группы эксенатида) [Таблица 7]. Из 634 пациентов, 536 (85,2% в группе ликсисенатида и 83,9% в группе эксенатида) имели по меньшей мере 169 дня (24 недель) лечения и 345 (55,0% в группе ликсисенатида и 53,8% в группе эксенатида) имели по меньшей мере 547 дня (18 месяцев) лечения. Замечено, что продолжительность лечения 5 пациентов (4 пациентов в группе ликсисенатида и 1 пациента в группе эксенатида) не была суммирована вследствие отсутствия дат завершения их лечения.
Для группы ликсисенатида, 295 (92,8%) пациентов и 293 (92,1%) пациентов были на намеченной общей суточной дозе, равной 20 мкг в конце 24-недельного периода лечения и в конце лечения, соответственно (Таблицы 8 и 9). Для группы эксенатида, 263 (83,2%) пациентов и 217 (68,7%) пациентов были на намеченной общей суточной дозе 20 мкг в конце 24-недельного периода лечения и в конце лечения, соответственно (Таблицы 8 и 9).
(N=318)
(N=316)
(N=318)
(N=316)
(N=318)
(N=316)
6.2 ЭФФЕКТИВНОСТЬ
Основная конечная точка эффективности
Основной анализ
Таблица 10 суммирует результаты основного параметра эффективности, изменения HbAlc по сравнению с исходным к 24 неделе (LOCF) с использованием анализ ANCOVA.
средние изменения LS HbA1c по сравнению с исходным к 24 неделе составили -0,79% для группы ликсисенатида и -0,96% для группы эксенатида (среднее различие LS по сравнению с эксенатидомом = 0,17%). На основании предварительно заданного основного анализа, не меньшая эффективность ликсисенатида по сравнению с эксенатидомом была продемонстрирована в виде верхней границы двустороннего 95% CI среднего различия LS было менее чем предварительно заданный предел не меньшей эффективности 0,4%. Превосходство ликсисенатида над эксенатидом не было продемонстрировано.
к 24 неделе - mITT популяция
(N=311)
(N=305)
а ковариационный анализ (ANCOVA) модели с группами лечения (Эксенатида и Ликсисенатида), рандомизационные страты, различающиеся уровнем HbA1c при скрининге (<8,0, ≥8,0%), рандомизационные страты скрининга BMI (<30, ≥30 кг/м2), и страна в качестве фиксированных эффектов и базовое значение HbA1c в качестве ковариата.
Примечание: анализ включал измерения, полученные перед введением резервного препарата и за 3 суток после инъекции последней дозы исследуемого препарата при 11 осмотре или перед ним (24 неделя), или на 169 день, если 11 осмотр (24 неделя) невозможен.
Включены пациенты с измерениями, как исходного уровня, так и 24 недели (LOCF).
Фиг.3 иллюстрирует Среднее (±SE) изменение HbA1c по сравнению с исходным на протяжении времени в течение периода всего лечения (показаны до 2 лет). Уменьшение HbAlc было относительно постоянным на протяжении времени за пределами 24 недель.
Дополнительный анализ
Таблица 11 суммирует соотношение пациентов с ответом на лечение HbAlc ≤6,5% или <7% на 24 неделе, соответственно. На 24 неделе, 28,5% пациентов, подвергавшихся лечению ликсисенатидом, и 35,4% пациентов, подвергавшихся лечению эксенатидом, достигли значений HbA1c 35,5%; 48,5% пациентов в группе ликсисенатида и 49,8% пациентов в группе эксенатида достигли значений HbA1c <7%.
(N=311)
(N=305)
6.2.2 Другие конечные точки эффективности
Таблица 12 и Таблица 13 приводит анализы ANCOVA FPG и массы тела, соответственно. Фиг.4 и Фиг.5 иллюстрируют Среднее (±SE) изменение FPG и массы тела по сравнению с исходным на протяжении времени в процессе всего периода лечения (показано до 2 лет).
Для FPG, LS средние изменения по сравнению с исходным к 24 неделе составляли -1,22 ммоль/л для группы ликсисенатида и -1,45 ммоль/л для группы эксенатида (LS среднее различие по сравнению с эксенатидом = 0,23 ммоль/л).
LS средняя убыль массы тела по сравнению с исходной на 24 неделе составила 2,96 кг для пациентов, подвергавшихся лечению ликсисенатидом, и 3,98 кг для пациентов, подвергавшихся лечению эксенатидом, (LS среднее различие по сравнению с эксенатидом = 1,02 кг). Масса тела продолжала снижаться после 24 недели основного периода лечения в обеих группах лечения (Фиг.5). Около 5,1% пациентов, подвергавшихся лечению ликсисенатидом, и 31,4% пациентов, подвергавшихся лечению эксенатидом, имели ≥5% убыли массы тела по сравнению с исходной, к 24 неделе (Таблица 14).
Процентные доли пациентов, нуждающихся в резервном варианте лечения на 24 неделе, были небольшими в двух группах (Таблица 15).
(N=311)
(N=305)
Примечание: анализ включал измерения, полученные перед введения резервного препарата и до 1 дня после инъекции последней дозы исследуемого препарата при 11 осмотре или перед ним (24 неделя), или на 169 день, если 11 Осмотр (24 неделя) невозможен. Включали пациентов с измерениями как исходного уровня, так и 24 недели (LOCF).
(N=311)
(N=305)
a Ковариационный анализ (ANCOVA) модели с группами лечения (Эксенатид и Ликсисенатид), рандомизационными стратами, различающимися уровнем HbA1c при скрининге (<8,0, ≥8,0%), рандомизационными стратами скрининга BMI (<30, ≥30 кг/м2), и страной в качестве фиксированных эффектов и исходный уровень массы тела в качестве ковариата.
Примечание: анализ включал измерения, полученные перед введением резервного препарата и за 3 суток после инъекции последней дозы исследуемого препарата при 11 осмотре или перед ним (24 неделя), или на 169 день, если 11 Осмотр (24 неделя) невозможен. Включали пациентов с измерениями, как исходного уровня, так и 24 недели (LOCF).
с исходной к 24 неделе - mITT популяция
(N=311)
(N=305)
(N=311)
(N=305)
6.3 БЕЗОПАСНОСТЬ
Обзор нежелательных явлений, наблюдаемых в течение периода терапии всего исследования, предоставлены в Таблице 16. Соотношение пациентов, переживших возникшие после начала лечения нежелательные явления, было в общем сравнимо между группами подвергавшихся лечению ликсисенатидом и подвергавшихся лечению эксенатидом. Шесть пациентов (3 пациента в каждой лечебной группе) имели SAE в течение периода терапии, приведшие к смерти. Сорок восемь тяжелых возникших после начала лечения нежелательных явления возникли в течение периода терапии всего исследования с одинаковой частотой встречаемости в каждой лечебной группе (8,2% для ликсисенатида и 7,0% для эксенатида). Процентная доля пациентов с возникшими после начала лечения нежелательными явлениями, приведшими к прекращению лечения, была одинаковой в обеих группах (14,2%). Таблицы 17, 18, и 19 приводят возникшие после начала лечения нежелательные явления, приведшие к смерти, тяжелые возникшие после начала лечения нежелательные явления, и возникшие после начала лечения нежелательные явления, приведшие к прекращению лечения, посредством основных SOC, HLGT, HLT и PT, соответственно. Наиболее частой TEAE, приведшей к прекращению лечения, была тошнота в обеих группах лечения (15 [4,7%] пациентов в группе ликсисенатида и 19 [6,0%] пациентов в группе эксенатида).
Таблица 29 в приложении представляет встречаемость Возникших после начала лечения нежелательных явлений в течение периода терапии всего исследования, встречающихся по меньшей мере у 1% пациентов в каждой лечебной группе. Тошнота была наиболее часто сообщаемой TEAE в группе ликсисенатида (91 пациентов [28,6%]). Более высокая процентная доля пациентов, подвергавшихся лечению эксенатидом, сообщала о тошноте (119 [37,7%] пациентов). Второй наиболее часто упоминаемой TEAE у пациентов, подвергавшихся лечению ликсисенатидом, была диарея (48 пациентов [15,1%]), после которой шла головная боль (46 пациентов [14,5%]). В группе эксенатида соответствующее количество пациентов (%) составило 54 (17,1%) для диареи и 31 (9,8%) для головной боли.
(N=318)
(N=316)
n (%) = количество и процентная доля пациентов по меньшей мере с одним нежелательным явление
HLGT: Групповой термин высокого уровня
HLT: Термин высокого уровня
Термин предпочтительного употребления
(N=318)
(N=316)
MedDRA version: 13.1
Примечание: Таблица разделена посредством SOC в международно согласованном порядке и HLGT, HLT, PT в алфавитном порядке.
HLGT: Групповой термин высокого уровня
HLT: Термин высокого уровня
Термин предпочтительного употребления
(N=318)
(N=316)
MedDRA version: 13.1
Примечание: Таблица разделена посредством SOC в международно-согласованном порядке и HLGT, HLT, PT в алфавитном порядке.
HLGT: Групповой термин высокого уровня
HLT: Термин высокого уровня
Термин предпочтительного употребления
(N=318)
(N=316)
ПАТОЛОГИЧЕСКИЕ СОСТОЯНИЯ
MedDRA version: 13.1
Примечание: Примечание: Таблица разделена посредством SOC в международно-согласованном порядке и HLGT, HLT, PT в алфавитном порядке.
Гипогликемия
Шестнадцать (5,0%) пациентов, подвергавшихся лечению ликсисенатидом, имели симптоматические гипогликемические события в протоколе определения в течение периода терапии в течение всего исследования, тогда как 46 (14,6%) пациентов, подвергавшихся лечению эксенатидом, сообщали о симптоматической гипогликемии в течение такого же периода (Таблица 20). Ни одно из симптоматических гипогликемических событий не было тяжелым по интенсивности.
Симптоматическая гипогликемия
Симптоматическая гипогликемия определяется как событие с клиническими симптомами, которые считаются обусловленными гипогликемическим эпизодом (например, потливость, учащенное сердцебиение, чувство голода, беспокойство, тревога, утомляемость, раздражительность, головная боль, потеря концентрации, сонливость, психические нарушения или нарушения зрения, транзиторные сенсорные или двигательные нарушения, спутанность сознания, судороги или кома), сопровождающимся глюкозой в плазме <60 мг/дл (3,3 ммоль/л) или связанным с быстрой нормализацией после перорального введения углевода, если не доступно значение глюкозы в плазме. О симптомах, связанных с глюкозой в плазме ≥60 мг/дл (3,3 ммоль/л), не следует сообщать, как о гипогликемии.
Симптоматическую гипогликемию следует рассматривать, как нежелательное явление. Дополнительную информацию необходимо собирать в дополнительной форме конкретного симптоматического гипогликемического события.
Тяжелая симптоматическая гипогликемия
Тяжелую симптоматическую гипогликемию определяют как событие с клиническими симптомами, которые считаются обусловленными гипогликемией, при которых пациенту требуется помощь другого человека, потому что пациент не может лечить его/ее вследствие острого неврологического дефицита, обусловленным непосредственно гипогликемическим событием, и являются одним из следующих:
• событие было связано с уровнем глюкозы в плазме ниже 36 мг/дл (2,0 ммоль/л).
• если значение глюкозы в плазме не доступно, тогда событие было, связанное с быстрой нормализацией после введения перорально углеводов, внутривенно глюкозы или глюкагона.
Определение тяжелой симптоматической гипогликемии включает все эпизоды, в которых неврологический дефицит был достаточно тяжелым для того, чтобы не допустить самолечения, и в которых соответственно были основания поместить пациентов в группу риска повреждения себя или других. Следует отметить, что "нуждается в помощи" означает, что пациент не может себе помочь. Когда, кто-либо, будучи добрым, спонтанно помогает пациенту, который в этом не нуждается, это не расценивается как "нуждается в помощи".
Тяжелая симптоматическая гипогликемия будет квалифицироваться как SAE, только если она соответствует критериям SAE.
(N=318)
(N=316)
b Подсчитано как (количество пациентов с событиями*100 разделенное на общее воздействие + 3 дня в пациенто-годах).
Примечание: Симптоматическая гипогликемия = симптоматическая гипогликемия, как определено в протоколе.
Термин предпочтительного употребления
(N=318)
(N=316)
В общей сложности в 42 случаях исследователи сообщили о заподозренных аллергических событиях в течение периода терапии всего исследования и отправили в ARAC для экспертизы. Из них тринадцать (у 6 (1,9%) пациентов, подвергавшихся лечению ликсисенатидом, и у 3 (0,9%) пациентов, подвергавшихся лечению эксенатидом) были расценены ARAC как аллергические реакции, но ни одно из событий не было расценено как возможно связанное с IP.
(N=318)
(N=316)
IP = Исследуемый препарат.
В течение периода терапии всего исследования, 5 (1,6%) пациентов, подвергавшихся лечению ликсисенатидом и 9 (2,8%) пациентов, подвергавшихся лечению эксенатидом, сообщали о случаях изменений в панкреатических ферментах, или липазы или амилазы, на специальной странице AE для "подозрения на панкреатит", идущего за рекомендациями протокола (Таблица 23). Пациенты по меньшей мере с одним значением липазы или амилазы ≥3 ULN были собраны в Таблице 24. Один пациент, подвергшийся лечению ликсисенатидом, который сообщил об одном повышении липазы и одном событии повышения панкреатических ферментов на специальной странице AE, имел значение липазы >3 ULN, а также значение амилазы >3 ULN в течение периода лечения. Случаев острого панкреатита в исследовании не наблюдалось.
В каждой лечебной группе с повышенной липазой (3ULN) наблюдалось одинаковое количество пациентов (11 [3,5%] пациентов в группе ликсисенатида и 11 [3,6%>] в группе эксенатида) [Таблица 24]. В группе ликсисенатида повышенную амилазу (3ULN) имели три (1,0%) пациента, и ни одного в группе эксенатида.
(N=318)
(N=316)
Исходный уровень
По критериям PCSA n/Nl (%)
(N=318)
(N=316)
*Независимо от исходного уровня.
Примечание: число (n) представляет подгруппу общего числа пациентов, которые по меньшей мере один раз соответсвуют интересующему критерию. Знаменателем (/Nl) для каждого параметра в пределах лечебной группы является количество пациентов для группы лечения, которые имели такой параметр, оцененный после исходного уровня посредством состояния PCSA исходного уровня. Только ухудшение самого плохого случая для каждого пациента отображается исходным состоянием.
Восемь пациентов (4 [1,3%] в каждой группе) сообщали о значении кальцитонина ≥20 нг/л на специальной странице AE для "повышенного кальцитонина" (Таблица 25). О значениях ≥50 нг/л не сообщалось.
Пять (1,8%) пациентов в группе ликсисенатида и 8 (3,0%) пациентов в группе эксенатида имели значение кальцитонина ≥20 нг/л в течение периода лечения (Таблица 26). Следует подчеркнуть, что измерения кальцитонина были добавлены в поправку протокола после того, как все пациенты были уже рандомизированы. Вследствие этого базовые значения для всех пациентов отсутствуют.
(N=318)
(N=316)
Состояние исходного уровня
После исходного уровня
(N=318)
(N=316)
Примечание: числитель представляет количество пациентов, которые находились в предварительно заданных категориях в категории каждого исходного уровня.
Знаменателем для каждого параметра в пределах лечебной группы является количество пациентов для группы лечения, которые имели такой параметр, оцененный после исходного уровня на исходное состояние.
Пациента считают только в самой плохой категории.
6.4 СВЯЗАННОЕ СО ЗДОРОВЬЕМ КАЧЕСТВО ЖИЗНИ (ОПРОСНИК PAGI-QOL)
Таблица 27 приводит результаты анализа ANCOVA общей оценки PAGI-QOL. LS средние изменения в общей оценке PAGI-QOL по сравнению с исходным к 24 неделе составило -0,09 для группы ликсисенатида и -0,06 для группы эксенатида (LS среднее различие по сравнению с эксенатидом =-0,03).
(N=311)
(N=305)
а ковариационный анализ (ANCOVA) модели с группами лечения (Эксенатида и Ликсисенатида), рандомизационными стратами, различающимися уровнем HbA1c при скрининге (<8,0, ≥8,0%), рандомизационными стратами скрининга BMI (<30, ≥30 кг/м2) и страны в качестве фиксированных эффектов и базовым значением HbA1c в качестве ковариата.
Примечание: анализ включал измерения, полученные перед введением резервного препарата и за 3 суток после инъекции последней дозы исследуемого препарата при 11 осмотре или перед ним (24 неделя), или на 169 день, если 11 Осмотр (24 неделя) невозможен. Включали пациентов с измерениями как исходного уровня, так и 24 недели (LOCF).
7. ДОПОЛНЕНИЕ
Временная точка
Примечание: анализ исключал измерения, полученные после введения резервного препарата и/или после прекращения лечения плюс 3 дня.
Для 24 недели (LOCF), анализ включал измерения, полученные за 3 суток после инъекции последней дозы исследуемого препарата при 11 осмотре или перед ним (24 неделя), или на 169 день, если 11 Осмотр (24 неделя) невозможен.
HLGT: Групповой термин высокого уровня
HLT: Термин высокого уровня
Термин предпочтительного употребления
(N=318)
(N=316)
MedDRA version: 13.1
Примечание: Таблица разделена посредством SOC в международно-согласованном порядке и HLGT, HLT, PT в алфавитном порядке.
Представлены только SOC по меньшей мере с одним PT ≥1% по меньшей мере в одной группе.

Группа изобретений относится к медицине, а именно к терапии и эндокринологии, и может быть использована для предотвращения тяжелой симптоматической гипогликемии, связанной с концентрацией глюкозы в плазме ниже 50 мг/дл, при сахарном диабете 2 типа. Для этого вводят desPro36Эксендин-4(1-39)-Lys6-NH2 (ликсисенатид) и/или его фармацевтически приемлемую соль, и метформин и/или его фармацевтически приемлемую соль, нуждающемуся в этом субъекту, при этом субъект, подлежащий лечению, имеет значение HbA1c по меньшей мере 8%. Также предложена фармацевтическая комбинация и применение комбинации. Группа изобретений обеспечивает контроль уровня глюкозы в крови и массы тела в течение периода, составляющего 24 недели, у страдающих ожирением пациентов с сахарным диабетом 2 типа моложе чем 50. 3 н. и 16 з.п. ф-лы, 5 ил., 29 табл.
1. Способ предотвращения тяжелой симптоматической гипогликемии, связанной с концентрацией глюкозы в плазме ниже 50 мг/дл, при сахарном диабете 2 типа, включающий введение
(a) desPro36Эксендин-4(1-39)-Lys6-NH2 и/или его фармацевтически приемлемой соли, и
(b) метформина и/или его фармацевтически приемлемой соли, нуждающемуся в этом субъекту, при этом субъект, подлежащий лечению, имеет значение HbA1c по меньшей мере 8%.
2. Способ по п. 1, в котором desPro36Эксендин-4(1-39)-Lys6-NH2 вводят посредством одной инъекции в день.
3. Способ по п. 1, в котором desPro36Эксендин-4(1-39)-Lys6-NH2 и/или его фармацевтически приемлемую соль вводят подкожно.
4. Способ по п. 1, в котором метформин вводят перорально.
5. Способ по п. 1, в котором desPro36Эксендин-4(1-39)-Lys6-NH2 и/или фармацевтически приемлемую соль вводят в виде дополнительной терапии к введению метформина.
6. Способ по п. 1, в котором субъект, подлежащий лечению, страдает ожирением.
7. Способ по п. 6, в котором субъект имеет индекс массы тела, равный по меньшей мере 30 кг/м2.
8. Способ по п. 1, в котором субъектом, подлежащим лечению, является взрослый субъект.
9. Способ по п. 1, в котором сахарный диабет 2 типа не регулируется должным образом только метформином.
10. Способ по п. 9, в котором лечение дозой, составляющей по меньшей мере 1,5 г/день, одного метформина в течение трех месяцев, не регулирует должным образом сахарный диабет 2 типа.
11. Способ по п. 1, в котором субъект, подлежащий лечению, имеет значение HbA1c до 10%.
12. Способ по п. 1, в котором гипогликемия связана с концентрацией глюкозы в плазме ниже 40 мг/дл или ниже 36 мг/дл.
13. Способ по п. 1, в котором гипогликемией является симптоматическая гипогликемия.
14. Способ по п. 13, в котором симптоматическая гипогликемия связана по меньшей мере с одним симптомом, выбранным из потливости, учащенного сердцебиения, чувства голода, беспокойства, тревоги, утомляемости, раздражительности, головной боли, потери концентрации, сонливости, психических расстройств, нарушений зрения, транзиторных сенсорных нарушений, транзиторных двигательных нарушений, спутанности сознания, судорог и комы.
15. Способ по п. 1, в котором гипогликемия связана с концентрацией глюкозы в плазме ниже 36 мг/дл.
16. Способ по п. 1 или 15, в котором тяжелая симптоматическая гипогликемия связана с острым неврологическим дефицитом.
17. Способ по п. 16, в котором острым неврологическим дефицитом является по меньшей мере острый неврологический дефицит, выбранный из сонливости, психических расстройств, нарушений зрения, транзиторных сенсорных нарушений, транзиторных двигательных нарушений, спутанности сознания, судорог и комы.
18. Фармацевтическая комбинация, содержащая
(а) desPro36Эксендин-4(1-39)-Lys6-NH2 и/или его фармацевтически приемлемую соль, и
(b) метформин и/или его фармацевтически приемлемую соль, для применения с целью предотвращения тяжелой симптоматической гипогликемии, связанной с концентрацией глюкозы в плазме ниже 50 мг/дл у пациента с сахарным диабетом 2 типа, при этом пациент, подлежащий лечению, имеет значение HbA1c, равное по меньшей мере 8%.
19. Применение комбинации
(a) desPro36Эксендин-4(1-39)-Lys6-NH2 и/или его фармацевтически приемлемой соли, и
(b) метформина и/или его фармацевтически приемлемой соли, для получения лекарства для предотвращения тяжелой симптоматической гипогликемии, связанной с концентрацией глюкозы в плазме ниже 50 мг/дл у пациентов с сахарным диабетом 2 типа, при этом пациент, подлежащий лечению, имеет значение НbА1c, составляющее по меньшей мере 8%.

