Настоящее изобретение относится к новым твердым формам лекарственного средства, к фармацевтическим композициям, содержащим их, и к способам их получения.
Желательно, чтобы в составе лекарственных композиций лекарственное вещество находилось в форме, в которой можно будет удобно обращаться с ним и его обрабатывать. Это важно не только с точки зрения получения коммерчески жизнеспособного способа изготовления, но также с точки зрения последующего изготовления фармацевтических композиций, содержащих активное соединение.
Химическая стабильность, стабильность твердого состояния и "срок хранения" активных ингредиентов также являются очень важными факторами. Лекарственное вещество и композиции, содержащие его, должны эффективно храниться без значительного изменения физико-химических характеристик активного компонента (например его химического состава, плотности, гигроскопичности и растворимости).
Кроме того, также желательно иметь возможность предлагать лекарственное средство в форме, в которой оно является настолько химически чистой, насколько это возможно.
Аморфные или полуаморфные вещества могут создавать значительные проблемы в этом отношении. Например, такие вещества обычно сложно обрабатывать и готовить в виде фармацевтического препарата, обеспечивают ненадежную растворимость и их часто находят нестабильными и химически загрязненными.
Специалисту очевидно, что, если лекарственное средство можно легко получать в стабильной кристаллической форме, то вышеуказанные проблемы могут быть решены.
Кроме того, было установлено, что кристаллические лекарственные соединения обеспечивают более достоверные и воспроизводимые профили концентраций в плазме после введения пациенту.
Таким образом, для изготовления коммерчески жизнеспособных и фармацевтически приемлемых композиций лекарственных средств желательно, где это возможно, предлагать лекарственное средство по существу в кристаллической и стабильной форме.
Следует отметить, однако, эта цель не всегда достижима. Действительно, обычно невозможно предсказать только по молекулярной структуре, какой будет характер кристаллизации соединения. Обычно это может быть определено только эмпирически.
В международной патентной заявке WO 2006/024823 раскрыт ряд производных пиримидинсульфонамида в качестве модуляторов хемокиновых рецепторов, включая конкретное соединение N-[2-[[(2,3-дифторфенил)метил]тио]-6-{[(1R,2S)-2,3-дигидрокси-1-метилпропил]окси}-4-пиримидинил]-1-азетидинсульфонамид (Пример 47):

также упоминаемое в данном описании изобретения как Соединение I.
В WO 2006/024823 раскрыт способ получения Соединения I, но не раскрыта какая-либо конкретная информация относительно кристаллических форм этого соединения.
Авторы изобретения обнаружили, что возможно получить стабильные кристаллические формы Соединения I или его фармацевтически приемлемой соли, такие кристаллические формы могут упоминаться в данном описании изобретения как "соединения по изобретению".
Согласно одному аспекту изобретения предлагается по существу кристаллическая форма Соединения I или его фармацевтически приемлемой соли (во избежание неопределенности, они представляют собой "соединения по изобретению").
В другом аспекте изобретения предлагается кристаллическая форма Соединения I или его фармацевтически приемлемая соль.
Согласно еще одному аспекту изобретения предлагается по существу кристаллическая ангидратная форма Соединения I (во избежание неопределенности, они представляют собой "соединения по изобретению"). В одном аспекте Соединение I не находится в форме соли. Кроме того, предпочтительно, чтобы оно не находилось в форме сольвата, то есть оно представляет собой "ансольват". Следовательно, термин "ангидрат" охватывает "ансольват".
Авторы изобретения обнаружили, что Соединение I может быть получено в формах, которые по своей сущности являются по существу кристаллическими. Хотя можно получать Соединение 1 в формах, которые являются кристаллическими более чем примерно на 90%, например более чем примерно на 95% (например более чем примерно на 98% кристаллическими и, особенно на 100%, или приблизительно на 100% кристаллическими), в "по существу кристаллические" авторы изобретения включают более чем примерно на 60%, в другом аспекте более чем примерно на 75% и в еще одном аспекте более чем примерно на 80% (например примерно на 90%) кристаллические. Степень (%) кристалличности может быть определена специалистом с использованием дифракции рентгеновских лучей на порошке (XRPD). Можно также использовать другие способы, такие как ЯМР твердого тела, FT-IR (инфракрасная спектроскопия с преобразованием Фурье), Рамановская спектроскопия, дифференциальная сканирующая калориметрия (DSC) микрокалориметрия и вычисления истинной плотности.
Подходящая кристаллическая модификация соединения по изобретению по существу не содержит других кристаллических модификаций соединения. Соответственно, описанная кристаллическая модификация соединения формулы I включает, например, менее 20%, 15%, 10%, 5%, 3% или особенно менее 1% масс. других кристаллических форм этого соединения.
Выше в данном описании изобретения указано, что Соединение I может быть получено в кристаллической форме, которая представляет собой ангидрат. Под этим термином авторы изобретения подразумевают, что кристаллическая форма содержит менее 10% (а) гидратной(ых) формы(м) (например моногидрата) Соединения I.
Две предпочтительные ангидратные кристаллические формы Соединения I могут быть охарактеризованы при помощи рентгеновской порошковой дифрактограммы с использованием длины волны рентгеновских лучей 1,5418 Å, содержащей следующие характеристические кристаллические пики с приблизительными величинами 2-тета (в градусах), а также указания относительной интенсивности этих пиков в скобках, где процент относительной интенсивности приблизительно 25-100% обозначен как "vs", приблизительно 10-25% обозначены как "s", приблизительно 3-10% обозначены как "m" и приблизительно 1-3% обозначены как "w":
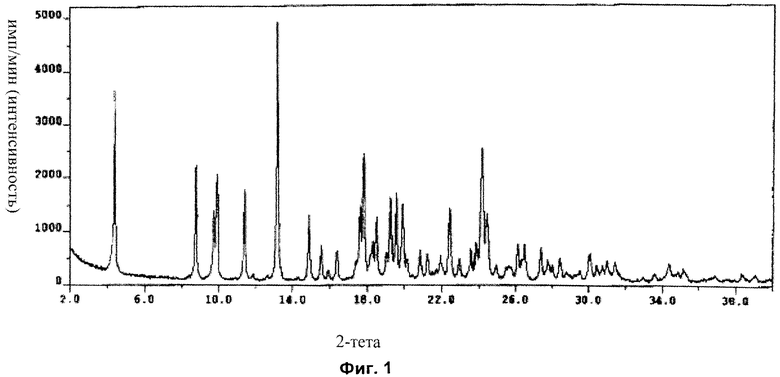
Форма А: характеристические кристаллические пики по меньшей мере с одним значением 2-тета (в градусах) около (то есть примерно или приблизительно) 14,8, 17,8 и/или 24,1. В одном аспекте характеристические кристаллические пики по меньшей мере с одним значением 2-тета (в градусах) 14,8, 17,8 и/или 24,1. В другом аспекте присутствуют все эти пики. В еще одном аспекте также содержится по меньшей мере один дополнительный кристаллический пик со значением 2-тета (в градусах) около (то есть примерно или приблизительно) 16,3, 15,5, 11,4, 9,9, 13,1 и/или 4,4. В еще одном аспекте присутствуют все вышеупомянутые пики. В еще одном аспекте также содержится по меньшей мере один дополнительный кристаллический пик со значением 2-тета (в градусах) 16,3, 15,5, 11,4, 9,9, 13,1 и/или 4,4. В другом аспекте присутствуют все вышеупомянутые пики.
В другом аспекте форма А имеет характеристические кристаллические пики по меньшей мере с одним из значений 2-тета (в градусах) около (то есть примерно или приблизительно) 14,8 (s), 17,8 (vs) и/или 24,1 (vs). В одном аспекте присутствуют все эти пики. В другом аспекте также содержится по меньшей мере один дополнительный кристаллический пик со значением 2-тета (в градусах) около (то есть примерно или приблизительно) 16,3 (s), 15,5 (s), 11,4 (vs), 9,9 (vs), 13,1 (vs) и/или 4,4 (vs). В другом аспекте присутствуют все эти пики. В одном аспекте форма содержит все характеристические пики (например с указанной относительной интенсивностью), как показано в Примере 1 ниже в данном описании изобретения и, следовательно, форма может быть охарактеризована рентгеновской порошковой дифрактограммой, которая является по существу такой, как показано на Фиг.1.
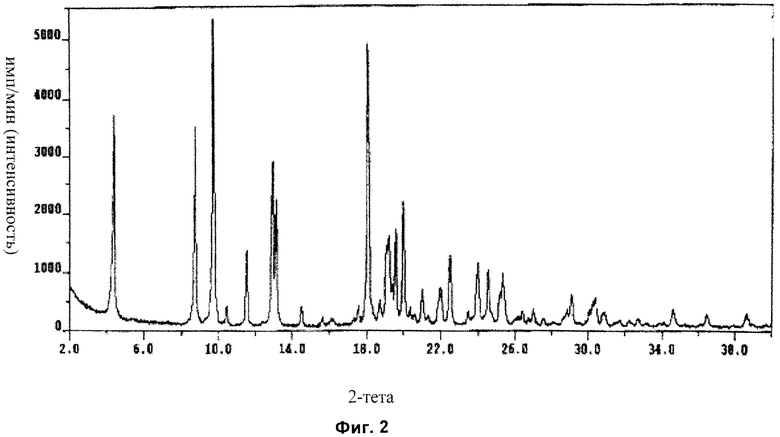
Форма D: характеристические кристаллические пики по меньшей мере с одним из значений 2-тета (в градусах) около (то есть примерно или приблизительно) 12,9, 18,0 и/или 21,0. В одном аспекте присутствуют все эти пики. В другом аспекте также содержится по меньшей мере один дополнительный кристаллический пик со значением 2-тета (в градусах) около (то есть примерно или приблизительно) 25,1, 25,3, 27,0 и/или 29,1. В еще одном аспекте присутствуют все эти пики.
В другом аспекте форма D имеет характеристические кристаллические пики по меньшей мере с одним из значений 2-тета (в градусах) около (то есть примерно или приблизительно) 12,9 (vs), 18,0 (vs) и/или 21,0 (s). В одном аспекте присутствуют все эти пики. В другом аспекте также содержится по меньшей мере один дополнительный кристаллический пик со значением 2-тета (в градусах) около (то есть примерно или приблизительно) 25,1 (s), 25,3 (s), 27,0 (s) и/или 29,1 (s). В одном аспекте присутствуют все эти пики, перечисленные для формы D. В другом аспекте форма содержит все характеристические пики (например с указанной относительной интенсивностью), как показано в Примере 2 ниже в данном описании изобретения, и, следовательно, форма может быть охарактеризована рентгеновской порошковой дифрактограммой, которая является по существу такой, как показано на Фиг.2.
В другом аспекте форма D характеризуется рентгеновской порошковой дифрактограммой, измеренной с использованием длины волны рентгеновских лучей 1,5418 Å и содержащей по меньшей мере 1 кристаллический пик со значением 2-тета (в градусах) 21,0, 28,8 и/или 29,1.
В другом аспекте форма D характеризуется рентгеновской порошковой дифрактограммой, измеренной с использованием длины волны рентгеновских лучей 1,5418 Å и содержащей по меньшей мере 2 кристаллических пика со значением 2-тета (в градусах) 21,0, 28,8 и/или 29,1.
В другом аспекте форма D характеризуется рентгеновской порошковой дифрактограммой, измеренной с использованием длины волны рентгеновских лучей 1,5418 Å и содержащей по меньшей мере 3 кристаллических пика со значением 2-тета (в градусах) 21,0, 28,8 и/или 29,1.
В другом аспекте форма D характеризуется рентгеновской порошковой дифрактограммой, измеренной с использованием длины волны рентгеновских лучей 1,5418 Å и содержащей дополнительный кристаллический пик со значением 2-тета (в градусах), выбранным из 12,9 и 18,0.
В другом аспекте форма D характеризуется рентгеновской порошковой дифрактограммой, измеренной с использованием длины волны рентгеновских лучей 1,5418 Å и содержащей кристаллические пики со значением 2-тета (в градусах) 12,9, 13,1, 18,0, 21,0, 22,5, 25,1, 25,3, 28,8, 29,1 и 30,4.
В одном аспекте соединения по изобретению являются по существу кристаллографически чистыми. В выражение "по существу кристаллографически чистый" авторы изобретения включают кристаллическую форму ангидрата Соединения I, насколько ее можно оценить посредством измерениям дифракции рентгеновских лучей на порошке (XRPD), которая содержит менее чем примерно 5%, в другом аспекте менее чем примерно 3% и в еще одном аспекте менее чем примерно 1% других кристаллических форм Соединения I (или другой ангидратной формы или иного, и оценивается по присутствию пиков XRPD таких других кристаллических форм).
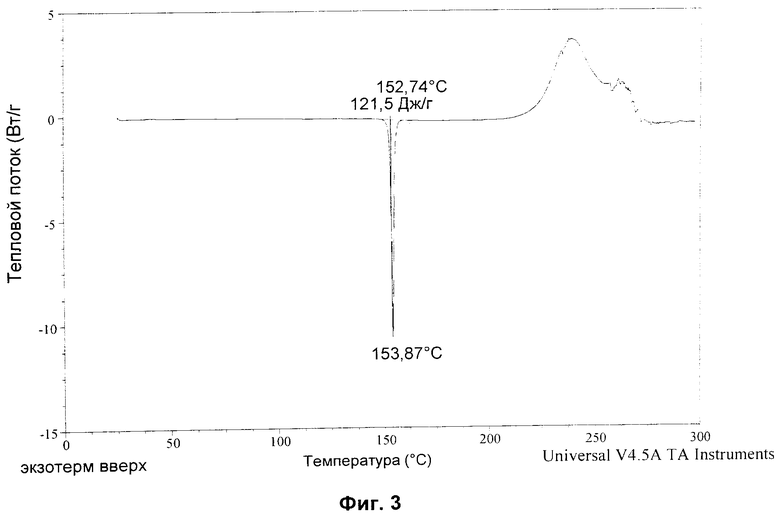
Анализ DSC показывает, что Форма D имеет начало плавления при 152,7°C. Термограмма DSC представлена на Фиг.3. В одном аспекте изобретение относится к кристаллической форме соединения формулы 1 с точкой плавления примерно 152,7°C (начало).
Авторы изобретения обнаружили, что для некоторых ангидратов Соединения I сушка растворителя (во время процесса кристаллизации) не является необходимой для того, чтобы обеспечить их образование. Однако для гарантии получения ангидрата, растворитель, из которого происходит кристаллизация, может быть высушен, либо до процесса кристаллизации, либо во время него, чтобы понизить содержание воды ниже критического уровня, который предпочтительно не следует превышать в ходе кристаллизации. Растворитель можно сушить во время процесса кристаллизации, например путем уменьшения содержания воды в смеси соединения, которое будет кристаллизоваться и подходящей системы органический растворитель/водный растворитель (например путем увеличения количества органического растворителя, который присутствует и/или путем удаления воды посредством образования азеотропной смеси с последующими перегонками).
Следовательно, ангидрат Соединения I может быть получен посредством кристаллизации из системы растворителей, которая по существу не содержит воды.
В выражение "по существу не содержит воды" авторы изобретения включают то, что содержание воды в системе растворителей ниже уровня, который приводит к образованию не более 10% моногидрата для любой конкретной системы растворителей и набора условий кристаллизации.
Кристаллическая Форма D ангидрата Соединения I может быть получена путем суспендирования Соединения I (например в аморфной форме или в другой кристаллической форме, такой как Форма А) в системе растворителей. Следовательно предлагается кристаллическая форма, получаемая таким способом преобразования (кристаллизацией). Специалисту понятно, что способ суспендирования представляет собой по существу способ "образования взвеси" или способ, который включает по меньшей мере частичное (но не полное) растворение в системе растворителей.
Следовательно, в одном аспекте изобретения предлагается превращение одной кристаллической формы (например одной ангидратной формы) Соединения I в другую. В частности, Форма А (также упоминаемая в данном описании изобретения как А-форма) может быть превращена в Форму D (также упоминаемую в данном описании изобретения как D-форма). Следовательно, опять же предлагается кристаллическая форма, получаемая таким способом преобразования (кристаллизацией).
Для получения D-формы, А-форма может быть суспендирована или образовать взвесь (или по меньшей мере частично раствориться) в системе растворителей, которая не содействует образованию сольватной формы Соединения I.
Термины "суспендированный" и "образующий взвесь" (или "частично растворенный") хорошо понятны специалисту. Например, для образования суспензии или взвеси, добавляют избыток твердого вещества относительно его растворимости в растворителе, так что (нерастворенное) твердое вещество присутствует в системе растворителей в течение процесса "суспендирования" или "образования взвеси". Поэтому его также называют в данном описании изобретения "частичным растворением".
Предпочтительные системы растворителей, используемые для получения D-формы посредством суспендирования или образования взвеси (то есть для достижения превращения, например, аморфного Соединения I или А-формы в D-форму), включают любой подходящий растворитель (также называемый в данном описании изобретения "растворителем для суспензии") или смесь растворителей, которые не приводят к образованию сольвата Соединения I. Предпочтительные системы растворителей могут включать системы, в которых Соединение I только частично (или по меньшей мере частично) растворимо. В одном аспекте система растворителей содержит (или, в другом аспекте, по существу состоит из) органические(их) растворители(ей), которые являются полярными, например спирты (такие как низшие алкильные спирты, например C1-6спирт). В другом аспекте система растворителей содержит (или, в другом аспекте, по существу состоит из) этанол(а) или, особенно, метанол(а). Следовательно, вышеупомянутые полярные органические растворители являются особенно предпочтительными растворителями для суспендирования, используемыми в системе растворителей (и в другом аспекте система растворителей состоит главным образом или по существу из таких растворителей для суспендирования). В одном аспекте растворитель для суспендирования (например спирт, такой как метанол) составляет по меньшей мере 90% масс./масс. (например по меньшей мере 95%, например приблизительно 100%) всей системы растворителей, которую применяют для получения D-формы. То есть растворитель для суспендирования может содержать вплоть до 10% масс./масс. (например вплоть до 5% или примерно 0%) других (нежелательных или менее желательных) растворителей.
Фазовое превращение в системе растворителей (включая растворитель для суспендирования, который описан выше в данном описании изобретения) для получения D-формы может занимать период длительностью несколько недель (например шесть недель; смотри Пример ниже в данном описании изобретения), но продолжительность времени можно уменьшать в зависимости от температуры способа (или его можно сделать продолжительнее, если выполнять при более низких температурах) и так далее. Однако специалист легко может определить продолжительность времени, необходимого для превращения в D-форму. Кроме того, D-форму можно получать, используя затравку, например, как описано ниже в данном описании изобретения.
А-форма Соединения I может кристаллизоваться из аморфной формы Соединения I в смеси, содержащей конкретный растворитель (например ацетонитрил), неорганическую кислоту (например фосфорную кислоту) и воду, где смесь может быть нагрета и затем охлаждена для содействия кристаллизации, как описано ниже в данном описании изобретения (смотри, например, Пример 2).
А-форма Соединения I может кристаллизоваться из N-[2-[[(2,3-дифторфенил)метил]тио]-6-[(1R)-1-[(4S)-2,2-диметил-1,3-диоксолан-4-ил]этокси]-4-пиримидинил]-1-азетидин-сульфонамида в смеси, содержащей конкретный растворитель (например ацетонитрил), неорганическую кислоту (например фосфорную кислоту) и воду, где смесь может быть нагрета и затем охлаждена для содействия кристаллизации, как описано ниже в данном описании изобретения (смотри например Пример 2).
Кристаллизации, описанные в данном описании изобретения, можно стимулировать добавлением затравочных кристаллов (если они доступны).
Например, D-форму можно также получать при помощи затравки, используя следующую процедуру: растворение Соединения I (например один "массовый" %) в растворителе (таком как спирт, например изопропанол; например 30 относительных объемов); перемешивание при повышенной температуре (например 70°C) в течение некоторого периода времени (который может составлять несколько часов) до достижения полного растворения; продолжение перемешивания при повышенной температуре (например 55°C; то есть при температуре ниже той, которая требуется для достижения растворения) в течение периода времени, например нескольких часов (например в течение ночи); использование затравки с Формой D Соединения I (например 0,1 масс.%); охлаждение до более низкой температуры (например 20°C); и фильтрование с получением D-формы.
Альтернативно, D-форму также можно получать при помощи затравки, используя следующую процедуру: растворение Соединения I (например один относительный "массовый" %) в растворителе (таком как спирт, например этанол; например 15 относительных объемов); перемешивание при повышенной температуре (например 65°C), например в течение периода времени (который может составлять несколько часов) до достижения полного растворения; продолжение перемешивания при повышенной температуре (например 55°C; то есть при температуре ниже той, которая требуется для достижения растворения) в течение некоторого периода времени, например нескольких часов (например в течение ночи); использование затравки с Формой D Соединения I (например 0,1 масс.%); охлаждение до более низкой температуры (например 20°C); и фильтрование с получением D-формы.
Для подтверждения того, что кристаллические формы, как описано в данном описании изобретения, получают в отсутствие других кристаллических форм, кристаллизации можно выполнять с использованием затравки с зародышами кристаллизации и/или затравочными кристаллами целевой кристаллической формы в отсутствие зародышей кристаллизации и/или затравочных кристаллов других кристаллических форм.
Специалисту очевидно, что концентрация в растворе соединения, которое следует кристаллизировать, и система растворителей, которую используют, может оказывать влияние на температуры кристаллизации и продолжительность кристаллизации.
Разные кристаллические формы могут иметь разную растворимость в разных органических растворителях при любой заданной температуре. В этом отношении вышеупомянутые или другие растворители могут быть использованы в качестве "антирастворителей" (то есть растворитель, в котором соединения по изобретению плохо растворимы, но который смешивается с другим растворителем, в котором соединения по изобретению более растворимы), и могут, таким образом, способствовать способу кристаллизации.
Специалисту очевидно, что получаемая кристаллическая форма зависит как от кинетики, так и от термодинамики кристаллизационного процесса. В определенных термодинамических условиях (система растворителей, температура, давление и концентрация соединения по изобретению) одна кристаллическая форма может быть более стабильной, чем другая (или даже любая другая). Однако другие кристаллические формы, которые могут иметь сравнительно относительно низкую термодинамическую стабильность, могут быть кинетически предпочтительными. Таким образом, дополнительно кинетические факторы, такие как время, состав примесей, взбалтывание, присутствие затравок и так далее, также могут оказывать влияние на то, какие формы появятся. Таким образом, способы, рассмотренные в данном описании изобретения, при необходимости могут быть адаптированы специалистом с целью получения конкретной кристаллической формы Соединения I (например А-формы или D-формы).
Кроме того, температура сушки и время сушки могут оказывать влияние на свойства твердого состояния и/или на твердую форму соединений по изобретению. Например, дегидратация может происходить при низкой влажности, и/или при повышенных температурах, и/или при пониженном давлении. Следовательно, кристаллические ангидраты соединений по изобретению также могут быть образованы посредством дегидратации гидрата.
Как указано выше в данном описании изобретения, предпочтительные соединения по изобретению также могут характеризоваться рентгеновской порошковой дифрактограммой, которая по существу соответствует показанной на прилагаемых Фиг.1 или Фиг.2, и/или такой, как представлено в Таблице 1 или Таблице 2 ниже в данном описании изобретения (смотри Примеры 1 и 2). Специалисту очевидно, что форма кристаллической ангидратной формы Соединения I показывает "по существу" такую же рентгеновскую порошковую дифрактограмму, как другая, когда этому специалисту очевидно из соответствующих диаграмм (то есть относительного расположение пиков, допускающего ошибку эксперимента, такого как предпочтительная ориентация образца и соответствующие настройки инструмента (например тип аппарата, стандартизация и/или калибровка)), что образовалась та же самая кристаллическая форма. Таким образом, может иметь место некоторая ошибка эксперимента для величины 2°тета, как может быть указано в данном описании изобретения (например изменение вплоть до ±0,5°2-тета).
Авторы изобретения обнаружили, что соединения по изобретению имеют неожиданно улучшенную физическую и/или химическую стабильность по сравнению с другими формами Соединения I, которые могли быть получены ранее.
Термин "стабильный", как определено в данном описании изобретения, включает химическую стабильность и стабильность твердого состояния.
В выражение "химическая стабильность" авторы изобретения включают то, что соединение можно хранить в выделенной твердой форме или в форме твердой композиции, в которой оно может быть представлено в смеси с фармацевтически приемлемыми носителями, разбавителями или вспомогательными веществами, в условиях нормального хранения, с незначительной степенью химического разрушения или разделения.
В выражение "стабильность твердого состояния" авторы изобретения включают то, что соединение можно хранить в выделенной твердой форме или в форме твердой композиции, в которой оно может быть представлено в смеси с фармацевтически приемлемыми носителями, разбавителями или вспомогательными веществами, в нормальных условиях хранения, с незначительной степенью изменения твердого состояния (например кристаллизации, рекристаллизации, потери кристалличности, твердофазного перехода, гидратации, дегидратации, сольватации или десольватации).
Примеры "нормальных условий хранения" включают температуры от минус 80 до плюс 50°C (в одном аспекте от 0 до 40°C, и в другом аспекте при температуре окружающей среды, такой как от 15 до 30°C), давления от 0,1 до 2 бар (от 104 до 2×105 Па) (в одном аспекте при атмосферном давлении) и/или влияние 460 люкс Уф/видимого света в течение длительных периодов (то есть больше или равных шести месяцам). При таких условиях можно обнаружить, что соединения по изобретение являются менее чем на примерно 15%, в одном аспекте менее чем на примерно 10% и в еще одном аспекте менее чем на примерно 5% химически разрушившимися/разложившимися, или имеют измененное твердое состояние, по обстоятельствам. Специалисту очевидно, что указанные выше верхний и нижний пределы температуры и давления означают предельные значения нормальных условий хранения и что некоторые комбинации этих предельных значений не будут встречаться во время нормального хранения (например температура 50°C и давление 0,1 бар (104 Па).
Термин "нормальные условия хранения" может также включать относительную влажность от 5 до 95% (в одном аспекте от 10 до 60%). Однако в случае некоторых кристаллических форм согласно изобретению изменения конформации или кристаллической структуры при гидратации и/или дегидратации могут происходить в результате продолжительного воздействия некоторых предельных значений относительной влажности при нормальных температурах/давлении.
Получение и характеристика соединений по изобретению описаны ниже в данном описании изобретения. Разные кристаллические формы соединений по изобретение могут быть легко охарактеризованы с использованием методов дифракции рентгеновских лучей на порошке (XRPD), например, как описано ниже в данном описании изобретения.
Соединения по изобретению могут быть выделены с использованием методов, которые хорошо известны специалисту в данной области техники, например декантирования, фильтрования и/или центрифугирования.
Авторы изобретения обнаружили что, применяя способы кристаллизации или превращения, описанные в данном описании изобретения, можно получать соединения по изобретению с высокой химической чистотой.
При получении соединений по изобретению, как описано в данном описании изобретения, полученное соединение находится в форме, которая имеет улучшенную химическую стабильность и стабильность твердого состояния, как указано выше в данном описании изобретения, а также улучшенные профили растворимости и гигроскопичности по сравнению с другими ранее известными формами.
Хотя в одном аспекте соединения по изобретению (то есть кристаллические формы) не находятся в форме солей, соли, которые могут быть указаны, включают соли присоединения кислоты и соли присоединения основания.
Фармацевтические препараты и медицинское применение
Соединения по изобретению являются полезными, так как они обладают фармакологической активностью. Следовательно, они указаны как фармацевтические средства.
В частности, соединения по изобретению находят применение в лечении заболеваний/состояний, где полезно модулирование активности хемокиновых рецепторов, особенно CXCR2.
Термин "модулирование" может относиться к любому измеримому снижению и/или предупреждению релевантной активности (активности в отношении хемокинового рецептора). Модулирование активности хемокинового рецептора можно измерять путем сравнения активности хемокинового рецептора в образце, содержащем соединение по изобретению, и в образце в отсутствие соединения по изобретению (как будет очевидно специалисту в данной области техники). Измеримое изменение может быть объективным (например измеримым при помощи какого-то теста или маркера, например в анализе или тесте in vitro или in vivo, таком как описано ниже в данном описании изобретения, или же посредством другого подходящего анализа или теста, известного специалисту в данной области техники) или субъективным (например субъект указывает на эффект или ощущает эффект).
Соединение формулы (1) или фармацевтически приемлемые соли могут быть полезными в лечении (терапевтическом или профилактическом) состояний/заболеваний человека и животных, не являющихся человеком, которые обостряются или вызываются избыточным или нерегулируемым продуцированном хемокинов. Примеры таких состояний/заболеваний включают (каждое взято независимо):
(1) дыхательные пути - обструктивные заболевания дыхательных путей, включая хроническую обструктивную болезнь легких (COPD); астму, такую как бронхиальная, аллергическая, наследственная, приобретенная и пылевая астма, в частности хроническая или застарелая астма (например поздняя астма и гиперактивность дыхательных путей); бронхит; острый, аллергический, атрофический ринит и хронический ринит, включающий хронический ринит с образованием кавеозных масс, гипертрофический ринит, гнойный ринит, сухой ринит и медикаментозный ринит; бронхоэктаз; мембранозный ринит, включающий крупозный, фибринозный и псевдомембранозный ринит, и скрофулезный ринит; сезонный ринит, включающий rhinitis nervosa (сенную лихорадку) и вазомоторный ринит; саркоидоз, аллергический альвеолит у сельскохозяйственных рабочих и родственные заболевания, пневмофиброз и идиопатическая интерстициальная пневмония;
(2) кости и суставы - ревматоидный артрит, серонегативные спондилоартропатии (включая анкилозирующий спондилит, псориатический артрит и синдром Рейтера), болезнь Бехчета, синдром Шегрена и системный склероз;
(3) кожа - псориаз, атопический дерматит, контактный дерматит и другие экзематозные дерматиты, себорейный дерматит, красный плоский лишай, обыкновенная пузырчатка, буллезная пузырчатка, буллезный эпидермолиз, крапивница, ангиодермит, васкулит, эритема, подкожная эозинофилия, увеит, очаговая алопеция и весенний конъюктивит;
(4) желудочно-кишечный тракт - глютеновая болезнь, проктит, эозинофильный гастроэнтерит, мастоцитоз, болезнь Крона, неспецифический язвенный колит, недифференцированный колит, микроскопический колит, воспалительная болезнь кишечника, синдром раздраженной толстой кишки, невоспалительная диарея, пищевые аллергии, которые оказывают воздействия, отдаленные от пищеварительного тракта, например мигрень, ринит и экзема;
(5) центральная и периферическая нервная система нейродегенеративные заболевания и дементные расстройства, например болезнь Альцгеймера, амиотрофический боковой склероз и другие заболевания двигательных нейронов, болезнь Крейтцфельдта-Якоба и другие прионные заболевания, ВИЧ-энцефалопатия (комплекс СПИД-деменция), заболевание Гентингтона, лобно-височная деменция, деменция с тельцами Леви и сосудистая деменция; полиневропатии, например синдром Гийена-Барре, хроническая воспалительная демиелинизирующая полирадикулоневропатия, мультифокальная моторная невропатия, плексопатии; демиелинизация ЦНС, например рассеянный склероз, острый диссеминированный/геморрагический энцефаломиелит, и подострый склерозирующий панэнцефалит; нейромышечные расстройства, например тяжелая псевдопаралитическая миастения и синдром Ламберта-Итона; спинальные расстройства, например тропический спастический парапарез и синдром мышечной скованности: паранеопластические синдромы, например мозжечковая дегенерация и энцефаломиелит; травма ЦНС; мигрень; и инсульт.
(6) другое тканевое и системное заболевание - атеросклероз, синдром приобретенного иммунодефицита (СПИД), эритематозная волчанка, системная волчанка, эритематоз, тиреоидит Хашимото, диабет 1 типа, нефротический синдром, эозинофилия, фасцит, синдром гипер-lgE, лепроматозная проказа и идиопатическая тромбоцитопеническая пурпура; послеоперационные спайки и сепсис.
(7) отторжение аллотрансплантата - острое и хроническое после, например, пересадки почки, сердца, печени, легкого, костного мозга, кожи и роговицы; и хроническая реакция «трансплантат против хозяина»;
(8) раковые заболевания - особенно немелкоклеточный рак легкого (NSCLC), злокачественная меланома, рак предстательной железы и сквамозная саркома, и метастазирование опухоли, немеланомный рак кожи и химиопрофилактика метастазов;
(9) заболевания - при которых ангиогенез ассоциирован с повышенными уровнями хемокинов CXCR2 (например NSCLC, диабетическая ретинопатия);
(10) муковисцидоз;
(11) ожоговые раны и хронические кожные язвы;
(12) заболевания репродуктивной системы- например расстройства овуляции, менструации и имплантации, преждевременные роды, эндометриоз;
(13) реперфузионное повреждение - в сердце, головном мозге, в периферических конечностей и других органах, ингибирование атеросклероза.
Таким образом, в настоящем изобретении предлагается соединение формулы (1) или фармацевтически приемлемая соль, как определено выше в данном описании изобретения, для применения в терапии.
Соединения по изобретению можно применять для лечения заболеваний, в которых хемокиновый рецептор представляет собой рецептор CXCR2,
Конкретные состояния, которые можно лечить соединениями по изобретению, представляют собой рак, заболевания, при которых ангиогенез ассоциирован с повышенными уровнями CXCR2 хемокинов, и воспалительные заболевания, такие как астма, аллергический ринит, COPD, бронхоэктаз, ревматоидный артрит, псориаз, воспалительная болезнь кишечника, остеоартрит или остеопороз.
В другом аспекте конкретные состояния, которые можно лечить соединениями по изобретению, представляют собой астму, COPD и бронхоэктаз.
В качестве еще одного аспекта настоящего изобретения соединение формулы (1) может применяться в качестве антагонистов рецептора CX3CR1. Предполагается, что такие соединения будут особенно полезными в лечении расстройств центральной и периферической нервной системы и других состояний, характеризующихся активацией микроглии и/или инфильтрацией лейкоцитов (например инсульт/ишемия и травма головы). В частности, соединения показаны для применения в лечении нейродегенеративных расстройств или демиелинизирующего заболевания у млекопитающих, включая человека. Более конкретно, соединения показаны для применения в лечении рассеянного склероза. Соединения также показаны как полезные в лечении боли, ревматоидного артрита, остеоартрита, инсульта, атеросклероза и артериальной легочной гипертензии.
В еще одном аспекте в настоящем изобретении предлагается соединение формулы (1) или фармацевтически приемлемая соль, как определено выше в данном описании изобретения, для применения в качестве лекарственного средства.
В еще одном аспекте в настоящем изобретении предлагается применение соединения формулы (1) или фармацевтически приемлемой соли, как определено выше в данном описании изобретения, для применения в качестве лекарственного средства для лечения заболеваний или состояний человека, для которых модулирование активности хемокинового рецептора является полезным.
В еще одном аспекте в настоящем. изобретении предлагается применение соединения формулы (1) или фармацевтически приемлемой соли, как определено выше в данном описании изобретения, для применения в качестве лекарственного средства для лечения астмы, аллергического ринита, ракового заболевания, COPD, ревматоидного артрита, псориаза, воспалительных заболеваний кишечника, остеоартрита или остеопороза.
В еще одном аспекте в настоящем изобретении предлагается применение соединения формулы (1) или фармацевтически приемлемой соли, как определено выше в данном описании изобретения, в изготовлении лекарственного средства для применения в терапии.
В еще одном аспекте в настоящем изобретении предлагается применение соединения формулы (1) или фармацевтически приемлемой соли, как определено выше в данном описании изобретения, в изготовлении лекарственного средства для лечения заболеваний или состояний человека, для которых модулирование активности хемокинового рецептора является полезным.
В еще одном аспекте в настоящем изобретении предлагается применение соединения формулы (1) или фармацевтически приемлемой соли, как определено выше в данном описании изобретения, в изготовлении лекарственного средства для лечения астмы, аллергического ринита, ракового заболевания, COPD, ревматоидного артрита, псориаза, воспалительных заболеваний кишечника, остеоартрита или остеопороза.
В контексте настоящего описания изобретения термин «терапия» также включает «профилактику», если нет конкретных указаний на обратное. Термины «терапевтический» и «терапевтически» следует истолковывать соответственно.
В изобретении также предлагается способ лечения опосредованного хемокинами заболевания, где хемокин связывается с хемокиновым рецептором (особенно CXCR2), включающий введение пациенту терапевтически эффективного количества соединения формулы (1) или фармацевтически приемлемой соли, как определено выше в данном описании изобретения.
В изобретении также предлагается способ лечения воспалительного заболевания, особенно астмы, аллергического ринита, COPD, ревматоидного артрита, псориаза, воспалительных заболеваний кишечника, остеоартрита или остеопороза, у пациента, страдающего от, или имеющего риск указанного заболевания, включающий введение пациенту терапевтически эффективного количества соединения формулы (1) или фармацевтически приемлемой соли, как определено выше в данном описании изобретения.
Более конкретно, соединения по изобретению могут быть полезными в лечении астмы, аллергического ринита, COPD, воспалительных заболеваний кишечника, синдрома раздраженной толстой кишки, остеоартрита, остеопороза, ревматоидного артрита, псориаза или рака.
Соединения по изобретению показаны как в терапевтическом, так и/или в профилактическом лечении вышеупомянутых состояний.
Специалисту в данной области техники понятно, что термин "атеросклероз" включают любое заболевание, характеризующееся накоплением холестерина, образованием пенистых клеток, воспалением и пролиферацией клеток в кровеносном сосуде, особенно в стенке артерии.
Согласно еще одному аспекту в изобретении предлагается способ лечения заболевания/состояния, для которого модулирование активности хемокинового рецептора является полезным (например конкретного заболевания/состояния, указанного в данном описании изобретения), включающий введение соединения по изобретению пациенту, нуждающемуся в таком лечении.
"Пациенты" включают млекопитающих пациентов (включая человека). Следовательно, способ лечения, рассмотренный выше, может включать лечение организма человека или животного.
Термин "эффективное количество" относится к количеству соединения, которое оказывает терапевтический эффект на лечащегося пациента. Этот эффект может быть объективным (например измеряемым посредством какого-либо теста или маркера) или субъективным (например человек сообщает о признаке или ощущениях эффекта).
Соединения по изобретению можно вводить перорально, внутривенно, подкожно, буккально, ректально, дермально, назально, трахеально, бронхиально, сублингвально, любым другим парентеральным путем или посредством ингаляции, в фармацевтически приемлемой лекарственной форме. Например, фармацевтические композиции можно вводить местно (например в легкие, и/или в дыхательные пути, или в кожу) в форме растворов, суспензий, гептафторалкановых аэрозолей и сухих порошковых композиций;
или системно, например посредством перорального введения в форме таблеток, капсул, сиропов, порошков или гранул, или посредством парентерального введения в форме растворов или суспензий, или посредством подкожного введения, или посредством ректального введения в форме суппозиториев или трансдермально. В одном аспекте соединения по изобретению вводят перорально.
Соединения по изобретению можно вводить сами по себе, но в одном аспекте изобретения их вводят в виде известных фармацевтических препаратов, включающих таблетки, капсулы или эликсиры для перорального введения, суппозитории для ректального введения, стерильные растворы или суспензии для парентерального или внутримышечного введения и тому подобное. Тип фармацевтической композиции может быть выбран с должным учетом предполагаемого пути введения и стандартной фармацевтической практики. Такие фармацевтически приемлемые носители могут быть химически инертными к активным соединениям и могут не иметь вредных побочных эффектов или токсичности в условиях применения.
Такие композиции можно получать в соответствии с стандартной и/или общепринятой фармацевтической практикой. Или же получение подходящих композиций может быть выполнено специалистом без изобретательского подхода, с применением обычных операций и/или в соответствии с стандартной и/или общепринятой фармацевтической практикой.
Таким образом, согласно еще одному аспекту изобретения предлагается фармацевтическая композиция, включающая соединение по изобретению, как определено выше в данном описании изобретения, в смеси с фармацевтически приемлемым вспомогательным веществом, разбавителем и/или носителем, Такие композиции можно вводить, как описано выше в данном описании изобретения. Соединение по изобретению (то есть кристаллическая форма), которая является активным ингредиентом фармацевтической композиции, может быть измельчена или размолота на более мелкие частицы.
В зависимости, например, от эффективности и физических характеристик соединения по изобретению (то есть активного ингредиента), фармацевтические композиции, которые можно упомянуть, включают те, в которых активный ингредиент (то есть соединение по изобретению) присутствует в количестве по меньшей мере 1% масс.(или по меньшей мере 10%, по меньшей мере 30% или по меньшей мере 50% масс.). То есть отношение активного ингредиента к другим компонентам (то есть добавление вспомогательного вещества, разбавителя и носителя) фармацевтической композиции составляет по меньшей мере 1:99 (или по меньшей мере 10:90, по меньшей мере 30:70 или по меньшей мере 50:50) по массе.
Количество соединения по изобретению в композиции будет зависеть от тяжести состояния и от пациента, подлежащего лечению, а также соединения(ий), которое(ые) применяют, но может быть определено специалистом без изобретательского подхода.
В изобретении также предлагается способ получения фармацевтической композиции, как определено выше в данном описании изобретения, включающий объединение соединения по изобретению, как определено выше в данном описании изобретения, с фармацевтически приемлемым вспомогательным веществом, разбавителем или носителем.
Соединения по изобретению можно также комбинировать с другими терапевтическими агентами, например с теми, которые также являются полезными в лечении заболевания/состояния, для которого модулирование активности хемокинового рецептора является полезным (например заболеваний/состояний, указанных в данном описании изобретения). Соединения по изобретению также можно комбинировать с другими терапиями.
Согласно еще одному аспекту изобретения предлагается комбинированный продукт, содержащий:
(A) соединение по изобретению, как определено выше в данном описании изобретения; и
(B) другой терапевтический агент, который является полезным в лечении заболевания/состояния, для которого модулирование активности хемокинового рецептора является полезным (например заболевание/состояние, описанное в данном описании изобретения), где каждый из компонентов (А) и (В) готовят в виде фармацевтического препарата в смеси с фармацевтически приемлемым вспомогательным веществом, разбавителем или носителем.
Такие комбинированные продукты предлагают для введения соединения по изобретению в сочетании с другим терапевтическим агентом, и они могут, таким образом, быть представлены либо в виде отдельных композиций, где по меньшей мере одна из этих композиций содержит соединение по изобретению, и по меньшей мере одна содержит другой терапевтический агент, либо они могут быть представлены (то есть изготовлены в виде препарата) в виде комбинированного препарата (то есть представлены в виде одной композиции, включающей соединение по изобретению и другой терапевтический агент). Таким образом, также предлагается:
(1) фармацевтическая композиция, включающая соединение по изобретению, как определено выше в данном описании изобретения, другой терапевтический агент, который является полезным в лечении заболевания/состояния, для которого модулирование активности хемокинового рецептора является полезным, и фармацевтически приемлемое вспомогательное вещество, разбавитель или носитель; и
(2) набор, содержащий компоненты:
(а) фармацевтическую композицию, включающую соединение по изобретению, как определено выше в данном описании изобретения, в смеси с фармацевтически приемлемым вспомогательным веществом, разбавителем или носителем; и
(б) фармацевтическую композицию, включающую другой терапевтический агент, который является полезным в лечении заболевания/состояния, для которого модулирование активности хемокинового рецептора является полезным, в смеси с фармацевтически приемлемым вспомогательным веществом, разбавителем или носителем, где каждый из компонентов (а) и (б) предлагается в форме, которая является подходящей для введения в сочетании с другой.
В изобретении также предлагается способ получения комбинированного продукта, как определено выше в данном описании изобретения, включающий объединение соединения по изобретению, как определено выше в данном описании изобретения, с другим терапевтическим агентом, который является полезным в лечении рака и/или пролиферативного заболевания, и по меньшей мере с одним фармацевтически приемлемым вспомогательным веществом, разбавителем или носителем.
Под "объединением" авторы изобретения подразумевают, что два компонента приводят в состояние, подходящее для введения в сочетании друг с другом.
Таким образом, в отношении способа получения набора компонентов, как определено выше в данном описании изобретения, под "объединением" двух компонентов друг с другом, авторы изобретения подразумевают, что эти два компонента набора могут быть:
(1) представлены в виде отдельных композиций (то есть независимо один от другого), которые затем берут вместе для применения в сочетании друг с другом в комбинированной терапии; или
(2) упакованы и представлены вместе в виде отдельных компонентов "комбинированной упаковки" для применения в сочетании друг с другом в комбинированной терапии.
В зависимости от расстройства и пациента, подлежащего лечению, а также пути введения, соединения по изобретению можно вводить в различных терапевтически эффективных дозах пациенту, нуждающемуся в этом. Однако доза, введенная млекопитающему, особенно человеку, в контексте настоящего изобретения, должна быть достаточной для того, чтобы вызвать терапевтический ответа у млекопитающего в пределах приемлемого периода времени. Специалист в данной области понимает, что на выбор точной дозы, композиции и наиболее подходящего режима доставки также будут оказывать влияние, среди прочего, фармакологические свойства композиции, природа и тяжесть состояния, подлежащего лечению, и физическое состояние и ясность ума пациента, а также эффективность конкретного соединения, возраст, состояние, масса тела, пол и ответ пациента, подлежащего лечению, и стадия/тяжесть заболевания.
Введение может быть непрерывным или прерывистым (например посредством болюсной инъекции). Дозировка также может определяться по времени и частоте введения. В случае перорального или парентерального введения дозировка может варьироваться от примерно 0,01 мг до примерно 1000 мг соединения по изобретению в сутки.
В любом случае практикующий врач или другой специалист, легко может определить фактическую дозировку, которая будет наиболее подходящей для отдельного пациента. Вышеуказанные дозировки являются примерными для усредненного случая; могут, конечно, быть отдельные случаи, когда полезны более высокие или более низкие диапазоны дозировок, и таковые входят в объем данного изобретения.
Где бы слово "примерно" не использовалось в данном описании изобретения, например в контексте количеств (например значений, масс, объемов, молей), температур, степеней кристалличности, степеней разрушений, степеней чистоты, степеней растворения и доз активных ингредиентов, будет очевидно, что такие переменные являются примерными и как таковые могут варьироваться на ±10%, например ±5%, ±2%, или ±1% от чисел, указанных в данном описании изобретения.
Соединения по изобретению обладают тем преимуществом, что они находятся в форме, которая обеспечивает улучшенное удобство в обращении, и могут быть получены в формах, которые имеют улучшенную химическую стабильность и стабильность твердого состояния по сравнению с формами, полученными ранее. Таким образом, соединения могут быть стабильными при хранении в течение продолжительных периодов. В частности, D-форма (смотри Пример 2 ниже в данном описании изобретения) может обладать улучшенной термодинамической стабильностью по сравнению с формами Соединения I, полученными ранее.
Соединения по изобретению также имеют улучшенные профили растворимости и гигроскопичности по сравнению с ранее доступными формами.
Соединения по изобретению могут также обладать тем преимуществом, что они могут быть получены с хорошими выходами, с более высокой чистотой, в течение меньшего времени, более удобно и с более низкой стоимостью, чем формы, полученные ранее.
Соединения по изобретению могут также обладать тем преимуществом, что они могут быть более эффективными, менее токсичными, иметь более пролонгированное действие, быть более эффективными, вызывать меньше побочных эффектов, легче абсорбироваться, и/или иметь лучший фармакокинетический профиль (например более высокую пероральную биодоступность и/или более низкий клиренс), и/или иметь другие полезные фармакологические, физические или химические свойства по сравнению с известными соединениями (например ранее известными формами Соединения I), как для применения в вышеуказанных показаниях, так и для иного.
Изобретение проиллюстрировано, но без ограничения ими, следующими примерами со ссылкой на прилагаемые фигуры, где:
На Фиг.1 показана рентгеновская порошковая дифрактограмма кристаллической формы А ангидрата Соединения I с использованием длины волны рентгеновских лучей 1,5418 Å, полученная посредством Примера 1 (величину импульсов/с (интенсивности) наносят на график в зависимости от значений °2-тета).
На Фиг.2 показана рентгеновская порошковая дифрактограмма кристаллической формы D ангидрата Соединения I с использованием длины волны рентгеновских лучей 1,5418 Å, полученная посредством Примера 2 (величину импульсов/с (интенсивности) наносят на график в зависимости от значений °2-тета).
На Фиг.3 показана DSC кристаллической формы ангидрата Соединения I, полученного посредством Примера 2.
Общая методика Описание способа дифракции рентгеновских лучей на порошке
Известно, что можно получить рентгеновскую порошковую дифрактограмму, которая имеет одну или более ошибок измерения в зависимости от условий измерения (таких как используемый прибор или устройство). В частности, общеизвестно, что интенсивности на рентгеновской порошковой дифрактограмме могут колебаться в зависимости от условий измерения. Следовательно, следует понимать, что Формы А и D по настоящему изобретению не ограничиваются кристаллами, которые демонстрируют рентгеновские порошковые дифрактограммы идентичные рентгеновской порошковой дифрактограмме, показанной на Фиг.1 и 2, и любые кристаллы, демонстрирующие рентгеновские порошковые дифрактограммы по существу такие же, как показано на Фиг.1 и 2, входят в объем настоящего изобретения. Специалист в области дифракции рентгеновских лучей на порошке способен сделать вывод об идентичности по существу рентгеновских порошковых дифрактограмм.
Специалист в области дифракции рентгеновских лучей на порошке понимает, что на относительную интенсивность пиков могут влиять, например, крупинки размером более 30 микрон и неодинаковые соотношения геометрических размеров, которые могут влиять на анализ образцов. Специалист также понимает, что на положение отражений может оказывать влияние точная высота, на которой образец помещен в дифрактометре и калибровка нуля дифрактометра. Планарность поверхности образца также может оказывать небольшое влияние. Следовательно, представленные данные дифрактограммы не следует принимать в качестве абсолютных величин.
Обычно ошибка измерения угла дифракции на рентгеновской порошковой дифрактограмме составляет примерно 5% или менее, в частности плюс или минус 0,5° 2-тета. Обычно плюс или минус 0,2° 2-тета. Такую степень ошибки измерения следует принимать во внимание, рассматривая рентгеновские порошковые дифрактограммы на Фиг.1 и 2 и просматривая Таблицы 1, 2 и 2А. Кроме того, следует понимать, что интенсивности могут меняться в зависимости от экспериментальных условий и приготовления образца (преимущественная ориентация).
Анализ дифракции рентгеновских лучей на порошке (XRPD) выполняли на образцах, приготовленных стандартными способами, например описанными в Giacovazzo, С. et al (1995), Fundamentals of Crystallography, Oxford University Press; Jenkins, R. and Snyder, R.L. (1996), Introduction to X-Ray Powder Diffractometry, John Wiley & Sons, New York; Bunn, C.W. (1948), Chemical Crystallography, Clarendon Press, London; or Klug, H.P. & Alexander, L.E. (1974), X-ray Diffraction Procedures, John Wiley and Sons, New York.
Анализы дифракции рентгеновских лучей выполняли, используя Then-no ARL X′TRA (длина волны рентгеновских лучей 1,5418 Å , источник излучения Cu, напряжение 45 кВ, эмиссия нити накаливания 44 мА) в течение 152 минут от 2 до 40°. Выполняли вычисление положений пиков (°2-тета), они могут варьироваться в диапазоне ±0,5° 2-тета. Однако данные, представленные в Таблице 2А для Формы D, получали, используя устройство Bruker D4 и длину волны 1,5418 Å.
Специалисту в данной области техники очевидно, что интенсивности XRPD могут варьироваться при измерении по существу той же самой кристаллической формы, например преимущественной ориентации.
Дифференциальная сканирующая калориметрия
Аналитический прибор: ТА Instruments Q1000 DSC. Обычно менее 5 мг вещества, содержащегося в 40 мкл алюминиевой кювете, оснащенной притертой крышкой, нагревали в диапазоне температур от 25°C до 300°C при постоянной скорости нагрева 10°C в минуту. Использовали азот в качестве продувочного газа - скорость потока 100 мл в минуту.
Ссылочный Пример 1
(R)-1-((S)-2,2-диметил-1,3-диоксолан-4-ол)этанол

1) 5,6-O-изопропилиден-L-аскорбиновой кислота
В смесь L-аскорбиновой кислоты (65 кг, 369 моль), ацетона (283 кг) и 2,2-диметоксипропана (46 кг, 443 моль) загружали пара-толуолсульфоновую кислоту (1,1 кг, 5,5 моль). Температуру регулировали до 25±5°C. Взвесь перемешивали в течение 2 часов, в течение которых азот часто пропускали через донный клапан для предотвращения оседания вещества на дно реактора. Последующий анализ посредством ЯМР (растворитель: D2O) показал 98,5% превращения.
Загружали гептаны (222 кг) и температуру регулировали до 5±5°C. Реакционную смесь перемешивали в течение по меньшей мере 30 минут перед фильтрацией. Остатки ацетонидного продукта в реакторе смывали в остаток на фильтре, используя маточные растворы. Остаток на фильтре промывали гептанами (111 кг) и сушили при 50°C с получением 5,6-O-изопропилиден-L-аскорбиновой кислоты (73 кг, 336 моль) в виде почти белого порошка. Выход: 91%.
1H ЯМР (400 МГц, d6-DMSO, с малеиновой кислотой и TFA (трифторуксусная кислота)) δ 4.71 (d, J=3,0 Гц, 1H), 4.28 (m, 1H), 4.11 (dd, J=7,0, 8,4 Гц, 1H), 3.90 (dd, J=6,3, 8,4 Гц, 1H), 1.27 (s, 6H).
2) (R)-Метил 2-((S)-2,2-диметил-1,3-диоксолан-4-ол)-2-гидроксиацетат
5,6-O-Изопропилиден-L-аскорбиновую кислоту (58,8 кг, 272 моль) загружали в раствор гидроксида натрия (27,5 кг, 50%, 340 моль), разбавляли водой (294 кг) и температуру регулировали до 30±5°C. Загружали бикарбонат натрия (57 кг, 680 моль) и смесь взбалтывали в течение 15 минут, затем температуру повышали до 40±5°C. Перекись водорода 35% (55 кг, 562 моль) добавляли к смеси при 35-60°C в течение периода времени более 60 минут. Реакционную смесь взбалтывали в течение двух часов перед ЯМР-анализом (растворитель: D2O), показавшим <1% оставшегося исходного вещества.
Сульфат натрия (4,2 кг, 33 моль) загружали в реактор и после перемешивания в течение 30 минут тест на перекиси был отрицательным.
После загрузки дополнительного количества бикарбоната натрия (34 кг, 408 моль), смесь нагревали до 70±5°C и взбалтывали в течение по меньшей мере одного часа до ЯМР-анализа (растворитель: D2O), показавшего 98,5% превращение в следующее промежуточное соединение, (2R)-[(4S)-2,2-диметил-1,3-диоксолан-4-ил](гидрокси)уксусную кислоту.
Приблизительно 150 л воды отделяли при пониженном давлении перед отфильтровыванием солей. Остаток на фильтре промывали водой (30 л).
NMP (N-метилпирролидон) (330 кг) загружали в объединенные маточные растворы/промывные воды и температуру регулировали до 30±5°C. Загружали метилйодид (83 кг, 585 моль) и реактор закрывали. Температуру регулировали до 55±5°C и реакционную смесь оставляли взаимодействовать в течение по меньшей мере 120 минут перед ЯМР-анализом (растворитель: D2O), показавшим 6% оставшегося промежуточного соединения, гидроксиуксусной кислоты.
Загружали сульфит натрия (56 кг, 446 моль), растворенный в воде (147 кг) и смесь взбалтывали в течение 30 минут. Раствор экстрагировали четыре раза в течение 10 минут при 30±10°C, используя 406 кг толуола в каждой экстракции. Объединенную органическую фазу концентрировали путем отделения растворителя при пониженном давлении и максимальной температуре 70°C до достижения остаточного объема приблизительно 350 л. Раствор охлаждали до температуры ниже 30°C и переносили в стальных бочонках на фильтр Millipore с получением раствора (R)-метил-2-((S)-2,2-диметил-1,3-диоксолан-4-ол)-2-гидроксиацетата в толуоле (359 кг, 9,4%, 177 моль). Выход: 65%.
1H ЯМР соответствует имеющемуся в продаже образцу указанного в подзаголовке продукта.
3) (R)-Метил 2-((S)-2,2-диметил-1,3-диоксолан-4-ол)-2-(тозилокси)ацетат
Из раствора (R)-метил-2-((S)-2,2-диметил-1,3-диоксолан-4-ол)-2-гидроксиацетата в толуоле (359 кг, 9,4%, 177 моль) отгоняют толуол при пониженном давлении и максимальной температуре 70°C до прекращения конденсации.
Загружали ацетонитрил (153 кг) и температуру регулировали до 25±5°C. Добавляли триэтиламин (41 кг, 405 моль), 4-(диметиламино)пиридин (1,12 кг, 9,2 моль) и потом, примерно через 30 минут, раствор пара-толуолсульфонилхлорида (52,5 кг, 276 моль) в ацетонитриле (146 кг) при 25±5°C. После перемешивания реакционной смеси в течение еще трех часов анализ ЯМР-анализ (растворитель: d6-DMSO) показал приемлемое превращение (94%).
Загружали МТВЕ (метил-трет-бутиловый эфир) (235 кг) и воду (326 кг) и двухфазную систему взбалтывали в течение примерно 3 часов, после чего анализ ВЭЖХ показал уровень пара-толуолсульфонилхлорида менее 0,1% от общей площади пика. Температуру регулировали до 25±5°C и затем смесь оставляли разделяться в течение 15 минут. Водную фазу отбирали и экстрагировали дополнительным МТВЕ (156 кг) перед отбрасыванием. Объединяли вместе 2 органические фазы и промывали водой (326 кг). Затем органическую фазу промывали 4 раза раствором хлорида натрия (каждая порция 16 кг) в воде (каждая порция 140 кг), каждый раз в течение 5-10 минут при 25±5°C. Затем органическую фазу промывали два раза водой (185 кг на порцию) каждый раз в течение 5-10 минут при 25±5°C. Затем ЯМР-анализ (растворитель: d6-DMSO) показал менее 2% NMP (оставшегося от исходного раствора) в молях относительно промежуточного сульфонатного эфира.
Загружали активированный уголь (6,0 кг) и взвесь взбалтывали в течение 15 минут при 25±5°C, затем отфильтровывали уголь на двух параллельных рукавных фильтрах. После рукавных фильтров использовали кассетный фильтр 0,6 мкм. Фильтры и трубы промывали МТВЕ (27 кг).
Маточные растворы и промывки объединяли и объем уменьшали путем отделения растворителя при пониженном давлении и максимальной температуре 50°C до прекращения конденсации. Загружали гептаны (106 кг) и объем раствор понижали еще раз путем отделения растворителя при пониженном давлении и максимальной температуре 50°C до прекращения конденсации, оставляя примерно 60 л раствора в реакторе. Загружали МТВЕ (185 кг), затем после регулирования температуры до 25±5°C добавляли гептаны (75 кг). Раствор охлаждали до 0-5°C в течение не менее 30 минут и добавляли гептаны (150 кг) в течение еще 20 минут. Взвесь взбалтывали в течение одного часа при 0-5°C и затем фильтровали. Остаток на фильтре промывали смесью МТВЕ (16 кг) и гептанов (30 кг). Влажный продукт загружали в вакуумную полочную сушилку и сушили при 35°C (давление менее 100 мбар (104 Па)) с получением (R)-метил-2-((S)-2,2-диметил-1,3-диоксолан-4-ол)-2-(тозилокси)ацетата (51,3 кг, 154 моль) в виде светло-коричневого порошка. Выход: 87%.
1H ЯМР (400 МГц, CDCL3) δ 7.83 (m, 2H), 7.35 (m, 2H), 4.84 (d, J=4,8 Гц, 1H), 4.46 (m, 1H), 4.04 (dd, J=6,6, 9,1 Гц, 1H), 3.97 (dd, J=5,2, 9,1 Гц, 1H), 3.70 (s, 3H), 2.45 (s, 3H), 1.30 (s, 3H), 1.29 (s, 3H).
4) (S)-2,2-Диметил-4-((R)-оксиран-2-ол)-1,3-диоксолан
(R)-Метил-2-((S)-2,2-диметил-1,3-диоксолан-4-ол)-2-(тозилокси)ацетат (76,1 кг, 221 моль) растворяли метаноле (57 кг) и дихлорметане (208 кг).
Метанол (14 кг), дихлорметан (53 кг) и одну треть исходного раствора вещества (74 моль) загружали в реактор. Раствор доводили до 10-15°C. Затем боргидрид натрия (6.3 кг, 169 моль) 18 порциями загружали в реактор, поддерживая температуру 8-15°C. Смесь перемешивали в течение получаса после завершения добавления. Загружали следующую одну треть исходного раствора вещества (74 моль) и снова боргидрид натрия (6,3 кг, 169 моль) с последующим получасовым перемешиванием, используя такой же способ, как раньше. Эту процедуру повторяли опять с последней одной третью исходного раствора вещества (74 моль) и дополнительным боргидридом натрия (6,3 кг, 169 моль). Затем анализ посредством ВЭЖХ показал более чем 99,9% превращение в промежуточное соединение, (S)-1-((S)-2,2-диметил-1,3-диоксолан-4-ол)-2-гидроксиэтил-4-метилбензолсульфонат.
Дихлорметан (200 кг) загружали в реакционную смесь. Раствор метилата натрия в метаноле (43 кг, 30%, 239 моль) добавляли порциями при 20-25°C в течение 60 минут. Приблизительно через полчаса анализ ВЭЖХ показал 99,7% расходование промежуточного спирта.
Раствор ацетата натрия (25 кг) в воде (230 л) загружали в реакционную смесь. Смесь перемешивали в течение 10-15 минут при 20-25°C. После разделения в течение 15 минут нижнюю органическую фазу удаляли. Верхнюю водную фазу экстрагировали дихлорметаном (376 кг). Нижнюю органическую фазу удаляли, объединяя с первой органической фазой, и водную фазу отбрасывали.
Воду (359 л) загружали в комбинированные органические фазы. После перемешивания в течение 10-15 минут при 20-25°C и отстаивания в течение 15 минут нижнюю органическую фазу переносили в реактор, содержащий сульфат натрия (63 кг).
Объем смеси уменьшали до 310 л путем отделения растворителя и затем отфильтровывали сульфат натрия. Остаток на фильтре промывали дихлорметаном (94 кг). Объединенные жидкости тщательно смешивали и затем выгружали в стальные барабаны посредством полипропиленового рукавного фильтра с получением раствора (S)-2,2-диметил-4-((R)-оксиран-2-ол)-1,3-диоксолана в DCM (467,5 кг, 6,2%, 203 моль) в виде прозрачной желтой жидкости. Выход: 91%.
Образец, не содержащий растворителей, можно было выделить в небольшом объеме путем выпаривания растворителя и затем перегонки под вакуумом.
1H ЯМР (выделенный образец, 400 МГц, d6-DMSO) δ 4.01 (dd, J=6,6, 8,2 Гц, 1H), 3.92 (m, 1H), 3.72 (dd, J=5,8, 8,2 Гц, 1H), 3.03 (ddd, J=2,6, 4,1, 5,2 Гц, 1H), 2.77 (dd, J=4,1, 5,0 Гц, 1H), 2.58 (dd, J=2,6, 5,0 Гц, 1H), 1.34 (s, 3H), 1.27 (s, 3H).
5) (R)-1-((S)-2,2-диметил-1,3-диоксолан-4-ол)этанол
Из раствора (S)-2,2-диметил-4-((R)-оксиран-2-ол)-1,3-диоксолана в дихлорметане (465 кг, 6,2%, 200 моль) отгоняли дихлорметан при 41-42°C и заменяли THF (тетрагидрофураном) (129 кг). Отгонку продолжали при 60°C до достижения заданного объема в реакторе (235 л). Раствор алюмогидрида лития (LAH) в THF (26,4 кг, 10%, 70 моль) добавляли порциями в реактор при 22°C, и после последующего перемешивания при 25°C в течение приблизительно одного часа ОС(газовая хроматография)-анализ показал более чем 99,9% расходование исходного вещества.
Небольшие порции воды добавляли через загрузочную воронку со скоростью, которую регулировали, чтобы контролировать температуру и образование пены. Всего было добавлено 2,6 литра воды (1 л на кг LAH). Раствор гидроксида натрия (2,6 кг, 15%,1 л на кг LAH) добавляли таким же образом, как описано для воды. Воду (7,9 л, 3 л на кг LAH) загружали еще раз через загрузочную воронку, используя такой же метод, как раньше.
Взвесь фильтровали и остаток на фильтре промывали THF (36 кг). Фильтрат концентрировали отделением THF при максимальной температуре 85°C до прекращения конденсации. Загружали 2-MeTHF (129 кг) в реактор и затем растворитель отгоняли до достижения объема раствора приблизительно 120 л. Анализ методом KF (Карла Фишера) показал менее 0,1% воды. Раствор выгружали посредством кассетного фильтра в барабан, выстеленный РЕ (полиэтиленом), с получением раствора (R)-1-((S)-2,2-диметил-1,3-диоксолан-4-ол)этанола (103 кг, 27%, 187 моль) в виде прозрачной светло-желтой жидкости. Выход: 94%.
Образец, не содержащий растворителей, можно было выделить в небольшом объеме путем выпаривания растворителя и последующей перегонки под вакуумом.
1H ЯМР (выделенный образец, 400 МГц, d6-DMSO) δ млн-1 4.77 (d, J=5,1 Гц, 1H), 3.95 (dd, J=8,0, 6,2 Гц, 1H), 3.76 (dd, 8,0, 6,0 Гц, 1H), 3.70 (m, 1H), 3.46 (m, 1H), 1.29 (s, 3H), 1.25 (s, 3H), 1.07 (d, J=6,2 Гц, 3H).
Пример 1: Форма А - ангидрат Соединения I
Соединение I, то есть N-[2-[[(2,3-дифторфенил)метил]тио]-6-{[(1R,2S)-2,3-дигидрокси-1-метилпропил]окси}-4-пиримидинил]-1-азетидин-сульфонамид (например полученное в соответствии с Примером 47 в WO 2006/024823) применяют в следующем способе.
1. Загружают Соединение I MAPI (43,94 г, 100% масс. = 40,0 г) и ацетонитрил (160 мл, 4,0 относительных объема) в сосуд 1 и перемешивают при 20°C.
2. Отфильтровывают смесь в сосуд 2 и промывают сосуд 1 и поточные линии ацетонитрилом (20 мл, 0,50 относительных объемов). Перемешивают и нагревают до 55°C.
- Использовали ватманскую фильтровальную бумагу, сорт 3 (размер пор 6 мкм, диаметр круговой области фильтра 32 мм).
- Фильтровальную бумагу можно заменять чистой (если скорость фильтрации с использованием аспирации замедляется).
- После фильтрации раствор взвешивали и обнаружили, что его масса меньше, чем предполагаемая. Его долили небольшой порцией ацетонитрила (10 мл, 0,25 относительных объемов) для корректировки очевидных потерь при испарении.
3. Загружают фосфорную кислоту (8,80 г, 0,22 относительной массы), затем воду (20 мл, 0,50 относительных объемов) в перемешиваемый раствор в сосуде 2, растворить и изменяют установленное значение на 51°C.
- Кислоту и воду можно загружать вместе, если это удобнее.
4. Отбирают пробу реакционной смеси для определения превращения через 10 и 35 минут.
- Важно разбавить образцы для ВЭЖХ-анализа как можно быстрее после их отбора, записав время, в которое разбавления были выполнены фактически.
- Превращение составляло 45, 81 и 93% через 10,5, 32 и 57 мин соответственно. Переходили к стадии 5 при t=135 мин.
5. Нагревают до 71°C.
Стадию 6 следует начинать как можно быстрее после достижения примерно 68°C.
6. Загружают воду (204 мл, 5,1 относительных объемов), достаточно медленно, чтобы сохранить температуру выше 67°C.
7. Охлаждают до 65°C.
- Важно, чтобы температура реакционной смеси была как можно ближе к заданному значению перед продолжением. Температура на 2-3°C ниже может привести к очень быстрой кристаллизации и связанным с этим проблемам. Температура на 3°C выше может выйти за пределы температуры прозрачности и из-за этого кристаллизация не сможет начаться.
8. Если необходимо, создают затравку для кристаллизации путем разбавления образца раствора (0,48 мл, 0,012 относительных объемов) водой (1,44 мл, 0,036 относительных объема). Хорошо смешивают и затем загружают жидкую смесь обратно в раствор.
9. Поддерживают при 65°C в течение 40 мин.
10. Охлаждают на 6°C со скоростью 3°C/ч (65-59°C в течение 2,0 часов), затем на 8°C со скоростью 4°C/ч (59-51°C) и наконец до 20°C со скоростью 6°C/ч (51-20°C в течение 5,2 ч).
11. Проверяют, что кристаллизация достигла соответствующего равновесия.
12. Фильтруют, промывают остаток два раза смесью 3:2 об.:об вода:ацетонитрил. (2×120 мл, 2×3,00 относительных объема), включая промывку сосуда 2 для каждого промывания, и затем сушат при 60°C до постоянной массы.
Альтернативно, Форма А может быть получена следующим образом:
1. Загружают N-[2-[[(2,3-дифторфенил)метил]тио]-6-[(1R)-1-[(4S)-2,2-диметил-1,3-диоксолан-4-ил]этокси]-4-пиримидинил]-1-азетидин-сульфонамид (например полученный в соответствии с Примером 47 в WO 2006/024823) (43,94 г, 100% масс. = 40,0 г) и ацетонитрил (160 мл, 4,0 относительных объемов) в сосуд 1 и перемешивают при 20°C.
2. Фильтруют смесь в сосуд 2 и промывают сосуд 1 и поточные линии ацетонитрилом (20 мл, 0,50 относительных объемов). Перемешивают и нагревают до 55°C.
- Использовали ватманскую фильтровальную бумагу сорт 3 (размер пор 6 мкм, диаметр круговой области фильтра 32 мм).
- Фильтровальную бумагу можно заменять чистой (если скорость фильтрации с использованием аспирации замедляется).
- После фильтрации раствор взвешивали и обнаружили, что масса меньше, чем предполагаемая. Его долили небольшой порцией ацетонитрила (10 мл, 0,25 относительных объемов) для корректировки очевидных потерь при испарении ацетонитрила.
3. Загружают фосфорную кислоту (8,80 г, 0,22 относительной массы), затем воду (20 мл, 0,50 относительных объема) в перемешиваемый раствор в сосуде 2 изменяют заданное значение на 51°C.
- Кислоту и воду можно загружать вместе, если это удобнее.
4. Отбирают пробу реакционной смеси для определения превращения через 10 и 35 минут.
- Важно разбавить образцы для ВЭЖХ-анализа как можно быстрее после их отбора, записав время, в которое разбавления были выполнены фактически.
- Превращение составляло 45, 81 и 93% через 10,5, 32 и 57 мин, соответственно. Переходят к стадии 5 при t=135 мин.
5. Нагревают до 71°C.
- Стадию 6 следует начинать как можно скорее после достижения примерно 68°C.
6. Загружают воду (204 мл, 5,1 относительных объемов) достаточно медленно, чтобы сохранить температуру выше 67°C.
7. Охлаждают до 65°C.
- Важно, чтобы температура реакционной смеси была как можно ближе к заданному значению до продолжения способа. Температура на 2-3°C ниже может привести к очень быстрой кристаллизации и связанным с этим проблемам. Температура на 3°C выше может выйти за пределы температуры прозрачности и из-за этого кристаллизация не сможет начаться.
8. Поддерживают при 65°C в течение 40 мин.
9. Охлаждают на 6°C со скоростью 3°C/ч (65-59°C в течение 2,0 часов), затем на 8°C со скоростью 4°C/ч (59-51°C) и наконец до 20°C со скоростью 6°C/ч (51-20°C в течение 5,2 ч).
10. Проверяют, что кристаллизация достигла соответствующего равновесия.
11. Фильтруют, промывают остаток два раза смесью 3:2 об.:об. вода:ацетонитрил (2×120 мл, 2×3,00 относительных объема), включая промывку сосуда 2 для каждого промывания, и затем сушат при 60°C до постоянной массы с получением Соединения I в виде Формы А.
Альтернативно, форму А можно получать растворением N-[2-[[(2,3-дифторфенил)метил]тио]-6-{[(1R,2S)-2,3-дигидрокси-1-метилпропил]окси}-4-пиримидинил]-1-азетидин-сульфонамида (полученного из N-[2-[[(2,3-дифторфенил)метил]тио]-6-[(1R)-1-[(4S)-2,2-диметил-1,3-диоксолан-4-ил]этокси]-4-пиримидинил]-1-азетидин-сульфонамида в соответствии с Примером 47 в WO 2006/024823) в воде (6,2 относительных объемов) и ацетонитриле (5 относительных объемов) при нагревании до 70°C. Как только эта температура достигнута и раствор стал прозрачным, раствор медленно охлаждают до 20°C. Кристаллизация обычно начинается при температуре около 60°C с получением Формы А.
Дифрактограмма XRPD Формы А с использованием длины волны 1,5418 А на устройстве Thermo ARL X′TRA XRD показана на Фиг.1 и представлена в Таблице 1 ниже.
приблизительно 25-100%="vs", приблизительно 10-25%="s", приблизительно 3-10%="m" и приблизительно 1-3%="w"
Эта ангидратная форма Соединения I была высококристаллической.
Пример 2: Форма D - ангидрат Соединения I
Форму А Соединения I, полученную, например, способом, описанным в Примере 1 выше, превращали в Форму D.
Форму А Соединения I сууспендировали в метаноле при 50°C в течение 6 недель с получением D-формы.
Также можно получить форму D путем супендирования продукта из Примера 47 в WO 2006/024823 в метаноле при 50°C в течение 6 недель.
Согласно DSC форма D имела точку плавления 152,7°C (начало). Смотри Фиг.3 ниже.
Дифрактограмма XRPD формы D с использованием длины волны 1,5418 Å, на устройстве Thermo ARL X′TRA XRD показана на Фиг.2 и представлена в Таблице 2 ниже.
приблизительно 25-100%="vs", приблизительно 10-25%="s", приблизительно 3-10%="m" и приблизительно 1-3%="w"
Дифрактограмма XRPD формы, полученной согласно Примеру 1 с использованием длину волны 1,5418 Å на устройстве Bruker D4 XRD, представлена в Таблице 2А ниже.
Эта ангидратная форма Соединения I также была высококристаллической и также была более термодинамически стабильной, чем А-форма, полученная посредством Примера 1 (смотри Пример 3 ниже).
Пример 3: Форма D - ангидрат Соединения I
Модификацию D можно получить, применяя кристаллизацию с антирастворителем, используя этанол в качестве растворителя и воду в качестве антирастворителя.
Получают насыщенный 80% раствор соединения в этаноле, например путем растворения 1 относительной массы соединения в 83 относительных объемах этанола при температуре 25°C (эквивалентно 12 мг AZD 5069 в 1 мл этанола). Раствор может быть получен из кристаллического вещества формы А или из аморфного вещества. К этому раствору добавляют 166 относительных объемов воды (то есть двойной объем воды относительно этанола), либо непрерывно в течение некоторого периода времени, либо несколькими аликвотами. При добавлении воды соединение кристаллизуется в виде модификации D и может быть выделено из взвеси посредством фильтрации.
Пример 4: Форма D - Соединение 1 ангидрат (метод использования затравки)
Форму D также получали с выходом 80% методом применения затравки, который включает растворение Соединения I (например одной относительной "массы") в растворителе (таком как спирт, например изопропанол; например 30 относительных объемов, или этанол; 15 относительных объемов); перемешивание при повышенной температуре (например 70°C), например в течение периода времени (который может составлять несколько часов) до достижения полного растворения; продолжение перемешивания при повышенной температуре (например 55°C; то есть при температуре ниже той, которая требуется для достижения растворения) в течение несколько часов (например в течение ночи); внесение затравки Формы D Соединения I (например 0,1 масс.%); охлаждение до более низкой температуры (например 20°C); и фильтрование.
Пример 5: Термодинамическая стабильность
Выполняли сравнительные испытания взвесей А-формы (полученной согласно Примеру 1) и D-формы (полученной согласно Примеру 2).
Сравниваемые взвеси, содержащие А-форму и D-форму в метаноле поддерживали при любых температурах в диапазоне от примерно 5°C до примерно 50°C. Обнаружили, что D-форма остается стабильной в этих условиях. Однако А-форма превращается в D-форму.
Полное превращение А-формы в D-форму происходит, если взвесь поддерживают при конкретной температуре в течение достаточного периода времени.
Это указывает на то, что D-форма является термодинамически более стабильной формой, чем А-форма, по меньшей мере в конкретном релевантном диапазоне температур, и следовательно D-форма может быть еще более предпочтительной, чем А-форма, для применения в качестве лекарственного средства.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ КРИСТАЛЛИЧЕСКИЕ ФОРМЫ НАТРИЕВОЙ СОЛИ(4-{ 4-[5-(6-ТРИФТОРМЕТИЛ-ПИРИДИН-3-ИЛАМИНО) ПИРИДИН-2-ИЛ] ФЕНИЛ} ЦИКЛОГЕКСИЛ) УКСУСНОЙ КИСЛОТЫ | 2011 |
|
RU2612556C2 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ ИНГИБИТОРА TLR7/TLR8 | 2019 |
|
RU2792005C2 |
| Замещенные производные бисфенилового эфира масляной кислоты в качестве ингибиторов NEP | 2019 |
|
RU2784522C2 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ БИЛАСТИНА И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 2016 |
|
RU2772222C2 |
| НОВЫЕ КРИСТАЛЛИЧЕСКИЕ ФОРМЫ СОЛИ МЕЗИЛАТА 2,3-ДИМЕТИЛ-8-(2, 6-ДИМЕТИЛБЕНЗИЛАМИНО)-N-ГИДРОКСИЭТИЛ-ИМИДАЗО[1, 2-a]ПИРИДИН-6-КАРБОКСАМИДА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКИЙ ПРЕПАРАТ, СОДЕРЖАЩИЙ ИХ, ИХ ПРИМЕНЕНИЕ И СПОСОБ ЛЕЧЕНИЯ | 2004 |
|
RU2376306C2 |
| ПРОИЗВОДНЫЕ ПИРРОЛО[2,3-d]ПИРИМИДИНА | 2009 |
|
RU2493157C2 |
| НОВЫЕ КРИСТАЛЛИЧЕСКИЕ ФОРМЫ | 2007 |
|
RU2446156C2 |
| НОВЫЕ КРИСТАЛЛИЧЕСКИЕ ФОРМЫ ТИОТРОПИЙБРОМИДА | 2006 |
|
RU2417224C2 |
| КРИСТАЛЛИЧЕСКИЕ СОЛЬВАТЫ ГИДРОХЛОРИДА 6-(ПИПЕРИДИН-4-ИЛОКСИ)-2Н-ИЗОХИНОЛИН-1-OHA | 2012 |
|
RU2619129C2 |
| ТВЕРДЫЕ ФОРМЫ (R)-1-(2, 2-ДИФТОРБЕНЗО[d][1, 3]ДИОКСОЛ-5-ИЛ)-N-(1-(2, 3-ДИГИДРОКСИПРОПИЛ)-6-ФТОР-2-(1-ГИДРОКСИ-2-МЕТИЛПРОПАН-2-ИЛ)-1H-ИНДОЛ-5-ИЛ)ЦИКЛОПРОПАНКАРБОКСАМИДА | 2011 |
|
RU2711481C2 |
Изобретение относится к новой кристаллической форме N-[2-[[(2,3-дифторфенил)метил]тио]-6-{[(1R,2S)-2,3-дигидрокси-1-метилпропил]окси}-4-пиримидинил]-1-азетидин-сульфонамида, имеющей рентгеновскую порошковую дифрактограмму, измеренную с использованием длины волны рентгеновских лучей 1,5418 Å и содержащую по меньшей мере один кристаллический пик со значением 2-тета (в градусах) 21,0, 28,8 и/или 29,1; либо содержащую по меньшей мере 2 кристаллических пика со значением 2-тета (в градусах) 21,0, 28,8 и/или 29,1; либо содержащую по меньшей мере 3 кристаллических пика со значением 2-тета (в градусах) 21,0, 28,8 и/или 29,1. Указанная кристаллическая форма может содержать дополнительные кристаллические пики со значением 2-тета (в градусах), выбранным из 12,9 и 18,0, полученные в вышеуказанных условиях. Более полно кристаллическая форма имеет рентгеновскую порошковую дифрактограмму, измеренную с использованием длины волны рентгеновских лучей 1,5418 Å, с кристаллическими пиками со значением 2-тета (в градусах) 12,9, 13,1, 18,0, 21,0, 22,5, 25,1, 25,3, 28,8, 29,1 и 30,4, и имеет точку плавления (начало) 152,7°С. 2 н. и 4 з.п. ф-лы, 3 ил.,2 табл., 5 пр.
1. Кристаллическая форма N-[2-[[(2,3-дифторфенил)метил]тио]-6-{[(1R,2S)-2,3-дигидрокси-1-метилпропил]окси}-4-пиримидинил]-1-азетидин-сульфонамида, отличающаяся рентгеновской порошковой дифрактограммой, измеренной с использованием длины волны рентгеновских лучей 1,5418 Å, содержащей по меньшей мере один кристаллический пик со значением 2-тета (в градусах) 21,0, 28,8 и/или 29,1.
2. Кристаллическая форма по п. 1, отличающаяся рентгеновской порошковой дифрактограммой, измеренной с использованием длины волны рентгеновских лучей 1,5418 Å, содержащей по меньшей мере 2 кристаллических пика со значением 2-тета (в градусах) 21,0, 28,8 и/или 29,1.
3. Кристаллическая форма по п. 2, отличающаяся рентгеновской порошковой дифрактограммой, измеренной с использованием длины волны рентгеновских лучей 1,5418 Å, содержащей по меньшей мере 3 кристаллических пика со значением 2-тета (в градусах) 21,0, 28,8 и/или 29,1.
4. Кристаллическая форма по п. 1, отличающаяся рентгеновской порошковой дифрактограммой, измеренной с использованием длины волны рентгеновских лучей 1,5418 Å, содержащей дополнительный кристаллический пик со значением 2-тета (в градусах), выбранным из 12,9 и 18,0.
5. Кристаллическая форма по п. 1, отличающаяся рентгеновской порошковой дифрактограммой, измеренной с использованием длины волны рентгеновских лучей 1,5418 Å, содержащей кристаллические пики со значением 2-тета (в градусах) 12,9, 13,1, 18,0, 21,0, 22,5, 25,1, 25,3, 28,8, 29,1 и 30,4.
6. Кристаллическая форма N-[2-[[(2,3-дифторфенил)метил]тио]-6-{[(1R,2S)-2,3-дигидрокси-1-метилпропил]окси}-4-пиримидинил]-1-азетидин-сульфонамида, которая имеет точку плавления (начало) 152,7°С.
| WO 2006024823 A1,09.03.2006 & RU2408587 | |||
| CAIRA M R,CRYSTALLINE POLYMORPHISM OF ORGANIC COMPOUNDS, TOPICS IN CURRENT CHEMISTRY, 1998, Vol:198, Page(s):163 - 208 | |||
| ПРОИЗВОДНЫЕ АРИЛСУЛЬФОНАМИДА ИЛИ ИХ СОЛИ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ПРОЯВЛЯЮЩАЯ АНГИОПРОТЕКТОРНОЕ, АНТИГИПЕРТЕНЗИВНОЕ И ВАЗОСПАЗМОЛИТИЧЕСКОЕ, В ЧАСТНОСТИ ПРОТИВОИШЕМИЧЕСКОЕ, ДЕЙСТВИЕ | 1992 |
|
RU2083567C1 |

