Настоящее изобретение касается модифицированных аминокислот, пептидов и пептидомиметиков, а также их использования в качестве цитолитических препаратов, которые особо широко используются в качестве противомикробных и противоопухолевых препаратов.
Инфекции, вызванные мультирезистентными бактериями, стали основной проблемой общества за последние 20-25 лет. В частности, эта проблема встала в больницах, где инфекции, вызванные метициллин-резистентным Staphylococcus aureus (MRSA), ванкомицин-резистентным Staphylococcus aureus (VRSA) и метициллин-резистентным Staphylococcus epidermidis (MRSE), могут привести к серьезным повреждениям, длительной госпитализации и смерти, особенно среди пациентов с ослабленной иммунной системой. Ванкомицин раньше был препаратом для приема в исключительных обстоятельствах, однако отчеты из больниц со всего мира демонстрируют, что ванкомицин используется в настоящее время намного чаще.
Как и для вышеуказанных грамположительных штаммов, врачи-клиницисты также сообщают о проблемах, возникающих вследствие грамотрицательных штаммов, включая Pseudomonas aeruginosa и Eschericha coli. Весьма желательно, чтобы препараты-антибиотики оказывали действие против широкого спектра бактерий, включая грамположительные и грамотрицательные штаммы.
Долгое время между крупнейшими фармацевтическими компаниями велась борьба за разработку новых классов противомикробных соединений, очевидно, вследствие огромных затрат на разработку и сравнительно ограниченную продолжительность лечения пациентов по сравнению с лечением хронических заболеваний. Однако потребность в новых противомикробных препаратах является насущной, поскольку количество смертей в США, вызванных инфекциями, приобретенными в больнице, в настоящее время превышает смертность от ВИЧ.
Многообещающий класс Противомикробных препаратов - это катионные противомикробные пептиды (AMP), также известные как иммунологические защитные пептиды. AMP характеризуются уникальным механизмом действия, фокусируя свое влияние на внутренние и/или внешние оболочки бактерий нерецепторно-специфическим образом. Подробный механизм разрыва оболочки AMP все еще не полностью изучен. Предлагаются различные модели для объяснения наблюдаемых эффектов.
Вследствие относительной схожести между компонентами липидной клеточной оболочки прокариотных и эвкариотных опухолевых клеток, селективная мембранная дестабилизация и вследствие этого литическая активность против опухолевых клеток также наблюдались для этих Противомикробных пептидов. Для обоих видов клеток-мишеней был идентифицирован эффективный класс амфипатических пептидов и пептидообразных молекул, имеющих чистый положительный заряд, и липофильная группа или группы. Хотя для первого поколения настоящих молекул предполагался порообразующий механизм действия, и они, как правило, включали десять или больше аминокислот, недавно было доказано, что намного меньшие молекулы могут поддерживать терапевтически существенные уровни активности и селективности (Strøm M.B. et al., J. Med. Chem. 2003, 46, 1567-1570).
Несмотря на это иногда противоречащие цели терапевтической активности, селективности, токсичности, стабильности in vivo и in vitro, стоимости производства и простоты приема означают, что существует постоянная потребность в разработке новых препаратов в настоящем общем классе молекул.
Настоящие изобретатели установили, что путем применения двузамещенной β-аминокислоты, можно создать новый класс цитолитических препаратов, которые обладают потенциально мощным действием и другими желаемыми характеристиками, включая широкий спектр антибактериальной активности, например, действие против грамположительных и грамотрицательных штаммов. Эти препараты, в основном, предназначаются для перорального приема.
Таким образом, в одном аспекте настоящее изобретение является пептидом, пептидомиметиком или (модифицированной) аминокислотой, имеющей чистый положительный заряд, по меньшей мере, +2, и включающей двузамещенную β-аминокислоту, каждую из замещающих групп в β-аминокислоте, которые могут быть одной и той же или разными, включает, по меньшей мере, 7 неводородных атомов, является липофильной или имеет, по меньшей мере, одну циклическую группу, одна или больше циклических групп в замещающей группе может быть связана или смешана с одной или больше циклических групп в другой замещающей группе, и если циклические группы смешаны таким образом, совокупное общее количество неводородных атомов двух замещающих групп, составляет, по меньшей мере, 12, для использования в качестве цитолитического терапевтического препарата. 2 замещающие группы в β-аминокислоте желательно одни и те же.
Не желая ограничиваться теорией, вероятно, что включение двузамещенный β-аминокислоты повышает стабильность препарата, и хотя принудительные конформационные расходы улучают амфипатичность препарата, а также способствуют мощному разрушающему действию на оболочку вследствие взаимному отталкиванию двух липофильных фрагментов двузамещенного остатка. Это оказывает цитолитическое действие, которое может быть цитотоксичным.
Цитотоксическое действие может быть противомикробным, предпочтительно антибактериальным действием и/или противоопухолевым действием, и такое медицинское применение является предпочитаемыми воплощениями настоящего изобретения. Таким образом, настоящее изобретение обеспечивает пептиды, пептидомиметики и модифицированные аминокислоты, как указано выше (и подробнее описано ниже) для использования в качестве цитолитических противомикробных или противоопухолевых препаратов. Рассматривая с альтернативной точки зрения, настоящее изобретение обеспечивает пептиды, пептидомиметики и модифицированные аминокислоты, как указано выше (и подробнее описано ниже) для использования в лечении микробной (в частности, бактериальной) инфекции или для лечения опухолевых клеток (в частности, солидных опухолей).
Микробы, против которых может быть направлено действие или лечение, включают бактерии (грамположительные и грамотрицательные), грибки, археи и протисты. Бактерии представляют особый интерес ввиду их способности инфицировать людей и животных, нанося вред их здоровью и угрожая жизни.
Предпочтительные бактериальные мишени включают грамположительные бактерии, в частности, Staphylococcus aureus, метициллин-резистентный Staphylococcus aureus (MRSA) и метициллин-резистентный Staphylococcus epidermidis (MRSE). Грамотрицательные штаммы, например, Pseudomonas aeruginosa и Eschericha coli, также могут подвергаться лечению. Хронические раны часто инфицируются грамположительными и грамотрицательными штаммами, а лечение пациента, который имеет или может иметь мультипатогенную инфекцию (например, в области хронической раны), является предпочтительным использованием в соответствии с настоящим изобретением.
Противомикробное действие также может использовать в нетерапевтических целях, например, в качестве дезинфицирующего агента. Еще один аспект настоящего изобретения обеспечивает ex vivo использование пептида, пептидомиметика или модифицированной аминокислоты, как указано и описано в настоящем документе в качестве цитолитического агента.
Липофильность может быть измерена на основе распределения препарата по двухфазной системе, например, «жидкость-жидкость», например, 1-октанол/вода. Широко известно, что полярные заместители, например, гидроксильные, карбоксильные, карбонильные, аминовые группы и эфиры снижают коэффициент разделения по двухфазной систем, например, 1-октанол/вода, поскольку они снижают липофильность; липофильные замещающие группы, таким образом, должны предпочтительно содержать не больше одной или двух, предпочтительно одну или совсем не содержать таких полярных групп.
β-аминокислота содержит аминогруппу, присоединенную β-атому углерода; генетически закодированные аминокислоты, в которых аминогруппы присоединены к атому углерода. Такая организация удлиняется на один атом на β-аминокислоту основу пептида, содержащего одну или более β аминокислот. При такой организации α и/или β атом углерода может быть замещен, α или β атом углерода может быть двузамещенным; если атом углерода - двузамещенный, получается β2,2 аминокислота, а если β атом углерода - двузамещенный, получается создается β3,3 аминокислота. Одна замещающая группа в каждом α или β атоме углерода приводит к формированию β2,3 аминокислоты, β2,2 и β3,3 двузамещенные аминокислоты предпочтительны для использования в соответствии с настоящим изобретением, причем β2,2 двузамещенные аминокислоты особенно предпочтительны.
β аминокислота замещается двумя группам, содержащими, по меньшей мере, 7 неводородных атомов. Предпочтительно одна, более предпочтительно обе из замещающих групп должны содержать, по меньшей мере 8, более предпочтительно, по меньшей мере, 10 неводородных атомов. Эти группы - липофильные по своей природе, и хотя они могут быть разными, предпочтительно они должны быть одинаковыми. Каждая из них содержит, по меньшей мере, одну циклическую группу, как правило, 6-членное кольцо, которое может быть алифатическим или ароматическим, предпочтительно, ароматическим, и может быть замещенным, а замещающие группы могут включать такие атомы, как кислород, азот, серу или галоген, в частности, фтор или хлор. Предпочтительные замещающие группы включают C1-C4 алкильные (в частности, t-бутил), метоксильные, фторовые и фторметиловые группы. Циклические группы могут быть гомо- или гетероциклические, предпочтительно, они являются гомоциклическим кольцом атомов углерода. Предпочтительные липофильные замещающие группы содержат две или три циклические группы, предпочтительно, две циклические группы, которые могут быть связаны или соединены, предпочтительно, соединены. Особо предпочтительно, чтобы замещающие группы включали нафталиновую группу.
Еще одна предпочтительная группа липофильных замещающих групп должна иметь одну замещенную или незамещенную циклическую группу, предпочтительно, фениловую или циклогексильную группу.
Циклическая группа или группы, как правило, отделены от пептидной основы (т.е. от α или β атомов углерода β-аминокислоты) цепью из 1-4, предпочтительно, из 1-3 атомов; эти связывающие атомы могут включать азот и/или кислород, однако, как правило, являются атомами углерода, предпочтительно, связывающие атомы - незамещенные. Эти разделители естественно являются частью замещающих групп, как указано выше.
Каждая замещающая функциональная группа двузамещенной β-аминокислоты, как правило, включает 7-20 неводородных атомов, предпочтительно - 7-13, более предпочтительно - 8-12, но наиболее предпочтительно - 9-11 неводородных атомов.
Молекулы для использования в соответствии с изобретением, предпочтительно, будут пептидами или пептидомиметиками 1 или 2-12 аминокислота или эквивалентных по длине подъединиц. Если контекстом не предусматривается иное, упоминание «аминокислот» в настоящем документе включает эквивалентную подъединицу в пептидомиметике. В Противомикробных целях, предпочтительные молекулы имеют 1-3 или 4 аминокислоты, в противоопухолевых целях, предпочтительные молекулы длиннее, т.е. 3-12 аминокислот, более предпочтительно, 5-12 аминокислот по длине. Как показано в Примерах, молекулы для использования в соответствии с изобретением могут включать только одну аминокислоту, однако это будет «модифицированная» аминокислота с целью выполнения требований заряда.
Отдельные аминокислоты, а также пептиды и пептидомиметики будут предпочтительно инкорпорированы в модифицированный С-терминал, С-терминал модифицирующая группа, как правило, приводит к обращению заряда, т.е. снимает отрицательный заряд карбоксильной группы и добавляет положительный заряд, например, посредством наличия аминогруппы. Такая модификация сама по себе, при условии, что N-терминал не модифицирован, обеспечит общий чистый заряд молекулы, равный +2. Если С-терминал модифицирован для обеспечения обращения заряда или просто для снятия отрицательного заряда карбоксильной группы, молекула, предпочтительно также должна содержать одну или больше катионных аминокислот. Таким образом, общий заряд молекулы может составлять +3, +4 или больше для больших молекул.
Подходящие C-терминальные группы, которые предпочтительно катионные по своей природе, будут, как правило, иметь максимальный размер 15 неводородных атомов. C-терминал предпочтительно амидированный, а амидная группа может быть дополнительно замещена для формирования N-алкильного или N,N-диалкильного амида. Первичные и вторичные амидные группы являются предпочтительными. Соответствующие группы для замещения амидной группы включают аминоалкил, например, аминоэтил или диметиламиноэтил; атом азота амидной группы может формировать часть циклической группы, например пиразолидин, пиперидин, имидазолидин и пиперазин, причем пиперазин является предпочтительным, а эти циклические группы могут сами по себе быть замещены, например, алкильной аминоалкильной группой.
Пептиды для использования в соответствии с изобретением предпочтительно должны включать одну или больше катионных аминокислот, лизин, аргинин, орнитин и гистидин предпочтительны, однако не должны содержать никаких негенетически кодированных или модифицированных аминокислот, несущих положительный заряд при pH 7,0.
Соответствующие негенетически кодированные катионные аминокислоты и модифицированные катионные аминокислоты включают аналоги лизина, аргинина и гистидина, например гомолизин, орнитин, диаминобутановую кислоту, диаминопимелиновую кислоту, диаминопропионовую кислоту и гомоаргинин, а также триметилизин и триметилорнитин, 4-аминопиперидин-4-карбоксильную кислоту, 4-амино-1-карбамимидоилпиперидин-4-карбоксильную кислоту и 4-гуанидинофенилаланил.
Дипептиды, как правило, включают одну катионную аминокислоту, а более длинные пептиды, как правило, будут содержать дополнительные катионные аминокислоты, поэтому пептиды из 4 или 5 аминокислот могут иметь 2 или 3 катионные аминокислоты, а пептиды из 6-9 аминокислот могут включать могут иметь 3-6 катионных аминокислот.
Предпочтительная группа молекул включает β2,2 двузамещенную аминокислоту, присоединенную к остатку C-терминального L-аргининамида, причем дипептиды с такой структурой являются особо предпочтительными.
Пептиды с тремя или больше аминокислотами, как правило, имеют одну или больше дополнительных липофильных аминокислот, т.е. аминокислот с липофильной R-группой. Как правило, липофильная R-группа имеет, по меньшей мере, одну, предпочтительно две циклические группы, которые могут быть слиты или соединены. Липофильная R-группа может содержать такие атомы, как О, N или S, однако, как правило, не больше одного гетероатома, который предпочтительно является азотом. Такая R-группа буте предпочтительно иметь не более 2 полярные группы, более предпочтительно вообще не иметь или иметь одну, наиболее предпочтительно - вообще не иметь.
Триптофан - это предпочтительная липофильная аминокислота, а пептиды предпочтительно включают 1-3 триптофановых остатков. Дополнительные генетически кодированные липофильные аминокислоты, которые могут быть инкорпорированы, - это фенилаланил и тирозин.
Липофильные аминокислоты могут быть негенетически кодированные, включая, генетически кодированные аминокислоты с модифицированной R-группой.
Пептидомиметик, как правило, характеризуется сохранением полярности, трехмерного размера и функциональности (биологического действия) своего пептидного эквивалента, однако если пептидная связь была замещена, как правило, более стабильными соединениями. Под «стабильными» понимается более устойчивые к ферментной деградации гидролитическими ферментами. Как правило, соединение, замещающее амидную связь (суррогат амидной связи), сохраняет множество свойств амидной связи, например, структуру, стерический объем, электростатический характер, возможность связывания с водородом и т.д. В главе 14 "Drug Design and Development", Krogsgaard, Larsen, Liljefors and Madsen (Eds) 1996, Horwood Acad. Pub, предоставляют общее описание способик дизайна и синтеза пептидомиметиком. В данном случае, где молекула вступает в реакцию с оболочкой, а не со специфическим активным участком фермента, некоторые описанные проблемы точного симулирования сродства и эффективности или функции субстрата не являются существенными, а пептидомиметик может быт легко подготовлен на основе данной пептидной структуры или мотива требуемой функциональной группы. Подходящие суррогаты амидной связи включают такие группы: N-алкилация (Schmidt, R. et al., Int. J. Peptide Protein Res., 1995, 46, 47), ретро-инверсный амид (Chorev, M and Goodman, M., Acc. Chem. Res, 1993, 26, 266), тиоамид (Sherman D.B. and Spatola, A.F. J. Am. Chem. Soc., 1990, 112, 433), тиоэфир, фосфанат, кетометилен (X Hoffman, R.V. and Kim, H.O. J. Org. Chem., 1995, 60, 5107), гидроксимтилен, фторвинил (Allmendinger, Т. et al., Tetrahydron Lett., 1990, 31, 7297), винил, метиленамино (Sasaki, Y and Abe, J. Chem. Pharm. Bull. 1997, 45, 13), метилентио (Spatola, A.F., Methods Neurosci, 1993, 13, 19), алкан (Lavielle, S. et al., Int. J. Peptide Protein Res., 1993, 42, 270) и сульфонамидо (Luisi, G. et al. Tetrahedron Lett. 1993, 34, 2391).
Молекулы настоящего изобретения содержат двузамещенную β-аминокислоту, а различные молекулы описаны в примерах, которые содержат только одну дополнительную аминокислоту, т.е. молекулы, имеющие 2 аминокислоты, соединенные амидной связью.
Такие молекулы могут считаться дипептидами ввиду амидной связи; однако амидная связь в этих молекулах фактически является нерасщепляющей ввиду распределения β-аминокислот и как таковые эти молекулы должны считаться пептидомиметиками. В целях настоящего изобретения, такие молекулы (и более крупные молекулы с большими аминокислотами) считаются пептидами (а не пептидомиметиками), ввиду наличия амидной связи. Это позволяет разъяснить номенклатуру без проведения испытаний того, является ли данная амидная связь расщепляющей и в какой степени. Другими словами, если все аминокислоты в молекуле соединены амидными связями, молекула считается пептидом, даже если одна или более амидных связей не являются расщепляющими.
Пептидомиметические соединения в настоящем изобретении, как правило, будут иметь идентифицируемые подъединицы, которые приблизительно эквивалентны по размеру и по функциям аминокислотам. Пептидомиметики, как правило, имеют группы, эквивалентные R-группам аминокислот, и описание подходящих R-групп и N и С-терминал модифицирующих групп применяется, mutatis mutandis, к пептидомиметическим соединениям.
Как описано в пособии, указанном выше, наряду с замещением амидных связей, пептидомиметики могут принимать участие в замещении больших структурных групп диортрипептидомиметическими структурами, и в таком случае, миметические группы, содержащие пептидную связь, например азольные производные миметики, могут использоваться в качестве дипептидных замещений. Пептидомиметик и, поэтому, основы пептидомиметиков, в которых были замещены только амидные связи, как описано выше, являются, однако, предпочтительными.
Подходящие пептидомиметики включают восстановленные пептиды, в которых амидная связь была восстановлена до метиленамина путем обработки таким восстанавливающим веществом, например бораном или гидридным реагентом, таким как алюмогидрид натрия. Такое восстановление обладает дополнительным преимуществом повышения общего содержания катионов молекул.
К другим пептидомиметикам входят сформированные пептиоиды, например, путем поэтапного синтеза амидно-функцианализированных полиглицинов. Некоторые основы пептидомиметиков будут доступными из их пептидных прекурсоров, например пептиды, которые были подвержены перметилированы. Соответствующие способы описаны Ostresh, J.M. et al. in Proc. Natl. Acad. Sci. USA (1994) 91, 11138-11142. Сильноосновные условия будут способствовать преобладанию M-метилации над O-метилацией и приведут к метилации некоторых или всех атомов азота в пептидной связи и N-терминальном азоте.
Предпочтительные заменители пептидной связи включают эфиры, поламины и производные, а также замещенные алканы и алкены, в частности аминометил и кетометилен. Пептидомиметики будут предпочтительно иметь N и C-терминалы, которые могут быть модифицированы, как указано в настоящей работе.
В следующем аспекте настоящего изобретения описан способ лечения или профилактики микробной инфекции, предпочтительно, бактериальной инфекции, который предусматривает введение пациенту пептида, пептидомиметика или модифицированной аминокислоты, как указано выше.
В следующем аспекте настоящего изобретения описан способ лечения опухолевых клеток или профилактики или подавления роста, распространения или метастаз опухоли, который предусматривает введение пациенту пептида, пептидомиметика или модифицированной аминокислоты, как указано выше.
Как и в настоящем изобретении, лечение микробной/бактериальной инфекции будет предпочтительно означать уменьшение количества жизнеспособных микробных/бактериальных клеток, но также может предусматривать бактериостатический механизм действия, при котором клетки удерживаются в тех количествах, которые менее вредны для пациента, чем если бы инфекции протекали без вмешательства. «Профилактика» предусматривает подавление роста микробных/бактериальных клеток таким образом, чтобы измеримая и/или вредная популяция не была установлена у пациента, получившего лечение.
Обработанные опухолевые клетки могут быть циркулирующие, однако, как правило, являются частью солидной опухоли; как и для микробных клеток, лечение будет предпочтительно предусматривать гибель клеток посредством клеточного лизиса. Клеточный лизис может привести к появлению антигенов опухолевых клеток и формированию приобретенного иммунитета, который может помешать или ингибировать развитие вторичных опухолей.
В следующем аспекте настоящего изобретения описан продукт, содержащий (а) пептид, пептидомиметик или модифицированную аминокислоту, как указано выше, и (б) дополнительный Противомикробный агент в виде комбинированного препарата для самостоятельного, одновременного или последовательного использования при лечении или профилактике микробных инфекций.
Еще в одном аспекте настоящего изобретения описан продукт, содержащий (а) пептид, пептидомиметик или модифицированную аминокислоту, как указано выше, и (б) дополнительный противоопухолевый агент в виде комбинированного препарата для самостоятельного, одновременного или последовательного использования при лечении опухолевых клеток.
В следующем аспекте настоящего изобретения описано использование пептида, пептидомиметика или модифицированной аминокислоты, как указано выше, при производстве медикаментов для лечения микробных инфекций, предпочтительно, бактериальных инфекций.
В следующем аспекте настоящего изобретения описано использование пептида, пептидомиметика или модифицированной аминокислоты, как указано выше, при производстве медикаментов для лечения опухолевых клеток или профилактики или замедления роста, распространения или метастаз опухолей.
Из вышеописанного класса молекул существует новая группа высокоэффективных молекул. Эти молекулы подходят для различных применений и способов, указанных в этом документе. Таким образом, в следующем аспекте настоящее изобретение является пептидом, пептидомиметиком или (модифицированной) аминокислотой, имеющей чистый положительный заряд, по меньшей мере, +2, который инкорпорирует группу формулы I
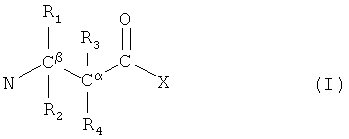
где любые 2 из R1, R2, R3 и R4 являются атомами водорода, а 2 - замещающими группами, которые могут быть одинаковыми или разными, включают, по меньшей мере, 7 неводородных атомов, липофильные и включают циклическую группу, причем вышеуказанная циклическая группа не присоединена непосредственно к α или β атому углерода, а выборочно связана или соединена с циклической группой в другой замещающей группе, где циклические группы соединены с совокупным общим количеством неводородных атомов для двух замещающих групп, по меньшей мере, 12, и где X отображает O, C, N или S,
однако за исключением соединений N-метил-L-фенилаланил-L-лизил-L-пропил-2,2-бис(фенилметил)-β-аланил-D-аргинин и N-метил-L-фенилаланил-L-лизил-L-пропил-D-циклогексилаланил-2,2-бис(фенилметил)-β-аланил-D-аргинин.
Используя номенклатуру Химической реферативной службы, а не систему IUPAC, вышеуказанные две молекулы будут называться D-аргинин, N2-[N-[1-[N2-(N-метил-L-фенилаланил)-L-лизил]-L-пропил]-2,2-бис(фенилметил)-β-аланил] и D-аргинин, N2-[N-[3-циклогексил-N-[1-[N2-(N-метил-L-фенилаланил)-L-лизил]-L-пропил]-D-аланил]-2,2-бис(фенилметил)-β-аланил] и их номера в соответствии с базой данных CAS-145149-42-4 и 145149-43-5, соответственно.
Аннулированные соединения описаны в WO 92/12168 как лиганды анафилатоксинового рецептора, полезные для лечения воспалительных болезненных состояний, и как таковой настоящий документ не описывает проблему, разрешаемую настоящим изобретением.
Желательно, чтобы минимальное количество 12 для комбинированного общего количества неводородных атомов из двух групп R1-4, кода циклические группы из каждого соединенного фрагмента, было достигнуто путем добавления минимального количества несоединенных групп (7+7=14) и вычитания 2, поскольку два неводородных атома эффективно способствуют формированию кольца в каждой группе. Предпочтительно, комбинированное общее количество неводородных атомов в двух группах R1-4, кода циклические группы из каждого фрагмента соединены, составляет 14. Сложные соединенные и связанные группы могут быть проанализированы, если две группы присоединены к Cα или Cβ может содержать более чем одну пару соединенных циклических групп, с или без дополнительного соединения между замещающими группами. Несмотря на это две замещающие группы, предпочтительно не соединенные и не связанные как молекулы, в которых эти группы характеризуются самой высокой гибкостью движения, предпочтительные.
Атом азота в группу из формулы (I) предпочтительно не присоединен к какому-либо атому группы R1-4, кроме, естественно, непрямо через Сβ или Сα. Предпочтительно 5 атомов в вышеуказанной основе (N-Cβ-Cα-C-X) связаны друг с другом только линейным, а не циклическим образом. Желательно, чтобы X и N в формуле (I) имели свои обычные валентности и поэтому были дополнительно замещены, поскольку они связаны с другими частями соединения, например с дополнительными аминокислотами или N- или C-терминал блокирующими группами.
Замещающие группы R1-4, как правило, липофильные по своей природе и предпочтительно не несут никакого заряда и предпочтительно имеют не более двух, более предпочтительно не более одной полярной группы. Одна или обе замещающие группы R1-4 предпочтительно содержат, по меньшей мере, 8, более предпочтительно, по меньшей мере, 9 или 10 неводородных атомов, например 7-13, 7-12, 8-12 или 9-11 неводородных атомов. Эти две замещающие группы предпочтительно одинаковые исключительно в целях простоты синтеза. Предпочтительно, две замещающие группы являются R1 и R2 или R3 и R4, R3 и R4 наиболее предпочтительны.
Как указывалось выше, циклическая группа R1-4 не присоединена непосредственно к α или β атому углерода, поскольку они отделены от нее цепью из 1-4, предпочтительно из 1-3 атомов; эти связывающие атомы могут включать азот и/или кислород, но, как правило, будут атомами углерода, предпочтительно, связывающие атомы - незамещенные. Предпочтительные разделяющие фрагменты показаны на Примерах и формируют часть замещающей группы R1-4, как указано в настоящем документе.
X может быть замещено или незамещено и предпочтительно должно являться N-атомом и предпочтительно замещенным. Если X - это N, X может формировать часть амидной связи в дополнительной аминокислоте, как показано в молекулах в Примере 1. Альтернативно, N-атом может быть замещенным, например аминоалкиловой группой, например, аминоэтилом или аминопропилом или диметиламиноэтилом. Эти молекулы показаны в Примере 2. В дальнейшей альтернативе N-атом может быть частью циклической группы, например, пиперазина, который может быть сам по себе замещен алкиловой или аминоалкиловой группой, что также показано в Примере 2.
Пептиды или пептидомиметики, содержащие группу формулы I, будут предпочтительно иметь модифицированный С-терминал, который предпочтительно амидирован и описан выше в связи с молекулами для использования в качестве цитолитических терапевтических препаратов.
В предыдущих абзацах, определяющих предпочтительные замещающие группы β-аминокислоты применяются, mutatis mutandis, две замещающие группы R1-4. Пептиды и пептидомиметики, содержащие группу формулы I, являются предпочтительным подклассом молекул, описанных ранее в данном применении для использования в качестве цитолитического терапевтического препарата, и поэтому все предыдущие абзацы, описывающие предпочтительные характеристики молекул, например, их длину и другие аминокислоты, которые они содержат, также применяются к этим молекулам, определяемым их инкорпорированием группы формулы I и наоборот. Особо предпочтительные молекулы - это молекулы длинной 1-7 или 8 (например 1-5), более предпочтительно 1, 2, 3 или 4 аминокислоты. Молекулы пептидомиметиков будут включать такое количество подъединиц, однако эти подъединицы будут, как правило, связаны имитаторами амидной связи; предпочтительные связи описаны ранее и включают эфиры и аминометил и кетометилен.
Примеры показывают, что структурный мотив настоящего изобретения в форме ди-пептидов (Пример 1) и отдельных модифицированных аминокислот и гепта-пептидов (Примеры 2 и 4). Молекулы в Примерах отображают предпочтительные молекулы и использование их в настоящем изобретении.
Пептиды, пептидомиметики и аминокислоты изобретения могут быть в форме соли, циклические или эстерифицированые, а предпочтительные амидированные производные описаны выше.
Предпочтительный класс молекул и их использование в настоящем изобретении - это производные β, предпочтительно β2,2-аминокислоты, которые имеют отдельную β2,2-аминокислоту, содержащую две липофильные боковые цепи, как описано выше, двузамещенную β-аминокислоту, окруженные двумя катионными группами. Как описано выше, две замещающие группы предпочтительно одинаковые и включают 6-членную циклическую группу и, по меньшей мере, 8, предпочтительно, по меньшей мере, 10 неводородных атомов. Эти молекулы особо подходят в качестве Противомикробных препаратов и приемлемы для перорального приема.
Молекулы, описанные в настоящем изобретении, могут быть синтезированы каким-либо удобным способом. Как правило, реактивные группы (например, аминовые, тиолевые и/или карбоксильные) будут защищены в течение всего процесса синтеза. Завершающий шаг в синтезе будет, таким образом, снятием защиты с защищенного производного изобретения. Способы синтеза соединений представляют собой еще один аспект настоящего изобретения. Например, в одном способе осуществления изобретения описан способ синтезирования пептида, пептидомиметика или модифицированной аминокислоты, имеющей чистый положительный заряд, по меньшей мере, +2, и которая инкорпорирует группу формулы I, как указано в настоящем документе, причем этом способ включает извлечение защитной группы из указанного пептида, пептидомиметика или модифицированной аминокислоты.
При построении пептида, можно, в принципе, начать с C-терминала или N-терминала, хотя процедура начала C-терминала предпочтительная.
Способы пептидного синтеза широко известны в науке, однако для настоящего изобретения может быть особо целесообразно осуществлять синтез на основе твердой фазы, и такие основы широко известны в науке.
Существует большое количество защитных групп для аминокислот, и соответствующие аминозащитные группы могут включать карбобензилокси (также именуемую Z) t-бутоксикарбонил (также именуемую Boc), 4-метокси-2,3,6-триметилбензол сульфонил (Mtr) и 9-фторэнилметокси-карбонил (также именуемая Fmoc). Желательно, чтобы при построении пептида на основе окончания C-терминала, присутствовала аминозащитная группа в α-аминогруппу каждого добавляемого нового остатка, которую необходимо извлечь выборочно до следующего этапа соединения.
Карбоксильные защитные группы, которые могут использоваться, включают, например, готовые расщепленные эфирные группы, как бензильные (Bzl), p-нитробензильные (ONb), или t-бутиловые (OtBu) группы, а также соединяющие группы на твердых основах, например амид Ринка, связанный с полистиролом.
Тиолевые защитные группы включают p-метоксибензил (Mob), тритил (Trt) и ацетамидометил (Acm).
Предпочтительные пептиды изобретения могут быть легко подготовлены с помощью t-бутилоксикарбонильной (Boc) защитной группы для амидных боковых цепей Lys, Orn, Dab и Dap, а также для защиты остатков триптофана индольного азота. Fmoc можно использовать для защиты альфа-амино групп. Для пептидов, содержащих Arg, 2,2,4,6,7-пентаметилдигидробензофуран-5-сульфонил можно использовать для защиты гуанидиновой боковой цепи.
Существует множество процедур для извлечения аминовых и карбоксильных защитных групп. Однако они должны соответствовать применяемым синтетическим стратегиям. Боковая цепь защитной группы должны быть стабильной относительно условий, применяемых при извлечении временной α-амино защитной группы до следующего этапа соединения.
Аминовые защитные группы, например, Boc, и карбоксильные защитные группы, например, tBu, следует извлечь одновременно путем кислотной обработки, например с помощью трифторуксусной кислоты. Тиолевые защитный группы, например, Trt, можно извлекать выборочно с помощью окисляющего агента, например йода.
Научная литература и методики синтеза пептидомиметических соединений описаны выше и известны опытным ученым.
Рецептуры, содержащие одно или больше соединений изобретения, смешанные с соответствующим растворителем, носителем или вспомогательным веществом, составляют следующий аспект настоящего изобретения. Такие рецептуры могут использоваться, среди прочего, в фармацевтических (включая ветеринарных) целях и поэтому подходящий растворитель, носитель или вспомогательное вещество будет предпочтительно приемлемое с фармацевтической точки зрения. Соответствующие растворители, вспомогательные вещества и носители известны опытным ученым.
Молекулы, описанные в настоящем документе, являются цитолитическими по своей природе и особо полезны в качестве противомикробного препарата, например антибактериального или противогрибкового препарата, причем использование в антибактериальных целях предпочтительно. Специфичность молекул также обеспечивает их пользу в качестве противоопухолевых препаратов. Таким образом, в следующем аспекте настоящего изобретения описан пептид, пептидомиметик или (модифицированная) аминокислота, имеющая чистый положительный заряд, по меньшей мере, +2, и инкорпорирующей группу формулы I
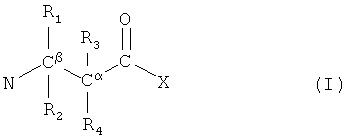
где любые 2 из R1, R2, R3 и R4 - это атомы водорода, а 2 - замещающие группы, которые могут быть одинаковыми или разными, включают, по меньшей мере, 7 неводородных атомов, липофильные и включают циклическую группу, причем такая циклическая группа не присоединена непосредственно к α или β атому углерода, однако выборочно связана или слита с циклической группой в другой замещающей группе, где циклические группы слиты, а совокупное общее количество неводородных атомов для двух замещающих групп составляет, по меньшей мере, 12, и где X отображает O, C, N или S,
однако за исключением соединения N-метил-L-фенилаланил-L-лизил-L-пропил-2,2-бис(фенилметил)-β-аланил-D-аргинин и N-метил-L-фенилаланил-L-лизил-L-пропил-D-циклогексилаланил-2,2-бис(фенилметил)-β-аланил-D-Аргинин, для использования в терапии.
Изобретение также представляет пептид, пептидомиметик или модифицированную аминокислоту, имеющую чистый положительный заряд, по меньшей мере, +2, которая инкорпорирует группу формулы I
где любые 2 из R1, R2, R3 и R4 - это атомы водорода, а 2 - замещающие группы, которые могут быть одинаковыми или разными, содержат, по меньшей мере, 7 неводородных атомов, липофильные и включают циклическую группу, причем такая циклическая группа не присоединена непосредственно к α или β атому углерода, но выборочно связана или слита с циклической группой в другой замещающей группе, где циклические группы слиты, а совокупное общее количество неводородных атомов двух замещающих групп составляет, по меньшей мере, 12, и где X отображает O, C, N или S, для использования в качестве цитолитического терапевтического препарата. Предпочтительные сферы использования - это противомикробные, особенно антибактериальные препараты, или противоопухолевые препараты.
В следующем аспекте настоящего изобретения описан способ лечения или профилактики микробной инфекции, предпочтительно, бактериальной инфекции, который предусматривает введение пациентам пептида, пептидомиметика или модифицированной аминокислоты, как указано выше.
В следующем аспекте настоящее изобретение описан способ лечения опухолевых клеток, или профилактики, или подавления роста, распространения или метастаз опухоли, который предусматривает введение пациентам пептида, пептидомиметика или модифицированной аминокислоты, как указано выше.
В следующем аспекте настоящего изобретения описано использование пептида, пептидомиметика или модифицированной аминокислоты, как указано выше, при производстве медикамента для лечения микробной инфекции, предпочтительно, бактериальной инфекции.
В следующем аспекте настоящего изобретения описано использование пептида, пептидомиметика или модифицированной аминокислоты, как указано выше, при производстве медикамента для лечения опухолевых клеток или профилактики или подавления роста, распространения или метастаз опухоли.
Микробные инфекции могут присутствовать или вызывать подозрения на мультипатогенные инфекции, а лечение таких инфекций (например, в очаге хронической раны), например инфекции, включающей как грамположительные, так и грамотрицательные бактериальные штаммы, представляют предпочтительные целевые использования и способы в соответствии с настоящим изобретением.
Количество введенного препарата должно быть эффективным с точки зрения гибели всех или части целевых клеток или профилактики или снижения скорости их размножения, или подавления метастаз или снижения вредного действия опухолей на пациента. Врач-клиницист или пациент должны увидеть улучшение по одному или нескольким параметрам или симптомам, связанным с опухолью. Введение также может быть профилактическим. Пациент, как правило, должен быть человеком, однако допускается также лечение нечеловекообразных животных, например домашних животных или домашнего скота.
В отличие от большинства препаратов, которые имеют потенциальные мишени, молекулы настоящего изобретения могут оказывать целенаправленное действие против широкого спектра видов рака. Предпочтительные виды рака - лимфомы, лейкемии, нейробластомы и глиобластомы (например, в головном мозге), карциномы и аденокарциномы (в частности, в молочных железах, толстом кишечнике, почках, печени, легких, яичниках, поджелудочной железе, простате и коже) и меланомы.
Композиции в соответствии с изобретением могут быть представлены, например, в форме, подходящей для перорального, местного, назального, парентерального, внутривенного, внутриопухолевого, ректального или очагового (например, изолированная перфузия конечностей) введения. Введение, как правило, осуществляется парентеральным путем, предпочтительно - путем инъекции подкожно, внутримышечно, внутрикапсулярно, интраспинально, внутриопухолево или внутривенно.
Активные соединения, описанные в настоящем документе, могут быть представлены в традиционных фармакологических формах введения, например в форме таблеток, таблеток, покрытых оболочкой, назальных спреев, растворов, эмульсий, липосомов, порошков, капсул или форм непрерывного высвобождения. Традиционные фармацевтические вспомогательные вещества, а также стандартные способы производства могут применяться при подготовке данных форм.
Также могут применяться органоспецифические системы-носители.
Растворы для инъекций могут, например, производиться традиционным способом, например, путем добавления консервантов, таких как p-гидроксибензоаты, или стабилизаторов, например, EDTA. Растворы после этого заполняются в флаконы или ампулы.
Предпочтительные рецептуры - это те, в которых пептиды растворены в соляном растворе. Такие рецептуры подходят для использования в предпочтительных способах введения, особенно при местном применении, т.е. внутриопухолевом, например, путем инъекции или перфузии/инфузии предпочтительно при изолированной (включая частично изолированной) конечности, участке тела или органе.
Единица дозы, содержащей активные молекулы, предпочтительно содержит 0,1-10 мг, например, 1-5 мг противоопухолевого агента. Фармацевтические композиции могут дополнительно содержать другие активные ингредиенты, включая другие цитотоксические препараты, например противоопухолевые пептиды. Другие активные ингредиенты могут включать различные виды цитокинов, например IFN-y, TNF, CSF, и факторы роста, иммуномодуляторы, химиотерапевтические, например, цисплатин или антитела или противораковые вакцины.
При применении таких композиций системно, активная молекула присутствует в количестве, достаточном для достижения уровня биоактивной молекулы в сыворотке, по меньшей мере, около 5 мкг/мл. В основном, уровни в сыворотке не должны превышать 500 мкг/мл. Предпочтительный уровень в сыворотке составляет около 100 мкг/мл. Такие уровни в сыворотке могут быть достигнуты при содержании биоактивной молекулы в композиции, которая вводится системно в дозе от 1 до приблизительно 10 мг/кг. Как правило, препарат(-ы) нельзя вводить в дозе, превышающей 100 мг/кг.
В следующем аспекте настоящего изобретения описан продукт, содержащий (а) пептид, пептидомиметик или модифицированную аминокислоту, имеющую чистый положительный заряд, по меньшей мере, +2 и содержащую группу формулы (I) как указано в настоящем документе, и (б) дополнительный противомикробный агент в качестве комбинированного препарата для самостоятельного, одновременного или последовательного применения или профилактики микробных инфекций.
Еще в одном аспекте настоящего изобретения описан продукт, содержащий (а) пептид, пептидомиметик или модифицированную аминокислоту, имеющую чистый положительный заряд, по меньшей мере, +2 и содержащую группу формулы (I) как указано в настоящем документе, и (б) дополнительный противоопухолевый агент в виде комбинированного препарата для самостоятельного, одновременного или последовательного использования при лечении опухолевых клеток.
Соединения изобретения и соединения, подходящие для способов и использований изобретения, включают соляные формы и соответствующие фармацевтически приемлемые соли для пептидов и подобных молекул, и широко известны опытным ученым.
Изобретение также описано в последующих Примерах, которые включают некоторые эталонные молекулы, выходящие за границы настоящего изобретения, и со ссылкам на фигуры.
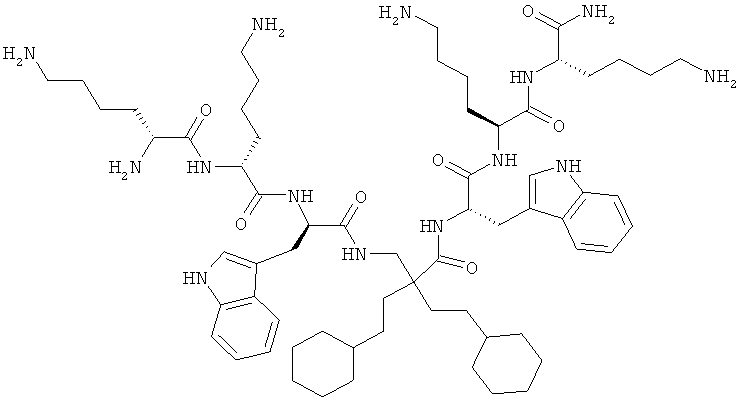
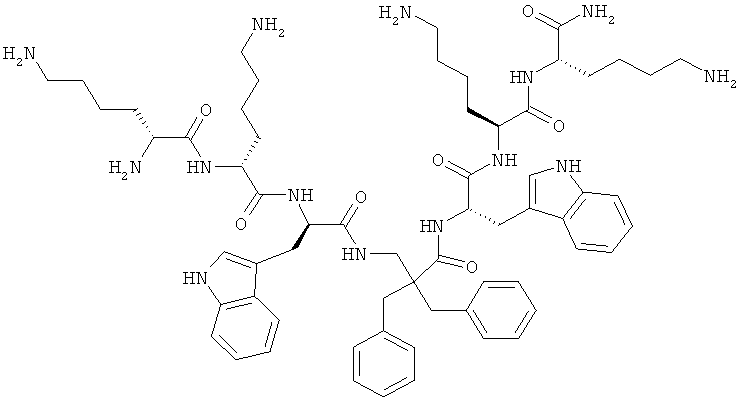
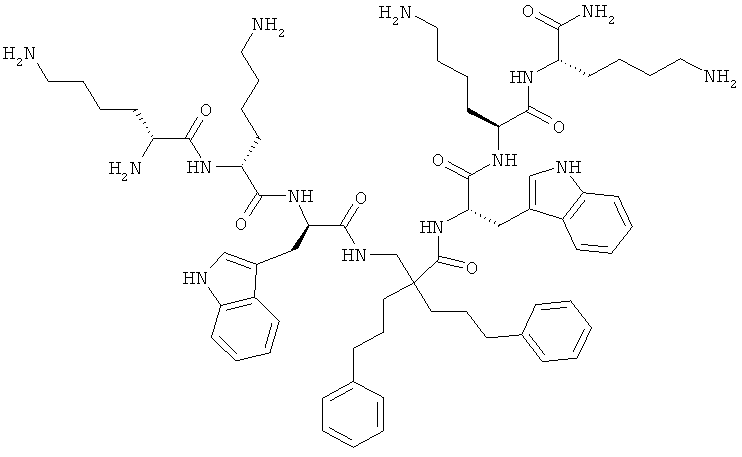
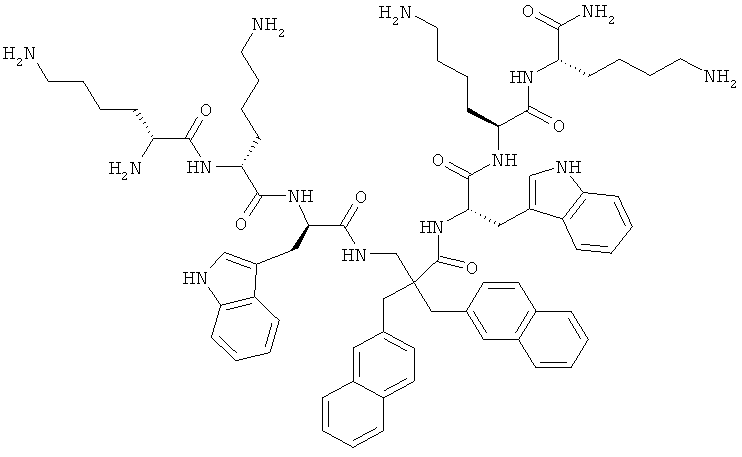
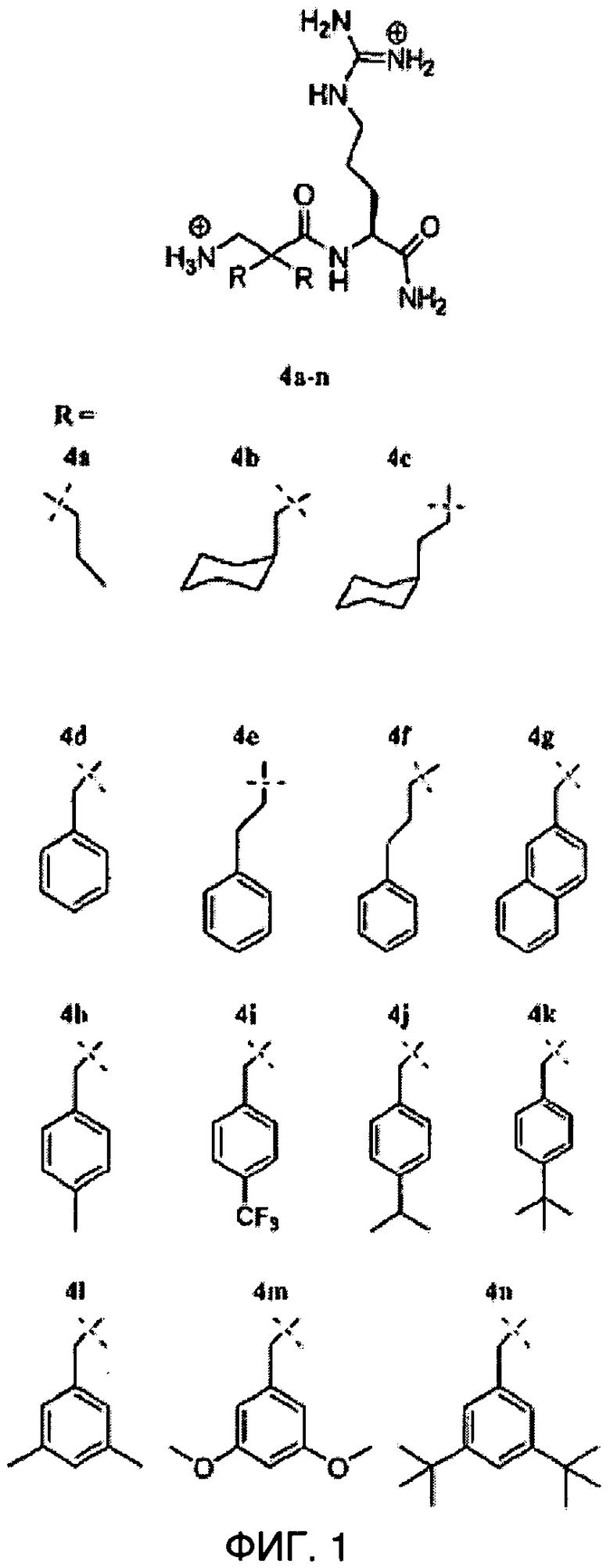
ФИГ.1 демонстрирует соединения 4a-n в Примере 1.
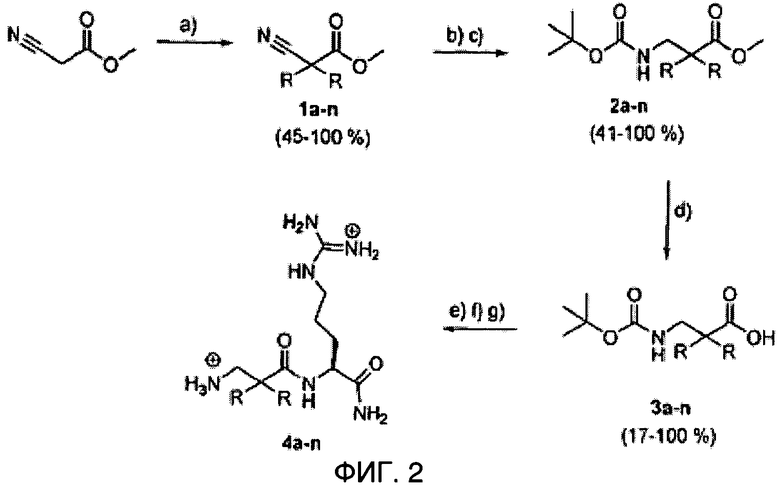
ФИГ.2 демонстрирует схематический обзор синтеза соединения 4a-n, как описано в Примере 1.
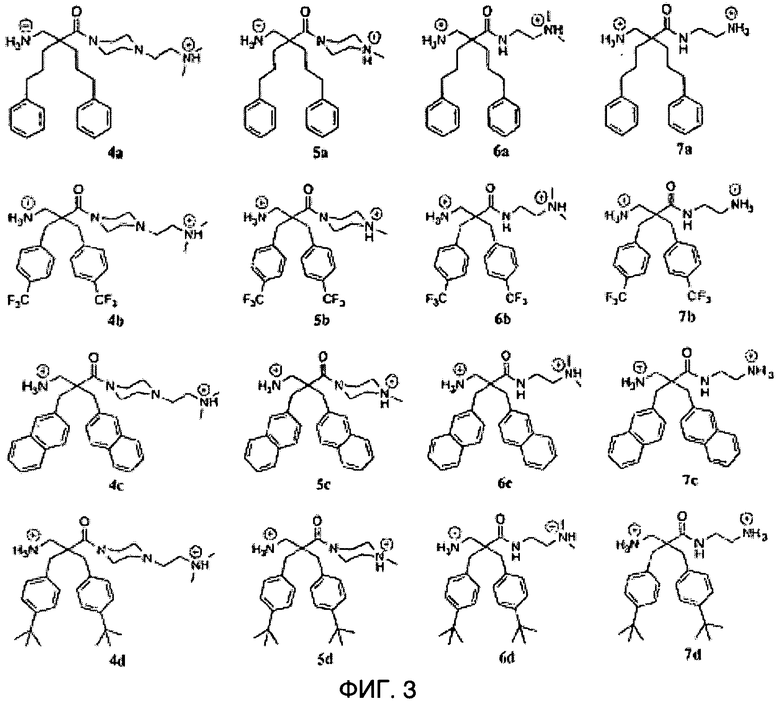
ФИГ.3 демонстрирует соединения 4a-d, 5a-d, 6a-d и 7a-d в Примере 4.
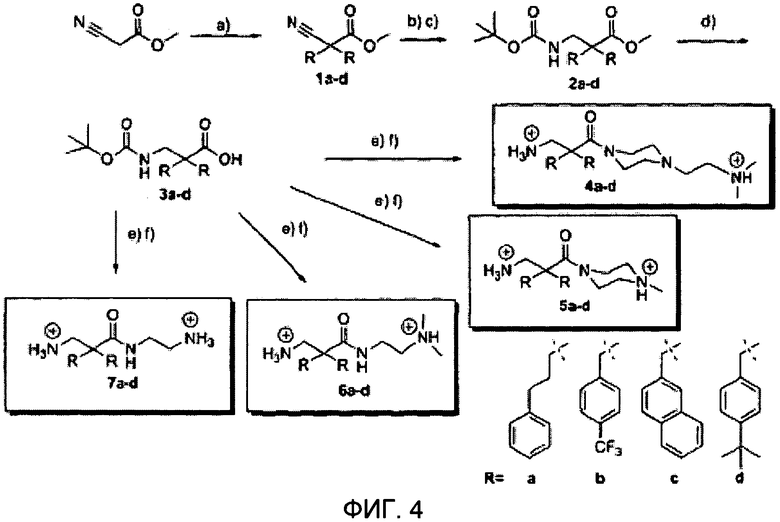
ФИГ.4 демонстрирует схематический обзор синтеза соединения в Примере 4; а) NaOMe (1 экв.), R-Br (1 экв.), проведено дважды, 78°C s: MeOH; b) Ra/Ni, H2(g), 45°C, 5 дней, s: MeOH, содержащий 2% уксусной кислоты; с) ТЭА, pH 8, 15 Вос2O (1.2 экв.), комнатная температура, 18 часов, s: Н2O: диоксан (1:5); d) LiOH (6 экв.), 18 часов, 100°C, s: H20: диоксан (1:3); е) ДИПЭА (3 экв.), TFFH (1.5 экв.), комнатная температура, 2 часа, после этого добавляют требуемый амин (2 экв.), в течение максимум 7 дней, s: ДМФ; f) ТФУК:TIS:Н2O (95:2.5:2.5), комнатная температура, 2 часа, s: ДХМ. Все производные β2,2-аминокислоты были изолированы в виде из ди-трифторацетатных солей.
Примеры
Пример 1
Последовательность молекул на основе остова ахиральной липофильной 3-амино-2,2-двузамещенной пропионовой кислоты (β2,2-аминокислота), соединенной с амидным остатком С-терминал L-аргинина, была создана и испытана на противомикробное действие. Эти ди-пептиды имеют функции боковой цепи три-пептида из-за производного β2,2-аминокислоты. Был исследован широкий спектр липофильных заместителей производных β2,2-аминокислоты, как показано на ФИГ.1. Обзор синтеза соединения 4a-n показано на ФИГ.2.
Реагенты и аналитические способы
Спектры 1H и 13C ЯМР были зафиксированы с помощью спектрометров Varian на 400 или 600 MHz. Масс-спектры были получены на Micromass Quattro LC (Micromass, Манчестер, Великобритания). Масс-спектры с высоким разрешением были получены на Waters Micromass LCT Premier (Micromass, Манчестер, Великобритания). Доступные в продаже соединения и растворители были приобретены у Sigma-Aldrich и использовались без дальнейшей очистки. Препаративная RP-ВЭЖХ была проведена на системе Waters, оснащенной колонкой RP BondaPak C18 125 Ǻ, 10 мкм, 25×100 мм, и элюирована с помощью ацетонитрила и воды, содержащих по 0,1% ТФУК. Аналитическая ВЭЖХ была проведена на системе Waters 2695 HPLC, оснащенной водяной колонкой RP-ВЭЖХ Delta Pak С18.100 Ǻ, 5 мкм, 3.9×150 мм и проанализирована при длине волны 214 нм с помощью детектора PDA с диапазоном волны от 210 до 310 нм. Все соединения были подготовлены с помощью карусели параллельных реакций системе Radleys®.
Общие процедуры диалкилирования метилцианоацетата (GP1) 1a-n.
Натрия метилат (20 ммоль) разбавили в метаноле (0,2 М) и добавили метилцианоацетат (20 ммоль). После 5 минут взбалтывания при комнатной температуре добавили требуемый бромистый бензил (20 ммоль), а раствор нагрели до дефлегмирования. Через 15 минут раствор охладили до комнатной температуры. Добавили вторую порцию метилата натрия (20 ммоль) и через 5 минут смешивания при комнатной температуре добавили вторую порцию требуемого бромистого бензила (20 ммоль), после чего снова провели дефлегмирование в течение 15 минут. Объем реакционной смеси был уменьшен до приблизительно 1/3 в вакууме и экстрагирован с помощью воды/этилацетата. Органическую фазу высушили над MgSO4 и испарили до сухости. Продукт использовали в последующем синтезе без какой-либо дополнительной очистки.
Общая процедура восстановления нитрилов в амины с помощью Ra/Ni и последующая защита Boc (GP2) 2a-n.
Ra/Ni (приблизительно 2 мл/г нитрила) промыли трижды с помощью метанола под аргоном, после чего добавили требуемый нитрил (3,5 ммоль), растворенный в метаноле (0,1 М), вместе с уксусной кислотой (приблизительно 1 мл/г нитрила). Реакционную смесь гидрогенировали при температуре 45°C в течение 5 дней под давлением Н2 1 бар. После этого реакционную смесь профильтровали через фильтр Celite для удаления Ra/Ni до его испарения до сухости. Сырой метиловый эфир β2,2-аминокислоты (0,35 ммоль) разбавили в смеси 1,4-диоксана и воды 5:1 (-0,35 М), а pH отрегулировали до уровня 8 с помощью ТЭА, Вос2O (0,42 ммоль) растворили в минимально возможном количестве 1,4-диоксана. Раствор взболтали при комнатной температуре в течение приблизительно 18 часов до его окисления до pH 2-3 с помощью 10% лимонной кислоты и экстрагировали трижды с помощью этилацетата. Органическую фазу высушили над MgSO4 испарили до сухости. Продукт использовали в последующем синтезе без какой-либо дополнительной очистки.
Общая процедура эфиргидролиза (GP3) 3a-n.
Вос-защищенный метиловый эфир β2,2-аминокислоты (0,35 ммоль) разбавили в смеси 1,4-диоксана и воды 3:1 (1,17 мМ) и добавили гидроксид лития (2,1 ммоль), разбавленный в минимально возможном количестве воды. Реакционную смесь взболтали в обратном холодильнике под N2 в течение 18 часов до уменьшения объема приблизительно до 1/5 под вакуумом. Воду (10 мл) добавили в реакционную смесь, а pH скорректировали покапельным способом до 1-2 с помощью 0,1 М HCl. Этот водный раствор экстрагировали трижды с помощью равного объема этилацетата. Органическую фазу высушили над MgSO4 и испарили до сухости. Продукт использовали в последующем синтезе без какой-либо дополнительной очистки.
Общая процедура соединения L-аргинина Вос-защищенных β2,2-аминокислот (GP4) 4a-n.
Вос-защищенную β2,2-аминокислоту (0,2 ммоль) разбавили в ДМФ (0,02 М) и добавили ДИПЭА (0,6 ммоль) вместе с TFFH (0,2 ммоль). Аминокислоту предварительно активировали за 2 часа до добавления H-Arg-NH2×2HCl (0,3 ммоль). Реакционную смесь взболтали при комнатной температуре в течение 7 дней до того, как разбавили этилацетатом и промыли соляным раствором. Органическую фазу высушили над MgSO4 и испарили до сухости. С сырого Вос-защищенного продукта сняли защиту путем разбавления его в ДХМ (-0,4 М) и добавления объема, эквивалентного ТФУК:TIS:вода (95:2.5:2.5). Смесь взболтали при комнатной температуре в течение 2 часов до того, как испарили до сухости. Сырой продукт очистили способом препаративной RP-ВЭЖХ. Чистоту пептидов проверили способом аналитической RP-ВЭЖХ перед тем, как раствор испарили до сухости, а остаток повторно растворили в воде и лиофилизировали. Все соединения характеризовались чистотой выше 95%.
Испытания в камере
Противомикробные испытания были проведены TosLab A/S (Тромсо, Норвегия). Каждое соединение было разбавлено до 1 мг/мл в воде и испытаны в двойном экземпляре при 200, 100, 50, 35, 15, 10, 5, 2,5, 1, 0,5 мкг/мл, кроме 4n, которое испытали при 50, 35, 15, 10, 5, 2,5, 1, 0,5 мкг/мл из-за проблем с растворимостью. Все испытанные соединения были ди-ТФУК солями.
Бактериальные штаммы выращивались в 2% Бакто-пептоне до достижения экспоненциального роста. MIC определили путем ночной инкубации в 1% Бакто-пептоне при температуре 37°C. Использовали бактериальную концентрацию 2×106 кое/мл. Оба отрицательных контроля (без пептида) и положительный контроль (гентамицин) использовались для всех бактериальных штаммов. Рост или отсутствие роста определялись на основе мутности в лунках. МВС определяли путем посева на агаровых пластинах всех концентраций при значениях MIC и выше, инкубируя при температуре 37°C в течение ночи и определяя рост или отсутствие роста.
Все соединения, демонстрировавшие значения MIC менее 50 мкг/мл, были испытаны в помощью той же процедуры.
Значения MIC и МВС показаны в Таблицах 1А и В.
Гемолитические испытания на эритроцитах человека проводились в Lytix Biopharma A/S (Тромсо, Норвегия). Каждое соединение было испытано на основе концентрации 1 мг/мл и ниже, кроме 4k и 4n, которые были испытаны только при максимум 0,5 мг/мл из-за проблем с растворимостью. 8 мл крови было получено от взрослых здоровых доноров-мужчин. Кровь поделили на равные порции и распределили по доступной в продаже пробирке, содержащей EDTA (BD вакутейнер, 7,2 мг К2 EDTA) и в реакционной пробирке объемом 10 мл, содержащей 40 мкл гепаринового раствора (1000 единиц/мл в 0,9% хлорида натрия). Через 30 минут определили гематокрит крови, обработанной EDTA. Гепаринизированную кровь центрифугировали в течение 10 минут при 1500 об/мин и удалили надосадочную жидкость. Впоследствии, RBC промыли предварительно разогретым PBS трижды и разбавили до 10% гематокрита. Соединения разбавили в PBS (в концентрации от 1 мкг/мл до 1000 мкг/мл), а эритроциты инкубировали при постоянном взбалтывании при температуре 37°C в течение одного часа. Добавили положительный контроль с конечной концентрацией 0,1% Triton Х-100 и негативный контроль, содержащий чистый PBS-буфер. Образцы центрифугировали (4000 об/мин) в течение 5 минут, а абсорбцию надосадочной жидкости измерили при 405 нм. Значения, поданные в Таблице 1А, соответствуют 50% гемолизу.
Анализ соответствия на основе α-химотрипсина
Испытания стабильности на основе α-химотрипсин провели путем разбавления требуемого соединения до 1 мг/мл в воде. α-химотрипсин разбавили до 0,1 мг/мл в 1 мМ HCl, содержащей 2 мМ CaCl2. Ферментное переваривание провели в 100 мМ TRIS HCl, содержащей 10 мМ CaCl2. Конечная ферментная концентрация составляла 2 мкг/мл, а конечная пептидная концентрация составляла 100 мкг/мл. Общий объем составлял 0,5 мл.
15 мкл образцов получили через 0, 15, 30, 60, 120 и 240 минут в добавок к образцам, собранным через 24 и 48 часов. К образцам добавили внешний стандарт (атенолола гидрохлорид) и 100 мкл 10% уксусной кислоты для прекращения переваривания до того, как разбавить до 1 мл водой.
Для каждого испытания проводили негативный контроль без фермента для обеспечения того, чтобы деградация происходила вследствие действия ферментов, а не других факторов. Сукцинил-ала-ала-про-фе-пара нитроанилин использовали в качестве положительного контроля. Все испытания проводили в тройном экземпляре.
Результаты
Подборка из шести соединений (4c, 4g, 4h, 4i, 4k и 4m) была исследована на протеолитическую стабильность на α-химотрипсин. Результаты продемонстрировали, что деградацию нельзя было установить ни для какого соединения в течение 48-часового периода. Также мы установили, что все испытуемые соединения были химически стабильными в водных растворах при pH 7,4, по меньшей мере, в 48 часов.
Результаты продемонстрировали сильную корреляцию между противомикробной мощностью и общей липофильностью подготовленных соединений. Это можно проиллюстрировать путем сравнения мощности соединений против S. aureus и время их удерживания (Rt) в аналитической колонке С18 для RP-ВЭЖХ, что продемонстрировало сродство соединения с гидрофобной стационарной фазой колонки (результаты не показаны).
Гемолитическое действие подготовленных соединений использовалось для измерения токсичности, и за исключением соединений 4g, 4j и 4n, соединения были негемолитическими в испытанном диапазоне концентраций (<1000 мкг/мл). Самое мощное гемолитическое действие было продемонстрировано соединением 4n, однако несмотря на это, эта концентрация EC50 была в 10-50 раз выше, чем значения MIC против грамположительных и грамотрицательных бактерий. Следует отметить, что соединения 4j и 4k, которые отражали силу противомикробного действия, сильно отличались относительно гемолитического действия. Это свидетельствовало о том, что гемолитическое действие и противомикробное действие не были определены на основе одинаковых структурных свойств.
Пример 2
Дополнительные препараты изобретения были изготовлены и испытаны на антибактериальное и противораковое действие.
Материалы и способы
Бактериальные штаммы
Staphylococcus aureus (ATTC 25923)
Метициллин-резистентный Staphylococcus aureus (ATCC 33591)
Метициллин-резистентный Staphylococcus epidermidis (ATCC 27626)
Escherichia coli (ATTC 25922)
Раковые клеточные линии
А20 или MethA
Клетки для исследований токсичности
MRC-5 и RBC человека
Самые высокие испытанные концентрации в различных образцах были:
Противомикробные образцы: 200 мкг/мл
Противораковые образцы: 500 мкг/мл
Образцы MRC-5: 500 мкг/мл
Образцы RBC: 1000 мкг/мл
Chem Draw Ultra, версия 11.0 использовали для подсчета физико-химических свойств Log P, tPSA и CLogP.
Синтез молекул
Набухание смолы: амидная смола Ринка МВНА (загрузка 0,64 ммоль/г) была помещена в лунки в 7 мл ДМФ на 1 час до того, как была промыта пять раз с помощью 7 мл ДМФ.
Извлечение Fmoc: 7 мл 20% пиперидина в ДМФ добавили в реакционные пробирки, а суспензию взболтали в течение 10 минут до извлечения раствора. Эту процедуру повторили дважды, взбалтывая в течение 1 минут до того, как смолу промыли пять раз с помощью 7 мл ДМФ.
Соединение аминокислот с незащищенной смолой: Fmoc-Lys(Boc)-OH или Fmoc-Trp(Вос)-ОН (4 экв.), гидрат HOBt (4 экв.) и HBTU (3,92 экв.) растворили в 5 мл ДМФ, добавили ДИПЭА (8 экв.), а смесь оставили для предварительной активации в течение 15 минут до того, как добавить ее в смолу. После взбалтывания в течение 1 часа соединенную смесь извлекли, а смолу промыли пять раз с помощью 7 мл ДМФ.
Соединение β-аминокислоты с незащищенной смолой: Fmoc-β-aa-OH (2 экв.) и TFFH (1,96 экв.) растворили в 5 мл ДМФ, добавили ДИПЭА (8 экв.), а смесь оставили для предварительной активации на 15 минут перед тем, как добавить ее в смолу. После взбалтывания в течение 48 часов соединенную смесь извлекли, а смолу промыли пять раз с помощью 7 мл ДМФ.
Соединение аминокислоты после соединения β-аминокислоты в незащищенной смолой: Fmoc-Trp(Boc)-OH (4 экв.) и TFFH (3.92 экв.) разбавили в 5 мл ДМФ, добавили ДИПЭА (8 экв.), а смесь оставили для предварительной активации в течение 15 минут перед тем, как добавить ее в смолу. После взбалтывания в течение 24 часов соединенную смесь извлекли, а смолу промыли пять раз с помощью 7 мл ДМФ.
После последнего соединения смолу промыли пять раз с помощью ДХМ перед тем, как оставить ее для высыхания на ночь.
Отщепления от смолы и извлечения Boc: 10 мл ТФУК:TIS:вода 95:2.5:2,5 добавили в реакционные, а суспензию взболтали в течение 2 часов перед тем, как собрали пептидный раствор. Данную процедуру повторили дважды при взбалтывании в течение 10 минут. Собранные пептидные растворы испарили до сухости под пониженным давлением, осадили диэтиловым эфиром и промыли диэтиловым эфиром. Пептиды очистили ВЭЖХ и лиофилизировали.
Испытание в камере
Соответствующие способы описаны в Примере 1.
Результаты
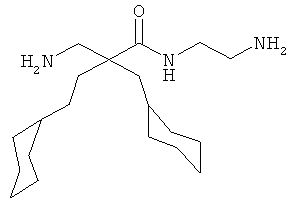
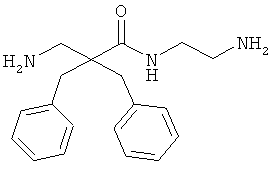
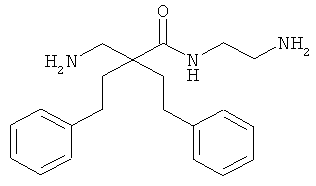
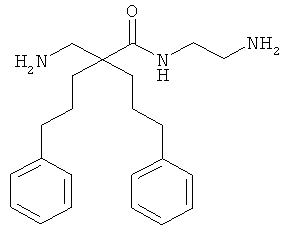
Точная масса: 311,1998 Молекулярный вес: 311,4213
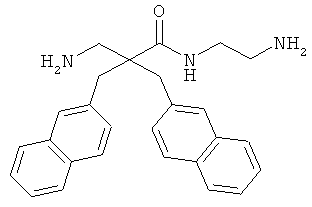
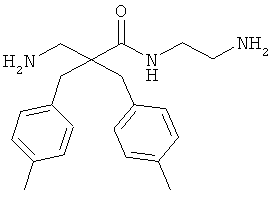
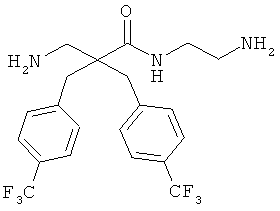
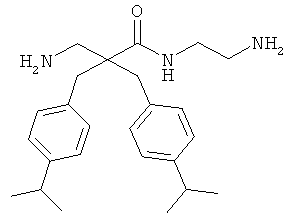
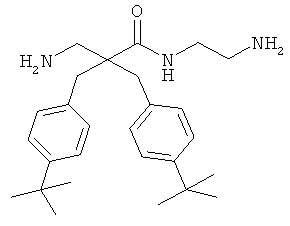
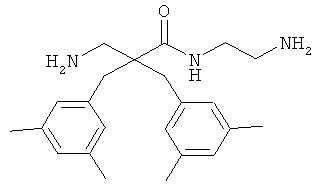
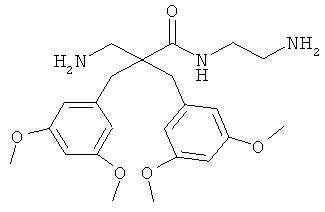
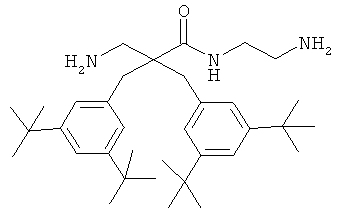
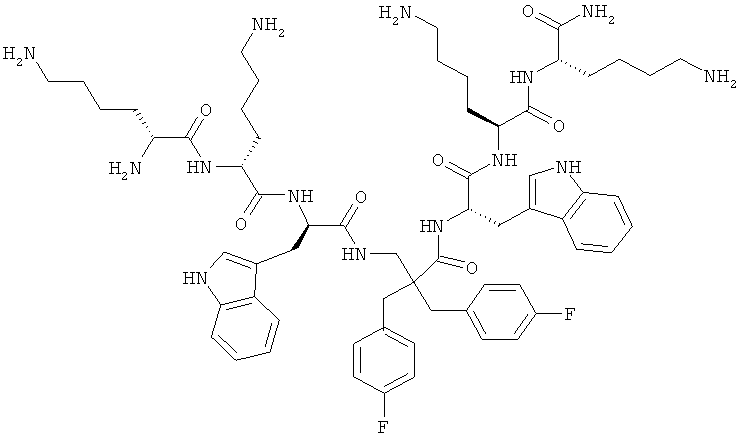
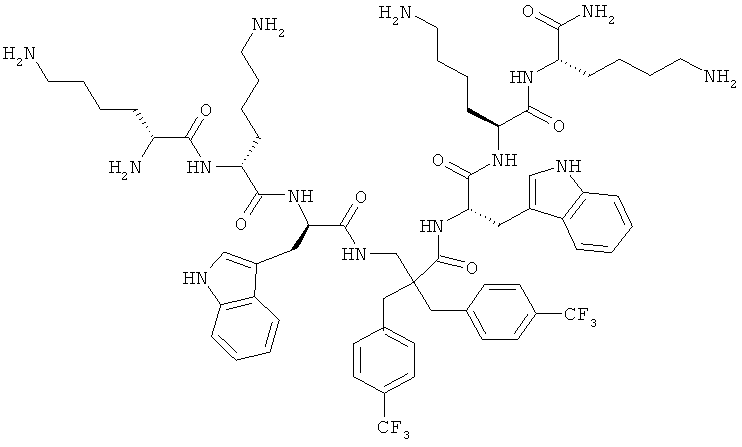
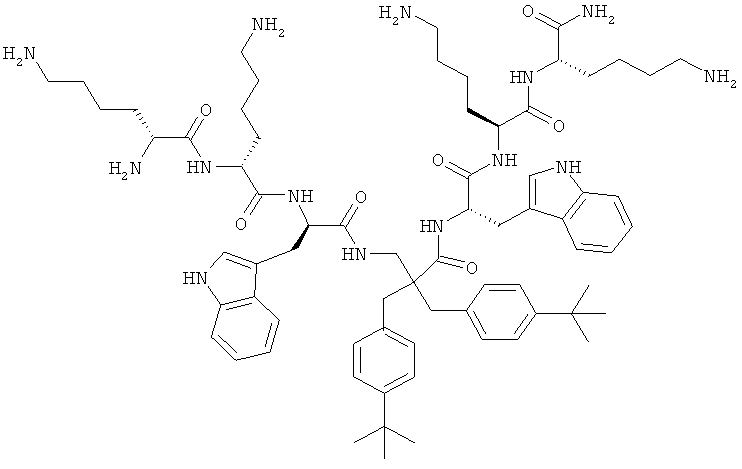
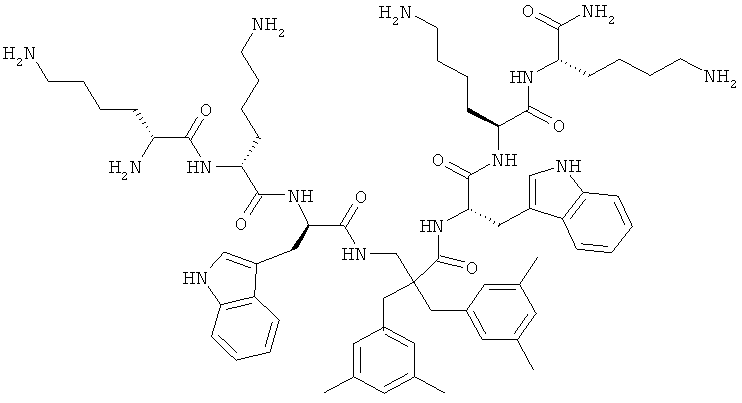
селективность
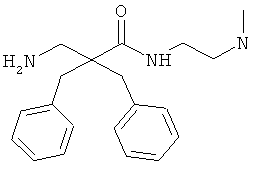
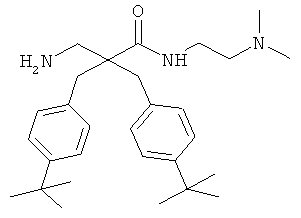
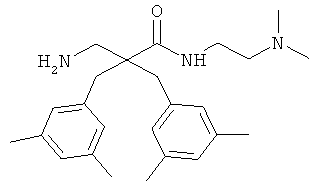
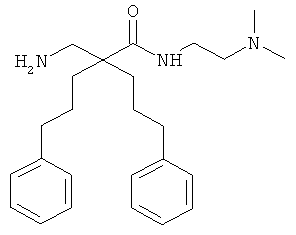
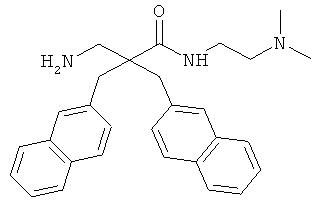
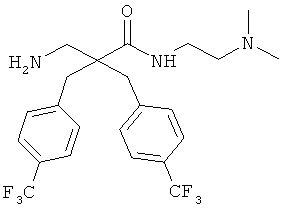
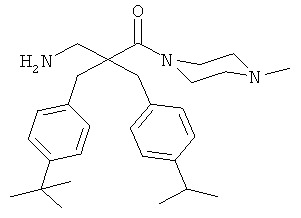
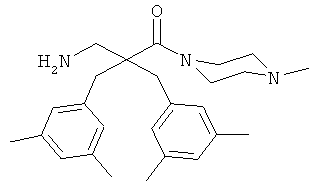
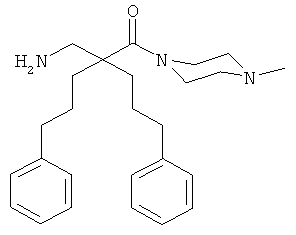
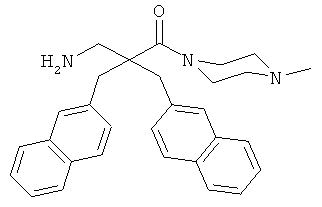
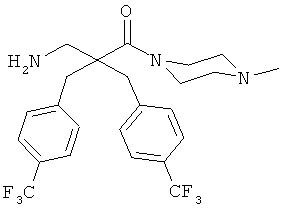
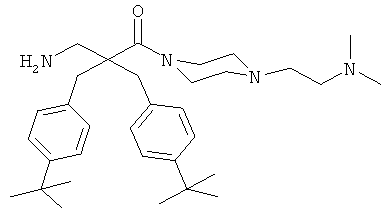
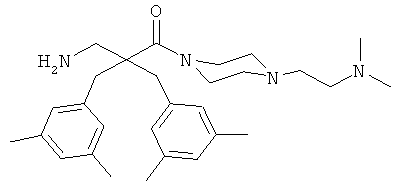
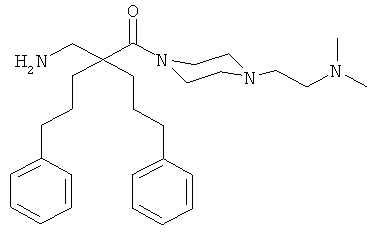
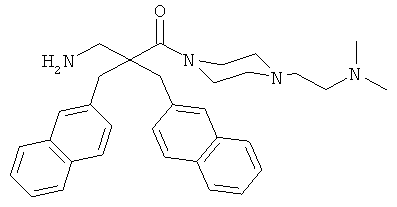
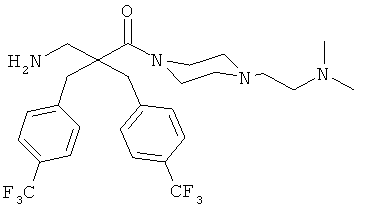
Пример 3
Соединение 7, описанное в Примере 2, проанализировали на двух нормальных клеточных линиях и девяти раковых клеточных линиях.
Все клеточные линии были человеческого происхождения. Базовый раствор соединения 7 растворили в среде образца, содержащей 10% ДМСО. Провели три параллельных эксперимента на каждой клеточной линии.
Предыдущие результаты для соединения 7 на раковых клетках А20 продемонстрировали IC50 0,7 мкг/мл, а на RBC ЕС50 292 мкг/мл (Таблица 2). Предыдущие испытания соединения 7 на клетках MRC-5 продемонстрировали IC50 13,5 и 14 мкг/мл, а скрининг-диагностика продемонстрировала IC50 8,21 мкг/мл.
Пример 4
Производные β2,2- аминокислоты изобретения были изготовлены и проанализированы на противомикробное действие и исследованы на их соответствие для перорального приема.
Синтез
Производные β2,2-аминокислоты (ФИГ.3) были синтезированы в соответствии со стратегиями, показанными на ФИГ.4. Чтобы ознакомиться с широким спектром способов подготовки β-аминокислот, см. Abele, S. et al. Eur. J. Org. Chem. [2000] 2000, 1-15. Вос-защищенные β2,2-аминокислоты 3а-d соединили с четырьмя различными C-терминальными катионными группами (ФИГ.4). Для улучшения результатов, 1.5 экв. соединяющего реагента TFFH использовали на окончательном этапе соединения, а β2,2-аминокислоты также были предварительно активированы в течение 2 часов с помощью TFFH вместо стандартных 10 минут. После реакций соединения провели масс-спектрометрический анализ и прекратили путем добавления водного Na2CO3. Однако ввиду стерических препятствий время соединения необходимо было увеличить до 7 дней для присоединения C-терминальных катионных групп.
Общая процедура синтеза 1a-d.
Синтез был основан на предыдущем синтезе, проведенным ранее Cronin et al. Anal. Biochem. [1982] 124, 139-149. Вкратце, метилат натрия (20 ммоль) растворили в метаноле для достижения приблизительной концентрации 0.2 М до того, как добавили метил цианоацетат (20 ммоль), а реакционную смесь взболтали в течение 5 минут при комнатной температуре. Добавили требуемый бромистый бензил (20 ммоль), а раствор нагрели до дефлегмирования в течение 15 минут, после чего раствор охладили до комнатной температуры и добавили вторую порцию метилата натрия (20 ммоль). После взбалтывания в течение 5 минут при комнатной температуре добавили еще одну порцию требуемого бромистого бензила (20 ммоль), после чего снова провели дефлегмирование в течение 15 минут. Приблизительно 2/3 метанола была извлечено in vacuo, после чего реакционную смесь разбавили этилацетатом и промыли водой. Органическую фазу высушили над MgSO4, профильтровали и испарили до сухости. Продукт 1a-d использовали в последующем синтезе без какой-либо дальнейшей очистки.
Общая процедура синтеза 2a-d.
Синтез был основан на предыдущем синтезе, проведенном Cronin et al. (см. выше) и Bodanszky et al. "The practice of peptide synthesis" Springe-Verlag 1994. Вкратце, Ra/Ni (как правило, 2 мл) промывали метанолом с целью удаления воды, после чего добавили требуемое производное метилцианоацетата (1a-d) (3,5 ммоль) вместе с уксусной кислотой (как правило, 1 мл) и метанолом для получения окончательной концентрации 1a-d, составляющей приблизительно 0,1 М. Реакционную смесь гидратировали при температуре 45°C с помощью обратного конденсатора, установленного на 5 дней при давлении Н2 1 бар, после чего Ra/Ni профильтровали, а реакционную смесь испарили до сухости. Сырой метиловый эфир β2,2-аминокислоты растворили в смеси 1,4-диоксан и воды 5:1 (0,35 М), а pH скорректировали до 8 с помощью ТЭА, после чего добавили Boc2O (1,5 экв.), растворенный в минимальном количестве 1,4-диоксана, раствор взболтали при комнатной температуре в течение 18 часов. Реакционную смесь окислили до pH 4-5 при помощи 0,1 М HCl и экстрагировали трижды с помощью этилацетата, после чего органическую фазу высушили над MgSO4, профильтровали и испарили до сухости. Сырой продукт 2a-d использовали в последующем синтезе без какой-либо дальнейшей очистки.
Общая процедура синтеза 3а-d.
Синтез был основан на публикации Seebach et al. Helv. Chim. Acta [1998] 81, 2218-2243. Вкратце, сырой Вос-защищенный метиловый эфир β2,2-аминокислоты 2a-d (0,35 ммоль) растворили в смеси 1,4-диоксана и воды 3:1 для достижения окончательной концентрации приблизительно 1,2 мМ, после чего гидроксид лития (2,1 ммоль) разбавили в минимальном количестве воды. Реакционную смесь нагрели для дефлегментирования и оставили на 18 часов, после чего объем уменьшился приблизительно на 1/5 in vacuo, a pH скорректировали до 1-2 путем медленного добавления 0,1 М HCl. Водный раствор экстрагировали трижды с помощью этилацетата, после чего органическую фазу высушили над MgSO4, профильтровали и испарили до сухости. Сырой продукт 3а-d использовали в последующем синтезе без какой-либо дальнейшей очистки.
Общая процедура синтеза 4a-d, 5a-d, 6a-d и 7a-d
Синтез был основан на пособии Chan and White, "Fmoc solid phase peptide synthesis: a practical approach", Oxford University Press 2000; p346. Вкратце, Boc-защищенную β2,2-аминокислоту (3а-d) (как правило, 0,2 ммоль) разбавляют в ДМФ (0,02 М), после чего добавляют ДИПЭА (3 экв.) вместе с TFFH (1 экв.). β2,2-аминокислоты активируют в течение 2 часов до добавления требуемого амина (2 экв.). После реакций добавляют MS, и протекает реакция в течение максимум 7 дней, после этого разбавляют этилацетатом и промывают соляным раствором. Органическую фазу высушили над MgSO4, профильтровали и испарили до сухости. Сняли защиту с производных Boc-защищенной β2,2-аминокислоты путем их растворения в ДХМ, добавления эквивалентного объема ТФУК:TIS:воды (95:2.5:2.5) и взболтали при комнатной температуре в течение 2 часов, после чего испарили до сухости. Сырые продукты очистили способом препаративной RP-ВЭЖХ и лиофилизировали. Чистоту соединения проверили способом аналитической ВЭЖХ с детекторов PDA в диапазоне от 210 нм до 310 нм. Все соединения обладали чистотой выше 95%.
Противомикробное действие
Каждое соединение испытали в двойном экземпляре при 200, 100, 50, 35, 15, 10, 5, 2,5, 1, 0,5 мкг/мл. Все испытанные соединения были солями ди-трифтороуксусной кислоты.
Гемолитическое действие
Фракции плазмы гепаринизированной крови человека сперва извлекли способом центрифугирования и трех дополнительных этапов промывания при температуре 37°C в предварительно нагретом фосфатно-буферном соляном растворе (PBS). После этого RBC человека разбавили до 10% гематокрита, а производные β2,2-аминокислоты растворили в PBS, обеспечивая концентрации в диапазоне от 1-1000 мкг/мл. Разбавленную RBC добавили в соединительные растворы для достижения окончательной концентрации эритроцитов 1% о/о. PBS и Triton Х-100, в окончательной концентрации 0,1% о/о, использовались как отрицательный и положительный контроль. Через 1 час после инкубации при постоянном взбалтывании при температуре 37°C образцы центрифугировали при 4000 об/мин в течение 5 минут. Высвобождение гемоглобина определялось измерением абсорбции надосадочной жидкости при 405 нм. Гемолитическое действие было подсчитано как коэффициент обработанного образца с производным β2,2-аминокислоты и образца, обработанного тенсидом в соответствии с формулой

Подобие лекарственным препаратам и пероральная абсорбция
Используя аппликатор Шредингера QikProp, который поставляется с программным обеспечением Шредингера Maestro v9.1, оценка подобия лекарственным препаратам производных β2,2-аминокислоты относительно «правила пяти» Упински была проведена вместе с оценкой процентной пероральной абсорбции у людей. Правило гласит, что активный препарат для перорального приема не должен нарушать больше чем один из таких четырех критериев; 1) логарифм P коэффициента разделения октанол-вода должен быть меньше 5, 2) молекулярная масса (молекулярный вес) не должна превышать 500 дальтон, 3) допускается максимум 5 групп водородных донорных связей (HBD), и 4) должно быть не более 10 групп водородных донорно-акцепторных связей (НВА). Результаты поданы в Таблице 10 ниже.
Результаты подсчетов демонстрируют, что все производные β2,2-аминокислоты соответствуют «Правилу пяти» Упински, поскольку ни одно соединение не нарушило более одного из четырех правил. По одному правилу нарушили 5 из 16 подготовленных производных β2,2-аминокислоты, три нарушения из которых возникли в результате того, что молекулярная масса превышала 500 (4b, 4с и 4d), а два других нарушения возникли в результате того, что значение логарифма Р превышало допустимое значение - 5 (5d и 6d).
Подсчет процента пероральной абсорбции у людей с помощью программного обеспечения продемонстрировал, что производные β2,2-аминокислоты могут достаточно хорошо абсорбироваться, причем подсчитанная пероральная абсорбция составляла от 62% до 89%. Самый большой процент пероральной абсорбции был подсчитан для производных β2,2-аминокислоты 5а, 5b, 5с, 6b и 6с, все из которых были в диапазоне 86%-89% пероральной абсорбции.
Проницаемость
На основе теоретических подсчетов, проницаемость производных β2,2-аминокислоты была также исследована с помощью недавно установленной барьерной модели на основе фосфолипидных пузырьков (Флатен и соавт., Eur. J. Pharm. Sci. [2006] 27, 80-90). Были исследованы четыре производных β2,2-аминокислоты (4с, 5с, 6с и 7с), которые демонстрировали подобную активность против MRSA и против Е. coli. На основе классификации абсорбции модели, все четыре соединения демонстрировали проницаемость, эквивалентную умеренной абсорбции у людей. С точки зрения экспериментальных значений проницаемости, соединение 5 с демонстрировало самую высокую концентрацию, после него - соединения 6с, 4с и 7с.
| название | год | авторы | номер документа |
|---|---|---|---|
| ТЕРАПЕВТИЧЕСКИЕ ПЕПТИДЫ | 2010 |
|
RU2676713C2 |
| НОНАПЕПТИД С ПРОТИВООПУХОЛЕВОЙ АКТИВНОСТЬЮ | 2009 |
|
RU2503685C2 |
| НОВЫЕ СИНТЕТИЧЕСКИЕ ПЕПТИДЫ И ИХ ПРИМЕНЕНИЕ | 2012 |
|
RU2596393C2 |
| АНТИБИОТИЧЕСКИЕ ПЕПТИДЫ | 2008 |
|
RU2472805C2 |
| ПЕПТИДЫ, ВЫДЕЛЕННЫЕ ИЗ ЧЕЛОВЕЧЕСКОГО ЛАКТОФЕРРИНА, И ИХ ПРИМЕНЕНИЕ | 2012 |
|
RU2593757C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПЕПТИДОВ, ИЛИ БЕЛКОВ, ИЛИ ПЕПТИДОМИМЕТИКОВ | 2020 |
|
RU2828026C2 |
| ЦИКЛИЧЕСКИЕ ПРОТИВОМИКРОБНЫЕ ПЕПТИДЫ | 2006 |
|
RU2463308C2 |
| АНАЛОГИ ГЛЮКАГОНА, ОБЛАДАЮЩИЕ ПОВЫШЕННОЙ РАСТВОРИМОСТЬЮ И СТАБИЛЬНОСТЬЮ В БУФЕРАХ С ФИЗИОЛОГИЧЕСКИМИ ЗНАЧЕНИЯМИ Ph | 2009 |
|
RU2560254C2 |
| ПЕПТИДЫ ЦИТОПЛАЗМАТИЧЕСКОГО ДОМЕНА БЕЛКА MUC-1 В КАЧЕСТВЕ ИНГИБИТОРОВ РАКОВЫХ ЗАБОЛЕВАНИЙ | 2009 |
|
RU2539832C2 |
| ПРОИЗВОДНАЯ ИМИДАЗОПИРИДИНА И ФАРМАЦЕВТИЧЕСКИЙ ПРЕПАРАТ, СОДЕРЖАЩИЙ ЕЕ В КАЧЕСТВЕ АКТИВНОГО ИНГРЕДИЕНТА | 2020 |
|
RU2810973C2 |
Настоящее изобретение относится к пептиду, пептидомиметику или производному аминокислоты, которые содержат двузамещенную β-аминокислоту, в которой каждая из замещающих групп в β-аминокислоте, которые могут быть одинаковыми или разными, включает по меньшей мере 7 неводородных атомов, является липофильной и включает по меньшей мере одну циклическую группу, причем одна или больше циклических групп в замещающей группе могут быть связаны или конденсированы с одной или большим числом циклических групп в другой замещающей группе, и когда циклические группы слиты таким образом, совокупное общее количество неводородных атомов для таких двух замещающих групп составляет по меньшей мере 12, где указанный пептид, пептидомиметик или производное аминокислоты состоят из 1-4 аминокислот или эквивалентных по длине субъединиц. 3 н. и 14 з.п. ф-лы, 4 ил., 10 табл., 4 пр.
1. Способ лечения микробной инфекции, включающий введение пациенту пептида, пептидомиметика или производного аминокислоты, имеющего чистый положительный заряд по меньшей мере +2 при рН 7.0 и содержащего двузамещенную β-аминокислоту, причем каждая из замещающих групп в β-аминокислоте, которые могут быть одинаковыми или разными, включает по меньшей мере 7 неводородных атомов, является липофильной и включает по меньшей мере одну циклическую группу, причем одна или больше циклических групп в замещающей группе могут быть связаны или конденсированы с одной или большим числом циклических групп в другой замещающей группе, и когда циклические группы слиты таким образом, совокупное общее количество неводородных атомов для таких двух замещающих групп составляет по меньшей мере 12, где указанный пептид, пептидомиметик или производное аминокислоты состоят из 1-4 аминокислот или эквивалентных по длине субъединиц, а введение осуществляют в дозе от 1 до 10 мг/кг.
2. Способ по п.1, при котором две замещающие группы одинаковые.
3. Способ по п.1, при котором β-аминокислота - это β2,2 или β3,3 двузамещенная аминокислота.
4. Способ по п.1, при котором каждая из липофильных замещающих групп включает возможно замещенную фенильную или циклогексильную группу.
5. Способ по п.4, при котором указанные фенильные или циклогексильные группы отделены от α или β атома углерода β-аминокислоты с помощью 1-4 связующих атомов.
6. Способ по п.1, при котором каждая липофильная замещающая группа содержит 8-12 неводородных атомов.
7. Способ по п.1, при котором С-конец пептида, пептидомиметика или производного аминокислоты амидируется и возможно замещается.
8. Способ по п.1, при котором пептид или пептидомиметик включает катионную аминокислоту, предпочтительно аргинин или лизин.
9. Пептид, пептидомиметик или производное аминокислоты, имеющее чистый положительный заряд по меньшей мере +2 при рН 7.0, которое включает группу формулы I
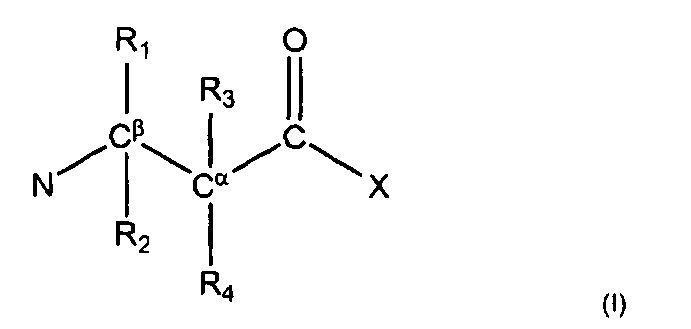
где любые 2 из R1, R2, R3 и R4 - это атомы водорода, а 2 - замещающие группы, которые могут быть одинаковыми или разными,
включает по меньшей мере 7 неводородных атомов, является липофильным и включает циклическую группу, причем указанная циклическая группа не присоединена непосредственно к α или β атому углерода, а возможно связана или конденсирована с циклической группой в другой замещающей группе, и когда циклические группы конденсированы, совокупное общее количество неводородных атомов для двух замещающих групп составляет по меньшей мере 12, и
где X обозначает О, С, N или S, где указанный пептид, пептидомиметик или производное аминокислоты состоят из 1-4 аминокислот или эквивалентных по длине субъединиц.
10. Пептид, пептидомиметик или производное аминокислоты по п.9, при котором две замещающие группы одинаковые.
11. Пептид, пептидомиметик или производное аминокислоты по п.9, при котором β-аминокислота - это β2,2 или β3,3 двузамещенная аминокислота.
12. Пептид, пептидомиметик или производное аминокислоты по п.9, при котором липофильная замещающая группа включает возможно замещенную фениловую или циклогексильную группу.
13. Пептид, пептидомиметик или производное аминокислоты по п.12, при котором указанные фениловые или циклогексильные группы отделены от α или β атома углерода β-аминокислоты с помощью 1-4 связующих атомов.
14. Пептид, пептидомиметик или производное аминокислоты по п.9, при котором каждая липофильная замещающая группа содержит 8-12 неводородных атомов.
15. Пептид, пептидомиметик или производное аминокислоты по п.9, при котором С-конец производного вещества пептида, пептидомиметика или аминокислоты амидируется и возможно замещается.
16. Пептид, пептидомиметик или производное аминокислоты по п.9, при котором пептид или пептидомиметик включает катионную аминокислоту, предпочтительно аргинин или лизин.
17. Фармацевтическая композиция, обладающая противомикробной активностью, включающая эффективное количество молекулы по любому из пп.9-16, и разбавитель, носитель или вспомогательное вещество.
| DE 102006018080 A1, 18.10.2007 | |||
| ЭЛЕКТРОВОДОНАГРЕВАТЕЛЬ | 1996 |
|
RU2105433C1 |
| WO 9212168 A1, 23.07.1992 | |||
| АНТИБАКТЕРИАЛЬНЫЕ ПЕПТИДЫ | 2007 |
|
RU2468033C2 |
| STROEM M B ET AL: "The pharmacophore of short cationic antibacterial peptides", JOURNAL OF MEDICINAL CHEMISTRY, AMERICAN CHEMICAL SOCIETY, 2003, vol | |||
| Способ изготовления звездочек для французской бороны-катка | 1922 |
|
SU46A1 |
| Разборный с внутренней печью кипятильник | 1922 |
|
SU9A1 |
| HANSEN TERKEL ET AL: "Antimicrobial activity of small | |||

